Область техники, к которой относится изобретение
Изобретение относится к производным 1,4-бензотиазепина и их применению для лечения нарушений и заболеваний, связанных с рецепторами рианодина (RyRs), которые регулируют кальциевый канал, функционирующий в клетках. В изобретении также описаны фармацевтические композиции, содержащие эти соединения, и их применения для лечения заболеваний и состояний, связанных с RyRs, в частности нарушений сердечной деятельности, костно-мышечных нарушений и нарушений центральной нервной системы (ЦНС).
Предпосылки создания изобретения
Саркоплазматический ретикулум (SR) представляет собой структуру в клетках, которая функционирует, в частности, в качестве специализированного внутриклеточного запаса кальция (Са2+). RyRs представляют собой каналы в SR, которые открываются и закрываются для регуляции высвобождения Са2+ из SR во внутриклеточную цитоплазму клетки. Высвобождение Са в цитоплазму из SR повышает концентрацию Са2+ в цитоплазме. Вероятность открытия RyRs относится к вероятности, что RyR открывается в любой данный момент и, следовательно, способен высвобождать Са2+ в цитоплазму из SR.
Существует три типа RyR, все они являются высоко гомологичными: RyR1, RyR2 и RyR3. RyR1 обнаружен главным образом в скелетных мышцах, а также в других тканях, RyR2 обнаружен главным образом в сердце, а также в других тканях, и RyR3 обнаружен в головном мозге, а также в других тканях. RyR представляет собой тетрамер. Часть RyR комплекса образована четырьмя RyR полипептидами в ассоциации с четырьмя FK506 связывающими белками (FKBPs) (калстабины), в особенности FKBP12 (калстабин 1) и FKBP12,6 (калстабин 2). Калстабин 1 связывается с RyR1 и RyR3, тогда как калстабин 2 связывается с RyR2. Калстабины связываются с RyR (одна молекула на RyR субъединицу), стабилизируют RyR функцию, способствуют сопряженному отпиранию между соседними RyRs и предотвращают анормальную активацию (Са2+ утечка) канала путем стабилизации закрытого состояния канала.
Рецептор рианодина 2 и сердечные заболевания
В поперечно-полосатых мышках сердца, RyR2 является основным каналом, высвобождающим Са2+, необходимым для сопряжения возбуждения-сокращения (ЕС) и сокращения мышц. При ЕС сопряжении, деполяризация мембраны сердечной мышечной клетки в фазе ноль действия потенциала активирует потенциалозависимые Са2+ каналы. Поступление Са2+ через открытые потенциалозависимые каналы, в свою очередь, инициирует высвобождение Са2+ из SR через RyR2. Этот процесс известен как Са2+-индуцированное высвобождение Са2+. Затем RyR2-опосредованное Са2+-индуцированное высвобождение Са2+ активирует сократительные белки в сердечной клетке, что приводит к сокращению сердечной мышцы.
Фосфорилирование RyR2 с помощью протеинкиназы А (PKA) является важной частью ответа "бей или беги", который повышает сердечное ЕС связанное усиление путем увеличения количества Са2+, высвобождаемого для данного триггера. Этот путь передачи сигналов обеспечивает механизм, с помощью которого активация симпатической нервной системы (SNS), в ответ на стресс, приводит к повышенному сердечному выбросу. Фосфорилирование RyR2 с помощью РКА приводит к частичной диссоциации калстабин 2 из канала, которая, в свою очередь, приводит к повышенной вероятности открытия и повышенному высвобождению Са2+ из SR во внутриклеточную цитоплазму.
Сердечная недостаточность (HF) характеризуется долгосрочным гиперадренергическим состоянием, при котором уровни катехоламина в сыворотке хронически повышены. Одним из последствий этого хронического гиперадренергического состояния является устойчивое PKA гиперфосфорилирование RyR2, таким образом, что 3-4 из четырех Ser 2808 в каждом гомотетрамерном RyR2 канале хронически фосфорилированы (Marx SO и др. Cell, 2000; 101(4): 365-376). В частности, хроническое PKA гиперфосфорилирование RyR2 связано с истощением канал-стабилизирующей субъединицы калстабин 2 из макромолекулярного комплекса RyR2 канала. Истощение калстабина приводит к диастолической SR Са "утечке" из RyR комплекса, что способствует ослабленной сокращаемости (Marx и др., 2000). Вследствие активации входящих деполяризующих токов, эта диастолическая SR Са2+ "утечка" также связана со смертельными сердечными аритмиями (Lehnart и др., J Clin Invest. 2008; 118(6): 2230-2245). Действительно, мыши, сконструированные с RyR2 с отсутствующим PKA сайтом фосфорилирования, защищены от HF прогрессии после инфаркта миокарда (MI) (Wehrens XH и др. Proc Natl Acad Sci USA. 2006; 103(3): 511-518). Дополнительно, хроническое PKA гиперфосфорилирование RyR2 в HF связано с ремоделированием RyR2 макромолекулярного комплекса, который включает истощение фосфатаз (Marx и др. 2000) РР1 и РР2а (ослабленное дефосфорилирование Ser 2808) и сАМР-специфический тип 4 фосфодиэстераз (PDE4D3) из RyR2 комплекса. Истощение PDE4D3 из RyR2 комплекса вызывает долговременное повышение локальных уровней cAMP (Lehnart SE и др., Cell 2005; 123(1): 25-35). Таким образом, диастолическая SR Са2+ утечка способствует HF прогрессированию и аритмиям. Кроме того, в недавнем отчете было показано, что у «нокин» мышей RyR2-S2808D+/+ (аспарагиновая кислота, заменяющая серии 2808) мимическое конститутивное PKA гиперфосфорилирование RyR2, проявляет истощение калстабин 2 и утечку RyR2. У мышей RyR2-S2808D+/+ развивается зависимая от возраста кардиомиопатия, проявляется повышенное RyR2 окисление и нитрозилирование, уменьшенное содержание SR Са2+ запасов и повышенная диастолическая SR Са2+ утечка. После инфаркта миокарда, у мышей RyR2-S2808D+/+ проявляется повышенная смертность по сравнению с WT однопометными животными. Лечение с помощью S107, производного 1,4-бензотиазепина, которое стабилизирует RyR2-калстабин 2 взаимодействия (WO 2007/024717), ингибирует RyR2-опосредованную диастолическую SR Са утечку и уменьшает HF прогрессирование, как у мышей WT, так и у мышей RyR2-S2808D+/+ (Shan и др., J Clin Invest. 2010 Dec 1; 120(12): 4375-87).
Кроме того, RyR2 содержит около 33 свободных тиольных остатков, придающих ему высокую чувствительность к окислительно-восстановительному состоянию в клетке. Окисление цистеина облегчает RyR открытие и SR Са2+ утечку. Shan и др., 2010, показали, что окисление и нитрозилирование RyR2 и диссоциация стабилизирующей субъединицы калстабин 2 из RyR2 индуцирует SR Са2+ утечку.
Катехоламинергическая полиморфная желудочковая тахикардия (CPVT) представляет собой наследственное нарушение у особей со структурно нормальными сердцами. Более чем 50 индивидуальных RyR2 мутаций были связаны с CPVT. У пациентов с CPVT наблюдается обморок и внезапная сердечная смерть (SCD) от подросткового возраста до совершеннолетия, и к 35 годам смертность составляет вплоть до 50%. У особей с CPVT наблюдаются желудочковые аритмии при нагрузках, но в состоянии покоя аритмии не развиваются. CPVT-ассоциированные RyR2 мутации приводят к "дающим утечку" RyR2 каналам вследствие снижения связывания субъединицы калстабин 2 (Lehnart и др., 2008). Мыши, гетерозиготные по R2474S мутации в RyR2 (RyR2-R2474S мыши), проявляют самопроизвольные генерализированные тонико-клонические пароксизмы (которые развиваются при отсутствии сердечных аритмий), вызванные физической нагрузкой желудочковые аритмии и SCD. Лечение с помощью S107 увеличивает связывание калстабин 2 с мутантным RyR2-R2474S каналом, ингибирует утечку канала, предотвращает сердечные аритмии и повышает судорожный порог (Lehnart и др., 2008).
Рецептор рианодина 1 и заболевания скелетных мышц
Сокращения скелетных мышц активируется SR Са2+, высвобождаемым с помощью RyR1. Деполяризация поперечной (Т)-канальцевой мембраны активирует потенциальный датчик дигидропиридинового рецептора (Cav1.1), который, в свою очередь, активирует RyR1 каналы посредством прямого белок-белкового взаимодействия, вызывая высвобождение SR Са2+ запасов. Са2+ связывается с тропонином С, предоставляя возможность происходить перекрестному связыванию актина-миозина и укорочению саркомера.
В условиях пролонгированного мышечного стресса (например, при беге на марафонные дистанции) или при заболевании, таком как сердечная недостаточность, которые оба характеризуются хронической активацией SNS, функционирование скелетных мышц нарушается, вероятно, вследствие изменения ЕС связывания. В частности, количество Са, высвобождаемое из SR при каждом сокращении мышцы, уменьшается, могут происходить явления аберрантного высвобождения Са2+ и замедляется обратное поглощение Са2+ (Reiken, S. и др. 2003. J. Cell Biol. 160: 919-928). Эти наблюдения свидетельствуют о том, что вредные эффекты хронической активации SNS на скелетные мышцы может быть обусловлена, по меньшей мере частично, дефектами в Са передаче сигналов.
RyR1 макромолекулярный комплекс состоит из тетрамера 560 кДа RyR1 субъединицы, которая образует каркас для белков, которые регулируют функционирование канала, включая PKA и фосфодиэстераза 4D3 (PDE4D3), протеинфосфатаза 1 (РР1) и калстабин 1. A-киназный якорный белок (mAKAP) нацеливает PKA и PDE4D3 на RyR1, в то время как синофилин нацеливает РР1 на канал (Marx и др. 2000; Brillantes и др., Cell, 1994, 77, 513-523; Bellinger и др. J. Clin. Invest. 2008, 118, 445-53). Каталитические и регуляторные субъединицы PKA, РР1, и PDE4D3 регулируют PKA-опосредованное фосфорилирование RyR1 на Ser2843 (Ser2844 у мышей). Было показано, что PKA-опосредованное фосфорилирование RyR1 при Ser2844 повышает чувствительность канала к цитоплазматическому Са, уменьшает связывающую способность калстабин 1 к RyR1 и дестабилизирует закрытое состояние канала (Reiken и др., 2003; Marx, S.O. и др., Science, 1998, 281: 818-821). Описано, что концентрации калстабин 1 в скелетных мышцах составляют приблизительно 200 нМ и что PKA фосфорилирование RyR1 уменьшает связывающую способность калстабин 1 к RyR1 от приблизительно 100-200 нМ до более чем 600 нМ. Таким образом, в физиологических условиях, уменьшение связывающей способности калстабин 1 к RyR1, возникающее вследствие PKA фосфорилирования RyR1 на Ser2843, достаточно для существенного уменьшения количества калстабин 1, присутствующего в RyR1 комплексе. Хроническое PKA гиперфосфорилирование RyR1 Hat Ser2843 (определяемое как PKA фосфорилирование 3 или 4 из 4 PKA Ser2843 сайтов, присутствующих в каждом RyR1 гомотетрамере) приводит к "дающим утечку" каналам (то есть каналы, подверженные открытию в состоянии покоя), что способствует нарушению функций скелетных мышц, которая связана с устойчивыми гиперадренергическими состояниями, такими как происходящие у особей с сердечной недостаточностью (Reiken и др., 2003).
Кроме того, было описано, что регуляция RyR1 посредством посттрансляционных модификаций, отличающихся от фосфорилирования, таких как нитрозилирование свободных сульфгидрильных групп на остатках цистеина (S-нитрозилирование), а также окисление канала повышает активность RyR1 канала. Было показано, что S-нитрозилирование и окисление RyR1 каждый уменьшается связывание калстабин1 связывание с RyR1.
Ранее Bellinger и др. (Proc. Natl. Acad. Sci. 2008, 105(6): 2198-2002) было показано, что при чрезмерных нагрузках у мышей и людей, RyR1 прогрессивно PKA-гиперфосфорилирован, S-нитрозилирован и истощен PDE4D3 и калстабин1, что приводит к "дающим утечку" каналам, которые вызывают снижение способности переносить физическую нагрузку у мышей. Лечение с помощью S107 предотвращает истощение калстабин 1 из RyR1 комплекса, улучшает создание силы и способность переносить физическую нагрузку и уменьшает активность Са2+ "зависимой нейтральной протеазы кальпаин и уровни креатинин-киназы в плазме.
Мышечная дистрофия Дюшенна (DMD) представляет собой одно из основных летальных генетических заболеваний у детей. DMD связана с X хромосомой, поражая 1 из 3,5 тыс. новорожденных особей мужского пола и типично приводит к смерти приблизительно в возрасте ~30 лет от дыхательной или сердечной недостаточности. Мутации в дистрофине, ассоциированные с DMD, приводят к полной потере дистрофинового белка, таким образом разрушая связь между субсарколеммным цитоскелетом и внеклеточным матриксом. Эта связь является необходимой для защиты и стабилизации мышцы от повреждения, вызванного сокращением. В настоящее время, отсутствует лечение DMD и большинство лечений являются паллиативными. Перспективные интервенции в Фазе I/II клинических исследований представляют собой перескакивание экзонов, ингибирование миостатина и повышенную регуляцию утрофина. Тем не менее, существуют проблемы с системной доставкой, поддерживанием перескакивания экзонов и повышенной регуляций утрофина. Дополнительно, в Фазе I/II клинических исследований, инактивация миостатина для повышения размера мышц не проявляет улучшения относительно мышечной силы или функции. Нестабильность сарколеммы вследствие мутаций в дистрофине имеет каскадный эффект. Одним из основных эффектов является повышение цитозольной концентрации Са2+, что приводит к активации Са2+- зависимых протеаз (кальпаинов). Другим эффектом является воспаление и повышение активности iNOS, что может вызывать окисление / нитрозилирование белков, липидов и ДНК. DMD патология мышц является прогрессирующей и значительно превышает нестабильность сарколеммы. Таким образом, патология согласуется с нестабильностью сарколеммы, повышая чувствительность к дальнейшему повреждению. Недавно было показано, что чрезмерное окисление или нитрозилирование RyR1 может разрушать взаимодействие калстабин 1 с RyR1 комплексом, приводя к RyR1 утечке и мышечной слабости на мышиной модели мышечной дистрофии (mdx) и что лечение с помощью S107 улучшает показатели функционирования мышц на этой мышиной модели (Bellinger, А. и др. 2009, Nature Medicine, 15: 325-330).
Возрастная потеря мышечной массы и силы (саркопения) способствует инвалидности и повышает смертность. Andersson, D. и др. (Cell Me tab. 2011 Aug 3; 14(2): 196-207) описали, что RyR1 от старых (24 месяцев) мышей окислена, цистеин-нитрозилирована и истощена на калстабин 1, по сравнению с RyR1 от более молодых (3-6 месяцев) взрослых особей. Ремоделирование этого комплекса RyR1 канала приводит к "дающим утечку" каналам с повышенной вероятностью открытия, что обуславливает внутриклеточную утечку кальция в скелетных мышцах. Лечение старых мышей с помощью S107 стабилизирует связывание калстабин 1 с RyR1, уменьшает внутриклеточную утечку кальция, снижает активные формы кислорода (ROS) и повышает тетаническое высвобождение Са2+, удельную мышечную силу и способность переносить физическую нагрузку.
В опубликованных РСТ международных патентных заявках WO 2005/094457, WO 2006/101496 и WO 2007/024717 описаны производные 1,4-бензотиазепина и их применение для лечения, в частности, сердечных нарушений, нарушений скелетных мышц и когнитивных нарушений.
Опубликованная РСТ международная патентная заявка WO 2008/060332 относится к применению производных 1,4-бензотиазепина для лечения мышечного утомления у субъектов, страдающих от таких патологий, как мышечная дистрофия, или у субъектов, страдающих от мышечного утомления в результате долговременной, длительной и/или энергичной нагрузки, или хронического стресса.
Опубликованная РСТ международная патентная заявка WO 2008/021432 относится к применению производных 1,4-бензотиазепина для лечения и/или предотвращения заболеваний, нарушений и состояний, поражающих нервную систему.
Опубликованная РСТ международная патентная заявка WO 2012/019076 относится к применению производных 1,4-бензотиазепина для лечения и/или предотвращения кардиального ишемического/реперфузионного повреждения. Fauconnier и др., Proc Natl Acad Sci USA, 2011, 108(32): 13258-63 описали, что RyR утечка, опосредованная активацией каспазы-8, приводит к повреждению левого желудочка после миокардиальной ишемии-реперфузии и что лечение с помощью S107 ингибирует SR Са2+ утечку, уменьшает желудочковые аритмии, размер инфаркта и реконструкцию левого желудочка на 15 дней после реперфузии.
Опубликованная РСТ международная патентная заявка WO 2012/019071 относится к применению производных 1,4-бензотиазепина для лечения и/или предотвращения саркопении.
Опубликованная РСТ международная патентная заявка WO 2012/037105 относится к применению производных 1,4-бензотиазепина для лечения и/или предотвращения индуцированных стрессом нарушений и заболеваний нейронов.
Существует потребность в идентификации новых соединений, эффективных для лечения нарушений и заболеваний, связанных с RyRs, включая нарушения и заболевания скелетных мышц и нарушения сердечной деятельности и кардиологические заболевания. Более предпочтительно, существует потребность в идентификации новых средств, которые можно использовать для лечения RyR-ассоциированных нарушений, например, путем репарации утечки в RyR каналах и усиления связывания калстабинов с PKA-фосфорилированными/окисленными/нитрозилированными RyRs и с мутантными RyRs, которые другим способом имеют уменьшенное сродство к или не связываются с калстабинами.
Сущность изобретения
Настоящее изобретение обеспечивает новые производные 1,4-бензотиазепина и их фармацевтически приемлемые соли. В некоторых вариантах осуществления соединения согласно настоящему изобретению представляют собой стабилизаторы рецептора рианодина (RyR) кальциевого канала, в некоторых источниках обозначаемые как "Rycals™." Настоящее изобретение дополнительно обеспечивает способы применения этих соединений для лечения нарушений и заболеваний, связанных с RyRs.
Соединения согласно настоящему изобретению выбирают из производных 1,4-бензотиазепина, описанных в WO 2007/024717. В WO 2007/024717 раскрыты структурно сходные соединения, тем не менее, как более подробно описано в настоящей заявке, было обнаружено, что эти соединения являются очень нестабильными и, следовательно, их терапевтическая активность в качестве лекарственных средств ограничена. Таким образом, задачей, лежащей в основе настоящего изобретения, является обеспечение альтернативных производных 1,4-бензотиазепина, которые являются не только фармакологически активными - но также имеют благоприятные свойства, такие как высокая метаболическая стабильность, и, следовательно, они являются пригодными в качестве лекарственных средств для лечения заболеваний и состояний, связанных с RyR, например, кардиальных нарушений, нарушений скелетных мышц и нарушений центральной нервной системы (ЦНС). Неожиданно было обнаружено, что соединения формулы (I) являются стабильными, а также фармакологически активными, таким образом обеспечивая техническое решение задачи, лежащей в основе настоящего изобретения.
Соединения согласно настоящему изобретению представлены структурой формулы (I):
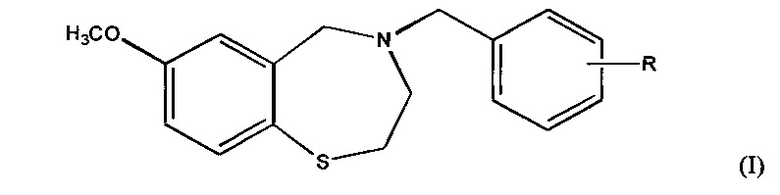
где R представляет собой COOH;
и его фармацевтически приемлемые соли.
Соединения формулы (I) могут быть представлены в форме соли с фармацевтически приемлемой кислотой или основанием. Такие соли предпочтительно выбирают из группы, включающей соли натрия, калия, магния, гемифумарат, гидрохлорид и гидробромид, с каждой возможностью представляя отдельный вариант осуществления настоящего изобретения. Одна предпочтительная сейчас соль представляет собой натриевую соль. Другая предпочтительная сейчас соль представляет собой гемифумаратную соль.
В некоторых специфических вариантах осуществления соединение выбирают из группы, включающей соединение 1, соединение 4 и соединение 6 и его фармацевтически приемлемые соли. Структуры этих соединений представлены в настоящей заявке ниже.
В предпочтительном варианте осуществления соединение представлено структурой соединения (1):
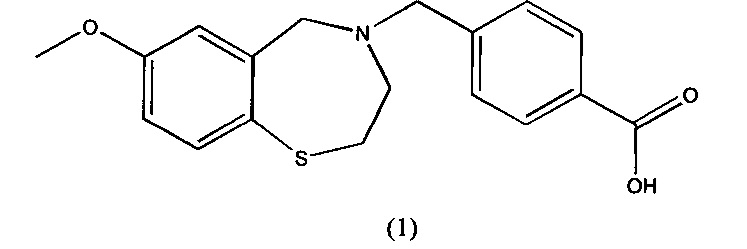
или его фармацевтически приемлемые соли.
В некоторых вариантах осуществления соединение 1 обеспечивается в виде исходного соединения. Тем не менее, в других вариантах осуществления соединение 1 обеспечивается в форме соли с фармацевтически приемлемой кислотой или основанием. Предпочтительно, такую соль выбирают из группы, включающей соли натрия, калия, магния, гемифумарат, гидрохлорид и гидробромид, с каждой возможностью представляя отдельный вариант осуществления настоящего изобретения. Одна предпочтительная сейчас соль представляет собой натриевую соль. Другая предпочтительная сейчас соль представляет собой гемифумаратную соль.
Настоящее изобретение также обеспечивает способы синтеза соединений согласно изобретению и их солей.
Настоящее изобретение также обеспечивает фармацевтические композиции, которые содержат одно или несколько соединений согласно изобретению и по меньшей мере одно вспомогательное вещество или наполнитель, например, заполнители, разбавители, связующие, агенты, вызывающие дезинтеграцию, буферные вещества, красители, эмульсификаторы, средства, улучшающие запах, гелеобразующие вещества, скользящие вещества, консерванты, солюбилизаторы, стабилизаторы, суспендирующие агенты, подсластители, вещества, регулирующие тоничность, смачивающие вещества, эмульсификаторы, диспергирующие вещества, агенты, вызывающие набухание, ретарданты, скользящие вещества, абсорбирующие вещества и средства, повышающие вязкость. Композиции могут быть представлены в дозированной форме в виде капсул, гранул, порошков, растворов, саше, суспензий, или таблеток.
Настоящее изобретение дополнительно обеспечивает способы лечения или предотвращения различных нарушений, заболеваний и состояний, связанных с RyRs, такие как кардиальные, мышечно-скелетные когнитивные, ЦНС и нейромышечные нарушения и заболевания, которые включают введение субъекту, нуждающемуся в таком лечении, количества соединения формулы (I) или его соли, эффективного для предотвращения или лечения нарушения или заболевания, связанного с RyR. Настоящее изобретение также обеспечивает способ предотвращения или лечения утечки в RyR (включая RyR1, RyR2 и RyR3) у субъекта, включающего введение субъекту количества соединения формулы (I) или его соли, эффективного для предотвращения или лечения утечки в RyR.
Дополнительно, настоящее изобретение обеспечивает способ модулирования связывания RyRs и калстабинов у субъекта, включающий введение субъекту количества соединения формулы (I) или его соли, эффективного для модуляции количества RyR-связанного калстабина.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) для приготовления лекарственного средства для лечения и/или предотвращения нарушений, заболеваний и состояний, связанных с RyRs, таких как кардиальные, мышечно-скелетные и когнитивные нарушения и заболевания/нарушения и заболевания ЦНС. В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (I) для приготовления лекарственного средства для предотвращения или лечения утечки в RyR. В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (I) для приготовления лекарственного средства для модуляции количества RyR-связанных калстабинов.
Способы согласно изобретению могут быть практически осуществлены на системах in vitro (например, культивируемые клетки или ткани) или in vivo (например, на животном, отличающемся от человека, или человеке).
В некоторых вариантах осуществления соединения согласно изобретению обеспечиваются в комбинации с терапией перескакивания экзона, например, антисмысловыми олигонуклеотидами (AOs), таким образом, чтобы усилить перескакивание экзона в мРНК, представляющей интерес, например, DMD ген, как в дальнейшем описано в настоящей заявке. Другие характерные особенности и преимущества настоящего изобретения будут понятны со следующих подробных описаний и фигур.
Краткое описание чертежей
Фигура 1A. Иммуноблот с антителом к калстабин 2, показывающий связывание калстабин 2 с PKA-фосфорилированным RyR2 при отсутствии (-) или в присутствии 100 нМ соединения 1. (+): калстабин связывается с не-PKA фосфорилированным RyR2. S36 (US 7,544,678), использовали в качестве положительного контроля.
Фигура 1B. Иммуноблот с антителом к калстабин 2, показывающий связывание калстабин 2 с PKA-фосфорилированным RyR2 при отсутствии (-) или в присутствии 100 нМ соединения 2, соединения 3 или соединения 4. (+): калстабин связывается с не-PKA фосфорилированным RyR2. S36 использовали в качестве положительного контроля.
Фигура 1C. Иммуноблот с антителом к калстабин 1, показывающий связывание калстабин 1 с PKA-фосфорилированным RyR1 при отсутствии (Отриц) или в присутствии указанных концентраций соединения 1 или соединения 4. (Полож): калстабин связывается с не-PKA фосфорилированным RyR1. S36 использовали в качестве положительного контроля.
Фигура 2. Фигура 2A: Иммуноблот с антителом к калстабин 1, который показывает уровни калстабин 1 в иммуноосажденных RyR1 комплексах из лизатов большеберцовой кости у мышей, которым вводили носитель (50:50 ДМСО/PEG), только изопротеренол (ISO) или изопротеренол совместно с указанными концентрациями соединения 1 в осмотических насосах. S36 использовали в качестве контроля при 3,6 мМ. Фигура 2B: определение количества % калстабин 1, который повторно связывается с RyR1.
Фигура 3. Модель хронической сердечной недостаточности у крыс, индуцируемая ишемически-реперфузионным (I/R) повреждением. Для I/R протокола, переднюю нисходящую ветвь (LAD) левой коронарной артерии окклюзировали в течение 1 ч.
Фигура 4. Объемы левого желудочка (LV) и фракции выброса (EF) у крыс, леченных с помощью соединения 1 в дозе 5 мг/кг/д (5 MK) или 10 мг/кг/д (10 MK) в питьевой воде отн. леченных с помощью наполнителя (H2O) и ложнооперированных животных. Хроническую сердечную недостаточность индуцировали путем ишемически-реперфузионного (I/R) повреждения. LAD артерию окклюзировали в течение 1 ч; лечение начинали через 1 неделю после реперфузии и продолжали в течение 3 месяцев. Эхокардиографические параметры получали через 1, 2 или 3 месяца после лечения. Фигура 4A: LV Конечный диастолический объем; Фигура 4B: LV Конечный систолический объем; Фигура 4C: EF. Фигуры 4A и 4B: § P<0,001 отн. ложн.; * P<0,05 отн. наполнителя; † P<0,001 отн. наполнителя. Фигура 4C: § P<0,001 отн. ложн., † P<0,001 отн. наполнителя.
Фигура 5. На фигурах 5A-С представлены вес тела (BW) (5A), размер инфаркта (5B) и вес LV (5C), и на фигуре 5D представлено содержание коллагена у крыс, леченных с помощью соединения 1 в дозе 5 мг/кг/д (5 MK) и 10 мг/кг/д (10 MK) в питьевой воде отн. леченных с помощью наполнителя(H2O) - и ложнооперированных животных. Хроническую сердечную недостаточность индуцировали путем ишемически-реперфузионного (I/R) повреждения. LAD артерию окклюзировали в течение 1 ч; лечение начинали через 1 неделю после реперфузии и продолжали в течение 3 месяцев. Параметры измеряли через 3 месяца после лечения. Фигуры 5A-C: не достоверны. Фигура 5D: ††† P<0,001 отн. ложн.; * P<0,05 отн. наполнителя.
Фигура 6. Инвазивные гемодинамики: Систолическое давление в левом желудочке (LV SP) (6A), dP/dtmax (6B); и dP/dtmin (6C) у крыс, леченных с помощью соединения 1 в дозе 5 мг/кг/д (5 MK) или 10 мг/кг/д (10 MK) в питьевой воде отн. леченных с помощью наполнителя (H2O) и ложнооперированных животных. Хроническую сердечную недостаточность индуцировали путем ишемически-реперфузионного (I/R) повреждения. LAD артерию окклюзировали в течение 1 ч; лечение начинали через 1 неделю после реперфузии и продолжали в течение 3 месяцев. Гемодинамические параметры измеряли через 3 месяца после лечения. Фигура 6A: не достоверны. Фигура 6B: § P<0,05 отн. ложн.; * P<0,05 отн. наполнителя. Фигура 6C: † P<0,01 отн. ложн.; * P<0,05 отн. наполнителя.
Фигура 7. Концентрации соединения 1 в плазме (мкМ) отн. времени суток.
Фигура 8. EF у крыс, леченных с помощью соединения 1 или соединения А в дозе 5 мг/кг/д (5 MK) в питьевой воде отн. леченных с помощью наполнителя (H2O) и ложнооперированных животных. LAD артерию окклюзировали в течение 1 ч; лечение начинали через 1 неделю после реперфузии и продолжали в течение 3 месяцев. Эхокардиографические параметры получали через 1, 2 или 3 месяца после лечения. § P<0,001 отн. ложн.; * P<0,05 отн. наполнителя; † P<0,001 отн. наполнителя.
Фигура 9. Влияние соединения 1 на самопроизвольную физическую активность mdx и WT мышей по сравнению с контролем, леченным с помощью наполнителя (H2O). P<0,001 для активности в дни 1-19 у мышей mdx, которым вводили дозу 10 и 50 мг/кг/сутки (целевая доза) в питьевой воде, по сравнению с контрольным наполнителем.
Фигура 10. Специфическое соотношение сила-частота EDL мышцы. (А) mdx мыши, леченные с помощью соединения 1 (5, 10 и 50 мг/кг/д (целевая доза)), которое вводили в питьевой воде, по сравнению с контролем, леченным с помощью наполнителя (H2O) (n=5). p<0,05, для дозы 50 мг/кг/д, при частотах 150 Гц и выше. (В) WT, C57BL/6, мыши, леченные с помощью соединения 1 (50 мг/кг/д (целевая доза), которое вводили в питьевой воде, по сравнению с контролями, леченными с помощью наполнителя (H2O) (n=4)
Фигура 11. Средний вес тела (12A) и среднее потребление воды (12B) для mdx и WT мышей, леченных с помощью наполнителя (H2O) или соединения 1 (50 мг/кг/д (целевая доза), которое вводили в питьевой воде.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Подразумевается, что подробное описание и специфические примеры, в то время как раскрывают различные варианты осуществления изобретения, представлены только с целью иллюстрации, при этом различные изменения и модификации в пределах сущности и объема изобретения станут очевидными для специалистов в данной области техники из этого подробного описания.
Как используется в настоящей заявке и приложенных пунктах формулы изобретения, формы единственного числа включают также формы множественного числа, если из контекста очевидно не следует иначе. Все публикации, патентные заявки, патенты и другие ссылки, упомянутые в настоящей заявке, полностью включены в качестве ссылки.
Термин "Rycals™" относится к стабилизаторам кальциевого канала рецептора рианодина, представленным соединениями общей Формулы (I) или (IA), как обеспечивается изобретением, а также специфическими соединениями, обозначенными цифровыми номерами, как обеспечивается изобретением, и в настоящей заявке собирательно обозначается как "соединение(я) согласно изобретению".
Соединения
В некоторых вариантах осуществления соединения согласно настоящему изобретению представлены структурой формулы (IA):
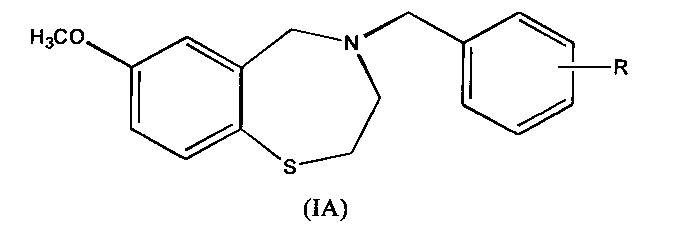
где R представляет собой COOH или его биоизостер, COOR1 или CN; и
R1 представляет собой C1-C4 алкил;
и его фармацевтически приемлемые соли.
В некоторых предпочтительных вариантах осуществления R в Формуле (IA) представляет собой карбоновую кислоту (COOH). В других предпочтительных вариантах осуществления R в Формуле (IA) представляет собой биоизостер карбоновой кислоты, например, тетразол. Альтернативно, биоизостер карбоновой кислоты может представлять собой кислый гетероцикл, такой как 1,2,4-оксадиазол-5(4H)-он, 1,2,4-тиадиазол-5(4H)-он, 1,2,4-оксадиазол-5(4H)-тион, 1,3,4-оксадиазол-2(3H)-тион, 4-метил-1H-1,2,4-триазол-5(4H)-тион, 5-фтороротовую кислоту и другие. Дополнительные биоизостеры карбоновых кислот описаны, например, в Hamada, Y. и др., Bioorg. Med. Chem. Lett. 2006; 16: 4354-4359; Herr, R.J. и др., Bioorg. Med. Chem. 2002; 10: 3379-3393; Olesen, P.H., Curr. Opin. Drug Discov. Devel. 2001; 4: 471; Patani. G.A. и др., J. Chem. Rev. 1996; 96: 3147; Kimura, T. и др. Bioorg. Med. Chem. Lett. 2006; 16: 2380-2386; и Kohara, Y. и др. Bioorg. Med. Chem. Lett. 1995; 5(17): 1903-1908. Содержание каждой из вышеуказанных ссылок включено в настоящую заявку в качестве ссылки.
В одном предпочтительно варианте осуществления соединения согласно настоящему изобретению представлено структурой формулы (IA), где R представляет собой COOH и его фармацевтически приемлемые соли (то есть соединение формулы (I)).
В других предпочтительных вариантах осуществления R в Формуле (IA) находится в положении 4 фенильного кольца (то есть положении 7 бензотиазепинового кольца). Каждая возможность представляет собой отдельный вариант осуществления настоящего изобретения. Соединения формулы (IA) или (I) могут быть представлены в форме соли с фармацевтически приемлемой кислотой или основанием. Такие соли предпочтительно выбирают из группы, включающей натрия, калия, магния, гемифумарат, гидрохлорид и гидробромид соли, с каждой возможностью представляя отдельный вариант осуществления настоящего изобретения. Одна предпочтительная сейчас соль представляет собой натриевую соль. Другая предпочтительная сейчас соль представляет собой гемифумаратную соль.
В некоторых специфических вариантах осуществления соединение выбирают из группы, включающей соединение 1, соединение 2, соединение 3, соединение 4, соединение 5, соединение 6, соединение 7, соединение 8, соединение 9, соединение 10, соединение 11 и соединение 12 и его фармацевтически приемлемые соли. Эти соединения представлены следующими структурами:
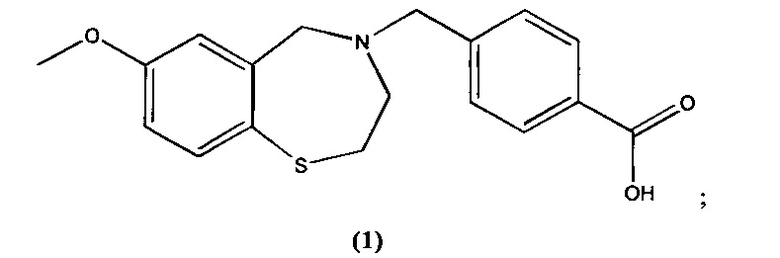
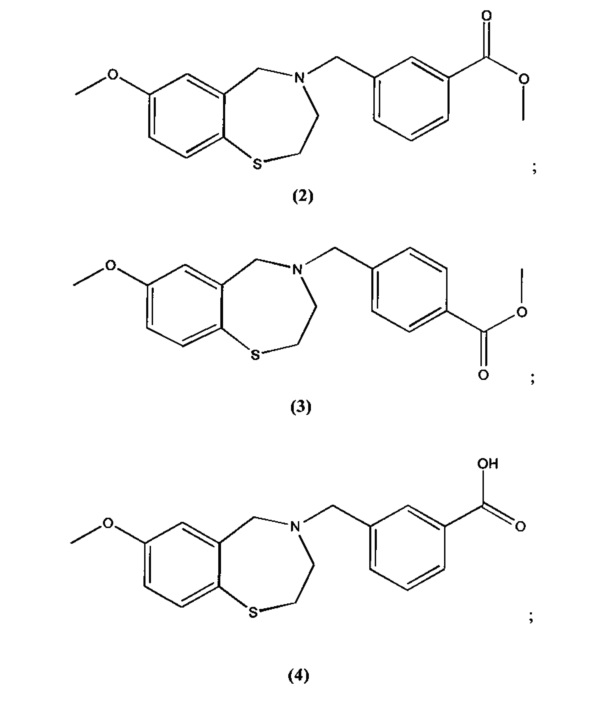
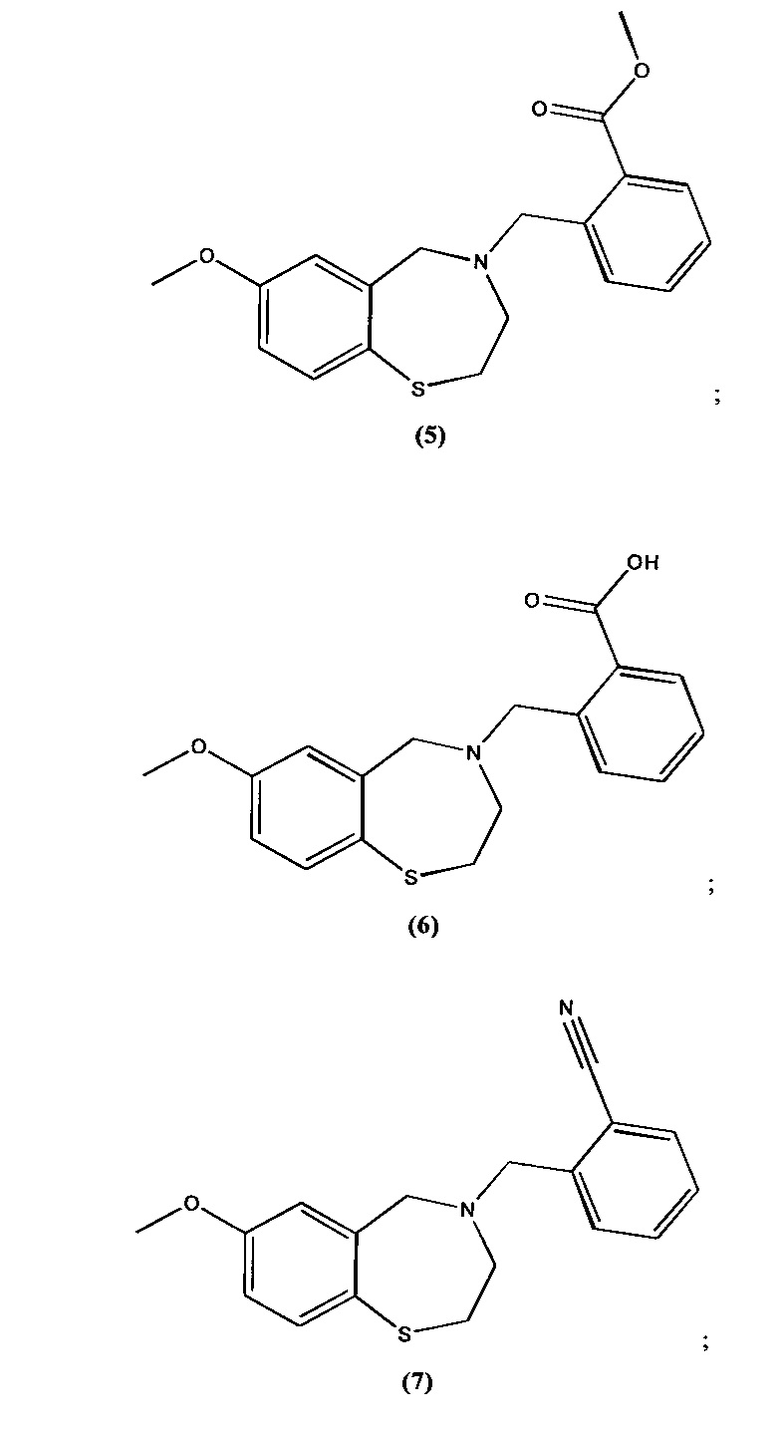
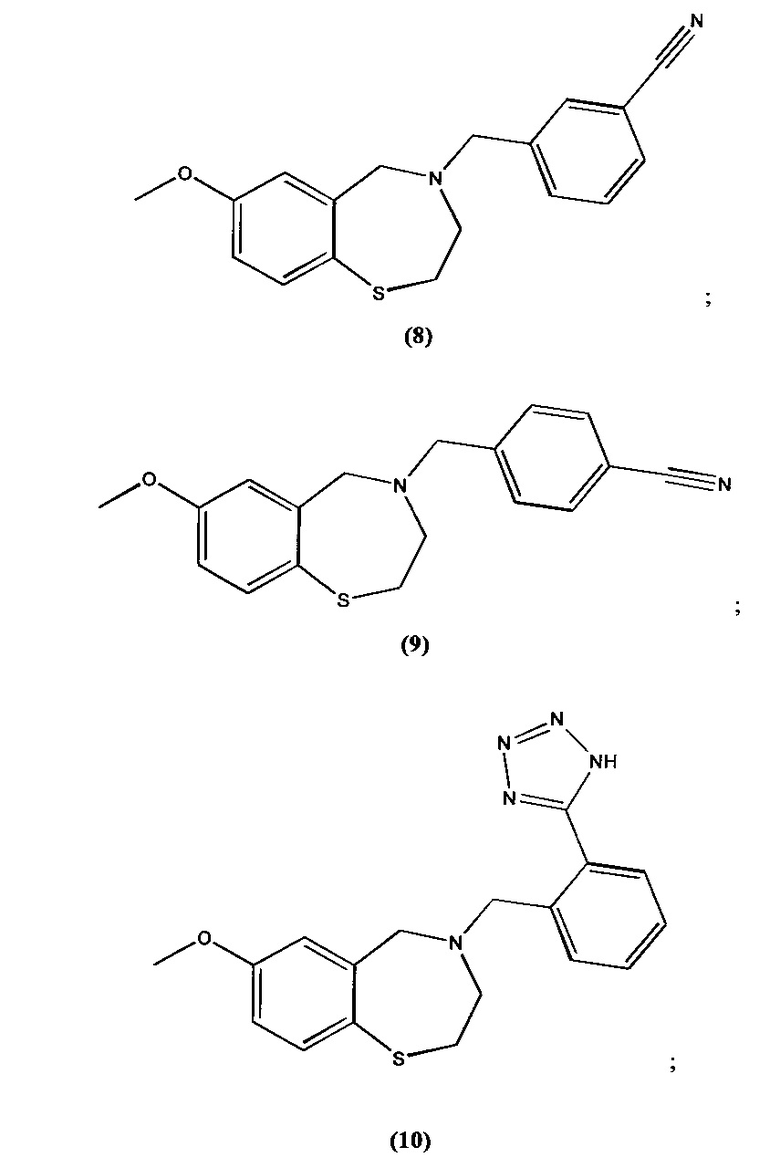
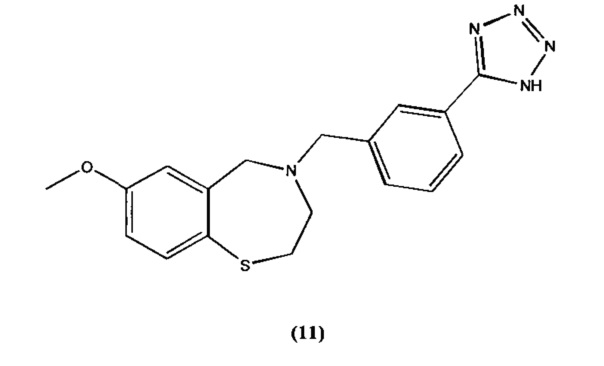 ; и
; и
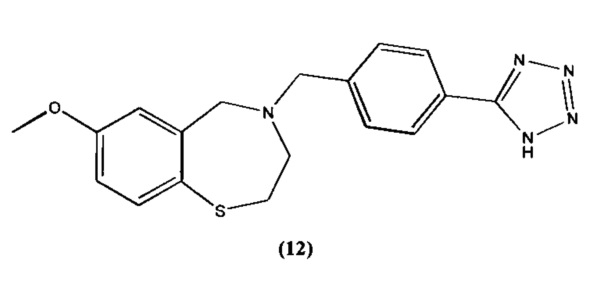
Химические определения
Термин "алкил", как используется в настоящей заявке, относится к линейному или разветвленному, насыщенному углеводороду, который имеет от 1 до 4 атомов углерода ("C1-C4 алкил"). Типичные алкильные группы включают, но не ограничиваясь только ими, метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил. Алкильная группа может быть незамещена или замещена одной или несколькими группами, выбранными из галогена, галоалкила, гидрокси, алкокси, галоалкокси, циклоалкила, арила, гетероциклила, гетероарила, амидо, алкиламидо, диалкиламидо, нитро, амино, циано, N3, оксо, алкиламино, диалкиламино, карбоксила, тио, тиоалкила и тиоарила.
Соединения согласно настоящему изобретению могут существовать в их таутомерной форме. Все такие таутомерные формы рассматриваются в данной заявке как часть настоящего изобретения.
Все стереоизомеры соединений согласно настоящему изобретению (например, те, которые могут существовать благодаря ассиметричным углеродам на различных заместителях), включая энантиомерные формы и диастереомерные формы, также охватываются объемом настоящего изобретения. Индивидуальные стереоизомеры соединения согласно изобретению, например, могут быть по существу свободными от других изомеров (например, в виде чистого или по существу чистого оптического изомера, имеющего удельную активность) или могут быть смешаны, например, в виде рацематов или в виде смесей, обогащенных одним стереоизомером. Хиральные центры согласно настоящему изобретению могут иметь S или R конфигурацию, как определено Рекомендациями ИЮПАК (IUPAC) 1974. Рацемические формы могут быть разделены физическими методами, такими как, например, фракционированная кристаллизация, разделение или кристаллизация производных диастереомеров или разделение с помощью хиральной колоночной хроматографии. Индивидуальные оптические изомеры могут быть получены из рацематов с помощью любого подходящего метода, включая, без ограничений, общепринятые методы, такие как, например, образование соли с оптически активной кислотой или основанием, с последующей кристаллизацией.
Соединения согласно настоящему изобретению, после их получения, предпочтительно выделяют и очищают, получая композицию, содержащую количество по весу, равное или больше чем приблизительно 90% соединения, приблизительно 95% соединения и даже более предпочтительно больше чем приблизительно 99% соединения ("по существу чистое" соединение), которое затем используют или приготавливают в виде фармацевтического препарата, как описано в настоящей заявке. Такие "по существу чистые" соединения согласно настоящему изобретению также рассматриваются в данной заявке как часть настоящего изобретения.
Терапевтическое применение
Настоящее изобретение обеспечивает соединения, которые способны лечить состояния, нарушения и заболевания, связанные с RyRs. Более предпочтительно, настоящее изобретение обеспечивает соединения, которые способны фиксировать утечку в RyR каналах, которые могут представлять собой RyR1, RyR2 и/или RyR3 каналы. В одном варианте осуществления соединения согласно изобретению усиливают ассоциацию и/или ингибируют диссоциацию RyR и калстабина (например, RyR1 и калстабин 1; RyR2 и калстабин 2; и RyR3 и калстабин 1). "Состояния, нарушения и заболевания, связанные с RyRs", обозначают нарушения и заболевания, которые можно лечить и/или предотвращать путем модуляции RyRs и включают, без ограничений, нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, нарушения и заболевания опорно-двигательного аппарата, нарушения и заболевания ЦНС, когнитивную дисфункцию, нейромышечные заболевания и нарушения, улучшение когнитивной функции, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти.
Таким образом, в одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, нарушения и заболевания опорно-двигательного аппарата, нарушения и заболевания ЦНС, когнитивную дисфункцию, нейромышечные заболевания и нарушения, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции, способ включает стадию введения субъекту, который в этом нуждается, терапевтически эффективного количества соединения формулы (I) или (IA), как описано в данной заявке, или его соли, для осуществления такого лечения. Данное предпочтительное соединение представляет собой соединение формулы (1).
В другом варианте осуществления настоящее изобретение относится к применению эффективного количества соединения формулы (I) или (IA), как описано в данной заявке, или его соли, для приготовления лекарственного средства для предотвращения или лечения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, скелетно-мышечные нарушения и заболевания, нарушения и заболевания ЦНС, нейромышечные заболевания и нарушения, когнитивную дисфункцию, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции. Данное предпочтительное соединение представляет собой соединение формулы (1).
В другом варианте осуществления настоящее изобретение относится к соединению формулы (I) или (IA), как описано в данной заявке, или его соли, для применения для приготовления лекарственного средства для предотвращения или лечения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, скелетно-мышечные нарушения и заболевания, нарушения и заболевания ЦНС, когнитивную дисфункцию, нейромышечные заболевания и нарушения, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции. Данное предпочтительное соединение представляет собой соединение формулы (1).
В одном варианте осуществления состояние, нарушение или заболевание связано с аномальной функцией RyR1. В другом варианте осуществления состояние, нарушение или заболевание связано с аномальной функцией RyR2. В другом варианте осуществления состояние, нарушение или заболевание связано с аномальной функцией RyR3. Каждая возможность представляет собой отдельный вариант осуществления настоящего изобретения.
Нарушения сердечной деятельности и кардиологические заболевания включают, но не ограничиваясь только ими, нарушения и заболевания, связанные с аритмией сердца, вызванные физической нагрузкой нарушения и заболевания, связанные с аритмией сердца, сердечную недостаточность, застойную сердечную недостаточность, хроническую сердечную недостаточность, острую сердечную недостаточность, систолическую сердечную недостаточность, диастолическую сердечную недостаточность, острую декомпенсированную сердечную недостаточность, сердечную ишемию / реперфузионное повреждение (I/R) (включая I/R повреждение после коронарной ангиопластики или после тромболизиса при инфаркте миокарда (MI)), хроническое обструктивное заболевание легких и повышенное кровяное давление. Нарушения и заболевания, связанные с аритмией сердца включают, но не ограничиваясь только ими предсердную и желудочковую аритмию, предсердную и желудочковую фибрилляцию, предсердную и желудочковую тахиаритмию, предсердную и желудочковую тахикардию, катехоламинергическую полиморфную желудочковую тахикардию (CPVT) и вызванные физической нагрузкой их варианты.
Соединения согласно изобретению также пригодны для лечения мышечного утомления, которое может быть обусловлено пролонгированной нагрузкой или нагрузкой высокой интенсивности, или может быть вызвано мышечно-скелетными заболеваниями. Примеры мышечных нарушений и заболеваний включают, но не ограничиваясь только ими, утомление скелетных мышц, поражение сердцевины мышечных волокон, вызванное физической нагрузкой утомление скелетных мышц, нарушения мочевого пузыря, недержание, возрастное мышечное утомление, саркопению, врожденные миопатии, миопатии и/или атрофии скелетных мышц, раковую кахексию, миопатию с сердцевинами и палочками, митохондриальные миопатии [например, синдром Kearns-Sayre, синдром MELAS (митохондриальную миопатию, энцефалопатию, лактацидоз и удар) и синдром MERRF (миоклоническая эпилепсия с разорванными красными волокнами)], эндокринные миопатии, болезни накопления гликогена в мышцах [например, болезнь Pompe, болезнь Andersen и болезнь Cori], миоглобинурии [например, болезнь McArdle, болезнь Tarui и болезнь DiMauro], дерматомиозиты, оссифицирующий миозит, семейный периодический паралич, полимиозит, миозит с тельцами включения, нейромиотонию, синдром мышечной скованности, злокачественную гипертермию, обыкновенные мышечные судороги, тетанию, миастению гравис, спинальную мышечную атрофию (SMA), спинальную и бульбарную мышечную атрофию (SBMA, также известную как спинобульбарную мышечную атрофия, бульбо-спинальную атрофию, X-связанную бульбоспинальную невропатию (XBSN), X-связанную спинальную мышечную атрофию 1 типа (SMAX1) и болезнь Kenneds (KD)) и мышечную дистрофию. Предпочтительные скелетно-мышечные нарушения включают, но не ограничиваясь только ими, вызванное физической нагрузкой утомление скелетных мышц, врожденную миопатию, мышечную дистрофию, старческое мышечное утомление, саркопению, поражение сердцевины мышечных волокон, раковую кахексию, нарушения мочевого пузыря и недержание.
Примеры мышечной дистрофии включают, но не ограничиваясь только ими, мышечную дистрофию Дюшенна (DMD), мышечную дистрофию Беккера (BMD), тазо-плечевую мышечную дистрофию (LGMD), врожденную мышечную дистрофию (CMD), дистальную мышечную дистрофию, плече-лопаточно-лицевую мышечную дистрофию, миотоническую мышечную дистрофию, мышечную дистрофию Эмери-Дрейфуса и окулофарингеальную мышечную дистрофию, где особенно предпочтительной является DMD.
Врожденная мышечная дистрофия, как используется в настоящей заявке, относится к мышечной дистрофии, которая является перинатальной. CMD классифицируется на основании генетических мутаций: 1) гены, кодирующие структурные белки базальной мембраны или внеклеточного матрикса волокон скелетных мышц; 2) гены, кодирующие предполагаемые или доказанные гликозилтрансферазы, которые, в свою очередь, оказывают влияние на гликозилирование дистрогликана, наружного мембранного белка базальной мембраны; и 3) другие. Примеры CMD включают, но не ограничиваясь только ими ламинин-α2-дефицитная CMD (MDC1A), Ullrich CMG (UCMDs 1, 2 и 3), синдром Walker-Warburg (WWS), заболевание мышцы-глаза-головной мозг (МЕВ), Fukuyama CMD (FCMD), CMD плюс вторичная недостаточность ламинина 1 (MDC1B), CMD плюс вторичная недостаточность ламинина 2 (MDC1C), CMD с умственной отсталостью и пахигирией (MDC1D) и ригидный позвоночник с мышечной дистрофией 1 типа (RSMD1).
Когнитивная дисфункция может быть связана или включать, но не ограничиваясь только ими, потерю памяти, связанную с возрастом потерю памяти, посттравматическое стрессовое расстройство (PTSD), синдром дефицита внимания с гиперактивностью (ADHD), расстройства аутистического спектра (ASD), генерализированное тревожное расстройство (GAD), обсессивно-компульсивное расстройство (OCD), шизофрению, биполярное расстройство или большую депрессию.
Нарушения и заболевания ЦНС включают, но не ограничиваясь только ими болезнь Альцгеймера (AD), невропатию, пароксизмы, болезнь Паркинсона (PD) и болезнь Хантингтона (HD).
Нейромышечные нарушения и заболевания включают, но не ограничиваясь только ими спинально-церебеллярную атаксию (SCA) и боковой амиотрофический склероз (ALS, болезнь Лу Герига).
В некоторых вариантах осуществления соединения согласно настоящему изобретению улучшают когнитивную функцию, которая может быть выбрана из кратковременной памяти, долговременной памяти, внимания, обучения и любой их комбинации.
В некоторых вариантах осуществления соединения согласно настоящему изобретению пригодны для лечения раковой кахексии, то есть мышечная слабость, которая связана со злокачественным новообразованием в целом, и предпочтительно мышечная слабость при метастатическом злокачественном новообразовании, таком как метастазы в костях. Мышечная слабость и мышечная атрофия (кахексия) являются распространенными паранеопластическими симптомами у пациентов со злокачественными новообразованиями. Эти состояния вызывают существенное утомление и значительное снижение качества жизни пациентов. Настоящее изобретение обеспечивает способ лечения и предотвращения мышечной слабости у пациента со злокачественным новообразованием, основанном, в частности, на открытии того, что при определенных типах злокачественных новообразований, например, при раке предстательной железы и молочной железы с метастазами в костях, RyR1 окисляется, что вызывает его "утечку". Дополнительно было обнаружено, что предотвращение утечки путем введения Rycal соединений улучшает мышечную функцию. Примеры злокачественных новообразований включают, но не ограничиваясь только ими, рак молочной железы, рак предстательной железы, рак костей, рак поджелудочной железы, рак легкого, рак ободочной кишки и рак желудочно-кишечного тракта.
Терапия перескакиванием экзона
В некоторых вариантах осуществления соединения согласно настоящему изобретению модулируют (например, усиливают) мРНК сплайсинг путем усиления антисмыслового опосредованного перескакивание экзона. Эта модуляция сплайсинга осуществляется в присутствии антисмысловых олигонуклеотидов (AOs), которые являются специфическими для сплайсинговых последовательностей, представляющих интерес. В некоторых вариантах осуществления согласно изобретению, соединение формулы (I) или (IA) и АО могут действовать синергетически, соединение формулы (I) или (IA) усиливает АО опосредованное перескакивание экзона. Таким образом, в некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции для применения для лечения или предотвращения любого из состояний, описанных в настоящей заявке, которые связаны с дающими утечку RyR, дополнительно включающие применение антисмыслового АО, который специфический для сплайсинговой последовательности в мРНК последовательности, для усиливающего экзона, пропущенного в мРНК, представляющей интерес.
Один предпочтительный вариант осуществления изобретения для усиления перескакивания экзона с помощью соединений согласно настоящему изобретению касается мышечной дистрофии Дюшенна (DMD). DMD представляет собой летальное X-связанное рецессивное заболевание, которое характеризуется прогрессирующей мышечной слабостью в течение периода жизни пациента. DMD главным образом вызывается выходящими из рамки мульти-экзоннными делециями в DMD гене, которые отсекают продукцию белка дистрофина. Потеря экспрессии дистрофина отдельно не раскрывает патофизиологию DMD. Разрушение комплекса дистрофин-гликопротеин (DGC) также приводит к окислительному стрессу, перегрузке митохондриального Са2+ и апоптозу, повышению протока Са2+ в мышцы и паталогической Са2+ передаче сигналов. Отсутствует эффективное лечение для DMD, и фармакологическое лечение было продемонстрировано только для кортикостероидов, которые могут пролонгировать способность передвигаться, но имеют существенные побочные действия. Опосредованное антисмысловым олигонуклеотидом перескакивание экзона является перспективным терапевтическим подходом, нацеленным на восстановление DMD рамки считывания и предоставляющим возможность экспрессии интактного комплекса дистрофина с гликопротеином. До настоящего времени, с помощью этого метода были получены низкие уровни белка дистрофина у людей. Kendall и др. (Sci Transl Med, 2012, 4(164), p. 164ra 160) описали, что определенные низкомолекулярные структуры, такие как Дантролен и другие RyR модуляторы, потенциируют опосредованное антисмысловым олигомеров перескакивание экзона для повышения перескакивания экзона для восстановления мРНК рамки считывания, сарколеммного белка дистрофина и комплекса дистрофина с гликопротеном в скелетных мышцах mdx мышей, мышиная модель DMD.
Таким образом, в одном варианте осуществления настоящее изобретение относится к способу лечения DMD, путем введения субъекту, который в этом нуждается, соединения формулы (I) или (IA) в соответствии с настоящим изобретением, в комбинации с антисмысловым олигонуклеотидом (АО), который специфический для сплайсинговой последовательности одного или нескольких экзонов DMD гена, например, экзон 23, 45, 44, 50, 51, 52 и/или 53 DMD гена. Предпочтительные AOs включают, но не ограничиваясь только ими, AOs нацеливающий DMD экзон 23, 50 и/или 51 DMD гена, такой как 2'-O-метил (2'ОМе) фосфоротиоат или фосфородиамидат морфолино (РМО) AOs. Примеры таких AOs включают, но не ограничиваясь только ими, Pro051/GSK2402968, AVI4658/Eteplirsen и РМО Е23 морфолино (5'-GGCCAAACCTCGGCTTACCTGAAAT-3').
Термин "эффективное количество", "достаточное количество" или "терапевтически эффективное количество" средства, как используется в настоящей заявке взаимозаменяемо, представляет собой количество, которого достаточно для осуществления благоприятных или желательных результатов, включая клинические результаты и, как таковые, эффективное количество" или его варианты зависят от контекста, в котором они применяется. Ответ в некоторых вариантах осуществления является профилактическим, в других терапевтическим и в других их комбинацией. Термин "эффективное количество" также включает количество соединения согласно изобретению, которое является "терапевтически эффективным" и которое исключает или существенно ослабляет нежелательные побочные эффекты.
Как используется в настоящей заявке и очевидно для специалиста в данной области техники, "лечение" представляет собой подход для получения благоприятных или желательных результатов, включая клинические результаты. Благоприятные или желательные клинические результаты могут включать, но не ограничиваясь только ими, ослабление или уменьшение одного или нескольких симптомов или состояний, снижение степени заболевания, стабилизации (то есть отсутствие ухудшения) стадии заболевания, предотвращение распространения заболевания, замедления или снижения прогрессирования заболевания, улучшение или облегчение степени заболевания и ремиссия (либо частичная, либо общая), либо обнаруживаемая, либо необнаруживаемая. "Лечение" также может обозначать пролонгирование выживания по сравнению с ожидаемым выживанием у субъектов, не получавших лечения.
Фармацевтические композиции
Соединения согласно изобретению приготавливаются в виде фармацевтических композиций для введения людям в биологически совместимой форме, подходящей для введения in vivo. В соответствии с другим аспектом настоящее изобретение обеспечивает фармацевтическую композицию, которая содержит соединения согласно изобретению в смеси с фармацевтически приемлемым разбавителем и/или носителем. Фармацевтически приемлемый носитель предпочтительно является "приемлемым" в смысле совместимости с другими компонентами композиции и не опасным для его реципиента.
Соединение можно вводить отдельно, но предпочтительно его вводят с одним или несколькими фармацевтически приемлемыми носителями. Фармацевтически приемлемый носитель, используемый в настоящей заявке, может быть выбран из различных органических или неорганических материалов, которые используются в качестве материалов для фармацевтических препаратов и которые инкорпорированы в качестве любого одного или нескольких из следующих: заполнители, разбавители, связующие, агенты, вызывающие дезинтеграцию, буферные вещества, красители, эмульсификаторы, средства, улучшающие запах, гелеобразующие вещества, скользящие вещества, консерванты, солюбилизаторы, стабилизаторы, суспендирующие агенты, подсластители, вещества, регулирующие тоничность, смачивающие вещества, эмульсификаторы, диспергирующие вещества, агенты, вызывающие набухание, ретарданты, скользящие вещества, абсорбирующие вещества и средства, повышающие вязкость.
Соединения согласно настоящему изобретению вводят человеку или животному с помощью известных процедур, включая, без ограничений, пероральное, сублингвальное, буккальное, парентеральное (внутривенное, внутримышечное или подкожное), трансдермальное, чрес- или транскожное, интраназальное, интра-вагинальное, ректальное, глазное и респираторное (путем ингаляционного введения). Соединения согласно изобретению также могут вводиться субъекту путем доставки в мышцы субъекта, включая, но не ограничиваясь только ими, сердечные или скелетные мышцы субъекта. В одном варианте осуществления соединение вводят субъекту путем целевой доставки в мышечные клетки сердца с помощью катетера, вставленного в сердце субъекта. В других вариантах осуществления соединения могут вводиться непосредственно в ЦНС, например, путем эндолюмбальной инъекции или внутрижелудочковой инъекции соединений непосредственно в цереброспинальную жидкость (CSF) или путем внутрижелудочкового, интратекального или интерстициального введения. Особенно предпочтительным является пероральное введение.
Фармацевтические композиции в соответствии с изобретением для твердого перорального введения включают, в особенности, таблетки или драже, подъязычные таблетки, саше, капсулы, включая желатиновые капсулы, порошки и гранулы и композиции для жидкого перорального, назального, буккального или глазного введения включают, в особенности, эмульсии, растворы, суспензии, капли, сиропы и аэрозоли. Соединения также можно вводить в виде суспензии или раствора с помощью питьевой воды или с пищей. Примеры приемлемых фармацевтических носителей включают, но не ограничиваясь только ими, производные целлюлозы, включая карбоксиметил целлюлозу, метил целлюлозу, гидроксипропил целлюлозу, гидроксипропилметилцеллюлозу, этил целлюлозу и микрокристаллическую целлюлозу; сахара, такие как, в частности, маннит, сахароза или лактоза; глицерин, гуммиарабик, стеарат магния, натрия стеарил фумарат, солевой раствор, альгинат натрия, крахмал, тальк и воду.
Фармацевтические композиции в соответствии с изобретением для парентеральных инъекций включают, в особенности, стерильные растворы, которые могут быть водными или неводными, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления инъекционных растворов или дисперсий. Соединения согласно изобретению можно комбинировать со стерильным водным раствором, который является изотоническим с кровью субъекта. Такой препарат приготавливают путем растворения твердого активного компонента в воде, содержащей физиологически совместимые вещества, такие как хлорид натрия, глицин и другие, и имеющей забуференное pH, совместимое с физиологическими условиями, таким образом, что получают водный раствор, и после этого указанному раствору придают стерильность. Препарат представлен в единичных или многодозовых контейнерах, таких как запечатанные ампулы или флаконы. Препарат доставляется с помощью любого способа инъекции, включая, без ограничений, эпифасциальный, внутрикасульный, внутричерепной, внутрикожный, интратекальный, внутримышечный, внутриорбитальный, внутрибрюшинный, интраспинальный, интрастернальный, внутрисосудистый, внутривенный, паренхиматозный, подкожный или подъязычный или с помощью катетера в сердце субъекта.
Фармацевтические композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории и композиции для черес- или транскожного введения включают, в особенности, порошки, аэрозоли, кремы, мази, гели и пластыри.
Для трансдермального введения соединения согласно изобретению комбинируют с усилителями проникновения через кожу, такими как пропиленгликоль, полиэтиленгликоль, изопропанол, этанол, олеиновая кислота, N-метилпирролидон и другими, которые повышают проницаемость кожи для соединений согласно изобретению и позволяют соединениям проникать через кожу и в кровоток. Композиции соединение/усилитель можно дополнительно комбинировать с полимерным веществом, таким как этилцеллюлоза, гидроксипропил целлюлоза, этилен/винилацетат, поливинил пирролидон и другими, обеспечивая композиции в форме геля, которую растворяют в растворителе, упаривают до желательной вязкости и затем наносят на материал-подложку, обеспечивая пластырь.
Фармацевтические препараты согласно настоящему изобретению приготавливают с помощью методов, хорошо известных в области фармацевтики, включая, но не ограничиваясь только ими, методы влажной и сухой грануляции, или путем прямого прессования. Выбор носителя определяется растворимостью и химической природой соединений, выбранным путем введения и стандартной фармацевтической практикой.
Фармацевтические композиции, упомянутые выше, иллюстрируют изобретение, но никоим образом его не ограничивают.
В соответствии со способами согласно настоящему изобретению любые из этих соединений могут вводиться субъекту (или контактировать с клетками субъекта) в количестве, эффективном для ограничения или предотвращения снижения уровня RyR-связанного калстабина у субъекта, предпочтительно в клетках субъекта. Это количество легко может быть определено квалифицированным специалистом в данной области техники, на основе известных процедур, включая анализы титрационных кривых, полученные in vivo, и способы и анализы, описанные в настоящей заявке. Подходящее количество соединений согласно изобретению эффективно для ограничения или предотвращения снижения уровня RyR-связанного калстабина у субъекта в диапазоне от приблизительно 0,01 мг/кг/сутки до приблизительно 100 мг/кг/сутки (например, 1, 2, 5, 10, 20, 25, 50 или 100 мг/кг/сутки), и/или представляет собой количество, достаточное для достижения уровней в плазме в диапазоне от приблизительно 300 нг/мл до приблизительно 5,000 нг/мл. Альтернативно, количество соединений согласно изобретению находится в диапазоне от приблизительно 1 мг/кг/сутки до приблизительно 50 мг/кг/сутки. Альтернативно, количество соединений согласно изобретению находится в диапазоне от приблизительно 10 мг/кг/сутки до приблизительно 20 мг/кг/сутки. Также охватываются количества от приблизительно 0,01 мг/кг/сутки или 0,05 мг/кг/сутки до приблизительно 5 мг/кг/сутки или приблизительно 10 мг/кг/сутки, которые можно вводить.
Способы синтеза
Настоящее изобретение обеспечивает, в дальнейшем аспекте, способы получения соединения согласно изобретению и его соли. Более предпочтительно, настоящее изобретение обеспечивает способы получения соединений формулы (I) или (IA), например соединения 1, соединения 2, соединения 3, соединения 4, соединения 5, соединения 6, соединения 7, соединения 8, соединения 9, соединения 10, соединения 11 и соединения 12, или их солей. Различные пути синтеза соединений описаны в примерах. Общий путь синтеза (ROS) представлен на Схеме 1 ниже:
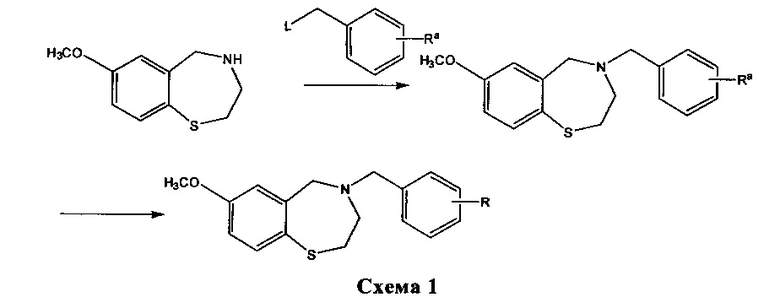
На Схеме 1, Ra представляет собой COOR1 или CN; R1 представляет собой C1-C4 алкил, и L представляет собой уходящую группу, которая представляет собой, например, галоген, сульфонат (OSO2R' где R' представляет собой алкил или арил, например, OMs (мезилат), OTs (тозилат)) и другие. Аминовое исходное вещество подвергают реакции с алкилирующим агентом (производным бензила, представленным выше), предпочтительно в присутствии основания, получая желательный продукт или его предшественник (R=Ra). Если это является желательным, то предшественник в дальнейшем можно подвергать реакции для превращения группы Ra в группу R, как подтверждено примерами в экспериментальном разделе в данной заявке ниже, или с помощью любого другого метода, известного специалисту в данной области техники. Например, сложноэфирный предшественник (Ra=COOR1 где R1 представляет собой C1-C4 алкил), можно превращать в соответствующую карбоновую кислоту (R=COOH) путем гидролиза в кислых или щелочных условиях в соответствии с известными способами. Альтернативно, нитрильный предшественник (Ra=CN) можно превращать в тетразол (изостер карбоновой кислоты) путем реакции с азидом натрия в подходящих условиях, или в карбоновую кислоту (R=COOH) путем гидролиза.
Аминовое исходное вещество может быть получено в соответствии с методами, описанными в WO 2009/111463 или WO 2007/024717, или с помощью любого другого метода, известного специалисту в данной области техники. Содержание всех вышеуказанных документов включено в настоящую заявку в качестве ссылки. Природа основания особо не ограничена. Предпочтительные основания включают, но не ограничиваясь только ими, гидриды (например, гидрид натрия или калия) и N,N-диизопропилэтиламин. Другие подходящие основания bases включают, но не ограничиваясь только ими, органическое основание, такое как третичный амин, выбранный из группы, включающей ациклические амины (например, триметиламин, триэтиламин, диметилфениламин диизопропилэтиламин и трибутиламин), циклические амины (например, N-метилморфолин) и ароматические амины (диметиланилин, диметиламинопиридин и пиридин).
Реакция может быть осуществлена в присутствии или отсутствии растворителя. Природа растворителя, если он используется, особо не ограничена, примеры включают растворители, такие как сложный эфир (например, этил ацетат), простой эфир (например, ТГФ), хлорированный растворитель (например, дихлорметан или хлороформ), диметилформамид (ДМФА) и другие растворители, такие как ацетонитрил или толуол или смеси этих растворителей друг с другом или с водой.
Соли соединений формулы (I), где R=COOH, могут быть приготовлены путем взаимодействия исходной молекулы с подходящим основанием, например, NaOH или KOH, получая соответствующие соли щелочных металлов, например, соли натрия или калия. Альтернативно, сложные эфиры (R=COOR1) могут быть непосредственно превращены в соли путем реакций с подходящими основаниями.
Соли соединений формулы (I) также могут быть приготовлены путем реакции исходной молекулы с подходящей кислотой, например HCl, фумаровой кислотой, или пара-толуолсульфоновой кислотой, получая соответствующие соли, например гидрохлорид, тозилат или геми-фумарат.
Примеры
Следующие примеры представлены в качестве иллюстрации некоторых предпочтительных вариантов осуществления изобретения.
Пример 1: Синтез
Приборы
ЯМР: Bruker AVANCE III 400 или Varian Mercury 300
ЖХ/МС: Waters Delta 600, оборудованный автоматическим пробоотборником 717 Plus, детектором с фотодиодной матрицей 2996 и масс-детектором 3100, или Shimadzu 210
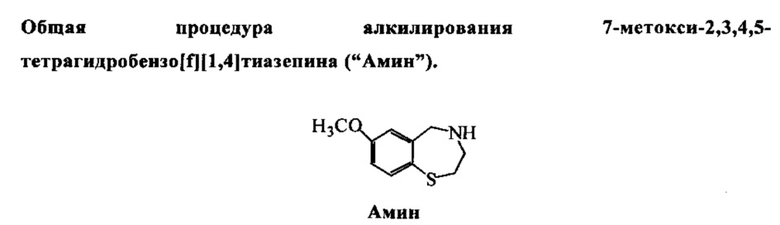
Амин (структура, представленная выше) (1 ммоль) растворяли в 3 мл дихлорметана. К раствору добавляли алкилирующий реагент (1 ммоль), после этого добавляли N,N-диизопропилэтиламин (0,34 мл, 2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Раствор загружали на колонку сразу и элюировали с гексаном /EtOAc (2:1, об./об.).
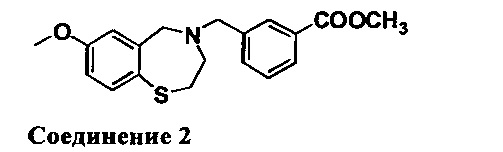
Метил 3-((7-метокси-2,3-дигидробензо[f][1,4]тиазепин-4(5H)-ил)метил)бензоат: 1НЯМР (300 МГц, CDCl3): 7,96 (m, 2Н), 7,46 (m, 3Н), 6,70 (dd, J=8,4 Гц, 3,0 Гц, 1Н), 6,50 (d, J=2,7 Гц, 1H), 4,09 (s, 2Н), 3,90 (s, 3Н), 3,72 (s, 3Н), 3,57 (s, 2Н), 3,35 (m, 2Н), 2,72 (m, 2Н). МС: 344 (M+1)
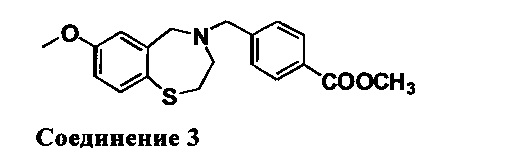
Метил 4-((7-метокси-2,3-дигидробензо[1][1,4]тиазепин-4(5Н)-ил)метил)бензоат: 1НЯМР (300 МГц, CDCl3): 7,99 (d, J=8,4 Гц, 2Н), 7,46 (d, J=8,4 Гц, 1Н), 7,37 (d, J=8,7 Гц, 2H), 6,70 (dd, J=8,4 Гц, 3,0 Гц, 1H), 6,50 (d, J=2,7 Гц, 1H), 4,09 (s, 2H), 3,90 (s, 3H), 3,72 (s, 3H), 3,57 (s, 2H), 3,35 (m, 2H), 2,72 (m, 2H). MC: 344 (M+1)
Метил 4-((7-метокси-2,3-дигидробензо[1][1,4]тиазепин-4(5Н)-ил)метил)бензоат: 1НЯМР (300 МГц, CDCl3): 7,99 (d, J=8,4 Гц, 2Н), 7,46 (d, J=8,4 Гц, 1Н), 7,37 (d, J=8,7 Гц, 2H), 6,70 (dd, J=8,4 Гц, 3,0 Гц, 1H), 6,50 (d, J=2,7 Гц, 1H), 4,09 (s, 2H), 3,90 (s, 3H), 3,72 (s, 3H), 3,57 (s, 2H), 3,35 (m, 2H), 2,72 (m, 2H). MC: 344 (M+1)
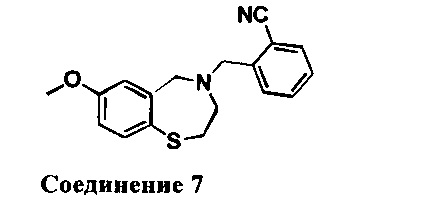
2-((7-Метокси-2,3-дигидробензо[f][1,4]тиазепин-4(5Н)-ил)метил)бензонитрил: 1НЯМР (300 МГц, CDCl3): 7,67-7,26 (m, 5Н), 6,73 (d, J=2,7 Гц, 1Н), 6,74 (dd, J=2,7,8,4 Гц, 1H), 4,14 (s, 2Н), 3,78(s, 3Н), 3,70 (s, 2Н), 3,36 (m, 2Н), 2,76 (m, 2Н). МС: 311 (M+1)
3-((7-Метокси-2,3-дигидробензо[1][1,4]тиазепин-4(5Н)-ил)метил)бензонитрил: 1НЯМР (300 МГц, CDCl3): 7,64-7,42 (m, 5Н), 6,74 (dd, J=2,7,8,4 Гц, 1Н), 6,48 (d, J=2,7 Гц, 1H), 4,08 (s, 2Н), 3,75(s, 3Н), 3,57 (s, 2Н), 3,36 (m, 2Н), 2,76 (m, 2Н). МС: 311 (M+1)
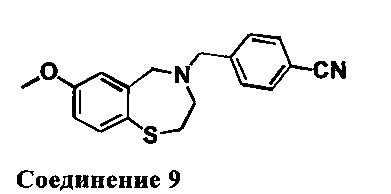
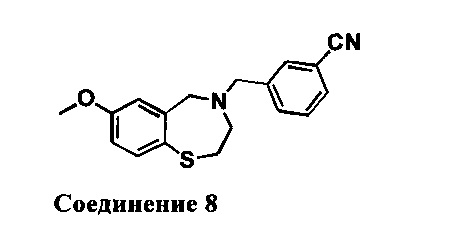
4-((7-Метокси-2,3-дигидробензо[1][1,4]тиазепин-4(5Н)-ил)метил)бензонитрил: 1НЯМР (300 МГц, CDCl3): 7,64 (d, J=7,2 Гц, 2Н), 7,42 (m, 3Н), 6,74 (dd, J=2,7,8,4 Гц, 1Н), 6,48 (d, J=2,7 Гц, 1H), 4,08 (s, 2Н), 3,75(s, 3Н), 3,58 (s, 2Н), 3,36 (m, 2Н), 2,76 (m, 2Н). МС: 311 (M+1)
Гидролиз сложного эфира (общая процедура)
Метиловый сложный эфир (3 ммоль) растворяли в 30 мл ТГФ/метанол/1 M NaOH (1:1:1, об./об.). Смесь перемешивали в течение 8 часов и ТСХ показала полное исчезновение сложного эфира. Добавляли 1 мл конц. HCl для доведения до кислого pH. Органический растворитель удаляли и образованное твердое вещество собирали путем фильтрации. Твердое вещество высушивали на воздухе.
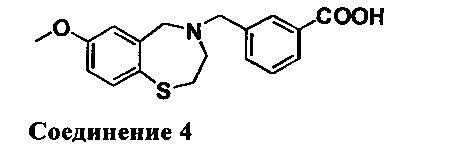
3-((7-Метокси-2,3-дигидробензо[f][1,4]тиазепин-4(5Н)-ил)метил)бензойная кислота: Ее получали путем экстрагирования с EtOAc в качестве растворителя. 1НЯМР (300 МГц, CDCl3): 8,10 (s, 1Н), 8,04 (d, J=8,4 Гц, 1Н), 7,80 (br, 1Н), 7,46 (m, 2Н), 6,80 (m, 2Н), 4,40 (s, 2Н), 3,90 (s, 2Н), 3,76 (s, 3Н), 3,42 (s, 2Н), 2,86 (s, 2Н). МС: 330 (M+1), 328 (M-1).
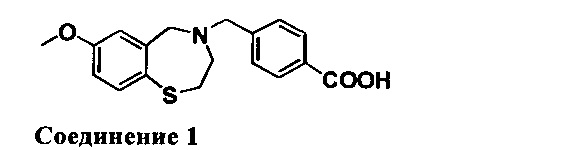
4-((7-Метокси-2,3-дигидробензо[1][1,4]тиазепин-4(5Н)-ил)метил)бензойная кислота: Ее получали путем экстрагирования с EtOAc в качестве растворителя. 1НЯМР (300 МГц, CDCl3): 8,02 (d, J=8,4 Гц, 2Н), 7,46 (d, J=8,4 Гц, 1Н), 7,42 (d, J=8,7 Гц, 2Н), 6,70 (dd, J=8,4 Гц, 3,0 Гц, 1Н), 6,50 (d, J=3,0 Гц, 1Н), 4,11 (s, 2Н), 3,72 (s, 3Н), 3,62 (s, 2Н), 3,35 (m, 2Н), 2,76 (m, 2Н). МС: 330 (M+1), 328 (M-1).
Соединение 1, натрия соль
Натриевую соль соединения 1 получали из исходной молекулы, используя 1 эквивалент NaOH в EtOH (tпл соли: > 290°C).
1НЯМР (ДМСО-D6, 600 МГц), δ (част. на млн): 7,77 (2Н, m), 7,41 (1Н, d), 7,13 (2Н, m), 6,75 (1Н, dd), 6,63 (1H, d), 4,00 (2Н, s), 3,70 (3Н, s), 3,49 (2Н, s), 3,18 (2Н, m), 2,70 (2Н, m).
Соединение 1, гемифумаратная соль
1,6 г соединения 1 (нейтральная форма) и 265 мг фумаровой кислоты вносили в круглодонную колбу. После добавления 18 мл ацетона и 2 мл воды, реакционную смесь нагревали в колбе с обратным холодильником. Наблюдали частичную солюбилизацию (но не полное осветление) с последующим осаждением. После этого реакционную смесь нагревали в колбе с обратным холодильником в течение ночи. После охлаждения оставшееся твердое вещество выделяли путем фильтрации, промывали 3 мл ацетона и высушивали в вакууме (40°C /10 мбар) в течение 4 часов.
1НЯМР (ДМСО-D6, 600 МГц), δ (част. на млн): 12,97 (2Н, bs), 7,90 (2Н, m), 7,43 (1H, d), 7,40 (2Н, m), 6,77 (1Н, dd), 6,64 (1Н, d), 6,62 (1H, s), 4,03 (2Н, s), 3,70 (3Н, s), 3,58 (2Н, s), 3,20 (2Н, m), 2,72 (2Н, m).
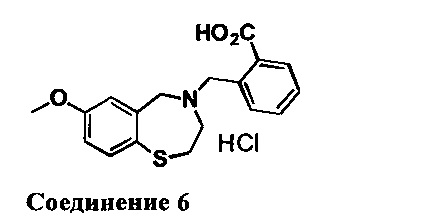
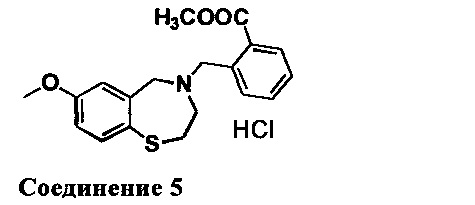
2-((7-Метокси-2,3-дигидробензо[f][1,4]тиазепин-4(5Н)-ил)метил)бензойная кислота: Соединение превращали в гидрохлоридную соль с 2М HCl в простом эфире. 1НЯМР (300 МГц, ДМСО-d6): 10,10 (br, 1Н), 8,08 (d, J=7,5 Гц, 1Н), 7,66-7,51 (m, 4Н), 7,17 (d, J=2,1 Гц, 1Н), 6,99 (dd, J=8,4, 2,1 Гц, 1Н), 4,80-4,40 (m, br, 4Н), 3,78 (s, 3Н), 3,46 (m, 2Н), 3,13 (m, 2Н). МС: 330 (M+1), 328 (M-1).
Синтез тетразола (общая процедура)
Нитрильный предшественник (3,22 ммоль), азид натрия (830 мг, 12,9 ммоль) и триэтиламин гидрохлорид (1,72 г, 12,9 ммоль) перемешивали в 40 мл безводного ДМФА при 100°C в течение 5 дней. ДМФА удаляли в высоком вакууме и остаток смешивали с водой. Водный раствор экстрагировали дихлорметаном (3 × 100 мл), Чистое соединение очищали путем колоночной хроматографии (EtOAc/метанол).
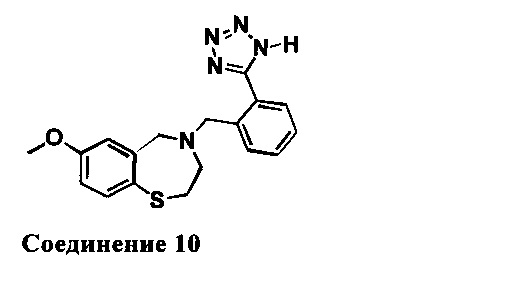
4-(2-(1Н-Тетразол-5-ил)бензил)-7-метокси-2,3,4,5-тетрагидробензо[f][1,4]тиазепин: 1НЯМР (300 МГц, CDCl3 и капля CD3OD): 8,30 (d, J=8,7 Гц, 1Н), 7,53 (m, 2Н). 7,14 (t, J=7,8 Гц, 1H), 7,20 (d, J=7,5 Гц, 1Н), 6,84 (dd, J=2,7,8,4 Гц, 1Н), 6,69 (d, J=2,7 Гц, 1H), 4,46 (s, 2H), 3,80 (s, 2H), 3,75 (s, 2H), 3,43 (m, 2H), 2,96 (m, 2H). MC: 354 (M+1), 352 (M-1)
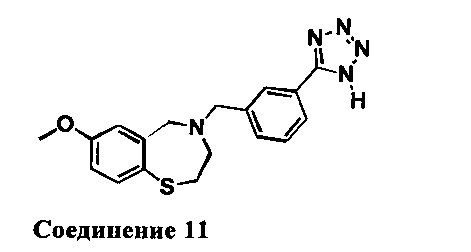
4-(3-(1Н-Тетразол-5-ил)бензил)-7-метокси-2,3,4,5-тетрагидробензо[f][1,4]тиазепин: 1НЯМР (300 МГц, CDCl3): 8,16 (s, 1Н), 7,90 (d, J=7,5 Гц, 1H), 7,40 (d, J=8,4 Гц, 1H),7,20 (m, 2H), 6,74 (dd, J=2,7,8,4 Гц, 1H), 6,58 (d, J=2,7 Гц, 1H), 4,18 (s, 2H), 3,75(s, 5H), 3,36 (m, 2H), 2,76 (m, 2H).). MC: 354 (M+1), 352 (M-1)
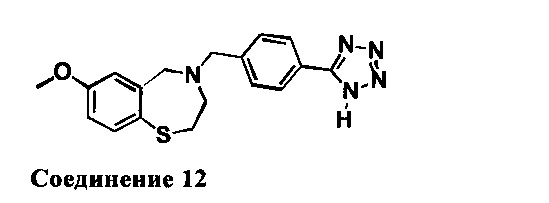
4-(4-(1Н-Тетразол-5-ил)бензил)-7-метокси-2,3,4,5-тетрагидробензо[f][1,4]тиазепин: 1НЯМР (300 МГц, CDCl3 и капля CD3OD): 7,99 (d, J=7,2 Гц, 2Н), 7,42 (m, 3Н), 6,74 (dd, J=2,7,8,4 Гц, 1Н), 6,53 (d, J=2,7 Гц, 1Н), 4,10 (s, 2Н), 3,71(s, 3Н), 3,58 (s, 2Н), 3,36 (m, 2Н), 2,76 (m, 2Н).). МС: 354 (M+1), 352 (M-1)
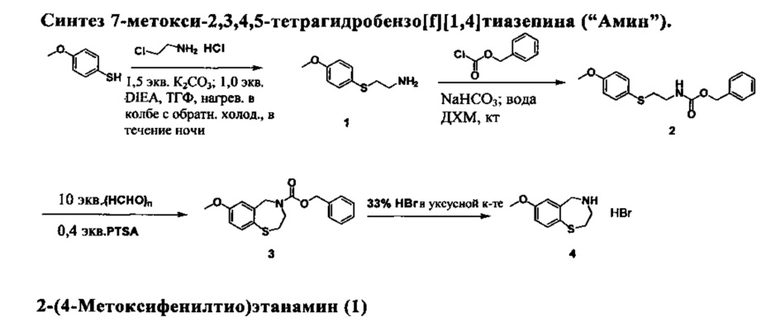
4-Метокситиофенол (50 г, 0,357 моль), 2-хлорэтиламин моногидрохлорид (39,8 г, 0,343 моль), K2CO3(78,8 г, 0,57 моль) и диизопропил этиламин (32 мл, 0,178 моль) смешивали в 200 мл ТГФ. Смесь дегазировали в течение 5 мин при пониженном давлении и нагревали в колбе с обратным холодильником в атмосфере аргона в течение ночи. Растворитель удаляли и в колбу добавляли воду (300 мл). Смесь экстрагировали дихлорметаном (3×200 мл). Органические компоненты собирали, удаляли дихлорметан и добавляли 50 мл конц. HCl, затем добавляли 200 мл воды. Раствор экстрагировали с помощью 1:1 EtOAc/гексан (3 × 200 мл). Водный слой доводили до pH 10 с помощью 2 М NaOH и экстрагировали с помощью дихлорметана (3×200 мл). Объединенный органический раствор высушивали над безводным сульфатом натрия. Удаление растворителя обеспечивает 61 г целевого соединения в виде бесцветной жидкости, с выходом 97%.
1Н-ЯМР (300 МГц, CDCl3): 7,35 (d, J=8,7 Гц, 2Н), 6,81 (d, J=8,7 Гц, 2Н), 3,77 (s, 3Н), 2,88-2,80 (m, 4Н), 1,44 (s, 2Н).
Бензил 2-(4-метоксифенилтио)этилкарбамат (2)
Первый способ
В колбу, содержащую соединение 1 (8,0 г, 43,7 ммоль), бикарбонат натрия (12,1 г, 144 ммоль), воду (100 мл) и дихлорметан (200 мл), добавляли бензил хлорформиат (8,2 г, 48,1 ммоль, разведенный в 100 мл дихлорметана) по каплям при 0°C. После добавления, смесь перемешивали при к.т. в течение 5 часов. Органический слой собирали и водный раствор экстрагировали с помощью 100 мл дихлорметана. Объединенный органический раствор высушивали над сульфатом натрия. Растворитель удаляли и полученное твердое вещество растирали в порошок с 200 мл ТГФ/гексан (1:10). Твердое вещество собирали и высушивали, получая целевой продукт (12,9 г) с выходом 93%.
Альтернативный способ
К раствору соединения 1 (10 г, 54,6 ммоль) и триэтиламина (15 мл, 106 ммоль) в 200 мл дихлорметана, добавляли бензил хлорформиат (7,24 мл, 51,5 ммоль, разведенный в 100 мл дихлорметана) по каплям при 0°C. После добавления, раствор перемешивали при к.т. в течение одного часа. Твердое вещество удаляли путем фильтрации. Раствор экстрагировали с помощью 100 мл 0,1 М HCl и 100 мл нас. карбоната натрия и высушивали над безводным сульфатом натрия. Удаление растворителя обеспечивает белое твердое вещество, которое перемешивали в 200 мл ТГФ/гексан (1:20) в течение трех часов. Твердое вещество собирали путем фильтрации, получая 14,2 г целевого соединения с выходом 87%.
1Н-ЯМР (300 МГц, CDCl3): 7,35(m, 7Н), 6,83 (d, J=8,7 Гц, 2Н), 5,07 (m, 3Н), 3,77 (s, 3Н), 3,10 (q, J=6,3 Гц, 2Н), 2,92 (t, J=6,3 Гц, 2Н).
Бензил 7-метокси-2,3-дигидробензо[f][1,4]тиазепин-4(5Н)-карбоксилат (3)
Смесь соединения 2 (7,3 г, 23 ммоль), параформальдегида (6,9 г 0,23 моль) и n-толуолсульфоновой кислоты (1,45 г, 7,6 ммоль) в 250 мл толуола перемешивали при 70°C в течение ночи. После охлаждения до к.т., твердое вещество отфильтровали. Раствор экстрагировали с помощью нас. карбоната натрия (100 мл) и органический слой высушивали над безводным сульфатом натрия. Получали целевой продукт (7,4 г) в виде жидкости после удаления растворителя с выходом 97%.
1Н-ЯМР (300 МГц, CDCl3): 7,44 (d, J=8,1 Гц, 0,77Н), 7,32 (m, 5,60Н), 7,07 (d, J=2,7 Гц, 0,33H), 6,68 (m, 1,30Н), 5,04 (s, 2Н), 4,59 (ss, 2Н), 3,96 (br, 1,80), 3,80 (ss, 1,23Н), 3,55 (s, 1,97Н), 2,76 (m, 2Н).
7-Метокси-2,3,4,5-тетрагидробензо[f][1,4]тиазепин гидробромид (Амин) (4 HBr соль)
Первый способ
Раствор HBr (33% в уксусной кислоте, 10 мл) добавляли к соединению 3 (4,2 г, 12,8 ммоль). После добавления, начинал выделяться углекислый газ и образовывалось белое твердое вещество. Смесь оставляли отстаиваться при к.т. в течение дополнительных 2 часов. К смеси добавляли простой диэтиловый эфир (150 мл) и ее перемешивали в течение 30 мин. Твердое вещество собирали путем фильтрации и промывали простым диэтиловым эфиром. Твердое вещество высушивали в вакууме, получая 3,40 г целевого соединения с выходом 91,8%.
1Н-ЯМР (300 МГц, ДМСО-d6): 9,02 (br, 2Н), 7,52 (d, J=8,1 Гц, 1Н), 7,27 (d, J=3,3 Гц, 1H), 6,92 (dd, J=8,4, 2,7 Гц, 1Н), 4,41 (s, 2Н), 3,77 (s, 3Н), 3,53 (m, 2Н), 2,96 (m, 2Н).
Альтернативный способ (свободное основание 4а)
Соединение 3 (10 г, 30 ммоль) смешивали с 50 мл конц. HCl, 50 мл воды и 30 мл диоксана. Смесь перемешивали при 100°C в течение ночи. После охлаждения до к.т., большинство растворителя и НС1 удаляли при пониженном давлении. К раствору добавляли воду (100 мл) и твердое вещество отфильтровали. Водный раствор экстрагировали с помощью EtOAc/гексан (1:1, 3 × 100 мл) и подщелачивали путем добавления 15 г NaOH. Смесь экстрагировали с помощью дихлорметана (3 × 150 мл). Объединенный раствор высушивали над безводным сульфатом натрия. Удаление растворителя обеспечивает жидкость, которая отвердевала после отстаивания при кт, получая 6,2 г целевого соединения.
1Н-ЯМР (300 МГц, CDCl3): 7,42 (d, J=8,1 Гц, 1Н), 6,78 (d, J=2,7 Гц, H), 6,68 (dd, J=2,7, 8,1 Гц, 1H), 4,08 (s, 2H), 3,96 (br, 1,80), 3,76 (s, 3H), 3,38 (m, 2H), 2,68 (m, 2H).
Пример 2: Связывание калстабин 2 с PKA-фосфорилированным RyR2
Мембраны кардиального SR приготавливали, как было описано ранее (Marx и др., 2000; Kaftan и др., Circ. Res., 1996, 78: 990-97). Иммуноблотинг микросом (50 мкг) осуществляли, как было описано ранее, с антителом к калстабину (1:1,000) (Jayaraman и др., J. Biol. Chem., 1992, 267: 9474-77) в течение 1 часа при комнатной температуре (Reiken и др., Circulation, 107: 2459-66, 2003). После инкубирования с HRP-меченным антикроликовым IgG (1:5,000 разведение; Transduction Laboratories, Lexington, Ky.), блоты формировали, используя ECL (Amersham Pharmacia, Piscataway, N.J.) и обнаруживали на рентгеновской пленке, или подвергали воздействию вторичных антител, меченных с помощью инфракрасного красителя и визуализировали на оборудовании от Li-Cor Biosciences (модель Odyssey). Если специально не указано иначе, соединения тестировали при концентрации 100 нМ. Репрезентативный анализ связывания калстабин 2 представлен ниже.
А. PKA фосфорилирование саркоплазматического ретикулума сердца (CSR)
Реакционную смесь вносили в микроцентрифужную пробирку объемом. 200 мкг кардиального SR добавляли к реакционной смеси киназного буфера, PKA и ATP до конечного объема 100 мкл (реакционная смесь ниже). ATP добавляли последним для инициирования реакции.
Реакционная смесь:
20 мкл = Проба (кардиальный SR, 2 или 10 мкг/мкл)
10 мкл = 10x Киназный буфер (80 мМ MgCl2, 100 мМ EGTA, 500 мМ Tris/PIPES), pH=7,0
20 мкл = PKA (2 ед./мкл) (Sigma # Р2645)
10 мкл = 10x АТР (1,0 мМ) (Sigma А 9187)
40 мкл = разведенный H2O
1. Пробирки инкубировали при 30°C в течение 30 минут.
2. После этого реакционную смесь переносили в толстостенные стеклянные пробирки объемом 0,5 мл.
3. Стеклянные пробирки, содержащие реакционную смесь, центрифугировали в течение 10 минут при 50,000 × g на центрифуге Sorvall RCM120EX, используя ротор S120AT3. Центрифугирование при 50,000 × g в течение 10 минут достаточно для выделения микросом.
4. Полученный осадок после центрифугирования промывали 4 раза со связывающим буфером (10 мМ Имидазол, 300 мМ Сахароза, pH=7,4). Каждый раз в пробирку добавляли 100 мкл 1x связывающего буфера для промывки осадка после центрифугирования. Осадок после центрифугирования ресуспендировали путем промывки вверх и вниз, используя микродозатор. После последнего центрифугирования добавляли 50 мкл связывающего буфера и осадок после центрифугирования со всех пробирок собирали. Реакцию хранили при -20°C.
5. Фосфорилирование подтверждали путем отделения приблизительно 10 мкг CSR путем гель-электрофореза на 6% полиациламиде (PAGE) и иммуноблоты анализировали относительно как общего RyR (5029 антитело, 1:3000 разведение или Моноклональное антитело от Affinity Bioreagents, Cat # МА3-916, 1:2000 разведение), так и фосфорилированного PKA RyR2 (Р2809 Ab, 1:10000 разведение).
6. Аликвоты можно хранить при -80С.
В. Анализ повторного связывания калстабина
1. PKA-фосфорилированный CSR (около 20 мкг) инкубировали с 250 нМ Калстабин 2 в 100 мкл связывающего буфера (как описано выше) с соединениями или без них.
2. Реакцию запускали в толстостенной стеклянной пробирке объемом 0,5 мл (оборудование Hitachi Centrifuge, № каталога В4105).
3. Калстабин 2 добавляли в качестве последнего реагента в реакционную смесь. Реакцию осуществляли при комнатной температуре в течение 30 минут.
4. После реакции, пробирки центрифугировали в течение 10 минут при 100,000 g. (Sorvall RCM120EX центрифуга с S120AT3 ротор).
5. Полученный осадок после центрифугирования промывали 4 раза в 1x связывающем буфере при 4°C. После каждой промывки пробирки центрифугировали при 50,000 g в течение 10 минут при 4°C.
6. После конечной промывки, супернатант отбрасывали
7. Добавляли 20 мкл буфера для образца (2x) [6x буфер для образца, описанный ниже] и осадок после центрифугирования ресуспендировали с верхушкой и/или путем короткого встряхивания. Суспензию переносили в микроцентрифужные пробирки объемом 1,5 мл.
8. Пробирки нагревали при 90°C в течение 4 мин.
9. Белки разделяли с помощью 15% SDS/PAGE.
10. Связывание калстабин 2 определяли с анти-FKBP (Jayaraman и др., J. Biol. Chem. 1992; 267: 9474-77, 1:2000) первичным антителом и подходящим вторичным антителм.
6x Буфер для образца
7,0 мл 4x Tris-HCl/SDS, pH 6,8
3,0 мл глицерина (30% конечная концентрация)
1,0 г SDS (10% конечная концентрация)
0,93 г DTT (0,6 М конечная)
1 мг бромфенолового синего (0,001% конечная концентрация)
Дистиллированная вода до конечного объема 10 мл.
Хранение в аликвотах 1 мл при -70°C.
Результаты
На фигуре 1A представлен иммуноблот с антителом к калстабин 2, показывающий связывание калстабин 2 с PKA-фосфорилированным RyR2 при отсутствии (-) или в присутствии 100 нМ соединения 1. (+): калстабин связывается с не-PKA фосфорилированным RyR2. S36, бензотиазепин, описанный в US 7544678, использовали в качестве контроля. Как показано, соединение 1, при концентрации 100 нМ, предотвращает диссоциацию калстабин 2 из PKA-фосфорилированного RyR2 и/или усиливает (повторное)связывание калстабин 2 с PKA-фосфорилированным RyR.
Как показано на Фигуре 1 В, также было обнаружено, что следующие репрезентативные соединения предотвращают диссоциацию калстабин 2 из PKA-фосфорилированный RyR2, и/или усиливают (повторное)связывание калстабин 2 с PKA-фосфорилированным RyR2 при тестировании в вышеуказанном анализе повторного связывания калстабин 2 при 100 нМ: соединение 2, соединение 3 и соединение 4.
Пример 3: Связывание калстабин 1 с PKA-фосфорилированным RyR1
SR мембраны из скелетных мышц приготавливали аналогично описанному в примере 2 и как дополнительно описано в опубликованной заявке на патент США №2004/0224368, содержание которой включено в настоящую заявку в качестве ссылки. Иммуноблотинг микросом (50 мкг) осуществляли, как описано, с антителом к калстабин 1 (Zymed) (1:1,000). Блоты развивали и количественно определяли, как описано в примере 2.
На фигуре 1C представлен иммуноблот с антителом к калстабин 1, показывающий связывание калстабин 1 с PKA фосфорилированным RyR1 при отсутствии (Отриц) или в присутствии указанных концентраций соединения 1 или соединения 4. (Полож): калстабин связывается с не-PKA фосфорилированным RyR1. S36 использовали в качестве контроля. Как показано, соединение 1 и соединение 4 предотвращает диссоциацию калстабин 1 из PKA фосфорилированного RyR1 и/или усиливает (повторное)связывание калстабин 1 с PKA-фосфорилированным RyR1 в зависимости от дозы, с оценочным ЕС50 приблизительно 100 нМ и 150 нМ, соответственно.
Пример 4: Повторное связывание Калстабин 1 с RyR1 у мышей, которым вводили изопротеренол
Изопротеренол, агонист бета-адренергического рецептора, индуцирует сердечную недостаточность у мышей путем сверхстимуляции бета-адренергического рецептора. Конкурирует с этим активация PKA, фосфорилирование RyR2 на SR и снижение взаимодействия калстабина 2 (FKBP12.6) с RyR2. Сходный каскад событий происходит в скелетных мышцах, где RyR1 фосфорилирован, что приводит к снижению связывания калстабина 1 (FKBP12) c RyR1.
Как более подробно описано в опубликованной международной заявке № WO 2008/064264, содержание которой включено в настоящую заявку в качестве ссылки, хроническое введение изопротеренола мышам дикого типа предоставляет быстрый и надежный способ индуцирования изменений в RyR биохимии, который легко можно количественно определить. Эти изменения включают повышение RyR фосфорилирования и сопутствующее снижение связывания калстабина.
Животные и реагенты
Мышей C57B1/6 содержали и исследовали в соответствии с установленными протоколами. Синтетический бета-адренергический агонист, изопротеренол (ISO) получали от Sigma (165627) и готовили в виде 100 мг/мл маточного раствора в воде. Лизирующий буфер готовили путем добавления сахарозы (1 мМ), дитиотреита (320 мМ) и 1 таблетки ингибитора протеазы (10X) к 10 мл маточного раствора (10 мМ HEPES, 1 мМ EDTA, 20 мМ NaF, 2 мМ Na3VO4).
Приготовление осмотического насоса и хирургическая имплантация
Мышам непрерывно инфузировали в течение пяти дней 10 мг/мл изопротеренола (1 мкл/hr) с помощью имплантированного подкожно осмотического инфузионного насоса (Alzet MiniOsmotic Pump, модель 2001, Durect Corporation, Cupertino, СА).
Для ввода лекарственного средства, осмотический насос удерживали вертикально и 200 мкл раствора лекарственного средства инъецировали в насос с помощью шприца 1 мл (присоединенного к канюле), который содержит избыток раствора лекарственного средства (~ 250-300 мкл). Раствор лекарственного средства инъецировали медленно наклонно, в то время как шприц медленно поднимали до тех пор, пока насос не был переполнен. Переливание через край жидкости при закрытии насоса подтверждал, что насос был заполнен правильно.
Загруженные осмотические насосы имплантировали подкожно с помощью следующих стадий. Мышь-реципиент анестезировали 1,5-2% изофлураном в O2, который вводили при 0,6 л/мин, и после этого ее вес измеряли и записывали. После этого мышь помещали на пенополистирол грудной клеткой вниз, лицом в носовую часть. Шерсть срезали на затылке, начиная сзади ушей и до верхушки головы. Участок аккуратно протирали 70% спиртом и осуществляли небольшой надрез по средней линии затылка голова/шей. Держатель шва смазывали спиртом, вставляли в разрез и открывали для освобождения кожи от нижерасположенной ткани. Для приспособления насоса, это отверстие удлиняли назад до задней части туловища. Загруженную помпу вставляли в отверстие с высвобождением ее участка, расположенного вдали от рассечения, и предоставляли возможность установиться под кожей с минимальным напряжением. Разрез зашивали с помощью 5,0 нейлоновых швов, для этого требуется приблизительно 5-6 швов, и участок тщательно протирали 70% спиртом. После хирургического вмешательства, мышей помещали в индивидуальные клетки для минимизации повреждения и возможной активации симпатической нервной системы.
Выделение скелетных мышц
Ткань скелетных мышц мышей выделяли следующим образом. На мышцах лапы срезали кожу на лодыжке и тянули вверх. Ткань поддерживали влажной с помощью буфера Тироде (10 мМ HEPES, 140 мМ NaCl, 2,68 мМ KCl, 0,42 мМ Na2HPO4, 1,7 мМ MgCl2, 11,9 мМ NaHCO3, 5 мМ глюкоза, 1,8 мМ CaCl2, приготовленного путем добавления 20 мг CaCl2 к 100 мл 1X буферу, полученному из 10X раствора без CaCl2). Следующие мышцы выделяли и замораживали в жидком азоте. Длинный разгибатель пальцев (EDL) выделяли путем вставления ножниц между латеральным сухожилием и X, образованным EDL и болыпеберцовыми сухожилиями, срезали вверх по направлению к коленям; разрезали малоберцовую мышцу для раскрытия веерообразного малоберцового сухожилия; вставляли зажимы под X и под мышцу для освобождения EDL сухожилия; срезали EDL сухожилие и натягивали мышцу и в завершение срезали ослабленную EDL. Камбаловидную мышцу выделяли путем удаления малоберцовой мышцы с верхушки икроножной мышцы путем разреза и поднятия вверх ахиллового сухожилия; срезали камбаловидную мышцу на верхушке мышцы позади колена; и в завершение натягивали камбаловидную мышцу и отрезали ее от икроножной мышцы. Большеберцовую кость выделяли путем разреза большеберцового сухожилия срезания с верхушки лодыжки, оттягивали сухожилие вверх и отрезали его от большеберцовой кости. Широкую мышцу (мышцу бедра) выделяли с обеих ног путем отрезания мышцы немного выше колена и удаляли мышечный пучок. Образцы замораживали в жидком азоте.
RyR1 иммунопреципитация с тканевых лизатов
RyR1 иммунопреципитатировали с образцов путем инкубирования 200-500 мкг гомогената с 2 мкл анти-RyR1 антитела (Zymed) в 0,5 мл модифицированного RIPA буфера (50 мМ Tris-HCl (pH 7,4), 0,9% NaCl, 5,0 мМ NaF, 1,0 мМ Na3VO4, 0,5% Triton-X100 и ингибиторы протеазы) при 4°C в течение 1,5 часа. После этого образцы инкубировали с шариками Белок А сефароза (Amersham Pharmacia Biotech, Piscatawy, NJ) при 4°C в течение 1 часа, после этого шарики промывали три раза с помощью ледяного RIPA. Образцы нагревали до 95°C и разделяли на фракции по размерам с помощью SDS-PAGE (15% SDS-PAGE для калстабины). Иммуноблоты развивали, используя анти-FKBP антитело (FKBP 12/12,6, Jayaraman и др., J. Biol. Chem. 1992; 267: 9474-77) при 1:2,000 разведении. Антитела разводили в 5% молоке или TBS-T (20 мМ Tris-HCl, pH 7,5, 0,5 М NaCl, 0,05% Tween® 20, 0,5% Triton Х-100).
Результаты
Осмотические насосы, содержащие изопротеренол с или без тестируемого соединения, имплантировали мышам, как описано выше. Мышам осмотически перфузировали в течение пяти дней либо только наполнитель (ДМСО/PEG), только изопротеренол (ISO) (0,5 мг/кг/ч), или комбинацию изопротеренола (0,5 мг/кг/ч) и соединения 1 в указанных концентрациях. В день 6 каждую мышь умертвляли и ткани скелетных мышц выделяли и использовали для анализа связывания калстабин 1 в RyR1 иммунопреципитатах.
Влияние соединения 1 на усиление связывания калстабин 1 с RyR1 в скелетных мышцах, выделенных у мышей, которым вводили изопротеренол, представлено на Фигурах 2A (иммуноблот) и 2B (графическое количественное описание). Как показано, соединение 1 усиливает уровни калстабин 1, связанного с RyR1, в мембранах скелетных мышц до уровня, которых сходный с уровнем, наблюдаемым при введении 3,6 мМ S36, другого производного бензотиазепина, которое использовали в качестве положительного контроля (WO 2008/064264). Сходные результаты получали для соединения 4 (данные не показаны).
Пример 5: Влияние соединения 1 на модели хронической пост-ишемической сердечной недостаточности у крыс
Задачи
Задачей этого исследования являлось тестирование способности соединения 1 уменьшать сердечную дисфункцию и ослабления реконструкции желудочка на модели сердечной недостаточности, индуцированной ишемией-реперфузией.
Методология
Хроническую сердечную недостаточность индуцировали у самцов крыс Вистар (224-240 г, в возрасте 10-11) путем ишемически-реперфузионного (I/R) повреждения. Для I/R протокола, переднюю нисходящую ветвь (LAD) левой коронарной артерии окклюзировали в течение 1 ч. Лечение с помощью лекарственного средства (5 мг/кг/д или 10 мг/кг/д в питьевой воде) начинали через 1 неделю после реперфузии и поддерживали в течение периода исследования 3 месяца. Эффективность соединения 1 оценивали путем эхокардиографии через один, два и три месяца после начала и с помощью инвазивной гемодинамики через 3 месяца путем сравнения с наполнителем и ложнооперированными животными. Образцы сердца также анализировали для оценки гипертрофии и содержания коллагена. Кровь собирали у каждой крысы после окончания исследования для оценки концентраций лекарственного средства в плазме крови, как показано на Фигуре 3. Схема исследования представлена на Фигуре 3. Эксперименты осуществляли слепым образом.
Статистические методы
Для параметров, измеренных в динамике, сравнение ложнооперированных относительно наполнителя и сравнение лечения лекарственным средством анализировали путем 2 факторного ANOVAs с повторными измерениями. Для параметров, измеренных при умерщвлении и морфометрии, сравнение ложнооперированных относительно наполнителя анализировали с помощью критерия Стьюдента и сравнения лечения лекарственным средством путем 1-факторного ANOVA с последующим критерием Даннета.
Результаты
Животные, леченные с помощью наполнителя I/R, по сравнению с ложнооперированными животными, проявили повышенный левожелудочковый (LV) конечный систолический (LV ESV) и конечный диастолический (LV EDV) объемы (Фигуры 4A и B), угнетение сердечной функции, что определяли по снижению фракции выброса (EF) (Фигура 4C) и повышенному интерстициальному содержанию коллагена (Фигура 5D). Соединение 1, которое вводили в дозе 5 и 10 мг/кг/д, существенно повышает EF, а также снижает как LVESV, так и LVEDV по сравнению с наполнителем, от одного до трех месяцев (Фигуры 4A-C), а также уменьшает интерстициальное содержание коллагена (Фигура 5D).
Инвазивное гемодинамическое исследование (через 3 месяца) показало сохранение LV dP/dt max и LV dP/dt min у животных, леченных с помощью соединения 1 в дозах 5 и 10 мг/кг/д по сравнению с наполнителем (Фигуры 6B и C), с статистически недостоверным изменением LV систолического давления при лечении (Фигура 6A).
Не наблюдали влияния на вес тела (BW), размер инфаркта или гипертрофию (вес LV) при лечении (Фигуры 5A-C). Концентрации лекарственного средства в плазме представлены на Фигуре 7.
Результаты указывают на то, что соединение 1, при концентрации всего лишь 5 мг/кг/д проявляет благоприятный эффект как на систолическую, так и диастолическую сердечную функцию на модели хронической пост-ишемической сердечной недостаточности у крыс.
Соединение 1 существенно и неожиданно более активно по сравнению с соединением А, структурно сходным производным бензотиазепина, описанным в WO 2007/024717. Как показано на Фигуре 8, соединение А, вводимое при концентрации 5 мг/кг/д в течение 3 месяцев, не способно улучшить систолическую и диастолическую сердечную функцию по сравнению с соединением 1 на модели хронической пост-ишемической сердечной недостаточности у крыс при окончании исследования. Таким образом, благоприятные эффекты соединения 1, но не соединения А, наблюдали в дозе 5 мг/кг/д после лечения в течение 3 месяцев на модели CHF у крыс.
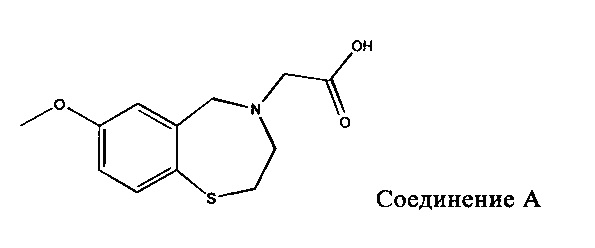
Пример 6: Влияние соединения 1 на мышечную функцию на модели мышечной дистрофии у мышей (mdx)
Задача
Задачей этой исследования является определение, будет ли лечение с помощью соединения 1 улучшать мышечную функцию на модели с дефицитом дистрофина у мышей (mdx).
Методология
C57BL/10ScSn-DMDmdx/J (сокращенно как mdx, n=5 на группу) мышей, в возрасте 6 недель и около 20 грамм в начале исследования, акклиматизировали в клетках на колесах в течение шести дней, перед рандомизацией на группы, получающие лечение либо с помощью наполнителя (H2O) или целевых доз 5 мг/кг/д, 10 мг/кг/д, или 50 мг/кг/д (действующие дозы: 7,9 мг/кг/д; 12,8 мг/кг/д; и 61,5 мг/кг/д, соответственно, определенных на основании еженедельно измеренного потребления раствора лекарственного средства, разделенного на вес тела) натриевой соли соединения 1 (на основании веса исходного лекарственного средства; натрия соль далее в этом настоящем изобретении обозначается как "соединение 1"), которую вводили в питьевой воде ad libitum в течение 4 недель. Мыши C57BL/6 (сокращенные как WT, n=4 на группу) в соответствующей возрастной группе, рандомизировали на группы, получающие лечение либо с помощью наполнителя (H2O) или целевой дозы 50 мг/кг/д (действующая доза: 67,7 мг/кг/д) натриевой соли соединения 1.
Произвольную активность на колесо, вес тела и среднее потребление воды измеряли в течение первых 3 недель. Удельную мышечную силу измеряли через 4 недели лечения, после окончания исследования.
Пройденное расстояние (км/день) в течение 24-ти часового периода анализировали в качестве показателя улучшенной функциональной активности (см. DMD_M.2.1.002 SOP на http://www.treat-nmd.eu/). По результатам исследования, мышцу длинный разгибатель пальцев (EDL) отделяли для анализа мышечной силы, как более подробно описано далее ниже. Кровь собирали от каждой мыши путем ретроорбитального спуска крови при окончании исследования (после окончания темного цикла - приблизительно 7 часов утра) для оценки концентрации лекарственного средства в плазме крови. Эксперименты были слепыми.
Измерения силы
После окончания исследования, EDL мышцу иссекали от задних конечностей для анализа изометрической силы, используя 407А Muscle Test System от Aurora Scientific (Aurora, Ontario, Canada). 6-0 швами привязывали к каждому сухожилию и цельную EDL мышцу, сухожилье к сухожилью, переносили в баню Ragnoti O2/CO2 (95%/5%), барботированную раствором Тироде (в мМ: NaCl 121, KCl 5,0, CaCl2 1,8, MgCl2, NaH2PO4, NaHCO3 24 и глюкоза 5,5). Используя швы, одну связку привязывали вертикально к крюку из нержавеющей стали, присоединенному к датчику силы Другую пришитую связку прижимали к движущемуся плечу на системе Aurora. EDL мышцу стимулировали для сокращения, используя электрическое поле между двумя платиновыми электродами. В начале каждого эксперимента, длину мышцы доводили для получения максимальной силы. Определяли соотношения сила-частота путем запускания сокращений, используя поэтапные стимулирующие частоты (5-250 Гц для 200 мс при сверхпороговом напряжении). Между стимуляциями мышце предоставляли возможность отдыхать ~3 мин. После окончания измерения силы, измеряли длину (Lo) мышцы EDL, в то время как она была привязана в Aurora системе, исключая связки. После этого EDL мышцу удаляли из системы и взвешивали после отсечения концов связок и снятия швов. Затем EDL мышцу замораживали в жидком азоте. Площадь поперечного сечения (мм) EDL мышцы рассчитывали путем деления веса EDL мышцы на длину EDL мышцы и постоянную плотности мышц млекопитающих 1,056 мг/м3 (Yamada, T. и др. Arthritis and rheumatism 60:3280-3289). Для определения удельной силы EDL (кН/м2), абсолютную судорожную силу разделяли на площадь поперечного сечения EDL мышцы.
Статистические методы
Для определения статистической достоверности, использовали критерий Стьюдента для сравнения между двумя группами. Все объединенные данные выражали в виде среднее значение +/- СКО.
Результаты
Тестировали способность соединения 1 улучшать произвольные упражнения у mdx мышей. После акклиматизации мышей к клетке с произвольными колесами, наблюдали за активностью мышей на произвольно движущихся колесах с помощью компьютера 24/7. Собранные данные пересчитывали в пройденное расстояние в день в течение 3 недель. Mdx мыши, которых лечили с помощью 10 и 50 мг/кг/д (целевая доза) соединения 1, перемещались на достоверно более длинные расстояния на колесах по сравнению с mdx мышами, которых лечили только с помощью наполнителя (H2O) (P<0,001 с дня 1 до дня 19). Эффект лечения наблюдали уже через 2-3 дня после начала лечения, и он продолжался в течение наблюдаемого периода активности. Не наблюдали эффекта соединения 1 на пройденное расстояние у WT мышей, которых лечили с помощью 50 мг/кг/д соединения 1 (Фигура 9). Дополнительно, как было определено с помощью измерений силы in vitro на EDL мышце (Фигура 10), лечение с помощью соединения 1 повышает удельную силу в mdx мышце в зависимости от дозы. При частоте стимуляции 150 Гц и выше, у mdx мышей, которых лечили с помощью 50 мг/кг/д, было обнаружено статистически достоверное повышение удельной мышечной силы (P<0,05). Не наблюдали эффекта лечения с помощью соединения 1 на удельную мышечную силу у мышей WT.
Как показано на Фигуре 11, лечение с помощью соединения 1 не оказывает влияния на тела. Не наблюдали дозо-зависимых эффектов на потребление воды. Утреннее экспонирование в крови для соединения 1 составляло (среднее значение ± СКО) 3,3±0,4 мкМ для дозы 5 мг/кг/д у mdx мышей, 10,7±0,9 мкМ для дозы 10 мг/кг/д у mdx мышей, 52,8±1,7 мкМ для дозы 50 мг/кг/д у mdx мышей и 72,8±7,0 мкМ для дозы 50 мг/кг/д у мышей WT. Взятые в совокупности, результаты показывают, что, по сравнению с контролями, которых лечили наполнителем, лечение с помощью соединения 1 в дозе 10 мг/кг/д и 50 мг/кг/д (целевая доза) повышает упражнения на произвольно движущихся колесах через 3 недели и удельную мышечную силу через 4 недели у mdx мышей, мышиная модель мышечной дистрофии Дюшенна (DMD), таким образом демонстрируя полезность соединения 1 и его аналогов, как заявлено в данной заявке, для лечения мышечной дистрофии.
Пример 7: Метаболическая стабильность
Метаболическую стабильность соединения 1, типичного Rycal™ в соответствии с настоящим изобретением, сравнивали с соединением В и соединением C, структурно сходными производными бензотиазепина, описанными в WO 2007/024717.
А. Метаболическая стабильность в микросомах печени человека
Методы
Солюбилизация соединения: Маточные растворы готовили в ДМСО и рабочие растворы - в воде, содержащей 1 мг/мл BSA.
Предсказания метаболической биодоступности
Предсказания метаболической биодоступности (MF%) основывались на измерениях метаболической стабильности in vitro с микросомами печени, предполагая общую абсорбцию. Вкратце, неизмененные лекарственные средства количественно определяли с помощью ЖХ-МС-МС с последующим инкубированием (10-7 М) с микросомами печени человека и крысы (0,33 мг белка/мл) после инкубирования в течение 0, 5, 15, 30 и 60 минут в присутствии NADPH (1 мМ). Ферментативную реакцию останавливали с помощью метанола (об./об.) и белки осаждали путем центрифугирования. Собственный клиренс in vitro (Clint_mic), выраженный в виде мл/мин/г белка, представлял собой наклон (после LN линеаризации) оставшейся концентрации неизмененного лекарственного средства относительно времени инкубирования. После этого in vitro Clint увеличивали масштаб до всего организма in vivo (vivoClint), используя 0,045 мг белка/кг печени и вес печени 11 г для крысы и 1,2 кг для человека. Затем in vivo Clint преобразовывали в клиренсы в печение (HepCl), используя тщательно перемешанную модель (HepCl=vivoClint*HBF/ (vivoClint+HBF), где HBF (ток крови в течение) принимали как 22 мл/минуту для крысы и 1500 мл/мин для человека. После этого MF% вычитали из коэффициента очищения с помощью следующего уравнения (MF%=1-HepCl/HBF). Результаты представлены в таблице 1:

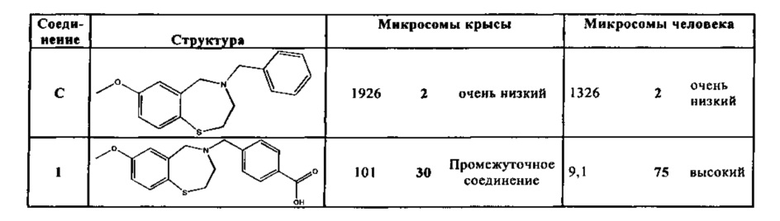
a) Clint_mic: собственный клиренс in vitro в мл/мин/мг белка
b) MF%: метаболическая биодоступность в %
В. Метаболическая стабильность в гепатоцитах печени крыс и человека
Солюбилизация соединения
Маточные растворы готовили в ДМСО и рабочие растворы - в среде Вильяма, содержащей 1/10 плазмы крысы или 1/4 плазы человека.
Определение метаболической стабильности
Соединения инкубировали при 10-7 М с выделенными гепатоцитами (6E + 5 клеток/мл для гепатоцитов крысы и 4E + 5 клеток/мл для гепатоцитов человека) при 37°C в плазме с одних и тех же видов, разведенных в среде Вильяма (1/10 разведение для крысы и ¼ разведение для человека). Отбор образцов осуществляли через 0, 10, 20, 30, 60 и 120 минут и ферментативную реакцию останавливали с помощью метанола (об./об.). Белки осаждали путем центрифугирования и супернатант анализировали с помощью ЖХ/МС/МС. Clint, выраженный в виде мл/мин/г белка, рассчитывали для микросом печени, используя соотношение 0,134 мг белка/мл для 4E+5 клеток/мл для человека и 0,201 мг белка/мл для 6E+5 клеток/мл для крысы. Присутствие препарата сравнения и потенциального метаболита проверяли с помощью ЖХ/МС/МС при исследовании в каждом образце. Результаты представлены в таблице 2:
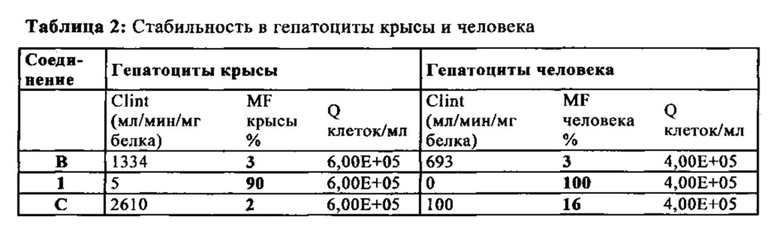
a) Clint_mic: in vitro собствнная стабильность в мл/мин/мг белка
b) MF%: метаболическая биодоступность в %
c) Q: количество клеток на мл
С. Метаболическая стабильность в микросомах мыши и крысы
Материалы и методы
Буфер для разведения: 0,1 М Tris HCl буфер при pH 7,4, содержащий 5 мМ EDTA.
Раствор кофактора NADPH: В пробирку фирмы Falcon объемом 50 мл, содержащей 2,79 мл буфера для разведения, добавляли 0,429 мл NADPH-регенерирующего раствора А и 0,079 мл NADPH-регенерирующего раствора В
Приготовление микросом: (1,5 мг/мл раствора)
В пробирку фирмы Falcon объемом 50 мл, содержащую 3,32 мл буфера для разведения, предварительно нагретого при 37°C в течение 15 мин (по меньшей мере 10 мин), добавляли 0,178 мл микросомы (24,6 мг/мл) к предварительно нагретому буферу для разведения. Концентрация белка в этом препарате микросом составляла 1,25 мг/мл.
Образец (Тестируемое соединение) - Исходный и промежуточный маточные растворы
Готовили 1 мг/ мл (0,5 мг/мл использовали для соединения 1) раствор тестируемого соединения в метаноле. 100 мкМ промежуточного раствора тестируемого соединения из исходного маточного раствора готовили с помощью буфера для разведения. 5 мкМ раствор готовили путем разведения 100 мкМ промежуточного раствора, используя буфер для разведения.
Эксперимент
(Эксперименты осуществляли в микроцентрифужных пробирках эппендорфа объемом 1,5 мл)
0 минут инкубирование. Процедура:
a) Добавляли 100 мкл предварительно нагретых микросом
b) Добавляли 50 мкл 5 мкМ раствора тестируемого соединения.
c) Добавляли 500 мкл холодного стоп-раствора (ледяной метанол)
d) Добавляли 100 мкл раствора кофактора NADPH в эппендорф.
а) Перемешивали вихрем эппендорф.
"t" минут инкубирование
b) Добавляли 100 мкл раствора кофактора NADPH в эппендорф.
c) Добавляли 50 мкл 5 мкМ раствора тестируемого соединения.
d) Добавляли 100 мкл предварительно нагретых микросом
e) Инкубировали эппендорф при 37°C 300 об/мин для 't' мин на термомиксере.
f) Удаляли эппендорф с термомиксера
g) Добавляли 500 мкл холодного стоп-раствора (ледяной метанол)
h) Перемешивали вихрем эппендорф.
Оба образца, инкубированных в течение '0' и 't' минут, центрифугировали при 15,000 rcf при 4°C в течение 15 минут. 500 мкл раствора супернатанта удаляли и его подвергали ЖХ/МС анализу (SIM - Контроль заданных ионов)
Результаты выражали в виде % оставшегося тестируемого соединения = (МС Площади пика образца 't' мин / МС Площади пика образца '0' мин) * 100. Площадь МС использовали в виде среднего для двойных инъекций.
Моменты времени = 0, 15, 30 и 60 мин для каждого тестируемого соединения
Положительный контроль
2 мкМ Имипрамин - 5 мин и 2 мкМ Имипрамин - 15 мин инкубирование использовали в качестве положительного контроля в экспериментах стабильности микросом печени у крыс и людей.
Результаты представлены в таблице 3:
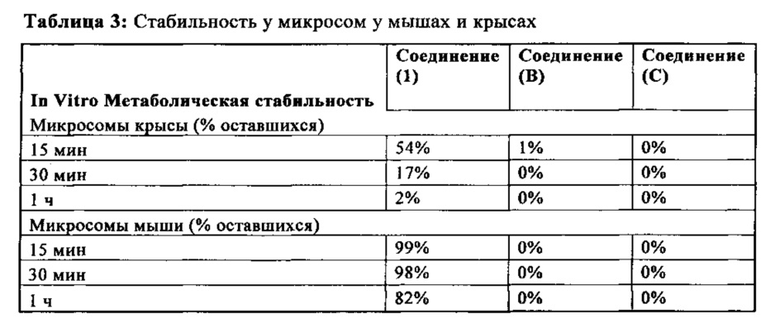
Неожиданно, как видно в таблицах 1-3, соединение 1 было существенно более стабильным в микросомах мыши, крысы и человека и в гепатоцитах крысы и человека по сравнению со структурными аналогами соединениями В и С, описанными в WO 2007/024717, оба из которых, как было обнаружено, обладают плохой метаболической стабильностью in-vitro в тестируемых системах, что делает эти соединения непригодными для разработок в качестве кандидатов для лекарственных средств. Неожиданно и непредсказуемо, вытеснение Н или OH компонентов в соединениях, известных из уровня техники, с помощью COOH компонента, приводит к соединению 1, которое проявляет высокую метаболическую стабильность во всех тестируемых системах. Повышение метаболической стабильности соединения 1 по сравнению с его структурными аналогами было действительно неожиданным и обосновывает неожиданные преимущества этого соединения по сравнению с соединениями, известными из уровня техники.
Все публикации, ссылки, патенты и патентные заявки, процитированные в настоящей заявке, полностью включены в качестве ссылки таким образом, как если бы каждая отдельная заявка, патент или патентная публикация были специфически и индивидуально включены полностью в качестве ссылки
Представленное выше описание специфических вариантов осуществления, таким образом, полностью раскрывает общую природу изобретения, что другие могут путем применения известных в настоящий период знаний легко модифицировать и/или адаптировать для различных применений такие специфические варианты осуществления без чрезмерных экспериментов и в пределах объема заявленного изобретения, и, следовательно, такие адаптации и модификации должны и предназначены включаться с объем и диапазон эквивалентов раскрытых вариантов осуществления изобретения. Подразумевается, что фразеология или терминология, используемая в настоящей заявке, представлена с целью иллюстрации и без ограничения. Средства, материалы и стадии осуществления различных раскрытых функций могут принимать альтернативные формы без отклонения от объема изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИАЗОЛЫ ДЛЯ РЕГУЛЯЦИИ ГОМЕОСТАЗА ВНУТРИКЛЕТОЧНОГО КАЛЬЦИЯ | 2017 |
|
RU2753509C2 |
| РЕКОМБИНАНТНЫЙ АДЕНОАССОЦИИРОВАННЫЙ ВИРУС, ЯВЛЯЮЩИЙСЯ ГИБРИДОМ СЕРОТИПОВ AAV9 И AAVrh74, С ПОНИЖЕННЫМ ТРОПИЗМОМ К ПЕЧЕНОЧНОЙ ТКАНИ | 2019 |
|
RU2812279C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА | 2017 |
|
RU2743424C2 |
| ИСПОЛЬЗОВАНИЕ МЕТАБОЛИТОВ ТРИПТОФАНА ДЛЯ ЛЕЧЕНИЯ МЫШЕЧНОЙ АТРОФИИ | 2017 |
|
RU2806346C2 |
| СВЯЗАННЫЕ С ПЕПТИДОМ МОРФОЛИНОВЫЕ АНТИСМЫСЛОВЫЕ ОЛИГОНУКЛЕОТИДЫ ДЛЯ ЛЕЧЕНИЯ МИОТОНИЧЕСКОЙ ДИСТРОФИИ | 2013 |
|
RU2662962C2 |
| УЛУЧШЕННЫЕ АНТИТЕЛА-АНТАГОНИСТЫ ПРОТИВ GDF-8 И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2630634C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ МЫШЕЧНОЙ АТРОФИИ | 2020 |
|
RU2814282C2 |
| Новая композиция, содержащая аминокислоты с разветвленными боковыми цепями | 2020 |
|
RU2817963C2 |
| Фармацевтически эффективные соединения, селективно ингибирующие изоформы миозина 2 | 2019 |
|
RU2817643C2 |
| ВЫДЕЛЯЮЩИЕ ОКСИД АЗОТА СОЕДИНЕНИЯ | 2007 |
|
RU2560144C2 |
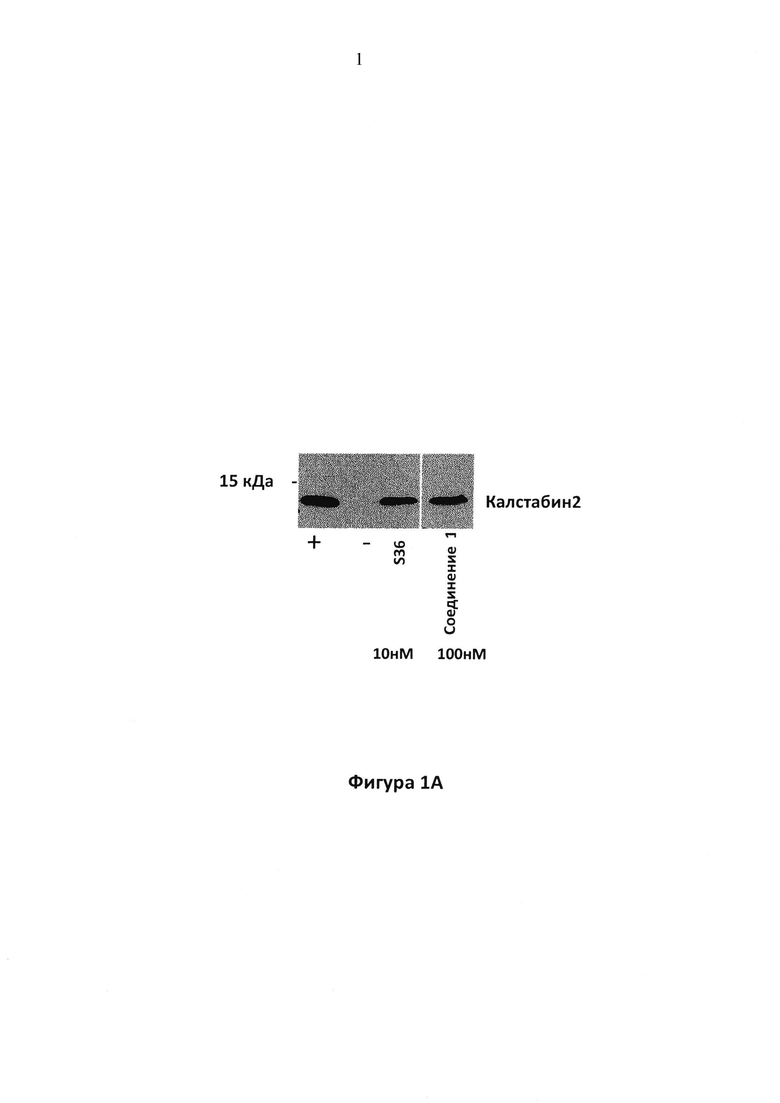

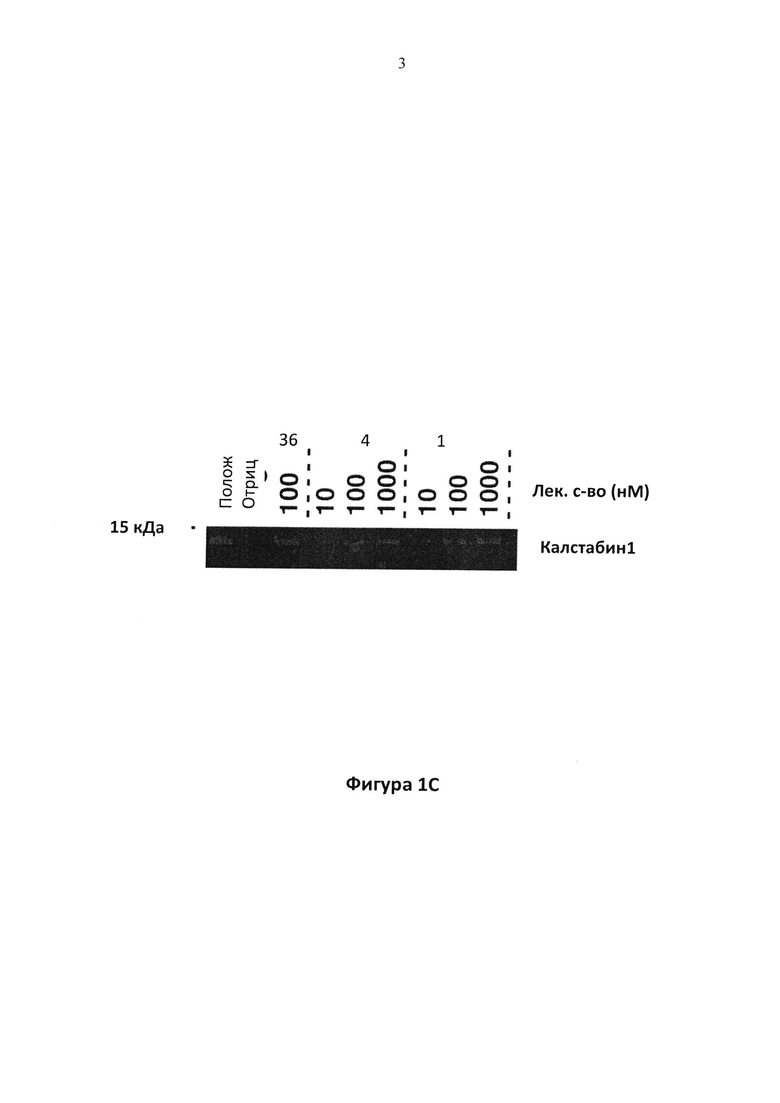
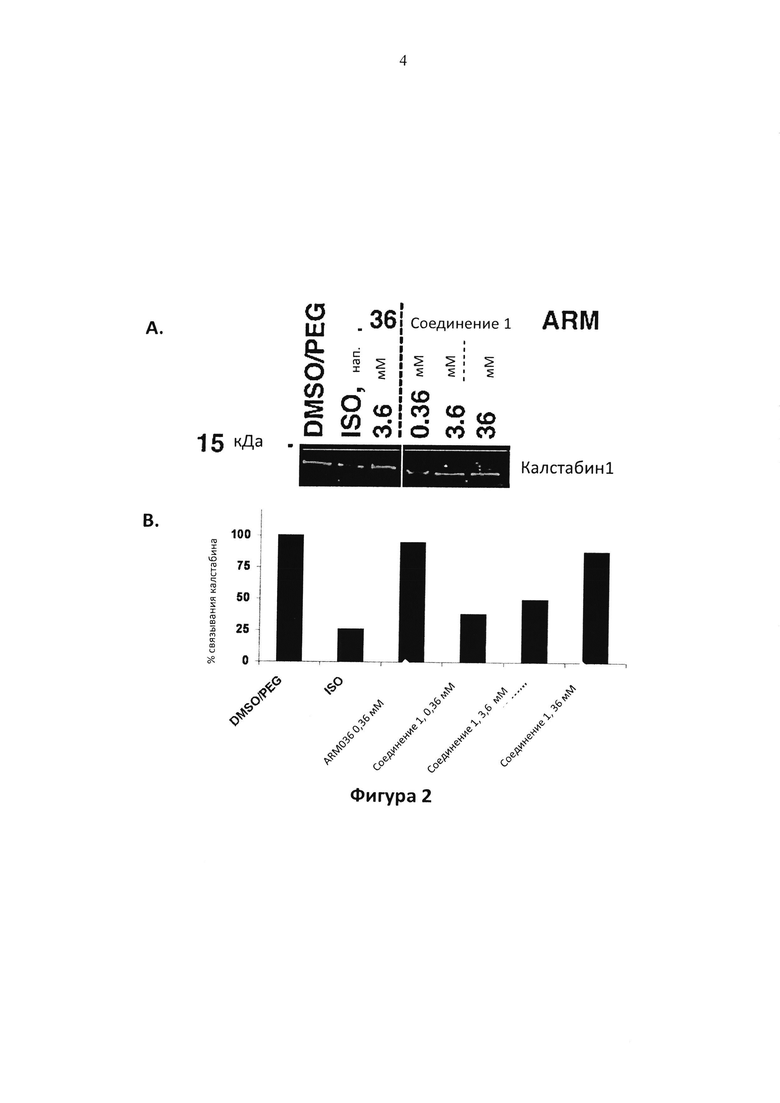

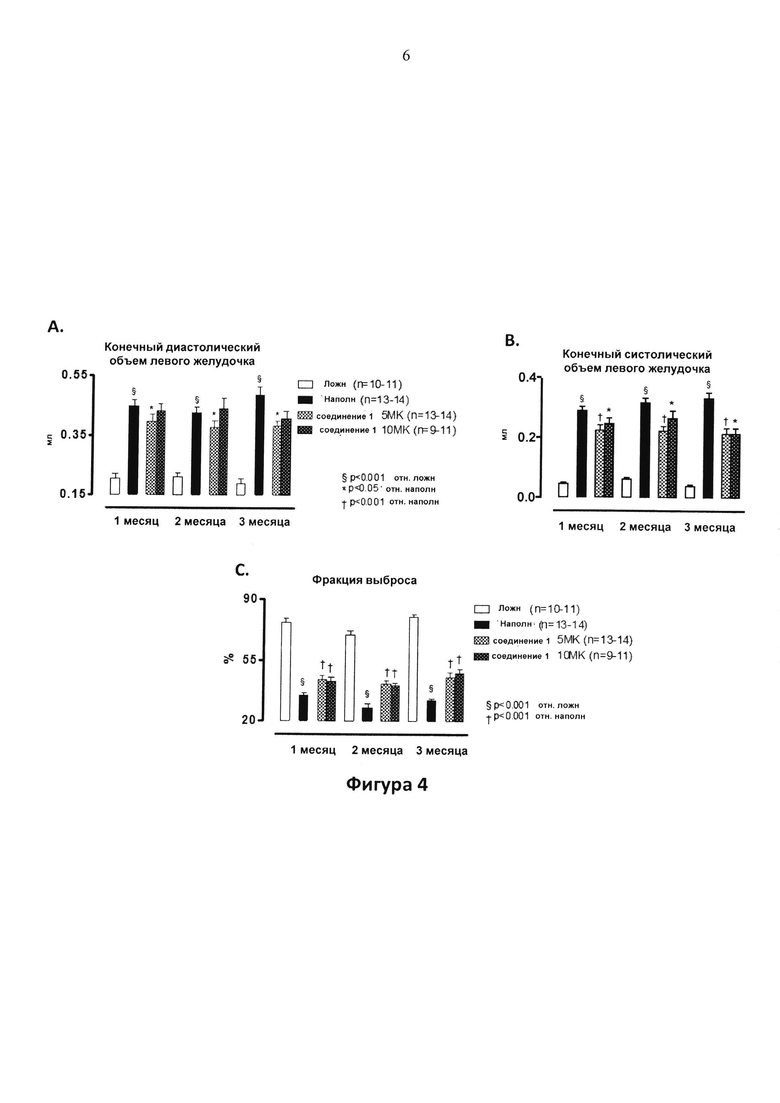
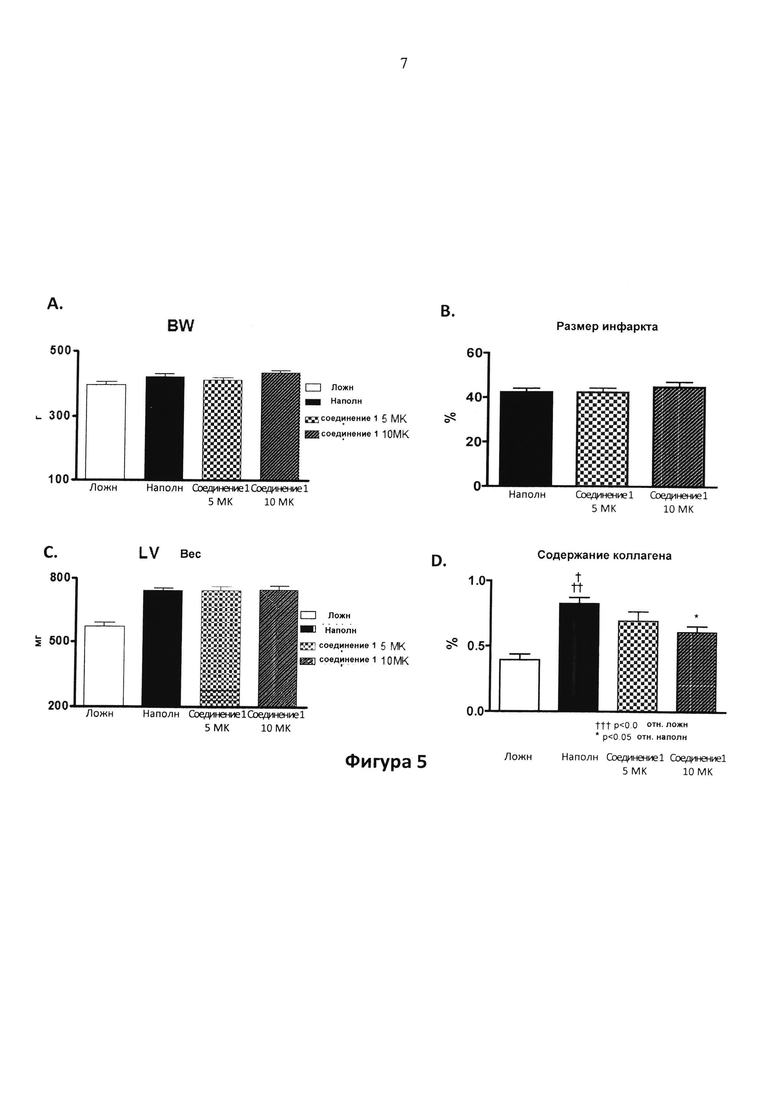
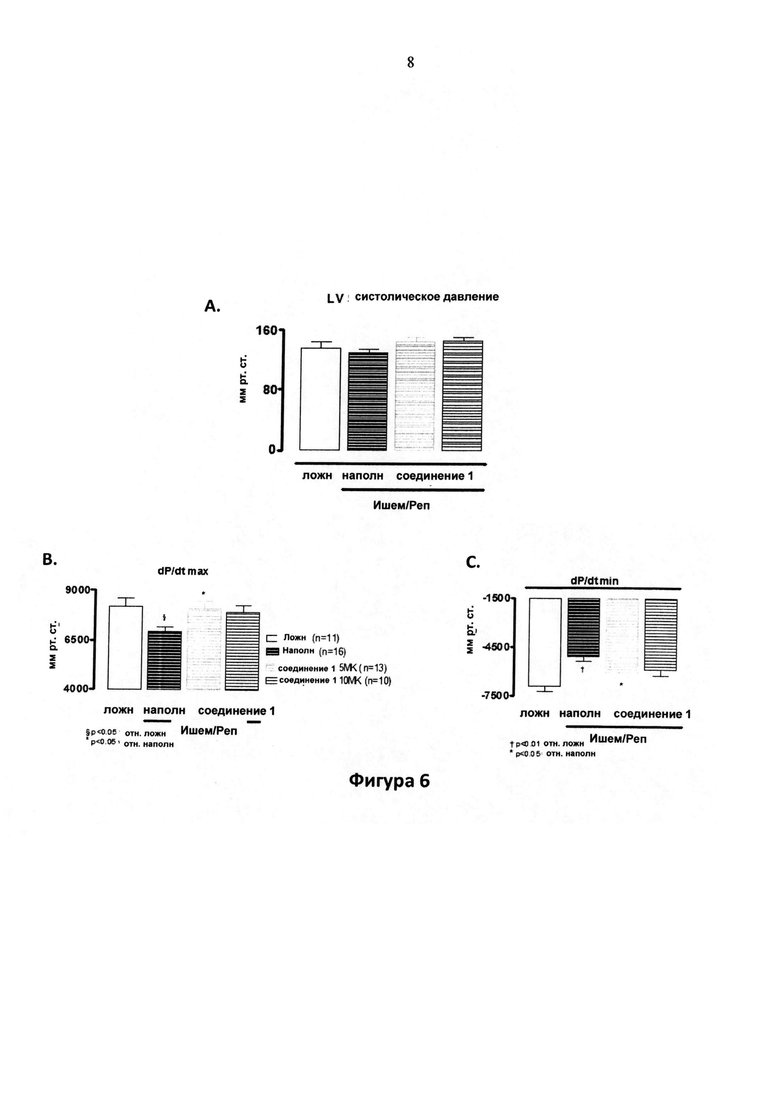
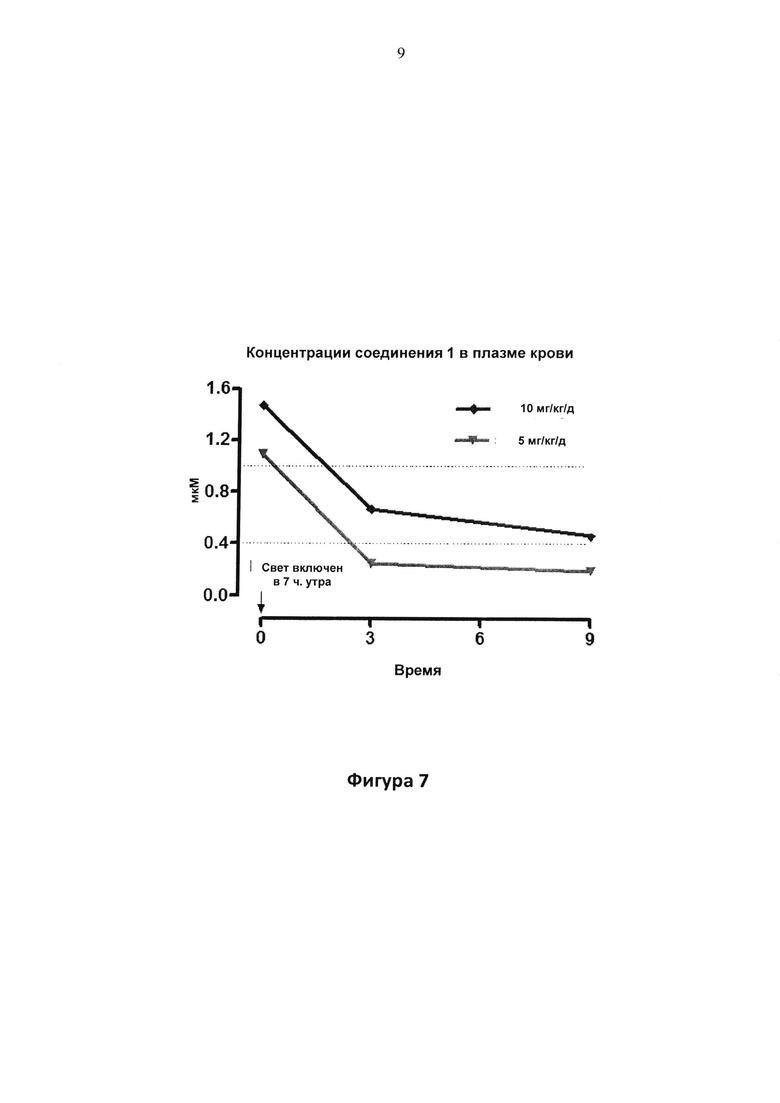
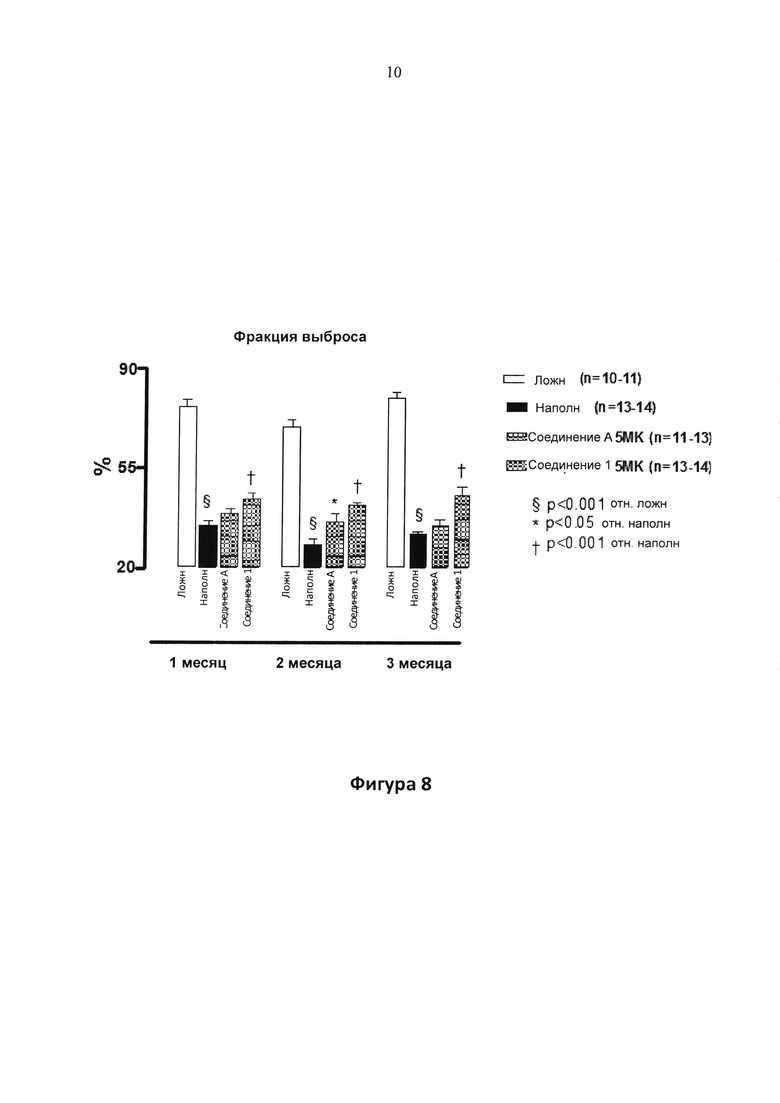
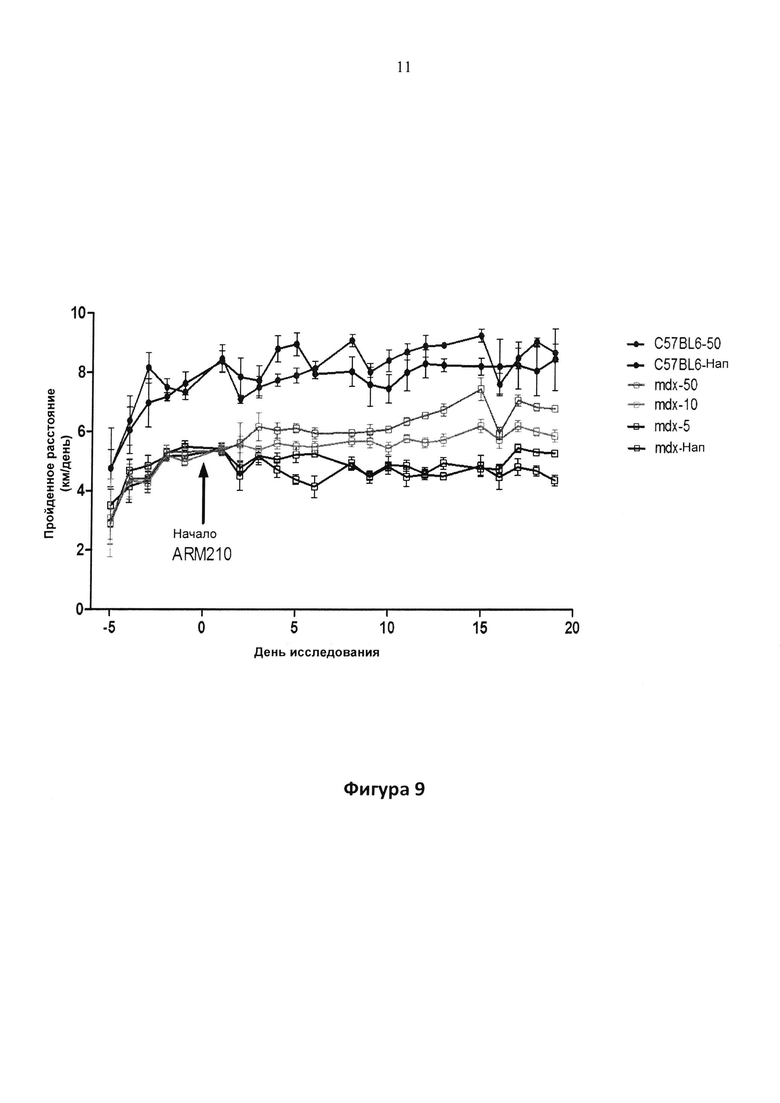
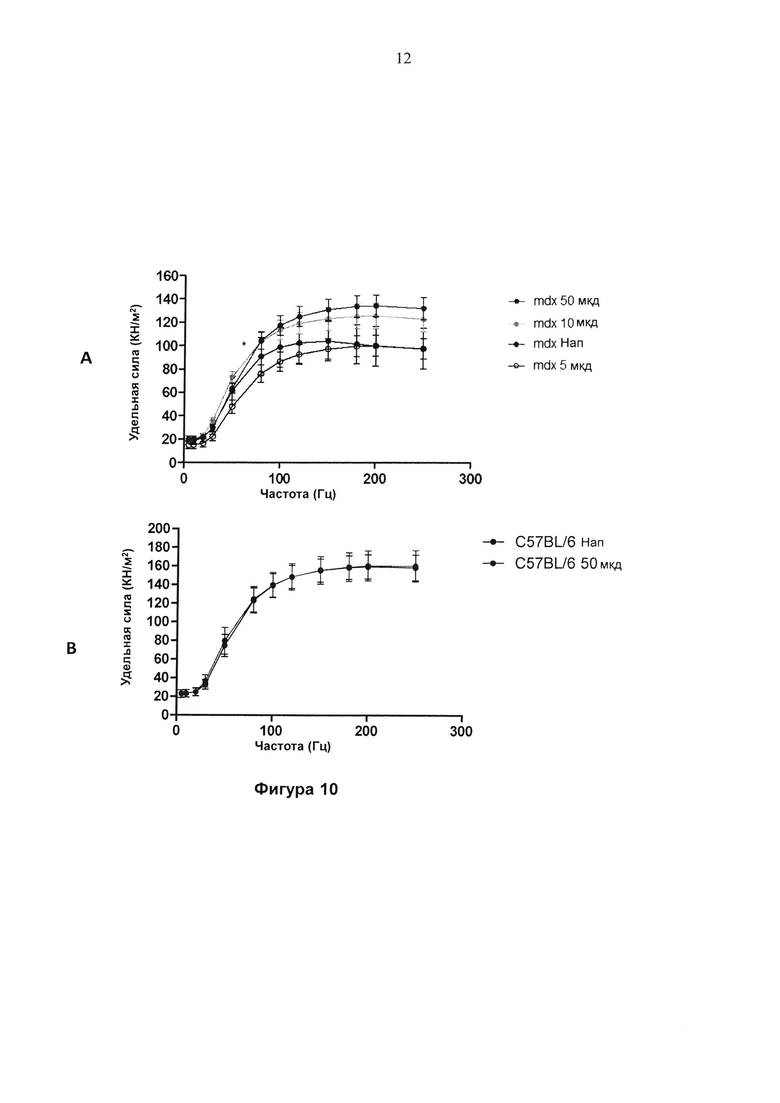
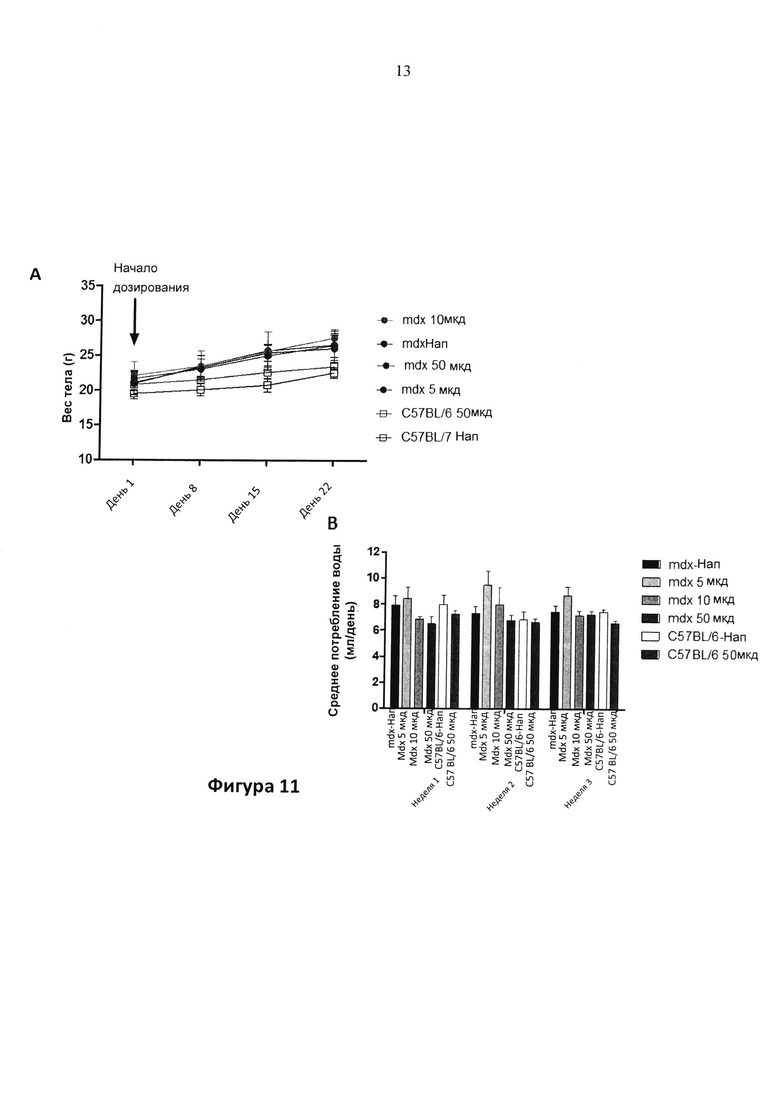
Изобретение относится к производным 1,4-бензотиазепина
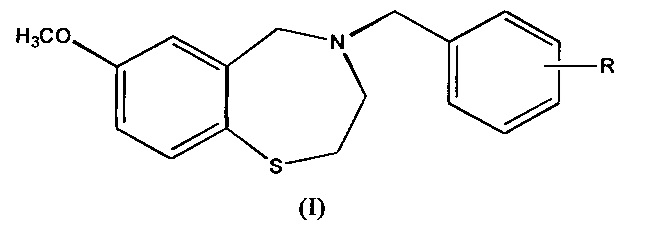 , а также к фармацевтическим композициям и применению. Технический результат: получены новые соединения, которые могут быть использованы для лечения заболеваний и состояний, связанных с RyRs, в частности нарушений сердечной деятельности, костно-мышечных нарушений и нарушений центральной нервной системы (ЦНС). 5 н. и 22 з.п. ф-лы, 11 ил., 3 табл.
, а также к фармацевтическим композициям и применению. Технический результат: получены новые соединения, которые могут быть использованы для лечения заболеваний и состояний, связанных с RyRs, в частности нарушений сердечной деятельности, костно-мышечных нарушений и нарушений центральной нервной системы (ЦНС). 5 н. и 22 з.п. ф-лы, 11 ил., 3 табл.
1. Соединение, представленное структурой формулы (I):
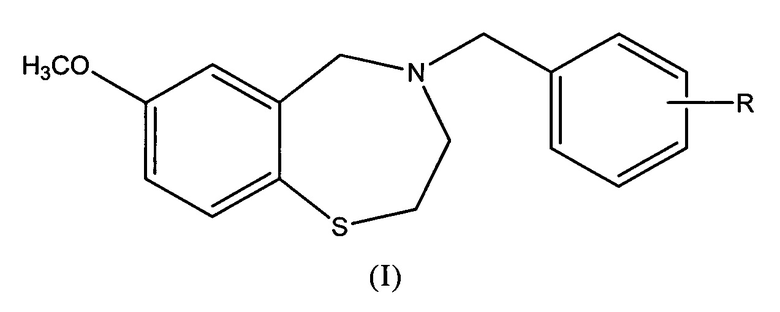
где
R представляет собой СООН;
и его фармацевтически приемлемые соли.
2. Соединение в соответствии с п. 1, в форме соли с фармацевтически приемлемой кислотой или основанием.
3. Соединение в соответствии с п. 2, где соль выбирают из группы, включающей соль натрия и гемифумарат.
4. Соединение в соответствии с п. 1, которое выбирают из группы, включающей:
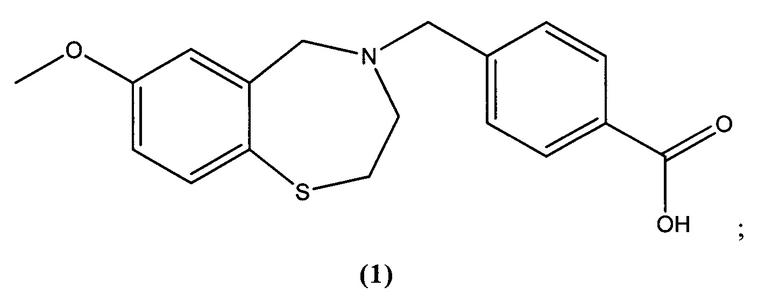
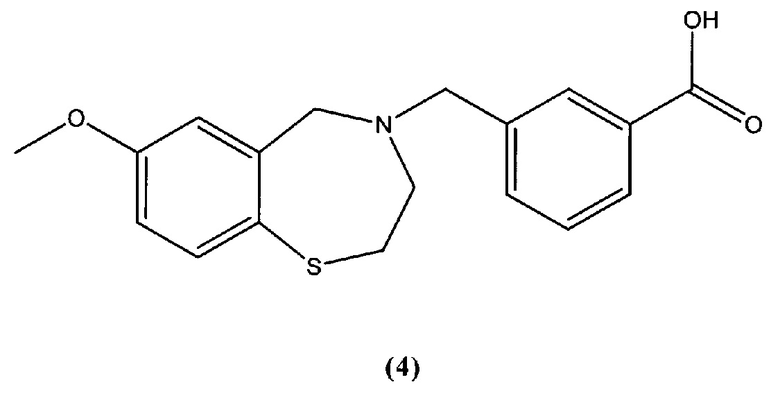 и
и
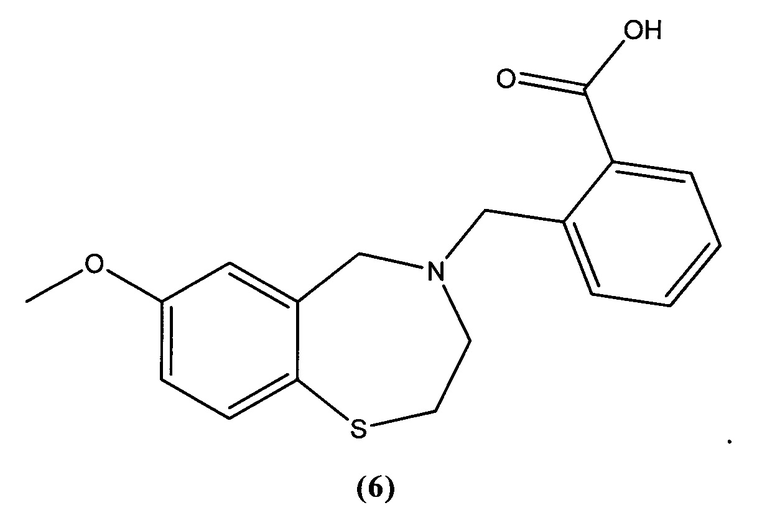
5. Соединение в соответствии с п. 1, которое представлено структурой формулы (1):
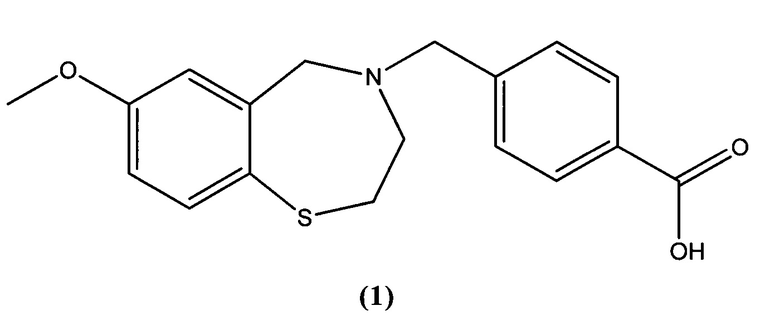
или его фармацевтически приемлемые соли.
6. Соединение в соответствии с п. 5 в форме соли с фармацевтически приемлемой кислотой или основанием.
7. Соединение в соответствии с п. 6, где соль выбирают из группы, включающей соль натрия и гемифумарат.
8. Соединение в соответствии с п. 7, где соль представляет собой натриевую соль.
9. Соединение в соответствии с п. 7, где соль представляет собой гемифумаратную соль.
10. Фармацевтическая композиция, обладающая активностью в отношении рецепторов рианодина (RyRs), которая содержит эффективное количество соединения в соответствии с любым из пп. 1-9, в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями или носителями.
11. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, нарушения и заболевания опорно-двигательного аппарата, нарушения и заболевания ЦНС, когнитивную дисфункцию, нейромышечные нарушения и заболевания, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции.
12. Способ лечения или предотвращения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, нарушения и заболевания опорно-двигательного аппарата, нарушения и заболевания ЦНС, когнитивную дисфункцию, нейромышечные нарушения и заболевания, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции, способ включает стадию введения субъекту, который в этом нуждается, терапевтически эффективного количества соединения в соответствии с любым из пп. 1-9, или фармацевтической композиции в соответствии с п. 10 для осуществления такого лечения.
13. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения состояния в соответствии с п. 11, или способа в соответствии с п. 12, где состояние связано с аномальной функцией рецептора рианодина 1 (RyR1), рецептора рианодина типа (RyR2), рецептора рианодина типа 3 (RyR3), или их комбинацией.
14. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения состояния в соответствии с п. 11, или способ в соответствии с п. 12, где нарушения сердечной деятельности и кардиологические заболевания выбирают из группы, включающей нарушения и заболевания, связанные с аритмией сердца, такие как нарушения и заболевания, связанные с аритмией сердца, выбранные из группы, включающей предсердную и желудочковую аритмию, предсердную и желудочковую фибрилляцию, предсердную и желудочковую тахиаритмию, предсердную и желудочковую тахикардию, катехоламинергическую полиморфную желудочковую тахикардию (CPVT), и вызванные физической нагрузкой их варианты; вызванные физической нагрузкой нарушения и заболевания, связанные с аритмией сердца; сердечную недостаточность; застойную сердечную недостаточность; хроническую сердечную недостаточность; острую сердечную недостаточность; систолическую сердечную недостаточность; диастолическую сердечную недостаточность; острую декомпенсированную сердечную недостаточность; сердечную ишемию/реперфузионное повреждение (I/R); хроническое обструктивное заболевание легких; I/R повреждение после коронарной ангиопластики или после тромболизиса для лечения инфаркта миокарда (MI); и повышенное кровяное давление.
15. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где мышечное утомление обусловлено заболеванием, нарушением или состоянием скелетных мышц.
16. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где скелетно-мышечное нарушение, заболевание или состояние выбирают из группы, включающей вызванное физической нагрузкой утомление скелетных мышц; вызванное физической нагрузкой мышечное утомление, обусловленное пролонгированной нагрузкой или нагрузкой высокой интенсивности; врожденную миопатию; мышечную дистрофию, такую как мышечную дистрофию Дюшенна (DMD), мышечную дистрофию Беккера (BMD), тазо-плечевую мышечную дистрофию (LGMD), плече-лопаточно-лицевую мышечную дистрофию, миотоническую мышечную дистрофию, врожденную мышечную дистрофию (CMD), дистальную мышечную дистрофию, мышечную дистрофию Эмери-Дрейфуса и окулофарингеальную мышечную дистрофию; спинальную мышечную атрофию (SMA), спинальную и бульбарную мышечную атрофию (SBMA), старческое мышечное утомление; саркопению, поражение сердцевины мышечных волокон; раковую кахексию; нарушения мочевого пузыря; и недержание.
17. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где нарушения и заболевания ЦНС выбирают из группы, включающей болезнь Альцгеймера (AD), невропатию, пароксизмы, болезнь Паркинсона (PD) и болезнь Хантингтона (HD); и нейромышечные нарушения и заболевания выбирают из группы, включающей спинально-церебеллярную атаксию (SCA) и боковой амиотрофический склероз (ALS, болезнь Лу Герига).
18. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где когнитивная дисфункция связана со стрессом или возрастом, или где когнитивная функция, подлежащая улучшению, представляет собой кратковременную память, долговременную память, внимание или обучение, или где когнитивная дисфункция связана с заболеванием или нарушением, выбранным из группы, включающей болезнь Альцгеймера (AD), синдром дефицита внимания с гиперактивностью (ADHD), расстройства аутистического спектра (ASD), генерализированное тревожное расстройство (GAD), обсессивно-компульсивное расстройство (OCD), болезнь Паркинсона (PD), посттравматическое стрессовое расстройство (PTSD), шизофрению, биполярное расстройство и большую депрессию.
19. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где состояние представляет собой раковую кахексию, предпочтительно вследствие злокачественного новообразования, имеющего метастазы в костях.
20. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где соединение используют в дозе, достаточной для восстановления или усиления связывания калстабин2 с RyR2.
21. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где соединение используют в дозе, достаточной для восстановления или усиления связывания калстабин1 с RyR1.
22. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, где соединение используют в дозе, достаточной для снижения Са2+ пика через RyR канал.
23. Соединение в соответствии с любым из пп. 1-9, или его соль, для применения для лечения или предотвращения состояния, выбранного из группы, включающей нарушения сердечной деятельности и кардиологические заболевания, мышечное утомление, нарушения и заболевания опорно-двигательного аппарата, нарушения и заболевания ЦНС, когнитивную дисфункцию, нервно-мышечные заболевания и нарушения, нарушения и заболевания костной системы, раковую кахексию, злокачественную гипертермию, диабет, внезапную сердечную смерть и синдром внезапной детской смерти, или для улучшения когнитивной функции.
24. Фармацевтическая композиция в соответствии с п. 10 для применения для лечения или предотвращения в соответствии с п. 11, или способ в соответствии с п. 12, дополнительно включающая(ий) применение антисмыслового олигонуклеотида (АО), который специфический для сплайсинговой последовательности в мРНК, представляющей интерес, для усиливающего экзона, пропущенного в указанной мРНК, представляющей интерес.
25. Фармацевтическая композиция в соответствии с п. 24 для применения для лечения мышечной дистрофии Дюшенна (DMD), где антисмысловой олигонуклеотид (АО) специфический для сплайсинговой последовательности по меньшей мере одного экзона DMD гена, предпочтительно сплайсинговой последовательности экзона 23, 45, 44, 50, 51, 52 и/или 53 DMD гена.
26. Способ лечения субъекта, который имеет мышечную дистрофию Дюшенна (DMD), который включает стадию введения указанному субъекту соединения в соответствии с любым из пп. 1-9, или фармацевтической композиции в соответствии с п. 10 в комбинации с антисмысловым олигонуклеотидом (АО), который специфический для сплайсинговой последовательности по меньшей мере одного экзона DMD гена, предпочтительно сплайсинговой последовательности экзона 23, 45, 44, 50, 51, 52 и/или 53 DMD гена.
27. Способ получения соединения в соответствии с любым из пп. 1-9, который включает стадию взаимодействия соединения формулы
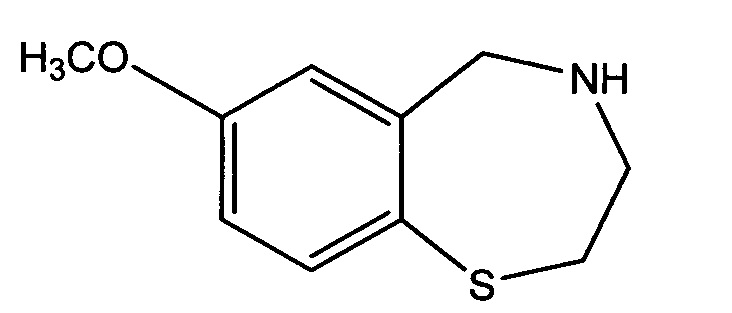
с соединением формулы
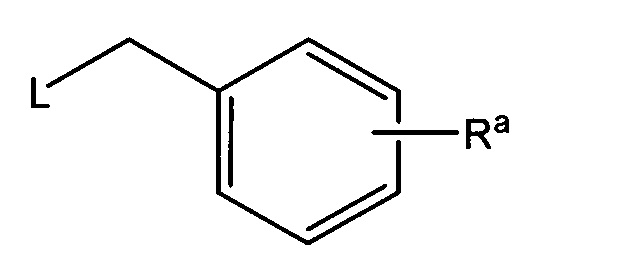
где Ra представляет собой COOR1 или CN; R1 представляет собой С1-С4 алкил и L представляет собой уходящую группу, получая соединение формулы:
 и
и
превращения группы Ra в группу R, таким образом получая соединение формулы (I).
| EA 200800665 A1, 29.08.2008 | |||
| WO 2008021432 A2, 21.02.2008 | |||
| Способ получения производных пергидротиазепина или их аддитивных солей с галоидводородными кислотами | 1986 |
|
SU1801110A3 |

