Настоящее изобретение относится к способу гидролиза эфиров хинолонкарбоновых кислот до хинолонкарбоновых кислот. Фторхинолонкарбоновые кислоты являются важными промежуточными продуктами для получения известных фармацевтически активных соединений из класса, состоящего из хинолонов. Конкретные примеры включают в себя: бенофлоксацин, бинфлоксацин, циноксацин, ципрофлоксацин, данофлоксацин, дифлоксацин, эноксацин, энрофлоксацин, флероксацин, ибафлоксацин, левофлоксацин, ломефлоксацин, марбофлоксацин, моксифлоксацин, норфлоксацин, офлоксацин, орбифлоксацин, пефлоксацин, пипемидовую кислоту темафлоксацин, тосуфлоксацин, сарафлоксацин, спарфлоксацин и прадофлоксацин.
Прадофлоксацин представляет собой высокоэффективный хинолоновый антибиотик в ветеринарии. Его антибактериальное действие и показания, формы применения и подходящие препараты приведены, например, в WO 97/31001 A1, WO 03/007995 A1, WO 03/101422 A1, WO 04/082658 A1, WO 05/018641 A1, WO 05/044271 А1 и WO 06/061156 А1.
Одна стадия в многоступенчатом синтезе прадофлоксацина представляет собой гидролиз этилхинолонкарбоксилата. Для гидролиза эфиров хинолонкарбоновых кислот, значение рН может быть понижено соляной кислотой или серной кислотой/уксусной кислотой. Недостаток способа с использованием соляной кислоты состоит в том, что реакционная смесь является очень коррозионной, и оборудование, предназначенное для проведения реакции, соответственно должно быть устойчивым к коррозии. Следствием этого являются высокие затраты. Кроме того, маточные растворы перед утилизацией необходимо нейтрализовать, что требует определенных затрат, так как возникает большое количество отходов, а процесс требует сравнительно большого количества стадий.
Способ с использованием серной кислоты/уксусной кислоты имеет то преимущество, что использованная уксусная кислота может быть регенерирована путем дистилляции, и что можно использовать менее коррозионные среды.
WO 98/26779 А1 раскрывает, в примере Z 22 описания, синтез 7-хлор-8-циано-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты. Для этого 3.8 г (0.1 моль) этил 7-хлор-8-циано-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоксилата нагревали с обратным холодильником в смеси 100 мл уксусной кислоты, 20 мл воды и 10 мл концентрированной серной кислоты в течение 3 часов. После охлаждения, смесь выливали в 100 мл ледяной воды, осажденный осадок отфильтровывали с отсасыванием, промывали водой и этанолом и сушили при 60°С в вакууме. В перерасчете на моль сложного эфира использовали 17.3 моль уксусной кислоты, 1.86 моль серной кислоты и 11 моль воды.
ЕР 0276700 А1 раскрывает в примере 1 описания гидролиз этил 7-хлор-8-циано-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоксилата. 1 г этого соединения нагревали вместе с 3,5 мл уксусной кислоты, 3 мл воды и 0,3 мл серной кислоты до 140-145°С в течение 4 часов. Затем смесь разбавляли водой и выделяли твердое вещество. Получали 0,7 г свободной карбоновой кислоты с температурой плавления 281-282°С. В перерасчете на моль сложного эфира использовали 20 моль уксусной кислоты, 1,89 моль серной кислоты и 55,8 моль воды.
ЕР 0169993 А2 раскрывает в примере А описания, что смесь 94 г этил-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоксилата, 600 мл ледяной уксусной кислота, 450 мл воды и 70 мл концентрированной серной кислоты нагревали с обратным холодильником в течение 1,5 часов. Горячую суспензию выливали на лед, осадок отфильтровывали с отсасыванием, промывали водой и сушили в вакууме при 100°С. Таким образом было получено 88,9 г 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты. В перерасчете на моль сложного эфира использовали 34,7 моль уксусной кислоты, 4,35 моль серной кислоты и 82,8 моль воды.
ЕР 1319656 А1 раскрывает в примере 2 реакцию 29,4 г этил 1-циклопропил-7-хлор-6-фтор-8-метокси-1,4-дигидро-4-оксо-3-хинолинкарбоксилата (0,088 моль), 160 мл уксусной кислоты, 100 мл воды и 18 мл концентрированной серной кислоты. Смесь перемешивали при 100-110°С в течение 40 минут. Полученную смесь охлаждали и фильтровали. Осадок перекристаллизовывали из хлороформ-этанола. Это давало 23,8 г свободной карбоновой кислоты. В перерасчете на моль сложного эфира использовали 31,8 моль уксусной кислоты, 3,84 моль серной кислоты и 61,1 моль воды.
ЕР 1236718 А1 раскрывает в примере 1, что сначала загружали 300 г этил 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоксилата, 106,8 г воды и 426 г уксусной кислоты и добавляли 3,8 г серной кислоты. Смесь нагревали с обратным холодильником в течение 3 часов. Затем 310 мл дистиллята отгоняли до достижения нижней температуры 109°С. Смесь охлаждали до 80°С и по каплям добавляли 157,5 г 4,8 масс. % раствора ацетата натрия. Затем значение рН устанавливали в диапазоне от 3 до 4. Затем смесь охлаждали до 20°С и твердое вещество отфильтровывали с отсасыванием. Твердое вещество промывали 200 мл воды и сушили при пониженном давлении при 50°С. Выделяли 270,3 г 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты, что соответствует выходу 99% от теоретического. В перерасчете на моль сложного эфира использовали 7,4 моль уксусной кислоты, 0,04 моль серной кислоты и 6,2 моль воды.
Задачей настоящего изобретения было обеспечить улучшенный способ гидролиза эфиров хинолонкарбоновых кислот, в котором образование побочных продуктов, подлежащих переработке, является как можно меньшим, и в котором полученная в результате хинолонкарбоновая кислота имеет максимально высокую возможную чистоту.
Задачу решают в соответствии с изобретением с помощью способа гидролиза эфиров хинолонкарбоновых кислот общей формулы (II) с получением хинолонкарбоновых кислот общей формулы (I):
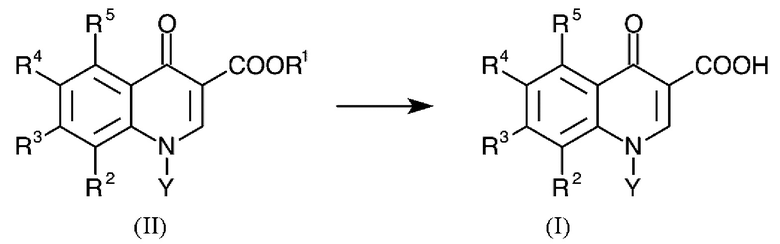
где в формуле (II) R1 представляет собой C1-C4-алкил и
соответственно в формулах (I) и (II):
R2 представляет собой водород, C1-C4-алкил, С1-С4-алкокси, галоген, нитро или циано,
R3 и R4 каждый представляет собой галоген,
R5 представляет собой водород, C1-C4-алкил, галоген или нитро, и
Y представляет собой C1-С6-алкил, циклопропил или фенил, каждый из которых может необязательно быть замещен галогеном,
где R2 и Y вместе могут также представлять собой -СН2-СН2-О- или -СН(СН3)-СН2-О- мостик, присоединенный к атому азота с помощью атома углерода, и
где по меньшей мере один из радикалов R2-R5 представляет собой фтор,
который включает стадию:
А) введения в реакцию соединений формулы (II) со смесью, которая включает уксусную кислоту серную кислоту и воду.
На стадии А), ≥30-≤40 моль уксусной кислоты, ≥0.3-≤1 моль серной кислоты и ≥0.9-≤2.5 моль воды используют в перерасчете на моль соединений формулы (II). Предпочтительным является использование ≥32-≤38 моль уксусной кислоты, ≥0.4-≤0.8 моль серной кислоты и ≥0.9-≤2.3 моль воды, более предпочтительно ≥33-≤35 моль уксусной кислоты, ≥0.4-≤0.6 моль серной кислоты и ≥0.9-≤2.2 моль воды в перерасчете на моль соединений формулы (II).
В способе в соответствии с изобретением уксусная кислота и серная кислота могут использоваться в водосодержащей или безводной форме. Описанные количественные данные относятся к 100% уксусной кислоте и 100% серной кислоте. Если используется уксусная кислота, содержащая воду, и/или серная кислота, содержащая воду, необходимо использовать меньше воды в зависимости от содержания в них воды. Уксусную кислоту предпочтительно используют в виде ледяной уксусной кислоты, серную кислоту предпочтительно используют в виде 96 - 100% серной кислоты.
Добавление воды, уксусной кислоты и серной кислоты предпочтительно осуществляют таким образом, что сначала загружают сложный эфир (II), уксусную кислоту и серную кислоту и затем добавляют воду. Также можно сначала загрузить сложный эфир (II), воду и уксусную кислоту, а затем добавить серную кислоту. Реакционную смесь предпочтительно нагревают в течение 10-25 часов, более предпочтительно в течение 12-22 часов, особенно предпочтительно в течение 16-20 часов.
На стадии А) предпочтительно не используются другие химически реактивные или каталитически активные соединения, кроме сложного эфира (II), уксусной кислоты, воды и серной кислоты.
Ниже описаны предпочтительные варианты осуществления способа согласно изобретению. Они могут быть скомбинированы друг с другом по желанию, если из контекста не очевидно противоположное.
В варианте осуществления способа на стадии А), ≥95 мол. % используемых соединений формулы (II) превращают в соединения формулы (I). Этот выход составляет предпочтительно ≥96 мол. % и более предпочтительно ≥99.5 мол. %.
В еще одном варианте осуществления способа, реакцию на стадии А) проводят при температуре от ≥90°С до ≤99°С. Эта температура предпочтительно находится в диапазоне от ≥92 до ≤97°С, более предпочтительно от ≥94 до ≤95°С. Было обнаружено, что более высокие температуры реакции на этой стадии приводят к повышенному содержанию примесей (см. аналитические данные, приведенные ниже для примеров и сравнительных примеров).
Нагревание реакционной смеси можно проводить при пониженном, атмосферном или повышенном давлении. Например, возможно давление в диапазоне от 0,5 до 3 бар. Если не указано иначе, все описанные в данном документе стадии способа обычно осуществляют при атмосферном давлении.
Одним из преимуществ способа согласно изобретению является то, что не требуется дистилляции, и что продукт можно выделить непосредственно из реакционной смеси с помощью фильтрации. Это более экономически выгодно по сравнению со способами со стадией дистилляции.
Образовавшаяся хинолонкарбоновая кислота может быть выделена из смеси, например, таким образом, чтобы присутствующий осадок затем фильтровали при отсасывании, промывали и сушили. Этанол предпочтительно используют для промывания осадка, в частности предпочтительно осадок сначала промывают уксусной кислотой и затем этанолом. Можно избежать промывания водой, в результате чего собранная уксусная кислота также может быть легче регенерирована. Выделенный продукт целесообразно многократно промыть, чтобы получить его достаточно свободным и практически без следов серной кислоты. Добавление основания на этом этапе в способе согласно изобретению не требуется. Это опять же экономит отходы и затраты.
В еще одном варианте осуществления способа, соответственно в формулах (I) и (II) принято, что:
R2 представляет собой водород, метил, метокси, фтор, хлор, нитро или циано,
R3 представляет собой фтор или хлор,
R4 представляет собой фтор,
R5 представляет собой водород, метил, фтор, хлор или нитро,
Y представляет собой метил, этил, изопропил, циклопропил, фторциклопропил, 4-фторфенил или 2,4-дифторфенил
и в формуле (I) R1 представляет собой метил или этил.
Также возможно, что соответственно в формулах (I) и (II):
R2 представляет собой водород, С1-С4-алкокси или циано,
R3 представляет собой галоген, в особенности хлор,
R4 представляет собой фтор,
R5 представляет собой водород,
Y представляет собой циклопропил,
и в формуле (I) R1 представляет собой метил или этил.
В еще одном варианте осуществления способа, формула (I) представляет собой 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-6,7-дифтор-8-циано-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-(2-фтор)циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-8-хлор-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-этил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту или 1-циклопропил-6-фтор-7-хлор-8-циано-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
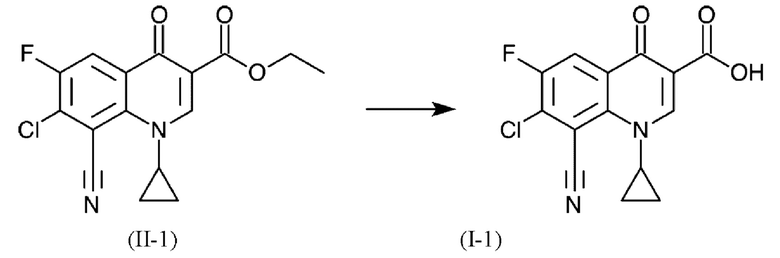
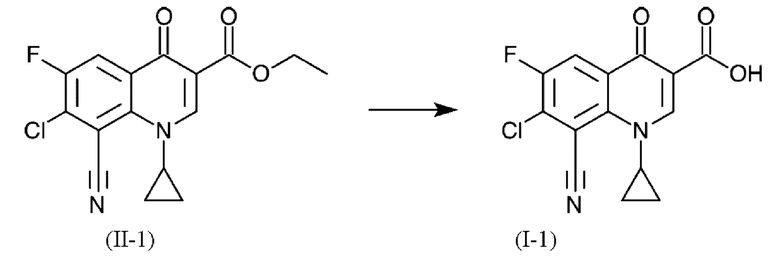
В еще одном варианте осуществления способа, формулы (II) и (I) имеют следующее определения в соответствии с формулами (11-1) и (I-1) соответственно:
В еще одном варианте осуществления способа, соединения формулы (II) получают посредством введения в реакцию соединений общей формулы (III):
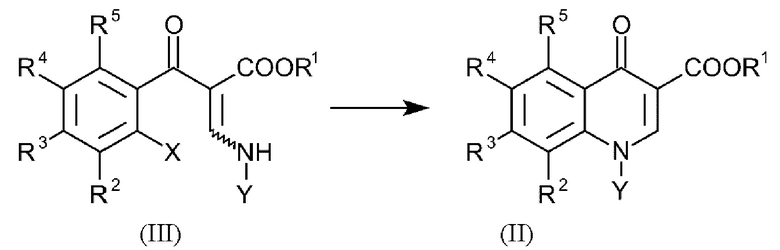
где R1-R5 и Y имеют вышеуказанные определения и X представляет собой галоген.
В еще одном варианте осуществления способа, соединения формулы (III) получают посредством введения в реакцию соединений общей формулы (IV):
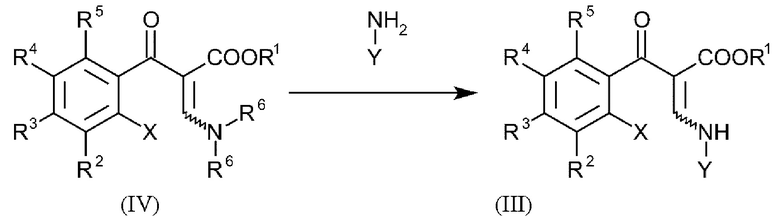
и где R1-R5, X и Y имеют вышеуказанные определения и R6 представляет собой C1-C4-алкил.
В еще одном варианте осуществления способа, соединения формулы (IV) получают посредством введения в реакцию соединений общей формулы (V):
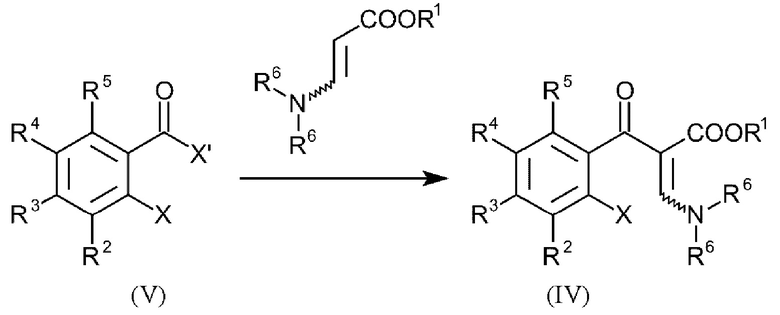
и где R1-R6, X и Y имеют вышеуказанные определения и X' представляет собой галоген.
Для предпочтительного варианта получения промежуточного соединения прадофлоксацина (I) следует осуществлять последовательность реакций:
(V)→(IV)→(III)→(II)→(I)
и во всех соответствующих соединениях этих общих формул R1 представляет собой этил, R2 представляет собой циано, R3 представляет собой хлор, R4 представляет собой фтор, R5 представляет собой водород, R6 представляет собой метил, X представляет собой хлор, X' представляет собой хлор и Y представляет собой циклопропил.
Примеры
Настоящее изобретение подробно поясняется примерами, которые приведены ниже, но не ограничивается ими. Схема синтеза показана на Фиг. 1.
Пример 1
Синтез этил 7-хлор-8-циано-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат (формула (II-2) на Фиг. 1)
К 11.8 кг толуола добавляли 2.70 кг (18.8 моль) этил (2Е)-3-(диметиламино)акрилата (бета-диметиламиноакрилат, β-DAASE) и 2.12 кг (20.9 моль) триэтиламина, нагретого при Твн.=45°С-55°С. Затем раствор 4.50 кг (17.8 моль) 2,4-дихлор-3-циано-5-фторбензоилхлорида (формула (V-1) на Фиг. 1) в 11.6 кг толуола отмеривали при Твн.=50°С. Реакционную смесь перемешивали при Твн.=50°С в течение 3 часов и охлаждали до Твн.=22°С. Суспензию фильтровали и осадок на фильтре промывали с помощью 3.4 кг толуола. К фильтрату (формула (IV-1) на Фиг. 1) отмеривали 1.23 кг (20.5 моль) уксусной кислоты и раствор 1.11 кг (19.4 моль) циклопропиламина в 2.40 кг толуола в течение 2 часов при Твн.=5-15°С и смесь затем перемешивали в течение 5 часов при Твн.=10°С. Затем добавляли 13.5 кг воды, смесь нагревали при Твн.=40°С и перемешивание продолжали при этой температуре в течение 30 минут. Затем фазы разделали при Твн.=40°С. Раствор 300 г карбоната натрия в 6.0 кг воды добавляли к органической фазе, смесь нагревали до Твн.=40°С, дополнительно перемешивали в течение 30 минут и фазы разделяли при Твн.=40°С.
Жидкость в количестве 22,6 л дистиллировали из органической фазы при пониженном давлении до температуры рубашки 60°С. Затем добавляли 19,2 кг N,N-диметилформамида, и смесь перемешивали в течение по меньшей мере 10 минут при 40°С. Затем смесь снова дистиллировали при пониженном давлении до температуры рубашки Tm=60°С до тех пор, пока дистиллят больше не перестанет образовываться, и остаток (формула (III-1) на Фиг. 1) охлаждали до комнатной температуры.
К остатку добавляли 2,22 кг (16,0 моль) поташа, суспензию нагревали до Твн.=55°С, и смесь перемешивали при этой температуре в течение 5 часов. Смесь охлаждали до Твн.=22°С и 16,5 л дистиллята дистиллировали при пониженном давлении до температуры рубашки 80°С. К остатку добавляли 18,0 кг воды и смесь затем перемешивали при Твн.=55°С в течение по меньшей мере 10 минут. Затем смесь охлаждали в течение 2 часов до Твн.=5°С и дополнительно перемешивали при этой температуре в течение 2 часов.
Полученный продукт отфильтровывали, дважды промывали 6,0 кг воды каждый раз и перемешивали с 9,9 кг этилацетата в течение по меньшей мере 3 часов при Твн.=22°С. Суспензию фильтровали и осадок на фильтре дважды промывали 4,8 кг этилацетата каждый раз и неочищенный продукт (формула (II-1) на Фиг. 1) сушили при 50°С при пониженном давлении в течение по меньшей мере 12 часов.
Выход: 5.08 кг; 85.1% от теоретического в перерасчете на 2,4-дихлор-3-циано-5-фторбензоилхлорид.
2,96 кг сырого продукта нагревали в 29,4 кг N,N-диметилформамида при 60°С и нерастворимые примеси отфильтровывали при этой температуре. 3.8 кг воды добавляли к фильтрату который затем перемешивали в течение 1,5 часов, и затем в течение 1,5 часов было отмерено 13,9 кг воды. Полученную суспензию охлаждали до Твн.=22°С, перемешивали 30 минут и твердое вещество отфильтровывали. Осадок на нутчфильтре промывали 3,1 кг воды, затем дважды по 2,8 кг этанола и сушили при 50°С при пониженном давлении в течение по меньшей мере 12 часов (формула (II-2) на Фиг. 1). Описанный в данном случае способ имеет преимущество, что сложный эфир (II-2) может быть получен с высокой чистотой. Это позволяет получать предпочтительные хинолонкарбоновые кислоты высокой чистоты. (Сложный эфир формулы (II-2) получают на стадии очистки из сложного эфира (II-1). Таким образом, обе формулы описывают одну и ту же химическую структуру. Различные функциональные формулы предназначены только для иллюстрации разной степени чистоты.)
Выход: 2.87 кг; прибл. 97% от теоретического в перерасчете на сырой продукт.
Пример 2
Гидролиз продукта Примера 1
К 20.0 г (59.8 ммоль) продукта из Примера 1 (формула (II-2) на Фиг. 1) добавляли 122.8 г (2.04 моль) уксусной кислоты, 3.37 г (33.0 ммоль) серной кислоты и 1.1 г (63.0 ммоль) воды. Смесь нагревали до 95°С и перемешивали при этой температуре в течение 18.5 ч. Суспензию охлаждали до 10°С, твердое вещество отфильтровывали с отсасыванием, промывали 48 мл уксусной кислоты и затем 48 мл этанола и сушили в течение ночи при 60°С в вакуумном сушильном шкафу.
В перерасчете на моль соединения (II-2) использовали: 34.11 моль уксусной кислоты, 0.55 моль серной кислоты и 1.05 моль воды.
Выход: 17.6 г; (96.1% от теоретического)
Данные о чистоте см. в таблице ниже.
Пример 3
Гидролиз продукта Примера 1
К 20.0 г (59.8 ммоль) продукта из Примера 1 (формула (II-2) на Фиг. 1) добавляли 122.8 г (2.04 моль) уксусной кислоты, 3.37 г (33.0 ммоль) серной кислоты и 2.3 г (126.0 ммоль) воды. Смесь нагревали до 95°С и перемешивали при этой температуре в течение 18.5 ч. Суспензию охлаждали до 10°С, твердое вещество отфильтровывали с отсасыванием, промывали 48 мл уксусной кислоты и затем 48 мл этанола и сушили в течение ночи при 60°С в вакуумном сушильном шкафу.
В перерасчете на моль соединения (II-2) использовали: 34.11 моль уксусной кислоты, 0.55 моль серной кислоты и 2.10 моль воды.
Выход: 17.8 г; (97.1% от теоретического)
Данные о чистоте см. в таблице ниже.
Сравнительный Пример 1
В соответствии с методикой из Примера 1 документа ЕР 1236718
К 30.0 г (89.6 ммоль) продукта из Примера 1 (формула (II-2) на Фиг. 1) добавляли 39.6 г (656.1 ммоль) уксусной кислоты, 0.20 мл (3.5 ммоль) серной кислоты и 9.9 г (549.5 ммоль) воды. Смесь нагревали с обратным холодильником в течение 3 ч. Затем 14.1 г дистиллята дистиллировали до достижения нижней температуры 109°С. Смесь охлаждали до 80°С и по каплям добавляли 40.1 г 4.8 масс. % раствора ацетата натрия. Затем значение рН устанавливали в диапазоне от 3 до 4. Смесь затем охлаждали до 20°С и твердое вещество отфильтровывали с отсасыванием. Твердое вещество промывали 50 мл воды и сушили при пониженном давлении при 50°С.
В перерасчете на моль соединения (II-2) использовали: 7.32 моль уксусной кислоты, 0.04 моль серной кислоты и 6.13 моль воды.
Выход: 26.7 г; (97.2% от теоретического)
Данные о чистоте см. в таблице ниже.
Сравнительный Пример 2
В соответствии с методикой из Примера 2 документа ЕР 1236718
К 30.0 г (89.6 ммоль) продукта из Примера 1 (формула (II-2) на Фиг. 1) добавляли 78.9 г (1.31 моль) уксусной кислоты, 0.51 мл (9.1 ммоль) серной кислоты и 2.25 г (124.9 ммоль) воды. Смесь нагревали с обратным холодильником в течение 4 ч. Затем 8.1 г дистиллята дистиллировали до достижения нижней температуры 109°С. Смесь охлаждали до 80°С и по каплям добавляли 75.3 г 4.8 масс. % раствора ацетата натрия. Затем значение рН устанавливали в диапазоне от 3 до 4. Смесь затем охлаждали до 20°С и твердое вещество отфильтровывали с отсасыванием. Твердое вещество промывали 50 мл воды и сушили при пониженном давлении при 50°С.
В перерасчете на моль соединения (II-2) использовали: 14.62 моль уксусной кислоты, 0.10 моль серной кислоты и 1.39 моль воды.
Выход: 27.0 г; (98.2% от теоретического)
Данные о чистоте см. в таблице ниже.
Сравнительный Пример 3
В соответствии с методикой из Примера Z 22 документа WO 98/26779
К 9.5 г (28.4 ммоль) продукта из Примера 1 (формула (II-2) на Фиг. 1) добавляли 29.5 г (490.9 ммоль) уксусной кислоты, 5.2 г (52.5 ммоль) серной кислоты и 5.6 г (310.2 ммоль) воды. Смесь нагревали с обратным холодильником в течение 3 ч и охлаждали до 20°С. Смесь затем добавляли к 28 г ледяной воды и твердое вещество отфильтровывали с отсасыванием. Твердое вещество промывали 100 мл воды и 10 мл этанола и сушили при пониженном давлении при 60°С.
В перерасчете на моль соединения (II-2) использовали: 17.28 моль уксусной кислоты, 1.85 моль серной кислоты и 10.92 моль воды.
Выход: 8.5 г; (97.7% от теоретического)
В следующей таблице приведены степени чистоты продуктов гидролиза формулы I-1, полученных в примерах и сравнительных примерах.
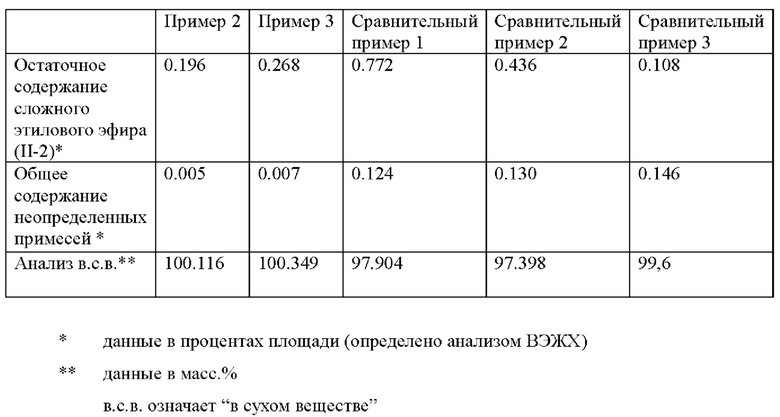
Очевидно, что в способе в соответствии с изобретением продукты получают с большей чистотой, чем в сравнительных способах, где не наблюдаются требуемые соотношения реагента (II-2), уксусной кислоты, серной кислоты и воды.
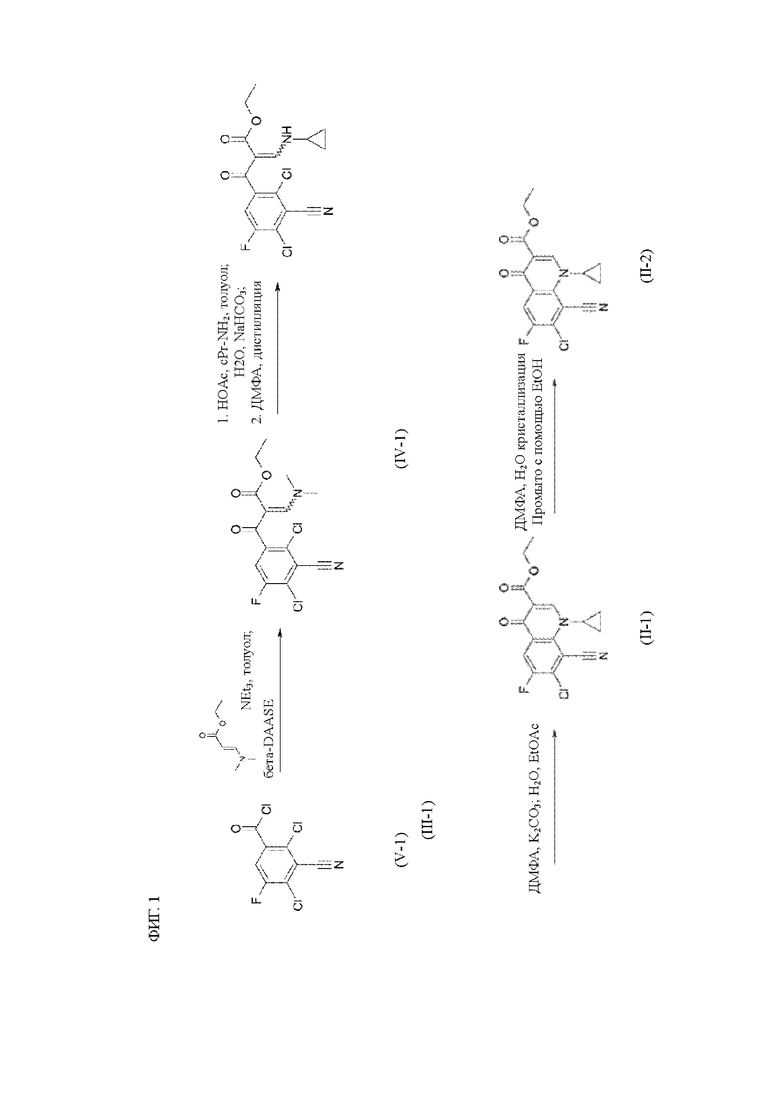
Изобретение относится к способу гидролиза эфиров хинолонкарбоновых кислот общей формулы (II) с получением хинолонкарбоновых кислот общей формулы (I):
где соответственно в формулах (I) и (II): R2 представляет собой водород, метил, метокси, фтор, хлор, нитро или циано, R3 представляет собой фтор или хлор, R4 представляет собой фтор, R5 представляет собой водород, метил, фтор, хлор или нитро и Υ представляет собой метил, этил, изопропил, циклопропил, фторциклопропил, 4-фторфенил или 2,4-дифторфенил, и в формуле (I) R1 представляет собой метил или этил, который включает стадию: А) введение в реакцию соединений формулы (II) со смесью, которая содержит уксусную кислоту, серную кислоту и воду, который отличается тем, что на стадии А) от 30 до 40 моль уксусной кислоты, от 0.3 до 1 моль серной кислоты и от 0.9 до 2.5 моль воды используют в перерасчете на моль соединений формулы (II). Технический результат: разработан улучшенный способ гидролиза эфиров хинолонкарбоновых кислот общей формулы (II) с получением хинолонкарбоновых кислот общей формулы (I), в котором образование побочных продуктов, подлежащих переработке, является как можно меньшим и в котором полученная в результате хинолонкарбоновая кислота имеет максимально высокую возможную чистоту. 7 з.п. ф-лы, 1 ил.
1. Способ гидролиза эфиров хинолонкарбоновых кислот общей формулы (II) с получением хинолонкарбоновых кислот общей формулы (I):
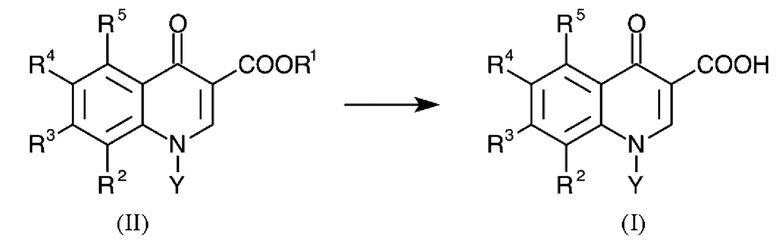 ,
,
где соответственно в формулах (I) и (II):
R2 представляет собой водород, метил, метокси, фтор, хлор, нитро или циано,
R3 представляет собой фтор или хлор,
R4 представляет собой фтор,
R5 представляет собой водород, метил, фтор, хлор или нитро, и
Υ представляет собой метил, этил, изопропил, циклопропил, фторциклопропил, 4-фторфенил или 2,4-дифторфенил,
и в формуле (I) R1 представляет собой метил или этил, который включает стадию:
А) введение в реакцию соединений формулы (II) со смесью, которая содержит уксусную кислоту, серную кислоту и воду,
который отличается тем, что
на стадии А) от 30 до 40 моль уксусной кислоты, от 0.3 до 1 моль серной кислоты и от 0.9 до 2.5 моль воды используют в перерасчете на моль соединений формулы (II).
2. Способ по п. 1, где на стадии А) ≥95 мол. % используемых соединений формулы (II) превращают в соединения формулы (I).
3. Способ по п. 1 или 2, где реакцию на стадии А) осуществляют при температуре ≥90-≤99°С.
4. Способ по любому из пп. 1-3, где формула (I) представляет собой 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-6,7-дифтор-8-циано-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-(2-фтор)циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-циклопропил-8-хлор-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту, 1-этил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту или 1-циклопропил-6-фтор-7-хлор-8-циано-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
5. Способ по любому из пп. 1-4, где формулы (II) и (I) имеют следующие определения в соответствии с формулами (II-1) и (I-1) соответственно:
6. Способ по любому из пп. 1-5, где соединения формулы (II) получают посредством введения в реакцию соединений общей формулы (III):
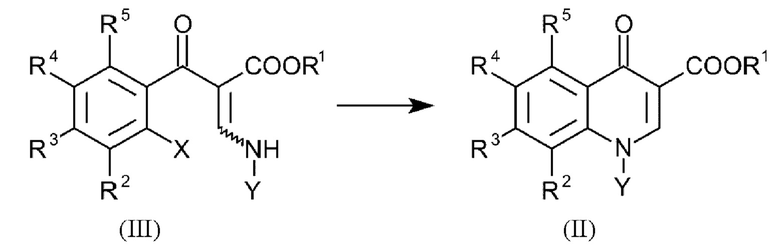
где R1-R5 и Υ имеют вышеуказанные определения и X представляет собой галоген.
7. Способ по п. 6, где соединения формулы (III) получают посредством введения в реакцию соединений общей формулы (IV):
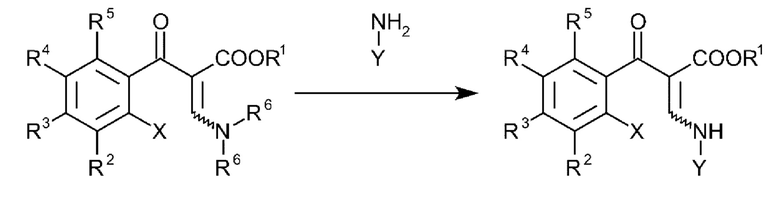

и где R1- R5, X и Υ имеют вышеуказанные определения и R6 представляет собой C1-С4-алкил.
8. Способ по п. 7, где соединения формулы (IV) получают посредством введения в реакцию соединений общей формулы (V):
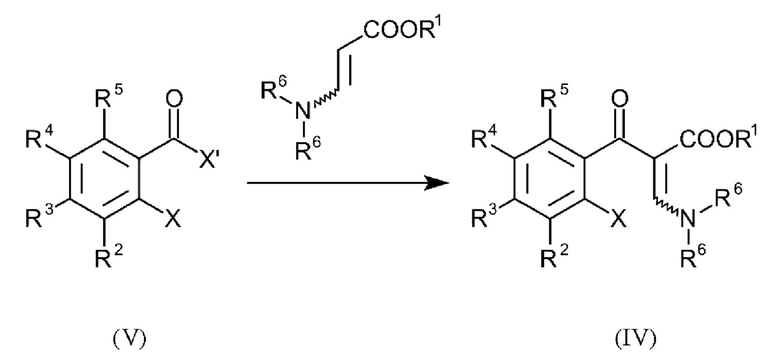
и где R1-R6 и X имеют вышеуказанные определения и X' представляет собой галоген.
| WO 9826779 A1, 25.06.1998 | |||
| EP 1319656 A1, 18.06.2003 | |||
| УСТРОЙСТВО для РАЗДЕЛЕНИЯ СВЯЗАННЫХ МЕЖДУ СОБОЙДЕТАЛЕЙ | 0 |
|
SU276700A1 |
| EP 1236718 A1, 04.09.2002 | |||
| RU 2005105337 A, 20.01.2006 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 1,8-НАФТИРИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2310654C1 |
