ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к о-аминогетероарилалкинил содержащему соединению, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Рецепторная тирозинкиназа (РТК) представляет собой класс трансмембранного фермент-связанного рецептора, сверхэкспрессия или сверхактивация которого тесно связана с возникновением и развитием опухолей. Рецепторы фактора роста фибробластов (РФРФ) и белок RET, кодируемый онкогенным RET (реаранжированный во время трансфекции), являются важными составляющими суперсемейства PTK и важными мишенями при терапии опухолей.
РФРФ в основном включают в себя четыре подтипа: РФРФ1, РФРФ2, РФРФ3 и РФРФ4 (Turner N., Grose R., Fibroblast growth factor signalling: From development to cancer, Nature Reviews Cancer. (2010) 10:116-129; Dieci M.V., Arnedos M., Andre F., Soria J.C., Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives, Cancer Discovery. (2013) 3:264-279.) Сверхэкспрессия или сверхактивация РФРФ посредством генной амплификации, мутации, слияния или индукции лиганда играют важную роль в стимулировании пролиферации, инвазии, миграции и ангиогенеза опухолевых клеток. В исследовании установлено, что РФРФ сверхэкспрессируются или сверхактивируются в различных опухолях, таких как немелкоклеточный рак легких, рак молочной железы, рак желудка, рак мочевого пузыря, рак эндометрия, рак простаты, рак шейки матки, рак толстой кишки, рак пищевода, кератинома, миелома, рабдомиосаркома и т. д. (Dieci MV, Arnedos M, Andre F, Soria JC: Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer discovery, 2013, 3, 264-79; Turner N, Grose R: Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer, 2010, 10, 116-29.) Например, гиперактивация сигнального пути РФРФ1 при плоскоклеточном раке немелкоклеточного рака легкого составляет до 20%; (Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer (vol 3, 66er5, 2011), Sci Transl Med. (2010); Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer, PLoS ONE. (2011) 6:e20351.) Сверхактивация сигнального пути РФРФ2 при раке желудка составляет 5-10% (Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I, Taniguchi H, Koizumi F, Yanagihara K, Sasaki H, Nishio K, Yamada Y: FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer, 2012, 106, 727-32.). Мутация РФРФ3 при раке мочевого пузыря составляет 50-60% (неинвазивный) и 10-15% (инвазивный). Различные подтипы РФРФ, например, РФРФ2, РФРФ3, РФРФ4 и т.д., сверхэкспрессируются и сверхактивируются при раке печени (Cheng AL, Shen YC, Zhu AX: Targeting Fibroblast Growth Factor Receptor Signaling in Hepatocellular Carcinoma. Oncology-Basel, 2011, 81, 372-80.).
RET также является членом семейства РТК, и его нормальные физиологические функции включают в себя развитие почек, развитие нервной системы, поддержание и обновление стволовых клеток спермы, дифференцировку миеломоноцитарных клеток, образование лимфоидных тканей и т.д. RET экспрессируется в клетках ганглия кишечника человека, нейробластоме, феохромоцитоме, медуллярном раке щитовидной железы, С клетках щитовидной железы и меланоцитах и т.д. В последние годы, основываясь на интенсивных исследованиях RET, было выявлено, что сверхактивация RET в опухолях значительно способствует пролиферации, выживанию, инвазии, метастазированию и воспалению различных опухолей (Maria Grazia Borrello, Induction of a proinflammolatory program in normal human thyrocytes by the RET/PTC1 oncogene, PNAS, October 11, 2005). Например, точечная мутация RET составляет до 95% у пациентов с медуллярной карциномой щитовидной железы; перестройка гена RET составляет от 20% до 40% у пациентов с папиллярным раком щитовидной железы; а также RET сверхэкспрессируется при аденокарциноме, раке толстой кишки, раке поджелудочной железы, раке молочной железы и остром лейкозе. (Lois M. Mulligan: RET revisited: Expanding the oncogenic portfolio, Nature Reviews Cancer 14, 173-186 (2014)).
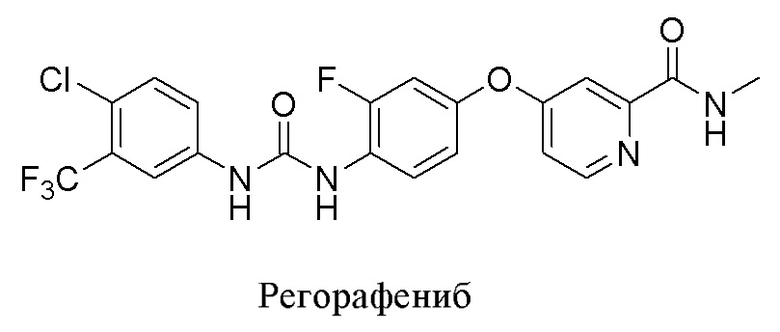
В настоящее время имеющиеся в продаже лекарственные средства в качестве многоцелевого ингибитора, обладающего ингибирующей активностью в отношении РФРФ и RET, например, Регорафениб, в основном нацелены на рецептор 2 фактора роста эндотелия сосудов (VEGFR2, также известный как KDR). В ходе исследований было выявлено, что сильное ингибирование KDR вызывает у больных раком сильные побочные эффекты со стороны сердечно-сосудистой системы, например, тромботическая микроангиопатия, гипертония, застойная сердечная недостаточность, коагулопатия, панкреатит и т.д. Текущие исследования содержат немного сообщений об ингибиторах RET с высокой селективностью. Между тем, неизбежная проблема лекарственной резистентности у других ингибиторов киназы также существует и у ингибиторов RET. Например, были обнаружены классические мутации области привратника - RET V804M и V804L. В настоящее время в ходе доклинических исследований было выявлено, что лишь немногие ингибиторы способны преодолеть резистентность.
Соединения, раскрытые в патенте Boral Sougato, должны содержать сульфон имин (US 2012196902 A1, WO 2013062843 A1, WO 2015089210 A1, WO 2015089220 A1) или тетразолий (US 2016102081 A1) в качестве предпочтительной структуры в мета-положении пиридинового кольца и фокусироваться на VEGFR. Соединения обладают низкой лекарственной способностью, низким воздействием in vivo и не способны достигать противоопухолевого эффекта in vivo.
Патент Kassoum Nacro предлагает серию амино-замещенных азотсодержащих гетероароматических колец (WO2015108490A2), мишенью которых является тирозинкиназа MNK. Однако, что касается кольца A, оно конкретно не раскрывает о-амино-замещенный гетероцикл.
CN 201310224333.8 раскрывает класс алкинилгетероциклических соединений и их применение. Однако о-амино-замещенное гетероарильное кольцо также не раскрывается.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено новое о-аминогетероарил-алкинил содержащее соединение, а также способ его получения и его применение.
Настоящее изобретение реализуется посредством следующих технических решений:
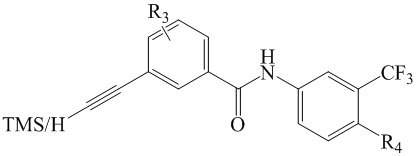
Соединение формулы (I), или дейтерированное соединение, или его фармацевтически приемлемая соль или пролекарство:
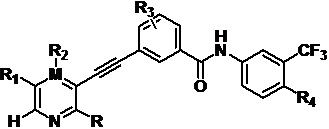 (I)
(I)
в котором:
R представляет собой амино, который заменен по выбору одним или несколькими алкилами или модифицированным алкилом;
М представляет собой С или N, а когда М представляет собой N, R2 отсутствует;
R1 выбран из -H, -N(Q1)(Q2), амино, галогена, гидроксила, циано, арила, гетероарила, алкила или модифицированного алкила;
R2 выбран из -H, -N(Q1)(Q2), амино, галогена, гидроксила, оксо, арила, гетероарила, алкила или модифицированного алкила;
R3 выбран из -H, галогена, циано, алкила или модифицированного алкила;
R4 выбран из -(CH2)nN(R7)(R8), -NHR9, -OR9 или модифицированного алкила;
R7 и R8 вместе с соседниматомом N образуют гетероарильное кольцо;
R9 выбран из -H, арила или гетероарила;
Q1 и Q2 каждый независимо выбран из -H, арила, алкила или модифицированного алкила, и по меньшей мере один из Q1 и Q2 представляет собой арил;
арил, гетероарил, гетероарильное кольцо каждый независимо и опционально замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, оксо, алкила и модифицированного алкила;
алкил представляет собой насыщенную алифатическую линейную или разветвленную алкильную группу, имеющую 1-6 атомов углерода;
модифицированный алкил представляет собой алкил, имеющий 1-6 атомов углерода, в котором любой углерод (первичная, вторичная, третичная или четвертичная углеродная группа) замещен одним или несколькими заместителями, выбранными из -O-, -OH, -(C=O)- , галогена, первичного амино, вторичного амино, третичного амино, циклоалкила, циклоалкилена, гетероциклила и гетероциклилена, а углерод-углеродная одинарная связь алкила по выбору и независимо заменена углерод-углеродной двойной связью или углерод-углеродной тройной связью;
каждый галоген независимо выбран из группы, состоящей из F, Cl, Br и I;
арил представляет собой 5-10-членное моноциклическое или конденсированное бициклическое кольцо;
гетероарил или гетероарильное кольцо представляет собой 5-10-членное ароматическое моноциклическое или конденсированное бициклическое кольцо, имеющее один или несколько гетероатомов, выбранных из N, O и S;
циклоалкил представляет собой насыщенное или ненасыщенное 3-10-членное моноциклическое или полициклическое алициклическое кольцо;
циклоалкилен представляет собой насыщенный или ненасыщенный 3-10-членный моноциклический или полициклический алифатический циклоалкилен;
гетероциклил представляет собой насыщенный или ненасыщенный 3-10-членный моноциклический или полициклический алифатический гетероцикл, содержащий один или несколько гетероатомов, выбранных из N, O и S;
гетероциклилен представляет собой насыщенный или ненасыщенный 3-10-членный моноциклический или полициклический алифатический гетероциклилен, содержащий один или несколько гетероатомов, выбранных из N, O и S;
n = 0-3;
предпочтительно, чтобы фармацевтически приемлемая соль содержала гидрохлорид, метансульфонат, малеат или подобное соединение, а пролекарство содержало сложный эфир, амид, карбоксамид или подобное вещество соединения формулы (I).
В вышеуказанном соединении формулы (I) или дейтерированном соединении или его фармацевтически приемлемой соли или пролекарстве предпочтительно
алкил представляет собой насыщенный алифатический линейный или разветвленный алкил, имеющий 1-6 атомов углерода, предпочтительно имеющий 1-4 атома углерода, более предпочтительно 1-3 атома углерода, и еще более предпочтительно представляет собой метил, этил, пропил, изопропил или трет-бутил;
модифицированный алкил представляет собой алкил, содержащий один или несколько заместителей, выбранных из группы, состоящей из -O-, -COO-, -CONH-, -CH=CH-,  , галогена, гидроксила, карбоксила, первичного амино, вторичного амино, третичного амино, циклоалкила, гетероциклила и гетероциклилена;
, галогена, гидроксила, карбоксила, первичного амино, вторичного амино, третичного амино, циклоалкила, гетероциклила и гетероциклилена;
арил представляет собой 6-10-членное и предпочтительно 6-8-членное моноциклическое или конденсированное бициклическое кольцо;
гетероарил или гетероарильное кольцо представляет собой 6-10-членное и предпочтительно 6-8-членное моноциклическое или сочлененное бициклическое кольцо, содержащее 1-3 гетероатома, выбранных из N, О и S;
циклоалкил представляет собой насыщенное или ненасыщенное 3-6-членное моноциклическое или полициклическое кольцо;
циклоалкилен представляет собой насыщенное или ненасыщенное 3-6-членное моноциклическое или полициклическое кольцо;
гетероциклил представляет собой 4-7-членный и предпочтительно 4-6-членный моноциклический или полициклический гетероцикл, содержащий 1-3 гетероатома, выбранных из N, O и S;
гетероциклилен представляет собой 4-7-членный и предпочтительно 4-6-членный моноциклический или полициклический гетероцикл, содержащий 1-3 гетероатома, выбранных из N, O и S;
n от 0 до 1.
В вышеуказанном соединении формулы (I) или дейтерированном соединении или его фармацевтически приемлемой соли или пролекарстве предпочтительно следующее:
R является амино;
М представляет собой С или N, а когда М представляет собой N, R2 отсутствует;
R1 выбран из -H, -N(Q1)(Q2), -N(Q1')(Q2'),галогена, гидроксила, циано, C1-C6 алкила (опционально замещенный 1-5 галогенами), амино C1-C6 алкила, метиламино C1-C6 алкила, диметиламино C1-C6 алкила, C1-C6 алкоксила, гидроксил C1-C6 алкила, C3-C6 циклоалкила, карбоксила, -C(=O)O(C1-C6 алкил), -C(=O)NH(C1-C6 алкил), C6-C10 арила, 5-8-членного гетероарила или 4-7-членного гетероциклила;
R2 выбран из -H, -N(Q1)(Q2), -N(Q1')(Q2'), галогена, гидроксила, оксо, C1-C6 алкила (опционально замещенный 1-5 галогенами), амино C1-C6 алкила, метиламино C1-C6 алкила, диметиламино C1-C6 алкила, C1-C6 алкокси, гидроксила C1-C6 алкила, C3-C6 циклоалкила, C6-C10 арила, 5-8-членного гетероарила или 4-7-членного гетероциклила;
Q1 и Q2 каждый независимо выбран из -H, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 алканоила, C1-C6 еноила или фенила и по меньшей мере один из Q1 или Q2 представляет собой фенил, где фенил опционально замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-C6 алкила, C1-C6галогеналкила и C1-C6 алкоксила;
Q1' и Q2' каждый независимо выбран из группы, состоящей из -H, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 алканоила и C1-C6 еноила;
R3 выбран из -H, галогена, циано, опционально галогенированного C1-C6 алкила, C1-C6 алкокси или C3-C6 циклоалкила;
R4 выбран из -(CH2)nN(R7')(R8'), -NHR9' или -OR9';
где n равно 0 или 1;
R7' и R8' каждый независимо выбран из -Н, опционально галогенированного C1-C6 алкила, C3-C6 циклоалкила; или R7' и R8' вместе с соседним атомом N образуют 5-10-членное гетероарильное кольцо или 4-10-членный гетероцикл;
R9' выбран из C6-C10 арила, 5-10-членного гетероарила или 4-7-членного гетероциклила;
C6-C10 арил, 5-10-членный гетероарил, 4-7-членный гетероциклил, 5-10-членное гетероарильное кольцо и 4-10-членный гетероцикл по выбору и независимо замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, оксо, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкокси;
каждый из 5-10-членного гетероарила, 4-7-членного гетероциклила, 5-10-членного гетероарильного кольца и 4-10-членного гетероцикла независимо содержит 1-3 гетероатома, выбранных из N, O и S;
C6-C10 арил предпочтительно по выбору замещен 1-5 заместителями, выбранными из группы, состоящей из галогена, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкоксила.
В вышеуказанном соединении формулы (I) или дейтерированном соединении или его фармацевтически приемлемой соли или пролекарстве предпочтительно следующее:
М представляет собой С или N, а когда М представляет собой N, R2 отсутствует;
R1 выбран из -H, -N(Q1)(Q2), -N(Q1')(Q2'), C1-C4 алкила (опционально замещен 1-3 галогенами), амино C1-C4 алкила, метиламино C1-C4 алкила, диметиламино C1-C4 алкила, C1-C4 алкокси, гидрокси C1-C4 алкила, C3-C6 циклоалкила, карбоксила, -C(=O)O(C1- C4 алкил), -C(=O)NH(C1-C4 алкил), C6-C10 арила, 5-6-членного гетероарила или 4-6-членного гетероциклила;
R2 выбран из -H, галогена, гидрокси, оксо, C1-C4 алкила (опционально замещен 1-3 галогенами), амино C1-C4 алкила, метиламино C1-C4 алкила, диметиламино C1-C4 алкила, C1-C4 алкокси, гидрокси C1-C4 алкила, C3-C6 циклоалкила, C6-C10 арила, 5-6-членного гетероарила или 4-6-членного гетероциклила;
Q1 и Q2 каждый независимо выбран из -H, C1-C4 алкила, C3-C6 циклоалкила, C1-C4 алканоила, C1-C4 еноила или фенила и по меньшей мере один из Q1 или Q2 представляет собой фенил, где фенил опционально замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-C4 алкила, C1-C4 галогеналкила и C1-C4 алкокси;
Q1' и Q2' каждый независимо выбран из группы, состоящей из -H, C1-C4 алкила, C3-C6 циклоалкила, C1-C4 алканоила и C1-C4 эноила;
R3 выбран из -H, галогена, циано, по выбору галогенированного C1-C4 алкила, C1-C4 алкокси или C3-C4 циклоалкила;
R4 выбран из -OR9', -CH2N (R7')(R8');
R7' и R8' каждый независимо выбран из -Н, опционально галогенированного C1-C6 алкила, или C3-C6 циклоалкила; или R7' и R8' вместе с соседним атомом N образуют 5-10-членное гетероарильное кольцо или 4-10-членный гетероцикл;
R9 выбран из C6-C10 арила, 5-10-членного гетероарила или 4-7-членного гетероциклила;
C6-C10 арил, 5-6-членный гетероарил и 4-6-членный гетероциклил каждый независимо и опционально замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-C4 алкила, C1-C4 галогеналкила и C1-C4 алкоксила;
5-10-членный гетероарил или гетероарильное кольцо и 4-10-членный гетероцикл каждый независимо и опционально замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, оксо, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкоксила;
каждый из 5-6-членного гетероарила, 4-6-членного гетероциклила, 5-10-членного гетероарила или гетероарильного кольца и 4-10-членного гетероцикла независимо содержит 1-3 гетероатома, выбранных из N, O и S;
предпочтительно C6-C10 арил опционально замещен 1-4 заместителями, выбранными из группы, состоящей из галогена, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкоксила.
В вышеуказанном соединении формулы (I) или его дейтерированном соединении или его фармацевтически приемлемой соли или пролекарстве предпочтительно следующее:
М представляет собой С или N, а когда М представляет собой N, R2 отсутствует;
R представляет собой амино;
R1 выбран из группы, состоящей из -H, галогена, гидроксила, циано, C1-4 алкила (опционально замещен галогеном, гидроксилом, C1-4 алкокси, трифторметоксилом, моно- или ди C1-4 алкиламино), C1-4 алкокси (опционально замещен галогеном, гидроксилом, C1-4 алкоксилом, амино, моно или диC1-4 алкиламино), амино, моно или ди C1-4 алкиламино, C1-4 алкиламидо, C3-6 циклоалкиламидо и C2-4 алкениламидо, опционально замещенный моно или ди C1-4 алкиламино; предпочтительно, R1 выбран из -H, галогена, гидроксила, циано, метила, трифторметила, метокси, трифторметокси, циклопропила, циклопропилокси, эпоксибутилокси, 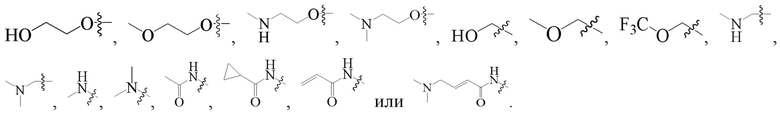
R2 выбран из -H, или галогена;
R3 выбран из группы, состоящей из -H, галогена, циано, опционально галогенированного C1-4 алкила и C1-4 алкокси; предпочтительно R3 представляет собой водород, хлор, фтор, метил, метоксил, циано или трифторметил;
R4 представляет собой C1-4 алкил или оксил, замещенный 5- или 6-членным алифатическим гетероциклилом, имеющим 1-2 атома N в кольце, где 5- или 6-членный алифатический гетероциклил опционально замещен C1-4 алкилом, и предпочтительно R4 представляет собой 4-метилпиперазин-1-илметил или 1-метилпиперидин-4-илоксил.
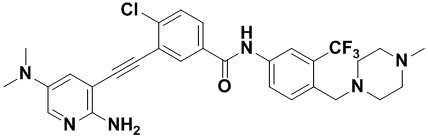
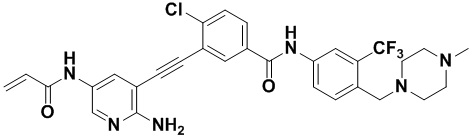
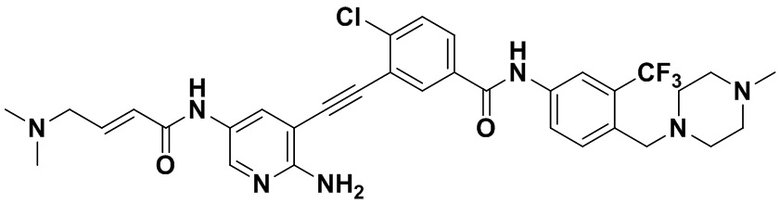
Предпочтительно, чтобы в вышеуказанном соединении формулы (I) или дейтерированном соединении, или его фармацевтически приемлемой соли или пролекарстве, предпочтительное соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль или пролекарство были выбраны из следующих соединений:

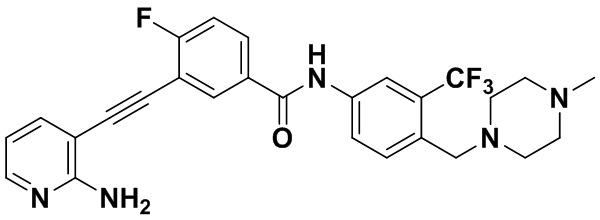
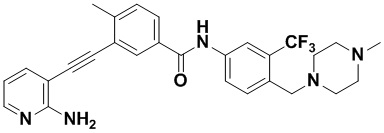
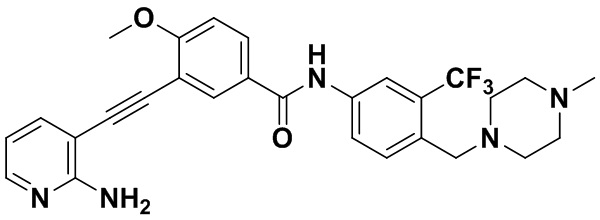
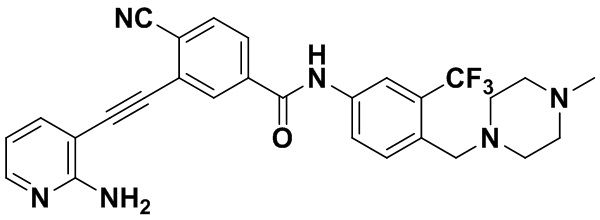
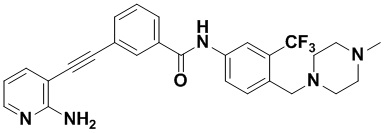
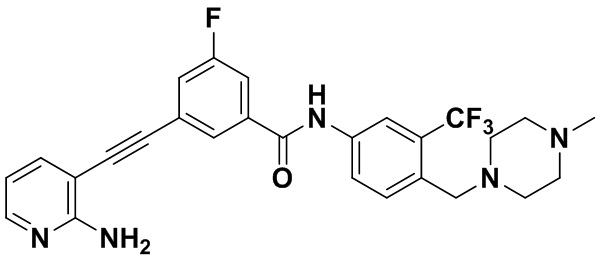

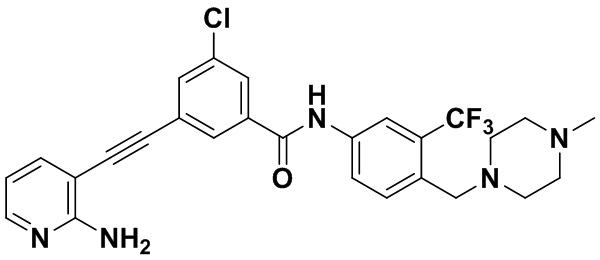
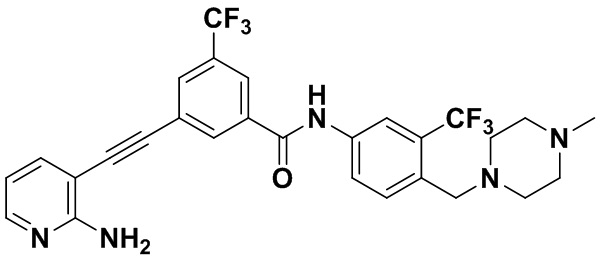

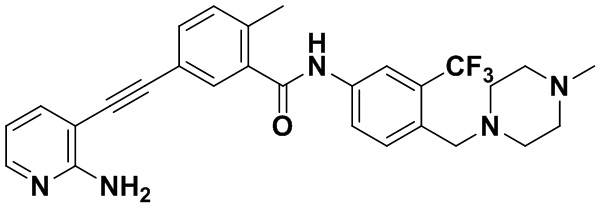
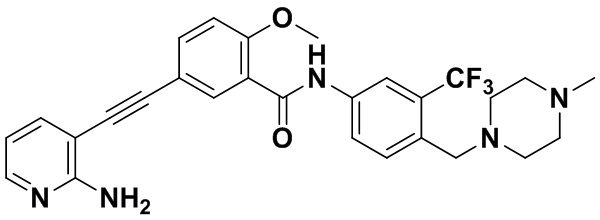
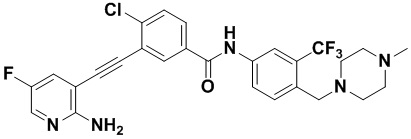
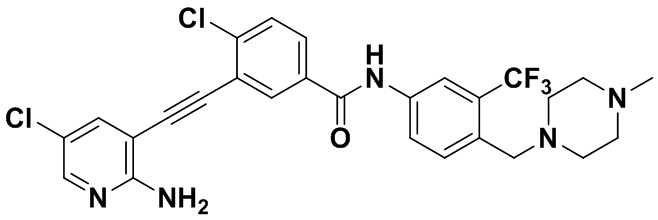
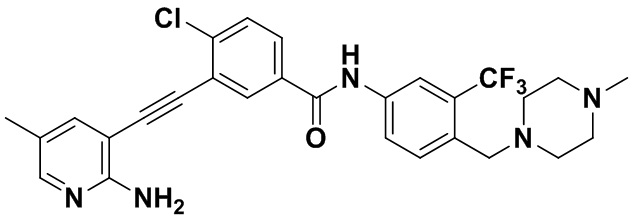
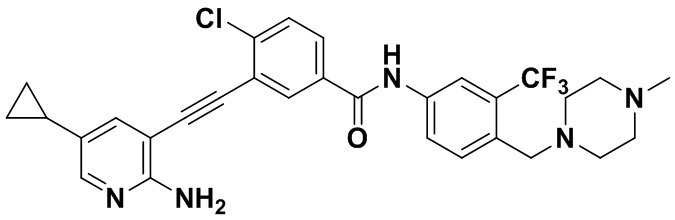
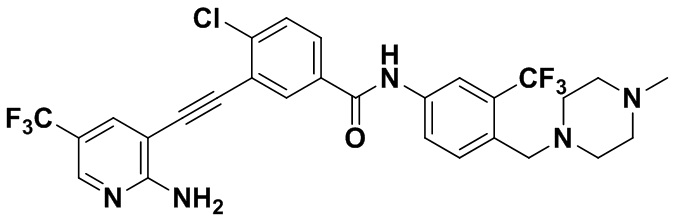
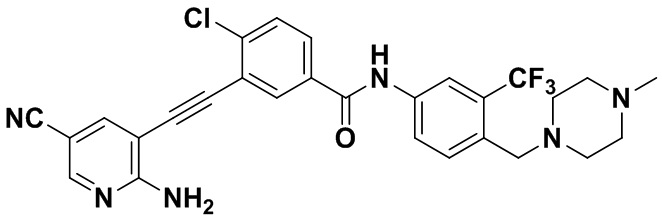 HuFGFR359
HuFGFR359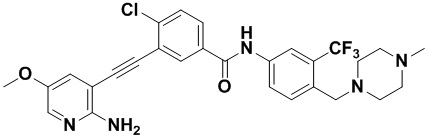 HuFGFR360
HuFGFR360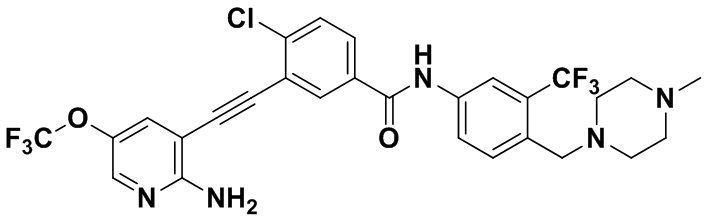


Настоящее изобретение также относится к способу получения соединения формулы (I) или дейтерированного соединения или его фармацевтически приемлемой соли или пролекарства, который содержит этап взаимодействия соединения формулы (1) с соединением формулы (2)
(1) (2)
где R и R1-R4 каждый независимо определен, как указано выше.
Предпочтительно он включает следующее: в присутствии катализатора на основе палладия и меди с переходным металлом и в щелочных условиях сочетание соединения формулы (1) с соединением формулы (2). Палладиевый катализатор предпочтительно содержит Pd(PPh3)2Cl2, Pd(OAc)2, и/или Pd(PPh3)4. Предпочтительно медный катализатор содержит CuI и/или CuCl. Предпочтительно основание, используемое для щелочных условий, содержит одно или несколько оснований, выбранных из CsF, Cs2CO3, K2CO3, триэтиламина, диизопропилэтиламина и ДМАП (диметиламинопиридина). Предпочтительно растворитель для реакции сочетания содержит один или несколько растворителей, выбранных из ацетонитрила, 1,4-диоксана и DMF.
Более предпочтительно, способ содержит этап взаимодействия соединения формулы (1) с соединением формулы (2) в присутствии фторида цезия, Pd(PPh3)2Cl2, CuI и триэтиламина и в ацетонитриле в качестве растворителя.
Более предпочтительно, способ содержит любую из следующих схем I или II:
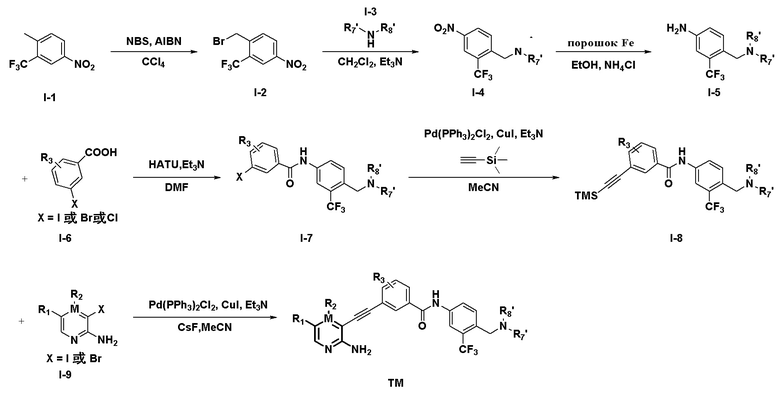
Схема I
Схема I включает следующие этапы:
Этап 1: соединение I-1, NBS, AIBN и CCl4 помещают в круглодонную колбу и проводят реакцию, нагревая на масляной бане при температуре реакции 100°C в течение 24 часов до завершения, и соединение I-2 получено очисткой; где эквивалентное соотношение соединения I-1: NBS: AIBN - 1:1,1:0,2.
Этап 2: соединение I-2, I-3, CH2Cl2 и Et3N добавляют в круглодонную колбу и реакцию проводят при комнатной температуре в течение 12 часов до завершения, и соединение I-4 получают очисткой; где эквивалентное соотношение соединения I-2 и соединения I-3: Et3N - 1:1,1:1,2.
Этап 3: соединение I-4, восстановитель (порошок Fe), EtOH и NH4Cl добавляют в круглодонную колбу, и реакцию проводят при нагревании на масляной бане при температуре реакции 100°C в течение 10 часов до завершения, и соединение I-5 получают очисткой; где эквивалентное соотношение соединения I-4 и восстанавливающего порошка Fe: NH4Cl - 1:4:2.
Этап 4: соединение I-6, HATU, Et3N, DMF помещают в круглодонную колбу, и смесь перемешивают при комнатной температуре в течение 30 минут, затем добавляют соединение I-5 и продолжают перемешивание при комнатной температуре в течение 12 часов; после завершения реакции соединение I-7 получают путем очистки; где эквивалентное соотношение соединения I-6: HATU: Et3N: соединение I-5 составляет 1:2:2:0,9.
Этап 5: соединение I-7, триметилсилилацетилен, Pd(PPh3)2Cl2, CuI и Et3N помещают в круглодонную колбу, при этом MeCN используют в качестве растворителя, смесь нагревают до 80°C на масляной бане в течение 12 часов до завершения реакции; соединение I-8 получают очисткой; где эквивалентное соотношение соединения I-7 и триметилсилилацетилена: Pd(PPh3)2Cl2:CuI:Et3N представляет собой 1:1,5:0,05:0,1:3;
Этап 6: соединение I-8, соединение I-9, фторид цезия, Pd(PPh3)2Cl2, CuI и Et3N добавляют в круглодонную колбу, при этом MeCN используют в качестве растворителя, и смесь нагревают до 80°C на масляной бане в течение 12 часов до завершения реакции, и соединение ТМ получают очисткой; где эквивалентное соотношение соединения I-8: соединения I-9: фторида цезия: Pd(PPh3)2Cl2:CuI:MeCN: Et3N представляет собой 1:1,5:4:0,05:0,1:3.

Схема II
Схема II содержит следующие этапы:
Этап 1: соединение II-2 и NaH добавляют в круглодонную колбу и DMF используют в качестве растворителя; смесь перемешивают на ледяной бане в течение 30 минут, затем добавляют соединение II-1 и реакцию проводят при комнатной температуре в течение 12 часов, соединение II-3 получают путем очистки; где эквивалентное соотношение соединения II-1: соединения II-2: NaH - 1:1,2:1,5.
Этап 2: соединение II -3, порошок Fe, AcOH и этанол добавляют в круглодонную колбу, смесь подвергают взаимодействию при 80°C в течение 12 часов до завершения, и соединение II-4 получают очисткой; где эквивалентное соотношение соединения II -3: порошок Fe: AcOH составляет 1:1,1:1,2.
Этап 3: соединение II-5, HATU, Et3N и DMF помещают в круглодонную колбу. После перемешивания при комнатной температуре в течение 30 минут добавляют соединение II-4 и смесь перемешивают при комнатной температуре в течение 12 часов до завершения реакции, соединение II-6 получают путем очистки; где эквивалентное соотношение соединения II-6: HATU: Et3N: соединение II-5 представляет собой 1:2:2:0,9.
Этап 5: соединение II-6, триметилсилилацетилен, Pd(PPh3)2Cl2, CuI и Et3N помещают в круглодонную колбу, при этом MeCN используют в качестве растворителя, смесь нагревают до 80°C на масляной бане в течение 12 часов до завершения реакции; соединение II-7 получают очисткой; где эквивалентное соотношение соединения ΙI-6 и триметилсилилацетилена: Pd(PPh3)2Cl2: CuI:Et3N - 1:1,5:0,05:0,1:3.
Этап 6: соединение ΙI-7, соединение ΙI-8, фторид цезия, Pd(PPh3)2Cl2, CuI и Et3N помещают в круглодонную колбу, при этом MeCN используют в качестве растворителя, и смесь нагревают до 80°C на масляной бане в течение 12 часов до завершения реакции, и соединение ТМ получают очисткой; где эквивалентное соотношение соединения II-7: соединения II-8: фторида цезия: Pd (PPh3) 2Cl2: CuI: MeCN: Et3N представляет собой 1:1,5:4:0,05:0,1:3.
Настоящее изобретение также относится к фармацевтической композиции, содержащей одно или несколько из указанных выше соединений формулы (I) или дейтерированное соединение, или его фармацевтически приемлемую соль или пролекарство, и фармацевтически приемлемый наполнитель.
Настоящее изобретение также предусматривает применение вышеуказанного соединения формулы (I) или дейтерированного соединения, или его фармацевтически приемлемой соли или пролекарства, или вышеуказанной фармацевтической композиции при приготовлении ингибитора киназы РФРФ, ингибитора киназы RET и/или ингибитора мутантной формы РФРФ или RET киназ.
Настоящее изобретение также относится к применению вышеуказанного соединения формулы (I) или дейтерированного соединения, или его фармацевтически приемлемой соли или пролекарства, или вышеуказанной фармацевтической композиции для приготовления лекарственного средства для лечения опухоли; опционально, опухоль подразумевает немелкоклеточный рак легких, рак молочной железы, рак щитовидной железы (медуллярный рак щитовидной железы, папиллярный рак щитовидной железы), рак желудка, рак мочевого пузыря, рак эндометрия, рак простаты, рак шейки матки, рак толстой кишки, рак пищевода, кератиному, миелому, рабдомиосаркому, острый лейкоз, рак печени, аденокарциному и рак поджелудочной железы.
Настоящее изобретение также относится к применению вышеуказанного соединения формулы (I) или его дейтерированного соединения, или его фармацевтически приемлемой соли или пролекарства, или вышеуказанной фармацевтической композиции для лечения опухоли. Опционально, опухоль подразумевает немелкоклеточный рак легких, рак молочной железы, рак щитовидной железы (медуллярный рак щитовидной железы, папиллярный рак щитовидной железы), рак желудка, рак мочевого пузыря, рак эндометрия, рак предстательной железы, рак шейки матки, рак толстой кишки, рак пищевода, кератиному, миелому, рабдомиосаркому, острый лейкоз, рак печени, аденокарциному и рак поджелудочной железы.
В соответствии с вариантом осуществления настоящего изобретения о-аминогетероарилалкинил содержащее соединение обладает преимуществом высокой ингибирующей активности двойной направленности в отношении РФРФ и RET.
Согласно другому варианту осуществления настоящего изобретения о-аминогетероарилалкинил содержащее соединение обладает преимуществом низкой активности KDR.
Согласно другому варианту осуществления настоящего изобретения о-аминогетероарилалкинил содержащее соединение проявляет значительное ингибирование активности пролиферации клеток при раке легкого человека NCI-H1581, линии клеток рака желудка SNU16 и RET-зависимой чувствительной линии клеток BaF3-CCDC6-Ret, а также их мутантных форм.
В соответствии с другим вариантом осуществления настоящего изобретения фармакокинетические данные указывают на то, что о-аминогетероарилалкинил содержащее соединение обладает хорошей лекарственной способностью и проявляет значительную ингибирующую активность в отношении роста опухоли в долгосрочной фармакодинамической модели на животных.
Согласно другому варианту осуществления настоящего изобретения, животное находится в хорошем состоянии (включая в себя отсутствие значительного снижения массы тела) при эффективной дозе, и значительной токсичности других многоцелевых ингибиторов РТК (отсутствие гибели и линьки животных) не наблюдается.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой вестерн-блот-диаграмму Примера 2 фармакологического эксперимента, на которой показано, что соединения HuFGFR267, HuFGFR293 и положительный контроль Понатиниб ингибируют фосфорилирование киназы RET и нижестоящие сигнальные пути в опухолевых клетках, где P-RET представляет собой фосфорилированную киназу RET, P-АКТ является фосфорилированной киназой AKT, P-ErK является фосфорилированной киназой ErK, GAPDH представляет собой глицеральдегид-3-фосфатдегидрогеназу, а ACTIN является актином.
Фиг. 2 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, на которой показано ингибирующее действие соединений HuFGFR267 и AZD4547 на рост ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей, где при t-критерии Стьюдента *** p <0,001 при сравнении с контрольной группой растворителя.
Фиг. 3 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, на которой показаны эффекты соединений HuFGFR267 и AZD4547 на массу тела мышей с опухолью рака легких человека NCI-H1581.
Фиг. 4 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, на которой показано ингибирующее действие соединений HuFGFR267 и AZD4547 на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей, где при критерии Стьюдента, *** p <0,001 при сравнении с контрольной группой растворителя.
Фиг. 5 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, на которой показаны эффекты соединений HuFGFR267 и AZD4547 на массу тела мышей с опухолью рака желудка человека SNU-16.
Фиг. 6 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, которая показывает ингибирующее действие сравнительных соединений HuFGFR1-117 и HuFGFR1-113 на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей.
Фиг. 7 представляет собой линейную диаграмму Примера 3 фармакологического эксперимента, на которой показано влияние сравнительных соединений HuFGFR1-117 и HuFGFR1-113 на массу тела мышей с опухолью рака желудка человека SNU-16.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Конкретные варианты осуществления настоящего изобретения будут подробно представлены ниже. Следует понимать, что конкретные варианты осуществления, представленные в настоящем документе, являются иллюстрацией изобретения и не предназначены для ограничения изобретения.
ПРИМЕР ПРИГОТОВЛЕНИЯ
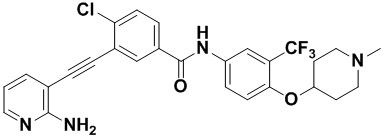
Пример 1. Приготовление HuFGFR267
Этап первый:
2-метил-5-нитробензотрифторид (1 г, 5 ммоль), NBS (980 мг, 5,5 ммоль), AIBN (164 мг, 1 ммоль) и CCl4 (20 мл) поместили в круглодонную колбу и проводили реакцию нагреванием на масляной бане при 100°C в течение 36 часов до ее завершения. Смесь охлаждали до комнатной температуры, и растворитель удаляли при пониженном давлении. После колоночной хроматографии получали продукт 2-трифторметил-4-нитробензилбромид (1,04 г, выход: 70%).
Этап второй:
2-трифторметил-4-нитробензилбромид (849 мг, 3 ммоль), N-метилпиперазин (330 мг, 3,3 ммоль), Et3N (364 мг, 3,6 ммоль) и CH2Cl2 (10 мл) поместили в круглодонную колбу, и реакцию проводили при комнатной температуре в течение 12 часов до ее завершения. Растворитель удаляли в вакууме. После колоночной хроматографии получили продукт 1-метил-4- (4-нитро-2-(трифторметил) бензил) пиперазин (901 мг, выход: 97%).
Этап третий:
1-метил-4- (4-нитро-2-(трифторметил) бензил) пиперазин (901 мг, около 3 ммоль), восстановительный порошок Fe (672 мг, 12 ммоль), NH4Cl (318 мг, 6 ммоль), EtOH (15 мл) поместили в круглодонную колбу; и реакцию проводили при 80°С на масляной бане в течение 10 часов до ее завершения. После фильтрации через слой целита фильтрат концентрировали при пониженном давлении. Продукт 4-((4-метил пиперазин-1-ил) метилен)-3-(трифторметил) анилин (754 мг, выход: 91%) был получен посредством колоночной хроматографии.
Этап четвертый:
3-йод-4-хлорбензойная кислота (1 г, 3,55 ммоль), Et3N (574 мг, 7,1 ммоль), HATU (2,7 г, 7,1 ммоль) DMF (50 мл) поместили в круглодонную колбу. После перемешивания при комнатной температуре в течение 0,5 часа поместили 4-((4-метилпиперазин-1-ил)метилен)-3- (трифторметил) анилин (776 мг, 2,84 ммоль). Реакцию проводили при комнатной температуре в течение 6 часов до завершения, и растворитель удаляли при пониженном давлении. 3-йод-4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил) бензамид (1,5 г, выход: 98%) был получен посредством колоночной хроматографии.
Этап пятый:
3-йод-4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил) бензамид (537 мг, 1 ммоль), триметилсилилацетилен (147 мг, 1,5 ммоль), Pd (PPh3)2Cl2 (60 мг, 0,05 ммоль), CuI (20 мг, 0,1 ммоль), Et3N (404 мг, 4 ммоль)) и MeCN (40 мл)поместили в круглодонную колбу, и смесь нагревали до 70°С на масляной бане, и проводили реакцию в течение ночи до завершения реакции. После колоночной хроматографии был получен продукт4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил)-3-((триметилсилил) этинил) бензамид (466 мг, выход: 92%).
Этап шестой:
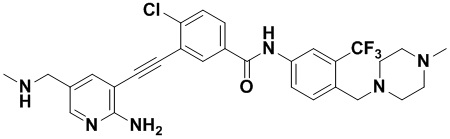
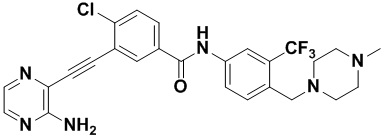
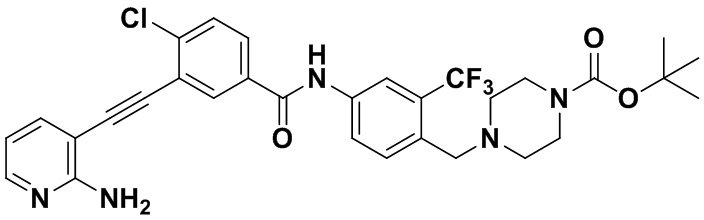
4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил)-3-((триметилсилил) этинил) бензамид (320 мг, 0,63 ммоль), 2-амино-3-йодпиридин (165 мг, 0,75 ммоль), Pd (PPh3)2Cl2 (22 мг, 0,032 ммоль), CuI (13 мг, 0,063 ммоль), CsF (383 мг, 2,52 ммоль), Et3N (254,5 мг, 2,52 ммоль) и MeCN (40 мл) поместили в круглодонную колбу, и смесь нагревали до 70°С на масляной бане, реакцию проводили в течение ночи до завершения реакции. После колоночной хроматографии был получен продукт 3-(2-амипиридин-3-этинил)-4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил) бензамид (HuFGFR267) (305 мг, выход: 92%).
1H ЯМР (400 МГц, CD3OD) δ 8,24 (d, J = 2,2 Гц, 1H), 8,13 (d, J=2,2 Гц, 1H), 8,00 (s, 1H), 7,93 (d, J=2,2 Гц, 1H), 7,91 (d, J=2,2 Гц, 1H), 7,76 (d, J=8,5 Гц, 1H), 7,70 (dd, J=7,5, 1,6 Гц, 1H), 7,65 (d, J=8,5 Гц, 1H)), 6,70 (dd, J=7,4, 5,1 Гц, 1H), 3,64 (d, J=18,7 Гц, 2H), 2,55 (s, 8H), 2,33 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 528 (M+1).
Пример 2. Приготовление HuFGFR302
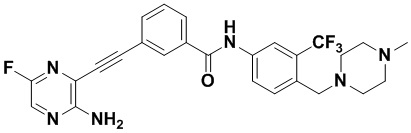
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-фторбензойная кислота.
1H ЯМР (400 МГц, CD3OD) δ 8,26 (dd, J=6,7, 2,3 Гц, 1H), 8,14 (d, J=2,2 Гц, 1H), 8,08-7,92 (m, 3H), 7,79 (d, J=8,5 Гц, 1H), 7,71 (dd, J=7,5, 1,8 Гц, 1H), 7,38 (t, J=8,9 Гц, 1H), 6,71 (dd, J=7,5, 5,1 Гц, 1H), 3,69 (s, 2H), 2,59 (s, 8H), 2,40 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 512 [M+1]+.
Пример 3. Приготовление HuFGFR301
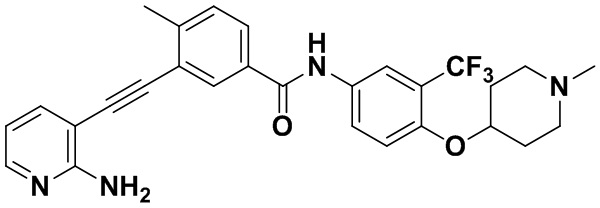
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-метилбензойная кислота.
1H ЯМР (400 МГц, CD3OD) δ 8,20-8,11 (m, 2H), 8,00-7,92 (m, 2H), 7,86 (dd, J=7,9, 2,0 Гц, 1H), 7,75 (d, J=8,5 Гц, 1H), 7,68 (dd, J=7,5, 1,8 Гц, 1H), 7,42 (d, J=8,1 Гц, 1H), 6,68 (dd, J=7,5, 5,1 Гц, 1H), 3,70 (s, 2H), 2,94 (s, 4H), 2,64 (d, J=13,5 Гц, 7H), 2,57 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 508 (M+1).
Пример 4. Приготовление HuFGFR321
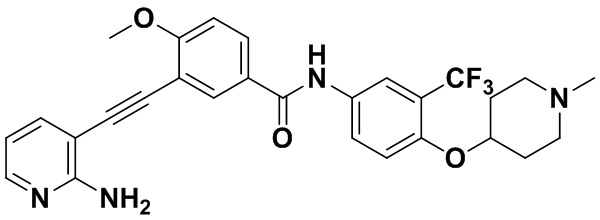
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-метоксибензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,07 (s, 1H), 8,39 (d, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,94 м, 2H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=15,0, 3,0 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 7,08 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,92 (s, 3H), 3,54 (s, 2H), 2,54-2,42 (м, 4H), 2,34 (td, J=10,1, 1,7 Гц, 6H), 2,18 (d, J=30,2 Гц, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 524 (M+1).
Пример 5. Приготовление HuFGFR322
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-цианобензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,07 (s, 1H), 8,57 (d, J=3,0 Гц, 1H), 8,07 (dt, J=5,9, 3,0 Гц, 2H), 7,97 (dd, J=15,0, 3,0 Гц, 1H), 7,80 (d, J=15,0 Гц, 1H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=15,0, 3,0 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,56-2,44 (m, 4H), 2,42 (s, 2H), 2,40-2,30 (m, 4H), 2,18 (d, J=30,2 Гц, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 519 (M+1).
Пример 6. Приготовление HuFGFR293
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йодбензойная кислота.
1H ЯМР (400 МГц, CD3OD) δ 8,17 (t, J=1,5 Гц, 1H), 8,15 (d, J=2,2 Гц, 1H), 7,99-7,92 (m, 3H), 7,79-7,73 (m, 2H) 7,66 (dd, J=7,5, 1,8 Гц, 1H), 7,53 (t, J=7,8 Гц, 1H), 6,66 (dd, J=7,5, 5,1 Гц, 1H), 3,67 (s, 2H), 2,64 (d, J=45,5 Гц, 8H), 2,43 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 494 (M+1).
Пример 7. Приготовление HuFGFR315
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-фторбензойная кислота.
1H ЯМР (400 МГц, CD3OD) δ 8,14 (d, J=2,1 Гц, 1H), 8,00 (t, J=1,4 Гц, 1H), 8,00-7,92 (m, 2H), 7,75 (d, J=8,5 Гц , 1H), 7,73-7,65 (m, 2H), 7,55 (ddd, J=8,9, 2,5, 1,3 Гц, 1H), 6,66 (dd, J=7,5, 5,1 Гц, 1H), 3,68 (s, 2H), 2,67 (d, J=60,6 Гц, 8H), 2,48 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 512 [M+1]+.
Пример 8. Приготовление HuFGFR314
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 2-фтор-5-йодбензойная кислота.
1H ЯМР (400 МГц, ДМСО) δ 10,83 (s, 1H), 8,17 (d, J=1,9 Гц, 1H), 8,05 (dd, J=6,8, 2,2 Гц, 1H), 8,02-7,92 (m, 2H) 7,86 (ddd, J=8,6, 4,9, 2,2 Гц, 1H), 7,73 (d, J=8,5 Гц, 1H), 7,61 (dd, J=7,5, 1,9 Гц, 1H), 7,45 (dd, J=9,8 , 8,7 Гц, 1Н), 6,58 (dd, J=7,5, 4,9 Гц, 1Н), 6,44 (s, 2Н), 3,61 (s, 2Н), 2,60 (s, 4Н), 2,51-2,39 (m, 4Н) 2,35 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 512 [M+1]+.
Пример 9. Приготовление HuFGFR327
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-хлорбензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,11 (s, 1H), 8,27 (t, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,97 (dd, J=15,0, 3,0 Гц, 1H), 7,84 (dt, J=8,0, 3,0 Гц, 2H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=15,0, 3,0 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,52-2,44 (m, 4H)), 2,42-2,26 (m, 4H), 2,21 (s, 2H), 2,18 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 528 (M+1).
Пример 10. Приготовление HuFGFR329
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-трифторметилбензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (s, 1H), 8,39 (t, J=3,0 Гц, 1H), 8,10 (ddd, J=17,4, 10,2, 3,0 Гц, 3H), 7,97 (dd, J=14,9, 3,0 Гц, 1H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=15,0, 2,9 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,57-2,43 (m, 4H), 2,39-2,29 (m, 4H), 2,10 (s, 5H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 562 (M+1).
Пример 11. Приготовление HuFGFR330
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-цианобензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,06 (s, 1H), 8,67 (t, J=3,0 Гц, 1H), 8,55 (t, J=3,0 Гц, 1H), 8,21-8,01 (m, 2H), 7,97 (dd, J=14,9, 3,1 Гц, 1H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=14,9, 2,9 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,54-2,43 (m, 4H), 2,38-2,28 (m, 4H), 2,15 (s, 2H), 2,13 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 519 (M+1).
Пример 12. Приготовление HuFGFR331
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-метилбензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (s, 1H), 8,29 (t, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,97 (dd, J=14,9, 3,1 Гц, 1H), 7,81 (dt, J=16,3, 3,0 Гц, 2H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,56 (dd, J=14,9, 2,9 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,54-2,44 (m, 4H), 2,39-2,29 (m, 7H), 2,14 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 508 (M+1).
Пример 13. Приготовление HuFGFR332
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-5-метоксибензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,14 (s, 1H), 8,07 (d, J=4,0 Гц, 2H), 7,97 (s, 1H), 7,88 (s, 1H), 7,70 (s, 1H), 7,56 (s, 1Н), 7,37 (s, 1Н), 6,75 (s, 1Н), 6,54 (s, 1Н), 3,79 (s, 3Н), 3,54 (s, 2Н), 2,48 (s, 4Н), 2,34 (s, 4Н), 2,18 (s, 2Н), 2,10 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 524 (M+1).
Пример 14. Приготовление HuFGFR333
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 2-метил-5-йодбензойная кислота.
1Н ЯМР (400 МГц, CDCl3) δ 8,77 (s, 1Н), 8,34 (s, 1Н), 8,06 (s, 1Н), 7,97 (s, 1Н), 7,80-7,51 (m, 3Н), 7,37 (s, 2H), 6,54 (s, 1H), 3,54 (s, 2H), 2,48 (s, 4H), 2,34 (s, 4H), 2,22 (s, 3H), 2,14 (s, 3H), 2,00 (s, 2H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 508 (M+1).
Пример 15. Приготовление HuFGFR334
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 2-метокси-5-йодбензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,49 (s, 1H), 8,39 (s, 1H), 8,06 (s, 1H), 7,97 (s, 1H), 7,69 (d, J=4,0 Гц, 2H), 7,56 (s, 1Н), 7,37 (s, 1Н), 7,08 (s, 1Н), 6,54 (s, 1Н), 3,93 (s, 3Н), 3,54 (s, 2Н), 2,48 (s, 4Н), 2,34 (s, 4Н), 2,21 (s, 2Н), 2,14 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 524 (M+1).
Пример 16. Приготовление HuFGFR355
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-фторпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CD3OD) δ 8,26 (d, J=2,2 Гц, 1H), 8,15 (d, J=2,1 Гц, 1H), 7,95 (dd, J=18,6, 12,3, 6,0 Гц, 3H), 7,76 (d, J=8,5 Гц, 1H), 7,64 (d, J=8,5 Гц, 1H), 7,52 (dd, J=8,3, 2,8 Гц, 1H), 3,72 (s, 2H), 2,83 (m, 11H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 546 (M+1).
Пример 17. Приготовление HuFGFR356
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-хлорпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,07 (s, 1H), 8,33 (s, 1H), 8,04 (d, J=12,5 Гц, 2H), 7,89 (d, J=12,0 Гц, 2H), 7,55 (d, J=8,0 Гц, 2H), 7,37 (s, 1H), 3,54 (s, 2H), 2,64 (s, 2H), 2,48 (s, 4H), 2,34 (s, 4H), 2,13 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 562 (M+1).
Пример 18. Приготовление HuFGFR357
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-метилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (s, 1H), 8,06 (s, 1H), 7,86 (d, J=20,0 Гц, 2H), 7,57 (d, J=4,0 Гц, 2H), 7,30 (d, J=8,0 Гц, 2H), 6,89 (s, 2H), 3,54 (s, 2H), 2,48 (s, 4H), 2,34 (s, 4H), 2,23 (s, 3H), 2,19 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 542 (M+1).
Пример 19. Приготовление HuFGFR358
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-циклопропилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,78 (d, J=2,9 Гц, 1H), 7,65-7,52 (m, 2H), 7,30 (dd, J=8,9, 7,4 Гц, 2H), 6,89 (s, 2H), 3,54 (s, 2H), 2,50 (ddd, J=24,7, 19,4, 10,9 Гц, 4H), 2,41-2,28 (m, 4H), 2,18 (d, J=30,1 Гц, 3H), 1,86-1,52 (m, 1H), 1,39 - 0,82 (m, 4H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 568 (M+1).
Пример 20. Приготовление HuFGFR307
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-трифторметилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CD3OD) δ 8,26 (d, J=2,1 Гц, 2H), 8,13 (d, J=2,0 Гц, 1H), 7,92 (dd, J=13,3, 4,8 Гц, 2H), 7,85 (d, J=2,2 Гц, 1H), 7,73 (d, J=8,5 Гц, 1H), 7,60 (d, J=8,4 Гц, 1H), 3,68 (s, 2H), 2,85 (s, 4H), 2,59 (d, J=28,5 Гц, 7H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 596 (M+1).
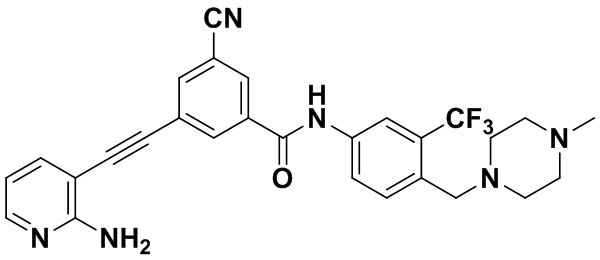
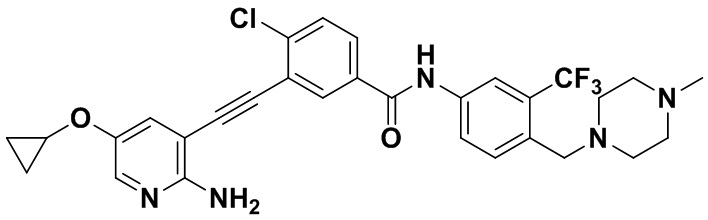
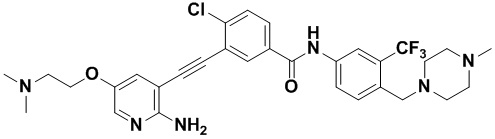
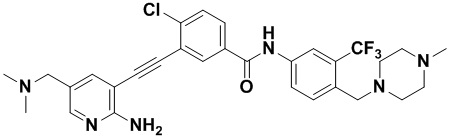
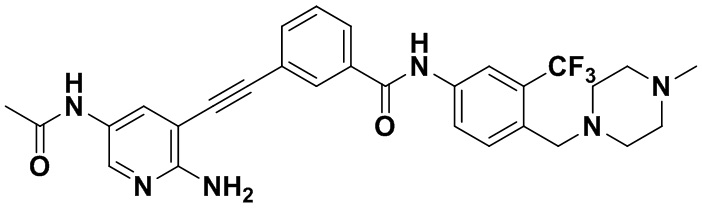
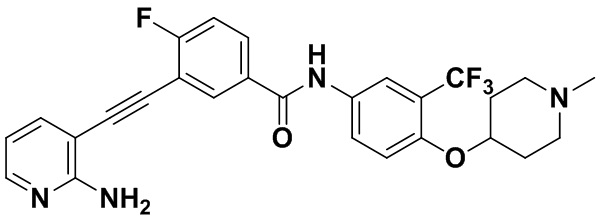
Пример 21. Приготовление HuFGFR359
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-цианопиридин использовался вместо 2-амино-3-йодопиридина.
ЯМР 8,400 (d, J=2,9 Гц, 1H), 7,95-7,83 (m, 2H), 7,64-7,49 (m, 2H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 3,54 (s, 2Н), 2,57-2,44 (m, 4Н), 2,41-2,31 (m, 4Н), 2,11 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 553 (M+1).
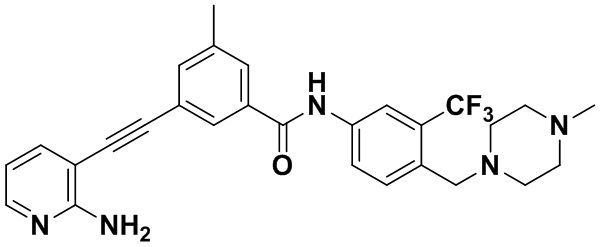
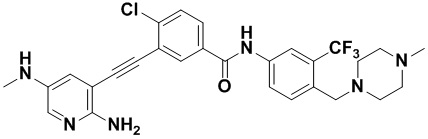
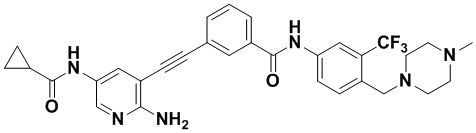
Пример 22. Приготовление HuFGFR360
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-метоксипиридин использовался вместо 2-амино-3-йодопиридина.
ЯМР 8,50 (s, 1H), 8,04 (s, 1H) (d, J=16,0 Гц, 2H), 3,91 (s, 3H), 3,53 (s, 2H), 2,47 (s, 3H), 2,33 (s, 3H), 2,13 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 558 (M+1).
Пример 23. Приготовление HuFGFR361
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-трифторметоксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,64 (d, J=2,9 Гц, 1H), 7,61-7,53 (m, 2H), 7,46 (d, J=3,1 Гц, 1H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 3,54 (s, 2Н), 2,54-2,43 (m, 4Н), 2,42-2,29 (m, 4Н), 2,18 (d, J=30,1 Гц, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 612 (M+1).
Пример 24. Приготовление HuFGFR362
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-циклопропилоксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,30 (d, J=2,9 Гц, 1H), 8,04 (d, J=2,9 Гц, 1H), 7,86 (dd, J=14,9, 2,9 Гц, 1H), 7,62 (d, J=3,1 Гц, 1H), 7,59-7,49 (m, 2H), 7,34 (d, J=2,9 Гц, 1H), 7,29 (d, J=15,0 Гц, 1H), 6,87 (s, 2H), 3,53 (s, 2Н), 3,44-3,21 (m, 1Н), 2,49 (ddd, J=24,6, 18,7, 12,1 Гц, 4Н), 2,39-2,26 (m, 4Н), 2,13 (s, 3Н), 0,71-0,28. (m, 2Н), 0,28-0,20 (m, 2Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 584 (M+1).
Пример 25. Приготовление HuFGFR363
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(3-оксетанил) оксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,58 (ddd, J=14,9, 13,4, 3,0 Гц, 3H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 4,04 (d, J=2,9 Гц, 1H), 3,54 (s, 2H) 2,50 (ddd, J=24,7, 19,4, 10,9 Гц, 4H), 2,38-2,27 (m, 4H), 2,11 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 600 (M+1).
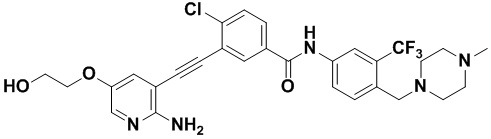
Пример 26. Приготовление HuFGFR377
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(2-гидроксиэтил) оксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,58 (ddd, J=14,9, 13,4, 2,9 Гц, 3H), 7,33 (dd, J=18,3, 9,0 Гц, 2H), 6,89 (s, 2H), 4,90 (s, 1H), 4,33 (td, J=14,5, 0,5 Гц, 2H), 3,68 (dd, J=21,5, 7,4 Гц, 2H), 3,54 (s, 2H), 2,59-2,40 (m, 4H), 2,40-2,28 (m, 4H), 2,10 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 588 (M+1).
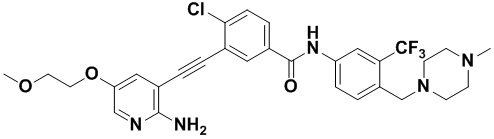
Пример 27. Приготовление HuFGFR378
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(2-метоксиэтил) оксипиридин использовался вместо 2-амино-3-йодопиридина.
ЯМР 8,400 (d, J=2,9 Гц, 1H), 1H), 7,57 (ddd, J=14,9, 13,4, 3,0 Гц, 3H), 7,32 (dd, J=17,7, 8,9 Гц, 2H), 6,88 (s, 2H), 4,30 (td, J=14,5, 0,7 Гц, 2H), 3,76 (td, J=14,6, 0,8 Гц, 2H), 3,53 (s, 2H), 3,40 (s, 3H), 2,59-2,43 (m, 4H), 2,43-2,25 (m, 4H), 2,19 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 602 (M+1).
Пример 28. Приготовление HuFGFR379
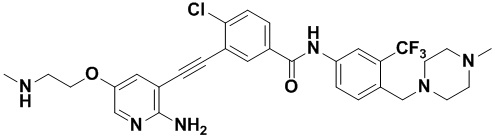
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(2-метиламиноэтил) оксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,58 (ddd, J=14,9, 13,4, 2,9 Гц, 3H), 7,33 (dd, J=14,4, 9,0 Гц, 2H), 6,89 (s, 2H), 4,13 (t, J=14,6 Гц, 2H), 3,54 (s, 2H), 3,26 (s, 3H), 3,01 (t, J=14,6 Гц, 2H), 2,60-2,43 (m, 4H), 2,42-2,26 (m, 4H), 2,14 (s, 3H), 1,84 (s, 1Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 601 (M+1).
Пример 29. Приготовление HuFGFR380
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(2-диметиламиноэтил) оксипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,64 (d, J=2,9 Гц, 1H), 7,62-7,57 (m, 1H), 7,57-7,53 (m, 1H), 7,36 (d, J=2,9 Гц, 1H), 7,31 (d, J=15,0 Гц, 1H) 6,89 (s, 2H), 4,07 (t, J=14,4 Гц, 2H), 3,54 (s, 2H), 2,72 (t, J=14,4 Гц, 2H), 2,52-2,44 (m, 4H), 2,38 - 2,30 (m, 4H), 2,27 (s, 6H), 2,14 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 615 (M+1).
Пример 30. Приготовление HuFGFR384
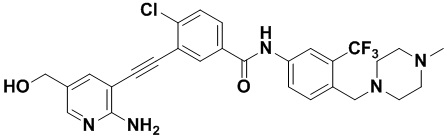
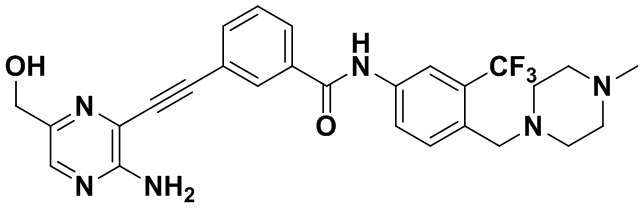
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-гидроксиметилпиридин использовался вместо 2-амино-3-йодопиридина.
1Н ЯМР (500 МГц, CD3OD) δ 8,33 (s, 1Н), 8,06 (s, 1Н), 7,89 (d, J=15,0 Гц, 2Н), 7,58-7,46 (m, 3Н), 7,37 (s, 1Н) 4,61 (s, 2Н), 3,54 (s, 2Н), 2,48 (s, 3Н), 2,34 (s, 3Н), 2,17 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 558 (M+1).
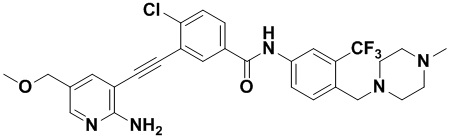
Пример 31. Приготовление HuFGFR385
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-метоксиметилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,91 (d, J=2,9 Гц, 1H), 7,90-7,85 (m, 1H), 7,62-7,53 (m, 2H), 7,50 (d, J=2,9 Гц, 1H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 4,80 (s, 2H) , 3,54 (s, 2H), 3,28 (s, 3H), 2,52-2,45 (m, 4H), 2,39-2,30 (m, 4H), 2,14 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 572 (M+1).
Пример 32. Приготовление HuFGFR386
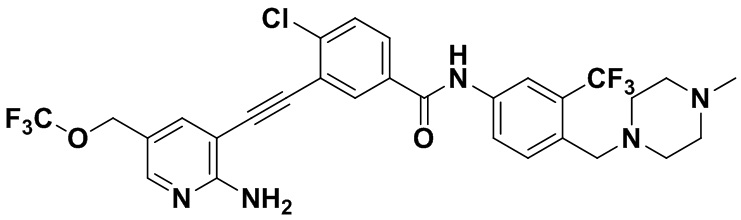
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-трифторметоксиметилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,30 (d, J=2,9 Гц, 1H), 8,03 (d, J=2,9 Гц, 1H), 7,91-7,82 (m, 2H), 7,59-7,45 (m, 3H) 7,28 (d, J=14,9 Гц, 1H), 6,87 (s, 2H), 4,78 (s, 2H), 3,53 (s, 2H), 2,53-2,41 (m, 4H), 2,40-2,25 (m, 4H), 2,17 (d, J=30,1 Гц, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 626 (M+1).
Пример 33. Приготовление HuFGFR387
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-метиламинометилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,96 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,61-7,45 (m, 3H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 3,76 (s, 2H), 3,54 (s, 2H) 3,26 (s, 3H), 2,58-2,44 (m, 4H), 2,44-2,26 (m, 4H), 2,14 (s, 3H), 1,98 (s, 1H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 571 (M+1).
Пример 34. Приготовление HuFGFR388
Синтез осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-диметиламинометилпиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,96 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,62-7,53 (m, 2H), 7,48 (d, J=3,1 Гц, 1H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 3,66 (s, 2Н), 3,54 (s, 2Н), 2,54-2,44 (m, 4Н), 2,38-2,29 (m, 4Н), 2,15 (d, J=8,1 Гц, 9Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 585 (M+1).
Пример 35. Приготовление HuFGFR389
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-метиламинопиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=2,9 Гц, 1H), 8,06 (d, J=3,1 Гц, 1H), 7,88 (dd, J=14,9, 2,9 Гц, 1H), 7,68-7,43 (m, 2Н), 7,31 (d, J=15,0 Гц, 1Н), 7,11 (dd, J=8,9, 3,0 Гц, 2Н), 6,89 (s, 2Н), 5,88 (s, 1Н), 3,54 (s, 2H), 2,68 (s, 3H), 2,58-2,41 (m, 4H), 2,40-2,28 (m, 4H), 2,14 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 557 (M+1).
Пример 36. Приготовление HuFGFR390
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-диметиламинопиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, ДМСО) δ 8,33 (s, 1H), 8,06 (s, 1H), 7,88 (s, 1H), 7,57 (d, J=4,0 Гц, 2H), 7,31 (s, 1H), 7,11 (d, J=11,3 Гц, 2H), 6,89 (s, 2H), 3,54 (s, 2H), 2,92 (s, 6H), 2,48 (s, 4H), 2,34 (s, 4H), 2,24 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 571 (M+1).
Пример 37. Приготовление HuFGFR392
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-ацетиламинопиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,10 (s, 1H), 8,38 (t, J=2,9 Гц, 1H), 8,05 (d, J=2,9 Гц, 1H), 7,98 (s, 1H), 7,92 (dt, J=14,6, 3,2 Гц, 1H), 7,81-7,69 (m, 2H), 7,63 (d, J=14,7 Гц, 1H), 7,59-7,47 (m, 2H), 7,36 (d, J=14,9 Гц, 1H), 3,53 (s, 2H), 2,52-2,43 (m, 4H), 2,40-2,31 (m, 4H), 2,23 (s, 2H), 2,14 (s, 3H), 2,06 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 551 (M+1).
Пример 38. Приготовление HuFGFR396
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(2-циклопропилацетил) аминопиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,10 (s, 1H), 8,39 (t, J=2,9 Гц, 1H), 8,30 (s, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,93 (dt, J=14,6, 3,2 Гц, 1H), 7,80-7,69 (m, 2H), 7,68-7,50 (m, 3H), 7,37 (d, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,55 - 2,42 (m, 4H), 2,41-2,30 (m, 4H), 2,23 (s, 1H), 2,22-2,00 (m, 4H), 1,02-0,40 (m, 4H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 577 (M+1).
Пример 39. Приготовление HuFGFR284
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-акриламидопиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,08 (s, 1H), 9,05 (s, 1H), 8,31 (d, J=3,0 Гц, 1H), 8,04 (d, J=3,0 Гц, 1H), 7,86 (dd, J=15,0, 3,0 Гц, 1H), 7,77 (d, J=3,0 Гц, 1H), 7,60-7,45 (m, 3H), 7,35 (d, J=15,0 Гц, 1H), 6,11 (m, 2H) 5,67 (dd, J=32,6, 5,2 Гц, 1H), 3,53 (s, 2H), 2,52-2,43 (m, 4H), 2,41-2,26 (m, 6H), 2,13 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 597 (M+1).
Пример 40. Приготовление HuFGFR411
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-(4-диметиламино-2-алкенилбутаноил) амипипиридин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,06 (d, J=9,5 Гц, 2H), 8,33 (d, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,88 (dd, J=15,0, 3,0 Гц, 1H), 7,79 (d, J=3,0 Гц, 1H), 7,60-7,49 (m, 3H), 7,37 (d, J=15,0 Гц, 1H), 6,79 (dt, J=30,2, 12,4 Гц, 1H), 5,57 (dt, J=30,2, 1,9 Гц, 1H), 3,54 (s, 2H), 3,02 (dd, J=12,4, 1,9 Гц, 2H), 2,75 (s, 6H), 2,55 - 2,44 (m, 4H), 2,43-2,26 (m, 6H), 2,14 (s, 3H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 654 (M+1).
Пример 41. Приготовление HuFGFR310
Этап первый:
Соединение 1-метил-4-пиперидинол (1,26 г, 11 ммоль) и NaH (240 мг, 12 ммоль) поместили в круглодонную колбу с использованием DMF в качестве растворителя/ Смесь перемешивали на бане с ледяной водой в течение 30 минут, добавили 2-фтор-5-нитротрифтортолуол (2,09 г, 10 ммоль), и реакцию проводили при комнатной температуре в течение 12 часов. Продукт 1-метил-4-(4-нитро-2-(трифторметил) фенилгидрокси) пиперидин (2,9 г, выход: 95%) был получен путем очистки.
Этап второй:
Соединение 1-метил-4-(4-нитро-2-(трифторметил) фенилгидрокси) пиперидин (304 мг, 1 ммоль), порошок Fe (280 мг, 5 ммоль), AcOH (1,2 г, 20 ммоль) и этанол (растворитель) поместили в круглодонную колбу, реакцию проводили при 80°С в течение 12 часов до ее завершения, и продукт 4-((1-метилпиперидинил-4-ил) гидрокси)-3-(трифторметил) анилин (261 мг, выход: 95%) был получен путем очистки.
Этап третий:
3-йод-4-фторбензойную кислоту (1 г, 3,55 ммоль), Et3N (574 мг, 7,1 ммоль) и HATU (2,7 г, 7,1 ммоль) поместили в круглодонную колбу, и последовательно добавили DMF (50 мл). После перемешивания при комнатной температуре в течение 0,5 часа добавили 4-((1-метилпиперидинил-4-ил) гидрокси)-3-(трифторметил) анилин (778 мг, 2,84), и реакцию проводили при комнатной температуре в течение 6 часов до ее завершения. Растворитель выпаривали досуха при пониженном давлении. После колоночной хроматографии был получен 4-хлор-3-йод-N-(4-((1-метилпиперидинил-4-ил) гидрокси)-3-(трифторметил) фенил) бензамид (1,77 г, выход: 93%).
Этап четвертый:
4-хлор-3-йод-N-(4-((1-метилпиперидин-4-ил) гидрокси)-3-(трифторметил) фенил) бензамид (538 мг, 1 ммоль)), триметилсилилацетилен (147 мг, 1,5 ммоль)), Pd (PPh3)2 Cl2 (60 мг, 0,05 ммоль), CuI (20 мг, 0,1 ммоль), Et3N (404 мг, 4 ммоль)) и MeCN (40 мл) поместили в круглодонную колбу, и реакцию проводили в течение ночи при 70°С на масляной бане до ее завершения. После колоночной хроматографии был получен продукт 4-хлор-N-(4-((1-метилпиперидин-4-ил) гидрокси)-3-(трифторметил) фенил-3-((триметилсилил) этинил) бензамид (477 мг, выход: 94%).
Этап пятый:
4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил)-3-((триметилсилил) этинил) бензамид (320 мг, 0,63 ммоль), 2-амино-3-йодпиридин (165 мг, 0,75 ммоль), Pd (PPh3)2⋅Cl2 (22 мг, 0,032 ммоль), CuI (13 мг, 0,063 ммоль), CsF (383 мг, 2,52 ммоль), Et3N (254,5 мг, 2,52 ммоль)) и MeCN (40 мл) поместили в круглодонную колбу, и реакцию проводили в течение ночи при 70°С на масляной бане до ее завершения. После колоночной хроматографии был получен продукт 3-(2-амипиридин-3-этинил)-4-хлор-N-(4-((4-метилпиперазин-1-ил) метилен)-3-(трифторметил) фенил)-3-((триметилсилил) этинил) бензамид (301 мг, выход: 90%).
1H ЯМР (400 МГц, CD3OD) δ 8,24 (d, J=2,2 Гц, 1H), 8,04 (d, J=2,6 Гц, 1H), 8,00 (dd, J=5,1, 1,7 Гц, 1H), 7,91 (dt, J=9,0, 2,0 Гц, 2H), 7,71 (dd, J=7,5, 1,8 Гц, 1H), 7,65 (d, J=8,5 Гц, 1H), 7,26 (d, J=9,1 Гц, 1H), 6,70 (dd, J=7,5, 5,1 Гц, 1H), 4,81 (s, 1H), 3,04 (dd, J=15,6, 6,3 Гц, 4H), 2,69-2,64 (m, 3H), 2,14 (ddd, J=51,1, 16,4, 11,2 Гц, 4H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 529 (M+1).
Пример 42. Приготовление HuFGFR313
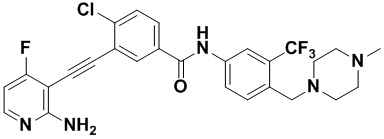
Способ синтеза осуществлялся, как показано в Примере 41, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-фторбензойная кислота.
1H ЯМР (400 МГц, CD3OD) δ 8,24 (dd, J=6,7, 2,4 Гц, 1H), 8,05 (d, J=2,6 Гц, 1H), 8,04-7,95 (m, 2H), 7,91 (dd, J=9,0, 2,7 Гц, 1H), 7,68 (dd, J=7,5, 1,8 Гц, 1H), 7,33 (t, J=8,9 Гц, 1H), 7,28 (d, J=9,1 Гц, 1H), 6,69 (dd, J=7,5, 5,1 Гц, 1Н), 4,89-4,84 (m, 1Н), 3,30-3,16 (m, 4Н), 2,81 (s, 3Н), 2,31-2,06 (m, 4Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 513 (M+1).
Пример 43. Приготовление HuFGFR402
Способ синтеза осуществлялся, как показано в Примере 41, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-метилбензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,03 (s, 1H), 8,34 (d, J=3,0 Гц, 1H), 8,03 (d, J=3,0 Гц, 1H), 7,97 (dd, J=15,0, 3,0 Гц, 1H), 7,82 (dd, J=15,0, 3,0 Гц, 1H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,55 (dd, J=15,0, 3,0 Гц, 1H), 7,37 (d, J=15,0 Гц, 1H), 6,81 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1H), 3,83 (р, J=14,7 Гц, 1H), 2,62-2,32 (m, 7Н), 2,29-2,03 (m, 7Н), 2,00-1,81 (m, 2Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 509 (M+1).
Пример 44. Приготовление HuFGFR403
Синтез осуществлялся, как показано в Примере 41, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йод-4-метоксибензойная кислота.
1H ЯМР (400 МГц, CDCl3) δ 9,00 (s, 1H), 8,39 (d, J=3,0 Гц, 1H), 8,03 (d, J=3,0 Гц, 1H), 7,94 (m, 2H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,55 (dd, J=15,0, 3,0 Гц, 1H), 7,08 (d, J=15,0 Гц, 1H), 6,81 (d, J=15,0 Гц, 1H), 6,54 (t, J=15,0 Гц, 1Н), 3,97-3,70 (m, 4Н), 2,63-2,33 (m, 4Н), 2,30 (s, 2Н), 2,23-2,03 (m, 5Н), 2,02-1,85 (m, 2H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 525 (M+1).
Пример 45. Приготовление HuFGFR312
Способ синтеза осуществлялся, как показано в Примере 41, за исключением того, что вместо 3-йод-4-хлорбензойной кислоты использовалась 3-йодбензойная кислота.
1Н ЯМР (400 МГц, CD3OD) δ 8,18 (t, J=1,5 Гц, 1Н), 8,09 (d, J=2,6 Гц, 1Н), 8,01-7,90 (m, 3Н), 7,82-7,75 (m, 1Н) 7,68 (dd, J=7,5, 1,8 Гц, 1H), 7,55 (t, J=7,8 Гц, 1H), 7,29 (d, J=9,1 Гц, 1H), 6,68 (dd, J=7,5, 5,1 Гц, 1H), 4,91-4,89 (m, 1H), 3,37-3,23 (m, 4H), 2,89 (s, 3H), 2,39-2,08 (m, 4H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 495 (M+1).
Пример 46. Приготовление HuFGFR268
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йодопиразин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CD3OD) δ 8,36 (d, J=2,2 Гц, 1H), 8,17 (d, J=2,1 Гц, 1H), 8,05 (d, J=2,4 Гц, 1H), 8,03-7,98 (m, 2H), 7,87 (s, 1H), 7,79 (d, J=8,5 Гц, 1H), 7,73 (d, J=8,5 Гц, 1H), 3,75 (s, 2H), 3,08 (s, 4H), 2,73 (s, 7Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 529 (M+1).
Пример 47. Приготовление HuFGFR463
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-фторпиразин использовали вместо 2-амино-3-йодпиридина, а 3-йодбензойную кислоту - вместо 3-йод-4-хлорбензойной кислоты.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (s, 1H), 8,39 (t, J=2,9 Гц, 1H), 8,25 (d, J=16,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,93 (dt, J=14,6, 3,2 Гц, 1H), 7,74 (dt, J=15,0, 3,2 Гц, 1H), 7,64 (d, J=14,7 Гц, 1H), 7,61-7,53 (m, 1H) 7,37 (d, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,69-2,43 (m, 4H), 2,41-2,30 (m, 4H), 2,18 (d, J= 30,2 Гц, 3H), 1,62 (s, 2Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 513 (M+1).
Пример 48. Приготовление HuFGFR464
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-5-гидроксиметилпиридин использовали вместо 2-амино-3-йодпиридина, а 3-йодбензойную кислоту - вместо 3-йод-4-хлорбензойной кислоты.
ЯМР 8,40 (s, 1Н), 8,26 (s, 1Н) (s, 1Н), 4,71 (s, 2Н), 3,51 (s, 2Н), 2,81 (s, 1Н), 2,46 (s, 3Н), 2,32 (s, 3Н), 2,12 (s, 3Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 525 (M+1).
Пример 49. Приготовление HuFGFR452
Способ синтеза осуществлялся, как показано в Примере 1, за исключением того, что 2-амино-3-йод-4-фторпиразин использовался вместо 2-амино-3-йодопиридина.
1H ЯМР (400 МГц, CDCl3) δ 9,08 (s, 1H), 8,33 (d, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,92 (ddd, J=17,9, 15,0, 6,4 Гц, 2H), 7,60-7,48 (m, 2H), 7,37 (d, J=15,0 Гц, 1H), 6,51 (dd, J=15,9, 15,1 Гц, 1H), δ-МС (электроспрей) м/з 546 (М+1).
Пример 50. Приготовление HuFGFR459
Синтез проводили, как показано в Примере 1, за исключением того, что вместо N-метилпиперазина использовали 1-трет-бутоксикарбонилпиперазин.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (d, J=3,0 Гц, 1H), 8,07-7,81 (m, 3H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,60-7,48 (m, 2H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 6,53 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 3,19 (t, J=10,4 Гц, 4H), 2,48 (t, J=10,4 Гц, 4H), 1,42 (s, 9H). (электроспрей) м/з 614 (M+1).
Пример 51. Приготовление HuFGFR472
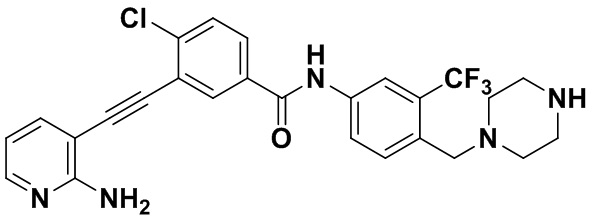
HuFGFR459 (1,0 г, 1,75 ммоль) растворяли в безводном дихлорметане (20 мл) и по каплям в раствор в условиях ледяной бани добавляли трифторуксусную кислоту (10 мл). Реакцию проводили на ледяной бане в течение 30 минут. После очистки был получен продукт HuFGFR472 (0,78 г, выход: 93%).
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=3,0 Гц, 1H), 8,11-7,78 (m, 3H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,62-7,43 (m, 2H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 6,53 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 2,68 (dd, J=15,4, 5,2 Гц, 4H), 2,33 (dd, J=15,4, 5,4 Гц, 4H), 1,75 (s, 1H). (электроспрей) м/з 514 (M+1).
Пример 52. Приготовление HuFGFR473
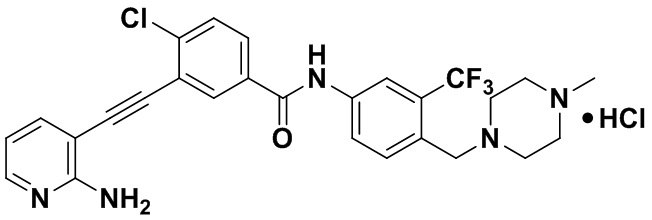
HuFGFR267 (1,0 г, 1,89 ммоль) растворяли в безводном метаноле и по каплям в раствор в условиях ледяной бани добавляли 1 М хлористого водорода в метаноле (1,89 мл). Реакцию проводили в течение 10 мин при комнатной температуре, растворитель выпаривали и получали HuFGFR459 (1,07 г, выход: 100%).
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,97 (dd, J=15,0, 3,0 Гц, 1H), 7,88 (dd, J=15,0, 3,0 Гц, 1H), 7,70 (dd, J=15,0, 3,0 Гц, 1H), 7,61-7,51 (m, 2H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 6,53 (t, J=15,0 Гц, 1H), 3,54 (s, 2H), 3,14-3,03 (m, 4H), 2,89-2,82 (m, 7H). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 528 (M+1).
Пример 53. Приготовление HuFGFR474
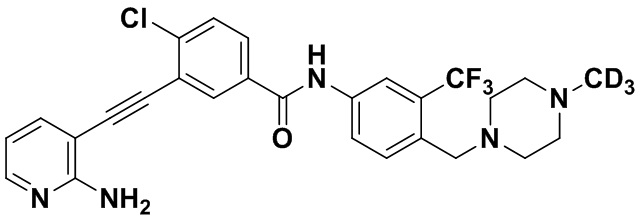
HuFGFR472 (1,0 г, 1,95 ммоль) растворяли в безводном DMF, последовательно добавляли карбонат калия (0,54 г, 3,9 ммоль), и затем добавляли дейтерированный йодметан (0,28 г, 1,95 ммоль) в условиях ледяной бани. Реакцию проводили в течение 1 ч на ледяной бане с получением HuFGFR474 (0,8 г, выход: 79%).
1H ЯМР (400 МГц, ДМСО) δ 8,33 (d, J=3,0 Гц, 1H), 8,06 (d, J=3,0 Гц, 1H), 7,97 (dd, J=15,0, 3,0 Гц, 1H), 7,88 (dd, J=15,0, 3,0 Гц, 1H), 7,79-7,46 (m, 3H), 7,31 (d, J=15,0 Гц, 1H), 6,89 (s, 2H), 6,53 (t, J=15,0 Гц, 1H) 3,54 (s, 2Н), 2,61-2,42 (m, 4Н), 2,40-2,20 (m, 4Н). Масс-спектрометр с низкой разрешающей способностью (электроспрей) м/з 531 (M+1).
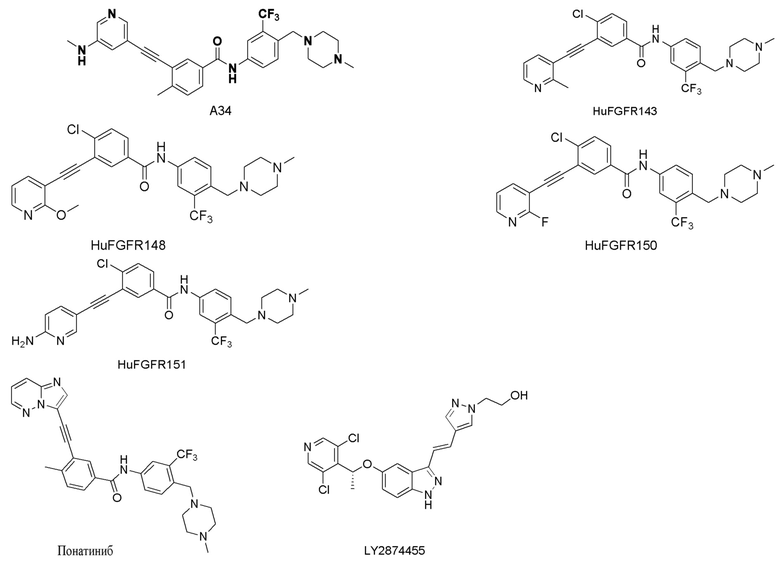
Структуры соединений A34, HuFGFR143, HuFGFR148, HuFGFR150, HuFGFR151, Понатиниб (или AP24534) и LY2874455 являются следующими:
 (2) Пример анализа биологической активности
(2) Пример анализа биологической активности
Пример испытания 1: ингибирование активности рецепторной тирозинкиназы на молекулярном уровне
Субстрат ферментативной реакции Poly (Glu, Tyr)4:1 разбавляли фосфатно-солевым буфером (ФСБ) без калия (10 мМ натрий-фосфатный буфер, 150 мМ NaCl, рН 7,2-7,4) до 20 мкг/мл. Планшет с ферментной меткой покрывали 125 мкл/лунку. Реакцию проводили при 37°С в течение 12-16 часов. После удаления жидкости из лунок планшет трижды промывали Т-ФСБ 200 мкл/лунку (ФСБ, содержащий 0,1% Твин-20), каждую в течение 5 минут. Планшет с ферментом сушили в сушилке при 37°С в течение 1-2 часов.
В каждую лунку поместили 50 мкл раствора АТФ, разбавленного буфером (50 мМ HEPES, pH 7,4, 50 мМ MgCl2, 0,5 мМ MnCl2, 0,2 мМ Na3VO4, 1 мМ DTT) до конечной концентрации 5 мкМ. Соединение разбавляли ДМСО до подходящей концентрации (1 мкл/лунка), или лунка содержала соответствующую концентрацию ДМСО (лунка отрицательного контроля). Затем поместили различные рекомбинантные киназные белки, разбавленные 49 мкл реакционного буфера, чтобы начать реакцию. Для каждого эксперимента требовалась двойная лунка контроля без ферментов. Реакцию проводили в течение 1 часа на шейкере при 37°С (100 об/мин). Планшет трижды промывали с T-ФСБ. Поместили разведение первичного антитела PY99 (100 мкл/лунку), и реакцию проводили в шейкере при 37°C в течение 0,5 часа. Планшет трижды промывали с T-ФСБ. Поместили разбавленное вторичное антитела козы к иммуноглобулину G мыши, коньюгированные с пероксидазой хрена (100 мкл/лунку), и реакцию проводили в шейкере при 37°С в течение 0,5 часа. Планшет трижды промывали с T-ФСБ. Был добавлен раствор ДОФ (дигидрохлорид орто-фенилендиамина) для окрашивания 2 мг/мл (разбавленный 0,1 М лимонно-кислым натрий-цитратным буфером, содержащим 0,03% H2O2 (pH = 5,4)) (100 мкл/лунку), реакция проводилась в течение 1-10 минут при 25°C без доступа света. Реакцию гасили 2М H2SO4 (50 мкл/лунку) и считывали данные при 490 нм с использованием настраиваемого микропланшетного ридера SPECTRA MAX 190.
Коэффициент ингибирования образца определяли по следующей формуле:

Значения IC50 были получены посредством четырехпараметрического регрессионного анализа с использованием программного обеспечения, поставляемого с устройством для считывания с микропластинок.
Данные по активности ферментов соединений, полученных в настоящем изобретении, соединения HuFGFR151, соединения HuFGFR117, положительного контроля Panatinib и положительного контроля LY2874455 в отношении трех ферментов РФРФ1, RET и KDR, приведены в таблице 1:
Таблица 1. Влияние соединений на активность тирозинкиназы
Примечание: Данные представлены в виде среднего отношения ингибирования соединения в двух независимых экспериментах по ингибированию фосфорилирования киназного субстрата.
Результаты эксперимента: Как видно из Таблицы 1, при оценке биологической активности, по настоящему изобретению, о-аминогетероарилалкинилсодержащие соединения обладают высокой активностью ингибирования РФРФ1 и RET-киназы в концентрации 10 мкМ, тогда как соединения, приготовленные в примерах настоящего изобретения обладают низкой активностью KDR. Активность этих соединений в отношении KDR значительно слабее, чем в отношении РФРФ1 или RET, что указывает на то, что эти соединения обладают четкой избирательностью, что полезно для решения технических проблем гепатотоксичности и кардиотоксичности панатиниба. По сравнению с соединениями по настоящему изобретению, соединение HuFGFR151 имеет другое положение аминокислот, что приводит к значительному снижению его ингибирующей активности в отношении киназ РФРФ1 и RET. По сравнению с соединением A34, HuFGFR143, HuFGFR148 и HuFGFR150, введение о-аминов в соединения по настоящему изобретению приводит к значительному увеличению активности в отношении киназ РФРФ1 и RET, а также селективности. Однако активность препаратов позитивного контроля (Panatinib и LY2874455) KDR является высокой.
Данные по ингибирующей активности соединений HuFGFR267 и HuFGFR293 в отношении RET-связанного мутантного фермента приведены в таблице 2, где Ret (V804M) представляет собой имеющийся в продаже рекомбинантный белок. Результаты показали, что HuFGFR267 и HuFGFR293 обладали значительной ингибирующей активностью в отношении Ret и Ret (V804M), особенно в отношении Ret и его мутантной киназы V804M.
Таблица 2. Значение IC50 для соединений против активности тирозинкиназы (нМ)
Примечание: Ингибирующее значение IC50 соединений при фосфорилировании киназного субстрата было независимо измерено дважды и представлено как среднее ± СО (стандартное отклонение).
Фармакологический эксперимент 2: Анализ рецепторноготирозинкиназ-зависимого ингибирования на клеточном уровне
Выявление влияния соединений на активацию сигнального пути RET в клетках TT и BaF3/CCDC6-RET посредством вестерн-блоттинга
Клетки высевали на 12-луночный планшет (250000/лунку). После инкубации в течение 18-24 часов соединения поместили для проведения реакции в течение 2 часов, а затем клетки собирали и сначала промывали один раз холодным ФСБ (содержащим 1 ммоль ванадата натрия); затем 1× гелевым загрузочным буфером SDS (50 ммоль трис-HCl (pH 6,8), 100 ммоль DTT, 2% SDS, 10% глицерин, 1 ммоль ванадат натрия, 0,1% бромфеноловый синий), и поместили к лизирующим клеткам. Клеточный лизат нагревали на кипящей водяной бане в течение 10 минут, а затем центрифугировали при 12 000 об/мин в течение 10 минут при 4°С.
Надосадочную жидкость отбирали для электрофореза в SDS-PAGE (Mini-PROTEAN 3 Cell, Bio-Rad, Hercules, CA, USA). После электрофореза белки переносили на нитроцеллюлозную мембрану с использованием полусухой системы электропереноса (Amersham Life Sciences, Арлингтон Хайтс, Иллинойс, США). Нитроцеллюлозную мембрану помещали в блокирующий раствор (5% сухого обезжиренного молока, разведенного в TBS, содержащем 1 ммоль ванадата натрия) на 2 часа при комнатной температуре, а затем мембрану помещали и реакцию проводили с первичным антителом при 4°С в течение ночи. Мембрану трижды промывали с TBS, содержащим 1 ммоль ванадата натрия, в течение 15 минут каждый раз. Мембрану помещали в раствор вторичного антитела и проводили реакцию в течение 1-2 часов при комнатной температуре. После трехкратной промывки мембраны, как описано выше, ее окрашивали с использованием реагента ECL (Picece, Рокфорд, Иллинойс), который использовали для окрашивания, а затем проявляли.
Результаты, показывающие, что соединение HuFGFR267 (267), HuFGFR293 (293) и позитивный контроль Panatinib ингибируют фосфорилирование RET и нижестоящий сигнальный путь в опухолевых клетках и линиях инструментальных клеток, показаны на рис. 1. Как видно из фиг.1, о-аминогетероарилалкинилсодержащее соединение по настоящему изобретению нацелено и значительно ингибирует активацию сигнального пути RET на клеточном уровне.
Структура AZD4547 выглядит следующим образом:
Фармакологический эксперимент 3: Оценка ингибирующего действия соединений на рост подкожных ксенотрансплантатов рака легких человека NCI-H1581 и рака желудка человека SNU-16 у голых мышей
1. Ингибирующая активность соединения HuFGFR267 в отношении роста подкожных ксенотрансплантатов рака легких человека NCI-H1581 и рака желудка человека SNU-16 у голых мышей
Опухолевая ткань в период интенсивного роста была разрезана на сегменты примерно 1,5 мм3 и инокулирована в асептических условиях подкожно в правую подмышечную область у голых мышей. Диаметр подкожно имплантированной опухоли у голых мышей измеряли посредством штангенциркуля. Когда средний объем составлял около 120 мм3, животные были случайным образом разделены на группы. В группе 50 мг/кг соединения HuFGFR267 соединение перед использованием было доведено 0,5% метилцеллюлозы (МЦ) до необходимой концентрации. Состав соединения готовили один раз в неделю и перорально вводили один раз в день в течение 14 дней. Препарат положительного контроля AZD4547 разбавляли перед использованием до необходимой концентрации водой для инъекций, содержащей 1% Твин 80. Состав AZD4547 готовили один раз в неделю и перорально вводили один раз в день в течение 14 дней. В контрольной группе с растворителем вводили равное количество воды для инъекций. Диаметр трансплантированной опухоли измеряли два раза в неделю в течение всего эксперимента, и взвешивали массу тела мышей. Объем опухоли (ОО) рассчитывали следующим образом: ОО = 1/2 x a x b2, где a и b - длина и ширина, соответственно. Относительный объем опухоли (ООО) рассчитывали на основе измеренных результатов; использовалась следующая формула: ООО = Vt/V0, где V0 представляет собой объем опухоли при разделении и введении мышам (то есть Д0), и Vt представляет собой объем опухоли при каждом измерении. Индекс оценки противоопухолевой активности представяет собой относительную скорость пролиферации опухоли T/C (%), и использовалась следующая формула: T/C (%)=(TOOO / COOO) x 100%, где TOOO - ООО в группе лечения; а COOO - ООО в группе отрицательного контроля.
Результаты ингибирующего действия соединения HuFGFR267 на рост ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей приведены в таблице 3 и на фиг. 2, при этом данные в таблице 3 соответствуют числовым точкам на кривой фиг. 2. Как видно из фиг. 2, в группе 50 мг/кг HuFGFR267 после перорального введения один раз в день в течение 14 дней рост подкожных ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей был значительно ингибирован, и T/C, измеренный на 14-й день, составил 3,77%. В группе положительного контроля 12,5 мг/кг AZD4547 его вводили так же, как описано выше, и рост подкожных ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей был значительно ингибирован, а T/C, полученный на 14-й день составил 24,03%. Во время эксперимента мыши не погибли, и у особей в каждой группе было хорошее состояние здоровья. Это видно из фиг. 3 и таблицы 4 (данные в таблице 4 соответствуют числовым точкам на кривой на фиг. 3), масса тела мышей с опухолью рака легкого человека NCI-H1581 в группе соединения HuFGFR267 не была существенно изменена. Таким образом, это указывало на то, что о-аминогетероарилалкинилсодержащее соединение по настоящему изобретению оказывало значительное ингибирующее действие на рост подкожных ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей, и его преимуществом была низкая токсичность.
Таблица 3. Влияние HuFGFR-267 на объем опухоли ксенотрансплантатов рака легкого человека NCI-H1581 у голых мышей
Примечание: Значение Р в сравнении с контролем растворителя
Таблица 4. Влияние HuFGFR-267 на массу тела мышей с опухолью рака легких человека NCI-H1581
Результаты ингибирующего действия соединения HuFGFR267 на рост ксенотрансплантатов рака желудка человека SNU-16 у голых мышей показаны на фиг.4, где данные таблицы 5 соответствуют числовым точкам на кривой фиг.4. В группах HuFGFR267 50 мг/кг и 25 мг/кг соединения вводили перорально один раз в день в течение 21 дня, и рост ксенотрансплантатов рака желудка человека SNU-16 у голых мышей был в значительной степени ингибирован. Значения T/C, полученные на 21 день, составили 11,66% и 18,55% соответственно. В группе положительного контроля AZD4547 12,5 мг/кг AZD4547 вводили так же, как описано выше, и рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей был значительно ингибирован. T/C, измеренный на 21 день, составил 18,46%. Во время эксперимента мыши не погибли, и у особей в каждой группе было хорошее состояние здоровья. Как видно из фиг. 5 (данные в таблице 6 соответствуют каждой числовой точке на кривой на фиг. 5), масса тела мышей с опухолью рака желудка человека SNU-16 в группе соединения HuFGFR267 не была существенно изменена. Таким образом, это указывало на то, что о-аминогетероарилалкинилсодержащее соединение по настоящему изобретению оказывало значительное ингибирующее действие на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей и его преимуществом была низкая токсичность.
Таблица 5. Влияние HuFGFR-267 на объем опухоли ксенотрансплантатов рака желудка человека SNU-16 у голых мышей
Таблица 6. Влияние HuFGFR-267 на массу тела мышей с опухолью желудка человека SNU-16
2. Ингибирующее действие сравнительных соединений HuFGFR1-117 и HuFGFR1-113 (структура показана ниже) на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей
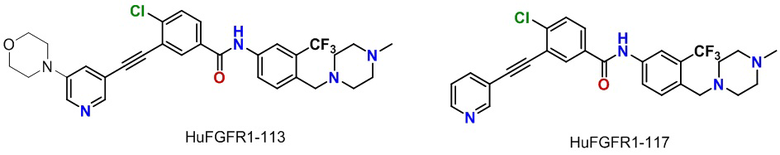
Опухолевая ткань в период интенсивного роста была разрезана на сегменты по 1,5 мм3 и инокулирована в асептических условиях подкожно в правую подмышечную область у голых мышей. Диаметр ксенотрансплантата у голых мышей измеряли посредством штангенциркуля. Животные были случайным образом разделены на группы, когда средний объем опухоли вырос приблизительно до 190 мм3. В группах HuFGFR1-113 и HuFGFR1-117 (100 мг/кг и 20 мг/кг) соединения вводили перорально один раз в день в течение 21 дня подряд. В группе положительного контроля препарата Panatinib 30 мг/кг Panatinib вводили перорально один раз в день в течение 21 дня. В контрольной группе растворителя мышам давали равное количество растворителя. Диаметр ксенотрансплантата измеряли два раза в неделю в течение всего эксперимента, и взвешивали массу тела мышей. Объем опухоли (ОО) рассчитывали следующим образом: ОО = 1/2 x a x b2, где a и b - длина и ширина, соответственно. Относительный объем опухоли (ООО) рассчитывали на основе измеренных результатов; использовалась следующая формула: ООО = Vt/V0, где V0 представляет собой объем опухоли при разделении и введении мышам (то есть Д0), и Vt представляет собой объем опухоли при каждом измерении. Индекс оценки противоопухолевой активности представлял собой относительную скорость пролиферации опухоли T/C (%), и использовалась следующая формула: T/C (%)=(TOOO / COOO) x 100%, где TOOO - ООО в группе лечения; а COOO - ООО в группе отрицательного контроля.
Результаты эксперимента представлены на фиг. 6 и 7 (данные в таблицах 7 и 8 соответствуют числовым значениям на кривых на фиг. 6 и 7 соответственно). В группе 100 мг/кг HuFGFR1-113 была отмечена значительная токсичность, и введение было прекращено из-за того, что состояние здоровья мышей ухудшилась, и на второй день понизилась температура. Одна мышь умерла на третий день. Две мыши умерли на пятый день. Соединение вводили снова, потому что на 20 день мышь выздоровела. В группе 20 мг/кг HuFGFR1-113 соединение вводили перорально один раз в день в течение 21 дня, и рост рака желудка человека SNU-16 у голых мышей был значительно ингибирован. T/C, измеренный на 21 день, составил 23,30%, но одна мышь умерла на 20 день. В группе 100 мг/кг соединения HuFGFR1-117 рост подкожного ксенотрансплантата рака желудка человека SNU-16 у голых мышей был значительно ингибирован. T/C, измеренный на 21-й день, составлял 14,38%, но одна мышь умерла на 9-й и одна на 10-й день соответственно, а у других мышей наблюдалась сухость кожи, линька и плохое состояние, поэтому введение было прекращено. Введение препарата было возобновлено, потому что мыши выздоровели, линька прошла, и на 17 день кожа вернулась в нормальное состояние. В группе 20 мг/кг HuFGFR1-117 препарат вводили перорально один раз в день, и рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей был слабо ингибирован. T/C, полученный на 21 день, составлял 47,88%, и степень ингибирования опухоли была низкой. В группе 30 мг/кг Panatiniba его вводили перорально один раз в день в течение 21 дня, и рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей был значительно ингибирован. T/C, измеренный на 21 день, составлял 36,20%.
Таблица 7. Влияние различных соединений на объем опухоли ксенотрансплантатов рака желудка человека SNU-16 у голых мышей
30 мг/кг
Примечание: Значение Р в сравнении с контролем растворителя
Таблица 8. Влияние различных соединений на массу тела мышей с опухолью желудка человека SNU-16
Экспериментальное заключение: HuFGFR267 по настоящему изобретению значительно ингибировал рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей, и T/C, полученный на 21 день, составлял 11,66% и 18,55% соответственно. Вес мышей с опухолями существенно не изменялся, и состояние здоровья мышей было хорошим. Соединение HuFGFR1-113 (морфолин, замещенный в мета-положении пиридина), которое структурно было сходным с соединением HuFGFR267 по настоящему изобретению, оказывало более слабое ингибирование на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей в дозе 20 мг/кг, чем HuFGFR267, и T/C, полученный на 21 день, составлял 23,30%, в то время как вес мышей с опухолями значительно снизился. Одна мышь умерла на 20 день. Дозировка 100 мг/кг была высокотоксичной. На второй день у мышей наблюдалось плохое состояние и низкая температура. Одна мышь умерла на третий день. Две мыши умерли на пятый день. Другое соединение HuFGFR1-117(без замены амино-веществ в о-положении пиридина), которое было структурно сходным с HuFGFR267 по настоящему изобретению, оказывает более слабое ингибирующее воздействие на рост подкожных ксенотрансплантатов рака желудка человека SNU-16 у голых мышей, чем у HuFGFR267; и T/C, полученные на 21-й день, составили 14,38% и 47,88%, в то время как вес мышей с опухолями значительно снизился. Одна мышь умерла на 9-й и одна - на 10-й день соответственно, а у других мышей появилась сухость кожи, линька, и состояние ухудшилось. Это указывает на то, что введение о-амино пиридина в настоящем изобретении может эффективно увеличивать ингибирующую активность соединений в отношении опухоли и демонстрировать значительное преимущество низкой токсичности.
Пример эксперимента 4: Фармакокинетический эксперимент на крысах
Panatinib (AP24534) вводили внутривенно (в/в) и перорально (п/о) крысам линии Спрег-Доули. В разные моменты времени были взяты образцы крови. Метод ЖХ/МС/МС использовался для определения концентрации соединения в плазме крыс после введения испытуемого соединения, и соответствующие фармакокинетические параметры рассчитывали для изучения пероральной биодоступности и фармакокинетических свойств соединения у крыс. Результаты представлены в таблице 3.
Таблица 3
(нг/мл)
Соединение HuFGFR267 вводили внутривенно и перорально крысам линии Спрег-Доули. В разные моменты времени были взяты образцы крови. Метод ЖХ/МС/МС использовался для определения концентрации соединения в плазме крыс после введения испытуемого соединения, и соответствующие фармакокинетические параметры рассчитывали для изучения пероральной биодоступности и фармакокинетических свойств соединения у крыс. Результаты представлены в таблице 4.
Таблица 4
В таблице 3 и таблице 4 представлены данные, что соединение HuFGFR267 превосходит по воздействию зарегистрированный для продажи препарат AP24534 и, следовательно, имеет больший потенциал для его разработки в качестве лекарства.
Таким образом, о-аминогетероарилалкинилсодержащие соединения в примерах настоящего изобретения обладают преимуществами высокой ингибирующей активности двойной направленности в отношении РФРФ и RET, а также относительно низкой активности KDR. Соединение формулы (I) проявляет сильную ингибирующую активность в отношении линии клеток рака легкого человека NCI-H1581 и линии клеток рака желудка SNU16, а также RET-зависимой чувствительной линии клеток BaF3-CCDC6-Ret и ее мутантных форм. Согласно фармакокинетическим данным, о-аминогетероарилалкинилсодержащее соединение обладает хорошей лекарственной способностью и оказывает значительное ингибирование на рост родственных опухолей в долгосрочной модели на животных, тогда как в эффективной дозировке у животного наблюдается хорошее состояние (включая отсутствие значительного снижения массы тела), а также не наблюдается существенной токсичности (гибель животных или линька).
Предпочтительные варианты осуществления настоящего изобретения были подробно описаны выше, и к ним прилагались чертежи. Однако настоящее изобретение не ограничено конкретными деталями вариантов осуществления, описанных выше. В техническую концепцию настоящего изобретения могут быть внесены различные простые модификации технических решений настоящего изобретения, и такие простые модификации оказываются в границах объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФТОРИРОВАННОЕ СОЕДИНЕНИЕ ЦИКЛОПРОПИЛАМИНА, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ИСПОЛЬЗОВАНИЕ | 2017 |
|
RU2746323C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2823875C1 |
| ПИРИМИДО[5,4-b]ИНДОЛИЗИНОВОЕ ИЛИ ПИРИМИДО[5,4-b]ПИРРОЛИЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2721774C1 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ПРОИЗВОДНОЕ ТРИПТОЛИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2753036C1 |
| КОНДЕНСИРОВАННЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПИРИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПРОИЗВОДСТВА И ПРИМЕНЕНИЕ | 2014 |
|
RU2668074C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, АКТИВИРУЮЩИЕ ФЕРМЕНТЫ | 2013 |
|
RU2652989C2 |
| 5-АРОМАТИЧЕСКОЕ АЛКИНИЛЗАМЕЩЕННОЕ БЕНЗАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2695371C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |


Изобретение относится к соединению формулы (I), его дейтерированному производному или его фармацевтически приемлемой соли, способу его получения и его применению для приготовления ингибитора киназы РФРФ и/или ингибитора киназы RET или для получения лекарственного средства для лечения опухоли, связанной со сверхэкспрессией или сверхактивацией киназы ФРФР или киназы RET. В общей формуле (I) R представляет собой амино; М представляет собой С или N, причем когда М представляет собой N, R2 отсутствует; R1 выбран из группы, состоящей из: -H, галогена, циано, амино, C3-6 циклоалкила; моно- или ди- C1-4 алкиламино; C1-4 алкила, не замещенного или замещенного одним гидроксилом, C1-4 алкокси, трифторметоксилом, моно- или ди- C1-4 алкиламино или 1-5 галогенами; C1-4 алкокси, не замещенного или замещенного одним гидроксилом, C1-4 алкоксилом, моно- или ди- C1-4 алкиламино или 1-5 галогенами; C1-4 алкиламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино; C3-6 циклоалкиламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино; C2-4 алкениламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино; R2 выбран из -H, галогена, С1 алкила; R3 выбран из -H, галогена, циано, C1-4 алкокси, C1-4 алкила, не замещенного или замещенного 1-5 галогенами; R4 выбран из -(CH2)nN(R7)(R8), -OR9; R7 и R8 вместе с соседним атомом N образуют 5-6-членный алифатический гетероцикл с 1-2 атомами N в кольце, не замещенный или замещенный C1-4 алкилом; R9 выбран из 5-6-членного алифатического гетероцикла с 1-2 атомами N в кольце, не замещенного или замещенного C1-4 алкилом; каждый галоген независимо выбран из группы, состоящей из F, Cl, Br и I; n=0-3. 5 н. и 13 з.п. ф-лы, 7 ил., 8 табл., 55 пр.
(I)
1. Соединение формулы (I), или дейтерированное соединение, или его фармацевтически приемлемая соль
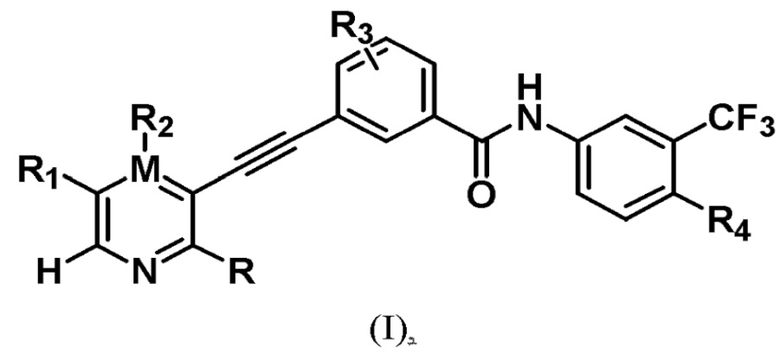
в котором:
R представляет собой амино;
М представляет собой С или N, причем когда М представляет собой N, R2 отсутствует;
R1 выбран из группы, состоящей из:
-H, галогена, циано, амино, C3-6 циклоалкила;
моно- или ди- C1-4 алкиламино;
C1-4 алкила, не замещенного или замещенного одним гидроксилом, C1-4 алкокси, трифторметоксилом, моно- или ди- C1-4 алкиламино или 1-5 галогенами;
C1-4 алкокси, не замещенного или замещенного одним гидроксилом, C1-4 алкоксилом, моно- или ди- C1-4 алкиламино или 1-5 галогенами;
C1-4 алкиламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино;
C3-6 циклоалкиламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино;
C2-4 алкениламидо, не замещенного или замещенного одним моно- или ди- C1-4 алкиламино;
R2 выбран из -H, галогена, С1 алкила;
R3 выбран из -H, галогена, циано, C1-4 алкокси, C1-4 алкила, не замещенного или замещенного 1-5 галогенами;
R4 выбран из -(CH2)nN(R7)(R8), -OR9;
R7 и R8 вместе с соседним атомом N образуют 5-6-членный алифатический гетероцикл с 1-2 атомами N в кольце, не замещенный или замещенный C1-4 алкилом;
R9 выбран из 5-6-членного алифатического гетероцикла с 1-2 атомами N в кольце, не замещенного или замещенного C1-4 алкилом;
каждый галоген независимо выбран из группы, состоящей из F, Cl, Br и I;
n=0-3;
или
соединение HuFGFR362

или дейтерированное соединение HuFGFR362, или фармацевтически приемлемая соль соединения HuFGFR362;
или
соединение HuFGFR363
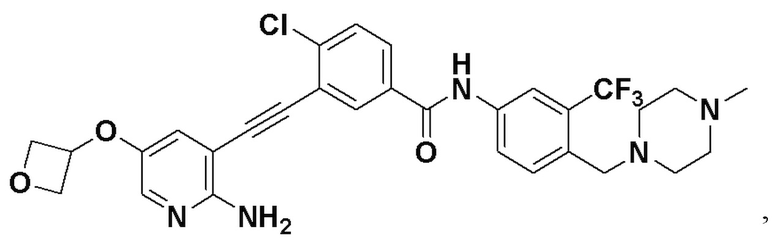
или дейтерированное соединение HuFGFR363, или фармацевтически приемлемая соль соединения HuFGFR363;
или
соединение HuFGFR459
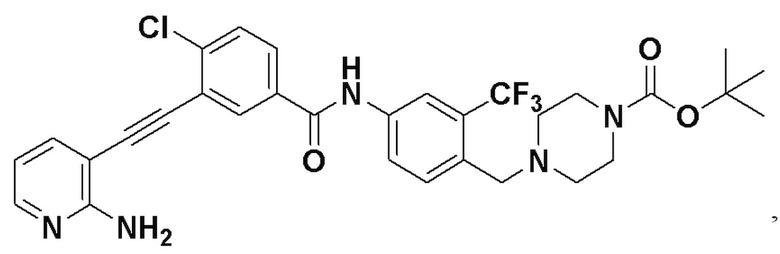
или дейтерированное соединение HuFGFR459, или фармацевтически приемлемая соль соединения HuFGFR459.
2. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п. 1, в котором n имеет значения от 0 до 1.
3. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п. 1, в котором:
М представляет собой С или N, причем когда М представляет собой N, R2 отсутствует;
R представляет собой амино;
R2 выбран из -H или галогена;
R3 выбран из группы, состоящей из -H, галогена, циано, незамещенного или галогенированного C1-4 алкила, C1-4 алкокси;
R4 представляет собой -(CH2)nN(R7)(R8), -OR9;
R7 и R8 вместе с соседним атомом N образуют 6-членный алифатический гетероцикл с 1-2 атомами N в кольце, не замещенный или замещенный C1-4 алкилом, при этом n=1;
R9 выбран из 6-членного алифатического гетероцикла с 1-2 атомами N в кольце, не замещенного или замещенного C1-4 алкилом.
4. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п.1, в котором фармацевтически приемлемая соль представляет собой гидрохлорид, метансульфонат или малеат.
5. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п.2, в котором:
C1-4 алкил представляет собой метил, этил, пропил, изопропил или трет-бутил;
алифатический гетероцикл представляет собой 6-членный моноциклический гетероцикл, содержащий 2 атома N.
6. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п.3, в котором:
R3 представляет собой -Н, хлор, фтор, метил, метоксил, циано или трифторметил; и
R4 представляет собой 4-метилпиперазин-1-илметил или 1-метилпиперидин-4-илоксил.
7. Соединение, или дейтерированное соединение, или его фармацевтически приемлемая соль по п.1, выбранное из следующих соединений:
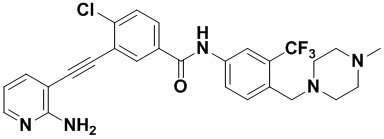
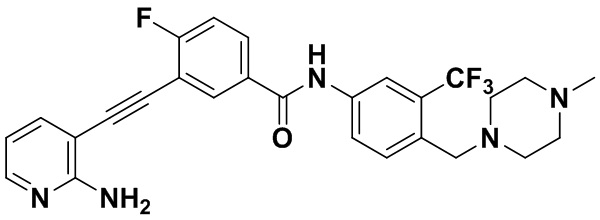
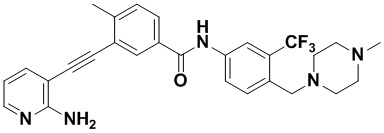
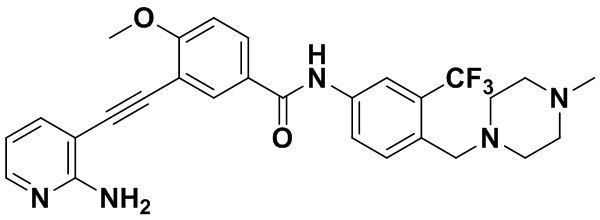

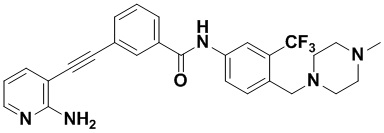
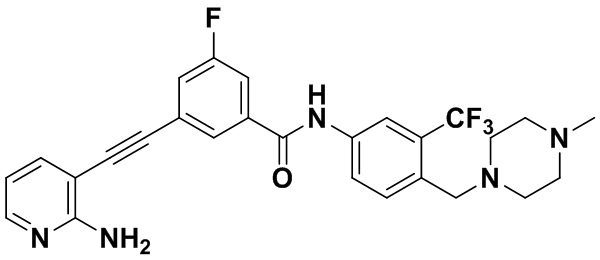
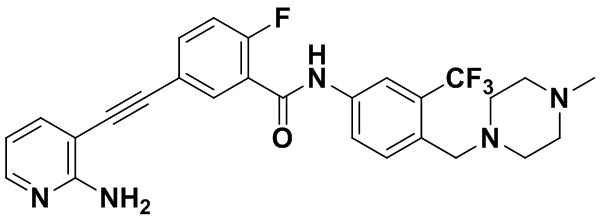
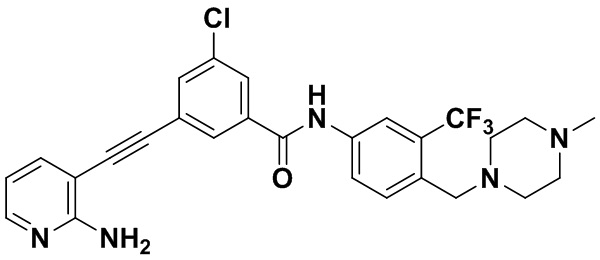
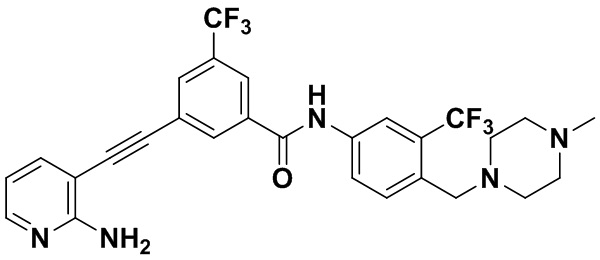
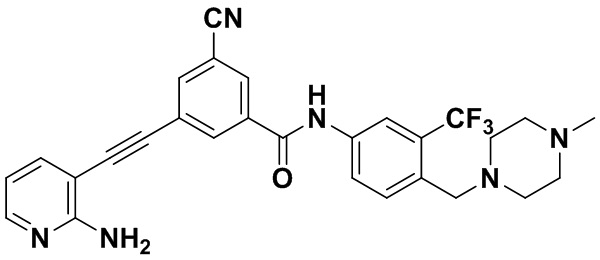
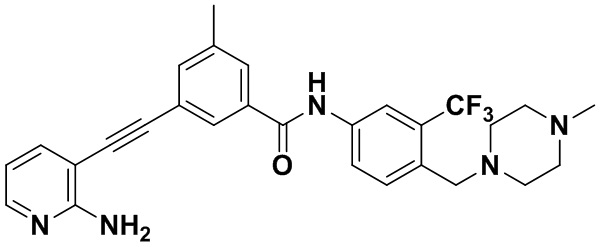

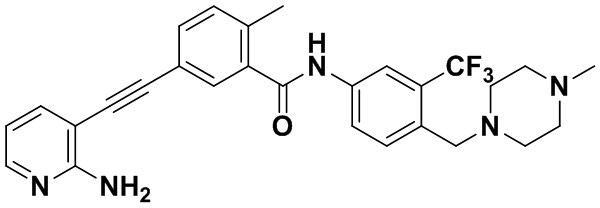
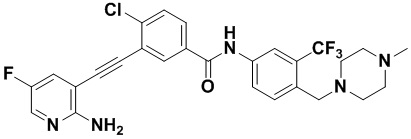
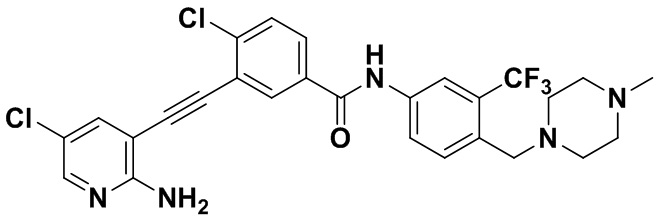
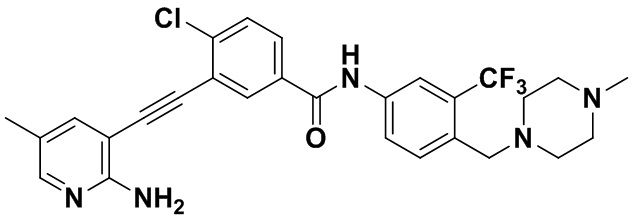
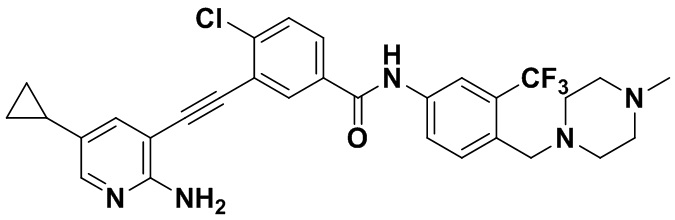
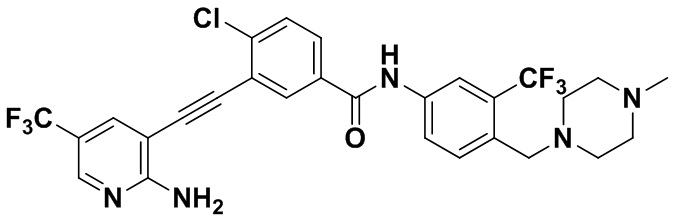
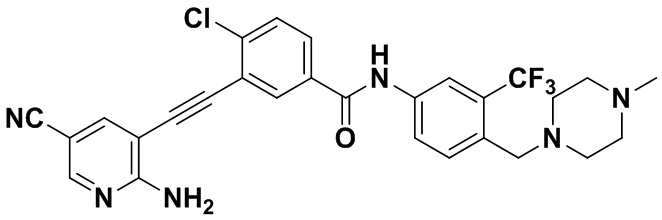 HuFGFR359
HuFGFR359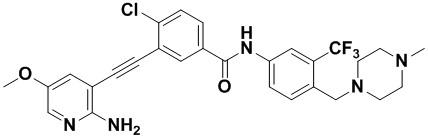 HuFGFR360
HuFGFR360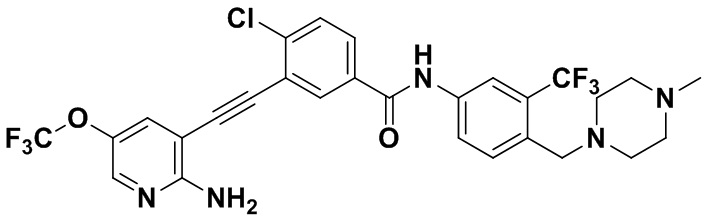
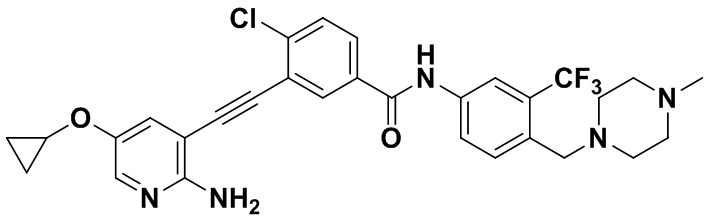
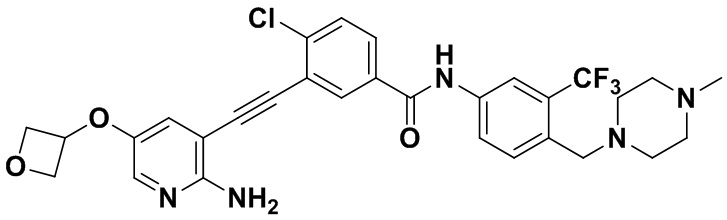
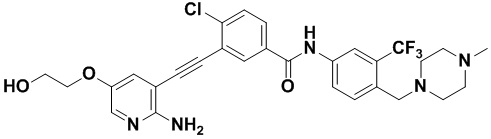

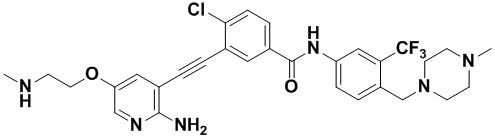
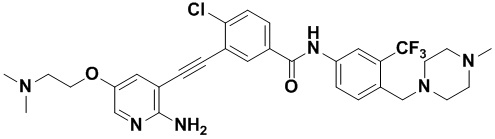
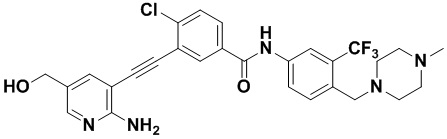
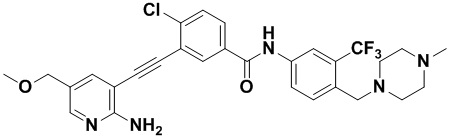
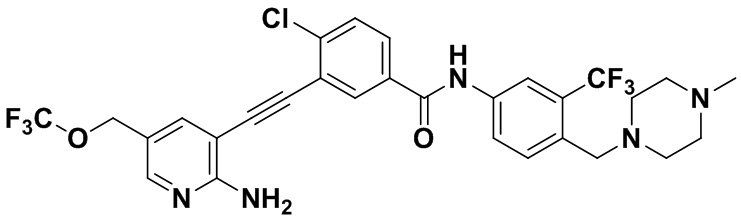
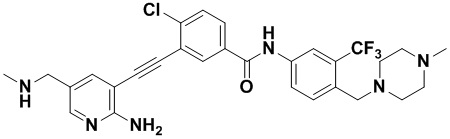
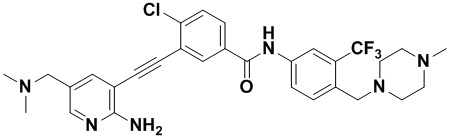
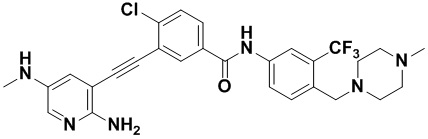
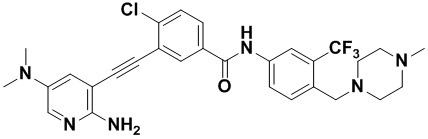
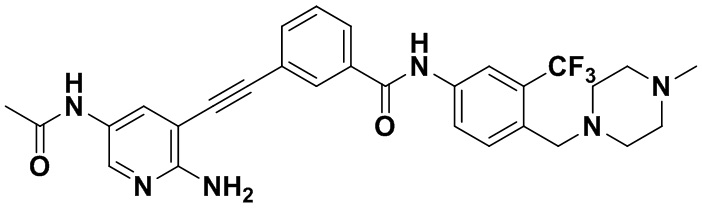
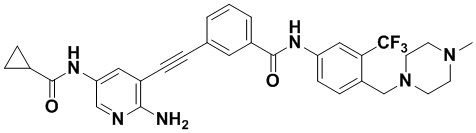
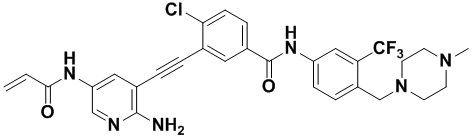

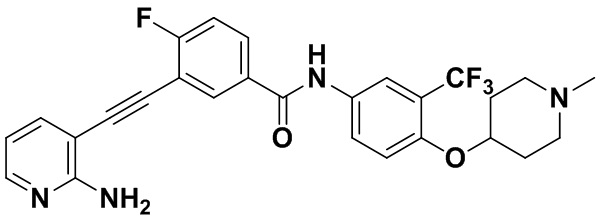
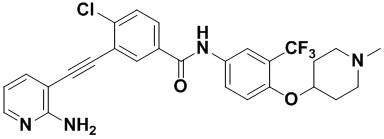
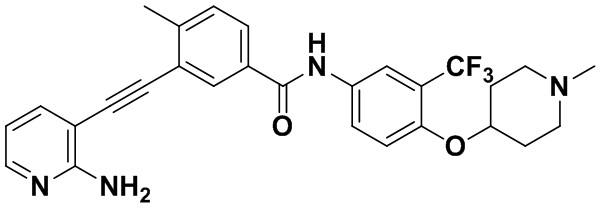
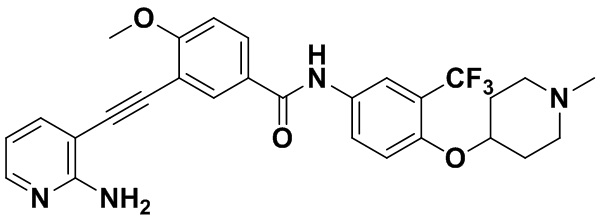



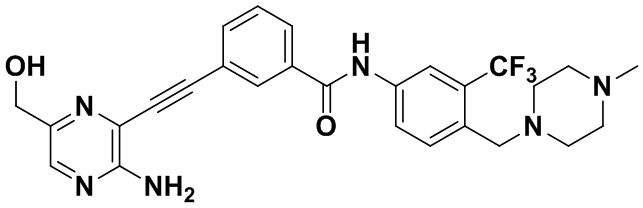
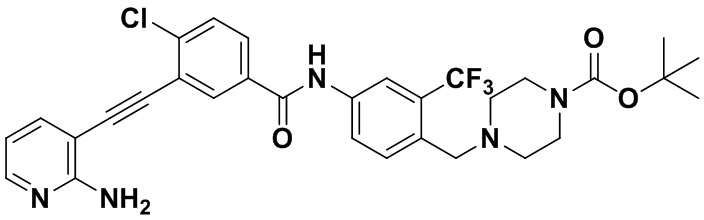

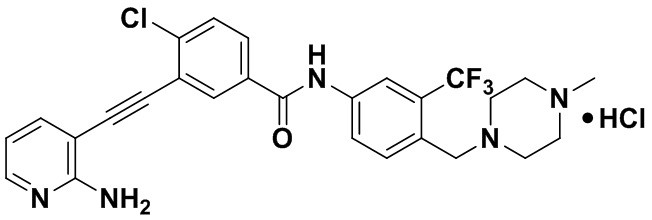
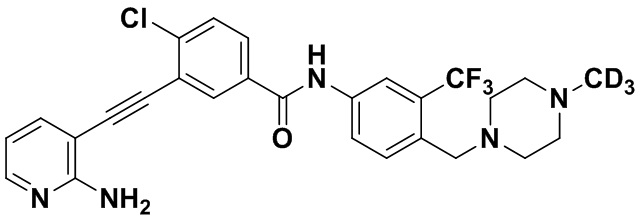
8. Способ получения соединения по п.1, который включает этап реакции соединения формулы (1) с соединением формулы (2)

где каждый из R, М и R1-R4 независимо определен в п.1 и Х представляет собой I или Br.
9. Способ получения по п.8, где сочетание соединения формулы (1) с соединением формулы (2) осуществляют в присутствии катализатора из палладия и меди с переходным металлом и в щелочных условиях.
10. Способ получения по п.9, где палладиевый катализатор представляет собой Pd(PPh3)2Cl2, Pd(OAc)2, и/или Pd(PPh3)4.
11. Способ получения по п.10, где медный катализатор представляет собой CuI и/или CuCl.
12. Способ получения по п.11, где основание, используемое для щелочных условий, включает одно или несколько оснований, выбранных из CsF, Cs2CO3, K2CO3, триэтиламина, диизопропилэтиламина и ДМАП.
13. Способ получения по п.12, где растворитель для реакции сочетания включает один или несколько растворителей, выбранных из ацетонитрила, 1,4-диоксана и DMF.
14. Способ получения по п.8, где способ включает этап взаимодействия соединения формулы (1) с соединением формулы (2) в присутствии фторида цезия, Pd(PPh3)2Cl2, CuI и триэтиламина и в ацетонитриле в качестве растворителя.
15. Фармацевтическая композиция, включающая соединение по п.1 в эффективной дозировке и фармацевтически приемлемый наполнитель, обладающая ингибирующей активностью в отношении киназы РФРФ или киназы RET.
16. Применение соединения по любому из пп.1-7 или фармацевтической композиции по п.15 для приготовления ингибитора киназы РФРФ и/или ингибитора киназы RET.
17. Применение соединения по любому из пп.1-7 или фармацевтической композиции по п.15 для приготовления лекарственного средства для лечения опухоли, связанной со сверхэкспрессией или сверхактивацией киназы ФРФР или киназы RET.
18. Применение по п.17, в котором опухоль представляет собой немелкоклеточный рак легких или рак желудка.
| Liu Y, Peng X, Guan X et al | |||
| "Discovery of novel Ponatinib analogues for reducing KDR activity as potent FGFRs inhibitors", European Journal of Medicinal Chemistry, Volume 126, 2017, Pages 122-132 | |||
| Xianming Deng et al | |||
| Способ очищения амида ортотолуолсульфокислоты | 1921 |
|
SU315A1 |

