[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №62/592721, поданной 30 ноября 2017 г., содержание которой во всей своей полноте включено в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Низкомолекулярные противоопухолевые лекарственные средства часто обладают малой широтой терапевтического воздействия («терапевтическим окном»), что ограничивает их клиническую эффективность. Эти низкомолекулярные соединения проявляют высокую тенденцию к проникновению в ткани организма путем диффузии, что приводит к равномерному биораспределению. Как следствие, только небольшие количества лекарственного средства достигают необходимого места воздействия, а в результате проникновения в здоровые ткани организма, указанные лекарственные средства вызывают существенные побочные эффекты.
[0003] Эти недостатки особенно существенны для тех препаратов, которые обладают высокой цитотоксичностью и имеют очень малую широту терапевтического воздействия. Ауристатины являются лекарственными средствами на основе тубулин-связывающих пептидов, а типичные их представители, такие как доластатин 10, доластатин 15, ауристатин PE, ауристатин E или ауристатин F, проявляют значительные цитотоксические эффекты. В клинических исследованиях I и II фазы доластатин 10, доластатин 15 и ауристатин PE проявили неприемлемую системную токсичность, в то время как их противоопухолевая активность отсутствовала, поэтому дальнейшие исследования были прекращены (E.A. Perez et al., Invest. New Drugs, 23:257-261 (2005); M. von Mehren et al., Sarcoma, 8:107-111 (2004); RS Marks et al., Am. J. Clin. Oncol., 26:336-337 (2003)).
[0004] Доставка лекарственных средств в онкологии является подходом, который может потенциально увеличить низкий терапевтический индекс высоко цитотоксических агентов. В большинстве систем доставки лекарств цитотоксическое лекарственное средство связывается с носителем через спейсер, который включает заранее определенную точку разрыва, позволяющую связанному лекарственному средству высвобождаться в клеточном целевом сайте (F. Kratz et al., ChemMedChem, 3:20-53 (2008)).
[0005] Альбумин или его лекарственные конъюгаты обладают заметно длительным периодом полураспада в системной циркуляции до 19 дней. Особенно перспективным подходом является разработка пролекарств высоко цитотоксических агентов, которые ковалентно связываются с циркулирующим сывороточным альбумином, служащим при введении макромолекулярным носителем. По причине 1.) повышенной проницаемости макромолекул через стенки сосудов злокачественной, инфицированной или воспаленной ткани в сочетании с интактной лимфодренажной системой и 2.) экспрессии альбумин-связывающих белков на эндотелии опухоли и в интерстиции опухоли, конъюгаты альбумина с лекарственными средствами транспортируют терапевтически эффективное вещество в целевой участок (т.е. опухоль), где высвобождается высоко цитотоксический агент (US 7387771; F. Kratz, J. Control. Release, 132:171-183 (2008); F. Kratz, U. Beyer, Drug Delivery, 5:281-299 (1998)).
[0006] Однако при разработке систем доставки лекарств с высоко цитотоксическими агентами необходимо соблюдать критический баланс между сохранением нацеливающих свойств носителя лекарственного средства при одновременном обеспечении контролируемого высвобождения цитотоксического лекарственного средства и предотвращения его преждевременного высвобождения в кровотоке или системно. Кислоточувствительные системы доставки лекарственных препаратов должны обладать достаточной стабильностью в кровотоке и в то же время обеспечивать эффективное высвобождение лекарственного средства в месте локализации опухоли в зависимости от рН (F. Kratz et al., ChemMedChem, 3: 20-53 (2008)).
[0007] Для высоко цитотоксических лекарственных средств со значениями IC50 против опухолевых клеток в пикомолярном диапазоне, таких как класс ауристатинов на основе низкомолекулярных пептидов, которые нельзя вводить из-за их нерастворимости в воде и очень узкого терапевтического окна, существует необходимость в эффективных системах доставки и высвобождения лекарственных средств для достижения более эффективной и контролируемой доставки и высвобождения таких сильнодействующих средств. Вследствие вышеизложенного, настоящее изобретение предлагает более эффективные и/или лучше переносимые фармацевтические композиции альбумин-связывающих пролекарств на основе производных ауристатина Е, которые можно использовать для лечения злокачественных заболеваний.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
[0008] В настоящем изобретении предложено соединение, имеющее структуру, описанную формулой I или II:
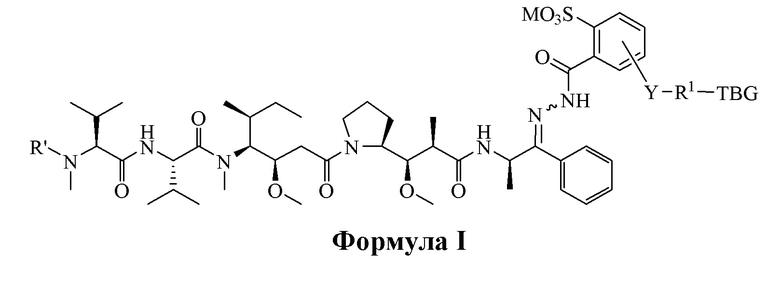
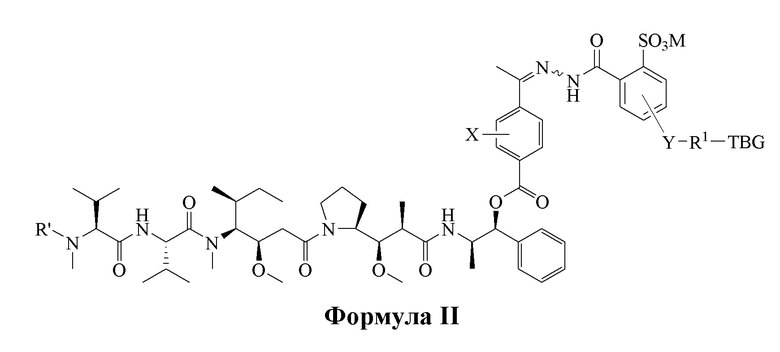
или его фармацевтически приемлемая соль, гидрат, сольват или изомер;
в которой:
R' является H или -CH3,
М является Н или фармацевтически приемлемым противоионом;
Y отсутствует или выбран из необязательно замещенного C1-C6 алкила, -NH-C(O)-, -C(O)-NH-, -NH-C(O)-NH-, -C(O)-O- и
-O-C(O)-;
R1 отсутствует или является необязательно замещенным C1-C18 алкилом, где необязательно до шести атомов углерода в указанном C1-C18 алкиле, каждый независимо, замещены -OCH2CH2-;
X является H или выбран из группы, включающей галоген (например, -F, -Cl, -Br или -I), -NO2, -NR2R3, -OR2, -NHCOR2 и -OCOR2, где R2 и R3, каждый независимо, выбран из H и C1-C4 алкила;
TBG является тиолсвязывающей группой, выбранной из следующего: необязательно замещенной малеимидной группы, необязательно замещенной галоацетамидной группы, необязательно замещенной галоацетатной группы, необязательно замещенной пиридилтиогруппы, необязательно замещенной изотиоцианатной группы, необязательно замещенной винилкарбонильной группы, необязательно замещенной азиридиновой группы, необязательно замещенной дисульфидной группы и необязательно замещенной ацетиленовой группы.
[0009] В некоторых вариантах воплощения R' является -CH3. В других вариантах воплощения R' является H.
[0010] В некоторых вариантах воплощения TBG выбран из необязательно замещенной малеимидной группы.
[0011] В некоторых вариантах воплощения TBG является малеимидной группой, имеющей формулу:
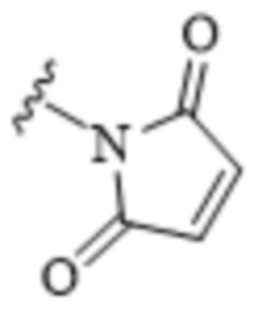 .
.
[0012] В некоторых вариантах воплощения Y является -NH-C(O)-.
[0013] В некоторых вариантах воплощения М является H+ или Na+.
[0014] В некоторых вариантах воплощения R1 является необязательно замещенным C1-C5 алкилом. В других вариантах воплощения R1 является C1-C5 алкилом.
[0015] В некоторых вариантах воплощения изобретение относится к соединению, имеющему структуру, описанную формулой III:
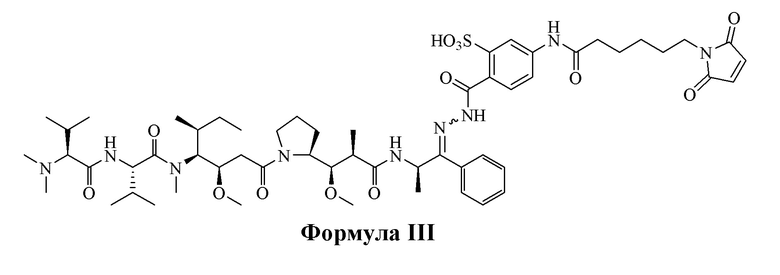
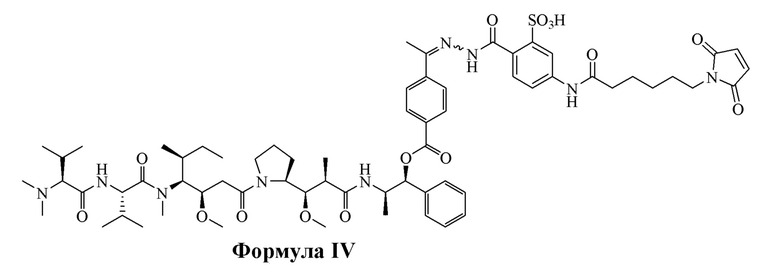
[0016] В других вариантах воплощения настоящее изобретение относится к соединению, имеющему структуру, описанную формулой IV:
[0017] Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, представленное в данном документе, и фармацевтически приемлемый носитель. В некоторых вариантах воплощения фармацевтически приемлемый носитель выбран из группы, включающей один или более из следующего: солюбилизирующий агент, инкапсулирующий агент и лиопротектор. В других вариантах воплощения фармацевтически приемлемый носитель содержит одно или более из следующего: диметил-β-циклодекстрин, гидроксиэтил-β-циклодекстрин, гидроксипропил-β-циклодекстрин и триметил-β-циклодекстрин.
[0018] В некоторых вариантах воплощения фармацевтическая композиция предназначена для внутривенного введения. В некоторых вариантах воплощения композиция при внутривенном введении пациенту ковалентно и избирательно быстро связывается in situ с эндогенным альбумином в кровотоке. В других вариантах воплощения композиция при внутривенном введении пациенту ковалентно и избирательно быстро связывается in situ с тиольной группой цистеина-34 эндогенного альбумина в кровотоке.
[0019] В настоящем изобретении также предложен способ лечения пациента, страдающего заболеванием, выбранным из следующего: рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами и другими микроорганизмами, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения или фармацевтической композиции способом, описанным в данном документе. В некоторых вариантах воплощения заболевание является раком и выбрано из следующего: карцинома, саркома, лейкоз, лимфома, множественная миелома и меланома. В некоторых вариантах воплощения способом введения является внутривенное введение.
[0020] Настоящее изобретение также относится к способу снижения цитотоксичности соединения, включающему введение нуждающемуся в этом пациенту соединения или фармацевтической композиции, как описано в настоящем документе, в котором введение приводит к снижению цитотоксичности по сравнению с эквивалентной дозой немодифицированного активного агента.
[0021] В настоящем изобретении дополнительно предложен способ увеличения концентрации метаболита соединения в опухоли, включающий описанное в настоящем документе введение соединения или фармацевтической композиции нуждающемуся в этом пациенту, в котором данное увеличение сравнивают с эквивалентной дозой немодифицированного активного агента.
[0022] Настоящее изобретение относится к предлагаемому в данном документе соединению для применения в качестве лекарственного средства.
[0023] В настоящем изобретении также предложено описанное в данном документе соединение для применения при лечении заболевания или состояния, выбранного из группы, включающей рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами или другими микроорганизмами.
[0024] В настоящем изобретении дополнительно предложено применение описанного в данном документе соединения или композиции для приготовления лекарственного средства для лечения заболевания или состояния, выбранного из следующего: рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами или другими микроорганизмами.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0025] На фиг. 1 показано сравнение стабильности AE-Keto-Sulf07 (панель (а)) и AE-Keto-EMCH (панель (b)) в буфере для восстановления (50 мМ натрий-фосфатный буфер pH 7,65, 5% сахароза (w/v) и 2% 2-гидроксипропил-β-циклодекстрин (2-HPβCD (w/v)).
[0026] На фиг. 2 показано сравнение стабильности AE-Эстер-Sulf07 (панель (а)) и AE-Эстер-EMCH (панель (b)) в буфере для восстановления (50 мМ натрий-фосфатный буфер, рН 7,65, 5% сахароза (w/v) и 4% 2-гидроксипропил-β-циклодекстрин (2-HPβCD, (w/v)).
[0027] На фиг. 3 показана конъюгация AE-Keto-Sulf07 с альбумином в человеческой плазме.
[0028] На фиг. 4 показана конъюгация AE-Эстер-Sulf07 с альбумином в человеческой плазме.
[0029] На фиг. 5 показана кинетика связывания AE-Keto-Sulf07 с альбумином в человеческой плазме.
[0030] На фиг. 6 показана кинетика связывания AE-Keto-Sulf07 с альбумином в мышиной плазме.
[0031] На фиг. 7 показана кинетика связывания AE-Keto-Sulf07 с альбумином в крысиной плазме.
[0032] На фиг. 8 показана кинетика связывания AE-Эстер-Sulf07 с альбумином в человеческой плазме.
[0033] На фиг. 9 показана кинетика связывания AE-Эстер-Sulf07 с альбумином в мышиной плазме до достижения предела количественного определения (LOQ) для лекарственного средства.
[0034] На фиг. 10 показана кинетика связывания AE-Эстер-Sulf07 с альбумином в крысиной плазме до достижения LOQ для лекарственного средства.
[0035] На фиг. 11 показано зависимое от pH высвобождение AE-Keto (pH 7,4 (панель (a)) и pH 4,1 (панель (b)) из связанного с человеческим альбумином AE-Keto-Sulf07.
[0036] На фиг. 12 показано зависимое от pH высвобождение AE-Эстер (pH 7,4 (панель (a)) и pH 4,1 (панель (b)) из связанного с человеческим альбумином AE-Эстер-Sulf07.
[0037] На фиг. 13 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на раковой модели злокачественной меланомы A375, среднее значение срединного начального объема опухоли ~134 мм3.
[0038] На фиг. 14 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на раковой модели злокачественной меланомы A375, среднее значение срединного начального объема опухоли ~332 мм3.
[0039] На фиг. 15 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на модели ксенотрансплантата NSCLC LXFA737, среднее значение срединного начального объема опухоли ~132 мм3.
[0040] На фиг. 16 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на модели ксенотрансплантата NSCLC LXFA737, среднее значение срединного начального объема опухоли ~330 мм3.
[0041] На фиг. 17 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на модели карциномы яичника человека A2780, среднее значение срединного начального объема опухоли ~148 мм3.
[0042] На фиг. 18 показан противоопухолевый эффект ауристатина E и AE-Keto-Sulf07 по сравнению с контрольной группой на модели карциномы яичника человека A2780, среднее значение срединного начального объема опухоли ~351 мм3.
[0043] На фиг. 19 показан противоопухолевый эффект ауристатина E и AE-Эстер-Sulf07 по сравнению с контрольной группой на раковой модели почечно-клеточного рака RXF631, начальный объем опухоли ~140 мм3.
[0044] На фиг. 20 показан противоопухолевый эффект ауристатина E и AE-Эстер-Sulf07 по сравнению с контрольной группой на раковой модели злокачественной меланомы A375, среднее значение срединного начального объема опухоли ~332 мм3.
[0045] На фиг. 21 показан противоопухолевый эффект ауристатина E и AE-Эстер-Sulf07 по сравнению с контрольной группой на модели ксенотрансплантата NSCLC LXFA737, среднее значение срединного начального объема опухоли ~132 мм3.
[0046] На фиг. 22 показан противоопухолевый эффект ауристатина E и AE-Эстер-Sulf07 по сравнению с контрольной группой на модели карциномы яичника человека A2780, начальный объем опухоли ~351 мм3.
[0047] На фиг. 23 показан противоопухолевый эффект AE-Эстер-Sulf07 по сравнению с контрольной группой на модели карциномы яичника человека A2780, начальный объем опухоли ~148 мм3.
[0048] На фиг. 24 показано ускоренное разложение лиофилизированных составов ауристатина для сравнения стабильности AE-Keto-Sulf07 и AE-Keto-EMCH.
[0049] На фиг. 25 показано ускоренное разложение лиофилизированных составов ауристатина для сравнения стабильности AE-Эстер-Sulf07 и AE-Эстер-EMCH.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0050] Если не указано иное, научные и технические термины, используемые в настоящей заявке, имеют значения, которые обычно понятны специалистам в данной области техники. Как правило, номенклатура, относящаяся к способам химии, молекулярной биологии, биологии клетки и рака, иммунологии, микробиологии, фармакологии и химии белка, описанным в данном документе, является хорошо известной и широко используемой в данной области.
[0051] Все публикации, патенты и опубликованные патентные заявки, упомянутые в этой заявке, специально включены в данный документ посредством ссылки. В случае конфликта настоящее описание, включая его конкретные определения, будет иметь преимущественную силу. Если не указано иное, следует понимать, что каждый вариант воплощения, представленный в данном документе, может использоваться отдельно или в комбинации с каким-либо одним или более из других вариантов воплощения, представленных в данном документе.
Определения
[0052] В данном описании слово «содержать» или его варианты, такие как «содержит» или «содержащий», следует понимать как подразумевающее включение указанного целого числа (или компонентов) или группы целых чисел (или компонентов), но не исключение какого-либо другого целого числа (или компонентов) или группы целых чисел (или компонентов).
[0053] Во всей заявке, где соединение или композиция описываются как имеющая, включающая или содержащая конкретные компоненты, предполагается, что такое соединение или композиция также может состоять по существу из или состоять из перечисленных компонентов. Аналогичным образом, когда способы или процессы описаны как имеющие, включающие в себя или содержащие конкретные этапы процесса, данные процессы также могут состоять по существу из или состоять из перечисленных этапов процесса. Кроме того, следует понимать, что порядок этапов или порядок выполнения определенных действий не имеет значения, поскольку соединения, композиции и способы, описанные в настоящем документе, остаются пригодными к использованию. Кроме того, два или более шагов или действий могут быть выполнены одновременно.
[0054] Формы в единственном числе включают и множественное число, если контекст явно не подразумевает иное.
[0055] Термин «включающий» имеет значение «включающий без ограничений». Термины «включающий» и «включающий без ограничений» используются взаимозаменяемо.
[0056] Используемый в данном документе термин «или» следует понимать как означающий «и/или», если контекст явно не указывает на иное.
[0057] Термины «лекарственное средство», «агент», «терапевтический агент», «терапевтически активный агент», «цитотоксический агент или лекарство», «высоко цитотоксический агент или лекарство» или «терапевтически эффективное вещество» используются для обозначения любого соединения, которое вызывает фармакологический эффект само по себе или после его превращения в рассматриваемом организме и, таким образом, также включает производные этих превращений. Фармакологический эффект лекарств композиции согласно описанию настоящего изобретения может быть только единичным, например, цитостатический и/или цитотоксический эффект, или широкий фармакологический спектр действия, такой как иммунодепрессивный и противовоспалительный эффект одновременно.
[0058] Термины «пациент», «субъект» или «индивидуум» используются взаимозаменяемо и относятся к человеку или животному, не являющемуся человеком. Эти термины включают млекопитающих, таких как люди, приматы, домашний скот (например, крупный рогатый скот, свинообразные), домашние питомцы (например, псовые, кошачьи) и грызуны (например, мыши и крысы). В определенных вариантах воплощения пациентом или субъектом является человек, имеющий состояние, нуждающееся в лечении.
[0059] Термин «фармацевтическая композиция» относится к композиции, предназначенной для фармацевтического применения у живого субъекта, включая людей и млекопитающих, например, в сочетании с одним или более фармацевтически приемлемыми носителями, наполнителями или растворителями. Такая композиция также может содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы, защитные средства и другие материалы, хорошо известные в данной области техники. В некоторых вариантах воплощения фармацевтическая композиция включает композицию, содержащую активный (е) ингредиент(ы) и инертный(е) ингредиент(ы), которые составляют наполнитель, носитель или разбавитель, а также любой продукт, который прямо или косвенно возникает в результате комбинации, комплексообразования или агрегации каких-либо двух или более ингредиентов, или в результате диссоциации одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции по настоящему изобретению включают любую композицию, полученную смешением соединения по настоящему изобретению и одного или более из фармацевтически приемлемых наполнителя(ей), носителя(ей) и/или разбавителя(ей).
[0060] Термин «фармацевтически приемлемый носитель» относится к нетоксичному носителю, который может быть введен пациенту вместе с терапевтически эффективным веществом, представленным в данном документе, и который не подавляет фармакологическую активность агента. Термин «наполнитель» относится к добавке в составе или композиции, которая не является фармацевтически активным ингредиентом. В определенных вариантах воплощения «фармацевтически приемлемое» вещество предназначено для использования при контакте с клетками, тканями или органами животных или людей без чрезмерной токсичности, раздражения, аллергического ответа, иммуногенности или других нежелательных реакций в количестве, используемом в лекарственной форме согласно режиму дозирования и соразмерно с разумным соотношением пользы/риска. В некоторых вариантах воплощения «фармацевтически приемлемое» вещество, которое является компонентом фармацевтической композиции, является, кроме того, совместимым с другим ингредиентом(ами) композиции. В некоторых вариантах воплощения термины «фармацевтически приемлемый наполнитель», «фармацевтически приемлемый носитель» и «фармацевтически приемлемый разбавитель» охватывают, без ограничения, фармацевтически приемлемые неактивные ингредиенты, материалы, композиции и носители, такие как жидкие наполнители, твердые наполнители, разбавители, наполнители, носители, растворители и герметизирующие материалы. Носители, разбавители и наполнители также включают все фармацевтически приемлемые дисперсионные среды, покрытия, буферы, изотонические агенты, стабилизаторы, агенты, замедляющие абсорбцию, антимикробные агенты, антибактериальные агенты, противогрибковые агенты, адъюванты и т.д. За исключением тех случаев, когда используются какие-либо обычные наполнители, носители или разбавители, несовместимые с активным ингредиентом, содержание настоящего изобретения включает использование обычных фармацевтических наполнителей, носителей и разбавителей в фармацевтических композициях. См., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins (Philadelphia, Pennsylvania, 2005); Handbook of Pharmaceutical Excipients, 5th Ed., Rowe et al., Eds., The Pharmaceutical Press and the American Pharmaceutical Association (2005); Handbook of Pharmaceutical Additives, 3rd Ed., Ash and Ash, Eds., Gower Publishing Co. (2007); and Pharmaceutical Preformulation and Formulation, Gibson, Ed., CRC Press LLC (Boca Raton, Florida, 2004).
[0061] Термины «фармацевтически эффективное количество», «терапевтически эффективное количество» или «терапевтически эффективная доза» относятся к количеству, эффективному для лечения заболевания у пациента, например, для осуществления полезного и/или желаемого изменения общего состояния здоровья пациента, страдающего от заболевания (например, рака), лечения, заживления, подавления или ослабления физиологического ответа или состояния и т.д. Полный терапевтический эффект необязательно возникает при введении одной дозы и может происходить только после введения серии доз. Таким образом, терапевтически эффективное количество может быть доставлено за одно или более введение. Точное эффективное количество, необходимое для субъекта, будет зависеть, например, от размеров тела, состояния и возраста субъекта, природы и степени заболевания, терапевтических средств или комбинации терапевтических средств, выбранных для введения, и от способа введения. Специалист в данной области сможет легко определить эффективное для данной ситуации количество путем рутинных экспериментов. Специалисту в данной области техники понятно, что лечение рака включает без ограничений уничтожение раковых клеток, предотвращение роста новых раковых клеток, инициирование регрессии опухоли (уменьшение размера опухоли), инициирование уменьшения метастазирования, улучшение жизненно важных функций организма пациента, улучшение самочувствия пациента, уменьшение болей, улучшение аппетита, улучшение веса пациента и любую их комбинацию. Термины «фармацевтически эффективное количество», «терапевтически эффективное количество» или «терапевтически эффективная доза» также относятся к количеству, необходимому для улучшения клинических симптомов у пациента. Терапевтические способы или способы лечения рака, описанные в данном документе, не должны толковаться или иным образом ограничиваться понятием «лечение» рака.
[0062] Используемый в данном документе термин «лечение» или «терапия» включает изменение, уменьшение или исчезновение симптомов, клинических признаков и патологических процессов, обуславливающих состояние пациента, с целью улучшения или стабилизации состояния субъекта. Используемый в данном документе и хорошо понятный специалисту в данной области техники, термин «лечение» обозначает способ получения благоприятных или оптимальных результатов, включая клинические результаты. Благоприятные или оптимальные клинические результаты могут включать, без ограничений, уменьшение или исчезновение одного или более симптомов, улучшение или замедление прогрессирования одного или более состояний, связанных с заболеванием, например, раком, уменьшение степени заболевания, стабилизированное (т.е. без ухудшения) состояние заболевания, задержку или замедление прогрессирования заболевания, улучшение или облегчение состояния при заболевании и ремиссию (частичную или полную), определяемую или не определяемую. Термин «лечение» также может означать увеличение выживаемости по сравнению с ожидаемой выживаемостью, если лечение не проводится. Примерные благоприятные клинические результаты описаны в данном документе.
[0063] «Введение» или «применение» вещества, соединения или агента субъекту может быть осуществлено с использованием одного из многих способов, известных специалистам в данной области. Например, соединение или агент можно вводить внутривенно, внутриартериально, внутрикожно, внутримышечно, внутрибрюшинно, подкожно, интраокулярно, сублингвально, перорально (путем приема внутрь), интраназально (путем ингаляции), интраспинально, интрацеребрально и трансдермально (путем абсорбции, например, через кожные протоки). Соединение или агент также можно соответствующим образом вводить с помощью перезаряжаемых или биоразлагаемых полимерных устройств или других устройств, например пластырей и насосов, или составов, которые обеспечивают пролонгированное, медленное или контролируемое высвобождение соединения или агента. Введение также может быть выполнено, например, один раз, множество раз и/или в течение одного или более продолжительных периодов. В некоторых аспектах введение включает как прямое введение, включая самостоятельное введение, так и косвенное введение, включая назначение лекарственного средства. Например, как указано в данном документе, врач, который инструктирует пациента самостоятельно вводить лекарство или обязывает другое лицо вводить лекарство пациенту и/или предоставляет пациенту рецепт на лекарство, является применяющим лекарство к пациенту. Когда способ является частью режима лечения, включающего более одного агента или способа лечения, в предлагаемом изобретении предполагается, что агенты могут вводиться в одно и то же или в разное время и одними и теми же или разными путями введения. Приемлемые способы введения вещества, соединения или агента субъекту также будут зависеть, например, от возраста субъекта, от того, является ли субъект активным или неактивным во время введения, нарушено ли у субъекта восприятие во время введения, от степени нарушения и химических и биологических свойств соединения или агента (например, растворимости, усвояемости, биодоступности, стабильности и токсичности).
[0064] Термин «замещенный» относится к фрагментам, имеющим заместители, замещающие водород в одном или более атомах углерода основной цепи химического соединения. Специалисту в данной области техники понятно, что термины «замещение» или «замещенный» включают в себя необязательное условие, что такое замещение соответствует допустимой валентности замещенного атома и заместителя, и что замещение приводит к образованию стабильного соединения, например, которое не подвержено самопроизвольной трансформации, такой как перегруппировка, циклизация, элиминация и т. д. Используемый в данном документе термин «замещенный» предполагает включение всех допустимых заместителей органических соединений. В широком аспекте допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Допустимые заместители могут быть одним или более и одинаковыми или разными для соответствующих органических соединений. В описании данного изобретения гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанных в данном документе, которые соответствуют валентности гетероатомов. Заместители могут включать любые заместители, описанные в данном документе, например, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как тиоэфир, тиоацетат или тиоформиат), алкоксил, алкилтио, ацилокси, фосфорил, фосфат, фосфонат, амино, амидо, амидин, имин, циано, нитро, азидо, сульфгидрил, алкилтио, сульфат, сульфонатную, сульфамоильную, сульфонамидо, сульфонильную, гетероциклильную, аралкильную или ароматическую (например, C6-C12 арил) или гетероароматическую (например, гетероарильную) группы.
[0065] Термины «необязательный(ая)» или «необязательно» означает, что описанное обстоятельство, возникающее впоследствии, может возникать или не возникать, таким образом, данная заявка включает в себя случаи, когда это обстоятельство возникает, и случаи, когда этого не происходит. Например, фраза «необязательно замещенный» означает, что неводородный заместитель может присутствовать или не присутствовать на данном атоме, и, таким образом, данная заявка включает структуры, в которых присутствует неводородный заместитель, и структуры, в которых неводородный заместитель не присутствует.
[0066] Если специально не указан термин «незамещенный», это означает, что в данном документе ссылки на химические группы включают замещенные варианты. Например, ссылка на «алкильную» группу или фрагмент косвенно включает как замещенные, так и незамещенные варианты. Примеры заместителей в химических группах включают, без ограничений, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как тиоэфир, тиоацетат или тиоформиат), алкоксил, алкилтио, ацилокси, фосфорил, фосфат, фосфонат, амино, амидо, амидин, имин, циано, нитро, азидо, сульфгидрил, алкилтио, сульфат, сульфонат, сульфамоильную, сульфонамидо, сульфонильную, гетероциклильную, аралкильную, арильную или гетероарильную группы.
[0067] Термин «арил» обозначает ароматическое углеродное кольцо, имеющее в кольце указанное число атомов углерода, например от 6 до 12 или от 6 до 10 атомов углерода. Арильные группы могут быть моноциклическими или полициклическими (например, бициклическими, трициклическими). В некоторых случаях оба кольца полициклической арильной группы являются ароматическими (например, нафтил). В других случаях полициклические арильные группы могут включать неароматическое кольцо (например, циклоалкил, циклоалкенил, гетероциклоалкил, гетероциклоалкенил), конденсированное с ароматическим кольцом, при условии, что полициклическая арильная группа связана с исходной структурой через атом в ароматическом кольце. Таким образом, 1,2,3,4-тетрагидронафталин-5-ильная группа (в которой фрагмент связан с исходной структурой через ароматический атом углерода) считается арильной группой, в то время как 1,2,3,4-тетрагидронафталин-1-ил (в которой фрагмент связан с исходной структурой через неароматический атом углерода) не считается арильной группой. Подобным образом, 1,2,3,4-тетрагидрохинолин-8-ильная группа (в которой фрагмент связан с исходной структурой через ароматический атом углерода) считается арильной группой, в то время как 1,2,3,4-тетрагидрохинолин-1-ильная группа (в которой фрагмент связан с исходной структурой через неароматический атом азота) не считается арильной группой. Однако термин «арил» не охватывается и не перекрывается термином «гетероарил», как определено в настоящем документе, независимо от точки присоединения (например, как хинолин-5-ил, так и хинолин-2-ил являются гетероарильными группами).
[0068] Термин «гетероарил» означает ароматическое кольцо, содержащее указанное число атомов кольца (например, 5-12 или 5-10-членный гетероарил), и состоящее из одного или более гетероатомов (например, 1, 2, 3 или 4 гетероатома), выбранных из N, O и S, и остальных атомов кольца, являющихся атомами углерода. 5-членный гетероарил является гетероарилом, имеющим 5 кольцевых атомов. 6-членный гетероарил является гетероарилом, имеющим 6 кольцевых атомов. Гетероарильные группы не содержат соседних атомов S и O. В некоторых вариантах воплощения общее количество атомов S и O в гетероарильной группе составляет не более 2. В некоторых вариантах воплощения общее количество атомов S и O в гетероарильной группе составляет не более 1. Если не указано иное, гетероарильные группы могут быть связаны с основной структурой атомом углерода или азота, если позволяет валентность. Например, термин «пиридил» включает 2-пиридильную, 3-пиридильную и 4-пиридильную группы, а термин «пирролил» включает 1-пирролильную, 2-пирролильную и 3-пирролильную группы. Если в гетероарильном кольце присутствует азот, он может, если позволяет природа соседних атомов и групп, существовать в окисленном состоянии (то есть, N+-O-). Кроме этого, если в гетероарильном кольце присутствует сера, она может, если позволяет природа соседних атомов и групп, существовать в окисленном состоянии (то есть, S+-O- или SO2). Гетероарильные группы могут быть моноциклическими или полициклическими (например, бициклическими, трициклическими).
[0069] В некоторых случаях гетероарильная группа является моноциклической. Примеры включают пиррол, пиразол, имидазол, триазол (например, 1,2,3-триазол, 1,2,4-триазол, 1,3,4-триазол), тетразол, фуран, изоксазол, оксазол, оксадиазол (например, 1, 2,3-оксадиазол, 1,2,4-оксадиазол, 1,3,4-оксадиазол), тиофен, изотиазол, тиазол, тиадиазол (например, 1,2,3-тиадиазол, 1,2,4-тиадиазол, 1,3,4-тиадиазол), пиридин, пиридазин, пиримидин, пиразин, триазин (например, 1,2,4-триазин, 1,3,5-триазин) и тетразин.
[0070] Термин «ацил» известен в данной области техники и относится к группе, представленной общей формулой гидрокарбил-C(O)-, например,
алкил-C(O)-.
[0071] Термин «алкил» относится к радикалу насыщенных алифатических групп, включая алкильные группы с прямой цепью и алкильные группы с разветвленной цепью. В некоторых вариантах воплощения алкил с прямой или разветвленной цепью имеет 30 или менее атомов углерода в своей основной цепи (например, C1-C30 для прямых цепей, C4-C30 для разветвленных цепей), а в других вариантах воплощения 20 или менее. В некоторых вариантах воплощения алкильные группы являются низшими алкильными группами, например, метил, этил, н-пропил, изопропил, н-бутил и н-пентил. Кроме того, термин «алкил», используемый в описании, примерах и формуле изобретения, используется для включения как «незамещенных алкилов», так и «замещенных алкилов», последние из которых относятся к алкильным группам, имеющим заместители, замещающие водород на одном или более атомах углерода углеводородного остова. В некоторых вариантах воплощения алкил с прямой или разветвленной цепью имеет 30 или менее атомов углерода в своей основной цепи (например, C1-C30 для прямых цепей, C3-C30 для разветвленных цепей). В некоторых вариантах воплощения цепь имеет десять или менее атомов углерода (C1-C10) в своей основной цепи. В других вариантах воплощения цепь имеет шесть или менее атомов углерода (C1-C6) в своей основной цепи.
[0072] Термины «гидразоновый фрагмент» или «гидразон» относятся к E- и/или Z-изомерам гидразонов, например,
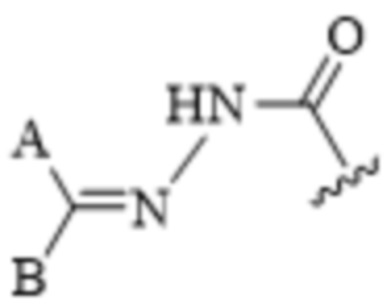 или
или 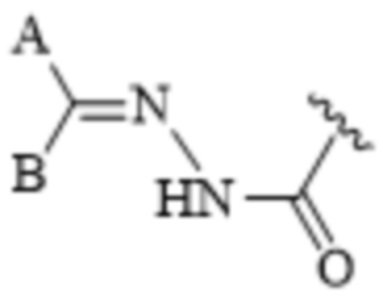 .
.
Стереохимия гидразонового фрагмента может включать E- или Z-изомеры. Термин «гидразон», используемый в данном документе, включает как E-, так и Z-изомеры. Гидразоновые фрагменты, представленные в данном документе, в общем, изображены в одной конфигурации, но очевидно, что описание в данном документе может включать как E-, так и/или Z-изомеры.
[0073] В различных пунктах описания настоящего изобретения заместители соединений по изобретению описаны в группах или в диапазонах. В частности, предполагается, что описание включает в себя каждую отдельную субкомбинацию членов таких групп и диапазонов. Например, термин «C1-C6 алкил» специально предназначен для описания в отдельности метила, этила, пропила, изопропила, н-бутила, втор-бутила, изобутила и т. д.
[0074] Термин «фармацевтически приемлемая соль» означает соль соединения, предназначенную для фармацевтического применения, включая, без ограничений, соли металлов (например, соли натрия, калия, магния, кальция и т.д.), соли присоединения кислоты (например, минеральных кислот, карбоновых кислот и т.д.) и соли присоединения основания (например, аммиака, органических аминов и т.д.). Кислотно-аддитивная соль соединения, которое встречается в свободной форме в виде основания, может быть получена путем обработки указанной формы свободного основания соответствующей кислотой, такой как неорганическая кислота, например, галогенводородная, включая соляную или бромистоводородную, серная, азотная. фосфорная кислота и тому подобное; или такой как органическая кислота, например, уксусная, гидроксиуксусная, пропионовая, молочная, пировиноградная, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, циклическая, салициловая, п-аминосалициловая, памоевая и тому подобное (см., например, заявку WO 01/062726. Некоторые фармацевтически приемлемые соли, перечисленные Berge et al., J. Pharm. Sci., 66: 1-19 (1977), включены в данный документ во всей полноте посредством ссылки). Соединения, содержащие кислотные протоны, могут быть преобразованы в их терапевтически активную, нетоксичную форму соли присоединения основания, например, соли металлов или аминов путем обработки соответствующими органическими и неорганическими основаниями. Соответствующие формы основных солей включают, например, соли аммония, соли щелочных и щелочноземельных металлов или ионов, например, соли лития, натрия, калия, магния, кальция и тому подобное, соли органических оснований, например, N-метил-D-глюкамина, соли гидрабамина, соли аминокислот, таких как, например, аргинина, лизина и тому подобное. И наоборот, указанные солевые формы можно превратить в свободные формы путем обработки соответствующим основанием или кислотой. Соединения и их соли могут быть в форме сольватов, что входит в объем настоящего изобретения. Такие сольваты включают, например, гидраты, алкоголяты и тому подобное (см., например, заявку WO 01/062726).
[0075] В настоящем изобретении также предложены фармацевтические композиции, содержащие одно или более соединений по изобретению вместе с фармацевтически приемлемым носителем или наполнителем. Соединения или фармацевтические композиции по изобретению могут быть использованы in vitro или in vivo.
[0076] Используемый в данном документе термин «изомер» включает, без ограничений, таутомеры, цис- и транс-изомеры (E (entgegen), Z (zusammen)), R- и S-энантиомеры (указанные обозначения R и S используются в соответствии с правилами, описанными в Pure Appl. Chem. (1976), 45, 11-30), диастереомеры, (D) -изомеры, (L) -изомеры, стереоизомеры, соответственно, их рацемические смеси и, соответственно, другие смеси. Все такие изомеры, а также их смеси предназначены для включения в объем данного изобретения. Таутомеры, хотя это явно не указано в формулах, описанных в настоящем документе, предназначены для включения в объем настоящего изобретения.
Соединения предлагаемого изобретения
[0077] В вариантах воплощения настоящего изобретения предложено соединение, имеющее структуру, описанную формулой I или формулой II:
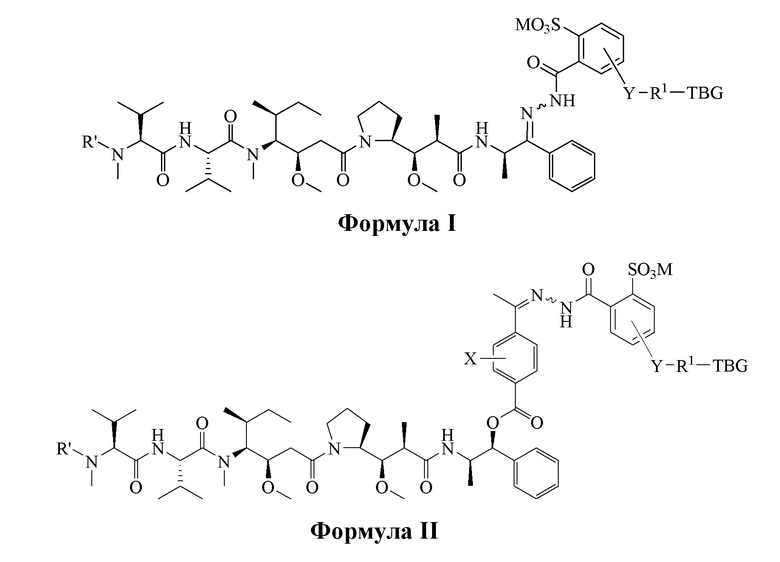
или его фармацевтически приемлемая соль, гидрат, сольват или изомер, где:
R' является -H или -CH3,
M выбран из H и фармацевтически приемлемого противоиона, такого как Na+, K+, NR4+ или NHR3+, где R выбран из H и C1-C4 алкила,
Y отсутствует или выбран из необязательно замещенного C1-C6 алкила, -NH-C(O)-, -C(O)-NH-, -NH-C(O)-NH-, -C(O)-O- и
-OC(O)-,
R1 отсутствует или является необязательно замещенным C1-C18-алкилом, где необязательно до шести атомов углерода в указанном C1-C18 алкиле, каждый независимо, замещен -OCH2CH2-,
X является H или выбран из галогеновых групп (например, -F, -Cl, -Br или -I), -NO2, -NR2R3, -OR2, -NHCOR2 и -OCOR2, где R2 и R3, каждый независимо, выбран из H и C1-C4 алкила, и
TBG является тиолсвязывающей группой, выбранной из следующего: необязательно замещенной малеимидной группы, необязательно замещенной галоацетамидной группы, необязательно замещенной галоацетатной группы, необязательно замещенной пиридилтиогруппы, необязательно замещенной изотиоцианатной группы, необязательно замещенной винилкарбонильной группы, необязательно замещенной азиридиновой группы, необязательно замещенной дисульфидной группы и необязательно замещенной ацетиленовой группы.
[0078] В некоторых вариантах воплощения соединение, имеющее структуру, описанную формулой I или II, составлено в виде фармацевтической композиции, содержащей необязательно фармацевтически приемлемый носитель, которая вводится в организм и ковалентно, селективно и быстро связывается in situ с тиольной группой цистеина- 34 эндогенного альбумина в кровотоке.
[0079] В некоторых вариантах воплощения R' является -CH3. В других вариантах воплощения R' является H.
[0080] В некоторых вариантах воплощения тиолсвязывающая группа (TBG) является малеимидной группой:
.
[0081] В некоторых вариантах воплощения лекарственное средство на основе высоко цитотоксического пептида является ауристатином Е, дериватизированным таким образом, что в результате он содержит карбонильную группу, обеспечивающую образование кислоточувствительной гидразонной связи с малеимидным водорастворимым линкером, содержащим гидразидную группу.
[0082] В некоторых вариантах воплощения настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описанное в данном документе, и, необязательно, фармацевтически приемлемый носитель, где фармацевтическая композиция вводится внутривенно и ковалентно, селективно и быстро связывается in situ с тиольной группой цистеина-34 эндогенного альбумина в кровотоке.
[0083] В некоторых вариантах воплощения, в представленных в данном документе альбуминсвязывающих соединениях, высоко цитотоксическое лекарственное средство является карбонилсодержащим пентапептидным производным ауристатина E, альбуминсвязывающий фрагмент является тиолсвязывающей группой (TBG), например, малеимидной группой, которая после введения быстро и избирательно связывается с цистеином-34 альбумина, а кислоточувствительная связь является дериватизированной бензоилгидразоновой связью соединения с общей формулой I и II:
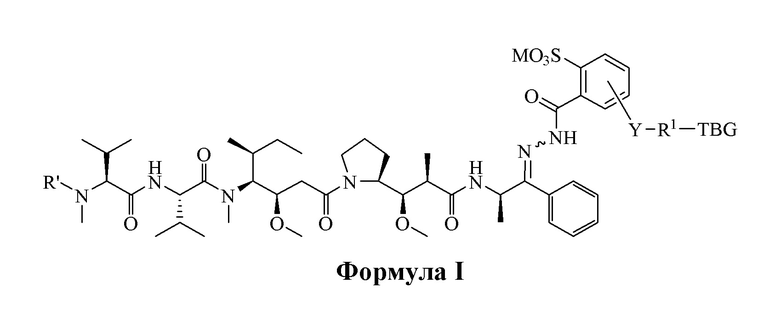
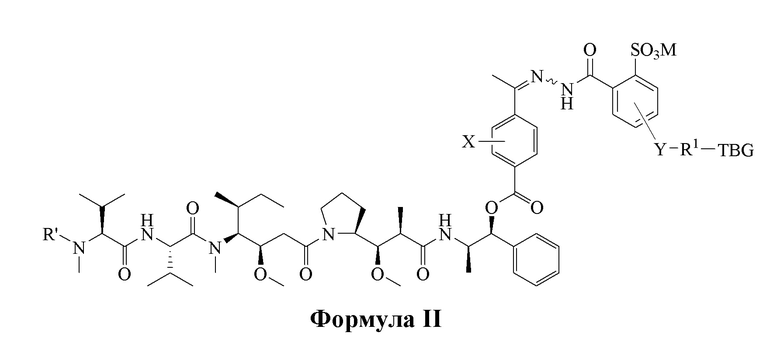
или его фармацевтически приемлемой соли, гидрата, сольвата или изомера, где:
R' является H или -CH3,
M выбран из H и фармацевтически приемлемого противоиона, такого как Na+, K+, NR4+ или NHR3, где R выбран из H и C1-C4 алкила,
Y отсутствует или выбран из необязательно замещенного C1-C6 алкила, -NH-C(O)-, -C(O)-NH-, -NH-C(O)-NH-, -C(O)-O- и
-OC(O)-,
R1 отсутствует или является необязательно замещенным C1-C18 алкилом, в котором необязательно до шести атомов углерода, каждый независимо, замещен
-OCH2CH2-,
X является H или выбран из галогеновых групп (например, -F, -Cl, -Br или -I), -NO2, -NR2R3, -OR2, -NHCOR2 и -OCOR2, где R2 и R3, каждый независимо, выбран из H и C1-C4 алкила,
TBG является тиолсвязывающей группой, выбранной из следующего: необязательно замещенной малеимидной группы, необязательно замещенной галоацетамидной группы, необязательно замещенной галоацетатной группы, необязательно замещенной пиридилтиогруппы, необязательно замещенной изотиоцианатной группы, необязательно замещенной винилкарбонильной группы, необязательно замещенной азиридиновой группы, необязательно замещенной дисульфидной группы и необязательно замещенной ацетиленовой группы.
[0084] В некоторых вариантах воплощения TBG замещена C1-C6 алкилом или галогеном. В некоторых вариантах воплощения TBG замещена метилом, -Cl или -Br.
[0085] Дисульфидная группа может быть активирована тионитробензойной кислотой (например, 5'-тио-2-нитробензойной кислотой) в качестве обменной группы. Малеимидная или пиридилдитогруппа может быть при необходимости замещена алкильной группой или вышеуказанными водорастворимыми группами. Обычно тиолсвязывающая группа обладает белково-связывающими свойствами, то есть она ковалентно связывается («ковалентная белково-связывающая группа») в физиологической среде с конкретными аминокислотами на поверхности белка. В некоторых вариантах воплощения малеимидная группа, галоацетамидная группа, галоацетатная группа, пиридилдитиогруппа, дисульфидная группа, винилкарбонильная группа, азиридиновая группа и/или ацетиленовая группа реагируют с тиольными (-SH) группами цистеинов. В некоторых вариантах воплощения белок-связывающая группа является малеимидной группой
,
которая связывается с цистеином-34 альбумина.
[0086] В некоторых вариантах воплощения система доставки лекарственного средства содержит кислоточувствительный расщепляемый гидразоновый фрагмент. Расщепление гидразонового фрагмента и полупериод высвобождения лекарственного средства варьируются в зависимости от структуры производного карбонила.
[0087] В некоторых вариантах воплощения полупериод высвобождения связанного с альбумином лекарственного средства в диапазоне рН 4,0-6,5 варьируется от примерно 1,5 часа до примерно 80 часов.
[0088] Не будучи связанными теорией, авторы полагают, что фенильное кольцо, содержащее одну электроноакцепторную группу, такую как сульфоновая кислота (-SO3H) или сульфонатная группа (-SO3-), присоединенная в орто-положении к гидразоновой связи, стабилизирует гидразоновый фрагмент, что приводит к медленному и длительному высвобождению препарата в кислой среде.
[0089] В некоторых вариантах воплощения R' является -CH3. В других вариантах воплощения R' является H.
[0090] В некоторых вариантах воплощения Y и/или R1 присутствуют. В некоторых вариантах воплощения Y отсутствует. В некоторых вариантах воплощения R1 отсутствует. В некоторых вариантах воплощения как Y, так и R1 отсутствуют.
[0091] В некоторых вариантах воплощения Y выбран из следующего: метил, этил, -NH-C(O)-, -C(O)-NH-, -C(O)-O- и -O-C(O)-.
[0092] В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором один атом углерода замещен -OCH2CH2-. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором два атома углерода замещены -OCH2CH2-. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором три атома углерода замещены -OCH2CH2-. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором четыре атома углерода замещены -OCH2CH2-. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором пять атомов углерода замещены -OCH2CH2-. В некоторых вариантах воплощения R1 является необязательно замещенным C1-C18 алкилом, в котором шесть атомов углерода замещены -OCH2CH2-.
[0093] В US6884869-B2 (заявка №:10/001191, поданная 01.11.2001) описаны конъюгаты антител с лекарственными средствами, в которых пентапептидные производные, имеющие химическую структуру, описанную формулой A и формулой B, изображены ниже:
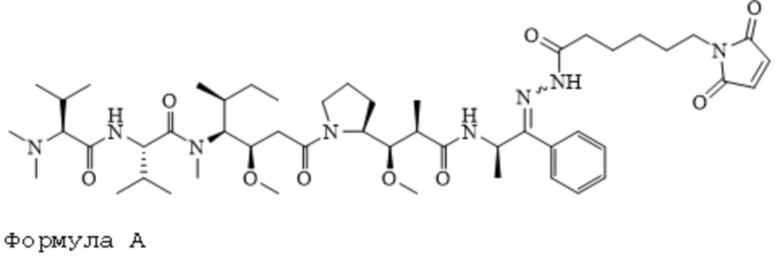
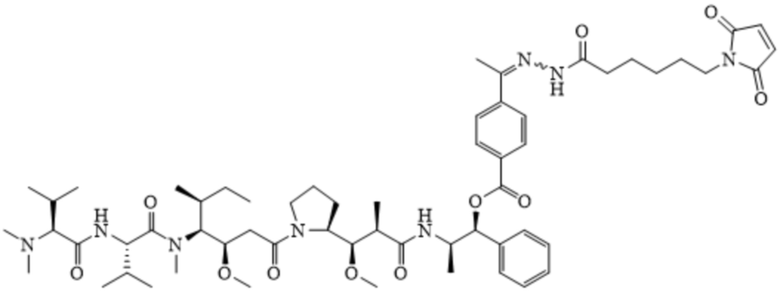
Формула B
[0094] Эти соединения были конъюгированы с тиолсодержащими антителами. Оба соединения содержат алифатический 6-малеимидокапроилгидразоновый фрагмент, обеспечивающий минимальную растворимость в воде для двух соединений, имеющих формулу A и формулу B, изображенных выше. Действительно, конъюгирование обоих указываемых выше соединений с антителами было достигнуто путем растворения соединений только с помощью органических растворителей, то есть смеси ацетонитрил:ДМСО в соотношении 9:1. Использование исключительно органических растворителей в рецептуре применяемой фармацевтической композиции для связывания in situ с цистеином-34 альбумина, циркулирующего в кровотоке, а именно подход к доставке лекарственного средства, описанный в данном документе, не представляется возможным. Соответственно, в некоторых вариантах воплощения композиции по настоящему изобретению не включают органический растворитель.
[0095] Таким образом, изобретение включает ароматические малеимидные линкеры, содержащие фрагмент сульфоновой кислоты, обеспечивающие достаточную растворимость в воде альбумин-связывающих пролекарств для приготовления фармацевтической композиции для внутривенного введения. Одним из таких линкеров является 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразинкарбонил)-бензолсульфоновая кислота, сокращенно обозначаемая Sulf07, имеющая химическую структуру, представленную формулой:
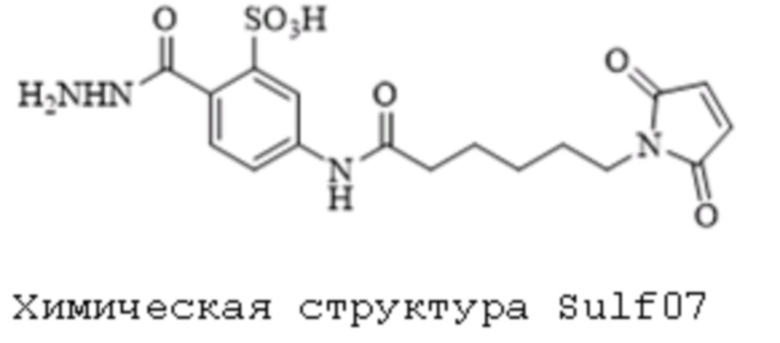
[0096] Линкер Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразинкарбонил)-бензолсульфоновая кислота, был получен в соответствии со способом синтеза A и/или способом синтеза B, как показано на следующих схемах путей синтеза:

[0097] Новый способ синтеза и характеристика нового линкера Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразинкарбонил)-бензолсульфоновой кислоты, который был получен способом синтеза А и В, описаны в Примере 1.
[0098] Линкер Sulf07 реагировал с:
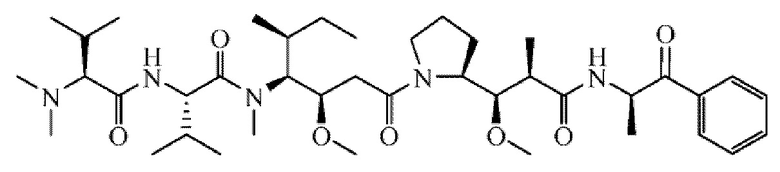
AE-Keto
или
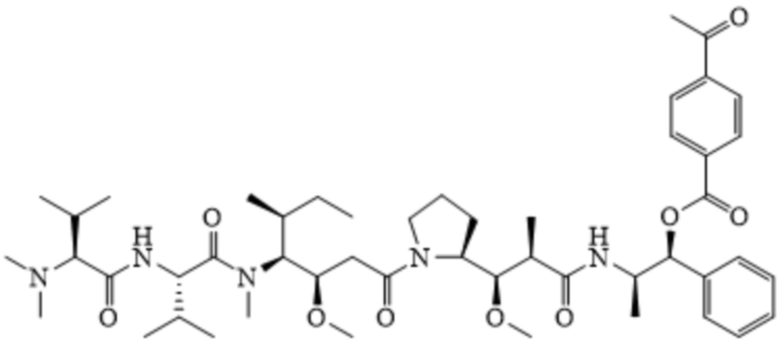
AE-Эстер для того, чтобы получить соединения, описанные формулой III (сокращенно AE-Keto-Sulf07) и формулой IV (сокращенно AE-Эстер-Sulf07), соответственно:
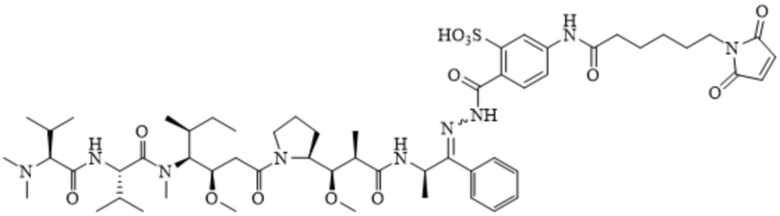
Формула III (AE-Keto-Sulf07)
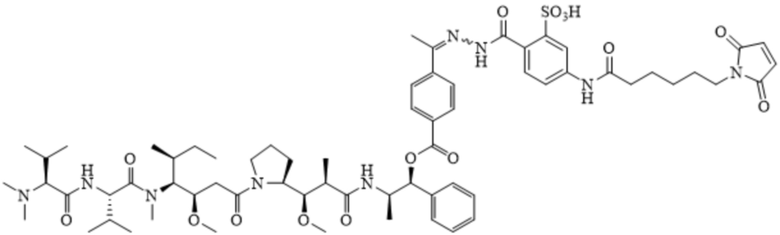
Формула IV (AE-Эстер-Sulf07)
[0099] Синтез и характеристика AE-Keto-Sulf07 (формула III) и AE-Эстер-Sulf07 (формула IV) описаны в примерах 2 и 3.
[00100] Анализ структур AE-Keto-Sulf07 и AE-Эстер-Sulf07 показывает, что в обеих молекулах две группы (группы -SO3H и -N(CH3)2) существуют в виде пары кислота-основание, образуя таким образом цвиттерионы, изображенные на схеме 3 и схеме 4.
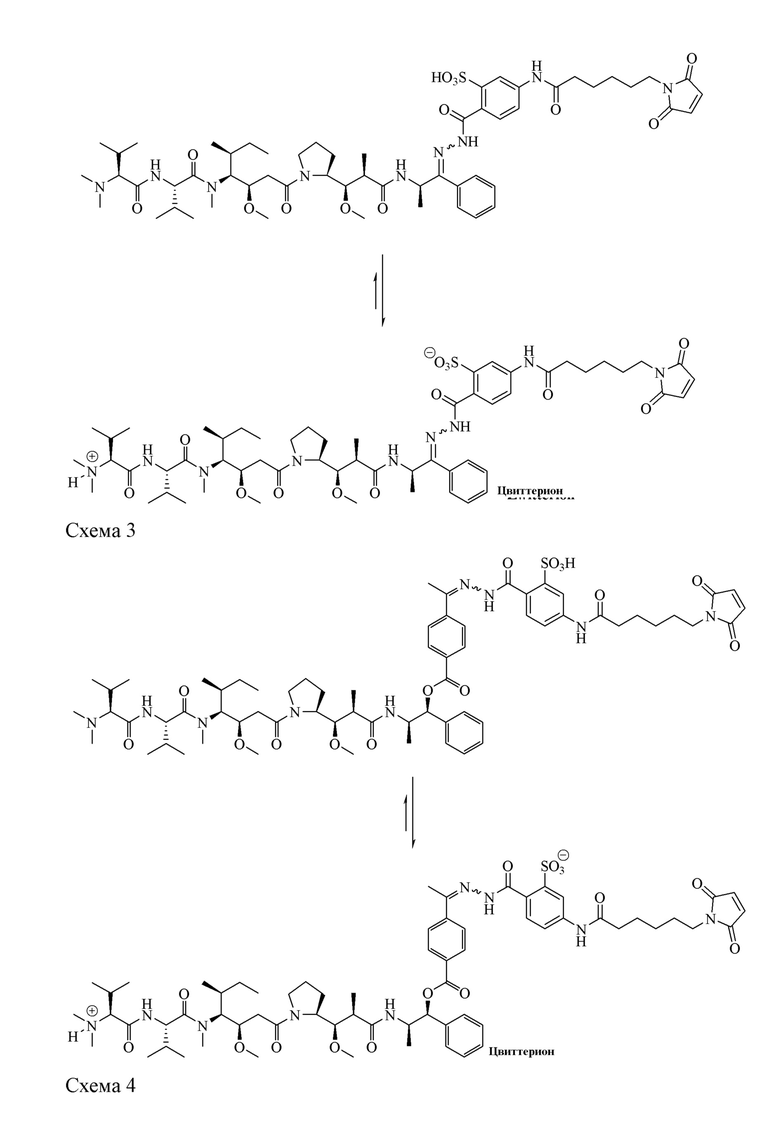
[00101] Фрагмент сульфоновой кислоты, интегрированный в линкер Sulf07, а также цвиттерионное свойство двух пролекарств ауристатина AE-Keto-Sulf07 и AE-Эстер-Sulf07, обеспечивают достаточную растворимость в воде для приготовления фармацевтической композиции и, что более важно, высокую стабильность малеимидной группы, которая значительно улучшена по сравнению с формулой A (AE-Keto-EMCH) и B (AE-Эстер-EMCH):
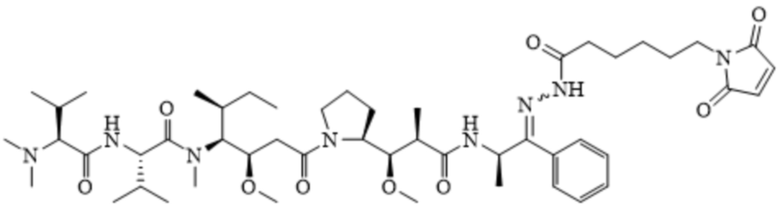
Формула А
Формула B
представлено в US 6884869-B2 (заявка №:10/001191, поданная 01.11.2001).
[00102] Для связывания in situ с цистеином-34 эндогенного альбумина стабильность малеимидной группы в физиологических условиях в диапазоне рН 7,4-7,6 является критерием для внутривенного введения и эффективного связывания с циркулирующим в кровотоке альбумином.
[00103] Как показано на фиг. 1 и фиг. 2, стабильность малеимидной группы по отношению к гидролизу как для AE-Keto-Sulf07, так и для AE-Эстер-Sulf07 значительно улучшена по сравнению с двумя соединениями, описанными в US6884869-B2, т.е. AE-Keto-EMCH (соединение, описанное формулой A) и AE-Эстер-EMCH (соединение, описанное формулой B). После 4 часов инкубации при комнатной температуре в восстановительном буфере (50 мМ фосфат натрия, рН 7,65, содержащий 4% HPβCD и 5% сахарозы), 2,2% малеимида AE-Keto-Sulf07 подверглись гидролизу по сравнению с 5,4% гидролиза малеимида AE-Keto -EMCH и аналогичным образом 2,5% малеимида AE-Эстер-Sulf07 подверглись гидролизу по сравнению с 11,0% для AE-Эстер-EMCH. Кроме того, композиции с активным фармацевтическим ингредиентом (API) производных Sulf07 выявили высокий уровень стабильности (фиг. 24 и фиг. 25) в условиях ускоренного разложения (например, при 55°C в течение до 264 часов), тогда как малеимидные группы производных EMCH быстро подверглись гидролизу. Минимальный уровень гидролиза малеимида необходим для разработки и производства продукта, чтобы обеспечить количественное связывание эндогенного альбумина и, таким образом, ограничить любое преждевременное свободное высвобождение лекарственного средства в кровотоке и максимизировать клиническую эффективность.
[00104] Еще одним преимуществом водных растворов AE-Keto-Sulf07 и AE-Эстер-Sulf07 является то, что они имеют физиологическое значение рН в диапазоне от 6,8 до 7,5.
[00105] Кроме того, растворимость и стабильность цвиттерионных API повышаются при использовании в комбинации с одобренными фармацевтическими носителями, такими как Tween 80, 2-гидроксипропил-β-циклодекстрин, и это может облегчить составление фармацевтической композиции.
[00106] Фармацевтические составы AE-Keto-Sulf07 и AE-Эстер-Sulf07 достигли очень быстрого связывания с альбумином в плазме [человека, мыши и крысы (фиг. 5-10)]. Для обоих терапевтических агентов также была продемонстрирована специфичность связывания с альбумином в плазме человека (фиг. 3-4).
Фармацевтические композиции
[00107] В некоторых вариантах воплощения настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описанное в данном документе.
[00108] Общее количество соединения в композиции для введения пациенту является одним из приемлемых для данного пациента. Специалисту в данной области техники будет понятно, что разным пациентам может потребоваться разное общее количество терапевтически эффективного вещества. В некоторых вариантах воплощения количество соединения является фармацевтически эффективное количеством. Специалист в данной области техники сможет определить количество соединения в композиции, необходимое для лечения пациента, на основе таких факторов, как, например, возраст, вес и физическое состояние пациента. Концентрация соединения зависит от его растворимости в растворе для внутривенного введения и от объема жидкости, который можно вводить. Например, концентрация соединения может составлять от примерно 0,1 мг/мл до примерно 50 мг/мл в композиции для инъекций. В некоторых вариантах воплощения концентрация соединения может находиться в диапазоне от примерно 0,1 мг/мл до примерно 40 мг/мл.
[00109] Фармацевтические композиции и наборы по настоящему изобретению также могут содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы, защитные средства и другие материалы, хорошо известные в данной области техники. Термин «фармацевтически приемлемый» означает нетоксичный материал, который не влияет на степень биологической активности активного(ых) ингредиента(ов). Характеристики носителя будут зависеть от способа введения.
[00110] Композиции можно вводить различными обычными способами. Типичные пути введения, которые можно использовать, включают пероральный, парентеральный, внутривенный, внутриартериальный, кожный, подкожный, внутримышечный, местный, внутричерепной, интраорбитальный, внутриглазной, интравитреальный, интравентрикулярный, интракапсулярный, интраспинальный, интрацистернальный, внутрибрюшинный, интраназальный, аэрозольный, введение в центральную нервную систему (ЦНС) или введение посредством суппозитория. В некоторых вариантах воплощения композиции подходят для парентерального введения. Эти композиции можно вводить, например, внутрибрюшинно, внутривенно или интратекально. В некоторых вариантах воплощения композиции вводят внутривенно. В некоторых вариантах воплощения повторно растворенную композицию можно приготовить путем повторного растворения лиофилизированного соединения композиции в жидкости для повторного растворения, содержащей, например, спирт, ДМСО и/или полиэтиленгликоль и воду и/или солевой буфер. Такое повторное растворение может включать добавление жидкости для повторного растворения и перемешивание, например, путем взбалтывания или встряхивания смеси. Затем повторно растворенную композицию можно сделать подходящей для инъекции путем смешивания, например, лактированного раствора Рингера, 5% раствора глюкозы, изотонического солевого раствора или подходящего солевого буфера с лекарственной формой для создания композиции, пригодной для инъекций. Специалисту в данной области техники будет понятно, что способ введения терапевтически эффективного состава или композиции вещества будет зависеть от таких факторов, как возраст, вес и физическое состояние пациента, подлежащего лечению, и заболевания или состояния, подлежащего лечению. Таким образом, квалифицированный специалист в данной области техники сможет выбрать оптимальный для пациента способ введения в каждом конкретном случае.
[00111] В некоторых вариантах воплощения соединения и композиции, представленные в данном документе, предназначены для применения при лечении рака, вирусного заболевания, аутоиммунного заболевания, острого или хронического воспалительного заболевания и заболевания, вызванного бактериями, грибами или другими микроорганизмами.
[00112] В некоторых вариантах воплощения соединение, представленное в данном документе, может быть использовано при изготовлении лекарственного средства для лечения заболевания, выбранного из следующего: рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами или другими микроорганизмами.
[00113] В некоторых вариантах воплощения рак является раком крови или раком, являющимся солидной опухолью. В некоторых вариантах воплощения рак выбран из карциномы, саркомы, лейкоза, лимфомы, множественной миеломы и меланомы.
[00114] В некоторых вариантах воплощения раком является аденокарцинома, увеальная меланома, острый лейкоз, акустическая неврома, ампулярная карцинома, анальная карцинома, астроцитома, базалиома, рак поджелудочной железы, опухоль соединительной ткани, рак мочевого пузыря, бронхиальная карцинома, немелкоклеточный бронхиальный рак, рак молочной железы, лимфома Беркитта, карцинома главной части органа, рак без выявленного первичного очага (синдром CUP), рак толстой кишки, рак тонкой кишки, рак яичников, рак эндометрия, рак желчного пузыря, карциномы желчного пузыря, рак матки, рак шейки матки, опухоли шеи, носа и ушей, гематологические новообразования, волосатоклеточный лейкоз, рак уретры, рак кожи, глиомы, рак яичка, саркома Капоши, рак гортани, рак кости, колоректальный рак, опухоли головы и/или шеи, рак толстой кишки, краниофарингеома, рак печени, лейкоз, рак легких, немелкоклеточный рак легкого, лимфома Ходжкина, неходжкинская лимфома, рак желудка, рак толстой кишки, медуллобластома, меланома, менингиома, рак почки, почечно-клеточные карциномы, олигодендроглиома, рак пищевода, остеолитические карциномы и остеопластические карциномы, остеосаркома, рак яичника, рак поджелудочной железы, рак полового члена, рак простаты, рак языка, рак яичника или рак лимфатического узла.
[00115] В некоторых вариантах воплощения настоящего изобретения, предложен набор, содержащий соединение, описанное в настоящем документе, и фармацевтически приемлемый наполнитель, носитель и/или разбавитель.
[00116] В некоторых вариантах воплощения в композицию могут быть включены одно или более вспомогательных веществ. Специалисту в данной области техники будет понятно, что выбор какого-либо одного вспомогательного вещества может влиять на выбор какого-либо другого вспомогательного вещества. Например, выбор вспомогательного вещества может препятствовать использованию одного или более из дополнительных вспомогательных веществ, поскольку комбинация вспомогательных веществ будет вызывать нежелательные эффекты. Специалист в данной области техники сможет эмпирически определить, какие вспомогательные вещества, если таковые имеются, следует включать в композиции. Вспомогательные вещества могут включать, без ограничений, сорастворители, солюбилизирующие агенты, буферы, агенты, регулирующие рН, наполнители, поверхностно-активные вещества, инкапсулирующие агенты, агенты, регулирующие тоничность, стабилизаторы, защитные вещества и модификаторы вязкости. В некоторых вариантах воплощения полезным может быть включение в композиции фармацевтически приемлемого носителя.
[00117] В некоторых вариантах воплощения в композиции может быть включен солюбилизирующий агент. Солюбилизирующие агенты могут использоваться для повышения растворимости какого-либо из компонентов композиции, включая соединение или наполнитель. Описанные в данном документе солюбилизирующие агенты не составляют исчерпывающий перечень, а перечислены только в качестве примеров солюбилизирующих агентов, которые можно использовать в композициях. В некоторых вариантах воплощения солюбилизирующие агенты включают, без ограничений, этиловый спирт, трет-бутиловый спирт, полиэтиленгликоль, глицерин, метилпарабен, пропилпарабен, полиэтиленгликоль, поливинилпирролидон, циклодекстрины, такие как диметил-β-циклодекстрин, гидроксиэтил-β-циклодекстрин, гидроксипропил-β-циклодекстрин и триметил-β-циклодекстрин и их комбинации, а также любые фармацевтически приемлемые соли и/или их комбинации.
[00118] pH композиций может быть каким-либо pH, обеспечивающим оптимальные свойства соединения или композиции. Оптимальные свойства могут включать, например, стабильность соединения, повышенную устойчивость соединения по сравнению с композициями при других значениях рН и улучшенную эффективность фильтрации. В некоторых вариантах воплощения значение рН композиций может составлять от приблизительно 3,0 до приблизительно 9,0, например, от приблизительно 5,0 до приблизительно 7,0. В конкретных вариантах воплощения значение рН композиций может составлять 5,5±0,1, 5,6±0,1, 5,7±0,1, 5,8±0,1, 5,9±0,1, 6,0±0,1, 6,1±0,1, 6,2±0,1, 6,3±0,1, 6,4±0,1, 6,5±0,1, 6,6±0,1, 6,7±0,1, 6,8±0,1, 6,9±0,1, 7,0±0,1, 7,1±0,1 и 7,2±0,1.
[00119] В некоторых вариантах воплощения может быть предпочтительно буферировать рН путем включения одного или более буферов в композиции. В некоторых вариантах воплощения буфер может иметь pKa, например, приблизительно 5,5, приблизительно 6,0 или приблизительно 6,5. Специалисту в данной области техники будет понятно, что подходящий буфер может быть выбран для включения в композиции на основе его pKa и других свойств. Буферы являются хорошо известными специалисту в данной области техники. Соответственно, описанные в данном документе буферы не составляют исчерпывающий перечень, а перечислены только в качестве примеров буферов, которые могут использоваться в составах или композициях по настоящему изобретению. В некоторых вариантах воплощения буфер включает, без ограничений, трис, трис-HCl, фосфат калия, фосфат натрия, цитрат натрия, аскорбат натрия, комбинации фосфата натрия и калия, трис/трис-HCl, бикарбонат натрия, фосфат аргинина, гидрохлорид аргинина, гидрохлорид гистидина, какодилат, сукцинат, 2-(N-морфолино)этансульфоновую кислоту (MES), малеат, бис-трис, фосфат, карбонат и какие-либо фармацевтически приемлемые соли и/или их комбинации.
[00120] В некоторых вариантах воплощения в композиции может быть включен агент, регулирующий рН. Изменение рН композиции может оказывать положительное воздействие, например, на стабильность или растворимость соединения, или может быть полезным при получении композиции, подходящей для парентерального введения. Агенты, регулирующие рН, являются хорошо известными специалисту в данной области техники. Соответственно, агенты, регулирующие рН, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров агентов, регулирующих рН, которые могут использоваться в композициях. Агенты, регулирующие рН, могут включать, например, кислоты и основания. В некоторых вариантах воплощения агент, регулирующий рН, включает, без ограничений, уксусную кислоту, соляную кислоту, фосфорную кислоту, гидроксид натрия, карбонат натрия и их комбинации.
[00121] В некоторых вариантах воплощения в композиции может быть включен наполнитель. Наполнители обычно используются в лиофилизированных композициях, чтобы придать композиции дополнительный объем и облегчить визуализацию композиции, особенно в тех случаях, когда без их использования лиофилизированный осадок будет трудно увидеть. Наполнители также могут помочь предотвратить выброс активного(ых) компонента(ов) фармацевтической композиции и/или помочь криозащите композиции. Наполнители являются хорошо известными специалисту в данной области техники. Соответственно, наполнители, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров наполнителей, которые можно использовать в композициях.
[00122] Типичные наполнители могут включать углеводы, моносахариды, дисахариды, полисахариды, сахарные спирты, аминокислоты и сахарные кислоты и их комбинации. Углеводные наполнители включают, без ограничений, моно-, ди- или поликарбоуглеводы, крахмалы, альдозы, кетозы, аминосахары, глицеральдегид, арабинозу, ликсозу, пентозу, рибозу, ксилозу, галактозу, глюкозу, гексозу, идозу, маннозу, талозу, гептозу, глюкозу, фруктозу, метил-α-D-глюкопиранозид, мальтозу, лактон, сорбозу, эритрозу, треозу, арабинозу, аллозу, альтрозу, гулозу, идозу, талозу, эритрулозу, рибулозу, ксилулозу, псикозу, тагатозу, глюкозамин, галактозамин, арабинаны, фруктаны, фуканы, галактаны, галактуронаны, глюканы, маннаны, ксиланы, инулин, леван, фукоидан, каррагинан, галактокаролозу, пектины, амилозу, пуллулан, гликоген, амилопектин, целлюлозу, пустулан, хитин, агарозу, кератин, хондроитин, дерматан, гиалуроновую кислоту, ксантиновую камедь, сахарозу, трегалозу, декстран и лактозу. Наполнители на основе сахарного спирта включают, без ограничений, альдиты, инозиты, сорбит и маннит. Наполнители на основе сахарных кислот включают, без ограничений, альдоновые кислоты, уроновые кислоты, альдаровые кислоты, глюконовую кислоту, изоаскорбиновую кислоту, аскорбиновую кислоту, глюкарную кислоту, глюкуроновую кислоту, глюконовую кислоту, глюкариновую кислоту, галактуроновую кислоту, маннуроновую кислоту, нейраминовую кислоту, пектиновые кислоты и альгиновую кислоту. Аминокислотные наполнители включают, без ограничений, глицин, гистидин и пролин.
[00123] В некоторых вариантах воплощения в композиции может быть включено поверхностно-активное вещество. Поверхностно-активные вещества, как правило, снижают поверхностное натяжение жидкой композиции. Это может обеспечить полезные свойства, такие как улучшенную простоту фильтрации. Поверхностно-активные вещества также могут действовать как эмульгирующие агенты и/или солюбилизирующие агенты. Поверхностно-активные вещества являются хорошо известными специалисту в данной области техники. Соответственно, поверхностно-активные вещества, описанные в данном документе, не составляют исчерпывающий перечень, а представлены только в качестве примеров поверхностно-активных веществ, которые могут быть использованы в составах или композициях по настоящему изобретению. Поверхностно-активные вещества, которые могут быть использованы, включают, без ограничений, сложные эфиры сорбита, такие как полисорбаты (например, полисорбат 20 и полисорбат 80), липополисахариды, полиэтиленгликоли (например, PEG 400 и PEG 3000), полоксамеры (то есть плуроники), этиленоксиды и полиэтиленоксиды (например, Triton X-100), сапонины, фосфолипиды (например, лецитин) и их комбинации.
[00124] В некоторых вариантах воплощения в композиции может быть включен инкапсулирующий агент. Инкапсулирующие агенты могут изолировать молекулы и способствовать их стабилизации или растворению. Инкапсулирующие агенты являются хорошо известными специалисту в данной области техники. Соответственно, инкапсулирующие агенты, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров инкапсулирующих агентов, которые могут использоваться в композициях. Инкапсулирующие агенты, которые могут быть включены в композиции, включают, без ограничений, α-циклодекстрины, β-циклодекстрины, γ-циклодекстрин и их комбинации (например, α-циклодекстрин, диметил-α-циклодекстрин, гидроксиэтил-α-циклодекстрин, гидроксипропил-α-циклодекстрин, триметил-α-циклодекстрин, β-циклодекстрин, диметил-β-циклодекстрин, гидроксиэтил-β-циклодекстрин, гидроксипропил-β-циклодекстрин, триметил-β-циклодекстрин, γ-циклодекстрин, диметил-γ-циклодекстрин, гидроксиэтил-γ-циклодекстрин, гидроксипропил-γ-циклодекстрин, триметил-γ-циклодекстрин и их комбинации.
[00125] В некоторых вариантах воплощения в композиции может быть включен агент, регулирующий тоничность. Тоничность жидкой композиции является важным фактором при введении композиции пациенту, например, путем парентерального введения. Таким образом, агенты, регулирующие тоничность, могут быть использованы для создания композиции, подходящей для введения. Агенты, регулирующие тоничность, являются хорошо известными специалисту в данной области техники. Соответственно, агенты, регулирующие тоничность, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров агентов, регулирующих тоничность, которые могут использоваться в композициях. Агенты, регулирующие тоничность, могут быть ионными или неионными и включают, без ограничений, неорганические соли, аминокислоты, углеводы, сахара, сахарные спирты и углеводы. Типичные неорганические соли могут включать хлорид натрия, хлорид калия, сульфат натрия и сульфат калия. Примером аминокислоты является глицин. Типичные сахара могут включать сахарные спирты, такие как глицерин, пропиленгликоль, глюкозу, сахарозу, лактозу, декстрозу и маннит.
[00126] В некоторых вариантах воплощения в композиции может быть включен стабилизирующий агент. Стабилизирующие агенты помогают увеличить стабильность соединения в композициях. Это может происходить, например, путем уменьшения разложения или предотвращения агрегации соединения. Не ограничиваясь какой-либо теорией, механизмы повышения стабильности могут включать в себя секвестрацию соединения из растворителя или ингибирование свободнорадикального окисления терапевтически эффективного вещества. Стабилизирующие агенты являются хорошо известными специалисту в данной области техники. Соответственно, стабилизирующие агенты, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров стабилизирующих агентов, которые могут использоваться в композициях. Стабилизирующие агенты могут включать, без ограничений, эмульгаторы и поверхностно-активные вещества.
[00127] В некоторых вариантах воплощения в композиции может быть включен защитный агент. Защитные агенты являются агентами, которые защищают фармацевтически активный ингредиент (например, терапевтически эффективное вещество или соединение) от нежелательного состояния (например, нестабильности, вызванной замораживанием, лиофилизацией или окислением). Защитные агенты могут включать, например, криопротекторы, лиопротекторы и антиоксиданты. Криопротекторы полезны для предотвращения потери активности активного фармацевтического ингредиента (например, антрациклинового соединения), когда композицию подвергают воздействию температуры ниже ее точки замерзания. Например, криопротектор может быть включен в восстановленную лиофилизированную композицию, так что композицию можно заморозить перед разбавлением для внутривенного введения. Криопротекторы являются хорошо известными специалисту в данной области техники. Соответственно, криопротекторы, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров криопротекторов, которые можно использовать в композициях. Криопротекторы включают, без ограничений, растворители, поверхностно-активные вещества, инкапсулирующие агенты, стабилизирующие агенты, модификаторы вязкости и их комбинации. Криопротекторы могут включать, например, дисахариды (например, сахарозу, лактозу, мальтозу и трегалозу), полиолы (например, глицерин, маннит, сорбит и дульцит), гликоли (например, этиленгликоль, полиэтиленгликоль и пропиленгликоль).
[00128] Лиопротекторы полезны для стабилизации компонентов композиции, подвергаемой лиофилизации. Например, терапевтически эффективное вещество может быть лиофилизировано с лиопротектором до восстановления. Лиопротекторы являются хорошо известными специалисту в данной области техники. Соответственно, описанные в данном документе лиопротекторы не предназначены для составления исчерпывающего перечня, а представлены только в качестве примеров лиопротекторов, которые можно использовать в композициях. Лиопротекторы включают, без ограничений, растворители, поверхностно-активные вещества, инкапсулирующие агенты, стабилизирующие агенты, модификаторы вязкости и их комбинации. Примерами лиопротекторов могут быть, например, сахара и полиолы. Трегалоза, сахароза, декстран и гидроксипропил-бета-циклодекстрин являются неограничивающими примерами лиопротекторов.
[00129] Антиоксиданты полезны для предотвращения окисления компонентов композиции. Окисление может привести к агрегации лекарственного продукта или другим вредным воздействиям на чистоту лекарственного продукта или его эффективность. Антиоксиданты являются хорошо известными специалисту в данной области техники. Соответственно, антиоксиданты, описанные в данном документе, не составляют исчерпывающий перечень, а перечислены только в качестве примеров антиоксидантов, которые можно использовать в композициях. Антиоксидантами могут быть, например, аскорбат натрия, цитрат, тиолы, метабисульфит и их комбинации.
[00130] В некоторых вариантах воплощения в композицию может быть включен модификатор вязкости. Модификаторы вязкости изменяют вязкость жидких композиций. Это может быть полезно, поскольку вязкость играет важную роль в легкости фильтрации жидкой композиции. Композиция может быть отфильтрована до лиофилизации и восстановления или после восстановления. Модификаторы вязкости являются хорошо известными специалисту в данной области техники. Соответственно, описанные в данном документе модификаторы вязкости не предназначены для составления исчерпывающего перечня, а представлены только в качестве примерных модификаторов вязкости, которые можно использовать в композициях. Модификаторы вязкости включают растворители, солюбилизирующие агенты, поверхностно-активные вещества и капсулирующие агенты. Типичные модификаторы вязкости, которые могут быть включены в композиции, включают, без ограничений, N-ацетил-DL-триптофан и N-ацетил-цистеин.
Противоопухолевая активность на мышиных ксеногенных моделях опухолей человека
[00131] Альбумин-связывающие пролекарства AE-Keto-Sulf07 и AE-Эстер-Sulf07 продемонстрировали исключительную противоопухолевую активность в опухолевых клеточных линиях с IC50 свободных лекарств AE-Keto и AE-Эстер в пикомолярном диапазоне (259 и 339 пМ соответственно по сравнению с IC50 130 пМ исходного соединения AE (см. пример 4), а также на некоторых ксеногенных моделях опухолей человека у голых мышей, вызывая частичную и полную ремиссии во всех ксеногенных моделях опухолей человека, подвергшихся оценке (см. примеры на фиг. 13-23). Эти случаи включали небольшие опухоли с начальными объемами в диапазоне приблизительно 130-170 мм3, а также крупные опухоли с начальными объемами до приблизительно 380 мм3. Кроме того, в большинстве случаев терапия альбумин-связывающими пролекарствами AE-Keto-Sulf07 и AE-Эстер-Sulf07 вызывала длительные ремиссии и уменьшение относительного объема опухоли (RTV). Исходное соединение ауристатин E (AE) в тестируемых моделях было принципиально неактивным или показывало лишь незначительное ингибирование опухоли. Ход эксперимента и результаты на мышиных моделях опухолей подробно описаны в примере 5 и на фиг. 13-23.
Способы лечения
[00132] Соединения и композиции, описанные в данном документе, могут быть использованы для различных клинических применений.
[00133] Соединения и композиции, описанные в настоящем документе, могут вызывать пролонгированное или отдаленное ингибирование роста опухоли. В некоторых вариантах воплощения пролонгированное или отдаленное ингибирование роста опухоли происходит без какой-либо потери массы тела или без какой-либо или только с незначительной токсичностью для костного мозга.
[00134] В некоторых вариантах воплощения, настоящее изобретение относится к способу лечения злокачественного заболевания, включающему введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции, содержащей соединение, описанное в настоящем документе. Например, некоторые варианты воплощения включают в себя способ лечения пациента, страдающего заболеванием, выбранным из группы, включающей рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами и другими микроорганизмами, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения согласно настоящему изобретению.
[00135] Настоящее изобретение относится к способам лечения состояния или заболевания у пациента, выбранного из группы, включающей рак, вирусное заболевание, аутоиммунное заболевание, острое или хроническое воспалительное заболевание и заболевание, вызванное бактериями, грибами или другими микроорганизмами, включающим введение пациенту соединения или фармацевтической композиции способом, описанным в данном документе.
[00136] В некоторых вариантах воплощения рак является раком крови или раком, являющимся солидной опухолью. В некоторых вариантах воплощения рак выбран из группы, включающей карциному, саркому, лейкоз, лимфому, множественную миелому или меланому.
[00137] В некоторых вариантах воплощения раком является аденокарцинома, увеальная меланома, острый лейкоз, акустическая неврома, ампулярная карцинома, анальная карцинома, астроцитома, базалиома, рак поджелудочной железы, опухоль соединительной ткани, рак мочевого пузыря, бронхиальная карцинома, немелкоклеточный бронхиальный рак, рак молочной железы, лимфома Беркитта, карцинома тела органа, рак без выявленного первичного очага (синдром CUP), рак толстой кишки, рак тонкой кишки, рак яичников, рак эндометрия, рак желчного пузыря, карциномы желчного пузыря, рак матки, рак шейки матки, опухоли шеи, носа и ушей, гематологические новообразования, волосатоклеточный лейкоз, рак уретры, рак кожи, глиомы, рак яичка, саркома Капоши, рак гортани, рак кости, колоректальный рак, опухоли головы и/или шеи, рак толстой кишки, краниофарингеома, рак печени, лейкоз, рак легких, немелкоклеточный рак легкого, лимфома Ходжкина, неходжкинская лимфома, рак желудка, рак толстой кишки, медуллобластома, меланома, менингиома, рак почки, почечно-клеточные карциномы, олигодендроглиома, рак пищевода, остеолитические карциномы и остеопластические карциномы, остеосаркома, рак яичника, рак поджелудочной железы, рак полового члена, рак простаты, рак языка, рак яичника или рак лимфатического узла.
[00138] Некоторые варианты воплощения включают способ увеличения концентрации метаболита соединения в опухоли, включающий введение соединения в соответствии с настоящим изобретением.
Варианты и модификации
[00139] Варианты, модификации и другие способы воплощения, описанные в данном документе, будут рассматриваться специалистами в данной области техники без отклонения от сущности и объема настоящего изобретения. Соответственно, настоящее изобретение не должно быть ограничено предшествующим описанием или следующими примерами.
Иллюстративный пример
[00140] Аспекты, в целом описанные в настоящем изобретении, будут более понятны при их рассмотрении со ссылками на следующие примеры, которые включены только в целях иллюстрации определенных признаков и вариантов воплощения настоящего изобретения, но не ограничиваются ими.
Эквиваленты
[00141] Специалисты в данной области техники распознают или смогут установить, используя не более чем обычные эксперименты, многочисленные эквиваленты соединений, композиций и способов их применения, описанных в данном документе. Такие эквиваленты считаются находящимися в рамках настоящего изобретения.
Примеры
Материалы и способы приготовления и анализа соединений
[00142] Все реакции проводили в атмосфере азота, если не указано иное. Коммерчески доступные реагенты использовали без дополнительной очистки, если не указано иное. Безводные растворители были приобретены в безводной форме (дихлорметан, диметилсульфоксид, N, N-диметилформамид, тетрагидрофуран и т.д.), и все другие используемые растворители были класса реагентов для высокоэффективной жидкостной хроматографии (ВЭЖХ) или жидкостной хроматографии в сочетании с масс-спектрометрией (LCMS).
[00143] Стеклянную посуду и мешалки обычно высушивали в печи при 110°С в течение по меньшей мере 12 часов и затем охлаждали в атмосфере азота перед использованием, где это было применимо. Все остальные реакции проводили в круглодонных колбах, герметизированных резиновыми перегородками. Пластиковые шприцы или стеклянные трубки использовались для переноса жидких реагентов. Реакционные смеси перемешивали с помощью магнитных мешалок с тефлоновым покрытием. Органические растворы концентрировали при пониженном давлении с использованием роторного испарителя KNF RC 600 и Heidolph Hei-VAP.
[00144] Колоночную флэш-хроматографию проводили с использованием предварительно упакованных картриджей для флэш-силикагеля Biotage® SNAP Ultra и SNAP Ultra C18, используя системы флэш-очистки Biotage® IsoleraTM One и Biotage® IsoleraTM SL (большого масштаба).
[00145] Значение рН раствора измеряли при комнатной температуре с использованием рН-метра WTW Inolab 7310 с электродами SenTix® mic-D.
[00146] Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили с использованием системы ВЭЖХ Shimadzu Nexera XR, оснащенной матричным фотодиодным детектором SPD-M20A, и проводили мониторинг при 220 нм, если не указано иное.
[00147] Масс-спектры низкого разрешения (LRMS) собирали с использованием жидкостной хроматографии в сочетании с масс-спектрометрией (LCMS) на преимущественных спектрометрах Bruker Amazon SL или Thermo Fisher LCQ (электрораспылительная ионизация, ESI). Масс-спектры высокого разрешения (HRMS) регистрировали на приборе micrOTOF от Bruker, используя жидкостную хроматографию, электрораспылительную ионизацию и масс-спектрометр времени пролета (ESI-TOF). Элементный анализ проводился на Leco TruSpec® CHNS Macro с ИК-детектором для C, H и S; и с детектором теплопроводности (TCD) для N.
[00148] Лиофилизацию проводили с использованием сублимационной сушилки Martin Christ Alpha или Epsilon 2-4 LSCplus.
[00149] Центрифугирование проводили с использованием центрифуги Eppendorf 5810 R, охлажденной, с ротором A-4-81, 230 В/50-60 Гц.
[00150] Содержание трифторуксусной кислоты (ТФК) измеряли с помощью ионной хроматографии, а содержание воды измеряли с помощью кулонометрии Карла Фишера (KF).
[00151] Спектры ядерного магнитного резонанса (ЯМР) регистрировали при температуре окружающей среды на спектрометре 400 МГц: Bruker Avance 400 Ultrashield (400 МГц в 1H, 100 МГц в 13C). Все значения химических сдвигов протонов приведены в частях на миллион (δ) и относятся к дейтерированным протонам в ДМСО-d6 (δ 2,50). Все значения химических сдвигов углерода приведены в частях на миллион (δ) и относятся к углеродным резонансам в ДМСО-d6 (δ 39,52). Данные ЯМР представлены следующим образом: химический сдвиг, кратность (s=синглет, d = дублет, t = триплет, q = квартет, p = пентет, m = мультиплет, br = широкий, dd = дублет дублетов, td = триплет дублетов), константа связи = J (Гц=Герц) и интегрирование.
Способы ВЭЖХ
[00152] Способ 1: способ ВЭЖХ для промежуточных линкеров и чистоты конечного продукта Sulf07. Колонка: Phenomenex Kinetex Polar C18 (150×4,6 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 95: 5 ацетат аммония, 10 мМ, pH 7,0: ацетонитрил, подвижная фаза B: 95: 5, ацетонитрил: ацетат аммония 10 мМ, рН 7,0. Градиент элюирования фазы B: 0-2,5 мин: 0%, 2,5-18 мин: 0-45%, 18-20 мин: 45-75%, 20-24 мин: 75%, 24-26 мин: 75% - 0%, 26-30 минут: 0%, 30 минут: конец способа, скорость потока = 1,0 мл/мин.
[00153] Способ 2: способ ВЭЖХ для примера 2 и примера 3. Колонка: Phenomenex Kinetex Polar C18 (150×4,6 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 95: 5 ацетат аммония, 10 мМ, pH 7,0: ацетонитрил, подвижная фаза B: 95: 5, ацетонитрил: ацетат аммония 10 мМ pH 7,0. Градиент элюирования фазы B: 0-0,5 мин: 30%, 0,5-9 мин: 30-95%, 9-11 мин: 95%, 11-12,5 мин: 95-30%, 12,5-15 мин: 30%, 15 минут: конец способа, скорость потока = 1,0 мл/мин.
[00154] Способ 3: способ LCMS для промежуточных линкеров и конечного продукта Sulf07. Колонка: Phenomenex Kinetex Polar C18 (150×2,1 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 95: 5 ацетат аммония 10 мМ, pH 7,0: ацетонитрил, подвижная фаза B: 95: 5, ацетонитрил: ацетат аммония 10 мМ, рН 7,0. Градиент элюирования фазы B: 0-2,5 мин: 0%, 2,5-18 мин: 0-45%, 18-20 мин: 45-75%, 20-24 мин: 75%, 24-26 мин: 75% - 0%, 26-30 минут: 0%, 30 минут: конец способа. Скорость потока = 0,4 мл/мин.
[00155] Способ 4: способ LCMS для примера 2 и примера 3. Колонка: Phenomenex Luna Omega Polar C18 (50×2,1 мм, 1,6 мкм, 100 Å), градиент: подвижная фаза A: 99,9: 0,1 вода: муравьиная кислота, подвижная фаза В: 99,9: 0,1 ацетонитрил: муравьиная кислота. Градиент элюирования фазы B: 0-0,5 мин: 20%, 0,5-3,5 мин: 20-100%, 3,5-5,0 мин: 100%, 5,0-5,5 мин: 100-20%, 5,5-7,0 мин: 20%, 7 минут: конец способа. Скорость потока = 0,4 мл/мин.
[00156] Способ 5: способ LCMS для экспериментов по связыванию в плазме. Колонка: Phenomenex Kinetex Polar C18 (50×2,1 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 95: 5 ацетат аммония, 10 мМ, pH 7,0: ацетонитрил, подвижная фаза B: 95: 5, ацетонитрил: ацетат аммония 10 мМ, рН 7,0. Градиент элюирования фазы B: 0-1,0 мин: 0%, 1,0-7,0 мин: 0-75%, 7,0-7,5 мин: 75-90%, 7,5-8,5 мин: 90-0%, 8,5-10,0 мин: 0%, 10 минут: конец способа, скорость потока = 0,4 мл/мин.
[00157] Способ 6: Способ хроматографии гидрофобного взаимодействия (HIC) для связывания в плазме. Колонка: MabPac HIC-20 (250×4,6 мм, 5 мкм, 100 Å), градиент: подвижная фаза A: 1,5М (NH4)2SO4, 20 мМ Na2HPO4, рН 8,0, подвижная фаза B: 20 мМ Na2HPO4, рН 8,0 и 20% изопропиловый спирт. Градиент элюирования фазы B: 0-2,0 мин: 0%, 2,0-10,0 мин: 0-50%, 10,0-13,0 мин: 50-60%, 13,0-17,0 мин: 60-100%, 17,0-20,0 мин: 100%, 20,0-25,0 мин: 100-0%, 25,0-30,0 мин: 0%, 30 минут: конец способа, скорость потока = 1 мл/мин.
[00158] Способ 7: способ ВЭЖХ для профиля стабильности рН конъюгата лекарственного средства с альбумином. Колонка: Phenomenex Aeris WP C18 (250×4,6 мм, 3,6 мкм, широкопористая форма), градиент: подвижная фаза A: 20 мМ Трис-буфер, pH 8,0, подвижная фаза B: 90% UHPLC ацетонитрила и 10% воды. Градиент элюирования фазы B: 0-0,5 мин: 25%, 0,5-2,5 мин: 25-35%, 2,5-16,0 мин: 35-85%, 16,0-17,0 мин: 85-95%, 17,0-20,0 мин: 95%, 20,0-25,0 мин: 95-25%, 25,0-30,0 мин: 25%, 30 минут: конец способа, скорость потока=1,0 мл/мин.
[00159] Способ 8: способ ВЭЖХ для ускоренного разложения составов API. Колонка: Phenomenex Kinetex Polar C18 (150×4,6 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 95: 5 ацетат аммония, 10 мМ, pH 7,0: ацетонитрил, подвижная фаза B: 95: 5, ацетонитрил: ацетат аммония 10 мМ, рН 7,0. Градиент элюции фазы B: 0-2,5 мин: 20%, 0,5-9 мин: 20-75%, 9-11 мин: 75%, 11-12,5 мин: 75-20%, 12,5-15 мин: 20%, 15 минут: конец способа, скорость потока=1,0 мл/мин.
[00160] Способ 9: способ ВЭЖХ для промежуточных линкеров и конечного продукта чистоты Sulf07, полученного соответственно пути синтеза С. Колонка: Phenomenex Kinetex Polar C18 (150×4,6 мм, 2,6 мкм, 100 Å), градиент: подвижная фаза A: 0,1% трифторуксусная кислота в H2O, подвижная фаза B: 0,1% трифторуксусная кислота в ацетонитриле. Градиент элюции фазы B: 0-5,5 мин: 1%, 5,5-20 мин: 1-40%, 20-22 мин: 40-65%, 22-24 мин: 65%, 24-27 мин: 65% - 1%, 27-30 минут: 1%, 30 минут: конец способа, скорость потока = 1,0 мл/мин.
[00161] Способ 10: способ ВЭЖХ для определения чистоты AE-Keto-Sulf07, полученного из линкера Sulf07 соответственно пути синтеза C. Колонка Kinetex Polar C18 (2,6 мкм, 100 Å, 150 мм × 4,6 мм), градиент: подвижная фаза A : 95: 5 ацетат аммония 5 мМ pH 7,0: метанол, подвижная фаза B: 95: 5 метанол: ацетат аммония 5 мМ pH 7,0. Градиент элюирования фазы B: 0-2,5 мин: 60%, 2,5-18 мин: 60-80%, 18-22 мин: 80-95%, 22-26 мин: 95%, 26-30 мин: конец способа. Скорость потока = 1,0 мл/мин. Термостат колонок: 37°C.
Пример 1
[00162] Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил) гексанамидо) -2-(гидразин-карбонил)бензолсульфоновую кислоту) получали, как описано ниже и показано на схеме 1, соответственно пути синтеза А.
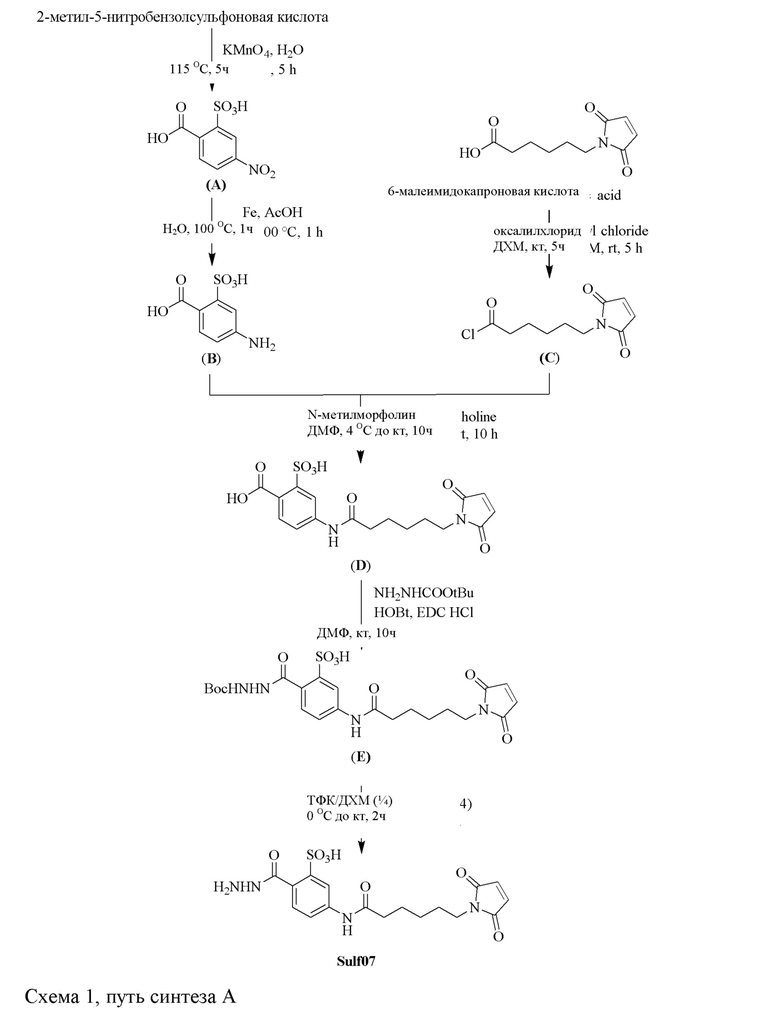
Синтез 4-нитро-2-сульфобензойной кислоты (продукт А)
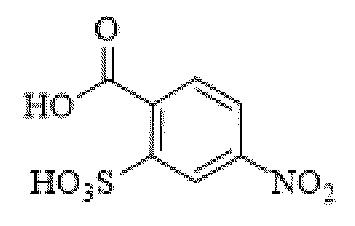
[00163] К перемешиваемому раствору перманганата калия (72 г, 460 ммоль, 4,5 экв., Sigma Aldrich) в воде Millipore (450 мл) в течение 10 секунд добавляли раствор гидрата 4-нитро-2-сульфоновой кислоты (26 г, 102 ммоль, 1,0 экв., Abcr) в воде Millipore (100 мл). Полученную смесь пурпурного цвета перемешивали при 115°C в течение 5 часов, по прошествии этого времени смесь становилась коричневой. ВЭЖХ (способ 1, 220 нм) подтвердил, что реакция завершилась через 5 часов. Реакционную смесь охлаждали до комнатной температуры. Коричневое твердое вещество, образовавшееся во время реакции, удаляли фильтрованием с отсасыванием на подушке из целита, промывали водой Millipore (300 мл) и раствор коричнево-желтого фильтрата концентрировали до ~125 мл при пониженном давлении и температуре 40°C, медленно подкисляли с помощью 5 М HCl до образования белой суспензии (~рН 1). Белую суспензию затем нагревали при 100°C до получения прозрачного раствора, который оставляли стоять на ледяной бане в течение 10 минут до образования белого твердого вещества. Белое твердое вещество получали фильтрованием с отсасыванием с использованием фильтра с фриттой (размер пор 4). Белое твердое вещество затем высушивали в высоком вакууме в течение 10 часов, получая продукт А. Выход: 18 г, 72%. Чистота по ВЭЖХ (способ 1, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C7H4NO7S [М-Н]- : 245,98. Результат: 245,83.
Синтез 4-амино-2-сульфобензойной кислоты (продукт B)
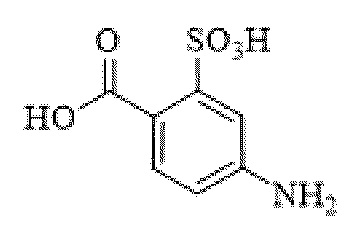
[00164] Перемешиваемую суспензию 4-нитро-2-сульфобензойной кислоты (продукт A) (13 г, 51 ммоль, 1,0 эквив.) в воде Millipore (75 мл) нагревали с обратным холодильником (120°C) до полного растворения. При этой температуре добавляли уксусную кислоту (7,2 мл, Sigma Aldrich), а затем порошок железа (9,5 г, 180 ммоль, 3,5 экв., Sigma Aldrich), который добавляли порциями (~ 1 г/1,0 мин) в течение 10 мин, чтобы избежать экзотермической реакции. Затем реакционную смесь оставляли перемешиваться с обратным холодильником в течение 1 часа. В течение этого времени образовывалось коричневое твердое вещество, и анализ ВЭЖХ (способ 1, 220 нм) подтвердил, что реакция завершилась. Коричневое твердое вещество удаляли фильтрованием с отсосом непосредственно на подушке из целита (когда она еще была горячей) и затем промывали горячей водой (3×50 мл). Фильтрат затем фильтровали через фильтровальную бумагу Whatman (11 мкм). Полученный фильтрат концентрировали при пониженном давлении и температуре 50°С до объема ~100 мл. Концентрированную HCl добавляли по каплям (~1 мл/2,0 мин) до достижения значения pH 1, в результате чего выпадало в осадок белое/желтое твердое вещество. Суспензию оставляли при 4°С на 1 час. Твердое вещество собирали фильтрованием с отсасыванием, используя фильтр с фриттой (размер пор 4), и сушили в высоком вакууме в течение 10 часов, получая продукт B в виде белого твердого вещества. Выход: 9 г, 81%. Чистота по ВЭЖХ (способ 1, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C7H4NO5S [М-Н]- : 216,00. Результат: 216,16.
Синтез 6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноилхлорида или EMC-Cl (продукт C)
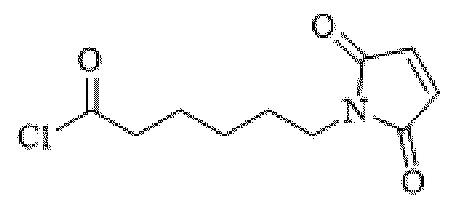
[00165] К перемешиваемому раствору 6-малеимидокапроновой кислоты (EMC) (33 г, 156 ммоль, 1,0 экв., Alfa Aesar) в сухом дихлорметане (150 мл) при комнатной температуре и в атмосфере азота добавляли в течение 30 минут (~1 мл/2 мин) оксалилхлорид (15 мл, 171 ммоль, 1,1 эквив., Sigma Aldrich) с использованием капельной воронки. Внимание: во время процесса добавления наблюдалось выделение газа. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Цвет реакционного раствора изменялся на темно-желтый в течение времени реакции, и анализ ВЭЖХ (способ 1, 220 нм) подтвердил, что реакция была завершена через 5 часов. Растворитель удаляли при пониженном давлении и температуре 40°С, получая масло, которое сушили в высоком вакууме в течение ночи, получая затвердевшее соединение. Полученное светло-коричневатое твердое вещество измельчали шпателем и сушили в течение еще 20 часов в высоком вакууме, получая продукт С в виде желтого микрокристаллического твердого вещества. Соединение использовали в следующей реакции без дополнительной очистки. Выход: 34 г, 95%. Чистота по ВЭЖХ (способ 1, 220 нм) более 95% в виде метилового эфира. LRMS-ESI (m/z) рассчит. для C11H16NO4 (в виде метилового эфира) [M+H]+: 226,10. Результат: 225,97.
Синтез 4-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-сульфобензойной кислоты (продукт D)
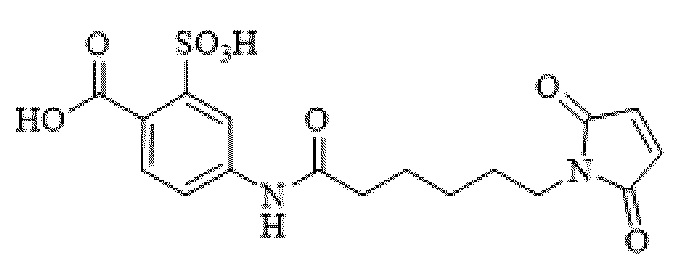
[00166] Продукт B (18,5 г, 85 ммоль, 1,0 эквив.) растворяли в безводном N, N-диметилформамиде (300 мл) в атмосфере азота. Раствор охлаждали на ледяной бане и оставляли перемешиваться в течение 10 мин до достижения 4°С. Затем 4-метилморфолин (18,69 мл, 170 ммоль, 2,0 экв., Sigma Aldrich) добавляли к охлажденному раствору по каплям (~1 мл/3,5 мин) в течение 1 ч, используя капельную воронку. После завершения добавления смесь становилась темно-коричневой, и к этой темно-коричневой смеси добавляли по каплям (~0,5 г/мин) в течение 1 часа, используя капельную воронку, раствор продукта С (29,28 г, 127 ммоль, 1,5 экв.) в безводном N, N-диметилформамиде (200 мл). Реакционной смеси давали постепенно нагреться до комнатной температуры в течение ночи и затем давали перемешиваться при комнатной температуре в течение 10 часов. После полного превращения реакции, что было определено с помощью ВЭЖХ (способ 1, 220 нм), реакционный раствор распределяли в пробирки Falcon объемом 8×50 мл и центрифугировали в течение 20 минут при 10°C и 4000 об/мин. Супернатанты удаляли и твердые вещества ресуспендировали в 10 мл N,N-диметилформамида на каждую пробирку и снова центрифугировали в течение 20 минут при 10°С и 4000 об/мин. Все супернатанты объединяли и концентрировали при пониженном давлении и температуре 50°С в течение 3 ч, получая светло-оранжевое твердое вещество (выход: 34 г, чистота по ВЭЖХ (способ 1, 220 нм) 66%). Это твердое вещество повторно суспендировали в метаноле (250 мл), переносили в пробирки Falcon 8×50 мл и центрифугировали в течение 20 минут при 10°C и 4000 об/мин. Супернатанты удаляли, и твердые вещества повторно суспендировали в 5 мл метанола на каждую пробирку и снова центрифугировали в течение 20 минут при 10°С и 4000 об/мин. Все твердые вещества объединяли и сушили в высоком вакууме в течение 24 часов, получая продукт D в виде твердого вещества кристаллического желтого цвета. Выход: 24 г, 37%. Чистота по ВЭЖХ (способ 1, 220 нм) 97%. LRMS-ESI (m/z) рассчит. для C17H17N2O8S [М-Н]- : 409,08. Результат: 409,13.
Синтез 2-(2-(трет-бутоксикарбонил)гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил) гексанамидо)бензолсульфоновой кислоты или BOC-защищенного линкера (продукт E)
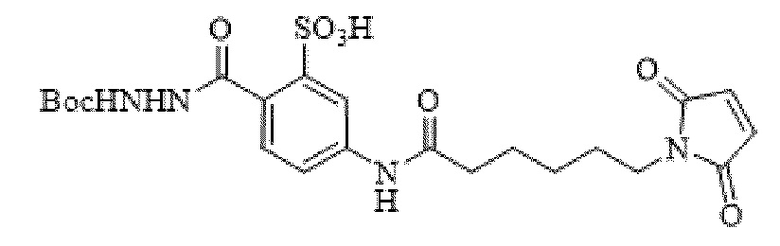
[00167] К раствору продукта D (17 г, 41,4 ммоль, 1,0 эквив.) в безводном N-N-диметилформамиде (350 мл) в атмосфере азота добавляли N-(3-диметиламинопропил) -N'-этилкарбодиимид-гидрохлорид (EDC-HCl) (8,72 г, 45,5 ммоль, 1,1 экв., Roth) и гидроксибензотриазол (HOBt) (6,15 г, 45,5 ммоль, 1,1 экв., Sigma Aldrich). Реакционную смесь оставляли перемешиваться в течение 30 мин при комнатной температуре, а затем добавляли трет-бутилкарбазат (7,12 г, 53,9 ммоль, 1,3 экв., Sigma Aldrich) и раствор становился прозрачно-желтым до красноватого цвета. Реакционную смесь перемешивали при комнатной температуре в течение ночи. По истечении этого времени полная конверсия реакции была подтверждена ВЭЖХ (способ 1, 220 нм). Растворитель удаляли при пониженном давлении и температуре 40°C и затем в высоком вакууме при комнатной температуре в течение 1 часа, получая пурпурно-коричневое масло, которое очищали с помощью системы быстрой очистки с двумя предварительно упакованными картриджами SNAP Ultra 340 g. Очистку осуществляли с использованием системы линейного градиента от 100% дихлорметана к соотношению 90%/10% дихлорметан/метанол в 60 объемах колонки. Пробирки, содержащие желаемый продукт, объединяли и сушили при пониженном давлении при 40°C в течение 1 часа и в высоком вакууме еще в течение 6 часов, чтобы получить продукт E в виде пенисто-желтого твердого вещества. Выход: 9 г, 17,1 ммоль, 42%. ВЭЖХ (способ 1, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C22H27N4O9S [М-Н]- : 523,16. Результат: 523,15.
Синтез линкера Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил) гексанамидо)-2-(гидразинкарбонил)бензолсульфоновой кислоты
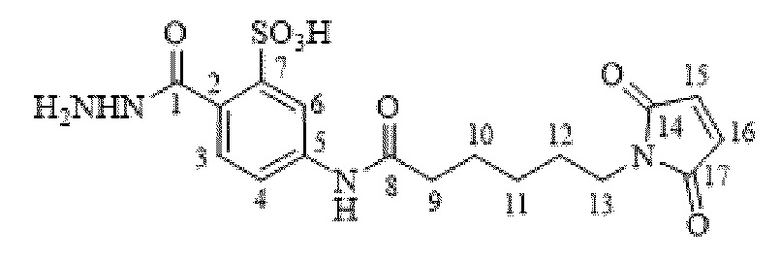
[00168] К раствору желтого цвета продукта Е (10,2 г, 19,4 ммоль, 1,0 эквив.) в безводном дихлорметане (30 мл), который охлаждался до 4-5°С, добавляли по каплям (~1 мл/2,0 мин) в течение 30 минут трифторуксусную кислоту (15 мл, Roth.). После добавления холодную баню убирали и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 3 часов. По истечении этого времени завершение реакции подтверждали ВЭЖХ (способ 1, 220 нм). Реакционную смесь выливали по каплям в 6 пробирок Falcon, каждая из которых содержала ~35 мл холодного диэтилового эфира. Белый осадок образовывался сразу. Пробирки оставляли стоять при 4°С в течение 3 часов. После центрифугирования пробирок Falcon в течение 20 минут при 10°C и 4000 об/мин супернатанты удаляли и твердые вещества повторно суспендировали в 5 мл диэтилового эфира на каждую пробирку и снова центрифугировали (4000 об/мин, 20 мин, 10°C). Супернатанты снова удаляли, и твердые вещества собирали и сушили в высоком вакууме в течение 20 часов, получая линкер Sulf07 в виде белого микрокристаллического твердого вещества. Выход: 10 г, 23,6 ммоль, 96%. ВЭЖХ (способ 1, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C17H21N4O7S [М+Н]+: 425,11. Результат: 425,07. LRMS-ESI (m/z) рассчит. для C17H19N4O7S [М-Н]- : 423,11. Результат: 423,12. HRMS-ESI (m/z) рассчит. для C17H21N4O7S [М+Н]+: 425,1125. Результат: 425,1125. LRMS-ESI (m/z) рассчит. для C17H19N4O7S [М-Н]-: 423,0978. Результат: 423,0980.
[00169] Структура 5-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил) гексаанамидо)-2-(гидразинкарбонил)бензолсульфоновой кислоты, Sulf07, была подтверждена с помощью 1H ЯМР: 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,94 (s, 1Н; С1-NH), 10,27 (s, 1Н; С8-NH), 8,01 (d, J=2,2 Гц, 1Н; С4-СН ), 7,93 (dd, J=8,5, 2,2 Гц, 1H; C6-CH), 7,68 (d, J=8,4 Гц, 1H; C7-CH), 7,00 (s, 2H; C15-CH, C16-CH) 3,40 (t, J=7,0 Гц, 2H; C13-CH2), 2,32 (t, J=7,4 Гц, 2H; C9-CH2), 1,60 (p, J=7,5 Гц, 2H; C10-CH2), 1,52 (p, J=7,2 Гц, 2H; C12-CH2), 1,26 (q, J=8,8 Гц, 2H; C11-CH2); 13C ЯМР (101 МГц, ДМСО-d6) δ 172,20 (С8), 171,54 (С14, С17), 167,59 (С1), 145,73 (С5), 142,09 (С3), 134,90 (С15, С16), 132,09 (С7), 123,79 (С2), 119,44 (С6), 117,47 (С4), 37,42 (С13), 36,65 (С9), 28,22 (С12), 26,21 (С11), 24,90 (С10). Элементный анализ вычисл. для C17H21N4O7S; С 45,71; Н 4,26; N 11,85; S 6,78. Найдено: С 46,2917; Н 4,4836; N 12,8879; S 6,7886. Содержание ТФК < 2% (w/w).
[00170] Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразин-карбонил) бензолсульфоновую кислоту, получали, как описано ниже, и показано на схеме 2 соответственно пути синтеза B.
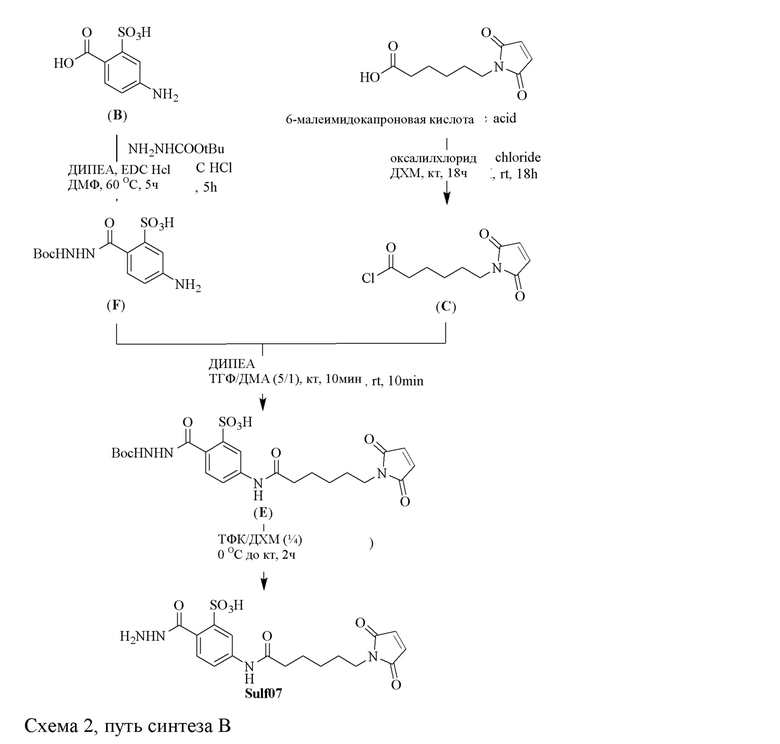
Синтез 5-амино-2-(2-(трет-бутоксикарбонил)гидразин-1-карбонил) бензолсульфоновой кислоты (продукт F)
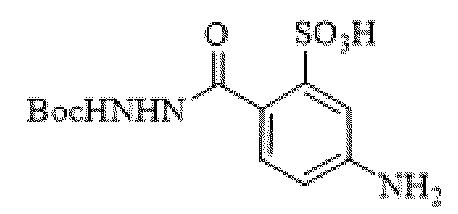
[00171] К суспензии 4-амино-2-сульфобензойной кислоты (продукт B) (50,00 г, 230,20 ммоль, 1,00 экв.) в безводном N, N-диметилформамиде (ДМФ, 1000 мл) добавляли N,N-диизопропилэтиламин (ДИПЕА, 40,20 мл, 29,75 г, 230,20 ммоль, 1,00 эквив.) в течение 5 минут. Затем добавляли EDC-HCl (48,54 мг, 253,21 ммоль, 1,10 эквив.) и смесь перемешивали при 23°C в течение 30 минут. Затем добавляли трет-бутилкарбазат (33,46 г, 253,18 ммоль, 1,10 экв.) и смесь нагревали до 70°С и перемешивали в течение 2 часов. Добавляли вторую порцию EDC-HCl (24,31 г, 126,68 ммоль, 0,55 экв.), трет-бутилкарбазат (16,76 мг, 126,68 ммоль, 0,55 экв.) и реакционную смесь дополнительно перемешивали при 70°C в течение 14 часов. Реакционную смесь охлаждали до 23°C и фильтровали через Celite® 545 (100 г). Celite® 545 дополнительно промывали 700 мл метанола. Фильтрат концентрировали при пониженном давлении и очищали с помощью системы быстрой очистки, используя семь предварительно упакованных картриджей SNAP Ultra 340 g. Очистку осуществляли с использованием системы линейного градиента от 100% дихлорметана до 70% дихлорметана/30% метанола с получением указанного в заголовке продукта F в виде белого твердого вещества. Выход: 32,50 г, 98,09 ммоль, 42,6%, ВЭЖХ (способ 1, 220 нм) более 97%. LRMS-ESI (m/z) рассчит. для C12H16N3O6S [М-Н]-: 330,08. Результат: 330,17.
Синтез N-этил-N-изопропилпропан-2-амин 2-(2-(трет-бутоксикарбонил) гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-)гексанамидо)бензолсульфоната (продукт E)
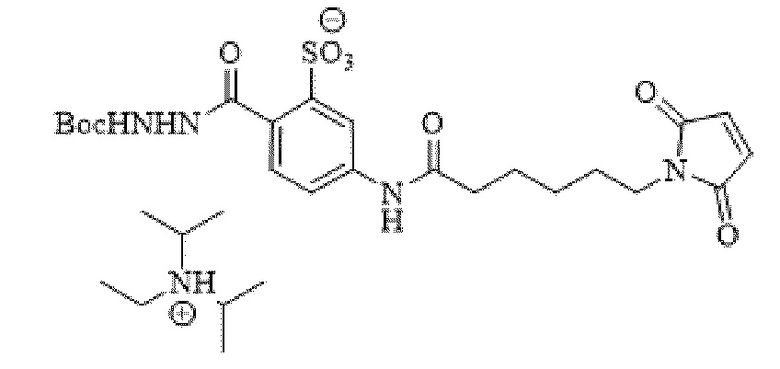
[00172] К раствору продукта F (26,97 г, 81,40 ммоль, 1,00 экв.) в смеси безводного диметилацетамида (110 мл) и безводного тетрагидрофурана (ТГФ, 275 мл) добавляли раствор хлорида 6-малеимидокапроновой кислоты (продукт C) (26,60 г, 115,82 ммоль, 1,42 экв.) в безводном тетрагидрофуране (165 мл) одной порцией при комнатной температуре. Прозрачный реакционный раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь добавляли при перемешивании в две 2-литровые конические колбы, каждая из которых содержала диизопропиловый эфир (1100 мл) и ДИПЕА (10,15 мл). После отстаивания масла надосадочную жидкость осторожно выливали и масло промывали диизопропиловым эфиром (2×100 мл), растворяли в метаноле, объединяли и растворитель удаляли в высоком вакууме. Полученное масло очищали с помощью системы флэш-очистки с использованием шести предварительно упакованных картриджей SNAP Ultra 340 г. Очистку осуществляли с использованием системы линейного градиента от 100% дихлорметана до 80% дихлорметана/20% метанола с получением указанного в заголовке соединения E в виде белого твердого вещества. Выход: 24,00 г, 36,71 ммоль, 45,1%, ВЭЖХ (способ 1, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C22H27N4O9S [М-Н]-: 523,15. Результат: 523,30.
Синтез 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразин-карбонил) бензолсульфоновой кислоты (Sulf07)
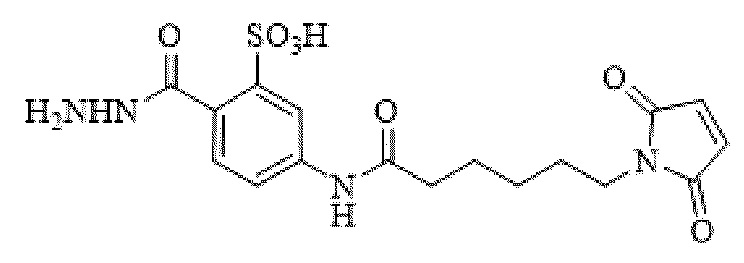
[00173] К холодному раствору (4°C) продукта E (21,00 г, 32,12 ммоль, 1,00 экв.) в дихлорметане (150 мл) добавляли трифторуксусную кислоту (25,00 мл, 37,25 г, 326,70 ммоль, 10,17 экв.) более 1 часа. Затем реакционной смеси давали постепенно нагреться (в течение 30 минут) до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь добавляли по каплям через делительную воронку в течение 1 часа в коническую колбу объемом 2 л, содержащую диизопропиловый эфир (1,4 л) при 0°С. Полученное белое твердое вещество отфильтровывали через воронку с пористостью 4 Å и последовательно промывали дихлорметаном (2×1000 мл) и диизопропиловым эфиром (2×1000 мл). Твердое вещество оставляли сушиться на воронке с фриттой в течение ночи при комнатной температуре. Дальнейшую сушку проводили в высоком вакууме при 25°С в течение 2 часов. Конечный продукт Sulf07 был получен в виде белого твердого вещества. Выход: 13,58 г, 32,00 ммоль, 99,6%, ВЭЖХ (способ 1, 220 нм)> 96%. LRMS-ESI (m/z) рассчит. для C17H19N4O7S [М-Н]-: 423,10. Найдено: 423,21.
[00174] Sulf07, 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразин-карбонил)бензолсульфокислоту получали, как описано ниже, и показано на схеме 2 соответственно пути синтеза C.
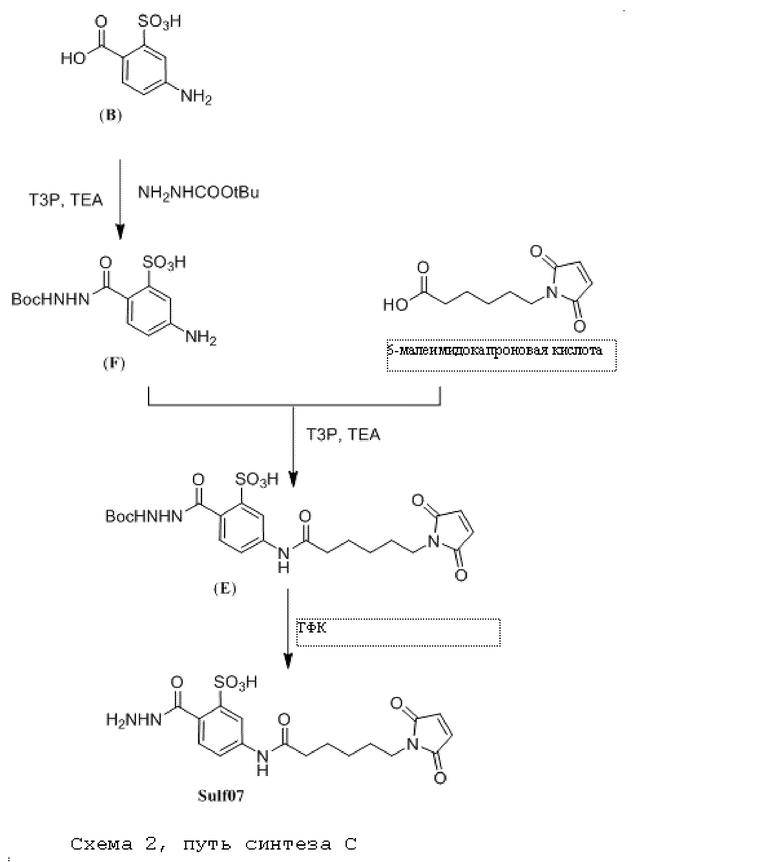
Синтез 5-амино-2-(2-(трет-бутоксикарбонил) гидразин-1-карбонил)бензолсульфоновой кислоты (продукт F)
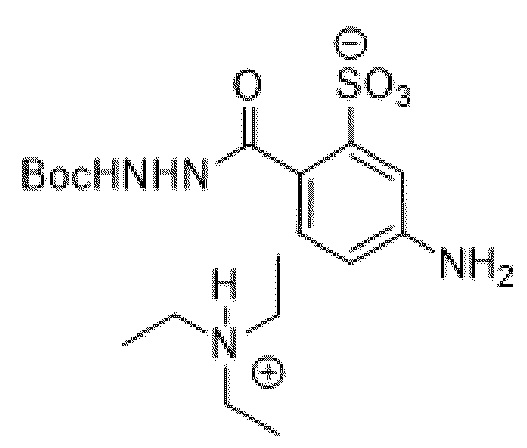
[00175] К суспензии продукта B (30,00 г, 138,12 ммоль, 1,00 экв.) в безводном ацетонитриле (600 мл) добавляли триэтиламин (41,93 г, 57,76 мл, 414,37 ммоль, 3,00 экв.) и смесь перемешивали в течение 10 минут. Затем добавляли трет-бутилкарбазат (27,38 г, 207,19 ммоль, 1,50 экв.) и смесь охлаждали до минус 35°С. При этой температуре по каплям в течение 1 часа добавляли раствор пропилфосфонового ангидрида, Т3Р (114,27 г, 106,79 мл, 179,56 ммоль, 50% раствор в этилацетате, 1,3 экв.). Реакционную смесь перемешивали при минус 35°С в течение 2 часов. Смеси давали нагреться до комнатной температуры и фильтровали через Celite® 545 (100 г). Celite ® дополнительно промывали ацетонитрилом (500 мл). Оба фильтрата объединяли и концентрировали до 250 мл. Раствор разделяли поровну на 6 порций и растворитель удаляли при пониженном давлении. Каждую порцию растворяли в дихлорметане, содержащем 1% Et3N (50 мл), и очищали способом флэш-хроматографии NP на системе быстрой очистки Biotage IsoleraTM One, с предварительно заполненной колонкой SNAP Ultra 340 г, используя ступенчатый градиент от 2% до 12% метанола (содержащий 1% NEt3) в ДХМ (содержащий 1% NEt3) на 7 объемах колонки. Затем очищенные фракции из всех порций объединяли, растворитель удаляли при пониженном давлении и твердое вещество сушили в высоком вакууме, получая указанное в заголовке соединение F в виде беловатого твердого вещества. Выход: 53,25 г, 108,0 ммоль, 78,2% (ЯМР в ДМСО-d6 показал присутствие 1,6 экв. триэтиламина). ВЭЖХ (способ 9, 220 нм) более 99%. LRMS-ESI (m/z) рассчит. для C12H16N3O6S [М-Н]-: 330,08. Результат: 330,08.
Синтез N-этил-N-изопропилпропан-2-амин 2-(2-(трет-бутоксикарбонил) гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-)гексанамидо) бензолсульфоната (продукт E)
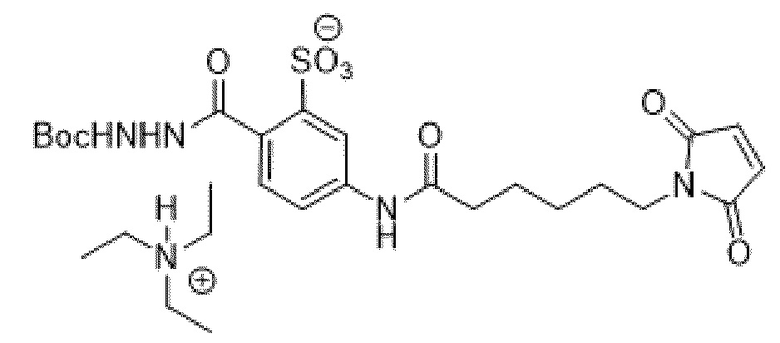
[00176] К смеси продукта F (45,00 г, 91,24 ммоль, 1,00 экв.) и 6-малеимидокапроновой кислоты (19,27 г, 91,24 ммоль, 1,00 экв.) добавляли одной порцией при комнатной температуре ацетонитрил (450 мл), триэтиламин (13,85 г, 19,08 мл, 136,86 ммоль) и T3P (43,55 г, 40,70 мл, 136,86 ммоль, 50% раствор в этилацетате). Раствор перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли при пониженном давлении. Затем неочищенный продукт очищали с помощью системы флэш-очистки, используя семь предварительно упакованных картриджей SNAP Ultra 340 г с линейным градиентом от 2% метанола до 15% метанола в дихлорметане, с получением указанного в заголовке соединения E в виде беловатого твердого вещества. Выход: 30,55 г, 53,5% (ЯМР в ДМСО-d6 показал присутствие 1,1 экв. триэтиламина). ВЭЖХ (способ 9, 220 нм) более 99%. LRMS-ESI (m/z) рассчит. для C22H27N4O9S [М-Н]-: 523,15. Результат: 523,26.
Синтез 5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-2-(гидразин-карбонил) бензолсульфоновой кислоты (Sulf07)
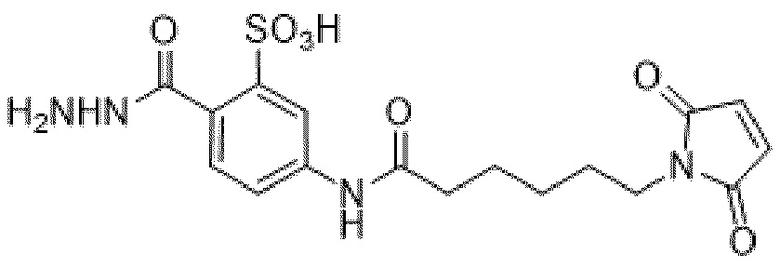
[00177] К холодной суспензии (4°С) продукта Е (10,00 г, 15,98 ммоль, 1,00 экв.) в дихлорметане (50 мл) добавляли трифторуксусную кислоту (18,22 г, 12,31 мл, 159,81 ммоль, 10,17 экв.) в течение 15 мин. Смесь дополнительно перемешивали при 4°С в течение 15 минут, а затем давали постепенно нагреться до комнатной температуры и перемешивали в течение 150 минут. Реакционную смесь добавляли по каплям через делительную воронку к перемешиваемому раствору метил-трет-бутилового эфира, МТВЕ (400 мл) и дихлорметана (200 мл). Полученное белое твердое вещество отфильтровывали через воронку с пористостью 4 Å и последовательно промывали дихлорметаном (2×150 мл) и МТВЕ (1×50 мл), МеОН (1×50 мл) и снова МТВЕ (2×150 мл). Твердое вещество оставляли сушиться на воронке с фриттой в ночной период при комнатной температуре в течение 10 минут. Дальнейшую сушку проводили в высоком вакууме при 25°С в течение 18 часов. Конечный продукт Sulf07 получали в виде желтого твердого вещества. Выход: 5,786 г, 13,63 ммоль, 98,7%, ВЭЖХ (способ 9, 220 нм) более 96%. LRMS-ESI (m/z) рассчит. для C17H19N4O7S [М-Н]- : 423,10. Результат: 422,95.
Пример 2
[00178] Соединение AE-Keto-Sulf07, 2-(2-((R)-2-((2R, 3R)-3-((S)-1-((3R, 4S, 5S)-4-((S)-2-((S)-2-(диметиламино)-3-метилбутанамидо)-N, 3-диметилбутанамидо) -3-метокси-5-метилгептаноил) пирролидин-2-ил) -3-метокси-2- метилпропанамидо) -1-фенилпропилиден) гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил) гексанамидо) бензолсульфоновую кислоту синтезировали, как описано ниже, и показано на схеме 5.
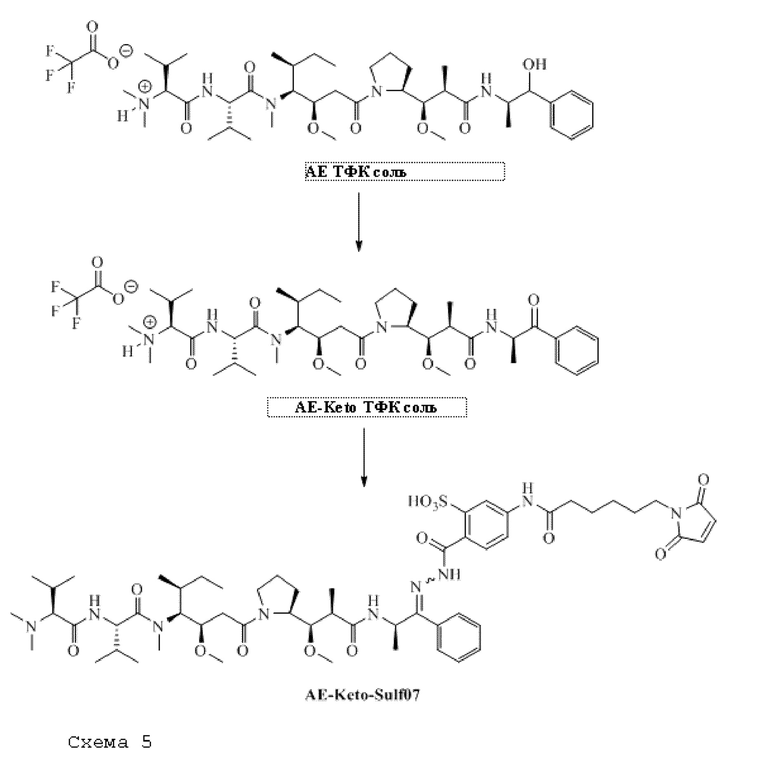
Синтез (S)-1-(((S)-1-(((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((R)-1-оксо-1-фенилпропан-2-ил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)(метил)амино)-3-метил-1-оксобутан-2-ил)амино)-N,N,3-триметил-1-оксобутан-2-амин 2,2,2-трифторацетата (AE-Keto ТФК соль)
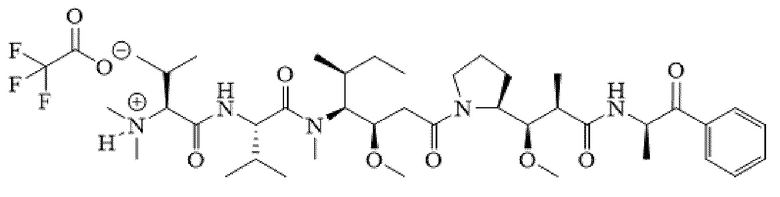
Путь синтеза A: Синтез в соответствии с улучшенной процедурой патента US6884869-B2.
[00179] Хлорхромат пиридиния (PCC) (382 мг, 1,772 ммоль, 1,5 экв., Sigma Aldrich) добавляли к раствору соли ТФК ауристатина E (1000 мг, 1,183 ммоль, Levena Biopharma) в безводном дихлорметане (30 мл) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 17 часов. LC-MS (способ 4, положительный режим) показала полную конверсию исходного материала. Реакционную смесь разбавляли 25 мл дихлорметана и промывали рассолом (3×20 мл). Водную фазу экстрагировали один раз дихлорметаном (20 мл). Объединенные органические фазы упаривали при пониженном давлении и температуре 40°С. Остаток растворяли в 7,5 мл ацетонитрила и очищали с помощью RP-флэш-хроматографии с предварительно упакованным 30 г картриджем SNAP Ultra C18, используя ступенчатый градиент от 2% до 40% ацетонитрила (+0,1% ТФК) в воде (+0,1% ТФК) в более 16 объемах колонки. Фракции чистого продукта объединяли, ацетонитрил удаляли при пониженном давлении и температуре 40°С и остаточный растворитель лиофилизировали с получением белого твердого вещества. Выход: 530 мг, 53%, RP-ВЭЖХ (способ 2, 220 нм): более 95%. LRMS-ESI (m/z) рассчит. для C40H68N5O7 [М+Н]+: 730,51. Результат: 730,34.
Путь синтеза B: синтез с использованием связанного с полимером окислителя.
[00180] IBX-полистирол (1,13 г, 4,5 экв., 1,34 ммоль, загрузка 1,18 ммоль/г, Merck Novabiochem) суспендировали в безводном дихлорметане (7,5 мл) и осторожно перемешивали в течение 15 минут при комнатной температуре. Добавляли соль AE ТФК (250 мг, 295,51 мкмоль, Sage Chemical Co.), растворенную в безводном дихлорметане (5 мл), и смесь осторожно перемешивали при комнатной температуре. Через 21 ч полная конверсия исходного материала в продукт наблюдалась с помощью ВЭЖХ (способ 2, 220 нм). Смолу удаляли фильтрацией на согнутой фильтровальной бумаге (класс 3, Sartorius), промывали дихлорметаном и объединенные фильтраты упаривали досуха при пониженном давлении при 40°C. Масляный остаток растирали с диэтиловым эфиром, образуя беловатый осадок, который сушили в высоком вакууме, получая пенистое беловатое твердое вещество. Выход: 171 мг, 69%, ВЭЖХ (способ 2, 220 нм) более 99%. LRMS-ESI (m/z) рассчит. для C40H68N5O7 [М+Н]+: 730,51. Результат: 730,69; рассчит. для C40H66N5O7 [М-Н]- : 728,50. Результат: 728,79.
Синтез 2- (2-((R)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-(диметиламино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-1-фенилпропилиден)гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)бензолсульфоновой кислоты (AE-Keto-Sulf07)
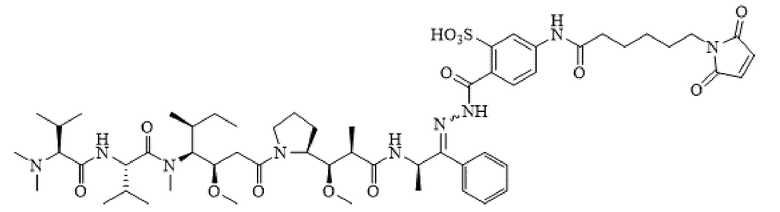
[00181] Синтез 1. В атмосфере азота соль AE-Keto ТФК (200 мг, 236,96 мкмоль) растворяли в безводном этаноле (6 мл) с последующим добавлением молекулярных сит (шарики 0,3 нм, 3А). Добавляли Sulf07 (201 мг, 473,92 мкмоль, 2 экв.), растворенный в безводном диметилсульфоксиде (1,5 мл). Желтый раствор перемешивали при комнатной температуре. Образцы для ВЭЖХ/LC-MS анализов (способы 2 и 4, 220 нм) отбирали через 24 и 44 часа. После конверсии (85% через 44 часа) смесь фильтровали через шприцевой фильтр с PVDF 0,2 мкм, большую часть этанола удаляли при пониженном давлении. 3 мл 200 мМ фосфата натрия pH 7,2 добавляли по каплям до тех пор, пока pH раствора не составлял ~6. Раствор (~5 мл) очищали с помощью RP-флэш-хроматографии на предварительно упакованном картридже SNAP Ultra C18 30 г, используя ступенчатый градиент от 2% до 50% ацетонитрила в воде на 16 объемов колонки. Фракции продукта анализировали с помощью ВЭЖХ (способ 2, 220 нм), чистые фракции объединяли, ацетонитрил удаляли при пониженном давлении и оставшийся раствор лиофилизировали с получением беловатого рыхлого твердого вещества. Выход: 113 мг, 42%, температура плавления: выше 105°C. ВЭЖХ (способ 2, 220 нм) более 95%. LRMS-ESI (m/z) рассчит. для C57H86N9O13 [М+Н]+: 1136,61. Результат: 1136,76; рассчит. для C57H84N9O13 [М-Н]-: 1134,59. Результат: 1135,09. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,21 (2 х s, 1H), 10,09 (2 х s, 1H), 8,66 (d, J=8,3 Гц, 1H), 8,20 (2 х d, J=8,4 Гц, 1H ), 7,84-7,69 (m, 2H), 7,50-7,41 (m, 1H), 7,35-7,21 (m, 5H), 6,98 (s, 2H), 4,96-4,84 (m, 1H), 4,77-4,60 (m, 1H), 4,56 (td, J=8,6, 2,2 Гц, 1H), 4,03-3,92 (m, 1H), 3,61-3,52 (m, 1H), 3,51-3,41 (m, 2H), 3,38 (t, J=7,0 Гц, 2Н), 3,35-3,32 (m, 1Н), 3,20 (m, 7Н), 3,02 (2 х s, 3Н), 2,61 (2 х s, 6Н), 2,47-2,37 (m, 1Н), 2,26 ( t, J=7,4 Гц, 2H), 2,22-2,04 (m, 3H), 2,04-1,91 (m, 1H), 1,88-1,63 (m, 3H), 1,62-1,43 (m, 7H), 1,37-1,27 (m, 4H), 1,27-1,19 (m, 2H), 1,06 (d, J=6,6 Гц, 2H), 0,98 (d, J=6,6 Гц, 1H), 0,96-0,88 (m, 8H), 0,87-0,81 (m, 5Н), 0,80-0,73 (m, 5Н). 13C ЯМР (101 МГц, ДМСО-d6) δ 172,67, 172,38, 172,26, 171,47, 171,45, 171,13, 171,10, 168,76, 168,52, 166,64, 166,23, 164,82, 164,61, 156,63, 155,75, 144,76, 144,68, 140, 27, 140,14, 134, 46,133,00, 131,89, 131,79, 128,78, 128,56, 128,40, 128,31, 127,66, 127,61, 127,10, 120,25, 118,66, 118,56, 116,47, 116,34, 85,45, 81,38, 77,69, 76,74, 71,91, 60,94, 60,20, 58,57, 58,16, 57,05 , 55,07, 54,48, 54,39, 49,25, 48,86, 47,18, 46,24, 43,58, 43,12, 41,50, 41,44, 37,15, 36,96, 36,15, 35,01, 31,83, 31,57, 30,04, 29,94, 27,77, 26,68, 26,64, 25,76, 25,39, 25,27 24,52, 24,50, 24,29, 23,17, 19,30, 19,28, 18,85, 18,62, 18,57, 18,48, 18,20, 17,17, 16,92, 15,58, 15,51, 15,23, 14,96, 10,43, 10,25. Примечание: некоторые сигналы 1H-ЯМР разделяются на два разных сигнала из-за присутствия конформеров в растворе. По этой причине число пиков в спектре 13C-ЯМР выше, чем количество атомов углерода, ожидаемых для AE-Keto-Sulf07.
[00182] Синтез 2. В круглодонной колбе объемом 25 мл в атмосфере азота AE-Keto (150 мг, 0,177 ммоль, 1,0 эквив.) и Sulf07, полученный по пути синтеза C (93 мг, 0,213 ммоль, 1,2 экв.), растворяли в смеси безводного EtOH (225 мкл) и безводного ДМСО (225 мкл). Полное растворение было достигнуто после 5 минут ультразвукового воздействия при комнатной температуре. Добавляли активированные молекулярные сита и полученную желтую суспензию перемешивали при комнатной температуре, и ход проверяли с помощью анализа ВЭЖХ. Через 22 ч реакционную смесь переносили в пробирку Эппендорфа объемом 2 мл, реакционный сосуд дважды промывали 600 мкл ДМСО-EtOH (1:1) и объединенные порции центрифугировали. Твердое вещество отделяли от супернатанта. Продукт осаждали добавлением 45 мкл МТВЕ в супернатант. Белую суспензию центрифугировали и полученный супернатант удаляли. Очистку целевого соединения осуществляли на системе Isolera One (Biotage AB, Упсала, Швеция), снабженной сферическим картриджем AQ C18 20-35 мкм 100A 40 гр с градиентом от 2% до 50% вода/ацетонитрил. После лиофилизации указанное в заголовке соединение получали в виде твердого вещества белого цвета. Выход 80 мг, 40% ВЭЖХ (способ 10, 220 нм) более 97%. Результаты ЯМР и LC-MS-анализа соответствовали описанным выше.
Пример 3
[00183] Соединение AE-Эстер-Sulf07, 2-(2-(1(4-(((1S,2R)-2)-((2R,3R)-3-((S)-1-((3R,4R,5S)-4-((S)-2-((R)-2-(диметиламино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-1-фенилпропокси)карбонил)фенил) этилиден)гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)бензолсульфоновую кислоту синтезировали, как описано ниже и показано на схеме 6.
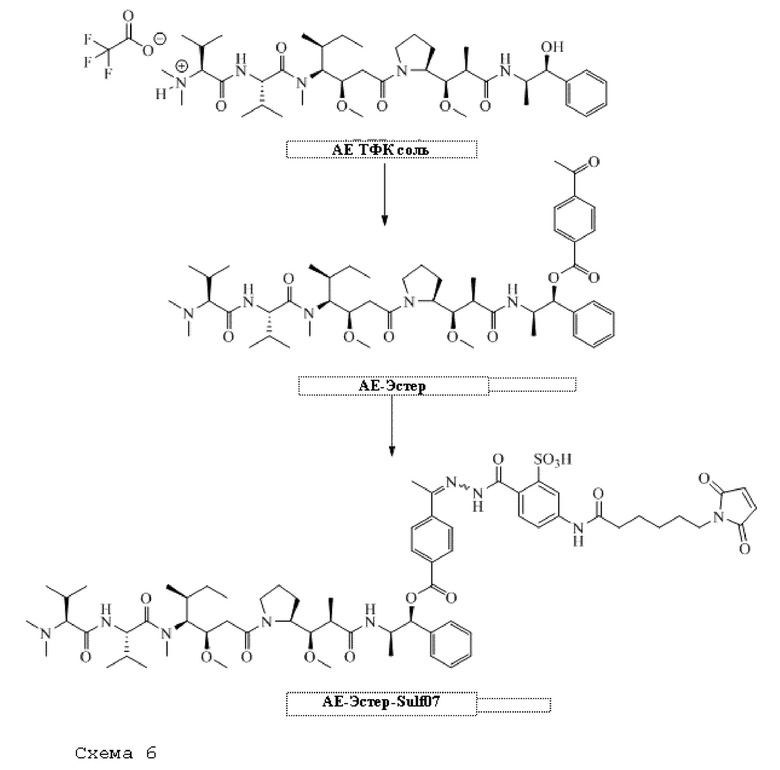
Синтез (1S,2R)-2-((3R)-3-((S)-1-((3R,4S,5R)-4-((S)-2-((S)-2-(диметиламино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-1-фенилпропил-4-ацетилбензоата (AE-Эстер)
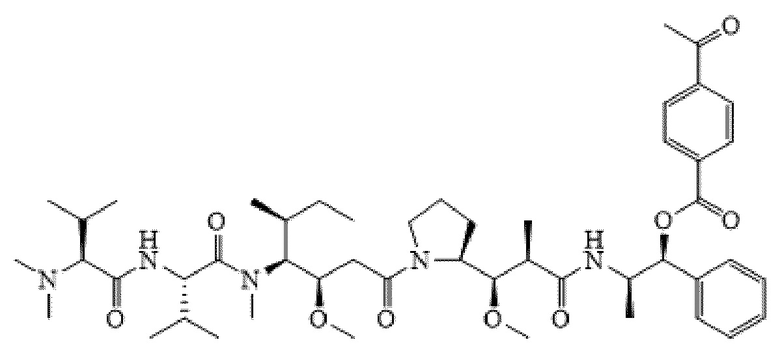
Синтез по усовершенствованному способу патента US6884869-B2
[00184] К раствору соли ТФК ауристатина E (1,0 г, 1,18 ммоль, 1,0 экв., Levena Biopharma) и 4-ацетилбензойной кислоты (198 мг, 1,42 ммоль, 1,2 экв., Acros Organics) в безводном дихлорметане (18 мл). в присутствии молекулярных сит (шарики 0,3 нм, 3 Å) добавляли 4-диметиламинопиридин (DMAP, 244 мг, 2,36 ммоль, 2,0 экв., Carbolution) и N, N'-диизопропилкарбодиимид (DIC, 186 мкл, 1,42 ммоль, 1,2 экв., Sigma Aldrich). Смесь перемешивали при комнатной температуре в течение 5 часов. Ход реакции контролировали с помощью ВЭЖХ (способ 2, 220 нм), показывая конверсию до желаемого продукта 81%. Затем добавляли DIC (155 мкл, 1,18 ммоль, 1,0 экв., Sigma Aldrich) и смесь оставляли перемешиваться при комнатной температуре в течение 18 часов, после чего ВЭЖХ показала 98% превращение реакции в желаемый продукт. Во время синтеза различных частей соединения было отмечено, что при использовании свежего DIC из вновь открытой склянки нет необходимости во втором добавлении. Молекулярные сита и мелкодисперсный осадок, побочный продукт DIC, отфильтровывали через сложенную фильтровальную бумагу (grade 3 hw, Sartorius) и полученный прозрачный желтый раствор промывали один раз HCl (0,1 М, 15 мл). Водный слой подвергали обратной экстракции один раз дихлорметаном (15 мл), затем органический слой объединяли, промывали один раз рассолом (15 мл) и сушили над сульфатом натрия. Раствор концентрировали при пониженном давлении при 40°С, получая желтое масло, которое затем растворяли в минимальном количестве дихлорметана (5 мл). Избыток диэтилового эфира (50 мл) добавляли для получения белого твердого вещества. Смесь оставляли при 2-4°С на 30 мин, а затем твердое вещество отделяли центрифугированием. Процедуру осаждения повторяли три раза, и белый порошок сушили при пониженном давлении. Выход: 706 мг, выход 68%. Чистота по ВЭЖХ (способ 2, 220 нм) 98%. LRMS-ESI (m/z) рассчитан для C49H75FN5O9 [M+H]+: 879,20. Результат: 878,70.
Синтез 2- (2-(1-(4-(((1S,2R)-2-((2R,3R)-3-((S)-1-((3R,4R,5S)-4-)-((S)-2-((R)-2-(диметиламино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-1-фенилпропокси)карбонил)фенил)этилиден)гидразин-1-карбонил)-5-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)бензолсульфоновой кислоты (AE-Эстер-Sulf07)
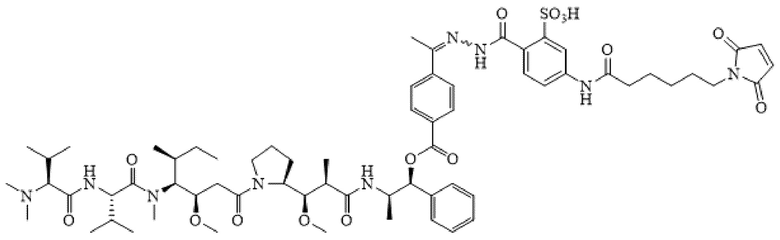
[00185] Раствор Sulf07 (164 мг, 0,31 ммоль, 1,1 эквив.) в безводном диметилсульфоксиде (2 мл) добавляли к раствору AE-Эстер (243 мг, 0,28 ммоль, 1,0 эквив.) в безводном этаноле ( 8 мл) в присутствии молекулярных сит (шарики 0,3 нм, 3 Å). Паратолуолсульфоновую кислоту (10,5 мг, 0,05 ммоль, 0,2 эквив.) добавляли для катализа реакции, и затем реакционную смесь перемешивали при комнатной температуре. ТФК также может быть использован вместо пара-толуолсульфокислоты. Реакцию контролировали с помощью ВЭЖХ (способ 2, 220 нм), показывая полную конверсию через 15 часов. Молекулярные сита и образовавшийся мелкий осадок удаляли из реакционного раствора фильтрованием через сложенную фильтровальную бумагу (grade 3 hw, Sartorius). Раствор осаждали избытком метил-трет-бутилового эфира (60 мл) и смесь оставляли при 2-4°С на 30 мин. Затем осадок отделяли центрифугированием, чтобы получить желтое липкое твердое вещество. Желтое липкое твердое вещество растворяли в смеси метанол/дихлорметан 3/7 (10 мл) и раствор впрыскивали в систему флэш-очистки с предварительно упакованным картриджем SNAP Ultra 25 g. Очистку проводили на нормальной фазе с использованием ступенчатого градиента от 100% дихлорметана до 60% дихлорметана/40% метанола в 17 объемах колонки. Пробирки, содержащие продукт, объединяли и сушили в высоком вакууме. Выход: 162 мг, выход 45%. Чистота с помощью ОФ-ВЭЖХ (способ 2, 220 нм) 96%. LRMS-ESI (m/z) рассчитан для C66H93N9O15S [М+Н]+: 1285,58. Результат: 1284,20. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,24 (d, J=5,3 Гц, 1H), 10,21 (s, 1H), 9,20 (2 × br.s, 1H), 8,32-7,90 (m, 8H), 7,74-7,71 (dd, J=8,5, 2,7 Гц, 1H), 7,41-7,27 (m, 5H), 7,00 (s, 2H), 6,13-6,02 (2 x d, J=5,0 Гц, 1H), 4,80-4,51 (m, 2H), 4,41-4,23 (m, 1H), 4,09-3,55 (m, 3H), 3,54-3,37 (t, J=7,0 Гц, 3H), 3,27-3,17 (m, 8H), 3,00 (s , 2H), 2,88-2,63 (m, 5H), 2,37-2,30 (m, 6H), 2,22-1,80 (m, 4H), 1,75-1,48 (m, 7H), 1,42-1,22 (m, 6H), 1,18. 1,11 (m, 3Н), 1,10-1,04 (m, 3Н), 1,00-0,74 (m, 20Н). 13C ЯМР (101 МГц, ДМСО) δ 172,93, 172,65, 171,44, 170,94, 168,61, 168,49, 164,59, 164,52, 164,24, 164,16, 149,20, 144,83, 143,05, 142,96, 140,71, 137,97, 137,78, 134,30, 132,25, 129,43, 129,38, 129,31, 129,21, 128, 39, 128, 31, 126, 49, 126,44, 126,35, 125,72, 118,77, 116,77, 85,20, 81,26, 77,65, 77,04, 76,84, 60,82, 60,12, 58,59, 58,03, 57,01, 48,35, 47,76, 47,15, 46,03, 43,60, 42,99, 36,89, 36,11, 31,64, 29,89, 29,73, 27,70, 26,56, 25,70, 25,33, 25,04, 24,42, 24,04, 23,03, 19,16, 18,63, 18,44, 18,37, 15,50, 15,47, 15,29, 15,14, 13,88, 13,84, 10,19 10.04. Примечание: некоторые сигналы 1H-ЯМР разделяются на два разных сигнала из-за присутствия конформеров в растворе. По этой причине число пиков в спектре 13C-ЯМР выше, чем количество атомов углерода, ожидаемых для AE-Эстер-Sulf07.
рН-зависимое высвобождение конъюгатов человеческого сывороточного альбумина AE-Keto-Sulf07 и AE-Эстер-Sulf07 при pH 7,4 и pH 4,1 (фиг. 11 и 12)
[00186] Альбумин-связывающие производные ауристатина-Sulf07 готовили в виде 2 мМ маточных растворов в безводном ДМСО.
[00187] Высвобождение при pH 7,4: 442,4 мкл 1000 мкМ восстановленного человеческого сывороточного альбумина (881 мкМ свободного цистеина-34) и 727,6 мкл буфера PBS (4 мМ фосфата натрия pH 7,4 и 150 мМ NaCl) добавляли в герметичный флакон для ВЭЖХ и инкубировали при 37°С в течение 30 минут. После 30 минут инкубации в предварительно инкубированный флакон добавляли 130 мкл соответствующего исходного раствора лекарственного средства ДМСО для получения 200 мкМ раствора ауристатина и 300 мкМ раствора альбумина (свободного цистеина-34). Смеси давали прореагировать в течение 10 минут при 37°С и затем анализировали с помощью ВЭЖХ (способ 7, 20 мкл инъекции). Инъекции повторяли через 1 час, а затем каждый час до 24 часов.
[00188] Высвобождение при рН 4,1: 408,5 мкл 1000 мкМ восстановленного человеческого сывороточного альбумина (881 мкМ свободного цистеина-34) и 539,4 мкл воды добавляли в герметичный флакон ВЭЖХ и инкубировали при 37°С в течение 30 минут. В отдельном флаконе 190 мкл натрий-ацетатного буфера 50 мМ рН 3,0 и 24,7 мкл 1 М HCl инкубировали при 37°С в течение 30 минут. Через 30 минут инкубации в предварительно инкубированный флакон добавляли 120 мкл соответствующего исходного раствора лекарственного средства ДМСО для получения конъюгата альбуминного лекарственного средства. После 10 минут инкубации добавляли 132 мкл буферного раствора ацетата натрия для получения 200 мкМ раствора производного ауристатина и 300 мкМ раствора альбумина (свободный цистеин-34). Полученный раствор анализировали непосредственно способом ВЭЖХ (способ 7). Инъекцию из флакона повторяли через 1 час, а затем каждый час до 24 часов.
[00189] Процент высвобожденного свободного лекарственного средства определяли с помощью ВЭЖХ с калибровочной кривой (200 мкМ, 100 мкМ, 50 мкМ, 25 мкМ и 12,5 мкМ) для свободных лекарств, т.е. AE-Keto для AE-Keto-Sulf07 и AE-Эстер для AE-Эстер-Sulf07. Исходные лекарственные растворы готовили с использованием ДМСО в качестве растворителя и разбавляли в 10 раз фосфатным буфером (4 мМ фосфата натрия, рН 7,4 и 150 мМ NaCl) перед анализом ВЭЖХ. Расчеты высвобождения лекарственного средства были основаны на AUC: для AE-Keto при 254 нм и AE-Эстер при 310 нм (локальные ультрафиолетовые максимумы). Процент высвобожденного AE-Эстер был выше, чем процент высвобожденного AE-Keto при изменении значения pH с 7,4 до 4,1 (сравните Фигуры 11 и 12).
Восстановление стабильности AE-Keto-Sulf07 и AE-Keto-EMCH (фиг. 1)
[00190] Альбумин-связывающие производные лекарственного средства AE-Keto восстанавливали в 50 мМ натрий-фосфатном буфере, рН 7,6, который содержал 5% сахарозы (вес/объем) и 2% 2-гидроксипропил-β-циклодекстрина (2-HPβCD). Оба препарата были восстановлены в концентрации 1277 мкМ (равной 4,5 мг/кг дозы мышиного ксенотрансплантата), растворение обоих препаратов было подтверждено ВЭЖХ (способ 2, 254 нМ). Стабильность восстановленных лекарственных средств контролировали с помощью ВЭЖХ при комнатной температуре каждые 15 минут в течение периода 240 минут для гидролиза малеимида и потери API. AE-Keto-Sulf07 был более стабильным, чем AE-Keto-EMCH (см. фиг. 1).
Восстановление стабильности AE-Эстер-Sulf07 и AE-Эстер-EMCH (фиг. 2)
[00191] Альбумин-связывающие производные AE-Эстер восстанавливали в 50 мМ натрий-фосфатном буфере, рН 7,6, который содержал 5% сахарозы (вес/объем) и 4% 2-HPβCD. Оба препарата были восстановлены в концентрации 655 мкМ (равной 2,4 мг/кг дозы мышиного ксенотрансплантата), растворение обоих препаратов было подтверждено ВЭЖХ (способ 2, 310 нм). Стабильность восстановленных лекарственных средств контролировали с помощью ВЭЖХ при комнатной температуре каждые 15 минут в периоде 240 минут для гидролиза малеимида и потери API. AE-Эстер-Sulf07 был более стабильным, чем AE-Эстер-EMCH (см. фиг. 2).
Кинетика связывания производных ауристатина-Sulf07 в плазме человека, плазме крысы и мышиной плазме CD1 (фиг. 5-10).
[00192] Мышиную плазму CD1 и плазму крыс Sprague Dawley извлекали из хранилища при минус 80°C и позволяли достичь комнатной температуры. Оттаявшую плазму центрифугировали при 13,6 кРПМ в течение 60 секунд, супернатант фильтровали через фильтрующую иглу (5 мкм) и затем через мембрану СА 0,45 мкм.
[00193] Цельную человеческую кровь брали у здорового волонтера (пробирка для сбора ЭДТА), плазму отделяли от эритроцитов, вращая ее в течение 3 минут при 3000 об/мин. Затем плазма использовалась напрямую в течение 2 часов после сдачи крови. 360 мкл соответствующей плазмы использовали для каждого эксперимента по связыванию.
[00194] Все эксперименты по связыванию повторяли троекратно. Плазму инкубировали при 37°C с использованием термомиксера Eppendorf C. Через 30 минут инкубации 40 мкл соответствующего альбумин-связывающего производного ауристатина-Sulf07 растворяли в растворе для восстановления и добавляли в плазму. Образцы (40 мкл) отбирали через 15 секунд, 2 минуты, 4 минуты, 8 минут и 15 минут (5 образцов).
[00195] Образцы немедленно добавляли к 160 мкл ацетонитрила (содержащего 4 мкг/мл ММАЕ в качестве внутреннего стандарта) и встряхивали в течение 1 минуты.
[00196] Растворы пипетировали в планшет для осаждения из плазмы Impact (встряхивали в течение 60 секунд) и фильтровали в вакууме на 96-луночный планшет. Оставшееся лекарственное средство определяли количественно с помощью LCMS (способ 5, инъекция 30 мкл) с использованием отрицательного режима MRM MS2 (мониторинг множественных реакций). Родительские ионы: ауристатин F (масса-744,8), AE-Keto-Sulf07 (масса-1135,1) и AE-Эстер-Sulf07 (масса-1283,2). Процент связывания определяли путем сравнения AUC для AE-Keto-Sulf07 и AE-Эстер-Sulf07 с калибровочными кривыми, полученными с помощью LCMS для каждого соединения.
Процедура: Специфичность связывания производных ауристатина-Sulf07 с цистеином-34 человеческого сывороточного альбумина (фиг. 3-4).
[00197] Альбумин-связывающие производные ауристатина-Sulf07 восстанавливали в 50 мМ натрий-фосфатном буфере с pH 7,6 и 5% сахарозе, которая содержала 2-HPβCD. Для восстановления AE-Keto-Sulf07 использовали 2% концентрацию HPβCD и 4% для AE-Эстер-Sulf07 соответственно. Оба препарата были восстановлены в концентрации 1 мг/мл (AE-Эстер-Sulf07 780 мкМ, AE-Keto-Sulf07 880 мкМ).
[00198] Плазму человека извлекали из хранилища при минус 80°С и давали возможность достичь комнатной температуры. Оттаявшую плазму центрифугировали при 13,6 кРПМ в течение 60 секунд, супернатант фильтровали через иглу фильтра (5 мкм) и затем через мембрану СА 0,45 мкм. 180 мкл соответствующей плазмы использовали для каждого эксперимента по связыванию. Плазму инкубировали при 37°C, используя термомиксер Eppendorf C. Через 30 минут инкубации добавляли 20 мкл соответствующего альбумин-связывающего производного ауристатина-Sulf07. Альбумин-связывающему препарату давали реагировать в плазме в течение 5 минут при 37°С. Через 5 минут раствор анализировали непосредственно с помощью ВЭЖХ с гидрофобным взаимодействием (HIC, способ 6, 250 нм), чтобы подтвердить сайт-специфическое конъюгирование с альбумином и образование альбуминового лекарственного конъюгата.
Ускоренная деградация лиофилизированных составов ауристатина: сравнение стабильности EMCH и Sulf07 (фиг. 24-25).
[00199] API растворяли в буфере для лиофилизации, который содержал 2-HPβCD, 10 мМ цитрата натрия, рН 6,2, 50% трет-бутиловый спирт (V/V). Для производных AE-Keto 2% (вес/объем) 2-HPβCD использовали для растворения API AE-Keto при концентрации 1,27 мМ; для производных AE-Эстер 4% (вес/объем) 2-HPβCD использовали для растворения API в концентрации 1,05 мМ. Растворенные API стерильно фильтровали с использованием шприцевого фильтра Acrodisc Fluorodyne II (0,2 мкм). Стерильные растворы пипеткой переносили во флаконы для лиофилизации, которые затем частично закрывали резиновой пробкой. Затем флаконы помещали в лиофилизатор для лиофилизации. Флаконы замораживали на полке сублимационной сушилки при минус 40°С в течение 2 часов, после 2 часов начинали основную сушку. Основная сушка проводилась в течение 26 часов, параметры: температура полки минус 20°С, вакуум 0,0048 мбар. После основной сушки начиналась окончательная сушка. Окончательная сушка проводилась в течение 16 часов, параметры: температура полки 20°C, вакуум 0,0047 мбар. По окончании окончательной сушки флаконы закрывали в вакууме.
[00200] Запечатанные флаконы инкубировали при 55°C, используя термомиксер Eppendorf C с пластинчатым вкладышем (Smartblock) и крышкой. В выбранные моменты времени (t: 0, 46, 94, 166 и 264 часа) образцы удаляли и растворяли в безводном ДМСО. Стабильность API определяли с помощью ВЭЖХ (способ 8, 254 нм для производных AE-Keto, 310 нм для производных AE-Эстер). Процент гидролиза малеимида в каждый момент времени определяли путем сравнения с AUC для каждого API в момент времени 0. AE-Keto-Sulf07 и AE-Эстер-Sulf07 были более стабильными, чем соответствующие производные EMCH AE-Keto-EMCH и AE-Эстер-EMCH (см. фиг. 24 и фиг. 25).
Пример 4
Общая процедура оценки ауристатина E, AE-Keto и AE-Эстер против панели линий опухолевых клеток
Определение IC50
[00201] Образцы были предоставлены Charles River Discovery Research Services Germany GmbH в виде замороженных исходных растворов в фармацевтической чистоте DMSO (Sigma-Aldrich, Taufkirchen, Germany). В каждый день эксперимента оттаивали замороженную аликвоту исходного раствора. Последующие серийные разведения осуществляли с помощью полной культуральной среды RPMI 1640 с использованием планшета для промежуточного разведения. 10 мкл, взятых из планшета для промежуточного разведения, затем переносили в 140 мкл/лунку планшета для культивирования клеток. Клетки обрабатывали испытуемыми соединениями в 10 концентрациях в трех повторениях с половинным логарифмическим шагом от 0,003 нМ до 100 нМ в течение 96 часов.
[00202] Соединения тестировали на панели из 6 отобранных линий раковых клеток человека с использованием анализа жизнеспособности клеток CellTiter-Blue® (Promega, Mannheim, Germany). Значения IC представлены в виде абсолютных значений IC50, полученных для тестируемого соединения, в качестве среднего геометрического значения IC50 для всех протестированных клеточных линий.
[00203] Использованными клеточными линиями были LXFL 1674L (созданный из ксенотрансплантата, полученного от пациента в Charles River Discovery Research Services Germany GmbH), SW-620 (любезно предоставленный NCI, Bethesda, MD, USA), CAL-27 (приобретенный у DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany), RKO, MDA-MB-468 и SK-OV-3 (от ATCC, American Type Culture Collection, Rockville, MD, США). Подлинность всех клеточных линий была доказана в DSMZ с помощью STR анализа (короткого тандемного повтора), методики ДНК-фингерпринта на основе ПЦР. Клеточные линии обычно пассировали один или два раза в неделю и выдерживали в культуре до 20 пассажей. Клетки выращивали при 37°C в увлажненной атмосфере с 5% CO2 в среде RPMI 1640 (25 мМ HEPES, с L-глутамином, Biochrom, Berlin, Germany) с добавлением 10% (по объему) фетальной сыворотки теленка (Sigma-Aldrich, Taufkirchen, Germany) и 0,1 мг/мл гентамицина (Life Technologies, Karlsruhe, Germany). Анализ жизнеспособности клеток CellTiter-Blue® использовали в соответствии с инструкциями производителя. Вкратце, клетки собирали из культур экспоненциальной фазы, подсчитывали и высевали в 96-луночные планшеты с плоским дном для микротитрования при плотности клеток 4000-10000 клеток на лунку в зависимости от скорости роста клеточной линии. После 24-часового периода восстановления, чтобы позволить клеткам возобновить экспоненциальный рост, добавляли 10 мкл культуральной среды (шесть контрольных лунок на планшет) или культуральной среды с тестируемым соединением. Соединения наносили в 10 концентрациях в трех экземплярах с половинным логарифмическим шагом до 100 нМ и клетки обрабатывали непрерывно в течение 96 часов. После четырехдневной обработки клеток добавляли 20 мкл на лунку реагента CellTiter-Blue®. После инкубационного периода до 4 ч флуоресценцию (FU) измеряли с использованием многомодового планшет-ридера Enspire (λ возбуждения = 531 нм, λ излучения = 615 нм). Значения IC50 определяли с помощью биоаналитического программного обеспечения GraphPad Prism (San Diego, CA, USA).
[00204] Зависимые от концентрации активности с сигмоидальными кривыми концентрация-эффект наблюдали в шести протестированных линиях опухолевых клеток. Среднегеометрические значения IC50 составляли 259±0,11 пМ для AE-Keto и 399±0,19 пМ для AE-Эстер. По сравнению с сильнодействующими лекарственными средствами MMAE (171±0,10 пМ) и AE (130±0,05 пМ), производные AE-Keto и AE-Эстер имеют сходную цитотоксичность в пикомолярном диапазоне.
Пример 5
Общая процедура оценки активности ауристатина Е и альбумин-связывающих производных ауристатина Е на ксеногенных моделях опухоли, полученных от пациента.
[00205] Голым мышам женского пола с иммунодефицитом NMRI от Charles River Discovery Research Services Germany GmbH были пересажены односторонние опухолевые имплантаты человеческого происхождения подкожно на левом боку, под изофлурановым наркозом, пока опухоли не прижились и не достигли желаемого объема.
[00206] Животных содержали в клетках, температуру внутри клеток поддерживали на уровне 25°С ± 1°С с относительной влажностью 45-65% и скоростью воздухообмена 60 раз в час. Мышей содержали в цикле искусственного освещения 14 часов света/10 часов темноты. Животных кормили по автоклавированной экструдированной диете с 19% протеином Teklad Global (T.2019S.12) от Envigo RMS SARL, они имели доступ к стерильной отфильтрованной и подкисленной (pH 2,5) водопроводной воде, которую меняли два раза в неделю. Животные были обеспечены кормом и водой ad libitum. До начала терапии животные были рандомизированы (7-8 мышей на группу) с учетом сравнимого медианного и среднего группового объема опухоли. Животных регулярно контролировали два раза в день в рабочие дни и ежедневно по субботам и воскресеньям. Начиная с 0-го дня животных взвешивали два раза в неделю. Относительную массу тела (RBW) отдельных животных рассчитывали путем деления индивидуальной абсолютной массы тела в день X (BWx) на индивидуальную массу тела в день рандомизации, умноженную на 100%. Объем опухоли определяли двумерным измерением с помощью штангенциркуля в день рандомизации (день 0) и затем дважды в неделю. Объемы опухолей рассчитывали по следующему уравнению:
Объем опухоли [мм3] = l [мм] × w2 [мм2] × 0,5, где «l» - длина, а «w» - ширина опухоли. Относительный объем отдельной опухоли в день X (RTVx) рассчитывали путем деления абсолютного индивидуального объема [мм3] соответствующей опухоли в день X (Tx) на абсолютный объем той же отдельной опухоли опухоли в день рандомизации, умноженный на 100%. Расчеты применялись в той степени, в которой это позволяет политика в области защиты животных. Уничтожение отдельных мышей проводили при объеме опухоли > 2000 мм3 (одностороннем). Для оценки статистической значимости противоопухолевой эффективности был проведен непараметрический критерий Крускала-Уоллиса с последующим способом Данна для парных сравнений, посредством чего отдельные RTV тестовой и контрольной групп сравнивались в дни, в которые минимальные значения T/C были достигнуты в тестовых группах, где T/C [%] = (медианная группа, обработанная RTVx/медианная контрольная группа RTVx) х 100. Статистический анализ проводился только в том случае, если по меньшей мере 50% первоначально рандомизированных животных в соответствующей группе были еще живы. Сравнения между тестовыми группами проводились за те же дни. По общему правилу, значения p≤0,05 указывали на значимость ингибирования опухоли. Статистические расчеты были выполнены с использованием биоаналитического программного обеспечения GraphPad Prism (San Diego California, USA, www.graphpad.com).
Общая процедура оценки эффективности ауристатина Е и альбумин-связывающих производных ауристатина Е в исследованиях по обнаружению дозы в ксеногенных моделях опухолей, полученных из клеточной линии.
[00207] Голым мышам женского пола с иммунодефицитом NMRI, Janvier, France, были подкожно инокулированны 5×106-107 раковых клеток в буфере/Matrigel (1:1) до тех пор, пока опухоли не были ощутимы и не достигли желаемого объема (для исследований ксенотрансплантата).
[00208] Животных содержали в клетках (проволочная сетка Macrolon Type-II) с температурой, поддерживаемой на уровне 22°С±1°С, относительной влажностью 50±10% и скоростью воздухообмена 60 раз в час. Мышей содержали в цикле искусственного освещения 12 часов света/12 часов темноты. Животных кормили в автоклаве Ssniff NM (Soest, Германия), они имели доступ к стерильной фильтрованной и подкисленной (pH 4,0) водопроводной воде, которую меняли два раза в неделю. Животные были обеспечены кормом и водой ad libitum. До начала терапии животные были рандомизированы (3-4 мыши на группу в исследованиях по выявлению дозы и 7-8 мышей в группе в исследованиях ксенотрансплантата) с учетом сравнимого медианного и среднего объема опухоли в группе. Состояние здоровья животных проверяли в начале эксперимента и дважды в день в течение эксперимента. Используемая идентификация: метка уха и ярлыки клетки. Начиная с дня 0, животных взвешивали два или три раза в неделю, и средний вес тела на группу был связан с начальным значением в процентах для расчета изменения веса тела (BWC). Диаметр опухоли определяли двумерным измерением с помощью штангенциркуля в день рандомизации (день 0), а затем два или три раза в неделю. Объемы опухолей рассчитывали по следующему уравнению:
Объем опухоли [мм3] = l [мм] × w2 [мм2] × 0,5, где «l» - длина, а «w» - ширина опухоли. Относительный объем отдельной опухоли в день X (RTVx) рассчитывали путем деления абсолютного индивидуального объема соответствующей опухоли [мм3] в день X (Tx) на абсолютный объем той же отдельной опухоли в день рандомизации, умноженный на 100%. Расчеты применялись в той степени, в которой это позволяет политика в области защиты животных. Уничтожение отдельных мышей проводили при объеме опухоли больше 1500 мм3 (одностороннем) или при наблюдении изъязвления. Статистическое сравнение было выполнено с применением U-теста Манна-Уитни.
Исследования по выявлению дозы ауристатина E и альбумин-связывающих производных ауристатина E AE-Keto-Sulf07 и AE-Эстер-Sulf07.
[00209] Исходные растворы готовили следующим образом:
1) 3 мыши на группу, 26 г среднего веса в день рандомизации: 0,3, 0,6 или 1,2 мг/кг соли ТФК ауристатина E (эквивалент AE), которую вводят один раз в неделю в течение 4 недель в день 0, 6, 13, 20 ≡ 0,35-1,41 мг/кг ≡ 7,1-28,3 мкг/20 г мыши. Подготовка образца: 8,5 мг взвешивали в 50 мл флаконе, растворяли в 30,0 мл 25/75 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 0,2-0,8 мл были разделены на аликвоты в 2 мл флаконах в зависимости от желаемой конечной дозы. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 0,8 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 3 мыши на группу, 26 г среднего веса в день рандомизации: 1,2 и 2,4 мг/кг AE-Keto-Sulf07 (эквивалент AE), которые вводили дважды в неделю в течение 4 недель ≡ 1,94-3,87 мг/кг ≡ 38,7 и 77,4 г/20 г мыши. Приготовление образца: 11,0 мг взвешивали в 30 мл флаконе, растворяли в 14,2 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, pH 7,0; 0,4-0,8 мл были разделены на аликвоты в 2 мл флаконах в зависимости от желаемой конечной дозы. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 0,8 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
3) 3 мыши на группу, 26 г среднего веса в день рандомизации: 1,2, 1,6 или 2,0 мг/кг AE-Эстер-Sulf07 (эквивалент AE), которые вводили дважды в неделю в течение 4 недель в день ≡ 2,18-3,64 мг/кг ≡ 43,7-72,8 мкг/20 г мыши. Приготовление образца: 19,2 мг взвешивали во флаконе объемом 30 мл, растворяли в 26,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; в 2 мл пробирках отбирали аликвоты по 0,48-0,8 мл в зависимости от желаемой конечной дозы. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 0,8 мл 50 мМ натрий-фосфатного буфера, 5% Tween 80, рН 7,6.
4) 4 мыши на группу, средний вес 23 г в день рандомизации: 2,0, 2,2 и 2,4 мг/кг AE-Эстер-Sulf07 (эквивалент AE), которые вводили дважды в неделю в течение 4 недель в день ≡ 3,64-4,37 мг/кг ≡ 72,8-87,3 мкг/20 г мыши и 4,0, 4,4 и 4,8 мг/кг AE-Эстер-Sulf07 (AE эквивалент), которые вводили один раз в неделю в течение 4 недель в день 0, 7, 14, 21≡7,28-8,73 мг/кг ≡ 145,6-174,7 мкг/20 г мыши. Приготовление образца: 56,0 мг взвешивали в 50 мл флаконе, растворяли в 32,0 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 0,33-0,8 мл были аликвотными в 2 мл флаконах в зависимости от желаемой конечной дозы. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 0,8 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
[00210] Дозы ауристатина Е 1,2 и 0,6 мг/кг показали высокую токсичность, и ни одно животное не выжило после первой и второй инъекции соответственно. Доза 0,3 мг/кг хорошо переносилась. В более поздних исследованиях было установлено, что АЕ переносился при 0,3 мг/кг, вводимых два раза в неделю в течение четырех недель (всего восемь инъекций), и поэтому эту дозу использовали в исследованиях ксенотрансплантата в качестве максимальной переносимой дозы (МПД).
[00211] Все дозы, проверенные на AE-Keto-Sulf07, вводимые восемь раз по двухнедельному графику, хорошо переносились. В более поздних исследованиях ксенотрансплантата доза для этого препарата была увеличена до 6,5 мг/кг при той же схеме дозирования, провоцируя при этой максимальной дозе потерю массы тела у одной мыши и общее плохое самочувствие у второй мыши, и лечение пришлось прекратить после пяти инъекций, а не восьми, как планировалось. Поэтому такая доза считалась выше MTD, и доза 4,5 мг/кг была выбрано в качестве безопасной дозы.
AE-Эстер-Sulf07 был первоначально испытан до 2,0 мг/кг два раза в неделю в течение четырех недель без каких-либо явных признаков токсичности. Поскольку раздражение кожи позже наблюдалось в исследованиях ксенотрансплантата, повторное исследование по определению дозы было повторено при дозах 2,0, 2,2 и 2,4 мг/кг два раза в неделю в течение четырех недель и сравнивалось с соответствующими удвоенными дозами, введенными 4,0, 4,4 и 4,8 мг/кг только один раз в неделю. Изменения массы тела не наблюдалось, но раздражение кожи появилось через 9-14 дней во всех группах. Раннее и более частое раздражение кожи наблюдалось при более высоких дозах. Побочный эффект казался обратимым, так как раздражения начали заживать после окончания лечения. Группа, получавшая 4,0 мг/кг один раз в неделю, показала наименьший побочный эффект, в то время как между другими группами не наблюдалось значительного различия.
Критерии оценки ауристатина Е и альбумин-связывающих производных ауристатина Е в моделях ксенотрансплантата опухоли
[00212] Эффективность каждого лекарственного средства определяли на основании уменьшения объема опухоли (% объема опухоли в выбранный день по сравнению с днем 0): полная ремиссия менее 10%; частичная ремиссия более 10-50%; незначительная ремиссия более 50-75%; стабильная болезнь более 75-125%; прогрессирующее заболевание более 125% по сравнению с исходным объемом опухоли.
Оценка эффективности ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 в модели злокачественной меланомы человека A375 - мелкие опухоли (фиг. 13).
[00213] Оценку ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 на модели злокачественной меланомы человека A375 проводили, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00214] Исходные растворы готовили следующим образом:
1) 7 мышей, средний вес 29 г, средний медианный объем опухоли 135 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (экв. AE) дважды в неделю в течение 3 недель в день 0, 6, 9, 13, 16, 19 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40 ° С и лиофилизировали при минус 20 ° С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средняя масса 28 г, средний медианный объем опухоли 137 мм3 в день рандомизации: 3,0 мг/кг AE-Keto-Sulf07 (экв. AE) дважды в неделю в течение 3 недель в день 0, 6, 9, 13, 16, 19 ≡ 5,32 мг/кг ≡ 106,3 мкг/20 г мыши. Приготовление образца: 19,2 мг взвешивали во флаконе объемом 30 мл, растворяли в 19,0 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
[00215] Развитие роста опухоли на модели ксенотрансплантата меланомы A 375 показало статистически значимую противоопухолевую эффективность соединения AE-Keto-Sulf07 в дозе 3,0 мг/кг по сравнению с дозой ауристатина E 0,3 мг/кг, обе вводили шестикратно два раза в неделю (р <0,01 с 27 по 37 день). Мыши, получавшие ауристатин Е, достигли состояния стабильного заболевания до 21 дня, после чего опухоль снова начала расти. В отличие от этого, мыши в группе, получавшей AE-Keto-Sulf07, достигли долгосрочной регрессии опухоли, демонстрируя полную ремиссию с 19 дня до конца исследования на 37 день (конечный объем опухоли 1 мм3).
[00216] Мыши в группе, получавшей AE-Keto-Sulf07, имели только незначительное снижение прироста массы тела по сравнению с контролем, но не было выявлено никаких побочных эффектов.
Оценка ауристатина E и альбумин-связывающего производного ауристатина E AE-Keto-Sulf07 в модели злокачественной меланомы человека A375 - крупные опухоли (фиг. 14).
[00217] Оценка ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 на модели злокачественной меланомы человека A375 была проведена, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00218] Исходные растворы готовили следующим образом:
1) 7 мышей, средний вес 28 г, средний медианный объем опухоли 310 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (экв. AE) дважды в неделю в течение 3 недель в день 1, 6, 9, 13, 16 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40 °С и лиофилизировали при минус 20 ° С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средняя масса 27 г, средний медианный объем опухоли 331 мм3 в день рандомизации: 6,5 мг/кг AE-Keto-Sulf07 (экв. AE) дважды в неделю в течение 3 недель в день 1, 6, 9, 13, 16 ≡ 11,52 мг/кг ≡ 230,4 мкг/20 г мыши. Приготовление образца: 28,0 мг взвешивали в 30 мл флаконе, растворяли в 12,2 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
[00219] Противоопухолевую эффективность соединения AE-Keto-Sulf07 также тестировали на мышах, имеющих большие злокачественные опухоли меланомы A375 (~320 мм3), чтобы доказать, что эффективность лекарственного средства не ограничивается небольшими опухолями. Мыши, получавшие AE-Keto-Sulf07 в дозе 6,5 мг/кг, показали статистически значимое улучшение противоопухолевого эффекта по сравнению с группой, получавшей ауристатин Е в дозе 0,3 мг/кг, обе вводили пять раз два раза в неделю (р <0,01 с 9-го дня по 33-й день). Ауристатин Е демонстрировал исходное стабильное заболевание до 14 дня, а затем стабильный рост, в то время как у мышей в группе, получавшей AE-Keto-Sulf07, отмечалась полная ремиссия с 19 дня до конца исследования на 37 день, достигая минимального объема опухоли 1 мм3 на 28-й день (12 дней после последнего лечения) и ~ 14 мм3 в конце исследования, что приводит к долгосрочному эффекту, несмотря на относительно большой средний начальный объем опухоли.
[00220] AE-Keto-Sulf07 вызывал кратковременную потерю массы тела (13% достигнуто на 21 день), и двух мышей пришлось умерщвлять из-за общего плохого состояния.
Оценка эффективности ауристатина E и альбумин-связывающего производного ауристатина E AE-Keto-Sulf07 на модели немелкоклеточного рака легких человека LXFA737- мелкие опухоли (фиг. 15).
[00221] Оценку эффективности ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 на модели немелкоклеточного рака легкого человека LXFA737 проводили, как описано в общей процедуре для моделей ксенотрансплантата, полученных от пациента.
[00222] Исходные растворы готовили следующим образом:
1) 7 мышей, средний вес 27 г, средний медианный объем опухоли 137 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (экв. AE) дважды в неделю в течение 4 недель в день 0, 4, 7, 11, 14, 18, 21, 25 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивают во флаконе объемом 30 мл, растворенном в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средний вес 26 г, средний медианный объем опухоли 128 мм3 в день рандомизации: 4,5 мг/кг AE-Keto-Sulf07 (эквивалент AE) дважды в неделю в течение 4 недель в день 0, 4, 7, 11, 14, 18, 21, 25 ≡ 7,98 мг/кг ≡ 159,5 мкг/20 г мыши. Приготовление образца: 38 мг взвешивали во флаконе объемом 30 мл, растворяли в 23,8 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали с помощью 1,2 мл 50 мМ натрий-фосфатного буфера, 2% 2-гидроксипропил-β-циклодекстрина 2-HPβCD, pH 7,6.
3) 7 мышей, средний вес 27 г, средний медианный объем опухоли 130 мм3 в день рандомизации: 4,5 мг/кг AE-Keto-Sulf07 (эквивалент AE), дважды в неделю в течение 4 недель в день 0, 4, 7, 11, 14, 18, 21, 25 ≡ 7,98 мг/кг ≡ 159,5 мкг/20 г мыши. Приготовление образца: 38 мг взвешивали во флаконе объемом 30 мл, растворяли в 23,8 мл 50/50 трет-бутанола/10 мМ цитратного буфера натрия, 5% сахарозы, pH 6; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
[00223] Развитие роста опухоли в модели ксенотрансплантата NSCLC LXFE737 показало превосходящую противоопухолевую эффективность соединения AE-Keto-Sulf07 при 4,5 мг/кг (в буферах Tween 80 и 2-HPβCD) по сравнению с ауристатином E в дозе 0,3 мг/кг восьмикратно два раза в неделю со статистической значимостью (р <0,01 на 56 и 63 день). Опухоли в группе, получавшей ауристатин Е, оставались только временно в состоянии стабильного заболевания (с 21 дня по 42 день) и выявляли значительный рост после окончания терапии. В отличие от этого, мыши, получавшие AE-Keto-Sulf07, достигли полной ремиссии опухоли на 28-й день, через 3 дня после окончания терапии, в результате чего объем опухоли снизился до неизмеримого уровня до конца исследования на 74-й день (независимо от используемого буфера), что привело к долгосрочному противоопухолевому эффекту.
[00224] Изменение массы тела по сравнению с контролем показало частичное увеличение токсичности в группе, получавшей AE-Keto-Sulf07. На 32-й день была зафиксирована максимальная потеря массы тела (-7%) без других признаков токсичности в случае 2-HPβCD, в то время как две мыши должны были быть принесены в жертву из-за общих плохих состояний, когда препарат вводился с Tween 80.
Оценка эффективности ауристатина E и альбумин-связывающего производного ауристатина E AE-Keto-Sulf07 на модели немелкоклеточного рака легкого человека LXFA737- крупные опухоли (фиг. 16).
[00225] Оценку ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 на модели немелкоклеточного рака легкого человека LXFA737 проводили, как описано в общей процедуре для моделей ксенотрансплантата, полученных от пациента.
[00226] Исходные растворы готовили следующим образом:
1) 8 мышей, средний вес 26 г, средний медианный объем опухоли 324 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE) дважды в неделю в течение 4 недель в день 1, 5, 8, 12, 15, 19, 22, 26 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 3 мг взвешивали во флаконе объемом 100 мл, растворяли в 43,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 2,4 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 2,4 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 8 мышей, средний вес 28 г, средний медианный объем опухоли 342 мм3 в день рандомизации: 4,5 мг/кг AE-Keto-Sulf07 (эквивалент AE), дважды в неделю в течение 4 недель в день 1, 5, 8, 12, 15, 19, 22, 26 ≡ 6,99 мг/кг ≡ 139,7 мкг/20 г мыши. Приготовление образца: 51 мг взвешивали во флаконе объемом 100 мл, растворенном в 36,5 мл 50/50 трет-бутанола/10 мМ цитратного буфера натрия, 3% 2-HPβCD, pH 6,2; 2,4 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 2,4 мл 50 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,6.
[00227] Развитие роста опухоли в модели ксенотрансплантата NSCLC LXFE737 показало превосходящую противоопухолевую эффективность соединения AE-Keto-Sulf07 в дозе 4,5 мг/кг по сравнению с ауристатином E в дозе 0,3 мг/кг, обе дозы вводили 8 раз два раза в неделю, со статистической значимостью (р < 0,01 на 28 день). Опухоли в группе, получавшей ауристатин Е, не проявляли какой-либо активности, в то время как мыши, обработанные AE-Keto-Sulf07, достигли частичной ремиссии опухоли на 15-й день до конца исследования на 59-й день, что привело к долгосрочному противоопухолевому эффекту.
[00228] Никаких побочных эффектов не было зарегистрировано.
Оценка эффективности ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 при карциноме яичника человека, модель A2780 - мелкие опухоли (фиг. 17).
[00229] Оценка ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 в модели карциномы яичника человека A2780 была проведена, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00230] Исходные растворы готовили следующим образом:
1) 7 мышей со средним весом 27 г, средним медианным объемом опухоли 147 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE) дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средний вес 27 г, средний медианный объем опухоли 153 мм3 в день рандомизации: 3,0 мг/кг AE-Keto-Sulf07 (эквивалент AE) дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 5,32 мг/кг ≡ 106,3 мкг/20 г мыши. Приготовление образца: 51,0 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,8 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 0,72 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
3) 7 мышей, средняя масса 25 г, средний медианный объем опухоли 141 мм3 в день рандомизации: 5,0 мг/кг AE-Keto-Sulf07 (эквивалент AE), дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 8,86 мг/кг ≡ 177,2 мкг/20 г мыши. Приготовление образца: 51,0 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,8 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
[00231] Развитие роста опухоли в модели карциномы яичника A2780 показало статистически значимую превосходящую противоопухолевую эффективность соединения AE-Keto-Sulf07 в дозах 3,0 и 5,0 мг/кг по сравнению с ауристатином E в дозе 0,3 мг/кг, вводимой восьмикратно два раза в неделю (р < 0,01 с 7 по 29 день). У мышей, получавших ауристатин Е, не был выявлен какой-либо противоопухолевый эффект, и опухоли росли сопоставимо с опухолями в контрольной группе. Однако терапия AE-Keto-Sulf07 вызывала частичную ремиссию с 9-го дня до конца исследования на 60-й день, с кратковременной полной ремиссией с 23-го по 35-й день для более низкой дозы и с 25-го по 51-й день для более высокой дозы, что приводило к длительной регрессии опухоли, независимо от дозы.
[00232] Группы, обработанные AE-Keto-Sulf07, выявили временную потерю массы тела ~2% после первой обработки. Одна мышь из группы с более высокой дозой была найдена мертвой на 35-й день, а одна из группы с более низкой дозой была умерщвлена на 42-й день из-за общего плохого состояния.
Оценка ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 при карциноме яичника человека, модель A2780 - крупные опухоли (фиг. 18).
[00233] Оценка ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Keto-Sulf07 на модели карциномы яичника человека A2780 была проведена, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00234] Исходные растворы готовили следующим образом:
1) 8 мышей, средний вес 28 г, средний медианный объем опухоли 341 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE), вводимой дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 3,7 мг взвешивали во флаконе объемом 100 мл, растворяли в 52,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,5 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,5 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, pH 7,0.
2) 8 мышей, средний вес 28 г, средний медианный объем опухоли 378 мм3 в день рандомизации: 4,5 мг/кг AE-Keto-Sulf07 (эквивалент AE), дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 6,99 мг/кг ≡139,7 мкг/20 г мыши. Приготовление образца: 40,0 мг взвешивали в 30 мл флаконе, растворяли в 28,6 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,5 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,5 мл 50 мМ натрий-фосфатного буфера, 2% 2-HPβCD, pH 7,6.
[00235] Противоопухолевую эффективность соединения AE-Keto-Sulf07 также тестировали на мышах с более крупными опухолями (~360 мм3) модели рака яичника человека A2780. Препарат показал статистически значимый превосходящий противоопухолевый эффект в дозе 4,5 мг/кг по сравнению с ауристатином Е, дозированным в дозе 0,3 мг/кг, оба препарата вводили восьмикратно два раза в неделю (р <0,001 с 22 по 40 день). У мышей, получавших ауристатин Е, не был получен какой-либо противоопухолевый эффект, в отличие от мышей, которых лечили AE-Keto-Sulf07, где отмечалась полная ремиссия с 26 дня до конца исследования на 103 день, и была достигнута долгосрочная регрессия опухоли, как уже наблюдалось при меньшем объеме опухоли.
[00236] Мыши, обработанные AE-Keto-Sulf07, имели только слегка уменьшенный прирост массы тела по сравнению с контролем.
Первоначальная оценка эффективности ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Эстер-Sulf07 на модели рака почки человека RXF631 - мелкие опухоли (фиг. 19).
[00237] Первоначальная оценка эффективности ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Эстер-Sulf07 на модели рака почки человека RXF631 была проведена, как описано в общей процедуре для модели ксенотрансплантата, полученной от пациента.
[00238] Для этого эксперимента in vivo исходные растворы готовили следующим образом:
1) 7 мышей, 25 г среднего веса, 138 мм3 среднего объема опухоли в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE), вводимой дважды в неделю в течение 4 недель в день 1, 5, 8, 12, 15, 19, 22, 26 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, 26 г среднего веса, 141 мм3 среднего объема опухоли в день рандомизации: 2,2-3,0 мг/кг AE-Эстер-Sulf07 (эквивалент AE), вводимых дважды в неделю в течение 4 недель в день 1, 5, 8, 12, 15, 19, 26 ≡ 4,00 мг/кг ≡ 80,1 мкг/20 г мыши. Приготовление образца: 15,7 мг взвешивали во флаконе объемом 30 мл, растворяли в 19,6 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
[00239] Развитие опухолевого роста в модели почечно-клеточного рака RXF631 показывает статистически значимую превосходящую противоопухолевую эффективность соединения AE-Эстер-Sulf07, дозированного по 2,2 мг/кг (доза была временно увеличена до 3,0 мг/кг на 15 и 19 день инъекции) по сравнению с ауристатином Е, дозированным по 0,3 мг/кг, вводимым восьмикратно и семикратно соответственно два раза в неделю (р <0,05). Ауристатин Е вызывал достижение состояния стабильного заболевания до приблизительно 40 дня, после чего стабильно рос до конца исследования. В отличие от этого, терапия AE-Эстер-Sulf07 вызывала полную ремиссию с 24 дня до 53 дня при достижении минимального объема опухоли 1 мм3 и долгосрочного эффекта.
[00240] AE-Эстер-Sulf07 вызывал кратковременную потерю массы тела с минимумом 9%, достигнутым на 31-й день. Начиная с 17-го дня также наблюдались раны от укусов и царапин. Две мыши умерли, а одну пришлось умерщвлять из-за плохого общего состояния (в день 20, 43 и 66).
Оценка эффективности ауристатина E и альбумин-связывающего производного ауристатина E AE-Эстер-Sulf07 на модели злокачественной меланомы человека A375 - крупные опухоли (фиг. 20).
[00241] Оценку ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Эстер-Sulf07 на модели злокачественной меланомы человека A375 проводили, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00242] На основании опыта дозирования в модели ксенотрансплантата RXF631 готовые растворы приготовляли следующим образом:
1) 7 мышей, средний вес 28 г, средний медианный объем опухоли 310 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE) дважды в неделю в течение 3 недель в день 1, 6, 9, 13, 16 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средний вес 29 г, средний медианный объем опухоли 371 мм3 в день рандомизации: 2,4 мг/кг AE-Эстер-Sulf07 (эквивалент AE) дважды в неделю в течение 3 недель в день 1, 6, 9, 13, 16 ≡ 4,37 мг/кг ≡ 87,3 мкг/20 г мыши. Приготовление образца: 12,6 мг взвешивали во флаконе объемом 30 мл, растворяли в 14,4 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 50 мМ натрий-фосфатного буфера, 2% Tween 80, рН 7,6.
3) 7 мышей, средний вес 31 г, средний медианный объем опухоли 355 мм3 в день рандомизации: 2,4 мг/кг AE-Эстер-Sulf07 (эквивалент AE) дважды в неделю в течение 3 недель в день 1, 6, 9, 13, 16 ≡ 4,37 мг/кг ≡ 87,3 мкг/20 г мыши. Приготовление образца: 12,6 мг взвешивали во флаконе объемом 30 мл, растворенном в 14,4 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали с помощью 1,2 мл 50 мМ натрий-фосфатного буфера, 4% 2-HPβCD, pH 7,6.
[00243] Развитие роста опухоли в модели ксенотрансплантата меланомы A375 демонстрирует статистически значимый противоопухолевый эффект соединения AE-Эстер-Sulf07, дозированного по 2,4 мг/кг (независимо от буфера для восстановления, 2% Tween 80 или 4% 2-HPβCD) по сравнению с ауристатином Е, дозированным в дозе 0,3 мг/кг, вводимым пятикратно два раза в неделю (р <0,01 с 9 по 33 день). Лечение ауристатином Е показало начальное стабильное заболевание до 14 дня, после чего опухоли неуклонно росли. Однако мыши, обработанные AE-Эстер-Sulf07, достигли почти полной ремиссии с 16-го дня до конца исследования на 37-й день, достигнув минимума 4 мм3 на 23-й день и 1 мм3 на 26-й день для восстановленного AE-Эстер-Sulf07 с 2% Tween 80 и 4% 2-HPβCD соответственно, несмотря на большой медианный объем опухоли в начале эксперимента.
[00244] Не было зарегистрировано значительного изменения массы тела по сравнению с контролем, но с 9-го дня наблюдались раны от укусов и царапин. Лечение бепантеном улучшало здоровье раненых мышей. Только одну мышь в группе, обработанной AE-Эстер-Sulf07 2% Tween 80, пришлось умерщвить из-за общего плохого состояния.
Оценка эффективности ауристатина E и альбумин-связывающего производного ауристатина E AE-Эстер-Sulf07 на модели немелкоклеточного рака легких человека LXFA737- небольшие опухоли (фиг. 21).
[00245] Оценку ауристатина Е и альбумин-связывающего производного ауристатина Е AE-Эстер-Sulf07 на модели немелкоклеточного рака легкого человека LXFA737 проводили, как описано в общей процедуре для моделей ксенотрансплантата, полученных от пациента.
[00246] Исходные растворы готовили следующим образом:
1) 7 мышей, средний вес 27 г, средний медианный объем опухоли 137 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE) дважды в неделю в течение 4 недель в день 0, 4, 7, 11, 14, 18, 21, 25 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 2 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,2 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,2 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, рН 7,0.
2) 7 мышей, средний вес 27 г, средний медианный объем опухоли 126 мм3 в день рандомизации: 2,4 мг/кг AE-Эстер-Sulf07 (эквивалент AE), дважды в неделю в течение 4 недель в день 0, 4, 7, 11, 14, 18, 21, 25 ≡ 4,37 мг/кг ≡ 87,3 мкг/20 г мыши. Приготовление образца: 31 мг взвешивали во флаконе объемом 30 мл, растворяли в 28,5 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 0,96 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали с помощью 1,2 мл 50 мМ натрий-фосфатного буфера, 4% 2-HPβCD, pH 7,6.
[00247] Развитие опухолевого роста в модели ксенотрансплантата NSCLC LXFE737 показало превосходящую противоопухолевую эффективность соединения AE-Эстер-Sulf07 в дозе 2,4 мг/кг по сравнению с ауристатином E в дозе 0,3 мг/кг, оба дозировались восьмикратно два раза в неделю, со статистической значимостью (p < 0,05 на 56 и 63 день). Опухоли в группе, получавшей ауристатин Е, оставались только временно в состоянии стабильного заболевания с 21 по 42 день и значительно увеличивались после окончания терапии. В отличие от этого, мыши, получавшие AE-Эстер-Sulf07, достигли полной ремиссии опухоли на 21 день, в результате чего объем опухоли снизился до неизмеримого уровня до конца исследования на 74 день, что привело к долгосрочному противоопухолевому эффекту.
[00248] В ходе исследования пять мышей были подвергнуты эвтаназии из-за сочетания потери массы тела и поражений кожи.
Оценка эффективности ауристатина E и альбумин-связывающего производного ауристатина E AE-Эстер-Sulf07 при карциноме яичника человека, модель A2780 - крупные опухоли (фиг. 22).
[00249] Оценка ауристатина E и альбумин-связывающего производного ауристатина E AE-Эстер-Sulf07 на модели карциномы яичника человека A2780 была проведена, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00250] Исходные растворы готовили следующим образом:
1) 8 мышей, средний вес 28 г, средний медианный объем опухоли 341 мм3 в день рандомизации: 0,3 мг/кг соли ТФК ауристатина E (эквивалент AE), вводимой дважды в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 0,35 мг/кг ≡ 7,1 мкг/20 г мыши. Приготовление образца: 3,7 мг взвешивали во флаконе объемом 100 мл, растворяли в 52,3 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,5 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,5 мл 10 мМ натрий-фосфатного буфера, 20% пропиленгликоля, pH 7,0.
2) 8 мышей, средний вес 27 г, средний медианный объем опухоли 403 мм3 в день рандомизации: 1,9 мг/кг AE-Эстер-Sulf07 (эквивалент AE) вводили один раз в неделю в течение 4 недель в день 1, 4, 8, 11, 15, 18, 22, 25 ≡ 3,46 мг/кг ≡ 69,2 мкг/20 г мыши. Приготовление образца: 36,5 мг взвешивали во флаконе объемом 30 мл, растворяли в 26,4 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы - рН 7,0; 0,75 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день° инъекции лиофилизированные образцы восстанавливали 1,5 мл 50 мМ натрий-фосфатного буфера, 2% 2-HPβCD, pH 7,6.
[00251] AE-Эстер-Sulf07, дозированный по 1,9 мг/кг, демонстрирует превосходную противоопухолевую эффективность в модели карциномы яичника A2780 по сравнению с ауристатином E, дозированным по 0,3 мг/кг, оба вводились восьмикратно два раза в неделю (р <0,001 с 37-го дня до день 40). Мыши, получавшие ауристатин Е, не выявили какой-либо противоопухолевый эффект. Однако у мышей, получавших AE-Эстер-Sulf07, наблюдалась частичная ремиссия с 19 дня до 51 дня и полная ремиссия с 54 дня до конца исследования на 103 день, что приводило к длительной регрессии опухоли.
[00252] Мыши, обработанные AE-Эстер-Sulf07, не имели признаков потери массы тела по сравнению с контролем, но четырех животных пришлось исключить из исследования из-за некроза опухоли.
Оценка эффективности альбумин-связывающих производных ауристатина Е - соединения AE-Эстер-Sulf07 для карциномы яичника человека, модель A2780 - небольшие опухоли (фиг. 23).
[00253] Оценку альбумин-связывающего производного ауристатина E - AE-Эстер-Sulf07 на модели карциномы яичника человека A2780 проводили, как описано в общей процедуре для моделей ксенотрансплантатов, полученных из клеточной линии.
[00254] Исходные растворы готовили следующим образом:
1) 8 мышей, средний вес 27 г, средний медианный объем опухоли 174 мм3 в день рандомизации: 3,8 мг/кг AE-Эстер-Sulf07 (эквивалент AE) один раз в неделю в течение 4 недель в день 1, 8, 15, 22 ≡ 6,67 мг/кг ≡ 133,4 мкг/20 г мыши. Приготовление образца: 25,6 мг взвешивали во флаконе объемом 30 мл, растворяли в 19,2 мл 50/50 трет-бутанола/10 мМ натрий-фосфатного буфера, 5% сахарозы, рН 7,0; 1,5 мл были разделены на аликвоты в 4 мл флаконах. Флаконы замораживали в течение 1 ч при минус 40°С и лиофилизировали при минус 20°С в течение примерно 36 ч, а затем закупоривали. В день инъекции лиофилизированные образцы восстанавливали 1,5 мл 50 мМ натрий-фосфатного буфера, 6% 2-HPβCD, pH 7,6.
[00255] AE-Эстер-Sulf07 также тестировали на той же модели карциномы яичника A2780 с более мелкими опухолями и другой схемой дозирования, чтобы уменьшить побочные эффекты лекарственного средства на коже мышей. AE-Эстер-Sulf07 дозировали по 3,8 мг/кг четырехкратно один раз в неделю, снова демонстрируя статистически значимое улучшение противоопухолевого эффекта по сравнению с контролем (р <0,01 с 7 по 14 день). У мышей, получавших AE-Эстер-Sulf07 в новой схеме дозирования, была достигнута частичная ремиссия с 12 дня до 35 дня и полная ремиссия с 39 дня до конца исследования на 70 день. Три мыши, обработанные AE-Эстер-Sulf07, показали потерю массы тела и их пришлось подвергнуть эвтаназии в день 59.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВ НА ОСНОВЕ МАЙТАНЗИНОИДА | 2018 |
|
RU2783076C2 |
| СИСТЕМЫ ДОСТАВКИ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2016 |
|
RU2748992C2 |
| СРЕДСТВА ВИЗУАЛИЗАЦИИ ДЛЯ МЕЧЕННОГО РАДИОАКТИВНЫМ ИЗОТОПОМ ЭКЗОГЕННОГО И ЭНДОГЕННОГО АЛЬБУМИНА | 2019 |
|
RU2819907C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ НАНОЧАСТИЦЫ, СОДЕРЖАЩИЕ НЕРАСТВОРИМОЕ СОЕДИНЕНИЕ НА ОСНОВЕ КАМПТОТЕЦИНА, ДЛЯ ЛЕЧЕНИЯ РАКА, И КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2021 |
|
RU2827894C1 |
| РАДИОАКТИВНО МЕЧЕНЫЕ ПЕПТИДЫ, СВЯЗЫВАЮЩИЕСЯ С HER2 | 2011 |
|
RU2592685C2 |
| КОНЪЮГАТ МОНОМЕТИЛ АУРИСТАТИНА Е ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАКА ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2019 |
|
RU2729192C1 |
| КОМПОЗИЦИИ ВАКЦИН | 2010 |
|
RU2581020C2 |
| ПРОТИВООПУХОЛЕВЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2015 |
|
RU2581022C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ САПОЗИНА С И СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2767199C1 |
| СТАБИЛЬНЫЙ ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ОСНОВЕ АНТИТЕЛА К PD-1 И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2731418C2 |
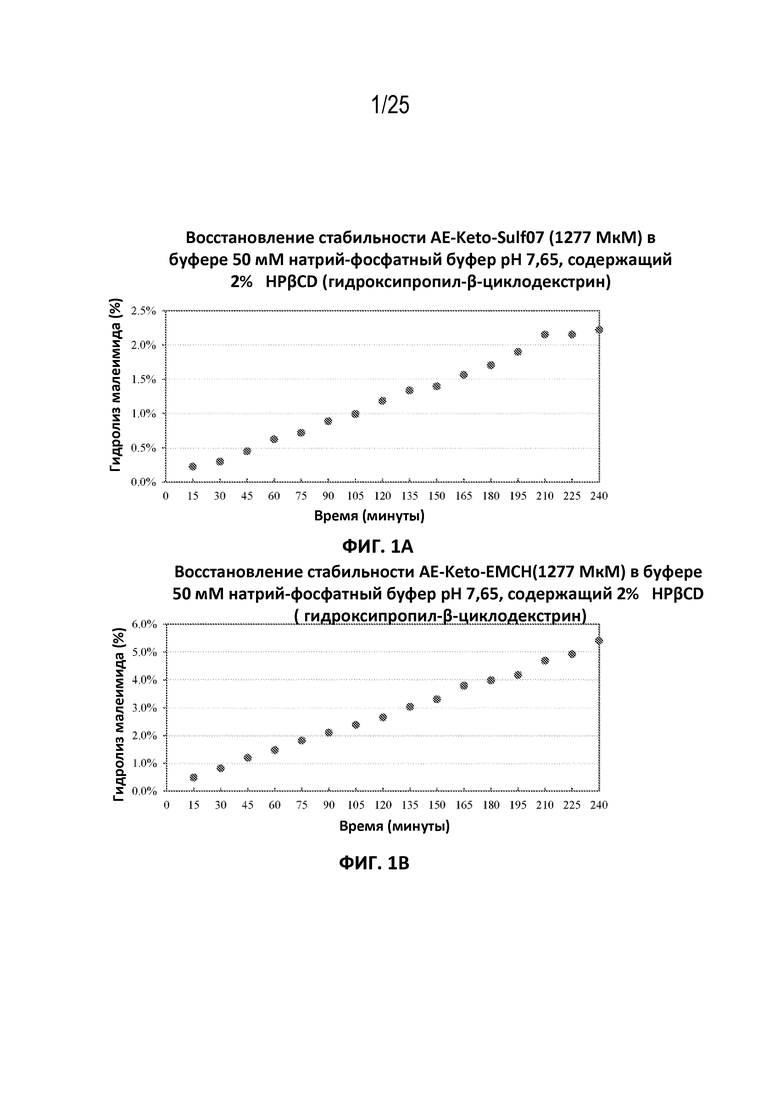
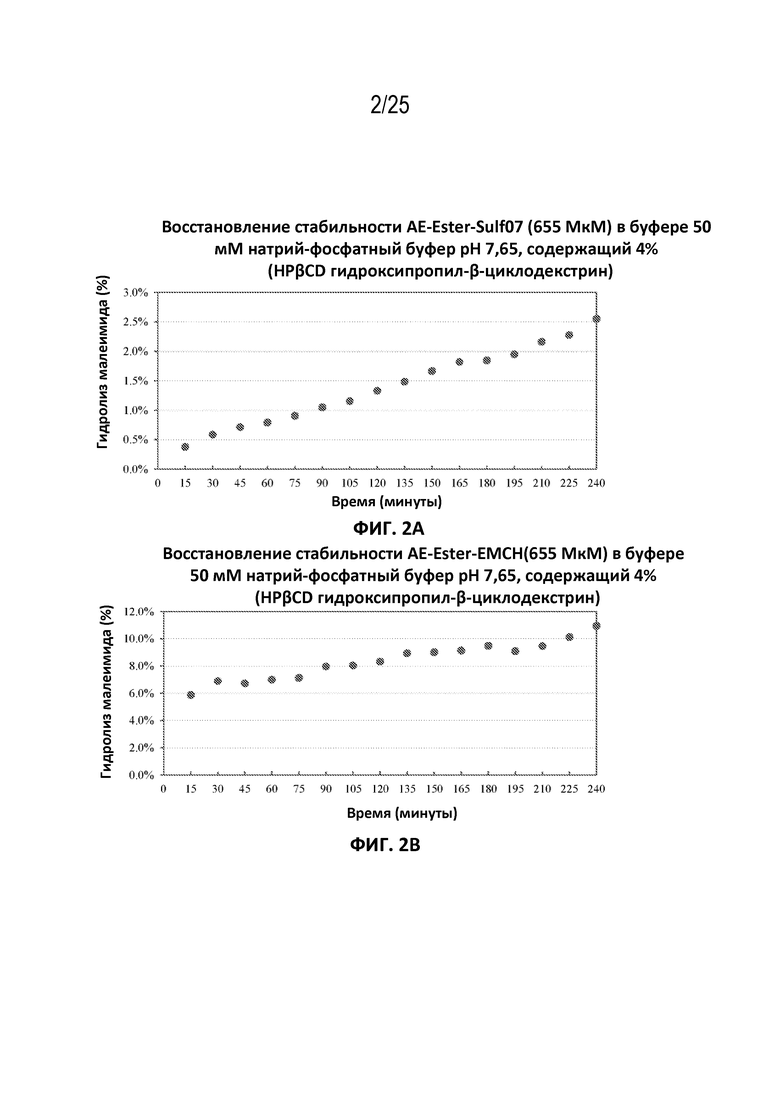
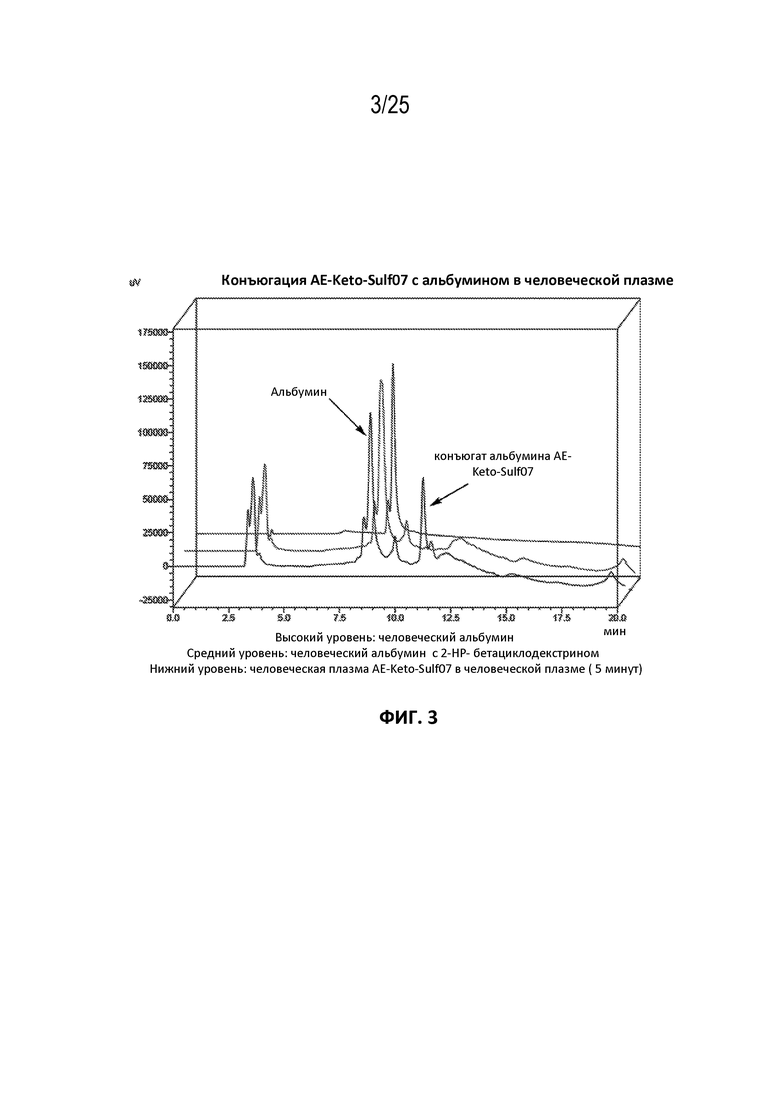
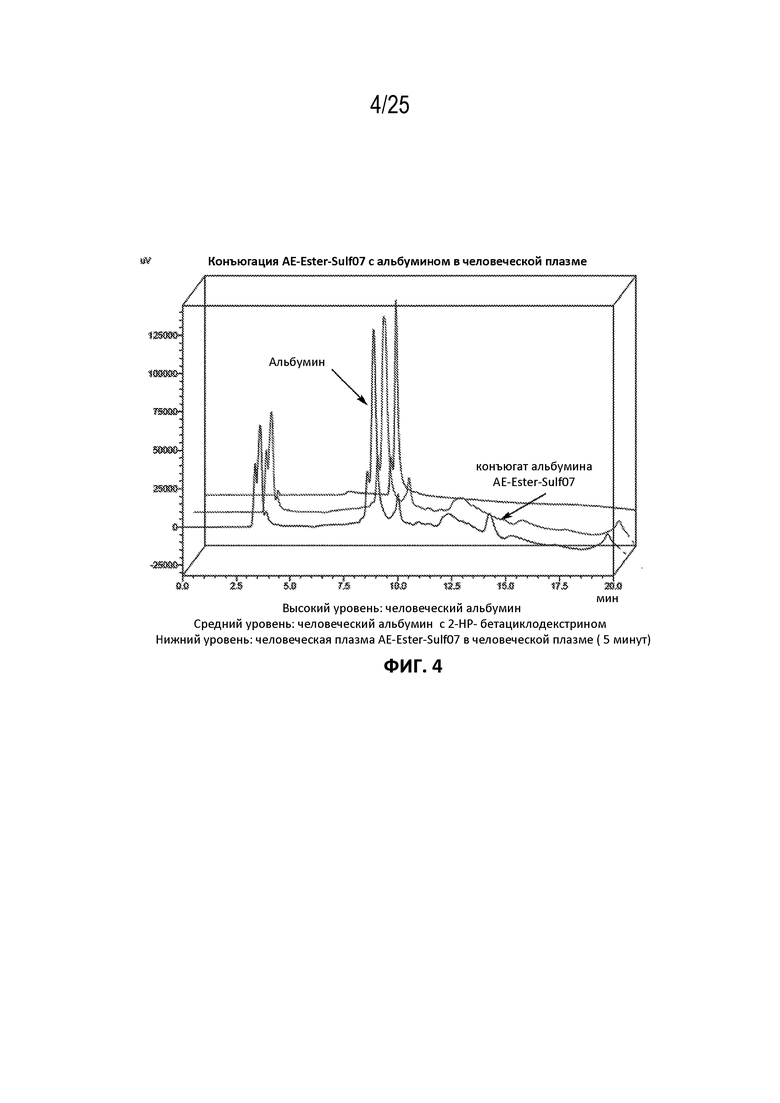
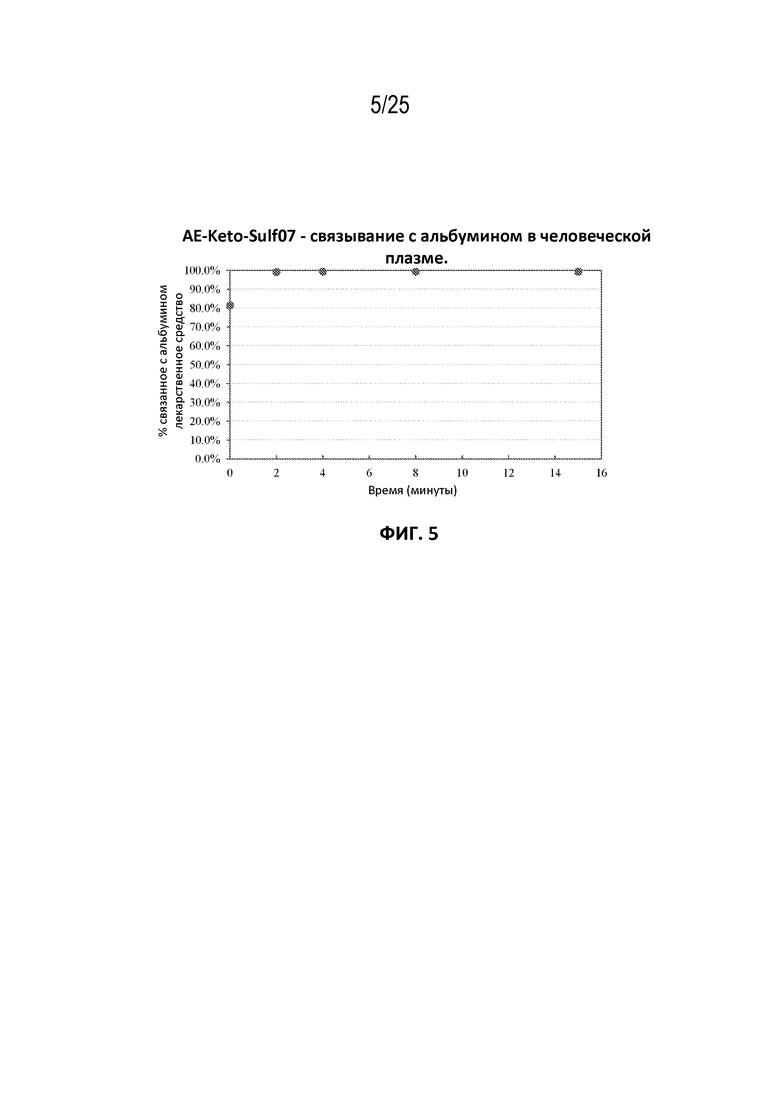
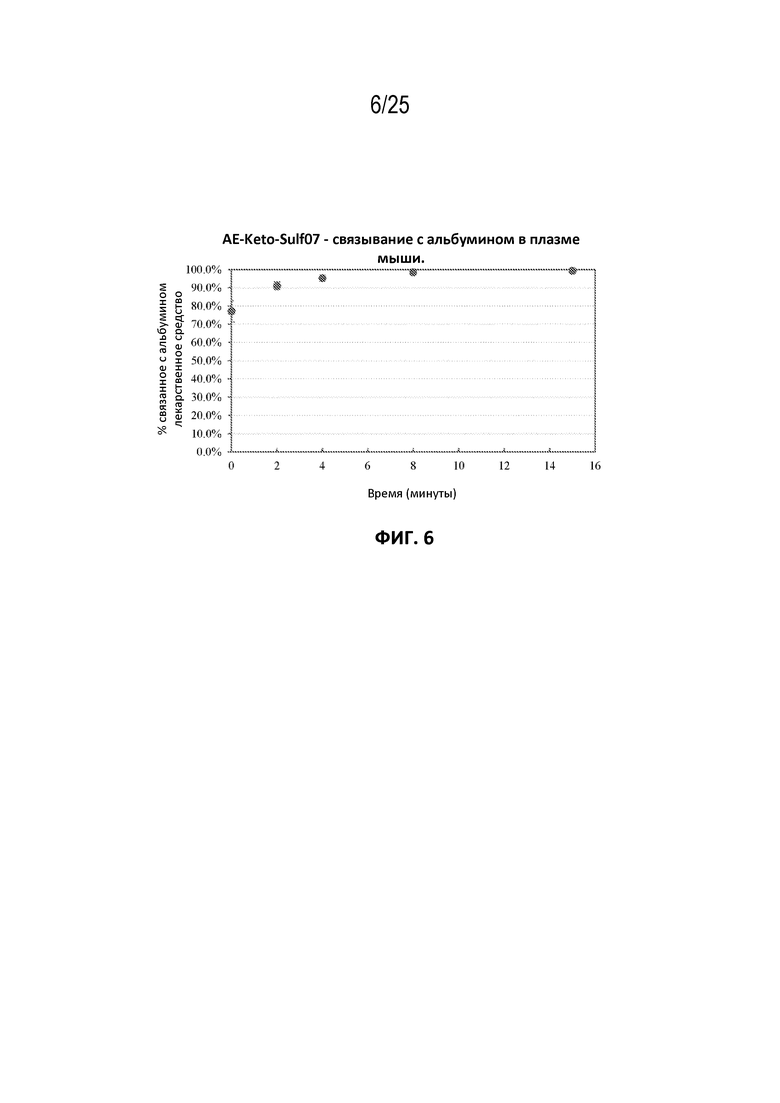


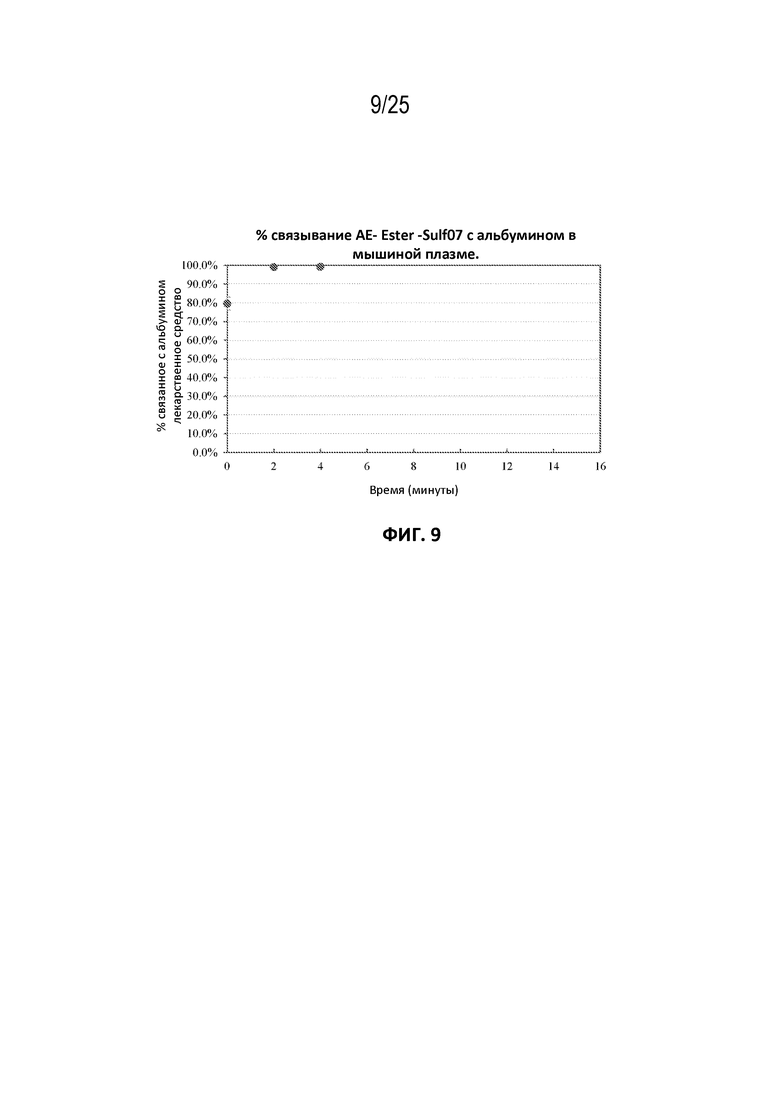
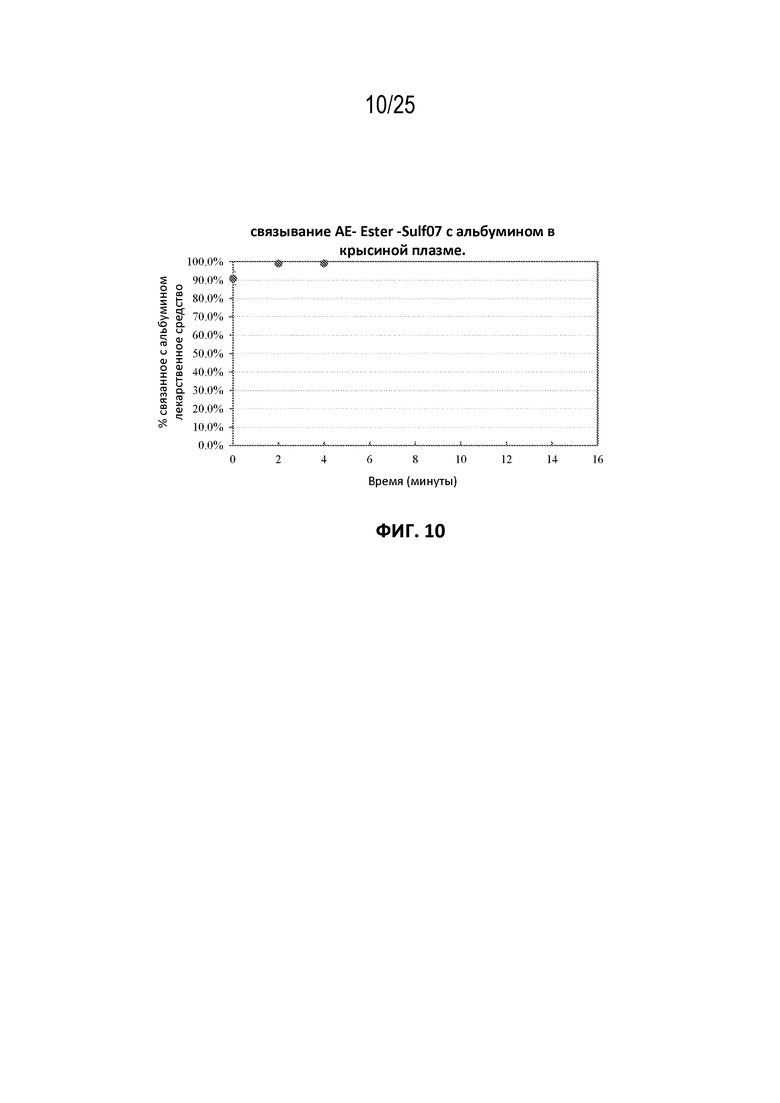
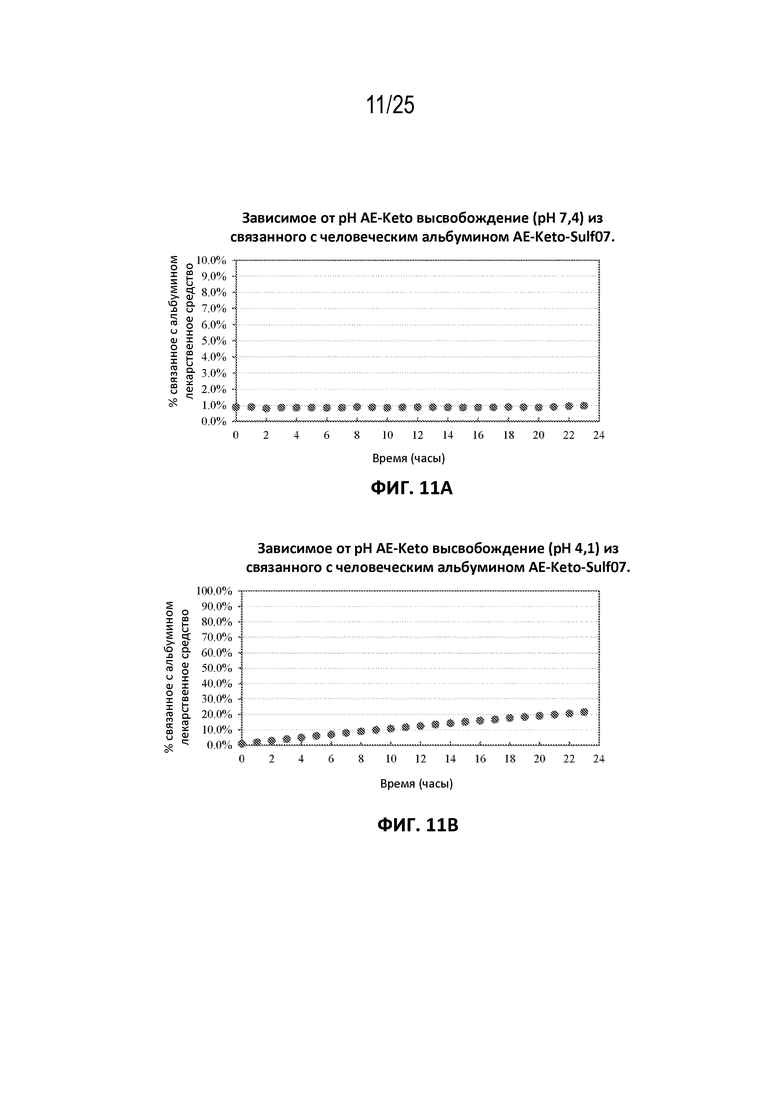
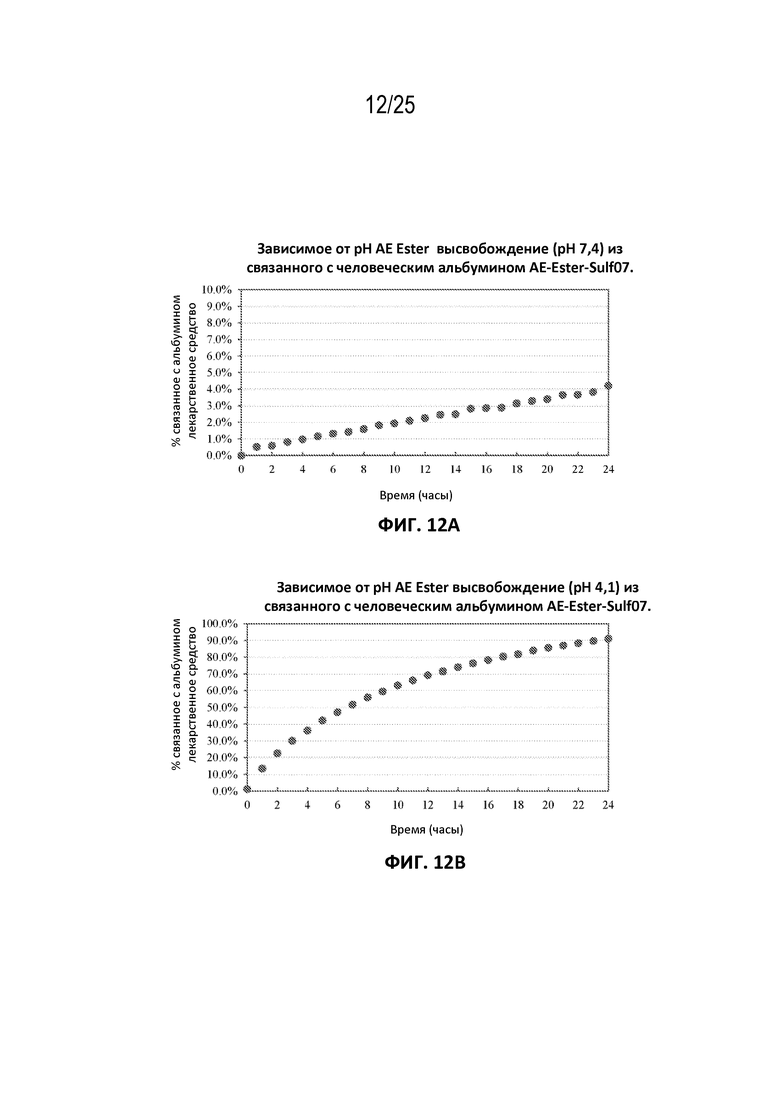
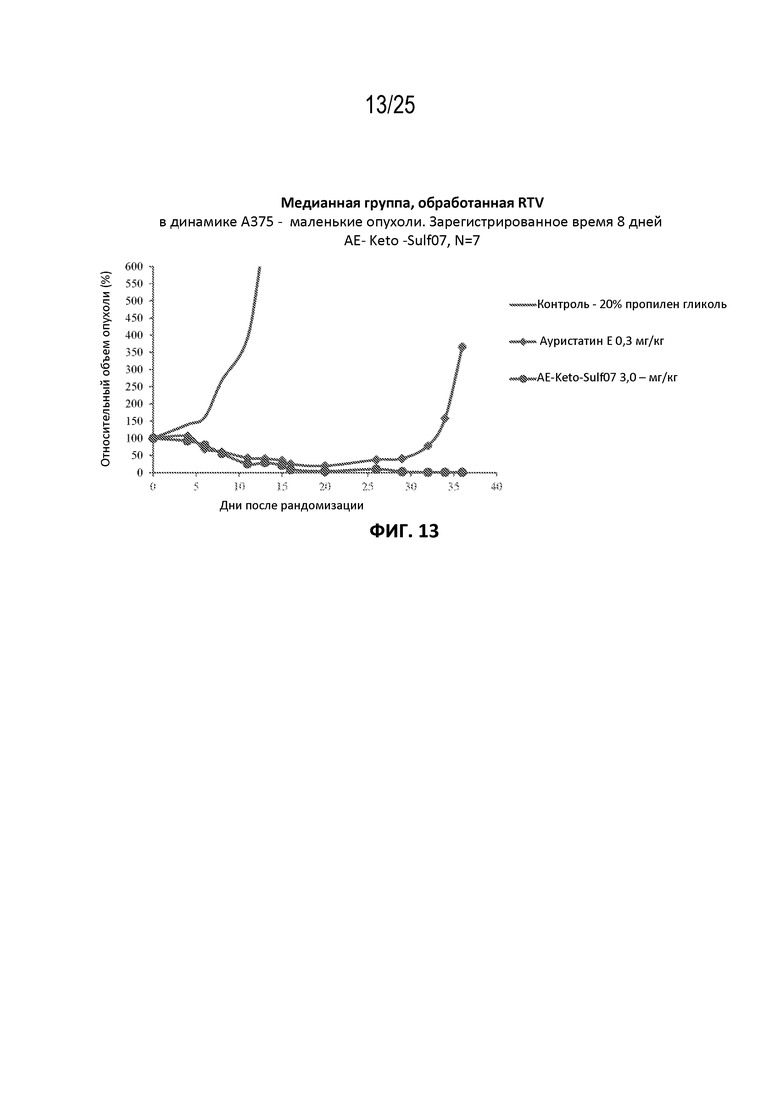
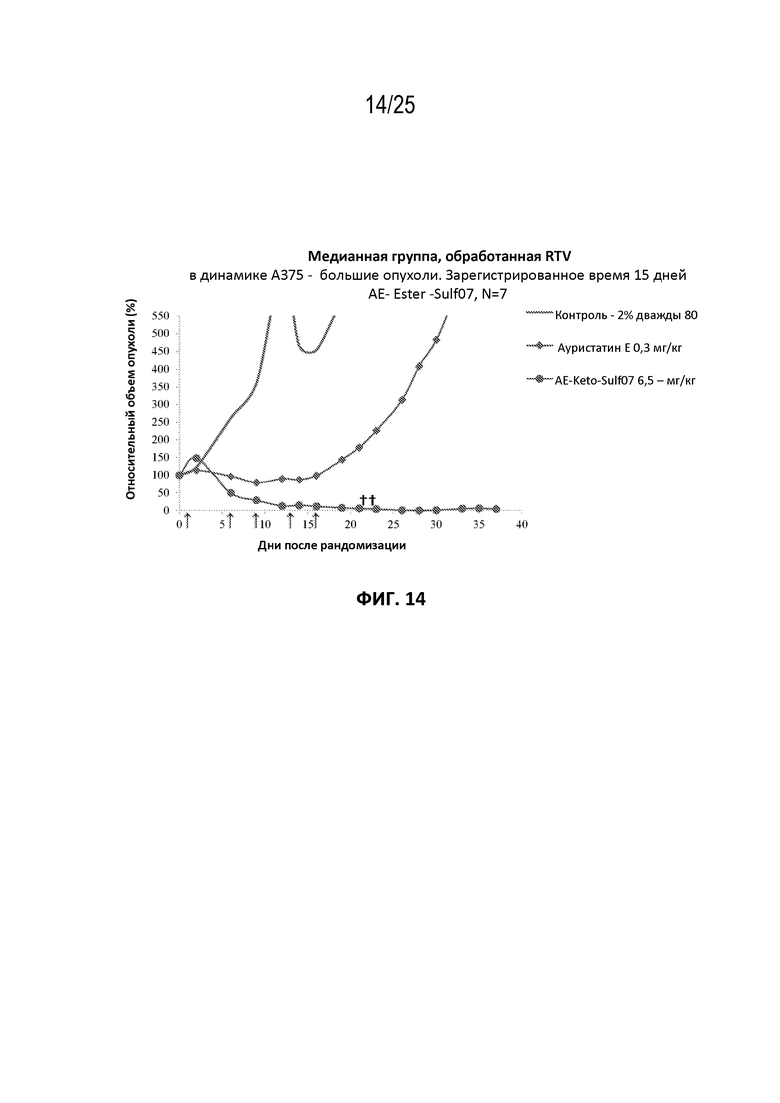
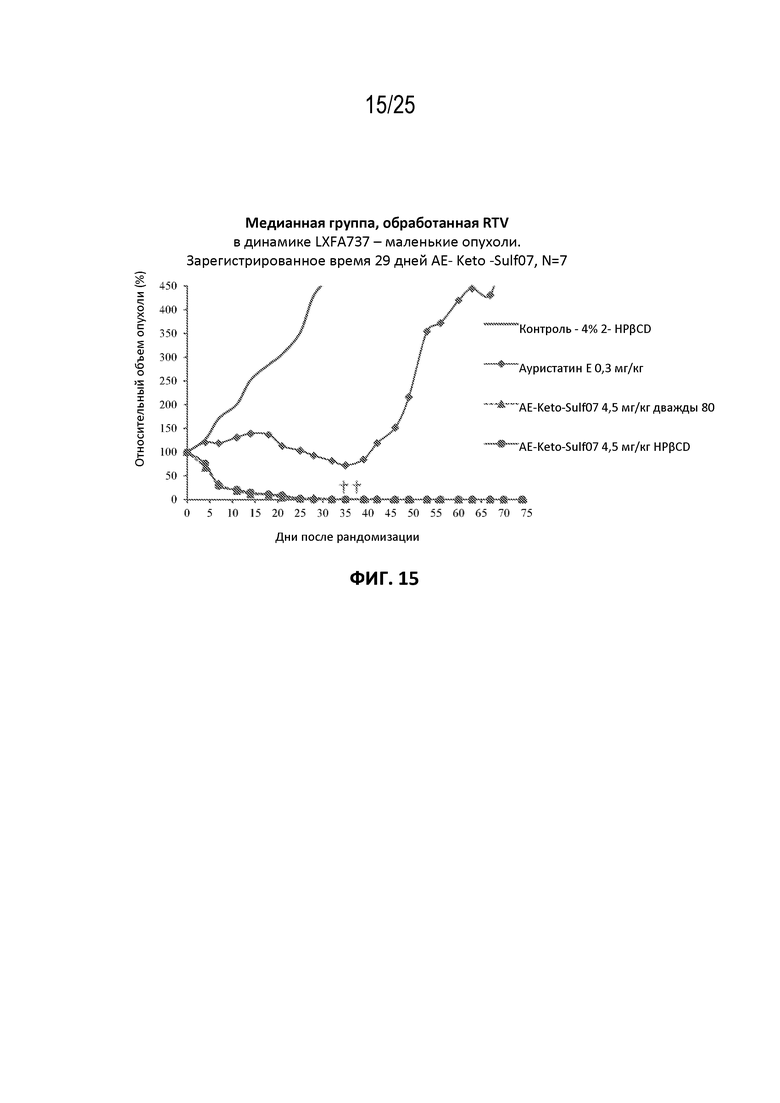
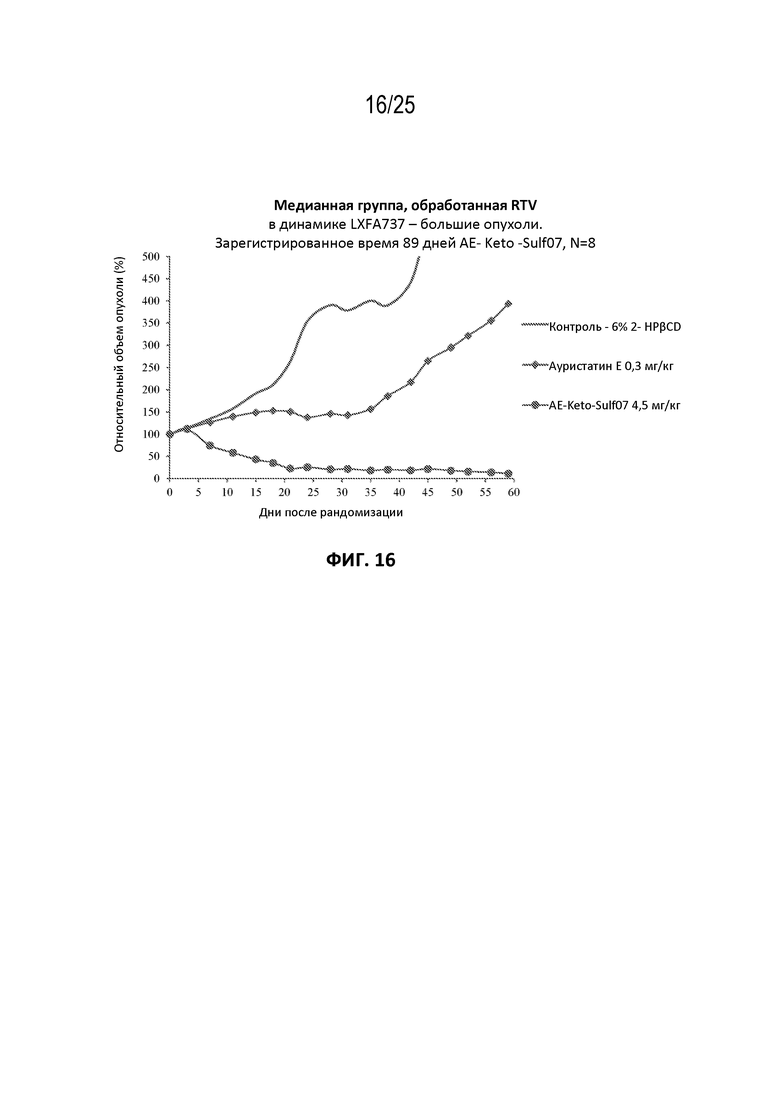

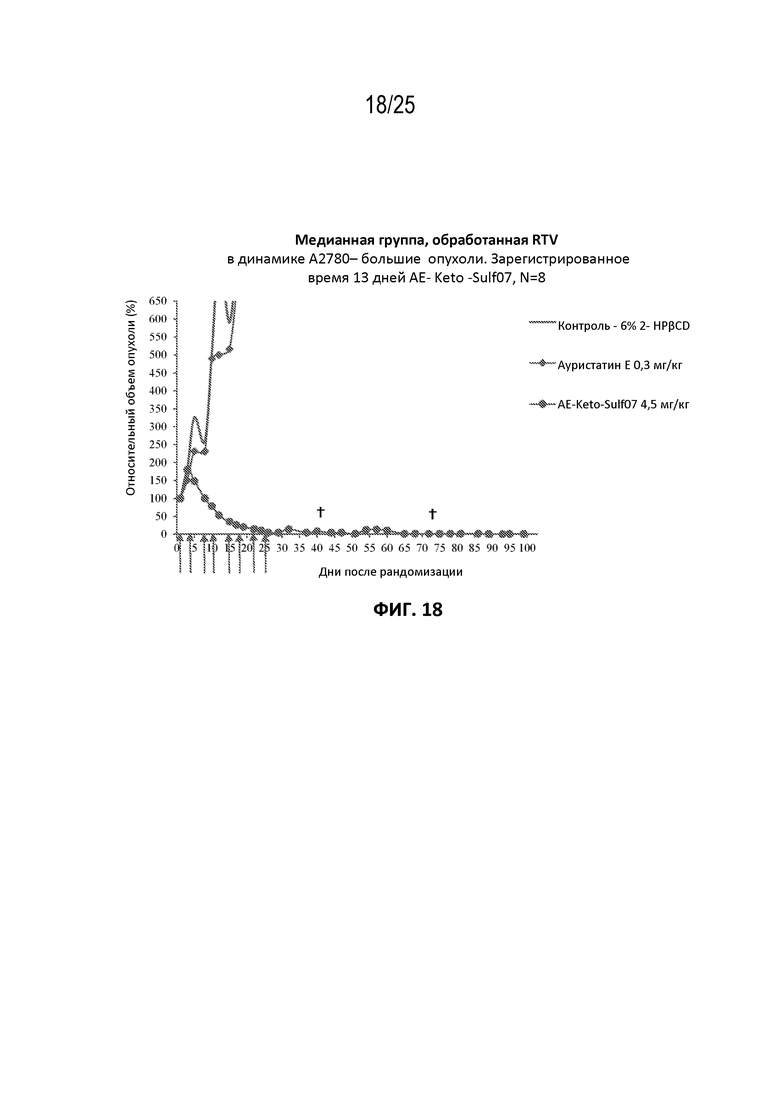
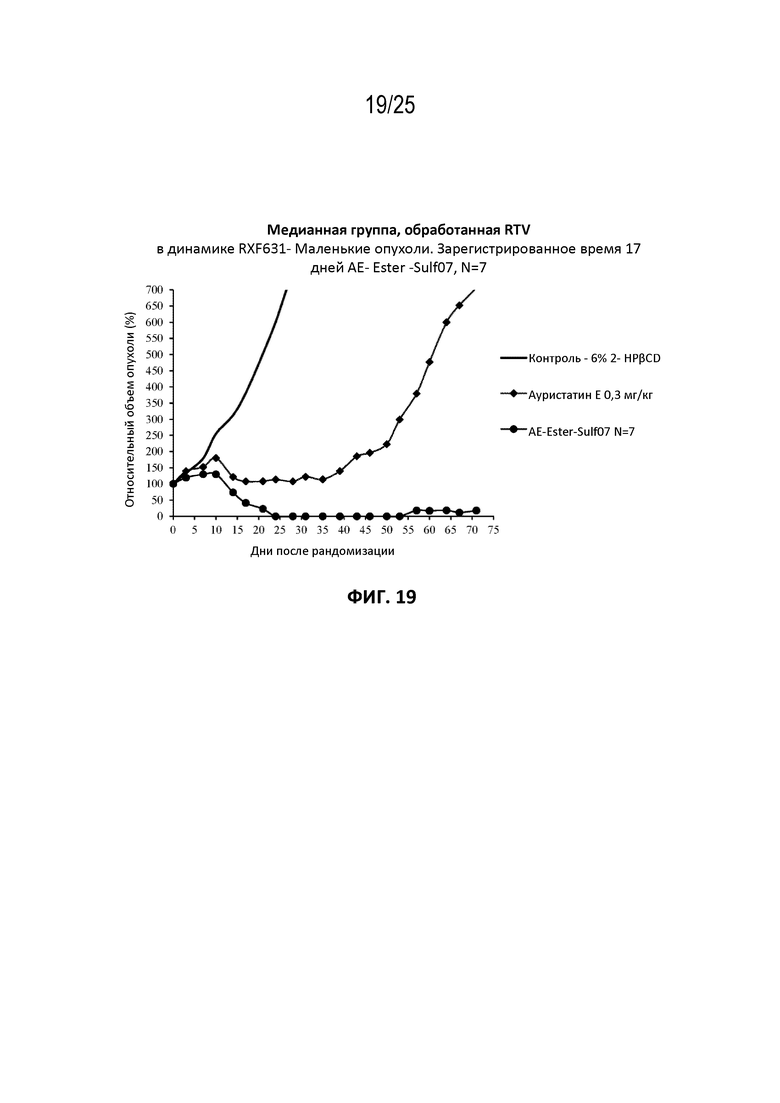
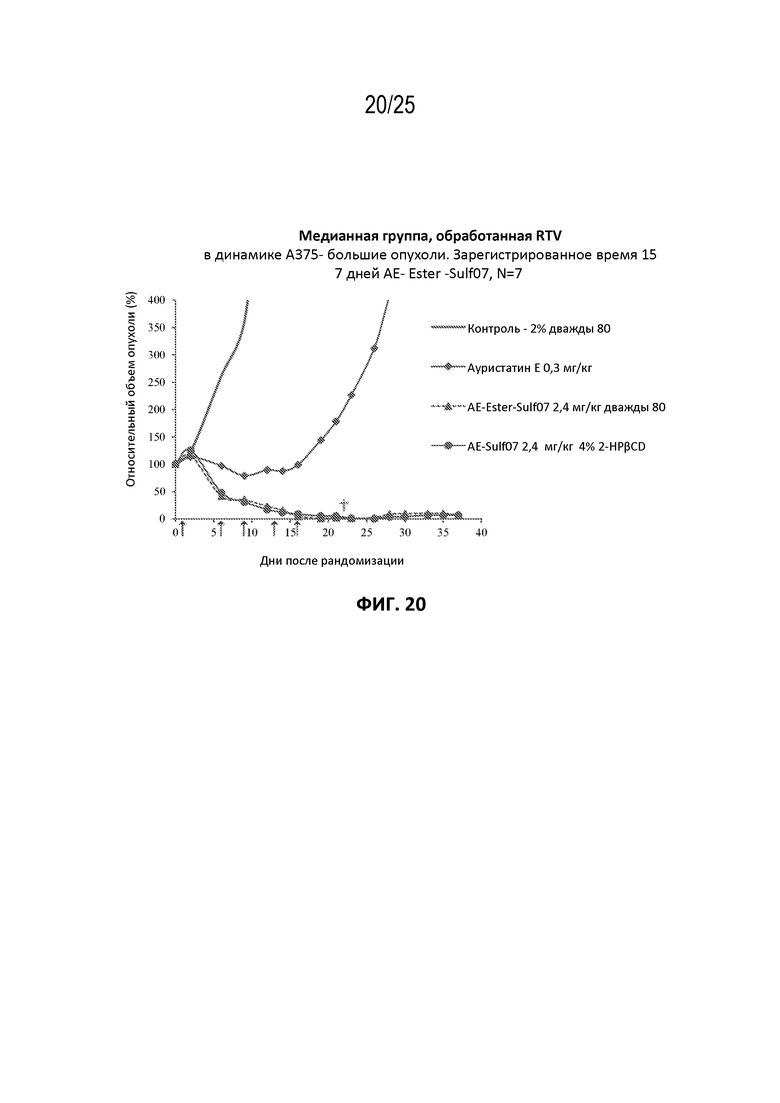
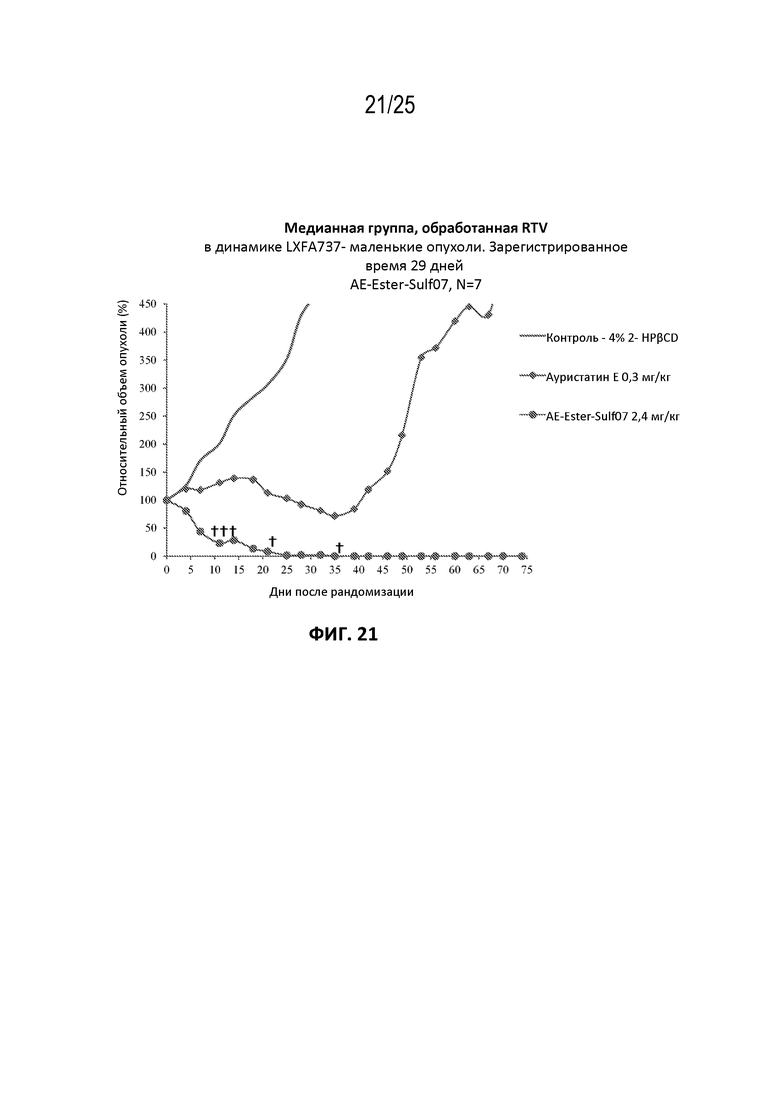

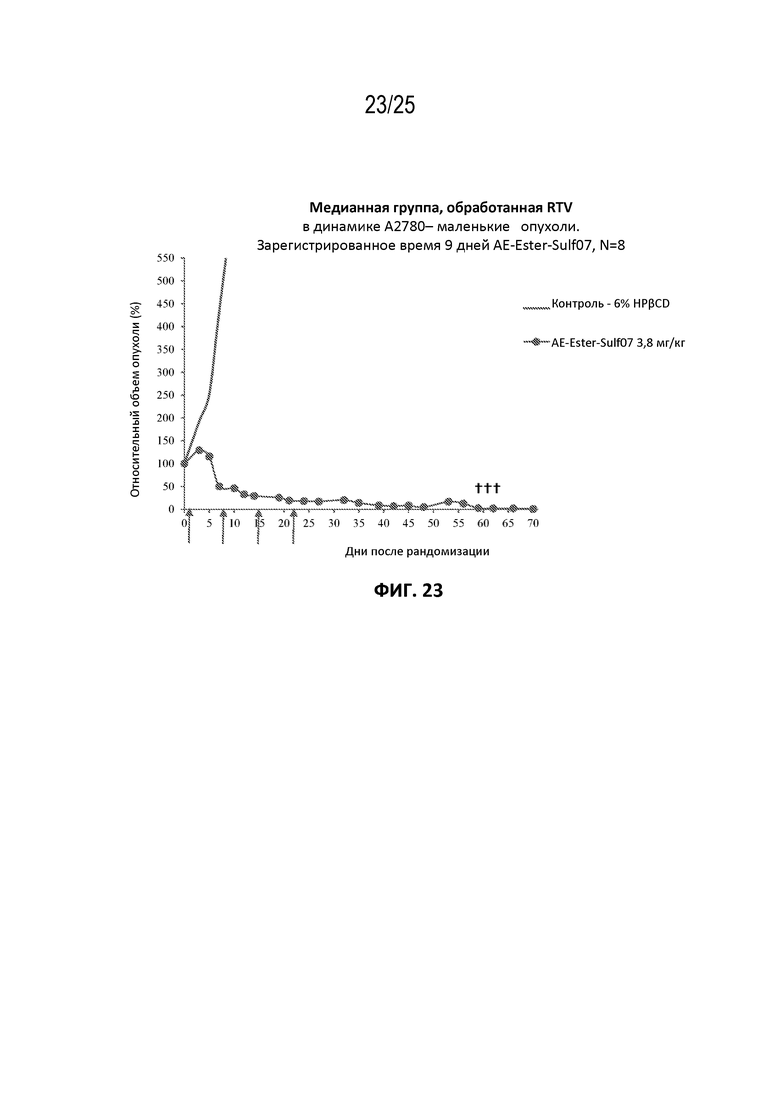


Изобретение относится к альбуминсвязывающим пролекарствам, производным ауристатина Е, которые могут быть использованы для лечения злокачественных заболеваний. Введение соединений настоящего изобретения приводит к снижению цитотоксичности по сравнению с эквивалентной дозой немодифицированного активного агента. 5 н. и 13 з.п. ф-лы, 25 ил., 5 пр.
1. Соединение, имеющее структуру, описанную формулами I или II:
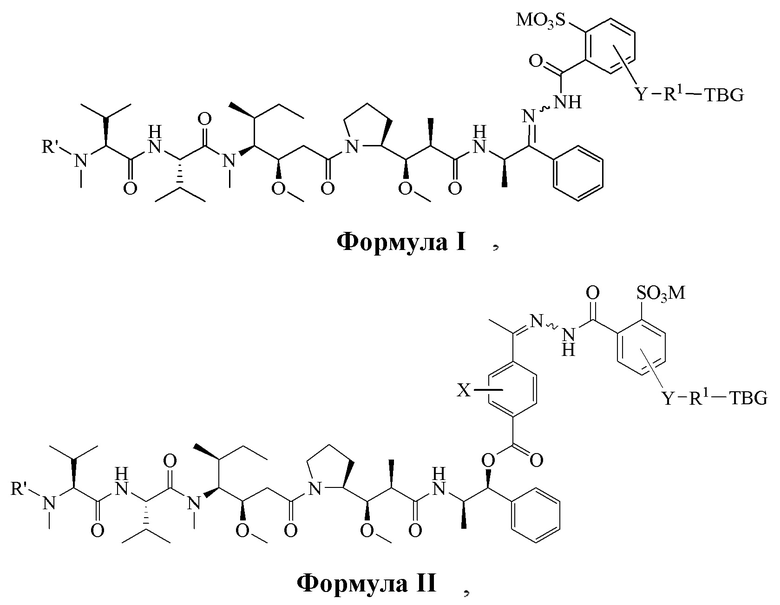
или его фармацевтически приемлемая соль;
в которой:
R’ представляет собой H или -CH3,
М представляет собой Н или Na+, K+, NR4+ или NHR3+, где R представляет собой H;
Y представляет собой -NH-C(O)-;
R1 отсутствует или представляет собой C1-C18 алкил;
Х представляет собой Н;
TBG представляет собой малеимидную группу.
2. Соединение по п.1, отличающееся тем, что R’ представляет собой -CH3.
3. Соединение по п.1 или 2, отличающееся тем, что
TBG представляет собой малеимидную группу, имеющую формулу
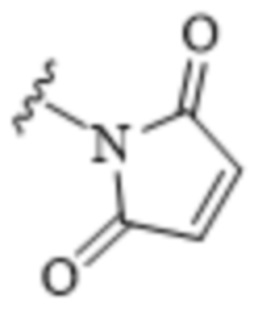 .
.
4. Соединение по любому из пп.1-3, отличающееся тем, что М представляет собой H+ или Na+.
5. Соединение по любому из пп.1-4, отличающееся тем, что
R1 представляет собой С1-С5 алкил.
6. Соединение по п.1, отличающееся тем, что имеет структуру, представленную формулой III
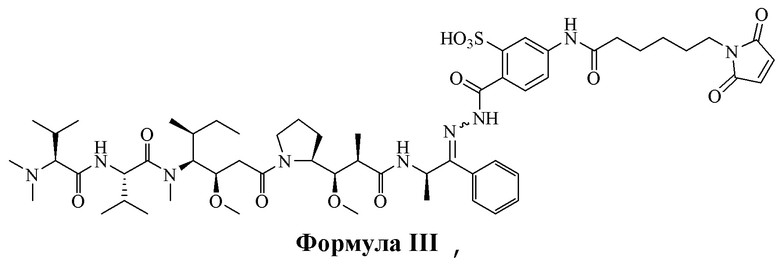
или его фармацевтически приемлемая соль.
7. Соединение по п.1, отличающееся тем, что имеет структуру, представленную формулой IV
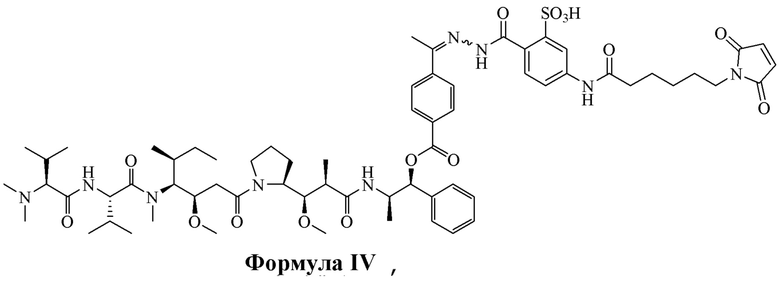
или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция для лечения рака у нуждающегося в этом пациента, содержащая терапевтически эффективное количество соединения по любому из пп.1-7 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
9. Фармацевтическая композиция по п.8, отличающаяся тем, что фармацевтически приемлемый носитель выбран из одного или более из следующего: солюбилизирующий агент, инкапсулирующий агент и лиопротектор.
10. Фармацевтическая композиция по п.9, отличающаяся тем, что фармацевтически приемлемый носитель содержит один или более из диметил-β-циклодекстрина, гидроксиэтил-β-циклодекстрина, гидроксипропил-β-циклодекстрина и триметил-β-циклодекстрина.
11. Фармацевтическая композиция по любому из пп.8-10, отличающаяся тем, что фармацевтическая композиция пригодна для внутривенного введения.
12. Фармацевтическая композиция по п. 11, отличающаяся тем, что композиция при внутривенном введении пациенту ковалентно и быстро избирательно связывается in situ с:
а) эндогенным альбумином в кровотоке; или
b) тиольной группой цистеина-34 эндогенного альбумина в кровотоке.
13. Способ лечения рака, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.8-12.
14. Способ по п.13, отличающийся тем, что рак выбирают из группы, включающей карциному, саркому, лейкоз, лимфому, множественную миелому и меланому.
15. Способ по п.14, где рак выбирают из карциномы яичника человека, почечно-клеточной карциномы, немелкоклеточного рака легкого и меланомы.
16. Способ по любому из пп.13-15, отличающийся тем, что способом введения является внутривенное введение.
17. Способ снижения цитотоксичности соединения, включающий введение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.8-12 нуждающемуся в этом пациенту, отличающийся тем, что введение приводит к снижению цитотоксичности по сравнению с эквивалентной дозой немодифицированного активного агента.
18. Способ увеличения концентрации метаболита соединения в опухоли, включающий введение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.8-12 нуждающемуся в этом пациенту, при этом увеличение сравнивается с эквивалентной дозой немодифицированного активного агента.
| WO 2002088172 A2, 07.11.2002 | |||
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| KAI TEMMING ET AL, "Evaluation of RGD-Targeted Albumin Carriers for Specific Delivery of Auristatin E to Tumor Blood Vessels", BIOCONJUGATE CHEMISTRY, 2006, vol | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| HAO CHEN ET AL, "Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy", MOLECULES, | |||

