РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка заявляет на приоритет предварительной заявки на патент США № 62/483044, поданной 7 апреля 2008 года. Полный объем идей вышеуказанной заявки включен в данный документ посредством ссылки.
Область техники
Данное изобретение относится к способам и промежуточным соединениям, пригодным для получения биологически активных молекул, применяемых в качестве модуляторов FXR или TGR5, особенно относится к производным желчных кислот и способам их получения и применения.
Уровень техники
Фарнезоидный Х-рецептор (FXR) представляет собой орфанный ядерный рецептор, первоначально идентифицированный из библиотеки к ДНК печени крысы (BM. Forman, et al., Cell, 1995, 81(5), 687-693), который наиболее тесно связан с экдизоновым рецептором насекомых. FXR является членом семейства ядерных рецепторов лиганд-активированных факторов транскрипции, которое включает рецепторы стероидных, ретиноидных и тиреоидных гормонов (DJ. Mangelsdorf, et al., Cell, 1995,83(6), 841-850). Соответствующими физиологическими лигандами FXR являются желчные кислоты (D. Parks et al., Science, 1999, 284(5418), 1362-1365). Одной из самых активных является хенодезоксихолевая кислота (CDCA), которая регулирует экспрессию нескольких генов, участвующих в гомеостазе желчных кислот. Фарнезол и его производные, которые вместе называются фарнезоидами, первоначально описаны для активации ортолога крысы в высокой концентрации, но они не активируют рецептор человека или мыши. FXR экспрессируется в печени по всему желудочно-кишечному тракту, включая пищевод, желудок, двенадцатиперстную кишку, тонкую кишку, толстую кишку, яичник, надпочечник и почку. Помимо контроля внутриклеточной экспрессии генов, FXR, по-видимому, также участвует в паракринной и эндокринной передаче сигналов путем повышения регуляции экспрессии цитокинов, фактора роста фибробластов (J. Holt et al., Genes Dev., 2003, 17(13), 1581-1591; T. Inagaki et al., Cell Metab., 2005, 2(4), 217-225).
Низкомолекулярные соединения, которые действуют как модуляторы FXR, были раскрыты в следующих публикациях: WO 2000/037077, WO 2002/072598, WO 2003/015771, WO 2003/099821, WO 2004/00752, WO 2004/048349, WO 2005/009387, WO 2005/082925, US 2005/0054634, WO 2007/052843, WO 2007/070796, WO 2007/076260, WO2007/092751, WO 2007/095174, WO 2007/140174, WO 2007/140183, US 2007/0142340, WO 2008/000643, WO 2008/002573, WO 2008/025539, WO 2008/025540, WO 2008/051942, WO 2008/073825, WO 2008/157270, US 2008/0299118, US 2008/0300235, WO 2009/005998, WO 2009/012125, WO 2009/027264, WO 2009/062874, WO 2009/127321, WO 2009/149795, US 2009/0131409, US 2009/0137554, US 2009/0163474, US 2009/0163552, US 2009/0215748, WO 2010/043513, WO 2011/020615, WO 2011/117163, WO 2012/087519, WO 2012/087520, WO 2012/087521, WO 2013/007387, WO 2013/037482, WO 2013/166176, WO 2013/192097, WO 2014/184271, US 2014/0186438, US 2014/0187633, WO 2015/017813, WO 2015/069666, WO 2016/073767, WO 2016/116054, WO 2016/103037, WO 2016/096116, WO 2016/096115, WO 2016/097933, WO 2016/081918, WO 2016/127924, WO 2016/130809, WO 2016/145295, WO 2016/173524, CN 106632294, CN 106588804, US 2017/0196893, WO 2017/062763, WO 2017/053826, CN 106518708, CN 106518946, CN 106478759, CN 106478447, CN 106478453, WO 2017/027396, WO 2017/049172, WO 2017/049173, WO 2017/049176, WO 2017/049177, WO 2017/118294, WO 2017/128896,WO 2017/129125, WO 2017/133521, WO 2017/147074, WO 2017/147174, WO 2017/145041 и WO 2017/156024 Al.
Недавно были рассмотрены другие низкомолекулярные FXR модуляторы (R. C. Buijsman et al. Curr. Med. Chem.2005, 12, 1017-1075).
Рецептор TGR5 представляет собой рецептор, связанный с G-белком, который был идентифицирован как рецептор клеточной поверхности, который чувствителен к желчным кислотам (BA). Было обнаружено, что первичная структура TGR5 и его чувствительность к желчным кислотам высоко консервативны в TGR5 у человека, крупного рогатого скота, кролика, крысы и мыши, и, таким образом, можно предположить, что TGR5 выполняет важные физиологические функции. Обнаружено, что TGR5 широко распространен не только в лимфоидных тканях, но и в других тканях. Высокие уровни мРНК TGR5 были обнаружены в плаценте, селезенке и моноцитах/макрофагах. Было показано, что желчные кислоты вызывают интернализацию слитого белка TGR5 из клеточной мембраны в цитоплазму (Kawamata et al., J. Bio. Chem., 2003, 278, 9435). Было обнаружено, что TGR5 идентичен hGPCR19, о котором сообщают Takeda et al., FEBS Lett. 2002, 520, 97-101.
TGR5 связан с внутриклеточным накоплением цАМФ, который широко экспрессируется в клетках разных типов. Хотя активация этого мембранного рецептора в макрофагах снижает выработку провоспалительных цитокинов, (Kawamata, Y., et al., J. Biol. Chem. 2003, 278, 9435-9440) стимуляция TGR5 BA в адипоцитах и миоцитах увеличивает расход энергии (Watanabe, M., et al. Nature. 2006, 439, 484-489). Этот последний эффект включает в себя цАМФ-зависимую индукцию иодтирониндеиодиназы 2-го типа (D2), которая путем локального превращения Т4 в Т3 вызывает повышение активности гормонов щитовидной железы. В соответствии с ролью TGR5 в контроле энергетического обмена у самок мышей, нокаутированных по TGR5, наблюдается значительное накопление жира с увеличением массы тела, когда им дают диету с высоким содержанием жиров, что указывает на то, что недостаток TGR5 снижает расход энергии и вызывает ожирение (Maruyama, T., et al., J. Endocrinol. 2006, 191, 197-205). Кроме того, и в соответствии с участием TGR5 в энергетическом гомеостазе, активация мембранных рецепторов желчных кислот также способствует выработке глюкагоноподобного пептида 1 (GLP-1) в мышиных энтероэндокринных клеточных линиях. (Katsuma, S., Biochem. Biophys. Res. Commun., 2005, 329, 386-390). На основании всех вышеуказанных наблюдений TGR5 является привлекательной мишенью для лечения заболеваний, например ожирения, диабета и метаболического синдрома.
В дополнение к применению агонистов TGR5 для лечения и профилактики метаболических заболеваний, соединения, которые модулируют модуляторы TGR5, также пригодны для лечения других заболеваний, например заболеваний центральной нервной системы, а также воспалительных заболеваний (WO 01/77325 и WO 02/84286). Модуляторы TGR5 также предлагают способы регуляции гомеостаза желчных кислот и холестерина, абсорбции жирных кислот и переваривания белков и углеводов.
Существует необходимость в разработке модуляторов FXR и/или TGR5 для лечения и профилактики заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способам получения соединений формулы (I) и соединений формулы (II):
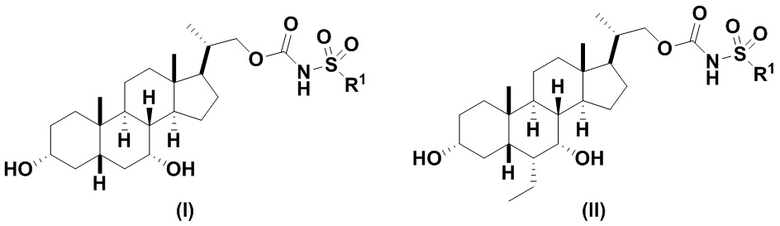
или их фармацевтически приемлемой соли, где:
R1 выбирают из группы, состоящей из:
1) замещенного или незамещенного -C1-C8 алкила;
2) замещенного или незамещенного -C2-C8 алкенила;
3) замещенного или незамещенного -C2-C8 алкинила;
4) замещенного или незамещенного -C3-C8 циклоалкила;
5) замещенного или незамещенного арила;
6) замещенного или незамещенного арилалкила;
7) замещенного или незамещенного 3-12- членного гетероциклоалкила;
8) замещенного или незамещенного гетероарила;
9) замещенного или незамещенного гетероарилалкила; а также
10) NRaRb; где Ra и Rb, каждый независимо, выбирают из водорода, замещенного или незамещенного -C1-C8 алкила, замещенного или незамещенного -C2-C8 алкенила, замещенного или незамещенного -C2-C8 алкинила, замещенного или незамещенного -C3-C8 циклоалкила. Альтернативно, Ra и Rb взяты вместе с атомом азота, к которому они присоединены, с образованием 3-12-членного гетероциклического кольца.
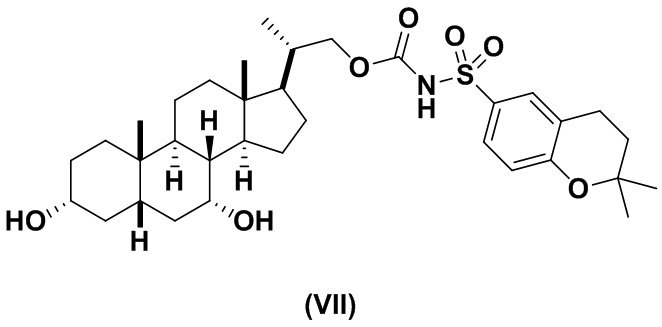
Предпочтительный вариант осуществления соединения формулы (I) представляет собой соединение (VII):
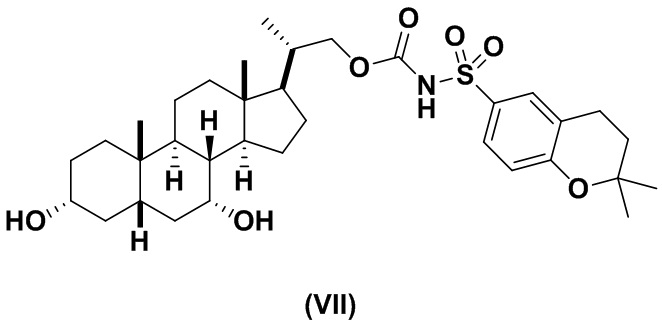
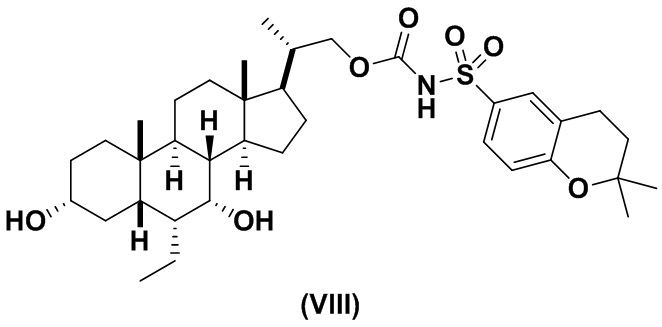
Предпочтительный вариант осуществления соединения формулы (II) представляет собой соединение (VIII):
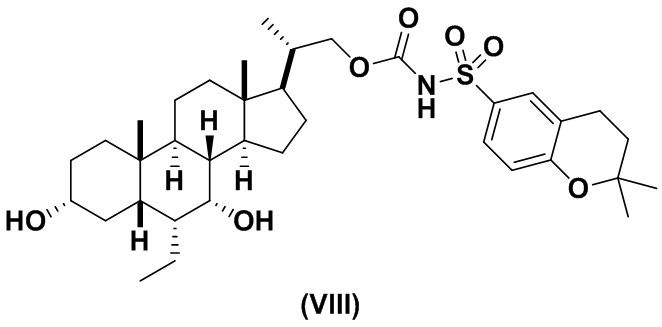
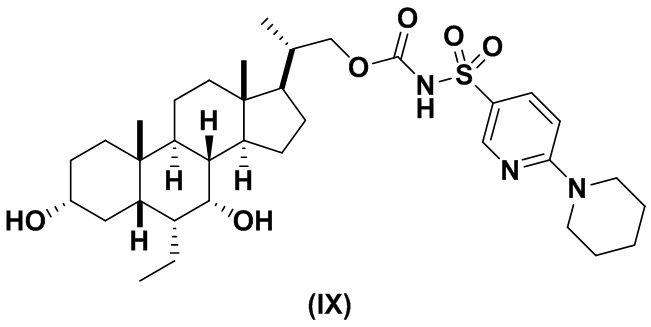
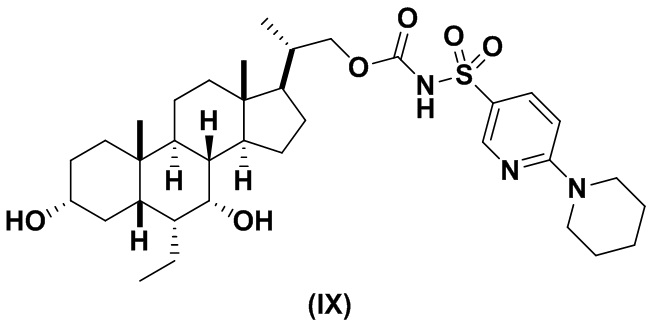
Другой предпочтительный вариант осуществления соединения формулы (II) представляет собой соединение (IX):
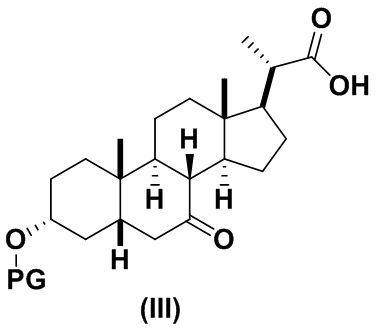
В определенных вариантах осуществления изобретения, данное изобретение относится к способам получения соединения формулы (III), которое представляет собой промежуточное соединение в синтезе соединений формулы (I) и формулы (II).
 ,
,
где PG представляет собой гидроксил-защитную группу такую как, но не ограничивающуюся ими, ацетильную, THP, MOM, MEM,SEM или силильную группу, такую как TBS, TES, TMS, TIPS или TBDPS. Предпочтительно, PG представляет собой TBS.
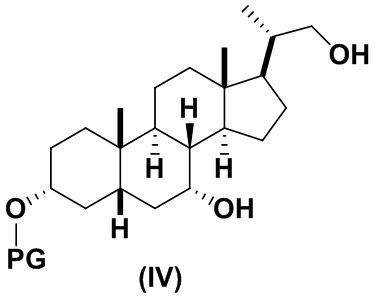
В определенных вариантах осуществления изобретения, данное изобретение относится к способам получения соединения формулы (IV), которое представляет собой промежуточное соединение в синтезе соединений формулы (I).
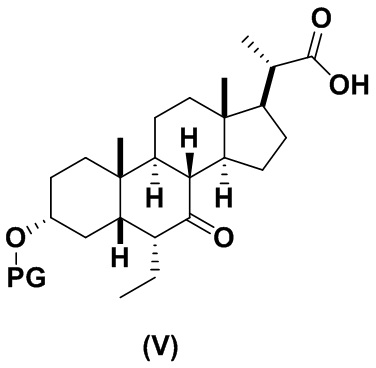
В определенных вариантах осуществления изобретения, данное изобретение относится к способам получения соединения формулы (V), которое представляет собой промежуточное соединение в синтезе соединений формулы (II).
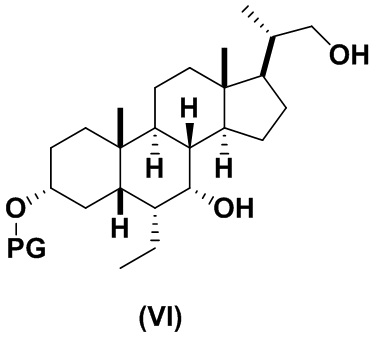
В определенных вариантах осуществления изобретения, данное изобретение относится к способам получения соединения формулы (VI), которое представляет собой подходящее промежуточное соединение в синтезе соединений формулы (II).
В одном варианте осуществления изобретения способ получения соединения формулы (I) включает стадии:
1(a) превращение соединения 1 (CDCA) в соединение формулы (III)
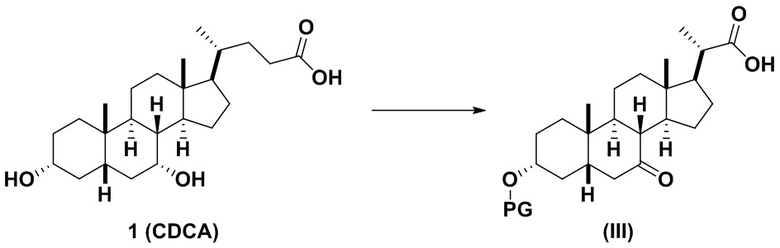 ;
;
2(a) превращение соединения формулы (III) в соединение формулы (IV)
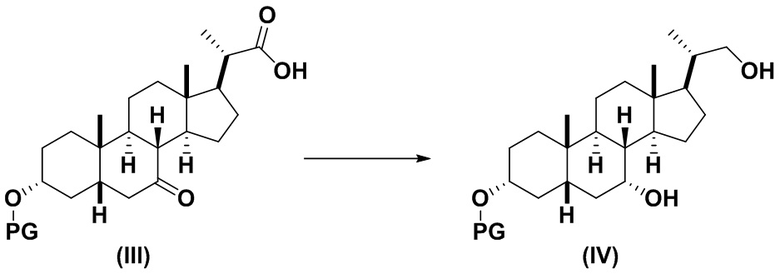 ;
;
3(a) превращение соединения формулы (IV) в соединение формулы (I)
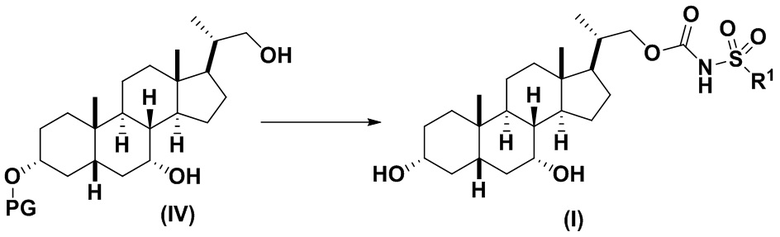 ,
,
где PG, и R1 соответствует определениям, описанным выше.
В одном варианте осуществления изобретения способ получения соединения формулы (II) включает стадии:
1(a) превращение соединения 1 (CDCA) в соединение формулы (III)
;
2(b) превращение соединения формулы (III) в соединение формулы (V)
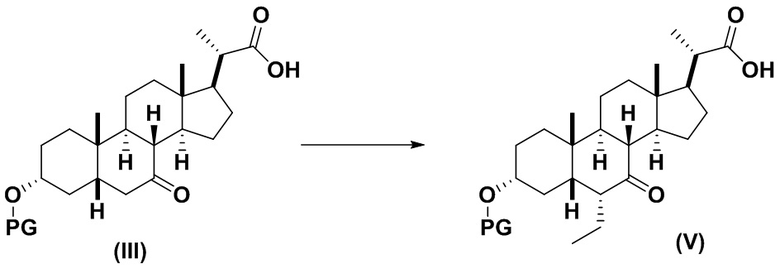 ;
;
3(b) превращение соединения формулы (V) в соединение формулы (VI)
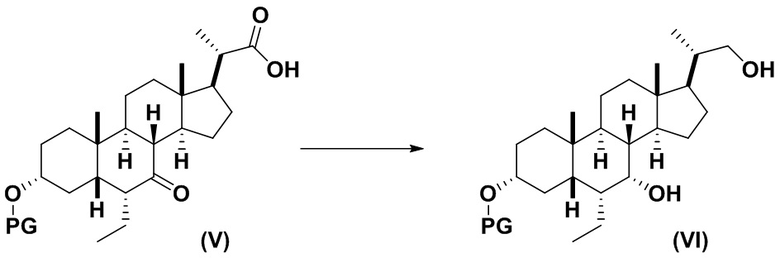 ;
;
4(b) превращение соединения формулы (VI) в соединение формулы (II)
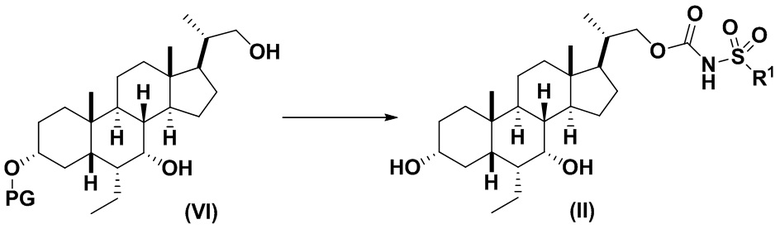 ,
,
где PG и R1 соответствуют определениям, указанным ранее.
Изобретение также относится к способам увеличения выхода продукта и уменьшения числа стадий процесса для промежуточного и крупномасштабного производства соединений формулы (I), формулы (II), формулы (III), формулы (IV), формулы (V) и формулы (VI).
Соединения формулы (I), формулы (II), соединения (VII), соединения (VIII) и соединения (IX) пригодны для лечения хронического заболевания печени, такого как заболевание, выбранное из группы, состоящей из билиарного первичного цирроза печени (PBC), церебротендинального ксантоматоза (CTX), первичного склерозирующего холангита (PSC), обусловленного лекарствами холестаза, внутрипеченочного холестаза при беременности, холестаза, связанного с парентеральным питанием (PNAC), холеостаза, связанного с чрезмерным развитием микрофлоры или сепсисом, аутоиммунного гепатита, хронического вирусного гепатита, алкогольного заболевания печени, неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), заболевания трансплантата печени, связанного с реакцией трансплантат против хозяина, регенерации трансплантата печени живого донора, врожденного фиброза печени, холедохолитиаза, гранулематозной болезни печени, внутри- или внепеченочная малигнизации, синдрома Шегрена, саркоидоза, болезни Вильсона, болезни Гоше, гемохроматоза и дефицита альфа-1-антитрипсина (WO2016/086218A1).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способам получения соединений формулы (I) и соединений формулы (II)
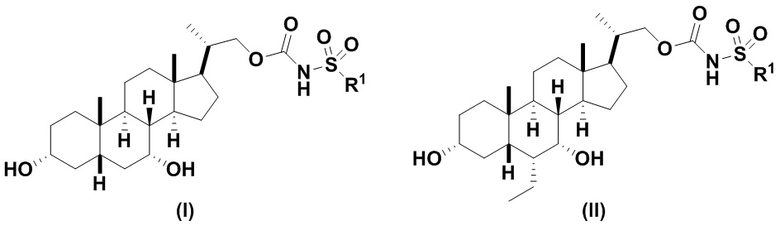
или фармацевтически приемлемой соли, где R1 соответствует определениям, описанным выше.
В определенных вариантах осуществления изобретения данное изобретение относится к способам получения соединений формулы (I) и соединений формулы (II), и их фармацевтически приемлемых солей, где R1 представляет собой замещенный или незамещенный арил; замещенный или незамещенный гетероарил; замещенный или незамещенный 3-12- членный гетероциклоалкил.
В определенных вариантах осуществления изобретения данное изобретение относится к способам получения соединений формулы (I) и соединений формулы (II), и их фармацевтически приемлемых соей, где R1 представляет собой замещенный или незамещенный фенил; или замещенный или незамещенный пиридил.
В определенных вариантах осуществления изобретения данное изобретение относится к способам получения соединений формулы (I) и соединений формулы (II), и их фармацевтически приемлемых солей, где R1 представляет собой замещенный или незамещенный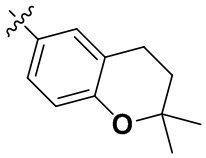 или замещенный или незамещенный
или замещенный или незамещенный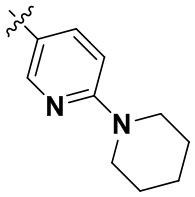 .
.
В другом варианте осуществления изобретения данное изобретение относится к способам получения соединения (VII).
В другом варианте осуществления изобретения данное изобретение относится к способам получения соединения (VIII).
В другом варианте осуществления изобретения данное изобретение относится к способам получения соединения (IX).
В одном варианте осуществления изобретения стадия 1(a) изложена на Схеме 1. Способ включает стадии 1(a)(i): эстерификация хенодезоксихолевой кислоты (CDCA, соединение 1) с C1-C6-алканолом, предпочтительно метанолом или этанолом, более предпочтительно метанолом, с получением соединения 2; 1(a)(ii): введение соединения 2 в реакцию с сильным основанием в присутствии подходящего гидроксил-защитного агента и электрофильного источника галогена с получением соединения 3; 1(a)(iii): отщепление группы R2 в соединении 3 с получением соединения 4; 1(a)(iv): снятие защитной группы в соединении 4 с получением соединения 5; 1(a)(v): введение защитной группы в соединение 5 с получением соединения 6; а также 1(a)(vi): окислительное расщепление и окисление соединения 6 с получением соединения (III).
Схема 1
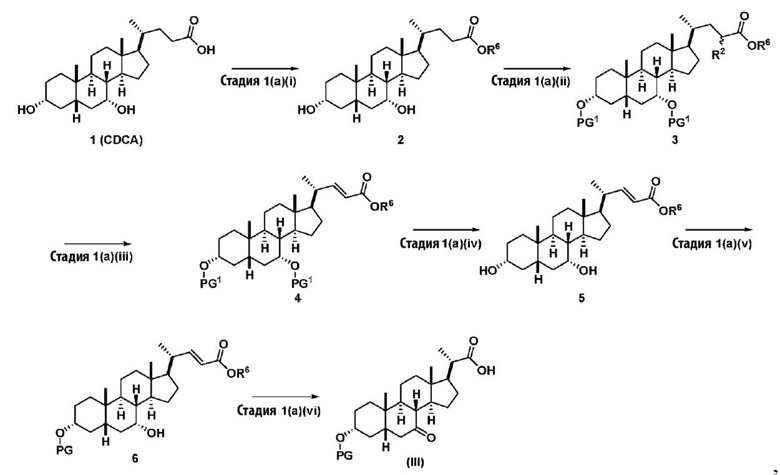 ,
,
где PG соответствует определениям, описанным ранее; PG1 представляет собой гидроксил-защитную группу предпочтительно выбранные из силильных групп, таких как, но не ограничивающихся ими, TMS, TES, TBS, TIPS и TBDPS; и R2 выбирают из Cl, Br и I. Предпочтительная PG1 представляет собой TMS. Предпочтительная PG представляет собой TBS.R6 представляет собой C1-C6-алкил, предпочтительно метил или этил и, более предпочтительно, метил.
Стадию 2(a) проводят, как изложено на Схеме 2. Так, на стадии 2(a)(i) соединение (III) вводят в реакцию с хлорформиатным реагентом R3OC(O)Cl для получения соединения ангидрида 7, с последующим восстановлением соединения 7 на стадии 2(a)(ii) с получением соединения (IV).
Схема 2
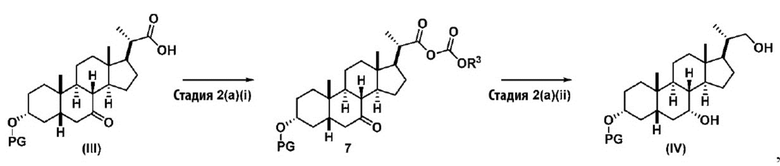 ,
,
где PG соответствует определениям, указанным ранее; R3 представляет собой алкильную группу, предпочтительно C1-C6-алкил, такой как, но не ограничивающийся ими, метил, этил, изопропил или изобутил. Предпочтительный R3 представляет собой изобутил.
Данное изобретение будет лучше понято в связи со стадиями 1(a)(i)-1(a)(vi) и 2(a)(i)-2(a)(ii), где PG, PG1, R2 и R3 соответствуют определениям, указанным ранее, если не указано иное. Для специалиста в данной области техники будет очевидно, что способ по данному изобретению может быть осуществлен путем замены соответствующих реагентов и что порядок самих стадий может варьироваться.
Стадия 1(a)(i), превращение соединения 1 в соединение 2:
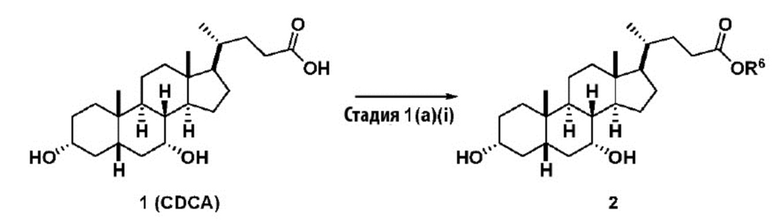
Стадия 1(a)(i) представляет собой эстерификацию CDCA алкиловым спиртом, предпочтительно C1-C6-алканолом и, более предпочтительно, метанолом, как известно в данной области техники, например, методики, описанные в Tetrahedron, 57(8), 1449-1481; 2001.
Стадия 1(a)(ii), превращение соединения 2 в соединение 3:
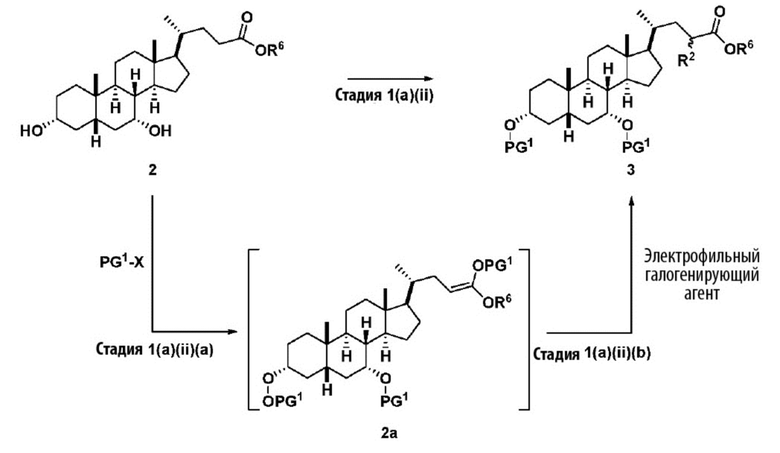
Стадия 1(a)(ii) представляет собой превращение соединения 2 в соединение 3 путем галогенирования промежуточного силилкетенацеталя 2a. Промежуточное соединение силилкетенацеталя 2a непосредственно получают in situ путем введения соединения 2 в реакцию с сильным основанием, таким как, но не ограничивающимся им, ЛДА в присутствии подходящего гидроксил-защитного агента PG1-X, где X выбирают из Cl, Br, I и OTf. Предпочтительный гидроксил-защитный агент представляет собой TMS-Cl. В одном аспекте температура составляет от -100°C до -50°C. В одном аспекте температура составляет от -90°C до -60°C. В одном аспекте температура составляет от -80°C до -70°C. Промежуточное соединение силилкетенацеталя 2а вводят в реакцию с электрофильным галогенирующим реагентом, таким как, но не ограничивающимся им, Br2, I2, I-Cl, I-Br, NBS, NIS и 1,3-дибром-5,5-диметилгидантоин с получением соединения 3. Предпочтительный электрофильный галогенирующий реагент представляет собой I2.
Стадия 1(a)(iii), превращение соединения 3 в соединение 4:
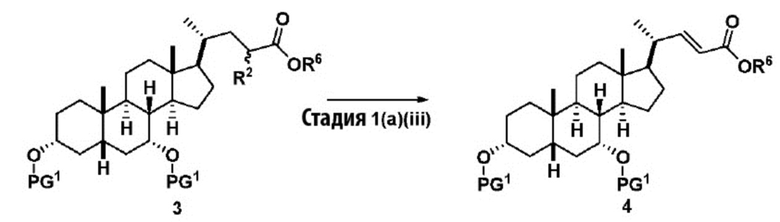
Стадия 1(a)(iii) представляет собой отщепление H-R2 из соединения 3 с образованием соединения 4. Соединение 3 обрабатывают подходящим органическим основанием, таким как, но не ограничивающимся им, DIPEA, Et3N, DBU, DBN или DABCO, в растворителе или смеси растворителей, такими как, но не ограничивающимися ими, ТГФ, ДХМ, ацетонитрил или толуол. Предпочтительное органическое основание представляет собой DBU. В предпочтительном аспекте растворитель представляет собой ТГФ. В предпочтительном аспекте соединение 3 со Стадии 1(a)(ii) используют непосредственно без дополнительной очистки. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Стадия 1(a)(iv), превращение соединения 4 в соединение 5:
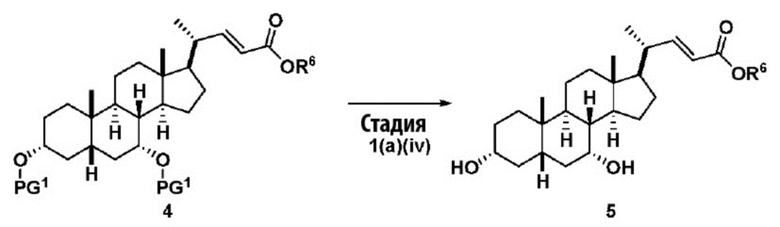
Стадия 1(a)(iv) представляет собой снятие защитной группы, PG1 из соединения 4 с образованием соединения 5. Защитная группа может быть удалена в подходящих условиях снятия защиты, которые известны в данной области техники. Например, PG1 может быть удалена с помощью реагента для снятия защиты, такого как, но не ограничивающегося им, TBAF, или кислотой, такой как HCl. Предпочтительно, соединение 4 обрабатывают кислотой в апротонном растворителе. В предпочтительном аспекте Стадии 1(a)(iv), соединение 4 со Стадии 1(a)(iii) используют непосредственно без дополнительной очистки. Предпочтительно, соединение 4 обрабатывают кислотой, такой как HCl, в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, 1,4-диоксан, MTBE, Et2O или смесь их двух. Предпочтительный растворитель представляет собой 1,4-диоксан. Реакция может быть проведена при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом предпочтительном аспекте температура реакции составляет около 25°C. В еще одном предпочтительном аспекте соединение 4 обрабатывают HCl в 1,4-диоксане при комнатной температуре с получением соединения 5. Соединение 5 может быть очищено с помощью колоночной хроматографии с получением соединения 5. Общий выход для превращения соединения 1 в соединение 5 составляет более 60% после очистки соединения 5.
Стадия 1(a)(v), превращение соединения 5 в соединение 6:
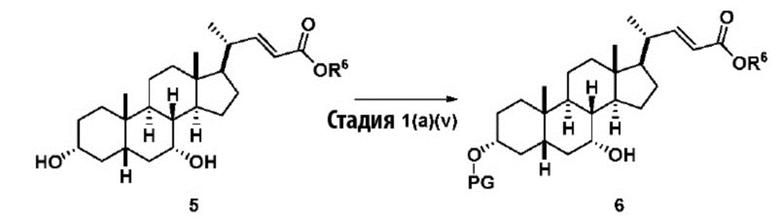
Стадия 1(a)(v) представляет собой защиту 3-гидроксила соединения 5 подходящим гидроксил-защитным агентом PG-X, где X представляет собой подходящую уходящую группу, например Cl, Br, I или OTf, в присутствии органического основания такого как, но не ограничивающегося им, имидазол, TEA, DIPEA с получением соединения 6. Предпочтительный гидроксил-защитный агент представляет собой TBS-Cl. Предпочтительное органическое основание представляет собой имидазол. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Стадия 1(a)(vi), превращение соединения 6 в соединение III:
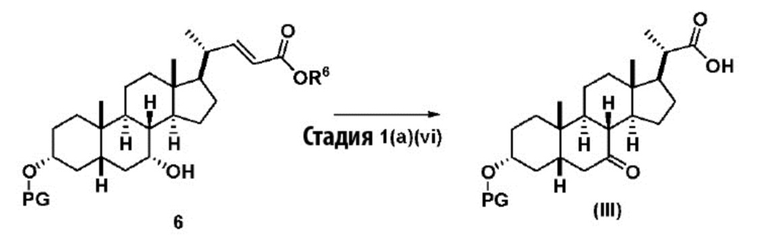
Стадия 1(a)(vi) представляет собой дигидроксилирование, окислительное расщепление и 7-OH окисление соединения 6 подходящим катализатором, таким как, но не ограничивающимся им, RuCl3 в присутствии стехиометрического количества окислителя, такого как, но не ограничивающегося им, NaIO4, n-Bu4N+IO4- и NMO, с получением соединения (III). Реакцию проводят в присутствии подходящего основания такого как, но не ограничивающегося им, K2CO3, Na2CO3 и 2,6-лутидин. Предпочтительный окислитель представляет собой NaIO4. Предпочтительное основание представляет собой K2CO3. Реакцию проводят в растворителе, таком как, но не ограничивающимся им, H2O, CCl4, CH3CN или EtOAc. Предпочтительный растворитель представляет собой смесь H2O, CH3CN и EtOAc. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C. Соединение (III) может быть перекристаллизовано из органического растворителя или смеси растворителей, такой как, но не ограничивающейся ею, смесь гексанов/EtOAc с получением соединения (III) с чистотой более чем 95%.
Стадия 2(a)(i), превращение соединения (III) в соединение 7:
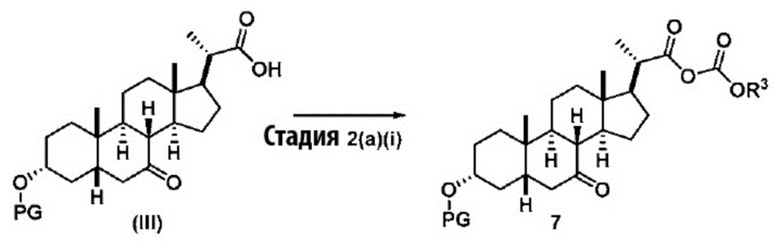
Стадия 2(a)(i) представляет собой реакцию соединения (III) с хлорформиатом R3OCOCl в присутствии основания, такого как, но не ограничивающегося им, TEA или DIPEA для получения смешанного ангидрида 7. Стадию 2(a)(i) предпочтительно проводят в апротонном растворителе, таком как, но не ограничивающимся им, ДХМ. Предпочтительный хлорформиат представляет собой изобутилхлорформиат, где R3 представляет собой изобутил. Предпочтительное органическое основание представляет собой TEA. Соединение 7 выделяют и используют на следующей стадии реакции без очистки.
Стадия 2(a)(ii), превращение соединения 7 в соединение (IV):
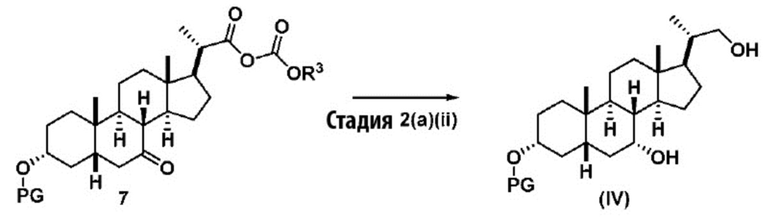
Стадия 2(a)(ii) представляет собой реакцию соединения 7 с подходящим восстанавливающим агентом, таким как, но не ограничивающимся им, NaBH4, LiBH4, LiAlH4 или DIBAL с получением соединения (IV). Стадию 2(a)(ii) предпочтительно проводят в смеси протонного и непротонного растворителя, такой как, но не ограничивающейся ею, смесь воды и ТГФ. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
В одном варианте осуществления изобретения Стадию 2(b) проводят, как изложено на Схеме 3. Способ включает стадии 2(b)(i): одновременные снятие TBS защиты и эстерификацию соединения (III) с получением соединения 8; 2(b)(ii): введение соединения 8 в реакцию с сильным основанием в присутствии подходящего гидроксил-защитного агента с получением соединения енольного эфира 9; 2(b)(iii): введение соединения 9 в реакцию с ацетальдегидом с получением соединения 10; 2(b)(iv): гидрирование соединения 10 с получением соединения 11; 2(b)(v): введение соединения 11 в реакцию с основанием в протонном растворителе или смеси протонного растворителя и непротонного растворителя с получением соединения (12); а также 2(b)(vi): введение защитной группы в соединение 12 с получением соединения (V).
Схема 3
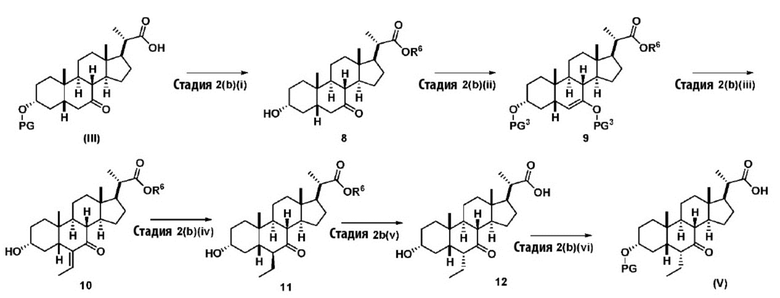
где PG соответствует определениям, описанным выше; PG3 представляет собой гидрокси-защитную группу, выбранную из силильных групп, таких как, но не ограничивающихся ими, TMS, TES, TBS, TIPS и TBDPS. Предпочтительная PG3 представляет собой TMS. R6 соответствует определениям, описанным выше.
Данное изобретение будет лучше понято в связи со Стадиями от 2(b)(i) до 2(b)(vi), где PG, PG3 и R3 соответствуют определениям, указанным ранее, если не указано иное. Для специалиста в данной области техники будет очевидно, что способ по данному изобретению может быть осуществлен путем замены соответствующих реагентов и что порядок самих стадий может варьироваться.
Стадия 2(b)(i), превращение соединения (III) в соединение 8:
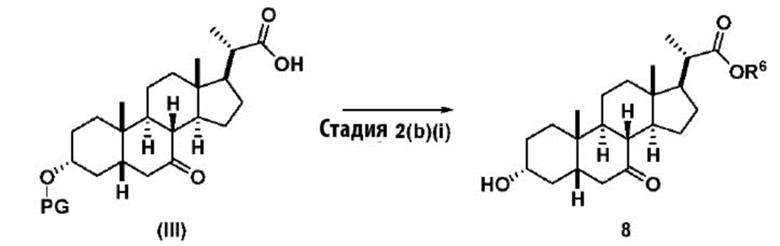
Стадия 2(b)(i) представляет собой эстерефикацию и снятие защитной группы PG из соединения (III) с образованием соединения 8. Предпочтительная защитная группа представляет собой TBS. Защитная группа может быть удалена в подходящих условиях снятия защиты, которые известны в данной области техники. Например, защитная группа может быть удалена реагентом для снятия защиты, таким как, но не ограничивающимся им, кислота, такая как HCl. Предпочтительно, соединение (III) обрабатывают кислотой в C1-C6-алканоле, предпочтительно MeOH или EtOH, более предпочтительно MeOH. Реакцию могут проводить при температуре в диапазоне от 25°C до 100°C. В предпочтительном аспекте температура реакции составляет от 35°C до 80°C. В другом аспекте температура реакции составляет около 50°C. Соединение 8 может быть очищено с помощью перекристаллизации с получением соединения 8 с чистотой более чем 95%.
Стадия 2(b)(ii), превращение соединения 8 в соединение 9:
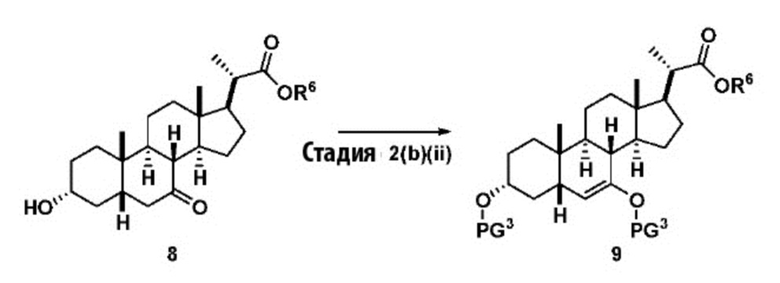
Стадия 2(b)(ii) представляет собой образование силилового эфирного соединения 9 путем введения в реакцию соединения 8 с силилирующим агентом в присутствии основания в апротонном растворителе, таком как, но не ограничивающимся им, ДХМ и ТГФ.
В одном аспекте стадии 2(b)(ii) силилирующий агент представляет собой TMSCl и основание представляет собой сильное органическое основание, такое как, но не ограничивающееся им, NaHMDS, LiHMDS или ЛДА и реакция протекает при низкой температуре, такой как около -78°C.
В другом предпочтительном аспекте стадии 2(b)(ii), силилирующий агент представляет собой TMSOTf и его используют вместе с органическим основанием, таким как, но не ограничивающимся им, TEA или DIPEA при температуре реакции в диапазоне от -20°C до 30°C. В предпочтительном аспекте температура реакции составляет от около -5°C до около 15°C. В другом аспекте температура составляет около 0°C. Молярное соотношение TMSOTf к соединению 8 предпочтительно находится в диапазоне от 3 до 12. В одном аспекте молярное соотношение составляет от 3 до 6. В одном аспекте молярное соотношение составляет от 4,5 до 5,5.
В предпочтительном аспекте соединение 9 может быть использовано непосредственно на стадии 2(b)(iii) без очистки.
Перед проведением Стадии 2(b)(iii) предпочтительно удалить остаточную воду в неочищенном соединении 9 со Стадии 2(b)(ii), чтобы контролировать разложение соединения 9.В одном аспекте соединение 9, полученное на Стадии 2(b)(ii), растворяют в апротонном растворителе, таком как, но не ограничивающимся им, ДХМ, гептан, гексан или толуол и интенсивно промывают водой для удаления остаточного количества основания. Содержание воды ограничено <0,5% (титрование по Карлу Фишеру) путем совместной перегонки с безводным апротонным растворителем, таким как ДХМ, гексан, гептан, толуол или ТГФ.
Стадия 2(b)(iii), превращение соединения 9 в соединение 10:
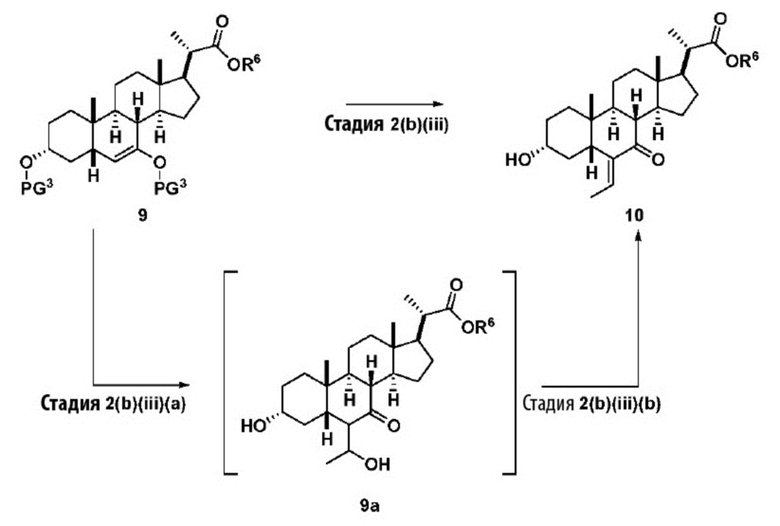
Стадия 2(b)(iii) представляет собой альдольную реакцию соединения 9 с ацетальдегидом с получением промежуточного соединения 9а с последующим образованием соединения 10 в присутствии кислоты Льюиса, такой как, но не ограничивающейся ею, BF3.Et2O или Ti(OiPr)4. В одном аспекте стадии 2(b)(iii) кислота Льюиса представляет собой BF3.Et2O. Реакцию предпочтительно проводят в апротонном растворителе, таком как, но не ограничивающимся им, ДХМ. Температура реакции составляет предпочтительно от около -78°C до 25 ºC. В одном аспекте температура реакции составляет от около -78°C до около -50°C. В другом предпочтительном аспекте температура реакции составляет около -60°C.
Следуя реакции соединения 9 с ацетальдегидом при около -78°C до около -50°C (стадия 2(b)(iii)(a)), соединение альдольного продукта 9а первоначально образуется в качестве основного продукта. Затем к реакционной смеси добавляют метанол, чтобы погасить реакцию и облегчить удаление с образованием олефинового соединения 10 (стадия 2(b)(iii)(b). В альтернативном варианте реакции оставляют протекать при более высокой температуре, такой как от -10°C до комнатной температуры, без добавления метанола для облегчения образования олефинов с получением соединения 10.
В одном аспекте стадии 2(b)(iii) соединение 10 представляет собой смесь E- и Z- олефиновых изомеров, проиллюстрированных структурами соединения 10A ниже. Соотношение E/Z может составлять от 1/1 до >9/1.
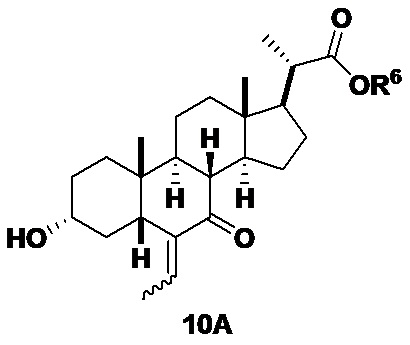
Соединение 10 может быть очищено с помощью колоночной хроматографии с получением соединения 10 с чистотой более чем 95%. В предпочтительном аспекте соединение 10 может быть использовано непосредственно на стадии 2(b)(iv) без очистки.
В одном аспекте стадии 2(b)(iii) E-изомер соединения 10 получают в качестве превалирующего изомера (E-изомера 10 более чем 80% и Z-изомера менее чем 20%). В другом аспекте E-изомера более чем 90% и Z-изомера менее чем 10%. В другом аспекте E-изомера более чем 95% и Z-изомера менее чем 5%.
В одном аспекте стадии 2(b)(iii) неочищенный продукт 10 содержит менее чем 5% кетона соединения 8. В другом аспекте неочищенный продукт 10 содержит менее чем 3% кетона соединения 8. В другом аспекте неочищенный продукт 10 содержит менее чем 2% кетона соединения 8.
Стадия 2(b)(iv), превращение соединения 10 в соединение 11:
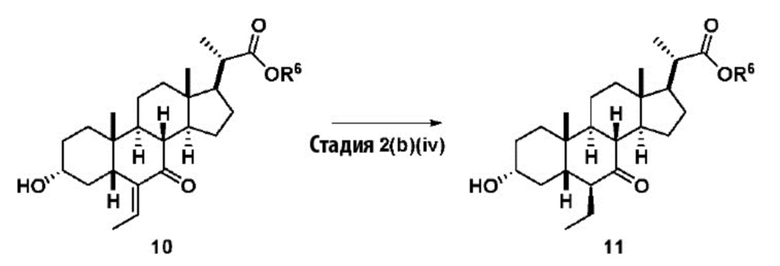
На Стадии 2(b)(iv), соединение 10 со Стадии 2(b)(iii) превращают в соединение 11 путем каталитического гидрирования для восстановления олефина. В одном аспекте стадии 2(b)(iv) соединение 10 со Стадии 2(b)(iii) очистили колоночной хроматографией. В предпочтительном аспекте Стадии 2(b)(iv) неочищенный продукт 10, полученный после обработки Стадии 2(b)(iii), используют непосредственно без очистки. В одном аспекте стадии 2(b)(iv) неочищенный продукт 10 содержал как E-, так и Z олефиновые изомеры (10A). Процентное содержание Z-изомера предпочтительно находится в диапазоне от 0% до 50%.
Каталитическое гидрирование проводят в присутствии катализатора, такого как, но не ограничивающегося им, палладий на угле (Pd/C), Pd(OAc)2, Pd(OH)2 и PtO2. Предпочтительный катализатор представляет собой Pd/C. Содержание палладия в указанном Pd/C может находиться в диапазоне от около 5% до около 10%. Количество катализатора может находиться в диапазоне от около 1 % моль до около 10 % моль. Источником водорода может быть, помимо прочего, газообразный водород и формиат аммония. Давление газообразного водорода находится в диапазоне от атмосферного давления до около 500 фунт. на кв. дюйм. В одном аспекте стадии 2(b)(iv) давление газообразного водорода равно атмосферному давлению. В одном аспекте давление газообразного водорода равно от около 50 до около 150 фунт. на кв. дюйм. Температура реакции предпочтительно находится в диапазоне от около 5°C до около 120°C. В одном аспекте температура реакции составляет от около 5°C до около 80°C. В одном аспекте стадии 2(b)(iv) температура реакции составляет от около 20°C до около 50°C. В одном аспекте температура реакции составляет около 25°C. Реакция может быть проведена в протонном или апротонном растворителе или смеси двух растворителей. Подходящие растворители включают, но не ограничиваются ими, метанол, этанол, изопропанол, трет-бутанол и ТГФ. В одном аспекте стадии 2(b)(iv) растворитель представляет собой смесь метанола и ТГФ. В еще одном аспекте стадии 2(b)(iv) смесь этанола и ТГФ используют в качестве растворителя.
В определенных вариантах осуществления изобретения соединение 11 получают в виде смеси 6α-этилового изомера и 6β-этилового изомера. В определенных вариантах осуществления изобретения 6β-этил изомер представляет собой превалирующий изомер в продукте. В одном аспекте стадии 2(b)(iv) неочищенное соединение 11 содержит менее чем 20% 6α-этилового изомера. В одном аспекте стадии 2(b)(iv) неочищенное соединение 11 содержит менее чем 10% 6α-этилового изомера. В одном аспекте стадии 2(b)(iv) неочищенное соединение 11 содержит менее чем 5% 6α-этилового изомера. Соединение 11 может быть использовано непосредственно на стадии 2(b)(v) без очистки.
Хотя соединение 11 проиллюстрировано выше в виде 6β-этилового изомера, в вариантах осуществления изобретения, в которых соединение представляет собой смесь 6-альфа и 6-бета-этиловые изомеры, оно может быть представлено в виде соединения 11A ниже.
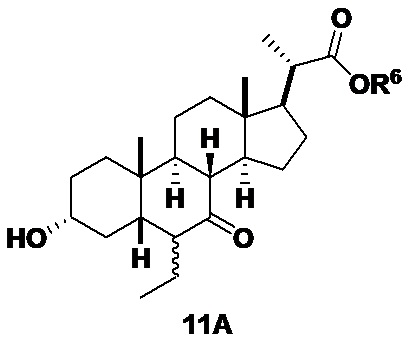
Стадия 2(b)(v), превращение соединения 11 в соединение 12:
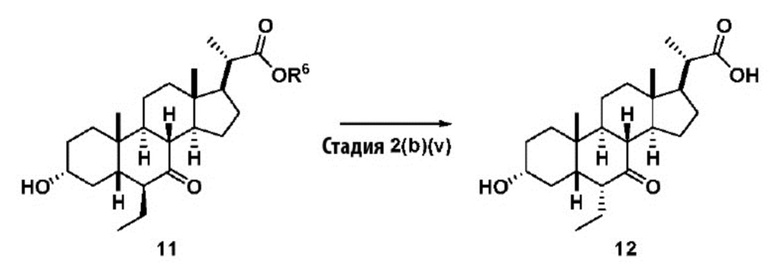
Стадия 2(b)(v) представляет собой эпимеризацию 6β-этилового изомера соединения 11 в 6α-этиловый изомер, соединение 12, в основных условиях. В одном аспекте стадии 2(b)(v) неочищенный продукт, полученный на Стадии 2(b)(iv), содержал как 6β-этил изомер, так и 6α-изомер, используют на Стадии 2(b)(vi) без дополнительной очистки.
Основание может представлять собой, но не ограничиваясь им, гидроксид натрия или гидроксид калия. В одном аспекте основание представляет собой водный раствор гидроксида натрия. В одном аспекте стадии 2(b)(vi), основание представляет собой 50% раствор гидроксида натрия в воде.
В одном аспекте стадии 2(b)(v) неочищенный продукт Стадии 2(b)(iv) непосредственно используют на Стадии 2(b)(v) после удаления катализатора, такого как Pd/C, путем фильтрации. В одном аспекте стадии 2(b)(v) неочищенный продукт 11 используют после удаления катализатора и растворителя.
Стадию 2(b)(v) предпочтительно проводят в протонном растворителе, таком как, но не ограничивающимся им, метанол или этанол, или смеси протонного и непротонного растворителя, такой как, но не ограничивающейся ею, смесь метанола или этанола и ТГФ.
В одном аспекте стадии 2(b(v) растворитель представляет собой этанол. В другом аспекте стадии 2(b)(v) растворитель представляет собой метанол. В другом аспекте стадии 2(b)(v) растворитель представляет собой смесь этанола и ТГФ. В другом аспекте стадии 2(b)(v) растворитель представляет собой смесь метанола и ТГФ. Соединение 12 может быть использовано непосредственно на стадии 2(b)(vi) без очистки.
Стадия 2(b)(vi), превращение соединения 12 в соединение (V):
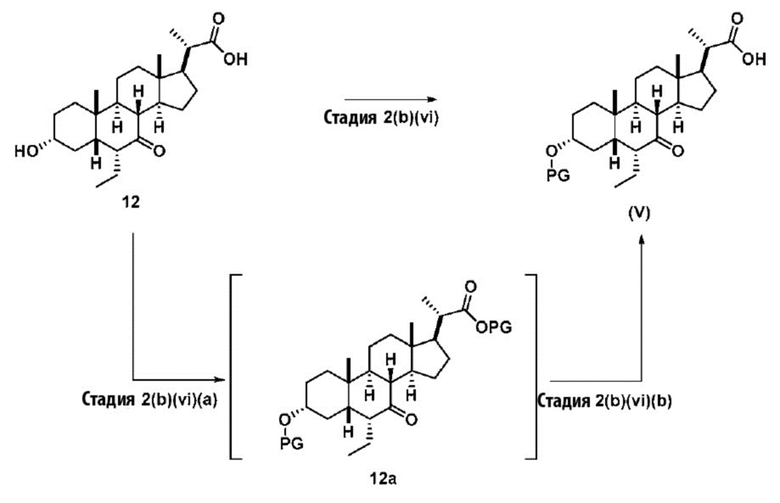
Стадия 2(b)(vi) представляет собой защиту 3-гидроксила соединения 12 подходящим гидроксил-защитным агентом PG-X, где X представляет собой подходящую уходящую группу, например Cl, Br, I или OTf, в присутствии органического основания, такого как, но не ограничивающегося им, имидазол, TEA, DIPEA с получением промежуточного соединения 12а, с последующим снятием защиты с карбоновой кислоты подходящим основанием в протонном растворителе для получения соединения (V). Предпочтительный гидроксил-защитный агент представляет собой TBS-Cl. Предпочтительное органическое основание представляет собой имидазол. Предпочтительное основание представляет собой K2CO3. Предпочтительный протонный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Соединение (V) может быть перекристаллизовано из органического растворителя или смеси органических растворителей, такой как, но не ограничивающейся ею, смесь гексанов/CH2Cl2 с получением соединения (V) с чистотой более чем 95%.
В одном варианте осуществления изобретения Стадию 3(b) проводят, как изложено на Схеме 4. Способ включает стадии 3(b)(i): введение соединения (V) в реакцию с подходящим ацилирующим реагентом с получением соединения 13; а также 3(b)(ii): восстановление соединения 13 с получением соединения (VI).
Схема 4
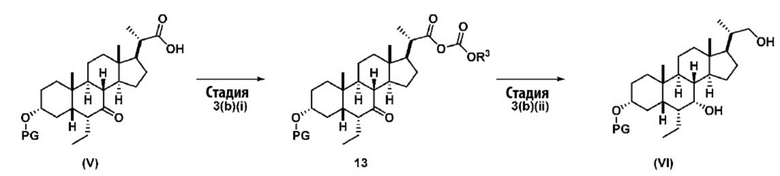
где R3 соответствует определениям, описанным выше.
Способ по данной заявки никогда не сообщался в данной области. Синтез соединения (VI) из обетихоловой кислоты в 6 стадий описан в заявке США 2016/0289262. Синтез обетихоловой кислоты был опубликован в US 2013/0345188, исходя из KLCA, в 6 стадий. В целом, известный способ получения соединения спирта (VI) включал 12-стадийный синтез из KLCA. Этот предыдущий способ включал в себя стадии с низкими выходами и требовал нескольких стадий очистки хроматографией на колонке, что является дорогостоящим и не подходит для коммерциализации в крупном масштабе. Дополнительно было использовано несколько токсичных и опасных реагентов. Способ получения соединения (V) по данному изобретению состоит из шести стадий, исходя из соединения (III), с хорошим общим выходом и требует только одной очистки хроматографией на колонке. Ключевые промежуточные соединения, такие как соединение 12, могут быть получены путем кристаллизации с высокой степенью чистоты. Также соединение (V) может быть получено путем кристаллизации с высокой степенью чистоты. Соединение (V) может быть превращено в соединение (VI) путем образования смешанного ангидрида с последующим восстановлением.
Стадия 3(b)(i), превращение соединения (V) в соединение 13:
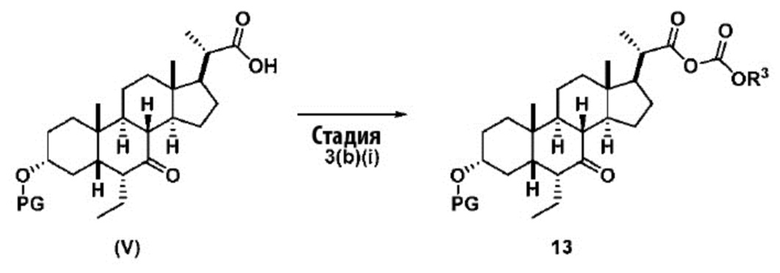
Стадия 3(b)(i) представляет собой реакцию соединения (V) с подходящим хлорформиатом R3OCOCl в присутствии основания, такого как, но не ограничивающегося им, TEA или DIPEA для получения смешанного ангидрида 13. Стадию 3(b)(i) проводят в апротонном растворителе, таком как, но не ограничивающимся им, ДХМ. Предпочтительный хлорформиат представляет собой изобутилхлорформиат, где R3 представляет собой изобутил. Предпочтительное органическое основание представляет собой TEA. Соединение 13 выделяют в неочищенном виде и используют без дальнейшей очистки.
Стадия 3(b)(ii), превращение соединения 13 в соединение (VI):
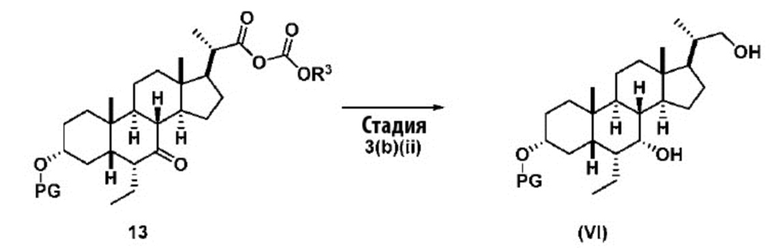
Стадия 3(b)(ii) представляет собой реакцию соединения 13 с подходящим восстанавливающим агентом, таким как, но не ограничивающимся им, NaBH4, LiBH4, LiAlH4 или DIBAL с получением соединения (VI). Стадию 3(b)(ii) предпочтительно проводят в смеси протонного и непротонного растворителя, такой как, но не ограничивающейся ею, смесь воды и ТГФ. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Соединение VI может быть очищено с помощью колоночной хроматографии с получением соединения VI с чистотой более чем 95%.
Способ получения соединения формулы (I)
Данное изобретение также включает способ получения соединения формулы (I), исходя из соединения (IV), проиллюстрированного на Схеме 5.
Схема 5
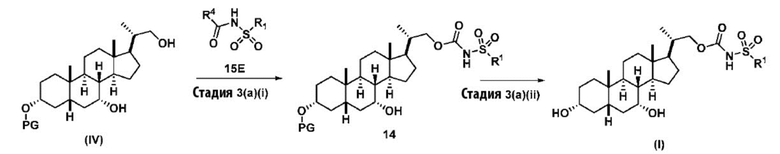
где R4 представляет собой имидазол-1-ил, алкил-O-, арил-O, Cl или CCl3; R1 соответствует определениям, описанным выше.
Стадия 3(a)(i), превращение соединения (IV) в соединение формулы 14:
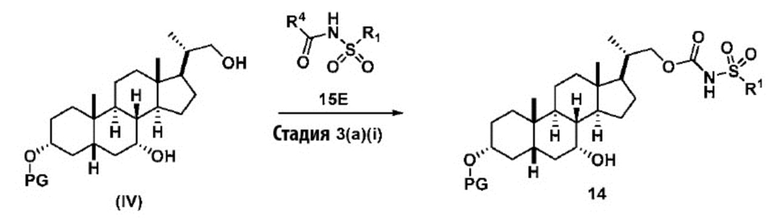
Стадия 3(a)(i) включает превращение соединения (IV) в соединение формулы 14, где R1 соответствует определениям, описанным выше, путем введения в реакцию соединения (IV) с соединением, представленным 15E, 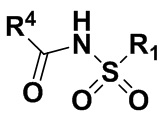 , где R4 представляет собой имидазол-1-ил, алкил-O-, арил-O, Cl или CCl3, и R1 соответствует определениям, описанным выше, в присутствии органического основания. Реакцию предпочтительно проводят в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин и диизопропилэтиламин. DMAP, в диапазоне от 1 % моль до 50 % моль, может быть добавлен для облегчения протекания реакции. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при около 50°C.
, где R4 представляет собой имидазол-1-ил, алкил-O-, арил-O, Cl или CCl3, и R1 соответствует определениям, описанным выше, в присутствии органического основания. Реакцию предпочтительно проводят в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин и диизопропилэтиламин. DMAP, в диапазоне от 1 % моль до 50 % моль, может быть добавлен для облегчения протекания реакции. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при около 50°C.
Предпочтительно, R4 в соединении 15E представляет собой имидазол-1-ил, MeO-, EtO- или PhO-. Более предпочтительно R4 представляет собой PhO-.
Стадия 3(a)(ii) превращение соединения формулы 14 в соединение формулы (I):
Стадия 3(a)(ii) представляет собой снятие защитной группы PG из соединения 14 с образованием соединения (I), где R1 соответствует определениям, описанным выше. Защитная группа PG может быть удалена в подходящих условиях снятия защиты, которые известны в данной области техники. Предпочтительная защитная группа PG представляет собой TBS. Предпочтительно защитную группу удаляют с помощью реагента для снятия защиты, такого как TBAF, или кислоты, такой как HCl. Предпочтительно, соединение 14 обрабатывают кислотой в протонном растворителе. Предпочтительно, соединение 14 обрабатывают кислотой, такой как HCl, в протонном растворителе, таком как, но не ограничивающемся им, MeOH, EtOH, i-PrOH, H2O или смесь их двух. Предпочтительный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Способ получения соединения (VII)
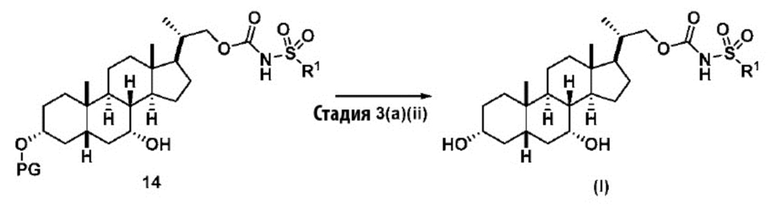
Способ по данному изобретению также включает способ получения соединения (VII), исходя из соединения (IV), следуя способу, описанному на Схеме 6.
Схема 6
 ,
,
Способ включает превращения соединения спирта(IV) в сульфонилкарбамат 16 с подходящим реагентом, таким как реагент, выбранный из 15А, 15В, 15С и 15D, в присутствии органического основания.
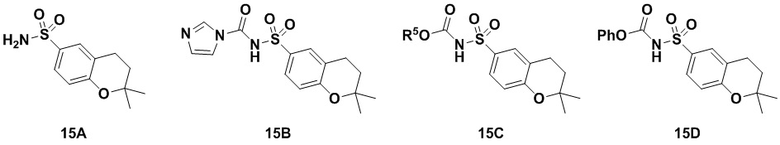
В одном аспекте реагент представляет собой 15B, который может быть образован в результате реакции сульфонамидного соединения 15A с CDI.
В другом аспекте реагент представляет собой соединение сульфонилкарбамата 15C, где R5 представляет собой алкил или арил, предпочтительно метил, этил или фенил. Предпочтительный реагент представляет собой 15D.
В одном аспекте соединение спирта (IV) вступает в реакцию 15D в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин, диизопропилэтиламин. DMAP, в диапазоне от 1 % моль до 50 % моль, может быть добавлен для облегчения протекания реакции. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при 50°C. Соединение 16 может быть очищено с помощью колоночной хроматографии с получением соединения 16 с чистотой более чем 95%.
В одном аспекте соединение 16 вводят в реакцию с реагентом для снятия защитной группы, таким как, но не ограничивающимся им, TBAF, или кислотой, такой как HCl. Предпочтительно, соединение 16 обрабатывают кислотой в протонном растворителе. Предпочтительно, соединение 16 обрабатывают кислотой, такой как HCl, в протонном растворителе, таком как, но не ограничивающимся им, MeOH, EtOH, i-PrOH, H2O или смесь их двух. Предпочтительный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C. Соединение (VII) может быть очищено с помощью колоночной хроматографии с получением соединения (VII) с чистотой более чем 95%.
Способ получения соединения формулы (II)
Данное изобретение также включает способ получения соединения формулы (II), исходя из соединения (VI), как проиллюстрировано на Схеме 7.
Схема 7
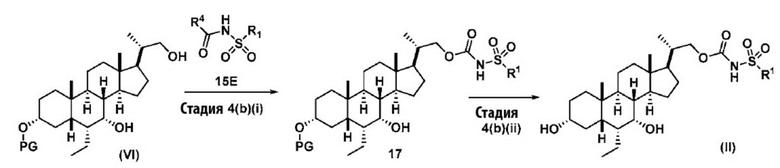
Стадия 4(b)(i), превращение соединения (VI) в соединение формулы 17:
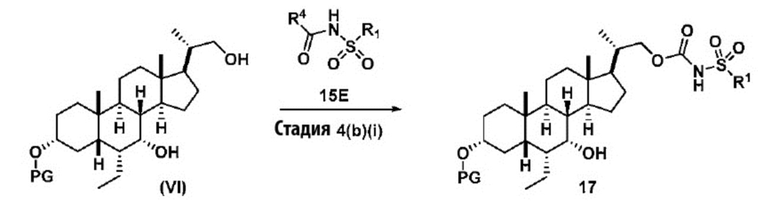
где R1 и R4 соответствуют определениям, указанным ранее. Предпочтительно, R4 в соединении 15E представляет собой имидазол-1-ил, MeO-, EtO- или PhO-. Более предпочтительно, R4 представляет собой PhO-.
Реакцию предпочтительно проводят в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин, диизопропилэтиламин и DMAP. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при около 50°C.
Стадия 4(b)(ii), превращение соединения формулы 17 в соединение формулы (II):

Стадия 4(b)(ii) представляет собой снятие защитной группы PG из соединения формулы 17 для образования соединения (II), где PG, и R1 соответствует определениям, описанным выше. Защитная группа PG может быть удалена в подходящих условиях снятия защиты, известных в данной области техники. Предпочтительная защитная группа PG представляет собой TBS. Предпочтительно защитную группу удаляют с помощью реагента для снятия защиты, такого как TBAF, или кислоты, такой как HCl. Предпочтительно, соединение формулы 17 обрабатывают кислотой в протонном растворителе. Предпочтительно, соединение формулы 17 обрабатывают кислотой, такой как HCl, в протонном растворителе, таком как, но не ограничивающимся им, MeOH, EtOH, i-PrOH, H2O или смесь их двух. Предпочтительный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C.
Способ получения соединения (VIII)
Способ по данному изобретению также включает способ получения соединения (VIII), исходя из соединения (VI), следуя способу, описанному на Схеме 8.
Схема 8
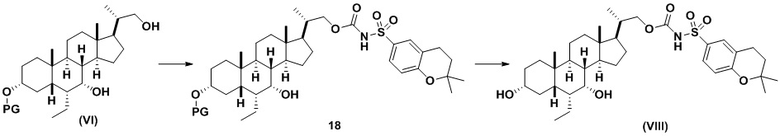
Данный способ включает превращение соединения спирта (VI) в соединение сульфонилкарбамата 18, с последующим снятием защиты с получением соединения (VIII). Условия для способа, описанного для Схемы 8, являются такими же, как были определены ранее для Схемы 7.
Способ включает превращение соединения спирта (VI) в сульфонилкарбамат 18 с подходящим реагентом (например, выбранным из 15A-15D) в присутствии органического основания.
В одном аспекте реагент представляет собой 15B, который может быть образован в результате реакции сульфонамидного соединения 15A с CDI.
В другом аспекте реагент может представлять собой соединение сульфонилкарбамата 15C, где R5 представляет собой алкил или арил, предпочтительно метил, этил или фенил. Предпочтительный реагент представляет собой 15D.
В одном аспекте соединения спирта (VI) вводят в реакцию с 15D в апротонном растворителе, таком как, но не ограничивающимся ими, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин и диизопропилэтиламин. DMAP, в диапазоне от 1 % моль до 50 % моль, может быть добавлен для облегчения протекания реакции. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при 50°C. Соединение 18 может быть очищено с помощью колоночной хроматографии с получением соединения 18 с чистотой более чем 95%.
В одном аспекте соединение 18 вводят в реакцию с реагентом для снятия защитной группы, таким как, но не ограничивающимся им, TBAF, или кислотой, такой как HCl. Предпочтительно, соединение 18 обрабатывают кислотой в протонном растворителе. Предпочтительно, соединение 18 обрабатывают кислотой, такой как HCl, в протонном растворителе, таком как, но не ограничивающимся им, MeOH, EtOH, i-PrOH, H2O или смесь их двух. Предпочтительный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C. Соединение (VIII) может быть очищено с помощью колоночной хроматографии с получением соединения (VIII) с чистотой более чем 95%.
Способ получения соединения (IX)
Способ по данному изобретению также включает способ получения соединения (IX), исходя из соединения (VI), следуя способу, описанному на Схеме 9.
Схема 9
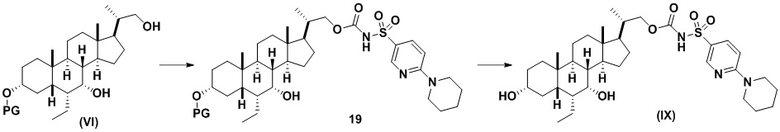
Способ включает превращение спирта в соединение (VI) или соединения (VIb) для соединения сульфонилкарбамата (19) с подходящим реагентом, например одним из соединений 20А, 20В, 20С или 20D, в присутствии органического основания.
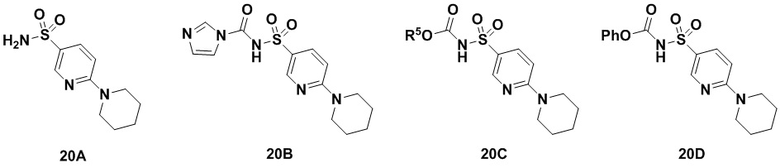
В одном аспекте реагент представляет собой 20B, который может быть образован в результате реакции сульфонамидного соединения 20A с CDI.
В другом аспекте реагент может представлять собой соединение сульфонилкарбамата 20C, где R5 представляет собой алкил или арил, предпочтительно метил, этил или фенил. Предпочтительный реагент представляет собой 20D.
В одном аспекте соединения спирта (VI) вводят в реакцию с 20D в апротонном растворителе, таком как, но не ограничивающимся им, ТГФ, ДХМ или толуол. В одном предпочтительном аспекте реакционный растворитель представляет собой ТГФ. Подходящие органические основания включают, но не ограничиваются ими, триэтиламин, диизопропилэтиламин и DMAP. Температура реакции предпочтительно находится в диапазоне от около 0°C до около 80°C. В одном аспекте реакцию проводят при около 0°C. В другом аспекте реакцию проводят при около комнатной температуре (около 25°C). В еще одном аспекте реакцию проводят при 50°C. Соединение 19 может быть очищено с помощью колоночной хроматографии с получением соединения 19 с чистотой более чем 95%.
В одном аспекте соединение 19 вводят в реакцию с реагентом для снятия защитной группы, таким как, но не ограничивающимся им, TBAF, или кислотой, такой как HCl. Предпочтительно, соединение 19 обрабатывают кислотой в протонном растворителе. Предпочтительно, соединение 19 обрабатывают кислотой, такой как HCl, в протонном растворителе, таком как, но не ограничивающимся им, MeOH, EtOH, i-PrOH, H2O или смеси их двух. Предпочтительный растворитель представляет собой MeOH. Реакцию могут проводить при температуре в диапазоне от -10°C до 50°C. В предпочтительном аспекте температура реакции составляет от 0°C до 30°C. В другом аспекте температура реакции составляет около 25°C. Соединение (IX) может быть очищено с помощью колоночной хроматографии с получением соединения (IX) с чистотой более чем 95%.
В другом варианте осуществления изобретения данное изобретение также включает способ получения соединения формулы (II), исходя из соединения (VIb), как проиллюстрировано на Схеме 10, и получение соединения (VIb) является таким, как проиллюстрировано на Схеме 11.
Схема 10
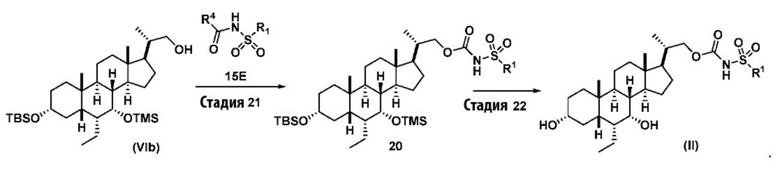 .
.
Схема 11
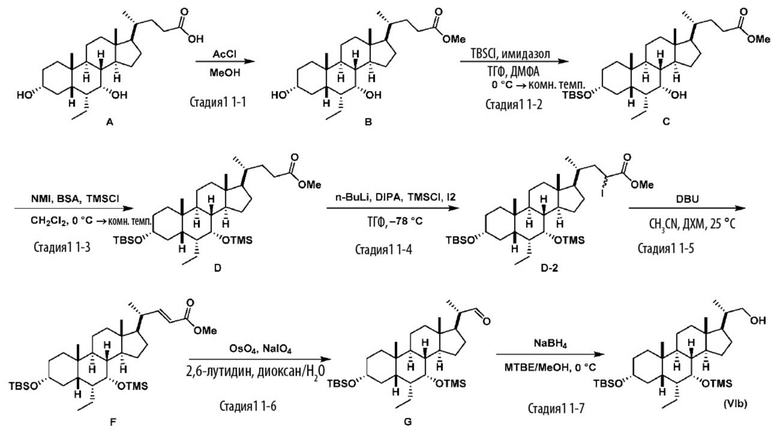 .
.
Определения
Ниже перечислены определения различных терминов, использующихся для описания настоящего изобретения. Данные определения относятся только к терминам, использующимся в настоящей заявке и формуле изобретения, если специально не указано иное, индивидуально или как часть более крупной группы.
Термин "алкил”, в контексте данного документа, относится к насыщенной одновалентной углеводородной группе с прямой или разветвленной цепью. Предпочтительные алкильные радикалы включают C1-C6 алкильные и C1-C8 алкильные радикалы. Примеры C1-C6 алкильных групп включают, но не ограничиваются ими, метильную, этильную, пропильную, изопропильную, н-бутильную, трет-бутильную, неопентильную, н-гексильную группы; и примеры C1-C8 алкильных групп включают, но не ограничиваются ими, метильную, этильную, пропильную, изопропильную, н-бутильную, трет-бутильную, неопентильную, н-гексильную, гептильную и октильную группы.
Термин “алкенил”, в контексте данного документа, обозначает одновалентную группу, полученную из углеводородного фрагмента путем удаления одного атома водорода, причем углеводородный фрагмент имеет по меньшей мере одну двойную углерод-углеродную связь. Предпочтительные алкенильные группы включают C2-C6 алкенильные и C2-C8 алкенильные группы. Алкенильные группы включают, но не ограничиваются ими, например, этенил, пропенил, бутенил, 1-метил-2-бутен-1-ил, гептенил, октенил и тому подобное.
Термин “алкинил”, в контексте данного документа, обозначает одновалентную группу, полученную из углеводородного фрагмента путем удаления одного атома водорода, причем углеводородный фрагмент имеет по меньшей мере одну тройную углерод-углеродную связь. Предпочтительные алкинильные группы включают C2-C6 алкинильные и C2-C8 алкинильные группы. Типичные алкинильные группы включают, но не ограничиваются ими, например, этинил, 1-пропинил, 1-бутинил, гептинил, октинил и тому подобное.
Термин “циклоалкил”, в контексте данного документа, относится к моноциклическому или полициклическому насыщенному карбоциклическому кольцу или би- или трициклической группе, конденсированной, мостиковой или спиро-системе, а атомы углерода могут быть необязательно оксозамещенными или необязательно замещенными экзоциклической олефиновой двойной связью. Предпочтительные циклоалкильные группы включают C3-C8 циклоалкильные и C3-C12 циклоалкильные группы. Примеры C3-C8-циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклопентил и циклооктил; а также примеры C3-C12-циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклооктил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил, спиро[2.5]октил, 3-метиленбицикло[3.2.1]октил, спиро[4.4]нонанил, бицикл[3.1.0]гексанил, спиро[2.3]гексанил, бицикл[3.1.1]гептанил, спиро[2.5]октанил, бицикл[4.1.0]гептанил, бицикл[3.1.0]гексан-6-ил, спиро[2.3]гексан-5-ил, бицикл[3.1.1]гептан-3-ил, спиро[2.5]октан-4-ил и бицикл[4.1.0]гептан-3-ил и тому подобное.
Термин “циклоалкенил”, в контексте данного документа, относится к моноциклическому или полициклическому карбоциклическому кольцу или би- или трициклической группе, конденсированной, мостиковой или спиро-системе, имеющей по меньшей мере одну двойную углерод-углеродную связь, а атомы углерода могут быть необязательно оксозамещенными или необязательно замещенными экзоциклической олефиновой двойной связью. Предпочтительные циклоалкенильные группы включают C3-C8 циклоалкенильные и C3-C12 циклоалкенильные группы. Примеры C3-C8-циклоалкенил включают, но не ограничиваются ими, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил и тому подобное; и примеры C3-C12-циклоалкенила включают, но не ограничиваются ими, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил, бицикло[2.2.1]гепт-2-енил, бицикло[3.1.0]гекс-2-енил, спиро[2.5]окт-4-енил, спиро[4.4]нон-1-енил, бицикло[4.2.1]нон-3-ен-9-ил и тому подобное.
Термин «гетероциклический» или «гетероциклоалкил» может использоваться взаимозаменяемо и относиться к неароматическому кольцу или би- или трициклической группе с конденсированной, мостиковой или спиро-системой, где (i) каждая кольцевая система содержит по меньшей мере один гетероатом, независимо выбранный от кислорода, серы и азота, (ii) каждая кольцевая система может быть насыщенной или ненасыщенной (iii) гетероатомы азота и серы могут быть необязательно окислены, (iv) гетероатом азота может быть необязательно кватернизован, (v) любое из вышеуказанных колец может быть конденсировано с ароматическим кольцом и (vi) остальные атомы кольца представляют собой атомы углерода, которые могут быть необязательно оксозамещенными или необязательно замещенными экзоциклической олефиновой двойной связью. Типичные гетероциклоалкильные группы включают, но не ограничиваются ими, [1.3]диоксолан, пирролидинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, пиперидинил, пиперазинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил, хиноксалинил, пиридазинонил, тетрагидрофурил, 2-азабицикло[2.2.1]гептил, 8-азабицикло[3.2.1]октил 5-азаспиро[2.5]октил, 1-окса-7-азаспиро [4.4]нонанил, 7-оксооксепан-4-ил и тетрагидрофурил. Такие гетероциклические группы могут быть дополнительно замещены. Гетероарильные или гетероциклические группы могут быть C-присоединенными или N-присоединенными (где это возможно).
Термин «арил», в контексте данного документа, относится к моно- или полициклической карбоциклической кольцевой системе, содержащей по меньшей мере одно ароматическое кольцо, включая, но не ограничиваясь этим, фенил, нафтил, тетрагидронафтил, инданил и инденил. Полициклический арил представляет собой полициклическую кольцевую систему, которая содержит по меньшей мере одно ароматическое кольцо. Полициклические арилы могут содержать конденсированные кольца, ковалентно присоединенные кольца или их комбинацию.
Термин «арилалкил», в контексте данного документа, относится к функциональной группе, в которой алкиленовая цепь присоединена к арильной группе, например, -CH2CH2-фенилу. Термин «замещенный арилалкил» означает функциональную арилалкильную группу, в которой арильная группа замещена. Примеры включают, но не ограничиваются ими, бензил, фенэтил и тому подобное. Предпочтительные арилалкильные группы включают арил-C1-C8-алкильные группы.
Термин «гетероарил», в контексте данного документа, относится к моно-, би- или трициклической группе, содержащей по меньшей мере одно 5- или 6-членное ароматическое кольцо, содержащее по меньшей мере один кольцевой атом, выбранный из S, O и N. Предпочтительные гетероарильные группы являются моноциклическими или бициклическими. Гетероарильные группы включают моноциклические группы, имеющие 5 или 6 кольцевых атомов, и конденсированные бициклические группы, содержащие от 8 до 10 кольцевых атомов. Гетероарильные группы включают, но не ограничиваются ими, пиридинил, пиразолил, пиразинил, пиримидинил, пирролил, пиразолил, имидазолил, тиазолил, тиенил, триазолил, изотиазолил, оксазолил, изоксазолил, тиадиазолил, оксадиазолил, тиофенил, фуранил, хинолинил, изохинолинил, бензимидазолил, бензоксазолил, бензотиенил, хиноксалил, индолил, индазолил, бензизоксазолил, бензофуранил, бензотриазолил, бензотиазолил и тому подобное.
Термин «гетероарилалкил», в контексте данного документа, относится к алкиленовой цепи, присоединенной к гетероарильной группе. Термин «замещенный гетероарилалкил» означает гетероарилалкильную функциональную группу, в которой гетероарильная группа замещена. Примеры включают, но не ограничиваются ими, пиридинилметил, пиримидинилэтил и тому подобное. Предпочтительные гетероарилалкильные группы включают гетероарил-C1-C8-алкильные группы.
Термин «биарил», в контексте данного документа, относится к группе, состоящей из двух арильных групп, двух гетероарильных групп или арильной группы и гетероарильной группы, причем две группы соединены одинарной связью. Замещенная биарильная группа представляет собой биарильную группу, в которой по меньшей мере одна из связанных групп имеет по меньшей мере один неводородный заместитель. Примеры биарильных групп включают бифенильную, пиридилфенильную, пиримидилфенильную, пиримидипиридильную и пиримидилоксадизолильную группы.
Термин «арилгетероциклил» относится к бициклической группе, включающей моноциклический арил или гетероарильную группу, связанную с гетероциклической группой одинарной связью. Примеры арилгетероциклильных групп включают фенилпиперидинильную и пиридилпиперидинильную группы.
В контексте данного документа термин «алкокси», используемый отдельно или в сочетании с другими терминами, означает, если не указано иное, алкильную группу, имеющую указанное число атомов углерода, связанных с остальной частью молекулы через атом кислорода, такой как, например, метокси, этокси, 1-пропокси, 2-пропокси (изопропокси) и высшие гомологи и изомеры. Предпочтительные алкокси представляют собой (C1-C3) алкокси.
Термин «замещенный» относится к замещению путем независимой замены одного, двух или трех или более атомов водорода заместителями, включая, но не ограничиваясь ими, -F, -Cl, -Br, -I, -OH, C1-C12-алкил; C2-C12-алкенил, C2-C12-алкинил, замещенный гидрокси, -NO2, -N3, -CN, -NH2, замещенный амино, оксо, тиоксо, -NH-C1-C12-алкил, -NH-C2-C8-алкенил, -NH-C2-C8-алкинил, -NH-C3-C12-циклоалкил, -NH-арил, -NH-гетероарил, -NH-гетероциклоалкил, -диалкиламино, -диариламино, -дигетероариламино, -O-C1-C12-алкил, -O-C2-C8-алкенил, -O-C2-C8-алкинил, -O-C3-C12-циклоалкил, -O-арил, -O-гетероарил, -O-гетероциклоалкил, -C(O)-C1-C12-алкил, -C(O)-C2-C8-алкенил, -C(O)-C2-C8-алкинил, -C(O)-C3-C12-циклоалкил, -C(O)-арил, -C(O)-гетероарил, -C(O)-гетероциклоалкил, -CONH2, -CONH-C1-C12-алкил, -CONH-C2-C8-алкенил, -CONH-C2-C8-алкинил, -CONH-C3-C12-циклоалкил, -CONH-арил, -CONH-гетероарил, -CONH-гетероциклоалкил, -OCO2-C1-C12-алкил, -OCO2-C2-C8-алкенил, -OCO2-C2-C8-алкинил, -OCO2-C3-C12-циклоалкил, -OCO2-арил, -OCO2-гетероарил, -OCO2-гетероциклоалкил, -CO2-C1-C12 алкил, -CO2-C2-C8 алкенил, -CO2-C2-C8 алкинил, CO2-C3-C12-циклоалкил, -CO2- арил, CO2-гетероарил, CO2-гетероциклоалкил, -OCONH2, -OCONH-C1-C12-алкил, -OCONH-C2-C8-алкенил, -OCONH-C2-C8-алкинил, -OCONH-C3-C12-циклоалкил, -OCONH-арил, -OCONH-гетероарил, -OCONH- гетероцикло-алкил, -NHC(O)H, -NHC(O)-C1-C12-алкил, -NHC(O)-C2-C8-алкенил, -NHC(O)-C2-C8-алкинил, -NHC(O)-C3-C12-циклоалкил, -NHC(O)-арил, -NHC(O)-гетероарил, -NHC(O)-гетероцикло-алкил, -NHCO2-C1-C12-алкил, -NHCO2-C2-C8-алкенил, -NHCO2- C2-C8-алкинил, -NHCO2-C3-C12-циклоалкил, -NHCO2-арил, -NHCO2-гетероарил, -NHCO2- гетероциклоалкил, -NHC(O)NH2, -NHC(O)NH-C1-C12-алкил, -NHC(O)NH-C2-C8-алкенил, -NHC(O)NH-C2-C8-алкинил, -NHC(O)NH-C3-C12-циклоалкил, -NHC(O)NH-арил, -NHC(O)NH-гетероарил, -NHC(O)NH-гетероциклоалкил, NHC(S)NH2, -NHC(S)NH-C1-C12-алкил, -NHC(S)NH-C2-C8-алкенил, -NHC(S)NH-C2-C8-алкинил, -NHC(S)NH-C3-C12-циклоалкил, -NHC(S)NH-арил, -NHC(S)NH-гетероарил, -NHC(S)NH-гетероциклоалкил, -NHC(NH)NH2, -NHC(NH)NH-C1-C12-алкил, -NHC(NH)NH-C2-C8-алкенил, -NHC(NH)NH-C2-C8-алкинил, -NHC(NH)NH-C3-C12-циклоалкил, -NHC(NH)NH-арил, -NHC(NH)NH-гетероарил, -NHC(NH)NH-гетероциклоалкил, -NHC(NH)-C1-C12-алкил, -NHC(NH)-C2-C8-алкенил, -NHC(NH)-C2-C8-алкинил, -NHC(NH)-C3-C12-циклоалкил, -NHC(NH)-арил, -NHC(NH)-гетероарил, -NHC(NH)-гетероциклоалкил, -C(NH)NH-C1-C12-алкил, -C(NH)NH-C2-C8-алкенил, -C(NH)NH-C2-C8-алкинил, -C(NH)NH-C3-C12-циклоалкил, -C(NH)NH-арил, -C(NH)NH-гетероарил, -C(NH)NH-гетероциклоалкил, -S(O)-C1-C12-алкил, -S(O)-C2-C8-алкенил, - S(O)-C2-C8-алкинил, -S(O)-C3-C12-циклоалкил, -S(O)-арил, -S(O)-гетероарил, -S(O)-гетероциклоалкил, -SO2NH2, -SO2NH-C1-C12-алкил, -SO2NH-C2-C8-алкенил, -SO2NH- C2-C8-алкинил, -SO2NH-C3-C12-циклоалкил, -SO2NH-арил, -SO2NH-гетероарил, -SO2NH- гетероциклоалкил, -NHSO2-C1-C12-алкил, -NHSO2-C2-C8-алкенил, - NHSO2-C2-C8-алкинил, -NHSO2-C3-C12-циклоалкил, -NHSO2-арил, -NHSO2-гетероарил, -NHSO2-гетероциклоалкил, -CH2NH2, -CH2SO2CH3, -арил, -арилалкил, -гетероарил, -гетероарилалкил, -гетероциклоалкил, -C3-C12-циклоалкил, полиалкоксиалкил, полиалкокси, -метоксиметокси, -метоксиэтокси, -SH, -S-C1-C12-алкил, -S-C2-C8-алкенил, -S-C2-C8-алкинил, -S-C3-C12-циклоалкил, -S-арил, -S-гетероарил, -S-гетероциклоалкил или метилтиометил. Понятно, что арилы, гетероарилы, алкилы, циклоалкилы и тому подобное могут быть дополнительно замещены. В некоторых случаях каждый заместитель в замещенном фрагменте дополнительно необязательно замещен одной или более группами, причем каждая группа независимо выбрана из -F, -Cl, -Br, -I, -OH, -NO2, -CN или -NH2.
Термин “необязательно замещенный”, в контексте данного документа, означает, что указанная группа может быть замещенной или незамещенной. В одном варианте осуществления изобретения указанная группа необязательно замещена нулевыми заместителями, т.е. указанная группа является незамещенной. В другом варианте осуществления изобретения указанная группа необязательно замещена одной или более дополнительными группами, индивидуально и независимо выбранными из групп, описанных в данном документе.
В соответствии с данным изобретением любой из арилов, замещенных арилов, гетероарилов и замещенных гетероарилов, описанных в данном документе, может представлять собой любую ароматическую группу. Ароматические группы могут быть замещенными или незамещенными.
Понятно, что любой алкил, алкенил, алкинил, циклоалкил и циклоалкенил, описанный в данном документе, также может представлять собой алифатическую группу, алициклическую группу или гетероциклическую группу. «Алифатическая группа» представляет собой неароматическую группу, которая может содержать любую комбинацию атомов углерода, атомов водорода, атомов галогена, кислорода, азота или других атомов и, необязательно, содержать одно или более ненасыщенных звеньев, например, двойные и/или тройные связи. Алифатическая группа может быть с линейной цепью, разветвленной или циклической и, кроме того, содержит от около 1 до около 24 атомов углерода, более типично от около 1 до около 12 атомов углерода. В дополнение к алифатическим углеводородным группам алифатические группы включают, например, полиалкоксиалкилы, такие как, например, полиалкиленгликоли, полиамины и полиимины. Такие алифатические группы могут быть дополнительно замещены. Понятно, что алифатические группы могут быть использованы вместо алкильной, алкенильной, алкинильной, алкиленовой, алкениленоввой и алкиниленовой групп, описанных в данном документе.
Термин “алициклический”, в контексте данного документа, обозначает одновалентную группу, полученную из соединения с моноциклическим или полициклическим насыщенным карбоциклическим кольцом путем удаления одного атома водорода. Примеры включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.1]гептил и бицикло[2.2.2]октил. Такие алициклические группы могут быть дополнительно замещены.
Очевидно, что в разнообразных вариантах осуществления изобретения замещенный или незамещенный алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкинил, арилалкил, гетероарилалкил и гетероциклоалкил предназначены для того, чтобы быть одновалентными или двухвалентными. Поэтому, алкиленовая, алкениленовая и алкиниленовая, циклоалкиленовая, циклоалкениленовая, циклоалкиниленовая, арилалкиленовая, гетероарилалкиленовая и гетероциклоалкиленовая группы должны быть включены в приведенные выше определения и применимы для обеспечения правильности валентности в формулах.
Термин “гидроксил-защитный агент”, в контексте данного документа, представляет собой соединение, представленное PG-X, PG1-X или PG3-X, где PG, PG1 и PG3 являются такими, как определено в данном документе, и X представляет собой подходящую уходящую группу, кроме галогена, алкилсульфоната или фторалкилсульфоната. Предпочтительно, X представляет собой Cl, Br, I или трифлат (OTf).Гидроксил-защитный агент, где PG, PG1 или PG3 представляет собой силильную группу, альтернативно упоминается в данном описании как «силилирующий агент».
Термины «галоген», в контексте данного документа, относятся к атому, выбранному из фтора, хлора, брома и иода.
Термин «водород» включает водород и дейтерий. Кроме того, формулировка атома включает другие изотопы данного атома, если получающееся соединение является фармацевтически приемлемым
Термин «гидроксил-защитная группа», в контексте данного документа, относится к лабильному химическому фрагменту, который известен в данной области техники для защиты гидроксильной группы от нежелательных реакций во время процедур синтеза. После указанной синтетической процедуры (процедур) гидроксильная защитная группа, описанная в данном документе, может быть селективно удалена. Гидроксил-защитные группы, известные в данной области техники, в общем описаны в T.H. Greene и P.G. M. Wuts, Protective Groups in Organic Synthesis, 3-е изд., John Wiley & Sons, Нью-Йорк (1999). Примеры гидроксил-защитные группы включают бензилоксикарбонил, 4-метоксибензилоксикарбонил, трет-бутоксикарбонил, изопропоксикарбонил, дифенилметоксикарбонил, 2,2,2-трихлорэтоксикарбонил, аллилоксикарбонил, ацетил, формил, хлорацетил, трифторацетил, метоксиацетил, феноксиацетил, бензоил, метил, т-бутил, 2,2,2-трихлорэтил, 2-триметилсилилэтил, аллил, бензил, трифенилметил (тритил), метоксиметил, метилтиометил, бензилоксиметил, 2-(триметилсилил)-этоксиметил, метансульфонил, триметилсилил, триизопропилсилил и тому подобные.
Термин «защищенный гидроксил», в контексте данного документа, относится к гидроксильной группе, защищенной гидроксил-защитной группой такой, как определено выше, включая, например, бензоильную, ацетильную, триметилсилильную, триэтилсилильную, метоксиметильную группы.
Когда соединения, описанные в данном документе, содержат один или более асимметричных центров, они приводят к появлению энантиомеров, диастереомеров и других стереоизомерных форм, которые могут быть определены в терминах абсолютной стереохимии как (R)- или (S)- или как (D)- или (L)- для аминокислот. Данное изобретение включает все такие возможные изомеры, а также их рацемические и оптически чистые формы. Оптические изомеры могут быть получены из их соответствующих оптически активных предшественников по методикам, описанным выше, или путем разделения рацемических смесей. Разделение может быть осуществлено в присутствии разделительного агента, с помощью хроматографии или повторной кристаллизации или с помощью некоторой комбинации данных методов, которые известны специалистам в данной области техники. Более подробную информацию о методах разделения можно найти в Jacques, et al., Enantiomers, Racemates, and Resolutions (John Wiley & Sons, 1981). Если описанные в данном документе соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иное, предполагается, что соединения включают и E, и Z геометрические изомеры. Подобным образом, подразумевается также, что включены все таутомерные формы. Конфигурация любой углерод-углеродной двойной связи, представленной в данном документе, выбрана только для удобства и не предназначена для обозначения конкретной конфигурации, если в тексте так не указано; таким образом, двойная углерод-углеродная связь, произвольно обозначенная в данном документе как транс, может представлять собой цис, транс или их смесь двух в любой пропорции.
В контексте данного документа термин «фармацевтически приемлемая соль» относится к тем солям соединений, полученных способом по данному изобретению, которые в рамках здравого медицинского заключения подходят для применения в контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергического ответа и тому подобного и являются соразмерными с разумным соотношением выгоды/риска. Фармацевтически приемлемые соли хорошо известны в данной области техники.
Berge, et al. подробно описывает фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19 (1977). Соли могут быть получены in situ во время конечного разделения и очистки соединений по данному изобретению или отдельно путем взаимодействия функции свободного основания с подходящей органической кислотой. Примерами фармацевтически приемлемых солей включают, но не ограничиваются ими, соли присоединения нетоксичных кислот, например, соли по аминогруппе, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или органическими кислотами, такими как уксусная кислота, малеиновая кислота, винная кислота, кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других методов, используемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают, но не ограничиваются ими, адипатную, альгинатную, аскорбатную, аспартатную, бензолсульфонатную, бензоатную, бисульфатную, боратную, бутиратную, камфоратную, камфорсульфонатную, цитратную, циклопентанпропионатную, диглюконатную, додецилсульфатную, этансульфонатную, формиатную, фумаратную, глюкогептонатную, глицерофосфатную, глюконатную, полусульфатную, гептаноатную, гексаноатную, гидроиодидную, 2-гидроксиэтансульфонатную, лактобионатную, лактатную, лауратную, лаурилсульфатную, малатную, малеатную, малонатную, метансульфонатную, 2-нафталинсульфонатную, никотинатную, нитратную, олеатную, оксалатную, пальмитатную, памоатную, пектинатную, персульфатную, 3-фенилпропионатную, фосфатную, пикратную, пивалатную, пропионатную, стеаратную, сукцинатную, сульфатную, тартратную, тиоцианатную, п-толуолсульфонатную, ундеканоатную, валератную соли и тому подобное. Типичные соли щелочных или щелочноземельных металлов включают натрий, литий, калий, кальций, магний и тому подобное. Другие фармацевтически приемлемые соли включают, когда это уместно, нетоксичные катионы аммония, четвертичного аммония и амина, образованные с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, алкил, имеющий 1-6 атомов углерода, сульфонат и арилсульфонат.
Термин «лечение», в контексте данного документа, означает облегчение, ослабление, уменьшение, устранение, модуляцию или улучшение, то есть обеспечение регрессии болезненного состояния или патологического состояния. Лечение может также включать в себя ингибирование, то есть прекращение развития существующего болезненного состояния или патологического состояния, и ослабление или улучшение, то есть обеспечение регрессии существующего болезненного состояния или патологического состояния, например, когда болезненное состояние или патологического состояние уже может присутствовать.
Термин «предотвращение», в контексте данного документа означает полную или почти полную остановку болезненного состояния или патологического состояния, возникающего у пациента или субъекта, особенно когда пациент или субъект предрасположен к такому или рискует заразиться болезненным состоянием или патологическим состоянием.
Кроме того, соединения по данному изобретению, например, соли данных соединений, могут существовать в гидратированной или негидратированной (безводной) форме или в виде сольватов с другими молекулами растворителя. Неограничивающие примеры гидратов включают моногидраты, дигидраты и т.д. Неограничивающие примеры сольватов включают сольваты этанола, сольваты ацетона и т.д.
Термин “кислота Льюиса” относится к веществу, которое принимает электронную пару от основания, образуя ковалентную связь с основанием. Определение также приведено в литературе, такой как “Advanced Organic Chemistry” Jerry March, 4-е изд., опубликованной Wiely Interscience.
«Сольваты» означают формы добавления растворителя, которые содержат либо стехиометрические, либо нестехиометрические количества растворителя. Некоторые соединения имеют тенденцию захватывать фиксированное молярное соотношение молекул растворителя в кристаллическом твердом состоянии, образуя таким образом сольват. Если растворителем является вода, образованный сольват представляет собой гидрат, когда растворителем является спирт, образованный сольват представляет собой алкоголят Гидраты образуются в результате сочетания одной или более молекул воды с одним из веществ, в которых вода сохраняет свое молекулярное состояние в виде H2O, причем такая комбинация способна образовывать один или более гидратов.
В контексте данного документа, термин «аналог» относится к химическому соединению, которое структурно сходно с другим, но несколько отличается по составу (как при замене одного атома атомом другого элемента, или в присутствии конкретной функциональной группы, так и при замене одной функциональной группы другой функциональной группой). Поэтому аналогом является соединение, которое аналогично или сопоставимо по функции и внешнему виду по отношению к эталонному соединению.
Термин «апротонный растворитель», в контексте данного документа, относится к растворителю, который относительно инертен к протонной активности, т.е. не действует как донор протонов. Примеры включают, но не ограничиваются ими, углеводороды, такие как гексан и толуол, например, галогенированные углеводороды, такие как, например, метиленхлорид, этиленхлорид, хлороформ и тому подобное, гетероциклические соединения, такие как, например, тетрагидрофуран и N-метилпирролидинон, и простые эфиры, такие как диэтиловый эфир, бис-метоксиметиловый эфир. Такие растворители хорошо известны специалистам в данной области техники, а индивидуальные растворители или их смеси могут быть предпочтительными для конкретных соединений и условий реакции, в зависимости от таких факторов, как, например, растворимость реагентов, реакционная способность реагентов и предпочтительные интервалы температур. Дальнейшие обсуждения апротонных растворителей можно найти в учебниках по органической химии или в специализированных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4-е изд., под редакцией John A. Riddick et al., том II, в Techniques of Chemistry Series, John Wiley & Sons, Нью-Йорк, 1986.
Термины «протогенный органический растворитель» или «протонный растворитель», в контексте данного документа, относится к растворителю, который имеет тенденцию давать протоны, такому как спирт, например, метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол и тому подобное. Такие растворители хорошо известны специалистам в данной области техники, а индивидуальные растворители или их смеси могут быть предпочтительными для конкретных соединений и условий реакции, в зависимости от таких факторов, как, например, растворимость реагентов, реакционная способность реагентов и предпочтительные интервалы температур. Дальнейшие обсуждения протогенных растворителей можно найти в учебниках по органической химии или в специализированных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4-е изд., под редакцией John A. Riddick et al., том II, в Techniques of Chemistry Series, John Wiley & Sons, Нью-Йорк, 1986.
Комбинации заместителей и переменных, предусмотренных данным описанием, представляют собой только такие, которые приводят к образованию стабильных соединений. Термин “стабильный”, в контексте данного документа, относится к соединениям, которые обладают стабильностью, достаточной для производства, и которая сохраняет целостность соединения в течение достаточного периода времени, чтобы быть подходящей для целей, подробно описанных в данном документе (например, терапевтическое или профилактическое введение субъекту).
Синтезированные соединения могут быть отделены от реакционной смеси и дополнительно очищены с помощью метода, такого как колоночная хроматография, высокоэффективная жидкостная хроматография или перекристаллизация. Кроме того, разнообразные стадии синтеза могут быть выполнены в альтернативной последовательности или для получения желаемых соединений. Кроме того, описанные в данном документе растворители, температуры, длительности реакций и т. д. приведены только в целях иллюстрации, и изменение условий реакции может давать желаемые изоксазольные продукты по данному изобретению. Синтетические химические превращения и методологии для защитных групп (защита и снятие защиты), подходящие для синтеза соединений, описанных в данном документе, включают, например, такие, которые описаны в R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene и P.G.M. Wuts, Protective Groups in Organic Synthesis, 2-е изд., John Wiley and Sons (1991); L. Fieser и M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); а также L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
Соединения по данному изобретению могут быть модифицированы путем добавления разнообразных функциональных групп с помощью синтетических средств, описанных в данном документе, для усиления селективных биологических свойств. Такие модификации включают модификации, которые увеличивают биологическое проникновение в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), увеличивают пероральную доступность, увеличивают растворимость, чтобы обеспечить возможность введения путем инъекции, изменяют метаболизм и изменяют скорость выведения.
СОКРАЩЕНИЯ
Сокращения, которые использовались в описаниях схем и следующих примерах:
Ac для ацетила;
AcOH для уксусной кислоты;
ACN для ацетонитрила;
водн. для водного;
BA для желчной кислоты;
Насыщенный водный раствор хлорида натрия для раствора хлорида натрия в воде;
n-BuLi для н-бутиллития;
цАМФ для циклического аденозинмонофосфата;
CDCA для хенодезоксихолевой кислоты;
CDI для карбонилдиимидазола;
CTX для церебросухожильного ксантоматоза;
D2 для иодтирониндеиодиназы 2-го типа;
DABCO для 1,4-диазабицикло[2.2.2]октана;
DBN для 1,5-диазабицикло[4.3.0]нон-5-ена;
DBU для 1,8-диазабицикло[5.4.0]ундец-7-ена;
ДХМ для дихлорметана;
DIBAL для диизобутилалюминийгидрида;
DIPEA или (i-Pr)2EtN для N,N-диизопропилэтиламина;
Периодинан Десса-Мартина для 1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензиодоксол-3-(1H)-она;
DMAP для 4-диметиламинопиридина;
ДМФА для N,N-диметилформамида;
ДМСО для диметилсульфоксида;
DPPA для дифенилфосфорилазида;
EtOAc для этилацетата;
EtOH для этанола;
Et2O для диэтилового эфира;
экв. для эквивалента;
FXR для фарнезоидного х рецептора;
GLP-1 для глюкагоноподобного пептида 1
ч для часов;
IBX для 2-иодоксибензойной кислоты;
KHMDS для бис(триметилсилил)амида калия;
KLCA для 7-кетолитохолевой кислоты;
OTf или трифлат для трифторметансульфоната;
Ph для фенила;
ЛДА для диизопропиламида лития;
LiHMDS бис(триметилсилил)амида лития;
мин для минут;
MOM для метоксиметила;
MEM для метоксиэтоксиметила;
NAFLD для неалкогольной жировой болезни печени;
NaHMDS для бис(триметилсилил)амида натрия;
NASH для неалкогольного стеатогепатита;
NBS для N-бромсукцинимида;
NIS для N-иодсукцинимида;
NMO для N-метилморфолин N-оксида;
В течение ночи для в течение ночи;
PBC для первичного билиарного цирроза;
PCC для пиридиния хлорохромата;
PDC для пиридиния дихромата;
Pd/C для палладия на угле;
PNAC дляхолестаза, свзанного с парентеральным питанием;
PSC для первичного склерозирующего холангита;
i-PrOAc для изопропилацетата;
фунт. на кв. дюйм для фунтов на квадратный дюйм;
комн. темп. для комнатной температуры;
насыщ. для насыщенного;
SEM для 2-триметилсилилэтоксиметила;
TBAF для тетрабутиламмоний фторида;
TBDPS: для трет-бутилдифенилсилила;
TBS для трет-бутилдиметилсилила;
TEA или Et3N для триэтиламина;
TES для триэтилсилила;
ТФК или CF3COOH для трифторуксусной кислоты;
ТГФ для тетрагидрофурана;
THP для тетрагидропиранила;
TIPS для триизопропилсилила;
TMS для триметилсилила;
TMSCl для триметилсилилхлорида;
TMSOTf для триметилсилилтрифторметансульфоната;
TBME или MTBE для трет-бутилметилового эфира;
TCX для тонкослойной хроматографии.
Все другие сокращения, используемые в данном документе, которые конкретно не определены выше, должны соответствовать значению, которое придаст специалист в данной области техники.
Все ссылки, цитируемые в данном документе, будь то печатные, электронные, машиночитаемые носители данных или другие формы, прямо включены посредством ссылки в полном объеме, включая, но не ограничиваясь этим, рефераты, статьи, журналы, публикации, тексты, трактаты, интернет-сайты, базы данных, патенты и патентные публикации.
Примеры
Соединения и способы по данному изобретению будут лучше поняты в связи со следующими примерами, которые предназначены только для иллюстрации, а не для ограничения объема изобретения. Разнообразные изменения и модификации раскрытых вариантов осуществления изобретения будут очевидны для специалистов в данной области техники, и такие изменения и модификации, включая, без ограничения, изменения, относящиеся к химическим структурам, заместителям, производным, составам и/или способам данного изобретения, могут быть сделаны без отклонения от сущности изобретения и объема прилагаемой формулы изобретения.
Пример 1. Получение соединения 2 из соединения 1.
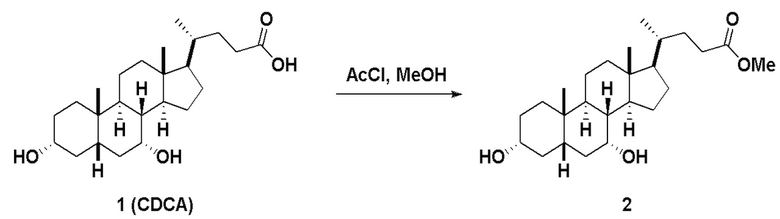
К раствору соединения 1 (101,2 г, 258 ммоль, 1,0 экв.) в MeOH (607 мл, 6v), в 2 л колбу загружали ацетилхлорид (2,0 г, 1,8 мл, 25,8 ммоль, 0,1 экв.), и реакционную смесь перемешивали в течение 18 ч. Реакционную смесь концентрировали при пониженном давлении и перегнали совместно с ТГФ (2 × 300 мл). Полученный остаток сушили в вакууме с получением 105 г неочищенного соединения 2 (ВЭЖХ чистота: 99,6%) в виде бесцветного аморфного твердого вещества, содержащего ТГФ. На следующей стадии этот материал используется без дополнительной очистки. 1H ЯМР (400 МГц, хлороформ-d) δ 3,84 (к, J = 3,1 Гц, 1 H), 3,65 (с, 3 H), 3,45 (тт, J = 11,1, 4,4 Гц, 1 H), 2,34 (ддд, J = 15,3, 10,1, 5,2 Гц, 1 H), 2,28 - 2,13 (компл., 2 H), 2,03 - 1,92 (компл., 3 H), 1,87 - 1,76 (компл., 4 H), 1,74 - 1,58 (компл., 5 H), 1,55 - 1,04 (компл., 10 H), 0,99 (дд, J = 14,2, 3,4 Гц, 1 H), 0,91 (д, J = 6,4 Гц, 3 H), 0,89 (с, 3 H), 0,65 (с, 3 H).
Пример 2. Получение соединения 3 из соединения 2.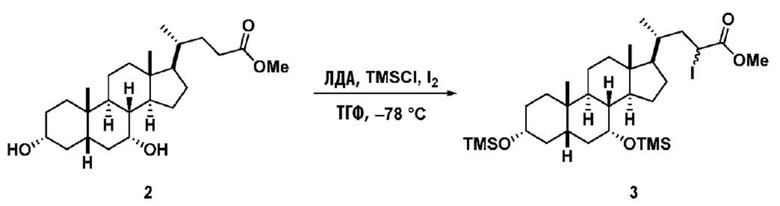
К раствору диизопропиламина (53,5 г, 75,0 мл, 529 ммоль, 4,1 экв.) в безводном ТГФ (143 мл, 2,7v) при -78°C по каплям загружали n-BuLi (206 мл 2,5M раствора в смеси гексанов, 516 ммоль, 4,0 экв.). Реакционную смесь перемешивали в течение 15 мин при -78°C, после чего прибавили TMSCl (84,0 г, 99,0 мл, 774 ммоль, 6,0 экв.). Раствор соединения 2 (52,5 г, 129 ммоль, 1,0 экв.) в безводном ТГФ (322 мл, 6,2v) по каплям прибавили при -78°C. Реакционную смесь нагревали до комн. темп. и перемешивали в течение 3 ч. Реакционную смесь охладили до -78°C и по каплям прибавили раствор I2 (45,8 г, 181 ммоль, 1,4 экв.) в безводном ТГФ (401 мл, 7,6v). Реакционную смесь перемешивали в течение 0,5 ч при -78°C. Реакционную смесь вылили в 10% водный раствор NH4Cl (600 мл) и разбавили MTBE (600 мл). Слои разделили, и водный слой экстрагировали MTBE (2 x 300 мл). Объединенные органические слои промывали водным раствором 10% Na2S2O3 (2 × 400 мл), насыщенным водным раствором хлорида натрия (400 мл), сушили (MgSO4), отфильтровали и концентрировали с получением 118 г неочищенного соединения 3 в виде желтой смолы. На следующей стадии этот материал используется без дополнительной очистки. Соединение 3 выделили в виде ~1:1 смеси диастереомеров: 1H ЯМР (400 МГц, хлороформ-d) δ 4,48 (дд, J = 12,3, 3,8 Гц, 0,5 H), 4,37 (дд, J = 11,2, 4,0 Гц, 0,5 H), 3,79 - 3,72 (компл., 6 H), 3,41 (тт, J = 10,2, 4,5 Гц, 1 H), 2,60 - 2,47 (м, 0,5 H), 2,32 (к, J = 13,1 Гц, 1 H), 2,18 (ддд, J = 14,4, 11,1, 2,7 Гц, 0,5 H), 1,99 - 1,71 (компл., 6 H), 1,70 - 0,95 (компл., 14 H), 0,92 (д, J = 6,4 Гц, 1,5 H), 0,89 (д, J = 6,5 Гц, 1,5 H), 0,86 (д, J = 5,1 Гц, 3 H), 0,67 (с, 1,5 H), 0,59 (с, 1,5 H), 0,16 (с, 9 H), 0,10 (с, 9 H).
Пример 3. Получение соединения 4 из соединения 3.
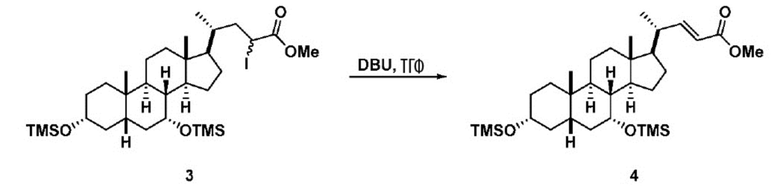
К раствору неочищенного соединения 3 (87 г, 129 ммоль, 1,0 экв.) в безводном ТГФ (1071 мл, 12,3v) загружали DBU (58,7 г, 58,1 мл, 386 ммоль, 3,0 экв.), и реакционную смесь перемешивали в течение 48 ч. Реакционную смесь погасили водным 10% NH4Cl (600 мл) и разбавили MTBE (600 мл). Слои разделили, и водный слой экстрагировали MTBE (2 x 300 мл). Объединенные органические слои сушили (MgSO4), отфильтровали и концентрировали с получением 80 г неочищенного соединения 4 в виде коричневой смолы. На следующей стадии этот материал используется без дополнительной очистки. 1H ЯМР (400 МГц, хлороформ-d) δ 6,84 (дд, J = 15,6, 9,0 Гц, 1 H), 5,72 (д, J = 15,6 Гц, 1 H), 3,76 (кд, J = 6,8, 6,0, 3,3 Гц, 1 H), 3,71 (с, 3 H), 3,40 (тт, J = 10,8, 4,6 Гц, 1 H), 2,39 - 2,18 (компл., 2 H), 2,00 - 1,11 (м, 18 H), 1,07 (д, J = 6,6 Гц, 3 H), 1,04 - 0,90 (компл., 2 H), 0,86 (с, 3 H), 0,65 (с, 3 H), 0,10 (с, 9 H), 0,07 (с, 9 H).
Пример 4. Получение соединения 5 из соединения 4.
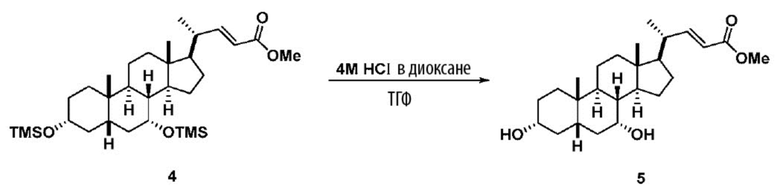
К раствору неочищенного соединения 4 (70,6 г, 129 ммоль, 1,0 экв.) в безводном ТГФ (334 мл, 4,7v) загрузили 4M HCl в 1,4-диоксане (33,4 мл, 0,47v), и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и совместно перегнали с ДХМ (1 × 400 мл). Полученную коричневую смолу очищали колоночной хроматографией, элюируя смесью гексанов/ацетоном (5% ацетона → 35% ацетона, колонка 2 × 330 г) с получением соединения 5 (32,2 г, 80,0 ммоль, выход 62% за 4 стадии. 1H ЯМР (400 МГц, хлороформ-d) δ 6,83 (дд, J = 15,6, 9,0 Гц, 1 H), 5,73 (дд, J = 15,6, 0,9 Гц, 1 H), 3,84 (к, J = 3,1 Гц, 1 H), 3,71 (с, 3 H), 3,51 - 3,40 (м, 1 H), 2,33 - 2,13 (компл., 2 H), 2,03 - 1,91 (компл., 2 H), 1,91 - 1,78 (компл., 2 H), 1,78 - 1,57 (компл., 4 H), 1,55 - 1,11 (компл., 11 H), 1,08 (д, J = 6,6 Гц, 3 H), 0,98 (тд, J = 14,1, 3,3 Гц, 1 H), 0,90 (с, 3 H), 0,69 (с, 3 H).
Пример 5. Получение соединения 6 из соединения 5.
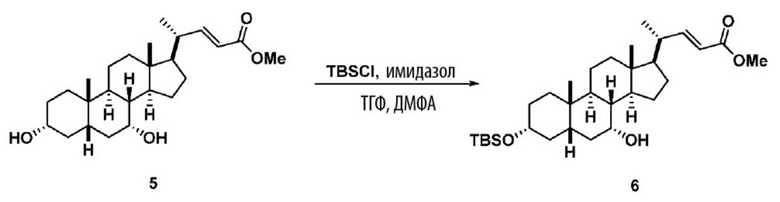
К раствору соединения 5 (32,2 г, 80,0 ммоль, 1,0 экв.) и имидазола (10,8 г, 159 ммоль, 2,0 экв.) в безводном ТГФ (166 мл, 5,2v) и ДМФА (33,2 мл, 1v) при 0°C загружали TBSCl (13,2 г, 88,0 ммоль, 1,2 экв.). Реакционную смесь нагревали до комн. темп. и перемешивали в течение 3 ч. При завершении реакции реакционную массу концентрировали для удаления большей части ТГФ. Разбавили MTBE (300 мл) и H2O (300 мл). Слои разделили, органический слой промывали 10% водным раствором лимонной кислоты (150 мл), H2O (150 мл), насыщенным водным NaHCO3 (150 мл), H2O (150 мл) и насыщенным водным раствором хлорида натрия (150 мл). Органический слой сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного соединения 6 (44 г) в виде бесцветного аморфного твердого вещества. На следующей стадии этот материал используется без дополнительной очистки. 1H ЯМР (500 МГц, хлороформ-d) δ 6,83 (дд, J = 15,6, 9,0 Гц, 1 H), 5,73 (д, J = 15,5 Гц, 1 H), 3,83-3,81 (м, 1 H), 3,71 (с, 3 H), 3,48 - 3,38 (м, 1 H), 2,26 (тд, J = 8,8, 6,2 Гц, 1 H), 2,19 (тд, J = 13,3, 11,1 Гц, 1 H), 2,02 - 1,10 (компл., 19 H), 1,08 (д, J = 6,6 Гц, 3 H), 0,98-0,91 (м, 1 H), 0,89 (с, 3 H), 0,87 (с, 9 H), 0,68 (с, 3 H), 0,04 (с, 6 H).
Пример 6. Получение соединения (III) из соединения 6.
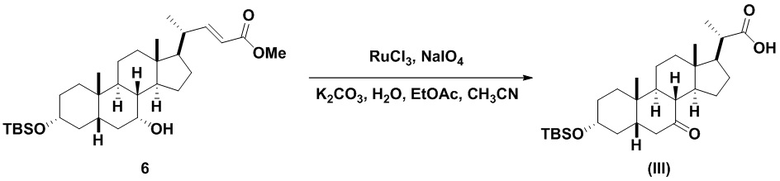
К раствору соединения 6 (41,3 г, 80,0 ммоль, 1,0 экв.) в EtOAc (227 мл, 5,4v) и CH3CN (227 мл, 5,4v) прибавили раствор K2CO3 (110 г, 796 ммоль, 10,0 экв.) в H2O (341 мл, 8,3v). Прибавили гидрат RuCl3 (0,90 г, 4,0 ммоль, 0,05 экв.), с последующим прибавлением NaIO4 (170 г, 796 ммоль, 10,0 экв.), и реакционную смесь интенсивно перемешивали в течение ночи до тех пор, пока не осталось исходного материала согласно ВЭЖХ. Реакционную смесь отфильтровали, и твердое вещество промыли EtOAc. Фильтрат погасили 10% водным раствором лимонной кислоты (600 мл). Слои разделили, и водный слой экстрагировали с помощью EtOAc (250 мл). Объединенные органические слои промывали водой (2 × 400 мл), насыщенным водным раствором хлорида натрия (400 мл), сушили (Na2SO4) и концентрировали до около 400 мл. Прибавили около 400 мл гептана и смесь медленно концентрировали при пониженном давлении при 38°C для уменьшения общего объема до около 100 мл. Полученную смесь охладили до комнатной температуры и отфильтровали, чтобы собрать твердое вещество, промывали смесью гексанов. Данную процедуру кристаллизации повторяли один раз для получения второй партии материала. В общем получили соединение (III) (12,0 г, 25,1 ммоль, выход 76%) в виде желто-коричневого твердого вещества. 1H ЯМР (400 МГц, хлороформ-d) δ 3,56 (тт, J = 10,4, 4,7 Гц, 1 H), 2,82 (дд, J = 12,3, 5,7 Гц, 1 H), 2,45 - 2,39 (м, 1 H), 2,39 - 2,32 (м, 1 H), 2,27 (дтт, J = 10,1, 7,2, 3,6 Гц, 1 H), 1,99 - 1,75 (м, 5 H), 1,67 - 1,25 (компл., 11 H), 1,23 (д, J = 6,8 Гц, 3 H), 1,17 (с, 3 H), 1,16 - 1,07 (м, 1 H), 0,99 (кд, J = 12,1, 6,3 Гц, 1 H), 0,86 (с, 9 H), 0,67 (с, 3 H), 0,03 (с, 6 H).
Пример 7. Получение соединения 8 из соединения (III).
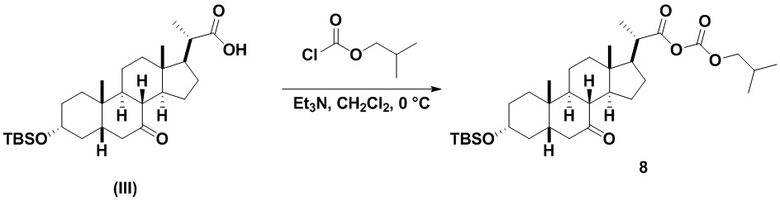
Изобутилхлорформиат (10,51 мл, 81 ммоль) по каплям прибавили к раствору соединения (III) (32,2 г, 67,5 ммоль) и Et3N (14,12 мл, 101 ммоль) в ДХМ (225 мл) при 0°C. Реакционную смесь перемешивали при 0 °C в течение 30 мин. Реакционную смесь разбавили MBTE (500 мл), промывали водой (2 x 500 мл), насыщенным водным раствором хлорида натрия (500 мл), сушили (MgSO4) и концентрировали с получением соединения 8 в виде оранжевой смолы, которую использовали непосредственно на следующей стадии. 1H ЯМР (400 МГц, хлороформ-d) δ 4,04 (д, J = 6,7 Гц, 2 H), 3,55 (тт, J = 10,4, 4,7 Гц, 1 H), 2,82 (дд, J = 12,6, 6,0 Гц, 1 H), 2,50 (дк, J = 10,3, 6,8 Гц, 1 H), 2,36 (т, J = 11,3 Гц, 1 H), 2,27 (дддд, J = 12,9, 10,1, 7,1, 3,2 Гц, 1 H), 2,11 - 1,32 (компл., 15 H), 1,31-1,24 (м, 1 H), 1,28 (д, J = 6,9 Гц, 3 H), 1,18 (с, 3 H), 1,17-1,09 (м, 1 H), 1,07 - 0,99 (м, 1 H), 1,00-0,94 (м, 1 H), 0,97 (д, J = 6,7 Гц, 6 H), 0,86 (с, 9 H), 0,68 (с, 3 H), 0,03 (с, 6 H).
Пример 8. Получение соединения (IV) из соединения 8.
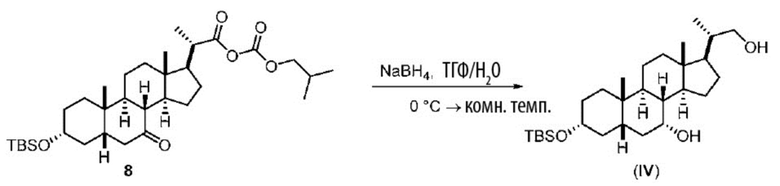
NaBH4 (5,11 г, 135 ммоль) прибавили к раствору соединения 8 (38,9 г, 67,5 ммоль) в ТГФ (270 мл)/H2O (67,5 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 ч и прибавили вторую порцию NaBH4 (5,11 г, 135 ммоль). Реакционную смесь перемешивали в течение ночи, медленно нагревая до комн. темп. Реакция завершилась согласно ТСХ и ВЭЖХ. Реакционную смесь охладили до 0°C, разбавили EtOAc (300 мл) и погасили 10% лимонной кислотой (300 мл). Слои разделили, и водную фазу экстрагировали с помощью EtOAc (2 x 150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (300 мл), сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии, элюируя смесью гексанов/ацетоном (0% ацетон → 25% ацетон, 330 г колонка) с получением соединения (IV) (26,2 г, 56,4 ммоль, выход 84 %) в виде бесцветного аморфного твердого вещества. 1H ЯМР (400 МГц, хлороформ-d) δ 3,83 (к, J = 3,1 Гц, 1 H), 3,64 (дд, J = 10,4, 3,3 Гц, 1 H), 3,48 - 3,38 (м, 1 H), 3,35 (дд, J = 10,5, 7,1 Гц, 1 H), 2,19 (тд, J = 13,3, 11,0 Гц, 1 H), 2,03 - 1,73 (компл., 5 H), 1,72 - 1,08 (компл., 15 H), 1,04 (д, J = 6,6 Гц, 3 H), 0,94 (тд, J = 14,5, 3,3 Гц, 1 H), 0,89 (с, 3 H), 0,88 (с, 9 H), 0,67 (с, 3 H), 0,04 (с, 6 H).
Пример 9. Синтез соединения 15D.
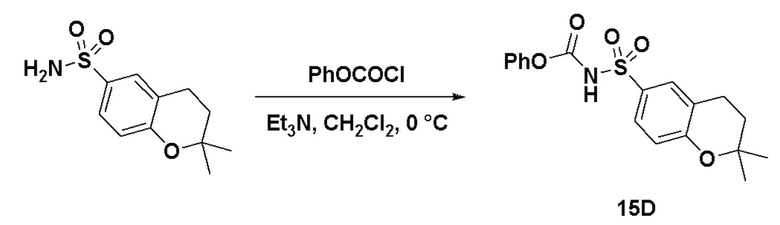
К суспензии 2,2-диметилхроман-6-сульфонамида (65 г, 269 ммоль, 1,0 экв.) и Et3N (82 г, 113 мл, 808 ммоль, 3,0 экв.) в безводном CH2Cl2 (673 мл, 10v) при 0°C загружали PhOCOCl (50,6 г, 40,6 мл, 323 ммоль, 1,2 экв.). Реакционную смесь перемешивали при 0°C в течение 3 ч. При завершении реакции реакционную смесь разбавили CH2Cl2 (400 мл). Смесь промывали холодным H2O (1000 мл), 10% холодным водным раствором лимонной кислоты (2 × 500 мл) и насыщенным водным раствором хлорида натрия (1000 мл). Органический слой сушили (Na2SO4), отфильтровали и концентрировали до около 500 мл общего объема. Прибавили смесь гексанов (500 мл) и раствор концентрировали до около 250 мл общего объема. Смесь охладили до комн. темп. Полученный осадок собирали фильтрацией, промывая смесью гексанов (3 × 100 мл) и сушили в вакууме с получением соединения 15D (72,5 г, ВЭЖХ, ELSD чистота: 98,8%, ВЭЖХ УФ240 чистота: 85,7%) в виде желто-коричневого твердого вещества. 1H ЯМР (400 МГц, хлороформ-d) δ 7,85 - 7,74 (м, 2H), 7,58 (с, 1H), 7,40 - 7,29 (м, 2H), 7,27 - 7,18 (м, 1H), 7,11 - 7,02 (м, 2H), 6,86 (т, J = 8,6 Гц, 1H), 2,82 (т, J = 6,7 Гц, 2H), 1,84 (т, J = 6,7 Гц, 2H), 1,36 (д, J = 2,1 Гц, 6H).
Пример 10. Получение соединения 17 из соединения (IV).
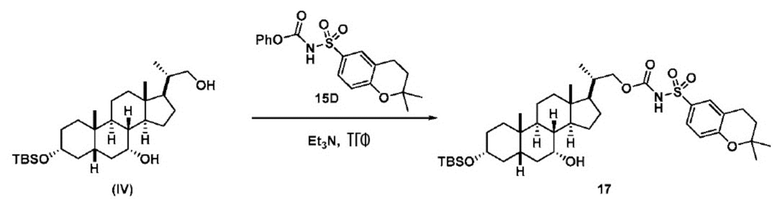
Соединение 15D (17,46 г, 48,3 ммоль) прибавили к раствору соединения (IV) (21,8 г, 46,9 ммоль) и триэтиламина (19,61 мл, 141 ммоль) в сухом ТГФ (188 мл) при комн. темп. Реакционная смесь вначале была прозрачным раствором. Примерно через час образовалось много твердого осадка. После перемешивания при комн. темп. в течение ~ 2 ч, реакционную смесь разбавили MTBE (150 мл) и перемешивали при 35°C в течение ~ 45 мин для обеспечения состаривания твердого осадка. Затем суспензионную смесь медленно концентрировали в вакууме для удаления ~ 80 мл растворителя. Оставшийся раствор с твердым осадком медленно охлаждали до комнатной температуры и состаривали в течение ночи. Затем твердое вещество собирали фильтрованием, промывали МТВЕ и сушили в высоком вакууме с получением ~ 30 г хорошего твердого соединения 17 в форме соли NEt3. Полученное соединение 17, соль NEt3 (~30 г) разделили между EtOAc 500 мл/10% лимонная кислота (300 мл). Органический слой отделили и промывали водой и насыщ. NaHCO3, насыщенным водным раствором хлорида натрия, сушили и концентрировали с получением соединения 17 (27,7 г, выход 87%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,75 - 7,64 (м, 2H), 7,36 (с, 1H), 6,82 (д, J = 8,6 Гц, 1H), 4,08 (м, 1H), 3,81 - 3,68 (м, 2H), 3,39 (м, 1H), 2,79 (т, J = 6,7 Гц, 2H), 2,14 (тд, J = 13,2, 11,1 Гц, 1H), 1,97 - 1,67 (м, 6H), 1,67 - 1,32 (м, 10H),1,31 (с, 6H), 1,31-0,99 (м, 6H),0,97 - 0,87 (м, 4H), 0,84 (с, 12H), 0,60 (с, 3H), 0,00 (с, 6H).
Пример 11. Получение соединения (VII) из соединения 17.
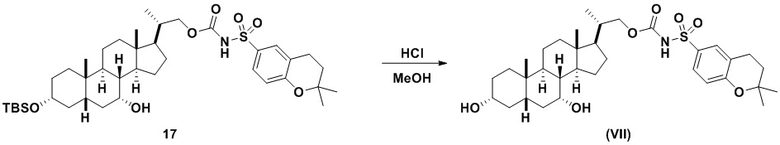
Предварительно смешанный раствор конц. HCl (294 мг, 2,98 ммоль) в MeOH (5 мл) по каплям прибавили к раствору соединения 17 (21,84 г, 29,8 ммоль) в MeOH (120 мл) при комн. темп. Реакционную смесь перемешивали при комн. темп. в течение ~ 30 мин, и контроль за ходом реакции осуществляли с помощью ТСХ и ВЭЖХ. При завершении реакции прибавили раствор 50% NaOH (239 мг, 2,98 ммоль) в воде (1 мл), и реакционную смесь концентрировали досуха. Неочищенное твердое вещество очищали хроматографией на силикагеле с (220 г SiO2, от 100% смеси гексанов до 50% ацетон/смесь гексанов, продукт выходил при около 50% ацетон/смесь гексанов) с получением соединения (VII) (15 г, выход 81%). ЖХ-МС (m/z, ES-): 616,33 [M-1]. 1H ЯМР (400 МГц, хлороформ-d) δ 7,79 - 7,68 (м, 2H), 7,32 (с, 1H), 6,87 (д, J = 8,7 Гц, 1H), 4,12 (дд, J = 10,5, 3,3 Гц, 1H), 3,87 - 3,74 (м, 2H), 3,48 (м, 1H), 2,83 (т, J = 6,7 Гц, 2H), 2,28 - 2,13 (м, 1H), 2,04 - 1,89 (м, 2H), 1,88-1,79 (м, 4H), 1,78 - 1,59 (м, 4H), 1,55 (с, 3H), 1,48 - 1,36 (м, 3H), 1,36 (с, 6H), 1,37 - 1,23 (м, 3H), 1,25 - 1,06 (м, 3H), 1,05 - 0,92 (м, 4H), 0,90 (с, 3H), 0,65 (с, 3H).
Пример 12. Получение соединения 9 из соединения (III).
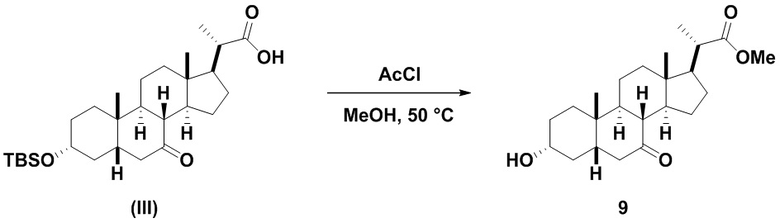
К суспензии соединения (III) (15,2 г, 31,9 ммоль, 1,0 экв.) в MeOH (91 мл, 6v) при 0°C по капли загружали AcCl (12,5 г, 11,3 мл, 159 ммоль, 5,0 экв.). Полученный раствор нагревали при 50°C в течение 24 ч. Реакционную смесь концентрировали, и полученное коричневое твердое вещество разделили между EtOAc (250 мл) и насыщ. водным NaHCO3 (250 мл). Слои разделили, органический слой промывали H2O (200 мл) и насыщенным водным раствором хлорида натрия (200 мл). Органический слой сушили (MgSO4), отфильтровали и концентрировали до около 100 мл. Прибавили смесь гексанов (150 мл) и смесь концентрировали при 40°C до около 100 мл общего объема. Полученную суспензию охладили до комнатной температуры и оставили созревать в течение ночи. Остаток отфильтровали, промывали смесью гексанов, с получением соединения 9 (8,9 г, 23,6 ммоль, выход 74%) в виде бесцветного твердого вещества. 1H ЯМР (500 МГц, хлороформ-d) δ 3,65 (с, 3H), 3,67-3,58 (м, 1H), 2,85 (ддд, J = 12,7, 6,1, 1,1 Гц, 1H), 2,48 - 2,33 (м, 2H), 2,22 (дддд, J = 13,0, 10,2, 7,2, 3,3 Гц, 1H), 2,01 - 1,66 (м, 7H), 1,66 - 1,53 (м, 3H), 1,54 - 1,45 (м, 3H), 1,45 - 1,11 (м, 10H), 1,04 - 0,92 (м, 1H), 0,92 - 0,80 (м, 1H), 0,67 (с, 3H).
Пример 13. Получение соединения 10 из соединения 9.
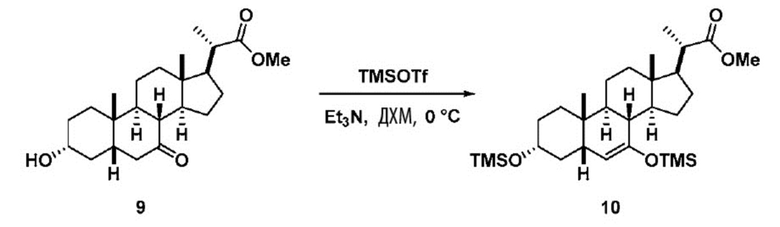
К раствору соединения 9 (8,7 г, 23,1 ммоль, 1,0 экв.) и Et3N (18,7 г, 25,8 мл, 185 ммоль, 8,0 экв.) в ДХМ (154 мл, 17v) при 0°C по каплям загружали TMSOTf (25,7 г, 20,9 мл, 116 ммоль, 5,0 экв.). Реакционную смесь перемешивали в течение 1 ч при 0°C, затем вылили в холодный насыщ. водный NaHCO3 (300 мл) и разбавили ДХМ (150 мл). Слои разделили, органический слой промывали водой (2 × 150 мл) и насыщенным водным раствором хлорида натрия (150 мл), сушили (MgSO4), отфильтровали и концентрировали при пониженном давлении. Остаток разделили между смесью гексанов (300 мл) и водой (150 мл). Слои разделили, органический слой промывали насыщенным водным раствором хлорида натрия (150 мл), сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного соединения 10 (11,1 г) в виде бесцветного аморфного твердого вещества. На следующей стадии этот материал используется без дополнительной очистки. CDCl3, использованный для H-ЯМР, предварительно обработали K2CO3, чтобы избежать кислотно-опосредованного разложения соединения 10. 1H ЯМР (400 МГц, хлороформ-d) δ 4,61 (дд, J = 5,9, 1,9 Гц, 1H), 3,53 (с, 3H), 3,44-3,35 (м, 1H), 2,31 (дк, J = 10,2, 6,9 Гц, 1H), 1,88 - 1,75 (м, 3H), 1,73 - 1,64 (м, 2H), 1,62-1,50 (м,3H), 1,48 - 1,37 (м, 3H), 1,37 - 1,02 (м, 7H), 1,08 (д, J = 6,8 Гц, 3H), 0,94 (тд, J = 14,2, 3,2 Гц, 1H), 0,72 (с, 3H), 0,58 (с, 3H), 0,04 (с, 9H), -0,00 (с, 9H).
Пример 14. Получение соединения 11 из соединения 10.
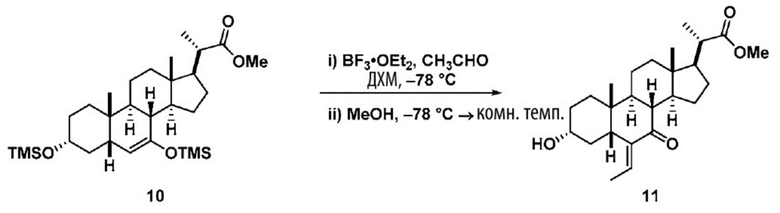
К раствору соединения 10 (11,1 г, 21,3 ммоль, 1,0 экв.) и ацетальдегида (2,3 г, 3,0 мл, 53,3 ммоль, 2,5 экв.) в ДХМ (107 мл, 10v) при -78°C по каплям загружали BF3⋅OEt2 (13,2 мл, 107 ммоль, 5,0 экв.). Реакционную смесь перемешивали при -78°C в течение 4 ч, после чего по каплям прибавили MeOH (40 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь охладили до 0°C и осторожно погасили с водным насыщ. NaHCO3 (250 мл) и разбавили ДХМ (150 мл). Слои разделяли, и водный слой экстрагировали ДХМ (1 × 150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (250 мл), сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного соединения 11 (8,7 г, ВЭЖХ чистота: 98,7%, смесь E/Z изомеров) в виде желтого аморфного твердого вещества. На следующей стадии этот материал используется без дополнительной очистки. ЖХ-МС (m/z, ES+): 403,29 [M+H].
Пример 15. Получение соединения 12 из соединения 11.
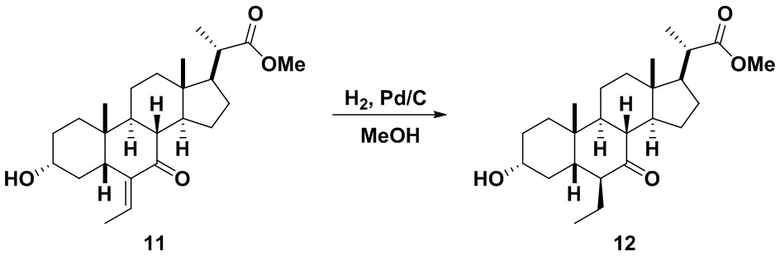
К раствору неочищенного соединения 11 (8,6 г, 21,3 ммоль, 1,0 экв.) в MeOH (71 мл, 12,4v) загружали 10% Pd/C (50% H2O, 2,3 г, 0,25 масс.). Из реакционной емкости откачали воздух и её снова заполнили H2 (3×). Реакционную смесь перемешивали в течение 72 ч, разбавили EtOAc и отфильтровали через Celite®. Фильтрат концентрировали с получением неочищенного соединения 12 (8,1 г, ВЭЖХ чистота: 98,0%) в виде бесцветного аморфного твердого вещества. На следующей стадии этот материал используется без дополнительной очистки. 1H ЯМР (400 МГц, хлороформ-d) δ 3,58 (с, 3H), 3,53-3,47 (м, 1H), 2,49 (дд, J = 12,0, 10,7 Гц, 1H), 2,35 (дк, J = 10,4, 6,8 Гц, 1H), 2,19 - 2,04 (м, 1H), 1,93 - 1,79 (м, 3H), 1,79 - 1,64 (м, 4H), 1,64-1,54 (м, 3H), 1,53 - 1,35 (м,4H), 1,32 - 1,15 (м, 3H), 1,15 (с, 3H), 1,13 (к, J = 7,2 Гц, 2H), 0,92-0,82 (м, 1H), 0,78 (т, J = 7,2 Гц, 3H), 0,61 (с, 3H).
Пример 16. Получение соединения 13 из соединения 12.
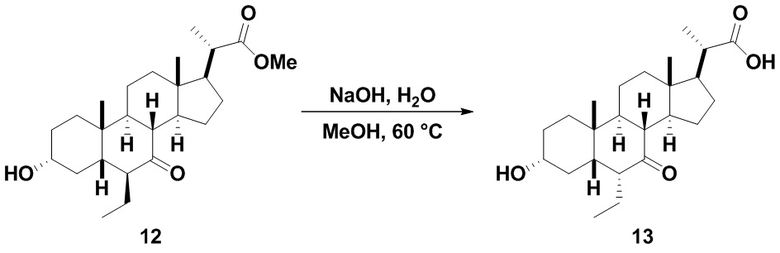
К раствору неочищенного соединения 12 (8,0 г, 19,8 ммоль, 1,0 экв.) в MeOH (99 мл, 12,3v) и H2O (24,7 мл, 3,1v) загружали 50% водный NaOH (7,9 мл, 98,9 ммоль, 5,0 экв.) и реакционную смесь перемешивали при 60°C в течение 24 ч. Реакционную смесь охладили до 0°C и сделали кислым (pH ~1-2) с помощью 1M HCl. Водную смесь экстрагировали с помощью EtOAc (3 x 200 мл). Объединенные органические слои сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного соединения 13 (7,5 г, ВЭЖХ чистота: 98,0%) в виде бледно-желтого аморфного твердого вещества. На следующей стадии этот материал используется без дополнительной очистки.1H ЯМР (400 МГц, хлороформ-d) δ 3,45-3,36 (м, 1H), 2,55 (к, J = 6,2 Гц, 1H), 2,34 - 2,16 (м, 1H), 2,14-2,05 (м, 1H), 1,85 - 1,52 (м, 8H), 1,36 (дддт, J = 23,7, 9,8, 5,5, 3,3 Гц, 3H), 1,29 - 1,18 (м, 1H), 1,17 - 1,06 (м, 9H), 1,06 - 0,91 (м, 2H), 0,90 - 0,72 (м, 2H), 0,66 (т, J = 7,4 Гц, 3H), 0,54 (с, 3H).
Пример 17. Получение соединения (V) из соединения 13.
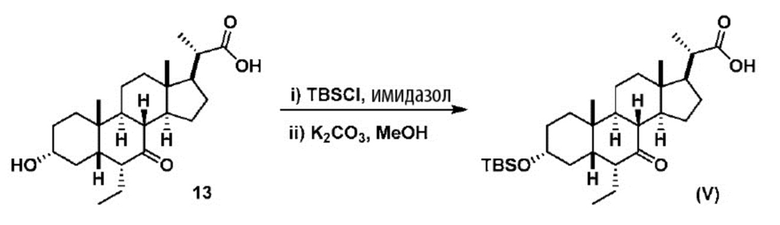
К раствору неочищенного соединения 13 (7,7 г, 19,8 ммоль, 1,0 экв.) в ТГФ (65,9 мл, 8,5v) и ДМФА (16,5 мл, 2,1v) при 0°C загружали имидазол (8,1 г, 119 ммоль, 6,0 экв.) и TBSCl (6,6 г, 43,5 ммоль, 2,2 экв.). Ледяную баню удалили и реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Прибавили метанол (16,5 мл, 2,1v) и K2CO3 (1,4 г, 9,9 ммоль, 0,5 экв.) и реакционную смесь перемешивали в течение 2 ч. Реакционную смесь осторожно подкислили 10% водным раствором лимонной кислоты (200 мл) и разбавили EtOAc (200 мл). Слои разделили, и водную фазу экстрагировали EtOAc (2 x 200 мл). Объединенные органические слои промывали H2O (3 × 100 мл) и насыщенным водным раствором хлорида натрия (100 мл), затем сушили (MgSO4), отфильтровали и концентрировали с получением желтого твердого вещества. Данное твердое вещество перекристаллизовали из CH2Cl2/смесь гексанов с получением соединения (V) (7,1 г, выход 71% за 5 стадий, ВЭЖХ чистота: 98,2%).1H ЯМР (500 МГц, хлороформ-d) δ 9,78 (уш.с., 1H), 3,50-3,43 (м, 1H), 2,63 (к, J = 6,3 Гц, 1H), 2,41-2,34 (м, 1H), 2,26-2,19 (м, 1H), 1,91-1,82 (м, 3H), 1,81 - 1,65 (м, 3H), 1,64 - 1,27 (м, 8H), 1,25-1,16 (7H), 1,14-1,07 (м, 2H), 1,01 - 0,86 (м, 3H), 0,83 (с, 9H), 0,78 (т, J = 7,4 Гц, 3H), 0,65 (с, 3H), -0,00 (с, 6H).
Пример 18. Получение соединения 15 из соединения (V).
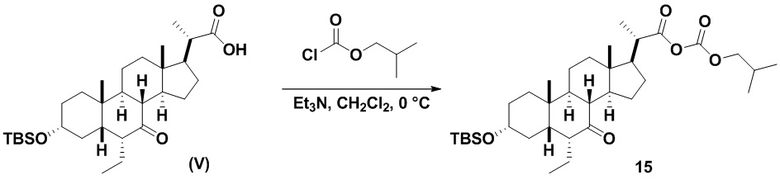
К раствору соединения (V) (7,1 г, 14,1 ммоль, 1,0 экв.) в ДХМ (46,9 мл, 6,6v) при 0°C загружали Et3N (2,1 г, 2,9 мл, 21,1 ммоль, 1,5 экв.) и изобутилхлорформиат (2,3 г, 2,2 мл, 16,9 ммоль, 1,2 экв.), и реакционную смесь перемешивали в течение 30 мин при 0°C. Реакционную смесь разбавили MBTE (150 мл) и промывали водой (2 × 100 мл), насыщенным водным раствором хлорида натрия (100 мл), сушили (MgSO4) и концентрировали с получением неочищенного соединения 15 (9,0 г, ВЭЖХ чистота: 99,1%) в виде оранжевого аморфного твердого вещества. Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Пример 19. Получение соединения (VI) из соединения 15.
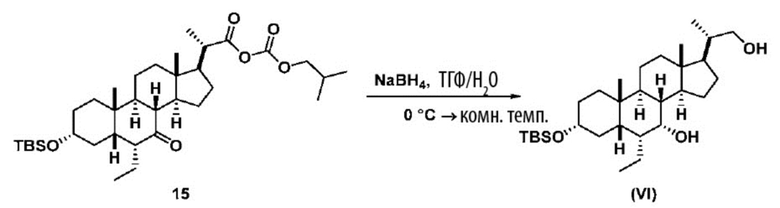
К раствору неочищенного соединения 15 (8,5 г, 14,1 ммоль, 1,0 экв.) в ТГФ (47 мл, 5,5v) и H2O (23 мл, 2,7v) при 0°C загружали NaBH4 (1,1 г, 28,1 ммоль, 2,0 экв.), и реакционную смесь перемешивали в течение 30 мин при 0°C. Прибавили NaBH4 (1,1 г, 28,1 ммоль, 2,0 экв.) и реакционную смесь перемешивали в течение ночи, медленно нагревали до комн. темп. Реакционную смесь охладили до 0°C, разбавили EtOAc (85 мл) и осторожно погасили 10% раствором лимонной кислоты (85 мл). Слои разделили, и водную фазу экстрагировали EtOAc (2 x 40 мл). Объединенные органические слои сушили (MgSO4), отфильтровали и концентрировали с получением неочищенного соединения (VI) (6,9 г, ВЭЖХ чистота: 95,8%) в виде бледно-желтого аморфного твердого вещества. 1H ЯМР (400 МГц, хлороформ-d) δ 3,71 - 3,52 (м, 2H), 3,44 - 3,20 (м, 2H), 1,91 (дт, J = 12,3, 2,9 Гц, 1H), 1,76 (дтд, J = 20,6, 10,0, 4,5 Гц, 4H), 1,62 (ддп, J = 9,8, 6,8, 3,8, 3,0 Гц, 2H), 1,58 - 1,27 (м, 8H), 1,27 - 1,05 (м, 5H), 1,00 (д, J = 6,6 Гц, 2H), 0,94 (дд, J = 14,3, 3,6 Гц, 1H), 0,90 - 0,85 (м, 4H), 0,84 (с, 9H), 0,63 (с, 3H), -0,00 (с, 6H).
Пример 20. Получение соединения 18b из соединения (VIb).
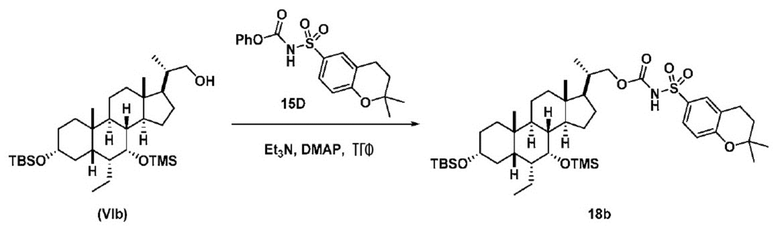
К перемешиваемому раствору (VIb) (22,33 г, 39,5 ммоль) и 15D (20 г, 55,3 ммоль) в ТГФ (180 мл) при комн. темп. прибавили TEA (8,26 мл, 59,3 ммоль), с последующим прибавлением DMAP (0,966 г, 7,91 ммоль). Полученную смесь нагревали до 50°C и перемешивали в течение 2 ч при 50°C, охладили до комн. темп., погасили 3% раствором лимонной кислоты и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и концентрировали в вакууме, и остаток очищали хроматографией на силикагеле, используя гексан/EtOAc (от 100/0 до 70/30, 15 мин) с получением продукта в виде белой пены. 32,2 г соединения 18b.1H ЯМР (500 МГц, хлороформ-d) δ 7,71 - 7,61 (м, 2H), 7,19 - 7,16 (м, 1H), 6,83 - 6,72 (м, 1H), 4,06 (дд, J = 10,3, 3,4 Гц, 1H), 3,65 (дд, J = 10,5, 8,1 Гц, 1H), 3,56 (с, 1H), 3,26 (дт, J = 10,9, 6,7 Гц, 1H), 2,75 (т, J = 6,7 Гц, 2H), 1,88 - 1,70 (м, 5H), 1,74 - 1,46 (м, 4H), 1,46 - 1,36 (м, 4H), 1,35 - 1,13 (м, 12H), 1,09 (м, 4H), 0,99 - 0,91 (м, 2H), 0,95 - 0,76 (м, 9H), 0,92 - 0,74 (м, 21H), 0,81 (с, 9H), 0,54 (с, 3H), 0,00 (с, 9H), -0,03(с, 6H).
Пример 21. Получение соединения (VIII) из соединения 18b.
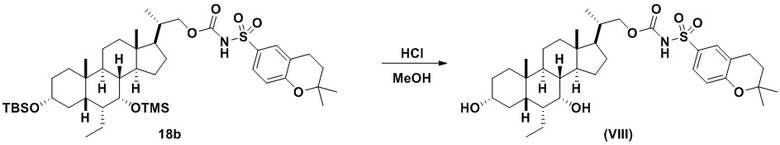
К перемешиваемому раствору 18b (32 г, 38,4 ммоль) в MeOH (180 мл) при комн. темп. прибавили HCl (0,316 мл, 3,84 ммоль). Смесь перемешивали при комн. темп. в течение 1 ч и затем погасили 0,6 мл 6N NaOH и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с использованием гексана/ацетона (от 100/0 до 50/50, 20 мин) с получением соединения (VIII) в виде белого твердого вещества (21 г, 85%). ЖХ-МС (m/z, ES-): 644,36 [M-1]. 1H ЯМР (400 МГц, хлороформ-d) δ 7,71 - 7,55 (м, 3H), 6,79 (д, J = 8,7 Гц, 1H), 4,04 (дд, J = 10,4, 3,3 Гц, 1H), 3,76 - 3,57 (м, 2H), 3,34 (тт, J = 10,5, 5,1 Гц, 1H), 2,75 (т, J = 6,7 Гц, 2H), 1,88 - 1,70 (м, 5H), 1,74 - 1,46 (м, 4H), 1,46 - 1,36 (м, 4H), 1,35 - 1,13 (м, 12H), 1,09 (м, 4H), 0,99 - 0,91 (м, 2H), 0,95 - 0,76 (м, 9H), 0,57 (с, 3H).
Пример 22. Синтез 20A
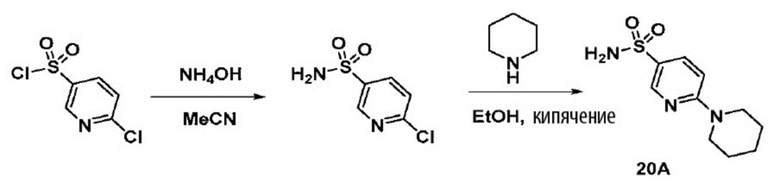
Раствор 6-хлорпиридин-3-сульфонилхлорида (50 г) в MeCN (75 мл) по каплям прибавили к водн. NH4OH (28-30%, 125 мл) при 0°C (ледяная баня) (экзотермическая). После добавления ледяную баню убрали и реакционную смесь перемешивали еще 45 мин и затем снова охлаждали до 0 °C, погасили водой и подкислили 37% HCl (120 мл) до pH 1-2. Смесь экстрагировали EtOAc, и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, отфильтровали и концентрировали. Неочищенный продукт (40 г) сушили в течение ночи в высоком вакууме и использовали непосредственно на следующей стадии.
Вначале неочищенный продукт, 6-хлорпиридин-3-сульфонамид (79г), суспендировали в EtOH (560 мл) и затем медленно прибавили (85 мл). Полученную смесь кипятили с обратным холодильником в течение 22 ч, охлаждали до комнатной температуры и осажденные твердые вещества (после 3 ч старения) собирали фильтрованием и промывали EtOH, и сушили. Твердые вещества дополнительно очищали путем смешивания с водой, фильтрации и промывки водой, а затем сушили с получением соединения 20A в виде белого твердого вещества (94 г, выход 95%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,42 (д, J = 2,5 Гц, 1H), 7,79 (дд, J = 9,2, 2,6 Гц, 1H), 7,15 (с, 2H), 6,91 (д, J = 9,1 Гц, 1H), 3,64 (дд, J = 6,5, 4,4 Гц, 4H), 1,64 (кд, J = 6,2, 5,8, 3,4 Гц, 2H), 1,53 (тк, J = 7,9, 4,8, 4,1 Гц, 4H).
Пример 23. Получение соединения 20D из 20A
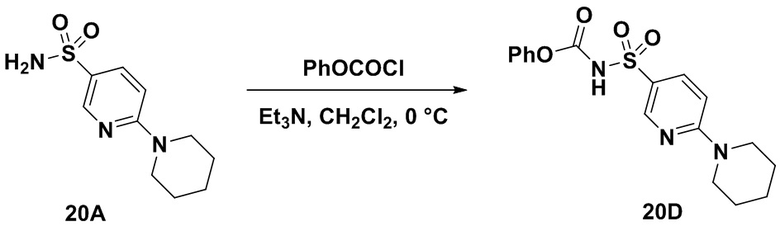
Фенилхлорформиат (24,95 мл, 199 ммоль) по каплям прибавили к суспензии 20A (40 г, 166 ммоль) и TEA (69,3 мл, 497 ммоль) в ДХМ (400 мл) при 0°C. Суспензия медленно становилась прозрачной при добавлении фенилхлорформиата (около половины пути), затем снова мутной. Реакционную смесь перемешивали при 0°C в течение 2 ч, затем разбавили ДХМ (700 мл), промывали водой (2×700 мл) и 10% лимонной кислотой (2×400 мл), органический слой быстро отделили (осадок образовывался быстро после второй промывки 10%-ной лимонной кислотой), а образовавшиеся твердые вещества (после 30 мин старения) отфильтровывали и промывали ДХМ и сушили, давая 29 г соединения 20D. Фильтрат концентрировали до 200 мл и твердые вещества отфильтровывали, промывали ДХМ и сушили с получением еще 19 г соединения 20D (всего 48 г, выход 80%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,71 (дд, J = 2,6, 0,7 Гц, 1H), 7,96 (дд, J = 9,3, 2,6 Гц, 1H), 7,66 (с, 1H), 7,40 - 7,30 (м, 2H), 7,27 - 7,18 (м, 1H), 7,13 - 7,05 (м, 2H), 6,58 (дд, J = 9,4, 0,7 Гц, 1H), 3,70 (т, J = 5,4 Гц, 4H), 1,77 - 1,59 (м, 6H).
Пример 24. Получение соединения 19b из соединения (VIb).
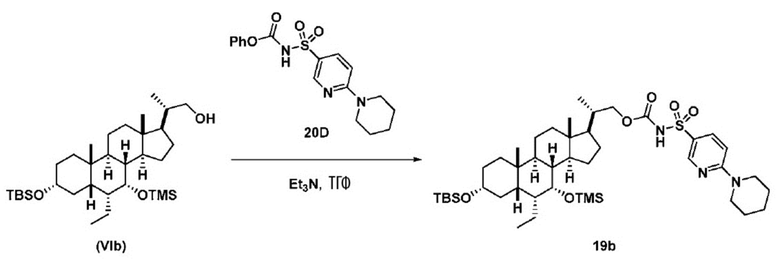
К раствору 20D (22,83 г, 63,2 ммоль) в ТГФ (354 мл) при комн. темп. прибавили соединение (VIb) (34 г, 60,2 ммоль), триэтиламин (25,2 мл, 181 ммоль) и DMAP (0,735 г, 6,02 ммоль). Полученную суспензию нагревали до 50°C и перемешивали в течение 4 ч (смесь была почти прозрачной), затем охладили до комн. темп. и разбавили EtOAc (1000 мл). Смесь промывали 3% лимонной кислотой и насыщенным водным раствором хлорида натрия, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографией на силикагеле с использованием гексана/ацетона (от 100/0 до 65/35, 25 мин) с получением соединения 19b (49 г, выход ~93%) в виде белой пены, содержащей небольшое количество фенола (побочный продукт). 1H ЯМР (400 МГц, хлороформ-d) δ 8,62 (д, J = 2,5 Гц, 1H), 7,99-7,94 (м, 1H), 7,19 - 7,14 (м, 1H), 6,62 (д, J = 9,4 Гц, 1H), 4,08 (дд, J = 10,4, 3,6 Гц, 1H), 3,69 (с, 3H), 3,73 - 3,61 (м, 1H), 3,56 (д, J = 2,6 Гц, 1H), 3,51 (с, 1H), 3,27 (тд, J = 10,8, 5,3 Гц, 1H), 1,94 - 1,58 (м, 12H), 1,54 - 1,45 (м, 2H), 1,44 (с, 1H), 1,38 (с, 1H), 1,39 - 1,25 (м, 2H), 1,28 - 1,11 (м, 3H), 1,14 - 0,93 (м, 3H), 0,97 - 0,84 (м, 4H), 0,79 (д, J = 10,3 Гц, 16H), 0,78-0,74 (м, 6H), 0,57 (д, J = 11,6 Гц, 1H), 0,55 (с, 2H), 0,00 (с, 9H), -0,03 (с, 6H).
Пример 25. Получение соединения (IX) из соединения 19b.
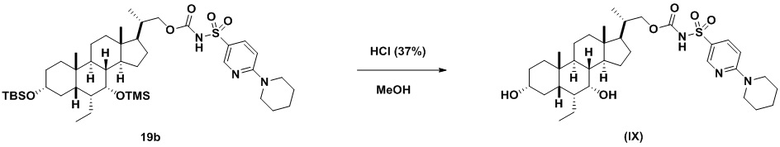
К раствору 19b (49 г, 58,9 ммоль) в MeOH (294 мл) при комн. темп. прибавили HCl (37%) (1,450 мл, 17,66 ммоль). Полученную смесь перемешивали в течение 3 ч, затем нейтрализовали гидроксидом натрия (50%) (0,933 мл, 17,66 ммоль) в 2 мл воды и концентрировали. Остаток очищали хроматографией на силикагеле, используя гексан/ацетон (от 100/0 до 50/50, 15 мин) с получением продукта в виде белой пены, который трижды промывали этанолом для удаления остаточного ацетона. Полученную белую пену лиофилизировали в течение 7 дней с получением соединения (IX) (32 г, 84%). ЖХ-МС (m/z, ES+): 646,39 [M+1]. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,70 (с, 1H), 8,46 (д, J = 2,6 Гц, 1H), 7,78 (дд, J = 9,3, 2,7 Гц, 1H), 6,93 (д, J = 9,3 Гц, 1H), 4,33 (д, J = 18,4 Гц, 1H), 4,05 (д, J = 5,1 Гц, 1H), 3,98 (дд, J = 10,6, 3,4 Гц, 1H), 3,74 (дд, J = 10,6, 7,0 Гц, 1H), 3,68 (т, J = 5,5 Гц, 4H), 3,52 - 3,41 (м, 2H), 3,14 (с, 1H), 1,89 - 1,74 (м, 2H), 1,74 - 1,60 (м, 6H), 1,61 - 1,51 (м, 6H), 1,44 (тд, J = 14,7, 8,4 Гц, 4H), 1,41 - 1,25 (м, 2H), 1,28 - 1,00 (м, 7H), 1,02 - 0,88 (м, 2H), 0,92 - 0,79 (м, 9H), 0,59 (с, 3H).
Хотя данное изобретение было изложено и описано со ссылками на его предпочтительные варианты осуществления, специалисты в данной области определят, что возможны различные изменения в форме и деталях без отступления от существа изобретения, охватываемого прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЖЕЛЧНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2707280C2 |
| ТЕТРАЗОЛ-СОДЕРЖАЩИЕ ИНГИБИТОРЫ РЕГУЛИРУЮЩЕЙ АПОПТОТИЧЕСКИЕ СИГНАЛЫ КИНАЗЫ 1 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2807545C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2018 |
|
RU2794124C2 |
| 8-АНИЛИНОИМИДАЗОПИРИДИНЫ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2008 |
|
RU2498985C2 |
| ХИНОКСАЛИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2008 |
|
RU2493160C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2544010C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ МЕК | 2009 |
|
RU2509078C2 |
| ПРОИЗВОДНЫЕ АРИЛАНИЛИНА В КАЧЕСТВЕ АГОНИСТОВ β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2005 |
|
RU2370490C2 |
| 5-АНИЛИНОИМИДАЗОПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2441004C1 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ IRAK1/4 ИНГИБИТОРОВ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2730016C2 |
Изобретение относится к области органической химии, а именно к способу получения соединения формулы (II), где значения заместителей формулы (II) представлены в формуле изобретения. Способ включает следующие стадии: (1ai) этерификация CDCA, (1aii) введение гидроксил-защитной группы в продукт (1ai) и взаимодействие полученного соединения с галогенирующим агентом, (1aiii) отщепление галогенводорода от продукта (1aii), (1aiv) снятие защитной группы с продукта (1aiii), (1av) введение гидроксил-защитной группы в продукт (1aiv), (1avi) окислительное расщепление и окисление продукта (1av), (2bi) этерификация продукта (1avi), (2bii) взаимодействие силилирующего агента в присутствии основания с продуктом (2bi), (2biii) взаимодействие продукта (2bii) с ацетальдегидом в присутствии кислоты Льюиса, (2biv) гидрирование продукта (2biii), (2bv) взаимодействие продукта (2biv) с основанием в протонном растворителе или в смеси протонного и непротонного растворителей, (2bvi) введение гидроксил-защитной группы в продукт (2bv), (3bi) взаимодействие подходящего хлорформиата с продуктом (2bvi), (3bii) восстановление продукта (3bi), (4bi) взаимодействие продукта (3bii) с подходящим сульфонилкарбаматом, (4bii) снятие защитной группы в продукте (4bi) с получением соединения формулы (II), где структурные формулы указанных промежуточных соединений представлены в формуле изобретения. Технический результат: усовершенствование способа получения соединения формулы (II) за счет увеличения выхода целевого продукта и уменьшения стадийности процесса. 6 з.п. ф-лы, 25 пр.
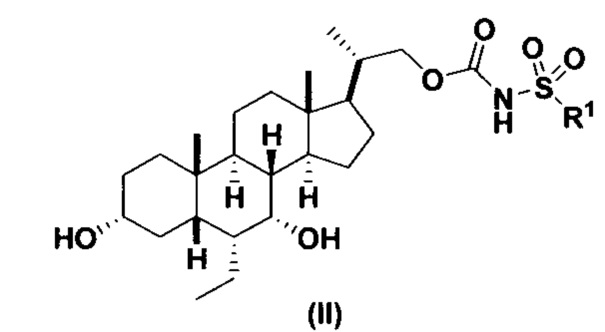
1. Способ получения соединения формулы (II)
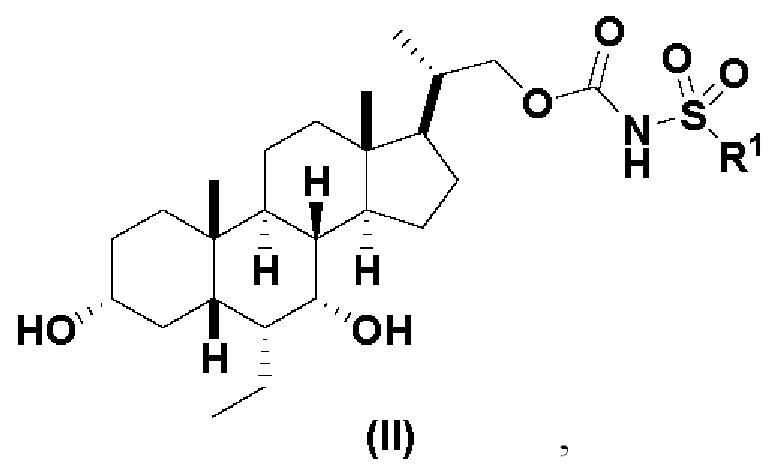
где R1 выбирают из группы, состоящей из:
1) С3-С8 циклоалкила;
2) незамещенного нафтила или фенила, необязательно замещенного гидроксилом, галогеном, C1-C6-алкилом, гидроксил C1-С6-алкилом, галоген C1-С6-алкилом, C1-C6-алкокси, галоген C1-C6-алкокси, С3-С8-циклоалкилом, 4-7-членным гетероциклоалкилом, в котором один или два атома в кольце выбраны из N и О и который необязательно замещен окси, C1-С6-алкилом, галогеном, 7-8-членным бициклическим гетероциклоалкилом, в котором один или два атома в кольце выбраны из N и О; фенила, конденсированного с 5- или 6-членным гетероциклоалкильным кольцом, имеющим атом в кольце, который представляет собой О и необязательно замещен одним или двумя C1-C6-алкилами;
3) 5-6-членного гетероциклоалкила, в котором два атома в кольце представляют собой N, О или S, необязательно замещенного оксигруппой;
4) 5-6-членного гетероарила, в котором 1-2 атома в кольце выбраны из S и N, необязательно замещенного C1-С6-алкилом; 4-6-членным гетероциклоалкилом, в котором 1-2 атома кольца выбраны из N и О; фенилом, необязательно замещенного галогеном, C1-С6-алкилом, галоген-C1-С6-алкокси, галоген-C1-С6-алкилом; 5-членным гетероарилом, в котором 2-4 атома в кольце выбраны из N и О, и 9-10-членным бициклическим гетероарилом, в котором 1-2 атома в кольце выбраны из N, О и S, и который может быть частично насыщенным; и
5) NRaRb, где Ra и Rb, каждый независимо, выбирают из водорода, незамещенного -C1-C8 алкила, или Ra и Rb, взятые вместе с атомом азота, к которому они присоединены, так чтобы образовать 4-6-членное гетероциклическое кольцо, необязательно замещенное C1-С6-алкилом или галогеном;
причем указанный способ включает стадии:
(1ai) введение соединения 1
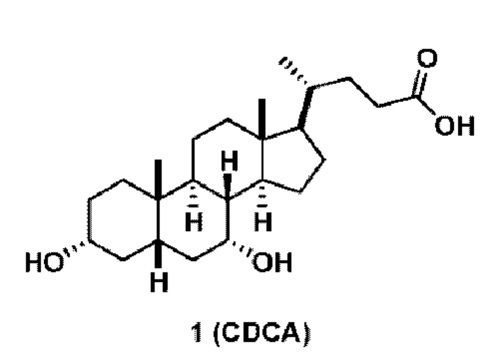
в реакцию с C1-С6-алканолом в условиях кислотного катализа с получением соединения 2
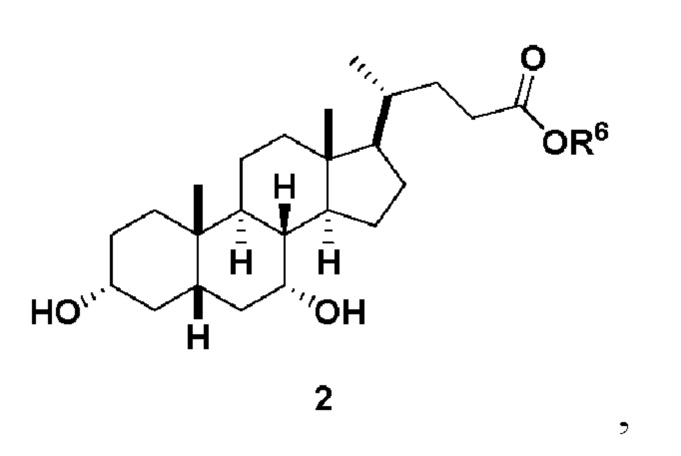
где R6 представляет собой C1-С6-алкил;
(1aii) введение соединения 2 в реакцию с сильным основанием в присутствии гидроксил-защитного агента с получением соединения формулы 2а
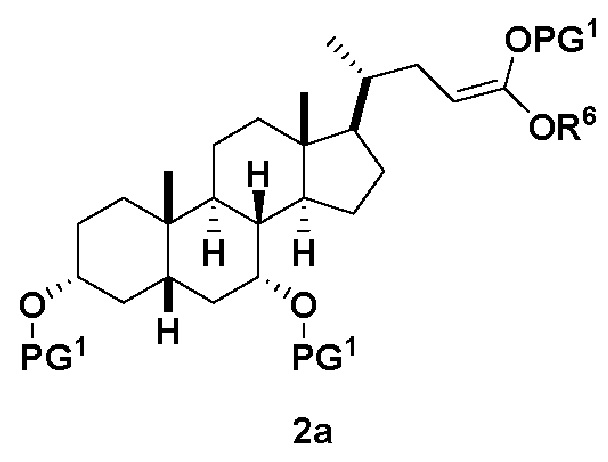
и введение соединения формулы 2а в реакцию с галогенирующим агентом с получением соединения 3
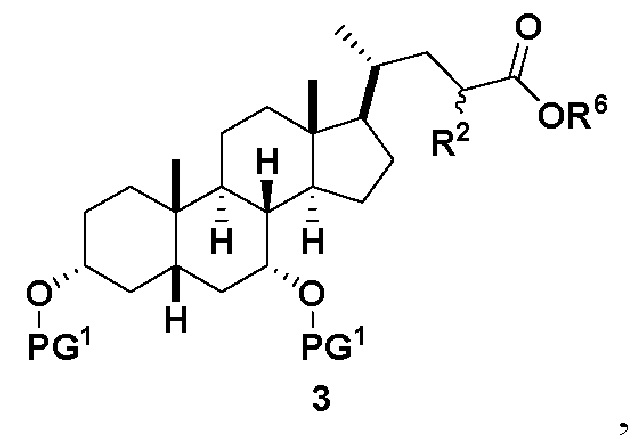
где PG1 представляет собой гидроксил-защитную группу; а также
R2 выбирают из Br, I и Cl;
(1aiii) введение соединения 3 в реакцию с органическим основанием для отщепления HR2 и получения соединения 4
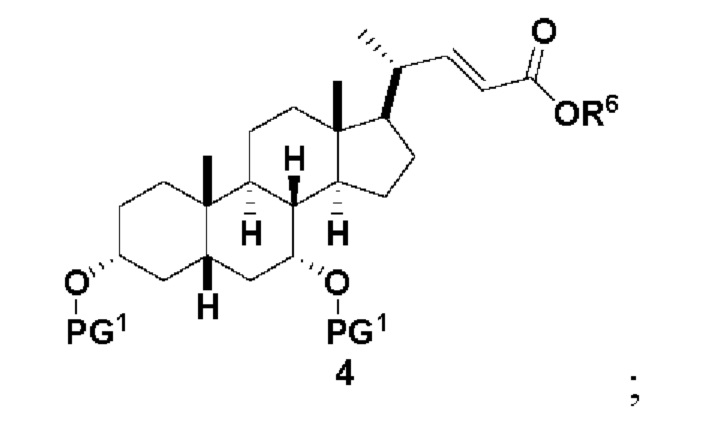
(1aiv) снятие защитной группы в соединении 4 с получением соединения 5
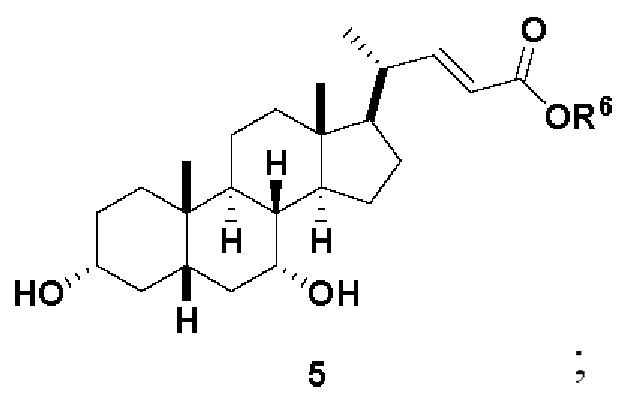
(1av) введение соединения 5 в реакцию с гидроксил-защитным агентом с получением соединения 6
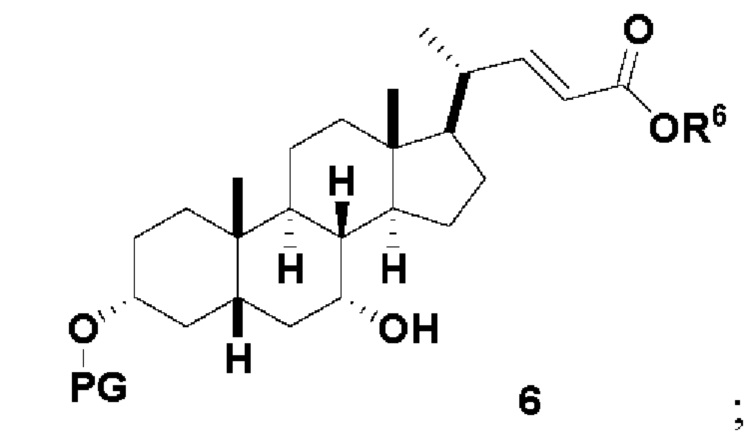 а также
а также
(1avi) окислительное расщепление и окисление соединения 6 в присутствии основания для получения соединения формулы (III)
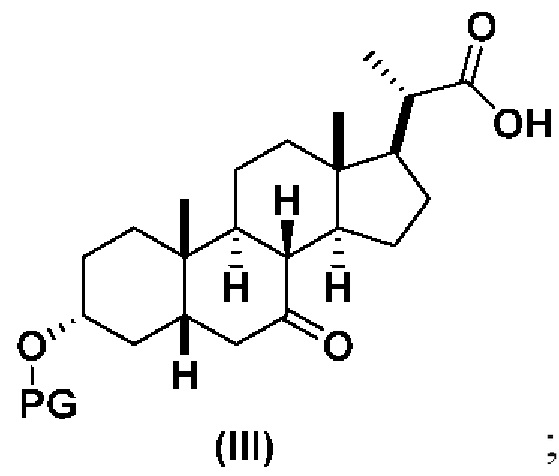
(2bi) введение соединения (III) в реакцию с C1-С6-алканолом в условиях кислотного катализа с получением соединения 8
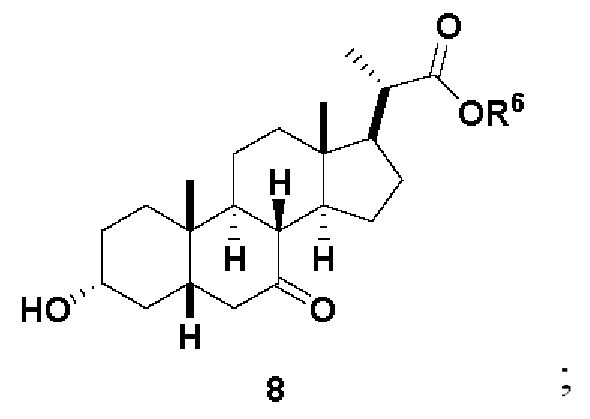
(2bii) введение соединения 8 в реакцию с силилирующим агентом в присутствии основания с получением соединения 9
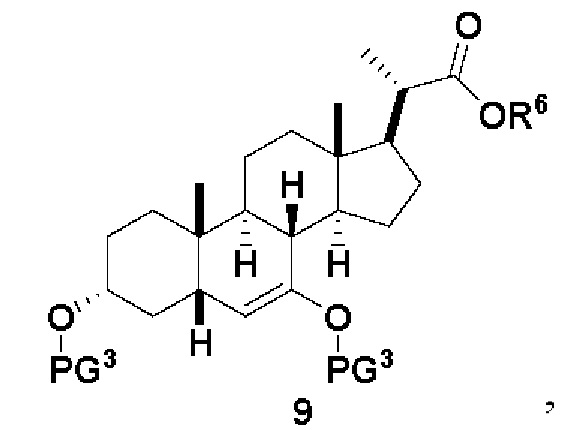
где PG3 представляет собой силильную группу;
(2biii) введение соединения 9 в реакцию с ацетальдегидом в присутствии кислоты Льюиса с получением соединения 10
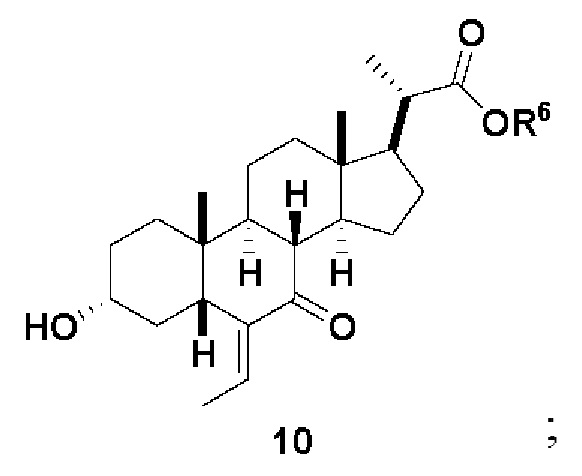
(2biv) гидрирование соединения 10 с получением соединения 11
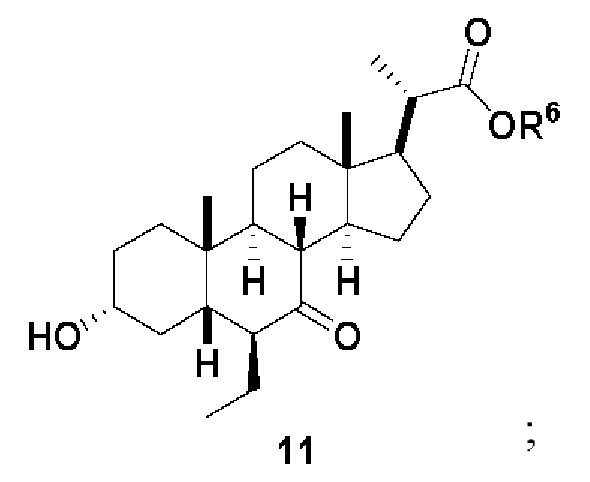
(2bv) введение соединения 11 в реакцию с основанием в протонном растворителе или смеси протонного растворителя и непротонного растворителя с получением соединения 12
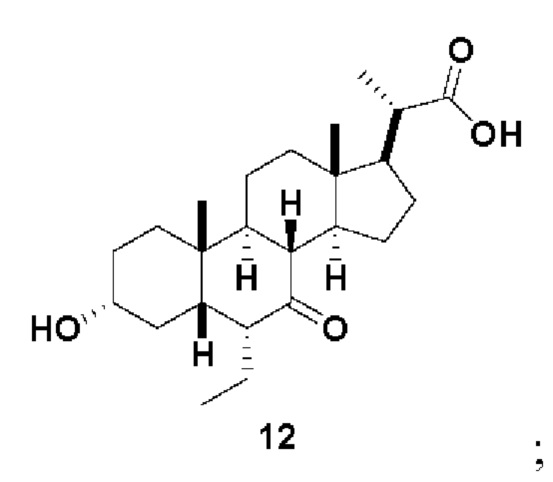 а также
а также
(2bvi) введение соединения 12 в реакцию с гидроксил-защитным агентом с получением соединения формулы (V)
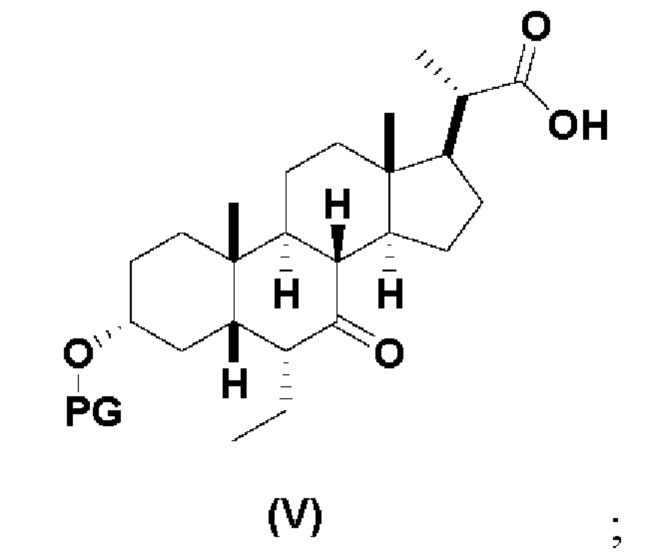
(3bi) введение соединения формулы (V) в реакцию с соединением формулы R3OC(O)Cl для получения соединения формулы 13
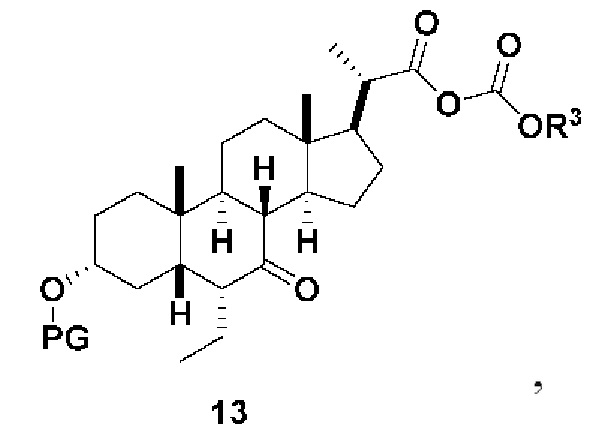
где R3 представляет собой алкильную группу; а также
(3bii) восстановление соединения 13 для получения соединения формулы (VI)
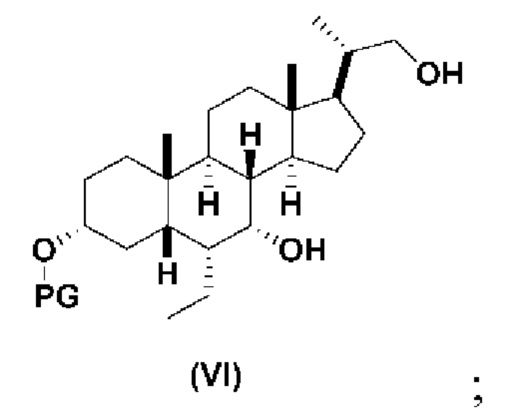
(4bi) введение соединения VI в реакцию с соединением формулы 15Е
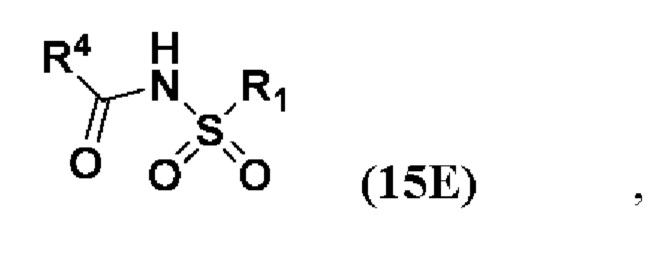
где R4 представляет собой имидазол-1-ил, алкил-О-, арил-О, Cl или -CCl3, в присутствии органического основания с получением соединения формулы 17
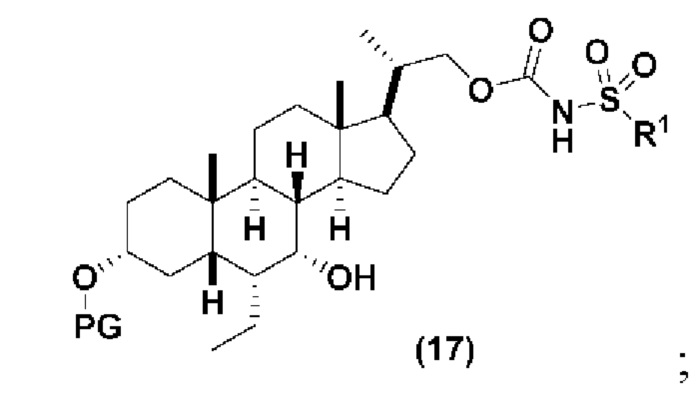 а также
а также
(4bii) снятие защитной группы в соединении формулы 17 для получения соединения формулы (II).
2. Способ по п. 1, где R1 представляет собой 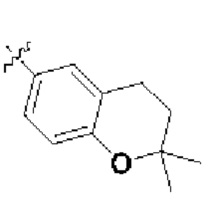 или
или 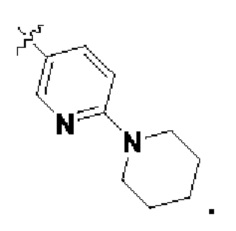
3. Способ по п. 1 или 2, отличающийся тем, что стадию (4bii) проводят в апротонном растворителе при температуре от около 0°С до около 80°С.
4. Способ по п. 2, отличающийся тем, что соединение формулы 15Е выбирают из соединений 15 В, 15С и 15D:
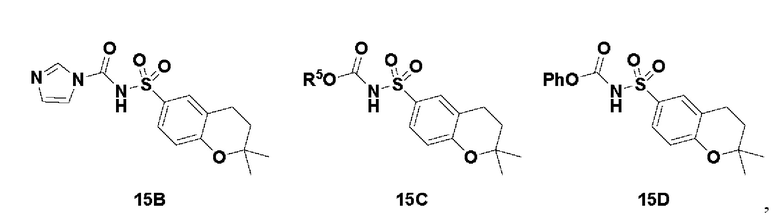
где R5 представляет собой алкил или арил.
5. Способ по п. 2, отличающийся тем, что соединение формулы 15Е выбирают из соединений 20В, 20С и 20D:
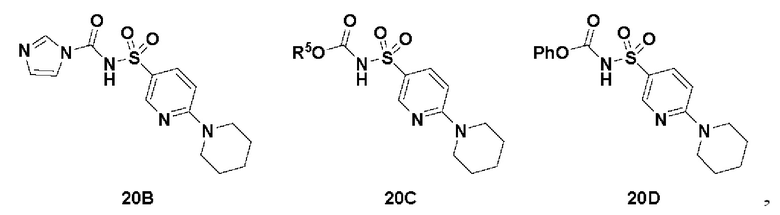
где R5 представляет собой алкил или арил.
6. Способ по любому одному из пп. 1-5, где PG представляет собой TBS.
7. Способ по любому одному из пп. 1-6, где R6 представляет собой метил.
| WO 2015181275 A1, 03.12.2015 | |||
| US 2014206657 A1, 24.07.2014 | |||
| US 2016289262 A1, 06.10.2016 | |||
| EA 201170276 A1, 31.10.2011. |

