Область техники, к которой относится изобретение
Настоящее изобретение относится к пирролидиновым соединениям, являющимся антагонистами αvβ6 интегрина, фармацевтическим композициям, содержащим такие соединения, и к их применению в терапии, особенно при лечении состояний, для которых требуется антагонист αvβ6 интегрина, к применению соединения при изготовлении лекарственного препарата для лечения состояний, при которых требуется антагонист αvβ6 интегрина, и способу лечения или профилактики заболеваний у человека, для которых требуется антагонист αvβ6 интегрина.
Предпосылки создания изобретения
Белки суперсемейства интегринов представляют собой поверхностные рецепторы гетеродимерных клеток, состоящих из альфа и бета субъединиц. Имеются сообщения, по меньшей мере, о 18 альфа и 8 бета субъединицах, которые наглядно показаны в форме 24 различных альфа/бета гетеродимеров. Каждая цепь состоит из большого внеклеточного домена (>640 аминокислот для бета субъединиц, >940 аминокислот для альфа субъединиц), с трансмембранной областью, охватывающей около 20 аминокислот в цепи, и, как правило, коротким хвостом цитоплазмы с 30-50 аминокислотами в цепи. Было показано, что различные интегрины участвуют в многочисленных клеточных биопроцессах, включая клеточную адгезию во внеклеточном матриксе, межклеточные взаимодействия и влияние на миграцию клеток, пролиферацию, дифференциацию и выживание клеток (Barczyk et al, Cell and Tissue Research, 2010, 339, 269).
Рецепторы интегрина взаимодействуют с белками связывания через короткие белок-белок связывающие интерфейсы. Семейство интегринов можно сгруппировать в подсемейства, которые принимают участие в аналогичных мотивах распознавания таких лигандов. Основное подсемейство представляет собой RGD-интегрины, которые распознают лиганды, содержащие RGD мотив (аргинин-глицин-аспарагиновая кислота) в их белковой последовательности. Существуют 8 интегринов в этом подсемействе, а именно, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, αIIbβ3, α5β1, α8β1, где система обозначений указывает на то, что αvβ1, αvβ3, αvβ5, αvβ6 и αvβ8 имеют общую долю αv субъединиц с различными β субъединицами, и αvβ1, α5β1 и α8β1 имеют общую долю β1 субъединиц с различными α субъединицами. β1 субъединицы показаны в паре с 11 различными α субъединицами, из которых только 3 из перечисленных выше обычно распознают пептидный мотив RGD (Humphries et al, Journal of Cell Science, 2006, 119, 3901).
8 RGD-связывающих интегринов обладают различной аффинностью связывания и специфичностью в отношении различных RGD-содержащих лигандов. Лиганды включают белки, такие как фибронектин, витронектин, остеопонтин, и латентно связанные пептиды (LAP) трансформирующего фактора роста β1 и β3 (TGFβ1 и TGFβ3). Связывание интегрина с LAP TGFβ1 и TGFβ3 показано в некоторых системах для включения активации биологической активности TGFβ1 и TGFβ3, и последующих TGFβ-зависящих биологических процессов (Worthington et al, Trends in Biochemical Sciences, 2011, 36, 47). Разнообразие таких лигандов в сочетании с экспрессией системы RGD-связывания интегринов, создает многочисленные возможности для вмешательства в ход заболевания. Такие заболевания включают фиброзные заболевания (Margadant et al, EMBO reports, 2010, 11, 97), воспалительные заболевания, раковые заболевания (Desgrosellier et al, Nature Reviews Cancer, 2010, 10, 9), рестеноз и другие заболевания с ангиогенным компонентом (Weis et al, Cold Spring. Harb. Perspect. Med. 2011, 1, a 006478).
Значительное число антагонистов αv интегрина (Goodman et al, Trends in Pharmacological Sciences, 2012, 33, 405) описаны в литературе, включая ингибирующие антитела, пептиды и малые молекулы. Для антител они включают пан-αv антагонисты интетумумаб и абитузумаб (Gras, Drugs of the Future, 2015, 40, 97), селективный αvβ3 антагонист этарацизумаб и селективный αvβ6 антагонист STX-100. Циленгитид представляет собой циклический пептидный антагонист, который ингибирует как αvβ3, так и αvβ5, и SB-267268 является примером соединения (Wilkinson-Berka et al, Invest. Ophthalmol. Vis. Sci., 2006, 47, 1600), которое ингибирует как αvβ3, так и αvβ5. Изобретение соединений для действия в качестве антагонистов различных комбинаций αv интегринов дает возможность для создания новых средств специально для указанных конкретных заболеваний.
Легочный фиброз представляет собой конечную стадию некоторых интерстициальных легочных заболеваний, включая идиопатические интерстициальные пневмонии, и характеризуется чрезмерным осаждением внеклеточного матрикса в легочной интерстиции. Среди идиопатических интерстициальных пневмоний, идиопатический легочной фиброз (IPF) является наиболее распространенным и наиболее фатальным состоянием, при котором выживание составляет от 3 до 5 лет после диагностирования. Фиброз в IPF, как правило, является прогрессирующим, трудно поддающимся лечению при текущем фармакологическом вмешательстве и неумолимо приводит к дыхательной недостаточности вследствие облитерации альвеолярных функциональных единиц. IPF затрагивает примерно 500000 человек в Европе и США.
Существуют иммуногистохимические данные in vitro экспериментов на животных и IPF пациентах для поддержки ключевой роли при активации TGFβ1 эпителиально ограниченного интегрина αvβ6. Экспрессия такого интегрина низка в нормальных эпителиальных тканях и значительно активирована в поврежденном и воспаленном эпителии, включая активированный эпителий в IPF. Таким образом, направленное взаимодействие этого интегрина уменьшает теоретическую возможность вмешательства с широкими гомеостатическими функциями TGFβ. Частичное ингибирование αvβ6 интегрина блокирующим антителом было показано для предотвращения легочного фиброза без воспалительного обострения (Horan GS et al Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 2008 177: 56-65). Помимо легочного фиброза, αvβ6 также считается важным активатором фиброзных заболеваний других органов, в том числе печени и почек (Reviewed in Henderson NC et al Integrin-mediated regulation of TGFβ in Fibrosis, Biochimica et Biophysica Acta-Molecular Basis of Disease 2013 1832:891-896), из чего можно заключить, что αvβ6 антагонист может быть эффективным при лечении фиброзных заболеваний во многих органах.
В соответствии с результатами недавних наблюдений, показывающих, что некоторые RGD-связывающие интегрины могут связывать и активировать TGFβ, различные αv интегрины вовлечены в фиброзное заболевание (Henderson NC et al Targeting of αv Integrin identifies a core molecular pathway that regulates fibrosis in several organs Nature Medicine 2013 Vol 19, Number 12: 1617-1627; Sarrazy V et al Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction Cardiovasc Res 2014 102:407-417; Minagawa S et al Selective targeting of TGF-β activation to treat fibroinflammatory airway disease Sci Transl Med 2014 Vol 6, Issue 241: 1-14; Reed NI et al. The αvβ1 Integrin plays a critical in vivo role in tissue fibrosis Sci Transl Med 2015 Vol 7, Issue 288: 1-8). Поэтому ингибиторы против конкретных членов семейства RGD-связывающих интегринов, или со специфической селективностью фингерпринтов в отношении семейства RGD-связывающих интегринов могут быть эффективными при лечении фиброзных заболеваний во многих органах.
Зависимость активности лекарственного вещества от структуры (SAR) серии антагонистов интегрина против αvβ3, αvββ5, αvβ6 и αvβ8 была описана (Macdonald, SJF et al. Structure activity relationships of αv Integrin antagonists for pulmonary fibrosis by variation in aryl substituents. ACS MedChemLett 2014, 5, 1207-1212. 19 Sept 2014).
Задачей настоящего изобретения является предоставление αvβ6 антагонистов, предпочтительно с активностью против остальных αv интегринов, таких как αvβ1, αvβ3, αvβ5 или αvβ8.
Сущность изобретения
В первом аспекте настоящего изобретения предоставлено соединение формулы (I) или его соль, более предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль:
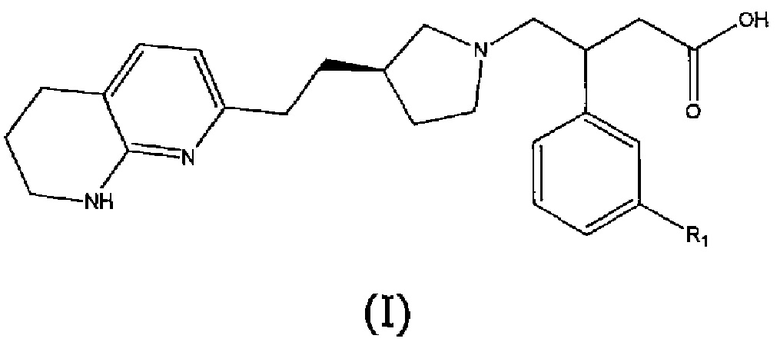
где R1 представляет собой пятичленный ароматический гетероцикл, выбранный из N- или C-связанного моно- или дизамещенного пиразола, N- или C-связанного, необязательно моно- или дизамещенного триазола или N- или C-связанного, необязательно дизамещенного имидазола, причем пятичленный ароматический гетероцикл может быть замещен одной или двумя группами, выбранными из атома водорода, метильной группы, этильной группы, атома фтора, гидроксиметильной группы, 2-гидроксипропан-2-ильной группы, трифторметильной группы, дифторметильной группы или фторметильной группы, за исключением случаев, когда R1 представляет собой N-связанный моно- или дизамещенный пиразол, R1 не является 3,5-диметил-1H-пиразол-1-илом, 5-метил-1H-пиразол-1-илом, 5-этил-3-метил-1H-пиразол-1-илом, 3,5-диэтил-1H-пиразол-1-илом, 4-фтор-3,5-диметил-1H-пиразол-1-илом, 3-метил-1H-пиразол-1-илом или 1H-пиразол-1-илом.
Соединения формулы (I) и их соли обладают антагонистической активностью в отношении αvβ6 интегрина, и считается, что они могут быть использованы при лечении или профилактике некоторых заболеваний. Термин αvβ6 антагонистическая активность в данном описании включает αvβ6 ингибирующую активность.
Во втором аспекте настоящего изобретения предоставлена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
В третьем аспекте настоящего изобретения предоставлено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии, в частности, при лечении заболевания или состояния, для которого требуется антагонист αvβ6 интегрина.
В четвертом аспекте настоящего изобретения предоставлен способ лечения или профилактики заболевания или состояния, для которого требуется антагонист αvβ6 интегрина, у человека, нуждающегося в этом, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В пятом аспекте настоящего изобретения предоставлено применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для лечения заболевания или состояния, для которого требуется антагонист αvβ6 интегрина.
В шестом аспекте настоящего изобретения предоставлено соединение формулы (XI):
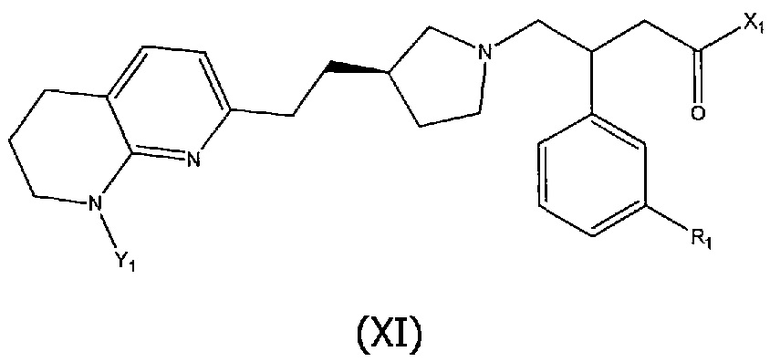
где R1 является таким, как определено выше,
X1 представляет собой гидроксил или фрагмент, который является гидролизуемым при метаболизме в организме человека с образованием соответствующего кислотного соединения формулы (I), в которой X1 представляет собой -OH;
Y1 представляет собой водород или фрагмент, который является гидролизуемым при метаболизме в организме человека с образованием соответствующего аминосоединения формулы (I), в которой Y1 представляет собой водород;
при условии, что когда X1 представляет собой гидроксил, тогда Y1 не является водородом.
Подробное описание изобретения
В первом аспекте настоящего изобретения предоставлено соединение формулы (I) или его соль, более предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль:
где R1 представляет собой пятичленный ароматический гетероцикл, выбранный из N- или C-связанного моно- или дизамещенного пиразола, N- или C-связанного, необязательно моно- или дизамещенного триазола или N- или C-связанного, необязательно дизамещенного имидазола, причем пятичленный ароматический гетероцикл может быть замещен одной или двумя группами, выбранными из атома водорода, метильной группы, этильной группы, атома фтора, гидроксиметильной группы, 2-гидроксипропан-2-ильной группы, трифторметильной группы, дифторметильной группы или фторметильной группы, за исключением случаев, когда R1 представляет собой N-связанный моно- или дизамещенный пиразол, R1 не является 3,5-диметил-1H-пиразол-1-илом, 5-метил-1H-пиразол-1-илом, 5-этил-3-метил-1H-пиразол-1-илом, 3,5-диэтил-1H-пиразол-1-илом, 4-фтор-3,5-диметил-1H-пиразол-1-илом, 3-метил-1H-пиразол-1-илом или 1H-пиразол-1-илом.
В одном из вариантов осуществления предоставлено соединение формулы (I) или его соль, более предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль:
где R1 представляет собой пятичленный ароматический гетероцикл, выбранный из N- или C-связанного моно- или дизамещенного пиразола, N- или C-связанного, необязательно моно- или дизамещенного триазола или N- или C-связанного, необязательно дизамещенного имидазола, причем пятичленный ароматический гетероцикл может быть замещен одной или двумя группами, выбранными из атома водорода, метильной группы, этильной группы, атома фтора, гидроксиметильной группы, 2-гидроксипропан-2-ильной группы, трифторметильной группы, дифторметильной группы или фторметильной группы, за исключением случаев, когда R1 представляет собой N-связанный моно- или дизамещенный пиразол, R1 не является 3,5-диметил-1H-пиразол-1-илом, 5-метил-1H-пиразол-1-илом, 5-этил-3-метил-1H-пиразол-1-илом, 3,5-диэтил-1H-пиразол-1-илом, 4-фтор-3,5-диметил-1H-пиразол-1-илом или 3-метил-1H-пиразол-1-илом.
В одном из вариантов осуществления R1 представляет собой C-связанный моно- или дизамещенный пиразол.
В другом варианте осуществления R1 представляет собой N-связанный моно- или дизамещенный пиразол.
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно моно- или дизамещенный триазол.
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол.
В одном из вариантов осуществления R1 представляет собой C-связанный моно- или дизамещенный пиразол, выбранный из 3-метил-1H-пиразол-5-ила и 1,4-диметил-1H-пиразол-5-ила.
В другом варианте осуществления R1 представляет собой N-связанный моно- или дизамещенный пиразол, выбранный из (2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ила, 3-(трифторметил)-1H-пиразол-1-ила, 3-(гидроксиметил)-5-метил-1H-пиразол-1-ила и 3-(фторметил)-5-метил-1H-пиразол-1-ила.
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно моно- или дизамещенный триазол, выбранный из 4H-1,2,4-триазол-4-ила, 3,5-диметил-1H-1,2,4-триазол-1-ила, 3-метил-4H-1,2,4-триазол-4-ила, 1H-1,2,3-триазол-1-ила, 1H-1,2,4-триазол-1-ила.
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол, выбранный из 1H-имидазол-1-ила и моно- или диметилимидазола, 1-метил-1H-имидазол-2-ила, 4-метил-1H-имидазол-2-ила, (1,4-диметил-1H-имидазол-2-ила) и (2,4-диметил-1H-имидазол-5-ила).
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол, выбранный из 1H-имидазол-1-ила и моно- или диметилимидазола, 1-метил-1H-имидазол-2-ила, 4-метил-1H-имидазол-2-ила и (1,4-диметил-1H-имидазол-2-ила).
В другом варианте осуществления R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол, выбранный из 1H-имидазол-1-ила, и (1,4-диметил-1H-имидазол-2-ила) и (2,4-диметил-1H-имидазол-5-ила).
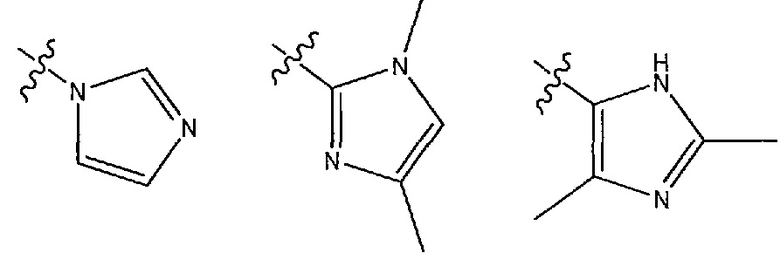
В одном из вариантов осуществления R1 выбран из следующих гетероциклов:



В некоторых вариантах осуществления соединение формулы (I) или его соль имеет структурную формулу (IA):
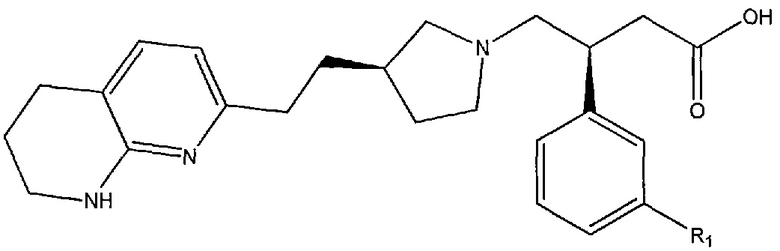
или ее фармацевтически приемлемую соль.
В других вариантах осуществления соединение формулы (I) или его соль имеет структурную формулу (IB):

или ее фармацевтически приемлемую соль.
Следует понимать, что настоящее изобретение охватывает все комбинации описанных выше особых и предпочтительных групп.
В некоторых вариантах осуществления R1 представляет собой 3-метил-1H-пиразол-5-ильную, 1,4-диметил-1H-пиразол-5-ильную, 3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ильную, 3-(трифторметил)-1H-пиразол-1-ильную, 3-(гидроксиметил)-5-метил-1H-пиразол-1-ильную, 3-(фторметил)-5-метил-1H-пиразол-1-ильную группу.
В других вариантах осуществления R1 представляет собой N- или C-связанный, необязательно моно- или дизамещенный триазол, выбранный из 4H-1,2,4-триазол-4-ила, 3,5-диметил-1H-1,2,4-триазол-1-ила, 3-метил-4H-1,2,4-триазол-4-ила, 1H-1,2,3-триазол-1-ила и 1H-1,2,4-триазол-1-ила.
В других вариантах осуществления R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол, выбранный из 1H-имидазол-1-ила и моно- или диметилимидазола, 1-метил-1H-имидазол-2-ила, 4-метил-1H-имидазол-2-ила, предпочтительно 1H-имидазол-1-ила, и (1,4-диметил-1H-имидазол-2-ила) и (2,4-диметил-1H-имидазол-5-ила).
В некоторых вариантах осуществления R1 представляет собой 3-метил-1H-пиразол-5-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 1,4-диметил-1H-пиразол-5-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3-(трифторметил)-1H-пиразол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3-(гидроксиметил)-5-метил-1H-пиразол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3-(фторметил)-5-метил-1H-пиразол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 4H-1,2,4-триазол-4-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3,5-диметил-1H-1,2,4-триазол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 3-метил-4H-1,2,4-триазол-4-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 1H-1,2,3-триазол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 1H-1,2,4-триазол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 1H-имидазол-1-ильную группу.
В некоторых вариантах осуществления R1 представляет собой 1,4-диметил-1H-имидазол-2-ильную группу.
В некоторых вариантах осуществления R1 представляет собой (2,4-диметил-1H-имидазол-5-ильную) группу.
В одном из вариантов осуществления данное соединение выбрано из:
3-(3-(3-метил-1H-пиразол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(3,5-диметил-1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(3-метил-4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)-3-(3-(3-(трифторметил)-1H-пиразол-1-ил)фенил)бутановой кислоты,
3-(3-(1H-1,2,3-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(1H-имидазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
3-(3-(1,4-диметил-1H-имидазол-2-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
(S)-3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты и
(S)-3-(3-(1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты,
или их фармацевтически приемлемой соли.
Соединения формулы (I) или (IA) или (IB) имеют как основную аминогруппу, так и группу карбоновой кислоты, и, следовательно, могут образовывать внутренние соли, т.е. цвиттер-ионные или внутримолекулярные соли. Поэтому, в одном из вариантов осуществления соединение формулы (I) находится в форме цвиттер-ионной соли. В другом варианте осуществления соединение формулы (IA) находится в форме цвиттер-ионной соли. В другом варианте осуществления соединение формулы (IB) находится в форме цвиттер-ионной соли.
Следует принимать во внимание, что настоящее изобретение предусматривает соединения формулы (I) в качестве родительского соединения и его соли, например, в форме его фармацевтически приемлемой соли. В одном из вариантов осуществления настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемой соли.
Для обзора подходящих солей см. Berge et al., J. Pharm. Sci., 66:1-19, (1977). Подходящие фармацевтически приемлемые соли перечислены авторами P H Stahl и C G Wermuth, в Handbook of Pharmaceutical Salts; Properties, Selection and Use, Weinheim/Zurich: Wiley-VCH/VHCA, 2002. Подходящие фармацевтически приемлемые соли могут включать кислотно-аддитивные соли с неорганическими кислотами, такими, например, как хлористоводородная кислота, бромистоводородная кислота, ортофосфорная кислота, азотная кислота, фосфорная кислота или серная кислота, или с органическими кислотами, такими, например, как метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, уксусная кислота, пропионовая кислота, молочная кислота, лимонная кислота, фумаровая кислота, яблочная кислота, янтарная кислота, салициловая кислота, малеиновая кислота, глицерофосфорная кислота, винная, бензойная, глутаминовая, аспарагиновая, бензолсульфоновая, нафталинсульфоновая, такая как 2-нафталинсульфоновая, гексановая кислота или ацетилсалициловая кислота.
Как правило, фармацевтически приемлемая соль может быть легко получена при использовании требуемой кислоты или основания, при необходимости. Полученную в результате соль можно осадить из раствора и отделить фильтрованием, или можно извлечь выпариванием растворителя.
Другие нефармацевтически приемлемые соли, например, формиаты, оксалаты или трифторацетаты, могут быть использованы, например, при выделении соединений формулы (I), и включены в объем настоящего изобретения.
Фармацевтически приемлемая аддитивная соль основания может быть получена взаимодействием соединения формулы (I) с подходящим органическим основанием, (например, триэтиламином, этаноламином, триэтаноламином, холином, аргинином, лизином или гистидином), необязательно в подходящем растворителе, с получением аддитивной соли основания, которую обычно выделяют, например, кристаллизацией или фильтрованием. Фармацевтически приемлемые соли неорганических оснований включают аммониевые соли, соли щелочных металлов, таких как натрий и калий, соли щелочноземельных металлов, таких как кальций и магний, и соли органических оснований, включая соли первичных, вторичных и третичных аминов, таких как изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин и N-метил-D-глюкамин.
В одном из вариантов осуществления соединение формулы (I) находится в форме родительского соединения.
Настоящее изобретение включает в свой объем все возможные стехиометрические и нестехиометрические формы солей соединений формулы (I).
Следует отметить, что многие органические соединения могут образовывать комплексы с растворителями, в которых они подвергаются взаимодействию или из которых их осаждают или кристаллизуют. Такие комплексы известны как ʺсольватыʺ. Например, комплекс с водой известен как ʺгидратʺ. Растворители с высокой точкой кипения и/или способные образовывать водородные связи, такие как вода, ксилол, N-метилпирролидинон, метанол и этанол, могут быть использованы для формирования сольватов. Способы идентификации сольватов включают, но, не ограничиваясь ими, ЯМР и микроанализы. Следует отметить, что кристаллические формы необязательно могут быть в сольватированной форме, например, фармацевтически приемлемых сольватов, таких как гидраты, которые могут быть стехиометрическими гидратами, а также соединениями, содержащими различные количества воды. Сольваты включают стехиометрические сольваты и нестехиометрические сольваты. Соединения формулы (I) могут существовать в сольватированной и несольватированной форме.
Соединения формулы (I) могут быть в кристаллической или аморфной форме. Более того, некоторые кристаллические формы соединений формулы (I) могут существовать в виде полиморфов, которые включены в объем настоящего изобретения. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и дифференцированы при использовании ряда общепринятых аналитических методов, включая, но, не ограничиваясь ими, дифрактограммы порошкового рентгеноструктурного анализа (XRPD), инфракрасный спектр (ИК), Романовский спектр, дифференциальную сканирующую калометрию (DSC), термогравиметрические анализы (TGA) и твердофазный ядерно-магнитный резонанс (SSЯМР).
Описанные в данном описании соединения содержат два асимметрических центра, таким образом, могут образовывать оптические изомеры, например, диастереоизомеры и энантиомеры. Соответственно, настоящее изобретение охватывает изомеры соединений формулы (I) или в виде изолированных индивидуальных изомеров, которые по существу свободны от других изомеров (т.е. чистые), или в виде смесей. Изолированный индивидуальный изомер, который по существу свободен от другого изомера (т.е. чистый), может быть выделен таким образом, что другой изомер будет присутствовать в количестве менее чем 10%, предпочтительно менее чем 1%, например, менее чем около 0,1%.
Специалисту в данной области техники будет понятно, что определенные диастереоизомеры могут быть менее активными, чем другие, и что активность индивидуального диастереоизомера может быть снижена ниже выбранного предела.
В одном из вариантов осуществления данное соединение представляет собой (IA):
или его фармацевтически приемлемую соль.
В другом варианте осуществления данное соединение представляет собой (IB):
или его фармацевтически приемлемую соль.
Разделение изомеров может быть осуществлено общепринятыми методиками, известными специалисту в данной области техники, например, фракционированной кристаллизацией, хроматографией, ВЭЖХ или комбинацией таких методик.
Соединения формулы (I) могут существовать в одной из различных таутомерных форм. Следует понимать, что настоящее изобретение охватывает все таутомеры соединений формулы (I) либо в виде индивидуальных таутомеров, либо в виде их смесей.
Из изложенного выше следует, что включенными в объем настоящего изобретения являются сольваты, изомеры и полиморфные формы соединений формулы (I) и их соли.
Определенные соединения формулы (I) могут быть помечены [18F] с образованием соединения, подходящего для применения в качестве лиганда PET для диагностики заболеваний, таких как идиопатический легочный фиброз. Меченные [18F] соединения включены в объем настоящего изобретения.
В шестом аспекте настоящего изобретения предоставлено соединение формулы (XI):
где R1 является таким, как определено выше,
X1 представляет собой гидроксил или фрагмент, который является гидролизуемым при метаболизме в организме человека с образованием соответствующего кислотного соединения формулы (I), в которой X1 представляет собой -OH;
Y1 представляет собой водород или фрагмент, который является гидролизуемым при метаболизме в организме человека с образованием соответствующего аминосоединения формулы (I), в которой Y1 представляет собой водород;
при условии, что когда X1 представляет собой гидроксил, тогда Y1 не является водородом.
В некоторых вариантах осуществления X1 может представлять собой фрагмент -ORa, так что соединение формулы (I) представляет собой сложный эфир.
Например, фрагмент Ra может быть выбран из C1-6алкила (с приведенными выше исключениями), такого как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил (и его изомеры) или гексил (и его изомеры); или из C1-6алкоксиалкила, такого как 2-метоксиэтил; или из C1-6алкиламиноалкила, такого как 2-(диметиламино)этил; или из C1-6циклических карбонатных групп, таких как (5-метил-2-оксо-1,3-диоксол-4-ил)метил; или C1-6ацилоксиалкила, такого как (пивалоилокси)метил.
Например, фрагмент Ra может быть выбран из арильных групп, таких как фенил, 5-инданил или L-тирозинил.
Например, фрагмент Ra может быть выбран из групп, содержащих аминогруппу или амидную группу, таких как C1-6 группы формулы: -(CH2)nNRbRc или -(CH2)nCONRbRc, где n равен 1-3, и Ra и Rb независимо представляют собой H или C1-6алкил, циклоалкил, гетероциклил, или Ra и Rb, вместе образуют циклическую группу, такую как морфолинил. Примеры таких фрагментов включают диметиламиноэтил, 2-(4-морфолино)этил и диметиламино-2-оксоэтил.
Например, фрагмент Ra может быть выбран из гидроксила, содержащего альфа-аминокислоту, такую как L-серин и L-треонин.
Например, фрагмент Ra может представлять собой циклический карбонат формулы:
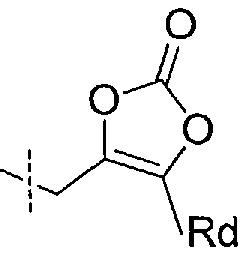
где Rd представляет собой водород, метил, этил или изопропил.
Например, фрагмент Ra может быть выбран из -CHRe-O-CO-Rf, где Re представляет собой водород или C1-3алкил, такой как метил, этил или изопропил, и Rf представляет собой C1-4алкил, такой как метил, этил, изопропил, трет-бутил или C5-6циклоалкил, или тетрагидропиранил.
Например, фрагмент Ra может быть выбран из -CH(Rg)-O-CO-O-Ri, где Rg представляет собой водород или C1-3алкил, такой как метил, этил или изопропил, и Ri представляет собой C1-4алкил, такой как метил, этил, трет-бутил или C5-6циклоалкил, или тетрагидропиранил.
В некоторых вариантах осуществления X1 может представлять собой фрагмент -NHRj, так что соединением формулы (I) является амид, где Rj может представлять собой, например, C1-6алкил. Например, соединение формулы (I) может являться амидом, полученным из аминокислоты, связанным с альфа-аминогруппой данной аминокислоты, например, существующей в природе L-протеиногенной аминокислоты, такой как глицин, аланин, фенилаланин, лейцин, валин, изолейцин, пролин, метионин, цистеин, серин, треонин, гистидин, тирозин, триптофан, лизин, аспарагин, глутамин, глутаминовая кислота, аспарагиновая кислота или аргинин, или дипептида приведенных выше протеиногенных аминокислот. Например, Rj может представлять собой фрагмент протеиногенной аминокислоты, такой как фрагмент L-лизина, связанный с боковой цепью эпсилон-аминогруппы данной аминокислоты, например, -(CH2)4CH(NH2)CO2H.
Например, Rj может представлять собой сульфонамидный фрагмент, такой как -SO2-Rk, где Rk представляет собой C1-6алкил, такой как метил, или -NRmRn, где Rm и Rn независимо представляют собой H или C1-6алкил, циклоалкил, гетероциклил, или Rm и Rn, вместе образуют циклическую группу, такую как морфолинил.
В некоторых вариантах осуществления Y1 может представлять собой водород.
В одном из вариантов осуществления (a) Y1 представляет собой C1-6алкил, замещенный C1-6 циклической карбонатной группой, например, (оксодиоксоленил)метильной группой, такой как:
,
где Rd представляет собой C1-3алкил, такой как метил, этил или изопропил.
В другом варианте осуществления (b) Y может представлять собой карбаматную группу, например,
 ,
,
где Rl представляет собой C1-6алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, н-пентил или н-гексил.
В другом варианте осуществления Rl представляет собой C1-6алкил, замещенный группой OH или HMe2, такой как -CH2CH2OH или -CH2CH2NMe2.
В другом варианте осуществления (c) Y1 может представлять собой группу общей структуры:
 ,
,
в которой Rm представляет собой водород, метил или изопропил, и Rn представляет собой C1-6алкил, например, метил, этил, изопропил, трет-бутил, циклоалкил, например, циклобутил, циклопентил, циклогексил, гетероциклил, например, 4-тетрагидропиранил, арил, например, фенил, замещенный фенил, гетероарил, например, 2-, 3- или 4-пиридил.
В другом варианте осуществления (d) Y1 может представлять собой группу общей структуры:
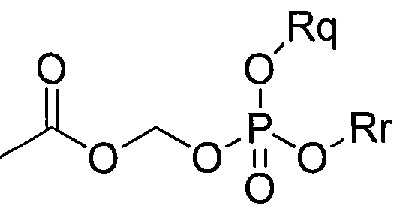 ,
,
где Rq и Rr независимо представляют собой водород, фенил, нафтил, алкил, Et2NCOCH2-, или Rq и Rr могут образовывать C1-6 кольцо, такое как салигенин.
В другом варианте осуществления соединения формулы (I) представляют собой двойные пролекарства, где X1 и Y1 являются такими, как определено выше, в любых комбинациях.
Настоящее изобретение относится ко всем пролекарствам соединений формулы (I) и их фармацевтически приемлемым солям, которые при введении реципиенту способны обеспечивать (прямо или косвенно) соединение формулы (I) или его фармацевтически приемлемую соль, или его активный метаболит или остаток. Другие подходящие пролекарства соединений формулы (I) очевидны специалисту в данной области техники (см., например, Burger's Medicinal Chemistry and Drug Discovery, 5th Edition, Vol 1: Principles and Practice, и J. Rautio et al (Nature Reviews Drug Discovery 2008, 7, 255-270).
Получение соединений
Соединения настоящего изобретения могут быть получены различными способами, включая стандартные химические. Любая определенная выше переменная будет и впредь иметь ранее определенный смысл, если не указано иное. Ниже приводятся иллюстративные общие синтетические способы, и затем получение конкретных соединений настоящего изобретения в рабочих примерах.
Соединения структурной формулы (I) могут быть получены способом, включающим сначала удаление защитных групп, т.е. расщепление сложноэфирной группы, с последующим преобразованием в соль соединения структурной формулы (II):

где R1 является таким, как определено выше, и
R2 представляет собой C1-C6алкильную группу, например, метил или трет-бутил.
Удаление защитных групп соединения структурной формулы (II), где R2 представляет собой метил, можно осуществить основным гидролизом с использованием, например, водного гидроксида натрия или гидроксида калия, в подходящем растворителе, таком как метанол, 1,4-диоксан.
Удаление защитных групп соединения структурной формулы (II), где R2 представляет собой трет-бутил, можно осуществить кислотным расщеплением при использовании, например, трифторуксусной кислоты или HCl, в подходящем растворителе, таком как дихлорметан, 1,4-диоксан или вода.
После расщепления сложноэфирной группы, образовавшийся в результате продукт может быть преобразован в требуемую соль способами, известными специалисту в данной области техники.
Соединения структурной формулы (II), где R1 представляет собой пятичленное гетероциклическое ароматическое кольцо, связанное через атом углерода, могут быть получены методами сочетания, включающими взаимодействие соединения структурной формулы (III):
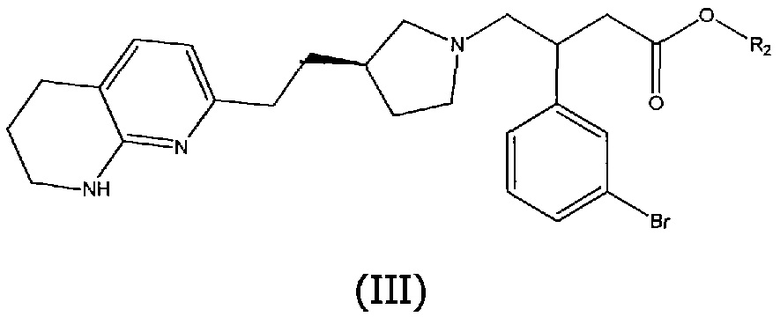
с боронатным эфиром или бороновой кислотой указанного ароматического гетероциклического соединения структурной формулы (IV):

где R3 представляет собой водород или циклический спирт, такой как пинакол. Соединения структурной формулы (IV) могут быть использованы в форме чистой бороновой кислоты (R3=H), или в форме сложного боронового эфира (R3=алкильная группа, например, пинаколил), который может быть преобразован in situ в бороновую кислоту в присутствии воды и основания, такого как гидроксид калия, и X, Y и Z представляют собой водород или алкильные группы, например, метил, в присутствии основания, такого как трифосфат калия, и катализатора, такого как хлор(динорбонилфосфино)(2'-диметиламино-1,1'-бифенил-2-ил)палладий (II), в подходящем растворителе, таком как водный этанол, и при повышенной температуре, например, 130°C, необязательно в микроволновом реакторе. В ходе процесса сочетания сложноэфирная метильная группа соединения (II) может гидролизоваться в основных реакционных условиях с получением соединения (I) непосредственно без необходимости проведения отдельной стадии гидролиза.
Соединения структурной формулы (III) могут быть получены методами сочетания, включающими взаимодействие соединения структурной формулы (V):
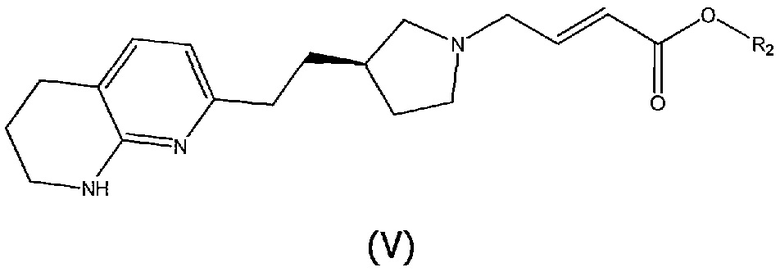
где R2 является таким, как определено выше, и геометрия двойной связи может быть в виде (E) или смеси (E) и (Z) изомеров, предпочтительно в виде чистого (E) изомера, с (3-бромфенил)бороновой кислотой (доступной от Aldrich) в присутствии подходящего катализатора, такого как димер хлор(1,5-циклооктадиен)родия(l) или тетрафторбората бис(норборнадиен)родия(I), необязательно в присутствии хирального лиганда, такого как (R)-BINAP, в подходящем растворителе, таком как 1,4-диоксан, и при повышенной температуре, например, 95°C.
Реакция сочетания в присутствии (R)-BINAP обеспечивает диастереоизомерную смесь с преобладающим изомером. Диастереоизомеры могут быть разделены различными методами разделения, включая кристаллизацию, хроматографию или предпочтительно препаративную хиральную ВЭЖХ на колонке Chiralpak или Chiralcel. Преобладающий диастереоизомер при использовании (R)-BINAP имеет (S) конфигурацию.
Соединения структурной формулы (V) могут быть получены реакцией алкилирования соединения структурной формулы (VI):

с (E)-метил 4-бромбут-2-еноатом, где R2 представляет собой метил, или с (E)-трет-бутил 4-бромбут-2-еноатом, где R2 представляет собой трет-бутил, и в присутствии основания, такого как диизопропилэтиламин, в подходящем растворителе, таком как дихлорметан.
Альтернативно, алкилирование соединения структурной формулы (VI) может быть осуществлено сочетанием соединения структурной формулы (VI) с (E)-метил 4-ацетоксибут-2-еноатом, где R2 представляет собой метил, или с трет-бутил (E)-трет-бутил 4-ацетоксибут-2-еноатом, где R2 представляет собой трет-бутил, в присутствии палладиевого катализатора, такого как 1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), в присутствии основания, такого как диизопропилэтиламин или триэтиламин, и в подходящем растворителе, таком как дихлорметан, при температуре окружающей среды.
Соединения структурной формулы (VI), могут быть получены отщеплением трет-бутоксикарбонильной защитной группы соединения формулы (VII):
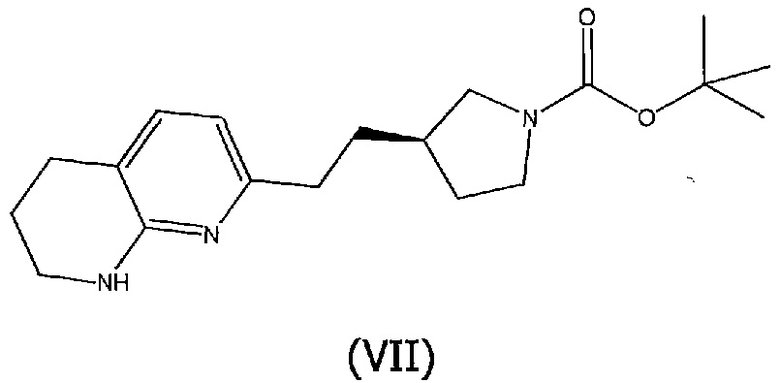
при использовании кислоты, такой как хлористый водород в 1,4-диоксане.
Соединения структурной формулы (VII) могут быть получены из соединения структурной формулы (VIII):
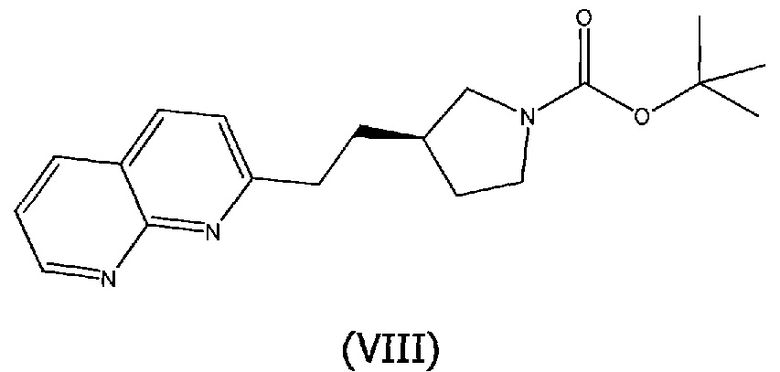
гидрированием над катализатором, таким как 5% родий на углероде, в растворителе, таком как этанол.
В альтернативном способе, соединения структурной формулы (II) могут быть получены из соединения формулы (V) путем взаимодействия с бороновой кислотой или сложным боронатным эфиром структурной формулы (IX):

Реакция сочетания в присутствии R-BINAP обеспечивает диастереоизомерную смесь с преобладающим изомером. Диастереоизомеры могут быть разделены различными методами разделения, включая кристаллизацию, хроматографию или предпочтительно препаративную хиральную ВЭЖХ. Соединения структурной формулы (IX) могут быть использованы в виде чистой бороновой кислоты (R3=H) или в виде эфира бороновой кислоты (R3=алкильная группа, например, пинакол), который может быть преобразован in situ в бороновую кислоту в присутствии воды и основания, такого как гидроксид калия. Сложноэфирная метильная группа соединения (II) может быть гидролизована в основных реакционных условиях в ходе процесса сочетания с получением соединения (I) непосредственно без необходимости проведения отдельной стадии гидролиза.
Случаи, при которых R1 представляет собой связанный через азот ароматический гетероцикл, как определено выше, и R3 представляет собой водород или циклический спирт, такой как пинакол.
Соединения структурной формулы (IX), где R3 представляет собой пинакол, могут быть получены из соединения структурной формулы (X):

с бис(пинаколато)дибороном (доступный от Aldrich), в присутствии палладиевого катализатора, такого как трис(дибензилиденацетон)дипалладий (доступный от Aldrich), и в присутствии фосфинового лиганда, такого как 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил (X-PHOS) (доступный от Aldrich), и в присутствии ацетата калия, в инертном растворителе, таком как 1,4-диоксан, при повышенной температуре, например, 110°C, и в инертной атмосфере, такой как азот. Добавление воды к реакционной смеси в конце реакции вызывает гидролиз полученного в результате сложного пинаколатового эфира, обеспечивая требуемую бороновую кислоту.
Соединения структурной формулы (X) могут быть получены способами, описанными в данном описании.
Соединение структурной формулы (VIII) [(R)-трет-бутил 3-(2-(1,8-нафтиридин-2-ил)этил)пирролидин-1-карбоксилат] может быть получено способами, приведенными на схеме 1.
Схема 1
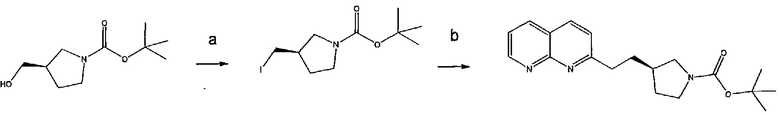
Реагенты и условия: (a) йод, имидазол, трифенилфосфин, ДХМ, 0°C; (b) 2-метил-[1,8]-нафтиридин, LiN(TMS)2, ТГФ, 0°C.
Следует отметить, что при любых путях синтеза, описанных выше, может быть выгодным защита одной или более функциональных групп. Примеры защитных групп и средств для их удаления можно найти в T. W. Greene 'Protective Groups in Organic Synthesis' (3rd edition, J. Wiley and Sons, 1999). Подходящие аминозащитные группы включают ацил (например, ацетил, карбамат (например, 2',2',2'-трихлорэтоксикарбонил, бензилоксикарбонил или трет-бутоксикарбонил) и арилалкил (например, бензил), которые могут быть удалены гидролизом (например, при использовании кислоты, такой как хлористоводородная кислота в диоксане или трифторуксусная кислота в дихлорметане) или восстановительно (например, гидрогенолизом бензильной или бензилоксикарбонильной группы или восстановительным удалением 2',2',2'-трихлорэтоксикарбонильной группы с использованием цинка в уксусной кислоте), при необходимости. Другие подходящие аминозащитные группы включают трифторацетил (-COCF3), который может быть удален основным каталитическим гидролизом.
Следует отметить, что при любых путях синтеза, описанных выше, точный порядок синтетических стадий, по которым различные группы и фрагменты вводятся в молекулу, могут варьироваться. Это будет находиться в компетенции практикующего специалиста в данной области техники для определения того, какие группы или фрагменты, вводимые на одной из стадий процесса, не повлияет на последующие преобразования и реакции, а также выбора порядка стадий синтеза, соответственно.
Абсолютную конфигурацию соединения (I) можно получить, следуя независимому энантиоселективному асимметричному синтезу из промежуточного соединения известной абсолютной конфигурации. Альтернативно, энантиомерно чистое соединение (I) может быть преобразовано в соединение, чья абсолютная конфигурация известна. В любом случае, сравнение спектроскопических данных, оптического вращения и времени удерживания на аналитической хиральной ВЭЖХ может быть использовано для подтверждения абсолютной конфигурации. Третьим вариантом, где это возможно, является определение абсолютной конфигурации из рентгеновской кристаллической структуры.
Некоторые соединения формул (II)-(X) также считаются новыми, и поэтому составляют еще один дополнительный аспект настоящего изобретения.
Способы применения
Считается, что соединения формулы (I) и их соли обладают антагонистической активностью в отношении αv интегрина, предпочтительно активностью в отношении рецептора αvβ6, и, таким образом, потенциальной полезностью при лечении заболеваний или состояний, для которых рекомендован антагонист αvβ6.
Настоящее изобретение, таким образом, предоставляет соединение формулы (I) или его фармацевтически приемлемую соль для применения в терапии. Соединение формулы (I) или его фармацевтически приемлемая соль могут применяться при лечении заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина.
Настоящее изобретение также предоставляет соединение формулы (I) или его фармацевтически приемлемую соль для применения при лечении заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина.
Также предоставлено применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для лечения заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина.
Также предоставлен способ лечения заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина, у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Подходящим субъектом, нуждающимся в этом, является млекопитающее, предпочтительно человек.
Как используется в данном описании, термин "эффективное количество" означает такое количество лекарственного средства или фармацевтического средства, которое вызовет биологический или медицинский ответ ткани, системы, животного или человека, которого добивается, например, исследователь или врач-клиницист. Кроме того, термин ʺтерапевтически эффективное количествоʺ означает любое количество, которое, по сравнению с соответствующим субъектом, который не получал такого количества, приводит в результате к улучшению лечения, здоровья, профилактики или уменьшению интенсивности заболевания, расстройства или побочного эффекта, или снижению темпов развития заболевания или расстройства. Термин также включает в пределах эффективных количеств усиление нормальной физиологической функции.
Фиброзные заболевания включают формирование избытка волокнистой соединительной ткани в органе или ткани в процессе репарации или реактивации. Считается, что антагонисты αvβ6, чтобы быть полезными при лечении таких различных заболеваний или состояний, должны включать такие, которые зависимы от функции αvβ6 интегрина и активации преобразования фактора роста бета через альфа v интегрины. Заболевания могут включать, но, не ограничиваясь ими, легочный фиброз (например, идиопатический легочный фиброз, неспецифическую интерстициальную пневмонию (NSIP), обычную интерстициальную пневмонию (UIP), синдром Германски-Пудлака, прогрессирующий массивный фиброз (осложненный у работников угольной промышленности пневмокониоз), связанный с болезнью соединительной ткани легочный фиброз, фиброзное заболевание дыхательных путей при астме и ХОБЛ, ОРДСВ, связанный с фиброзом, острое повреждение легких, вызванный радиацией фиброз, семейный легочный фиброз, легочную гипертензию); фиброз почек (диабетическую нефропатию, IgA нефропатию, волчаночный нефрит, фокальный сегментарный гломерулосклероз (FSGC), трансплантатную нефропатию, аутоиммунную нефропатию, вызванную лекарством нефропатию, связанную с гипертензией нефропатию, нефрогенный системный фиброз); фиброз печени (вирусно-индуцированный фиброз (например, гепатиты C или B), аутоиммунный гепатит, первичный билиарный цирроз, алкогольную болезнь печени, безалкогольное ожирение печени, включая неалкогольный стеатогепатит (NASH), врожденный фиброз печени, первичный склерозирующий холангит, вызванный лекарством гепатит, цирроз печени); фиброз кожи (гипертрофические рубцы, склеродерму, келоидные рубцы, дерматомиозит, эозинофильный фасцит, контрактуру Дюпюитрена, синдром Элерса-Данлоса, болезнь Пейрони, дистрофический буллезный эпидермолиз, пероральный субмукозный фиброз); глазной фиброз (возрастную макулярную дегенерацию (AMD), диабетический макулярный отек, сухость глаз, глаукому), рубцы на роговице, повреждение роговицы и заживление ран на роговице, предупреждение образования пузырьков при рубцевании, рубцы после операции трабекулэктомии; кардиафиброз (застойную сердечную недостаточность, атеросклероз, инфаркт миокарда, эндомиокардиальный фиброз, гипертрофическую кардиомиопатию (HCM)) и другие различные фиброзные состояния (средостенный фиброз, миелофиброз, ретроперитонеальный фиброз, болезнь Крона, нейрофиброматоз, лейомиому матки (миома), хроническое отторжение органом трансплантата. Это может являться дополнительным преимуществом для дополнительного ингибирования αvβ1, αvβ5 или αvβ8 интегринов.
Кроме того, также могут подвергаться лечению предраковые поражения или раковые заболевания, связанные с αvβ6 интегринами, (они могут включать, но, не ограничиваясь ими, эндометрии, базальных клеток, печени, толстой кишки, шейки матки, ротовой полости, поджелудочной железы, груди и яичников, саркому Капоши, опухоли гигантских клеток и связанной с раком стромы). Преимущественными состояниями могут быть состояния, при которых можно получить эффект от воздействия на ангиогенез (например, при солидных опухолях).
Термин ʺзаболевание или состояние, для которого рекомендован αvβ6 антагонистʺ, предназначен для включения любого или всех указанных выше болезненных состояний.
В одном из вариантов осуществления заболеванием или состоянием, для которого рекомендован αvβ6 антагонист, является идиопатический легочный фиброз.
В другом варианте осуществления заболевание или состояние, для которого рекомендован αvβ6 антагонист, выбрано из рубцов на роговице, повреждения роговицы и заживления ран на роговице.
Композиции
При возможности применения в терапии, соединение формулы (I), а также его фармацевтически приемлемые соли, можно вводить в виде родительского химического вещества, при этом для предоставления активного ингредиента общим является фармацевтическая композиция.
Настоящее изобретение, таким образом, в еще одном аспекте предоставляет фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей и/или эксципиентов. Соединения формулы (I) и их фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми, в смысле быть совместимыми с другими ингредиентами данной композиции и не наносить вреда реципиенту.
Согласно другому аспекту, настоящее изобретение также обеспечивает способ получения фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Фармацевтическая композиция может применяться при лечении любых состояний, описанных выше.
Далее представлена фармацевтическая композиция для лечения заболеваний или состояний, для которых рекомендован антагонист αvβ6 интегрина, содержащая соединение формулы (I) или его фармацевтически приемлемую соль.
Далее представлена фармацевтическая композиция, содержащая 0,01-3000 мг соединения формулы (I) или его фармацевтической соли и 0,1-2 г одного или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Поскольку соединения формулы (I) предназначены для применения в фармацевтической композиции, будет легко понятно, что они, предпочтительно каждое, предоставляется в по существу чистой форме, например, по меньшей мере, с 60% чистотой, более подходяще, по меньшей мере, с 75% чистотой и предпочтительно, по меньшей мере, с 85% чистотой, особенно, по меньшей мере, с 98% чистотой (% по массе от основного массы состава).
Фармацевтические композиции могут присутствовать в разовых дозированных формах, содержащих предопределенное количество активного ингредиента на разовую дозу. Предпочтительными разовыми дозированными композициями являются такие, которые содержат дневную дозу или субдозу или их соответствующую долю активного ингредиента. Такие разовые дозы, поэтому, могут вводиться более чем один раз в день. Предпочтительной разовой дозированной композицией является такая, которая содержит дневную дозу или субдозу (для введения более чем раз в день), как описано выше, или соответствующую долю активного ингредиента.
Фармацевтические композиции могут быть адаптированы для введения любым подходящим путем, например, пероральным (включая буккальный или подъязычный), ректальным, ингаляционным, интраназальным, местным (включая буккальный, подъязычный или трансдермальный), вагинальным, глазным или парентеральным (включая подкожный, внутримышечный, внутривенный или внутрикожный) путем введения. Такие композиции могут быть получены любым способом, известным в фармацевтической области, например, путем приведения во взаимосвязь активного ингредиента с носителем(ями) или эксципиентом(ами).
В одном из вариантов осуществления фармацевтическая композиция адаптирована для перорального введения.
Фармацевтические композиции, адаптированные для перорального введения, могут присутствовать в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобной пены или мусса; или жидких эмульсий масло-в-воде или вода-в-масле.
Например, для перорального введения в форме таблетки или капсулы, активный лекарственный компонент может быть скомбинирован с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Порошки, подходящие для включения в таблетки или капсулы, могут быть получены измельчением данного соединения до размера, подходящего для тонкодисперсных частиц (например, путем микронизации) и смешиванием с аналогичным образом полученным фармацевтическим носителем, таким как съедобный углевод, например, крахмал или маннит. Ароматизатор, консервант, диспергирующий агент и краситель также могут присутствовать.
Капсулы могут быть получены в виде порошковой смеси, как описано выше, и заполнением сформированных желатиновых капсул. Глиданты и лубриканты, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены в порошковую смесь перед операцией заполнения. Дезинтегрирующий или солюбилизирующий агент, такой как агарагар, карбонат кальция или карбонат натрия, также может быть добавлен для улучшения доступности медицинского препарата при проглатывании капсулы.
Кроме того, когда требуется или необходимо, в данную смесь могут быть включены подходящие связующие, глиданты, лубриканты, подсластители, ароматизаторы, дезинтегрирующие агенты и красители. Подходящие связующие включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и тому подобное.
Лубриканты, используемые в таких дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное.
Дезинтеграторы включают, но без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Таблетки формулируют, например, путем получения порошковой смеси, гранулирования или суспендирования, добавления лубриканта и дезинтегранта, и прессованием в таблетки. Порошковую смесь получают путем смешивания, надлежащим образом измельчают с разбавителем или основой, как описано выше, и необязательно со связующим, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем ресорбции, таким как четвертичная соль, и/или абсорбирующий агент, такой как бентонит, каолин или дикальцийфосфат. Порошковая смесь может быть гранулирована путем пропитки связующим, таким как сироп, крахмальная паста, гуммиарабик акадиа или растворы целлюлозных или полимерных материалов, и пропусканием через сито. В качестве альтернативы гранулированию, порошковую смесь можно пропустить через таблетирующую машину, и полученные в результате не полностью сформированные бруски разбить в гранулы. Полученные гранулы можно смазать для предотвращения риска слипания в таблетки путем добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Смазанную смесь затем спрессовывают в таблетки. Соединения настоящего изобретения также можно комбинировать со свободно текущим инертным носителем и спрессовывать в таблетки непосредственно без проведения стадий гранулирования или суспендирования. Можно снабдить прозрачным или непрозрачным защитным покрытием, состоящим из герметичного слоя шеллака, покрывающего сахара или полимерного материала, и отполировать покрытие воском. К указанным покрытиям могут быть добавлены красители, чтобы отличать различные разовые дозы.
Пероральные жидкости, такие как растворы, сиропы и эликсиры, можно получить в единичной дозированной форме таким образом, чтобы предоставить количественному содержанию предопределенное количество данного соединения. Сиропы могут быть получены растворением соединения в подходящем ароматизированном водном растворе, в то время как эликсиры получают при использовании нетоксичных спиртовых растворителей. Суспензии могут быть сформированы диспергированием данного соединения в нетоксичном растворителе. Солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые полиоксиэтиленовые эфиры сорбитола, консерванты, ароматизирующие добавки, такие как масло мяты перечной, или природные заменители сахара или сахарин или другие искусственные подсластители и тому подобное, также могут быть добавлены.
При необходимости, дозированные единицы композиций для перорального введения могут быть микроинкапсулированными. Данная рецептура может быть также получена для пролонгированного или непрерывного высвобождения, например, путем покрытия или встраивания частиц вещества в полимеры, воск или тому подобное.
Соединения настоящего изобретения также могут вводиться в форме липосомной системы доставки, такой как малые однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть сформированы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Фармацевтические композиции, адаптированные для трансдермального введения, могут присутствовать в виде дискретных патчей, предназначенных для того, чтобы оставаться в тесном контакте с эпидермисом пациента в течение длительного периода времени.
Фармацевтические композиции, адаптированные для местного введения, могут быть сформулированы в форме мази, крема, суспензии, лосьона, порошка, раствора, пасты, геля, спрея, аэрозоля или масла.
Для лечения глаз или других внешних тканей, например, ротовой полости и кожи, настоящие композиции применяют местно, предпочтительно в виде мази или крема. При формулировании мази, активный ингредиент может использоваться либо со смешиваемой парафиновой или водной мазевой основой. Альтернативно, активный ингредиент может быть сформулирован в виде крема с кремовой основой масло-в-воде или вода-в-масле. Соединения настоящего изобретения можно вводить в виде местных глазных капель. Соединения настоящего изобретения можно вводить конъюнктивальным, интракамерольным или интравитреальным путем введения, что потребует интервалы введения, которые длиннее, чем ежедневное введение.
Фармацевтические составы, адаптированные для местного введения в глаза, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе. Составы для введения в глаза должны иметь офтальмологически совместимый pH и осмоляльность. В композицию настоящего изобретения может включаться один или более офтальмологически приемлемых средств для регулирования pH и/или буферирующих агентов, включая кислоты, такие как уксусная, борная, лимонная, молочная, фосфорная и хлористоводородная кислоты; основания, такие как гидроксид натрия, фосфат натрия, борат натрия, цитрат натрия, ацетат натрия и лактат натрия; и буферы, такие как цитрат/декстроза, бикарбонат натрия и хлорид аммония. Такие кислоты, основания и буферы могут быть включены в количестве, требуемом для поддержания pH композиции в офтальмологически приемлемых пределах. В композицию может быть включена одна или более офтальмологически приемлемых солей в количестве, достаточном для приведения осмоляльности композиции в офтальмологически приемлемый предел. Указанные соли включают такие, которые имеют катионы натрия, калия или аммония, и анионы цитрата, аскорбата, бората, фосфата, бикарбоната, сульфата, тиосульфата или бисульфита.
Средства для глазной доставки могут быть разработаны для контролируемого высвобождения одного или более терапевтических средств с множественными определенными скоростями высвобождения и устойчивой кинетикой дозирования и проницаемости. Контролируемое высвобождение может быть достигнуто путем разработки полимерных матриц, включающих биоразлагаемые/биоразрушаемые полимеры в различных вариациях и с различными свойствами (например, поли(этиленвинил)ацетат (EVA), сверх гидролизованный PVA), гидроксиалкилцеллюлозу (HPC), метилцеллюлозу (MC), гидроксипропилметилцеллюлозу (HPMC), поликапролактон, поли(гликолевую) кислоту, поли(молочную) кислоту, полиангидрид, с полимерной молекулярной массой, полимерной кристалличностью, соотношением сополимеров, условиями обработки, защитным покрытием, геометрией, добавленным эксципиентом и полимерными покрытиями, которые будут усиливать диффузию лекарственного средства, эрозию, растворимость и осмос.
Составы для доставки лекарственного средства при использовании глазных средств могут объединять один или более активных средств и адъювантов, подходящих для указанного пути введения. Например, активные вещества могут быть смешаны с любым фармацевтически приемлемым эксципиентом, лактозой, сахарозой, порошком крахмала, целлюлозными эфирами алкановых кислот, стеариновой кислотой, тальком, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, аравийской камедью, желатином, альгинатом натрия, поливинилпирролидином и/или поливиниловым спиртом, таблетированы или инкапсулированы для общепринятого введения. Альтернативно, соединения можно растворить в полиэтиленгликоле, пропиленгликоле, коллоидных растворах карбоксиметилцеллюлозы, этаноле, кукурузном масле, арахисовом масле, хлопковом масле, кунжутном масле, трагаканте и/или различных буферах. Соединения также можно смешать с композицией как биоразлагаемых, так и небиоразлагаемых полимеров и носителем или разбавителем, которые обладают свойством временной задержки. Иллюстративные примеры биоразлагаемых композиций могут включать альбумин, желатин, крахмал, целлюлозу, декстраны, полисахариды, поли(D,L-лактид), поли(D,L-лактид-ко-гликолид), поли(гликолид), поли(гидроксибутират), поли(алкилкарбонат) и сложные поли(ортоэфиры), и их смеси. Иллюстративные примеры небиоразлагаемых полимеров могут включать EVA сополимеры, силиконовый каучук и поли(метилакрилат), и их смеси.
Фармацевтические композиции для глазной доставки также включают in situ желатинируемую водную композицию. Такая композиция содержит желирующий агент в концентрации, эффективной для промотирования гелеобразования после контакта с глазной или слезной жидкостью. Подходящие желирующие агенты включают, но, не ограничиваясь ими, термореактивные полимеры. Термин "in situ желатинируемая", как используется в данном описании, включает не только жидкости с низкой вязкостью, которые формируют гели после контакта с глазной или слезной жидкостью, но и включает более вязкие жидкости, такие как полужидкие и тиксотропные гели, которые демонстрируют значительное увеличение вязкости или гелевую жесткость после введения в глаз. См., например, Ludwig (2005) Adv. Drug Deliv. Rev. 3; 57:1595-639, которая включена в данное описание посредством ссылки, являющейся основополагающим примером полимеров, применяемых для доставки глазного лекарственного средства.
Фармацевтические композиции, адаптированные для местного введения в ротовую полость, включают таблетки для рассасывания, пастилки и средства для полоскания рта.
Фармацевтические композиции, адаптированные для ректального введения, могут присутствовать в виде суппозиториев или в виде клизмы.
Дозированные формы для назального или ингаляционного введения удобно могут быть сформулированы в виде аэрозолей, растворов, суспензий, гелей или сухих порошков.
Для композиций, подходящих и/или адаптированных для ингаляционного введения, желательно, чтобы соединения настоящего изобретения находились в форме с уменьшенным размером частиц, и более предпочтительно, чтобы указанная форма с уменьшенным размером частиц была получена или получаема путем микронизации. Предпочтительный размер частиц соединения или его соли с уменьшенным размером частиц (например, микронизированным) определяется значением D50 примерно от 0,5 до около 10 микрон (например, как измеряется с помощью лазерной дифракции).
Аэрозольные составы, например, для ингаляционного введения, могут состоять из растворов или тонкой суспензии активного вещества в фармацевтически приемлемом водном или неводном растворителе. Аэрозольные составы могут быть представлены в разовых или многодозовых количествах в стерильной форме в запечатанном контейнере, который может принимать форму картриджа или пополняемого баллончика для использования с ингалятором или устройством для распыления. Альтернативно, запечатанный контейнер может представлять собой унитарное распределительное устройство, такое как назальный ингалятор с разовой дозой или аэрозольный ингалятор с дозирующим клапаном (дозирующий ингалятор), который предназначен для удаления после того, как содержимое контейнера исчерпано.
В случае, когда дозированная форма предусматривает аэрозольный дозатор, предпочтительно содержание подходящего пропеллента под давлением, такого как сжатый воздух, диоксид углерода или органический пропеллент, такой как гидрофторуглерод (HFC). Подходящие HFC пропелленты включают 1,1,1,2,3,3,3-гептафторпропан и 1,1,1,2-тетрафторпентан. Аэрозольные дозированные формы могут также принимать форму насосного распылителя. Аэрозоль под давлением может содержать раствор или суспензию активного соединения. Это может потребовать включения дополнительных эксципиентов, например, сорастворителей и/или поверхностно-активных веществ, для улучшения дисперсионных характеристик и гомогенности суспендированных составов. Также может потребоваться добавление в раствор составленных композиций сорастворителей, таких как этанол. Могут быть также включены другие эксципиенты-модификаторы для улучшения, например, стабильности и/или вкусовых качеств и/или массовых характеристик тонкодисперсных частиц (количество и/или профиль) сформулированного состава.
Для фармацевтических композиций, подходящих и/или адаптированных для ингаляционного введения, фармацевтическая композиция может представлять собой сухой порошок ингалируемой композиции. Такая композиция может содержать порошковую основу, такую как лактоза, глюкоза, трегалоза, маннит или крахмал, соединение формулы (I) или его соль (предпочтительно в форме с уменьшенным размером частиц, например, в микронизированной форме), и необязательно модификатор активности, такой как L-лейцин или другая аминокислота и/или металлическая соль стеариновой кислоты, такая как стеарат магния или кальция. Предпочтительно, сухой порошок ингалируемой композиции содержит сухой порошок смешанного с лактозой соединения формулы (I) или его соли. Лактозой предпочтительно является гидрат лактозы, например, моногидрат лактозы, и/или предпочтительно лактоза ингаляционного класса и/или лактоза высшего сорта. Предпочтительно, размер частиц лактозы определяется содержанием 90% или более (по массе или по объему) лактозных частиц, составляющих менее 1000 микрон (микрометров) (например, 10-1000 мкм, 30-1000 мкм) в диаметре, и/или 50% или более лактозных частиц меньше чем 500 микрон (например, 10-500 мкм) в диаметре. Более предпочтительно, размер частиц лактозы определяется содержанием 90% или более лактозных частиц, составляющих менее 300 микрон (например, 10-300 микрон, например, 50-300 микрон) в диаметре, и/или 50% или более лактозных частиц, составляющих менее 100 микрон в диаметре. Необязательно, размер частиц лактозы определяется содержанием 90% или более лактозных частиц, составляющих менее 100-200 микрон в диаметре, и/или 50% или более лактозных частиц, составляющих менее 40-70 микрон в диаметре. Самое главное, предпочтительным является, чтобы примерно от 3 до примерно 30% (например, около 10%) (по массе или по объему) частиц составляло менее чем 50 микрон или менее чем 20 микрон в диаметре. Например, но без ограничения, подходящей лактозой ингаляционного класса является лактоза E9334 (10% мелкодисперности) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands).
Необязательно, в частности, для сухого порошка вдыхаемой композиции, фармацевтическая композиция для ингаляционного введения может быть включена в запечатанные многодозовые контейнеры (например, содержащие сухой порошок композиции), расположенные продольно в полосы или ленты внутри подходящего ингаляционного устройства. Контейнер представляет собой камеры с разрывающимися или расслаиваемыми по требованию межкамерными перегородками, и дозу, например, сухой порошковой композиции, можно вводить при вдыхании через устройства, такие как устройство DISKUS ТМ, продаваемое фирмой GlaxoSmithKline. DISKUS TM устройство для ингаляции описано, например, в GB 2242134 A, и в таком устройстве по меньшей мере один контейнер для фармацевтической композиции в порошковой форме (контейнер или контейнеры, предпочтительно, контейнеры с множественными запечатанными дозами, расположенные продольно в полосы или ленты), установленные между двумя удаляемыми частями, фиксированными относительно друг друга; устройство включает: средство для установки открытого состояния для указанного контейнера или контейнеров; средство для удаления предохранительного покрытия помимо частей в открытом состоянии для открытия контейнера; и выходное отверстие для связывания с открытым контейнером, через который пользователь может вдыхать фармацевтическую композицию в порошковой форме из открытого контейнера.
Соединения настоящего изобретения могут быть сформулированы для ингаляционного или интраназального введения в виде жидкого препарата для доставки из жидкостного дозатора, например, дозатора, имеющего распределительное сопло или выпускное отверстие, через которое распыляется отмеренная доза жидкого препарата при применении пользователем силы к насосному механизму жидкостного дозатора. Такие жидкостные дозаторы обычно снабжены многодозовым резервуаром для жидкого препарата, из которого отмеряются разовые дозы при последовательном срабатывании насоса. Сопло или отверстие дозатора может быть выполнено с возможностью вставки в ноздри пользователя для распыления дозированной жидкости препарата в полость носа. Жидкостной дозатор приведенного выше типа описан и проиллюстрирован в WO-A-2005/044354, содержание которого, таким образом, включено в данное описание посредством ссылки во всей своей полноте. Дозатор имеет корпус, в котором размещено устройство для выпуска жидкости, имеющее компрессор, установленный на контейнер для содержания жидкого состава. Корпус имеет по меньшей мере один управляемый пальцем нажимной боковой рычаг, который внутри движется относительно корпуса сверху к камере контейнера в корпусе, приводя насос к сжатию и нагнетанию струи отмеренной дозы препарата из насоса через назальное сопло корпуса. Особенно предпочтительным жидкостным дозатором является дозатор общего типа, проиллюстрированный на фигурах 30-40 WO-A-2005/044354.
Композиции для ингаляционного или интраназального введения также могут быть введены в легкие и другие области дыхательных путей путем распыления. Такие композиции могут представлять собой водные растворы или суспензии. Растворы для ингаляции распылением могут быть сформулированы при добавлении агентов, таких как кислота или щелочь, буферные соли, агенты для регулирования изотоничности, поверхностно-активные вещества или антимикробные препараты, такие как бензалконийхлорид (BAC). Композиции могут быть стерильными и не содержащими антимикробного консерванта. Их можно стерилизовать, например, фильтрованием или нагреванием в автоклаве. Они могут присутствовать в виде нестерильного раствора. Отдельную единичную дозу терапевтически эффективного количества соединения настоящего изобретения можно предоставить в виде предварительно смешанного, отмеренного препарата в одном контейнере.
Фармацевтические композиции, адаптированные для вагинального введения, могут быть предоставлены в виде вагинальных суппозиториев, тампонов, кремов, гелей, пасты, пены или спрея.
Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и солюты, которые приводят композицию к изотоничности с кровью пациента, и водные и неводные стерильные суспензии, которые включают суспендирующие агенты и сгущающие агенты. Композиции могут быть предоставлены в однодозовом или многодозовом контейнере, например, в запаянных ампулах и флаконах, и могут храниться в высушенном вымораживанием (лиофилизированном) состоянии, требуя только добавления стерильной жидкости носителя, например, воды для инъекций, непосредственно перед применением. Импровизированные инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Терапевтически эффективное количество соединения настоящего изобретения будет зависеть от ряда факторов, включая, например, возраст и массу пациента, конкретное состояние, требующее лечения, и его тяжесть, природу препарата и пути его введения, и, в конечном итоге, от компетенции лечащего врача или ветеринара. В фармацевтической композиции, каждая дозированная единица для перорального или парентерального введения предпочтительно составляет от 0,01-3000 мг, 0,1-2000 мг или более конкретно от 0,5 до 1000 мг соединения настоящего изобретения, рассчитанная на родительское соединение.
Каждая дозированная единица для назального или ингаляционного введения предпочтительно составляет от 0,001 до 50 мг, более предпочтительно 0,01-5 мг, еще более предпочтительно от 10 до 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, рассчитанная на свободное основание.
Для введения распыляемого раствора или суспензии, дозированная единица обычно составляет от 1 до 15 мг, например, от 2 мг до 10 мг, или от 4 мг до 6 мг, которое может должным образом осуществляться один раз в день, дважды в день или более чем два раза в день. Соединение настоящего изобретения может быть предоставлено в форме сухого или лиофилизированного порошка для растворения в аптеке или пациентом, или может быть, например, предоставлено в водном физиологическом растворе.
Каждая дозированная единица для назального или ингаляционного введения предпочтительно составляет от 0,001 до 50 мг, более предпочтительно 0,01-50 мг, еще более предпочтительно от 10 до 50 мг соединения формулы (I) или его фармацевтически приемлемой соли, рассчитанная на свободное основание.
Фармацевтически приемлемые соединения настоящего изобретения можно вводить в дневной дозе (для взрослого пациента), например, в пероральной или парентеральной дозе, составляющей от 0,01 мг до 3000 мг в день или 0,5-1000 мг в день, или в назальной или ингаляционной дозе 0,001-50 мг в день, или 0,01-50 мг в день, или 10-50 мг соединения формулы (I) или его фармацевтически приемлемой соли, рассчитанной на свободное основание. Это количество может предоставляться одной дозой в день или более, обычно в количестве (таком как две, три, четыре, пять или шесть) поддоз в день, таким образом, чтобы общая дневная доза была такой же. Эффективное количество соли данного соединения может быть определено как доля эффективного количества соединения формулы (I) как такового.
Соединения настоящего изобретения можно применять само по себе или в комбинации с другими терапевтическими средствами. Комбинированная терапия согласно настоящему изобретению также включает введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли, и применение по меньшей мере одного другого фармацевтически активного средства. Предпочтительно, комбинированная терапия согласно настоящему изобретению включает введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного другого фармацевтически активного средства. Соединение(я) настоящего изобретения и другое фармацевтически активное средство(а) могут вводиться вместе в единой фармацевтической композиции или раздельно, и при раздельном введении, это может происходить одновременно или последовательно в любом порядке. Количества соединения(й) настоящего изобретения и другого фармацевтически активного средства(в) и относительные сроки введения будут выбираться для того, чтобы добиться желаемого комбинированного терапевтического эффекта.
Таким образом, в дополнительном аспекте предоставлена комбинация, содержащая соединение настоящего изобретения и по меньшей мере одно другое фармацевтически активное средство.
Таким образом, в одном из аспектов соединение и фармацевтические композиции согласно настоящему изобретению могут применяться в комбинации с или при включении одного или более других терапевтических средств, включая средства для лечения аллергических заболеваний, воспалительных заболеваний, аутоиммунных заболеваний, антифиброзные средства лечения и средства лечения обструктивного заболевания дыхательных путей, диабетических глазных заболеваний и рубцов на роговице, средства лечения повреждений роговицы и средства при заживлении ран на роговице.
Противоаллергические средства лечения включают средства антиген-иммунотерапии (такие как компоненты и фрагменты пчелиного яда, пыльцы, молока, арахиса, CpG мотивов, коллагена, другие компоненты внеклеточного матрикса, которые могут вводиться в виде пероральных или подъязычных антигенных препаратов), антигистамины (такие как цетиризин, лоратадин, акривастин, фексофенидин, хлорфенамин) и кортикостероиды (такие как флутиказона пропионат, флютиказона фуроат, беклометазона дипропионат, будесонид, циклезонид, мометазона фуроат, триамцинолон, флюнизолид, преднизолон, гидрокортизон).
Противовоспалительные средства лечения включают NSAID (такие как аспирин, ибупрофен, напроксен), модуляторы лейкотриенов (такие как монтелукаст, зафирлукаст, пранлукаст) и другие противовоспалительные средства лечения (такие как ингибиторы iNOS, ингибиторы триптазы, ингибиторы IKK2, ингибиторы p38 (лосмапимод, дилмапимод), ингибиторы эластазы, бета2 агонисты, DP1 антагонисты, DP2 антагонисты, ингибиторы pI3K дельта, ингибиторы ITK, ингибиторы LP (лизофосфатидный) или ингибиторы FLAP (5-липоксигеназу активирующий белок) (такие как 3-(3-(трет-бутилтио)-1-(4-(6-этоксипиридин-3-ил)бензил)-5-((5-метилпиридин-2-ил)метокси)-1H-индол-2-ил)-2,2-диметилпропаноат) натрия; агонисты аденозин a2a (такие как аденозин и регаденозон), хемокиновые антагонисты (такие как CCR3 антагонисты или CCR4 антагонисты), ингибиторы медиаторов высвобождения.
Средства лечения аутоиммунных заболеваний включают DMARDS (такие как метотрексат, лефлюномид, азатиоприн), биофармацевтические средства лечения (такие как анти-IgE, анти-TNF, анти-интерлейкины (такие как анти-IL-1, анти-IL-6, анти-IL-12, анти-IL-17, анти-IL-18), рецепторные средства лечения (такие как этанерцепт и аналогичные средства); иммуноантиген неспецифические средства лечения (такие как интерферон или другие цитокины/хемокины, модуляторы рецепторов цитокина/хемокина, агонисты или антагонисты цитокина, TLR агонисты и подобные средства).
Другие антифиброзные средства лечения включает синтетические ингибиторы TGFβ (такие как пирфенидон), ингибиторы тирозинкиназы, направленные на сосудистый эндотелиальный фактор роста (VEGF), тромбоцитарный фактор роста (PDGF) и киназный рецептор фактора роста фибробластов (FGF) (такой как нинтеданиб (BIBF-1120) и иматиниба мезилат (Gleevec)), антагонисты рецепторов эндотелина (такие как амбризентан или мацитентан), антиоксиданты (такие как N-ацетилцистеин (NAC); антибиотики широкого спектра (такие как котримоксазолом, тетрациклины (миноциклина гидрохлорид)), ингибиторы Фосфодиэстеразы 5 (PDE5) (такие как силденафил), анти-αvβx антитела и лекарственные средства (такие как анти-αvβ6 моноклональные антитела, такие как описанные в WO2003100033A2, могут быть использованы в комбинации, интетумумаб, циленгитид) могут быть использованы в комбинации.
Средства лечения обструктивных заболеваний дыхательных путей включают бронходилататоры, такие как β2-агонисты короткого действия (такие как сальбутамол), β2-агонисты длительного действия (такие как салметерол, формотерол и вилантерол), мускариновые антагонисты короткого действия (такие как ипратропия бромид), мускариновые антагонисты длительного действия (такие как тиотропий, умеклидиний).
В некоторых вариантах осуществления лечение может также включать комбинацию соединения настоящего изобретения с другими существующими средствами лечения, например, существующими средствами лечения диабетических глазных заболеваний, такими как анти VEGF средства лечения, например, Lucentis®, Avastin® и Aflibercept®, и стероиды, например, триамцинолон, и стероидные имплантаты, содержащие флуоцинолона ацетонид.
В некоторых вариантах осуществления лечение может также включать комбинацию соединения настоящего изобретения с другими существующими средствами лечения, например, существующими средствами для лечения рубцов на роговице, повреждений роговицы или средствами для заживление ран на роговице, таких как Gentel®, экстракт крови телят, Levofloxacin® и Ofloxacin®.
Соединения и композиции настоящего изобретения можно использовать для лечения различных типов рака, такого как монотерапия или в комбинации со средствами лечения рака, включающими химиотерапию, радиотерапию, препараты направленного действия, иммунотерапию и клеточную или генную терапию.
Специалисту в данной области техники будет очевидно, что, при необходимости, другой терапевтический ингредиент(ы) может(гут) быть использован(ы) в форме солей, например, солей щелочного металла или аминных солей, или в форме кислотно-аддитивных солей, или пролекарственных средств, или в форме сложных эфиров, например, низших алкиловых эфиров, или в форме сольватов, например, гидратов, для оптимизации активности и/или стабильности и/или физических характеристик, таких как растворимость терапевтического ингредиента. Также будет очевидно, что, при необходимости, терапевтические ингредиенты должны использоваться в оптически чистой форме.
Приведенные выше комбинации удобно могут быть представлены для применения в форме фармацевтической композиции, и такие фармацевтические композиции, содержащие комбинацию, как описано выше, вместе с фармацевтически приемлемым разбавителем или носителем, составляют следующий аспект настоящего изобретения. Индивидуальные соединения таких комбинаций могут вводиться одновременно или последовательно в отдельной или комбинированной фармацевтической композиции. Предпочтительно, индивидуальные соединения будут вводиться одновременно в комбинированной фармацевтической композиции. Соответствующие дозы известных терапевтических средств будут легко оценены квалифицированными специалистами в данной области техники.
Следует отметить, что при введении соединения настоящего изобретения в комбинации с одним или более другими терапевтическими активными средствами, если индивидуальное соединение нормально вводится ингаляцией, внутривенно, перорально, интраназально, местным глазным или другим путем введения, то результирующая фармацевтическая композиция может вводиться таким же самым путем. Альтернативно, индивидуальные компоненты композиции могут вводиться различными путями.
Настоящее изобретение будет проиллюстрировано посредством следующих примеров.
Аббревиатуры
В следующем списке приведены определения некоторых сокращений, как используется в данном описании. Следует отметить, что список не является исчерпывающим, но смысл сокращений, приведенных ниже, но не представленных в данном списке, будут очевидны специалисту в данной области техники.
Ac (ацетил)
BCECF-AM (сложный ацетоксиметиловый эфир 2',7'-бис-(2-карбоксиэтил)-5-(и -6)-карбоксифлуоресцеина)
BEH (мостиковая этилен-гибридная технология)
Bu (бутил)
CHAPS (3-[(3-холамидопропил)диметиламмонио]-1-пропансульфонат)
Chiralcel OD-H (трис(3,5-диметилфенилкарбамат)целлюлоза с покрытием на 5 мкм силикагелем)
Chiralpak AD-H (трис(3,5-диметилфенилкарбамат)амилоза с покрытием на 5 мкм силикагелем)
Chiralpak ID (трис(3-хлорфенилкарбамат)амилоза с иммобилизованным на 5 мкм силикагелем)
Chiralpak AS (трис((S)-альфа-метилбензилкарбамат)амилоза с покрытием на 5 мкм силикагелем)
CSH (гибридная технология с заряженной поверхностью)
CV (объем колонки)
ДХМ (дихлорметан)
DIPEA (диизопропилэтиламин)
ДМФА (N,N-диметилформамид)
ДМСО (диметилсульфоксид)
Et (этил)
EtOH (этанол)
EtOAc (этилацетат)
ч (час/часы)
HCl (хлористоводородная кислота)
HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота)
ЖХМС (жидкостная хроматография-масс-спектрометрия)
LiHMDS (гексаметилдисилазид лития)
MDAP (масс-направленная автоматическая-препаративная ВЭЖХ)
Me (метил)
MeCN (ацетонитрил)
MeOH (метанол)
мин минута/минуты
МС (масс-спектр)
PdCl2(dppf)-CH2Cl2 комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном
Ph (фенил)
iPr (изопропил)
(R)-BINAP (R)-(+)-2,2′-бис(дифенилфосфино)-1,1′-бинафталин
Si (кремний)
SPE (твердофазная экстракция)
TBME (трет-бутилметиловый эфир)
TEA (триэтиламин)
TFA (трифторуксусная кислота)
ТГФ (тетрагидрофуран)
ТСХ (тонкослойная хроматография)
Все ссылки на насыщенный раствор соли относится к насыщенному водному раствору хлорида натрия.
Подробности экспериментов
Аналитическая ЖХМС
Аналитическую ЖХМС проводили на одной из следующих систем A-C.
УФ детекция для всех систем представляла собой усредненный сигнал при длине волны от 220 нм до 350 нм, и масс-спектры записывали на масс-спектрометре при использовании альтернативного сканирования в режиме распыления при положительной и отрицательной ионизации.
Экспериментальные подробности ЖХМС систем A-C, приведенных в данном описании, были следующими:
Система A
Колонка: 50 мм×2,1 мм ВД, 1,7 мкм Acquity UPLC BEH, C18 колонка.
При скорости потока: 1 мл/мин.
Температура: 40°C.
Растворители: A: 10 мМ бикарбонат аммония в воде, доведенный до pH10 добавлением раствора аммиака.
B: Ацетонитрил
Система B
Колонка: 50 мм×2,1 мм ВД, 1,7 мкм Acquity UPLC BEH C18 колонка.
При скорости потока: 1 мл/мин.
Температура: 40°C.
Растворители: A: 0,1% об./об. раствор муравьиной кислоты в воде.
B: 0,1% об./об. раствор муравьиной кислоты в ацетонитриле.
Система C
Колонка: 50 мм×2,1 мм ВД, 1,7 мкм Acquity UPLC CSH C18 колонка.
При скорости потока: 1 мл/мин.
Температура: 40°C.
Растворители: A: 10 мМ бикарбонат аммония в воде доведенный до pH10 добавлением раствора аммиака.
B: Ацетонитрил
Система D
Колонка: 50 мм×2,1 мм ВД, 1,7 мкм Acquity UPLC CSH C18 колонка.
При скорости потока: 1 мл/мин.
Температура: 40°C.
Растворители: A: 0,1% об./об. раствор муравьиной кислоты в воде.
B: 0,1% об./об. муравьиной кислоты в ацетонитриле.
Масс-направленная автоматическая-препаративная ВЭЖХ
Неочищенные продукты очищали при помощи MDAP ВЭЖХ следующим методом A. Время выполнения составляло 15 мин, если не указано иное. УФ детектирование для всех методов представляло собой усредненный сигнал при длине волны от 210 нм до 350 нм, и масс-спектры записывали на масс-спектрометре при использовании альтернативного сканирования в режиме распыления при положительной и отрицательной ионизации.
Метод A:
Метод A проводили на колонке XBridge C18 (конкретно, 100 мм×30 мм вн.д. 5 мкм диаметр сорбента) при температуре окружающей среды. Используемыми растворителями были:
A=10 мМ водный бикарбонат аммония, доведенный до pH 10 добавлением раствора аммиака;
B=ацетонитрил.
Применяемый градиент:
Получение промежуточных продуктов
Промежуточное соединение 1: (R)-трет-бутил 3-(йодметил)пирролидин-1-карбоксилат
5 л Реакционный сосуд с вакуумной рубашкой (Radley's LARA) загружали ДХМ (2 л), с последующим добавлением трифенилфосфина (339 г, 1,29 моль) и имидазола (88 г, 1,29 моль), и температуру снижали до 0°C. Затем порциями добавляли йод (328 г, 1,29 моль) в течение 30 мин, поддерживая реакционную температуру при 0-5°C для управления выделения тепла. В ходе добавления образовывался коричневый осадок. Полученному осадку давали возможность нагреться до комнатной температуры в течение 15 минут и затем перемешивали при комнатной температуре в течение дополнительных 30 минут. Порциями добавляли раствор (R)-трет-бутил 3-(гидроксиметилпирролидин-1-карбоксилата (200 г, 994 ммоль) (доступный от Flourochem или BePharm Ltd) в ДХМ (200 мл) в течение 15 минут, поддерживая реакционную температуру около 24-30°C. Реакционную смесь перемешивали в течение 2 часов, затем разбавляли TBME (8 л) и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток (700 г) растирали в диэтиловом эфире (2 л) не бане лед-вода, с получением 333 г неочищенного продукта. Порцию 27 г неочищенного продукта очищали хроматографией на силикагелевом картридже (100 г) при градиентном элюировании смесью 0-50% этилацетат-циклогексан в течение 30 минут. Соответствующие фракции объединяли и упаривали в вакууме, с получением указанного в заголовке соединения (16,33 г, 5%) в виде желтого масла. Оставшееся неочищенное вещество (~306 г) очищали хроматографией на силикагелевом картридже (1,5 кг) при градиентном элюировании смесью 0-30% этилацетат-циклогексан сверх 9,5 колоночных объемов. Соответствующие фракции объединяли и упаривали в вакууме, с получением указанного в заголовке соединения (233,94 г, 76%) в виде бледно-желтого масла: ЖХМС (система A) RT=1,19 мин, 100%, ES+ve m/z 312 (M+H)+; [α]D20=+23 (c 1,00 в EtOH).
Промежуточное соединение 2: (R)-трет-бутил 3-(2-(1,8-нафтиридин-2-ил)этил)пирролидин-1-карбоксилат
Перемешиваемый раствор 2-метил-1,8-нафтиридина (57,5 г, 399 ммоль) (доступный от Manchester Organics) и (R)-трет-бутил 3-(йодметил)пирролидин-1-карбоксилата (124,2 г, 399 ммоль) (промежуточное соединение 1) в ТГФ (1 л) охлаждали до 0°C и обрабатывали в атмосфере азота раствором бис(триметилсилил)амида лития в ТГФ (1M, 399 мл, 399 ммоль) около 20 минут, и реакционную смесь перемешивали при 0°C в течение 3 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (500 мл) и воды (500 мл), и добавляли этилацетат (1 л). Слои разделяли, и затем водную фазу экстрагировали этилацетатом (1 л). Объединенные органические слои сушили (MgSO4), фильтровали и упаривали в вакууме. Остаточное коричневое масло (162 г) очищали хроматографией на силикагелевом картридже (750 г) при градиентном элюировании смесью 0-100% [этилацетат в (5% MeOH-95% этилацетат)] сверх 8 колоночных объемов. Соответствующие фракции объединяли и упаривали в вакууме, с получением указанного в заголовке соединения (46,65 г, 36%) в виде оранжевого твердого вещества: ЖХМС (система A) RT=0,99 мин, 97%, ES+ve m/z 328 (M+H)+, [α]D20=+22 (c 1,00 в EtOH).
Промежуточное соединение 3: (R)-трет-бутил 3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-карбоксилат
Раствор (R)-трет-бутил 3-(2-(1,8-нафтиридин-2-ил)этил)пирролидин-1-карбоксилата (промежуточное соединение 2) (4,0 г, 12 ммоль) в EtOH (20 мл) гидрировали над влажным 5% Rh/C катализатором (1,2 г) при комнатной температуре в течение ночи. Катализатор отделяли фильтрованием через целит, и фильтрат концентрировали в вакууме, с получением указанного в заголовке соединения (4,0 г, 99%) в виде коричневой смолы: ЖХМС (система A) RT=1,20 мин, 95,5%, ES+ve m/z 332 (M+H)+.
Промежуточное соединение 4: (R)-7-(2-(пирролидин-3-ил)этил)-1,2,3,4-тетрагидро-1,8-нафтиридин
К раствору (R)-трет-бутил 3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-карбоксилата (промежуточное соединение 3) (18,92 г, 57,1 ммоль) в ДХМ (120 мл) на бане с холодной водой в атмосфере азота по каплям добавляли 4M HCl в 1,4-диоксане (57,1 мл, 228 ммоль). После завершения добавления, водяную баню удаляли. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи и затем концентрировали в вакууме. Полученный остаток (15,5 г) очищали несколькими партиями на SCX картриджах, промывая сначала метанолом и затем элюируя 2M аммиаком в метаноле, с получением указанного в заголовке соединения (8,94 г, 68%) в виде оранжевого масла: ЖХМС (система A) RT=0,70 мин, 100% ES+ve m/z 232 (M+H)+.
Промежуточное соединение 5: (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноат
К раствору (R)-7-(2-(пирролидин-3-ил)этил)-1,2,3,4-тетрагидро-1,8-нафтиридина (промежуточное соединение 4) (10,6 г, 45,8 ммоль) в ДХМ (200 мл) добавляли DIPEA (14,40 мл, 82 ммоль) в атмосфере азота.
Полученную реакционную смесь охлаждали до 0°C и по каплям добавляли (E)-метил 4-бромбут-2-еноат (5,39 мл, 45,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,75 часов, и затем реакционную смесь разбавляли водой (250 мл). Органический слой отделяли, и водный слой дополнительно экстрагировали ДХМ (2×100 мл). Объединенные органические фракции сушили (MgSO4) и упаривали в вакууме. Полученный остаток (13,94 г) очищали хроматографией на силикагелевом картридже (330 г), элюируя 0-100% EtOAc-(3:1 EtOAc-EtOH), содержащей 1% Et3N.
Соответствующие фракции объединяли и упаривали, с получением указанного в заголовке соединения (7,66 г, 51%) в виде желтого масла, которое затвердевало при стоянии в холодильнике. ЖХМС (система A) RT=1,02 мин, 100%, ES+ve m/z 330 (M+H)+.
Промежуточное соединение 6: Метил 3-(3-бромфенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат
Раствор (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 5) (2,8 г, 8,5 ммоль) в 1,4-диоксане (60 мл) обрабатывали (3-бромфенил)бороновой кислотой (доступна от Aldrich) (5,97 г, 29,7 ммоль), (R)-BINAP (0,529 г, 0,850 ммоль), водным раствором KOH (3,8M, 4,47 мл, 17,00 ммоль) и димером хлор(1,5-циклооктадиен)родия(l) (210 мг, 0,425 ммоль), и полученную смесь нагревали при 95°C в течение 1 часа. Полученной реакционной смеси давали охладиться и концентрировали в вакууме. Полученный остаток распределяли между водой (50 мл) и ДХМ (50 мл). К водным фракциям добавляли насыщенный раствор соли (50 мл) и экстрагировали дополнительно ДХМ (50 мл). Объединенные органические растворы промывали насыщенным раствором соли (50 мл), пропускали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток очищали хроматографией на аминопропиловой колонке (70 г) при градиентном элюировании смесью 0-100% EtOAc-циклогексан и затем хроматографией с обращенной фазой на Biotage SNAP картридже (120 г) при градиентном элюировании смесью 65-95% ацетонитрил-(10 мМ водный бикарбонат аммония, доведенный до pH 10 добавлением раствора аммиака). Соответствующие фракции объединяли и концентрировали в вакууме, с получением (400 мг) указанного в заголовке соединения в виде диастереоизомерной смеси. ЖХМС (система A) RT=1,39 мин, ES+ve m/z 486, 488 (M+H)+. Диастереоизомеры разделяли препаративной хиральной ВЭЖХ на колонке Chiralcel OJ-H (30 мм×25 см), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 30 мл/мин, детектирование при 215 нм. Соответствующие фракции объединяли и упаривали, с получением указанного в заголовке соединения:
Изомер 1. (S)-метил 3-(3-бромфенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат (650 мг, 16%): ЖХМС (система B) RT=0,46 мин, ES+ve m/z 486, 488 (M+H)+. Аналитическая хиральная ВЭЖХ RT=17,9 мин, на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм.
Изомер 2. (R)-метил 3-(3-бромфенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат (103 мг, 2%): аналитическая хиральная ВЭЖХ RT=14,3 мин, на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм.
Промежуточное соединение 7: 1-(3-бромфенил)-1H-имидазол
Перемешиваемую суспензию 1-бром-3-йодбензола (доступный от Apollo) (2,252 мл, 17,67 ммоль), 1H-имидазола (2,166 г, 31,8 ммоль), йодида меди(I) (0,673 г, 3,53 ммоль) и карбоната цезия (11,52 г, 35,3 ммоль) в MeCN (70 мл) дегазировали (3 раза) и затем кипятили с обратным холодильником в течение ночи в атмосфере азота. Полученной реакционной смеси давали охладиться до комнатной температуры, фильтровали, и фильтрат концентрировали в вакууме. Полученный остаток очищали хроматографией на картридже KP-silica (100 г), элюируя смесью 0-100% EtOAc-циклогексан. Соответствующие фракции объединяли и концентрировали в вакууме, с получением указанного в заголовке соединения (3,55 г, 90%) в виде бесцветного масла: ЖХМС (система C) RT=0,92 мин, 99%, ES+ve m/z 223, 225 (M+H)+.
Промежуточное соединение 8: 1-(3-(4,4,4,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазол
К раствору 1-(3-бромфенил)-1H-имидазола (промежуточное соединение 7) (1,0 г, 4,5 ммоль) в 1,4-диоксане (25 мл) добавляли бис(пинаколато)диборон (доступный от Aldrich) (1,25 г, 4,93 ммоль), дициклогексил(2',4',6'-триизопропил[1,1'-бифенил]-2-ил)фосфин (0,103 г, 0,215 ммоль), Pd2dba3 (0,062 г, 0,067 ммоль), ацетат калия (1,1 г, 11 ммоль). Полученную реакционную смесь нагревали при 110°C. Реакционную смесь концентрировали в вакууме и хранили в холодильнике в течение выходных. Полученный остаток распределяли между ДХМ (50 мл) и водой (50 мл). К водному слою добавляли насыщенный раствор соли (30 мл) и дополнительно экстрагировали ДХМ (30 мл). Объединенные органические фракции промывали насыщенным раствором соли (30 мл), пропускали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток очищали хроматографией с обращенной фазой на картридже C18 Biotage SNAP (60 г), элюируя смесью 30-60% ацетонитрил (содержащий 0,1% муравьиную кислоту)-вода (содержащая 0,1% муравьиной кислоты) (6 CV). Соответствующие фракции объединяли и концентрировали в вакууме, с получением указанного в заголовке соединения (594 мг, 49%) в виде бледно-желтой смолы: ЖХМС (система D) RT=0,33 мин, ES+ve m/z 189 (M+H)+.
Промежуточное соединение 9: (1-(3-бромфенил)-5-метил-1H-пиразол-3-ил)метанол
К перемешиваемому раствору этил 1-(3-бромфенил)-5-метил-1H-пиразолe-3-карбоксилата (описанный в WO2004092140) (500 мг, 1,62 ммоль) в ТГФ (10 мл) добавляли DIBAL-H (1M раствор в ТГФ) (7,12 мл, 7,12 ммоль) при 0°C, и полученную в результате смесь перемешивали при температуре окружающей среды в течение 16 часов. Полученную реакционную смесь гасили добавлением воды (10 мл) и разбавляли этилацетатом (10 мл). Органическую фазу концентрировали, и остаток очищали колоночной хроматографией на силикагеле (100-200 mesh), элюируя 50% этилацетатом в гексане, с получением указанного в заголовке соединения (300 мг, 66%) в виде не совсем белого твердого вещества:
1H ЯМР (400 МГц, CDCl3) 7,65 (1H, с), 7,50 (1H, д, J=7,5 Гц), 7,42-7,35 (2H, м), 6,20 (1H, с), 4,68 (2H, м), 2,35 (3H, с).
Промежуточное соединение 10: (5-метил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-пиразол-3-ил)метанол
К перемешиваемому раствору (1-(3-бромфенил)-5-метил-1H-пиразол-3-ил)метанола (промежуточное соединение 9) (4,0 г, 15 ммоль), бис(пинаколато)диборона (3,8 г, 15 ммоль), ацетата калия (4,41 г, 45 ммоль) в 1,4-диоксане (40 мл) добавляли аддукт PdCl2(dppf)-CH2Cl2 (1,22 г, 1,5 ммоль), и полученную в результате смесь перемешивали при 90°C в течение 6 часов. Полученную реакционную смесь охлаждали до комнатной температуры и фильтровали через рыхлый слой целита. Фильтрат концентрировали в вакууме, и остаток растворяли в этилацетате (100 мл) и фильтровали через целит. Фильтрат концентрировали в вакууме, и остаток очищали колоночной хроматографией на флоризил-силикагеле при градиентном элюировании 0-5% этилацетатом в петролейном эфире. Соответствующие фракции концентрировали в вакууме, с получением указанного в заголовке соединения (1,4 г, 23%) в виде коричневого масла: МС ES+ve m/z 315 (M+H)+.
Промежуточное соединение 11: 1-(3-бромфенил)-3,5-диметил-1H-1,2,4-триазол
N-ацетилацетамид (2,0 г, 20 ммоль), (3-бромфенил)гидразингидрохлорид (8,84 г, 39,6 ммоль) и пиридин (4 мл) помещали в 30 мл микроволновую пробирку (Anton Paar). Пробирку облучали в течение 2 минут (300 В, 200°C) при перемешивании. К полученной реакционной смеси добавляли этилацетат (15 мл) для осаждения остатка гидразингидрохлорида, и затем фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью 15% EtOAc-гексаны, с получением указанного в заголовке соединения (2,4 г, 43%) в виде бледно-желтого твердого вещества:
1H ЯМР (400 MГц, CDCl3) 7,65 (1H, ушир.с), 7,55 (1H, м), 7,38-7,34 (2H, м), 2,50 (3H, с), 2,41 (3H, с).
Промежуточное соединение 12: 3,5-диметил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-1,2,4-триазол получали из промежуточного соединения 11 (1,5 г, 5,9 ммоль) способом, аналогично описанному для промежуточного соединения 10, с получением указанного в заголовке соединения (936 мг, 37%): МС ES+ve m/z 300 (M+H)+.
Промежуточное соединение 13: 4-(3-бромфенил)-3-метил-4H-1,2,4-триазол
Смесь (3-бромфенил)бороновой кислоты (доступна от Apollo Scientific) (9,43 г, 46,9 ммоль), 3-метил-4H-1,2,4-триазола (доступный от Chemimpex) (3,0 г, 36 ммоль), Cs2CO3 (23,53 г, 72,2 ммоль), йодида меди(I) (688 мг, 3,61 ммоль) в ДМФА (30 мл) запечатывали в трубке и нагревали до 100°C в течение 16 часов. Реакционную смесь разбавляли водой (150 мл) и экстрагировали EtOAc (3×50 мл). Органический слой промывали водой (2×50 мл), сушили (Na2SO4) и упаривали в вакууме. Полученный остаток очищали препаративной ТСХ, элюируя смесью 20% EtOAc-гексан, с получением указанного в заголовке соединения (1,3 г, 15%) в виде белого твердого вещества. ТСХ RF=0,5 (подвижная фаза: 30% EtOAc-гексан).
Промежуточное соединение 14: 3-метил-4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4H-1,2,4-триазол получали из промежуточного соединения 13 (1,2 г, 5 ммоль) способом, аналогично описанному для промежуточного соединения 10, с получением указанного в заголовке соединения (700 мг, 44%): МС ES+ve m/z 286 (M+H)+.
Промежуточное соединение 15: 2-(1-(3-бромфенил)-5-метил-1H-пиразол-3-ил)пропан-2-ол
Суспензию этил 1-(3-бромфенил)-5-метил-1H-пиразолe-3-карбоксилата (описанный в WO2004092140) (3,1 г, 10,03 ммоль) в ТГФ (7 мл) по каплям обрабатывали метилмагнийбромидом (60,2 мл, 60,2 ммоль) при 0°C и перемешивали в течение 2 часов. Полученную реакционную смесь по каплям обрабатывали 1M KHSO4 (20 мл) и концентрировали при пониженном давлении. Полученный остаток растворяли в этилацетате и промывали водой (20 мл). Органический слой концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (100-200), элюируя 15% этилацетатом в петролейном эфире. Соответствующие фракции концентрировали при пониженном давлении, с получением указанного в заголовке соединения (2,45 г, 79%) в виде бледно-желтой жидкости: МС ES+ve m/z 295 (M+H)+.
Промежуточное соединение 16: 2-(5-метил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-пиразол-3-ил)пропан-2-ол получали из промежуточного соединения 15 (300 мг, 1 ммоль) способом, аналогично описанному для промежуточного соединения 10, с получением указанного в заголовке соединения (120 мг, 32%): МС ES+ve m/z 343 (M+H)+.
Промежуточное соединение 17: 1-(3-бромфенил)-3-(трифторметил)-1H-пиразол
Смесь 1-бром-3-йодбензола (доступный от Apollo Scientific) (1,351 мл, 10,60 ммоль), 3-(трифторметил)-1H-пиразола (доступный от Aldrich) (2,164 г, 15,91 ммоль), Cs2CO3 (10,37 г, 31,8 ммоль) и CuI (404 мг, 2,121 ммоль) в ацетонитриле (28 мл) нагревали до 106°C.
Полученной реакционной смеси давали охладиться до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме, и остаток очищали хроматографией на колонке с силикагелем (100 г), элюируя смесью 0-100% EtOAc-циклогексан, с получением указанного в заголовке соединения (2,67 г, 87%) в виде бледно-желтого масла: ЖХМС (система C) RT=1,37 мин, 96%, ES+ve m/z 291, 293 (M+H)+;
1H ЯМР (600 MГц, ДМСО-d6) δ 8,75 (дд, J=2,6, 1,1 Гц, 1H), 8,10-8,05 (м, 1H), 7,88 (ддд, J=8,3, 2,2, 0,9 Гц, 1H), 7,56-7,50 (м, 1H), 7,44 (т, J=8,1 Гц, 1H), 6,97 (д, J=2,9 Гц, 1H).
Промежуточное соединение 18:. 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3-(трифторметил)-1H-пиразол
К раствору 1-(3-бромфенил)-3-(трифторметил)-1H-пиразола (промежуточное соединение 17) (1,32 г, 4,54 ммоль) в 1,4-диоксане (24 мл), добавляли дициклогексил(2',4',6'-триизопропил-[1,1'-бифенил]-2-ил)фосфин (0,104 г, 0,218 ммоль), Pd2dba3 (62 мг, 0,068 ммоль), ацетат калия (1,113 г, 11,34 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,267 г, 4,99 ммоль). Полученную реакционную смесь кипятили с обратным холодильником при 110°C в течение 3 часов. После охлаждения, полученную реакционную смесь концентрировали в вакууме, и остаток распределяли между водой (30 мл) и EtOAc (30 мл). К водной фракции добавляли насыщенный раствор соли (30 мл), и полученное дополнительно экстрагировали EtOAc (40 мл). Объединенные органические фракции промывали насыщенным раствором соли (30 мл), пропускали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией с обращенной фазой на картридже C18 Biotage SNAP (60 г), элюируя смесью 40-85% MeCN-10 мМ бикарбонат аммония (содержащий 0,1% аммиак) (14 CV). Соответствующие фракции объединяли и концентрировали в вакууме, с получением указанного в заголовке соединения (700 мг): ЖХМС (система D) RT=1,49 мин, ES+ve m/z 339 (M+H)+.
Промежуточное соединение 19: 4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4H-1,2,4-триазол получали из 4-(3-бромфенил)-4H-1,2,4-триазола (доступный от Fluorochem) (3,0 г, 13 ммоль), способом, аналогично описанному для получения промежуточного соединения 10, с получением указанного в заголовке соединения (1,3 г, 36%) в виде белого твердого вещества:
1H ЯМР (400 MГц, ДМСО-d6) 9,13 (2H, с), 7,84-7,80 (2H, м), 7,58 (1H, ушир.д, J=7,5 Гц), 7,59 (1H, ушир.т, J=7,5 Гц), 1,32 (12H, с).
Промежуточное соединение 20: 1-(3-бромфенил)-1H-1,2,3-триазол
Раствор 1,3-дибромбензол (20 г, 85 ммоль) в ДМФА (200 мл) обрабатывали 1H-1,2,3-триазолом (6,97 г, 101 ммоль), йодидом меди(I) (1,62 г, 8,48 ммоль), Fe (III) ацетилацетонатом (6,78 г, 25,4 ммоль) и Cs2CO3 (55,2 г, г, 170 ммоль). Полученную реакционную смесь нагревали при 120°C в течение 18 часов, фильтровали через целит, промывали EtOAc (3×200 мл) и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, элюируя 20% EtOAc в гексане. Соответствующие фракции концентрировали при пониженном давлении, с получением указанного в заголовке соединения (4 г, 17%) в виде бледно-желтого твердого вещества: МС ES+ve m/z 224, 226 (M+H)+.
Промежуточное соединение 21: 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-1,2,3-триазол получали из 1-(3-бромфенил)-1H-1,2,3-триазола (промежуточное соединение 20) способом, аналогично описанному для промежуточного соединения 10, с получением указанного в заголовке соединения (1,5 г, 30%) в виде белого твердого вещества: МС ES+ve m/z 272 (M+H)+.
Промежуточное соединение 22: (R,E)-трет-бутил 7-(2-(1-(4-метокси-4-оксобут-2-ен-1-ил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилат
Раствор промежуточного соединения 5 (270 мг, 0,820 ммоль) и ди-трет-бутилдикарбоната (247 мкл, 1,065 ммоль) в ТГФ при 0°C в атмосфере азота по каплям обрабатывали LHMDS (1065 мкл, 1,065 ммоль) в течение 5 минут. Полученный раствор перемешивали при 0°C в течение 30 минут. Полученный раствор обрабатывали водным NH4Cl (10%, 5 мл) и экстрагировали 2-MeТГФ (5 мл). Органическую фазу непосредственно вносили в силикагелевый картридж (10 г), элюируемый смесью толуол-этанол-аммиак (80-10-1), с получением указанного в заголовке соединения (302 мг, 86%) в виде желтой смолы: ЖХМС (система C) RT=1,27 мин, ES+ve m/z 430 (M+H)+.
Промежуточное соединение 23: Смесь трет-бутил 7-(2-((R)-1-((S)-2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата и трет-бутил 7-(2-((R)-1-((R)-2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (9:1)
Раствор (5-метил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-пиразол-3-ил)метанола (промежуточное соединение 10) (549 мг, 1,746 ммоль) в диоксане (1 мл) обрабатывали водным KOH (0,404 мл, 1,536 ммоль) с последующим добавлением раствора (R,E)-трет-бутил 7-(2-(1-(4-метокси-4-оксобут-2-ен-1-ил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (промежуточное соединение 22) (300 мг, 0,698 ммоль) в диоксане (1 мл). Полученный раствор дегазировали в атмосфере азота/вакууме и обрабатывали R-BINAP (43,5 мг, 0,070 ммоль) и димером хлор(1,5-циклооктадиен)родия(l) (17,22 мг, 0,035 ммоль). Полученную смесь снова дегазировали в атмосфере азота/вакууме и нагревали при 50°C в течение 3 часов. Охлажденную смесь добавляли к смеси вода/насыщенный раствор соли (30 мл; 1:1) и экстрагировали EtOAc (2×25 мл). Высушенный экстракт (MgSO4) упаривали, и остаток очищали на силикагелевом картридже (20 г), элюируя смесью 0-100% [(3:1) EtOAc-изопропанол]-EtOAc, с получением указанного в заголовке соединения (205 мг, 47%) в виде бледно-коричневой смолы. Стереоизомерное соотношение при бензиловом центре не представлялось очевидным по данным ЯМР, но было определено по веществу с удаленными защитными группами (пример 10) как 9:1: ЖХМС (система C) RT=1,30 мин, ES+ve m/z 618 (M+H)+.
Промежуточное соединение 24: трет-бутил 7-(2-((3R)-1-(2-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилат (9:1 смесь изомер 1:изомер 2)
Раствор метансульфонилхлорида (0,018 мл, 0,227 ммоль) в ДХМ (0,1 мл) добавляли к раствору трет-бутил 7-(2-((3R)-1-(2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (промежуточное соединение 23) (140 мг, 0,227 ммоль) и триэтиламина (0,063 мл, 0,453 ммоль) в ДХМ (1,5 мл). Полученный раствор перемешивали в течение 1 часа, и данные анализа показали преимущественное завершение частичной замены хлорида мезилом. Полученный раствор обрабатывали ацетонитрилом (2 мл), фторидом калия (13,17 мг, 0,227 ммоль) и KryptofixTM (5,6-бензо-4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]гексакос-5-ен) (85 мг, 0,227 ммоль) и нагревали до 60°C в течение 1 часа. Анализ показал только присутствие хлорида, а не перемещение фтора. Полученную смесь распределяли между водным NaHCO3 (5 мл) и EtOAc (2×5 мл), и высушенный экстракт (MgSO4) упаривали. Полученный остаток растворяли в ДМФА (1,5 мл), обрабатывали 4 эквивалентами KF и KryptofixTM и нагревали в запаянной микроволновой пробирке при 120°C в течение 30 минут. Полученный раствор добавляли к воде (10 мл) и экстрагировали EtOAc (2×5 мл). Высушенный экстракт (MgSO4) упаривали, и остаток непосредственно использовали на следующей стадии (пример 11): ЖХМС (система C) RT=1,46 мин, ES+ve m/z 620 (M+H)+.
Промежуточное соединение 25: 2-(3-бромфенил)-4-метил-1H-имидазол
Раствор 3-бромбензимидамида (доступный от Fluorochem) (22 г, 111 ммоль), 1-хлорпропан-2-она (16,36 г, 177 ммоль) и хлорида аммония (21,88 г, 409 ммоль) в ТГФ (200 мл) обрабатывали в атмосфере азота гидроксидом аммония (176 мл, 4532 ммоль), и реакционную смесь перемешивали при 80°C в течение 12 часов. Полученную реакционную смесь концентрировали в вакууме, и остаток (11,5 г) очищали колоночной хроматографией на силикагеле (100-200 меш), элюируя 30% этилацетатом в петролейном эфире. Соответствующие фракции концентрировали, и полученное в результате бледно-желтое твердое вещество растирали в диэтиловом эфире, с получением указанного в заголовке соединения (5,2 г, 20%) в виде коричневого твердого вещества:
1H ЯМР (400 MГц, CDCl3) 9,2 (1H, шир.), 7,95 (1H, с), 7,70 (1H, д), 7,42 (1H, д), 7,27 (1H, м), 6,83 (1H, с), 2,35 (3H, с).
Промежуточное соединение 26: 2-(3-бромфенил)-1,4-диметил-1H-имидазол
Раствор 2-(3-бромфенил)-4-метил-1H-имидазола (промежуточное соединение 25) (5 г, 21 ммоль) и трет-бутоксида калия (2,366 г, 21,09 ммоль) в ТГФ (100 мл) обрабатывали раствором 18-кроун-6 (0,557 г, 2,109 ммоль) и йодметана (1,319 мл, 21,09 ммоль) в ТГФ (20 мл), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь выливали в насыщенный раствор соли и экстрагировали этилацетатом (100 мл). Органическую фазу отделяли, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток (5,2 г) очищали колоночной хроматографией на силикагеле (100-200), элюируя 20% этилацетатом в петролейном эфире. Соответствующие фракции упаривали, с получением указанного в заголовке соединения (2,0 г, 38%) в виде бледно-желтой жидкости:
1H ЯМР (400 МГц, ДМСО-d6) 7,85 (1H, с), 7,69 (1H, д, J=7,5 Гц), 7,60 (1H, д, J=7,5 Гц), 7,40 (1H, т, J=7,5 Гц), 6,97 (1H, с), 3,70 (3H, с), 2,15 (3H, с).
Промежуточное соединение 27: 1,4-диметил-2-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазол
Указанное в заголовке соединения получали из промежуточного соединения 26 (1 г, 4 ммоль) способом, аналогично описанному для промежуточного соединения 8, с получением (420 мг, 83%):
1H ЯМР (400 MГц, CDCl3) 8,04 (1H, ушир.с), 7,82 (1H, ушир.д, J=7,5 Гц), 7,70 (1H, ушир.д, J=7,5 Гц), 7,45 (1H, т, J=7,5 Гц), 6,66 (1H, с), 3,66 (3H, с), 2,26 (3H, с), 1,35 (12H, с).
Промежуточное соединение 28: 5-(3-Бромфенил)-2,4-диметил-1H-имидазол
К раствору (E)-1-бром-3-(2-нитропроп-1-ен-1-ил)бензола (U. Jana et. al. Europ. J. Org. Chem. 2013, (22), 4823) (20 г, 83 ммоль) в этаноле (200 мл) добавляли гидрохлорид ацетамидина (7,81 г, 83 ммоль), карбонат калия (11,42 г, 83 ммоль) и индий(III)оксид (1,147 г, 4,13 ммоль), и реакционную смесь перемешивали в атмосфере азота при 70°C в течение 6 часов. Реакционную смесь отслеживали ТСХ.
ТСХ Подвижная фаза: 10% MeOH в ДХМ, значение Rf: 0,5, детекция: УФ. Обработка: этанол удаляли при пониженном давлении. Неочищенную реакционную смесь разбавляли водой (150 мл) и экстрагировали EtOAc (3×200 мл), промывали насыщенным раствором соли (250 мл). Органический слой отделяли, сушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток растирали в диэтиловом эфире (50 мл), твердое вещество отделяли фильтрованием и сушили, с получением указанного в заголовке соединения (4,4 г, 21%) в виде белого твердого вещества ES+ve m/z 251, 253 (M+H)+.
Промежуточное соединение 29: трет-бутил 5-(3-бромфенил)-2,4-диметил-1H-имидазол-1-карбоксилат
Суспензию 5-(3-бромфенил)-2,4-диметил-1H-имидазола (4 г, 16 ммоль) (промежуточное соединение 28) в ДХМ (40 мл) обрабатывали TEA (5,55 мл, 39,8 ммоль), ди-трет-бутилдикарбонатом (5,55 мл, 23,9 ммоль), DMAP (0,195 г, 1,593 ммоль) при 0°C, и полученную смесь перемешивали в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 2 часов, разбавляли ДХМ и промывали водой, раствором лимонной кислоты, насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией, с получением указанного в заголовке соединения. Анализ ЖХМС: ES+ve m/z 351 (M+H)+.
Промежуточное соединение 30: трет-бутил 2,4-диметил-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазол-1-карбоксилат
К перемешиваемому раствору трет-бутил 5-(3-бромфенил)-2,4-диметил-1H-имидазол-1-карбоксилата (4,5 г, 12,8 ммоль) в 1,4-диоксане (50 мл) добавляли ацетат калия (2,51 г, 25,6 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (3,90 г, 15,37 ммоль). Полученный раствор продували газообразным аргоном в течение 15 минут. Добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II)-дихлорметан (0,937 г, 1,281 ммоль). Снова продували газообразным аргоном в течение 15 минут. Реакционную смесь перемешивали при 90°C в течение 16 часов. Полученную реакционную смесь фильтровали через рыхлый слой целита, промывали EtOAc (3×100 мл) и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, элюируя 10% EtOAc в гексане. Требуемые фракции концентрировали при пониженном давлении, с получением указанного в заголовке соединения (4,0 г, 78%) в виде белого твердого вещества: ЖХМС ES+ve m/z 399 (M+H)+.
Промежуточное соединение 31: 2,4-диметил-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазол
Раствор трет-бутил 2,4-диметил-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазол-1-карбоксилата (промежуточное соединение 30) (3,9 г, 9,79 ммоль) в 1,4-диоксане (40 мл) охлаждали в атмосфере азота до 0°C и затем по каплям обрабатывали 4M HCl в 1,4-диоксане (40 мл) около 10 минут. Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли при пониженном давлении, и остаток обрабатывали диэтиловым эфиром (50 мл). Полученное в результате твердое вещество отделяли фильтрованием и промывали диэтиловым эфиром (50 мл). Твердое вещество растворяли в ДХМ (200 мл) и промывали раствором NaHCO3 (100 мл). Органический слой отделяли, сушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток растирали в пентане (50 мл), с получением указанного в заголовке соединения (1,27 г, 37%) в виде белого твердого вещества: ЖХМС ES+ve m/z 299 (M+H)+.
Промежуточное соединение 32: Метил 3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат
Смесь 2,4-диметил-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазола (промежуточное соединение 31) (543 мг, 1,821 ммоль), (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 5) (200 мг, 0,607 ммоль), (R)-BINAP (41,6 мг, 0,067 ммоль) и тетрафторбората бис(норборнадиен)родия(I) (22,70 мг, 0,061 ммоль) добавляли в микроволновую пробирку. Смесь растворяли в 1,4-диоксане (10 мл) с последующим добавлением 3,8M раствора KOH (0,320 мл, 1,21 ммоль). Полученную реакционную смесь сразу нагревали при 95°C в течение 45 минут с использованием микроволновой системы Biotage. Образец разбавляли MeOH и загружали на колонку SCX (10 г). Промывали MeOH (2 CV) и затем элюировали 2M NH3 в MeOH. Растворитель удаляли при пониженном давлении, остаток (504 мг) растворяли в смеси 1:1 ДМСО:MeOH, загружали на колонку с обращенной фазой (C18) (12 г) и элюировали с использованием 30-85% градиента смеси 10 мм водный раствор бикарбоната аммония-MeCN над 12CV. Соответствующие фракции объединяли и растворитель выпаривали при пониженном давлении, с получением указанного в заголовке соединения в виде смеси диастереоизомеров (101 мг, 33%). ЖХМС (система C) RT=1,16 мин, 95%, ES+ve m/z 502 (M+H)+. Изомеры разделяли препаративной хиральной ВЭЖХ на колонке Chiralpak IC (250 мм×30 мм), элюируя смесью 10% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 30 мл/мин, детектирование при 215 нм с получением:
изомера 1 (2 мг) [(R)-метил 3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат]. Аналитическая хиральная ВЭЖХ RT=26,3 мин, 100% на колонке Chiralpak IC (250 мм×4,6 мм), элюируя смесью 10% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм;
изомера 2 (18 мг) [(S)-метил 3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат]. Аналитическая хиральная ВЭЖХ RT=29,4 мин, 100% на колонке Chiralpak IC (250 мм×4,6 мм), смесью 10% EtOH (содержащим смесь 0,2% изопропиламин-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм.
Промежуточное соединение 33: 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-1,2,4-триазол
К раствору 1-(3-бромфенил)-1H-1,2,4-триазола (WO2001090108, стр. 62) (27 г, 121 ммоль) в 1,4-диоксане (600 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (30,6 г, 121 ммоль) и ацетат калия (23,65 г, 241 ммоль), и реакционную смесь перемешивали в течение 15 минут перед добавлением аддукта PdCl2(dppf)-CH2Cl2 (9,84 г, 12,05 ммоль). Полученную смесь нагревали до 100°C в течение 18 часов, затем давали охладиться до комнатной температуры и фильтровали через целит. Твердое вещество промывали 1,4-диоксаном (50 мл), и объединенный и промытый фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в ДХМ (30 мл), абсорбировали на силикагеле (100-200 меш) (30 г), и колонку элюировали смесью 5% этилацетат-петролейный эфир. Соответствующие фракции объединяли и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (10,3 г, 31%) в виде окрашенного в бледно-коричневый цвет твердого вещества: ES+ve m/z 272 (M+H)+; ВЭЖХ RT=5,0 мин, 98,9% на колонке Zorbax CN (250 мм×4,6 мм) с изократическим элюированием 20% EtOH в н-гексане, при скорости потока 1 мл/мин.
Промежуточное соединение 34: (S)-метил 3-(3-(1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноат
Раствор (3-(1H-1,2,4-триазол-1-ил)фенил)бороновой кислоты (промежуточное соединение 33) (344 мг, 1,82 ммоль), (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 5) (200 мг, 0,607 ммоль), (R)-BINAP (41,6 мг, 0,067 ммоль) и тетрафторбората бис(норборнадиен)родия(I) (22,7 мг, 0,061 ммоль) в 1,4-диоксане (4 мл) добавляли в микроволновую пробирку и затем добавляли 3,8M раствор KOH (0,320 мл, 1,214 ммоль), и полученную реакционную смесь нагревали при 95°C в течение 60 минут в микроволновой системе Biotage. Полученную смесь разбавляли MeOH и загружали на колонку SCX (10 г). Содержимое промывали MeOH (2CV) и затем элюировали 2M NH3 в MeOH. Содержащие аммиак фракции упаривали при пониженном давлении, и полученное в результате оранжевое масло (472 мг) растворяли в 1:1 ДМСО-MeOH (2,5 мл) и очищали при помощи MDAP (метод A). Соответствующие фракции упаривали в атмосфере азота в блоке с набегающим потоком, с получением указанного в заголовке соединения (64 мг, 22%) в виде желтого масла. ЖХМС (система C) RT=1,15 мин, 82%, ES+ve m/z 475 (M+H)+. Продукт (60 мг) растворяли в EtOH (1 мл), и основной диaстереоизомер отделяли препаративной хиральной ВЭЖХ на колонке Chiralpak AD-H (250 мм×30 мм), элюируя смесью 40% EtOH (содержащий 0,2% изопропиламин)-гептан (содержащий 0,2% изопропиламин), при скорости потока 30 мл/мин, детектирование при 215 нм, с получением (S)-метил-3-(3-(1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноата (15 мг): аналитическая хиральная ВЭЖХ RT=20,0 мин, 100%, на колонке Chiralpak AD-H (4,6 мм×250 мм), элюируя смесью 40% EtOH-гептан, содержащей 0,2% изопропиламин, при скорости потока 1 мл/мин, детектирование при 215 нм.
Промежуточное соединение 35: (E)-трет-бутил 4-бромбут-2-еноат
Газообразный изобутилен (363 мл, 3,82 моль) барботировали через перемешиваемый раствор (E)-4-бромбут-2-еновой кислоты (Tetrahedron Asymmetry, 2010, 21, 1574) (210 г, 1,27 ммоль) и концентрированной H2SO4 (20,35 мл, 382 ммоль) в диэтиловом эфире (1 л) при -40°C в течение 30 минут в стальном автоклаве. Полученную смесь герметизировали в автоклаве, и смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь охлаждали до 0°C, затем подщелачивали триэтиламином (250 мл) и экстрагировали ДХМ (3×200 мл). Органический слой сушили и концентрировали в вакууме. Полученный остаток растирали в н-пентане (200 мл), с получением указанного в заголовке соединения (140 г, 50%) в виде коричневого сиропа:
1H ЯМР δ (CDCl3, 400 МГц) 6,89 (дт, J=15, 7,5 Гц, 1H), 5,95 (дт, J=15, 1 Гц, 1H), 3,99 (дд, J=7,5, 1 Гц, 2H), 1,48 (с, 9H).
Промежуточное соединение 36: (E)-трет-бутил 4-ацетоксибут-2-еноат
Перемешиваемый раствор (E)-трет-бутил 4-бромбут-2-еноата (промежуточное соединение 35) (280 г, 1,27 моль) в ацетонитриле (1,2 л) обрабатывали ацетатом калия (186 г, 1,9 моль) при комнатной температуре. Полученную смесь перемешивали при 60°C в течение 4 часов, и реакцию отслеживали ТСХ (10% диэтиловый эфир в петролейном эфире, Rf=0,4, детектирование при помощи УФ). Полученную реакционную смесь охлаждали до комнатной температуры, твердое вещество удаляли фильтрованием и промывали диэтиловым эфиром (600 мл). Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя 10% диэтиловым эфиром в петролейном эфире. Соответствующие фракции объединяли и упаривали, с получением указанного в заголовке соединения (148 г, 58% выход) в виде бесцветной жидкости:
1H ЯМР δ (CDCl3, 400 МГц) 6,80 (дт, J=15,5, 5 Гц, 1H), 5,93 (дт, J=15,5, 2 Гц, 1H), 4,70 (дд, J=5, 2 Гц, 2H), 2,09 (с, 3H), 1,46 (с, 9H).
Промежуточное соединение 37: (R,E)-трет-бутил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноат
Смесь (R)-7-(2-(пирролидин-3-ил)этил)-1,2,3,4-тетрагидро-1,8-нафтиридина (промежуточное соединение 4) (1,305 г, 5,64 ммоль), (E)-трет-бутил 4-ацетоксибут-2-еноата (промежуточное соединение 36) (1,13 г, 5,64 ммоль), 1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,207 г, 0,282 ммоль) и DIPEA (2,96 мл, 16,92 ммоль) в ДХМ (20 мл) перемешивали в течение 3 часов. Полученную реакционную смесь распределяли между раствором хлорида аммония (50 мл) и ДХМ (50 мл). Водный слой дополнительно экстрагировали ДХМ (50 мл). Объединенные органические слои пропускали через гидрофобную фритту и концентрировали в вакууме. Неочищенный остаток очищали хроматографией (KPNH, 110 г, 0-100% TBME-циклогексан), элюируя в течение 60 минут. Содержащие продукт фракции объединяли и концентрировали, с получением указанного в заголовке соединения (1,65 г, 79%) в виде желтого масла (E:Z соотношение 7,5:1).
1H ЯМР (400 MГц, CDCl3) 7,06 (д, J=7,3 Гц, 1H), 6,89 (дт, J=15,6, 6,2 Гц, 1H), 6,35 (д, J=7,3 Гц, 1H), 5,90 (дт, J=15,6, 1,6 Гц, 1H), 4,66-4,80 (м, 1H), 3,38-3,44 (м, 2H), 3,20 (ддд, J=6,2, 4,8, 1,8 Гц, 2H), 2,87 (дд, J=8,4, 7,4 Гц, 1H), 2,66-2,74 (м, 3H), 2,50-2,57 (м, J=8,2, 4,0, 4,0 Гц, 2H), 2,41-2,50 (м, J=8,7, 8,7, 6,0 Гц, 1H), 1,98-2,26 (м, J=8,6 Гц, 3H), 1,87-1,96 (м, J=11,7, 6,0, 6,0 Гц, 2H), 1,74 (кв, J=7,6 Гц, 2H), 1,50 (с, 9H).
Промежуточное соединение 38: (R,E)-трет-бутил7-(2-(1-(4-(трет-бутокси)-4-оксобут-2-ен-1-ил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилат
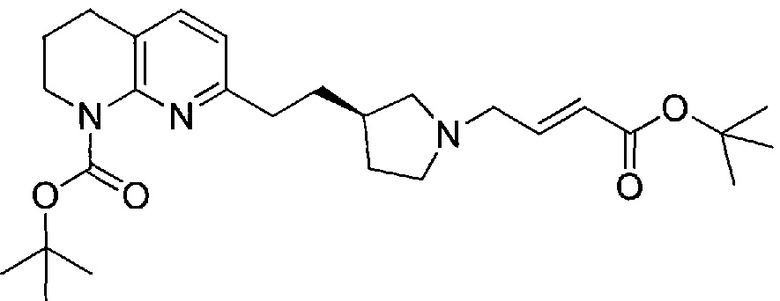
Раствор (R,E)-трет-бутил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 37) (1,20 г, 3,23 ммоль) и ди-трет-бутилдикарбоната (0,890 мл, 3,88 ммоль) в ТГФ (10 мл) при 0°C обрабатывали раствором LiHMDS (1,0M, 3,23 мл, 3,23 ммоль) и перемешивали в течение 0,5 часа. Реакцию гасили добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали ДХМ (2×15 мл). Объединенные органические слои пропускали через гидрофобную фритту и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (KPNH, 11 г), элюируя смесью 0-25% EtOAc-циклогексан около 40 минут. Соответствующие фракции объединяли и концентрировали в вакууме, с получением указанного в заголовке соединения (1,29 г, 85%) в виде желтой смолы: ЖХМС (система C) RT=1,46 мин, 95%, ES+ve m/z 472 (M+H)+.
Промежуточное соединение 39: трет-бутил 7-(2-((R)-1-((S)-4-(трет-бутокси)-2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилат

Смесь (R,E)-трет-бутил 7-(2-(1-(4-(трет-бутокси)-4-оксобут-2-ен-1-ил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (5,1 г, 10,8 ммоль), 5-метил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-пиразол-3-ил)метанола (промежуточное соединение 10) (6,21 г, 20 ммоль), KOH (5,69 мл, 21,6 ммоль) растворяли в 1,4-диоксане (20 мл). Колбу продували азотом в течение 5 минут, и затем добавляли (R)-BINAP (0,673 г, 1,08 ммоль) и [Rh(COD)Cl]2 (0,267 г, 0,541 ммоль). Полученную реакционную смесь нагревали до 90°C в течение 1 часа. Растворитель удаляли в вакууме, и остаток распределяли между ДХМ и водой. Органический слой пропускали через гидрофобную фритту и снова концентрировали в вакууме. Полученный остаток очищали хроматографией с обращенной фазой на колонке C-18 (400 г) при градиентном элюировании смесью 50-90% (0,1% водный NH3 в MeCN)-водный 10 мМ NH4HCO3, над 15CV. Соответствующие фракции объединяли и концентрировали в вакууме, с получением продукта в виде диастереоизомеров (4,11 г, 58%). ЖХМС (система C) RT=1,45 мин, 98%, ES+ve m/z 660 (M+H)+. Два диастереоизомера разделяли препаративной хиральной ВЭЖХ на колонке Chiralcel OD-H (30 мм×250 мм), элюируя смесью 10% EtOH-гептан, при скорости потока 30 мл/мин, детектирование при 215 нм, с получением указанного в заголовке соединения (3,26 г, 46%) (основной диастереоизомер): аналитическая хиральная ВЭЖХ RT=16,6 мин, 99,8% на колонке Chiralcel OD-H (4,6 мм×250 мм), элюируя смесью 10% EtOH-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, и трет-бутил 7-(2-((R)-1-((R)-4-(трет-бутокси)-2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилат (несущественный диастереоизомер) (260 мг, 4%): аналитическая хиральная ВЭЖХ RT=12,4 мин, 100%.
Примеры получения
Пример 1. (S)-3-(3-(3-метил-1H-пиразол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
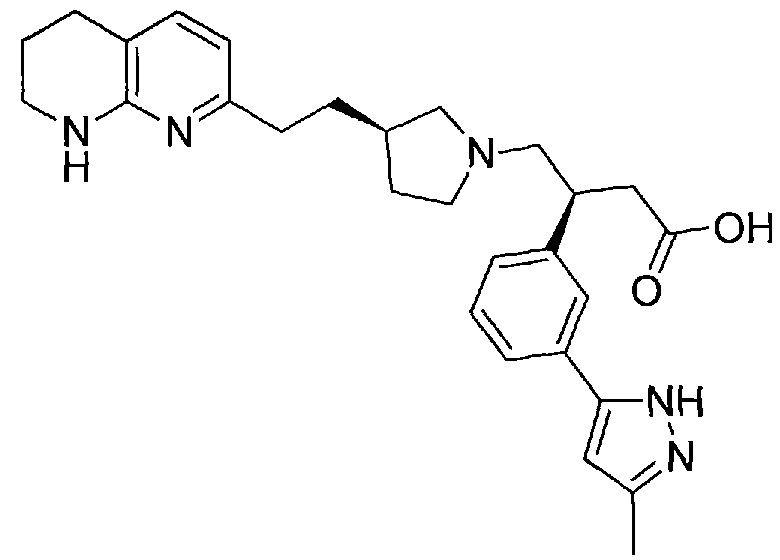
Смесь 3-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (доступный от CombiPhos) (43 мг, 0,21 ммоль), (S)-метил-3-(3-(бромфенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноата (промежуточное соединение 6) (50 мг, 0,10 ммоль), трифосфата калия (65,5 мг, 0,31 ммоль), хлор(динорбонилфосфино)(2'-диметиламино-1,1'-бифенил-2-ил)палладия (II) (6 мг, 0,01 ммоль) поглощали этанолом (0,5 мл) и водой (0,2 мл) и нагревали при 130°C в течение 30 минут в микроволновом реакторе (Anton Paar, 600 W). Охлажденную реакционную смесь очищали MDAP (способ А), с получением указанного в заголовке соединения (17,7 мг, 33%): ЖХМС (система B) RT=0,52 мин, ES+ve m/z 474 (M+H)+;
1H ЯМР (400 MГц, ДМСО-d6) 7,61 (с, 1H), 7,56 (ушир.д, J=7,5 Гц, 1H), 7,29 (т, J=7,5 Гц, 1H), 7,14 (ушир.д, J=7,5 Гц, 1H), 7,01 (д, J=7,5 Гц, 1H), 6,44 (с, 1H), 6,32 (ушир.с, 1H), 6,25 (д, J=7,5 Гц, 1H), 3,19-3,26 (м, 3H), 2,88-2,93 (м, 1H), 2,78-2,85 (м, 2H), 2,70-2,76 (м, 1H), 2,51-2,62 (м, 4H), 2,33-2,46 (м, 4H), 2,24 (с, 3H), 1,98-2,07 (м, 1H), 1,88-1,95 (м, 1H), 1,74 (квин., J=6,0 Гц, 2H), 1,55-1,67 (м, 2H), 1,31-1,39 (м, 1H).
Пример 2. (S)-3-(3-(1,4-диметил-1H-пиразол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота

Получали из (S)-метил 3-(3-бромфенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноата (промежуточное соединение 6) (98,4 мг, 0,202 ммоль) и 1,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (доступный от CombiPhos) (0,192 мл, 0,809 ммоль) способом, аналогично методике получения по примеру 1, с получением указанного в заголовке соединения (13,3 мг, 13%): ЖХМС (система A) RT=0,80 мин, 97%, ES+ve m/z 488 (M+H)+;
1H ЯМР (400 MГц, ДМСО-d6) 7,50 (т, J=7,5 Гц, 1H), 7,43 (ушир.д, J=7,5 Гц, 1H), 7,39 (ушир.с, 1H), 7,34 (с, 1H), 7,32 (ушир.д, J=7,5 Гц, 1H), 7,20 (ушир.д, J=7,0 Гц, 1H), 6,36 (ушир.д, J=7,0 Гц, 1H), 3,71 (с, 3H), 2,83 (дд, J=16,5, 6,0 Гц, 1H), 2,62-2,66 (м, 2H), 2,59 (дд, J=16,5, 7,5 Гц, 1H), 2,15-2,24 (м, 1H), 2,02-2,10 (м, 1H), 1,98 (с, 3H), 1,74-1,80 (м, 2H), 1,63-1,70 (м, 2H), 1,49-1,59 (м, 1H).
Пример 3. (S)-3-(3-(4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
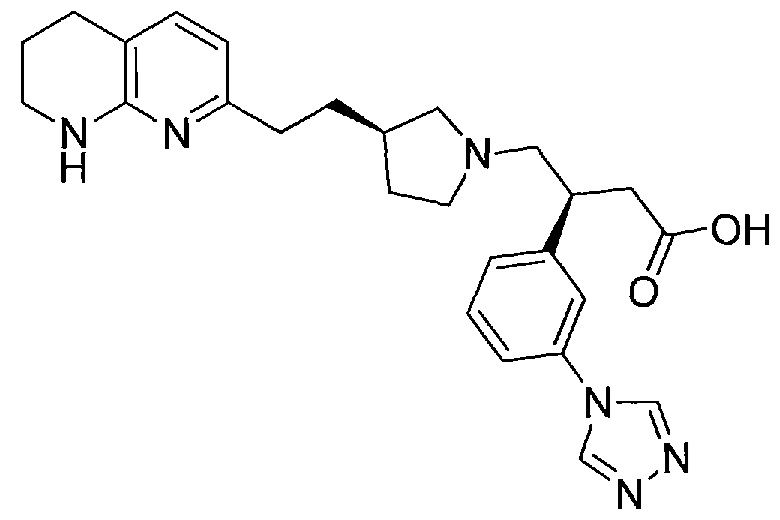
К дегазированной смеси (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 6) (170 мг, 0,516 ммоль), 4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4H-1,2,4-триазола (промежуточное соединение 19) (420 мг, 1,55 ммоль), (R)-BINAP (32,1 мг, 0,052 ммоль) и водного KOH (3,8M, 0,407 мл) в 1,4-диоксане (24 мл) добавляли димер хлор(1,5-циклооктадиен)родия(l) (12,72 мг, 0,026 ммоль). Полученную реакционную смесь нагревали до 100°C в течение 2 часов и затем помещали в SCX картридж (50 г), промывали MeOH (2CV) и затем элюировали 2M аммиаком в MeOH (4CV). Основные фракции концентрировали, и остаток очищали колоночной хроматографией (20 г) при градиентном элюировании смесью 0-100% EtOAc-циклогексан. Соответствующие фракции концентрировали при пониженном давлении, и остаток (66,8 мг) растворяли в MeCN (4 мл), обрабатывали водным NaOH (2M, 1,0 мл) и нагревали в микроволновом реакторе при 80°C в течение 2 часов. Полученную реакционную смесь нейтрализовали при помощи водного раствора 2M HCl и концентрировали в вакууме. Полученный остаток помещали в SCX картридж (10 г), промывали MeOH (1 CV) и элюировали 2M аммиаком в метаноле (2CV). Основные фракции концентрировали в вакууме, с получением двух диастереоизомеров указанного в заголовке соединения (80 мг). Изомеры разделяли препаративной хиральной ВЭЖХ на колонке Chiralpak ID (30 мм×25 см), элюируя смесью 50% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 30 мл/мин, детектирование при 215 нм, с получением после упаривания соответствующих фракций указанного в заголовке соединения: (40 мг, 16%) в виде смолы: ЖХМС (система D) RT=0,34 мин, 98%, ES+ve m/z 461 (M+H)+;
1H ЯМР (500 MГц, CDCl3) 8,48 (с, 2H), 7,45 (т, J=7,5 Гц, 1H), 7,34 (д, J=7,5 Гц, 1H), 7,27 (с, 1H), 7,20 (д, J=7,5 Гц, 1H), 7,17 (д, J=7,0 Гц, 1H), 6,27 (ушир.д, J=7,0 Гц, 1H), 3,60-3,67 (м, 1H), 3,52-3,60 (м, 1H), 3,38-3,45 (м, 2H), 2,87-2,98 (м, 2H), 2,75-2,84 (м, 1H), 2,38-2,48 (м, 2H), 2,22-2,33 (м, 1H), 1,46-1,58 (м, 1H), 1,34-1,45 (м, 1H);
аналитическая хиральная ВЭЖХ RT=13,5 мин, на колонке Chiralpak ID (4,6 мм в.д.×25 см), элюируя смесью 50% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1,0 мл/мин, детектирование при 215 нм.
Примеры 4-9
Соединения следующих примеров получали в определенном порядке по следующей общей методике.
Раствор (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (100 мг, 0,304 ммоль) в 1,4-диоксане (1 мл) и подходящего сложного боронатного эфира (0,607 ммоль), выбранного из: 3,5-диметил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-1,2,4-триазола (промежуточное соединение 12), 3-метил-4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4H-1,2,4-триазола (промежуточное соединение 14), 2-(5-метил-1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-пиразол-3-ил)пропан-2-ола (промежуточное соединение 16), 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3-(трифторметил)-1H-пиразола (промежуточное соединение 18), 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-1,2,3-триазола (промежуточное соединение 21) и 1-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазола (промежуточное соединение 8), обрабатывали (R)-BINAP (9,45 мг, 0,015 ммоль), димером хлор(1,5-циклооктадиен)родия(l) (14,97 мг, 0,030 ммоль) и водным гидроксидом калия (0,160 мл, 0,608 ммоль), полученную смесь нагревали до 80°C в течение 5 часов, и затем выстаивали при комнатной температуре в течение 18 часов. Смесь фильтровали для удаления нерастворимых веществ, разбавляли ДМФА (1 мл) и очищали MDAP (способ А). Растворитель удаляли в токе азота в пластинчатом аппарате для продувки, с получением продуктов в виде диастереоизомерных смесей. Диастереоизомеры разделяли препаративной хиральной ВЭЖХ на колонке Chiralcel OJ-H (30 мм×250 мм), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 30 мл/мин, детектирование при 215 нм с получением:
Пример 4. 3-(3-(3,5-диметил-1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты
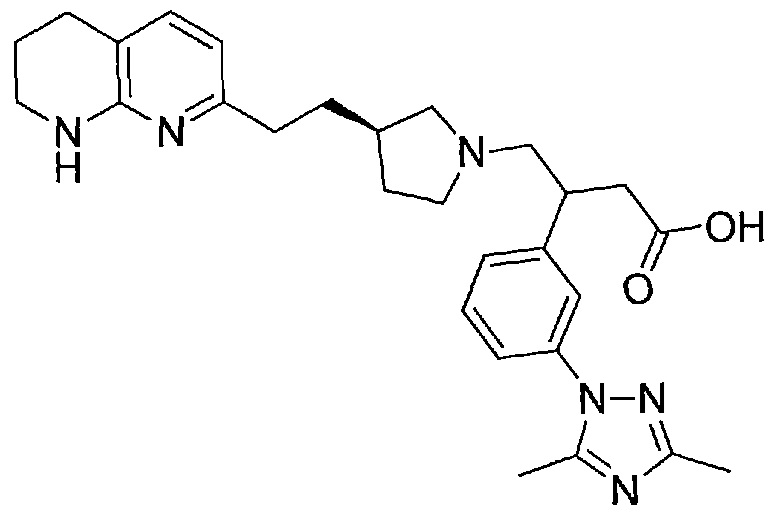
Изомер 1 (S)-3-(3-(3,5-диметил-1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (20 мг, 62%): ЖХМС (система C) RT=0,75 мин, ES+ve m/z 489 (M+H)+; аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм: RT=22,7 мин, 99,5%;
1H ЯМР δ (500 MГц, ДМСО-d6) 7,42-7,47 (м, 1H), 7,41 (с, 1H), 7,31-7,38 (м, 2H), 7,01 (д, J=7,0 Гц, 1H), 6,21-6,28 (м, 2H), 3,19-3,25 (м, 2H), 2,40 (с, 3H), 2,26 (с, 3H), 1,94-2,05 (м, 1H), 1,83-1,93 (м, 1H), 1,69-1,78 (м, 2H), 1,53-1,65 (м, 2H), 1,28-1,38 (м, 1H).
Изомер 2 (R)-3-(3-(3,5-диметил-1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (10 мг, 31%): аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, RT=13,1 мин, 99,5%.
Пример 5. (S)-3-(3-(3-метил-4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
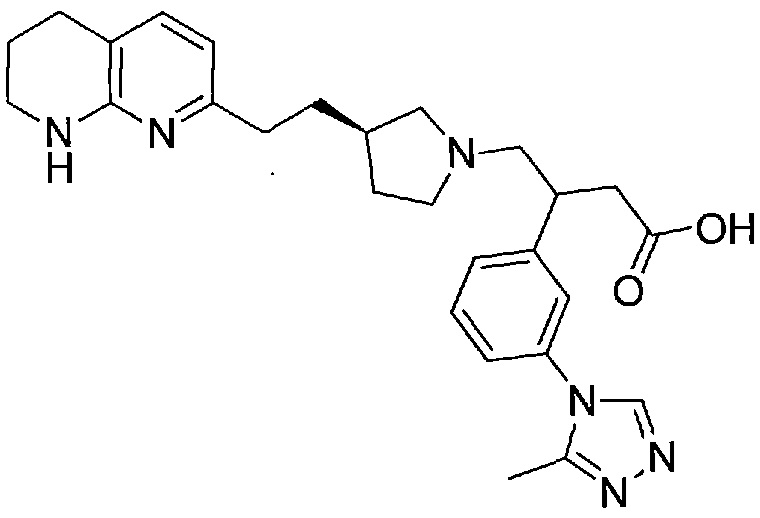
Изомер 1 (S)-3-(3-(3-метил-4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (9 мг): ЖХМС (система C) RT=0,77 мин, ES+ve m/z 475 (M+H)+; аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм: RT=18,1 мин, 99,4%;
1H ЯМР (500 MГц, ДМСО) 9,12 (с, 1H), 7,69 (с, 1H), 7,63 (ушир.д, J=7,5 Гц, 1H), 7,43 (т, J=7,5 Гц, 1H), 7,25 (ушир.д, J=7,5 Гц, 1H), 7,00 (ушир.д, J=7,0 Гц, 1H), 6,21-6,28 (м, 2H), 3,19-3,25 (м, 2H), 2,36 (с, 3H), 1,95-2,06 (м, 1H), 1,83-1,94 (м, 1H), 1,69-1,78 (м, 2H), 1,53-1,66 (м, 2H), 1,28-1,38 (м, 1H).
Изомер 2 (R)-3-(3-(3-метил-4H-1,2,4-триазол-4-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (4 мг): аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, RT=14,0 мин, 99,4%.
Пример 6. (S)-3-(3-(3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота

Изомер 1 (S)-3-(3-(3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (6 мг): ЖХМС (система D) RT=0,82 мин, ES+ve m/z 532 (M+H)+;
1H ЯМР (500 MГц, ДМСО) 7,36-7,42 (м, 1H), 7,29 (ушир.д, J=7,5 Гц, 1H), 7,25 (ушир.д, J=7,5 Гц, 1H), 7,00 (ушир.д, J=7,0 Гц, 1H), 4,86 (ушир.с, 1H), 2,28 (с, 3H), 1,94-2,06 (м, 1H), 1,83-1,94 (м, 1H), 1,69-1,79 (м, 2H), 1,53-1,66 (м, 2H), 1,43 (с, 6H), 1,28-1,36 (м, 1H);
аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, RT=22,5 мин, 99,5%.
Изомер 2 (R)-3-(3-(3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (4 мг): аналитическая хиральная ВЭЖХ RT=11,5 мин, 99,5%.
Пример 7. 4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)-3-(3-(3-(трифторметил)-1H-пиразол-1-ил)фенил)бутановая кислота
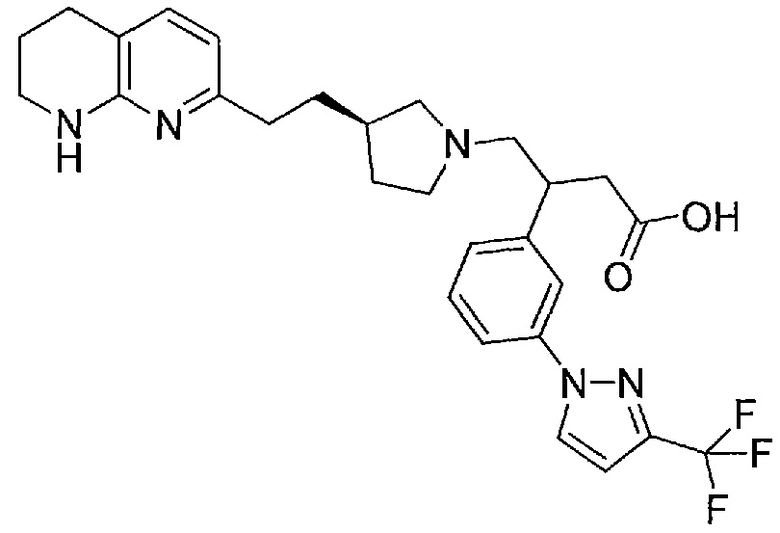
Изомер 1 (25 мг): ЖХМС (система C), RT=1,01 мин, ES+ve m/z 528 (M+H)+;
1H ЯМР (500 MГц, ДМСО-d6) 8,72 (ушир.с, 1H), 7,75 (ушир.с, 1H), 7,70 (д, J=7,5 Гц, 1H), 7,45 (т, J=7,5 Гц, 1H), 7,30 (д, J=7,5 Гц, 1H), 7,03 (ушир.с, 1H), 7,00 (д, J=7,0 Гц, 1H), 6,26 (ушир.с, 1H), 6,23 (д, J=7,0 Гц, 1H), 1,95-2,05 (м, 1H), 1,83-1,94 (м, 1H), 1,69-1,78 (м, 2H), 1,53-1,67 (м, 2H), 1,28-1,38 (м, 1H);
аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 25% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, RT=14,0 мин, 99,5%.
Изомер 2 (23 мг): аналитическая хиральная ВЭЖХ, RT=8,5 мин, 99,3%.
Пример 8. (S)-3-(3-(1H-1,2,3-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
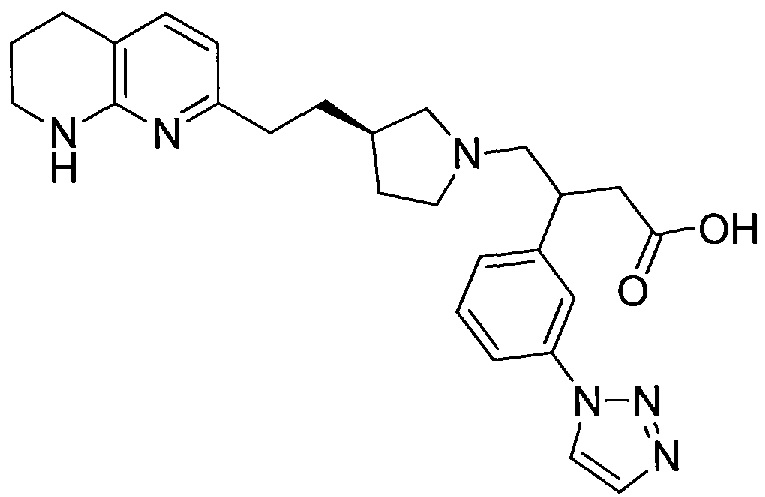
Изомер 1 (S)-3-(3-(1H-1,2,3-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (16 мг): ЖХМС (система C), RT=0,76 мин, ES+ve m/z 461 (M+H)+;
1H ЯМР (ДМСО-d6) 8,81 (с, 1H), 7,96 (с, 1H), 7,79 (ушир.с, 1H), 7,73 (ушир.д, J=7,5 Гц, 1H), 7,50 (ушир.т, J=7,5 Гц, 1H), 7,36 (ушир.д, J=7,5 Гц, 1H), 7,00 (ушир.д, J=7,0 Гц, 1H), 6,20-6,28 (м, 2H), 3,22 (ушир.с, 2H), 2,39 (ушир.т, J=7,5 Гц, 2H), 2,23-2,32 (м, 1H), 1,95-2,06 (м, 1H), 1,83-1,94 (м, 1H), 1,68-1,78 (м, 2H), 1,53-1,66 (м, 2H), 1,27-1,38 (м, 1H);
аналитическая хиральная ВЭЖХ на колонке Chiralcel AD-H (4,6 мм в.д.×25 см), элюирование смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм, RT=19,5 мин, 98,8%.
Изомер 2 (R)-3-(3-(1H-1,2,3-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (5 мг): аналитическая хиральная ВЭЖХ RT=16,0 мин, 99,5%.
Пример 9. 3-(3-(1H-имидазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
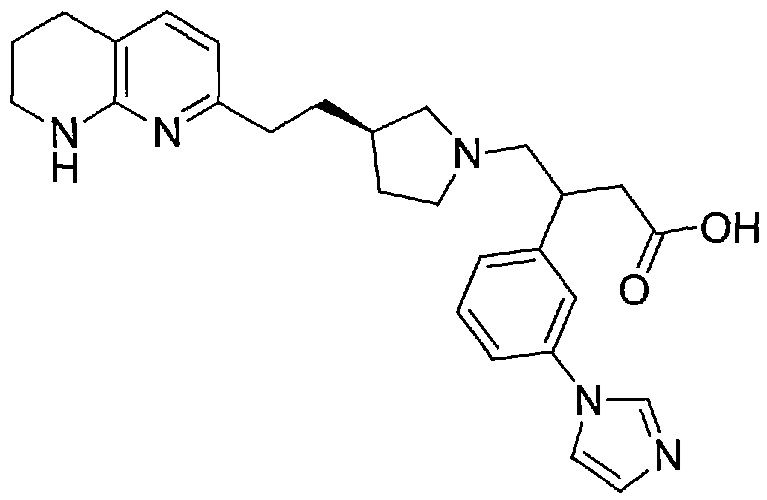
Соединение примера 9 выделяли в виде смеси диастереоизомеров (2,6 мг): ЖХМС (система C) RT=0,76 мин, 100%, ES+ve m/z 460 (M+H)+;
1H ЯМР (500 MГц, ДМСО-d6) 8,25 (ушир.с, 1H), 7,74 (ушир.с, 1H), 6,94-7,68 (м, 6H+), 6,15-6,40 (м, 1H+), 3,25-3,33 (м, 1H), 3,22 (ушир.с, 2H), 2,34-2,42 (м, 2H), 2,12-2,28 (м, 1H), 1,95-2,04 (м, 1H), 1,82-1,93 (м, 1H), 1,69-1,78 (м, 2H), 1,54-1,65 (м, 2H), 1,27-1,38 (м, 1H);
(спектр показал смесь диастереоизомеров, которые сгруппированы вместе с некоторыми вариациями в суммарных значениях).
Пример 10. 3-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота, в виде 9:1 смеси изомер 1:изомер 2
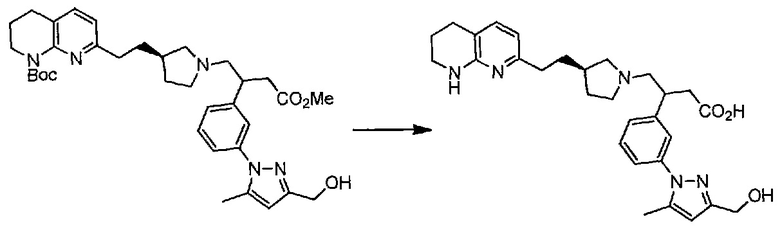
Раствору трет-бутил 7-(2-((3R)-1-(2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (промежуточное соединение 23) (20 мг, 0,032 ммоль) в TFA (0,5 мл, 6,49 ммоль) давали перемешиваться в течение 2 часов. Полученный раствор упаривали в токе азота, остаток растворяли в метаноле (0,5 мл), обрабатывали NaOH (0,5 мл, 1,0 ммоль) и перемешивали в течение 15 часов. Полученную смесь упаривали в токе азота для удаления метанола и подкисляли 2Н HCl (0,7 мл). Полученный раствор сушили вымораживанием, и остаточное твердое вещество обрабатывали 1:1 ДМСО-MeOH (1 мл). Полученную суспензию фильтровали, и полученный раствор очищали при помощи MDAP (способ А), с получением, после сушки вымораживанием, указанного в заголовке соединения (9,5 мг, 58%) в виде 9:1 смеси диастереоизомеров при помощи ЯМР: ЖХМС (система C) RT=0,77 мин, ES+ve m/z 504 (M+H)+;
1H ЯМР (CD3OD) 7,51 (т, J=7,5 Гц, 1H), 7,32-7,43 (м, 3H), 7,15 (д, J=7,0 Гц, 1H), 6,39 (д, J=7,0 Гц, 1H), 6,30 (с, 1H), 3,45-3,63 (м, 2H), 3,35-3,40 (м, 2H), 2,86 (дд, J=16,5, 10,0 Гц, 1H), 2,66-2,73 (м, 2H), 2,61-2,67 (м, 1H), 2,53-2,59 (м, 2H), 2,32 (с, 3H), 2,15-2,26 (м, 1H), 1,83-1,91 (м, 2H), 1,74-1,83 (м, 2H), 1,63-1,72 (м, 1H).
Диастереоизомеры (62 мг), полученные из другого эксперимента, разделяли препаративной хиральной ВЭЖХ на колонке Chiralcel OJ-H (30 мм×250 мм), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан (содержащий 0,2% изопропиламин), при скорости потока 30 мл/мин, детектирование при 215 нм, с получением указанного в заголовке соединения в виде изомеров:
изомер 1 (20 мг). ЖХМС (система C) RT=0,76 мин, ES+ve m/z 504 (M+H)+;
1H ЯМР (500 MГц, ДМСО-d6) δ 7,44-7,39 (1H, м), 7,34 (1H, с), 7,31 (1H, д, J=8,0 Гц), 7,27 (1H, д, J=7,4 Гц), 7,02 (1H, д, J=7,1 Гц), 6,28-6,21 (3H, м), 4,43 (2H, с), 3,50-3,27 (3H, м), 2,91-2,75 (3H, м), 2,74-2,64 (1H, м), 2,64-2,53 (4H, м), 2,48-2,36 (3H, м), 2,35-2,23 (4H, м), 2,08-1,96 (1H, м), 1,95-1,83 (1H, м), 1,79-1,70 (2H, м), 1,66-1,55 (2H, м), 1,40-1,28 (1H, м);
аналитическая хиральная ВЭЖХ RT=11,9 мин, 99,5% на колонке Chiralcel OJ-H (4,6 мм в.д.×250 мм), элюируя смесью 20% EtOH (содержащий 0,2% изопропиламин)-гептан, при скорости потока 1 мл/мин, детектирование при 215 нм;
изомер 2: аналитическая хиральная ВЭЖХ RT=7 мин, 99,5%.
Пример 11. 3-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота (9:1 смесь изомер 1:изомер 2)
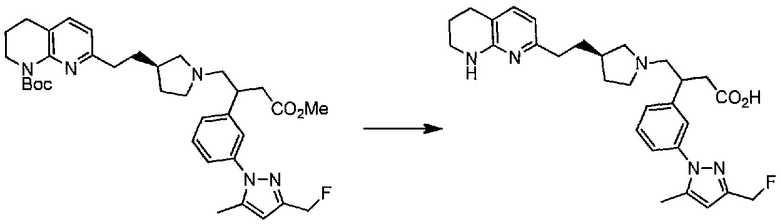
Раствор трет-бутил 7-(2-((3R)-1-(2-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-метокси-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (промежуточное соединение 24) (42 мг, 0,068 ммоль) в TFA (0,5 мл, 6,5 ммоль) перемешивали при комнатной температуре в течение 2 часов. TFA удаляли в токе азота, и остаток в метаноле (0,7 мл) обрабатывали NaOH (1 мл, 2,0 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Полученный раствор подкисляли водным HCl (2Н, 1,3 мл), и метанол удаляли в токе азота. Полученный водный раствор сушили вымораживанием, и остаток обрабатывали ДМСО/MeOH (1:1; 1 мл), фильтровали и очищали при помощи MDAP (способ А), с получением указанного в заголовке соединения (20 мг, 59%): ЖХМС (система C) RT=0,89 мин, ES+ve m/z 506 (M+H)+;
1H ЯМР (400 MГц, CD3OD) 7,49-7,56 (м, 1H), 7,35-7,45 (м, 3H), 7,14 (д, J=7,5 Гц, 1H), 6,36-6,42 (м, 2H), 5,33 (д, J=48,5 Гц, 2H), 2,86 (ушир.дд, J=16,0, 10,0 Гц, 1H), 2,56 (т, J=7,5 Гц, 2H), 2,34 (с, 3H), 1,82-1,91 (м, 2H), 1,74-1,83 (м, 2H), 1,61-1,71 (м, 1H) (перечисленные пики ассоциированы с основным изомером).
[18F]3-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота может быть получена для применения в качестве лиганда позитронно-эмиссионной томографии (PET) для диагностических целей.
Альтернативное получение соединения примера 11
(S)-3-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота
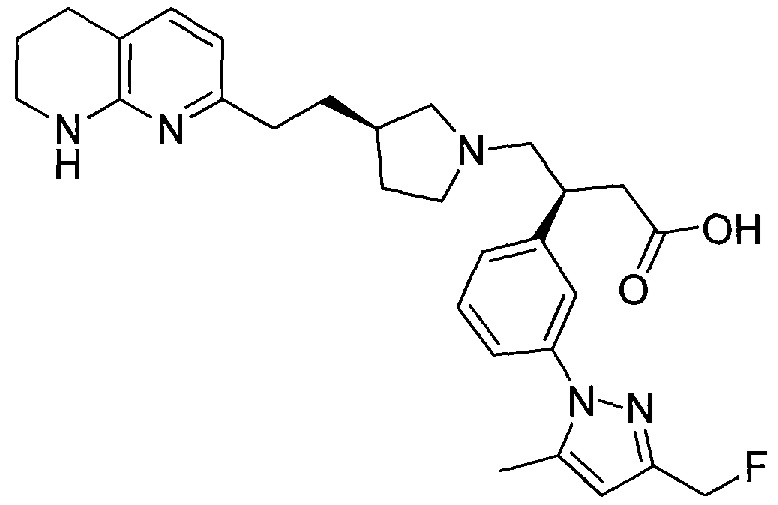
К раствору трет-бутил 7-(2-((R)-1-((S)-4-(трет-бутокси)-2-(3-(3-(гидроксиметил)-5-метил-1H-пиразол-1-ил)фенил)-4-оксобутил)пирролидин-3-ил)этил)-3,4-дигидро-1,8-нафтиридин-1(2H)-карбоксилата (промежуточное соединение 39) (100 мг, 0,152 ммоль) в ацетонитриле (3 мл) при комнатной температуре добавляли триэтиламин (0,127 мл, 0,909 ммоль) и метансульфонилхлорид (0,059 мл, 0,758 ммоль). Полученную реакционную смесь нагревали до 65°C. Спустя 30 минут, ЖХМС показала преобразование в алкилхлоридное промежуточное соединение. Полученную реакционную смесь распределяли между этилацетатом и раствором бикарбоната натрия. Органический слой сушили над MgSO4 и концентрировали в вакууме. Остаток светло-оранжевого масла растворяли в ДМФА (5,0 мл) и добавляли фторид калия (35,2 мг, 0,606 ммоль) и 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]гексаоксан (доступный от Aldrich) (228 мг, 0,606 ммоль), полученный в результате раствор нагревали в микроволновом реакторе при 120°C в течение 0,5 часа, ЖХМС показала замену хлорида фторидом. Полученную реакционную смесь распределяли между водой и этилацетатом. Органический слой сушили (MgSO4) и концентрировали в вакууме, с получением коричневого масла. Неочищенное масло растворяли в ДХМ (1 мл) и обрабатывали TFA (2 мл), и перемешивали в течение 2 часов. Полученный раствор концентрировали в токе азота, с получением оранжевой смолы, которую очищали при помощи MDAP (способ А), соответствующие фракции собирали и концентрировали, с получением (S)-3-(3-(3-(фторметил)-5-метил-1H-пиразол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (39,8 мг, 52%) в виде бесцветной смолы: ЖХМС (система C) RT=0,97 мин, 100%, ES+ve m/z 506 (M+H)+.
Пример 12. 3-(3-(1,4-диметил-1H-имидазол-2-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановая кислота

Дегазированную смесь (R,E)-метил 4-(3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бут-2-еноата (промежуточное соединение 5) (100 мг, 0,304 ммоль), 1,4-диметил-2-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1H-имидазола (промежуточное соединение 27) (272 мг, 0,911 ммоль), (R)-BINAP (9,45 мг, 0,015 ммоль) и водного 3,8M KOH (0,240 мл, 0,911 ммоль) в 1,4-диоксане (1 мл) обрабатывали димером хлор(1,5-циклооктадиен)родия(l) (14,97 мг, 0,030 ммоль). Полученную реакционную смесь нагревали при 100°C в течение 3,5 часов. Добавляли водный раствор KOH (0,240 мл, 0,911 ммоль), и полученную смесь нагревали при 100°C в течение 30 минут. Полученную смесь фильтровали для удаления нерастворимых веществ, разбавляли ДМФА (1 мл) и очищали при помощи MDAP (способ А). Соответствующие фракции упаривали в токе азота в аппарате для продувки. Полученный остаток (35 мг) растворяли в EtOH (1 мл), и диастереоизомеры разделяли хиральной ВЭЖХ на колонке Chiralcel OJ-H (30 мм×250 мм), элюируя смесью 25% EtOH (содержащий 0,2% изопропиламин)-гептан (содержащий 0,2% изопропиламин), при скорости потока 30 мл/мин, детектирование при 215 нм. После упаривания получали соответствующие фракции двух изомеров указанного в заголовке соединения:
изомер 1 (S)-3-(3-(1,4-диметил-1H-имидазол-2-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (16 мг): ЖХМС (система C) RT=0,81 мин, 100%, ES+ve m/z 488 (M+H)+; аналитическая хиральная ВЭЖХ на колонке Chiralcel OJ-H (4,6 мм в.д.×25 см), элюирование смесью 25% EtOH (содержащий 0,2% изопропиламин)-гептан (содержащий 0,2% изопропиламин), при скорости потока 1 мл/мин, детектирование при 215 нм, RT=13,2 мин, 99,5%;
1H ЯМР (500 MГц, ДМСО-d6) δ 7,48 (1H, с), 7,44 (1H, д, J=7,7 Гц), 7,35 (1H, т, J=7,6 Гц), 7,26 (1H, д, J=7,7 Гц), 7,01 (1H, д, J=6,6 Гц), 6,92 (1H, с), 6,32-6,21 (2H, м), 3,30-3,21 (4H, м), 3,18 (3H, с), 2,85-2,76 (2H, м), 2,75-2,63 (2H, м), 2,63-2,54 (3H, м), 2,46-2,30 (3H, м), 2,23 (1H, т, J=8,0 Гц), 2,12 (3H, с), 2,04-1,92 (1H, м), 1,92-1,82 (1H, м), 1,79-1,69 (2H, м), 1,67-1,53 (2H, м), 1,37-1,27 (1H, м);
изомер 2 (R)-3-(3-(1,4-диметил-1H-имидазол-2-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты (6 мг): аналитическая хиральная ВЭЖХ RT=7,6 мин, 99,3%.
Пример 13. Натриевая соль (S)-3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты

NaOH (2M в MeOH) (108 мкл, 0,215 ммоль) добавляли к перемешиваемому раствору метил 3-(3-(2,4-диметил-1H-имидазол-5-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноата (промежуточное соединение 32, изомер 2) (18 мг, 0,036 ммоль) в метаноле (40 мкл) и ДХМ (320 мкл). Полученному раствору давали перемешиваться при комнатной температуре в закрытой пробкой колбе в течение ночи. Растворитель выдували в атмосфере азота, с получением не совсем белого твердого вещества (47 мг). Образец растворяли в смеси 1:1 ДМСО:MeOH и очищали при помощи MDAP (способ А). Растворитель удаляли из соответствующих фракций, с получением указанного в заголовке соединения (8,49 мг, 49%) в виде бесцветной смолы: ЖХМС (система C) RT=0,77 мин, 99%, ES+ve m/z 488 (M+H)+.
1H ЯМР (ДМСО-d6, 400 МГц) 7,47-7,30 (м, 2H), 7,30-7,23 (м, 1H), 7,01 (д, J=7,3 Гц, 1H), 6,25 (д, J=7,3 Гц, 1H), 3,25-3,20 (м, 3H), 2,90-2,67 (м, 4H), 2,60 (т, J=6 Гц, 2H), 2,55-2,52 (м, 2H), 2,45-2,37 (м, 3H), 2,35-2,27 (м, 3H), 2,26 (с, 3H), 2,08-1,99 (м, 1H), 1,96-1,86 (м, 1H), 1,77-1,70 (м, 2H), 1,65-1,57 (м, 2H), 1,39-1,29 (м, 1H).
Пример 14. Натриевая соль (S)-3-(3-(1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутановой кислоты
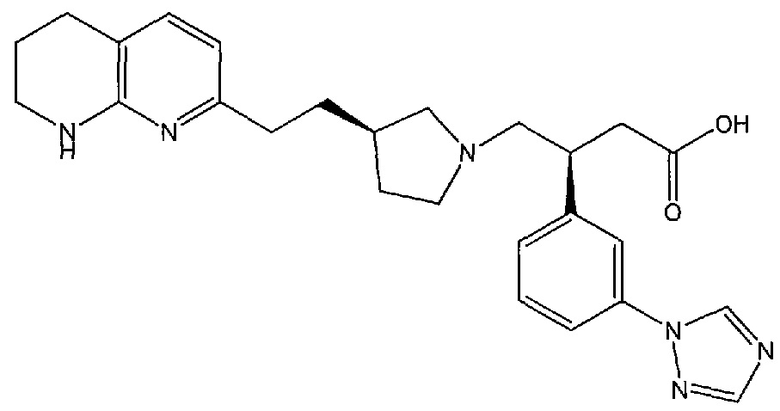
NaOH (2M в MeOH) (31,6 мкл, 0,063 ммоль) добавляли к перемешиваемому раствору (S)-метил 3-(3-(1H-1,2,4-триазол-1-ил)фенил)-4-((R)-3-(2-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)этил)пирролидин-1-ил)бутаноата (15 мг, 0,032 ммоль) в MeOH (60 мкл) и ДХМ (0,32 мл). Полученному раствору давали перемешиваться при комнатной температуре в закрытой пробкой колбе в течение 6 часов. Растворитель упаривали в блоке с набегающим потоком в атмосфере азота в течение ночи. Оставшееся белое твердое вещество растворяли в смеси 1:1 ДМСО-MeOH (0,8 мл) и очищали при помощи MDAP (способ А). Растворитель удаляли из соответствующих фракций, с получением указанного в заголовке соединения (12 мг, 82%) в виде не совсем белого твердого вещества. ЖХМС (система C) RT=0,75 мин, 100%, ES+ve m/z 461 (M+H)+.
1H ЯМР (ДМСО-d6, 400 МГц) δ 9,29 (с, 1H), 8,23 (с, 1H), 8,15 (с, 1H), 7,77 (м, 1H), 7,70 (ушир.д, J=8,5 Гц, 1H), 7,48 (т, J=7,8 Гц, 1H), 7,32 (д, J=7,8 Гц, 1H), 7,02 (д, J=7,3 Гц, 1H), 6,32 (шир, 1H), 6,25 (д, J=7,3 Гц, 1H), 3,36-3,29 (м, 1H), 3,23 (шир, 3H), 2,92 (д, J=12,1 Гц, 1H), 2,86-2,81 (м, 2H), 2,75-2,69 (м, 1H), 2,65 (м, 1H), 2,60 (м, 3H), 2,47-2,45 (м, 1H, скрыт ДМСО), 2,41 (т, J=7,7 Гц, 2H), 2,37-2,32 (м, 1H), 2,07-2,00 (м, 1H), 1,91 (м, 1H), 1,75 (м, 2H), 1,66-1,58 (м, 2H), 1,40-1,32 (м, 1H).
Биологические анализы
Анализ адгезии клеток
Использованные реагенты и способы были такими, как описано в [Ludbrook et al, Biochem. J. 2003, 369, 311), со следующими уточнениями. Использовали следующие линии клеток, с лигандами в скобках: K562-α5β1 (фибронектин), K562-αvβ3 (LAP-b1), K562-αvβ5 (витронектин), K562-αvβ6 (LAP-b1), K562-αvβ8 (LAP-b1). Двухвалентным катионом, используемым для облегчения адгезии, был 2 мМ MgCl2. Адгезию количественно определяли путем маркировки клеток флуоресцентным красителем BCECF-AM (Life Technologies), где суспензии клеток при 3×106 клеток/мл инкубировали с 0,33 мл/мл 30 мМ BCECF-AM при 37°C в течение 10 минут, перед распределением в планшет для анализа. По завершении количественного анализа прилипших клеток, их лизировали с использованием 50 мкл/лунка 0,5% Тритона X-100 в H2O для высвобождения флуоресценции. Интенсивность флуоресценции детектировали с помощью планшет-ридера Envision® (Perkin Elmer). Для активных антагонистов в количественном анализе данные соответствовали 4 параметрам логистического уравнения для определения значений IC50.
Средняя аффинность связывания (pIC50) соединения примера 1 в клеточных адгезионных анализах была для αvβ6 pIC50=8,5; αvβ3 pIC50=7,2; αvβ5 pIC50=8,1; αvβ8 pIC50=8,2.
Средняя аффинность связывания (pIC50) соединения примера 2 в клеточных адгезионных анализах была для αvβ6 pIC50=8,0; αvβ3 pIC50=5,8; αvβ5 pIC50=7,2; αvβ8 pIC50=7,9.
Средняя аффинность связывания (pIC50) соединения примера 3 в клеточных адгезионных анализах была для αvβ6 pIC50=8,7; αvβ3 pIC50=6,3; αvβ5 pIC50=7,3; αvβ8 pIC50=8,2; αvβ1 pIC50=7,6.
Средняя аффинность связывания (pIC50) соединения примера 4 в клеточных адгезионных анализах была для αvβ6 pIC50=8,5; αvβ3 pIC50=5,9; αvβ5 pIC50=6,8; αvβ8 pIC50=7,9; αvβ1 pIC50=7,3.
Средняя аффинность связывания (pIC50) соединения примера 5 в клеточных адгезионных анализах была для αvβ6 pIC50=8,3; αvβ3 pIC50=6,4; αvβ5 pIC50=7,5; αvβ8 pIC50=8,0; αvβ1 pIC50=7,1.
Средняя аффинность связывания (pIC50) соединения примера 6 в клеточных адгезионных анализах была для αvβ6 pIC50=7,9; αvβ3 pIC50=5,4; αvβ5 pIC50=6,7; αvβ8 pIC50=7,3.
Средняя аффинность связывания (pIC50) соединения примера 7 в клеточных адгезионных анализах была для αvβ6 pIC50=8,2; αvβ3 pIC50=6,2; αvβ5 pIC50=7,3; αvβ8 pIC50=7,6.
Средняя аффинность связывания (pIC50) соединения примера 8 в клеточных адгезионных анализах была для αvβ6 pIC50=8,5; αvβ3 pIC50=6,6; αvβ5 pIC50=7,5; αvβ8 pIC50=8,2; αvβ1 pIC50=7,3.
Средняя аффинность связывания (pIC50) соединения примера 9 в клеточных адгезионных анализах была для αvβ6 pIC50=8,2; αvβ3 pIC50=5,9; αvβ5 pIC50=7,3; αvβ8 pIC50=7,6.
Средняя аффинность связывания (pIC50) соединения примера 10 в клеточных адгезионных анализах была для αvβ6 pIC50=8,6; αvβ3 pIC50=6,2; αvβ5 pIC50=6,8; αvβ8 pIC50=8,0
Средняя аффинность связывания (pIC50) соединения примера 11 в клеточных адгезионных анализах была для αvβ6 pIC50=8,5; αvβ3 pIC50=5,9; αvβ5 pIC50=6,8; αvβ8 pIC50=7,8; αvβ1 pIC50=7,2.
Средняя аффинность связывания (pIC50) соединения примера 12 в клеточных адгезионных анализах была для αvβ6 pIC50=8,6; αvβ3 pIC50=5,6; αvβ5 pIC50=6,7; αvβ8 pIC50=8,0.
Средняя аффинность связывания (pIC50) соединения примера 13 в клеточных адгезионных анализах была для αvβ6 pIC50=8,6; αvβ3 pIC50=6,5; αvβ5 pIC50=7,0; αvβ8 pIC50=8,0; αvβ1 pIC50=7,0.
Средняя аффинность связывания (pIC50) соединения примера 14 в клеточных адгезионных анализах была для αvβ6 pIC50=8,9; αvβ3 pIC50=6,6; αvβ5 pIC50=7,5; αvβ8 pIC50=8,2; αvβ1 pIC50=7,0.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
Изобретение относится к органической химии, а именно к соединению формулы (I) или его фармацевтически приемлемой соли, где R1 представляет собой пятичленный ароматический гетероцикл, выбранный из N- или C-связанного моно- или дизамещенного пиразола, N- или C-связанного, необязательно моно- или дизамещенного триазола или N- или C-связанного, необязательно дизамещенного имидазола, причем пятичленный ароматический гетероцикл может быть замещен одной или двумя группами, выбранными из атома водорода, метильной группы, этильной группы, атома фтора, гидроксиметильной группы, 2-гидроксипропан-2-ильной группы, трифторметильной группы, дифторметильной группы или фторметильной группы, за исключением случаев, когда R1 представляет собой N-связанный моно- или дизамещенный пиразол, R1 не является 3,5-диметил-1H-пиразол-1-илом, 5-метил-1H-пиразол-1-илом, 5-этил-3-метил-1H-пиразол-1-илом, 3,5-диэтил-1H-пиразол-1-илом, 4-фтор-3,5-диметил-1H-пиразол-1-илом, 3-метил-1H-пиразол-1-илом или 1H-пиразол-1-илом. Также описываются способы профилактики или лечения указанных заболеваний, фармацевтическая композиция на основе соединения формулы (I) и его применение. Технический результат: получены новые гетероциклические соединения, полезные в качестве антагонистов αvβ6. 9 н. и 9 з.п. ф-лы, 5 табл., 15 пр.

1. Соединение формулы (I) или его фармацевтически приемлемая соль:
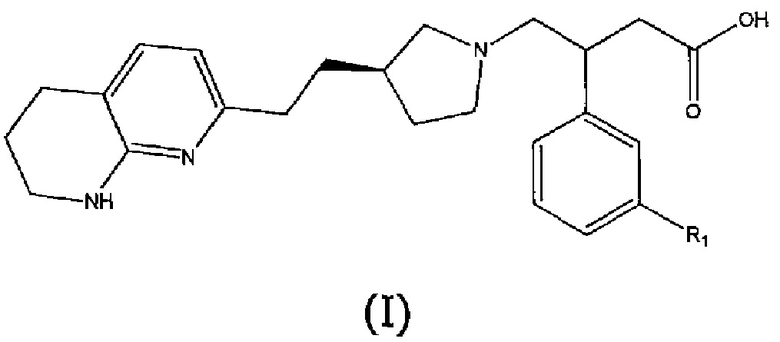
где R1 представляет собой пятичленный ароматический гетероцикл, выбранный из N- или C-связанного моно- или дизамещенного пиразола, N- или C-связанного, необязательно моно- или дизамещенного триазола или N- или C-связанного, необязательно дизамещенного имидазола, причем пятичленный ароматический гетероцикл может быть замещен одной или двумя группами, выбранными из атома водорода, метильной группы, этильной группы, атома фтора, гидроксиметильной группы, 2-гидроксипропан-2-ильной группы, трифторметильной группы, дифторметильной группы или фторметильной группы, за исключением случаев, когда R1 представляет собой N-связанный моно- или дизамещенный пиразол, R1 не является 3,5-диметил-1H-пиразол-1-илом, 5-метил-1H-пиразол-1-илом, 5-этил-3-метил-1H-пиразол-1-илом, 3,5-диэтил-1H-пиразол-1-илом, 4-фтор-3,5-диметил-1H-пиразол-1-илом, 3-метил-1H-пиразол-1-илом или 1H-пиразол-1-илом.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 представляет собой группу, выбранную из:


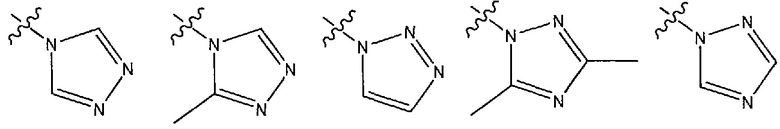

3. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 представляет собой C-связанный моно- или дизамещенный пиразол, выбранный из 3-метил-1H-пиразол-5-ила и 1,4-диметил-1H-пиразол-5-ила.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 представляет собой N-связанный моно- или дизамещенный пиразол, выбранный из -(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ила, 3-(трифторметил)-1H-пиразол-1-ила, 3-(гидроксиметил)-5-метил-1H-пиразол-1-ила и 3-(фторметил)-5-метил-1H-пиразол-1-ила.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 представляет собой N- или C-связанный, необязательно моно- или дизамещенный триазол, выбранный из 4H-1,2,4-триазол-4-ила, 3,5-диметил-1H-1,2,4-триазол-1-ила, 3-метил-4H-1,2,4-триазол-4-ила, 1H-1,2,3-триазол-1-ила и 1H-1,2,4-триазол-1-ила.
6. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 представляет собой N- или C-связанный, необязательно дизамещенный имидазол, выбранный из 1H-имидазол-1-ила и моно- или диметилимидазола, 1-метил-1H-имидазол-2-ила, 4-метил-1H-имидазол-2-ила, (1,4-диметил-1H-имидазол-2-ила), предпочтительно 1H-имидазол-1-ила, и (1,4-диметил-1H-имидазол-2-ила) и (2,4-диметил-1H-имидазол-5-ила).
7. Соединение по любому из пп. 1-6 находится в форме фармацевтически приемлемой соли.
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль для лечения или профилактики заболеваний, для которых рекомендован антагонист αvβ6 интегрина.
9. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль для применения при лечении заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина.
10. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль для применения при лечении идиопатического легочного фиброза.
11. Способ лечения заболеваний, при которых антагонизм рецептора αvβ6 благотворно влияет на человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
12. Способ профилактики заболеваний, при которых антагонизм рецептора αvβ6 благотворно влияет на человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
13. Способ лечения фиброзного заболевания у человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7, обладающего антагонистической активностью в отношении αvβ6 интегрина, или его фармацевтически приемлемой соли.
14. Способ профилактики фиброзного заболевания у человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7, обладающего антагонистической активностью в отношении αvβ6 интегрина, или его фармацевтически приемлемой соли.
15. Способ лечения идиопатического легочного фиброза у человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7, обладающего антагонистической активностью в отношении αvβ6 интегрина, или его фармацевтически приемлемой соли.
16. Способ профилактики идиопатического легочного фиброза у человека, включающий введение человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-7, обладающего антагонистической активностью в отношении αvβ6 интегрина, или его фармацевтически приемлемой соли.
17. Фармацевтическая композиция, обладающая антагонистической активностью в отношении αvβ6 интегрина, содержащая эффективное количество соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
18. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для лечения заболевания или состояния, для которого рекомендован антагонист αvβ6 интегрина.
| WO 2005082889 A1, 09.09.2005 | |||
| WO 2001024797 A1, 12.04.2001 | |||
| ПРОИЗВОДНЫЕ НАФТИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2002 |
|
RU2296764C2 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЕВЫХ КАНАЛОВ | 2008 |
|
RU2481347C2 |

