Область техники, к которой относится изобретение
Настоящее изобретение относится к новым кристаллическим формам ингибитора дипептидилпептидазы-IV. Более конкретно, настоящее изобретение относится к новым кристаллическим формам (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина, который представляет собой сильнодействующий, имеющий долговременное действие ингибитор дипептидилпептидазы-IV. Эти новые кристаллические формы являются пригодными при лечении и предотвращении заболеваний и состояний, для которых показан ингибитор дипептидилпептидазы-IV, в частности, диабета 2 типа, тучности и высокого давления крови. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим новые кристаллические формы по настоящему изобретению, пригодным для использования при лечении диабета 2 типа, тучности и высокого давления крови, а также к способам получения таких форм и их фармацевтических композиций.
Уровень техники
Ингибирование дипептидилпептидазы-IV (DP-IV), фермента, который дезактивирует как глюкозазависимый инсулинотропный пептид (GIP), так и глюкагоноподобный пептид 1 (GLP-1), представляет собой новый подход к лечению и предотвращению диабета 2 типа, также известного как не-инсулинзависимый сахарный диабет (NIDDM). Терапевтический потенциал ингибиторов DP-IV для лечения диабета 2 типа рассмотрен в C.F. Deacon and J.J. Hoist, "Dipeptidyl paptidase IV inhibition as an approach to treatment and prevention of Type 2 diabetes: historical perspective," Biochem. Biophys. Res. Commun., 294: 1-4 (2000); K. Augustyns, et al., "Dipeptidyl paptidase IV inhibitors as new therapeutic agents for the treatment of Type 2 diabetes," Expert. Opin. Ther. Patents. 13: 499-510 (2003); D.J. Drucker, "Therapeutic potential dipeptidyl paptidase IV inhibitors for the treatment of Type 2 diabetes," Expert Opin. Investig. Drugs. 12: 87-100 (2003); и M.A. Nauck et al., "Incretins and Their Analogues as New Antidiabetic Drugs," Drug News Perspect. 16: 413-422 (2003).
WO 2010/056708 (опубликованная 20 мая 2010), Merck & Co., описывает класс аминотетрагидропиранов, которые представляют собой сильнодействующие ингибиторы DP-IV и по этой причине пригодны для лечения диабета 2 типа. Конкретно в WO 2010/056708 описывается (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин.
Однако авторы обнаружили теперь новые кристаллические формы (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина (Соединение I).
Сущность изобретения
Настоящее изобретение относится к новым кристаллическим формам ингибитора дипептидилпептидазы-IV (DP-IV) (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина (Соединение I). Определенные кристаллические формы имеют преимущества при получении фармацевтических композиций (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина, таких как простота обработки и кристаллизации, манипуляций, стабильность в стрессовых условиях и дозировании. В частности, они демонстрируют улучшенные физико-химические свойства, такие как стабильность в стрессовых условиях, которые делают их особенно пригодными для использования при изготовлении разнообразных фармацевтических дозированных форм. Настоящее изобретение также относится к фармацевтическим композициям, содержащим их новые формы, а также к способам их использования в качестве ингибиторов DP-IV, в частности, для предотвращения или лечения диабета 2 типа, тучности и высокого давления крови.
В определенных вариантах осуществления, в настоящем документе описаны фармацевтические композиции, содержащие кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин и фармацевтически приемлемый носитель.
Краткое описание фигур
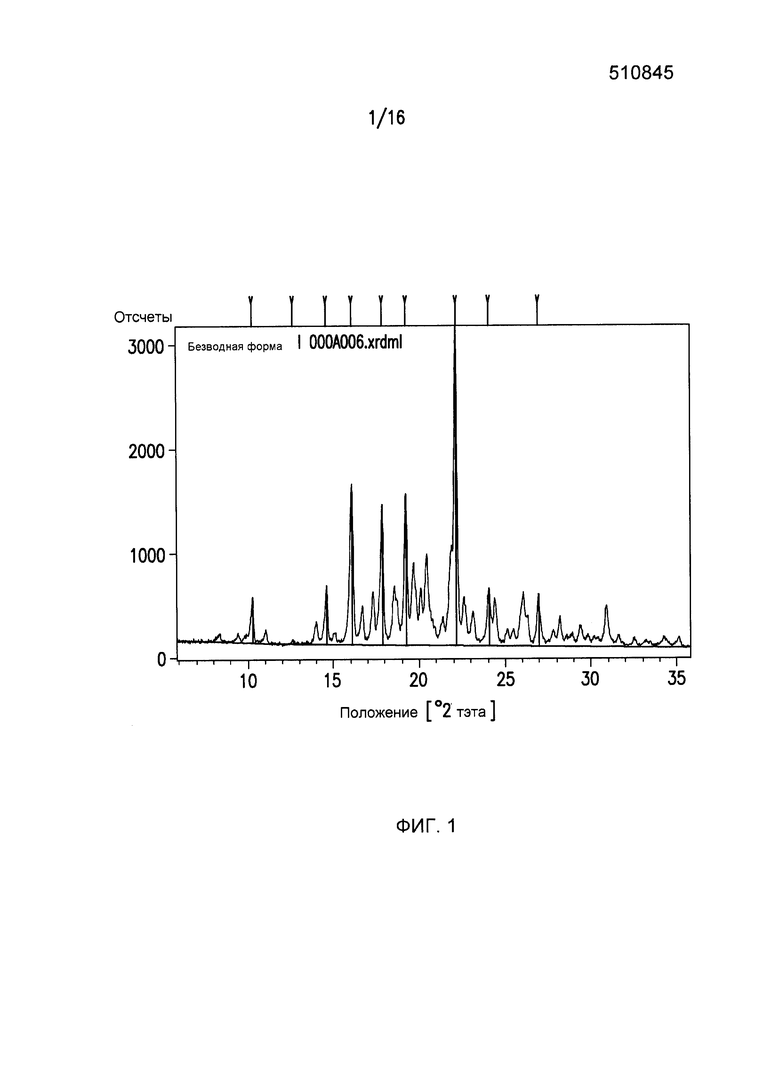
Фиг.1 представляет собой картину дифракции рентгеновского излучения для кристаллической формы I Соединения I.
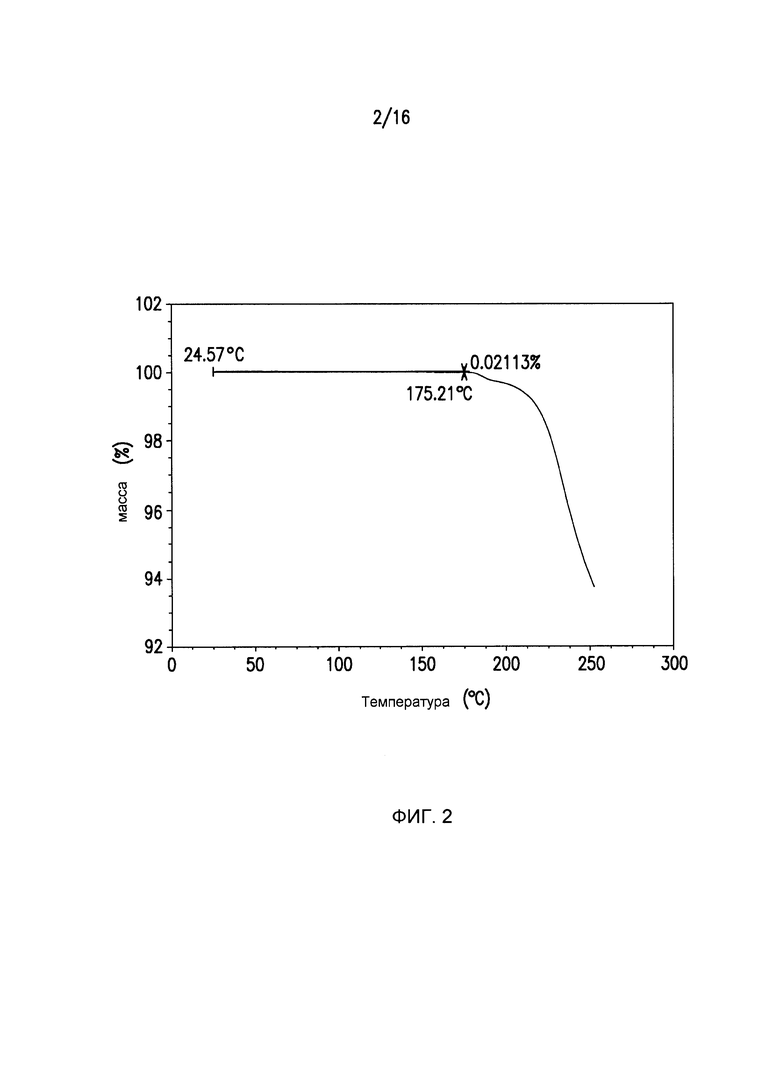
Фиг.2 представляет собой кривую термогравиметрического анализа (TGA) для кристаллической формы I Соединения I.
Фиг.3 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для кристаллической формы I Соединения I.
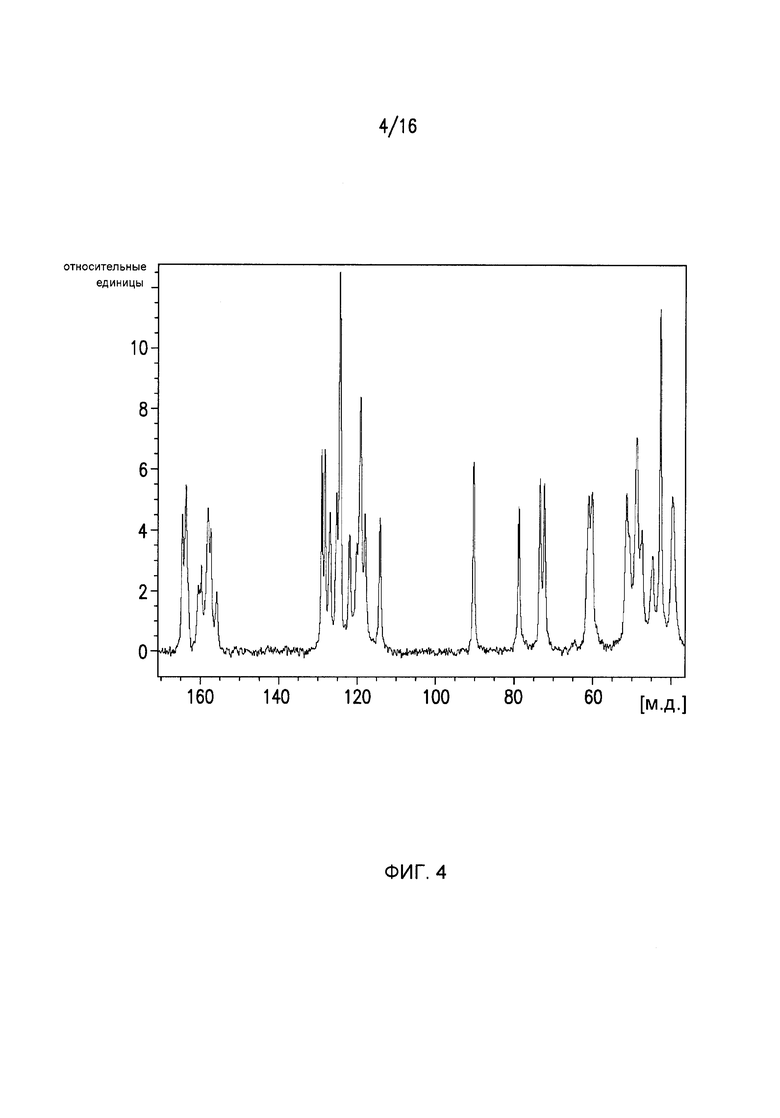
Фиг.4 представляет собой спектр твердотельного ЯМР для кристаллической формы I Соединения I.
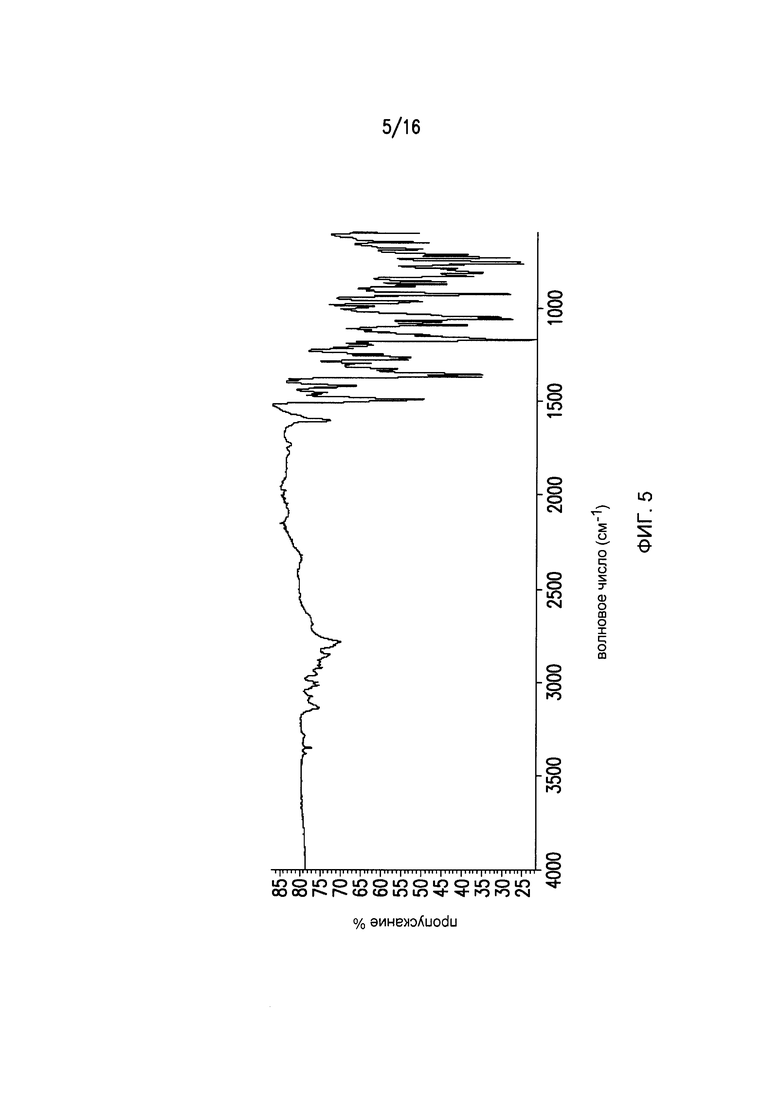
Фиг.5 представляет собой ИК-спектры для кристаллической формы II Соединения I.
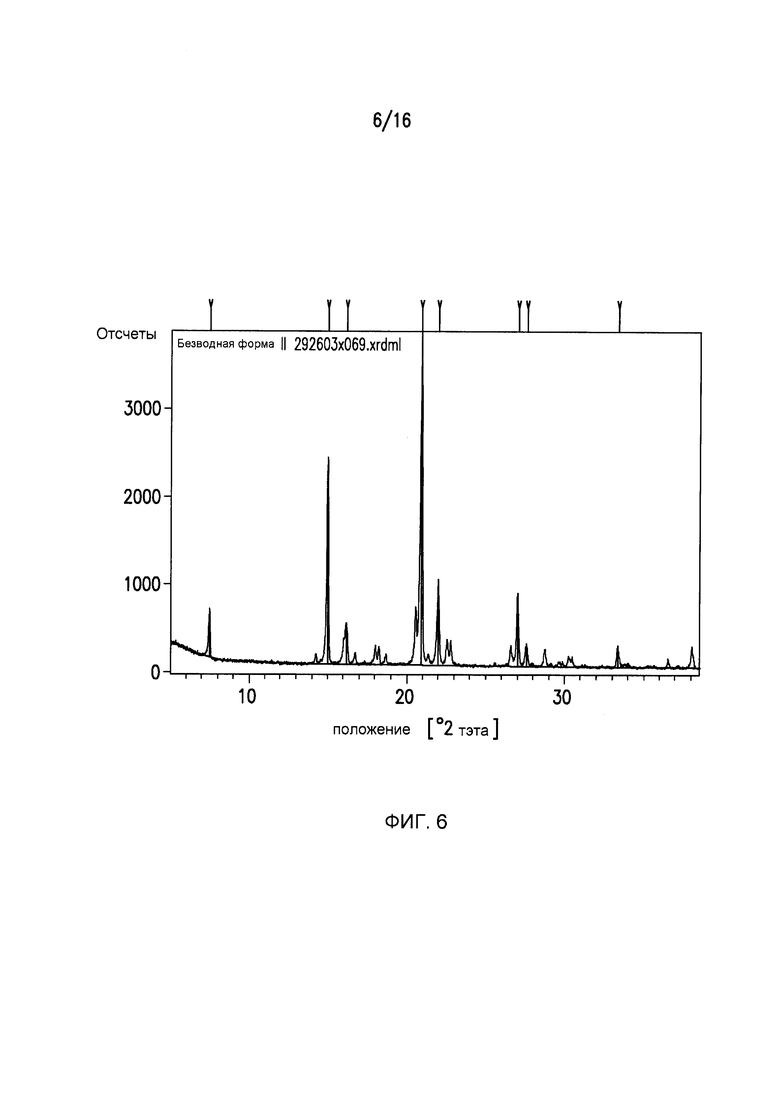
Фиг.6 представляет собой картину дифракции рентгеновского излучения для кристаллической формы II Соединения I.
Фиг.7 представляет собой кривую термогравиметрического анализа (TGA) для кристаллической формы II Соединения I.
Фиг.8 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для кристаллической формы II Соединения I.
Фиг.9 представляет собой спектр твердотельного ЯМР для кристаллической формы II Соединения I.
Фиг.10 представляет собой ИК спектр кристаллической формы II Соединения I.
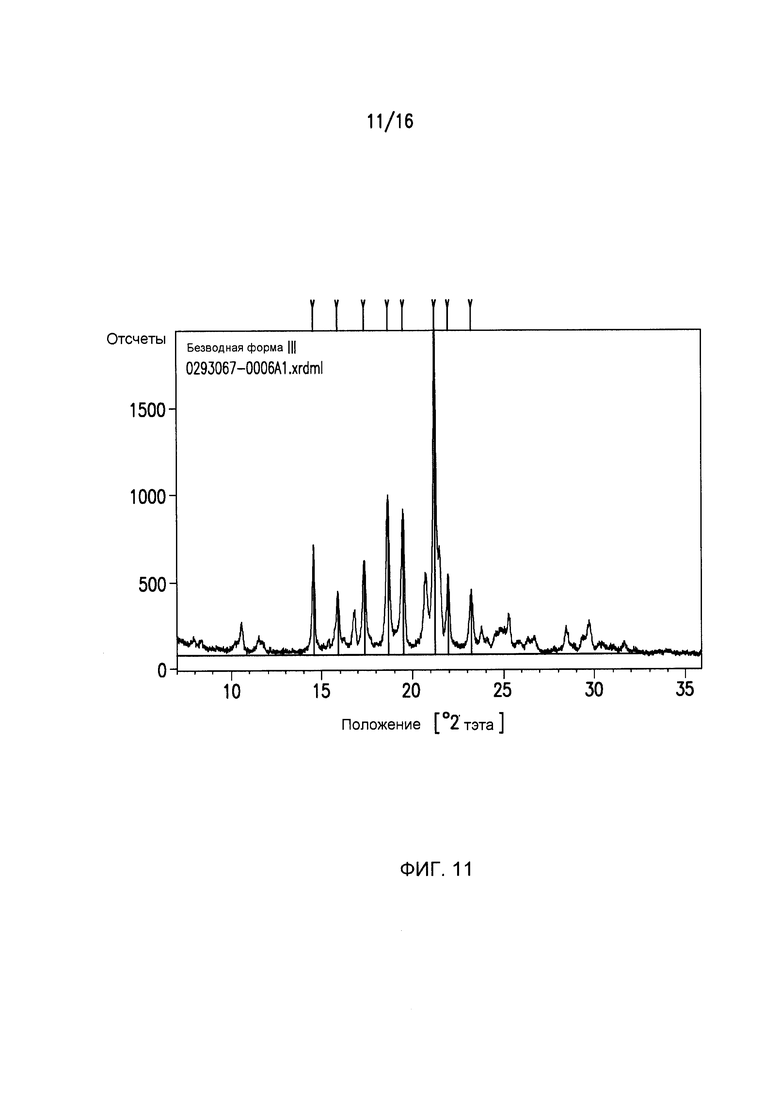
Фиг.11 представляет собой картину дифракции рентгеновского излучения для кристаллической формы ΙII Соединения I.
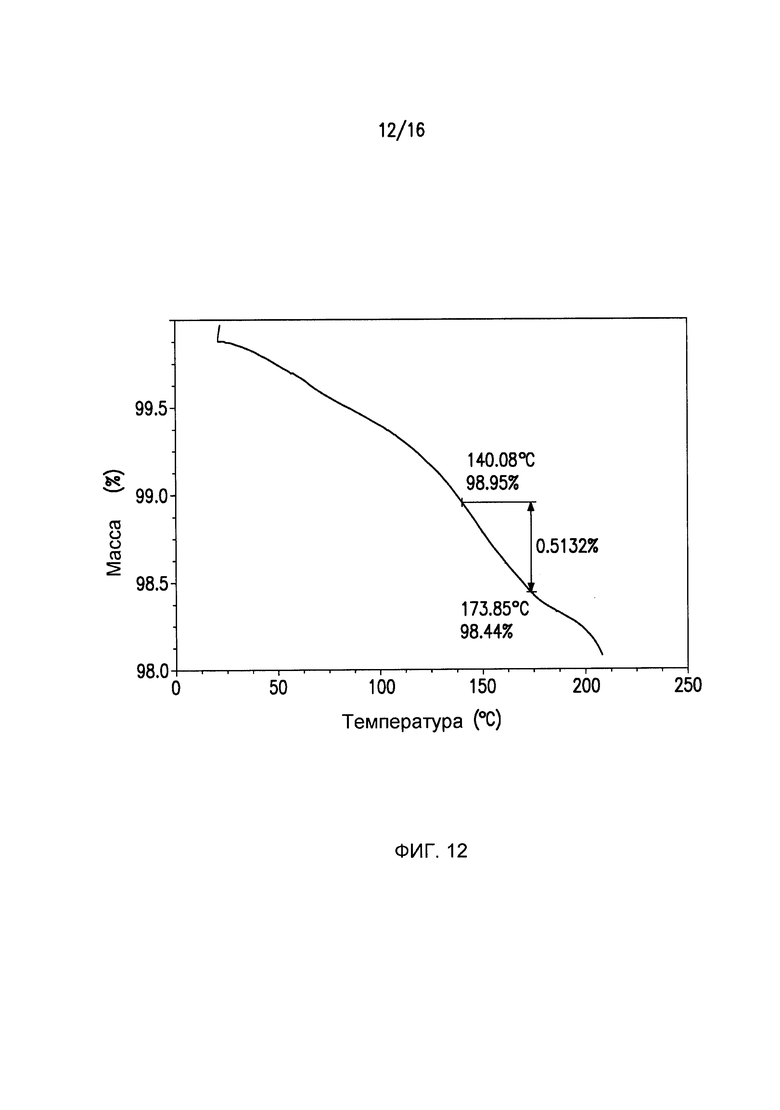
Фиг.12 представляет собой кривую термогравиметрического анализа (TGA) для кристаллической формы III Соединения I.
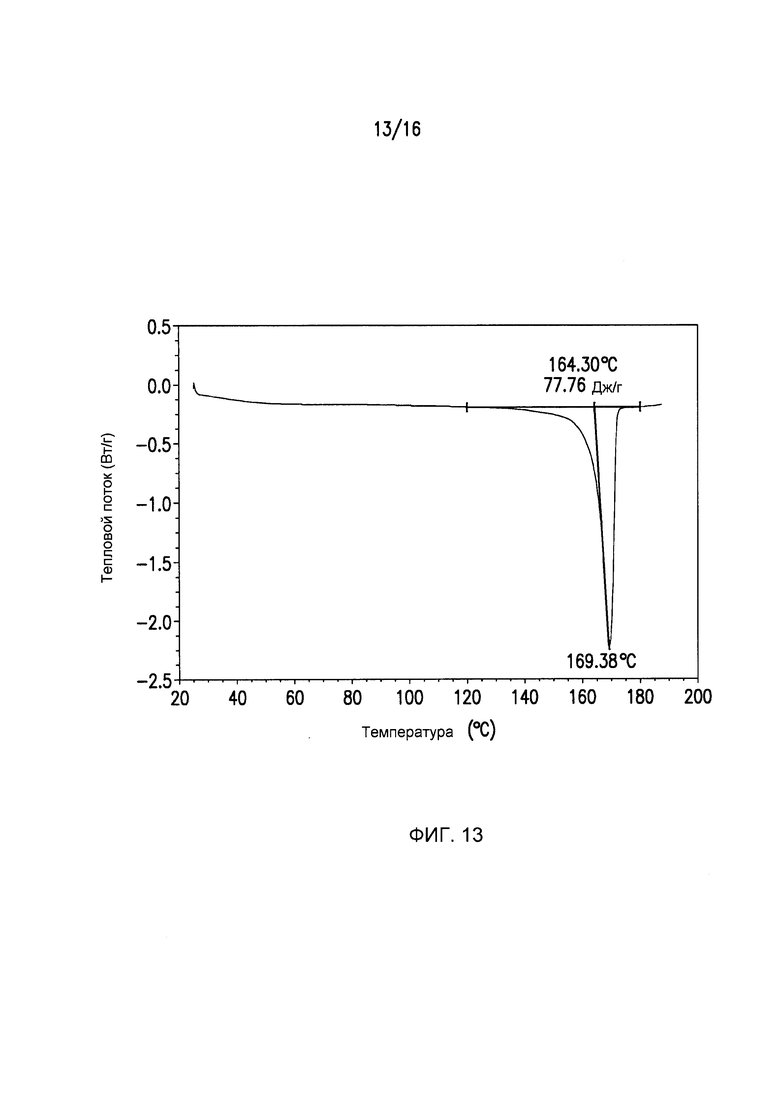
Фиг.13 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для кристаллической формы III Соединения I.
Фиг.14 представляет собой картину дифракции рентгеновского излучения для кристаллической формы IV Соединения I.
Фиг.15 представляет собой кривую термогравиметрического (TGA) для кристаллической формы IV Соединения I.
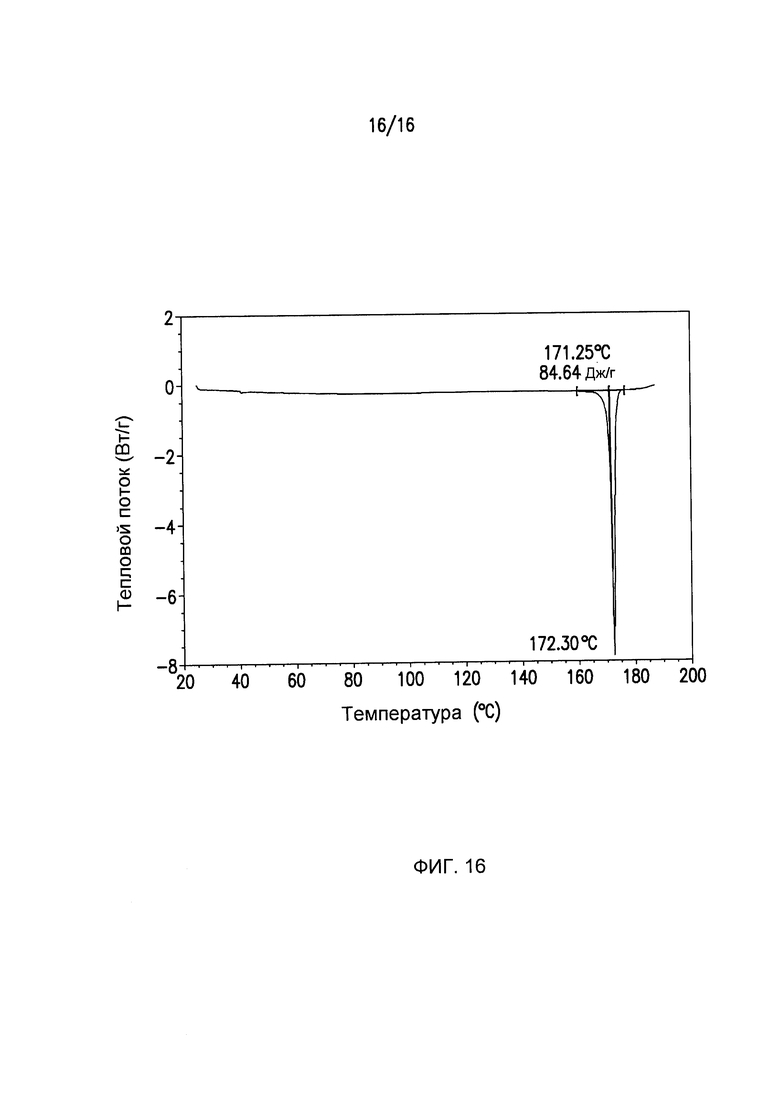
Фиг.16 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для кристаллической формы IV Соединения
Подробное описание изобретения
Настоящее изобретение относится к кристаллическому (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амину Соединения I:

Если не приводится конкретного обозначения формы, термин "кристаллический (2R,35',5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин" относится ко всем кристаллическим формам (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина, описанного в настоящем документе. Кристаллические формы, описанные в настоящем документе, существуют в виде безводного свободного основания (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина.
Один из вариантов осуществления кристаллических форм, описанных в настоящем документе, представляет собой (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма I). Форма I дополнительно описывается ниже.
Другой вариант осуществления кристаллических форм, описанных в настоящем документе, представляет собой (2R,3S,5R)-2-(2,5- дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма II). Форма II дополнительно описывается ниже.
Еще один вариант осуществления кристаллических форм, описанный в настоящем документе, представляет собой (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма III). Форма III дополнительно описывается ниже.
Еще один вариант осуществления кристаллических форм, описанный в настоящем документе, представляет собой (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма IV). Форма IV дополнительно описывается ниже.
Другой вариант осуществления настоящего изобретения предлагает конкретное лекарственное вещество, которое содержит, по меньшей мере, одну из кристаллических форм, описанных в настоящем документе. Под "лекарственным веществом" подразумевается активный фармацевтический ингредиент. Количество кристаллической формы в лекарственном веществе может количественно определяться посредством использования физических методов, таких как дифракция рентгеновского излучения на порошках, спектроскопия твердотельного ядерного магнитного резонанса фтора-19 с вращением образца под магическим углом (MAS), спектроскопия твердотельного ядерного магнитного резонанса углерода-13 с вращением образца под магическим углом и кросс-поляризацией (CPMAS), твердотельная инфракрасная спектроскопия с Фурье-преобразованием и Рамановская спектроскопия.
В некотором классе этого варианта осуществления, кристаллическая форма по настоящему изобретению составляет примерно от 5% примерно до 100% масс. от лекарственного вещества. Во втором классе этого варианта осуществления, кристаллическая форма по настоящему изобретению составляет примерно от 10% примерно до 100% масс. лекарственного вещества. В третьем классе этого варианта осуществления, кристаллическая форма по настоящему изобретению составляет примерно от 25% примерно до 100% масс. лекарственного вещества. В четвертом классе этого варианта осуществления, кристаллическая форма по настоящему изобретению составляет примерно от 50% примерно до 100% масс. лекарственного вещества. В пятом классе этого варианта осуществления, кристаллическая форма по настоящему изобретению составляет примерно от 75% примерно до 100% масс. лекарственного вещества. В шестом классе этого варианта осуществления, по существу все лекарственное вещество находится в кристаллической форме по настоящему изобретению, то есть, лекарственное вещество представляет собой по существу фазово-чистый кристалл.
В другом классе этого варианта осуществления, по меньшей мере, 5% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 10% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 15% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 20% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 25% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 30% масс лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 35% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 40% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 45% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 50% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 55% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 60% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 65% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 70% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 75% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 80% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 85% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 90% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. В другом классе этого варианта осуществления, по меньшей мере, 95% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению. Еще в одном классе этого варианта осуществления, по меньшей мере, 100% масс. лекарственного вещества представляет собой кристаллическую форму по настоящему изобретению.
Кристаллические формы по настоящему изобретению демонстрируют фармацевтические преимущества по сравнению с аморфным свободным основанием Соединения I, как описано в WO 2010/056708, при получении продукта фармацевтического лекарственного препарата, содержащего фармакологически активный ингредиент. В частности, улучшенная химическая и физическая стабильность кристаллических форм составляют преимущественные свойства при получении твердых фармацевтических дозированных форм, содержащих фармакологически активный ингредиент.
Кристаллические формы по настоящему изобретению, которые демонстрируют имеющие продолжительное действие, сильнодействующие свойства ингибирования DP-IV, являются особенно пригодными для использования при предотвращении или лечении диабета 2 типа, тучности и высокого давления крови.
Другой аспект настоящего изобретения предлагает способ предотвращения или лечения клинических состояний, для которых показан ингибитор DP-IV, этот способ включает введение пациенту, нуждающемуся в таком предотвращении или лечении, профилактически или терапевтически эффективного количества кристаллической формы по настоящему изобретению или его гидрата. Такие клинические состояние включают диабет, в частности, диабет 2 типа, гипергликемию, стойкость к инсулину и тучность.
Настоящее изобретение также предлагает использование кристаллической формы Соединения I по настоящему изобретению для предотвращения или лечения у млекопитающих клинических состояний, для которых показан ингибитор DP-IV, в частности, диабета 2 типа, гипергликемии, стойкости к инсулину и тучности.
Настоящее изобретение также предлагает использование кристаллической формы Соединения I по настоящему изобретению для получения лекарственного препарата для предотвращения или лечения у млекопитающих клинических состояний, для которых показан ингибитор DP-IV, в частности, диабета 2 типа, гипергликемии, стойкости к инсулину и тучности.
Настоящее изобретение также предлагает фармацевтические композиции, содержащие кристаллическую форму, описанную в настоящем документе, в ассоциации с одним или несколькими фармацевтически приемлемыми носителями или наполнителями. В одном из вариантов осуществления фармацевтическая композиция содержит терапевтически эффективное количество активного фармацевтического ингредиента в смеси с фармацевтически приемлемыми наполнителями, где активный фармацевтический ингредиент содержит детектируемое количество кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина.
Во втором варианте осуществления фармацевтическая композиция содержит терапевтически эффективное количество активного фармацевтического ингредиента в смеси с фармацевтически приемлемыми наполнителями, где активный фармацевтический ингредиент составляет примерно от 1% примерно до 100% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В одном из классов этого второго варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет примерно от 5% примерно до 100% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)- ил]тетрагидро-2H-пиран-3-амина. Во втором классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет примерно от 10% примерно до 100% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В третьем классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет примерно 25% примерно до 100% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В четвертом классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет примерно от 50% примерно до 100% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина.
В третьем варианте осуществления фармацевтическая композиция содержит терапевтически эффективное количество активного фармацевтического ингредиента в смеси с фармацевтически приемлемыми наполнителями, где активный фармацевтический ингредиент составляет, по меньшей мере, 1% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В одном из классов этого второго варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет примерно 5% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. Во втором классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет, по меньшей мере, 10% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В третьем классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет, по меньшей мере, 25% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина. В четвертом классе этого варианта осуществления, активный фармацевтический ингредиент в таких композициях составляет, по меньшей мере, 50% масс. кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина.
Композиции в соответствии с настоящим изобретением являются удобными в стандартных дозированных формах, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные растворы или суспензии, отмеряемые аэрозольные или жидкие спреи, капли, ампулы, аутоинжекторные устройства или суппозитории. Композиции предназначены для перорального, парентерального, интраназального, сублингвального или ректального введения, или для введения посредством ингаляции или инсуфляции. Приготовление композиций в соответствии с настоящим изобретением может быть удобным осуществлять с помощью способов, известных в данной области, например, как описано в Remington's Pharmaceutical Sciences. 17th ed., 1995.
Режим дозирования выбирают в соответствии с разнообразными факторами, включая тип, вид, возраст, массу тела, пол и медицинское состояние пациента; тяжесть состояния, которое должно лечиться; способ введения и функционирование почек и печени пациента. Обычный врач, ветеринар или клиницист может легко определить и прописать эффективное количество лекарственного средства, необходимое для предотвращения, противодействия или приостановки развития состояния.
Пероральные дозировки по настоящему изобретению, когда их используют для указанных воздействий, будут находиться в пределах примерно от 0,01 мг на кг массы тела в день (мг/кг/день) примерно до 100 мг кг/день, предпочтительно, от 0,01 до 10 мг/кг/день, а наиболее предпочтительно, от 0,1 до 5,0 мг кг/день. Для перорального введения, композиции предпочтительно предусматриваются в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100 и 500 миллиграмм активного ингредиента, для симптоматического установления дозы для пациента, который должен лечиться. Лекарственный препарат, как правило, содержит примерно от 0,01 мг примерно до 500 мг активного ингредиента, предпочтительно, примерно от 1 мг примерно до 200 мг активного ингредиента. При внутривенном введении, наиболее предпочтительные дозы будут находиться в пределах примерно от 0,1 примерно до 10 мг/кг/минут при постоянной скорости вливания. Кристаллические формы по настоящему изобретению могут вводиться в виде одной ежедневной дозы, или общая ежедневная доза может вводиться в разделенных дозах - два, три или четыре раза в день. Однако, (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин представляет собой ингибитор DPP-IV, имеющий продолжительное действие. Преимущественно, кристаллические формы по настоящему изобретению могут вводиться в виде одной еженедельной дозы.
Кроме того, кристаллические формы по настоящему изобретению могут вводиться в интраназальной форме посредством местного применения соответствующих интраназальных носителей, или посредством трансдермальных способов, с использованием таких форм трансдермальных кожных пластырей, которые хорошо известны специалистам в данной области. При введении в форме трансдермальной системы доставки, введение дозы будет, разумеется, непрерывным, а не периодическим в течение всего режима дозирования.
В способах по настоящему изобретению, кристаллические формы, описанные в настоящем документе, могут образовывать активный фармацевтический ингредиент, и, как правило, вводятся в смеси с соответствующими фармацевтическими разбавителями, наполнителями или носителями (совместно упоминаемыми в настоящем документе как материалы 'носители'), соответствующим образом выбираемыми в соответствии с предполагаемой формой введения, то есть, как пероральные таблетки, капсулы, эликсиры, сиропы, и тому подобное, и в соответствии с обычной фармацевтической практикой.
Например, для перорального введения в форме таблетки или капсулы, компонент активного лекарственного средства может объединяться с пероральным нетоксичным, фармацевтически приемлемым, инертным носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния, дикальций фосфат, сульфат кальция, маннитол, сорбитол, и тому подобное; для перорального введения в жидкой форме, компонент перорального лекарственного средства может объединяться с любым пероральным нетоксичным, фармацевтически приемлемым инертным носителем, таким как этанол, глицерол, вода, и тому подобное. Кроме того, когда это желательно или необходимо, соответствующие связующие вещества, смазывающие вещества, разрыхляющие агенты и окрашивающие агенты также могут включаться в смесь. Соответствующие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как смола акации, трагаканта или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски, и тому подобное. Смазывающие вещества, используемые в этих дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия, и тому подобное. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую смолу, и тому подобное.
Кристаллические формы Соединения I по настоящему изобретению, как обнаружено, обладают относительно высокой растворимостью в воде (примерно 2 мг/мл), что делает их особенно пригодными для приготовления препаратов, в особенности, интраназальных и внутривенных препаратов, которые требуют относительно концентрированных водных растворов активного фармацевтического ингредиента.
В другом аспекте, настоящее изобретение предлагает способ лечения и/или предотвращения клинических состояний, для которых показан ингибитор DP-IV, этот способ включает введение пациенту, нуждающемуся в таком предотвращении или лечении, профилактически или терапевтически эффективного количества кристаллической формы Соединения I, как определено выше, в сочетании с другим агентом, пригодным для лечения диабета 2 типа, тучности и высокого давления крови.
Соединения, описанные в настоящем документе, могут существовать как таутомеры, такие как кето-енольные таутомеры. Индивидуальные таутомеры, а также их смеси охватываются соединениями структурной формулы I.
Термин "% энантиомерный избыток" (сокращенно "ee") должен означать, что % главного энантиомера уменьшает % дополнительного энантиомера. Таким образом, 70% энантиомерный избыток соответствует образованию 85% одного энантиомера и 15% другого. Термин "энантиомерный избыток" является синонимом термина "оптическая чистота".
Соединение I может быть получено с помощью следующих способов:
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1
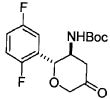
трет-Бутил [(2R,3S)-5-оксо-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
Стадия A: трет-Бутил (1-[метокси(метил)амино]-1-оксопент-4-ин-2-ил)карбамат
В емкость с инертным газом загружают сложный этиловый эфир N,N-дифенил глицина (105,45 кг, 394,5 моль), тетрабутиламмоний бромид (14 кг, 43,4 моль) и пропаргилбензолсульфонат (94,45 кг, 481 моль), а затем MTBE (750 кг). Затем добавляют карбонат цезия (мелкодисперсный сорт, 390 кг, 1197 моль), и реакционную смесь перемешивают при 50-60°C в течение 1 дня. Затем загрузку охлаждают до 0-5°C, и медленно добавляют воду (422 кг). Далее, добавляют простой трет-бутилметиловый эфир (170 кг), и загрузку концентрируют до 473-578 л. Затем добавляют 462 кг раствора HCl (43 кг конц. HCl в 420 кг воды) для установления pH 1-2 при температуре ниже комнатной температуры. После 7 часов перемешивания, pH составляет 1,5, и органический слой отделяют и сливают.
Затем водный слой охлаждают до 5-10°C и медленно добавляют 28% водный раствор NaOH (151 кг) до достижения pH 13. Затем добавляют раствор Boc2O (136 кг, 624 моль в 243 кг простого трет-бутилметилового эфира) при 5-10°C. Затем раствор перемешивают при комнатной температуре в течение 4 часов (pH 8), и медленно добавляют 17% водный раствор NaOH (126 кг), а затем дополнительный раствор Boc2O (30,7 кг, 141 моль в 60 кг простого трет-бутилметилового эфира). Затем раствор перемешивают при комнатной температуре в течение 4 часов (pH 9), и медленно добавляют 17% водный раствор NaOH (98 кг) (pH 13) и перемешивают в течение дополнительных 12 часов (pH 10), а затем добавляют дополнительный Boc2O (11 кг, 50 моль). После 4 часов перемешивания при комнатной температуре, слои разделяют (оставляют водный слой), и органические вещества экстрагируют 3% водным раствором NaOH (136 кг). Водные слои объединяют и добавляют к простому трет-бутилметиловому эфиру (338 кг). Затем добавляют 17% водный раствор HCl (362 кг) до тех пор, пока не достигнут pH 2. Слои разделяют, и водный слой экстрагируют простым трет-бутилметиловым эфиром (420 кг). Объединенные органические слои промывают 10% соляным раствором (139 кг), сушат с помощью Na2SO4, фильтруют и концентрируют до 105-158 л. Отгонка при постоянном объеме с помощью простого трет-бутилметилового эфира продолжается до достижения KF=0,4%.
К этому раствору добавляют карбонилдиимидазол (90 кг, 548 моль), и перемешивают в течение 2 часов при комнатной температуре. Затем добавляют (MeO)MeNH2Cl (48 кг, 492 моль), и реакционную смесь перемешивают в течение 6 часов. Затем загрузку охлаждают до 0-5°C, и добавляют воду (80 кг). Затем загрузку затравливают с помощью 100 г затравки, и добавляют воду (450 кг). Суспензию перемешивают при 0-5°C в течение 3 часов, а затем фильтруют. Осадок на фильтре сушат в вакууме при 45-60°C в течение 2 дней с получением трет-бутил-(1-[метокси(метил)амино]-1-оксопент-4-ин-2-ил)карбамата.
Стадия B: трет-Бутил [1-(2,5-дифторфенил)-1-оксопент-4-ин-2-ил]карбамат
В емкость с инертным газом загружают дихлорметан (866 кг), и охлаждают ее до -20 - -10°C. Затем медленно добавляют раствор изопропилмагния хлорида в ТГФ (2M, 326,1 кг, 669 моль), а затем 1-бром-2,5-дифторбензол (120,1 кг, 622 моль). После 2 часов при этой температуре, медленно добавляют дополнительную загрузку изопропилмагния хлорида в ТГФ растворе (2M, 58,65 кг, 121 моль), и реакционную смесь состаривают в течение 1 часа. Затем осуществляют добавление по каплям дихлорметанового раствора трет-бутил(1-[метокси(метил)амино]-1-оксопент-4-ин-2-ил)карбамата (70,8 кг, 276 моль в 292 кг дихлорметана) в течение 2 часов при температуре от -20 до -20°C. Затем смесь нагревают до комнатной температуры и перемешивают в течение 10 часов. Затем осуществляют медленное гашение обратной реакции в водном растворе хлорида аммония а(175,6 кг в 1550 кг воды) при 5-10°C. Затем pH раствора доводят до ~7 посредством добавления 68 кг конц. HCl. Затем слои разделяют, и водный слой экстрагируют дихлорметаном (414 кг). Затем объединенные органические слои сушат с помощью Na2SO4, фильтруют, обрабатывают активированным углем (10 кг), фильтруют и концентрируют до 71-141 л. Затем осуществляют замену растворителя вакуумной отгонки при постоянном объеме (71-141 л) на н-гептан для кристаллизации продукта. Затем суспензию охлаждают до 0°C и перемешивают 2 часа. Суспензию фильтруют, и осадок на фильтре промывают н-гептаном, 2-пропанолом, а затем водой. Твердые продукты сушат в вакууме при 40-50°C в течение ночи с получением трет-бутил[1-(2,5-дифторфенил)-1-оксопент-4-ин-2-ил]карбамата.
Стадия C: трет-Бутил[(1S,2S)-1-(2,5-дифторфенил)-1-гидроксипент-4-ин-2-ил]карбамат
В перемешиваемую емкость при продувке азотом загружают трет-бутил[1-(2,5-дифторфенил)-1-оксопент-4-ин-2-ил]карбамат (35,0 кг, 113 моль), 1,4-диазабицикло[2,2,2]октан (38,0 кг, 339 моль) и ТГФ (465 кг). После растворения, добавляют хлор{[(1R,2R)-(-)-2-амино-1,2-дифенилэтил](пентафторфенил-сульфонил)амидо}-(п-цимен)рутений (II) (410 г, 576 ммоль). Емкость продувают вакуумом и обратно заполняют азотом, три раза. Затем добавляют муравьиную кислоту (26,7 кг, 580 моль), и реакционную смесь нагревают до 45°C в течение ночи.
Затем смесь концентрируют в вакууме до 210-280 л, а затем добавляют простой трет-бутилметиловый эфир (210 кг). После охлаждения до 0-10°C, добавляют 0,4% водный раствор HCl (52 кг) до достижения pH 4-6. После перемешивания и разделения слоев, водный слой опять экстрагируют простым трет-бутилметиловым эфиром (87 кг). Затем объединенные органические слои промывают 4% водным раствором NaHCO3 (291 кг), а затем соляным раствором (216 кг). Полученные органические вещества сушат над Na2SO4, фильтруют через слой диоксида кремния и концентрируют до 70-105 л. Затем добавляют простой трет-бутилметиловый эфир (132 кг), с последующим дополнительным концентрированием загрузки до достижения KF (содержания воды по Карлу Фишеру)=0,1%. Далее, добавляют ДМФ (133 кг), и загрузку дополнительно концентрируют до 70-105 л. Полученный раствор в ДМФ составляет 165,6 кг, он содержит 19,4% трет-бутил[(1S,2S)-1-(2,5-дифторфенил)-1-гидроксипент-4-ин-2-ил]карбамата (диастереомерное отношение 8,1/1 и ee 97,9%).
Стадия D: трет-Бутил[(1S,2R)-1-(2,5-дифторфенил)-1-гидроксипент-4-ин-2-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия C.
Стадия E: трет-Бутил [(1R,2R)-1-(2,5-дифторфенил)-1-гидроксипент-4-ин-2-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия D.
Стадия F: трет-Бутил [(1R,2R)-1-(2,5-дифторфенил)-1- гидроксипент-4-ин-2-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия E.
Стадия G: трет-Бутил [(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамат
К 165,6 кг раствора трет-бутил[(1S,2S)-1-(2,5-дифторфенил)-1-гидроксипент-4-ин-2-ил]карбамата (19,4% масс./масс. в ДМФ, 103 моль) добавляют ДМФ (70 кг), 1-гидроксипирролидин-2,5-дион (5,95 кг, 51 моль), тетрабутиламмоний гексафторфосфат (5,20 кг, 13 моль) и NaHCO3 (4,50 кг, 54 моль). Полученную реакционную смесь продувают вакуумом с обратным заполнением азотом, три раза, а затем перемешивают в течение 30-40 мин. Затем добавляют хлор(циклопентадиенил)бис(трифенилфосфин)рутений (II) (823 г, 1,13 моль) и трифенилфосфин (892 г, 3,40 моль), и реакционную смесь продувают вакуумом с обратным заполнением азотом, три раза. Затем реакционную смесь нагревают до 75-85°C в течение ночи. Для завершения реакции, добавляют дополнительный хлор(циклопентадиенил)бис-(трифенилфосфин)рутений (II) (826 г, 1,14 моль) и трифенилфосфин (892 г, 3,40 моль), и реакционную смесь нагревают при 75-85°C в течение дополнительных 12-16 часов.
После охлаждения до комнатной температуры, добавляют воду (250 кг) и простой трет-бутилметиловый эфир (210 кг). После перемешивания, слои отделяют и полученный водный слой экстрагируют простым трет-бутилметиловым эфиром (2 x 150 кг). Объединенные органические слои промывают соляным раствором (4 x 220 кг). Затем органические вещества сушат с помощью Na2SO4, фильтруют и концентрируют. Сырой продукт пропускают через слой диоксида кремния с использованием простого трет-бутилметилового эфира и н-гептана. Затем в полученном растворе осуществляют замену растворителя с помощью вакуумной отгонки и введения н-гептана в суспензию, 64-128 л в н-гептане. Эту суспензию нагревают для растворения при 90-110°C. Затем ее охлаждают в течение 2-3 часов до 0-10°C. Затем суспензию фильтруют, и полученный влажный осадок на фильтре сушат при 40-50°C и в вакууме с получением трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамата.
Стадия H: трет-Бутил [(2R,3R)-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия G.
Стадия I: трет-Бутил [(2S,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия H.
Стадия J: трет-Бутил [(2S,3R)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия I.
Стадия K: трет-Бутил [(2S,3R)-2-(2,5-дифторфенил)-5-гидрокситетрагидро-2H-пиран-3-ил]карбамат
К 64,0 кг (206 моль) трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-ил]карбамата в перемешиваемой емкости добавляют простой трет-бутилметиловый эфир (500 кг). После растворения, раствор охлаждают до 0-5°C и добавляют 10M раствор комплекса боран-диметилсульфид (39 кг, 515 моль). После 1-3 часов перемешивания при этой температуре, медленно добавляют воду (35 кг), и раствор перемешивают в течение 2 часов при 0-10°C. Затем добавляют 3% водный раствор NaHCO3 (900 кг) и 1% водный раствор NaOH (582 кг). Далее, добавляют порциями NaBO3∙4H2O (115,6 кг, 751 моль) в течение 1 часа при 0-10°C. После перемешивания реакционной смеси в течение ночи при комнатной температуре, добавляют порциями дополнительный NaBO3∙4H2O (25,7 кг, 167 моль) в течение 1 часа при 0-10°C. Затем реакционную смесь перемешивают в течение дополнительных 6 часов при комнатной температуре.
Затем реакционную смесь экстрагируют этилацетатом (230 кг), и полученные органические вещества промывают 3% водным раствором NaHCO3 (500 кг), а затем соляным раствором (376 кг). Объединенные водные слои дополнительно экстрагируют этилацетатом (2×325 кг). Затем органические вещества обрабатывают активированным углем (14,4 кг) в течение 2 часов при 50-60°C. После фильтрования, органические вещества концентрируют и осуществляют замену растворителя на н-гептан для образования суспензии кристаллов. Затем эту суспензию фильтруют, и осадок на фильтре промывают н-гептаном. Затем этот влажный осадок на фильтре растворяют в этилацетате (99 кг) при 50-60°C. Затем добавляют н-гептан (251 кг), и загрузку охлаждают до 0°C. Затем полученную суспензию фильтруют, и осадок на фильтре промывают н-гептаном. Затем твердые продукты сушат при 40-50°C в вакууме с получением трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-5-гидрокситетрагидро-2H-пиран-3-ил]карбамата.
Стадия L: трет-Бутил [(2R,3R)-2-(2,5-дифторфенил)-5-гидрокситетрагидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия K.
Стадия M: трет-Бутил [(2S,3R)-2-(2,5-дифторфенил)-5- гидрокситетрагидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия L.
Стадия N: трет-Бутил [(2S,3S)-2-(2,5-дифторфенил)-5- гидрокситетрагидро-2H-пиран-3-ил]карбамат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия M.
Стадия O: трет-Бутил [(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2H-пиран-3-ил]карбамат
К 46,8 кг (142 моль) трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-5-гидрокситетрагидро-2H-пиран-3-ил]карбамата в перемешиваемой емкости добавляют ацетонитрил (150 кг), уксусную кислоту (50 кг) и воду (25 кг). После растворения при комнатной температуре, раствор охлаждают до 0°C, и в воду добавляют RuCl3∙3H2O (250 г, 956 ммоль) (50 кг) в атмосфере азота. Затем добавляют NaBrO3 (11,7 кг, 77,5 моль) в шести порциях, каждые 1,5 часа, в атмосфере азота. После перемешивания при 0°C в течение 6 часов, добавляют 2-пропанол (31 кг) в течение 30 мин при 0°C. Затем при этой температуре добавляют воду (720 кг) в течение 5 часов. Полученную суспензию перемешивают в течение ночи, фильтруют, и осадок на фильтре промывают водой. Затем твердые продукты сушат в вакууме при 40-60°C с получением трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2H-пиран-3-ил]карбамата.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2
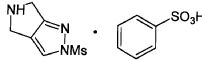
2-(метилсульфонил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-5-ий бензолсульфонат
Стадия A: трет-Бутил (3Z)-3-[(диметиламино)метилен]-4-оксопирролидин-1-карбоксилат
Раствор трет-бутил 3-оксопирролидин-1-карбоксилата (53,4 кг, 288 моль) в ТГФ (133 кг) обрабатывают ДМФ-DMA (103 кг, 864 моль) в ТГФ (472 кг) и нагревают при 65-70°C в атмосфере азота в течение 20 часов. Раствор охлаждают, выпаривают при пониженном давлении, и осуществляют замену растворителя на циклогексан при отгонке. Затем полученную суспензию фильтруют, осадок на фильтре промывают циклогексаном, а затем водой. Затем твердые продукты сушат в вакууме при 35-40°C с получением трет-бутил(3Z)-3-[(диметиламино)метилен]-4-оксопирролидин-1-карбоксилата.
Стадия B: трет-Бутил 6a-гидрокси-3a,4,6,6a-тетрагидропирроло[3,4-c]пиразол-5(1H)-карбоксилат
К раствору трет-бутил(3Z)-3-[(диметиламино)метилен]-4-оксопирролидин-1-карбоксилата (58,2 кг, 242 моль) в толуоле (251 кг) при 35-45°C добавляют гидразин гидрат (14,6 кг, 290 моль) посредством добавления по каплям в течение дополнительных 2 часов. Затем смесь перемешивают в течение 10 часов при этой температуре. Затем загрузку охлаждают до 0-10°C, и суспензию перемешивают в течение 6 часов. Затем эту суспензию фильтруют, и осадок на фильтре промывают н-гептаном. Затем твердые продукты сушат в вакууме в течение ночи при 35-50°C с получением трет-бутил 6a-гидрокси-3a,4,6,6a-тетрагидропирроло[3,4-c]пиразол-5(1H)-карбоксилата.
Стадия C: трет-Бутил 4,6-дигидропирроло[3,4-c]пиразол-5(1H)-карбоксилат
К раствору трет-бутил 6a-гидрокси-3a,4,6,6a-тетрагидропирроло[3,4-c]пиразол-5(1H)-карбоксилата (47,0 кг, 207 моль) в дихлорметане (669 кг) при 0°C добавляют по каплям метанольный раствор моногидрата толуол-4-сульфоновой кислоты (3,7 кг, 20 моль в 38 кг MeOH) в течение 2 часов. Затем реакционную смесь состаривают в течение 4 часов при этой температуре. Затем добавляют 5% водный раствор NaHCO3 (91 кг) и перемешивают при комнатной температуре в течение 30 мин. Затем слои разделяют, и водный слой экстрагируют дихлорметаном (312 кг). Объединенные органические слои промывают 5% соляным раствором (190 кг, а затем 483 кг), обрабатывают активированным углем (2,7 кг) и фильтруют. Полученные органические вещества сушат с помощью Na2SO4, фильтруют и концентрируют до 71-118 л. Затем добавляют н-гептан (238 кг), и загрузку дополнительно концентрируют до 188-235 л. Суспензию охлаждают до 10-20°C, фильтруют, и осадок на фильтре промывают н-гептаном. Твердые продукты сушат в вакууме при 40-50°C в течение ночи с получением трет-бутил 4,6-дигидропирроло[3,4-c]пиразол-5(1H)-карбоксилата.
Стадия D: трет-Бутил 2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат
Раствор трет-бутил 4,6-дигидропирроло[3,4-c]пиразол-5(1H)-карбоксилата (30,0 кг, 143 моль) в 2-метилтетрагидрофуране (384 мг) продувают вакуумом и обратно заполняют азотом, три раза. Добавляют триэтиламин (25,0 кг, 247 моль), и загрузку охлаждают до -10 - 5°C. Затем в течение 2 часов медленно добавляют метансульфонилхлорид (21,4 кг, 187 моль). После перемешивания в течение 1 часа при комнатной температуре, добавляют по каплям воду (150 кг) при 5-15°C. После этого следует добавление 1 н раствора HCl до достижения pH 7. Полученные слои разделяют и водный слой экстрагируют 2-метилтетрагидрофураном (106 кг). Объединенные органические слои промывают насыщенным соляным раствором (2×150 кг), сушат с помощью Na2SO4, фильтруют и концентрируют до 60-90 л.
Полученный сырой продукт растворяют в 2-метилтетрагидрофуране (381 кг) и загружают раствором трет-бутоксида калия в ТГФ (805 г в 6,6 кг ТГФ). После перемешивания в течение 1 часа при комнатной температуре в атмосфере азота, добавляют дополнительный трет-бутоксид калия в ТГФ (329 г в 3,0 кг ТГФ) и перемешивают в течение 1 часа. Аналитический анализ показывает, что трет-бутил 2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат представляет собой главный региоизомер, так что затем добавляют насыщенный соляной раствор (154 кг). После короткого перемешивания, слои разделяют и органические вещества промывают насыщенным соляным раствором (2×155 кг). Затем объединенные водные слои отходов экстрагируют 2-метилтетрагидрофураном (103 кг). Объединенные органические слои обрабатывают активированным углем (8,75 кг), фильтруют и сушат с помощью Na2SO4. Затем их фильтруют и концентрируют до 60-90 л. Затем эту суспензию нагревают для растворения твердых продуктов при 40-50°C и добавляют н-гептан (34 кг). После охлаждения до комнатной температуры в течение 2-4 часов, добавляют н-гептан (156 кг), и затем суспензию состаривают в течение 2-4 часов при 0-5°C. Суспензию фильтруют, и осадок на фильтре промывают н-гептаном. Твердые продукты сушат в вакууме при 45-55°C с получением трет-бутил 2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата.
Стадия E: трет-Бутил 1-(метилсульфонил)-4,6-дигидропирроло[3,4-c]пиразол-5(1H)карбоксилат
Это соединение получают, следуя такому же способу, как описано для Промежуточного соединения 1, Стадия D.
Стадия F: 2-(метилсульфонил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-5-иний бензолсульфонат
К раствору трет-бутил 2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (32,1 кг, 111 моль) в изопропилацетате (289 кг) добавляют бензолсульфоновую кислоту (35,35 кг, 223 моль). Реакционную смесь перемешивают в течение 3 дней при комнатной температуре, а затем охлаждают до 0-10°C и перемешивают дополнительно 1 час. Полученную суспензию фильтруют, и осадок на фильтре промывают изопропилацетатом. Твердые продукты сушат в течение ночи в вакууме при комнатной температуре с получением 2-(метилсульфонил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-5-ия бензолсульфоната.
[(2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин
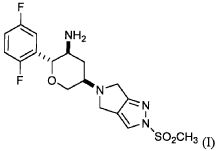
Стадия A: трет-Бутил {(2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-ил}карбамат
Емкость загружают Ν,Ν-диметилацетамидом (520,6 кг), 2-(метилсульфонил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-5-ием бензолсульфонатом (промежуточное соединение 2, 30,0 кг, 86,8 моль) и трет-бутил[(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2H-пиран-3-ил]карбаматом (промежуточное соединение 1, 31,2 кг, 95,3 моль). После растворения при комнатной температуре, раствор охлаждают до 0-10°C и добавляют натрий триацетоксиборогидрид (24 кг, 1 13 моль) в четырех равных порциях, каждые 40 мин. Затем реакционной смеси позволяют нагреться до комнатной температуры и перемешивают в течение дополнительных 5 часов. Затем раствор охлаждают до 5-15°C и добавляют воду (672 кг) в течение 1-2 часов. Полученную суспензию фильтруют, и осадок на фильтре промывают последовательно N,N-диметилацетамидом, дважды водой, а затем н-гептаном. Твердые продукты сушат в вакууме при 40-60°C с получением трет-бутил {(2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-ил}карбамата.
Стадия B: {(2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин
Бензолсульфоновую кислоту (32,95 кг, 271 моль) растворяют в дихлорметане (1020 кг) в атмосфере азота. Затем добавляют 880 г воды, так что KF раствора составляет 0,2%. Далее добавляют трет-бутил {(2R,35,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-ил}карбамат (38,4 кг, 100 моль) в трех равных порциях в течение 30 мин. Затем реакционную смесь состаривают в течение ночи при комнатной температуре. Далее, добавляют воду (733 кг) в течение 1 часа и реакционную смесь быстро перемешивают в течение 1 часа. Затем слои разделяют, выливая полученный слой органических веществ. В водный слой загружают дихлорметан (510 кг), а затем триэтиламин (22,4 кг, 592 моль). После перемешивания, слои разделяют, и водный слой экстрагируют дихлорметаном (510 кг). Объединенные органические слои промывают с помощью 7% водного раствора NaHCO3 (2×410 кг) и 5% соляным раствором (386 кг). Затем органические вещества сушат с помощью Na2SO4, фильтруют и обрабатывают активированным углем (6,2 кг C-941). Уголь отфильтровывают, и фильтрат концентрируют в вакууме до 154-193 л. Затем этот раствор нагревают до 30-35°C для растворения твердых продуктов (может добавляться дополнительный дихлорметан для растворения твердых продуктов). Далее, добавляют изопропилацетат (338 кг), и раствор перемешивают при комнатной температуре в течение 1,5 часа. Затем н-гептан (159 кг) загружают в емкость по каплям и перемешивают в течение 3 часов. Затем суспензию фильтруют, и осадок на фильтре промывают н-гептаном. Затем этот влажный осадок на фильтре опять перекристаллизовывают посредством растворения его в дихлорметане и добавления изопропилацетата и н-гептана, как перед этим, фильтрования и промывки н-гептаном. Твердые продукты сушат в вакууме при 40-50°C в течение ночи с получением кристаллического (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амина, который промывают холодным EtOAc/гексаном, 2:1, с получением указанного в заголовке соединения в виде беловатого твердого продукта. 1H ЯМР (500 МГц, CD3OD): 1,71 (кв, 1H, J=12 Гц), 2,56-2,61 (м, 1H), 3,11-3,18 (м, 1H), 3,36-3,40 (м, 1H), 3,48 (т, 1H, J=12 Гц), 3,88-3,94 (м, 4H), 4,30-4,35 (м, 1H), 4,53 (д, 1H, J=12 Гц), 7,14-7,23 (м, 2H), 7,26-7,30 (м, 1H), 7,88(с, 1H). LC-MS: 399,04 [M+1].
Форма I
Форму I получают посредством непосредственной кристаллизации аморфного свободного основания Соединения I в этилацетате. Результаты характеризации для XRPD, ssNMR, DSC, TGA и IR показаны ниже.
Форма II
Кристаллическую Форму II получают посредством перекристаллизации Формы I в изопропилацетате и гептане, 1:1, при комнатной температуре. Форму II характеризуют с использованием XRPD, ssNMR, DSC, TGA и IR. Преобразование Формы II в Форму I является медленным, но наблюдается во всех циклах экспериментов с затравкой, 50-50, содержащей DCM-гептан, при 25°C в течение двух дней, IPAc, при 25°C в течение 17 часов, IPAc, при 60°C в течение одного дня, H2O, при 60°C в течение двух недель, трех дней, NMP-воду, 1-1, при 35°C в течение трех дней. Соотношение между Формой I и Формой II является энантиотропным, при этом Форма I является наиболее стабильной фазой при температуре выше 13°C.
Форма III
Форму III получают посредством растворения Формы I в MeOH и выпаривания растворителя, с последующим нагревом до 140°C и выдерживанием при этой температуре в течение 10 мин. Эта фаза является метастабильной по отношению к Форме I и II, и ее характеризация ограничена доступным количеством образца. Форму III анализируют с помощью XRPD и DSC.
Форма IV
Форма IV получают посредством растворения Формы I в ТГФ-воде 1:1 и выпаривания растворителя. Безводная Форма IV является метастабильной по отношению к Форме I и II, и по этой причине ее характеризация ограничена доступным количеством образца. Форму IV анализируют с использованием XRPD, DSC и TGA.
Дифракция рентгеновского излучения на порошках
Исследования дифракции рентгеновского излучения на порошках широко используются для характеризации молекулярных структур, кристалличности и полиморфизма. Картины дифракции рентгеновского излучения на порошках для твердых фаз, для кристаллических форм Соединения I, генерируются на Philips Analytical X'Pert PRO X-ray Diffraction System с приставкой PW3040/60. K-альфа излучение рентгеновской трубки PW3373/00 с керамической Cu LEF используют в качестве источника. Положения дифракционных пиков сравнивают с кремнием (внутренний стандарт), который имеет значение 2 тэта 28,443 градуса. Эксперименты анализируют при условиях окружающей среды.
Кристаллические формы, описанные в настоящем документе, имеют фазовую чистоту, по меньшей мере, примерно 5% формы с указанными выше физическими характеристиками дифракции рентгеновского излучения на порошках и DSC. В одном из вариантов осуществления чистота фазы составляет, по меньшей мере, примерно 10% для формы с указанными выше физическими характеристиками твердого состояния. Во втором варианте осуществления чистота фазы составляет, по меньшей мере, примерно 25% для формы с указанными выше физическими характеристиками твердого состояния. В третьем варианте осуществления чистота фазы составляет, по меньшей мере, примерно 50% для формы с указанными выше физическими характеристиками твердого состояния. В четвертом варианте осуществления чистота фазы составляет, по меньшей мере, примерно 75% для формы с указанными выше физическими характеристиками твердого состояния. В пятом варианте осуществления чистота фазы составляет, по меньшей мере, примерно 90% для формы с указанными выше физическими характеристиками твердого состояния. В шестом варианте осуществления кристаллические формы по настоящему изобретению представляют собой по существу чистые фазовые формы с указанными выше физическими характеристиками твердого состояния. Под термином "фазовая чистота" подразумевается чистота твердого состояния конкретной формы по отношению к конкретной кристаллической форме, как определено с помощью твердотельных физических методов, описанных в настоящей заявке.
Фиг.1 представляет собой картину дифракции рентгеновского излучения на порошках (XRPD) для Формы I Соединения I с выбранными d - расстояниями между слоями, приведенными в Таблице 1.
XRPD: Форма I Соединения I
Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма I) характеризуется тем, что он имеет, по меньшей мере, четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 10,3±0,1 2θ, 12,7±0,1 2θ, 14,6±0,1 2θ, 16,1±0,1 2θ, 17,8±0,1 2θ, 19,2±0,1 2θ, 22,2±0,1 2θ, 24,1±0,1 2θ и 26,9±0,1 2θ. Кристаллическая форма 1 может характеризоваться с помощью следующих четырех пиков в ее картине дифракции рентгеновского излучения на порошках 17,8±0,1 2θ, 19,2±0,1 2θ, 22,2±0,1 2θ и 24,1±0,1 2θ. Кристаллическая форма 1 может характеризоваться с помощью следующих четырех пиков в ее картине дифракции рентгеновского излучения на порошках на Фиг.3.
Фиг.6 представляет собой картину дифракции рентгеновского излучения на порошках (XRPD) для Формы II Соединения I с выбранными d - расстояниями между слоями, приведенными в Таблице 2.
Дифракция рентгеновского излучения на порошках: Форма II Соединения I
Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма II) может характеризоваться тем, что он имеет, по меньшей мере, четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 7,5±0,1 2θ, 15,0±0,1 2θ, 16,2±0,1 2θ, 20,9±0,1 2θ, 22,0±0,1 2θ, 27,0±0,1 2θ, 27,6±0,1 2θ, 33,3±0,1 2θ. Кристаллическая форма II может характеризоваться с помощью следующих четырех пиков в ее картине дифракции рентгеновского излучения на порошках 20,9±0,1 2θ, 22,0±0,1 2θ, 27,0±0,1 2θ и 27,6±0,1 2θ. Кристаллическая форма II может характеризоваться с помощью картины дифракции рентгеновского излучения на порошках на Фиг.6.
Фиг.11 представляет собой картину дифракции рентгеновского излучения на порошках (XRPD) для Формы III Соединения I с выбранными d - расстояниями между слоями, приведенными в Таблице 3.
Дифракция рентгеновского излучения на порошках: Форма III Соединения I
Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма III) может характеризоваться тем, что он имеет, по меньшей мере, четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 14,5±0,1 2θ, 15,9±0,1 2θ, 17,3±0,1 2θ, 18,7±0,1 2θ, 19,5±0,1 2θ, 19,5±0,1 2θ, 21,2±0,1 2θ, 22,0±0,1 2θ и 23,2±0,1 2θ. Кристаллическая форма III может характеризоваться с помощью следующих четырех пиков в ее картине дифракции рентгеновского излучения на порошках 19,5±0,1 2θ, 21,2±0,1 2θ, 22,0±0,1 2θ и 23,2±0,1 2θ. Кристаллическая форма III может характеризоваться с помощью картины дифракции рентгеновского излучения на порошках на Фиг.11.
Фиг.14 представляет собой картину дифракции рентгеновского излучения на порошках (XRPD) для Формы IV Соединения I с выбранными d - расстояниями между слоями, приведенными в Таблице 4.
Дифракция рентгеновского излучения на порошках: безводная Форма IV Соединения I
Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-ил]тетрагидро-2H-пиран-3-амин (Форма IV) может характеризоваться тем, что он имеет, по меньшей мере, четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 8,1±0,1 2θ, 10,6±0,1 2θ, 16,0±0,1 2θ, 16,9±0,1 2θ, 19,5±0,1 2θ, 21,3±0,1 2θ, 23,3±0,1 2θ и 25,4±0,1 2θ.
Кристаллическая форма IV может характеризоваться с помощью следующих четырех пиков в ее картине дифракции рентгеновского излучения на порошках 16,9±0,1 2θ, 19,5±0,1 2θ, 21,3±0,1 2θ и 23,3±0,1 2θ. Кристаллическая форма IV может характеризоваться с помощью картины дифракции рентгеновского излучения на порошках на Фиг.14.
Спектры ssNMR
Спектр ядерного магнитного резонанса твердотельного углерода-13 регистрируют на ЯМР-спектрометре Bruker AV400 с использованием датчика Bruker 4 мм H/F/X BB двойного резонанса CPMAS. Спектр получают с использованием кросс-поляризации с переменной амплитудой протон/углерод-13 (VACP) при 10 кГц, при времени контакта 3 мсек. Другие экспериментальные параметры, используемые для получения данных, представляют собой 90-градусный импульс протона 100 кГц, развязку SPINAL64 на 100 кГц, задержку импульса 5 сек и усреднение сигнала по 1024 сканированиям. Скорость вращения под магическим углом (MAS) устанавливают на 10 кГц. Лоренцево уширение линии 10 Гц используют для спектра перед преобразованием Фурье. Химические сдвиги выражаются по сравнению с TMS, используя карбонильный углерод глицина (176,70 м.д.) в качестве вторичного эталона.
Кристаллическая форма I может дополнительно характеризоваться с помощью спектров ядерного магнитного резонанса (ЯМР) на Фиг.4, Фиг.4 представляет собой спектры ssNMR для Формы I Соединения I с выбранными пиками, приведенными в Таблице 5.
Выбранные пики ssNMR для Формы I Соединения I
Кристаллическая форма II может дополнительно характеризоваться с помощью спектров ядерного магнитного резонанса (ЯМР) на Фиг.9. Фиг.9 представляет собой спектр ssNMR для Формы Π Соединения I с выбранными пиками, приведенными в Таблице 6.
Выбранные пики ssNMR для Формы II Соединения I
ИК-Спектры
Инфракрасный спектр получают с использованием затухающего полного внутреннего отражения (ATR). Образец помещают непосредственно в устройство для отбора образцов для ATR-FTIR и регистрируют инфракрасный спектр с использованием спектрометра FTIR Nicolet Nexus 670.
Фиг.5 представляет собой ИК-спектр формы I Соединения I. Кристаллическая форма I может дополнительно характеризоваться с помощью ИК спектра на Фиг.5.
Фиг.10 представляет собой ИК-спектр формы II Соединения I. Кристаллическая форма II может дополнительно характеризоваться с помощью ИК спектра на Фиг.10.
В дополнение к картинам дифракции рентгеновского излучения на порошках, описанных выше, кристаллические формы Соединения I по настоящему изобретению дополнительно характеризуются посредством их кривых дифференциальной сканирующей калориметрии (DSC) и их кривых термогравиметрического анализа (TGA).
DSC
Данные дифференциальной сканирующей калориметрии получают с использованием DSC 2910 или DSC2000, TA Instruments. Образец, в пределах между 2 и 6 мг, отвешивают в поддон и прикрывают. Затем этот поддон закрывают и помещают в положение для образца в ячейке калориметра. Пустой поддон помещают в положение для эталона. Ячейку калориметра закрывают, и через ячейку пропускают поток азота. Программу нагрева устанавливают для нагрева образца при скорости нагрева 10°C/мин до температуры приблизительно 250°C. Данные анализируют с использованием Universal Analysis 2000 Version 3,9A. Термические события интегрируют между фоновыми температурными точками, которые находятся выше и ниже диапазона температур, в котором наблюдают термическое событие. Регистрируемые данные представляют собой температуру начала события, пиковую температуру и энтальпию.
Кристаллическая форма I может дополнительно характеризоваться с помощью кривой дифференциальной сканирующей калориметрии (DSC) на Фиг.3. Кристаллическая форма II может дополнительно характеризоваться с помощью кривой дифференциальной сканирующей калориметрии (DSC) на Фиг.8. Кристаллическая форма III может дополнительно характеризоваться с помощью кривой дифференциальной сканирующей калориметрии (DSC) на Фиг.13. Кристаллическая форма IV может дополнительно характеризоваться с помощью кривой дифференциальной сканирующей калориметрии (DSC) на Фиг.16.
TGA
Термогравиметрические данные регистрируют с использованием Perkin Elmer, модель TGA 7. Эксперименты осуществляют в потоке азота и с использованием скорости нагрева 10°C/мин до максимальной температуры приблизительно 250°C. После автоматического тарирования весов, на платиновый поддон добавляют 5-20 мг образца, включают печь и запускают программу нагрева. Данные масса/температура собираются инструментом автоматически. Анализы результатов осуществляют, выбирая функцию Delta Y в программном обеспечении инструмента и выбирая температуры, между которыми должна вычисляться потеря массы. Потери массы регистрируются до наступления разложения/испарения. Кристаллическая форма I может дополнительно характеризоваться с помощью кривой термогравиметрического анализа (TGA) на Фиг.2. Кристаллическая форма II может дополнительно характеризоваться с помощью кривой термогравиметрического анализа (TGA) на Фиг.7. Кристаллическая форма III может дополнительно характеризоваться с помощью кривой термогравиметрического анализа (TGA) на Фиг.12. Кристаллическая форма IV может дополнительно характеризоваться с помощью кривой термогравиметрического анализа (TGA) на Фиг.15.
Репрезентативный образец формы I анализируют с помощью DSC и TGA в соответствии со способами, описанными выше. Форма 1 демонстрирует одну эндотерму (плавление Формы I подтверждается с помощью микроскопии на горячем столике) при Tonset=173,48°C, Tpeak=175,32°C и ΔΗ=82,28 Дж/г (Фиг.3). Термогравиметрический анализ демонстрирует незначительные потери массы между комнатной температурой и температурой плавления Формы I (Фиг.2).
Репрезентативный образец формы II анализируют с помощью DSC (Фиг.8) и TGA (Фиг.7) в соответствии со способами, описанными выше. Первая эндотерма на кривой DSC связана с плавлением Формы II при Tonset=144,75°C, Tpeak=147,59°C и ΔΗ=23,41 Дж/г (Фиг.11). После первой эндотермы следует событие перекристаллизации с получением Формы I при ~150°C и, наконец, плавление Формы I при Tonset=170,18°, 172,95°C и ΔΗ=57,45 Дж/г. TG анализ демонстрирует минимальные потери массы (захваченный растворитель) между комнатной температурой и плавлением Формы I.
DSC Формы III (Фиг.13) демонстрируют одну эндотерму, связанную с плавлением Формы III при Tonset=164,30°C, Tpeak=169,38°C и ΔΗ=23,41 Дж/г. Термогравиметрический анализ (Фиг.12) показывает ~1% масс./масс. остаточного растворителя в исходном материале, который удаляется посредством нагрева при 140°C и выдерживания в течение 10 мин.
DSC Формы IV (Фиг.16) демонстрируют одну эндотерму, связанную с плавлением Формы IV, при Tonset=171,25°C, Tpeak=172,30°C и ΔΗ=84,64 Дж/г. При использовании TGA наблюдают менее 1% потери массы при плавлении (Фиг.15).
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ИНГИБИТОРЫ ЦИСТЕИНПРОТЕАЗ КАТЕПСИНОВ | 2014 |
|
RU2692799C2 |
| ЗАМЕЩЕННЫЕ АМИНО ШЕСТИЧЛЕННЫЕ НАСЫЩЕННЫЕ ГЕТЕРОАЛИЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ DPP-IV ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2016 |
|
RU2720488C2 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ПРОНИЦАЕМЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2707292C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2625308C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |







Изобретение относится к новой кристаллической форме (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-с]пиразол-5(4Н)-ил]тетрагидро-2Н-пиран-3-амина соединения I, характеризующегося тем, что имеет по меньшей мере четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 10,3+0,1 2θ, 12,7±0,1 2θ, 14,6±0,1 2θ, 16,1±0,1 2θ, 17,8±0,1 2θ, 19,2±0,1 2θ, 22,2±0,1 2θ, 24,1±0,1 2θ и 26,9±0,1 2θ, который является сильнодействующим ингибитором дипептидилпептидазы-IV. Изобретение также относится к применению этого соединения при лечении неинсулинзависимого сахарного диабета (Типа 2), также к фармацевтическим композициям на его основе. 4 н. и 8 з.п. ф-лы, 16 ил., 6 табл.
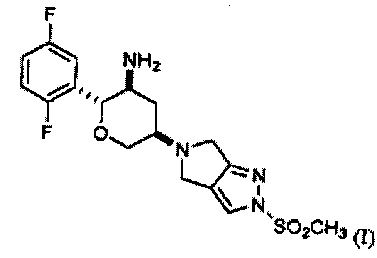
1. Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-с]пиразол-5(4Н)-ил]тетрагидро-2Н-пиран-3-амин Соединения I:

характеризующийся тем, что имеет по меньшей мере четыре пика в его картине дифракции рентгеновского излучения на порошках, выбранных из группы, состоящей из 10,3+0,1 2θ, 12,7±0,1 2θ, 14,6±0,1 2θ, 16,1±0,1 2θ, 17,8±0,1 2θ, 19,2±0,1 2θ, 22,2±0,1 2θ, 24,1±0,1 2θ и 26,9±0,1 2θ.
2. Кристаллическая форма по п. 1, отличающаяся следующими четырьмя пиками в ее картине дифракции рентгеновского излучения на порошках: 17,8±0,1 2θ, 19,2±0,1 2θ, 22,2±0,1 2θ и 24,1±0,1 2θ.
3. Кристаллическая форма по п. 2, отличающаяся кривой дифференциальной сканирующей калориметрии (DSC), которая показывает максимальный эндотермальный пик при или около 175,32°С.
4. Применение кристаллической формы по п. 1, в качестве активного ингредиента, для получения лекарственного средства для лечения диабета 2 типа у млекопитающего.
5. Фармацевтическая композиция, являющаяся ингибитором дипептидилпептидазы-IV (DPP-IV), содержащая лекарственное вещество, которое содержит кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-с]пиразол-5(4Н)-ил]тетрагидро-2Н-пиран-3-амин по п. 1 и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п. 5, где в лекарственном веществе присутствует по меньшей мере 50% масс. кристаллической формы.
7. Фармацевтическая композиция по п. 5, где в лекарственном веществе присутствует по меньшей мере 5% масс. кристаллической формы.
8. Кристаллическая форма по п. 1 или 2, отличающаяся кривой термогравиметрического анализа (TGA), характеризующейся незначительной потерей массы между комнатной температурой и температурой плавления.
9. Кристаллический (2R,3S,5R)-2-(2,5-дифторфенил)-5-[2-(метилсульфонил)-2,6-дигидропирроло[3,4-с]пиразол-5(4Н)-ил]тетрагидро-2Н-пиран-3-амин Соединения I:
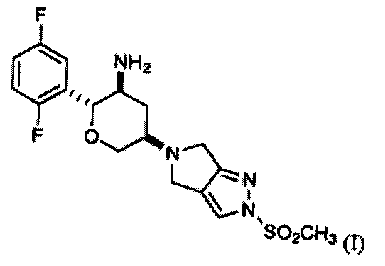
характеризующийся тем, что имеет спектр твердотельного ядерного магнитного резонанса углерода-13 (ЯМР), имеющий химические сдвиги при 163,6, 157,9, 128,9, 124,3 и 119,0 м.д.
10. Кристаллическая форма по п. 9, дополнительно отличающаяся спектром твердотельного ядерного магнитного резонанса углерода-13 (ЯМР), имеющим химические сдвиги при 163,6, 157,9, 128,9, 124,3, 119,0, 90,1, 73,2, 59,9, 48,6 и 42,6 м.д.
11. Кристаллическая форма по п. 9 или 10, дополнительно отличающаяся кривой дифференциальной сканирующей калориметрии (DSC), которая показывает максимальный эндотермальный пик при или около 175,32°С.
12. Кристаллическая форма по п. 9 или 10, отличающаяся кривой термогравиметрического анализа (TGA), характеризующейся незначительной потерей массы между комнатной температурой и температурой плавления.
| US 20100120863 A1, 13.05.2010 | |||
| US 2005003804 A1, 10.02.2005 | |||
| US 5041442 A, 20.08.1991 | |||
| Колосник для топочных решеток | 1940 |
|
SU63013A1 |

