Настоящая заявка относится к новым соединениям и их применению в качестве антагонистов Н4-гистаминового рецептора. Соединения, описанные в настоящем документе, могут быть применимы при лечении или предупреждении заболеваний, опосредованных Н4-рецепторами. Заявка также относится к фармацевтическим композициям, содержащим эти соединения, а также к производству и применению этих соединений и композиций при предупреждении или лечении заболеваний, опосредованных Н4-рецепторами.
УРОВЕНЬ ТЕХНИКИ
Гистамин представляет собой биогенный амин короткого действия, вырабатываемый в тучных клетках, где он накапливается в цитозольных гранулах и высвобождается в ответ на различные иммунологические и неиммунологические стимулы. Высвобождение гистамина из тучных клеток традиционно связывают с признаками и симптомами от легкой до тяжелой степени, которые характеризуют реакции гиперчувствительности, включая эритему, крапивницу, зуд, тахикардию, гипотензию, фибрилляцию желудочков, бронхоспазм, а также остановку сердца и дыхания. На сегодняшний день идентифицировано множество дополнительных источников, включая базофилы, нейроны и раковые клетки. Помимо модуляции широкого спектра физиологических процессов, гистамин связан с патологическими состояниями, включая аллергию и анафилаксию, астму и хроническое воспаление, аутоиммунные, сердечнососудистые, нейропсихиатрические и эндокринные расстройства, а также рак.
Гистамин проявляет свое плейотропное действие в основном за счет связывания с четырьмя типами рецепторов, связанных с G-белками (GPCR), называемых Н1-Н4, которые дифференциально экспрессируются в различных типах клеток и демонстрируют значительные видовые различия. Н2-рецептор отвечает за секрецию желудочного сока; Н3-рецептор контролирует высвобождение гистамина и других нейромодуляторов в ЦНС, а H1-рецептор связан с нарушением сна и воспалительной реакцией.
Обнаруженный в 2000 году высокоаффинный Н4-рецептор проявляет конститутивную активность и экспрессируется в основном, но не исключительно, на клетках иммунной системы, включая тучные клетки, моноциты, дендритные клетки, эозинофилы, базофилы, нейтрофилы и Т-клетки. Это открытие привело к появлению привлекательных перспектив создания новой лекарственной мишени с терапевтическим потенциалом при остром и хроническом воспалении, аутоиммунном заболевании, иммунной защите организма и невропатической боли.
H4R имеет только 40% гомологии со своим ближайшим соседом H3R, и ни для антагонистов Н2, ни для антагонистов H1 не была показана возможность ингибирования индуцированного гистамином хемотаксиса эозинофилов. Было показано, что гистамин ингибирует индуцированные форсколином ответы цАМФ чувствительным к коклюшному токсину (РТх) образом, что позволяет предположить, что H4R осуществляет сингализацию через гетеротримерные белки Gαi/o. Временная экспрессия H4R в гетерологичных клеточных системах (например, клетках HEK293) является широко используемым методом измерения сигнализации и связывания лиганда Н4 для получения оценок функциональной активности и аффинности к рецептору, соответственно.
Открытие антагонистов H4R с использованием этих методов и их изучение на различных моделях заболеваний на животных, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит, подтвердило, что антагонизм H4R приводит к значительному противовоспалительному эффекту, и обосновало терапевтическую пользу при нацеливании на этот рецептор. Уже проведено первое клиническое исследование фазы 2а антагониста H4R у пациентов, страдающих атопическим дерматитом от средней до тяжелой степени тяжести, которое еще раз подтвердило, что Н4 является поддающийся воздействию лекарственных средств мишенью у пациентов.
Несмотря на ряд известных лигандов H4R, остается потребность в разработке новых антагонистов H4R, которые являются хорошими лекарственными средствами-кандидатами. Эти антагонисты должны демонстрировать превосходную активность в низком наномолярном диапазоне и аффинность при полной селективности к рецепторам Н1-Н3. Они не должны проявлять агонистической активности из-за рисков, связанных с индукцией провоспалительных реакций, и в идеале должны иметь схожий фармакологический профиль у разных видов, чтобы подтверждать ФК (фармакокинетику)/ФД (фармакодинамику) в различных животных моделях заболеваний. Они должны быть метаболически стабильными, иметь отличную ФК, быть нетоксичными и демонстрировать отличную специфичность в отношении Н4 при исследовании безопасности на широкой скрининговой панели.
Человеческий ген специфических калиевых каналов сердца (hERG) кодирует порообразующую субъединицу быстро активируемого калиевого канала задержанного выпрямления (IKr), который играет важную роль в реполяризации желудочков и в определении интервала QT электрокардиограммы, где интервал QT представляет собой время между деполяризацией и реполяризацией желудочков. Общепризнано, что hERG очень чувствителен к ингибированию широким спектром структурно разнообразных соединений. Когда способность каналов проводить электрический ток через клеточную мембрану подавляется или нарушается при применении лекарственных средств, это может привести к потенциально смертельному заболеванию, называемому синдромом QT. Ряд клинически успешных лекарственных средств на рынке имели тенденцию ингибировать hERG и создавали сопутствующий риск внезапной смерти в качестве побочного действия, что сделало ингибирование hERG важной анти-мишенью, которой следует избегать при разработке лекарственных средств.
Соединения согласно изобретению являются антагонистами Н4-рецептора. Некоторые соединения характеризуются низким ингибированием hERG, что делает их особенно полезными.
ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, обладающим активностью антагонистов Н4-рецептора. Более конкретно, изобретение относится к соединениям, которые сочетают антагонизм Н4-рецептора с низкой активностью в отношении hERG.
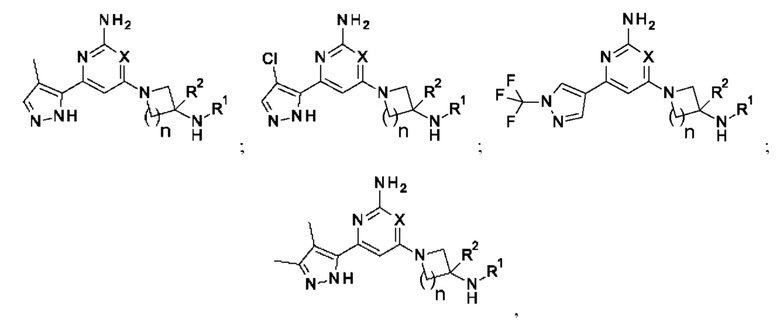
Соответственно, в одном из вариантов осуществления изобретение относится к соединению формулы (1)
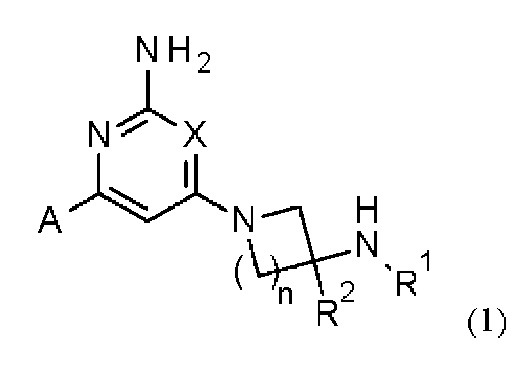
или его соли, где:
X представляет собой СН или N;
n равно 1 или 2;
R1 выбран из Н или С1-3 алкила, где С1-3 алкильная группа может быть циклизована с кольцом, к которому присоединен NHR1, с образованием второго кольца;
R2 представляет собой H или метил; и
А представляет собой необязательно замещенное пиразольное кольцо.
Кольцо А может представлять собой необязательно замещенное пиразольное кольцо, которое связано с кольцом, содержащим X, углерод-углеродной связью.
Конкретные соединения включают соединение формулы (1а):
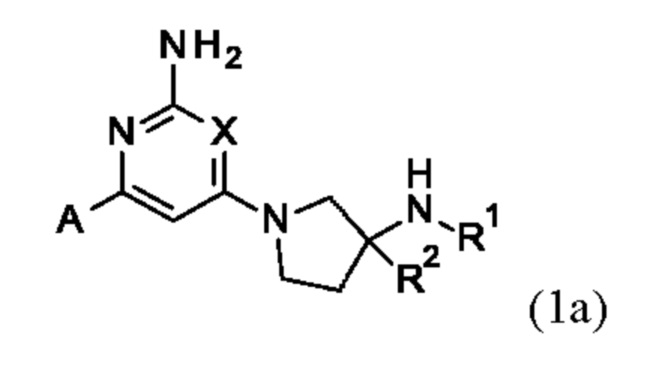
или его соли, где А, X, R1 и R2 являются такими, как определено выше.
Конкретные соединения также включают соединения формулы (2а), (2b) и (2с):
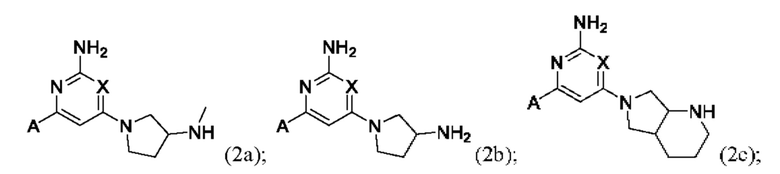
или их соль, где А и X и являются такими, как определено выше.
Конкретные изомеры включают соединения формулы (3а), (3b) и (3с):
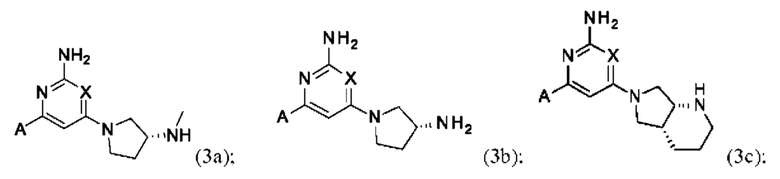
или их соль, где А и X и являются такими, как определено выше.
Конкретные соединения включают соединение формулы (1b):
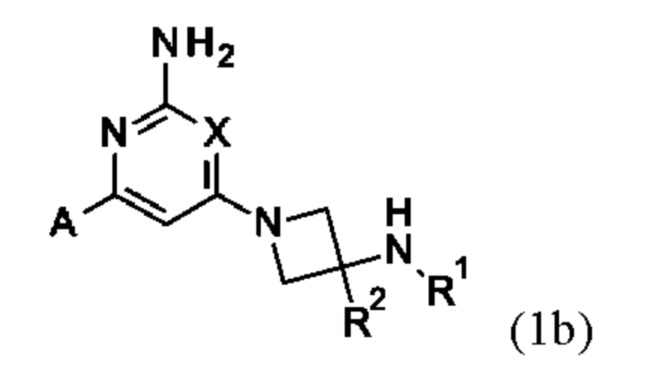
или его соль, где А, X, R1 и R2 являются такими, как определено выше.
Конкретные соединения также включают соединения формулы (2d) и (2е):
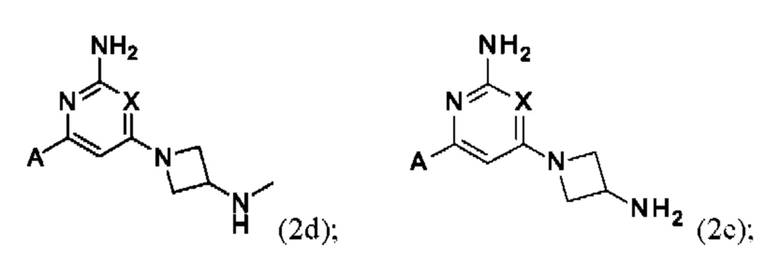
или их соль, где А и X и являются такими, как определено выше.
В приведенных в настоящем документе соединениях R1 может представлять собой Н или С1-3 алкил.
В приведенных в настоящем документе соединениях R1 может представлять собой метил, этил, пропил, изопропил или циклопропил.
В приведенных в настоящем документе соединениях R1 может представлять собой C1-3 алкил, где С1-3 алкильная группа циклизована с кольцом, к которому присоединен NHR1, с образованием второго кольца.
В приведенных в настоящем документе соединениях R2 может представлять собой Н или метил.
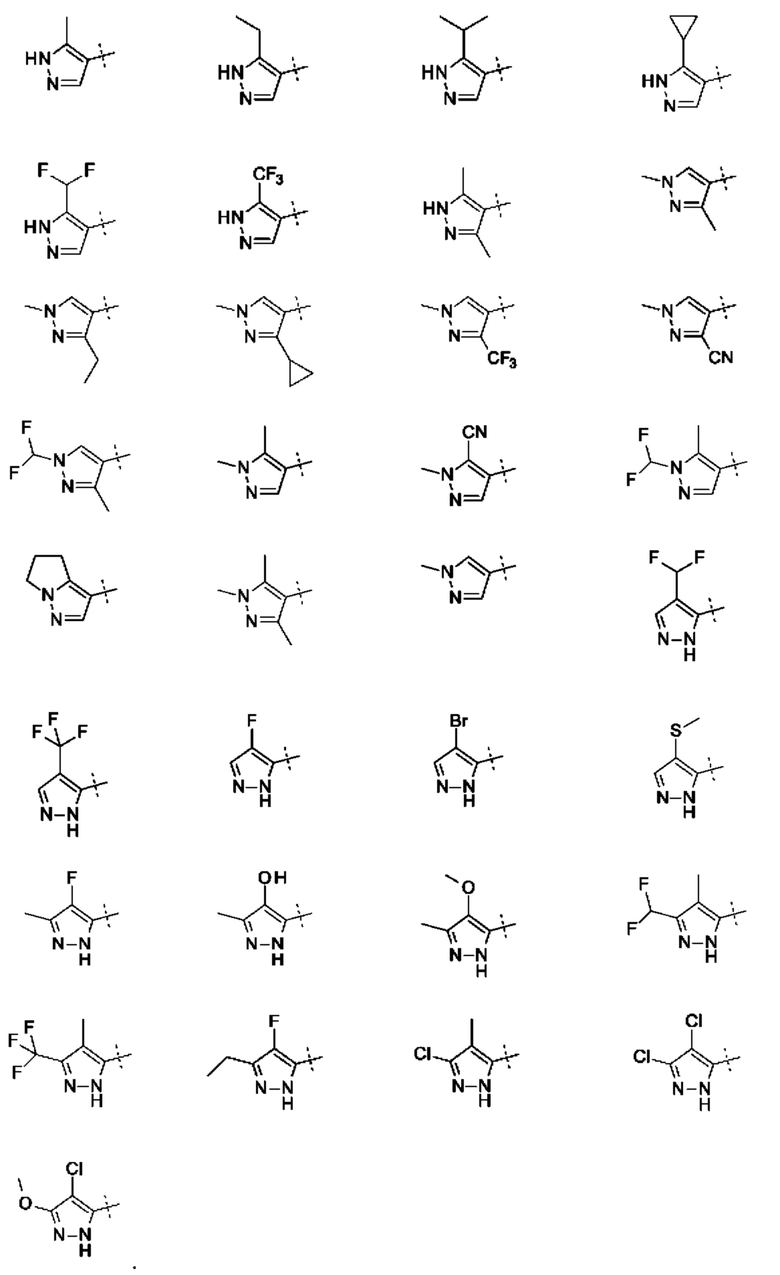
Кольцо А представляет собой необязательно замещенное пиразольное кольцо.
Кольцо А может представлять собой кольцо, выбранное из:
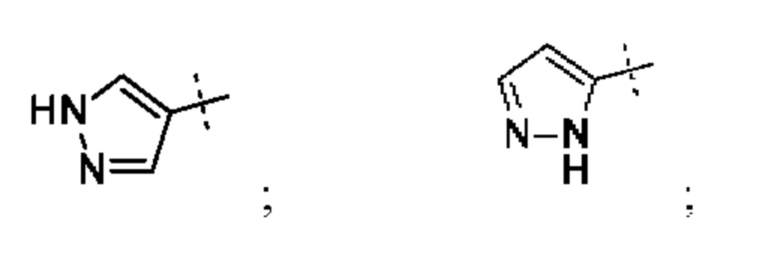
или его таутомер.
Кольцо А может представлять собой кольцо, выбранное из:
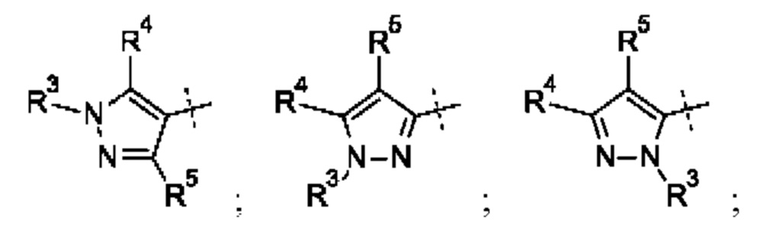
где R3 выбран из Н; С1-6 не ароматической углеводородной группы, необязательно замещенной 1-6 атомами фтора; (CH2)mR6, где m равно от 1 до 3 и R6 выбран из CN, ОН, C1-С3 алкокси и группы SR8 или ее окисленных форм, где R8 представляет собой С1-С3 алкил; необязательно замещенного 4-6-членного насыщенного гетероциклического кольца, содержащего 1 гетероатом, выбранный из О и N, где необязательный заместитель представляет собой CO2R7, где R7 представляет собой С1-3 алкил; где R4 и R5 независимо выбраны из: С1-6 неароматической углеводородной группы, необязательно замещенной 1-6 атомами фтора; (CH2)pR9, где р равно от 0 до 3 и R9 выбран из CN, галогена, ОН, С1-С3 алкокси и группы SR8 или ее окисленных форм, где R8 представляет собой С1-С3 алкил; или R4 и R5 могут быть необязательно соединены с образованием конденсированного 5-или 6-членного кольца; или R4 и R3 могут быть необязательно соединены с образованием конденсированного 5- или 6-членного кольца.
Соединения могут применяться в качестве антагонистов Н4-рецептора. Соединения могут применяться при производстве лекарственных средств. Соединения или лекарственные средства могут применяться при лечении, предупреждении, облегчении, контроле или снижении риска развития воспалительных расстройств, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к новым соединениям. Изобретение также относится к применению новых соединений в качестве антагонистов Н4-рецептора. Изобретение также относится к применению новых соединений при производстве лекарственных средств для применения в качестве антагонистов Н4-рецептора или для лечения системной дисфункции Н4. Изобретение также относится к соединениям, композициям и лекарственным средствам, которые являются селективными антагонистами Н4-рецептора.
Изобретение также относится к соединениям, композициям и лекарственным средствам, применимым для лечения острого и хронического воспаления, аутоиммунного заболевания, расстройств, связанных с иммунной защитой организма, и нейропатической боли.
Соединения согласно изобретению включают соединения формулы (1)
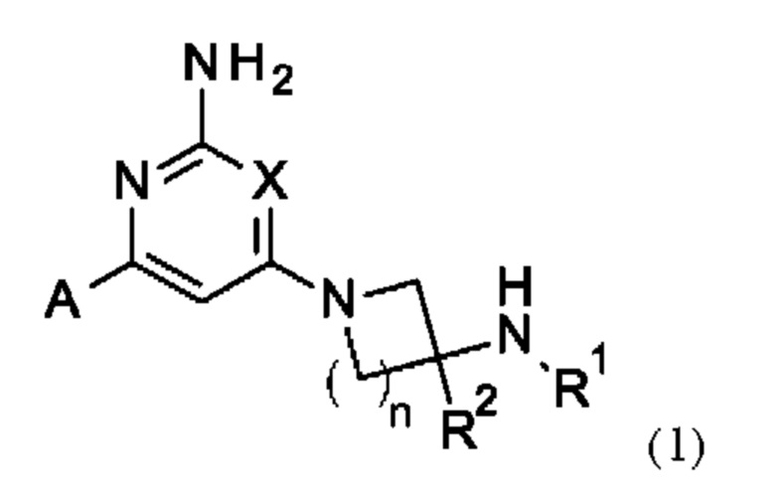
или их соль, где:
X представляет собой СН или N;
n равно 1 или 2;
R1 выбран из Н или С1-3 алкила, где C1-3 алкильная группа может быть циклизована с кольцом, к которому присоединен NHR1, с образованием второго кольца;
R2 представляет собой Н или метил; и
А представляет собой необязательно замещенное пиразольное кольцо, которое связано с кольцом, содержащим X, углерод-углеродной связью.
В приведенных в настоящем документе соединениях X может представлять собой СН или N. X может представлять собой СН. X может представлять собой N.
В приведенных в настоящем документе соединениях n может быть равно 1 или 2.n может быть равно 1.n может быть равно 2.
В приведенных в настоящем документе соединениях R1 может представлять собой Н или C1-3 алкил. С1-3 алкильная группа может быть циклизована с кольцом, к которому присоединен NHR1, с образованием второго кольца.
В приведенных в настоящем документе соединениях R2 может представлять собой Н или метил. R2 может представлять собой Н. R2 может представлять собой метил.
Примеры соединений могут включать
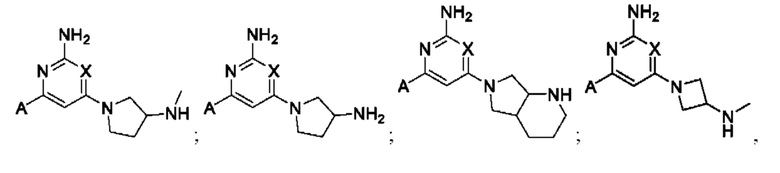
где А представляет собой необязательно замещенное пиразольное кольцо.
В приведенных в настоящем документе соединениях А может быть выбрано из:

или их таутомера.
Кольцо А может представлять собой кольцо, выбранное из:
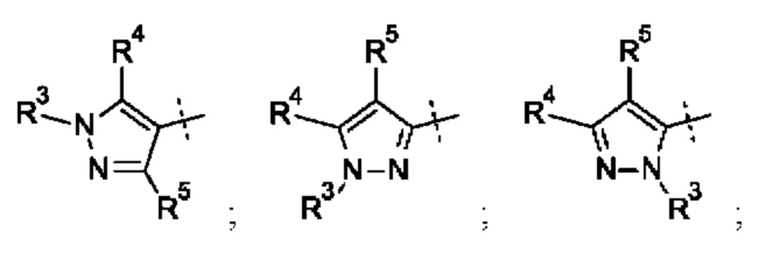
где R3 выбран из Н; С1-6 не ароматической углеводородной группы, необязательно замещенной 1-6 атомами фтора; (CH2)mR6, где m равно от 1 до 3 и R6 выбран из CN, ОН, С1-С3 алкокси и группы SR8 или ее окисленных форм, где R8 представляет собой С1-С3 алкил; необязательно замещенного 4-6-членного насыщенного гетероциклического кольца, содержащего 1 гетероатом, выбранный из О и N, где необязательный заместитель представляет собой CO2R7, где R7 представляет собой C1-3 алкил; где R4 и R5 независимо выбраны из: С1-6 неароматической углеводородной группы, необязательно замещенной 1-6 атомами фтора; (CH2)pR9, где р равно от 0 до 3 и R9 выбран из CN, галогена, ОН, C1-C3 алкокси и группы SR8 или ее окисленных форм, где R8 представляет собой C1-С3 алкил; или R4 и R5 могут быть необязательно соединены с образованием конденсированного 5-или 6-членного кольца; или R4 и R3 могут быть необязательно соединены с образованием конденсированного 5- или 6-членного кольца.
Конкретные заместители для кольца А включают один или более из следующих: метил, этил, изопропил, дифторметил, трифторметил, 1,1,1-трифторэтил, 1-гидроксиэтил, циклопропил, циклобутил, фтор, хлор, бром, циано, гидроксил, метокси, тиометил, 1-метоксиэтил, цианометил, 1-цианоэтил, оксетан, пиперидин или конденсированное кольцо. Конденсированное кольцо может представлять собой 6-членное ароматическое кольцо. Конденсированное кольцо может представлять собой 5- или 6-членное алифатическое кольцо. Заместитель пиперидина может представлять собой 3 N-этилкарбоксилат. Если А замещено двумя или тремя группами, все заместители могут быть одинаковыми или разными.
В приведенных в настоящем документе соединениях R3 может быть выбран из Н, метила, CF3, CF2H, этила, циклопропила, циклобутила, CH2CF3, СН2СН2ОН, СН2СН2ОСН3, CH2CH2CN, CH2CN, оксетана, этилпиперидинкарбоксилата, или R4 и R3 могут быть соединены с образованием конденсированного 5-членного кольца. R4 и R3 могут быть соединены с образованием конденсированного 5-членного алифатического кольца.
В приведенных в настоящем документе соединениях R4 или R5 может быть выбран из метила, этила, циклопропила, циклобутила, пропила, изопропила, CF3, CF2H, фтора, хлора, брома, циано, гидрокси, метокси, тиометила, или R4 и R5 соединены с образованием конденсированного 5- или 6-членного кольца. R4 и R5 могут быть соединены с образованием конденсированного 5- или 6-членного алифатического или ароматического кольца. R4 и R5 могут быть соединены с образованием конденсированного 5- или 6-членного алифатического кольца. R4 и R5 могут быть соединены с образованием конденсированного 5- или 6-членного ароматического кольца.
В приведенных в настоящем документе соединениях А может быть выбрано из группы, состоящей из:
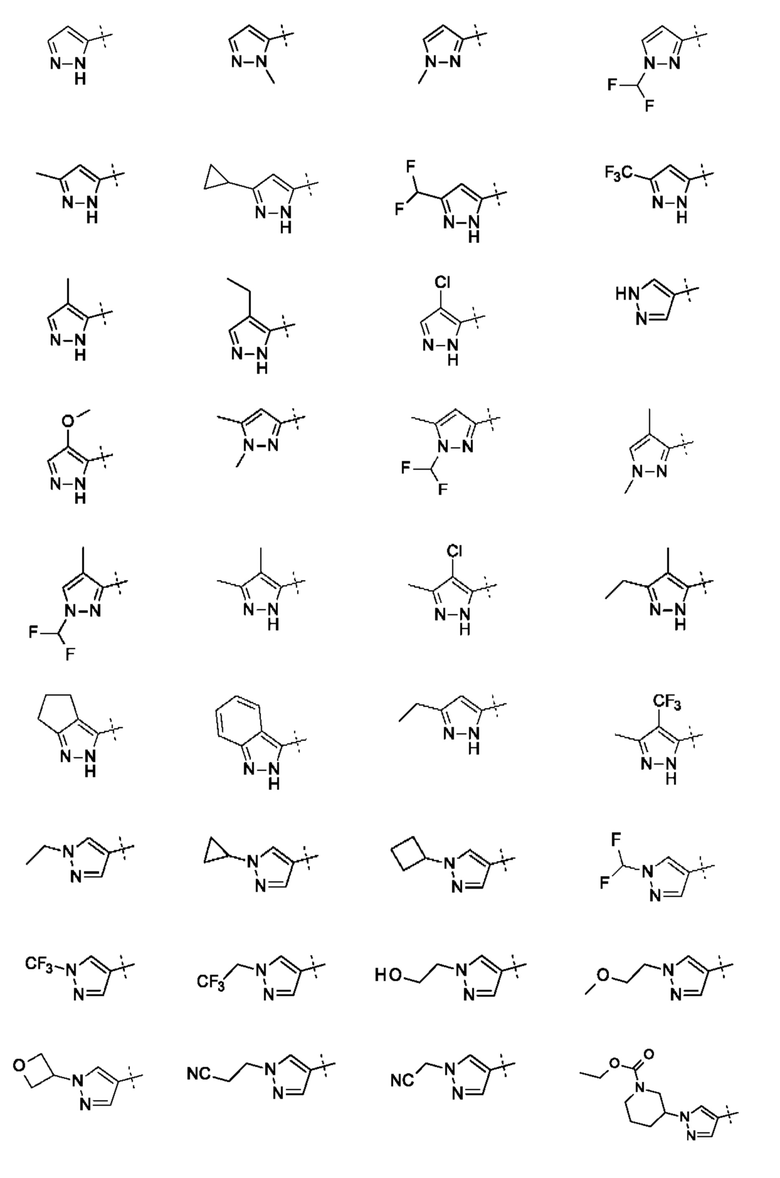
Примеры соединений могут включать
где n равно 1 или 2;
R1 выбран из Н или С1-3 алкила, где С1-3 алкильная группа может быть циклизована с кольцом, к которому присоединен NHR1, с образованием второго кольца; и
R2 представляет собой Н или метил.
Соединение может быть выбрано из группы, состоящей из:
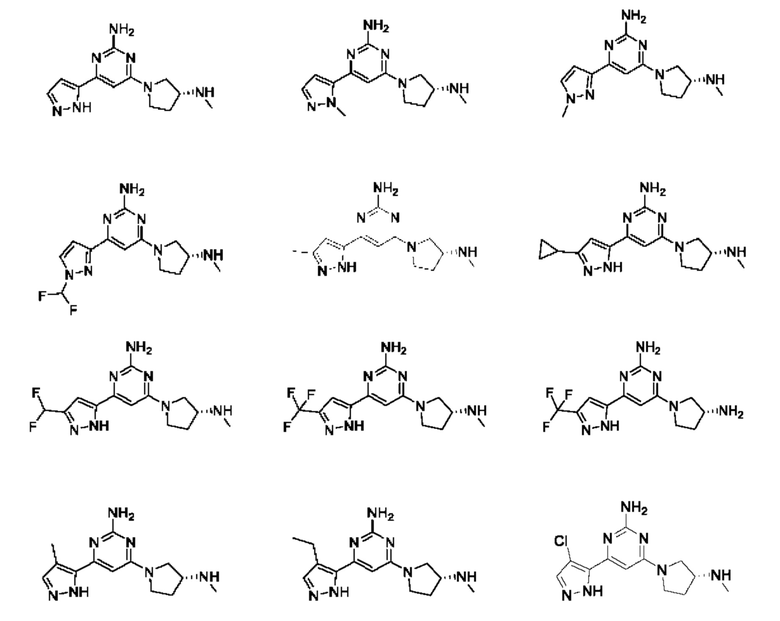
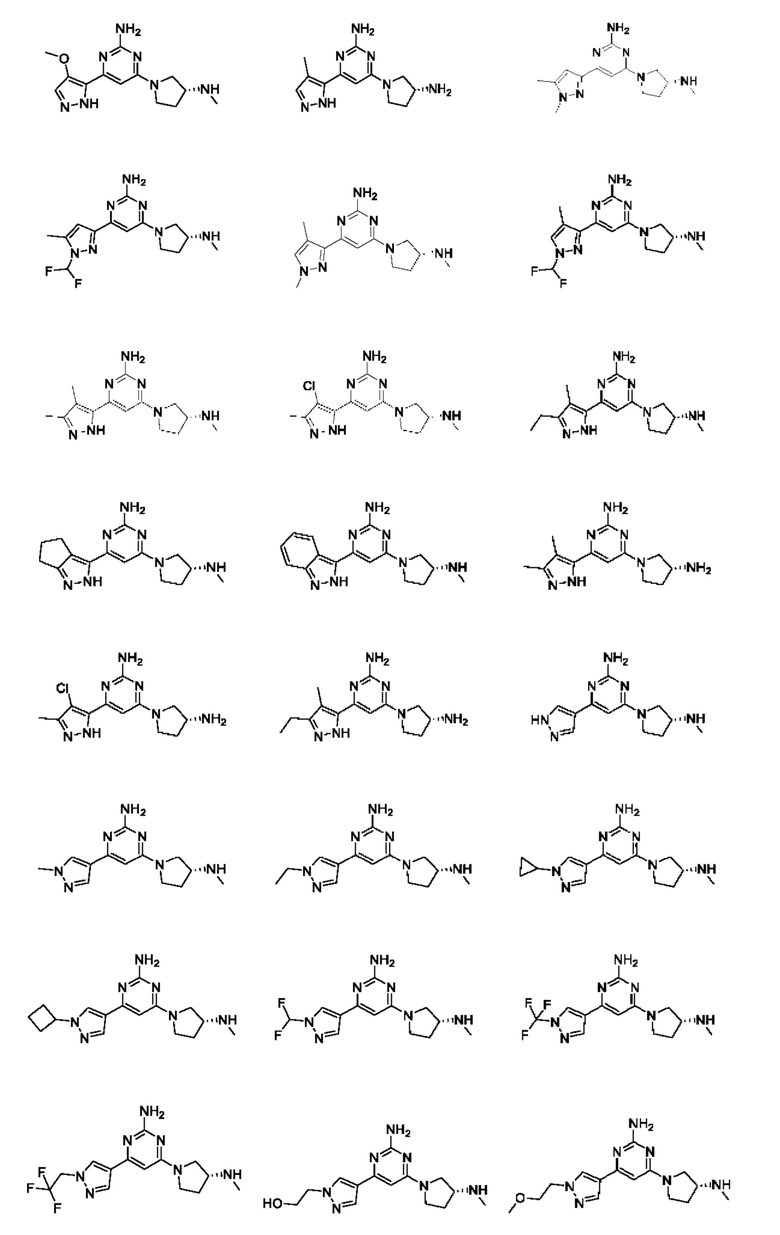
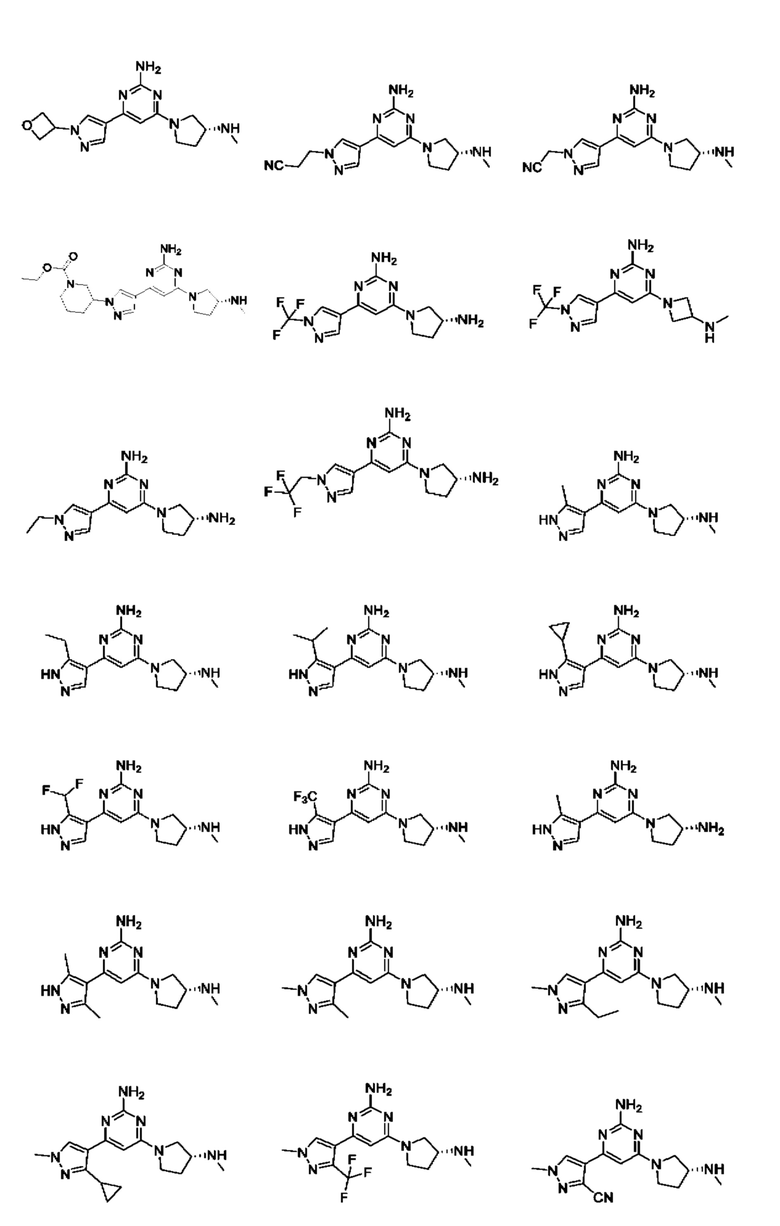
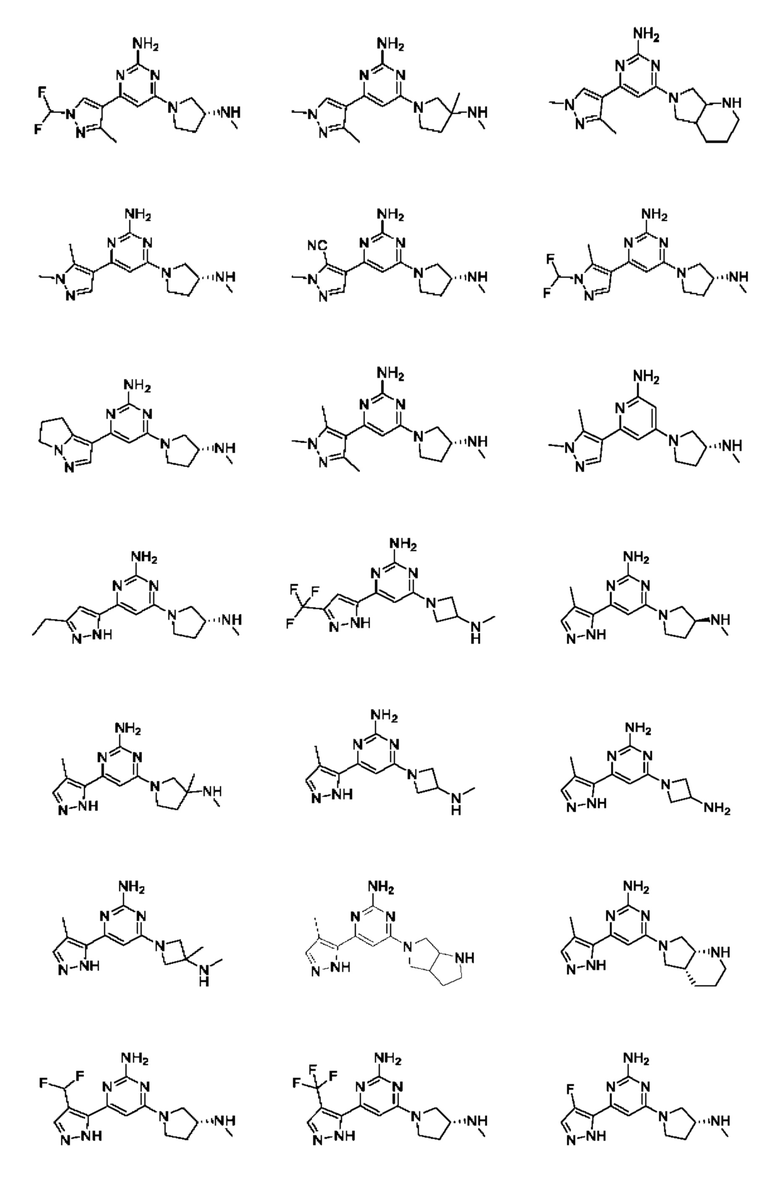
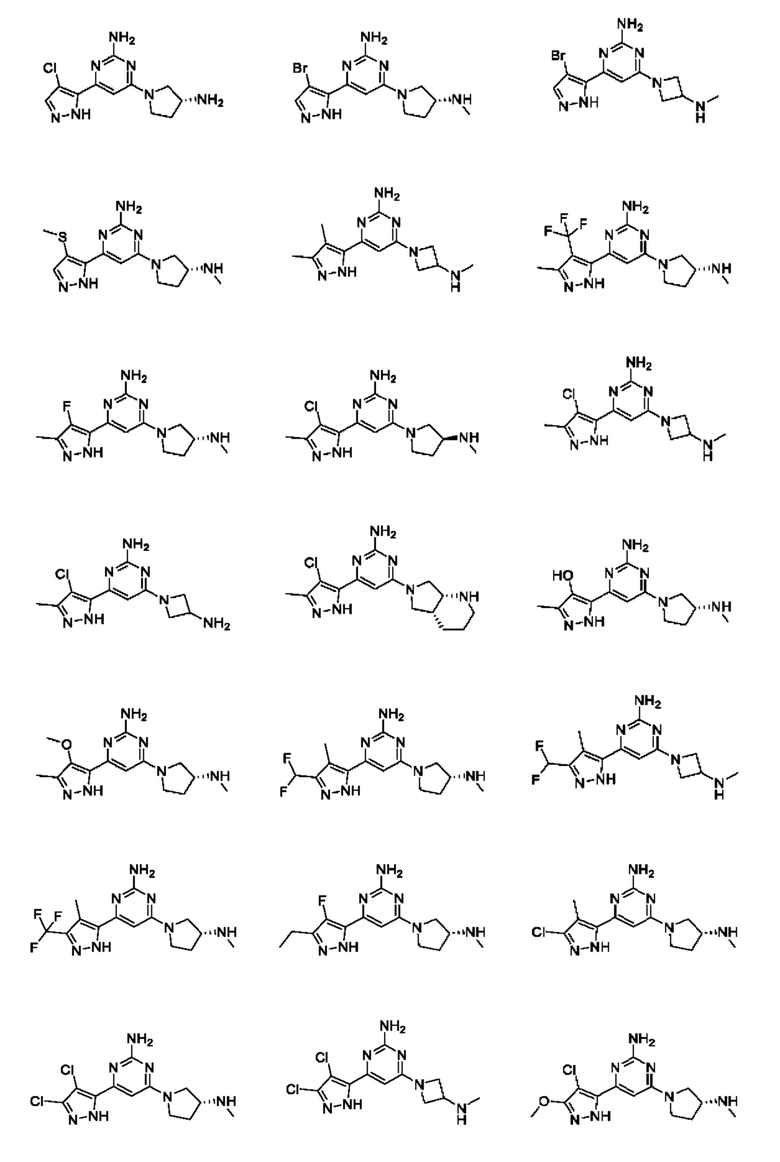
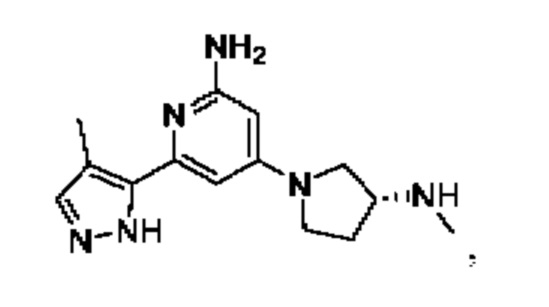
и их солей.
Конкретные примеры соединений включают соединения с низкой активностью в отношении hERG.
Конкретные соединения включают:
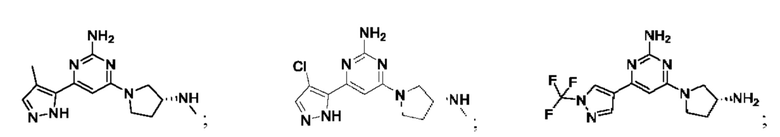

Определения
В этой заявке применяются следующие определения, если не указано иное.
Термин «лечение» применительно к применению любого из описанных в настоящем документе соединений, включая соединения формулы (1) или формулы (1а), используется для описания любой формы вмешательства, при которой соединение вводят субъекту, страдающему от рассматриваемого заболевания или расстройства, или подверженного риску их развития, или потенциально подверженного риску их развития. Таким образом, термин «лечение» охватывает как превентивное (профилактическое) лечение, так и лечение, при котором проявляются измеримые или обнаружимые симптомы заболевания или расстройства.
Термин «эффективное терапевтическое количество» в контексте настоящего документа (например, применительно к способам лечения заболевания или состояния) относится к количеству соединения, которое является эффективным для достижения желаемого терапевтического эффекта. Например, если состояние представляет собой боль, то эффективное терапевтическое количество представляет собой количество, достаточное для обеспечения желаемого уровня облегчения боли. Желаемый уровень облегчения боли может представлять собой, например, полное устранение боли или уменьшение ее силы.
Термин «неароматическая углеводородная группа», как в выражении «С1-6 неароматическая углеводородная группа», относится к группе, состоящей из атомов углерода и водорода и не содержащей ароматических колец. Углеводородная группа может быть полностью насыщенной или может содержать одну или более двойных углерод-углеродных связей или тройных углерод-углеродных связей, или смеси двойных и тройных связей. Углеводородная группа может представлять собой группу с прямой или разветвленной цепью или может состоять из циклической группы или содержать ее. Таким образом, термин «неароматический углеводород» включает алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкенилалкил и т.п.
Термин «насыщенный» относится к углеводородной группе, не содержащей двойных или тройных углерод-углеродных связей. Таким образом, насыщенная углеводородная группа может представлять собой алкильную группу, циклоалкильную группу, циклоалкилалкильную группу, алкилциклоалкильную группу или алкилциклоалкилалкильную группу. Примеры насыщенных углеводородных групп включают циклопропил, циклобутил и циклопропилметил.
Примеры 4-6-членных насыщенных гетероциклических колец, содержащих 1 гетероатом, выбранный из О и N, включают оксетан, азетидин, тетрагидрофуран, пирролидин, тетрагидропиран и пиперидин.
В той степени, в которой любое из описанных соединений имеет хиральные центры, настоящее изобретение распространяется на все оптические изомеры таких соединений, будь то в форме рацематов или разделенных энантиомеров. Изобретение, описанное в настоящем документе, относится ко всем кристаллическим формам, сольватам и гидратам любого из раскрытых соединений, каким бы способом они ни были получены. В той степени, в которой любое из соединений, раскрытых в настоящем документе, имеет кислотные или основные центры, такие как карбоксилаты или аминогруппы, в настоящее изобретение включены все солевые формы указанных соединений. В случае фармацевтического применения соль следует рассматривать как являющуюся фармацевтически приемлемой солью.
Соли или фармацевтически приемлемые соли, которые можно упомянуть, включают соли присоединения кислот и соли присоединения оснований. Такие соли могут быть образованы обычными способами, например, посредством реакции соединения в форме свободной кислоты или свободного основания с одним или несколькими эквивалентами подходящей кислоты или основания, необязательно в растворителе или в среде, в которой соль является нерастворимой, с последующим удалением указанного растворителя или указанной среды с использованием стандартных методик (например, в вакууме, посредством лиофилизации или посредством фильтрации). Соли также могут быть получены путем обмена противоиона соединения в форме соли на другой противоион, например, с использованием подходящей ионообменной смолы.
Примеры фармацевтически приемлемых солей включают соли присоединения кислот, полученные из неорганических кислот и органических кислот, и соли, полученные из металлов, таких как натрий, магний, калий и кальций.
Примеры солей присоединения кислот включают соли присоединения кислот, образованные с уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, арилсульфоновыми кислотами (например, бензолсульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой и п-толуолсульфоновой), аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензойной, 4-ацетамидобензойной, бутановой, (+) камфорной, камфорсульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, глюконовой (например, D-глюконовой), глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, соляной, йодистоводородной, изэтионовой, молочной (например, (+)-L-молочной и (±)-DL-молочной), лактобионовой, малеиновой, яблочной (например, (-)-L-яблочной), малоновой, (±)-DL-миндальной, метафосфорной, метансульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, винной (например, (+)-L-винной), тиоциановой, ундециленовой и валериановой кислотами.
Также включены любые сольваты соединений и их соли. Предпочтительные сольваты представляют собой сольваты, образованные включением в твердую структуру (например, кристаллическую структуру) соединений согласно изобретению молекул нетоксичного фармацевтически приемлемого растворителя (называемого ниже сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем перекристаллизации соединений согласно изобретению с растворителем или смесью растворителей, содержащей сольватирующий растворитель. Определить, образовался ли сольват в каком-либо конкретном случае, можно, подвергнув кристаллы соединения анализу с использованием хорошо известных и стандартных методик, таких как термогравиметрический анализ (ТГА), дифференциальная сканирующая калориметрия (ДСК) и рентгеновская кристаллография.
Сольваты могут представлять собой стехиометрические или нестехиометрические сольваты. Отдельные сольваты могут представлять собой гидраты, и примеры гидратов включают полугидраты, моногидраты и дигидраты. Более подробное обсуждение сольватов и способов, используемых для их получения и определения их характеристик, см. в Bryn et al., Solid-State Chemistry of Drags, Second Edition, опубликовано SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Термин «фармацевтическая композиция» в контексте данного изобретения означает композицию, содержащую активный агент и дополнительно содержащую один или более фармацевтически приемлемых носителей. Композиция может дополнительно содержать ингредиенты, выбранные, например, из разбавителей, адъювантов, эксципиентов, носителей, консервантов, наполнителей, дезинтегрирующих агентов, смачивающих агентов, эмульгирующих агентов, суспендирующих агентов, подсластителей, вкусоароматических веществ, отдушек, антибактериальных агентов, противогрибковых агентов, смазывающих агентов и диспергирующих агентов, в зависимости от характера способа введения и лекарственных форм. Композиции могут иметь форму, например, таблеток, драже, порошков, эликсиров, сиропов, жидких препаратов, включая суспензии, спреев, лекарственных форм для ингаляции, таблеток, пастилок, эмульсий, растворов, облаток, гранул, капсул и суппозиториев, а также жидких препаратов для инъекций, в том числе липосомальных препаратов.
Соединения согласно изобретению могут содержать одно или более изотопных замещений, и ссылка на конкретный элемент включает в себя все изотопы этого элемента. Например, ссылка на водород включает в себя 1Н, 2Н (D) и 3Н (Т). Точно так же ссылки на углерод и кислород включают в себя соответственно 12С, 13С и 14С, и 16О и 18O. Аналогичным образом, ссылка на конкретную функциональную группу также включает в себя изотопные вариации, если из контекста не следует иное. Например, ссылка на алкильную группу, такую как этильная группа, или алкоксигруппу, такую как метоксигруппа, также охватывает варианты, в которых один или более атомов водорода в группе находятся в форме изотопа дейтерия или трития, например, как в этильной группе, в которой все пять атомов водорода находятся в изотопной форме дейтерия (пердейтероэтильная группа), или в метоксигруппе, в которой все три атома водорода находятся в изотопной форме дейтерия (тридейтерометоксигруппа). Изотопы могут быть радиоактивными или нерадиоактивными.
Терапевтические дозировки могут варьироваться в зависимости от потребностей пациента, тяжести состояния, лечение которого осуществляют, и применяемого соединения. Определение правильной дозировки в конкретной ситуации находится в компетенции специалиста в данной области техники. Обычно лечение начинают с более низких доз, которые меньше оптимальной дозы соединения. После этого дозу увеличивают небольшими приращениями, пока не будет достигнут оптимальный эффект в данных обстоятельствах. Для удобства общая суточная доза может быть разделена и при желании вводиться частями в течение дня.
Величина эффективной дозы соединения, безусловно, будет варьироваться в зависимости от характера тяжести состояния, подлежащего лечению, а также от конкретного соединения и способа его введения. Выбор подходящих дозировок находится в пределах компетенции специалиста в данной области техники и не требует излишних усилий. В общем случае суточный диапазон доз может составлять от около 10 мкг до около 30 мг на кг массы тела человека и животного, не являющегося человеком, предпочтительно от около 50 мкг до около 30 мг на кг массы тела человека и животного, не являющегося человеком, например, от около 50 мкг до около 10 мг на кг массы тела человека и животного, не являющегося человеком, например, от около 100 мкг до около 30 мг на кг массы тела человека и животного, не являющегося человеком, например, от около 100 мкг до около 10 мг на кг массы тела человека и животного, не являющегося человеком, и наиболее предпочтительно от около 100 мкг до около 1 мг на кг массы тела человека и животного, не являющегося человеком.
Способы получения соединений формулы (1)
Соединения формулы (1) могут быть получены в соответствии со способами синтеза, хорошо известными специалисту и описанными в настоящем документе.
Соответственно, в еще одном варианте осуществления изобретение относится к способу получения соединения, как определено в формуле (1) выше, включающему:
(А) взаимодействие соединения формулы (10):
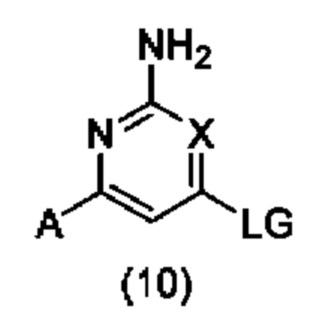
с соединением формулы (11):
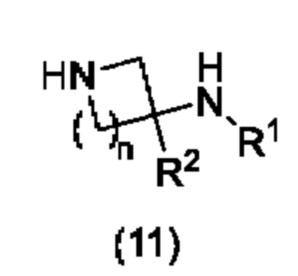
в условиях SNAr (ароматического нуклеофильного замещения) или в условиях сочетания, катализируемого переходными металлами; где A, R1, R2, X и n являются такими, как определено в формуле (1) выше, и LG представляет собой подходящую уходящую группу; или
(B) взаимодействие соединения формулы (12):
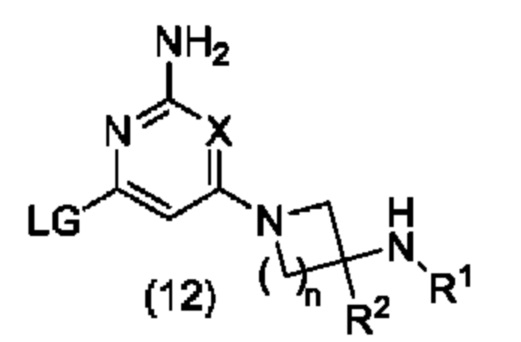
с соединением формулы (13):
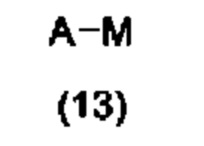
в условиях сочетания, катализируемого переходными металлами, или в условиях SNAr; где A, R1, R2, X и n являются такими, как определено в формуле (1) выше, LG представляет собой подходящую уходящую группу, и М, который может присутствовать или отсутствовать, представляет собой подходящим образом замещенный металл или неметалл; или
(C) превращение одного соединения формулы (1) в другое соединение формулы (1).
В варианте способа (А) соединение формулы (10) может быть подвергнуто взаимодействию с соединением формулы (11) в условиях SNAr. Реакцию SNAr обычно проводят с использованием либо избытка соединения формулы (11), либо стехиометрического количества соединения формулы (11) в присутствии основания, которое может представлять собой третичное аминовое основание, такое как TEA (триэтиламин) или DIPEA (N,N-диизопропилэтиламин), или неорганическое основание, такое как K2CO3, Cs2CO3 или NaHCO3, необязательно в подходящем растворителе, таком как Н2О, MeCN, 1,4-диоксан, ТГФ (тетрагидрофуран), МеОН, EtOH, IPA (изопропиловый спирт), BuOH, ДМФА (N,N-диметилформамид), NMP (N-метилпирролидон) или ДМСО (диметилсульфоксид), или комбинации подходящих растворителей, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления, необязательно в присутствии добавки, такой как KF или соль серебра. Необязательно соединение формулы (11) может присутствовать в реакции в виде кислой соли, такой как соль с HCl, HBr или TFA (трифторуксусная кислота), необязательно в присутствии третичного основания, такого как TEA или DIPEA. Уходящая группа LG в соединении формулы (10) может представлять собой галоген, такой как F, Cl или Br; алкоксигруппу, такую как ОМе; арилоксигруппу, такую как пентафторфенокси; сульфенильную группу, такую как SMe, сульфинильную группу, такую как SOMe, сульфонильную группу, такую как SO2Me, сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf; или уходящую группу, образованную реакцией гидроксигруппы с конденсирующим реагентом для образования пептидной связи, таким как ВОР (бензотриазолилокси-трис(диметиламино)фосфония гексафторфосфат), РуВОР (бензотриазол-1-илокси-трис(пирролидино)фосфония гексафторфосфат) или HATU (O-(7-азабензотриазол-1-ил)-N,N.N',N'-тетраметилурония гексафторфосфат).
В качестве альтернативы, в варианте способа (А) соединение формулы (10) может быть подвергнуто взаимодействию с соединением формулы (11) в условиях сочетания, катализируемого переходными металлами. Реакцию сочетания, катализируемого переходными металлами, обычно проводят с использованием соединения формулы (11) в присутствии неорганического основания, такого как NaOtBu, KOtBu, K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, ТГФ, DME (диметоксиэтан) или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PBu3, PtBu3, XPhos, Xantphos или BINAP, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (10) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
Соединения формулы (10) могут быть получены реакцией, показанной на схеме 1 ниже:
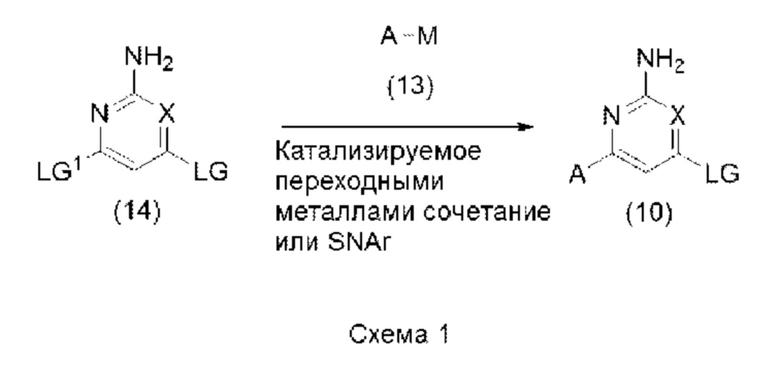
Таким образом, соединение формулы (14), где X является таким, как определено в формуле (1) выше, и LG и LG1 могут быть одинаковыми или разными и представляют собой подходящие уходящие группы, может быть подвергнуто взаимодействию с соединением формулы (13), где А является таким, как определено в формуле (1) выше, и М, который может присутствовать или отсутствовать, представляет собой подходящим образом замещенный металл или неметалл, в условиях сочетания, катализируемого переходными металлами, или в условиях SNAr, с образованием соединения формулы (10). Реакцию сочетания, катализируемого переходными металлами, или реакцию SNAr обычно проводят, как описано ниже в варианте способа (В), и соединения формулы (13) и формулы (14) могут быть коммерчески доступными или могут быть легко получены стандартными способами, описанными в опубликованных источниках, из простых исходных веществ, известных специалисту. Иногда из-за их нестабильности может возникнуть необходимость в получении соединений формулы (13), где присутствует М, in situ при низких температурах, например, от около -78°С до комнатной температуры, и их дальнейшем взаимодействии в реакции сочетания, катализируемого переходными металлами, без их предварительного выделения. Подробности таких способов известны из опубликованных источников, например, как описано в Oberli и Buchwald в Org. Lett., 2012, Vol. 14, No. 17, с. 4606.
В качестве альтернативы, соединения формулы (10), где X представляет собой N, а LG представляет собой Cl, обычно могут быть получены с помощью последовательности реакций, показанной на схеме 2 ниже:
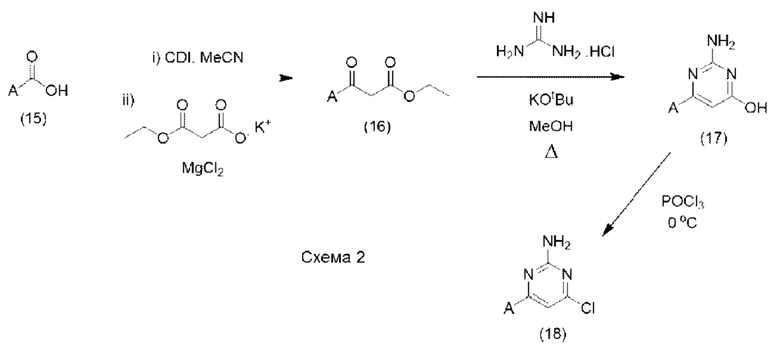
Таким образом, карбоновая кислота формулы (15) может быть гомологирована до соответствующего бета-кетоэфира (16), сначала посредством ее активации с помощью ряда стандартных способов, известных специалисту, например, путем взаимодействия с CDI (N,N'-карбонилдиимидазол) в подходящем растворителе, таком как MeCN, а затем подвергания взаимодействию с производным малоновой кислоты, таким как 3-этокси-3-оксопропаноат калия, в присутствии кислоты Льюиса, такой как MgCl2. После образования бета-кетоэфир (16) может быть циклизован до аналога аминогидроксипиримидина (17) посредством взаимодействия с гуанидином или подходящей солью гуанидина в присутствии подходящего основания, такого как KOtBu, в подходящем растворителе, таком как МеОН. Образованный таким образом аналог аминогидроксипиримидина (17) затем может быть подвергнут взаимодействию с POCl3 в присутствии или в отсутствие подходящего растворителя с образованием соединения формулы (18). Соединения формулы (15) могут быть коммерчески доступными или могут быть легко получены стандартными способами, описанными в опубликованных источниках, из простых исходных веществ, известных специалисту.
Соединения формулы (11) могут быть коммерчески доступными или могут быть легко получены стандартными способами, описанными в опубликованных источниках, из простых исходных веществ, известных специалисту.
В варианте способа (В) соединение формулы (12) может быть подвергнуто взаимодействию с соединением формулы (13) в условиях сочетания, катализируемого переходными металлами. Реакцию сочетания, катализируемого переходными металлами, обычно проводят с использованием соединения формулы (13), в котором присутствует М. Например, когда М представляет собой бороновую кислоту -В(ОН)2 или сложный эфир бороновой кислоты, такой как -В(ОМе)2, -B(OiPr)2 или Bpin, или триалкилборат лития, такой как -B(OiPr)3Li, реакцию сочетания, катализируемого переходными металлами, обычно проводят в присутствии неорганического основания, такого как NaHCO3, Na2CO3, K2CO3, Cs2CO3 или K3PO4, в подходящем растворителе, таком как H2O, MeCN, 1,4-диоксан, ТГФ, Et2O, DME, EtOH, IPA, ДМФА, NMP или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2, Pd(PPh3)4, или предкатализатора на основе переходного металла, такого как XPhos Pd G2, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PtBu3, РСу3 или XPhos, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs или OTf.
В качестве альтернативы, когда М представляет собой соль трифторбората BF3-, реакцию сочетания, катализируемого переходными металлами, обычно проводят в присутствии неорганического основания, такого как Na2CO3, K2CO3, Cs2CO3 или K3PO4, в подходящем растворителе, таком как H2O, MeCN, 1,4-диоксан, ТГФ, МеОН или EtOH, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, РСу3 или RuPhos, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
В качестве альтернативы, когда М представляет собой группу триалкилолова, такую как SnMe3 или SnBu3, реакцию сочетания, катализируемого переходными металлами, обычно проводят в подходящем растворителе, таком как 1,4-диоксан, ТГФ, ДМФА или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии неорганического основания, такого как K2CO3 или CsF, необязательно в присутствии добавки, такой как LiCl, CuI, Bu4NBr или Et4NCl, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
В качестве альтернативы, когда М отсутствует, реакцию сочетания, катализируемого переходными металлами, обычно проводят в присутствии неорганического основания, такого как NaOtBu, KOtBu, K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, ТГФ, DME или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PBu3, PtBu3, XPhos, Xantphos или BINAP, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
В качестве альтернативы, когда М отсутствует, реакцию сочетания, катализируемого переходными металлами, обычно проводят в присутствии неорганического основания, такого как K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, ДМФА, ДМСО или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как CuI, необязательно в присутствии субстехиометрического количества амина, такого как (S)-пролин или транс-N1,N2-диметилциклогексан-1,2-диамин, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
В качестве альтернативы, когда М отсутствует, реакцию сочетания, катализируемого переходными металлами, обычно проводят в присутствии органического основания, такого как nBu4OAc, в подходящем растворителе, таком как 1,4-диоксан, в присутствии субстехиометрического количества предкатализатора на основе переходного металла, такого как XPhos Pd G2, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как XPhos, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl.
В качестве альтернативы, в варианте способа (В) соединение формулы (12) может быть подвергнуто взаимодействию с соединением формулы (13) в условиях SNAr. Реакцию SNAr обычно проводят с использованием соединения формулы (13), где М отсутствует, в присутствии третичного аминового основания, такого как TEA или DIPEA, или неорганического основания, такого как K2CO3, Cs2CO3, KOtBu или NaH, в подходящем растворителе, таком как THF, ДМФА, H2O, ДМСО или NMP, или комбинации подходящих растворителей, при температуре от около комнатной температуры до около 200°С, с использованием общепринятого нагревания или, необязательно, путем нагревания с помощью микроволнового излучения, в открытом сосуде или, необязательно, в герметичном сосуде, необязательно под давлением выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как F, Cl или Br; алкоксигруппу, такую как ОМе; арилоксигруппу, такую как пентафторфенокси; сульфенильную группу, такую как SMe, сульфинильную группу, такую как SOMe, сульфонильную группу, такую как SO2Me, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
Соединение формулы (12) может быть получено с помощью последовательности реакций, показанной на схеме 3 ниже:
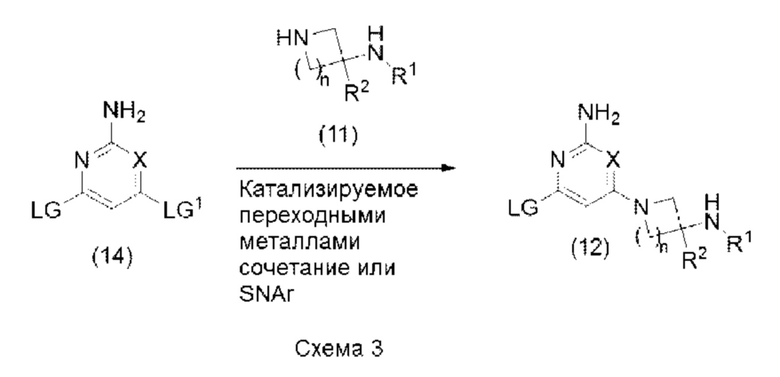
Таким образом, соединение формулы (14), где X является таким, как определено в формуле (1) выше, и LG и LG1 могут быть одинаковыми или разными и представляют собой подходящие уходящие группы, может быть подвергнуто взаимодействию с соединением формулы (11), где R1, R2 и n являются такими, как определено в формуле (1) выше, в условиях SNAr или в условиях сочетания, катализируемого переходными металлами, с образованием соединения формулы (12). Реакцию SNAr или реакцию сочетания, катализируемого переходными металлами, обычно проводят, как описано выше в варианте способа (А).
В варианте способа (С) одно соединение формулы (1) может быть превращено в другое соединение формулы (1) способами, хорошо известными специалисту. Примеры способов синтеза для превращения одной функциональной группы в другую функциональную группу изложены в стандартных руководствах, таких как March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition, Michael B. Smith, John Wiley, 2013, (ISBN: 978-0-470-46259-1), Organic Syntheses, Online Edition, www.orgsyn.org, (ISSN 2333-3553) и Fiesers' Reagents for Organic Synthesis, Volumes 1-17, John Wiley, под ред. Mary Fieser (ISBN: 0-471-58283-2).
Во многих реакциях, описанных выше, может быть необходимо защитить одну или несколько групп, чтобы предотвратить протекание реакции в нежелательном месте молекулы. Примеры защитных групп и способы защиты функциональных групп и снятия защиты с них можно найти в Greene's Protective Groups in Organic Synthesis, Fifth Edition, Editor: Peter G.M. Wuts, John Wiley, 2014, (ISBN: 9781118057483). В частности, подходящая защитная группа для работы с соединениями формулы (10) или формулы (12) включает группу 2,5-диметил-1H-пиррола; подходящие защитные группы для работы с соединениями формулы (11) или формулы (12) включают ВОС (трет-бутоксикарбонильная группа) и CBZ (бензилоксикарбонильная группа); и подходящие защитные группы для работы соединениями формулы (13) включают SEM ([2-(триметилсилил)этокси]метильная группа) и ТНР (2-тетрагидропиранильная группа).
Соединения, полученные указанными выше способами, могут быть выделены и очищены любым из множества способов, хорошо известных специалистам в данной области техники, и примеры таких способов включают перекристаллизацию и хроматографические методы, такие как колоночная хроматография (например, флэш-хроматография), ВЭЖХ и SFC.
Фармацевтические препараты
Хотя можно вводить активное соединение в чистом виде, предпочтительно представлять его в виде фармацевтической композиции (например, препарата).
Соответственно, в еще одном варианте осуществления изобретения предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (1), как определено выше, вместе с по меньшей мере одним фармацевтически приемлемым эксципиентом.
Композиция может представлять собой композицию в виде таблетки.
Композиция может представлять собой композицию в виде капсулы.
Фармацевтически приемлемый эксципиент(ы) может быть выбран, например, из носителей (например, твердого, жидкого или мягкого носителя), адъювантов, разбавителей (например, твердых разбавителей, таких как наполнители или объемообразующие агенты; и жидких разбавителей, таких как растворители и сорастворители), гранулирующих агентов, связующих, агентов для повышения текучести, покрывающих агентов, регуляторов высвобождения (например, полимеров или восков, замедляющих или отсрочивающих высвобождение), связывающих агентов, разрыхлителей, буферных агентов, смазывающих веществ, консервантов, противогрибковых и антибактериальных агентов, антиоксидантов, буферных агентов, агентов, регулирующих тоничность, загустителей, вкусоароматических веществ, подсластителей, пигментов, пластификаторов, агентов, маскирующих вкус, стабилизаторов или любых других эксципиентов, обычно используемых в фармацевтических композициях.
Термин «фармацевтически приемлемый» в контексте настоящего документа означает соединения, материалы, композиции и/или лекарственные формы, которые, в рамках здравого медицинского заключения, подходят для применения в контакте с тканями субъекта (например, человека) без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерно с разумным соотношением «польза/риск». Каждый эксципиент также должен быть «приемлемым» в смысле совместимости с другими ингредиентами препарата.
Фармацевтические композиции, содержащие соединения формулы (1), могут быть приготовлены в соответствии с известными методами, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания, США.
Фармацевтические композиции могут быть в любой форме, подходящей для перорального, парентерального, местного, интраназального, внутрибронхиального, сублингвального, офтальмологического, внутриушного, ректального, интравагинального или трансдермального введения.
Фармацевтические лекарственные формы, подходящие для перорального введения, включают таблетки (с покрытием или без покрытия), капсулы (с твердой или мягкой оболочкой), каплеты, пилюли, пастилки, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, облатки или пластыри, такие как буккальные пластыри.
Композиции в виде таблеток могут содержать стандартную дозу активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например, лактоза, сахароза, сорбит или маннит; и/или разбавителем, не являющимся производным сахара, таким как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производное, такое как микрокристаллическая целлюлоза (МКЦ), метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты, как связующие и гранулирующие агенты, такие как поливинилпирролидон, разрыхлители (например, набухающие сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие агенты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, БГТ (бутилированный гидрокситолуол)), буферные агенты (например, фосфатные или цитратные буферы) и шипучие агенты, такие как смеси цитрат/бикарбонат. Такие эксципиенты хорошо известны и не нуждаются в подробном описании в настоящем документе.
Таблетки могут быть предназначены для высвобождения лекарственного средства либо при контакте с желудочными жидкостями (таблетки с немедленным высвобождением), либо для высвобождения контролируемым образом (таблетки с контролируемым высвобождением) в течение длительного периода времени или в определенной области желудочно-кишечного тракта.
Фармацевтические композиции обычно содержат от приблизительно 1% (мас./мас.) до приблизительно 95%, предпочтительно % (мас./мас.) активного ингредиента и от 99% (мас./мас.) до 5% (мас./мас.) фармацевтически приемлемого эксципиента (например, как определено выше) или комбинации таких эксципиентов. Предпочтительно композиции содержат от приблизительно 20% (мас./мас.) до приблизительно 90% (мас./мас.) активного ингредиента и от 80% (мас./мас.) до 10% фармацевтического эксципиента или комбинации эксципиентов. Фармацевтические композиции содержат от приблизительно 1% до приблизительно 95%, предпочтительно от приблизительно 20% до приблизительно 90% активного ингредиента. Фармацевтические композиции согласно изобретению могут быть, например, в форме стандартной дозы, такой как форма ампул, флаконов, суппозиториев, предварительно заполненных шприцев, драже, порошков, таблеток или капсул.
Таблетки и капсулы могут содержать, например, 0-20% разрыхлителей, 0-5% смазывающих веществ, 0-5% добавок для повышения текучести и/или 0-99% (мас./мас.) наполнителей или объемообразующих агентов (в зависимости от дозы лекарственного средства). Они также могут содержать 0-10% (мас./мас.) полимерных связующих, 0-5% (мас./мас.) антиоксидантов, 0-5% (мас./мас.) пигментов. Таблетки с замедленным высвобождением, кроме того, обычно содержат 0-99% (мас./мас.) контролирующих (например, замедляющих) высвобождение полимеров (в зависимости от дозы). Пленочные оболочки таблетки или капсулы обычно содержат 0-10% (мас./мас.) полимеров, 0-3% (мас./мас.) пигментов и/или 0-2% (мас./мас.) пластификаторов.
Препараты для парентерального введения обычно содержат 0-20% (мас./мас.) буферов, 0-50% (мас./мас.) сорастворителей и/или 0-99% (мас./мас.) воды для инъекций (WFI) (в зависимости от дозы и наличия лиофилизации). Препараты для внутримышечных депо могут также содержать 0-99% (мас./мас.) масел.
Фармацевтические препараты могут быть представлены пациенту в «упаковках для пациентов», содержащих полный курс лечения в одной упаковке, обычно в блистерной упаковке.
Соединения формулы (1) обычно будут представлены в стандартной лекарственной форме и, как таковые, обычно будут содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, препарат может содержать от 1 нанограмма до 2 граммов активного ингредиента, например, от 1 нанограмма до 2 миллиграммов активного ингредиента. В пределах этих диапазонов конкретные поддиапазоны соединения составляют от 0,1 миллиграмма до 2 граммов активного ингредиента (чаще от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 500 миллиграммов) или от 1 микрограмма до 20 миллиграммов (например, от 1 микрограмма до 10 миллиграммов, например от 0,1 миллиграмма до 2 миллиграммов активного ингредиента).
Для пероральных композиций стандартная лекарственная форма может содержать от 1 миллиграмма до 2 граммов, чаще от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 1 грамма, например, от 100 миллиграммов до 1 грамма активного соединения.
Активное соединение будет вводиться нуждающемуся в этом пациенту (например, пациенту-человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта (эффективном количестве). Точные количества вводимого соединения могут быть определены лечащим врачом в соответствии со стандартными процедурами.
ПРИМЕРЫ
Далее изобретение будет проиллюстрировано, без ограничения, со ссылкой на конкретные варианты осуществления, описанные в следующих примерах.
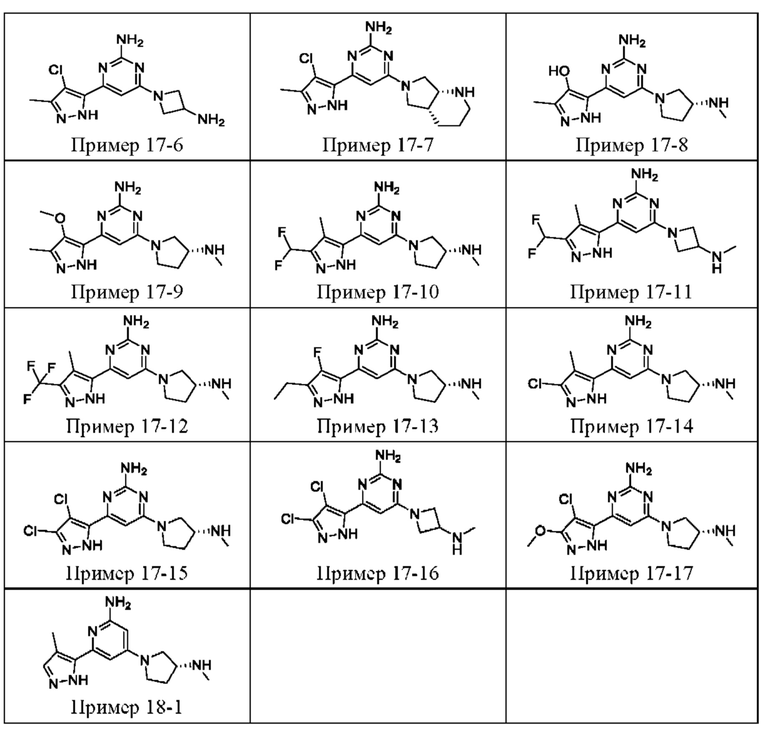
ПРИМЕРЫ С 1-1 ПО 18-1
Были получены соединения примеров с 1-1 по 18-1, показанные в таблице 1 ниже. Их свойства, определенные с помощью ЯМР (ЯМР-спектроскопия) и ЖХМС (жидкостная хроматография с тандемной масс-спектрометрией), а также способы, использованные для их получения, приведены в таблице 3. Исходные вещества для каждого из примеров перечислены в таблице 2.
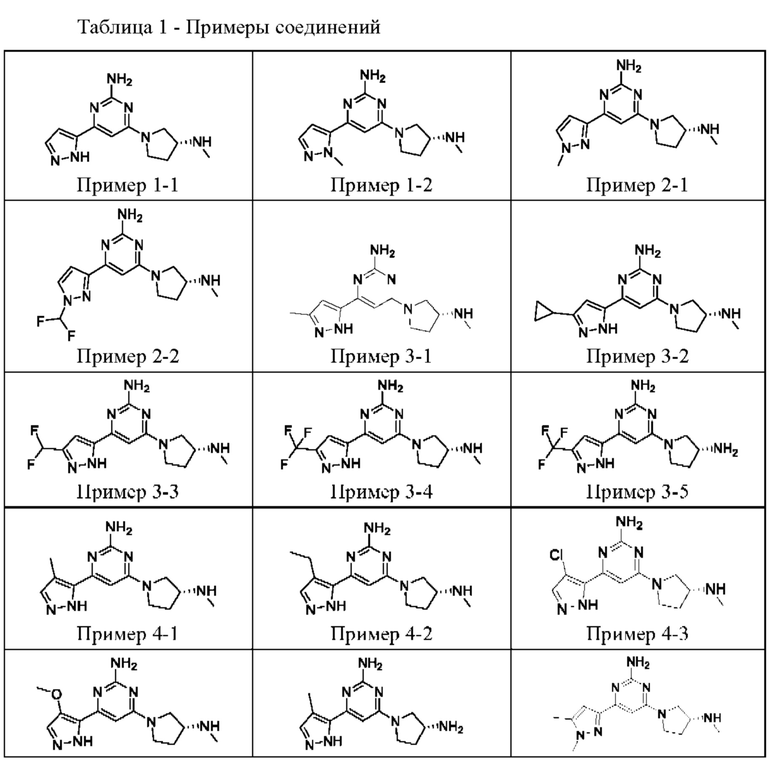
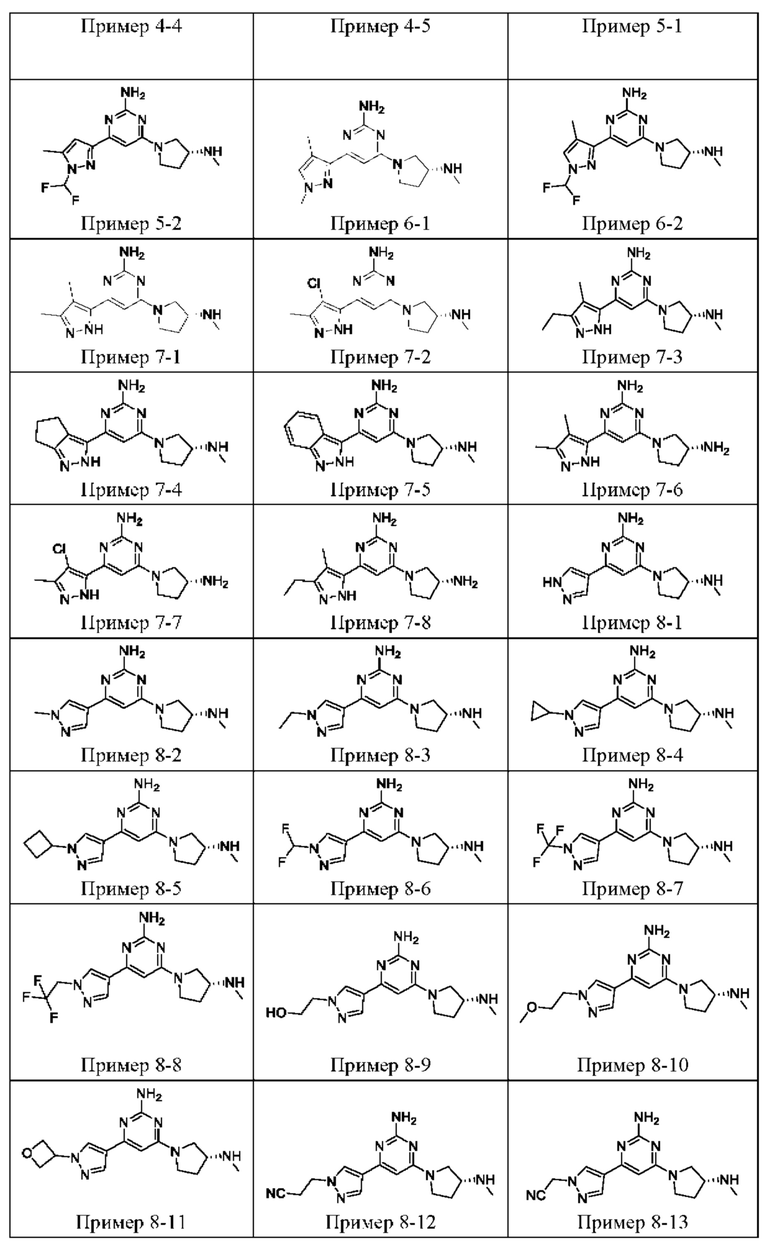
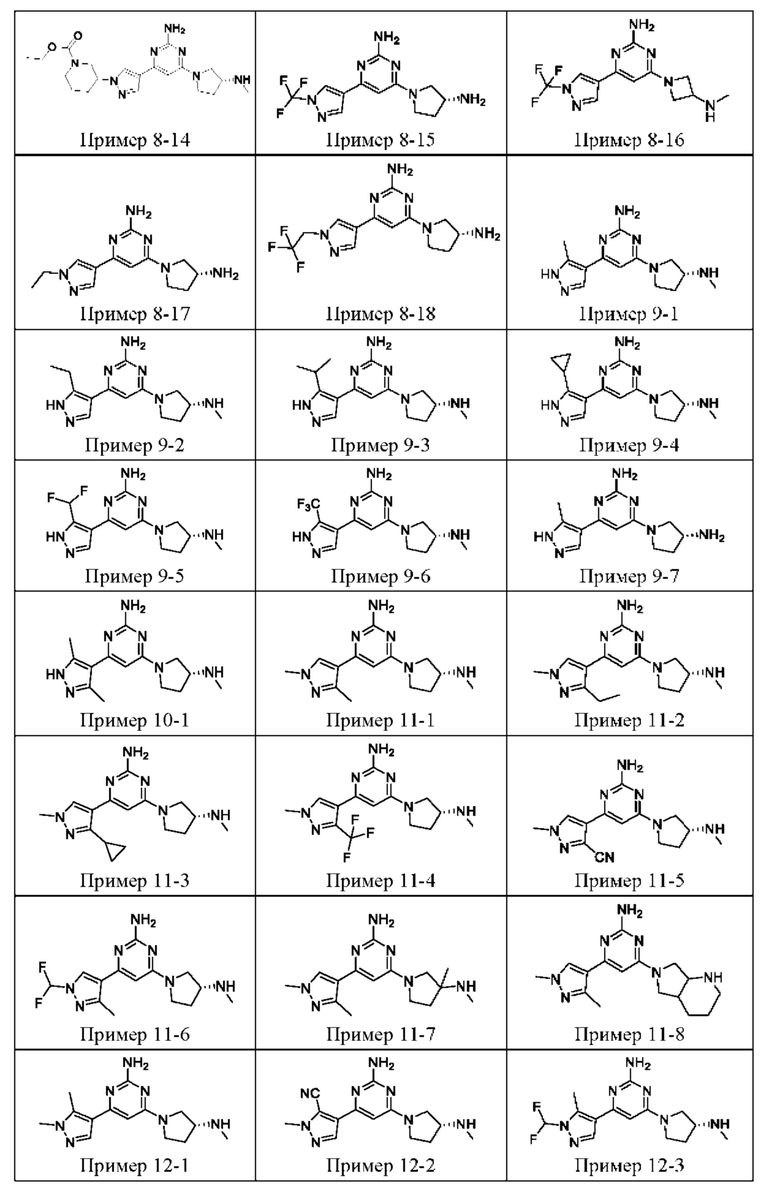
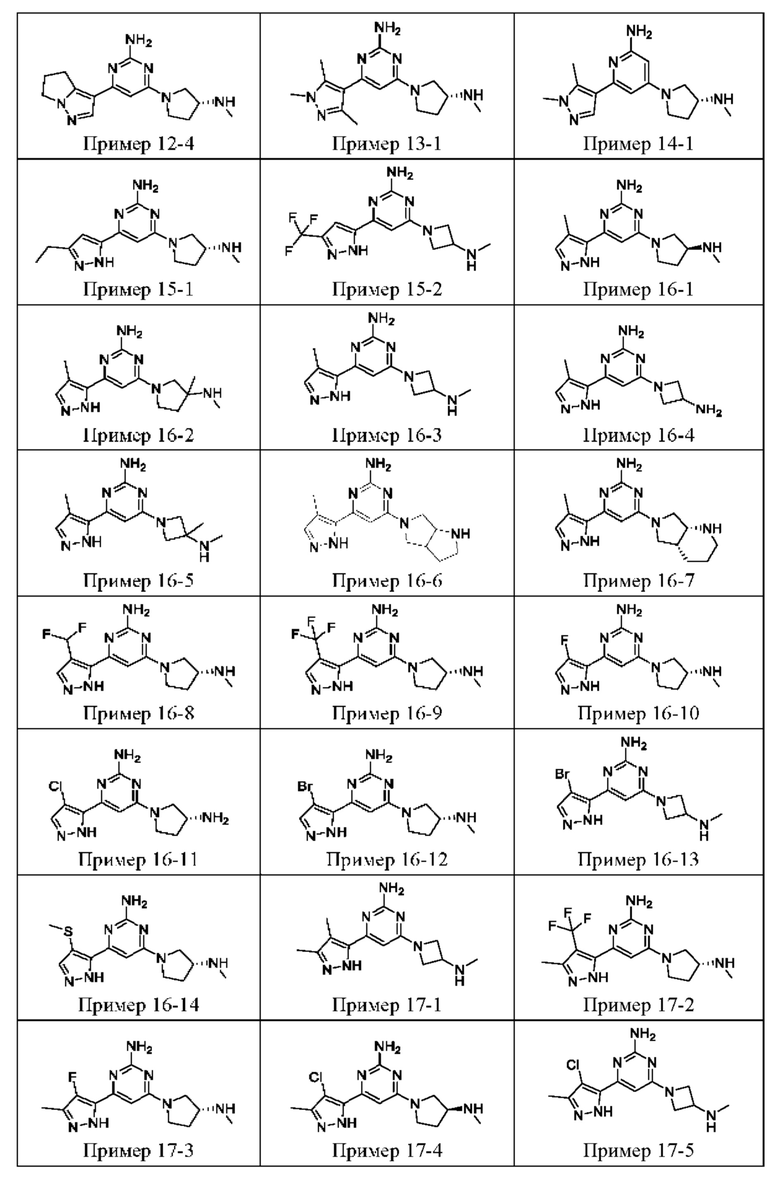
Общие методики
Если способы получения не указаны, соответствующее промежуточное соединение является коммерчески доступным. Коммерчески доступные реагенты использовали без дополнительной очистки. Комнатная температура (комн. т.) относится к приблизительно 20-27°С. Спектры 1Н ЯМР записывали при 400 МГц на приборе Bruker или Jeol. Значения химического сдвига выражены в миллионных долях (млн-1), т.е. (δ)-значениях. Для обозначения кратности сигналов ЯМР используются следующие сокращения: s = синглет, br = широкий, d = дублет, t = триплет, q = квартет, quint = квинтет, td = триплет дублетов, tt = триплет триплетов, qd = квартет дублетов, ddd = дублет дублетов дублетов, ddt = дублет дублетов триплетов, m = мультиплет. Константы взаимодействия указаны как значения J, измеренные в Гц. Результаты ЯМР и масс-спектроскопии были скорректированы с учетом фоновых пиков. Хроматография относится к колоночной хроматографии, проведенной с использованием силикагеля 60-120 меш и в условиях давления азота (флэш-хроматография). Колоночная хроматография, проводимая с использованием «основного диоксида кремния», относится к использованию силикагеля Biotage® KP-NH. Колоночная хроматография, проводимая в обращенно-фазовых условиях с использованием «диоксида кремния С18», относится к использованию силикагеля Biotage® KP-С18. ТСХ для контроля протекания реакций относится к ТСХ, проведенной с использованием указанной подвижной фазы и силикагеля F254 в качестве стационарной фазы от Merck. Реакции под воздействием микроволнового излучения проводили в микроволновых реакторах Biotage Initiator или СЕМ Discover.
ЖХМС-анализ
ЖХМС-анализ соединений проводили в условиях ионизации распылением в электрическом поле с использованием инструментов и методов, приведенных в таблицах ниже:
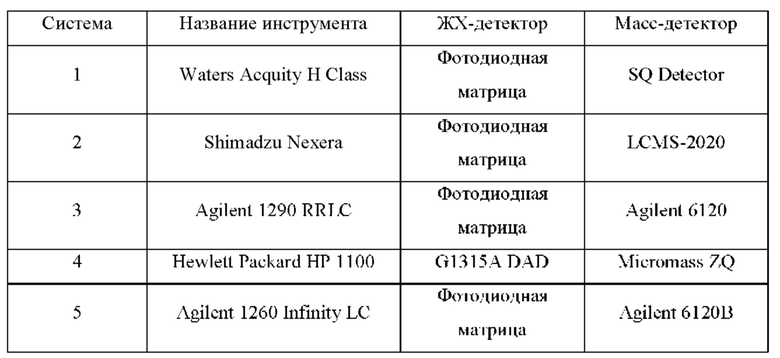
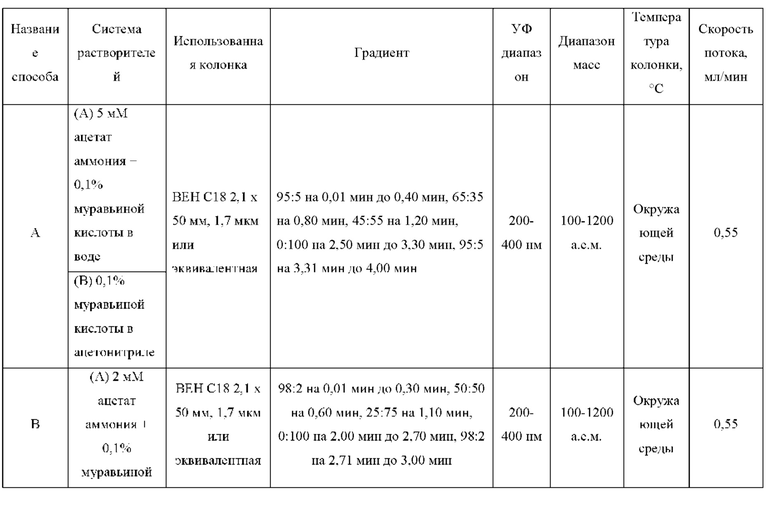
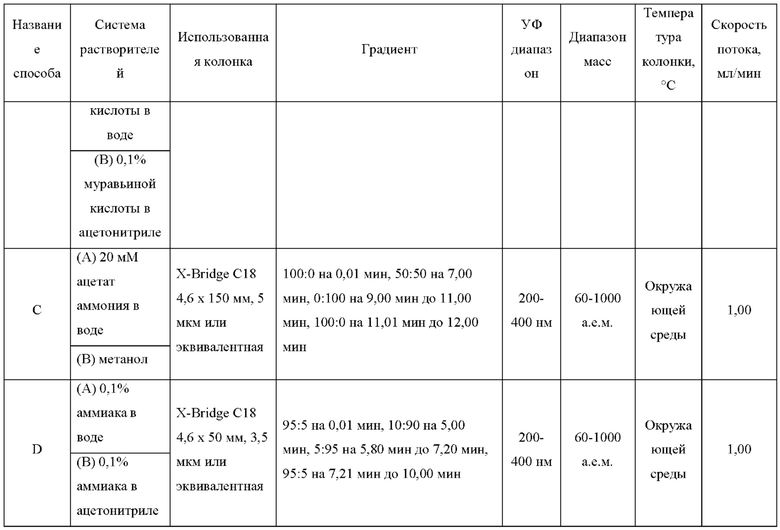
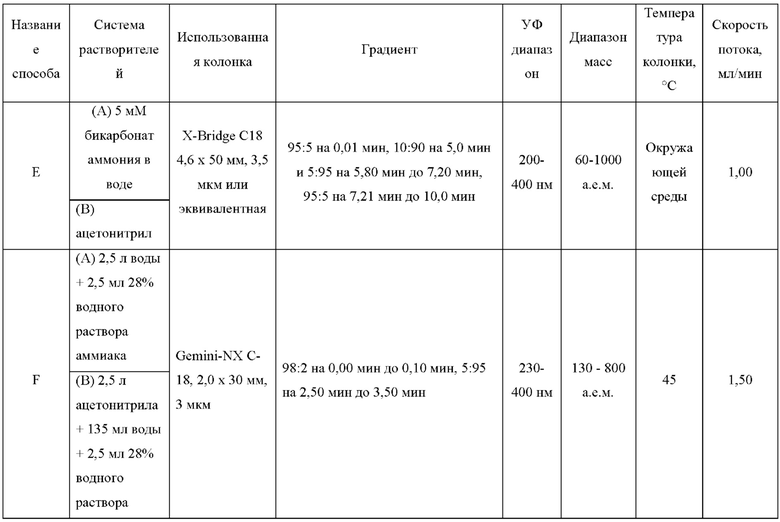
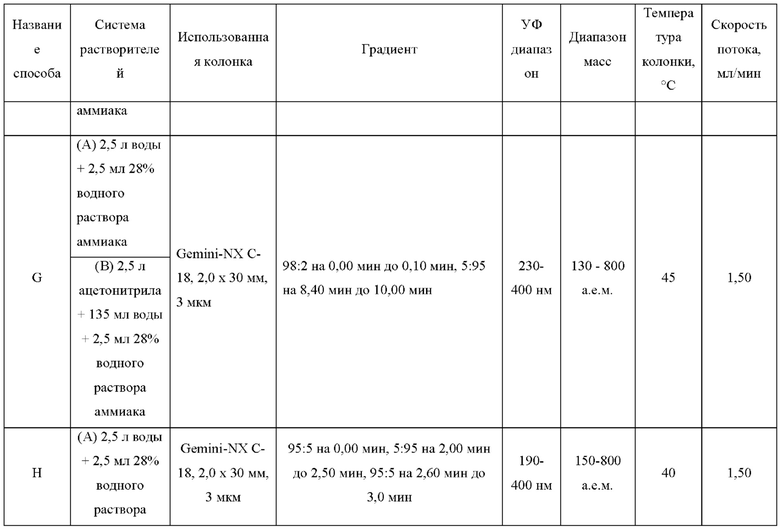
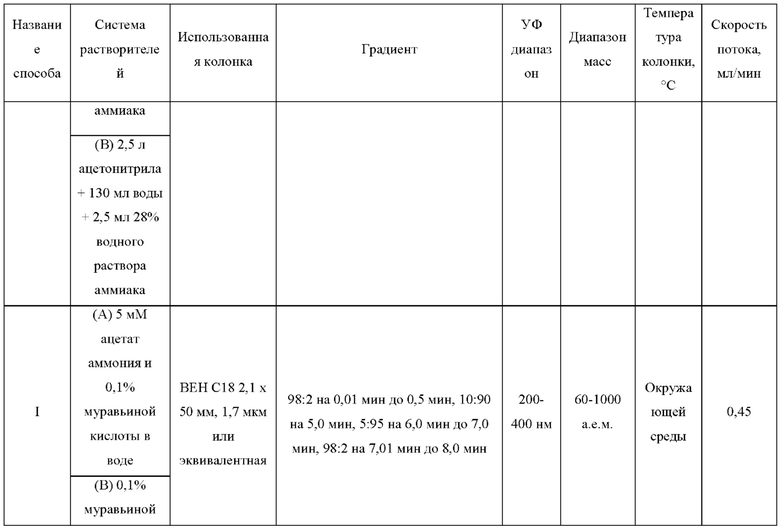
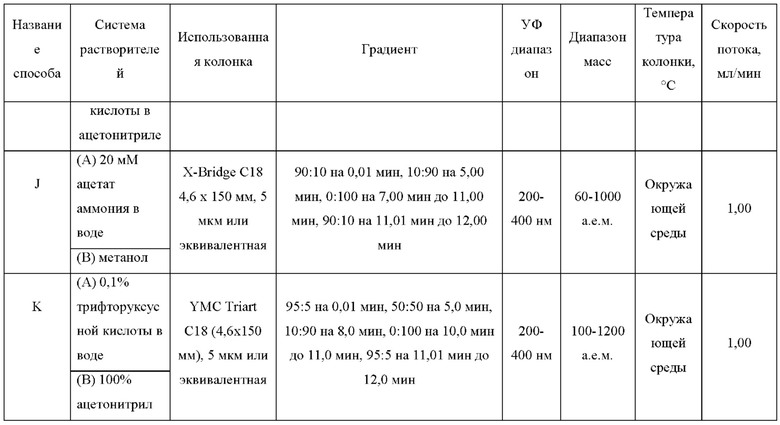
Данные ЖХМС в экспериментальном разделе и в таблицах 2 и 3 представлены в следующем формате: (Инструментальная система, способ): Масса иона, время удерживания, длина волны УФ-детектирования.
Очистка соединений
Окончательную очистку соединений проводили с помощью препаративной обращенно-фазовой ВЭЖХ, хиральной ВЭЖХ или хиральной SFC с использованием инструментов и методов, подробно описанных ниже, где данные представлены в следующем формате: Метод очистки: [фаза (описание колонки, длина колонки × внутренний диаметр, размер частиц), скорость потока растворителя, градиент - указан как % подвижной фазы B в подвижной фазе А (во времени), подвижная фаза (А), подвижная фаза (В)].
Очистка с помощью препаративной ВЭЖХ:
Бинарная система Shimadzu LC-20AP с УФ-детектором SPD-20A
Полупрепаративная ВЭЖХ система Gilson с насосом 321, жидкостным манипулятором GX-271 и Gilson 171 DAD, управляемыми с помощью программного обеспечения Gilson Trilution
Очистка с помощью хиральной ВЭЖХ:
Бинарная система Shimadzu LC-20AP с УФ-детектором SPD-20A
Очистка с помощью хиральной SFC:
Waters SFC 200
Способ очистки А
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 250 × 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 50% (в течение 18 мин), 100% (в течение 2 мин), 100% - 0% (в течение 3 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде (В): ацетонитрил: метанол (50:50)].
Способ очистки В
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 150 × 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 15% (в течение 21 мин), 15% - 15% (в течение 3 мин), 100% (в течение 2 мин), 100% - 0% (в течение 2 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде (В): 100% ацетонитрил].
Способ очистки С
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 40% - 60% (в течение 8,7 мин), 60% (в течение 0,5 мин), 60% - 100% (в течение 0,2 мин), 100% (в течение 1 мин), 100% - 40% (в течение 0,2 мин), 40% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки D
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 250 × 50 мм, 5 мкм), 65 мл/мин, градиент 0% - 25% (в течение 30 мин), 25% - 25% (в течение 1 мин), 100% (в течение 2 мин), 100% - 0% (в течение 5 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде (В): 100% ацетонитрил].
Способ очистки Е
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 60% - 100% (в течение 8,7 мин), 100% (в течение 1,7 мин), 100% - 60% (в течение 0,2 мин), 60% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки F
Препаративная ВЭЖХ: [обращенно-фазовая (Kromasil eternity С-18, 250 × 21,2 мм, 5 мкм), 15 мл/мин, градиент 7% - 20% (в течение 27 мин), 100% (в течение 2 мин), 100% - 7% (в течение 3 мин), подвижная фаза (А): 0,1% трифторуксусной кислоты в воде, (В): 100% ацетонитрил].
Способ очистки G
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-8, 150 × 19 мм, 5 мкм), 16 мл/мин, градиент 0% - 25% (в течение 20 мин), 25% - 25% (в течение 3 мин), 100% (в течение 2 мин), 100% - 0% (в течение 5 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде (В): 100% ацетонитрил].
Способ очистки Н
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 40% - 70% (в течение 8,7 мин), 70% (в течение 0,5 мин), 70% - 100% (в течение 0,2 мин), 100% (в течение 1 мин), 100% - 40% (в течение 0,2 мин), 40% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки I
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 5% - 95% (в течение 8,7 мин), 95% (в течение 0,5 мин), 95% - 100% (в течение 0,2 мин), 100% (в течение 1 мин), 100% - 5% (в течение 0,2 мин), 5% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки J
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 5% - 35% (в течение 8,7 мин), 35% (в течение 0,5 мин), 35% - 100% (в течение 0,2 мин), 100% (в течение 1 мин), 100% - 5% (в течение 0,2 мин), 5% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки K
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 60% - 100% (в течение 8,7 мин), 100% (в течение 1,7 мин), 100% - 60% (в течение 0,2 мин), 60% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Способ очистки L
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 250 × 19 мм, 5 мкм), 10 мл/мин, градиент 0% - 20% (в течение 30 мин), 20% - 20% (в течение 9 мин), 100% (в течение 3 мин), 100% - 0% (в течение 8 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде (В): 100% ацетонитрил].
Способ очистки М
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 150 × 19 мм, 5 мкм), 13 мл/мин, градиент 0% - 35% (в течение 18 мин), 100% (в течение 3 мин), 100% - 0% (в течение 4 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде, (В): 100% ацетонитрил].
Способ очистки N
Хиральная ВЭЖХ: [нормально-фазовая (CHIRALPAK IG, 250 × 21 мм, 5 мкм), 18 мл/мин, изократический (А:В) 70:30 (в течение 40 мин), подвижная фаза (А): 0,1% диэтиламина в гексане, (В): 0,1% диэтиламина в изопропаноле : метаноле (50:50)].
Способ очистки О
Препаративная ВЭЖХ: [обращенно-фазовая (X-BRIDGE С-18, 150 × 19 мм, 5 мкм), 15 мл/мин, градиент 10% - 35% (в течение 20 мин), 35% (в течение 3 мин), 100% (в течение 2 мин), 100% - 10% (в течение 3 мин), подвижная фаза (А): 5 мМ бикарбонат аммония + 0,1% аммиака в воде, (В): ацетонитрил : метанол (1:1)].
Способ очистки Р
SFC: [(CHIRALPAK 1С, 250 × 21 мм, 5 мкм), 80 мл/мин, изократический (А:В) 65:35 (в течение 23 мин), подвижная фаза (А): 100% жидкий СО2, (В): 0,1% диэтиламина в смеси изопропанол : ацетонитрил (50:50)].
Способ очистки Q
Препаративная ВЭЖХ: [обращенно-фазовая (Gemini-NX С-18, 100 × 30 мм, 5 мкм), 30 мл/мин, градиент 30% - 60% (в течение 8,7 мин), 60% (в течение 0,5 мин), 60% - 100% (в течение 0,2 мин), 100% (в течение 1 мин), 100% - 30% (в течение 0,2 мин), 30% (в течение 0,9 мин), подвижная фаза (А): 2,5 л воды + 5 мл 28% раствора аммиака в воде, (В): 100% ацетонитрил].
Сокращенные обозначения
CDI = карбонилдиимидазол
DAST = трифторид диэтиламиносеры
ДХМ = дихлорметан
DIPEA = N,N-диизопропилэтиламин
ESI = ионизация распылением в электрическом поле
EtOAc = этилацетат
ч = час(ы)
H2O = вода
HCl = хлороводород, соляная кислота
ВЭЖХ = высокоэффективная жидкостная хроматография
IPA = пропан-2-ол
ЖХ = жидкостная хроматография
MeCN = ацетонитрил
МеОН = метанол
мин = минута(ы)
МС = масс-спектрометрия
нм = нанометр(ы)
ЯМР = ядерный магнитный резонанс
POCl3 = оксихлорид фосфора
Комн. т. = комнатная температура
насыщ. = насыщенный
SFC = сверхкритическая жидкостная хроматография
TEA = триэтиламин
TFA = трифторуксусная кислота
ТГФ = тетрагидрофуран
ТСХ = тонкослойная хроматография.
Синтез промежуточных соединений:
Путь 1
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 12, 5-бром-3-(дифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола
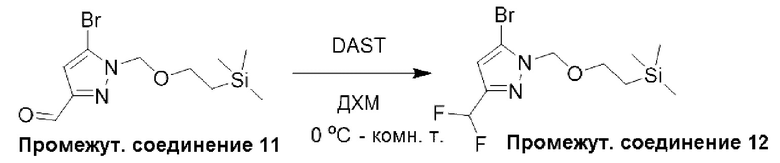
К раствору 5-бром-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-карбальдегида (промежуточное соединение 11) (800 мг, 2,60 ммоль), растворенного в ДХМ (8,7 мл) и охлажденного до 0°С, по каплям добавляли трифторид диэтиламиносеры (0,86 мл, 6,51 ммоль). Затем реакционную смесь перемешивали при 0°С в течение 23 часов, давая ей медленно нагреться до комнатной температуры. Затем реакцию останавливали при 0°С, добавляя насыщенный раствор бикарбоната натрия, и полученную смесь экстрагировали с использованием ДХМ (х 2). Объединенные органические фазы фильтровали через фазовый сепаратор и концентрировали при пониженном давлении. Неочищенный продукт затем очищали с помощью колоночной хроматографии (диоксид кремния, 0-50% дихлорметана в петролейном эфире) с получением 5-бром-3-(дифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола (промежуточное соединение 12) (716 мг, 84%).
Данные для промежуточного соединения 12 приведены в таблице 2.
Путь 2
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 15, 3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
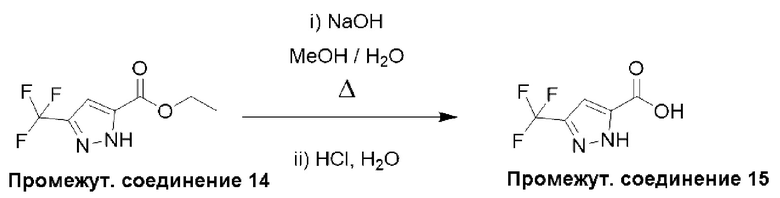
Этил-3-(трифторметил)-1Н-пиразол-5-карбоксилат (промежуточное соединение 14) (1,50 г, 7,21 ммоль) растворяли в МеОН (15 мл) и по каплям добавляли водный NaOH (2 М, 10 мл). Полученную реакционную смесь перемешивали при 70°С в течение 14 часов, затем концентрировали в вакууме. Остаток растворяли в воде (5 мл), подкисляли водной HCl (1 М) до рН = 2-3 и экстрагировали этилацетатом (3×15 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который растирали с пентаном (декантируя растворитель) и сушили в высоком вакууме с получением 3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты (промежуточное соединение 15) (1,30 г, 100%) в виде твердого вещества.
Данные для промежуточного соединения 15 приведены в таблице 2.
Путь 3
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 22, 4-этил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола
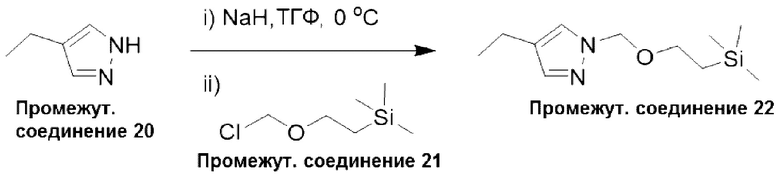
Суспензию гидрида натрия в минеральном масле (60%, 624 мг, 15,6 ммоль) добавляли небольшими порциями к раствору 4-этил-1Н-пиразола (промежуточное соединение 20) (1,0 г, 10,4 ммоль) в ТГФ (5,2 мл), предварительно охлажденного до 0°С. Реакционную смесь перемешивали при 0°С в течение 45 минут перед добавлением по каплям (2-(хлорметокси)этил)триметилсилана (промежуточное соединение 21) (2,0 мл, 11,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем останавливали реакцию при 0°С добавлением воды и экстрагировали этилацетатом. Водный слой дополнительно экстрагировали этилацетатом (х 2), и объединенные органические фазы промывали рассолом, фильтровали через фазовый сепаратор и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (диоксид кремния, 0-10% этилацетата в петролейном эфире) с получением 4-этил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола (промежуточное соединение 22), (1,50 г, 63%).
Данные для промежуточного соединения 22 приведены в таблице 2.
Путь 4
Типичная процедура получения пиримидинов, показанная на примере получения промежуточного соединения 26, трет-бутил-(R)-(1-(6-хлор-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата
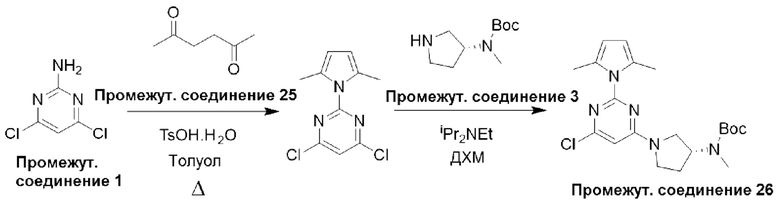
Смесь 4,6-дихлорпиримидин-2-амина (промежуточное соединение 1) (18,54 г, 113 ммоль), гексан-2,5-диона (промежуточное соединение 25) (26,5 мл, 226 ммоль) и моногидрата п-толуолсульфоновой кислоты (215 ммоль, 1,13 ммоль) в сухом толуоле (500 мл) нагревали с обратным холодильником в условиях Дина-Старка в течение 17 часов (в течение ночи). Реакционную смесь охлаждали до комнатной температуры и промывали насыщенным раствором бикарбоната натрия. Водный слой экстрагировали EtOAc, и объединенные органические фазы промывали водой и рассолом, фильтровали через фазовый сепаратор и концентрировали. Затем остаток фильтровали через слой диоксида кремния, промывали ДХМ и концентрировали с получением 4,6-дихлор-2-(2,5-диметил-1H-пиррол-1-ил)пиримидина (24,9 г, 91%).
1Н ЯМР (400 МГц, хлороформ-d) δ 7,19 (s, Ш), 5,91 (s, 2Н), 2,42 (s, 6Н).
К раствору 4,6-дихлор-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидина (3,0 г, 12,4 ммоль), растворенного в ДХМ (20 мл), добавляли N,N-диизопропилэтиламин (6,48 мл, 37,2 ммоль), затем трет-бутил-(R)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) (2,61 г, 13,0 ммоль), растворенный в ДХМ (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, затем останавливали реакцию добавлением водного раствора HCl (1 М) и экстрагировали с использованием ДХМ (х 2). Объединенные органические фазы фильтровали через фазовый сепаратор и концентрировали при пониженном давлении. Затем остаток очищали с помощью колоночной хроматографии (диоксид кремния, 0-25% этилацетат в петролейном эфире) с получением трет-бутил-(R)-(1-(6-хлор-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (промежуточное соединение 26) (3,87 г, 77%).
Данные для промежуточного соединения 26 приведены в таблице 2.
Путь 5
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 31, 1,5-диметил-1Н-пиразол-3-карбоновой кислоты
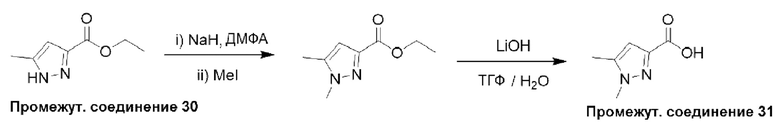
Этил-5-метил-1H-пиразол-3-карбоксилат (промежуточное соединение 30) (2,0 г, 0,01 моль) растворяли в ДМФА (15 мл) и добавляли суспензию гидрида натрия в минеральном масле (60%, 1,5 г, 0,03 моль) по частям в атмосфере азота при 0°С. Смесь перемешивали в течение 1 часа, затем по каплям добавляли метилиодид (3,6 г, 0,02 моль) в атмосфере азота и перемешивали полученную смесь при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали, и остаток разделяли между Н2О (25 мл) и EtOAc (15 мл). Водный слой дополнительно экстрагировали EtOAc (3×15 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, от 0 до 3% МеОН в ДХМ) с получением этил-1,5-диметил-1H-пиразол-3-карбоксилата (2,0 г, 96%) в виде смолы.
ЖХМС (система 1, способ В): m/z 169 (М+Н)+ (ESI +ve), при 1,42 мин, 230 нм.
Этил-1,5-диметил-1Н-пиразол-3-карбоксилат (2,0 г, 0,01 моль) и LiOH⋅H2O (1,4 г, 0,03 моль) помещали в ТГФ (5 мл) и воду (2 мл) и перемешивали при 0°С в течение 1 часа. Реакционную смесь разделяли между H2O (25 мл) и EtOAc (15 мл), и органический экстракт отбрасывали. Водный слой подкисляли до рН 1-2, используя водный раствор HCl (1 М), и полученную смесь повторно экстрагировали EtOAc (3×15 мл). Объединенные экстракты сушили (Na2SO4) и удаляли растворитель в вакууме с получением 1,5-диметил-1H-пиразол-3-карбоновой кислоты (промежуточное соединение 31) (1,3 г, 81%) в виде смолы.
Данные для промежуточного соединения 31 приведены в таблице 2.
Путь 6
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 36, 1-(дифторметил)-4-метил-1Н-пиразол-3-карбоновой кислоты
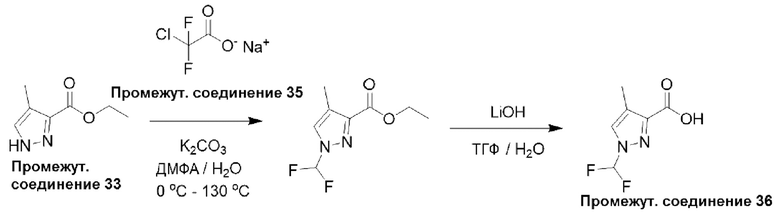
Этил-4-метил-1H-пиразол-3-карбоксилат (промежуточное соединение 33) (1,0 г, 6,49 ммоль) растворяли в ДМФА : H2O (9,0 мл : 1,0 мл) и K2CO3 (3,58 г, 25,9 ммоль) и добавляли 2-хлор-2,2-дифторацетат натрия (промежуточное соединение 35) (3,94 г, 25,9 ммоль) при 0°С, а затем смесь нагревали при 130°С в течение 20 мин. Реакционную смесь охлаждали до комнатной температуры и добавляли ледяную воду. Водный слой экстрагировали EtOAc (3×50 мл), и объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 25% EtOAc в гексанах) с получением этил-1-(дифторметил)-4-метил-1Н-пиразол-3-карбоксилата (325 мг, 25%) в виде твердого вещества.
ЖХМС (система 3, способ D): m/z 205 (М+Н)+ (ESI +ve), при 3,77 мин, 202 нм.
Этил-1-(дифторметил)-4-метил-1Н-пиразол-3-карбоксилат (325 мг, 1,59 ммоль) растворяли в МеОН : H2O (9:1, 10 мл), добавляли LiOH⋅H2O (334 мг, 7,96 моль) при 0°С и перемешивали реакционную смесь при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и добавляли ледяную воду. Смесь нейтрализовали разбавленным водным раствором HCl и экстрагировали водный слой EtOAc (3×50 мл). Объединенные органические экстракты промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением 1-(дифторметил)-4-метил-1Н-пиразол-3-карбоновой кислоты (промежуточное соединение 36) (251 мг, 96%) в виде твердого вещества.
Данные для промежуточного соединения 36 приведены в таблице 2.
Путь 7
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 63, этил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилата
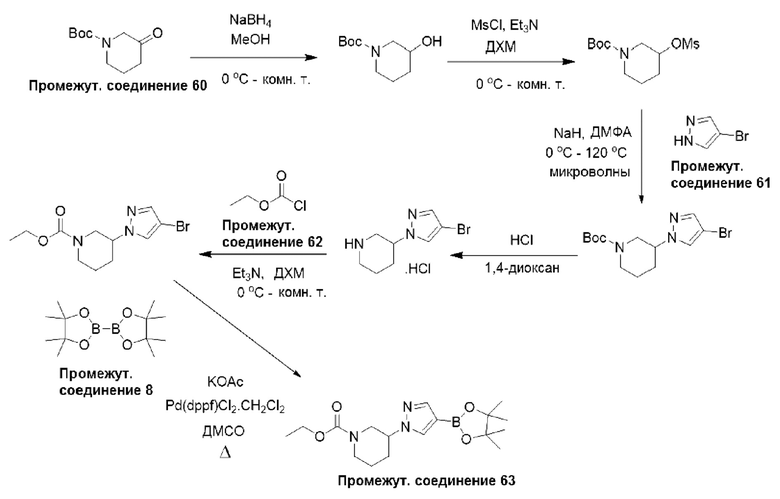
Трет-бутил-3-оксопиперидин-1-карбоксилат (промежуточное соединение 60) (1,30 г, 6,53 ммоль) растворяли в метаноле (20 мл) и по частям добавляли NaBH4 (750 мг, 19,6 ммоль) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 3 часов, затем разделяли между H2O (50 мл) и EtOAc (20 мл). Водный слой дополнительно экстрагировали EtOAc (2×20 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта. Остаток очищали с помощью колоночной хроматографией (нормально-фазовой с силикагелем 60-120 меш, 0-50% EtOAc в гексанах) с получением трет-бутил-3-гидроксипиперидин-1-карбоксилата (1,00 г, 76%) в виде твердого вещества.
ЖХМС (система 1, способ В): m/z 202 (М+Н)+ (ESI +ve), при 1,50 мин, 202 нм.
Трет-бутил-3-гидроксипиперидин-1-карбоксилат (1,00 г, 4,98 ммоль) и TEA (2,1 мл, 14,9 ммоль) растворяли в ДХМ (15 мл) при 0°С, добавляли метансульфонилхлорид (850 мг, 7,45 ммоль) по каплям при 0°С и перемешивали полученную смесь при комнатной температуре в течение 3 часов. Затем реакционную смесь разделяли между Н2О (50 мл) и ДХМ (20 мл), и водный слой дополнительно экстрагировали ДХМ (2×20 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографией (нормально-фазовой с силикагелем 60-120 меш, 0-30% EtOAc в гексанах) с получением трет-бутил-3-((метилсульфонил)окси)пиперидин-1-карбоксилата (1,03 г, 94%) в виде смолы.
ЖХМС (система 1, способ В): m/z 280 (М+Н)+ (ESI +ve), при 1,61 мин, 202 нм.
4-Бром-1Н-пиразол (промежуточное соединение 61) (526 мг, 3,58 ммоль) растворяли в ДМФА (10 мл), добавляли суспензию гидрида натрия в минеральном масле (60%, 260 мг, 6,45 ммоль) при 0°С и перемешивали полученную смесь в течение 30 мин. Трет-бутил-3-((метилсульфонил)окси)пиперидин-1-карбоксилат (1,00 г, 3,58 ммоль) в виде раствора в ДМФА (5 мл) добавляли по каплям при 0°С и перемешивали смесь при 120°С в течение 1 часа с использованием нагревания с помощью микроволнового излучения. Реакционную смесь разделяли между Н2О (50 мл) и EtOAc (20 мл), и водный слой дополнительно экстрагировали EtOAc (2×20 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-3% МеОН в ДХМ) с получением трет-бутил-3-(4-бром-1Н-пиразол-1-ил)пиперидин-1-карбоксилата (1,10 г, 93%) в виде смолы.
ЖХМС (система 1, способ В): m/z 274/276 (М-56+Н)+ (ESI +ve), при 1,82 мин, 230 нм.
Трет-бутил-3-(4-бром-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (700 мг, 2,12 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 М, 15 мл) при 0°С и перемешивали полученную смесь при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, а затем остаток растирали с диэтиловым эфиром (2×10 мл) с получением соли 3-(4-бром-1Н-пиразол-1-ил)пиперидина гидрохлорида (400 мг, 71%) в виде твердого вещества.
ЖХМС (система 2, способ Е): m/z 230/232 (М+Н)+ (ESI +ve), при 2,54 мин, 230 нм.
Соль 3-(4-бром-1Н-пиразол-1-ил)пиперидина гидрохлорид (500 мг, 2,17 ммоль) и TEA (0,90 мл, 6,52 ммоль) растворяли в ДХМ (15 мл) при 0°С и по каплям добавляли этилхлорформиат (промежуточное соединение 62) (350 мг, 3,26 ммоль) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 3 часов, затем разделяли между Н2О (20 мл) и ДХМ (10 мл). Водный слой дополнительно экстрагировали ДХМ (2×10 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-2% МеОН в ДХМ) с получением этил-3-(4-бром-1Н-пиразол-1-ил)пиперидин-1-карбоксилата (400 г, 61%) в виде смолы.
ЖХМС (система 1, способ В): m/z 302/304 (М+Н)+ (ESI +ve), при 1,67 мин, 233 нм.
Этил-3-(4-бром-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (400 мг, 1,32 ммоль), бис(пинаколато)диборон (промежуточное соединение 8) (400 мг, 1,59 ммоль) и ацетат калия (450 мг, 4,63 ммоль) растворяли в ДМСО (5 мл) в атмосфере азота, и полученный раствор дегазировали в течение 15 мин. Добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (CAS: 95464-05-4) (378 мг, 0,46 ммоль), и нагревали смесь при 90°С в течение 16 часов. Затем реакционную смесь разделяли между H2O (25 мл) и EtOAc (15 мл), и водный слой дополнительно экстрагировали EtOAc (2×15 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-2% МеОН в ДХМ) с получением этил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилата (промежуточное соединение 63) (200 г, 43%) в виде смолы.
Данные для промежуточного соединения 63 приведены в таблице 2.
Путь 8
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 71, 4-бром-3-этил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразола
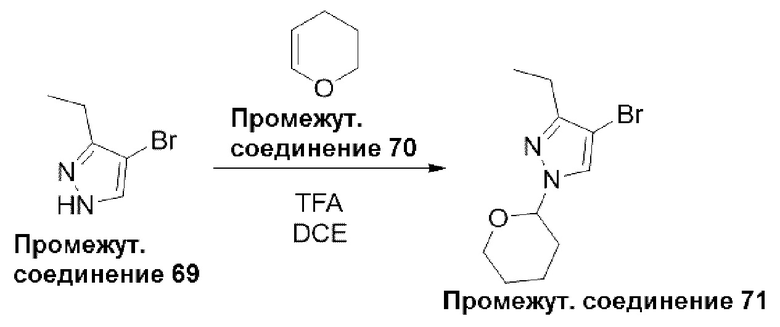
4-Бром-3-этил-1H-пиразол (промежуточное соединение 69) (500 мг, 2,8 ммоль) растворяли в 1,2-дихлорэтане (5 мл) и добавляли 3,4-дигидропиран (промежуточное соединение 70) (482 мг, 5,7 ммоль). Затем добавляли трифторуксусную кислоту (2-3 капли) и перемешивали полученную смесь при комнатной температуре в течение 24 часов. Растворитель выпаривали и остаток разделяли между этилацетатом (25 мл) и водой (15 мл). Органический слой отделяли, сушили (Na2SO4) и упаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (силикагель 60-120 меш, 0-20% этилацетата в гексане) с получением 4-бром-3-этил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразола (промежуточное соединение 71) (700 мг, 97%) в виде смолы.
Данные для промежуточного соединения 71 приведены в таблице 2.
Путь 9
Типичная процедура получения пирролидинов, показанная на примере получения промежуточного соединения 88, бензилметил-(3-метилпирролидин-3-ил)карбамата гидрохлорида
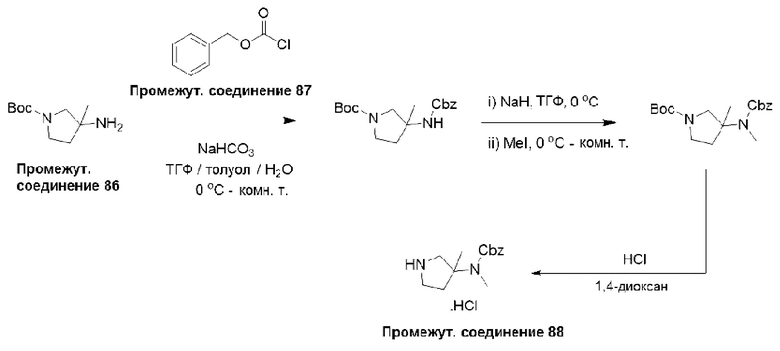
Трет-бутил-3-амино-3-метилпирролидин-1-карбоксилат (промежуточное соединение 86) (600 мг, 3,00 ммоль) растворяли в ТГФ (8 мл) и добавляли раствор NaHCO3 (504 мг, 6,00 ммоль) в воде (8 мл). Смесь охлаждали до 0°С и добавляли бензилхлорформиат (промежуточное соединение 87) в виде раствора в толуоле (50%, 1,1 мл, 3,30 ммоль), и перемешивали полученную смесь при 25°С в течение 2 часов. Затем реакционную смесь разделяли между H2O (30 мл) и этилацетатом (20 мл), и водный слой дополнительно экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали растиранием с пентаном с получением трет-бутил-3-(((бензилокси)карбонил)амино)-3-метилпирролидин-1-карбоксилата (900 мг, 90%) в виде смолы.
ЖХМС (система 3, способ D): m/z 333 (М-Н)- (ESI -ve), при 4,68 мин, 202 нм.
Трет-бутил-3-(((бензилокси)карбонил)амино)-3-метилпирролидин-1-карбоксилат (900 мг, 2,69 ммоль) растворяли в ТГФ (15 мл) и охлаждали раствор до 0°С. Добавляли суспензию гидрида натрия в минеральном масле (60%, 323 мг, 8,08 ммоль) и перемешивали реакционную смесь при 0°С в течение 30 мин. Добавляли метилиодид (573 мг, 4,04 ммоль) при 0°С и перемешивали полученную реакционную смесь при 25°С в течение 4 часов. Затем смесь разделяли между H2O (40 мл) и EtOAc (25 мл), и водный слой дополнительно экстрагировали EtOAc (2×25 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали растиранием с пентаном с получением трет-бутил-3-(((бензилокси)карбонил)(метил)амино)-3-метилпирролидин-1-карбоксилата (910 мг, 97%) в виде смолы.
ЖХМС (система 3, способ D): m/z 349 (М+Н)+ (ESI +ve), при 5,05 мин, 202 нм.
Трет-бутил-3-(((бензилокси)карбонил)(метил)амино)-3-метилпирролидин-1-карбоксилат (900 мг, 2,59 ммоль) растворяли в 1,4-диоксане (5 мл) и охлаждали до 0°С. В атмосфере азота добавляли раствор HCl в 1,4-диоксане (4 М, 10 мл) и перемешивали полученную смесь при комнатной температуре в течение 6 часов. Реакционную смесь концентрировали и очищали неочищенный продукт в виде соли растиранием с пентаном (2×2 мл) с получением бензилметил-(3-метилпирролидин-3-ил)карбамата гидрохлорида (промежуточное соединение 88) (640 мг, 100%) в виде смолы.
Данные для промежуточного соединения 88 приведены в таблице 2.
Путь 10
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 111, 4-(дифторметил)-1-(4-метоксибензил)-1Н-пиразол-3-карбоновой кислоты
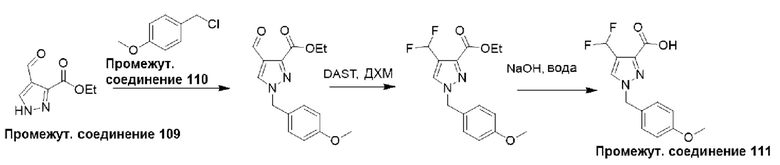
Этил-4-формил-1Н-пиразол-3-карбоксилат (промежуточное соединение 109) (1 г, 5,95 ммоль) растворяли в ДМФА (10 мл) с последующим добавлением 1-(хлорметил)-4-метоксибензола (промежуточное соединение 110) (1,02 г, 6,54 ммоль) при комнатной температуре. Затем к этому добавляли карбонат калия (904 мг, 6,54 ммоль) и иодид калия (10 мг) и перемешивали реакционную смесь при 80°С в течение 16 часов. Реакционную смесь разделяли между Н2О (250 мл) и EtOAc (500 мл), и водный слой дополнительно экстрагировали EtOAc (2×150 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный продукт очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-50% EtOAc в гексане) с получением этил-4-формил-1-(4-метоксибензил)-1Н-пиразол-3-карбоксилата (1,0 г, 58%).
ЖХМС (система 1, способ В): m/z 289 (М+Н)+ (ESI +ve), при 1,61 мин, 275 нм.
Этил-4-формил-1-(4-метоксибензил)-1Н-пиразол-3-карбоксилат (0,8 г, 2,77 ммоль) растворяли в ДХМ (8 мл). Реакционную смесь охлаждали до -70°С и затем к ней по каплям добавляли трифторид диэтиламиносеры (1,11 г, 6,94 ммоль). Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. Реакционную смесь разделяли между насыщенным водным раствором NaHCO3 (250 мл) и EtOAc (500 мл). Водный слой дополнительно экстрагировали EtOAc (2×150 мл), и объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный продукт очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-18% EtOAc в гексане) с получением этил-4-(дифторметил)-1-(4-метоксибензил)-1Н-пиразол-3-карбоксилата (0,8 г, 93%).
ЖХМС (система 1, способ В): m/z 311 (М+Н)+ (ESI +ve), при 1,71 мин, 230 нм.
Этил-4-(дифторметил)-1-(4-метоксибензил)-1Н-пиразол-3-карбоксилат (0,8 г, 2,58 ммоль) растворяли в ТГФ (4 мл) и МеОН (4 мл). К этому добавляли водный раствор NaOH (2М, 6,45 мл, 12,9 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Органический растворитель удаляли в вакууме и охлаждали полученный раствор до 10°С. Реакционную смесь подкисляли до рН 2, используя водный 6М раствор HCl, и полученный осадок собирали фильтрованием и сушили в вакууме с получением 4-(дифторметил)-1-(4-метоксибензил)-1Н-пиразол-3-карбоновой кислоты (промежуточное соединение 111) (0,7 г, 96%).
Данные для промежуточного соединения 111 приведены в таблице 2.
Путь 11
Типичная процедура частичного снятия защиты с пиримидинов, показанная на примере получения промежуточного соединения 118, трет-бутил-(R)-(1-(2-амино-6-(4-фтор-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата
Смесь трет-бутил-(R)-(1-(2-(2,5-диметил-1Н-пиррол-1-ил)-6-(4-фтор-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (промежуточное соединение 117) (226 мг, 0,39 ммоль), гидрохлорида гидроксиламина (268 мг, 3,86 ммоль) и триэтиламина (0,06 мл, 0,42 ммоль) в этаноле (8 мл) и воде (4 мл) нагревали при 100°С в течение ночи. Реакционную смесь разбавляли водой и экстрагировали EtOAc (х 3). Объединенные органические экстракты промывали рассолом, пропускали через фазовый сепаратор и концентрировали с получением трет-бутил-(R)-(1-(2-амино-6-(4-фтор-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (промежуточное соединение 118) (189 мг, 96%) в виде смолы.
Данные для промежуточного соединения 118 приведены в таблице 2.
Путь 12
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 121, 4-(метилтио)-1Н-пиразол-3-карбоновой кислоты
Этил-4-амино-1Н-пиразол-3-карбоксилат (промежуточное соединение 120) (4,00 г, 2,57 ммоль) растворяли в ACN (40,0 мл), затем добавляли изопентилнитрит (10,39 мл) с последующим добавлением диметилдисульфида (6,87 мл, 7,73 ммоль) по каплям в атмосфере азота при 0°С, и перемешивали в течение 1 часа. Затем реакционную смесь нагревали при 80°С при перемешивании в течение 16 часов. После полного израсходования исходного вещества реакционную смесь охлаждали до около 15°С и разделяли между Н2О (100 мл) и EtOAc (50 мл), водный слой дополнительно экстрагировали EtOAc (2×50 мл); все органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографией на силикагеле (60-120 меш) и градиентом 0-50% EtOAc в гексанах. Отгоняли растворитель с получением этил-4-(метилтио)-1H-пиразол-3-карбоксилата (3,0 г, 62,5%) в виде желтой смолы.
ЖХМС (система 1, способ В): m/z 187 (М+Н)+ (ESI +ve), при 1,39 мин, 230 нм.
Этил-4-(метилтио)-1Н-пиразол-3-карбоксилат (3,5 г, 1,87 ммоль) растворяли в метаноле (25 мл) с последующим добавлением по каплям 2 н. водного раствора NaOH (28 мл, 5,63 ммоль) и перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали, разбавляли ледяной водой (небольшим количеством), подкисляли разбавленной HCl и перемешивали полученную суспензию еще 20-30 мин. Твердое соединение собирали фильтрацией. Твердое вещество сушили при пониженном давлении с получением 4-(метилтио)-1Н-пиразол-3-карбоновой кислоты (2,5 г, 84,17%) в виде белого твердого вещества.
Данные для промежуточного соединения 121 приведены в таблице 2.
Путь 13
Типичная процедура получения пиразолов, показанная на примере получения промежуточного соединения 127, 4-метокси-5-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-3-карбоновой кислоты.
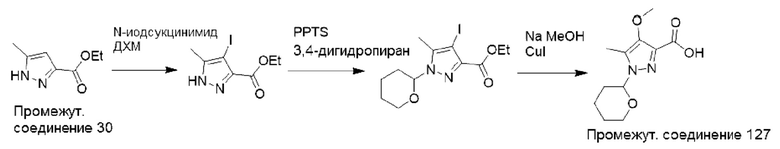
Этал-5-метил-1Н-пиразол-3-карбоксилат (промежуточное соединение 30) (4,00 г, 25,9 ммоль) растворяли в ДХМ (100 мл) с последующим добавлением N-иодсукцинимида (7,09 г, 31,1 ммоль) по частям, и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разделяли между Н2О (60 мл) и EtOAc (30 мл), водный слой дополнительно экстрагировали EtOAc (2×30 мл); объединенные органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель 60-120 меш, 0-4% метанола в ДХМ) с получением этил-4-иод-5-метил-1H-пиразол-3-карбоксилата (6,80 г, 93,53%) в виде бесцветной смолы.
ЖХМС (система 1, способ В): m/z 281 (М+Н)+ (ESI +ve), при 1,49 мин, 229 нм.
Этил-4-иод-5-метил-1Н-пиразол-3-карбоксилат (3,10 г, 11,1 ммоль) и 3,4-дигидро-2Н-пиран (1,39 г, 16,6 ммоль) растворяли в ДХМ (50,0 мл), после чего добавляли п-толуолсульфонат пиридиния (0,28 г, 1,11 ммоль) по частям и перемешивали в течение 16 часов при 40°С. Реакционную смесь разделяли между Н2О (50 мл) и EtOAc (20 мл), водный слой дополнительно экстрагировали EtOAc (2×20 мл), все органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель 60-120 меш, 0-2% метанола в ДХМ) с получением этил-4-иод-5-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-3-карбоксилата (3,20 г, 79,40%) в виде белого твердого вещества.
ЖХМС (система 1, способ В): m/z 365 (М+Н)+ (ESI +ve), при 1,73 мин, 235 нм.
Этил-4-иод-5-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-3-карбоксилат (3,20 г, 8,80 ммоль) и CuI (0,50 г, 2,64 ммоль) добавляли к свежеприготовленному раствору метилата натрия (30,0 мл) и перемешивали при комнатной температуре в течение 16 часов при 80°С.Реакционную смесь фильтровали через целит и фильтрат концентрировали. Концентрированную реакционную смесь выливали в воду (20 мл), подкисляли добавлением 1 н. раствора HCl (рН около 4,0) и экстрагировали 10% МеОН в ДХМ (3×30 мл), все органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме с получением 4-метокси-5-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-3-карбоновой кислоты (2,45 г, 100% мас./мас.) в виде желтой смолы.
Данные для промежуточного соединения 127 приведены в таблице 2.
Общие методики синтеза:
Путь А
Типичная процедура получения пиримидинов, показанная на примере получения примера 1-1, (R)-4-(3-(метиламино)пирролидин-1-ил)-6-(1H-пиразол-5-ил)пиримидин-2-амина
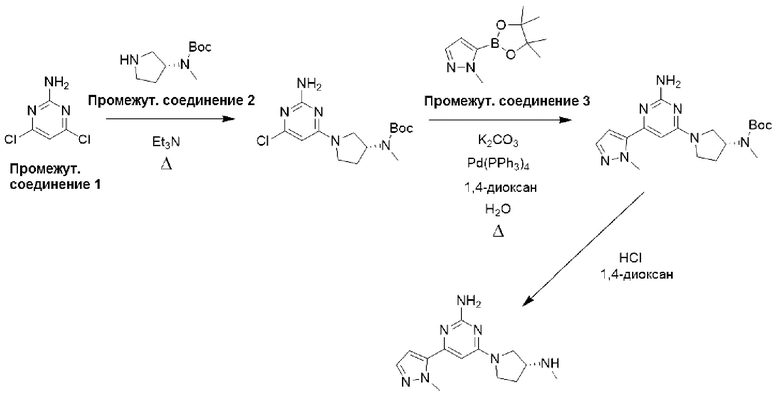
4,6-Дихлорпиримидин-2-амин (промежуточное соединение 1) (250 мг, 1,52 ммоль), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (промежуточное соединение 2) (354 мг, 1,82 ммоль) и K3PO4 (970 мг, 4,50 ммоль) растворяли в 1,4-диоксане (5 мл) и воде (0,5 мл) в атмосфере азота и дегазировали в течение 20 минут. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (CAS: 95464-05-4) (124 мг, 0,15 ммоль), в атмосфере азота, и перемешивали полученную смесь при 90°С в течение 16 часов. Реакционную смесь разделяли между H2O (25 мл) и EtOAc (15 мл), и водный слой дополнительно экстрагировали EtOAc (2×15 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-6% МеОН в ДХМ) с получением 4-хлор-6-(1Н-пиразол-5-ил)пиримидин-2-амина (75 мг, 25%) в виде твердого вещества.
ЖХМС (система 1, способ В): m/z 196 (М+Н)+ (ESI +ve), при 1,38 мин, 240 нм.
4-Хлор-6-(1Н-пиразол-5-ил)пиримидин-2-амин (75 мг, 0,38 ммоль) и трет-бутил-(11)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) (76 мг, 0,38 ммоль) растворяли в триэтиламине (3 мл) и перемешивали при 90°С в течение 16 часов. Реакционную смесь концентрировали и затем разделяли между H2O (25 мл) и EtOAc (15 мл). Водный слой дополнительно экстрагировали EtOAc (2×15 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-3% МеОН в ДХМ) с получением трет-бутил-(R)-(1-(2-амино-6-(1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (75 мг, 54%) в виде твердого вещества.
ЖХМС (система 1, способ В): m/z 360 (М+Н)+ (ESI +ve), при 1,44 мин, 220 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (75 мг, 0,20 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 М, 2 мл) в атмосфере азота при 0°С и перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали и растирали с диэтиловым эфиром (2×5 мл) с получением неочищенного продукта, который очищали способом очистки А с получением (R)-4-(3-(метиламино)пирролидин-1-ил)-6-(1Н-пиразол-5-ил)пиримидин-2-амина, примера 1-1 (21 мг, 39%), в виде бесцветной смолы.
Данные для примера 1-1 приведены в таблице 3.
Путь В
Типичная процедура получения пиримидинов, показанная на примере получения примера 1-2, (R)-4-(1-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дигидрохлорида
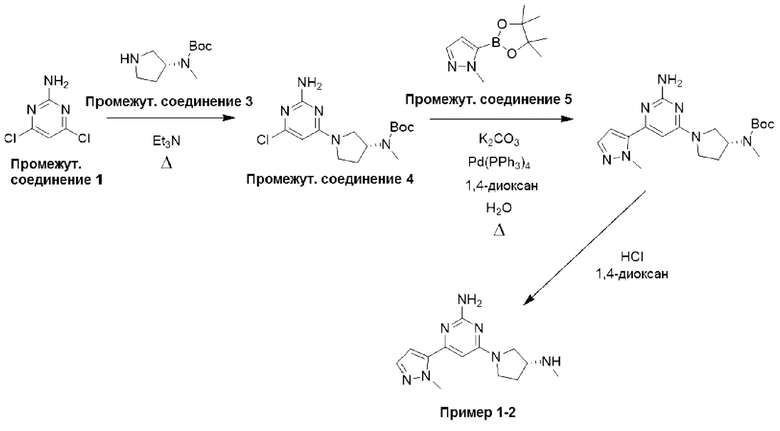
4,6-дихлорпиримидин-2-амин (промежуточное соединение 1) (5,5 г, 33,5 ммоль) и трет-бутил-(R)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) (7,3 г, 40,2 ммоль) растворяли в триэтиламине (13 мл) и перемешивали полученный раствор при 90°С в течение 3 часов. В процессе реакции продукт выпадал в осадок, его отфильтровывали, промывали водой и сушили в вакууме с получением трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (промежуточное соединение 4) (10,1 г, 92%) в виде грязно-белого твердого вещества.
Данные для промежуточного соединения 4 приведены в таблице 2.
Трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (150 мг, 0,46 ммоль), 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (промежуточное соединение 5) (115 мг, 0,55 ммоль) и K2CO3 (126 мг, 0,92 ммоль) растворяли в 1,4-диоксане (5 мл) и воде (2 мл) в атмосфере азота и дегазировали в течение 20 минут. В атмосфере азота добавляли тетракис(трифенилфосфин)палладий (0) (CAS: 95464-05-4) (26 мг, 0,02 ммоль) и перемешивали полученную смесь при 90°С в течение 16 часов. Реакционную смесь разделяли между H2O (25 мл) и EtOAc (15 мл), и водный слой дополнительно экстрагировали EtOAc (2×15 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с силикагелем 60-120 меш, 0-3% МеОН в ДХМ) с получением трет-бутил-(R)-(1-(2-амино-6-(1-метил-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (100 мг, 58%) в виде смолы.
ЖХМС (система 1, способ A): m/z 374 (М+Н)+ (ESI +ve), при 1,40 мин, 296 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1-метил-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (100 мг, 0,27 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 М, 4 мл) в атмосфере азота и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь концентрировали и затем растирали с диэтиловым эфиром (2×10 мл) с получением (R)-4-(1-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дигидрохлорида, примера 1-2 (59 мг, 81%), в виде твердого вещества.
Данные для примера 1-2 приведены в таблице 3.
Путь С
Типичная процедура получения пиримидинов, показанная на примере получения примера 2-1, (R)-4-(1-метил-1Н-пиразол-3-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
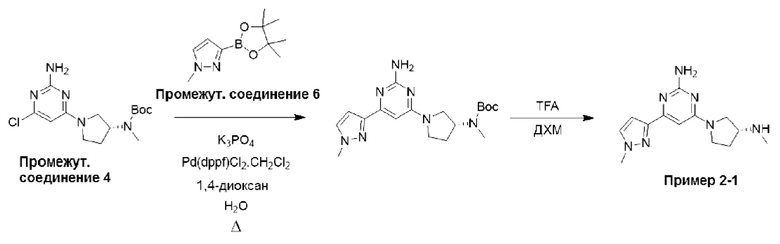
Трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (150 мг, 0,45 ммоль), 1-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (промежуточное соединение 6) (114 мг, 0,54 ммоль) и K3PO4 (291 мг, 0,13 ммоль) растворяли в 1,4-диоксане (12 мл) и воде (3 мл) в атмосфере азота и дегазировали в течение 20 минут. Затем в атмосфере азота добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (CAS: 95464-05-4) (37 мг, 0,04 ммоль), и перемешивали полученную смесь при 90°С в течение 16 часов. Реакционную смесь разделяли между Н2О (40 мл) и EtOAc (25 мл), и водный слой дополнительно экстрагировали EtOAc (3×25 мл). Органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с активированным оксидом алюминия, 2-4% МеОН в ДХМ) с получением трет-бутил-(R)-(1-(2-амино-6-(1-метил-1Н-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (169 мг, 99%) в виде твердого вещества.
ЖХМС (система 2, способ Е): m/z 374 (М+Н)+ (ESI +ve), при 3,31 мин, 254 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1-метил-1H-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (169 мг, 0,45 ммоль) растворяли в смеси TFA (2 мл) и ДХМ (4 мл) в атмосфере азота и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали, а затем растирали с пентаном (2×2 мл) с получением неочищенного продукта, который очищали способом очистки В с получением (R)-4-(1-метил-1H-пиразол-3-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 2-1 (94 мг, 76%), в виде твердого вещества.
Данные для примера 2-1 приведены в таблице 3.
Путь D
Типичная процедура получения пиримидинов, показанная на примере получения примера 2-2, (R)-4-(1-(дифторметил)-1Н-пиразол-3-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дигидрохлорида
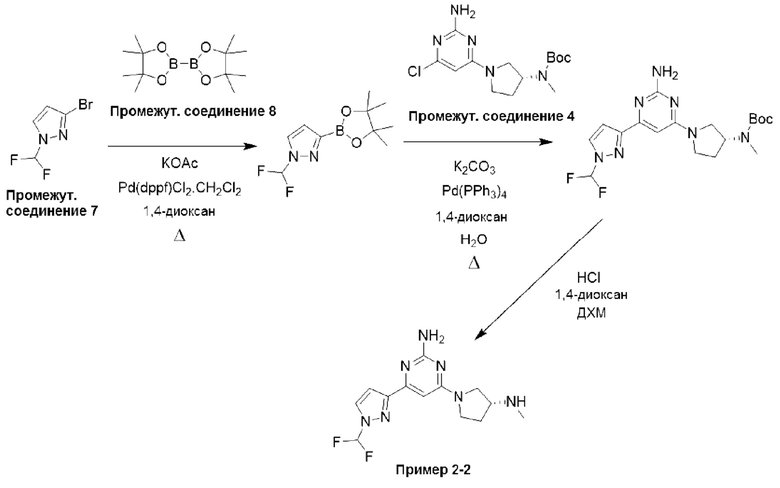
Смесь [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекс с дихлорметаном (CAS: 95464-05-4) (61 мг, 0,08 ммоль), бис(пинаколато)диборона (промежуточное соединение 8) (267 мг 1,05 ммоль), 3-бром-1-(дифторметил)-1H-пиразола (промежуточное соединение 7) (148 мг, 0,75 ммоль) и ацетата калия (294 мг, 3 ммоль) в 1,4-диоксане (2,5 мл) нагревали до 110°С и выдерживали при этой температуре в течение ночи. Реакционную смесь концентрировали, и продукт использовали непосредственно на следующей стадии синтеза без дополнительного выделения или очистки. Предполагаемый выход 100%.
ЖХМС (система 4, способ F): m/z 245 (М+Н)+ (ESI +ve), при 0,14 мин, 254 нм.
Смесь карбоната калия (138 мг, 1,0 ммоль), тетракис(трифенилфосфин)палладия (0) (CAS: 95464-05-4) (58 мг, 0,05 ммоль), 1-(дифторметил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразола (183 мг, 0,75 ммоль, предполагаемый выход с предыдущей стадии) и трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (промежуточное соединение 4) (164 мг, 0,50 ммоль) в 1,4-диоксане (2,2 мл) и воде (0,26 мл) нагревали до 110°С и выдерживали при этой температуре в течение ночи. Затем реакционную смесь разделяли между EtOAc (5 мл) и водой (5 мл), и фазы разделяли. Водную фазу дополнительно экстрагировали EtOAc (3×5 мл), и все органические фазы объединяли и концентрировали с получением неочищенного продукта, который очищали способом очистки C с получением трет-бутил-(R)-(1-(2-амино-6-(1-(дифторметил)-1Н-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (83 мг, 41%) в виде твердого вещества.
ЖХМС (система 4, способ F): m/z 410 (М+Н)+ (ESI +ve), при 2,06 мин, 254 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1-(дифторметил)-1Н-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (83 мг, 0,20 ммоль) растворяли в ДХМ (2 мл), добавляли раствор HCl в 1,4-диоксане (4 М, 0,25 мл, 1,01 ммоль) и перемешивали полученную смесь при комнатной температуре в течение ночи. По прошествии этого времени выделяли белый осадок с получением 4-[1-(дифторметил)пиразол-3-ил]-6-[(3R)-3-(метиламино)пирролидин-1-ил]пиримидин-2-амина дигидрохлорида, примера 2-2 (69 мг, 98%).
Данные для примера 2-2 приведены в таблице 3.
Путь Е
Типичная процедура получения пиримидинов, показанная на примере получения примера 3-1, (R)-4-(3-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
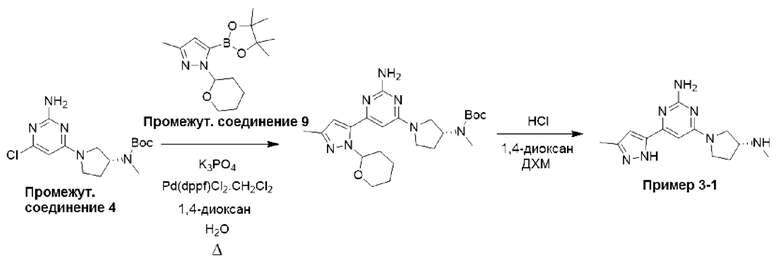
Трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (1,0 мг, 3,0 ммоль), 3-метил-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (промежуточное соединение 9) (1,06 мг, 3,63 ммоль) и K3PO4 (1,90 мг, 9,0 ммоль) растворяли в 1,4-диоксане (16 мл) и воде (4 мл) в атмосфере азота и дегазировали в течение 20 минут. Затем в атмосфере азота добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (CAS: 95464-05-4) (245 мг, 0,3 ммоль), и перемешивали полученную смесь при 90°С в течение 16 часов. Реакционную смесь разделяли между Н2О (50 мл) и EtOAc (30 мл), и водный слой дополнительно экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с нейтральным оксидом алюминия, 9% МеОН в ДХМ) с получением трет-бутил-((3R)-1-(2-амино-6-(3-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (1,2 мг, 86%) в виде твердого вещества.
ЖХМС (система 2, способ Е): m/z 458 (М+Н)+ (ESI +ve), при 3,96 мин, 313 нм.
Трет-бутил-((3R)-1-(2-амино-6-(3-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (1,2 г, 0,26 ммоль) растворяли в ДХМ (20 мл) и охлаждали до 0°С. По каплям добавляли раствор HCl в 1,4-диоксане (4 М, 25 мл) и перемешивали полученную реакционную смесь при 25°С в течение 2 часов. Растворители удаляли в вакууме и остаток упаривали совместно с толуолом (2×30 мл) с получением неочищенного продукта, который очищали способом очистки D с получением (R)-4-(3-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 3-1 (520 мг, 73%), в виде твердого вещества.
Данные для примера 3-1 приведены в таблице 3.
Путь F
Типичная процедура получения пиримидинов, показанная на примере получения примера 3-3, (R)-4-(3-(дифторметил)-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
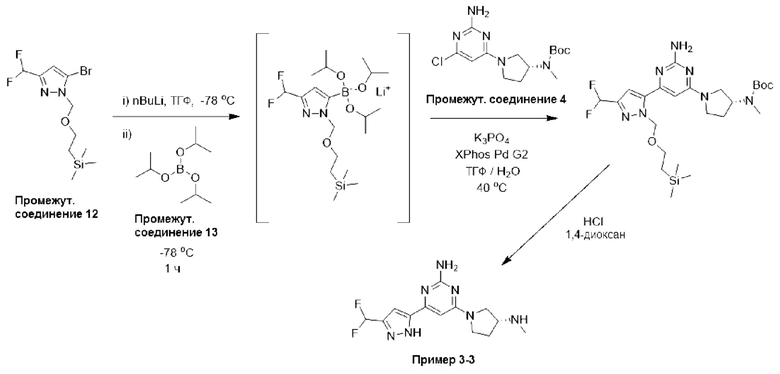
В продуваемый азотом сосуд для микроволновой обработки добавляли 5-бром-3-(дифторметил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол (промежуточное соединение 12) (100 мг, 0,31 ммоль), растворенный в ТГФ (0,40 мл), и охлаждали раствор до -78°С в атмосфере азота. Затем к раствору по каплям добавляли раствор н-бутиллития в гексанах (2,5 М, 0,13 мл, 0,34 ммоль) перед добавлением по каплям триизопропилбората (промежуточное соединение 13) (0,08 мл, 0,34 ммоль). Затем реакционную смесь перемешивали при -78°С в течение 1 часа. Затем к реакционной смеси добавляли водный раствор K3PO4 (0,5 М, 0,79 мл, 0,40 ммоль), а затем трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (70 мг, 0,21 ммоль) и предкатализатор XPhos Pd G2 (CAS: 1310584-14-5) (7 мг, 0,009 ммоль). Затем сосуд для микроволновой обработки герметично закрывали и нагревали до 40°С (обычное нагревание) при перемешивании в течение 18 часов. Реакционную смесь добавляли к раствору воды (20 мл) и насыщенного водного раствора NH4Cl (0,4 мл) и экстрагировали этилацетатом. Затем водный слой повторно экстрагировали этилацетатом (2 раза). Объединенные органические экстракты фильтровали через фазовый сепаратор и концентрировали при пониженном давлении, а остаток очищали с помощью колоночной хроматографии (основный диоксид кремния, 0-50% этилацетата в петролейном эфире) с получением неочищенного продукта (37 мг) в виде твердого вещества. Твердое вещество дополнительно очищали способом очистки Е с получением трет-бутил-(R)-(1-(2-амино-6-(3-(дифторметил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (9 мг, 5%).
ЖХМС (система 4, способ F): m/z 540 (М+Н)+ (ESI +ve), при 2,70 мин, 254 нм.
К раствору трет-бутил-(R)-(1-(2-амино-6-(3-(дифторметил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (8 мг, 0,01 ммоль), растворенного в 1,4-диоксане (0,55 мл), добавляли раствор HCl в 1,4-диоксане (4 М, 0,05 мл, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов, затем концентрировали при пониженном давлении и остаток упаривали совместно с толуолом. Затем неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (диоксид кремния С18, 0-10% MeCN в 0,2% NH3 в воде) с получением (R)-4-(3-(дифторметил)-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 3-3 (2 мг, 47%).
Данные для примера 3-3 приведены в таблице 3.
Путь G
Типичная процедура получения пиримидинов, показанная на примере получения примера 3-4, (R)-4-(3-(дифторметил)-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дитрифторацетата
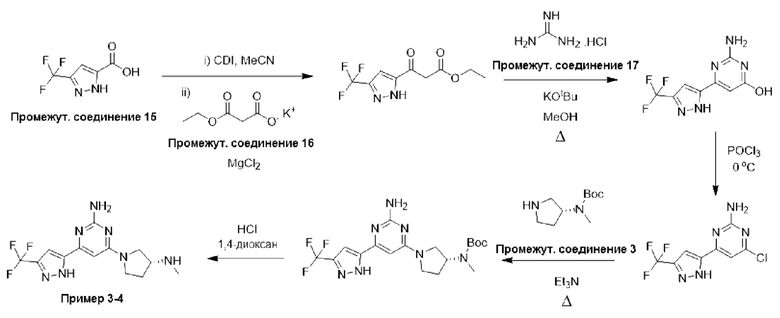
3-(Трифторметил)-1Н-пиразол-5-карбоновую кислоту (промежуточное соединение 15) (1,30 г, 7,20 ммоль) растворяли в ацетонитриле (20 мл), по частям добавляли CDI (1,40 г, 8,66 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 2 часов. Затем добавляли 3-этокси-3-оксопропаноат калия (промежуточное соединение 16) (1,22 г, 7,20 ммоль) и MgCl2 (823 мг, 7,20 ммоль) и перемешивали полученную реакционную смесь при комнатной температуре в течение 14 часов. Смесь концентрировали в вакууме, остаток разделяли между H2O (40 мл) и EtOAc (30 мл) и слои разделяли. Водный слой дополнительно экстрагировали EtOAc (2×30 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Неочищенный продукт очищали растиранием с пентаном (декантируя растворитель) и сушили в высоком вакууме с получением этил-3-оксо-3-(3-(трифторметил)-1Н-пиразол-5-ил)пропаноата (1,20 г, 67%) в виде смолы.
ЖХМС (система 2, способ Е): m/z 249 (М-Н)- (ESI -ve), при 4,47 мин, 241 нм.
Этил-3-оксо-3-(3-(трифторметил)-1Н-пиразол-5-ил)пропаноат (1,20 г, 4,80 ммоль) и гидрохлорид гуанидина (промежуточное соединение 17) (1,37 г, 14,4 ммоль) растворяли в метаноле (20 мл) в атмосфере азота при 0°С и перемешивали в течение 10 мин. Медленно добавляли трет-бутилат калия (806 мг, 7,20 ммоль) в атмосфере азота и перемешивали полученную реакционную смесь при 60°С в течение 16 часов. Органический растворитель удаляли в вакууме с получением неочищенного продукта, который очищали растиранием с пентаном (декантируя растворитель) и сушили в высоком вакууме с получением 2-амино-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-4-ола (2,0 г, неочищенный) в виде смолы.
ЖХМС (система 2, способ Е): m/z 246 (М+Н)+ (ESI +ve), при 3,47 мин, 237 нм.
Смесь 2-амино-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-4-ола (2,0 г, 8,16 ммоль) и POCI3 (5 мл) перемешивали при 0°С в течение 18 часов. Реакционную смесь выливали в смесь льда и водного раствора NaHCO3, затем разделяли между Н2О (50 мл) и EtOAc (40 мл) и разделяли фазы. Водную фазу дополнительно экстрагировали EtOAc (2×40 мл), все органические слои объединяли, сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с активированным оксидом алюминия, 20-30% МеОН в ДХМ) с получением 4-хлор-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-2-амина (350 мг, 16%) в виде смолы.
ЖХМС (система 2, способ Е): m/z 264/266 (М+Н)+ (ESI +ve), при 4,57 мин, 239 нм.
4-Хлор-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-2-амин (200 мг, 0,76 ммоль) растворяли в триэтиламине (5 мл) и добавляли трет-бутил-(И)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) (228 мг, 1,14 ммоль). Полученную реакционную смесь перемешивали при 90°С в течение 6 часов, затем разделяли между H2O (40 мл) и EtOAc (30 мл) и разделяли фазы. Водный слой дополнительно экстрагировали EtOAc (2×30 мл), объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с активированным оксидом алюминия, 5-10% МеОН в EtOAc) с получением трет-бутил-(R)-(1-(2-амино-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (315 мг, 97%) в виде смолы.
ЖХМС (система 2, способ Е): m/z 428 (М+Н)+ (ESI +ve), при 4,13 мин, 243 нм.
Трет-бутил-(R)-(1-(2-амино-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (310 мг, 0,73 ммоль) растворяли в 1,4-диоксане (3 мл) и охлаждали раствор до 0°С. Добавляли раствор HCl в 1,4-диоксане (4 М, 8 мл) и полученную реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Реакционную смесь концентрировали в вакууме и растирали остаток с пентаном (2×3 мл) с получением неочищенного продукта в виде соли с HCl. Неочищенную соль с HCl очищали способом очистки F с получением соли (R)-4-(3-(метиламино)пирролидин-1-ил)-6-(3-(трифторметил)-1Н-пиразол-5-ил)пиримидин-2-амина дитрифторацетата, примера 3-4 (60 мг, 19%), в виде смолы.
Данные для примера 3-4 приведены в таблице 3.
Путь Н
Типичная процедура получения пиримидинов, показанная на примере получения примера 4-1, (R)-4-(4-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
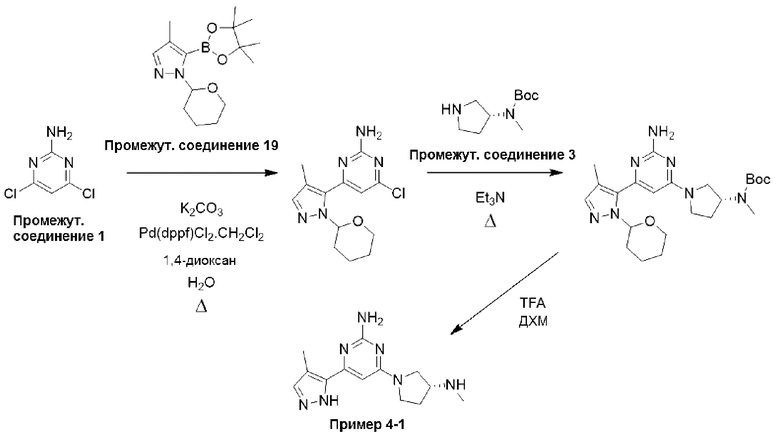
4,6-Дихлорпиримидин-2-амин (промежуточное соединение 1) (250 мг, 1,52 ммоль), 4-метил-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (промежуточное соединение 19) (443 мг, 1,52 ммоль) и K2CO3 (629 мг, 4,56 ммоль) растворяли в 1,4-диоксане (5 мл) и воде (5 мл) в атмосфере азота и дегазировали в течение 20 минут. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (CAS: 95464-05-4) (124 мг, 0,15 ммоль), в атмосфере азота, и полученную смесь перемешивали при 90°С в течение 16 часов. Реакционную смесь разделяли между Н2О (40 мл) и EtOAc (25 мл), и водный слой дополнительно экстрагировали EtOAc (3×25 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с активированным Al2O3, 30% этилацетата в гексанах) с получением 4-хлор-6-(4-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-2-амина (255 мг, 57%) в виде твердого вещества.
ЖХМС (система 2, способ Е): m/z 294 (М+Н)+ (ESI +ve), при 3,53 мин, 234 нм.
4-Хлор-6-(4-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-2-амин (255 мг, 0,87 ммоль) и трет-бутил-(R)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) растворяли в TEA (4 мл) в атмосфере азота и полученную реакционную смесь нагревали до 130°С в микроволновой печи от СЕМ и перемешивали при этой температуре в течение 12 часов. Затем реакционную смесь разделяли между Н2О (25 мл) и EtOAc (15 мл), и водный слой дополнительно экстрагировали EtOAc (2×15 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (нормально-фазовой с нейтральным активированным оксидом алюминия, 1-2% МеОН в ДХМ) с получением трет-бутил-((3R)-1-(2-амино-6-(4-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (101 мг, 25%) в виде смолы.
ЖХМС (система 2, способ Е): m/z 458 (М+Н)+ (ESI +ve), при 3,98 мин, 278 нм.
Трет-бутил-((3R)-1-(2-амино-6-(4-метил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (100 мг, 0,22 ммоль) растворяли в ДХМ (5 мл), добавляли TFA (0,5 мл) при 0°С в атмосфере азота и перемешивали полученную смесь при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали и растирали остаток с пентаном (2×2 мл) с получением неочищенного продукта, который очищали способом очистки G с получением (R)-4-(4-метил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 4-1 (17 мг, 28%), в виде твердого вещества.
Данные для примера 4-1 приведены в таблице 3.
Путь I
Типичная процедура получения пиримидинов, показанная на примере получения примера 4-2, (R)-4-(4-этил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
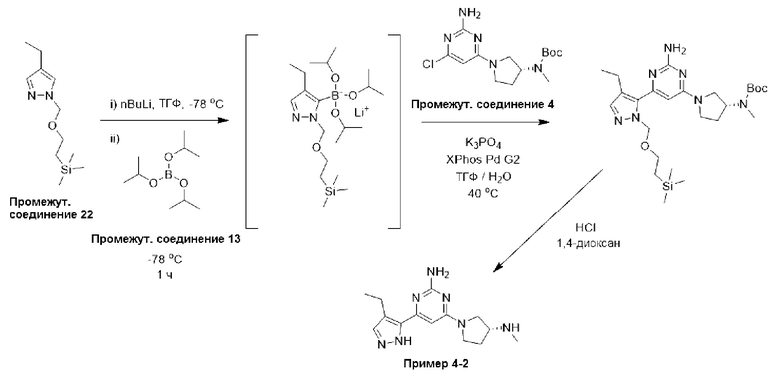
В продуваемый азотом сосуд для микроволновой обработки добавляли 4-этил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол (промежуточное соединение 22) (500 мг, 2,21 ммоль), растворенный в ТГФ (2,9 мл), и охлаждали раствор до -78°С. Затем к этому раствору добавляли раствор н-бутиллития в гексанах (2,5 М, 0,97 мл, 2,43 ммоль) по каплям в течение 10 минут, а затем добавляли триизопропилборат (промежуточное соединение 13) (0,56 мл, 2,43 ммоль) аналогичным образом по каплям. Реакционную смесь перемешивали при -78°С в течение 1 часа, затем добавляли водный раствор K3PO4 (0,5 М, 5,74 мл, 2,87 ммоль), а затем трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (217 мг, 0,66 ммоль) и предкатализатор XPhos Pd G2 (CAS: 1310584-14-5) (52 мг, 0,04 ммоль). Затем сосуд для микроволновой обработки герметично закрывали и нагревали до 40°С (обычное нагревание) при перемешивании в течение 19 часов. Реакционную смесь добавляли к раствору воды (49 мл) и насыщенного водного раствора NH4Cl (1 мл) и экстрагировали этилацетатом. Затем водный слой повторно экстрагировали этилацетатом (2×50 мл). Затем объединенные органические экстракты фильтровали через фазовый сепаратор, концентрировали при пониженном давлении и очищали остаток с помощью колоночной хроматографии (диоксид кремния, 0-100% этилацетата в петролейном эфире) с получением трет-бутил-(R)-(1-(2-амино-6-(4-этил-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (87 мг, 25%).
ЖХМС (система 4, способ F): m/z 518 (М+Н)+ (ESI +ve), при 2,58 мин, 254 нм.
К раствору трет-бутил-(R)-(1-(2-амино-6-(4-этил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (87 мг, 0,17 ммоль), растворенного в 1,4-диоксане (4 мл), добавляли раствор HCl в 1,4-диоксане (4 М, 1,26 мл, 5,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин, затем концентрировали при пониженном давлении и остаток упаривали совместно с толуолом. Остаток очищали способом очистки Н с получением (R)-4-(4-этил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 4-2 (16 мг, 33%).
Данные для примера 4-2 приведены в таблице 3.
Путь J
Типичная процедура получения пиримидинов, показанная на примере получения примера 4-3, (R)-4-(4-хлор-1H-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
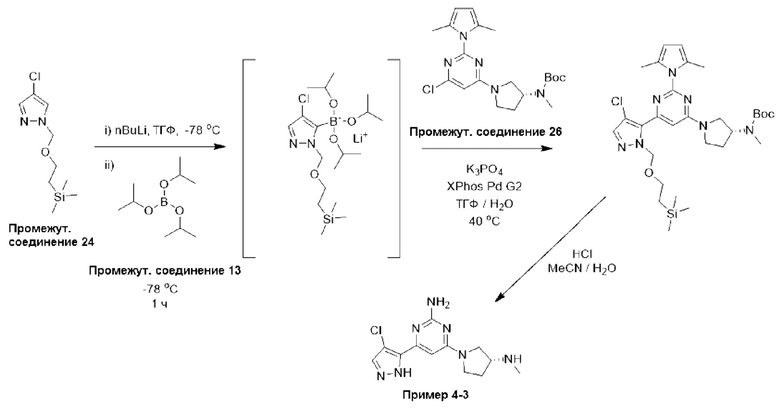
В продуваемый азотом сосуд для микроволновой обработки добавляли 4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (промежуточное соединение 24) (646 мг, 2,78 ммоль), растворенный в ТГФ (3,7 мл). Раствор охлаждали до -78°С и по каплям добавляли раствор н-бутиллития в гексанах (2,5 М, 1,22 мл, 3,05 ммоль) в течение 10 минут перед добавлением триизопропилбората (промежуточное соединение 13) (0,7 мл, 3,05 ммоль), добавленного аналогичным образом по каплям. Реакционную смесь перемешивали при -78°С в течение 1 часа. Затем к реакционной смеси добавляли водный раствор K3PO4 (0,5 М, 7,22 мл, 3,61 ммоль), а затем трет-бутил-(R)-(1-(6-хлор-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 26) (338 мг, 0,83 ммоль) и предкатализатор XPhos Pd G2 (CAS: 1310584-14-5) (66 мг, 0,08 ммоль). Затем сосуд для микроволновой обработки герметично закрывали и нагревали до 40°С посредством обычного нагревания при перемешивании в течение 1,5 часов. Реакционную смесь добавляли к раствору воды (49 мл) и насыщенного водного раствора NH4Cl (1 мл) и экстрагировали этилацетатом (50 мл). Водный слой дополнительно экстрагировали этилацетатом (2×50 мл), и объединенные органические фазы фильтровали через фазовый сепаратор и концентрировали при пониженном давлении. Затем остаток очищали с помощью колоночной хроматографии (диоксид кремния, 0-25% этилацетата в петролейном эфире) с получением трет-бутил-(R)-(1-(6-(4-хлор-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)-2-(2,5-диметил-1H-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (440 мг, 87%).
ЖХМС (система 4, способ F): m/z 602/604 (М+Н)+ (ESI +ve), при 3,13 мин, 254 нм.
К раствору трет-бутил-(R)-(1-(6-(4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (50 мг, 0,08 ммоль), растворенного в MeCN (0,83 мл), добавляли водный раствор HCl (4 М, 1,25 мл, 5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов и при 40°С в течение 2 часов. Параллельно проводили идентичную реакция в том же масштабе, где реакционную смесь перемешивали при комнатной температуре в течение ночи. Две реакционные смеси объединяли и концентрировали при пониженном давлении. Остаток упаривали совместно с толуолом для удаления следов воды, а затем очищали способом очистки I с получением (R)-4-(4-хлор-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 4-3 (3,7 мг, 8%).
Данные для примера 4-3 приведены в таблице 3.
Путь K
Типичная процедура получения пиримидинов, показанная на примере получения примера 4-4, (R)-4-(4-метокси-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
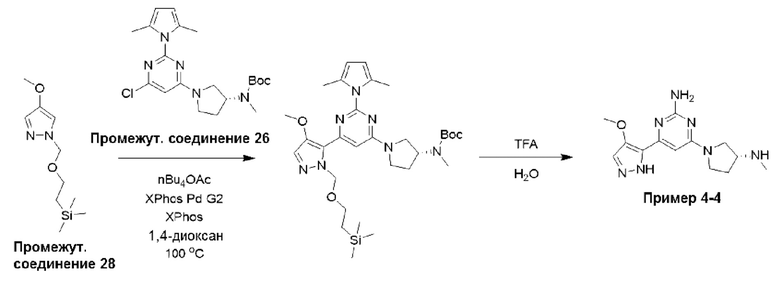
1,4-Диоксан дегазировали, пропуская через жидкость поток азота в течение 15 мин. Сосуд для микроволновой обработки на 5 мл, содержащий якорь магнитной мешалки, продували потоком азота в течение 5 мин, а затем закрывали пробкой. В сосуд для микроволновой обработки добавляли (в указанном порядке): трет-бутил-(R)-(1-(6-хлор-2-(2,5-диметил-1Н-пиррол-1-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 26) (107 мг, 0,26 ммоль), 4-метокси-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол (промежуточное соединение 28) (102 мг, 0,45 ммоль), ацетат тетрабутиламмония (198 мг, 0,66 ммоль) (очень гигроскопичен!), XPhos (CAS: 564483-18-7) (13 мг, 0,03 ммоль) и предкатализатор XPhos Pd G2 (CAS: 1310584-14-5) (9 мг, 0,01 ммоль). Сосуд снова недолго продували потоком азота и добавляли дегазированный 1,4-диоксан (3 мл). Сосуд герметично закрывали и нагревали при перемешивании при 100°С на нагревательной плите в течение 66 часов. Реакцию повторяли в аналогичном масштабе, и два реакционных раствора объединяли, используя этилацетат, и концентрировали на флэш-диоксиде кремния (10 мл) в вакууме. Полученный порошок очищали с помощью флэш-хроматографии (SiO2, 20%-60% EtOAc в изогексане) с получением трет-бутил-(R)-(1-(2-(2,5-диметил-1Н-пиррол-1-ил)-6-(4-метокси-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (197 мг, 63%) в виде масла.
ЖХМС (система 5, способ Н): m/z 598 (М+Н)+ (ESI+ve), при 2,21 мин, 205 нм.
Готовили смесь трифторуксусной кислоты (2,7 мл) и воды (0,3 мл) и добавляли к трет-бутил-(R)-(1-(2-(2,5-диметил-1Н-пиррол-1-ил)-6-(4-метокси-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамату (197 мг, 0,33 ммоль) с получением раствора, который перемешивали при комнатной температуре в атмосфере азота в течение 24 часов. Темно-красный/черный раствор разбавляли равным объемом толуола и концентрировали в вакууме. Остаток упаривали совместно с толуолом с получением темного масла, которое медленно затвердевало при стоянии с образованием красно-черного твердого вещества. Твердое вещество очищали способом очистки J с получением (R)-4-(4-метокси-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 4-4 (17 мг, 17%), в виде твердого вещества.
Данные для примера 4-4 приведены в таблице 3.
Путь L
Типичная процедура получения пиримидинов, показанная на примере получения примера 7-1, (R)-4-(3,4-диметил-1Н-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дигидрохлорида
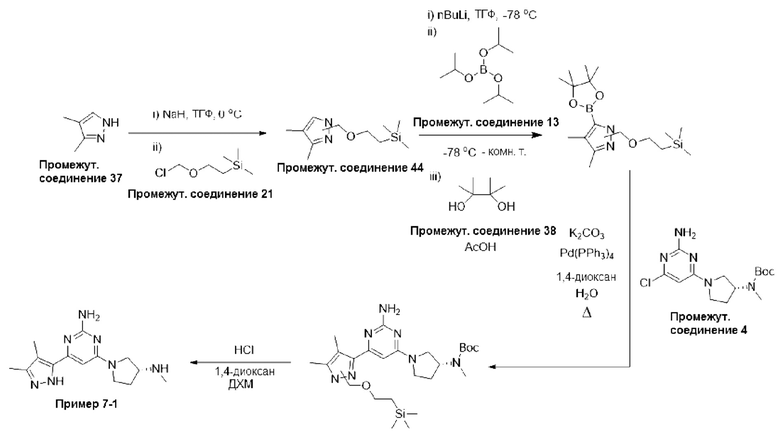
3,4-Диметил-1Н-пиразол (496 мг, 5,0 ммоль) растворяли в ТГФ (20 мл), добавляли суспензию гидрида натрия в минеральном масле (60%, 400 мг, 10 ммоль) и перемешивали реакционную смесь при 0°С в течение 1 часа. Добавляли (2-(хлорметокси)этил)триметилсилан (промежуточное соединение 21) (1,15 мл, 6,5 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь разделяли между водой (25 мл) и EtOAc (40 мл), и водную фазу дополнительно экстрагировали EtOAc (3 × 50 мл). Объединенные органические фазы концентрировали и очищали остаток с помощью колоночной флэш-хроматографии (нормально-фазовой с SiO2, 0-100% EtOAc в изогексане) с получением смеси 3,4-диметил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола и 4,5-диметил-1-((2-(триметилсилил)этокси)метил)-1H-пиразола в соотношении приблизительно 1:1 (промежуточное соединение 44) (1100 мг, 97%) в виде масла.
Данные для промежуточного соединения 44 приведены в таблице 2.
Раствор смеси 3,4-диметил-1-((2-(триметилсилил)этокси)метил)-1H-пиразола и 4,5-диметил-1-((2-(триметилсилил)этокси)метил)-1H-пиразола в соотношении приблизительно 1:1 (промежуточное соединение 44) (453 мг, 2,0 ммоль), растворенной в ТГФ (10 мл), охлаждали до -78°С. К этому раствору добавляли раствор н-бутиллития в гексанах (2,5 М, 2,0 мл, 5,0 ммоль) и перемешивали реакционную смесь при -78°С в течение 1 часа. Затем к реакционной смеси добавляли триизопропилборат (промежуточное соединение 13) (1,21 мл, 6,0 ммоль) в виде раствора в ТГФ (1 мл) при -78°С и перемешивали полученную смесь в течение 1 часа, затем давали нагреться до комнатной температуры в течение ночи. Добавляли 2,3-диметилбутан-2,3-диол (промежуточное соединение 38) (355 мг, 3,0 ммоль), затем через 10 минут добавляли уксусную кислоту (0,34 мл, 6,0 ммоль) и полученную смесь перемешивали в течение еще 10 минут. Реакционную смесь фильтровали через целит и концентрировали фильтрат с получением региоизомерной смеси 3,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола и 4,5-диметил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола в виде масла, которую использовали непосредственно в следующей реакции.
ЖХМС (система 4, способ F): m/z 252 (бороновал кислота - 18)+ (ES+), при 2,72 мин, 254 нм.
Смесь карбоната калия (276 мг, 2,0 ммоль), тетракис(трифенилфосфин)палладия (0) (CAS: 95464-05-4) (116 мг, 0,10 ммоль), региоизомерную смесь 3,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола и 4,5-диметил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола (352 мг, 1,0 ммоль) и трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (328 мг, 1,0 ммоль) в 1,4-диоксане (2,2 мл) и воде (0,10 мл) нагревали до 110°С и выдерживали при этой температуре в течение ночи. Затем реакционную смесь разделяли между ДХМ (5 мл) и водой (5 мл), и водную фазу дополнительно экстрагировали ДХМ (3 × 5 мл). Объединенные органические фазы концентрировали и очищали остаток способом очистки K с получением трет-бутил-(R)-(1-(2-амино-6-(4,5-диметил-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата или трет-бутил-(R)-(1-(2-амино-6-(3,4-диметил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата, или смеси обоих изомеров (6 мг, 1%).
ЖХМС (система 4, способ F): m/z 518 (М+Н)+ (ES+), при 2,55 мин, 254 нм.
Трет-бутил-(R)-(1-(2-амино-6-(4,5-диметил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-3-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат или трет-бутил-(R)-(1-(2-амино-6-(3,4-диметил-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат, или смесь обоих изомеров (6 мг, 0,01 ммоль) растворяли в ДХМ (2 мл) и добавляли раствор НС1 в 1,4-диоксане (4 М, 0,01 мл, 0,0400 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и удаляли образовавшийся осадок фильтрованием с получением (R)-4-(3,4-диметил-1H-пиразол-5-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина дигидрохлорида, примера 7-1 (3 мг, 72%).
Данные для примера 7-1 приведены в таблице 3.
Путь М
Типичная процедура получения пиримидинов, показанная на примере получения примера 11-1, ((R)-4-(1,3-диметил-1Н-пиразол-4-ил)-6-(3-(метиламино)пирролидин)-1-ил)пиримидин-2-амина
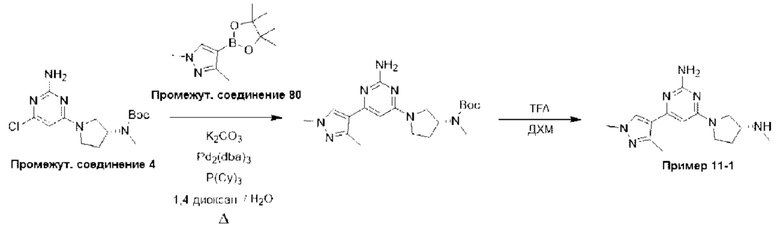
Трет-бутил-(R)-(1-(2-амино-6-хлорпиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (промежуточное соединение 4) (1,0 мг, 3,00 ммоль), 1,3-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (промежуточное соединение 80) (0,87 г, 3,90 ммоль), К2СО3 (1,65 г, 12,0 ммоль) и воду (4,0 мл) растворяли в 1,4-диоксане (16,0 мл) в атмосфере азота и дегазировали в течение 20 минут. В атмосфере азота добавляли трициклогексилфосфин (0,12 г, 0,4 ммоль) и трис(дибензилиденацетон)дипалладий (0) (CAS: 51364-51-3) (274 мг, 0,32 ммоль) и перемешивали смесь при 90°С в течение 12 часов. Реакционную смесь разделяли между Н2О (40 мл) и EtOAc (25 мл), и водный слой дополнительно экстрагировали EtOAc (3 × 25 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с активированным Al2O3, 0-10% МеОН в ДХМ) с получением трет-бутил-(R)-(1-(2-амино-6-(1,3-диметил-1H-пиразол-4-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (1,0 г, 86%) в виде твердого вещества.
ЖХМС (система 3, способ D): m/z 388 (М+Н)+ (ESI+ve), при 3,53 мин, 202 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (1,0 г, 2,58 ммоль) растворяли в ДХМ (20 мл), добавляли TFA (5 мл) при 0°С и перемешивали смесь в течение 1 часа при комнатной температуре. Смесь концентрировали и растирали остаток с пентаном (2 × 10 мл). Остаток очищали способом очистки L с получением ((R)-4-(1,3-диметил-1Н-пиразол-4-ил)-6-(3-(метиламино)пирролидин)-1-ил)пиримидин-2-амина, примера 11-1 (500 мг, 68%), в виде твердого вещества.
Данные для примера 11-1 приведены в таблице 3.
Путь N
Типичная процедура получения пиримидинов, показанная на примере получения примера 11-7, 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(3-метил-3-(метиламино)пирролидин-1-ил)пиримидин-2-амина
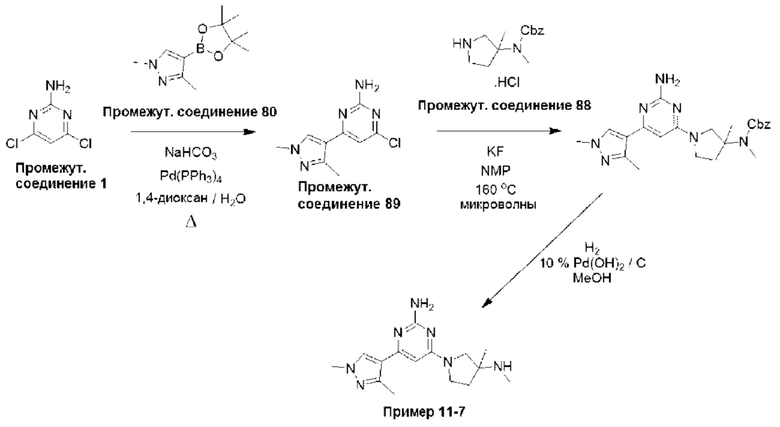
4,6-Дихлорпиримидин-2-амин (промежуточное соединение 1) (500 мг, 3,06 ммоль), 1,3-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (промежуточное соединение 80) (0,68 г, 3,07 ммоль) и NaHCO3 (0,967 г, 9,20 ммоль) растворяли в смеси 1,4-диоксана (10 мл) и воды (2 мл) в атмосфере азота, и дегазировали полученную смесь в течение 20 мин. В атмосфере азота добавляли тетракис(трифенилфосфин)палладий (0) (CAS: 95464-05-4) (0,355 г, 0,306 ммоль) и перемешивали полученную смесь при 50-70°С в течение 12 часов. Затем реакционную смесь разделяли между Н2О (40 мл) и EtOAc (40 мл), и водный слой дополнительно экстрагировали EtOAc (3 × 20 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографией (нормально-фазовой с активированным Al2O3, 20% этилацетата в гексане) с получением 4-хлор-6-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-2-амина (промежуточное соединение 89) (250 мг, 22%) в виде твердого вещества.
Данные для промежуточного соединения 89 приведены в таблице 2.
Бензилметил-(3-метилпирролидин-3-ил)карбамата гидрохлорид (промежуточное соединение 88) (222 мг, 0,78 ммоль) и 4-хлор-6-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-2-амин (промежуточное соединение 89) растворяли в N-метил-2-пирролидоне (8 мл) в атмосфере азота и добавляли фторид калия (156 мг, 2,68 ммоль). Полученную реакционную смесь перемешивали при 160°С в течение 4 часов, используя микроволновую печь от СЕМ. Затем смесь разделяли между H2O (35 мл) и EtOAc (25 мл), и водный слой дополнительно экстрагировали EtOAc (2 × 25 мл). Объединенные органические слои сушили (Na2SO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии (нормально-фазовой с активированным Al2O3, 2-6% МеОН в EtOAc) с получением бензил-(1-(2-амино-6-(1,3-диметил-1Н-пиразол)-4-ил)пиримидин-4-ил)-3-метилпирролидин-3-ил)(метил)карбамата (190 мг, 49%) в виде твердого вещества.
ЖХМС (система 3, способ Е): m/z 436 (М+Н)+ (ESI+ve), при 3,83 мин, 247 нм.
Бензил-(1-(2-амино-6-(1,3-диметил-1Н-пиразол)-4-ил)пиримидин-4-ил)-3-метилпирролидин-3-ил)(метил)карбамат (190 мг, 0,44 ммоль) растворяли в МеОН (15 мл) и добавляли 10% гидроксид палладия на угле (влажность 50%, 100 мг). Затем сосуд продували водородом и перемешивали в атмосфере водорода при 25°С в течение 6 часов. Реакционную смесь фильтровали через целит, промывая катализатор МеОН, и концентрировали фильтрат в вакууме с получением неочищенного продукта, который растирали с пентаном (2 × 2 мл) для удаления неполярных примесей. Продукт очищали способом очистки М, а затем способом очистки N, с получением 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(3-метил-3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 11-7, изомера 1 (19 мг, 15%) в виде твердого вещества и 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(3-метил-3-(метиламино)пирролидин-1-ил)пиримидин-2-амина, примера 11-7, изомера 2 (20 мг, 15%) в виде твердого вещества.
Данные для примера 11 -7, изомера 2 приведены в таблице 3.
Путь О
Типичная процедура получения пиримидинов, показанная на примере получения примера 11-8, 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(октагидро-6Н-пирроло[3,4-b]пиридин-6-ил)пиримидин-2-амина
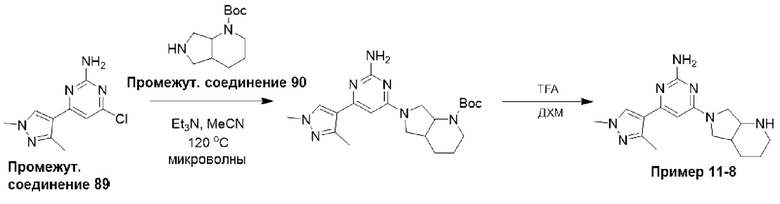
4-Хлор-6-(1,3-Диметил-1Н-пиразол-4-ил)пиримидин-2-амин (промежуточное соединение 89) (150 мг, 0,672 ммоль) растворяли в MeCN : TEA (1:1, 10 мл) и добавляли трет-бутилоктагидро-1Н-пирроло[3,4-b]пиридин-1-карбоксилат (промежуточное соединение 90) (228 мг, 1,01 ммоль) при комнатной температуре. Смесь перемешивали при 120°С в течение 6 часов, используя микроволновую печь от СЕМ. Реакционную смесь концентрировали в вакууме, и остаток очищали с помощью колоночной хроматографии (с нейтральным Al2O3, 0-10% МеОН : ДХМ) с получением трет-бутил-6-(2-амино-6-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-4-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-карбоксилата в виде твердого вещества (150 мг, 54%).
ЖХМС (система 3, способ D): m/z 414 (М+Н)+ (ESI+ve), при 3,75 мин, 254 нм.
Трет-бутил-6-(2-амино-6-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-4-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-карбоксилат (150 мг, 3,63 ммоль) растворяли ДХМ (10 мл) и добавляли TFA (2 мл) при 0°С. Полученную смесь перемешивали в течение 1 часа при комнатной температуре, затем концентрировали в вакууме и растирали остаток с пентаном (2 × 10 мл) с получением неочищенного продукта. Неочищенный продукт очищали способом очистки О, а затем способом очистки Р, с получением 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(октагидро-6Н-пирроло[3,4-b]пиридин-6-ил)пиримидин-2-амина, примера 11-8, изомера 1 (20 мг, 18%), и 4-(1,3-диметил-1Н-пиразол-4-ил)-6-(октагидро-6Н-пирроло[3,4-b]пиридин-6-ил)пиримидин-2-амина, примера 11-8, изомера 2 (10 мг, 9%).
Данные для примера 11 -7, изомера 1 и изомера 2 приведены в таблице 3.
Путь Р
Типичная процедура получения пиридинов, показанная на примере получения примера 14-1, (R)-6-(l,5-диметил-1Н-пиразол-4-ил)-4-(3-(метиламино)пирролидин-1-ил)пиридин-2-амина дигидрохлорида
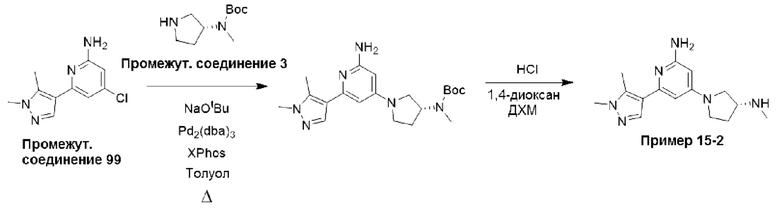
В продуваемый азотом сосуд для микроволновой обработки, содержащий XPhos (CAS: 564483-18-7) (93 мг, 0,19 ммоль), 4-хлор-6-(1,5-диметил-1Н-пиразол-4-ил)пиридин-2-амин (промежуточное соединение 99) (210 мг, 0,94 ммоль), трис(дибензилиденацетон)дипалладий (0) (CAS: 51364-51-3) (86 мг, 0,09 ммоль) и (R)-метил(пирролидин-3-ил)карбамат (промежуточное соединение 3) (208 мг, 1,04 ммоль) добавляли толуол (5 мл). Реакционный сосуд продували азотом и добавляли трет-бутилат натрия (272 мг, 2,83 ммоль). Затем сосуд герметично закрывали и нагревали обычным способом при 110°С в течение 16 часов. Реакционную смесь разделяли между EtOAc (5 мл) и водой (5 мл), и водную фазу дополнительно экстрагировали EtOAc (3 × 5 мл). Объединенные органические фазы концентрировали и очищали остаток с помощью колоночной флэш-хроматографии (нормально-фазовой с SiO2, 0-10% МеОН в ДХМ) с получением неочищенного продукта, который дополнительно очищали способом очистки Q с получением трет-бутил-(R)-(1-(2-амино-6-(1,5-диметил-1H-пиразол-4-ил)пиридин-4-ил)пирролидин-3-ил)(метил)карбамата (5 мг, 1%) в виде масла.
ЖХМС (система 4, способ F): m/z 387 (М+Н)+ (ES+), при 2,07 мин, 254 нм.
Трет-бутил-(R)-(1-(2-амино-6-(1,5-диметил-1H-пиразол-4-ил)пиридин-4-ил)пирролидин-3-ил)(метил)карбамат (5 мг, 0,010 ммоль) растворяли в ДХМ (2 мл), добавляли раствор HCl в 1,4-диоксане (4 М, 0,01 мл, 0,06 ммоль) и перемешивали полученную смесь при комнатной температуре в течение ночи. По прошествии этого времени белый осадок отфильтровывали с получением (R)-6-(1,5-диметил-1Н-пиразол-4-ил)-4-(3-(метиламино)пирролидин-1-ил)пиридин-2-амина дигидрохлорида, примера 14-1 (3 мг, 78%).
.
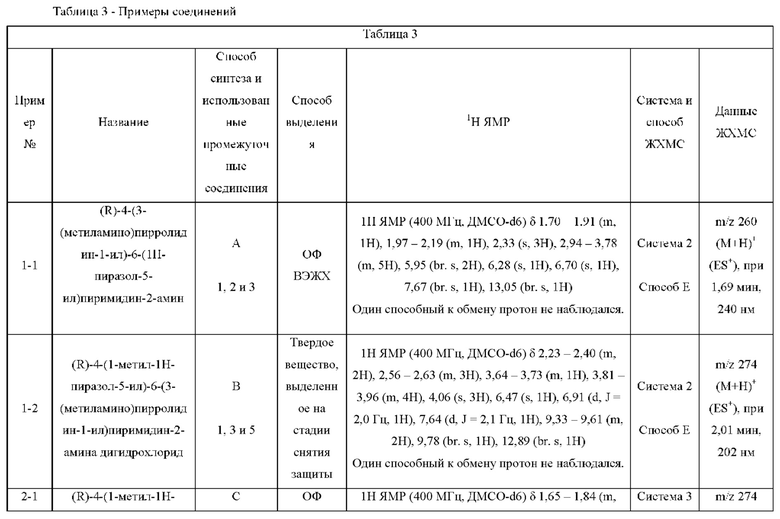
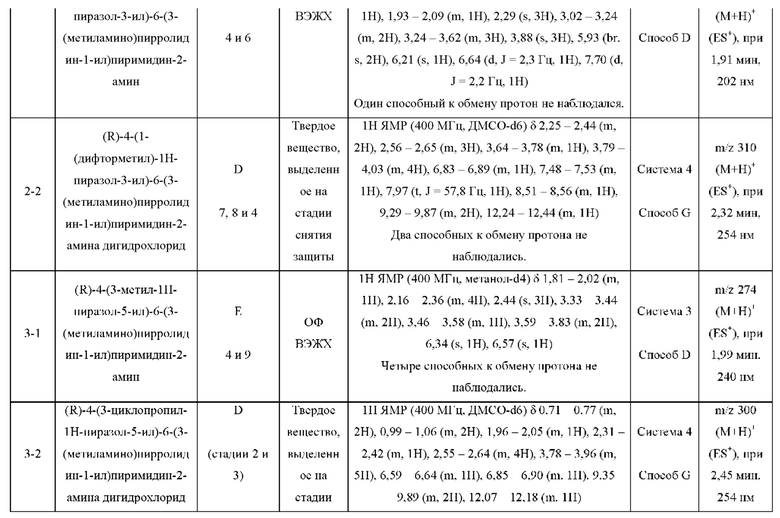
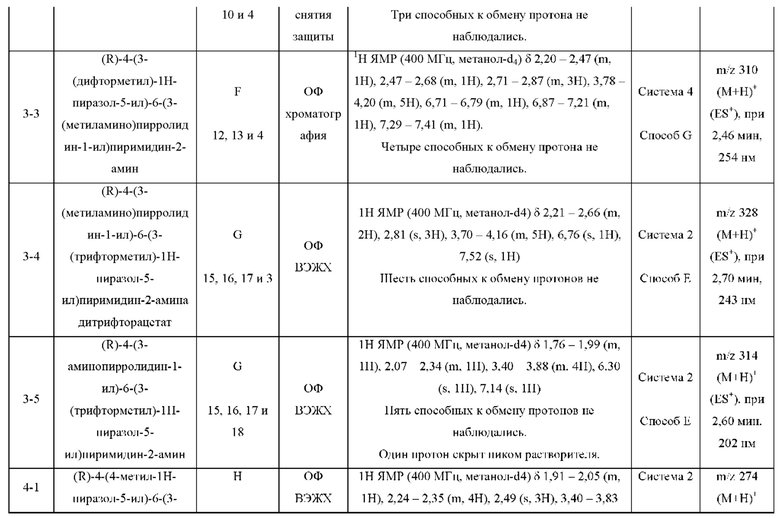
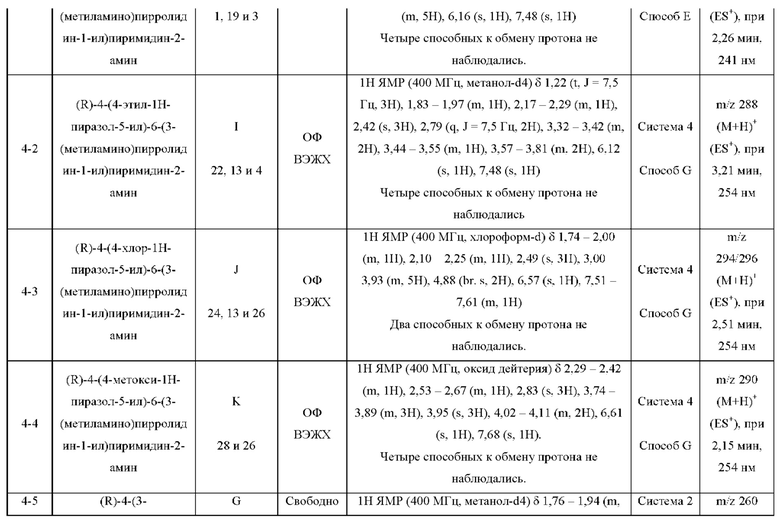
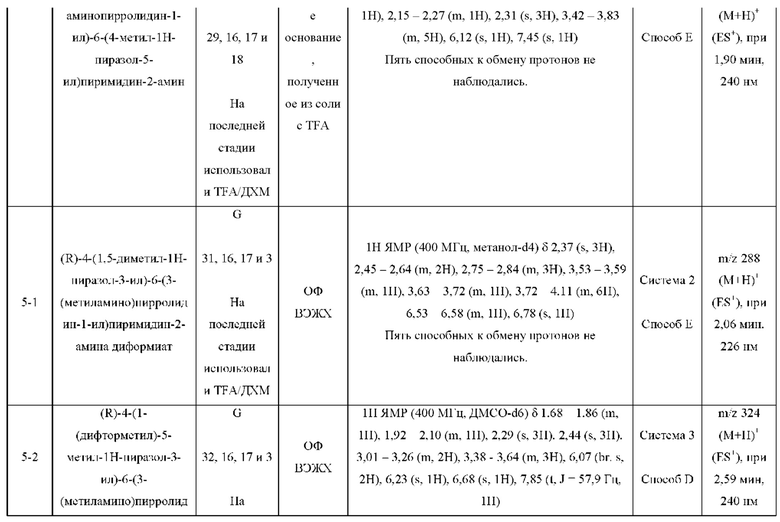
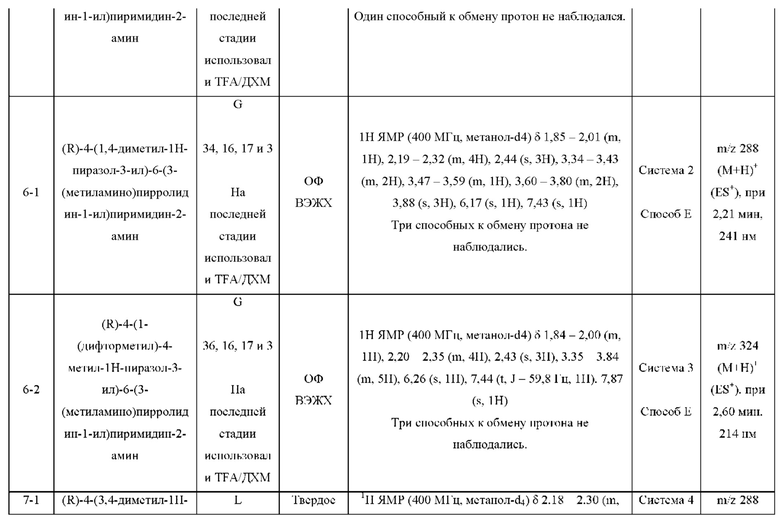
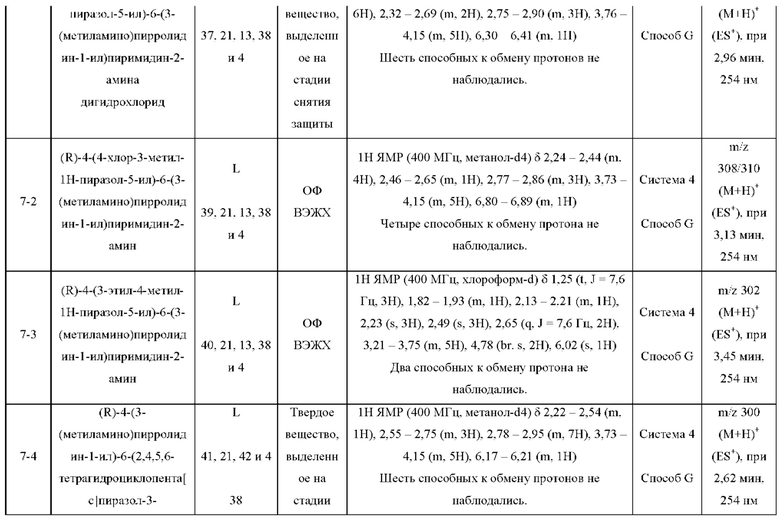
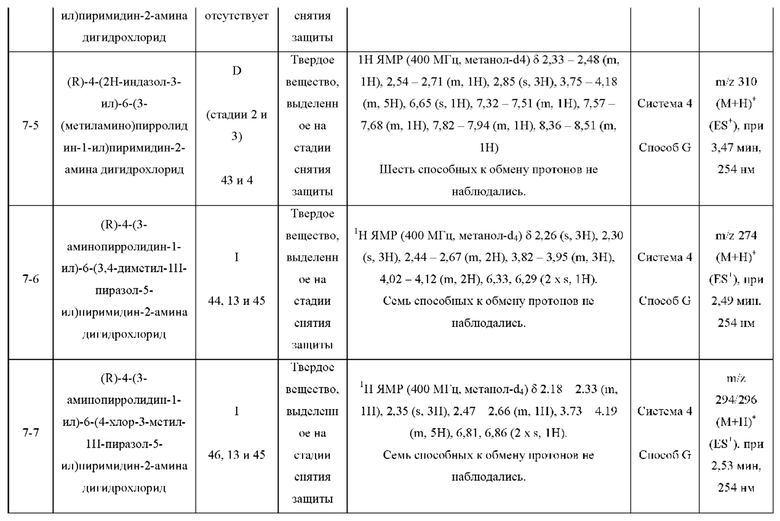
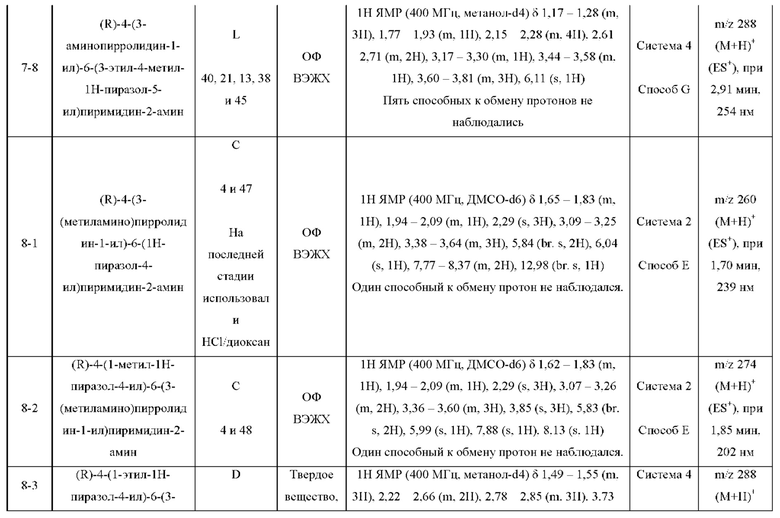
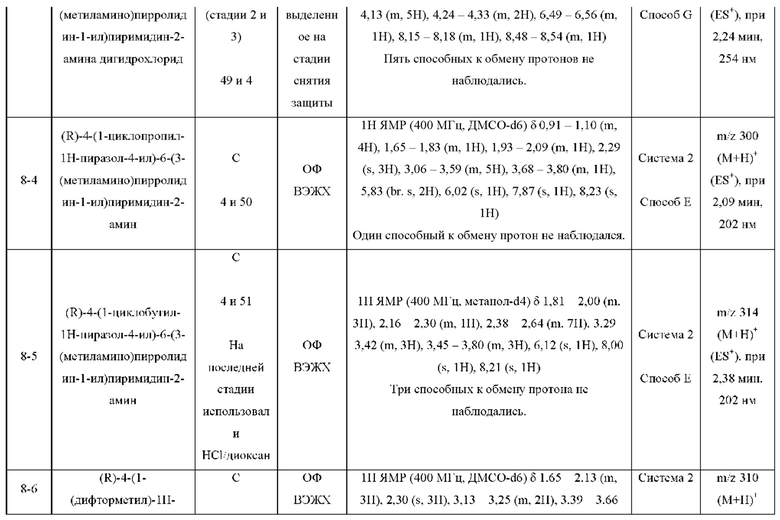
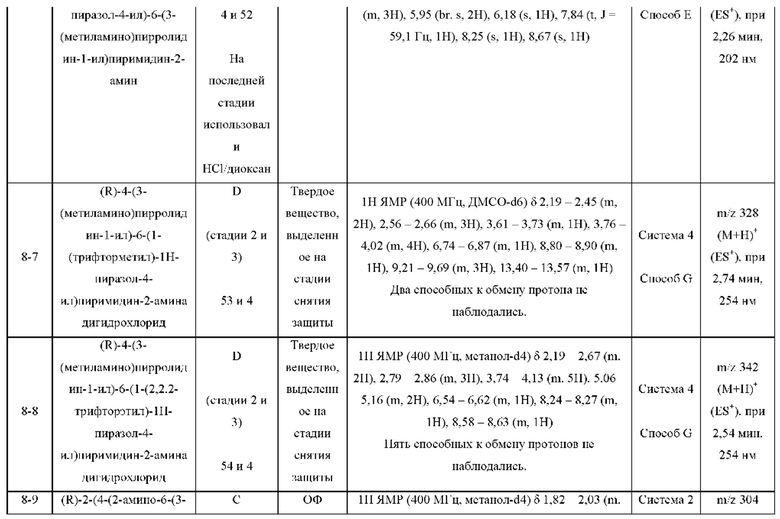
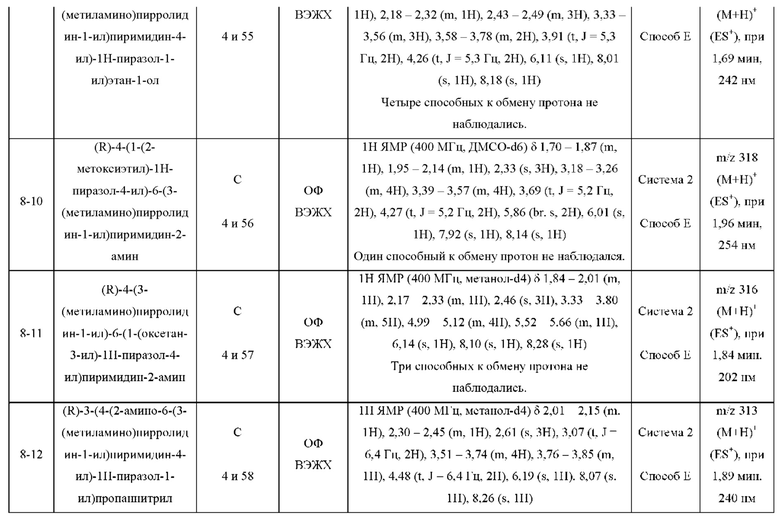
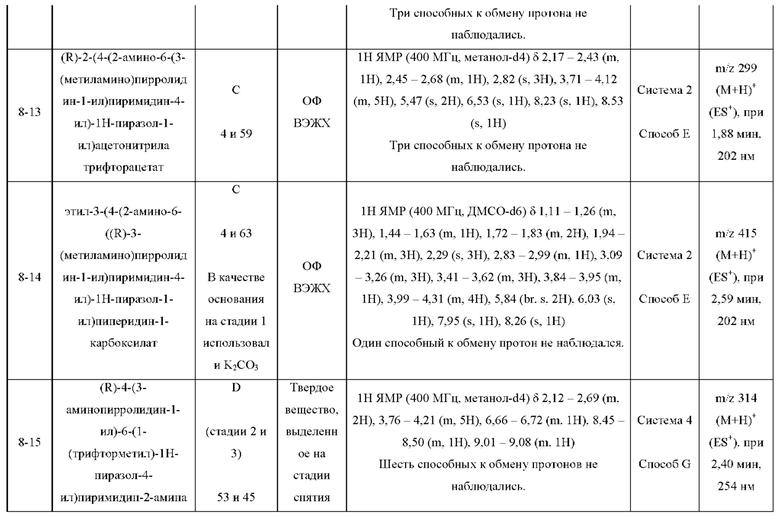
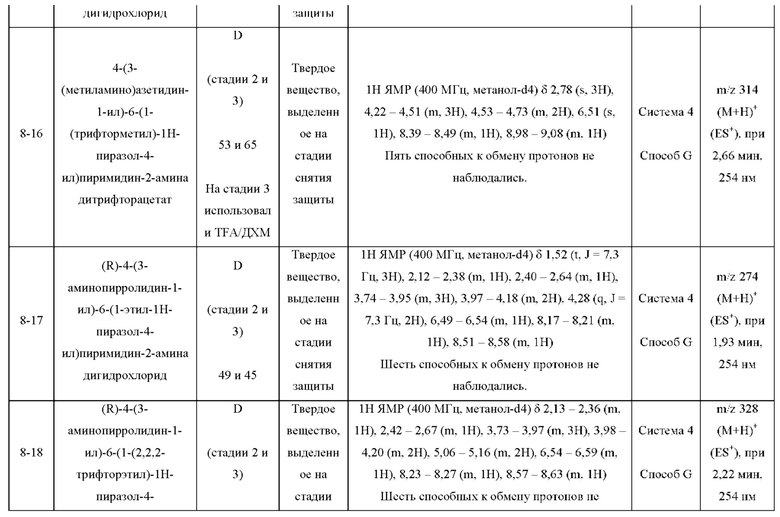
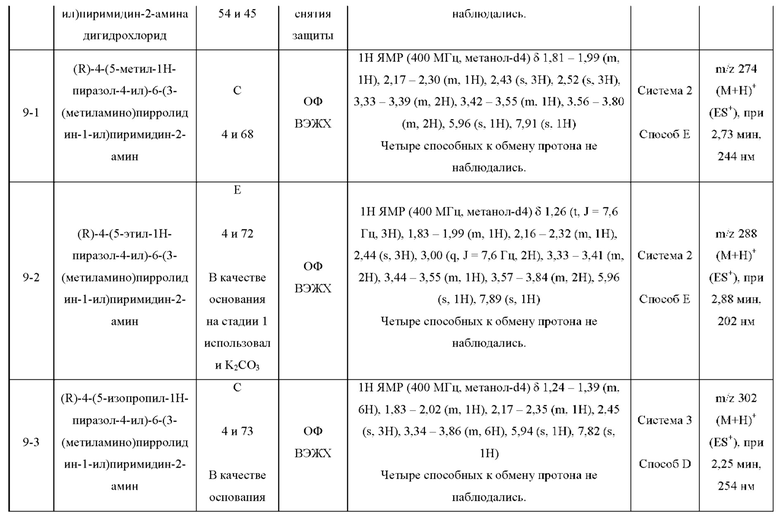
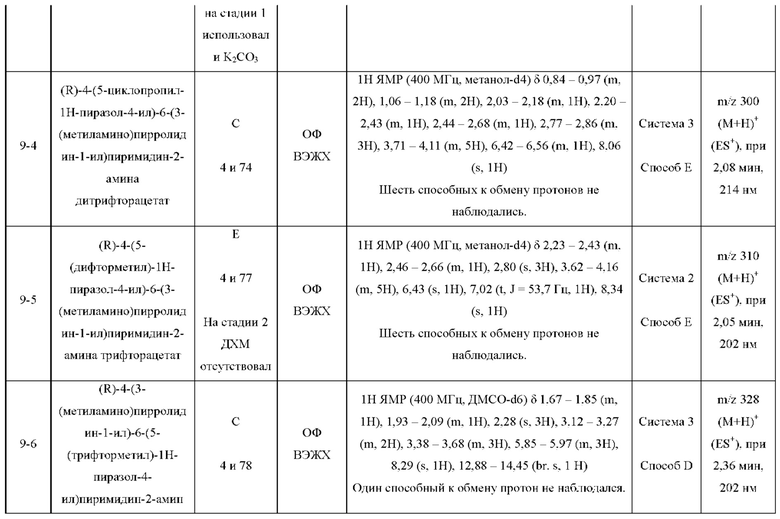
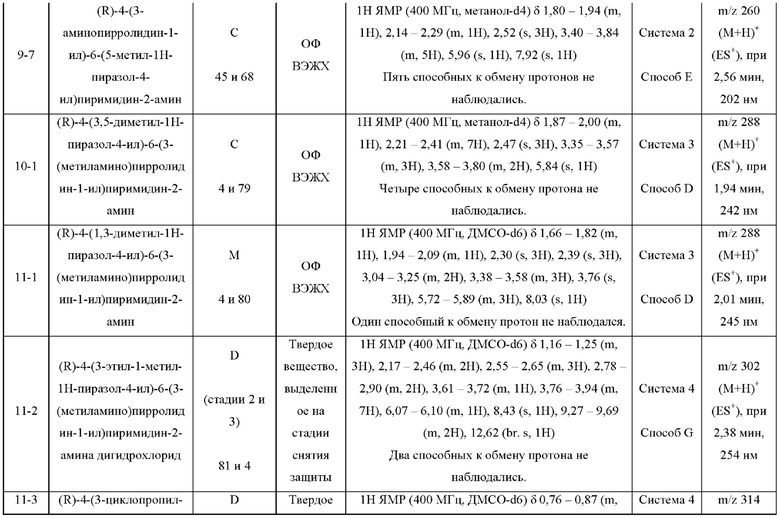
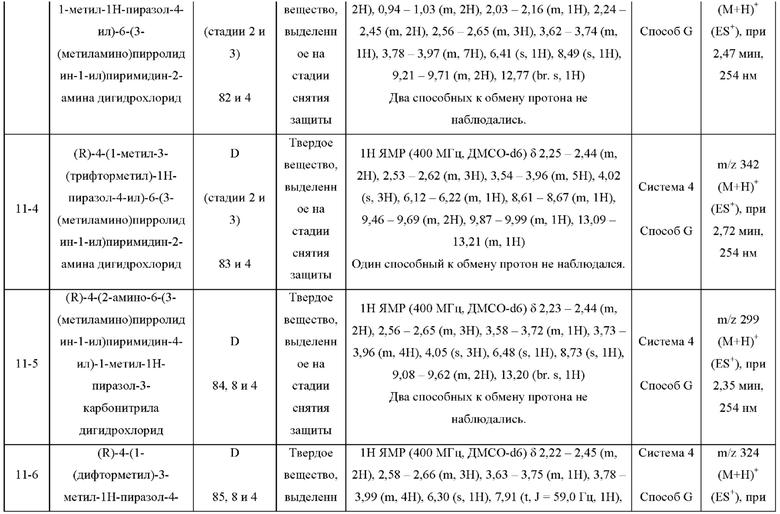
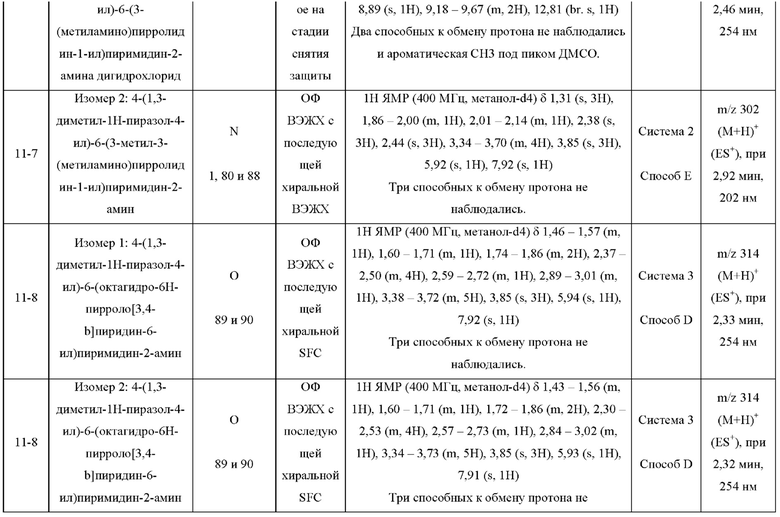
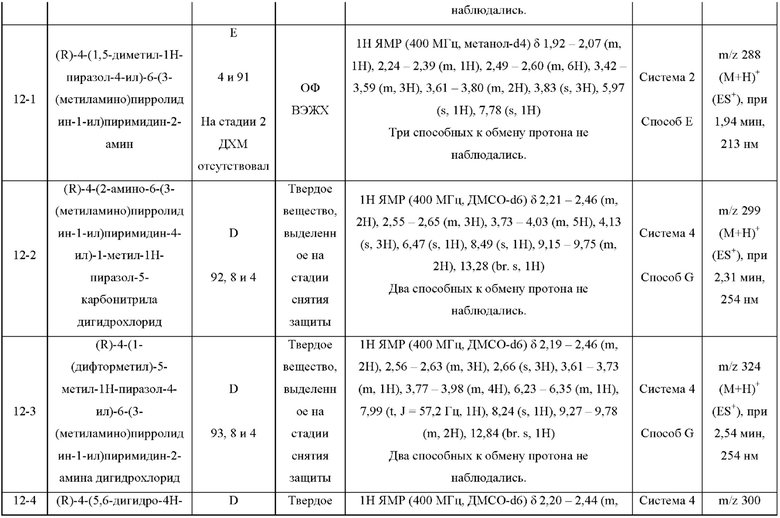
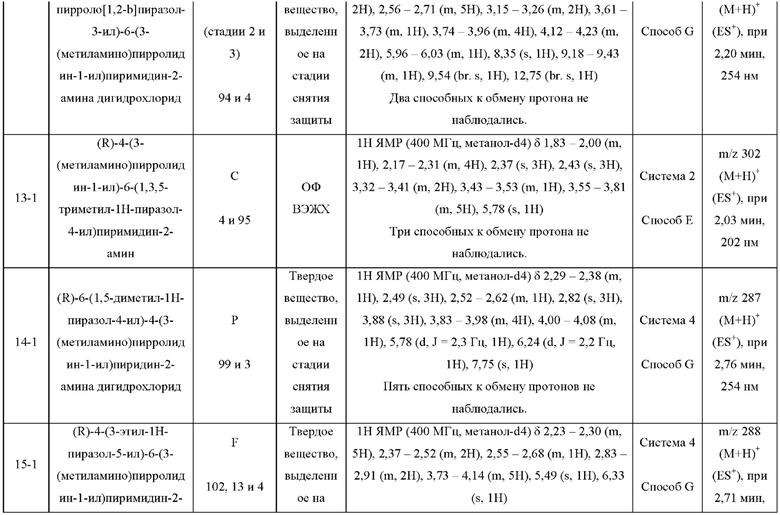
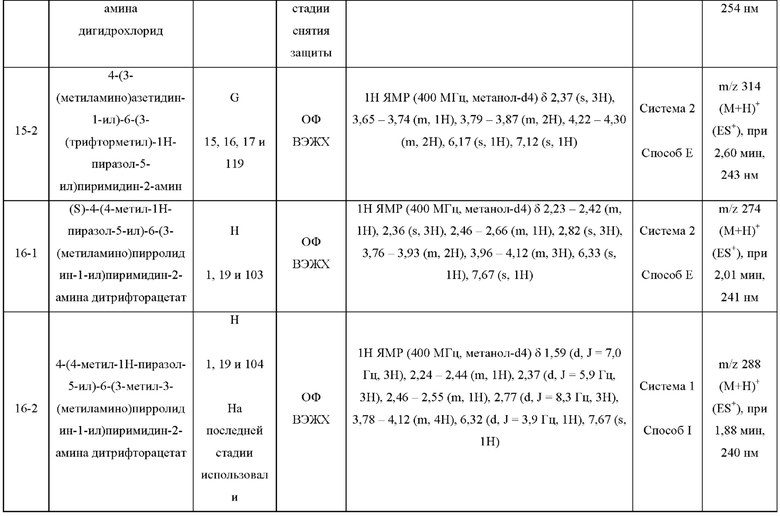
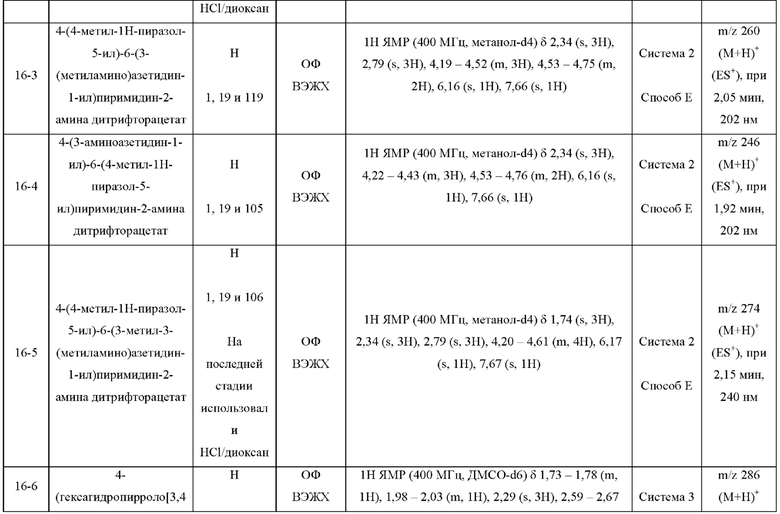
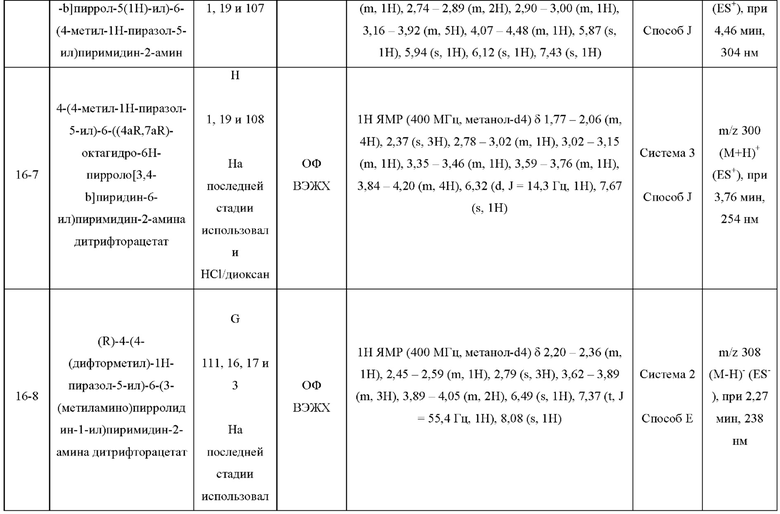
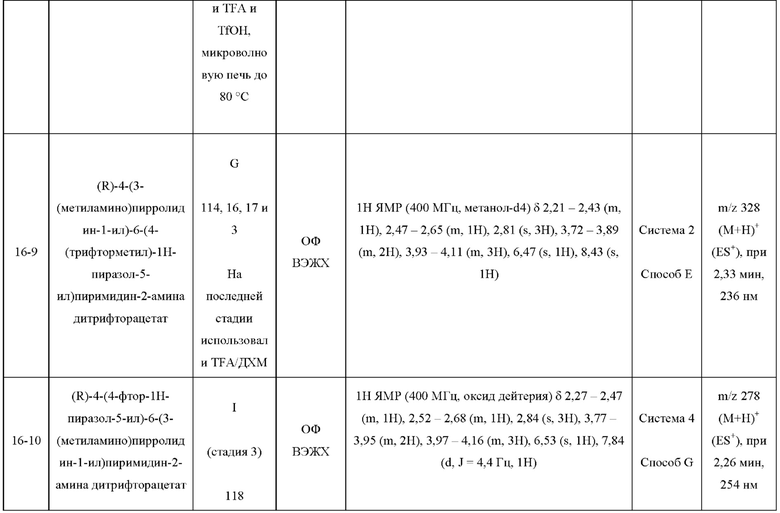
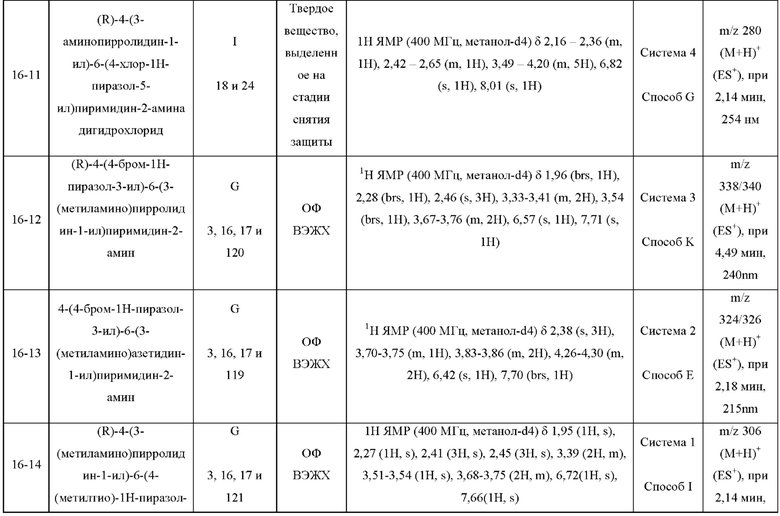
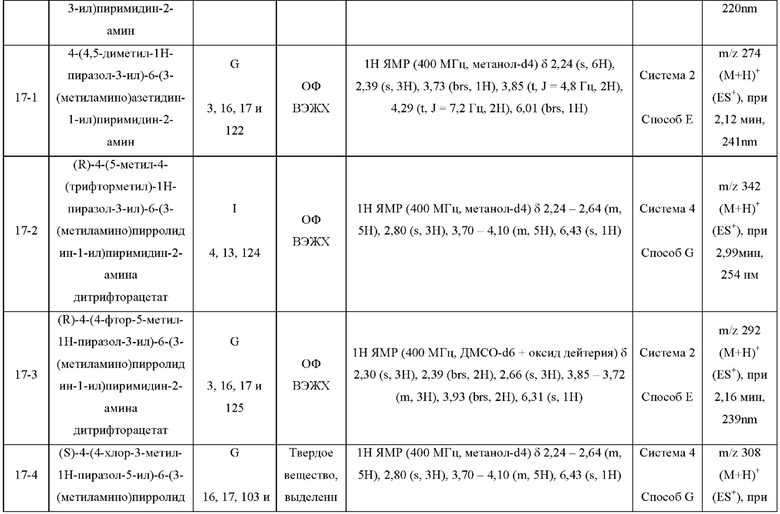
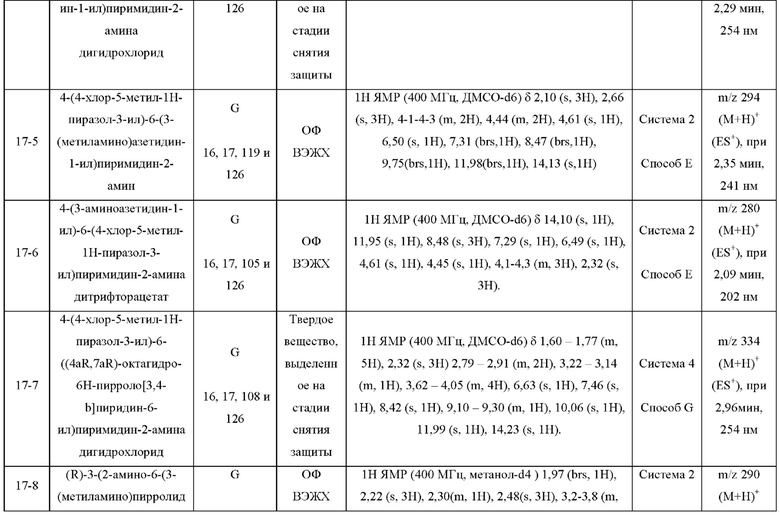

БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
ПРИМЕР А
Функциональный цАМФ Gi анализ антагонистов Н4
Клетки HEKf инфицировали в течение ночи с использованием бакуловируса, экспрессирующего человеческий Н4-рецептор, затем центрифугировали при 1200 об/мин в течение 5 минут, замораживали в среде для замораживания клеток (Sigma) и хранили при -150°С. В день анализа клетки размораживали и ресуспендировали в HBSS с 500 нМ IBMX для достижения плотности 1500 клеток/лунку. Готовили растворы лигандов Н4 в ДМСО и отмеряли с помощью акустического дозирования LabCyte ECHO в объеме 25 нл в малообъемные планшеты. 10 мкл/лунку клеток высевали в присутствии 1 мкМ форсколина, подвергали центрифугированию при 1200 об/мин в течение 1 мин и инкубировали в течение 30 мин перед добавлением реагентов для обнаружения цАМФ от Cisbio до общего объема 20 мкл/лунку. Для анализа антагонистов клетки предварительно инкубировали с лигандами-антагонистами Н4 в течение 30 мин перед добавлением гистамина в концентрации ЕС80 и последующей 30-минутной инкубацией. После добавления реагента для обнаружения и встряхивания при комнатной температуре в течение 60 мин измеряли накопление цАМФ с помощью HTRF (гомогенная флуоресценция с временным разрешением) на считывающем устройстве для планшетов PheraStar. Значения ЕС50 были получены с использованием 4-параметрического уравнения логистической аппроксимации для количественной оценки эффективности агонистов. Значения аффинности в функциональном анализе антагонистов были получены с использованием уравнения Ченга-Прусова для расчета значения pKb с использованием данных анализа антагонистов.
Функциональный анализ антагонистов Н4 по динамическому перераспределению массы
Клетки HEKf инфицировали с помощью бакуловируса, экспрессирующего человеческий Н4-рецептор, высевали в покрытые фибронектином планшеты EPIC с плотностью 10000 клеток/лунку и инкубировали в течение ночи при 37°С. Среду для клеток заменяли на 30 мкл HBSS с 20 мМ HEPES на лунку и добавляли 30 нл ДМСО на лунку с помощью акустического дозирования LabCyte ECHO. После 2-часового уравновешивания при комнатной температуре отмеряли 30 нл лигандов Н4, приготовленных в ДМСО, с помощью акустического дозирования LabCyte ECHO в засеянные планшеты EPIC, и отслеживали динамическое перераспределение клеточной массы с помощью считывающего устройства для планшетов Corning EPIC. После 45-минутного измерения добавляли 30 нл/лунку гистамина при ЕС80 и отслеживали, чтобы получить данные анализа антагонистов. Максимальные ответы с поправкой на исходный уровень в пМ использовали для построения кривых концентрация-ответ. Значения ЕС50 были получены с использованием 4-параметрического уравнения логистической аппроксимации для количественной оценки эффективности агонистов. Значения аффинности в функциональном анализе антагонистов были получены с использованием уравнения Ченга-Прусова для расчета значения pKb с использованием данных анализа антагонистов.
Анализ hERG
Данные анализа hERG были определены Metrion Biosciences, Кембридж, Великобритания, с использованием протоколов эксперимента, описанных ниже:
Клеточную линию яичника китайского хомячка (СНО), стабильно экспрессирующую человеческий ген специфических калиевых каналов сердца, выращивали и пассировали в стандартных условиях культивирования. Клетки были подготовлены для анализов с использованием протоколов диссоциации, разработанных для оптимизации здоровья клеток, выхода, контакта и качества и анализа. Тестируемые образцы были представлены в виде 10 мМ исходных растворов в 100% ДМСО. Все манипуляции с образцами и серийные разведения проводили с использованием стеклянных контейнеров и остеклованных планшетов. Наивысшую рабочую концентрацию 30 мкМ получали из 10 мМ исходного раствора образца с использованием 1:333-кратного разведения во внешнем растворе для регистрации (0,3% ДМСО об./об.). В анализе с одной концентрацией тестируемые образцы подвергали скринингу при концентрации 30 мкМ с как минимум тремя отдельными клетками. В анализе pIC50 тестируемые образцы подвергали скринингу при концентрации 1, 3, 10 и 30 мкМ с как минимум тремя отдельными клетками. Каждую четырехточечную кривую концентрация-ответ строили с использованием кумулятивного двойного добавления образца каждой концентрации в одну и ту же клетку.
Все эксперименты проводили на автоматизированной платформе фиксации потенциала QPatch с гигаомным контактом. Состав внешних и внутренних растворов для измерения для экспериментов в QPatch приведен в таблице А ниже. Все растворы фильтровали (0,2 мкм) перед каждым экспериментом.

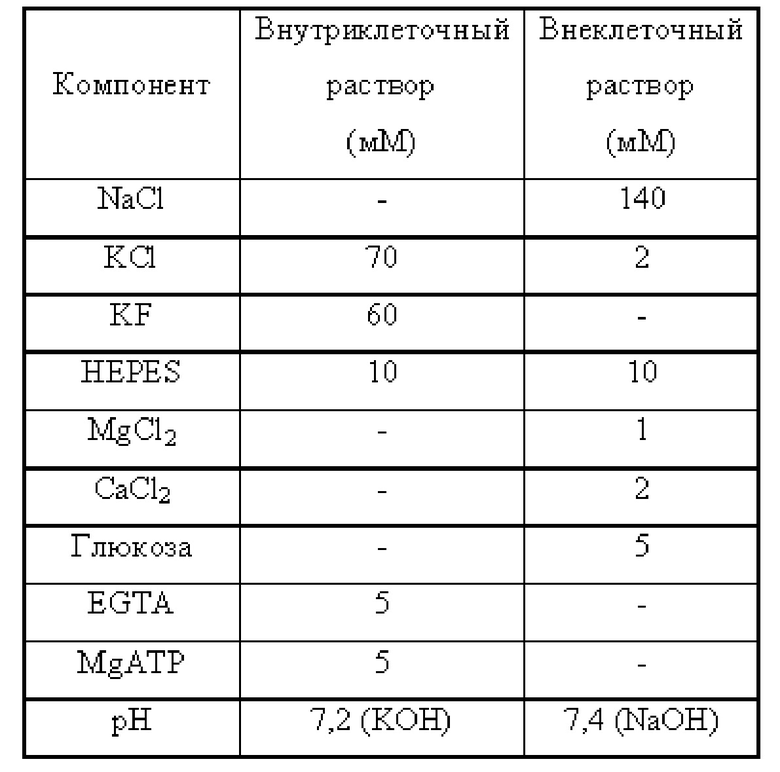
Все измерения были сделаны в стандартной конфигурации целой клетки и проводились при комнатной температуре (приблизительно 21°С) с использованием стандартных чипов с одним отверстием (Rchip 1,5-4 МОм). Последовательное сопротивление (4-15 МОм) было компенсировано на >80%. Токи были получены из поддерживающего потенциала -90 мВ с использованием протокола изменения напряжения промышленного стандарта «+40 / -40», как показано на фиг. А; его применяли при частоте стимула 0,1 Гц.
После достижения конфигурации целой клетки к каждой клетке применяли носитель (0,3% ДМСО об./об. во внешнем растворе для измерения) двумя болюсными добавлениями с двухминутным периодом измерения между каждым добавлением, чтобы обеспечить стабильные показания. По истечении периода носителя осуществляли одно из двух:
i) для анализа с одной концентрацией применяли одну концентрацию тестируемого образца 30 мкМ в виде пяти болюсных добавлений на тестируемую концентрацию с двухминутными интервалами; или
ii) для анализа pIC50 применяли четыре концентрации тестируемого образца от 1 мкМ до 30 мкМ в виде двух болюсных добавлений на тестируемую концентрацию с двухминутными интервалами;
и затем измеряли влияние на амплитуду хвостового тока hERG в течение четырехминутного периода измерения. Для каждой развертки протокола напряжения мембранный ток и пассивные свойства отдельных клеток регистрировали с помощью программного обеспечения для анализа QPatch (версия 5.0). Пиковая амплитуда выходящего хвостового тока, вызванного во время тестового импульса до -40 мВ, была измерена относительно мгновенного тока утечки, измеренного во время начального предымпульсного этапа до -40 мВ. Для целей контроля качества минимальная амплитуда тока для анализа составляет >200 пА пикового выходящего тока, измеренного в конце периода носителя. Программное обеспечение для анализа QPatch вычисляет средний пиковый ток для последних трех разверток в конце каждого периода применения концентрации, а данные экспортируются в Excel и запрашиваются с помощью пакета для биоинформатики, разработанного в Pipeline Pilot (Biovia, США). Шаблон рассчитывает процент ингибирования для периода применения каждой тестируемой концентрации как уменьшение среднего пикового тока или заряда относительно значения, измеренного в конце контрольного периода (т.е. периода носителя). Значения процента ингибирования для каждой клетки используют для построения кривых концентрация-ответ с использованием четырехпараметрической логистической аппроксимации с уровнями ингибирования 0 и 100%, фиксированными при очень низких и очень высоких концентрациях, соответственно, и свободном угловом коэффициенте Хилла. Затем определяют IC50 (50% ингибирующая концентрация) и коэффициент Хилла, но включены только данные для клеток с коэффициентами Хилла в пределах 0,5>nH<2,0. Данные IC50, представленные ниже, представляют собой среднее значение для по меньшей мере трех отдельных клеток (N≥3). Считается, что тестируемый образец, не обеспечивающий блокирование >40% при максимальной концентрации, дает неоднозначное значение IC50 из-за плохой или неограниченной аппроксимации. В этом случае возвращается произвольное значение IC50, которое на 0,5 логарифмических единиц выше максимальной протестированной концентрации. Например, если образец не демонстрирует среднее ингибирование >40% блокирования при максимальной концентрации 30 мкМ, то сообщается значение IC50, равное 100 мкМ, то есть pIC50≤4,0.
Для соединений, содержащих пирролидиновый амин, подавляющее большинство примеров было получено в виде отдельных энантиомеров с (R)-стереохимией. Тем не менее некоторые соединения были получены в виде рацематов, а затем энантиомеры были разделены с использованием методов хиральной ВЭЖХ или хиральной SFC. Для этих соединений определение изомера (изомер 1, изомер 2) основано на времени удерживания соединения с использованием методики разделения, которая была использована на последней стадии хирального разделения. Как следствие, это может быть время удерживания хиральной ВЭЖХ или хиральной SFC, и оно будет варьироваться от соединения к соединению.
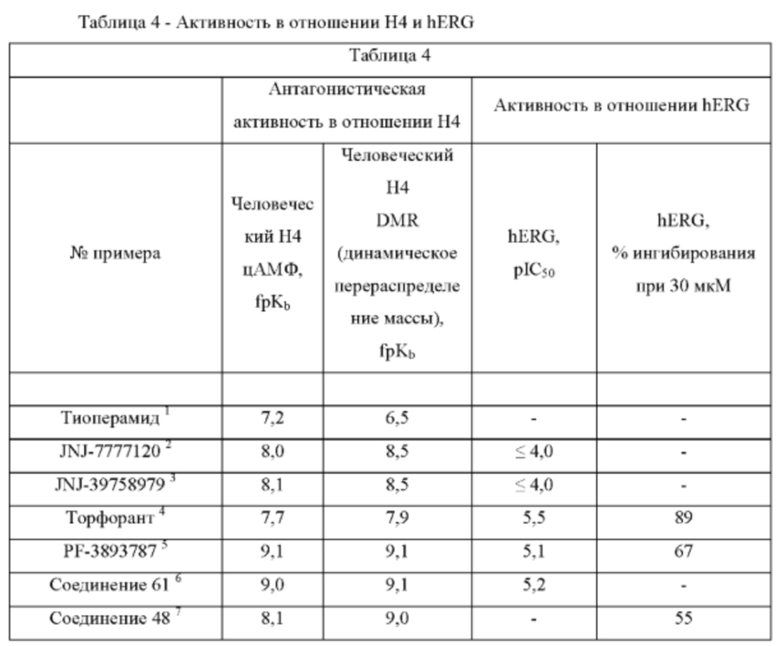
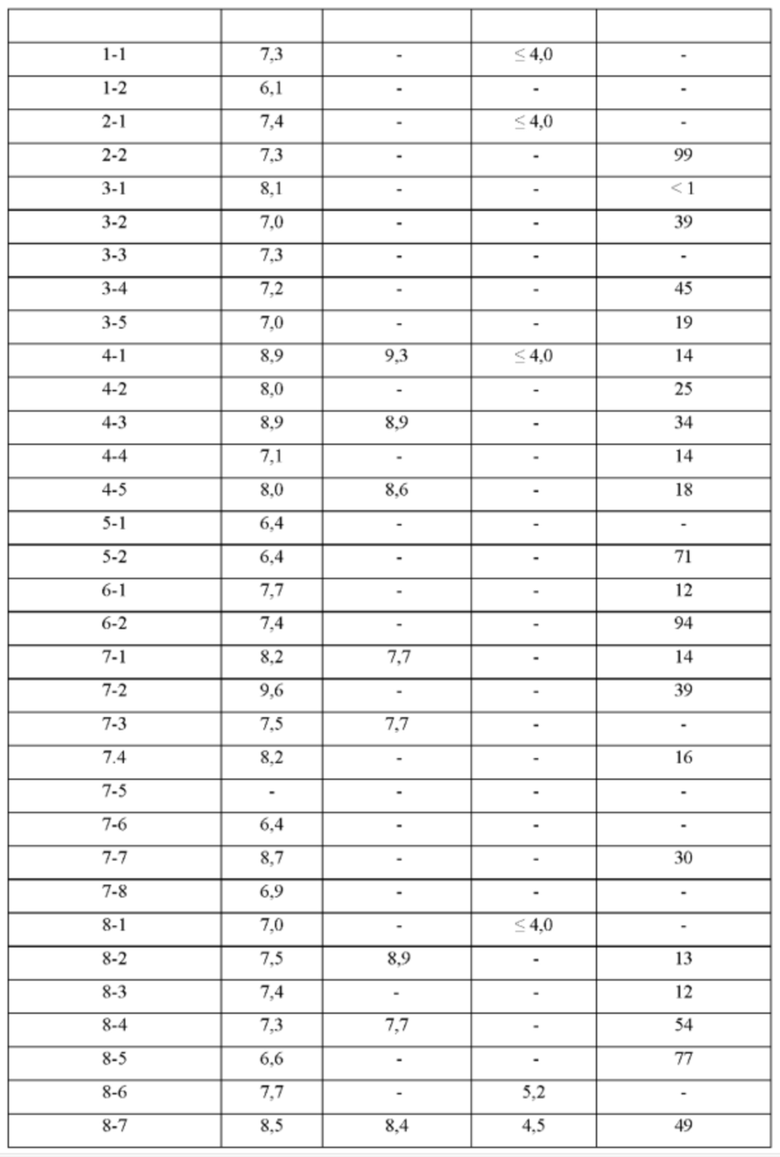
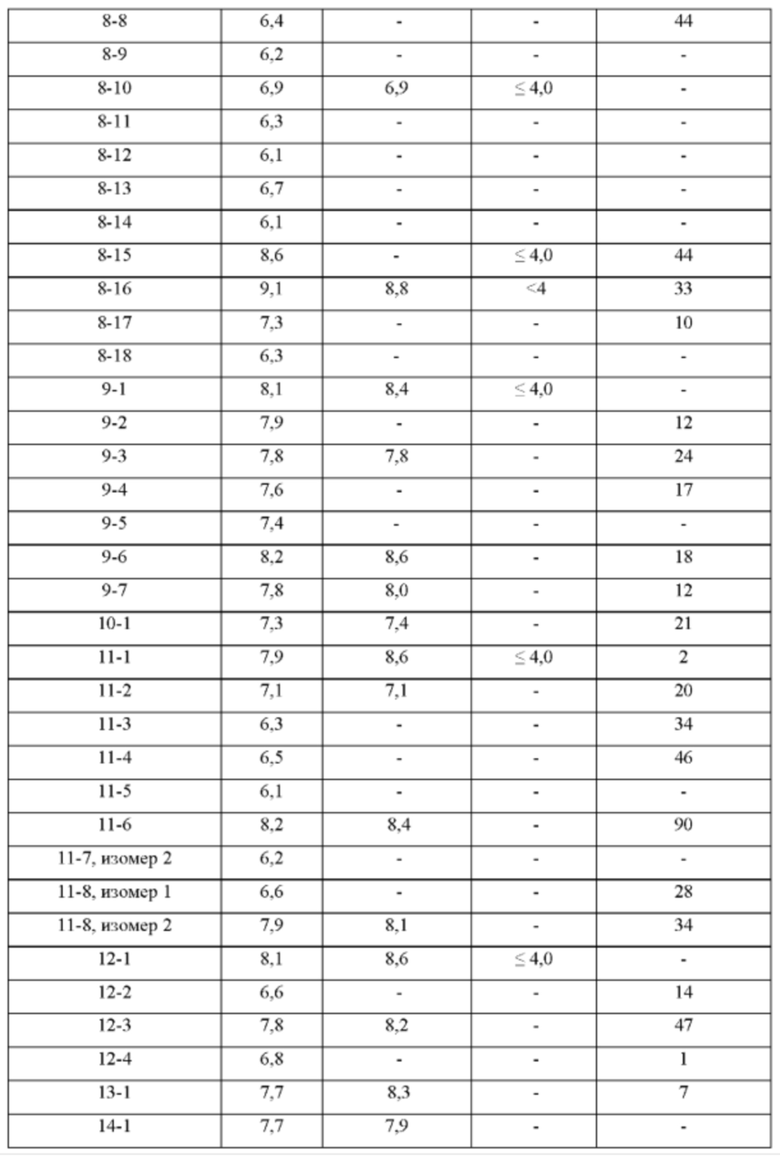
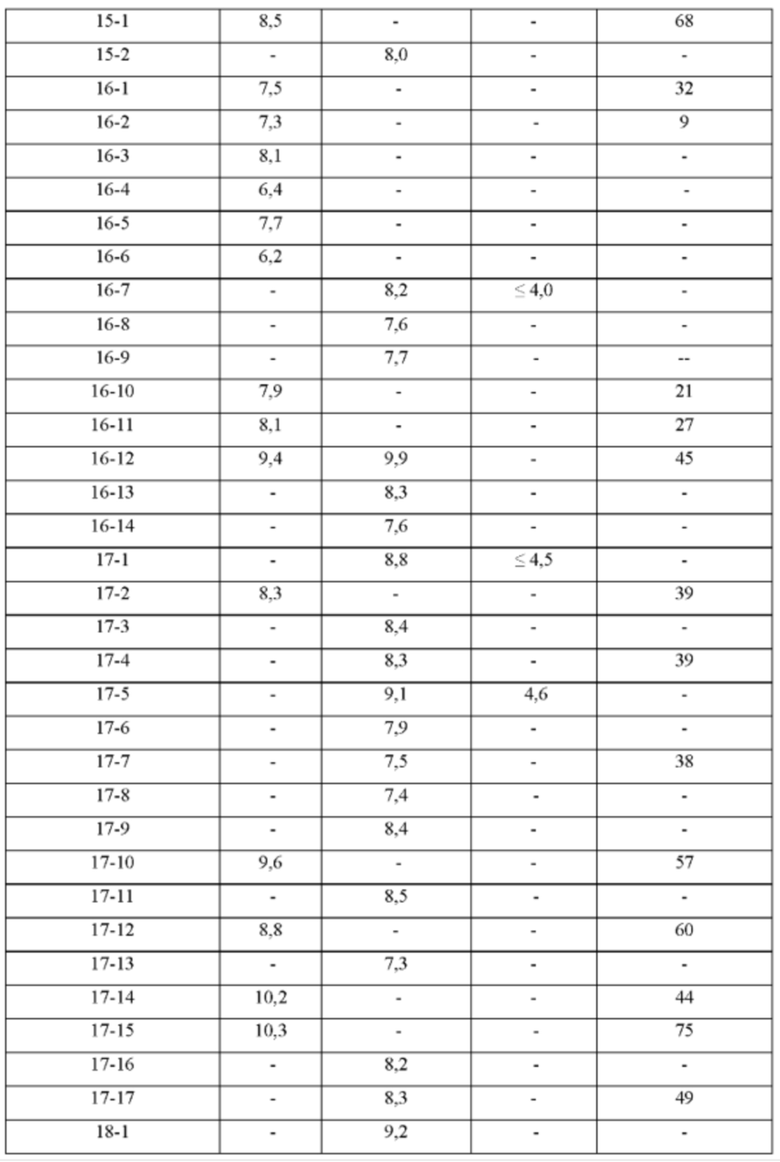
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2691389C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| СОЕДИНЕНИЯ ПИРРОЛИДИНИЛМОЧЕВИНЫ И ПИРРОЛИДИНИЛТИОМОЧЕВИНЫ КАК ИНГИБИТОРЫ КИНАЗЫ TrkA | 2012 |
|
RU2606131C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| ИНГИБИТОРЫ P14KIII БЕТА | 2019 |
|
RU2789127C2 |
| СОЕДИНЕНИЯ N-ПИРРОЛИДИНИЛМОЧЕВИНЫ, N'-ПИРАЗОЛИЛМОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRKA | 2013 |
|
RU2677667C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ МОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2664541C2 |
Изобретение относится к соединению формулы (1) или его фармацевтически приемлемой соли, которые обладают активностью антагониста рецептора Н4 и могут найти применение для лечения связанных с такой активностью заболеваний. В формуле (1) X представляет собой СН или N; n равно 2; R1 выбран из Н или С1-3 алкила; R2 представляет собой Н или метил и А представляет собой необязательно замещенное пиразольное кольцо, которое связано с кольцом, содержащим X, углерод-углеродной связью, где (i) А представляет собой необязательно замещенное пиразольное кольцо, выбранное из указанных ниже, где R3 выбран из Н; C1-6 неароматической углеводородной группы, выбранной из алкила, необязательно замещенного 1-6 атомами фтора, или циклоалкила, необязательно замещенного 1-6 атомами фтора; (CH2)mR6, где m равно от 1 до 3 и R6 выбран из CN, ОН, С1-С3 алкокси и необязательно замещенного 4-6-членного насыщенного гетероциклического кольца, содержащего 1 гетероатом, выбранный из О и N, где необязательный заместитель представляет собой CO2R7, где R7 представляет собой С1-3 алкил; где R4 и R5 независимо выбраны из C1-6 неароматической углеводородной группы, выбранной из алкила, необязательно замещенного 1-6 атомами фтора, или циклоалкила, необязательно замещенного 1-6 атомами фтора; и (CH2)pR9, где р равно от 0 до 3 и R9 выбран из CN, галогена, ОН, С1-С3 алкокси и SR8, где R8 представляет собой С1-С3 алкил; или R4 и R5 могут быть необязательно соединены с образованием конденсированного 5- или 6-членного кольца; или R4 и R3 могут быть необязательно соединены с образованием конденсированного 5-членного алифатического кольца; или где (ii) А выбрано из группы, состоящей из 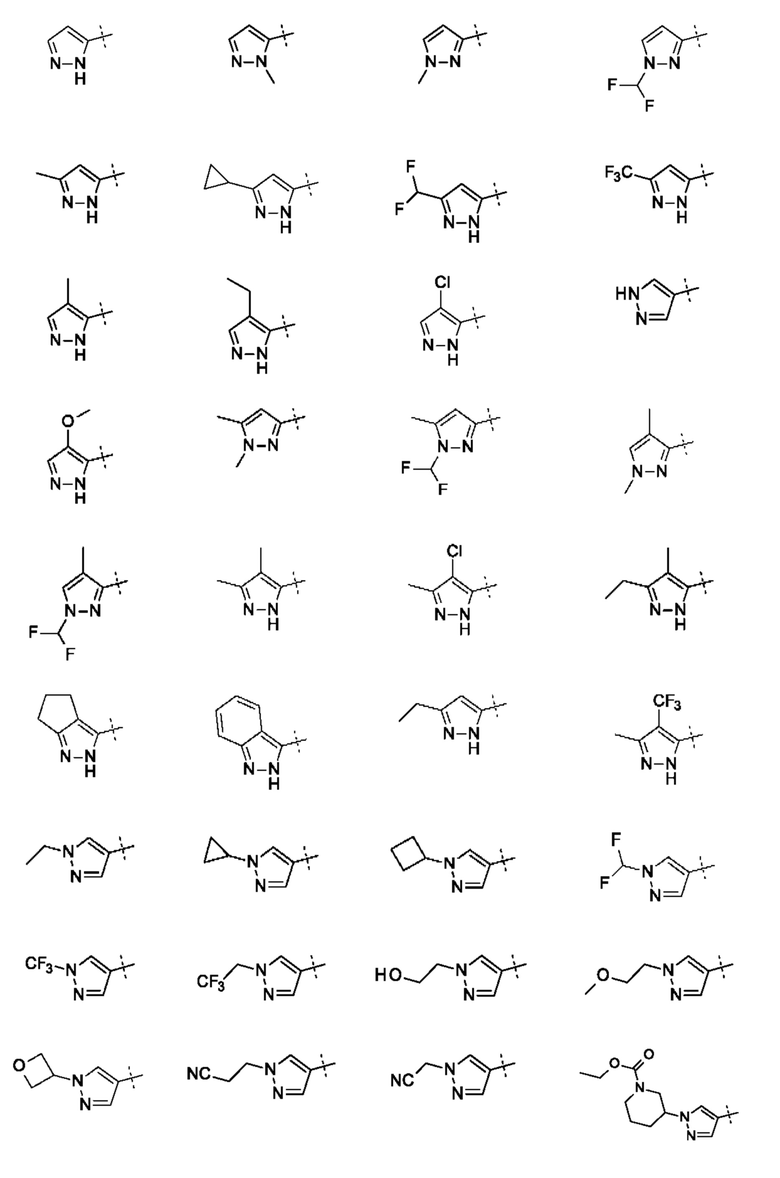 , и ряда других значений, приведенных в формуле изобретения. Изобретение относится также к фармацевтической композиции, содержащей указанное соединение. 2 н. и 17 з.п. ф-лы, 1 ил., 4 табл., 100 пр.
, и ряда других значений, приведенных в формуле изобретения. Изобретение относится также к фармацевтической композиции, содержащей указанное соединение. 2 н. и 17 з.п. ф-лы, 1 ил., 4 табл., 100 пр.
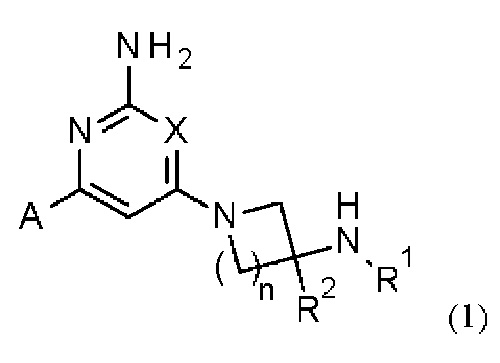
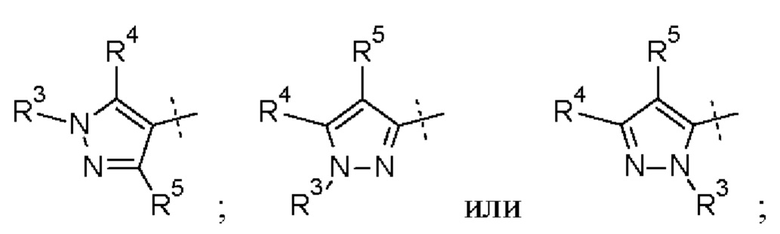
1. Соединение формулы (1)
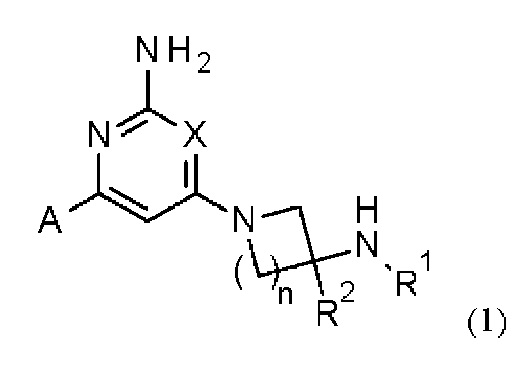
или его фармацевтически приемлемая соль, где
X представляет собой СН или N;
n равно 2;
R1 выбран из Н или С1-3 алкила;
R2 представляет собой Н или метил и
А представляет собой необязательно замещенное пиразольное кольцо, которое связано с кольцом, содержащим X, углерод-углеродной связью, где
(i) А представляет собой необязательно замещенное пиразольное кольцо, выбранное из
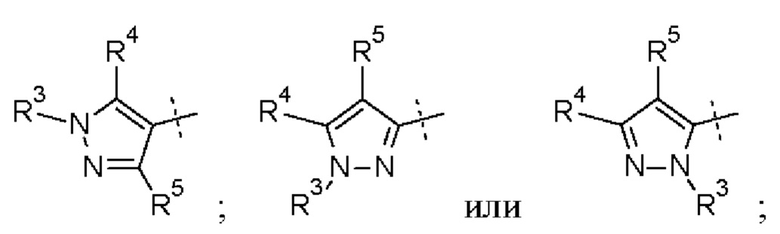
где R3 выбран из Н; C1-6 неароматической углеводородной группы, выбранной из алкила, необязательно замещенного 1-6 атомами фтора, или циклоалкила, необязательно замещенного 1-6 атомами фтора; (CH2)mR6, где m равно от 1 до 3 и R6 выбран из CN, ОН, С1-С3 алкокси и необязательно замещенного 4-6-членного насыщенного гетероциклического кольца, содержащего 1 гетероатом, выбранный из О и N, где необязательный заместитель представляет собой CO2R7, где R7 представляет собой С1-3 алкил; где R4 и R5 независимо выбраны из C1-6 неароматической углеводородной группы, выбранной из алкила, необязательно замещенного 1-6 атомами фтора, или циклоалкила, необязательно замещенного 1-6 атомами фтора; и (CH2)pR9, где р равно от 0 до 3 и R9 выбран из CN, галогена, ОН, С1-С3 алкокси и SR8, где R8 представляет собой С1-С3 алкил; или R4 и R5 могут быть необязательно соединены с образованием конденсированного 5- или 6-членного кольца; или R4 и R3 могут быть необязательно соединены с образованием конденсированного 5-членного алифатического кольца; или
(ii) А выбрано из группы, состоящей из
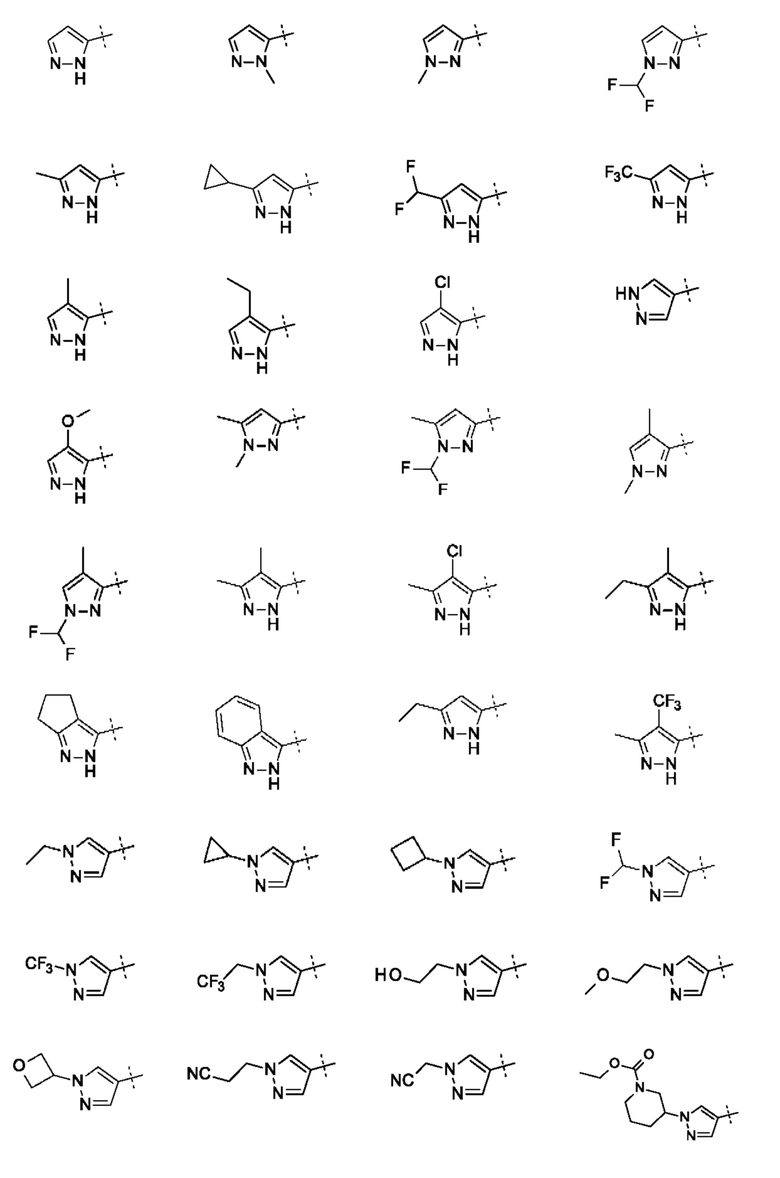
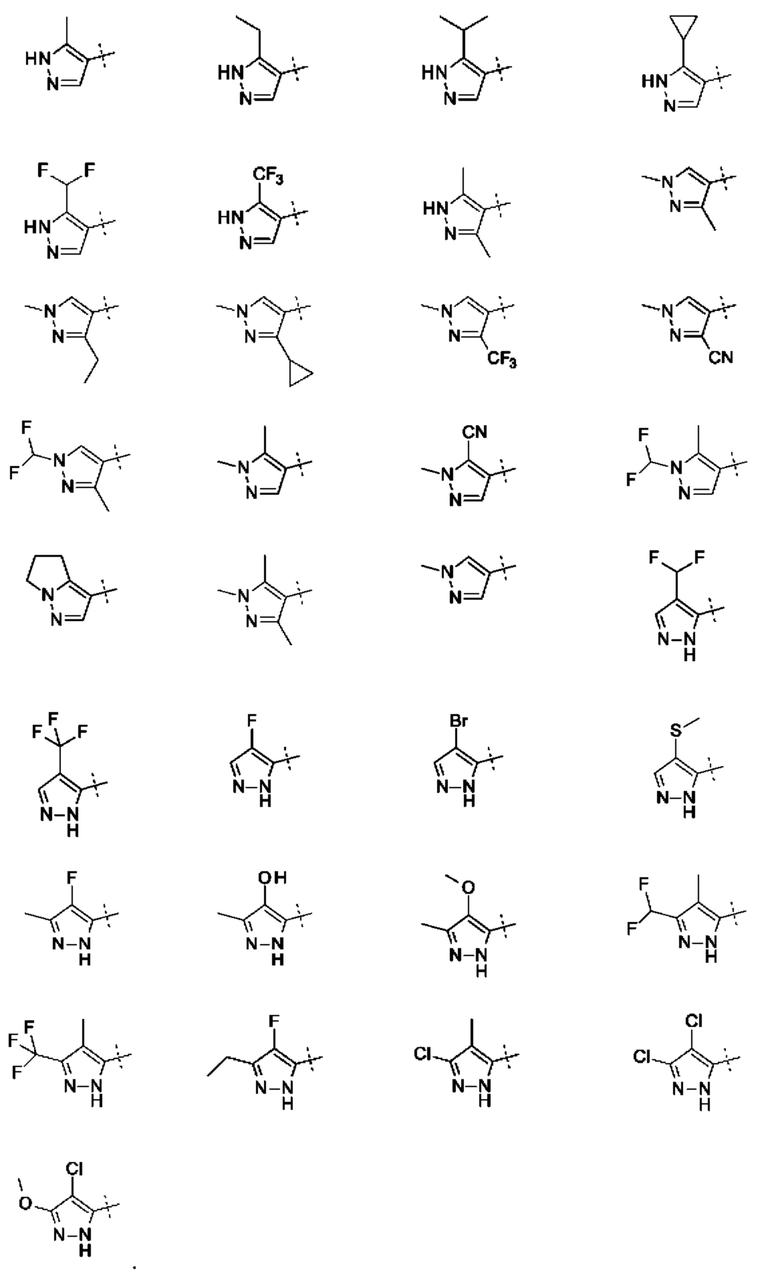
2. Соединение по п. 1, где X представляет собой N.
3. Соединение по п. 1 или 2, где R1 представляет собой Н или метил.
4. Соединение по любому из пп. 1-3, где R2 представляет собой Н.
5. Соединение по п. 1, представляющее собой соединение формулы (2а) или (2b)
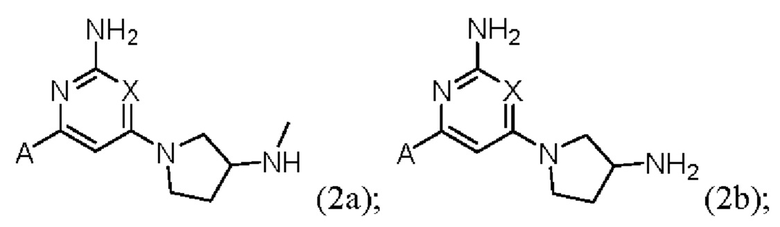
или его фармацевтически приемлемая соль.
6. Соединение по п. 5, представляющее собой соединение формулы (3а) или (3b)
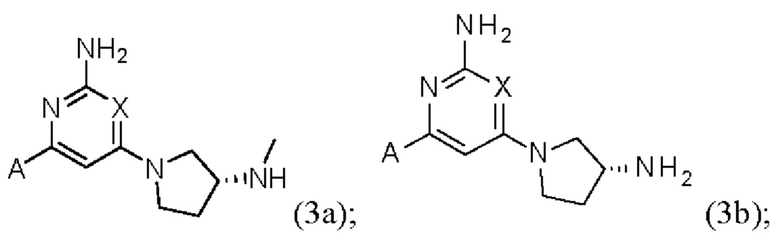
или его фармацевтически приемлемая соль.
7. Соединение по любому из пп. 1-6, где R3 выбран из Н, метила, CF3, CF2H, этила, циклопропила, циклобутила, CH2CF3, СН2СН2ОН, СН2СН2ОСН3, CH2CH2CN, CH2CN, оксетана, этилпиперидинкарбоксилата, или R4 и R3 соединены с образованием конденсированного 5-членного алифатического кольца.
8. Соединение по любому из пп. 1-7, где R4 или R5 выбран из метила, этила, циклопропила, циклобутила, пропила, изопропила, CF3, CF2H, фтора, хлора, брома, циано, метокси, или R4 и R5 соединены с образованием конденсированного 5- или 6-членного кольца, или R4 и R3 соединены с образованием конденсированного 5-членного алифатического кольца.
9. Соединение по любому из пп. 1-8, где А представляет собой
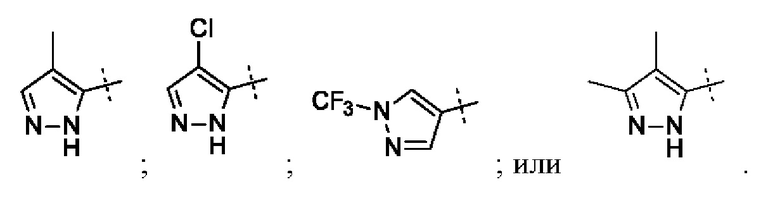
10. Соединение по п. 1, выбранное из группы, состоящей из
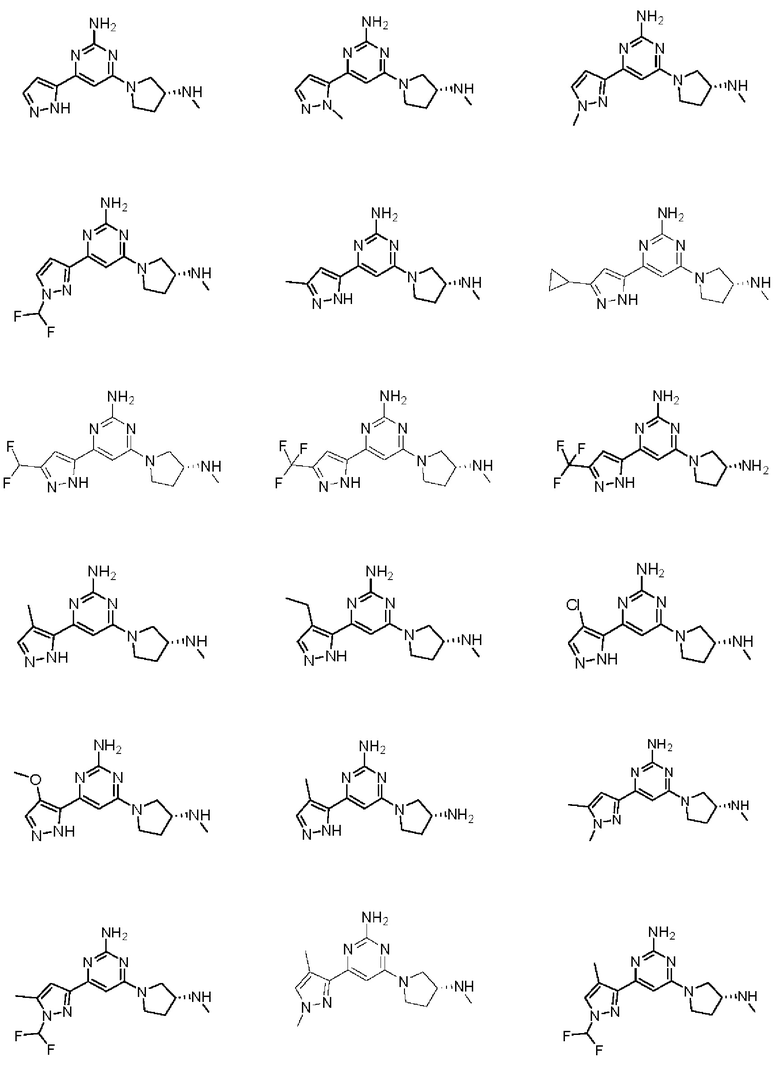
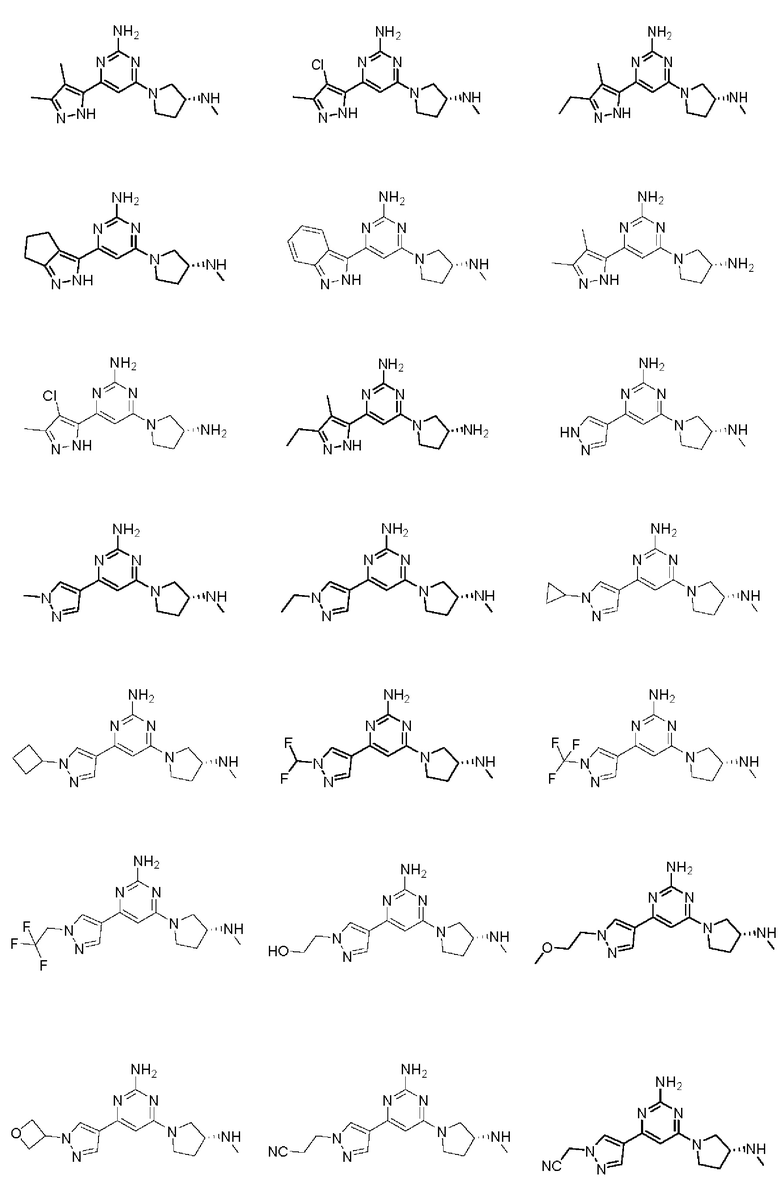
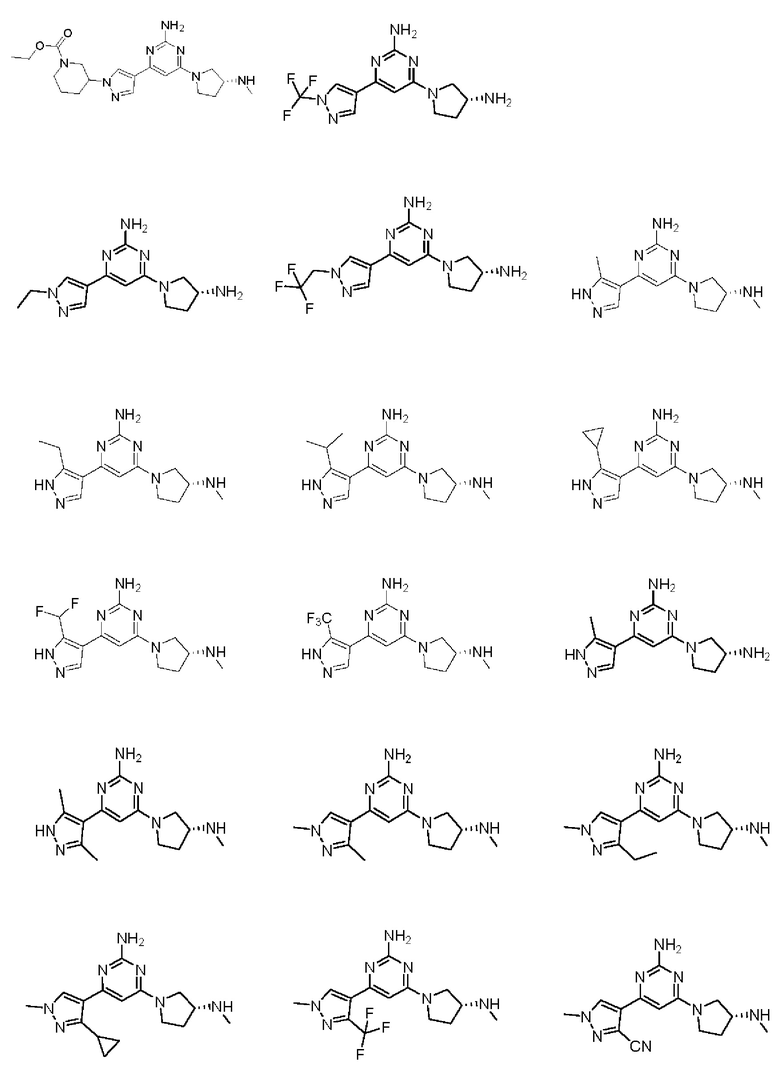
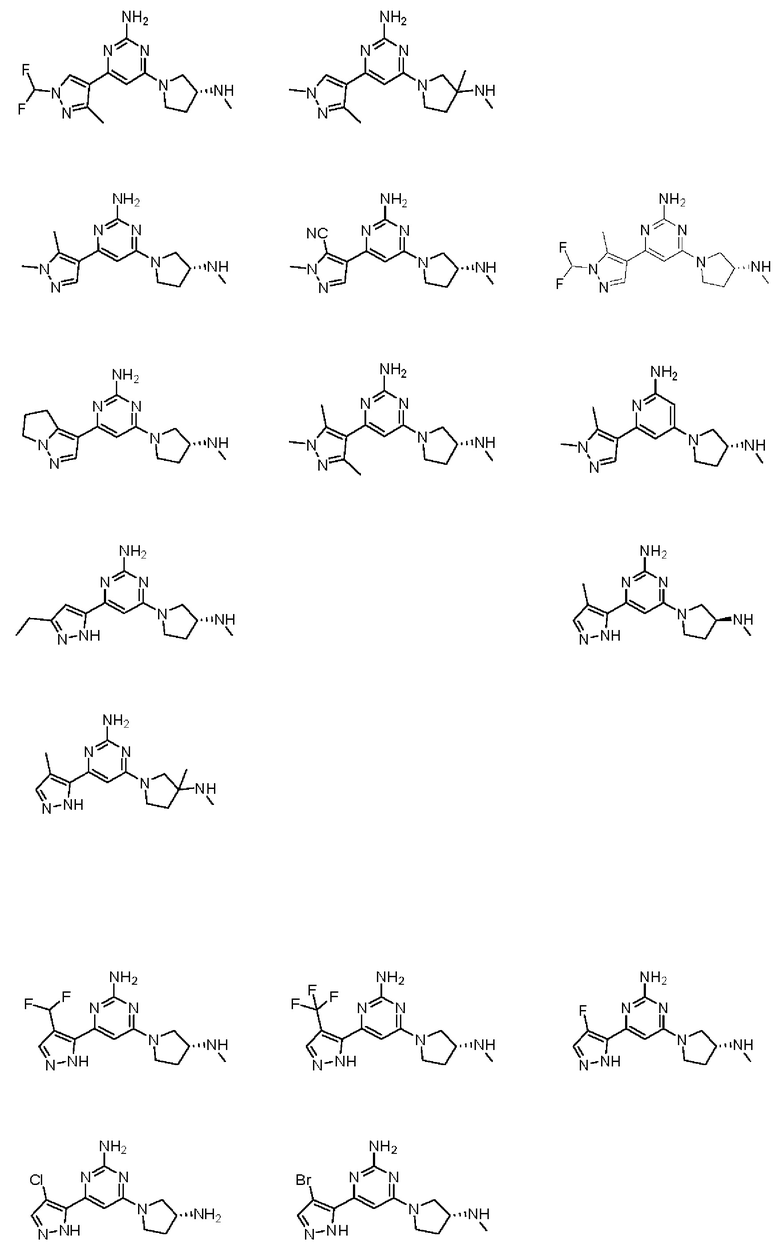
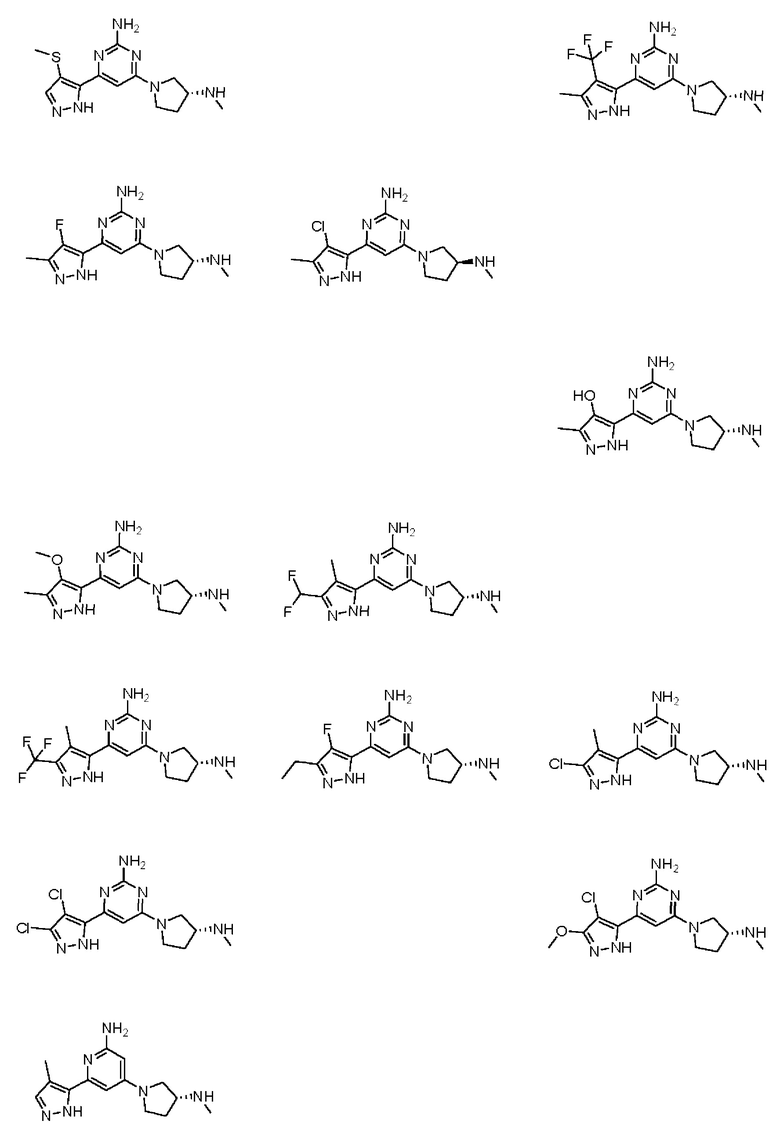
или его фармацевтически приемлемая соль.
11. Соединение по п. 1, представляющее собой
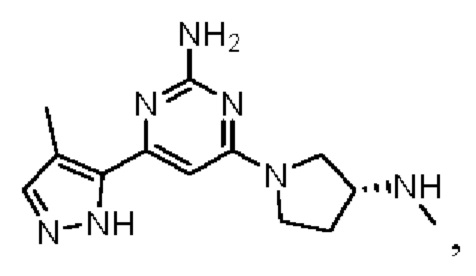
или его фармацевтически приемлемая соль.
12. Соединение по п. 1, представляющее собой
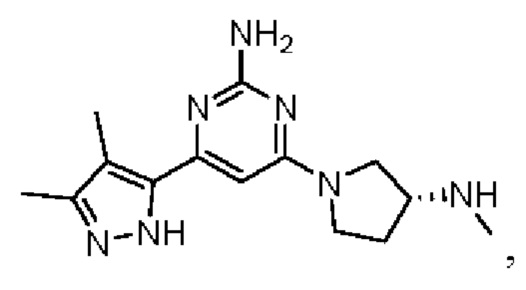
или его фармацевтически приемлемая соль.
13. Соединение по п. 1, представляющее собой
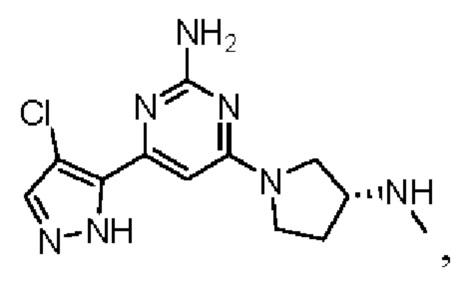
или его фармацевтически приемлемая соль.
14. Соединение по п. 1, представляющее собой
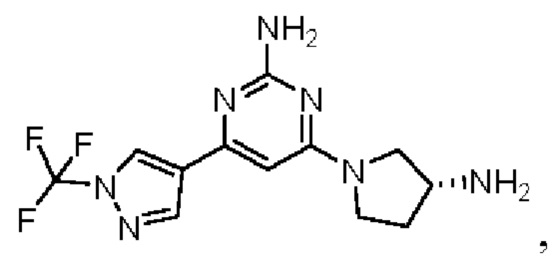
или его фармацевтически приемлемая соль.
15. Соединение по любому из пп. 1-14, обладающее активностью в отношении Н4-рецептора.
16. Соединение по п. 15, проявляющее низкую активность в отношении hERG.
17. Фармацевтическая композиция, имеющая активность антагониста рецептора Н4, где указанная фармацевтическая композиция содержит эффективное количество соединения по любому из пп. 1-16 и фармацевтически приемлемый эксципиент.
18. Соединение или композиция по любому из пп. 1-17 для применения в лечении аутоиммунного заболевания, расстройств, связанных с иммунной защитой организма, и нейропатической боли.
19. Соединение или композиция по любому из пп. 1-17 для применения при лечении воспалительных расстройств, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| R.J | |||
| ALTENBACH et al., Structure-Activity Studies on a Series of a 2-Aminopyrimidine-Containing Histamine H4 Receptor Ligands, J | |||
| MED | |||
| CHEM., 2008, vol | |||
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| КРИСТАЛЛИЧЕСКИЙ ДЕТЕКТОР | 1927 |
|
SU6571A1 |
| EP 1103551 A1, 30.05.2001 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-с]ПИРИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА ГИСТАМИНА 4 (hH4R) | 2012 |
|
RU2628074C2 |

