Область техники
[0001] Настоящее изобретение относится к дозировке и введению антисмыслового олигомера, способного вызывать пропуск экзона 53 гена дистрофина человека, и к фармацевтической композиции, содержащей олигомер.
Уровень техники
[0002] Мышечная дистрофия Дюшенна (МДД) представляет собой наиболее часто встречающуюся наследственную прогрессирующую мышечную атрофию, и МДД поражает примерно одного из 3500 мальчиков при рождении. В младенчестве у пациентов с МДД имеют место двигательные функции, практически аналогичные таковым у здоровых людей, но мышечная слабость начинает наблюдаться в возрасте примерно 4 или 5 лет. Затем мышечная слабость прогрессирует, и большинство из них к 12 годам уже не может передвигаться. Пациенты умирают в результате сердечной или дыхательной недостаточности в возрасте 20 лет. Следовательно, МДД представляет очень тяжелое заболевание. В настоящее время отсутствуют эффективные способы лечения МДД, и, следовательно, возникла большая потребность в разработке нового терапевтического средства.
[0003] Известно, что причиной МДД является мутация гена дистрофина. Ген дистрофина представляет огромный по размеру ген, состоящий из ДНК из 2200000 пар нуклеотидов, который находится на Х-хромосоме. ДНК транскрибируется с образованием мРНК-предшественника, и затем интроны удаляются посредством сплайсинга, в результате синтезируется мРНК, содержащая 79 экзонов. Из данной мРНК транслируется 3685 аминокислот, с образованием в результате белка дистрофина. Белок дистрофин ассоциирован с поддержанием стабильности мембран мышечных клеток и необходим для предупреждения разрушения мышечных клеток. Поскольку ген дистрофина у пациентов с МДД имеет мутацию, то белок дистрофин, функционирующий в мышечных клетках, практически не экспрессируется. Таким образом, в организме пациентов с МДД структура мышечных клеток не может поддерживаться, и большое количество ионов кальция проникает в мышечные клетки. В результате возникает реакция, похожая на воспаление, прогрессирует фиброз и в силу этого мышечные клетки не регенерируются.
[0004] Причиной мышечной дистрофии Беккера (МДБ) также является мутация гена дистрофина. Симптомы МДБ также проявляются в мышечной слабости за счет атрофии мышц, но симптомы мышечной слабости обычно слабее, чем при МДД, и мышечная слабость прогрессирует медленно. В большинстве случаев МДБ развивается в зрелом возрасте. Полагается, что такие различия в клинических симптомах между МДД и МДБ вызваны тем, насколько разрушается рамка считывания для аминокислот в результате мутации или сохраняется, когда мРНК дистрофина транслируется в белок дистрофина (непатентный документ 1). То есть при МДД происходит мутация сдвига рамки считывания для аминокислот, и поэтому функциональные белки дистрофины практически не экспрессируются. С другой стороны, при МДБ, хотя некоторые экзоны подвергаются делеции в результате мутации, рамка считывания аминокислот сохраняется, и, таким образом, генерируются функциональные белки дистрофины, хотя их функция является недостаточной.
[0005] Ожидается, что способ пропуска экзонов будет служить способом лечения МДД. В соответствии с данным способом аминокислотная рамка считывания мРНК дистрофина восстанавливается за счет модификации сплайсинга, и индуцируется экспрессия белка дистрофина, имеющего частично восстановленную функцию (непатентный документ 2). Часть аминокислотной последовательности, которая является мишенью для пропуска экзона, теряется. Таким образом, белок дистрофин, экспрессируемый в результате такого лечения, становится короче, чем нормальный белок дистрофин. Однако, поскольку рамка считывания для аминокислот сохраняется, то функция стабилизации мышечных клеток частично сохраняется. Следовательно, предполагается, что в результате пропуска экзона при МДД будут проявляться симптомы, аналогичные симптомам более легкой МДБ. Способ пропуска экзона исследовали в экспериментах на животных с использованием мышей или собак, и в настоящее время клинические исследования проводятся с включением пациентов с МДД.
[0006] Пропуск экзона можно индуцировать связыванием антисмысловых нуклеиновых кислот, которые нацелены либо на один, либо на оба 5'- и 3'-сайта сплайсинга, либо на сайты внутри экзона. Экзон включается в мРНК только тогда, когда оба сайта сплайсинга распознаются комплексом сплайсосомы. Следовательно, посредством нацеливания антисмысловых нуклеиновых кислот на сайты сплайсинга можно индуцировать пропуск экзона. Более того, для распознавания экзона с помощью механизма сплайсинга считается необходимым, чтобы белок SR связывался с экзонным энхансером сплайсинга (ESE), и пропуск экзона также может индуцироваться в результате нацеливания на ESE.
[0007] Мутация гена дистрофина различается у отдельных пациентов с МДД. Таким образом, необходимы адаптированные антисмысловые нуклеиновые кислоты в зависимости от положения или типа генетической мутации. На сегодняшний день Steve Wilton et al. в Университете Западной Австралии получили антисмысловые нуклеиновые кислоты, которые вызывают пропуск всех 79 экзонов (непатентный документ 3), и также Annemieke Aartsma-Rus et al. в Нидерландах получили антисмысловые нуклеиновые кислоты, которые индуцируют пропуск 39 экзонов (непатентный документ 4).
[0008] Считается, что примерно 10% всех пациентов с МДД можно лечить посредством пропуска экзона 53 (далее именуемого «экзоном 53»). В последние годы многочисленные исследовательские организации сообщили о проведении исследований, касающихся пропуска экзона 53 гена дистрофина (патентные документы 1-4; и непатентные документы 5 и 6).
Список цитированных ссылок
Патентные документы
[0009] Патентный документ 1: публикация международной заявки WO 2006/000057
Патентный документ 2: публикация международной заявки WO 2004/048570
Патентный документ 3: выложенная публикация патента США № 2010/0168212
Патентный документ 4: публикация международной заявки WO 2010/048586.
Непатентные документы
[0010] Непатентный документ 1: Monaco A. P. et al., Genomics 1988; 2: pp. 90-95
Непатентный документ 2: Matsuo M., Brain Dev 1996; 18: pp. 167-172
Непатентный документ 3: Wilton S. D. et al., Molecular Therapy 2007: 15: pp. 1288-96
Непатентный документ 4: Annemieke Aartsma-Rus et al., (2002) Neuromuscular Disorders 12: S71-S77
Непатентный документ 5: Linda J. Popplewell et al., (2010) Neuromuscular Disorders, vol. 20, no. 2, pp. 102-10
Непатентный документ 6: Bladen C. L. et al., Human Mutation (2015) 36: 395-402.
Сущность изобретения
[0011] Настоящее изобретение заключается в следующем, не ограничиваясь этим.
1. Фармацевтическая композиция для лечения пациента-человека с мышечной дистрофией Дюшенна, где фармацевтическая композиция содержит антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль, или его гидрат, где
лечение включает внутривенное введение пациенту-человеку антисмыслового олигомера или его фармацевтически приемлемой соли или его гидрата в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/неделю включительно.
2. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят внутривенно пациенту-человеку в дозе 40 мг/кг/неделю.
3. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно в дозе 80 мг/кг/неделю.
4. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где у пациента-человека имеется мутация, которая приводит к дефициту любого экзона, выбранного из группы, состоящей из экзонов 43-52, 45-52, 47-52, 48-52, 49-52, 50-52 или 52 в гене дистрофина.
5. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где экспрессия белка дистрофина у пациента-человека до лечения составляет 1% или ниже по сравнению с таковой у здорового субъекта, как измерено вестерн-блоттингом или масс-спектрометрией.
6. Фармацевтическая композиция в соответствии с вышеуказанным п.5, где экспрессия белка дистрофина не обнаруживается у пациента-человека до лечения.
7. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где нуклеотидная последовательность антисмыслового олигомера состоит из последовательности, показанной в SEQ ID NO: 3.
8. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где антисмысловой олигомер или его фармацевтически приемлемая соль, или его гидрат представляет собой вилтоларсен или его эквивалент.
9. Фармацевтическая композиция в соответствии с вышеуказанным п.1, содержащая антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат, в концентрации от 2,5 мг/мл включительно до 500 мг/мл включительно или от 10 мг/мл включительно до 100 мг/мл включительно.
10. Фармацевтическая композиция в соответствии с вышеуказанным п.1, содержащая антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат в концентрации 25 мг/мл.
11. Фармацевтическая композиция в соответствии с вышеуказанным п.1, содержащая антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат, в концентрации 50 мг/мл.
12. Фармацевтическая композиция в соответствии с вышеуказанным п.1, дополнительно содержащая, по меньшей мере, один компонент, выбранный из группы, состоящей из агента, регулирующего тоничность, регулятора pH и растворителя.
13. Фармацевтическая композиция в соответствии с вышеуказанным п.12, где агент, регулирующий тоничность, представляет, по меньшей мере, один, выбранный из группы, состоящей из хлорида натрия, хлорида калия, глюкозы, фруктозы, мальтозы, сахарозы, лактозы, маннита, сорбита, ксилита, трегалозы и глицерина.
14. Фармацевтическая композиция в соответствии с вышеуказанным пп.12 или 13, где регулятор pH представляет, по меньшей мере, один, выбранный из группы, состоящей из соляной кислоты, гидроксида натрия, лимонной кислоты, молочной кислоты, фосфата (гидрофосфата натрия, дигидрофосфата натрия, дигидрофосфата калия) и моноэтаноламина.
15. Фармацевтическая композиция в соответствии с любым из вышеуказанных пп.12-14, где растворитель представляет воду.
16. Фармацевтическая композиция в соответствии с вышеуказанным п.1, которая содержит антисмысловой олигомер в концентрации от 2,5 мг/мл включительно до 500 мг/мл включительно, или от 10 мг/мл включительно до 100 мг/мл включительно, и хлорид натрия в концентрации от 8 мг/мл включительно до 10 мг/мл включительно, и которая представляет водный раствор с pH от 7,2 до 7,4.
17. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где лечение обеспечивает, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) среднее значение уровня экспрессии белка дистрофина в скелетных мышцах пациента увеличивается в 9 или более раз по сравнению с исходным уровнем после введения фармацевтической композиции в течение 24 недель;
(2) изменение скорости, полученной по времени вставания из лежачего положения на спине (TTSTAND), составляет -0,055 раз/с или более по сравнению с исходным уровнем ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(3) изменение скорости, полученной по времени бега/ходьбы на 10 метров (TTRW), составляет -0,025 м/с или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(4) изменение скорости, полученной по времени подъема на 4 ступени (TTCLIMB), составляет -0,060 раз/с или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(5) изменение балла по шкале амбулаторного обследования North Star (NSAA) составляет -2,2 балла или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель; и
(6) изменение показателя в тесте 6-минутной ходьбы (6MWT) составляет -7,5 м или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель.
18. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где, когда проводится лечение пациентов в возрасте от 7 до 9 лет с мышечной дистрофией Дюшенна в течение 84 недель, то обеспечивается, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) процент пациентов, которые теряют способность подниматься, составляет менее 20% ко времени недели 85 после начала лечения;
(2) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 10% ко времени недели 85 после начала лечения;
(3) процент пациентов, которые теряют способность к самостоятельному передвижению, составляет менее 10% ко времени недели 85 после начала лечения;
(4) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(5) снижение показателя подъема на 4 ступени с возрастом не наблюдается ко времени недели 85 после начала лечения; и
(6) снижение скорости подъема с возрастом не наблюдается ко времени недели 85 после начала лечения.
19. Фармацевтическая композиция в соответствии с вышеуказанным п.1, где, когда проводится лечение пациентов в возрасте от 10 до 12 лет с мышечной дистрофией Дюшенна в течение 84 недель, то обеспечивается, по меньшей мере, один эффект выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) процент пациентов, теряющих способность подниматься, составляет менее 60% ко времени недели 85 после начала лечения;
(2) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 50% ко времени недели 85 после начала лечения;
(3) процент пациентов, теряющих способность к самостоятельному передвижению, составляет менее 50% ко времени недели 85 после начала лечения;
(4) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(5) наблюдается период, в течение которого увеличивается скорость подъема на 4 ступени ко времени недели 85 после начала лечения; и
(6) наблюдается период, в течение которого увеличивается скорость вставания ко времени недели 85 после начала лечения.
20. Способ лечения мышечной дистрофии Дюшенна, включающий внутривенное введение фармацевтической композиции, содержащей антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях от 36 до 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль, или его гидрат, пациенту-человеку один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/неделю антисмыслового олигомера, или его фармацевтически приемлемой соли или его гидрата.
20-1. Способ лечения по вышеуказанному п.20, где способом лечения обеспечивается, по меньшей мере, один из эффектов в соответствии с п.17, по меньшей мере, один из эффектов в соответствии с п.18 или, по меньшей мере, один из эффектов в соответствии с п.19.
21. Антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемая соль, или его гидрат, для применения в способе лечения пациента-человека с мышечной дистрофией Дюшенна, где:
антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/ неделю включительно.
21-1. Антисмысловой олигомер в соответствии с вышеуказанным п.21 или его фармацевтически приемлемая соль, или его гидрат, где обеспечивается, по меньшей мере, один из эффектов в соответствии с п.17, по меньшей мере, один из эффектов в соответствии с п.18 или, по меньшей мере, один из эффектов в соответствии с п.19 введением антисмыслового олигомера или его фармацевтически приемлемой соли, или его гидрата.
22. Применение антисмыслового олигомера, состоящего из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека или его фармацевтически приемлемой соли, или его гидрата, для производства фармацевтической композиции для лечения пациента-человека с мышечной дистрофией Дюшенна, где:
антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/ неделю включительно.
[0012] Кроме того, в качестве еще одного аспекта настоящее изобретение заключается в следующем, не ограничиваясь этим.
1. Способ лечения субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения субъекту NS-065/NCNP-01 в дозе примерно 40 мг/кг/неделю.
2. Способ лечения субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения субъекту NS-065/NCNP-01 в дозе примерно 80 мг/кг/неделю.
3. Способ лечения субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения субъекту NS-065/NCNP-01 в дозе 40 мг/кг/неделю или более и 80 мг/кг/неделю или менее.
4. Способ в соответствии с вышеуказанным п.1, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
5. Способ в соответствии с вышеуказанным п.2, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
6. Способ в соответствии с вышеуказанным п.3, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
7. Способ индукции генерации белка дистрофина у субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения субъекту NS-065/NCNP-01 в дозе примерно 40 мг/кг/неделю.
8. Способ индукции генерации белка дистрофина у субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения субъекту NS-065/NCNP-01 в дозе примерно 80 мг/кг/ неделю.
9. Способ индукции генерации белка дистрофина у субъекта с мышечной дистрофией Дюшенна, поддающейся лечению, включающему пропуск экзона 53, где способ включает стадию внутривенного введения NS-065/NCNP-01 субъекту в дозе 40 мг/кг/неделю или более и 80 мг/кг/неделю или менее.
10. Способ в соответствии с вышеуказанным п.7, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
11. Способ в соответствии с вышеуказанным п.8, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
12. Способ в соответствии с вышеуказанным п.9, где вводят водный раствор, содержащий:
NS-065/NCNP-01 в концентрации от 2,5 мг/мл или более до 500 мг/мл или менее; и
хлорид натрия в качестве агента, регулирующего тоничность, в концентрации от 8,55 мг/мл или более до 9,45 мг/мл или менее, и
где водный раствор имеет значение pH примерно 7,3.
В вышеприведенных пунктах 1-12 NS-065/NCNP-01 (который также относится в настоящем описании в «вилтоларсену») также может представлять его эквивалент. Кроме того, в вышеприведенных пунктах 1- 12 субъект также может быть пациентом-человеком.
[0013] Настоящее изобретение относится к фармацевтической композиции для применения в лечении мышечной дистрофии Дюшенна, которая имеет стабильную композицию вилтоларсена. Более того, что касается фармацевтической композиции, содержащей вилтоларсен, то обеспечиваются дозировка и способ введения вилтоларсена, при которых проявляются высокие терапевтические эффекты в отношении мышечной дистрофии Дюшенна, и которые находятся в безопасном диапазоне для пациентов-людей. Используя фармацевтическую композицию, можно эффективно уменьшить симптомы мышечной дистрофии Дюшенна с низкими побочными эффектами.
Краткое описание фигур
[0014] На фиг. 1 представлен дизайн фазы 2 клинического исследования по подбору дозы NS-065/NCNP-01-201, проведенного в США.
На фиг. 2 приведены результаты анализа уровня de novo экспрессии белка дистрофина в скелетных мышцах по данным метода вестерн-блоттинга.
На фиг. 3 представлен дизайн фазы 2 клинического исследования NS-065/NCNP-01-201 по подбору дозы, проведенного в США/Канаде.
На фиг. 4 показаны сравнения изменений относительно исходного уровня результатов временных функциональных тестов, проведенных в течение 24 недель. На пяти графиках термин «вилтоларсен» означает международное непатентованное название (МНН) NS-065/NCNP-01.
На фиг. 5 приведены результаты оценки стабильности вилтоларсена в буфере Бриттона-Робинсона (pH от 3 до 11).
На фиг. 6 приведены результаты оценки стабильности вилтоларсена в калий-фосфат-боратном буфере (pH от 6 до 9).
На фиг. 7 приведены результаты тестирования регуляторов pH при 121°C.
На фиг. 8 приведены графики, показывающие изменения результатов тестов оценки двигательной функции у отдельных пациентов по сравнению с исходным уровнем.
На фиг. 9 представлен график, показывающий результаты теста 6-минутной ходьбы, проведенного с включением отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг) на время недели 85 (через 84 недели после первоначального введения препарата).
На фиг. 10 представляет график, показывающий результаты амбулаторного обследования North Star, проведенного с включением отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг) на время недели 85 (через 84 недели после первоначального введения препарата).
На фиг. 11 представлен график, показывающий результаты теста с определением скорости по времени вставания, проведенного с включением отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг) на время недели 85 (через 84 недели после первоначального введения препарата).
На фиг. 12 представлен график, показывающий результаты теста с определением скорости по времени подъема на 4 ступени, проведенного с включением отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг) на время недели 85 (через 84 недели после первоначального введения препарата).
На фиг. 13 представлен график, показывающий результаты теста с определением скорости по времени для бега/ходьбы на 10 метров, проведенного с включением отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг) на время недели 85 (через 84 недели после первоначального введения препарата).
На фиг. 14 представлен график, показывающий корреляцию между изменением уровня экспрессии дистрофина относительно исходного уровня при количественном определении дистрофина с использованием WB, и изменением скорости по времени в тесте вставания относительно исходного уровня у отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг), на время недели 49 (через 48 недель после первоначального введения препарата).
На фиг. 15 представлен график, показывающий корреляцию между изменением уровня экспрессии дистрофина относительно исходного уровня при количественном определении дистрофина с использованием WB, и изменением скорости по времени в тесте подъема на 4 ступени относительно исходного уровня, у отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг), на время недели 49 (через 48 недель после первоначального введения препарата).
На фиг. 16 представлен график, показывающий корреляцию между изменением уровня экспрессии дистрофина относительно исходного уровня при количественном определении дистрофина с использованием WB, и изменением скорости по времени бега/ходьбы на 10 метров относительно исходного уровня, у отдельных исследуемых пациентов из групп, которым вводили вилтоларсен (группа с дозой 40 мг/кг и группа с дозой 80 мг/кг), на время недели 49 (через 48 недель после первоначального введения препарата).
Описание вариантов осуществления
[0015] Далее настоящее изобретение будет описано подробно. Следующие ниже варианты осуществления представлены в качестве примеров для пояснения настоящего изобретения и, таким образом, не предназначены для ограничения настоящего изобретения только этими вариантами осуществления. Настоящее изобретение может быть осуществлено в различных вариантах осуществления, не отступая от сущности изобретения.
Все публикации и патентные публикации, такие как выложенные публикации патентов или заявки на патенты, цитированные в настоящем описании, в полном объеме включены в настоящий документ посредством ссылки. Кроме того, настоящее описание включает содержание, описанное в спецификациях и на чертежах предварительной заявки на патент США (US 62/690270), поданной 26 июня 2018 г., и предварительной заявки на патент США (US 62/739 386), поданной 1 октября 2018 г. к которым настоящая заявка испрашивает приоритет.
[0016] I. Первый вариант осуществления
В первом варианте осуществления настоящее изобретение относится к фармацевтической композиции для лечения мышечной дистрофии Дюшенна. В частности, фармацевтическая композиция по настоящему изобретению представляет фармацевтическую композицию для лечения пациента-человека с мышечной дистрофией Дюшенна, где фармацевтическая композиция содержит антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях от 36 до 56 от 5'-конца экзона 53 гена дистрофина человека (далее также относящегося к «олигомеру по настоящему изобретению»), или его фармацевтически приемлемую соль, или его гидрат, где
лечение включает внутривенное введение пациенту-человеку антисмыслового олигомера или его фармацевтически приемлемой соли или его гидрата в дозе от 40 мг/кг/неделю включительно до 80 мг/ кг/неделю включительно.
[0017] Экзон 53 гена дистрофина человека
В настоящем изобретении термин «ген» включает кДНК, мРНК-предшественник и мРНК, а также геномный ген. Ген предпочтительно является мРНК-предшественником, а именно пре-мРНК.
В геноме человека ген дистрофина человека находится в локусе гена Xp21.2. Ген дистрофина человека имеет размер 3,0 млн.п.н., и это самый большой ген среди известных генов человека. Однако размер кодирующей области гена дистрофина человека составляет всего 14 т.п.н., и кодирующая область рассредоточена в виде 79 экзонов в гене дистрофина (Roberts R.G. et al., Genomics, 16: 536-538 (1993)). Пре-мРНК как продукт транскрипции гена дистрофина человека генерирует зрелую мРНК размером 14 т.п.н. в результате сплайсинга. Последовательность оснований зрелой мРНК гена дистрофина человека дикого типа известна (идентификационный номер в GenBank NM_004006).
Нулеотидная последовательность экзона 53 гена дистрофина человека дикого типа показана в SEQ ID NO: 1.
[0018] Фармацевтическая композиция по настоящему изобретению включает антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека (олигомер по настоящему изобретению), или его фармацевтически приемлемую соль, или его гидрат.
В данном случае олигомер по настоящему изобретению получают с целью модификации белка, кодированного геном дистрофина МДД-типа, с образованием белка дистрофина МДБ-типа посредством пропуска экзона 53. Следовательно, экзон 53 гена дистрофина в качестве мишени для пропуска экзона с использованием олигомера по настоящему изобретению включает не только экзон 53 дикого типа, но также экзон 53 мутантного типа.
[0019] Конкретным примером такого экзона 53 мутантного типа гена дистрофина человека может быть полинуклеотид, имеющий идентичность на уровне 90% или более, 91% или более, 92% или более, 93% или более, 94% или более, 95% или больше, 96% или более, 97% или более, 98% или более, 99% или более, 99,1% или более, 99,2% или более, 99,3% или более, 99,4% или более, 99,5% или более, 99,6% или более, 99,7% или более, 99,8% или более, или 99,9% или более, с нуклеотидной последовательностью, показанной в SEQ ID NO: 1. В настоящем описании термин «полинуклеотид» означает ДНК или РНК.
[0020] Кроме того, идентичность нуклеотидных последовательностей можно определить с помощью алгоритма BLAST (Basic Local Alignment Search Tool) Carlin and Arthur (Proc. Natl. Acad. Sci. USA, 872264-2268, 1990; Proc. Natl. Acad. Sci. USA, 90: 5873, 1993). На основе алгоритма BLAST были разработаны программы, называемые BLASTN или BLASTX (Altschul S.F. et al.: J. Mol. Biol., 215: 403, 1990). Когда нуклеотидную последовательность анализируют с использованием алгоритма BLASTN, то устанавливают параметры, например, балл=100 и длина слова=12. Когда используются программы BLAST и Gapped BLAST, то используются параметры по умолчанию для каждой отдельной программы.
[0021] В настоящем описании «комплементарная нуклеотидная последовательность» не ограничивается нуклеотидной последовательностью, которая образует пару оснований согласно правилу Уотсона-Крика с нуклеотидной последовательностью-мишенью, но также включает нуклеотидную последовательность, которая образует нестабильную пару оснований. В данном документе термин «пара оснований согласно правилу Уотсона-Крика» означает пару оснований, в которой водородные связи образуются между основаниями аденин-тимин, аденин-урацил и гуанин-цитозин, тогда как термин «нестабильная пара оснований» означает пару оснований, в которой водородные связи образуются между основаниями гуанин-урацил, инозин-урацил, инозин-аденин и инозин-цитозин. Кроме того, «комплементарная нуклеотидная последовательность» может не иметь комплементарности на уровне 100% относительно нуклеотидной последовательности-мишени, и, например, комплементарная нуклеотидная последовательность может содержать 1, 2, 3, 4 или 5 некомплементарных оснований относительно нуклеотидной последовательности-мишени. Кроме того, комплементарная нуклеотидная последовательность также может представлять нуклеотидную последовательность, которая короче нуклеотидной последовательности-мишени на 1, 2, 3, 4 или 5 нуклеотидов.
[0022] Примеры последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 (SEQ ID NO: 2) и нуклеотидной последовательности, комплементарной вышеуказанной последовательности (SEQ ID NO: 3), приведены в следующей таблице. 1.
[0023]
[0024] В данном случае тимин «Т» и урацил «U» могут взаимно обмениваться друг с другом. Даже если основание представляет собой «Т» или «U», то это не оказывает существенного влияния на активность пропуска экзона олигомера по настоящему изобретению. Следовательно, в настоящей заявке, даже если «T» в нуклеотидной последовательности, имеющей определенный порядковый номер, представляет «U», то она показана с тем же порядковым номером. Следовательно, последовательность, раскрытая в настоящей заявке, неизбежно включает как последовательность с «Т», так и последовательность с «U».
[0025] С учетом вышеизложенного, нуклеотидная последовательность олигомера по настоящему изобретению может состоять из последовательности, показанной в SEQ ID NO: 3. Более того, олигомер по настоящему изобретению может не иметь нуклеотидной последовательности, которая на 100% комплементарна с последовательностью-мишенью, при условии, что она обеспечивает пропуск экзона 53 гена дистрофина человека. Например, олигомер по настоящему изобретению может содержать 1, 2, 3, 4 или 5 некомплементарных оснований относительно последовательности SEQ ID NO: 2, которая является последовательностью-мишенью. Альтернативно олигомер по настоящему изобретению может представлять нуклеотидную последовательность, которая короче нуклеотидной последовательности-мишени на 1, 2, 3, 4 или 5 нуклеотидов.
[0026] Имел место или нет пропуск экзона 53 гена дистрофина человека, можно подтвердить посредством: введения олигомера по настоящему изобретению в клетки, экспрессирующие дистрофин (например, клетки рабдомиосаркомы человека), амплификации периферической области экзона 53 мРНК гена дистрофина человека из общей РНК вышеуказанных клеток, экспрессирующих дистрофин, с помощью ОТ-ПЦР; и затем выполнения гнездовой ПЦР или анализа последовательности на амплифицированном продукте ПЦР. В качестве альтернативы, имел место или нет такой пропуск, можно также подтвердить посредством измерения количества экзона 53 таким методом, как ОТ-ПЦР, вестерн-блоттинг или масс-спектрометрия в образце, полученном от пациента, которому был введен олигомер по настоящему изобретению.
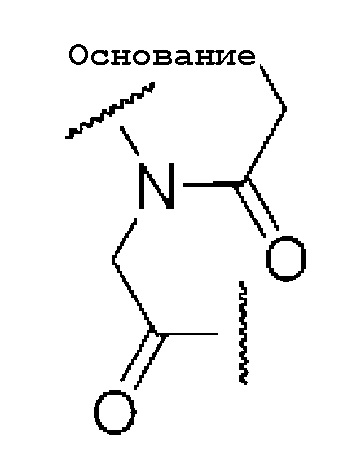
Эффективность пропуска можно определить выделением мРНК гена дистрофина человека из тестируемых клеток, затем измерением уровня полинуклеотида «А» полосы, включающей пропуск экзона 53, и уровня полинуклеотида «В» полосы, не включающей пропуск экзона 53 в мРНК, и затем вычислением эффективности пропуска в соответствии со следующим уравнением на основе измеренных значений «A» и «B».
Эффективность пропуска (%) = A/(A+B) × 100
[0027] Олигомер по настоящему изобретению может включать олигонуклеотид, морфолино-олигомер и олигомер пептидной нуклеиновой кислоты (PNA). Олигомер по настоящему изобретению предпочтительно представляет собой морфолино-олигомер.
[0028] Вышеописанный олигонуклеотид (в дальнейшем именуемый «олигонуклеотидом по настоящему изобретению») представляет олигомер по настоящему изобретению, содержащий нуклеотид в качестве структурной единицы, и такой нуклеотид может представлять любой из рибонуклеотида, дезоксирибонуклеотида или модифицированного нуклеотид.
Модифицированный нуклеотид означает рибонуклеотид или дезоксирибонуклеотид, в котором модифицированы все или часть азотистых оснований, остатков сахара и фосфатных связей, которые составляют рибонуклеотид или дезоксирибонуклеотид.
[0029] Примеры азотистого основания могут включать аденин, гуанин, гипоксантин, цитозин, тимин, урацил и их модифицированный нуклеотид. Пример такого модифицированного нуклеотида может включать, не ограничиваясь этим, псевдоурацил, 3-метилурацил, дигидроурацил, 5-алкилцитозини (например, 5-метилцитозин), 5-алкилурацил (например, 5-этилурацил), 5-галогенурацил (5-бромурацил), 6-азапиримидин, 6-алкилпиримидин (6-метилурацил), 2-тиоурацил, 4-тиоурацил, 4-ацетилцитозин, 5-(карбоксигидроксиметил)урацил, 5'-карбоксиметиламинометил-2-тиоурацил, 5-карбоксиметиламинометилурацил, 1-метиладенин, 1-метилгипоксантин, 2,2-диметилгуанин, 3-метилцитозин, 2-метиладенин, 2-метилгуанин, N6-метиладенин, 7-метилгуанин, 5-метоксиаминометил-2-тиоурацил, 5-метиламинометилурацил, 5-метилкарбонилметилурацил, 5-метилоксиурацил, 5-метил-2-тиоурацил, 2-метилтио-N6-изопентениладенин, урацил-5-оксиуксусную кислоту, 2-тиоцитозин, пурин, 2,6-диаминопурин, 2-аминопурин, изогуанин, индол, имидазол, ксантин.
[0030] Примеры модификация остатка сахара могут включать, например, модификации в 2'-положении рибозы и модификации других положений сахара. Модификация в 2'-положении рибозы включает замену ОН-группы в 2'-положении рибозы на OR, R, R', OR, SH, SR, NH2, NHR, NR2, N3, CN, F, Cl, Br и I. В данном случае R представляет алкил или арил. R' представляет алкилен.
Примеры модификации других положений остатка сахара могут включать, помимо прочего, замену О в 4'-положении рибозы или дезоксирибозы на S, и сшивание между 2'-положением и 4'-положением остатка сахара, например, LNA (запертая нуклеиновая кислота) или ENA (нуклеиновые кислоты с 2'-O, 4'-C-этиленовым мостиком).
[0031] Примерами модификации фосфатной связи может быть модификация с заменой фосфодиэфирной связи на фосфоротиоатную связь, фосфородитиоатную связь алкилфосфонатную связь, фосфороамидатную связь или боранофосфатную связь (Enya et al.: Bioorganic & Medicinal Chemistry, 2008, 18, 9154-9160) (см., например, повторные публикации международных заявок РСТ 2006/129594 и 2006/038608).
[0032] В качестве алкила предпочтительным является линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода. Конкретные примеры такого алкила могут включать метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-пентил, н-гексил и изогексил. Алкил может быть замещенным. Примеры такого заместителя могут включать атом галогена, алкокси, циано и нитро. Алкил может быть замещен 1-3 указанными заместителями.
[0033] В качестве циклоалкила предпочтительным является циклоалкил, содержащий от 5 до 12 атомов углерода. Конкретные примеры такого циклоалкила могут включать циклопентил, циклогексил, циклогептил, циклооктил, циклодецил и циклододецил.
[0034] Примеры галогена могут включать атомы фтора, хлора, брома и йода.
[0035] Примеры алкокси могут включать линейный или разветвленный алкокси, содержащий от 1 до 6 атомов углерода, такой как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, н-гексилокси и изогексилокси. Среди прочего предпочтительным является алкокси, содержащий от 1 до 3 атомов углерода.
[0036] В качестве арила предпочтительным является арил, содержащий от 6 до 10 атомов углерода. Конкретные примеры такого арила могут включать фенил, α-нафтил и β-нафтил. Среди прочего предпочтительным является фенил. Арил может быть замещенным. Примеры такого заместителя могут включать алкил, атом галогена, алкокси, циано и нитро. Арил может быть замещен 1-3 указанными заместителями.
[0037] В качестве алкилена предпочтительным является линейный или разветвленный алкилен, содержащий от 1 до 6 атомов углерода. Конкретные примеры такого алкилена могут включать метилен, этилен, триметилен, тетраметилен, пентаметилен, гексаметилен и 2-(этил)триметилен, 1-(метил)тетраметилен.
[0038] Примеры ацила могут включать линейный или разветвленный алканоил и ароил. Примеры алканоила могут включать формил, ацетил, 2-метилацетил, 2,2-диметилацетил, пропионил, бутирил, изобутирил, пентаноил, 2,2-диметилпропионил и гексаноил. Примеры ароила могут включать бензоил, толуоил и нафтоил. Такой ароил может быть замещен в замещаемом положении и может быть замещен алкилом.
[0039] В аспекте, в котором олигомер по настоящему изобретению представляет олигонуклеотид, то олигонуклеотид может предпочтительно включать в качестве структурной единицы группу, представленную следующей общей формулой, в которой ОН-группа в 2'-положении рибозы замещена метокси, и фосфатная связь представляет фосфоротиоатную связь:
Формула 1
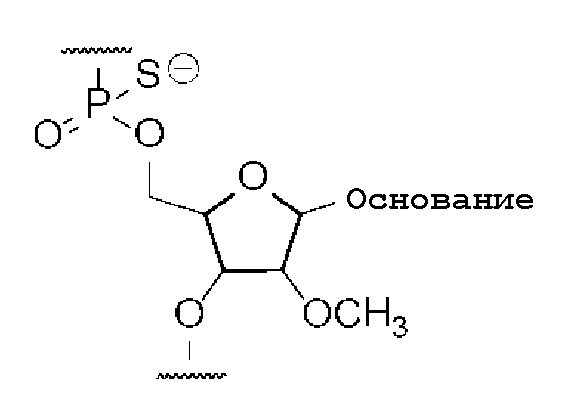
где основание представляет азотистое основание.
[0040] Такой олигонуклеотид можно легко синтезировать с использованием различных типов автоматических синтезаторов (например, AKTA oligopilot plus 10/100 (GE Healthcare)). В противном случае олигонуклеотид также может быть получен передачей его синтеза сторонней организации (например, Promega или Takara) и т. д.
[0041] Когда олигомер по настоящему изобретению представляет собой морфолино-олигомер, то морфолино-олигомер может включать в качестве структурной единицы группу, представленную следующей общей формулой:
Формула 2
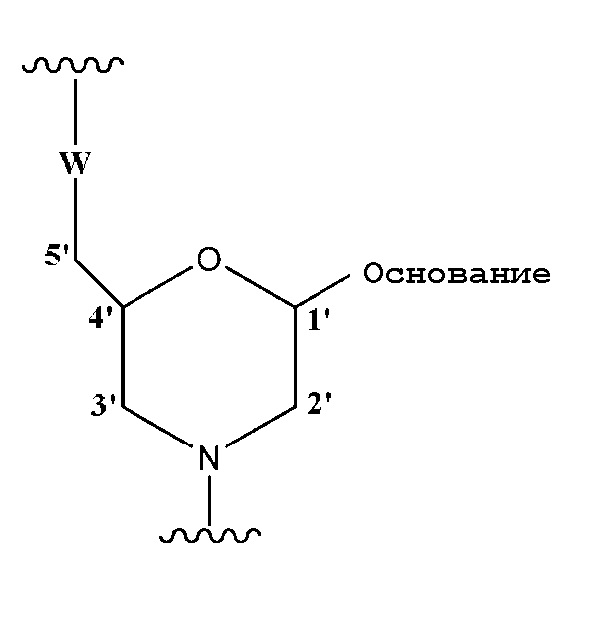
где основание имеет значение, определенное выше; и
W представляет группу, представленную любой из следующих формул:
Формула 3
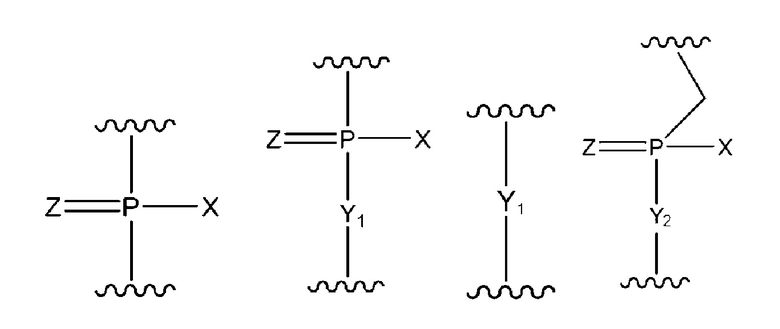
где X представляет -CH2R1, -O-CH2R1, -S-CH2R1, -NR2R3 или F;
R1 представляет H или алкил;
R2 и R3, которые являются одинаковыми или разными, и каждый представляет H, алкил, циклоалкил или арил;
Y1 представляет O, S, CH2 или NR1;
Y2 представляет O, S или NR1; и
Z представляет O или S.
[0042] Морфолино-олигомер предпочтительно представляет олигомер, содержащий в качестве структурной единицы группу, представленную следующей формулой (т.е. морфолино-фосфородиамидатный олигомер (далее именуемый «PMO»)):
Формула 4

где основание, R2 и R3 имеют значения, определенные выше.
[0043] Морфолино-олигомер можно получить, например, в соответствии с публикацией международной заявки WO 1991/009033 или публикацией международной заявки WO 2009/064471. В частности, PMO можно получить в соответствии со способом, описанным в публикации международной заявки WO 2009/064471, или в соответствии со способом, описанным ниже.
[0044] В одном аспекте PMO может включать, например, соединение, представленное следующей общей формулой (I) (далее именуемое «PMO (I)»):
Формула 5
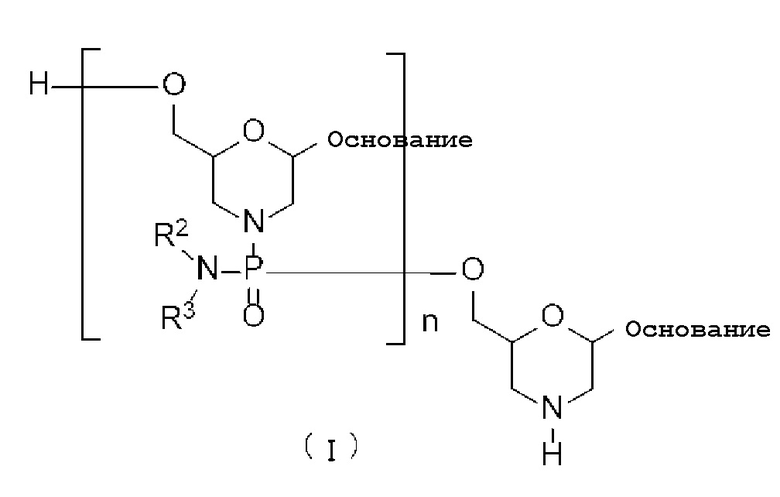
где каждое основание, R2 и R3 имеют значения, определенные выше; и
n представляет любое заданное целое число в диапазоне от 1 до 99 и предпочтительно представляет любое заданное целое число в диапазоне от 18 до 28.
[0045] PMO (I) можно получить в соответствии с известным способом, и соединения и реагенты, используемые для получения PMO (I), особым образом не ограничиваются, при условии, что они обычно используются в получении PMO. Кроме того, получение может осуществить жидкофазным методом или твердофазным методом (в котором используются руководства или коммерчески доступные твердофазные автоматические синтезаторы). Когда PMO получают твердофазным методом, то желательно использовать способ с автоматическим синтезатором с точки зрения упрощения операционных процедур и точности синтеза.
[0046] Пептидная нуклеиновая кислота представляет олигомер по настоящему изобретению, содержащий в качестве структурной единицы группу, представленную следующей общей формулой:
Формула 6
где основание имеет значение, определенное выше.
[0047] Пептидную нуклеиновую кислоту можно получить, например, согласно методам, описанным в следующих публикациях:
1) P. E. Nielsen, M. Egholm, R. H. Berg, O. Buchardt, Science, 254, 1497 (1991)
2) M. Egholm, O. Buchardt, P. E. Nielsen, R. H. Berg, Jacs., 114, 1895 (1992)
3) K. L. Dueholm, M. Egholm, C. Behrens, L. Christensen, H. F. Hansen, T. Vulpius, K. H. Petersen, R. H. Berg, P. E. Nielsen, O. Buchardt, J. Org. Chem., 59, 5767 (1994)
4) L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M. Egholm, O. Buchardt, P. E. Nielsen, J. Coull, R. H. Berg, J. Pept. Sci., 1, 175 (1995)
5) T. Koch H., F. Hansen, P. Andersen, T. Larsen, H. G. Batz, K. Otteson, H. Orum, J. Pept. Res., 49, 80 (1997).
[0048] Кроме того, 5'-конец олигомера по настоящему изобретению может представлять группу, представленную любой из следующих химических формул (1)-(3). Предпочтительно это ОН-группа, показанная в (3).
Формула 7
В дальнейшем группы, представленные вышеприведенными формулами (1), (2) и (3), называются «группой (1)», «группой (2)» и «группой (3)» соответственно.
[0049] Примеры фармацевтически приемлемой соли олигомера по настоящему изобретению могут включать: соли щелочных металлов, такие как соли натрия, соли калия или соли лития; соли щелочноземельных металлов, такие как соли кальция или соли магния; соли металлов, такие как соли алюминия, соли железа, соли цинка, соли меди, соли никеля или соли кобальта; соли аммония; соли органических аминов, такие как соли трет-октиламина, соли дибензиламина, соли морфолина, соли глюкозамина, соли сложного алкилового эфира фенилглицина, соли этилендиамина, соли N-метилглюкамина, соли гуанидина, соли диэтиламина, соли триэтиламина, соли дициклогексиламина, соли N, N'-дибензилэтилендиамина, соли хлорпрокаина, соли прокаина, соли диэтаноламина, соли N-бензилфенэтиламина, соли пиперазина, соли тетраметиламмония или соли трис(гидроксиметил)аминометана; соли гидрогалогеновой кислоты, такие как гидрофторид, гидрохлорид, гидробромид или гидроиодид; соли неорганических кислот, такие как нитрат, перхлорат, сульфат или фосфат; сульфонаты низших алканов, такие как метансульфонат, трифторметансульфонат или этансульфонат; арилсульфонаты, такие как бензолсульфонат или п-толуолсульфонат; соли органических кислот, такие как ацетат, малат, фумарат, сукцинат, цитрат, тартрат, оксалат или малеат; и соли аминокислот, такие как соли глицина, соли лизина, соли аргинина, соли орнитина, глутамат или аспартат. Данные соли можно получить известным способом. В противном случае олигомер по настоящему изобретению может быть в форме его гидрата.
[0050] В еще одном аспекте олигомер по настоящему изобретению может представлять вилтоларсен или его эквивалент.
Вилтоларсен представляет международное непатентованное название (МНН) NS-065/NCNP-01. В настоящем описании NS-065/ NCNP-01 также упоминается как NS-065/NCNP-01 (вилтоларсен) или вилтоларсен, а также как NS-065/NCNP-01.
[0051] NS-065/NCNP-01 (вилтоларсен) представляет лекарственную субстанцию на основе антисмыслового олигонуклеотида для лечения пациентов с мышечной дистрофией Дюшенна (МДД), поддающейся лечению, включающему пропуск экзона 53. NS-065/NCNP-01 (вилтоларсен) представляет соединение, описанное как «PMO № 8» в патенте США № 9079934 B2. Последовательность оснований NS-065/NCNP-01 (вилтоларсен) показана в SEQ ID NO: 35 (5'-CCTCCGGTTC TGAAGGTGTTC-3'; SEQ ID NO: 3 в настоящем описании), и его 5'-конец представляет ОН-группу. Содержание патента США № 9079934 B2 в полном объеме включено в настоящее описание посредством ссылки. Кроме того, в патенте США № 9079934 В2 раскрывается способ синтеза PMO № 8, а именно NS-065/NCNP-01 (вилтоларсена).
[0052] NS-065/NCNP-01 (вилтоларсен) имеет морфолиновый остов, который, как ожидается, обеспечивает более высокую безопасность, чем фосфоротиоатный олигонуклеотид. Например, разработка фосфоротиоатного олигонуклеотида, дрисаперсена (производства BioMarin), была приостановлена за счет возникших проблем с безопасностью. С другой стороны, морфолино-олигонуклеотид, этеплирсен (Exondys51® производства Sarepta), был одобрен FDA. Дрисаперсен и этеплирсен предназначены для пациентов с МДД, которые поддаются лечению, включающему пропуск экзона 51. Этеплирсен раскрыт в патенте США № 9506058 B2, и содержание которого в полном объеме включено в настоящее описание посредством ссылки.
[0053] Как раскрывается в патенте США № 9079934 B2, NS-065/NCNP-01 (вилтоларсен) был разработан с целью проявления специфической активности пропуска экзона 53 и в результате продукции функционального белка дистрофина у пациентов с МДД со специфическим дефицитом экзонов, включая экзоны 43-52, 45-52, 47-52, 48-52, 49-52, 50-52 или 52. Примеры мутаций, вызывающих МДД, теоретически излечимых посредством пропуска указанного экзона, приведены в таблице 3 Aartsma-Rus et al., 2002. Содержание публикации Aartsma-Rus et al. полностью включено здесь посредством ссылки (Annemieke Aartsma-Rus, Mattie Bremmer-Bout, Anneke A.M. Janson, Johan T. den Dunnen, Gert-Jan B. van Ommen и Judith C.T. van Deutekom, «Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy», Neuromuscular Disorders, Vol. 12, pp. S71-S77 (2002)).
[0054] «Эквивалент» вилтоларсена представляет соединение, которое является общим лекарственным средством с вилтоларсеном или его активным ингредиентом. Такие эквиваленты получили разрешение на производство и продажу в соответствии с Законом о фармацевтической продукции, основываясь на безопасности и эффективности, которые были подтверждены результатами клинических испытаний вилтоларсена, без прохождения клинических испытаний самих эквивалентов, и ожидается, что эквиваленты будут иметь активность пропуска экзона 53, аналогичную активности вилтоларсена. В определенном аспекте «эквивалент» вилтоларсена имеет ту же нуклеотидную последовательность, что и вилтоларсен, и понятие «эквивалент» вилтоларсена включает эквиваленты, в которых все или часть оснований нуклеиновой кислоты, остатков сахара и фосфатных связей эквивалента модифицированы таким же образом, как и в вилтоларсене, или модифицированы иначе, чем в вилтоларсене. Аспект такой модификации является таким же, как вышеописанный аспект. Кроме того, «эквивалент» вилтоларсена может находиться в форме свободного основания, фармацевтически приемлемой соли или гидрата.
[0055] 2. Композиция фармацевтического продукта
Фармацевтическая композиция по настоящему изобретению также может находиться в форме водного раствора. Фармацевтическая композиция по настоящему изобретению может содержать олигомер по настоящему изобретению или его фармацевтически приемлемую соль, или его гидрат в концентрации от 2,5 до 500 мг/мл, от 5 до 450 мг/мл, от 10 до 400 мг/мл, от 15 до 350 мг/мл, от 20 до 300 мг/мл, от 20 до 250 мг/мл, от 20 до 200 мг/мл, от 20 до 150 мг/мл, от 20 до 100 мг/мл, от 20 до 50 мг/мл мл, от 20 до 40 мг/мл, от 20 до 30 мг/мл, от 23 до 27 мг/мл, от 24 до 26 мг/мл или 25 мг/мл. В противном случае фармацевтическая композиция по настоящему изобретению может содержать олигомер по настоящему изобретению или его фармацевтически приемлемую соль, или его гидрат в концентрации от 10 до 100 мг/мл, от 15 до 95 мг/мл, от 20 до 80 мг/мл, от 25 до 75 мг/мл, от 30 до 70 мг/мл, от 35 до 65 мг/мл, от 40 до 60 мг/мл, от 45 до 55 мг/мл, от 47 до 53 мг/мл, от 48 до 52 мг/мл, от 49 до 51 мг/мл или 50 мг/мл.
[0056] В фармацевтической композиции по настоящему изобретению можно изменять концентрацию вилтоларсена в водном растворе. Для приготовления водного раствора вилтоларсена, например, 250 мг вилтоларсена можно смешать с 0,5-100 мл воды (что соответствует концентрации вилтоларсена от 2,5 мг/мл до 500 мг/мл), более предпочтительно с 1-50 мл воды (соответствует концентрации вилтоларсена от 5 мг/мл до 250 мг/мл) и наиболее предпочтительно с 5-10 мл воды (соответствует концентрации вилтоларсена от 25 мг/мл до 50 мг/мл).
[0057] Форма введения фармацевтической композиции по настоящему изобретению представляет внутривенное введение. Возможная лекарственная форма фармацевтической композиции по настоящему изобретению представляет, например, раствор для инъекций (включая жидкость для капельниц).
[0058] Фармацевтическая композиция по настоящему изобретению может дополнительно содержать, по меньшей мере, один компонент, выбранный из агента, регулирующего тоничность, регулятора pH и растворителя.
[0059] Агент, регулирующий тоничность, входящий в состав фармацевтической композиции по настоящему изобретению, может представлять, по меньшей мере, один, выбранный из хлорида натрия, хлорида калия, глюкозы, фруктозы, мальтозы, сахарозы, лактозы, маннита, сорбита, ксилита, трегалозы и глицерина.
[0060] 10 мл водного раствора, содержащего 250 мг вилтоларсена, подходящего для инъекций, могут содержать в качестве агента, регулирующего тоничность, 72,0 мг или более и 108,0 мг или менее хлорида натрия (что соответствует концентрации хлорида натрия от 7,2 мг/мл до 10,8 мг/мл), более предпочтительно от 81,0 мг или более до 99,0 мг или менее хлорида натрия (что соответствует концентрации хлорида натрия от 8,1 мг/мл до 9,9 мг/мл) и наиболее предпочтительно от 85,5 мг или более до 94,5 мг или менее хлорида натрия (что соответствует концентрации хлорида натрия от 8,55 мг/мл до 9,45 мг/мл).
[0061] В качестве агента, регулирующего тоничность, можно использовать фосфатный буфер. Примеры такого фосфатного буфера могут включать цитратный буфер, лактатный буфер и ацетатный буфер. Кроме того, в качестве агента, регулирующего тоничность, также могут использоваться сахара (другие, чем глюкоза). Примеры такого сахара могут включать сорбит и маннит. Когда готовят композицию, содержащую вилтоларсен, то также можно использовать множество агентов, регулирующих тоничность.
[0062] Регулятор pH, включенный в фармацевтическую композицию по настоящему изобретению, может представлять, по меньшей мере, один, выбранный из соляной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, гидроксида натрия, гидроксида калия, триэтаноламина, лимонной кислоты, молочной кислоты, фосфата (гидрофосфата натрия, дигидрофосфата натрия и дигидрофосфата калия) и моноэтаноламина.
[0063] В случае использования фосфатного буфера для олигомера по настоящему изобретению, например, для вилтоларсена, концентрация фосфатного буфера предпочтительно составляет менее 100 мМ. Соответственно, концентрация фосфатного буфера в фармацевтической композиции по настоящему изобретению может быть доведена до 90 мМ или ниже, 80 мМ или ниже, 70 мМ или ниже, 60 мМ или ниже, 50 мМ или ниже, 40 мМ или ниже, 30 мМ или ниже, 20 мМ или ниже, 10 мМ или ниже, или 5 мМ или ниже, или фармацевтическая композиция по настоящему изобретению может не содержать фосфатный буфер.
[0064] Растворителем, входящим в фармацевтическую композицию по настоящему изобретению, может быть вода.
[0065] Значение pH водного раствора, содержащего вилтоларсен, подходящего для инъекции, может составлять 6,0 или выше или 8,5 или ниже, более предпочтительно 6,5 или выше или 8,0 или ниже, наиболее предпочтительно 7,0 или выше или 7,5 или ниже.
Олигомер по настоящему изобретению демонстрирует стабильность в широком диапазоне значений pH. Значение pH фармацевтической композиции по настоящему изобретению предпочтительно доводят до pH 7,0-7,5, 7,0-7,4, 7,1-7,5, 7,1-7,4, 7,2-7,5, 7,2-7,4, 7,3-7,5, 7,3-7,4 или 7,3.
[0066] Кроме того, фармацевтическая композиция по настоящему изобретению может находиться в форме водного раствора, содержащего олигомер по настоящему изобретению в концентрации от 2,5 мг/мл включительно до 500 мг/мл включительно или от 10 мг/мл включительно до 100 мг/мл включительно, и хлорид натрия в концентрации от 8 мг/мл включительно до 10 мг/мл включительно и имеющий значение pH от 7,2 до 7,4.
Альтернативно, фармацевтическая композиция по настоящему изобретению может находиться в форме водного раствора, содержащего олигомер по настоящему изобретению в концентрации 25 мг/мл и имеющего значение рН, доведенное до рН 7,3, без содержания буфера. В этой фармацевтической композиции значение pH регулируется с помощью соляной кислоты и/или гидроксида натрия.
[0067] В качестве примера фармацевтической композиции по настоящему изобретению в следующей таблице 2 показана композиция, содержащая 250 мг вилтоларсена для применения в инъекциях, которая была использована в клинической программе фазы 2 в США/Канаде. Композиция, представленная в таблице 2 ниже, обозначена как «NS-065/NCNP-01 (вилтоларсен) для инъекций 250 мг».
[0068]
«NS-065/NCNP-01 (вилтоларсен) для инъекций 250 мг»
Композиция (250 мг в 10 мл или эквивалентно 25 мг/мл)
(USP=Фармакопея США)
(на ампулу)
(вилтоларсен)
[0069] Кроме того, в композиции «NS-065/NCNP-01 (вилтоларсен) для инъекций 250 мг» количество воды для инъекций и хлорида натрия, используемого в качестве агента, регулирующего тоничность, может быть установлено примерно на половине от каждого количества, и тогда общий объем устанавливается на 5 мл. Фармацевтическая композиция, содержащая вилтоларсен с вышеуказанной композицией в концентрации 50 мг/мл, также включается в настоящее изобретение.
[0070] Объем водного раствора, содержащего вилтоларсен, может быть увеличен или уменьшен при условии, пока концентрации NS-065/NCNP-01 и используемого агента, регулирующего тоничность, а также массовое соотношение между вилтоларсеном и используемым агентом, регулирующим тоничность, поддерживаются на одном уровне, как описано выше.
[0071] Композиция, содержащая вилтоларсен, также может содержать носитель, способствующий доставке вилтоларсена в мышечные ткани. Такой носитель особым образом не ограничивается, при условии, что он является фармацевтически приемлемым носителем. Примеры такого носителя могут включать катионные носители (например, катионные липосомы и катионные полимеры) и носители, в которых используется вирусная оболочка. Примеры катионных липосом могут включать липосомы, содержащие в качестве основных компонентов 2-O-(2-диэтиламиноэтил)карбамоил-1,3-O-диолеоилглицерин и фосфолипид, такой как Oligofectamine® (производства Thermo Fisher Scientific), Lipofectin® (производства Thermo Fisher Scientific), Lipofectamine® (производства Thermo Fisher Scientific), Lipofectamine® 2000 (производства Thermo Fisher Scientific), DMRIE-C (производства Thermo Fisher Scientific), GeneSilencer® (производства Gene Therapy Systems), TransMessenger® (производства QIAGEN) и TransIT-TKO®. Примеры катионных полимеров могут включать JetSI® (производства GeneX India Bioscience) и Jet-PEI® (полиэтиленимин производства GeneX India Bioscience). Примером носителей, использующих вирусную оболочку, может быть GenomeOne® (липосома HVJ-E производства ISHIHARA SANGYO KAISHA, LTD.).
[0072] Кроме того, фармацевтическая композиция по настоящему изобретению может содержать вспомогательное средство для эмульгирования (например, жирную кислоту, содержащую от 6 до 22 атомов углерода, или ее фармацевтически приемлемую соль, альбумин и декстран) и стабилизатор (например, холестерин и фосфатидную кислоту).
[0073] В фармацевтической композиции по настоящему изобретению, содержащей вилтоларсен и носитель, массовое соотношение между вилтоларсеном и носителем (т.е. носитель/вилтоларсен) можно изменять в зависимости от типа используемого носителя. Подходящее массовое соотношение находится в диапазоне от 0,1 до 100, предпочтительно в диапазоне от 1 до 50 и более предпочтительно в диапазоне от 10 до 20.
[0074] Водный раствор, содержащий вилтоларсен, можно вводить пациентам внутривенной капельной инфузией или капельным вливанием.
[0075] 3. Показания/эффекты и применение/дозировка
Когда фармацевтическая композиция по настоящему изобретению используется для лечения, то олигомер по настоящему изобретению или его фармацевтически приемлемую соль, или его гидрат вводят внутривенно пациенту-человеку в дозе от 40 мг/кг/неделю включительно и 80 мг/кг/неделю включительно (в дальнейшем именуемые «дозировка и способ введения по настоящему изобретению»). Альтернативно, дозировка и способ введения по настоящему изобретению могут быть следующими: олигомер по настоящему изобретению, или его фармацевтически приемлемую соль, или его гидрат вводят внутривенно пациенту-человеку в дозе 40 мг/кг/неделю. Альтернативно, дозировка и способ введения по настоящему изобретению могут быть следующими: олигомер по настоящему изобретению или его фармацевтически приемлемую соль, или его гидрат вводят внутривенно пациенту-человеку в дозе 80 мг/кг/неделю. В данном случае числитель «мг» в разовой дозе «мг/кг» указывает количество олигомера по настоящему изобретению, или его фармацевтически приемлемой соли, или его гидрата, которое обозначено единицей «миллиграмм», тогда как знаменатель «кг» указывает в расчете на 1 килограмм массы тела пациента.
[0076] NS-065/NCNP-01 (вилтоларсен) для инъекций 250 мг был разработан для применения внутривенной капельной инфузией, которая проводится один раз в неделю с целью лечения пациентов с МДД, поддающейся лечению, включающему пропуск экзона 53. Клиническая программа фазы 2, проведенная в США/Канаде, была разработана для оценки применения вилтоларсена в двух уровнях доз, а именно в дозе 40 мг/кг/неделю и дозе 80 мг/кг/неделю. В данном случае кг представляет единицу измерения массы тела пациента. Например, когда вилтоларсен вводят в дозе 40 мг/кг/неделю пациенту с массой тела 40 кг, это означает, что пациенту вводят 1600 мг (= 40 мг × 40) вилтоларсена один раз в неделю.
[0077] 4. Заболевание
МДД представляет мышечное заболевание, вызванное мутацией, приводящей к потере функции гена дистрофина, и данное заболевание вызывает потерю белка дистрофина в мышцах пациентов (Hoffman et al., 1987). Ген дистрофина находится на Х-хромосоме и проявляет высокую частоту спонтанных мутаций. Во всех исследованных популяциях по всему миру заболеваемость МДД составляет 1 на 5000 выживших при рождении мальчиков. Согласно недавней оценке, 10,1% из 7149 пациентов с МДД, оцененных в глобальной базе данных пациентов, поддаются лечению, включающему пропуск экзона 53 (Bladen et al., 2015). Клинические симптомы обычно проявляются в младшем школьном возрасте (к 4-6 годам), когда больные мальчики испытывают трудности, выражающиеся в отставании в физическом развитии от сверстников, за счет слабости проксимальных мышц (например, трудности при подъеме по лестнице или беге). Затруднения при вставании с пола наблюдаются у большинства пациентов, которые для достижения положения стоя используют типичный компенсаторный прием Говерса (а именно, используют опору для рук на ногах, коленях и бедрах для достижения положения стоя) (Hoffman E.P., Brown R.H. and Kunkel L.M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919-928 и Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T., Guergueltcheva V., Chan S., Korngut L., Campbell C., Dai Y., Wang J.,  , Brabec P., Lahdetie J., Walter M.C., Schreiber-Katz O., Karcagi V., Garami M., Viswanathan V., Bayat F., Buccella F., Kimura E., Koeks Z., van den Bergen J.C., Rodrigues M., Roxburgh R., Lusakowska A., Kostera-Pruszczyk A., Zimowski J., Santos R., Neagu E., Artemieva S., Rasic V.M., Vojinovic D., Posada M., Bloetzer C., Jeannet P.Y., Joncourt F., Díaz-Manera J., Gallardo E., Karaduman A.A., Topaloğlu H., El Sherif R., Stringer A., Shatillo A.V., Martin A.S., Peay H.L., Bellgard M.I., Kirschner J., Flanigan K.M., Straub V., Bushby K., Verschuuren J., Aartsma-Rus A., Béroud C., Lochmüller H. (2015). The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 36(4): 395-402).
, Brabec P., Lahdetie J., Walter M.C., Schreiber-Katz O., Karcagi V., Garami M., Viswanathan V., Bayat F., Buccella F., Kimura E., Koeks Z., van den Bergen J.C., Rodrigues M., Roxburgh R., Lusakowska A., Kostera-Pruszczyk A., Zimowski J., Santos R., Neagu E., Artemieva S., Rasic V.M., Vojinovic D., Posada M., Bloetzer C., Jeannet P.Y., Joncourt F., Díaz-Manera J., Gallardo E., Karaduman A.A., Topaloğlu H., El Sherif R., Stringer A., Shatillo A.V., Martin A.S., Peay H.L., Bellgard M.I., Kirschner J., Flanigan K.M., Straub V., Bushby K., Verschuuren J., Aartsma-Rus A., Béroud C., Lochmüller H. (2015). The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 36(4): 395-402).
[0078] Мышечные ткани пациентов с МДД демонстрируют хроническое воспаление с приступами мышечной дегенерации и регенерации, ведущими к истощению мышц, инвалидности и ранней смерти. Пациенты обычно теряют способность передвигаться во втором десятилетии жизни и нуждаются в помощи во многих аспектах повседневной жизни к третьему десятилетию. Данное заболевание обычно приводит к смерти в подростковом или раннем взрослом возрасте, хотя использование аппаратов для искусственной вентиляции легких иногда может продлить жизнь до четвертого десятилетия, а иногда и до пятого десятилетия жизни.
[0079] Мутацию гена DMD у субъекта можно выявить с помощью мультиплексной амплификации зонда, зависимой от лигирования (MLPA) (Murugan et al., 2010). Содержание данной статьи Murugan et al. в полном объеме включено здесь посредством ссылки (Sakthivel Murugan S.M., Arthi Chandramohan and Bremadesam Raman Lakshmi, «Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis», Indian Journal of Medical Research, Vol. 132, pp. 303-311 (September 2010)).
[0080] Пациент-человек, представляющий интерес для лечения с использованием фармацевтической композиции по настоящему изобретению (далее именуемый «пациентом, представляющим интерес по настоящему изобретению»), особым образом не ограничивается, при условии, что врачом диагностировано наличие у пациента МДД. В определенном аспекте у пациента может быть мутация, которая приводит к дефициту любого экзона, выбранного из группы, состоящей из экзонов 43-52, 45-52, 47-52, 48-52, 49-52, 50-52 или 52, в гене дистрофина.
[0081] Кроме того, представляющий интерес пациент по настоящему изобретению может быть охарактеризован тем, что экспрессия белка дистрофина до лечения с использованием фармацевтической композиции по настоящему изобретению или олигомера по настоящему изобретению, составляет 1% или ниже по сравнению с экспрессией у здорового субъекта (100%), по данным анализа вестерн-блоттингом или масс-спектрометрией. В данном случае «здоровый субъект» представляет человека, у которого отсутствует заболевание, связанное с белком дистрофина. Уровень экспрессии белка дистрофина у здорового субъекта может представлять общеизвестное значение или значение, полученное у отдельного здорового субъекта. Кроме того, представляющего интерес пациента по настоящему изобретению также можно охарактеризовать тем, что экспрессия белка дистрофина не наблюдается до лечения с использованием фармацевтической композиции по настоящему изобретению или олигомера по настоящему изобретению. Выражение «экспрессия белка дистрофина не наблюдается» означает, что экспрессия белка дистрофина находится почти на том же уровне экспрессии, что и у отрицательного контроля по данным анализа вестерн-блоттингом или масс-спектрометрией, или что экспрессия белка дистрофина находится ниже нижнего предела определения. Кроме того, вестерн-блоттинг и масс-спектрометрия особым образом не ограничиваются, при условии, что они представляют методы, обычно используемые в данной области техники. В качестве примеров вестерн-блоттинга и масс-спектрометрии можно указать экспериментальные методы, применяемые в настоящих примерах.
[0082] 5. Терапевтическое обоснование
МДД представляет тяжелое наследственное мышечное заболевание. Данное заболевание чаще всего возникает, когда трансляция аминокислот вне рамки считывания возникает в результате делеции одного или нескольких экзонов из гена дистрофина. Функциональный белок дистрофин, который важен для проявления мышечной функции, не экспрессируется у пациента в результате такой трансляции аминокислот вне рамки считывания. Менее тяжелая форма заболевания, мышечная дистрофия Беккера (МДБ), чаще всего возникает, когда отсутствие одного или нескольких экзонов в гене дистрофина приводит к трансляции аминокислот внутри рамки считывания из оставшихся экзонов. Пациенты с МДБ обычно имеют более медленное прогрессирование заболевания и более низкую степень инвалидности. Медикаментозное лечение пациентов с МДД обычно включает терапию глюкокортикоидами для замедления проявления симптомов в мышцах конечностей или в мышцах, поддерживающих дыхание. Как указывается в финальном руководстве FDA за февраль 2018 г., существует значительная нерешенная потребность в лечении пациентов с МДД «Duchenne Muscular Dystrophy and Related Dystrophinopathies: Developing Drugs for Treatment Guidance for Industry» (доступно на сайте https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM450229.pdf.).
[0083] Одним из терапевтических подходов к лечению пациентов с МДД является применение стратегии «пропуска экзонов» для продукции функционального белка дистрофина, который может обеспечить у пациентов переход с фенотипа МДД на фенотип BMD. Пропуск экзона позволяет восстановить рамку считывания для аминокислот в результате индуцированного пропуска экзона, следующего за отсутствующим экзоном (Cirak et al., 2011; Voit et al., 2014; Yokota et al., 2012). При пропуске экзона 53 экспрессируется белок дистрофин, который немного короче, чем обычный белок, но который сохраняет частичную функциональную активность. Ожидается, что инъекция NS-065/NCNP-01 (вилтоларсена) 250 мг приведет к смещению фенотипа МДД на более легкое заболевание МДБ, при котором прогрессирование болезни у пациентов замедляется, и качество жизни пациентов улучшается (Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J., Dickson G., Wood M.J., Wilton S.D., Straub V., Kole R., Shrewsbury S.B., Sewry C., Morgan J.E., Bushby K., Muntoni F. (2011). Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378: 595-605., Voit T., Topaloglu H., Straub V., Muntoni F., Deconinck N., Campion G., De Kimpe S.J., Eagle M., Guglieri M., Hood S., Liefaard L., Lourbakos A., Morgan A., Nakielny J., Quarcoo N., Ricotti V., Rolfe K., Servais L., Wardell C., Wilson R., Wright P., Kraus J.E. (2014). Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 13(10): 987-96, и Yokota T., Nakamura A., Nagata T., Saito T., Kobayashi M., Aoki Y., Echigoya Y., Partridge T., Hoffman E.P., Takeda S. (2012). Extensive and Prolonged Restoration of Dystrophin Expression with Vivo-Morpholino-Mediated Multiple Exon Skipping in Dystrophic Dogs. Nucleic Acid Ther 22(5): 306-15).
[0084] Следовательно, фармацевтическую композицию по настоящему изобретению вводят пациенту-человеку с МДД в соответствии с вышеуказанными дозировкой и способом введения по настоящему изобретению, тем самым обеспечивая лечение МДД.
Термин «лечить» используется здесь для обозначения уменьшения симптомов МДД у пациента.
[0085] Лечение с использованием фармацевтической композиции по настоящему изобретению в соответствии с дозировкой и способом введения по настоящему изобретению может обеспечить, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6) (где среднее значение ± стандартное отклонение показано в скобках):
(1) среднее значение уровня экспрессии белка дистрофина в скелетных мышцах пациента увеличивается в 9 или более раз по сравнению с исходным уровнем после введения фармацевтической композиции в течение 24 недель;
(2) изменение скорости, полученной по времени вставания из лежачего положения на спине (TTSTAND), составляет -0,055 раз/с или более, или 0,024±0,075 раз/с или более по сравнению с исходным уровнем на время недели 25 после введения фармацевтической композиции в течение 24 недель;
(3) изменение скорости, полученной по времени бега/ходьбы на 10 метров (TTRW), составляет -0,025 м/с или более, или 0,227±0,251 м/с или более по сравнению с исходным уровнем на время недели 25 после введения фармацевтической композиции в течение 24 недель;
(4) изменение скорости, полученной по времени подъема на 4 ступени (TTCLIMB), составляет -0,060 раз/с или более или 0,032±0,088 раз/с или более по сравнению с исходным уровнем на время недели 25 после введения фармацевтической композиции в течение 24 недель;
(5) изменение балла по шкале амбулаторного обследования North Star (NSAA) составляет -2,2 балла или более, или 0,8±2,9 балла или более по сравнению с исходным уровнем на время недели 25 после введения фармацевтической композиции в течение 24 недель; и
(6) изменение показателя в тесте 6-минутной ходьбы (6MWT) составляет -7,5 м или более, или 28,9±36,3 м по сравнению с исходным уровнем на время недели 25 после введения фармацевтической композиции в течение 24 недель.
[0086] В отношении вышеописанных эффектов (1)-(6), то термин «исходный уровень» означает среднее значение в группе пациентов, не подвергавшихся лечению. Кроме того, термин «изменение», используемый в отношении эффектов (2)-(6), означает изменение средних значений.
[0087] В определенном варианте осуществления, когда фармацевтическую композицию по настоящему изобретению вводят пациентам в возрасте от 7 до 9 лет с мышечной дистрофией Дюшенна в соответствии с вышеуказанными дозировкой и способом введения по настоящему изобретению в течение 84 недель, то, по меньшей мере, может обеспечиваться один эффект, выбранный из группы, состоящей из следующих эффектов (7)-(12):
(7) процент пациентов, которые теряют способность вставать, составляет менее 20% ко времени недели 85 после начала лечения;
(8) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 10% ко времени недели 85 после начала лечения;
(9) процент пациентов, которые теряют способность к самостоятельному передвижению, составляет менее 10% ко времени недели 85 после начала лечения;
(10) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(11) снижение показателя подъема на 4 ступени с возрастом не наблюдается ко времени недели 85 после начала лечения; и
(12) снижение скорости вставания с возрастом не наблюдается ко времени недели 85 после начала лечения.
Неделя, на которой было начато лечение, определяется как первая неделя, и вышеописанные эффекты (7)-(12) могут быть обеспечены на любую временную точку, начиная с недели 1 по неделю 85, с недели 1 по неделю 80, с недели 1 по неделю 75, с недели 1 по неделю 70, с недели 1 по неделю 65 и с недели 1 по неделю 60.
[0088] В определенном варианте осуществления, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (13)-(18), обеспечивается введением фармацевтической композиции по настоящему изобретению пациентам-людям в возрасте от 10 до 12 лет с мышечной дистрофией Дюшенна в соответствии с вышеуказанной дозировкой и способом введения по настоящему изобретению:
(13) процент пациентов, теряющих способность вставать, составляет менее 60% ко времени недели 85 после начала лечения;
(14) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 50% ко времени недели 85 после начала лечения;
(15) процент пациентов, теряющих способность к самостоятельному передвижению, составляет менее 50% ко времени недели 85 после начала лечения;
(16) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(17) наблюдается период, в течение которого скорость подъема на 4 ступени увеличивается ко времени недели 85 после начала лечения; и
(18) наблюдается период, в течение которого скорость вставания увеличивается ко времени недели 85 после начала лечения.
Неделя, на которой было начато лечение, определяется как первая неделя, и вышеописанные эффекты (13)-(18) могут быть обеспечены на любой временной точке, начиная с недели 1 по неделю 85, с недели 1 по неделю 80, с недели 1 по неделю 75, с недели 1 по неделю 70, с недели 1 по неделю 65 и с недели 1 по неделю 60.
[0089] То есть, фармацевтическая композиция по настоящему изобретению может обеспечивать, по меньшей мере, один из вышеописанных эффектов (1)-(18).
[0090] II. Второй вариант осуществления
Во втором варианте осуществления настоящее изобретение обеспечивает способ лечения мышечной дистрофии Дюшенна, включающий внутривенное введение фармацевтической композиции, содержащей антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль, или его гидрат, пациенту-человеку один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/неделю включительно антисмыслового олигомера или его фармацевтически приемлемой соли, или его гидрата (в дальнейшем именуемое «способом лечения по настоящему изобретению»).
Значения отдельных конфигураций, касающихся способа лечения по настоящему изобретению, такие же, как и значения конфигураций, касающихся фармацевтической композиции по настоящему изобретению, которые уже пояснялись в разделе «I. Первый вариант осуществления».
[0091] III. Третий вариант осуществления
Кроме того, в третьем варианте осуществления настоящее изобретение обеспечивает антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях от 36 до 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль или его гидрат для применения в способе лечения пациента-человека с мышечной дистрофией Дюшенна, где:
антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/ кг/неделю включительно (в дальнейшем именуемое «вариант настоящего изобретения с ограниченным применением»).
Значения отдельных конфигураций, касающиеся варианта осуществления с ограниченным применением по настоящему изобретению, такие же, как и значения конфигураций, касающихся фармацевтической композиции по настоящему изобретению, которые уже пояснялись в разделе «I. Первый вариант осуществления».
Кроме того, вариант осуществления настоящего изобретения с ограниченным применением может обеспечить, по меньшей мере, один эффект из эффектов (1)-(18), которые пояснялись в подразделе «5. Терапевтическое обоснование» раздела «I. Первый вариант осуществления».
[0092] IV. Четвертый вариант осуществления
Кроме того, в четвертом варианте осуществления настоящее изобретение обеспечивает применение антисмыслового олигомера, состоящего из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемой соли или его гидрата для производства фармацевтической композиции для лечения пациента-человека с мышечной дистрофией Дюшенна, где:
антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/ неделю включительно (в дальнейшем именуемое «применением по настоящему изобретению «швейцарского типа»).
Значения отдельных конфигураций, касающихся применения по настоящему изобретению «швейцарского типа», такие же, как и значения конфигураций, касающихся фармацевтической композиции по настоящему изобретению, которые уже пояснялись в разделе «I. Первый вариант осуществления».
Кроме того, применение по настоящему изобретению «швейцарского типа» может обеспечить, по меньшей мере, один эффект из эффектов (1)-(18), которые пояснялись в подразделе «5. Терапевтическое обоснование» раздела «I. Первый вариант осуществления».
[0093] Далее настоящее изобретение будет описано более подробно в следующих примерах. Однако настоящее изобретение не ограничивается объемом изобретения, показанным в этих примерах.
Примеры
[0094] Пример получения
(1) Получение 4-{[(2S, 6R)-6-(4-бензамид-2-оксопиримидин-1-ил)-4-тритилморфолин-2-ил]метокси}-4-оксобутановой кислоты на носителе из аминометилполистироловой смолы
Стадия 1. Получение 4-{[(2S,6R)-6-(4-бензамид-2-оксопиримидин-1(2H)-ил)-4-тритилморфолин-2-ил]метокси}-4-оксобутановой кислоты
В атмосфере аргона 22,0 г N-{1-[(2R, 6S)-6-(гидроксиметил)-4-тритилморфолин-2-ил]-2-оксо-1,2-дигидропиримидин-4-ил} бензамида и 7,04 г 4-диметиламинопиридина (4-DMAP) суспендировали в 269 мл дихлорметана, затем к суспензии добавляли 5,76 г янтарного ангидрида, и затем смесь перемешивали при комнатной температуре в течение 3 ч. После этого к реакционному раствору добавляли 40 мл метанола, и затем полученный раствор концентрировали при пониженном давлении. Остаток подвергали экстракции с использованием этилацетата и 0,5 М водного раствора дигидрофосфата калия. Полученный органический слой последовательно промывали 0,5 М водным раствором дигидрофосфата калия, водой и насыщенным раствором соли. Полученный органический слой высушивали над сульфатом натрия и затем концентрировали при пониженном давлении с получением 25,9 г продукта, представляющего интерес.
[0095] Стадия 2. Получение 4-{[(2S,6R)-6-(4-бензамид-2-оксопиримидин-1-ил)-4-тритилморфолин-2-ил]метокси}-4-оксобутановой кислоты на носителе из аминометилполистироловой смолы
4-{[(2S,6R)-6-(4-Бензамид-2-оксопиримидин-1(2H)-ил)-4-тритилморфолин-2-ил]метокси}-4-оксобутановую кислоту (23,5 г) растворяли в 336 мл пиридина (обезвоженного), и затем к раствору добавляли 4,28 г 4-DMAP и 40,3 г 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида. Затем к смеси добавляли 25,0 г аминометилполистироловой смолы, сшитой 1% DVB (производства Tokyo Chemical Industry Co., Ltd., A1543), и 24 мл триэтиламина, и затем полученную таким образом смесь встряхивали при комнатной температуре в течение 4 суток. После завершения реакции смолу собирали фильтрованием. Полученную смолу промывали пиридином, метанолом и дихлорметаном в указанном порядке, и затем высушивали при пониженном давлении. К полученной смоле добавляли 150 мл тетрагидрофурана (обезвоженного), 15 мл уксусного ангидрида и 15 мл 2,6-лутидина, и затем полученную смесь встряхивали при комнатной температуре в течение 2 ч. Смолу собирали фильтрованием и затем промывали пиридином, метанолом и дихлорметаном в указанном порядке с последующим высушиванием при пониженном давлении с получением 33,7 г продукта, представляющего интерес.
В отношении количества загруженного представляющего интерес продукта, то молярное количество тритила на грамм смолы определяли измерением УФ-поглощения при 409 нм в соответствии с известным методом. Количество, нанесенное на смолу, составило 397,4 мкмоль/г.
[0096] Условия УФ-измерения
Прибор: U-2910 (Hitachi, Ltd.)
Растворитель: метансульфоновая кислота.
Длина волны: 265 нм
Значение ε: 45000
[0097] (2) Получение PMO № 8
PMO № 8 нацелен на последовательность в положениях «от 36 до 56» в экзоне 53, группа на его 5'-конце представляет «группу (3)», и нулеотидная последовательность является такой, как показано в SEQ ID NO: 3.
4-{[(2S,6R)-6-(4-бензамид-2-оксопиримидин-1(2H)-ил)-4-тритилморфолин-2-ил]метокси}-4-оксобутановую кислоту (ссылочный пример 1) (2 г (800 мкмоль)), нанесенную на аминометилполистироловую смолу, переносили в реакционный сосуд и затем добавляли 30 мл дихлорметана, после чего полученную смесь выдерживали в течение 30 мин. Затем реакционную смесь дважды промывали 30 мл дихлорметана и начинали следующие синтетические циклы. Желаемое морфолино-мономерное соединение добавляли в каждом цикле с получением нуклеотидной последовательности представляющего интерес соединения.
[0098]
Следует отметить, что смесь трифторуксусной кислоты (2 экв) и триэтиламина (1 экв), растворенная в дихлорметановом растворе, содержащем 1% (об./об.) этанола и 10% (об./об.) 2,2,2-трифторэтанола с получением концентрации 3% (мас./об.), использовали в качестве деблокирующего раствора. В качестве нейтрализующего раствора использовали N, N-диизопропилэтиламин, растворенный в дихлорметановом растворе, содержащем 25% (об./об.) 2-пропанола, с получением концентрации 5% (об./об.). В качестве раствора для сочетания A использовали морфолино-мономерное соединение, растворенное в 1,3-диметил-2-имидазолидиноне, содержащем 10% (об./об.) N, N-диизопропилэтиламина, с получением концентрации 0,15 М. В качестве раствора для сочетания B использовали N, N-диизопропилэтиламин, растворенный в 1,3-диметил-2-имидазолидиноне с получением концентрации 10% (об./об.). В качестве кэппирующего раствора использовали смесь 20% (об./оОб.) уксусного ангидрида и 30% (об./об.) 2,6-лутидина, растворенную в дихлорметане.
[0099] Синтезированный таким образом PMO на носителе из аминометилполистироловой смолы извлекали из реакционного сосуда и затем высушивали при комнатной температуре при пониженном давлении в течение 2 ч или дольше. PMO, находящийся на высушенной аминометилполистироловой смоле, помещали в реакционный сосуд, и затем в него добавляли 200 мл 28% водно-этанольного раствора аммиака (1/4) с последующим перемешиванием полученной смеси при 55°С в течение 15 ч. После этого аминометилполистироловую смолу собирали фильтрованием и затем промывали 50 мл смеси вода-этанол (1/4). Полученный фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в 100 мл смешанного растворителя (4/1) из 20 мМ буфера уксусная кислота-триэтиламин (буфер TEAA) и ацетонитрила, и затем полученный раствор фильтровали через мембранный фильтр. Полученный фильтрат очищали с помощью обращенно-фазовой ВЭЖХ. Используемые условия были следующими.
[0100]
Каждую фракцию анализировали, и представляющий интерес продукт выделяли с использованием 100 мл смеси ацетонитрил-вода (1/1). К нему добавляли 200 мл этанола, и затем полученную смесь концентрировали при пониженном давлении. Полученный продукт дополнительно высушивали при пониженном давлении с получением белого твердого вещества. К полученному твердому веществу добавляли 300 мл 10 мМ водного раствора фосфорной кислоты, так что твердое вещество суспендировалось в нем. Затем к суспензии добавляли 10 мл 2 M водного раствора фосфорной кислоты и затем полученную смесь перемешивали в течение 15 мин. Затем к реакционной смеси для нейтрализации дополнительно добавляли 15 мл 2 М водного раствора гидроксида натрия. Затем к реакционной смеси дополнительно добавляли 15 мл 2 М водного раствора гидроксида натрия для подщелачивания смеси, и затем смесь фильтровали через мембранный фильтр (0,45 мкм). Полученный продукт полностью промывали 100 мл 10 мМ водного раствора гидроксида натрия с получением представляющего интерес продукта в виде водного раствора.
Полученный водный раствор, содержащий продукт, представляющий интерес, очищали на колонке с анионообменной смолой. Используемые условия были следующими.
[0101]
Каждую фракцию анализировали (ВЭЖХ), и представляющий интерес продукт получали в виде водного раствора. К полученному водному раствору добавляли 225 мл 0,1 М фосфатного буфера (pH 6,0) для нейтрализации. Полученную смесь фильтровали через мембранный фильтр (0,45 мкм). Затем проводили ультрафильтрацию в следующих условиях опреснения.
[0102]
Фильтрат концентрировали с получением примерно 250 мл водного раствора. Полученный водный раствор фильтровали через мембранный фильтр (0,45 мкм). Полученный водный раствор лиофилизировали с получением 1,5 г представляющего интерес соединения в форме белого хлопьевидного твердого вещества.
Расчетное значение ESI-TOF-MS: 6924,82
Найденное значение: 6923,54.
[0103] Пример 1. Исследование фазы 2, проведенное в США
Исследование фазы 2 в США по подбору дозы, «Исследование NS-065/NCNP-01-201», было начато в декабре 2016 г. под шифром «IND127474». Данное исследование было проведено под номером NCT02740972 на сайте ClinicalTrials.gov под названием «Исследование безопасности и определение дозы NS-065/NCNP-01 у мальчиков с мышечной дистрофией Дюшенна (МДД)». Протоколы проведения настоящего исследования доступны на сайте https://www.clinicaltrials.gov/ct2/show/study/NCT02740972, и их содержание в полном объеме включено в настоящее описание посредством ссылки. Настоящее исследование было в основном направлено на оценку безопасности NS-065/NCNP-01 (вилтоларсена), который вводили в виде внутривенной капельной инфузии в высокой дозе (80 мг/кг) и в низкой дозе (40 мг/кг) пациентам с мышечной дистрофией Дюшенна (МДД), поддающейся лечению пропуском экзона 53. Кроме того, дальнейшие цели настоящего исследования включают оценку переносимости, мышечной функции и мышечной силы, фармакокинетики и фармакодинамики.
[0104] В частности, настоящее исследование представляло собой фазу 2 многоцентрового, двухпериодного, рандомизированного, плацебо-контролируемого исследования по подбору доз, и NS-065/NCNP-01 (вилтоларсен) вводили капельной инфузией один раз в неделю в течение 24 недель мальчикам в возрасте от 4 лет и старше и младше 10 лет с МДД, для которых не требовался постельный режим. Были включены две когорты по уровням доз. Период 1 исследования проводили двойным слепым методом. Рандомизированные пациенты получали еженедельные внутривенные капельные инфузии NS-065/NCNP-01 (вилтоларсена) или плацебо в течение первых 4 недель их участия (период 1), и затем внутривенные капельные инфузии NS-065/NCNP-01 (вилтоларсена) в течение 5-24 недель (20 недель активного лечения - Период 2). Анализ данных по безопасности в периоде 1 для когорты с дозой 40 мг/кг был завершен до включения пациентов в когорту с дозой 80 мг/кг. Пациенты, завершившие 24-недельное исследование, были допущены к участию в открытом расширенном исследовании.
[0105] Клиническую эффективность оценивали при регулярных посещениях с целью обследования. Всем пациентам проводили мышечную биопсию двуглавой мышцы в исходном периоде и вторую мышечную биопсию проводили на неделе 24.
[0106] Безопасность оценивали сбором данных о нежелательных явлениях (НЯ), результатов лабораторных анализов крови и мочи, электрокардиограмм (ЭКГ), витальных показателей и физических обследований на протяжении всего исследования. Серийные образцы крови отбирали во время четырех визитов для исследования фармакокинетики NS-065/NCNP-01 (вилтоларсена).
[0107] Тип исследования: интервенционное (клиническое испытание)
- Фактическое включение: 16 участников.
- Распределение: рандомизированное.
- Модель вмешательства: параллельное назначение.
- Маскировка: четверка (участник, поставщик медицинских услуг, исследователь, эксперт по оценке результатов).
- Основная цель: лечение.
- Фактическая дата начала исследования: декабрь 2016 г.
- Дата первичного завершения: март 2018 г.
[0108] Группы пациентов и вмешательства
- Экспериментальный препарат: NS-065/NCNP-01 (вилтоларсен) 40 мг/кг.
Шести пациентам с подтвержденным МДД с генетическими делециями, поддающимися лечению, включающему пропуск экзона 53, проводили внутривенную капельную инфузию NS-065/NCNP-01 (вилтоларсена) в дозе 40 мг/кг один раз в неделю в течение 24 недель.
- Экспериментальный препарат: NS-065/NCNP-01 (вилтоларсен) 80 мг/кг.
Шести пациентам с подтвержденным МДД с генетическими делециями, поддающимися лечению, включающему пропуск экзона 53, проводили внутривенную капельную инфузию NS-065/NCNP-01 (вилтоларсена) в дозе 80 мг/кг один раз в неделю в течение 24 недель.
- Компаратор плацебо: плацебо
Двум или трем пациентам в каждой из групп вводили плацебо в виде внутривенной капельной инфузии один раз в неделю в течение 4 недель с последующими 20 неделями открытого лечения.
[0109] Критерии включения
- Мальчики в возрасте ≥ 4 лет и <10 лет.
- Подтвержденные мутации МДД в гене дистрофина, которые поддаются лечению, включающему пропуск экзона 53 для восстановления рамки считывания мРНК дистрофина.
- Возможность самостоятельно передвигаться без вспомогательных приспособлений.
- Пациенты, которые способны соответствовать показателям тестов оценки времени вставания, времени бега/ходьбы и времени подъема.
- Стабильная доза глюкокортикоидов не менее чем в течение 3 месяцев.
[0110] Критерии исключения
- Острое заболевание в течение 4 недель до первой дозы исследуемого лекарства.
- Признаки симптоматической кардиомиопатии. (Бессимптомная сердечная аномалия при обследовании не является исключением.)
- Сильная аллергия или гиперчувствительность к лекарственным препаратам.
- Тяжелые поведенческие или когнитивные проблемы, препятствующие, по мнению исследователя, участвовать в исследовании.
- Предшествующее или текущее заболевание, история болезни, физические данные или лабораторные отклонения, которые, по мнению исследователя, могут повлиять на безопасность, сделать маловероятным правильное завершение лечения и последующее наблюдение или нарушить оценку результатов исследования.
- Пациент принимал любой другой исследуемый препарат в настоящее время или в течение 3 месяцев до начала исследуемого лечения.
- Пациенту была сделана операция в течение 3 месяцев до первого предполагаемого введения NS-065/NCNP-01 (вилтоларсена), или операция была запланирована на любое время в течение всего периода исследования.
- Пациент ранее участвовал в данном исследовании или любом другом исследовании, в ходе которого вводили NS-065/NCNP-01 (вилтоларсен).
[0111] В качестве резюме: исследование представляло 24-недельное, двухкогортное исследование, в котором тестировали дозы 40 мг/кг/ неделю и 80 мг/кг/неделю у 16 пациентов мужского пола, поддающихся лечению, включающему пропуск экзона 53 и стабильную дозу глюкокортикоидов в течение более 3 месяцев. Пациенты из группы плацебо в каждой когорте имели начальный 4-недельный период рандомизации в качестве контроля нежелательных явлений (исходы безопасности). После этого пациенты, получавшие плацебо и активное лечение, продолжали участие в исследовании в течение 20-недельного периода лечения. В исследовании NS-065/NCNP-01-201 оценивали влияние инъекции NS-065/NCNP-01 (вилтоларсена) на de novo экспрессию белка дистрофина после 20-24 недель введения в двух группах доз: низкая доза 40 мг/кг/неделю и высокая доза 80 мг/кг/неделю (как показано на фиг. 1). Целью исследования NS-065/NCNP-01-201 было определение безопасной и эффективной дозы на основе экспрессии de novo белка дистрофина в скелетных мышцах, измеренной с использованием методологии вестерн-блоттинга (WB) (первичная суррогатная конечная точка). Вторичные суррогатные конечные точки включали иммунофлуоресцентное окрашивание (IF) и масс-спектрометрическое детектирование экспрессии de novo белка дистрофина, а также определение уровней мРНК дистрофина de novo с помощью ОТ-ПЦР. Несколько функциональных конечных точек также были включены в качестве вторичных конечных точек в исследование фазы 2. Конечными точками исследования NS-065/NCNP-01-201 были:
[0112] Первичные конечные точки
- Безопасность и переносимость низкой дозы (40 мг/кг/ неделю) и высокой дозы (80 мг/кг/неделю) при внутривенном (в/в) введении NS-065/NCNP-01 (вилтоларсена).
- Эффекты низкой и высокой внутривенной дозы NS-065/NCNP-01 (вилтоларсена) для инъекций определяли как индукцию белка дистрофина в мышцах после 20-24 недель лечения, измеренного с помощью вестерн-блоттинга.
[0113] На фиг. 1 временная шкала для «Высокой дозы: 80 мг/кг/неделю», представленная в нижней половине, смещена на более позднее время от временной шкалы для «Низкой дозы: 40 мг/кг/неделю», представленной в верхней половине для указания того, что введение высокой дозы было начато только после того, как была подтверждена безопасность при введении низкой дозы.
[0114] Содержание измерений:
1. Индукция мРНК дистрофина в мышцах, измеренная с помощью полимеразной цепной реакции в режиме реального времени (ОТ-ПЦР) для анализа мРНК. (Срок: 20-24 недели лечения)
С помощью ОТ-ПЦР определяют измененный сплайсинг РНК дистрофина. В данном методе РНК выделяют из замороженного среза биопсии мышцы и обратно транскрибируют в кДНК. Праймеры для ПЦР конструируют, фланкируя сайт экзона 53 в мРНК дистрофина. Полосы ОТ-ПЦР, соответствующие конкретным вариантам сплайсированной мРНК дистрофина, визуализируют с помощью гель-электрофореза и сравнивают количества различных изоформ мРНК. Если лекарственное средство успешно связывается с мишенью РНК, то экзон 53 исключается из полученных транскриптов мРНК.
2. Индукция белка дистрофина в мышцах по данным вестерн-блоттинга для анализа белка. (Временной интервал: 20-24 недели лечения)
[0115] Первичным показателем биохимического результата является измерение индуцированного лекарственным средством увеличения продукции дистрофина с помощью иммуноблоттинга (вестерн-блоттинг). В иммуноблоттинге дистрофина используются криосрезы биопсийного материала солюбилизированных мышц с фракционированием белков по молекулярной массе с использованием гель-электрофореза (SDS-PAGE), электроблоттингом на нитроцеллюлозу и затем инкубацией нитроцеллюлозы с антителами для детектирования белка дистрофина. Затем сигнал иммуноблоттинга дистрофина из биопсии пациента сравнивали с сигналом стандартной кривой дистрофина на том же геле (смешанный DMD и нормальный контроль). Это обеспечивает полуколичественную оценку содержания дистрофина в мышце.
[0116] Исследование фазы 2 в США: ЭФФЕКТИВНОСТЬ
Показатель исхода 1
Результаты, полученные по показателю исхода 1 (т.е. детектированию уровней мРНК дистрофина de novo с помощью ОТ-ПЦР), были следующими.
[0117]
[0118] Показатель исхода 2
Результаты, полученные по показателю исхода 2 (т.е. измерению de novo экспрессии белка дистрофина в скелетных мышцах с использованием методологии вестерн-блоттинга), показали увеличение в 19,0 раз (для дозы 40 мг/кг/неделю в течение 24 недель) и в 9,8 раза (для дозы 80 мг/кг/неделю в течение 24 недель) от исходного уровня, по результатам сравнения средних значений исходного уровня и средних значений во время лечения, и увеличение в 27,2 раза (для каждой дозы 40 и 80 мг/кг/неделю в течение 24 недель), которое рассчитывали в виде среднего значения увеличения для каждого пациента. Полученные данные обобщены в следующей таблице 8 и на фиг. 2.
[0119]
(0,1-0,4, 0,1)
(3,2-10,3, 2,4)
(0,1-2,6, 0,8)
(1,1-14,4, 4,5)
1) Сравнение среднего значения исходного уровня и среднего значения во время лечения
2) Среднее значение степени повышения у каждого пациента
[0120] Степень восстановления дистрофина с помощью NS-065/ NCNP-01 (вилтоларсена) (40 или 80 мг/кг/неделю, 24 недели) составляла примерно в 3-7 раз или 8,8-9,7 раз выше, чем ранее сообщалось для Exondys 51® (этеплирсена) (в дозе 30 мг/кг/ неделю, 48 и 180 недель), которая поддается лечению, включающему пропуск экзона 51 по умеренной оценке, которое было инициировано Sarepta Therapeutics в 2016 году. В частности, для целей сравнения ниже представлены соответствующие результаты вестерн-блоттинга для Exondys 51® (этеплирсена). У 3 из 16 пациентов, которым вводили NS-065/NCNP-01 (вилтоларсен) в течение 24 недель, уровень дистрофина увеличился более чем на 10% от исходного уровня. Повышение уровня дистрофина на 3% или более наблюдали у 12 из 16 пациентов, которым вводили NS-065/NCNP-01 (вилтоларсен) в течение 24 недель. Hoffman et al. сообщали, что: «Среди пациентов с синдромом Дюшенна (<3% дистрофина от нормального уровня) или дистрофией Беккера и аномальным фенотипом дистрофина наблюдалась четкая корреляция между тяжестью клинического фенотипа и результатами опредления уровня дистрофина». (Eric P. Hoffman et al., «Characterization of Dystrophin in Muscle-Biopsy Specimens from Patients with Duchenne's or Becker's Muscular Dystrophy», N. Engl. J. Med., 318: 1363-1368 (1988).
[0121] - Вестерн-блоттинг с этеплирсеном: увеличение в 2,8 раза, 0,16% → 0,44% (30 мг/кг/неделю в течение 48 недель). («У 12 пациентов с поддающимися оценке результатами уровень дистрофина до лечения составлял 0,16±0,12% (среднее значение±стандартное отклонение) от уровня дистрофина у здорового субъекта и 0,44± 0,43% после 48 недель лечения EXONDYS 51 (p <0,05)») (доступно на сайте https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/PediatricAdvisoryCommittee/UCM557917.pdf.)
- Вестерн-блоттинг с этеплирсеном: увеличение в 3,1 раза, 0,3% → 0,93% (30 мг/кг/неделю в течение 180 недель). («В вестерн-блотах из биопсии короткого разгибателя пальцев (EDB) уровни дистрофина в среднем составляли примерно 0,3% от нормы, но варьировались от неопределяемых до ≈ 1% от нормы или несколько выше». «По результатам вестерн-блоттинга, наиболее точного количественного метода, использованного авторами, средний уровень дистрофина после ~ 3,5 лет лечения этеплирсеном составлял 0,93±0,84% от нормы (среднее значение±стандартное отклонение)») (доступно на сайте https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/peripheralandcentralnervoussystemdrugsadvisorycommittee/ucm497063.pdf.).
- Вестерн-блоттинг с этеплирсеном: 0,93% на неделе 180, пределы значений 0-2,47% (30 мг/кг/неделю в течение 180 недель) (Kenji Rowel Q. Lim et al., «Eteplirsen in the treatment of Duchenne muscular dystrophy», Drug Des. Devel. Ther., 11: 533-545 (2017); https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5338848/).
- Также: «Для этеплирсена в проведенных исследованиях на неделе 180 не было отмечено различий в зависимости доза-ответная реакция от количества дистрофина, продуцированного при введении еженедельной дозы 30 мг/кг и 50 мг/кг, что позволяет предположить, что данный подход не может быть успешным для других экзонов» (Kenji Rowel Q. Lim, et al., «Eteplirsen in the treatment of Duchenne muscular dystrophy», Drug Des. Devel. Ther., 11: 533-545 (2017); https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5338848/).
[0122] Также было показано, что с использованием вилтоларсена были достигнуты лучшие эффекты по сравнению с SRP-4053 (голодирсен; Sarepta), который представляет морфолино-олигонуклеотид, для пациентов с МДД, поддающихся лечению, включающему пропуск экзона 53, в частности, в отношении наблюдаемого повышенного уровня дистрофина. В частности, для целей сравнения ниже приведены соответствующие данные вестерн-блоттинга для SRP-4053 (голодирсен), которые были получены после 48 недель введения, а не через 24 недели введения вилтоларсена.
[0123] - Вестерн-блоттинг с голодирсеном: 0,095% → 1,019% (30 мг/кг в течение 48 недель). («Среднее значение белка дистрофина увеличилось до 1,019% от нормы по сравнению со средним исходным уровнем 0,095% от нормы (p <0,001), измеренным с помощью вестерн-блоттинга, с основной биологической конечной точкой в исследовании, представляющей 10,7-кратное увеличение от исходного уровня» (доступно на сайте http://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-positive-results-its-study).
- Вестерн-блоттинг с голодирсеном: минимум 0,09%, максимум 4,30% (30 мг/кг/неделю в течение 48 недель). Повышение уровня дистрофина на 3% и более наблюдали у 2 из 25 пациентов (22nd International Congress of the World Muscle Society (3-7 October 2017, St. Malo, France)
(http://www.google.co.jp/url?sa=t&source=web&cd=2&ved=2ahUKEwid7ZTQ9OrbAhUETLwKHX3xDk8QFjABegQIARAB&url=http%3A%2F%2Finvestorrelations.sarepta.com%2Fstatic-files%2F64d8d897-2e4a-4119-80b4-115cbae17993&usg=AOvVaw1Amh1Mq1VauvLkPvYWfVdR).
[0124] Исследование фазы 2 в США: БЕЗОПАСНОСТЬ
В отношении данных по безопасности для 16 пациентов с МДД, получавших высокую и низкую дозы NS 065/NCNP-01 (вилтоларсена) (40 мг/кг/неделю и 80 мг/кг/неделю), каких-либо серьезных проблем при проведении исследования не возникло в отношении качества данных или безопасности для участников. Ни один участник не прекратил лечение по истечении 36 недель. В частности, отсутствовали серьезные нежелательные явления, нежелательные явления, приведшие к прекращению приема, и нежелательные явления, связанные с лекарственными препаратами. Все нежелательные явления (НЯ) были легкими или умеренными.
[0125] Пример 2: исследование фазы 2 в США и Канаде
Исследование фазы 2 в США по подбору дозы, «Исследование NS-065/NCNP-01-201», было начато в декабре 2016 г. под кодом «IND127474». Данное исследование было проведено под номером NCT02740972 на сайте ClinicalTrials.gov под названием «Исследование безопасности и определения дозы NS-065/NCNP-01 у мальчиков с мышечной дистрофией Дюшенна (МДД)». Клинический протокол настоящего исследования доступен на сайте https://www.clinicaltrials.gov/ct2/show/study/NCT02740972, и его содержание в полном объеме включено здесь посредством ссылки. Настоящее исследование было в основном направлено на оценку безопасности NS-065/NCNP-01 (вилтоларсена), который вводили в виде внутривенной капельной инфузии в высокой дозе (80 мг/кг) и в низкой дозе (40 мг/кг) пациентам с мышечной дистрофией Дюшенна (МДД), поддающимся лечению с пропуском экзона 53. Кроме того, дальнейшие цели настоящего исследования включают оценку переносимости, мышечной функции и мышечной силы, фармакокинетики и фармакодинамики.
Более конкретно, настоящее исследование представляло собой многоцентровое, двухпериодное, рандомизированное, плацебо-контролируемое исследование по подбору доз, и NS-065/NCNP-01 (вилтоларсен) вводили капельной инфузией один раз в неделю в течение 24 недель мальчикам в возрасте от 4 лет и старше и младше 10 лет с МДД, для которых не требовался постельный режим. Были включены две когорты по уровням доз. Период 1 исследования проводили двойным слепым методом. Рандомизированные пациенты получали еженедельные внутривенные капельные инфузии NS-065/NCNP-01 (вилтоларсена) или плацебо в течение первых 4 недель их участия (период 1), и затем внутривенные капельные инфузии NS-065/NCNP-01 (вилтоларсена) в течение 5-24 недель (20 недель активного лечения - Период 2). Анализ данных по безопасности в периоде 1 для когорты с дозой 40 мг/кг был завершен до включения пациентов в когорту с дозой 80 мг/кг. Пациенты, завершившие 24-недельное исследование, были допущены к участию в открытом расширенном исследовании.
[0126] Клиническую эффективность оценивали при регулярных посещениях с целью обследования. Всем пациентам проводили мышечную биопсию двуглавой мышцы в исходном периоде и вторую мышечную биопсию проводиди на неделе 24.
[0127] Безопасность оценивали сбором данных о нежелательных явлениях (НЯ), результатов лабораторных анализов крови и мочи, электрокардиограмм (ЭКГ), витальных показателей и физических обследований на протяжении всего исследования. Для исследования фармакокинетики NS-065/NCNP-01 (вилтоларсена) во время четырех визитов отбирали серийные образцы крови.
[0128] Тип исследования: интервенционное (клиническое испытание)
- Фактическое включение: 16 участников.
- Распределение: рандомизированное.
- Модель вмешательства: параллельное назначение.
- Маскировка: четверка (участник, поставщик медицинских услуг, исследователь, эксперт по оценке результатов).
- Основная цель: лечение.
- Фактическая дата начала исследования: декабрь 2016 г.
- Дата первичного завершения: март 2018 г.
[0129]
Группы пациентов и вмешательства
- Экспериментальный препарат: NS-065/NCNP-01 (вилтоларсен) 40 мг/кг.
Шести пациентам с подтвержденным дианозом МДД с генетическими делециями, поддающимися лечению, включающему пропуск экзона 53, проводили внутривенную капельную инфузию NS-065/NCNP-01 (вилтоларсен) в дозе 40 мг/кг один раз в неделю в течение 24 недель.
- Экспериментальный препарат: NS-065/NCNP-01 (вилтоларсен) 80 мг/кг.
Пяти пациентам с подтвержденным дианозом МДД с генетическими делециями, поддающимися лечению, включающему пропуск экзона 53, проводили внутривенную капельную инфузию NS-065/NCNP-01 (вилтоларсен) в дозе 80 мг/кг один раз в неделю в течение 24 недель.
- Компаратор плацебо: плацебо
Каждому из двух пациентов в группе с дозой 40 мг/кг и трех пациентов в группе с дозой 80 мг/кг вводили плацебо в виде внутривенной капельной инфузии один раз в неделю в течение 4 недель с последующими 20 неделями открытого лечения.
[0130] Критерии включения
- Мальчики в возрасте ≥ 4 лет на время получения согласия и <10 лет на время первой внутривенной капельной инфузии.
- Подтвержденные мутации МДД в гене дистрофина, которые поддаются лечению, включающему пропуск экзона 53 для восстановления рамки считывания мРНК дистрофина.
- Способность самостоятельно передвигаться без вспомогательных приспособлений.
- Пациенты, способные участвовать в оценке времени вставания (TSTAND), времени бега/ходьбы на 10 м (TTRW) и времени подъема на 4 ступени (TTCLIMB), которые оценивались экспертом по оценке двигательной функции.
- Стабильная доза глюкокортикоидов в течение не менее 3 месяцев.
[0131] Критерии исключения
- Острое заболевание в течение 4 недель до первой дозы исследуемого лекарственного средства.
- Признаки симптоматической кардиомиопатии. (Бессимптомные сердечные аномалии при обследовании не являются исключением.)
- Сильная аллергия или гиперчувствительность на лекарственные средства.
- Тяжелые поведенческие или когнитивные нарушения, препятствующие, по мнению исследователя, участию в исследовании.
- Предшествующее или текущее заболевание, история болезни, физические данные или лабораторные отклонения, которые, по мнению исследователя, могут повлиять на безопасность, сделать маловероятным правильное завершение лечения и последующее наблюдение или нарушить оценку результатов исследования.
- Пациент принимает любой другой исследуемый препарат в настоящее время или принимал в течение 3 месяцев до начала исследуемого лечения.
- Пациенту была сделана операция в течение 3 месяцев до первого предполагаемого введения NS-065/NCNP-01 (вилтоларсена), или операция была запланирована на любое время в течение всего периода исследования.
- Пациент ранее участвовал в данном исследовании или в любом другом исследовании, в ходе которого вводили NS-065/NCNP-01 (вилтоларсен).
[0132] В качестве резюме: исследование представляло 24-недельное, двухкогортное исследование, в котором тестировались дозы 40 мг/кг/неделю и 80 мг/кг/неделю у 16 пациентов мужского пола, поддающихся лечению, включающему пропуск экзона 53 и стабильную дозу глюкокортикоидов в течение более 3 месяцев. Пациенты из группы плацебо в каждой когорте имели начальный 4-недельный период рандомизации в качестве контроля нежелательных явлений (исходы по безопасности). Затем пациенты, получавшие плацебо и активное лечение, продолжали исследование в течение 20-недельного периода лечения. В исследовании NS-065/NCNP-01-201 оценивали влияние NS-065/NCNP-01 (вилтоларсена) для инъекций 250 мг на de novo экспрессию белка дистрофина после 20-24 недель введения в двух уровнях доз: низкая доза 40 мг/кг/неделю и высокая доза 80 мг/кг/неделю (как показано на фиг. 3). Целью исследования NS-065/NCNP-01-201 было определение безопасной и эффективной дозы на основе de novo экспрессии белка дистрофина в скелетных мышцах, измеренной с использованием методологии вестерн-блоттинга (WB) (первичная суррогатная конечная точка). Вторичные суррогатные конечные точки включали иммунофлуоресцентное окрашивание (IF) и масс-спектрометрическое детектирование de novo экспрессии белка дистрофина, а также определение уровней мРНК дистрофина de novo с помощью ОТ-ПЦР. Несколько функциональных конечных точек также были включены в качестве вторичных конечных точек в исследование фазы 2. Конечными точками исследования NS-065/NCNP-01-201 были:
[0133] Первичные конечные точки
- Безопасность и переносимость низкой дозы (40 мг/кг/неделю) и высокой дозы (80 мг/кг/неделю) при внутривенном (в/в) введении NS-065/NCNP-01 (вилтоларсена) для инъекций 250 мг.
- Влияние низкой и высокой внутривенных доз NS-065/NCNP-01 (вилтоларсена) для инъекций 250 мг на индукцию белка дистрофина в мышцах после 20-24 недель лечения, измеренное с помощью вестерн-блоттинга.
- Концентрация препарата в крови NS-065/NCNP-01 (вилтоларсена)
[0134] Вторичные конечные точки
- Оценка влияния низкой и высокой доз при внутривенном введении NS-065/NCNP-01 (вилтоларсена) для инъекций 250 мг на индукцию мРНК дистрофина и белка в мышцах после 20-24 недель лечения, как измерено с помощью ОТ-ПЦР для анализа мРНК и иммунофлуоресцентного окрашивания и масс-спектрометрии для анализа белков.
- Исследование влияния низкой и высокой доз при внутривенном введении NS-065/NCNP-01 (вилтоларсена) для инъекций 250 мг после 20-24 недель лечения на мышечную силу, подвижность и функциональную способность к физическим нагрузкам, которые оценивали по времени вставания (TTSTAND), времени бега/ходьбы на 10 метров (TTRW), времени подъема на 4 ступени (TTCLIMB), результатов амбулаторного обследования North Star (NSAA), теста 6-минутной ходьбы (6MWT) и количественному мышечному тестированию (QMT) по сравнению с соответствующей контрольной группой с естественным течением болезни без активного вмешательства.
[0135] На фиг. 3 временная схема для «Высокой дозы: 80 мг/кг/неделю», представленная в нижней половине, смещена на более позднее время по сравнению с временной схемой для «Низкой дозы: 40 мг/кг/неделю», представленной в верхней половине для указания на то, что введение в высокой дозе было начато только после подтверждения безопасности при введении в низкой дозе.
[0136] Вторичные конечные точки исследования фазы 2 в США/ Канаде: сравнение временных функциональных тестов с внешней контрольной группой с естественным течением болезни без лечения (Международная исследовательская группа по изучению нервно-мышечных заболеваний (CINRG) Исследование естественного течения болезни Дюшенна (DNHS))
Пациентов, включенных в исследование NS-065/NCNP-01-201 фазы 2 в США/Канаде, клинически оценивали в отношении мышечной функции в исходном периоде, на неделе 13 и 25. Корректировку на четырехнедельный период с плацебо не проводили. Данные оценки включали несколько типов временных функциональных тестов: время вставания из положения, лежа на спине (TTSTAND); время бега/ходьбы на 10 метров (TTRW); время подъема на 4 ступени (TTCLIMB); 6-минутная ходьба (6MWT); и амбулаторное обследование North Star (NSAA). Изменения во времени сравнивали с течением заболевания у соответствующих пациентов, которые исследовались в CINRG DNHS.
[0137] CINRG DNHS представляет исследование, в котором за каждым из 440 пациентов с МДД наблюдали в течение нескольких лет (с общей продолжительностью исследования примерно 10 лет). CINRG расшифровывается как Международная исследовательская группа по изучению нервно-мышечных заболеваний и «представляет собой консорциум медицинских и научных исследователей из академических и исследовательских центров, которые разделяют общую цель, заключающуюся в желании положительно повлиять на жизнь пациентов с нервно-мышечными заболеваниями и их семей посредством проведения хорошо контролируемых клинических исследований» (с http://www.cinrgresearch.org/). DNHS расшифровывается как Исследование естественного течения болезни Дюшенна и является «крупнейшим на сегодняшний день проспективным многоцентровым исследованием естественного течения мышечной дистрофии Дюшенна (МДД)», учрежденным CINRG (на сайте http://www.cinrgresearch.org/duchenne-natural-history/). (McDonald C.M., Henricson E.K., Abresch R.T., Han J.J., Escolar D.M., Florence J.M., Duong T., Arrieta A., Clemens P.R., Hoffman E.P. и Cnaan A., CINRG Investigators. The cooperative international neuromuscular research group Duchenne natural history study - a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used," Muscle Nerve., 48(1), 32-54 (2013)., Henricson E.K., Abresch R.T., Cnaan A., Hu F., Duong T., Arrieta A., Han J., Escolar D.M., Florence J.M., Clemens P.R., Hoffman E.P. и McDonald C.M., "CINRG Investigators. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures," Muscle Nerve, 48(1), 55-67 (2013), и McDonald C.M., Henricson E.K., Abresch R.T., Duong T., Joyce N.C., Hu F., Clemens P.R., Hoffman E.P., Cnaan A. and Gordish-Dressman H., "CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study," Lancet, 391(10119), 451-461 (2018)).
[0138] Исследование NS-065/NCNP-01-201 проводили в клинических центрах, участвующих в сети CINRG. Стандартные операционные процедуры (клинические руководства) и протоколы обучения клинических экспертов были очень похожи для клинических испытаний NS-065/NCNP-01-201 и CINRG DNHS. Сопоставление пациентов, включенных в NS-065/NCNP-01-201, и пациентов, включенных в CINRG DNHS, проводили с использованием следующего набора критериев.
- Мальчики в возрасте от 4 до <10,5 лет в исходном периоде.
- Пациенты с данными временных функциональных тестов за, по меньшей мере, 12 месяцев.
- Географический регион: Северная Америка (США и Канада).
- Прием стероидов в течение не менее 3 месяцев и непрерывный прием стероидов в течение 12/24 месяцев наблюдения.
- Невозможность принимать участие в другом клиническом исследовании.
[0139] Запрос в базе данных CINRG DNHS в отношении пациентов, сопоставимых с пациентами, включенными в клиническое исследование NS-065/NCNP-01-201, без указания мутации дистрофина, поддающейся лечению с пропуском экзона 53, но исключая пациентов с делециями экзона 3-7 и тех, у кого имеет место делеция дистрофина, поддающаяся лечению, включающему пропуск экзона 44, привели к 69 субъектам, как показано в таблице 9 ниже. Пациенты с делецией экзона 3-7 и пациенты с делецией дистрофина, поддающейся лечению, включающему пропуск экзона 44, были исключены, поскольку у этих пациентов, как сообщалось, наблюдали сравнительно слабые симптомы.
[0140]
Пациенты, сопоставимые из CINRG DNHS
n=6
n=63
n=69
(4,5-10,3)
(4,0-10,5)
(4,0-10,5)
(16,6-39,1)
(15,1-50,6)
(15,1-50,6)
[0141] В верхнем ряду таблицы 9 «Пропуск экзона 53» означает пациентов с МДД, подлежащих лечению, включающему пропуск экзона 53, тогда как «Без пропуска экзона 53» означает всех других пациентов (но с исключением указанных выше). В нижней строке таблицы 9 «Без крупной делеции/дупликации» включаются другие категории, как точечные мутации (которые не поддаются лечению, включающему пропуск экзона).
[0142] Количество пациентов в CINRG DNHS, у которых имелись данные по тесту 6MWT, было меньше, чем количество пациентов по другим исходам. Это связано с тем, что тест 6MWT был добавлен позднее в протокол CINRG DNHS, что привело к ограниченному объему данных по сравнению с другими временными функциональными тестами.
[0143] Сравнение течения заболевания в период 24 недель между 16 пациентами, включенными в исследование NS-065/NCNP-01-201, и 69 пациентами, включенными в CINRG DNHS, показало, что пациенты в сравнительной группе с естественным течением болезни имели снижение показателей функциональных тестов в течение 24-недель. Напротив, пациенты, включенные в NS-065/NCNP-01-201, показали среднее улучшение временных функциональных тестов за тот же период времени, составляющий 24 недели. Три показателя улучшения достигли статистической значимости: TTRW (на неделе 13 и на неделе 25); TTSTAND (на неделе 25); и 6MWT (на неделе 25). Статистически значимой разницы между дозами 40 мг/кг/неделю и 80 мг/кг/неделю NS 065 NCNP-01 (вилтоларсена) не наблюдали.
[0144] На фиг. 4 (в целом пять графиков и одна таблица) показано сравнение изменений относительно исходного уровня в показателях временных функциональных тестов за 24-недельный период у пациентов, включенных в исследование NS-065/NCNP-01-201 (обозначены как «вилтоларсен» синей сплошной линией), и соответствующих пациентов в CINRG DNHS (обозначены как «DNHS» красной пунктирной линией).
[0145] На пяти графиках на фиг. 4 «вилтоларсен» представляет международное непатентованное название (МНН) NS-065/NCNP-01. Также на всех графиках изменения относительно исходного уровня сравнивали между группами пациентов, которым вводили вилтоларсен, и группами пациентов CINRG DNHS с использованием смешанной модели для анализа повторных измерений (MMRM) на основе ограниченного максимального правдоподобия (REML).
[0146] (1) Временные функциональные тесты
Временные функциональные тесты: TTSTAND, TTRW, TTCLIMB и 6MWT. TTSTAND и TTRW были заранее определены как разные и отдельные показатели исходов и оценивались на основе времени и по 6-балльной шкале. TTSTAND и TTRW также входят в качестве компонентов NSAA. Таким образом, показатели были измерены один раз, и данные использовали в виде отдельно стоящей конечной точки, так и в виде части комбинированного теста NSAA. TTSTAND и TTRW описаны ниже в контексте объединенной шкалы NSAA. TTCLIMB использовали для оценки времени (в секундах), которое требовалось пациенту, чтобы подняться на 4 ступеньки. Следует отметить, что клинический протокол, указанный в вышеуказанном «исследовании фазы 2 США/Канады» некорректно описывает тест TTCLIMB как проводимый в виде компонента NSAA.
[0147] 6MWT представляет широко используемый и признанный тест для многих заболеваний; в настоящем исследовании использовалась версия, адаптированная для применения при МДД. Данный тест считается простым, стандартизированным, несложным и экономичным средством клинической оценки: 1) функционального моторного статуса; и 2) интегральных и глобальных ответов на нагрузки. Для выполнения настоящего теста 2 точки (конусы) устанавливали на расстоянии 25 м друг от друга, и пациентов просили быстро и безопасно передвигаться назад и вперед между конусами в течение 6 мин. Регистрировали общее расстояние в метрах, которое пациент прошел за 6 мин. Клинический эксперт(и) измерял количество шагов, сделанных пациентом на первых 50 м, и общее количество шагов, пройденных пациентом за 6 мин (Craig M. McDonald M.D., Erik K. Henricson M.P.H., Jay J. Han M.D., R. Ted Abresch M.S., Alina Nicorici B.S., Gary L. Elfring M.S., Leone Atkinson M.D., PhD, Allen Reha B.S., Samit Hirawat M.D. and Langdon L. Miller M.D., «The 6-minute Walk Test as a New Outcome Measure in Duchenne Muscular Dystrophy,» Muscle & Nerve, Vol. 41, pp. 500-510, April 2010, Wiley Periodicals, Inc. и Craig M. McDonald MD, Erik K. Henricson M.P.H., R. Ted Abresch M.S., Julaine Florence, PhD, Michelle Eagle, PhD, Eduard Gappmaier, PhD, Allan M. Glanzman D.P.T., Robert Spiegel, MD, Jay Barth, MD, Gary Elfring, MS, Allen Reha, MS, and Stuart W. Peltz, PhD, "The 6-minute Walk Test and Other Clinical Endpoints in Duchenne Muscular Dystrophy: Reliability, Concurrent Validity, and Minimal Clinically Important Differences from a Multicenter Study, Muscle & Nerve, Vol. 48, pp. 357-368, September 2013, Wiley Periodicals, Inc.).
[0148] Количественное мышечное тестирование (QMT) предназначено для измерения генерации мышечной силы во время изометрического сокращения и является хорошо зарекомендовавшим себя методом измерения мышечной слабости при нервно-мышечных заболеваниях. В указанных методах использовали количественную систему оценки мышечной силы CINRG (CQMS). CQMS включает процесс аудиовизуальной обратной связи, который повышает комплаенс детей с МДД. Пациентов помещали на стол для осмотра с системой поддержки спины, чтобы исключить необходимость в ручной стабилизации спины. После однократного практического применения каждый пациент прошел оценку QMT (выполнение 2 тестов; при анализе данных использовали большее из двух значений). QMT проводили посредством регистрации силы в фунтах через прямой компьютерный интерфейс с тензодатчиком. Положения тестирования и порядок проведения испытаний были стандартизированы. Было проведено двустороннее тестирование нижеперечисленных групп мышц (Mayhew J.E., Florence J.M., Mayhew T.P., Henricson E.K., Leshner R.T., Mccarter R.J. et al., «Reliable surrogate outcome measures in multicenter clinical trials of Duchenne muscular dystrophy» Muscle & Nerve, 2007; 35(1): 36-42. Epub 2006/09/14):
- Мышцы груди.
- Локтевые сгибатели (двуглавая мышца).
- Локтевые разгибатели (трехглавая мышца).
- Сгибатели коленного сустава (подколенные мышцы).
- Разгибатели коленного сустава (четырехглавая мышца).
[0149] В QMT наблюдали небольшое среднее снижение силы по всем параметрам в течение 24-недельного периода лечения, за исключением локтевых разгибателей. Локтевые разгибатели показали небольшое увеличение (улучшение) силы. Ни одно из этих изменений от исходного уровня не было статистически значимым на неделе 25.
[0150] Силовые и функциональные тесты выполняли в следующем порядке: TTSTAND, TTRW, TTCLIMB, NSAA, 6MWT и QMT.
[0151] (2) Амбулаторное обследование North Star (NSAA)
NSAA представляет функциональную шкалу, состоящую из 17 пунктов, разработанную для мальчиков с МДД, способных передвигаься не менее чем на 10 м (E.S. Mazzone S. Messina, G. Vasco, M. Main, M. Eagle, A. D'Amico, L. Doglio, L. Politano, F. Cavallaro, S. Frosini, L. Bello, F. Magri, A. Corlatti, E. Zucchini, B. Brancalion, F. Rossi, M. Ferretti, M.G. Motta, M.R. Cecio, A. Berardinelli, P. Alfieri, T. Mongini, A. Pini, G. Astrea, R. Battini, G. Comi, E. Pegoraro, L. Morandi, M. Pane, C. Angelini, C. Bruno, M. Villanova, G. Vita, M.A. Donati, E. Bertini, and E. Mercuri, «Reliability of the North Star Ambulatory Assessment in a multicentric setting», Neuromuscular Disorders, Vol. 19, Issue 7, pp. 458-461, Jul 2009, Elsevier B.V.). С использованием данного оценочного инструмента оценивают функциональную активность, включая стояние, вставание с пола, согласованные шаги, прыжки и бег. Оценка проводится по трехбалльной оценочной шкале: 2=способность нормально выполнять тест; 1=измененный метод или оказание помощи в выполнении теста; и 0=невозможность выполнения теста. Таким образом, общий балл по этим оценкам может варьироваться от 0 (полностью не передвигается) до 34 (нарушения отсутствуют). Регистрировали баллы по отдельным тестам и общий балл. TTSTAND и TTRW применялись в рамках NSAA.
- TTSTAND обычно выполняется во время стандартных клинических обследований пациентов с МДД. Тест использовали для оценки времени, которое требуется пациенту, чтобы подняться лежачего положения на полу в положени стоя. Регистрировали количество секунд, необходимых для выполнения теста, и использовали 6-балльную оценочную шкалу для того, как пациент достиг положения стоя.
- TTRW использовали для оценки времени (в секундах), которое потребовалось пациенту, чтобы пробежать/пройти 10 м (включая 6-балльную шкалу оценки качества бега/ходьбы).
[0152] Исследование фазы 2, проведенное в США/Канаде: безопасность
В отношении данных по безопасности, полученных с включением 16 пациентов с МДД, получавших высокую и низкую дозу NS 065/NCNP-01 (вилтоларсена) (40 мг/кг/неделю или 80 мг/кг/неделю), то какие-либо серьезных проблем при проведении исследования не возникало в отношении качества данных или безопасности для участников. Ни один участник не прекратил лечение по истечении 48 недель. В частности, отсутствовали TEAE (побочные эффекты, возникающие при лечении), которые требовали отмены или снижения дозы NS 065/NCNP-01 (вилтоларсен). Все нежелательные явления (НЯ) были легкими или умеренными.
[0153] Пример 3: влияние pH на стабильность вилтоларсена
(A) Оценка стабильности при pH (1)
Для исследования взаимосвязи между свойствами раствора и стабильностью вилтоларсена, для оценки стабильности использовали буфер Бриттона-Робинсона (pH 3, 4, 5, 6, 7, 8, 9, 10 или 11) вилтоларсена в растворах с различными значениями pH.
[0154] Условия испытания
Концентрация лекарственного препарата в растворе: вилтоларсен 2 мг/мл.
Раствор: буфер Бриттона-Робинсона (pH 3, 4, 5, 6, 7, 8, 9, 10 или 11).
Условия хранения: (1) при 121°C (обработка в автоклаве) в течение 10, 30 или 60 мин; или (2) при 80°C в термостате в течение 4-7 суток.
Конечные точки: внешний вид (подтверждение визуальным наблюдением), pH (pH-метр), тест определения чистоты (метод ВЭЖХ), уровень извлечения (уровень сохранения) (метод ВЭЖХ).
Условия ВЭЖХ:
Скорость потока: 1,0 мл/мин.
Детектор: ультрафиолетовый абсорбциометр (длина волны измерения: 264 нм)
Колонка: трубка из нержавеющей стали с внутренним диаметром 4,6 мм и длиной 15 см, заполненная 3,5 мкм октадецилсилилированным силикагелем для жидкостной хроматографии (Waters, X-bridge C18).
Температура колонки: 60°C
Мобильная фаза:
- 1,42 г гидрофосфата натрия растворяли примерно в 750 мл воды и затем добавляли водный раствор гидроксида натрия для доведения значения pH полученного раствора до 12,0. После этого к раствору добавляли воду с получением объема 1000 мл (раствор P).
- 300 мл ацетонитрила добавляли к 700 мл раствора P, и затем в полученном растворе растворяли 16,12 г бромида тетрабутиламмония (подвижная фаза A).
- 300 мл ацетонитрила добавляли к 300 мл раствора P и затем растворяли в полученном растворе 9,67 г бромида тетрабутиламмония (подвижная фаза B).
- Подача подвижных фаз: подвижные фазы A/B изменяли от (100 об.%/0 об.%) на (0 об.%/100 об.%) в течение 30 мин.
В настоящем примере и следующих примерах процент площади основного пика вилтоларсена (площадь основного пика (%)) показан как значение теста определения чистоты, когда сумма детектированных общих площадей пиков, содержащих примеси, была установлена на 100.
[0155] Результаты испытания
Результаты испытания приведены в таблице 10 и на фиг.5. «ST» означает водный раствор вилтоларсена, разбавленный очищенной водой в той же концентрации лекарственного препарата в растворе, и его готовили в каждом анализе (и не хранили). В результате было обнаружено, что вилтоларсен нестабилен в растворе с кислой средой, но относительно стабилен в нейтральном или слабощелочном растворе.
[0156]
Результаты оценки стабильности вилтоларсена в буфере Бриттона-Робинсона (pH от 3 до 11)
[0157] (B) Оценка стабильности pH (2)
Для дальнейшего исследования области pH, в которой вилтоларсен стабилен, для оценки стабильности вилтоларсена в растворах использовали калий-фосфат-боратный буфер (pH 6,0, 6,5, 7,0, 7,5, 8,0, 8,5, 9,0 или 9,2) с различными значениями pH (от 6 до 9).
[0158] Условия испытания
Концентрация лекарственного препарата в растворе: вилтоларсен 2 мг/мл.
Раствор: калий-фосфат-боратный буфер (pH 6,0, 6,5, 7,0, 7,5, 8,0, 8,5, 9,0 или 9,2).
Условия хранения: (1) при 121°C (обработка в автоклаве) в течение 10, 30 или 60 мин; или (2) при 80°C в термостате в течение 1, 4, 7 или 14 суток.
Конечные точки: внешний вид (подтверждение визуальным наблюдением), pH (pH-метр), тест на чистоту (метод ВЭЖХ), уровень извлечения (уровень сохранения) (метод ВЭЖХ).
Условия ВЭЖХ:
Такие же, как в предыдущем пункте (A), за исключением того, что температура колонки была установлена на уровне 50°C.
[0159] Результаты испытания
Результаты испытания приведены в таблице 11 и на фиг. 6. В результате было установленор, что вилтоларсен был наиболее стабильным в растворе с pH от 7 до 7,5.
[0160]
Результаты оценки стабильности вилтоларсена в калий-фосфат-боратном буфере (pH от 6 до 9)
[0161]
Пример 4: влияние регулятора pH на стабильность состава
Настоящий пример направлен на выбор регулятора pH для доведения раствора вилтоларсена для инъекций до стабильного диапазона значений pH (pH от 7 до 7,5). Различные типы регуляторов pH (10 мМ KH2PO4, 10 мМ Na2HPO4, 10 мМ KH2PO4-Na2HPO4 или 100 мМ KH2PO4-Na2HPO4) добавляли к раствору вилтоларсена с концентрацией 10 мг/мл для доведения pH раствора вилтоларсена до стабильного значения. Затем оценивали стабильность растворов вилтоларсена с концентрацией 10 мг/мл.
[0162] Условия испытания
Концентрация лекарственного препарата в растворе: вилтоларсен 10 мг/мл.
Состав раствора лекарственного препарата: как показано в следующей таблице 12.
Условия хранения: при 121°C (обработка в автоклаве) от 30 до 60 мин.
Конечные точки: внешний вид (подтверждение визуальным наблюдением), pH (pH-метр), тест на чистоту (метод ВЭЖХ), уровень извлечения (уровень сохранения) (метод ВЭЖХ).
[0163]
Состав раствора лекарственного препарата
(HCl/NaOH)
KH2PO4
Na2HPO4
KH2PO4-
Na2HPO4
KH2PO4-
Na2HPO4
[0164] Результаты испытания
Результаты испытания приведены в таблице 13 и на фиг. 7. В результате было установлено, что даже без использования буферов вилтоларсен был стабильным на уровне, эквивалентном или выше чем в случае использования фосфатного буфера. В частности, без использования буферов вилтоларсен был более стабильным, чем в фосфатном буфере с высокой концентрацией солей (100 мМ KH2PO4-Na2HPO4 в данном примере).
[0165]
Результаты исследования регуляторов рН
KH2PO4
Na2HPO4
KH2PO4
Na2HPO4
KH2PO4
Na2HPO4
[0166] Пример 5: взаимосвязь между буфером и образованием мультимеров
Вилтоларсен может образовывать мультимеры в зависимости от условий хранения, что, в свою очередь, может привести к снижению чистоты мономеров. В данном примере 0,9% хлорид натрия добавляли в качестве агента, регулирующего тоничность, к раствору лекарственного препарата вилтоларсена с концентрацией 50 мг/мл, и затем к нему дополнительно добавляли различные типы буферов. Затем определяли образование мультимеров.
[0167] Условия испытания
Концентрация лекарственного препарата в растворе: вилтоларсен 50 мг/мл.
Условия хранения: при 60°С в термостате в течение 5 суток.
Конечные точки: мультимеры (метод ВЭЖХ, колонка SEC).
Условия анализа:
Детектор: ультрафиолетовый абсорбциометр (длина волны измерения: 260 нм)
Колонка: трубку из нержавеющей стали, имеющая внутренний диаметр 7,8 мм и длину 30 см, заполняли 7 мкм гелем винилового полимера на основе стирола для жидкостной хроматографии; и две такие колонки последовательно соединяли (TSK gel G3000 PWXL, 7 мкм, 7,8 мм × 30 см, Tosoh Corporation).
Температура колонки: 25°C
Подвижная фаза: 15,6 г дигидрофосфата натрия дигидрата растворяли в 750 мл воды и затем добавляли раствор образца гидроксида натрия для доведения значения pH раствора до pH 7,3. Затем к раствору добавляли воду с получением объема 1000 мл. После этого к 800 мл полученного раствора добавляли 200 мл ацетонитрила.
Скорость потока: скорость потока регулировали таким образом, чтобы время удерживания мономеров вилтоларсена составляло примерно 21 мин.
Диапазон измерения площадей: до пика мономеров вилтоларсена.
[0168] Результаты испытания
Результаты испытания приведены в таблице 14. В результате было установлено, что образование мультимеров увеличивалось при добавлении 50 мМ фосфатного буфера (дигидрофосфат натрия дигидрат-гидрофосфат натрия: NaH2PO4∙2H2O-Na2HPO4) или цитратного буфера (цитрат натрия дигидрат).
[0169]
Результаты оценки образования мульмеров (хранили при 60°C)
(мМ)
[0170] Пример 6: исследования растворимости (100 мг/мл)
Исследовали повышение концентрации инъекционного раствора.
[0171] Условия испытания
Раствор лекарственного препарата: следующие 2 типа
[0172]
Раствор лекарственного препарата (2 типа)
(HCl, NaOH)
KH2PO4-Na2HPO4
Фильтр для фильтрации: (Millex GV, 0,22 мкм, 33 мм, Millipore)
Конечные точки: внешний вид (подтверждение визуальным наблюдением), pH (pH-метр), тест поередления чистоты (метод ВЭЖХ), уровень извлечения после фильтрования (метод ВЭЖХ).
[0173] Результаты испытания
Результаты испытания приведены в таблице 16. Был приготовлен раствор вилтоларсена для инъекций 100 мг/мл. Приготовленный раствор вилтоларсена для инъекций не имел проблем с растворимостью или фильтрованием с использованием фильтра для стерилизации, и это был прозрачный и бесцветный раствор. После хранения раствора в холодном месте раствор стал вязким, но внешний вид не изменился. На основании этих результатов было обнаружено, что можно готовить раствор для инъекций с концентрацией 100 мг/мл.
[0174]
Результаты исследования высококонцентрированного инъекционного раствора
Без буфера
10 мМ фосфатный буфер
[0175] Пример 7: исследование растворимости (50 мг/мл)
Оценивали растворимость лекарственного препарата в растворе с концентрацией 50 мг/мл. Готовили раствор лекарственного препарата и оценивали его растворимость и фильтрование с использованием фильтра для стерилизации.
[0176] Условия испытания
Раствор лекарственного препарата:
[0177]
(HCl, NaOH)
Фильтр для фильтрования: изготовлен из PVDF (Millidisk картриджный фильтр, 0,22 мкм, MCGL40S03/one+MCGL40S03/two, Merck)
Конечные точки: внешний вид (подтверждение визуальным наблюдением), pH (pH-метр), тест определения чистоты (метод ВЭЖХ), количественное определение (метод ВЭЖХ).
[0178] Результаты испытания
Результаты испытания приведены в таблице 18. В результате раствор лекарственного препарата 50 мг/мл был прозрачным и бесцветным раствором, и не было изменений в содержании до и после фильрования. На основе этих результатов было установлено, что можно приготовить раствор для инъекций с концентрацией 50 мг/мл, и какие-либо проблемы с масштабированием отсутствуют.
[0179]
Результаты оценки
(родственные соединения)
RRT=0,85
RRT=0,91
RRT=1,07
RRT=1,24
Другие индивидуальные соединения
Общее количество родственных соединений
1,74%
0,72%
2,09%
0,31%
0,13%
5,39%
1,74%
0,70%
1,99%
0,30%
0,12%
5,26%
[0180] В следующих примерах 8-10 проводили испытания образцов, полученных от пациентов, которые участвовали в клиническом исследовании, описанном в примере 2, или самих пациентов, которые участвовали в клиническом исследовании, описанном в примере 2.
Пример 8: количественное определение белка дистрофина масс-спектрометрией
(Экспериментальные методы)
Протокол: биологический метод анализа
Образец подвергали электрофорезу в SDS-PAGE для отделения белка, и затем отделенный белок подвергали расщеплению в геле с использованием трипсина с получением пептидных фрагментов. Пептидные фрагменты экстрагировали из среза геля и затем высушивали. Затем пептиды повторно растворяли, и после этого проводили идентификацию и количественную оценку белка дистрофина с помощью ВЭЖХ-МС/МС с использованием обращенно-фазовой колонки. Когда количество белка дистрофина в нормальном контроле устанавливали на 100%, то диапазон калибровочной кривой составлял от 1% до 25%. Концентрацию рассчитывали по соотношению площадей пиков с использованием филамина С в качестве стандартизованного белка. Для уравнения регрессии применяли метод наименьших квадратов и получали уравнение: y=mx+b (y: соотношение площадей пиков, x:% дистрофина).
[0181] Расщепление в геле
Образец, подлежащий анализу с использованием ЖХ-МС/МС, получали разделением белка с помощью SDS-PAGE (SDS: додецилсульфат натрия, PAGE: электрофорез в полиакриламидном геле) в зависимости от молекулярной массы и отделенный белок подвергали расщеплению в геле. В каждом геле было 11 образцов, а именно: аналиты, использованные для построения калибровочной кривой (0,0%, 1,0%, 3,0%, 10,0% и 25,0% дистрофина), 12,5 мкг белка (SILAC), экстрагированного из клеток миотуб человека, в которые были добавлены стабильные, меченые изотопами аминокислоты и которые затем культивировали (SILAC), холостой образец и 4 образца для клинических испытаний. В геле было 12 дорожек, и оставшуюся одну дорожку использовали для маркера молекулярной массы. Аналит получали смешиванием белковых экстрактов из 5 типов биопсийного материала мышц без МДД и 2 типов биопсийного материала мышц при МДД друг с другом. Биопсийный материал мышц при МДД получали из Бингемтонского университета, и его этическая экспертиза была проведена. Перед выполнением настоящего примера количество дистрофина в каждом биопсийном материале мышцы было предварительно определено вестерн-блоттингом. Используя устройство для подготовки криосрезов, из биопсийного материала мышцы получали 70 последовательных срезов толщиной 10 мкм каждый. Срез мышцы переносили в микропробирку, предварительно охлажденную на сухом льду. Используя буфер RIPA, содержащий ингибитор протеаз/ фосфатаз Thermo Scientific, из мышечного среза экстрагировали белок. Концентрацию белка в экстракте определяли количественно с использованием набора для анализа белка BCA (Pierce). Каждый образец для электрофореза содержал 50 мкг белка и был приготовлен добавлением 12,5 мкг SILAC. К аналиту и клиническому образцу добавляли экстракт SILAC в качестве внутреннего стандарта для получения % дистрофина. С использованием NuPAGE 3-8% Tris-Acetate геля проводили электрофорез при 150 В в течение 75 мин. Гель-электрофорез выполняли дважды, используя два геля (гель А и гель В) для одного образца. Гель после электрофореза иммобилизовали с использованием смеси метанол:вода:уксусная кислота (50:45:5) в течение 30 мин с последующим обменом с водой и затем дважды проводили регидратацию. После этого проводили окрашивание кумасси синим в течение 1 ч. Обесцвечивание геля проводили при 4°C в течение ночи. Гель, в котором присутствовал белок дистрофин в диапазоне от 460 кДа до 268 кДа в соответствии с маркером молекулярной массы, вырезали и затем дважды промывали водой:ацетонитрилом (50:50). Расщепление в геле выполняли с использованием трипсина (Gold для масс-спектрометрии, Promega Corporation) с получением пептидных фрагментов, и затем полученные пептидные фрагменты высушивали вакуумным центрифугированием. Пептидные фрагменты дистрофина хранили при -80°C, и затем использовали в анализе согласно Q Exactive Nano-LC-MS/MS.
[0182] Тестируемый образец
В клиническом исследовании приняли участие шестнадцать пациентов. Образцы биопсии мышц получали от каждого из 16 пациентов до и после введения препарата. Шестьдесят четыре образца готовили из отобранных 32 образцов в результате дублирования и затем анализировали. Кроме того, поскольку 8 образцов, полученных из 4 образцов в результате дублирования, были подвергнуты повторному анализу, то в целом было анализировано 72 образца.
[0183] Анализ ЖХ-МС/МС
Полученные пептидные фрагменты анализировали методом жидкостной хроматографии-масс-спектрометрии (LC-MS/MS), в котором систему для высокоэффективной жидкостной хроматографии Dionex Ultimate 3000 RSLCnano (Thermo Fisher Scientific) объединяли с масс-спектрометрическим устройством Q Exactive Plus (HRMS: масс-спектрометр высокого разрешения) (Thermo Fisher Scientific).
Высушенные пептидные фрагменты повторно растворяли в 2% ацетонитриле (ACN) + 0,1% трифторуксусной кислоте (TFA). Для введения образца в систему ЖХ-МС/МС использовали петлю для ввода 5 мкл, петлю для образца Dinoex nanoViper (Thermo Fisher Scientific).
[0184] Жидкостную хроматографию проводили в следующих условиях.
Колонка для анализа: колонка с обращенной фазой, Acclaim Pepmap RSLC C18, 15 см × 75 мкм, размер частиц: 3 мкм, EASY SPRAY (Thermo Fisher)
Температура аналитической колонки: 50°C
Подвижная фаза A: 1% муравьиная кислота (HCOOH); подвижная фаза B: 0,1% муравьиной кислоты в ACN
Скорость потока: 0,500 мкл/мин (NC)
Режим ввода образца: неполная петля (размер петли: 5 мкл)
Объем вводимой пробы: 1,00-4,00 мкл
Температура автосамплера: 10°C
Продолжительность: 40,00 мин
[0185]
Установка насоса для градиента NC
[0186] Устройство для масс-спектрометрии использовали в следующих условиях.
Источник ионов: Thermo Fisher EASY-Spray
Ионный режим: катионный
Сканирование: мониторинг параллельных реакций
Ширина хроматографического пика (полная ширина на половине максимума): 10 сек
Минимальное общее время цикла: 40 мин
Разрешение: 17500
Автоматическая регулировка усиления мишени: 1e5
Максимальное время ввода пробы: 50 миллисекунд
Ширина разделения квадрупольного масс-спектрометра: 1,0 м/z ед.
Данные спектра: профиль
Допуск по массе: 10 ppm
Типичные настраиваемые параметры (файл настройки дистрофин)
Напряжение распыления: 1,8 кВ
Капиллярная температура: 275°C
S-Lens RF-Уровень: 70
[0187]
Перечень отбора масс
старт (мин)
стоп (мин)
[0188]
Прогнозируемое время удерживания
[0189]
Порядок инжектирования образцов, подвергнутых электрофорезу в том же геле, в ЖХ-МС/МС
Образец, исключенный из анализа: SILAC, разбавленный холостым образцом.
[0190] Данные собирали с хроматограмм с использованием программного обеспечения LC Quan версии 3.0 производства Thermo Scientific. Допуск по массе устанавливали на уровне 20 ppm, и алгоритм интегрирования был установлен на ICIS. Когда площадь пика дистрофина в аналите 1% или других аналитах составляла 10000 или меньше, то обращали внимание на следующее. То есть, подтверждали, что как для дистрофина, так и для филамина C соотношение площадей пиков между полученными пептидами и мечеными пептидами является надежным цифровым значением, которое не нарушает % дистрофина по сравнению с аналитом 0%.
Площадь пика дистрофина и филамина С рассчитывали суммированием площадей пиков, соответствующих ион-продуктам. Площадь пика дистрофина получали из одного типа пептидного фрагмента дистрофина (т.е. аминокислотной последовательности DYST2: IFLTEQPLEGLEK (SEQ ID NO: 4)). Площадь пика филамина C была установлена на среднем значении для двух типов пептидных фрагментов филамина C (т.е. аминокислотная последовательность FILC1: VAVGQEQAFSVNTR (SEQ ID NO: 6) и аминокислотная последовательность FILC2: SPFVVNVAPPLDLSK (SEQ ID NO: 8)).
[0191]
(SEQ ID NO: 4)
(SEQ ID NO: 5)
(SEQ ID NO: 6)
[0192] Уровень белка дистрофина (соотношение площадей пиков дистрофина и филамина С) в аналитах и индивидуальных клинических образцах рассчитывали согласно следующему уравнению. Поскольку дистрофин детектировался даже в аналите 0%, то корректировку выполняли вычитанием цифрового значения уровня белка дистрофина в аналите 0% из цифрового значения уровня белка дистрофина в каждом аналите. Используя шаблон Excel, получали линию регрессии цифровых значений % дистрофина и уровней белка дистрофина из аналитов для каждого геля, и затем получали % дистрофина в клинических образцах из цифровых значений уровней белка дистрофина в клинических образцах.
Уравнение 1
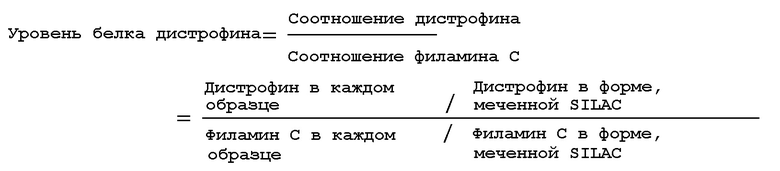
[0193]
(Рассчитано из цифрового значения уровня белка дистрофина, полученного делением соотношения площадей пиков Dyst 2 и меченного Dyst 2 на соотношение площадей пиков FilC и меченного FilC, и линейной регрессией % дистрофина в аналитах)
[0194] Результаты
% Дистрофина в анализированных индивидуальных образцах определяли как приемлемый или нет в соответствии с заранее определенными критериями приемлемости. Среди измеренных 72 образцов 60 образцов удовлетворяли указанным критериям. Результаты измерений представлены в таблицах 25 и 26. Индивидуальные образцы анализировали с использованием дублированных образцов. Однако в отношении 4 образцов, полученных от двух пациентов в группе с дозой 40 мг/кг (пациенты E и F в таблице 25) до и после введения, поскольку один образец (гель B) не удовлетворял критериям, измеренное значение было только одним из другого образца (гель А). Иными словами, в группе с дозой 40 мг/кг в 16 образцах, полученных из 8 образцов до введения препарата, можно было количественно оценить 14 образцов. Один образец показал 1% дистрофина, и в 11 образцах был опредлен уровень, который был ниже нижнего предела количественного определения. Кроме того, среди 16 образцов, полученных из 8 образцов, на неделе 25 можно было количественно оценить 14 образцов. Один образец показал результат ниже нижнего предела количественного определения для обоих из двух измерений. Кроме этого, было обнаружено 1% или более дистрофина.
В случае группы, получавшей дозу 80 мг/кг, то все 16 образцов, которые были получены из 8 образцов до введения, оказались ниже нижнего предела количественного определения. Кроме того, во всех 16 образцах из 8 образцов на неделе 25 дистрофин был обнаружен на уровне, превышающем предел количественного определения, и обнаруженное количество в среднем составляло 4,2%.
Из вышеприведенных результатов было подтверждено, что экспрессия белка дистрофина восстанавливается при введении вилтоларсена в дозах 40 мг/кг и 80 мг/кг.
[0195]
Концентрации белка дистрофина в экстрактах образцов в клинических испытаниях из биопсий мышц человека, рассчитанные с использованием DYST_2
Когорта 1 (группа с дозой 40 мг/кг)
(гель A)
(гель B)
BLQ: ниже нижнего предела количественного определения (1%) .
[0196]
Концентрации белка дистрофина в экстрактах образцов в клинических испытаниях из биопсий мышц человека, рассчитанные с использованием DYST_2
Когорта 2 (группа с дозой 80 мг/кг)
(гель A)
(гель B)
*: поскольку значение площади пика аналита дистрофина 1% было низким, то нижний предел количественного определения стал ниже 3%.
[0197] Пример 9
Тестирование двигательной функции (на неделе 13 и 25 (с момента первоначального введения прошло 12 недель и 24 недели)
(Экспериментальный метод)
Двигательную функцию для данной группы лекарственного средства оценивали ее сравнением с группой естественного течения болезни, использованной в качестве контроля. Группа с естественным течением болезни была выбрана на основе базы данных из исследования под названием «Естественное течение болезни Дюшенна» (CINRG DNHS), проведенного Международной исследовательской группой по изучению нервно-мышечных заболеваний (CINRG), которая является сетью клинических исследований мышечной дистрофии в США.
CINRG DNHS представляет продольное исследование естественного течения болезни, в котором каждый из 440 пациентов мужского пола с МДД наблюдался по целевымх показателям в течение нескольких лет, данные собирались в период с 2006 по 2016 гг., и пациенты приходили в больницу в исходном периоде, четыре раза в первый год, два раза во второй год, и затем один раз в год в течение максимального периода, составляющего 10 лет. При каждом посещении больницы пациенты проходили временной функциональный тест, тест на мышечную силу, функциональный тест с использованием вопросника, тест на функцию легких и оценку качества жизни. Был проведен 201 тест в организациях, относящихся к CINRG, и стандартные операционные процедуры (СОП) и протоколы обучения клинических экспертов были согласованы между обоими испытаниями.
[0198] В качестве контроля в настоящем исследовании были отобраны пациенты из CINRG DNHS, которые удовлетворяли следующим критериям, включая основные критерии регистрации, такие как возраст, статус использования стероидов и место проведения 201 исследования.
- Наличие данных временных функциональных тестов в течение не менее 12 месяцев [требовались данные по времени подъема из лежачего положения (TTSTAND), времени подъема на 4 ступеньки (TTCLIMB) и времени бега/ходьбы на 10 м (TTRW) в исходном периоде].
- Пациенты в возрасте от 4 до <10 лет в исходном периоде.
- Географический регион: Северная Америка (США и Канада).
- Принимали кортикостероиды в течение не менее 3 месяцев и непрерывно применяли кортикостероиды в течение 12-24 месяцев наблюдения.
- Пациенты, не зарегистрированные одновременно в участии в других клинических исследованиях в отношении других агентов, связанных с пропуском экзонов.
- Пациенты удовлетворяют следующим генетическим критериям приемлемости.
Критерии включения:
- Пациенты с результатами генетических тестов
- Пациенты с дупликационной мутацией
- Пациенты с несмысловой или minute мутацией, вызывающей сдвиг рамки считывания
Критерии исключения:
- Пациенты, имеющие мутацию между промотором и экзоном 8 (эти пациенты были исключены, поскольку сообщалось о медленном прогрессировании заболевания у них ((1) Hum. Mutat., 2018; 39: 1193-1202 и (2) Hum. Mutat., 2008; 29(5)): 728-37).
- Пациенты, поддающиеся лечению, включающему пропуск экзона 44 (эти пациенты были исключены, поскольку сообщалось о медленном прогрессировании заболевания у них ((1) Hum. Mutat., 2018; 39: 1193-1202).
- Пациенты, имеющие мутацию внутри рамки считывания.
[0199] Следовательно, 65 пациентов мужского пола с МДД соответствовали описанным выше критериям. Среди пациентов 9 пациентов с МДД подверглись лечению с пропуском экзона 53 (группа с пропуском экзона 53), и 56 пациентов с МДД не поддавались лечению с пропуском экзона 53 (группа без пропуска экзона 53).
[0200]
Параметры группы, получавшей вилтоларсен, и контрольной группы с естественным течением болезни CINRG (DNHS)
n=16
n=56
n=65
Среднее значение
(мин.-макс.)
(4,3-9,8)
(4,5-7,8)
(4,2-9,6)
(4,2-9,6)
Среднее значение
(мин.-макс.)
(14,9-35,4)
(16,6-28,1)
(14,8-38,7)
(14,8-38,7)
[0201] Результаты
Изменения показателей временных функциональных тестов [например, тесте 6-минутной ходьбы (6MWT), оценки скорости по времени вставания (TTSTAND), скорости по времени подъема на 4 ступени (TTCLIMB) и скорости по времени бега/ходьбы на 10 м (TTRW)] и амбулаторном обследовании North Star (NSAA) на неделе 13 и 25 относительно периода до введения или исходного уровня сравнивали между 16 субъектами, получавшими вилтоларсен (группа, принимавшая вилтоларсен), и 65 пациентами в группе естественного течения болезни CINRG DNHS (группа DNHS).
В методе анализа, использованном в настоящем примере, «значение до введения» или исходный уровень устанавливали в виде ковариаты, которая является исходным фактором, влияющим на результаты, данные, полученные на неделе 13 и на неделе 25, использовали в качестве значений, повторно измеренных от конкретных субъектов (повторные измерения), и проводили анализ MMRM. В результате группа, получавшая вилтоларсен по настоящему примеру, имела достоверные улучшения в тесте 6-минутной ходьбы и скорости по времени бега/ходьбы на 10 м. Более того, группа с введением данного лекарственного средства имела улучшения в большей степени, чем группа с естественным течением, даже с точки зрения всех других тестов оценки двигательной функции.
[0202] На фиг. 8 представлены графики, показывающие изменения результатов тестов оценки двигательной функции у отдельных пациентов в группе, получавшей вилтоларсен, и в контрольной группе естественного течения болезни CINRG (DNHS) на неделе 13 введения (прошло 12 недель после первоначального введения) и на неделе 25 введения (прошло 24 недели от первоначального введения) относительно каждого исходного уровня. Измененная величина указывает наименьший средний квадрат на неделе 13 или на неделе 25, измененной относительно исходного уровня, линия ошибки указывает стандартное отклонение, и значение P было рассчитано с использованием MMRM.
[0203] Количество образцов из группы, получавшей вилтоларсен, и группы DNHS на отдельные временные точки было следующим.
[0204]
0205] Пример 10
Тесты оценки двигательной функции (на неделе 85 (прошло 84 недели с момента первоначального введения))
(Экспериментальный метод)
В отношении показателей временных функциональных тестов [т.е. теста 6-минутной ходьбы (6MWT), времени вставания (TTSTAND), времени подъема на 4 ступени (TTCLIMB) и времени бега/ходьбы на 10 м (TTRW)] и амбулаторного обследования North Star (NSAA), то измерение проводили за 1 неделю до первоначального введения и на каждую временную точку, соответствующую каждым 12 неделям с момента первоначального введения (неделя 1) (т.е. на временные точки: неделя 13, 25, 37, 49, 61, 73 и 85). 201 испытание было проведено в организациях, относящихся к системе CINRG, и тесты оценки двигательных функций проводили в соответствии со стандартными операционными процедурами (SOP) и протоколами обучения клинических экспертов, используемыми в CINRG DNHS. Поскольку исследуемыми субъектами были дети, то некоторые задания нельзя было выполнить в некоторых тестах, потому что, например, пациенты не были заинтересованы в выполнении тестов.
[0206] Результаты
Результаты приведены на фиг. 9-16. В амбулаторном обследовании North Star многие пациенты в возрасте 5 лет или старше в группе, получавшей вилтоларсен, сохранили или улучшили свои оценки. Согласно публикации, касающейся исследований, в которых критерии включения пациентов, такие как критерии клинических исследований, были установлены на основе данных истории естественного течения болезни в Италии и Англии, и изучались изменения в оценках NSAA через 1 или 2 года, то средние изменения в течение курса лечения в течение 1 года у целевых пациентов с пропуском экзона 53 составили -4,1 балла (J. Neurol. Neurosurg. Psychicary, 87, 149-55, 2016). Измененное значение на неделе 48 у пациентов из группы, получавшей вилтоларсен, которые соответствовали критериям включения в публикацию, составило +1,3 балла. Согласно публикации, в которой сообщалось о результатах DHNS (Muscle Nerve, 48, 55-67, 2013), 20% пациентов с МДД в возрасте от 7 до 9 лет теряют способность вставать, но таких пациентов не было в группе, получавшей вилтоларсен. Кроме того, 10% пациентов с МДД в возрасте от 7 до 9 лет теряют способность подниматься на 4 ступеньки, но такие пациенты отсутствовали в группе, получавшей вилтоларсен. Скорость уменьшается с возрастом, но увеличение скорости наблюдали в группе, получавшей вилтоларсен, в целом. Кроме того, 10% пациентов с МДД в возрасте от 7 до 9 лет теряют способность к самостоятельному передвижению, но в NSP таких пациентов не было. Скорость относительно времени бега/ходьбы на 10 м уменьшается с возрастом, но увеличение скорости наблюдали в группе, получавшей вилтоларсен, в целом. В отношении изменения уровня экспрессии дистрофина по сравнению с исходным уровнем по данным количественного определения дистрофина с использованием WB, и изменений скорости по времени в тесте оценки вставания, времени в тесте подъема на 4 ступени и времени бега/ходьбы 10 м на неделе 48 от исходного уровня, наличие или отсутствие корреляции и уравнение линейной регрессии рассчитывали с использованием Excel. В отношении теста оценки времени вставания и теста оценки времени бега/ходьбы на 10 м, то была выявлена значимая корреляция между уровнем экспрессии дистрофина и изменением скорости (P <0,05), и было высказано предположение, что изменение скорости двигательной функции возрастает с увеличением уровня дистрофина.
Промышленная применимость
[0207] Согласно настоящему изобретению обеспечивается фармацевтическая композиция для применения в лечении мышечной дистрофии Дюшенна, которая имеет стабильную композицию NS-065/ NCNP-01 (вилтоларсен). Кроме того, в отношении фармацевтической композиции, содержащей NS-065/NCNP-01 (вилтоларсен), обеспечивается дозировка и способ введения, которые демонстрируют эффективное лечение МДД и находятся в безопасном диапазоне для пациентов-людей. Используя фармацевтическую композицию, можно эффективно уменьшить симптомы мышечной дистрофии Дюшенна с небольшими побочными эффектами.
--->
Список последовательностей
<110> NIPPON SHINYAKU CO., LTD.
<120> КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТИСМЫСЛОВОЙ ОЛИГОНУКЛЕОТИД, И ЕЕ
ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ МЫШЕЧНОЙ ДИСТРОФИИ ДЮШЕННА
<130> G2329
<150> US 62/690,270
<151> 2018-06-26
<150> US 62/739,386
<151> 2018-10-01
<160> 6
<170> PatentIn version 3.5
<210> 1
<211> 212
<212> ДНК
<213> Homo sapiens
<400> 1
ttgaaagaat tcagaatcag tgggatgaag tacaagaaca ccttcagaac cggaggcaac 60
agttgaatga aatgttaaag gattcaacac aatggctgga agctaaggaa gaagctgagc 120
aggtcttagg acaggccaga gccaagcttg agtcatggaa ggagggtccc tatacagtag 180
atgcaatcca aaagaaaatc acagaaacca ag 212
<210> 2
<211> 21
<212> ДНК
<213> Homo sapiens
<400> 2
gaacaccttc agaaccggag g 21
<210> 3
<211> 21
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетическая нуклеиновая кислота
<400> 3
cctccggttc tgaaggtgtt c 21
<210> 4
<211> 13
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 4
Ile Phe Leu Thr Glu Gln Pro Leu Glu Gly Leu Glu Lys
1 5 10
<210> 5
<211> 14
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 5
Val Ala Val Gly Gln Glu Gln Ala Phe Ser Val Asn Thr Arg
1 5 10
<210> 6
<211> 15
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 6
Ser Pro Phe Val Val Asn Val Ala Pro Pro Leu Asp Leu Ser Lys
1 5 10 15
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2702424C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2730681C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2695430C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2825834C2 |
| АНТИСМЫСЛОВАЯ НУКЛЕИНОВАЯ КИСЛОТА | 2016 |
|
RU2724554C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2011 |
|
RU2567664C2 |
| СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ, ВОВЛЕКАЮЩИХ МОДУЛЯЦИЮ РЕЦЕПТОРОВ РИАНОДИНА | 2013 |
|
RU2644350C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2012 |
|
RU2619184C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2018 |
|
RU2681470C1 |
| ИНДУКТОР СЧИТЫВАНИЯ И ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ МУТАЦИЯМИ ТИПА НОНСЕНС-МУТАЦИЙ | 2011 |
|
RU2571060C2 |
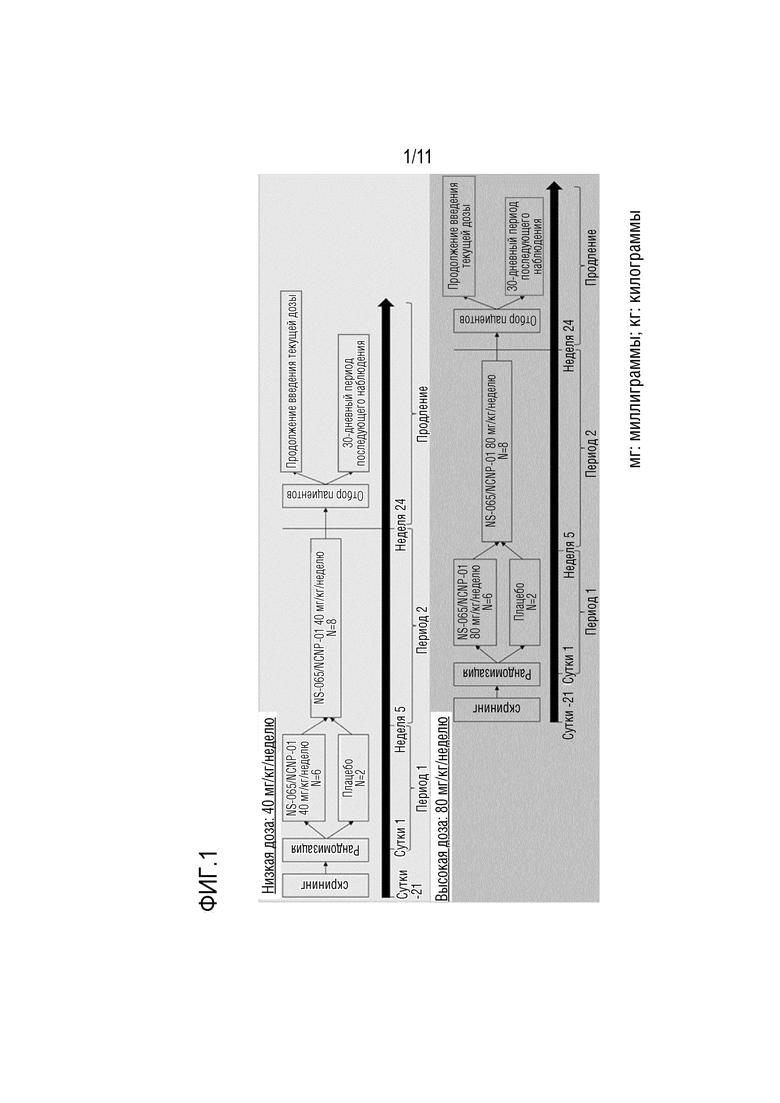
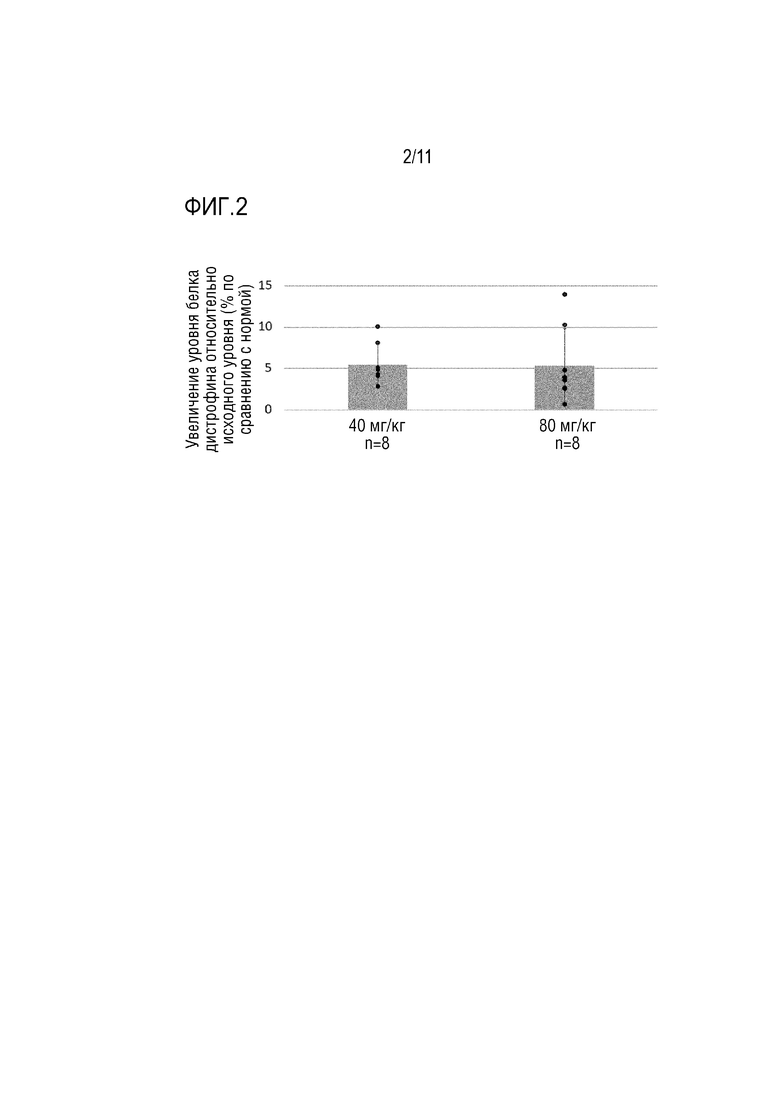

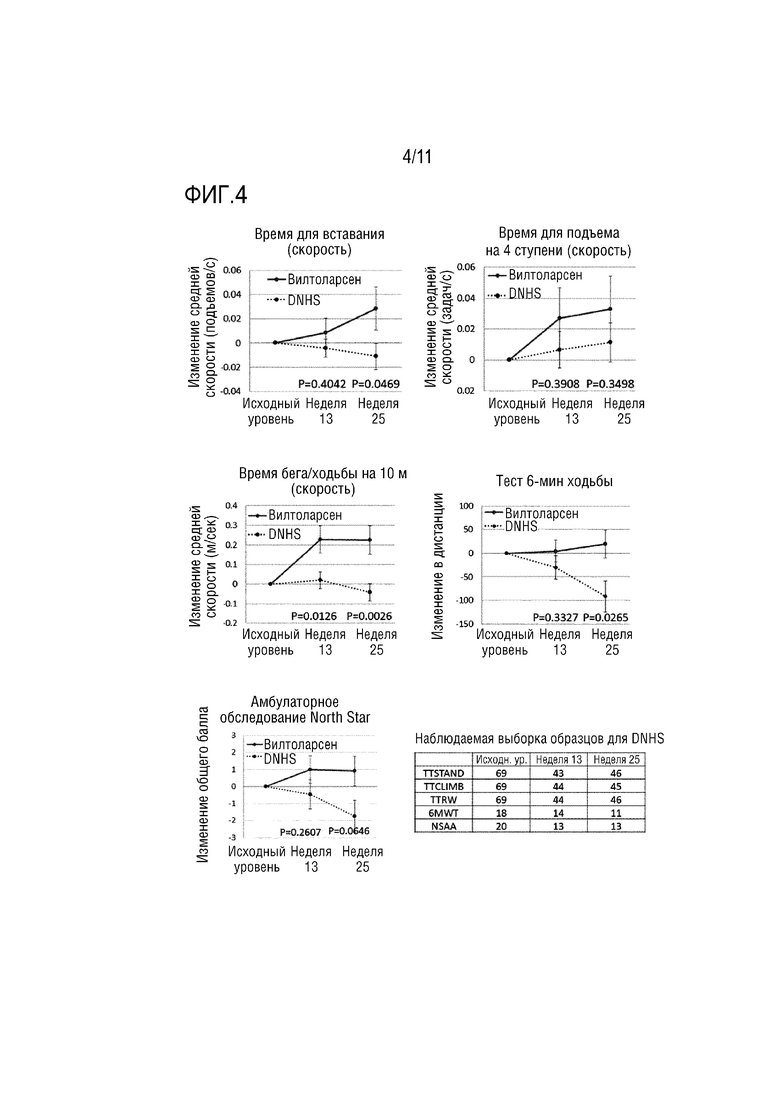
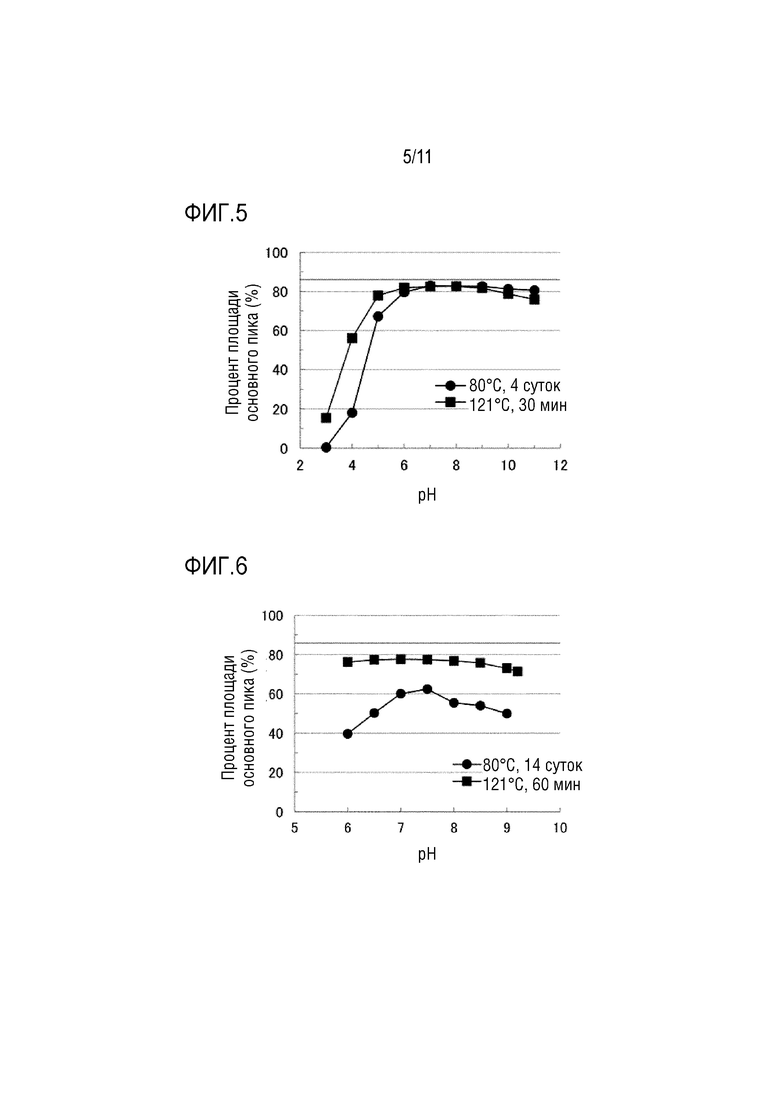
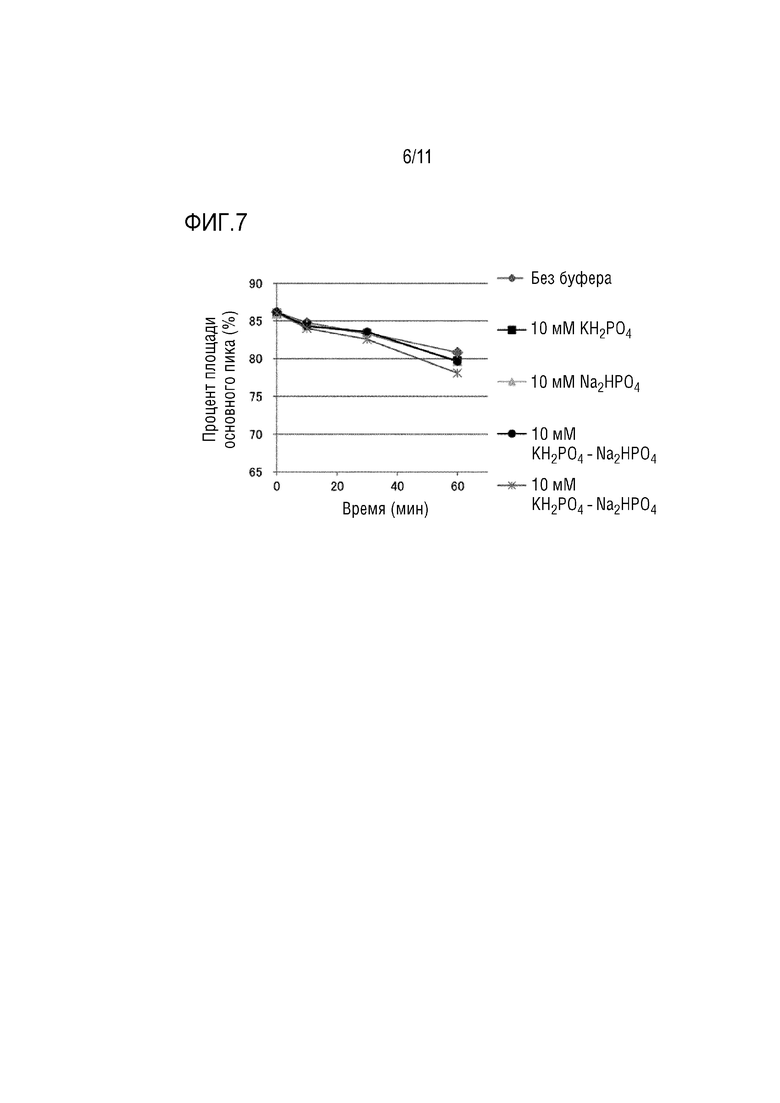

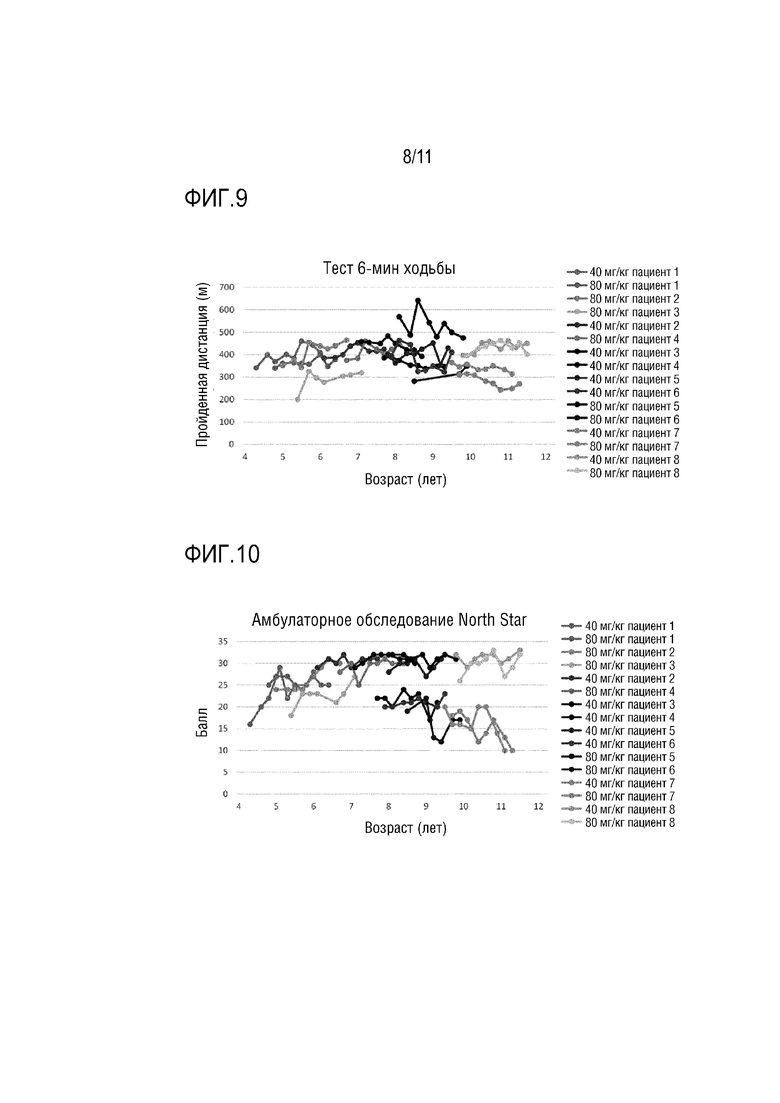


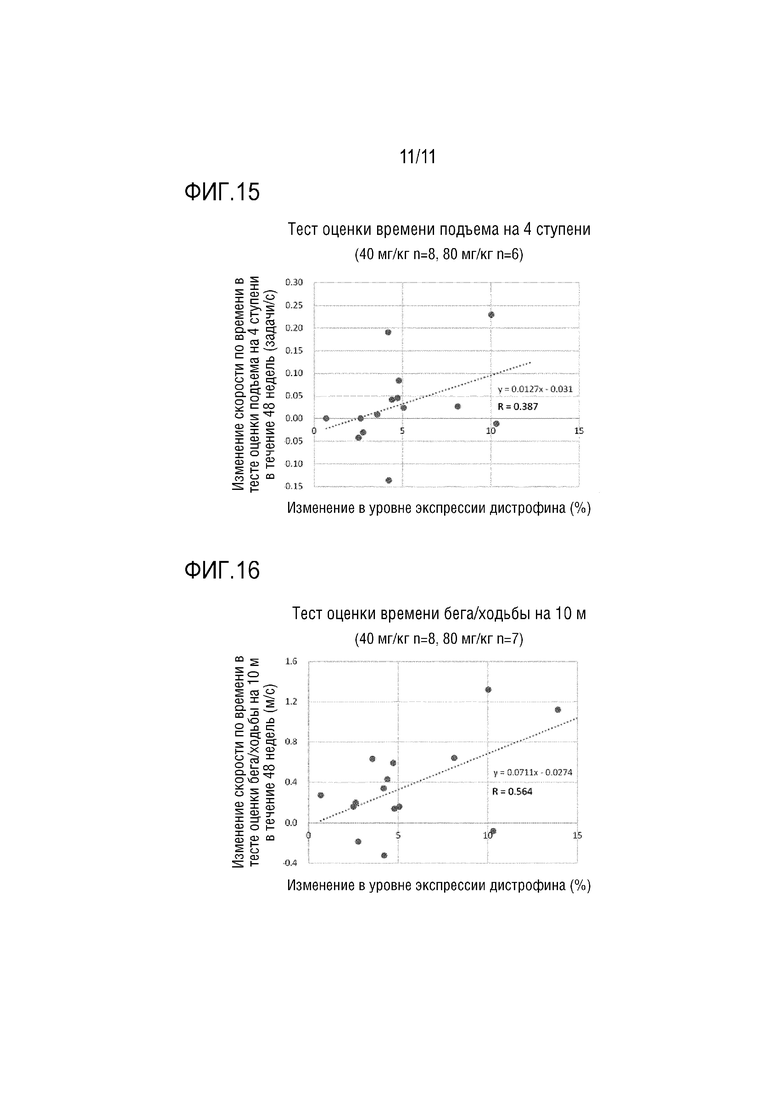
Изобретение относится к области биотехнологии, а именно к способу лечения мышечной дистрофии Дюшенна, включающему внутривенное введение фармацевтической композиции, содержащей антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях от 36 до 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль, или его гидрат. При этом антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/ неделю включительно. Изобретение эффективно для лечения мышечной дистрофии Дюшенна. 2 н. и 23 з.п. ф-лы, 16 ил., 29 табл., 10 пр.
1. Способ лечения мышечной дистрофии Дюшенна, включающий внутривенное введение фармацевтической композиции, содержащей антисмысловой олигомер, состоящий из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях от 36 до 56 от 5'-конца экзона 53 гена дистрофина человека, или его фармацевтически приемлемую соль, или его гидрат, пациенту-человеку один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/неделю антисмыслового олигомера, или его фармацевтически приемлемой соли или его гидрата.
2. Способ по п.1, в котором антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят внутривенно пациенту-человеку в дозе 40 мг/кг/неделю.
3. Способ по п.1, в котором антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно в дозе 80 мг/кг/неделю.
4. Способ по п.1, в котором у пациента-человека имеется мутация, которая приводит к дефициту любого экзона, выбранного из группы, состоящей из экзонов 43-52, 45-52, 47-52, 48-52, 49-52, 50-52 или 52 в гене дистрофина.
5. Способ по п.1, в котором экспрессия белка дистрофина у пациента-человека до лечения составляет 1% или ниже по сравнению с таковой у здорового субъекта, как измерено вестерн-блоттингом или масс-спектрометрией.
6. Способ по п.5, в котором экспрессия белка дистрофина не обнаруживается у пациента-человека до лечения.
7. Способ по п.1, в котором нуклеотидная последовательность антисмыслового олигомера состоит из последовательности, показанной в SEQ ID NO: 3.
8. Способ по п.1, в котором антисмысловой олигомер или его фармацевтически приемлемая соль, или его гидрат представляет собой вилтоларсен или его эквивалент, где эквивалент представляет собой антисмысловой олигонуклеотид или его фармацевтически приемлемую соль, или его гидрат, имеющий ту же последовательность оснований, что и у вилтоларсена.
9. Способ по п.1, в котором фармацевтическая композиция содержит антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат, в концентрации от 2,5 мг/мл включительно до 500 мг/мл включительно или от 10 мг/мл включительно до 100 мг/мл включительно.
10. Способ по п.1, в котором фармацевтическая композиция содержит антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат в концентрации 25 мг/мл.
11. Способ по п.1, в котором фармацевтическая композиция содержит антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат, в концентрации 50 мг/мл.
12. Способ по п.1, в котором фармацевтическая композиция дополнительно содержит по меньшей мере один компонент, выбранный из группы, состоящей из агента, регулирующего тоничность, регулятора pH и растворителя.
13. Способ по п.12, в котором агент, регулирующий тоничность, представляет, по меньшей мере, один выбранный из группы, состоящей из хлорида натрия, хлорида калия, глюкозы, фруктозы, мальтозы, сахарозы, лактозы, маннита, сорбита, ксилита, трегалозы и глицерина.
14. Способ по п.12 или 13, в котором регулятор pH представляет по меньшей мере один выбранный из группы, состоящей из соляной кислоты; гидроксида натрия; лимонной кислоты; молочной кислоты; фосфата, выбранного из группы, состоящей из гидрофосфата натрия, дигидрофосфата натрия, дигидрофосфата калия; и моноэтаноламина.
15. Способ по любому из пп.12-14, в котором растворитель представляет воду.
16. Способ по п.1, в котором фармацевтическая композиция содержит антисмысловой олигомер в концентрации от 2,5 мг/мл включительно до 500 мг/мл включительно, или от 10 мг/мл включительно до 100 мг/мл включительно, и хлорид натрия в концентрации от 8 мг/мл включительно до 10 мг/мл включительно, и которая представляет водный раствор с pH от 7,2 до 7,4.
17. Способ по п.1, в котором лечение обеспечивает, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) среднее значение уровня экспрессии белка дистрофина в скелетных мышцах пациента увеличивается в 9 или более раз по сравнению с исходным уровнем после введения фармацевтической композиции в течение 24 недель;
(2) изменение скорости, полученной по времени вставания из лежачего положения на спине (TTSTAND), составляет -0,055 раз/с или более по сравнению с исходным уровнем ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(3) изменение скорости, полученной по времени бега/ходьбы на 10 метров (TTRW), составляет -0,025 м/с или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(4) изменение скорости, полученной по времени подъема на 4 ступени (TTCLIMB), составляет -0,060 раз/с или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель;
(5) изменение балла по шкале амбулаторного обследования North Star (NSAA) составляет -2,2 балла или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель; и
(6) изменение показателя в тесте 6-минутной ходьбы (6MWT) составляет -7,5 м или более по сравнению с исходным уровнем, ко времени недели 25 после введения фармацевтической композиции в течение 24 недель.
18. Способ по п.1, в котором, когда проводится лечение пациентов в возрасте от 7 до 9 лет с мышечной дистрофией Дюшенна в течение 84 недель, то обеспечивается, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) процент пациентов, которые теряют способность подниматься, составляет менее 20% ко времени недели 85 после начала лечения;
(2) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 10% ко времени недели 85 после начала лечения;
(3) процент пациентов, которые теряют способность к самостоятельному передвижению, составляет менее 10% ко времени недели 85 после начала лечения;
(4) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(5) снижение показателя подъема на 4 ступени с возрастом не наблюдается ко времени недели 85 после начала лечения; и
(6) снижение скорости подъема с возрастом не наблюдается ко времени недели 85 после начала лечения.
19. Способ по п.1, в котором, когда проводится лечение пациентов в возрасте от 10 до 12 лет с мышечной дистрофией Дюшенна в течение 84 недель, то обеспечивается, по меньшей мере, один эффект, выбранный из группы, состоящей из следующих эффектов (1)-(6):
(1) процент пациентов, теряющих способность подниматься, составляет менее 60% ко времени недели 85 после начала лечения;
(2) процент пациентов, которые теряют способность подниматься на 4 ступени, составляет менее 50% ко времени недели 85 после начала лечения;
(3) процент пациентов, теряющих способность к самостоятельному передвижению, составляет менее 50% ко времени недели 85 после начала лечения;
(4) снижение скорости бега/ходьбы на 10 метров с возрастом не наблюдается ко времени недели 85 после начала лечения;
(5) наблюдается период, в течение которого увеличивается скорость подъема на 4 ступени ко времени недели 85 после начала лечения; и
(6) наблюдается период, в течение которого увеличивается скорость вставания ко времени недели 85 после начала лечения.
20. Применение антисмыслового олигомера, состоящего из нуклеотидной последовательности, комплементарной последовательности, состоящей из нуклеотидов в положениях с 36 по 56 от 5'-конца экзона 53 гена дистрофина человека или его фармацевтически приемлемой соли, или его гидрата, для производства фармацевтической композиции для лечения пациента-человека с мышечной дистрофией Дюшенна, где:
антисмысловой олигомер или его фармацевтически приемлемую соль, или его гидрат вводят пациенту-человеку внутривенно один раз в неделю в дозе от 40 мг/кг/неделю включительно до 80 мг/кг/ неделю включительно.
21. Способ по любому из пп.1-3, в котором фармацевтическая композиция не содержит фосфатный буфер.
22. Способ по п.21, отличающийся тем, что фармацевтическая композиция не содержит буфер.
23. Способ по п.22, в котором фармацевтическая композиция содержит антисмысловой олигомер в концентрации 50 мг/мл и хлорид натрия в концентрации 9 мг/мл, и фармацевтическая композиция представляет собой водный раствор с pH 7,0-7,5.
24. Способ по п.23, отличающийся тем, что pH составляет 7,2-7,4.
25. Способ по п.23, отличающийся тем, что pH составляет 7,3.
| СПОСОБ ПРОИЗВОДСТВА ВИСКИ | 2016 |
|
RU2612917C1 |
| WO2017213854 A1, 14.12.2017 | |||
| WO2017059131 А1, 06.04.2017 | |||
| ЗОТОВА Е.Д | |||
| и др., Анализ фенотипических проявлений делеций в гене дистрофина в контексте эффективности пропуска экзонов как метода терапии наследственных дистрофинопатий, ВЕСТНИК РГМУ, 2016, 3. | |||
