Перекрестная ссылка на родственные заявки
По настоящей заявки испрашивается приоритет на основании предварительной заявки на патент США №62/506226, озаглавленной “КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ”, поданной 15 мая 2017 года, содержание которой включено в настоящее описание посредством ссылки во всей своей полноте.
Участие правительства
Настоящее изобретение было осуществлено при поддержке правительства США в рамках гранта Национального института по проблемам старения (подразделение Национального института здравоохранения) под номером U01AG047059. Правительство США имеет определенные права на настоящее изобретение.
Сущность изобретения
Различные варианты осуществления предоставляют новые соединения, фармацевтические композиции, содержащие такие соединения, и способы ингибирования или устранения потери синапсов в нейронах, модулирования изменения мембранного переноса в нейронах, и лечения снижения когнитивных функций и нейродегенеративных заболеваний и нарушений.
Некоторые варианты осуществления настоящего изобретения относятся к соединению формулы I или его фармацевтически приемлемой соли:

где:
каждый Ra, Rb, Rc, Rd и Re независимо выбран из группы, состоящей из Н, гидроксила, галогена, алкила, алкокси, CF3, SO2CH3 и морфолино;
R1 выбран из группы, состоящей из водорода, алкила, фенила или –CH=C(CH3)2; и
R2 представляет собой необязательно замещенную циклическую аминогруппу.
Некоторые варианты осуществления настоящего изобретения относятся к соединению, выбранному из группы, состоящей из


Варианты осуществления в настоящем документе описывают фармацевтическую композицию, содержащую: соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, или его фармацевтически приемлемую соль; и фармацевтически приемлемый носитель или разбавитель.
В некоторых вариантах осуществления описан способ лечения болезни Альцгеймера (AD), включающий введение субъекту терапевтически эффективного количества соединения или фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления описан способ ингибирования снижения когнитивных функций у субъекта, демонстрирующего снижение когнитивных функций или подверженного риску снижения когнитивных функций, включающий введение терапевтически эффективного количества соединения или фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления описан способ ингибирования эффекта бета–амилоида в отношении нейрона, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления описан способ лечения умеренного когнитивного нарушения при болезни Альцгеймера у субъекта, нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления описано применение соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, при производстве лекарственного средства для лечения болезни Альцгеймера.
В некоторых вариантах осуществления описано соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, для применения в лечении болезни Альцгеймера.
В некоторых вариантах осуществления описано соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, для применения в лекарственной терапии.
В некоторых вариантах осуществления описана фармацевтическая композиция, содержащая терапевтически эффективное количество соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, и его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или разбавитель.
Подробное описание
Настоящее изобретение не ограничивается конкретными описанными способами, композициями или методами, поскольку они могут изменяться. Терминология, используемая в описании, предназначена только для описания конкретных версий или вариантов осуществления и не предназначена для ограничения объема настоящего изобретения. Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно понимаются специалистом в данной области техники. Все публикации, указанные в настоящем документе, включены посредством ссылки во всей своей полноте. Никакую часть настоящего документа не следует считать признанием того, что авторы настоящего изобретения могут претендовать на более раннюю дату настоящего описания путем отсылки к более раннему изобретению.
Определения
В случае, когда предоставлен диапазон значений, подразумевается, что каждое промежуточное значение между верхним и нижним пределом этого диапазона и любым другим указанным или промежуточным значением в указанном диапазоне охватывается описанием. Например, если указан диапазон от 1 до 8 мкм, предполагается, что 2 мкм, 3 мкм, 5 мкм, 6 мкм и 7 мкм также раскрыты явным образом.
По настоящему описанию, заместители соединений по изобретению представлены в группах или в диапазонах. В частности, предполагается, что варианты осуществления изобретения включают все и каждую отдельную субкомбинацию членов таких групп и диапазонов. Например, термин “C1–6 алкил”, в частности, независимо описывает, например, метил (C1 алкил), этил (C2 алкил), пропил (C3 алкил), бутил (C4 алкил), пентил (C5 алкил) и гексил (C6 алкил) а также, например, C1–C2 алкил, C1–C3 алкил, C1–C4 алкил, C2–C3 алкил, C2–C4 алкил, C3–C6 алкил, C4–C5 алкил и C5–C6 алкил.
Используемые в настоящем документе существительные в форме единственного числа соответствуют “один или более” или “по меньшей мере один”, если не указано иное. То есть ссылка на любой элемент настоящего изобретения в форме единственного числа не исключает возможности того, что присутствует больше одного элемента.
Используемый в настоящем документе термин “около” означает плюс или минус 10% численного значения количества, с которым оно используется. Соответственно, около 50 мл означает “в диапазоне 45–55 мл”.
“Бета–амилоиды” или “Aβ” включают компоненты, содержащие растворимый амилоидный пептид, например, бета–амилоидные мономеры, бета–амилоидные олигомеры или комплексы бета–амилоидного пептида (в мономерной, димерной или полимерной форме) с другими растворимыми пептидами или белками, а также другие растворимые бета–амилоидные структуры, включая любой обработанный продукт белка–предшественника амилоида. Известно, что растворимые Aβ олигомеры являются нейротоксичными. Известно, что даже Aβ1–42 димеры нарушают синаптическую пластичность в срезах гиппокампа мышей. В соответствии с одной из теорий, известной в данной области, нативные мономеры Aβ1–42 считаются нейропротективными, и для нейротоксичности необходима самоассоциация Aβ–мономеров в растворимые бета–амилоидные олигомеры. Тем не менее некоторые мутантные Aβ–мономеры (арктическая мутация (E22G), как сообщается, связаны с наследственной болезнью Альцгеймера).
Если не указано специально, термин “активный ингредиент” следует понимать как относящийся к соединению по любому описанному в настоящем документе варианту осуществления.
“Применение” или “введение” и тому подобное, когда они используются в сочетании с соединениями по настоящему изобретению, относится к предоставлению соединений или фармацевтических композиций в соответствии с любым из описанных в настоящем документе вариантов осуществления субъекту, нуждающемуся в лечении. Предпочтительно субъект является млекопитающим, более предпочтительно человеком. Настоящее изобретение включает введение фармацевтической композиции по изобретению отдельно или в сочетании с другим терапевтическим средством. Когда фармацевтическую композицию по изобретению вводят в сочетании с другим терапевтическим средством, фармацевтическую композицию по изобретению и другое терапевтическое средство можно вводить одновременно или в разные моменты времени.
Термин “агонист” относится к соединению, присутствие которого приводит к биологической активности рецептора, которая является такой же, как биологическая активность, обусловленная присутствием встречающегося в природе лиганда для рецептора.
Используемый в настоящем документе термин “алканоил” или “алкилкарбонил” относится к алкильной группе, присоединенной к карбонильному радикалу. Примером алканоила является 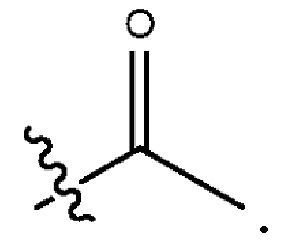
Используемый в настоящем документе термин “алкил” обозначает насыщенную углеводородную группу с прямой или разветвленной цепью. Примеры алкильных групп включают, но ими не ограничиваются, метил (Me), этил (Et), пропил (например, н–пропил и изопропил), бутил (например, н–бутил, изобутил, трет–бутил), пентил (например, н–пентил, изопентил, неопентил) и тому подобное. Алкильная группа может содержать от 1 до около 20, от 2 до около 20, от 1 до около 10, от 1 до около 8, от 1 до около 6, от 1 до около 4 или от 1 до около 3 атомов углерода. Предполагается, что “C1–C10 алкил” или “C1–10 алкил” включает C1, C2, C3, C4, C5, C6, C7, C8, C9 и C10 алкильные группы. Кроме того, например, “C1–C6 алкил” или “C1–6 алкил” обозначает алкил, имеющий от 1 до 6 атомов углерода. Термин “алкилен” относится к двухвалентной алкилсвязывающей группе. Примером алкилена является метилен (СН2).
Используемый в настоящем документе термин “алкенил” включает углеводородные цепи прямой или разветвленной конфигурации с одной или более, предпочтительно от одной до трех, двойными углерод–углеродными связями, которые могут встречаться в любой устойчивой точке вдоль цепи. Например, “C2–C6 алкенил” или “C2–6 алкенил” (или алкенилен) включает C2, C3, C4, C5 и C6 алкенильные группы. Примеры алкенила включают, но ими не ограничиваются, этенил, 1–пропенил, 2–пропенил, 2–бутенил, 3–бутенил, 2–пентенил, 3–пентенил, 4–пентенил, 2–гексенил, 3–гексенил, 4–гексенил, 5–гексенил, 2–метил–2–пропенил и 4–метил–3–пентенил.
Термин “алкокси” или “алкилокси” относится к –O–алкильной группе. “C1–C6 алкокси” или “C1–6 алкокси” (или алкилокси) включает C1, C2, C3, C4, C5 и C6 алкоксигруппы. Примеры алкоксигрупп включают, но ими не ограничиваются, метокси, этокси, пропокси (например, н–пропокси и изопропокси) и трет–бутокси.
Термин “алкоксилкокси” относится к алкоксигруппе, присоединенной к алкоксигруппе. Пример алкоксигруппы включает –O–(CH2)2–OCH3.
Используемый в настоящем документе термин “алкинил” включает углеводородные цепи с прямой или разветвленной конфигурацией, имеющие одну или более, предпочтительно от одной до трех, тройных углерод–углеродных связей, которые могут встречаться в любой устойчивой точке вдоль цепи. Например, “C2–C6 алкинил” включает C2, C3, C4, C5 и C6 алкинильные группы; такие как этинил, пропинил, бутинил, пентинил и гексинил.
Используемый в настоящем документе термин “бета–амилоидный эффект”, например, “нелетальный бета–амилоидный эффект” или “эффект бета–амилоидного олигомера”, относится к воздействию, в частности, нелетальному воздействию, в отношении клетки, которая контактирует с бета–амилоидами. Например, было обнаружено, что в случае, когда нейрон связывается с растворимым бета–амилоидным (“бета–амилоид”) олигомером, олигомеры связываются с разновидностью синапсов на субпопуляции нейронов in vitro. Это связывание может быть определено количественно в анализе, измеряющем, например, связывание бета–амилоидного олигомера in vitro. Другим документально подтвержденным эффектом бета–амилоидов является уменьшение количества синапсов, которое, как сообщается, соответствует около 18% в гиппокампе человека (Scheff et al, 2007), и может быть количественно определено (например, в анализе определения количества синапсов). В качестве другого примера, было обнаружено, что, когда нейрон связывается с бета–амилоидным (“Абета”) олигомером, модулируется мембранный перенос и происходит его изменение. Эта патология может быть визуализирована посредством различных анализов, включая, но им не ограничиваясь, МТТ–анализ. Например, желтые соли тетразолия эндоцитируются клетками и восстанавливаются до нерастворимого фиолетового формазана в эндосомальном пути. Уровень фиолетового формазана является отражением количества активно метаболизирующих клеток в культуре, а уменьшение количества формазана принимается в качестве меры гибели клеток или метаболической токсичности в культуре. Когда с помощью микроскопа наблюдаются клетки, которые контактируют с желтой солью тетразолия, фиолетовый формазан впервые виден во внутриклеточных везикулах, которые заполняют клетку. Со временем везикулы подвергаются экзоцитозу, и формазан осаждается в виде игольчатых кристаллов на внешней поверхности плазматической мембраны при воздействии водной среды на нерастворимый формазан. Другие эффекты бета–амилоидов включают снижение когнитивных функций, например, снижение способности формировать новые воспоминания и потеря памяти, что можно определить в анализах in vivo на животных моделях. В некоторых вариантах осуществления бета–амилоидное воздействие выбрано из индуцированной бета–амилоидным олигомером синаптической дисфункции, например, как видно из анализа in vitro, такого как анализ мембранного переноса, или анализ потери синапсов, или опосредованная бета–амилоидным олигомером сигма–2 рецепторная активация каспазы–3 или вызванная бета–амилоидом нейроннальная дисфункция, опосредованное бета–амилоидом снижение длительной потенциации (LTP) или снижение когнитивных функций в поведенческом анализе или у пациента, нуждающегося в таком исследовании.
В некоторых вариантах осуществления исследуемое соединение считается эффективным для лечения снижения когнитивных функций или заболевания, связанного с ним, когда оно может ингибировать эффект, ассоциированный с растворимыми видами бета–амилоидных олигомеров, в отношении нейрона более чем около на 10%, предпочтительно более чем около на 15% и предпочтительно более чем около на 20% по сравнению с негативным контролем. В некоторых вариантах осуществления исследуемое вещество считается эффективным, когда оно может ингибировать эффект, опосредованный продуктом процессинга белка–предшественника амилоида, более чем около на 10%, предпочтительно более чем на 15% и предпочтительно более чем на 20% по сравнению с положительным контролем. Хотя в настоящем описании основное внимание уделяется ингибированию нелетальных эффектов бета–амилоидов, таких как нарушения метаболизма нейронов и уменьшение количества синапсов, показано, что они коррелируют с когнитивной функцией и, кроме того, ожидается, что со временем это приведет к ослаблению (по сравнению с необработанными субъектами) последующих определяемых симптомов амилоидной патологии, в частности клинических симптомов, таких как 1) накопление фибрилл или бляшек, измеренное визуализирующими амилоид агентами, такими как фторбетапир, PittB или любой другой визуализирующий агент, 2) потеря синапса или гибель клеток, определяемая по гипометаболизму глюкозы, детектируемому с помощью FDG–PET, 3) изменения экспрессии белка или количества метаболита в головном мозге или организме, обнаруживаемое путем визуализации или обнаружения белков/метаболитов в спинномозговой жидкости, биопсии головного мозга или в плазме, полученной от пациентов, посредством ELISA (например изменения уровней и/или соотношений бета–амилоида 42, фосфорилированного тау белка, общего тау, измеренного посредством ELISA, или паттерны изменений экспрессии белка, обнаруживаемые на панели ELISA), 4) сосудистые нарушения головного мозга, определяемые по наличию сосудистой отечности или микрогеморрагии, обнаруживаемой посредством МРТ, и любых других симптомов, обнаруживаемых методами визуализации, и 5) утрата когнитивных функций, определяемая любым контрольным тестом для оценки когнитивных функций, таким как ADAS–Cog, MMSE, CBIC или любым другим инструментом когнитивного тестирования.
Используемый в настоящем документе термин “животное” включает, но ими не ограничивается, людей и не принадлежащих к человеческому роду позвоночных, таких как дикие, экспериментальные, домашние и сельскохозяйственные животные, и домашние питомцы.
Термин “антагонист” относится к объекту, например, соединению, антителу или фрагменту, присутствие которого приводит к снижению величины биологической активности рецептора. В некоторых вариантах осуществления присутствие антагониста приводит к полному ингибированию биологической активности рецептора. Используемый в настоящем документе термин “антагонист сигма–2–рецептора” используется для описания соединения, которое действует как “функциональный антагонист” в отношении сигма–2–рецептора в том смысле, что оно блокирует эффекты бета–амилоида, например индуцированную бета–амилоидным олигомером синаптическую дисфункцию, наблюдаемую, например, в анализе in vitro, таком как анализ мембранного переноса, анализ потери синапсов, или опосредованной бета–амилоидным олигомером сигма–2-рецепторной активации каспазы–3, или в поведенческом анализе, или у пациента, нуждающегося в таком ингибировании. Функциональный антагонист может действовать непосредственно путем ингибирования связывания, например, бета–амилоидного олигомера с сигма–2–рецептором, или косвенно, путем нарушения передачи нисходящего сигнала от бета–амилоидного олигомера, связывающего сигма–2–рецептор.
Используемый в настоящем документе термин “арил” относится к моноциклическим или полициклическим (например, имеющим 2, 3 или 4 конденсированных кольца) ароматическим углеводородам, таким как, например, фенил, нафтил, антраценил, фенантренил, инданил, инденил и тому подобное. В некоторых вариантах осуществления арильные группы имеют от 6 до около 20 атомов углерода. В некоторых вариантах осуществления арильные группы имеют от 5 до около 10 атомов углерода.
Используемый в настоящем документе термин “арилалкил” относится к арильной группе, присоединенной к алкильному радикалу. В предпочтительных вариантах осуществления алкил представляет собой C1–6 алкил.
Используемый в настоящем документе термин “ароил” или “арилкарбонил” относится к арильной группе, присоединенной к карбонильному радикалу. Примеры ароила включают, но им не ограничиваются, бензоил.
Используемый в настоящем документе термин “способность проникать в головной мозг” относится к способности лекарственного средства, антитела или фрагмента преодолевать гематоэнцефалический барьер. В некоторых вариантах осуществления исследование фармакокинетики (pK) на животных, например, исследование фармакокинетики/гематоэнцефалического барьера на мышах, может использоваться для определения или прогнозирования проникновения в головной мозг. В некоторых вариантах осуществления могут вводиться различные концентрации соединения или фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, например, в количестве 3, 10 и 30 мг/кг, например, перорально в течение 5 дней, и определяться различные pK свойства, например, на животной модели. В некоторых вариантах осуществления определяют дозозависимые уровни в плазме и мозге. В некоторых вариантах осуществления, Cmax мозга > 100, 300, 600, 1000, 1300, 1600 или 1900 нг/мл. В некоторых вариантах осуществления эффективное проникновение в головной мозг определяется как соотношение мозг/плазма, составляющее >0,1, >0,3, >0,5, >0,7, >0,8, >0,9, предпочтительно >1 и более предпочтительно >2, >5 или >10. В других вариантах осуществления эффективное проникновение в головной мозг определяется как более чем около 0,1%, 1%, 5%, более чем около 10% и предпочтительно более чем около 15% от введенной дозы, пересекающей ГЭБ через заранее определенное время. В определенных вариантах осуществления дозу вводят перорально (p.o,). В других вариантах осуществления перед измерением pK свойств дозу вводят внутривенно (i.v.). Фармакокинетические анализы и проникновение в головной мозг описаны в примере 7.
Используемый в настоящем документе термин “снижение когнитивных функций” может соответствовать любому негативному изменению когнитивной функции животного. Например, снижение когнитивных функций включает, но ими не ограничивается, потерю памяти (например, потерю поведенческой памяти), неспособность приобретать новые воспоминания, спутанность сознания, нарушение суждения, изменения личности, дезориентацию или любое их сочетание. Таким образом, соединение, которое эффективно в лечении снижения когнитивных функций, может быть эффективным посредством восстановления долговременной нейрональной потенциации (LTP) или долговременной нейрональной депрессии (LTD) или баланса определяемой электрофизиологически синаптической пластичности; ингибирования, лечения и/или ослабления нейродегенерации; ингибирования, лечения и/или ослабления общего амилоидоза; ингибирования, лечения, ослабления интенсивности одного или более процессов – продукции амилоидов, амилоидной сборки, амилоидной агрегации и связывания амилоидных олигомеров; ингибирования, лечения и/или ослабления нелетального эффекта одного или более бета–амилоидов в отношении нейрона (например, потеря или дисфункция синапса и патологический мембранный перенос); и любого их сочетания. Кроме того, это соединение может также быть эффективным при лечении нейродегенеративных заболеваний, связанных с бета–амилоидом, включая, но этим не ограничиваясь, деменцию, включая, но этим не ограничиваясь, болезнь Альцгеймера (AD), включая легкую форму болезни Альцгеймера, синдром Дауна, сосудистую деменцию (церебральная амилоидная ангиопатия и инсульт), деменцию с тельцами Леви, ВИЧ–деменция, легкое когнитивное расстройство (MCI); возрастное ухудшение памяти (AAMI); возрастное снижение когнитивных функций (ARCD), доклиническую болезнь Альцгеймера (PCAD); и когнитивные нарушения без деменции (CIND).
Используемый в настоящем документе термин “контактирование” относится к совместному сведению или объединению молекул (или молекулы со структурой более высокого уровня, такой как клетка или клеточная мембрана), так чтобы они находились на расстоянии, которое допускает межмолекулярные взаимодействия, например, нековалентное взаимодействие между двумя пептидами, или одним белком и другим белком или другой молекулой, такой как малая молекула. В некоторых вариантах осуществления контактирование происходит в растворе, в котором объединенные или контактирующие молекулы смешаны в общем растворителе и могут свободно связываться. В некоторых вариантах осуществления контактирование может осуществляться внутри или иным образом внутри клетки или в бесклеточной среде. В некоторых вариантах осуществления бесклеточная среда представляет собой лизат, полученный из клетки. В некоторых вариантах осуществления клеточный лизат может представлять собой цельноклеточный лизат, ядерный лизат, цитоплазматический лизат и их сочетание. В некоторых вариантах осуществления бесклеточный лизат представляет собой лизат, полученный в результате экстракции и выделения ядра, причем ядра клеточной популяции удаляются из клеток и затем лизируются. В некоторых вариантах осуществления ядра не лизируются, но все еще считаются бесклеточной средой. Молекулы могут быть объединяться путем смешивания, например, перемешиванием с завихрением, встряхиванием и тому подобное.
Используемый в настоящем документе термин “циклический амино” или “циклическая аминогруппа” представляет собой гетероциклоалкильную или гетероарильную группу, содержащую азотсодержащий радикал, что позволяет связываться через атом азота. Группа может быть представлена формулой:
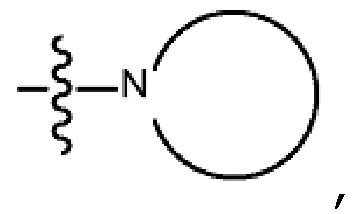 где
где 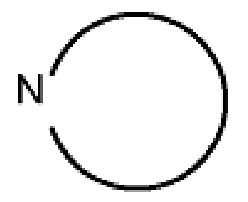 представляет собой любое гетероциклическое или гетероароматическое кольцо, содержащее 0–3 дополнительных гетероатома, выбранных из азота, серы и кислорода.
представляет собой любое гетероциклическое или гетероароматическое кольцо, содержащее 0–3 дополнительных гетероатома, выбранных из азота, серы и кислорода.
Используемый в настоящем документе термин “циклоалканоил” или “циклоалкилкарбонил” предназначен для описания циклоалкильной группы, присоединенной к карбонильному радикалу. Примеры циклоалканоила включают, но ими не ограничиваются,
 и
и 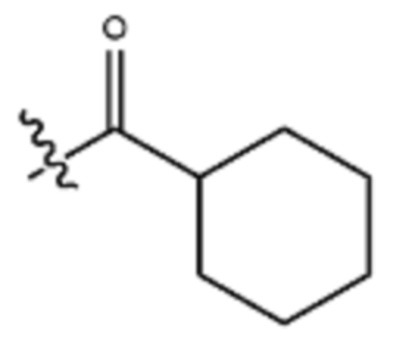 .
.
Используемый в настоящем документе термин “циклоалкил” относится к неароматическим циклическим углеводородам, включая циклизованные алкильные, алкенильные и алкинильные группы, которые содержат до 20 образующих кольцо атомов углерода. Циклоалкильные группы могут включать моно– или полициклические (например, имеющие 2, 3 или 4 конденсированных кольца) кольцевые системы, а также спироциклические системы. Циклоалкильная группа может содержать от 3 до около 15, от 3 до около 10, от 3 до около 8, от 3 до около 6, от 4 до около 6, от 3 до около 5 или от 5 до около 6 образующих кольцо атомов углерода. Кольцевые атомы углерода циклоалкильной группы могут быть необязательно замещены оксо или сульфидо. Примеры циклоалкильных групп включают, но ими не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, норборнил, норпинил, норкарнил, адамантил и тому подобное. В определение циклоалкила также включены фрагменты, в которых одно или более ароматических колец конденсированы с (т.е. имеют общую связь с) циклоалкильным кольцом, например, бензо– или тиенильные производные циклопентана, циклопентена, циклогексана и тому подобное (например, 2,3–дигидро–1H–инден–1–ил или 1H–инден–2(3H)–он–1–ил). Предпочтительно, “циклоалкил” относится к циклизованным алкильным группам, которые содержат до 20 образующих кольцо атомов углерода. Примеры циклоалкила предпочтительно включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, адамантил и тому подобное.
Термин “циклоалкилалкил” относится к циклоалкильной группе, присоединенной к алкильному радикалу. В предпочтительных вариантах осуществления алкил представляет собой C1–6 алкил.
Термин “лекарствоподобные свойства” используется в настоящем документе для описания фармакокинетических характеристик и характеристик стабильности соединения при введении; включая проникновение в головной мозг, метаболическую стабильность и/или плазменную стабильность.
Используемый в настоящем документе термин “эффективное количество” относится к количеству, которое приводит к заметному ингибированию, по меньшей мере, одного симптома или параметра конкретного расстройства или патологического процесса. Например, количество раскрываемого соединения в соответствии с любым из вариантов осуществления, описанным в настоящем документе, которое обеспечивает заметно более низкое снижение количества синапсов в присутствии бета–амилоидного олигомера, определяется как эффективное количество, поскольку оно снижает интенсивность патологического процесса, даже если не изменяются, по крайней мере, сразу, клинические симптомы амилоидной патологии.
Используемый в настоящем документе термин “галоген” или “галоген” включает фтор, хлор, бром и йод.
Используемый в настоящем документе термин “галогеналкокси” представляет собой галогеналкильную группу, как определено в данном документе, с определенным количеством атомов углерода, присоединенным через кислородный мостик. Например, “C1–C6 галогеналкокси” или “C1–6 галогеналкокси” включает C1, C2, C3, C4, C5 и C6 галогеналкоксигруппы. Примером галогеналкоксигруппы является OCF3. Используемый в настоящем документе термин “тригалогенметокси” относится к метоксигруппе, имеющей три галогеновых заместителя. Примеры тригалогенметоксигрупп включают, но ими не ограничиваются, –OCF3, –OCClF2, –OCCl3 и тому подобное.
Используемый в настоящем документе термин “галогеналкил” включает насыщенные алифатические углеводородные группы с разветвленной и прямой цепью, имеющие определенное количество атомов углерода, замещенные одним или более галогенами. Примеры галогеналкильных групп включают, но ими не ограничиваются, CF3, C2F5, CHF2, CCl3, CHCl2, C2Cl5, CH2CF3 и тому подобное.
Используемый в настоящем документе термин “гетероарильные” группы относится к ароматическому гетероциклу, имеющему до 20 образующих кольцо атомов и имеющему, по меньшей мере, один член гетероатомного кольца (образующий кольцо атом), такой как сера, кислород или азот. В некоторых вариантах осуществления гетероарильная группа имеет, по меньшей мере, один или более атомов, образующих гетероатомное кольцо, каждый из которых независимо выбран из серы, кислорода и азота. Гетероарильные группы включают моноциклические и полициклические (например, имеющие 2, 3 или 4 конденсированных кольца) системы. Примеры гетероарильных групп включают, без ограничения, пиридил (также известный как пиридинил), пиримидинил, пиразинил, пиридазинил, триазинил, фурил, хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пиррил (также известный как пирролил), оксазолил, бензофурил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, триазолил, тетразолил, индазолил, 1,2,4–тиадиазолил, изотиазолил, бензотиенил, пуринил, карбазолил, бензимидазолил, индолинил и тому подобное. В некоторых вариантах осуществления гетероарильная группа имеет от 1 до около 20 атомов углерода, а в дополнительных вариантах осуществления от около 1 до около 5, от около 1 до около 4, от около 1 до около 3, от около 1 до около 2 атомов углерода в качестве образующих кольцо атомов. В некоторых вариантах осуществления гетероарильная группа содержит от 3 до около 14, от 3 до около 7 или от 5 до 6 образующих кольцо атомов. В некоторых вариантах осуществления гетероарильная группа имеет от 1 до около 4, от 1 до около 3 или от 1 до 2 гетероатомов.
Используемый в настоящем документе термин “гетероциклоалкокси” относится к –О–гетероциклоалкильной группе. Примером гетероциклоалкоксигруппы является 
Используемый в настоящем документе термин “гетероциклоалкил” или “гетероциклил” относится к неароматической гетероциклильной группе, имеющей до 20 образующих кольцо атомов, включая циклизованные алкильные, алкенильные и алкинильные группы, где один или более атомов углерода, образующих кольцо, заменены на гетероатом, такой как атом O, N или S. Гетероциклоалкильные группы могут быть моно– или полициклическими (например, как конденсированные, так и спиросистемы). Так, например, “гетероциклоалкильные” группы включают морфолино, тиоморфолино, пиперазинили, тетрагидрофуранил, тетрагидротиенил, 2,3–дигидробензофурил, 1,3–бензодиоксол, бензо–1,4–диоксан, пиперидинил, пирролидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, оксазолидинил, тиазолидинил, имидазолидинил, пирролидин–2–он–3–ил и тому подобное. Образующие кольцо атомы и гетероатомы гетероциклоалкильной группы могут быть необязательно замещены оксо или сульфидо. Например, образующий кольцо атом S может быть замещен 1 или 2 оксо (то есть образует S(O) или S(O)2). Например, образующий кольцо атом C может быть замещен оксо (т.е. образует карбонил). Определение гетероциклоалкила также охватывает фрагменты, в которых одно или более ароматических колец конденсированы с (т.е. имеют общую связь с) неароматическим гетероциклическим кольцом, например пиридинил, тиофенил, фталимидил, нафталимидил и бензо производные гетероциклов, например индолин, изоиндолин, изоиндолин–1–он–3–ил, 4,5,6,7–тетрагидротиено[2,3–с]пиридин–5–ил, 5,6–дигидротиено[2,3–с]пиридин–7(4H)–он–5–ил и 3,4–дигидроизохинолин–1(2H)–он–3–ильные группы. Образующие кольцо атомы углерода и гетероатомы гетероциклоалкильной группы могут быть необязательно замещены оксо или сульфидо. В некоторых вариантах осуществления гетероциклоалкильная группа имеет от 2 до около 20 атомов углерода или от 3 до 20 атомов углерода. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 3 до около 14, от 3 до около 7 или от 5 до 6 образующих кольцо атомов. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 1 до 4 гетероатомов. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 0 до 3 двойных связей. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 0 до 2 тройных связей.
В настоящей заявке, термин “высокое сродство” относится к соединению, которое имеет значение Ki меньше 600 нМ, 500 нМ, 400 нМ, 300 нМ, 200 нМ, меньше 150 нМ, меньше 100 нМ, меньше 80 нМ, меньше 60 нМ или предпочтительно меньше 50 нМ в анализе связывания с сигма–рецептором, например, относительно [3H]–DTG, как описано в статье Weber et al., Proc. Natl. Acad. Sci (USA) 83: 8784–8788 (1986), включенной в настоящий документ посредством ссылки, который определяет аффинность связывания соединений относительно рецепторных участков как сигма–1, так и сигма–2. Особенно предпочтительные соединения показывают значения Ki меньше чем около 150 нМ, предпочтительно меньше чем 100 нМ, меньше чем около 60 нМ, меньше чем около 10 нМ или меньше чем около 1 нМ относительно [3H]–DTG.
Термины “гидроксил” и “гидрокси” используются взаимозаменяемо для обозначения группы ОН.
Термин “улучшает” означает, что предмет изобретения изменяет характеристики и/или физические свойства ткани, для которой оно предусмотрено, применяется или вводится. Термин “улучшает” также может использоваться в связи с болезненным состоянием, и, таким образом, что когда болезненное состояние “улучшается”, симптомы или физические характеристики, ассоциированные с болезненным состоянием, ослабевают, уменьшаются, устраняются, их появление задерживается во времени или они предотвращается.
Термин “ингибирование” включает блокирование, предотвращение определенного результата или процесса, или восстановление обратного результата или процесса. С точки зрения профилактики или лечения путем введения соединения по настоящему изобретению, “ингибирование” включает защиту (частичную или полную) от симптомов или задержку их появления, ослабление симптомов или защиту от болезни, состояния или нарушения или их уменьшение или устранение.
Термин “ингибирование дефицита переноса” относится к способности блокировать индуцированный растворимым Aβ–олигомером дефицит переноса через мембраны в клетках, предпочтительно нейронах. Соединение, способное ингибировать дефицит переноса, имеет ЕС50 <20 мкМ, меньше 15 мкМ, меньше 10 мкМ, меньше 5 мкМ и предпочтительно меньше 1 мкМ согласно анализу переноса через мембрану и, кроме того, на по меньшей мере 50%, предпочтительно по меньшей мере 60%, более предпочтительно по меньшей мере 70% максимальное ингибирование эффектов бета–амилоидного олигомера при вызнанном растворимыми бета–амилоидными олигомерами дефиците переноса через мембраны, например, как описано в примере 6.
Термин "log P" означает коэффициент распределения соединения. Коэффициент распределения представляет собой соотношение концентрации неионизированного соединения в каждой из двух жидких фаз, например, октаноле и воде. Для измерения коэффициента распределения ионизируемых растворенных соединений рН водной фазы регулируют таким образом, чтобы преобладающая форма соединения была неионизированной. Логарифм соотношения значений концентрации неионизированного растворенного соединения в растворителях называется log P. Log P является мерой липофильности. Например,
log Рокт./вода=log([раств. вещ.]октанол/[раств. вещ.]неионизиров., вода).
Используемый в настоящем документе термин “метаболическая стабильность” относится к способности соединения сохраняться после пресистемного метаболизма (деградация или конъюгация лекарственного средства, вводимого перорально, в кишечнике и печени). Эта стабильность может быть определена, например, in vitro путем экспозиции соединений микросомам печени мыши или человека. В некоторых вариантах осуществления хорошая метаболическая стабильность соответствует t1/2 >5 минутам, >10 минутам, >15 минутам, >20 минутам и предпочтительно >30 минутам при экспозиции соединения микросомам печени мыши или человека. В некоторых вариантах осуществления хорошая метаболическая стабильность относится к скорости собственного клиренса (Clint), составляющей <300 мкл/мин/мг, предпочтительно <200 мкл/мин/мг и более предпочтительно <100 мкл/мин/мг.
Термин “n–членный”, где n представляет собой целое число, обычно относится к количеству образующих кольцо атомов во фрагменте, в котором количество атомов, образующих кольцо, равно n. Например, пиридин является примером 6–членного гетероарильного кольца, а тиофен является примером 5–членной гетероарильной группы.
Используемый в настоящем документе термин “природный лиганд” относится к лиганду, присутствующему у субъекта, который может связываться с белком, рецептором, мембранным липидом или другим партнером по связыванию in vivo или который реплицируется in vitro. Природный лиганд может иметь синтетическое происхождение, но также должен присутствовать в природе и без человеческого вмешательства в организм субъекта. Например, известно, что бета–амилоидные олигомеры существуют в организме человека. Таким образом, бета–амилоидные олигомеры, обнаруживаемые у субъекта, должны рассматриваться как природные лиганды. Связывание бета–амилоидных олигомеров с партнером по связыванию может реплицироваться in vitro с применением технологий рекомбинации или синтеза, однако бета–амилоидный олигомер все равно считается природным лигандом независимо от того, каким образом бета–амилоидные олигомер получен или произведен. Синтетическая малая молекула, которая также может связываться с тем же партнером по связыванию, не является природным лигандом, если она не существует в организме субъекта. Например, описанные в настоящем документе соединения, обычно отсутствуют у субъекта и поэтому не могут считаться природными лигандами.
Используемый в настоящем документе термин “нейрон” может применяться в отношении к отдельной клетке или популяции клеток. В некоторых вариантах осуществления нейрон является первичным нейроном. В некоторых вариантах осуществления нейрон является иммортализованным или трансформированным нейроном или стволовой клеткой. Первичный нейрон является нейроном, который не может видоизменяться в другие типы нейронов, такие, как глиальные клетки. Стволовая клетка представляет собой клетку, которая может видоизменяться в нейроны и другие типы нейронов, такие как глиоцит. В некоторых вариантах осуществления в анализах применяют композицию, включающую, по меньшей мере, один нейрон и не содержащую глиальные клетки. В некоторых вариантах осуществления композиция включает меньше, чем примерно 30%, 25%, 20%, 15%, 10%, 5% или 1% глиальных клеток, которые поглощают и накапливают бета–амилоид. Первичный нейрон может быть взят из любого участка головного мозга животного. В некоторых вариантах осуществления нейрон представляет собой гиппокампальную или корковую клетку. Присутствие глиальных клеток может определяться любым способом. В некоторых вариантах осуществления глиальные клетки обнаруживают по присутствию GFAP, а нейроны могут обнаруживаться путем положительного окрашивания антителами, направленными против МАР2.
Используемый в настоящем документе термин “необязательно замещенный” означает, что замещение является необязательным и поэтому включает как незамещенные, так и замещенные атомы и фрагменты. “Замещенный” атом или фрагмент означает, что любой водород в указанном атоме или фрагменте может быть заменен вариантом из указанной группы заместителей при условии, что нормальная валентность указанного атома или фрагмента не превышена, и что замещение обеспечивает стабильное соединение. Например, если метильная группа (то есть СН3) является необязательно замещенной, то до 3 атомов водорода на атоме углерода могут быть заменены группами заместителей. Группы заместителей включают, но ими не ограничиваются, алканоил, алкокси, алкоксиалкил, (алкокси)алкоксиалкил, алкоксикарбонил, алкил, арилокси, арилоил, циклоалканоил, замещенный или незамещенный С3–С10 циклоалкил, –OC(O)NCH(CH3)2, (N, N–диметиламино)пиридинил, (N, N–диметиламино)сульфонил, галоген, гетероциклил, (гетероциклил)алкоксиалкил, гетероциклоалкил, гидроксил, гидроксиалкил, метилпиперидинил, метилсульфонил, метилсульфонилфенил, морфолинилпиридинил, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, перфторалкил, фенил, пиперидинил, пирролидинилпиридинил, тетрагидропиранил, CF3. Замещенная алкильная группа, например, указывает, что один или более атомов водорода в алкильной группе заменены замещающей группой, выбранной, но ими не ограничиваясь, из галогена, гидроксила, алкокси, гетероциклоалкокси, алкоксиалкокси, C(O)OMe и C(O)OEt. Например, замещенная арильная группа указывает на то, что один или более атомов водорода в арильной группе заменены группой заместителей, выбранной из, но ими не ограничиваясь, –SO2Me или фенильной группы. Замещенная гетероарильная группа, например, указывает, что один или более атомов водорода в гетероарильной группе заменены группой заместителей, выбранным из, но ими не ограничиваясь гетероциклоалкила, гетероарила, N, N–диметиламино. Замещенная гетероциклоалкильная группа, например, указывает на то, что один или более атомов водорода в гетероциклоалкильной группе заменены группой заместителей, выбранной из, но ими не ограничиваясь, гетероциклалкила, гетероарила, N,N–диметиламино, гидроксила, алкокси, алкоксикарбонила и алкила, арила, сульфонила, диметиламиносульфонила, ароила, циклоалканоила, алканоила и –OC(O)NCH(CH3)2. В некоторых случаях два атома водорода на одном и том же атоме углерода, например, гетероциклильной или алкильной группы, заменены группой с образованием спиросоединения, выбранного из, но ими не ограничиваясь, например, 
Термин “частичный агонист” означает соединение, присутствие которого обеспечивает в результате биологическую активность рецептора, которая относится к тому же типу, что и биологическая активность, являющаяся результатом присутствия встречающегося в природе лиганда для рецептора, но меньшей величины.
Фраза “фармацевтически приемлемый” относится к молекулярным веществам и композициям, которые обычно признаются безопасными и нетоксичными. В частности, фармацевтически приемлемые носители, разбавители или другие эксципиенты, используемые в фармацевтических композициях по настоящему изобретению, являются физиологически переносимыми, совместимыми с другими ингредиентами и обычно не вызывают аллергическую или подобную неблагоприятную реакцию (например, расстройство желудка, головокружение и тому подобное) при введении пациенту. Предпочтительно, используемый в настоящем документе термин “фармацевтически приемлемый” означает одобренный управляющим органом федерального или местного правительства штата или внесенный в перечень Фармакопеи США или другой общепризнанной фармакопеи для применения на животных и, в частности, человеке.
Используемая в настоящем документе фраза “фармацевтически приемлемая(ые) соль(и)”, включает соли соединений по изобретению, которые безопасны и эффективны для применения у млекопитающих и которые обладают желаемой биологической активностью. Фармацевтически приемлемые соли включают соли кислотных или основных групп, присутствующие в соединениях по изобретению или в соединениях, определенных в соответствии со способами по изобретению. Фармацевтически приемлемые соли присоединения кислоты включают, но ими не ограничиваются, следующие соли: гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п–толуолсульфонат и памоат (то есть 1,1'–метилен–бис–(2–гидрокси–3–нафтоат)). Некоторые соединения по настоящему изобретению могут образовывать фармацевтически приемлемые соли с различными аминокислотами. Подходящие основные соли включают, но ими не ограничиваются, соли алюминия, кальция, лития, магния, калия, натрия, цинка, железа и диэтаноламина. Фармацевтически приемлемые соли присоединения оснований также образуются с аминами, такими как органические амины. Примерами подходящих аминов являются N, N'–дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, дициклогексиламин, этилендиамин, N–метилглюкамин и прокаин.
Используемый в настоящем документе термин “фармацевтически приемлемый носитель” включает любой из стандартных фармацевтических носителей, таких как забуференный фосфатом физиологический раствор, воду, эмульсии, такие как эмульсия типа “масло/вода” или “вода/масло”, и различные типы смачивающих веществ. Термин также охватывает любые вещества, одобренные управляющим органом федерального правительства США или внесенный в перечень Фармакопеи США или другой общепризнанной фармакопеи для применения на животных и, в частности, на человеке.
Термин “селективность” или “селективный” означает разницу в аффинности связывания соединения (Ki) для сигма–рецептора, например, сигма–2–рецептора, по сравнению с другим отличным от сигма рецептором. Соединение обладает высокой селективностью к сигма–рецептору в синаптических нейронах. Ki для сигма–2 рецептора, или для обоих сигма–2- и сигма–1-рецепторов, сравнивают с Ki для другого отличного от сигма-рецептора. В некоторых вариантах осуществления соединение представляет собой селективный антагонист сигма–2-рецептора или лиганд сигма–1-рецептора и имеет, по меньшей мере, в 10, 20, 30, 50, 70, 100 или 500 и более раз большую аффинность связывания с сигма–рецептором по сравнению со значением отличного от сигма-рецептора, что определяется сравнением значений константы диссоциации связывания Ki или значений IC50 или константы связывания на различных рецепторах. Для оценки значений Ki или IC50 для различных рецепторов может быть использован любой известный протокол анализа, например, путем наблюдения вытеснения из рецепторов радиоактивно–меченного соединения с известной константой диссоциации, например, с применением способа Cheng и Prusoff (1973) (Biochem. Pharmacol. 22, 3099–3108) или способа, конкретно описанного в настоящем документе.
Используемый в настоящем документе термин “стабильность в плазме” означает распад соединений в плазме, например, с помощью ферментов, таких как гидролаза и эстераза. Могут применяться любые из различных in vitro анализов. Исследуемое соединения инкубируют в плазме в течение различных периодов времени. Процентное содержание исходного соединения (анализируемого вещества), остающегося в каждый момент времени, показывает стабильность плазмы. Низкие характеристики стабильности могут соответствовать низкой биодоступности. Cтабильность может определяться как хорошая, если количество остающегося в плазме анализируемого вещества превышает 50% через 30 мин, превышает 50% через 45 минут, предпочтительно превышает 50% через 60 минут.
“Сигма–2–лиганд” относится к соединению, которое связывается с сигма–2–рецептором и включает агонисты, антагонисты, частичные агонисты, обратные агонисты и просто конкуренты за другие лиганды этого рецептора или белка.
Термин “соединение антагониста сигма–2–рецептора” означает молекулу, которая связывается с сигма–2–рецептором в измеримом количестве и действует как функциональный антагонист по отношению к вызываемой действием бета–амилоидного олигомера синаптической дисфункции в результате связывания с сигма–2–рецептором.
Термины “субъект”, “индивидуум” или “пациент” используются взаимозаменяемо и, как они используются в настоящем документе, предназначены для обозначения человека и других животных, не относящихся к человеческому роду. Животные, не относящиеся к человеческому роду, включают всех позвоночных животных, например, млекопитающих и немлекопитающих, таких как нечеловекообразные приматы, овцы, собаки, кошки, коровы, лошади, цыплята, земноводные и рептилии, хотя предпочтительными являются млекопитающие, такие как нечеловекообразные приматы, овцы, собаки, кошки, коровы и лошади. Предпочтительные субъекты включают пациентов–людей. Способы являются особенно подходящими для лечения пациентов–людей, имеющих заболевание или расстройство, описанное в настоящем документе.
“Анализируемое соединение” представляет собой соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, которое исследуется в любом анализе. Анализы включают любые анализы in vivo или in vitro, компьютерную модель или симуляцию, виртуальные исследования лекарственных средств, методы анализа стволовых клеток и генетические анализы, неинвазивные методы визуализации и тому подобное.
Используемый в настоящем документе термин “терапевтический” означает средство, используемое для лечения, борьбы с нежелательным состоянием или заболеванием, уменьшения его интенсивности, защиты от него или его регресса у субъекта.
“Терапевтически эффективное количество” соединения, его фармацевтически приемлемой соли или фармацевтической композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, представляет собой количество, достаточное для вызывания определенного эффекта, по меньшей мере, в отношении одного симптома или параметра конкретного заболевания или расстройства. Терапевтический эффект может быть объективным (т.е. измеряемым при помощи какого–либо теста или маркера) или субъективным (т.е. субъект сам характеризует или ощущает эффект или физически наблюдает изменения). Терапевтически эффективное количество соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, может изменяться в широких пределах от 0,01 до около 500 мг/кг, от около 0,01 до около 250 мг/кг, от около 0,01 до около 25 мг/кг, от около 0,05 мг/кг до около 20 мг/кг, от около 0,1 мг/кг до около 400 мг/кг, от около 0,1 мг/кг до около 200 мг/кг, от около 0,1 мг/кг до около 25 мг/кг, от около 0,1 до около 10 мг/кг, от около 0,2 до около 5 мг/кг, от около 1 мг/кг до около 300 мг/кг, от около 10 мг/кг до около 100 мг/кг массы тела. Эффект, рассматриваемый в настоящем документе, включает как медицинское терапевтическое, так и/или профилактическое лечение, в зависимости от ситуации. Конкретная доза соединения, вводимая в соответствии с настоящим изобретением для получения терапевтических и/или профилактических эффектов, определяется в соответствии с конкретными обстоятельствами данного случая, включая, например, вводимое соединение, способ введения, совместное введение других активных ингредиентов, состояние, подвергаемое лечению, активность используемого конкретного соединения, конкретную используемую композицию, возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, способ введения и скорость выведения конкретного используемого соединения и продолжительность лечения. Вводимое терапевтически эффективное количество будет определяться врачом с учетом вышеуказанных соответствующих обстоятельств и по результатам медицинской оценки. Терапевтически эффективное количество соединения согласно изобретению является таким количеством, при введении которого в композиции с физиологически переносимым эксципиентом оно является достаточным для достижения эффективной системной концентрации или местной концентрации в ткани. Общая суточная доза соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе, вводимых человеку или другому животному одной или разделенными дозами, может составлять, например, от около 0,01 мг/кг до около 500 мг/кг, от около 0,01 до около 250 мг/кг, от около 0,01 до около 25 мг/кг, от около 0,05 до около 20 мг/кг, от около 0,1 до около 400 мг/кг, от около 0,1 до около 200 мг/кг, от около 0,1 до около 25 мг/кг, от около 0,1 до около 10 мг/кг, от около 0,2 до около 5 мг/кг, от около 1 до около 300 мг/кг, от около 10 мг/кг до около 100 мг/кг массы тела в сутки. Однодозные фармацевтические композиции по любому из вариантов осуществления, описанного в настоящем документе, могут содержать ее частичные количества или дольные единицы, которые вместе составляют суточную дозу. Например, соединения в соответствии с любым из вариантов осуществления, описанным в настоящем документе, могут вводиться в режиме от 1 до 4 раз в сутки, например, один, два, три раза или четыре раза в сутки. В некоторых вариантах осуществления терапевтически эффективное количество соединения в соответствии с любым из вариантов осуществления, описанным в настоящем документе, может составлять от около 0,01 до около 25 мг/кг/сутки. В некоторых вариантах осуществления терапевтически эффективное количество находится между нижним пределом, составляющим около 0,01 мг/кг массы тела, около 0,1 мг/кг массы тела, около 0,2 мг/кг массы тела, около 0,3 мг/кг массы тела, около 0,4 мг/кг массы тела, около 0,5 мг/кг массы тела, около 0,60 мг/кг массы тела, около 0,70 мг/кг массы тела, около 0,80 мг/кг массы тела, около 0,90 мг/кг массы тела, около 1 мг/кг массы тела, около 2,5 мг/кг массы тела, около 5 мг/кг массы тела, около 7,5 мг/кг массы тела, около 10 мг/кг массы тела, около 12,5 мг/кг массы тела, около 15 мг/кг массы тела, около 17,5 мг/кг массы тела, около 20 мг/кг массы тела, около 22,5 мг/кг массы тела и около 25 мг/кг массы тела; и верхним пределом, составляющим 25 мг/кг массы тела, около 22,5 мг/кг массы тела, около 20 мг/кг массы тела, около 17,5 мг/кг массы тела, около 15 мг/кг массы тела, около 12,5 мг/кг массы тела, около 10 мг/кг массы тела, около 7,5 мг/кг массы тела, около 5 мг/кг массы тела, около 2,5 мг/кг массы тела, около 1 мг/кг массы тела, около 0,9 мг/кг массы тела, около 0,8 мг/кг массы тела, около 0,7 мг/кг массы тела, около 0,6 мг/кг массы тела, около 0,5 мг/кг массы тела, около 0,4 мг/кг массы тела, около 0,3 мг/кг массы тела, около 0,2 мг/кг массы тела, около 0,1 мг/кг массы тела и около 0,01 мг/кг массы тела. В некоторых вариантах осуществления терапевтически эффективное количество составляет от около 0,1 мг/кг/сутки до около 10 мг/кг/сутки; в некоторых вариантах осуществления терапевтически эффективное количество составляет от около 0,2 до около 5 мг/кг/сутки. В некоторых вариантах осуществления схемы лечения по настоящему изобретению включают введение пациенту, нуждающемуся в таком лечении, обычно от около 1 до около 5000 мг, от около 10 до около 2000 мг, от около 10 до около 200 мг, около до 20 до около 1000 мг, от около 20 до около 500 мг, от около 20 до около 400 мг, от около 40 до около 800 мг, от около 50 до около 500 мг, от около 80 до около 1600 мг и около 50 мг соединения в соответствии с любым из вариантов осуществления, описанным в настоящем документе, или его фармацевтически приемлемой соли в сутки в однократной или многократных дозах. В некоторых вариантах осуществления терапевтически эффективное количество соответствует общей суточной дозе от 50 до 500 мг. В некоторых вариантах осуществления суточная доза находится между нижним пределом, составляющим около 50 мг, около 55 мг, около 60 мг, около 65 мг, около 70 мг, около 75 мг, около 80 мг, около 85 мг, около 90 мг, около 95 мг, около 100 мг, около 105 мг, около 110 мг, около 115 мг; около 120 мг, около 125 мг, около 130 мг, около 135 мг, около 140 мг, около 145 мг, около 150 мг, около 155 мг, около 160 мг, около 165 мг, около 170 мг, около 175 мг, около 180 мг, около 185 мг, около 190 мг, около 195 мг, около 200 мг, около 205 мг, около 210 мг, около 215 мг; около 220 мг, около 225 мг, около 230 мг, около 235 мг, около 240 мг, около 245 мг, около 250 мг, около 255 мг, около 260 мг, около 265 мг, около 270 мг, около 275 мг, около 280 мг, около 285 мг, около 290 мг, около 295 мг, 300 мг, около 305 мг, около 310 мг, около 315 мг; около 320 мг, около 325 мг, около 330 мг, около 335 мг, около 340 мг, около 345 мг, около 350 мг, около 355 мг, около 360 мг, около 365 мг, около 370 мг, около 375 мг, около 380 мг, около 385 мг, около 390 мг, около 395, около 400 мг, около 405 мг, около 410 мг, около 415 мг; около 420 мг, около 425 мг, около 430 мг, около 435 мг, около 440 мг, около 445 мг, около 450 мг, около 455 мг, около 460 мг, около 465 мг, около 470 мг, около 475 мг, около 480 мг, около 485 мг, около 490 мг, около 495 мг и около 500 мг и верхним пределом, составляющим около 500 мг, около 495 мг, около 490 мг, около 485 мг, около 480 мг, около 475 мг, около 470 мг, около 465 мг, около 460 мг, около 455 мг, около 450 мг, около 445 мг, около 440 мг, около 435 мг, около 430 мг, около 425 мг, около 420 мг, около 415 мг, около 410 мг, около 405 мг, около 400 мг, около 395 мг, около 390 мг, около 385 мг, около 380 мг, около 375 мг, около 370 мг, около 365 мг, около 360 мг, около 355 мг, около 350 мг, около 345 мг, около 340 мг, около 335 мг, около 330 мг, около 325 мг, около 320 мг, около 315 мг, около 310 мг, около 305 мг, около 300 мг, около 295 мг, около 290 мг, около 285 мг, около 280 мг, около 275 мг, около 270 мг около 265 мг, около 260 мг, около 255 мг, около 250 мг, около 245 мг, около 240 мг, около 235 мг, около 230 мг, около 225 мг, около 220 мг, около 215 мг, около 210 мг, около 205 мг, около 200 мг, около 195 мг, около 190 мг, около 185 мг, около 180 мг, около 175 мг, около 170 мг, около 165 мг, около 160 мг, около 155 мг, около 150 мг, около 145 мг, около 140 мг, около 135 мг, около 130 мг, около 125 мг, около 120 мг, около 115 мг, около 110 мг, около 105 мг, около 100 мг, около 95 мг, около 90 мг; около 85 мг, около 80 мг, около 75 мг, около 70 мг, около 65 мг, около 60 мг, около 55 мг и около 50 мг соединения в соответствии с любым из вариантов осуществления настоящего изобретения. В некоторых вариантах осуществления общая суточная доза составляет примерно от 50 до 150 мг. В некоторых вариантах осуществления общая суточная доза составляет примерно от 50 до 250 мг. В некоторых вариантах осуществления общая суточная доза составляет примерно от 50 до 350 мг. В некоторых вариантах осуществления общая суточная доза составляет примерно от 50 до 450 мг. В некоторых вариантах осуществления общая суточная доза составляет около 50 мг. Понятно, что фармацевтические композиции по настоящему изобретению необязательно должны содержать полное количество соединения, которое эффективно при лечении расстройства, поскольку такие эффективные количества могут быть достигнуты путем введения множества разделенных доз таких фармацевтических композиций. Соединения могут вводиться по схеме от 1 до 4 раз в сутки, например, один, два, три или четыре раза в сутки.
Термин “терапевтический фенотип” применяется для описания характера активности соединений в in vitro анализах, прогнозирующих поведенческую эффективность. Соединение, которое (1) селективно связывается с высокой аффинностью с сигма–2–рецептором и (2) действует как функциональный антагонист по отношению к вызванному бета–амилоидными олигомерами эффектами в нейронах, определяется как имеющее “терапевтический фенотип”, если оно (i) блокирует или уменьшает Аβ–индуцированный дефицит переноса через мембраны; (ii) блокирует или уменьшает Аβ–индуцированную потерю синапса и (iii) не влияет на перенос или количество синапсов в отсутствие бета–амилоидного олигомера. Этот характер активности в in vitro анализах называется “терапевтическим фенотипом” и может прогнозировать поведенческую эффективность.
Термин “терапевтический профиль” применяется для описания соединения, соответствующего терапевтическому фенотипу и также обладающего высокой способностью проникновения в головной мозг (способностью к преодолению гематоэнцефалического барьера), высокой стабильностью в плазме и высокой метаболической стабильностью.
Термин “ткань” относится к любой агрегации клеток подобной специализации, которые объединятся при осуществлении определенной функции.
Используемые в настоящем описании термины “лечить”, “поддающийся лечению” или “лечение” означают как терапевтическое лечение, так и профилактические или превентивные меры, причем задача заключается в защите (частичной или полной) или замедлении (например, уменьшении или задержке возникновения) нежелательного физиологического состояния, нарушения или болезни, или в достижении благоприятных или желательных клинических результатов, таких, как частичное или полное восстановление или ингибирование снижения параметра, значения, функции или результата, которые стали или смогли бы стать патологическими. С точки зрения настоящего изобретения благоприятными или желательными клиническими результатами, помимо прочих, могут быть облегчение симптомов; уменьшение степени или силы или скорости развития состояния, нарушения или заболевания; стабилизация (т.е. отсутствие ухудшения) состояния, нарушения или заболевания; задержка возникновения или замедление прогрессирования состояния, нарушения или болезни; устранение состояния, нарушения или заболевания; и ремиссия (частичная или полная), независимо от того, ведет ли это к непосредственному уменьшению фактических клинических симптомов или усилению или улучшению состояния, нарушения или заболевания. Цель лечения заключается в достижении клинически значимого ответа без возникновения чрезмерного уровня побочных эффектов. Лечение также включает увеличение продолжительности жизни по сравнению с ожидаемыми показателями при отсутствии лечения.
Антагонисты бета-амилоида и сигма–2 человека
Избыточная выработка и накопление бета–амилоида является патологической особенностью болезни Альцгеймера. Бета–амилоид человека (Abeta) является главным компонентом нерастворимых отложений амилоидных бляшек, находящихся в головном мозге пациентов с болезнью Альцгеймера. Бляшки состоят из фибриллярных агрегатов бета–амилоида. Бета–амилоидные фибриллы связаны с поздними стадиями болезни Альцгеймера.
Когнитивным признаком ранней стадии болезни Альцгеймера (БА) является чрезвычайная неспособность к запоминанию новой информации. Ранняя потеря памяти считается дисфункцией синапса, вызванной растворимыми бета–амилоидными олигомерами. Эти олигомеры блокируют долговременную потенциацию, классическую экспериментальную парадигму для синантической пластичности, и их содержание заметно повышается в ткани головного мозга при БА и в трансгенных моделях БА. Предполагается, что ранняя потеря памяти до гибели нейронов возникает в результате дисфункции синапса, а эта дисфункция синапса является следствием действия растворимых бета–амилоидных олигомеров, но не фибрилл. Lacor et al., Synaptic targeting by Alzheimer’s–related amyloid β oligomers, J. Neurosci. 2004, 24(45):10191–10200.
Бета–амилоид является продуктом расщепления интегрального мембранного белка, белка–предшественника амилоида (АРР), который в концентрированной форме содержится в синапсе нейронов. Растворимые формы бета–амилоида присутствуют в головном мозге и тканях пациентов с болезнью Альцгеймера, и их присутствие коррелирует с развитием болезни. Yu et al., 2009, Structural characterization of a soluble amyloid beta–peptide oligomer, Biochemistry, 48(9):1870–1877. Было показано, что растворимые β–амилоидные олигомеры вызывают изменения в нейронных синапсах и блокируют способность приобретать навыки и память.
Меньшие растворимые Аβ–олигомеры блокируют ряд сигнальных путей, которые являются основными для нормальной синаптической пластичности, что в конечном итоге приводит к потере шипика и синапсов. Selkoe et al., 2008, Soluble oligomers of the amyloid beta–protein impair synaptic plasticity and behavior, Behav Brain Res 192(1): 106–113. Болезнь Альцгеймера начинается и сохраняется в виде нарушения синаптической пластичности.
Полагают, что присутствием растворимых Аβ–олигомеров обусловлено раннее снижение когнитивных функций в головном мозге до болезни Альцгеймера. Известно, что бета–амилоидные олигомеры связываются в нейронных синапсах, а сигма–2–рецепторы присутствуют в значительном количестве в нейронах и глиальных клетках.
Сигма–рецепторы являются полифункциональными адапторными/шаперонными белками, участвующими в нескольких отдельных белковых сигнальных комплексах в зависимости от ткани и состояния. Сигма–2–рецептор экспрессируется в головном мозге и различных периферических тканях на низких уровнях (Walker et al., 1990 Sigma receptors: biology and function. Pharmacol. Rev. 42:355–402). Сигма–2–рецепторы присутствуют в гиппокампе и коре головного мозга человека. Сигма–2–рецептор также ранее был подтвержден как биомаркер пролиферации опухолевых клеток. (Mach et al., Sigma–2 receptors as potential biomarkers of proliferation in breast cancer. Cancer Res. 57:156–161, 1997).
Сигма–2–рецепторы задействованы во многих сигнальных путях, таких, как связывание гема, метаболизм Цитохрома Р450, синтез холестерина, сигнал прогестерона, апоптоз и мембранный перенос. К сигналу олигомера при БА имеет отношение только подгруппа сайтов связывания/сигнальных путей сигма–рецептора. В настоящее время отсутствуют нокауты по сигма–2–рецептору, а человеческие мутации в последовательности сигма–2 не исследовались в контексте нейродегенерации.
В последнее время сигма–2–рецептор был определен как мембранный компонент прогестеронового рецептора 1 (PGRMC1) в печени крыс при использовании фотоаффинного зонда WC–21, который необратимо метит сигма–2–рецепторы в печени крыс. См. публикацию Xu et al. Identification of the PGRMC1 protein complex as the putative sigma–2 receptor binding site. Nature Communications 2, article number 380, July 5, 2011, включенную в настоящее описание посредством ссылки. PGRMC1 (мембранный компонент прогестеронового рецептора 1) был определен как основной с массой 25 кДа компонент активности сигма–2–рецептора в августе 2011 г. группой Xu et al. PGRMC1 является одиночным трансмембранным белком без гомологии с белком сигма–1; к членам семейства относятся PGRMC2 и нейдезин. PGRMC1 содержит гем–связывающий домен цитохрома b5. PGRMC1 является одиночным трансмембранным белком без гомологии с белком S1; к членам семейства относятся PGRMC2 и нейдезин. PGRMC1 содержит гем–связывающий домен цитохрома b5. К эндогенным лигандам PGRMC1 относятся прогестерон/стероиды, метаболиты холестерина, глюкокортикоиды и гем. PGRMC1 функционирует как шаперон/адаптор, связанный с разными белковыми комплексами на разных субклеточных участках (Cahill 2007. Progesterone receptor membrane component 1: an integrative review. J. Steroid Biochem. Mol. Biol. 105:16–36). PGRMC1 связывает гем со сниженной активностью, образует комплекс с белками CYP450 (регулируемые окислительно–восстановительные реакции), ассоциируется с PAIRBP1 и опосредует блокировку прогестерона при апоптозе и ассоциируется с Insig–1 и SCAP для вызывания связанной с SRE транскрипции гена в ответ на низкий уровень холестерина. Гомолог VEM1 С. elegans ассоциируется с UNC–40/DCC для опосредования аксонального наведения. PGRMC1 содержит две последовательности–мишени SH2, последовательность–мишень SH3, сайт тирозинкиназы, два ацидофильных сайта киназы (CK2), и сайты консенсусного связывания для ERK1 и PDK1. PGRMC1 содержит несколько последовательностей ITAM, задействованных в мембранном переносе (транспорт везикул, клатрин–зависимый эндоцитоз содержащих кальвеолин ямок).
Не ограничиваясь теорией, предполагается, что сигма–2–рецептор является рецептором бета–амилоидного олигомера в нейронах. В литературе описаны различные рецепторы для растворимых бета–амилоидных олигомеров, включая прионный белок, рецептор инсулина, бета–адренергический рецептор и RAGE (рецептор для продвинутых конечных продуктов гликирования). Laurén, J. et al., 2009, Nature, 457(7233): 1128–1132; Townsend, M. et al., J. Biol. Chem. 2007, 282:33305–33312; Sturchler, E. et al., 2008, J. Neurosci. 28(20):5149–5158. Действительно, многие исследователи полагают, что бета–амилоидный олигомер может связываться с более чем одним рецепторным белком. Не ограничиваясь теорией, на основе представленных свидетельств, авторы настоящего изобретения предположили, что дополнительный рецептор для бета–амилоидного олигомера находится (не обязательно исключительно) в нейронах.
Не ограничиваясь теорией, бета–амилоидные олигомеры относятся к агонистам сигма–рецептора, которые связываются с сигма–белковыми комплексами и вызывают нарушение переноса и потерю синапса. В настоящем документе показано, что описанные здесь соединения, которые противодействуют этому взаимодействию и/или функции сигма–рецептора в нейронах, будут конкурировать или иным образом препятствовать бета–амилоидным олигомерам и возвращать нейронные ответы к норме. Такие соединения считаются функциональными антагонистами сигма–2-рецептора.
В некоторых вариантах осуществления соединение по любому варианту осуществления, описанному в настоящем документе, может действовать в качестве функционального антагониста в нейроне в отношении ингибирования потери синапса, вызванного растворимым Aβ–олигомером, и ингибирования дефицита, вызванного растворимым Aβ–олигомером, в анализе мембранного переноса; демонстрирует высокое сродство к сигма–2–рецептору; а также обладает высокой селективностью в отношении одного или более сигма–рецепторов по сравнению с любым другим отличным от сигма-рецептором; и демонстрирует хорошие подобные лекарственным свойства.
В некоторых вариантах осуществления соединение в соответствии с любым из вариантов осуществления, описанным в настоящем документе, которое действует как функциональный антагонист, отвечающий определенным критериям анализа in vitro, подробно описанным в настоящем документе, будет демонстрировать поведенческую эффективность или обладать прогнозируемой поведенческой эффективностью в одной или более соответствующих поведенческих моделях на животных. В некоторых вариантах осуществления поведенческая эффективность определяется при пероральной дозе 10 мг/кг или меньше.
Платформы для анализа in vitro, прогнозирующие поведенческую эффективность, используемую в настоящем изобретении, известны в данной области техники, в частности, описаны в патенте США 9796672, который включен в настоящий документ посредством ссылки полностью. В соответствии с платформой анализа in vitro, соединения по любому из вариантов осуществления, описанных в настоящем документе, могут с высокой аффинностью связываться с сигма–2-рецептором; действует как функциональный антагонист в отношении эффектов, индуцированных бета–амилоидным олигомером, в нейроне; ингибирует индуцированную бета–амилоидным олигомером потерю синапса в центральном нейроне, или уменьшает связывание бета–амилоидного олигомера с нейронами с ингибированием потери синапса; и не оказывает влияние на перенос или количество синапсов в отсутствие бета–амилоидного олигомера. Этот характер активности в in vitro анализах называется “терапевтическим фенотипом”. Способность соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, блокировать эффекты бета–амилоидных олигомеров в зрелых нейронах без нарушения нормальной функции в отсутствие бета–амилоидных олигомеров отвечает критериям терапевтического фенотипа. Соединения по любому из вариантов осуществления, описанных в настоящем документе, имеющие терапевтический фенотип, могут блокировать синаптическую дисфункцию, индуцированную бета–амилоидным олигомером.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, демонстрирует активность антагониста сигма–2, высокое сродство к сигма–2–рецептору и способность блокировать связывание растворимого бета–амилоидного олигомера или синаптическую дисфункцию, индуцированную бета–амилоидным олигомером.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, предназначено для усиления способности преодолевать гематоэнцефалический барьер.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, блокирует связывание между растворимыми бета–амилоидными олигомерами и сигма–2–рецептором.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, демонстрирует высокое сродство к сигма–2–рецептору.
Варианты осуществления изобретения относятся к соединениям в соответствии с любым вариантом осуществления, описанным в настоящем документе, которые могут быть использованы для лечения нейродегенеративного заболевания и снижения когнитивных функций, фармацевтическим композициям, содержащим такие соединения и фармацевтически приемлемые носители, эксципиенты или разбавители, и способам лечения нейродегенеративного заболевания и снижения когнитивных функций путем введения таких соединений и фармацевтических композиций в фармацевтически приемлемом количестве.
Соединения по изобретению
Различные варианты осуществления относятся к соединению формулы I:
или его фармацевтически приемлемой соли.
Каждый из заместителей Ra, Rb, Rc, Rd и Re формулы I независимо выбран из групп, состоящих из, H, гидроксила, галогена, алкила, алкокси, CF3, SO2CH3 и морфолино.
Заместитель R1 формулы I выбран из группы, состоящей из водорода, алкила, фенила или –CH=C(CH3)2.
Заместитель R2 формулы I представляет собой необязательно замещенную циклическую аминогруппу.
В некоторых вариантах осуществления каждый из заместителей Ra, Rb, Rc, Rd и Re формулы I независимо выбран из группы, состоящей из, H, гидроксила, Cl, F, метила, –OCH3, –OC(CH3)3, O–CH(CH3)2, CF3, SO2CH3 и морфолино.
В некоторых вариантах осуществления каждый из заместителей Ra, Rb, Rc, Rd и Re формулы I независимо выбран из группы, состоящей из, H, Cl, F и CF3.
В некоторых вариантах осуществления каждый из заместителей Ra, Rb, Rd и Re формулы I независимо представляет собой Н и Rc, выбран из группы, состоящей из Н, гидроксила, галогена, алкила, алкокси, CF3, SO2CH3 и морфолино.
В некоторых вариантах осуществления каждый из заместителей Ra, Rb, Rd и Re формулы I независимо представляет собой Н и Rc, выбран из группы, состоящей из Н, гидроксила, Cl, F, метила, –OCH3, –OC(CH3)3, O–CH(CH3)2, CF3, SO2CH3 и морфолино.
В некоторых вариантах осуществления каждый из заместителей Ra, Rb, Rd и Re формулы I независимо представляет собой Н и Rc, выбран из группы, состоящей из Н, Cl, F и CF3.
В различных вариантах осуществления R2 представляет собой любой гетероциклоалкил или гетероарил, содержащий атом азота в кольце, который связан с алифатической цепью формулы I через атом азота. В некоторых вариантах, например, R2 представляет собой необязательно замещенную циклическую аминогруппу, выбранную из:

и тому подобное, где каждый содержащий азот гетероциклоалкил или гетероарил может быть необязательно замещен одним или более заместителями, выбранными из гидроксила, галогена, CF3, алкокси, арилокси, необязательно замещенного C1–C10 алкила, необязательно замещенного C5–C10 арила, необязательно замещенного C3–C10 гетероарила, замещенного или незамещенного C3–C10 циклоалкила или гетероциклоалкила.
В различных вариантах осуществления R2 выбран из группы, состоящей из необязательно замещенного азиридинила, необязательно замещенного пирролидинила, необязательно замещенного имидизолидинила, необязательно замещенного пиперидинила, необязательно замещенного пиперазинила, необязательно замещенного оксопиперазинила и необязательно замещенного морфолинила.
В некоторых вариантах осуществления, когда R2 представляет собой замещенный циклический амино, один или более атомов водорода в циклической аминогруппе заменены группой, выбранной из алканоила, алкокси, алкоксиалкила, (алкокси)алкоксиалкила, алкоксикарбонила, алкила, арилокси, арилоила, циклоалканоила, –OC(O)NCH(CH3)2, (N,N–диметиламино)пиридинила, (N,N–диметиламино)сульфонила, галогена, гетероциклила, (гетероциклил)алкоксиалкила, гидроксила, гидроксиалкила, метилпиперидинила, метилсульфонила, метилсульфонилфенила, морфолинилпиридинила, перфторалкила, фенила, пиперидинила, пирролидинилпиридинила, тетрагидропиранила и CF3. В некоторых вариантах осуществления два атома водорода на одном и том же атоме углерода циклической аминогруппы заменены соединением, выбранным из
 и
и  с образованием спиросоединения.
с образованием спиросоединения.
В некоторых вариантах осуществления R2 представляет собой пирролидинил или замещенный пирролидинил, замещенный одним или более заместителями, выбранными из группы, состоящей из алкоксиалкила, алкоксикарбонила, алкила, гидроксила и гидроксиалкила. В некоторых вариантах осуществления R2 представляет собой замещенный пирролидинил, замещенный одним заместителем, выбранным из группы, состоящей из алкоксиалкила, алкоксикарбонила, алкила, гидроксила и гидроксиалкила. В некоторых вариантах осуществления R2 представляет собой замещенный пирролидинил, замещенный одним заместителем, выбранным из группы, состоящей из гидроксила, гидроксиметила, метоксиметила, метоксикарбонила и метила.
В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный одним или более заместителями, выбранными из группы, состоящей из алкокси, алкоксиалкила, (алкокси)алкоксиалкила, алкоксикарбонила, алкила, арилокси, –OC(O)NCH(CH3)2, (N, N–диметиламино)пиридинила, галогена, гетероциклила, (гетероциклил)алкоксиалкила, гидрокси, гидроксиалкила, метилпиперидинила, метилсульфонилфенила, морфолинилпиридинила, перфторалкила, фенила, пиперидинила, пирролидинилпиридинила, тетрагидропиранила и CF3. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный одним заместителем, выбранным из группы, состоящей из алкокси, алкоксиалкила, (алкокси)алкоксиалкила, алкоксикарбонила, алкила, арилокси, –OC(O)NCH(CH3)2, (N, N–диметиламино)пиридинила, галогена, гетероциклила, (гетероциклил)алкоксиалкила, гидроксила, гидроксиалкила, метилпиперидинила, метилсульфонилфенила, морфолинилпиридинила, перфторалкила, фенила, пиперидинила, пирролидинилпиридинила, тетрагидропиранила и CF3. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный одним заместителем, выбранным из группы, состоящей из алкокси, алкоксиалкила, (алкокси)алкоксиалкила, алкоксикарбонила, алкила, арилокси, –OC(O)NCH(CH3)2, (N, N–диметиламино)пиридинила, галогена, гетероциклила, (гетероциклил)алкоксиалкила, гидроксила, гидроксиалкила, метилпиперидинила, метилсульфонилфенила, морфолинилпиридинила, перфторалкила, фенила, пиперидинила, пирролидинилпиридинила, тетрагидропиранила и CF3. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный одним заместителем, выбранным из группы, состоящей из метила, изопропила, изобутила, CF3, гидроксиметила, гидроксиэтила, (изопропилокси)этила, –(CH2)2O(CH2)2OCH3, –(CH2)3OCH3, –C(O)OMe, –C(O)OEt, гидроксила, метокси, изопропилокси, фенилокси, F, этокси, фенила,
 .
.
В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный в 4 положении пиперидинила одним заместителем, выбранным из группы, состоящей из алкокси, алкоксиалкила, (алкокси)алкоксиалкила, алкоксикарбонила, алкила, арилокси, –OC(O)NCH(CH3)2, (N, N–диметиламино)пиридинила, галогена, гетероциклила, (гетероциклил)алкоксиалкила, гидроксила, гидроксиалкила, метилпиперидинила, метилсульфонилфенила, морфолинилпиридинила, перфторалкила, фенила, пиперидинила, пирролидинилпиридинила, тетрагидропиранила и CF3. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный в 4 положении пиперидинила одним заместителем, выбранным из группы, состоящей из метила, изопропила, изобутила, CF3, гидроксиметила, гидроксиэтила, (изопропилокси)этила, –(CH2)2O(CH2)2OCH3, –(CH2)3OCH3, –C(O)OMe, –C(O)OEt, гидроксила, метокси, изопропилокси, фенилокси, F, этокси, фенила, 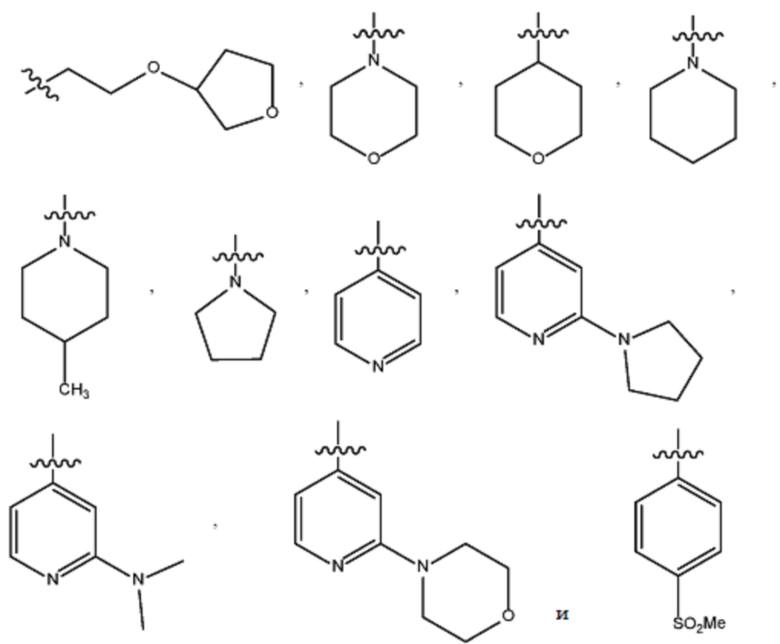 .
.
В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный двумя группами заместителей на одном и том же атоме углерода пиперидинила, независимо выбранными из группы, состоящей из алкоксиалкила, алкила, –OC(O)NCH(CH3)2, гидроксила и фенила. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный двумя группами заместителей в 4 положении пиперидинила, независимо выбранными из группы, состоящей из алкоксиалкила, алкила, –OC(O)NCH(CH3)2, гидроксила и фенила. В некоторых вариантах осуществления R2 представляет собой пиперидинил или замещенный пиперидинил, замещенный двумя заместителями в 4 положении, выбранными из группы, состоящей из гидроксила и метила; гидроксил и этил; гидроксил и –(CH2)2OCH3; гидроксил и фенил; метил и фенил; метил и –OC(O)NCH(CH3)2; и бутил и –OC(O)NCH(CH3)2. В некоторых вариантах осуществления два атома водорода на одном и том же атоме углерода пиперидинила заменены соединением, выбранным из 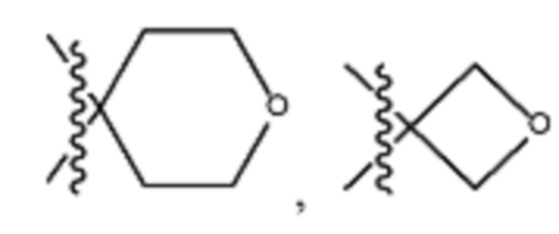 и
и 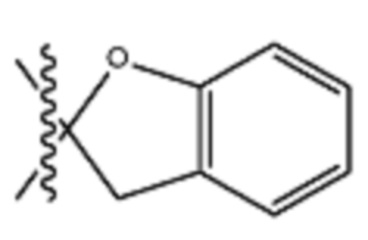 с образованием спиросоединения. В некоторых вариантах осуществления два атома водорода в 4 положении пиперидинила заменены соединением, выбранным из
с образованием спиросоединения. В некоторых вариантах осуществления два атома водорода в 4 положении пиперидинила заменены соединением, выбранным из 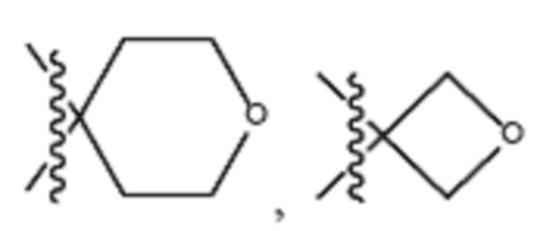 и с образованием спиросоединения.
и с образованием спиросоединения.
В некоторых вариантах осуществления R2 представляет собой пиперазинил или замещенный пиперазинил, замещенный одним или более заместителями, выбранными из группы, состоящей из алканоила, алкоксикарбонила, арилоила, циклоалканоила, (N, N–диметиламино)сульфонила, гетероциклила, метилсульфонила и фенила. В некоторых вариантах осуществления R2 представляет собой замещенный пиперазинил, замещенный одним заместителем, выбранным из группы, состоящей из алканоила, алкоксикарбонила, арилоила, циклоалканоила, (N, N–диметиламино)сульфонила, гетероциклила, метилсульфонила и фенила. В некоторых вариантах осуществления R2 представляет собой замещенный пиперазинил, замещенный одним заместителем, выбранным из группы, состоящей из –C(O)OC(CH3)3, –C(O)OCH2CH(CH3)2, –C(O)OCH2CH3, –C(O)OCH3, фенила, –C(O)CH3, –C(O)Ph, –SO2Me, –SO2N(CH3)2,  . В некоторых вариантах осуществления R2 представляет собой замещенный пиперазинил, замещенный одним заместителем в 4 положении, выбранным из группы, состоящей из –C(O)OC(CH3)3, –C(O)OCH2CH(CH3)2, –C(O)OCH2CH3, –C(O)OCH3, фенила, –C(O)CH3, –C(O)Ph, –SO2Me, –SO2N(CH3)2,
. В некоторых вариантах осуществления R2 представляет собой замещенный пиперазинил, замещенный одним заместителем в 4 положении, выбранным из группы, состоящей из –C(O)OC(CH3)3, –C(O)OCH2CH(CH3)2, –C(O)OCH2CH3, –C(O)OCH3, фенила, –C(O)CH3, –C(O)Ph, –SO2Me, –SO2N(CH3)2,  .
.
В некоторых вариантах осуществления R2 представляет собой замещенный пипердинил формулы:
 ,
,
где R3 представляет собой водород или C1–C8 алкил, и R4 представляет собой водород, гидроксил, галоген, CF3, алкокси, арилокси, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 гетероциклоалкил.
В некоторых вариантах осуществления R2 представляет собой
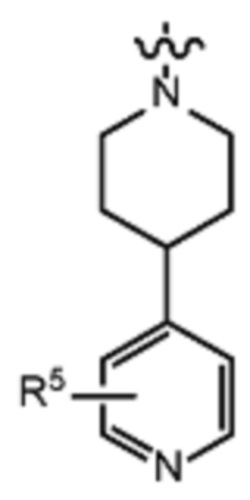 или
или  ,
,
где каждый из R5 и R6 независимо представляет собой водород, гидроксил, сульфонил, диалкиламино, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 гетероциклоалкил. В некоторых вариантах осуществления R5 представляет собой водород, диалкиламино или C3–C10 гетероциклоалкил. В некоторых вариантах осуществления R5 представляет собой водород, диалкиламино, пирролидинил или морфолинил. В некоторых вариантах R6 представляет собой сульфонил. В некоторых вариантах осуществления R6 представляет собой метилсульфонил.
В некоторых вариантах осуществления R2 представляет собой:


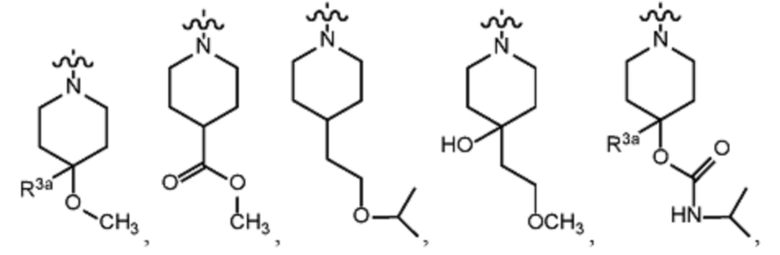
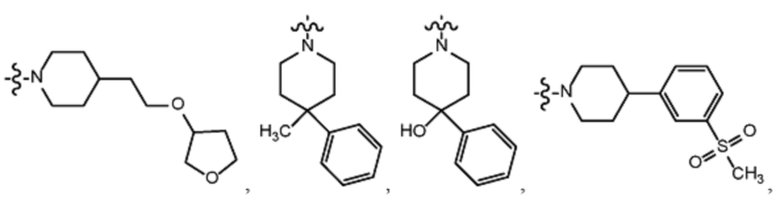




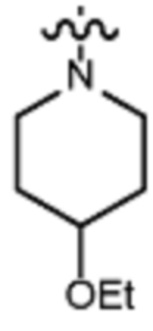 или
или 
где R3a выбран из группы, состоящей из водорода и C1–C8 алкила; и n является целым числом, выбранным из 0, 1 и 2.
В некоторых вариантах осуществления R2 представляет собой
 или
или 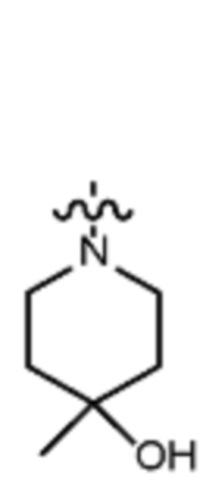 .
.
В некоторых вариантах осуществления R2 представляет собой необязательно замещенный морфолинил. В некоторых вариантах осуществления R2 представляет собой морфолинил.
В некоторых вариантах осуществления R2 представляет собой необязательно замещенный пиперазинил формулы
 ,
,
где R7 представляет собой водород, гидроксил, сульфонил, диалкиламиносульфонил, алкоксикарбонил, ацил, бензоил, циклоалкилкарбонил, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 циклоалкил или С10 гетероциклоалкил. В некоторых вариантах осуществления R7 представляет собой сульфонил, диалкиламиносульфонил, алкоксикарбонил, ацил, бензоил, циклоалкилкарбонил, C5–C10 арил или необязательно замещенный C3–C10 гетероциклоалкил.
В некоторых вариантах осуществления R2 представляет собой

 и
и 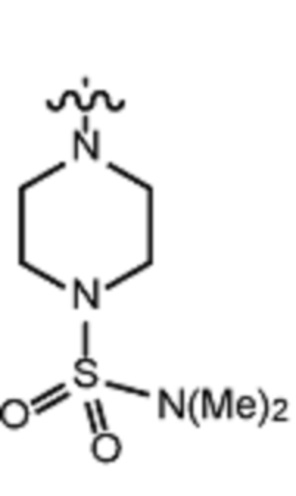 .
.
В различных вариантах осуществления R2 представляет собой необязательно замещенный пирролидинил:
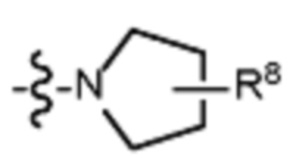 ,
,
где R8 представляет собой водород, гидроксил, сульфонил, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 гетероциклоалкил. В некоторых вариантах осуществления R8 представляет собой водород, гидроксил или необязательно замещенный C1–C10 алкил.
В некоторых вариантах осуществления R2 представляет собой:
 и
и 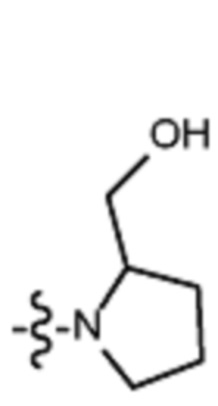 .
.
В некоторых вариантах осуществления R2 представляет собой необязательно замещенное бициклическое кольцо или необязательно замещенное конденсированное кольцо. Например, в некоторых вариантах осуществления R2 выбран из группы, состоящей из:
 и
и  ,
,
где R9 представляет собой водород, гидроксил, сульфонил, необязательно замещенный C1–C10 алкил, необязательно замещенный C5–C10 арил, необязательно замещенный C3–C10 гетероарил, необязательно замещенный C3–C10 циклоалкил или необязательно замещенный C3–C10 гетероциклоалкил.
В некоторых вариантах осуществления R2 представляет собой
 ,
,
где каждый R11a, R11b, R11c и R11d независимо выбран из водорода, гидрокси, сульфонила, необязательно замещенного C1–C10 алкила, необязательно замещенного C5–C10 арила, необязательно замещенного C3–C10 гетероарила, необязательно замещенного C3–C10 циклоалкила или необязательно замещенного C3–C10 гетероциклоалкила. В конкретных вариантах осуществления R2 представляет собой


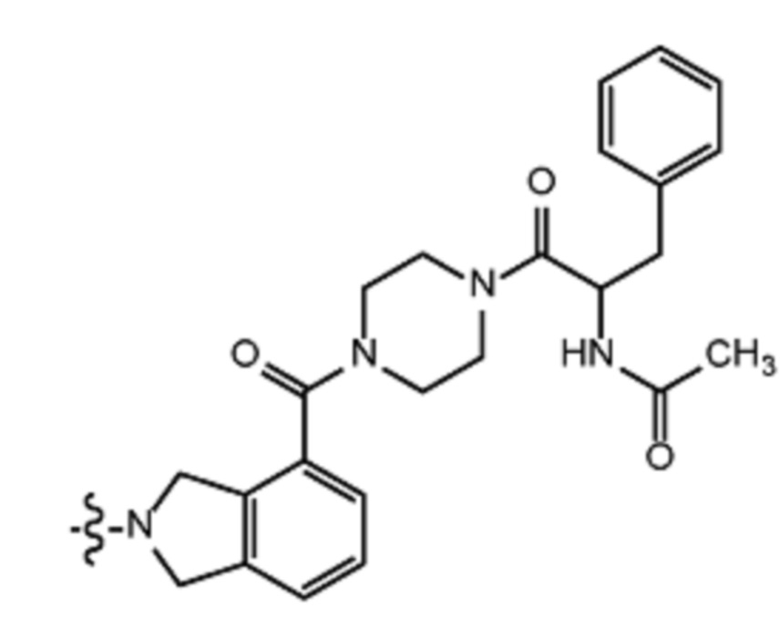 или
или  .
.
Некоторые варианты осуществления, описанного в настоящем документе, описывают соединение, в котором каждый Ra, Rb, Rc, Rd и Re выбран из любого варианта осуществления, описанного в настоящем документе, для каждого из Ra, Rb, Rc, Rd и Re; R1 выбран из любого варианта осуществления, описанного в настоящем документе для R1; R2 выбран из любого варианта осуществления, описанного в настоящем документе для R2.
Некоторые варианты осуществления относятся к соединению, выбранному из






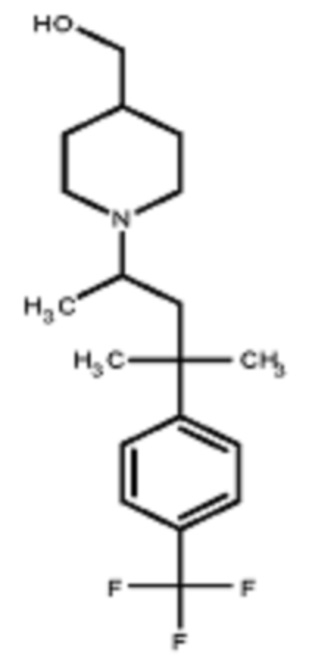










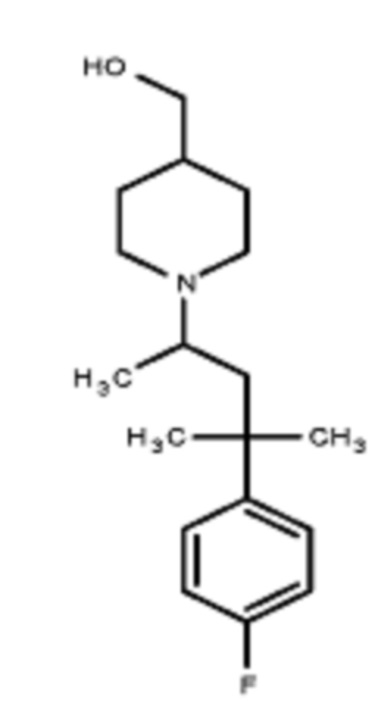



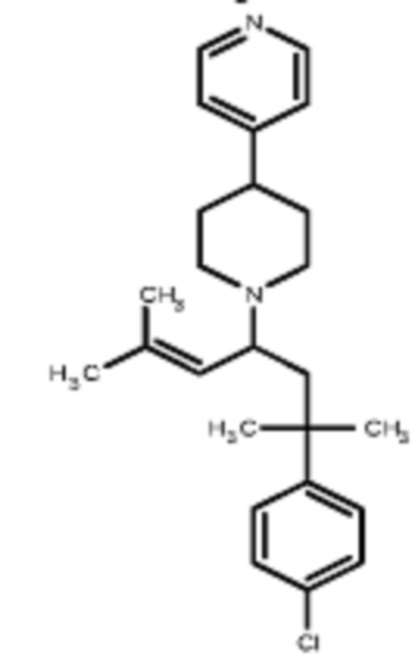



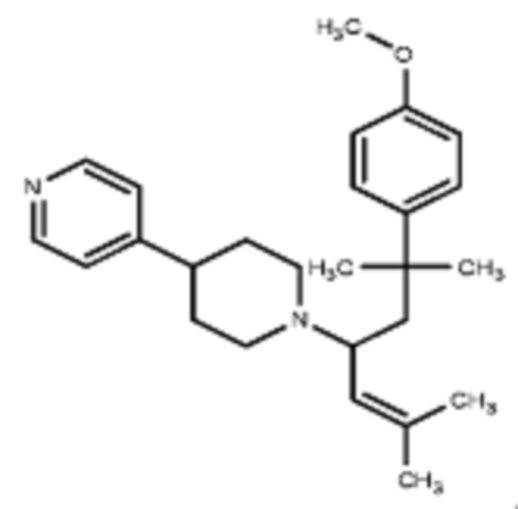




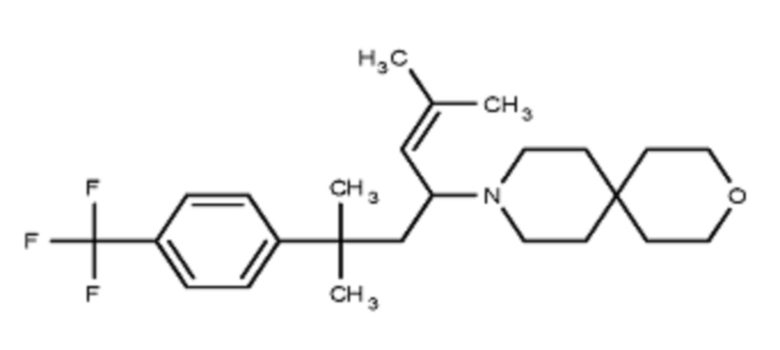
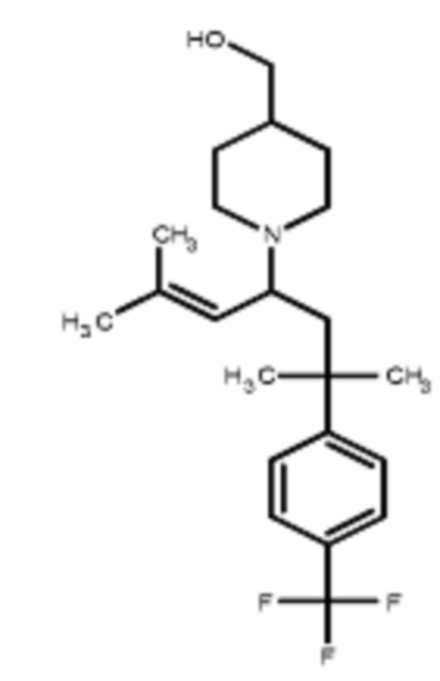


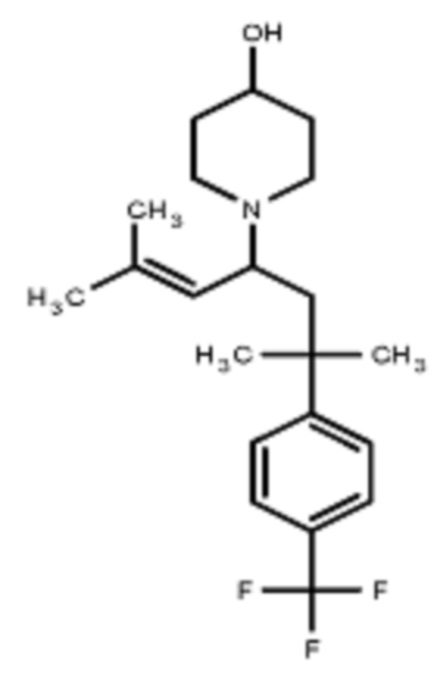




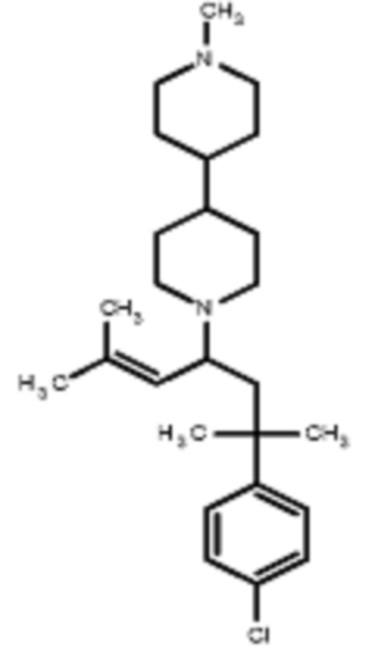


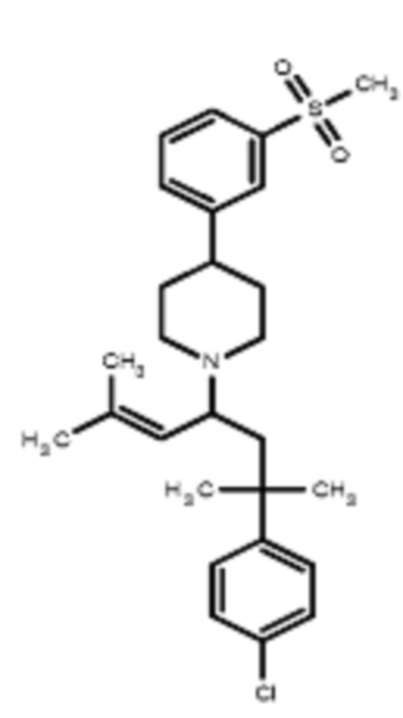








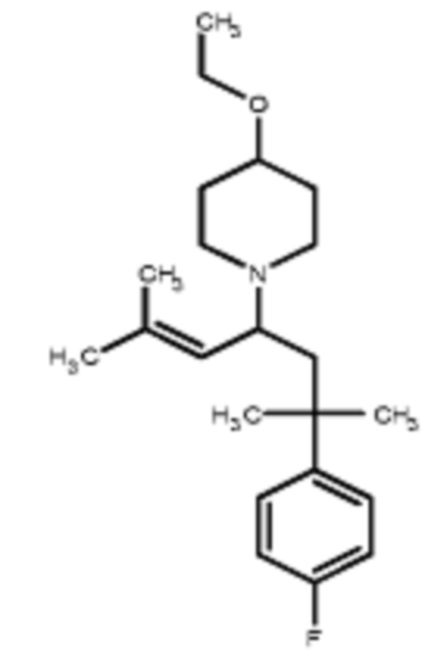
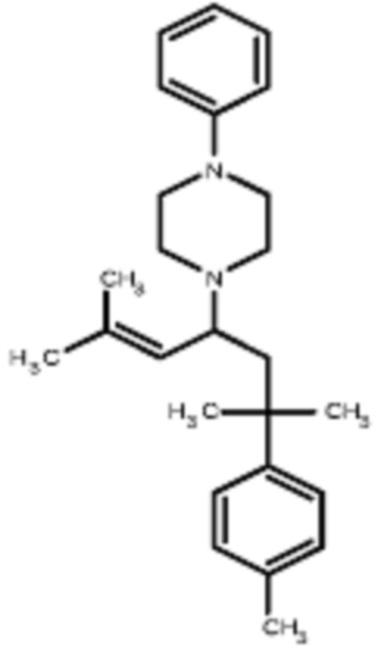
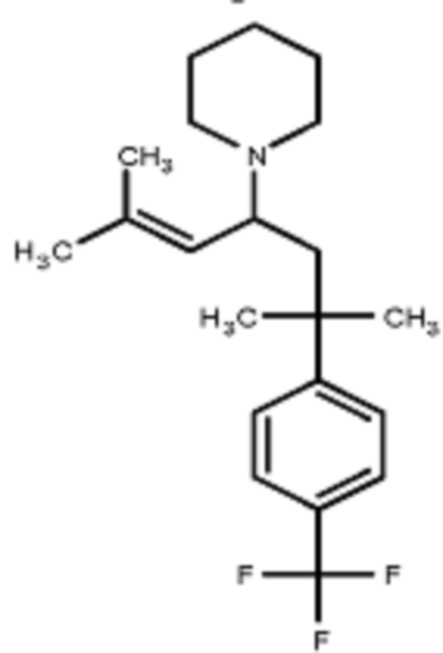

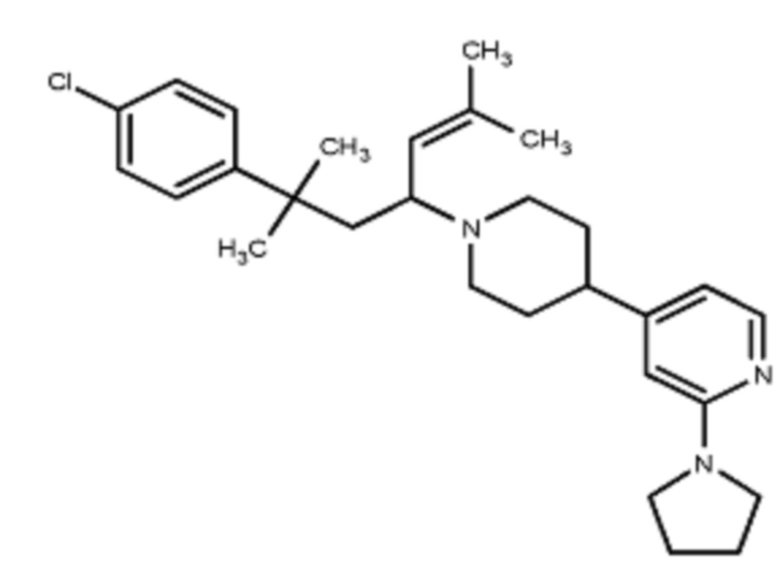

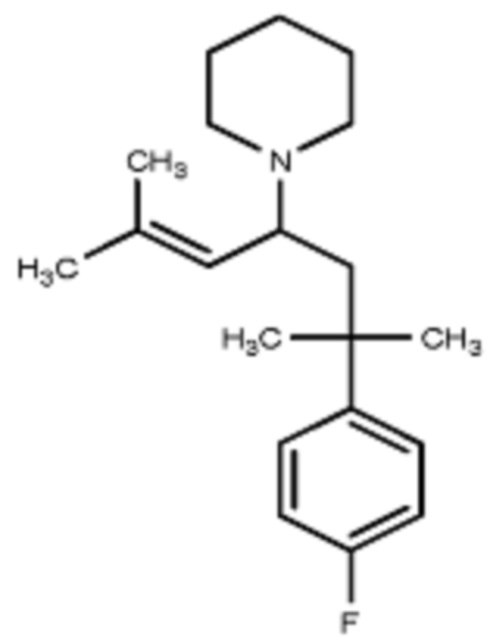

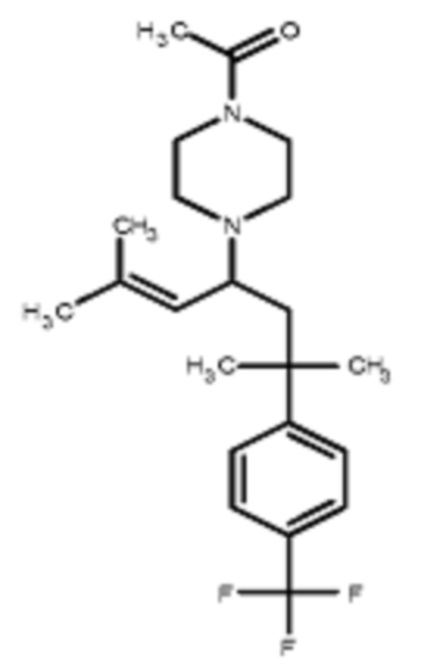












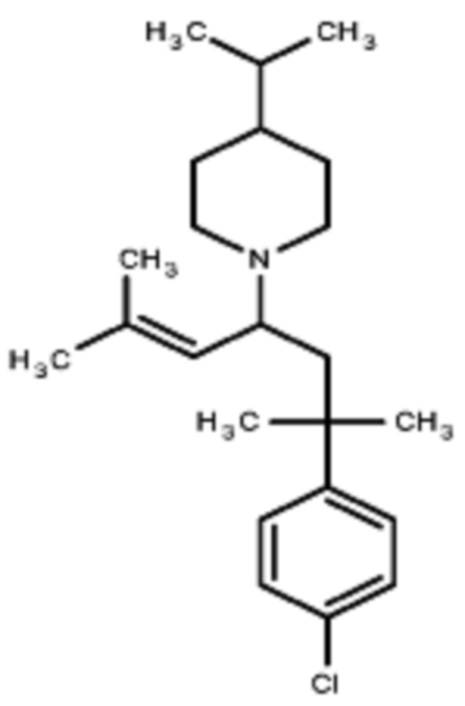




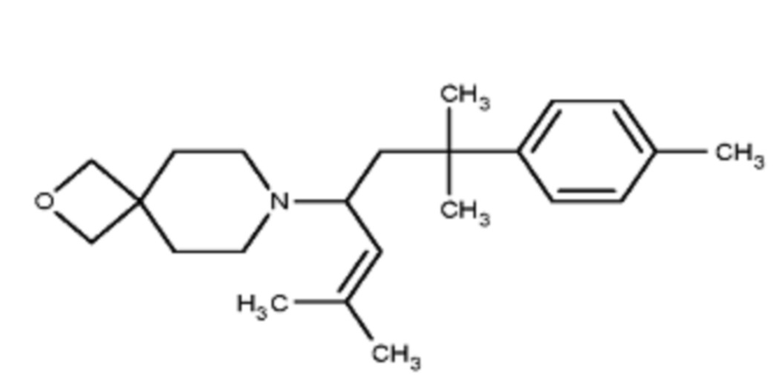

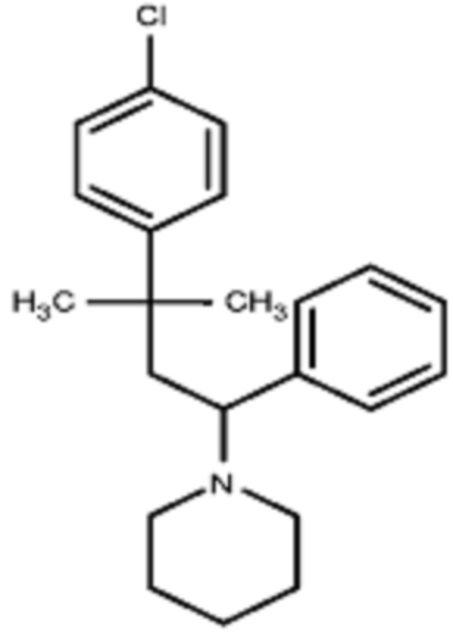
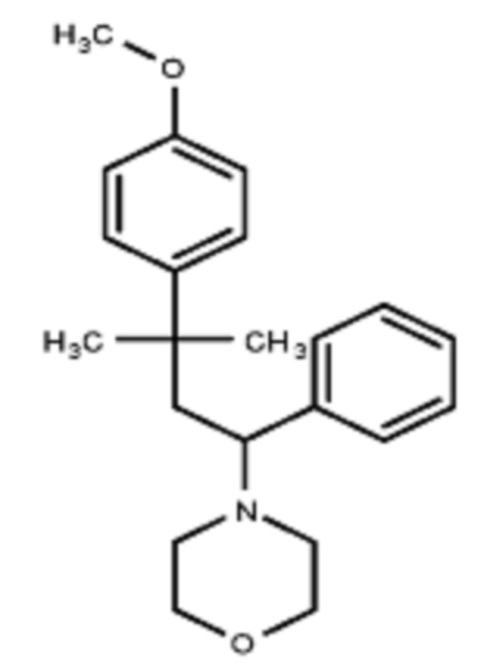



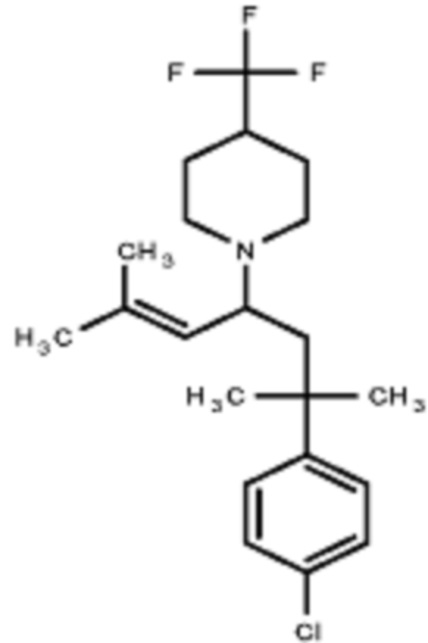

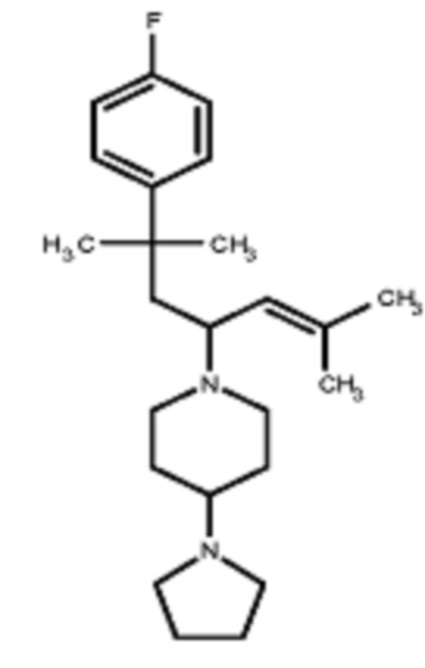

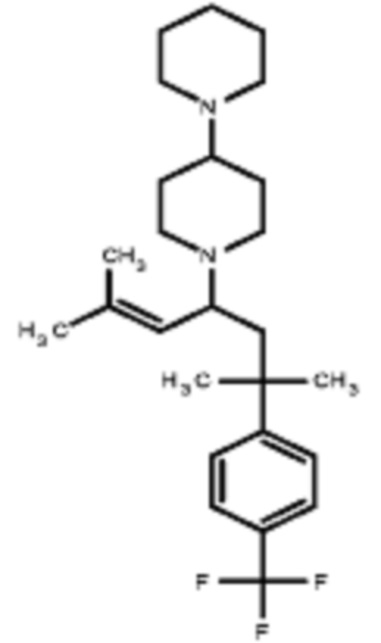
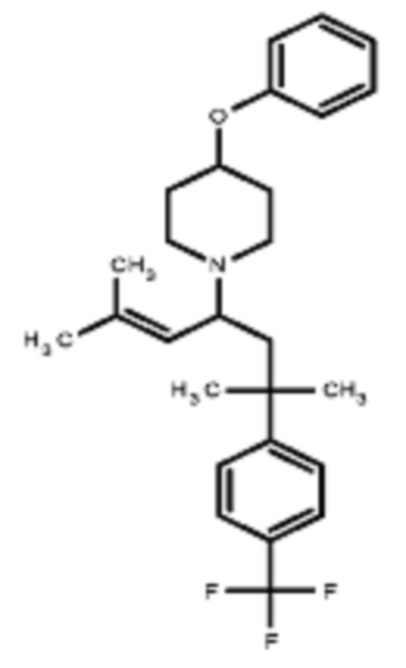
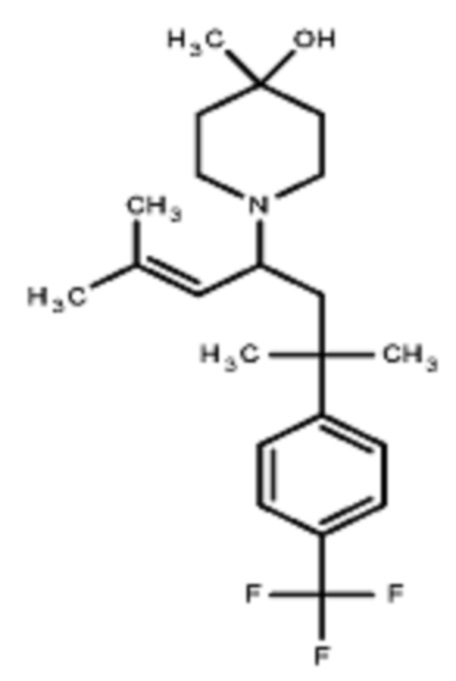

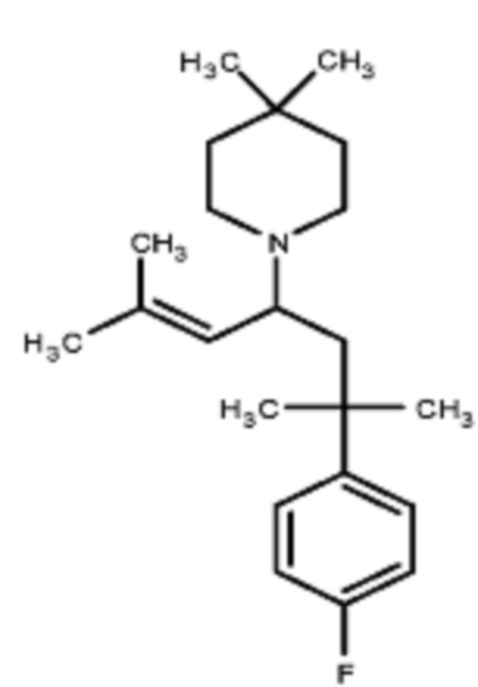


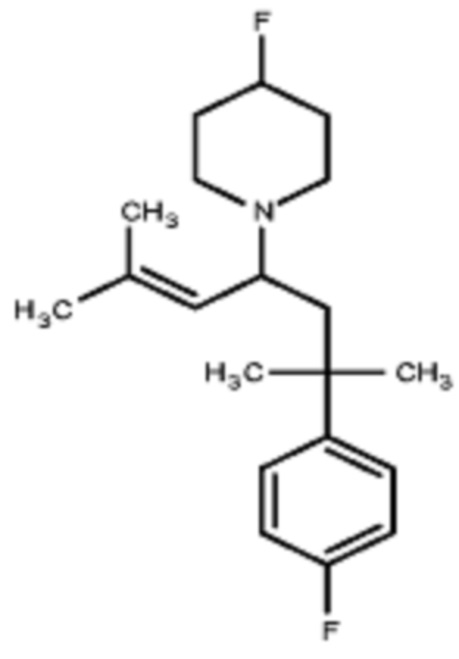


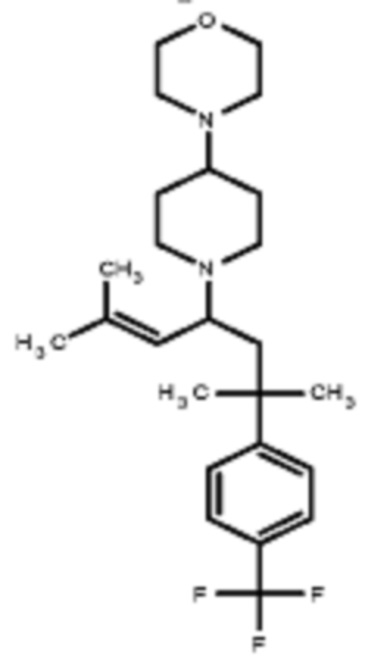


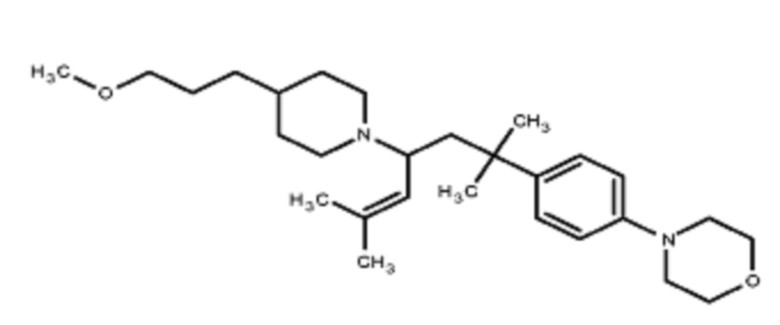
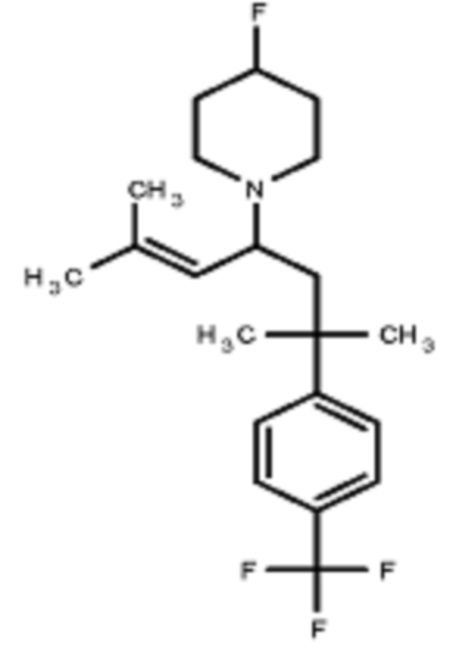





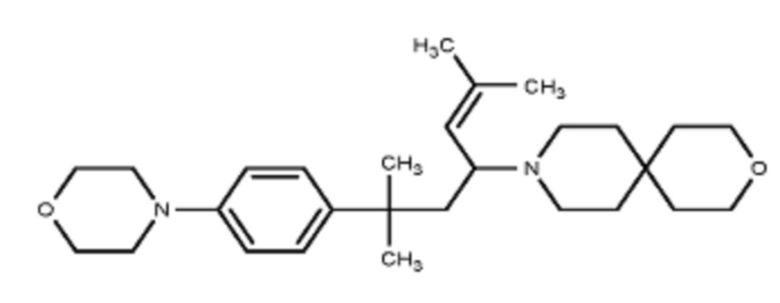

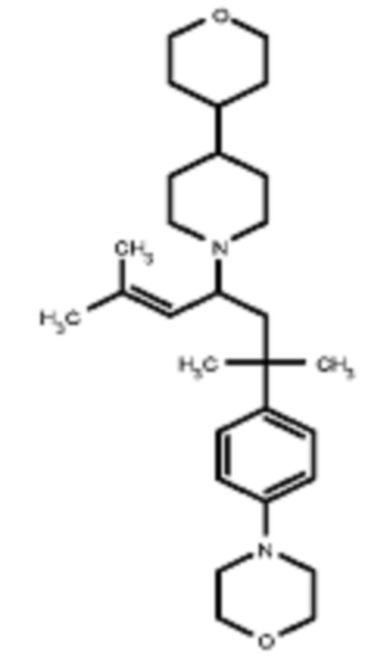

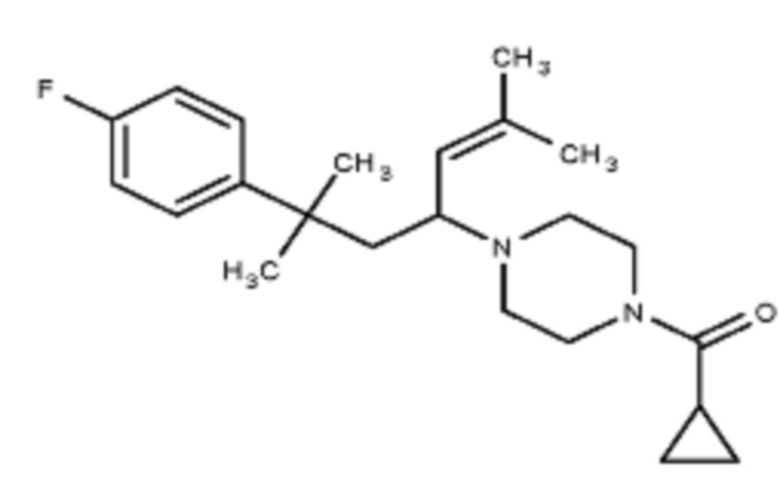











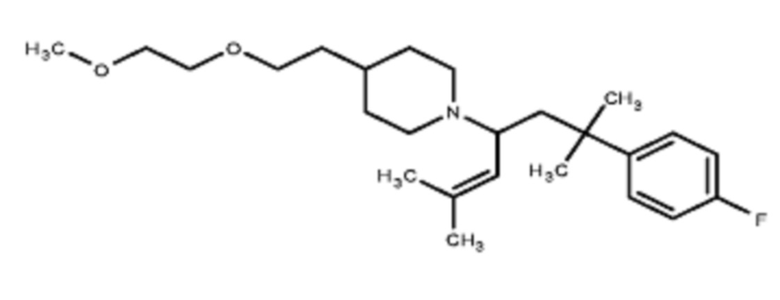
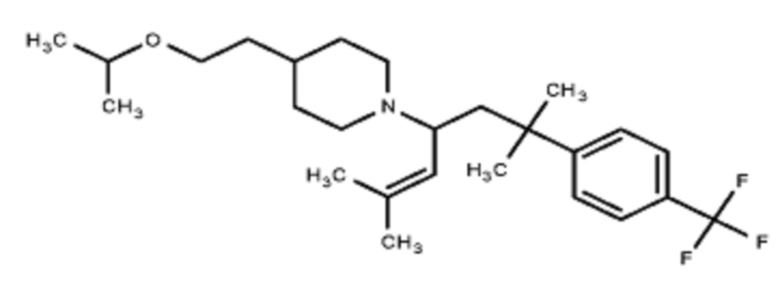


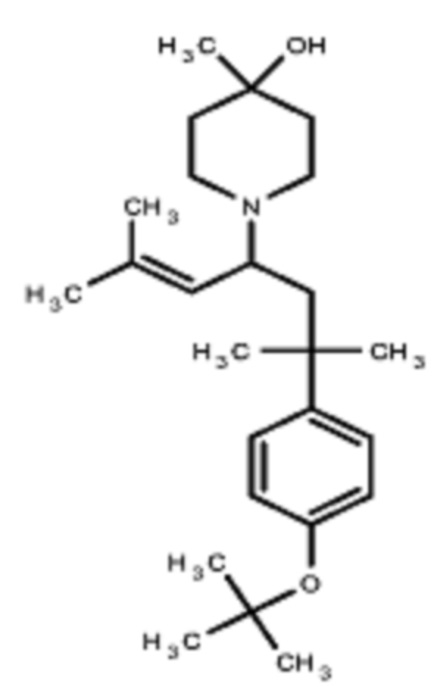

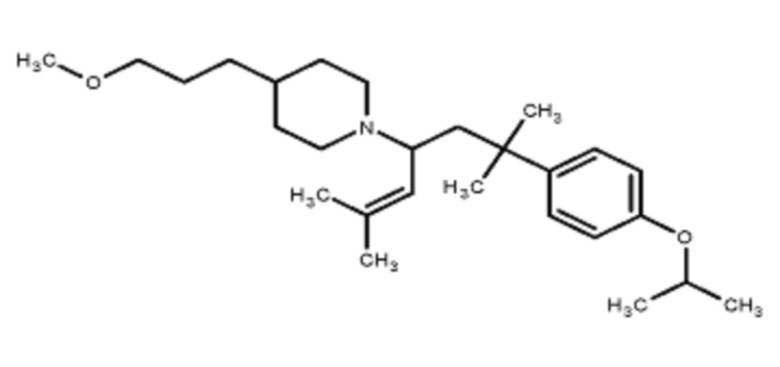

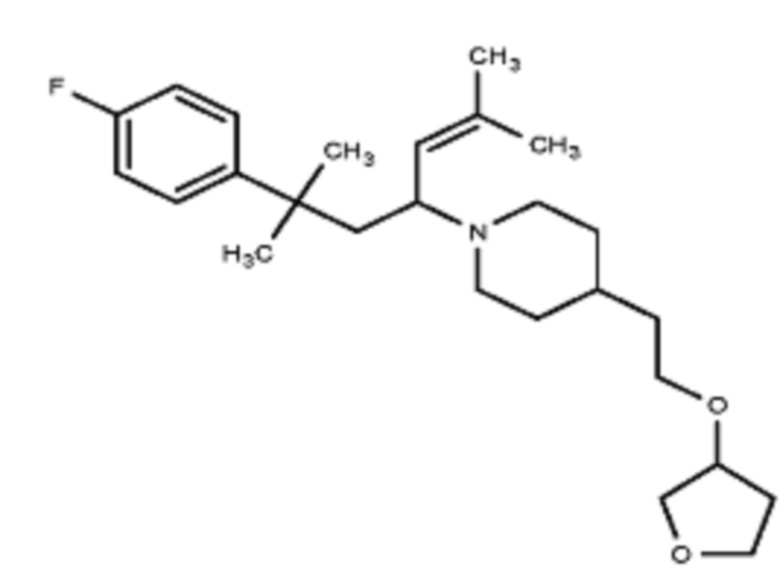

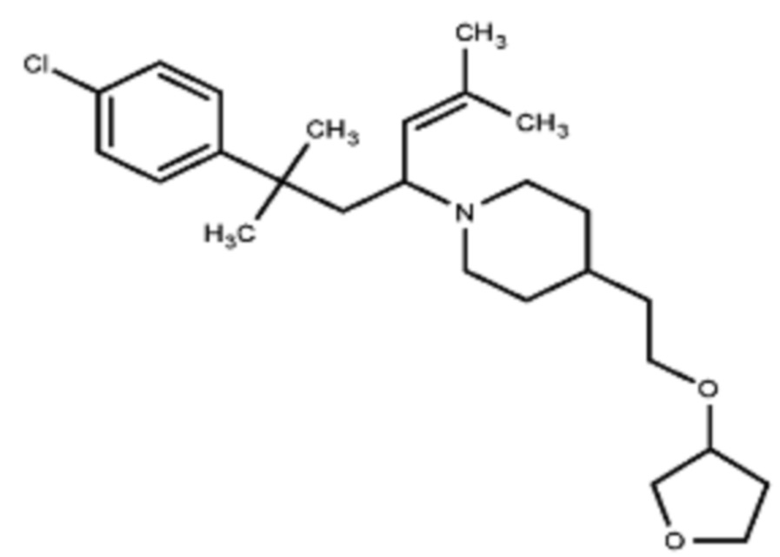


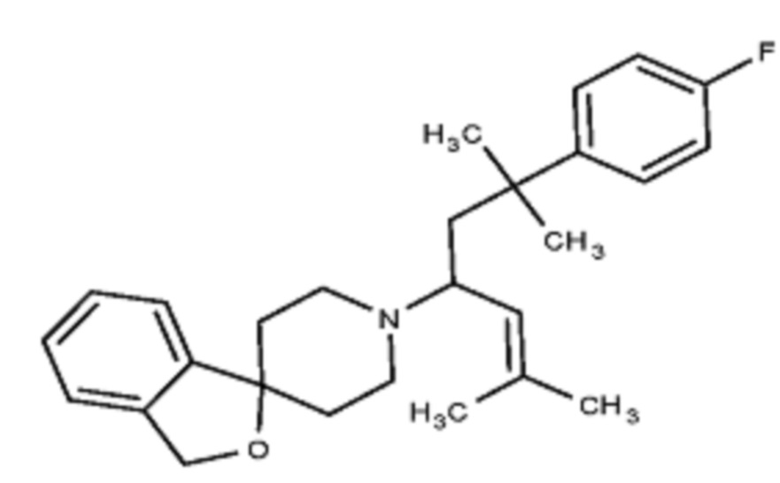
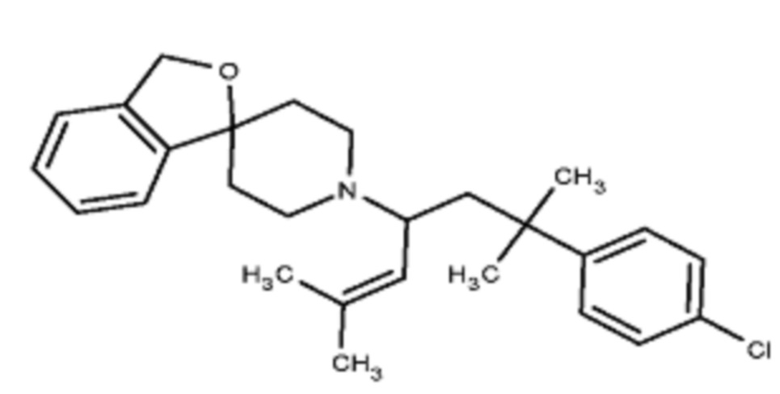

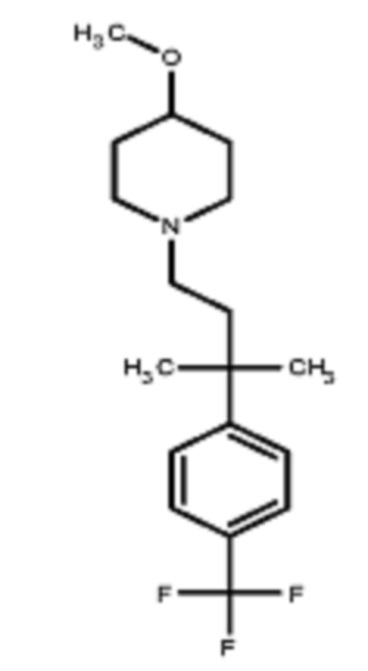


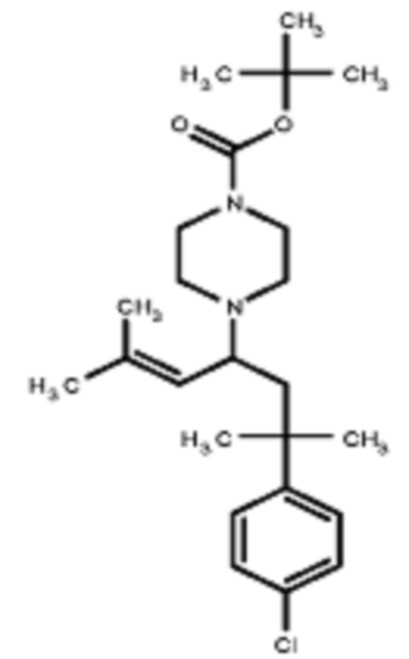


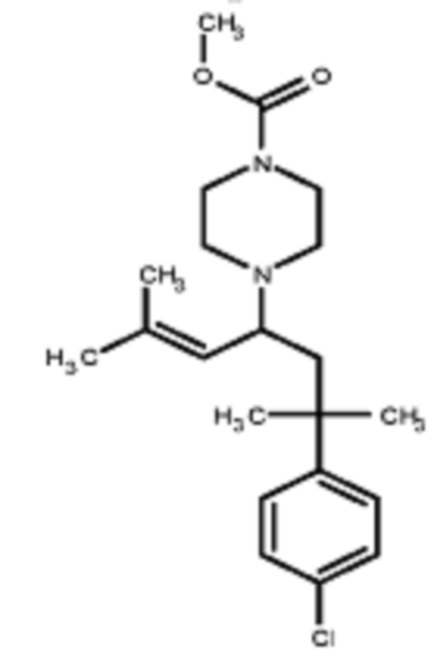


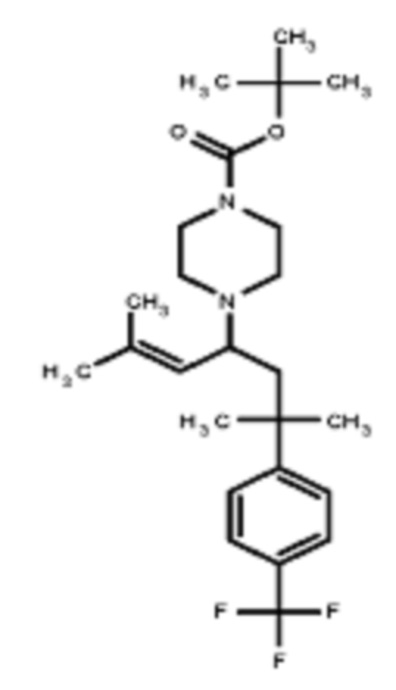



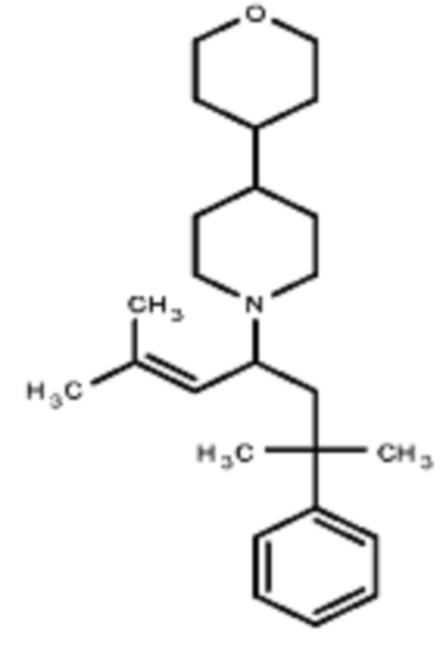

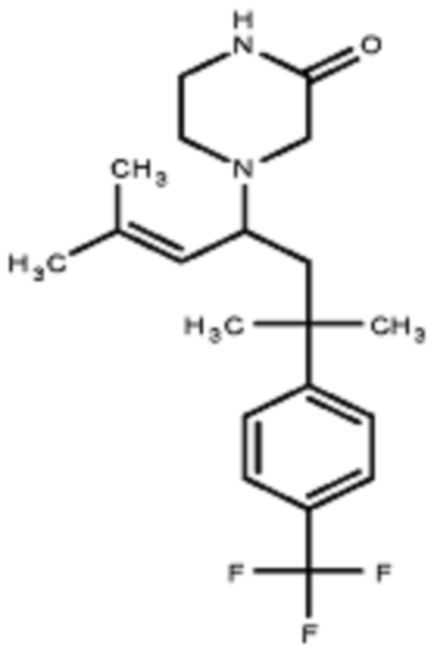

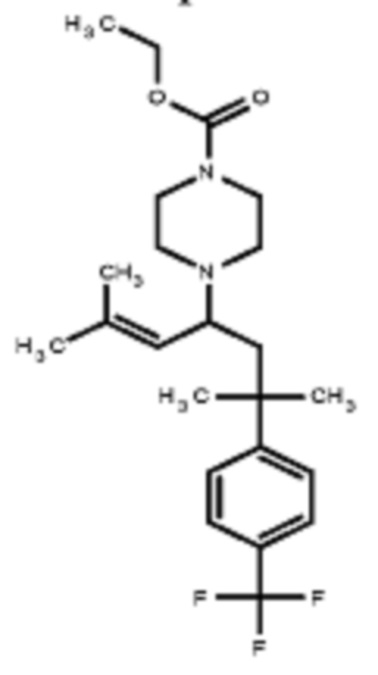


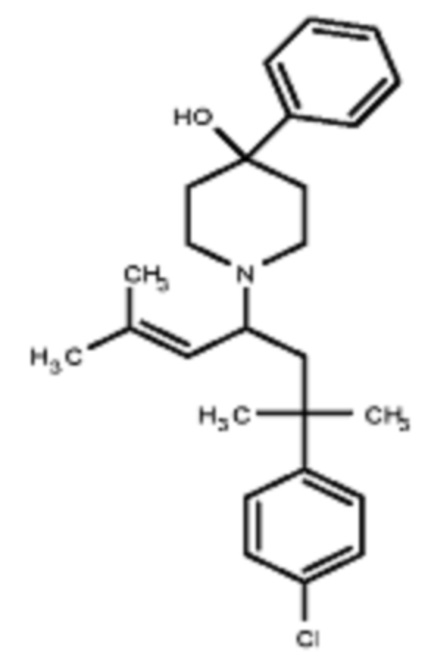
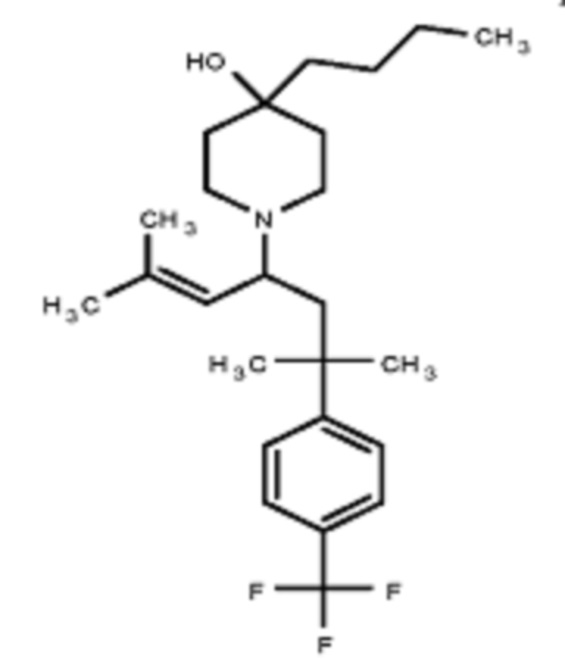

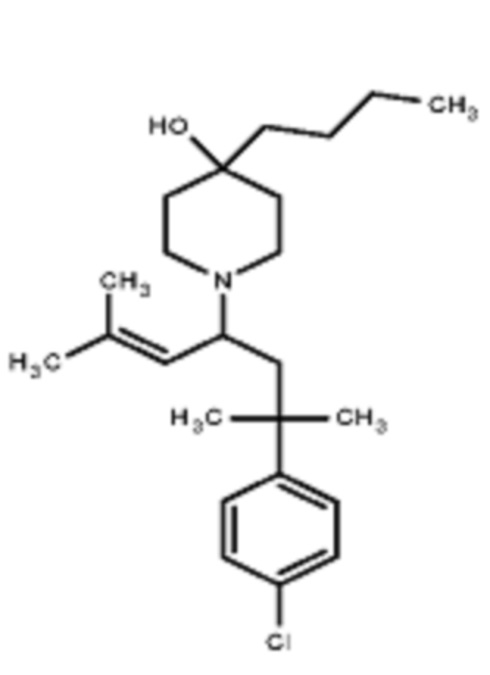








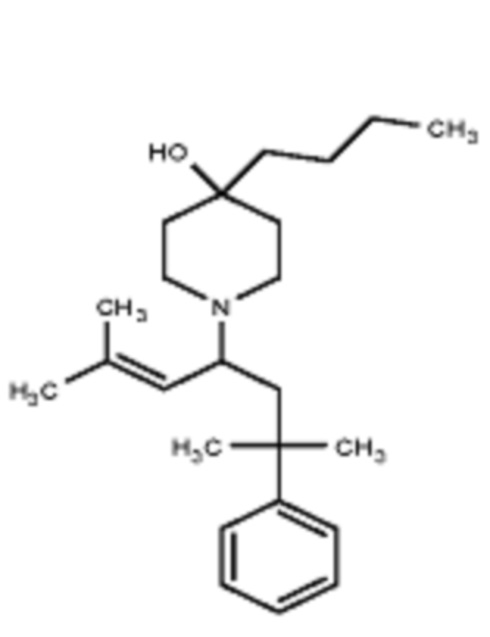


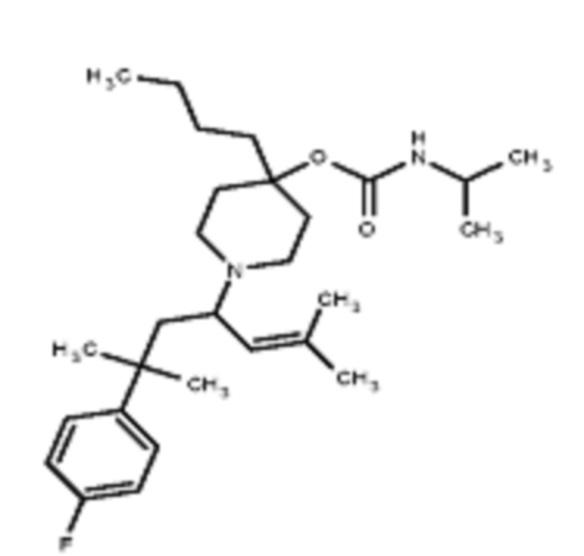






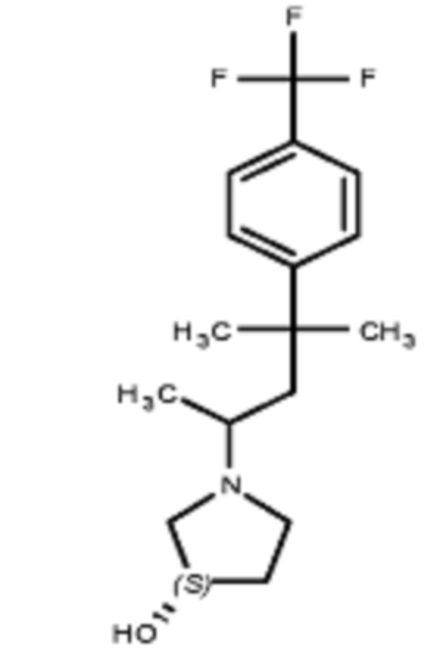

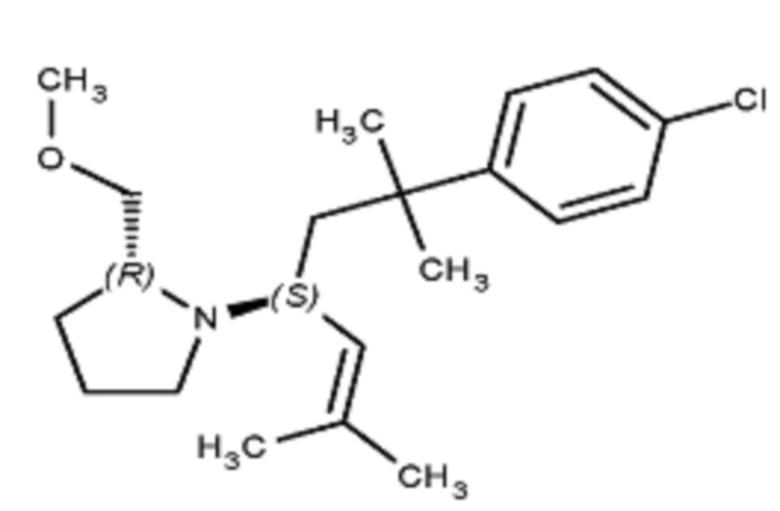












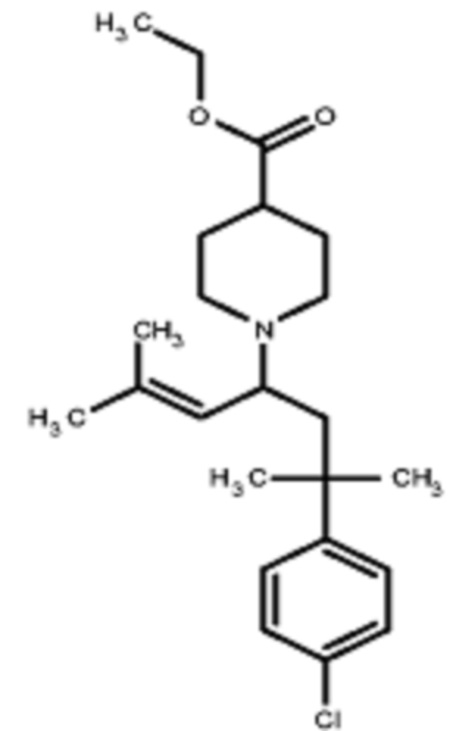
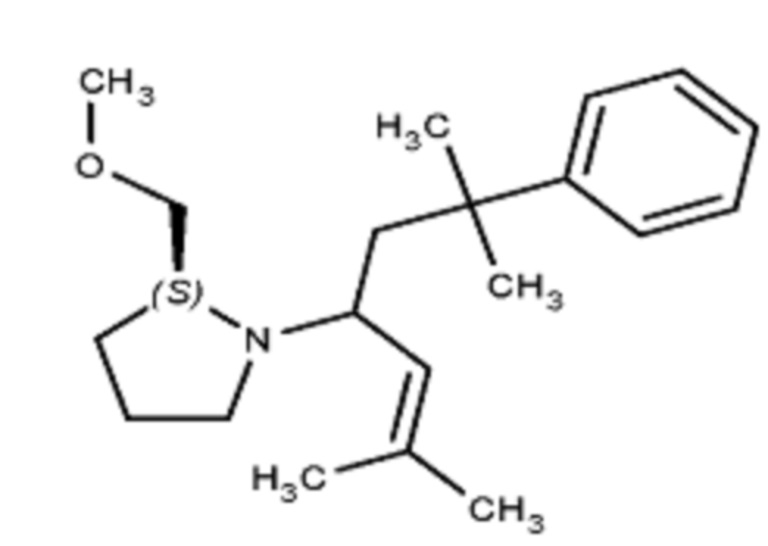



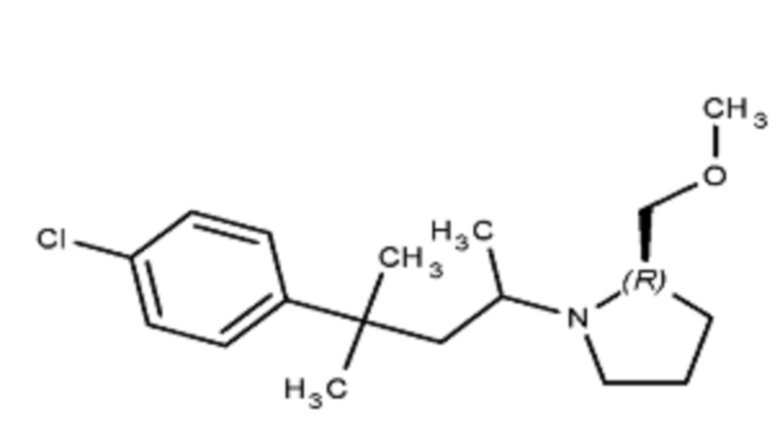
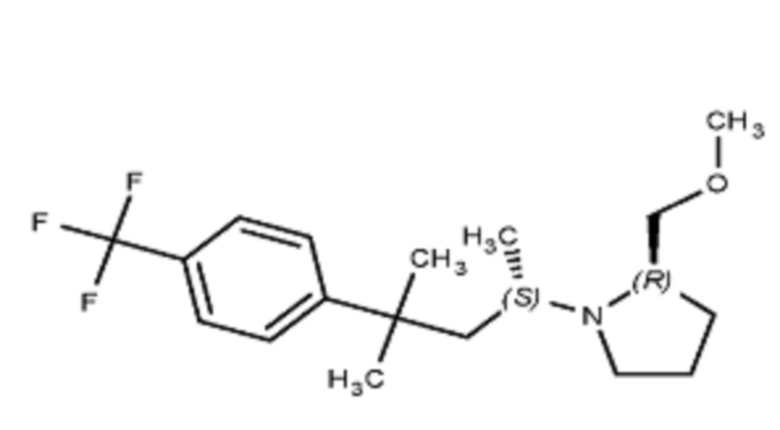




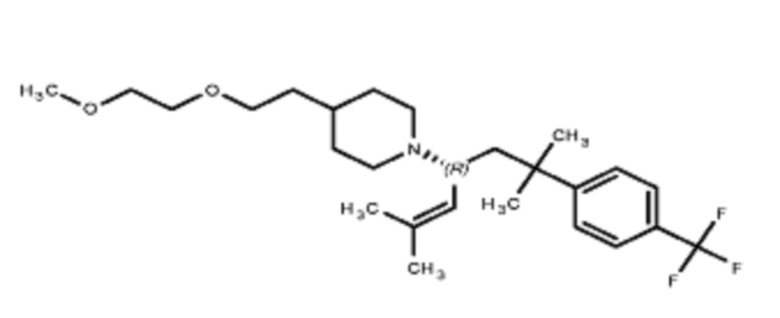

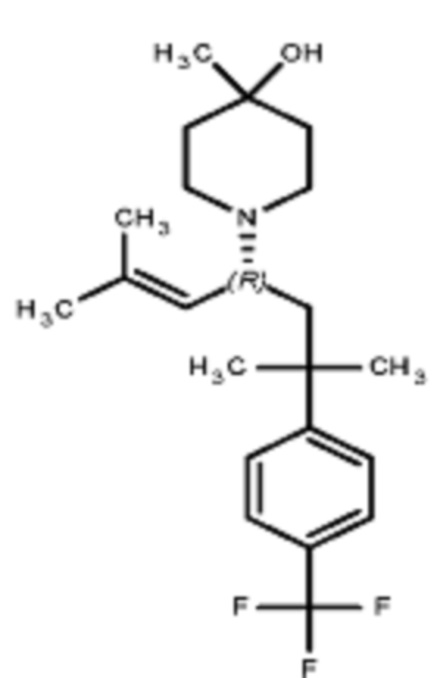


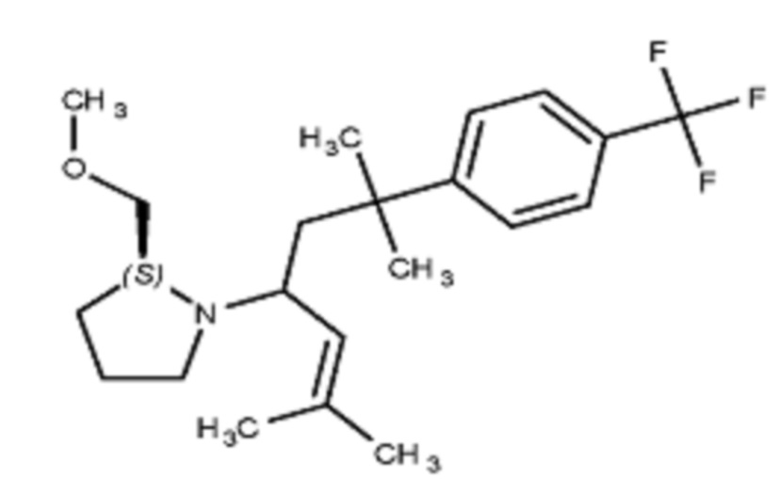













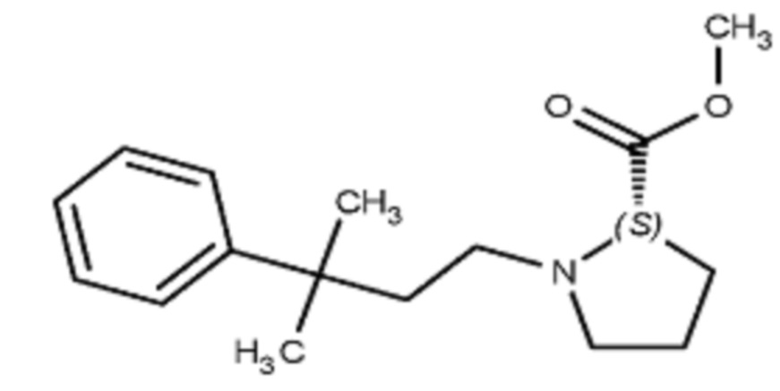

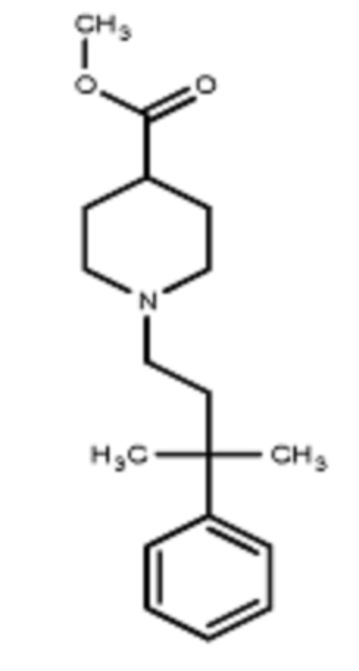







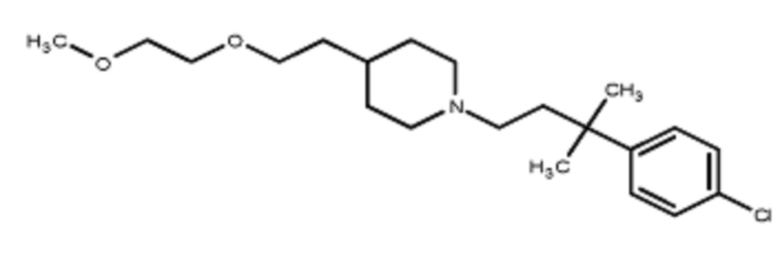
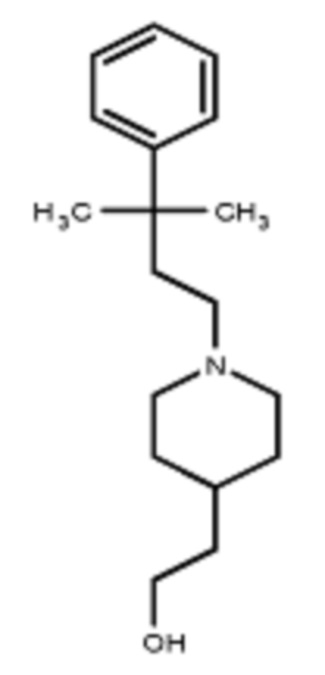




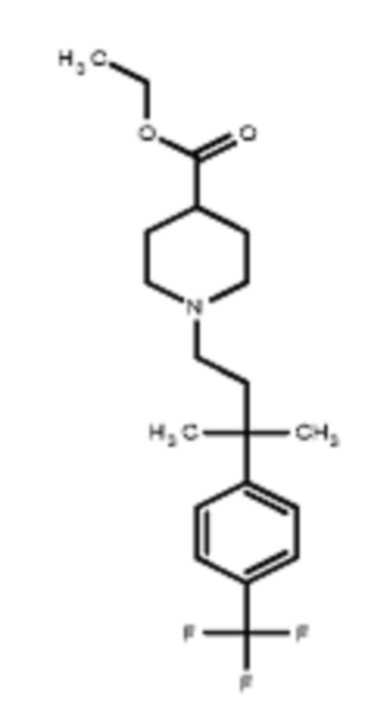







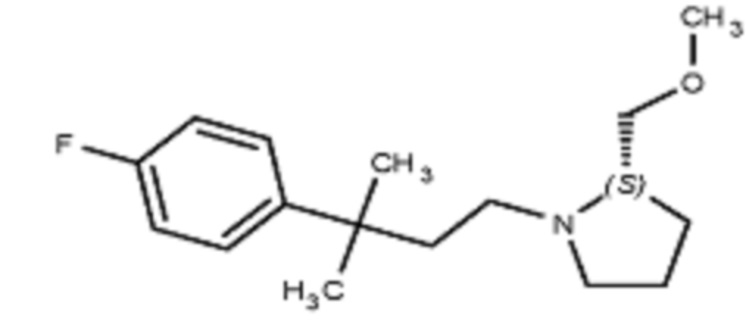
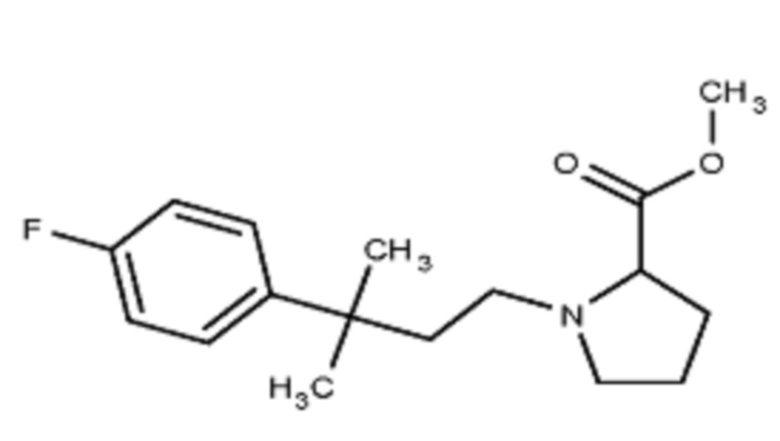




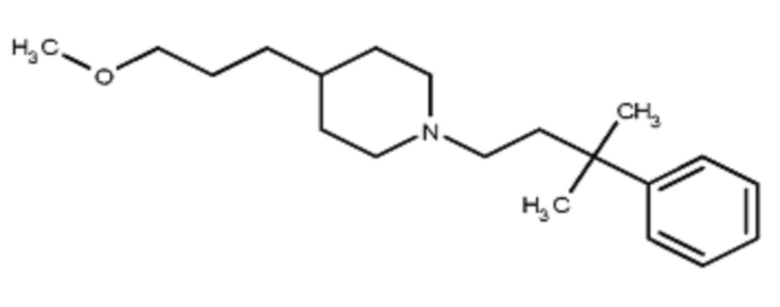

Некоторые варианты осуществления относятся к соединению, выбранному из группы, состоящей из
 ,
,
Пример 14 ,
,
Пример 17 и
и
Пример 9
Пример 262
Дополнительные варианты осуществления относятся к соединениям формулы II или их фармацевтически приемлемой соли:
 II.
II.
Каждый из заместителей Rf, Rg, Rh, Ri и Rj формулы II независимо выбран из групп, состоящих из, H, гидроксила, галогена, алкила, алкокси, CF3, SO2CH3 и морфолино.
Заместитель R10 формулы II представляет собой необязательно замещенную циклическую аминогруппу, и m представляет собой целое число от 0 до 3.
В некоторых вариантах осуществления каждый из заместителей Rf, Rg, Rh, Ri и Rj формулы II независимо выбран из группы, состоящей из, H, гидроксила и алкокси. В некоторых воплощениях каждый из заместителей Rf, Rg, Rh, Ri и Rj формулы II независимо выбран из группы, состоящей из, H, гидроксила и метокси. В некоторых вариантах осуществления каждый из заместителей Rf, Rg и Rj представляет собой H, и каждый из Rg и Rh независимо выбран из гидроксила или метокси.
В некоторых вариантах осуществления R10 представляет собой необязательно замещенный азиридинил, необязательно замещенный пирролидинил, необязательно замещенный имидизолидинил, необязательно замещенный пиперидинил, необязательно замещенный пиперазинил, необязательно замещенный оксопиперазинил или необязательно замещенный морфолинил и любой из отдельного замещенного или незамещенного пиперидинила, замещенного или незамещенного морфолинила, замещенного или незамещенного пиперазинила, замещенного или незамещенного пирролидинила, замещенных или незамещенных бициклических или замещенных или незамещенных конденсированных колец, описанных выше в отношении формулы I.
В некоторых вариантах осуществления R10 представляет собой необязательно замещенное конденсированное кольцо, такое как:
 ,
,
где каждый из R11e, R11f, R11g и R11h независимо выбран из водорода, гидрокси, сульфонила, необязательно замещенного C1–C10 алкила, необязательно замещенного C5–C10 арила, необязательно замещенного C3–C10 гетероарила, необязательно замещенного C3–C10 циклоалкила или необязательно замещенного С3–С10 гетероциклоалкила. В некоторых вариантах осуществления R10 не представляет собой
когда m равно 2.
В некоторых вариантах осуществления R10 представляет собой








 и
и  .
.
В некоторых вариантах осуществления описано соединение формулы IIa:
 IIa.
IIa.
Каждый из заместителей Rk и R1 независимо выбран из группы, состоящей из Н, гидроксила, галогена, алкила, алкокси, CF3, SO2CH3 и морфолино.
Заместитель R12 выбран из группы, состоящей из арилокси, алкенилокси, алкокси, аминоалкила, N, N–диметиламиноалкила, пирролидинила, н–метилпирролидинила, N–ацилпирролидинила, карбоксиаминоалкила, гидроксиалкила, –O(CH2)2OC(O)CH3, 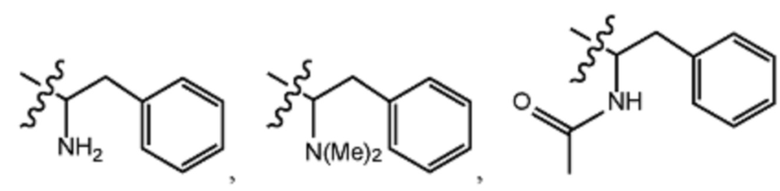 и
и  .
.
В некоторых вариантах осуществления каждый из заместителей Rk и R1 независимо выбран из группы, состоящей из H, гидроксила и метокси. В некоторых вариантах осуществления Rl представляет собой метоксигруппу и Rk представляет собой гидроксил.
В некоторых вариантах осуществления заместитель R12 выбран из группы, состоящей из фенилокси, –OCH2CH=CH2, метокси, –CH2NH2, –CH(NH2)CH3, –CH2N(Me)2, –CH(CH3)N(Me)2, –CH2NHC(O)CH3, –CH(OH)CH3, –O(CH2)2OC(O)CH3, 

 и
и  .
.
В некоторых вариантах осуществления описано соединение, выбранное из группы, состоящей из













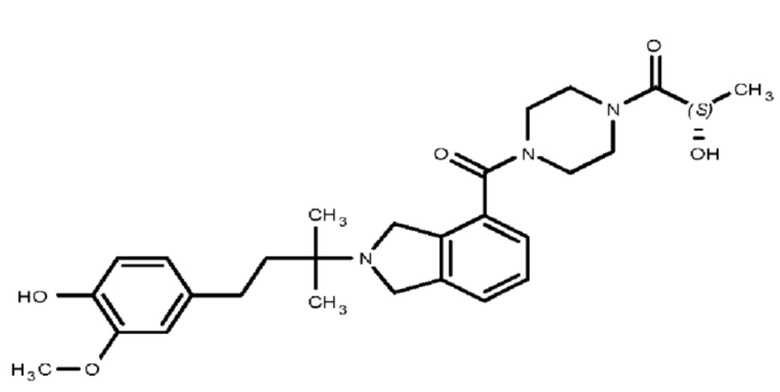

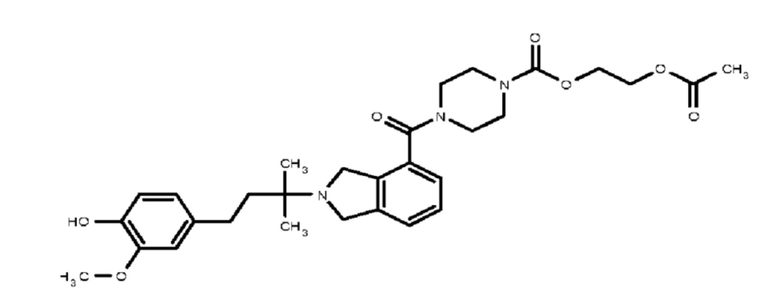

В некоторых вариантах осуществления описано соединение, выбранное из группы, состоящей из
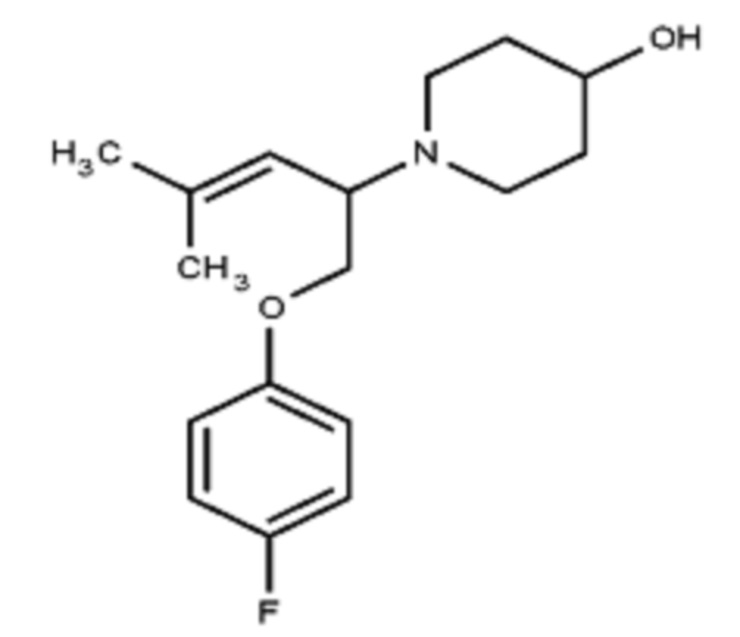
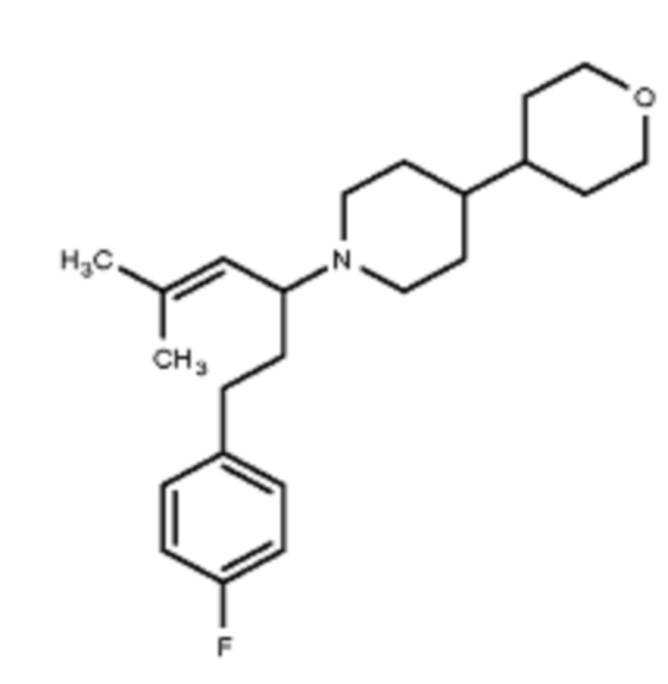


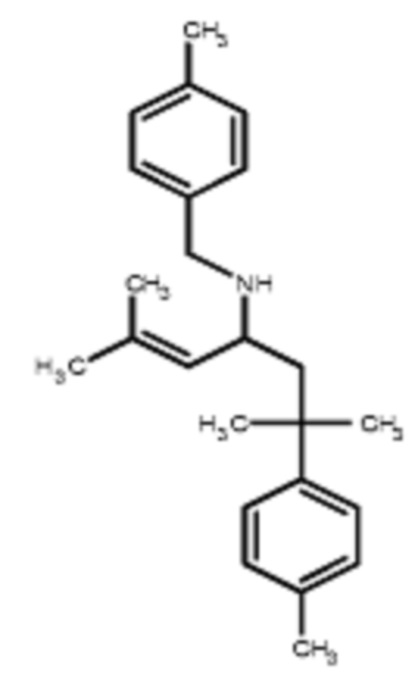




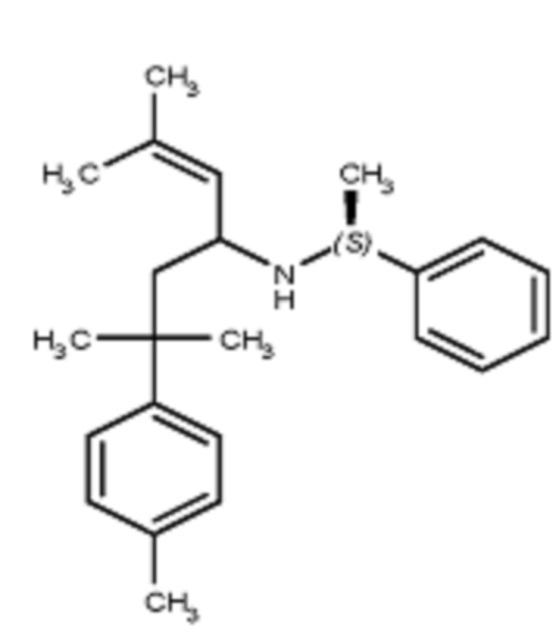
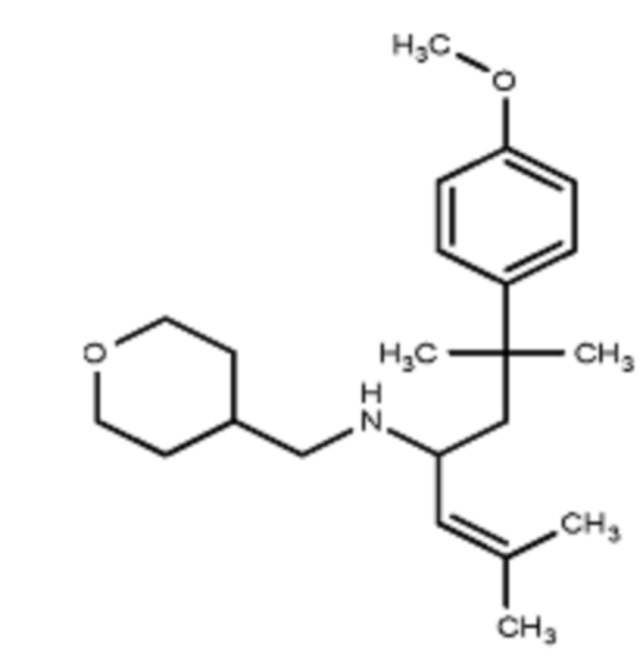
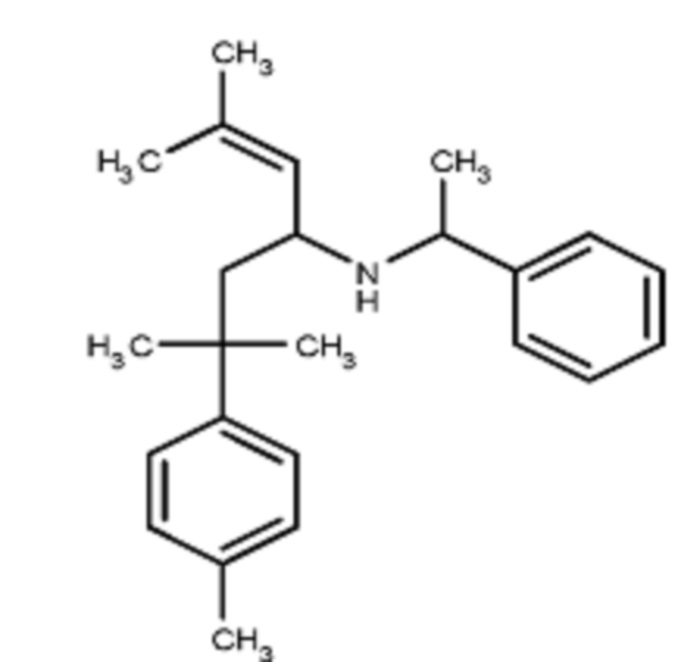










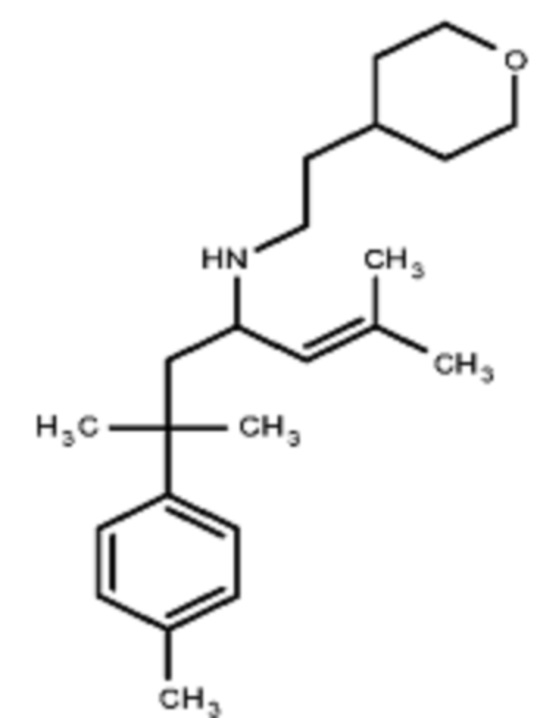
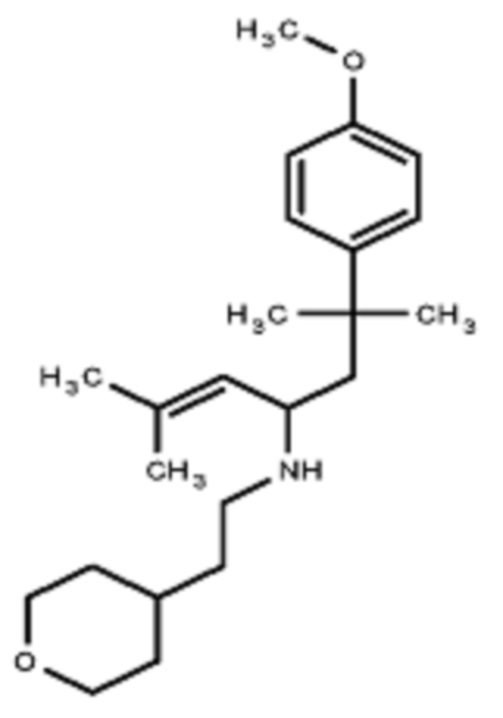

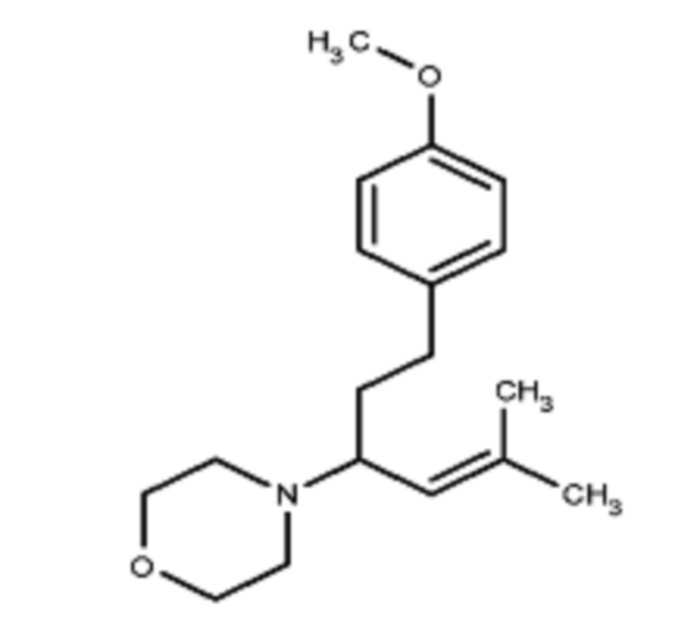



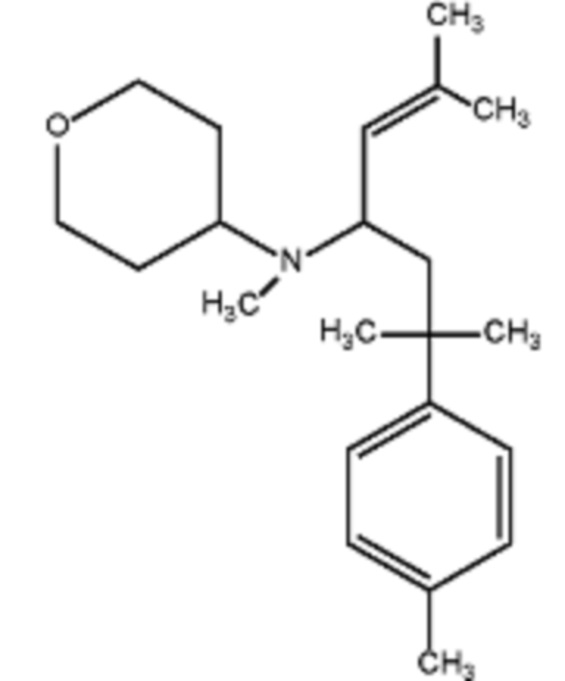
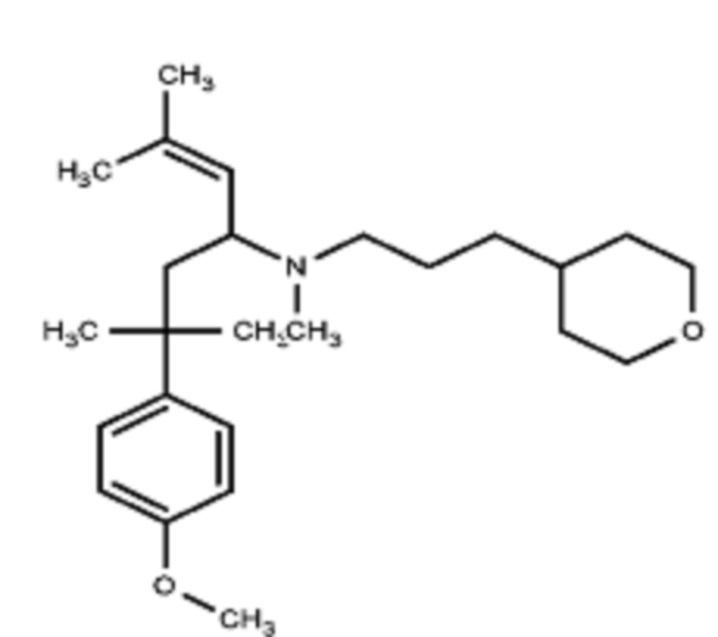







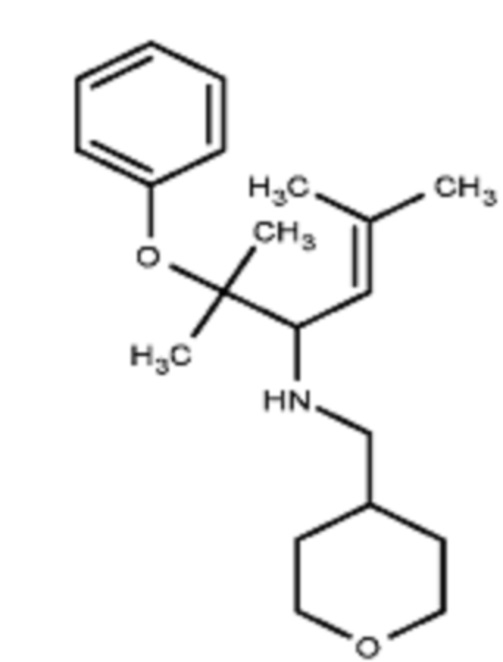





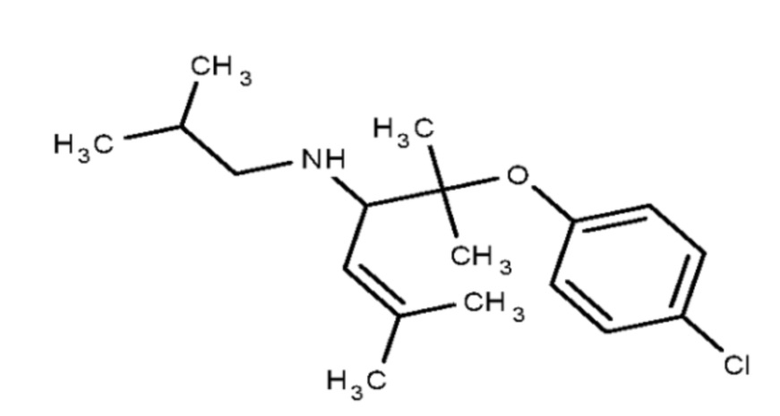




Дополнительные варианты осуществления включают соли, сольваты, стереоизомеры, пролекарства и активные метаболиты соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе.
Некоторые варианты осуществления относятся к формам свободных оснований соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе. Другие варианты осуществления включают соли таких соединений, включая, например, фармацевтически приемлемые соли присоединения кислоты или фармацевтически приемлемые соли присоединения свободных оснований. Примеры фармацевтически приемлемых солей присоединения кислоты включают, но ими не ограничиваются, соли, полученные из азотной, фосфорной, серной или бромистоводородной, иодистоводородной, фтористоводородной, фосфорной кислот, а также соли, полученные из нетоксичных органических кислот, таких как алифатические моно– и дикарбоновые кислоты, фенил–замещенные алкановые кислоты, гидроксиалкановые кислоты, алкандикарбоновые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты и уксусная, малеиновая, янтарная или лимонная кислоты. Неограничивающие примеры таких солей могут включать нападизилат, безилат, сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрогенфосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, трифтороацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себацинат, фумарат, малеат, манделат, бензоат, хлоробензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат и т.п. Дополнительные солевые формы соединений, описанных выше, включают соли аминокислот, таких как аргинат и тому подобные соли, и глюконат, галактуронат (см., например, Berge, et al. “Pharmaceutical Salts”, J. Pharma. Sci. 1977; 66:1).
Фармацевтически приемлемые соли присоединения основания образуются с металлами или аминами, такими как щелочные и щелочноземельные металлы или органические амины. Примерами металлов, используемых в качестве катионов, являются натрий, калий, магний, кальций и тому подобное. Примеры подходящих аминов включают N,N'–дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, дициклогексиламин, этилендиамин, N–метилглюкамин и прокаин. Соли присоединения оснований указанных кислотных соединений получают путем контакта формы свободной кислоты с достаточным количеством желаемого основания для получения соли обычным способом. Форма свободной кислоты может быть регенерирована путем контакта солевой формы с кислотой и выделения свободной кислоты.
Различные варианты осуществления включают полные и частичные соли, то есть соли с 1, 2 или 3, предпочтительно 2 эквивалентами основания на моль кислоты соединения или соли, описанной выше, с 1, 2 или 3 эквивалентами, предпочтительно 1 эквивалентом кислоты на моль основания соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе. Обычно фармацевтически приемлемая соль соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть легко получена, в зависимости от требований, путем использования желаемой кислоты или основания. Соль может осаждаться из раствора и собираться фильтрацией или может быть извлечена путем упаривания растворителя. Например, водный раствор кислоты, такой как соляная кислота, может добавляться к водной суспензии соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, и полученную смесь упаривают досуха (лиофилизируют) с получением соли присоединения кислоты в виде твердого вещества. Альтернативно, соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть растворено в подходящем растворителе, например, спирте, таком как изопропанол, и кислота может добавляться в том же самом растворителе или другом подходящем растворителе. Затем полученную соль соединения кислоты можно осаждать непосредственно или путем добавления менее полярного растворителя, такого как диизопропиловый эфир или гексан, и выделять путем фильтрации.
Множество органических соединений могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они осаждаются или кристаллизуются. Эти комплексы известны как “сольваты”. Например, комплекс с водой известен как “гидрат”. Различные варианты осуществления включают сольваты соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе. В некоторых вариантах осуществления соли этих соединений могут образовывать сольваты.
Дополнительные варианты осуществления включают N–оксиды соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе. N–оксиды включают гетероциклы, содержащие незамещенный в других условиях атом sp2 N. Примеры таких N–оксидов включают пиридил–N–оксиды, пиримидил–N–оксиды, пиразинил–N–оксиды и пиразолил–N–оксиды.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут иметь один или более хиральных центров и, в зависимости от природы отдельных заместителей, они также могут иметь геометрические изомеры. Таким образом, варианты осуществления включают стереоизомеры, диастереомеры и энантиомеры соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе. Хиральное соединение может существовать в виде отдельного энантиомера или в виде смеси энантиомеров. Смесь, содержащая равные количества энантиомеров, называется “рацемической смесью”. Смесь, содержащая неравные количества энантиомеров, описывается как имеющая “энантиомерный избыток” (ее) R– или S–соединения. Избыток одного энантиомера в смеси часто описывается как процентный показатель энантиомерного избытка. Соотношение энантиомеров также может определяться “оптической чистотой”, где степень, при которой смесь энантиомеров вращает плоскополяризованный свет, сравнивается с отдельными оптически чистыми R– и S–соединениями. Соединения также могут быть по существу чистым (+) или (–) энантиомером соединений, описанных в настоящем документе. В некоторых вариантах осуществления композиция может включать по существу чистый энантиомер, который включает, по меньшей мере, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% одного энантиомера. В некоторых вариантах осуществления композиция может включать по существу чистый энантиомер, который включает, по меньшей мере, 99,5% одного энантиомера.
Вышеприведенное описание охватывает все отдельные изомеры соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе, и обозначение или название конкретного соединения в описании и формуле изобретения включает как отдельные энантиомеры, так и их смеси. Способы определения стереохимии и расщепления или стереотаксического синтеза стереоизомеров хорошо известны в данной области техники. Диастереомеры отличаются как по физическим свойствам, так и по химической реакционной способности. Смесь диастереомеров может быть разделена на энантиомерные пары на основе растворимости, фракционной кристаллизации или хроматографических свойств, например, тонкослойной хроматографией, колоночной хроматографией или ВЭЖХ. Для очистки сложных смесей диастереомеров до энантиомеров обычно требуется два этапа. На первом этапе смесь диастереомеров разделяется на энантиомерные пары, как описано выше. На втором этапе энантиомерные пары подвергают дополнительной очистке до композиций, обогащенных одним или другим энантиомером, или, что более предпочтительно, разделяют на композиции, содержащие чистые энантиомеры. Разделение энантиомеров обычно требует реакции или молекулярного взаимодействия с хиральным агентом, например, растворителем или матриксом колонки. Разделение может достигаться, например, путем преобразования смеси энантиомеров, например, рацемической смеси, в смесь диастереомеров путем взаимодействия с чистым энантиомером второго агента, то есть разделяющего агента. После этого могут быть разделены два полученных в результате диастереомерных продукта. Затем разделенные диастереомеры повторно преобразуют в чистые энантиомеры путем обращения первичной химической трансформации.
Разделение энантиомеров также может быть осуществлено по различиям в их нековалентном связывании с хиральным веществом, например, путем хроматографии на гомохиральных адсорбентах. Нековалентное связывание между энантиомерами и хроматографическим адсорбентом приводит к образованию диастереомерных комплексов, приводящих к дифференциальному разделению в подвижном и связанном состояниях в хроматографической системе. Таким образом, два энантиомера проходят через хроматографическую систему, например, колонку, с разными скоростями, что обечивает возможность их разделения.
Дополнительные варианты осуществления включают пролекарства соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе, то есть соединения, которые высвобождают активное соединение в соответствии с любым из вариантов осуществления, описанных в настоящем документе, in vivo при введении субъекту–млекопитающему. Пролекарство представляет собой фармакологически активное или, более типично, неактивное соединение, которое превращается в фармакологически активное вещество путем метаболического превращения. Пролекарства соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, получают путем модификации функциональных групп, присутствующих в соединении, в соответствии с любым вариантом осуществления, описанным в настоящем документе, таким образом, что модификации могут быть расщеплены in vivo для высвобождения исходного соединения. In vivo пролекарство легко подвергается химическим изменениям в физиологических условиях (например, гидролизуется или подвергается действию естественного(ых) фермента(ов)), что приводит к высвобождению фармакологически активного агента. Пролекарства включают соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, в которых гидроксильная, амино или карбоксигруппа связана с любой группой, которая может быть расщеплена in vivo для регенерации свободной гидроксильной, амино или карбоксильной группы соответственно. Примеры пролекарств включают, но ими не ограничиваются, сложные эфиры (например, производные ацетата, формиата и бензоата) соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе, или любое другое производное, которое после приведения к физиологическому уровню Н или под действием фермента превращается в активное исходное лекарственное вещество. Обычные процедуры отбора и получения подходящих препаратов–предшественников описаны в данной области (см., например, Bundgaard. Design of Prodrugs. Elsevier, 1985).
Изобретение также включает выделенные соединения. Выделенное соединение относится к соединению, которое составляет, по меньшей мере, 10%, предпочтительно, по меньшей мере, 20%, более предпочтительно, по меньшей мере, 50% и наиболее предпочтительно, по меньшей мере, 80% соединения, присутствующего в смеси.
В некоторых вариантах осуществления один или более атомов водорода соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, заменены дейтерием. Хорошо известно, что дейтерирование физиологически активных соединений обеспечивает преимущество в сохранении фармакологического профиля их водородных аналогов, в то же время оказывая положительное влияние на их метаболический выход. Селективная замена одного или более атомов водорода дейтерием в соединении в соответствии с любым вариантом осуществления, описанным в настоящем документе, может повысить безопасность, переносимость и эффективность соединения по сравнению с его водородным аналогом.
Методы включения дейтерия в соединения хорошо являются известными. С использованием исследований метаболизма, установленных в данной области, соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть исследовано на предмет определения участков для селективного размещения изотопа дейтерия, на которых изотоп не будет подвергаться метаболизму. Кроме того, эти исследования определяют участки метаболизма как место для размещения атома дейтерия.
Фармацевтические композиции
В некоторых вариантах осуществления описана фармацевтическая композиция, содержащая: соединение в соответствии с любым вариантом осуществления, описанным в данном документе, его фармацевтически приемлемую соль, его сольват, его стереоизомер, его пролекарство или его активные метаболиты; и фармацевтически приемлемый носитель или разбавитель. Фармацевтические композиции могут быть получены способом, хорошо известным в области фармацевтики и могут вводиться различными способами, в зависимости от того, требуется ли местное или системное лечение, и в зависимости от участка, подлежащего лечению.
Если существует возможность введения соединения, описанное в любом из вариантов осуществления настоящего изобретения, в виде нерасфасованного вещества, предпочтительно включение соединения в фармацевтическую композицию, например, в которой активное вещество находится в смеси с выбранным фармацевтически приемлемым носителем, выбранным с учетом предполагаемого способа введения и стандартной фармацевтической практики.
В частности, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество, по меньшей мере, одного соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, и, необязательно, фармацевтически приемлемый носитель.
Комбинации
Для фармацевтических композиций и способов по настоящему изобретению, соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно использовать в комбинации с другими видами терапевтических средств и/или активных веществ.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, может объединяться с одним или более ингибиторами холинэстеразы, антагонистом глутаматного рецептора типа N–метил–D–аспартата (NMDA), ингибитор бета–секретазы 1 (ВАСЕ1, фермент 1, расщепляющий по бета–участку белок–предшественник амилоида), модулятор фактора некроза опухоли альфа (TNF–альфа), внутривенный иммуноглобулин (IVIG) или антагонист прионного белка. В некоторых вариантах осуществления соединение объединяют с ингибитором холинэстеразы, выбранным из такрина (COGNEX®; Sciele), донепезила (ARICEPT®; Pfizer), ривастигмина (EXELON®; Novartis) или галантамина (RAZADYNE®; Ortho–McNeil–Janssen). В некоторых вариантах осуществления соединение объединяют с модулятором TNF–альфа, который представляет собой периспинальный этанерцепт (ENBREL®, Amgen/Pfizer). В некоторых вариантах осуществления соединение объединяют со специфичным к бета–амилоиду антителом, выбранным из бапинейзумаба (Pfizer), соланезумаба (Lilly), PF–04360365 (Pfizer), GSK933776 (GlaxoSmithKline), Gammagard (Baxter) или Octagam (Octapharma). В некоторых вариантах осуществления соединение объединяют с антагонистом рецептора NMDA, которым является мемантин (NAMENDA®; Forest). В некоторых вариантах осуществления ингибитором BACE1 является MK–8931 (Merck). В некоторых вариантах осуществления соединение объединяют с IVIG, как описано в публикации Magga et al., J Neuroinflam 2010, 7:90, Human intravenous immunoglobulin provides protection against Ab toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease, and Whaley et al., 2011, Human Vaccines 7:3, 349–356, Emerging antibody products and Nicotiana manufacturing; каждая из которых включена в настоящее описание посредством ссылки. В некоторых вариантах осуществления соединение объединяют с антагонистом прионного белка, как описывается в публикации Strittmatter et al., US 2010/0291090, которая включена в настоящее изобретение посредством ссылки.
Соответственно, изобретение в еще одном аспекте обеспечивает фармацевтическую композицию, содержащую, по меньшей мере, одно соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, или его фармацевтически приемлемое производное; второе активное вещество; и, необязательно, фармацевтически приемлемый носитель.
При объединении в одной композиции следует учитывать, что два или больше соединений должны быть стабильными и совместимыми друг с другом и другими компонентами композиции. При раздельном получении лекарственной формы, они могут быть предусмотрены в любой удобной композиции, известным в данной области способом, предусмотренным для таких соединений.
Консерванты, стабилизаторы, красители и ароматизаторы могут быть предусмотрены в любой фармацевтической композиции, описанной в настоящем документе. Примеры консервантов включают бензоат натрия, аскорбиновую кислоту и сложные эфиры п–гидроксибензойной кислоты. Также могут быть использованы антиоксиданты и суспендирующие вещества.
В отношении комбинаций, включающих биологические препараты, такие как моноклональные антитела или фрагменты, будут использоваться подходящие эксципиенты для предотвращения агрегации и стабилизации антитела или фрагмента в растворе с низким содержанием эндотоксина, как правило, для парентерального введения, например, для внутривенного введения. Например, см. Formulation and Delivery Issues for Monoclonal Antibody Therapeutics, Daugherty et al., in Current Trends in Monoclonal Antibody Development and Manufacturing, Part 4, 2010, Springer, New York, pp. 103–129.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут быть измельчены с использованием известных процедур размола, таких как мокрый размол, для получения размера частиц, подходящего для формирования таблеток и для других типов лекарственных форм. Тонко диспергированные (в форме наночастиц) препараты соединений могут быть получены способами, известными в данной области, например, см. WO 02/00196 (SmithKline Beecham).
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, или их фармацевтически приемлемые соли, их сольваты, их стереоизомеры, их пролекарства или их активные метаболиты могут быть получены в лекарственной форме для любого способа введения.
Способы введения и стандартные лекарственные формы
Способы введения (доставки) включают, но ими не ограничиваются, один или более из следующих способов: пероральный (например, в виде таблетки, капсулы или в виде проглотываемого раствора), местный, мукозальный (например, в форме назального спрея или аэрозоля для ингаляции), парентеральный (например, с применением инъекционной формы), желудочно–кишечный, интраспинальный, внутрибрюшинный, внутримышечный, внутривенный, интрацеребровентрикулярный или другой способ, обеспечивающий замедленное введение и т.д.
Следовательно, фармацевтические композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают композиции в форме, специально полученной для способа введения. В некоторых вариантах осуществления фармацевтические композиции по изобретению получены в форме, которая подходит для пероральной доставки. В некоторых вариантах осуществления соединение представляет собой биодоступное пероральное соединение, подходящее для пероральной доставки. В других вариантах осуществления фармацевтические композиции по изобретению получены в форме, которая подходит для парентеральной доставки.
Соединениям в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть придана лекарственная форма для введения любым удобным способом для применения в медицине или ветеринарии, и, следовательно, изобретение включает в свой объем фармацевтические композиции, содержащие соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, адаптированные для применения в медицине или ветеринарии. Такие фармацевтические композиции могут быть представлены для применения обычным способом с помощью одного или более подходящих носителей. Приемлемые носители для терапевтического применения хорошо известны в области фармацевтики и описаны, например, в публикации Remington’s Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro edit. 1985). Выбор фармацевтического носителя может быть осуществлен с учетом предполагаемого способа введения и стандартной фармацевтической практики. Фармацевтические композиции могут включать, помимо носителя, любое(ые) подходящее(ие) связующее(ие), смазывающее(ие) вещество(а), суспендирующее(ие) вещество(а), покрывающее(ие) вещество(а) и/или солюбилизирующее(ие) вещество(а).
Могут быть предусмотрены различные требования к фармацевтической композиции/лекарственной форме в зависимости от разных систем доставки. Следует понимать, что не все соединения необходимо вводить одним и тем же способом. Аналогично, если фармацевтическая композиция содержит больше одного активного компонента, тогда эти компоненты могут вводиться различными способами. В качестве примера, фармацевтическая композиция по настоящему изобретению может быть получена в форме для доставки с использованием мининасоса или мукозальным способом, например, в виде назального спрея или аэрозоля для ингаляции, или проглатываемого раствора, или парентерально, причем фармацевтическую композицию получают в инъекционной форме для доставки, например, внутривенным, внутримышечным или подкожным путем. Альтернативно, композиция может быть разработана для доставки различными способами.
Комбинации соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, и молекул антитела или фрагмента антитела может быть придана лекарственная форма, которую вводят любым из многих способов и в концентрации, которая является терапевтически эффективной при соответствующем показании или с соответствующей целью. Для достижения этой цели, антителам может быть придана лекарственная форма с использованием множества приемлемых эксципиентов, известных в данной области техники. Обычно антитела вводят посредством инъекции, например, внутривенной инъекции. Способы осуществления этого введения известны специалистам в данной области. Например, в публикации Gokarn et al., 2008, J Pharm Sci 97(8):3051–3066, включенной в настоящее описание посредством ссылки, описаны различные высококонцентрированные самобуферирующиеся композиции антител. Например, моноклональные антитела в самобуферирующейся композиции, например, при 50 мг/мл mAb в 5,25% сорбита, рН 5,0; или 60 мг/мл mAb в 5% сорбита, 0,01% полисорбата 20, рН 5,2; или обычных буферных композициях, например, 50 мг/мл mAb1 в 5,25% сорбита, 25 или 50 мМ ацетата, глутамата или сукцината, при рН 5,0; или 60 мг/мл в 10 мМ ацетата или глутамата, 5,25% сорбита, 0,01% полисорбата 20, рН 5,2; могут применяться другие низкоконцентрированные композиции, известные специалистам в данной области.
Поскольку некоторые соединения по настоящему изобретению преодолевают гематоэнцефалический барьер, их можно вводить различными способами, включая, например, системный (например, внутривенно, подкожно, перорально, мукозально, трансдермально) или локализованными методами (например, внутричерепно). Когда соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, должно доставляться через слизистую оболочку желудочно–кишечного тракта, оно должно оставаться стабильным в процессе переноса через желудочно–кишечный тракт; например, оно должно быть устойчивым к протеолитической деградации, стабильным при кислотном уровне pH и устойчивым к детергентному воздействию желчи. Например, соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, приготовленные для перорального введения, могут быть покрыты слоем энтеросолюбильного покрытия. Вещество слоя энтеросолюбильного покрытия может быть диспергировано или растворено либо в воде, либо в подходящем органическом растворителе. В качестве полимеров слоя энтеросолюбильного покрытия могут быть использованы, по отдельности или в комбинации, один или более из следующих веществ; например, растворы или дисперсии сополимеров метакриловой кислоты, ацетатфталата целлюлозы, ацетатбутирата целлюлозы, фталата гидроксипропилметилцеллюлозы, сукцинатацетата гидроксипропилметилцеллюлозы, фталата поливинилацетата, ацетат–тримеллитата целлюлозы, карбоксиметилэтилцеллюлозы, шеллака или другого(их) подходящего(их) полимера(ов) для слоя энтеросолюбильного покрытия. В некоторых вариантах осуществления слой водного энтеросолюбильного покрытия представляет собой сополимер метакриловой кислоты.
В соответствующих случаях, фармацевтические композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно вводить путем ингаляции, путем применения кожного пластыря, перорально в форме таблеток, содержащих эксципиенты, такие как крахмал или лактоза, или в капсулах или суппозиториях, отдельно или в смеси с эксципиентами, или в форме эликсиров, растворов или суспензий, содержащих ароматизаторы или красители, или их можно вводить парентерально, например, внутривенно, внутримышечно или подкожно. Для трансбуккального или сублингвального введения, фармацевтические композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно вводить в форме таблеток или пастилок, которые могут быть получены общепринятым способом.
Если фармацевтическая композиция в соответствии с любым вариантом осуществления, описанным в настоящем документе, предназначена для парентерального введения, такое введение включает, без ограничения: внутривенное, внутриартериальное, интратекальное, интравентрикулярное, внутричерепное, внутримышечное или подкожное введение соединения по настоящему изобретению; и/или применение способов инфузии. Антитела или фрагменты, обычно, вводят парентерально, например, внутривенно.
Фармацевтические композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, подходящие для инъекции или инфузии, могут быть в форме стерильного водного раствора, дисперсии или стерильного порошка, который содержит активный ингредиент, отрегулированный, при необходимости, для приготовления такого стерильного раствора или дисперсии, подходящих для инфузии или инъекции. Этот препарат может быть необязательно инкапсулирован в липосомы. Во всех случаях конечный препарат должен быть стерильным, жидким и стабильным в условиях производства и хранения. Для улучшения стабильности при хранении такие препараты могут также содержать консервант для предотвращения роста микроорганизмов. Предотвращение действия микроорганизмов может быть достигнуто путем добавления различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола или аскорбиновой кислоты. Во многих случаях рекомендуются изотонические вещества, например, сахара, буферы и хлорид натрия, для обеспечения осмотического давления, подобного показателям жидкостей организма, в частности крови. Пролонгированная абсорбция таких инъецируемых смесей может быть достигнута путем введения задерживающих абсорбцию веществ, таких как моностеарат алюминия или желатин.
Дисперсии могут быть приготовлены в жидком носителе или промежуточном соединении, таком как глицерин, жидкие полиэтиленгликоли, триацетиновые масла и их смеси. Жидкий носитель или промежуточное соединение может представлять собой растворитель или жидкую диспергирующую среду, которая содержит, например, воду, этанол, полиол (например, глицерин, пропиленгликоль или тому подобное), растительные масла, нетоксичные эфиры глицерина и их подходящие смеси. Соответствующая текучесть может поддерживаться путем образования липосом, включения частиц соответствующего размера в случае дисперсий или путем добавления поверхностно–активных веществ.
Для парентерального введения, соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, предпочтительно применяют в форме стерильного водного раствора, который может содержать другие вещества, например, достаточное количество солей или глюкозу для обеспечения изотоничности раствора с кровью. Водные растворы, при необходимости, должны быть подходящим образом забуферены (предпочтительно до рН от 3 до 9). Приготовление подходящих композиций для парентерального введения в стерильных условиях легко выполняется с применением стандартных фармацевтических методов, хорошо известных специалистам в данной области.
Стерильные инъецируемые растворы могут быть получены путем смешивания соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, с подходящим растворителем и одним или более из вышеуказанных носителей с последующей стерильной фильтрацией. В случае стерильных порошков, подходящих для использования при получении стерильных растворов для инъекций, предпочтительные способы получения включают сушку в вакууме и лиофилизацию, которые обеспечивают порошкообразные смеси соединений и необходимые эксципиенты для последующего приготовления стерильных растворов.
Соединениям в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть придана лекарственная форма для применения в медицине или ветеринарии путем инъекций (например, путем внутривенной болюсной инъекции или инфузии или внутримышечным, подкожным или интратекальным путями) и могут быть представлены в стандартной лекарственной форме, в ампулах или других контейнерах для единичной стандартной лекарственной формы, или в многодозных контейнерах, при необходимости с добавлением консерванта. Фармацевтические композиции для инъекций могут быть в форме суспензий, растворов или эмульсий в масляных или водных носителях и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие, солюбилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может находиться в форме стерильного порошка для восстановления перед применением подходящим носителем, например, стерильной апирогенной водой.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно вводить в форме таблеток, капсул, пастилок, суппозиториев, эликсиров, растворов или суспензий для применений с немедленным, отсроченным, модифицированным, замедленным, импульсным или контролируемым высвобождением.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут быть представлены для медицинского или ветеринарного применения в форме, подходящей для перорального или буккального введения, например, в форме растворов, гелей, сиропов или суспензий, или сухого порошка для восстановления перед применением водой или другим подходящим носителем. Также могут применяться твердые композиции, такие, как таблетки, капсулы, пастилки, лепешки, пилюли, шарики, порошки, пасты, гранулы, сферы или предварительно приготовленные смеси. Твердые и жидкие композиции для перорального применения получают в соответствии со способами, хорошо известными специалистам в данной области. Такие композиции также могут содержать один или более фармацевтически приемлемых носителей и эксципиентов, которые могут быть в твердой или жидкой форме.
Таблетки могут содержать эксципиенты, такие, как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин, разрыхлители, такие, как крахмал (предпочтительно кукурузный, картофельный или тапиоковый крахмал), натрий крахмал гликолят, кроскармеллоза натрия, и некоторые сложные силикаты и связующие вещества для гранулирования, такие, как поливинилпирролидон, гидроксипропилметилцеллюлоза (НPMC), гидроксипропилцеллюлоза (НРС), сахароза, желатин и камедь.
Кроме того, могут быть включены лубриканты, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк.
Фармацевтические композиции в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно вводить перорально в форме таблеток с быстрым или контролируемым высвобождением, микрочастиц, минитаблеток, капсул, саше и растворов для перорального введения, или суспензий или порошков для их получения. Пероральные препараты могут необязательно включать различные стандартные фармацевтические носители и эксципиенты, такие как связующие вещества, наполнители, буферы, смазывающие вещества, вещества, способствующие скольжению, красители, дезинтегранты, отдушки, подсластители, поверхностно–активные вещества, антиадгезивы для форм, антиадгезивные вещества и покрывающие вещества. В фармацевтических композициях некоторые эксципиенты могут иметь несколько функций, например, действовать в качестве как связующих, так и разрыхлителей.
Примеры фармацевтически приемлемых разрыхлителей для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, крахмал, предварительно желатинизированный крахмал, крахмалгликолят натрия, карбоксиметилцеллюлозу натрия, кроскармеллозу натрия, микрокристаллическую целлюлозу, альгинаты, смолы, поверхностно–активные вещества, шипучие композиции, водные алюмосиликаты и сшитый поливинилпирролидон.
Примеры фармацевтически приемлемых связующих для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, камедь; производные целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза или гидроксиэтилцеллюлоза; желатин, глюкоза, декстроза, ксилит, полиметакрилаты, поливинилпирролидон, сорбит, крахмал, предварительно желатинизированный крахмал, трагакант, ксантиновая смола, альгинаты, алюмосиликат магния, полиэтиленгликоль или бентонит.
Примеры фармацевтически приемлемых наполнителей для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, лактозу, ангидролактозу, моногидрат лактозы, сахарозу, декстрозу, маннит, сорбит, крахмал, целлюлозу (в частности, микрокристаллическую целлюлозу), дигидро– или безводный фосфат кальция, карбонат кальция и сульфат кальция.
Примеры фармацевтически приемлемых смазывающих веществ, используемых в фармацевтических композициях в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, стеарат магния, тальк, полиэтиленгликоль, полимеры этиленоксида, лаурилсульфат натрия, лаурилсульфат магния, олеат натрия, стеарилфумарат натрия и коллоидный диоксид кремния.
Примеры подходящих фармацевтически приемлемых отдушек для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, синтетические ароматизаторы и натуральные ароматические масла, такие как экстракты масел, цветов, фруктов (например, банана, яблока, вишни, персика) и их комбинации, а также похожие ароматизаторы. Их применение зависит от многих факторов, наиболее важным из которых является органолептическая приемлемость для субъектов, которое будет принимать указанные фармацевтические композиции.
Примеры подходящих фармацевтически приемлемых красителей для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, синтетические и натуральные красители, такие как диоксид титана, бета–каротин и экстракты кожуры грейпфрута.
Примеры подходящих фармацевтически приемлемых покрытий для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, обычно используемых для облегчения глотания, изменения свойств высвобождения, улучшения внешнего вида и/или маскировки вкуса фармацевтических композиций, включают, но ими не ограничиваются, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу и акрилатметакрилатные сополимеры.
Подходящие примеры фармацевтически приемлемых подсластителей для пероральных фармацевтических композиций в соответствии с любым вариантом осуществления, описанным в настоящем документе, включают, но ими не ограничиваются, аспартам, сахарин, сахарин натрия, цикламат натрия, ксилит, маннит, сорбит, лактозу и сахарозу.
Подходящие примеры фармацевтически приемлемых буферов включают, но ими не ограничиваются, лимонную кислоту, цитрат натрия, бикарбонат натрия, двухосновный фосфат натрия, оксид магния, карбонат кальция и гидроксид магния.
Подходящие примеры фармацевтически приемлемых поверхностно–активных веществ включают, но ими не ограничиваются, лаурилсульфат натрия и полисорбаты.
Твердые композиции подобного типа также могут быть применяться в качестве наполнителей в желатиновых капсулах. Предпочтительные эксципиенты в этом отношении включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров вещество может сочетаться с различными подсластителями или ароматизаторами, пигментами или красителями, с эмульгаторами и/или суспендирующими веществами и с разбавителями, такими, как вода, этанол, пропиленгликоль и глицерин и их комбинации.
Как указано, соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут вводиться интраназально или путем ингаляции и удобно доставляться в форме сухого порошка для ингаляции или аэрозоля из баллона под давлением, насоса, пульверизатора или распылителя с использованием соответствующего газа–вытеснителя, например, дихлородифторометана, трихлорофторметана, дихлоротетрафторэтана, гидрофторалкана, такого, как 1,1,1,2–тетрафторэтан (НРА 134АТ) или 1,1,1,2,3,3,3–гептафторпропан (HFA 227EA), диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением единичная доза может определяться при помощи клапана, для доставки отмеренного количества. Баллон под давлением, насос, пульверизатор или распылитель может содержать раствор или суспензию активного соединения, например, с использованием смеси этанола и газа–вытеснителя в качестве растворителя, который также может содержать смазывающее вещество, например, сорбиттриолеат.
Капсулы и картриджи (выполненные, например, из желатина) для применения в ингаляторе или инсуфляторе, могут быть составлены таким образом, чтобы содержать порошковую смесь соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, и соответствующую порошковую основу, такую, как лактоза или крахмал.
Для местного введения путем ингаляции соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут доставляться для применения в медицине или ветеринарии через распылитель.
Фармацевтические композиции по изобретению могут содержать от 0,01 до 99% по массе на объем активного вещества. Например, для местного введения композиция обычно содержит 0,01–10%, более предпочтительно 0,01–1% активного вещества.
Соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, также можно вводить в форме систем доставки липосом, таких как малые моноламеллярные везикулы, большие моноламеллярные везикулы и полиламеллярные везикулы. Липосомы могут быть образованы из разных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Фармацевтическую композицию или стандартную лекарственную форму в соответствии с любым вариантом осуществления, описанным в настоящем документе, можно вводить в соответствии с режимом дозировки и введения, определяемым путем типового исследования с учетом представленных выше рекомендаций с целью достижения оптимальной активности при минимизации токсичности или побочных эффектов для конкретного пациента. Дозы соединений или стандартная лекарственная форма может изменяться в зависимости от множества факторов, таких как таких, как основные болезненные состояния, состояние здоровья субъекта, вес, пол и возраст и режим введения. Точное количество, которое должно вводиться пациенту, будет меняться в зависимости от состояния и тяжести расстройства и физического состояния пациента. Измеримое устранение любого симптома или параметра может определяться специалистом в данной области или сообщаться пациентом врачу. Следует понимать, что любое клинически или статистически значимое снижение или устранение любого симптома или параметра охватывается объемом изобретения. “Клинически значимое” снижение или устранение означает “ощутимое” для пациента и/или врача.
В некоторых вариантах осуществления количество вводимого соединения может составлять от около 0,01 до около 25 мг/кг/сутки. Обычно уровни дозировки от 0,01 до 25 мг/кг массы тела ежедневно вводят пациенту, например, человеку. В некоторых вариантах осуществления терапевтически эффективное количество находится между нижним пределом, составляющим около 0,01 мг/кг массы тела, около 0,1 мг/кг массы тела, около 0,2 мг/кг массы тела, около 0,3 мг/кг массы тела, около 0,4 мг/кг массы тела, около 0,5 мг/кг массы тела, около 0,60 мг/кг веса тела, около 0,70 мг/кг веса тела, около 0,80 мг/кг веса тела, около 0,90 мг/кг веса тела, около 1 мг/кг веса тела, около 2,5 мг/кг массы тела, около 5 мг/кг массы тела, около 7,5 мг/кг массы тела, около 10 мг/кг массы тела, около 12,5 мг/кг массы тела, около 15 мг/кг массы тела, около 17,5 мг/кг массы тела, около 20 мг/кг массы тела, около 22,5 мг/кг массы тела и около 25 мг/кг массы тела; и верхним пределом, составляющим 25 мг/кг массы тела, около 22,5 мг/кг массы тела, около 20 мг/кг массы тела, около 17,5 мг/кг массы тела, около 15 мг/кг массы тела, около 12,5 мг/кг веса тела, около 10 мг/кг веса тела, около 7,5 мг/кг веса тела, около 5 мг/кг веса тела, около 2,5 мг/кг веса тела, около 1 мг/кг веса тела. масса тела, около 0,9 мг/кг массы тела, около 0,8 мг/кг массы тела, около 0,7 мг/кг массы тела, около 0,6 мг/кг массы тела, около 0,5 мг/кг массы тела, около 0,4 мг/кг массы тела, около 0,3 мг/кг массы тела, около 0,2 мг/кг массы тела, около 0,1 мг/кг массы тела и около 0,01 мг/кг массы тела. В некоторых вариантах осуществления терапевтически эффективное количество составляет от около 0,1 мг/кг/сутки до около 10 мг/кг/сутки. В некоторых вариантах осуществления терапевтически эффективное количество составляет около 0,2 и около 5 мг/кг/сутки. Понятно, что фармацевтические композиции по настоящему изобретению необязательно должны содержать полное количество соединения, которое эффективно при лечении расстройства, поскольку такие эффективные количества могут быть достигнуты путем введения множества разделенных доз таких фармацевтических композиций. Соединения могут вводиться по схеме от 1 до 4 раз в сутки, например, один, два, три или четыре раза в сутки.
В некоторых вариантах осуществления изобретения соединению в соответствии с любым вариантом осуществления, описанным в настоящем документе, придают лекарственную форму капсулы или таблетки, обычно содержащих от около 10 до около 200 мг соединений. В некоторых вариантах осуществления капсула или таблетка содержат количество между нижним пределом, составляющим около 10 мг, около 15 мг, около 20 мг, около 25 мг, около 30 мг, около 35 мг, около 40 мг, около 45 мг, около 50 мг, около 55 мг, около 60 мг, около 65 мг, около 70 мг, около 75 мг, около 80 мг, около 85 мг, около 90 мг, около 95 мг, около 100 мг, около 105 мг, около 110 мг, около 115 мг; около 120 мг, около 125 мг, около 130 мг, около 135 мг, около 140 мг, около 145 мг, около 150 мг, около 155 мг, около 160 мг, около 165 мг, около 170 мг, около 175 мг, около 180 мг, около 185 мг, около 190 мг, около 195 мг и около 200 мг, и верхним пределом, составляющим около 200 мг, около 195 мг, около 190 мг, около 185 мг, около 180 мг, около 175 мг, около 170 мг, около 165 мг, около 160 мг, около 155 мг, около 150 мг, около 145 мг, около 140 около 135 мг, около 130 мг, около 125 мг, около 120 мг, около 115 мг, около 110 мг, около 105 мг, около 100 мг, около 95 мг, около 90 мг; около 85 мг, около 80 мг, около 75 мг, около 70 мг, около 65 мг, около 60 мг, около 55 мг, около 50 мг, около 45 мг, около 40 мг, около 35 мг, около 30 мг, около 25 мг, около 20 мг, около 15 мг и около 10 мг соединения в соответствии с любым вариантом осуществления настоящего изобретения.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления настоящего изобретения вводят пациенту в общей суточной дозе от 50 до 500 мг. В некоторых вариантах осуществления суточная доза находится между нижним пределом, составляющим около 50 мг, около 55 мг, около 60 мг, около 65 мг, около 70 мг, около 75 мг, около 80 мг, около 85 мг, около 90 мг, около 95 мг, около 100 мг, около 105 мг, около 110 мг, около 115 мг; около 120 мг, около 125 мг, около 130 мг, около 135 мг, около 140 мг, около 145 мг, около 150 мг, около 155 мг, около 160 мг, около 165 мг, около 170 мг, около 175 мг, около 180 мг, около 185 мг, около 190 мг, около 195 мг, около 200 мг, около 205 мг, около 210 мг, около 215 мг; около 220 мг, около 225 мг, около 230 мг, около 235 мг, около 240 мг, около 245 мг, около 250 мг, около 255 мг, около 260 мг, около 265 мг, около 270 мг, около 275 мг, около 280 мг, около 285 мг, около 290 мг, около 295 мг, 300 мг около 305 мг, около 310 мг, около 315 мг; около 320 мг, около 325 мг, около 330 мг, около 335 мг, около 340 мг, около 345 мг, около 350 мг, около 355 мг, около 360 мг, около 365 мг, около 370 мг, около 375 мг, около 380 мг, около 385 мг, около 390 мг, около 395, около 400 мг, около 405 мг, около 410 мг, около 415 мг; около 420 мг, около 425 мг, около 430 мг, около 435 мг, около 440 мг, около 445 мг, около 450 мг, около 455 мг, около 460 мг, около 465 мг, около 470 мг, около 475 мг, около 480 мг, около 485 мг, около 490 мг, около 495 мг и около 500 мг и верхним пределом, составляющим около 500 мг, около 495 мг, около 490 мг, около 485 мг, около 480 мг, около 475 мг, около 470 мг, около 465 мг, около 460 мг, около 455 мг, около 450 мг, около 445 мг, около 440 мг, около 435 мг, около 430 мг, около 425 мг, около 420 мг, около 415 мг, около 410 мг, около 405 мг, около 400 мг, около 395 мг, около 390 мг, около 385 мг около 380 мг, около 375 мг, около 370 мг, около 365 мг, около 360 мг, около 355 мг, около 350 мг, около 345 мг, около 340 мг, около 335 мг, около 330 мг, около 325 мг, около 320 мг, около 315 мг, около 310 мг, около 305 мг, около 300 мг, около 295 мг, около 290 мг, около 285 мг, около 280 мг, около 275 мг, около 270 мг, около 265 мг, около 260 мг, около 255 мг, около 250 мг, около 245 мг, около 240 мг, около 235 мг, около 230 мг, около 225 мг, около 220 мг, коло 215 мг, около 210 мг, около 205 мг, 200 мг, около 195 мг, около 190 мг, около 185 мг, около 180 мг, около 175 мг, около 170 мг, около 165 мг, около 160 мг, около 155 мг, около 150 мг, около 145 мг, около 140 мг, около 135 мг, около 130 мг, около 125 мг, около 120 мг, около 115 мг, около 110 мг, около 105 мг, около 100 мг, около 95 мг, около 90 мг; около 85 мг, около 80 мг, около 75 мг, около 70 мг, около 65 мг, около 60 мг, около 55 мг и около 50 мг соединения в соответствии с любым вариантом осуществления настоящего изобретения. В некоторых вариантах осуществления общая суточная доза составляет около от 50 до 150 мг. В некоторых вариантах осуществления общая суточная доза составляет от 50 до 250 мг. В некоторых вариантах осуществления общая суточная доза составляет около от 50 мг до 350 мг. В некоторых вариантах осуществления общая суточная доза составляет около от 50 мг до 450 мг. В некоторых вариантах осуществления общая суточная доза составляет около 50 мг.
Фармацевтическая композиция для парентерального введения содержит от около 0,01% до около 100% по массе активного соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, от 100% по массе всей фармацевтической композиции.
Как правило, трансдермальные лекарственные формы содержат от около 0,01% до около 100% по массе активного соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, от 100% общей массы лекарственной формы.
Фармацевтическую композицию или стандартную лекарственную форму можно вводить в однократной суточной дозе, или общую суточную дозу можно вводить в разделенных дозах. Кроме того, может быть желательным совместное введение или последовательное введение другого соединения для лечения расстройства. Для этой цели комбинированным активным веществам придают простую дозированную лекарственную форму.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут быть получены общими способами, описанными, например, в WO2013/029057, включенном в настоящий документ посредством ссылки, или как описано в далее в указанных способах, составляющих дополнительный аспект изобретения.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут быть синтезированы в соответствии с общими способами, приведенными в настоящем документе, и конкретными примерами синтеза.
Специалистам в данной области будет понятно, что может возникнуть потребность в применении защищенных производных промежуточных соединений, используемых при получении соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе. Присоединение и снятие защитных групп с функциональных групп может быть выполнено способами, известными специалистам в данной области (см., например, Green and Wuts Protective Groups in Organic Synthesis. John Wiley and Sons, New York, 1999). Гидрокси или аминогруппы могут быть защищены любой гидрокси или аминозащитной группой. Аминозащитные группы могут быть удалены обычными способами. Например, ацильные группы, такие как алканоильные, алкоксикарбонильные и ароильные группы, могут быть удалены путем сольволиза, например, путем гидролиза в кислотных или основных условиях. Арилметоксикарбонильные группы (например, бензилоксикарбонил) могут быть расщеплены путем гидрогенолиза в присутствии катализатора, такого как палладий на угле.
Синтез целевых соединений осуществляется удалением любых защитных групп, которые могут присутствовать в предпоследних промежуточных соединениях, с использованием стандартных способов, которые хорошо известны специалистам в данной области. Затем конечные продукты со снятой защитой очищают, при необходимости, с использованием стандартных способов, таких как хроматография на силикагеле, ВЭЖХ на силикагеле и т.п., или перекристаллизацией.
Способы применения
В некоторых вариантах осуществления изобретение относится к способам ингибирования снижения количества синапсов или нарушений мембранного переноса, ассоциированных с воздействием бета–амилоидов на нейрон, путем введения соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления изобретение также относится к способам лечения снижения когнитивных функций и/или нейродегенеративного заболевания, например, болезни Альцгеймера, или умеренного когнитивного нарушения (MCI) у пациента, включающим введение пациенту соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления нейродегенеративное заболевание выбрано из возрастных нарушений памяти (AAMI), возрастного снижения когнитивных функций (ARCD), агитационных синуклеинопатий, болезни Альцгеймера (AD), деменции при боковом амиотрофическом склерозе (ALS), аутосомно–доминантной болезни Паркинсона, когнитивного нарушения, не слабоумия (CIND), деменции, болезни диффузных телец Леви (DLBD), также известной как деменция с тельцами Леви (DLB), расстройств или состояний, характеризующихся присутствием телец Леви, синдрома Дауна, дискинезии, ВИЧ–деменции, болезни Хантингтона, случайной болезни телец Леви (LBD), наследственной болезни телец Леви (LBD), дисфагии при болезни телец Леви, умеренного когнитивного нарушения (MCI), рассеянного склероза, множественной системной атрофией (MSA), оливопонтоцеребеллярной атрофии, болезни Паркинсона (PD), доклинической болезни Альцгеймера (PCAD), психоза, идиопатической ортостатической гипотензии, синдрома Шая–Дрейджера, стрионигральной дегенерации, синуклеинопатии, комбинированной болезни Альцгеймера и Паркинсона и/или MS, васкулярной деменции, заболеваний, расстройств или состояний, ассоциированных с патологической экспрессией, стабильностью, активностями и/или клеточным преобразованием α–синуклеина, заболевания, расстройства или состояния, характеризующиеся присутствием телец Леви, и их сочетаний.
В некоторых вариантах осуществления способ ингибирования или лечения снижения когнитивных функций и/или нейродегенеративного заболевания, например, болезни Альцгеймера, включает ингибирование или лечение одного или более симптомов снижения когнитивных функций, выбранных из группы, состоящей из потери памяти, спутанности сознания, нарушения суждения, изменения личности, дезориентация и потеря языковых навыков. В некоторых вариантах осуществления способ включает ингибирование или лечение заболеваний, расстройств или состояний, опосредованных или ассоциированных с бета–амилоидными олигомерами.
В некоторых вариантах осуществления способ ингибирования или лечения снижения когнитивных функций и/или нейродегенеративного заболевания, например болезни Альцгеймера, включает один или более элементов: (i) восстановление долговременной потенциации (LTP), длительной депрессии (LTD) или синаптической пластичности, которые обнаруживают путем электрофизиологических измерений любого из других неблагоприятных изменений в когнитивной функции, упомянутых в определении указанного термина; и/или (ii) ингибирование или лечение нейродегенерации; и/или (iii) ингибирование или лечение общего амилоидоза; и/или (iv) ингибирование или лечение одного или более состояний, к которым относятся выработка амилоидов, амилоидная сборка, агрегация амилоидов и связывание амилоидных олигомеров и отложение амилоидов; и/или (v) ингибирование, лечение и/или ослабление действия, в частности, нелетального эффекта одного или более бета–амилоидных олигомеров в отношении нейрона.
В некоторых вариантах осуществления способ ингибирования, лечения и/или уменьшения снижения когнитивных функций и/или нейродегенеративного заболевания, например, болезни Альцгеймера, включает ингибирование, лечение и/или уменьшение одного или более состояний, к которым относится продукция амилоидов, амилоидов сборка, активность/эффект одного или более бета–амилоидных олигомеров в отношении нейрона, агрегация амилоидов, связывание амилоидов и отложение амилоидов.
В некоторых вариантах осуществления способ ингибирования, лечения и/или уменьшения снижения когнитивных функций и/или нейродегенеративного заболевания, например, болезни Альцгеймера, включает ингибирование, лечение и/или уменьшение одного или более состояний, к которым относится активность/эффект одного или более бета–амилоидных олигомеров в отношении нейрона.
В некоторых вариантах осуществления активность/эффект одного или более бета–амилоидных олигомеров в отношении нейрона, агрегация амилоидов и связывание амилоидов является эффектом бета–амилоидных олигомеров в отношении мембранного переноса или количества синапсов. В некоторых вариантах осуществления соединение согласно любому варианту осуществления, описанному в настоящем документе, ингибирует действие бета–амилоидного олигомера на мембранный перенос или количество синапсов или связывание бета–амилоидного олигомера.
В некоторых вариантах осуществления изобретение относится к способу лечения протеопатического заболевания, ассоциированного с токсичностью бета–амилоидного олигомера, в частности нелетальных эффектов бета–амилоидного олигомера. В некоторых вариантах осуществления способ включает контакт субъекта, страдаюзего таким протеопатическим заболеванием, с соединением в соответствии с любым вариантом осуществления, описанным в настоящем документе, или фармацевтической композицией, содержащей его, которая связывается с сигма–2-рецептором.
В некоторых вариантах осуществления протеопатическое заболевание представляет собой протеопатию ЦНС, которая характеризуется повышением бета–амилоидного белка, например, MCI, синдром Дауна, дегенерация желтого пятна или болезнь Альцгеймера и т.п.
В некоторых вариантах осуществления изобретение относится к способу лечения одного или более умеренных когнитивных нарушений (MCI) или деменции путем введения соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе. В некоторых вариантах осуществления изобретение относится к способам лечения MCI и деменции.
В некоторых вариантах осуществления болезнь относится к способу лечения болезни Альцгеймера путем введения соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления изобретение относится к способу лечения индивидуума с помощью соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, для восстановления, частично или полностью, клеток субъекта до нормального фенотипа с точки зрения функций, на которые неблагоприятно влияют бета–амилоиды, такие как бета–амилоидные олигомеры. Примерами могут быть такие отклонения, как снижение количества синапсов и нарушения мембранного переноса, которые могут определяться различными способами, включая описанные в настоящем документе анализы. Нормальным фенотипом может быть, например, нормальный мембранный перенос. В некоторых вариантах осуществления нормальным фенотипом является нормальная когнитивная способность. “Нормальный” фенотип может быть определен сравнением результатов субъекта с образцами нормальных субъектов. Образец может представлять всего 1 субъекта или 1 образец может представлять более 10 образцов или субъектов, и нормой является среднее значение, которое рассчитывается на основе показателей нескольких субъектов.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, обычно ингибирует эффект бета–амилоида в отношении нейронов. В некоторых вариантах осуществления описанные выше соединения имеют показатель IC50, составляющий меньше чем около 100 мкМ, около 50 мкМ, около 20 мкМ, около 15 мкМ, около 10 мкМ, около 5 мкМ, около 1 мкМ, около 500 нМ, около 100 нМ, около 50 нМ или около 10 нМ, в отношении ингибирования бета–амилоидного эффекта в отношении нейронов (например, нейронов головного мозга), сборки амилоидов или их разрушения и связывания амилоидов (включая амилоидный олигомер) и отложение амилоидов. В некоторых вариантах осуществления соединение согласно любому варианту осуществления, описанному в настоящем документе, может иметь показатель IC50, составляющий меньше чем около 100 мкМ, около 50 мкМ, около 20 мкМ, около 15 мкМ. около 10 мкМ, около 5 мкМ, около 1 мкМ, около 500 нМ, около 100 нМ, около 50 нМ или около 10 нМ, в отношении ингибирования активности/эффекта бета–амилоидов, таких как олигомеры, в отношении нейронов (например, нейронов центральной нервной системы).
Соединение в соответствии с любым вариантом осуществления описанным в настоящем документе, может ингибировать бета–амилоидный эффект путем специфического связывания с сигма–2–рецептором. Хотя соединение способно связываться как с сигма–1, так и сигма–2–рецептором, соединение может определяться как “специфичное” к сигма–2–рецептору, если оно связывается с аффинностью связывания, которая является по меньшей мере на 10% большей, чем к сигма–1–рецептору. Соединения таких вариантов осуществления могут демонстрировать специфичность, по меньшей мере, на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500% или на 1000% больше в отношении сигма–2–рецептора, чем сигма–1–рецептора.
В некоторых вариантах осуществления выраженное в процентах ингибирование соединением в соответствии с любым вариантом осуществления, описанным в настоящем документе, одного или более эффектов бета–амилоидов, таких как олигомеры, в отношении нейронов (таких как нейроны головного мозга), таких как связывание амилоидов (включая амилоидный олигомер) с синапсами, и отклонения в мембранном переносе, опосредуемые бета–амилоидным олигомером, могут составлять от около 1% до около 20%, от около 20% до около 50%, от около 1% до около 50% или от около 1% до около 80%, что определено при концентрации от 10 нМ до 10 мкМ. Например, ингибирование можно оценить путем определения количества синапсов нейрона до и после воздействия бета–амилоидов или определения количества синапсов в присутствии как соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, так и бета–амилоидов, причем воздействие соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, является одновременным, или предшествует, или следует за воздействием бета–амилоидов. В качестве другого примера, ингибирование может быть оценено путем определения мембранного переноса и сравнения одного или более параметров, которые измеряют скорость и степень экзоцитоза, скорость и степень эндоцитоза или другие показатели клеточного метаболизма в присутствии и отсутствии бета–амилоидов и в присутствии и отсутствии соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе.
В некоторых вариантах осуществления изобретение относится к способу определения ассоциированного с бета–амилоидами когнитивных функций у животного с использованием меченого соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе. В некоторых вариантах осуществления способ включает контакт животного с меченым соединением в соответствии с любым вариантом осуществления, описанным в настоящем документе, и определение активности или экспрессии сигма–2. В некоторых вариантах осуществления способ включает сравнение активности или экспрессии сигма–2 в организме животного с показателем животного с вызванным бета–амилоидами снижением когнитивных функций. Если активность или экспрессия не отличается от показателя животного с вызванным бета–амилоидами снижением когнитивных функций, животное считается имеющим аналогичный уровень снижения когнитивных функций. Животных можно классифицировать по сходству в известной активности или экспрессии различных стадий индуцированного бета–амилоидами снижения когнитивных функций. Любое из соединений в соответствии с любым вариантом осуществления, описанным в настоящем документе, может быть меченым, таким образом, чтобы меченое соединение могло быть использовано in vivo.
В некоторых вариантах осуществления анализ используется для определения того, может ли соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, связываться с сигма–2–рецептором. В некоторых вариантах осуществления способ дополнительно включает определение того, действует ли соединение, которое связывается с сигма–2–рецептором, в качестве функционального антагониста сигма–2–рецептора путем ингибирования нейротоксичности, индуцированной растворимым Aβ–олигомером, относительно ингибирования потери синапсов, индуцируемых растворимым Aβ–олигомером, и ингибирования дефицита, индуцированного растворимым олигомером, в анализе мембранного переноса.
Для практического осуществления способов скрининга и анализов согласно изобретению может применяться любая форма β–амилоида, включая β–амилоидные мономеры, олигомеры, фибрилы, а также β–амилоиды, ассоциированные с белками (“белковыми комплексами”) и в целом сборки β–амилоидов. Например, в способах скрининга могут применяться различные формы растворимых β–амилоидных олигомеров, как, описано, например, в патентной заявке США под регистрационным номером 13/021872; патентной публикации США 2010/0240868; международной патентной заявке WO/2004/067561; международной патентной заявке WO/2010/011947; патентной публикации США 20070098721; патентной публикации США 20100209346; международной патентной заявке WO/2007/005359; патентной публикации США 20080044356; патентной публикации США 20070218491; WO/2007/126473; патентной публикации США 20050074763; международной патентной заявке WO/2007/126473, международной патентной заявке WO/2009/048631 и патентной публикации США 20080044406, патенте США №7902328 и патенте США №6218506, каждая из которых включена в настоящее описание посредством ссылки.
Формы β–амилоидов, включая мономеры или олигомеры β–амилоидов, могут быть получены из любого источника. Например, в некоторых вариантах осуществления коммерчески доступные β–амилоидные мономеры и/или β–амилоидные олигомеры могут применяться в водном растворе, а в других вариантах осуществления β–амилоидные мономеры и/или β–амилоидные олигомеры, которые применяются в водном белковом растворе, могут быть выделены и очищены специалистами в данной области с применением любого количества известных способов. Обычно, β–амилоидные мономеры и/или β–амилоидные олигомеры, применяемые при приготовлении водного раствора белков и β–амилоида согласно различным вариантам осуществления, могут быть растворимыми в водном растворе. Таким образом, и белки водного раствора и β–амилоид могут быть растворимыми.
Добавляемый β–амилоид может представлять любую изоформу. Например, в некоторых вариантах осуществления β–амилоидные мономеры могут быть β–амилоидами 1–42, а в других вариантах осуществления β–амилоидные мономеры могут быть β–амилоидами 1–40. В других вариантах осуществления β–амилоид может быть β–амилоидом 1–39 или β–амилоидом 1–41. Таким образом, β–амилоиды различных вариантов осуществления могут охватывать любую С–концевую изоформу β–амилоида. Другие варианты осуществления включают β–амилоид, в котором N–конец был расщеплен, и в некоторых вариантах осуществления N–конец любого из вышеописанных С–концевых изомеров β–амилоидов может быть аминокислотой 2, 3, 4, 5 или 6. Например, β–амилоид 1–42 может включать β–амилоид 2–42, β–амилоид 3–42, β–амилоид 4–42 или β–амилоид 5–42 и их смеси, и, подобным образом, β–амилоид 1–40 может включать β–амилоид 2–40, β–амилоид 3–40, β–амилоид 4–40 или β–амилоид 5–40.
Формы β–амилоидов, применяемые в различных вариантах осуществления, могут относиться к дикому типу, т.е. имеющему аминокислотную последовательность, идентичную аминокислотной последовательности β–амилоида, синтезируемого in vivo большинством популяции, или в некоторых вариантах осуществления β–амилоид может быть мутантным β–амилоидом. Варианты осуществления не ограничиваются какой–либо конкретной разновидностью мутантного β–амилоида. Например, в некоторых вариантах осуществления β–амилоид, включенный в водный раствор, может включать известную мутацию, такую, как, например, β–амилоид, имеющий “Dutch” (E22Q) мутацию или “Arctic” (E22G) мутацию. Такие мутантные мономеры могут включать встречающиеся в природе мутации, такие, как, например, формы β–амилоида, выделенные из популяций субъектов, предрасположенных, например, к болезни Альцгеймера, наследственные формы β–амилоида. В других вариантах осуществления мутантные β–амилоидные мономеры могут быть получены синтетически путем применения молекулярных технологий для получения мутанта β–амилоида со специфической мутацией. В других вариантах осуществления мутантные β–амилоидные мономеры могут включать ранее не распознанные мутации, например, мутанты, находящиеся в случайно образованных мутантах β–амилоида. Термин “β–амилоид” в контексте настоящего описания охватывает как формы β–амилоида дикого типа, так и любые мутантные формы β–амилоида.
В некоторых вариантах осуществления β–амилоид в водном белковом растворе может представлять одну изоформу. В других вариантах осуществления различные С–концевые изоформы β–амилоида и/или различные N–концевые изоформы β–амилоида объединяют с образованием β–амилоидных смесей, которые могут быть предусмотрены в водном белковом растворе. В других вариантах осуществления β–амилоид может быть производным белка–предшественника амилоида (АРР), который добавляют к белку, содержащему водный раствор, и расщепляют in situ, и такие варианты осуществления, различные изоформы β–амилоида могут содержаться в растворе. Расщепление N–конца и/или удаление С–концевых аминокислот может осуществляться в водном растворе после добавления β–амилоида. Таким образом, водные растворы, полученные, способами, описанными в настоящем документе, могут включать различные изоформы β–амилоида даже если изначально к раствору добавляют одну изоформу.
Добавляемые к водному раствору β–амилоидные мономеры могут быть выделены из природного источника, такого, как живая ткань, а в других вариантах осуществления β–амилоид может быть получен из синтетического источника, такого, как трансгенные мыши или культивированные клетки. В некоторых вариантах осуществления формы β–амилоидов, включая мономеры, олигомеры или их комбинации, выделяют из организма нормальных субъектов и/или пациентов, у которых диагностированы снижение когнитивных функций или ассоциированные с ним заболевания, к которым, но им не ограничиваясь, относится болезнь Альцгеймера. В некоторых вариантах осуществления β–амилоидными мономерами, олигомерами или их комбинациями являются бета–амилоидные сборки, выделенные из организма нормальных субъектов или пациентов. В некоторых вариантах осуществления бета–амилоидные сборки являются высокомолекулярными, например, имеющими массу больше чем 100 кДа. В некоторых вариантах осуществления бета–амилоидные сборки имеют среднюю молекулярную массу, например, от 10 до 100 кДа. В некоторых вариантах осуществления бета–амилоидные сборки имеют молекулярную массу меньше 10 кДа.
В соответствии с некоторыми вариантами осуществления β–амилоидные олигомеры могут состоять из любого количества β–амилоидных мономеров, соответствующих общепринятому определению “олигомер”. Например, в некоторых вариантах осуществления β–амилоидные олигомеры могут включать от около 2 до около 300, от около 2 до около 250, от около 2 до около 200 β–амилоидных мономеров, а в других вариантах осуществления β–амилоидные олигомеры могут включать от около 2 до около 150, от около 2 до около 100, от около 2 до около 50 или от около 2 до около 25 β–амилоидных мономеров. В некоторых вариантах осуществления β–амилоидные олигомеры могут включать 2 или больше мономеров. В соответствии с различными вариантами осуществления β–амилоидные олигомеры могут отличаться от β–амилоидных фибрилл и β–амилоидных протофибрилл, исходя из подтверждения мономеров. В частности, β–амилоидные мономеры β–амилоидных олигомеров обычно являются глобулярными и состоят из β–складчатых листов, тогда как вторичная структура β–амилоидных мономеров фибрилл и протофибрилл представляет собой параллельные β–листы.
ПРИМЕРЫ
В примерах 1 и 2 описаны препараты бета–амилоидных олигомеров, которые можно использовать для описанных ниже экспериментов. Конкретные препараты, используемые в анализах мембранного переноса и связывания олигомеров/уменьшения количества синапсов, а также препараты, которые используются в описанных ниже in vivo анализах, описаны в примере, к которому они относятся.
Пример 1: Получение β–амилоидных олигомеров
Условия, в которых β–амилоид может олигомеризироваться в нервной ткани, среду водорастворимых белков, с которыми они могут ассоциироваться, создавали для распознавания более связанного с заболеванием структурного состояния β–амилоидных олигомеров и фибрилл. Водорастворимые белки получали из головного мозга крыс путем ультрацентрифугирования. В частности, 5 объемов TBS–буфера (20 мМ Tris–HCL, pH 7,5, 34 мМ NaCl и полную смесь ингибиторов протеаз (Santa Cruz) на грамм ткани мозга добавляли к тканям мозга крыс на льду. Гомогенизацию Даунса осуществляли с использованием плотно подогнанного пестика. Затем гомогенизированные ткани головного мозга центрифугировали при 150000×g в течение 1 часа при 4°С (40000 об/мин Ту65). Далее инфранатант (под всплывающим миелином и полсантиметра над осадком) удаляли и аликвоты замораживали при –75°С. Затем осадок ресуспендировали в TBS до пepвoнaчального объема и замораживали в аликвотном количестве при –75°С. Синтетический мономерный человеческий β–амилоид 1–42 добавляли к этой смеси для обеспечения конечной концентрации 1,5 мкМ β–амилоида и раствор инкубировали в течение 24 часов при 4°С. Для удаления фибриллярных образований осуществляли центрифугирование смеси при 5800g в течение 10 минут, а затем осуществляли иммунопреципитацию с применением спин–колонок с конъюгированной агарозой 6Е10 (Pierce Chemical Company) в течение 24 часов при 4°С. Потом элюированные β–амилоидные олигомеры подвергали анализу с применением время–пролетной масс–спектрометрии с лазерной ионизацией и десорбцией из жидкой матрицы для определения содержимого образца.
В содержащем белок растворе β–амилоид самоассоциировался с образованием сборок субъединиц 22599 Да 5–субъединичные пентамеры и 31950 Да 7–субъединичные 7–меры. Другой пик при 49291 Да может представлять 12–субъединичные 12–меры, хотя это может быть неточная молекулярная масса для β–амилоидных 12–меров. Следует заметить, что пики не наблюдаются при 4518 Да или 9036 Да, которые представляют β–амилоидные мономеры и димеры. Однако пики при 9882 Да и 14731 Да могут представлять β–амилоидные димеры, ассоциированные с 786 Да (или 2×393 Да) липидами или белками, и β–амилоидные тримеры, ассоциированные с 3×393 Да липидами или белками, соответственно. Кроме того, присутствие пиков при 19686 Да указывает на состояние сборки, которое может включать тримерный комплекс и крысиный β–амилоидный фрагмент 4954 Да. Соответственно, эти данные могут отражать ассоциацию малых липидов или белков с димерами и тримерами β–амилоида, которые могут контролировать сборку конформационных состояний, уникальных для физиологических систем.
Пример 2: Получение бета–амилоидных олигомеров
Раствор 1,5 мкМ мономерного человеческого β–амилоида 1–42 в смеси растворимых белков головного мозга крыс инкубировали в течение 24 часов при 4°С, как описано в примере 1. Затем этот раствор обрабатывали трифтороэтанолом (TFE) перед получением спектров. В TFE собранные белковые структуры и нековалентно связанные белковые комплексы диссоциируются в денатурированные белки, и пики, ассоциированные с собранными олигомерами, должны исчезать. Большинство белковых пиков, наблюдаемых в примере 1, исчезало, включая идентифицированные ранее пики 9822 Да, 14731 Да, 31950 Да и 49291 Да. Однако наблюдали лишний пик при 4518 Да, который представляет собой пик β–амилоидного мономера. Пик при 4954,7 является выраженным и может представлять более длинный бета–амилоид–фрагмент, аналогичный β–амилоиду 1–46. При 7086 Да наблюдали дополнительный пик, который отсутствовал в препарате, описанном в примере 1, и может представлять β–амилоидные мономеры, ассоциированные с 2550 Да ковалентно связанным белком.
Пример 3: Выделение бета–амилоидных олигомеров из ткани головного мозга человека с БА.
Растворимые в TBS экстракты: образцы взятых в ходе патологоанатомического исследования тканей головного мозга людей, характеризуемые путем гистопатологического анализа как соответствующие болезни Альцгеймера (БА) стадии V/VI по Брааку, получали из больничного банка тканей головного мозга. Подобранные по возрасту и полу образцы ткани с БА и нормальной ткани разбавляли до 0,15 мг ткани/мл в 20 мМ Tris–HCL, 137 мМ NaCl, pH 7,6 содержащем 1 мМ EDTA и 1 мг/мл полной смеси ингибиторов протеаз (Sigma Р8340) и гомогенизировали. Ультрацентрифугирование гомогенатов ткани осуществляли при 105000g в течение 1 часа в ультрацентрифуге Beckman Optima XL–80K. Полученные в результате растворимые в TBS фракции подвергали иммуноистощению с применением агарозных колонок на белке–А и белке–G (Pierce Chemical), а затем фракционировали по размеру при помощи фильтров Amicon Ultra 3, 10 и 100 кДа NMWCO (Millipore Corporation).
Иммунопреципитация: фракционированные по размеру и иммуноистощенные растворимые в TBS экстракты концентрировали до около 200 мкл в соответствующих фильтрах NMWCO Amicon Ultra. Концентрированные растворимые в TBS экстракты разбавляли до 400 мкл TBS–буфером для образца (Pierce Chemical) и центрифугировали в течение 10 минут при 5800g для удаления фибрилл. Затем образовавшийся в результате супернатант подвергали иммунопреципитации с 6Е10–конъюгированными агарозными шариками в течение суток при 4°С с последующим антигенным элюированием с применением высокоосмотических элюирующих буферов Gentle (Pierce Chemical) для выделения содержащих бета–амилоид видов белка.
MALDI–масс–спектрометрия: иммуноизолированный бета–амилоид подвергали масс–спектроскопическому анализу с применением устройства Applied Biosystems (ABI) Voyager DE–Pro MALDI–Tof. Образцы анализировали с использованием различных типов матрицы, таких как α–циано–4–гидроксикоричная кислота (СНСА), синаповая кислота (SA) или 6–аза–2–тиотимин (АТТ), в зависимости от заданного диапазона молекулярной массы для анализа. Устройство работало в линейном режиме определения положительных ионов с изменяемой задержкой экстракции. Ненакопленные спектры представляли 100 снимков “горячей точки” на каждое обнаружение, тогда как накопленные спектры были представлены 12 отдельными участками каждой точки с 200 лазерными снимками на каждое обнаружение.
Анализ данных: получение и анализ данных выполняли с применением пакета программ Voyager’s Data Explorer. Стандартная обработка масс–спектров включала функции сглаживания спектра и вычитание исходного показателя в дополнение к изменениям соотношения сигнала и шума.
ИФА–анализ для количественного определения антитела: подвергнутые иммунопреципитации растворимые в TBS фракции анализировали на “общий” показатель бета–амилоид и концентрацию бета–амилоидного олигомера с применением модифицированного сэндвич–варианта ИФА. Вкратце, покрытые 6Е10 и 4G8 96–луночные планшеты Nunc MaxiSorp инкубировали с содержащими бета–амилоид образцами, а затем зондировали обнаруживающим биотинилированный 4G8 антителом. Инкубация со стрептавидином–HRP (Rockland) с последующей проявкой тетраметилбензидинового (ТМВ) субстрата обеспечивала возможность колориметрического обнаружения (OD 450) бета–амилоида на устройстве для считывания планшетов BioTEk Synergy HT. Мономерный бета–амилоид 1–42 использовали для построения стандартной кривой, и вместе с программой GEN 5 он позволял определять уровень Бета–амилоид в подвергнутых иммунопреципитации образцах.
Пример 4: Анализы рецепторного связывания
Некоторые соединения испытывали на взаимодействие с несколькими рецепторами путем блокирования связывания или действия их агонистов или антагонистов. Некоторые соединения испытывают, чтобы проверить, взаимодействуют ли они непосредственно с известными клеточными рецепторными или сигнальными белками. Соединения могут быть исследованы на предмет их способности к смещению связывания известных агонистов или антагонистов данного рецептора человека, который был сверхэкспрессирован в клеточных линиях или выделен из ткани. Соединения также могут быть исследованы на предмет их способности блокировать передачу сигналов вниз по течению, индуцированную агонистами или антагонистами данного человеческого рецептора. Соединения могут быть исследованы на действие в отношении 100 известных рецепторов, и предпочтительно, чтобы специфическая активность возникала только при небольшой субпопуляции ЦНС–релевантных рецепторов.
Используя аналогичный протокол, некоторые соединения, для которых данные мембранного переноса приведены в таблице 1, исследовали на распознавание сигма–2–рецептора. Некоторые соединения предпочтительно связываются с сигма–2–рецептором.
Анализ 1 конкурентного радиолигандного связывания
Для связывания сигма–1 применяли различные концентрации исследуемых соединений от 100 мкМ до 1 нМ для смещения 8 нМ [3Н](+)пентазоцина с эндогенных рецепторов на мембранах клеток Юркат (Ganapathy ME et al., 1991, J Pharmacol. Exp. Ther. 289:251–260). Для определения неспецифического связывания использовали 10 мкМ галоперидола. Для сигма–2–рецепторов применяли различные концентрации исследуемых соединений от 100 мкМ до 1 нМ для смещения 5 нМ [3Н] 1,3–ди–(2–толил)гуанидина с эндогенных рецепторов на мембранах из коры головного мозга крыс в присутствии 300 нМ (+)пентазоцина для маскировки сигма–1–рецепторов. (Bowen WD, et al. 1993, Mol. Neuropharmcol 3:117–126). Для определения неспецифического связывания использовали 10 мкМ галоперидола. Реакции прекращали путем быстрой фильтрации через фильтры Whatman GF/C с применением сборщика клеток Brandel 12R с последующими двумя промываниями ледяным буфером. Радиоактивность на высушенных дисках фильтрата измеряли с применением жидкостно–сцинтилляционного анализатора (Tri–Carb 2900TR; PerkinElmer Life and Analytical Sciences). Строили кривые смещения, и значения Ki испытуемых лигандов для подтипов рецептора определяли при помощи GraphPad Prism (GraphPad Software Inc., San Diego, СА). Процент специфического связывания определяли путем деления разницы между общим связыванием (распадов в минуту) и неспецифическим связыванием (распадов в минуту) на общее связывание (распадов в минуту).
Данные аффинности к сигма–1 и сигма–2–рецепторам обычно получают из опубликованных исследований с использованием гомогенатов мозговой ткани с [3H](+)пентазоцином для измерения смещения с сигма–1–рецепторов и [3Н] 1,3–ди–(2–толил)гуанидином в присутствии 300 нМ (+)пентазоцина для измерения смещения с сигма–2–рецепторов.
Анализ 2 конкурентного радиолигандного связывания.
Аффинность исследуемых соединений в сигма–1 и сигма–2–рецепторах также определяли путем смещения различных известных лигандов сигма–2 или сигма–1. Анализы фильтрации осуществляли в соответствии с ранее опубликованной процедурой (Xu, et al., 2005). Исследуемые соединения растворяли в N, N–диметилформамиде (ДМФА), диметилсульфоксиде (ДМСО) или этаноле, а затем разбавляли в 50 мМ буфера Tris–HCl, pH 7,4, содержащем 150 мМ NaCl и 100 мМ EDTA. Гомогенаты мембран получали из головного мозга морской свинки для анализа связывания сигма–1 и печени крыс для анализа связывания сигма–2. Гомогенаты мембран разбавляли 50 мМ буфера Tris–HCl, pH 8,0, и инкубировали при 25°С в общем объеме 150 мкл в 96–луночных планшетах с радиолигандом и исследуемыми соединениями с концентрацией от 0,1 нМ до 10 мкМ. После завершения инкубации, реакции завершали путем добавления 150 мкл ледяного промывочного буфера (10 мМ Tris HCl, 150 мМ NaCl, pH 7,4) с применением 96–канальной пипетки для переноса (Fisher Scientific, Pittsburgh, PA) и образцы собирали и быстро фильтровали с помощью 96–луночного планшета со стекловолоконным фильтром (Millipore, Billerica, MA), предварительно пропитанным 100 мкл 50 мМ буфера Tris–HCl. Каждый фильтр промывали четыре раза 200 мкл ледяного промывочного буфера (10 мМ Tris–HCl, 150 мМ NaCl, pH 7,4). Применяли жидкостный сцинтилляционный счетчик Wallac 1450 MicroBeta (Perkin Elmer, Boston, MA) для количественного определения связанной радиоактивности.
Анализы рецепторного связывания сигма–1 осуществляли с использованием гомогенатов мембран головного мозга морской свинки (~300 мкг белка) и ~5 нМ [3Н](+)–пентазоцина (34,9 Ки/ммоль, Perkin Elmer, Boston, MA), время инкубации составляло 90 мин при комнатной температуре. Неспецифическое связывание определяли по образцам, которые содержали 10 мкМ холодного галоперидола.
Анализы рецепторного связывания сигма–2 выполняли с использованием только гомогенатов мембран печени крыс (~300 мкг белка) и ~2 нМ высокоселективного радиолиганда сигма–2 [3H]RHM–1 (без других блокаторов) (America Radiolabeled Chemicals Inc. St. Louis, МО), ~10 нМ [3H]DTG (58,1 Ки/ммоль, Perkin Elmer, Boston, MA) или ~10 нМ [3Н]галоперидола (America Radiolabeled Chemicals Inc., St. Louis, МО) в присутствии 1 мкМ (+)–пентазоцина для блокирования сайтов сигма–1, время инкубации составляло 6 минут для [3H]RHM–l, 120 мин для [3H]DTG и [3H]галоперидола при комнатной температуре. Неспецифическое связывание определяли по образцам, которые содержат 10 мкМ холодного галоперидола.
Данные экспериментов конкурентного ингибирования моделировали с применением анализ нелинейной регрессии для определения концентрации ингибитора, которая ингибирует 50% специфического связывания радиолиганда (значение IC50). Аффинность связывания, значения Ki определяли с применением способа Cheng and Prusoff. Значение Kd, используемое для [3H](+)–пентазоцина в головном мозге морской свинки, составляло 7,89 нМ для [3H]RHM–1 и [3H]DTG в печени крыс составляло 0,66 нМ и 30,73 нМ, соответственно. Стандартное соединение галоперидол использовали для гарантии качества. Данные об аффинности в сигма–2–рецепторе для типичных соединений показаны в таблице 1.
В некоторых вариантах осуществления соединения в соответствии с любым вариантом осуществления настоящего изобретения или их фармацевтически приемлемые соли демонстрируют аффинность Ki к сигма–2–рецептору не больше 1000 нМ, не больше 750 нМ, не больше 500 нМ, не больше 250 нМ, не больше 100 нМ, не больше 50 нМ, не больше 25 нМ или не больше 10 нМ при тестировании в соответствии с протоколом анализа связывания сигма–2–рецептора, предусмотренным в настоящем документе.
Пример 5: Ингибирование эффекта бета–амилоидного олигомера в отношении нейронов в исследовании мембранного переноса
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, исследовали на их способность ингибировать бета–амилоидный эффект в отношении клеток. Соединения, обычно, были способны ингибировать бета–амилоидный эффект, измеренный с помощью мембранного анализа переноса/экзоцитоза (МТТ–анализа). Результаты приведены в таблице 1. Обоснование этого анализа было следующим:
Поскольку на самых ранних стадиях болезни Альцгеймера преобладают синаптические дефекты и нарушения памяти, а не обширная гибель клеток, анализы с измерением этих изменений являются особенно эффективными для обнаружения низкомолекулярных ингибиторов активности олигомеров. Анализ МТТ часто применяют в качестве меры токсичности в культурах. Желтые тетразолиевые соли эндоцитозируются клетками и восстанавливаются до нерастворимого пурпурного формазана в эндосомальном пути. Уровень пурпурного формазана показывает количество активно метаболизирующихся клеток в культуре, а уменьшение количества формазана является мерой гибели клеток или метаболической токсичности в культуре. При наблюдении с помощью микроскопа, пурпурный формазан сначала виден во внутриклеточных везикулах, заполняющих клетку. Со временем везикулы экзоцитозируются, и формазан осаждается в виде игольчатых кристаллов на внешней поверхности цитоплазматической мембраны при воздействии на нерастворимый формазан окружающей водной среды. Liu and Schubert ('97) обнаружили, что клетки отвечают на сублетальные уровни бета–амилоидных олигомеров выборочным ускорением экзоцитоза восстановленного формазана, при этом скорость эндоцитоза остается неизменной. Авторы изобретения воспроизвели эти наблюдения в зрелых первичных нейронах в культуре и подсчитали эти морфологические сдвиги при помощи автоматизированной микроскопии и обработки изображений. В этих обстоятельствах не наблюдается общих изменений в общем количестве восстановленного формазана, а просто сдвиг в его морфологии, отражающий изменения скорости его образования и/или вытеснения из клетки. Авторы изобретения подтвердили более ранние выводы о том, что этот анализ чувствителен к низкому уровню олигомеров, которые не вызывают гибель клеток (Liu and Schubert '04, Hong et al., '07). Действительно, низкое количество олигомеров, которые приводят к ингибированию LTP, не ведет к гибели клеток (Tong et al., '04) и не должно вызывать изменений в общем количестве формазана в культуре (или в срезах головного мозга).
Свидетельства, приведенные другими исследователями, показывают, что опосредованное бета–амилоидным олигомером снижение экспрессии рецептора поверхности нейронов, опосредованное мембранным переносом, является основанием для олигомерное ингибирование электрофизиологических показателей синаптической пластичности (LTP) и, таким образом, обучения и памяти (Kamenetz et al., '03, Hseih et al., '06). Изменения показателей скорости мембранного переноса, индуцированное олигомерами из–за морфологических сдвигов формазана, использовали в линиях клеток для обнаружения блокирующих бета–амилоидный олигомер лекарственных средств (Maezawa et al., '06, Liu and Schubert '97, '04, '06, Rana et al., '09, Hong et al., '08), которые снижают уровень бета–амилоидов в головном мозге грызунов in vivo (Hong et al., '09). Аналогичные процедуры анализов экзоцитоза/МТТ–анализов описаны в литературе. См., например, Liu Y, et. al., Detecting bioactive amyloid beta peptide species in Alzheimer's disease. J Neurochem. 2004 Nov;91(3):648–56; Liu Y, and Schubert D. “Cytotoxic amyloid peptides inhibit cellular 3–(4,5–dimethylthiazol–2–yl)–2,5–diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis.” J Neurochem. 1997 Dec;69(6):2285–93; и Liu Y, and Schubert D. “Treating Alzheimer's disease by inactivating bioactive amyloid beta peptide” Curr. Alzheimer Res. 2006 Apr; 3(2):129–35. Таким образом, этот подход является эффективным.
Представленный анализ экзоцитоза адаптировали для применения со зрелыми первичными культурами нейронов, выращиваемых в течение 3 недель in vitro. См. заявку WO 2011/106785, которая включена в настоящее описание посредством ссылки полностью. Бета–амилоидные олигомеры вызывают зависимое от дозы снижение количества внутриклеточных везикул (точек), заполненных восстановленным пурпурным формазаном, согласно измерению путем обработки изображений с применением системы автоматизированной микроскопии Cellomics VTI. На микрофотоснимках культивированных нейронов, подвергнутых воздействию только индифферентной основы, видны везикулы, заполненные формазаном; причем на микрофотоснимке нейрона, подвергнутого воздействию носителя плюс бета–амилоидного олигомера, видно значительно меньшее количество везикул, заполненных формазаном, при этом можно увидеть экзоцитозированный формазан, который при наличии во внеклеточной среде, осаждается в форме кристаллов. Увеличение количества бета–амилоидных олигомеров в итоге приводит к выраженной токсичности. Таким образом, концентрация нейроактивных бета–амилоидных олигомеров, используемых в анализе, является намного меньшей, чем та, которая вызывает гибель клеток. Авторы изобретения подтвердили эффективность анализа, продемонстрировав, что действие бета–амилоидного олигомера блокируется после добавления анти–бета–амилоидного антитела, однако само по себе антитело не имеет воздействия (данные не показаны). При такой конфигурации анализ позволяет обнаруживать соединения, которые ингибируют нелетальные эффекты бета–амилоидного олигомера, независимо от того, действуют ли эти соединения через разрушение олигомеров, ингибирование связывания олигомеров с нейронами или противодействие механизмам действия трансдукции сигнала, вызванной связыванием олигомеров.
Для достижения результатов применяли способы, представленные ниже в описании анализа мембранного переноса/экзоцитоза (МТТ).
Первичные гиппокампальные нейроны эмбрионов крыс линии Е18 Sprague–Dawley высевали в оптимизированной концентрации в 384–луночные планшеты в NB–среде (Invitrogen). Нейроны выдерживали в культурах в течение 3 недель с подпитыванием NB–среды два раза в неделю путем добавления N2 (Invitrogen). Эти нейроны экспрессируют полный комплемент синаптических белков, характерных для нейронов в зрелом головном мозге, и демонстрируют сложную систему зависимого от активности электрического сигнала. Нейроны и глиальные клетки в таких культурах имеют системы молекулярного сигнала, демонстрирующие превосходное совпадение с интактной сетью головного мозга, и поэтому уже более двадцати лет используются в качестве модельной системы обучения и памяти (См. например Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1(5):2406–15. Epub 2007 Jan 11; Смотри также Craig AM, Graf ER, Linhoff MW. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006 Jan;29(1):8–20. Epub 2005 Dec 7. Review).
Исследуемое соединение добавляли к клеткам в концентрации от 100 мкМ до 0,001 нМ с последующим добавлением препаратов носителя или бета–амилоидного олигомера (общая концентрация бета–амилоидного белка 3 мкМ) и инкубировали в течение 1–24 ч при 37°С в 5% СO2. Реагент МТТ бромида (3–(4,5–диметилтиазол–2–ил)–2,5–дифенилтетразолия) (Roche Molecular Biochemicals) восстанавливали в фосфатно–буферном растворе до 5 мг/мл. В каждую лунку добавляли 10 мкл МТТ–метящего реагента и инкубировали при 37°С в течение 1 ч, и затем визуализировали. Экзоцитоз определяли путем автоматизированной микроскопии и обработки изображений для подсчета количества эндоцитозированного и экзоцитозированного формазана.
Каждый аналитический планшет форматировали таким образом, чтобы соединения подвергались исследованию с бета–амилоидным олигомером и без него на каждом планшете. Этот план предусматривает удаление токсичных или метаболически активных соединений на ранней стадии каскада скрининга (на первичном уровне). Восстановленный формазан сначала был видимым во внутриклеточных везикулах. Последующий экзоцитоз формазана ускоряли пoд действием бета–амилоидных олигомеров.
В присутствии эффективной концентрации активного исследуемого соединения изменения мембранного переноса блокируются, и клетка не отличается от обработанного носителем нейрона. Кроме того, в некоторых случаях этот эффект исследуемого соединения оказывается не зависящим от того, добавляется ли исследуемое соединение до или после воздействия на клетки бета–амилоидного олигомера, что указывает на терапевтический, а также профилактический эффект. Достаточная концентрация активного исследуемого соединения блокирует эффект мембранного переноса бета–амилоидного олигомера, наблюдаемый в этом анализе. Повышаемые дозы селективных высокоаффинных соединений антагонистов сигма–2–рецептора останавливают действия олигомера, в результате чего культуры выглядят более похожими на обработанные носителем культуры.
Соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, которые являются селективными высокоаффинными агонистами сигма–2–рецептора, которые эффективны для ингибирования токсичности бета–амилоидного олигомера, являются перспективными в качестве терапевтических и профилактических средств при связанном с токсичностью бета–амилоидного олигомера снижении когнитивных функций, например, наблюдаемых при болезни Альцгеймера.
При анализе мембранного переноса, вводили дозы синтетических бета–амилоидных олигомеров и они демонстрировали ЕС50 820 нМ. Каждую концентрацию бета–амилоида исследовали относительно нескольких концентраций каждого исследуемого соединения. Активные соединения вызывали сдвиг вправо EC50 почти на два порядка величины. При согласовании данных с классическими линейными и нелинейными моделями, данные были линейными согласно анализу Шилда (угловой коэффициент Хилла nH 1), что указывает на то, что соединения сигма–2–рецептора демонстрируют действительную фармакологическую конкуренцию между олигомерами и соединением за мишени, которые опосредуют мембранный перенос.
Бета–амилоид–олигомеры, полученные из головного мозга пациентов с болезнью Альцгеймера, могут дозироваться в сравнении с исследуемыми соединениями, и ожидается, что в результате воздействия соединения также должен происходить сдвиг вправо. В частности, при эффективных дозах, активные исследуемые соединения демонстрируют фармакологическую конкуренцию как с синтетическими, так и с полученными из организма человека с болезнью Альцгеймера олигомерами.
Экспериментальный контроль
Бета–амилоидные 1–42 олигомеры, полученные в соответствии с опубликованными способами, использовали в качестве положительного контроля. [см., например Dahlgren et al., “Oligomeric and fibrillar species of amyloid–beta peptides differentially affect neuronal viability” J Biol Chem. 2002 Aug 30;277(35):32046–53. Epub 2002 Jun 10.; LeVine H 3rd. “Alzheimer's beta–peptide oligomer formation at physiologic concentrations” Anal Biochem. 2004 Dec 1; 335(1):81–90; Shrestha et.al, “Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons” Mol Cell Neurosci. 2006 Nov;33(3):274–82. Epub 2006 Sep 8; Puzzo et al., “Amyloid–beta peptide inhibits activation of the nitric oxide/cGMP/cAMP–responsive element–binding protein pathway during hippocampal synaptic plasticity” J Neurosci. 2005 Jul 20;25(29):6887–97; Barghorn et al., “Globular amyloid beta–peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer's disease” J Neurochem. 2005 Nov;95(3):834–47. Epub 2005 Aug 31; Johansson et al., Physiochemical characterization of the Alzheimer's disease–related peptides A beta 1–42 Arctic and A beta 1–42wt. FEBS J. 2006 Jun;2 73(12):2618–30], а также бета–амилоидные олигомеры головного мозга (см. например Walsh et al., Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long–term potentiation in vivo. Nature (2002). 416, 535–539; Lesne et al., A specific amyloid–beta protein assembly in the brain impairs memory. Nature. 2006 Mar 16;440(7082):352–7; Shankar et al, Amyloid–beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008 Aug;14(8):837–42. Epub 2008 Jun 22). Следует заметить, что любая композиция бета–амилоидного олигомера может использоваться в указанном анализе или в качестве контроля, включая композиции, описанные в патентной литературе, приведенной выше и включенной посредством ссылки полностью.
Было продемонстрировано, что разнообразные различные препараты бета–амилоидного олигомера вызывают эффект бета–амилоида в анализе мембранного переноса, включая, в частности, олигомерные препараты, выделенные из мозга пациентов с болезнью Альцгеймера.
Олигомеры выделяли из гиппокампа или префронтальной коры человека в процессе посмертного вскрытия без применения детергентов, и они ингибировали мембранный перенос в зависимости от дозы при Kd 6 пМ. Взятые у пациента с болезнью Альцгеймера бета–амилоидные олигомеры (137 пМ) обеспечивают статистически значимое ингибирование мембранного переноса по сравнению с носителем. Соединение 4–(3(4–(трифторметил)бензиламинобутил)–2–метоксифенол устраняет дефицит переноса через мембраны, вызванный бета–амилоидными олигомерами, взятыми из головного мозга пациентов с БА, но не оказывает влияние на перенос при дозировании в отсутствие бета–амилоида.
Хотя эффективность различных препаратов бета–амилоидных олигомеров различна (например, нативные изоляты болезни Альцгеймера являются более эффективными, чем любой из протестированных синтетических препаратов – данные не представлены), качественно результаты являются аналогичными: соединения в соответствии с любым вариантом осуществления настоящего изобретения, которые действуют как функциональные антагонисты сигма–2–рецептора, противодействуют патологиям, опосредованным олигомерами.
Первичные культуры нейронов
Оптимальную плотность клеток определяли на основе клеточной реакции на бета–амилоидные олигомеры с применением анализа экзоцитоза для считывания данных и иммуногистохимического анализа соотношения глиальных клеток с нейронами в культурах. Культуры наблюдали еженедельно посредством иммуногистохимии и количественного определения на основе обработки изображений для отслеживания процента культур с соотношением нейронов и глиальных клеток. Отбраковывали культуры, содержащие более 20% глиальных клеток (положительные на GFAP) относительно нейронов (положительно окрашенные (куриными поликлональными) антителами (Millipore) против МАР2 при 1:5000 (переменная концентрации)) в возрасте отбора 21 день in vitro (21 DIV).
Препараты бета–амилоидных олигомеров
Человеческий амилоидный пептид 1–42 получали от многих коммерческих поставщиков, таких, как California Peptide, с выбором партии в зависимости от результатов анализа контроля качества. Контроль качества олигомерных композиций включал Вестерн–блоты для определения диапазона размеров олигомеров и относительной концентрации и МТТ–анализа для подтверждения ускорения экзоцитоза без токсичности. Токсичность контролировали в каждом анализе на основе изображений путем количественной оценки ядерной морфологии, которая визуализируется ДНК–связывающим синим красителем DAPI (Invitrogen). Фрагментированные ядра рассматривали как находящиеся на поздней стадии апоптоза (Majno and Joris '95), и тест отбраковывали. Партии пептидов, создающие необычный диапазон размеров пептидов или значительную токсичность при стандартной концентрации 1,5 мкМ на нейронах, также отбраковывали.
Контроль планшетов – Оптимизация анализа считалась завершенной, если переформатированные планшеты достигали минимума статистически значимого двухкратного разделения между обработанными носителем и бета–амилоидным олигомером нейронами (р<0,01, t–критерий Стьюдента, неодинаковая дисперсия) на традиционной основе, с коэффициентом вариации между планшетами не более 10%.
Статистическая программа и анализ
Обработку и анализ данных выполняли при помощи программы для анализа изображений Cellomics VTI и программы STORE для автоматизированной обработки базы данных. Из–за низкого динамического диапазона и различий между нейронами в разных лунках после трех недель культивирования осуществляли статистические сравнения с применением попарного анализа Тьюки–Крамера для определения значимости отделения между соединением+бета–амилоидными олигомерами от бета–амилоида в отдельности и между соединением в отдельности от носителя. Способность зрелых первичных нейронов к большему приближению к электрофизиологически опосредованной системе трансдукции сигнала взрослого головного мозга оправдывает такую методику скрининга. Анализ мощности был предусмотрен для нескольких копий скрининговых лунок, что минимизировало ложноотрицательные результаты (например, N=4). Исследуемые соединения по изобретению в значительной степени реверсировали действия бета–амилоидных олигомеров на мембранный перенос, но сами не оказывали влияние на нейронный метаболизм.
Отобранные соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, дозировали в МТТ–анализе, описанном в настоящем документе перед добавлением бета–амилоидного олигомера и, было показано, что они блокируют индуцированный бета–амилоидными олигомерами дефицит мембранного переноса с указанным ЕС50. В частности, эти результаты указывают на то, что соединения блокируют/ослабляют активност/действие бета–амилоидного олигомера на мембранный перенос нейронов при микромолярных концентрациях. Было показано, что некоторые соединения в таблице 1 блокируют вызванное бета–амилоидным олигомером ускорение экзоцитоза с указанным ЕС50. Соответственно, соединения в таблице 1 значительно блокировали опосредованные бета–амилоидными олигомерами изменения в мембранном переносе. Эти результаты показывают, что соединения блокируют/ослабляют активность/эффект бета–амилоидного олигомера в отношении нейронов и что соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, могут применяться для блокирования индуцированных бета–амилоидным олигомером нарушений мембранного переноса.
В некоторых вариантах осуществления соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, ингибируют индуцированный бета–амилоидным олигомером дефицит мембранного переноса, с ЕС50 не больше 20 мкМ, не больше 15 мкМ, не больше 10 мкМ, не больше 5 мкМ, не больше 1 мкМ, не больше 0,5 мкМ при испытаниях в соответствии с протоколом анализа мембранного переноса, приведенным в настоящем документе.
Пример 6: Исследование фармакокинетической и метаболической стабильности.
Первое фармакокинетическое исследование выполняли на микросомах печени мышей, (MLM). Исследования выполняли в соответствии с публикацией Obach, R.S et al. (1997) J. Pharmacol. Exp. Ther., 283: 46–58, которая включена в настоящее описание посредством ссылки. Показатели периода полувыведения (t1/2) соединений в анализе MLM представлены в Таблице 1.
В некоторых вариантах осуществления соединение в соответствии с любым вариантом осуществления, описанным в настоящем документе, или его фармацевтически приемлемые соли демонстрируют период полувыведения (t1/2) в анализе на микросомах печени мыши (MLM), как предусмотрено в настоящем документе, составляющем, по меньшей мере 5 минут, по меньшей мере, 10 минут, по меньшей мере, 25 минут, по меньшей мере 50 минут, по меньшей мере 100 минут или по меньшей мере 200 минут.
Результаты показывают, что некоторые из исследованных соединений имели значительно более длительный период полувыведения в микросомах печени мыши. Этот результат предполагает большую биодоступность после перорального введения для этих соединений. Те же соединения исследовали посредством описанного выше анализа мембранного переноса и по их активности, как указано в настоящем документе.
Если скорость собственного клиренса испытуемого соединения была высокой, это указывает на значительный пресистемный метаболизм. Для улучшения фармакокинетических свойств предусматривались соединения, повышающие метаболическую стабильность и улучшающие подобные лекарственным свойства. Эксперименты с микросомальной стабильностью и эксперименты со стабильностью в плазме выполняли для определения метаболической и печеночной стабильности потенциально эффективных соединений. В некоторых вариантах осуществления in vitro микросомальную устойчивость нормализовали до стандартного соединения 4–(3–(4–(трифторметил)бензиламинобутил)–2–метоксифенола, соединения–прототипа, которое имело t1/2 микросом печени мыши, составляющее 16 (мин). Соединения по изобретению имеют более высокие показатели, чем указанное соединение–прототип.
Второе фармакокинетическое исследование может быть проведено in vivo, и оно включает измерение уровня в плазме и уровня в головном мозге исследуемых соединений, вводимых различными способами и в неотложном или длительном режиме, как указано ниже:
Оптимизация ВЭЖХ–МС
Раствор каждого исследуемого соединения приготавливали и вводили путем инфузии в источник спектрометра TSQ Quantum (Fisher Thermo Scientific) через шприцевой насос с постоянной скоростью. Осуществляли МС (масс–спектроскопический) анализ с полным сканированием и генерировали хроматограммы общего ионного тока и соответствующие масс–спектры для каждого исследуемого соединения как в положительном, так и в отрицательном режимах ионизации. Родительские ионы для МС/МС выбирали из положительного или отрицательного масс–спектра в зависимости от соответствующей распространенности ионов. Кроме того, выполняли МС/МС анализ дочерних ионов с целью определения соответствующей выбранной реакции фрагментации для применения в количественном анализе. Конечные параметры контроля реакции выбирали для обеспечения максимальной возможности определения количества исследуемого соединения при наличии в сложной смеси компонентов. После распознавания специфического SRM–перехода, применяемого для каждого исследуемого соединения, параметры обнаружения оптимизировали с применением автоматизированного протокола в рабочей среде TSQ Quantum Compound Optimization. И наконец, хроматографические условия, применяемые для ЖХ–МС–анализа, определяли путем впрыскивания и отделения анализируемого вещества на соответствующей колонке для жидкостной хроматографии и, в случае необходимости, выполняли регулирование градиентных условий.
Композиция для внутривенного введения:
Растворимость исследуемого соединения в фосфатно–буферном растворе, рН 7,4 (PBS), сначала определяли путем визуального наблюдения. PBS использовали в качестве носителя, если соединение являлось растворимым в заданной концентрации (могут оцениваться другие носители, совместимые с внутривенным введением, если соединение не является полностью растворимым в PBS. К таким носителям, помимо прочих, относятся ДМСО, полиэтиленгликоль (PEG 400), Solutol HS 15 и Cremophor EL). В описанных в настоящем документе экспериментах внутривенно вводится одноразовую дозу, 10 мг/кг, исследуемого соединения.
Композиция для перорального введения: Сначала определяли растворимость исследуемого соединения в PBS. PBS использовали в качестве носителя, если соединение являлось растворимым в заданной концентрации (применяют ДМСО/Solutol HS 15/PBS (5/5/90, объем/объем/объем) или ДМСО/1% метилцеллюлозы (5/95, объем/объем), если исследуемое соединение не являлось полностью растворимым в PBS в соответствующей концентрации).
Линейность в плазме
В аликвоты плазмы в качестве аналитического вещества добавляли исследуемые соединения в указанных концентрациях. Образцы с аналитическим веществом обрабатывали, применяя осаждение ацетонитрилом, и анализировали посредством ВЭЖХ–МС или ВЭЖХ–МС/МС. Строили градуировочную кривую зависимости площади под пиком от концентрации. Определяли регистрируемый линейный диапазон анализа, вместе с нижним пределом количественного определения (LLQ).
Количественный биоанализ образцов плазмы
Образцы плазмы обрабатывали, применяя осаждение ацетонитрилом, и анализировали путем ВЭЖХ–МС или ВЭЖХ–МС/МС. Строили градуировочную кривую плазмы. В аликвоты плазмы, не содержащей лекарственное средство, в качестве аналитического вещества добавляли исследуемое соединение в указанной концентрации. Образцы плазмы с аналитическим веществом обрабатывали вместе с неизвестными образцами плазмы с применением одной и той же процедуры. Обычно обработанные образцы плазмы (высушенные экстракты) хранят замороженными (–20°С) до анализа ВЭЖХ–МС или ВЭЖХ–МС/МС. Высушенные экстракты восстанавливали в подходящем растворителе и после центрифугирования анализировали с помощью ВЭЖХ–МС или ВЭЖХ–МС/МС. Регистрировали показатели площади под пиками и определяли концентрацию исследуемого соединения в неизвестных образцах плазмы, используя соответствующую градуировочную кривую. Определяли регистрируемый линейный диапазон анализа, а также нижний предел количественного определения (LLQ).
В исследовании типично использовали животных – самцов мышей линии C57BL/6, каждый массой 20–30 г, или самцов крыс линии Sprague–Dawley массой 180–250 г. Трех животных обрабатывали для каждого из условий введения и каждого момента времени, таким образом, чтобы у каждого животного лишь один раз отбирали образец крови. Подкожное введение соединения осуществляли путем внутрибрюшинной инъекции. Пероральное введение осуществляли через желудочный зонд. Внутривенное введение осуществляли через яремный катетер.
После введения соединение в различных концентрациях собирали образцы плазмы, например, через 10, 30, 60, 120, 240, 360, 480 и 1440 мин.
Сбор образцов плазмы у мышей и крыс
Животных усыпляли посредством общей ингаляционной анестезии (3% изофлуран) для сбора образцов крови путем пункции сердца (мыши) или через яремный катетер (крысы). Аликвоты крови (300–400 мкл) собирали в пробирки, покрытые литий–гепарином, осторожно смешивали и выдерживали на льду и центрифугировали при 2500×g в течение 15 минут при 4°С в течение 1 часа после сбора. Затем образцы плазмы собирали и хранили замороженными при –20°С до дальнейшей обработки.
Режим дозирования для животных – In vivo PK – Неканюлированные животные, без ограничений в пище
Группа 1: SC, n=3 животных на каждый момент времени (всего 24 животных) или
IV, n=3 животных на каждый момент времени (всего 24 животных)
Группа 2: РО, n=3 животных на каждый момент времени (всего 24 животных)
Группа 3: Контрольные животные (для крови без лекарственного средства), n=5 мышей
У каждого животного отбирали один образец крови и один образец головного мозга.
Взятие образцов головного мозга у животных
Сразу после взятия образца крови, животных обезглавливали и головной мозг быстро извлекали, промывали холодным солевым раствором (0,9% NaCl, г/мл), отделяли поверхностные сосуды, насухо промакивали марлей, взвешивали, выдерживали на льду до дальнейшей обработки в течение одного часа после сбора. Каждый образец головного мозга гомогенизировали в 1,5 мл холодного фосфатно–буферного раствора, рН 7,4 (мыши=1,5 мл, крысы =), в течение 10 секунд на льду с использованием Power Gen 125. Затем гомогенат каждого образца головного мозга хранили при –20°С до дальнейшей обработки.
Линейность в образцах головного мозга
К аликвотам гомогената головного мозга в качестве аналитического вещества добавляли исследуемое соединение в указанных концентрациях. К каждому определенному количеству головного мозга добавляли один и тот же объем охлажденного 26% (г/мл) раствора нейтрального декстрана (средняя молекулярная масса 65000–85000 от Sigma, каталожный номер D–1390) для получения конечной концентрации декстрана 13%. Гомогенат центрифугировали при 54000×g в течение 15 минут при 4°С. Затем супернатанты обрабатывали, применяя осаждение ацетонитрилом, и анализировали с применением ВЭЖХ–МС/МС. Строили градуировочную кривую зависимости пика от концентрации. Определяли регистрируемый линейный диапазон анализа, а также нижний предел количественного определения (LLQ).
Количественный анализ образцов головного мозга
К каждой аликвоте гомогената головного мозга добавляли один и тот же объем охлажденного 26% (г/мл) раствора нейтрального декстрана (средняя молекулярная масса 65000–85000 от Sigma, каталожный номер D–1390) для получения конечной концентрации декстрана 13%. Гомогенат центрифугировали при 54000×g в течение 15 минут при 4°С. Затем супернатанты обрабатывали, применяя осаждение ацетонитрилом, и анализировали с применением ВЭЖХ–МС/МС. Строили градуировочную кривую для образцов головного мозга. К аликвотам не содержащего лекарственное средство гомогената головного мозга в качестве аналитического вещества добавляли исследуемое соединение при заданных уровнях концентрации. Образцы гомогената головного мозга с аналитическим веществом обрабатывали вместе с неизвестными образцами гомогената головного мозга с использовнием аналогичной процедуры. Обработанные образцы головного мозга хранили при –20°С до проведения анализа ЖХ–МС/МС, в тоже время регистрировали значения площади под пиками, и концентрацию исследуемого соединения в неизвестных образцах головного мозга определяли с применением соответствующей градуировочной кривой. Регистрируемый линейный диапазон анализа определяли вместе с нижним пределом количественного определения (LLQ).
Способность проникать в головной мозг
Концентрацию исследуемого соединения в головном мозге (нг/г ткани) и в плазме (нг/мл), а также соотношение концентрации в головном мозге и концентрации в плазме в каждый момент времени определяли при помощи ЖХ–МС/МС и представляли, так, как описано выше.
Фармакокинетика
Строили графики зависимости концентрации соединения в плазме от времени. Получали основные фармакокинетические параметры соединения после перорального и подкожного введения дозы (AUClast, AUCINF, Т1/2, Tmax и Cmax) в результате некомпартментного анализа (NCA) данных плазмы с применением WinNonlin (Pharsight). Некомпартментный анализ не требует принятия конкретной некомпартментной модели для лекарственного средства или метаболита. NCA допускает применение правила трапеций для измерений площади под кривой зависимости концентрации в плазме от времени (Gabrielsson, J. and Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications. Swedish Pharmaceutical Press. 1997).
Определение приведенных терминов
Площадь под кривой (AUC) – мера общего количества лекарственного средства, которое без изменений достигает большого круга кровообращения. Площадь под кривой представляет собой геометрический показатель, который рассчитывают путем построения графика зависимости концентрации от времени и суммирования возрастающих площадей каждой трапеции.
WinNonlin включает два способа вычисления площади: линейный метод трапеций и линейно–логарифмический метод трапеций. Поскольку линейный метод трапеций может давать результаты с погрешностью на нисходящей части кривой зависимости концентрации от времени и завышать AUC, WinNonlin обеспечивает линейно–логарифмический вариант вычисления AUC. Следуя общему правилу, применяют логарифмически–линейный метод трапеций для определения площади через Tmax для остальной части кривой зависимости концентрации в плазме от времени.
AUClast: площадь под кривой с момента времени введения дозы до момента времени последнего наблюдения, которая превышала предел количественного определения.
AUCINF: площадь под кривой с момента введения дозы, экстраполированная в бесконечность.
Cmax – максимальная концентрация лекарственного средства в плазме, достигнутая после перорального или отличного от внутривенного введения лекарственного средства между временем введения и конечным наблюдаемым моментом времени.
Tmax – время при максимальной наблюдаемой концентрации в плазме (Сmax), указываемое в минутах после введения лекарственного средства.
T1/2 – период полувыведения в конечной фазе при внутривенном и отличном от внутривенного введении.
Где лямбда Z (z) является константой скорости первого порядка, связанной с конечной (логарифмически–линейной) частью кривой зависимости концентрации в плазме от времени. Z определяли по линейной регрессии времени в зависимости от логарифмической концентрации.
Ожидается, что результаты покажут, что некоторые исследуемые соединения демонстрируют хорошую биодоступность и хорошее проникновение в головной мозг при введении в дозах от 0,1 до 0,5 мг/кг в неотложном или длительном режиме (ежедневно в течение 5 дней). Выбранные исследуемые соединения таким способом оценивали на пероральную биодоступность.
Пример 8: исследование ингибирования hERG in vitro
Исследование ингибирования hERG in vitro проводили в стандартном анализе (см: Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, Escande D, Franz M, Malik M, Moss A and Shah R. (2000) Eur Heart J 21 (15); 1216–31). Результаты для исследуемых соединений для ингибирования hERG (IC50, нМ) показаны в таблице 1. В некоторых вариантах осуществления соединения, в соответствии с любым вариантом осуществления, описанным в настоящем документе, или их фармацевтически приемлемые соли демонстрируют минимальное ингибирование hERG с IC50 больше 300 нМ, больше 500 нМ, больше 1000 нМ, больше 3000 нМ, больше 5000 нМ, больше 10000 или больше 20000 нМ. В конкретных вариантах осуществления соединения в соответствии с любым вариантом осуществления, описанным в настоящем документе, или их фармацевтически приемлемые соли демонстрируют минимальное ингибирование hERG и демонстрируют IC50 больше 5000 нМ, больше 10000 или больше 20000 нМ.
Объединеннные результаты для конкретных соединений, описанных в настоящем документе, в отношении log P, мембранного переноса (мкМ), аффинности к сигма–2–рецептору, аффинности к сигма–1–рецептору микросомальной стабильности в микросомах печени мыши (MLM) (t1/2, мин), t1/2, нормализованному к CT010914, и токсичности калиевого канала hERG in vitro (IC50, нМ) представлены в таблице 1:
г/моль
[MH]+
Бета–амилоид (мкM)
*нормализованный к 4–(3–(4–(трифторметил)бензиламино)бутил)–2–метоксифенолу.
Примеры синтеза
Пример синтеза 1: Получение соединения примера 9:
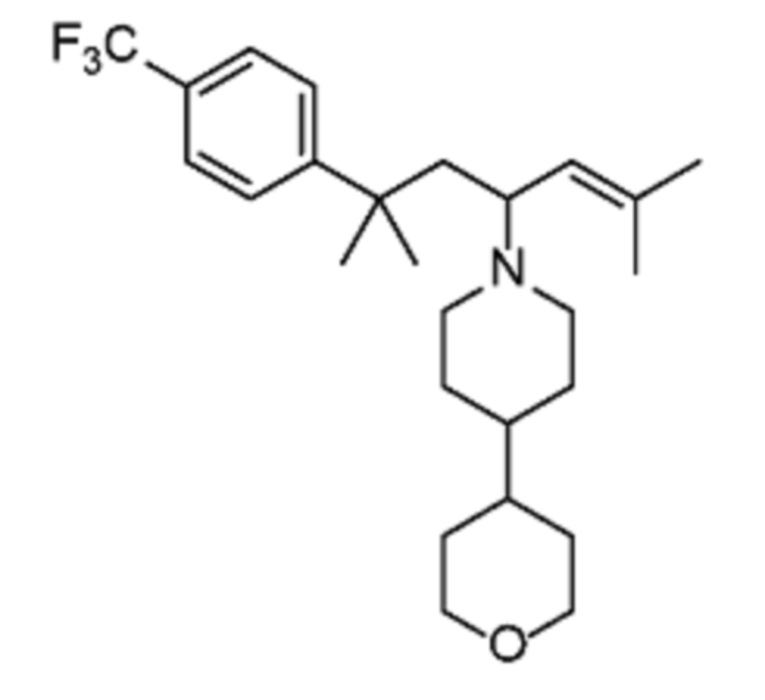
Пример 9

Общая процедура получения соединения S1–2

В трехгорлую колбу помещали магний (9,39 г, 391,09 ммоль, 1,1 экв.) и гранулы иода. Затем в смесь добавляли 15 объемных процентов соединения S1–1 (80 г, 355,54 ммоль, 1,0 экв.) в ТГФ (800 мл) в атмосфере азота, и перемешиваемую смесь нагревали до 70°C до исчезновения желто–коричневого цвета, и затем перемешивали при этой температуре в течение еще 6 ч с получением раствора соединения S1–2 (0,44 М) в ТГФ, который использовали непосредственно на следующей стадии.
Общая процедура получения соединения S1–4
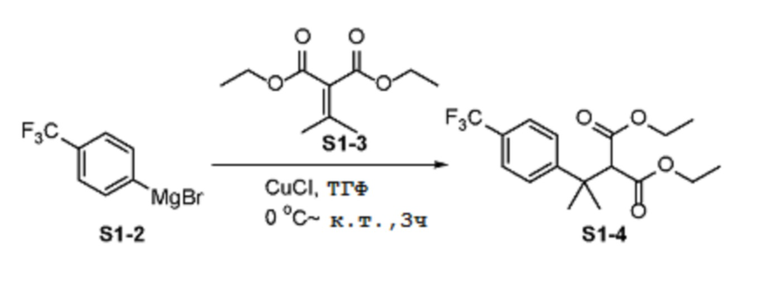
К раствору соединения S1–2 (320 ммоль, 1,0 экв.) в ТГФ (500 мл) добавляли CuCl (3,17 г, 32 ммоль, 0,1 экв.). Когда раствор охлаждался до 0°С, по каплям добавляли соединение S1–3 (64 г, 320 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, гасили насыщенным раствором NH4Cl, экстрагировали этилацетатом. Органический слой сушили над Na2SO4, концентрировали в вакууме. Остаток очищали колоночной хроматографией (PE/EA, 50:1~10:1) с получением указанного в заголовке соединения S1–4 (51 г, 46%). 1H ЯМР (400 МГц, CDCl3) δ 7,57–7,49 (м, 4H), 4,08–4,03 (м, 4H), 3,80 (д, J=2,4 Гц, 1H) 1,62–1,58 (м, 6H), 1,13–1,09 (м, 6H).
ТСХ: PE/EA=10:1, УФ 254 нм
Rf (соединение S1–4) = 0,5
Общая процедура получения соединения S1–5

К раствору соединения S1–4 (51 г, 147,25 ммоль,1,0 экв.) в MeOH (300 мл) добавляли 1н водный раствор NaOH (736 мл, 736,25 ммоль, 5,0 экв.). Реакционную смесь перемешивали при 90 °C в течение 3 ч, и затем охлаждали до комнатной температуры. Смесь подкисляли 1 н раствором HCl до pH 4–5 и экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и концентрировали в вакууме с получением соединения S1–5 (40 г, 94%), которое непосредственно использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,57–7,55 (м, 2H), 7,50–7,48 (м, 2H), 3,86 (с, 1H), 1,59 (с, 6H);
ТСХ: DCM/MeOH=10:1, УФ 254 нм
Rf (соединение S1–5) = 0,5
Общая процедура получения соединения S1–6
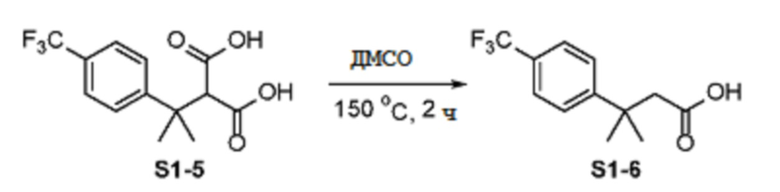
Раствор соединения S1–5 (40 г, 137,82 ммоль, 1,0 экв.) в ДМСО (200 мл) перемешивали при 150°C в течение 2 ч, разбавляли этилацетатом, промывали водой, насыщенным солевым раствором. Затем органическую фазу сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединения S1–6 (30 г, 88%), которое непосредственно использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,57–7,55 (м, 2H), 7,48–7,45 (м, 2H), 2,67 (с, 2H), 1,49–1,46 (м, 6H);
ТСХ: DCM/MeOH=10:1, УФ 254 нм
Rf (соединение S1–5) = 0,4
Rf (соединение S1–6) = 0,8
Общая процедура получения соединения S1–8

К раствору соединения S1–6 (30 г, 121,84 ммоль, 1,0 экв.) в ТГФ (500 мл) добавляли HOBt (19,8 г, 146,2 ммоль, 1,2 экв.), EDCI (28 г, 146,21 ммоль, 1,2 экв.), TEA (37 г, 365,52 ммоль, 3,0 экв.) и соединение S1–7 (23,77 г, 243,68 ммоль, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи, и затем гасили водой, экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE/EA, 10:1~3:1) с получением соединения S1–8 (31 г, 88%). 1H ЯМР (400 МГц, CDCl3) δ 7,56–7,54 (м, 2H), 7,50–7,47 (м, 2H), 3,59 (с, 3H), 3,06 (с, 3H), 2,78 (с, 2H), 1,49 (с, 6H);
ТСХ: PE/EA=3:1, УФ 254 нм
Rf (соединение S1–6) = 0,4
Rf (соединение S1–8) = 0,8
Общая процедура получения соединения S1–10

В трехгорлую колбу помещали магний (8,0 г, 331,39 ммоль, 1,1 экв.) и гранулы иода. В смесь в атмосфере азота добавляли 15 объемных процентов соединения S1–9 (40,67 г, 301,26 ммоль, 1,0 экв.) в ТГФ (300 мл), и перемешиваемую смесь нагревали до 70°C до исчезновения желто–коричневого цвета, и затем раствор перемешивали при этой температуре еще 6 ч с получением раствора соединения S1–10 (1,0 M) в ТГФ, которое непосредственно использовали на следующей стадии.
Общая процедура получения соединения S–11

К раствору соединения S1–8 (31 г, 107,16 ммоль, 1,0 экв.) в ТГФ (200 мл) при 0°С добавляли соединение S1–10 (1,0 M, 215 мл, 214,32 ммоль, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Затем смесь гасили насыщенным раствором NH4Cl, экстрагировали эфиром и органический слой сушили над Na2SO4, концентрировали в вакууме с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE) с получением сырого соединения S–11 (28,9 г, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,56–7,54 (м, 2H), 7,47–7,45 (м, 2H), 7,75 (м, 1H), 2,75 (с, 2H), 2,01–1,99 (м, 3H), 1,74–1,73 (м, 3H), 1,44–1,42 (м, 6H);
ТСХ: PE/EA=10:1, УФ 254 нм
Rf (соединение S1–8) = 0,5
Rf (соединение S1–11) = 0,9
Общая процедура получения примера 9

Смесь соединения S1–11 (2,4 г, 8,44 ммоль, 1,0 экв.), соединения S1–12 (1,43 г, 8,44 ммоль, 1,0 экв.) и Ti(EtO)4 (7,7 г, 33,76 ммоль, 4,0 экв.) в ТГФ (100 мл) перемешивали при 70°C в течение ночи в атмосфере азота. Затем смесь оставляли охлаждаться до комнатной температуры, добавляли NaBH4 (1,23 г, 33,76 ммоль, 4,0 экв.). После завершения добавления, смесь перемешивали при комнатной температуре в течение 3 ч. Потом добавляли воду и экстрагировали этилацетатом, фильтровали. Органическую фазу сушили над Na2SO4, концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (PE/EA, 10:1~1:1) с получением соединения примера 9, которое растворяли в HCl/EA (2,0 M, 10 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали с получением примера 9 HCl (600 мг, 15%) в виде масла. 1H ЯМР (400 МГц, CD3OD) δ 7,60–7,52 (м, 4H), 4,70–4,67 (м, 1H), 3,95–3,92 (м, 2H), 3,79–3,73 (м, 1H), 3,61–3,59 (м, 1H), 2,81–2,73 (м, 2H), 2,38–2,22 (м, 2H), 1,98 (с, 6H), 1,64–1,61 (м, 2H), 1,46–1,43 (м, 12H), 1,31–1,28 (м, 6H);
MS: [M+H]+= 438,5
Пример синтеза 2: Получение соединения примера 262:

Пример 262

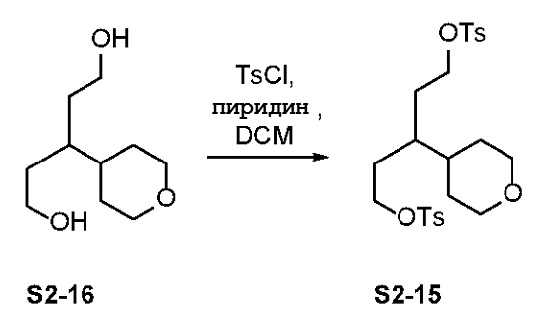
Общая процедура получения соединения S2–7

К раствору соединения S2–6 (450 г, 1,83 моль, 1,0 экв.) в безводном ТГФ (4,5 л) охлажденном до 0 oC, добавляли по каплям раствор 1M BH3 (2,75 л, 2,745 моль, 1,5 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию контролировали с помощью ТСХ. Бесцветную гомогенную реакционную смесь охлаждали до 0°C и осторожно добавляли MeOH (2 л), затем воду (1 л). Потом MeOH и ТГФ удаляли в вакууме. Смесь экстрагировали DCM (3 × 1 л), объединенные органические экстракты промывали насыщенным солевым раствором (1 л), сушили над MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением сырого соединения S2–7 (340 г, 80%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3): δ 7,57 (д, J=8,0 Гц, 2H), 7,47 (д, J=8,4 Гц, 2H), 3,47 (т, J=7,2 Гц, 2H), 1,98 (т, J=7,2 Гц, 2H), 1,29 (с, 6H).
Общая процедура получения соединения S2–9

Раствор соединения S2–7 (296 г, 1,27 моль, 1,0 экв.) растворяли в CH3CN (3,6 л), NMI (10,3 г, 127 ммоль, 0,1 экв.), TEMPO (9,8 г, 63 ммоль, 0,05 экв.) и BPy (9,7 г, 63 ммоль, 0,05 экв.) добавляли, затем добавляли Cu(PF6)(MeCN)4 (23,5 г, 63 ммоль, 0,05 экв.). Смесь перемешивали в атмосфере кислорода в течение 4 ч. Реакцию контролировали с помощью ТСХ. Смесь выливали в воду (2 л), экстрагировали DCM (3 × 2 л). Объединенные экстракты промывали водой, сушили над Na2SO4 и концентрировали с получением соединения S2–9 (281 г, 96%) в виде масла темно–зеленого цвета. 1H ЯМР (400 МГц, CDCl3): δ 9,54 (с, 1H), 7,61 (д, J=7,6 Гц, 2H), 7,51 (д, J=7,6 Гц, 2H), 2,66 (с, 2H), 1,49 (с, 6H).
ТСХ: PE/EA=20:1, УФ 254 нм
Rf (соединение S2–7) = 0,1
Rf (S2–9) = 0,8
Общая процедура получения соединения S2–11R

К раствору соединения S2–9 (150 г, 652,5 ммоль, 1,0 экв) в ТГФ (750 мл), добавляли соединение S2–10R (118,96 г, 978,77 ммоль, 1,5eq) и Ti (OEt)4 (74,4 г, 326,2 ммоль, 0,5 экв.). Смесь перемешивали при комнатной температуре в течение ночи, а затем гасили водой (24 г, 1,3 моль), фильтровали через пад из целита и разбавляли этилацетатом, промывали насыщенным солевым раствором, водой. Органический слой сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE/EA, 5:1) с получением соединения S2–11R (165 г, 75,9%).
ТСХ: PE/EA=3:1, УФ 254 нм
Rf (соединение S2–9) = 0,6
Rf (соединение S2–11R) = 0,5
Общая процедура получения соединения S2–12

В трехгорлую колбу помещали магний (31,9 г, 1,3 моль, 1,1 экв.) и гранулы иода. В смесь добавляли 15 объемных процентов соединения S2–31 (149 г, 1,2 моль, 1,0 экв.) в ТГФ (1,5 л) в атмосфере азота и перемешиваемую смесь нагревали до 70°C до исчезновения желто–коричневого цвета. Далее оставшийся раствор добавляли по каплям и перемешивали в течение еще 6 ч с получением раствора соединения S2–12 (1,0 M) в ТГФ, которое непосредственно использовали на следующей стадии.
Общая процедура получения соединения S2–13R

К раствору соединения S2–11R (120 г, 359,9 ммоль, 1,0 экв.) в ТГФ (80 мл) добавляли соединение S2–12 (1080 мл, 539,9 ммоль, 1,50 экв., 0,5 M) при 0–5℃. После перемешивания при комнатной температуре в течение ночи, реакционную смесь гасили NH4Cl, экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE/EA, 10:1~3:1) с получением соединения S2–13R (69,1 г, 49,3%).
Соединение S2–13R (69,1 г) перекристаллизовывали из смеси эфир/гексан (1/1) (30 мл) при температуре 4~8°C в течение двух дней с получением твердого вещества белого цвета (25,6 г, 37%).), которое использовали для получения примера 262 c 99% э.и. Исходный раствор перекристаллизовывали из смеси эфир/гексан (1/1) (20 мл) с получением твердого вещества белого цвета (15,1 г).
ТСХ: PE/EA=3:1, УФ 254 нм
Rf (соединение S2–11R) = 0,5
Rf (соединение S2–13R) = 0,3
Общая процедура получения соединения S2–14R

В круглодонную колбу добавляли соединение S2–13R (34,6 г, 88,8 ммоль, 1,0 экв.). Затем добавляли HCl в EtOAc (90 мл, 2 н, 2 экв.). После перемешивания в течение двух часов, около 80 мл растворителя удаляли в вакууме. Смесь разбавляли эфиром (20 мл), твердое вещество белого цвета отфильтровывали в виде соединения S2–14R гидрохлорида. Гидрохлорид растворяли в H2O (30 мл), подщелачивали насыщенным водным раствором Na2CO3 до pH 10. Затем экстрагировали эфиром (30 мл × 3), сушили над (Na2SO4), концентрировали с получением бесцветного масла (20,2 г, 79,7%).
ТСХ: PE/EA=3:1, УФ 254 нм
Rf (соединение S2–13R) = 0,3
Rf (соединение S2–14R) = 0,1
Общая процедура получения соединения примера 262

К раствору соединения S2–14R (4,8 г, 16,8 ммоль, 1,0 экв.) в ACN (48 мл) добавляли соединение S2–15 (8,35 г, 16,8 ммоль, 1,0 экв.) и K2CO3 (4,46 г, 33,6 ммоль, 2,0 экв.). После перемешивания при 85°C в течение ночи, реакционную смесь гасили H2O (50 мл) и экстрагировали этилацетатом (40 мл × 3). Органический слой сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE/EA, 10:1~3:1) с получением соединения примера 262 (5,1 г 69%) в виде свободного основания. Посредством хиральной ВЭЖХ определяли энантиомерный избыток в %, соответствующий 99%. Свободное основание растворяли в HCl/EA (1,3 M, 50 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали с получением примера 262 HCl (5,5 г, 69%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CD3OD) δ 7,60–7,52 (м, 4H), 4,70–4,67 (м, 1H), 3,95–3,92 (м, 2H), 3,79–3,73 (м, 1H), 3,61–3,59 (м, 1H), 2,81–2,73 (м, 2H), 2,38–2,22 (м, 2H), 1,98 (с, 6H), 1,64–1,61 (м, 2H), 1,46–1,43 (м, 12H), 1,31–1,28 (м, 6H);
MS: [M+H]+= 438,5
Общая процедура получения соединения 2–15
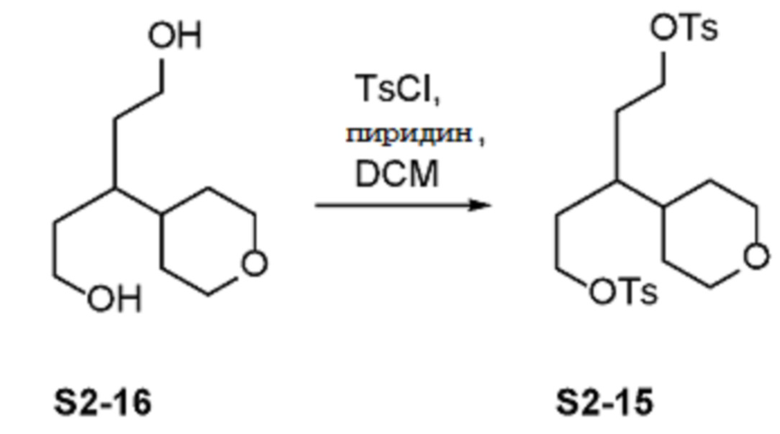
К раствору соединения S2–16 (5,0 г, 26,6 ммоль, 1,0 экв.) в DCM (50 мл) добавляли соединение TsCl (20,2 г, 106,2 ммоль, 4,0 экв.) и пиридин (8,4 г, 106,2 ммоль, 4,0 экв.). После перемешивания при комнатной температуре в течение ночи, реакционную смесь гасили H2O (100 мл) и экстрагировали этилацетатом (40 мл × 3). Органический слой сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали хроматографией на колонке с силикагелем (PE/EA, 10:1~3:1) с получением соединения S2–15 (5,4 г 40,9%).
Пример синтеза 3: Получение соединения примера 14:

Пример 14

Общая процедура получения соединения примера 14

К смеси соединения S3–9 (60,0 г, 0,26 моль, 1,0 экв.) и соединения S3–10 (30,0 г, 0,26 моль, 1,0 экв.) в MeOH (600 мл) добавляли NaBH4 (39,6 г, 1,04 моль, 4,0 экв.). Смесь перемешивали при комнатной температуре в течение 3 ч в атмосфере азота. Затем добавляли насыщенный солевой раствор, и раствор экстрагировали этилацетатом. Органическую фазу объединяли, сушили над Na2SO4, концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением соединения примера 14 (45,1 г, 52%).
ТСХ: DCM: MeOH =10:1
Rf (пример 14) = 0,2
Общая процедура получения соединения примера 14 HCl

Смесь примера 14 (65 г, 0,2 моль) в HCl/EA (2,5 M, 160 мл) перемешивали при комнатной температуре в течение 2 ч. Непрозрачную смесь концентрировали с получением примера 14 HCl (68 г, 94%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CD3OD): δ м.д. 7,67–7,61 (м, 4H), 3,31–3,3 (м, 3H), 3,15–3,11 (м, 2H), 2,85–2,79 (м, 2H), 2,17–2,12 (м, 2H), 1,82–1,76 (м, 2H), 1,43 (с, 6H), 1,27 (с, 3H).
MS: [M+H]+=330,20
Пример синтеза 4: Получение соединения примера 17:
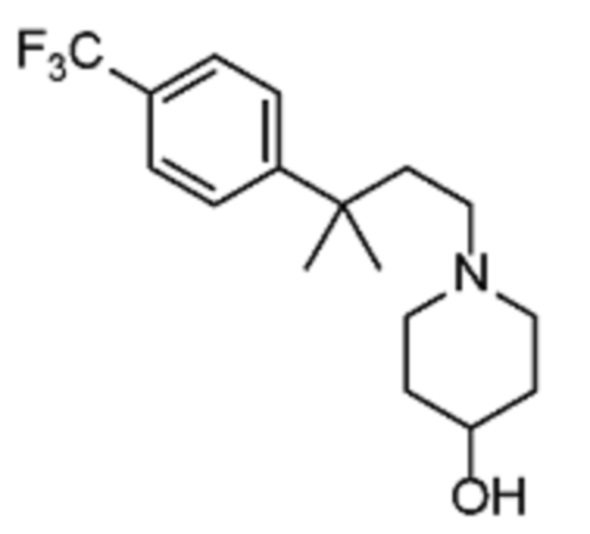
Пример 17

Смесь соединения S4–11 (300 мг, 1,3 ммоль, 1,0 экв.), соединения S4–12 (150 мг, 1,3 ммоль, 1,0 экв.) и NaBH4 (197 мг, 5,2 ммоль, 4,0 экв.) в MeOH (10 мл) перемешивали при комнатной температуре в течение 3 ч в атмосфере азота. Затем добавляли водный раствор NaCl и экстрагировали этилацетатом, фильтровали. Органическую фазу сушили над Na2SO4, концентрировали в вакууме. Остаток очищали препаративной ТСХ с получением соединения примера 17, которое растворяли в HCl/EA (1,3 M, 10 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали с получением примера 17 HCl (144,1 мг, 31,4%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,75–7,60 (м, 4H), 4,07–4,03 (м, 1H), 3,63–3,57 (м, 1H), 3,34–3,30 (м, 6H), 3,16–3,09 (м, 2H), 2,87–2,83 (м, 2H), 2,19–2,14 (м, 2H), 1,43 (с, 6H);
MS: [M+H]+= 316,4
Пример синтеза 5: Получение соединения примера 307:
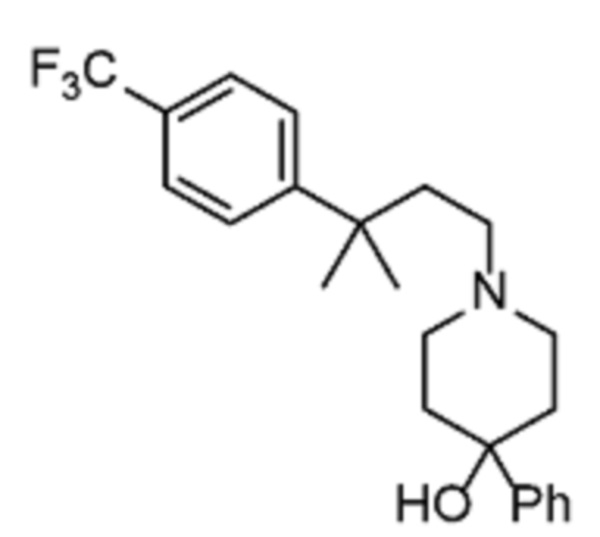
Пример 307

К раствору соединения S5–9 (3,54 г, 15,38 ммоль, 1,0 экв.) в MeOH (60 мл) добавляли соединение S5–10 (3,0 г, 16,9 ммоль, 1,1 экв.) и 2 капли AcOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли NaBH4 (2,33 г, 61,55 ммоль, 4,0 экв.) и затем реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили H2O (20 мл), фильтровали, экстрагировали EA (20 мл × 3) и концентрировали с получением остатка, который очищали колоночной хроматографией (PE:EA=1:1) с получением соединения примера 307 (1,3 г, 21,6%).
ТСХ: DCM:EA:MeOH=1:1:0,1
Rf (соединение S5–9) = 0,9
Rf (соединение примера 307) = 0,3
Общая процедура получения примера 307 HCl

К раствору соединения примера 307 (1,9 г, 4,85 ммоль, 1,0 экв.) в EA (5 мл) добавляли EA/HCl (2,5 M, 6 мл, 3,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали с получением примера 307 HCl (2,1 г, 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,62–7,60 (м, 2H), 7,50–7,47 (м, 4H), 7,46–7,31 (м, 2H), 7,28 (м, 2H), 3,32 (м, 2H), 3,13 (м, 2H), 2,64 (м, 2H), 2,32 (м, 2H), 1,67 (м, 3H), 1,39 (м, 6H);
MS: [M+H]+= 392,6
Пример синтеза 6: Получение соединения примера 191:

Пример 191

К раствору соединения S6–9 (5,4 г, 23,68 ммоль, 1,0 экв.) в MeOH (100 мл) добавляли соединение S6–12 (3,0 г, 26 ммоль, 1,1 экв.) и 2 капли AcOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли NaBH4 (3,58 г, 94,71 ммоль, 4,0 экв.) и затем реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили H2O (30 мл), фильтровали, экстрагировали EA (30 мл × 3) и концентрировали с получением остатка, который очищали колоночной хроматографией (PE:EA=1:1) с получением примера 191 (3,4 г, 44%).
ТСХ: DCM:EA:MeOH=1:1:0,1
Rf (соединение S6–9) = 0,9
Rf (Пример 191) = 0,3
Общая процедура получения примера 191 HCl

К раствору примера 191 (3,4 г, 10,33 ммоль, 1,0 экв.) в EA (10 мл) добавляли EA/HCl (2,5 M, 8,9 мл, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали с получением примера 191 HCl (3,5 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 7,60–7,58 (м, 2H), 7,49–7,47 (м, 2H), 3,56 (с, 1H), 3,25 (с, 3H), 3,19 (м, 2H), 2,80 (м, 2H), 2,59 (м, 2H), 2,39 (м, 2H), 2,30 (м, 2H), 1,93 (м, 2H), 1,37 (с, 6H);
MS: [M+H]+= 330,2
Пример синтеза 7: Получение соединения примера 317:
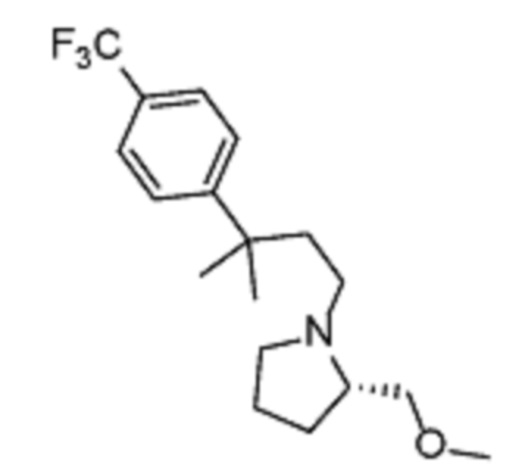
Пример 317

К раствору соединения S7–9 (5,46 г, 23,7 ммоль, 1,0 экв.) в MeOH (100 мл) добавляли соединение S7–14 (3,0 г, 26 ммоль, 1,1 экв.) и 2 капли AcOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли NaBH4 (3,58 г, 94,63 ммоль, 4,0 экв.) и затем реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили H2O (30 мл), фильтровали, экстрагировали EA (30 мл × 3) и концентрировали с получением остатка, который очищали колоночной хроматографией (PE:EA=1:1) с получением примера 317 (2,1 г, 27%).
ТСХ: DCM:EA:MeOH=1:1:0,1
Rf (соединение S7–9) = 0,9
Rf (пример 317) = 0,3
Общая процедура получения примера 317 HCl
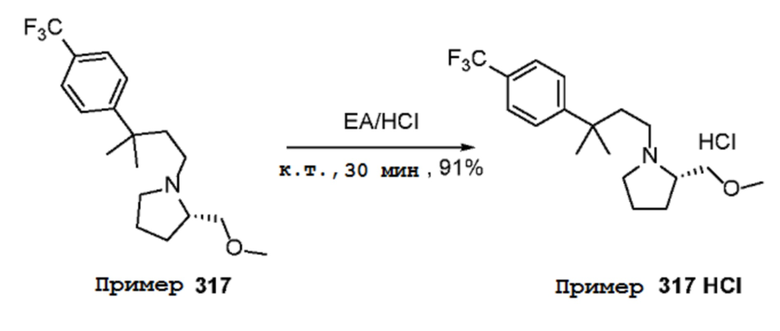
К раствору примера 317 (2,1 г, 6,37 ммоль, 1,0 экв.) в EA (10 мл) добавляли EA/HCl (2,5 M, 6 мл, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали с получением примера 317 HCl (2,1 г, 91%). 1H ЯМР (400 МГц, CDCl3) δ 12,17 (м, 1H), 7,61–7,59 (м, 2H), 7,50–7,48 (м, 2H), 4,16–4,13 (м, 1H), 3,90–3,86 (м, 1H), 3,55–3,53 (м, 1H), 3,33 (м, 1H), 3,23 (с, 3H), 2,63–2,59 (м, 2H), 2,51 (м, 1H), 2,23 (м, 1H), 2,19–1,90 (м, 4H), 1,39–1,37 (м, 6H);
MS: [M+H]+= 330,6
Пример синтеза 8: Получение соединения примера 306:
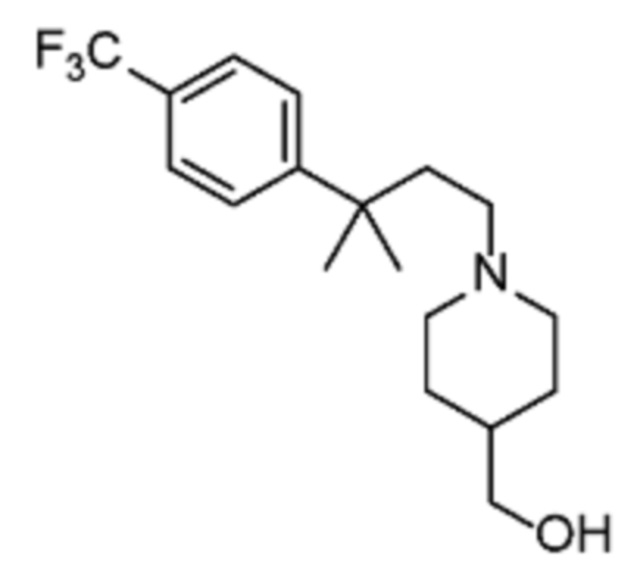
Пример 306

К раствору соединения S8–9 (3,08 г, 13,3 ммоль, 1,0 экв.) в MeOH (80 мл) добавляли соединение S8–16 (2,0 г, 17,36 ммоль, 1,1 экв.) и 2 капли AcOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли NaBH4 (2,02 г, 53,4 ммоль, 4,0 экв.) и затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакцию гасили водой (30 мл), фильтровали, экстрагировали EA (30 мл × 3) и концентрировали с получением остатка, который очищали колоночной хроматографией (PE:EA=1:1) с получением примера 306 (1,2 г, 27%).
ТСХ: DCM:EA:MeOH=1:1:0,1
Rf (соединение S8–9) = 0,9
Rf (пример 306) = 0,3
Общая процедура получения примера 306 HCl
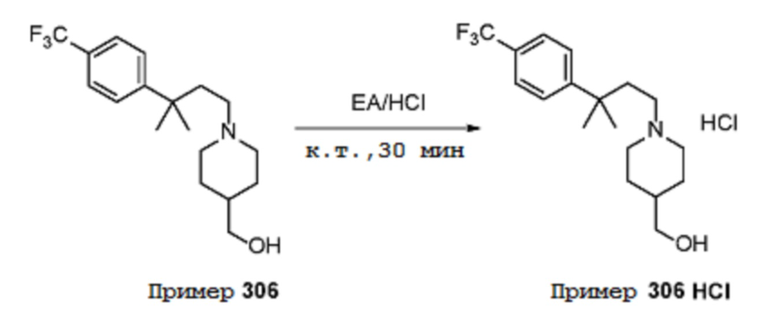
К раствору примера 306 (2,0 г, 26,07 ммоль, 1,0 экв.) в EA (5 мл) добавляли EA/HCl (2,5 M, 5 мл, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали с получением примера 306 HCl (2,28 г, 100%). 1H ЯМР (400 МГц, CDCl3) δ 12,75 (м, 1H), 7,60–7,58 (м, 2H), 7,49–7,47 (м, 2H), 3,53–3,51 (м, 2H), 2,63–2,60 (м, 2H), 2,53–2,50 (м, 2H), 2,34–2,30 (м, 2H), 2,05 (м, 4H), 2,00 (м, 1H), 1,97–1,88 (м, 2H), 1,38 (с, 6H);
MS: [M+H]+= 330,3.
Группа изобретений относится к фармацевтической химии, а именно к соединению формулы (I) и конкретным соединениям, указанным в формуле изобретения, их фармакологически приемлемым солям, фармацевтическим композициям, применениям и способам ингибирования и лечения когнитивных нарушений и болезни Альцгеймера. В формуле (I) Ra, Rb, Rc, Rd и Re независимо выбран из группы, состоящей из Н и CF3; R1 выбран из группы, состоящей из водорода или –CH=C(CH3)2; и R2 и его заместители выбран из группы заместителей, приведённых в формуле изобретения. Технический результат: полученные соединения обладают способностью ингибировать бета-амилоидный эффект в отношении нейрона. 9 н. и 8 з.п. ф-лы, 1 табл., 342 пр.

1. Соединение формулы I или его фармацевтически приемлемая соль или стереоизомер:
 I
I
где:
каждый из Ra, Rb, Rc, Rd и Re независимо выбран из группы, состоящей из Н и CF3;
R1 выбран из группы, состоящей из водорода или –CH=C(CH3)2; и
R2 выбран из группы, состоящей из
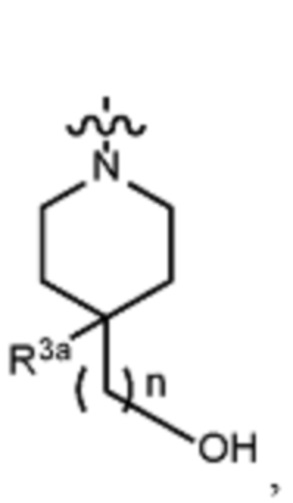

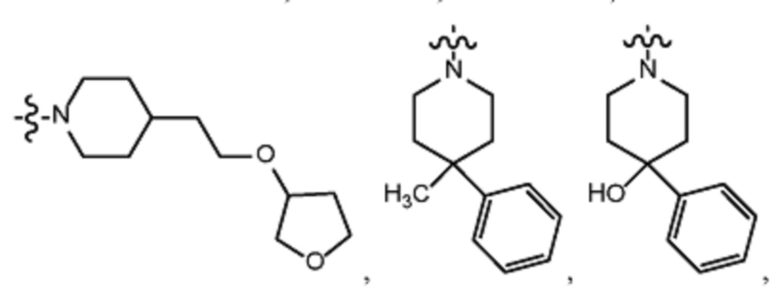
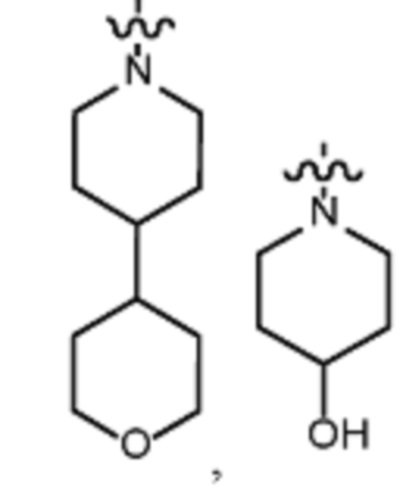 ,
,  , и
, и 
где R3a представляет собой водород, и n равно 1.
2. Соединение по п.1, выбранное из групп, состоящих из:
 ,
, ,
,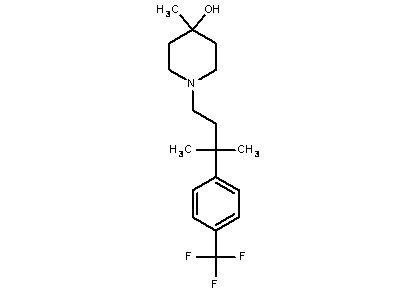 ,
, ,
, ,
, ,
,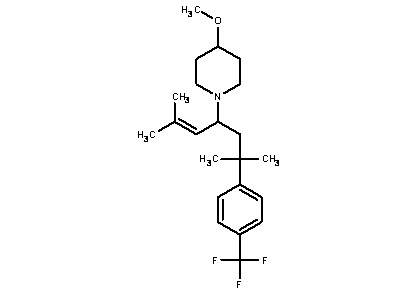 ,
,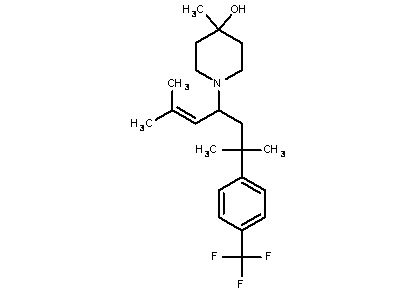 ,
, ,
, ,
, ,
,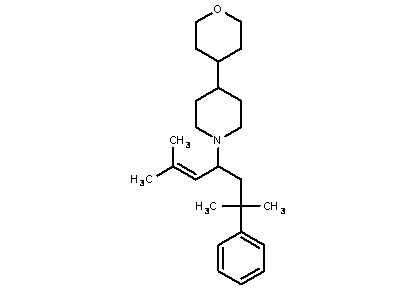 ,
,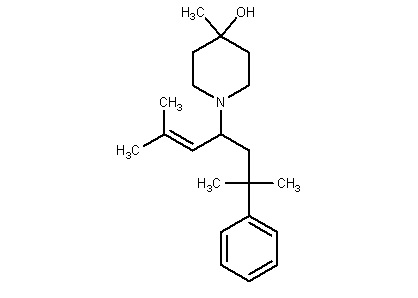 ,
, ,
, ,
, ,
, ,
, ,
,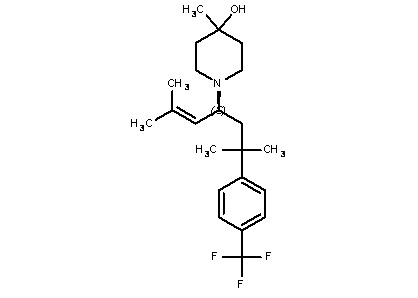 ,
, ,
, ,
, ,
, ,
, ,
, ,
,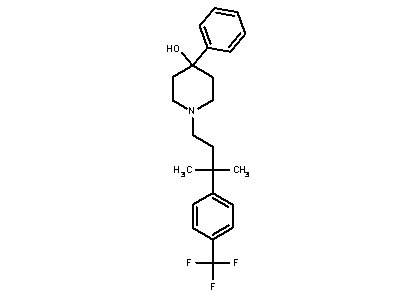 ,
, ,
, ,
, ,
,
 ;
;
или его фармацевтически приемлемая соль.
3. Соединение по п.1, выбранное из группы, состоящей из

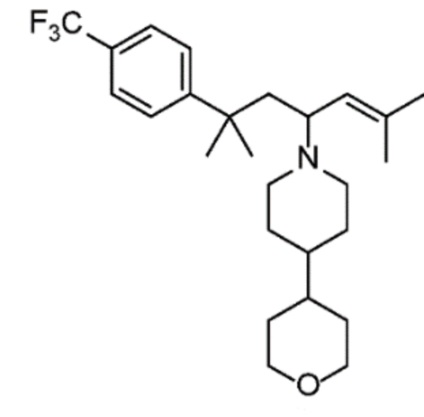 и
и  ;
;
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, обладающая способностью ингибировать бета–амилоидный эффект в отношении нейрона, содержащая терапевтически эффективное количество соединения по любому из пп.1–3 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
5. Фармацевтическая композиция по п.4, отличающаяся тем, что соединение выбрано из группы, состоящей из

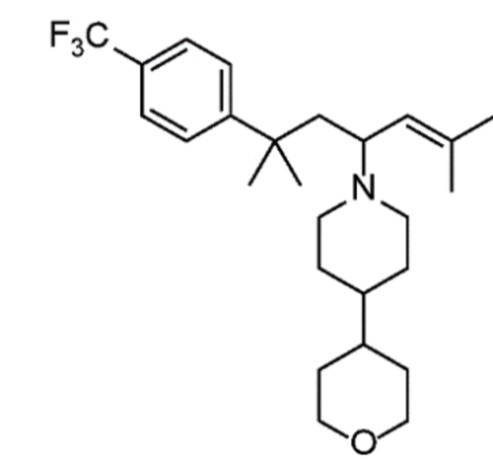 и
и  ;
;
или его фармацевтически приемлемой соли.
6. Способ лечения болезни Альцгеймера, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции по п.4 или 5.
7. Способ ингибирования снижения когнитивных функций у субъекта, демонстрирующего снижение когнитивных функций или подверженного риску снижения когнитивных функций, включающий введение терапевтически эффективного количества фармацевтической композиции по п.4 или 5.
8. Способ ингибирования бета–амилоидного эффекта в отношении нейрона, включающий введение субъекту, нуждающемуся в таком ингибировании, терапевтически эффективного количества фармацевтической композиции по п.4 или 5.
9. Способ лечения умеренного когнитивного нарушения при болезни Альцгеймера у субъекта, нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества фармацевтической композиции по п.4 или 5.
10. Применение соединения по любому из пп.1–3 в производстве лекарственного средства для лечения болезни Альцгеймера.
11. Применение по п.10, отличающееся тем, что соединение выбрано из группы, состоящей из

 и
и  ;
;
или его фармацевтически приемлемой соли.
12. Соединение по любому из пп.1–3 для лечения болезни Альцгеймера.
13. Соединение по п.12, выбранное из группы, состоящей из
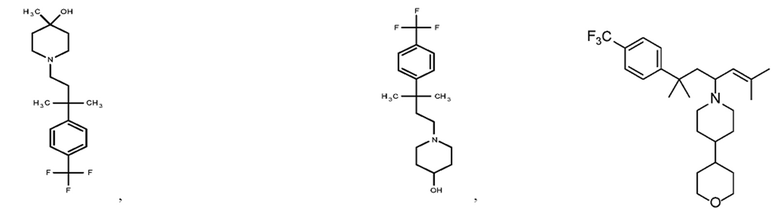 и
и  ;
;
или его фармацевтически приемлемая соль.
14. Соединение по любому из пп.1–3 для лекарственной терапии.
15. Соединение по п.14, выбранное из группы, состоящей из
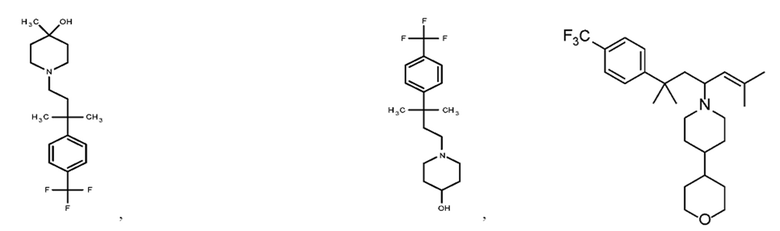 и
и  ;
;
или его фармацевтически приемлемая соль.
16. Фармацевтическая композиция для лечения болезни Альцгеймера, содержащая терапевтически эффективное количество соединения по любому из пп.1–3 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
17. Применение соединения по любому из пп.1–3 для лечения болезни Альцгеймера.
| КРИСТАЛЛОГЕНЕРАТОР НЕПРЕРЫВНОГО ДЕЙСТВИЯ | 2002 |
|
RU2215789C1 |
| DATABASE CAS on STN Online MARTIROSYAN, G.T.; KAZARYAN, A.TS.; ASATRYAN, G.G.; BABAYAN, A.T.: "Synthesis and reactions of.beta.gamma.-unsaturated amines | |||
| X.Condensation of.beta.gamma.-unsaturated amines with aromatichydrocarbons in the presence of aluminum chloride", ARMYANSKII KHIMICHESKII ZHURNAL, vol | |||
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |

