Перекрестная ссылка на родственные заявки
[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США № 62/574544, поданной 19 октября 2017; содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
Декларация государственного интереса
[0002] Настоящее изобретение реализовано при государственной поддержке из федеральных фондов управления перспективных биомедицинских исследований и разработок, канцелярии помощника секретаря по вопросам готовности и реагирования, канцелярии секретаря, министерства здравоохранения и социального обеспечения по контракту № HHSO100201000046C. Правительство имеет определенные права на настоящее изобретение.
Область техники
[0003] Настоящее изобретение относится к новым способам получения антибактериальных аминогликозидных соединений, а также к родственным промежуточным соединениям и кристаллическим формам промежуточных соединений, подходящим для применения в таких способах.
Уровень техники
[0004] В настоящее время по меньшей мере 30% всех госпитализированных пациентов получают один или более курсов терапии антибиотиками, и миллионы потенциально смертельных инфекций были вылечены. Указанные фармацевтические агенты стали одними из самых неправильно применяемых среди доступных практикующему врачу. Тем не менее, одним из результатов широкого применения противомикробных агентов стало появление резистентных к антибиотикам патогенов, что, в свою очередь, привело к возникновению постоянно возрастающей потребности в новых лекарственных средствах.
[0005] При первом испытании противомикробной активности нового агента обычно определяют профиль чувствительности и устойчивости. К сожалению, указанный спектр активности может впоследствии измениться в значительной степени из-за появления у микроорганизмов множества оригинальных изменений, которые позволяют им выживать в присутствии антибиотиков. Механизм резистентности к лекарственным средствам варьируется от микроорганизма к микроорганизму и от лекарственного средства к лекарственному средству.
[0006] Усилия по разработке новых аминогликозидных антибиотиков, обладающих активностью в отношении грам-отрицательных бактерий с лекарственной мультирезистентностью, привели к аналогам сизомицина и неомицина, модифицированных по положениям 6’- и N-1. Известные некоторые способы синтеза селективной функциональности положения 1-N. Селективную модификацию положения 1-N обычно осуществляют в многостадийной процедуре с применением защитных групп. Сперва положения 1-N и 3”N аминогликозидного остова блокируют путем образования комплекса переходного металла с ионом двухвалентного металла (обычно цинка, никеля, меди или кобальта). Затем все другие положения (6’- 2’- и 3-N) защищают с применением стандартных азотзащитных групп (обычно указанные амины защищают в виде карбаматов или ацетатов). После защиты положений 6’- 2’- и 3-N комплексы переходного металла удаляют и положение 1-N модифицируют с высокой селективностью. При этом положения 6’- 2’- и 3-N обычно защищены, и единственным оставшимся свободным амином является вторичный 3”N амин, который реагирует медленнее, чем 1-N амин, в типичных реакциях ацилирования или алкилирования.
[0007] Напротив, селективная функционализация положения 6’-N остается серьезной проблемой. Положение 6’-N является наиболее реакционноспособным положением в множестве условий реакций, и предыдущие способы основывались на указанной сравнительно высокой реакционной способности для функционализации или защиты 6’-N амина. Тем не менее, разница в реакционной способности между положением 6’-N и другими положениями (в частности, 2’-, 3- и 1-N) является небольшой. В результате попытки непосредственно функционализировать положение 6’-N осложняются образованием изомерных побочных продуктов и побочных продуктов избыточного протекания реакции (дифункционализированных или трифункционализированных аминогликозидных производных). Образование побочных продуктов в больших количествах требует осуществления процедур очистки, которые увеличивают время и стоимость производства функционализированных по положению 6’-N аминогликозидных производных, таких как описанные в патентах США № 8383596; № 8822424; № 9266919, № 9688711; и публикации США № 2012-0214759.
[0008] Для ускорения процесса обнаружения и разработки лекарственных средств необходимы новые способы синтеза аминогликозидных антибиотиков для обеспечения множества соединений, которые являются потенциальными новыми лекарственными средствами для лечения бактериальных инфекций. Настоящее изобретение может удовлетворить указанные потребности и обеспечить дополнительные сопутствующие преимущества.
Краткое описание изобретения
[0009] Вкратце, настоящее изобретение относится к новым способам получения антибактериальных аминогликозидных соединений и новым промежуточным соединениям и кристаллическим формам некоторых промежуточных соединений, применяемым в новых способах.
[0010] В настоящем изобретении предложены способы получения соединений формулы (9), которые включают плазомицин, которые являются масштабируемыми, воспроизводимыми в промышленном масштабе и обладают хорошими выходами. Указанные способы включают реакции, которые могут обеспечивать новые промежуточные соединения, получаемые в результате экспериментов и разработки новых комбинаций условий реакций. Способы также могут включать кристаллизацию некоторых промежуточных соединений. Кристаллизация указанных конкретных промежуточных соединений неожиданно способствует улучшению очистки (например, снижению содержания примесей) и может упрощать очистку по сравнению с предыдущими способами очистки.
[0011] Один из аспектов настоящего изобретения относится к способу получения соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера, включающему: (a) приведение соединения формулы (1):
 (1),
(1),
или его энантиомера, или его диастереомера в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt) с получением соединения формулы (2):
 (2),
(2),
или его соли, или его сольвата, или его энантиомера, или его диастереомера; где  представляет собой одинарную связь или двойную связь; R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил. В некоторых таких вариантах реализации стадию (a) осуществляют в присутствии растворителя, выбранного из группы, состоящей из дихлорметана, метанола и их комбинации. В некоторых вариантах реализации PNZ-Bt присутствует в количестве от примерно 1,0 до 1,2 молярного эквивалента в расчете на соединение формулы (1), или его энантиомер, или его диастереомер.
представляет собой одинарную связь или двойную связь; R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил. В некоторых таких вариантах реализации стадию (a) осуществляют в присутствии растворителя, выбранного из группы, состоящей из дихлорметана, метанола и их комбинации. В некоторых вариантах реализации PNZ-Bt присутствует в количестве от примерно 1,0 до 1,2 молярного эквивалента в расчете на соединение формулы (1), или его энантиомер, или его диастереомер.
[0012] В некоторых вариантах реализации изложенного выше или далее представляет собой одинарную связь или двойную связь. В некоторых вариантах реализации изложенного выше или далее представляет собой одинарную связь. В других вариантах реализации изложенного выше или далее представляет собой двойную связь. В некоторых вариантах реализации изложенного выше или далее R1, R2 или R3 представляют собой H.
[0013] Другой аспект настоящего изобретения относится к способу, дополнительно включающему стадию (b1) или (b2): (b1), где, если R1, R2 и R3 представляют собой H, приведение соединения формулы (2) в контакт с реагентом для введения Boc-защиты приводит к получению соединения формулы (3):
 (3),
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера; или (b2), где, если один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил, сперва удаление указанного C1-C3 алкила, а затем приведение соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты приводит к получению соединения формулы (3), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации реагент для введения Boc-защиты представляет собой Boc2O или Boc-ONb. В некоторых вариантах реализации стадию (b1) или (b2) осуществляют в присутствии кислоты Льюиса. В некоторых таких вариантах реализации Кислота Льюиса представляет собой Zn(OAc)2, ZnCl2 или Zn(OPiv)2. В некоторых вариантах реализации Кислота Льюиса содержит ион меди или ион никеля. В некоторых вариантах реализации стадию (b1) или (b2) осуществляют в присутствии триэтиламина. В некоторых вариантах реализации стадию (b1) или (b2) осуществляют в присутствии метанола.
[0014] Один из аспектов настоящего изобретения относится к способу, дополнительно включающему: (c) приведение соединения формулы (3), или его соли, или его сольвата, или его энантиомера, или его диастереомера, в контакт с

с получением соединения формулы (4):
 (4),
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации стадию (c) осуществляют в присутствии активирующего реагента и реагента для образования пептидной связи. В некоторых таких вариантах реализации активирующий реагент представляет собой HOBt. В некоторых таких вариантах реализации активирующий реагент присутствует в количестве от примерно 0,05 до 1,0 молярного эквивалента в расчете на
.
В некоторых вариантах реализации реагент для образования пептидной связи представляет собой EDAC или PyBOP. В некоторых таких вариантах реализации реагент для образования пептидной связи присутствует в количестве от примерно 1,0 до 1,4 молярного эквивалента в расчете на
.
[0015] В некоторых вариантах реализации стадию (c) осуществляют в кислотных условиях. В некоторых таких вариантах реализации кислотные условия представляют собой pH от примерно 4 до 7. В некоторых таких вариантах реализации кислотные условия представляют собой pH примерно 5.
[0016] Один из аспектов настоящего изобретения относится к способу, дополнительно включающему получение кристаллической формы соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации способ дополнительно включает выделение соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
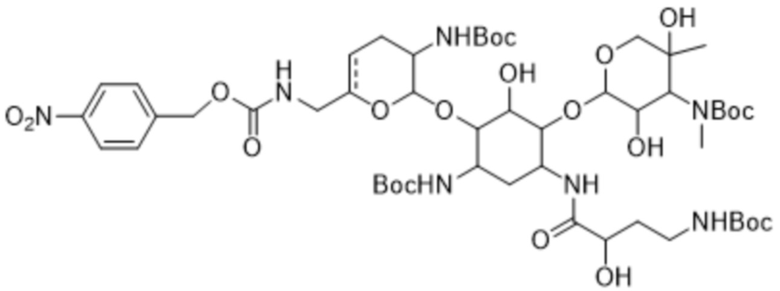
[0017] Другой аспект настоящего изобретения относится к способу, дополнительно включающему: (d) приведение соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты с получением соединения формулы (5):
 (5),
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации реагент для введения Boc-защиты представляет собой Boc2O. В некоторых вариантах реализации стадию (d) осуществляют в присутствии спирта. В некоторых таких вариантах реализации спирт представляет собой метанол. В некоторых вариантах реализации стадию (d) осуществляют при температуре вплоть до примерно 60°C.
[0018] Один из аспектов настоящего изобретения относится к способу, дополнительно включающему: (e) приведение соединения формулы (5), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия PNZ-защиты с получением соединения формулы (6):
 (6),
(6),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации реагент для снятия PNZ-защиты представляет собой дитионит натрия.
[0019] Другой аспект настоящего изобретения относится к способу, дополнительно включающему получение кристаллической формы соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации способ дополнительно включает выделение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[0020] В некоторых вариантах реализации способ дополнительно включает (f) приведение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с 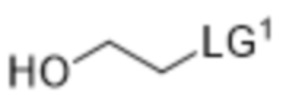 , где LG1 представляет собой уходящую группу, с получением соединения формулы (7):
, где LG1 представляет собой уходящую группу, с получением соединения формулы (7):
 (7),
(7),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации уходящая группа представляет собой йод-. В некоторых вариантах реализации присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6). В некоторых вариантах реализации стадию (f) осуществляют в по существу безводных условиях. В некоторых вариантах реализации стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации. В некоторых вариантах реализации стадию (f) осуществляют в присутствии NaHCO3. В некоторых вариантах реализации стадию (f) осуществляют при температуре от примерно 30°C до 40°C. В некоторых вариантах реализации стадия (f) дополнительно включает добавление к реакционной смеси 1,4-диазабицикло[2.2.2]октана (DABCO).
[0021] Один из аспектов настоящего изобретения относится к способу, дополнительно включающему получение кристаллической формы соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера. Другой аспект настоящего изобретения относится к способу, дополнительно включающему выделение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[0022] Другой аспект настоящего изобретения относится к способу, дополнительно включающему: (g) приведение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия Boc-защиты с получением соединения формулы (8):
 (8),
(8),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации стадии (g) реагент для снятия Boc-защиты представляет собой ТФК, что приводит к получению ТФК соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации способ дополнительно включает удаление ТФК соли с получением соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[0023] Один из аспектов настоящего изобретения относится к способу, дополнительно включающему: (h) осуществление образования соли с кислотой с получением соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации кислота на стадии (h) представляет собой серную кислоту, что приводит к получению сульфатной соли соединения формулы (9):
• x H2SO4 (9),
или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5.
[0024] В некоторых вариантах реализации любого изложенного выше или далее стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3” и 4” в формулах (1)-(3) указана, как в формуле (X), где  обозначает место прикрепления к водороду или фрагменту:
обозначает место прикрепления к водороду или фрагменту:
 (X).
(X).
[0025] В некоторых вариантах реализации любого изложенного выше или далее стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3”, 4” и 1-z в формулах (4)-(9) указана, как в формуле (Y), где обозначает место прикрепления к водороду или фрагменту:
 (Y).
(Y).
[0026] Один из аспектов настоящего изобретения относится к способу получения соединения формулы (5):
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера, включающему: (a) приведение соединения формулы (4):
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты где представляет собой одинарную связь или двойную связь. В некоторых вариантах реализации соединение формулы (4), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (3):
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с
.
[0027] В других вариантах реализации соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем (b1) приведения соединения формулы (2a):
 (2a),
(2a),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты; или (b2) удаления C1-C3 алкила в соединении формулы (2):
(2),
или его соли, или его сольвате, или его энантиомере, или его диастереомере; где R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил, и где один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил; с последующим приведением соединения формулы (2) в контакт с реагентом для введения Boc-защиты.
[0028] В некоторых вариантах реализации соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (1):
(1),
или его энантиомера, или его диастереомера в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt). В некоторых вариантах реализации реагент для введения Boc-защиты на стадии (a), (b1) или (b2) представляет собой Boc2O. В некоторых вариантах реализации стадию (a), (b1) или (b2) осуществляют в присутствии спирта. В некоторых таких вариантах реализации спирт представляет собой метанол. В некоторых вариантах реализации стадию (a), (b1) или (b2) осуществляют при температуре вплоть до примерно 60°C.
[0029] Один из аспектов настоящего изобретения относится к способу получения соединения формулы (7):
(7),
или его соли, или его сольвата, или его энантиомера, или его диастереомера, включающему: (f) приведение соединения формулы (6),
(6),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с , где LG1 представляет собой уходящую группу, и где представляет собой одинарную связь или двойную связь. В некоторых таких вариантах реализации уходящая группа представляет собой йод-. В некоторых вариантах реализации присутствует в количестве от примерно 1,0 до 1.5 молярного эквивалента в расчете на соединение формулы (6). В некоторых вариантах реализации стадию (f) осуществляют в по существу безводных условиях. В некоторых вариантах реализации стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации. В некоторых вариантах реализации стадию (f) осуществляют в присутствии NaHCO3. В некоторых вариантах реализации стадию (f) осуществляют при температуре от примерно 30°C до 40°C. В некоторых вариантах реализации стадия (f) дополнительно включает добавление к реакционной смеси 1,4-диазабицикло[2.2.2]октана (DABCO).
[0030] Другой аспект настоящего изобретения относится к способу, в котором соединение формулы (6), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (5):
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия PNZ-защиты.
[0031] Один из аспектов настоящего изобретения относится к соединению формулы (4):
 (4),
(4),
или его соли, или его сольвату, или его энантиомеру, или его диастереомеру. В некоторых таких вариантах реализации соединение формулы (4) представляет собой соединение формулы:
 (4a),
(4a),
или его соль, или его сольват.
[0032] Другой аспект настоящего изобретения относится к кристаллическому трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамату формулы (4a) или его сольвату.
[0033] Один из аспектов настоящего изобретения относится к способу получения кристаллического трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамата формулы (4a) или его сольвата, включающему:
(a) обработку соединения формулы (4a), или его соли, или его сольвата ацетонитрилом с получением раствора;
(b) нагревание раствора со стадии (a);
(c) добавление воды к нагретому раствору со стадии (b);
(d) охлаждение раствора со стадии (c);
(e) внесение в раствор со стадии (d) затравочного кристалла; и
(f) выделение полученных твердых веществ с получением кристаллического соединения формулы (4a) или его сольвата.
[0034] Другой аспект настоящего изобретения относится к кристаллическому трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамату формулы (6a) или его сольвату.
[0035] Один из аспектов настоящего изобретения относится к способу получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) или его сольвата, включающему:
(a) обработку соединения формулы (6), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление воды к раствору со стадии (a) с получением смеси;
(c) добавление дихлорметана к смеси со стадии (b) с получением смеси;
(d) внесение в смесь со стадии (c) затравочного кристалла;
(e) выделение полученных твердых веществ с получением кристаллического соединения формулы (6a) или его сольвата.
В некоторых таких вариантах реализации стадию (d) осуществляют при низкой температуре.
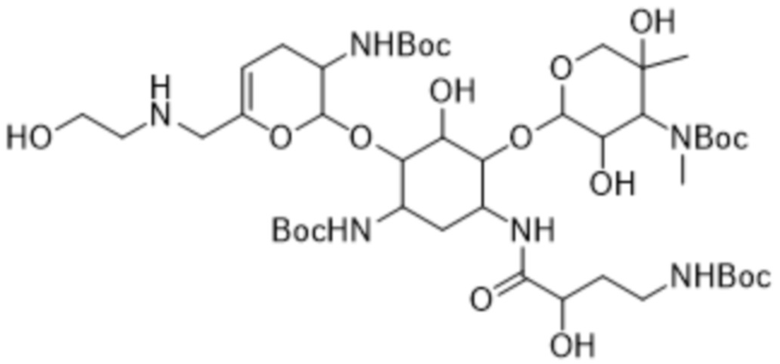
[0036] Другой аспект настоящего изобретения относится к соединению формулы (7):
 ,
,
или его соли, или его сольвату, или его энантиомеру, или его диастереомеру. В некоторых таких вариантах реализации соединение формулы (7) представляет собой соединение формулы:
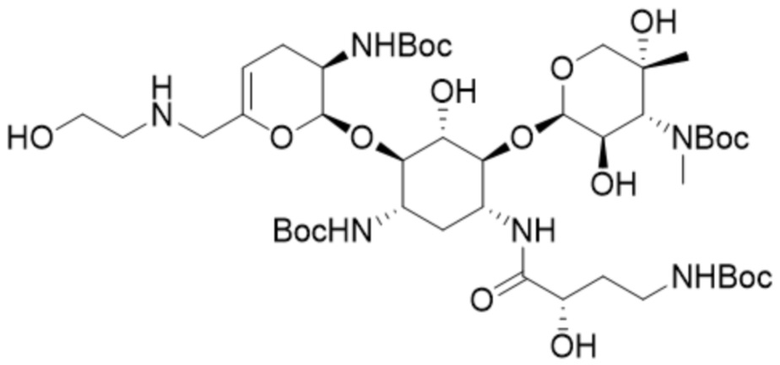 (7a),
(7a),
или его соль, или его сольват.
[0037] Один из аспектов настоящего изобретения относится к кристаллическому трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамату формулы (7a) или его сольвату.
[0038] Другой аспект настоящего изобретения относится к способу получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) или его сольвата, включающему:
(a) обработку соединения формулы (7a), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление ацетонитрила к раствору со стадии (a) с получением смеси;
(c) внесение в смесь со стадии (b) затравочного кристалла;
(d) выделение полученных твердых веществ с получением кристаллического соединения формулы (7a) или его сольвата.
[0039] Детали настоящего изобретения представлены в прилагаемом ниже описании. Хотя способы и материалы, подобные или эквивалентные описанным в настоящем изобретении, можно применять при реализации на практике или испытании настоящего изобретения, далее описаны иллюстративные способы и материалы. Другие отличительные признаки, объекты и преимущества настоящего изобретения будут очевидны из описания и пунктов формулы изобретения. В описании и пунктах прилагаемой формулы изобретения формы единственного числа также включают формы множественного числа, если в контексте явно не указано иное. Если не указано иное, все технические и научные термины, применяемые в настоящем описании, имеют то же значение, которое обычно понимают специалисты в области техники, к которой относится настоящее изобретение. Все патенты и публикации, процитированные в настоящем описании, включены в настоящее описание во всей полноте посредством ссылки.
[0040] Каждый из вариантов реализации, описанных в настоящей заявке, можно использовать индивидуально или в комбинации с любым одним или более другими вариантами реализации.
Краткое описание чертежей
[0041] Различные аспекты настоящего изобретения проиллюстрированы со ссылкой на прилагаемые чертежи.
[0042] На ФИГ. 1 представлен XRPD спектр соединения 4a.
[0043] На ФИГ. 2 представлен ТГА профиль соединения 4a.
[0044] На ФИГ. 3 представлен ДСК профиль соединения 4a.
[0045] На ФИГ. 4 представлен XRPD спектр соединения 6a.
[0046] На ФИГ. 5 представлен ТГА профиль соединения 6a.
[0047] На ФИГ. 6 представлен ДСК профиль соединения 6a.
[0048] На ФИГ. 7 представлен XRPD спектр соединения 7a.
[0049] На ФИГ. 8 представлен ТГА профиль соединения 7a.
[0050] На ФИГ. 9 представлен ДСК профиль соединения 7a.
Подробное описание изобретения
[0051] Настоящее изобретение относится к новым способам получения антибактериальных аминогликозидных соединений, а также к родственным промежуточным соединениям, подходящим для применения в таких способах, и кристаллическим формам конкретных промежуточных соединений.
[0052] Как обсуждалось выше, в настоящем изобретении предложены способы получения соединений формулы (9), которые включают сульфат плазомицина, которые являются масштабируемыми и воспроизводимыми в промышленном масштабе с хорошими выходами. Способы включают комбинации реакций и условий, которые могут обеспечить получение конкретных новых промежуточных соединений. В конкретных вариантах реализации способы также включают кристаллизацию конкретных промежуточных соединений, для которых кристаллизация неожиданно способствует процессу очистки (например, путем очистки от примесей и, следовательно, снижения содержания примесей), таким образом обеспечивая более простую очистку по сравнению с предыдущими способами очистки.
[0053] В одном аспекте настоящее изобретение относится к способу получения соединений формулы (2):
(2),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь; R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил.
[0054] В другом аспекте настоящее изобретение относится к способу получения соединений формулы (3):
(3),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0055] В одном аспекте настоящее изобретение относится к способу получения соединений формулы (4):
(4),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0056] В другом аспекте настоящее изобретение относится к способу получения соединений формулы (5):
(5),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0057] В одном аспекте настоящее изобретение относится к способу получения соединений формулы (6):
(6),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0058] В другом аспекте настоящее изобретение относится к способу получения соединений формулы (7):
(7),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0059] В некоторых аспектах настоящее изобретение относится к способу получения соединений формулы (8):
(8),
или их солей, или их сольватов, или их энантиомеров, или их диастереомеров, где представляет собой одинарную связь или двойную связь.
[0060] В другом аспекте настоящее изобретение относится к способу получения сульфатной соли соединения формулы (9):
• x H2SO4 (9),
или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5, и где представляет собой одинарную связь или двойную связь.
[0061] В некоторых вариантах реализации стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3” и 4” в формулах (1)-(3) указана, как в формуле (X), где обозначает место прикрепления к водороду или фрагменту:
(X).
[0062] В некоторых вариантах реализации стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3”, 4” и 1-z в формулах (4)-(9) указана, как в формуле (Y), где обозначает место прикрепления к водороду или фрагменту:
(Y).
[0063] В одном аспекте настоящее изобретение относится к соединению формулы (4):
(4),
или его соли, или его сольвату, или его энантиомеру, или его диастереомеру.
[0064] В некоторых вариантах реализации соединение формулы (4) представляет собой соединение формулы:
(4a),
или его соль, или его сольват.
[0065] В некоторых вариантах реализации настоящее изобретение также относится к кристаллическому трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамату формулы (4a) или его сольвату.
[0066] В некоторых вариантах реализации настоящее изобретение также относится к кристаллическому трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамату формулы (6a):
 (6a)
(6a)
или его сольвату.
[0067] В одном аспекте настоящее изобретение относится к соединению формулы (7):
,
или его соли, или его сольвату, или его энантиомеру, или его диастереомеру.
[0068] В некоторых вариантах реализации соединение формулы (7) представляет собой соединение формулы:
(7a),
или его соль, или его сольват.
[0069] В некоторых вариантах реализации настоящее изобретение относится к кристаллическому трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамату формулы (7a) или его сольвату.
Термины и сокращения
[0070] Формы единственного числа (соответствующие англ. артиклям “a” и “an”), применяемые в настоящем описании, могут относиться к одному или более, чем одному (т.е. к по меньшей мере одному), грамматическим объектам. Например, “элемент ” обозначает один элемент или более одного элемента.
[0071] Применяемый в настоящем описании “и/или” может обозначать “и” ии “или”, если не указано иное.
[0072] Применяемый в настоящем описании может относиться к одинарной связи или двойной связи.
[0073] “Алкил” может относиться к насыщенному углеводороду с прямой или разветвленной цепью. C1-C3 алкильные группы содержат от 1 до 3 атомов углерода. Примеры C1-C3 алкильных групп включают, но не ограничиваются ими, метил, этил и пропил.
[0074] “Реагент для введения Boc-защиты” может относиться к реагенту, который можно применять для прикрепления защитной группы Boc к аминогруппе. Примеры реагентов для введения Boc-защиты включают, но не ограничиваются ими, Boc-ангидрид (Boc2O), N-трет-бутоксикарбонилимидазол, 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил, 2-(трет-бутоксикарбонилтио)-4,6-диметилпиримидин, 1-трет-бутоксикарбонил-1,2,4-триазол, трет-бутилфенилкарбонат, N-(трет-бутоксикарбонилокси)фталимид, трет-бутил-2,4,5-трихлорфенилкарбонат и трет-бутил-((4R,7S)-1,3-диоксо-1,3,3a,4,7,7a-гексагидро-2H-4,7-метаноизоиндол-2-ил)карбонат (Boc-ONb).
[0075] “Реагент для снятия Boc-защиты” может относиться к реагенту, который можно применять для отщепления защитной группы Boc от аминогруппы. Примеры реагентов для снятия Boc-защиты включают, но не ограничиваются ими, ТФК, водный раствор фосфорной кислоты, метансульфоновую кислоту (МСК или MsOH), SnCl4, HCl/диоксан и HCl/MeOH. Другие примеры реагентов для снятия Boc-защиты включают HCl, H2SO4 и PTSA (п-толуолсульфоновую кислоту или тозиловую кислоту).
[0076] “Кислота Льюиса” может относиться к соединению или ионным фрагментам, которые акцептируют электронную пару от соединения-донора. Примеры кислот Льюиса включают, но не ограничиваются ими, Zn(OAc)2, ZnCl2, Zn(OPiv)2. Кроме того, в качестве кислот Льюиса могут выступать катионы другиметаллов, такие как катионы меди и никеля.
[0077] “Реагент для введения PNZ-защиты” может относиться к реагенту, который можно применять для прикрепления п-нитробензилоксикарбонильной защитной группы к аминогруппе. Примеры реагентов для введения PNZ-защиты могут включать, но не ограничиваются ими, 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазол и 4-нитробензил-1H-бензо[d]имидазол-1-карбоксилат.
[0078] Термины “1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазол” или “PNZ-Bt” могут относиться к соединениям формулы:
 .
.
[0079] Термин “4-нитробензил-1H-бензо[d]имидазол-1-карбоксилат” может относиться к соединениям формулы:
 .
.
[0080] “Реагент для снятия PNZ-защиты” может относиться к реагенту, который можно применять для отщепления п-нитробензилоксикарбонильной защитной группы от аминогруппы. Примеры агентов для снятия PNZ-защиты могут включать, но не ограничиваются ими, дитионит натрия и гидрирование при помощи H2 и Pd/C или PtO2.
[0081] Термин “защитная группа”, применяемый в настоящем описании, может относиться к подвижному химическому фрагменту, который известен в данной области техники для защиты реакционноспособных групп, включая без ограничений, гидроксильные и аминогруппы, от нежелательных реакций во время процедур синтеза. Гидроксильные и аминогруппы, которые защищены защитной группой, в настоящем описании называют “защищенными гидроксильными группами” и “защищенными аминогруппами”, соответственно. Защитные группы обычно применяют селективно и/или ортогонально для защиты участков во время реакций на других реакционноспособных участках, и затем их можно удалять для оставления незащищенной группы в исходном виде или доступной для дальнейших реакций. Защитные группы, известные в данной области техники, обычно описаны в Greene and Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Группы можно селективно встраивать в аминогликозиды, описанные в настоящей заявке, как предшественники. Например, аминогруппа может быть расположена в соединении, описанном в настоящей заявке, как азидогруппа, которую можно химически превращать в аминогруппу в желаемый момент синтеза. Как правило, группы защищены или присутствуют в качестве предшественника, который является инертным к реакциям, которые модифицируют другие участки исходной молекулы для превращения их в конечные группы в подходящее время. Кроме того, иллюстративные защитные группы или группы-предшественники описаны в Agrawal, et al., Protocols for Oligonucleotide Conjugates, Eds, Humana Press; New Jersey, 1994; Vol. 26 pp. 1-72. Примеры “групп для защиты гидроксильных групп” включают, но не ограничиваются ими, трет-бутил, трет-бутоксиметил, метоксиметил, тетрагидропиранил, 1-этоксиэтил, 1-(2-хлорэтокси)этил, 2-триметилсилилэтил, п-хлорфенил, 2,4-динитрофенил, бензил, 2,6-дихлорбензил, дифенилметил, п-нитробензил, трифенилметил, триметилсилил, триэтилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил (TBDPS), трифенилсилил, бензоилформиат, ацетат, хлорацетат, трихлорацетат, трифторацетат, пивалоат, бензоат, п-фенилбензоат, 9-флуоренилметилкарбонат, мезилат и тозилат. Примеры “групп для защиты аминогрупп” включают, но не ограничиваются ими, 2-триметилсилилэтоксикарбонил (Teoc), 1-метил-1-(4-бифенилил)этоксикарбонил (Bpoc), трет-бутоксикарбонил (Boc), аллилоксикарбонил (Alloc), 9-флуоренилметилoxyкарбонил (Fmoc), бензилоксикарбонил (Cbz), п-нитробензилоксикарбонил (PNZ), формил, ацетил, тригалогенацетил (например, трифторацетил), бензоил, нитрофенилацетил, 2-нитробензолсульфонил, фталимидо и дитиасукциноил.
Получение соединения формулы (9) и его промежуточных соединений:
[0082] Настоящее изобретение включает процессы, способы, реагенты и промежуточные соединения для синтеза соединения формулы (9), которое имеет структуру:

или его сольвата, или его энантиомера, или его диастереомера, где представляет собой одинарную связь или двойную связь, и где x составляет от 1 до 5.
[0083] В некоторых вариантах реализации соединение формулы (9) представляет собой сульфат плазомицина, который имеет структуру:

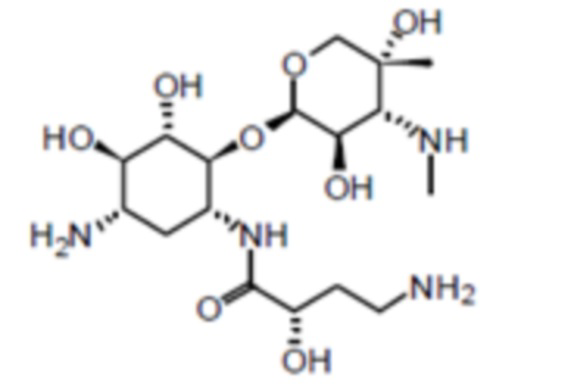
или его сольват, где x составляет от 1 до 5.
[0084] Сульфат плазомицина также может упоминаться, как сульфат (2”R,3”R,4”R,5”R)-2”-[(1S,2S,3R,4S,6R)-4-амино-6-[(2’”S)-4’”-амино-2’”-гидроксибутанамидо)амино]-3-[(2’S,3’R)-3’-амино-6’-((2-гидроксиэтиламино)метил)-3’,4’-дигидро-2H-пиран-2’-илокси]-2-гидроксициклогексилокси]-5”-метил-4”-(метиламино)тетрагидро-2H-пиран-3”,5”-диола. Плазомицин также может упоминаться, как:
6'-(гидроксиэтил)-1-(HABA)-сизомицин;
6′-(2-гидрокси-этил)-1-(4-амино-2(S)-гидрокси-бутирил)-сизомицин;
(2S)-4-амино-N-[(1R,2S,3S,4R,5S)-5-амино-4-[[(2S,3R)-3-амино-6-[(2-гидроксиэтиламино)метил]-3,4-дигидро-2H-пиран-2-ил]окси]-2-[(2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)оксан-2-ил]окси-3-гидроксициклогексил]-2-гидроксибутанамид;
бутанамид, 4-амино-N-[(1R,2S,3S,4R,5S)-5-амино-4-[[(2S,3R)-3-амино-3,4-дигидро-6-[[(2-гидроксиэтил)амино]метил]-2H-пиран-2-ил]окси]-2-[[3-дезокси-4-C-метил-3-(метиламино)-β-L-арабинопиранозил]окси]-3-гидроксициклогексил]-2-гидрокси-, (2S)-;
D-стрептамин, O-2-амино-2,3,4,6-тетрадезокси-6-[(2-гидроксиэтил)амино]-a-D-глицеро-гекс-4-енопиранозил-(1→4)-O-[3-дезокси-4-C-метил-3-(метиламино)-b-L-арабинопиранозил-(1→6)]-N1-[(2S)-4-амино-2-гидрокси-1-оксобутил]-2-дезокси-;
O-2-амино-2,3,4,6-тетрадезокси-6-[(2-гидроксиэтил)амино]-a-D-глицеро-гекс-4-енопиранозил-(1→4)-O-[3-дезокси-4-C-метил-3-(метиламино)-b-L-арабинопиранозил-(1→6)]-N1-[(2S)-4-амино-2-гидрокси-1-оксобутил]-2-дезокси-D-стрептамин; и
(2S)-4-амино-N-[(1R,2S,3S,4R,5S)-5-амино-4-{[(2S,3R)-3-амино-6-{[(2-гидроксиэтил)амино]метил}-3,4-дигидро-2H-пиран-2-ил]окси}-2-{[3-дезокси-4-C-метил-3-(метиламино)-β-L-арабинопиранозил]окси}-3-гидроксициклогексил]-2-гидроксибутанамид.
[0085] Обычная нумерация атомов в плазомицине представлена ниже:
 .
.
[0086] Способ получения соединения формулы (9) и конкретных промежуточных соединений, полученных при получении соединения формулы (9), представлен на схеме 1 ниже и более подробно описан в настоящей заявке.
Схема 1
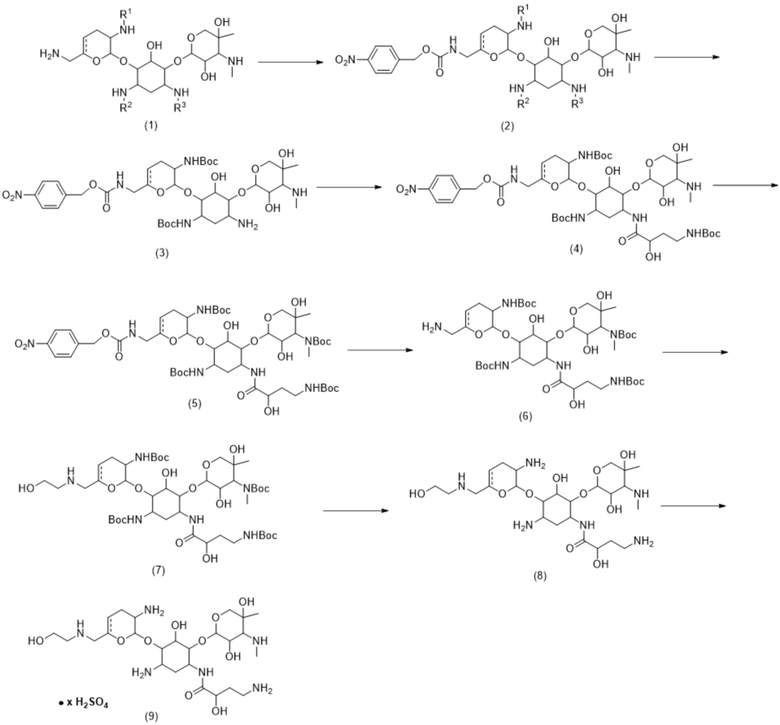
[0087] Как отмечалось выше, в настоящем соединении предложены способы получения соединений формулы (9), которые включают сульфат плазомицина, которые не только масштабируются до промышленных масштабов, но также являются надежно воспроизводимыми от партии к партии в таком промышленном масштабе и характеризуются хорошими выходами. Таким образом, в способах синтеза и процессах очистки, описанных в настоящей заявке, описан масштабируемый, экономически выгодный способ получения соединений формулы (9) и их промежуточных соединений, который не основывается на дорогостоящих и/или сложных стадиях во время получения, таким образом, делая указанную методологию особенно подходящей для крупномасштабного производства антибиотиков.
[0088] Способы включают реакции, которые могут приводить к получению новых промежуточных соединений посредством комбинирования условий и стадий реакций. Например, селективное функционализирование положения 6’-N являлось сложной задачей. Тем не менее, как описано в настоящей заявке, взаимодействие соединения формулы (6) с соединением формулы (7), как описано в настоящей заявке, неожиданно приводит к получению продуктов моноалкилирования, тогда как при нормальных обстоятельствах ожидают, что такие реакции будут приводить к избыточному алкилированию.
[0089] Способы также могут включать кристаллизацию конкретных промежуточных соединений. Как описано в настоящей заявке, кристаллизация конкретных промежуточных соединений способствует процессу очистки (например, снижению содержания примесей) и упрощает способ очистки по сравнению с предыдущими способами очистки. Например, предыдущие способы очистки включали осаждение, которое может обеспечивать более низкую чистоту и/или худшие свойства выделенного соединения. Стадии осаждения также могут снижать выход и приводить к большей изменчивости содержания примесей от партии к партии. Кристаллизацию, как описано в настоящей заявке, можно применять для очистки от примесей. Кроме того, хотя для первоначального определения подходящих условий кристаллизации требуются значительные усилия, после определения условий кристаллизации процесс становится простым и воспроизводимым. Например, образцы кристаллов можно собирать из предшествующего процесса кристаллизации для применения в качестве затравочных кристаллов для последующих кристаллизаций. Как обсуждается ниже, соединения формул (4), (6) и (7) можно подвергать кристаллизации.
[0090] Соединения, описанные в настоящей заявке, и способы получения соединений могут включать соли соединений, описанных в настоящей заявке. Типичные соли включают, но не ограничиваются ими, например, водорастворимые соли и не растворимые в воде соли, такие как ацетат, амсонат (4,4-диаминостильбен-2,2-дисульфонат), бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, кальций, эдетат кальция, камсилат, карбонат, хлорид, цитрат, клавулариат, дигидрохлорид, эдетат, эдизилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глютамат, гликоллиларсанилат, гексафторфосфат, гексилрезоцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изетионат, лактат, лактобионат, лаурат, магний, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, аммониевую соль N-метилглюкамина, 3-гидрокси-2-нафтоат, олеат, оксалат, пальмитат, памоат (1,1-метен-бис-2-гидрокси-3-нафтоат, эйнбонат), пантотенат, фосфат/дифосфат, пикрат, полигалактуронат, пропионат, п-толуолсульфонат, салицилат, стеарат, субацетат, сукцинат, сульфат, сульфосалицилат, сурамат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат.
[0091] Соль также может включать соль присоединения кислоты. “Соль присоединения кислоты” может относиться к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются биологически или иным образом нежелательными, и которые образуются с неорганическими кислотами, такими как, но не ограничиваясь ими, хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органическими кислотами, такими как, но не ограничиваясь ими, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламиновая кислота, додецилсульфоновая кислота, этан-1,2-дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксоглутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муциновая кислота, нафталин-l,5-дисульфоновая кислота, нафталин-2-сульфоновая кислота, l-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и т.п.
[0092] Соединения, описанные в настоящей заявке, и способ получения соединений могут включать сольваты соединений, описанных в настоящей заявке. Термин “сольват” может относиться к комплексу с переменной стереохимией, образованному растворенным веществом и растворителем. Такие растворители для целей настоящего изобретения могут не препятствовать биологической активности растворенного вещества. Примеры подходящих растворителей могут включать, но не ограничиваются ими, воду, MeOH, EtOH и AcOH. Сольваты, в которых молекула растворителя представляет собой воду, обычно называют гидратами. Гидраты могут включать композиции, содержащие стехиометрические количества воды, а также композиции, содержащие переменные количества воды.
[0093] Специалистам в данной области техники понятно, присутствует ли стереоцентр в каком-либо из соединений, описанных в настоящей заявке, и способе получения соединений. Соответственно, настоящее изобретение включает оба возможных стереоизомера (если стереохимия не указана в настоящем описании) и включает не только рацемические соединения, но также отдельные энантиомеры и/или диастереомеры. Кроме того, специалистам в данной области техники понятно, существует ли позиционный или геометрический изомер для соединения, описанного в настоящей заявке. Соответственно, настоящее изобретение включает все возможные позиционные или геометрические изомеры (если изомер не указан в настоящем описании). В структурах, представленных в настоящей заявке, в которых стереохимия любого конкретного атома не указана, или геометричесий или позиционный изомер не указан, все стереоизомеры и геометрические или позиционные изомеры рассматриваются и включаются в соединения, описанные в настоящей заявке, и способ получения соединений. Если стереохимия или геометрический или позиционный изомер указаны, указанная стереохимия или геометрический или позиционный изомер указываются и определяются таким образом.
[0094] Термин “стереизомеры” может относиться к набору соединений, которые имеют одинаковое количество и тип атомов и имеют одинаковый порядок связей между атомами, но отличаются трехмерной структурой. Термин “стереоизомер” может относиться к любому члену указанного набора соединений. Например, стереоизомер может представлять собой энантиомер или диастереомер. Соединения, описанные в настоящей заявке, и способ получения соединений могут включать стереоизомеры.
[0095] Термин “энантиомеры” может относиться к паре стереоизомеров, которые являются несовместимыми зеркальными отражениями друг друга. Термин “энантиомер” может относиться к одном члену указанной пары стереоизомеров. Термин “рацемический” может относиться к 1:1 смеси пары энантиомеров. Соединения, описанные в настоящей заявке, и способ получения соединений могут включать энантиомеры. Каждое соединение, описанное в настоящей заявке, может включать все энантиомеры, которые соответствуют общей структуре соединения (если энантиомер не указан в настоящем описании). Соединения могут находиться в рацемической или энантиомерно чистой форме или в любой другой форме с точки зрения стереохимии (если стереохимия не указана в настоящем описании). В некоторых вариантах реализации соединения представляют собой (S)-энантиомер. В других вариантах реализации соединения представляют собой (R)-энантиомер. В других вариантах реализации соединения представляют собой (+) или (-) энантиомеры. В некоторых вариантах реализации соединения, описанные в настоящей заявке, могут быть обогащены для обеспечения преимущественного содержания одного из энантиомеров соединения, описанного в настоящей заявке. Смесь с преимущественным содержанием одного из энантиомеров может содержать, например, по меньшей мере 60 мол. процентов одного из энантиомеров или более предпочтительно по меньшей мере 75, 80, 85, 90, 95, 96, 97, 98, 99, 99,5 или даже 100 мол. процентов. В некоторых вариантах реализации соединение, описанное в настоящей заявке, с преимущественным содержанием одного из энантиомеров, может по существу не содержать другого энантиомера, где “по существу не содержит” означает, что рассматриваемое вещество составляет менее 10%, или менее 5%, или менее 4%, или менее 3%, или менее 2%, или менее 1% по сравнению с количеством другого энантиомера, например, в смеси соединений. Например, если смесь соединений содержит 98 граммов первого энантиомера и 2 грамма второго энантиомера, говорят, что она содержит 98 мол. процентов первого энантиомера и только 2 мол. процента второго энантиомера.
[0096] Термин “диастереомеры” может относиться к набору стереоизомеров, которые нельзя наложить друг на друга путем вращения вокруг одинарных связей. Например, цис- и транс-двойные связи, эндо- и экзозамещение в бициклических кольцевых системах и соединения, содержащие несколько стереогенных центров с различными относительными конфигурациями, относят к диастереомерам. Термин “диастереомер” может относиться к любому члену указанного набора соединений. В некоторых представленных примерах способ синтеза может приводить к получению одного диастереомера или смеси диастереомеров. Соединения, описанные в настоящей заявке, и способ получения соединений могут включать диастереомеры. В некоторых вариантах реализации соединения, описанные в настоящей заявке, могут быть обогащены для обеспечения преимущественного содержания одного из диастереомеров соединения, описанного в настоящей заявке. Смесь с преимущественным содержанием одного из диастереомеров может содержать, например, по меньшей мере 60 мол. процентов одного из диастереомеров или более предпочтительно по меньшей мере 75, 99, 95, 96, 97, 98, 99 или даже 100 мол. процентов.
[0097] Кроме того, соединения, описанные в настоящей заявке, и способ получения соединений включает все геометрические и позиционные изомеры. Например, если соединение, описанное в настоящей заявке, содержит двойную связь или конденсированное кольцо, в объем настоящего изобретения могут быть включены цис- и транс-формы, а также смеси. Если соединение содержит двойную связь, заместитель может иметь E- или Z-конфигурацию (если конфигурация не указана в настоящем описании). Если соединение содержит дизамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию (если конфигурация не указана в настоящем описании).
[0098] Соединения, описанные в настоящей заявке, могут дополнительно включать все соединения с изотопными метками. Соединение с “изотопной” или “радиоактивной” меткой представляет собой соединение, в котором один или более атомов заменены или замещены атомов, атомная масса или массовое число которого отличается от атомной массы или массового числа атома, обычно обнаруживаемого в природе (т.е. встречающегося в природе). Например, в некоторых вариантах реализации в соединениях, описанных в настоящей заявке, атомы водорода могут быть заменены или замещены одним или более атомами дейтерия или трития. Некоторые соединения с изотопными метками согласно настоящему изобретению, например, соединения, которые содержат радиоактивный изотоп, могут являться полезными для применения в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы трития, т.е. 3H, и углерода-14, т.е. 14C, особенно полезны для указанной цели благодаря легкости их включения и имеющихся средств для детектирования. Замена более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечивать определенные терапевтические преимущества с результате более высокой метаболической стабильности, например, увеличенного периода полувыведения in vivo или пониженных требований к дозировке, и, следовательно, может являться предпочтительной в некоторых случаях. Подходящие изотопы, которые можно включать в соединения, описанные в настоящей заявке, могут включать, но не ограничиваются ими, 2H (также обозначается, как D в случае дейтерия), 3H (также обозначается, как T в случае трития), 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl , 82Br, 75Br, 76Br, 77Br, 123I, 124I, 125I и 131I. Замена на позитронно-активные изотопы, такие как 11C, 18F, 15O и 13N, может являться полезной в исследованиях путем позитронно-эмиссионной томографии (ПЭТ).
[0099] Соединения любой из формул, описанных в настоящей заявке, можно получать при помощи способов, известных в области органического синтеза, которые частично изложены в следующих схемах синтеза и примерах в сочетании с руководством, приведенным в настоящем документе. В схемах, представленных ниже, подразумевают, что защитные группы для чувствительных или реакционноспособных групп можно применять при необходимости в соответствии с общими принципами или химией в соответствии с руководством, приведенным в настоящем документе. Защитными группами можно управлять в соответствии со стандартными способами органического синтеза (T. W. Greene and P. G. M. Wuts, “Protective Groups in Organic Synthesis”, Third edition, Wiley, New York 1999). Указанные группы можно удалять на удобной стадии синтеза соединения с применением способов, которые очевидны для специалистов в данной области техники, на основе подробного описания, приведенного в настоящем документе. Способы отбора, а также условия реакции и порядок их выполнения должны соответствовать настоящему изобретению.
[00100] На следующих схемах 2-5 также представлен синтез соединения формулы (9) и его промежуточных соединений.
Схема 2: Синтез соединения формулы (4)
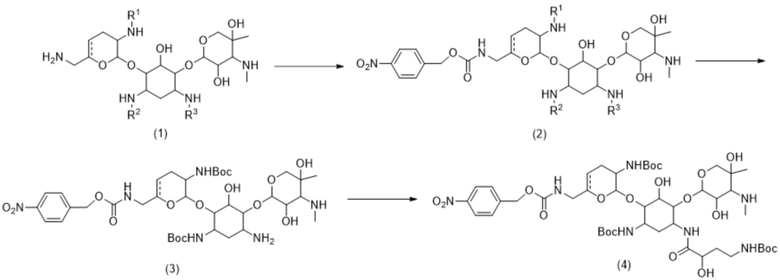
[00101] На схеме 2 представлен синтез соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера. На схеме 2 R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил; и представляет собой одинарную связь или двойную связь.
Синтез соединения формулы (2)
[00102] Со ссылкой на схему 2 в некоторых вариантах реализации реакции, подробно описанные на схеме 2, проводили при температуре вплоть до примерно 60°C. Соединение формулы (1), или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для введения PNZ-защиты с получением соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации реагент для введения PNZ-защиты выбран из 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазола (PNZ-Bt) и 4-нитробензил-1H-бензо[d]имидазол-1-карбоксилата. В некоторых вариантах реализации реагент для введения PNZ-защиты представляет собой PNZ-Bt.
[00103] В некоторых таких вариантах реализации реагент для введения PNZ-защиты может присутствовать в реакции в количестве от примерно 1,0 до 1,2 молярного эквивалента в расчете на соединение формулы (1), или его энантиомер, или его диастереомер. В некоторых вариантах реализации реакцию соединения формулы (1), или его энантиомера, или его диастереомера с реагентом для введения PNZ-защиты можно проводить в присутствии растворителя, выбранного из группы, состоящей из дихлорметана, спирта и их комбинации. В некоторых таких вариантах реализации спирт представляет собой метанол. Присутствие метанола может увеличивать селективность реакции, когда R1, R2 и/или R3 представляют собой H. В других вариантах реализации реакцию соединения формулы (1), или его энантиомера, или его диастереомера с реагентом для введения PNZ-защиты можно проводить в присутствии растворителя, выбранного из группы, состоящей из дихлорметана, этанола и их комбинации.
[00104] В некоторых вариантах реализации соединение формулы (2) можно применять в следующей реакции без существенной очистки.
[00105] В некоторых вариантах реализации R1, R2 и R3 в соединении формулы (1), или его энантиомере, или его диастереомере представляют собой H. В некоторых таких вариантах реализации соединение формулы (2) представляет собой соединение формулы (2a):
(2a);
или его соль, или его сольват, или его энантиомер, или его диастереомер. В некоторых таких вариантах реализации представляет собой двойную связь.
[00106] В некоторых вариантах реализации соединение формулы (1), которое приводят в контакт с реагентом для введения PNZ-защиты, может представлять собой сизомицин в виде свободного основания (представлен ниже):
 .
.
В некоторых таких вариантах реализации соединение формулы (2) представляет собой соединение формулы (2b):
 ,
,
или его соль, или его сольват.
Синтез соединения формулы (3)
[00107] Со ссылкой на схему 2 соединение формулы (3):
(3),
или его соль, или его сольват, или его энантиомер, или его диастереомер можно получать из соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации представляет собой двойную связь.
[00108] Если R1, R2 и R3 представляют собой H, соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для введения Boc-защиты (трет-бутилоксикарбонил) с получением соединения формулы (3), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В других вариантах реализации, если один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил, одну или более C1-C3 алкильных групп удаляют, и соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для введения Boc-защиты с получением соединения формулы (3), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00109] В некоторых вариантах реализации реагент для введения Boc-защиты представляет собой ди-трет-бутил-дикарбонат, N-(трет-бутоксикарбонилокси)-5-норборнен-эндо-2,3-дикарбоксимид, N-трет-бутоксикарбонилимидазол, 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил, 2-(трет-бутоксикарбонилтио)-4,6-диметилпиримидин, 1-трет-бутоксикарбонил-1,2,4-триазол, трет-бутил-фенилкарбонат, N-(трет-бутоксикарбонилокси)фталимид или трет-бутил-2,4,5-трихлорфенилкарбонат. В некоторых вариантах реализации реагент для введения Boc-защиты представляет собой Boc2O (Boc-ангидрид; ди-трет-бутил-дикарбонат) или Boc-ONb (N-(трет-бутоксикарбонилокси)-5-норборнен-эндо-2,3-дикарбоксимид). В некоторых таких вариантах реализации реагент для введения Boc-защиты представляет собой Boc2O. В некоторых таких вариантах реализации реагент для введения Boc-защиты представляет собой Boc-ONb.
[00110] Реакцию между соединением формулы (2), или его солью, или его сольватом, или его энантиомером, или его диастереомермо и реагентом для введения Boc-защиты можно проводить в присутствии кислоты Льюиса. В некоторых таких вариантах реализации кислота Льюиса представляет собой Zn(OAc)2, ZnCl2 или Zn(OPiv)2. Альтернативно, реакцию между соединением формулы (2), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты также можно проводить в присутствии кислоты Льюиса, содержащей ион меди или ион никеля.
[00111] Кроме того, реакцию между соединением формулы (2), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты можно проводить в присутствии амина. В некоторых вариантах реализации амин представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), пиридин, пиперидин, 4-диметиламинопиридин (DMAP), 2,6-лутидин, диметиланилин, N-метилпирролидон, N-диизопропилэтиламин, N-метилимидазол, N-этилдиметиламин, триметиламин или триэтиламин. В некоторых вариантах реализации амин представляет собой триэтиламин.
[00112] Кроме того, реакцию между соединением формулы (2), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты можно проводить в присутствии спирта. В некоторых таких вариантах реализации спирт представляет собой метанол.
[00113] В некоторых вариантах реализации соединение формулы (3) можно применять в следующей реакции без существенной очистки.
[00114] В некоторых вариантах реализации соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (3a):
 ,
,
или его соль, или его сольват, полученный путем приведения соединения формулы (2b), или его соли, или его сольвата в контакт с реагентом для введения Boc-защиты.
Синтез соединения формулы (4)
[00115] Со ссылкой на схему 2 соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с
с получением соединения формулы (4):
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации представляет собой двойную связь.
[00116] Реакцию между соединением формулы (3), или его солью, или его сольватом, или его энантиомером, или его диастереомером и
можно проводить в присутствии активирующего реагента и реагента для образования пептидной связи.
[00117] Активирующий реагент относится к реагенту, который превращает карбонильную группу карбоновой кислоты в группу, которая более восприимчива к нуклеофильной атаке. В некоторых вариантах реализации активирующий реагент представляет собой HATU, HOOBt, HOSu, HOAt, DMAP, BOP, PyBOP, PyBrOP, PyAOP, PyOxim, DEPBT, TBTU, HBTU, HCTU, HDMC, COMU, CDI или HOBt. В некоторых таких вариантах реализации активирующий реагент представляет собой HOBt. В некоторых вариантах реализации активирующий реагент присутствует в количестве от примерно 0,05 до 1,0 молярного эквивалента в расчете на
.
[00118] В некоторых вариантах реализации реагент для образования пептидной связи представляет собой DCC, EDC, DIC, WSC, EDAC или PyBOP. В некоторых вариантах реализации реагент для образования пептидной связи представляет собой EDAC или PyBOP. В некоторых таких вариантах реализации реагент для образования пептидной связи представляет собой EDAC. В некоторых вариантах реализации реагент для образования пептидной связи присутствует в количестве от примерно 1,0 до 1,4 молярного эквивалента в расчете на
.
В некоторых вариантах реализации реагент для образования пептидной связи присутствует в количестве примерно 1,0; 1,1; 1,2; 1,3 или 1,4 молярного эквивалента в расчете на
.
[00119] Реакцию между соединением формулы (3), или его солью, или его сольватом, или его энантиомером, или его диастереомером и
можно проводить в кислотных условиях. В некоторых таких вариантах реализации кислотные условия представляют собой pH от примерно 4 до 7. В некоторых таких вариантах реализации кислотные условия представляют собой pH примерно 5.
[00120] Реакцию между соединением формулы (3), или его солью, или его сольватом, или его энантиомером, или его диастереомером и
можно проводить в присутствии спирта. В некоторых таких вариантах реализации спирт представляет собой метанол.
[00121] Соединение формулы (4), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (4a):
 ,
,
или его соль, или его сольват, полученный путем приведения соединения формулы (3a), или его соли, или его сольвата в контакт с .
[00122] В настоящем изобретении также предложен способ, включающий получение кристаллической формы соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В настоящем изобретении также предложен способ, включающий выделение соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера, например, как описано ниже в примере 1.
[00123] Как отмечено в настоящем описании, определенные стадии кристаллизации при определенных условиях могут способствовать очистке путем удаления примесей. И после определения условий кристаллизации применение кристаллизации в качестве одного из способов очистки может приводить как к хорошим выходам, так и к меньшей изменчивости содержания примесей от партии к партии по сравнению с предыдущими способами очистки.
Схема 3: Синтез соединения формулы (6)

[00124] На схеме 3 представлен синтез соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера. представляет собой одинарную связь или двойную связь.
Синтез соединения формулы (5)
[00125] Со ссылкой на схему 3 соединение формулы (4), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для введения Boc-защиты с получением соединения формулы (5), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации представляет собой двойную связь.
[00126] Для указанного превращения можно применять стандартный реагент для введения Boc-защиты, включая, но не ограничиваясь ими, N-трет-бутоксикарбонилимидазол, 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил, 2-(трет-бутоксикарбонилтио)-4,6-диметилпиримидин, 1-трет-бутоксикарбонил-1,2,4-триазол, трет-бутил-фенилкарбонат, N-(трет-бутоксикарбонилокси)фталимид, трет-бутил-2,4,5-трихлорфенилкарбонат, Boc2O и Boc-ONb. В некоторых вариантах реализации реагент для введения Boc-защиты представляет собой Boc2O.
[00127] В некоторых вариантах реализации реакцию между соединением формулы (4), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты можно проводить при температуре вплоть до примерно 60°C. В некоторых вариантах реализации реакцию между соединением формулы (4), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты можно проводить в присутствии спирта. В некоторых таких вариантах реализации спирт представляет собой метанол или этанол. В некоторых таких вариантах реализации спирт представляет собой метанол. Применение спирта в реакции между соединением формулы (4), или его солью, или его сольватом, или его энантиомером, или его диастереомером и реагентом для введения Boc-защиты может способствовать дальнейшему использованию в следующей реакции.
[00128] В некоторых вариантах реализации соединение формулы (5) можно применять в следующей реакции без существенной очистки.
[00129] Соединение формулы (5), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (5a):
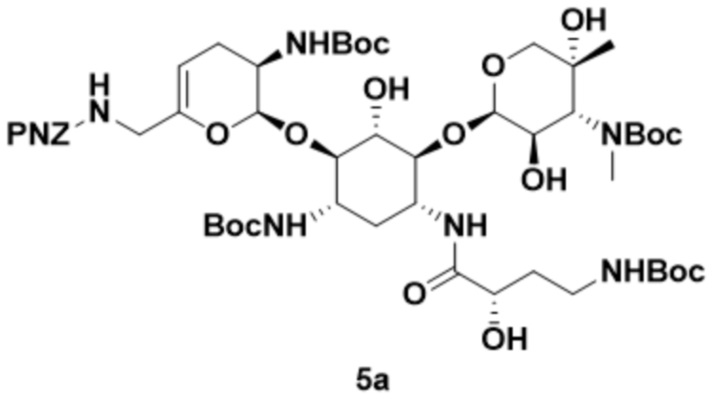 ,
,
или его соль, или его сольват, полученный путем приведения соединения формулы (4a), или его соли, или его сольвата в контакт с реагентом для введения Boc-защиты.
Синтез соединения формулы (6)
[00130] Со ссылкой на схему 3 соединение формулы (5), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для снятия PNZ-защиты с получением соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации представляет собой двойную связь.
[00131] В некоторых вариантах реализации реагент для снятия PNZ-защиты представляет собой дитионит натрия. В других вариантах реализации реакция снятия PNZ-защиты может представлять собой гидрирование при помощи H2 и катализатора, такого как Pd/C или PtO2. Дитионит натрия может являться более предпочтительным по сравнению с реакциями снятия защиты, требующими гидрирования, поскольку его легки применять в большем масштабе, он может являться хемоселективным и менее опасным и может не требовать специального оборудования для использования.
[00132] Соединение формулы (6), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (6a):
 ,
,
или его соль, или его сольват, полученный путем приведения соединения формулы (5a), или его соли, или его сольвата в контакт с реагентом для снятия PNZ-защиты.
[00133] В настоящем изобретении также предложен способ получения кристаллической формы соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В настоящем изобретении также предложен способ, включающий выделение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера, например, как описано ниже в примере 2.
[00134] Таким образом, как отмечалось ранее, указанная кристаллизация может способствовать процессу очистки (например, снижению содержания примесей) и упрощать процесс очистки по сравнению с предыдущими способами очистки, что потенциально может приводить к большей воспроизводимости всего процесса.
Схема 4: Синтез соединения формулы (7)

[00135] На схеме 4 представлен синтез соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера. представляет собой одинарную связь или двойную связь. В некоторых вариантах реализации представляет собой двойную связь.
[00136] Протекание реакции превращения соединения формулы (6) в соединение формулы (7) является неожиданным в отношении алкилирования. При нормальных обстоятельствах первичный амин, такой как группа в соединении формулы (6), имеет тенденцию к алкилированию более одного раза. Таким образом, неожиданно, что соединение формулы (7) содержит вторичный амин (например, алкилированный одним -CH2CH2OH). Как обсуждается ниже, условия реакции, включая выбор растворителя и применение реагента для предотвращения избыточного алкилирования, могут обеспечивать протекание моноалкилирования в соответствующем месте.
Синтез соединения формулы (7)
[00137] Со ссылкой на схему 4 соединение формулы (6), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с , где LG1 представляет собой уходящую группу, с получением соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых таких вариантах реализации уходящая группа представляет собой йод-, бром- или хлор-. В некоторых таких вариантах реализации уходящая группа представляет собой йод-. В некоторых вариантах реализации присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6).
[00138] Реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и можно проводить в по существу безводных условиях. В некоторых вариантах реализации реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и можно проводить в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации. Ацетон может содействовать селективности реакции, способствуя моноалкилированию. Ацетонитрил можно применять для удаления воды путем образования азеотропа.
[00139] В некоторых вариантах реализации реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и можно проводить в присутствии NaHCO3, карбоната лития, карбоната натрия, карбоната калия, карбоната цезия, сульфата натрия, DIPEA, фосфата натрия, триметилортоформиата и гексаметилдисилана. В некоторых таких вариантах реализации реакцию осуществляют в присутствии NaHCO3. В некоторых вариантах реализации реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и можно проводить при температуре от примерно 30°C до 40°C. В некоторых таких вариантах реализации температура составляет примерно 35°C. Температуры выше 50°C могут приводить к образованию диалкилированного побочного продукта.
[00140] Реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и можно гасить путем добавления 1,4-диазабицикло[2.2.2]октана (DABCO) к реакционной смеси. DABCO можно применять для по существу остановки реакции и предотвращения избыточного алкилирования. Альтернативно, реакцию между соединением формулы (6), или его солью, или его сольватом, или его энантиомером, или его диастереомером и также можно гасить путем добавления 1-пропиламина, пиперидина, диэтиламина, N-этилдиметиламина, триэтиламина, ДБУ, MeOH, карбонатного буфера, диметиламина, цистеина, диэтаноламина или NaOH.
[00141] В некоторых вариантах реализации соединение формулы (7), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (7a):
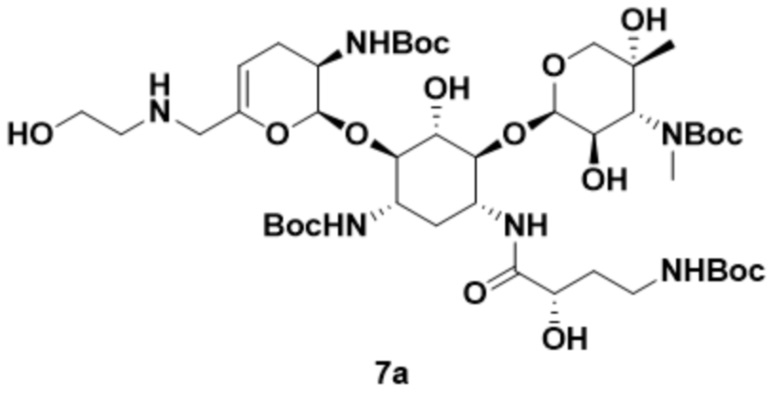 ,
,
или его соль, или его сольват, полученный путем приведения соединения формулы (6a), или его соли, или его сольвата в контакт с .
[00142] В настоящем изобретении также предложен способ получения кристаллической формы соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В настоящем изобретении также предложен способ, включающий выделение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера, например, как описано ниже в примере 3.
[00143] Указанная кристаллизация может способствовать процессу очистки (например, снижению содержания примесей) и упрощать очистку по сравнению с предыдущими способами очистки. Кристаллизация также может способствовать удалению примесей.
Схема 5: Синтез соединения формулы (9)

[00144] На схеме 5 представлен синтез соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера. представляет собой одинарную связь или двойную связь, и x составляет от 1 до 5. В некоторых вариантах реализации представляет собой двойную связь. В некоторых вариантах реализации x составляет от 2 до 3.
Синтез соединения формулы (8)
[00145] Со ссылкой на схему 5 соединение формулы (7), или его соль, или его сольват, или его энантиомер, или его диастереомер можно приводить в контакт с реагентом для снятия Boc-защиты с получением соединения формулы (8), или его соли, или его сольвата, или его энантиомера, или его диастереомера. В некоторых вариантах реализации реагент для снятия Boc-защиты представляет собой ТФК, MsOH (метансульфоновую кислоту или CH3SO3H), PTSA (п-толуолсульфоновую кислоту или тозиловую кислоту), H2SO4 или HCl. В некоторых вариантах реализации реагент для снятия Boc-защиты представляет собой ТФК или MsOH. В некоторых вариантах реализации реагент для снятия Boc-защиты представляет собой ТФК, H2SO4 или HCl.
[00146] В некоторых вариантах реализации реагент для снятия Boc-защиты представляет собой ТФК, что приводит к получению ТФК соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера. Поскольку реакция снятия защиты должна являться безводной, для указанного превращения обычно применяют ТФК.
[00147] Стадия удаления Boc-группы может приводить к гидролизу субстрата и присутствию соединения (IMP-1) сразу после реакции (например, перед любой очисткой).
 (IMP-1)
(IMP-1)
В некоторых вариантах реализации стадия удаления Boc-группы не приводит к значительному гидролизу, и присутствие соединения (IMP-1) сразу после реакции (например, перед любой очисткой) минимизировано. В некоторых вариантах реализации присутствие или количество соединения (IMP-1) можно определять путем ВЭЖХ. В некоторых вариантах реализации соединение IMP-1 может присутствовать в количестве от 0 до 7%, таком как примерно 0, 0,2, 0,4, 0,6, 0,8, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5 или 7%, сразу после реакции (например, перед любой очисткой). Если количество IMP-1 составляет 0%, указанное количество указывает, что указанная примесь может присутствовать на уровнях ниже уровня детектирования типичных аналитических способов, известных и обычно применяемых специалистами в данной области техники (например, ВЭЖХ).
[00148] В некоторых вариантах реализации кислота представляет собой ТФК или MsOH, и количество соединения IMP-1 может составлять от примерно 0 до 7% сразу после реакции (например, перед любой очисткой). В некоторых случаях кислота представляет собой ТФК или MsOH, и количество соединения IMP-1 может составлять от примерно 0 до 2,5% сразу после реакции (например, перед любой очисткой). В некоторых случаях кислота представляет собой ТФК, и количество соединения IMP-1 может составлять от примерно 0 до 2,5%, такое как примерно 0, 0,2, 0,4, 0,6, 0,8, 1, 1,5, 2 или 2,5%, сразу после реакции (например, перед любой очисткой). В некоторых случаях кислота представляет собой MsOH, и количество соединения IMP-1 может составлять от примерно 0,2 до 2.5%, такое как примерно 0,2, 0,4, 0,6, 0,8, 1, 1,5, 2 или 2,5%, сразу после реакции (например, перед любой очисткой).
[00149] В некоторых случаях кислота представляет собой HCl, и количество соединения IMP-1 может составлять от примерно 0 до 1%, такое как примерно 0, 0,2, 0,4, 0,6, 0,8 или 1%, сразу после реакции (например, перед любой очисткой).
[00150] В некоторых случаях кислота представляет собой H2SO4, и количество соединения IMP-1 может составлять от примерно 0 до 7%, такое как примерно 0, 0,2, 0,4, 0,6, 0,8, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5 или 7%, сразу после реакции (например, перед любой очисткой).
[00151] Если соединение формулы (7), или его соль, или его сольват, или его энантиомер, или его диастереомер приводят в контакт с реагентами для снятия Boc-защиты, количества IMP-1 могут варьироваться в зависимости от природы реагента для снятия Boc-защиты. В таблице ниже представлены количества IMP-1 в реакциях соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера с различными реагентами для снятия Boc-защиты.
0,81%
0,80%
2,50%
4,61%
6,61%
3,80%
5,52%
0,74%
(при различных условиях реакции)
[00152] В некоторых вариантах реализации соединение формулы (8) можно применять в следующей реакции без существенной очистки.
[00153] В некоторых вариантах реализации соединение формулы (8), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой соединение формулы (8a):
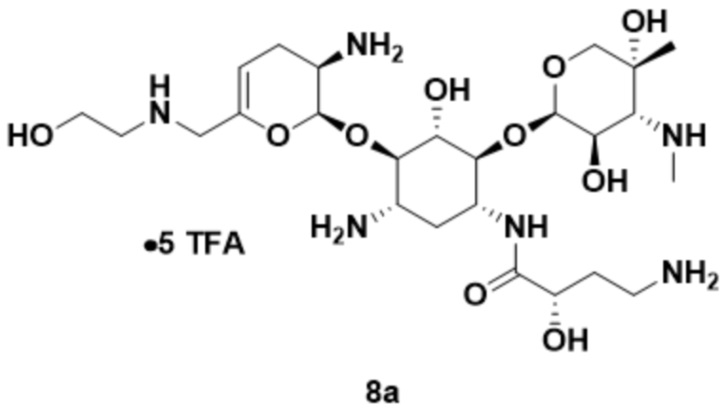 ,
,
или его сольват, полученный путем приведения соединения формулы (7a), или его соли, или его сольвата в контакт с реагентом для снятия Boc-защиты.
Синтез соединения формулы (9)
[00154] Образование соли с кислотой можно проводить с получением соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера. Если соединение формулы (8) уже представляет собой соль, такую как ТФК соль, соль можно удалять с получением соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера перед получением другой соли.
[00155] В некоторых вариантах реализации кислота на стадии образования соли представляет собой серную кислоту, что приводит к получению сульфатной соли соединения формулы (9),

или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5. В некоторых вариантах реализации x составляет от 2 до 3. Сульфатная соль соединения формулы (9) может обладать улучшенной стабильностью по сравнению с другими солями. В некоторых вариантах реализации соединение формулы (9), или его соль, или его сольват, или его энантиомер, или его диастереомер может представлять собой сульфат плазомицина:

или его сольват, полученный путем образования соли с соединением формулы (8a), или его солью, или его сольватом, где x составляет от 1 до 5. В некоторых вариантах реализации x составляет от 2 до 3.
Кристаллические соединения и их получение и характеризация:
[00156] В одном аспекте настоящее изобретение относится к промежуточным соединениям в способе синтеза, который можно применять для получения соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера. Указанные промежуточные соединения могут находиться в кристаллической форме. В настоящем изобретении предложены способы получения кристаллических промежуточных соединений в способе синтеза, который можно применять для получения соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера. Кристаллические промежуточные соединения согласно настоящему изобретению можно характеризовывать путем порошковой рентгеновской дифрактографии (XRPD), дифференциальной сканирующей калориметрии (ДСК) и термогравиметрического анализа (ТГА). Способы сбора данных XRPD, ДСК и ТГА и свойства кристаллических промежуточных соединений согласно настоящему описанию дополнительно проиллюстрированы в приведенных ниже примерах.
Соединения 4 и 4a
[00157] В некоторых вариантах реализации промежуточное соединение в способе получения соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера представляет собой соединение формулы (4):
(4),
или его соль, или его сольват, или его энантиомер, или его диастереомер. В некоторых таких вариантах реализации соединение формулы (4) представляет собой соединение формулы:
(4a),
или его соль, или его сольват. В некоторых таких вариантах реализации соединение формулы (4a) представляет собой кристаллический трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамат или его сольват.
[00158] В некоторых вариантах реализации кристаллический трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамат или его сольват характеризуют по XRPD спектру, в котором положения пиков по существу соответствует показанным на ФИГ. 1.
[00159] В некоторых вариантах реализации кристаллический трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамат или его сольват характеризуют по следу на графике ТГА, показанному на ФИГ. 2. В некоторых вариантах реализации кристаллический трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамат или его сольват характеризуют по ДСК профилю, показанному на ФИГ. 3.
[00160] В настоящем изобретении предложен способ получения кристаллического трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамата формулы (4a) или его сольвата, включающий:
(a) обработку соединения формулы (4a), или его соли, или его сольвата ацетонитрилом с получением раствора;
(b) нагревание раствора со стадии (a);
(c) добавление воды к нагретому раствору со стадии (b);
(d) охлаждение раствора со стадии (c);
(e) внесение в раствор со стадии (d) затравочного кристалла; и
(f) выделение полученных твердых веществ с получением кристаллического соединения формулы (4a) или его сольвата.
[00161] В некоторых вариантах реализации способа получения кристаллического трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамата для кристаллизации можно применять ацетонитрил с 1,5% деионизированной (DI) воды при 75 ± 3°C со внесением затравки и последующим охлаждением. Кроме того, кристаллизацию можно проводить при 65 ± 3°C или 70 ± 3°C с последующим охлаждением. В некоторых вариантах реализации для кристаллизации трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамата можно применять 1-пропанол, с деионизированной (DI) водой или без нее, вместо ацетонитрила. Испытанные условия кристаллизации приведены в примере 1 и таблице 5.
Соединение 6a
[00162] В некоторых вариантах реализации промежуточное соединение в способе получения соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера представляет собой соединение формулы (6a):
,
или его соль, или его сольват. В некоторых таких вариантах реализации в настоящем изобретении предложен кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (6a) или его сольват.
[00163] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по XRPD спектру, в котором положения пиков по существу соответствует показанным на ФИГ. 4 и в таблице 1. В некоторых вариантах реализации наблюдается изменчивость примерно ± 0,2°2θ относительно углов дифракции в XRPD спектрах, например, как представлено в таблице 1.
Таблица 1: данные XRPD для кристаллического соединения формулы (6a)
[00164] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по одному или более значениям 2θ при примерно 6,10, 10,66, 12,17, 17,65 и 18,55 градусах при порошковой рентгеновской дифрактографии, где наблюдается изменчивость примерно ± 0,2°2θ относительно углов дифракции в XRPD спектрах.
[00165] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по следу на графике ТГА, показанному на ФИГ. 5. Основной экзотермический эффект согласно ДСК кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата или его сольвата наблюдается с ограничением температуры слева на уровне 231,8°C и с энергией -52,9 кДж/кг (ФИГ. 6).
[00166] В настоящем изобретении предложен способ получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) или его сольвата, включающий:
(a) обработку соединения формулы (6), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление воды к раствору со стадии (a) с получением смеси;
(c) добавление дихлорметана к смеси со стадии (b) с получением смеси;
(d) внесение в смесь со стадии (c) затравочного кристалла;
(e) выделение полученных твердых веществ с получением кристаллического соединения формулы (6a) или его сольвата.
[00167] В некоторых вариантах реализации стадию (d) осуществляют при низкой температуре. В некоторых вариантах реализации стадию (d) осуществляют при примерно 15-25°C, например, примерно 15-20°C или примерно 20-25°C.
[00168] В некоторых вариантах реализации способа получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата можно применять смесь дихлорметан/изопропилацетат (50/50 об./об.) с 1% воды, смесь изопропилацетат/дихлорметан (71/29 об./об.) с 2% воды или смесь изопропилацетат/дихлорметан (71/29 об./об.) с 8% воды. В некоторых таких вариантах реализации можно применять смесь дихлорметан/изопропилацетат (50/50 об./об.) с 1% воды. Изопропилацетат не требуется для кристаллизации, но может присутствовать в качестве экстрагирующего растворителя. Фильтрование может являться более легким при выделении твердых веществ из смеси дихлорметан/изопропилацетат (50/50 об./об.) с 1% воды в виде липких твердых веществ, и наблюдается некоторое разжижение твердых веществ, выделяемых из смеси изопропилацетат/дихлорметан (71/29 об./об.) с 2% и 8% воды. Кроме того, материал, выделяемый из смеси дихлорметан/изопропилацетат (50/50 об./об.) с 1% воды, является кристаллическим. Тем не менее, материал, выделяемый из смеси изопропилацетат/дихлорметан (71/29 об./об.) с 2% воды, является неоднородным, и материал, выделяемый из смеси изопропилацетат/дихлорметан (71/29 об./об.) с 8% воды, может содержать небольшое количество рентгеноаморфного материала. Испытанные условия кристаллизации приведены в примере 2.
Соединения 7 и 7a
[00169] В некоторых вариантах реализации промежуточное соединение в способе получения соединения формулы (9), или его соли, или его сольвата, или его энантиомера, или его диастереомера представляет собой соединение формулы (7):
,
или его соль, или его сольват, или его энантиомер, или его диастереомер. В некоторых таких вариантах реализации соединение формулы (7) представляет собой соединение формулы:
(7a),
или его соль, или его сольват. В некоторых таких вариантах реализации соединение формулы (7a) представляет собой кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват.
[00170] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по XRPD спектру, в котором положения пиков по существу соответствует показанным на ФИГ. 7 и в таблице 2. В некоторых вариантах реализации наблюдается изменчивость примерно ± 0,2°2θ относительно углов дифракции в XRPD спектрах, например, как представлено в таблице 2.
Таблица 2: данные XRPD для кристаллического соединения формулы (7a)
[00171] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по одному или более значениям 2θ при примерно 5,17, 7,39, 10,85 и 12,17 градусах при порошковой рентгеновской дифрактографии, где наблюдается изменчивость примерно ± 0,2°2θ относительно углов дифракции в XRPD спектрах.
[00172] В некоторых вариантах реализации кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат или его сольват характеризуют по следу на графике ТГА, показанному на ФИГ. 8. ДСК профиль кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата или его сольвата представлен на ФИГ. 9.
[00173] В настоящем изобретении предложен способ получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) или его сольвата, включающий:
(a) обработку соединения формулы (7a), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление ацетонитрила к раствору со стадии (a) с получением смеси;
(c) внесение в смесь со стадии (b) затравочного кристалла;
(d) выделение полученных твердых веществ с получением кристаллического соединения формулы (7a) или его сольвата.
[00174] В некоторых вариантах реализации способа получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата можно применять 10 об.%/об.% смесь IPAc/АЦН с добавлением 0,75-2 масс.%/масс.% воды и со внесением затравки. Кроме того, длительное время выдерживания при затравочной температуре (например, >2 часов при температуре от 60°C до 65°C) и медленное охлаждение (например, от 60°C до 0°C при 5-10°C/час) можно применять для получения подвижной суспензии. Содержание воды можно использовать для управления стабильностью результатов кристаллизации. Кристаллизация с добавлением 0,5-2% воды может являться более воспроизводимой при постоянном значении извлечения (например, обычно более 80%) и может обеспечивать повышение чистоты. Указанная процедура может по существу удалять непрореагировавшее соединение формулы (6a), пента-Boc примесь (например, 63% удаление суммарно) и диалкилированный побочный продукт (например, 36% удаление) и может обеспечивать получение легко фильтруемого и промываемого твердого продукта. Испытанные условия кристаллизации приведены в примере 3 и в таблицах 7-11.
Приведенные в качестве примера варианты реализации
[00175] Некоторые варианты реализации настоящего изобретения представляют собой вариант реализации I, как описано ниже:
[00176] Вариант реализации I-1. Способ получения соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера, включающий:
(a) приведение соединения формулы (1):
(1),
или его энантиомера, или его диастереомера в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt) с получением соединения формулы (2):
(2),
или его соли, или его сольвата, или его энантиомера, или его диастереомера;
где представляет собой одинарную связь или двойную связь;
R1 представляет собой H или C1-C3 алкил;
R2 представляет собой H или C1-C3 алкил; и
R3 представляет собой H или C1-C3 алкил.
[00177] Вариант реализации I-2. Способ по варианту реализации I-1, отличающийся тем, что стадию (a) осуществляют в присутствии растворителя, выбранного из группы, состоящей из дихлорметана, метанола и их комбинации.
[00178] Вариант реализации I-3. Способ по варианту реализации I-1 или I-2, отличающийся тем, что PNZ-Bt присутствует в количестве от примерно 1,0 до 1,2 молярного эквивалента в расчете на соединение формулы (1), или его энантиомер, или его диастереомер.
[00179] Вариант реализации I-4. Способ по любому из вариантов реализации от I-1 до I-3, дополнительно включающий стадию (b1) или (b2):
(b1) где, если R1, R2 и R3 представляют собой H, приведение соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты приводит к получению соединения формулы (3):
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера; или
(b2) где, если один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил, сперва удаление указанного C1-C3 алкила, а затем приведение соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты приводит к получению соединения формулы (3):
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00180] Вариант реализации I-5. Способ по варианту реализации I-4, отличающийся тем, что реагент для введения Boc-защиты представляет собой Boc2O или Boc-ONb.
[00181] Вариант реализации I-6. Способ по варианту реализации I-4 или I-5, отличающийся тем, что стадию (b1) или (b2) осуществляют в присутствии кислоты Льюиса.
[00182] Вариант реализации I-7. Способ по варианту реализации I-6, отличающийся тем, что кислота Льюиса представляет собой Zn(OAc)2, ZnCl2 или Zn(OPiv)2.
[00183] Вариант реализации I-8. Способ по варианту реализации I-6, отличающийся тем, что кислота Льюиса содержит ион меди или ион никеля.
[00184] Вариант реализации I-9. Способ по любому из вариантов реализации от I-4 до I-8, отличающийся тем, что стадию (b1) или (b2) осуществляют в присутствии триэтиламина.
[00185] Вариант реализации I-10. Способ по любому из вариантов реализации от I-4 до I-8, отличающийся тем, что стадию (b1) или (b2) осуществляют в присутствии метанола.
[00186] Вариант реализации I-11. Способ по любому из вариантов реализации от I-4 до I-10, дополнительно включающий:
(c) приведение соединения формулы (3), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с
с получением соединения формулы (4):
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00187] Вариант реализации I-12. Способ по варианту реализации I-11, отличающийся тем, что стадию (c) осуществляют в присутствии активирующего реагента и реагента для образования пептидной связи.
[00188] Вариант реализации I-13. Способ по варианту реализации I-12, отличающийся тем, что активирующий реагент представляет собой HOBt.
[00189] Вариант реализации I-14. Способ по варианту реализации I-13, отличающийся тем, что активирующий реагент присутствует в количестве от примерно 0,05 до 1,0 молярного эквивалента в расчете на
.
[00190] Вариант реализации I-15. Способ по варианту реализации I-12, отличающийся тем, что реагент для образования пептидной связи представляет собой EDAC или PyBOP.
[00191] Вариант реализации I-16. Способ по варианту реализации I-15, отличающийся тем, что реагент для образования пептидной связи присутствует в количестве от примерно 1,0 до 1,4 молярного эквивалента в расчете на
.
[00192] Вариант реализации I-17. Способ по любому из вариантов реализации от I-11 до I-16, отличающийся тем, что стадию (c) осуществляют в кислотных условиях.
[00193] Вариант реализации I-18. Способ по варианту реализации I-17, отличающийся тем, что кислотные условия представляют собой pH от примерно 4 до 7.
[00194] Вариант реализации I-19. Способ по варианту реализации I-17, отличающийся тем, что кислотные условия представляют собой pH примерно 5.
[00195] Вариант реализации I-20. Способ по любому из вариантов реализации от I-11 до I-19, дополнительно включающий получение кристаллической формы соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00196] Вариант реализации I-21. Способ по любому из вариантов реализации от I-11 до I-20, дополнительно включающий выделение соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00197] Вариант реализации I-22. Способ по любому из вариантов реализации от I-11 до I-21, дополнительно включающий:
(d) приведение соединения формулы (4), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты с получением соединения формулы (5):
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00198] Вариант реализации I-23. Способ по варианту реализации I-22, отличающийся тем, что реагент для введения Boc-защиты представляет собой Boc2O.
[00199] Вариант реализации I-24. Способ по варианту реализации I-22 или I-23, отличающийся тем, что стадию (d) осуществляют в присутствии спирта.
[00200] Вариант реализации I-25. Способ по варианту реализации I-24, отличающийся тем, что спирт представляет собой метанол.
[00201] Вариант реализации I-26. Способ по любому из вариантов реализации от I-22 до I-25, отличающийся тем, что стадию (d) осуществляют при температуре вплоть до примерно 60°C.
[00202] Вариант реализации I-27. Способ по любому из вариантов реализации от I-22 до I-26, дополнительно включающий:
(e) приведение соединения формулы (5), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия PNZ-защиты с получением соединения формулы (6):
(6),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00203] Вариант реализации I-28. Способ по варианту реализации I-27, отличающийся тем, что реагент для снятия PNZ-защиты представляет собой дитионит натрия.
[00204] Вариант реализации I-29. Способ по варианту реализации I-27 или I-28, дополнительно включающий получение кристаллической формы соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00205] Вариант реализации I-30. Способ по любому из вариантов реализации от I-27 до I-29, дополнительно включающий выделение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00206] Вариант реализации I-31. Способ по любому из вариантов реализации от I-27 до I-30, дополнительно включающий:
(f) приведение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с , где LG1 представляет собой уходящую группу, с получением соединения формулы (7):
(7),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00207] Вариант реализации I-32. Способ по варианту реализации I-31, отличающийся тем, что уходящая группа представляет собой йод-.
[00208] Вариант реализации I-33. Способ по варианту реализации I-31, отличающийся тем, что присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6).
[00209] Вариант реализации I-34. Способ по любому из вариантов реализации от I-31 до I-33, отличающийся тем, что стадию (f) осуществляют в по существу безводных условиях.
[00210] Вариант реализации I-35. Способ по любому из вариантов реализации от I-31 до I-34, отличающийся тем, что стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации.
[00211] Вариант реализации I-36. Способ по любому из вариантов реализации от I-31 до I-35, отличающийся тем, что стадию (f) осуществляют в присутствии NaHCO3.
[00212] Вариант реализации I-37. Способ по любому из вариантов реализации от I-31 до I-36, отличающийся тем, что стадию (f) осуществляют при температуре от примерно 30°C до 40°C.
[00213] Вариант реализации I-38. Способ по любому из вариантов реализации от I-31 до I-37, дополнительно включающий добавление 1,4-диазабицикло[2.2.2]октана (DABCO) к реакционной смеси.
[00214] Вариант реализации I-39. Способ по любому из вариантов реализации от I-31 до I-38, дополнительно включающий получение кристаллической формы соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00215] Вариант реализации I-40. Способ по любому из вариантов реализации от I-31 до I-39, дополнительно включающий выделение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00216] Вариант реализации I-41. Способ по любому из вариантов реализации от I-31 до I-40, дополнительно включающий:
(g) приведение соединения формулы (7) в контакт с реагентом для снятия Boc-защиты с получением соединения формулы (8):
(8),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00217] Вариант реализации I-42. Способ по варианту реализации I-41, отличающийся тем, что реагент для снятия Boc-защиты представляет собой ТФК, что приводит к получению ТФК соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00218] Вариант реализации I-42a. Способ по варианту реализации I-41 или I-42, отличающийся тем, что соединение (IMP-1)
(IMP-1)
присутствует сразу после реакции (например, перед любой очисткой) в количестве менее 7%.
[00219] Вариант реализации I-42b. Способ по варианту реализации I-41 или I-42, отличающийся тем, что соединение (IMP-1)
(IMP-1)
присутствует сразу после реакции (например, перед любой очисткой) в количестве менее 2,5%.
[00220] Вариант реализации I-43. Способ по варианту реализации I-42, дополнительно включающий удаление ТФК соли с получением соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00221] Вариант реализации I-44. Способ по варианту реализации I-41 или I-43, дополнительно включающий:
(h) осуществление образования соли с кислотой с получением соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00222] Вариант реализации I-45. Способ по варианту реализации I-44, отличающийся тем, что кислота на стадии (h) представляет собой серную кислоту, что приводит к получению сульфатной соли соединения формулы (9):

или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5.
[00223] Вариант реализации I-46. Способ по любому из вариантов реализации от I-1 до I-3, отличающийся тем, что R1, R2 или R3 представляют собой H.
[00224] Вариант реализации I-47. Способ по любому из вариантов реализации от I-1 до I-46, отличающийся тем, что представляет собой двойную связь.
[00225] Вариант реализации I-48. Способ по любому из вариантов реализации от I-1 до I-10, отличающийся тем, что стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3” и 4” в формулах (1)-(3) указана, как в формуле (X), где обозначает место прикрепления к водороду или фрагменту:
(X).
[00226] Вариант реализации I-49. Способ по любому из вариантов реализации от I-11 до I-47, отличающийся тем, что стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3”, 4” и 1-z в формулах (4)-(9) указана, как в формуле (Y), где обозначает место прикрепления к водороду или фрагменту:
(Y).
[00227] Вариант реализации I-50. Способ получения соединения формулы (5):
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера,
включающий:
(a) приведение соединения формулы (4):
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты, где представляет собой одинарную связь или двойную связь.
[00228] Вариант реализации I-51. Способ по варианту реализации I-50, отличающийся тем, что соединение формулы (4), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (3):
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с
.
[00229] Вариант реализации I-52. Способ по варианту реализации I-51, отличающийся тем, что соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем:
(b1) приведения соединения формулы (2a):
(2a),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты; или
(b2) удаления C1-C3 алкила из соединения формулы (2):
(2),
или его соли, или его сольвата, или его энантиомера, или его диастереомера;
где R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил, и где один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил;
с последующим приведением соединения формулы (2), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты.
[00230] Вариант реализации I-53. Способ по варианту реализации I-52, отличающийся тем, что соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (1):
(1),
или его энантиомера, или его диастереомера в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt).
[00231] Вариант реализации I-54. Способ по любому из вариантов реализации от I-50 до I-53, отличающийся тем, что реагент для введения Boc-защиты представляет собой Boc2O.
[00232] Вариант реализации I-55. Способ по любому из вариантов реализации от I-50 до I-54, отличающийся тем, что стадию (a), (b1) или (b2) осуществляют в присутствии спирта.
[00233] Вариант реализации I-56. Способ по варианту реализации I-55, отличающийся тем, что спирт представляет собой метанол.
[00234] Вариант реализации I-57. Способ по любому из вариантов реализации от I-50 до I-56, отличающийся тем, что стадию (a), (b1) или (b2) осуществляют при температуре вплоть до примерно 60°C.
[00235] Вариант реализации I-58. Способ по любому из вариантов реализации от I-50 до I-57, дополнительно включающий:
(e) приведение соединения формулы (5), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия PNZ-защиты с получением соединения формулы (6):
(6),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00236] Вариант реализации I-59. Способ по варианту реализации I-58, отличающийся тем, что реагент для снятия PNZ-защиты представляет собой дитионит натрия.
[00237] Вариант реализации I-60. Способ по любому из вариантов реализации от I-50 до I-59, дополнительно включающий получение кристаллической формы соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00238] Вариант реализации I-61. Способ по любому из вариантов реализации от I-50 до I-60, дополнительно включающий выделение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00239] Вариант реализации I-62. Способ по любому из вариантов реализации от I-50 до I-61, дополнительно включающий:
(f) приведение соединения формулы (6), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с , где LG1 представляет собой уходящую группу, с получением соединения формулы (7):
(7),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00240] Вариант реализации I-63. Способ по варианту реализации I-62, отличающийся тем, что уходящая группа представляет собой йод-.
[00241] Вариант реализации I-64. Способ по варианту реализации I-62, отличающийся тем, что присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6).
[00242] Вариант реализации I-65. Способ по любому из вариантов реализации от I-62 до I-64, отличающийся тем, что стадию (f) осуществляют в по существу безводных условиях.
[00243] Вариант реализации I-66. Способ по любому из вариантов реализации от I-62 до I-65, отличающийся тем, что стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации.
[00244] Вариант реализации I-67. Способ по любому из вариантов реализации от I-62 до I-66, отличающийся тем, что стадию (f) осуществляют в присутствии NaHCO3.
[00245] Вариант реализации I-68. Способ по любому из вариантов реализации от I-62 до I-67, отличающийся тем, что стадию (f) осуществляют при температуре от примерно 30°C до 40°C.
[00246] Вариант реализации I-69. Способ по любому из вариантов реализации от I-62 до I-68, дополнительно включающий добавление 1,4-диазабицикло[2.2.2]октана (DABCO) к реакционной смеси.
[00247] Вариант реализации I-70. Способ по любому из вариантов реализации от I-62 до I-69, дополнительно включающий получение кристаллической формы соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00248] Вариант реализации I-71. Способ по любому из вариантов реализации от I-62 до I-70, дополнительно включающий выделение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00249] Вариант реализации I-72. Способ по любому из вариантов реализации от I-62 до I-71, дополнительно включающий:
(g) приведение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия Boc-защиты с получением соединения формулы (8):
(8),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00250] Вариант реализации I-73. Способ по варианту реализации I-72, отличающийся тем, что реагент для снятия Boc-защиты представляет собой ТФК, что приводит к получению ТФК соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00251] Вариант реализации I-74. Способ по варианту реализации I-73, дополнительно включающий удаление ТФК соли с получением соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00252] Вариант реализации I-75. Способ по варианту реализации I-72 или I-74, дополнительно включающий:
(h) осуществление образования соли с кислотой с получением соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00253] Вариант реализации I-76. Способ по варианту реализации I-75, отличающийся тем, что кислота на стадии (h) представляет собой серную кислоту, что приводит к получению сульфатной соли соединения формулы (9):
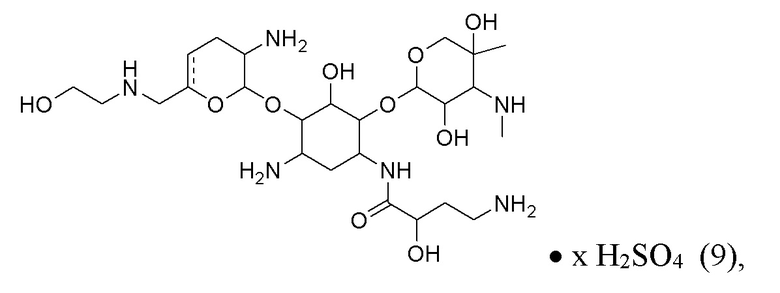
или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5.
[00254] Вариант реализации I-77. Способ получения соединения формулы (7):
(7),
или его соли, или его сольвата, или его энантиомера, или его диастереомера, включающий:
(f) приведение соединения формулы (6),
(6),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с , где LG1 представляет собой уходящую группу, и где представляет собой одинарную связь или двойную связь.
[00255] Вариант реализации I-78. Способ по варианту реализации I-77, отличающийся тем, что соединение формулы (6), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (5):
(5),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия PNZ-защиты.
[00256] Вариант реализации I-79. Способ по варианту реализации I-78, отличающийся тем, что соединение формулы (5), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (4):
(4),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты.
[00257] Вариант реализации I-80. Способ по варианту реализации I-79, отличающийся тем, что соединение формулы (4), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (3):
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с .
[00258] Вариант реализации I-81. Способ по варианту реализации I-80, отличающийся тем, что соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем:
(b1) приведения соединения формулы (2a):
(2a),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты; или
(b2) удаления C1-C3 алкила из соединения формулы (2):
(2),
или его соли, или его сольвата, или его энантиомера, или его диастереомера;
где R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил, и где один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил;
с последующим приведением соединения формулы (2) в контакт с реагентом для введения Boc-защиты.
[00259] Вариант реализации I-82. Способ по варианту реализации I-81, отличающийся тем, что соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (1):
(1),
или его энантиомера, или его диастереомера в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt).
[00260] Вариант реализации I-83. Способ по любому из вариантов реализации от I-77 до I-82, отличающийся тем, что уходящая группа представляет собой йод-.
[00261] Вариант реализации I-84. Способ по варианту реализации I-83, отличающийся тем, что присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6).
[00262] Вариант реализации I-85. Способ по любому из вариантов реализации от I-77 до I-84, отличающийся тем, что стадию (f) осуществляют в по существу безводных условиях.
[00263] Вариант реализации I-86. Способ по любому из вариантов реализации от I-77 до I-85, отличающийся тем, что стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации.
[00264] Вариант реализации I-87. Способ по любому из вариантов реализации от I-77 до I-86, отличающийся тем, что стадию (f) осуществляют в присутствии NaHCO3.
[00265] Вариант реализации I-88. Способ по любому из вариантов реализации от I-77 до I-87, отличающийся тем, что стадию (f) осуществляют при температуре от примерно 30°C до 40°C.
[00266] Вариант реализации I-89. Способ по любому из вариантов реализации от I-77 до I-88, дополнительно включающий добавление 1,4-диазабицикло[2.2.2]октана (DABCO) к реакционной смеси.
[00267] Вариант реализации I-90. Способ по любому из вариантов реализации от I-77 до I-89, дополнительно включающий получение кристаллической формы соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00268] Вариант реализации I-91. Способ по любому из вариантов реализации от I-77 до I-90, дополнительно включающий выделение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00269] Вариант реализации I-92. Способ по любому из вариантов реализации от I-77 до I-91, дополнительно включающий:
(g) приведение соединения формулы (7), или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для снятия Boc-защиты с получением соединения формулы (8):
(8),
или его соли, или его сольвата, или его энантиомера, или его диастереомера.
[00270] Вариант реализации I-93. Способ по варианту реализации I-92, отличающийся тем, что реагент для снятия Boc-защиты представляет собой ТФК, что приводит к получению ТФК соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00271] Вариант реализации I-94. Способ по варианту реализации I-93, дополнительно включающий удаление ТФК соли с получением соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00272] Вариант реализации I-95. Способ по варианту реализации I-92 или I-94, дополнительно включающий:
(h) осуществление образования соли с кислотой с получением соли соединения формулы (8), или его сольвата, или его энантиомера, или его диастереомера.
[00273] Вариант реализации I-96. Способ по варианту реализации I-95, отличающийся тем, что кислота на стадии (h) представляет собой серную кислоту, что приводит к получению сульфатной соли соединения формулы (9):

или его сольвата, или его энантиомера, или его диастереомера, где x составляет от 1 до 5.
[00274] Вариант реализации I-97. Способ по любому из вариантов реализации от I-50 до I-96, отличающийся тем, что представляет собой двойную связь.
[00275] Вариант реализации I-98. Соединение формулы (4):
(4),
или его соль, или его сольват, или его энантиомер, или его диастереомер.
[00276] Вариант реализации I-99. Соединение по варианту реализации I-98, отличающееся тем, что соединение формулы (4) представляет собой соединение формулы:
(4a),
или его соль, или его сольват.
[00277] Вариант реализации I-100. Кристаллический трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамат формулы (4a) или его сольват.
[00278] Вариант реализации I-101. Способ получения кристаллического трет-бутил-((2S,3R)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-3-ил)карбамата формулы (4a) или его сольвата, включающий:
(a) обработку соединения формулы (4a), или его соли, или его сольвата ацетонитрилом с получением раствора;
(b) нагревание раствора со стадии (a);
(c) добавление воды к нагретому раствору со стадии (b);
(d) охлаждение раствора со стадии (c);
(e) внесение в раствор со стадии (d) затравочного кристалла; и
(f) выделение полученных твердых веществ с получением кристаллического соединения формулы (4a) или его сольвата.
[00279] Вариант реализации I-102. Кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (6a) или его сольват.
[00280] Вариант реализации I-103. Способ получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) или его сольвата, включающий:
(a) обработку соединения формулы (6a), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление воды к раствору со стадии (a) с получением смеси;
(c) добавление дихлорметана к смеси со стадии (b) с получением смеси;
(d) внесение в смесь со стадии (c) затравочного кристалла;
(e) выделение полученных твердых веществ с получением кристаллического соединения формулы (6a) или его сольвата.
[00281] Вариант реализации I-104. Способ по варианту реализации I-103, отличающийся тем, что стадию (d) осуществляют при низкой температуре.
[00282] Вариант реализации I-105. Соединение формулы (7):
,
или его соль, или его сольват, или его энантиомер, или его диастереомер.
[00283] Вариант реализации I-106. Соединение по варианту реализации I-105, отличающееся тем, что соединение формулы (7) представляет собой соединение формулы:
(7a),
или его соль, или его сольват.
[00284] Вариант реализации I-107. Кристаллический трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (7a) или его сольват.
[00285] Вариант реализации I-108. Способ получения кристаллического трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) или его сольвата, включающий:
(a) обработку соединения формулы (7a), или его соли, или его сольвата изопропилацетатом (IPAc) с получением раствора;
(b) добавление ацетонитрила к раствору со стадии (a) с получением смеси;
(c) внесение в смесь со стадии (b) затравочного кристалла;
(d) выделение полученных твердых веществ с получением кристаллического соединения формулы (7a) или его сольвата.
Примеры
[00286] Настоящее изобретение дополнительно проиллюстрировано следующими примерами, которые не следует считать ограничивающими объем и сущность настоящего изобретения конкретными процедурами, описанными в настоящей заявке. Следует понимать, что примеры представлены для иллюстрации определенных вариантов реализации, и что они не накладывают каких-либо ограничений на объем настоящего изобретения. Также следует понимать, что могут быть реализованы различные другие варианты реализации, модификации и их эквиваленты, которые могут быть предложены специалистам в данной области техники без отступления от сущности настоящего изобретения и/или объема пунктов прилагаемой формулы изобретения.
[00287] Если не указано иное, исходные компоненты можно получать из источников, таких как Sigma Aldrich, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI и Fluorochem USA и т.д., или синтезированы в соответствии с источниками, известными специалистам в данной области техники (см., например, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)), или получены, как описано в настоящей заявке.
[00288] Сизомицин в виде свободного основания представляет собой ферментированный гликозид, и его получали в Zhejiang Zhenyuan Pharmaceutical Co. Ltd. Boc-(S)-HABA получали в Senn Chemicals AG или Porton Fine Chemicals Inc. PNZ-Bt получали в Luxembourg BioTechnologies LTD (KINSY S.L.) или Porton Fine Chemicals Inc. 2-Йодэтанол получали в Dona Chemicals, Польша.
[00289] Следующие сокращения имеют следующие значения, если не указано иное, и любые другие сокращения, применяемые в настоящем описании и не определенные, имеют свое стандартное общепринятое значение:
пл.%/пл.%: процент, нормализованный по площади
Ac: ацетат
АЦН: ацетонитрил
Boc: трет-бутоксикарбонил
BocO2: ди-трет-бутил-дикарбонат или Boc-ангидрид
Boc-ONb: трет-бутил-((4R,7S)-1,3-диоксо-1,3,3a,4,7,7a-гексагидро-2H-4,7-метаноизоиндол-2-ил) карбонат
DABCO: 1,4-диазабицикло[2.2.2]октан
ДБУ: 1,8-диазабицикло[5.4.0]ундец-7-ен
ДХМ: дихлорметан
DI: деионизированная вода
DIPE: диизопропиловый эфир
DIPEA: N, N-диизопропилэтиламин
ДМФА: диметилформамид,
ДСК: дифференциальная сканирующая калориметрия
EDAC: N-(3-диметиламинопропил)-N′-этилкарбодиимида гидрохлорид
EtOH: этанол
ГХ: газовая хроматография
ч: час(ы)
HABA: 4-амино-2-гидроксибутановая кислота
HCl: хлористоводородная кислота
HOBt: 1-гидроксибензотриазола гидрат
ВЭЖХ: высокоэффективная жидкостная хроматография
ИПС: изопропиловый спирт
IPAc: изопропилацетат
ЖХ/МС: жидкостная хроматография/масс-спектрометрия
MeCN: ацетонитрил
MeOH: метанол
мин: минута(ы)
МТБЭ: метил-трет-бутиловый эфир
NaOH: гидроксид натрия
PNZ-Bt: 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазол
ppm: миллионная доля
PrOH: пропанол
PyBOP: (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат
КТ: комнатная температура
TBDMS: трет-бутилдиметилсилил
ТЭА: триэтиламин
ТФК: трифторуксусная кислота
ТГА: термогравиметрический анализ
ТГФ: тетрагидрофуран
СВЭЖХ: сверхвысокоэффективная жидкостная хроматография
УФ: ультрафиолет
об./об.: объемное отношение
об.: объем(ы)
масс.%/масс.%: процентное массовое отношение
масс.: масса
XRPD: порошковая рентгеновская дифрактография
Zn(OPiv)2: пивалат цинка
Порошковая рентгеновская дифрактография
[00290] XRPD спектры получали на дифрактометре PANalytical X’Pert PRO MPD с применением падающего пучка Cu-излучения, полученного с использованием источника с длинным тонким фокусом Optix. Эллиптически градуированное многослойное зеркало использовали для фокусировки рентгеновских лучей Cu Kα через образец и на детекторе. Перед анализом проводили анализ образца кремния (NIST SRM 640d) для подтверждения того, что наблюдаемое положение пика Si 111 соответствует положению, сертифицированному NIST. Фрагмент образца помещали между пленками толщиной 3 мкм и анализировали в геометрии пропускания. Для минимизации фона, создаваемого воздухом, использовали поглотитель пучка, короткий антирассеивающий удлинитель и антирассеивающий коллиматор. Щели Соллера для падающего и отраженного пучков использовали для минимизации расширения в результате осевой расходимости. Дифрактограммы получали с применением сканирующего позиционно-чувствительного детектора (X’Celerator), расположенного в 240 мм от образца, и программного обеспечения Data Collector software v. 2.2b.
Термический анализ
[00291] Дифференциальную сканирующую калориметрию (ДСК) проводили с применением оборудования TA Instruments DSC с изменением температуры от 0°C до 300°C со скоростью 10°C/минута. Использовали стандартные алюминиевые тигели.
[00292] Термогравиметрический анализ (ТГА) проводили с применением термогравиметрического анализатора TA Instruments 2950. Калибровку температуры проводили с использованием никеля и AlumeI™. Каждый образец помещали в платиновый тигель и вносили в печь для термогравиметрии. Печь нагревали при продувке азотом.
Пример 1: Схема синтеза соединения 4a
[00293] Ниже подробно описана общая схема синтеза соединения 4a.

[00294] Сизомицин в виде свободного основания (1,0 кг ± 1%, 2,23 моль), затем MeOH (3,96 кг ± 5% или 5 л ± 5%), а затем ДХМ (6,64 кг ± 5% или 5 л ± 5%) загружали в реактор. Затем температуру стабилизировали на уровне 15 ± 5°C и смесь перемешивали до достижения полного растворения. Температуру можно повышать до примерно 30°C для содействия растворению. После растворения смесь охлаждали до температуры 15 ± 5°C. В отдельной смесительной емкости PNZ-Bt [(0,696 кг) ± 1%, 2,33 моль)] растворяли в ДХМ (18,59 кг ± 5% или 14 л ± 5%). Раствор PNZ-Bt загружали в реактор в течение периода времени от примерно 1 до примерно 4 часов, при этом поддерживая температуру партии на уровне 15 ± 5ºC. При получении раствора PNZ-Bt кожух загрузочной емкости не нагревали до температуры, превышающей или равной примерно 35ºC, для облегчения растворения PNZ-Bt. Полное растворение PNZ-Bt не требуется.
[00295] Загрузочную систему, применяемую для загрузки раствора PNZ-Bt, промывали ДХМ (1,33 кг ± 5% или 1 л ± 5%) и смывочный раствор загружали в реактор (TBatch=15 ± 5°C). Партию перемешивали при 15 ± 5°C и содержимое отбирали для определения завершения реакции. Реакцию считали завершенной, когда содержание сизомицина не превышало или равнялось примерно 2,0% площади, что оценивали путем ВЭЖХ анализа (см. таблицу 3 для применяемого способа ВЭЖХ). Первый образец отбирали после периода от примерно 15 минут до 12 часов после завершения загрузки PNZ-Bt. В некоторых вариантах реализации первый образец отбирали через примерно 30 минут после завершения загрузки PNZ-Bt. По мере необходимости для завершения реакции загружали дополнительное количество PNZ-Bt в растворе ДХМ. Количество дополнительного загруженного PNZ-Bt рассчитывали по следующей формуле: Загрузка=P1 * D1/(100 - D1) ± 1%, где P1=количество первоначально загруженного PNZ-Bt в кг, и D1=количество непрореагировавшего сизомицина в % площади согласно ВЭЖХ. После завершения реакции партию концентрировали в вакууме при температуре кожуха, не превышающей или равной примерно 40°C (например, от 10 до 40°C), до остаточного объема 5 л ± 5%.
[00296] Затем в реактор, содержащий соединение 2b, вносили MeOH (5,54 кг ± 5% или 7,00 л ± 5%). Смесь можно поддерживать при температуре, не превышающей или равной примерно 25°C (например, от 0 до 25°C), в течение периода времени, не превышающего или равного примерно 48 часам (например, от 0 до 48 часов), перед проведением следующей реакции (часть 2).
Таблица 3: способ ВЭЖХ для части 1 примера 1
0,25 М раствор NH4OH в метаноле
Часть 2: синтез соединения 3a
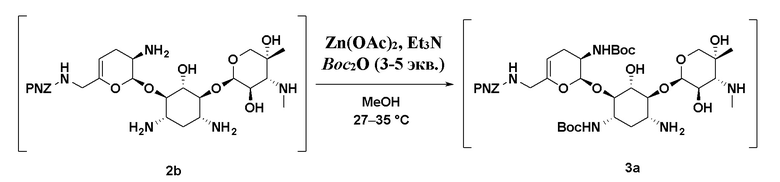
[00297] Триэтиламин [(1,48 л) ± 2% или (1,08 кг ± 2%)] загружали в реактор, содержащий соединение 2b, упомянутое в конце части 1, при этом поддерживая температуру партии на уровне от примерно 27°C до примерно 35°C (целевое значение примерно 33°C). Загрузочную систему, применяемую для загрузки триэтиламина, промывали MeOH (2,38 кг ± 5% или 3,00 л ± 5%) и смывочный раствор добавляли в реакционную смесь. Температуру партии стабилизировали на уровне от примерно 27°C до примерно 35°C (целевое значение примерно 33°C). Zn(OAc)2•2H2O (1,63 кг ± 2%) загружали в партию и загрузочную систему, применяемую для загрузки дигидрата ацетата цинка, промывали MeOH (2,38 кг ± 5% или 3,00 л ± 5%) и смывочный раствор добавляли в реакционную смесь. Смесь перемешивали в течение периода времени от примерно 30 минут до 12 часов при температуре от примерно 27°C до примерно 35°C (целевое значение примерно 33°C). В некоторых вариантах реализации смесь перемешивали в течение периода времени, не превышающего или равного примерно 60 минутам, при температуре от примерно 27°C до примерно 35°C (целевое значение примерно 33°C).
[00298] В отдельной смесительной емкости готовили раствор Boc2O [(2,55 кг) ± 2%, 11,7 моль] в MeOH (1,58 кг ± 5% или 2,00 л ± 5%). Полученный раствор Boc2O загружали в реактор в течение периода времени от примерно 15 минут до 12 часов, при этом поддерживая температуру партии на уровне от примерно 27°C до примерно 35°C (целевое значение примерно 33°C). В некоторых вариантах реализации полученный раствор Boc2O загружали в реактор в течение периода времени, не превышающего или равного примерно 1 часу, при этом поддерживая температуру партии на уровне от примерно 27°C до примерно 35°C (целевое значение примерно 33°C). Загрузочную систему, применяемую для раствора Boc2O, промывали метанолом (0,16 кг ± 5% или 0,20 л ± 5%) и смывочный раствор добавляли в реактор.
[00299] Партию выдерживали при целевой температуре от примерно 20 до 40°C в течение периода времени от примерно 3 до 24 часов и образец содержимого отбирали для определения завершения реакции. В некоторых вариантах реализации партию выдерживали при целевой температуре примерно 33°C в течение периода времени, не превышающего или равного примерно 5 часам, и образец содержимого отбирали для определения завершения реакции. Реакцию считали завершенной, когда содержание промежуточного моно-Boc-соединения 2b,
 ,
,
относительно соединения 3a равнялось или составляло не более чем примерно 2,0% площади согласно анализу ВЭЖХ (см. таблицу 4 для применяемого способа ВЭЖХ). Первый образец отбирали для проверки завершения реакции через период времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации первый образец отбирали для проверки завершения реакции через период времени, равный или превышающий примерно 5 часов, и последующие образцы (при необходимости) отбирали с интервалами примерно 3 часа. При этом температура партии не отклонялась за пределы диапазона примерно 27-35°C. В некоторых случаях для завершения реакции загружали дополнительное количество Boc2O. Загрузку осуществляли с той же концентрацией Boc2O, которую применяли для полученного выше раствора Boc2O. Количество загружаемого Boc2O рассчитывали по следующей формуле: Загрузка=P2 * 2 * D2 / [100 - (2 * D20)] ± 2%, где: P2=количество первоначально загруженного Boc2O в кг, и D2=количество оставшегося моно-Boc-соединения 2b в % площади согласно ВЭЖХ.
[00300] После завершения реакции реакционную смесь концентрировали в вакууме при температуре кожуха, не превышающей или равной примерно 40°C (например, от 20 до 40°C), до остаточного объема 12 л ± 5%. Загружали аммиак в количестве примерно 25 масс.%/масс.% (5,46 кг ± 5% или 6,00 л ± 5%), при этом поддерживая температуру партии на уровне от примерно 20°C до примерно 30°C, и выдерживали в течение периода времени от примерно 15 минут до 12 часов при указанном диапазоне температур. В некоторых вариантах реализации загружали аммиак в количестве примерно 25 масс.%/масс.% (5,46 кг ± 5% или 6,00 л ± 5%), при этом поддерживая температуру партии на уровне от примерно 20°C до примерно 30°C, и выдерживали в течение периода времени, не превышающего или равного примерно 1 часу, при указанном диапазоне температур. Добавление является экзотермическим.
[00301] ДХМ (13,28 кг ± 5% или 10,00 л ± 5%) загружали в партию и содержимое перемешивали в течение периода времени от примерно 12 минут до 12 часов при 25 ± 5°C. В некоторых вариантах реализации ДХМ (13,28 кг ± 5% или 10,00 л ± 5%) загружали в партию и содержимое перемешивали в течение периода времени, не превышающего или равного примерно 30 минутам, при 25 ± 5°C. Содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 45 минутам. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе.
[00302] Органическую фазу возвращали в реактор. Аммиак в количестве примерно 25 масс.%/масс.% (2,28 кг ± 5% или 2,50 л ± 5%) и DI воду (2,5 кг ± 5% или 2,5 л ± 5%), смешанные в смесительной емкости, загружали в органическую фазу и перемешивали в течение периода времени от примерно 15 минут до 12 часов при 25 ± 5°C. В некоторых вариантах реализации аммиак в количестве примерно 25 масс.%/масс.% (2,28 кг ± 5% или 2,50 л ± 5%) и DI воду (2,5 кг ± 5% или 2,5 л ± 5%), смешанные в смесительной емкости, загружали в органическую фазу и перемешивали в течение периода времени, не превышающего или равного примерно 30 минутам, при 25 ± 5°C. Содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 45 минутам. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе.
[00303] Органическую фазу возвращали в реактор, MeOH (0,79 кг ± 5% или 1,00 л ± 5%) загружали в органическую фазу и перемешивали в течение периода времени от примерно 15 до примерно 30 минут при 25 ± 5°C. DI воду (5,0 кг ± 5% или 5,0 л ± 5%) загружали в смесь и перемешивали в течение периода времени от примерно 15 минут до 12 часов при 25 ± 5°C. В некоторых вариантах реализации DI воду (5,0 кг ± 5% или 5,0 л ± 5%) загружали в смесь и перемешивали в течение периода времени, не превышающего или равного примерно 30 минутам, при 25 ± 5°C. Содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 1 часу. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе.
[00304] Органическую фазу возвращали в реактор. MeOH (1,98 кг ± 5% или 2,50 л ± 5%) загружали в органическую фазу и перемешивали в течение периода времени от примерно 15 до примерно 30 минут при 25 ± 5°C. DI воду (5,0 кг ± 5% или 5,0 л ± 5%) загружали в смесь и перемешивали в течение периода времени от примерно 15 минут до 12 часов при 25 ± 5°C. В некоторых вариантах реализации DI воду (5,0 кг ± 5% или 5,0 л ± 5%) загружали в смесь и перемешивали в течение периода времени, не превышающего или равного примерно 30 минутам, при 25 ± 5°C. Содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 2 часам. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе. Партию выдерживали под вакуумом при температуре кожуха, не превышающей или равной примерно 40°C (например, от 20 до 40°C), до остаточного объема примерно 9 л ± 5%. Смесь можно поддерживать при температуре, не превышающей или равной примерно 25°C (например, от 0 до 25°C), в течение периода времени, не превышающего или равного примерно 48 часам (например, от 0 до 48 часов).
Таблица 4: способ ВЭЖХ для частей 2 и 3 примера 1
0,25 М раствор NH4OH в метаноле
Часть 3: синтез соединения 4a

[00305] DI воду (0,5 кг ± 5% или 0,5 л ± 5%) загружали в смесь из части 2, содержащей соединение 3a, при этом поддерживая температуру реакции на уровне 20 ± 5°C. Boc-(S)-HABA [(0,512 кг) ± 2%, 2,34 моль] загружали в реакционную смесь и температуру поддерживали в диапазоне 20 ± 5°C. Загрузочную систему, применяемую для загрузки Boc-(S)-HABA, промывали ДХМ (0,27 кг ± 5% или 0,20 л ± 5%) и смывочный раствор добавляли в реакционную смесь. Смесь перемешивали при температуре партии 20 ± 5°C.
[00306] Загружали 1-гидроксибензотриазола моногидрат (HOBt•H2O, 0,057 кг ± 2%, 0,42 моль), при этом поддерживая температуру реакции на уровне 20 ± 5°C. Загрузочную систему, применяемую для загрузки HOBt•H2O, промывали ДХМ (0,27 кг ± 5% или 0,2 л ± 5%) и смывочный раствор добавляли в реакционную смесь.
[00307] Значение pH партии доводили до 5,0 ± 0,2, при этом поддерживая температуру в диапазоне 20 ± 5°C. Значение pH партии регулировали путем добавления 2 М раствора HCl (по необходимости). Требуемый кислотный раствор готовили в отдельной подходящей емкости путем добавления концентрированной HCl (1,392 кг ± 5% или 1,18 л ± 5%) к DI воде (6,00 кг ± 5% или 6,00 л ± 5%). Для регулирования pH обычно требуется загрузка в количестве от примерно 3,5 до примерно 4,5 л/кг. В некоторых случаях, если значение pH падало ниже примерно 4,8 (например, примерно 5,0 ± 0,2), добавляли 2 М раствор NaOH (по необходимости) для доведения значения pH партии до указанного спектра 5,0 ± 0,2. В некоторых случаях, при необходимости, в подходящей емкости готовили щелочной раствор из NaOH (0,56 кг ± 5%) и DI воды (7,0 кг ± 5% или 7,0 л ± 5%).
[00308] EDAC [(0,447 кг) ± 2%, 2,33 моль] загружали в партию, при этом поддерживая температуру в диапазоне 20 ± 5°C. Загрузочную систему, применяемую для загрузки EDAC, промывали ДХМ (0,27 кг ± 5% или 0,20 л ± 5%) и смывочный раствор добавляли в реакционную смесь. Смесь перемешивали при температуре реакции 20 ± 5°C в течение периода времени от примерно 15 минут до 6 часов и отбирали образцы для определения завершения реакции. В некоторых вариантах реализации смесь перемешивали при температуре реакции 20 ± 5°C в течение периода времени, не превышающего или равного примерно 1 часу, и отбирали образцы для определения завершения реакции. Реакцию считали завершенной, когда содержание соединения 3a относительно соединения 4a равнялось или составляло не более, чем примерно 1,0% площади согласно анализу ВЭЖХ (способ приведен в таблице 4). Значение pH проверяли при каждом отборе образцов и корректировали по мере необходимости, поддерживая значение pH в диапазоне от примерно 4,8 до примерно 6,0. В некоторых случаях, при необходимости, добавляли растворы HCl или NaOH, как описано выше. Первый образец отбирали после времени выдерживания от примерно 15 минут до 6 часов. В некоторых вариантах реализации первый образец отбирали после времени выдерживания примерно 1 час. В некоторых вариантах реализации дополнительные образцы отбирали с интервалами примерно 3 часа. Если после отбора двух образцов реакция не завершалась, загружали дополнительное количество EDAC и Boc-(S)-HABA для завершения реакции, определяемое по следующей формуле: Загрузка=P3*D3/(100-D3), где: P3=количество первоначально загруженного EDAC или Boc-(S)-HABA в кг, и D3=количество непрореагировавшего соединения 3a в пл.%/пл.%.
[00309] После завершения реакции в реакционную смесь загружали MeOH (1,58 кг ± 5% или 2,00 л ± 5%), при этом поддерживая температуру реакции на уровне 20 ± 5°C. Затем в реакционную смесь загружали ДХМ (15,94 кг ± 5% или 12,00 л ± 5%), при этом поддерживая температуру партии на уровне 20 ± 5°C. Затем в реакционную смесь загружали DI воду (5,0 кг ± 5% или 5,0 л ± 5%), при этом поддерживая температуру партии на уровне 20 ± 5°C. Значение pH партии регулировали до значения от примерно 9,0 до примерно 10,0 путем добавления 2 М раствора NaOH (по необходимости), при этом поддерживая температуру на уровне 25 ± 5°C. Регулирование значения pH обычно требует загрузки 2 М раствора NaOH в количестве от примерно 3,5 до примерно 4,5 л/кг. Если значение pH партии превышало 10,0, загружали 2 М раствор HCl для достижения указанного диапазона. Смесь перемешивали при температуре от примерно 20°C до примерно 38°C в течение периода времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации смесь перемешивали при температуре от примерно 20°C до примерно 38°C в течение периода времени, не превышающего или равного примерно 30 минутам. Затем содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое затем оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 1 часу. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе. Органическую фазу возвращали в реактор, а затем загружали MeOH (2,38 кг ± 5% или 3,00 л ± 5%), при этом поддерживая температуру партии на уровне от примерно 20°C до примерно 38°C. Затем в смесь загружали DI воду (7,0 кг ± 5% или 7,0 л ± 5%) и смесь перемешивали при температуре от примерно 20°C до примерно 38°C в течение периода времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации затем в смесь загружали DI воду (7,0 кг ± 5% или 7,0 л ± 5%) и смесь перемешивали при температуре от примерно 20°C до примерно 38°C в течение периода времени, не превышающего или равного примерно 30 минутам. Содержимое оставляли осаждаться и отделяли в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации содержимое оставляли осаждаться и отделяли в течение периода времени, не меньшего или равного примерно 1 часу. Органическую фазу (нижнюю фазу) переносили в приемник и водную фазу отбрасывали для утилизации. Продукт находился в органической фазе. Партию концентрировали в вакууме при температуре кожуха, не превышающей или равной примерно 40°C (например, от 20 до 40°C), до остаточного объема примерно 10 л ± 5%.
Часть 4: кристаллизация соединения 4a
[00310] Ацетонитрил (7,87 кг ± 5% или 10,00 л ± 5%) загружали в партию из части 3 в течение периода времени от примерно 5 минут до 4 часов. В некоторых вариантах реализации ацетонитрил (7,87 кг ± 5% или 10,00 л ± 5%) загружали в партию из части 3 в течение периода времени, не меньшего или равного примерно 10 минутам. Смесь концентрировали в вакууме при температуре кожуха, не превышающей или равной примерно 40°C (например, от 20 до 40°C), с получением конечного остаточного объема примерно 10 л ± 5%. Ацетонитрил (7,87 кг ± 5% или 10,00 л ± 5%) загружали в партию и партию концентрировали в вакууме при температуре кожуха, не превышающей или равной примерно 40°C (например, от 20 до 40°C), с получением конечного остаточного объема примерно 10 л ± 5%. Затем загружали ацетонитрил (переменное количество) для достижения конечного объема партии примерно 25 л ± 5%. Содержимое реактора представляло собой густую непрозрачную суспензию белых твердых веществ.
[00311] Затем партию нагревали до температуры обратной конденсации (~82°C) и выдерживали в течение периода времени от примерно 5 минут до 12 часов. В некоторых вариантах реализации затем партию нагревали до температуры обратной конденсации (~82°C) и выдерживали в течение периода времени, не превышающего или равного примерно 15 минутам. Ожидали, что в течение периода выдерживания не все твердые вещества растворятся. В смесь загружали DI воду (0,375 кг ± 5% или 0,375 л ± 5%), при этом поддерживая при температуре обратной конденсации. Партию перемешивали в течение периода времени от примерно 30 до примерно 60 минут до получения однородного раствора. Если однородный раствор не получали за примерно 30-60 минут, загружали дополнительные порции DI воды (0,125 кг ± 5% или 0,125 л ± 5%) для растворения оставшихся твердых веществ. Добавление большего количества воды обычно оказывает незначительное положительного влияние на качество, но может приводить к снижению выхода. После получения однородного раствора партию охлаждали до температуры 75 ± 3°C в течение периода времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации после получения однородного раствора партию охлаждали до температуры 75 ± 3°C в течение периода времени, не превышающего или равного примерно 1 часу.
[00312] В партию загружали затравку соединения 4a (0,01 кг ± 2%), при этом поддерживая температуру на уровне 75 ± 3°C. Емкость для загрузки суспензии затравки и трубки промывали ацетонитрилом (0,08 кг ± 5% или 0,10 л ± 5%) и смывочный раствор добавляли в партию. Затем партию перемешивали в течение периода времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации затем партию перемешивали в течение периода времени, не меньшего или равного примерно 30 минутам. Партию охлаждали до температуры 50 ± 5°C в течение периода времени от 30 минут до 12 часов и дополнительно выдерживали в течение примерно 2-12 часов при умеренном перемешивании с получением густой суспензии. В некоторых вариантах реализации партию охлаждали до температуры 50 ± 5°C в течение периода времени, не меньшего или равного примерно 2 часам, и дополнительно выдерживали в течение примерно 2-12 часов при умеренном перемешивании с получением густой суспензии. Партию охлаждали до температурного диапазона от примерно -5 до примерно 5°C в течение периода времени от примерно 1 до 24 часов и дополнительно выдерживали в течение примерно 4-12 часов. В некоторых вариантах реализации партию охлаждали до температурного диапазона от примерно -5 до примерно 5°C в течение периода времени, не меньшего или равного примерно 1 часу, и дополнительно выдерживали в течение примерно 4-12 часов. Партию фильтровали и обезвоживали и слой фильтрата промывали ацетонитрилом (0,79 кг ± 5% или 1,00 л ± 5%) с температурой 0 ± 5°C и обезвоживали.
[00313] Продукт сушили под вакуумом при температуре, не превышающей или равной примерно 55°C (например, от 0 до 55°C). Сушку считали завершенной, когда потери при сушке равнялись или составляли не более, чем примерно 1 масс.%/масс.%. Продукт сушили под потоком сухого азота. После сушки продукт можно протирать при помощи подходящей операции просеивания. Выход после трех стадий превращения сизомицина в виде свободного основания в соединение 4a составлял примерно 65%.
[00314] Кристаллизацию с затравкой в ацетонитриле с 1,5% DI воды при 75 ± 3°C с последующим охлаждением можно выбирать из-за высокого выхода ~70%, высокой чистоты, не меньшей или равной примерно 88,08% (чистота материала до кристаллизации составляла примерно 77,75%), и простоты фильтрования смеси. Тем не менее, были испытаны несколько процедур кристаллизации до выбора ацетонитрила с 1,5% DI воды при 75 ± 3°C с последующим охлаждением. Они подробно описаны в таблице 5. Применение воды (примерно 1,5%) в качестве сорастворителя ацетонитрила может снижать обрабатываемый объем, необходимый для кристаллизации.
Таблица 5: испытанные условия кристаллизации для соединения 4a
(об./об.)
a Чистота материала до кристаллизации составляла 77,75%.
bВсе указанные концентрации, температуры, выходы (по общей массе) и времена являются примерными.
Часть 5: характеристические данные для соединения 4a
[00315] XRPD спектр соединения 4a представлен на ФИГ. 1. ТГА соединения 4a соответствовал безводному/несольватированному материалу, т.к. наблюдали только 0,1 масс.% изменение от 25 до 189°C, что указывает на низкое содержание легколетучих соединений (ФИГ. 2). ДСК профиль соединения 4a представлен на ФИГ. 3.
Пример 2: Схема синтеза соединения 6a
[00316] Ниже подробно описана общая схема синтеза соединения 6a.

Часть 1: синтез соединения 5a

[00317] Соединение 4a (1,0 кг ± 1%) и MeOH (7,92 кг ± 5% или 10,00 л ± 5%) загружали в реактор. Смесь нагревали до температуры 50 ± 5°C. Раствор Boc2O (0,255 кг ± 2%) в метаноле (0,20 кг ± 5% или 0,25 л ± 5%) добавляли в реактор, при этом поддерживая температуру на уровне 50 ± 5°C. Загрузочную систему, применяемую для загрузки раствора Boc2O с метанолом (0,20 кг ± 5% или 0,25 л ± 5%), промывали и смывочный раствор добавляли в реакционную смесь, при этом поддерживая температуру в диапазоне 50 ± 5°C. Партию выдерживали при 50 ± 5°C до завершения реакции. Реакцию считали завершенной, когда содержание соединения 4a составляло менее или равнялось примерно 3,0% площади согласно ВЭЖХ (см. таблицу 6 для применяемого способа ВЭЖХ). Первый образец для анализа отбирали через время реакции от примерно 15 минут до 12 часов. В некоторых вариантах реализации первый образец для анализа отбирали через 3 часа протекания реакции и последующие образцы, при необходимости, собирали с интервалами примерно 3 часа. Если требовалось завершение реакции, к реакционной смеси добавляли дополнительное количество раствора Boc2O в метаноле. Добавляемое количество рассчитывали по следующей формуле: Загрузка=P1 × (A2/[100-A2]) ± 1%, где: P1=количество Boc2O (кг), загруженного ранее в реакцию, и A2=количество непрореагировавшего соединения 4a в пл.%/пл.% в последнем образце.
[00318] После завершения реакции температуру партии стабилизировали на уровне 20 ± 5°C. Партию можно выдерживать в указанном диапазоне температур в течение периода времени вплоть до примерно 55 часов (например, от 0 до 55 часов).
Таблица 6: способ ВЭЖХ для частей 1 и 2 примера 2
0,25 М раствор NH4OH в ацетонитриле
Часть 2: синтез соединения 6a

[00319] Деионизированную воду (7,78 кг ± 5% или 7,78 л ± 5%) загружали в реактор и добавляли твердый гидроксид натрия (0,39 кг ± 1%), а затем смесь перемешивали до визуального растворения. Часть деионизированной воды можно загружать после загрузки гидроксида натрия и/или после загрузки дитионита натрия и применять для промывания загрузочного устройства, применяемого для загрузки указанных материалов. Температуру стабилизировали на уровне примерно 0-5°C. К щелочному водному раствору добавляли дитионит натрия (1,196 кг ± 1%), при этом поддерживая температуру раствора в диапазоне от примерно 0°C до примерно 5°C. Более высокие температуры в процессе добавления могут приводить к снижению чистоты продукта. Смесь перемешивали в течение периода времени от примерно 5 минут до 12 часов при примерно 0-5°C. В некоторых вариантах реализации смесь перемешивали в течение периода времени, не превышающего или равного примерно 15 минутам, при примерно 0-5°C. Раствор дитионита следует применять в течение примерно 0-4 часов после загрузки Na2S2O4. В некоторых вариантах реализации раствор дитионита следует применять в течение примерно 90 минут после загрузки Na2S2O4.
[00320] Реакционную смесь из части 1 добавляли к щелочному раствору дитионита натрия в течение примерно 1-8 часов (например, 1-4 часов), при этом поддерживая температуру смеси на уровне от примерно ниже -5°C до примерно ниже 15°C. В некоторых вариантах реализации реакционную смесь из части 1 добавляли к щелочному раствору дитионита натрия в течение примерно 1-4 часов, при этом поддерживая температуру смеси на уровне примерно ниже 10°C. Добавление является экзотермическим, и более быстрое добавление приводит к получению вязких твердых веществ. Загрузочную систему, применяемую для загрузки реакционной смеси, промывали метанолом (0,40 кг ± 5% или 0,50 л ± 5%) и смывочный раствор добавляли в партию. Партию нагревали до температуры от примерно 25°C до примерно 30°C в течение периода времени от примерно 1 до 4 часов. В некоторых вариантах реализации партию нагревали до температуры от примерно 25°C до примерно 30°C в течение примерно 2 часов. Как правило, максимальная температура кожуха составляла от примерно 25 до 35ºC, и поддерживали ΔT между температурами кожуха и партии, не превышающую или равную примерно 10°C (например, от 0 до 10ºC). В некоторых вариантах реализации максимальная температура кожуха составляла примерно 30ºC, и поддерживали ΔT между температурами кожуха и партии, не превышающую или равную примерно 10°C. Партию перемешивали при температуре примерно 25-30°C до завершения реакции. Более высокие температуры реакции могут приводить к увеличению количества примесей.
[00321] Реакцию считали завершенной, когда величина пика с относительным временем удерживания 0,43,
 ,
,
составляла меньше или равнялась примерно 3,0 пл.%/пл.% согласно ВЭЖХ (тот же способ, который подробно описан в таблице 6). В некоторых вариантах реализации первый образец отбирали в конце периода нагревания и последующие образцы отбирали с интервалами примерно 3 часа.
[00322] После завершения реакции смесь перегоняли в вакууме при температуре кожуха от примерно 20 до 40°C до получения конечного объема 13 л ± 5%. В некоторых вариантах реализации после завершения реакции смесь перегоняли в вакууме при температуре кожуха, не превышающей или равной примерно 35°C, до получения конечного объема 13 л ± 5%. В партию загружали IPAc (3,49 кг ± 5% или 4,00 л ± 5%), при этом поддерживая температуру на уровне примерно 25-40°C. Затем в партию загружали деионизированную воду (7,0 кг ± 5% или 7,0 л ± 5%), при этом поддерживая температуру на уровне примерно 25-40°C. Смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре примерно 30-40°C. В некоторых вариантах реализации смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре примерно 30-40°C. Полное растворение солей не требовалось, поскольку разделение фаз можно осуществлять без проблем, связанных с присутствием некоторого количества нерастворенных солей.
[00323] Перемешивание прекращали и слои оставляли разделяться в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 1 часу. Водную фазу (водная фаза 1) переносили в приемник. Продукт с преимущественным содержанием органической фазы (органическая фаза 1) переносили в другой приемник. В водную фазу 1 загружали IPAc (1,74 кг ± 5% или 2,00 л ± 5%), поддерживая температуру на уровне примерно 25-40°C. Смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре 35 ± 5°C. В некоторых вариантах реализации смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре 35 ± 5°C. Соли могут выпадать в осадок при температуре ниже примерно 30°C. Перемешивание прекращали и слои оставляли разделяться в течение периода времени от примерно 30 минут до 12 часов. В некоторых вариантах реализации перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 1 часу. Нижнюю водную фазу (водная фаза 2) отправляли в отходы.
[00324] Органические фазы 1 и 2 объединяли в подходящем приемнике. Примерно 6,5 масс.%/масс.% водный раствор бикарбоната натрия получали путем растворения NaHCO3 (0,42 кг ± 5%) в деионизированной воде (6,00 кг ± 5% или 6,00 л ± 5%). Температуру указанного раствора стабилизировали на уровне 25 ± 5°C. Примерно от 2 до 6 л 6,5 масс.%/масс.% раствора NaHCO3 загружали в объединенные органические фазы. В некоторых вариантах реализации примерно 3 л 6,5 масс.%/масс.% раствора NaHCO3 загружали в объединенные органические фазы. Раствор перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре 25 ± 5°C. В некоторых вариантах реализации раствор перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре 25 ± 5°C. Затем перемешивание прекращали и слои оставляли разделяться в течение периода времени от 0 до 48 часов. В некоторых вариантах реализации затем перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 1 часу. Нижнюю водную фазу отправляли в отходы. Органическую фазу (органическая фаза 3) можно поддерживать при температуре, не превышающей или равной примерно 25°C (например, от 0 до 25°C), в течение периода времени от 0 до 48 часов В некоторых вариантах реализации нижнюю водную фазу отправляли в отходы. Органическую фазу (органическая фаза 3) можно поддерживать при температуре, не превышающей или равной примерно 25°C, в течение периода времени вплоть до 24 часов.
[00325] Примерно от 2 до 6 л 6,5 масс.%/масс.% раствора NaHCO3 загружали в органическую фазу 3. В некоторых вариантах реализации примерно 3 л 6,5 масс.%/масс.% раствора NaHCO3 загружали в органическую фазу 3. Смесь перемешивали в течение периода времени от примерно 30 минут до 12 часов при температуре 25 ± 5°C. В некоторых вариантах реализации смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре 25 ± 5°C. Затем перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 1 часу. Нижнюю водную фазу отправляли в отходы. Органическую фазу (органическая фаза 4) можно поддерживать при температуре, не превышающей или равной примерно 20-25°C, в течение периода времени вплоть до 24 часов. В некоторых вариантах реализации органическую фазу (органическая фаза 4) можно поддерживать при температуре, не превышающей или равной примерно 25°C, в течение периода времени вплоть до 24 часов.
[00326] Органическую фазу 4 перегоняли в вакууме при температуре кожуха, не превышающей или равной примерно 50°C (например, от 20 до 50°C), до получения конечного объема 3 л ± 5%. IPAc (2,62 кг ± 5% или 3,00 л ± 5%) загружали в партию. Смесь перегоняли в вакууме при температуре кожуха, не превышающей или равной примерно 50°C (например, от 20 до 50°C), до получения конечного объема 3 л ± 5%. IPAc (2,62 кг ± 5% или 3,00 л ± 5%) загружали в партию. Затем смесь перегоняли в вакууме при температуре кожуха, не превышающей или равной примерно 50°C (например, от 20 до 50°C), до получения конечного объема 4,5 л ± 5%. Затем в партию загружали деионизированную воду (0,135 кг ± 5% или 0,135 л ± 5%), при этом поддерживая температуру на уровне примерно 35-40°C. Температуру партии регулировали до уровня от примерно 15 до примерно 30°C. В партию загружали ДХМ (5,98 кг ± 5% или 4,50 л ± 5%), при этом поддерживая температуру на уровне примерно 15-30°C. В некоторых случаях смесь оставалась мутной даже при достижении полного растворения соединения 6a.
[00327] В раствор партии загружали затравку соединения 6a (0,02 кг ± 2%), при этом поддерживая температуру на уровне примерно 20-25°C. Наблюдали кристаллизацию, тем не менее, скорость кристаллизации могла являться низкой. Смесь перемешивали при температуре примерно 20-25°C в течение примерно 1-2 часов, и образовывалась суспензия. Если суспензия не наблюдалась, смесь охлаждали до температуры примерно 15-20°C и в раствор партии добавляли дополнительное количество затравки соединения 6a (0,02 кг ± 2%), при этом поддерживая температуру на уровне примерно 15-20°C, в течение периода времени от примерно 1 до примерно 2 часов. Затем образовывалась суспензия. Более низкие температуры во время и после внесения затравки могут приводить к снижению чистоты, но увеличению выхода.
[00328] Партию охлаждали до примерно 15-25°C и перемешивали в указанном диапазоне температур в течение примерно 18-24 часов. Партию охлаждали до температуры от примерно 5°C до примерно -5°C в течение периода времени от примерно 1 до 12 часов и выдерживали при перемешивании в указанном диапазоне температур в течение примерно 6-12 часов. В некоторых вариантах реализации партию охлаждали до температуры от примерно 5°C до примерно -5°C в течение примерно 2 часов и выдерживали при перемешивании в указанном диапазоне температур в течение примерно 6-12 часов. Указанное время состаривания важно для выхода. Партию выгружали на подходящий фильтр и обезвоживали. Сырой слой фильтрата промывали раствором ДХМ (1,33 кг ± 5% или 1,00 л ± 5%) с температурой от примерно 5°C до -5°C, а затем обезвоживали. Продукт сушили под вакуумом при температуре, не превышающей или равной примерно 45°C (например, от 0 до 45°C). Сушку завершали, когда потери при сушке равнялись или составляли не более, чем примерно 1 масс.%/масс.%. Продукт можно сушить в вакууме про продувке азотом. После сушки продукт можно просеивать. Выход после двух стадий превращения соединения 4a в соединение 6a составлял примерно 85%. Регулирование некоторых переменных, таких как конечный объем после перегонки, количество ДХМ, содержание воды, температуры внесения затравки и состаривания и время состаривания после внесения затравки, может обеспечивать типичную кристаллизацию с типичными чистотами. Вода может влиять на выход, но, похоже, не имеет отношения к качеству продукта.
Часть 3: оптимизация условий кристаллизации соединения 6a
[00329] Растворимость соединения 6a в смеси дихлорметан/изопропилацетат (50/50 об./об.) с примерно 1% воды, смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 2% воды и смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 8% воды демонстрирует сильную зависимость от температуры. При повышенных температурах в смеси дихлорметан/изопропилацетат (50/50 об./об.) с примерно 1% воды можно наблюдать ограниченную растворимость и в смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 2% воды и смеси изопропилацетат/дихлорметан (71/29 об./об.) с 8% воды можно наблюдать промежуточную растворимость. Все температуры помутнения наблюдается, когда реактор достигает самых низких температур, составляющих примерно 6-8°C, что, возможно, свидетельствует о большой ширине метастабильной зоны при любых условиях. При примерно 7,9 мг/мл в смеси дихлорметан/изопропилацетат (50/50 об./об.) с примерно 1% воды температуры помутнения не наблюдаются до выдерживания в течение периода времени от примерно 3 до 8 часов при примерно 6-8°C. Указанные результаты указывают на то, что без применения затравки можно ожидать нестабильности с точки зрения времени основного события нуклеации (самопроизвольной нуклеации). Кроме того, нуклеацию стабильно наблюдают при высоких уровнях перенасыщения, что иногда приводит к плохой кристалличности, дефектам кристаллов, таким как включения растворителя, и/или образованию мелких частиц.
[00330] Отобранные образцы для каждой из систем растворителей из экспериментов по определению растворимости и ширины метастабильной зоны выделяли путем вакуумного фильтрования. При выделении твердых частиц из смеси дихлорметан/изопропилацетат (50/50 об./об.) с примерно 1% воды не наблюдали никаких проблем с фильтрованием. Тем не менее, в случае твердых частиц, выделяемых из смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 2% и примерно 8% воды, наблюдали липкие твердые вещества и некоторое разжижение. Материал, выделенный из смеси дихлорметан/изопропилацетат (50/50 об./об.) с примерно 1% воды, являлся кристаллическим. Материал, выделенный из смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 2% воды, являлся неоднородным, и материал, выделенный из смеси изопропилацетат/дихлорметан (71/29 об./об.) с примерно 8% воды, мог содержать небольшое количество рентгеноаморфного материала.
Часть 4: характеристические данные для соединения 6a
[00331] XRPD спектр соединения 6a представлен на ФИГ. 4, и положения пиков по существу соответствуют перечисленным в таблице 1. ТГА соединения 6a демонстрировали 4,5 масс.% потерю в интервале 24,1-78,6°C (ФИГ. 5). Основной экзотермической эффект согласно ДСК соединения 6a наблюдали с ограничением температуры слева на уровне 231,8°C и с энергией -52,9 кДж/кг (ФИГ. 6).
Пример 3: Схема синтеза соединения 7a
[00332] Ниже подробно описана общая схема синтеза соединения 7a.
Часть 1: синтез соединения 7a

[00333] Соединение 6a (1,0 кг ± 1%) и ацетонитрил (3,94 кг ± 5% или 5,00 л ± 5%) загружали в реактор. Температуру стабилизировали на уровне от примерно 15°C до примерно 30°C при перемешивании. Раствор мог являться мутным. Смесь перегоняли в вакууме при максимальной температуре примерно 45°C (например, от 20 до 45°C) до получения конечного объема 2 л ± 5%.
[00334] Отбирали образец для определения содержания воды по методу Карла Фишера. Если содержание воды составляло менее или равнялось примерно 0,30 масс.%/масс.%, переходили к следующей стадии реакции. В противном случае в реактор вносили ацетонитрил (2,36 кг ± 5% или 3,00 л ± 5%) и перегонку повторяли до получения содержания воды в растворе после перегонки менее или равного примерно 0,30 масс.%/масс.% по методу Карла Фишера.
[00335] Реакционную смесь охлаждали до температуры от примерно 30°C до примерно 15°C и в реактор вносили ацетон (3,94 кг ± 5% или 5,00 л ± 5%). Обратите внимание, что часть ацетона можно загружать до загрузки бикарбоната натрия и/или 2-йодэтанола и применять для промывания загрузочного устройства, применяемого для указанных загрузок.
[00336] Реакционную смесь нагревали до температуры от примерно 33°C до примерно 37°C с целевой температурой примерно 35°C. В реактор загружали бикарбонат натрия (0,177 кг ± 2%), при этом поддерживая температуру реакционной смеси от примерно 33°C до примерно 37°C с целевой температурой примерно 35°C. В реакционную смесь загружали 2-йодэтанол (0,226 кг ± 2% или 0,102 л ± 2%), при этом поддерживая температуру реакционной смеси от примерно 33°C до примерно 37°C с целевой температурой примерно 35°C. Реакционную смесь перемешивали при температуре от примерно 33°C до примерно 37°C с целевой температурой примерно 35°C до получения содержания соединения 6a относительно соединения 7a менее или равного примерно 2,5% площади согласно ВЭЖХ. Если требовалось завершение реакции, к реакционной смеси добавляли дополнительное количество 2-йодэтанола. Количество дополнительного 2-йодэтанола в кг рассчитывали с применением следующей формулы: [P2 x D/(100-D)] ± 2%, где: P2 представляет собой количество 2-йодэтанола, первоначально загруженного в реакцию, в кг, и D представляет собой содержание непрореагировавшего соединения 6a в % площади согласно ВЭЖХ в последнем образце производственного контроля. Процент площади определяли путем ВЭЖХ-УФ: Zorbax SB-CN, 3,5 мкм, 150×4,6 мм, A: 25 мМ K2HPO4; B: ацетонитрил. Градиент: 5-80% B в течение 25 минут, выдерживали в течение 5 минут; повторно уравновешивали в течение 5 минут, скорость потока: 1,0 мл/мин, УФ: 210 нм, темп. колонки: 30°C.
[00337] После завершения реакции реакционную смесь охлаждали до температуры от примерно 25°C до примерно 20°C. В реакционную смесь загружали 1,4-диазабицикло[2.2.2]октан (DABCO, 0,24 кг ± 2%), при этом поддерживая температуру на уровне от примерно 20°C до примерно 25°C, и смесь перемешивали в указанном диапазоне температур до содержания 2-йодэтанола, определяемого путем ГХ, меньше потерь при сушке согласно способу (< 0,003 масс.%/масс.%). В способе ГХ применяли 50 см DB-1 колонку с внутренним диаметром 0,32 мм. Начальная температура термостата составляла примерно 70°C с выдерживанием в течение примерно 1 минуты с последующим нагреванием со скоростью примерно 10°C/минута до примерно 250°C). К смеси добавляли деионизированную воду (5,00 кг ± 5% или 5,00 л ± 5%), поддерживая температуру на уровне от примерно 15°C до примерно 30°C. Указанное добавление является экзотермическим. Добавляли изопропилацетат (4,36 кг ± 5% или 5,00 л ± 5%), при этом поддерживая температуру на уровне от примерно 15°C до примерно 30°C, и смесь перемешивали в течение периода времени от примерно 5 минут до 12 часов при температуре от примерно 15°C до примерно 30°C. В некоторых вариантах реализации добавляли изопропилацетат (4,36 кг ± 5% или 5,00 л ± 5%), при этом поддерживая температуру на уровне от примерно 15°C до примерно 30°C, и смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре от примерно 15°C до примерно 30°C. Затем перемешивание прекращали и слои оставляли разделяться в течение примерно 30 минут до 12 часов. В некоторых вариантах реализации затем перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 30 минутам. Водную фазу 1 (нижняя фаза) отбрасывали и органическую фазу 1 переносили в приемник. Продукт находился в органической фазе.
[00338] Изопропилацетат (2,62 кг ± 5% или 3,00 л ± 5%) добавляли в водную фазу 1 и смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре от примерно 15°C до примерно 30°C. В некоторых вариантах реализации изопропилацетат (2,62 кг ± 5% или 3,00 л ± 5%) добавляли в водную фазу 1 и смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре от примерно 15°C до примерно 30°C. Перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 30 минутам (например, от 30 минут до 12 часов). Водную фазу 2 (нижняя фаза) отбрасывали для утилизации. Органическую фазу 2 переносили в приемник. Продукт находился в органической фазе, и органические фазы 1 и 2 объединяли.
[00339] Водный раствор хлорида натрия получали путем растворения хлорида натрия (технического) (2,00 кг ± 1%) в деионизированной воде (5,80 кг ± 5% или 5,80 л ± 5%). От примерно 2 до 6 л водного раствора хлорида натрия загружали в объединенные органические фазы и смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре от примерно 15°C до примерно 30°C. В некоторых вариантах реализации примерно 3 л водного раствора хлорида натрия загружали в объединенные органические фазы и смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре от примерно 15°C до примерно 30°C. Перемешивание прекращали и слои оставляли разделяться в течение периода времени от примерно 15 минут до 12 часов. В некоторых вариантах реализации перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 30 минутам. Водную фазу 3 (нижняя фаза) отбрасывали для утилизации. Органическую фазу 3, содержащую продукт, переносили в приемник.
[00340] Примерно от 2 до 6 л водного раствора хлорида натрия загружали в объединенные органические фазы и смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов при температуре от примерно 15°C до примерно 30°C. В некоторых вариантах реализации примерно 3 л водного раствора хлорида натрия загружали в объединенные органические фазы и смесь перемешивали в течение периода времени, не меньшего или равного примерно 20 минутам, при температуре от примерно 15°C до примерно 30°C. Перемешивание прекращали и слои оставляли разделяться в течение периода времени, не меньшего или равного примерно 30 минутам (например, от 30 минут до 12 часов). Водную фазу 4 (нижняя фаза) отбрасывали для утилизации. Органическую фазу 4, содержащую продукт, переносили в приемник.
[00341] Органическую фазу перегоняли в вакууме при максимальной температуре кожуха примерно 20-50°C (например, 50°C) до получения конечного объема 2 л ± 5%. В смесь загружали ацетонитрил (8,03 кг ± 5% или 10,20 л ± 5%) и смесь перегоняли в вакууме при максимальной температуре кожуха примерно 20-50°C (например, 50°C) до получения конечного объема 7,2 л ± 5%. В раствор вносили изопропилацетат (0,70 кг ± 5% или 0,80 л ± 5%) и деионизированную воду (0,095 кг ± 2% или 0,095 л ± 2%) и раствор нагревали до температуры от примерно 70°C до примерно 80°C для обеспечения полного растворения соединения 7a.
[00342] Раствор соединения 7a охлаждали до температуры от примерно 65°C до примерно 60°C. Если кристаллизация наблюдается до добавления затравки, смесь можно повторно нагревать до температуры от примерно 70°C до примерно 80°C до полного растворения. Передо добавлением затравки смесь охлаждали до температуры от примерно 65°C до примерно 60°C. Затравку соединения 7a (0,02 кг ± 5% кг) загружали в раствор соединения 7a, при этом поддерживая температуру на уровне от примерно 60°C до примерно 65°C. Загрузочную систему, применяемую для загрузки затравки соединения 7a, промывали ацетонитрилом (0,04 кг ± 5% или 0,05 л ± 5%) и смывочный раствор добавляли в реакционную смесь. Смесь перемешивали при температуре от примерно 60°C до примерно 65°C в течение периода времени от примерно 4 до примерно 6 часов. Образовывалась очень густая суспензия.
[00343] Смесь охлаждали до температуры от примерно 5°C до примерно 0°C в течение примерно 12 часов и перемешивали в указанном диапазоне температур в течение периода времени от примерно 1 до примерно 2 часов. Целевая скорость охлаждения составляла примерно 5-10°C/час. Продукт фильтровали и промывали ацетонитрилом (2,83 кг ± 5% или 3,60 л ± 5%), предварительно охлажденным до температуры от примерно 0°C до примерно 5°C. Затем продукт сушили в вакууме при температуре, меньшей или равной примерно 45°C (например, от 0 до 45°C), до потерь при сушке, меньших или равных примерно 1 масс.%/масс.%. Продукт сушили под потоком азота. Продукт можно просеивать во время или после сушки. Выход соединения 7a составлял примерно 90%.
Часть 2: оптимизация алкилирования амина в положении 6’ при помощи галогенэтанола
[00344] Были испытаны несколько условий алкилирования амина в положении 6’ до выбора азеотропной перегонки АЦН с последующим добавлением ацетона, 2-йодэтанола и бикарбоната натрия и применением DABCO для гашения реакции.
[00345] Первоначально были исследованы два подхода для алкилирования амина в положении 6’ при помощи галогенэтанола. В первом подходе амин в положении 6’ активировали с применением о-нитробензолсульфонилхлорида (нозил-Cl) с получением соответствующей нозилатной группы на амине в положении 6’. Последующее алкилирование нозилата с применением 2-йод- или 2-бромэтанола, с гидроксильной защитой TBDMS или без нее, не являлось оптимальным и незначительно улучшалось за счет изменения растворителя (например, ТГФ, АЦН или ДМФА), основания (например, DIPEA или K2CO3) и температуры (например, от температуры окружающей среды до примерно 80°C). Второй подход, прямое алкилирование амина в положении 6’ с применением 2-бромэтанола в присутствии NaI и Na2CO3 при температуре окружающей среды, возможно обеспечивает более полное превращение и более чистый неочищенный продукт. Условия в виде 2-бромэтанола, йодида натрия и карбоната натрия были выбраны для дальнейшей оценки и разработки.
[00346] Было обнаружено, что от примерно 2 до примерно 3 молярных эквивалентов 2-бромэтанола улучшают условия реакции, при этом немного меньше диалкилированного побочного продукта образуется при примерно 2 эквивалентах. Диапазон степеней превращения составлял примерно 51-68%, при этом для достижения степени превращения >65% требовалось примерно 1-2 эквивалента NaI. Тем не менее, бромэтанол и йодид натрия идентифицировали, как возможные существенные источники воды для реакционной смеси. Вода в реакционной смеси не являлась оптимальной для результата и, возможно коррелировала с более высоким содержанием диалкилированного побочного продукта. Соединение 6a также потенциально вносит значительное количество воды в партию, но ее можно удалять путем азеотропной перегонки с применением ацетонитрила. Другие азеотропные растворители являлись потенциально менее эффективными, такие как этанол, или вызывали последующее осаждение продукта (изопропанол). Ацетон идентифицировали, как возможный предпочтительный растворитель для реакции алкилирования. На основе данных ЖХ/МС предполагали промежуточность потенциальных ацетониминиевых фрагментов, которые, не ограничиваясь теорией, могут предотвращать избыточное алкилирование. Не ограничиваясь теорией, возможно, что вода в партии ингибировала образование или дестабилизацию указанного предположительного имина, что позволяло образовываться диалкилированному побочному продукту. Не ограничиваясь теорией, роль карбоната натрия в качестве осушающего агента может являться значительной, поскольку он обеспечивает более высокие выходы по сравнению с карбонатом лития, карбонатом калия, карбонатом сезия, карбонатом натрия, сульфатом натрия, фосфатом натрия и DIPEA. Температура реакции составляла примерно 35°C по сравнению с примерно -10°C, примерно 0°C и примерно 23°C, что обеспечивало наиболее высокую относительную степень превращения с наименьшим относительным количеством диалкилированного побочного продукта. Более высокие температуры реакции (например, примерно 50°C) могут приводить к образованию большего количества диалкилированного побочного продукта.
[00347] Тем не менее, поскольку бромэтанол и йодид натрия являлись возможными основными источниками воды для реакционной смеси, что приводило к образованию побочного продукта, и их применяли для получения 2-йодэтанола in situ, такжы испытывали 2-йодэтанол. Бромэтанол также приводил к намного более медленному протеканию реакции по сравнению с 2-йодэтанолом. Применение примерно 1,2 молярного эквивалента 2-йодэтанола в ацетоне являлось достаточным для получения примерно 86% выхода соединения 7a за примерно 24 часа при примерно 35°C с примерно только 2% соединения 6a и примерно 2,6% диалкилированной примеси. Применение примерно 1,1 молярного эквивалента 2-йодэтанола приводило к получению примерно 86% выхода соединения 7a с примерно 6,9% соединения 6a и примерно 1,5% диалкилированной примеси, и применение примерно 1,5 молярного эквивалента 2-йодэтанола приводило к получению примерно 87% выхода соединения 7a с примерно 1,9% соединения 6a и примерно 2,3% диалкилированной примеси. Более длительное время реакции (примерно 41 или примерно 48 часов) может вызывать снижение количества соединения 7a без дальнейшего расходования соединения 6a. NaHCO3 испытывали в качестве основания для реакции, и он может являться предпочтительным по сравнению с Na2CO3, Na2SO4, триметиламином, триметилортоформиатом и гексметилдисилазаном. Na2CO3 приводил к относительно более высокому конечному значению pH и более высоким количествам диалкилированного побочного продукта в конце реакции по сравнению с NaHCO3 и Na2SO4, и Na2SO4 приводил к появлению новой примеси в выделенном продукте. DABCO может являться предпочтительным агентом для гашения реакции для избытка 2-йодэтанола, поскольку другие испытанные нейтрализаторы, такие как диметиламин, триметиламин, диэтаноламин, водный раствор гидроксида натрия, ДБУ, цистеин и MeOH, демонстрировали увеличение количества диалкилированного побочного продукта на примерно 63-1030% между уровнями в процессе обработки и в выделенном продукте, что свидетельствует о том, что остаточный 2-йодэтанол возможно не был устранен.
[00348] Было обнаружено, что азеотропная сушка соединения 6a в примерно 5 объемах АЦН хорошо подходит для реакции алкилирования амина с применением ацетона, 2-йодэтанола и бикарбоната натрия с последующим гашением реакции при помощи DABCO. Объединенное условие реакции, включающее применение примерно 5 объемов ацетона, NaHCO3 и примерно 1,2 молярного эквивалента 2-йодэтанола с температурой реакции от примерно 20 до 40°C, позволяло достичь < 2% соединения 6a при низких уровнях диалкилированного побочного продукта. В некоторых вариантах реализации объединенное условие реакции, включающее применение примерно 5 объемов ацетона, NaHCO3 и примерно 1,2 молярного эквивалента 2-йодэтанола с температурой реакции примерно 35°C, позволяло достичь < 2% соединения 6a при низких уровнях диалкилированного побочного продукта. Кроме того, было обнаружено, что качество соединения 7a, полученного с применением упрощенной обработки при помощи солевого раствора и без кислотных и щелочных экстракций, может быть лучше по сравнению с более длительной обработкой с применением кислотных и щелочных экстракций. Кроме того, замена промывки водой на промывку солевым раствором коррелировала с увеличением молярного выхода на примерно 15 процентных пунктов без потери качества продукта.
Часть 3: оптимизация условий кристаллизации соединения 7a
[00349] Были испытаны несколько процедур кристаллизации до выбора кристаллизации неочищенного соединения 7a из примерно 8 объемов 10 об.%/об.% смеси IPAc/АЦН с примерно 0,75-2 масс.%/масс.% воды с внесением затравки (см. таблицы 7-11). Кроме того, длительное время выдерживания при затравочной температуре (>2 часов при температуре от примерно 60°C до примерно 65°C) и медленное охлаждение (от примерно 60°C до примерно 0°C при примерно 5-10°C/час) могут являться наилучшими для получения подвижной суспензии. Указанная процедура позволяет по существу удалять непрореагировавшее соединение 6a, пента-Boc примесь (примерно 63% удаление суммарно) и диалкилированный побочный продукт (примерно 36% удаление) и может обеспечивать получение легко фильтруемого и промываемого твердого продукта. Обезвоженные свойства кристаллического соединения 7a из смеси IPAc/АЦН могут улучшать удаление примесей в маточных растворах, поскольку продукт образует игольчатые или стержнеобразные кристаллы. Напротив, эксперименты с IPAc и н-гептаном приводили к получению аморфного материала, который медленно фильтровался и обезвоживался, оставляя примеси в продукте.
Таблица 7: ранние попытки кристаллизации соединения 7a (без затравки)
общее
Выделения
(a) Густая, неперемешиваемая суспензия. Добавляли дополнительное количество растворителя (обычно 5 объемов) для обеспечения возможности фильтрования.
Таблица 8: кристаллизация с применением различных комбинаций растворитель/антирастворитель
тель(и)
общее
да/нет
15 ч
(a) Суспензия. (b) Ацетонитрил и гептан не смешиваются при RT. (c) Густая, неперемешиваемая суспензия. Добавляли дополнительное количество растворителя (обычно 5 объемов) для обеспечения возможности фильтрования. (d) Добавляли твердое соединение 7a, но оно не выступало в качестве затравки. (e) Кристаллизацию с образованием кристаллов проводили в течение 4 ч, а затем продукт нагревали почти до температуры обратной конденсации для растворения твердых веществ. Раствор охлаждали до КТ и перемешивали 15 ч перед сбором твердых веществ. В случае медленного охлаждения процесса кристаллизации получаемый продукт имел тонкоигольчатую морфологию.
Таблица 9: кристаллизация с затравкой с применением различных систем растворителей АЦН/IPAc в большем масштабе
рителей
да/нет
% загрузки
(a) Твердая масса в 1 л реакторе.
Таблица 10: кристаллизация с затравкой с содержанием воды, доведенным до уровня 0,5-1%
общее
(a) Содержание воды доводили до уровня 0,5%. (b) Содержание воды доводили до уровня 1%. (c) Содержание воды доводили до уровня 2,7%. (d) Три температурных цикла: 65-40, 70-40, 70-0; образовавшиеся кристаллы являлись “стержнеобразными”. (e) Применяли сухую затравку. (f) Вносили затравку соединения 7a в виде “стержней”, а не иголок.
Таблица 11: процесс превращения соединения 6a в соединение 7a путем кристаллизации с применением АЦН-IPAc
да/нет
7a (соединения 6a)
(a) Содержание воды доводили до уровня примерно 0,3%. (b) Содержание воды доводили до уровня примерно 0,5%. (c) Низкое извлечение, вероятно в результате неправильного регулирования содержания воды. (d) Содержание воды доводили до уровня 3,5%. В указанном случае применяли промывку солевым раствором вместо обработки кислотой/основанием для снижения потерь продукта и удаления реагентов. В других случаях, указанных в таблице, применяли только процедуру промывки водой вместо обработки кислотой/основанием.
[00350] Содержание воды может являться важным для контроля устойчивости кристаллизации. Наблюдали изменчивую метастабильную зону при добавлении примерно 0-2% воды. “Безводный” эксперимент с добавлением примерно 0% воды (содержание воды примерно 0,2-0,3%) приводил к широкой метастабильной зоне с температурой прозрачности примерно 55-60°C и температуре помутнения ниже температуры окружающей среды (примерно 8-15°C). При добавлении примерно 0,5-2% воды метастабильная зона значительно сужалась при более высокой температуре прозрачности. Наблюдение показало, что определенное содержание воды в смеси (первоначально примерно 0,5-1%) может делать кристаллизацию более воспроизводимой и устойчивой. Определение воды по методу Карла Фишера после замены растворителя при азеотропной сушке с IPAc на АЦН в процессе превращения соединения 6a в соединение 7a показало, что раствор являлся почти сухим (~200 ppm воды), таким образом, кристаллизация, осуществляемая после азеотропной сушки, проходила в безводном режиме с очень широкой метастабильной зоной. Это может приводить к очень медленному разбавлению и увеличению вероятности самопроизвольной нуклеации. Кристаллизация с добавлением примерно 0,5-2% воды может являться более воспроизводимой с постоянным значением извлечения (обычно более примерно 80%) и повышенной чистотой.
Часть 4: характеристические данные для соединения 7a
[00351] XRPD спектр соединения 7a представлен на ФИГ. 7 и и положения пиков по существу соответствуют перечисленным в таблице 2. ТГА профиль соединения 7a представлен на ФИГ. 8. ДСК профиль соединения 7a представлен на ФИГ. 9.
Пример 4: синтез сульфата плазомицина

Часть 1: синтез соединения 8a
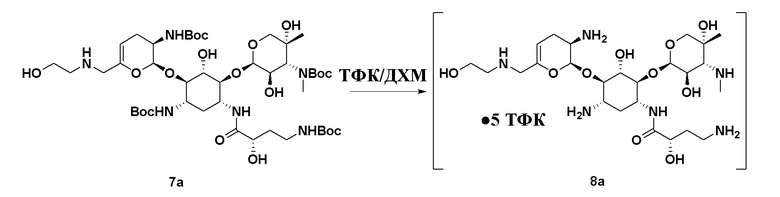
[00352] Соединение 7a (1,0 кг ± 1%, 1,00 моль) загружали в реактор. Загружали дихлорметан (5,0 л ± 5%) и смесь охлаждали до примерно 0-5°C. Трифторуксусную кислоту (ТФК, 3,0 л ± 5%, 39,5 моль) загружали в смесь со скоростью, позволяющей поддерживать температуру на уровне примерно 0-5°C. Затем смесь нагревали до примерно 20-25°C и перемешивали в течение примерно 1-2 часов. Затем смесь перегоняли в вакууме, поддерживая температуру кожуха реактора от примерно 20 до 40°C, с получением конечного объема 3 л ± 5%. В некоторых вариантах реализации затем смесь перегоняли в вакууме, поддерживая температуру кожуха реактора примерно 30°C, с получением конечного объема 3 л ± 5%. Затем смесь охлаждали до примерно 20-25°C, а затем перемешивали в течение примерно 1-4 часов перед охлаждением до примерно 0-10°C. Затем полученный раствор загружали в реактор, содержащий воду (примерно 2,5 л, предварительно охлажденной до примерно 0-5°C), поддерживая температуру примерно 0-10°C.
[00353] В реактор загружали изопропилацетат (IPAc, 3,0 л ± 5%) и смесь перемешивали в течение примерно 20-30 минут, а затем перемешивание прекращали и слои оставляли разделяться. Нижний водный слой (водная фаза 1) отбрасывали, органическую фазу два раза экстрагировали водой (примерно 0,5 л ± 5%) и смесь перемешивали в течение периода времени от примерно 15 минут до 12 часов, а затем перемешивание прекращали и слои оставляли разделяться. В некоторых вариантах реализации нижний водный слой (водная фаза 1) отбрасывали, органическую фазу два раза экстрагировали водой (примерно 0,5 л ± 5%) и смесь перемешивали в течение примерно 30 минут, а затем перемешивание прекращали и слои оставляли разделяться. Все нижние водные слои (водные фазы 2 и 3) собирали и объединяли с водной фазой 1. Затем объединенные водные фазы три раза промывали IPAc для удаления ТФК путем загрузки IPAc (3,0 л ± 5%) и перемешивали в течение примерно 20-30 минут, а затем перемешивание прекращали и слои оставляли разделяться. Слой нижней водной фазы отбрасывали и измеряли pH для подтверждения pH > 2,0. Если pH≤2,0, промывку IPAc повторяли до получения pH более, чем примерно 2,0.
Часть 2: синтез сульфата плазомицина
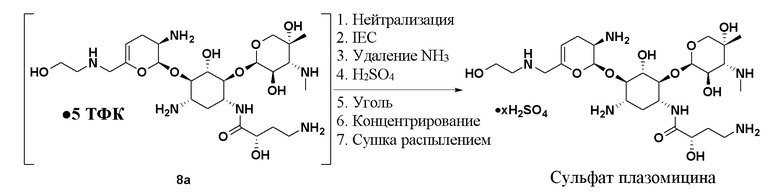
[00354] Затем водную фазу из части 1 разбавляли водой с получением конечного объема 9,0 л ± 5% и обрабатывали водным раствором аммиака (полученным путем разбавления примерно 25% аммиака (4 кг ± 5%) в воде (96 кг ± 5%)) до получения pH от примерно 5,8 до примерно 6,2. Измеряли электропроводность для подтверждения электропроводности, не меньшей или равной примерно 20 мСм/см (например, от 0 до 20 мСм/см). Если электропроводность превышала 20 мСм/см, добавляли дополнительное количество воды для получения электропроводности, меньшей или равной примерно 20 мСм/см (например, от 0 до 20 мСм/см).
[00355] Раствор неочищенной ТФК соли соединения 8a очищали путем ионообменной хроматографии, загружая раствор в ионообменную колонку, содержащую смолу Amberlite CG-50 (тип 1) (2,17 кг), предварительно превращенную в аммиачную форму. Содержимое колонки элюировали водой (1 объем колонки ± 5%) для элюирования трифторацетата аммония, а затем элюировали водным раствором аммиака (полученным путем разбавления примерно 25% аммиака (2,0 кг ± 5%) в воде (96,0 кг ± 5%)). Хроматографию контролировали по УФ-поглощению. Фракции собирали и анализировали путем СВЭЖХ, объединяя фракции с более, чем примерно 95% (по площади) соединения 8a в виде свободного основания (соединения без ТФК соли).
[00356] Объединенные хроматографические фракции пропускали через фильтр (пористость≤5 микрон) и фильтр промывали водой (1 л ± 5%). Полученный раствор концентрировали путем нанофильтрования через мембрану Dow Filmtec XLE с получением конечного объема 16 л ± 5%, поддерживая температуру на уровне примерно 0-10°C. Затем аммиак удаляли путем диафильтрования через мембрану Dow Filmtec XLE.
[00357] Раствор соединения 8a в виде свободного основания охлаждали до примерно 0-5°C и обрабатывали водным раствором серной кислоты (полученным путем растворения чистой серной кислоты (0,51 кг ± 5%) в DI воде (0,72 л ± 5%)) до pH примерно 6,0-6,5. Полученный раствор сульфата плазомицина пропускали через фильтр с активированным углем (R55SP) и фильтр промывали водой.
[00358] Раствор концентрировали путем нанофильтрования с применением мембраны Dow Filmtec XLE, а затем фильтровали через 0,22-микронный фильтр. Затем полученный раствор подавали в распылительную сушилку, поддерживая температуру на выходе из распылительной сушилки примерно 60-100°C, для сбора сульфата плазомицина в виде аморфного твердого вещества.
Пример 5: пример способа получения 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) из сизомицина.

Пример 5a
[00359] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали сизомицин в виде свободного основания (300,0 г, 0,670 моль, KF=5,65), а затем метанол (1500 мл) и дихлорметан (1500 мл). После растворения температуру реакции стабилизировали на уровне 15°C. Раствор п-нитробензилбензотриазолкарбамата (207 г, 0,694 моль, 1,04 экв.) в дихлорметане (4200 мл) добавляли в реактор в течение трех часов, поддерживая температуру реакции на уровне примерно 15°C. Добавление завершали промывкой дихлорметаном (300 мл). Через 30 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление сизомицина). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 1500 мл. В реакционную смесь загружали метанол (4500 мл) и проводили вторую концентрацию в вакууме до целевого объема 4500 мл.
[00360] В реакционную смесь загружали метанол (900 мл) и температуру стабилизировали на уровне 27,5°C. Последовательно загружали триэтиламин (442 мл, 3,189 моль, 4,76 экв.) и дигидрат ацетата цинка (487 г, 2,219 моль, 3,31 экв.) и смесь перемешивали в течение 1 ч. Предварительно полученный раствор ди-трет-бутилдикарбоната (759 г, 3,478 моль, 5,19 экв.) в метаноле (600 мл) добавляли в реакционный раствор в течение 70 минут. Перенос завершали промывкой метанолом (60 мл). После добавления реакционную смесь перемешивали в течение 9,5 ч при 27,5°C, а затем из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-амино-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата и моно-Boc-защищенного промежуточного 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 3600 мл. Температуру стабилизировали на уровне от 20 до 30°C и реакцию гасили путем добавления 25% аммиака (1800 мл) в течение 1,5 ч (осторожно: экзотермическое добавление, скорость добавления контролировали для поддержания температуры реакции на уровне от 20 до 30°C). Добавляли дихлорметан (3000 мл) и двухфазную смесь перемешивали в течение 20 минут, после чего перемешивание прекращали и слои оставляли разделяться. Нижний органический слой с преимущественным содержанием продукта (OP1) переносили в приемник, тогда как верхний водный слой с малым содержанием продукта (AQ1) отбрасывали. OP1 возвращали в реактор, в который загружали воду (750 мл) и 25% аммиака (750 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP2) переносили в приемник, тогда как верхний водный слой (AQ2) отбрасывали. OP2 возвращали в реактор, в который загружали метанол (300 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP3) переносили в приемник, тогда как верхний водный слой (AQ3) отбрасывали. OP3 возвращали в реактор, в который загружали метанол (300 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP4) переносили в приемник, тогда как верхний водный слой (AQ4) отбрасывали. Конечную промытую органическую фазу (OP4) возвращали в реактор и концентрировали в вакууме до объема 2700 мл.
[00361] В реактор добавляли воду (150 мл), Boc-(S)-HABA (153 г, 0,698 моль, 1,04 экв.) и 1-гидроксибензотриазол (17 г, 0,125 моль, 0,19 экв.). После загрузки каждого из твердых веществ (Boc-(S)-HABA и 1-гидроксибензотриазол) дихлорметан (60 мл) применяли для промывки загрузочной системы. Значение pH реакционной смеси регулировали до 5,5 с применением 2 М раствора хлористоводородной кислоты (1250 мл). После регулирования pH в реакционную смесь загружали 1-этил-3-(3’-диметиламинопропил)карбодиимида гидрохлорид (133 г, 0,694 моль, 1,03 экв.). Дихлорметан (60 мл) применяли для промывки после загрузки твердого вещества. Через 70 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ (потребление 4-нитробензил-(((2S,3R)-2-(((1R,2R,3S,4R,6S)-4-амино-6-((трет-бутоксикарбонил)амино)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-6-ил)мета). В реакционную смесь загружали метанол (600 мл), а затем дихлорметан (3600 мл) и воду (1500 мл). Значение pH регулировали до 9,5 с применением гидроксида натрия (1200 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP5) переносили в приемник, тогда как верхний водный слой (AQ5) отбрасывали. OP5 возвращали в реактор, а затем добавляли воду (2100 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP6) переносили в приемник, тогда как верхний водный слой (AQ6) отбрасывали. OP6 возвращали в реактор и концентрировали в вакууме с получением конечного объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реакционную смесь загружали ацетонитрил (примерно 4500 мл) до конечного целевого объема 7500 мл.
[00362] Смесь нагревали до температуры обратной конденсации (83°C) и последовательно добавляли две порции воды (75 мл и 38 мл) с получением раствора. Раствор охлаждали до 75°C, после чего в реакционную смесь загружали затравку 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (3 г, 0,0029 моль, 0,004 экв.). Вновь образовавшуюся суспензию охлаждали до 3°C в течение 5 ч и перемешивали при 0-5°C в течение дополнительных 4 ч. Смесь фильтровали, промывали ацетонитрилом (две порции по 300 мл) и сушили в вакууме с получением 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (345,19 г, 50% молярный выход).
Пример 5b
[00363] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали сизомицин в виде свободного основания (300,0 г, 0,670 моль, 1 экв., KF=5,65), а затем метанол (1500 мл) и дихлорметан (1500 мл). После растворения температуру реакции стабилизировали на уровне 15°C. Раствор п-нитробензилбензотриазолкарбамата (207 г, 0,694 моль, 1,04 экв.) в дихлорметане (4200 мл) добавляли в реактор в течение трех часов, поддерживая температуру реакции на уровне примерно 15°C. Добавление завершали промывкой дихлорметаном (300 мл). Через 30 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление сизомицина). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 1500 мл.
[00364] В реакционную смесь загружали метанол (4500 мл) и температуру стабилизировали на уровне 20°C. Последовательно загружали триэтиламин (498 мл, 3,593 моль, 5,36 экв.) и дигидрат ацетата цинка (425 г, 1,935 моль, 2,89 экв.) и смесь перемешивали в течение 1 ч. Предварительно полученный раствор ди-трет-бутилдикарбоната (894 г, 4,096 моль, 6,11 экв.) в метаноле (600 мл) добавляли в реакционный раствор в течение 80 минут. Перенос завершали промывкой метанолом (60 мл). После добавления реакционную смесь перемешивали в течение 5 ч при 20°C, а затем из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-амино-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата и моно-Boc-замещенного промежуточного 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 3000 мл. Температуру стабилизировали на уровне от 20 до 30°C и реакцию гасили путем добавления 25% аммиака (1500 мл) в течение 1,5 ч (осторожно: экзотермическое добавление, скорость добавления контролировали для поддержания температуры реакции на уровне от 20 до 30°C). Добавляли дихлорметан (3000 мл) и двухфазную смесь перемешивали, после чего перемешивание прекращали и слои оставляли разделяться. Нижний органический слой с преимущественным содержанием продукта (OP1) переносили в приемник, тогда как верхний водный слой с малым содержанием продукта (AQ1) отбрасывали. OP1 возвращали в реактор, в который загружали воду (750 мл) и 25% аммиака (750 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP2) переносили в приемник, тогда как верхний водный слой (AQ2) отбрасывали. OP2 возвращали в реактор, в который загружали метанол (225 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP3) переносили в приемник, тогда как верхний водный слой (AQ3) отбрасывали. OP3 возвращали в реактор, в который загружали метанол (225 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP4) переносили в приемник, тогда как верхний водный слой (AQ4) отбрасывали. Конечную промытую органическую фазу (OP4) возвращали в реактор и концентрировали в вакууме до объема 2700 мл.
[00365] В реактор добавляли воду (150 мл), Boc-(S)-HABA (153 г, 0,698 моль, 1,04 экв.) и 1-гидроксибензотриазол (17 г, 0,126 моль, 0,19 экв.). После загрузки каждого из твердых веществ (Boc-(S)-HABA и 1-гидроксибензотриазола) дихлорметан (60 мл) применяли для промывки загрузочной системы. Значение pH реакционной смеси регулировали до 6,2 с применением 2 М раствора хлористоводородной кислоты (1098 мл). После регулирования pH в реакционную смесь загружали 1-этил-3-(3’-диметиламинопропил)карбодиимида гидрохлорид (133 г, 0,694, 1,03 экв.). Дихлорметан (60 мл) применяли для промывки после загрузки твердого вещества. Через 2 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ (потребление 4-нитробензил-(((2S,3R)-2-(((1R,2R,3S,4R,6S)-4-амино-6-((трет-бутоксикарбонил)амино)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-6-ил)мета). В реакционную смесь загружали метанол (525 мл), а затем дихлорметан (3600 мл) и воду (1500 мл). Значение pH регулировали до 8,5 с применением гидроксида натрия (365 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP5) переносили в приемник, тогда как верхний водный слой (AQ5) отбрасывали. OP5 возвращали в реактор, а затем добавляли воду (2100 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP6) переносили в приемник, тогда как верхний водный слой (AQ6) отбрасывали. OP6 возвращали в реактор и концентрировали в вакууме с получением конечного объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реакционную смесь загружали ацетонитрил (примерно 6000 мл) до конечного целевого объема 9000 мл.
[00366] Смесь нагревали до температуры обратной конденсации (83°C) и последовательно добавляли две порции воды (75 мл и 38 мл) с получением раствора. Раствор охлаждали до 75°C, после чего в реакционную смесь загружали затравку 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (3 г, 0,0029 моль, 0,004 экв.). Вновь образовавшуюся суспензию охлаждали до 3°C в течение 5 ч и перемешивали при 0-5°C в течение дополнительных 4 ч. Смесь фильтровали, промывали ацетонитрилом (две порции по 300 мл) и сушили в вакууме с получением 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (340,68 г, 49% молярный выход).
Пример 5c
[00367] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали сизомицин в виде свободного основания (300,0 г, 0,670 моль, 1 экв., KF=5,65), а затем метанол (1500 мл) и дихлорметан (1500 мл). После растворения температуру реакции стабилизировали на уровне 15°C. Раствор п-нитробензилбензотриазолкарбамата (189 г, 0,664 моль, 0,99 экв.) в дихлорметане (4200 мл) добавляли в реактор в течение трех часов, поддерживая температуру реакции на уровне примерно 15°C. Добавление завершали промывкой дихлорметаном (300 мл). Через 29 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление сизомицина). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 1500 мл. В реакционную смесь загружали метанол (4500 мл) и проводили вторую концентрацию в вакууме до целевого объема 4500 мл.
[00368] В реакционную смесь загружали метанол (900 мл) и температуру стабилизировали на уровне 35°C. Последовательно добавляли триэтиламин (385 мл, 2,777 моль, 4,14 экв.) и дигидрат ацетата цинка (549 г, 2,501 моль, 3,73 экв.) и смесь перемешивали в течение 1 ч. Предварительно полученный раствор ди-трет-бутилдикарбоната (620 г, 2,841 моль, 4,24 экв.) в метаноле (600 мл) добавляли в реакционный раствор в течение 60 минут. Перенос завершали промывкой метанолом (60 мл). После добавления реакционную смесь перемешивали в течение 5 ч при 35°C, а затем из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-амино-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата и моно-Boc-защищенного промежуточного 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 4200 мл. Температуру стабилизировали на уровне от 20 до 30°C и реакцию гасили путем добавления 25% аммиака (2100 мл) в течение 1 ч и 27 мин (осторожно: экзотермическое добавление, скорость добавления контролировали для поддержания температуры реакции на уровне от 20 до 30°C). Добавляли дихлорметан (3000 мл) и двухфазную смесь перемешивали в течение 20 минут, после чего перемешивание прекращали и слои оставляли разделяться. Нижний органический слой с преимущественным содержанием продукта (OP1) переносили в приемник, тогда как верхний водный слой с малым содержанием продукта (AQ1) отбрасывали. OP1 возвращали в реактор, в который загружали воду (750 мл) и 25% аммиака (750 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP2) переносили в приемник, тогда как верхний водный слой (AQ2) отбрасывали. OP2 возвращали в реактор, в который загружали метанол (375 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP3) переносили в приемник, тогда как верхний водный слой (AQ3) отбрасывали. OP3 возвращали в реактор, в который загружали метанол (375 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP4) переносили в приемник, тогда как верхний водный слой (AQ4) отбрасывали. Конечную промытую органическую фазу (OP4) возвращали в реактор и концентрировали в вакууме до объема 2700 мл.
[00369] В реактор добавляли воду (150 мл), Boc-(S)-HABA (153 г, 0,698 моль, 1,04 экв.) и 1-гидроксибензотриазол (17 г, 0,126 моль, 0,19 экв.). После загрузки каждого из твердых веществ (Boc-(S)-HABA и 1-гидроксибензотриазола) дихлорметан (60 мл) применяли для промывки загрузочной системы. Значение pH реакционной смеси регулировали до 4,8 с применением 2 М раствора хлористоводородной кислоты (1250 мл). После регулирования pH в реакционную смесь загружали 1-этил-3-(3’-диметиламинопропил)карбодиимида гидрохлорид (133 г, 0,694 моль, 1,03 экв.). Дихлорметан (60 мл) применяли для промывки после загрузки твердого вещества. Через 70 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ (потребление 4-нитробензил-(((2S,3R)-2-(((1R,2R,3S,4R,6S)-4-амино-6-((трет-бутоксикарбонил)амино)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-6-ил)мета). В реакционную смесь загружали метанол (675 мл), а затем дихлорметан (3600 мл) и воду (1500 мл). Значение pH регулировали до 10,5 с применением гидроксида натрия (1264 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP5) переносили в приемник, тогда как верхний водный слой (AQ5) отбрасывали. OP5 возвращали в реактор, а затем добавляли воду (2100 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP6) переносили в приемник, тогда как верхний водный слой (AQ6) отбрасывали. OP6 возвращали в реактор и концентрировали в вакууме с получением конечного объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реакционную смесь загружали ацетонитрил (примерно 3000 мл) до конечного целевого объема 6000 мл.
[00370] Смесь нагревали до температуры обратной конденсации (83°C) и последовательно добавляли две порции воды (75 мл и 38 мл) с получением раствора. Раствор охлаждали до 75°C, после чего в реакционную смесь загружали затравку 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (3 г, 0,0029 ммоль, 0,004 экв.). Вновь образовавшуюся суспензию охлаждали до 3°C в течение 5 ч и перемешивали при 0-5°C в течение дополнительных 4 ч. Смесь фильтровали, промывали ацетонитрилом (две порции по 300 мл) и сушили в вакууме с получением 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (347,03 г, 50% выход).
Пример 5d
[00371] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, вносили сизомицин в виде свободного основания (90,0 г, KF=5,65), а затем метанол (450 мл) и дихлорметан (450 мл). После растворения температуру реакции стабилизировали на уровне 15°C. Раствор п-нитробензилбензотриазолкарбамата (62 г) в дихлорметане (1260 мл) добавляли в реактор в течение трех часов, поддерживая температуру реакции на уровне примерно 15°C. Добавление завершали промывкой дихлорметаном (90 мл). Через 32 минуты из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление сизомицин). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 450 мл.
[00372] В реакционную смесь загружали метанол (1350 мл) и реакционную смесь дополнительно концентрировали в вакууме до конечного целевого объема 1350 мл. В реакционную смесь загружали вторую порцию метанола (270 мл) и температуру стабилизировали на уровне 27,5°C. Последовательно загружали триэтиламин (132 мл, 0,952 моль, 4,74 экв.) и дигидрат ацетата цинка (146 г, 0,665 моль, 3,31 экв.) и смесь перемешивали в течение 1 ч. Предварительно полученный раствор ди-трет-бутилдикарбоната (228 г, 1,045 моль, 5,19 экв.) в метаноле (180 мл) добавляли в реакционный раствор в течение 70 минут. Перенос завершали промывкой метанолом (18 мл). После добавления реакционную смесь перемешивали в течение 13 ч при 27,5°C, а затем из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-амино-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата и моно-Boc-защищенного промежуточного 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 1080 мл. Температуру стабилизировали на уровне от 20 до 30°C и реакцию гасили путем добавления 25% аммиака (540 мл) в течение 37 мин (осторожно: экзотермическое добавление, скорость добавления контролировали для поддержания температуры реакции на уровне от 20 до 30°C). Добавляли дихлорметан (900 мл) и двухфазную смесь перемешивали в течение 20 минут, после чего перемешивание прекращали и слои оставляли разделяться. Нижний органический слой с преимущественным содержанием продукта (OP1) переносили в приемник, тогда как верхний водный слой с малым содержанием продукта (AQ1) отбрасывали. OP1 возвращали в реактор, в который загружали воду (225 мл) и 25% аммиака (225 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP2) переносили в приемник, тогда как верхний водный слой (AQ2) отбрасывали. OP2 возвращали в реактор, в который загружали метанол (90 мл) и воду (450 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP3) переносили в приемник, тогда как верхний водный слой (AQ3) отбрасывали. OP3 возвращали в реактор, в который загружали метанол (90 мл) и воду (450 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP4) переносили в приемник, тогда как верхний водный слой (AQ4) отбрасывали. Конечную промытую органическую фазу (OP4) возвращали в реактор и концентрировали в вакууме до объема 810 мл.
[00373] В реактор добавляли воду (45 мл), Boc-(S)-HABA (46 г, 0,210 моль, 1,04 экв.) и 1-гидроксибензотриазол (5 г, 0,037 моль, 0,18 экв.). После загрузки каждого из твердых веществ (Boc-(S)-HABA и 1-гидроксибензотриазола) дихлорметан (18 мл) применяли для промывки загрузочной системы. Значение pH реакционной смеси регулировали до 5,5 с применением 2 М раствора хлористоводородной кислоты (380 мл). После регулирования pH в реакционную смесь загружали 1-этил-3-(3’-диметиламинопропил)карбодиимида гидрохлорид (40 г, 0,209 моль, 1,04 экв.). Дихлорметан (18 мл) применяли для промывки после загрузки твердого вещества. Через 2 ч и 40 мин из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ (потребление 4-нитробензил-(((2S,3R)-2-(((1R,2R,3S,4R,6S)-4-амино-6-((трет-бутоксикарбонил)амино)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-6-ил)мета). В реакционную смесь загружали метанол (180 мл), а затем дихлорметан (1080 мл) и воду (450 мл). Значение pH регулировали до 9,5 с применением гидроксида натрия (400 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP5) переносили в приемник, тогда как верхний водный слой (AQ5) отбрасывали. OP5 возвращали в реактор, а затем добавляли воду (630 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP6) переносили в приемник, тогда как верхний водный слой (AQ6) отбрасывали. OP6 возвращали в реактор и концентрировали в вакууме с получением конечного объема 3000 мл. В реактор загружали ацетонитрил (900 мл) и смесь концентрировали в вакууме до конечного целевого объема 900 мл. В реактор загружали ацетонитрил (900 мл) и смесь концентрировали в вакууме до конечного целевого объема 900 мл. В реакционную смесь загружали ацетонитрил (примерно 1350 мл) до конечного целевого объема 2250 мл.
[00374] Смесь нагревали до температуры обратной конденсации (примерно 80°C) и последовательно добавляли две порции воды (23 мл и 11 мл) с получением раствора. Раствор охлаждали до 75°C, после чего в реакционную смесь загружали затравку 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (1 г, 0,0010 моль, 0,005 экв.). Вновь образовавшуюся суспензию охлаждали до 3°C в течение 5 ч и перемешивали при 0-5°C в течение дополнительных 4 ч. Смесь фильтровали, промывали ацетонитрилом (две порции по 300 мл) и сушили в вакууме с получением 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (105,43 г, 51% молярный выход).
Пример 5e
[00375] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали сизомицин в виде свободного основания (300,0 г, 0,670 моль, 1 экв., KF=5,65), а затем метанол (1500 мл) и дихлорметан (1500 мл). После растворения температуру реакции стабилизировали на уровне 15°C. Раствор п-нитробензилбензотриазолкарбамат (226 г, 0,758 моль, 1,13 экв.) в дихлорметане (4200 мл) добавляли в реактор в течение трех часов, поддерживая температуру реакции на уровне примерно 15°C. Добавление завершали промывкой дихлорметаном (300 мл). Через 30 минут из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление сизомицина). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 1500 мл. В реакционную смесь загружали метанол (4500 мл) и проводили вторую концентрацию в вакууме до целевого объема 4500 мл.
[00376] В реакционную смесь загружали метанол (900 мл) и температуру стабилизировали на уровне 20°C. Последовательно загружали триэтиламин (498 мл, 3,593 моль, 5,36 экв.) и дигидрат ацетата цинка (425 г, 1,936 моль, 2,89 экв.) и смесь перемешивали в течение 1 ч. Предварительно полученный раствор ди-трет-бутилдикарбоната (894 г, 4,096 моль, 6,11 экв.) в метаноле (600 мл) добавляли в реакционный раствор в течение 60 минут. Перенос завершали промывкой метанолом (60 мл). После добавления реакционную смесь перемешивали в течение 9 ч при 20,0°C, а затем из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-амино-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата и моно-Boc-защищенного промежуточного 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-4,6-диамино-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата). Неочищенную реакционную смесь концентрировали в вакууме с получением конечного объема 3000 мл. Температуру стабилизировали на уровне от 20 до 30°C и реакцию гасили путем добавления 25% аммиака (1500 мл) в течение 109 мин (осторожно: экзотермическое добавление, скорость добавления контролировали для поддержания температуры реакции на уровне от 20 до 30°C). Добавляли дихлорметан (3000 мл) и двухфазную смесь перемешивали в течение 20 минут, после чего перемешивание прекращали и слои оставляли разделяться. Нижний органический слой с преимущественным содержанием продукта (OP1) переносили в приемник, тогда как верхний водный слой с малым содержанием продукта (AQ1) отбрасывали. OP1 возвращали в реактор, в который загружали воду (750 мл) и 25% аммиака (750 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP2) переносили в приемник, тогда как верхний водный слой (AQ2) отбрасывали. OP2 возвращали в реактор, в который загружали метанол (225 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP3) переносили в приемник, тогда как верхний водный слой (AQ3) отбрасывали. OP3 возвращали в реактор, в который загружали метанол (225 мл) и воду (1500 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP4) переносили в приемник, тогда как верхний водный слой (AQ4) отбрасывали. Конечную промытую органическую фазу (OP4) возвращали в реактор и концентрировали в вакууме до объема 2700 мл.
[00377] В реактор добавляли воду (150 мл), Boc-(S)-HABA (153 г, 0,698 моль, 1,04 экв.) и 1-гидроксибензотриазол (17 г, 0,126 моль, 0,19 экв.). После загрузки каждого из твердых веществ (Boc-(S)-HABA и 1-гидроксибензотриазола) дихлорметан (60 мл) применяли для промывки загрузочной системы. Значение pH реакционной смеси регулировали до 6,2 с применением 2 М раствора хлористоводородной кислоты (1122 мл). После регулирования pH в реакционную смесь загружали 1-этил-3-(3’-диметиламинопропил)карбодиимида гидрохлорид (133 г, 0,694 моль, 1,03 экв.). Дихлорметан (60 мл) применяли для промывки после загрузки твердого вещества. Через 4 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ (потребление 4-нитробензил-(((2S,3R)-2-(((1R,2R,3S,4R,6S)-4-амино-6-((трет-бутоксикарбонил)амино)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-6-ил)мета). В реакционную смесь загружали метанол (525 мл), а затем дихлорметан (3600 мл) и воду (1500 мл). Значение pH регулировали до 8,5 с применением гидроксида натрия (430 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP5) переносили в приемник, тогда как верхний водный слой (AQ5) отбрасывали. OP5 возвращали в реактор, а затем добавляли воду (2100 мл). Слои смешивали путем перемешивания, а затем оставляли разделяться, после чего нижний органический слой с преимущественным содержанием продукта (OP6) переносили в приемник, тогда как верхний водный слой (AQ6) отбрасывали. OP6 возвращали в реактор и концентрировали в вакууме с получением конечного объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реактор загружали ацетонитрил (3000 мл) и смесь концентрировали в вакууме до конечного целевого объема 3000 мл. В реакционную смесь загружали ацетонитрил (примерно 6000 мл) до конечного целевого объема 9000 мл.
[00378] Смесь нагревали до температуры обратной конденсации (83°C) и последовательно добавляли две порции воды (75 мл и 38 мл) с получением раствора. Раствор охлаждали до 80°C, после чего в реакционную смесь загружали затравку 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (3 г, 0,0029 моль, 0,004 экв.). Вновь образовавшуюся суспензию охлаждали до 3°C в течение 5 ч и перемешивали при 0-5°C в течение дополнительных 4 ч. Смесь фильтровали, промывали ацетонитрилом (две порции по 300 мл) и сушили в вакууме с получением 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a) (364,8 г, 53% молярный выход).
Пример 6: пример способа получения трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) из 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a).
Пример 6a
[00379] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамат формулы (4a) (320 г, 0,311 моль, 1 экв.). В реактор загружали метанол (3200 мл) и температуру стабилизировали на уровне от 45 до 55°C (47°C). Предварительно полученный раствор ди-трет-бутилдикарбоната (81,60 г, 0,374 моль, 1,20 экв.) в метаноле (80 мл) добавляли в реакционный раствор в течение 3 минут. Загрузку завершали промывкой метанолом (80 мл). Через 3 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a)).
[00380] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (2810 г) и гидроксид натрия (131 г, 3,275 моль, 10,52 экв.). Указанный щелочной раствор охлаждали до 10 °C. В раствор загружали литионит натрия (402 г) и температуру стабилизировали на уровне 12,5°C.
[00381] Реакционную смесь, содержащую промежуточный трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат (реактор A), переносили в смесь дитионита натрия (реактор B) в течение 1 ч, при этом поддерживая температуру реакционной смеси дитионита на уровне 12,5°C. Перенос завершали загрузкой метанола (160 мл). После завершения добавления реакционную смесь грели при 20°C в течение 3 ч. Через 14 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(((((4-аминобензил)окси)карбонил)амино)метил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата). Реакционную смесь концентрировали в вакууме при максимальной температуре кожуха 35°C с получением конечного объема 4160 мл. В реакционную смесь загружали изопропилацетат (1280 мл) и воду (1920 мл). Слои смешивали путем перемешивания при 35°C, а затем оставляли разделяться, после чего нижний водный слой (AP1) и верхний органический слой (OP1) переносили в приемники. AP1 возвращали в реактор и дополнительно экстрагировали другой порцией изопропилацетата (640 мл) с получением водного слоя 2 (AP2) и органического слоя (OP2). AP2 отбрасывали, тогда как OP2 объединяли с OP1 в реакторе. Указанный объединенный органический раствор с преимущественным содержанием продукта промывали двумя порциями 6,5% раствора бикарбоната натрия (2 порции по 960 мл). Промытые органические фазы концентрировали в вакууме до целевого объема 960 мл. В реактор загружали изопропилацетат (960 мл) и проводили вторую азеотропную перегонку до целевого объема 960 мл. В реактор загружали дополнительную порцию изопропилацетата (960 мл) и проводили третью азеотропную перегонку до целевого объема 1120 мл. Температуру стабилизировали на уровне 37,5°C и в смесь загружали воду (17 мл) с достижением значения по KF 1,5 масс.%/масс.%. В реактор загружали дихлорметан (1120 мл) и температуру стабилизировали на уровне 10,7°C. В смесь вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (6,4 г, 0,0067 моль, 0,02 экв.). Порцию дихлорметана (32 мл) применяли для смывания затравки. Смесь с затравкой перемешивали в течение 2 ч при 10°C и охлаждали до 0°C в течение 2 ч. Суспензию дополнительно перемешивали в течение 3,5 ч, после чего фильтровали и промывали дихлорметаном (320 мл). Выделенный материал сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (259,66 г, 88% молярный выход).
Пример 6b
[00382] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамат формулы (4a) (320 г, 0,311 моль, 1 экв.). В реактор загружали метанол (3200 мл) и температуру стабилизировали на уровне от 45 до 55°C (50°C). Предварительно полученный раствор ди-трет-бутилдикарбонат (81,60 г, 0,374 моль, 1,20 экв.) в метаноле (80 мл) добавляли в реакционный раствор в течение 5 минут. Загрузку завершали промывкой метанолом (80 мл). Через 5 ч в реакционный раствор добавляли дополнительную порцию предварительно полученного раствора ди-трет-бутилдикарбоната (1,92 г) в метаноле (1,9 мл). Через 1 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a)).
[00383] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (2490 г) и гидроксид натрия (125 г, 3,125 моль, 10,04 экв.). Указанный щелочной раствор охлаждали до 5°C. В раствор загружали дитионит натрия (383 г, 2,200 моль, 7,07 экв.) и температуру стабилизировали на уровне 7,5°C.
[00384] Реакционную смесь, содержащую промежуточный трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат (реактор A), переносили в смесь дитионита натрия (реактор B) в течение 1 ч, при этом поддерживая температуру реакционной смеси дитионита на уровне 7,5°C. Перенос завершали загрузкой метанола (160 мл). После завершения добавления реакционную смесь грели при 27,5°C в течение 2 ч. Через 7 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(((((4-аминобензил)окси)карбонил)амино)метил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата). Реакционную смесь концентрировали в вакууме при максимальной температуре кожуха 35°C с получением конечного объема 4160 мл. В реакционную смесь загружали изопропилацетат (1280 мл) и воду (1920 мл). Слои смешивали путем перемешивания при 35°C, а затем оставляли разделяться, после чего нижний водный слой (AP1) и верхний органический слой (OP1) переносили в приемники. AP1 возвращали в реактор и дополнительно экстрагировали другой порцией изопропилацетата (640 мл) с получением водного слоя 2 (AP2) и органического слоя (OP2). AP2 отбрасывали, тогда как OP2 объединяли с OP1 в реакторе. Указанный объединенный органический раствор с преимущественным содержанием продукта промывали двумя порциями 6,5% раствора бикарбоната натрия (2 порции по 960 мл). Промытые органические фазы концентрировали в вакууме до целевого объема 960 мл. В реактор загружали изопропилацетат (960 мл) и проводили вторую азеотропную перегонку до целевого объема 960 мл. В реактор загружали дополнительную порцию изопропилацетата (960 мл) и проводили третью азеотропную перегонку до целевого объема 1440 мл. Температуру стабилизировали на уровне 37,5°C и в смесь загружали воду (42 мл) с достижением значения по KF 2,9 масс.%/масс.%. В реактор загружали дихлорметан (1440 мл) и температуру стабилизировали на уровне 22,5°C. В смесь вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (6,4 г, 0,0067 моль, 0,02 экв.). Порцию дихлорметана (32 мл) применяли для смывания затравки. Смесь с затравкой перемешивали в течение 2 ч при 22,5°C и охлаждали до 0°C в течение 2 ч. Суспензию дополнительно перемешивали в течение 3,5 ч, после чего фильтровали и промывали дихлорметаном (320 мл). Выделенный материал сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (257,77 г, 87% выход).
Пример 6c
[00385] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамат формулы (4a) (320 г, 0,311 моль, 1 экв.). В реактор загружали метанол (3200 мл) и температуру стабилизировали на уровне от 45 до 55°C (50°C). Предварительно полученный раствор ди-трет-бутилдикарбоната (81,60 г, 0,394 моль, 1,20 экв.) в метаноле (80 мл) добавляли в реакционный раствор в течение 3 минут. Загрузку завершали промывкой метанолом (80 мл). Через 3 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a)).
[00386] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (2170 г) и гидроксид натрия (118 г, 2,950 моль, 9,48 экв.). Указанный щелочной раствор охлаждали до 0°C. В раствор загружали дитионит натрия (364 г, 2,091 моль, 6,72 экв.) и температуру стабилизировали на уровне 2,5°C.
[00387] Реакционную смесь, содержащую промежуточный трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат (реактор A), переносили в смесь дитионита натрия (реактор B) в течение 5 ч, при этом поддерживая температуру реакционной смеси дитионита на уровне 2,5°C. Перенос завершали загрузкой метанола (160 мл). После завершения добавления реакционную смесь грели при 35°C в течение 1 ч. Через 3 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(((((4-аминобензил)окси)карбонил)амино)метил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата). Реакционную смесь концентрировали в вакууме при максимальной температуре кожуха 35°C с получением конечного объема 4160 мл. В реакционную смесь загружали изопропилацетат (1280 мл) и воду (1920 мл). Слои смешивали путем перемешивания при 35°C, а затем оставляли разделяться, после чего нижний водный слой (AP1) и верхний органический слой (OP1) переносили в приемники. AP1 возвращали в реактор и дополнительно экстрагировали другой порцией изопропилацетата (640 мл) с получением водного слоя 2 (AP2) и органического слоя (OP2). AP2 отбрасывали, тогда как OP2 объединяли с OP1 в реакторе. Указанный объединенный органический раствор с преимущественным содержанием продукта промывали двумя порциями 6,5% раствора бикарбоната натрия (2 порции по 960 мл). Промытые органические фазы концентрировали в вакууме до целевого объема 960 мл. В реактор загружали изопропилацетат (960 мл) и проводили вторую азеотропную перегонку до целевого объема 960 мл. В реактор загружали дополнительную порцию изопропилацетата (960 мл) и проводили третью азеотропную перегонку до целевого объема 1760 мл. Температуру стабилизировали на уровне 37,5°C и в смесь загружали воду (71,2 мл) с достижением значения по KF 3,9 масс.%/масс.%. В реактор загружали дихлорметан (1760 мл) и температуру стабилизировали на уровне 35°C. В смесь вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (6,4 г, 0,0067 моль, 0,02 экв.). Порцию дихлорметана (32 мл) применяли для смывания затравки. Смесь с затравкой перемешивали в течение 2 ч при 35°C и охлаждали до 0°C в течение 2 ч. Суспензию дополнительно перемешивали в течение 3,5 ч, после чего фильтровали и промывали дихлорметаном (320 мл). Выделенный материал сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (103,5 г, 0,109 моль, 35% молярный выход).
Пример 6d
[00388] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамат формулы (4a) (320 г, 0,311 моль, 1 экв.). В реактор загружали метанол (3200 мл) и температуру стабилизировали на уровне от 45 до 55°C (47°C). Предварительно полученный раствор ди-трет-бутилдикарбоната (81,60 г, 0,374 моль, 1,20 экв.) в метаноле (80 мл) добавляли в реакционный раствор в течение 3 минут. Загрузку завершали промывкой метанолом (80 мл). Через 6 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a)).
[00389] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (2810 г) и гидроксид натрия (131,2 г, 3,280 моль, 10,54 экв.). Указанный щелочной раствор охлаждали до 10°C. В раствор загружали дитионит натрия (401,9 г, 2,308 моль, 7,42 экв.) и температуру стабилизировали на уровне 12,5°C.
[00390] Реакционную смесь, содержащую промежуточный трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат (реактор A), переносили в смесь дитионита натрия (реактор B) в течение 1 ч, при этом поддерживая температуру реакционной смеси дитионита на уровне 12,5°C. Перенос завершали загрузкой метанола (160 мл). После завершения добавления реакционную смесь грели при 20°C в течение 3 ч. Через 18 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(((((4-аминобензил)окси)карбонил)амино)метил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата). Реакционную смесь концентрировали в вакууме при максимальной температуре кожуха 35°C с получением конечного объема 4160 мл. В реакционную смесь загружали изопропилацетат (1280 мл) и воду (1920 мл). Слои смешивали путем перемешивания при 35°C, а затем оставляли разделяться, после чего нижний водный слой (AP1) и верхний органический слой (OP1) переносили в приемники. AP1 возвращали в реактор и дополнительно экстрагировали другой порцией изопропилацетата (640 мл) с получением водного слоя 2 (AP2) и органического слоя (OP2). AP2 отбрасывали, тогда как OP2 объединяли с OP1 в реакторе. Указанный объединенный органический раствор с преимущественным содержанием продукта промывали двумя порциями 6,5% раствора бикарбоната натрия (2 порции по 960 мл). Промытые органические фазы концентрировали в вакууме до целевого объема 960 мл. В реактор загружали изопропилацетат (960 мл) и проводили вторую азеотропную перегонку до целевого объема 960 мл. В реактор загружали дополнительную порцию изопропилацетата (960 мл) и проводили третью азеотропную перегонку до целевого объема 1280 мл. Температуру стабилизировали на уровне 37,5°C и в смесь загружали воду (26 мл) с достижением значения по KF 2,1 масс.%/масс.%. В реактор загружали дихлорметан (1280 мл) и температуру стабилизировали на уровне 15,1°C. В смесь вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (6,4 г, 0,0067 моль, 0,02 экв.). Порцию дихлорметана (32 мл) применяли для смывания затравки. Смесь с затравкой перемешивали в течение 2 ч при 15°C и охлаждали до 0°C в течение 2 ч. Суспензию дополнительно перемешивали в течение 3,5 ч, после чего фильтровали и промывали дихлорметаном (320 мл). Выделенный материал сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (257,5 г, 0,271 моль, 87% молярный выход).
Пример 6e
[00391] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамат формулы (4a) (320 г, 0,311 моль, экв.). В реактор загружали метанол (3200 мл) и температуру стабилизировали на уровне от 45 до 55°C (47°C). Предварительно полученный раствор ди-трет-бутилдикарбоната (81,60 г, 0,374 моль, 1,20 экв.) в метаноле (80 мл) добавляли в реакционный раствор в течение 3 минут. Загрузку завершали промывкой метанолом (80 мл). Через 8 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление 4-нитробензил-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-2-(((1R,2S,3S,4R,6S)-6-((трет-бутоксикарбонил)амино)-4-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2R,3R,4R,5R)-3,5-дигидрокси-5-метил-4-(метиламино)тетрагидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,4-дигидро-2H-пиран-6-ил)метил)карбамата формулы (4a)).
[00392] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (2170 г) и гидроксид натрия (118,4 г, 2,960 моль, 9,51 экв.). Указанный щелочной раствор охлаждали до 0°C. В раствор загружали дитионит натрия (363,5 г, 2,088 моль, 6,71 экв.) и температуру стабилизировали на уровне 2,5°C.
[00393] Реакционную смесь, содержащую промежуточный трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((((4-нитробензил)окси)карбонил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат (реактор A), переносили в смесь дитионита натрия (реактор B) в течение 5 ч, при этом поддерживая температуру реакционной смеси дитионита на уровне 2,5°C. Перенос завершали загрузкой метанола (160 мл). После завершения добавления реакционную смесь грели при 35°C в течение 1 ч. Через 4 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(((((4-аминобензил)окси)карбонил)амино)метил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата). Реакционную смесь концентрировали в вакууме при максимальной температуре кожуха 35°C с получением конечного объема 4160 мл. В реакционную смесь загружали изопропилацетат (1280 мл) и воду (1920 мл). Слои смешивали путем перемешивания при 35°C, а затем оставляли разделяться, после чего нижний водный слой (AP1) и верхний органический слой (OP1) переносили в приемники. AP1 возвращали в реактор и дополнительно экстрагировали другой порцией изопропилацетата (640 мл) с получением водного слоя 2 (AP2) и органического слоя (OP2). AP2 отбрасывали, тогда как OP2 объединяли с OP1 в реакторе. Указанный объединенный органический раствор с преимущественным содержанием продукта промывали двумя порциями 6,5% раствора бикарбоната натрия (2 порции по 960 мл). Промытые органические фазы концентрировали в вакууме до целевого объема 960 мл. В реактор загружали изопропилацетат (960 мл) и проводили вторую азеотропную перегонку до целевого объема 960 мл. В реактор загружали дополнительную порцию изопропилацетата (960 мл) и проводили третью азеотропную перегонку до целевого объема 1600 мл. Температуру стабилизировали на уровне 37,5°C и в смесь загружали воду (29 мл) с достижением значения по KF 3,5 масс.%/масс.%. В реактор загружали дихлорметан (1600 мл) и температуру стабилизировали на уровне 30°C. В смесь вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (6,4 г, 0,0067 моль, 0,02 экв.). Порцию дихлорметана (32 мл) применяли для смывания затравки. Смесь с затравкой перемешивали в течение 2 ч при 30°C и охлаждали до 0°C в течение 2 ч. Суспензию дополнительно перемешивали в течение 3,5 ч, после чего фильтровали и промывали дихлорметаном (320 мл). Выделенный материал сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a) (259,50 г, 0,273 моль, 88% молярный выход).
Пример 7: пример способа получения трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) из трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a).
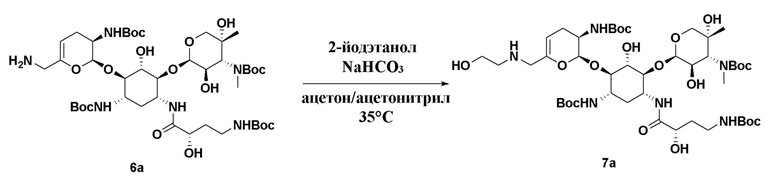
Пример 7a
[00394] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (6a) (250 г, 0,263 моль, 1 экв.). В реактор ацетонитрил загружали (1250 мл) и температуру стабилизировали на уровне от 15 до 30°C (27,4°C). Смесь концентрировали в вакууме до конечного целевого объема 500 мл. Из раствора отбирали пробу для определения содержания воды по методу Карла Фишера, который демонстрировал результат 0,27 масс.%/масс.%. В реактор загружали дополнительную порцию ацетонитрила (750 мл) и проводили вторую азеотропную перегонку до целевого объема 500 мл. Из смеси отбирали пробу для метода Карла Фишера и получали результат 0,08 масс.%/масс.%. Температуру реакции стабилизировали на уровне 29,9°C и в смесь загружали ацетон (1250 мл). Реакционную смесь грели и температуру стабилизировали на уровне 40°C. В реакционную смесь загружали бикарбонат натрия (44,25 г, 0,527 моль, 2 экв.), а затем 2-йодэтанол (65,2 г, 29,6 мл, 0,379 моль, 1,44 экв.). Через 23 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a)). Реакционную смесь охлаждали до 22,7°C и загружали 1,4-диазабицикло[2.2.2]октан (60,0 г, 0,535 моль, 2,03 экв.) в виде твердого вещества. Разрушение 2-йодэтанола отслеживали при помощи способа ГХ и через 18 ч гашение указанного реагента считали завершенным. В реакционную смесь загружали воду (1250 мл) и изопропилацетат (1250 мл). Содержимое реактора перемешивали в течение 25 мин и слои оставляли разделяться. Нижний водный слой (AP1) и верхний органический слой (OP1) объединяли в приемниках. AP1 возвращали в реактор и загружали вторую порцию изопропилацетата (750 мл). Содержимое реактора перемешивали в течение 30 минут и слои оставляли разделяться. Нижний водный слой (AP2) и верхний органический слой (OP2) объединяли в приемниках. OP1 и OP2 объединяли в реакторе и экстрагировали двумя порциями насыщенного раствора хлорида натрия (750 мл, полученного путем растворения 100 г NaCl в 290 мл воды). Промытую органическую фазу (OP4) концентрировали в вакууме до целевого объема 500 мл. В реактор загружали ацетонитрил (2550 мл). Проводили вторую азеотропную вакуумную перегонку до целевого объема 2050 мл. В смесь загружали изопропилацетат (200 мл). В смесь загружали воду (35,4 мл) до получения значения по KF 2,0%. Содержимое реактора нагревали до 75°C, после чего получали раствор. Реакционную смесь охлаждали до 65°C и вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (5 г, 0,0050 моль, 0,02 экв.). Перемешивание продолжали при 65°C в течение 8 ч, в течение которых образовывалась густая суспензия. Смесь охлаждали от 65°C до 2,5°C в течение 12 ч. Суспензию фильтровали, промывали ацетонитрилом (900 мл) и сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (203,8 г, 0,205 моль, 78% выход).
Пример 7b
[00395] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (6a) (250 г, 0,263 моль, 1 экв.). В реактор загружали ацетонитрил (1250 мл) и температуру стабилизировали на уровне от 15 до 30°C (30°C). Смесь концентрировали в вакууме до конечного целевого объема 500 мл. Из раствора отбирали пробу для определения содержания воды по методу Карла Фишера, который демонстрировал результат 0,18 масс.%/масс.%. В реактор загружали дополнительную порцию ацетонитрила (750 мл) и проводили вторую азеотропную перегонку до целевого объема 500 мл. Из смеси отбирали пробу для метода Карла Фишера и получали результат 0,075 масс.%/масс.%. Температуру реакции стабилизировали на уровне 29,9°C и в смесь загружали ацетон (1250 мл). Реакционную смесь грели и температуру стабилизировали на уровне 40°C. В реакционную смесь загружали бикарбонат натрия (44,25 г, 0,527 моль, 2,0 экв.), а затем 2-йодэтанол (56,6 г, 25,7 мл, 0,329 моль, 1,25 экв.). Через 56 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a)). Реакционную смесь охлаждали до 22,7°C и загружали 1,4-диазабицикло[2.2.2]октан (60,0 г, 0,535 моль, 2,0 экв.) в виде твердого вещества. Разрушение 2-йодэтанола отслеживали при помощи способа ГХ и через 18 ч гашение указанного реагента считали завершенным. В реакционную смесь загружали воду (1250 мл) и изопропилацетат (1250 мл). Содержимое реактора перемешивали в течение 25 мин и слои оставляли разделяться. Нижний водный слой (AP1) и верхний органический слой (OP1) объединяли в приемниках. AP1 возвращали в реактор и загружали вторую порцию изопропилацетата (750 мл). Содержимое реактора перемешивали в течение 30 минут и слои оставляли разделяться. Нижний водный слой (AP2) и верхний органический слой (OP2) объединяли в приемниках. OP1 и OP2 объединяли в реакторе и экстрагировали двумя порциями насыщенного раствора хлорида натрия (750 мл, полученного путем растворения 100 г NaCl в 290 мл воды). Промытую органическую фазу (OP4) концентрировали в вакууме до целевого объема 500 мл. В реактор загружали ацетонитрил (2550 мл). Проводили вторую азеотропную вакуумную перегонку до целевого объема 1800 мл. В смесь загружали изопропилацетат (200 мл). В смесь загружали воду (22,8 мл) до получения значения по KF 1,46%. Содержимое реактора нагревали до 75°C, после чего получали раствор. Реакционную смесь охлаждали до 62,5°C и вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (5 г, 0,0050 моль, 0,02 экв.). Перемешивание продолжали при 62,5°C в течение 5 ч, в течение которых образовывалась густая суспензия. Смесь охлаждали от 65°C до 2,5°C в течение 12 ч. Суспензию фильтровали, промывали ацетонитрилом (900 мл) и сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (208,1 г, 0,220 моль, 83% молярный выход).
Пример 7c
[00396] В стеклянный реактор с кожухом, снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (6a) (250 г, 0,263 моль, 1 экв.). В реактор загружали ацетонитрил (1250 мл) и температуру стабилизировали на уровне от 15 до 30°C (24,3°C). Смесь концентрировали в вакууме до конечного целевого объема 500 мл. Из раствора отбирали пробу для определения содержания воды по методу Карла Фишера, который демонстрировали результат 0,22 масс.%/масс.%. В реактор загружали дополнительную порцию ацетонитрила (750 мл) и проводили вторую азеотропную перегонку до целевого объема 500 мл. Из смеси отбирали пробу для метода Карла Фишера и получали результат 0,097 масс.%/масс.%. Температуру реакции стабилизировали на уровне 29,9°C и в смесь загружали ацетон (1250 мл). Реакционную смесь грели и температуру стабилизировали на уровне 29,9°C. В реакционную смесь загружали бикарбонат натрия (44,25 г, 0,527, 2,0 экв.), а затем 2-йодэтанол (44,4 г, 20,14 мл, 0,258 моль, 0,98 экв.). Через 43 ч к реакционной смеси добавляли дополнительную порцию 2-йодэтанола (0,25 мл). Через 9,5 часов к реакционной смеси добавляли третью порцию 2-йодэтанола (0,3 мл). Через дополнительные 2 ч из реакционной смеси отбирали пробу и определяли завершение реакции путем ВЭЖХ анализа (потребление трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-3-(((2S,3R)-6-(аминометил)-3-((трет-бутоксикарбонил)амино)-3,4-дигидро-2H-пиран-2-ил)окси)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (6a)). Реакционную смесь охлаждали до 22,7°C и загружали 1,4-диазабицикло[2.2.2]октан (60,0 г, 0,535 моль, 2,03 экв.) в виде твердого вещества. Разрушение 2-йодэтанола отслеживали при помощи способа ГХ и через 10 ч гашение указанного реагента считали завершенным. В реакционную смесь загружали воду (1250 мл) и изопропилацетат (1250 мл). Содержимое реактора перемешивали в течение 25 мин и слои оставляли разделяться. Нижний водный слой (AP1) и верхний органический слой (OP1) объединяли в приемниках. AP1 возвращали в реактор и загружали вторую порцию изопропилацетата (750 мл). Содержимое реактора перемешивали в течение 30 минут и слои оставляли разделяться. Нижний водный слой (AP2) и верхний органический слой (OP2) объединяли в приемниках. OP1 и OP2 объединяли в реакторе и экстрагировали двумя порциями насыщенного раствора хлорида натрия (750 мл, полученного путем растворения 100 г NaCl в 290 мл воды). Промытую органическую фазу (OP4) концентрировали в вакууме до целевого объема 500 мл. В реактор загружали ацетонитрил (2550 мл). Проводили вторую азеотропную вакуумную перегонку до целевого объема 1550 мл. В смесь загружали изопропилацетат (200 мл). В смесь загружали воду (10,7 мл) до получения значения по KF 0,85%. Содержимое реактора нагревали до 75°C, после чего получали раствор. Реакционную смесь охлаждали до 57°C и вносили затравку трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (5 г, 0,0050 моль, 0,02 экв.). Перемешивание продолжали при 57°C в течение 2 ч, в течение которых образовывалась густая суспензия. Смесь охлаждали от 65°C до 2,5°C в течение 12 ч. Суспензию фильтровали, промывали ацетонитрилом (900 мл) и сушили в вакуумном сушильном шкафу с получением трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) (218,1 г, 0,220 моль, 83% молярный выход).
Пример 8: пример способа получения плазомицина из трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамата формулы (7a) и регенерации слоя смолы CG-50.

Пример 8a
[00397] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (7a) (150 г, 0,151 моль, 1 экв.) и дихлорметан (750 мл). Смесь перемешивали при температуре от 0 до 5°C (0,6°C). К реакционной смеси медленно добавляли трифторуксусную кислоту, поддерживая температуру на уровне от 0 до 5°C [отмечали экзотермическое добавление]. После добавления реакционный раствор нагревали до 22,5°C и выдерживали при указанной температуре в течение 2 ч. Реакционную смесь концентрировали путем перегонки в вакууме до конечного целевого объема 450 мл. Концентрированную реакционную смесь выдерживали при 22,5°C в течение дополнительных 2 ч, после чего температуру стабилизировали на уровне от 0 до 10°C (3,0°C).
[00398] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (375 мл), которую охлаждали до температуры от 0 до 5°C (2,5°C). Содержимое реактора A переносили в содержимое реактора B в течение 45 минут, поддерживая температуру на уровне от 0 до 10°C [отмечали экзотермическое добавление]. В реакционную смесь загружали изопропилацетат с получением двухфазной смеси. Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP1) и верхний органический слой (OP1) разделяли. OP1 возвращали в реактор и в смесь загружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP2) и верхний органический слой (OP2) собирали. OP2 возвращали в реактор и загружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP3) и верхний органический слой (OP3) собирали. Три водных слоя с преимущественным содержанием продукта (AP1, AP2 и AP3) возвращали в реактор и объединяли. Измеренное значение pH объединенного органического слоя составляло 0,15. В объединенные водные фазы загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP4) и верхний органический слой (OP4) собирали. Измеренное значение pH AP4 составляло 0,73. AP4 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP5) и верхний органический слой (OP5) собирали. Измеренное значение pH AP5 составляло 1,78. AP5 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP6) и верхний органический слой (OP6) собирали. Измеренное значение pH AP6 составляло 2,95. AP6 возвращали в реактор и значение pH доводили до уровня от 5,8 до 6,2 (конечное значение pH 6,01) с применением 1% раствора аммиака (34,7 мл). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 10°C. В реакционную смесь загружали воду (примерно 820 мл) до получения конечного объема 1350 мл. Снова измеряли значение pH (5,67) и доводили до уровня от 5,8 до 6,2 (6,01) с применением 1% раствора аммиака (11,7 мл). Измеренное значение электропроводности раствора составляло 15,36 мСм/см.
[00399] Указанный раствор неочищенного плазомицина загружали в колонку XK-50/100, содержащую смолу CG-50 (325,5 г) в амонийной форме, с пространством над жидкостью, заполненным водой. Температуру колонки стабилизировали на уровне 13°C. Неочищенный раствор загружали в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (примерно 9,2 мл/мин). Затем неочищенный раствор загружали на слой смолы. Загружали воду (1 объем колонки, 1620 мл) в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (9,2 мл/мин). После загрузки воды в колонку загружали 0,43% раствор аммиака (приготовленный из 86,7 г 25% аммиака в 5 л воды) в режиме нисходящего потока до элюирования плазомицина из колонки, что определяли по следам УФ-поглощения при 210 нм. После определения завершения элюирования плазомицина колонку дополнительно промывали 5% раствором аммиака в режиме нисходящего потока. Фракции с преимущественным содержанием продукта (#36-58) объединяли с получением 6520 мл очищенного раствора плазомицина в виде свободного основания. Содержание аммиака в растворе, измеряемое путем ионной хроматографии, составляло 2720 мкг/мл, и концентрация плазомицина в виде свободного основания, измеряемая путем СВЭЖХ, составляла 1,17 масс.%/об.%. Раствор подвергали обратному осмосу с применением мембраны XLE в режиме диафильтрования до получения отношения аммиака (мкг/мл) к плазомицину (масс.%/об.%) менее 20. Для достижения указанного критерия использовали 6 диаобъемов воды. В процессе удаления аммиака при помощи процесса диафильтрования температуру поддерживали на уровне ниже 10°C. После процесса удаления аммиака значение pH очищенного раствора плазомицина доводили до 6,0 с применением 6 М раствора серной кислоты (57,5). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 5°C. Нейтрализованный раствор пропускали через предварительно промытый угольный картридж Zetacarbon R55 при скорости потока 72 л/ч*м2 (18 мл/мин). После фильтрования картридж промывали водой (1068 мл) и смывочный раствор объединяли с отфильтрованным раствором плазомицина. Объединенные фильтрат и смывочный раствор концентрировали до 540 мл путем обратного осмоса с применением мембраны XLE в режиме нанофильтрования. Концентрированный раствор пропускали через 0,22-микронный фильтр и выделяли путем сушки распылением с применением лабораторной распылительной сушилки Buchi (89,05 г, 0,106 моль, 70% молярный выход).
Пример 8b - Регенерация смолы
[00400] Колонку XK-50/100, содержащую смолу CG-50 (326 г), регенерировали при помощи следующих стадий. Загружали 4% раствор гидроксида натрия (0,49 кг NaOH в 11,8 л воды) в режиме восходящего потока до достижения электропроводности эффлюента 110 мСм/см. Затем загружали воду в режиме восходящего потока до достижения электропроводности эффлюента 1,5 мСм/см. Загружали 2% раствор серной кислоты (приготовленный из 0,10 кг концентрированной серной кислоты в 4,9 л воды) в режиме восходящего потока до достижения электропроводности эффлюента мСм/см. Загружали воду (4782 мл) в режиме восходящего потока до достижения конечной электропроводности 4,8 мСм/см. Загружали 2% раствор аммиака (приготовленный из 400 г 25% аммиака в 9,6 л воды) в режиме восходящего потока. Конечная электропроводность эффлюента составляла 970 мкСм/см (pH 12,04). Смолу оставляли отстояться без потока в течение 1 ч. Загружали воду (3419 мл) в режиме нисходящего потока (13,1 мл/мин).
[00401] Упаковку слоя смолы испытывали путем импульсного введения трифторацетата аммония. Раствор трифторацетата аммиака получали путем растворения трифторацетата аммиака (1,397 г) в воде (106 мл). Указанный 0,1 М раствор трифторацетата аммиака (27,5 мл) загружали непосредственно над поверхностью слоя смолы CG-50 в водокольцевом режиме. Загружали воду в режиме нисходящего потока до исчезновения УФ-поглощения, соответствующего импульсному введению трифторацетата аммония, при 210 нм. Слой резины считали упакованным и подходящим для применения для очистки неочищенного плазомицина.
Пример 8c
[00402] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (7a) (150 г, 0,151 моль, 1 экв.) и дихлорметан (750 мл). Смесь перемешивали при температуре от 0 до 5°C (0,0°C). К реакционной смеси медленно добавляли трифторуксусную кислоту, поддерживая температуру на уровне от 0 до 5°C [отмечали экзотермическое добавление]. После добавления реакционный раствор нагревали до 22,5°C и выдерживали при указанной температуре в течение 1,5 ч. Реакционную смесь концентрировали путем перегонки в вакууме до конечного целевого объема 450 мл. Концентрированную реакционную смесь выдерживали при 22,5°C в течение дополнительных 8 ч, после чего температуру стабилизировали на уровне от 0 до 10°C (5,0°C).
[00403] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (375 мл), которую охлаждали до температуры от 0 до 5°C (0,3°C). Содержимое реактора A переносили в содержимое реактора B в течение 19 минут, поддерживая температуру на уровне от 0 до 10°C [отмечали экзотермическое добавление]. В реакционную смесь загружали изопропилацетат с получением двухфазной смеси. Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP1) и верхний органический слой (OP1) разделяли. OP1 возвращали в реактор и в смесь загружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP2) и верхний органический слой (OP2) собирали. OP2 возвращали в реактор и заружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP3) и верхний органический слой (OP3) собирали. Три водных слоя с преимущественным содержанием продукта (AP1, AP2 и AP3) возвращали в реактор и объединяли. Измеренное значение pH объединенного органического слоя составляло 0,49. В объединенные водные фазы загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP4) и верхний органический слой (OP4) собирали. Измеренное значение pH AP4 составляло 0,85. AP4 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP5) и верхний органический слой (OP5) собирали. Измеренное значение pH AP5 составляло 1,72. AP5 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP6) и верхний органический слой (OP6) собирали. Измеренное значение pH AP6 составляло 2,86. AP6 возвращали в реактор и значение pH доводили до уровня от 5,8 до 6,2 (конечное значение pH 5,99) с применением 1% раствора аммиака (21 мл). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 10°C. В реакционную смесь загружали воду (примерно 820 мл) до получения конечного объема 1350 мл. Снова измеряли значение pH (5,54) и доводили до уровня от 5,8 до 6,2 (6,01) с применением 1% раствора аммиака (10 мл). Измеренное значение электропроводности раствора составляло 14,92 мСм/см.
[00404] Указанный раствор неочищенного плазомицина загружали в колонку XK-50/100, содержащую смолу CG-50 (325,5 г) в амонийной форме, с пространством над жидкостью, заполненным водой. Температуру колонки стабилизировали на уровне 20°C. Неочищенный раствор загружали в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (примерно 9,2 мл/мин). Затем неочищенный раствор загружали на слой смолы. Загружали воду (1 объем колонки, 1620 мл) в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (9,2 мл/мин). После загрузки воды в колонку загружали 0,50% раствор аммиака (приготовленный из 102 г 25% аммиака в 5 л воды) в режиме нисходящего потока до элюирования плазомицина из колонки, что определяли по следам УФ-поглощения при 210 нм. Загружали раствор аммиака при линейной скорости потока 38 см/ч (12,4 мл/мин). Фракции с преимущественным содержанием продукта (#34-50) объединяли с получением 4800 мл очищенного раствора плазомицина в виде свободного основания. Содержание аммиака в растворе, измеряемое путем ионной хроматографии, составляло 3200 мкг/мл, и концентрация плазомицина в виде свободного основания, измеряемая путем СВЭЖХ, составляла 1,52 масс.%/об.%. Раствор подвергали обратному осмосу с применением мембраны XLE в режиме диафильтрования до получения отношения аммиака (мкг/мл) к плазомицину (масс.%/масс.%) менее 20. Для достижения указанного критерия использовали 9 диаобъемов воды. В процессе удаления аммиака при помощи процесса диафильтрования температуру поддерживали на уровне ниже 10°C. После процесса удаления аммиака значение pH очищенного раствора плазомицина доводили до 6,47 с применением 6 М раствора серной кислоты (52,5). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 5°C. Нейтрализованный раствор пропускали через предварительно промытый угольный картридж Zetacarbon R55 при скорости потока 72 л/ч*м2 (15 мл/мин). После фильтрования картридж промывали водой (1068 мл) и смывочный раствор объединяли с отфильтрованным раствором плазомицина. Объединенные фильтрат и смывочный раствор концентрировали до 540 мл путем обратного осмоса с применением мембраны XLE в режиме нанофильтрования. Концентрированный раствор пропускали через 0,22-микронный фильтр и выделяли путем сушки распылением с применением лабораторной распылительной сушилки Buchi (88,42 г, 0,106 моль, 70% молярный выход).
Пример 8d
[00405] В стеклянный реактор с кожухом (реактор A), снабженный мешалкой с верхним приводом, загружали трет-бутил-((2R,3R,4R,5R)-2-(((1S,2S,3R,4S,6R)-4-((трет-бутоксикарбонил)амино)-6-((S)-4-((трет-бутоксикарбонил)амино)-2-гидроксибутанамидо)-3-(((2S,3R)-3-((трет-бутоксикарбонил)амино)-6-(((2-гидроксиэтил)амино)метил)-3,4-дигидро-2H-пиран-2-ил)окси)-2-гидроксициклогексил)окси)-3,5-дигидрокси-5-метилтетрагидро-2H-пиран-4-ил)(метил)карбамат формулы (7a) (150 г, 0,151 моль, 1 экв.) и дихлорметан (750 мл). Смесь перемешивали при температуре от 0 до 5°C (0,9°C). К реакционной смеси медленно добавляли трифторуксусную кислоту, поддерживая температуру на уровне от 0 до 5°C [отмечали экзотермическое добавление]. После добавления реакционный раствор нагревали до 22,5°C и выдерживали при указанной температуре в течение 1,5 ч. Реакционную смесь концентрировали путем перегонки в вакууме до конечного целевого объема 450 мл. Концентрированную реакционную смесь выдерживали при 22,5°C в течение дополнительных 6 ч, после чего температуру стабилизировали на уровне от 0 до 10°C (6,5°C).
[00406] Во второй стеклянный реактор с кожухом (реактор B), снабженный мешалкой с верхним приводом, загружали воду (375 мл), которую охлаждали до температуры от 0 до 5°C (3,5°C). Содержимое реактора A переносили в содержимое реактора B в течение 7 минут, поддерживая температуру на уровне от 0 до 10°C [отмечали экзотермическое добавление]. В реакционную смесь загружали изопропилацетат с получением двухфазной смеси. Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP1) и верхний органический слой (OP1) разделяли. OP1 возвращали в реактор и в смесь загружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP2) и верхний органический слой (OP2) собирали. OP2 возвращали в реактор и загружали воду (75 мл). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP3) и верхний органический слой (OP3) собирали. Три водных слоя с преимущественным содержанием продукта (AP1, AP2 и AP3) возвращали в реактор и объединяли. Измеренное значение pH объединенного органического слоя составляло 0,47. В объединенные водные фазы загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP4) и верхний органический слой (OP4) объединяли. Измеренное значение pH AP4 составляло 0,83. AP4 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP5) и верхний органический слой (OP5) собирали. Измеренное значение pH AP5 составляло 1,90. AP5 возвращали в реактор. В реактор загружали изопропилацетат (450). Содержимое реактора смешивали при помощи мешалки с верхним приводом, а затем перемешивание прекращали, позволяя слоям разделиться. Нижний водный слой с преимущественным содержанием продукта (AP6) и верхний органический слой (OP6) собирали. Измеренное значение pH AP6 составляло 3,1. AP6 возвращали в реактор и значение pH доводили до уровня от 5,8 до 6,2 (конечное значение pH 6,04) с применением 1% раствора аммиака (35 мл). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 10°C. В реакционную смесь загружали воду (примерно 820 мл) до получения конечного объема 1350 мл. Снова измеряли значение pH (5,83) и доводили до уровня от 5,8 до 6,2 (6,01) с применением 1% раствора аммиака (5 мл). Измеренное значение электропроводности раствора составляло 15,29 мСм/см.
[00407] Указанный раствор неочищенного плазомицина загружали в колонку XK-50/100, содержащую смолу CG-50 (325,5 г) в амонийной форме, с пространством над жидкостью, заполненным водой. Температуру колонки стабилизировали на уровне 27°C. Неочищенный раствор загружали в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (примерно 9,2 мл/мин). Затем неочищенный раствор загружали на слой смолы. Загружали воду (1 объем колонки, 1620 мл) в режиме нисходящего потока при линейной скорости потока от 10 до 20 см/ч (9,2 мл/мин). После загрузки воды в колонку загружали 0,50% раствор аммиака (приготовленный из 102 г 25% аммиака в 5 л воды) в режиме нисходящего потока до элюирования плазомицина из колонки, что определяли по следам УФ-поглощения при 210 нм. Загружали раствор аммиака при линейной скорости потока 38 см/ч (12,4 мл/мин). Фракции с преимущественным содержанием продукта (#31-45) объединяли с получением 4072 мл очищенного раствора плазомицина в виде свободного основания. Содержание аммиака в растворе, измеряемое путем ионной хроматографии, составляло 4060 мкг/мл, и концентрация плазомицина в виде свободного основания, измеряемая путем СВЭЖХ, составляла 1,52 масс.%/об.%. Раствор подвергали обратному осмосу с применением мембраны XLE в режиме диафильтрования с получения отношения аммиака (мкг/мл) к плазомицину (масс.%/об.%) менее 20. Для достижения указанного критерия использовали 5 диаобъемов воды. В процессе удаления аммиака при помощи процесса диафильтрования температуру поддерживали на уровне ниже 10°C. После процесса удаления аммиака значение pH очищенного раствора плазомицина доводили до 6,74 с применением 6 М раствора серной кислоты (56). Температуру в процессе регулирования pH поддерживали на уровне от 0 до 5°C. Нейтрализованный раствор пропускали через предварительно промытый угольный картридж Zetacarbon R55 при скорости потока 72 л/ч*м2 (15 мл/мин). После фильтрования картридж промывали водой (1068 мл) и смывочный раствор объединяли с отфильтрованным раствором плазомицина. Объединенные фильтрат и смывочный раствор концентрировали до 540 мл путем обратного осмоса с применением мембраны XLE в режиме нанофильтрования. Концентрированный раствор пропускали через 0,22-микронный фильтр и выделяли путем сушки распылением с применением лабораторной распылительной сушилки Buchi (95,86 г, 0,114 моль, 76% выход).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2023 |
|
RU2826177C1 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ПРОИЗВОДНЫЕ КАЛИХЕАМИЦИНА И ИХ КОНЪЮГАТЫ "АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО" | 2018 |
|
RU2732568C1 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |

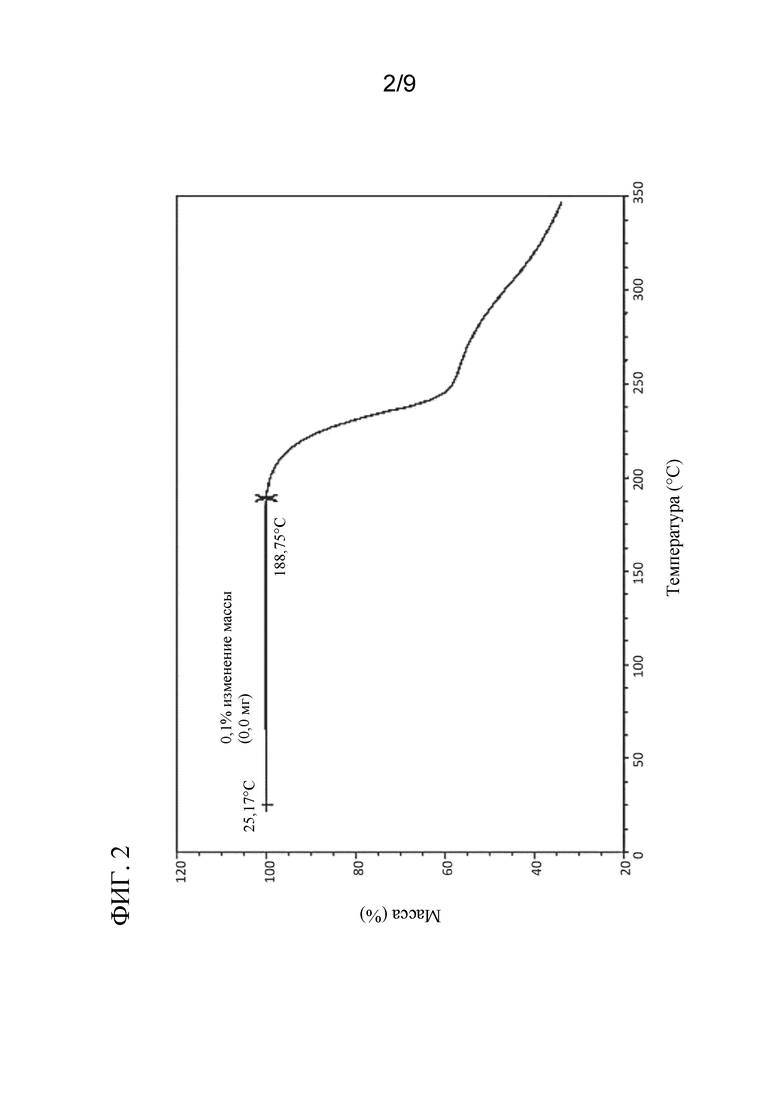
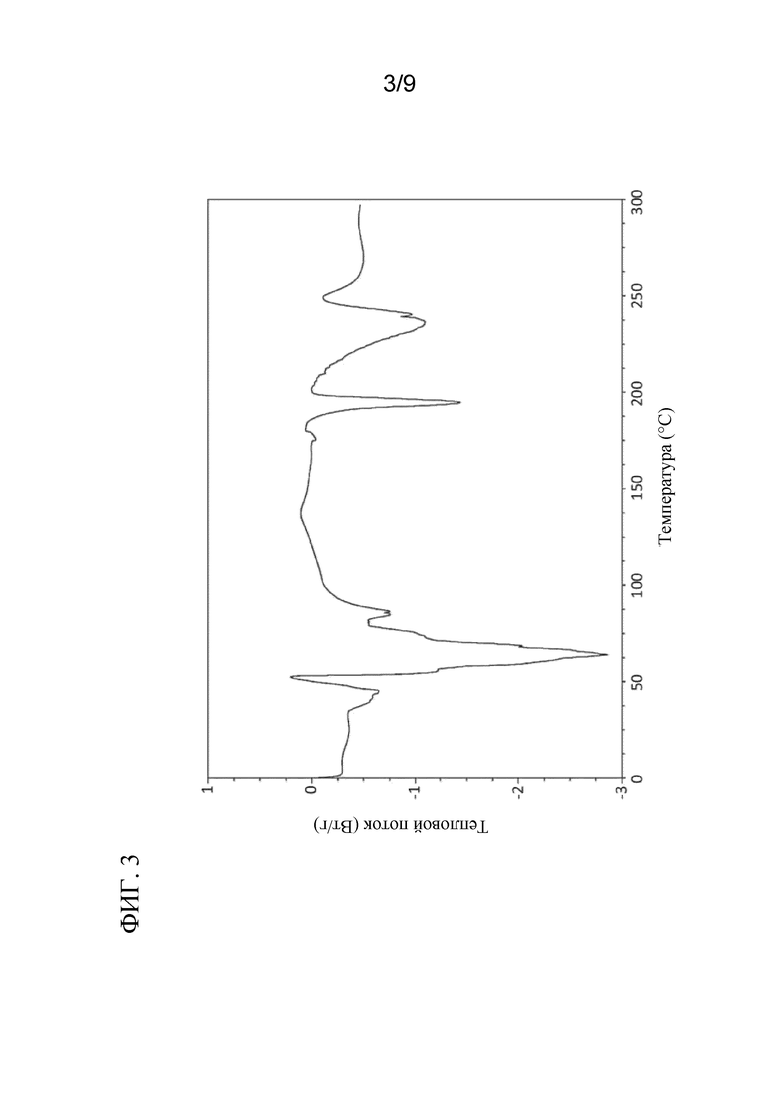
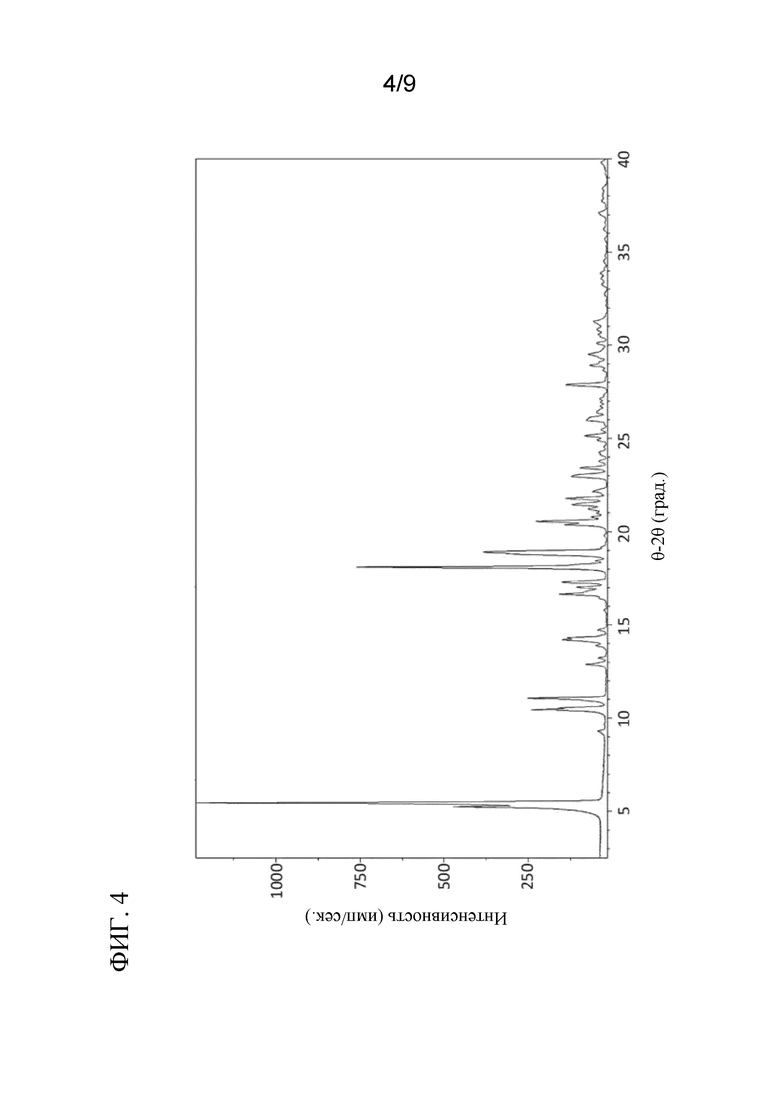





Группа изобретений относится к области органической химии и фармакологии и направлена на получение новых промежуточных соединений, используемых в синтезе антибактериальных аминогликозидных соединений. Раскрывается способ получения соединения формулы (7) включающий: (f) приведение соединения формулы (6) в контакт с  , где LG1 представляет собой уходящую группу и где
, где LG1 представляет собой уходящую группу и где  представляет собой одинарную связь или двойную связь; при этом необязательно соединение формулы (6) получают путем приведения соединения формулы (5) в контакт с реагентом для снятия PNZ-защиты, где дополнительно необязательно соединение формулы (5) получают путем приведения соединения формулы (4) в контакт с реагентом для введения Boc-защиты. Кроме того, раскрываются соединения формул (4) и (7). Группа изобретений обеспечивает эффективное получение промежуточных соединений, используемых в синтезе антибактериальных аминогликозидных соединений. 3 н. и 7 з.п. ф-лы, 9 ил., 11 табл., 8 пр.
представляет собой одинарную связь или двойную связь; при этом необязательно соединение формулы (6) получают путем приведения соединения формулы (5) в контакт с реагентом для снятия PNZ-защиты, где дополнительно необязательно соединение формулы (5) получают путем приведения соединения формулы (4) в контакт с реагентом для введения Boc-защиты. Кроме того, раскрываются соединения формул (4) и (7). Группа изобретений обеспечивает эффективное получение промежуточных соединений, используемых в синтезе антибактериальных аминогликозидных соединений. 3 н. и 7 з.п. ф-лы, 9 ил., 11 табл., 8 пр.
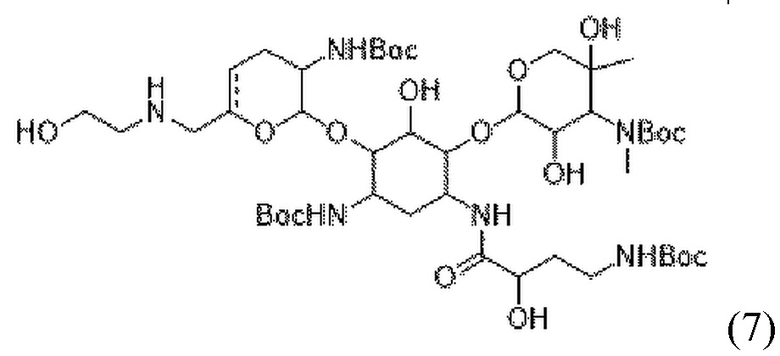


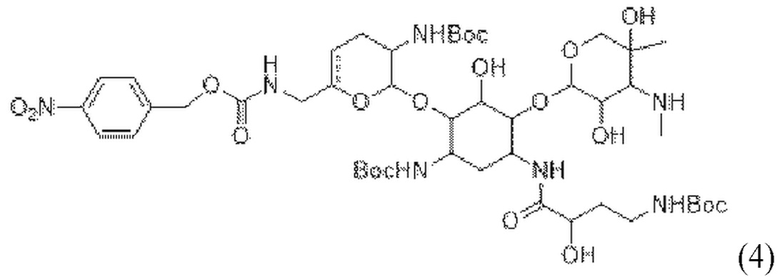
1. Способ получения соединения формулы (7)
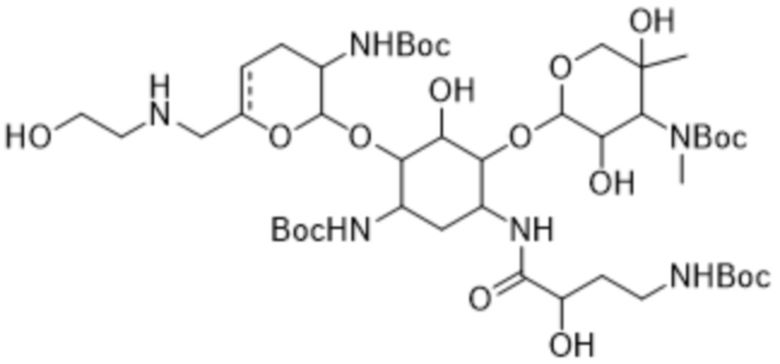 (7),
(7),
включающий:
(f) приведение соединения формулы (6)
 (6)
(6)
в контакт с  , где LG1 представляет собой уходящую группу, и где
, где LG1 представляет собой уходящую группу, и где  представляет собой одинарную связь или двойную связь;
представляет собой одинарную связь или двойную связь;
при этом необязательно соединение формулы (6) получают путем приведения соединения формулы (5)
 (5)
(5)
в контакт с реагентом для снятия PNZ-защиты,
где дополнительно необязательно соединение формулы (5) получают путем приведения соединения формулы (4)
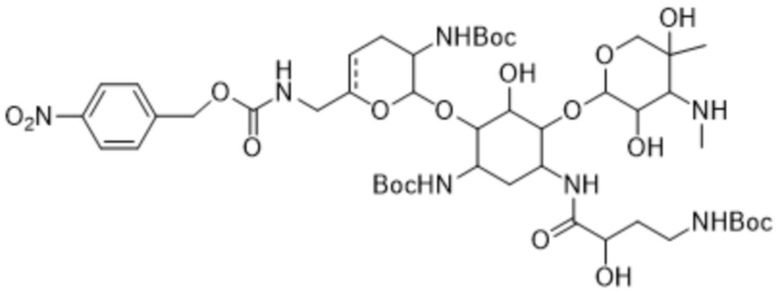 (4)
(4)
в контакт с реагентом для введения Boc-защиты.
2. Способ по п. 1, отличающийся тем, что соединение формулы (4), или его энантиомер, или его диастереомер получают путем приведения соединения формулы (3)
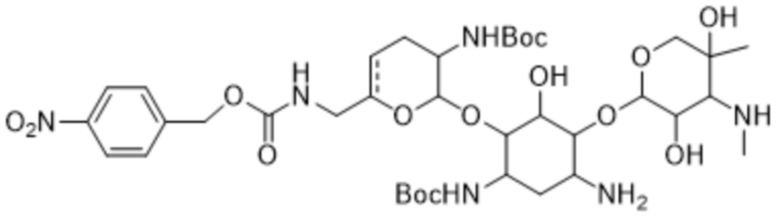 (3),
(3),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с  ;
;
при этом необязательно соединение формулы (3), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем:
(b1) приведения соединения формулы (2a)
 (2a),
(2a),
или его соли, или его сольвата, или его энантиомера, или его диастереомера в контакт с реагентом для введения Boc-защиты, при этом необязательно реагент для введения Boc-защиты представляет собой Boc2O; или
(b2) удаления C1-C3 алкила из соединения формулы (2)
 (2),
(2),
или его соли, или его сольвата, или его энантиомера, или его диастереомера;
где R1 представляет собой H или C1-C3 алкил; R2 представляет собой H или C1-C3 алкил; и R3 представляет собой H или C1-C3 алкил и где один или более из R1, R2 или R3 независимо представляют собой C1-C3 алкил; и
с последующим приведением соединения формулы (2) в контакт с реагентом для введения Boc-защиты, при этом необязательно реагент для введения Boc-защиты представляет собой Boc2O,
при этом необязательно соединение формулы (2), или его соль, или его сольват, или его энантиомер, или его диастереомер получают путем приведения соединения формулы (1)
 (1),
(1),
или его энантиомера, или его диастереомера, в контакт с 1-{[(п-нитробензил)окси]карбонил}-1H-бензотриазолом (PNZ-Bt).
3. Способ по п. 1 или 2, отличающийся тем, что стадию (b1) или (b2) осуществляют в присутствии спирта,
при этом необязательно спирт представляет собой метанол, и/или где стадию (b1) или (b2) осуществляют при температуре вплоть до примерно 60°C.
4. Способ по любому из пп. 1-3, отличающийся тем, что уходящая группа представляет собой йод-;
при этом необязательно присутствует в количестве от примерно 1,0 до 1,5 молярного эквивалента в расчете на соединение формулы (6).
5. Способ по любому из пп. 1-4, отличающийся тем, что:
(i) стадию (f) осуществляют в по существу безводных условиях; и/или
(ii) стадию (f) осуществляют в присутствии растворителя, выбранного из группы, состоящей из ацетонитрила, ацетона и их комбинации; и/или
(iii) стадию (f) осуществляют в присутствии NaHCO3; и/или
(iv) стадию (f) осуществляют при температуре от примерно 30°C до 40°C.
6. Способ по любому из пп. 1-5, дополнительно включающий:
(i) добавление 1,4-диазабицикло[2.2.2]октана (DABCO) к реакционной смеси.
7. Способ по любому из пп. 1-6, отличающийся тем, что представляет собой двойную связь.
8. Способ по любому из пп. 1-6, отличающийся тем, что:
(i) стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3” и 4” в формулах (1)-(3) указана, как в формуле (X), где  обозначает место прикрепления к водороду или фрагменту:
обозначает место прикрепления к водороду или фрагменту:
 (X);
(X);
и/или
(ii) стереохимия у атомов углерода 1, 3, 4, 5, 6, 1’, 2’, 1”, 2”, 3”, 4” и 1-z в формулах (4)-(9) указана, как в формуле (Y), где обозначает место прикрепления к водороду или фрагменту:
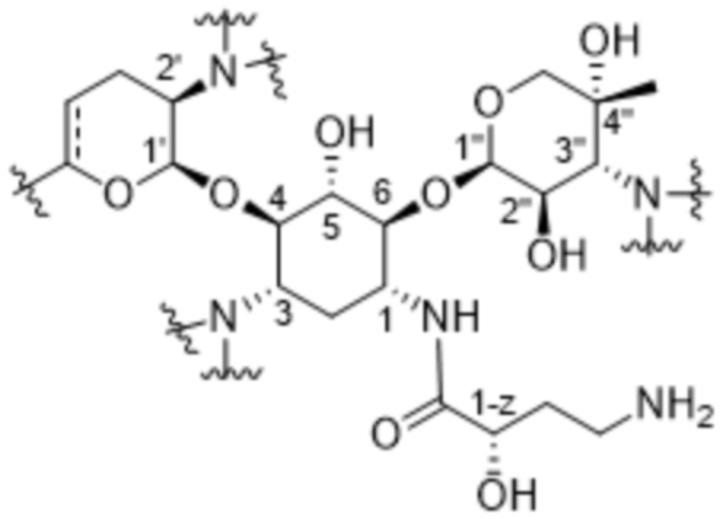 (Y).
(Y).
9. Соединение, выбранное из
соединения формулы (4)
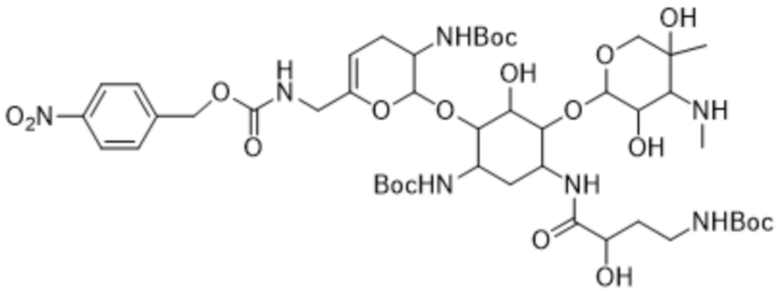 (4).
(4).
10. Соединение, выбранное из
соединения формулы (7)
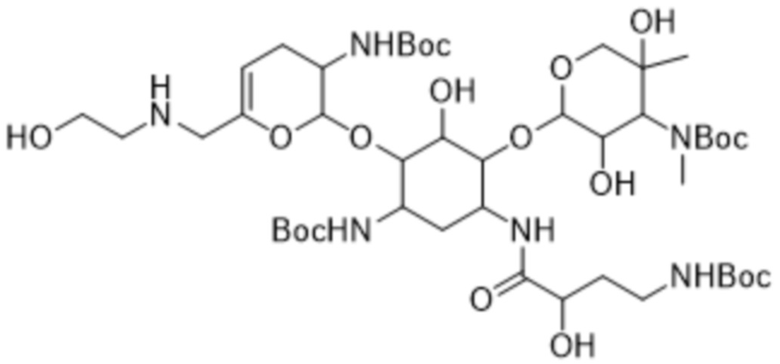 .
.
| WO 2011143497 A1, 17.11 | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| WO 2014145713 A2, 18.09.2014 | |||
| WO 2010132770 A1, 18.11.2010 | |||
| WO 2010132777 A2, 18.11.2010 | |||
| WO 2010147836 A1, 23.12.2010 | |||
| US 4388233 А, 14.06.1983 | |||
| ПРИБОР ДЛЯ ОПРЕДЕЛЕНИЯ ТВЕРДОСТИ МАТЕРИАЛОВ | 1929 |
|
SU17824A1 |
