ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к гетероциклическому амидному соединению или его фармацевтически приемлемой соли, способу его получения и применению.
УРОВЕНЬ ТЕХНИКИ
В последние годы разработка иммунотерапии продемонстрировала потенциал для изменения подходов к терапии злокачественных новообразований. Противоопухолевые Т-клетки высвобождаются и действуют, блокируя иммунные контрольные точки, что вызывает значимый эффект при не поддающихся лечению опухолях. Одним из основных направлений увеличения эффективности иммунотерапии является поиск методов иммунологической трансформации «холодных» опухолей в «горячие», тем самым преодолевая локальные иммуносупрессивные механизмы и повышая эффективность таких методов, как иммуноанализ. Эти методы включают использование иммуностимулирующих моноклональных антител (например, анти-OX40, агонистическое моноклональное антитело к рецептору OX40 или BB1, нацеленное на CD74) для прямой стимуляции активности Т-клеток и онколитического вируса, который может избирательно и специфически лизировать опухолевые клетки для создания воспалительного микроокружения. Одна из стратегий местной иммунной стимуляции заключается в активации пути STING, ключевой сенсорной системы, которая позволяет врожденной иммунной системе реагировать на инфекцию и рост опухоли и координирует иммунный ответ.
В цитоплазме циклическая ГМФ-АМФ-синтаза (cGAS) связывается с дцДНК из патогенных или поврежденных клеток хозяина и производит низкомолекулярные лиганды - циклический ГМФ-АМФ (цГАМФ). После связывания цГАМФ с димером STING в эндоплазматическом ретикулуме (ЭР), STING фосфорилируется SER/THR-киназой - TANK-связывающей киназой 1 (TBK1). Фосфорилированный STING связывается с фактором транскрипции, регуляторным фактором интерферона 3 (IRF3), и способствует фосфорилированию IRF3 под действием TBK1, что приводит к димеризации IRF3. IRF3 димеризуется и затем транслоцируется в ядро, где связывается с промотором ИФН-β, что запускает продукцию ИФН-β и других цитокинов.
После открытия STING было установлено, что он играет важную роль при инфекционных, онкологических и аутоиммунных заболеваниях. Поэтому STING постепенно стали рассматривать как потенциальную мишень для иммунотерапии. Прямая активация STING бактериальным CDN была подтверждена с помощью рентгеновской кристаллографии (Burdette DL et al. Nature Immunolog, 2013(14): 19-26). Установлено, что новая молекула трансдукции сигнала CDN цГАМФ активирует STING, и ее взаимодействие со STING также было подтверждено с помощью рентгеновской кристаллографии (Cai X et al. Molecular Cell, 2014(54):289-296). Синтетические агонисты STING с хорошими перспективами разработки лекарственных препаратов на их основе могут быть использованы в качестве адъювантов, противоопухолевых или других иммунотерапевтических препаратов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Цель изобретения. Целью настоящего изобретения является создание гетероциклического амидного соединения с новой структурой и его фармацевтически приемлемой соли.
Другой целью настоящего изобретения является представление фармацевтической композиции.
Еще одной целью настоящего изобретения является способ получения гетероциклического амидного соединения и его фармацевтически приемлемой соли.
Последней целью настоящего изобретения является способ применения гетероциклического амидного соединения и его фармацевтически приемлемой соли.
В первом аспекте настоящего изобретения представлено гетероциклическое амидное соединение или его фармацевтически приемлемая соль,
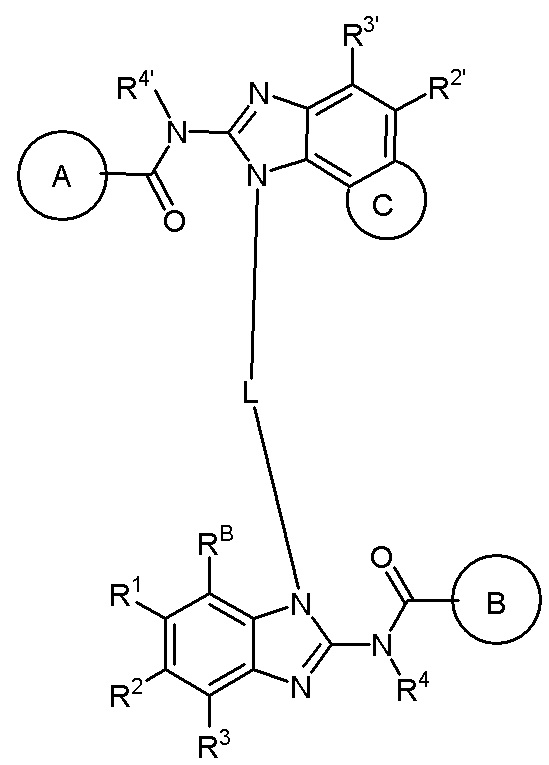 формула I,
формула I,
где:
кольцо A выбирают из 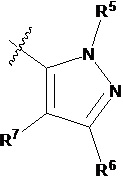 ; кольцо B выбирают из
; кольцо B выбирают из  ; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца опциональнозамещенного 0-4 Ri;
; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца опциональнозамещенного 0-4 Ri;
R4 и R4' каждый выбирают из водорода, C1-C6 алкила или галогена;
R5 и R5' каждый выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R6 и R7 каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6 и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R6' и R7' каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6' и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R1, R2, R3, R2' и R3' каждый выбирают из водорода, -ORd, -NRdRe, галогена, CN, C(O)ORd, C1-C6 алкила, галогенированного C1-C6 алкила, -SO2NH2 или -C(O)NRaRb;
Ra, Rb, Rd и Re каждый выбирают из водорода или C1-C6 алкила;
RB выбирают из водорода, галогена, C1-C6 алкила, замещенного 0-4 Rh, -ORf, -NRfRg, -C(O)Rf, -CO2Rf, -C(O)NRfRg, -NRfC(O)Rg или -NRfC(O)R; RB и R1 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
Rf и Rg каждый выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино, галогенированного C1-C6 алкила или циано;
Rj и Rk выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкилена, замещенного 0-4 Rn, C4-C6 алкенилена, замещенного 0-4 Rn, или C4-C6 алкинилена, замещенного 0-4 Rn; атомы углерода в C4-C6 алкилене, C4-C6 алкенилене или C4-C6 алкинилене замещены -O-, -S- или -NRm-;
Rm выбирают из водорода или C1-C6 алкила;
Rn выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино или галогенированного C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле (I) или его фармацевтически приемлемая соль имеет следующие характеристики:
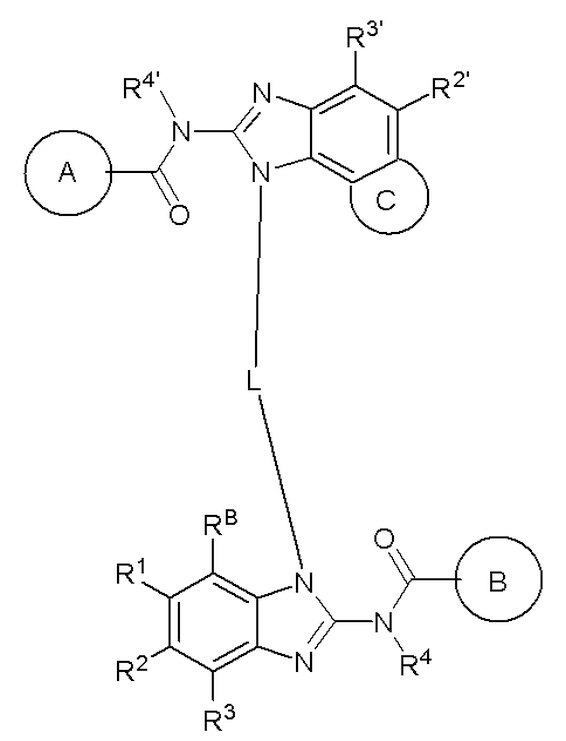 формула I,
формула I,
где:
кольцо A выбирают из ; кольцо B выбирают из 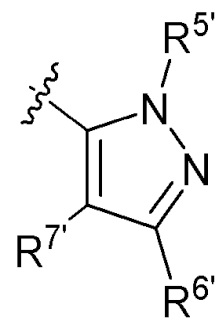 ; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца, замещенного 0-4 Ri;
; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца, замещенного 0-4 Ri;
R4 и R4' каждый выбирают из водорода, C1-C6 алкила или галогена;
R5 и R5' каждый независимо друг от друга выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила, галогенированного C1-C6 алкила или -(CH2)s-L3; где s равно 1, 2 или 3, а L3 выбирают из C3-C6 циклоалкила или 3-6-членного гетероциклила;
R6 и R7 каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6 и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R6' и R7' каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6' и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R1, R2, R3, R2' и R3' каждый выбирают из водорода, -ORd, -NRdRe, галогена, CN, C(O)ORd, C1-C6 алкила, галогенированного C1-C6 алкила, -SO2NH2, -C(O)NRaRb, или -L1-L2-C(O)NRaRb;
Ra, Rb, Rd и Re каждыйвыбирают из водорода или C1-C6 алкила;
L1 выбирают из -(CH2)t-, -C(O)-, -SO2- или -C(O)-; где t равно 0, 1, 2 или 3;
L2 выбирают из C1-C6 алкила, C2-C6 алкинила, C2-C6 алкенила, C3-C6 циклоалкила или 3-6-членного гетероциклила;
RB выбирают из водорода, галогена, C1-C6 алкила, замещенного 0-4 Rh, -ORf, -NRfRg, -C(O)Rf, -CO2Rf или -C(O)NRfRg; или RB и R1 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
Rf и Rg выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино, галогенированного C1-C6 алкила или циано;
Rj и Rk каждый выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкилена, замещенного 0-4 Rn, C4-C6 алкенилена, замещенного 0-4 Rn, или C4-C6 алкинилена, замещенного 0-4 Rn; атомы углерода в C4-C6 алкилене, C4-C6 алкенилене или C4-C6 алкинилене могут быть замещены -O-, -S- или -NRm-;
Rm выбирают из водорода или C1-C6 алкила;
Rn выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино или галогенированного C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения R5 и R5' каждый независимо друг от друга выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, -CH2-C3-C6 циклоалкила; предпочтительно - R5 и R5' каждый независимо друг от друга представляют собой метил, этил, пропил, бутил, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3, аллил или  .
.
В другом предпочтительном варианте осуществления настоящего изобретения C представляет собой насыщенный или ненасыщенный фурил или тетрагидрофурил, замещенный 0-4 Ri, где Ri выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила.
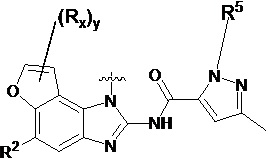
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле I или его фармацевтически приемлемая соль, где кольцо A и кольцо B одинаковые или разные, каждое из них независимо друг от друга представляет собой 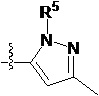 ,
, 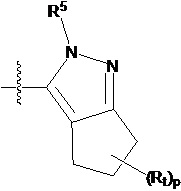 или
или 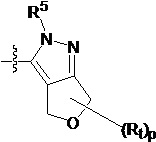 , а каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси; p равно 0, 1, 2, 3 или 4;
, а каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси; p равно 0, 1, 2, 3 или 4;
R5 определен выше.
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле I или его фармацевтически приемлемая соль, где кольцо A и кольцо B одинаковые, каждое из них независимо друг от друга представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения кольцо A и кольцо B одинаковы, и каждое из них независимо друг от друга представляет собой , где R5 выбирают из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
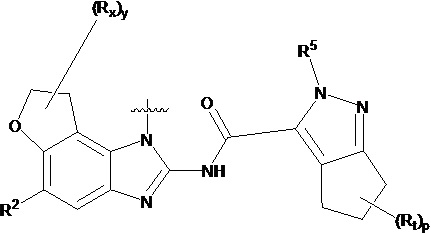
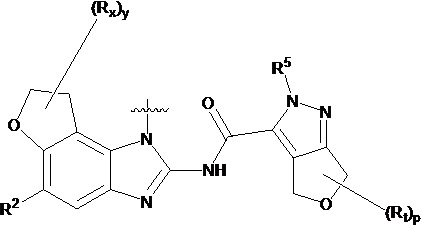
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле I или его фармацевтически приемлемая соль, в которой фрагмент 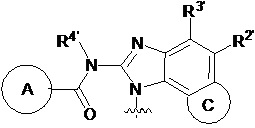 выбирают из
выбирают из 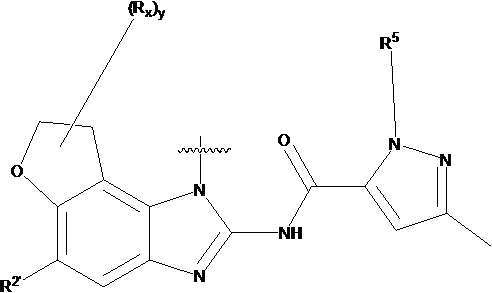 ,
,  ,
,  ,
, 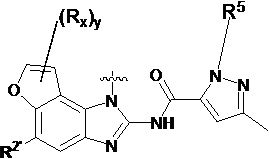 ,
,  или
или 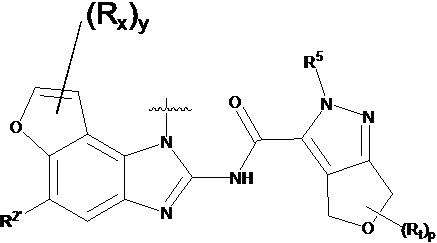 ,
,
где y равно 0, 1, 2, 3 или 4;
p равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила,
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси,
R5 и R2' определены выше.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент представляет собой или ;
где:
y равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила;
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2' независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb;
где Ra и Rb независимо выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент представляет собой 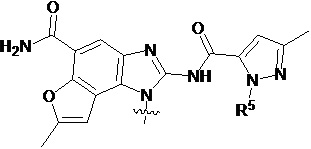 , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
, где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
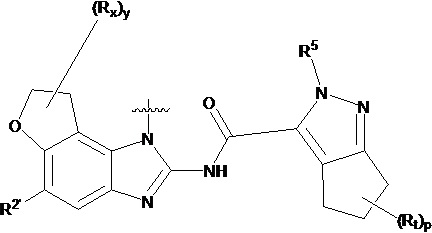
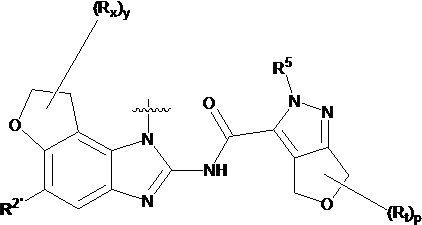
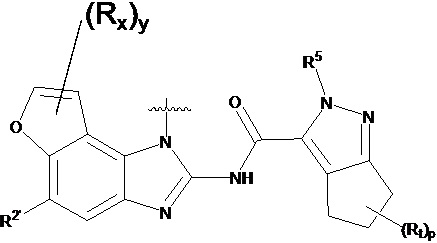
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле I, его фармацевтически приемлемая соль, в которой фрагмент 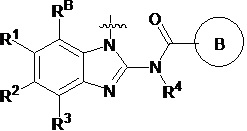 выбирают из
выбирают из 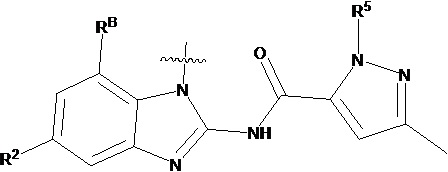 ,
, 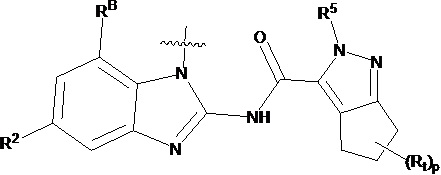 ,
, 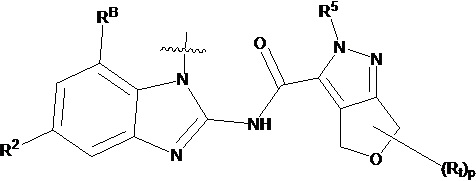 ,
,  ,
,  ,
,  ,
, 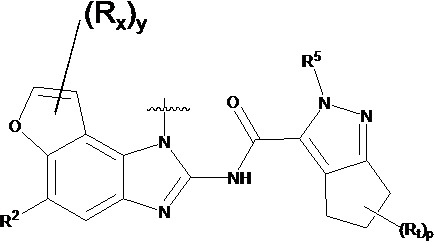 или
или 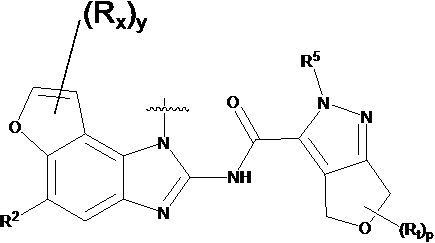 , где
, где
y равно 0, 1, 2, 3 или 4;
p равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила;
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2 независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb;
где Ra и Rb независимо друг от друга выбирают из водорода или C1-C6 алкила;
RB выбирают из водорода и -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk независимо друг от друга выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2 независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb;
где Ra и Rb независимо друг от друга выбирают из водорода или C1-C6 алкила;
RB выбирают из водорода и -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент представляет собой ; где R2 независимо представляет собой -C(O)NH2;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила;
RB выбирают из водорода или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент и фрагмент одинаковые или разные.
В другом предпочтительном варианте осуществления настоящего изобретения фрагмент и фрагмент одинаковы, и каждый из них независимо друг от друга представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
В другом предпочтительном варианте осуществления настоящего изобретения L выбирают из C4-C6 алкенилена, замещенного 0-4 Rn, или C4-C6 алкилена, замещенного 0-4 Rn, где Rn выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино или галогенированного C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле (I) или его фармацевтически приемлемая соль, где RB выбирают из водорода или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение по формуле (I) или его фармацевтически приемлемая соль, где RB выбирают из H, метокси, 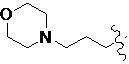 ,
, 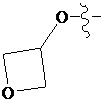 ,
, 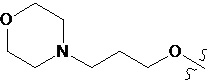 ,
, 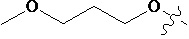 или
или 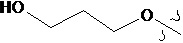 .
.
В другом предпочтительном варианте осуществления настоящего изобретения R4 и R4' представляют собой Н.
В другом предпочтительном варианте осуществления настоящего изобретения R1, R3, R1' и R3' представляют собой Н.
В другом предпочтительном варианте осуществления настоящего изобретения R4 и R4' представляют собой водород;
R5 и R5' каждый выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R6 и R7 каждый выбирают из водорода или C1-C6 алкила, или R6' и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R6' и R7' каждый выбирают из водорода или C1-C6 алкила, или R6' и R7' вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R1, R3 и R3' каждый выбирают из водорода, C1-C6 алкила или -ORd;
R2 и R2' каждый выбирают из водорода, -SO2NH2 или -C(O)NRaRb;
Ra и Rb каждый выбирают из водорода или C1-C6 алкила;
Rd выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
RB выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкенилена или C4-C6 алкилена.
В другом предпочтительном варианте осуществления настоящего изобретения R6 и R7' независимо друг от друга выбирают из водорода или C1-C6 алкила;
R6' и R7' каждый независимо друг от друга выбирают из водорода или C1-C6 алкила;
R1, R3, и R3' каждый независимо друг от друга выбирают из водорода или C1-C6 алкила;
R2 и R2' каждый независимо друг от друга выбирают из водорода, -SO2NH2 или -C(O)NH2;
RB выбирают из водорода или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
В другом предпочтительном варианте осуществления настоящего изобретения L представляет собой 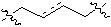 , где
, где  обозначает одинарную или двойную связь.
обозначает одинарную или двойную связь.
В другом предпочтительном варианте осуществления настоящего изобретения A, B, C, L, R1, R2, R3, R4, RB, R2', R3' и R4' являются определенными группами, соответствующими каждому определенному соединению в Примерах.
В другом предпочтительном варианте осуществления настоящего изобретения гетероциклическое амидное соединение или его фармацевтически приемлемая соль имеет одну из следующих структур:
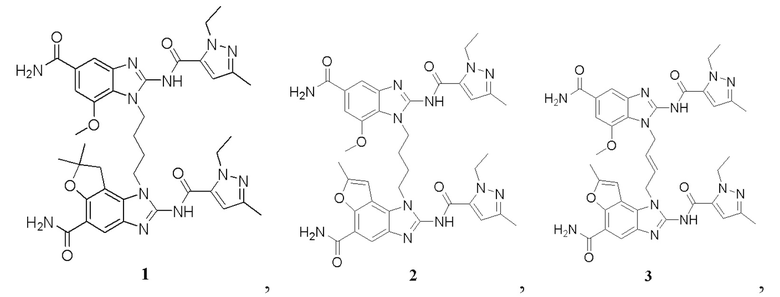
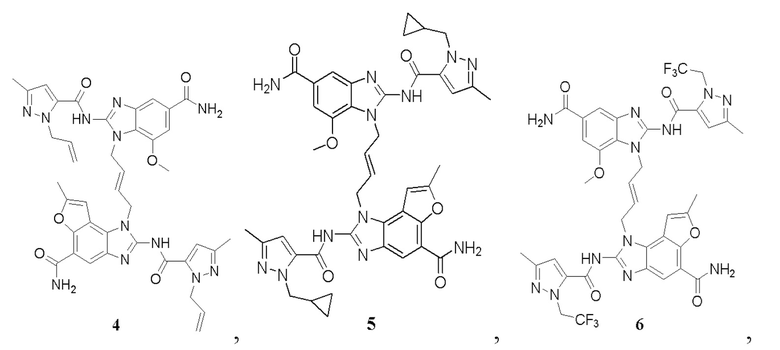
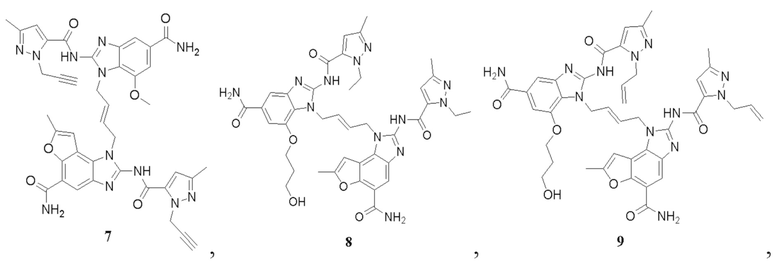
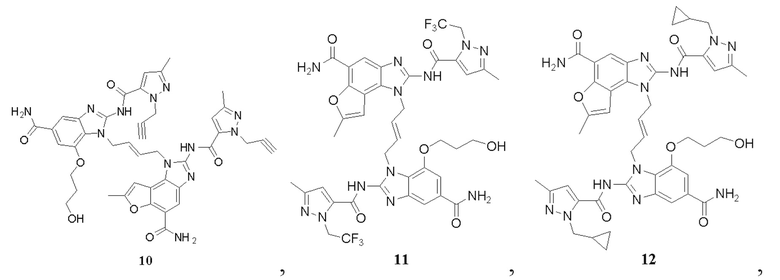
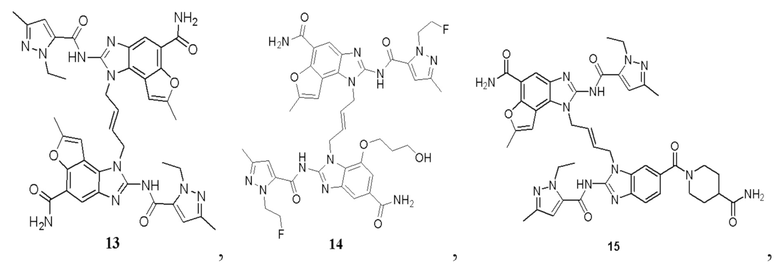
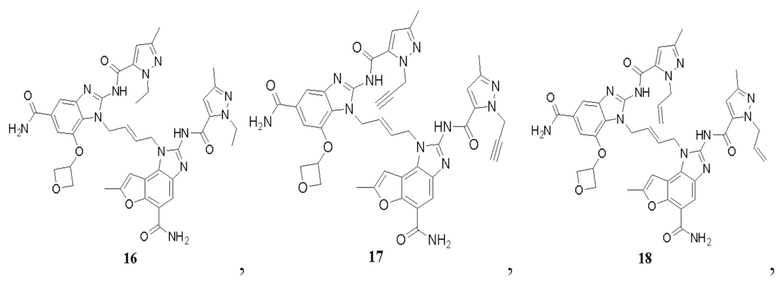
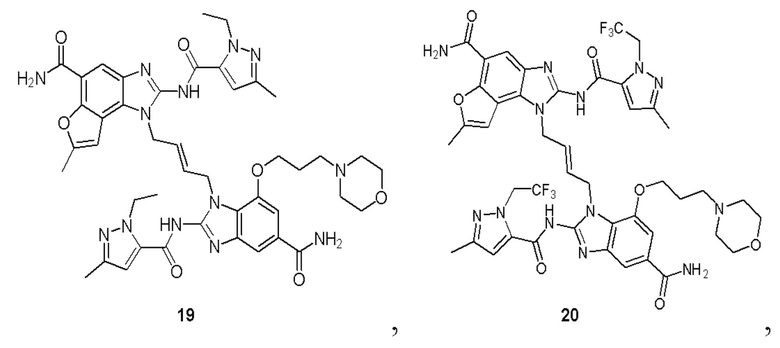
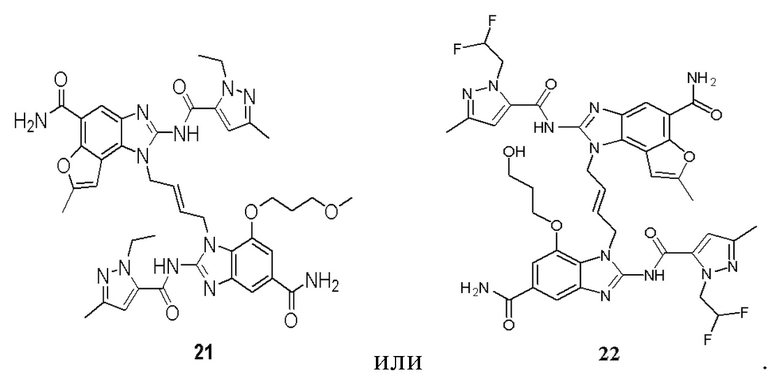
В другом предпочтительном варианте осуществления настоящего изобретения соединение представляет собой соединение, показанное в Примере.
Во втором аспекте настоящего изобретения представлена фармацевтическая композиция, включающая терапевтически эффективное количество одного или более гетероциклических амидных соединений по формуле (I) или их фармацевтически приемлемых солей по первому аспекту, а также фармацевтически приемлемый носитель.
Во третьем аспекте настоящего изобретения представлена фармацевтическая композиция, включающая терапевтически эффективное количество одного или болеегетероциклических амидных соединений по формуле (I) или их фармацевтически приемлемых солей по первому аспекту, а также фармацевтически приемлемое вспомогательное вещество.
В четвертом аспекте настоящего изобретения представлен способ получения гетероциклического амидного соединения по формуле (I) или его фармацевтически приемлемой соли по первому аспекту, который включает следующие этапы:
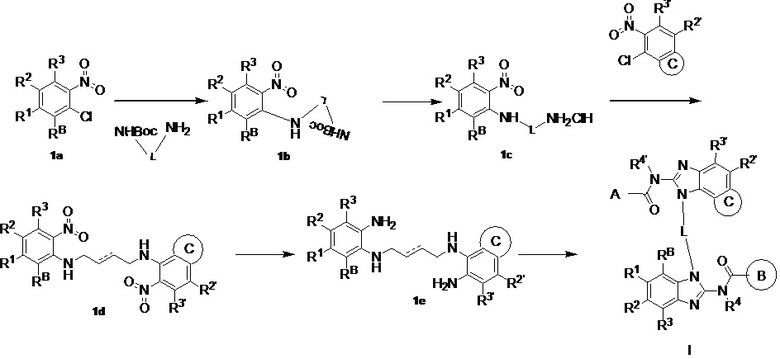
(i) в инертном растворителе в присутствии основания сырье 1a вступает в реакцию нуклеофильного замещения с моно-БОК-защищенным диамином 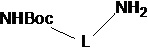 с образованием соединения 1b;
с образованием соединения 1b;
(ii) в инертном растворителе в присутствии кислоты снимается БОК-защита из соединения 1b с образованием промежуточного соединения 1c;
(iii) в инертном растворителе в присутствии основания промежуточное соединение 1c вступает в реакцию замещения с 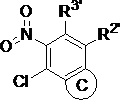 с образованием промежуточного соединения 1d;
с образованием промежуточного соединения 1d;
(iv) в инертном растворителе в присутствии восстановителя промежуточное соединение 1d реагирует с образованием соединения 1e;
(v) в инертном растворителе в присутствии катализатора соединение 1e реагирует с 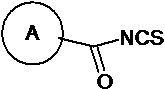 или
или 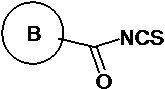 с образованием целевого соединения I;
с образованием целевого соединения I;
в данной формуле A, B, L, R1, R2, R3, R4, RB, R2', R3' и R4' определены выше.
В пятом аспекте настоящего изобретения представлено применение гетероциклического амидного соединения по формуле (I) или его фармацевтически приемлемой соли по первому аспекту для получения иммунного адъюванта.
В шестом аспекте настоящего изобретения представлено применение гетероциклического амидного соединения по формуле (I) или его фармацевтически приемлемой соли по первому аспекту для получения лекарственного препарата для активации STING.
В седьмом аспекте настоящего изобретения представлено применение гетероциклического амидного соединения по формуле (I) или его фармацевтически приемлемой соли по первому аспекту для получения лекарственного препарата для лечения заболевания, связанного с активностью STING; где заболевание, связанное с активностью STING, представляет собой одно или несколько из заболеваний, связанных с воспалением, аутоиммунное заболевание, инфекционное заболевание, онкологическое заболевание или предраковый синдром.
В другом предпочтительном варианте осуществления настоящего изобретения заболевание, связанное с активностью STING, включает воспалительные, аллергические и аутоиммунные заболевания, инфекционные заболевания, онкологические заболевания или предраковый синдром.
В другом предпочтительном варианте осуществления настоящего изобретения онкологическое заболевание выбирают из группы, включающей рак толстой кишки, рак молочной железы (например, инвазивный протоковый рак, неинвазивный протоковый рак и воспалительный рак молочной железы), рак легких (например, немелкоклеточный рак легкого, мелкоклеточный рак легкого и злокачественная мезотелиома) и рак яичников (например, эпителиальная карцинома яичника, герминогенная опухоль яичника, опухоль яичника с низким потенциалом злокачественности), рак головного мозга, фибросаркома и плоскоклеточная карцинома, меланома и др.
В другом предпочтительном варианте осуществления настоящего изобретения воспалительное заболевание выбирают из группы, включающей обыкновенные угри, бронхиальную астму, целиакию, хронический простатит, гломерулонефрит, воспалительные заболевания кишечника, воспалительные заболевания органов малого таза, реперфузионное повреждение, ревматоидный артрит, саркоидоз, васкулит, воспаление дыхательных путей, вызванное клещами домашней пыли, и интерстициальный цистит.
В другом предпочтительном варианте осуществления настоящего изобретения инфекционное заболевание представляет собой инфекционное заболевание, вызванное вирусом, выбранным из группы, включающей вирус гепатита В, вирус папилломы человека и вирус Эпштейна - Барр, ассоциированный с назофарингеальной карциномой.
В другом предпочтительном варианте осуществления настоящего изобретения воспалительное заболевание и аутоиммунное заболевание в значительной степени совпадают.
В другом предпочтительном варианте осуществления настоящего изобретения воспалительное заболевание представляет собой бронхиальную астму.
В восьмом аспекте настоящего изобретения представлен способ индуцирования иммунного ответа у субъекта, где способ включает введение субъекту терапевтически эффективного количества соединения по первому аспекту или фармацевтической композиции по второму аспекту.
В другом предпочтительном варианте осуществления настоящего изобретения субъект является млекопитающим, предпочтительно человеком.
В девятом аспекте настоящего изобретения представлен способ активации STING, включающий этап контакта соединения или его фармацевтически приемлемой соли по первому аспекту настоящего изобретения с соматической клеткой (или тканью) с последующей активацией STING.
В другом предпочтительном варианте осуществления настоящего изобретения соматическая клетка (или ткань) получена от грызуна (например, мышь и крыса) или приматов (например, человек).
В десятом аспекте настоящего изобретения представлен способ лечения заболевания, связанного с активностью STING, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения по первому аспекту или фармацевтической композиции по второму аспекту.
Заболевание, связанное с активностью STING, определенное в настоящем изобретении, представляет собой заболевание, при котором STING играет роль в патогенезе заболевания. Заболевание, связанное с активностью STING, включает воспалительные, аллергические и аутоиммунные заболевания, инфекционные заболевания, онкологические заболевания, предраковый синдром и др.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показана противоопухолевая активность in vivo соединения 19 в модели рака толстой кишки CT26.
На фигуре 2 показана противоопухолевая активность in vivo соединения 19 в модели ортотопической трансплантированной опухоли рака молочной железы EMT6.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
После обширных и глубоких исследований впервые случайно было разработано гетероциклическое амидное соединение с новой структурой, и результаты экспериментов показали, что соединение настоящего изобретения обладает хорошим активирующим действием на STING, и может быть использовано для получения лекарственного препарата для лечения заболеваний, связанных с активацией STING или активностью STING. На этой основе было создано настоящее изобретение.
Термины
В настоящем изобретении, если не указано иное, используемые термины имеют общие значения, известные специалистам в данной области.
Если заместитель описывается обычной химической формулой, написанной слева направо, он также включает химически эквивалентный заместитель, полученный путем написания структурной формулы справа налево. Например, -CH2O- эквивалентен -OCH2-.
Термин «алкил» сам по себе или в составе другого заместителя относится к углеводородной группе с прямой или разветвленной цепью, имеющей определенное число атомов углерода (например, C1-C6 обозначает группу, содержащую 1, 2, 3, 4, 5 или 6 атомов углерода). Примеры алкила включают, помимо прочего, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил и подобные алкилы. В настоящей заявке термин «алкил» также включает замещенные алкилы, т. е. одно или несколько положений в алкиле являются замещенными, в частности 1-4 заместителями, которые могут находиться в любом положении.
Термин «алкенил» означает углеводородную группу с прямой или разветвленной цепью, которая содержит одну или несколько двойных связей и обычно имеет длину от 2 до 20 атомов углерода (или C2-C8). Например, в настоящем изобретении «C2-C6 алкенил» включает алкенил, имеющий 2, 3, 4, 5 или 6 атомов углерода, а также «C3-C6 алкенил». Примеры алкенила включают, помимо прочего, винил, пропенил, бутенил, 1-метил-2-бутен-1-ил и др. В настоящем изобретении алкенил включает также замещенный алкенил.
Термин «алкинил» означает углеводородную группу с прямой или разветвленной цепью, которая содержит одну или несколько тройных связей и обычно имеет длину от 2 до 20 атомов углерода (или C2-C8). В настоящем изобретении термин «C2-C6 алкинил» включает алкинилы с прямой или разветвленной цепью, имеющие 2, 3, 4, 5 или 6 атомов углерода, включая, помимо прочего, этинил, пропинил и др. В настоящем изобретении алкинил также включает замещенный алкинил, и заместитель может быть галогенированным, гидроксильным, циано, нитро и др.
Термин «алкиламино» относится к алкилу, связанному с остальной частью молекулы через аминогруппу. В настоящем изобретении «C1-C6 алкиламино» имеет структуру C1-C6алкил-NH-.
Термин «алкилен» сам по себе или в составе другого заместителя относится к двухвалентной группе, полученной из алкана, такой как -CH2CH2CH2CH2-. Алкил (или алкилен) обычно имеет 1-24 атома углерода, в рамках настоящего изобретения предпочтительно те группы, которые имеют 10 или менее атомов углерода (например, C4-C6 алкилен). Аналогичным образом термины «алкенилен» или «алкинилен» относятся к ненасыщенной форме «алкилена» с двойными или тройными связями соответственно. Примеры «алкенилена» или «алкинилена» включают, помимо прочего, пропенилен,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 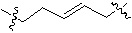 и др.
и др.
В настоящем изобретении «C1-C6 алкокси» относится к алкоксигруппам с прямой, разветвленной или циклической цепью (например, C3-C6 циклоалкокси), имеющим от 1 до 6 атомов углерода; типичные примеры включают, помимо прочего, метоксил, этоксил, пропоксил, изопропоксил, бутоксил и др. Предпочтительна C1-C3 алкоксигруппа.
Термин «циклоалкил» относится к циклическим алкилам, которые включают насыщенные моноциклические, бициклические (например, конденсированные бициклические или спиробициклические) или полициклические кольца. В настоящем изобретении «3-6-членный циклоалкил» относится к циклическому алкилу, содержащему 3, 4, 5 или 6 атомов углерода. Типичные циклоалкилы в рамках настоящего изобретения включают, помимо прочего, циклопропил, циклобутил, циклопентил, циклогексил и др. Следует понимать, что замещенные или незамещенные циклоалкилы, такие как разветвленные циклоалкилы (например, 1-метилциклопропил и 2-метилциклопропил), включены в определение «циклоалкил».
Термин «гетероциклоалкил» относится к циклоалкилу, содержащему от одного до пяти гетероатомов, выбранных из N, O и S, где атомы азота и серы могут быть окислены, а атомы азота могут быть кватернизированы. Гетероциклоалкилы могут представлять собой моноциклические, бициклические или полициклические системы. В настоящем изобретении «3-6-членный гетероциклоалкил» относится к группе, в которой 1 или 2 кольцевых атома С в C3-C6 циклоалкиле замещены гетероатомами, выбранными из N, O и S. Примеры гетероциклоалкилов включают, помимо прочего, пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиапиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и др. Гетероциклоалкил может быть присоединен к остальной части молекулы через циклический углерод или гетероатом.
Термин «гетероциклил» относится к насыщенной или частично насыщенной циклической группе с гетероатомом, выбранным из N, S и O, которая может иметь моноциклическую или бициклическую форму, например, мостиковое или спирокольцо. Предпочтителен 3-8-членный гетероциклил, более предпочтителен 4-6-членный гетероциклил, еще более предпочтителен 5-6-членный гетероциклил. Примеры гетероциклилов включают, помимо прочего, оксетан, азетидин, тетрагидро-2H-пиранил, пиперидинил, пиперазинил, тетрагидрофурил, морфолинил, пирролидил и др.
Термин «гетероарил» относится к циклической ароматической группе, имеющей 1-3 гетероатома, выбранных из N, S и O, которая может представлять собой моноциклическое или конденсированное кольцо. В настоящем изобретении предпочтительным является 5-6-членный гетероарил. Примеры гетероарилов включают, помимо прочего, пиридил, пиридазинил, пиримидинил, пиразинил, триазинил, пирролил, пиразолил, имидазолил, (1,2,3)-триазолил и (1,2,4)-триазолил, тетразолил, фурил, тиенил, изоксазол, тиазолил или оксазолил и др.
В настоящем изобретении «карбоциклический или гетероциклический» отдельно или в составе других групп относится к моноциклическим или бициклическим насыщенным, частично насыщенным или ароматическим карбоциклическим (например, циклоалкил, циклоалкенил, фенил и др., как описано выше), или моноциклический или бициклический насыщенный, частично насыщенный или ароматический гетероцикл (например, гетероалкил, гетероциклил, гетероарил и др., как описано выше), где 4-6-членное карбоциклическое или гетероциклическое кольцо относится к карбоциклическому или гетероциклическому кольцу, содержащему кольца из 4-6 атомов; предпочтительно 5-6-членное карбоциклическое или гетероциклическое кольцо. Примеры карбоциклического или гетероциклического кольца включают, помимо прочего, циклопропил, циклобутил, циклопентил, циклогексил, циклопентенил, циклогексенил, циклопентадиенил, циклогексадиенил, оксетан, азетидин, тетрагидро-2H-пиранил, пиперидинил, пиперазинил, тетрагидрофурил, морфолинил, пирролидил, пиридил, пиридазинил, пиримидинил, пиразинил, триазинил, пирролил, пиразолил, имидазолил, (1,2,3)-триазолил и (1,2,4)-триазолил, тетразолил, фурил, тиенил, изоксазол, тиазолил или оксазолил и др.
В настоящем изобретении «амино» относится к группе со структурой -N(R)(R'), где R и R' могут независимо друг от друга представлять собой водород, алкил или замещенный алкил, циклоалкил или замещенный циклоалкил, арил или замещенный арил, гетероцикл или замещенный гетероцикл, как определено выше. R и R' могут быть одинаковыми или разными в диалкиламиновом фрагменте.
Термины, используемые в настоящем документе, подразумевают, что каждая из вышеупомянутых групп алкила, алкокси, циклоалкила, гетероарила, циклогетероалкила, алкенила, алкинила, гетероциклила, карбоциклила или гетероциклила и другие могут быть замещенными или незамещенными.
Если не указано иное, предполагается, что любой гетероатом с более низким валентным состоянием имеет достаточно атомов водорода для восполнения своего валентного состояния.
В настоящем изобретении термин «замещенный» относится к замещению одного или нескольких атомов водорода на определенной группе определенным заместителем. Определенными заместителями являются те, которые описаны в предыдущем параграфе, или те, которые указаны в каждом примере. Если не указано иное, замещенная группа может иметь заместитель, выбранный из определенной группы, в любом замещаемом положении группы, причем заместитель может быть одним и тем же или разным в каждом положении. Специалисты в данной области должны понимать, что комбинации заместителей, предусмотренные настоящим изобретением, представляют собой стабильные или химически достижимые комбинации. Типичные заместители включают, помимо прочего, одного или несколько примеров из следующих групп: водород, дейтерий, галоген (например, моногалогенированный заместитель или полигалогенированные заместители, среди последних такие как трифторметил или алкил, содержащий Cl3), циано, нитро, оксо (= O), трифторметил, трифторметокси, циклоалкил, алкенил, алкинил, гетероциклическое, ароматическое кольцо, ORa, SRa, S(=O)Re, S(=O)2Re, P(=O)2Re, S(=O)2ORe, P(=O)2ORe, NRbRc, NRbS(=O)2Re, NRbP(=O)2Re, S(=O)2NRbRc, P(=O)2NRbRc, C(=O)ORd, C(=O)Ra, C(=O)NRbRc, OC(=O)Ra, OC(=O)NRbRc, NRbC(=O)ORe, NRdC(=O)NRbRc, NRdS(=O)2NRbRc, NRdP(=O)2NRbRc, NRbC(=O)Ra or NRbP(=O)2Re; где Ra может независимо от других заместителей представлять собой водород, дейтерий, алкил, циклоалкил, алкенил, алкинил, карбоциклил или гетероциклил; Rb, Rc и Rd могут независимо от других заместителей представлять собой водород, дейтерий, алкил, циклоалкил, карбоциклил или гетероциклил; или Rb и Rc вместе с атомом N образуют гетероциклил; Re может независимо от других заместителей представлять собой водород, алкил, циклоалкил, алкенил, алкинил, гетероциклил или ароматическое кольцо. Замещение вышеуказанными типичными заместителями, такими как алкил, циклоалкил, алкенил, циклоалкенил, алкинил, карбоциклическое или гетероциклическое кольцо, необязательно. Заместители могут включать, помимо прочего, следующие: галоген, гидрокси, циано, карбокси (-COOH), C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, 3-12-членный гетероциклил, арил, гетероарил, C1-C8 альдегид, C2-C10 ацил, C2-C10 сложный эфир, амино, C1-C6 алкокси, C1-C10 сульфонил, C1-C6 урамидо и др.
Термин «галогенированный» или «галоген» включает фтор, хлор, бром и йод.
Термин «гидроксил» обозначает группу -OH.
В настоящем изобретении термин «циклизованный в R6 и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо» относится к R6 и R7 вместе с атомами С, с которыми они соединены с образованием конденсированного кольца; аналогичным образом «циклизованный» в R6' и R7', и RB и R1 имеют то же самое значение. Где 4-6-членное карбоциклическое или гетероциклическое кольцо описано выше и включает в себя замещенные или незамещенные 4-6-членные карбоциклические или гетероциклические кольца; «замещенный» означает замещенный одним или более (1, 2, 3 или 4) заместителями, выбранными из группы, состоящей из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино, галогенированного C1-C6 алкила или циано.
В настоящем изобретении C(O)- в -C(O)NRaRb, -C(O)OR, -C(O)R или -NRC(O)R представляет собой карбонильную группу (-C=O-), образованную двумя атомами углерода и кислорода через двойную связь.
Действующее вещество
В настоящей заявке термин «соединение по настоящему изобретению» относится к соединению, представленному по формуле I, и дополнительно включает стереоизомер или оптический изомер, фармацевтически приемлемую соль, пролекарство или сольват соединения по формуле I.
Соединение по формуле I настоящего изобретения имеет следующую структуру:
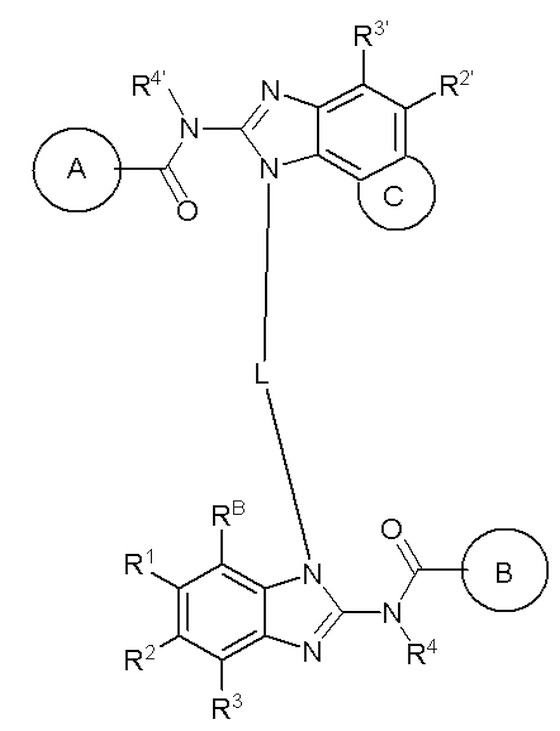 формула I,
формула I,
где:
кольцо A выбирают из ; кольцо B выбирают из  ; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца, замещенного 0-4 Ri;
; C выбирают из насыщенного или ненасыщенного 4-6-членного карбоциклического или гетероциклического кольца, замещенного 0-4 Ri;
R4 и R4' каждый выбирают из водорода, C1-C6 алкила или галогена;
R5 и R5' каждый выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R6 и R7 каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6 и R7 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R6' и R7' каждый выбирают из водорода, C1-C6 алкила, нитро, галогена или галогенированного C1-C6 алкила, или R6' и R7' вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
R1, R2, R3, R2' и R3' каждый выбирают из водорода, -ORd, -NRdRe, галогена, CN, C(O)ORd, C1-C6 алкила, галогенированного C1-C6 алкила, -SO2NH2 или -C(O)NRaRb;
Ra, Rb, Rd и Re каждый выбирают из водорода или C1-C6 алкила;
RB выбирают из водорода, галогена, C1-C6 алкила, замещенного 0-4 Rh, -ORf, -NRfRg, -C(O)Rf, -CO2Rf, -C(O)NRfRg или -NRfC(O)Rg; или RB и R1 вместе с атомами, с которыми они соединены, циклизованы в насыщенное или ненасыщенное 4-6-членное карбоциклическое или гетероциклическое кольцо;
Rf и Rg каждый выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино, галогенированного C1-C6 алкила или циано;
Rj и Rk каждый выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкилена, замещенного 0-4 Rn, C4-C6 алкенилена, замещенного 0-4 Rn, или C4-C6 алкинилена, замещенного 0-4 Rn; атомы углерода в C4-C6 алкилене, C4-C6 алкенилене или C4-C6 алкинилене могут быть замещены -O-, -S- или -NRm-;
Rm выбирают из водорода или C1-C6 алкила;
Rn выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино или галогенированного C1-C6 алкила.
В настоящем изобретении соединение по формуле I также может иметь следующую структуру:
формула I,
где:
кольцо A выбирают из ; кольцо B выбирают из ;
R1, R2, R3, R2' и R3' каждый выбирают из водорода, -ORd, -NRdRe, галогена, CN, C(O)ORd, C1-C6 алкила, галогенированного C1-C6 алкила, -SO2NH2, -C(O)NRaRb, или -L1-L2-C(O)NRaRb;
Ra, Rb, Rd и Re каждый выбирают из водорода или C1-C6 алкила;
L1 выбирают из -(CH2)t-, -C(O)-, -SO2- или -C(O)-; где t равно 0, 1, 2 или 3;
L2 выбирают из C1-C6 алкила, C2-C6 алкинила, C2-C6 алкенила, C3-C6 циклоалкила или 3-6-членного гетероциклила;
R5 и R5' каждый независимо друг от друга выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила, галогенированного C1-C6 алкила или -(CH2)s-L3; где s равно 1, 2 или 3, а L3 выбирают из C3-C6 циклоалкила или 3-6-членного гетероциклила. R5 и R5' предпочтительно независимо друг от друга представляют собой водород, C1-C6 алкил, C3-C6 алкенил, галогенированный C1-C6 алкил, -CH2-C3-C6 циклоалкил; более предпочтительно, R5 и R5' независимо друг от друга представляют собой метил, этил, пропил, бутил, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3, аллил или ;
A, B, C, L, R4, RB, R4 ', R6, R6', R7, R7' определены выше.
Предпочтительно -L1-L2-C(O)NRaRb представляет собой  .
.
C предпочтительно представляет собой насыщенный или ненасыщенный фурил или тетрагидрофурил, замещенный 0-4 Ri, где Ri выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила.
Предпочтительно кольцо A и кольцо B одинаковые или разные, каждое из них независимо друг от друга представляет собой , или , а каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси; p равно 0, 1, 2, 3 или 4;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила.
Предпочтительно в указанном выше соединении кольцо A и кольцо B одинаковы, и каждый из них независимо друг от друга представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила, предпочтительно R5 выбирают из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
Предпочтительно в указанном выше соединении фрагмент выбирают из , , , , или ,
где y равно 0, 1, 2, 3 или 4; p равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила,
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси,
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2' независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb; где Ra и Rb независимо друг от друга выбирают из водорода или C1-C6 алкила; Предпочтительно в указанном выше соединении фрагмент представляет собой или ;
где:
y равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила;
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2' независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb;
где Ra и Rb каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
Предпочтительно в указанном выше соединении фрагмент представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
Предпочтительно в указанном выше соединении фрагмент выбирают из , , , , , , или , предпочтительно ; где
y равно 0, 1, 2, 3 или 4;
p равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из галогена, гидроксила, амино, C1-C6 алкила или галогенированного C1-C6 алкила;
каждый Rt независимо выбирают из C1-C6 алкила, галогенированного C1-C6 алкила или C1-C6 алкокси;
R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила, галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила;
R2' независимо выбирают из водорода, -SO2NH2 или -C(O)NRaRb, предпочтительно -C(O)NH2;
где Ra и Rb независимо друг от друга выбирают из водорода или C1-C6 алкила;
RB выбирают из водорода или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила.
Предпочтительно фрагмент и фрагмент одинаковые или разные.
Предпочтительно фрагмент и фрагмент одинаковы, и каждый из них независимо друг от друга представляет собой , где R5 выбирают из водорода, C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила, предпочтительно - из метила, этила, пропила, бутила, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3 или аллила.
Предпочтительно L выбирают из C4-C6 алкенилена, замещенного 0-4 Rn, C4-C6 алкилена, замещенного 0-4 Rn, где Rn выбирают из галогена, гидроксила, амино, C1-C6 алкила, C1-C6 алкокси, C1-C6 алкиламино или галогенированного C1-C6 алкила.
Предпочтительно в указанном выше соединении RB выбирают из водорода или -ORf;
Rf выбирают из водорода, C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Rh выбирают из галогена, -ORj, -NRjRk, -C(O)Rj, -CO2Rk, -C(O)NRjRk, -NRjC(O)Rk, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, замещенного 0-4 Ri;
Ri выбирают из галогена, гидроксила, амино или C1-C6 алкила;
Rj и Rk каждый независимо друг от друга выбирают из водорода или C1-C6 алкила; более предпочтительно RB выбирают из H, метокси, , , , или .
Соли, которые могут быть образованы соединением по настоящему изобретению, также входят в объем настоящего изобретения. Если не указано иное, соединение по настоящему изобретению включает его соли. Термин «соль», используемый в настоящем документе, относится к соли, образованной в форме кислоты или основания из неорганической или органической кислоты и основания, а также случаи, когда соединение по настоящему изобретению содержит фрагмент основания, который включает, помимо прочего, пиридин или имидазол, когда оно содержит фрагмент кислоты, который включает, помимо прочего, карбоновую кислоту. Потенциально образующийся цвиттер-ион («внутренняя соль») также включен в термин «соль». Фармацевтически приемлемая (т.е. нетоксичная, физиологически приемлемая) соль является предпочтительной, хотя другие соли также полезны и могут быть использованы, например, на этапах разделения или очистки в процессе производства. Соединение по настоящему изобретению может образовывать соль, например, соединение I реагирует с определенным количеством (например, эквивалентным количеством) кислоты или основания, и осаждается в среде, или лиофилизируется в водном растворе.
Фрагмент основания, содержащийся в соединениях по настоящему изобретению, включает, помимо прочего, амины или пиридиновые или имидазольные кольца и может образовывать соль с органической или неорганической кислотой. Типичные кислоты, которые могут образовывать соли, включают ацетат (например, ацетат или тригалогенированную уксусную кислоту, например, трифторуксусную кислоту), адипат, альгинат, аскорбат, аспартат, бензоат, бензолсульфонат, дисульфат, борат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диэтиленгликолят, лаурилсульфат, этансульфонат, фумарат, глюцептат, глицерофосфат, гемисульфат, энантат, капроат, гидрохлорид, гидробромид, гидриодат, изетионат (например, 2-гидрокси-этэсульфонат), лактат, малеат, мезилат, нафталинсульфонат (например, 2-нафталинсульфонат), никотинат, нитрат, оксалат, пектат, персульфат, фенилпропионат (например, 3-фенилпропионат), фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат (например, образованный серной кислотой), сульфонат, тартрат, тиоцианат, толуолсульфонат (например, тозилат), додеканоат и др.
Некоторые соединения по настоящему изобретению могут содержать кислотные фрагменты, включая, помимо прочего, карбоновую кислоту, которая может образовывать соли с различными органическими или неорганическими основаниями. Соли, образованные типичными основаниями, включают аммониевую соль, соль щелочного металла (например, соли натрия, лития и калия), соль щелочноземельного металла (например, соли кальция и магния), и соль, образованную органическими основаниями (например, органическими аминами), такими как бензатин, дициклогексиламин, гидрабамин (соль, образованная N,N-бис(дегидроабиетил)этилендиамином), N-метил-D-глюканамин, N-метил-D-глюкоамид, трет-бутиламин, и соль, образованная аминокислотами, такими как аргинин, лизин и др. Основные азотсодержащие группы могут образовывать четвертичные аммониевые соли с галогенидами, такими как низкомолекулярные алкилгалогениды (например, хлориды, бромиды и йодиды метила, этила, пропила и бутила), диалкилсульфаты (например, диметил-, диэтил-, дибутил- и дипентилсульфаты), длинноцепочечные галогениды (например, хлориды, бромиды и йодиды децила, додецила, тетрадецила и тетрадецила), аралкилгалогениды (например, бромиды бензила и фенила) и др.
Пролекарство и сольват соединения по настоящему изобретению также входят в объем настоящего изобретения. Термин «пролекарство» в настоящем документе относится к соединению, полученному в результате химического преобразования в метаболическом или химическом процессе для получения соединения, соли или сольвата по настоящему изобретению для лечения соответствующего заболевания. Соединения по настоящему изобретению включают сольваты, такие как гидраты.
Соединение, соль или сольват по настоящему изобретению могут присутствовать в таутомерных формах, таких как амид и иминоэфир. Все эти таутомеры входят в объем настоящего изобретения.
Стереизомеры всех соединений (например, асимметричные атомы углерода, которые могут присутствовать вследствие различных замещений), включая их энантиомерные формы и неэнантиомерные формы, также включены в объем правовой охраны настоящего изобретения. Независимый стереоизомер в настоящем изобретении может не сосуществовать с другими изомерами (например, как чистый или практически чистый оптический изомер с особой активностью), или может быть смесью (например, рацематом), или смесью, образованной со всеми другими стереоизомерами или их частью. Хиральный центр соединения по настоящему изобретению имеет две конфигурации - S или R, которые определены Международным союзом теоретической и прикладной химии (IUPAC), основанным в 1974 году. Форма рацемизации может быть решена физическими методами, такими как фракционная кристаллизация, или разделительная кристаллизация путем разделения на диастереомеры, или разделение с помощью хиральной колоночной хроматографии. Индивидуальный оптический изомер может быть получен из рацемата соответствующими методами, включая, помимо прочего, традиционные методы, такие как перекристаллизация после образования солей с оптически активными кислотами.
Массовое содержание соединения по настоящему изобретению, полученного путем синтеза, разделения и очистки поочередно, равно или больше 90 %, например, равно или больше 95 %, равно или больше 99 % («очень чистое» соединение), и указано в текстовом описании. «Очень чистое» соединение по настоящему изобретению также является частью настоящего изобретения.
В объем изобретения входят все конфигурационные изомеры соединения по настоящему изобретению, как в смеси, так и в чистом или очень чистом виде. Определение соединения настоящего изобретения включает цис (Z) и транс (E) изомеры олефинов, а также цис и транс изомеры карбоциклических и гетероциклических соединений.
Во всей спецификации группы и заместители могут быть выбраны для получения стабильных фрагментов и соединений.
Специфические функциональные группы и определения химических терминов подробно представлены ниже. Для целей настоящего изобретения химические элементы соответствуют Периодической таблице элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed. Также приведены определения конкретных функциональных групп. Кроме того, основные принципы органической химии, а также специфические функциональные группы и реакционная способность описаны в издании Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito: 1999, все содержание которого включено в настоящий документ посредством ссылки.
Некоторые соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение включает все соединения, включая их цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, изомеры (D) типа, изомеры (L) типа, рацемические смеси и другие смеси. Кроме того, асимметричный атом углерода может входить в состав заместителя, например, алкила. Все изомеры и их смеси включены в настоящее изобретение.
Согласно изобретению, смеси изомеров могут содержать различные соотношения изомеров. Например, смеси, содержащие только два изомера, могут иметь следующие комбинации: 50:50, 60:40, 70:30, 80:20, 90:10, 95:5, 96:4, 97:3, 98:2, 99:1 или 100:0, все соотношения изомеров входят в объем настоящего изобретения. Сходные соотношения, понятные специалистам в данной области, и соотношения для смесей более сложных изомеров также входят в объем настоящего изобретения.
Изобретение также включает соединения, меченные изотопами, которые эквивалентны исходным соединениям, раскрытым в настоящем изобретении. Однако на самом деле это обычно происходит, когда один или несколько атомов заменяют на атомы с другим атомным весом или массовым числом. Примеры изотопов соединений, которые могут быть указаны в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H,13C, 11C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl. Соединение, или энантиомер, диастереомер, изомер, фармацевтически приемлемая соль или сольват вышеуказанного соединения, содержащий изотопы или другие изотопные атомы, входят в объем изобретения. В настоящее изобретение также включены некоторые соединения, меченные изотопами, такие как радиоактивные изотопы 3H и 14C, которые могут использоваться в экспериментах по изучению распределения лекарственных препаратов и субстратов в тканях. Тритий (3H) и углерод-14 (14C), которые относительно легко получить и обнаружить, и являются предпочтительным выбором. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т. е. 2H, имеют преимущества в некоторых видах терапии благодаря своей хорошей метаболической стабильности, например, увеличенный период полураспада или уменьшенная доза in vivo, и поэтому могут быть предпочтительными в определенных ситуациях. Меченные изотопами соединения могут быть получены обычными методами путем замены легкодоступных меченных изотопами реактивов на не содержащие изотопов реактивы, которые могут быть получены по раскрытой схеме, представленной в Примере.
Если необходимо разработать способ синтеза конкретного энантиомера соединения по настоящему изобретению, его можно получить путем асимметрического синтеза или дериватизировать хиральным вспомогательным реагентом, разделить полученную диастереомерную смесь и удалить хиральный вспомогательный реактив для получения чистого энантиомера. Кроме того, если молекула содержит функциональную группу основания, такую как аминокислота, или функциональную группу кислоты, такую как карбоксильная группа, соль диастереомера может быть образована с подходящими оптически активными кислотами или основаниями, которые могут быть разделены традиционными средствами, такими как разделительная кристаллизация или хроматография, для получения чистого энантиомера.
Как описано в настоящем документе, соединение по настоящему изобретению может быть замещено любым количеством заместителей или функциональных групп для расширения его области применения. В целом, присутствует ли термин «замещенный» до или после термина «по выбору», общая формула, включающая заместители в соединение настоящего изобретения, означает замещение указанного структурного заместителя на водородный радикал. Когда несколько положений в конкретной структуре замещаются несколькими конкретными заместителями, они могут быть одинаковыми или разными во всех положениях. Термин «замещенный», используемый в настоящем документе, включает все заместители, которые могут встречаться в органических соединениях. В широком смысле допустимые заместители включают нециклические, циклические, разветвленные, неразветвленные, карбоциклические и гетероциклические, ароматические кольца и неароматические органические соединения. В настоящем изобретении, например, имеется гетероатомный азот, его валентное состояние может быть дополнено водородным заместителем или любым допустимым органическим соединением, описанным выше. Кроме того, изобретение непреднамеренно ограничивается любым образом замещенными органическими соединениями. В настоящем изобретении считается, что сочетание заместителей и вариабельных групп в форме стабильных соединений хорошо подходит для лечения заболеваний. Термин «стабильный» в настоящем документе относится к соединению, которое достаточно стабильно для сохранения целостности своей структуры в течение достаточно длительного времени, предпочтительно эффективному в течение достаточно длительного времени при использовании для вышеуказанных целей, описанных в настоящем изобретении.
Метаболиты соединений и их фармацевтически приемлемых солей по настоящей заявке, а также пролекарства, которые могут быть преобразованы in vivo в соединения и их фармацевтически приемлемые соли по настоящей заявке, также включены в формулу изобретения.
Способ получения соединения
Способы получения соединения по формуле I описаны в следующих схемах и примерах. Сырье и промежуточные продукты закупают из коммерческих источников, их производство осуществляется известными способами или описано иным образом. В некоторых случаях последовательность этапов для выполнения схемы реакции может быть изменена, чтобы облегчить реакцию или избежать образования нежелательных побочных продуктов реакции.
Как правило, в процессе получения каждую реакцию обычно проводят в инертном растворителе при температуре от комнатной до температуры обратного холодильника, например, от 0°C до 150°C, предпочтительно от 10°C до 100°C. Время реакции обычно составляет 0,1-60 часов, предпочтительно 0,5-48 часов.
Предпочтительно соединение по формуле I настоящего изобретения может быть получено следующим образом:
(i) в инертном растворителе (например, н-бутанол), в присутствии основания (N,N-диизопропилэтиламина), сырье 1a реагирует с моно-БОК-защищенным диамином посредством реакции нуклеофильного замещения с образованием соединения 1b;
(ii) в инертном растворителе (например, этилацетат) в присутствии кислоты (хлористоводородной кислоты) снимается БОК-защита из соединения 1b с образованием промежуточного соединения 1c;
(iii) в инертном растворителе (например, н-бутанол) в присутствии основания (N,N-диизопропилэтиламина) промежуточное соединение 1c вступает в реакцию замещения с с образованием промежуточного соединения 1d;
(iv) в инертном растворителе (метанол) в присутствии восстановителя (натрия дитионит) промежуточное соединение 1d реагирует с образованием соединения 1e;
(v) в инертном растворителе (например N,N-диметилформамид и/или диоксан), в присутствии катализатора (например, EDCI) соединение 1e реагирует с или с образованием целевого соединения I;
в данной формуле A, B, L, R1, R2, R3, R4, RB, R2', R3' и R4' определены выше.
Предпочтительно соединение по настоящему изобретению может быть получено следующим образом:
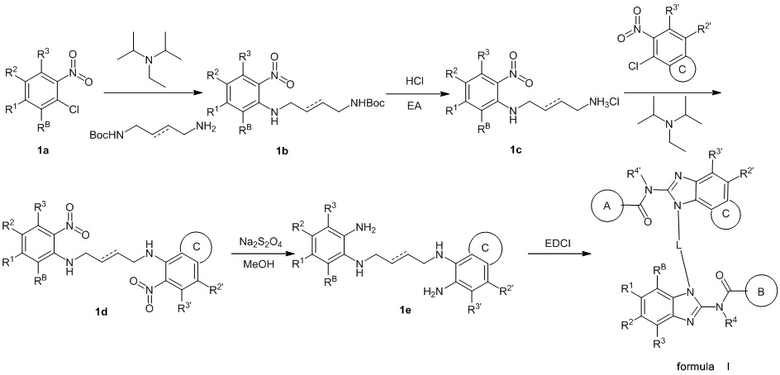
формула I
Сырье 1a реагирует с моно-БОК-защищенным бутандиамином или транс-бутен-1,4-диамином посредством реакции нуклеофильного замещения в присутствии N,N-диизопропилэтиламина в н-бутаноле с образованием соединения 1b, затем снимается БОК-защита в растворе хлористоводородной кислоты в этилацетате с образованием промежуточного соединения 1c, которое затем реагирует с  посредством реакции замещения в присутствии N,N-диизопропилэтиламина в н-бутаноле с образованием промежуточного соединения 1d, которое восстанавливается натрия дитионитом в метаноле с образованием соединения 1e, а затем конденсируется с различными изотиоцианатами ( или ) в присутствии EDCI с получением целевого соединения;
посредством реакции замещения в присутствии N,N-диизопропилэтиламина в н-бутаноле с образованием промежуточного соединения 1d, которое восстанавливается натрия дитионитом в метаноле с образованием соединения 1e, а затем конденсируется с различными изотиоцианатами ( или ) в присутствии EDCI с получением целевого соединения;
где L представляет собой , а представляет собой одинарную или двойную связь;
A, B, R1, R2, R3, R4, RB, C, R2', R3' и R4' определены выше.
Фармацевтическая композиция и способ применения
Поскольку соединения по настоящему изобретению обладают превосходной агонистической активностью в отношении STING киназы, соединение по настоящему изобретению, его стереоизомер или оптический изомер, фармацевтически приемлемая соль, пролекарства или его сольваты, и фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве основных действующих веществ, могут быть использованы для профилактики и/или лечения (стабилизации, облегчения симптомов или излечения) заболеваний связанных с киназой STING, таких как воспалительные заболевания (угри обыкновенные, бронхиальная астма, целиакия, хронический простатит, гломерулонефрит, воспалительные заболевания кишечника, воспалительные заболевания органов малого таза, реперфузионное повреждение, ревматоидный артрит, саркоидоз, васкулит, воспаление дыхательных путей, вызванное клещами домашней пыли, и интерстициальный цистит), аутоиммунные заболевания, инфекционные заболевания (например, инфекционные заболевания, вызванные вирусом гепатита В, вирусом папилломы человека, вирусом Эпштейна-Барр, ассоциированным с назофарингеальной карциномой и др.), онкологические заболевания (например, рак толстой кишки, рак головного мозга, рак молочной железы, фибросаркома и плоскоклеточная карцинома, меланома, рак молочной железы, рак толстой кишки, рак легких и рак яичников), заболевания, связанные с предраковым синдромом.
Фармацевтическая композиция настоящего изобретения включает безопасное и эффективное количество соединения по настоящему изобретению и фармацевтически приемлемое вспомогательное вещество или носитель. В данном случае «Безопасное и эффективное количество» относится к такому количеству соединения, которого достаточно для существенного улучшения состояния без появления серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения по настоящему изобретению/доза, а предпочтительно содержит 10-200 мг соединения по настоящему изобретению/доза. Предпочтительно «одна доза» содержится в капсуле или таблетке.
«Фармацевтически приемлемый носитель» относится к одному или нескольким совместимым твердым или жидким наполнителям или гелеобразным веществам, которые подходят для использования на людях и которые должны иметь достаточную чистоту и достаточно низкую токсичность. Термин «совместимость» в настоящем изобретении относится к способности каждого компонента композиции смешиваться с соединением по настоящему изобретению и смешиваться друг с другом без заметного снижения эффективности соединения. Примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза натрия, ацетат целлюлозы и др.), желатин, тальк, твердый смазочный материал (такой как стеариновая кислота или магния стеарат), кальция сульфат, растительное масло (такое как соевое масло, кунжутное масло, арахисовое масло, оливковое масло и др.), полиол (такой как пропиленгликоль, глицерин, маннит, сорбит и др.), эмульгатор (такой как Твин®), смачивающий агент (такой как натрия лаурилсульфат), краситель, ароматизатор, стабилизатор, антиоксидант, консервант или свободная от пирогенов вода и др.
Для соединения или фармацевтических композиций по настоящему изобретению не существует специальных ограничений по способу введения, и репрезентативный способ введения включает, помимо прочего, пероральное и парентеральное (внутривенное, внутримышечное или подкожное) введение.
Твердые лекарственные формы для приема внутрь включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах действующие вещества смешивают как минимум с одним традиционным инертным вспомогательным веществом (или носителем), таким как натрия цитрат или дикальцийфосфат, или смешивают с любым из следующих компонентов: (а) наполнители или вещества для улучшения совместимости, такие как крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие вещества, например, гидроксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и аравийская камедь; (с) увлажнители, такие как глицерин; (d) разрыхлители, такие как агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (e) агенты, замедляющие растворение, такие как парафин; (f) ускорители абсорбции, например, четвертичные аммониевые соединения; (g) смачивающие агенты, такие как цетиловый спирт и глицерилмоностеарат; (h) адсорбенты, например, каолин; и (i) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смесь. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные вещества.
Твердые лекарственные формы, такие как таблетки, сахарные пилюли, капсулы, таблетки и гранулы, могут быть получены с использованием материалов покрытия и оболочки, таких как кишечнорастворимая оболочка и любые другие материалы, известные в данной области. Они могут содержать замутнитель. Высвобождение действующих веществ или соединений в композициях может происходить замедленно в определенной части пищеварительного тракта. Примеры компонентов состава включают полимеры и воски. При необходимости действующие вещества и одно или несколько вышеуказанных вспомогательных веществ могут образовывать микрокапсулы.
Жидкие лекарственные формы для приема внутрь включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. В дополнение к действующим веществам жидкие лекарственные формы могут содержать любые традиционные инертные разбавители, известные в данной области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, или их комбинации.
Помимо этих инертных разбавителей, композиция может содержать такие добавки, как смачивающие агенты, эмульгаторы и суспендирующий агент, подсластитель, ароматизаторы и отдушки.
Помимо действующих веществ, суспензия может содержать суспендирующий агент, например, этоксилированный изооктадеканол, полиоксиэтилен сорбитол и эфиры сорбитана, микрокристаллическую целлюлозу, метанол алюминий и агар, или их комбинацию.
Композиции для парентерального введения могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, которые могут быть повторно растворены в стерильных растворах или дисперсиях для инъекций. Подходящие водные и неводные носители, разбавители, растворители или наполнители включают в себя воду, этанол, многоатомные спирты и любые их подходящие смеси.
Соединения по настоящему изобретению можно применять отдельно или в комбинации с другими фармацевтически приемлемыми соединениями (например, агонистами STING).
При комбинированном применении фармацевтическая композиция дополнительно включает одно или более (2, 3, 4 или более) других фармацевтически приемлемых соединений (например, агонисты STING). Одно или несколько (2, 3, 4 или более) других фармацевтически приемлемых соединений могут быть использованы одновременно, отдельно или последовательно с соединением по настоящему изобретению для профилактики и/или лечения заболеваний, связанных с активностью или экспрессией киназы STING.
При использовании фармацевтических композиций безопасное и эффективное количество соединения по настоящему изобретению вводят млекопитающему (например, человеку), нуждающемуся в нем, при этом вводимая доза является фармацевтически эффективной. Для человека с массой тела 60 кг суточная доза обычно составляет 1-2000 мг, предпочтительно 20-500 мг. Конечно, конкретная доза также должна зависеть от различных факторов, таких как способ введения, состояние здоровья пациента, оценку которого может произвести опытный врач.
Основные преимущества настоящего изобретения включают в себя:
1. Соединение по настоящему изобретению имеет новую структуру и обладает высокой способностью активировать киназу STING.
2. Из соединения по настоящему изобретению можно получить агонист STING, который обладает более высокой эффективностью и лучшими фармакокинетическими характеристиками.
3. Соединение, полученное по настоящему изобретению, обладает лучшим или эквивалентным активирующим действием на STING, чем положительные контроли (ADU-S100, соединение GSK 3 или соединение GSK 1) в макрофагах (таких как клетки THP1-Dual, клетки Raw-lucia и др.), предоставляя новый вариант для клинического скрининга и/или получения лекарственных препаратов для лечения заболеваний, связанных с активностью STING.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что эти примеры предназначены только для иллюстрации настоящего изобретения, а не для ограничения объема настоящего изобретения. Экспериментальные способы без конкретных условий в последующих примерах обычно предусматривают стандартные условия или условия, предложенные производителем. Если не указано иное, доли и проценты рассчитаны по массе.
Материалы и реагенты, используемые для проведения экспериментов в следующих примерах, могут быть получены из коммерческих источников, если нет специальных инструкций.
ПРИМЕР
Структуру соединения определяют с помощью ядерного магнитного резонанса (1H-ЯМР) или масс-спектрометрии (МС). ЯМР-спектр регистрируют с использованием прибора для ядерного магнитного резонанса типа BrukerAV-300. Растворитель для определения - дейтерированный диметилсульфоксид (ДМСО-D6) или дейтерированный хлороформ (CDCl3), внутренний стандарт - TMS.
Получение соединения по изобретению.
Синтез промежуточного соединения 1: 1-этил-3-метил-1Н-пиразол-5-карбонилизотиоцианат
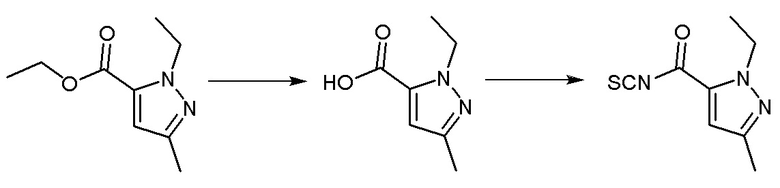
Этил 1-этил-3-метил-1H-пиразол-5-карбоксилат (1 г, 5,49 ммоль) растворяли в смеси метанола (10 мл) и воды (5 мл). После перемешивания при комнатной температуре в течение десяти минут к вышеуказанному реакционному раствору добавляли лития гидроксид (0,263 г, 10,98 ммоль). Реакция в реакционном растворе продолжалась при комнатной температуре в течение 3 часов, после чего проводили перегонку метанола для удаления при пониженном давлении, а pH доводили до 5-6 с использованием разбавленной хлористоводородной кислоты, фильтровали с использованием аспирации для получения твердого вещества белого цвета, которое не очищали, а непосредственно использовали в следующей реакции.
1-Этил-3-метил-1H-пиразол-5-карбоновую кислоту (0,8 г, 5,19 ммоль) растворяли в растворе дихлорметана (5 мл), по каплям добавляли 3 капли N,N-диметилформамида, перемешивали в течение 10 минут на ледяной бане. Оксалилхлорид (0,527 мл, 6,23 ммоль), разбавленный в дихлорметане (5 мл), медленно добавляли в вышеуказанный реакционный раствор. После перемешивания на ледяной бане в течение десяти минут ледяная баню удаляли, и реакционная смесь реагировала при комнатной температуре в течение двух часов. Реакцию контролировали методом ТСХ до ее завершения, затем реакционный раствор концентрировали перегонкой при пониженном давлении, а остаток использовали для следующей реакции. Калия тиоцианат (0,61 г, 6,26 ммоль) растворяли в растворе ацетона (10 мл) на ледяной бане и перемешивали на ледяной бане в течение 10 минут. Остаток, разбавленный ацетоном (5 мл), медленно добавляли в вышеуказанный реакционный раствор, и реакцию продолжали в течение 30 минут на ледяной бане. Реакцию контролировали методом ТСХ до завершения, к вышеуказанному реакционному раствору добавляли н-гексан (15 мл), объем раствора перегоняли и концентрировали до 1/2 под пониженным давлением, затем добавляли н-гексан (15 мл), объем раствора перегоняли и концентрировали до 1/2 под пониженным давлением. Раствор фильтровали путем аспирации при пониженном давлении, фильтрат собирали для получения осадка, отделяли и очищали колоночной хроматографией (петролейный эфир:этилацетат = 6:1) с получением бесцветного прозрачного маслянистого соединения - промежуточного соединения 1. МС (ИЭР) m/z = 196[M+H]+; 1H-ЯМР (300 МГц, CDCl3) δ 6,76 (синглет, 1H), 4,52 (синглет, 2H), 2,32 (синглет, 3H), 1,42 (синглет, 3H).
Синтез промежуточного соединения 2: 1-пропаргил-3-метил-1Н-пиразол-5-карбонилизотиоцианат
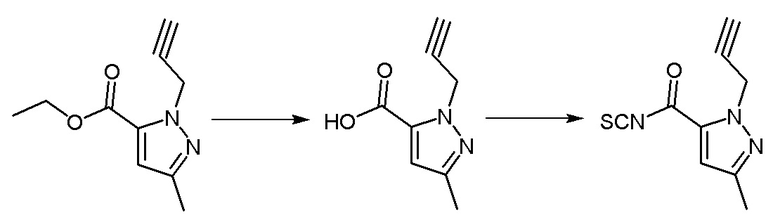
Операционные процедуры и условия реакции были такими же, как для промежуточного соединения 1, за исключением того, что исходный материал представлял собой этил-1-пропаргил -3-метил-1H-пиразол-5-карбоксилат, МС (ИЭР) m/z = 206[M+H]+.
Синтез промежуточного соединения 3: 1-аллил-3-метил-1Н-пиразол-5-карбонилизотиоцианат
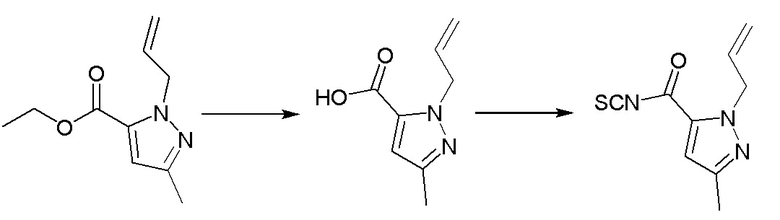
Операционные процедуры и условия реакции были такими же, как для промежуточного соединения 1, за исключением того, что исходный материал представлял собой этил-1-аллил-3-метил -1Н-пиразол-5-карбоксилат, МС (ИЭР) m/z = 208[M+H]+.
Синтез промежуточного соединения 4: 1-(циклопропилметил)-3-метил-1Н-пиразол-5-карбонил-изотиоцианат
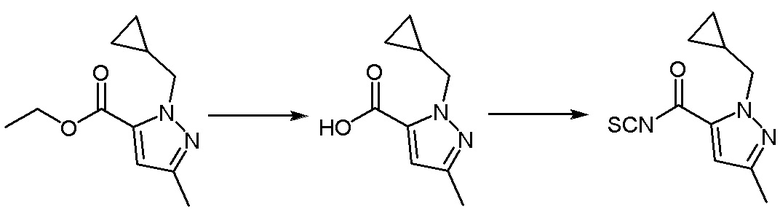
Операционные процедуры и условия реакции были такими же, как для промежуточного соединения 1, за исключением того, что исходный материал представлял собой этил-1-(циклопропилметил)-3-метил-1Н-пиразол-5-карбоксилат, МС (ИЭР) m/z = 222[M+H]+.
Синтез промежуточного соединения 5: 3-метил-1-(2,2,2-трифторэтил)-1Н-пиразол-5-карбонилизотиоцианат
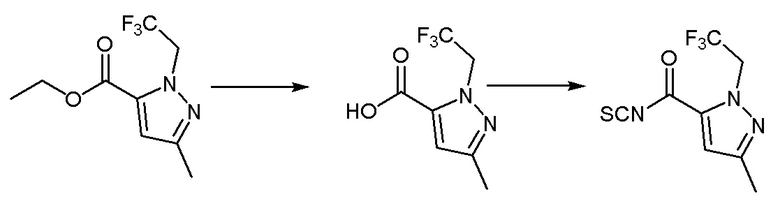
Операционные процедуры и условия реакции были такими же, как для промежуточного соединения 1, за исключением того, что исходный материал представлял собой этил-3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-5-карбоксилат, МС (ИЭР) m/z = 250[M+H]+.
Синтез промежуточного соединения 6: 1-(2-фторэтил)-3-метил-1Н-пиразол-5-карбонилизотиоцианат
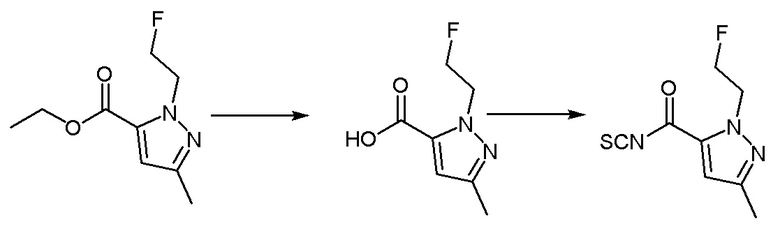
Операционные процедуры и условия реакции были такими же, как для промежуточного соединения 1, за исключением того, что исходный материал представлял собой этил-1-(2-фторэтил)-3-метил-1Н-пиразол-5-карбоксилат, МС (ИЭР) m/z = 214[M+H]+.
Синтез промежуточного соединения 7: 4-хлор-3-метокси-5-нитробензамид
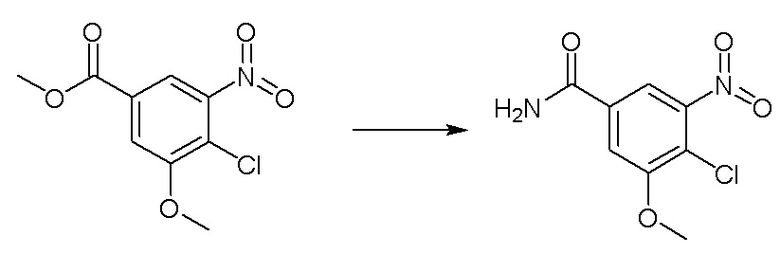
Метил-4-хлор-3-метокси-5-нитробензоат (4 г, 16,3 ммоль) диспергировали в водном растворе аммиака (20 мл), нагревали до 50°C и проводили реакцию в течение ночи. На следующий день проверяли завершение реакции методом ТСХ. Реакционную смесь фильтровали путем аспирации, промывали чистой водой (10 мл) и получали промежуточное соединение 7 в виде твердого вещества желтоватого цвета. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,31 (синглет, 1H), 8,05 (дублет, J = 2,1 Гц, 1H), 7,85 (дублет, J = 21,0 Гц, 2H), 4,02 (дублет, J = 2,1 Гц, 3H).
Синтез промежуточного соединения 8: 4-хлор-3-гидрокси-5-нитробензамид
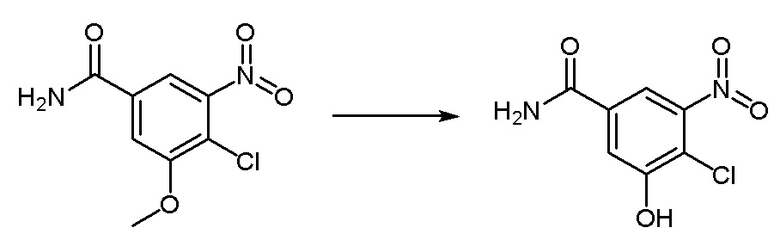
4-Хлор-3-метокси-5-нитробензамид (4 г, 8,67 ммоль) диспергировали в растворе дихлорметана (30 мл), перемешивали при комнатной температуре в течение 10 минут. 1 моль/л раствор бора трибромида в растворе дихлорметана (52 мл, 52 ммоль) медленно добавляли в вышеуказанный реакционный раствор и продолжали перемешивать при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Вышеуказанный реакционный раствор медленно добавляли в ледяную воду (100 мл). После добавления реакционный раствор продолжали перемешивать в течение двух часов, фильтровали путем аспирации с получением промежуточного соединения 8 в виде твердого вещества серо-белого цвета. 1H-ЯМР (300 МГц, ДМСО-d6) δ 11,51 (синглет, 1H), 8,16 (синглет, 1H), 7,92 (синглет, 1H), 7,72 (синглет, 1H), 7,65 (синглет, 1H).
Синтез промежуточного соединения 9: 3-(3-((трет-бутилдиметилсилил)окси)пропокси)-4-хлор-5-нитробензамид
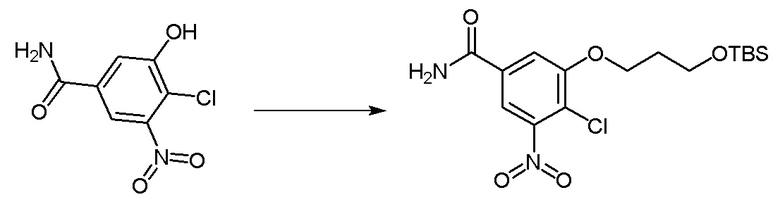
4-Хлор-3-гидрокси-5-нитробензамид (1 г, 4,62 ммоль) растворяли в растворе N-диметилформамида (10 мл), затем к указанному раствору добавляли (3-бромпропокси)(трет-бутил)диметилсилан (1,29 мл, 5,54 ммоль) и калия карбонат (0,83 г, 6,0 ммоль) соответственно. Реакционный раствор перемешивали при комнатной температуре в течение десяти минут, затем нагревали до 80°C на масляной бане и проводили реакцию в течение ночи. Завершение реакции контролировали методом ТСХ. Вышеуказанный реакционный раствор добавляли в воду (20 мл), а затем добавляли этилацетат (20 мл). Органическую фазу отделяли, дважды экстрагировали этилацетатом (20 мл), органические фазы объединяли и высушивали над безводным натрия сульфатом. Его концентрировали при пониженном давлении для получения осадка, отделяли и очищали колоночной хроматографией (петролейный эфир:этилацетат = 8:1-3:1) с получением промежуточного соединения 9 в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, ДМСО) δ 8,30 (синглет, 1H), 8,09-7,98 (мультиплет, 1H), 7,88 (триплет, J = 9,0 Гц, 1H), 7,79 (синглет, 1H), 4,30 (триплет, J = 5,9 Гц, 2H), 3,80 (двойной дублет, J = 7,8, 4,2 Гц, 2H), 1,97 (двойной дублет, J = 12,0, 6,0 Гц, 2H), 0,84 (синглет, 9H), 0,07-0,04 (мультиплет, 6H).
Синтез промежуточного соединения 10: 4-хлор-3-нитро-5-(оксетан-3-илокси)бензамид
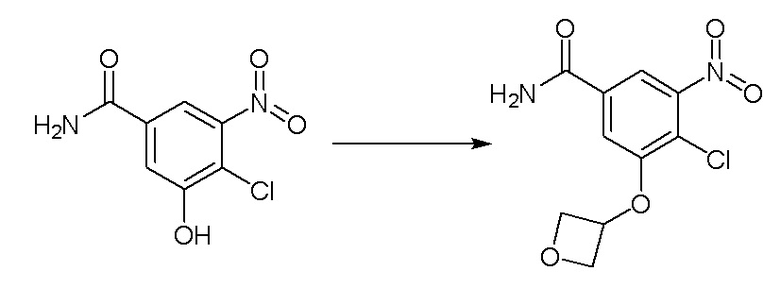
4-Хлор-3-гидрокси-5-нитробензамид (1 г, 4,62 ммоль) растворяли в растворе N,N-диметилформамида (10 мл), затем к указанному раствору добавляли 3-йодоксетан (0,48 мл, 5,54 ммоль) и калия карбонат (1,28 г, 9,23 ммоль) соответственно. После перемешивания при комнатной температуре в течение 10 минут реакционный раствор переносили на масляную баню для нагревания до 80°C и проводили реакцию в течение ночи. Завершение реакции контролировали методом ТСХ. Вышеуказанный реакционный раствор добавляли в воду (20 мл), затем добавляли этилацетат (20 мл), органическую фазу отделяли, дважды экстрагировали этилацетатом (20 мл), органические фазы объединяли и высушивали над безводным натрия сульфатом. Его концентрировали при пониженном давлении для получения осадка, отделяли и очищали колоночной хроматографией (петролейный эфир:этилацетат = 8:1-3:1) с получением промежуточного соединения 10 в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, ДМСО) δ 8,29 (синглет, 1H), 8,11 (дублет, J = 1,7 Гц, 1H), 7,89-7,78 (мультиплет, 1H), 7,45 (дублет, J = 1,6 Гц, 1H), 5,56 (двойной триплет, J = 10,3, 5,2 Гц, 1H), 5,00 (двойной дублет, J = 13,4, 7,4 Гц, 2H), 4,63 (двойной дублет, J = 7,4, 4,6 Гц, 2H).
Синтез промежуточного соединения 11: 4-хлор-2-гидрокси-5-нитробензамид
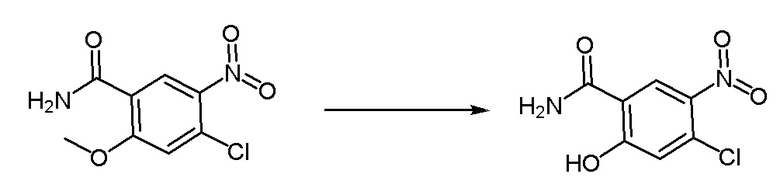
4-Хлор-2-гидрокси-5-нитробензамид (4 г, 8,67 ммоль) диспергировали в растворе дихлорметана (30 мл), перемешивали при комнатной температуре в течение 10 минут. 1 моль/л раствор бора трибромида в растворе дихлорметана (52 мл, 52 ммоль) медленно добавляли в вышеуказанный реакционный раствор и продолжали перемешивать при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Вышеуказанный реакционный раствор медленно добавляли в ледяную воду (100 мл). После добавления реакционный раствор продолжали перемешивать в течение двух часов, фильтровали путем аспирации с получением промежуточного соединения 11 в виде твердого вещества серо-белого цвета. 1H-ЯМР (400 МГц, ДМСО) δ 14,11 (синглет, 1H), 8,77 (синглет, 1H), 8,69 (синглет, 1H), 8,33 (синглет, 1H), 7,27 (синглет, 1H).
Синтез промежуточного соединения 12: 4-хлор-2-метил-5-нитробензофуран-7-формамид
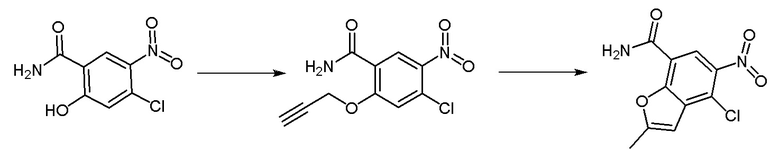
4-Хлор-2-гидрокси-5-нитробензамид (2,4 г, 11,08 ммоль) растворяли в растворе N,N-диметилформамида (10 мл), перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли 3-бромпропин (1,04 мл, 13,3 ммоль) и калия карбонат (3,06 г, 22,16 ммоль). Реакционный раствор переносили на масляную баню для нагревания до 80°C и проводили реакцию в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционный раствор охлаждали до комнатной температуры, затем реакционный раствор добавляли в воду (50 мл), перемешивали при комнатной температуре и фильтровали путем аспирации с получением твердого вещества светло-желтого цвета, которое использовали для следующей реакции. 1H-ЯМР (400 МГц, ДМСО) δ 8,42 (синглет, 1H), 7,91 (синглет, 1H), 7,76 (синглет, 1H), 7,57 (синглет, 1H), 5,16 (синглет, 2H), 3,79 (дублет, J = 2,1 Гц, 1H).
2-(Пропаргилокси)-4-хлор-5-нитробензамид (0,7 г, 2,75 ммоль) растворяли в растворе N,N-диэтиланилина (10 мл), добавляли цезия фторид (0,63 г, 4,12 ммоль), перемешивали при комнатной температуре в течение 10 минут, а затем переносили на масляную баню для нагревания до 220°C. После того, как реакционный раствор прореагировал в течение 4 часов, методом ТСХ подтверждено завершение реакции. Реакционный раствор охлаждали до комнатной температуры, добавляли этилацетат (30 мл) для разбавления реакционного раствора, затем промывали несколько раз 1 моль/л разбавленным раствором хлористоводородной кислоты и высушивали над безводным натрия сульфатом. Реакционный раствор перегоняли при пониженном давлении, концентрировали до получения осадка и очищали колоночной хроматографией (петролейный эфир:этилацетат = 5:1-1:1) с получением промежуточного соединения 12 в виде твердого вещества светло-желтого цвета. 1H-ЯМР (400 МГц, ДМСО) δ 8,29 (синглет, 1H), 8,07 (синглет, 1H), 7,95 (синглет, 1H), 6,97 (синглет, 1H), 2,57 (синглет, 3H).
Синтез промежуточного соединения 13: 4-хлор-2-метил-5-нитробензофуран-7-формамид
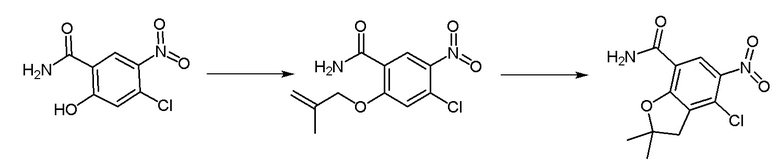
4-Хлор-2-гидрокси-5-нитробензамид (2,4 г, 11,08 ммоль) растворяли в растворе N,N-диметилформамида (10 мл), перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли 3-бром-2-метилпропилен (1,34 мл, 13,3 ммоль) и калия карбонат (3,06 г, 22,16 ммоль), переносили на масляную баню, нагревали до 80°C и проводили реакцию в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционный раствор охлаждали до комнатной температуры, затем добавляли воду (50 мл), перемешивали при комнатной температуре и фильтровали путем аспирации с получением твердого вещества светло-желтого цвета, которое использовали для следующей реакции. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,53 (синглет, 1H), 7,83 (синглет, 2H), 6,53 (синглет, 1H), 5,20 (синглет, 1H), 5,00 (синглет, 1H), 4,50 (синглет, 2H), 3,76 (синглет, 3H), 1,81 (синглет, 3H).
4-Хлор-2-((2-метилаллил)окси)-5-нитробензамид (0,7 г, 2,59 ммоль) растворяли в растворе N,N-диэтиланилина (10 мл), добавляли цезия фторид (0,59 г, 3,88 ммоль), перемешивали при комнатной температуре в течение 10 минут, затем переносили на масляную баню и нагревали до 220°C. После того, как реакционный раствор прореагировал в течение 4 часов, методом ТСХ подтверждено завершение реакции. Реакционный раствор охлаждали до комнатной температуры, добавляли этилацетат (30 мл) для разбавления реакционного раствора, затем промывали несколько раз 1 моль/л разбавленным раствором хлористоводородной кислоты и высушивали над безводным натрия сульфатом. Реакционный раствор перегоняли при пониженном давлении, концентрировали до получения осадка и очищали колоночной хроматографией (петролейный эфир:этилацетат = 5:1-1:1) с получением промежуточного соединения 13 в виде твердого вещества светло-желтого цвета. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,52 (синглет, 1H), 7,48 (синглет, 2H), 3,81-3,72 (мультиплет, 3H), 2,89 (синглет, 2H), 1,49 (дублет, J = 1,8 Гц, 6H).
Пример 1
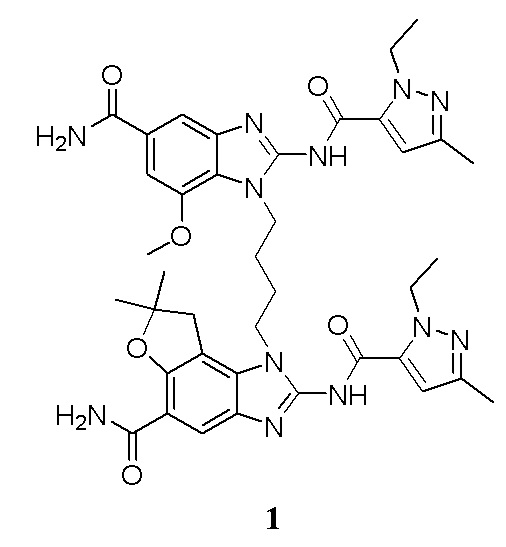
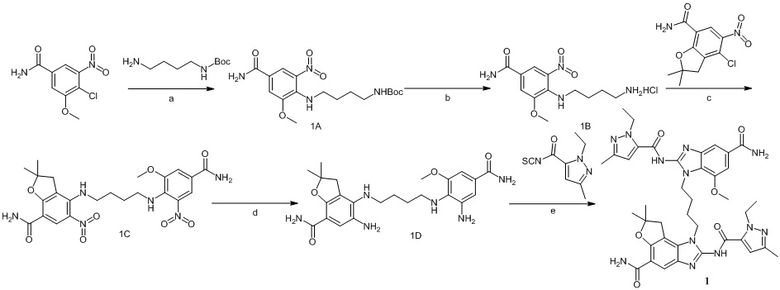
Этап a: синтез трет-бутил(4-((4-карбамоил-2-метокси-6-нитрофенил)амино)бутил)карбамата
4-Хлор-3-метокси-5-нитробензамид (1 г, 4,34 ммоль) растворяли в растворе н-бутанола (10 мл), перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли N-(трет-бутоксикарбонил)-1, 4-диаминобутан (1,1 мл, 5,64 ммоль) и N,N-диизопропилэтиламин (2,15 мл, 13,0 ммоль). Температуру в запечатанной пробирке повышали до 110°C, реакционный раствор реагировал в течение 12 часов. На следующий день реакционный раствор фильтровали путем аспирации и промывали соответствующим количеством этилацетата для получения трет-бутил(4-((4-карбамои-2-метокси-6-нитрофенил)амино)бутилкарбамата (соединение 1A) в виде твердого вещества светло-желтого цвета с выходом 89 %. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,23-8,17 (мультиплет, 1H), 8,02 (синглет, 1H), 7,72 (дублет, J = 5,9 Гц, 1H), 7,55 (синглет, 1H), 7,33 (синглет, 1H), 6,81 (синглет, 1H), 3,92-3,86 (мультиплет, 3H), 3,46 (дублет, J = 6,3 Гц, 2H), 2,89 (дублет, J = 6,4 Гц, 2H), 1,49 (синглет, 4H), 1,37-1,32 (мультиплет, 9H).
Этап b: синтез 4-((4-аминобутил)амино)-3-метокси-5-нитробензамида гидрохлорида
Соединение 1А (2 г, 5,23 ммоль) диспергировали в растворе этилацетата (20 мл), перемешивали на ледяной бане в течение 10 минут, и свежеприготовленный газообразный HCl вводили в указанный раствор до насыщения. После продолжения реакции в течение 10 минут на ледяной бане последнюю удаляли, и реакционный раствор реагировал при комнатной температуре в течение 2 часов. Завершение реакции контролировали методом ТСХ. Этилацетат удаляли путем перегонки при пониженном давлении. К остатку добавляли эфир (15 мл), перемешивали при комнатной температуре в течение двух часов и фильтровали путем аспирации с получением 4-((4-аминобутил)амино)-3-метокси-5-нитробензамида гидрохлорида (соединение 1B) в виде твердого вещества красно-коричневого цвета с выходом 93 %. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,21 (двойной дублет, J = 3,9, 2,0 Гц, 1H), 8,04 (синглет, 1H), 7,73 (синглет, 1H), 7,57 (дублет, J = 3,0 Гц, 1H), 7,34 (синглет, 1H), 3,89 (дублет, J = 4,0 Гц, 3H), 3,50 (синглет, 2H), 2,78 (синглет, 2H), 1,61-1,51 (мультиплет, 4H).
Этап c: синтез 4-((4-(4-карбамоил-2-метокси-6-нитрофенил)амино) бутил)амино)-2,2-диметил-5-нитро-2,3-дигидробензофуран-7-формамида
Соединение 4-((4-аминобутил)амино)-3-метокси-5-нитробензамида гидрохлорид (1 г, 3,14 ммоль) растворяли в растворе н-бутанола (10 мл) и перемешивали при комнатной температуре. Затем в вышеуказанный реакционный раствор добавляли 4-хлор-2,2-диметил-5-нитро-2,3-дигидробензофуран-7-формамид (1,02 г, 3,76 ммоль) и N,N-диизопропилэтиламин (1,56 мл, 9,41 ммоль). Температуру в запечатанной пробирке повышали до 110°C, и реакционный раствор реагировал в течение 12 часов. На следующий день реакционный раствор фильтровали путем аспирации и промывали соответствующим количеством этилацетата для получения 4-((4-((4-карбамоил-2-метокси-6-нитрофенил)амино)бутил)амино)-2,2-диметил-5-нитро-2,3-дигидробензофуран-7-формамид (соединение 1C) в виде твердого вещества красновато-коричневого цвета с выходом 81 %. 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,56 (синглет, 1H), 8,35 (синглет, 1H), 8,17 (синглет, 1H), 8,02 (синглет, 1H), 7,78 (синглет, 1H), 7,61 (синглет, 1H), 7,53 (синглет, 1H), 7,33 (синглет, 1H), 7,14 (синглет, 1H), 3,87 (синглет, 3H), 3,53 (синглет, 4H), 3,31 (синглет, 2H), 1,58 (дублет, J = 7,2 Гц, 4H), 1,50 (синглет, 6H).
Этап d: синтез 5-амино-4-((4-((2-амино-4-карбамоил-6-метоксифенил)амино)бутил)амино)-2,2-диметил-2,3-дигидробензофуран-7-формамида
Соединение 1C (1 г, 1,94 ммоль) диспергировали в растворе метанола (30 мл), добавляли натрия дитионит (3,37 г, 19,36 ммоль) и аммиак (2 мл) к указанному выше реакционному раствору, перемешивали при комнатной температуре в течение 10 минут. Затем реакционный раствор переносили на масляную баню для нагревания до 60°C и проводили реакцию в течение 4 часов. Завершение реакции контролировали методом ТСХ. Реакционный раствор фильтровали с помощью аспирации, а остаток на фильтре промывали метанолом. Собранные фильтраты объединяли. Объединенный фильтрат перегоняли при пониженном давлении и концентрировали для получения осадка, отделяли и очищали колоночной хроматографией (дихлорметан:метанол = 30:1-7:1) с получением 5-амино-4-((4-((2-амино-4-карбамоил-6-метоксифенил)амино)бутил)амино)-2,2-диметил-2,3-дигидробензофуран-7-формамида (соединение 1D) в виде твердого вещества светло-желтого цвета с выходом 63 %. 1H-ЯМР (300 МГц, ДМСО-d6) δ 7,61 (синглет, 1H), 6,97 (дублет, J = 15,0 Гц, 4H), 6,85 (синглет, 1H), 6,78 (синглет, 1H), 4,64 (синглет, 2H), 4,05 (синглет, 2H), 3,73 (синглет, 3H), 3,20 (синглет, 2H), 3,05 (синглет, 2H), 2,94 (синглет, 2H), 1,49 (синглет, 4H), 1,39 (дублет, J = 6,0 Гц, 6H),
Этап e: синтез 1-(4-(5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метокси-1H-бензо[d]имидазол-1-ил)бутил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7,7-диметил-7,8-дигидро-1H-бензофуран[4,5-d]имидазол-5-формамида
Соединение 1D (0,55 г, 1,2 ммоль) растворяли в растворе N,N-диметилформамида (5 мл), добавляли 0,4 М раствор 1-этил-3-метил-1H-пиразол-5-карбонилизотиоцианата в диоксане (4 мл, 1,6 ммоль) и перемешивали на ледяной бане в течение 10 минут, добавляли еще один 0,4 М раствор 1-этил-3-метил-1H-пиразол-5-карбонилизотиоцианата в диоксане (1 мл, 0,4 ммоль), и после перемешивания в течение 15 минут на ледяной бане добавляли еще 0,4 М раствор 1-этил-3-метил-1H-пиразол-5-карбонилизотиоцианата в диоксане (1 мл, 0,4 ммоль), и реакцию продолжали на ледяной баней в течение получаса, затем добавляли 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (0,58 мг, 3,01 ммоль) и триэтиламин (1 мл, 7,23 ммоль). Реакционный раствор продолжали перемешивать в течение 10 минут на ледяной бане, а затем выдерживали при комнатной температуре для реакции в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционный раствор медленно добавляли в воду (20 мл), и выпадал нерастворимый осадок вещества белого цвета. Его фильтровали путем аспирации, осадок на фильтре собирали, высушивали, отделяли и очищали колоночной хроматографией (дихлорметан:метанол = 20:1-5:1) с получением 1-(4-(5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метокси-1H-бензо[d]имидазол-1-ил)бутил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7,7-диметил-7,8-дигидро-1H-бензофуран[4,5-d]имидазол-5-формамида (пример соединения 1) в виде твердого вещества серо-белого цвета с выходом 78 %. ИЭР-МС (m/z): 779,37[M+H]+; 1H-ЯМР (300 МГц, ДМСО-d6) δ 12,84 (синглет, 1H), 12,66 (синглет, 1H), 8,01 (синглет, 1H), 7,81 (синглет, 1H), 7,63 (синглет, 2H), 7,36 (синглет, 3H), 6,59 (синглет, 2H), 4,57 (синглет, 4H), 4,39 (синглет, 2H), 4,18 (синглет, 2H), 3,89 (синглет, 3H), 3,27 (синглет, 2H), 2,09 (синглет, 6H), 1,89 (синглет, 2H), 1,77 (синглет, 2H), 1,45 (синглет, 6H), 1,30 (синглет, 6H).
Пример 2
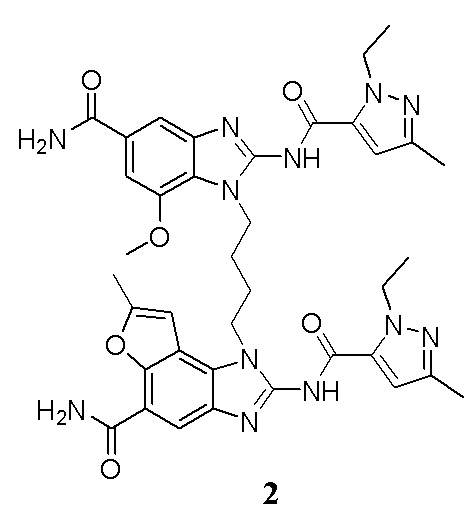
Операционные процедуры и условия реакции были такими же, как в примере 1, за исключением того, что исходный материал на этапе c представлял собой 4-хлор-2-метил-5-нитробензофуран-7-формамид в виде твердого вещества светло-желтого цвета с выходом 68 %. ИЭР-МС (m/z): 763,34[M+H]+; 1H-ЯМР (300 МГц, ДМСО-d6) δ 12,85 (синглет, 1H), 12,70 (синглет, 1H), 7,98 (дублет, J = 18,0 Гц, 2H), 7,82 (синглет, 2H), 7,65 (синглет, 1H), 7,54 (синглет, 1H), 7,39 (синглет, 1H), 7,30 (синглет, 1H), 6,90 (синглет, 1H), 6,61 (синглет, 1H), 6,54 (синглет, 1H), 4,56 (синглет, 4H), 4,39 (синглет, 2H), 4,31 (синглет, 2H), 3,81 (синглет, 3H), 2,10 (дублет, J = 10,4 Гц, 6H), 1,88 (синглет, 4H), 1,30 (дублет, J = 8,0 Гц, 6H).
Пример 3
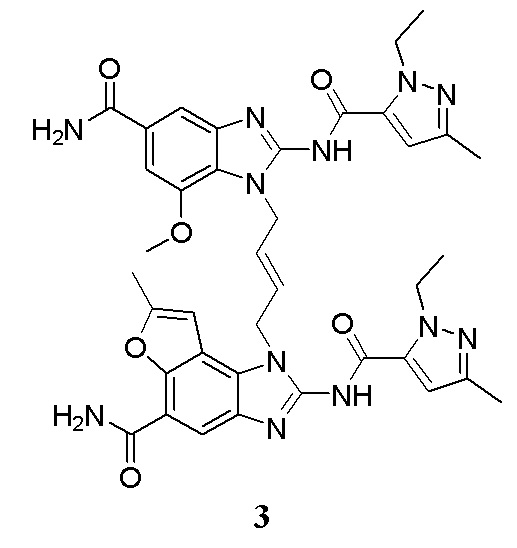
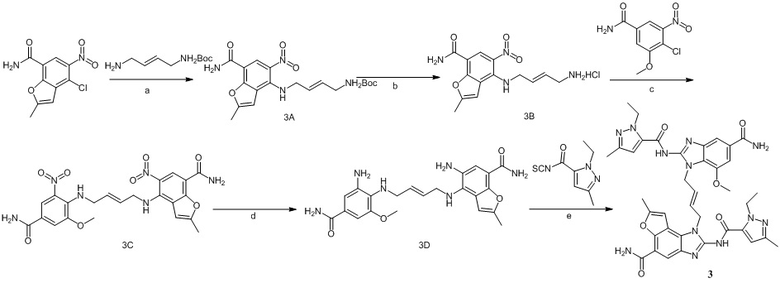
Этап a: синтез трет-бутил(E)-(4-((7-карбамоил-2-метил-5-нитробензофуран-4-ил)амино)бут-2-ен-1-ил)карбамата
Соединение 4-хлор-2-метил-5-нитробензофуран-7-формамид (1 г, 3,93 ммоль) растворяли в растворе н-бутанола (10 мл) и перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли трет-бутил-(E)-(4-аминобутан-2-ен-1-ил)карбамат (0,88 г, 4,71 ммоль) и N,N-диизопропилэтиламин (2,06 мл, 11,78 ммоль). Температуру в запечатанной пробирке повышали до 110°C, реакционный раствор реагировал в течение 12 часов. На следующий день реакционный раствор фильтровали путем аспирации и промывали соответствующим количеством этилацетата для получения трет-бутил-(E)-(4-((7-карбамоил-2-метил-5-нитробензофуран-4-ил)амино)бут-2-ен-1-ил)карбамата (соединение 3A) в виде твердого вещества красновато-коричневого цвета с выходом 69 %. 1H-ЯМР (300 МГц, ДМСО) δ 9,08 (синглет, 1H), 8,53 (синглет, 1H), 7,63 (дублет, J = 52,0 Гц, 2H), 7,04 (синглет, 2H), 5,83-5,62 (мультиплет, 2H), 4,36 (синглет, 2H), 4,11 (синглет, 2H), 2,48 (синглет, 3H), 1,37 (синглет, 9H).
Этап b: синтез (E)-4-((4-аминобутан-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамида гидрохлорида
Соединение 1А (1,6 г, 3,93 ммоль) диспергировали в растворе этилацетата (20 мл), перемешивали на ледяной бане в течение 10 минут, и свежеприготовленный газообразный HCl вводили в указанный раствор до насыщения. После продолжения реакции в течение 10 минут на ледяной бане последнюю удаляли, и выдерживали для протекания реакции при комнатной температуре в течение 2 часов. Завершение реакции контролировали методом ТСХ. Этилацетат удаляли путем перегонки при пониженном давлении. К остатку добавляли эфир (15 мл), перемешивали при комнатной температуре в течение двух часов и фильтровали путем аспирации с получением (E)-4-((4-аминобутан-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамид гидрохлорида (соединение 1B) в виде твердого вещества красно-коричневого цвета с выходом 93 %. 1H-ЯМР (300 МГц, ДМСО) δ 9,13 (синглет, 1H), 8,54 (синглет, 1H), 7,95 (синглет, 3H), 7,66 (дублет, J = 53,1 Гц, 2H), 7,02 (синглет, 1H), 6,09 (дублет, J = 15,5 Гц, 1H), 5,82-5,64 (мультиплет, 1H), 4,43 (синглет, 2H), 3,18 (синглет, 2H), 2,49 (синглет, 3H).
Этап c: синтез (E)-4-((4-((4-карбамоил-2-метокси-6-нитрофенил)амино)бутил-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамида
Соединение 3B (1 г, 2,93 ммоль) растворяли в растворе н-бутанола (10 мл), перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли 4-хлор-3-метокси-5-нитробензамид (0,812 г, 3,52 ммоль) и N,N-диизопропилэтиламин (1,54 мл, 8,8 ммоль). Температуру в запечатанной пробирке повышали до 110°C, и реакционный раствор реагировал в течение 12 часов. На следующий день реакционный раствор фильтровали путем аспирации и промывали соответствующим количеством этилацетата для получения (E)-4-((4-((4-карбамоил-2-метокси-6-нитрофенил)амино)бут-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамида (соединение 3C) в виде твердого вещества красновато-коричневого цвета с выходом 69 %. 1H-ЯМР (400 МГц, ДМСО) δ 9,06 (триплет, J = 5,9 Гц, 1H), 8,51 (синглет, 1H), 8,15 (синглет, 1H), 8,03 (синглет, 1H), 7,76 (триплет, J = 5,8 Гц, 1H), 7,70 (синглет, 1H), 7,51 (синглет, 2H), 7,34 (синглет, 1H), 6,91 (синглет, 1H), 5,81-5,73 (мультиплет, 2H), 4,32 (синглет, 2H), 4,13 (синглет, 2H), 3,81 (синглет, 3H), 2,41 (синглет, 3H).
Этап d: синтез (E)-5-амино-4-((4-((2-амино-4-карбамоил-6-метоксифенил)амино)бут-2-ен-1-ил)амино)-2-метилбензофуран-7-формамида
Соединение 3C (1 г, 2,01 ммоль) диспергировали в растворе метанола (30 мл), добавляли натрия дитионит (3,49 г, 20,06 ммоль) и аммиак (2 мл) к указанному выше реакционному раствору, перемешивали при комнатной температуре в течение 10 минут. Затем реакционный раствор переносили на масляную баню для нагревания до 60°C и проводили реакцию в течение 4 часов. Завершение реакции контролировали методом ТСХ. Реакционный раствор фильтровали с помощью аспирации, а остаток на фильтре промывали метанолом. Собранные фильтраты объединяли. Объединенный фильтрат перегоняли при пониженном давлении и концентрировали для получения осадка, отделяли и очищали колоночной хроматографией (дихлорметан:метанол = 30:1-7:1) с получением (E)-5-амино-4-((4-((2-амино-4-карбамоил-6-метоксифенил)амино)бут-2-ен-1-ил)амино)-2-метилбензофуран-7-формамида (соединение 3D) в виде твердого вещества светло-желтого цвета с выходом 62 %. 1H-ЯМР (400 МГц, ДМСО) δ 7,62 (синглет, 1H), 7,28 (синглет, 1H), 7,15 (синглет, 1H), 7,10 (синглет, 1H), 6,97 (синглет, 1H), 6,86 (дублет, J = 1,8 Гц, 1H), 6,78 (дублет, J = 1,8 Гц, 1H), 6,61 (дублет, J = 1,0 Гц, 1H), 5,76 (синглет, 2H), 5,08 (триплет, J = 5,9 Гц, 1H), 4,65 (синглет, 2H), 4,56 (синглет, 1H), 4,10 (дублет, J = 5,2 Гц, 1H), 3,98 (синглет, 2H), 3,71 (синглет, 3H), 3,55 (синглет, 2H), 3,37 (синглет, 1H), 2,38 (синглет, 3H).
Этап e: синтез (E)-1-(4-(5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метокси-1H-бензо[d]имидазол-1-ил)бут-2-ен-1-ил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метил-1H-бензофуран[4,5-d]имидазол-5-формамида
Операционные процедуры и условия реакции были такими же, как в примере 1, за исключением того, что исходный материал на этапе c представлял собой соединение 3D в виде твердого вещества светло-желтого цвета с выходом 70 %. ИЭР-МС (m/z): 761,32[M+H]+; 1H-ЯМР (300 МГц, ДМСО-d6) δ 12,91 (синглет, 1H), 12,82 (синглет, 1H), 8,00 (синглет, 1H), 7,88 (синглет, 1H), 7,81 (синглет, 1H), 7,64 (синглет, 1H), 7,53 (синглет, 1H), 7,38 (синглет, 1H), 7,24 (синглет, 1H), 6,67 (синглет, 1H), 6,58 (синглет, 1H), 6,44 (синглет, 1H), 5,83 (синглет, 2H), 4,97 (синглет, 2H), 4,85 (синглет, 2H), 4,52 (синглет, 4H), 3,60 (синглет, 3H), 2,29 (синглет, 3H), 2,11 (дублет, J = 13,0 Гц, 6H), 1,26 (синглет, 6H).
Пример 4
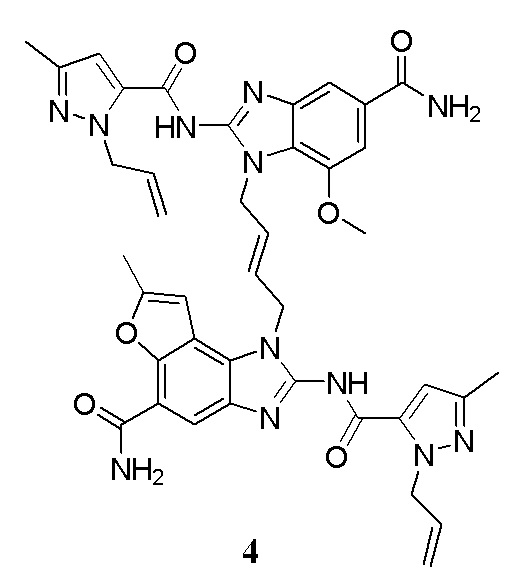
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал представлял собой 1-аллил-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества светло-желтого цвета с выходом 66 %. ИЭР-МС (m/z): 785,4[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,84 (дублет, J = 37,3 Гц, 2H), 7,99 (синглет, 1H), 7,90 (синглет, 1H), 7,79 (синглет, 1H), 7,65 (синглет, 1H), 7,53 (синглет, 1H), 7,35 (синглет, 1H), 7,25 (синглет, 1H), 6,66 (дублет, J = 16,1 Гц, 2H), 6,50 (синглет, 1H), 6,04-5,89 (мультиплет, 3H), 5,85 (дублет, J = 2,6 Гц, 2H), 5,17 (двойной дублет, J = 25,3, 4,2 Гц, 4H), 5,01 (двойной дублет, J = 10,5, 1,4 Гц, 4H), 4,88 (синглет, 4H), 3,61 (синглет, 3H), 2,31 (синглет, 3H), 2,15 (синглет, 3H), 2,11 (синглет, 3H).
Пример 5
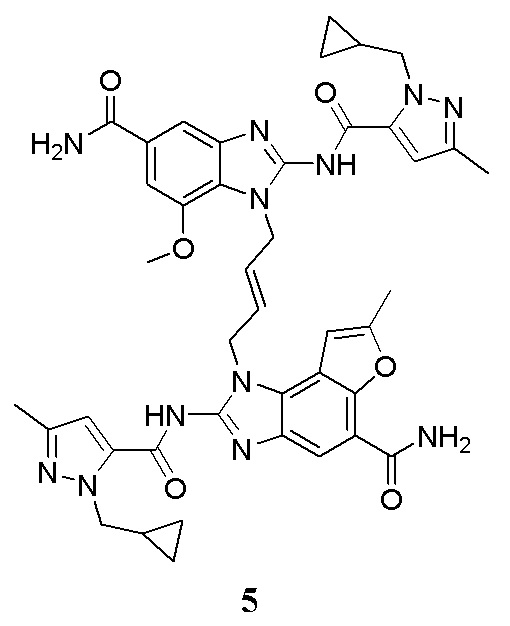
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал представлял собой 1-(циклопропилметил)-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества светло-желтого цвета с выходом 55 %. ИЭР-МС (m/z): 813,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,84 (дублет, J = 37,5 Гц, 2H), 7,97 (синглет, 1H), 7,89 (синглет, 1H), 7,78 (синглет, 1H), 7,64 (синглет, 1H), 7,51 (синглет, 1H), 7,34 (синглет, 1H), 7,24 (синглет, 1H), 6,67 (синглет, 1H), 6,62 (синглет, 1H), 6,47 (синглет, 1H), 5,85 (дублет, J = 4,1 Гц, 2H), 5,00 (синглет, 2H), 4,87 (синглет, 2H), 4,44 (дублет, J = 6,8 Гц, 2H), 4,37 (дублет, J = 6,7 Гц, 2H), 3,59 (синглет, 3H), 2,29 (синглет, 3H), 2,14 (синглет, 3H), 2,10 (синглет, 3H), 1,31 (двойной дублет, J = 15,0, 6,7 Гц, 2H), 0,38 (триплет, J = 8,1 Гц, 4H), 0,30 (двойной дублет, J = 13,9, 4,1 Гц, 4H).
Пример 6
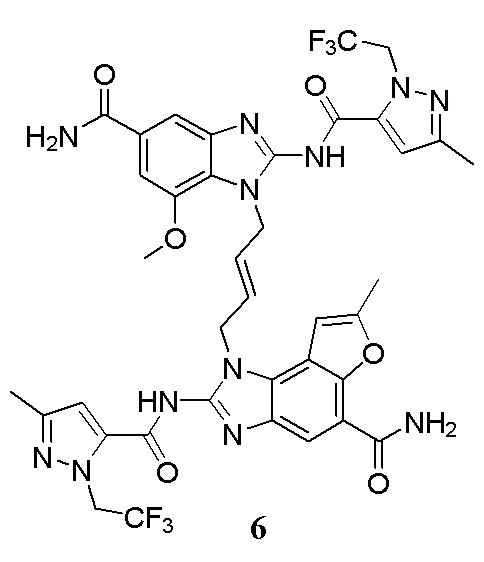
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал представлял собой 3-метил-1-(2,2,2-трифторэтил)-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 72 %. ИЭР-МС (m/z): 867,3[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,93 (синглет, 1H), 12,83 (синглет, 1H), 7,99 (синглет, 1H), 7,90 (синглет, 1H), 7,81 (синглет, 1H), 7,67 (дублет, J = 1,0 Гц, 1H), 7,53 (синглет, 1H), 7,37 (синглет, 1H), 7,26 (синглет, 1H), 6,77 (синглет, 1H), 6,66 (синглет, 1H), 6,57 (синглет, 1H), 5,88 (синглет, 2H), 5,60 (двойной дублет, J = 17,8, 8,8 Гц, 2H), 5,56-5,48 (мультиплет, 2H), 5,02 (синглет, 2H), 4,89 (синглет, 3H), 3,60 (синглет, 3H), 2,28 (синглет, 3H), 2,18 (синглет, 3H), 2,12 (синглет, 3H).
Пример 7
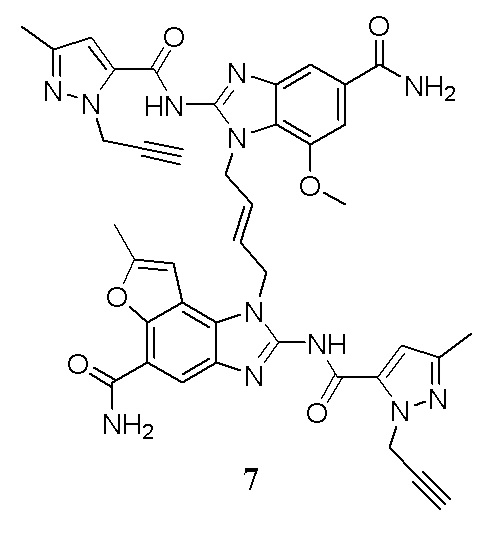
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал представлял собой 1-пропаргил-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 71 %. ИЭР-МС (m/z): 782,4[M+H]+; 1H-ЯМР (300 МГц, ДМСО) δ 12,92 (дублет, J = 27,5 Гц, 1H), 8,04 (синглет, 1H), 7,92 (синглет, 1H), 7,84 (синглет, 1H), 7,68 (синглет, 1H), 7,56 (синглет, 1H), 7,41 (синглет, 1H), 7,25 (синглет, 1H), 6,69 (синглет, 2H), 6,53 (синглет, 1H), 5,91 (синглет, 2H), 5,43 (дублет, J = 21,7 Гц, 4H), 4,99 (дублет, J = 36,9 Гц, 4H), 3,60 (синглет, 3H), 3,27 (дублет, J = 7,2 Гц, 2H), 2,30 (синглет, 3H), 2,19 (синглет, 3H), 2,14 (синглет, 3H).
Пример 8
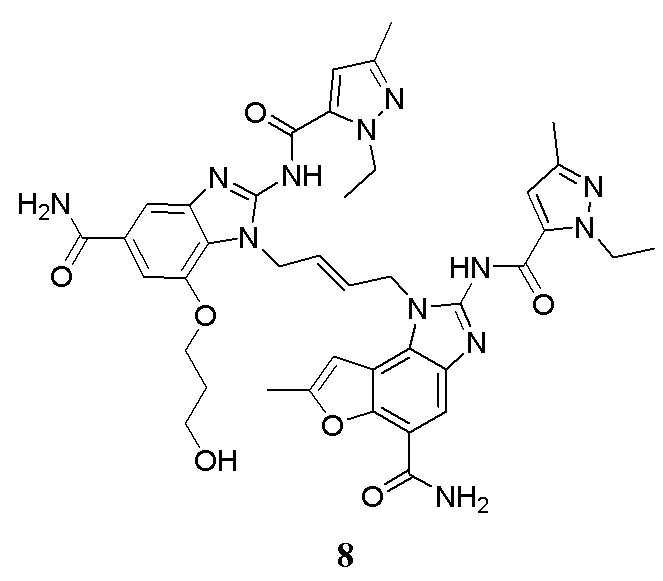
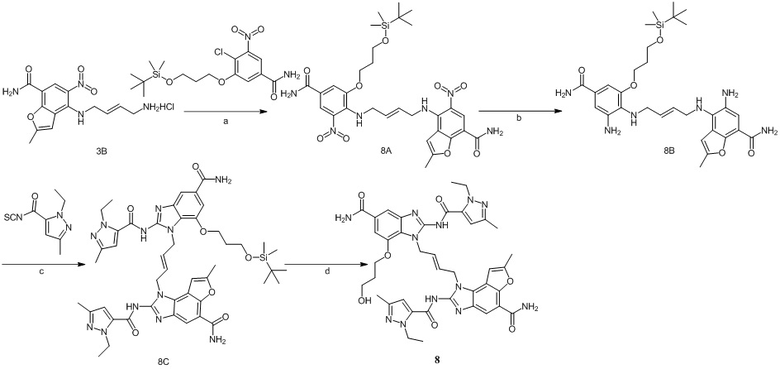
Этап a: синтез (E)-4-((4-((2-(3-((трет-бутилдиметилсилил)окси)пропокси)-4-карбамоил-6-нитрофенил)амино)бутил-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамида
Соединение 3B (1 г, 2,93 ммоль) растворяли в растворе н-бутанола (10 мл), перемешивали при комнатной температуре, затем к указанному реакционному раствору добавляли 3-(3-((трет-бутилдиметилсилил)окси)пропокси)-4-хлор-5-нитробензамид (1,37 г, 3,52 ммоль) и N,N-диизопропилэтиламин (1,46 мл, 8,8 ммоль). Температуру в запечатанной пробирке повышали до 110°C, и реакционный раствор реагировал в течение 12 часов. На следующий день реакционный раствор фильтровали путем аспирации и промывали соответствующим количеством этилацетата для получения (E)-4-((4-((2-((3-(трет-бутилдиметилсилил)окси)пропокси)-4-карбамоил-6-нитрофенил)амино)бут-2-ен-1-ил)амино)-2-метил-5-нитробензофуран-7-формамид (соединение 8A) в виде твердого вещества красновато-коричневого цвета с выходом 66 %. МС (ИЭР) m/z = 657[M+H]+.
Этап b: Синтез (E)-5-амино-4-((4-((2-амино-6-(3-((трет-бутилдиметилсилил)окси)пропокси)-4-карбамоилфенил)амино)бут-2-ен-1-ил)амино)-2-метилбензофуран-7-формамида
Соединение 8A (1 г, 1,52 ммоль) диспергировали в растворе метанола (30 мл), добавляли натрия дитионит (2,65 г, 15,2 ммоль) и аммиак (2 мл) к указанному выше реакционному раствору, перемешивали при комнатной температуре в течение 10 минут. Затем реакционный раствор переносили на масляную баню для нагревания до 60°C и проводили реакцию в течение 4 часов. Завершение реакции контролировали методом ТСХ. Реакционный раствор фильтровали с помощью аспирации, а остаток на фильтре промывали метанолом. Собранные фильтраты объединяли. Объединенный фильтрат перегоняли при пониженном давлении и концентрировали для получения осадка, отделяли и очищали колоночной хроматографией (дихлорметан:метанол = 30:1-7:1) с получением (E)-5-амино-4-((4-((2-амино-6-(3-((трет-бутилдиметилсилил)окси)пропокси)-4-карбамоилфенил)амино)бут-2-ен-1-ил)амино)-2-метилбензофуран-7-формамид (соединение 8B) в виде твердого вещества светло-желтого цвета с выходом 61 %. МС (ИЭР) m/z = 597[M+H]+.
Этап c: синтез ((E)-1-(4-(7-(3-((трет-бутилдиметилсилил)окси)пропокси)-5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-1H-бензо[d]имидазол-1-ил)бут-2-ен-1-ил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метил-1H-бензофуран[4,5-d]имидазол-5-формамида
Операционные процедуры и условия реакции были такими же, как в примере 1, за исключением того, что исходный материал на этапе c представлял собой соединение 8B в виде твердого вещества светло-желтого цвета с выходом 58 %. МС (ИЭР) m/z = 919[M+H]+.
Этап d: синтез (E)-1-(4-(5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-(3-гидроксипропокси)-1H-бензо[d]имидазиперазин-1-ил)бут-2-ен-1-ил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метил-1H-бензофуран[4,5-d]имидазол-5-формамида
Соединение 8C (0,5 г, 0,54 ммоль) диспергировали в 4 М растворе HCl в диоксане (5 мл) и перемешивали при комнатной температуре в течение 30 минут. Завершение реакции контролировали методом ТСХ. Растворитель удаляли путем перегонки при пониженном давлении. Добавляли эфир (5 мл) и перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор фильтровали путем аспирации, осадок на фильтре собирали и высушивали, получая (E)-1-(4-(5-карбамоил-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-(3-гидроксипропокси)-1H-бензо[d]имидазиперазин-1-ил)бут-2-ен-1-ил)-2-(1-этил-3-метил-1H-пиразол-5-формамидо)-7-метил-1H-бензофуран[4,5-d]имидазол-5-формамид (пример соединения 8) в виде твердого вещества серо-белого цвета с выходом 89 %. ИЭР-MС (m/z): 805,4[M+H]+ 1H-ЯМР (400 МГц, ДМСО) δ 8,02 (синглет, 1H), 7,90 (синглет, 1H), 7,81 (синглет, 1H), 7,66 (синглет, 1H), 7,53 (синглет, 1H), 7,34 (синглет, 1H), 7,26 (синглет, 1H), 6,68 (синглет, 1H), 6,62 (синглет, 1H), 6,46 (синглет, 1H), 5,86 (синглет, 2H), 5,00 (синглет, 2H), 4,91 (синглет, 2H), 4,61-4,46 (мультиплет, 6H), 3,89 (триплет, J = 6,1 Гц, 2H), 3,58 (синглет, 1H), 2,30 (синглет, 3H), 2,15 (синглет, 3H), 2,11 (синглет, 3H), 1,59-1,54 (мультиплет, 2H), 1,33-1,28 (мультиплет, 6H).
Пример 9
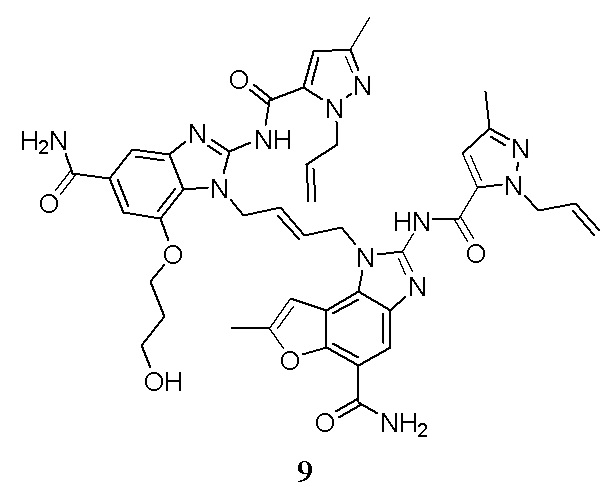
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 1-аллил-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 88 %. ИЭР-МС (m/z): 829,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 8,02 (синглет, 1H), 7,90 (синглет, 1H), 7,81 (синглет, 1H), 7,66 (синглет, 1H), 7,53 (синглет, 1H), 7,38 (дублет, J = 10,0 Гц, 1H), 7,27 (синглет, 1H), 6,67 (дублет, J = 3,6 Гц, 2H), 6,50 (синглет, 1H), 6,02-5,93 (мультиплет, 2H), 5,85 (синглет, 2H), 5,19 (дублет, J = 3,9 Гц, 2H), 5,12 (дублет, J = 3,8 Гц, 2H), 5,06-4,98 (мультиплет, 5H), 4,90 (дублет, J = 10,4 Гц, 5H), 3,89 (синглет, 2H), 3,80 (синглет, 1H), 3,38 (дублет, J = 8,6 Гц, 5H), 2,29 (синглет, 3H), 2,15 (синглет, 3H), 2,11 (синглет, 3H), 1,59-1,52 (мультиплет, 2H).
Пример 10
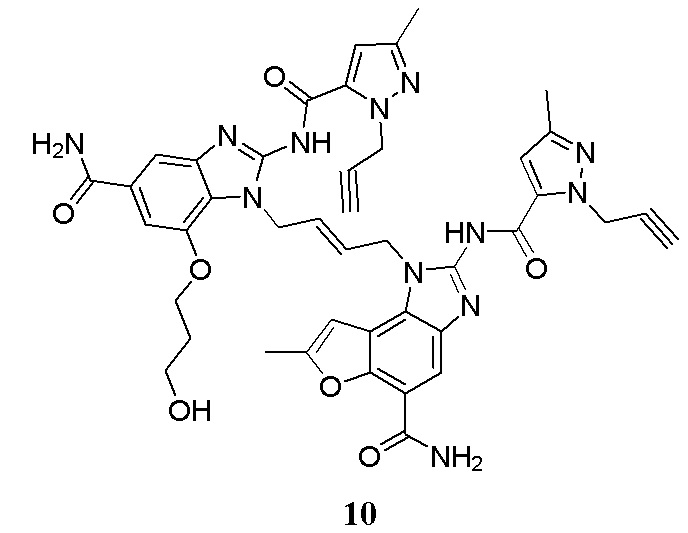
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 1-пропаргил-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 84 %. ИЭР-МС (m/z): 825,4[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 8,09-7,97 (мультиплет, 1H), 7,90 (синглет, 1H), 7,81 (синглет, 1H), 7,66 (синглет, 1H), 7,52 (синглет, 1H), 7,34 (синглет, 1H), 7,24 (синглет, 1H), 6,66 (дублет, J = 9,1 Гц, 2H), 6,51 (синглет, 1H), 5,88 (синглет, 2H), 5,42 (синглет, 2H), 5,36 (синглет, 2H), 5,03 (синглет, 2H), 4,93 (синглет, 2H), 3,86 (дублет, J = 6,0 Гц, 2H), 3,57 (синглет, 1H), 3,37 (дублет, J = 5,9 Гц, 2H), 3,24 (дублет, J = 2,0 Гц, 2H), 2,27 (синглет, 3H), 2,16 (синглет, 3H), 2,12 (синглет, 3H), 1,55-1,51 (мультиплет, 2H).
Пример 11
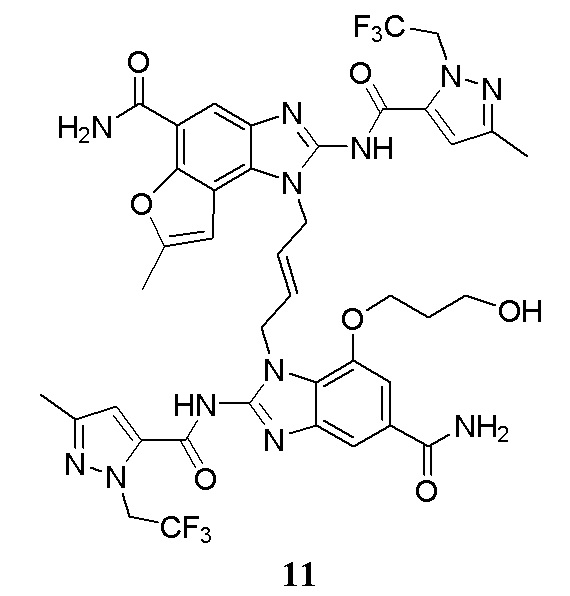
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 3-метил-1-(2,2,2-трифторэтил)-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 84 %. ИЭР-МС (m/z): 911,3[M-H]-; 1H-ЯМР (400 МГц, ДМСО) δ 12,85 (дублет, J = 41,7 Гц, 2H), 8,02 (синглет, 1H), 7,89 (синглет, 1H), 7,79 (синглет, 1H), 7,65 (синглет, 1H), 7,53 (синглет, 1H), 7,34 (синглет, 1H), 7,23 (синглет, 1H), 6,75 (синглет, 1H), 6,61 (синглет, 1H), 6,55 (синглет, 1H), 5,85 (синглет, 2H), 5,63-5,46 (мультиплет, 4H), 4,99 (синглет, 2H), 4,89 (синглет, 2H), 4,58 (триплет, J = 5,1 Гц, 1H), 3,85 (триплет, J = 6,4 Гц, 2H), 3,51 (дублет, J = 0,8 Гц, 2H), 2,26 (синглет, 3H), 2,17 (синглет, 3H), 2,12 (синглет, 3H), 1,58-1,48 (мультиплет, 2H).
Пример 12
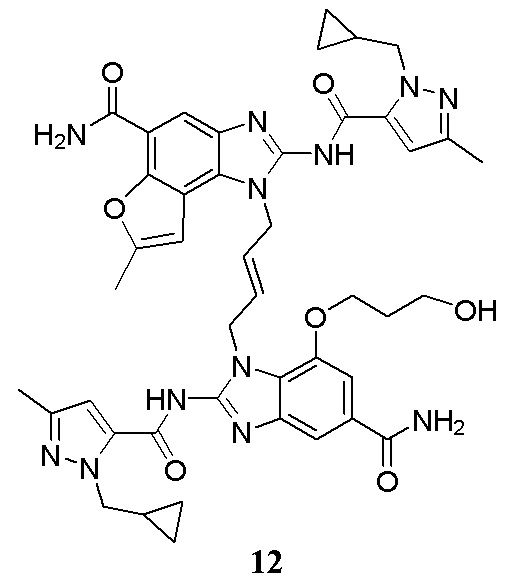
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 1-(циклопропилметил)-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 82 %. ИЭР-МС (m/z): 855,4[M-H]-; 1H-ЯМР (400 МГц, ДМСО) δ 12,83 (дублет, J = 33,7 Гц, 2H), 8,01 (синглет, 1H), 7,90 (синглет, 1H), 7,79 (синглет, 1H), 7,65 (синглет, 1H), 7,51 (дублет, J = 6,3 Гц, 1H), 7,33 (синглет, 1H), 7,24 (синглет, 1H), 6,64 (дублет, J = 12,9 Гц, 2H), 6,49 (синглет, 1H), 5,85 (синглет, 2H), 5,00 (синглет, 2H), 4,89 (синглет, 2H), 4,52 (дублет, J = 21,5 Гц, 1H), 4,44 (дублет, J = 6,6 Гц, 2H), 4,38 (дублет, J = 6,9 Гц, 2H), 3,86 (синглет, 4H), 2,29 (синглет, 3H), 2,15 (синглет, 3H), 2,11 (синглет, 3H), 1,59-1,47 (мультиплет, 2H), 1,30 (двойной дублет, J = 12,2, 7,3 Гц, 2H), 0,39 (синглет, 4H), 0,31 (двойной дублет, J = 9,6, 4,3 Гц, 4H).
Пример 13
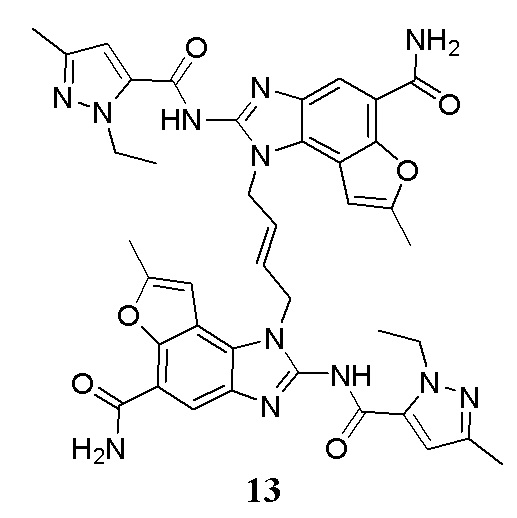
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал на этапе c представлял собой 4-хлор-2-метил-5-нитробензофуран-7-формамид в виде твердого вещества серо-белого цвета с выходом 61 %. ИЭР-МС (m/z): 785,4[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,91 (синглет, 2H), 7,91 (синглет, 2H), 7,79 (синглет, 2H), 7,53 (синглет, 2H), 6,56 (дублет, J = 3,3 Гц, 4H), 5,92 (синглет, 2H), 4,95 (синглет, 4H), 4,56 (дублет, J = 7,0 Гц, 4H), 2,21 (синглет, 6H), 2,15 (синглет, 6H), 1,31 (триплет, J = 7,0 Гц, 7H).
Пример 14
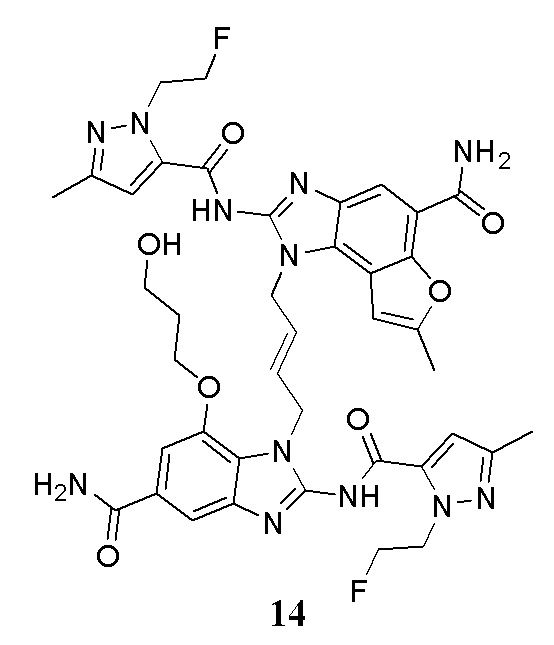
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 1-(2-фторэтил)-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества серо-белого цвета с выходом 76 %. ИЭР-МС (m/z): 839,4[M-H]-; 1H-ЯМР (300 МГц, ДМСО) δ 8,01 (синглет, 1H), 7,90 (синглет, 1H), 7,80 (синглет, 1H), 7,65 (синглет, 1H), 7,52 (синглет, 1H), 7,34 (синглет, 1H), 7,24 (синглет, 1H), 6,66 (дублет, J = 4,7 Гц, 2H), 6,51 (синглет, 1H), 5,84 (синглет, 2H), 4,99 (синглет, 2H), 4,85 (двойной дублет, J = 13,3, 9,0 Гц, 10H), 4,70-4,63 (мультиплет, 2H), 3,39-3,34 (мультиплет, 2H), 2,28 (синглет, 3H), 2,13 (дублет, J = 12,2 Гц, 6H), 1,53 (синглет, 2H).
Пример 15
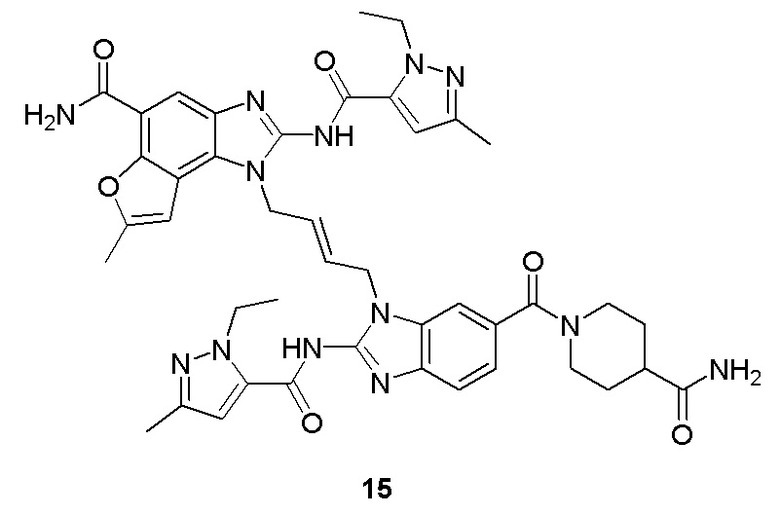
Операционные процедуры и условия реакции были такими же, как в примере 2, за исключением того, что исходный материал представлял собой 1-(4-хлор-3-нитробензоил)пиперидин-4-формамид в виде твердого вещества белого цвета с выходом 78 %. ИЭР-МС (m/z): 842,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,76 (синглет, 2H), 7,88 (синглет, 1H), 7,79 (синглет, 1H), 7,52 (дублет, J = 9,1 Гц, 2H), 7,42-7,32 (мультиплет, 2H), 7,09 (дублет, J = 8,1 Гц, 1H), 6,84 (синглет, 1H), 6,75 (синглет, 1H), 6,61 (дублет, J = 9,6 Гц, 1H), 6,48 (синглет, 1H), 6,04 (дублет, J = 16,1 Гц, 1H), 5,80 (дублет, J = 21,4 Гц, 1H), 5,01 (синглет, 2H), 4,88-4,73 (мультиплет, 2H), 4,62-4,43 (мультиплет, 4H), 2,90 (синглет, 4H), 2,38 (дублет, J = 16,7 Гц, 3H), 2,14 (триплет, J = 11,7 Гц, 6H), 1,91 (синглет, 1H), 1,73 (синглет, 2H), 1,56-1,42 (мультиплет, 2H), 1,23 (дублет, J = 10,5 Гц, 6H).
Пример 16
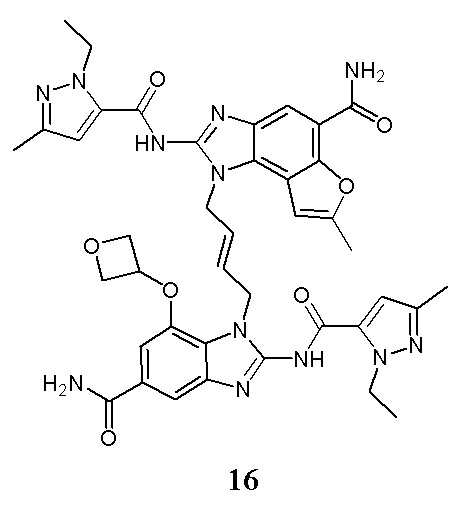
Операционные процедуры и условия реакции были такими же, как в примере 3, за исключением того, что исходный материал на этапе c представлял собой 4-хлор-3-нитро-5-(оксетан-3-илокси)бензамид в виде твердого вещества серо-белого цвета с выходом 69 %. ИЭР-МС (m/z):803,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,88 (дублет, J = 16,9 Гц, 2H), 8,06-7,85 (мультиплет, 2H), 7,80 (синглет, 1H), 7,69 (дублет, J = 7,2 Гц, 1H), 7,54 (синглет, 1H), 7,38 (синглет, 1H), 6,82 (дублет, J = 7,7 Гц, 1H), 6,73 (дублет, J = 8,2 Гц, 1H), 6,59 (дублет, J = 7,7 Гц, 1H), 6,48 (дублет, J = 7,6 Гц, 1H), 5,90 (синглет, 2H), 5,00 (триплет, J = 21,9 Гц, 4H), 4,77-4,63 (мультиплет, 2H), 4,53 (dq, J = 14,6, 7,3 Гц, 4H), 4,44-4,31 (мультиплет, 2H), 3,08 (дублет, J = 7,5 Гц, 1H), 2,30 (дублет, J = 7,1 Гц, 3H), 2,24-2,06 (мультиплет, 6H), 1,31-1,24 (мультиплет, 6H).
Пример 17
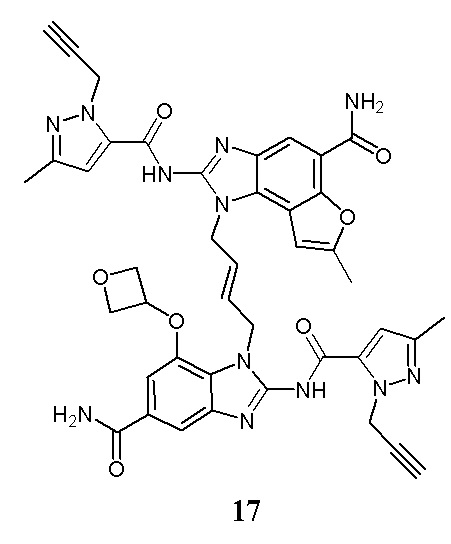
Операционные процедуры и условия реакции были такими же, как в примере 16, за исключением того, что исходный материал представлял собой 3-метил-1-(проп-2-ин-1-ил)-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества светло-желтого цвета с выходом 82 %. ИЭР-МС (m/z): 823,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,90 (синглет, 2H), 7,93 (дублет, J = 17,4 Гц, 2H), 7,73 (дублет, J = 29,6 Гц, 2H), 7,35 (синглет, 1H), 7,19 (синглет, 1H), 6,81 (синглет, 1H), 6,68 (синглет, 1H), 6,64 (синглет, 1H), 6,52 (синглет, 1H), 5,93 (синглет, 2H), 5,40 (дублет, J = 20,5 Гц, 4H), 5,01 (дублет, J = 14,1 Гц, 5H), 4,67 (синглет, 2H), 4,35 (синглет, 2H), 3,21 (синглет, 2H), 2,27 (синглет, 3H), 2,18-2,10 (мультиплет, 6H).
Пример 18
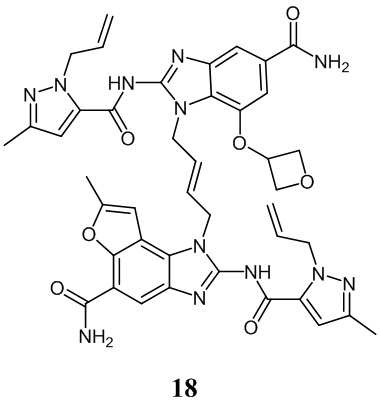
Операционные процедуры и условия реакции были такими же, как в примере 16, за исключением того, что исходный материал представлял собой 1-аллил-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества светло-желтого цвета с выходом 78 %. ИЭР-МС (m/z): 849,5[M+Na]+; 1H-ЯМР (400 МГц, ДМСО) δ 12,87 (дублет, J = 16,5 Гц, 2H), 7,93 (дублет, J = 19,0 Гц, 2H), 7,80 (синглет, 1H), 7,69 (синглет, 1H), 7,53 (синглет, 1H), 7,38 (синглет, 1H), 6,81 (синглет, 1H), 6,71 (синглет, 1H), 6,63 (синглет, 1H), 6,52 (синглет, 1H), 5,93 (дублет, J = 20,1 Гц, 4H), 5,18 (дублет, J = 18,0 Гц, 4H), 5,02 (дублет, J = 15,5 Гц, 5H), 4,91 (двойной дублет, J = 25,2, 14,8 Гц, 4H), 4,69 (синглет, 2H), 4,36 (синглет, 2H), 2,29 (синглет, 3H), 2,13 (дублет, J = 9,7 Гц, 6H).
Пример 19
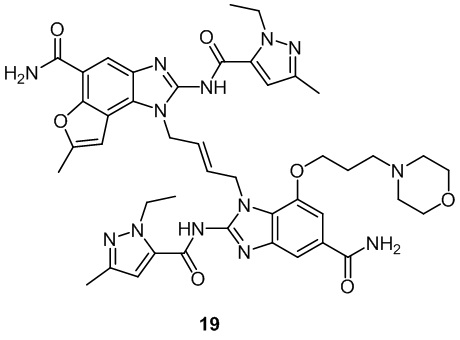
Операционные процедуры и условия реакции были такими же, как в примере 2, за исключением того, что исходный материал представлял собой 4-хлор-3-(3-морфолинопропокси)-5-нитробензамид в виде твердого вещества белого цвета с выходом 78 %. ИЭР-МС (m/z): 874,3450[M+H]+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,90 (дублет, J = 29,2 Гц, 2H), 7,99 (синглет, 1H), 7,91 (синглет, 1H), 7,82 (синглет, 1H), 7,67 (синглет, 1H), 7,56 (синглет, 1H), 7,36 (синглет, 1H), 7,16 (синглет, 1H), 6,64 (синглет, 1H), 6,59 (дублет, J = 9,7 Гц, 1H), 6,52 (синглет, 1H), 5,80 (дублет, J = 22,4 Гц, 2H), 4,96 (дублет, J = 13,1 Гц, 2H), 4,89 (синглет, 2H), 4,56 (p, J = 7,8, 7,3 Гц, 4H), 3,71 (синглет, 4H), 3,61-3,49 (мультиплет, 2H), 3,07 (двойной триплет, J = 13,3, 5,9 Гц, 4H), 2,26 (синглет, 2H), 2,15 (дублет, J = 5,1 Гц, 9H), 1,31 (q, J = 6,5 Гц, 8H).
Пример 20
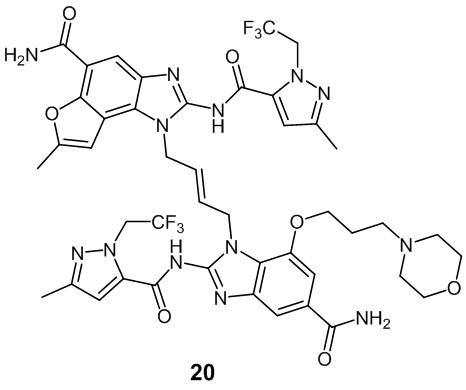
Операционные процедуры и условия реакции были такими же, как в примере 19, за исключением того, что исходный материал представлял собой 3-метил-1-(2,2,2-трифторэтил)-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества белого цвета с выходом 78 %. ИЭР-МС (m/z): 982,3543[M+H]+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,93 (дублет, J = 32,4 Гц, 2H), 8,00 (синглет, 1H), 7,91 (синглет, 1H), 7,84 (синглет, 1H), 7,69 (синглет, 1H), 7,58 (синглет, 1H), 7,39 (синглет, 1H), 7,21 (синглет, 1H), 6,83-6,72 (мультиплет, 1H), 6,62 (синглет, 2H), 5,99 (синглет, 2H), 5,65-5,51 (мультиплет, 4H), 5,00 (синглет, 2H), 4,91 (синглет, 2H), 4,12 (синглет, 2H), 3,75 (синглет, 4H), 3,12-3,06 (мультиплет, 4H), 2,39-1,92 (мультиплет, 11H), 1,77 (синглет, 2H).
Пример 21
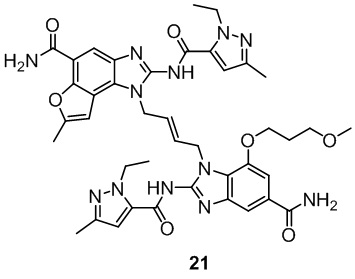
Операционные процедуры и условия реакции были такими же, как в примере 2, за исключением того, что исходный материал представлял собой 4-хлор-3-(3-метоксипропокси)-5-нитробензамид в виде твердого вещества белого цвета с выходом 78 %. ИЭР-МС (m/z): 819,5[M+H]+; 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,87 (дублет, J = 34,2 Гц, 2H), 8,00 (синглет, 1H), 7,90 (синглет, 1H), 7,81 (синглет, 1H), 7,65 (синглет, 1H), 7,55 (дублет, J = 15,0 Гц, 1H), 7,35 (дублет, J = 10,1 Гц, 2H), 6,68 (дублет, J = 10,7 Гц, 1H), 6,58 (синглет, 1H), 6,47 (дублет, J = 16,6 Гц, 1H), 5,83 (синглет, 2H), 4,91 (двойной дублет, J = 43,4, 19,8 Гц, 4H), 4,54 (dq, J = 14,8, 7,7, 7,0 Гц, 4H), 3,80 (дублет, J = 6,7 Гц, 2H), 3,20 (дублет, J = 6,4 Гц, 2H), 3,12 (синглет, 3H), 2,29 (синглет, 3H), 2,13 (дублет, J = 8,0 Гц, 6H), 1,71-1,53 (мультиплет, 2H), 1,29 (q, J = 9,7, 8,1 Гц, 6H).
Пример 22
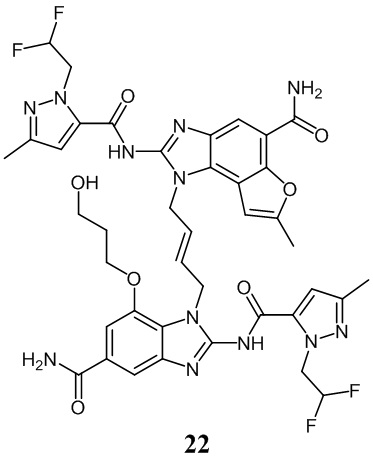
Операционные процедуры и условия реакции были такими же, как в примере 8, за исключением того, что исходный материал представлял собой 1-(2,2-дифторэтил)-3-метил-1Н-пиразол-5-карбонилизотиоцианат в виде твердого вещества светло-желтого цвета с выходом 78 %.
Пример 23. Активирующий эффект соединения на путь интерферона I типа (ISG), продуцируемого клетками МКПК линии мононуклеарных клеток периферической крови человека
В этом эксперименте функцию агониста STING оценивали путем выявления изменений интерферона I типа ИФН-β, продуцируемого линией мононуклеарных клеток периферической крови человека МКПК под действием соединения, стимулирующего клетки (AllCells, кат. №: PB005F). Перед использованием криоконсервированные МКПК размораживали в среде RPMI1640 и восстанавливали жизнеспособность в течение ночи. Исследуемое соединение растворяли в ДМСО и получали исходный раствора с концентрацией 10 ммоль/л. Перед анализом соединение разбавляли до в 100 раз для получения рабочей концентрации (рабочая концентрация составляла 30, 10, 3,33, 1,11, 0,370, 0,123, 0,0412, 0,0137 мкмоль/л соответственно). МКПК собирали и доводили концентрацию клеток до 5 × 105/мл. ЛПС добавляли к МКПК в конечной концентрации 10 нг/мл. В 96-луночный планшет для культивирования клеток в каждую лунку добавляли по 200 мкл МКПК (1 ×105 клеток на лунку). Затем добавляли 2 мкл разбавленного соединения на лунку (лунки в двух повторностях, конечная концентрация ДМСО в каждой лунке составляла 1 %). Затем с помощью пипетки тщательно перемешивали клетки и распределяли соединение равномерно. В два отрицательного контроля добавляли по 2 мкл ДМСО (конечная концентрация 1 %). Клетки культивировали в течение ночи в инкубаторе при 37°C. Супернатант клеточной культуры собирали для определения концентрации интерферона I типа ИФН-β. Концентрацию ИФН-β определяли с помощью набора ИФА с твердофазным антителом R&D-MAB8144-100 и детектирующим антителом R&D-MAB8143-100 (биотинилированным), пересчитывали в концентрацию IP10 по стандартной кривой, а значение EC50 рассчитывали с помощью GraphPad 5.0, подбирая калибровочную кривую. Соединение по настоящему изобретению может оказывать мощное стимулирующее действие на человеческие МКПК клетки, запуская высвобождение ИФН-β, и значение EC50 составляло менее 10 мкмоль/л. Результаты эксперимента представлены в таблице 1.
Таблица 1. Перечень результатов эксперимента с клетками
Результаты эксперимента показывают, что соединение, полученное по настоящему изобретению, оказывает активирующее действие на STING и может быть использовано для получения лекарственного средства для активации STING или лекарственного средства для лечения заболеваний, связанных с активностью STING.
Пример 24. Активирующий эффект соединения на путь интерферона I типа (ISG) в человеческих клетках THP1-Dual
В этом эксперименте способность соединений активировать STING оценивали путем выявления способности соединений стимулировать выработку ИФН-β в линии мононуклеарных клеток периферической крови человека THP1-Dual . Клетки THP1-Dual для эксперимента были приобретены у компании InvivoGen. Соединение растворяли в 10 ммоль/л растворе ДМСО. В каждую лунку 96-луночного планшета для культивирования клеток добавляли по 20 мкл соединения, разбавленного ДМСО/физиологическим раствором, в концентрации 100 мкмоль/л и положительного контроля ADU-S100 в концентрации 100 мкмоль/л. В контрольную группу без препарата добавляли 20 мкл физиологического раствора, содержащего 1 % ДМСО. Каждый раствор вносили в три лунки. Подсчитывали число клеток THP1-Dual, и их концентрацию корректировали до 1 × 106/мл, и 180 мкл суспензии клеток добавляли в каждую лунку для инкубации. Таким образом, конечный объем раствора в каждой лунке в эксперименте составлял 200 мкл, концентрация ДМСО - 0,1 %, а концентрация испытуемого соединения - 10 мкмоль/л. Конечная концентрация положительного контроля ADU-S100 составляла 10 мкмоль/л, и инкубировали в течение 24 часов. Кроме того, в группу холостых образцов добавляли по 200 мкл культурального раствора. Через 24 часа отбирали по 20 мкл культурального раствора из каждой лунки и помещали в новый 96-луночный планшет с прозрачным дном, добавляли по 50 мкл реактива для обнаружения люциферазы QUANTI-Luc™ и регистрировали значение флуоресценции. Соединение 3 описанное в литературе (Nature.2018Dec; 564(7736):439-443), и соединение 1, представленное в WO2019069275, были использованы в качестве положительных контрольных соединений. Данные описывали по соотношению флуоресценции с испытуемым соединением к флуоресценции с холостым контрольным образцом. Результаты эксперимента представлены в таблице 2.
Соединение GSK 1
Соединение GSK 3
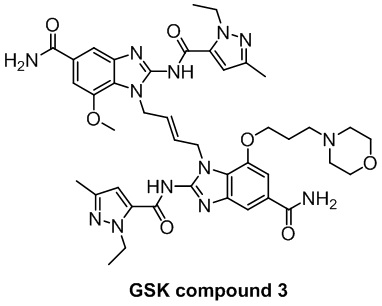
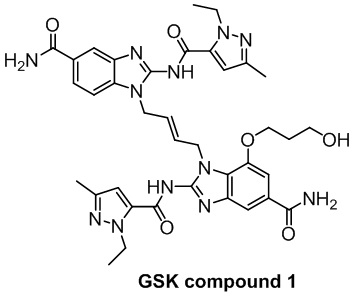
Таблица 2. Перечень результатов эксперимента с клетками
Результаты эксперимента свидетельствуют о том, что активирующий эффект в отношении STING соединения, полученного по настоящему изобретению, эквивалентен или превышает эффект положительного контроля (ADU-S100, соединение GSK 3 или соединение GSK 1), особенно высокой активностью обладают соединения 16, 18 и 20. По сравнению с положительным контролем ADU-S100, их активирующий эффект на STING в 2-3 и более раз превышает таковой положительного контроля, а по сравнению с соединением GSK 3 их активирующий эффект в отношении STING более чем в 2 раза превышает таковой положительного контроля. Поэтому соединение по настоящему изобретению может быть использовано для получения лекарственного препарата для активации STING или лекарственного препарата для лечения заболеваний, связанных с активностью STING.
Пример 25. Исследование эффективности соединения по настоящему изобретению в модели подкожной трансплантированной опухоли рака толстой кишки CT26.
1. Экспериментальный способ
В этом эксперименте использовали самок мышей Balb/C в возрасте 4-6 недель, и клетки CT26 (2,5 × 105) вводили подкожно в подмышечную область каждой мыши. Наблюдался рост опухоли у мышей. Группировку проводили, когда объем опухоли составлял 50-100 мм3. Вводили соединение 19 по настоящему изобретению и отрицательный контроль. В дни 1, 4 и 7 после группировки вводили соединение 19 по настоящему изобретению и отрицательный контроль. Доза соединения 19 в каждой группе составляла 0,1 мг/кг и 0,3 мг/кг соответственно. Результаты экспериментов оценивали в виде кривых роста объема опухоли, они показаны на фигуре 1.
2. Экспериментальные результаты
В модели опухоли рака толстой кишки CT26 на мышах Balb/C соединение 19 по настоящему изобретению обладает значимым противоопухолевым эффектом по сравнению с отрицательным контролем.
Пример 26. Исследование эффективности соединения по настоящему изобретению в модели ортотопической трансплантированной опухоли рака молочной железы EMT6.
1. Экспериментальный способ
В этом эксперименте использовали самок мышей Balb/C в возрасте 4-6 недель, и клетки EMT6 (2,5 × 105) вводили подкожно в подмышечную область каждой мыши. Наблюдался рост опухоли у мышей. Группировку проводили, когда объем опухоли составлял 50-100 мм3. В дни 1, 4 и 9 после группировки вводили соединение 19 по настоящему изобретению и отрицательный контроль. Доза соединения 19 по настоящему изобретению составляла 1 мг/кг. Результаты экспериментов оценивали в виде кривых роста объема опухоли, они показаны на фигуре 2.
2. Экспериментальные результаты
В модели ортотопической трансплантированной опухоли рака молочной железы EMT6 на мышах Balb/C соединение 19 по настоящему изобретению обладает значимым противоопухолевым эффектом по сравнению с отрицательным контролем.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ДЕЙСТВУЮЩЕЕ КАК ИНГИБИТОР КИНАЗЫ | 2021 |
|
RU2826488C1 |
| ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ НА ОСНОВЕ КЕТОАМИДА | 2021 |
|
RU2819346C1 |
| БЕНЗОКСАЗИН-ОКСАЗОЛИДИНОНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ АЗОТСОДЕРЖАЩИМ ГЕТЕРОЦИКЛОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2744784C1 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2158739C2 |
| СОЕДИНЕНИЕ 3,4-ДИГИДРОИЗОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2825312C1 |
| О-АМИНОГЕТЕРОАРИЛАЛКИНИЛСОДЕРЖАЩЕЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2797694C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ЗАМЕЩЕННОГО ТИАЗОЛЬНОГО АРОМАТИЧЕСКОГО КОЛЬЦА И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2830426C1 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ/АКТИВАЦИИ PD1/PD-L1 | 2019 |
|
RU2777980C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| СОЕДИНЕНИЯ ТИЕНИЛ[3, 2-d]ПИРИМИДИН-4-ОН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2624021C2 |
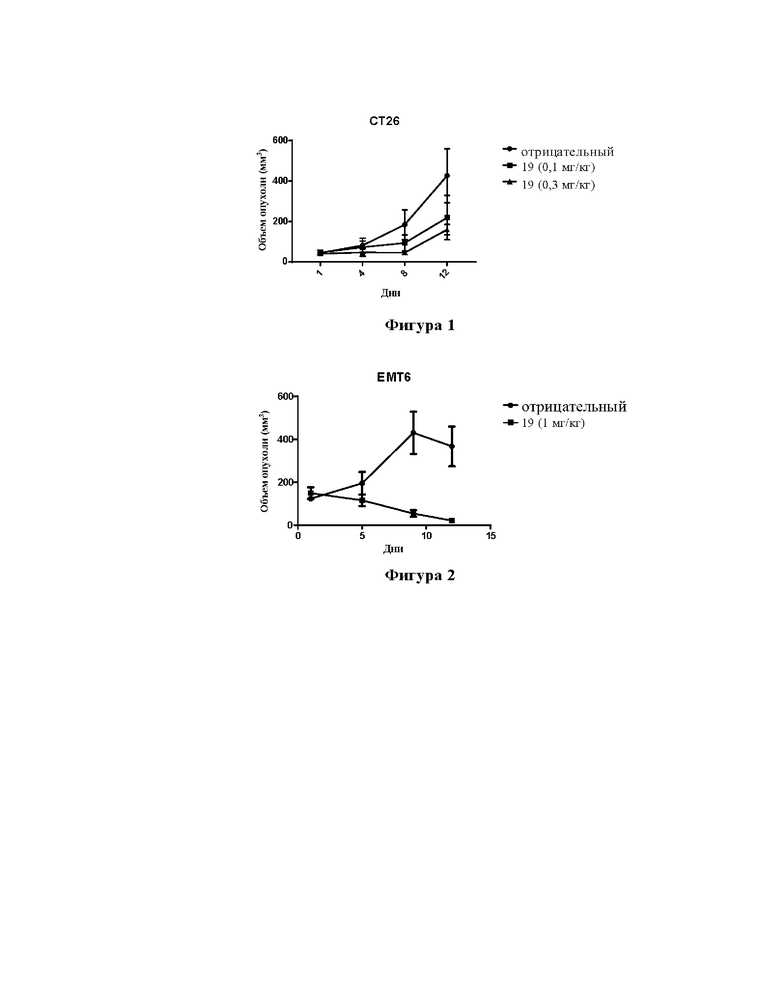
Изобретение относится к гетероциклическому амидному соединению общей формулы I, где A, B, C, R1-R4, R2’-R4’, RB определены в формуле изобретения, и его фармацевтически приемлемой соли, способу его получения, а также применению соединения для получения препарата для лечения заболеваний, связанных с активностью STING, для получения иммунологического адъюванта и для получения препарата для активации STING. 8 н. и 8 з.п. ф-лы, 2 ил., 2 табл., 26 пр.
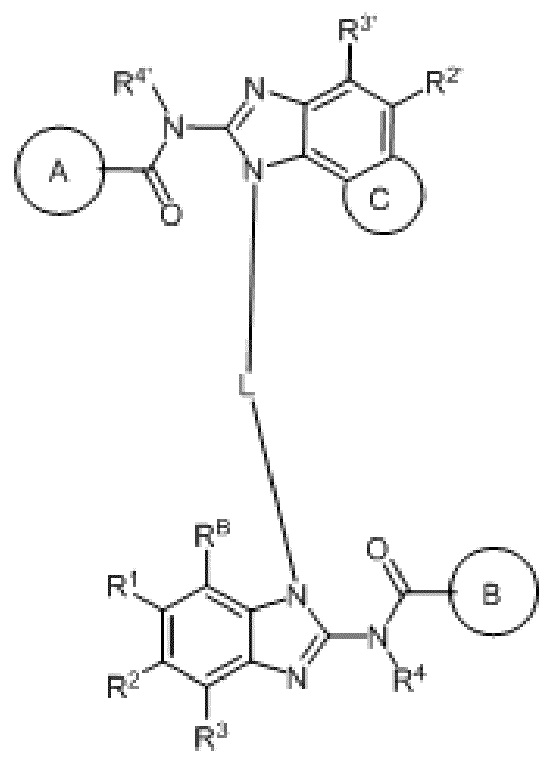 Формула I
Формула I
1. Гетероциклическое амидное соединение формулы (I) или его фармацевтически приемлемая соль:
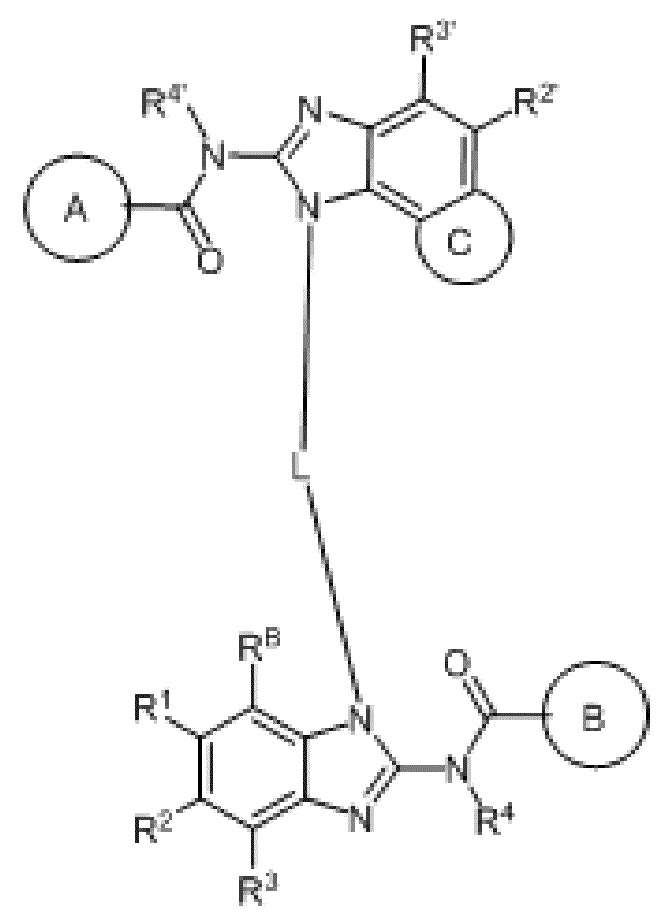
Формула I,
где кольцо A выбирают из 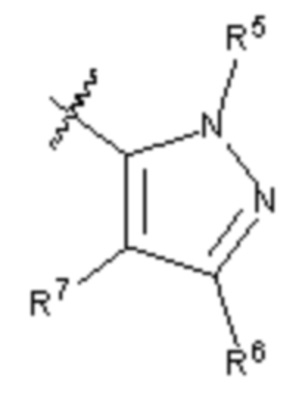 ;
;
кольцо B выбирают из 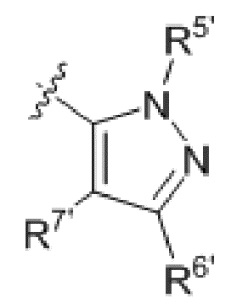 ;
;
C выбирают из насыщенного или ненасыщенного 4-6-членного гетероциклического кольца, содержащего гетероатом O, замещенного 0-4 Ri;
R4 и R4' каждый выбирают из водорода или C1-C6 алкила;
R5 и R5' каждый выбирают из C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R6 и R7 каждый выбирают из водорода, C1-C6 алкила, галогена или галогенированного C1-C6 алкила;
R6' и R7' каждый выбирают из водорода, C1-C6 алкила, галогена или галогенированного C1-C6 алкила;
R1, R2, R3, R2' и R3' каждый выбирают из водорода- или -C(O)NRaRb;
Ra, Rb каждый выбирают из водорода или C1-C6 алкила;
RB представляет собой -ORf или RB и R1 вместе с атомами, с которыми они соединены, циклизованы в ненасыщенное 4-6-членное гетероциклическое кольцо, содержащее гетероатом O;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила или галогенированного C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкилена, C4-C6 алкенилена или C4-C6 алкинилена;
фрагмент 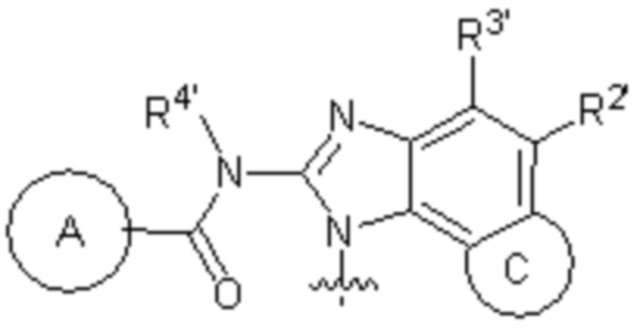 выбирают из
выбирают из 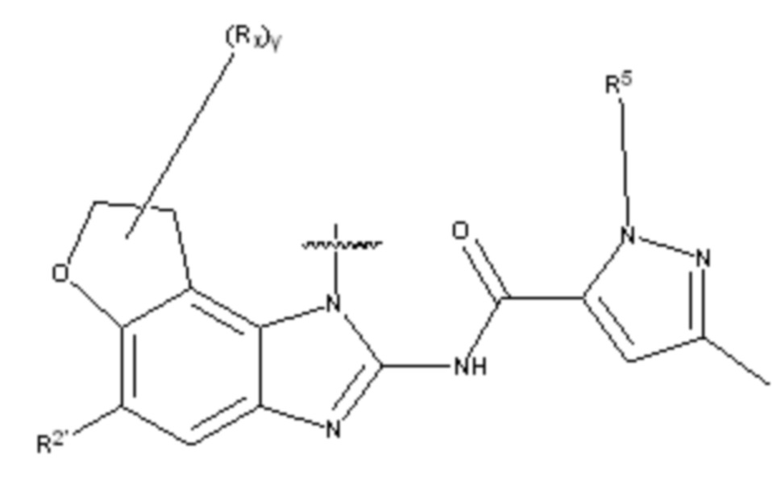 или
или 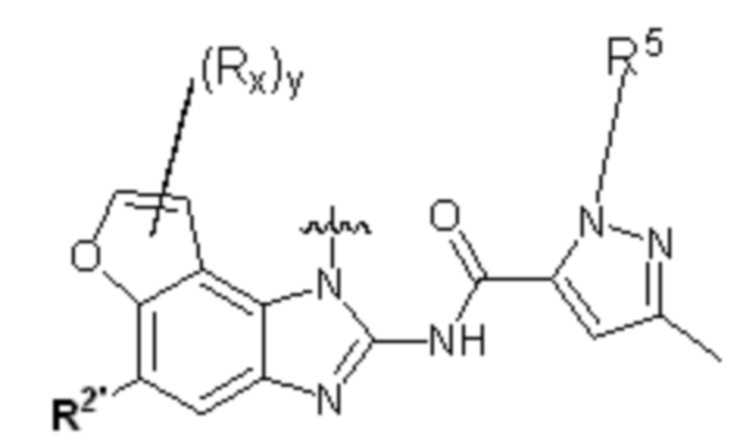 ,
,
где y представляет собой 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из C1-C6 алкила или галогенированного C1-C6 алкила.
2. Гетероциклическое амидное соединение формулы (I) или его фармацевтически приемлемая соль:
Формула I,
где кольцо A выбирают из 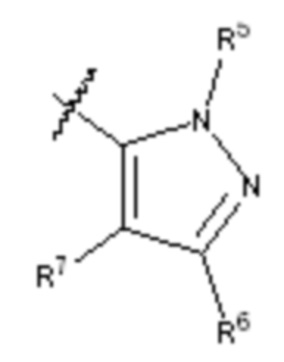 ;
;
кольцо B выбирают из ;
C выбирают из насыщенного или ненасыщенного 4-6-членного гетероциклического кольца, содержащего гетероатом O, замещенного 0-4 Ri;
R4 и R4' каждый выбирают из водорода или C1-C6 алкила;
R5 и R5' каждый независимо друг от друга выбирают из C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила, галогенированного C1-C6 алкила или -(CH2)s-L3, где s равно 1, 2 или 3, а L3 выбирают из C3-C6 циклоалкила;
R6 и R7 каждый выбирают из водорода, C1-C6 алкила, галогена или галогенированного C1-C6 алкила;
R6' и R7' каждый выбирают из водорода, C1-C6 алкила, галогена или галогенированного C1-C6 алкила;
R1, R2, R3, R2' и R3' каждый выбирают из водорода или -C(O)NRaRb;
Ra, Rb каждый выбирают из водорода или C1-C6 алкила;
RB представляет собой -ORf или RB и R1 вместе с атомами, с которыми они соединены, циклизованы в ненасыщенное 4-6-членное гетероциклическое кольцо, содержащее гетероатом O;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила или галогенированного C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкилена, C4-C6 алкенилена или C4-C6 алкинилена;
фрагмент  выбирают из
выбирают из 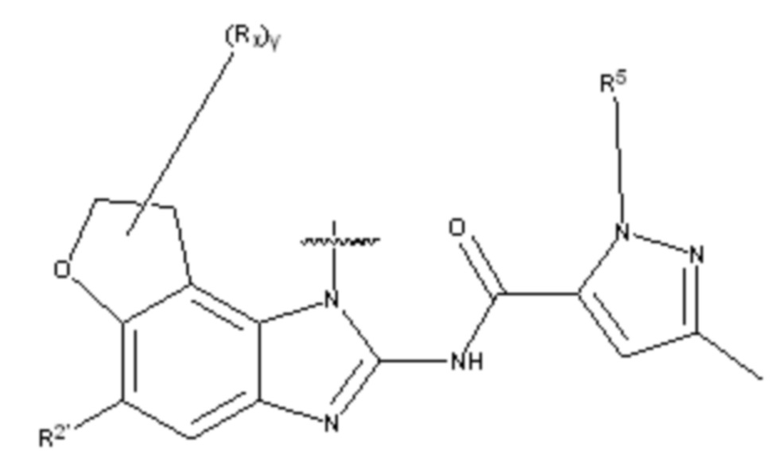 или
или 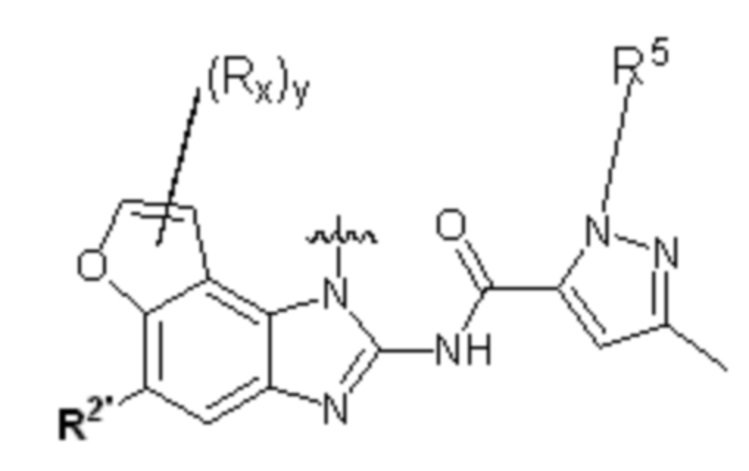 ,
,
где y представляет собой 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из C1-C6 алкила или галогенированного C1-C6 алкила.
3. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где кольцо A и кольцо B одинаковые или разные, каждое из них независимо друг от друга представляет собой  ,
,
R5 определен в п. 1.
4. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где кольцо A и кольцо B одинаковые, каждое из них независимо друг от друга представляет собой  , где R5 выбирают из C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила.
, где R5 выбирают из C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила.
5. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где фрагмент 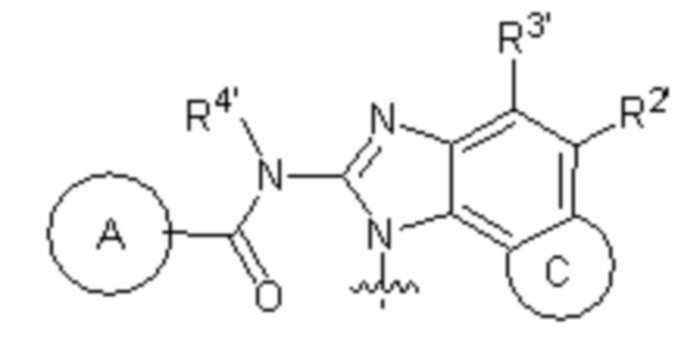 представляет собой
представляет собой 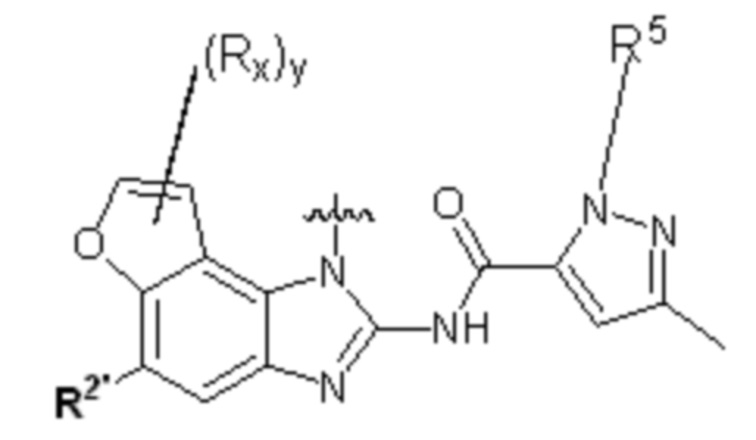 ,
,
где y равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из C1-C6 алкила или галогенированного C1-C6 алкила,
R5 и R2' определены в п. 1.
6. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где фрагмент 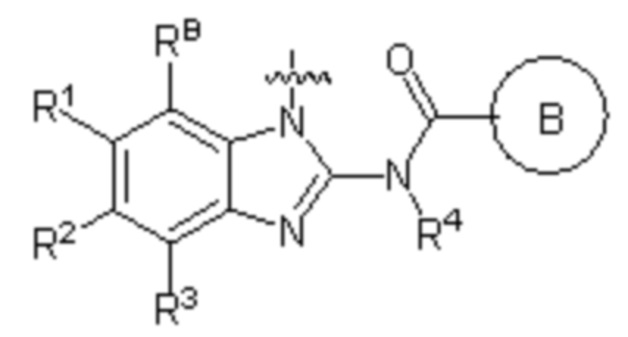 выбирают из
выбирают из  или
или 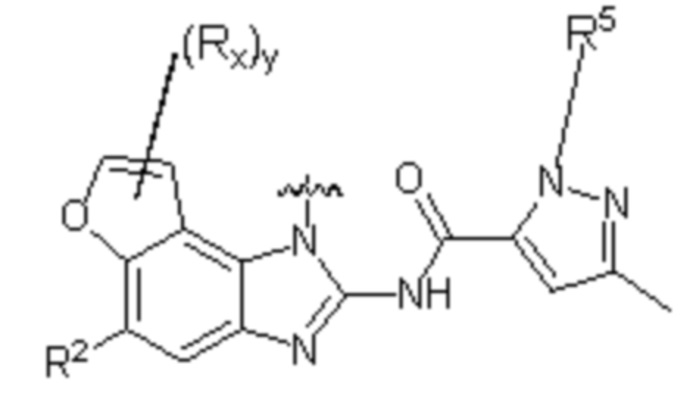 ,
,
где y равно 0, 1, 2, 3 или 4;
каждый Rx независимо выбирают из C1-C6 алкила или галогенированного C1-C6 алкила;
R5 выбирают из C1-C6 алкила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R2 независимо выбирают из водорода или -C(O)NRaRb;
где Ra и Rb каждый независимо друг от друга выбирают из водорода или C1-C6 алкила;
RB выбирают из -ORf;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила.
7. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где RB выбирают из -ORf;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила.
8. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где RB выбирают из метокси,  ,
,  ,
,  или
или  .
.
9. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где:
R4 и R4' каждый представляют собой водород;
R5 и R5' каждый выбирают из C1-C6 алкила, C3-C6 циклоалкила, C3-C6 алкинила, C3-C6 алкенила или галогенированного C1-C6 алкила;
R6 и R7 каждый выбирают из водорода или C1-C6 алкила;
R6' и R7' каждый выбирают из водорода или C1-C6 алкила;
R1, R3 и R3' каждый выбирают из водорода;
R2 и R2' каждый выбирают из -C(O)NRaRb;
Ra и Rb каждый выбирают из водорода или C1-C6 алкила;
RB выбирают из -ORf;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила;
L выбирают из C4-C6 алкенилена или C4-C6 алкилена.
10. Гетероциклическое амидное соединение формулы I или его фармацевтически приемлемая соль по п. 1 или 2, где:
R6' и R7' каждый независимо друг от друга выбирают из водорода или C1-C6 алкила;
R6' и R7' каждый независимо друг от друга выбирают из водорода или C1-C6 алкила;
R1, R3, и R3' представляют собой водород;
R2 и R2' представляют собой -C(O)NH2;
RB выбирают из -ORf;
Rf выбирают из C1-C6 алкила, замещенного 0-4 Rh, 3-6-членного циклоалкила, замещенного 0-4 Ri, или 3-6-членного гетероциклоалкила, содержащего гетероатом O;
Rh выбирают из -ORj, 3-6-членного циклоалкила или 3-6-членного гетероциклоалкила, содержащего гетероатомы, выбранные из N, O;
Ri выбирают из C1-C6 алкила;
Rj выбирают из водорода или C1-C6 алкила.
11. Гетероциклическое амидное соединение или его фармацевтически приемлемая соль, где соединение представляет собой одно из следующих соединений:
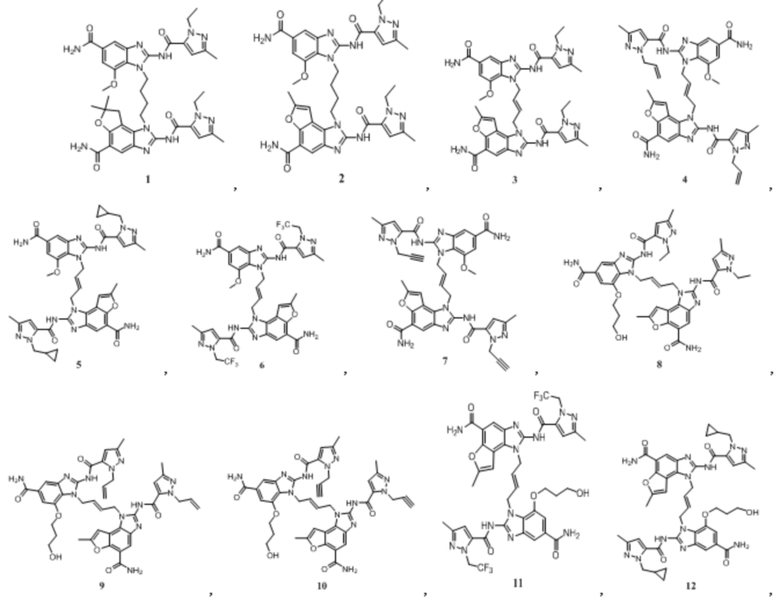
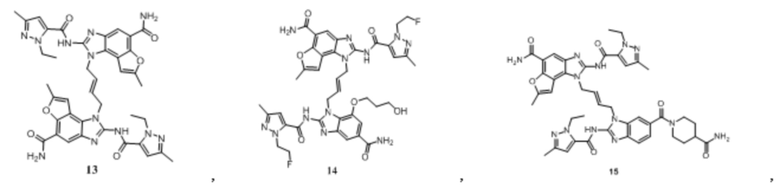
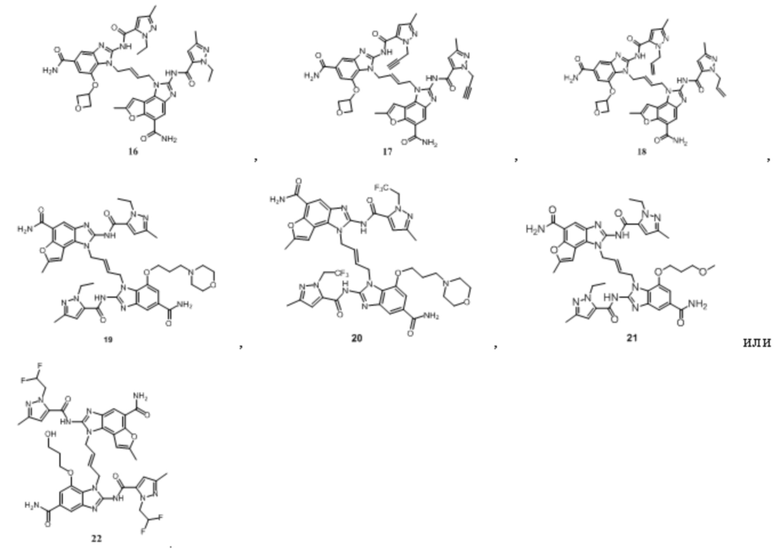
12. Фармацевтическая композиция для профилактики и/или лечения заболевания, связанного с киназой STING, включающая терапевтически эффективное количество одного или более гетероциклических амидных соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-11, а также фармацевтически приемлемый носитель.
13. Фармацевтическая композиция для профилактики и/или лечения заболевания, связанного с киназой STING, включающая терапевтически эффективное количество одного или более гетероциклических амидных соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-11, а также фармацевтически приемлемый адъювант.
14. Способ получения гетероциклического амидного соединения формулы (I) или его фармацевтически приемлемой соли по п. 1, который включает следующие этапы:
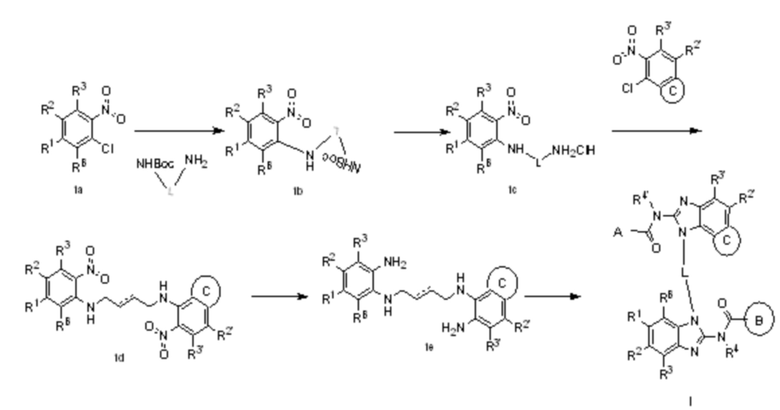
(i) в инертном растворителе в присутствии основания сырье 1a вступает в реакцию нуклеофильного замещения с моно-БОК-защищенным диамином 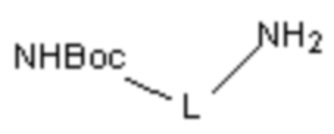 с образованием соединения 1b;
с образованием соединения 1b;
(ii) в инертном растворителе в присутствии кислоты снимается БОК-защита из соединения 1b с образованием промежуточного соединения 1c;
(iii) в инертном растворителе в присутствии основания промежуточное соединение 1c вступает в реакцию замещения с 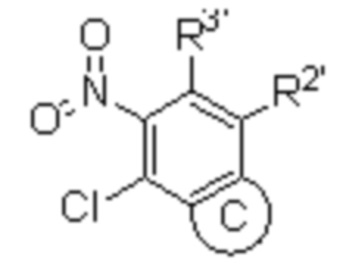 с образованием промежуточного соединения 1d;
с образованием промежуточного соединения 1d;
(iv) в инертном растворителе в присутствии восстановителя промежуточное соединение 1d реагирует с образованием соединения 1e;
(v) в инертном растворителе в присутствии катализатора соединение 1e реагирует с 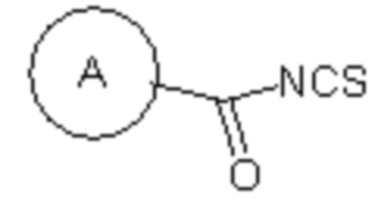 или
или 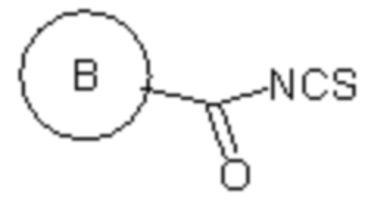 с образованием целевого соединения I;
с образованием целевого соединения I;
где A, B, L, R1, R2, R3, R4, RB, R2 ', R3' и R4' определены в п. 1.
15. Применение гетероциклического амидного соединения формулы (I) или его фармацевтически приемлемой соли по п. 1-11 для получения иммунного адъюванта или лекарственного препарата для активации STING.
16. Применение гетероциклического амидного соединения формулы (I) или его фармацевтически приемлемой соли по п. 1-11 для получения лекарственного препарата для лечения заболевания, связанного с активностью STING; где заболевание, связанное с активностью STING, представляет собой одно или более из заболеваний, связанных с воспалением, аутоиммунное заболевание, инфекционное заболевание, онкологическое заболевание или предраковый синдром.
| WO 2020028566 A1, 06.02.2020 | |||
| WO 2019069275 A1, 11.04.2019 | |||
| EA 201892424 A1, 30.04.2019 | |||
| WO 2017175147 A1, 12.10.2017. |

