Настоящее изобретение относится к способу изготовления бумаги или картона, включающему стадии добавления конечного полимера А к первой водной суспензии волокнистого материала, обезвоживания полученной второй водной суспензии волокнистого материала, содержащей конечный полимер А, на водопроницаемой подложке с получением мокрой бумажной структуры и обезвоживания мокрой бумажной структуры с получением бумаги или картона. Полученная бумага или картон обладает хорошей прочностью в сухом состоянии. Другими объектами настоящего изобретения являются бумага или картон, получаемые этим способом, и конечный полимер А и исходный полимер V. Исходный полимер V является синтетическим предшественником конечного полимера А.
Тенденции, характерные для современной бумажной промышленности, иногда оказывают существенное неблагоприятное влияние на прочность бумаги или картона в сухом состоянии. Так, например, продолжает возрастать количество повторно используемой бумажной макулатуры. Это сопровождается ухудшением качества волокна. Наблюдается использование более коротких целлюлозных волокон, обладающих ухудшенными характеристиками набухания и шероховатостью. Использование недорогого сырья обычно является привлекательным даже, если это на практике связано с использованием более коротких целлюлозных волокон. Постоянной задачей является уменьшение плотности бумаги или картона с целью экономии сырья. Постоянно уменьшается водооборот в бумагоделательных машинах. Поэтому представляют интерес способы изготовления бумаги или картона, которые обеспечивают хорошую прочность полученных бумаги или картона в сухом состоянии.
В DE 4328975 А в качестве объекта изобретения раскрыты полимеры, предназначенные для изготовления бумаги, в которых доля 2-аминодигидропиррольных структурных звеньев составляет от 20 до 90 мол. %. Для получения полимеров, соответствующих примерам, сначала проводят реакцию радикальной полимеризации N-винилформамида и акрилонитрила и получают исходный полимер. После завершении реакции полимеризации этот исходный полимер образуется в виде суспензии в воде. После фильтрования исходный полимер обрабатывают концентрированной хлористоводородной кислотой и нагревание примерно до 100°С приводит к амидированию. Полученный таким образом конечный полимер осаждают с использованием ацетона и сушат. олученный конечный полимер "F", для которого исходный полимер получают по реакции радикальной полимеризации 50 мол. % N-винилформамида 50 мол. % акрилонитрила, указан, как единственный полимер, содержание лактама составляет, в частности, 1 мол. %:
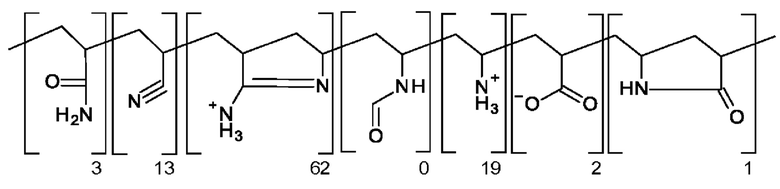
Конечные полимеры, в частности, добавляют к суспензии волокнистого материала. Бумагу изготавливают с помощью длинносеточной бумагоделательной машины, соответствующей стандарту TAPPI (TAPPI=техническое общество целлюлозно-бумажной промышленности). Для бумаги, изготовленной с использованием конечного полимера "F", определяют содержание золы. Также определяют прочность бумаги, изготовленной с использованием других конечных полимеров, путем определения значения показателя сопротивления продавливанию. Полученные конечные полимеры "G", "К", "Q" и "R" не содержат 2-аминодигидропиррольные структурные звенья или содержат лишь небольшое их количество и поэтому они не соответствуют этому изобретению, и в примерах практического осуществления они обеспечивают худшие результаты.
В ЕР 0528409 А в качестве объекта изобретения раскрыты полимеры, которые соответствуют описанным в указанном выше документе DE 4328975 А, в качестве флокулянта. В разделе "Примеры" также приведен конечный полимер "F", описанный в DE 4328975 А, обозначенный, как конечный полимер "Р". В примерах конечные полимеры добавляют к осадку для улучшения фильтруемости. По аналогии с DE 4328975 А конечные полимеры, не соответствующие этому изобретению, не содержат 2-аминодигидропиррольные структурные звенья или содержат лишь небольшое их количество.
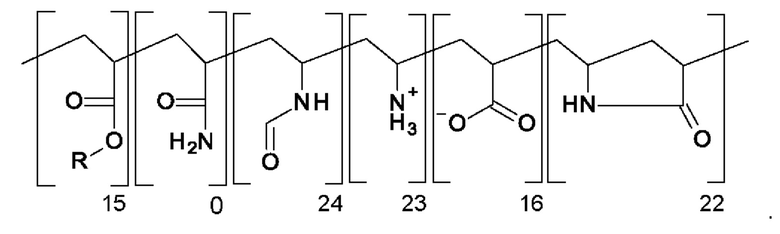
В DE 4441940 А в качестве объекта изобретения раскрыты полимеры, которые в качестве структурных звеньев содержат 5-членные лактамы (=пирролидин-2-оновые структурные звенья), доля которых составляет от 20 до 100 мол. %. В разделе "примеры" показана улучшенная термостойкость конечных полимеров. Конечные полимеры рекомендуют применять в качестве модификаторов для термопластичной смолы, полимерной добавки для третичной методики добычи нефти, смазывающих веществ, детергентов-диспергирующих средств, ингибиторов образования отложений, полимеров, добавляемых для быстрого завершения реакции, загустителей для бурового раствора, приспособленных для передвижения по трубопроводу загустителей, связующих и т.п. ля получения полимеров, соответствующих примерам, сначала проводят реакцию радикальной полимеризации N-винилформамида и акриламида, и в одном случае N-винилформамида, акриламида и акриламид-2-метилпропансульфоновой кислоты, и в другом случае N-винилформамида и метакриламида, в каждом случае получают исходный полимер. В случае использования N-винилформамида и акриламида исходный полимер осаждают с использованием метанола и в двух других случаях исходный полимер отфильтровывают в виде полимерного геля. Полученные таким образом исходные полимеры обрабатывают водным раствором хлористоводородной кислоты. В смеси проводят осаждение путем добавления ацетона или метанола и затем ее сушат. В соответствующих случаях определяют растворимость в воде и уменьшенную вязкость. Для полученного конечного полимера "С", для которого исходный полимер получают по реакции радикальной полимеризации 50 мол. % N-винилформамида 50 мол. % акриламида, указан следующих состав:
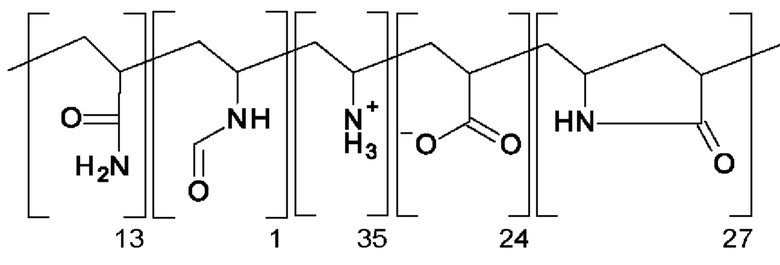
Для полученного конечного полимера "М", для которого исходный полимер получают по реакции радикальной полимеризации 40 мол. % N-винилформамида, 40 мол. % акриламида и 20 мол. % акриламид-2-метилпропансульфоновой кислоты, указан следующих состав:

Для полученного конечного полимера "N", для которого исходный полимер получают по реакции радикальной полимеризации N-винилформамида и метилакриламида, указан следующих состав:
В US 4898915 в качестве объекта изобретения раскрыты полимеры, которые включают структурные звенья, содержащие ароматическую или алифатическую аминогруппу, и структурные звенья, содержащие по меньшей мере один следующий заместитель: нитрил, альдегид, карбоновая кислота или эфир карбоновой кислоты. В примерах исходные полимеры получают путем полимеризации мономеров, содержащих защищенные аминогруппы, и эфиров акриловой кислоты в толуоле, указанную реакцию полимеризации катализируют кислотой Льюиса. Неочищенные исходные полимеры отделяют декантацией и путем добавления метанола, фильтруют в растворенном в хлороформе виде и осаждают путем повторного добавления метанола. Для получения конечных полимеров полученные таким образом исходные полимеры обрабатывают гидразином в хлороформе для образования первичных аминогрупп. Точнее, в примере 3 метилакрилат полимеризуют с N-винилфталимидом, реакцию катализируют этилсесквихлоридом алюминия. В примере 6 этот исходный полимер растворяют в хлороформе и обрабатывают гидразином. Указано, что полученный конечный полимер содержит метоксигруппы, аминогруппы и лактамные звенья. В примере 7 конечный полимер, полученный в примере 6, обрабатывают водным раствором гидроксида калия при 70°С, при этом получают полимер, содержащий чередующиеся аминогруппы и карбоксигруппы. Конечные полимеры рекомендуют применять в качестве антистатических агентов или в качестве загустителей при добыче нефти.
В публикации "A novel synthetic procedure for N-vinylformamide and free radical polymerization", S. Sawayama et al., Mitsubishi Kasei R&D Review, 1993, Vol. 7, p. 55-61, в разделе 3.5 и с помощью фиг. 4 описана сополимеризация N-винилформамида с акриламидом и сополимеризация N-винилформамида со стиролом, в каждом случае при разных отношениях количества молей.
В публикации "Alternating copolymerization of methyl acrylate with donor monomers having a protected amine Group", R. N. Majumdar et al., Journal of Polymer Science, 1983, Vol. 21, p. 1717-1727, в частности, описан пример 6, приведенный в указанном выше документе US 4898915, и назван гидразинолизом чередующегося сополимера метилакрилата с N-винилфталимидом. На фиг. 4 показан полученный с помощью 13С-ЯМР (ядерный магнитный резонанс) спектр сополимера метилакрилата с N-винилфталимидом и полученный с помощью 13С-ЯМР спектр чередующегося сополимера метилакрилата с N-винилфталимидом.
В публикации "Amine functional polymers based on N-ethenylformamide", R. K. Pinschmidt et al., Progress in Organic Coatings, 1996, 27, p.209-218, в разделе 2.1 описана полимеризация 32 мол. % N-винилформамида, 38 мол. % бутилакрилата и 30 мол. % метилметакрилата, проводимая в растворителе, например, в спирте, кетоне или смеси спирт/толуол, с использованием радикального инициатора Vazo 52. В разделе 2.2 описано, что проводимый в щелочной среде гидролиз сополимеров и тройных сополимеров (мет)акрилат/N-винилформамид с использованием гидроксида калия в содержащем спирт растворителе протекает быстро. В случае использования исходного полимера, полученного путем полимеризации смеси акрилат: N-винилформамид = 1:1, осаждается содержащий лактам полимер, который известен из указанного выше документа US 4898915. В разделе 3.4 и с помощью схемы 3 описан гидролиз и образование лактама для сополимеров N-винилформамида с (мет)акрилатами. Высокое содержание лактама приводит к нерастворимости в обычных растворителях.
В публикации "N-vinylformamide - building block for novel polymer structures", R. K. Pinschmidt et al., Pure Applied Chemistry, 1997, A34(10), p. 1885-1905, в частности, описано, что гидролиз N-винилформамида с (мет)акрилатами или акрилонитрилом в кислой среде протекает легко и с обеспечением высокого выхода. Это связано с отсутствием сильного отталкивания зарядов виниламиновых звеньев, содержащихся в этих обладающих высокой степенью чередования чередующихся сополимеров. При нейтрализации или при гидролизе в щелочной среде очень быстро образуется лактам. Это схематически представлено на фиг. 9 и лактамная структура названа нерастворимой.
GB 752290 в качестве объекта изобретения раскрыты полимеры, которые в качестве структурных звеньев содержат 5-членные лактамы (=пирролидин-2-оновые структурные звенья). Для получения полимеров, соответствующих примерам, сначала проводят реакцию полимеризации акрилоилхлорида с получением исходного полимера. Этот исходный полимер растворяют в диметилформамиде и вводят в реакцию с азидом натрия или гидроксиламином. После фильтрования и добавления ацетона конечный полимер осаждается, его растворяют в воде и осаждают путем добавления хлористоводородной кислоты. В частности, описаны конечный полимер, содержащий 70 мол. % лактамных структурных звеньев, 23 мол. % кислотных групп и 7 мол. % аминогрупп, и конечный полимер, содержащий 63 мол. % лактамных структурных звеньев, 24,5 мол. % кислотных групп и 12,5 мол. % аминогрупп. Конечные полимеры рекомендуют применять, в частности, в качестве пленкообразователей и в фотографических слоях.
В публикации "Determination of the sequence distribution and ionization constant of poly(acrylic acid-co-vinylamine) by 13С-ЯМР", C. Chang et al., Journal of Polymer Science, Polymer Symposium, 1986, 74, p. 17-30, конечные полимеры, которые получают по реакции Шмидта, проводимой с использованием полиактриловой кислоты и азотоводородной кислоты, и которые содержат первичную аминогруппу и карбоксигруппы, исследуют с помощью ядерного магнитного резонанса. Описано образование лактама в случае исследованных конечных полимеров, в которых 12 или 30, или 52% карбоксигрупп превращены в аминогруппы.
В публикации "Polymers and group interaction. IV. Hofmann reaction on polyvinylamides", M. Mullier et al., Journal of Polymer Science, 1957, XXIII, p. 915-930, в частности, исследуют продукты, полученные при перегруппировки Гофмана, проводимой с использованием полиакриламида и полиметакриламида. В примерах, использующиеся в качестве исходных полимеров полиакриламиды вводят в реакцию с гипохлоритом натрия, при этом в качестве конечных полимеров получают полимеры, содержащие аминогруппы. В этих конечных полимерах содержится большая доля 5-членных лактамных структурных звеньев. Приведенный ниже полимер приведен в таблице 1, как конечный полимер "полимер I", который получают по реакции Гофмана с использованием полиакриламида и 1 экв. гипохлорита натрия:

Приведенный ниже полимер приведен в таблице 1, как конечный полимер "полимер II", который получают по реакции Гофмана с использованием полиметакриламида и 1 экв. гипохлорита натрия:

В приведенном в JP 2016-186023 А примере 1 описана реакция радикальной полимеризации 43 мол. % N-винилформамида и 57 мол. % метилметакрилата в метилэтилкетоне. В примере 2 описана реакция радикальной полимеризации 24 мол. % N-винилформамида и 76 мол. % метилметакрилата в метилэтилкетоне. Полученные полимеры представляют интерес для применения для оптических линз и т.п.
В приведенном в JP 2017-061602 А примере 3 описана реакция радикальной полимеризации 32 мол. % N-винилформамида и 68 мол. % метилметакрилата в метилэтилкетоне. Полученный полимер представляет интерес для применения для оптических компонентов.
В приведенном в JP 2017-039867 А примере 4 описана реакция радикальной полимеризации 20 мол. % N-винилформамида и 80 мол. % метилметакрилата в метилэтилкетоне. В примере 5 описана реакция радикальной полимеризации 32 мол. % N-винилформамида и 68 мол. % метилметакрилата в метилэтилкетоне. Полученные полимеры представляют интерес для применения для оптических компонентов.
В приведенном в JP 2017-039868 А примере 2 описана реакция радикальной полимеризации 32 мол. % N-винилформамида и 68 мол. % метилметакрилат в метилэтилкетоне. В примере 3 описана реакция радикальной полимеризации 48 мол. % N-винилформамида и 52 мол. % метилметакрилата в метилэтилкетоне. В примере 4 описана реакция радикальной полимеризации 20 мол. % N-винилформамида и 80 мол. % метилметакрилата в метилэтилкетоне. Полученные полимеры представляют интерес для применения для оптических компонентов.
Необходимы дополнительные способы изготовления бумаги или картона, в которых полученная бумага или картон обладает хорошей прочностью в сухом состоянии. Если в этих способах используют добавки, то дополнительно предпочтительно, если их получение можно провести в промышленном масштабе и как можно более простым образом.
Согласно изобретению разработан способ изготовления бумаги или картона, включающий стадии
(А) добавления конечного полимера А к первой водной суспензии волокнистого материала с получением таким образом второй водной суспензии волокнистого материала, содержащей конечный полимер А,
где конечный полимер А представляет собой получаемый путем
- радикальной полимеризации следующих мономеров:
(i) от 30 до 90 мол. % мономера формулы I
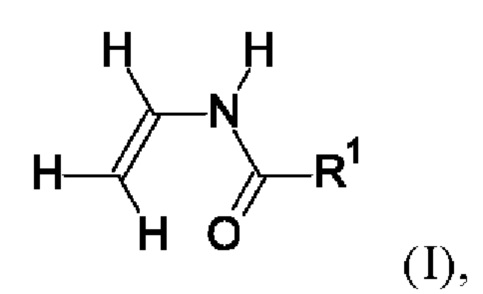
в которой R1=Н или обозначает фиг. C1-С6-алкил,
(ii) от 3 до 60 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 45 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 35 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы, где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм,
(B) обезвоживания второй водной суспензии волокнистого материала, содержащей конечный полимер А, на водопроницаемой подложке с получением мокрой бумажной структуры,
(C) обезвоживания мокрой бумажной структуры с получением таким образом бумаги или картона.
На стадии (А) первая водная суспензия волокнистого материала означает композицию, включающую (а-а) воду и (а-b) волокнистый материал, содержащий целлюлозные волокна. Альтернативным термином для "суспензии волокнистого материала" является "пульпа".
Для получения первой водной суспензии волокнистого материала можно использовать механические и/или химические методики. Так, например, размол водной суспензии волокнистого материала является механической методикой для получения более коротких волокон и, в случае целлюлозных волокон, также для дефибриллирования волокон. Степень обезвоживания водной суспензии волокна определяется обеспеченной степенью помола. Одной методикой определения степени помола суспензии волокнистого материала является определение кинетики обезвоживания в соответствии с методикой Шоппера-Риглера и она выражена в градусах Шоппера-Риглера (°ШР).
В качестве волокнистого материала можно использовать натуральные и/или регенерированные волокна. Можно использовать все волокна древесины или однолетних растений, обычно использующихся в бумажной промышленности. Однолетними растениями, подходящими для получения волокнистых материалов, являются, например, рис, пшеница, сахарный тростник и кенаф. Древесный материал, полученный, например, из древесины мягких пород или древесины твердых пород, включают древесную массу, термомеханическую древесную массу (ТММ), химикотермомеханическую древесную массу (ХТММ), получаемую под давлением древесную массу, полухимическую древесную массу, целлюлозу с высоким выходом и рафинерную механическую древесную массу (РММ). Древесный материал грубого помола обычно обладает степенью помола, равной 40-60°ШР, в отличие от древесного материала стандартного помола, обладающего степенью помола, равной 60-75°ШР, и древесного материала тонкого помола, обладающего степенью помола, равной 70-80°ШР. Пульпы, полученные, например, из древесины мягких пород или древесины твердых пород, включают химически обработанную сульфатную, сульфитную или натронную целлюлозу. Кроме того, целлюлоза также может являться беленой или небеленой. Предпочтительной является небеленая целлюлоза, также известная, как небеленая крафт-целлюлоза. Неразмолотая пульпа обычно обладает степенью помола, равной 13-17°ШР, в отличие от пульпы грубого и среднего помола, обладающей степенью помола, равной 20-40°ШР, и пульпы тонкого помола, обладающей степенью помола, равной 50-60°ШР. Регенерированные волокна могут быть получены, например, из бумажной макулатуры. Бумажную макулатуру необязательно можно предварительно очистить от краски. Смешанная макулатура обычно обладает степенью помола, равной примерно 40°ШР, в отличие от макулатуры, очищенной от краски, обладающей степенью помола, равной примерно 60°ШР. Регенерированные волокна, полученные из макулатуры, можно использовать по отдельности или в смеси с другими, в особенности, с натуральными волокнами.
Предпочтительным способом является такой, в котором первая водная суспензия волокнистого материала обладает степенью помола, определенной по кинетике обезвоживания по методике Шоппера-Риглера, составляющей от 13 до 70°ШР, более предпочтительно от 20 до 60°ШР и особенно предпочтительно от 30 до 50°ШР.
Первую водную суспензию волокнистого материала можно получить, например, путем переработки имеющихся бумаги или картона, например, путем механической обработки макулатуры в дефибрере вместе с водой до обеспечения необходимой консистенции водной суспензии волокнистого материала. Другим примером объединения волокон, полученных из двух источников, является смешивание первичной суспензии волокнистого материала с переработанными отходами бумаги с покрытием, полученной с использованием первичной суспензии волокнистого материала.
Первая водная суспензия волокнистого материала может содержать не только воду (а-а) и волокнистый материал (а-b), но также другие компоненты, которые в соответствующих случаях специально добавляют к суспензии волокнистого материала, или которые в соответствующих случаях содержатся вследствие использования макулатуры или имеющейся бумаги.
В настоящем изобретении "содержание сухих веществ" означает отношение массы образца после сушки к массе образца до сушки, выраженное с массовых процентах. Предпочтительно, если содержание сухих веществ определяют путем сушки при 105°С до постоянной массы. Для этого проводят сушку при 105°С (±2°С) в сушильном шкафу до обеспечения постоянной массы. В соответствии с настоящим изобретением постоянная масса обеспечена, если при содержании сухих веществ, составляющем от 1 до 100%, больше не изменяется округленное до первого десятичного разряда выраженное в процентах значение, и при содержании сухих веществ, составляющем от 0 до менее 1%, больше не изменяется округленное до второго десятичного разряда выраженного в процентах значение. Сушку проводят при давлении окружающей среды, необязательно при 101,32 кПа, без внесения поправки на какое-либо отклонение вследствие погодных условий или высоты над уровнем моря. В разделе "Примеры" в параграфе под заголовком "Определение содержания сухих веществ" приведено описание практического осуществления.
При содержании сухих веществ, составляющем от более 1,5 до 6 мас. %, предпочтительно от 2,0 до 4,0 мас. % в пересчете на массу первой водной суспензии волокнистого материала (соответствует концентрации волокнистого материала, равной примерно от более 15 до 60 г/л, если содержится практически только волокнистый материал), в настоящем изобретении смесь называется густой массой. В контексте настоящего изобретения следует отметить то отличие, что при содержании сухих веществ, составляющем от 0,1 до 1,5 мас. %, предпочтительно от 0,3 до 1,4 мас. % в пересчете на массу водной суспензии волокнистого материала (соответствует концентрации волокнистого материала, равной примерно от 1 до 15 г/л, если содержится практически только волокнистый материал), смесь обычно называется жидкой массой. Содержание сухих веществ или масса водной суспензии волокнистого материала в сухом состоянии означает содержание всех компонентов, которые являются нелетучими или, предпочтительно, нелетучими при определении содержания сухих веществ, проводимого путем сушки при 105°С до постоянной массы.
Предпочтительно, если содержание сухих веществ в первой водной суспензии волокнистого материала составляет от 0,1 до 6 мас. %, более предпочтительно от 0,12 до 5 мас. %, особенно предпочтительно от 0,15 до 4 мас. %, еще более предпочтительно от более 1,5 до 4,0 мас. % и наиболее предпочтительно от 2,0 до 4,0 мас. %.
Предпочтительным является способ, в котором на стадии (А) первая водная суспензия волокнистого материала обладает содержанием сухих веществ, составляющим от 0,1 до 6 мас. %.
На стадии (А) конечный полимер А добавляют к первой суспензии волокнистого материала, в которой содержание сухих веществ составляет от более 1,5 и вплоть до 6,0 мас. %. Более предпочтительно, если полученную вторую суспензию волокнистого материала, содержащую конечный полимер А, затем разбавляют до обеспечения содержания сухих веществ, составляющего от 0,1 и вплоть до 1,5 мас. %. Предпочтительно, если на стадии (А) конечный полимер А добавляют к первой суспензии волокнистого материала, в которой содержание сухих веществ составляет от 0,1 и вплоть до 1,5 мас. %.
Предпочтительным является способ, в котором на стадии (А) конечный полимер А добавляют к первой водной суспензии волокнистого материала, в которой в момент добавления содержание сухих веществ составляет от более 1,5 до 6,0 мас. %.
После добавления конечного полимера А к первой водной суспензии волокнистого материала предпочтительно подождать с проводимым на стадии (В) обезвоживанием в течение от 0,5 с до 2 ч, более предпочтительно в течение от 1,0 с до 15 мин и особенно предпочтительно в течение от 2 до 20 с. Это обеспечивает необходимую продолжительность реакции конечного полимера А.
Предпочтительно, если количество добавленного конечного полимера А составляет от 0,01 до 3,0 мас. % в пересчете на содержание сухих веществ в первой водной суспензии волокнистого материала. В настоящем изобретении количество конечного полимера А рассчитывают, как содержание полимера. Содержание полимера означает выраженное в массовых процентах содержание конечного полимера А в водном растворе без противоионов, т.е. противоионы не учитывают. аким образом, содержание полимера означает сумму выраженных в массовых процентах количеств всех структурных звеньев конечного полимера А в граммах, которые содержатся в 100 г водной дисперсии или водного раствора конечного полимера А. В разделе "Примеры" в параграфе под заголовком "Содержание полимера" приведено описание практического осуществления. Более предпочтительным является количество, составляющее от 0,02 до 1,0 мас. %, особенно предпочтительным от 0,06 до 0,8 мас. %, более предпочтительным от 0,09 до 0,6 мас. %, еще более предпочтительным от 0,12 до 0,5 мас. %, еще более предпочтительным от 0,15 до 0,5 мас. % и наиболее предпочтительным от 0,2 до 0,4 мас. %.
Предпочтительным является способ, в котором на стадии (А) конечный полимер добавляют в количестве, составляющем от 0,2 до 0,5 мас. % в пересчете на массу первой суспензии волокнистого материала, где содержание сухих веществ в первой суспензии волокнистого материала составляет от более 1,5 и вплоть до 6,0 мас. %. Более предпочтительно, если полученную вторую суспензию волокнистого материала, содержащую конечный полимер А, затем разбавляют до обеспечения содержания сухих веществ, составляющего от 0,1 и вплоть до 1,5 мас. %.
Синтетическим предшественником конечного полимера А является исходный полимер V, который представляет собой получаемый по реакции радикальной полимеризации мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v).
Для полимеризации мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v) с получением исходного полимера V подходящими являются полимеризация в растворе, с осаждением, обращенная суспензионная или эмульсионная полимеризация. Предпочтительной является полимеризация в растворе, проводимая в водных средах. Подходящими водными средами являются вода и смеси воды и по меньшей мере одного смешивающегося с водой растворителя, например, спирта. Примерами спиртов являются метанол, этанол, н-пропанол, этиленгликоль или пропиленгликоль. Полимеризацию проводят посредством радикалов, например, с использованием инициаторов радикальной полимеризации, например, пероксидов, гидропероксидов, которые известны, как окислительно-восстановительные катализаторы, или с использованием азосоединений, которые распадаются с образованием радикалов. Примерами пероксидов являются пероксосульфаты щелочных металлов или аммония, диацетилпероксид, дибензоилпероксид, сукцинилпероксид, ди-трет-бутилпероксид, трет-бутилпербензоат, трет-бутилперпивалат, трет-бутилперокси-2-этилгексаноат, трет-бутилпермалеат, гидропероксид кумола, диизопропилпероксидикарбамат, бис(о-толуоил)пероксид, дидеканоилпероксид, диоктаноилпероксид, дилауроилпероксид, трет-бутилперизобутират, трет-бутилперацетат или ди-трет-амилпероксид. Примером гидропероксида является трет-бутилгидропероксид. Примерами азосоединений, которые распадаются с образованием радикалов, являются азобис-изобутиронитрил, 2,2'-азобис(2-метилпропионамидин)дигидрохлорид или 2,2'-азобис-(2-метилбутиронитрил). Примерами так называемых окислительно-восстановительных катализаторов являются аскорбиновая кислота/сульфат железа (II)/пероксодисульфат натрия, трет-бутилгидропероксид/дисульфит натрия, трет-бутилгидропероксид/гидроксиметансульфинат натрия или H2O/CuI.
Полимеризацию проводят, например, в воде или в смеси, содержащей воду в качестве растворителя, при температуре, находящейся в диапазоне от 30 до 150°С, предпочтительно от 40 до 110°С, причем реакцию можно провести при давлении окружающей среды, при пониженном или повышенном давлении. Для полимеризации в растворе выбирают растворимый в воде инициатор реакции полимеризации, например, 2,2'-азобис(2-метилпропионамидин)дигидрохлорид. Предпочтительно, если радикальную полимеризацию мономеров проводят в воде или в содержащей воду смеси растворителей. Более предпочтительной является вода или содержащая воду смесь растворителей, содержащая не менее 50 мас. % воды в пересчете на полное количество смеси растворителей. Особенно предпочтительной является вода или содержащая воду смесь растворителей, содержащая не менее 80 мас. % воды, более предпочтительно не менее 90 мас. % и еще более предпочтительно не менее 95 мас. % воды. Предпочтительно, если полимеризацию проводят в воде или в содержащей воду смеси растворителей, значение рН которой равно более 6, более предпочтительно значение рН которой равно от 6,1 до 9 и особенно предпочтительно значение рН которой равно от 6,2 до 6,8. Соответствующее значение рН можно обеспечить, например, путем добавления кислоты и/или основания, необязательно с добавлением буфера.
Предпочтительным является способ, в котором радикальную полимеризацию мономеров проводят в воде или в содержащей воду смеси растворителей.
При полимеризации мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v) с получением исходного полимера V к реакционной смеси можно добавить регуляторы полимеризации. Обычно используют количества, составляющие от 0,001 до 5 мол. % в пересчете на суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v). Регуляторы полимеризации хорошо известны из литературы и ими являются, например, соединения серы, гипофосфит натрия, муравьиная кислота или трибромхлорметан. Отдельными примерами соединений серы являются меркаптоэтанол, 2-этилгексилтиогликолят, тиогликолевая кислота и додецилмеркаптан.
Предпочтительно, если исходный полимер V обладает среднемассовой молекулярной массой Mw, равной от 75000 до 5000000 Да. Более предпочтительно, если исходный полимер V обладает среднемассовой молекулярной массой Mw, равной от 100000 до 4500000 Да, особенно предпочтительно от 180000 до 2500000 Да, еще более предпочтительно от 210000 до 1500000 Да и наиболее предпочтительно от 250000 до 1000000 Да. Среднемассовую молекулярную массу можно определить с помощью статического светорассеяния, например, при значении рН, равном 7,0, в растворе NaN03.
Примерами мономеров (i) формулы I являются N-винилформамид (R1=Н), N-винилацетамид (R1=C1-алкил), N-винилпропионамид (R1=С2-алкил) и N-винилбутирамид (R1=С3-алкил). С3-С6-Алкилы могут являться линейными или разветвленными. Примером C1-С6-алкила является метил, этил, н-пропил, 1-метилэтил, н-бутил, 2-метилпропил, 3-метилпропил, 1,1-диметилэтил, н-пентил, 2-метилбутил, 3-метилбутил, 2,2-диметилпропил или н-гексил. Предпочтительно, если R1 обозначает Н или С1-С4-алкил, более предпочтительно Н или С1-С2-алкил, более предпочтительно Н или C1-алкил, особенно предпочтительно Н, т.е. мономером (i) является N-винилформамид. Хотя один мономер формулы I может являться единственным, также включена смесь разных мономеров формулы I, использующаяся в качестве мономера (i). Предпочтительно, если доля мономера, в котором R1=Н, в полном количестве всех мономеров (i) формулы I составляет от 85 до 100%, более предпочтительно от 90 до 100%, особенно предпочтительно от 95 до 100% и наиболее предпочтительно от 99 до 100%.
Предпочтительным является способ, в котором мономером (i) является N-винилформамид, т.е. в формуле I R1=Н.
Предпочтительно, если полное количество всех мономеров (i) составляет от 50 до 89 мол. % в пересчете на количество всех мономеров, полимеризованных с получением исходного полимера V, т.е. всех мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), более предпочтительно, если оно составляет от 58 до 83 мол. %, особенно предпочтительно от 60 до 83 мол. % и наиболее предпочтительно от 65 до 80 мол. %. Условием остается то, что суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %.
Примерами мономеров (ii) являются метилакрилат, этилакрилат, н-пропилакрилат, изопропилакрилат, н-бутилакрилат, втор-бутилакрилат, трет-бутилакрилат, метилметакрилат, этилметакрилат, н-пропилметакрилат, изопропилметакрилат, н-бутилметакрилат, втор-бутилметакрилат и трет-бутилметакрилат. Хотя один мономер (ii) может являться единственным, в объем настоящего изобретения также входит смесь разных мономеров (ii), использующаяся в качестве мономера (i). Предпочтительными являются С1-С4-алкиловые эфиры акриловой кислоты и C1-алкиловые эфиры метакриловой кислоты, более предпочтительными являются C1-С3-алкиловые эфиры акриловой кислоты и C1-алкиловые эфиры метакриловой кислоты, особенно предпочтительными являются C1-С3-алкиловые эфиры акриловой кислоты, еще более предпочтительными являются С1-С2-алкиловые эфиры акриловой кислоты и наиболее предпочтительными являются С2-алкиловые эфиры акриловой кислоты (=этилакрилат). Предпочтительно, если доля С2-алкилового эфира акриловой кислоты в полном количестве всех мономеров (ii) составляет от 30 до 100%, более предпочтительно от 50 до 100%, особенно предпочтительно от 80 до 100% и наиболее предпочтительно от 95 до 100%. В случае использования С1-С4-алкилового эфира метакриловой кислоты предпочтительно, если также содержится С1-С4-алкиловый эфир акриловой кислоты, более предпочтительно при отношении количества С1-С4-алкилового эфира метакриловой кислоты к количеству С1-С4-алкилового эфира акриловой кислоты, составляющем не менее 1:1.
Предпочтительным является способ, в котором мономером (ii) является C1-С3-алкиловый эфир акриловой кислоты или C1-алкиловый эфир метакриловой кислоты.
Предпочтительным является способ, в котором мономером (ii) является C1-С2-алкиловый эфир акриловой кислоты.
Предпочтительным является способ, в котором мономером (ii) является этилакрилат.
Предпочтительно, если полное количество всех мономеров (ii) составляет от 5 до 45 мол. % в пересчете на количество всех мономеров, полимеризованных с получением исходного полимера V, т.е. всех мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), более предпочтительно, если оно составляет от 8 до 39 мол. %, особенно предпочтительно от 8 до 30 мол. %, еще более предпочтительно от 8 до 25 мол. % и наиболее предпочтительно от 8 до 21 мол. %. Условием остается то, что суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %.
В контексте настоящего изобретения "этиленовоненасыщенный мономер" означает мономер, содержащий по меньшей мере одно звено С2, атомы углерода которого соединены углерод-углеродной двойной связью. В случае, если единственными заместителями являются атомы водорода, им является этилен. В случае замещения с помощью 3 атомов водорода содержится производное винила. В случае замещения с помощью 2 атомов водорода содержится E/Z-изомер или производное этен-1,1-диила. В контексте настоящего изобретения "моноэтиленовоненасыщенный мономер" означает мономер, в котором содержится ровно одно звено С2.
В случае, если в определенной молекуле или классе молекул содержится катионная группа, то солевая форма означает, что соответствующий анион обеспечивает нейтральный заряд. Такими анионами являются, например, хлорид, бромид, гидросульфат, сульфат, гидрофосфат, метилсульфат, ацетат или формиат. Предпочтительным является хлорид, формиат или гидросульфат, особенно предпочтительным является хлорид или формиат. В случае, если в определенной молекуле или классе молекул содержится анионная группа, то солевая форма означает, что соответствующий катион обеспечивает нейтральный заряд. Такими катионами являются, например, катионы щелочных металлов, щелочноземельных металлов, аммиака, алкиламинов или алканоламинов. Предпочтительными являются Li + , Na + , K + , Rb + , Cs + , Mg2 + , Ca2 + , Sr2 + , Ba2 + или NH4 + . Более предпочтительными являются Li + , Na + , K + , Mg2 + , Ca2 + или NH4 + , особенно предпочтительными являются Na + , K + , Ca2 + или NHV", еще более предпочтительными являются Na + , K + или NH4 + , еще более предпочтительными являются Na + или К + и наиболее предпочтительным является Na + .
Мономер (iii) также включает смесь отдельных мономеров, относящихся к группе мономеров (iii).
Примерами мономера (iii), которым является моноэтиленовоненасыщенная карбоновая кислота или ее солевая форма, являются моноэтиленовоненасыщенные С3-С8-моно- или дикарбоновые кислоты или их солевые формы. Примерами являются акриловая кислота, акрилат натрия, метакриловая кислота, метакрилат натрия, диметакриловая кислота, метакриловая кислота, малеиновая кислота, фумаровая кислота, итаконовая кислота, мезаконовая кислота, цитраконовая кислота, метиленмалоновая кислота, аллилуксусная кислота, винилуксусная кислота или кротоновая кислота.
Примерами мономера (iii), которым является моноэтиленовоненасыщенная сульфоновая кислота или ее солевая форма, являются винилсульфоновая кислота, акриламидо-2-метилпропансульфоновая кислота, метакриламидо-2-метилпропансульфоновая кислота, аллилсульфоновая кислота, металлилсульфоновая кислота, сульфоэтилакрилат, сульфоэтилметакрилат, сульфопропилакрилат, сульфопропилметакрилат, 2-гидрокси-3-метакрилоксипропилсульфоновая кислота или стиролсульфоновая кислота.
Примерами мономера (iii), которым является моноэтиленовоненасыщенная фосфоновая кислота или ее солевая форма, являются винилфосфоновая кислота, монометиловый эфир винилфосфоновой кислоты, аллилфосфоновая кислота, монометиловый эфир аллилфосфоновой кислоты, акриламидометилпропилфосфоновая кислота или акриламидометиленфосфоновая кислота.
Предпочтительно, если мономером (iii) является моноэтиленовоненасыщенная карбоновая кислота или моноэтиленовоненасыщенная сульфоновая кислота, или их солевые формы. Предпочтительно, если мономером (iii) является моноэтиленовоненасыщенная С3-С8-моно- или дикарбоновая кислота, моноэтиленовоненасыщенная сульфоновая кислота или винилфосфоновая кислота, или их солевые формы. Более предпочтительно, если мономером (iii) является моноэтиленовоненасыщенная С3-C8-моно- или дикарбоновая кислота, винилсульфоновая кислота, акриламидо-2-метилпропансульфоновая кислота, метакриламидо-2-метилпропансульфоновая кислота или винилфосфоновая кислота, или их солевые формы. Более предпочтительными являются моноэтиленовоненасыщенные C3-C8-моно- или дикарбоновые кислоты или их солевые формы. Особенно предпочтительными являются акриловая кислота, метакриловая кислота, винилсульфоновая кислота или акриламидо-2-метилпропансульфоновая кислота, или их солевые формы. Еще более предпочтительными являются акриловая кислота или метакриловая кислота, или их солевые формы. Наиболее предпочтительными являются акриловая кислота, акрилат натрия, метакриловая кислота или метакрилат натрия. Предпочтительно, если доля акриловой кислоты и метакриловой кислоты или их солевых форм в полном количестве всех мономеров (iii) составляет от 30 до 100%, более предпочтительно от 50 до 100%, особенно предпочтительно от 80 до 100% и наиболее предпочтительно от 95 до 100%.
Предпочтительным является способ, в котором мономером (iii) является моноэтиленовоненасыщенная карбоновая кислота или моноэтиленовоненасыщенная сульфоновая кислота, или их солевые формы.
Предпочтительным является способ, в котором мономером (iii) является акриловая кислота, метакриловая кислота, винилсульфоновая кислота или 2-акриламидо-2-метилпропансульфоновая кислота, или их солевые формы.
Предпочтительно, если полное количество всех мономеров (iii) составляет от 0 до 40 мол. % в пересчете на количество всех мономеров, полимеризованных с получением исходного полимера V, т.е. всех мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), более предпочтительно, если оно составляет от 0 до 30 мол. %, особенно предпочтительно от 0 до 25 мол. %, еще более предпочтительно от 1 до 25 мол. %, еще более предпочтительно от 2 до 23 мол. %, еще более предпочтительно от 3 до 21 мол. % и наиболее предпочтительно от 5 до 18 мол. %. Условием остается то, что суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %. ??? Согласно изобретению неожиданно было установлено, что некоторые исходные полимеры V обеспечивают преимущество для промышленного производства при проведении гидролиза в щелочной среде с получениям конечного полимера А. Если исходный полимер V содержит мономер (iii), то в случае проведения гидролиза в щелочной среде с получениям конечного полимера А отсутствует или по меньшей мере уменьшается пик вязкости, возникающий во время гидролиза в щелочной среде. Результаты для наличия, уменьшения или отсутствия пика вязкости представлены на фиг. 1 и в таблице А-4-1, приведенных в разделе "Примеры". Наблюдение уменьшенной или даже обращенной вихревой воронки на вале мешалки во время исследований протекания гидролиза, приведенных в разделе "Примеры", является показателем наличия пика вязкости и его количественной оценки. В соответствии с настоящим изобретением такие результаты оценки, как отсутствующий, минимальный, небольшой и умеренный, указанные в разделе "Примеры", означают умеренное повышение вязкости, которое все же является приемлемым и регулируемым в случае промышленного производства (в увеличенном масштабе). Предпочтительными являются такие результаты оценки, как отсутствующий, минимальный и небольшой, тогда как более предпочтительными являются отсутствующий и минимальный. Соответственно, исходный полимер V, который содержит мономер (iii), является предпочтительным для применения в способе.
Предпочтительным является способ, в котором мономер (iii) используют в количестве, составляющем от 1 до 25 мол. %.
Предпочтительно, если полное количество всех мономеров (iv) составляет от 0 до 7 мол. % в пересчете на количество всех мономеров, полимеризованных с получением исходного полимера V, т.е. всех мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), более предпочтительно, если оно составляет от 0 до 5 мол. %, особенно предпочтительно от 0 до 3 мол. %, еще более предпочтительно от 0,5 до 2 мол. % и наиболее предпочтительно от 1 до 1,5 мол. %. Условием остается то, что суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %.
Мономер (v) также включает смесь отдельных мономеров, относящихся к группе мономеров (v).
Примерами мономеров (v) являются
(v-1) моноэтиленовоненасыщенный мономер, который не обладает зарядом при рН=7,
(v-2) диэтиленовоненасыщенный мономер, который не обладает зарядом при рН=7 и содержит две сопряженные этиленовые двойные связи,
(v-3) моноэтиленовоненасыщенный мономер, который содержит по меньшей мере одну первичную, вторичную или третичную аминогруппу, и который обладает положительным зарядом при рН=7, или его солевая форма,
(v-4) диаллилзамещенный амин, атом азота которого не является кватернизованным,
(v-5) моноэтиленовоненасыщенный мономер, который содержит по меньшей мере единичный постоянный положительный заряд,
(v-6) мономер, который содержит по меньшей мере две этиленовоненасыщенные двойные связи, которые не являются сопряженными, и который отличается от диаллилзамещенного амина.
В случае мономеров (v), которые обладают зарядом, в их число также соответственно включены их солевые формы. Постоянный положительный заряд всегда содержится в виде положительного заряда, независимо от значения рН.
Примерами мономера (v-1) являются моноэфиры α,β-этиленовоненасыщенных монокарбоновых кислот, образованные с С5-С18-алканолами, моноэфиры α,β-этиленовоненасыщенных монокарбоновых кислот, образованные с С2-С18-алкандиолами, диэфиры α,β-этиленовоненасыщенных дикарбоновых кислот, образованные с С1-С18-алканолами или С2-С18-алкандиолами, первичные амиды α,β-этиленовоненасыщенных монокарбоновых кислот, N-алкиламиды α,β-этиленовоненасыщенных монокарбоновых кислот, N,N-диалкиламиды α,β-этиленовоненасыщенных монокарбоновых кислот, динитрилы α,β-этиленовоненасыщенных дикарбоновых кислот, эфиры, образованные из винилового спирта с С1-С18-монокарбоновыми кислотами, эфиры, образованные из аллилового спирта с C1-С30-монокарбоновыми кислотами, N-виниллактамы, не содержащие атом азота гетероциклы, содержащие α,β-этиленовоненасыщенную двойную связь, винилароматические соединения, винилгалогениды, винилиденгалогениды или С2-С8-моноолефины.
Моноэфирами α,β-этиленовоненасыщенных монокарбоновых кислот, образованными с С5-С18-алканолами, являются, например, н-гексилакрилат, н-гексилметакрилат, н-октилакрилат, н-октилметакрилат, 1,1,3,3-тетраметилбутилакрилат, 1,1,3,3-тетраметилбутилметакрилат или 2-этилгексилакрилат.
Моноэфирами α,β-этиленовоненасыщенных монокарбоновых кислот, образованными с С2-С18-алкандиолами, являются, например, 2-гидроксиэтилакрилат, 2-гидроксиэтилметакрилат, 2-гидроксиэтилэтакрилат, 2-гидроксипропилакрилат, 2-гидроксипропилметакрилат, 3-гидроксипропилакрилат, 3-гидроксипропилметакрилат, 3-гидроксибутилакрилат, 3-гидроксибутилметакрилат, 4-гидроксибутилакрилат, 4-гидроксибутилметакрилат, 6-гидроксигексилакрилат или 6-гидроксигексилметакрилат.
Первичными амидами α,β-этиленовоненасыщенных монокарбоновых кислот являются, например, амид акриловой кислоты или амид метакриловой кислоты.
N-Алкиламидами α,β-этиленовоненасыщенных монокарбоновых кислот являются, например, N-метилакриламид, N-метилметакриламид, N-изопропилакриламид, N-изопропилметакриламид, N-этилакриламид, N-этилметакриламид, N-(н-пропил)акриламид, N-(н-пропил)метакриламид, N-(н-бутил)акриламид, N-(н-бутил) метакриламид, N-(трет-бутил)акриламид, N-(трет-бутил)метакриламид, N-(н-октил)акриламид, N-(н-октил)метакриламид, N-(1,1,3,3-тетраметилбутил)акриламид, N-(1,1,3,3-тетраметилбутил)метакриламид, N-(2-этилгексил)акриламид или N-(2-этилгексил)метакриламид.
N,N-Диалкиламидами α,β-этиленовоненасыщенных монокарбоновых кислот являются, например, N,N-диметилакриламид или N,N-диметилметакриламид.
Эфирами, образованными из винилового спирта с С1-С30-монокарбоновыми кислотами, являются, например, винилформиат, винилацетат или винилпропионат.
N-Виниллактамами являются, например, N-винилпирролидон, N-винилпиперидон, N-винилкапролактам, N-винил-5-метил-2-пирролидон, N-винил-5-этил-2-пирролидон, N-винил-6-метил-2-пиперидон, N-винил-6-этил-2-пиперидон, N-винил-7-метил-2-капролактам или N-винил-7-этил-2-капролактам.
Винилароматическими соединениями, являются, например, стирол или метилстирол. Винилгалогенидами являются, например, винилхлорид или винилфторид. Винилиденгалогенидами являются, например, винилиденхлорид или винилиденфторид. С2-С8-Моноолефинами являются, например, этилен, пропилен, изобутилен, 1-бутен, 1-гексен или 1-октен.
Предпочтительным мономером (v-1) является 2-гидроксиэтилакрилат, 2-гидроксиэтилметакрилат, 2-гидроксиэтилэтакрилат, 2-гидроксипропилакрилат, 2-гидроксипропилметакрилат, винилпирролидон или винилацетат.
Примерами мономера (v-2) являются С4-С10-олефины, содержащие ровно две двойные связи, которые являются сопряженными, например, бутадиен или изопрен.
Примерами мономера (v-3) являются эфиры α,β-этиленовоненасыщенных монокарбоновых кислот, образованные с аминоспиртами, моно- и диэфиры α,β- этиленовоненасыщенных дикарбоновых кислот, образованные с аминоспиртами, амиды α,β-этиленовоненасыщенных монокарбоновых кислот, образованные с диалкилированными диаминами, N-винилимидазол или винилпиридин.
Кислотным компонентом эфиров α,β-этиленовоненасыщенных монокарбоновых кислот, образованных с аминоспиртами, предпочтительно является акриловая кислота или метакриловая кислота. Аминоспирты, предпочтительно С2-С12-аминоспирты, могут быть моноалкилированными с помощью C1-C8 или диалкилированными с помощью C1-C8 по аминному атому азота. Примерами являются диалкиламиноэтилакрилаты, диалкиламиноэтилметакрилаты, диалкиламинопропилакрилаты или диалкиламинопропилметакрилаты. Отдельными примерами являются N-метиламиноэтилакрилат, N-метиламиноэтилметакрилат, N,N-диметиламиноэтилакрилат, N,N-диметиламиноэтилметакрилат, N,N-диэтиламиноэтилакрилат, N,N-диэтиламиноэтилметакрилат, N,N-диметиламинопропилакрилат, N,N-диметиламинопропилметакрилат, N,N-диэтиламинопропилакрилат, N,N-диэтиламинопропилметакрилат, N,N-диметиламиноциклогексилакрилат или N,N-диметиламиноциклогексилметакрилат.
Кислотным компонентом моно- и диэфиров α,β-этиленовоненасыщенных дикарбоновых кислот, образованных с аминоспиртами, предпочтительно является фумаровая кислота, малеиновая кислота, монобутилмалеат, итаконовая кислота или кротоновая кислота. Аминоспирты, предпочтительно С2-С12-аминоспирты, могут быть моноалкилированными с помощью C1-C8 или диалкилированными с помощью C1-C8 по аминному атому азота.
Амидами, α,β-этиленовоненасыщенных монокарбоновых кислот, образованными с диалкилированными диаминами, являются, например, диалкиламиноэтилакриламиды, диалкиламиноэтилметакриламиды, диалкиламинопропилакриламиды или диалкиламинопропилметакриламиды. Отдельными примерами являются N-[2-(диметиламино)этил]акриламид, N-[2-(диметиламино)этил]метакриламид, N-[3-(диметиламино)пропил]акриламид, N-[3-(диметиламино)пропил]метакриламид, N-[4-(диметиламино)бутил]акриламид, N-[4-(диметиламино)бутил]метакриламид, N-[2-(диэтиламино)этил]акриламид или N-[2-(диэтиламино)этил]метакриламид.
Примерами мономера (v-4) являются диаллиламин или метилдиаллиламин.
Примерами мономера (v-5) являются диаллиламины, которые кватернизованы по атому азота, солевая форма N-алкил-N'-винилимидазолия, солевая форма N-алкилированного винилпиридиния, солевая форма акриламидоалкилтриалкиламмония или солевая форма метакриламидоалкилтриалкиламмония. Диаллиламином, который кватернизован по атому азота, является, например, диаллилдиметиламмонийхлорид, диаллилдиэтиламмонийхлорид, диаллилдипропиламмонийхлорид или диаллилдибутиламмонийхлорид. Солевой формой N-алкил-N'-винилимидазолия является, например, 1-метил-3-винилимидазол-1-ийхлорид, 1-метил-3-винилимидазол-1-ийметилсульфат или 1-этил-3-винилимидазол-1-ийхлорид. Солевой формой N-алкилированного винилпиридиния является, например, 1-метил-4-винилпиридин- 1-ийхлорид, 1-метил-3-винилпиридин-1-ийхлорид, 1-метил-2-винилпиридин- 1-ийхлорид или 1-этил-4-винилпиридин-1-ийхлорид. Солевой формой акриламидоалкилтриалкиламмония является, например, акриламидоэтилтриметиламмонийхлорид (триметил-[2-(проп-2-еноиламино)этил]аммонийхлорид), акриламидоэтилдиэтилметиламмонийхлорид (диэтилметил-[3-(проп-2-еноиламино)этил] аммонийхлорид), акриламидопропилтриметиламмонийхлорид (триметил-[3-(проп-2-еноиламино)пропил]аммонийхлорид) или акриламидопропилдиэтилметиламмонийхлорид (диэтилметил-[3-(проп-2-еноиламино)пропил]аммонийхлорид). Солевой формой метакриламидоалкилтриалкиламмония является, например, метакриламидоэтилтриметиламмонийхлорид (триметил-[2-(2-метилпроп-2-еноиламино)этил] аммонийхлорид), метакриламидоэтилдиэтилметиламмонийхлорид (диэтилметил-[3-(2-метилпроп-2-еноиламино)этил] аммонийхлорид), метакриламидопропилтриметиламмонийхлорид (триметил-[3-(2-метилпроп-2-еноиламино)пропил]аммонийхлорид) или метакриламидопропилдиэтилметиламмонийхлорид (диэтилметил-[3-(2-метилпроп-2-еноиламино)пропил]аммонийхлорид).
Примером мономера (v-6) является тетрааллиламмонийхлорид, триаллиламин, метиленбисакриламид, гликольдиакрилат, гликольдиметакрилат, глицеринтриакрилат, пентаэритриттриаллиловый эфир, N,N-дивинилэтиленмочевина, полиалкиленгликоли или полиолы, этерифицированные по меньшей мере дважды акриловой кислотой и/или метакриловой кислотой, такие как пентаэритрит, сорбит и глюкоза.
Предпочтительным является мономер (v), который не является эфиром акриловой кислоты или метакриловой кислоты. Более предпочтительным является мономер (v), который не является эфиром этиленовоненасыщенной карбоновой кислоты.
Предпочтительно, если доля мономеров (v-1) составляет от 50 до 100% от полного количества всех мономеров (v). Особенно предпочтительно, если она составляет от 80 до 100%; наиболее предпочтительно от 95 до 100%. Для приведенной выше доли от полного количества всех мономеров (v) особенно предпочтительными являются следующие мономеры (v-1): 2-гидроксиэтилакрилат, 2-гидроксиэтилметакрилат, 2-гидроксиэтилэтакрилат, 2-гидроксипропилакрилат, 2-гидроксипропилметакрилат, винилпирролидон или винилацетат.
Предпочтительно, если доля мономеров (v-3), (v-4) и (v-5) составляет от 50 до 100% от полного количества всех мономеров (v). Особенно предпочтительно, если она составляет от 80 до 100%; наиболее предпочтительно от 95 до 100%.
Предпочтительно, если доля мономеров (v-3), (v-4) и (v-5) составляет от 50 до 100% от полного количества всех мономеров (v). Особенно предпочтительно, если она составляет от 80 до 100%; наиболее предпочтительно от 95 до 100%.
Предпочтительно, если полное количество всех мономеров (v) составляет от 0 до 25 мол. % в пересчете на количество всех мономеров, полимеризованных с получением исходного полимера V, т.е. всех мономеров (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), более предпочтительно, если оно составляет от 0 до 24 мол. %, особенно предпочтительно от 0 до 19 мол. %, еще более предпочтительно от 0,01 до 15 мол. %, еще более предпочтительно от 0,1 до 8 мол. %, еще более предпочтительно от 0,2 до 4 мол. % и наиболее предпочтительно от 0,4 до 2 мол. %. Условием остается то, что суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %.
В случае акриламида, как типичного мономера (v-1), предпочтительно, если количество акриламида составляет от 0 до 6 мол. %, где указанные в мол. % количества приведены в пересчете на суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v), и суммарное количество всех мономеров составляет 100 мол. %. Более предпочтительно, если количество акриламида составляет от 0 до 5 мол. %, особенно предпочтительно от 0 до 3 мол. %, еще более предпочтительно от 0 до 2 мол. %, еще более предпочтительно от 0 до 1 мол. %, и наиболее предпочтительно, если акриламид совсем не содержится.
Способ, в котором мономеры (v) содержат акриламид в количестве, составляющем от 0 до 6 мол. %, указанные в мол. % количества приведены в пересчете на суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v), и суммарное количество всех мономеров составляет 100 мол. %.
Мономер (v-б) действует, как сшивающий реагент. Если сшивающий реагент используют, то предпочтительно, если использующееся количество составляет от 0,001 до 1 мол. % в пересчете на суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v), и суммарное количество всех мономеров составляет 100 мол. %, более предпочтительно, если оно составляет от 0,01 до 0,5 мол. % и особенно предпочтительно от 0,015 до 0,1 мол. %. Предпочтительно, если для радикальной полимеризации не используют мономер (v-6).
Предпочтительно, если исходный полимер V находится в виде водной дисперсии или водного раствора. Более предпочтительно, если содержание воды в водной дисперсии или водном составляет от 75 до 95 мас. % и содержание исходного полимера V составляет от 5 до 25 мас. %, где содержание исходного полимера V определено, как содержание твердых веществ. Определение содержания твердых веществ описано в экспериментальном разделе. Предпочтительно, если водная дисперсия обладает значением рН, равным более 6, более предпочтительно значением рН, равным от 6,1 до 9, и особенно предпочтительно значением рН, равным 6,2 до 6,8. Соответствующее значение рН можно обеспечить, например, путем добавления кислоты и/или основания, необязательно с добавлением буфера.
Предпочтительным является способ, в котором для радикальной полимеризации используют следующие мономеры:
(i) от 50 до 89 мол. % мономера формулы I,
(ii) от 5 до 45 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 30 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
Предпочтительным является способ, в котором для радикальной полимеризации используют следующие мономеры:
(i) от 58 до 83 мол. % мономера формулы I,
(ii) от 8 до 39 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
Предпочтительным является способ, в котором для радикальной полимеризации используют следующие мономеры:
(i) от 60 до 83 мол. % N-винилформамида,
(ii) от 8 до 25 мол. % этилакрилата,
(iii) от 3 до 21 мол. % акриловой кислоты или метакриловой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 24 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
Предпочтительным является способ, в котором для радикальной полимеризации используют следующие мономеры:
(i) от 60 до 83 мол. % N-винилформамида,
(ii) от 8 до 21 мол. % этилакрилата,
(iii) от 3 до 21 мол. % акриловой кислоты или метакриловой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 24 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
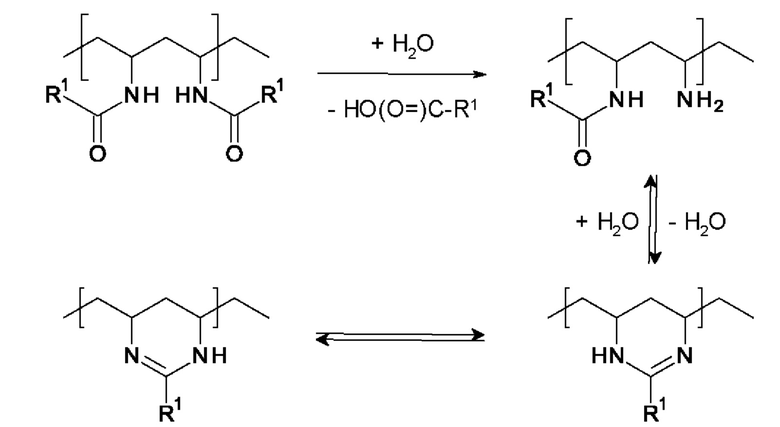
Конечный полимер А получают путем частичного или полного гидролиза исходного полимера V. Как известно, например, из ЕР 0438744 А1, стр. 8/строки 26-34, амидную группу, содержащуюся в звеньях мономеров (i), полимеризованных с получением исходного полимера, т.е. группу N-C(=O)R1, содержащуюся в мономере формулы (I), можно по меньшей мере частично гидролизовать, что приводит к образованию первичных аминогрупп. При отщеплении карбоновой кислоты, например, муравьиной кислоты или формиата в случае R1=Н, образуется первичная аминогруппа. Если гидролизованы не все амидные группы, то известно, что вследствие конденсации первичной аминогруппы с соседней амидной группой в конечном полимере А образуется циклическая 6-членная амидиновая группа, вероятно, в соответствии с приведенной ниже схемой реакции.
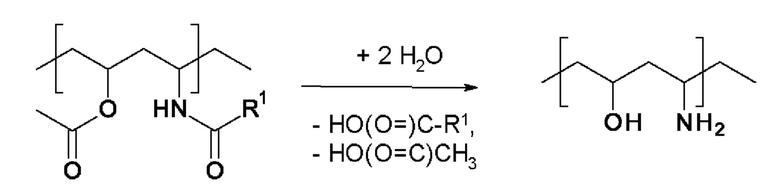
В случае полимеризации производных этилена, замещенных непосредственно по этиленовой функциональной группе цианогруппой, например, мономера (iv), исходный полимер V дополнительно содержит цианогруппы. Известно, что первичная аминогруппа, содержащаяся в конечном полимере А, образовавшаяся вследствие гидролиза, может вступать в реакцию с одной из этих цианогрупп с образованием циклического 5-членного амидина. В этом случае гидролиз амидной группы приводит к образованию в конечном полимере А 5-членной амидиновой группы в соответствии с приведенной ниже схемой реакции. На приведенной ниже схеме реакции замещенное цианогруппой производное этилена в этом случае представляет собой полимеризованный акрилонитрил.

В обоих описанных случаях гидролиз амидной группы, содержащейся в мономере формулы I, приводит к образованию первичной аминогруппы или амидиновой группы. Первичная аминогруппа или амидиновая группа обладает положительным зарядом при рН=7 и это обеспечивает наличие катионного заряда в конечном полимере А.
Условия проведения гидролиза амидных групп, которые происходят из мономеров формулы I, с получением конечного полимера А, также могут привести к гидролизу других групп, содержащихся в исходном полимере V, которые легко подвергаются гидролизу при этих условиях. Так, например, из ЕР 0216387 А2, столбец 6/строки 7-43, или из WO 2016/001016 А1, стр. 17/строки 1-8, известно, что гидролизуются ацетатные группы, содержащиеся в исходном полимере V, которые происходят из винилацетата, использующегося в качестве полимеризованного мономера (v-1). Соответственно, в конечном полимере А образуется вторичная гидроксигруппа, как это показано ниже в настоящем изобретении.
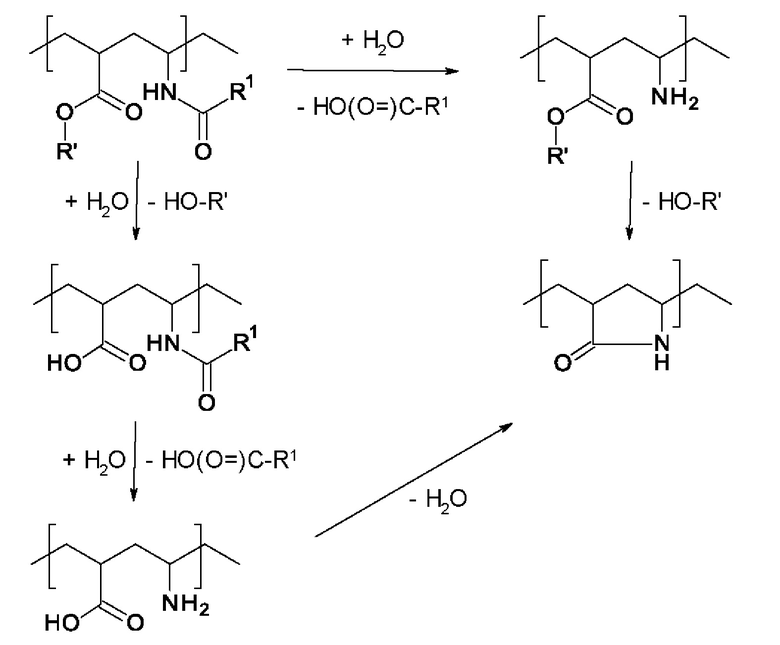
Наличие мономеров (ii) означает, что в исходном полимере V содержатся сложноэфирные группы. При проведении гидролиза амидных групп, содержащихся в конечном полимере А, которые происходят из мономеров формулы I, в кислой или щелочной среде наблюдается по меньшей мере частичное превращение сложноэфирых групп. Одной реакцией превращения является образование 5-членного лактамного структурного звена, содержащего полученную аминогруппу. Другой реакцией превращения является образование карбоксигруппы. На приведенной ниже схеме реакции представлены некоторые пути протекания реакции.
Количество звеньев мономеров формулы (I), которые полимеризуют с получением исходного полимера V, и которые гидролизуют с получением конечного полимера А, можно определить экспериментально путем определения количества карбоновых кислот HOC(=O)R1, отщепленных от групп N-C(=O)R1. В случае R1=Н можно определить количество выделившейся муравьиной кислоты или формиата, например, путем ферментативного анализа с использованием набора для исследования, выпускающегося фирмой Boehringer, Mannheim. Количество гидролизованных групп N-C(=O)R1, содержащихся в полимеризованных звеньях формулы I, отнесенное к количеству всех полимеризованных звеньев формулы I, умноженное на 100 мол. %, дает степень гидролиза (=СГ). Предпочтительно, если гидролизовано по меньшей мере от 50 до 100 мол. % мономеров (i), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (i), полимеризованных с получением исходного полимера V. Более предпочтительно, если гидролизовано по меньшей мере от 65 до 100%, особенно предпочтительно от 70 до 100%, еще более предпочтительно от 72 до 100%, еще более предпочтительно от 85 до 99,9%, еще более предпочтительно от 94 до 99,5% и наиболее предпочтительно от 94 до 99% этих мономеров.
Предпочтительным является способ, в котором гидролизовано по меньшей мере от 50 до 100 мол. % мономеров (i), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (i), полимеризованных с получением исходного полимера V.
Предпочтительным является способ, в котором гидролизовано не менее 70 и не более 99,5% полимеризованных мономеров (i) в пересчете на количество всех мономеров (i), полимеризованных с получением исходного полимера V
Количество звеньев мономеров (ii), которые полимеризуют с получением исходного полимера V, и которые подвергают превращению с получением конечного полимера А, можно определить экспериментально путем определения количества спиртов, отщепленных от сложноэфирных групп. Для определения количества отщепленного спирта подходящей является газовая хроматография или жидкостная хроматография высокого давления. Количество подвергнутых превращению сложноэфирных групп, содержащихся в полимеризованных мономерах (ii), отнесенное к количеству всех полимеризованных мономеров (ii), умноженное на 100 мол. %, дает степень превращения (=СП). Предпочтительно, если подвергается превращению по меньшей мере от 50 до 100 мол. % мономеров (ii), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (ii), полимеризованных с получением исходного полимера V. Более предпочтительно, если подвергается превращению по меньшей мере от 70 до 100%, особенно предпочтительно от 86 до 100%, еще более предпочтительно от 90 до 100%, еще более предпочтительно от 95 до 99,9%, еще более предпочтительно от 98 до 99,5% и наиболее предпочтительно 100% этих мономеров.
Предпочтительным является способ, в котором подвергается превращению по меньшей мере от 50 до 100 мол. % мономеров (ii), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (ii), полимеризованных с получением исходного полимера V.
Предпочтительным является способ, в котором подвергается превращению не менее 90 и не более 99,5% полимеризованных мономеров (ii) в пересчете на количество всех мономеров (ii), полимеризованных с получением исходного полимера V.
Предпочтительным является способ, в котором гидролизовано по меньшей мере от 70 до 100 мол. % мономеров (i), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (i), полимеризованных с получением исходного полимера V, и подвергается превращению по меньшей мере от 90 до 100% мономеров (ii), полимеризованных с получением исходного полимера V, в пересчете на количество всех мономеров (ii), полимеризованных с получением исходного полимера V.
Предпочтительно, если исходный полимер V подвергают гидролизу в щелочной, кислой среде или ферментативному гидролизу, боле предпочтительно гидролизу в щелочной или кислой среде и особенно предпочтительно гидролизу в щелочной среде. В случае гидролиза в кислой среде аминогруппы, содержащиеся в конечном полимере А, находятся в солевой форме. Обеспеченная степень гидролиза (=СГ) и обеспеченная степень превращения (=СП) зависят от кислоты или основания, использующегося количества кислоты или основания, использующейся температуры и продолжительности проведения реакции. Предпочтительно, если гидролиз проводят при температурах, равных от 20 до 170°С, более предпочтительно находящихся в диапазоне от 50 до 140°С. Гидролиз можно провести при нормальном давлении, при пониженном давлении или при повышенном давлении, т.е. при давлении, находящемся в диапазоне от 100 мбар до 16 бар. Предпочтительным является гидролиз при нормальном давлении. Для проведения гидролиза в щелочной среде подходящими являются гидроксиды металлов первой и второй главной группы Периодической системы элементов, например, гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид кальция, и аммиак и производные аммиака, например, триэтиламин, моноэтаноламин, диэтаноламин, триэтаноламин или морфолин. Предпочтительными являются гидроксиды металлов первой и второй главной группы Периодической системы элементов, более предпочтительными являются гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид кальция, особенно предпочтительными являются гидроксид натрия или гидроксид калия и наиболее предпочтительным является гидроксид натрия. Для проведения гидролиза в кислой среде подходящими являются неорганические кислоты, такие как галогениды водорода, серная кислота, азотная кислота и фосфорная кислота, а также органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, бензосульфоновая кислота, алкилсульфоновая кислота и фосфоновые кислоты. Предпочтительными являются хллристоводородная кислота и серная кислота. Предпочтительно использовать 0,2-2,0 экв. кислоты или основания в пересчете на сумму мольных доль N-виниламидов и эфиров (мет)акриловой кислоты, содержащихся в исходном полимере V. Более предпочтительными являются количества, составляющие от 0,5 до 1,5 экв., и особенно предпочтительно составляющие 0,7-1,2 экв. Предпочтительно, если основание или кислоту добавляют к исходному полимеру V в количестве, составляющем от 30 до 150 мол. % в пересчете на количество мономеров (i), полимеризованных с получением исходного полимера V. Предпочтительно, если количество составляет от 90 до 150 мол. %, особенно предпочтительно от 100 до 140 мол. % и наиболее предпочтительно от 110 до 130 мол. %. Предпочтительно, если основание или кислоту добавляют в количестве, составляющем от 30 до 130 мол. % в пересчете на количество всех мономеров (i), (ii), (iii), (iv) и (v). Предпочтительно, если гидролиз проводят в водном растворе, более предпочтительно в водном растворе, обладающем содержанием воды, составляющим от 40 до 95 мас. % в пересчете на полную массу водного раствора, особенно предпочтительно составляющим от 60 до 94 мас. % и наиболее предпочтительно составляющим от 75 до 93 мас. %.
Предпочтительным является способ, в котором исходный полимер V подвергают гидролизу в щелочной среде с получением конечного полимера А.
Как описано выше, исходные полимеры V, содержащие мономер (iii), обладают благоприятными характеристиками в случае проведения гидролиза в щелочной среде.
Предпочтительным является способ, в котором для проводимого путем радикальной полимеризации получения исходного полимера V используют от 1 до 25 мол. % мономера (iii) и исходный полимер V подвергают гидролизу в щелочной среде с получением конечного полимера А.
Предпочтительно, если конечный полимер А содержит 5-членные лактамные структурные звенья. Структурными звеньями, содержащимися в конечном полимере А, являются, с одной стороны, все мономеры (i), (ii), необязательно (iii), необязательно (iv) и необязательно (v), полимеризованные с получением исходного полимера V. Кроме того, ими также являются структурные звенья, которые могут быть образованы вследствие гидролиза. Они включают указанные выше 6-членные амидины, указанные выше 5-членные амидины, указанные выше этиленовые звенья, содержащие вторичные гидроксигруппы, указанные выше 5-членные лактамы и указанные выше эфиры акриловой кислоты или метакриловой кислоты, гидролизов анные с образованием карбоновой кислоты. При образовании некоторых из этих структурных звеньев полностью расходуются два полимеризованных мономера, содержащихся в исходном полимере V. Таким образом, полное количество всех структурных звеньев, содержащихся в конечном полимере А, представляет собой полное количество всех мономеров (i), (ii), (iii), (iv) и (v), которые полимеризованы с образованием исходного полимера V, за вычетом поправочного значения, соответствующего количеству тех структурных звеньев, которые образовались из двух полимеризованных мономеров. В качестве примера ниже в настоящем изобретении это представлено с помощью формулы (II)

в которой R2=Н или обозначает C1-алкил и R3=Н или обозначает С1-алкил, а, b, с, d и е обозначают выраженные в процентах мольные доли (=мол. %) структурных звеньев,
f обозначает выраженную в процентах мольную долю (=мол. %) по меньшей мере одного полимеризованного дополнительного структурного звена (не показано в формуле (II)), и сумма a, b, с, d, е и f равна 100 мол. %.
Предпочтительным является конечный полимер А формулы (II). Более предпочтительным является конечный полимер А формулы (II), где в формуле (II) а равно от 0,1 до 20 мол. %, b равно от 0 до 10 мол. %, с равно от 25 до 85 мол. %, d равно от 1 до 50 мол. %, е равно от 1 до 50 мол. %, и f равно от 0 до 40 мол. %, и где сумма количеств всех структурных звеньев а, b, с, d, е и f равна 100 мол. %. Особенно предпочтительно, если в формуле (II) а равно от 0,1 до 20 мол. %, b равно от 0 до 20 мол. %, с равно от 25 до 85 мол. %, d равно от 1 до 50 мол. %, и е равно от 1 до 50 мол. %, где сумма количеств всех структурных звеньев а, b, с, d и е равна 100 мол. %. Еще более предпочтительно, если в формуле (II) R2=R3=Н, а равно от 0,1 до 20 мол. %, b равно от 0 до 20 мол. %, с равно от 25 до 85 мол. %, d равно от 1 до 50 мол. %, е равно от 1 до 50 мол. %, и количество любых других структурных звеньев f равно от 0 до 40 мол. %, где сумма количеств всех структурных звеньев а, b, с, d, е и f равна 100 мол. %.
Наиболее предпочтительно, если в формуле (II) R2=R3=Н, а равно от 0,1 до 20 мол. %, b равно от 0 до 20 мол. %, с равно от 25 до 85 мол. %, d равно от 1 до 50 мол. %, и е равно от 1 до 50 мол. %, где сумма количеств всех структурных звеньев а, b с, d и е равна 100 мол. %.
Более предпочтительно, если содержание лактамных структурных звеньев составляет от 10 до 60 мол. %, где выраженное в процентах значение приведено в пересчете на полное количество всех структурных звеньев, содержащихся в конечном полимере А. Особенно предпочтительно, если содержание составляет от 15 до 50 мол. %, еще более предпочтительно от 17 до 35 мол. %. Особенно предпочтительно, если указанные выше содержания применимы для конечного полимеру А, находящегося в водной среде при значении рН, равном от 3,5 до 9, и особенно предпочтительно при значении рН, равном 3,5.
Предпочтительным является способ, в котором по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы.
Предпочтительно, если конечный полимер А обладает среднемассовой молекулярной массой Mw, равной от 8000 до 8000000 Да. Более предпочтительно, если конечный полимер А обладает среднемассовой молекулярной массой Mw, равной от 16000 до 4000000 Да, особенно предпочтительно от 80000 до 3600000 Да, еще более предпочтительно от 150000 до 2000000 Да и наиболее предпочтительно от 170000 до 1200000 Да. Среднемассовую молекулярную массу можно определить с помощью статического светорассеяния.
Предпочтительно, если конечный полимер А является катионогенным, более предпочтительно амфотерным-катионогенным. Конечный полимер А является катионогенным, если полное количество всех положительных зарядов, содержащихся в конечном полимере А, превышает полное количество всех отрицательных зарядов, содержащихся в конечном полимере А, при заданном значении рН, предпочтительно при значении рН, равном 7. При этом учитывают соответствующие обладающие зарядом структурные звенья и их заряды при номинальном значении рН, равном 7. Конечный полимер А является амфотерным-катионогенным, если полное количество всех положительных зарядов, содержащихся в конечном полимере А, превышает полное количество всех отрицательных зарядов, содержащихся в конечном полимере А, и в то же время в конечном полимере А содержатся отрицательные заряды при заданном значении рН, предпочтительно при значении рН, равном 7. Это также является справедливым при рассмотрении обладающих зарядом структурных звеньев при номинальном значении рН, равном 7. Количество мономеров (i). полимеризованных с получением исходного полимера V, и степень их гидролиза с образованием конечного полимера А являются наиболее важными параметрами для обеспечения положительных зарядов в конечном полимере А. Для этого возможно, что мономеры (v) обеспечивают положительный заряд в исходном полимере V и что этот положительный заряд все еще существует в конечном полимере А также после гидролиза, проводимого с получением конечного полимера А.
Предпочтительно, если конечный полимер А обладает плотностью положительного заряда. Более предпочтительно, если плотность заряда определяют путем титрования полиэлектролита поливинилсульфонатом калия. Более предпочтительно, если плотность заряда определяют при значении рН, равном 3,5, в водной среде. Особенно предпочтительно, если плотность заряда определяют путем титрования полиэлектролита поливинилсульфонатом калия при значении рН, равном 3,5, в водной среде. Предпочтительно, если плотность положительного заряда составляет от 2 до 16 ммоль/г, где 1 г относится к содержанию полимера в конечном полимере А. Более предпочтительно, если она составляет от 4 до 14 ммоль/г, особенно предпочтительно от 5 до 12 ммоль/г.
Предпочтительно, если конечный полимер А находится в виде водной дисперсии или водного раствора. Более предпочтительно, если содержание воды в водной дисперсии или водном растворе составляет от 75 до 95 мас. % и содержание конечного полимера А составляет от 5 до 25 мас. %, где содержание конечного полимера А определено, как содержание полимера. Предпочтительно, если водная дисперсия или водный раствор обладает значением рН, равным более 5, более предпочтительно значением рН, равным от 6 до 9, особенно предпочтительно значением рН, равным от 6 до 8, и наиболее предпочтительно значением рН, равным от 6,1 до 6,8. Соответствующее значение рН можно обеспечить, например, путем добавления кислоты и/или основания. Предпочтительно, если плотность положительного заряда конечного полимера А, который находится в виде водной дисперсии или водного раствора, составляет от 20 до 120 ммоль/100 г, где 100 г относится водной дисперсии или водному раствору конечного полимера А. Более предпочтительно, если она составляет от 30 до 100 ммоль/100 г, особенно предпочтительно от 35 до 90 ммоль/100 г.
Более предпочтительным является способ, в котором конечный полимер А представляет собой получаемый путем - радикальной полимеризации следующих мономеров:
(i) от 58 до 83 мол. % мономера формулы I

в которой R1=Н или обозначает C1-C6-алкил,
(ii) от 8 до 39 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы, где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм.
Предпочтительно, если конечный полимер А добавляют к первой водной суспензии волокнистого материала в виде водной дисперсии или водного раствора конечного полимера, обладающего А значением рН, равным более 5, более предпочтительно значением рН, равным от 6 до 9, особенно предпочтительно значением рН, равным от 6 до 8, и особенно предпочтительно значением рН, равным от 6,1 до 6,8.
Предпочтительным является способ, в котором на стадии (А) конечный полимер А добавляют в виде водной дисперсии или водного раствора, обладающего значением рН, равным от 5 до 9, к первой водной суспензии волокнистого материала.
Вторая водная суспензия волокнистого материала, содержащая конечный полимер А, включает
(а-а) воду,
(а-b) волокнистый материал,
(а-с) конечный полимер А.
Возможным дополнительным компонентом второй водной суспензии волокнистого материала является (a-d) органический полимер, который отличается от волокнистого материала и от конечного полимера А. Органический полимер (a-d) может являться нейтральным, катионогенным или анионогенным.
Нейтральный органический полимер (a-d) может являться незаряженным-нейтральным, поскольку он не включает полимерные звенья, содержащие функциональную группу, которая содержит по меньшей мере единичный заряд при значении рН, равном 7. Примерами нейтрального органического полимера (a-d), который не включает никакие полимерные звенья, содержащие функциональную группу, которая обладает зарядом при значении рН, равном 7, являются полиакриламид, сополимер акриламида с акрилонитрилом, поли(виниловый спирт) или сополимер винилового спирта с винилацетатом.
Нейтральный органический полимер (a-d) также может являться амфотерным-нейтральным, поскольку он включает полимерные звенья, содержащие функциональную группу, которая обладает отрицательным зарядом по меньшей мере при значении рН, равном 7, а также полимерные звенья, содержащие функциональную группу, которая обладает положительным зарядом по меньшей мере при значении рН, равном 7, где, кроме того, в функциональных группах количество всех отрицательных зарядов и количество всех положительных зарядов уравновешивает друг друга.
Катионогенный органический полимер (a-d) может являться полностью катионогенным, т.е. он включает полимерные звенья, содержащие функциональную группу, которая обладает положительным зарядом по меньшей мере при значении рН, равном 7, но не включает никакие полимерные звенья, содержащие функциональную группу, которая обладает отрицательным зарядом по меньшей мере при значении рН, равном 7. Примерами полностью катионогенного органического полимера (a-d) являются поли(аллиламин), поли(диаллиламин), поли(диаллилдиметиламмонийхлорид), сополимер акриламида с диаллилдиметиламмонийхлоридом или сополимер акриламида с 2-(N,N,N-триметиламмоний)этилакрилатхлоридом. Катионогенный органический полимер (a-d) также может являться амфотерным-катионогенным, т.е. он включает полимерные звенья, содержащие функциональную группу, которая обладает положительным зарядом по меньшей мере при значении рН, равном 7, и полимерные звенья, содержащие функциональную группу, которая обладает отрицательным зарядом по меньшей мере при значении рН, равном 7, и в функциональных группах количество всех положительных зарядов превышает количество всех отрицательных зарядов.
Анионогенный органический полимер (a-d) может являться полностью анионогенным, т.е. он включает полимерные звенья, содержащие функциональную группу, которая обладает отрицательным зарядом по меньшей мере при значении рН, равном 7, но не включает полимерные звенья, содержащие функциональную группу, которая обладает положительным зарядом по меньшей мере при значении рН, равном 7. Примерами полностью анионогенного органического полимера (a-d) являются поли(акриловая кислота), сополимер стирола с н-бутилакрилатом и с акриловой кислотой или сополимер акриламида с акрилонитрилом и с акриловой кислотой.
Анионогенный органический полимер (a-d) также может являться амфотерным-анионогенным, т.е. он включает полимерные звенья, содержащие функциональную группу, которая обладает отрицательным зарядом по меньшей мере при значении рН, равном 7, и полимерные звенья, содержащие функциональную группу, которая обладает положительным зарядом по меньшей мере при значении рН, равном 7, и в функциональных группах количество всех отрицательных зарядов превышает количество всех положительных зарядов.
Органические полимеры (a-d) также можно разделить на линейные, разветвленные и сшитые. Сшивку можно обеспечить, например, путем добавления сшивающего реагента уже во время проведения полимеризации исходных мономеров или путем добавления сшивающего реагента после завершения полимеризации, в особенности, непосредственно перед добавлением органического полимера (a-d) ко второй водной суспензии волокнистого материала. Так, например, полиакриламид можно сшить уже во время проведения полимеризации путем добавления к акриламиду сшивающего реагента - метиленбисакриламида или путем добавления сшивающего реагента, такого как глиоксаль, после проведения полимеризации. При необходимости можно объединить обе методики сшивки. Следует особо отметить сшитый органический полимер, который обладает высокой степенью сшивки, обычно уже во время проведения полимеризации мономеров. Он содержится во второй водной суспензии волокнистого материала, содержащей конечный полимер А, в виде частиц, в особенности, в виде так называемых органических микрочастиц.
Органические полимеры (a-d) также можно разделить на натуральные, модифицированные натуральные или синтетические. Натуральный органический полимер обычно является полимером природного происхождения, при этом при необходимости проводят соответствующие стадии выделения, однако не проводят никаких специальных модификаций путем химического синтеза. Примером натурального органического полимера (a-d) является немодифицированный крахмал. Целлюлоза не является примером натурального органического полимера (a-d), которая в настоящем изобретении является волокнистым материалом (а-b). Модифицированный натуральный органический полимер представляет собой полимер, модифицированный путем проведения стадий химического синтеза. Примером модифицированного натурального органического полимера (a-d) является катионогенный крахмал. Синтетический органический полимер (a-d) получают путем химического синтеза из отдельных мономеров. Примером синтетического органического полимера (a-d) является полиакриламид.
Предпочтительным является способ, в котором на стадии (А) органический полимер (a-d) добавляют к первой суспензии волокнистого материала или ко второй суспензии волокнистого материала, содержащей конечный полимер А. Более предпочтительно, если добавляют органический полимер (a-d), который является модифицированным натуральным органическим полимером. Особенно предпочтительно, если органическим полимером (a-d) является катионогенный крахмал. Наиболее предпочтительно, если катионогенный крахмал является единственным органическим полимером (a-d), который добавляют на стадии (А) к первой суспензии волокнистого материала в дополнение к конечному полимеру А или ко второй суспензии волокнистого материала, содержащей полимер А.
Возможным дополнительным компонентом водной суспензии волокнистого материала, содержащей конечный полимер А, является (а-е) наполнитель. Наполнитель (а-е) представляет собой неорганические частицы, в частности, неорганический пигмент. В качестве неорганических пигментов можно использовать все пигменты на основе оксидов, силикатов и/или карбонатов металлов, которые обычно используют в бумажной промышленности, в частности, пигменты, выбранные из группы, состоящей из следующих: карбонат кальция, который можно использовать в виде молотой извести, мела, мрамора (РКК (размолотый карбонат кальция)) или осажденного карбоната кальция (ОКК), тальк, каолин, бентонит, сатинит, сульфат кальция, сульфат бария и диоксид титана. Неорганическими частицами также является коллоидный раствор поликремниевых кислот, в котором частицы кремниевой кислоты обычно обладают размером, равным от 5 до 150 нм.
В контексте настоящего изобретения наполнитель (а-е) также включает два или большее количество разных наполнителей. Соответственно, наполнитель (а-е), использующийся в качестве возможного дополнительного компонента водной суспензии волокнистого материала, разделяют на первый наполнитель (а-е-1), второй наполнитель (а-е-2) и т.п.
Предпочтительно, если используют неорганические пигменты, обладающие средним размером частиц (средним объемным размером), составляющим ≤10 мкм, более предпочтительно, равным от 0,3 до 5 мкм, особенно предпочтительно от 0,5 до 2 мкм. Определение среднего размера частиц (среднего объемного размера) неорганических пигментов, а также частиц порошкообразной композиции в контексте настоящего изобретения обычно проводят по методике квазиупругого светорассеяния (DIN-ISO 13320-1), например, с использованием прибора Mastersizer 2000, выпускающегося фирмой Malvern Instruments Ltd.
Предпочтительным является способ, в котором на стадии (А) наполнитель (а-е) добавляют к первой суспензии волокнистого материала или ко второй суспензии волокнистого материала, содержащей полимер А.
Предпочтительно, если полное количество наполнителя (а-е) составляет от 0 до 40 мас. % в пересчете на массу полученной бумаги или картона и в пересчете на содержание сухих веществ в наполнителе (а-е), составляющее 100 мас. %, и на содержание сухих веществ в бумаге или картоне, составляющее 100 мас. %. Более предпочтительно, если полное количество наполнителя (а-е) составляет от 5 до 30 мас. %, особенно предпочтительно от 15 до 25 мас. % и наиболее предпочтительно от 15 до 20 мас. %.
Предпочтительно, если полученная бумага или картон содержит наполнитель (а-е) при полном количестве, составляющем от 5 до 30 мас. %. Такой бумагой является, например, бумага без содержания древесной массы. Предпочтительно, если полученная бумага или картон содержит наполнитель (а-е) при полном количестве, составляющем от 5 до 20 мас. %. Такую бумагу в основном используют в качестве упаковочной бумаги. Предпочтительно, если полученная бумага или картон содержит наполнитель (а-е) при полном количестве, составляющем от 5 до 15 мас. %. Такую бумагу в основном используют для печатания газет. Предпочтительно, если полученная бумага или картон содержит наполнитель (а-е) при полном количестве, составляющем от 25 до 40 мас. %. Такой бумагой является, например, СК (суперкаландрированная) бумага.
Предпочтительно, если на стадии (А) конечный полимер А добавляют к первой водной суспензии волокнистого материала до добавления наполнителя (а-е). Более предпочтительно, если конечный полимер А добавляют до добавления наполнителя (а-е) и до добавления органического полимера (a-d), за исключением случая добавления катионогенного крахмала. Особенно предпочтительно, если конечный полимер А добавляют к первой водной суспензии волокнистого материала до добавления наполнителя (а-е), до добавления органического полимера (a-d), отличающегося от катионогенного крахмала, и до добавления любой другой вспомогательной добавки для бумаги (a-f).
На стадии (А) любой добавленный наполнитель (а-е) предпочтительно добавляют ко второй суспензии волокнистого материала, содержащей конечный полимер А, которая обладает содержанием сухих веществ, составляющим от 0,1 до 1,5 мас. %. Это добавление соответствует так называемому добавлению к жидкой массе. Вторая суспензия волокнистого материала, содержащая конечный полимер А, уже имеется в наличии и обладает этим содержанием сухих веществ или ее предварительно разбавляют до обеспечения содержания сухих веществ, составляющего от 0,1 до 1,5 мас. %, исходя из содержания сухих веществ, составляющего от более 0,15 и вплоть до 6,0 мас. %.
На стадии (А) любой добавленный наполнитель (а-е) предпочтительно добавляют ко второй суспензии волокнистого материала, содержащей конечный полимер А, где первую часть полного количества наполнителя (а-е), которое необходимо добавить, добавляют к суспензии волокнистого материала, содержащей конечный полимер А, которая обладает содержанием сухих веществ, составляющим от более 0,15 до 6,0 мас. %, и вторую часть полного количества наполнителя (а-е), которое необходимо добавить, добавляют к суспензии волокнистого материала, содержащей конечный полимер А, после разбавления до обеспечения содержания сухих веществ, составляющего от 0,1 до 1,5 мас. %. Необходимо добавить первую часть и вторую часть полного количества наполнителя (а-е). Отношение массы первой части к массе второй части составляет от 5 до 0,2.
Возможным дополнительным компонентом водной суспензии волокнистого материала, содержащей конечный полимер А, является (a-f) другая вспомогательная добавка для бумаги. Другая вспомогательная добавка для бумаги (a-f) отличается от указанных выше компонентов: (а-b), конечного полимера А, использующего в качестве компонента (а-с), (a-d) и (а-е). Другой вспомогательной добавкой для бумаги (a-f) является, например, проклеивающий агент для проклеивания в массе, растворимая в воде соль, образованная с катионом трехвалентного металла, противовспениватель, не являющийся полимером агент, придающий прочность во влажном состоянии, биоцид, оптический отбеливатель или краситель для бумаги. Примерами проклеивающих агентов являются димеры алкилкетонов (ДАК), алкенилянтарные ангидриды (АЯА) и смоляной клей. Примерами растворимых в воде солей, образованных с катионом трехвалентного металла, являются соли алюминия(III), в особенности, AlCl3, такая как AlCl3-6H2O, Al2(SO4)3, такая как Al2(SO4)31⋅8H2O, или KAl(SO4)2⋅12H2O. Другие вспомогательные добавки для бумаги (a-f) предпочтительно можно добавлять в обычных количествах.
Предпочтительно, если другую вспомогательную добавки для бумаги (a-f) добавляют ко второй суспензии волокнистого материала, содержащей конечный полимер А, которая обладает содержанием сухих веществ, составляющим от 0,1 до 1,5 мас. %. Это добавление соответствует так называемому добавлению к жидкой массе. Вторая суспензия волокнистого материала, содержащая конечный полимер А, уже имеется в наличии и обладает этим содержанием сухих веществ или ее предварительно разбавляют до обеспечения содержания сухих веществ, составляющего от 0,1 до 1,5 мас. %, исходя из содержания сухих веществ, составляющего от более 0,15 и вплоть до 6,0 мас. %.
В контексте настоящего изобретения другая вспомогательная добавка для бумаги (a-f) также включает две или большее количество разных других вспомогательных добавок для бумаги. Соответственно, другую вспомогательную добавку для бумаги (a-f), использующуюся в качестве возможного дополнительного компонента второй водной суспензии волокнистого материала, содержащей конечный полимер А, разделяют на первую другую вспомогательную добавку для бумаги (а-f-l), вторую другую вспомогательную добавку для бумаги (a-f-2) и т.п.
Во время изготовления бумаги к водной суспензии волокнистого материала часто добавляют более, чем один органический полимер (a-d), и более, чем один наполнитель (а-е). В случае органического полимера (a-d) это осуществляют, например, для оказания воздействия на технические характеристики самого способа изготовления бумаги или на технические характеристики изготовленной бумаги. Таким образом, используют удерживающие средства, обезвоживающие средства, средства, придающие прочность во влажном состоянии, или другие средства, придающие прочность в сухом состоянии.
Примерами удерживающих средств являются катионогенные, амфотерные или анионогенные полимеры (a-d). Примерами являются анионогенный полиакриламид, катионогенный полиакриламид, катионогенный крахмал, катионогенный полиэтиленимин или катионогенный поливиниламин. Кроме того, в качестве удерживающих средств также можно добавить неорганические наполнители (а-е), которые действуют, как так называемые анионогенные микрочастицы. Они включают, в частности, коллоидный диоксид кремния или бентонит.Возможны комбинации приведенных выше веществ. В частности, возможной комбинацией является двухкомпонентная система, которая состоит из катионогенного полимера и анионогенных микрочастиц или анионогенного полимера и катионогенных микрочастиц. Предпочтительным удерживающим средством является синтетический органический полимер (a-d) или двухкомпонентная система. В случае использования в качестве удерживающего средства двухкомпонентной системы, например, катионогенный первый органический полимер (а-d-1) содержится в комбинации с анионогенными неорганическими микрочастицами, например, подходящим бентонитом, использующимся в качестве первого наполнителя (а-е-1).
Примерами другого средства, придающего прочность в сухом состоянии, являются синтетический органический полимер (a-d), такой как поливиниламин, полиэтиленимин, полиакриламид или глиоксилированный полиакриламид, натуральный органический полимер (a-d), такой как немодифицированный крахмал, или модифицированный натуральный органический полимер (a-d), такой как катионогенно модифицированный крахмал или подвергнутый окислительному или ферментативному разложению крахмал. Предпочтительно, если другое средство, придающее прочность в сухом состоянии, добавляют к первой водной суспензии волокнистого материала или ко второй водной суспензии пульпы, содержащей конечный полимер А, которые обе обладают содержанием сухих веществ, составляющим от более 1,5 до 6,0 мас. %. Можно провести добавление к первой водной суспензии волокнистого материала или ко второй водной суспензии волокнистого материала, содержащей конечный полимер А, каждая из которых обладает содержанием сухих веществ, составляющим от 0,1 до 1,5 мас. %.
На стадии (В) вторую водную суспензию волокнистого материала, содержащую конечный полимер А, наносят на водопроницаемую подложку. Водопроницаемая подложка содержит верхнюю и нижнюю сторону и мелкие отверстия, которые позволяют воде протекать сквозь нее, но в основном не позволяют проходить сквозь нее волокнистым компонентам. Вторую водную суспензию волокнистого материала, содержащую конечный полимер А, равномерно наносят на водопроницаемую подложку. Верхняя сторона водопроницаемой подложки в момент нанесения является в основном плоской поверхностью, т.е. за исключением мелких отверстий или других связанных с природой материала неровностей и любого определенного радиуса кривизны. Это обеспечивает возможность получения равномерно тонкого, как можно более однородного мокрого полотна волокнистого материала или мокрой бумажной структуры, или мокрого бумажного листа. После нанесения второй водной суспензии волокнистого материала, содержащей конечный полимер А, часть воды (а-а) стекает через мелкие отверстия, при этом на верхней стороне происходит образования листа, таким образом образуется мокрая бумажная структура. Полученная таким образом мокрая бумажная структура является плоской, т.е. она обладает крайне малой высотой по сравнению с ее длиной и шириной. Волокнистый материал, содержащаяся во второй суспензии волокнистого материала, содержащей конечный полимер А и возможные другие компоненты, которые должны содержаться в конечной изготовленной бумаге или картоне, например, наполнитель (а-е), в идеальном случае полностью остаются или по меньшей мере в основном остаются в мокрой бумажной структуре, которая образуется. В этом способе эффективным является использование возможных дополнительных компонентов во второй водной суспензии волокнистого материала, содержащей конечный полимер А, которые добавляют для содействия удерживанию других компонентов, для содействия обезвоживанию или для содействия образованию однородного листа, например, органического полимера (a-d). В большинстве случаев эти возможные дополнительные компоненты, содержащиеся в суспензии волокнистого материала, также полностью остаются или по меньшей мере в основном остаются в полученном полотне волокнистого материала. Часть мокрой бумажной структуры, которая определяет содержание сухих веществ в мокрой бумажной структуре, содержит удержанные компоненты волокнистого материала, возможные другие компоненты, которые должны содержаться в конечной изготовленной бумаге, и возможные дополнительные компоненты. В зависимости от характеристик удерживания этих компонентов ими могут являться, например, указанный волокнистый материал, органические полимеры, наполнители и другие вспомогательные добавки для бумаги. После завершения стадии (В) мокрая бумажная структура является достаточно прочной, чтобы ее можно было удалить с водопроницаемой подложки.
Предпочтительно, если использующейся на стадии (В) водопроницаемой подложкой является сетка. Сетка, которая содержит нижнюю сторону сетки и верхнюю сторону сетки, представляет собой сетку с мелкими отверстиями. Сетка включает, например, металлическую или пластмассовую сетку. В случае бумагоделательной машины более предпочтительно, если сеткой является бесконечная сетка. После отделения полученной мокрой бумажной структуры от бесконечной сетки бесконечная сетка движется обратно для нанесения материала, где новую вторую суспензию волокнистого материала, содержащую конечный полимер А, наносят на движущуюся бесконечную сетку. Более предпочтительно, если сеткой является бесконечная сетка, движущаяся вокруг нескольких цилиндров.
Содержание сухих веществ в мокрой бумажной структуре, которую получают на стадии (В), предпочтительно составляет от 15 до 25 мас. %, более предпочтительно от 18,7 до 24 мас. %, особенно предпочтительно от 18,8 до 23 мас. %, еще более предпочтительно от 18,9 до 22 мас. %, еще более предпочтительно от 19,0 до 21 мас. % и наиболее предпочтительно от 19,0 до 20,7 мас. %.
Предпочтительным является способ, в котором на стадии (В) мокрая бумажная структура обладает содержанием сухих веществ, составляющим от 18,5 до 25 мас. %.
Предпочтительным является способ, в котором на стадии (А) конечный полимер А добавляют к первой водной суспензии волокнистого материала, которая в момент добавления обладает содержанием сухих веществ, составляющим от более 1,5 и вплоть до 6 мас. %, и в котором на стадии (В) мокрая бумажная структура обладает содержанием сухих веществ, составляющим от 18,5 до 25 мас. %.
На стадии (С) мокрую бумажную структуру, полученную на стадии (В), обезвоживают с получением бумаги или картона. Проводимое на стадии (С) обезвоживание включает следующие стадии
(С-1) обезвоживание мокрой бумажной структуры путем прессования с получением таким образом влажного бумажного листа, (С-2) обезвоживание влажного бумажного листа путем подачи тепла с получением таким образом бумаги или картона.
Проводимое на стадии (С-1) прессование мокрой бумажной структуры приводит к дополнительному обезвоживанию и соответствующему увеличению содержания сухих веществ. В случае обезвоживания прессованием к мокрой бумажной структуре прикладывают механическое давление. Удаление воды путем приложения механического давления является более энергоэффективным, чем сушка путем подачи тепла. При помещении мокрой бумажной структуры на адсорбирующий воду лист или ленту, например, войлокообразное полотно, обезвоживанию способствует адсорбция отпрессованной воды. Для оказания давления на слоистый композит подходящим является цилиндр. В частности, подходящим является пропускание слоистого композита через два цилиндра, по возможности с остановкой на адсорбирующей воду ленте. Поверхность цилиндра изготовлена, например, из стали, гранита или твердой резины. На поверхность цилиндра может быть нанесено покрытие из адсорбирующего воду материала. Адсорбирующие воду материалы обладают высокой впитывающей способностью, пористостью, прочностью и эластичностью. После завершения стадии (С-1) получают влажный бумажный лист.После завершения стадии (С-1) влажный бумажный лист является достаточно прочным, чтобы его можно было направить на следующую стадию (С-2) без использования механической опоры. Влажный бумажный лист предпочтительно обладает содержанием сухих веществ, составляющим от 35 до 65 мас. %, более предпочтительно от 37 до 60 мас. %, особенно предпочтительно от 38 до 55 мас. %, наиболее предпочтительно от 40 до 50 мас. %.
На стадии (С-2) проводят дополнительное обезвоживание влажного бумажного листа, полученного на стадии (С-1), таким образом получают бумагу или картон. Подачу тепла к влажному бумажному листу обеспечивают, например, с помощью нагреваемых пластин, на которые помещают влажный бумажный лист, с помощью нагреваемых цилиндров, над которыми пропускают влажный бумажный лист, с помощью источников ИК (инфракрасное излучение), с помощью теплого воздуха, пропускаемого над влажным бумажным листом или с использованием комбинации двух, трех или всех этих методик.
Полученная бумага или картон обладает наиболее высокой прочностью в сравнении с прочностью мокрой бумажной структуры или мокрого бумажного листа. Предполагают, что при содержании сухих веществ, составляющем 80 мас. %, гидроксигруппы, содержащиеся в целлюлозных волокнах, все в большей степени связаны водородными связями, это обеспечивает предварительное механическое свойлачивание волокон. Одним показателем прочности полученной бумаги или картона является, например, внутренняя прочность.
Предпочтительно, если содержание сухих веществ в полученной бумаге или картоне составляет не менее 88 мас. %. Более предпочтительно, если содержание сухих веществ в полученной бумаге или картоне составляет от 89 до 100 мас. %, особенно предпочтительно от 90 до 98 мас. % и наиболее предпочтительно от 91 до 96 мас. %.
В зависимости от основной массы в пересчете на единицу площади, которая также называется поверхностной плотностью или плотностью, меняется название плоского формованного изделия, полученного из второй суспензии волокнистого материала, содержащей конечный полимер А. В настоящем изобретении высушенное формованное изделие, обладающее массой в пересчете на единицу площади, равной от 7 до 225 г/м2, называется бумагой и обладающее массой в пересчете на единицу площади, равной от 225 г/м2, называется картоном. Плотность бумаги или картона предпочтительно равна от 20 до 400 г/м2, более предпочтительно от 40 до 280 г/м2, особенно предпочтительно от 60 до 200 г/м2, еще более предпочтительно от 80 до 160 г/м2, еще более предпочтительно от 90 до 140 г/м2 и наиболее предпочтительно от 100 до 130 г/м2.
Предпочтительно, если полученной бумагой или картоном является упаковочная бумага, более предпочтительно гофрированная бумага.
Предпочтения, указанные для способа изготовления бумаги или картона, также применимы к другим объектам настоящего изобретения.
Другим объектом настоящего изобретения является бумага или картон, получаемые способом, включающим стадии
(А) добавления конечного полимера А к первой водной суспензии волокнистого материала с получением таким образом второй водной суспензии волокнистого материала, содержащей конечный полимер А,
где конечный полимер А представляет собой получаемый путем
- радикальной полимеризации следующих мономеров:
(i) от 30 до 90 мол. % мономера формулы I

в которой R1=Н или обозначает C1-C6-алкил,
(ii) от 3 до 60 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 45 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 35 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы,
где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм,
(B) обезвоживания второй водной суспензии волокнистого материала, содержащей конечный полимер А, на водопроницаемой подложке с получением мокрой бумажной структуры,
(C) обезвоживания мокрой бумажной структуры с получением таким образом бумаги или картона.
Бумага или картон предпочтительно обладает внутренней прочностью, равной от 165 до 400 Дж/м2, более предпочтительно от 190 до 350 Дж/м2, особенно предпочтительно от 200 до 300 Дж/м2 и наиболее предпочтительно от 220 до 280 Дж/м2, где внутренняя прочность соответствует определенной в соответствии со стандартной методикой TAPPI Т833 pm-94.
Другим объектом настоящего изобретения является конечный полимер А, получаемый путем
- радикальной полимеризации следующих мономеров:
(i) от 58 до 83 мол. % мономера формулы I

в которой R1=Н или обозначает C1-С6-алкил,
(ii) от 8 до 39 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы, где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм.
Предпочтительным является конечный полимер А, где для радикальной полимеризации с получением исходного полимера используют следующие мономеры:
(i) от 60 до 83 мол. % N-винилформамида,
(ii) от 8 до 21 мол. % этилакрилата,
(iii) от 2 до 21 мол. % акриловой кислоты или метакриловой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 24 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
Другим объектом настоящего изобретения является исходный полимер V, получаемый путем радикальной полимеризации следующих мономеров:
(i) от 58 до 83 мол. % мономера формулы I
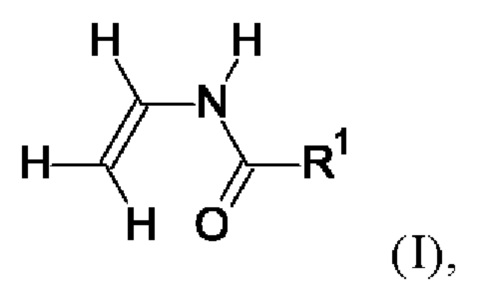
в которой R1=Н или обозначает C1-C6-алкил,
(ii) от 8 до 39 мол. % С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол. % моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол. %.
Предпочтительным является исходный полимер V, для получения которого путем радикальной полимеризации используют следующие мономеры:
(i) от 60 до 83 мол. % N-винилформамида,
(ii) от 8 до 21 мол. % этилакрилата,
(iii) от 2 до 21 мол. % акриловой кислоты или метакриловой кислоты, или их солевых форм,
(iv) от 0 до 9 мол. % акрилонитрила или метакрилонитрила,
(v) от 0 до 24 мол. % одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
На фиг. 1 с помощью кривой А схематически представлена зависимость вязкости (в мПа⋅с) от времени (ч) при проведении гидролиза первого исходного полимера, полученного из 70 мол. % N-винилформамида и 30 мол. % метилакрилата, в щелочной среде. С помощью кривой В схематически представлена зависимость вязкости (в мПа⋅с) от времени (ч) при проведении гидролиза второго исходного полимера, полученного из 70 мол. % N-винилформамида, 20 мол. % метилакрилата и 10 мол. % акрилата натрия, в щелочной среде.
Примеры
Приведенные в примерах выраженные в процентах значения являются массовыми, если не указано иное.
А) Добавки
А-1) Методики характеризации полимеров
Содержание твердых веществ в растворе полимера определяли путем помещения от 0,5 до 1,5 г раствора полимера в металлическую крышку диаметром 4 см и последующей сушки в сушильном шкафу с циркуляцией воздуха при 140°С в течение двух часов (=2 ч). Отношение массы образца после сушки при указанных выше условиях к массе исходного отвешенного образца, умноженное на 100, дает содержание твердых веществ в полимере, выраженное в мас. %.
Степень гидролиза N-винилформамидных звеньев (=СГ) означает выраженное в мол. % отношение количества гидролизованных N-винилформамидных звеньев к количеству N-винилформамидных звеньев, изначально содержащихся в полимере. Степень гидролиза определяли путем ферментативного анализа муравьиной кислоты или формиата, выделяющихся во время гидролиза (набор для исследования, выпускающийся фирмой Boehringer, Mannheim).
Степень превращения (мет)акрилатных звеньев (=СП) означает выраженное в мол. % отношение подвергнутых превращению (мет)акрилатных звеньев к количеству (мет)акрилатных звеньев, изначально содержащихся в полимере. В настоящем изобретении термин "превращение" означает отщепление сложноэфирного звена, например, вследствие гидролиза, с образованием (мет)акрилатного звена или его соответствующей солевой формы, протекающее по реакции с соседней аминогруппой с образованием таким образом лактама. Степень превращения можно определить путем определения количества спирта, выделившегося в ходе превращения. Это можно определить, например, с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) или газовой хроматографии, в зависимости от выделившегося спирта.
Содержание полимера означает выраженное в массовых процентах содержание полимера в растворе без противоионов, т.е. противоионы не учитывают.Содержание полимера означает сумму выраженных в массовых процентах количеств всех структурных звеньев полимера в граммах, которые содержатся в 100 г водного раствора. Его определяли путем расчета. При этом включали возможно обладающие зарядом структурные звенья в заряженной форме, т.е., например, аминогруппы в протонированной форме и кислотные группы в депротонированной форме. Противоионы заряженных структурных звеньев, такие как катион натрия, хлорид-ион, фосфат-ион, формиат-ион, ацетат-ион и т.п., не учитывали. Расчет можно провести таким образом, что для смеси, исходя из количеств использующихся мономеров и в соответствующих случаях принимая во внимание степень гидролиза (СГ) и степень превращения (СП), определяли молярные количества структурных звеньев полимера, содержащихся после завершения реакции, и их пересчитывали в массовые доли с использованием молярных масс структурных звеньев. Сумма массовых долей давала полное количество полимера в этой смеси. Содержанием полимера являлось отношение полного количества полимера к полной массе смеси.
Значения K определяли в соответствии с публикацией Н. Fikentscher, Cellulosechemie ("Cellulose Chemistry"), volume 13, 48-64 and 71-74, при указанных в ней условиях. Значения в скобках означают концентрацию раствора, рассчитанную с учетом содержания полимера и растворителя.
Плотности зарядов определяли путем титрования полиэлектролита поливинилсульфонатом калия при значении рН, равном 3,5 (см. публикацию D. Horn, Progress in Colloid & Polymer Science, 65 (1978), pages 251-264).
Если не указано иное, для получения полимеров использовали только полностью обессоленную воду.
Аббревиатуры для мономеров:
ЭА: этилакрилат
МА: метилакрилат
ВФА: N-винилформамид
Акрилат Na: натриевая соль акриловой кислоты
Метакрилат Na: натриевая соль метакриловой кислоты
АМПС-Na: натриевая соль 2-акриламидо-2-метилпропансульфоновой кислоты
Винилсульфонат Na: натриевая соль винилсульфоновой кислоты
ДАДМАХ: диаллилдиметиламмонийхлорид
АПТАХ: (3-акриламидопропил)триметиламмонийхлорид
AM: акриламид
Для выяснения того, возникает ли пик вязкости во время проведения гидролиза, наблюдали воронку, образующуюся при вращении лопастной мешалки (стеклянная мешалка с изготовленной из тефлона лопастью округлой формы диаметром 7,0 см и высотой 2,5 см), и ее оценивали следующим образом:

Состав конечных полимеров формулы III в соответствии с расчетом:

где а, b с, d и е обозначают выраженные в процентах мольные доли (=мол. %) структурных звеньев и сумма а, b, с, d и е равна 100 мол. %.
(1)
а = амидиний / (амидиний + ВФА + виниламмоний + акрилат-анион + лактам) × 100
b = ВФА / (амидиний + ВФА + виниламмоний + акрилат-анион + лактам) ×100
с = виниламмоний / (амидиний + ВФА + виниламмоний + акрилат-анион + лактам) × 100
d = акрилат-анион / (амидиний + ВФА + виниламмоний + акрилат-анион + лактам) × 100
е = лактам / (амидиний + ВФА + виниламмоний + акрилат-анион + лактам) × 100 (2)
ВФА [ммоль/100 г]: концентрация структурных звеньев ВФА, содержащихся в конечном продукте
акрилат-анион [ммоль/100 г]: концентрация структурных звеньев акрилат-анионов, содержащихся в конечном продукте
виниламмоний [ммоль/100 г]: концентрация виниламмониевых структурных звеньев, содержащихся в конечном продукте
амидиний [ммоль/100 г]: концентрация амидиниевых структурных звеньев, содержащихся в конечном продукте
лактам [ммоль/100 г]: концентрация лактамных структурных звеньев, содержащихся в конечном продукте
В настоящем изобретении конечный продукт означает раствор полимера, который получен при проведении гидролиза.
(3)
При степени превращения, СП, равной 100 мол. %, обеспечены следующие результаты:
амидиний = (ВФА0 - ФА) × ПАД/(ПФА + ПАД)
ВФА = (ВФА0 - ФА) × ПФА/(ПФА + ПАД)
виниламмоний = ФА - лактам - амидиний
акрилат-анион = AK-Na0 + МА0 + ЭА0 - ФА + ПЗ
лактам=ФА - ПЗ
(4)
ФА [ммоль/100 г]: содержание формиата в конечном продукте
ПЗ [ммоль/100 г]: плотность заряда в конечном продукте (альтернативно:
[мэкв./100 г])
ПФА: интегрируемая площадь полученного с помощью 13С-ЯМР пика для атома углерода, содержащегося в карбонильной группе структурного звена ВФА, содержащегося в полимере, в диапазоне от 164 до 168 част./млн ПАД: интегрируемая площадь полученного с помощью 13С-ЯМР пика для иминного атома углерода амидиниевого структурного звена, содержащегося в полимере при 152 част./млн
ВФА0 [ммоль/100 г]: концентрация звеньев ВФА, которые содержались бы в конечно продукте, если бы не происходила дополнительная реакция полимеризованных мономеров; рассчитана в начале полимеризации
АК-Na0 [ммоль/100 г]: концентрация звеньев акрилата Na, которые содержались бы в конечно продукте, если бы не происходила дополнительная реакция полимеризованных мономеров; рассчитана в начале полимеризации
МА0, ЭА0 [ммоль/100 г]: концентрация метил- или этилакрилатных звеньев, которые содержались бы в конечно продукте, если бы не происходила дополнительная реакция полимеризованных мономеров; рассчитана в начале полимеризации
А-2) Получение исходных полимеров по реакции полимеризации
Исходный полимер VE1: Сополимер (ВФА/МА=70 мол. %/30 мол. %)
В качестве загрузки 1 использовали 150,4 г ВФА (99%).
В качестве загрузки 2 использовали 77,3 г МА.
В качестве загрузки 3 использовали 1,13 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при комнатной температуре (=КТ) в 112,1 г воды.
В качестве загрузки 4 использовали 0,67 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 67,2 г воды.
В качестве загрузки 5 использовали 187,3 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 782,6 г воды и 2,8 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин (=оборотов в минуту), добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 187,3 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали светло-желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 18,8%. Значение K сополимера было равно 84 (0,5 мас. % в воде).
Исходный полимер VE2: Сополимер (ВФА/МА=70 мол. %/30 мол. %)
В качестве загрузки 1 использовали 150,4 г ВФА (99%).
В качестве загрузки 2 использовали 77,3 г МА.
В качестве загрузки 3 использовали 1,13 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 112,1 г воды.
В качестве загрузки 4 использовали 0,67 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 67,2 г воды.
В качестве загрузки 5 использовали 176,6 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 782,6 г воды и 2,5 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 176,6 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,1%. Значение K сополимера было равно 84 (0,5 мас. % в воде).
Исходный полимер VE3: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/29 мол. %/1 мол. %)
В качестве загрузки 1 использовали смесь 9,3 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,4, 158,2 г ВФА (99%) и 210,0 г воды.
В качестве загрузки 2 использовали 78,6 г МА.
В качестве загрузки 3 использовали 1,19 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 117,5 г воды при КТ.
В качестве загрузки 4 использовали 0,71 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 70,5 г воды при КТ.
В качестве загрузки 5 использовали 172,7 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 547,4 г воды и 2,5 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 172,7 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,6%. Значение K тройного сополимера было равно 90 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE4: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/28 мол. %/2 мол. %)
В качестве загрузки 1 использовали смесь 18,5 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,4, 158,0 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 75,8 г МА.
В качестве загрузки 3 использовали 1,18 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 117,1 г воды при КТ.
В качестве загрузки 4 использовали 0,71 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 70,3 г воды при КТ.
В качестве загрузки 5 использовали 184,0 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 551,7 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 184,0 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,4%. Значение K тройного сополимера было равно 90 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE5: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 46,1 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 157,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 67,4 г МА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116,1 г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,7 г воды при КТ.
В качестве загрузки 5 использовали 196,6 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 534,7 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,2 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 196,6 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,4%. Значение K тройного сополимера было равно 93 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE6: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 43,0 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 147,0 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 62,9 г МА.
В качестве загрузки 3 использовали 0,33 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 32,5 г воды при КТ.
В качестве загрузки 4 использовали 1,42 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 140,9 г воды при КТ.
В качестве загрузки 5 использовали 164,8 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 565,7 г воды и 2,4 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 60°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 60°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 60°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 60°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 280 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 164,8 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 13,9%. Значение K тройного сополимера было равно 138 (0,1 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE7: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 91,6 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 156,7 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 53,9 г МА.
В качестве загрузки 3 использовали 1,15 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 114,3 г воды при КТ.
В качестве загрузки 4 использовали 0,69 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 68,6 г воды при КТ.
В качестве загрузки 5 использовали 184,4 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 506,5 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,2 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 184,4 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,7%. Значение K тройного сополимера было равно 94 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE8: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/15 мол. %/15 мол. %)
В качестве загрузки 1 использовали смесь 136,7 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 155,9 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 40,0 г МА.
В качестве загрузки 3 использовали 1,14 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 112,6 г воды при КТ.
В качестве загрузки 4 использовали 0,68 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 67,5 г воды при КТ.
В качестве загрузки 5 использовали 227,5 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 478,7 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,2 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 227,5 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,9%. Значение K тройного сополимера было равно 99 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE9: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/10 мол. %/20 мол. %)
В качестве загрузки 1 использовали смесь 181,4 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 155,0 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 26,6 г МА.
В качестве загрузки 3 использовали 1,12 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 110,8 г воды при КТ.
В качестве загрузки 4 использовали 0,67 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 66,5 г воды при КТ.
В качестве загрузки 5 использовали 200,5 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 451,1 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 200,5 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 20,2%. Значение K тройного сополимера было равно 102 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE10: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 55,9 г 30 мас. % раствора метакрилата Na, значение рН которого обеспечивали равным 6,5, 156,1 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 66,8 г МА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116Д г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,7 г воды при КТ.
В качестве загрузки 5 использовали 185,7 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 526,7 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 68°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 68°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 68°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 68°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 185,7 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,2%. Значение K тройного сополимера было равно 94 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE11: Тройной сополимер (ВФА/МА/АМПС-Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 66 г 50 мас. % водного раствора АМПС-Na, значение рН которого обеспечивали равным 6,5, 144,6 г ВФА (99%) и 210,0 г воды.
В качестве загрузки 2 использовали 61,9 г МА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116,1 г воды при КТ.
В качестве загрузки 4 использовали 0,71 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,8 г воды при КТ.
В качестве загрузки 5 использовали 186,7 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 532,8 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 186,7 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 20,0%. Значение K тройного сополимера было равно 89 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE12: Тройной сополимер (ВФА/MA/Na винилсульфонат=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 79,6 г 25 мас. % водного раствора винилсульфоната Na, значение рН которого обеспечивали равным 6,5, 153,9 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 65,9 г МА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116,2 г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,7 г воды при КТ.
В качестве загрузки 5 использовали 164,5 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 506,1 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 65°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 65°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 65°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 65°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 164,5 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 20,7%. Значение K тройного сополимера было равно 87 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE13: Тройной сополимер (ВФА/МА/ДАДМАХ=65 мол. %/30 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 138,7 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 76,8 г МА.
В качестве загрузки 3 использовали 1,16 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 115,2 г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,2 г воды при КТ.
В качестве загрузки 5 использовали 174,4 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 554,6 г воды, и 37,0 г 65 мас. % водного раствора ДАДМАХ и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,3 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 67°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и при дальнейшем проведении полимеризации азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 67°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 67°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 67°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 330 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 174,4 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,8%. Значение K тройного сополимера было равно 82 (0,5 мас. % в воде).
Исходный полимер VE14: Тройной сополимер (ВФА/МА/АПТАХ=65 мол. %/30 мол. %/5 мол. %)
В качестве загрузки 1 использовали 134,9 г ВФА (99%).
В качестве загрузки 2 использовали 74,7 г МА.
В качестве загрузки 3 использовали смесь 39,8 г 75 мас. % водного раствора АПТАХ и 200 г воды.
В качестве загрузки 4 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 115,3 г воды при КТ.
В качестве загрузки 5 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,2 г воды при КТ.
В качестве загрузки 6 использовали 170,9 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 557,5 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,3 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 4 загрузок 1-4. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 4 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 330 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 170,9 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,6%. Значение K тройного сополимера было равно 87 (0,5 мас. % в воде).
Исходный полимер VE15: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/15 мол. %/15 мол. %)
В качестве загрузки 1 использовали смесь 133,1 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 151,7 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 45,3 г ЭА.
В качестве загрузки 3 использовали 1,14 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 112,7 г воды при КТ.
В качестве загрузки 4 использовали 0,68 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 67,6 г воды при КТ.
В качестве загрузки 5 использовали 537,8 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 481,0 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 72°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 72°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 72°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 72°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 340 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 137,8 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 15,1%. Значение K тройного сополимера было равно (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE16: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 55,3 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 94,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 37,6 г ЭА.
В качестве загрузки 3 использовали 0,72 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 71,6 г воды при КТ.
В качестве загрузки 4 использовали 0,43 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 43,0 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 612,8 г воды и 1,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 2,4 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 65°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 65°С, сначала в течение 3 мин добавляли 10% загрузки 1 и недолго перемешивали. Затем начинали одновременное добавление оставшейся части загрузки 1 (90%) и загрузок 2 и 3. Оставшуюся часть загрузки 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 65°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и температуру реакционной смеси повышали до 70°С.Смесь выдерживали при 70°С в течение 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 340 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 114,1 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 15,2%. Значение K тройного сополимера было равно 99 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE17: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 55,3 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 94,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 37,6 г ЭА.
В качестве загрузки 3 использовали 0,72 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 71,6 г воды при КТ.
В качестве загрузки 4 использовали 0,43 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 43,0 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 612,8 г воды и 1,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 2,4 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 64°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 64°С, сначала в течение 3 мин добавляли 10% загрузки 1 и недолго перемешивали. Затем начинали одновременное добавление оставшейся части загрузки 1 (90%) и загрузок 2 и 3. Оставшуюся часть загрузки 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 64°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и температуру реакционной смеси повышали до 70°С.Смесь выдерживали при 70°С в течение 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 340 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 138,7 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 15,6%. Значение K тройного сополимера было равно 103 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE18: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 55,3 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 94,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 37,6 г ЭА.
В качестве загрузки 3 использовали 0,72 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 71,6 г воды при КТ.
В качестве загрузки 4 использовали 0,43 г 2,2'-азобис(2-метилпропионамидин)дигидрохлор ида, который растворяли в 43,0 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 612,8 г воды и 1,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 2,6 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 65°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 65°С, сначала в течение 3 мин добавляли 10% загрузки 1 и недолго перемешивали. Затем начинали одновременное добавление оставшейся части загрузки 1 (90%) и загрузок 2 и 3. Оставшуюся часть загрузки 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 65°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и температуру реакционной смеси повышали до 70°С.Смесь выдерживали при 70°С в течение 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 340 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 126,7 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 15,4%. Значение K тройного сополимера было равно 101 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE19: Сополимер (ВФА/МА=70 мол. %/30 мол. %)
В качестве загрузки 1 использовали 150,4 г ВФА (99%).
В качестве загрузки 2 использовали 77,3 г МА.
В качестве загрузки 3 использовали 1,13 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 112,1 г воды.
В качестве загрузки 4 использовали 0,67 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 67,2 г воды.
В качестве загрузки 5 использовали 168,4 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 784,9 г воды и 2,8 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 70°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и при дальнейшем проведении полимеризации азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 70°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 70°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 70°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 168,4 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 18,6%. Значение K сополимера было равно 82 (0,5 мас. % в воде).
Исходный полимер VE20: Сополимер (ВФА/МА=60 мол. %/40 мол. %)
В качестве загрузки 1 использовали 126,4 г ВФА (99%).
В качестве загрузки 2 использовали 101,0 г МА.
В качестве загрузки 3 использовали 1,13 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 112,0 г воды при КТ.
В качестве загрузки 4 использовали 0,68 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 67,2 г воды.
В качестве загрузки 5 использовали 188,5 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 785,2 г воды и 2,5 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 67°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 67°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 67°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 67°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 188,5 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 18,7%. Значение K сополимера было равно 84 (0,5 мас. % в воде).
Исходный полимер VE21: Сополимер (ВФА/МА=80 мол. %/20 мол. %)
В качестве загрузки 1 использовали 175,4 г ВФА (99%).
В качестве загрузки 2 использовали 52,6 г МА.
В качестве загрузки 3 использовали 1,13 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 112,0 г воды при КТ.
В качестве загрузки 4 использовали 0,68 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли при КТ в 67,2 г воды.
В качестве загрузки 5 использовали 163,6 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 784,7 г воды и 2,5 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 3,9 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и при дальнейшем проведении полимеризации азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 310 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 163,6 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,0%. Значение K сополимера было равно 84 (0,5 мас. % в воде).
Исходный полимер VE22: Тройной сополимер (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 46,1 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 157,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 67,4 г МА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116,1 г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,7 г воды при КТ.
В качестве загрузки 5 использовали 552,6 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 534,7 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,2 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 74°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 74°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 74°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 74°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 152,6 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый немного вязкий раствор, обладающий содержанием твердых веществ, составляющим 14,5%. Значение K тройного сополимера было равно 81 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE23: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 44,0 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 150,6 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 186,9 г ЭА.
В качестве загрузки 3 использовали 1,17 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 116,1 г воды при КТ.
В качестве загрузки 4 использовали 0,70 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 69,7 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 536,0 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 67°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 67°С, сначала в течение 3 мин добавляли 10% загрузки 1 и недолго перемешивали. Затем начинали одновременное добавление оставшейся части загрузки 1 (90%) и загрузок 2 и 3. Оставшуюся часть загрузки 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 67°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4. Реакционную смесь выдерживали при 67°С в течение 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 320 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 186,9 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 19,9%. Значение K тройного сополимера было равно 90 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE24: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 88,4 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 151,1 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 60,2 г ЭА.
В качестве загрузки 3 использовали 1,16 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 114,4 г воды при КТ.
В качестве загрузки 4 использовали 0,69 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 68,6 г воды при КТ.
В качестве загрузки 5 использовали 158,4 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 508,6 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 67°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 67°С, начинали одновременное добавление загрузок 1, 2 и 3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 67°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 67°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 158,4 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 20,1%. Значение K тройного сополимера было равно 99 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE25: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/10 мол. %/20 мол. %)
В качестве загрузки 1 использовали смесь 178,2 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 152,3 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 30,3 г ЭА.
В качестве загрузки 3 использовали 1,12 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 111,0 г воды при КТ.
В качестве загрузки 4 использовали 0,67 г 2,2''-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 66,5 г воды при КТ.
В качестве загрузки 5 использовали 185,7 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 453,2 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 68°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 68°С, начинали одновременное добавление загрузок 1, 2 и 3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 68°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 68°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 310 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 185,74 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 20,3%. Значение K тройного сополимера было равно 101 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE26: Тройной сополимер (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В качестве загрузки 1 использовали смесь 55,3 г 32 мас. % водного раствора акрилата Na, значение рН которого обеспечивали равным 6,5, 94,5 г ВФА (99%) и 200,0 г воды.
В качестве загрузки 2 использовали 37,6 г ЭА.
В качестве загрузки 3 использовали 0,72 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 71,6 г воды при КТ.
В качестве загрузки 4 использовали 0,43 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 43,0 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 612,8 г воды и 1,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 2,4 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 65°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 65°С, сначала в течение 3 мин добавляли 10% загрузки 1 и недолго перемешивали. Затем начинали одновременное добавление оставшейся части загрузки 1 (90%) и загрузок 2 и 3. Оставшуюся часть загрузки 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 65°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и температуру реакционной смеси повышали до 70°С. Смесь выдерживали при 70°С в течение 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 300 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 120,5 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали немного мутный желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 15,1%. Значение K тройного сополимера было равно 102 (0,5 мас. % в 5 мас. % водном растворе NaCl).
Исходный полимер VE27: Тройной сополимер (ВФА/МА/АМ=70 мол. %/25 мол. %/5 мол. %)
В качестве загрузки 1 использовали смесь 22,6 г 50 мас. % водного раствора AM, 159,9 г ВФА (99%) и 210,0 г воды.
В качестве загрузки 2 использовали 68,5 г МА.
В качестве загрузки 3 использовали 1,19 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 117,9 г воды при КТ.
В качестве загрузки 4 использовали 0,71 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 70,7 г воды при КТ.
В качестве загрузки 5 использовали 189,6 г воды.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 541,8 г воды и 2,6 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 4,1 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 69°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 69°С, начинали одновременное добавление 3 загрузок 1-3. Загрузку 1 добавляли в течение 3 ч, загрузку 2 добавляли в течение 3,5 ч и загрузку 3 добавляли в течение 4 ч. После завершения добавления загрузки 3 смесь выдерживали при 69°С в течение еще 1 ч. Затем в течение 5 мин добавляли загрузку 4 и реакционную смесь выдерживали при 69°С в течение еще 1,5 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 310 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 189,6 г воды. Затем вакуумирование прекращали путем подачи воздуха, добавляли загрузку 5 и реакционную смесь охлаждали до КТ.
Получали желтый вязкий раствор, обладающий содержанием твердых веществ, составляющим 21,9%. Значение K тройного сополимера было равно 89 (0,5 мас. % в воде).
Исходный полимер VV1: Сополимер (ВФА/акрилат Na=70 мол. %/30 мол. %)
В качестве загрузки 1 использовали смесь 316,7 г 32 мас. % водного раствора акрилата Na, 180,5 г ВФА (99%) и 141,0 г воды.
В качестве загрузки 2 использовали 1,79 г 2,2'-азобис(2-метилпропионамидин)дигидрохлорида, который растворяли в 176,9 г воды при КТ.
В стеклянный аппарат объемом 2 л, снабженный якорной мешалкой, обратным холодильником, термометром для измерения внутренней температуры и патрубком для подачи азота, помещали 573,4 г воды и 3,0 г 75 мас. % раствора фосфорной кислоты. Реактор помещали в водяную баню, снабженную модулем нагревания-охлаждения, с помощью которого автоматически регулировали внутреннюю температуру. При скорости перемешивания, равной 100 об/мин, добавляли примерно 5,2 г 25 мас. % раствора гидроксида натрия, таким образом обеспечивали значение рН, равное 6,5. Затем приемник нагревали в течение 30 мин до 80°С и в это же время в аппарат подавали азот (20 л/ч) с целью замены кислорода. Затем подачу азота прекращали и азот продолжали подавать только через обратный холодильник, чтобы предотвратить проникновение кислорода. При постоянной внутренней температуре, равной 80°С, начинали одновременное добавление загрузок 1 и 2. Загрузку 1 добавляли в течение 1,5 ч и загрузку 2 добавляли в течение 2,5 ч. После завершения добавления загрузки 2 смесь выдерживали при 80°С в течение еще 1 ч. Затем обратный холодильник заменяли на нисходящий холодильник и внутреннее давление медленно уменьшали до равного примерно 460 мбар с помощью водоструйного насоса, при этом содержимое реактора начинало кипеть. При этих условиях отгоняли 178,7 г воды. Затем вакуумирование прекращали путем подачи воздуха и реакционную смесь охлаждали до КТ.
Получали желтоватый вязкий раствор, обладающий содержанием твердых веществ, составляющим 24,1%. Значение K сополимера было равно 88 (0,5 мас. % в 5% водном растворе NaCl).
А-3) Получение конечных полимеров путем гидролиза исходных полимеров
Конечный полимер АЕ1: Гидролизованный в кислой среде исходный полимер VE1 (ВФА/МА=70 мол. %/30 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 150,1 г раствора полимера, полученного с использованием исходного полимера VE1, смешивали с 1,3 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 30,0 г 37 мас. % раствора хлористоводородной кислоты (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 64,8 г 25 мас. % раствора гидроксида натрия.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 8,3%. Степень гидролиза, СГ, составляла 98 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ2: Гидролизованный в щелочной среде исходный полимер VE2 (ВФА/МА=70 мол. %/30 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 170,5 г раствора полимера, полученного с использованием исходного полимера VE2, смешивали 1,5 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 56,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 20,1 г 37 мас. % раствора хлористоводородной кислоты и 1,3 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,9%. Степень гидролиза, СГ, составляла 96 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ3: Гидролизованный в щелочной среде исходный полимер VE3 (ВФА/МА/акрилат Na=70 мол. %/29 мол. %/1 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 173,4 г раствора полимера, полученного с использованием исходного полимера VE3, смешивали 1,6 г 40 мас. % водного раствора бисульфита натрия и 55,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 59,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 21,7 г 37 мас. % раствора хлористоводородной кислоты и 9,4 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,7%. Степень гидролиза, СГ, составляла 99 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ4: Гидролизованный в щелочной среде тройной сополимер VE4 (ВФА/МА/акрилат Na=70 мол. %/28 мол. %/2 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 174,1 г раствора полимера, полученного с использованием исходного полимера VE4, смешивали 1,6 г 40 мас. % водного раствора бисульфита натрия и 54,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 58,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 22,5 г 37 мас. % раствора хлористоводородной кислоты и 7,0 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,7%. Степень гидролиза, СГ, составляла 98 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ5: Гидролизованный в щелочной среде тройной сополимер VE5 (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 173,6 г раствора полимера, полученного с использованием исходного полимера VE5, смешивали 1,6 г 40 мас. % водного раствора бисульфита натрия и 62,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 58,5 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 23,8 г 37 мас. % раствора хлористоводородной кислоты.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,6%. Степень гидролиза, СГ, составляла 99 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ6: Гидролизованный в щелочной среде тройной сополимер VE6 (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 149,9 г раствора полимера, полученного с использованием исходного полимера VE6, смешивали 1,0 г 40 мас. % водного раствора бисульфита натрия и 136,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 36,2 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 13,7 г 37 мас. % раствора хлористоводородной кислоты и 7,5 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 4,5%. Степень гидролиза СГ, составляла 93 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ7: Гидролизованный в щелочной среде тройной сополимер VE7 (ВФА/МА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 170,4 г раствора полимера, полученного с использованием исходного полимера VE7, смешивали 1,6 г 40 мас. % водного раствора бисульфита натрия и 57,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 58,90 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 25,1 г 37 мас. % раствора хлористоводородной кислоты и 4,5 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,5%. Степень гидролиза, СГ, составляла 99 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ8: Гидролизованный в щелочной среде тройной сополимер VE8 (ВФА/МА/акрилат Na=70 мол. %/15 мол. %/15 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 171,0 г раствора полимера, полученного с использованием исходного полимера VE8, смешивали 1,6 г 40 мас. % водного раствора бисульфита натрия и 63,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 57,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 27,5 г 37 мас. % раствора хлористоводородной кислоты.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,5%. Степень гидролиза, СГ, составляла 94 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ9: Гидролизованный в щелочной среде тройной сополимер VE9 (ВФА/МА/акрилат Na=70 мол. %/10 мол. %/20 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 177,9 г раствора полимера, полученного с использованием исходного полимера VE9, смешивали 1,7 г 40 мас. % водного раствора бисульфита натрия и 65,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 61,5 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 31,3 г 37 мас. % раствора хлористоводородной кислоты и 1,8 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,2%. Степень гидролиза, СГ, составляла 99 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ10: Гидролизованный в щелочной среде тройной сополимер VE10 (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 170,2 г раствора полимера VE10, полученного выше, смешивали с 1,5 г 40 мас. % водного раствора бисульфита натрия и 50,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 56,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 22,2 г 37 мас. % раствора хлористоводородной кислоты и 8,4 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,2%. Степень гидролиза, СГ, составляла 97 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ11: Гидролизованный в щелочной среде тройной сополимер VE11 (ВФА/МА/АМПС-Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 172,1 г раствора полимера, полученного с использованием исходного полимера VE11, смешивали с 1,5 г 40 мас. % водного раствора бисульфита натрия и 65,5 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 55,9 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 22,7 г 37 мас. % раствора хлористоводородной кислоты и 7,8 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,5%. Степень гидролиза, СГ, составляла 94 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ12: Гидролизованный в щелочной среде тройной сополимер VE12 (ВФА/MA/Na винилсульфонат=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 178,5 г раствора полимера, полученного с использованием исходного полимера VE12, смешивали с 1,7 г 40 мас. % водного раствора бисульфита натрия и 75,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 62,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 25,4 г 37 мас. % раствора хлористоводородной кислоты и 5,6 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,7%. Степень гидролиза, СГ, составляла 98 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ13: Гидролизованный в щелочной среде тройной сополимер VE13 (ВФА/МА/ДАДМАХ=65 мол. %/30 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 177,6 г раствора полимера, полученного с использованием исходного полимера VE13, смешивали с 1,5 г 40 мас. % водного раствора бисульфита натрия и 70,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревают до 80°С. Затем добавляли 53,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 1 ч. Полученный продукт больше не поддавался перемешиванию. Эксперимент прекращали.
Конечный полимер АЕ14: Гидролизованный в щелочной среде тройной сополимер VE14 (ВФА/МА/АПТАХ=65 мол. %/30 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 178,0 г раствора полимера, полученного с использованием исходного полимера VE14, смешивали с 1,4 г 40 мас. % водного раствора бисульфита натрия и 60,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 51,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 18,1 г 37 мас. % раствора хлористоводородной кислоты и 19,1 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,5%. Степень гидролиза, СГ, составляла 95 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер АЕ15: Гидролизованный в щелочной среде тройной сополимер VE15 (ВФА/ЭА/акрилат Na=70 мол. %/15 мол. %/15 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 222,5 г раствора полимера, полученного с использованием исходного полимера VE15, смешивали с 1,5 г 40 мас. % водного раствора бисульфита натрия и 10,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 56,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 25,6 г 37 мас. % раствора хлористоводородной кислоты и 1,1 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,5%
Содержание формиата, ФА: 91,4 ммоль/100 г
Степень гидролиза, СГ: 98 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 64,0 ммоль/100 г
Вязкость (20 об/мин, РВ (ротационный вискозиметр), шпиндель №3): 185 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,11
ПФА (13С-ЯМР, 164-167 част./млн): 0,82
ВФА0: 93,7 ммоль/100 г
ЭА0: 20,0 ммоль/100 г
AK-Na0: 20,0 ммоль/100 г.
Конечный полимер АЕ16: Гидролизованный в щелочной среде тройной сополимер VE16 (ВФА/МА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником,652,7 г раствора полимера, полученного с использованием исходного полимера VE16, смешивали с 4,5 г 40 мас. % водного раствора бисульфита натрия и 185,3 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 165,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 6 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 70,2 г 37 мас. % раствора хлористоводородной кислоты и 12,7 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 6,6%
Содержание формиата, ФА: 74,0 ммоль/100 г
Степень гидролиза, СГ: 94 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 51,3 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 268 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,86 ПФА
(13С-ЯМР, 164-167 част./млн): 2,78
ВФА0: 79,5 ммоль/100 г
ЭА0: 22,7 ммоль/100 г
AK-Na0: 11,4 ммоль/100 г.
Конечный полимер АЕ17: Гидролизованный в щелочной среде тройной сополимер VE17 (ВФА/МА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 249,5 г раствора полимера, полученного с использованием исходного полимера VE17, смешивали с 1,8 г 40 мас. % водного раствора бисульфита натрия и 20,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 53,9 г 25 мас. % водного раствора гидроксида натрия (100 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 6 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 20,7 г 37 мас. % раствора хлористоводородной кислоты.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 8,4%
Содержание формиата, ФА: 83,4 ммоль/100 г
Степень гидролиза, СГ: 85 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 56,7 ммоль/100 г
Вязкость (50 об/мин, РВ, шпиндель №3): 1172 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 0,90
ПФА (13С-ЯМР, 164-167 част./млн) 3,82
ВФА0: 98,3 ммоль/100 г
ЭА0: 28,1 ммоль/100 г
AK-Na0: 14,0 ммоль/100 г.
Конечный полимер АЕ18: Гидролизованный в щелочной среде тройной сополимер VE18 (ВФА/МА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 248,8 г раствора полимера, полученного с использованием исходного полимера VE18, смешивали с 1,7 г 40 мас. % водного раствора бисульфита натрия и 20,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 50°С. Затем добавляли 63,7 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 50°С в течение 24 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 27,9 г 37 мас. % раствора хлористоводородной кислоты.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 8,2%
Содержание формиата, ФА: 88,2 ммоль/100 г
Степень гидролиза, СГ: 91 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 67,7 ммоль/100 г
Вязкость (50 об/мин, РВ, шпиндель №3): 866 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 0,77
ПФА (13С-ЯМР, 164-167 част./млн): 3,14
ВФА0: 97,7 ммоль/100 г
ЭА0: 27,9 ммоль/100 г
AK-Na0: 14,0 ммоль/100 г.
Конечный полимер АЕ19: Гидролизованный в щелочной среде сополимер VE19 (ВФА/МА=70 мол. %/30 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 121,3 г раствора полимера, полученного с использованием исходного полимера VE19, смешивали с 1,1 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 39,5 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 14,5 г 37 мас. % раствора хлористоводородной кислоты.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,9%
Содержание формиата, ФА: 97,5 ммоль/100 г
Степень гидролиза, СГ: 99 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 64,3 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 794 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 10,0
ПФА (13С-ЯМР, 164-167 част./млн): <0,01
ВФА0: 98,8 ммоль/100 г
МА0: 42,3 ммоль/100 г.
Конечный полимер АЕ20: Гидролизованный в щелочной среде сополимер VE20 (ВФА/МА=60 мол. %/40 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 180,0 г раствора полимера, полученного с использованием исходного полимера VE20, смешивали с 1,3 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 51,4 г 25 мас. % водного раствора гидроксида натрия (125 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 14,2 г 37 мас. % раствора хлористоводородной кислоты и 10,4 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 8,3%
Содержание формиата, ФА: 76,5 ммоль/100 г
Степень гидролиза, СГ: 94 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 34,0 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 2320 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 5,1
ПФА (13С-ЯМР, 164-167 част./млн): 0,9
ФА0: 98,8 ммоль/100 г
МА0: 42,3 ммоль/100 г.
Конечный полимер АЕ21: Гидролизованный в щелочной среде сополимер VE21 (ВФА/МА=80 мол. %/20 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 197,6 г раствора полимера, полученного с использованием исходного полимера VE21, смешивали с 2,1 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 73,8 г 25 мас. % водного раствора гидроксида натрия (116 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 32,5 г 37 мас. % раствора хлористоводородной кислоты и 130,2 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,0%
Содержание формиата, ФА: 105,8 ммоль/100 г
Степень гидролиза, СГ: 98 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 79,5 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 755 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 10,0
ПФА (13С-ЯМР, 164-167 част./млн): 2,9
ВФА0: 108 ммоль/100 г
МА0: 42,3 ммоль/100 г.
Конечный полимер АЕ22: Гидролизованный в щелочной среде тройной сополимер VE22 (ВФА/МА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 265,8 г раствора полимера, полученного с использованием исходного полимера VE22, смешивали с 1,8 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 67,1 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 26,0 г 37 мас. % раствора хлористоводородной кислоты и 3,3 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,7%
Содержание формиата, ФА: 94,8 ммоль/100 г
Степень гидролиза, СГ: 98 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 66,0 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 325 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,90
ПФА (13С-ЯМР, 164-167 част./млн): 2,80
ВФА0: 96,7 ммоль/100 г
МА0: 34,6 ммоль/100 г
AK-Na0: 6,9 ммоль/100 г.
Конечный полимер АЕ23: Гидролизованный в щелочной среде тройной сополимер VE23 (ВФА/ЭА/акрилат Na=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 174,4 г раствора полимера, полученного с использованием исходного полимера VE23, смешивали с 1,6 г 40 мас. % водного раствора бисульфита натрия и 64,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 50°С. Затем добавляли 57,5 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 50°С в течение 24 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 22,7 г 37 мас. % раствора хлористоводородной кислоты и 6,5 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,8%
Содержание формиата, ФА: 89,0 ммоль/100 г
Степень гидролиза, СГ: 97 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 66,9 ммоль/100 г
Вязкость (50 об/мин, РВ, шпиндель №3): 715 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 2,0
ПФА (13С-ЯМР, 164-167 част./млн): 2,8
ВФА0: 92,5 ммоль/100 г
ЭА0: 33,0 ммоль/100 г
AK-Na0: 6,6 ммоль/100 г.
Конечный полимер АЕ24: Гидролизованный в щелочной среде тройной сополимер VE24 (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 173,1 г раствора полимера, полученного с использованием исходного полимера VE24, смешивали с 2,6 г 40 мас. % водного раствора бисульфита натрия и 65,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 58,1 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 6 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 24,6 г 37 мас. % раствора хлористоводородной кислоты и 6,0 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,7%
Содержание формиата, ФА: 87,9 ммоль/100 г
Степень гидролиза, СГ: 96 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 55,0 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 735 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,93
ПФА (13С-ЯМР, 164-167 част./млн): 2,65
ВФА0: 92,85 ммоль/100 г
AK-Na0: 13,3 ммоль/100 г
ЭА0: 26,5 ммоль/100 г.
Конечный полимер АЕ25: Гидролизованный в щелочной среде тройной сополимер VE25 (ВФА/ЭА/акрилат Na=70 мол. %/10 мол. %/20 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 185,3 г раствора полимера, полученного с использованием исходного полимера VE25, смешивали с 1,7 г 40 мас. % водного раствора бисульфита натрия и 65,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 63,2 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 6 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 31,2 г 37 мас. % раствора хлористоводородной кислоты и 1,3 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,3%
Содержание формиата, ФА: 92,0 ммоль/100 г
Степень гидролиза, СГ: 99 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 70,1 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 535 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 2,18
ПФА (13С-ЯМР, 164-167 част./млн): 2,20
ВФА0: 92,85 ммоль/100 г
ЭА0: 26,5 ммоль/100 г
AK-Na0: 13,3 ммоль/100 г.
Конечный полимер АЕ26: Гидролизованный в щелочной среде тройной сополимер VE26 (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 169,1 г раствора полимера, полученного с использованием исходного полимера VE26, смешивали с 1,2 г 40 мас. % водного раствора бисульфита натрия и 20,0 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 50°С. Затем добавляли 29,0 г 25 мас. % водного раствора гидроксида натрия (82 мол. % в пересчете на ВФА). Смесь выдерживали при 50°С в течение 24 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 10,7 г 37 мас. % раствора хлористоводородной кислоты и 5,3 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,9%
Содержание формиата, ФА: 63,2 ммоль/100 г
Степень гидролиза, СГ: 72 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 39,8 ммоль/100 г
Вязкость (50 об/мин, РВ, шпиндель №3): 594 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 4,1
ПФА (13С-ЯМР, 164-167 част./млн): 4,0
ВФА0: 88,4 ммоль/100 г
ЭА°: 25,3 ммоль/100 г
AK-Na0: 12,6 ммоль/100 г.
Конечный полимер АЕ27: Гидролизованный в щелочной среде тройной сополимер VE18 (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В стальном выдерживающем давление реакторе объемом 2 л, снабженном перемешивающим устройством, термометром для измерения внутренней температуры, нагревательной/охлаждающей рубашкой, монометром, клапаном сброса давления, обратным холодильником и выдерживающим давление питающим сосудом, 1006,2 г раствора полимера, полученного с использованием исходного полимера VE18, при перемешивании смешивали с 126,4 г воды и нагревали до 107°С. Обеспечивали давление, равное 2,8 бар. В питающий сосуд помещали 256,8 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). При давлении, равном 5 бар, в реактор подавали раствор гидроксида натрия и смесь перемешивали. Обеспечивали температуру, равную 100°С, и ее поддерживали в течение 60 мин. Затем реактор как можно быстрее охлаждали до КТ. Получали 306,9 г продукта и его значение рН устанавливали равным 6,0 путем добавления 26,4 г 37 мас. % раствора хлористоводородной кислоты и 3,7 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,3%
Содержание формиата, ФА: 90,1 ммоль/100 г
Степень гидролиза, СГ: 94 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 66,5 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 1030 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,79
ПФА (13С-ЯМР, 164-167 част./млн): 1,46
ВФА0: 97,2 ммоль/100 г
ЭА0: 27,8 ммоль/100 г
AK-Na0: 13,9 ммоль/100 г.
Конечный полимер АЕ28: Гидролизованный в щелочной среде тройной сополимер VE18 (ВФА/ЭА/акрилат Na=70 мол. %/20 мол. %/10 мол. %)
В стальном выдерживающем давление реакторе объемом 2 л, снабженном перемешивающим устройством, термометром для измерения внутренней температуры, нагревательной/охлаждающей рубашкой, монометром, клапаном сброса давления, обратным холодильником и выдерживающим давление питающим сосудом, 990,2 г раствора полимера, полученного с использованием исходного полимера VE18, при перемешивании смешивали с 126,4 г воды и нагревали до 125°С. Обеспечивали давление, равное 4 бар. В питающий сосуд помещали 126,4 г 50 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). При давлении, равном 6 бар, в реактор подавали раствор гидроксида натрия и смесь перемешивали. Обеспечивали температуру, равную 120°С, и ее поддерживали в течение 30 мин. Затем реактор как можно быстрее охлаждали до КТ. Получали 295,8 г продукта и его значение рН устанавливали равным 6,0 путем добавления 26,1 г 37 мас. % раствора хлористоводородной кислоты и 2,9 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера.
Содержание полимера: 7,2%
Содержание формиата, ФА: 94,7 ммоль/100 г
Степень гидролиза, СГ: 97,4 мол. %
Степень превращения, СП: 100 мол. %
Плотность заряда, ПЗ: 68,8 ммоль/100 г
Вязкость (20 об/мин, РВ, шпиндель №3): 940 мПа⋅с
ПАД (13С-ЯМР, 152,3 част./млн): 1,31
ПФА (13С-ЯМР, 164-167 част./млн): 1,01
ВФА0: 97,2 ммоль/100 г
ЭА0: 27,8 ммоль/100 г
AK-Na0: 13,9 ммоль/100 г.
Конечный полимер АЕ29: Гидролизованный в щелочной среде тройной сополимер VE27 (ВФА/МА/AM=70 мол. %/25 мол. %/5 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 156,0 г раствора полимера, полученного с использованием исходного полимера VE27, смешивали с 1,6 г 40 мас. % водного раствора бисульфита натрия и 72,9 г воды при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 60,3 г 25 мас. % водного раствора гидроксида натрия (120 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 6,0 путем добавления 24,1 г 37 мас. % раствора хлористоводородной кислоты и 7,5 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 7,9%. Степень гидролиза, СГ, составляла 93 мол. % и степень превращения, СП, составляла 100 мол. %.
Конечный полимер AV1: Гидролизованный в щелочной среде сополимер VV1 (ВФА/акрилат Na=70 мол. %/30 мол. %)
В четырехгорлой колбе объемом 500 мл, снабженной лопастью для перемешивания, термометром для измерения внутренней температуры, капельной воронкой и обратным холодильником, 206,1 г раствора полимера, полученного с использованием исходного полимера VV1, смешивали с 2,3 г 40 мас. % водного раствора бисульфита натрия при скорости перемешивающего устройства, равной 80 об/мин, и затем нагревали до 80°С. Затем добавляли 77,0 г 25 мас. % водного раствора гидроксида натрия (110 мол. % в пересчете на ВФА). Смесь выдерживали при 80°С в течение 5 ч. Полученный продукт охлаждали до КТ и значение рН устанавливали равным 8,5 путем добавления 32,3 г 37 мас. % раствора хлористоводородной кислоты и 9,6 г воды.
Получали немного мутный желтоватый и вязкий раствор полимера, обладающий содержанием полимера, составляющим 9,9%. Степень гидролиза, СГ, составляла 100 мол. %.
А-4) Подробное описание полученных полимеров
Подробное описание полученных полимеров приведено в таблицах А-4-1 и А-4-2.
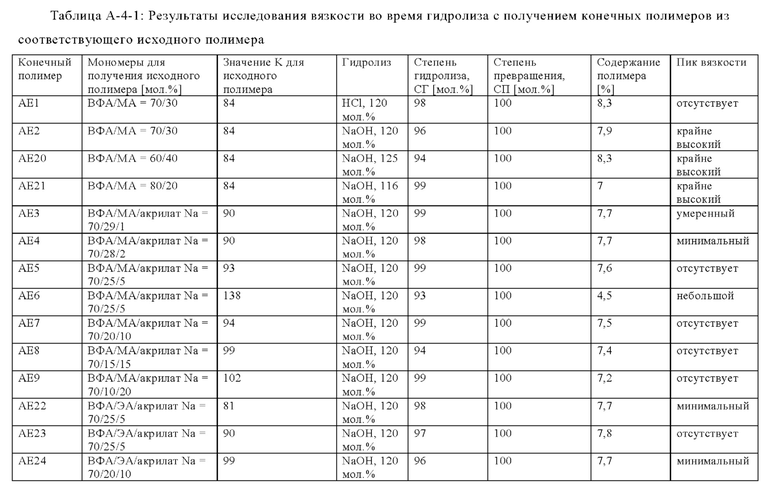
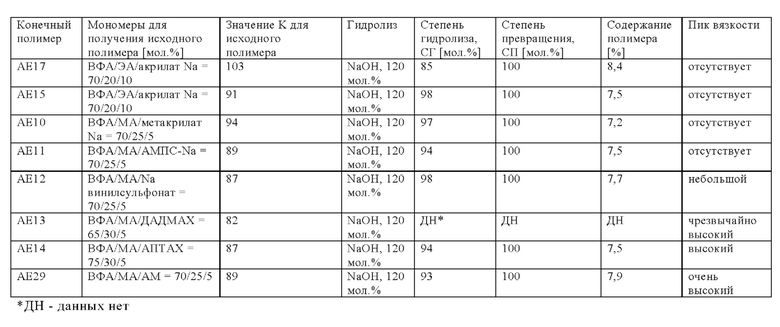
Примечание:
Исходный полимер VE1, предназначенный для получения конечного полимера АЕ1, получали практически так же, как исходный полимер VE2, предназначенный для получения конечного полимера АЕ2. При гидролизе исходного полимера VE1 в кислой среде с получением конечного полимера АЕ1 пик вязкости отсутствует, тогда как при гидролизе исходного полимера VE2 в щелочной среде с получением конечного полимера АЕ2 наблюдается крайне высокий пик вязкости. Наличие полимеризованного анионогенного мономера в исходном полимере V обеспечивает уменьшение или предотвращение возникновения пика вязкости при проведении гидролиза в щелочной среде, протекающего с получением конечных полимеров АЕЗ, АЕ4, АЕ5, АЕ6, АЕ7, АЕ8, АЕ9, АЕ10, АЕ11, АЕ12, АЕ15, АЕ17, АЕ22, АЕ23 и АЕ24. Наличие полимеризованного диаллилдиметиламмонийхлорида (ДАДМАХ), (3-акриламидопропил)триметиламмонийхлорида (АПТАХ) или акриламида (AM) в исходном полимере V, предназначенного для получения конечных полимеров АЕ13, АЕ14 и АЕ29, не обеспечивает такой эффект.

В) Бумага
В-1) Получение бумажного материала
В качестве бумажного материала для изготовления бумаги использовали пульпу, полученную путем обработки бумажного полотна в дефибрере. Бумажным полотном являлась упаковочная необработанная бумага марки "Testliner 2", обладающая поверхностной плотностью, равной 120 г/м2, выпускающейся фирмой Thurpapier, находящейся в Weinfelden (Switzerland). Пульпу получали путем растворения в питьевой воде и механической обработки бумажного полотна в дефибрере при содержании твердых веществ, составляющем примерно 3,5 мас. %. При этом пульпа обычно обладала степенью помола, составляющей примерно 50°ШР (Шоппера-Риглера).
В-2) Обработка бумажного материала конечными полимерами
Обработку конечными полимерами проводили или путем обработки "густой массы" при содержании твердых веществ в пульпе, составляющем примерно 3,5%, или путем обработки "жидкой массы" при содержании сухих веществ в пульпе, составляющем примерно 0,8%.
В случае "обработки густой массы" 500 г пульпы помещали в большой стеклянный стакан. Затем при перемешивании добавляли 2% водный раствор конечного полимера. Указанное выраженное в процентах количество означает содержание полимера в растворе конечного полимера. Пульпу обрабатывали с помощью 1,315 г 2% водного раствора конечного полимера или с помощью 2,63 г 2% водного раствора конечного полимера, т.е. с помощью 1,315 г или 2,63 г. в пересчете на 500 г пульпы. Это соответствовало обработке конечным полимером при его содержании, составляющем 0,15% или 0,3% в пересчете на количество сухого бумажного материала. Затем 100 г обработанной пульпы переносили в другой стеклянный сосуд и затем разбавляли питьевой водой до обеспечения содержания твердых веществ, составляющего 0,8%.
В случае "обработки жидкой массы" 114,3 г пульпы помещали в большой стеклянный стакан. Затем пульпу разбавляли питьевой водой до обеспечения содержания твердых веществ, составляющего 0,8%. При перемешивании добавляли добавки в виде 2% водного раствора конечного полимера. Указанное выраженное в процентах количество означает содержание полимера в растворе конечного полимера. Разбавленную пульпу обрабатывали с помощью 0,3 г 2% водного раствора конечного полимера или с помощью 0,6 г 2% водного раствора конечного полимера. Это соответствовало обработке конечным полимером при его содержании, составляющем 0,15% или 0,3% в пересчете на количество сухого бумажного материала.
В-3) Изготовление бумажных листов
Задачей являлось изготовление бумажных листов, обладающих поверхностной плотностью, равной 120 г/м2, с использованием в качестве сырья бумажного материала, обработанного конечными полимерами, обладающего содержанием твердых веществ, составляющим 0,8%. Бумажные листы изготавливали с помощью динамической листоформовочной машины, выпускающейся фирмой TechPap (France). Суспензию бумажного материала, т.е. бумажный материал, при необходимости обработанный конечным полимером, распыляли на сетку. Сетку закрепляли в вертикально расположенном быстро вращающем барабане. В этой системе обезвоживание и образование листа определяется не только структурой листа, но и главным образом центробежными силами внутри вращающего барабана. Путем изменения скорости вращения барабана можно менять центробежную силу, воздействующую на полученную структуру листа. Результатом является разная степень обезвоживания листа, что приводит к разному содержанию сухих веществ в полученной мокрой бумажной структуре. В настоящем изобретении это означает содержание сухих веществ в мокрой бумажной структуре непосредственно после ее снятия с сетки, закрепленной в барабане динамической листоформовочной машины. Скорость вращения барабана можно менять в 5 стадий от равного 600 до равного 1100 об/мин, таким образом можно было регулировать содержания сухих веществ в диапазоне от 15 до 21 мас. %. Небольшую часть еще мокрой бумажной структуры использовали для определения содержания сухих веществ непосредственно после снятия мокрой бумажной структуры с сетки динамической листоформовочной машины.
После извлечения мокрых бумажных структур из барабана динамической листоформовочной машины их накрывали с двух сторон промокательной бумагой и обезвоживали с помощью неподвижного пресса при давлении, равном 6 бар, в течение 30 с, таким образом из бумажной структуры получали влажный бумажный лист.После этого содержание сухих веществ в мокром бумажном листе обычно составляло от 41 до 43 мас. %. Если содержание сухих веществ является существенно меньшим, чем более низкое значение, то для обеспечения значения, находящегося в указанном выше диапазоне, можно увеличить толщину промокательной бумаги или количество наносимых слоев.
Затем полученный влажный бумажный лист повторно накрывали с двух сторон чистой промокательной бумагой и затем закрепляли в сушильном цилиндре и выдерживали в течение 10 мин. Температура поверхности сушильного цилиндра составляла примерно 100°С.Получали сухой бумажный лист.После сушки высушенные бумажные листы помещали в камеру с кондиционированием воздуха для кондиционирования.
В-4) Определение содержания сухих веществ в бумажном образце и внутренней прочности высушенных бумажных листов
Для определения содержания сухих веществ (СВ) в бумажном образце определяли массу влажного образца (MB) с использованием влажного бумажного образца с помощью откалиброванных содержащих расположенную сверху чашку весов, с помощью которых можно обеспечить точность взвешивания, составляющую 0,01 г. Предпочтительно, если влажный бумажный образец обладал площадью, равной не менее 10 см×10 см. Затем влажный бумажный образец помещали в откалиброванную сушильную камеру, в которой можно поддерживать установленную температуру с отклонением, составляющим ±2°С, и сушили при установленной температуре, равной 105°С, до постоянной массы. Обычно постоянная масса обеспечивалась через 90 мин. Затем еще теплый высушенный бумажный образец переносили в эксикатор, который содержал подходящий осушающий реагент, такой как силикагель. После охлаждения до комнатной температуры с помощью указанных выше весов определяли массу сухого бумажного образца (МС). Затем содержание сухих веществ в бумажном образце, рассчитанное в соответствии с формулой СВ=100×МС/МВ, указывали в мас. %. Выраженное в процентах значение часто указано с одним десятичным знаком. Если это выраженное в процентах значение, округленное до первого десятичного разряда, не изменяется, то это указывает на то, что обеспечена постоянная масса в случае содержаний сухих веществ, составляющих от 1 до 100 мас. %. В случае содержаний сухих веществ, составляющих от 0 до менее 1 мас. %, на это соответственно указывает не изменяющееся округленное до второго десятичного разряда выраженное в процентах значение. Сушку проводили при давлении окружающей среды, необязательно при 101,32 кПа, без внесения поправки на какое-либо отклонение вследствие погодных условий или высоты над уровнем моря. Во время сушки поддерживали обычно преобладающее давление воздуха, другими словами, теоретически равное 101,32 кПа. Не вносили поправку на незначительные изменения давления воздуха вследствие погодных условий или высоты над уровнем моря. В случае, если влажный образец еще не обладал консистенцией листа, например, представлял собой суспензию волокнистого материала или пульпу, то влажный образец сушили на соответствующем лотке, обладающем большой площадью поверхности.
Для определения внутренней прочности высушенного бумажного листа этот лист хранили в камере с кондиционированием воздуха при постоянных температуре, равной 23°С, и при влажности, равной 50%, в течение 12 ч. Внутреннюю прочность определяли по методике, соответствующей стандартной методике TAPPI Т833 pm-94. В этой методике отрезали 10 полосок бумаги шириной 2,5 см и длиной 12,7 см от двух бумажных листов, полученных и затем высушенных, как это описано выше. Каждый отдельный бумажный образец присоединяли с помощью двухсторонней клейкой ленты к отдельной базовой пластине и металлическому кронштейну. По металлическому кронштейну ударяли с помощью маятника, при этом исследуемый бумажный образец разрывался в направлении, параллельном поверхности бумаги. Определяли количество энергии, необходимой для осуществления этой процедуры. Использовавшимся для исследования прибором являлся Internal-Bond-Test-Station, выпускающийся фирмой TMI (Testing Machines Inc. Islandia, New York USA). Двухсторонняя клейкая лента представляла собой продукт фирмы 3 М (ширина 25,4 мм, типа Scotch №140). С помощью измерительного прибора определяли энергию, необходимую для разрыва, в пересчете на стандартизированную площадь поверхности, выраженную в Дж/м2. Значение внутренней прочности являлось средним значением 10 отдельных измерений.
В-5) Изготовленные высушенные бумажные листы и результаты Для получения высушенных бумажных листов из необработанного бумажного материала, использовавшегося в качестве эталонного образца (RB), получали три образца мокрой бумажной структуры, обладающие содержаниями сухих веществ, составляющими 15,7 мас. %, 17,4 мас. % и 20,4 мас. %. Затем образцы мокрой бумажной структуры прессовали и сушили. В случае каждого конечного полимера из бумажных материалов, обработанных конечным полимером, получали образцы мокрой бумажной структуры, обладающие двумя разными значениями содержания сухих веществ, составляющими от 16,5 до 21 мас. %, причем одно значение содержания сухих веществ составляло менее 18,5 мас. % и одно значение содержания сухих веществ составляло более 18,5 мас. %. В таблице В-5-1 приведены использовавшиеся конечные полимеры и полученные результаты.

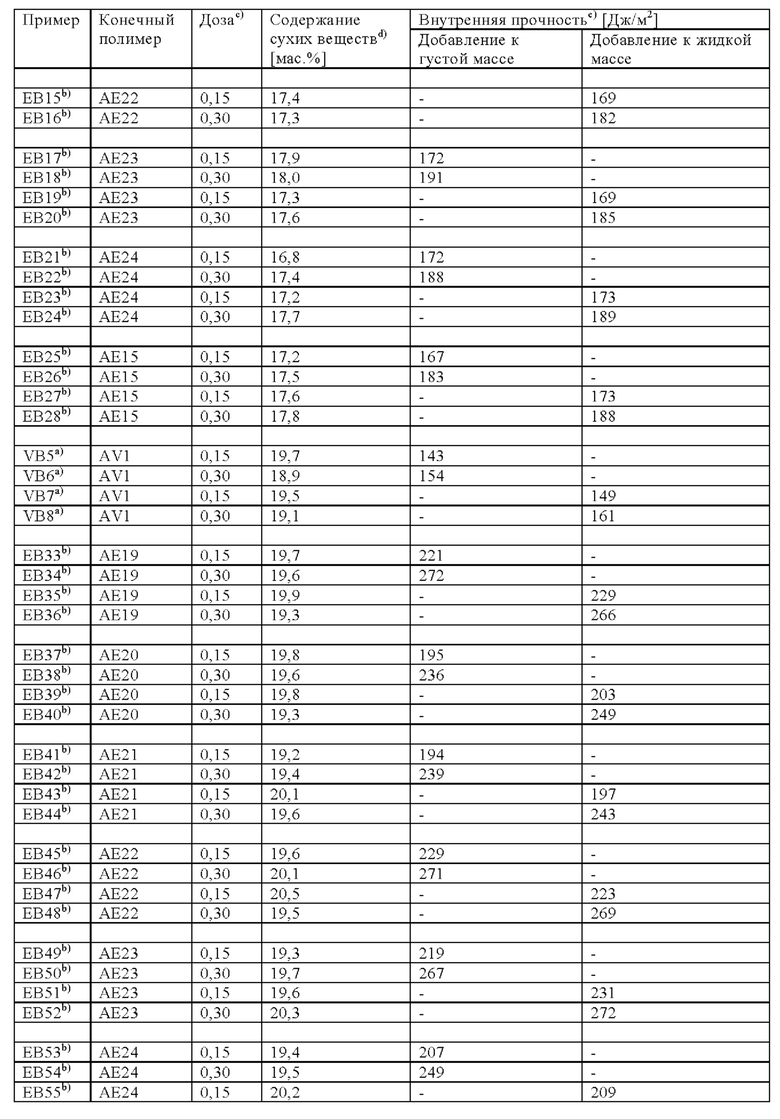
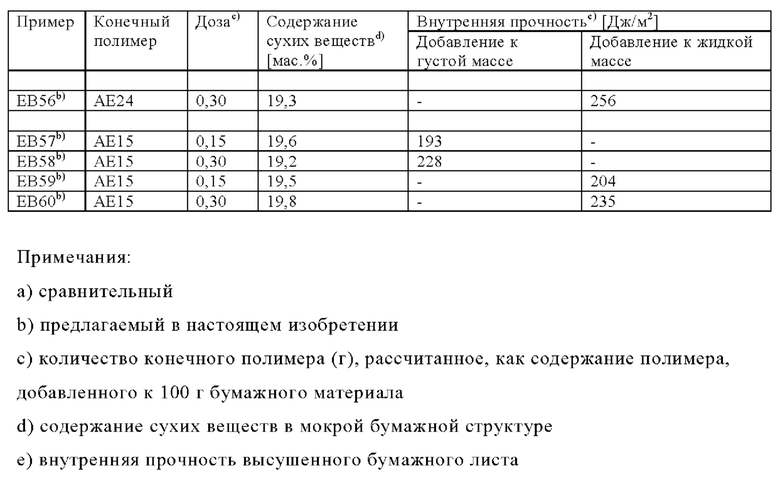
В-6) Краткое описание полученных результатов
Значения внутренней прочности для эталонных образцов (RB1-RB3, без добавления конечного полимера) составляют примерно 125 Дж/м2. Различия значений внутренней прочности высушенных бумажных листов, для изготовления которых использовали мокрую бумажную структуру, обладающую содержанием сухих веществ, составляющим от 15,3 до 20,2 мас. %, являются незначительными.
При добавляемом количестве полимеров сравнительных примеров (VB1, VB3, VB5, VB7), составляющем 0,15 г/100 г, внутренняя прочность увеличивается примерно на 20 Дж/м по сравнению с прочностью эталонных образцов независимо от того, проводят ли добавление к густой массе или к жидкой массе, и независимо от содержания сухих веществ. При добавляемом количестве полимеров сравнительных примеров (VB2, VB4, VB6, VB8), составляющем 0,3 г/100 г, внутренняя прочность увеличивается примерно на 30 Дж/м2 независимо от того, проводят ли добавление к густой массе или к жидкой массе, и независимо от содержания сухих веществ.
При добавляемом количестве полимеров примеров, предлагаемых в настоящем изобретении, составляющем 0,15 г/100 г, и при содержании сухих веществ, составляющем <18,5 мас. % (в каждом случае нечетные номера примеров ЕВ1-ЕВ28), внутренняя прочность увеличивается примерно на 40 Дж/м2 по сравнению с прочностью эталонных образцов независимо от того, проводят ли добавление к густой массе или к жидкой массе. При добавляемом количестве полимеров примеров, предлагаемых в настоящем изобретении, составляющем 0,3 г/100 г, и при содержании сухих веществ, составляющем <18,5 мас. % (в каждом случае четные номера примеров ЕВ1-ЕВ28), внутренняя прочность увеличивается примерно на 55 Дж/м2 по сравнению с прочностью эталонных образцов независимо от того, проводят ли добавление к густой массе или к жидкой массе.
При добавляемом количестве полимеров примеров, предлагаемых в настоящем изобретении, составляющем 0,15 г/100 г, и при содержании сухих веществ, составляющем >18,5 мас. % (в каждом случае нечетные номера примеров ЕВЗ3-ЕВ60), по сравнению с прочностью эталонных образцов внутренняя прочность увеличивается по меньшей мере на 70 Дж/м в случае добавления к густой массе и по меньшей мере на 50 Дж/м2 в случае добавления к жидкой массе. При добавляемом количестве полимеров примеров, предлагаемых в настоящем изобретении, составляющем 0,3 г/100 г, и при содержании сухих веществ, составляющем >18,5 мас. % (в каждом случае четные номера примеров ЕВЗ3-ЕВ60), по сравнению с прочностью эталонных образцов внутренняя прочность увеличивается по меньшей мере на 90 Дж/м2 в случае добавления к густой массе и по меньшей мере на 70 Дж/м2 в случае добавления к жидкой массе.
При сопоставлении образцов, предлагаемых в настоящем изобретении, при содержании сухих веществ в мокрой бумажной структуре, составляющем <18,5 мас. % (ЕВ1-ЕВ28), и образцов, предлагаемых в настоящем изобретении, при содержании сухих веществ в мокрой бумажной структуре, составляющем >18,5 мас. % (ЕВ33-ЕВ60), видно, что при добавлении одного и того же конечного полимера и использовании одинакового добавляемого количества внутренние прочности по меньшей мере на 20 Дж/м2 выше при более высоком содержании сухих веществ в мокрой бумажной структуре.
Конечный полимер AVI, использующийся в сравнительных примерах, формально состоит из 70 мол. % содержащих аминогруппу этиленовых звеньев и 30 мол. % содержащих карбоксигруппу этиленовых звеньев. Конечные полимеры АЕ15, АЕ19, АЕ22, АЕ23 и АЕ24, использующиеся в примерах, предлагаемых в настоящем изобретении, также формально состоят из примерно 70 мол. % содержащих аминогруппу этиленовых звеньев и 30 мол. % содержащих карбоксигруппу этиленовых звеньев. Степень гидролиза, СГ, составляет примерно 98 мол. % в случае АЕ15, 99 мол. % в случае АЕ19, 98 мол. % в случае АЕ22, 97 мол. % в случае АЕ23 и 96 мол. % в случае АЕ24. На полученную внутреннюю прочность бумаги использующиеся конечные полимеры оказывают разное воздействие в зависимости от того, используют ли для получения содержащих карбоксигруппу звеньев конечного полимера только звенья акрилата натрия, предварительно полимеризованные с получением исходного полимера, или используют по меньшей мере дополнительно или исключительно метиловый или этиловый эфир акриловой кислоты, предварительно полимеризованный с получением исходного полимера. Полагают, что существуют разные механизмы включения мономеров и, таким образом, разные степени чередования мономерных звеньев, которые полимеризованы. При увеличении степени чередования можно ожидать изменение количества возможных 5-членных лактамных звеньев. N-Винилформамид представляет собой богатый электронами мономер, тогда как эфир акриловой кислоты, наоборот, представляет собой мономер, обладающий низким содержанием электронов. В отличие от этого, забуференная акриловая кислота, обладающая значением рН, равным от 6 до 7, представляет собой мономер, обладающий высоким содержанием электронов. Другим отличием эфира акриловой кислоты и акрилата является растворимость.
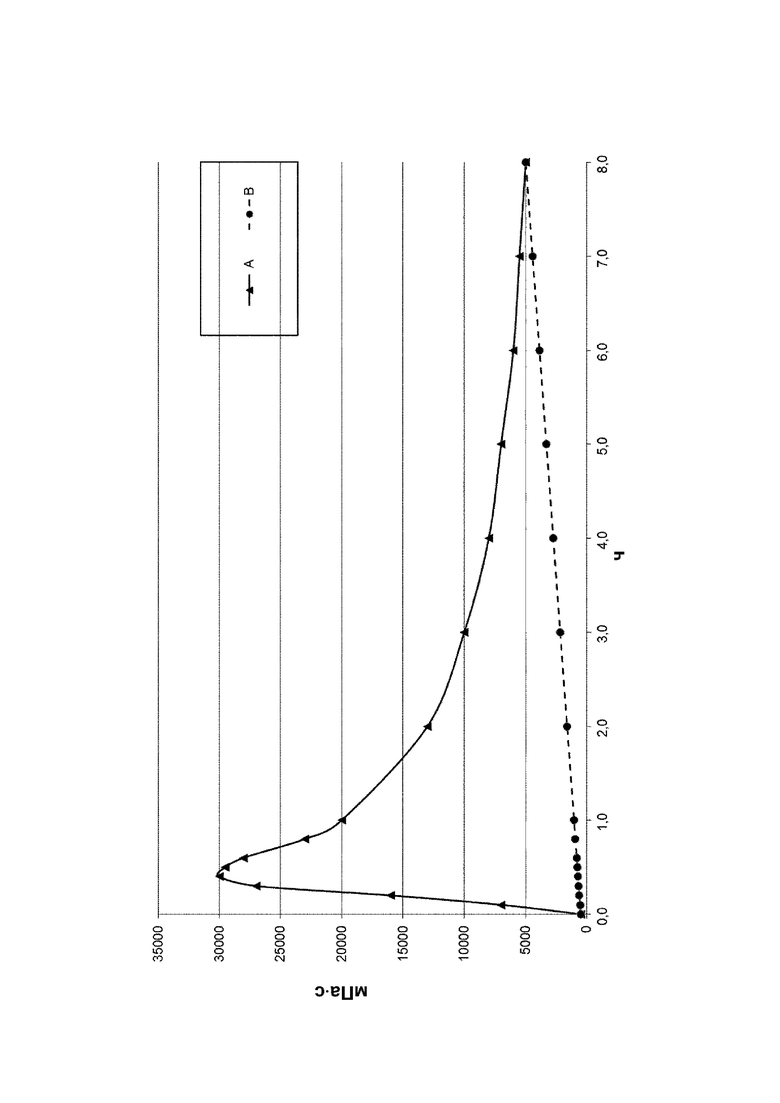
Изобретение относится к способу изготовления бумаги или картона. Способ включает стадии добавления конечного полимера А к первой водной суспензии волокнистого материала с получением второй водной суспензии волокнистого материала, содержащей конечный полимер А, обезвоживания второй водной суспензии волокнистого материала, содержащей конечный полимер А, на водопроницаемой подложке с получением мокрой бумажной структуры, обезвоживания мокрой бумажной структуры с получением бумаги или картона. При этом конечный полимер А представляет собой полимер, получаемый путем радикальной полимеризации: (i) 30-90 мол.% N-винилформамида, (ii) 3-60 мол.% С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты, (iii) 0-45 мол.% моноэтиленовоненасыщенных карбоновой, сульфоновой или фосфоновой кислот или их солевых форм, (iv) 0-9 мол.% акрилонитрила или метакрилонитрила, (v) 0-35 мол.% этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv), с получением исходного полимера V, и гидролиза исходного полимера V с получением конечного полимера А. Обеспечивается улучшение прочности в сухом состоянии. 2 н. и 8 з.п. ф-лы, 1 ил., 4 табл.
1. Способ изготовления бумаги или картона, включающий стадии (А) добавления конечного полимера А к первой водной суспензии волокнистого материала с получением таким образом второй водной суспензии волокнистого материала, содержащей конечный полимер А, где конечный полимер А представляет собой получаемый путем
- радикальной полимеризации следующих мономеров:
(i) от 30 до 90 мол.% мономера формулы I

в которой R1=Н,
(ii) от 3 до 60 мол.% С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 45 мол.% моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол.% акрилонитрила или метакрилонитрила,
(v) от 0 до 35 мол.% одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол.%, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы,
где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм,
(B) обезвоживания второй водной суспензии волокнистого материала, содержащей конечный полимер А, на водопроницаемой подложке с получением мокрой бумажной структуры,
(C) обезвоживания мокрой бумажной структуры с получением таким образом бумаги или картона.
2. Способ по п. 1, в котором для радикальной полимеризации используют
(i) от 50 до 89 мол.% мономера формулы I,
(ii) от 5 до 45 мол.% С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 30 мол.% моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол.% акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол.% одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
3. Способ по п. 1, в котором для радикальной полимеризации используют
(i) от 58 до 83 мол.% мономера формулы I,
(ii) от 8 до 39 мол.% С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол.% моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол.% акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол.% одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
4. Способ по п. 1, в котором мономер (iii) используют в количестве, составляющем от 1 до 25 мол.%.
5. Способ по п. 1, в котором мономером (i) является N-винилформамид, при этом в формуле I R1=Н.
6. Способ по п. 1, в котором мономер (ii) выбирают из С1-С3-алкилового эфира акриловой кислоты или C1-алкилового эфира метакриловой кислоты; этилакрилата; или их комбинаций.
7. Способ по п. 1, в котором мономер (iii) выбирают из моноэтиленовоненасыщенной карбоновой кислоты или моноэтиленовоненасыщенной сульфоновой кислоты, или их солевых форм; акриловой кислоты, метакриловой кислоты, винилсульфоновой кислоты или 2-акриламидо-2-метилпропансульфоновой кислоты, или их солевых форм; или их комбинаций.
8. Способ по п. 1, в котором для радикальной полимеризации используют следующие мономеры:
(i) от 60 до 83 мол.% N-винилформамида,
(ii) от 8 до 21 мол.% этилакрилата,
(iii) от 2 до 21 мол.% акриловой кислоты или метакриловой кислоты, или их солевых форм,
(iv) от 0 до 9 мол.% акрилонитрила или метакрилонитрила,
(v) от 0 до 24 мол.% одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv).
9. Способ по любому из пп. 1-8, в котором:
- на стадии (А) первая водная суспензия волокнистого материала обладает содержанием сухих веществ, составляющим от 0,1 до 6 мас.%;
- на стадии (В) мокрая бумажная структура обладает содержанием сухих веществ, составляющим от 18,5 до 25 мас.%.
10. Конечный полимер А, который представляет собой получаемый путем
- радикальной полимеризации следующих мономеров:
(i) от 58 до 83 мол.% мономера формулы I

в которой R1=Н,
(ii) от 8 до 39 мол.% С1-С4-алкилового эфира акриловой кислоты или С1-С4-алкилового эфира метакриловой кислоты,
(iii) от 0 до 25 мол.% моноэтиленовоненасыщенной карбоновой кислоты, моноэтиленовоненасыщенной сульфоновой кислоты или моноэтиленовоненасыщенной фосфоновой кислоты, или их солевых форм,
(iv) от 0 до 9 мол.% акрилонитрила или метакрилонитрила,
(v) от 0 до 25 мол.% одного или большего количества этиленовоненасыщенных мономеров, которые отличаются от мономера (i), (ii), (iii) и (iv),
где суммарное количество всех мономеров (i), (ii), (iii), (iv) и (v) составляет 100 мол.%, с получением исходного полимера V, и
- гидролиза исходного полимера V с получением конечного полимера А, где по меньшей мере часть групп N-C(=O)R1, содержащихся в мономерах (i) формулы (I), полимеризованных с получением исходного полимера V, гидролизована и при этом образованы первичные аминогруппы, где по меньшей мере часть сложноэфирных групп, содержащихся в мономерах (ii), полимеризованных с получением исходного полимера V, подвергается превращению и по меньшей мере частью превращения является образование 5-членных лактамных структурных звеньев, содержащих полученные первичные аминогруппы, или образование карбоксигрупп или их солевых форм.
| Токарный резец | 1924 |
|
SU2016A1 |
| DE 4241117 A1, 09.06.1994 | |||
| RU 2002114071 A, 27.03.2004 | |||
| МОДИФИЦИРОВАННЫЕ СОДЕРЖАЩИЕ ВИНИЛАМИН ПОЛИМЕРЫ И ИХ ПРИМЕНЕНИЕ ПРИ ИЗГОТОВЛЕНИИ БУМАГИ | 2015 |
|
RU2678672C2 |
