Настоящее изобретение относится к соединениям, которые способны к модуляции ERAP1. Соединения имеют потенциальные терапевтические применения при лечении различных нарушений, включая пролиферативные, вирусные, иммунные и воспалительные заболевания.
УРОВЕНЬ ТЕХНИКИ
ERAP1 (Аминопептидаза эндоплазматического ретикулума 1; также называемая APPILS или ARTS1) представляет собой аминопептидазу, важную для генерации некоторых антигенов и неоантигенов в качестве компонента пути презентации антигенов [1]. Путь презентации антигена начинается с расщепления белков протеасомой с образованием пептидов. Эти пептиды транспортируются в эндоплазматический ретикулум, в котором некоторые из них процессируются ERAP1 перед связыванием с главным комплексом гистосовместимости класса I (MHC Class I) [1]. Затем антигены, связанные с MHC класса I, транспортируются на поверхность клетки, где презентируются CD8+ T-клеткам и распознаются как свои или чужие. Неоантигены являются антигенами, специфичными для рака, и могут распознаваться иммунной системой как чужие, что приводит к разрушению раковых клеток. Неоантигены образуются либо в результате соматических мутаций в ДНК раковых клеток, приводящих к образованию мутантных белков, либо в результате непрямых последствий соматических мутаций, затрагивающих процессинг и экспрессию белков. Злокачественные опухоли с более высокой частотой мутаций и, соответственно, более высокими уровнями неоантигенов имеют гораздо более высокие показатели ответа на иммунотерапию ингибиторами контрольных точек, такими как антитела против PD-1 (например, пембролизумаб, ниволумаб), против PD-L1 (например, атезолизумаб, авелумаб, дурвалумаб) и против CTLA4 (например, ипилимумаб, тремелимумаб), по сравнению со злокачественными опухолями, несущими меньшее количество неоантигенов [2, 3].
Роль ERAP1 в пути презентации антигенов заключается в уменьшении некоторых пептидов посредством своей аминопептидазной активности, с получением антигенов и неоантигенов оптимальной длины для связывания с MHC класса I. ERAP1 также разрезает некоторые неоантигены, предотвращая их связывание с MHC класса I и презентацию на клеточной поверхности [4]. Было показано, что блокирование активности ERAP1 изменяет репертуар антигенов и неоантигенов, что приводит к увеличению презентации некоторых антигенов/неоантигенов, а также к презентации совершенно новых антигенов/неоантигенов [5]. Кроме того, блокирование ERAP1 вызывает CD8+ T-клеточно-зависимое отторжение опухоли в моделях рака у мышей [4]. Таким образом, модуляторы активности ERAP1 могут применяться для лечения рака, отдельно или в комбинации с существующими средствами для противораковой иммунотерапии, включая ингибиторы контрольных точек, поскольку они изменяют антигены и неоантигены, презентируемые на поверхности раковых клеток, и делают их более заметными для иммунной системы, что приводит к атаке и разрушению опухолей.
Также было показано, что нокдаун ERAP1 снижает уровни регуляторных T-клеток и усиливает уничтожение раковых клеток естественными киллерными клетками [6, 7]. Это указывает на то, что модуляторы активности ERAP1 могут быть эффективными средствами для лечения рака, которые будут не только модулировать заметность раковых клеток, но и формировать более эффективный противотуморогенный иммунный ответ. Роль ERAP1-опосредованного процессинга пептидов в презентации антигенов также может быть применена в случае инфекционного вирусного заболевания.
Настоящее изобретение направлено на предоставление соединений, которые способны к модуляции ERAP1. Такие соединения имеют потенциальные терапевтические применения при лечении различных нарушений, включая пролиферативные нарушения, иммунные нарушения и воспалительные нарушения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Первый аспект изобретения относится к соединениям формулы (Ia) или их фармацевтически приемлемой соли или гидрату,
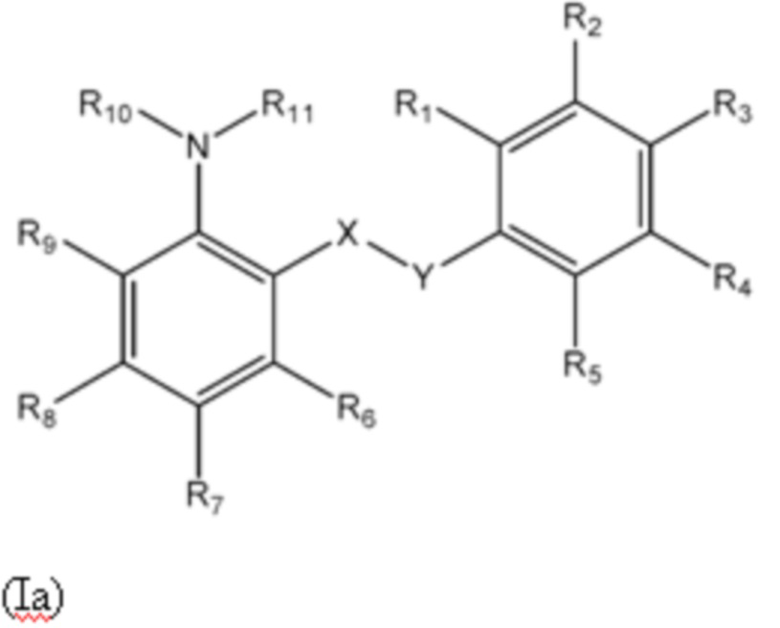
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, алкинила, алкенила, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 и R11, вместе с азотом, к которому они присоединены, образуют азепанильную группу, где: (a) указанная азепанильная группа замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила, или (b) один или два углерода в указанной азепанильной группе замещены группой, выбранной из O, NH, S и CO, и указанная азепанильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют азетидинильную, пирролидинильную или пиперидинильную группу, где: (a) указанная азетидинильная, пирролидинильная или пиперидинильная группа замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила, или (b) один или два углерода в указанной азетидинильной, пирролидинильной или пиперидинильной группе замещены группой, выбранной из NH, S и CO; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
Второй аспект изобретения относится к соединениям формулы (Ib) или их фармацевтически приемлемой соли или гидрату,
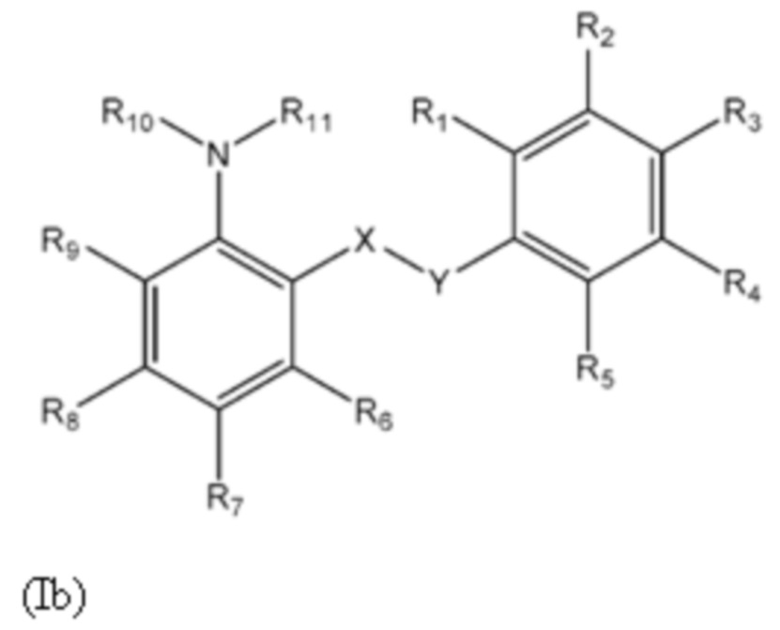
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 представляет собой тетразолильную группу;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
Третий аспект изобретения относится к соединениям формулы (Ic) или их фармацевтически приемлемой соли или гидрату,
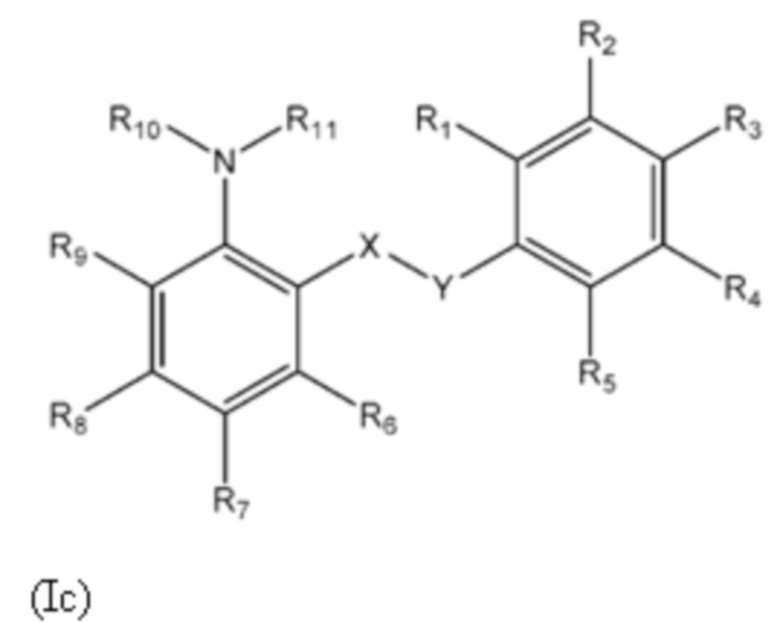
где:
X представляет собой SO2;
Y представляет собой NH;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH, и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
Четвертый аспект изобретения относится к соединениям формулы (Id) или их фармацевтически приемлемой соли или гидрату,
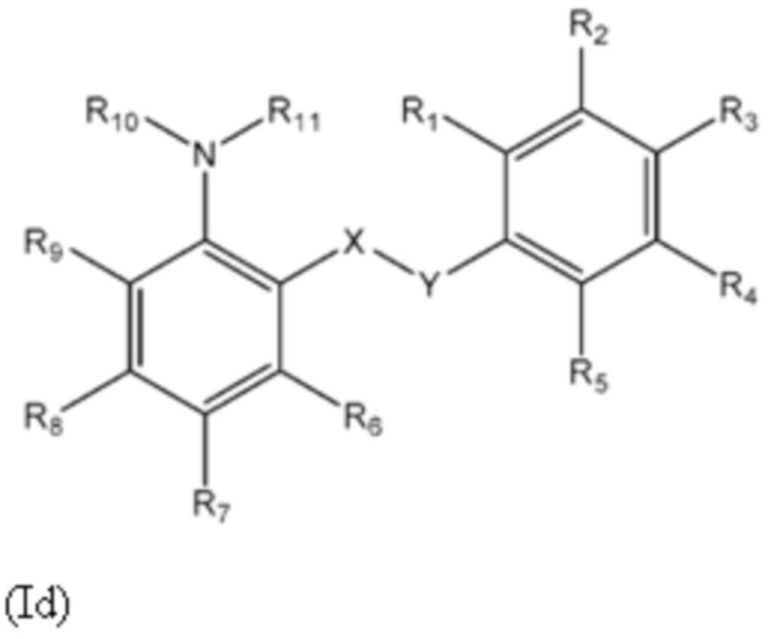
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 представляет собой CN, SO2-алкильную, SO2NR13R14 или гетероарильную группу, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH, и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
Предпочтительно заявленные в настоящем документе соединения способны модулировать ERAP 1, что делает данные соединения представляющими терапевтический интерес для лечения различных нарушений, например, в области онкологии и иммуноонкологии.
Пятый аспект изобретения относится к фармацевтической композиции, включающей по меньшей мере одно соединение, как описано выше, и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
Шестой аспект изобретения относится к соединению, как описано выше, для применения в медицине.
Седьмой аспект изобретения относится к соединению, как описано выше, для применения в лечении или предупреждении нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
Восьмой аспект изобретения относится к применению соединения, как описано выше, в изготовлении лекарственного средства для лечения или предупреждения нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
Девятый аспект изобретения относится к соединению, как описано выше, для применения в предупреждении или лечении нарушения, вызванного, связанного с или сопровождаемого какой-либо патологической активностью ERAP1.
Десятый аспект изобретения относится к применению соединения, как описано выше, в изготовлении лекарственного средства для предупреждения или лечения нарушения, вызванного, связанного с или сопровождаемого патологической активностью ERAP1.
Одиннадцатый аспект изобретения относится к способу лечения млекопитающего, имеющему патологическое состояние, облегчаемое при модуляции ERAP1, где способ включает введение млекопитающему терапевтически эффективного количества соединения, как описано выше.
Двенадцатый аспект изобретения относится к соединению, как описано выше, для применения при лечении или предупреждении патологического состояния, облегчаемого при модуляции ERAP1.
Тринадцатый аспект изобретения относится к применению соединения, как описано выше, в изготовлении лекарственного средства для лечения или предупреждения патологического состояния, облегчаемого при модуляции ERAP1.
Четырнадцатый аспект изобретения относится к способу лечения или предупреждения нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и нарушения у субъекта, где способ включает введение субъекту терапевтически эффективного количества соединения, как описано выше.
Пятнадцатый аспект изобретения относится к соединениям формулы (I) или их фармацевтически приемлемой соли или гидрату,
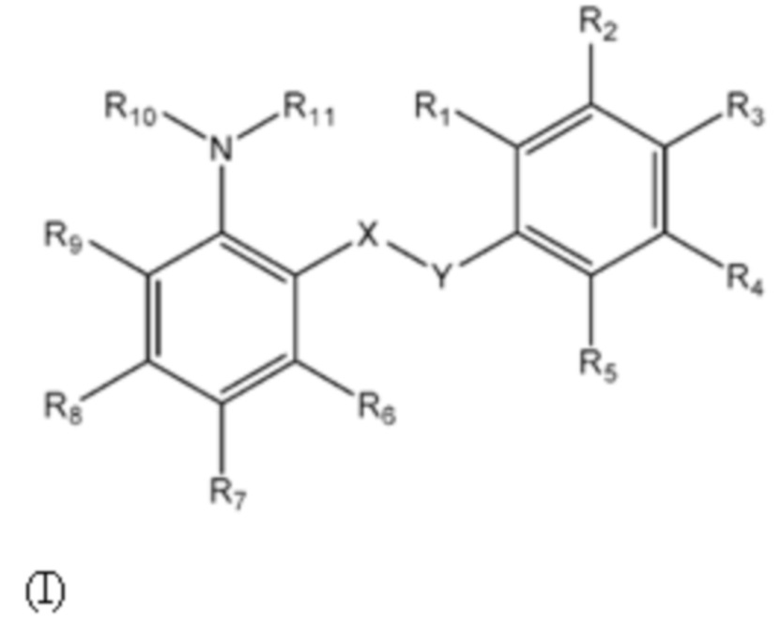
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил;
для применения при лечении или предупреждении нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
ПОДРОБНОЕ ОПИСАНИЕ
Настоящее изобретение относится к бис-арилсульфонамидным соединениям, которые способны модулировать ERAP1. Предпочтительно соединения селективно модулируют ERAP1.
"Алкил" определен в настоящем документе как алкильный радикал с нормальной или разветвленной цепью, предпочтительно C1-20 алкил, более предпочтительно C1-12 алкил, еще более предпочтительно C1-10 алкил или C1-6 алкил или C1-3-алкил. Примеры подходящих алкильных групп включают, без ограничения перечисленными, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил.
"Циклоалкил" определен в настоящем документе как моноциклическое алкильное кольцо, предпочтительно C3-7-циклоалкил, более предпочтительно C3-6-циклоалкил. Предпочтительные примеры включают циклопропил, циклобутил, циклопентил, циклогексил или циклогептил или конденсированную бициклическую кольцевую систему, такую как норборнан.
"Галоген" определен в настоящем документе как хлор, фтор, бром или иод.
При использовании в настоящем документе термин "арил" относится к C6-12 ароматической группе, которая может быть бензоконденсированной, например, фенилу или нафтилу.
"Гетероарил" определен в настоящем документе как моноциклическое или бициклическое C2-12 ароматическое кольцо, включающее один или более гетероатомов (которые могут быть одинаковыми или разными), таких как кислород, азот или сера. Примеры подходящих гетероарильных групп включают тиенил, фуранил, пирролил, пиридинил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тетразолил, тиадиазолил и т.д. и их бензопроизводные, такие как бензофуранил, бензотиенил, бензимидазолил, индолил, изоиндолил, индазолил и т.д.; или пиридил, пиразинил, пиримидинил, пиридазинил, триазинил и т.д. и их бензопроизводные, такие как хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, хиноксалинил, нафтиридинил и т.д. Особенно предпочтительные гетероарильные группы включают 1H-имидазол-5-ил, 1H-имидазол-4-ил, 1H-имидазол-2-ил, 1H-пиррол-1-ил, 1H-пиррол-2-ил, 1H-пиррол-3-ил, 1H-пиррол-4-ил, 1H-пиррол-5-ил, 1H-пиразол-1-ил, 1H-пиразол-5-ил, 1H-пиразол-3-ил, 1H-пиразол-4-ил, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, 1H-1,2,4-триазол-3-ил, 1H 1,2,4 триазол 5-ил, 1H-1,2,4-триазол 1-ил, 1H-1,2,3-триазол 4-ил, 1H-1,2,3-триазол-5-ил, 1H-1,2,3-триазол-1-ил, тиазол-5-ил, тиазол-4-ил, тиазол-2-ил, 1H-1,2,3,4-тетразол-4-ил, 2H-1,2,3,4-тетразол-5-ил, оксазол-5-ил, оксазол-4-ил, оксазол-2-ил, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, пиридазин-3-ил, пиридазин-4-ил, пиразинил, 1,3,4-оксадиазол-2-ил, 1,3,4-оксадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,5-оксадиазол-4-ил, 1,2,3-оксадиазол-4-ил, 1,2,3-оксадиазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, изоксазол-5-ил, изоксазол-4-ил и изоксазол-3-ил.
"Гетероциклоалкил" относится к циклической алифатической группе, содержащей один или более гетероатомов, выбранных из азота, кислорода и серы, которая необязательно прервана одной или более группами -(CO)- в кольце, и/или необязательно содержащей одну или больше двойных связей в кольце. Предпочтительно гетероциклоалкильная группа является моноциклической или бициклической. Предпочтительно гетероциклоалкильная группа представляет собой C3-7 гетероциклоалкил, более предпочтительно C3-6 гетероциклоалкил. Также гетероциклоалкильная группа представляет собой C4-7 гетероциклоалкил, более предпочтительно C4-6 гетероциклоалкил. Предпочтительные гетероциклоалкильные группы включают, без ограничения, пиперазинил, пиперидинил, морфолинил, тиоморфолинил, пирролидинил, тетрагидрофуранил и тетрагидропиранил. Предпочтительно гетероциклоалкильная группа является полностью насыщенной.
"Азепанил" относится к 7-членному насыщенному гетероциклическому кольцу, содержащему шесть атомов углерода и один атом азота. "Пиперидинил" относится к 6-членному насыщенному гетероциклическому кольцу, содержащему пять атомов углерода и один атом азота. "Пирролидинил" относится к 5-членному насыщенному гетероциклическому кольцу, содержащему четыре атома углерода и один атом азота. "Азетидинил" относится к 4-членному насыщенному гетероциклическому кольцу, содержащему три атома углерода и один атом азота.
Соединения формулы (Ia)
Один аспект изобретения относится к соединениям формулы (Ia), как описано выше.
В одном предпочтительном варианте осуществления R1 представляет собой H или Me, более предпочтительно H.
В одном предпочтительном варианте осуществления R2 представляет собой COOH.
В одном предпочтительном варианте осуществления X-Y представляет собой NH-SO.
В одном предпочтительном варианте осуществления R5 выбран из алкила, алкенила, алкинила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси.
В одном предпочтительном варианте осуществления R5 выбран из H, Me, CF3, CHF2, SO2-Me, Cl, этинила, MeO, OH, CH2OH, SME, циклопропила, триазолила, оксетанила и CN. Более предпочтительно R5 выбран из H, CN, Me, SO2-Me, CF3 и CHF2, CH2OH, SME, циклопропила, 3,4-триазол-1-ил, оксетан-3-ила. Более предпочтительно R5 выбран из H, CN, Me, SO2-Me, CF3 и CHF2.
В другом предпочтительном варианте осуществления R5 выбран из OMe, Me, Et, Pr, этинила и Cl, более предпочтительно OMe, Me, Et, Pr и Cl, и является более предпочтительно OMe или Et.
В одном предпочтительном варианте осуществления R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила.
В одном предпочтительном варианте осуществления R7 выбран из H, CN, CF3, CHF2, Cl, F, SO2-Me, SO2NH2, гетероарила и Me. Более предпочтительно R7 выбран из H, CN, Me, SO2-Me, тетразолила, CF3 и CHF2.
В одном предпочтительном варианте осуществления R7 представляет собой CF3.
В одном предпочтительном варианте осуществления R7 представляет собой CN.
В другом предпочтительном варианте осуществления R7 представляет собой SO2-алкил, более предпочтительно SO2-Me.
В одном предпочтительном варианте осуществления R7 представляет собой SO2NR13R14, более предпочтительно SO2NH2,
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, необязательно замещенную одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из пиридинила, тиенила, имидазолила, пиримидинила, пиразолила, пиразинила, пиридазинила, тиазолила, изотиазолила, триазинила, пирролила, фуранила, оксазолила, изоксазолила, оксадиазолила, тетразолила и триазолила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из имидазолила, пиразолила, пиразинила, пиридазинила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, тетразолила и триазолила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из 1H-имидазол-5-ила, 1H-имидазол-4-ила, 1H-имидазол-2-ила, 1H-пиррол-1-ила, 1H-пиррол-2-ила, 1H-пиррол-3-ила, 1H-пиррол-4-ила, 1H-пиррол-5-ила, 1H-пиразол-1-ила, 1H-пиразол-5-ила, 1H-пиразол-3-ила, 1H-пиразол-4-ила, оксазол-2-ила, оксазол-4-ила, оксазол-5-ила, 1H 1,2,4 триазол-3-ила, 1H-1,2,4-триазол-5-ила, 1H-1,2,4-триазол-1-ила, 1H-1,2,3-триазол-4-ила, 1H-1,2,3-триазол-5-ила, 1H-1,2,3-триазол-1-ила, тиазол-5-ила, тиазол-4-ила, тиазол-2-ила, 1H-1,2,3,4-тетразол-4-ила, 2H-1,2,3,4-тетразол-5-ила, оксазол-5-ила, оксазол-4-ила, оксазол-2-ила, изоксазол-3-ила, изоксазол-4-ила, изоксазол-5-ила, изотиазол-3-ила, изотиазол-4-ила, изотиазол-5-ила, пиридазин-3-ила, пиридазин-4-ила, пиразинила, 1,3,4-оксадиазол-2-ила, 1,3,4-оксадиазол-5-ила, 1,2,5-оксадиазол-3-ила, 1,2,5-оксадиазол-4-ила, 1,2,3-оксадиазол-4-ила, 1,2,3-оксадиазол-5-ила, 1,2,4-оксадиазол-3-ила, 1,2,4-оксадиазол-5-ила, изоксазол-5-ила, изоксазол-4-ила и изоксазол-3-ила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, CN, алкокси, галогеналкила и OH.
В одном крайне предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из 1H-пиразола-5-ила, 1H-пиразол-3-ила, 1H-пиразол-4-ила, оксазол-2-ила, 1H-1,2,3-триазол-4-ила, 1H-1,2,3-триазол-5-ила, тиазол-5-ила, 1H-1,2,3,4-тетразол-4-ила, 2H-1,2,3,4-тетразол-5-ила, изоксазол-4-ила, изоксазол-5-ила, изотиазол-5-ила, пиридазин-3-ила, пиридазин-4-ила, пиразинил и 1,3,4-оксадиазол-2-ила, каждый из которых необязательно замещен одним или более заместителями, выбранными из Me, F, Cl, CN и MeO.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, которая необязательно замещена одной или более алкильными группами, предпочтительно одной или более группами Me.
В одном предпочтительном варианте осуществления R7 представляет собой галогеналкил или гетероарил, более предпочтительно тетразолил.
В одном предпочтительном варианте осуществления R7 представляет собой галогеналкил, более предпочтительно CF3.
В одном предпочтительном варианте осуществления R8 представляет собой H или галогеналкил, более предпочтительно H или CF3, еще более предпочтительно H.
В одном предпочтительном варианте осуществления R8 выбран из H, Me, CF3, Cl, Br и F.
В другом предпочтительном варианте осуществления R8 выбран из H, галогеналкила и Cl.
В одном предпочтительном варианте осуществления R9 представляет собой H, Me или F, более предпочтительно H или F, более предпочтительно H.
В одном предпочтительном варианте осуществления все R1, R3, R4, R6, R8 и R9 представляют собой H.
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 выбран из OMe, Me, Et, Pr и Cl, и более предпочтительно представляет собой OMe;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 представляет собой галогеналкил, более предпочтительно, CF3.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют азепанильную группу, где: (a) указанная азепанильная группа замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, галогена и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из галогена и алкила, или (b) один или два углерода в указанной азепанильной группе замещены группой, выбранной из O, NH, S и CO, и указанная азепанильная группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, галогена и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из галогена и алкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют азетидинильную, пирролидинильную или пиперидинильную группу, где: (a) указанная азетидинильная, пирролидинильная или пиперидинильная группа замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из галогена и алкила, или (b) один или два углерода в указанной азетидинильной, пирролидинильной или пиперидинильной группе замещены группой, выбранной из NH, S и CO.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют азетидинильную, пирролидинильную или пиперидинильную группу, где указанная азетидинильная, пирролидинильная или пиперидинильная группа замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из галогена и алкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют азетидинильную группу, которая замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пирролидинильную группу, которая замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, которая замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, OH и галогена.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную мостиковую бициклическую гетероциклоалкильную группу, где один или два углерода в мостиковом бициклическом гетероциклоалкильном кольце необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, OH и галогена.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, которая необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, OH и галогена, и где два несмежных углерода в кольце указанной пиперидинильной группы связаны друг с другом алкиленовым мостиком, содержащим 3 углерода или 2 углерода.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе необязательно замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, CN, галогена и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой. Предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют 7-12-членную бициклическую группу, содержащую спироциклический атом углерода.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют бициклическую группу, содержащую спироциклический атом углерода, которая имеет следующую формулу (Z):
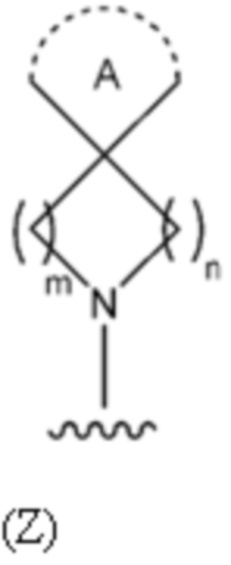
где:
m равно 1 или 2;
n равно 1, 2 или 3; и
кольцо A представляет собой 3, 4, 5 или 6-членную циклоалкильную или гетероциклоалкильную группу.
В одном предпочтительном варианте осуществления кольцо A представляет собой 3-членную циклоалкильную или гетероциклоалкильную группу.
В одном предпочтительном варианте осуществления кольцо A представляет собой 4-членную циклоалкильную или гетероциклоалкильную группу.
В одном предпочтительном варианте осуществления кольцо A представляет собой 5-членную циклоалкильную или гетероциклоалкильную группу.
В одном предпочтительном варианте осуществления кольцо A представляет собой 6-членную циклоалкильную или гетероциклоалкильную группу.
В одном предпочтительном варианте осуществления m равно 1, а n равно 1.
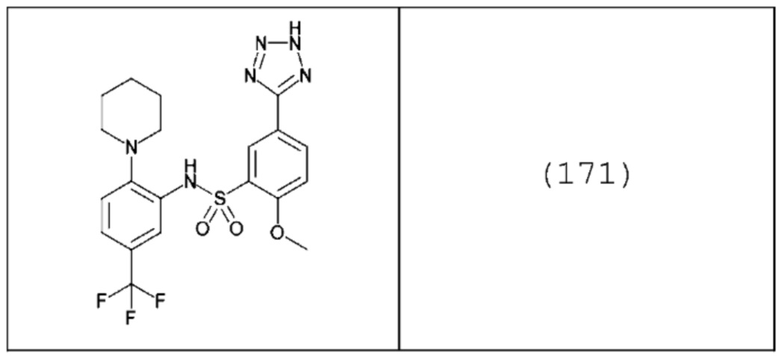
В одном предпочтительном варианте осуществления m равно 1, а n равно 2.
В одном предпочтительном варианте осуществления m равно 2, а n равно 2.
В одном предпочтительном варианте осуществления m равно 2, а n равно 3.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 7-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 8-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 9-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 10-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 11-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют бициклическую группу, включающую кольцевую систему, выбранную из конфигурации спиро[3.3]гептана, спиро[3.4]октана, спиро[3.5]нонана, спиро[4.4]нонана, спиро[4,5]декана, спиро[3.6]декана, спиро[5.5]ундекана и спиро[5,6]додекана, где в каждой из вышеуказанных бициклических групп азот группы NR10R11 формирует одного члена кольцевой системы, а еще один углерод в кольцевой системе необязательно замещен O, и указанная бициклическая группа необязательно замещена одной или более группами (более предпочтительно одной или двумя группами), выбранными из алкила, галогена и гетероарила.
В одном предпочтительном варианте осуществления NR10R11 выбран из следующего:
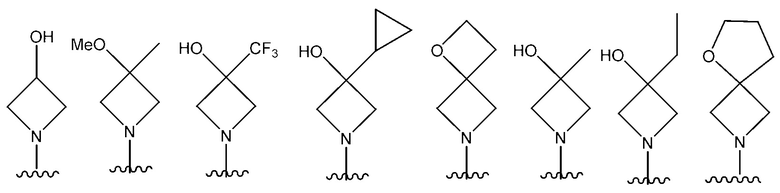
В одном предпочтительном варианте осуществления NR10R11 выбран из следующего:
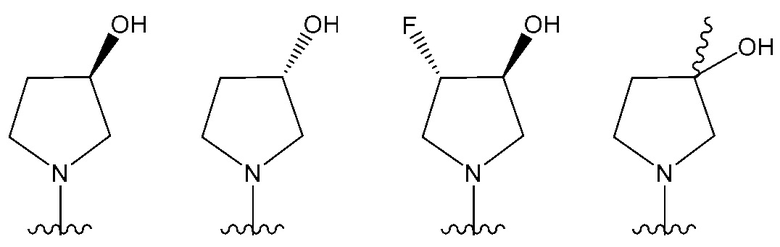
В одном предпочтительном варианте осуществления NR10R11 выбран из следующего:
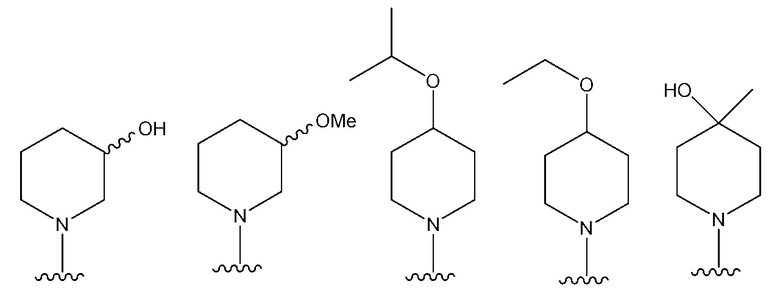
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой циклопропил;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CN, галогеналкила, гетероарила и SO2-алкила; и
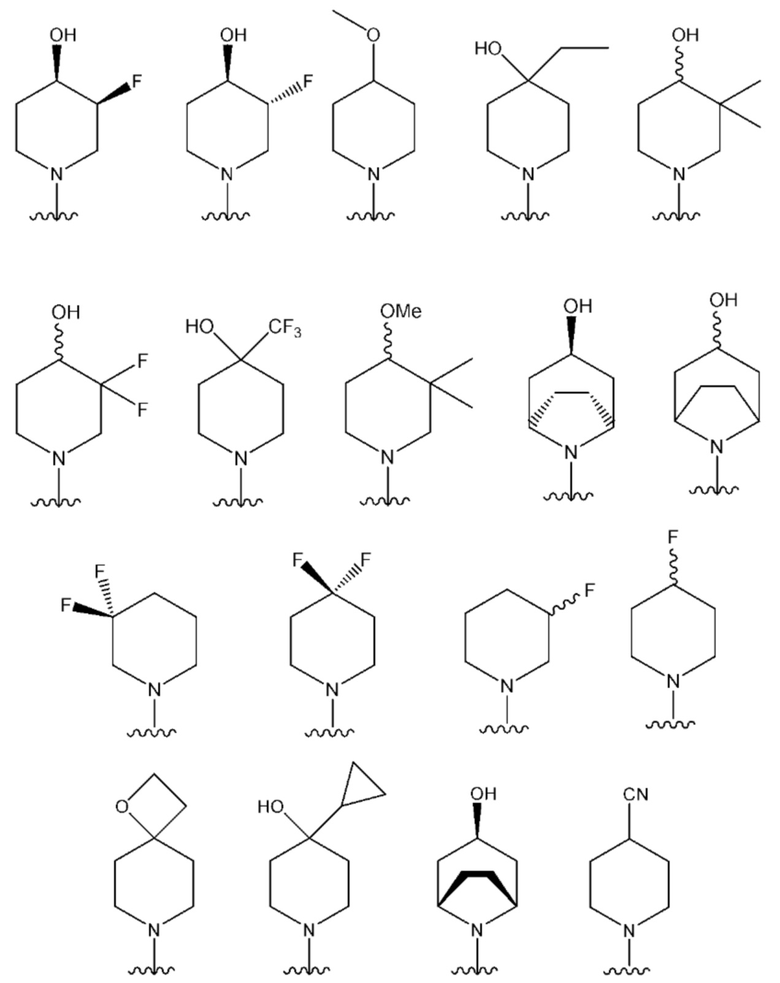
NR10R11 выбран из следующего:
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой циклопропил;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CN, CF3, тетразолила и SO2-Me, более предпочтительно CN и SO2-Me;
NR10R11 выбран из следующего:
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой этил;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CN, галогеналкила, гетероарила и SO2-алкила; и
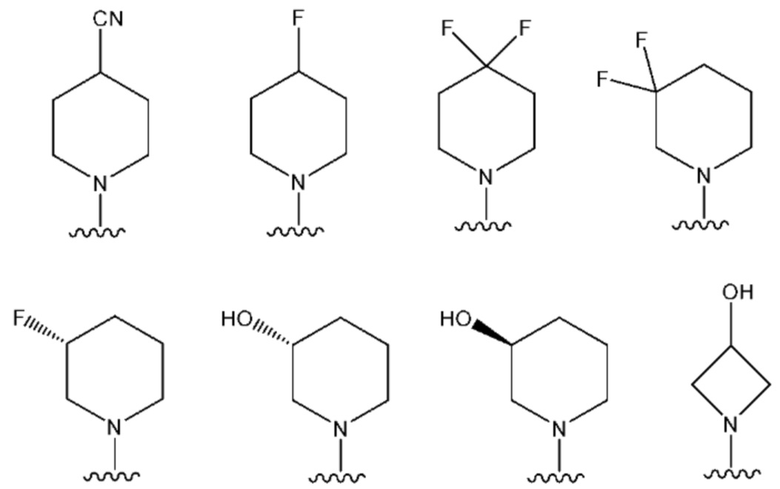
NR10R11 выбран из следующего:
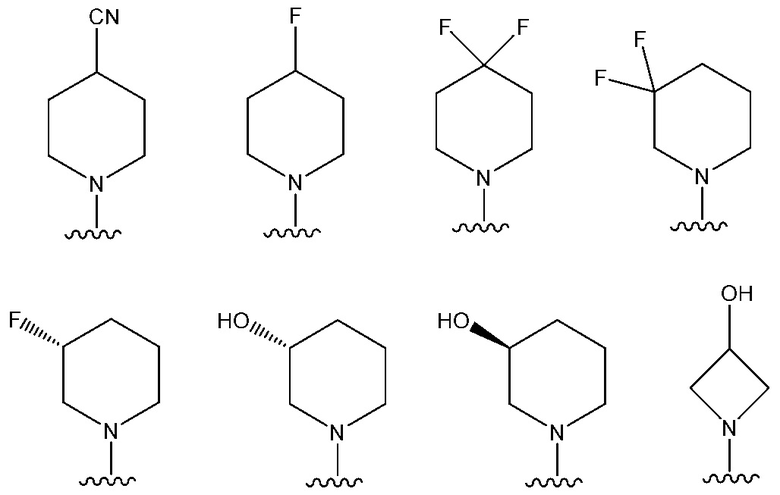
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой этил;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CN и CF3; и
NR10R11 представляет собой:
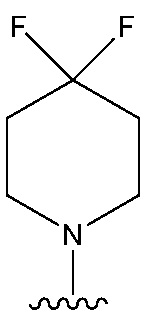
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой OMe;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CN, галогеналкила, гетероарила и SO2-алкила; и
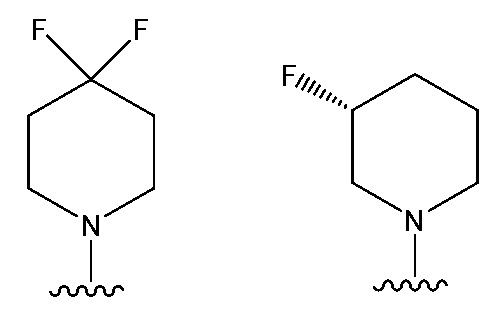
NR10R11 выбран из следующего:
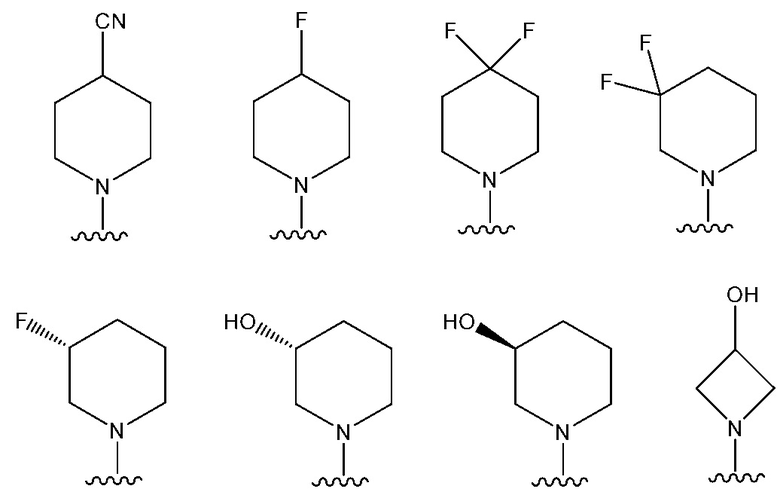
В одном предпочтительном варианте осуществления:
R2 представляет собой COOH;
X-Y представляет собой NH-SO2;
R5 представляет собой OMe;
все R1, R3, R4, R6, R8 и R9 представляют собой H; и
R7 выбран из CF3 и SO2-Me; и
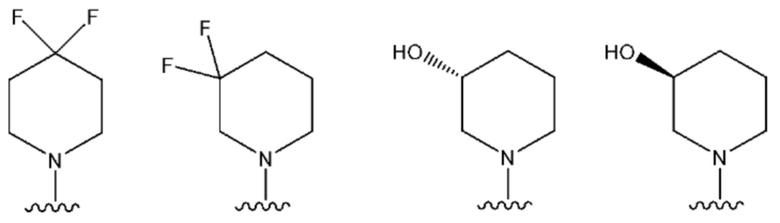
NR10R11 выбран из следующего:
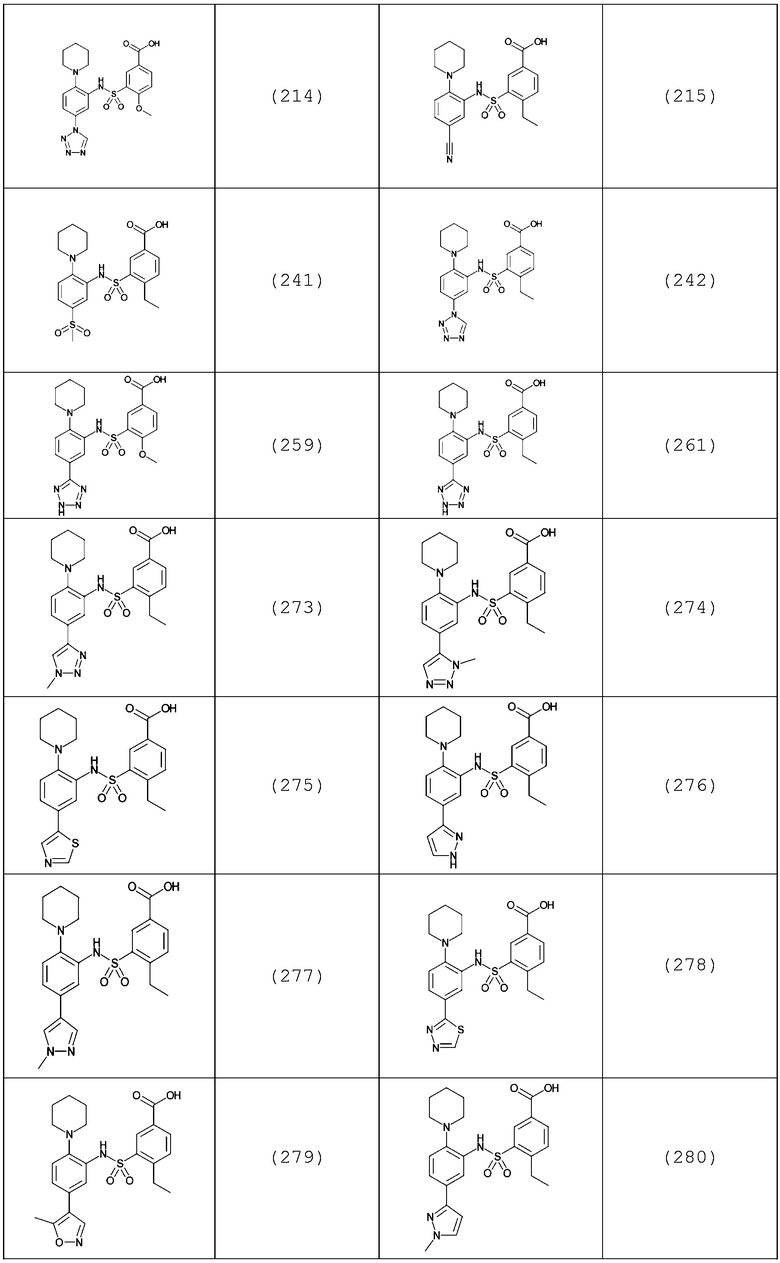
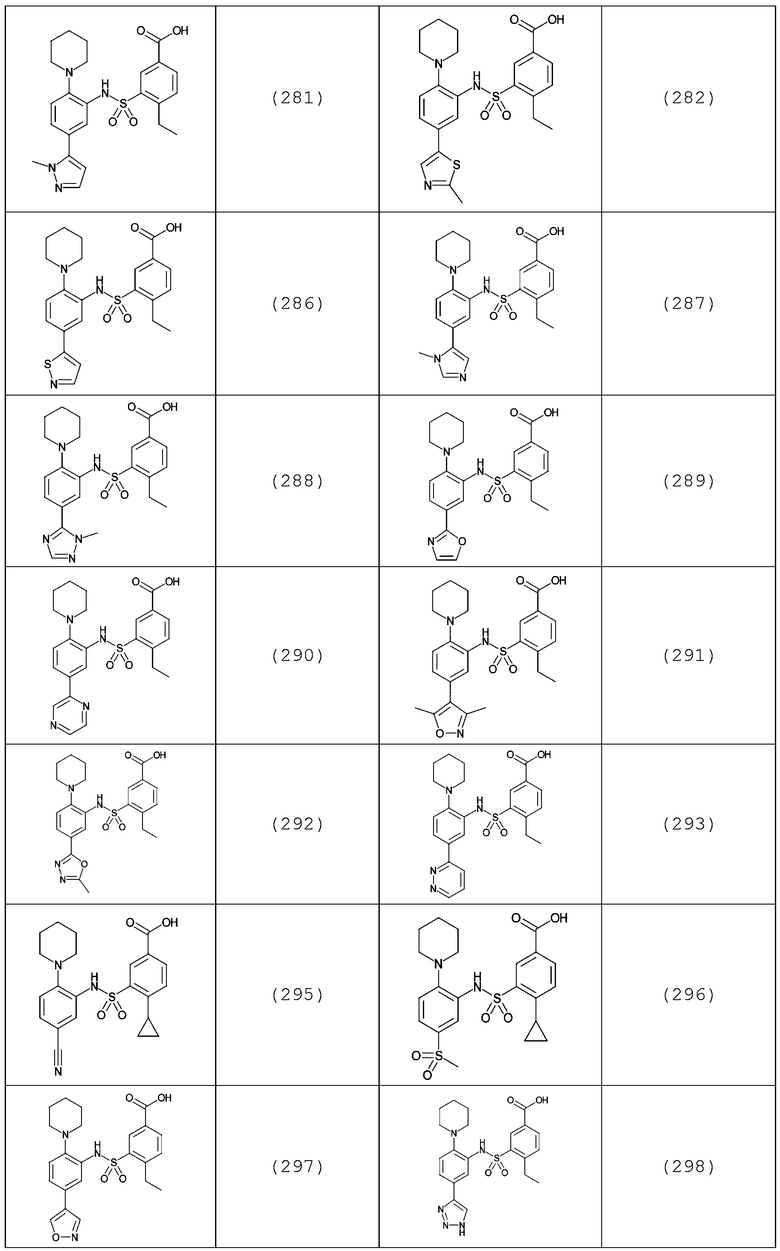
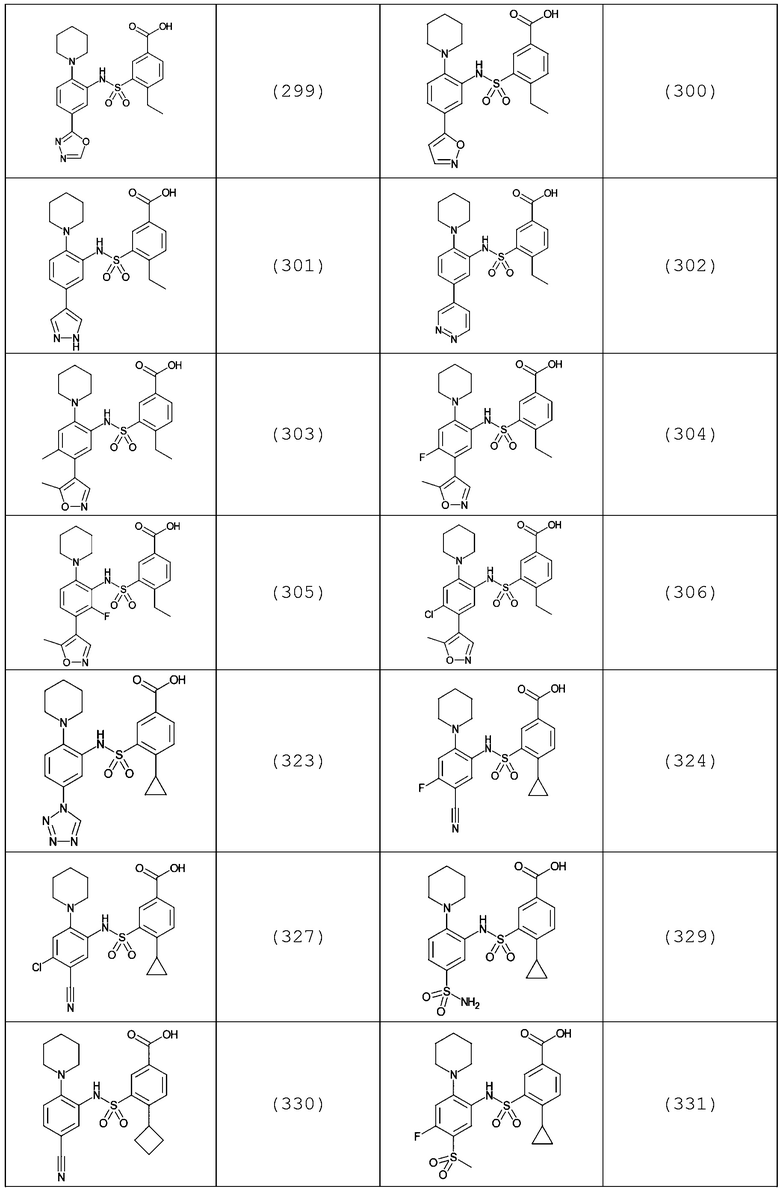
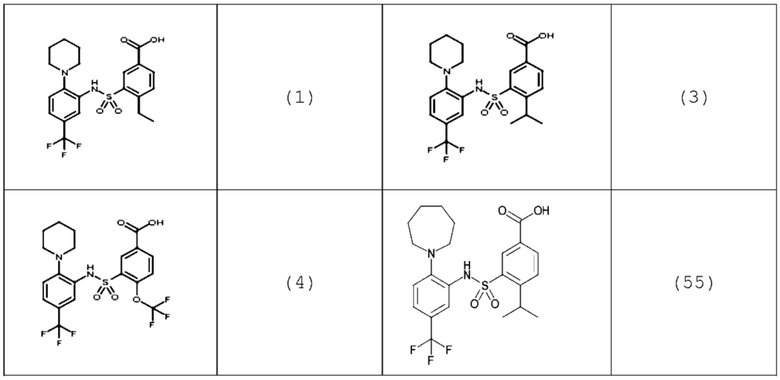
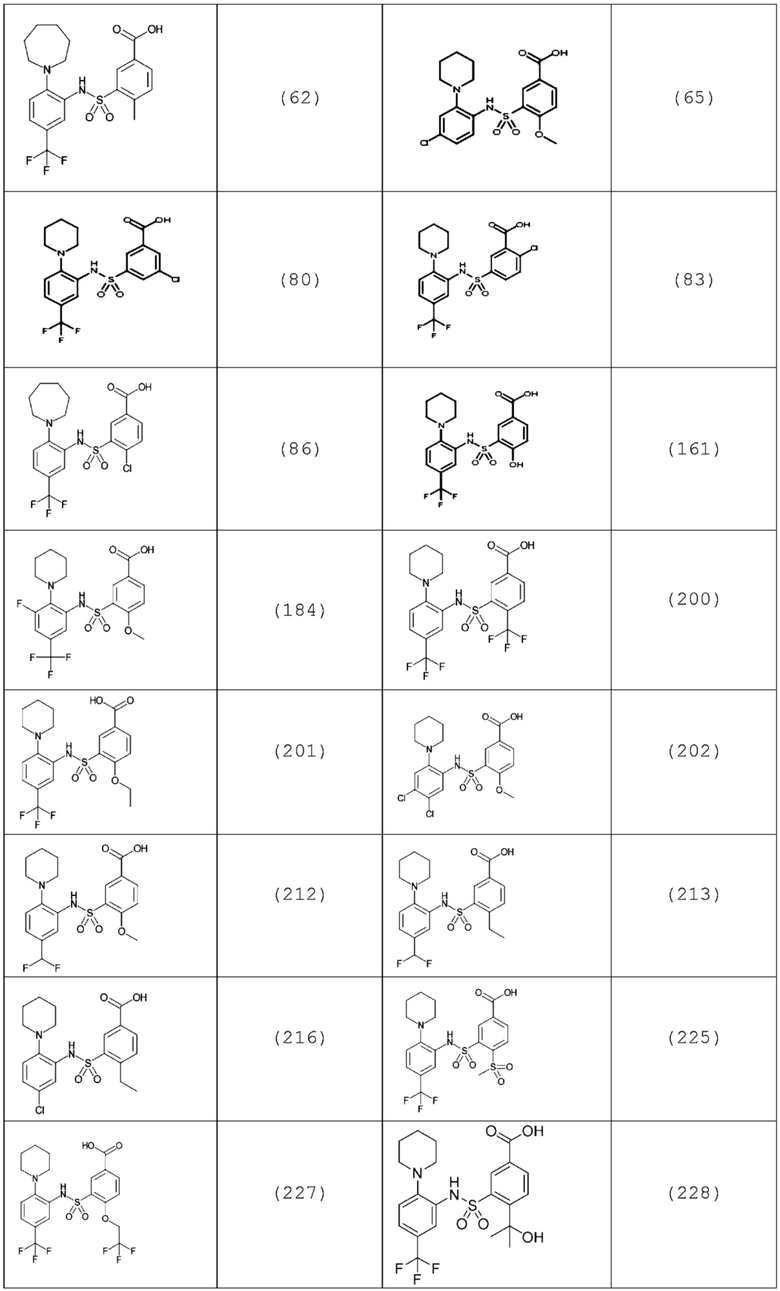
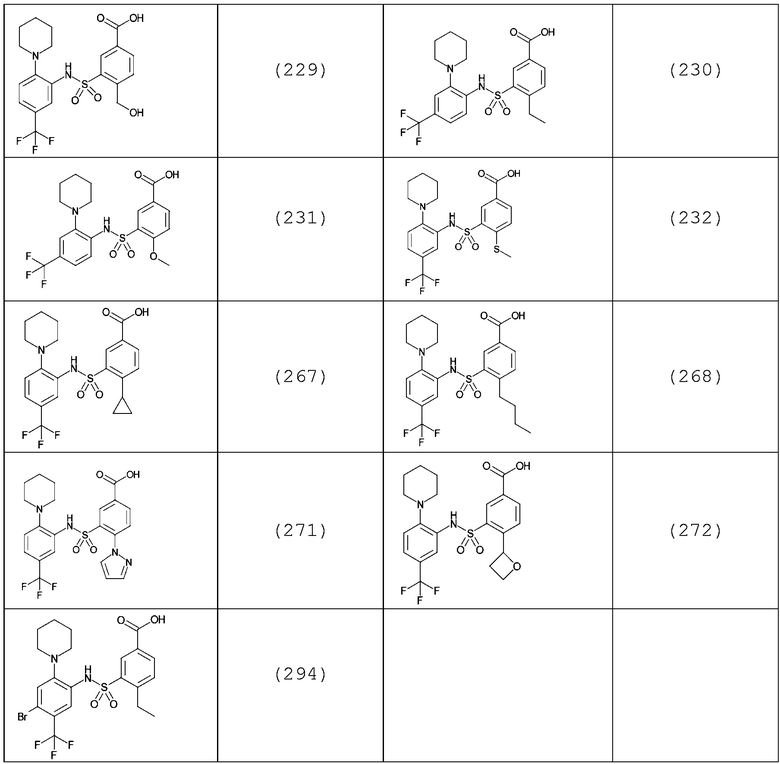
В одном предпочтительном варианте осуществления соединение формулы (Ia) выбрано из следующего:
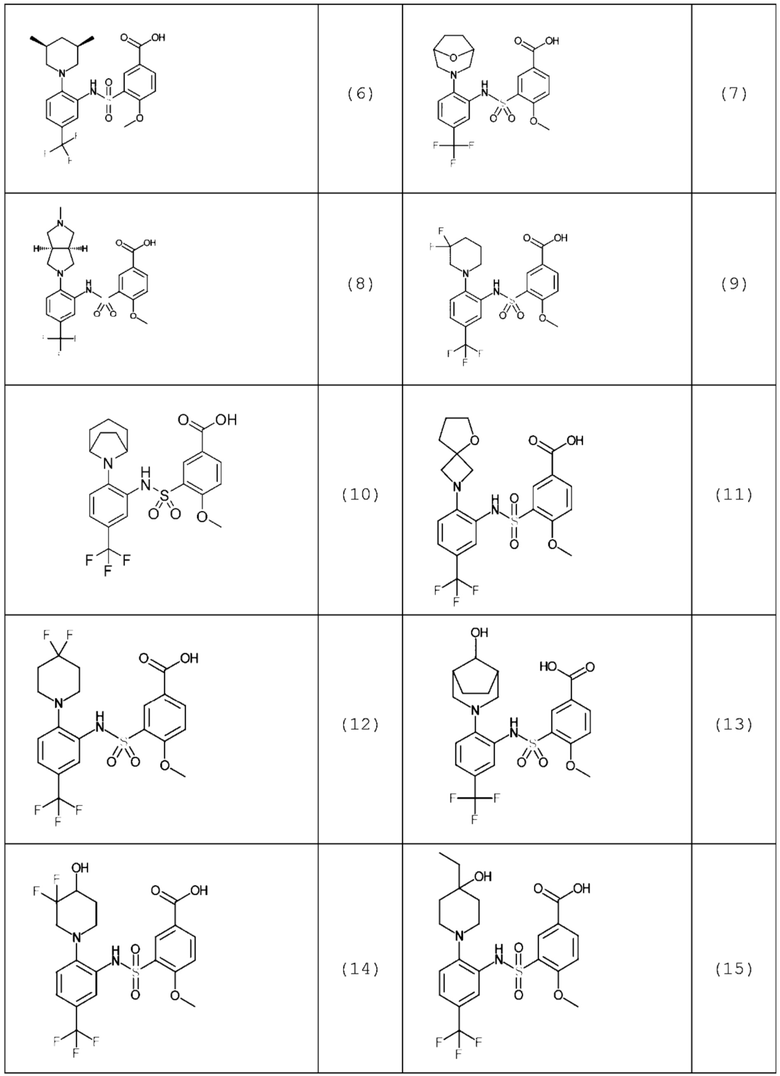
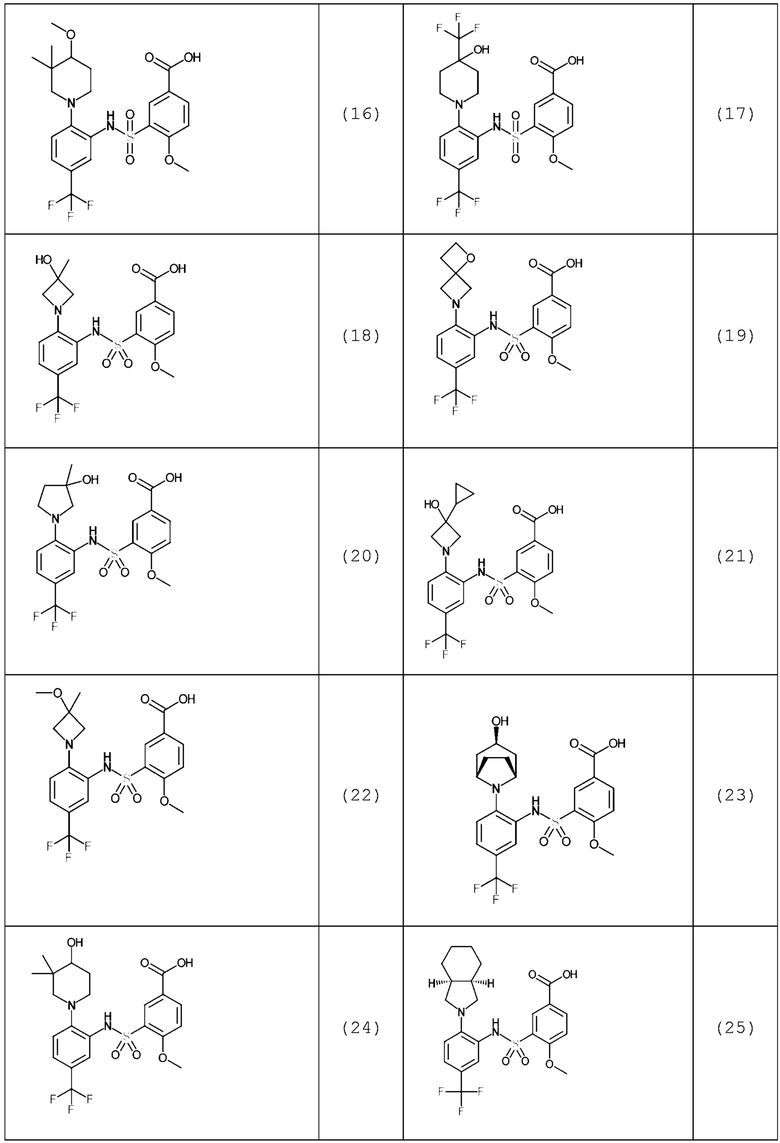
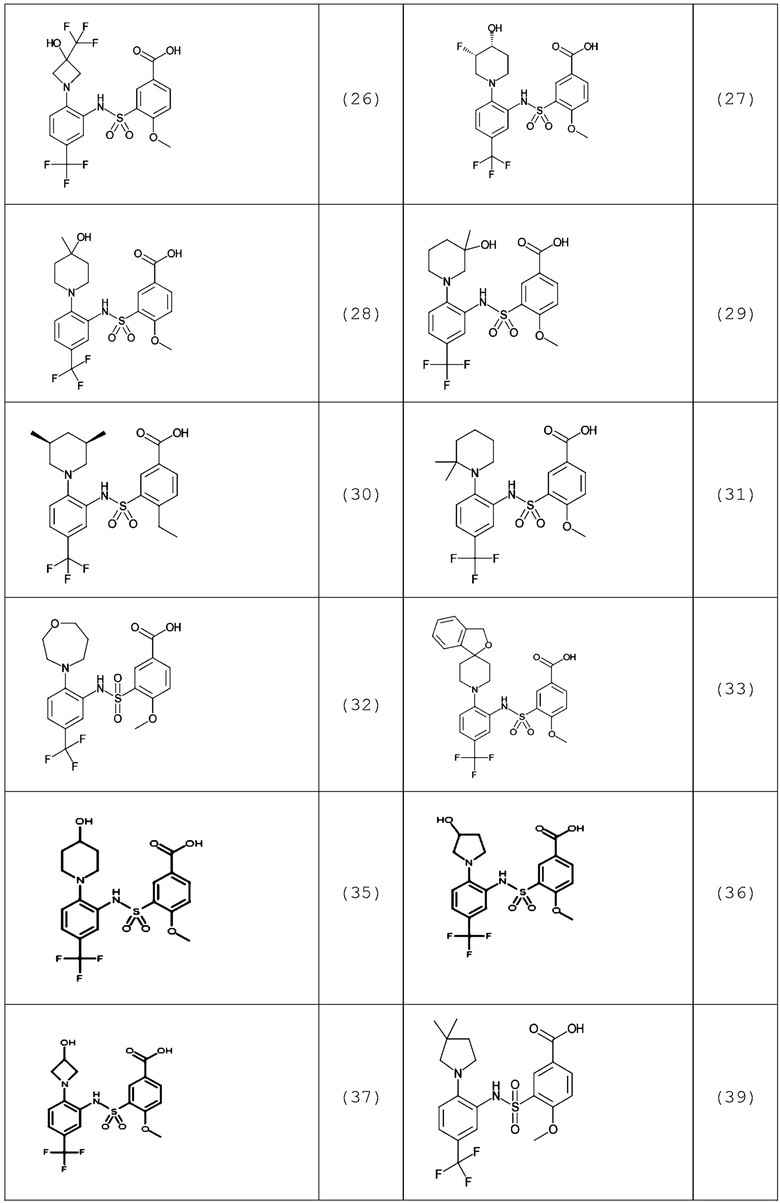
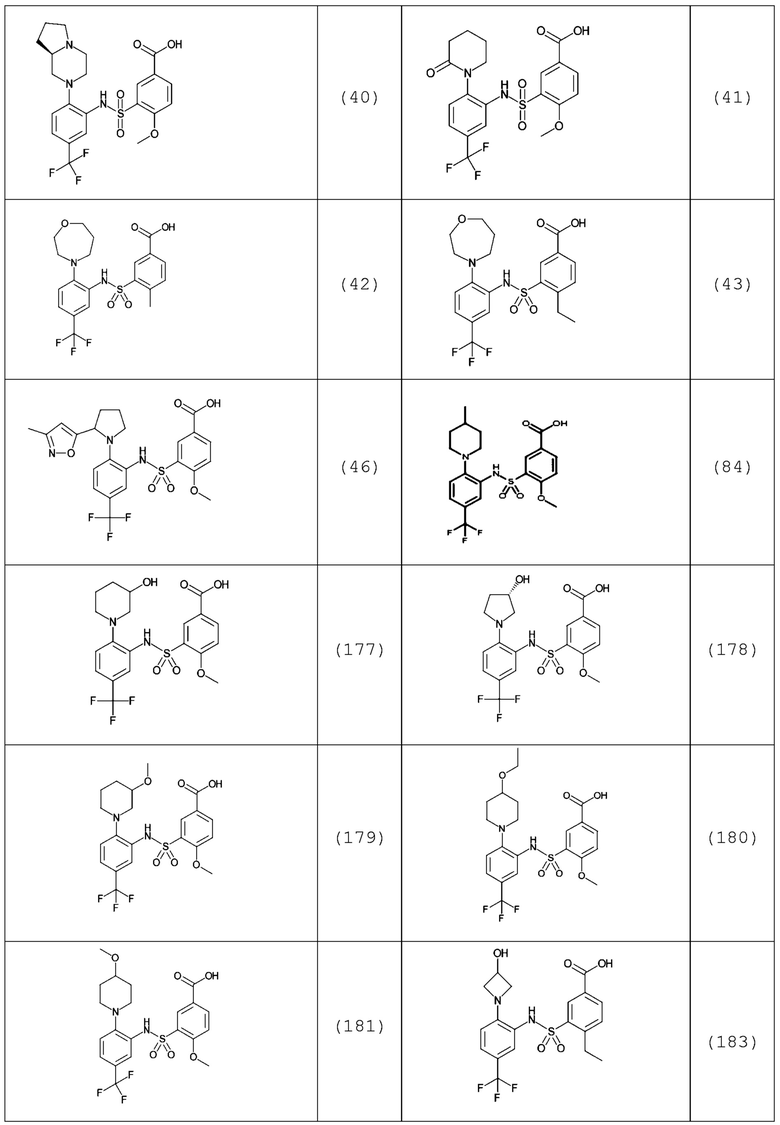
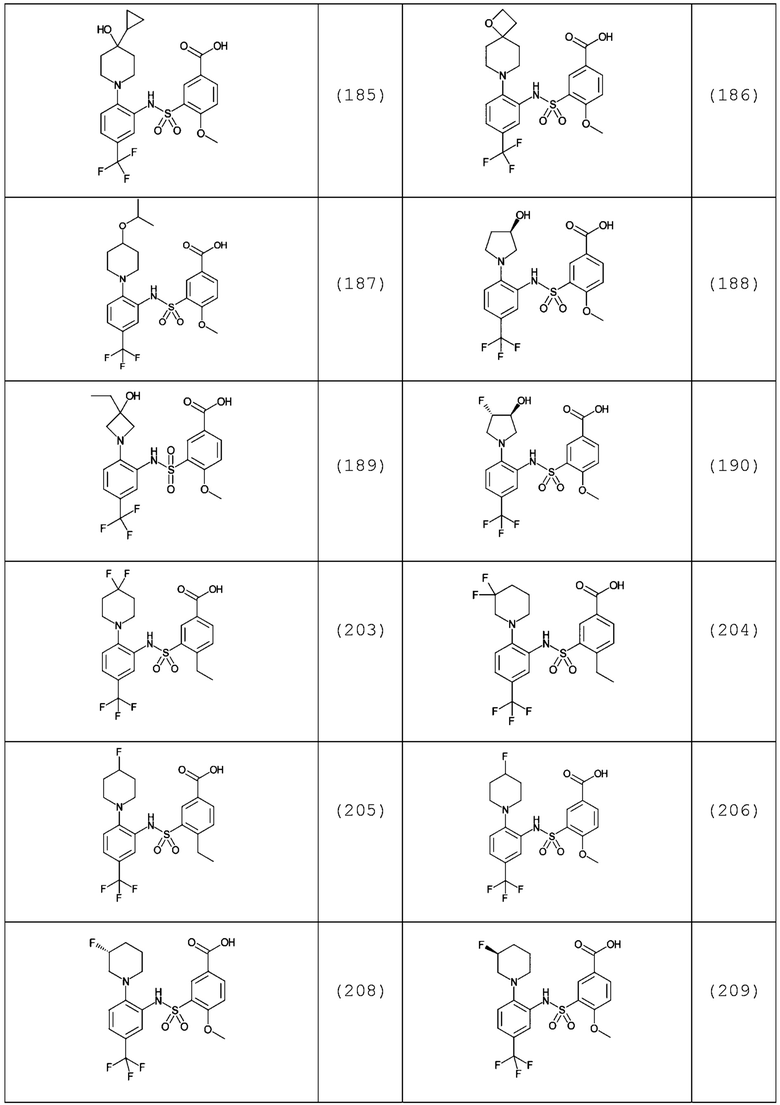
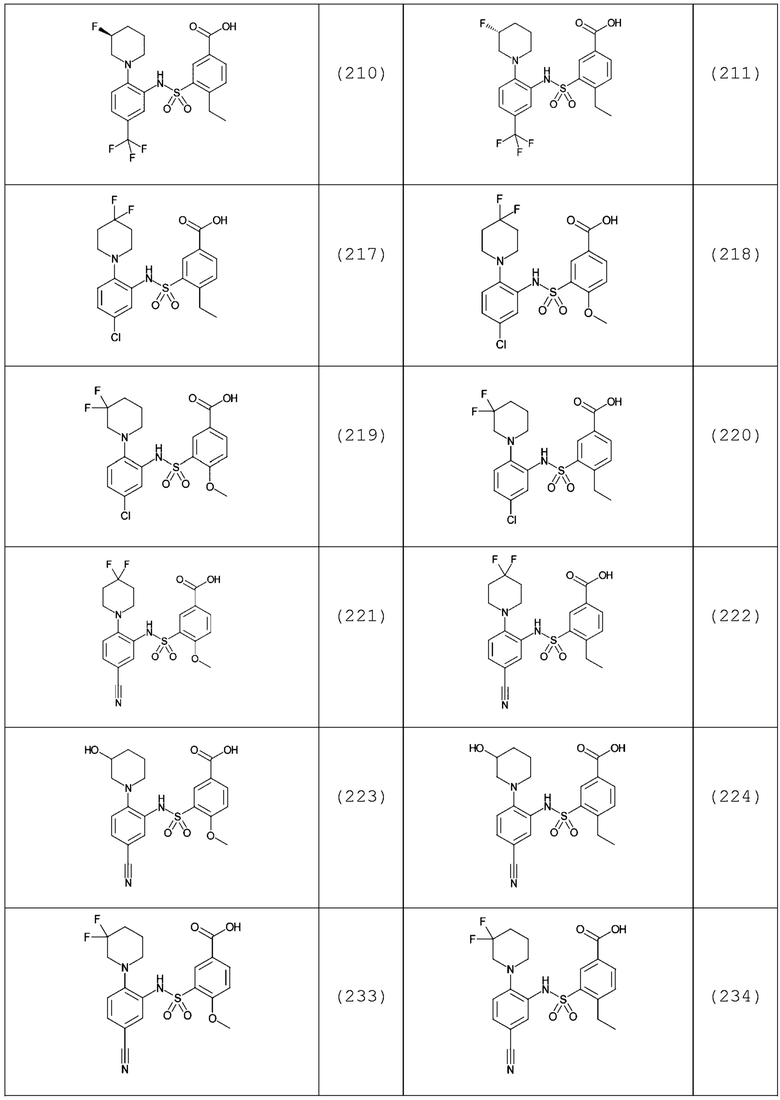
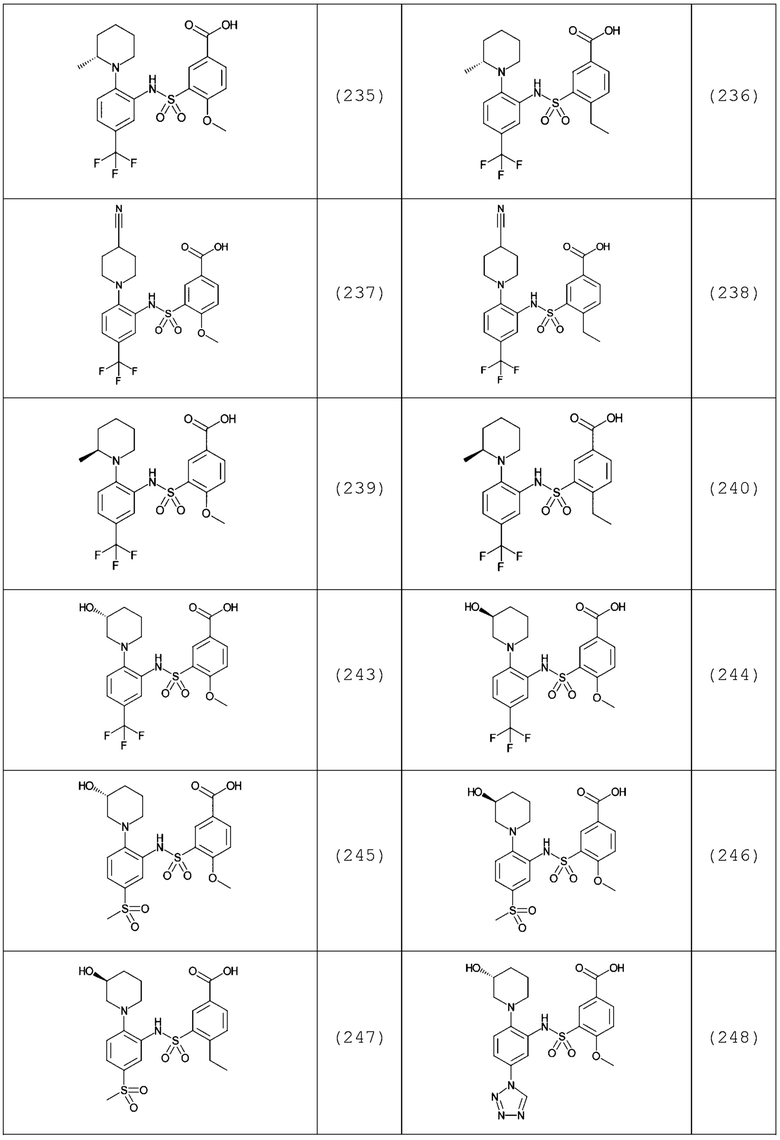
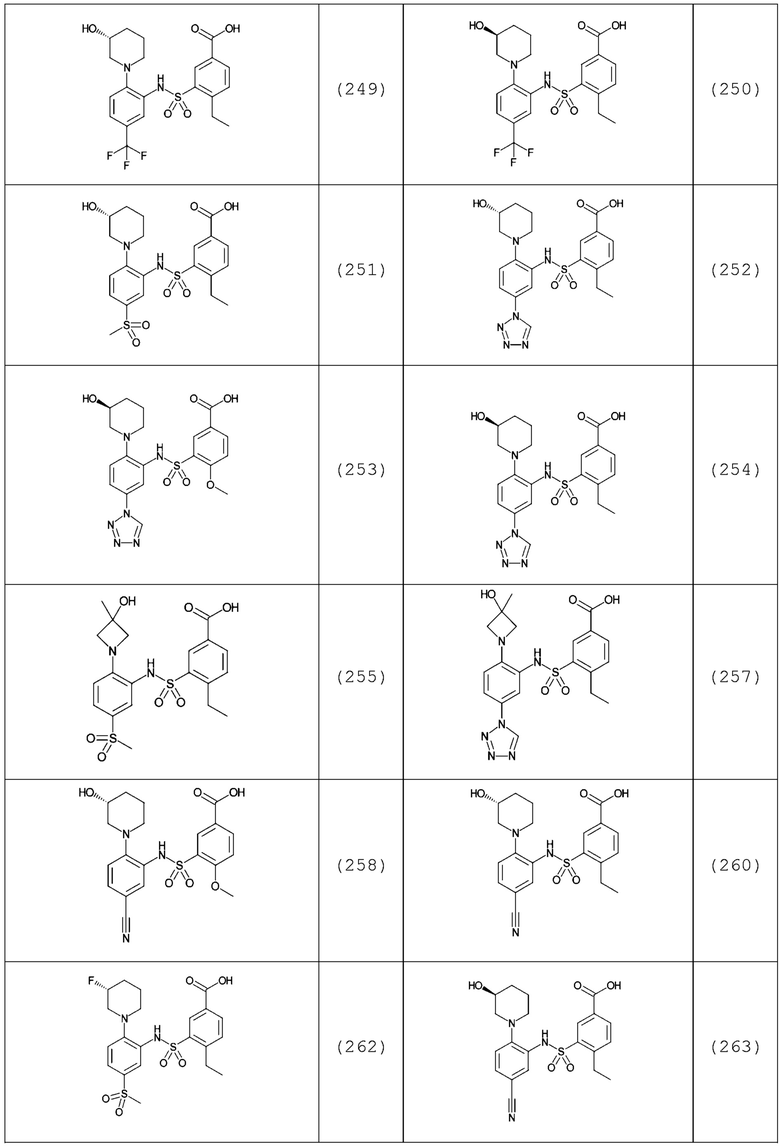
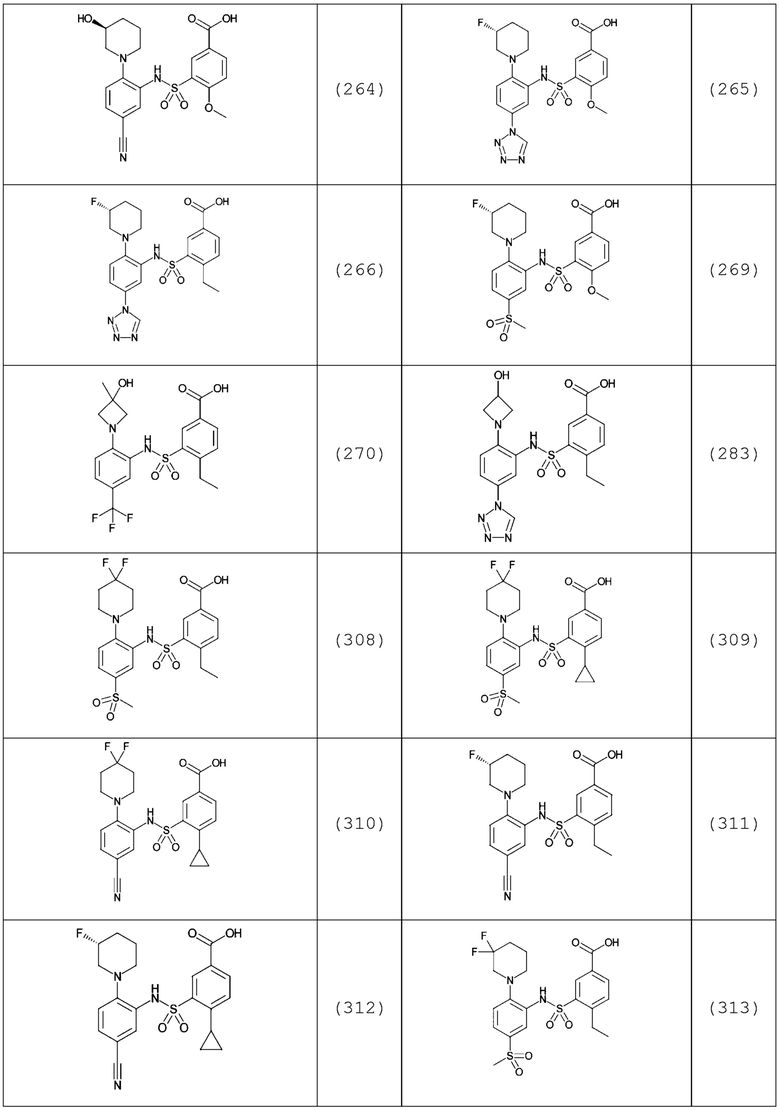
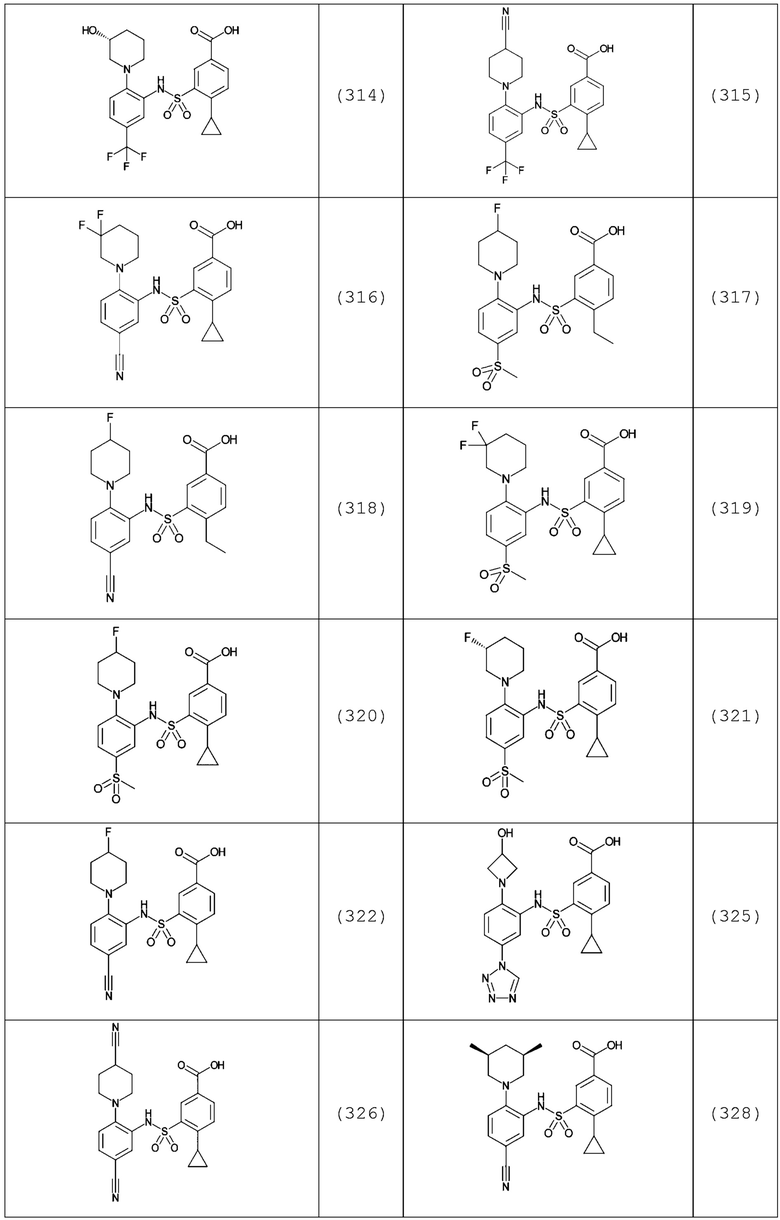
и их фармацевтически приемлемых солей и гидратов.
Соединения формулы (Ib)
Другой аспект изобретения относится к соединениям формулы (Ib) или их фармацевтически приемлемой соли или гидрату,
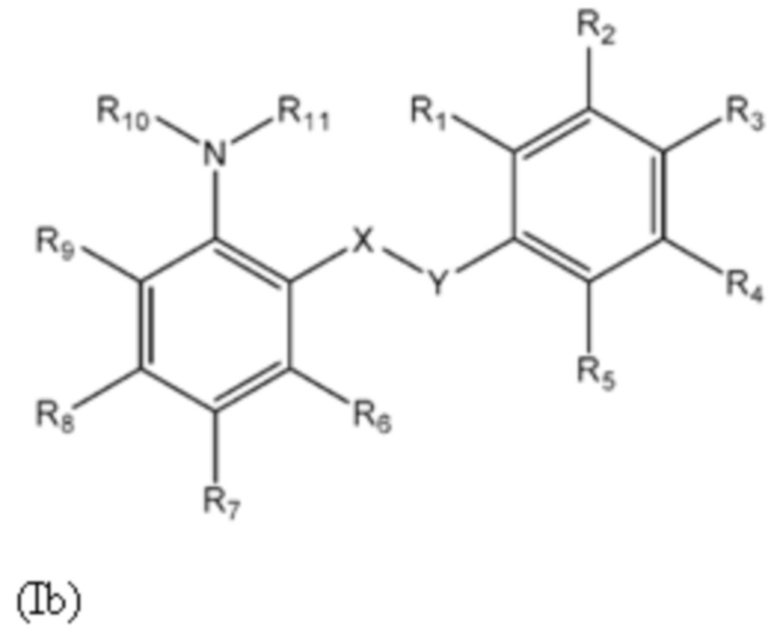
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 представляет собой тетразолильную группу;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H;
R9 представляет собой H, C1-C3-алкил или галоген;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную, пирролидинильную, азепанильную или азетидинильную группу, каждая из которых необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная пиперидинильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. В одном крайне предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют незамещенную пиперидинильную или пирролидинильную группу, более предпочтительно незамещенный пиперидинил.
Другие предпочтительные определения для групп R1, R3-11, X и y соответствуют изложенному выше для соединений формулы (Ia) и применяются с необходимыми изменениями к соединениям формулы (Ib).
В одном предпочтительном варианте осуществления соединение формулы (Ib) представляет собой:
или его фармацевтически приемлемую соль или гидрат.
Соединения формулы (Ic)
Другой аспект изобретения относится к соединениям формулы (Ic) или их фармацевтически приемлемой соли или гидрату,
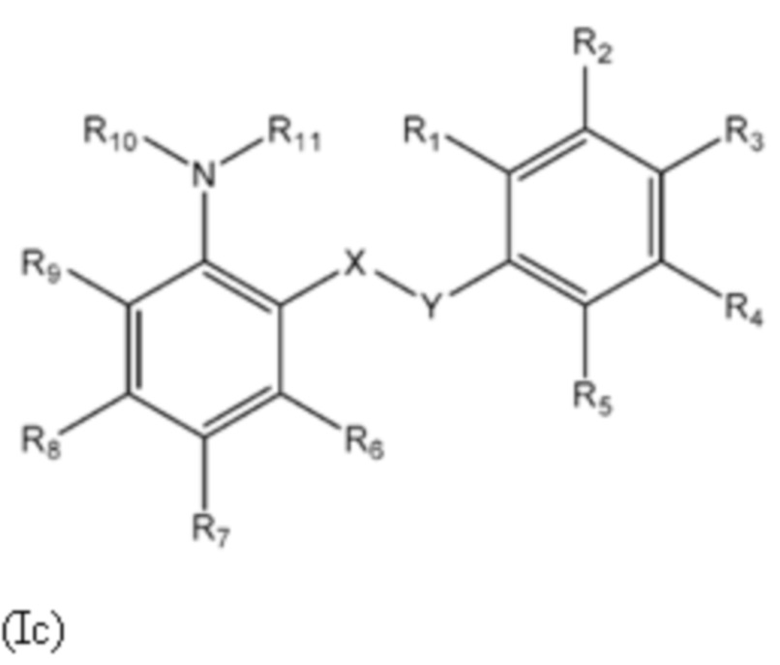
где:
X представляет собой SO2;
Y представляет собой NH;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную, пирролидинильную, азепанильную или азетидинильную группу, каждая из которых необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная пиперидинильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. В одном очень предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют незамещенную пиперидинильную или пирролидинильную группу, более предпочтительно, незамещенный пиперидинил.
Другие предпочтительные определения для групп R1-11 соответствуют изложенному выше для соединений формулы (Ia) и применяются с необходимыми изменениями к соединениям формулы (Ic).
В одном варианте осуществления соединение формулы (Ic) выбрано из следующего:
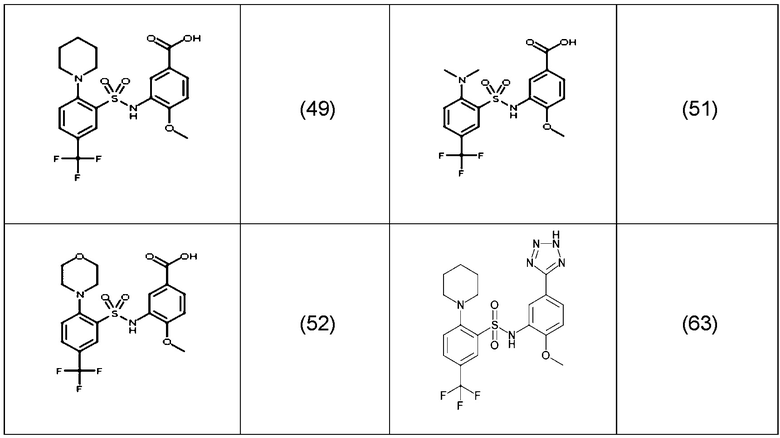
и их фармацевтически приемлемых солей и гидратов.
Соединения формулы (Id)
Другой аспект изобретения относится к соединениям формулы (Id) или их фармацевтически приемлемым солям или гидратам,
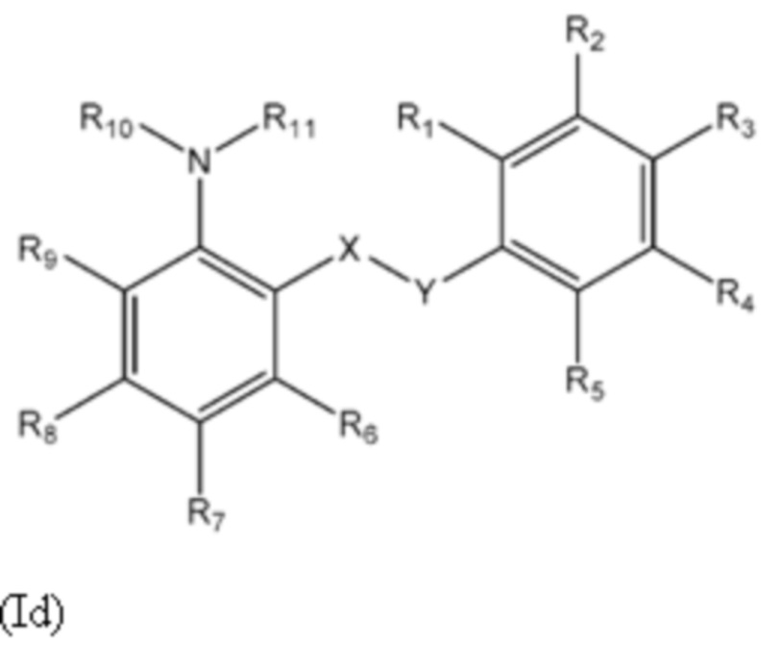
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 представляет собой CN, SO2-алкил, SO2NR13R14 или гетероарильную группу, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкил и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил.
Предпочтительные определения для заместителей X, Y, R1-6 и R8-11 соответствуют указанным выше для соединений формулы (Ia) и применяются с необходимыми изменениями к соединениям формулы (Id).
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную, пирролидинильную, азепанильную или азетидинильную группу, каждая из которых необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, выбранную из пиперидинила, морфолинила, тиоморфолинила и пиперазинила, каждый из которых необязательно замещен одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена и галогеналкила.
В одном предпочтительном варианте осуществления R10 и R11, вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, которая необязательно замещена одной или более группами, выбранными из алкила, CN, циклоалкила, OH, алкокси, галогена, галогеналкила и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила. Более предпочтительно R10 и R11, вместе с азотом, к которому они присоединены, образуют незамещенную пиперидинильную группу.
В одном предпочтительном варианте осуществления R7 представляет собой CN.
В другом предпочтительном варианте осуществления R7 представляет собой SO2-алкил, более предпочтительно SO2-Me.
В одном предпочтительном варианте осуществления R7 представляет собой SO2NR13R14, более предпочтительно SO2NH2.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, которая необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из пиридинила, тиенила, имидазолила, пиримидинила, пиразолила, пиразинила, пиридазинила, тиазолила, изотиазолила, триазинила, пирролила, фуранила, оксазолила, изоксазолила, оксадиазолила, тетразолила и триазолила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из имидазолила, пиразолила, пиразинила, пиридазинила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, тетразолила и триазолила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из 1H-имидазола-5-ила, 1H-имидазол-4-ила, 1H-имидазол-2-ила, 1H-пиррол-1-ила, 1H-пиррол-2-ила, 1H-пиррол-3-ила, 1H-пиррол-4-ила, 1H-пиррол-5-ила, 1H-пиразол-1-ила, 1H-пиразол-5-ила, 1H-пиразол-3-ила, 1H-пиразол-4-ила, оксазол-2-ила, оксазол-4-ила, оксазол-5-ила, 1H-1,2,4-триазол-3-ила, 1H-1,2,4-триазол-5-ила, 1H-1,2,4-триазол-1-ила, 1H-1,2,3-триазол-4-ила, 1H-1,2,3-триазол-5-ила, 1H-1,2,3-триазол-1-ила, тиазол-5-ила, тиазол-4-ила, тиазол-2-ила, 1H-1,2,3,4-тетразол-4-ила, 2H-1,2,3,4-тетразол-5-ила, оксазол-5-ила, оксазол-4-ила, оксазол-2-ила, изоксазол-3-ила, изоксазол-4-ила, изоксазол-5-ила, изотиазол-3-ила, изотиазол-4-ила, изотиазол-5-ила, пиридазин-3-ила, пиридазин-4-ила, пиразинил, 1,3,4-оксадиазол-2-ила, 1,3,4-оксадиазол-5-ила, 1,2,5-оксадиазол-3-ила, 1,2,5-оксадиазол-4-ила, 1,2,3-оксадиазол-4-ила, 1,2,3-оксадиазол-5-ила, 1,2,4 оксадиазол-3-ила, 1,2,4 оксадиазол-5-ила, изоксазол-5-ила, изоксазол-4-ила и изоксазол-3-ила, каждый из которых необязательно замещен одним или более заместителями, выбранными из алкила, галогена, CN, алкокси, галогеналкила и OH.
В одном крайне предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, выбранную из 1H-пиразола-5-ила, 1H-пиразол-3-ила, 1H-пиразол-4-ила, оксазол-2-ила, 1H-1,2,3-триазол-4-ила, 1H-1,2,3-триазол-5-ила, тиазол-5-ила, 1H-1,2,3,4-тетразол-4-ила, 2H-1,2,3,4-тетразол-5-ила, изоксазол-4-ила, изоксазол-5-ила, изотиазол-5-ила, пиридазин-3-ила, пиридазин-4-ила, пиразинила и 1,3,4-оксадиазол-2-ила, каждый из которых необязательно замещен одним или более заместителями, выбранными из Me, F, Cl, CN и MeO.
В одном предпочтительном варианте осуществления R7 представляет собой гетероарильную группу, которая необязательно замещена одной или более алкильными группами, предпочтительно одной или более группами Me.
В одном крайне предпочтительном варианте осуществления соединение формулы (Id) выбрано из следующего:
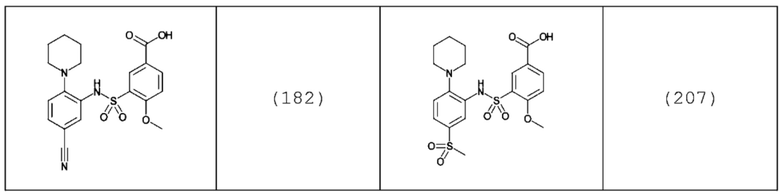
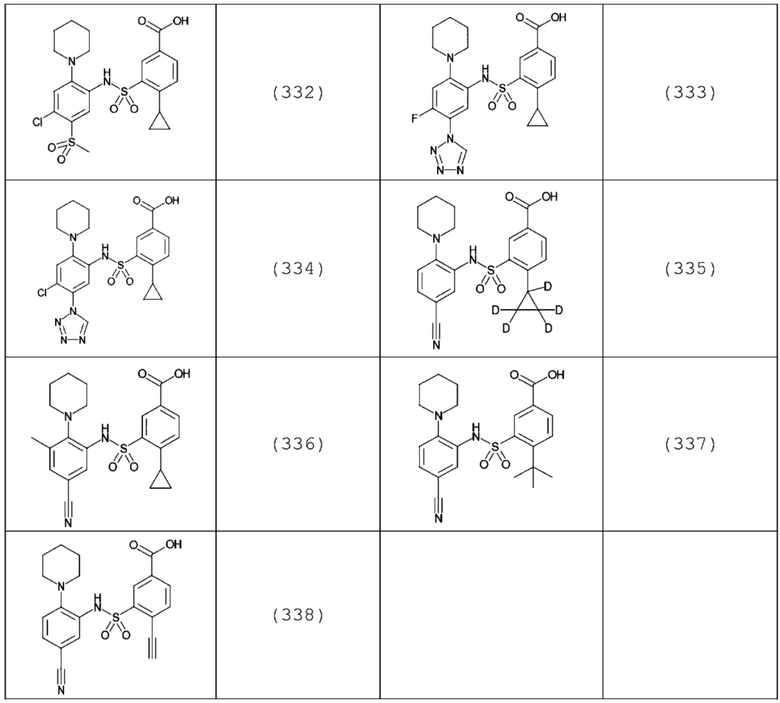
и их фармацевтически приемлемых солей и гидратов.
Другой аспект изобретения относится к соединению, выбранному из следующего:
и его фармацевтически приемлемым солям и гидратам.
ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ
Другой аспект изобретения относится к соединениям, описанным в настоящем документе, для применения в медицине. Соединения имеют конкретное применение в области онкологии и иммуноонкологии, как более подробно описано ниже.
Еще один аспект изобретения относится к соединениям, описанным в настоящем документе, для применения при лечении или предупреждении нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, воспалительного нарушения и вирусного нарушения.
В предпочтительном варианте осуществления соединение изобретения модулирует ERAP1. Более предпочтительно соединение модулирует активность процессинга клеточных антигенов ERAP1.
В одном варианте осуществления соединение ингибирует активность ERAP1. Более предпочтительно соединение ингибирует активность процессинга клеточных антигенов ERAP1.
В альтернативном варианте осуществления соединение повышает активность ERAP1.
В одном варианте осуществления соединение согласно изобретению может менять репертуар презентированных антигенов.
Один аспект изобретения относится к соединению, описанному в настоящем документе, для применения при лечении пролиферативного нарушения. Предпочтительно пролиферативным нарушением является рак или лейкоз.
Рак может быть выбран из базальноклеточной карциномы, рака желчных протоков; рака мочевого пузыря; рака кости; рака головного мозга и центральной нервной системы; рака молочной железы; рака брюшины; рака шейки матки; хориокарциномы; рака толстой и прямой кишки; рака соединительной ткани; рака пищеварительной системы; рака эндометрия; рака пищевода; рака глаза; рака головы и шеи; рака желудка (включая рак желудочно-кишечного тракта); глиобластомы; карциномы печени; гепатомы; внутриэпителиальной неоплазии; рака почки; рака гортани; лейкоза; рака печени; рака легкого (например, мелкоклеточного рака легкого, немелкоклеточного рака легкого, аденокарциномы легкого и плоскоклеточной карциномы легкого); меланомы; миеломы; нейробластомы; рака полости рта (губ, языка, полости рта и глотки); рака яичников; рака поджелудочной железы; рака предстательной железы; ретинобластомы; рабдомиосаркомы; рака прямой кишки; рака дыхательной системы; карциномы слюнной железы; саркомы; рака кожи; плоскоклеточного рака; рака желудка; рака яичка; рака щитовидной железы; рака матки или эндометрия; рака мочевыводящей системы; рака вульвы; лимфомы, включая лимфому Ходжкина и неходжкинскую лимфому, а также В-клеточную лимфому (включая низкозлокачественную/фолликулярную неходжкинскую лимфому (НХЛ); мелкоклеточную лимфоцитарную (SL) НХЛ; среднезлокачественную/фолликулярную НХЛ; диффузную НХЛ средней степени злокачественности; высокозлокачественную иммунобластную НХЛ; высокозлокачественную лимфобластную НХЛ; высокозлокачественную мелкоклеточную НХЛ с нерасщепленными ядрами; НХЛ с массивным поражением лимфоузлов; мантийноклеточную лимфому; лимфому, ассоциированную со СПИДом; и макроглобулинемии Вальденстрема; хронического лимфоцитарного лейкоза (ХЛЛ); острого лимфобластного лейкоза (ОЛЛ); волосатоклеточного лейкоза; хронического миелобластного лейкоза; а также других карцином и сарком; и посттрансплантационного лимфопролиферативного нарушения (ПТЛН), а также аномальной пролиферации сосудов, связанной с факоматозами, отеками (например, связанной с опухолями головного мозга), и синдрома Мейгса.
Без ограничения теорией, следует понимать, что модуляторы ERAP1 способны изменять по меньшей мере 10% репертуара антигенов и неоантигенов раковых клеток, при измерении с помощью иммунопептидомики и масс-спектрометрического анализа. Примерно 50% этого изменения вызвано апрегуляцией презентации некоторых антигенов и неоантигенов, тогда как остальные 50% представляют собой совершенно новые антигены и неоантигены. Оба изменения приводят к увеличению заметности опухоли для иммунной системы, что приводит к измеримым изменениям репертуара CD8+ T-клеток и статуса активации CD8+ T-клеток. Такое изменение ответа CD8+ T-клеток приводит к иммуноопосредованному устранению опухоли и потенциально может быть усилено при комбинированном применении с противоопухолевыми средствами, такими как антитела-ингибиторы контрольных точек (например, антитела против PD-1).
Без ограничения теорией, следует понимать, что модуляторы ERAP1 вызывают уничтожение раковых клеток естественными киллерами (NK-клетками) из-за нарушения взаимодействия между Ig-подобными рецепторами клеток-киллеров (KIR) или лектин-подобным рецептором CD94-NKG2A на NK-клетках с классическими или неклассическими комплексами пептидов MHC-I (pMHC-I) на раковых клетках.
В одном предпочтительном варианте осуществления нарушением является рак, и соединение увеличивает заметность раковых клеток для иммунной системы путем изменения репертуара антигенов и неоантигенов, презентируемых иммунной системе.
Другой аспект изобретения относится к способу увеличения заметности раковых клеток для иммунной системы у субъекта путем изменения репертуара антигенов и неоантигенов, презентируемых иммунной системе, где указанный способ включает введение субъекту соединения формулы (I), (Ia), (Ib), (Ic) или (Id).
В одном предпочтительном варианте осуществления соединение увеличивает ответ CD8+ T-клеток в отношении раковой клетки.
В одном предпочтительном варианте осуществления соединение согласно изобретению предназначено для применения при лечении заболевания, связанного с неконтролируемым ростом, пролиферацией и/или выживанием клеток, нарушенным клеточным иммунным ответом или нарушенным клеточным воспалительным ответом, в частности, при котором неконтролируемый рост, пролиферация и/или выживание клеток, нарушенный клеточный иммунный ответ или нарушенный клеточный воспалительный ответ подвергаются модуляции по пути ERAP1.
В одном предпочтительном варианте осуществления заболевание, связанное с неконтролируемым ростом, пролиферацией и/или выживанием клеток, нарушенным клеточным иммунным ответом или нарушенным клеточным воспалительным ответом выбрано из гемобластной опухоли, солидной опухоли и/или их метастазов.
Более предпочтительно соединение предназначено для применения при лечении нарушения, выбранного из лейкозов и миелодиспластического синдрома, злокачественных лимфом, опухолей головы и шеи, включая опухоли головного мозга и метастазы в мозг, опухоли грудной клетки, включая немелкоклеточные и мелкоклеточные опухоли легкого, желудочно-кишечные опухоли, эндокринные опухоли, опухоли молочной железы и другие гинекологические опухоли, урологические опухоли, включая опухоли почки, мочевого пузыря и предстательной железы, опухоли кожи, а также саркомы, и/или их метастазы.
Соединение может убивать раковые клетки, снижать количество пролиферирующих клеток при раке и/или уменьшать объем или размер опухоли, включающей раковые клетки. Соединение может снижать количество метастазирующих раковых клеток.
В одном варианте осуществления соединение может применяться при лечении рака у субъекта, у которого ранее уже был рак. Соединение может применяться для уменьшения вероятности рецидива рака или вероятности дальнейшего развития рака. Соединение может индуцировать неоантиген при рецидивирующем или развившемся раке, на который субъект уже обладает существующим иммунным ответом. В таком качестве соединение может увеличивать или усиливать иммунный ответ против рака.
В одном варианте осуществления соединение предназначено для применения при предупреждении рака. Соединение может применяться для профилактики против развития рака. То есть соединение может стимулировать иммунный ответ, такой как ответ после вакцинации, против рака, который может возникнуть в будущем. Соединение может стимулировать у субъекта иммунный ответ, направленный на неоантиген. Как только у субъекта развивается рак, его могут снова лечить с применением такого соединения (или другого соединения), чтобы стимулировать развитие такого же неоантигена, индуцировав, таким образом, уже существующий иммунный ответ у субъекта на указанный неоантиген, с целью лечения или предупреждения рака.
Такое же или другое соединение могут применять до и после развития рака у субъекта.
В одном варианте осуществления соединение может применяться для предупреждения рака.
В одном варианте осуществления субъект мог ранее иметь рак, может иметь случай рака в семье, может иметь высокий риск развития рака, может иметь генетическую предрасположенность к развитию рака или может подвергаться воздействию канцерогенного агента. В одном из вариантов осуществления субъект может иметь ремиссию рака.
В одном варианте осуществления предложены полученные ex vivo антигенпрезентирующие клетки, такие как дендритные клетки (ДК). Антигенпрезентирующие клетки могут быть получены ex vivo для презентации неоантигенов, таких как неоантигены, образующиеся с помощью соединения согласно настоящему изобретению. Соединение может применяться в способе получения ex vivo антигенпрезентирующей клетки, которая презентирует неоантиген, где клетка может применяться в качестве вакцины против рака.
Антигенпрезентирующую клетку, такую как дендритную клетку, могут сенсибилизировать или нагружать неоантигеном или генетически модифицировать (путем переноса ДНК или РНК) для экспрессии одного, двух или больше неоантигенов. Способы получения вакцин из дендритных клеток известны в уровне техники.
Неоантиген может быть получен из нормальной ткани субъекта, в которой ERAP1 модулируют соединением согласно изобретению. Источниками нормальной ткани могут быть фибробласты или B-клетки, например, которые можно легко размножать in vitro. альтернативе могут использовать РНК из рака, суммарную или мРНК обогащенную поли A+ РНК. Поли A+ РНК также могут амплифицировать с получением достаточного количества антигена для нагрузки ДК и, таким образом, лимитировать этап культивирования ex vivo.
В одном варианте осуществления дендритная клетка, которая была обработана соединением, как описано выше, может применяться для лечения субъекта. Дендритную клетку могут подвергать контакту с соединением ex vivo, после чего дендритную клетку могут вводить субъекту. Соединение, таким образом, может применяться in vitro или in vivo, например, либо для лечения in situ, либо для лечения ex vivo, с последующим введением обработанных клеток субъекту.
Другой аспект изобретения относится к соединению, как описано выше, для применения при лечении иммунного нарушения или для модуляции иммунного ответа. В одном предпочтительном варианте осуществления иммунное нарушение является аутоиммунным нарушением, таким как опосредованное T-клетками аутоиммунное нарушение.
Примеры аутоиммунных нарушений включают, без ограничения перечисленными: ревматоидный артрит (РА), тяжелую миастению (ТМ), рассеянный склероз (РС), системную красную волчанку (СКВ), аутоиммунный тиреоидит (тиреоидит Хашимото), болезнь Грейвса, воспалительное заболевание кишечника, аутоиммунный увеоретинит, полимиозит и некоторые типы диабета, системный васкулит, полимиозит-дерматомиозит, системный склероз (склеродермию), синдром Шегрена, анкилозирующий спондилит и связанные с ним спондилоартропатии, ревматизм, гиперчувствительный пневмонит, аллергический бронхолегочный аспергиллез, неорганические пылевые пневмокониозы, саркоидоз, аутоиммунную гемолитическую анемию, иммунологические тромбоцитарные нарушения, криопатии, такие как криофибриногенемию, псориаз, болезнь Бехчета, birdshot (от англ. выстрел дробью) хориоретинопатию и аутоиммунные полиэндокринопатии.
Полиморфизмы в гене ERAP1, влияющие на ферментативную активность ERAP1, сильно связаны с повышенным риском аутоиммунитета, в том числе таких заболеваний, как анкилозирующий спондилит, псориаз, болезнь Бехчета и birdshot хориоретинопатия [11]. Варианты ERAP1, которые снижают ферментативную активность ERAP1, защищают от развития таких заболеваний, тогда как варианты, которые по имеющимся сообщениям повышают активность, связаны с увеличенным риском заболевания [12]. Это дает основание предполагать, что модуляция активности ERAP1 могла бы являться эффективным лечением аутоиммунных заболеваний.
Таким образом, в одном предпочтительном варианте осуществления иммунное нарушение выбрано из анкилозирующего спондилита, псориаза, болезни Бехчета и birdshot хориоретинопатии.
В одном предпочтительном варианте осуществления иммунным нарушением является анкилозирующий спондилит. Анкилозирующий спондилит (АС) является типом артрита, при котором присутствует длительное воспаление суставов позвоночника. Как правило, также затронуты суставы, где позвоночник соединен с тазом. Иногда поражаются другие суставы, такие как плечи или бедра. Заболевание поражает от 0,1% до 1,8% людей, причем заболевание начинает проявляться, как правило, в молодом возрасте. Несмотря на то, что причина анкилозирующего спондилита неизвестна, он включает комбинацию наследственных факторов и факторов внешней среды. Больше чем 90% больных имеют специфический человеческий лейкоцитарный антиген, известный как антиген HLA-B27 [13]. Кроме того, некоторые варианты ERAP1, в сочетании с HLA-B27, четко связаны либо с повышенным, либо с пониженным риском заболевания, подтверждая явную роль модулируемой презентации антигенов при заболевании [18]. Полного излечения анкилозирующего спондилита добиться не удается, и существующие методы лечения служат лишь для облегчения симптомов и предотвращения ухудшения. Лекарственные средства, применяемые в настоящее время, включают НПВС, стероиды, БПРП, такие как сульфасалазин, а также биологические средства, такие как инфликсимаб.
В одном предпочтительном варианте осуществления иммунным нарушением является болезнью Бехчета (ББ). Болезнь Бехчета (ББ) является типом воспалительного заболевания, которое поражает различные части тела. Наиболее распространенные симптомы включают болезненные язвы в полости рта, генитальные язвы, воспаление частей глаза и артрит. Причина точно не определена, и хотя факторы внешней среды играют некую роль, генетические исследования выявили повышенный риск появления болезни у пациентов, несущих HLA-B51, в сочетании с определенными вариантами ERAP1 [19]. Эта болезнь, прежде всего, характеризуется аутовоспалением кровеносных сосудов, поэтому иногда ее классифицируют как аутовоспалительное заболевание. В настоящее время лечения болезни Бехчета не существует, однако симптомы можно контролировать лекарственными препаратами, которые уменьшают воспаление в пораженных частях тела, например, кортикостероидами, иммунодепрессантами или биопрепаратами, которые воздействуют на биологические процессы, вовлеченные в процесс воспаления. В одном предпочтительном варианте осуществления иммунным нарушением является birdshot хориоретинопатия. Birdshot хориоретинопатия, также известная как Birdshot увеит или HLA-A29 увеит, является редкой формой двустороннего заднего увеита, поражающего глаза. Это заболевание вызывает тяжелое, прогрессирующее воспаление сосудистой оболочки и сетчатки глаза. Симптомы включают "мушки" в глазах, нечеткое зрение, фотопсию (яркие вспышки в глазах), потерю цветного видения и никталопию. Birdshot хориоретинопатию считают аутоиммунным заболеванием. Это заболевание имеет четкую связь с человеческим лейкоцитарным антигеном (HLA) гаплотипа A29. Это указывает на роль T-лимфоцитов в патогенезе. Birdshot хориоретинопатия связана с IL-17, специфическим цитокином клеток TH17, играющих важную роль в аутоиммунитете [15, 16]. В полногеномном исследовании ассоциаций определили HLA-A29:02 в качестве основного фактора риска и идентифицировали, что с birdshot хориоретинопатией связан как ERAP1, так и ERAP2 [17, 20]. Генетические варианты в локусах ERAP1 и ERAP2 модулируют ферментную активность, а также экспрессию мРНК и белка. ERAP2 является аминопептидазой, которая, вместе с ERAP1, отрезает пептиды в эндоплазматическом ретикулуме и загружает эти пептиды на молекулы HLA для презентации T-клеткам иммунной системы.
В одном предпочтительном варианте осуществления иммунным нарушением является псориаз. Псориаз является хроническим кожным заболеванием, при котором клетки кожи быстро нарастают на поверхности кожи, образуя чешуйки и красные пятна, которые сопровождаются зудом и иногда являются болезненными. Причина четко не определена, но включает факторы внешней среды и наследственные факторы. С риском заболевания сильно ассоциирован HLA-C06, а также варианты в ERAP1, возможно в сочетании с HLA-C06 [21]. Полного излечения псориаза добиться не удается, и современное лечение служит лишь для облегчения симптомов и предотвращения ухудшения. Лекарственные средства, применяемые в терапии, включают стероиды, метотрексат, сульфасалазин и биопрепараты, такие как этанерцепт.
Другой аспект изобретения относится к соединению, как описано выше, для применения при лечении или предупреждении вирусного нарушения. Модуляторы ERAP1, такие как соединения, описанные в настоящем документе, способны менять антигенный репертуар различных вирусов, приводя к распознаванию и разрушению инфицированных вирусом клеток. Таким образом, модуляторы ERAP1 имеют потенциальные терапевтические применения при лечении вирусной инфекции и заболеваний. ERAP1 модулирует некоторые вирусные антигены, включая антигены вируса папилломы человека (ВПЧ), цитомегаловируса человека (ЦМВ), гепатита С (ВГС) и вируса иммунодефицита человека (ВИЧ) [8, 9, 10]. Кроме того, нокдаун ERAP1 в инфицированных ВПЧ клетках изменяет репертуар презентируемых антигенов ВПЧ, приводя к повышению распознавания CD8+ T-клетками [8].
В одном предпочтительном варианте осуществления вирусным нарушением является вирусное заболевание или вирусная инфекция, выбранные из ВИЧ, ВПЧ, ЦМВ и ВГС.
В одном предпочтительном варианте осуществления вирусным нарушением является ВИЧ.
В одном предпочтительном варианте осуществления вирусным нарушением является ВПЧ.
В одном предпочтительном варианте осуществления вирусным нарушением является ЦМВ.
В одном предпочтительном варианте осуществления вирусным нарушением является ВГС.
Другой аспект изобретения относится к соединению, как описано выше, для применения при лечении или предупреждении артериальной гипертензии.
Другой аспект относится к соединению, как описано в настоящем документе, для применения в предупреждении или лечении нарушения, вызванного, ассоциированного с или сопровождаемого нарушением активности ERAP1.
Другой аспект относится к соединению, как описано в настоящем документе, для применения в предупреждении или лечении ERAP1-ассоциированного заболевания или нарушения.
Еще один аспект относится к применению соединения, как описано в настоящем документе, в изготовлении лекарственного средства для предупреждения или лечения нарушения, вызванного, ассоциированного с или сопровождаемого любым нарушением активности ERAP1.
При использовании в настоящем документе фраза "изготовление лекарственного средства" включает применение компонентов согласно изобретению непосредственно в качестве лекарственного средства в дополнение к их применению в любой стадии изготовления такого лекарственного средства.
Другой аспект относится к применению соединения, как описано выше, в изготовлении лекарственного средства для лечения или предупреждения нарушения, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
Еще один аспект относится к применению соединения, как описано в настоящем документе, в изготовлении лекарственного средства для предупреждения или лечения ERAP1-ассоциированного заболевания или нарушения.
Другой аспект изобретения относится к способу лечения ERAP1-ассоциированного заболевания или нарушения у субъекта. Способ согласно данному аспекту настоящего изобретения осуществляют путем введения нуждающемуся в этом субъекту терапевтически эффективного количества соединения согласно настоящему изобретению, как описано выше, либо непосредственно, либо, что более предпочтительно, в качестве части фармацевтической композиции, в смеси, например, с фармацевтически приемлемым носителем, как подробно описано ниже.
Еще один аспект изобретения относится к способу лечения субъекта с патологическим состоянием, которое облегчается при модуляции ERAP1, где способ включает введение субъекту терапевтически эффективного количества соединения согласно изобретению.
Другой аспект относится к способу лечения патологического состояния, которое облегчается при модуляции ERAP1, где способ включает введение субъекту терапевтически эффективного количества соединения согласно изобретению.
Предпочтительно субъектом является млекопитающее, более предпочтительно человек.
Термин "способ" относится к методам, путям, методикам и процедурам для выполнения данной задачи, в том числе, но без ограничения, к таким методам, путям, методикам и процедурам, которые известны или с легкостью могут быть разработаны на основе известных методов, путей, методик и процедур практикующими специалистами в области химических, фармакологических, биологических, биохимических и медицинских наук.
В настоящем документе термин "лечение" включает устранение, существенное ингибирование, замедление или обратное изменение направления прогрессирования заболевания или нарушения, существенное улучшение клинических симптомов заболевания или нарушения или по существу предотвращение появления клинических симптомов заболевания или нарушения.
В настоящем документе термин "предупреждение" относится к способу предотвращения первичного приобретения организмом нарушения или заболевания.
Термин "терапевтически эффективное количество" относится к такому количеству вводимого соединения, которое приведет к облегчению в некоторой степени одного или более симптомов заболевания или нарушения, подвергаемого лечению.
Для любого соединения, применяемого в настоящем изобретении, терапевтически эффективное количество, также именуемое в настоящем документе терапевтически эффективной дозой, может быть первоначально оценено на основе анализов на культурах клеток. Например, доза может быть установлена в моделях на животных с достижением диапазона циркулирующих концентраций, который включает значения IC50 или IC100, определенные в культуре клеток. Такая информация может использоваться для более точного определения полезных доз у человека. Начальные дозы также могут оценивать на основе данных in vivo. При использовании этих первоначальных рекомендаций средний специалист в данной области сумеет определить эффективную дозу у человека.
Кроме того, токсичность и терапевтическая эффективность соединений, описанных в настоящем документе, можно определить с помощью стандартных фармацевтических процедур на культурах клеток или экспериментальных животных, например, путем определения LD50 и ED50. Соотношение доз между токсическим и терапевтическим действием является терапевтическим индексом и может быть выражено как отношение между LD50 и ED50. Соединения с высокими терапевтическими индексами являются предпочтительными. Данные, полученные в результате таких анализов на культурах клеток и исследований на животных, можно использовать для определения диапазона доз, которые являются нетоксичными для применения у человека. Доза таких соединений предпочтительно находится в границах диапазона циркулирующих концентраций, который включает ED50 с низкой токсичностью или отсутствием токсичности. Доза может изменяться в этом диапазоне в зависимости от применяемой лекарственной формы и используемого пути введения. Точный состав, путь введения и дозу может подбирать лечащий врач с учетом состояния пациента (см., например, Fingl et al, 1975, The Pharmacological Basis of Therapeutics, глава 1, страница 1).
Величину доз и интервал между введением можно регулировать индивидуально, чтобы обеспечить уровни активного соединения в плазме крови, достаточные для поддержания терапевтического эффекта. Обычные дозы для пациентов при пероральном введении находятся в диапазоне от приблизительно 50-2000 мг/кг/день, обычно от приблизительно 100-1000 мг/кг/день, предпочтительно от приблизительно 150-700 мг/кг/день и наиболее предпочтительно от приблизительно 250-500 мг/кг/день. Предпочтительно терапевтически эффективные уровни в сыворотке будут достигаться при ежедневном введении нескольких доз. В случаях местного введения или селективного поглощения эффективная локальная концентрация лекарственного средства может быть не связана с концентрацией в плазме. Специалист в данной области сумеет оптимизировать терапевтически эффективные дозы при местном применении без ненужных экспериментов. При использовании в настоящем документе термин "заболевание или нарушение, связанное с ERAP1" относится к заболеванию или нарушению, характеризуемому нарушенной активностью ERAP1. Нарушенная активность относится либо к повышению, либо к снижению активности ERAP1 по сравнению с ERAP1 дикого типа (номер в Uniprot Q9NZ08), вызванному изменением последовательности белка ERAP1, при измерении с помощью ферментного или клеточного анализа. Нарушенная активность также может быть связана с повышенной экспрессией ERAP1 в пораженной ткани по сравнению со здоровой окружающей тканью.
Предпочтительные заболевания или нарушения, при предотвращении которых могут применяться соединения, описанные в настоящем документе, включают пролиферативные нарушения, вирусные нарушения, иммунные нарушения и воспалительные нарушения, как описано выше.
Таким образом, в настоящем изобретении также предложено применение соединений, как определено в настоящем документе, для производства лекарственных средств для лечения заболеваний, при которых желательна модуляция ERAP1. Такие заболевания включают пролиферативные нарушения, вирусные нарушения, иммунологические нарушения и воспалительные нарушения, как описано выше.
В одном предпочтительном варианте осуществления соединение активирует превращение (L)-лейцин-7-амидо-4-метилкумарина (L-AMC) при воздействии ERAP1 в (L)-лейцин и флуоресцентную молекулу 7-амино-4-метилкумарин. Тогда как такой же анализ также позволяет идентифицировать ингибиторы расщепления ERAP1 амидной связи в L-AMC, в рамках настоящей заявки данный анализ именуется "анализом активатора L-AMC". Активность любого активатора вычисляют и выражают как концентрацию активатора, требуемую для повышения ферментной активности ERAP1 на 50% по сравнению с ее исходным уровнем (т.е. EC50).
В одном предпочтительном варианте осуществления соединение демонстрирует значение EC50 в анализе активатора L-AMC меньше чем приблизительно 25 мкМ. Более предпочтительно соединение демонстрирует значение EC50 в анализе активатора L-AMC меньше чем приблизительно 10 мкМ, более предпочтительно меньше чем приблизительно 5 мкМ, еще более предпочтительно меньше чем приблизительно 1 мкМ, еще более предпочтительно меньше чем приблизительно 0,1 мкМ, еще более предпочтительно меньше чем приблизительно 0,01 мкМ.
В одном предпочтительном варианте осуществления соединение ингибирует способность ERAP1 гидролизовать декапептидный субстрат WRVYEKCdnpALK. Этот пептид обладает минимальной флуоресценцией, поскольку флуоресценцию N-концевого остатка триптофана в пептиде тушит остаток динитрофенола (DNP). Однако при гидролизе ERAP1 N-концевой амидной связи триптофан высвобождается, снимая внутреннее тушение, и реакцию контролируют по увеличению флуоресценции триптофана в течение анализа. В рамках настоящей заявки данный анализ именуется "анализом ингибирования 10мера", при этом активность соединения вычисляют и выражают как IC50, как будет известно специалисту в данной области.
В одном предпочтительном варианте осуществления соединение демонстрирует значение IC50 в анализе 10мера меньше чем приблизительно 25 мкМ. Более предпочтительно соединение демонстрирует значение IC50 в анализе 10мера меньше чем приблизительно 10 мкМ, более предпочтительно меньше чем приблизительно 5 мкМ, еще более предпочтительно меньше чем приблизительно 1 мкМ, еще более предпочтительно меньше чем приблизительно 0,1 мкМ, еще более предпочтительно меньше чем приблизительно 0,01 мкМ.
Терапевтическое применение соединений Формулы I
Другой аспект изобретения относится к соединениям формулы (I) или их фармацевтически приемлемой соли или гидрату,
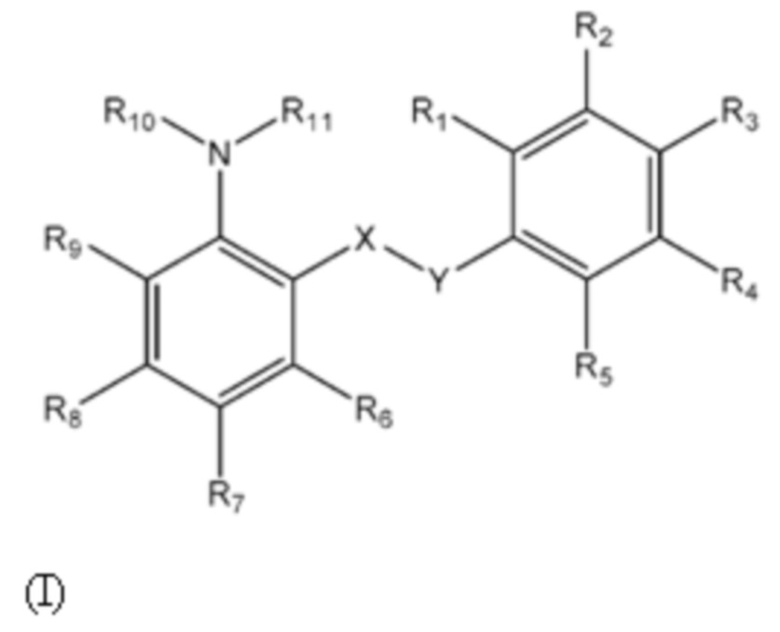
где:
группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H или алкил;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, алкила, алкинила, алкенила, галогеналкила, SO2-алкила, Cl, алкокси, OH, CN, гидроксиалкила, алкилтио, гетероарила, циклоалкила, гетероциклоалкила и галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, галогеналкила, Cl, F, SO2-алкила, SO2NR13R14, гетероарила и алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из алкила, галогена, алкокси, CN, галогеналкила и OH;
R8 выбран из H, алкила, галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой H или алкил;
R11 представляет собой алкил, который необязательно замещен одним или более заместителями, выбранными из NH2, OH и NHCO2R12, где R12 представляет собой алкил; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один или два углерода в моноциклической гетероциклоалкильной группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN, OH, галогена и гетероарила, где указанная гетероарильная группа в свою очередь необязательно дополнительно замещена одной или более группами, выбранными из галогена и алкила; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 8, 9 или 10-членную бициклическую гетероциклоалкильную группу, где один или два углерода в бициклическом гетероциклоалкильном кольце необязательно заменены группой, выбранной из O, NH, S и CO, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из алкила, CN и галогена; или
R10 и R11, вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один или два углерода в бициклической группе необязательно замещены группой, выбранной из O, NH, S и CO, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из алкила, CN, галогена и гетероарила, или указанная бициклическая группа необязательно конденсирована с 5 или 6-членной арильной или гетероарильной группой; и
каждый R13 и R14 независимо представляет собой H или алкил;
для применения при лечении или предупреждении нарушения, выбранного из пролиферативного нарушения, аутоиммунного нарушения, вирусного нарушения и воспалительного нарушения.
Предпочтительные определения для групп X, Y и R1-11 соответствуют определениям, представленным выше для соединений формулы (Ia), и применяются с необходимыми изменениями к соединениям формулы (I). Детали подходящих пролиферативных нарушений, аутоиммунных нарушений, вирусных нарушений и воспалительных нарушений соответствуют представленному выше под заголовком "Терапевтические применения".
В одном предпочтительном варианте осуществления соединение формулы (I) для применения, как описано выше, выбрано из следующего:
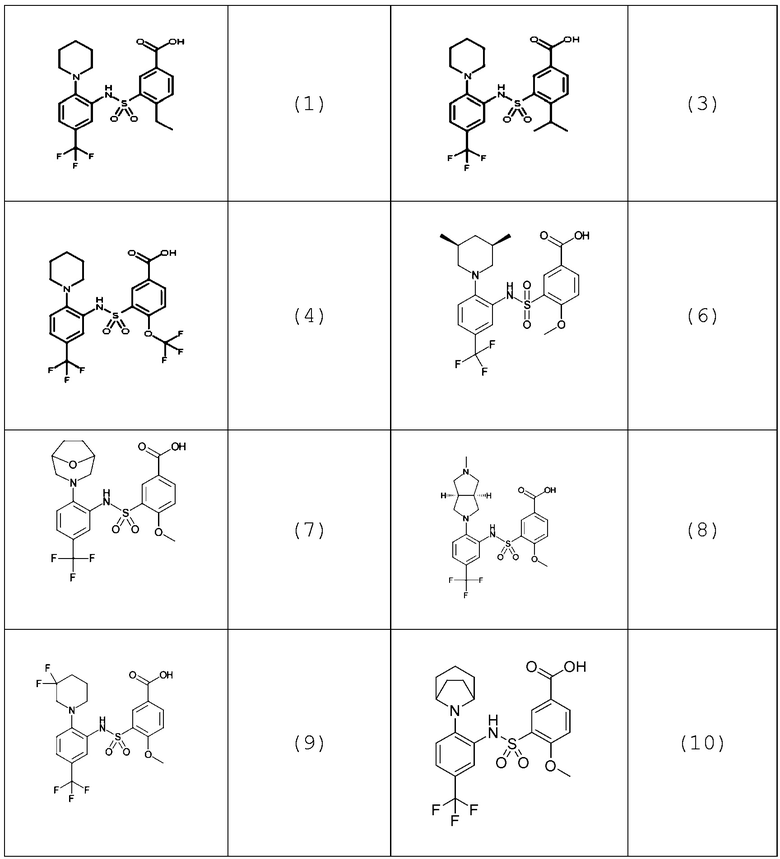
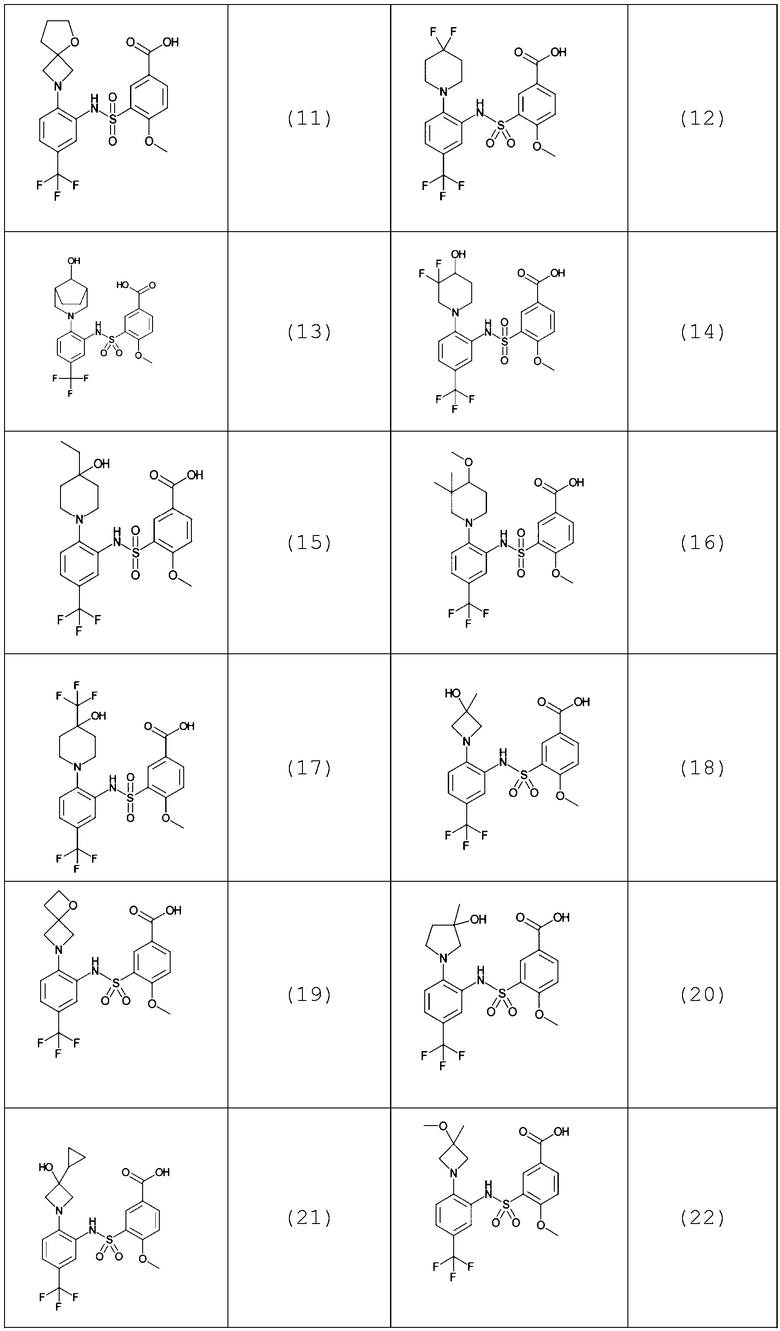
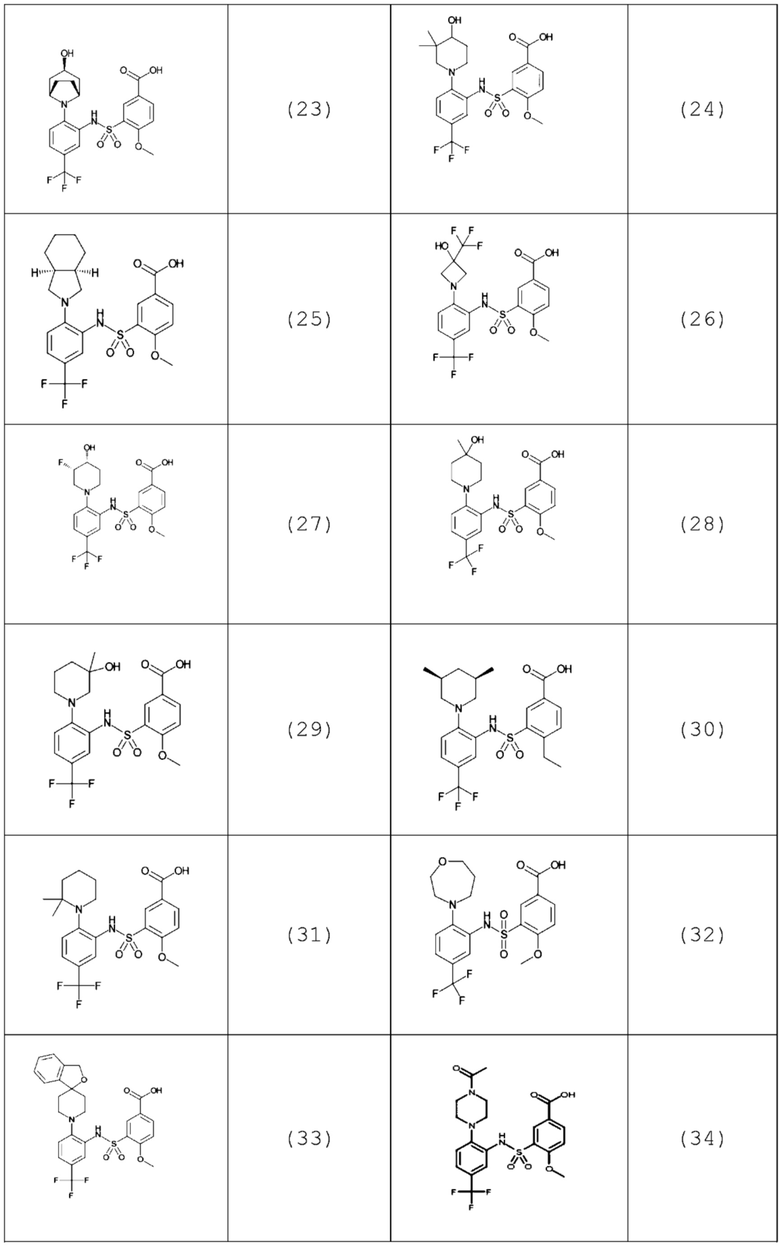
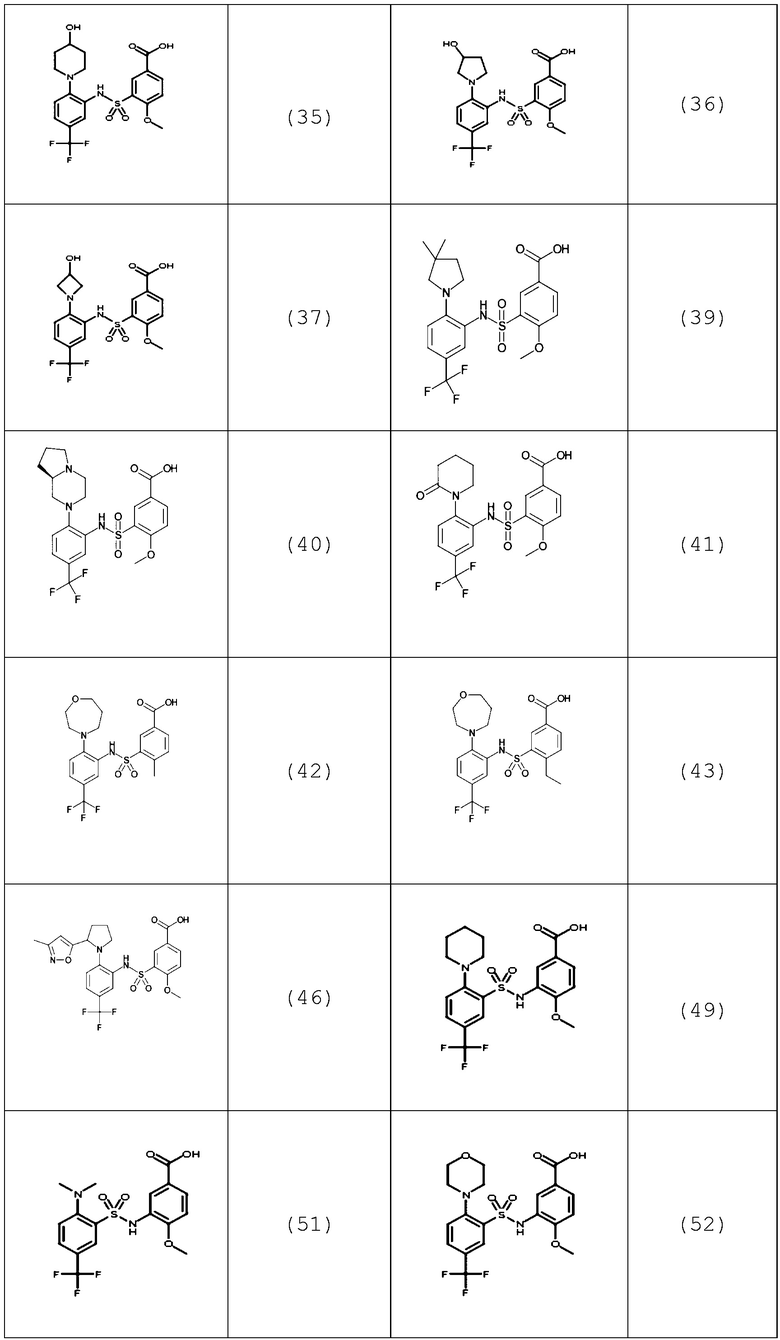
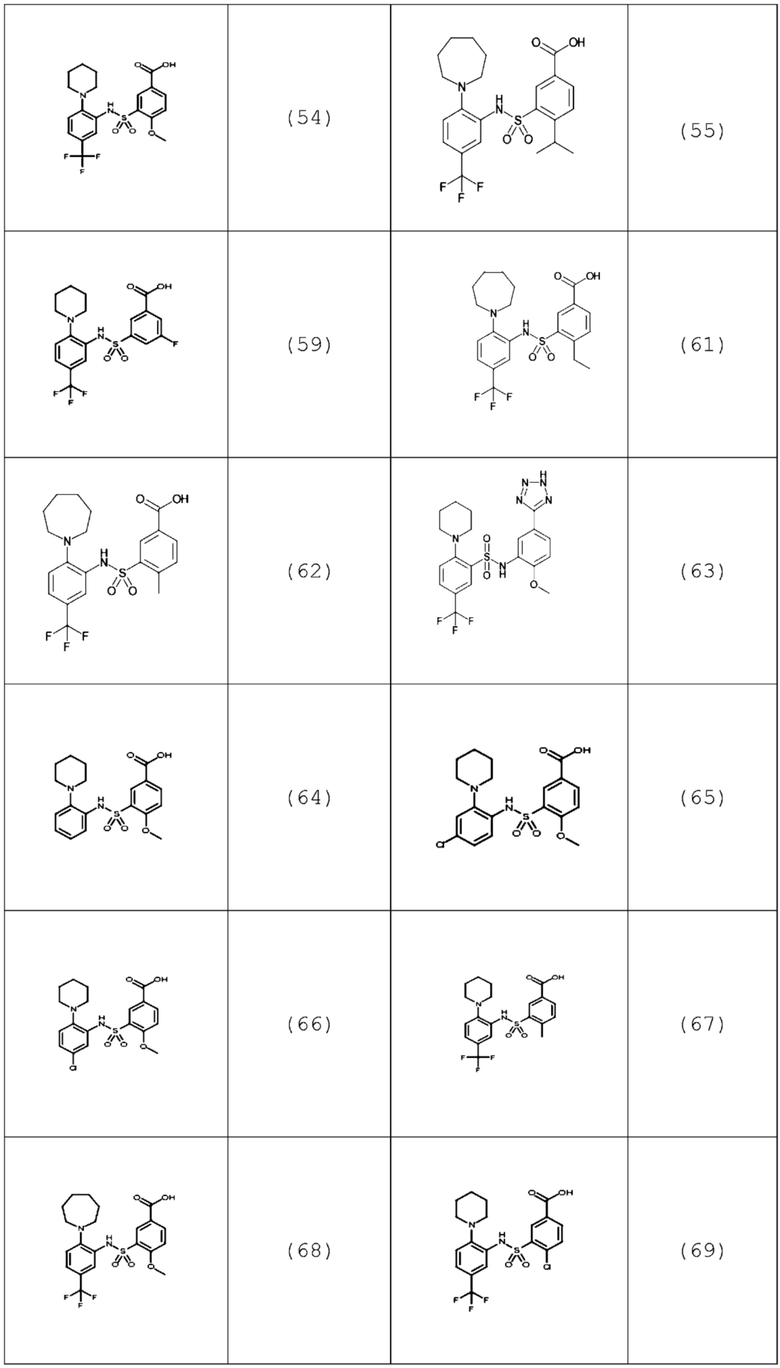
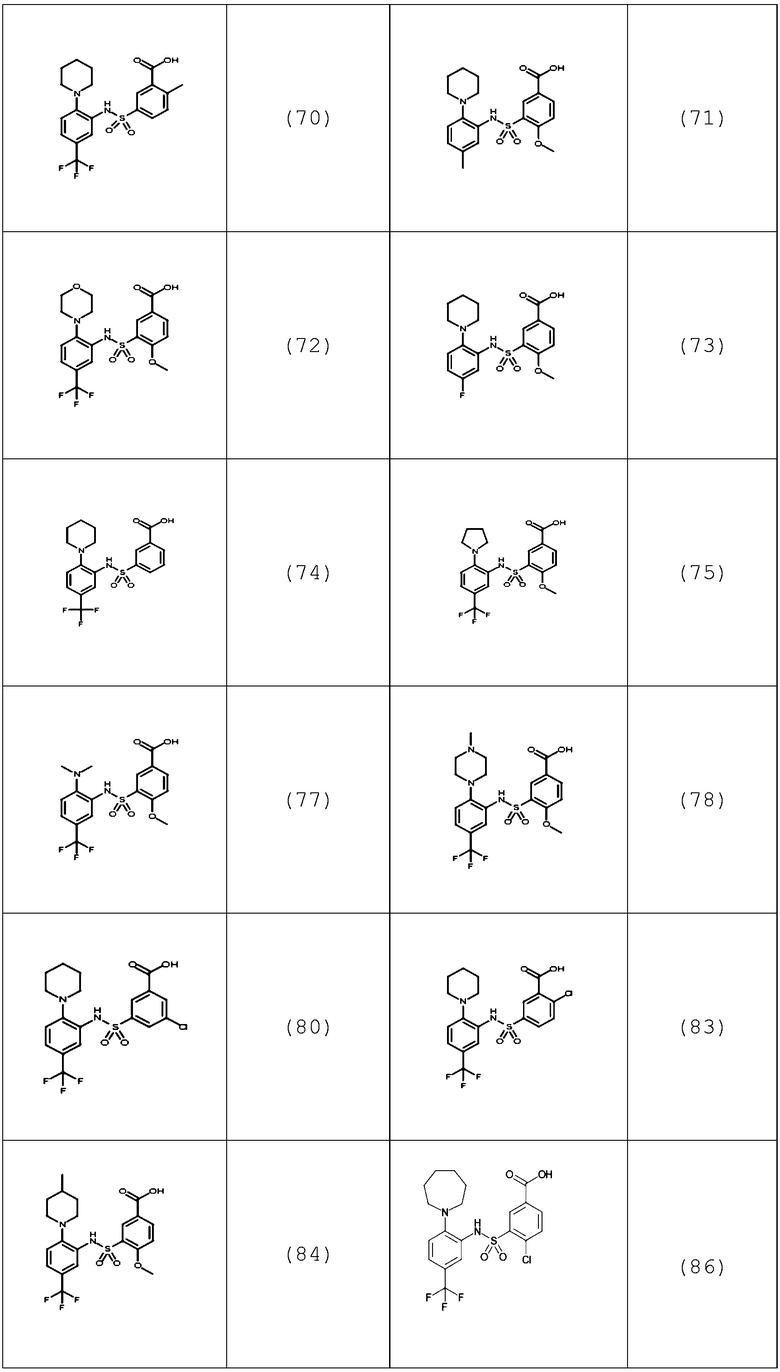
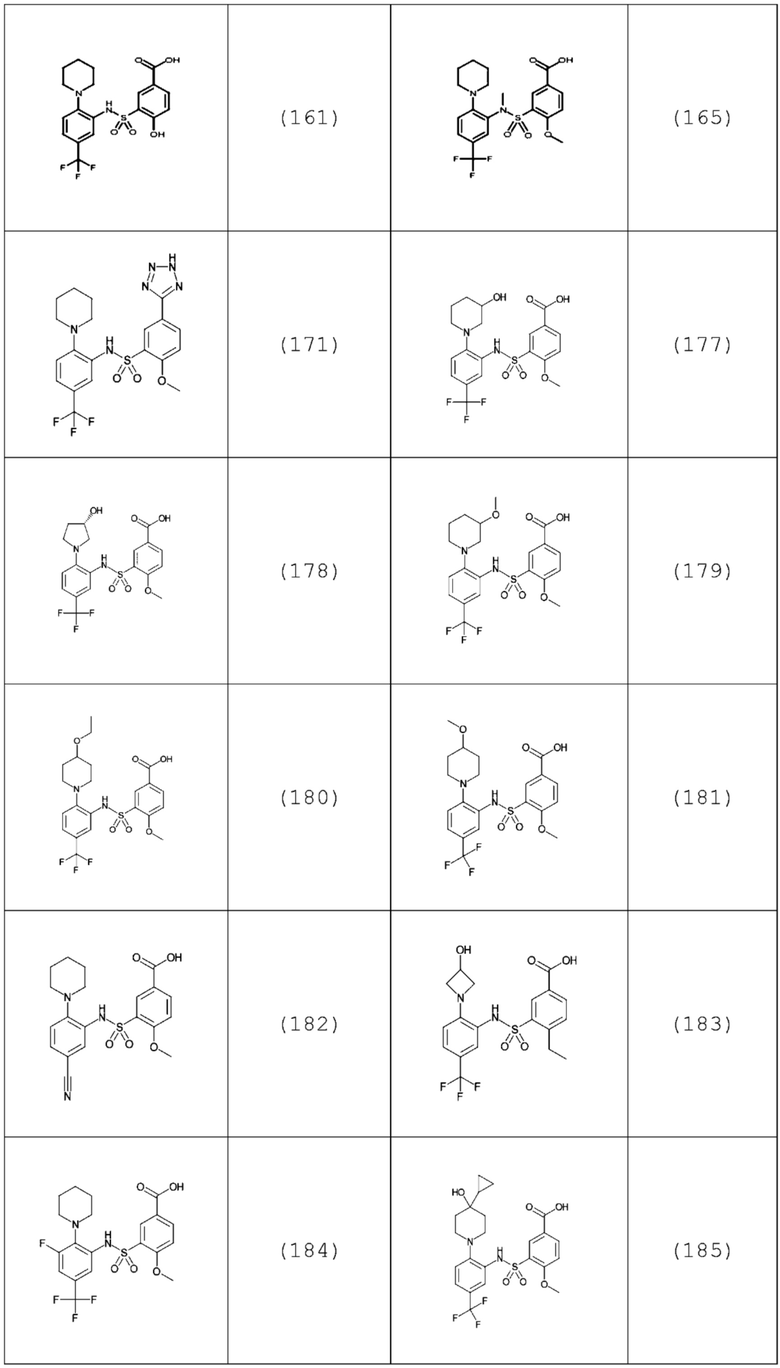
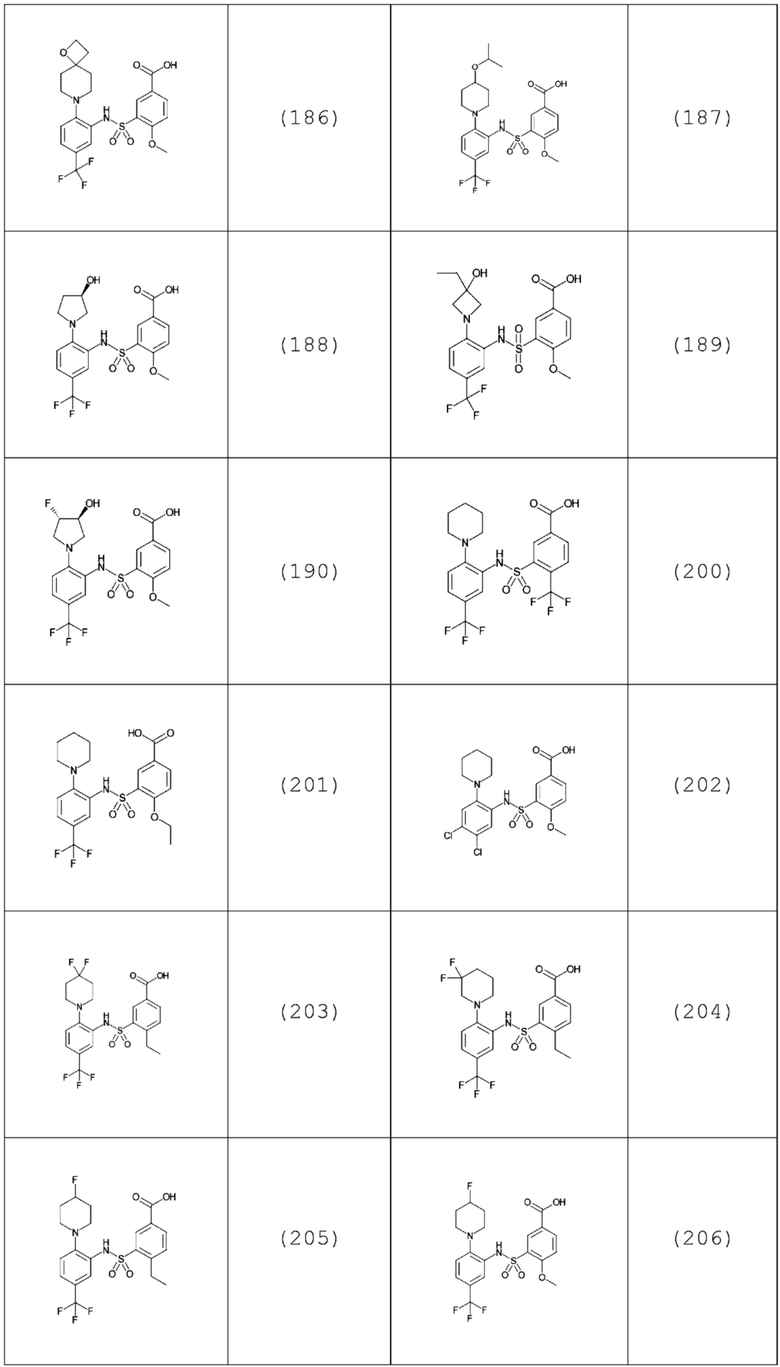
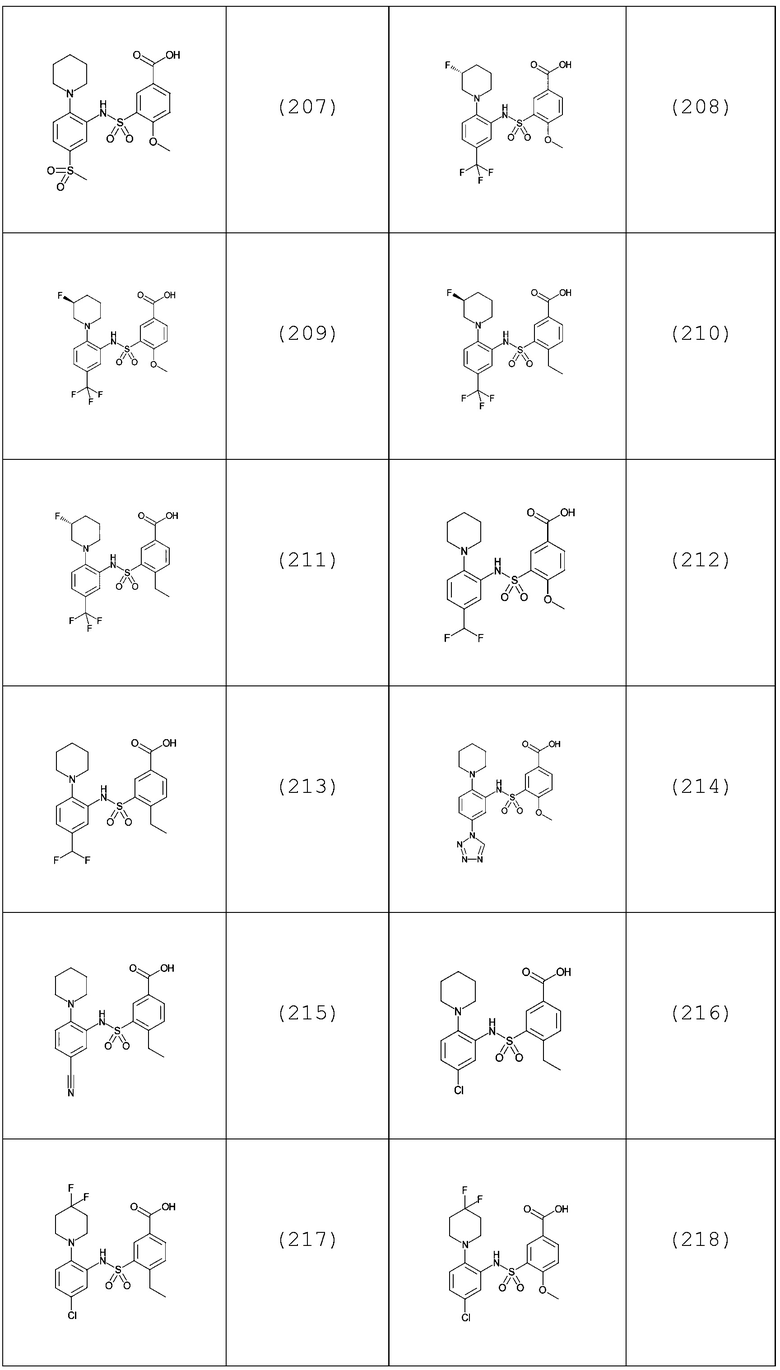
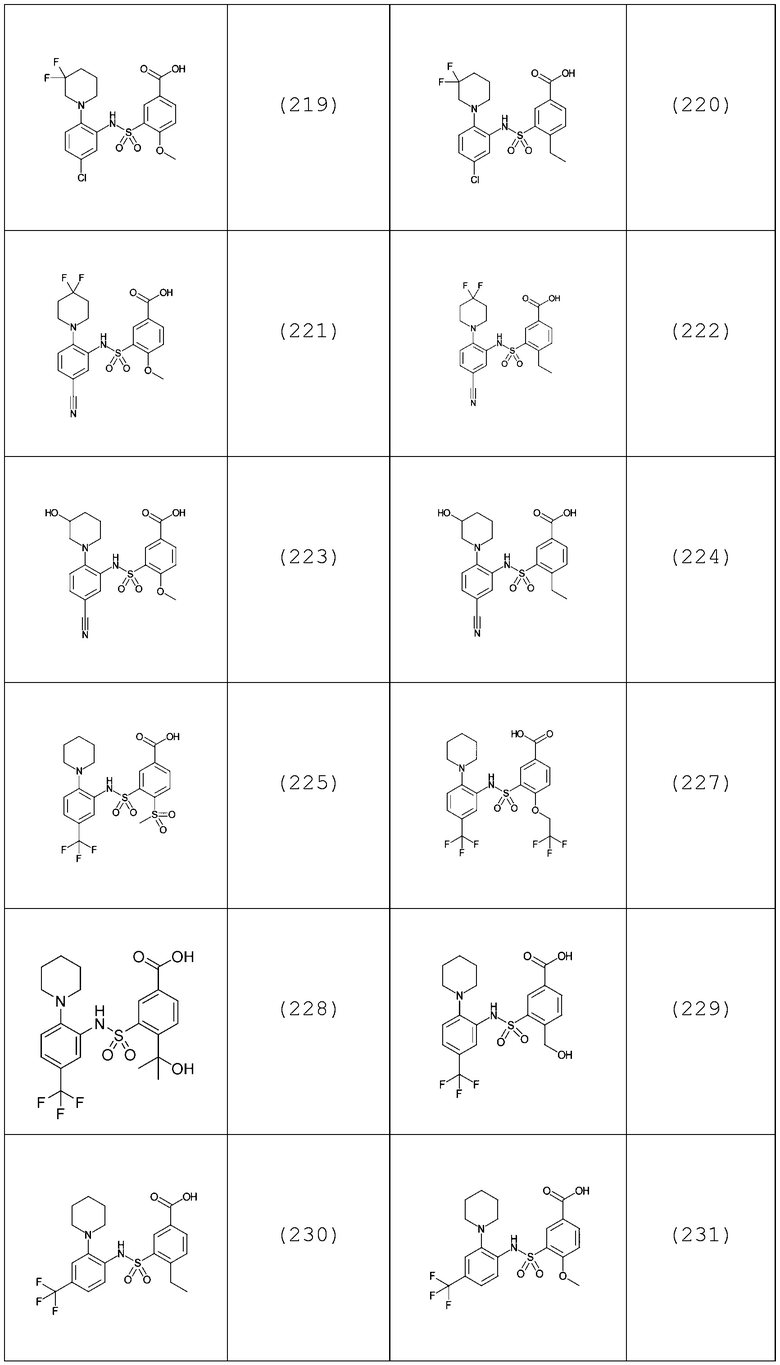
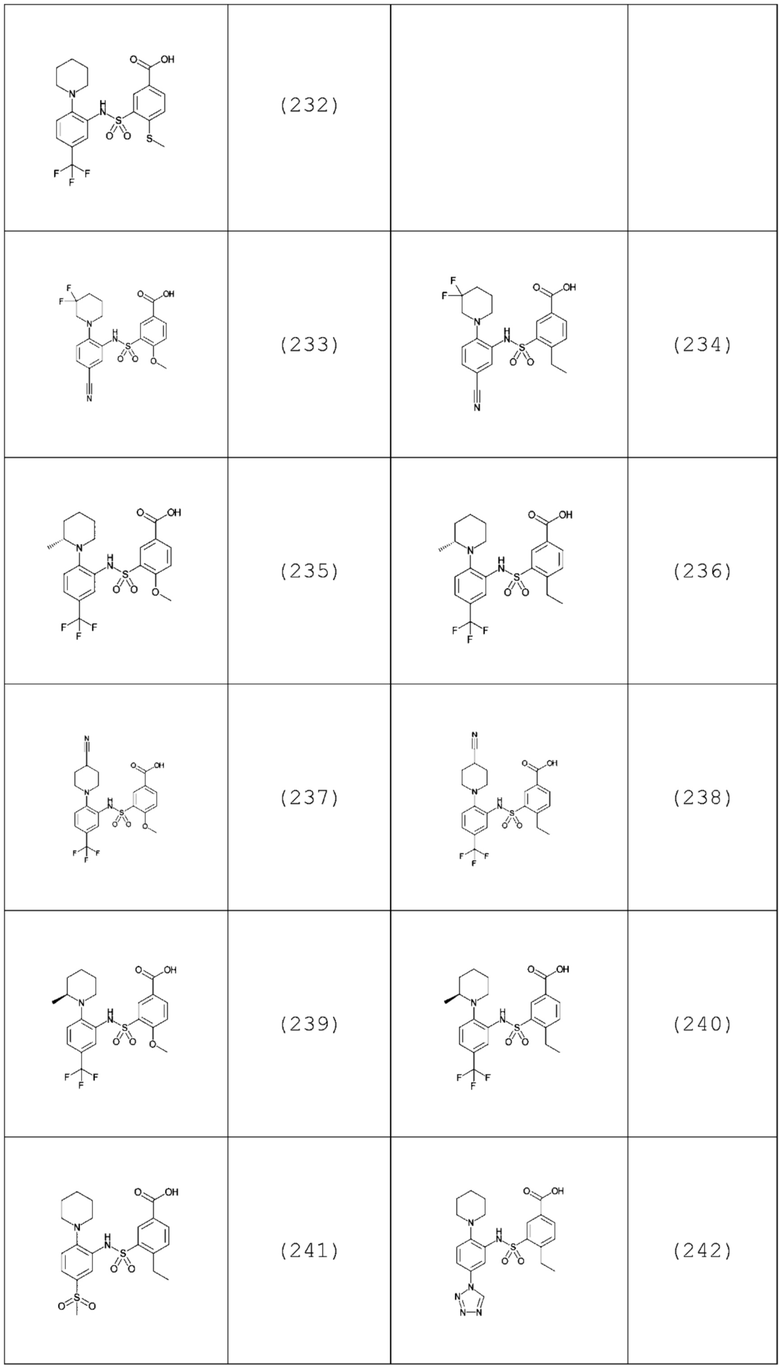
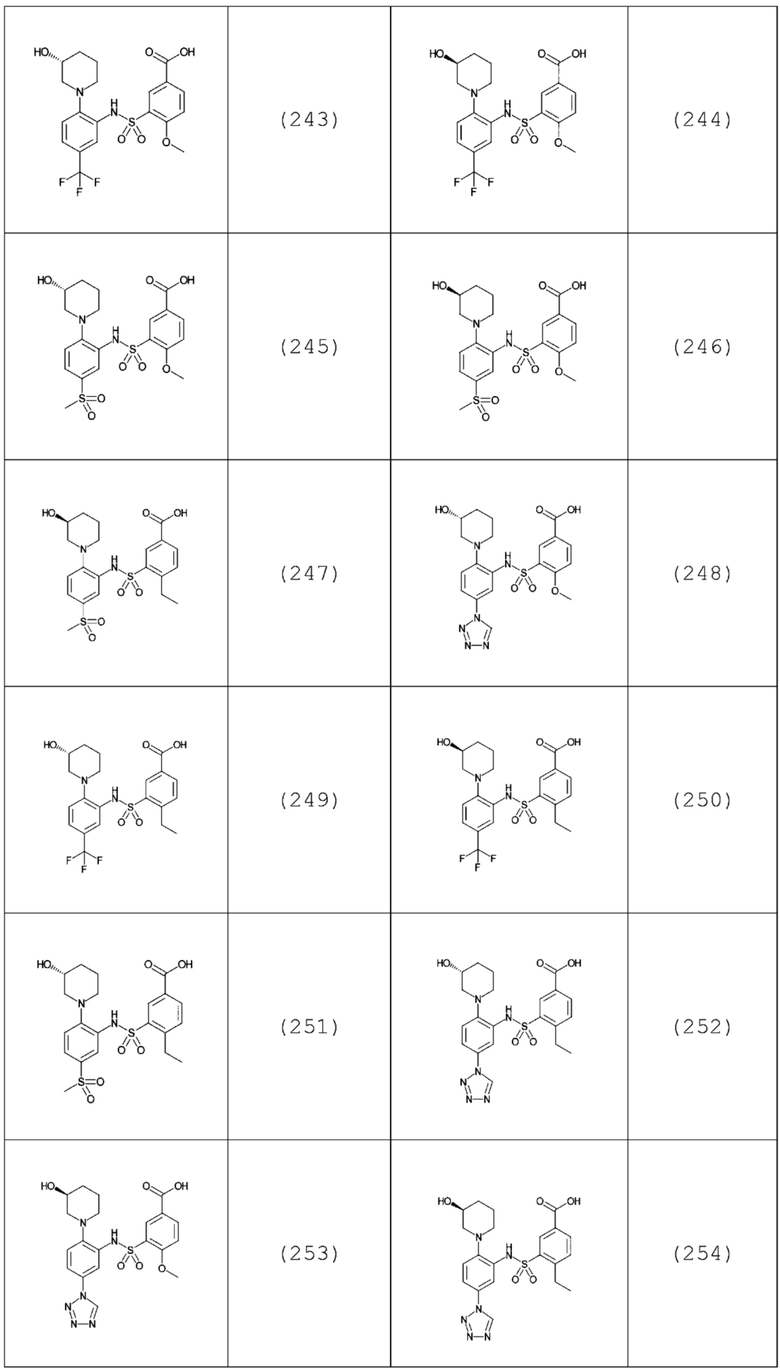
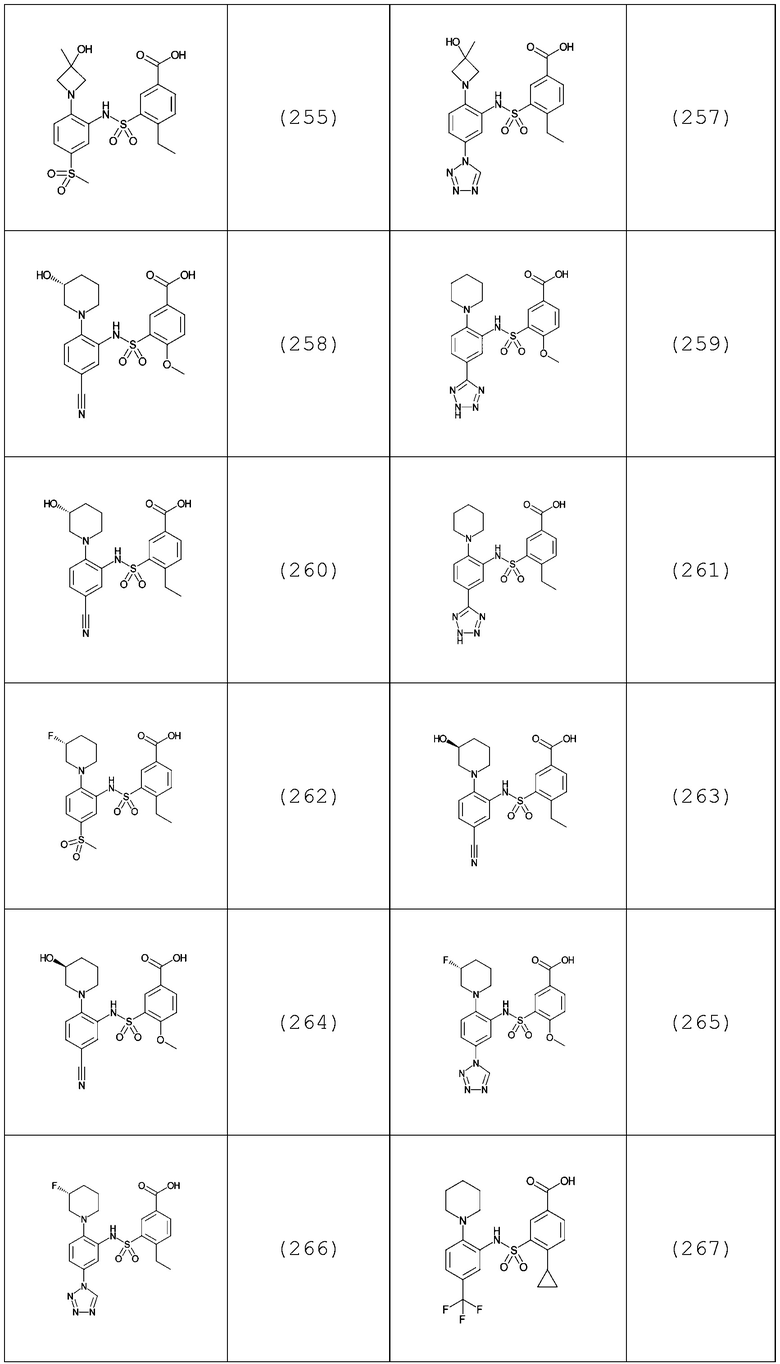
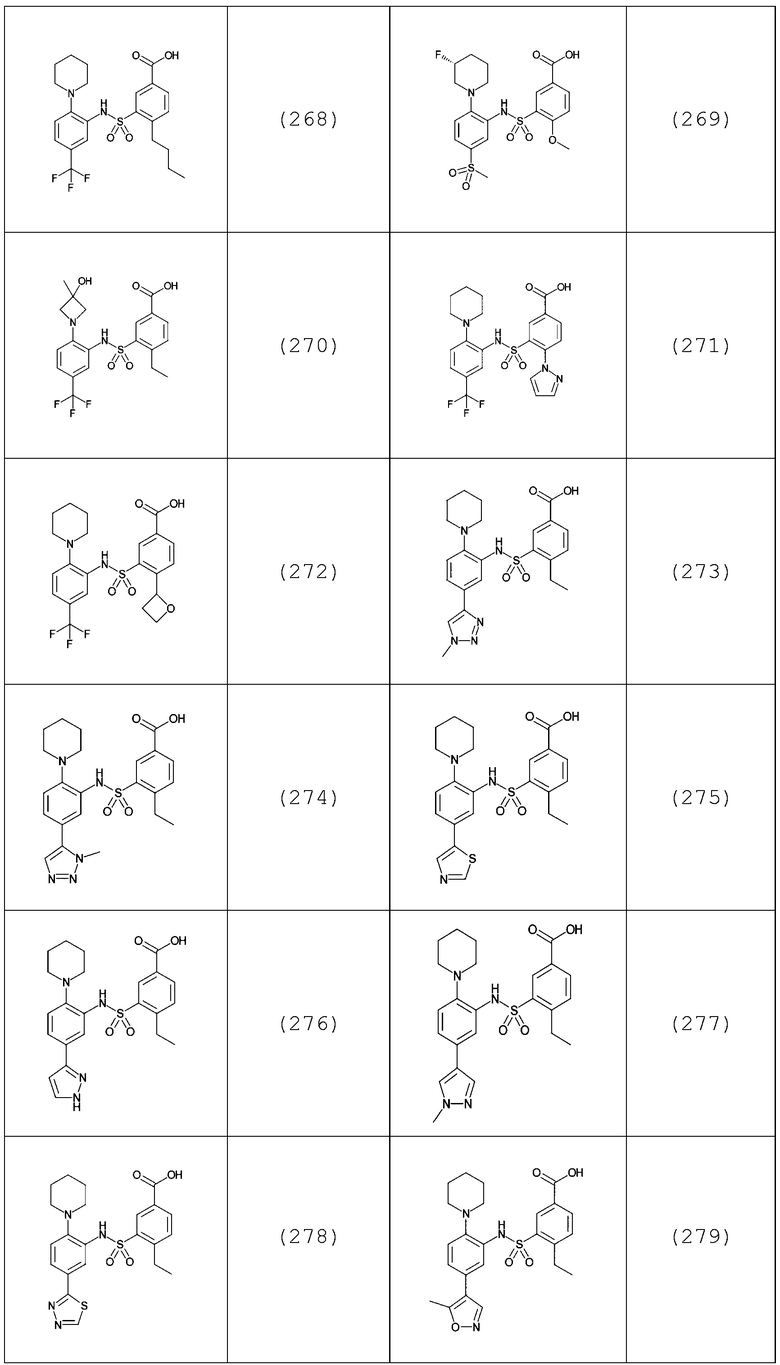
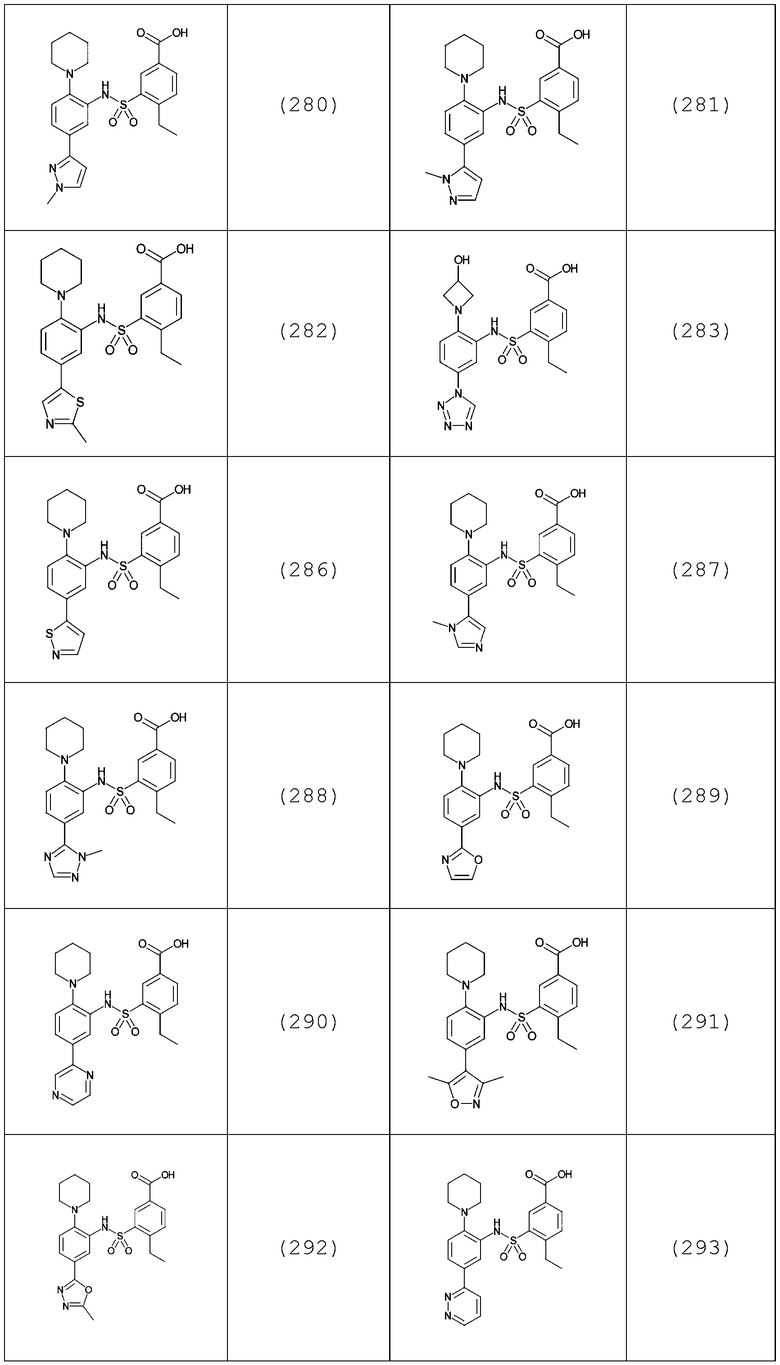
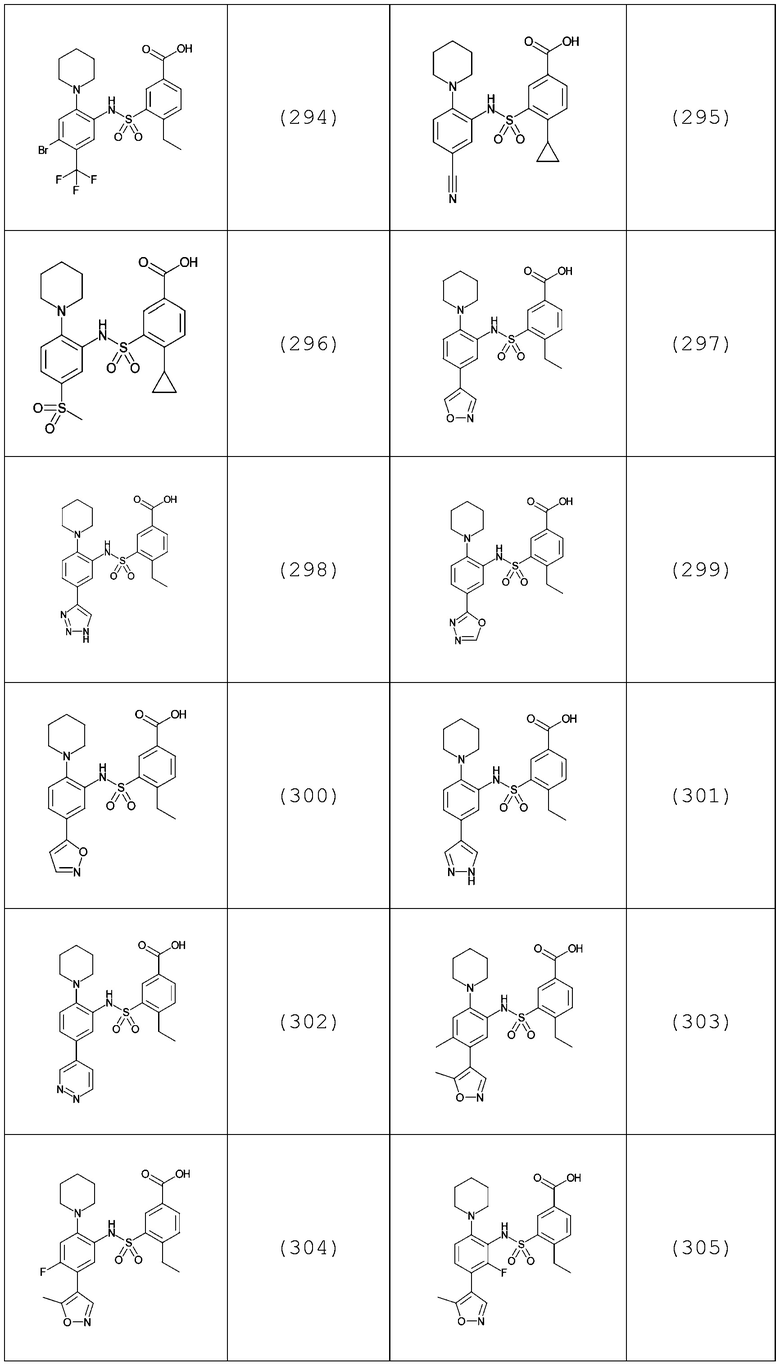
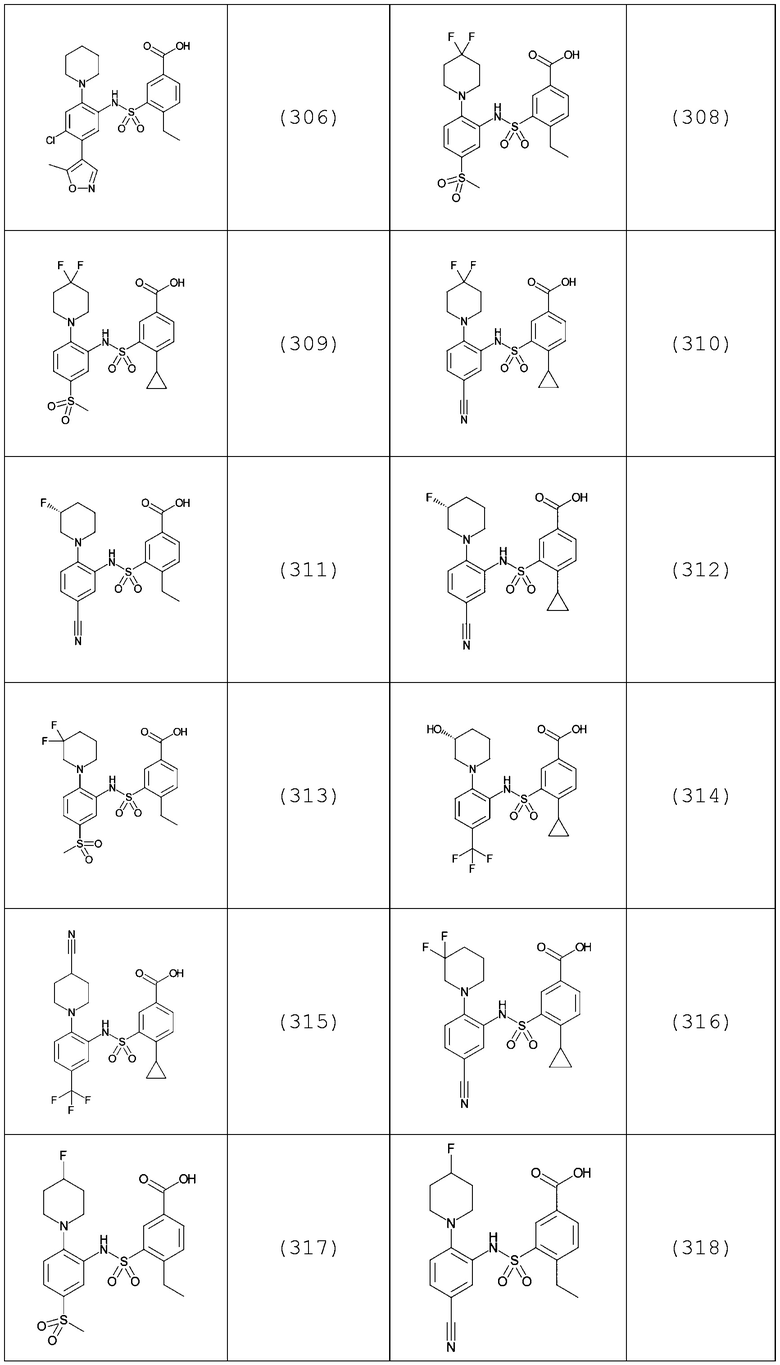
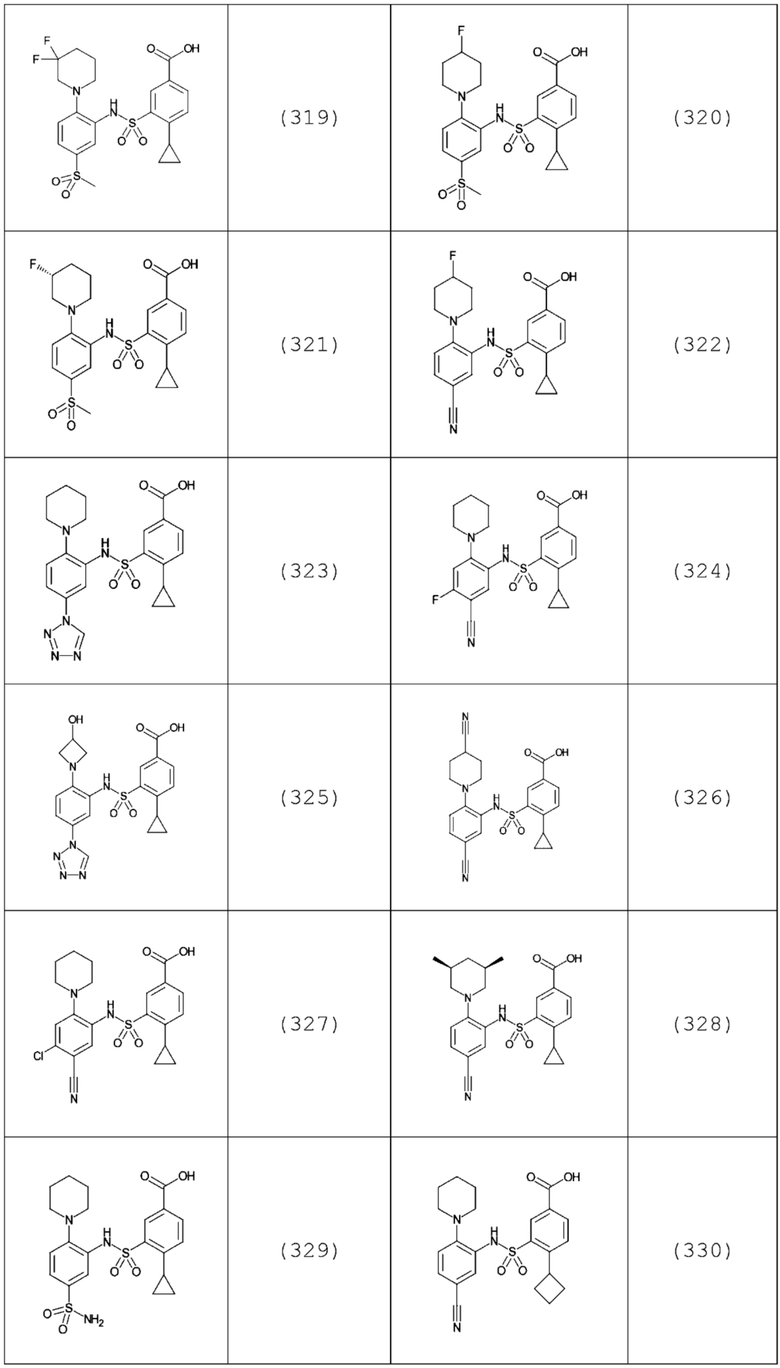
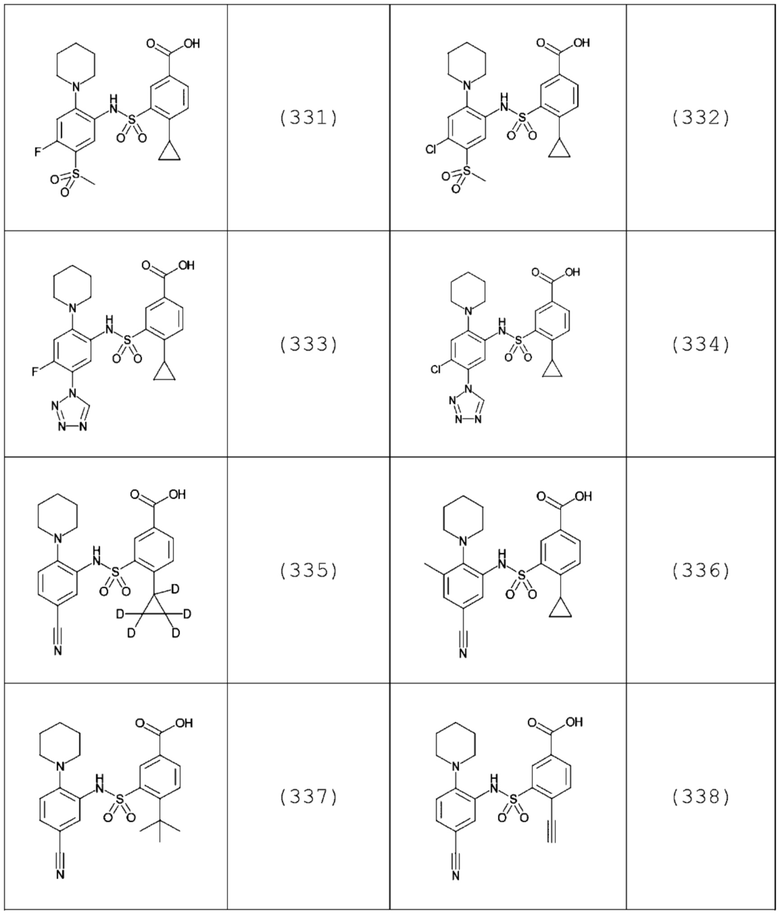
и их фармацевтически приемлемых солей и гидратов.
Другой аспект изобретения относится к соединениям формулы (I), как определено выше, кроме соединений (54), (64), (69), (71), (72), (73), (74), (78) и (165).
Еще один аспект относится к соединениям формулы (I), как определено выше, кроме соединений (54), (64), (69), (71), (72), (73), (74), (78) и (165), для применения, как определено выше.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Для применения согласно настоящему изобретению, соединения или их физиологически приемлемая соль, сложный эфир или другое физиологически функциональное производное, описанные в настоящем документе, могут быть представлены в форме фармацевтического состава, включающего соединения или их физиологически приемлемую соль, сложный эфир или другое физиологически функциональное производное вместе с одним или более фармацевтически приемлемыми носителями и, необязательно, другими терапевтическими и/или профилактическими компонентами. Носитель(и) должен быть приемлемым в том смысле, что он должен быть совместимым с другими компонентами состава и не токсичным для реципиента. Фармацевтические композиции могут быть предназначены для медицинского или ветеринарного применения в медицине и ветеринарии.
Примеры таких подходящих вспомогательных веществ для различных форм фармацевтических композиций, описанных в настоящем документе, можно найти в "Handbook of Pharmaceutical Excipients", 2nd Edition, (1994), Edited by A Wade and PJ Weller. Носитель или, если присутствует больше одного из них, каждый из носителей должен быть приемлемым в том смысле, что он должен быть совместимым с другими компонентами состава и не токсичным для реципиента.
Приемлемые носители или разбавители для терапевтического применения известны в области фармацевтики и описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985).
Примеры подходящих носителей включают лактозу, крахмал, глюкозу, метилцеллюлозу, стеарат магния, маннит, сорбит и т.п. Примеры подходящих разбавителей включают этанол, глицерин и воду.
Выбор фармацевтического носителя, вспомогательного вещества или разбавителя может быть сделан в зависимости от предполагаемого пути введения и стандартной фармацевтической практики. Фармацевтические композиции могут включать в качестве носителя, вспомогательного вещества или разбавителя или в дополнение к ним любое подходящее связующее вещество(а), смазывающее вещество(а), суспендирующее вещество(а), вещество(а) для получения покрытия, солюбилизирующее вещество(а), буфер(ы), ароматизирующее вещество(а), поверхностно-активное вещество(а), загуститель(и), консервант(ы) (включая антиоксиданты) и т.п., и вещества, добавляемые с целью сделать состав изотоническим с кровью предполагаемого реципиента.
Примеры подходящих связующих веществ включают крахмал, желатин, натуральные сахара, такие как глюкозу, безводную лактозу, сыпучую лактозу, бета-лактозу, подсластители из кукурузы, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлозу и полиэтиленгликоль.
Примеры подходящих смазывающих веществ включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п.
Консерванты, стабилизаторы, красители и даже ароматизаторы могут быть представлены в фармацевтической композиции. Примеры консервантов включают бензоат натрия, сорбиновую кислоту и сложные эфиры п-гидроксибензойной кислоты. Также могут использоваться антиоксиданты и суспендирующие вещества.
Фармацевтические составы включают составы, подходящие для перорального, наружного (включая кожное, трансбуккальное и подъязычное), ректального или парентерального (включая подкожное, кожное, внутримышечное и внутривенное), назального и пульмонального введения, например, путем ингаляции. Состав при необходимости может быть удобно представлен в дискретных единицах дозы и может быть изготовлен любым способом, известным в области фармации. Все способы включают этап объединения активного соединения с жидкими носителями и/или тонко измельченными твердыми носителями и затем, при необходимости, формование продукта в форме требуемого состава.
Фармацевтические составы, подходящие для перорального введения, в которых носитель является твердым, наиболее предпочтительно представлены в виде составов с единичной дозой, таких как болюсы, капсулы или таблетки, каждая из которых содержит заданное количество активного соединения. Таблетка может быть изготовлена путем прессования или формования, необязательно с одним или более вспомогательными компонентами. Прессованные таблетки могут быть изготовлены прессованием в подходящем аппарате активного соединения в сыпучей форме, такой как порошок или гранулы, необязательно смешиваемого со связующим веществом, смазывающим, инертным разбавителем, смазывающим веществом, поверхностно-активным веществом или диспергирующим веществом. Формованные таблетки могут быть получены путем формования активного соединения с инертным жидким разбавителем. Таблетки могут быть необязательно покрыты оболочкой и, если они не имеют оболочки, необязательно могут иметь риску. Капсулы можно быть изготовлены путем заполнения оболочки капсул активным соединением либо отдельно, либо в смеси с одним или больше вспомогательными компонентами, с их последующим герметичным закрытием обычным способом. Облатки аналогичны капсулам, в которых активное соединение вместе с любым дополнительным ингредиентом(ами) запечатывают в оболочке из рисовой бумаги. Активное соединение также могут изготавливать в форме диспергируемых гранул, которые, например, могут суспендировать в воде перед введением или посыпать ими пищу. Гранулы могут быть упакованы, например, в саше. Составы, подходящие для перорального введения, в которых носитель представляет собой жидкость, могут быть представлены в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии типа масло в воде.
Составы для перорального введения включают лекарственные формы с контролируемым высвобождением, например, таблетки, в которых активное соединение включено в подходящую регулирующую высвобождение матрицу или покрыто подходящей регулирующей высвобождение пленкой. Такие составы могут быть особенно удобными для профилактиктического применения.
Фармацевтические составы, подходящие для ректального введения, в которых носитель является твердым, наиболее предпочтительно представлены в виде суппозиториев единичной дозы. Подходящие носители включают масло какао и другие материалы, обычно используемые в данной области. Суппозитории могут быть удобно сформированы путем смешивания активного соединения с размягченным или расплавленным носителем(ями), с последующим охлаждением и формованием в формах. Фармацевтические составы, подходящие для парентерального введения, включают стерильные растворы или суспензии активного соединения в водных или масляных растворителях.
Препараты для инъекций могут быть предназначены для струйного введения или непрерывной инфузии. Такие препараты обычно представлены в контейнерах с единичной дозой или множеством доз, которые герметично закрывают после введения состава, пока они не потребуются для применения. В альтернативе активное соединение может быть в форме порошка, который перед применением смешивают с подходящим растворителем, таким как стерильная апирогенная вода.
Активное соединение может также быть изготовлено в форме депо-препаратов длительного действия, которые можно вводить путем внутримышечной инъекции или имплантации, например, подкожно или внутримышечно. Депо-препараты могут включать, например, подходящие полимерные или гидрофобные материалы или ионообменные смолы. Такие составы длительного действия особенно удобны для профилактического применения.
Составы, подходящие для пульмонального введения через полость рта, представлены в такой форме, что частицы, содержащие активное соединение и предпочтительно имеющие диаметр в пределах 0,5-7 микронов, доставляются в бронхиальное дерево реципиента.
В качестве одного из возможных вариантов такие составы находятся в виде тонко измельченных порошков, которые могут быть удобно представлены либо в форме прокалываемой капсуле, препочтительно, например, из желатина, для применения в ингаляционном устройстве, либо, в альтернативном варианте, в форме самораспыляемого состава, содержащего активное соединение, подходящий жидкий или газообразный пропеллент и, необязательно, другие компоненты, такие как поверхностно-активное вещество и/или твердый разбавитель. Подходящие жидкие пропелленты включают пропан и хлорфторуглероды, а подходящие газообразные пропелленты включают диоксид углерода. Также могут применяться самораспыляемые составы, в которых активное соединение дозируется в форме капель раствора или суспензии.
Такие самораспыляемые составы аналогичны составам, известным в уровне техники, и могут быть изготовлены согласно установленным процедурам. Предпочтительно они представлены в контейнере, который снабжен ручным или автоматически функционирующим клапаном, имеющим требуемые характеристики распыления; предпочтительно клапан относится к дозирующему типу и обеспечивает доставку фиксированного объема, например 25-100 микролитров, при каждом нажатии.
В качестве дополнительной возможности активное соединение может быть в форме раствора или суспензии для применения в распылителе или небулайзере, в котором ускоренный поток воздуха или ультразвуковое встряхивание используется для получения аэрозоля из тонких капель для ингаляции.
Составы, подходящие для назального введения, включают препараты, в целом аналогичные описанным выше для пульмонального введения. При дозировании такие составы предпочтительно должны иметь диаметр частиц в пределах от 10 до 200 микрон, чтобы обеспечить удерживание в носовой полости; при необходимости это может быть достигнуто при использовании порошка с подходящим размером частиц или путем подбора подходящего клапана. Другие подходящие составы включают грубые порошки с диаметром частиц в пределах от 20 до 500 микрон, для введения путем быстрого вдыхания через носовой проход из контейнера, удерживаемого близко к носу, а также назальные капли, содержащие от 0,2 до 5% в/об активного соединения в водном или масляном растворе или суспензии.
Фармацевтически приемлемые носители хорошо известны специалистам в данной области и включают, без ограничения этим, 0,1 М, и предпочтительно 0,05 М, фосфатный буфер или 0,8% раствор хлорида натрия. Кроме того, такие фармацевтически приемлемые носители могут быть водными или неводными растворами, суспензиями и эмульсиями. Примерами неводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и органические сложные эфиры для инъекций, такие как этилолеат. Водные носители включают воду, спиртовые/водные растворы, эмульсии или суспензии, включая раствор хлорида натрия и забуференные среды. Парентеральные носители включают раствор хлорида натрия, раствор Рингера с декстрозой, раствор декстрозы и хлорида натрия, лактированный раствор Рингера или нелетучие масла. Также могут присутствовать консерванты и другие добавки, такие как, например, противомикробные средства, антиоксиданты, хелатообразующие вещества, инертные газы и т.п.
Составы, подходящие для наружного применения, могут быть представлены, например, в форме гелей, кремов или мазей. Такие препараты можно наносить, например, на рану или язву, при этом они либо непосредственно распространяются по поверхности раны или язвы, либо нанесены на подходящую подложку, такую как повязка, марля, сетка и т.п., которую можно накладывать на обрабатываемую область.
Могут быть также предоставлены жидкие или порошковые составы, которые можно распылять или орошать ими непосредственно участок, подлежащий обработке, например, рану или язву. В альтернативе такой носитель, как повязка, марля, сетка и т.п. может опрыскивать или орошать составом и затем накладывать на обрабатываемый участок.
Согласно другому аспекту изобретения предложен способ изготовления фармацевтической или ветеринарной композиции, как описано выше, включающий объединение активного соединения(й) с носителем, например, путем смешивания.
Как правило, составы изготавливают путем однородного и тщательного объединения действующего вещества с жидкими носителями и/или тонко измельченными твердыми носителями, и затем, при необходимости, формования продукта. Изобретение распространяется на способы изготовления фармацевтической композиции, включающей объединение соединения, описанного в настоящем документе, с фармацевтически или ветеринарно приемлемым носителем или растворителем.
СОЛИ/СЛОЖНЫЕ ЭФИРЫ
Соединения согласно изобретению могут присутствовать в виде солей или сложных эфиров, в частности, фармацевтически и ветеринарно приемлемых солей или сложных эфиров.
Фармацевтически приемлемые соли соединений согласно изобретению включают подходящие соли присоединения кислот или оснований. Обзор подходящих фармацевтических солей можно найти в Berge et al, J Pharm Sci, 66, 1-19 (1977). Соли получают, например, с сильными неорганическими кислотами, такими как, например, галогенводородные кислоты, такие как гидрохлорид, гидробромид и гидроиодид, серная кислота, сульфат фосфорной кислоты, бисульфат, гемисульфат, тиоцианат, персульфат и сульфоновые кислоты; с сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты из 1-4 атомов углерода, которыми не замещены или замещены (например, галогеном), такие как уксусная кислота; с насыщенными или ненасыщенными дикарбоновыми кислотами, например, щавелевой, малоновой, янтарной, малеиновой, фумаровой, фталевой или терефталевой; с гидроксикарбоновыми кислотами, например аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной кислотой; с аминокислотами, например аспарагиновой кислотой или глутаминовой кислотой; с бензойной кислотой; или с органическими сульфокислотами, такими как (C1-C4)-алкил- или арилсульфоновые кислоты, которые не замещены или замещены (например, галогеном), такие как метан- или п-толуолсульфоновая кислота. Соли, которые не являются фармацевтически или ветеринарно приемлемыми, могут быть все же ценными в качестве промежуточных соединений.
Предпочтительные соли включают, например, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат, пантотенат, адипат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентанат, глюкогептанат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат, памоат, пектинат, 3-фенилпропионат, пикрат, пивалат, пропионат, тартрат, лактобионат, пивалат, камфорат, ундеканоат и сукцинат, органические сульфокислоты, такие как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, камфорсульфонат, 2-нафталнсульфонат, бензолсульфонат, п-хлорбензолсульфонат и п-толуолсульфонат; и неорганические кислоты, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, бисульфат, гемисульфат, тиоцианат, персульфат, фосфорная и сульфоновая кислоты.
Сложные эфиры образуются либо при использовании органических кислот, либо спиртов/гидроксидов, в зависимости от функциональной группы, подвергаемой этерификации. Органические кислоты включают карбоновые кислоты, такие как алканкарбоновые кислоты из 1-12 атомов углерода, которые не замещены или замещены (например, галогеном), такие как уксусная кислота; с насыщенной или ненасыщенной дикарбоновой кислотой, например щавелевой, малоновой, янтарной, малеиновой, фумаровой, фталевой или терефталевой; с оксикарбоновыми кислотами, например аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной кислотой; с аминокислотами, например, аспарагиновой кислотой или глутаминовой кислотой; с бензойной кислотой; или с органическими сульфокислотами, такими как (C1-C4)-алкил- или арилсульфоновые кислоты, которые не замещены или замещены (например, галогеном), такие как метан- или п-толуолсульфоновая кислота. Подходящие гидроксиды включают неорганические гидроксиды, такие как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия. Спирты включают алканолы из 1-12 атомов углерода, которые могут быть незамещенными или замещенными, например, галогеном).
ЭНАНТИОМЕРЫ/ТАУТОМЕРЫ
Во всех аспектах настоящего изобретения, обсуждаемых выше, изобретение в соответствующих случаях включает все энантиомеры, диастереоизомеры и таутомеры соединений согласно изобретению. Специалист в данной области определит соединения, которые обладают оптическими свойствами (один или больше хиральных атомов углерода) или таутомерными характеристиками. Соответствующие энантиомеры и/или таутомеры могут быть выделены/получены способами, известными в уровне техники.
Энантиомеры характеризуются абсолютной конфигурацией их хиральных центров и описываются по правилам последовательности при определении R- и S-конфигурации Кана, Ингольда и Прелога. Такие правила известны в уровне техники (например, см. 'Advanced Organic Chemistry', 3rd edition, ed. March, J., John Wiley and Sons, New York, 1985).
Соединения согласно изобретению, содержащие хиральный центр, могут использоваться в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическую смесь могут разделять с помощью известных методов, и могут использовать один отдельный энантиомер.
СТЕРЕО И ГЕОМЕТРИЧЕСКИЕ ИЗОМЕРЫ
Некоторые соединения согласно изобретению могут существовать в виде стереоизомеров и/или геометрических изомеров, например, они могут обладать одним или больше асимметрическими и/или геометрическими центрами и, таким образом, могут существовать в двух или больше стереоизомерных и/или геометрических формах. Настоящее изобретение предусматривает применение всех отдельных стереоизомеров и геометрических изомеров таких соединений, а также их смесей. Термины, используемые в формуле изобретения, охватывают такие формы, если указанные формы сохраняют соответствующую функциональную активность (хотя и не обязательно в той же степени).
Настоящее изобретение также включает все подходящие изотопные варианты соединения или их фармацевтически приемлемую соль. Изотопный вариант соединения согласно настоящему изобретению или его фармацевтически приемлемая соль определяют как вариант, в котором по меньшей мере один атом заменен атомом, имеющим такой же атомный номер, но другую атомную массу, которая отличается от атомной массы, существующей в природе. Примеры изотопов, которые могут быть включены в средство, и его фармацевтически приемлемые соли включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F и 36Cl соответственно. Некоторые изотопные варианты средства и их фармацевтически приемлемые соли, например, в которые включен радиоактивный изотоп, такой как 3H или 14C, могут применяться в исследованиях распределения лекарственных средств и/или субстратов в тканях. Тритированные, т.е. содержащие 3H, и углерод 14, т.е. 14C, изотопы особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение изотопами, такими как дейтерий, т.е. 2H, может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным периодом полувыведения in vivo или необходимостью введения меньшей дозы, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Например, изобретение включает соединения общей формулы (I), в которых любой атом водорода заменен атомом дейтерия. Изотопные варианты средства согласно настоящему изобретению и их фармацевтически приемлемые соли согласно настоящему изобретению обычно могут быть получены с помощью стандартных методик с использованием соответствующих изотопных вариантов подходящих реагентов.
АТРОПОИЗОМЕРЫ
Некоторые соединения согласно изобретению могут существовать в виде атропоизомеры. Атропоизомеры представляют собой стереоизомеры, возникающими из-за затрудненного вращения вокруг одинарной связи, когда различия в энергии из-за стерического напряжения или другие факторы создают барьер для вращения, которое достаточно высок, что позволяет выделить отдельные конформеры. Изобретение охватывает все такие атропоизомеры.
ПРОЛЕКАРСТВА
Изобретение также включает соединения согласно настоящему изобретению в форме пролекарства, то есть ковалентно связанные соединения, которые высвобождают активное исходное лекарственное соединение in vivo. Такие пролекарства в целом представляют собой соединения согласно настоящему изобретению, в которых одна или больше подходящих групп были модифицированы таким образом, что модификация может быть снята при введении человеку или млекопитающему. Такое обратное превращение обычно выполняет фермент, обычно присутствующий в организме такого субъекта, хотя возможно введение второго средства вместе с таким пролекарством, чтобы выполнить обратное превращение in vivo. Примеры таких модификаций включают сложный эфир (например, любой из описанных выше), в котором обратное превращение может осуществлять эстераза и т.д. Другие такие системы будут хорошо известны специалистам в данной области.
СОЛЬВАТЫ
Настоящее изобретение также включает сольватированные формы соединений согласно настоящему изобретению. Термины, используемые в формуле изобретения, охватывают такие формы.
ПОЛИМОРФНЫЕ МОДИФИКАЦИИ
Изобретение также относится к соединениям настоящего изобретения в различных кристаллических формах, полиморфных формах и (де)гидратированных формах. В фармацевтической промышленности хорошо известно, что химические соединения могут быть выделены в любой из таких форм при небольшом изменении способа очистки и/или при выделении из растворителей, используемых в синтезе таких соединений.
ВВЕДЕНИЕ
Фармацевтические композиции согласно настоящему изобретению могут быть предназначены для ректального, назального, внутрибронхиального, наружного (в том числе трансбуккального и подъязычного), вагинального или парентерального (в том числе подкожного, внутримышечного, внутривенного, внутриартериального и внутрикожного), внутрибрюшинного или интратекального введения. Предпочтительно состав представляет собой состав для перорального введения. Составы могут быть с удобством представлены в виде единичной лекарственной формы, то есть в форме дискретных частей, содержащих стандартную дозу, или множества или субединицы единичной дозы. Например, составы могут быть в форме таблеток и капсул с замедленным высвобождением и могут быть изготовлены любым способом, хорошо известным в области фармации.
Составы для перорального введения в настоящем изобретении могут быть представлены в форме: дискретных единиц, таких как капсулы, желатиновые капсулы, капли, облатки, пилюли или таблетки, каждая из которых содержит заданное количество действующего вещества; в виде порошка или гранул; в виде раствора, эмульсии или суспензии действующего вещества в водной жидкости или неводной жидкости; или в виде жидкой эмульсии типа масло в воде или жидкой эмульсии типа вода в масле; или в виде болюса и т.д. Предпочтительно такие композиции содержат от 1 до 250 мг и более предпочтительно от 10 до 100 мг действующего вещества на дозу.
В отношении композиций для перорального введения (например, таблеток и капсул) термин "приемлемый носитель" включает разбавители, такие как обычные вспомогательные вещества, например, связующие вещества, например сироп, гуммиарабик, желатин, сорбит, трагакант, поливинилпирролидон (повидон), метилцеллюлозу, этилцеллюлозу, карбоксиметилцеллюлозу натрия, гидроксипропилметилцеллюлозу, сахарозу и крахмал; наполнители и носители, например, кукурузный крахмал, желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, фосфат дикальция, хлорид натрия и альгиновую кислоту; и смазывающие вещества, такие как стеарат магния, стеарат натрия и другие стеараты металлов, глицерилстеарат, стеариновая кислота, силиконовая жидкость, тальковые воски, масла и коллоидный диоксид кремния. Также могут использоваться ароматизаторы, такие как перечная мята, масло березы вишневой, вишневый аромат и т.п. Может потребоваться добавить краситель, чтобы лекарственную форму можно было легко идентифицировать. Таблетки также могут быть покрыты оболочкой хорошо известными в данной области способами.
Таблетка может быть изготовлена прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть изготовлены путем прессования в подходящем аппарате действующего вещества в сыпучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим веществом. Формованные таблетки могут быть изготовлены путем формования в подходящем аппарате смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки необязательно могут быть покрыты оболочкой или снабжены риской и могут иметь такой состав, чтобы обеспечивать медленное или контролируемое высвобождение действующего вещества.
Другие составы, подходящие для приема внутрь, включают таблетки для рассасывания, включающие действующее вещество в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, включающие действующее вещество в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и ополаскиватели для полости рта, включающие действующее вещество в подходящем жидком носителе.
Другие лекарственные формы включают растворы или эмульсии, которые можно вводить внутривенно, внутриартериально, интратекально, подкожно, внутрикожно, внутрибрюшинно или внутримышечно, и которые изготовлены из стерильных или стерилизуемых растворов. Инъекционные формы, как правило, содержат от 10 до 1000 мг, предпочтительно от 10 до 250 мг, действующего вещества на дозу.
Фармацевтические композиции настоящего изобретения могут быть также в форме суппозиториев, пессариев, суспензий, эмульсий, лосьонов, мазей, кремов, гелей, спреев, растворов или присыпок.
Альтернативный способ трансдермального введения осуществляют путем применения кожного пластыря. Например, действующее вещество может быть включено в крем, состоящий из водной эмульсии полиэтиленгликолей или жидкого парафина. Действующее вещество также может быть включено в концентрации 1-10% по весу в мазь, состоящую из белого воска или белой мягкой парафиновой основы вместе с такими стабилизаторами и консервантами, которые могут потребоваться.
ДОЗИРОВКА
Средний специалист в данной области сумеет легко определить подходящую дозу одной из настоящих композиций для введения субъекту без излишних экспериментов. Обычно врач определяет фактическую дозировку, которая будет наиболее подходящей для отдельного пациента, при этом она будет зависеть от множества факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и длительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, режим и время введения, скорость выведения, комбинацию лекарственных средств, тяжесть конкретного состояния и индивида, подвергаемого терапии. Дозировки, раскрытые в настоящем документе, являются типичными для среднего случая. Конечно, могут быть отдельные случаи, когда необходимы более высокие или более низкие диапазоны доз, и такие дозы входят в объем настоящего изобретения.
Величину дозы будут дополнительно изменять в соответствии со способом введения соединения. Например, для достижения "эффективного количества" в случае интенсивной терапии обычно предпочтительно парентеральное введение соединения. Внутривенная инфузия соединения в 5% растворе декстрозы в воде или физиологическом растворе, или аналогичный состав с подходящими вспомогательными веществами, является наиболее эффективной, хотя также может применяться внутримышечная болюсная инъекция. Обычно парентеральная доза составляет от приблизительно 0,01 до приблизительно 100 мг/кг; предпочтительно от 0,1 до 20 мг/кг, чтобы поддерживать концентрацию лекарственного вещества в плазме на уровне, эффективном для модуляции ERAP1. Соединения могут вводить от одного до четырех раз в день до достижения общей суточной дозы от приблизительно 0,4 до приблизительно 400 мг/кг/день. Точное количество соединения согласно изобретению, которое является терапевтически эффективным, и путь, которым такое соединение лучше всего вводить, легко определит средний специалист в данной области при сравнении уровня средства в крови с концентрацией, требуемой для терапевтического эффекта.
Соединения настоящего изобретения также могут вводить пациенту перорально, таким образом, чтобы концентрация лекарственного средства была достаточной для достижения одного или больше терапевтических показаний, раскрытых в настоящем документе. Как правило, фармацевтическую композицию, содержащую соединение, вводят в дозе для приема внутрь от приблизительно 0,1 до приблизительно 50 мг/кг, таким образом, который соответствует состоянию пациента. Предпочтительно доза для приема внутрь составляет от приблизительно 0,5 до приблизительно 20 мг/кг.
Никаких неприемлемых токсикологических эффектов не ожидается, когда соединения настоящего изобретения вводят в соответствии с настоящим изобретением. Соединения согласно настоящему изобретению, которые могут обладать хорошей биодоступностью, могут быть исследованы в одном из нескольких биологических анализов для определения концентрации соединения, которая требуется для получения данного фармакологического эффекта.
КОМБИНАЦИИ
В особо предпочтительном варианте осуществления одно или больше соединений изобретения вводят в комбинации с одним или более дополнительными действующими веществами, например, существующими препаратами, доступными на рынке. Таким образом, другой аспект изобретения относится к комбинации, включающей соединение, как описано в настоящем документе, и одно или более дополнительных действующих веществ. В одном предпочтительном варианте осуществления соединения согласно изобретению могут вводить последовательно, одновременно или поочередно с одним или больше другими действующими веществами.
Лекарственные средства в целом являются более эффективными в случае их применения в комбинации. В частности, комбинированная терапия предпочтительна для предотвращения наложения основных токсических эффектов, механизма действия и механизма(ов) резистентности. Кроме того, также желательно вводить большинство лекарственных средств в их максимальных переносимых дозах с минимальными интервалами между такими дозами. Основные преимущества комбинирования химиотерапевтических средств состоят в том, что оно может вызвать дополнительные или возможные синергические эффекты посредством биохимических взаимодействий, а также может уменьшить возникновение резистентности.
Полезные комбинации могут быть предложены при изучении активности исследуемых соединений со средствами, которые, как известно или ожидается, будут полезными при лечении конкретного нарушения. Эта процедура также может использоваться для определения порядка введения средств, т.е. до, одновременно или после доставки. Такой подбор схемы введения может быть особенностью всех действующих веществ, идентифицированных в настоящем документе.
В одном предпочтительном варианте осуществления дополнительное действующее вещество является иммунотерапевтическим средством, более предпочтительно средством для иммунотерапии рака. "Иммунотерапевтическое средство" относится к лечению, в котором собственная иммунная система субъекта используется для борьбы с заболеваниями, такими как рак.
В одном предпочтительном варианте осуществления соединение согласно изобретению ингибирует активность ERAP1, причем соединение вводят в комбинации с иммунотерапией.
Соединение может повышать чувствительность раковых клеток к иммунотерапии. Иммунотерапия может быть опосредована T-клетками. В одном варианте осуществления соединение может увеличивать количество CD8+ T-клеток в опухоли.
В одном варианте осуществления соединение может применяться для лечения таких форм рака, которые плохо отвечают или не отвечают на иммунотерапии.
В одном предпочтительном варианте осуществления дополнительным действующим веществом является молекула, способная к вмешательству в иммунную контрольную точку, костимулирующее антитело, химиотерапевтическое средство, радиотерапевтическое средство, средство для направленной терапии или антитело, в частности моноклональное антитело.
В одном предпочтительном варианте осуществления дополнительное действующее вещество является молекулой, способной к вмешательству в иммунную контрольную точку.
Молекулы иммунных контрольных точек включают CTLA-4, PD-1, VISTA, B7-H2, B7-H3, PD-L1, B7-H4, B7-H6, ICOS, HVEM, PD-L2, CD160, gp49B, PIR-B, рецепторы семейства KIR, TIM-1, TIM-3, TIM-4, LAG-3, GITR, 4-IBB, OX-40, BTLA, SIRP, CD47, CD48, 2B4, B7.1, B7.2, ILT-2, ILT-4, TIGIT, HHLA2, IDO, CD39, CD73, A2aR и бутирофилины.
Молекулы иммунных контрольных точек включают ингибирующие и активирующие молекулы, при этом вмешательство может относиться к молекуле одного или обоих типов.
Например, ингибиторы иммунных контрольных точек включают, без ограничения, ингибиторы PD-1, ингибиторы PD-L1, ингибиторы LAG-3, ингибиторы TIM-3, ингибиторы TIGIT, ингибиторы BTLA и ингибиторы CTLA-4. Костимулирующие антитела доставляют положительные сигналы через иммуннорегуляторные рецепторы, включающие, без ограничения перечисленными, ICOS, CD137, CD27, OX-40 и GITR.
В одном крайне предпочтительном варианте осуществления дополнительное действующее вещество является антителом-ингибитором контрольной точки. Подходящие примеры антител-ингибиторов контрольных точек включают, без ограничения перечисленными, антитела против PD-1, антитела против PD-L1 и антитела против CTLA4.
В одном предпочтительном варианте осуществления антитело-ингибитор контрольной точки является антителом против PD-1, более предпочтительно выбранным из пембролизумаба, цемиплимаба и ниволумаба.
В одном предпочтительном варианте осуществления антитело-ингибитор контрольной точки является антителом против PD-L1, более предпочтительно выбранным из атезолизумаба, авелумаба и дурвалумаба.
В одном предпочтительном варианте осуществления антитело-ингибитор контрольной точки является антителом против CTLA4, более предпочтительно выбранным из ипилимумаба и тремелимумаба.
В одном предпочтительном варианте осуществления иммунотерапия является противораковой вакциной или вирусом, таким как онколитический вирус.
В одном предпочтительном варианте осуществления иммунотерапия является клеточной терапией. В одном варианте осуществления клеточная терапия может быть T-клеточной терапией, такой как адоптивная T-клеточная терапия или терапия CAR-T-клетками.
Адоптивная клеточная иммунотерапия может включать следующее: облученные аутологичные или аллогенные опухолевые клетки, лизаты опухолей или апоптотические опухолевые клетки, иммунотерапию на основе антигенпрезентирующих клеток, иммунотерапию на основе дендритных клеток, адоптивный перенос T-клеток, адоптивную CAR-T-клеточную терапию, аутологичную иммуностимулирующую терапию (AIET), противораковые вакцины и/или антигенпрезентирующие клетки. Такие клеточные иммунотерапии могут быть также модифицированы для экспрессии одного или более продуктов генов, чтобы дополнительно модулировать иммунные ответы, например, путем экспрессии цитокинов, таких как ГМ-КСФ, и/или экспрессии опухолеассоциированных антигенов (TAA), такие как Mage-1, gp-100, пациентспецифичных неоантигенных вакцин и т.п.
В другом варианте осуществления иммунотерапия может включить иммунотерапию не на основе клеток. В одном варианте осуществления могут использоваться композиции, включающие антигены с или без усиливающих вакцину адъювантов. Такие композиции существуют во многих известных формах, таких как пептидные композиции, онколитические вирусы и слитые белки, включающие рекомбинантные антигены.
В альтернативном варианте осуществления могут использоваться иммуномодулирующие интерлейкины, такие как IL-2, IL-6, IL-7, IL-12, IL-17, IL-23, а также их модуляторы (например, блокирующие антитела или более активные или более длительно действующие формы). Также могут использоваться иммуномодулирующие цитокины, такие как интерфероны, Г-КСФ, имихимод, ФНО-альфа и т.п., а также их модуляторы (например, блокирующие антитела или более активные или более длительно действующие формы). В другом варианте осуществления могут использоваться иммуномодулирующие хемокины, такие как CCL3, CCL26 и CXCL7, и т.п., а также их модуляторы (например, блокирующие антитела или более активные или более длительно действующие формы). В другом варианте осуществления могут использоваться иммуномодулирующие молекулы, направленные против иммуносупрессии, такие как модуляторы сигнализации STAT3, модуляторы сигнализации FkappaB и модуляторы иммунных контрольных точек.
В другом варианте осуществления могут использоваться иммуномодулирующие средства, такие как иммуноцитостатические средства, глюкокортикоиды, цитостатики, иммунофилины и их модуляторы (например, рапамицин, ингибитор кальциневрина, такролимус, циклоспорин, пимекролимус, абетимус, гусперимус, ридафоролимус, эверолимус, темсиролимус, зотаролимус и т.д.), гидрокортизон (кортизол), кортизона ацетат, преднизон, преднизолон, метилпреднизолон, дексаметазон, бетаметазон, триамцинолон, беклометазон, флудрокортизона ацетат, дезоксикортикостерона ацетат (doca) альдостерон, неглюкокортикоидный стероид, ингибитор синтеза пиримидинов, лефлуномид, терифлуномид, аналог фолиевой кислоты, метотрексат, антитимоцитарный глобулин, антилимфоцитарный глобулин, талидомид, леналидомид, пентоксифиллин, бупропион, куркумин, катехин, опиоид, ингибитор EVIPDH, микофеноловая кислота, мириоцин, финголимод, ингибитор NF-xB, ралоксифен, дротрекогин альфа, деносумаб, ингибиторе сигнального каскада F-xB, дисульфирам, олмесартан, дитиокарбамат, ингибиторе протеасомы, бортезомиб, MG132, Prol, PI-0052, куркумин, генистеин, ресвератрол, партенолид, талидомид, леналидомид, флавопиридол, нестероидные противовоспалительные препараты (НПВС), триоксид мышьяка, дегидроксиметилэпоксихиномицин (DHMEQ), I3C (индол-3-карбинол)/DIM (ди-индолметан) (13C/DIM), Bay 1 1-7082, лутеолин, проникающий в клетки пептид SN-50, оверэкспрессия суперрепрессора IKBa, FKB олигодезоксинуклеотид-ловушка (ODN), или производное или аналог любого из них.
В еще одном варианте осуществления могут использоваться иммуномодулирующие антитела или белок. Например, антитела, которые связываются с CD40, Toll-подобным рецептором (TLR), OX40, GITR, CD27 или с 4-1BB, T-клеточно-биспецифичные антитела, антитело против рецептора IL-2, антитело против CD3, OKT3 (муромонаб), отеликсизумаб, теплизумаб, висилизумаб, антитело против CD4, кленоликсимаб, келиксимаб, занолимумаб, антитело против CD11, эфализумаб, антитело против CD18, эрлизумаб, ровелизумаб, антитело против CD20, афутузумаб, окрелизумаб, офатумумаб, пасколизумаб, ритуксимаб, антитело против CD23, лимиликсимаб, антитело против CD40, тенеликсимаб, торализумаб, антитело против CD40L, руплизумаб, антитело против CD62L, аселизумаб, антитело против CD80, галиксимаб, антитело против CD147, гавилимомаб, ингибирующее антитело к стимулятору B-лимфоцитов (BLyS), белимумаб, слитый белок CTLA4-Ig, абатацепт, белатацепт, антитело против CTLA4, ипилимумаб, тремелимумаб, антитело против эотаксина 1, бертилимумаб, антитело против a4-интегрина, натализумаб, антитело против IL-6R, тоцилизумаб, антитело против LFA-1, одулимомаб, антитело против CD25, базиликсимаб, даклизумаб, инолимомаб, антитело против CD5, золимомаб, антитело против CD2, сиплизумаб, нерелимомаб, фаралимомаб, атлизумаб, аторолимумаб, цеделизумаб, дорлимомаб аритокс, дорликсизумаб, фонтолизумаб, гантенерумаб, гомиликсимаб, лебрилизумаб, маслимомаб, моролимумаб, пекселизумаб, реслизумаб, ровелизумаб, тализумаб, телимомаб аритокс, вапаликсимаб, вепалимомаб, афлиберцепт, алефацепт, рилонацепт, антагонист рецептора IL-1, анакинра, антитело против IL-5, меполизумаб, ингибитор IgE, омализумаб, тализумаб, ингибитор IL12, ингибитор IL23, устекинумаб.
В одном варианте осуществления субъект может подвергаться или ранее подвергался лечению химиотерапевтическим средством. Примеры химиотерапевтических средств включают, без ограничения перечисленным, алкилирующие средства, такие как тиотепа и ЦИТОКСАН циклофосфамид; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилоломеламин; ацетогенины (например, буллатацин и буллатацинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; CC-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (например, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги KW-2189 и CB 1-TM1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, холофосфамид, эстрамустин, ифосфамид, мехлорэтамин, мехлорэтамина оксида гидрохлорид, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урациловый иприт; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как эндииновые антибиотики (например, калихеамицин, в особенности калихеамицин гамма II и калихеамицин омега II (см., например, Agnew. Chem. Intl. Ed. Engl., 33: 183-186 (1994)); динемицин, включая динемицин А; бисфосфонаты, такие как клодронат; эсперамицин; а также хромофор неокарциностатина и родственные хромофоры хромопротеин ендииновых антибиотиков), аклациномицины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, каминомицин, карцинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, Адриамицин доксорубицин (включая морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, потфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-ФУ); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пуринов, например, флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидинов, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; антагонисты гормонов надпочечников, такие как аминоглутетимид, митотан, трилостан; восполнители фолиевой кислоты, такие как фолиновая кислота; ацеглатон; гликозид альдофосфамид; аминолевулиновая кислота; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатрексат; демеколцин; диазиквон; эфлорнитин; эллиптиния ацетат; эпотилон; этоглюцид; галлия нитрат; гидроксимочевину; лентинан; лонидаинин; майтанзиноиды, например, майтанзин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; PSK полисахаридный комплекс (JHS Natural Products, Eugene, Oreg.); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2"-трихлортриэтиламин; трихотецены (например, Т-2 токсин, верракурин А, роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-С"); циклофосфамид; тиотепа; таксоиды, например, Таксол (паклитаксел; Bristol-Myers Squibb Oncology, Princeton, N.J.), Абраксан, не содержащий Кремофор препарат стабилизированных альбумином наночастиц паклитаксела (American Pharmaceutical Partners, Schaumberg, Ill.) и Таксотер доцетаксел (Rhone-Poulenc Rorer, Antony, France); хлорамбуцил; Гемзар гемцитабин; 6-тиогуанин; меркаптопурин; метотрексат; платиновые аналоги, например, цисплатин и карбоплатин; винбластин; платина; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; Навелбин винорелбин; новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; Кселоду; ибандронат; иринотекан (Камптозар, СРТ-11) (включая схему лечения иринотеканом с 5-ФУ и лейковорином); ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; капецитабин; комбретастатин; лейковорин (LV); оксалиплатин, включая схему лечения оксалиплатином (FOLFOX); лапатиниб (Тайверб); ингибиторы PKC-a, Raf, H-Ras, EGFR (например, эрлотиниб (Тарцева)) и VEGF-A, которые уменьшают пролиферацию клеток, а также фармацевтически приемлемые соли, кислоты или производные любого из перечисленных выше. Кроме того, способы лечения могут дополнительно включать применение радиации. Кроме того, способы лечения могут дополнительно включать применение фотодинамической терапии.
Далее настоящее изобретение описано посредством следующих неограничивающих примеров и со ссылкой на следующие фигуры, где:
На Фигуре 1 показан клеточный эффект репрезентативных соединений 1 и 242 согласно изобретению в отношении презентации антигена, при измерении путем оценки их действия на презентацию овальбумин-специфического пептида (SIINFEKL). В частности, на Фигуре 1 показана репрезентативная кривая IC50 для примерных соединений согласно изобретению. Данные были нормализованы по сигналу, полученному в отсутствие соединения (высокий) и в отсутствие антигена (низкий), и представлены как среднее ±STD (n=2).
На Фигуре 2 показаны обобщенные данные IC50, полученные для примерных соединений 1 и 242 согласно изобретению, как определено с помощью вышеуказанного анализа презентации OVA антигена. Данные представлены как среднее ±SEM (n=6).
На Фигуре 3 показано действие соединения 1 согласно изобретению на общий процессинг антигенов, как определено при использовании линии объективного протеомного исследования. В частности, на Фигуре 3 показаны эффекты ингибирования ERAP1 миРНК и соединения (в концентрации 1 и 10 мкМ) по сравнению с контролем в отношении иммунопептидома клеток SiHa, при измерении эффекта в отношении общего процента пептидов из 8, 9, 10, 11, 12 и 13 аминокислот.
ПРИМЕРЫ
Если получение исходных соединений не описано, они доступны в продаже, известны в литературе или могут быть с легкостью получены специалистами в данной области при использовании стандартных методик. Если указано, что соединения были получены аналогично предыдущим примерам или промежуточным соединениям, для специалиста будет очевидно, что время реакции, число эквивалентов реагентов, растворитель, концентрации и температуры можно изменить для каждой конкретной реакции, и что может быть необходимо или потребуется использовать другие методы выделения или очистки.
Общие схемы
Сокращения
Ниже показан перечень некоторых общих сокращений, где другие используемые сокращения, которые не перечислены, будут понятны специалисту в данной области.
водн.: водный; ш: широкий; прибл.: приблизительно; д: дуплет; ДХМ: дихлорметан; диоксан: 1,4-диоксан; DMAP: 4-диметиламинопиридин; ДМФА: диметилформамид; EDC: гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида; Et3N: триэтиламин; EtOAc: этилацетат; EtOH: этанол; ч: часы; ВЭЖХ: высокоэффективная жидкостная хроматография; ИПС, изопропанол; ЖХ: жидкостная хроматография; м: мультиплет; M: молярный, молекулярный ион; MeCN: ацетонитрил; MeOH: метанол; мин.: минуты; МС: масс-спектрометрия; ЯМР: ядерный магнитный резонанс; PDA: фотодиодная матрица; к: квартет; КТ: комнатная температура (прибл. 20°C); RT: время удерживания; с: синглет; тв: твердый; т.: триплет; МТБЭ: метил-трет-бутиловый эфир; ТФУ: трифторуксусная кислота; ТГФ: тетрагидрофуран; СВЭЖХ: сверхвысокоэффективная жидкостная хроматография; УФ: ультрафиолетовый; колич.: количественный; SEM: [2-(триметилсилил)этокси]метилацеталь; dppf: 1,1'-ферроцендиил-бис(дифенилфосфин); NBS: N-бромсукцинимид; XantPhos-Pd-G3: метансульфонат [(4,5-бис(дифенилфосфино)-9,9-диметилксантен)-2-(2'-амино-1,1'-бифенил)]палладия (II) (CAS: 1445085-97-1); XPhos Pd G3: метансульфонат (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия (II) (CAS: 1445085-55-1); Pd-174: трифлат аллил(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)палладия (II) (CAS: 1798782-25-8); TBAF: фторид тетра-н-бутиламмония.
Другие сокращения должны иметь общепринятое значение.
Схема 1
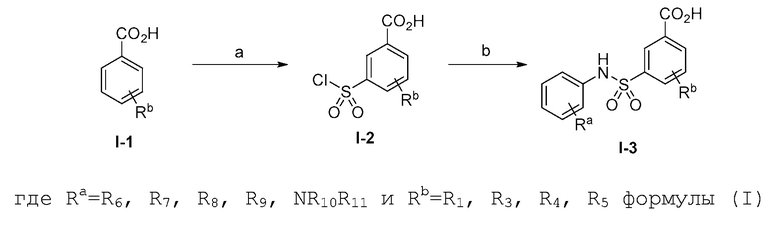
Реагенты: (a) ClSO3H, 100°C; (b) Амин, пиридин, ДХМ, КТ
Хлорсульфонилирование I-1 с использованием хлоросульфоновой кислоты давало сульфонилхлорид I-2. Его подвергали реакции с соответствующим амином в присутствии пиридина с получением сульфонамида I-3.
Схема 2
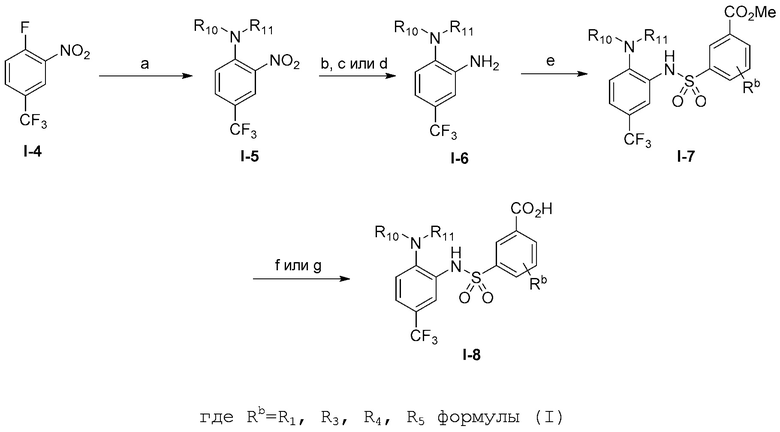
Реагенты: (a) Амин, ДХМ; (b) H2, 10% Pd/C, EtOH; (c) Fe, NH4Cl, ИПС, вода; (d) NH4OH (водн.), Na2S2O4, ТГФ, H2O, КТ; (e) сульфонилхлорид, пиридин, ДХМ, КТ; (f) LiOH (водн.), ТГФ, MeOH; (g) LiOH (водн.), диоксан.
Фтор-2-нитро-4-(трифторметил)бензол (I-4) подвергали реакции с подходящим амином в реакции нуклеофильного замещения с последующим восстановлением полученного нитросоединения I-5 до анилина I-6. Анилин подвергали реакции с соответствующим сульфонилхлоридом с получением сульфонамида I-7. Гидролиз сложного эфира давал соответствующую карбоновую кислоту I-8.
Схема 3
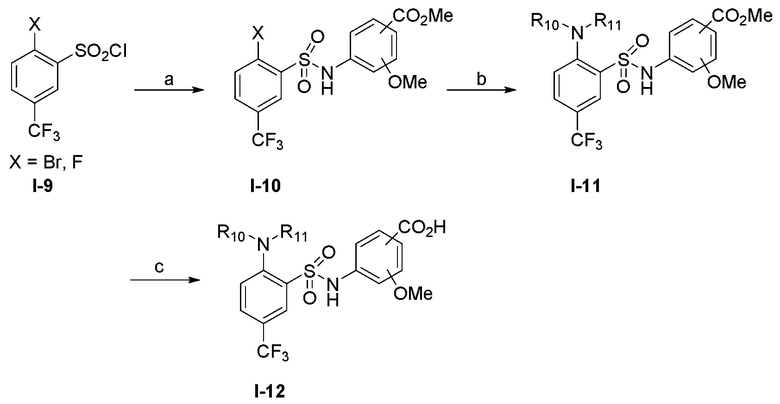
Реагенты: (a) Анилин, пиридин, ДХМ, КТ; (b) амин, ТГФ, 60°C; (c) LiOH (водн.), ТГФ, 50°C.
Сульфонилхлорид I-9 подвергали реакции с подходящим анилином с получением сульфонамида I-10. Нуклеофильное замещение соответствующим амином давало I-11, который гидролизовали с получением соответствующей карбоновой кислоты I-12.
Схема 4
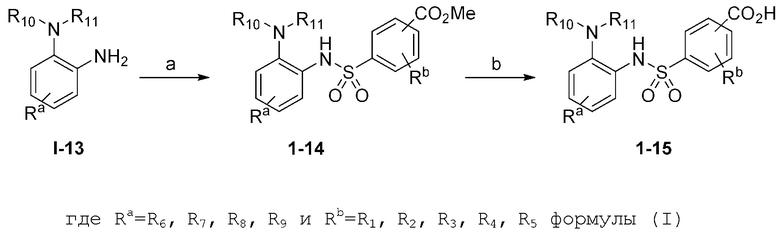
Реагенты: (a) Сульфонилхлорид, пиридин, ДХМ, КТ; (b) LiOH (водн.), диоксан или ТГФ, КТ.
Сульфонамид I-14 получали в реакции анилина I-13 с соответствующим сульфонилхлоридом. Гидролиз сложного эфира давал соответствующую карбоновую кислоту I-15.
Схема 5
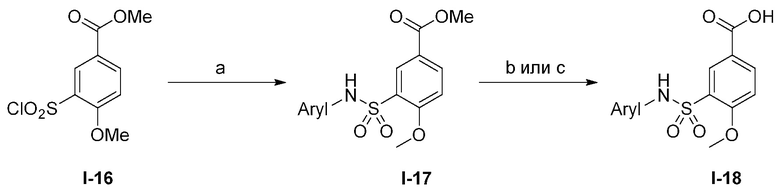
Реагенты: (a) Анилин, пиридин, ДХМ, КТ; (b) NaOH (водн.), MeOH, H2O, КТ; (c) LiOH (водн.), ТГФ, КТ.
Сульфонамид I-17 получали в реакции сульфонилхлорида I-16 с подходящим анилином. Гидролиз сложного эфира давал соответствующую карбоновую кислоту I-18.
Схема 6
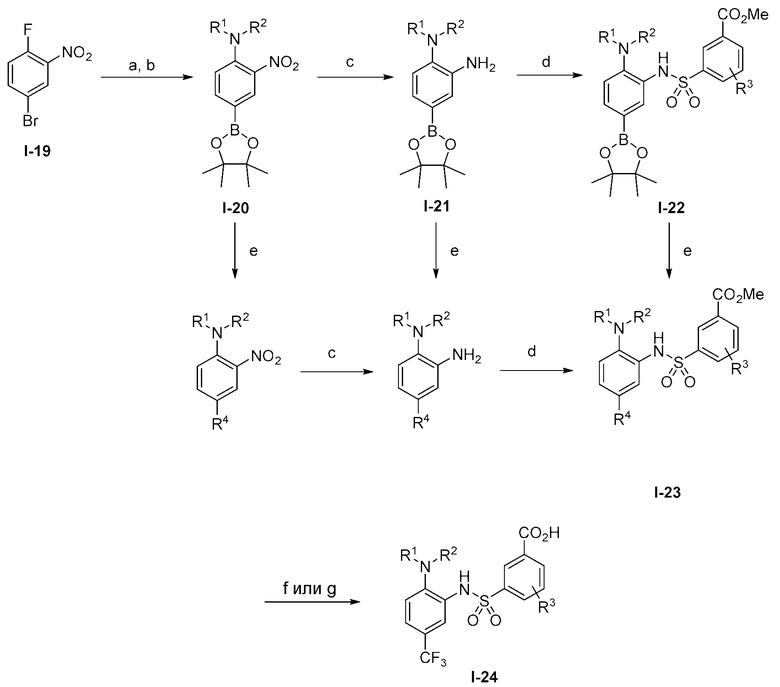
Реагенты: (a) Амин, MeCN; (b) Бис(пинаколато)дибор, PdCl2(dppf)∙ДХМ, KOAc, диоксан; (c) H2, Pd/C, MeOH; (d) сульфонилхлорид, пиридин, ДХМ, КТ; (e) Арилгалогенид, Xphos Pd G3, K3PO4, диоксан, вода; (f) LiOH (водн.), ТГФ, MeOH; (g) HCl, диоксан.
4-Бром-1-фтор-2-нитробензол (I-19) подвергали реакции с подходящим амином в реакции нуклеофильного замещения с последующим превращением полученного арилбромида в боронатный эфир I-20. Затем нитрогруппу восстанавливали с получением соответствующего анилина I-21. Его подвергали реакции с подходящим сульфонилхлоридом с получением сульфонамида I-22. Оставшийся заместитель вводили с помощью сочетания Сузуки перед гидролизом сложного эфира с получением соответствующей карбоновой кислоты I-24. В альтернативе стадии могут быть выполнены в другой последовательности, как указано.
Общие условия экспериментов
Все исходные соединения и растворители получали из коммерческих источников или получали в соответствии с литературными источниками. Реакционные смеси перемешивали на магнитной мешалке, а реакции проводили при комнатной температуре (прибл. 20°C), если не указано иное. Колоночную хроматографию выполняли на автоматической системе флэш-хроматографии, такой как система CombiFlash Rf, с использованием предварительно набитых картриджей с силикагелем (40 мкм), если не указано иное. Спектры 1H-ЯМР регистрировали с использованием спектрометра Bruker Avance III HD при 500 МГц, оборудованного Bruker 5 мм SmartProbe™. Химические сдвиги выражены в милионных долях с использованием в качестве стандарта центральных пиков остаточного протонного растворителя, либо внутреннего стандарта тетраметилсилана. Спектры регистрировали при 298 K, если не указано иное. Аналитические эксперименты СВЭЖХ-МС для определения времени удерживания и связанных масс ионов проводили с использованием системы Waters ACQUITY UPLC® H-Class, оборудованной детектором ACQUITY PDA и масс-детектором ACQUITY QDa, с использованием одного из аналитических методов, описанных ниже. Аналитические эксперименты ЖХ-МС для определения времени удерживания и связанных масс ионов проводили с использованием системы ВЭЖХ Agilent 1200 серии, сопряженной с одноквадрупольным масс-спектрометром Agilent серии 1956, 6100 или 6120, при использовании одного из аналитических методов, описанных ниже. Очистки с помощью препаративной ВЭЖХ проводили либо при использовании колонки Waters X-Select CSH C18, 5 мкм, 19×50 мм в градиенте MeCN и воды, которые содержали 0,1% об/об муравьиной кислоты в качестве модификатора, либо на колонке Waters X-Bridge BEH C18, 5 мкм, 19×50 мм при использовании градиента MeCN и 10 мМ бикарбоната аммония (водн.). Фракции собирали с последующей УФ-детекцией при одной длине волны, при измерении детектором с переменной длиной волны. Номенклатура структур была создана с использованием преобразования "Структуры в название" из ChemDraw® Professional 17 (PerkinElmer).
Аналитические методы
Метод 1 - Кислотный 3 мин метод
Колонка: Waters ACQUITY СВЭЖХ® CSH C18, 1,7 мкм, 2,1×30 мм при 40°C
Детекция: УФ при 254 нм, если не указано иное, МС с электрораспылительной ионизацией
Растворители: A: 0,1% об/об муравьиной кислоты в воде, B: 0,1% об/об муравьиной кислоты в MeCN
Градиент:
Метод 2 - Основный 3 мин метод
Колонка: Waters ACQUITY СВЭЖХ® BEH C18, 1,7 мкм, 2,1×30 мм при 40°C
Растворители: A: 10 мМ бикарбонат аммония (водн.), B: MeCN
(другие параметры такие же, как в Методе 1)
Метод 3 - Кислотный 4 мин метод
Колонка: Waters X-Select CSH C18, 2,5 мкм, 4,6×30 мм при 40°C
Детекция: УФ при 254 нм, если не указано иное, МС с электрораспылительной ионизацией
Растворители: A: 0,1% об/об муравьиной кислоты в воде, B: 0,1% об/об муравьиной кислоты в MeCN
Градиент:
Метод 4 - Основный 4 мин метод
Колонка: Waters X-Bridge BEH C18, 2,5 мкм, 4,6×30 мм при 40°C
Растворители: A: 10 мМ бикарбонат аммония (водн.), B: MeCN
(другие параметры такие же, как в Методе 3)
Пример 1: 4-Этил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензойная кислота
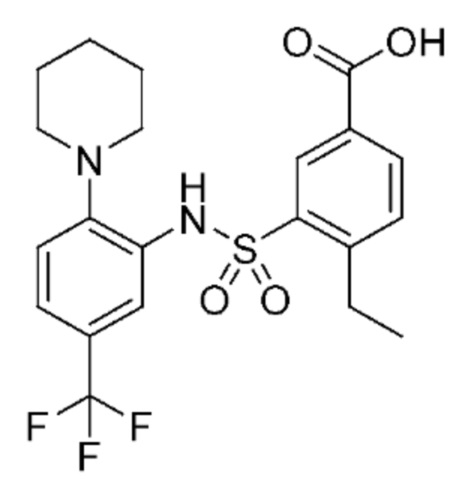
Стадия 1: 3-(хлорсульфонил)-4-этилбензойная кислота: 4-этилбензойную кислоту (1 г, 6,66 ммоль) в хлоросульфоновой кислоте (10 мл, 149 ммоль) нагревали при 100°C в течение ночи. Смесь охлаждали и осторожно добавляли в перемешиваемый лед. Образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (1,58 г, 6,04 ммоль, выход 91%, чистота 95%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,34 (д, J=1,9 Гц, 1H), 7,82 (дд, J=7,9, 2,0 Гц, 1H), 7,32 (д, J=7,9 Гц, 1H), 3,08 (к, J=7,5 Гц, 2H), 1,19 (т, J=7,5 Гц, 3H). Один обмениваемый протон не наблюдали.
Стадия 2: 4-Этил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензойная кислота: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0,200 г, 0,819 ммоль) в пиридине (3 мл, 37,1 ммоль) обрабатывали продуктом из стадии 1 выше (0,244 г, 0,983 ммоль) и перемешивали при КТ в течение 24 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексаны, затем 0-50% EtOAc/ДХМ) с получением указанного в заголовке соединения (36,3 мг, 0,076 ммоль, выход 9,23%, чистота 97%) в виде рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 457,4 (M+H)+, 455,2 (M-H)- через 1,87 мин 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,28 (шс, 1H), 9.44 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,42 (дд, J=8,4, 2,2 Гц, 1H), 7,29 (д, J=2,1 Гц, 1H), 7,25 (д, J=8,4 Гц, 1H), 3,04 (к, J=7,4 Гц, 2H), 2,72 (т, J=4,9 Гц, 4H), 1,57 (п, J=5,0 Гц, 4H), 1,50-1,45 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 3: 4-изопропил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
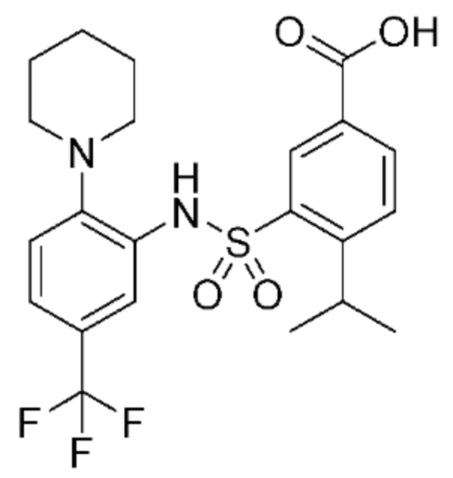
Стадия 1: 3-(хлорсульфонил)-4-изопропилбензойная кислота: 4-изопропилбензойную кислоту (1 г, 6,09 ммоль) в хлоросульфоновой кислоте (5 мл, 74,7 ммоль) нагревали при 100°C в течение ночи. Смесь охлаждали и осторожно добавляли в перемешиваемый лед. Образовавшийся осадок собирали с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (1,28 г, 4,63 ммоль, выход 76%, чистота 95%) в виде рыжевато-коричневого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 12,50 (шс, 1H), 8,36 (д, J=1,9 Гц, 1H), 7,83 (дд, J=8,1, 1,9 Гц, 1H), 7,44 (д, J=8,1 Гц, 1H), 4,20 (септет, J=6,8 Гц, 1H), 1,16 (д, J=6,9 Гц, 6H).
Стадия 2: 4-изопропил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0,070 г, 0,287 ммоль) в ДХМ (1 мл) и пиридине (0,139 мл, 1,720 ммоль) добавляли в раствор продукта из стадии 1 выше (0,090 г, 0,344 ммоль) в ДХМ (1 мл) и перемешивали при КТ в течение 16 ч. Растворитель удаляли в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/ДХМ) с получением указанного в заголовке соединения (14,3 мг, 0,029 ммоль, выход 10%, чистота 95%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,4 (M+H)+, 469,3 (M-H)- через 1,93 мин 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (шс, 1H), 9,40 (шс, 1H), 8,45 (д, J=1,9 Гц, 1H), 8,13 (дд, J=8,3, 1,9 Гц, 1H), 7,76 (д, J=8,2 Гц, 1H), 7,42 (дд, J=8,2, 1,9 Гц, 1H), 7,27 (д, J=8,3 Гц, 1H), 7,19 (д, J=1,9 Гц, 1H), 3,86 (септет, J=6,8 Гц, 1H), 2,78 (т, J=5,2 Гц, 4H), 1,58 (п, J=5,5 Гц, 4H), 1,51-1,45 (м, 2H), 1,24-1,10 (м, 6H).
Пример 4: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(трифторметокси)бензойная кислота
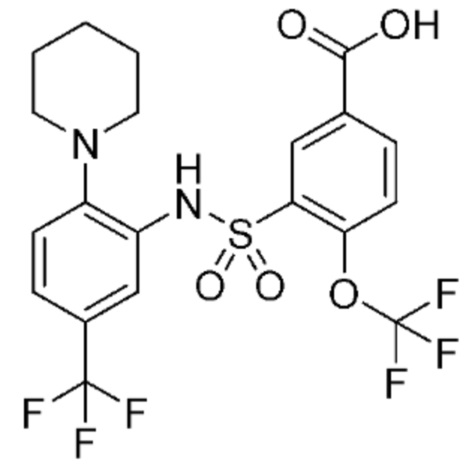
Стадия 1: 3-(хлорсульфонил)-4-(трифторметокси)бензойная кислота: 4-(трифторметокси)бензойную кислоту (1 г, 4,85 ммоль) в хлоросульфоновой кислоте (5 мл, 74,7 ммоль) нагревали при 100°C в течение ночи. Смесь охлаждали и осторожно добавляли в перемешиваемый лед. Образовавшийся осадок собирали с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (0,770 г, 2,28 ммоль, выход 46,9%, чистота 90%) в виде кремового твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 12,50 (шс, 1H), 8,40 (д, J=2,2 Гц, 1H), 8,00 (дд, J=8,5, 2,2 Гц, 1H), 7,41 (дк, J=8,5, 1,8 Гц, 1H).
Стадия 2: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(трифторметокси)бензойная кислота: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0,070 г, 0,287 ммоль) в ДХМ (1 мл) и пиридине (0,139 мл, 1,72 ммоль) добавляли в раствор продукта из стадии 1 выше (0,105 г, 0,344 ммоль) в ДХМ (1 мл) и перемешивали при КТ в течение 16 ч. Растворитель удаляли в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/ДХМ) с получением указанного в заголовке соединения (5,6 мг, 10,4 мкмоль, выход 3,6%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 513,3 (M+H)+, 511,1 (M-H)- через 1,94 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,68 (шс, 1H), 9,50 (шс, 1H), 8,45 (д, J=1,7 Гц, 1H), 8,27 (дд, J=8,2, 1,5 Гц, 1H), 7,68 (д, J=8,7 Гц, 1H), 7,46-7,44 (м, 2H), 7,27 (д, J=8,2 Гц, 1H), 2,71 (т, J=5,0 Гц, 4H), 1,62-1,34 (м, 6H).
Пример 6: 3-(N-(2-(цис-3,5-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
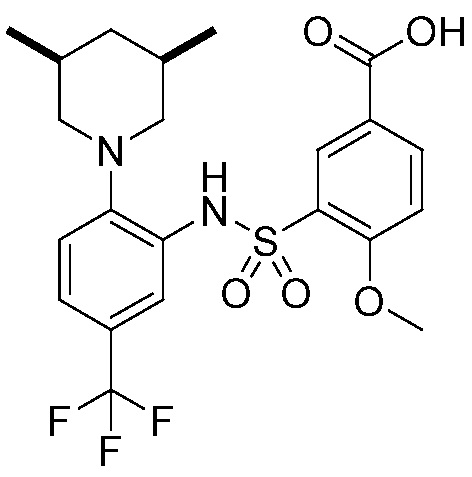
Стадия 1: цис-3,5-диметил-1-(2-нитро-4-(трифторметил)-фенил)пиперидин: Et3N (0,5 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и цис-3,5-диметилпиперидина (211 мг, 1,87 ммоль) в ДХМ (6 мл), и полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc в изогексанах, затем 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (356 мг, 1,12 ммоль, выход 78%, чистота 95%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 303,4 (M+H)+ через 2,01 мин.
Стадия 2: 2-(цис-3,5-диметилпиперидин-1-ил)-5-(трифторметил)анилин: Продукт из Стадии 1 выше (150 мг, 0,496 ммоль) растворяли в EtOH (9,9 мл), и реакционную смесь гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (133 мг, 0,479 ммоль, выход 96%, чистота 98%) в виде бледно-коричневого масла. СВЭЖХ-МС (Метод 2) m/z 273,3 (M+H)+, 271,1 (M-H)- через 2,00 мин.
Стадия 3: метил-3-(N-(2-(цис-3,5-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из Стадии 2 выше (51,4 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (50 мкл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь наносили непосредственно и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (63 мг, 0,120 ммоль, выход 63,3%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 501,4 (M+H)+, 498,9 (M-H)- через 2,05 мин.
Стадия 4: 3-(N-(2-(цис-3,5-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из Стадии 3 выше (61 мг, 0,122 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (443 мкл, 0,487 ммоль). По каплям добавляли MeOH до образования прозрачного раствора. Реакционную смесь нагревали при 40°C в течение 24 ч, затем охлаждали до КТ в течение ночи. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и разбавляли полученный водный раствор водой (5 мл). По каплям добавляли 1М HCl (водн.) до приблизительно pH 6. Полученный белый осадок собирали с помощью фильтрации и промывали водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (55 мг, 0,113 ммоль, выход 88%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,4 (M+H)+, 485,2 (M-H)- через 1,89 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (с, 1H), 8,82 (с, 1H), 8,34 (д, J=2,3 Гц, 1H), 8,15 (дд, J=8,7, 2,3 Гц, 1H), 7,47 (д, J=2,1 Гц, 1H), 7,36 (дд, J=8,5, 2,1 Гц, 1H), 7,30 (д, J=8,7 Гц, 1H), 7,29 (д, J=8,5 Гц, 1H), 3,84 (с, 3H), 2,89-2,80 (м, 2H), 2,14 (т, J=11,0 Гц, 2H), 1,82-1,65 (м, 3H), 0,81 (д, J=6,4 Гц, 6H), 0,67-0,59 (м, 1H).
Пример 7: 3-(N-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
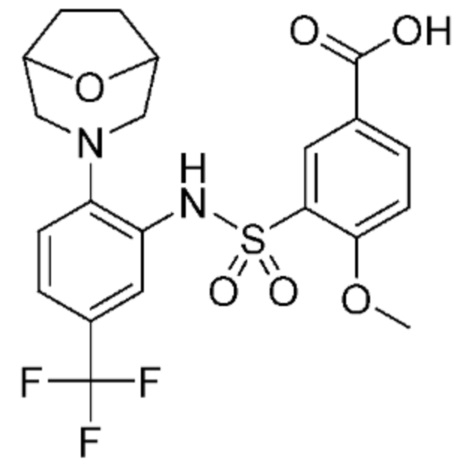
Стадия 1: 3-(2-нитро-4-(трифторметил)фенил)-8-окса-3-азабицикло[3.2.1]октан: Et3N (0,583 мл, 4,18 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,167 мл, 1,20 ммоль) и гидрохлорида 8-окса-3-азабицикло[3.2.1]октана (221 мг, 1,44 ммоль) в ДХМ (5 мл) и перемешивали полученный раствор при КТ в течение 2 ч. Добавляли 1 М HCl (водн.) (2 мл), органическую фазу отделяли при пропускании через сепаратор фаз и выпаривали в вакууме с получением указанного в заголовке соединения (384 мг, 1,08 ммоль) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 303,2 (M+H)+ через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,13-8,08 (м, 1H), 7,84 (дд, J=8,9, 2,3 Гц, 1H), 7,46 (д, J=8,9 Гц, 1H), 4,39-4,32 (м, 2H), 3,16-3,11 (м, 2H), 3,02-2,97 (м, 2H), 1,89-1,77 (м, 4H).
Стадия 2: 2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)анилин: Продукт из Стадии 1 выше (323 мг, 1,07 ммоль) растворяли в EtOH (21,2 мл) и реакционную смесь гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 4 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (8 мл) с получением указанного в заголовке соединения (310 мг, 1,059 ммоль, выход 100%, чистота 93%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 273,3 (M+H)+ через 1,43 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,05 (д, J=8,2 Гц, 1H), 7,02 (д, J=2,2 Гц, 1H), 6,86 (дд, J=8,2, 2,2 Гц, 1H), 5,01 (br s, 2H), 4,36-4,31 (м, 2H), 2,88-2,82 (м, 2H), 2,79 (дд, J=11,5, 2,0 Гц, 2H), 2,09-2,03 (м, 2H), 1,88-1,80 (м, 2H).
Стадия 3: метил-3-(N-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (58 мкл, 0,72 ммоль) добавляли к раствору продукта из стадии 2 выше (66,5 мг, 0,239 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) в ДХМ (2 мл) при КТ. Полученный раствор перемешивали при 40°C в течение 4 ч, после чего добавляли дополнительное количество метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) и пиридин (58 мкл, 0,718 ммоль), и перемешивали смесь при 40°C в течение еще 19 ч. Реакционную смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (25 г картридж, 0-80% EtOAc/изогексаны) с получением указанного в заголовке соединения (88,3 мг, 0,173 ммоль, выход 72,3%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 501,3 (M+H)+, 499,2 (M-H)- через 1,59 мин.
Стадия 4: 3-(N-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1 М LiOH (водн.) (0,699 мл, 0,699 ммоль) добавляли к раствору продукта из стадии 3 выше (87,4 мг, 0,175 ммоль) в ТГФ (1,4 мл) при КТ, и полученный раствор перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме, а остаток снова растворяли в воде (3 мл) и поокисляли с использованием 1 М HCl (водн.) до pH 4-5. Осадок отделяли с помощью фильтрации, а затем сушили с получением указанного в заголовке соединения (74 мг, 0,152 ммоль, выход 87%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,1 (M-H)- через 1,45 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,13 (шс, 1H), 8,88 (шс, 1H), 8,24 (д, J=2,2 Гц, 1H), 8,18 (дд, J=8,7, 2,2 Гц, 1H), 7,46-7,34 (м, 2H), 7,27 (д, J=8,5 Гц, 1H), 6,98 (д, J=2,1 Гц, 1H), 4,40-4,33 (м, 2H), 3,95 (с, 3H), 3,01 (д, J=11,2 Гц, 2H), 2,95 (дд, J=11,6, 2,0 Гц, 2H), 2,13-2,05 (м, 2H), 1,92-1,84 (м, 2H).
Пример 8: 4-метокси-3-(N-(2-(цис-5-метилгексагидро-пирроло[3,4-c]пиррол-2-(1H)-ил)-5-(трифторметил)фенил)-сульфамоил)бензойная кислота
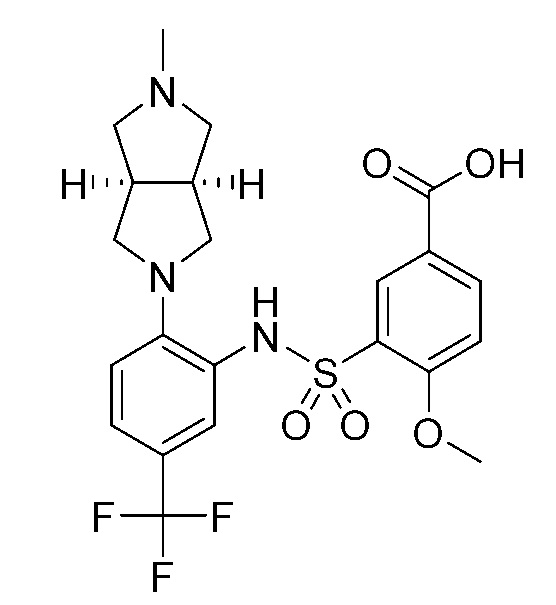
Стадия 1: цис-2-метил-5-(2-нитро-4-(трифторметил)фенил)-октагидропирроло[3,4-c]пиррол: Et3N (0,417 мл, 2,99 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,167 мл, 1,20 ммоль) и цис-2-метилоктагидропирроло[3,4-c]пиррола (187 мг, 1,44 ммоль) в ДХМ (5 мл) при КТ и перемешивали полученный раствор при КТ в течение 2 ч. Добавляли 1М HCl (водн.) (2 мл), органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (402 мг, 1,20 ммоль, колич. выход, чистота 93%) в виде оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 316,3 (M+H)+ через 1,40 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,04-8,01 (м, 1H), 7,72 (дд, J=9,1, 2,4 Гц, 1H), 7,22 (д, J=9,0 Гц, 1H), 3,49-3,42 (м, 2H), 3,13 (дд, J=10,8, 3,4 Гц, 2H), 2,94-2,85 (м, 2H), 2,53-2,44 (м, 4H), 2,24 (с, 3H). Сигнал при 2,49 м.д. перекрыт сигналом ДМСО.
Стадия 2: 2-(цис-5-метилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (376 мг, 1,19 ммоль) растворяли в EtOH (23,9 мл), и реакционную смесь гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (12 мл) с получением указанного в заголовке соединения (355 мг, 1,17 ммоль, выход 98%, чистота 94%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) 286,3 (M+H)+ через 1,24 мин.
Стадия 3: метил-4-метокси-3-(N-(2-(цис-5-метилгексагидро-пирроло[3,4-c]пиррол-2(1H)-ил)-5-(трифторметил)фенил)-сульфамоил)бензоат: Пиридин (58 мкл, 0,72 ммоль) добавляли к суспензии продукта из стадии 2 выше (72,6 мг, 0,239 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) в ДХМ (2 мл) при КТ. Полученный раствор перемешивали при 40°C в течение 4 ч, после чего добавляли дополнительное количество метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) и пиридин (0,058 мл, 0,718 ммоль) и перемешивали смесь при 40°C в течение еще 19 ч. Реакционную смесь выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (25 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (158 мг, 0,193 ммоль, выход 81%, чистота 63%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 514,4 (M+H)+, 512,2 (M-H)- через 1,26 мин.
Стадия 4: 4-метокси-3-(N-(2-(цис-5-метилгексагидро-пирроло[3,4-c]пиррол-2(1H)-ил)-5-(трифторметил)фенил)-сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,23 мл, 1,23 ммоль) добавляли к раствору продукта из стадии 3 выше (158 мг, 0,308 ммоль) в ТГФ (2,5 мл) при КТ и перемешивали раствор при КТ в течение 26 ч. Реакционную смесь выпаривали в вакууме, остаток повторно растворяли в воде (3 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Осадок выделяли с помощью фильтрации и затем сушили в вакууме с получением указанного в заголовке соединения (63,5 мг, 0,127 ммоль, выход 41,3%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 500,3 (M+H)+, 498,3 (M-H)- через 0,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,22 (д, J=2,2 Гц, 1H), 8,13 (дд, J=8,7, 2,2 Гц, 1H), 7,31 (д, J=8,7 Гц, 1H), 7,28-7,24 (м, 1H), 6,97-6,91 (м, 2H), 3,90 (с, 3H), 3,36 (дд, J=9,8, 6,5 Гц, 2H), 3,22 (дд, J=10,0, 2,7 Гц, 2H), 2,86-2,80 (м, 2H), 2,75-2,69 (м, 2H), 2,64-2,59 (м, 2H), 2,38 (с, 3H). Два обмениваемых протона не наблюдали.
Пример 9: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
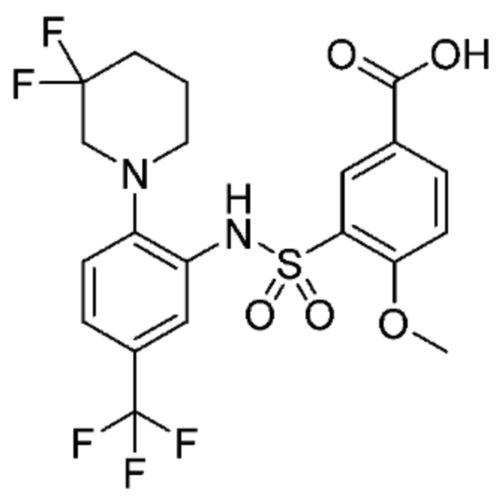
Стадия 1: 3,3-дифтор-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и гидрохлорида 3,3-дифторпиперидина (271 мг, 1,72 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли воду (3 мл) и разделяли фазы с помощью фазового сепаратора. Водную фазу экстрагировали ДХМ (2×3 мл), органические фазы объединяли, сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (399 мг, 1,26 ммоль, выход 87,8%, чистота >98%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 309,0 (M-H)- через 1,64 мин.
Стадия 2: 2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)-анилин: Продукт из стадии 1 выше (156 мг, 0,503 ммоль) растворяли в EtOH (10,1 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (119 мг, 0,408 ммоль, выход 81%, чистота 96%) в виде бесцветного масла. СВЭЖХ-МС (Метод 2) m/z 280,8 (M+H)+ через 1,64 мин.
Стадия 3: метил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (53,0 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60,0 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 4 дней. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (39,9 мг, 0,075 ммоль, выход 39,4%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 509,4 (M+H)+, 507,2 (M-H)- через 1,75 мин.
Стадия 4: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (38 мг, 0,075 ммоль) растворяли в ТГФ (2 мл), обрабатывали 1,1М LiOH (водн.) (272 мкл, 0,299 ммоль) и по каплям добавляли MeOH, пока смесь не становилась раствором. Реакционную смесь перемешивали при 30°C в течение 4 дней. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл), и нейтрализовали до приблизительно pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора. Белый осадок собирали с помощью фильтрации, промывали водой, и твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (34 мг, 0,065 ммоль, выход 87%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 495,1 (M+H)+, 493,1 (M-H)- через 1,59 мин, чистота 98% (254 нм). 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (шс, 1H), 8,60 (шс, 1H), 8,37 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,41-7,36 (м, 3H), 7,32 (д, J=8,8 Гц, 1H), 3,91 (с, 3H), 3,17 (т, J=11,1 Гц, 2H), 2,95 (т, J=5,3 Гц, 2H), 2,13-2,00 (м, 2H), 1,88-1,84 (м, 2H).
Пример 10: 3-(N-(2-(8-азабицикло[3.2.1]октан-8-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
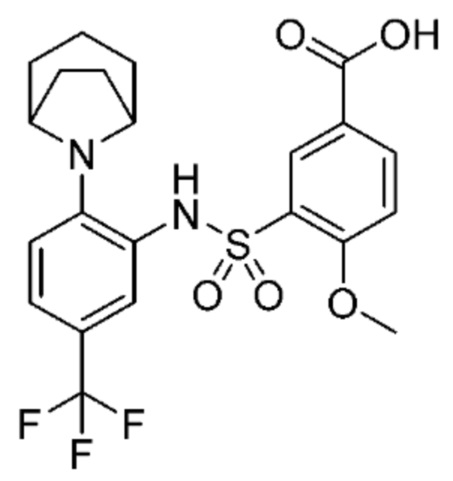
Стадия 1: 8-(2-нитро-4-(трифторметил)фенил)-8-азабицикло[3.2.1]октан: Et3N (0,236 мл, 1,69 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,095 мл, 0,677 ммоль) и гидрохлорида 8-азабицикло[3.2.1]октана (100 мг, 0,677 ммоль) в ДХМ (2 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли воду (3 мл) и разделяли фазы с помощью фазового сепаратора. Водную фазу экстрагировали ДХМ (2×3 мл) и органические фазы объединяли, сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (183 мг, 0,597 ммоль, выход 88,2%, чистота 98%). СВЭЖХ-МС (Метод 2) m/z 301,3 (M+H)+ через 1,85 мин.
Стадия 2: 2-(8-азабицикло[3.2.1]октан-8-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (134 мг, 0,446 ммоль) растворяли в EtOH (8,9 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (104 мг, 0,366 ммоль, выход 82%, чистота 95%) в виде бесцветного масла. СВЭЖХ-МС (Метод 2) m/z 271,3 (M+H)+ через 1,83 мин.
Стадия 3: метил-3-(N-(2-(-8-азабицикло[3.2.1]октан-8-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (51,1 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60,0 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 4 дней. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (32,1 мг, 0,061 ммоль, выход 32,4%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 499,3 (M+H)+, 497,2 (M-H)- через 1,90 мин.
Стадия 4: 3-(N-(2-(-8-азабицикло[3.2.1]октан-8-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (30 мг, 0,060 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1М LiOH (водн.) (219 мкл, 0,241 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 4 дней. Реакционную смесь разбавляли водой (3 мл), упаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл), и нейтрализовали до прибл. pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, осадок собирали с помощью фильтрации и промывали водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (9,0 мг, 0,018 ммоль, выход 29,3%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,2 (M+H)+, 483,3 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,12 (шс, 1H), 8,96 (шс, 1H), 8,21 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,8, 2,2 Гц, 1H), 7,34 (д, J=8,7 Гц, 1H), 7,27 (дд, J=8,7, 2,3 Гц, 1H), 7,01 (д, J=8,7 Гц, 1H), 6,95-6,92 (м, 1H), 4,29 (с, 2H), 3,93 (с, 3H), 1,91-1,86 (м, 2H), 1,79-1,68 (м, 6H), 1,55-1,46 (м, 1H), 1,45-1,37 (м, 1H).
Пример 11: 3-(N-(2-(5-окса-2-азаспиро[3.4]октан-2-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
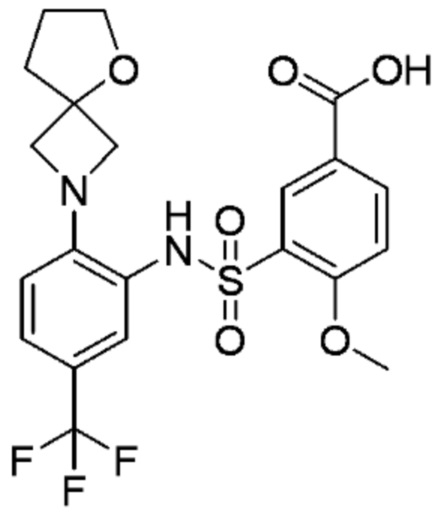
Стадия 1: 2-(2-нитро-4-(трифторметил)фенил)-5-окса-2-азаспиро[3.4]октан: Et3N (500 мкл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и гемиоксалата 5-окса-2-азаспиро[3.4]октана (349 мг, 2,21 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (438 мг, 1,44 ммоль, выход 100%, чистота 99%) в виде светло-желтого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 303,3 (M+H)+ через 1,59 мин.
Стадия 2: 2-(5-окса-2-азаспиро[3.4]октан-2-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (217 мг, 0,718 ммоль) растворяли в EtOH (14,4 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (198 мг, 0,691 ммоль, выход 96%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 273,3 (M+H)+ через 1,37 мин.
Стадия 3: метил-3-(N-(2-(5-окса-2-азаспиро[3.4]октан-2-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (0,073 г, 0,268 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,087 мл, 1,07 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (0,085 г, 0,321 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (93,7 мг, 0,178 ммоль, выход 70,0%, чистота 95%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 501,4 (M+H)+, 498,8 (M-H)- через 1,54 мин.
Стадия 4: 3-(N-(2-(5-окса-2-азаспиро[3.4]октан-2-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (92 мг, 0,184 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1М LiOH (водн.) (668 мкл, 0,735 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 3 дней. Реакционную смесь разбавляли водой (3 мл), упаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл), и нейтрализовали до прибл. pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора. Белый осадок собирали с помощью фильтрации, промывали водой и твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (3 мг, 5,98 мкмоль, выход 3,25%, чистота 97%) в виде рыхлого белого вещества. СВЭЖХ-МС (Метод 1) m/z 487,0 (M+H)+, 485,2 (M-H)- через 1,37 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,33 (д, J=2,2 Гц, 1H), 8,29 (дд, J=8,7, 2,2 Гц, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,30 (дд, J=8,6, 2,2 Гц, 1H), 6,70 (д, J=2,1 Гц, 1H), 6,55 (д, J=8,6 Гц, 1H), 4,21 (д, J=9,0 Гц, 2H), 4,08 (д, J=9,0 Гц, 2H), 4,02 (с, 3H), 3,88 (т, J=7,0 Гц, 2H), 2,20 (т, J=7,0 Гц, 2H), 2,00 (п, J=7,0 Гц, 2H). Два обмениваемых протона не наблюдали.
Пример 12: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
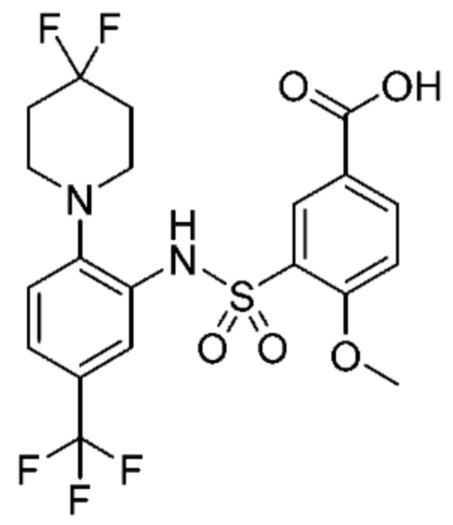
Стадия 1: 4,4-дифтор-1-(2-нитро-4-(трифторметил)-фенил)пиперидин: Et3N (0,47 мл, 3,37 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,188 мл, 1,34 ммоль) и 4,4-дифторпиперидина (196 мг, 1,62 ммоль) в ДХМ (5 мл) и перемешивали полученный раствор при КТ в течение 19 ч. Добавляли воду (2,5 мл), органическую фазу отделяли с помощью фазового сепаратора и выпаривали в вакууме с получением указанного в заголовке соединения (434 мг, 1,04 ммоль, 77% выход, 74% чистота) в виде оранжевого масла. СВЭЖХ (Метод 2) 1,67 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,20 (д, J=2,3 Гц, 1H), 7,89 (дд, J=8,9, 2,4 Гц, 1H), 7,53 (д, J=8,8 Гц, 1H), 3,28-3,23 (м, 4H), 2,16-2,06 (м, 4H).
Стадия 2: 2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)-анилин: Продукт из стадии 1 выше (180 мг, 0,580 ммоль) растворяли в EtOH (23,2 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 21°C, скорость потока 1 мл/мин, 1 цикл). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (8 мл) с получением указанного в заголовке соединения (159 мг, 0,545 ммоль, выход 94%, чистота 96%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 281,3 (M+H)+ через 1,63 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,04 (д, J=8,1 Гц, 1H), 6,97 (д, J=2,2 Гц, 1H), 6,82 (дд, J=8,2, 2,1 Гц, 1H), 5,27 (с, 2H), 2,93 (шт, J=5,5 Гц, 4H), 2,24-2,09 (м, 4H).
Стадия 3: метил-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,058 мл, 0,718 ммоль) добавляли к раствору продукта из стадии 2 выше (69,8 мг, 0,239 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) в ДХМ (2,0 мл) при КТ. Реакционную смесь перемешивали и нагревали при 40°C в течение 18 ч. Добавляли дополнительное количество метил-3-(хлорсульфонил)-4-метоксибензоата (33 мг, 0,120 ммоль) и перемешивали полученный раствор при 40°C еще в течение 3 ч. Реакционную смесь выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-30% EtOAc/изогексаны) с получением указанного в заголовке соединения (107 мг, 0,196 ммоль, выход 82%, чистота 93%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 509,3 (M+H)+, 507,2 (M-H)- через 1,72 мин.
Стадия 4: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,632 мл, 0,632 ммоль) добавляли к раствору продукта из стадии 3 выше (107 мг, 0,210 ммоль) в ТГФ (1,26 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 20 ч. Добавляли дополнительное количество 1М LiOH (водн.) (0,211 мл, 0,211 ммоль) и перемешивали раствор еще в течение 1 ч. Реакционную смесь выпаривали в вакууме, а остаток повторно растворяли в воде (3 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Осадок растворяли в ДХМ (10 мл) и разделяли фазы. Водную фазу экстрагировали ДХМ (2×3 мл). Объединенные органические фазы сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-3,5% MeOH/ДХМ) с получением почти белого твердого вещества (40,1 мг). Продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (19 мг, 0,038 ммоль, выход 18,3%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 495,3 (M+H)+, 493,2 (M-H)- через 1,61 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,15 (шс, 1H), 9,30 (шс, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,3 Гц, 1H), 7,48-7,44 (м, 1H), 7,41-7,35 (м, 1H), 7,35 (д, J=8,5 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 3,87 (с, 3H), 2,96-2,86 (м, 4H), 2,18-2,08 (м, 4H).
Пример 13: 3-(N-(2-(8-гидрокси-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
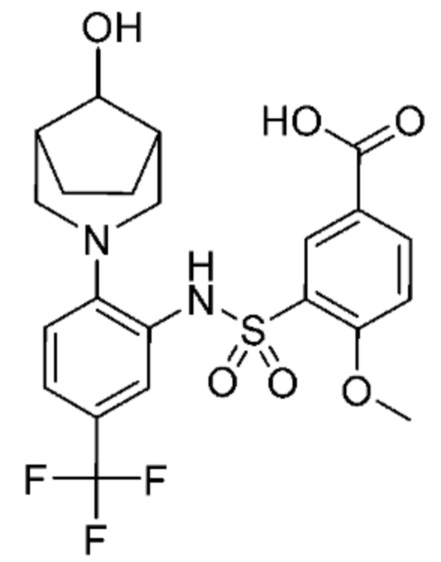
Стадия 1: 3-(2-нитро-4-(трифторметил)фенил)-3-азабицикло[3.2.1]октан-8-ол: Et3N (500 мкл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и гидрохлорида 3-азабицикло[3.2.1]октан-8-ола (250 мг, 1,53 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (468 мг, 1,44 ммоль, выход 100%, чистота 97%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 315,1 (M-H)- через 1,53 мин.
Стадия 2: 3-(2-амино-4-(трифторметил)фенил)-3-азабицикло[3.2.1]октан-8-ол: Продукт из стадии 1 выше (227 мг, 0,718 ммоль) растворяли в EtOH (14,4 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, полный водород, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (186 мг, 0,585 ммоль, выход 81%, чистота 90%) в виде светло-розового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 287,3 (M+H)+, 285,2 (M-H)- через 1,38 мин.
Стадия 3: метил-3-(N-(2-(8-гидрокси-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (63,1 мг, 0,220 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (71,3 мкл, 0,882 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (70 мг, 0,264 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (51 мг, 0,087 ммоль, выход 39,6%, чистота 88%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 515,4 (M+H)+, 513,2 (M-H)- через 1,60 мин.
Стадия 4: 3-(N-(2-(8-гидрокси-3-азабицикло[3.2.1]октан-3-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (49 мг, 0,095 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1М LiOH (водн.) (346 мкл, 0,381 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до прибл. pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до прибл. 2 мл. Осадок собирали с помощью фильтрации и промывали водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл) и выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (21,9 мг, 0,042 ммоль, выход 44,6%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 501,3 (M+H)+, 499,2 (M-H)- через 1,42 мин, 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,17 (с, 1H), 8,69 (с, 1H), 8,34 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,40-7,32 (м, 3H), 7,17 (д, J=1,6 Гц, 1H), 5,07 (с, 1H), 3,93 (с, 3H), 3,90-3,82 (м, 1H), 3,33-3,31 (м, 2H), 2,61 (дд, J=10,7, 3,6 Гц, 2H), 2,01-1,97 (м, 2H), 1,86-1,73 (м, 4H).
Следующие примеры были получены способами, аналогичными Примеру 13, с заменой подходящими исходными соединениями и промежуточными соединениями при необходимости:
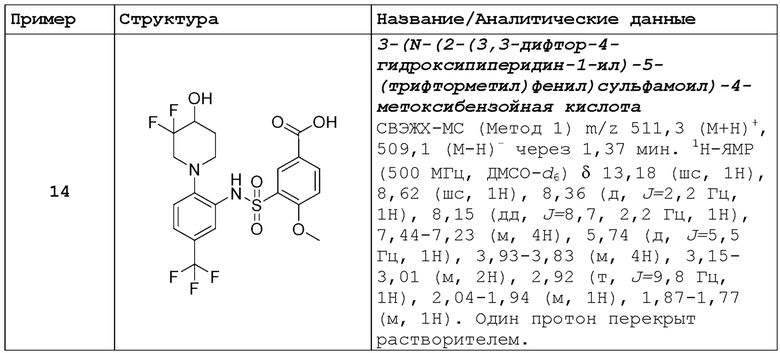
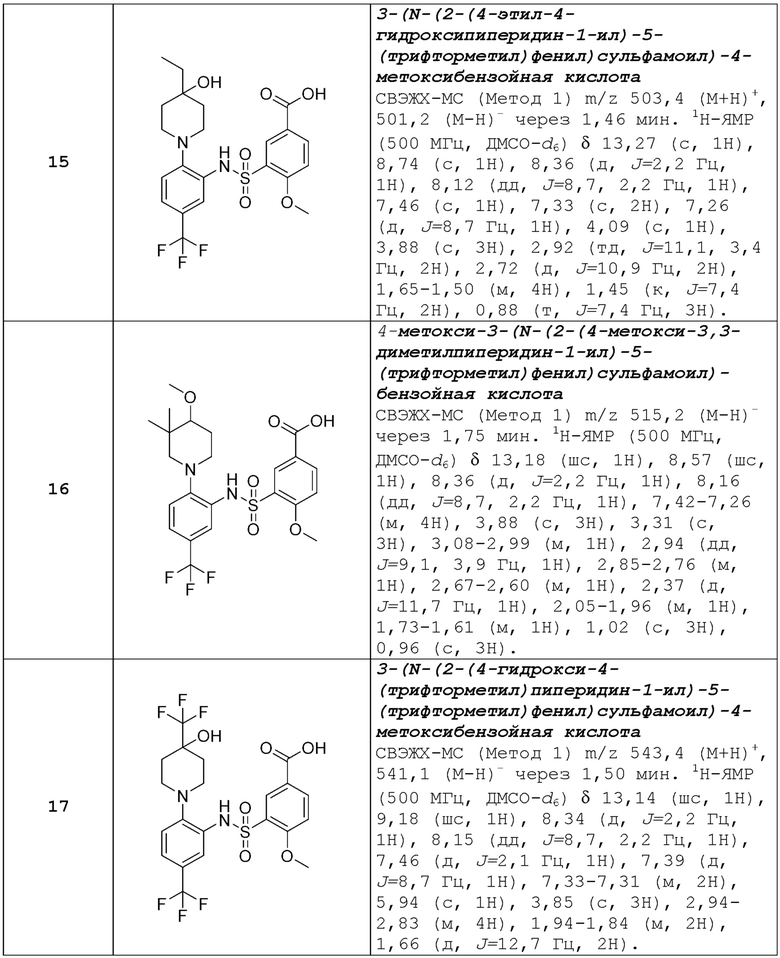
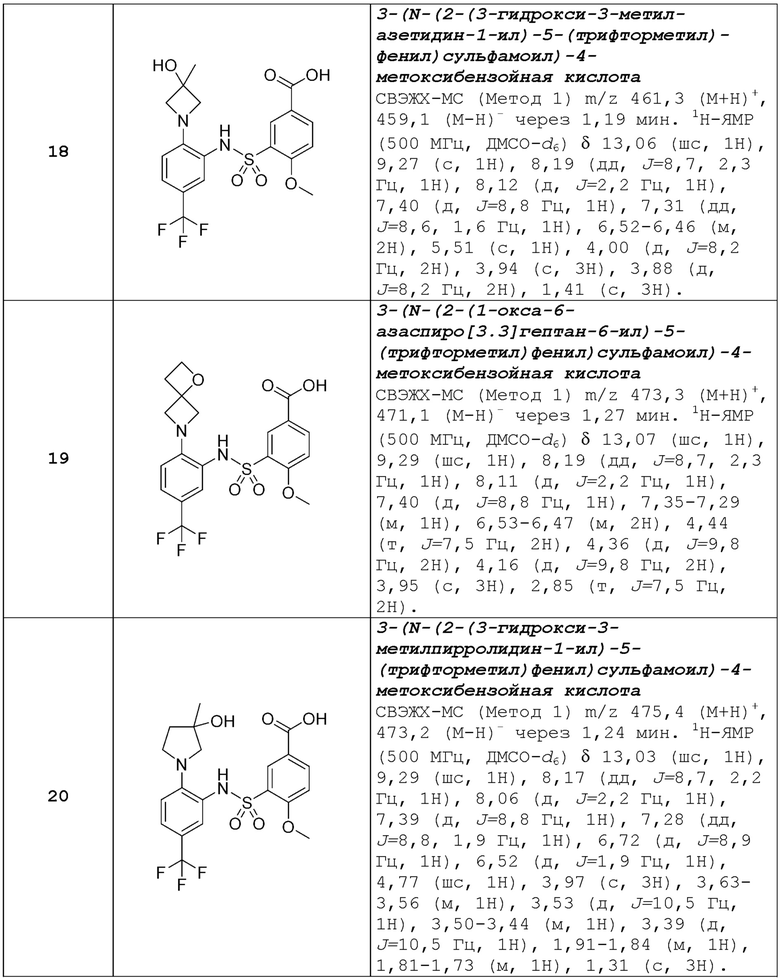
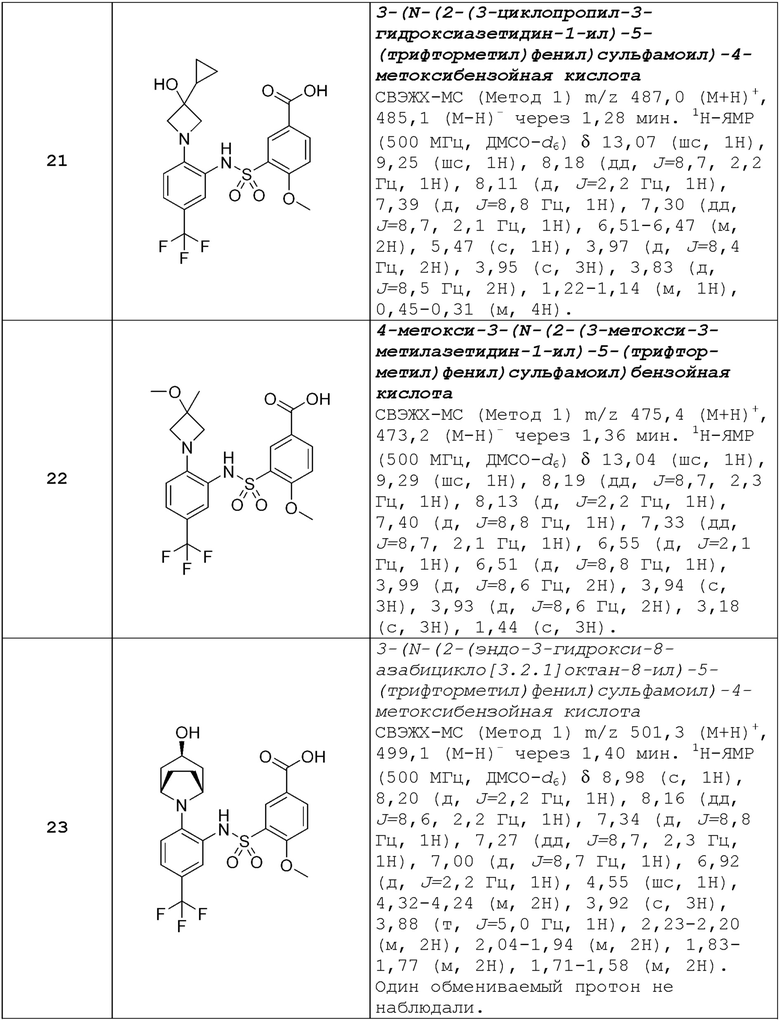
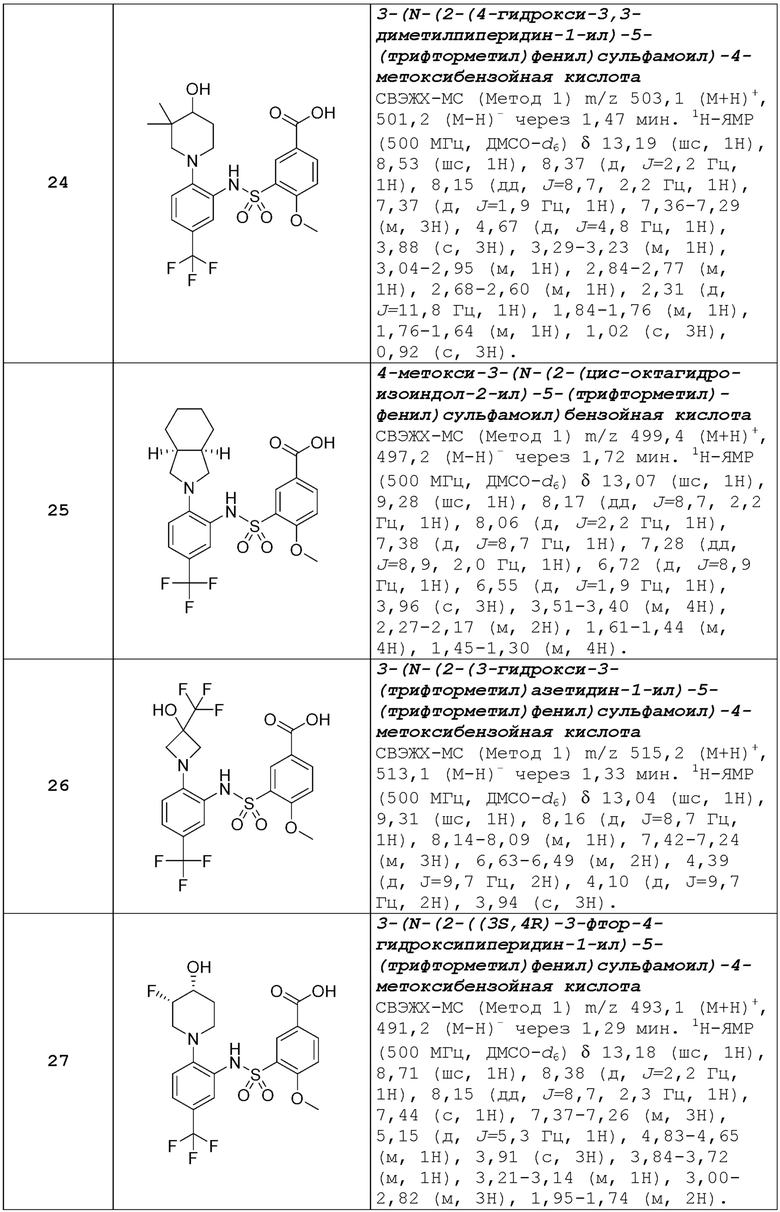
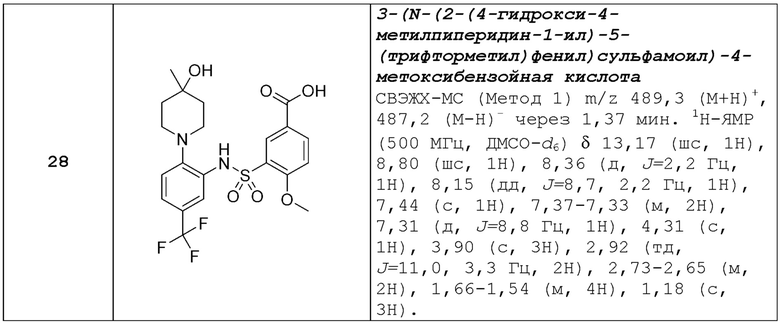
Пример 29: 3-(N-(2-(3-гидрокси-3-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
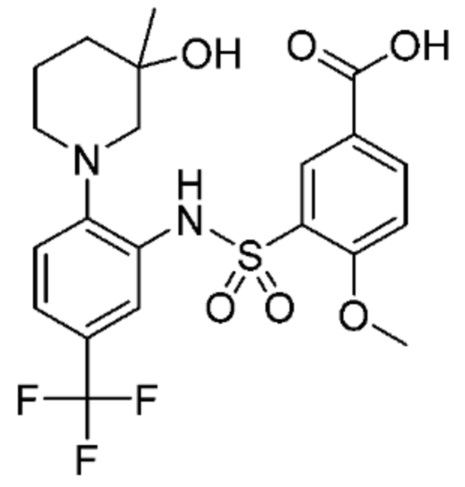
Стадия 1: 3-метил-1-(2-нитро-4-(трифторметил)-фенил)пиперидин-3-ол: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и 3-метилпиперидин-3-ола (198 мг, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 17 ч. Органическую фазу промывали 1М HCl (водн.) (3 мл) и сушили органическую фазу при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (452 мг, 1,35 ммоль, выход 94%, чистота 91%) в виде красно-оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 305,2 (M+H)+ через 1,49 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,07 (д, J=2,3 Гц, 1H), 7,76 (дд, J=9,0, 2,4 Гц, 1H), 7,44 (д, J=8,9 Гц, 1H), 4,51 (с, 1H), 3,16 (ддд, J=13,2, 6,1, 3,7 Гц, 1H), 3,08 (ддд, J=12,8, 8,3, 3,2 Гц, 1H), 3,00 (д, J=12,6 Гц, 1H), 2,90 (д, J=12,7 Гц, 1H), 1,87-1,76 (м, 1H), 1,60-1,55 (м, 2H), 1,55-1,48 (м, 1H), 1,10 (с, 3H).
Стадия 2: 1-(2-амино-4-(трифторметил)фенил)-3-метилпиперидин-3-ол: 5% Pd/C (50% об/об воды) Тип 87L (50 мг, 0,012 ммоль) в EtOH (0,5 мл) добавляли к раствору продукта из стадии 1 выше (224 мг, 0,670 ммоль) в EtOH (3,0 мл) при КТ. Реакционную смесь гидрировали под давлением 4 бара при КТ в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Органическую фазу выпаривали в вакууме, а остаток повторно растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (112 мг, 0,404 ммоль, выход 60,3%, чистота 99%) в виде бледно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 275,3 (M+H)+ через 1,42 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,94 (д, J=8,1 Гц, 1H), 6,92 (д, J=1,8 Гц, 1H), 6,81 (дд, J=8,1, 1,8 Гц, 1H), 5,27 (шс, 2H), 4,58 (с, 1H), 2,91-2,81 (м, 1H), 2,73-2,67 (м, 1H), 2,60-2,51 (м, 2H), 1,95-1,84 (м, 1H), 1,60-1,50 (м, 2H), 1,47-1,38 (м, 1H), 1,15 (с, 3H).
Стадия 3: метил-3-(N-(2-(3-гидрокси-3-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,075 мл, 0,933 ммоль) добавляли к мутному раствору продукта из стадии 2 выше (64,6 мг, 0,233 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (78 мг, 0,280 ммоль) в ДХМ (2,0 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 20 ч. Реакционную смесь выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 30-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (98,5 мг, 0,196 ммоль, выход 84%, чистота 100%) в виде почти белой пены. СВЭЖХ-МС (Метод 1) m/z 503,4 (M+H)+, 501,2 (M-H)- через 1,66 мин.
Стадия 4: 3-(N-(2-(3-гидрокси-3-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,784 мл, 0,784 ммоль) добавляли к раствору продукта из стадии 3 выше (98,5 мг, 0,196 ммоль) в ТГФ (1,57 мл) при КТ. Раствор перемешивали при КТ в течение 18 ч, а затем выпаривали в вакууме. Остаток повторно растворяли в воде (3 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Осадок выделяли с помощью фильтрации, а затем повторно растворяли в EtOAc (5 мл). Органическую фазу промывали водой (3 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (64 мг, 0,130 ммоль, выход 73,4%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,4 (M+H)+, 487,3 (M-H)- через 1,49 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,14 (шс, 1H), 9,44 (шс, 1H), 8,41 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,54 (д, J=2,1 Гц, 1H), 7,30 (дд, J=8,4, 1,7 Гц, 1H), 7,27 (д, J=8,8 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 5,02 (шс, 1H), 3,78 (с, 3H), 2,93-2,85 (м, 1H), 2,63 (тд, J=11,1, 2,4 Гц, 1H), 2,56-2,52 (м, 1H), 2,52-2,48 (м, 1H), 2,03-1,90 (м, 1H), 1,62-1,55 (м, 1H), 1,54-1,46 (м, 1H), 1,37 (тд, J=12,6, 4,5 Гц, 1H), 1,02 (с, 3H).
Пример 30: 3-(N-(2-(цис-3,5-диметилпиперидин-1-ил)-5-(трифторметил)фенилсульфамоил)-4-этилбензойная кислота
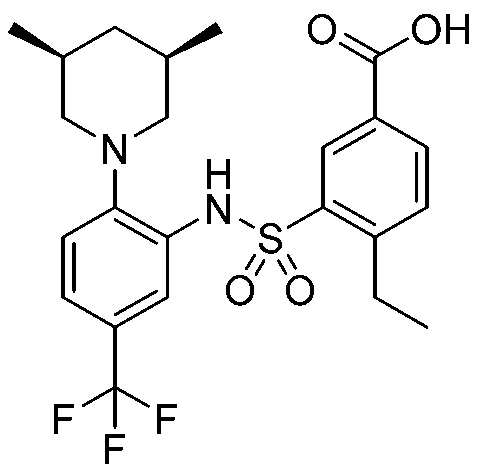
Раствор продукта из примера 6, стадии 2, (72 мг, 0,264 ммоль) в ДХМ (1 мл) и пиридине (0,128 мл, 1,59 ммоль) добавляли к суспензии продукта из примера 1, стадии 1 (79 мг, 0,317 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 4 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ). Продукт хроматографии делили между изогексанами (3 мл) и MeCN (3 мл). Фазы разделяли, фазу MeCN промывали изогексанами (2×3 мл) и выпаривали в вакууме. Продукт наносили на слой силикагеля в минимальном количестве ДХМ, колонку элюировали ДХМ (5 мл), изогексанами (5 мл), 5% MeOH в EtOAc (5 мл), затем 5% MeOH в EtOAc (5 мл), с получением указанного в заголовке соединения (26,7 мг, 0,052 ммоль, выход 19,80%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,4 (M+H)+, 483,3 (M-H)- через 2,06 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,54 (д, J=1,8 Гц, 1H), 8,15 (дд, J=8,0, 1,8 Гц, 1H), 7,59 (д, J=2,0 Гц, 1H), 7,55 (д, J=8,0 Гц, 1H), 7,36-7,27 (м, 2H), 3,07 (к, J=7,5 Гц, 2H), 2,79-2,72 (м, 2H), 2,18 (т, J=11,1 Гц, 2H), 1,89-1,76 (м, 3H), 1,28 (т, J=7,5 Гц, 3H), 0,90 (д, J=6,5 Гц, 6H), 0,75-0,64 (м, 1H). Два обмениваемых протона не наблюдали.
Пример 31: 3-(N-(2-(2,2-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
Стадия 1: 2,2-диметил-1-(2-нитро-4-(трифторметил)-фенил)пиперидин: Et3N (0.500 мл, 3,59 ммоль) добавляли к раствору 2,2-диметилпиперидина (195 мг, 1,72 ммоль) и 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 96 ч. Добавляли дополнительное количество 2,2-диметилпиперидина (75 мг, 0,663 ммоль) и перемешивали реакцию при КТ в течение 1 дня. Добавляли воду (3 мл) и разделяли фазы, после чего водную фазу экстрагировали ДХМ (2×3 мл). Органические фазы объединяли, сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (163 мг, 0,512 ммоль, выход 35,7%, чистота 95%) в виде темно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 303,3 (M+H)+ через 1,96 мин.
Стадия 2: 2-(2,2-диметилпиперидин-1-ил)-5-(трифторметил)-анилин: Порошок железа (297 мг, 5,33 ммоль) добавляли в раствор продукта из стадии 1 выше (161 мг, 0,533 ммоль) и хлорида аммония (34,2 мг, 0,639 ммоль) в ИПС (5 мл) и воде (2,5 мл) при КТ. Полученную суспензию нагревали и перемешивали при 90°C в течение 1 ч, затем охлаждали до КТ в течение ночи. Добавляли дополнительную порцию порошка железа (297 мг, 5,33 ммоль) и нагревали реакцию при 90°C в течение еще 2 ч, после чего охлаждали до КТ. Реакционную смесь фильтровали через Celite®, промывали избытком MeOH (100 мл) и выпаривали в вакууме. Остаток повторно растворяли в ДХМ (25 мл) и промывали водой (5 мл). Водную фазу экстрагировали ДХМ (2×5 мл) и объединенные органические фазы промывали солевым раствором (10 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (78 мг, 0,215 ммоль, выход 40,3%, чистота 75%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 2) m/z 273,3 (M+H)+ через 1,95 мин.
Стадия 3: метил-3-(N-(2-(2,2-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (51,4 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Реакционную смесь наносили непосредственно на силикагель и очищали с помощью колоночной хроматографии (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (64 мг, 0,121 ммоль, выход 64,3%, чистота 100%) в виде белого клейкого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 501,4 (M+H)+, 499,1 (M-H)- через 1,95 мин.
Стадия 4: 3-(N-(2-(2,2-диметилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (62 мг, 0,124 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1М LiOH (водн.) (450 мкл, 0,495 ммоль). Реакцию перемешивали при КТ в течение 1 дня. По каплям добавляли MeOH, пока смесь не становилась раствором, реакционную смесь нагревали при 40°C в течение 4 ч и затем охлаждали до КТ в течение ночи. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (5 мл). По каплям добавляли 1М HCl (водн.) до прибл. pH 6. Полученный белый осадок собирали с помощью фильтрации, промывали водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (57 мг, 0,111 ммоль, выход 90%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,2 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 8,96 (с, 1H), 8,41 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,59 (с, 1H), 7,47 (д, J=8,3 Гц, 1H), 7,33-7,27 (м, 2H), 3,93 (с, 3H), 1,73-1,55 (м, 6H), 1,32-0,62 (м, 8H).
Пример 32: 3-(N-(2-(1,4-оксазепан-4-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота
Стадия 1: 4-(2-нитро-4-(трифторметил)фенил)-1,4-оксазепан: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и гидрохлорида 1,4-оксазепана (237 мг, 1,72 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 7 дней. Добавляли воду (3 мл) и разделяли фазы с помощью фазового сепаратора. Водную фазу экстрагировали ДХМ (2×3 мл), и органические фазы объединяли, сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения в виде вязкого оранжевого масла (429 мг, 1,14 ммоль, выход 98%, чистота 95%). СВЭЖХ-МС (Метод 2) m/z 290,8 (M+H)+ через 1,48 мин.
Стадия 2: 2-(1,4-оксазепан-4-ил)-5-(трифторметил)анилин: Порошок железа (822 мг, 14,71 ммоль) добавляли в раствор продукта из стадии 1 выше (427 мг, 1,471 ммоль) и хлорида аммония (94 мг, 1,765 ммоль) в ИПС (5 мл) и воде (2,5 мл) при КТ. Полученную суспензию нагревали и перемешивали при 90°C в течение 1 ч, затем охлаждали до КТ. Реакционную смесь фильтровали через Celite®, промывали избытком MeOH (100 мл) и выпаривали в вакууме. Остаток повторно растворяли в ДХМ (25 мл) и промывали водой (5 мл). Водную фазу экстрагировали ДХМ (2×5 мл), и объединенные органические фазы промывали солевым раствором (10 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (186 мг, 0,700 ммоль, выход 47,6%, чистота 98%) в виде темно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 261,3 (M+H)+ через 1,39 мин.
Стадия 3: метил-3-(N-(2-(1,4-оксазепан-4-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (54,8 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Реакционную смесь наносили непосредственно на силикагель и очищали с помощью колоночной хроматографии (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (66 мг, 0,132 ммоль, выход 70,1%, чистота 98%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,2 (M-H)- через 1,59 мин.
Стадия 4: 3-(N-(2-(1,4-оксазепан-4-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (64 мг, 0,131 ммоль) растворяли в ТГФ (2 мл), обрабатывали 1,1М LiOH (водн.) (476 мкл, 0,524 ммоль) и перемешивали при КТ в течение 1 дня. По каплям добавляли MeOH, пока смесь не становилась раствором, реакционную смесь нагревали при 40°C в течение 4 ч, затем охлаждали при КТ в течение ночи. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученнем водного раствора, разбавляли водой (прибл. до 5 мл) и нейтрализовали до прибл. pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, белый осадок собирали с помощью фильтрации и промывали водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (60 мг, 0,120 ммоль, выход 92%, чистота 95%) в виде бледно-серого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 473,3 (M-H)- через 1,43 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,12 (с, 1H), 9,11 (с, 1H), 8,23 (с, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,38-7,32 (м, 2H), 7,23 (д, J=8,5 Гц, 1H), 7,10 (с, 1H), 3,93 (с, 3H), 3,76-3,70 (м, 4H), 3,29-3,20 (м, 4H), 1,91 (т, J=5,8 Гц, 2H).
Пример 33: 3-(N-(2-(спиро[изобензофуран-1,4'-пиперидин]-1'-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
Стадия 1: 1'-(2-нитро-4-(трифторметил)фенил)-спиро[изобензофуран-1,4'-пиперидин]: Et3N (0,417 мл, 2,99 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,167 мл, 1,20 ммоль) и гидрохлорида спиро[изобензофуран-1,4'-пиперидина] (324 мг, 1,44 ммоль) в ДХМ (6 мл) при КТ и перемешивали реакционную смесь при КТ в течение 68 ч. Добавляли воду (2 мл) и разделяли фазы. Водную фазу экстрагировали ДХМ (2×3 мл) и объединенные органические фазы сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (536 мг, 0,907 ммоль, выход 76%, чистота 64%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 379,2 (M+H)+ через 1,91 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,17 (д, J=1,6 Гц, 1H), 7,86 (дд, J=8,9, 2,3 Гц, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,34-7,27 (м, 4H), 5,04 (с, 2H), 3,39-3,29 (м, 4H), 2,06 (дт, J=17,4, 5,8 Гц, 2H), 1,74 (дд, J=13,9, 2,5 Гц, 2H).
Стадия 2: 2-(спиро[изобензофуран-1,4'-пиперидин]-1'-ил)-5-(трифторметил)анилин: Порошок железа (335 мг, 6,00 ммоль) добавляли в раствор продукта из стадии 1 выше (227 мг, 0,600 ммоль) и хлорида аммония (38,5 мг, 0,720 ммоль) в ИПС (3,5 мл) и воде (1,25 мл) и нагревали до 90°C в течение 2 ч. Реакционную смесь охлаждали до КТ, фильтровали и промывали избытком MeOH (100 мл). Фильтрат выпаривали в вакууме, повторно растворяли в ДХМ (25 мл) и промывали водой (5 мл). Водную фазу экстрагировали ДХМ (2×5 мл), и объединенные органические фазы промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-35% EtOAc/изогексаны) с получением указанного в заголовке соединения (144 мг, 0,401 ммоль, выход 66,8%, чистота 97%) в виде оранжевого порошка. СВЭЖХ-МС (Метод 1) m/z 349,2 (M+H)+ через 1,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,36-7,24 (м, 4H), 7,07 (д, J=8,1 Гц, 1H), 6,98 (д, J=2,1 Гц, 1H), 6,85 (дд, J=8,2, 2,1 Гц, 1H), 5,22 (с, 2H), 5,03 (с, 2H), 3,12-3,01 (м, 2H), 2,91 (тд, J=12,0, 2,3 Гц, 2H), 2,18 (тд, J=12,9, 4,5 Гц, 2H), 1,79-1,67 (м, 2H).
Стадия 3: метил-3-(N-(2-(спиро[изобензофуран-1,4'-пиперидин]-1'-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,058 мл, 0,718 ммоль) добавляли к раствору продукта из стадии 2 выше (86 мг, 0,239 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,287 ммоль) в ДХМ (2 мл) при КТ. Реакционную смесь перемешивали и нагревали при 40°C в течение 18 ч. Добавляли дополнительное количество метил-3-(хлорсульфонил)-4-метоксибензоата (33 мг, 0,120 ммоль) и перемешивали реакционную смесь при 40°C еще в течение 3 ч. Реакционную смесь выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (25 г картридж, 0-45% EtOAc/изогексаны) с получением указанного в заголовке соединения (117 мг, 0,187 ммоль, выход 78%, чистота 92%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 577,4 (M+H)+, 575,2 (M-H)- через 1,89 мин.
Стадия 4: 3-(N-(2-(спиро[изобензофуран-1,4'-пиперидин]-1'-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,812 мл, 0,812 ммоль) добавляли к раствору продукта из стадии 3 выше (117 мг, 0,203 ммоль) в ТГФ (1,6 мл) при КТ. Раствор перемешивали при КТ в течение 25 ч, после чего выпаривали в вакууме. Остаток повторно растворяли в воде (3 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Осадок выделяли с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (92 мг, 0,164 ммоль, выход 81%, чистота 94%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 563,3 (M+H)+, 561,1 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,02 (шс, 1H), 8,39 (д, J=2,3 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,51 (д, J=1,7 Гц, 1H), 7,39-7,27 (м, 7H), 5,02 (с, 2H), 3,88 (с, 3H), 3,01 (т, J=11,9 Гц, 2H), 2,96-2,90 (м, 2H), 2,19-2,08 (м, 2H), 1,73-1,65 (м, 2H). Один обмениваемый протон не наблюдали.
Следующие примеры были получены способами, аналогичными Примеру 33, с заменой подходящими исходными соединениями и промежуточными соединениями при необходимости:
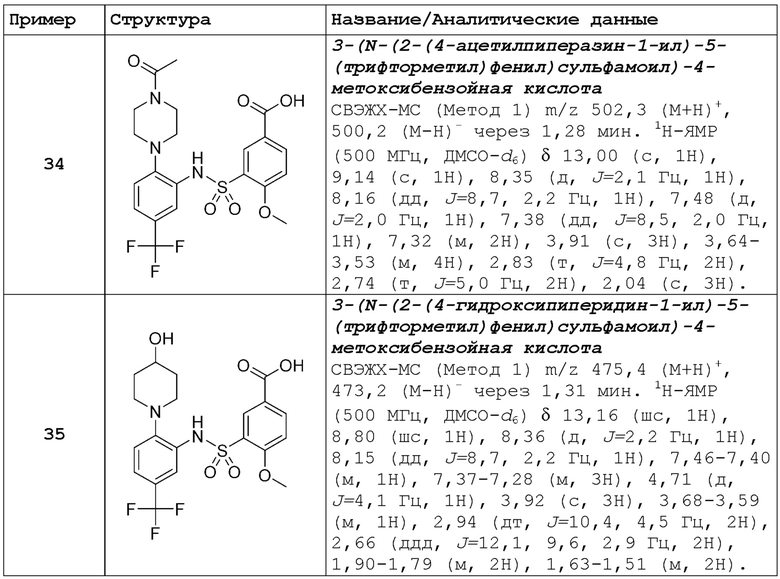
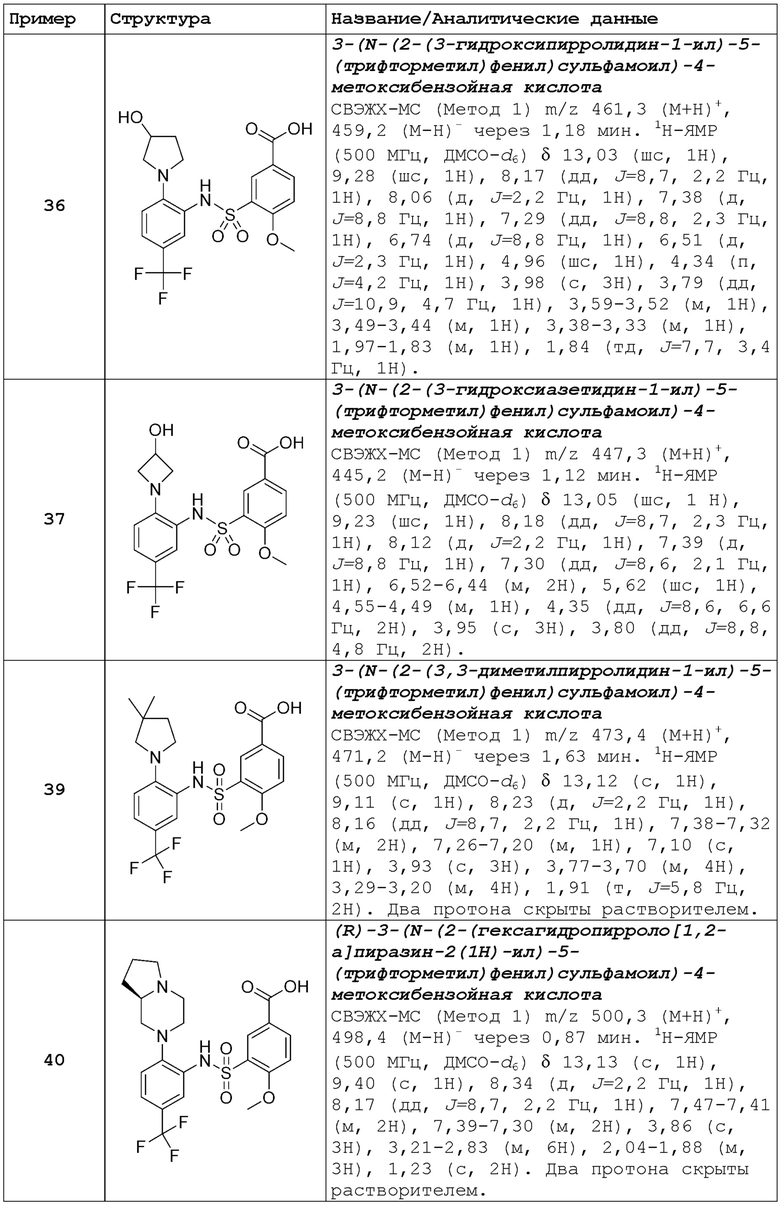
Пример 41: 4-метокси-3-(N-(2-(2-оксопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
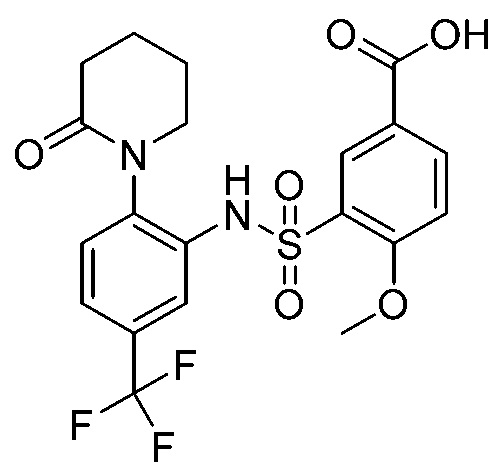
Стадия 1: 1-(2-нитро-4-(трифторметил)фенил)пиперидин-2-он: NaH (63,1 мг, 1,58 ммоль, 60% об/об в минеральном масле) добавляли к раствору пиперидин-2-она (142 мг, 1,44 ммоль) в безводном ДМФА (3 мл) при 0°C под N2. Реакцию перемешивали при этой температуре в течение 10 мин, после чего по каплям добавляли раствор 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) в безводном ДМФА (3 мл) при 0°C. Реакцию перемешивали при КТ в течение ночи. Реакционную смесь разбавляли EtOAc (100 мл) и последовательно промывали водой (50 мл) и солевым раствором (2×50 мл). Органическую фазу отделяли, сушили над MgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (245 мг, 0,808 ммоль, выход 56,3%, чистота 100%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 289,5 (M+H)+ через 1,23 мин.
Стадия 2: 1-(2-амино-4-(трифторметил)фенил)пиперидин-2-он: Порошок железа (508 мг, 9,09 ммоль) добавляли к суспензии продукта из стадии 1 выше (131 мг, 0,455 ммоль) и хлорида аммония (29,2 мг, 0,545 ммоль) в пропан-2-оле (5 мл) и воде (2,5 мл) при КТ. Полученную суспензию нагревали и перемешивали при 90°C в течение 2 ч. Реакцию фильтровали через Celite®, промывали избытком MeOH (100 мл) и выпаривали в вакууме. Остаток повторно растворяли в ДХМ (25 мл) и последовательно промывали водой (10 мл) и солевым раствором (10 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (22 мг, 0,076 ммоль, выход 16,7%, чистота 89%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 259,3 (M+H)+ через 1,07 мин.
Стадия 3: метил-4-метокси-3-(N-(2-(2-оксопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (22 мг, 0,085 ммоль) растворяли в смеси ДХМ (0,5 мл) и пиридина (22,5 мкл, 0,279 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (27,1 мг, 0,102 ммоль) в ДХМ (0,5 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Добавляли еще метил-3-(хлорсульфонил)-4-метоксибензоат (11,3 мг, 0,043 ммоль) и пиридин (6,89 мкл, 0,085 ммоль) и перемешивали реакционную смесь при КТ в течение 1 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (18,6 мг, 0,037 ммоль, выход 43,5%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,6 (M+H)+, 484,8 (M-H)- через 1,40 мин.
Стадия 4: 4-метокси-3-(N-(2-(2-оксопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (18,6 мг, 0,038 ммоль) растворяли в ТГФ (1 мл) и обрабатывали 1,1М LiOH (водн.) (139 мкл, 0,153 ммоль). Реакцию перемешивали при КТ в течение 1 дня, затем по каплям добавляли MeOH, пока смесь не становилась раствором, и нагревали реакционную смесь в 40°C в течение 20 ч, после чего охлаждали до КТ. Реакционную смесь разбавляли водой (3 мл), упаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл), и нейтрализовали до ~pH 6 1М HCl. Белый осадок собирали с помощью фильтрации, промывали водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (17,1 мг, 0,034 ммоль, выход 90%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,0 (M+H)+, 471,1 (M-H)- через 1,23 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,12 (с, 1H), 9,70 (с, 1H), 8,33 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,65 (с, 1H), 7,54-7,39 (м, 2H), 7,31 (д, J=8,8 Гц, 1H), 3,80 (с, 3H), 3,09-3,23 (м, 2H), 2,44-2,22 (м, 2H), 1,90-1,70 (м, 4H).
Пример 42: 3-(N-(2-(1,4-оксазепан-4-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метилбензойная кислота
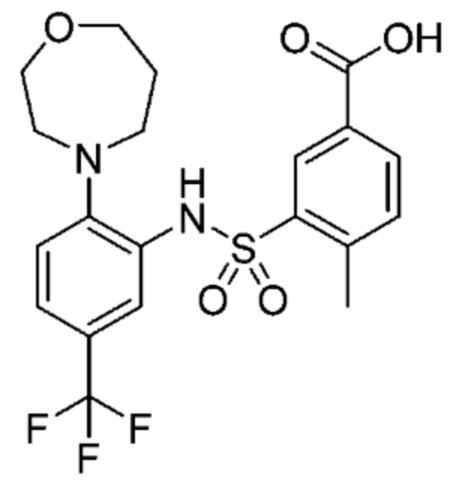
Раствор продукта из примера 32, стадии 2 выше 62 мг, 0,238 ммоль) в ДХМ (1 мл) и пиридине (0,116 мл, 1,43 ммоль) добавляли к раствору 3-(хлорсульфонил)-4-метилбензойной кислоты (67,1 мг, 0,286 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 4 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением кремового твердого вещества (23 мг). 8 мг этого неочищенного продукта наносили на слой силикагеля в минимальном количестве ДХМ, колонку элюировали ДХМ (5 мл), изогексанами (5 мл), 5% MeOH в EtOAc (5 мл), затем 20% MeOH в EtOAc (5 мл), с получением указанного в заголовке соединения (7,0 мг, 0,015 ммоль, выход 6,09%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,4 (M+H)+, 457,3 (M-H)- через 1,64 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,58 (д, J=2,1 Гц, 1H), 8,48 (с, 1H), 8,00 (дд, J=7,9, 2,1 Гц, 1H), 7,52-7,42 (м, 3H), 3,97 (т, J=6,2 Гц, 2H), 3,94-3,89 (м, 2H), 3,28-3,22 (м, 4H), 2,79 (с, 3H), 2,15-2,07 (м, 2H). Два обмениваемых протона не наблюдали.
Пример 43: 3-(N-(2-(1,4-оксазепан-4-ил)-5-(трифторметил)-фенил)сульфамоил)-4-этилбензойная кислота
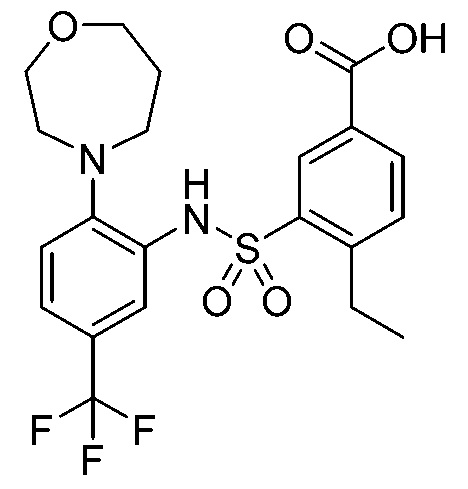
Раствор продукта из примера 32, стадии 2 выше (62 мг, 0,238 ммоль) в ДХМ (1 мл) и пиридине (0,116 мл, 1,429 ммоль) добавляли к раствору продукта из примера 1, стадии 1 выше (71,1 мг, 0,286 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 4 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением кремового твердого вещества. Его наносили на слой силикагеля в минимальном количестве ДХМ, колонку последовательно элюировали ДХМ (5 мл), изогексанами (5 мл), 5% MeOH в EtOAc (5 мл), затем 5% MeOH в EtOAc (5 мл), с получением указанного в заголовке соединения (11,7 мг, 0,024 ммоль, выход 9,87%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,2 (M-H)- через 1,61 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,53 (д, J=1,8 Гц, 1H), 8,17 (дд, J=8,0, 1,8 Гц, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,34-7,28 (м, 3H), 3,90 (т, J=5,9 Гц, 2H), 3,86-3,81 (м, 2H), 3,23-3,16 (м, 4H), 3,08 (к, J=7,5 Гц, 2H), 2,02 (п, J=5,8 Гц, 2H), 1,29 (т, J=7,5 Гц, 3H). Два обмениваемых протона не наблюдали.
Пример 46: 4-метокси-3-(N-(2-(2-(3-метилизоксазол-5-ил)пирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
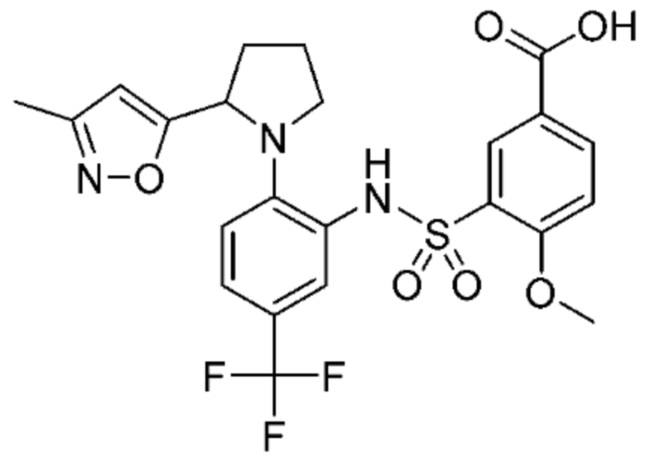
Стадия 1: 3-метил-5-(1-(2-нитро-4-(трифторметил)фенил)-пирролидин-2-ил)изоксазол: Et3N (302 мг, 2,99 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,167 мл, 1,20 ммоль) и 3-метил-5-(пирролидин-2-ил)изоксазола (218 мг, 1,44 ммоль) в ДХМ (5 мл) и перемешивали полученный раствор при КТ в течение 19 ч. Добавляли воду (2,5 мл), и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (489 мг, 1,19 ммоль, выход 99%, чистота 83%) в виде желтого масла. СВЭЖХ-МС (Метод 2) m/z 342,4 (M+H)+ через 1,61 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,07 (м, 1H), 7,71 (дд, J=9,1, 2,0 Гц, 1H), 7,17 (д, J=9,1 Гц, 1H), 6,18 (с, 1H), 5,34 (т, J=7,3 Гц, 1H), 3,55-3,50 (м, 1H), 3,02-2,98 (м, 1H), 2,54-2,51 (м, 1H), 2,16 (с, 3H), 2,08-2,02 (м, 1H), 2,02-1,89 (м, 2H).
Стадия 2: 2-(2-(3-метилизоксазол-5-ил)пирролидин-1-ил)-5-(трифторметил)анилин: Гидроксид аммония (28% водн. раствор) (0,319 мл, 2,30 ммоль) и дитионит натрия (1,18 г, 5,74 ммоль) добавляли к раствору продукта из стадии 1 выше (236 мг, 0,574 ммоль) в ТГФ (2,5 мл) и воде (2,5 мл) при КТ, а затем перемешивали при КТ в течение 2 ч. Реакционную смесь выпаривали в вакууме, а остаток повторно растворяли в ДХМ (10 мл) и промывали водой (5 мл). Водную фазу экстрагировали ДХМ (2×5 мл), и органические фазы объединяли, промывали солевым раствором (5 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (95 мг, 0,302 ммоль, выход 52,6%, чистота 99%) в виде красного/коричневого масла. СВЭЖХ-МС (Метод 1) m/z 312,1 (M+H)+ через 1,52 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,03 (д, J=8,2 Гц, 1H), 6,92 (д, J=2,2 Гц, 1H), 6,74 (дд, J=8,3, 2,1 Гц, 1H), 6,05 (с, 1H), 5,17 (с, 2H), 4,98 (дд, J=7,9, 5,9 Гц, 1H), 3,72-3,65 (м, 1H), 2,76-2,68 (м, 1H), 2,45-2,37 (м, 1H), 2,10 (с, 3H), 2,08-1,99 (м, 1H), 1,98-1,87 (м, 2H).
Стадия 3: метил-4-метокси-3-(N-(2-(2-(3-метилизоксазол-5-ил)пирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Пиридин (0,069 мл, 0,852 ммоль) добавляли к раствору продукта из стадии 2 выше (88 мг, 0,284 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (95 мг, 0,341 ммоль) в ДХМ (2,5 мл) при КТ. Реакционную смесь перемешивали при КТ в течение 65 ч, а затем при 40°C в течение 5 ч. Неочищенную реакционную смесь фильтровали и отфильтрованный продукт повторно растворяли в MeCN (10 мл) и выпаривали в вакууме с получением указанного в заголовке соединения (69 мг, 0,123 ммоль, выход 43,2%, чистота 96%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 540,3 (M+H)+, 538,2 (M-H)- через 1,58 мин.
Стадия 4: 4-метокси-3-(N-(2-(2-(3-метилизоксазол-5-ил)пирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,384 мл, 0,384 ммоль) добавляли к суспензии продукта из стадии 3 выше (69 мг, 0,128 ммоль) в ТГФ (0,768 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 20 ч. Добавляли дополнительное количество 1М LiOH (водн.) (0,128 мл, 0,128 ммоль) и перемешивали раствор еще в течение 1 ч. Реакционную смесь выпаривали в вакууме, а остаток повторно растворяли в воде (2 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Осадок растворяли в ДХМ (10 мл) и разделяли фазы. Водную фазу экстрагировали ДХМ (2×3 мл), и объединенные органические фазы сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (47,9 мг, 0,091 ммоль, выход 71,3%, чистота 97%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 526,3 (M+H)+, 524,2 (M-H)- через 1,46 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,08 (шс, 1H), 9,39 (шс, 1H), 8,17 (дд, J=8,7, 2,3 Гц, 1H), 8,10 (д, J=2,2 Гц, 1H), 7,38 (д, J=8,8 Гц, 1H), 7,27 (дд, J=8,8, 2,3 Гц, 1H), 6,78 (д, J=8,8 Гц, 1H), 6,67 (д, J=2,3 Гц, 1H), 6,02 (с, 1H), 5,35 (т, J=6,4 Гц, 1H), 4,01 (кажущ. дт, J=9,7, 7,0 Гц, 1H), 3,95 (с, 3H), 3,45 (ддд, J=9,9, 7,3, 5,2 Гц, 1H), 2,40-2,35 (м, 1H), 2,13 (с, 3H), 2,01-1,85 (м, 3H).
Пример 49 Метиловый сложный эфир: метил-4-метокси-3-((2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфонамидо)бензоат
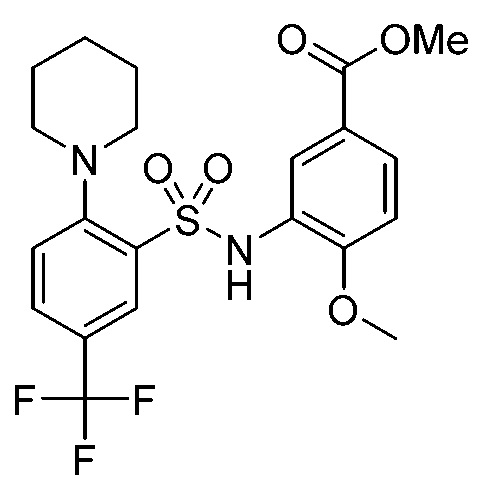
Стадия 1: метил-3-(2-бром-5-(трифторметил)-фенилсульфонамидо)-4-метоксибензоат: Смесь 2-бром-5-(трифторметил)бензол-1-сульфонилхлорида (230 мкл, 1,32 ммоль), метил-3-амино-4-метоксибензоата (200 мг, 1,10 ммоль) и пиридина (268 мкл, 3,31 ммоль) в ДХМ (4 мл) перемешивали при КТ в течение выходных. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (510 мг, 1,07 ммоль, выход 97%, чистота 98%) в виде бледно-бежевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 468,0/470,0 (M/M+2)+ через 1,43 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 10,26 (с, 1H), 8,12 (д, J=8,3 Гц, 1H), 8,10 (д, J=2,2 Гц, 1H), 7,92 (дд, J=8,3, 2,2 Гц, 1H), 7,83-7,77 (м, 2H), 7,08 (д, J=8,6 Гц, 1H), 3,80 (с, 3H), 3,56 (с, 3H).
Стадия 2: метил-4-метокси-3-(2-(пиперидин-1-ил)-5-(трифторметил)фенилсульфонамидо)бензоат: Смесь продукта из стадии 1 выше (100 мг, 0,214 ммоль) и пиперидина (25 мкл, 0,253 ммоль) в ТГФ (1 мл) нагревали до 60°C и перемешивали в течение ночи. Добавляли дополнительную порцию пиперидина (25 мкл, 0,253 ммоль) и продолжали перемешивание при 60°C в течение 7 ч. Добавляли дополнительную порцию пиперидина (25 мкл, 0,253 ммоль) и продолжали перемешивание при 60°C в течение ночи. После охлаждения до КТ смесь выпаривали в вакууме, а остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (82 мг, 0,165 ммоль, выход 78%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 473,3 (M+H)+ через 1,84 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,04 (с, 1H), 8,05 (с, 1H), 7,93 (д, J=8,4 Гц, 1H), 7,87 (с, 1H), 7,67 (д, J=8,7 Гц, 1H), 7,59 (д, J=8,4 Гц, 1H), 7,05 (д, J=8,7 Гц, 1H), 3,78 (с, 3H), 3,73 (с, 3H), 2,92 (т, J=5,3 Гц, 4H), 1,77-1,65 (м, 4H), 1,57-1,51 (м, 2H).
Пример 50: 4-метокси-3-((2-(пиперидин-1-ил)-5-(трифтор-метил)фенил)сульфонамидо)бензойная кислота
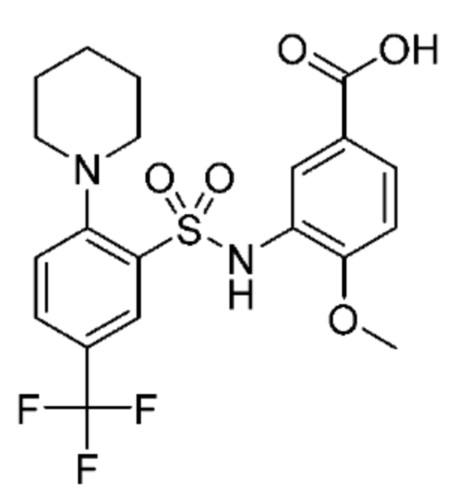
Смесь продукта из примера 49 метиловый сложный эфир, стадии 2 выше 70 мг, 0,148 ммоль) в ТГФ (1,25 мл) и 2М LiOH (водн.) (0,25 мл, 0,500 ммоль) перемешивали при 50°C в течение ночи. Добавляли дополнительную порцию 2М LiOH (водн.) (0,25 мл, 0,500 ммоль) и продолжали перемешивание при 50°C в течение 5 ч. Смесь разбавляли H2O (5 мл), подкисляли до прибл. pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×10 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), пропускали через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-10% MeOH/ДХМ) и растирали с МТБЭ с получением указанного в заголовке соединения (44,3 мг, 0,093 ммоль, выход 62,6%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 459,3 (M+H)+, 457,2 (M-H)- через 1,19 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 12,71 (с, 1H), 8,99 (с, 1H), 8,05 (д, J=2,3 Гц, 1H), 7,93 (дд, J=8,5, 2,3 Гц, 1H), 7,89 (д, J=2,1 Гц, 1H), 7,64 (дд, J=8,7, 2,1 Гц, 1H), 7,60 (д, J=8,5 Гц, 1H), 7,02 (д, J=8,7 Гц, 1H), 3,71 (с, 3H), 2,92 (т, J=5,1 Гц, 4H), 1,76-1,65 (м, 4H), 1,59-1,48 (м, 2H).
Общее соединение A: 4-метокси-2-((2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфонамидо)бензойная кислота
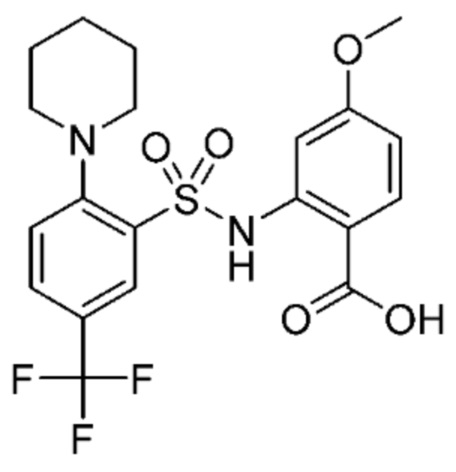
Стадия 1: метил-2-(2-фтор-5-(трифторметил)фенил-сульфонамидо)-4-метоксибензоат: Смесь 2-фтор-5-(трифторметил)-бензол-1-сульфонилхлорида (87 мг, 0,331 ммоль), метил-2-амино-4-метоксибензоата (50 мг, 0,276 ммоль) и пиридина (0,067 мл, 0,828 ммоль) в ДХМ (2 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексаны) с получением указанного в заголовке соединения (98 мг, 0,180 ммоль, выход 65,4%, чистота 75%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) 405,5 (M-H)- через 1,67 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 11,13 (с, 1H), 8,24-8,12 (м, 2H), 7,87 (д, J=8,9 Гц, 1H), 7,73 (т, J=9,5 Гц, 1H), 6,94 (д, J=2,5 Гц, 1H), 6,83-6,76 (м, 1H), 3,79 (с, 3H), 3,77 (с, 3H).
Стадия 2: метил-4-метокси-2-(2-(пиперидин-1-ил)-5-(трифторметил)фенилсульфонамидо)бензоат: Смесь продукта из стадии 1 выше (98 мг, 0,180 ммоль) и пиперидина (0,06 мл, 0,606 ммоль) в ТГФ (2 мл) перемешивали при 60°C в течение 6 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (52 мг, 0,109 ммоль, выход 60,4%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 473,3 (M+H)+ через 2,01 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 11,11 (с, 1H), 8,28 (д, J=2,3 Гц, 1H), 8,06-7,95 (м, 1H), 7,85 (д, J=8,9 Гц, 1H), 7,59 (д, J=8,5 Гц, 1H), 6,73 (д, J=2,5 Гц, 1H), 6,63 (дд, J=8,9, 2,5 Гц, 1H), 3,84 (с, 3H), 3,66 (с, 3H), 2,84 (т, J=5,3 Гц, 4H), 1,74-1,64 (м, 4H), 1,58-1,49 (м, 2H).
Стадия 3: 4-метокси-2-(2-(пиперидин-1-ил)-5-(трифторметил)-фенилсульфонамидо)бензойная кислота: Смесь продукта из стадии 2 выше (52 мг, 0,109 ммоль) и 2М LiOH (водн.) (250 мкл, 0,500 ммоль) в ТГФ (1,25 мл) перемешивали при 50°C в течение ночи. Смесь разбавляли H2O (2 мл) и подкисляли до прибл. pH 4 1М HCl. Смесь экстрагировали EtOAc (3×15 мл), объединенные органические экстракты промывали солевым раствором, пропускали через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (14,1 мг, 0,030 ммоль, выход 27,1%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 459,3 (M+H)+, 457,2 (M-H)- через 1,22 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,63 (с, 1H), 11,65 (с, 1H), 8,29 (д, J=2,3 Гц, 1H), 7,99 (дд, J=8,5, 2,3 Гц, 1H), 7,83 (д, J=8,9 Гц, 1H), 7,58 (д, J=8,5 Гц, 1H), 6,65 (д, J=2,4 Гц, 1H), 6,57 (дд, J=8,9, 2,4 Гц, 1H), 3,64 (с, 3H), 2,86 (т, J=5,1 Гц, 4H), 1,77-1,66 (м, 4H), 1,60-1,46 (м, 2H).
Следующие примеры были получены способами, аналогичными Общему соединению A, с заменой подходящими исходными соединениями и промежуточными соединениями в случае необходимости:
Пример 53 Метиловый сложный эфир: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат
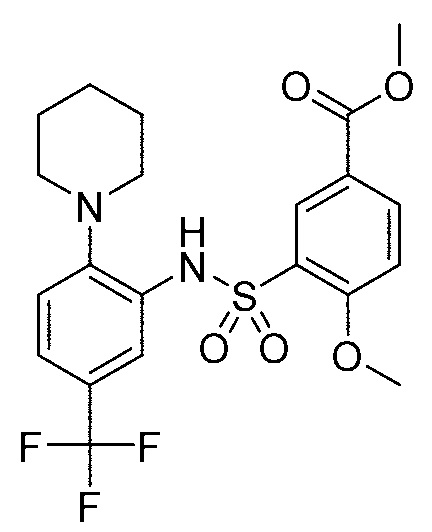
Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0.100 г, 0,409 ммоль) в ДХМ (1 мл) и пиридине (0,1 мл, 1,236 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,130 г, 0,491 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 23 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексаны) с получением оранжевого масла. Его повторно очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (0,143 г, 0,294 ммоль, выход 71,7%, чистота 97%) в виде бледно-желтого, медленно кристаллизующегося масла. СВЭЖХ-МС (Метод 2) m/z 473,2 (M+H)+, 471,1 (M-H)- через 1,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,81 (шс, 1H), 8,37 (д, J=2,3 Гц, 1H), 8,19 (дд, J=8,7, 2,3 Гц, 1H), 7,45 (д, J=2,0 Гц, 1H), 7,40-7,30 (м, 3H), 3,94 (с, 3H), 3,86 (с, 3H), 2,78-2,75 (м, 4H), 1,68-1,64 (м, 4H), 1,56-1,52 (м, 2H).
Пример 54: 4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
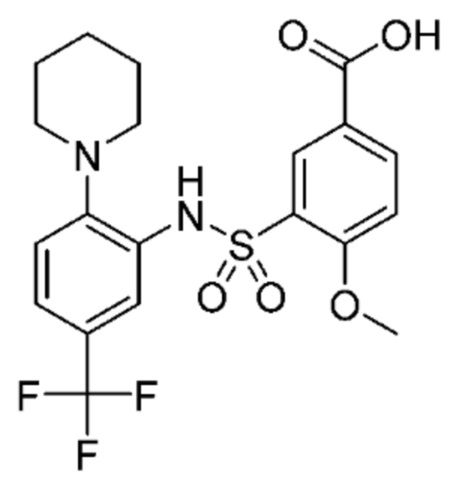
1М LiOH (водн.) (3 мл, 3,00 ммоль) добавляли к раствору продукта из примера 54 метиловый сложный эфир (0,068 г, 0,144 ммоль) в диоксане (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток повторно растворяли в воде (5 мл) и экстрагировали EtOAc (3×5 мл). Водную фазу подкисляли 1М HCl (водн.) и продукт экстрагировали EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,047 г, 0,100 ммоль, выход 69,8%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 459,2 (M+H)+, 457,0 (M-H)- через 1,15 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (с, 1H), 8,76 (с, 1H), 8,37 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,45 (д, J=1,9 Гц, 1H), 7,38-7,30 (м, 3H), 3,93 (с, 3H), 2,76 (т, J=5,3 Гц, 4H), 1,67 (п, J=5,3 Гц, 4H), 1,55 (п, J=5,3 Гц, 2H).
Пример 55: 3-(N-(2-(азепан-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-изопропилбензойная кислота
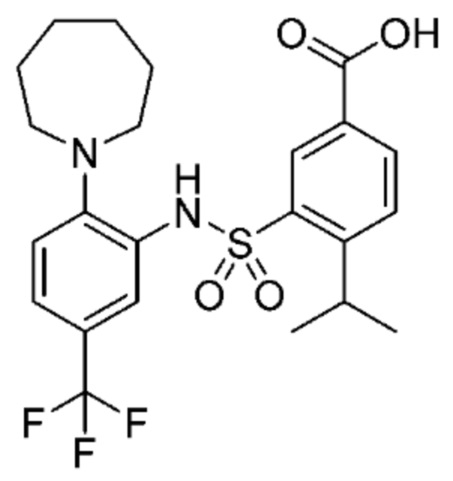
Раствор 2-(азепан-1-ил)-5-(трифторметил)анилина (50 мг, 0,194 ммоль) в ДХМ (1 мл) и пиридине (0,094 мл, 1,16 ммоль) добавляли к раствору 3-(хлорсульфонил)-4-изопропилбензойной кислоты (61,0 мг, 0,232 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 4 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением светло-желтого твердого вещества (11,1 мг). 9 мг этого продукта наносили на силикагель в минимальном количестве ДХМ, колонку элюировали ДХМ (5 мл), изогексанами (5 мл), 5% MeOH в EtOAc (5 мл), затем 5% MeOH в EtOAc (5 мл), с получением указанного в заголовке соединения (5,4 мг, 10,6 мкмоль, выход 5,47%, чистота 95%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 485,4 (M+H)+, 483,1 (M-H)- через 1,99 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,58 (д, J=1,8 Гц, 1H), 8,20 (дд, J=8,2, 1,8 Гц, 1H), 7,70 (д, J=8,2 Гц, 1H), 7,33-7,21 (м, 3H), 3,90 (септет, J=6,8 Гц, 1H), 3,20-3,13 (м, 4H), 1,86-1,77 (м, 4H), 1,76-1,71 (м, 4H), 1,24 (д, J=6,7 Гц, 6H). Два обмениваемых протона не наблюдали.
Следующие примеры были получены способами, аналогичными Примеру 55, с заменой подходящими исходными соединениями и промежуточными соединениями в случае необходимости:
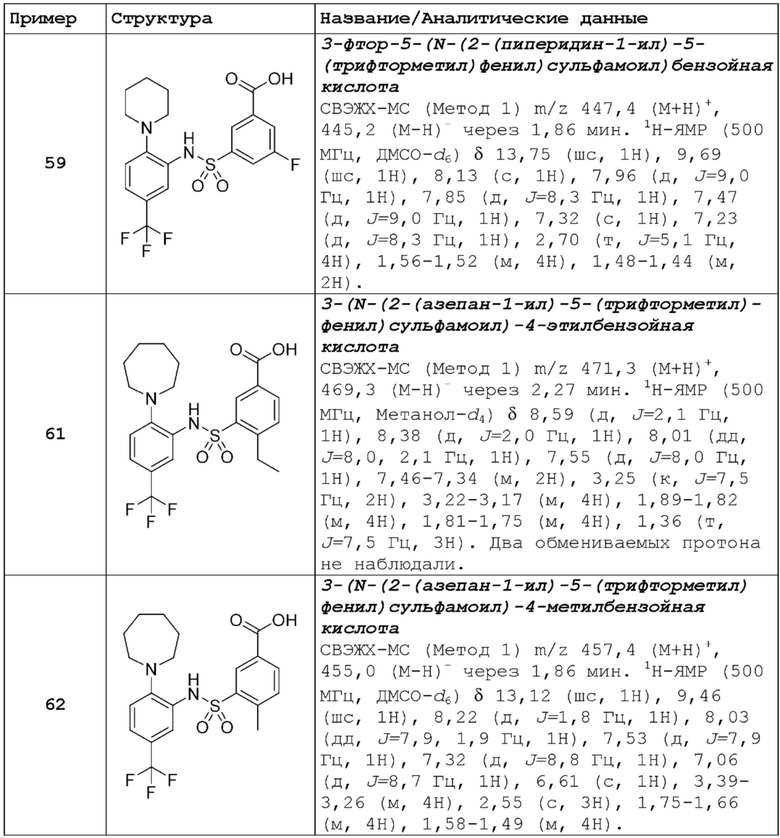
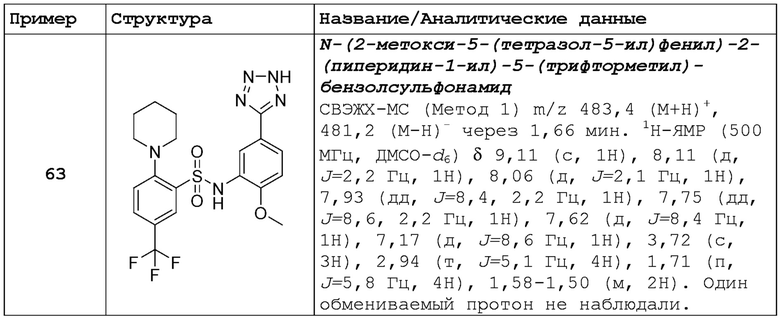
Пример 64: 4-метокси-3-(N-(2-(пиперидин-1-ил)фенил)-сульфамоил)бензойная кислота
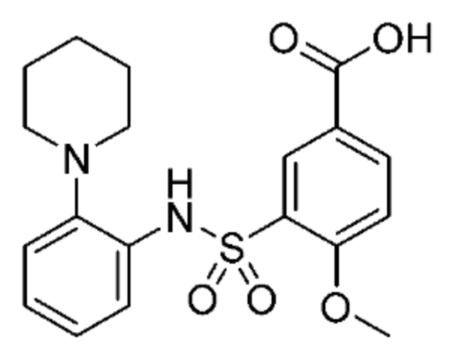
Стадия 1: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)фенил)-сульфамоил)бензоат: Раствор гидрохлорида 2-(пиперидин-1-ил)анилина (0,050 г, 0,235 ммоль) в ДХМ (1 мл) и пиридине (0,114 мл, 1,410 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,075 г, 0,282 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 96 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (0,095 г, 0,169 ммоль, выход 71,9%, 72% чистота) в виде бледно-желтого, медленно кристаллизующегося масла. СВЭЖХ-МС (Метод 2) m/z 405,2 (M+H)+, 403,4 (M-H)- через 1,69 мин.
Стадия 2: 4-метокси-3-(N-(2-(пиперидин-1-ил)фенил)-сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,470 мл, 0,470 ммоль) добавляли к раствору продукта из стадии 1 выше (0,095 г, 0,235 ммоль) в диоксане (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток повторно растворяли в воде (5 мл) и экстрагировали EtOAc (3×5 мл). Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-70% EtOAc/изогексаны) с получением указанного в заголовке соединения (40 мг, 0,097 ммоль, выход 41,4%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 391,3 (M+H)+, 389,3 (M-H)- через 1,41 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,23 (шс, 1H), 8,60 (с, 1H), 8,39 (д, J=2,3 Гц, 1H), 8,14 (дд, J=8,7, 2,3 Гц, 1H), 7,31 (д, J=8,8 Гц, 1H), 7,25 (дд, J=7,4, 2,1 Гц, 1H), 7,22 (дд, J=7,5, 2,2 Гц, 1H), 7,12-6,85 (м, 2H), 3,96 (с, 3H), 2,75-2,63 (м, 4H), 1,69 (п, J=5,5 Гц, 4H), 1,58-1,52 (м, 2H).
Пример 65: 3-(N-(4-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
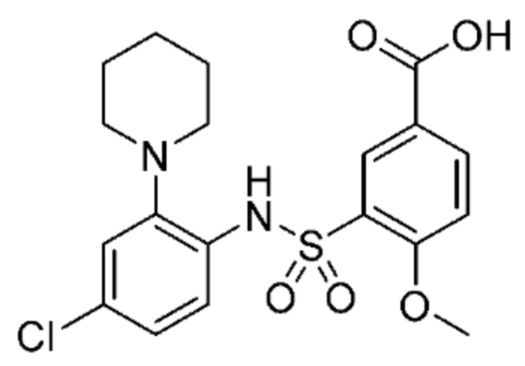
Стадия 1: метил-3-(N-(4-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензоат: Раствор 4-хлор-2-(пиперидин-1-ил)анилина (0,050 г, 0,237 ммоль) в ДХМ (1 мл) и пиридине (0,115 мл, 1,42 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,075 г, 0,285 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 96 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (0,093 г, 0,165 ммоль, выход 69,6%) в виде бледно-желтого, медленно кристаллизующегося масла. СВЭЖХ-МС (Метод 2) m/z 439,3 (M+H)+, 437,2 (M-H)- через 1,81 мин.
Стадия 2: 3-(N-(4-хлор-2-(пиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,424 мл, 0,424 ммоль) добавляли к раствору продукта из стадии 1 выше (0,093 г, 0,212 ммоль) в диоксане (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток повторно растворяли в воде (5 мл) и экстрагировали EtOAc (3×5 мл). Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-80% EtOAc/изогексаны) с получением указанного в заголовке соединения (34 мг, 0,076 ммоль, выход 35,9%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 425,3 (M+H)+, 423,2 (M-H)- через 1,69 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,24 (шс, 1H), 8,58 (с, 1H), 8,36 (д, J=2,3 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 7,28-7,14 (м, 2H), 7,07 (дд, J=8,8, 2,4 Гц, 1H), 3,96 (с, 3H), 2,72-2,68 (м, 4H), 1,66 (п, J=5,5 Гц, 4H), 1,56-1,50 (м, 2H).
Пример 66: 3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
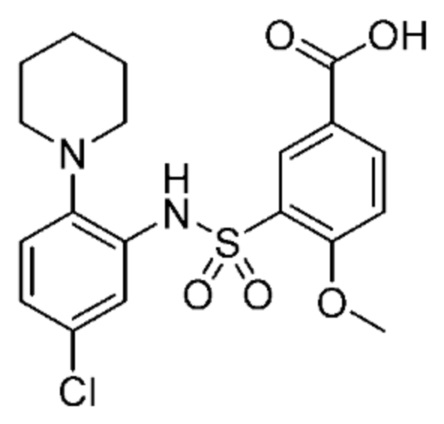
Стадия 1: метил-3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензоат: Раствор гидрохлорида 5-хлор-2-(пиперидин-1-ил)анилина (0,050 г, 0,202 ммоль) в ДХМ (1 мл) и пиридине (0,098 мл, 1,21 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,064 г, 0,243 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 96 ч. Растворитель удаляли в вакууме, неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексаны) с получением соединения, указанного в заголовке (0,066 г, 0,143 ммоль, выход 70,6%, чистоте 95%), в виде бледно-желтого, медленно кристаллизующегося масла. СВЭЖХ-МС (Метод 2) m/z 439,3 (M+H)+, 437,3 (M-H)- через 1,81 мин.
Стадия 2: 3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,301 мл, 0,301 ммоль) добавляли к раствору продукта из стадии 1 выше (0,066 г, 0,150 ммоль) в диоксане (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток повторно растворяли в воде (5 мл) и экстрагировали EtOAc (3×5 мл). Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-70% EtOAc/изогексаны) с получением указанного в заголовке соединения (22 мг, 0,049 ммоль, выход 32,7%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 425,1 (M+H)+, 423,2 (M-H)- через 1,67 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,25 (шс, 1H), 8,69 (шс, 1H), 8,38 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,34 (д, J=8,8 Гц, 1H), 7,25 (д, J=2,5 Гц, 1H), 7,24 (д, J=8,5 Гц, 1H), 7,05 (дд, J=8,5, 2,5 Гц, 1H), 3,96 (с, 3H), 2,75-2,61 (м, 4H), 1,67 (п, J=5,5 Гц, 4H), 1,56-1,50 (м, 2H).
Пример 67: 4-метил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
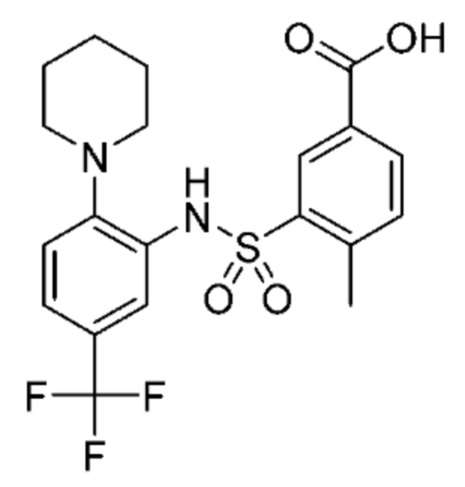
Стадия 1: метил-4-метил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: 2-(пиперидин-1-ил)-5-(трифторметил)анилин (50 мг, 0,205 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метилбензоата (52 мг, 0,209 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Добавляли дополнительное количество метил-3-(хлорсульфонил)-4-метилбензоата (15 мг, 0,060 ммоль) и перемешивали реакцию еще в течение 24 ч при КТ. Реакционную смесь наносили непосредственно на силикагель (12 г картридж, 0-50% EtOAc/изогексаны) и очищали с получением указанного в заголовке соединения (73 мг, 0,155 ммоль, выход 76%, чистота 97%) в виде бесцветного масла, которое кристаллизовалось при стоянии. СВЭЖХ-МС (Метод 1) m/z 457,1 (M+H)+, 455,3 (M-H)- через 1,95 мин.
Стадия 2: 4-метил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (71 мг, 0,151 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (499 мкл, 0,549 ммоль). Добавляли MeOH с получением прозрачного раствора, который оставляли при КТ. Через 2 дня раствор разбавляли водой (2 мл) и оставляли стоять при КТ еще на 24 ч. Затем раствор снова разбавляли водой (2 мл) и выпаривали в вакууме. Полученную водную суспензию разбавляли водой (2 мл) и фильтровали при промывании водой (1 мл). Полученный раствор нейтрализовали 1М HCl (водн.) (0,4 мл) и обрабатывали ультразвуком, после чего доводили до прибл. pH 6 1М HCl (водн.) (2 капли). Полученный почти белый осадок собирали с помощью фильтрации, промывая водой. Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (55 мг, 0,122 ммоль, выход 81%, чистота 98%) в виде рыжевато-коричневого порошка. СВЭЖХ-МС (Метод 1) m/z 443,3 (M+H)+ 441,3 (M-H)- через 1,81 мин.
Пример 68: 3-(N-(2-(азепан-1-ил)-5-(трифторметил)фенил)-сульфамоил)-4-метоксибензойная кислота
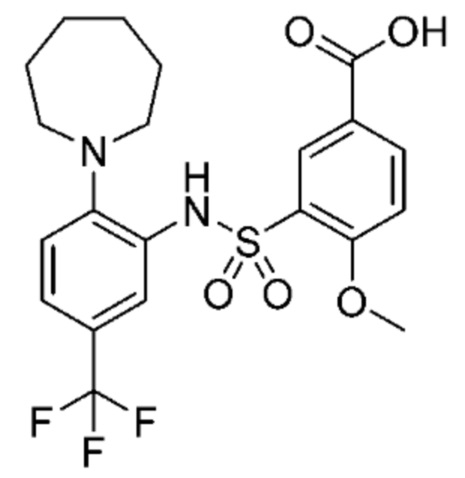
Стадия 1: метил-3-(N-(2-(азепан-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензоат: 2-(азепан-1-ил)-5-(трифторметил)анилин (48,8 мг, 0,189 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (60 мг, 0,227 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Реакционную смесь наносили непосредственно на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-70% EtOAc/изогексаны) с получением указанного в заголовке соединения (44 мг, 0,084 ммоль, выход 44,5%, чистота 93%) в виде клейкого светло-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,4 (M+H)+, 485,2 (M-H)- через 1,91 мин.
Стадия 2: 3-(N-(2-(азепан-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 1 выше (42 мг, 0,086 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (235 мкл, 0,259 ммоль). Реакционную смесь перемешивали при КТ в течение 2 дней. Добавляли дополнительную порцию 1,1 М LiOH (водн.) (78 мкл, 0,086 ммоль) и нагревали реакцию до 30°C в течение 18 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл) и нейтрализовали 1М HCl (водн.) (0,4 мл). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора и нейтрализовали до прибл. pH 6 1М HCl. Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-70% EtOAc/изогексаны) с получением указанного в заголовке соединения (2,2 мг, 4,42 мкмоль, выход 5,12%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,1 (M-H)- через 1,79 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,13 (шс, 1H), 8,76 (шс, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,44 (д, J=1,9 Гц, 1H), 7,38-7,26 (м, 3H), 3,91 (с, 3H), 2,92 (д, J=11,4 Гц, 2H), 2,67-2,57 (м, 2H), 1,72-1,65 (м, 1H), 1,55-1,43 (м, 1H), 1,34-1,20 (м, 3H), 0,97 (д, J=6,5 Гц, 3H).
Пример 69: 4-хлор-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
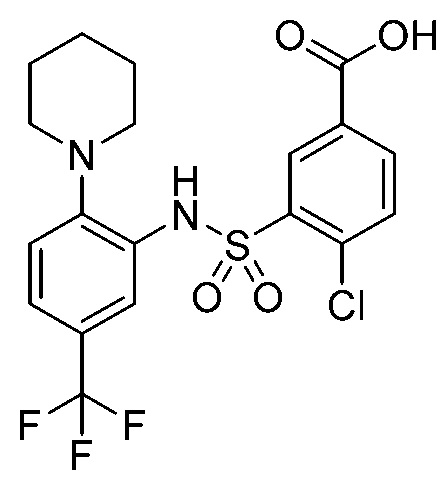
Стадия 1: метил-4-хлор-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: 2-(пиперидин-1-ил)-5-(трифторметил)анилин (45,4 мг, 0,186 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,05 мл, 0,618 ммоль) и обрабатывали раствором метил-4-хлор-3-(хлорсульфонил)бензоата (60 мг, 0,223 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Реакционную смесь наносили непосредственно на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-70% EtOAc/изогексаны) с получением указанного в заголовке соединения (45,5 мг, 0,094 ммоль, выход 50,3%, чистота 98%) в виде рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,3 (M+H)+, 475,1 (M-H)- через 2,00 мин.
Стадия 2: 4-хлор-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (43 мг, 0,090 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (328 мкл, 0,361 ммоль). Реакцию перемешивали при КТ в течение 2 дней. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (прибл. до 5 мл) и нейтрализовали 1М HCl (водн.) (0,4 мл). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутного раствора и нейтрализовали до прибл. pH 6 1М HCl (водн.). Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (20,5 мг, 0,042 ммоль, выход 46,7%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 463,3 (M+H)+, 461,2 (M-H)- через 1,88 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,48 (шс, 1H), 9,54 (шс, 1H), 8,44 (д, J=2,0 Гц, 1H), 8,12 (дд, J=8,3, 2,1 Гц, 1H), 7,80 (д, J=8,3 Гц, 1H), 7,42 (д, J=8,3 Гц, 1H), 7,34 (с, 1H), 7,29 (д, J=8,4 Гц, 1H), 2,77 (т, J=5,1 Гц, 4H), 1,58-1,51 (м, 4H), 1,50-1,43 (м, 2H).
Следующие примеры были получены способами, аналогичными Примеру 69, с заменой подходящими исходными соединениями и промежуточными соединениями в случае необходимости:
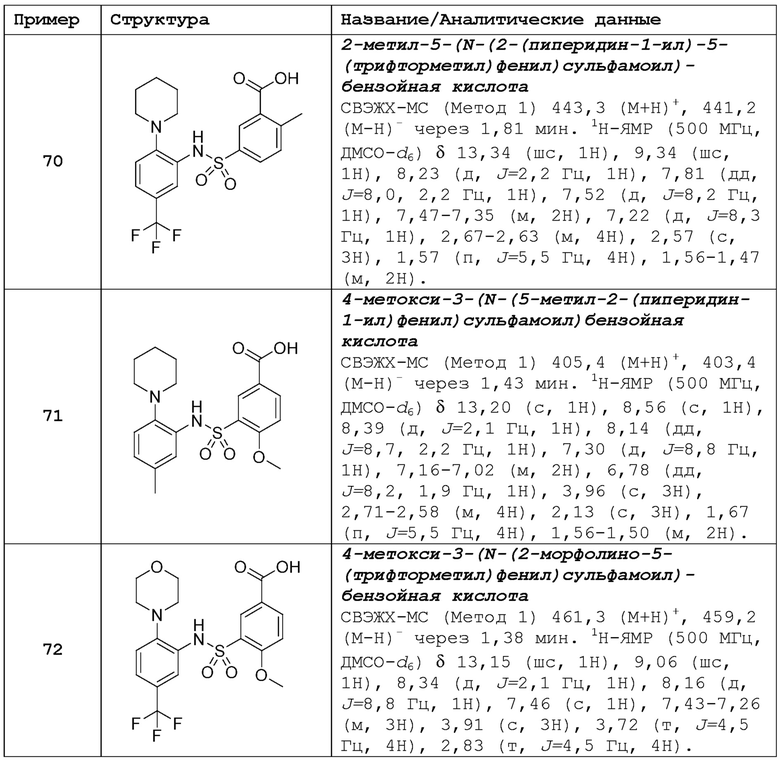
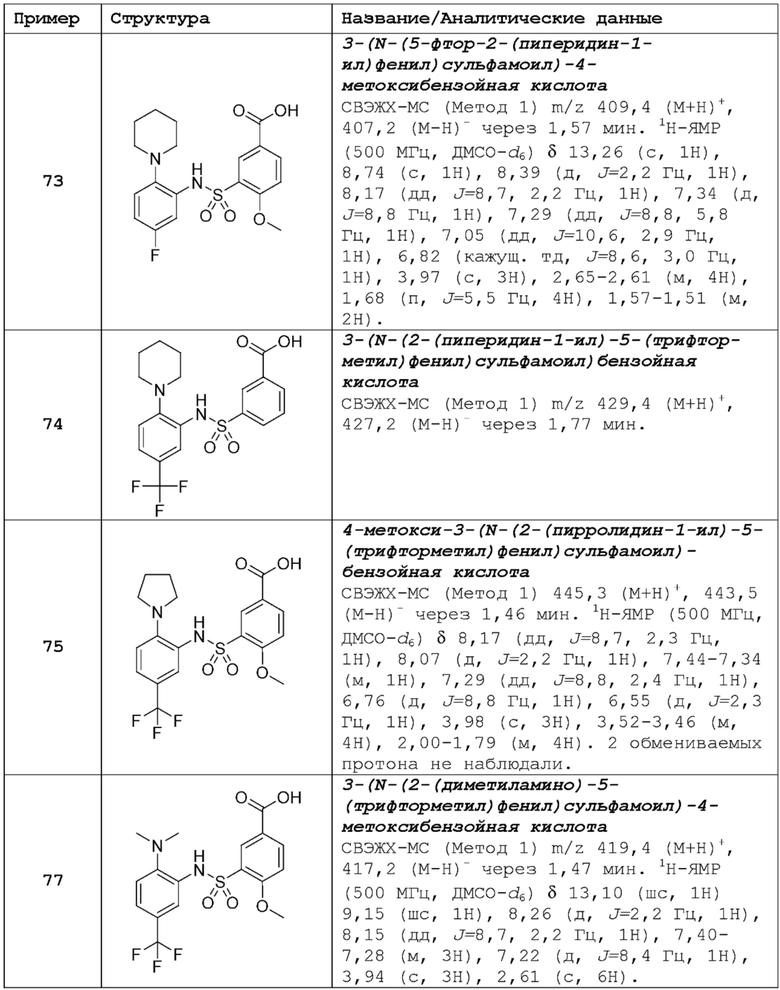
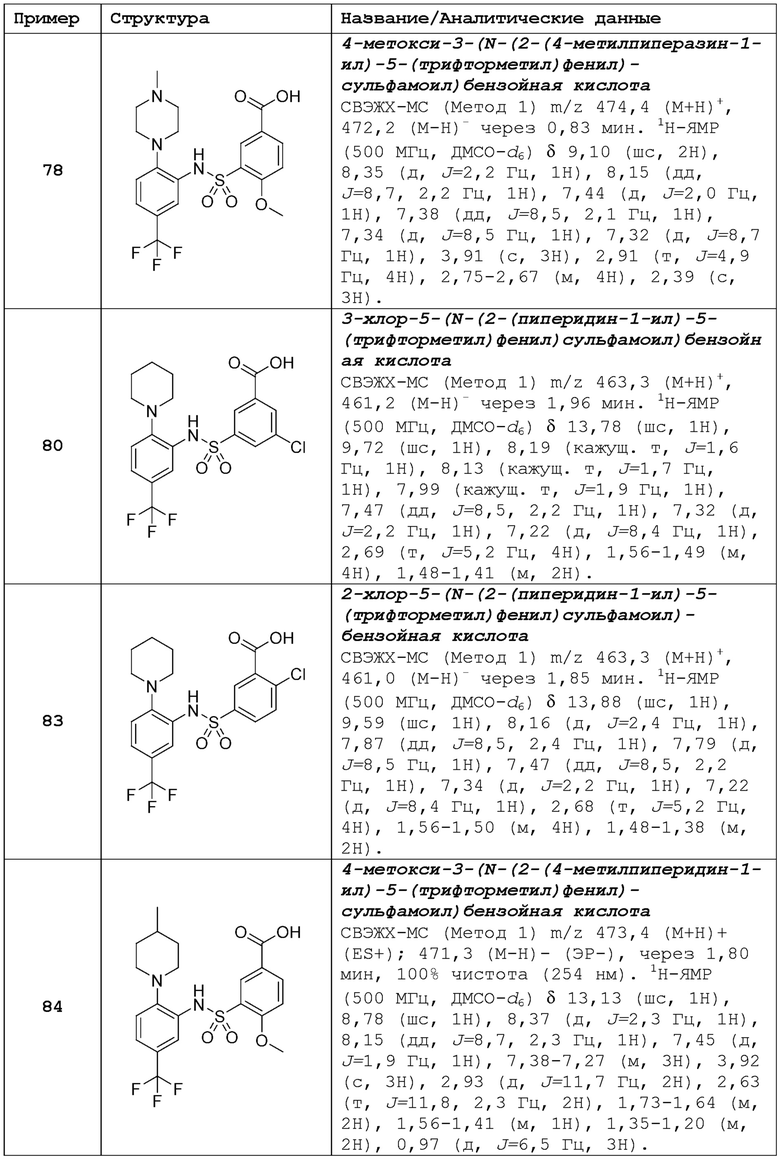
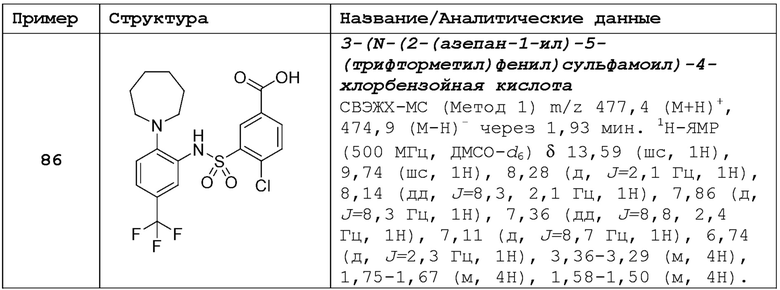
Пример 161: 4-гидрокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
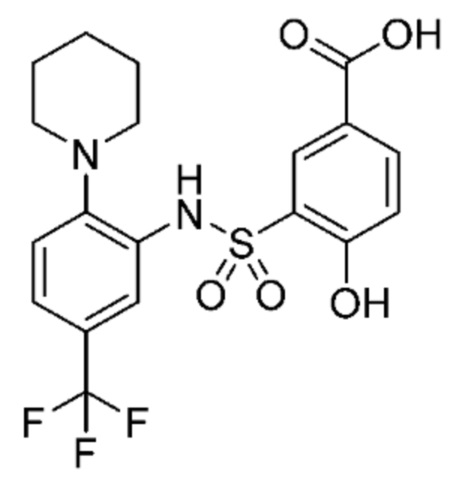
Стадия 1: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилин (0,130 г, 0,532 ммоль) в ДХМ (1 мл) и пиридине (0,258 мл, 3,19 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,169 г, 0,639 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 16 ч. Растворитель удаляли в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/ДХМ) с получением указанного в заголовке соединения (0,230 г, 0,433 ммоль, выход 81%, чистота 89%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,3 (M-H)- через 1,86 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,81 (с, 1H), 8,37 (д, J=2,3 Гц, 1H), 8,19 (дд, J=8,7, 2,3 Гц, 1H), 7,45 (д, J=2,0 Гц, 1H), 7,41-7,28 (м, 3H), 3,94 (с, 3H), 3,86 (с, 3H), 2,84-2,69 (м, 4H), 1,66 (п, J=5,6 Гц, 4H), 1,57-1,51 (м, 2H).
Стадия 2: метил-4-гидрокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 1 выше (0,230 г, 0,438 ммоль) в ДХМ (10 мл) обрабатывали 1,0М BBr3 в ДХМ (0,166 мл, 1,75 ммоль) и перемешивали раствор при КТ в течение 16 ч. Растворитель удаляли в вакууме с получением указанного в заголовке соединения в виде желтого масла (0,200 г, 0,393 ммоль, выход 90%, чистота 90%). СВЭЖХ-МС (Метод 1) m/z 459 (M+H)+ через 1,7 мин.
Стадия 3: 4-гидрокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,31 мл, 1,31 ммоль) добавляли к раствору продукта из стадии 2 выше (0,2 г, 0,436 ммоль) в MeOH (10 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток повторно растворяли в воде (5 мл) и экстрагировали EtOAc (3×5 мл). Водную фазу подкисляли 1М HCl (водн.) и экстрагировали продукт EtOAc (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/ДХМ) с получением указанного в заголовке соединения (60 мг, 0,128 ммоль, выход 29,4%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 445,3 (M+H)+, 443,2 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 12,96 (шс, 1H), 8,29 (д, J=2,3 Гц, 1H), 7,98 (дд, J=8,6, 2,3 Гц, 1H), 7,52 (д, J=1,8 Гц, 1H), 7,37-7,33 (м, 2H), 7,04 (д, J=8,6 Гц, 1H), 2,75 (т, J=5,2 Гц, 4H), 1,68 (п, J=5,5 Гц, 4H), 1,58-1,51 (м, 2H). 2 обмениваемых протона не наблюдали.
Пример 165: 4-метокси-3-(N-метил-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
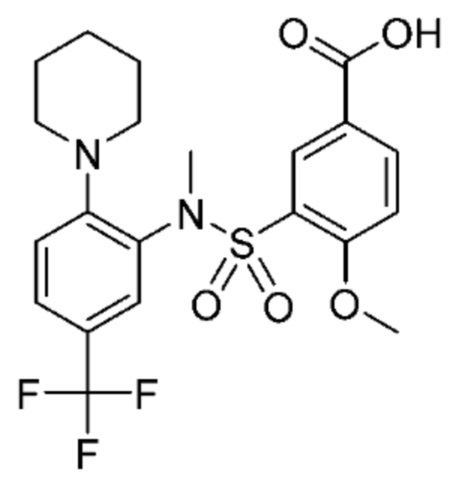
Стадия 1: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь 2-(пиперидин-1-ил)-5-(трифторметил)анилина (100 мг, 0,409 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (130 мг, 0,491 ммоль) и пиридина (100 мкл, 1,24 ммоль) в ДХМ (1,5 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (189 мг, 0,384 ммоль, выход 94%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 473,3 (M+H)+ через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,80 (с, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,18 (дд, J=8,8, 2,2 Гц, 1H), 7,44 (д, J=2,0 Гц, 1H), 7,40-7,29 (м, 3H), 3,93 (с, 3H), 3,85 (с, 3H), 2,76 (т, J=5,2 Гц, 4H), 1,70-1,61 (м, 4H), 1,59-1,49 (м, 2H).
Стадия 2: метил-4-метокси-3-(N-метил-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: К суспензии гидрида натрия (12 мг, 0,500 ммоль) в ТГФ (1 мл) при 0°C добавляли продукт из стадии 1 выше (189 мг, 0,384 ммоль) в ТГФ (1 мл). Смесь нагревали до КТ и перемешивали в течение 30 мин, после чего добавляли иодметан (30 мкл, 0,480 ммоль) и перемешивали смесь при КТ в течение ночи. В смесь вливали H2O (10 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (15 мл), пропускали через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (172 мг, 0,283 ммоль, выход 73,7%, чистота 80%) в виде прозрачного бесцветного масла. СВЭЖХ-МС (Метод 2) m/z 487,3 (M+H)+ через 1,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,25 (дд, J=8,7, 2,2 Гц, 1H), 8,22 (д, J=2,2 Гц, 1H), 7,54 (дд, J=8,6, 2,2 Гц, 1H), 7,48 (д, J=8,7 Гц, 1H), 7,21 (д, J=8,6 Гц, 1H), 7,02 (д, J=2,2 Гц, 1H), 4,00 (с, 3H), 3,83 (с, 3H), 3,27 (с, 3H), 3,06 (т, J=5,1 Гц, 4H), 1,64-1,57 (м, 4H), 1,57-1,50 (м, 2H).
Стадия 3: 4-метокси-3-(N-метил-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 2 выше (170 мг, 0,349 ммоль) и 2М LiOH (водн.) (0,35 мл, 0,700 ммоль) в ТГФ (1,5 мл) перемешивали при 50°C в течение ночи. Смесь разбавляли H2O (5 мл), подкисляли до прибл. pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×10 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), пропускали через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (66,1 мг, 0,134 ммоль, выход 38,3%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 473,3 (M+H)+, 471,2 (M-H)- через 1,17 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,10 (с, 1H), 8,22 (м, 2H), 7,53 (дд, J=8,5, 2,3 Гц, 1H), 7,48-7,41 (м, 1H), 7,20 (д, J=8,5 Гц, 1H), 7,01 (д, J=2,2 Гц, 1H), 3,99 (с, 3H), 3,28 (с, 3H), 3,09-3,02 (м, 4H), 1,65-1,57 (м, 4H), 1,57-1,48 (м, 2H).
Пример 171: 2-метокси-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-5-(тетразол-5-ил)бензолсульфонамид
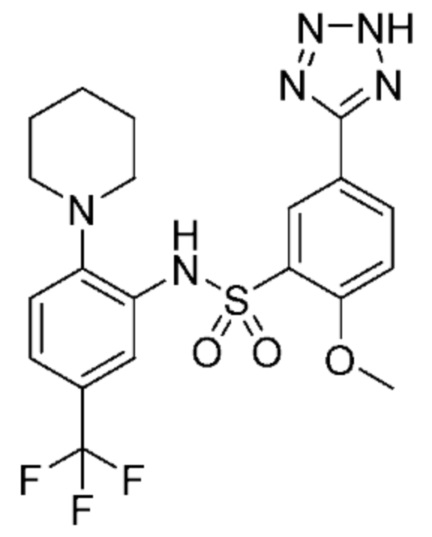
Стадия 1: 5-циано-2-метокси-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)бензолсульфонамид: 2-(пиперидин-1-ил)-5-(трифторметил)анилин (200 мг, 0,819 ммоль) растворяли в смеси ДХМ (2 мл) и пиридина (0,15 мл, 1,86 ммоль) и обрабатывали раствором 5-циано-2-метоксибензолсульфонилхлорида (237 мг, 1,02 ммоль) в ДХМ (1 мл). Полученный раствор оставляли при КТ на 18 ч, затем разбавляли водой (прибл. 0,1 мл) и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексаны) с получением указанного в заголовке соединения (325 мг, 0,717 ммоль, выход 88%, чистота 99%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 440,4 (M+H)+, 438,1 (M-H)- через 1,82 мин.
Стадия 2: 2-метокси-N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)-5-(тетразол-5-ил)бензолсульфонамид: Продукт из стадии 1 выше (100 мг, 0,228 ммоль) объединяли с азидом натрия (74,0 мг, 1,14 ммоль) и бромидом цинка (102 мг, 0,455 ммоль) в ИПС (1 мл) и воде (0,3 мл). Полученную смесь нагревали при 80°C в течение ночи, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексаны, затем 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (7,9 мг, 0,016 ммоль, выход 6,84%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,4 (M+H)+, 481,2 (M-H)- через 1,67 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,79 (с, 1H), 8,55 (д, J=2,2 Гц, 1H), 8,27 (дд, J=8,7, 2,2 Гц, 1H), 7,51 (с, 1H), 7,44 (д, J=8,8 Гц, 1H), 7,37-7,33 (м, 2H), 3,94 (с, 3H), 2,78 (т, J=5,3 Гц, 4H), 1,70-1,65 (м, 4H), 1,57-1,50 (м, 2H). Один обмениваемый протон не наблюдали.
Пример 177: 3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
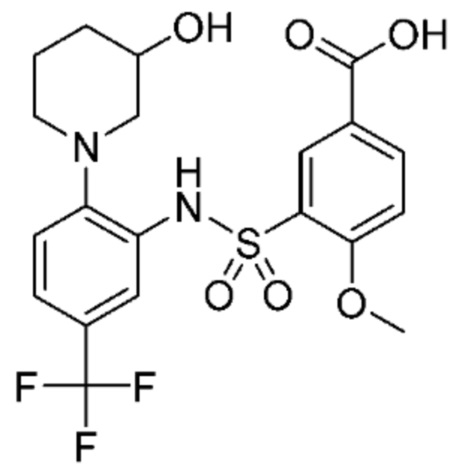
Стадия 1: 1-(2-нитро-4-(трифторметил)фенил)пиперидин-3-ол: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и пиперидин-3-ола (174 мг, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 17 ч. Органическую фазу промывали 1М HCl (3 мл) и сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (468 мг, 1,40 ммоль, выход 98%, чистота 87%) в виде красно-оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 291,5 (M+H)+ через 1,39 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,12-8,07 (м, 1H), 7,80 (дд, J=9,0, 2,4 Гц, 1H), 7,41 (д, J=8,9 Гц, 1H), 4,91 (д, J=4,3 Гц, 1H), 3,65-3,57 (м, 1H), 3,26 (дд, J=12,4, 3,9 Гц, 1H), 3,21 (дт, J=13,0, 4,5 Гц, 1H), 2,98-2,91 (м, 1H), 2,75 (дд, J=12,3, 8,5 Гц, 1H), 1,93-1,85 (м, 1H), 1,81-1,73 (м, 1H), 1,56-1,46 (м, 1H), 1,40-1,30 (м, 1H).
Стадия 2: 1-(2-амино-4-(трифторметил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об воды) Тип 87L (50 мг, 0,012 ммоль) в EtOH (0,5 мл) добавляли к раствору продукта из стадии 1 выше (234 мг, 0,701 ммоль) в EtOH (3,0 мл) при КТ. Реакционную смесь гидрировали (4 бара) при КТ в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite®, промывая MeOH (15 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в MeOH (10 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением белого твердого вещества. Добавляли MeCN (10 мл) и полученную суспензию снова сушили с использованием большого избытка MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (153 мг, 0,576 ммоль, выход 82%, чистота 98%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 261,4 (M+H)+ через 1,29 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,96 (д, J=8,1 Гц, 1H), 6,94 (д, J=2,2 Гц, 1H), 6,84-6,80 (м, 1H), 5,14 (с, 2H), 4,79 (д, J=5,4 Гц, 1H), 3,74-3,66 (м, 1H), 3,04-2,96 (м, 1H), 2,92-2,85 (м, 1H), 2,58-2,50 (м, 1H), 2,49-2,41 (м, 1H), 1,86-1,75 (м, 2H), 1,65-1,55 (м, 1H), 1,37-1,28 (м, 1H).
Стадия 3: метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,075 мл, 0,933 ммоль) добавляли к мутному раствору продукта из стадии 2 выше (62,0 мг, 0,233 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (78 мг, 0,280 ммоль) в ДХМ (2,0 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 20 ч и выпаривали реакционную смесь в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (88,1 мг, 0,177 ммоль, выход 76%, чистота 98%) в виде желтого масла. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,2 (M-H)- через 1,59 мин.
Стадия 4: 3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,707 мл, 0,707 ммоль) добавляли к раствору продукта из стадии 3 выше (88,1 мг, 0,177 ммоль) в ТГФ (1,4 мл) при КТ. Реакционную смесь перемешивали при КТ в течение 18 ч и затем выпаривали в вакууме. Остаток растворяли в воде (3 мл) и подкисляли с использованием 1М HCl до pH 4-5. Осадок выделяли с помощью фильтрации и затем растворяли в EtOAc (5 мл). Органическую фазу промывали водой (3 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (50 мг, 0,104 ммоль, выход 59%, чистота 99%) в виде бледно-розового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 473,1 (M-H)- через 1,38 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,14 (шс, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,46 (д, J=1,8 Гц, 1H), 7,33-7,27 (м, 2H), 7,25 (д, J=8,3 Гц, 1H), 5,09 (шс, 1H), 3,89 (с, 3H), 3,79-3,73 (м, 1H), 2,87-2,79 (м, 2H), 2,74-2,68 (м, 1H), 2,67-2,62 (м, 1H), 1,93-1,85 (м, 1H), 1,77-1,69 (м, 1H), 1,60-1,51 (м, 1H), 1,51-1,43 (м, 1H).
Пример 178: (S)-3-(N-(2-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
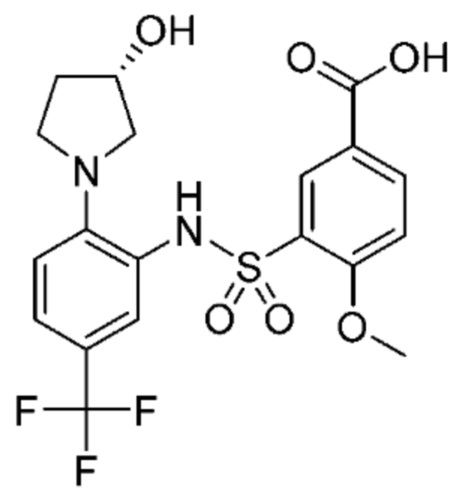
Стадия 1: (S)-1-(2-нитро-4-(трифторметил)фенил)пирролидин-3-ол: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и (S)-пирролидин-3-ола (0,139 мл, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 17 ч. Органическую фазу промывали 1М HCl (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (445 мг, 1,37 ммоль, выход 95%, чистота 85%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 277,2 (M+H)+ через 1,33 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,06-8,03 (м, 1H), 7,72 (дд, J=9,1, 2,3 Гц, 1H), 7,19 (д, J=9,1 Гц, 1H), 5,05 (д, J=3,4 Гц, 1H), 4,41-4,36 (м, 1H), 3,50 (кажущ. тд, J=9,8, 6,8 Гц, 1H), 3,41 (дд, J=11,1, 4,3 Гц, 1H), 3,25-3,19 (м, 1H), 2,85-2,80 (м, 1H), 2,04-1,96 (м, 1H), 1,94-1,88 (м, 1H).
Стадия 2: (S)-1-(2-амино-4-(трифторметил)фенил)пирролидин-3-ол: 5% Pd/C (50% об/об воды) Тип 87L (50 мг, 0,012 ммоль) в EtOH (0,5 мл) добавляли к раствору продукта из стадии 1 выше (220 мг, 0,677 ммоль) в EtOH (3,0 мл) при КТ. Реакционную смесь гидрировали (4 бара) при КТ в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite®, промывая MeOH (20 мл). Органическую фазу выпаривали в вакууме, а остаток растворяли в ДХМ (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (134 мг, 0,522 ммоль, выход 77%, чистота 96%) в виде темно-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 247,3 (M+H)+ через 1,08 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,92 (д, J=1,8 Гц, 1H), 6,88 (д, J=8,2 Гц, 1H), 6,80 (дд, J=8,2, 1,5 Гц, 1H), 4,97 (шс, 2H), 4,86 (д, J=4,9 Гц, 1H), 4,35-4,28 (м, 1H), 3,31-3,22 (м, 2H), 2,99 (ддд, J=9,1, 7,9, 5,0 Гц, 1H), 2,90 (дд, J=10,0, 3,0 Гц, 1H), 2,12-2,04 (м, 1H), 1,79-1,71 (м, 1H).
Стадия 3: (S)-метил-3-(N-(2-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,075 мл, 0,933 ммоль) добавляли к мутному раствору продукта из стадии 2 выше (60,5 мг, 0,233 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (78 мг, 0,280 ммоль) в ДХМ (2,0 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 20 ч, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (96,7 мг, 0,196 ммоль, выход 84%, чистота 96%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 473, (M-H)- через 1,35 мин.
Стадия 4: (S)-3-(N-(2-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,783 мл, 0,783 ммоль) добавляли к раствору продукта из стадии 3 выше (96,7 мг, 0,196 ммоль) в ТГФ (1,6 мл) при КТ. Реакционную смесь перемешивали при КТ в течение 20 ч, после чего выпаривали в вакууме. Остаток растворяли в воде (3 мл) и подкисляли с использованием 1М HCl до pH 4-5. Осадок выделяли с помощью фильтрации и затем растворяли в EtOAc (5 мл). Органическую фазу промывали водой (3 мл), сушили над MgSO4 и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (22,3 мг, 0,046 ммоль, выход 26,3%, чистота 96%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 461,3 (M+H)+, 459,2 (M-H)- через 1,17 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,06 (шс, 1H), 9,30 (шс, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 8,06 (д, J=2,2 Гц, 1H), 7,39 (д, J=8,8 Гц, 1H), 7,29 (дд, J=8,8, 2,4 Гц, 1H), 6,74 (д, J=8,9 Гц, 1H), 6,50 (д, J=2,3 Гц, 1H), 4,96 (шс, 1H), 4,37-4,31 (м, 1H), 3,99 (с, 3H), 3,79 (дд, J=11,0, 4,8 Гц, 1H), 3,59-3,52 (м, 1H), 3,49-3,43 (м, 1H), 3,38-3,34 (м, 1H), 1,96-1,88 (м, 1H), 1,87-1,81 (м, 1H).
Пример 179: 4-метокси-3-(N-(2-(3-метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
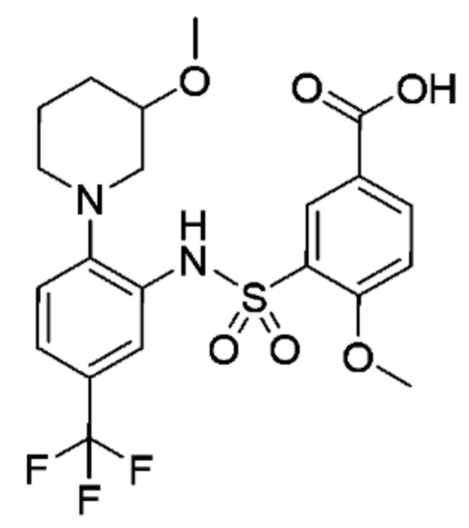
Стадия 1: 3-метокси-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (0.500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и 3-метоксипиперидина (198 мг, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 16 ч. Органическую фазу промывали 1М HCl (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (438 мг, 1,41 ммоль, выход 98%, чистота 98%) в виде оранжевого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,13-8,10 (м, 1H), 7,81 (дд, J=8,9, 2,4 Гц, 1H), 7,43 (д, J=8,9 Гц, 1H), 3,42-3,33 (м, 2H), 3,24 (с, 3H), 3,19 (кажущ. дт, J=12,9, 4,7 Гц, 1H), 3,03-2,96 (м, 1H), 2,86 (дд, J=12,2, 7,5 Гц, 1H), 2,00-1,93 (м, 1H), 1,82-1,73 (м, 1H), 1,57-1,47 (м, 1H), 1,47-1,38 (м, 1H).
Стадия 2: 2-(3-метоксипиперидин-1-ил)-5-(трифторметил)-анилин: 5% Pd/C (50% об/об воды) Тип 87L (50 мг, 0,012 ммоль) в EtOH (0,5 мл) добавляли к раствору продукта из стадии 1 выше (214 мг, 0,689 ммоль) в EtOH (3,0 мл) при КТ. Реакционную смесь гидрировали (4 бара) при КТ в течение 18 ч. Катализатор удаляли с помощью фильтрации через слой Celite®, промывая EtOH (15 мл). Фильтрат выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (151 мг, 0,484 ммоль, выход 70%, чистота 88%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 275,3 (M+H)+ (ЭР+), в 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,99 (д, J=8,1 Гц, 1H), 6,95 (д, J=2,2 Гц, 1H), 6,83 (дд, J=8,1, 1,5 Гц, 1H), 5,12 (шс, 2H), 3,46-3,40 (м, 1H), 3,29 (с, 3H), 3,17-3,09 (м, 1H), 2,99-2,93 (м, 1H), 2,57-2,46 (м, 2H), 1,98-1,90 (м, 1H), 1,80-1,73 (м, 1H), 1,67-1,58 (м, 1H), 1,40-1,29 (м, 1H).
Стадия 3: метил-4-метокси-3-(N-(2-(3-метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Пиридин (0,081 мл, 1,01 ммоль) добавляли к мутному раствору продукта из стадии 2 выше (79 мг, 0,252 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (2,0 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 18 ч, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (84 мг, 0,167 ммоль, выход 66%, чистота 100%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 503,4 (M+H)+, 501,2 (M-H)- через 1,77 мин.
Стадия 4: 4-метокси-3-(N-(2-(3 метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,669 мл, 0,669 ммоль) добавляли к раствору продукта из стадии 3 выше (84 мг, 0,167 ммоль) в ТГФ (1,3 мл) при КТ. Реакционную смесь перемешивали при КТ в течение 18 ч, после чего выпаривали в вакууме. Остаток растворяли в воде (3 мл) и промывали EtOAc (5 мл). Водную фазу подкисляли 1М HCl до pH 4-5, и экстрагировали продукт EtOAc (5 мл ×3). Объединенные органические фазы сушили над MgSO4 и выпаривали в вакууме с получением указанного в заголовке соединения (63 мг, 0,128 ммоль, выход 77%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,1 (M-H)- через 1,60 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (шс, 1H), 9,06 (шс, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,47 (д, J=1,6 Гц, 1H), 7,35-7,23 (м, 3H), 3,89 (с, 3H), 3,47-3,41 (м, 1H), 3,35 (с, 3H), 2,98-2,88 (м, 2H), 2,78-2,71 (м, 2H), 1,85-1,70 (м, 2H), 1,69-1,55 (м, 2H).
Пример 180: 3-(N-(2-(4-этоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
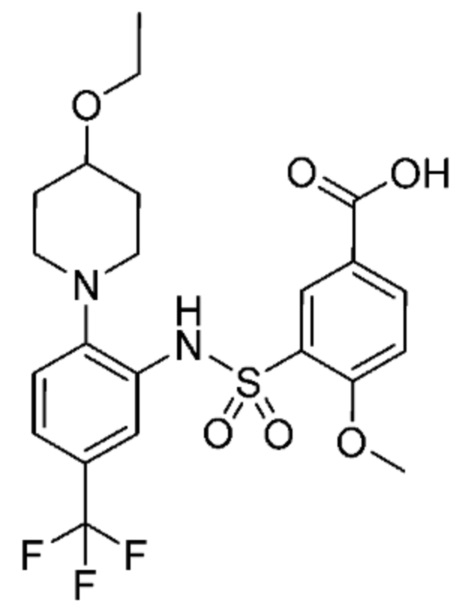
Стадия 1: 4-этокси-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (0,500 мл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и 4-этоксипиперидина (222 мг, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 16 ч. Органическую фазу промывали 1М HCl (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (471 мг, 1,435 ммоль, выход 100%, чистота 97%) в виде оранжевого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,13-8,10 (м, 1H), 7,81 (дд, J=8,9, 2,4 Гц, 1H), 7,42 (д, J=8,8 Гц, 1H), 3,55-3,45 (м, 3H), 3,30-3,25 (м, 2H), 3,03-2,97 (м, 2H), 1,95-1,87 (м, 2H), 1,59-1,51 (м, 2H), 1,12 (т, J=7,0 Гц, 3H).
Стадия 2: 2-(4-этоксипиперидин-1-ил)-5-(трифторметил)-анилин: 5% Pd/C (50% об/об воды) Тип 87L (50 мг, 0,012 ммоль) в EtOH (0,5 мл) добавляли к раствору продукта из стадии 1 выше (228 мг, 0,695 ммоль) в EtOH (3,0 мл) при КТ. Реакционную смесь гидрировали (4 бара) при КТ в течение 18 ч. Катализатор удаляли с помощью фильтрации через слой Celite®, промывая EtOH (15 мл). Фильтрат выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (179 мг, 0,559 ммоль, выход 80%, чистота 90%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 289,3 (M+H)+ через 1,66 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,99 (д, J=8,1 Гц, 1H), 6,95 (д, J=2,2 Гц, 1H), 6,82 (дд, J=8,2, 1,6 Гц, 1H), 5,10 (шс, 2H), 3,48 (к, J=7,0 Гц, 2H), 3,45-3,38 (м, 1H), 3,06-2,99 (м, 2H), 2,65-2,57 (м, 2H), 1,99-1,91 (м, 2H), 1,68-1,59 (м, 2H), 1,12 (т, J=7,0 Гц, 3H).
Стадия 3: метил-3-(N-(2-(4-этоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,081 мл, 1,01 ммоль) добавляли к мутному раствору продукта из стадии 2 выше (81 мг, 0,252 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (2,0 мл) при КТ. Полученный прозрачный раствор перемешивали при КТ в течение 18 ч, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (92,5 мг, 0,159 ммоль, выход 63%, чистота 89%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 517,4 (M+H)+, 515,2 (M-H)- через 1,80 мин.
Стадия 4: 3-(N-(2-(4-этоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (0,634 мл, 0,634 ммоль) добавляли к раствору продукта из стадии 3 выше (92 мг, 0,159 ммоль) в ТГФ (1,3 мл) при КТ. Реакционную смесь перемешивали при КТ в течение 18 ч, после чего выпаривали в вакууме. Остаток растворяли в воде (3 мл) и промывали EtOAc (2×5 мл). Водную фазу подкисляли 1М HCl до pH 4-5 и экстрагировали продукт EtOAc (3×5 мл). Объединенные органические фазы сушили над MgSO4 и выпаривали в вакууме с получением указанного в заголовке соединения (61 мг, 0,118 ммоль, выход 74%, чистота 97%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 503,3 (M+H)+, 501,3 (M-H)- через 1,63 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (шс, 1H), 8,87 (шс, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,44 (д, J=1,7 Гц, 1H), 7,36 (дд, J=8,4, 1,6 Гц, 1H), 7,34-7,30 (м, 2H), 3,91 (с, 3H), 3,52-3,42 (м, 3H), 2,99-2,91 (м, 2H), 2,72-2,64 (м, 2H), 1,98-1,90 (м, 2H), 1,67-1,58 (м, 2H), 1,14 (т, J=7,0 Гц, 3H).
Пример 181: 4-метокси-3-(N-(2-(4-метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
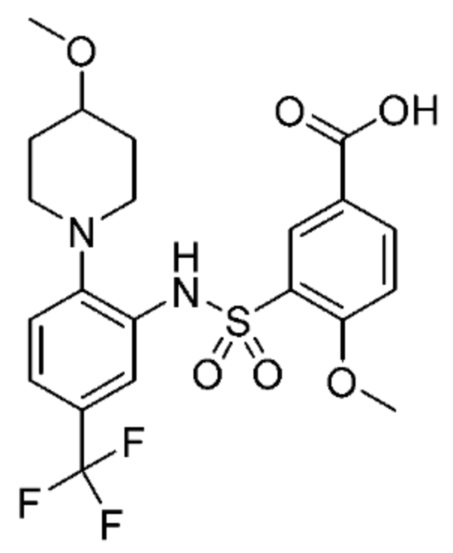
Стадия 1: 4-метокси-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (318 мкл, 2,28 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (128 мкл, 0,912 ммоль) и 4-метоксипиперидина (105 мг, 0,912 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (277 мг, 0,912 ммоль, выход 100%, чистота 100%) в виде светло-оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 305,6 (M+H)+ через 1,60 мин.
Стадия 2: 2-(4-метоксипиперидин-1-ил)-5-(трифторметил)-анилин: Продукт из стадии 1 выше (277 мг, 0,912 ммоль) растворяли в EtOH (14,2 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (239 мг, 0,854 ммоль, выход 94%, чистота 98%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 275,3 (M+H)+, 273,3 (M-H)- через 1,53 мин.
Стадия 3: метил-4-метокси-3-(N-(2-(4-метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (69,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 4 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (52,8 мг, 0,103 ммоль, выход 40,9%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 503,4 (M+H)+ (ЭР+); 501,2 (M-H)- (ЭР-), через 1,71 мин.
Стадия 4: 4-метокси-3-(N-(2-(4-метоксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (50 мг, 0,100 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (362 мкл, 0,398 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной смеси. Мутную смесь упаривали в вакууме до ~2 мл. Образовавшийся осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (38,8 мг, 0,078 ммоль, выход 78%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,2 (M+H)+, 487,1 (M-H)- через 1,53 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (с, 1H), 8,88 (с, 1H), 8,35 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,43 (д, J=2,0 Гц, 1H), 7,38-7,28 (м, 3H), 3,91 (с, 3H), 3,35-3,28 (м, 1H), 3,27 (с, 3H), 2,98-2,90 (м, 2H), 2,71-2,62 (м, 2H), 1,99-1,90 (м, 2H), 1,67-1,57 (м, 2H).
Пример 182: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
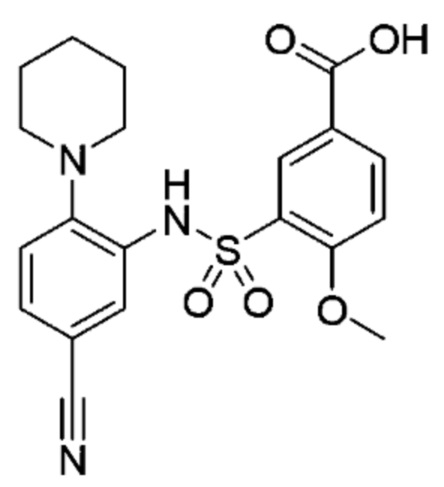
Стадия 1: 3-нитро-4-(пиперидин-1-ил)бензонитрил: Смесь 4-фтор-3-нитробензонитрила (300 мг, 1,81 ммоль), пиперидина (0,2 мл, 2,02 ммоль) и Et3N (0,65 мл, 4,66 ммоль) в ДХМ (6 мл) перемешивали при КТ в течение ночи. Смесь промывали водой (10 мл), пропускали через фазовый сепаратор, концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (400 мг, 1,73 ммоль, выход 96%, чистота 100%) в виде бледно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 232,1 (M+H)+ через 1,60 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,28 (д, J=2,1 Гц, 1H), 7,84 (дд, J=8,9, 2,1 Гц, 1H), 7,34 (д, J=8,9 Гц, 1H), 3,18-3,10 (м, 4H), 1,65-1,54 (м, 6H).
Стадия 2: 3-амино-4-(пиперидин-1-ил)бензонитрил: Раствор продукта из стадии 1 выше (398 мг, 1,72 ммоль) в EtOH (35 мл) гидрировали в проточном реакторе ThalesNano H-cube® (10% Pt/C, 30×4 мм, режим полной подачи водорода, 25°C, скорость потока 1 мл/мин, 1 цикл). Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (118 мг, 0,542 ммоль, выход 32%, чистота 93%) в виде густого красного масла. СВЭЖХ-МС (Метод 2) m/z 202,2 (M+H)+ через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,96-6,95 (м, 3H), 5,07 (с, 2H), 2,79 (т, J=5,1 Гц, 4H), 1,71-1,63 (м, 4H), 1,57-1,48 (м, 2H).
Стадия 3: метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензоат: Смесь продукта из стадии 2 выше (118 мг, 0,542 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (172 мг, 0,651 ммоль) и пиридина (130 мкл, 1,61 ммоль) в ДХМ (5 мл) оставляли с перемешиванием при КТ на выходные. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (172 мг, 0,394 ммоль, выход 73%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 430,2 (M+H)+, 428,1 (M-H)- через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,94 (с, 1H), 8,33 (д, J=2,3 Гц, 1H), 8,20 (дд, J=8,7, 2,3 Гц, 1H), 7,50 (дд, J=8,5, 2,0 Гц, 1H), 7,41-7,35 (м, 2H), 7,24 (д, J=8,5 Гц, 1H), 3,94 (с, 3H), 3,86 (с, 3H), 2,82 (т, J=5,3 Гц, 4H), 1,65-1,57 (м, 4H), 1,55-1,46 (м, 2H).
Стадия 4: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: Смесь продукта из стадии 3 выше (170 мг, 0,390 ммоль) и LiOH (40 мг, 1,67 ммоль) в ТГФ/H2O (4:1, 4 мл) перемешивали при КТ в течение 1 ч и затем при 35°C в течение ночи. Смесь разбавляли H2O (10 мл) и EtOAc (15 мл) и подкисляли до ~pH 4 1М HCl. Фазы разделяли и водную фазу экстрагировали EtOAc (2×15 мл). Объединенные органические экстракты промывали солевым раствором (15 мл), пропускали через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (105 мг, 0,243 ммоль, выход 62%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 416,2 (M+H)+, 413,7 (M-H)- через 1,47 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (с, 1H), 8,88 (с, 1H), 8,33 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,50 (дд, J=8,3, 2,0 Гц, 1H), 7,39 (д, J=2,0 Гц, 1H), 7,34 (д, J=8,7 Гц, 1H), 7,24 (д, J=8,3 Гц, 1H), 3,92 (с, 3H), 2,81 (т, J=5,2 Гц, 4H), 1,67-1,56 (м, 4H), 1,56-1,45 (м, 2H).
Пример 183: 4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
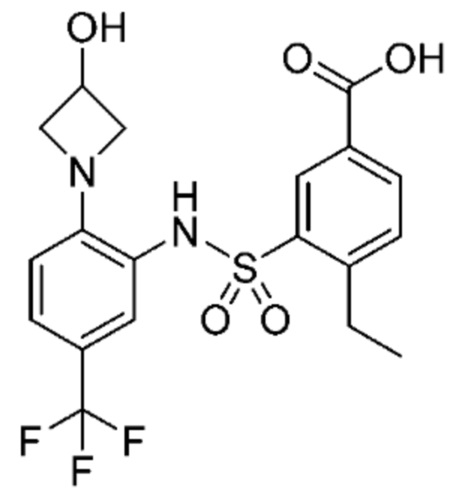
Стадия 1: метил-3-(хлорсульфонил)-4-этилбензоат: Тионилхлорид (5 мл, 68,5 ммоль) порциями добавляли к продукту из примера 1, стадии 1, 3-((хлорсульфонил)-4-этилбензойной кислоте) (0,888 г, 3,57 ммоль) при КТ. Смесь нагревали до 75°C в течение 1 ч. Раствор охлаждали до КТ и выпаривали в вакууме. Остаток растворяли в ДХМ (5 мл), обрабатывали MeOH (0,144 мл, 3,57 ммоль), затем Et3N (0,536 мл, 3,93 ммоль), и перемешивали при КТ в течение ночи. Смесь разбавляли ДХМ (50 мл), промывали водой (50 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (0,450 г, 1,37 ммоль, выход 38%, чистота 80%) в виде светло-коричневого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,73 (д, J=1,8 Гц, 1H), 8,32 (дд, J=8,1, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 3,99 (с, 3H), 3,28 (к, J=7,3 Гц, 2H), 1,40 (т, J=7,4 Гц, 3H).
Стадия 2: 1-(2-нитро-4-(трифторметил)фенил)азетидин-3-ол: Et3N (0,700 мл, 5,02 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (0,201 мл, 1,44 ммоль) и гидрохлорида азетидин-3-ола (189 мг, 1,72 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 16 ч. Органическую фазу промывали 1М HCl (3 мл), затем органическую фазу сушили на гидрофобном фарфоровом фильтре и выпаривали в вакууме с получением указанного в заголовке соединения (461 мг, 1,39 ммоль, выход 97%, чистота 79%) в виде оранжевого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,09-8,05 (м, 1H), 7,73 (дд, J=9,0, 2,3 Гц, 1H), 6,90 (д, J=8,9 Гц, 1H), 5,79 (д, J=6,3 Гц, 1H), 4,55-4,49 (м, 1H), 4,19 (ддд, J=9,7, 6,7, 1,4 Гц, 2H), 3,77 (ддд, J=9,7, 4,1, 1,3 Гц, 2H).
Стадия 3: 1-(2-амино-4-(трифторметил)фенил)азетидин-3-ол: Продукт из стадии 2 выше (455 мг, 1,37 ммоль) растворяли в EtOH (27,4 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 1 цикл). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (12 мл) с получением указанного в заголовке соединения (395 мг, 1,37 ммоль, выход 100%, чистота 81%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z 233,3 (M+H)+ через 1,00 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,86 (д, J=2,1 Гц, 1H), 6,83-6,79 (м, 1H), 6,50 (д, J=8,1 Гц, 1H), 5,52 (д, J=6,5 Гц, 1H), 4,74 (шс, 2H), 4,46 (секстет, J=6,2 Гц, 1H), 4,19-4,13 (м, 2H), 3,45-3,40 (м, 2H).
Стадия 4: метил-4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Пиридин (0,072 мл, 0,896 ммоль) добавляли к раствору продукта из стадии 3 выше (65 мг, 0,224 ммоль) и продукта из стадии 1 выше (92 мг, 0,280 ммоль) в ДХМ (2,0 мл) при КТ. Полученный мутный раствор перемешивали при КТ в течение 21 ч. Реакционную смесь выпаривали в вакууме, и неочищенный продукт очищали с помощью хроматографии на силикагеле (10 г картридж, 0-65% EtOAc/изогексан) с получением указанного в заголовке соединения (51 мг, 0,102 ммоль, выход 46%, чистота 92%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 459,4 (M+H)+, 457,2 (M-H)- через 0,66 мин.
Стадия 5: 4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,409 мл, 0,409 ммоль) добавляли к раствору продукта из стадии 4 выше (51 мг, 0,102 ммоль) в ТГФ (0,82 мл) при КТ. Раствор перемешивали при КТ в течение 17 ч, после чего выпаривали в вакууме. Остаток растворяли в воде (3 мл) и промывали EtOAc (5 мл). Водную фазу подкисляли 1М HCl до pH 4-5, и продукт экстрагировали EtOAc (3×5 мл). Органические фазы объединяли, сушили над MgSO4 и выпаривали в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (7,3 мг, 0,016 ммоль, выход 16%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 445,3 (M+H)+, 443,2 (M-H)- через 1,32 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,24 (шс, 1H), 9,55 (шс, 1H), 8,25 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,5 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,31 (шд, J=8,7 Гц, 1H), 6,51 (д, J=8,6 Гц, 1H), 6,24 (шс, 1H), 5,63 (шд, J=5,9 Гц, 1H), 4,58-4,48 (м, 1H), 4,40-4,33 (м, 2H), 3,82 (дд, J=8,7, 4,8 Гц, 2H), 2,94 (к, J=7,4 Гц, 2H), 1,17 (т, J=7,4 Гц, 3H).
Пример 184: 3-(N-(3-фтор-2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
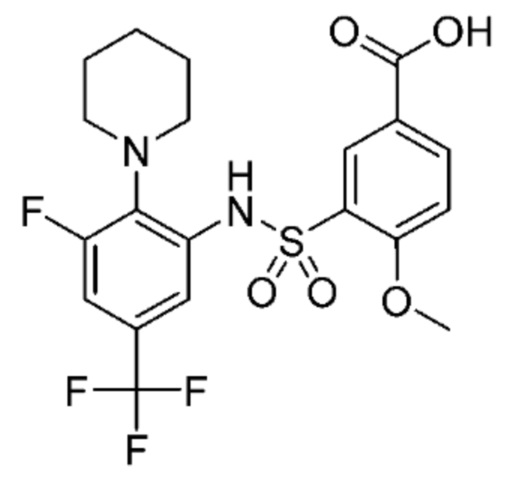
Стадия 1: 1-(2-фтор-6-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (0,767 мл, 5,50 ммоль) добавляли к раствору 1,2-дифтор-3-нитро-5-(трифторметил)бензола (500 мг, 2,20 ммоль) и пиперидина (0,261 мл, 2,64 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 23 ч. Органическую фазу промывали 1М HCl (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (676 мг, 2,20 ммоль, выход 100%, чистота 98%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 293,5 (M+H)+ через 1,93 мин.
Стадия 2: 3-фтор-2-(пиперидин-1-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (0,642 г, 2,20 ммоль) растворяли в EtOH (44 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Неочищенный продукт выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (12 мл) с получением указанного в заголовке соединения (0,543 г, 1,97 ммоль, выход 90%, чистота 95%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z 263,3 (M+H)+ через 1,89 мин.
Стадия 3: метил-3-(N-(3-фтор-2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,139 мл, 1,72 ммоль) добавляли к раствору продукта из стадии 2 выше (0,15 г, 0,572 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (0,189 г, 0,715 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, и неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,372 г, 0,546 ммоль, выход 95%, чистота 72%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 491,3 (M+H)+, 489,2 (M-H)- через 1,96 мин.
Стадия 4: 3-(N-(3-фтор-2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (3,28 мл, 3,28 ммоль) добавляли к раствору продукта из стадии 3 выше (0,268 г, 0,547 ммоль) в ТГФ (12 мл) и MeOH (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и экстрагировали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали продукт МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор. Растворитель удаляли в вакууме с получением указанного в заголовке соединения (0,184 г, 0,378 ммоль, выход 69%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,3 (M+H)+, 474,9 (M-H)- через 1,81 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (с, 1H), 8,99 (с, 1H), 8,38 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,37-7,31 (м, 3H), 3,94 (с, 3H), 2,91-2,81 (м, 4H), 1,69-1,62 (м, 4H), 1,58-1,51 (м, 2H).
Следующие примеры были получены способами, аналогичными Примеру 184, с заменой подходящими исходными соединениями и промежуточными соединениями в случае необходимости:
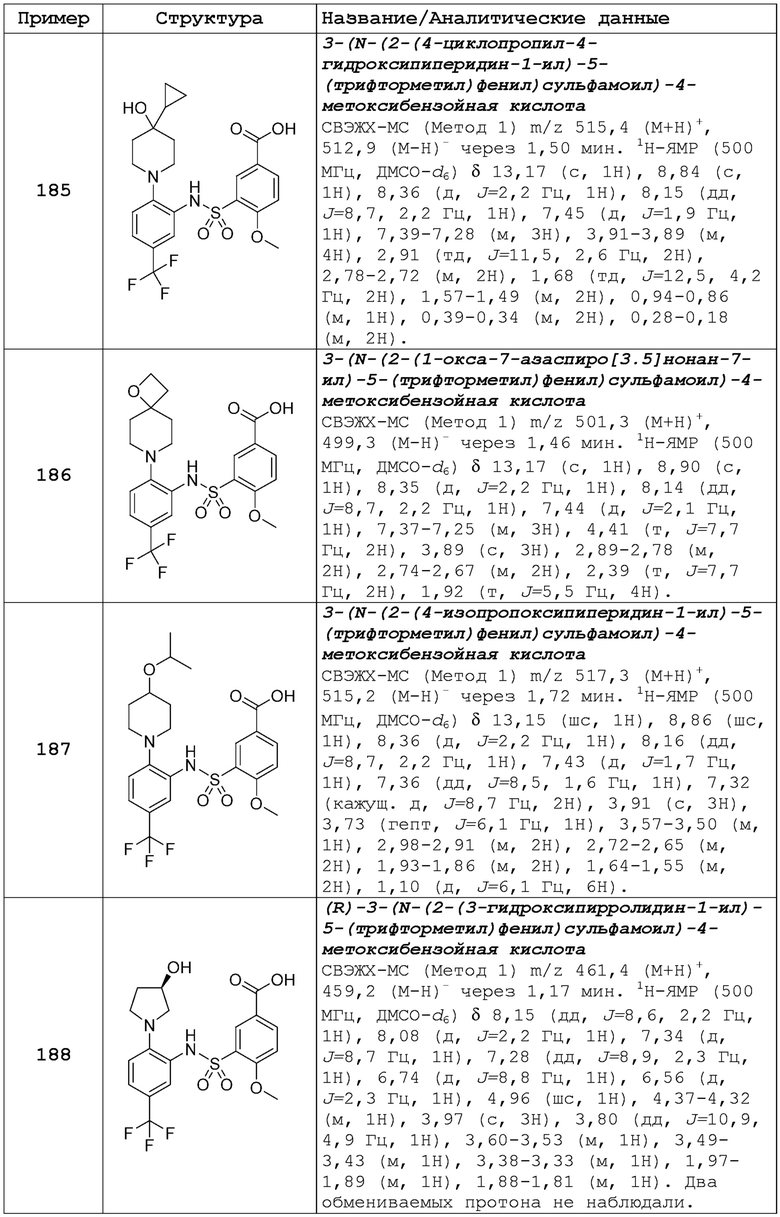
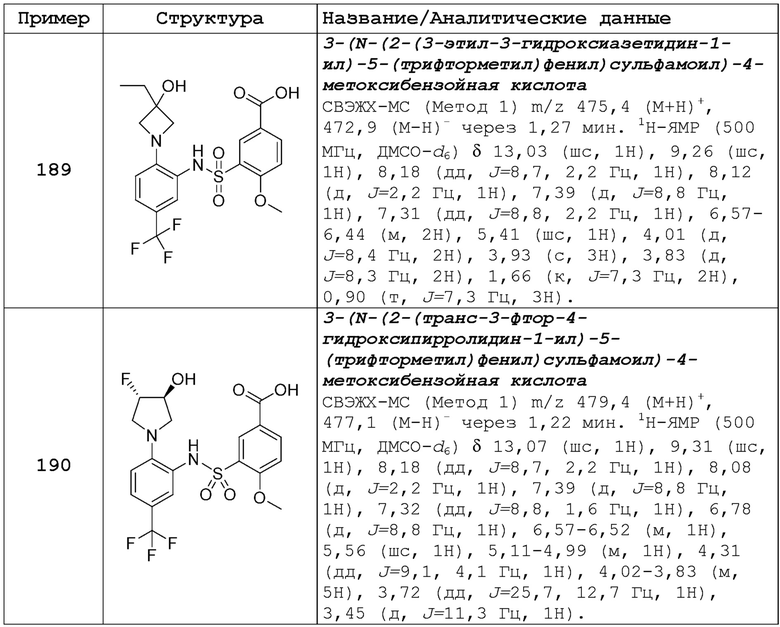
Пример 200: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(трифторметил)бензойная кислота
Стадия 1: метил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(трифторметил)бензоат: Смесь 2-(пиперидин-1-ил)-5-(трифторметил)анилина (75 мг, 0,307 ммоль), метил-3-(хлорсульфонил)-4-(трифторметил)бензоата (101 мг, 0,335 ммоль) и пиридина (75 мкл, 0,927 ммоль) в ДХМ (4 мл) перемешивали при КТ в течение ночи, а затем при 35°C в течение 11 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (91 мг, 0,178 ммоль, выход 58,1%, чистота 100%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 511,2 (M+H)+, 509,0 (M-H)- через 1,99 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,73 (с, 1H), 8,46 (с, 1H), 8,34 (д, J=8,2 Гц, 1H), 8,19 (д, J=8,2 Гц, 1H), 7,50 (д, J=8,4 Гц, 1H), 7,37 (д, J=2,2 Гц, 1H), 7,26 (дд, J=8,4, 2,2 Гц, 1H), 3,89 (с, 3H), 2,71-2,65 (м, 4H), 1,48-1,36 (м, 6H).
Стадия 2: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-сульфамоил)-4-(трифтор-метил)бензойная кислота: Смесь продукта из стадии 1 выше (91 мг, 0,178 ммоль) и LiOH (17 мг, 0,710 ммоль) в ТГФ/MeOH/воде (4:1:1, 2,4 мл) перемешивали при 35°C в течение ночи. Смесь разбавляли водой (10 мл) и EtOAc (15 мл) и подкисляли до ~pH 4 1М HCl (водн.). Фазы разделяли и водную фазу экстрагировали EtOAc (2×15 мл). Органические экстракты объединяли и промывали солевым раствором (15 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток растирали с изогексаном/МТБЭ (5:1) с получением указанного в заголовке соединения (33,4 мг, 0,066 ммоль, выход 37,0%, чистота 98%) в виде бежевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 497,2 (M+H)+, 495,1 (M-H)- через 1,92 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,89 (с, 1H), 9,69 (с, 1H), 8,48 (д, J=1,6 Гц, 1H), 8,32 (дд, J=8,2, 1,6 Гц, 1H), 8,16 (д, J=8,2 Гц, 1H), 7,49 (дд, J=8,5, 2,2 Гц, 1H), 7,35 (д, J=2,2 Гц, 1H), 7,26 (д, J=8,5 Гц, 1H), 2,73-2,64 (м, 4H), 1,49-1,35 (м, 6H).
Пример 201: 4-этокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
Стадия 1: метил-4-этокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0.100 г, 0,409 ммоль) в ДХМ (5 мл) и пиридине (0,199 мл, 2,46 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-этоксибензоата (0,114 г, 0,409 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 24 ч. Растворитель удаляли в вакууме, и неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,160 г, 0,326 ммоль, выход 80%, чистота 99%) в виде кремового воскообразного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,4 (M+H)+, 485,2 (M-H)- через 1,93 мин.
Стадия 2: 4-этокси-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,024 г, 0,987 ммоль) добавляли к раствору продукта из стадии 1 (0,160 г, 0,329 ммоль) в ТГФ (5 мл) и перемешивали раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме до воды. Доводили pH до pH 6 1М HCl (водн.) с образованием осадка, который отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл) с получением указанного в заголовке соединения (0,151 г, 0,304 ммоль, выход 92%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,2 (M-H)- через 1,78 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 8,55 (шс, 1H), 8,40 (д, J=2,2 Гц, 1H), 8,13 (дд, J=8,7, 2,2 Гц, 1H), 7,47 (д, J=2,0 Гц, 1H), 7,39-7,34 (м, 1H), 7,32-7,30 (м, 2H), 4,22 (к, J=7,0 Гц, 2H), 2,76 (т, J=5,3 Гц, 4H), 1,62 (п, J=5,5 Гц, 4H), 1,52 (п, J=6,3 Гц, 2H), 1,27 (т, J=7,0 Гц, 3H).
Пример 202: 3-(N-(4,5-дихлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
Стадия 1: метил-3-(N-(4,5-дихлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензоат: Пиридин (0,166 мл, 2,06 ммоль) добавляли к раствору 4,5-дихлор-2-(пиперидин-1-ил)анилина (0,168 г, 0,685 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (0,227 г, 0,857 ммоль) в ДХМ (10 мл). Раствор перемешивали при КТ в течение 18 ч, а затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,257 г, 0,543 ммоль, выход 79%, чистота 81%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 472,8 (M-H)- через 1,75 мин.
Стадия 2: 3-(N-(4,5-дихлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (3,26 мл, 3,26 ммоль) добавляли к раствору продукта из стадии 1 выше (0,257 г, 0,543 ммоль) в ТГФ (13 мл) и MeOH (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Объединенные органические фазы сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,229 г, 0,494 ммоль, выход 91%, чистота 97%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,3/461,3 (M+H)+, 457,2/459,2 (M-H)- через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,22 (с, 1H), 8,75 (с, 1H), 8,35 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,42 (с, 1H), 7,38 (с, 1H), 7,34 (д, J=8,8 Гц, 1H), 3,94 (с, 3H), 2,70-2,64 (м, 4H), 1,67-1,56 (м, 4H), 1,56-1,42 (м, 2H).
Пример 203: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензойная кислота
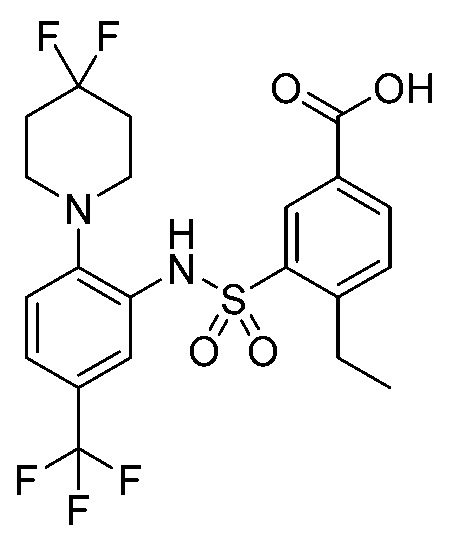
Стадия 1: 3-(хлорсульфонил)-4-этилбензойная кислота: 4-этилбензойную кислоту (7 г, 46,6 ммоль) в хлоросульфоновой кислоте (20 мл, 299 ммоль) нагревали при 100°C в течение 5 ч. Смесь охлаждали и осторожно добавляли в перемешиваемую воду со льдом (200 мл). Образовавшийся осадок собирали с помощью фильтрации, промывали водой (100 мл) и сушили в вакууме с получением указанного в заголовке соединения (10,9 г, 41,5 ммоль, выход 89%, чистота 95%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,65 (шс, 1H), 8,34 (д, J=1,9 Гц, 1H), 7,82 (дд, J=7,9, 2,0 Гц, 1H), 7,32 (д, J=7,9 Гц, 1H), 3,08 (к, J=7,5 Гц, 2H), 1,18 (т, J=7,5 Гц, 3H).
Стадия 2: метил-3-(хлорсульфонил)-4-этилбензоат: Тионилхлорид (10 мл, 137 ммоль) порциями добавляли к продукту из стадии 1 выше (4 г, 16,1 ммоль) при КТ. Смесь нагревали до 75°C в течение 2 ч, охлаждали до КТ, выпаривали в вакууме и подвергали азеотропной перегонке с толуолом. Твердое вещество растворяли в ДХМ (10 мл) и обрабатывали MeOH (0,716 мл, 17,7 ммоль), затем Et3N (2,41 мл, 17,7 ммоль), и перемешивали при КТ в течение ночи. Смесь разбавляли ДХМ (50 мл), промывали водой (50 мл), сушили (MgSO4) и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (3,60 г, 13,02 ммоль, выход 81%, чистота 95%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,74 (д, J=1,8 Гц, 1H), 8,32 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 3,99 (с, 3H), 3,28 (к, J=7,5 Гц, 2H), 1,41 (т, J=7,5 Гц, 3H).
Стадия 3: метил-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензоат: Пиридин (0,069 мл, 0,856 ммоль) добавляли к раствору продукта из примера 12, стадии 2 (0,08 г, 0,285 ммоль) и продукта из стадии 2 выше (0,094 г, 0,357 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, и неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,137 г, 0,227 ммоль, выход 80%, чистота 84%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 507,4 (M+H)+, 505,2 (M-H)- через 1,90 мин.
Стадия 4: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (1,35 мл, 1,35 ммоль) добавляли к раствору продукта из стадии 3 выше (0,137 г, 0,225 ммоль) в ТГФ (6 мл) и MeOH (1,3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,105 г, 0,209 ммоль, выход 93%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 493,3 (M+H)+, 490,9 (M-H)- через 1,76 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (с, 1H), 9,85 (с, 1H), 8,37 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,44 (дд, J=8,5, 2,1 Гц, 1H), 7,36-7,31 (м, 2H), 3,03 (к, J=7,4 Гц, 2H), 2,89-2,80 (м, 4H), 2,13-2,00 (м, 4H), 1,18 (т, J=7,4 Гц, 3H).
Пример 204: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензойная кислота
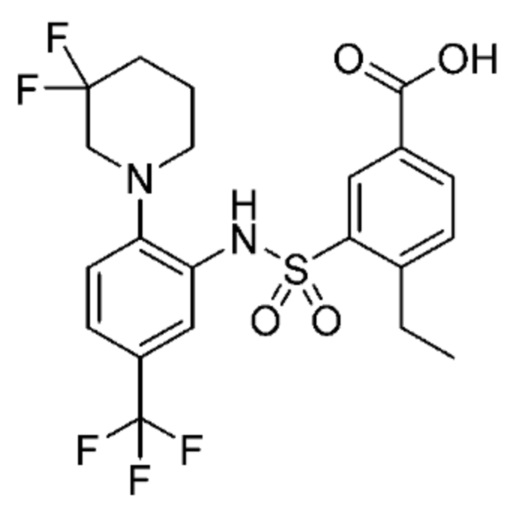
Стадия 1: метил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензоат: Пиридин (0,052 мл, 0,642 ммоль) добавляли к раствору продукта из примера 9, стадии 2 (60 мг, 0,214 ммоль) и продукта из примера 203, стадии 2 (70 мг, 0,268 ммоль) в ДХМ (10 мл). Раствор перемешивали при КТ в течение 18 ч, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,057 г, 0,113 ммоль, выход 52,6%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 507,7 (M+H)+, 505,2 (M-H)- через 1,89 мин.
Стадия 2: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (0,675 мл, 0,675 ммоль) добавляли к раствору продукта из стадии 1 выше (0,057 г, 0,113 ммоль) в ТГФ (8 мл) и MeOH (2 мл). Раствор перемешивали при КТ в течение ночи, а затем выпаривали в вакууме. Остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли, сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (0,056 г, 0,110 ммоль, выход 98%, чистота 97%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 493,7 (M+H)+, 491,1 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,30 (с, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,8 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,45 (дд, J=8,5, 2,1 Гц, 1H), 7,33 (д, J=8,5 Гц, 1H), 7,12 (д, J=2,1 Гц, 1H), 3,21 (т, J=11,4 Гц, 2H), 3,00 (к, J=7,4 Гц, 2H), 2,98-2,94 (м, 2H), 2,08-1,96 (м, 2H), 1,84-1,75 (м, 2H), 1,19 (т, J=7,4 Гц, 3H).
Пример 205: 4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
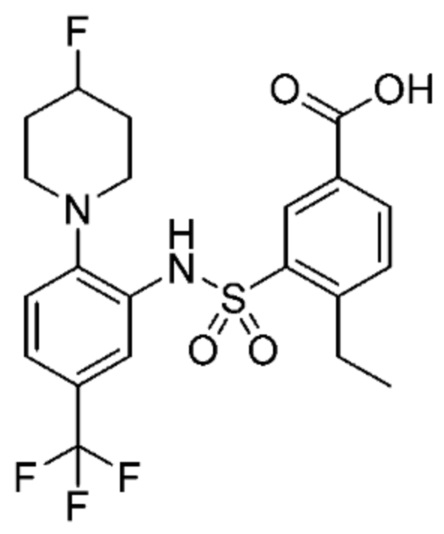
Стадия 1: 4-фтор-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (500 мкл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и 4-фторпиперидина (192 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 3 дней. Добавляли 1М HCl (водн.) (2 мл) и отделяли органическую фазу при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (419 мг, 1,44 ммоль, выход 100%, чистота 100%) в виде бледно-желтого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 293,3 (M+H)+ через 1,62 мин.
Стадия 2: 2-(4-фторпиперидин-1-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (419 мг, 1,44 ммоль) растворяли в EtOH (28,8 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (371 мг, 1,27 ммоль, выход 89%, чистота 90%) в виде прозрачного вязкого масла. СВЭЖХ-МС (Метод 2) m/z 263,3 (M+H)+ через 1,59 мин.
Стадия 3: метил-4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (66,5 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали суспензией продукта из примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (61 мг, 0,112 ммоль, выход 44,3%, чистота 90%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,2 (M-H)- через 1,87 мин.
Стадия 4: 4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (59 мг, 0,121 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (439 мкл, 0,483 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Образовавшийся осадок собирали с помощью фильтрации и промывали водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (38,4 мг, 0,078 ммоль, выход 64,3%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 473,2 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (шс, 1H), 9,68 (шс, 1H), 8,34 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,46-7,40 (м, 1H), 7,31-7,24 (м, 2H), 4,85-4,70 (м, 1H), 3,03 (к, J=7,4 Гц, 2H), 2,89 (т, J=9,9 Гц, 2H), 2,74-2,67 (м, 2H), 2,00-1,87 (м, 2H), 1,85-1,73 (м, 2H), 1,19 (т, J=7,4 Гц, 3H).
Пример 206: 3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
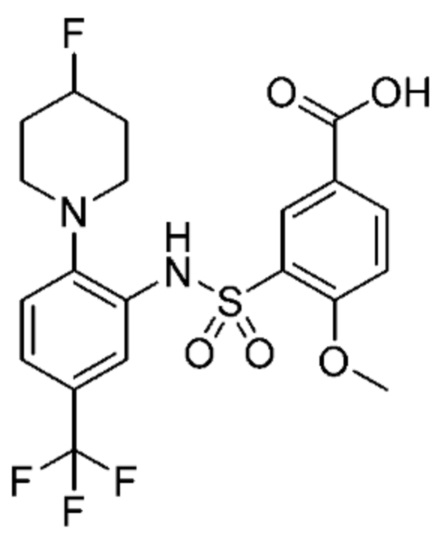
Стадия 1: метил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из Примера 205, стадии 2 (66,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (88 мг, 0,161 ммоль, выход 64,1%, чистота 90%) в виде клейкого кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 491,4 (M+H)+, 489,1 (M-H)- через 1,73 мин.
Стадия 2: 3-(N-(2-(4-фторпиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 1 выше (86 мг, 0,175 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (638 мкл, 0,701 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Образовавшийся осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (52,7 мг, 0,108 ммоль, выход 61,8%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,3 (M+H)+, 475,1 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,00 (шс, 1H), 8,35 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,44 (д, J=2,0 Гц, 1H), 7,40-7,29 (м, 3H), 4,93-4,75 (м, 1H), 3,90 (с, 3H), 2,94 (т, J=9,8 Гц, 2H), 2,79-2,73 (м, 2H), 2,10-1,94 (м, 2H), 1,93-1,79 (м, 2H).
Пример 207: 4-метокси-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
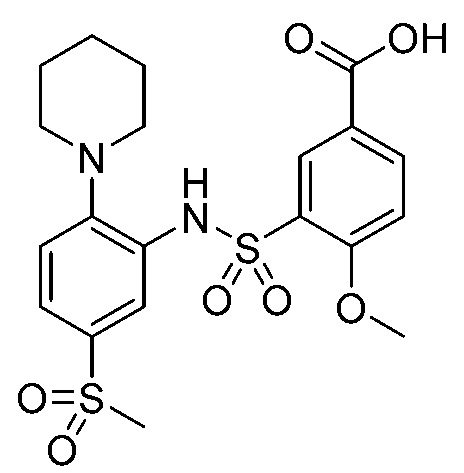
Стадия 1: 1-(4-(метилсульфонил)-2-нитрофенил)пиперидин: Et3N (0,795 мл, 5,70 ммоль) добавляли к раствору 1-фтор-4-(метилсульфонил)-2-нитробензола (500 мг, 2,28 ммоль) и пиперидина (0,226 мл, 2,28 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 23 ч. Органическую фазу промывали 1М HCl (водн.) (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (0,676 г, 2,28 ммоль, выход 100%, чистота 100%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 285,2 (M+H)+ через 1,32 мин.
Стадия 2: 5-(метилсульфонил)-2-(пиперидин-1-ил)анилин: Продукт из стадии 1 выше (0,676 г, 2,38 ммоль) растворяли в EtOH (44 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Реакционную смесь выпаривали в вакууме, а затем подвергали азеотропной перегонке с MeOH (12 мл) с получением указанного в заголовке соединения (0,615 г, 2,370 ммоль, выход 100%, чистота 98%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z 255,3 (M+H)+ через 1,20 мин.
Стадия 3: метил-4-метокси-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Пиридин (0,143 мл, 1,77 ммоль) добавляли к раствору продукта из стадии 2 выше (0,15 г, 0,590 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (0,195 г, 0,737 ммоль) в ДХМ (10 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Раствор выпаривали в вакууме, и неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,201 г, 0,412 ммоль, выход 69,9%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,3 (M+H)+, 481,0 (M-H)- через 1,49 мин.
Стадия 4: 4-метокси-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (2,47 мл, 2,47 ммоль) добавляли к раствору продукта из стадии 3 выше (0,199 г, 0,412 ммоль) в ТГФ (10 мл) и MeOH (2,5 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор. Растворитель удаляли в вакууме с получением указанного в заголовке соединения (0,176 г, 0,372 ммоль, выход 90%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,4 (M+H)+, 467,0 (M-H)- через 1,36 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,17 (с, 1H), 8,82 (с, 1H), 8,35 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,65 (д, J=2,2 Гц, 1H), 7,55 (дд, J=8,4, 2,2 Гц, 1H), 7,34 (д, J=8,4 Гц, 1H), 7,33 (д, J=8,7 Гц, 1H), 3,94 (с, 3H), 3,00 (с, 3H), 2,85-2,78 (м, 4H), 1,71-1,60 (м, 4H), 1,58-1,50 (м, 2H).
Пример 208: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
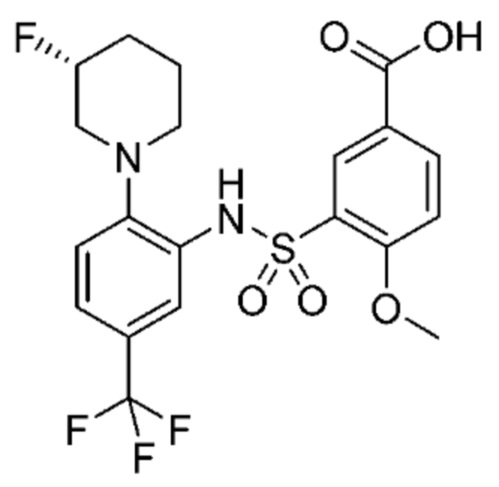
Стадия 1: (R)-3-фтор-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (500 мкл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (300 мг, 1,44 ммоль) и (R)-3-фторпиперидина (250 мг, 2,42 ммоль) в ДХМ (6 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу отделяли при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (488 мг, 1,44 ммоль, выход 100%, чистота 86%) в виде бледно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 293,0 (M+H)+ через 1,59 мин.
Стадия 2: (R)-2-(3-фторпиперидин-1-ил)-5-(трифторметил)-анилин: Продукт из стадии 1 выше (419 мг, 1,44 ммоль) растворяли в EtOH (28,8 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (457 мг, 1,394 ммоль, выход 97%, чистота 80%) в виде геля кремового цвета. СВЭЖХ-МС (Метод 2) m/z 263,3 (M+H)+ через 1,59 мин.
Стадия 3: (R)-метил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 3 выше (66,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (72,4 мг, 0,118 ммоль, выход 46,9%, чистота 80%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 491,3 (M+H)+, 489,1 (M-H)- через 1,73 мин.
Стадия 4: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (69 мг, 0,141 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (512 мкл, 0,563 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (55,2 мг, 0,110 ммоль, выход 78%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,4 (M+H)+, 475,1 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (шс, 1H), 8,75 (шс, 1H), 8,38 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,44 (с, 1H), 7,38-7,33 (м, 2H), 7,31 (д, J=8,8 Гц, 1H), 4,95-4,79 (м, 1H), 3,91 (с, 3H), 3,09-2,86 (м, 3H), 2,85-2,75 (м, 1H), 1,95-1,75 (м, 3H), 1,74-1,63 (м, 1H).
Пример 209: (S)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
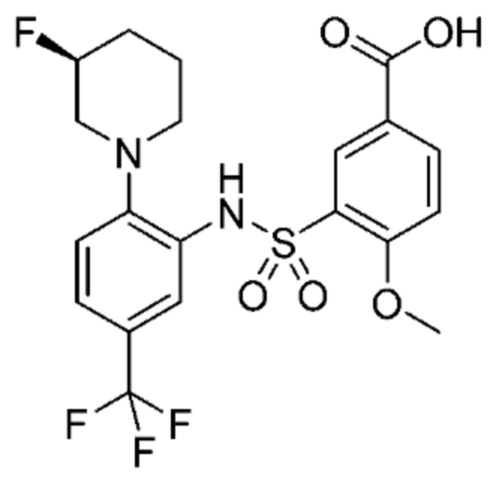
Стадия 1: (S)-3-фтор-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Et3N (500 мкл, 3,59 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (300 мг, 1,44 ммоль) и (S)-3-фторпиперидина (250 мг, 2,42 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу отделяли и выпаривали в вакууме с получением указанного в заголовке соединения (461 мг, 1,44 ммоль, выход 100%, чистота 91%) в виде бледно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 293,1 (M+H)+ через 1,60 мин.
Стадия 2: (S)-2-(3-фторпиперидин-1-ил)-5-(трифторметил)-анилин: Продукт из стадии 1 выше (419 мг, 1,44 ммоль) растворяли в EtOH (28,8 мл). Реакционную смесь гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (475 мг, 1,43 ммоль, выход 100%, чистота 79%) в виде геля кремового цвета. СВЭЖХ-МС (Метод 2) m/z 263,3 (M+H)+ через 1,59 мин.
Стадия 3: (S)-метил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (66,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (81,4 мг, 0,133 ммоль, выход 52,7%, чистота 80%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 491,4 (M+H)+, 489,3 (M-H)- через 1,74 мин.
Стадия 4: (S)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (78 мг, 0,159 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (578 мкл, 0,636 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Его промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (55,2 мг, 0,110 ммоль, выход 69,2%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,3 (M+H)+, 475,2 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (шс, 1H), 8,74 (шс, 1H), 8,37 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,44 (с, 1H), 7,39-7,27 (м, 3H), 4,95-4,79 (м, 1H), 3,91 (с, 3H), 3,08-2,86 (м, 3H), 2,83-2,76 (м, 1H), 1,95-1,75 (м, 3H), 1,74-1,63 (м, 1H).
Пример 210: (S)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
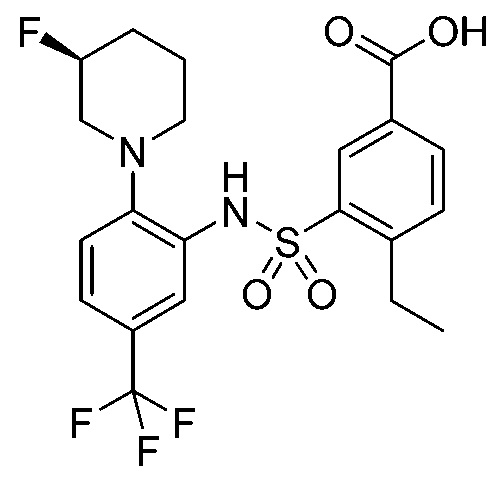
Стадия 1: (S)-метил-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 209, стадии 2 (67 мг, 0,255 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (83 мкл, 1,02 ммоль) и обрабатывали суспензией продукта из примера 203, стадии 2 (124 мг, 0,307 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (65,9 мг, 0,108 ммоль, выход 42,2%, чистота 80%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,4 (M+H)+, 487,2 (M-H)- через 1,89 мин.
Стадия 2: (S)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (63 мг, 0,129 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 LiOH (водн.) (469 мкл, 0,516 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (35,9 мг, 0,072 ммоль, выход 55,7%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 473,2 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (шс, 1H), 9,36 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,1 Гц, 1H), 7,42 (дд, J=8,4, 2,1 Гц, 1H), 7,33-7,24 (м, 2H), 1,98-1,85 (м, 1H), 3,13-2,97 (м, 3H), 2,89-2,79 (м, 2H), 2,71 (тд, J=8,1, 4,0 Гц, 1H), 1,98-1,85 (м, 1H), 1,82-1,71 (м, 1H), 1,71-1,55 (м, 2H), 1,19 (т, J=7,4 Гц, 3H).
Пример 211: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
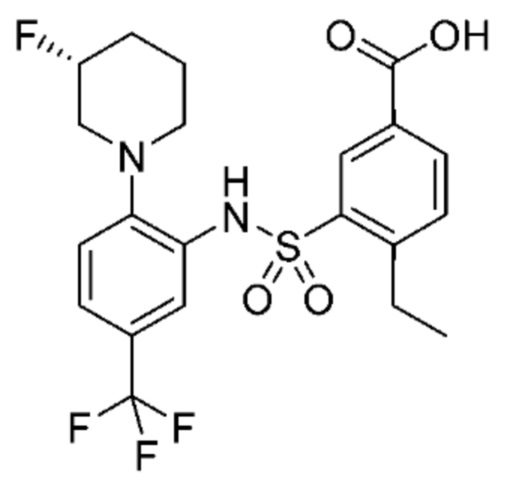
Стадия 1: (R)-метил-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 208, стадии 2 (67 мг, 0,255 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (83 мкл, 1,02 ммоль) и обрабатывали суспензией продукта из примера 203 стадии 2 (124 мг, 0,307 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (76,7 мг, 0,126 ммоль, выход 49,2%, чистота 80%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,2 (M-H)- через 1,89 мин.
Стадия 2: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (74 мг, 0,151 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (551 мкл, 0,606 ммоль). По каплям добавляли MeOH с получением раствора, который перемешивали при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl. Комковатую суспензию обрабатывали ультразвуком с получением мутного раствора, который выпаривали в вакууме до ~2 мл. Образовавшийся осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (43,5 мг, 0,087 ммоль, выход 57,5%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 473,2 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (шс, 1H), 9,36 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,45-7,38 (м, 1H), 7,33-7,25 (м, 2H), 4,84-4,68 (м, 1H), 3,13-2,96 (м, 3H), 2,89-2,79 (м, 2H), 2,75-2,67 (м, 1H), 1,98-1,85 (м, 1H), 1,82-1,72 (м, 1H), 1,70-1,54 (м, 2H), 1,19 (т, J=7,4 Гц, 3H).
Пример 212: 3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
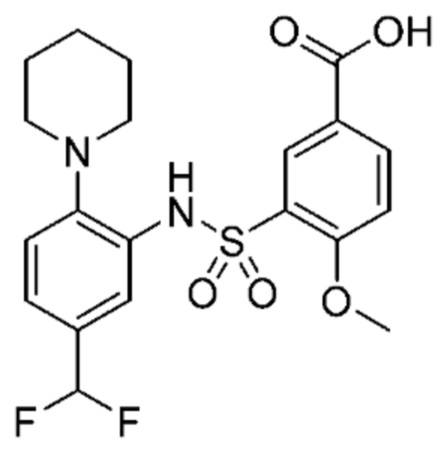
Синтез 3-(хлорсульфонил)-4-метоксибензойной кислоты
4-метоксибензойную кислоту (6,5 г, 42,7 ммоль) порциями добавляли к хлоросульфоновой кислоте (30 мл, 448 ммоль) при КТ. Смесь нагревали до 80°C в течение 2 ч, затем охлаждали до КТ и осторожно добавляли в воду со льдом (300 мл), после чего перемешивали в течение 1 ч. Твердое вещество собирали, промывали водой (200 мл) и сушили в вакууме с получением указанного в заголовке соединения (7,92 г, 30,0 ммоль, выход 70,3%, чистота 95%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (шс, 1H), 8,30 (д, J=2,3 Гц, 1H), 7,90 (дд, J=8,6, 2,4 Гц, 1H), 7,07 (д, J=8,6 Гц, 1H), 3,83 (с, 3H).
Синтез 3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота
Стадия 1: 1-(4-(дифторметил)-2-нитрофенил)пиперидин: Et3N (547 мкл, 3,92 ммоль) добавляли к раствору 4-(дифторметил)-1-фтор-2-нитробензола (300 мг, 1,57 ммоль) и пиперидина (202 мкл, 2,04 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и отделяли органическую фазу при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (402 мг, 1,57 ммоль, выход 100%, чистота 100%) в виде желтого вязкого масла. СВЭЖХ-МС (Метод 1) m/z 257,3 (M+H)+ через 1,63 мин.
Стадия 2: 5-(дифторметил)-2-(пиперидин-1-ил)анилин: Продукт из стадии 1 выше (402 мг, 1,57 ммоль) растворяли в EtOH (14,4 мл). Реакционную смесь гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, 40°C, скорость потока 1 мл/мин, 2 цикла). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (6 мл) с получением указанного в заголовке соединения (324 мг, 1,403 ммоль, выход 89%, чистота 98%) в виде светло-желтого масла. СВЭЖХ-МС (Метод 1) m/z 227,3 (M+H)+ через 1,58 мин.
Стадия 3: 3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 2 выше (60,2 мг, 0,266 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (86 мкл, 1,06 ммоль) и обрабатывали суспензией 3-(хлорсульфонил)-4-метоксибензойной кислоты (80 мг, 0,319 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Реакционную смесь фильтровали и очищали фильтрат непосредственно с помощью хроматографии на силикагеле (12 г картридж, 100% изогексан, затем 0-100% 10% MeOH в EtOAc/изогексане). Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (18 мг, 0,040 ммоль, выход 14,9%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 441,2 (M+H)+, 439,1 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,15 (шс, 1H), 8,66 (шс, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,42 (с, 1H), 7,30 (д, J=8,9 Гц, 1H), 7,29 (д, J=8,2 Гц, 1H), 7,18 (д, J=8,2, 1H), 6,89 (т, J=55,9 Гц, 1H), 3,93 (с, 3H), 2,71 (т, J=5,2 Гц, 4H), 1,67 (п, J=5,5 Гц, 4H), 1,57-1,50 (м, 2H).
Пример 213: 3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота
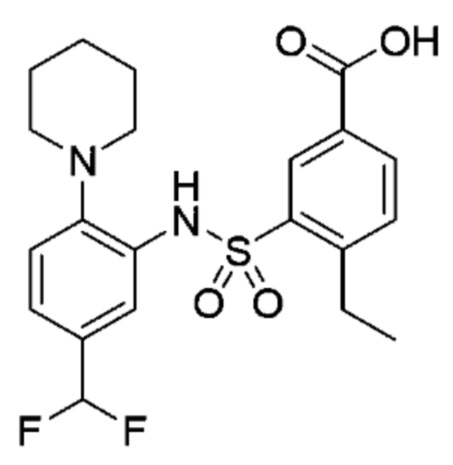
Стадия 1: метил-3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Продукт Примера 212, стадии 2 (57,4 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали суспензией продукта Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (54 мг, 0,119 ммоль, выход 47,0%, чистота 100%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 453,4 (M+H)+, 451,1 (M-H)- через 1,91 мин.
Стадия 2: 3-(N-(5-(дифторметил)-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: Конц. HCl (2,2 мл, 72,4 ммоль) добавляли в воду (0,737 мл) и добавляли этот раствор к раствору продукта из стадии 1 выше (52 мг, 0,115 ммоль) в диоксане (2,2 мл). Реакционную смесь нагревали при 50°C в течение 2 дней. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% 10% MeOH в EtOAc/пентане) с получением указанного в заголовке соединения (18 мг, 0,039 ммоль, выход 33,9%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 439,4 (M+H)+, 437,3 (M-H)- через 1,75 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,28 (шс, 1H), 9,21 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,07 (дд, J=8,0, 1,8 Гц, 1H), 7,59 (д, J=8,1 Гц, 1H), 7,32-7,18 (м, 3H), 7,02-6,08 (м, 1H), 3,03 (к, J=7,4 Гц, 2H), 2,67-2,61 (м, 4H), 1,59-1,50 (м, 4H), 1,49-1,42 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 214: 4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)фенил)сульфамоил)бензойная кислота
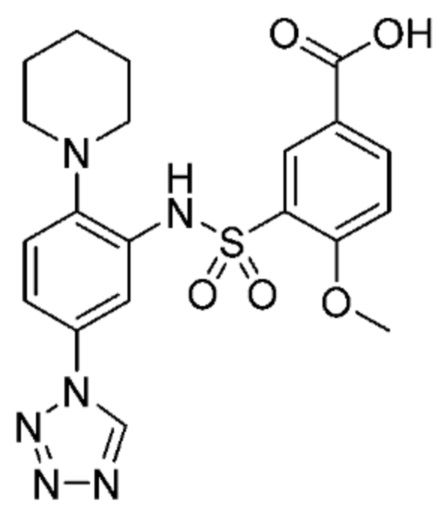
Стадия 1: 1-(4-фтор-3-нитрофенил)тетразол: Триметилсилил-азид (1,70 мл, 12,8 ммоль) добавляли к 4-фтор-3-нитроанилину (0,4 г, 2,56 ммоль) и триэтилортоформиату (2,13 мл, 12,8 ммоль) в уксусной кислоте (9,97 мл) при 0°C. Полученную смесь перемешивали в течение 30 мин, затем нагревали до 80°C в течение 1 ч и перемешивали в течение 20 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (0,469 г, 2,220 ммоль, выход 87%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z без ионизации, через 1,75 мин. 1H-ЯМР (500 МГц, CDCl3) δ 9,16 (с, 1H), 8,50 (дд, J=6,1, 2,8 Гц, 1H), 8,15-8,09 (м, 1H), 7,61 (кажущ. т, J=9,4 Гц, 1H).
Стадия 2: 1-(2-нитро-4-(1H-тетразол-1-ил)фенил)пиперидин: Et3N (0,781 мл, 5,61 ммоль) добавляли к раствору продукта из стадии 1 выше (0,469 г, 2,24 ммоль) и пиперидина (0,266 мл, 2,69 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 23 ч. Органическую фазу промывали 1М HCl (водн.) (3 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (0,609 г, 2,20 ммоль, выход 98%, чистота 99%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 1,39 мин 1H-ЯМР (500 МГц, CDCl3) δ 8,98 (с, 1H), 8,12 (д, J=2,7 Гц, 1H), 7,78 (дд, J=9,0, 2,7 Гц, 1H), 7,27 (д, J=9,0 Гц, 1H), 3,22-3,05 (м, 4H), 1,81-1,71 (м, 4H), 1,69-1,61 (м, 2H).
Стадия 3: 2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)анилин: Продукт из стадии 2 выше (0,609 г, 2,22 ммоль) растворяли в EtOH (48 мл) и гидрировали реакционную смесь в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Реакционную смесь выпаривали в вакууме и подвергали азеотропной перегонке с MeOH (12 мл) с получением указанного в заголовке соединения (0,531 г, 2,15 ммоль, выход 97%, чистота 99%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z без 1,18 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,91 (с, 1H), 7,14 (д, J=2,5 Гц, 1H), 7,05 (д, J=8,4 Гц, 1H), 6,98 (дд, J=8,3, 2,6 Гц, 1H), 5,21 (с, 2H), 2,91-2,69 (м, 4H), 1,75-1,62 (м, 4H), 1,60-1,47 (м, 2H).
Стадия 4: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)фенил)сульфамоил)бензоат: Пиридин (0,199 мл, 2,46 ммоль) добавляли к раствору продукта из стадии 3 выше (200 мг, 0,819 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (260 мг, 0,982 ммоль) в ДХМ (8 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,221 г, 0,468 ммоль, выход 57,1%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+, 471,3 (M-H)- через 1,52 мин.
Стадия 5: 4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (2,81 мл, 2,81 ммоль) добавляли к раствору продукта из стадии 4 выше (0,221 г, 0,468 ммоль) в ТГФ (11 мл) и MeOH (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,171 г, 0,369 ммоль, выход 79%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,3 (M+H)+, 457,2 (M-H)- через 1,40 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (с, 1H), 9,95 (с, 1H), 8,83 (с, 1H), 8,41 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,80 (д, J=2,5 Гц, 1H), 7,53 (дд, J=8,6, 2,5 Гц, 1H), 7,45 (д, J=8,6 Гц, 1H), 7,33 (д, J=8,8 Гц, 1H), 3,95 (с, 3H), 2,77-2,68 (м, 4H), 1,74-1,61 (м, 4H), 1,60-1,49 (м, 2H).
Пример 215: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота
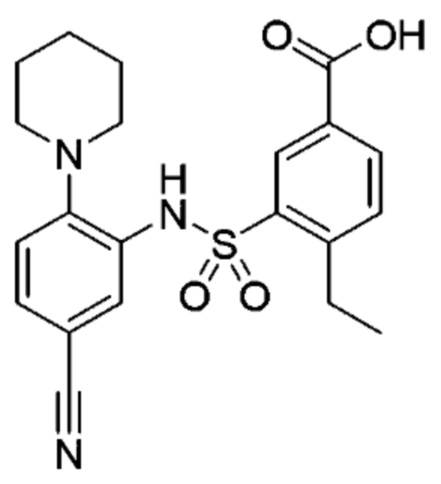
Стадия 1: метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензоат: Смесь продукта из Примера 182, стадии 2 (90 мг, 0,443 ммоль), продукта из Примера 203, стадии 2 (174 мг, 0,664 ммоль) и пиридина (150 мкл, 1,86 ммоль) в ДХМ (3 мл) перемешивали при 35°C в течение 2 дней. Смесь концентрировали на силикагеле, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-25% EtOAc/изогексан) с получением указанного в заголовке соединения (129 мг, 0,272 ммоль, выход 61,3%, чистота 90%) в виде бледно-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 428,2 (M+H)+, 426,2 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,62 (с, 1H), 8,30 (д, J=1,9 Гц, 1H), 8,12 (дд, J=8,0, 1,9 Гц, 1H), 7,65 (д, J=8,0 Гц, 1H), 7,56 (дд, J=8,4, 2,0 Гц, 1H), 7,26 (д, J=2,0 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 3,86 (с, 3H), 3,02 (к, J=7,4 Гц, 2H), 2,82-2,73 (м, 4H), 1,53-1,39 (м, 6H), 1,21 (т, J=7,4 Гц, 3H).
Стадия 2: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 1 выше (129 мг, 0,272 ммоль) и LiOH (26,0 мг, 1,09 ммоль) в ТГФ/MeOH/воде (4:1:1, 3,6 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и EtOAc (15 мл) и подкисляли до ~pH 4 1М HCl (водн.). Фазы разделяли и водную фазу экстрагировали EtOAc (2×15 мл). Органические экстракты объединяли, промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток растирали гексаном/МТБЭ (2:1) с получением указанного в заголовке соединения (50,3 мг, 0,121 ммоль, выход 44,4%, чистота 99%) в виде бежевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 414,2 (M+H)+, 412,0 (M-H)- через 1,65 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,58 (с, 1H), 8,30 (д, J=1,9 Гц, 1H), 8,10 (дд, J=8,0, 1,9 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,56 (дд, J=8,4, 2,0 Гц, 1H), 7,26 (д, J=2,0 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 3,01 (к, J=7,4 Гц, 2H), 2,84-2,69 (м, 4H), 1,54-1,39 (м, 6H), 1,21 (т, J=7,4 Гц, 3H).
Пример 216: 3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота
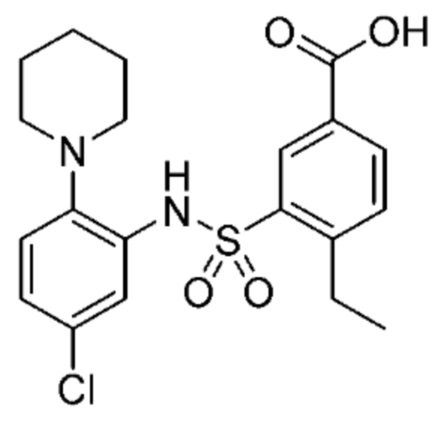
Стадия 1: метил-3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этилбензоат: Смесь 5-хлор-2-(пиперидин-1-ил)анилина (212 мг, 0,956 ммоль), продукта из Примера 203, стадии 2 (300 мг, 1,14 ммоль) и пиридина (0,34 мл, 4,20 ммоль) в ДХМ (7 мл) перемешивали при 35°C в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (326 мг, 0,671 ммоль, выход 70,2%, чистота 90%) как темно-фиолетового масла. СВЭЖХ-МС (Метод 1) m/z 437,2 (M+H)+, 435,2 (M-H)- через 2,01 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,29 (с, 1H), 8,34 (д, J=1,9 Гц, 1H), 8,11 (дд, J=8,0, 1,9 Гц, 1H), 7,64 (д, J=8,0 Гц, 1H), 7,19-7,10 (м, 3H), 3,86 (с, 3H), 3,06 (к, J=7,4 Гц, 2H), 2,54 (т, J=5,3 Гц, 4H), 1,59-1,48 (м, 4H), 1,47-1,37 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Стадия 2: 3-(N-(5-хлор-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 1 выше (326 мг, 0,671 ммоль) и 2М LiOH (водн.) (0,336 мл, 0,671 ммоль) в ТГФ/MeOH/воде (4:1:1, 8,4 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (10 мл) и EtOAc (25 мл) и подкисляли до ~pH 4 1М HCl. Фазы разделяли и водную фазу экстрагировали EtOAc (2×25 мл). Объединенные органические экстракты промывали солевым раствором (15 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (89,7 мг, 0,207 ммоль, выход 30,8%, чистота 98%) в виде светло-серого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 423,3 (M+H)+, 421,2 (M-H)- через 1,86 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (с, 1H), 9,23 (с, 1H), 8,34 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,19-7,09 (м, 3H), 3,05 (к, J=7,4 Гц, 2H), 2,55 (т, J=5,3 Гц, 4H), 1,60-1,50 (м, 4H), 1,48-1,38 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 217: 3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)-фенил)сульфамоил)-4-этилбензойная кислота
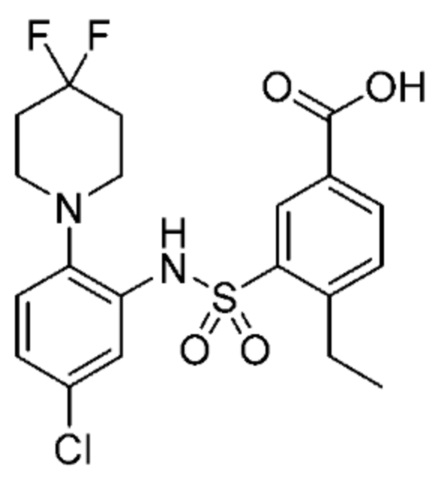
Стадия 1: 1-(4-хлор-2-нитрофенил)-4,4-дифторпиперидин: Et3N (0,992 мл, 7,12 ммоль) добавляли к раствору 4-хлор-1-фтор-2-нитробензола (500 мг, 2,85 ммоль) и 4,4-дифторпиперидина (414 мг, 3,42 ммоль) в ДХМ (6 мл) при КТ. Прозрачный раствор перемешивали при КТ в течение 23 ч. Органическую фазу промывали 1М HCl (водн.) (3 мл), отделяли при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (803 мг, 2,76 ммоль, выход 97%, чистота 95%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 277,2 (M+H)+ через 1,67 мин.
Стадия 2: 5-хлор-2-(4,4-дифторпиперидин-1-ил)анилин: Порошок железа (1,62 г, 28,9 ммоль) добавляли к суспензии продукта из стадии 1 выше (400 мг, 1,45 ммоль) и хлорида аммония (93 мг, 1,74 ммоль) в ИПС (10 мл) и воде (5 мл) при КТ. Полученную суспензию нагревали и перемешивали при 90°C в течение 2 ч. Реакцию фильтровали через Celite®, промывали MeOH (100 мл) и выпаривали в вакууме. Остаток растворяли в ДХМ (25 мл) и промывали водой (10 мл) и солевым раствором (10 мл), сушили (MgSO4) и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,152 г, 0,592 ммоль, выход 40,9%, чистота 96%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 247,3 (M+H)+ через 1,58 мин.
Стадия 3: метил-3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Пиридин (0,074 мл, 0,912 ммоль) добавляли к раствору продукта из стадии 2 выше (0,075 г, 0,304 ммоль) и продукта из Примера 203, стадии 2 (0.100 г, 0,380 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,096 г, 0,189 ммоль, выход 62,1%, чистота 93%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,0 (M+H)+, 470,9 (M-H)- через 1,89 мин.
Стадия 4: 3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (1,13 мл, 1,13 ммоль) добавляли к раствору продукта из стадии 3 выше (0,089 г, 0,189 ммоль) в ТГФ (4,5 мл) и MeOH (1,1 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,073 г, 0,156 ммоль, выход 82%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,6 (M+H)+, 456,9 (M-H)- через 1,76 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,34 (с, 1H), 9,70 (с, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,26-7,19 (м, 2H), 7,14 (дд, J=8,5, 2,5 Гц, 1H), 3,04 (к, J=7,4 Гц, 2H), 2,69 (т, J=5,6 Гц, 4H), 2,10-1,97 (м, 4H), 1,19 (т, J=7,4 Гц, 3H).
Пример 218: 3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)-фенил)сульфамоил)-4-метоксибензойная кислота
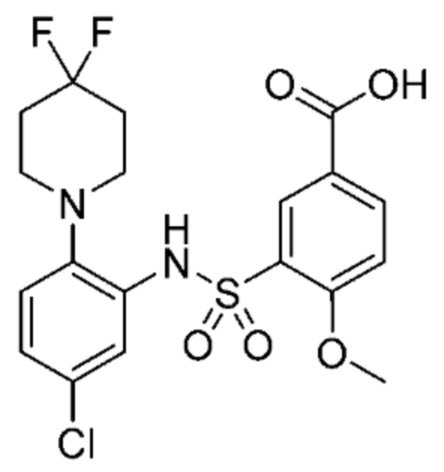
Стадия 1: Метил-3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,074 мл, 0,912 ммоль) добавляли к раствору продукта из Примера 217, стадии 2 (0,075 г, 0,304 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (0,101 г, 0,380 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,113 г, 0,193 ммоль, выход 63,4%, чистота 81%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,4 (M+H)+, 472,8 (M-H)- через 1,75 мин.
Стадия 2: 3-(N-(5-хлор-2-(4,4-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (1,16 мл, 1,16 ммоль) добавляли к раствору продукта из стадии 1 выше (0,092 г, 0,193 ммоль) в ТГФ (4,5 мл) и MeOH (1,1 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,078 г, 0,166 ммоль, выход 86%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 461,0 (M+H)+, 459,0 (M-H)- через 1,59 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (с, 1H), 9,13 (с, 1H), 8,37 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,33 (д, J=8,7 Гц, 1H), 7,29-7,23 (м, 2H), 7,07 (дд, J=8,5, 2,5 Гц, 1H), 3,90 (с, 3H), 2,78 (т, J=5,6 Гц, 4H), 2,18-2,06 (м, 4H).
Пример 219: 3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)-фенил)сульфамоил)-4-метоксибензойная кислота
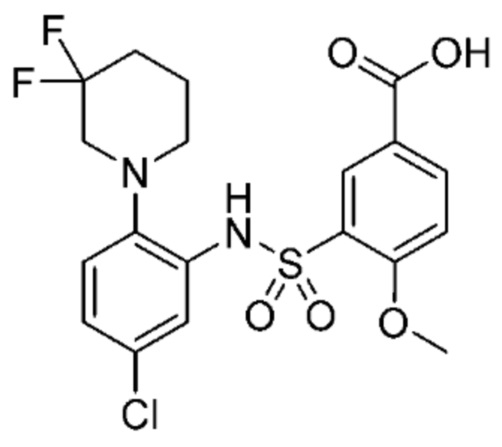
Стадия 1: 1-(4-хлор-2-нитрофенил)-3,3-дифторпиперидин: Et3N (1,79 мл, 12,8 ммоль) добавляли к раствору 4-хлор-1-фтор-2-нитробензола (0,335 мл, 2,85 ммоль) и гидрохлорида 3,3-дифторпиперидина (539 мг, 3,42 ммоль) в ДХМ (6 мл) при КТ. Смесь перемешивали при КТ в течение 23 ч. Растворитель удаляли в вакууме, а остаток растворяли в ТГФ (6 мл) и нагревали до 50°C в течение 18 ч. Добавляли ДМФА (6 мл) и нагревали смесь до 90°C в течение 18 ч. Растворители удаляли в вакууме и добавляли ДХМ (6 мл) и 1М HCl (водн.) (3 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (0,490 г, 1,68 ммоль, выход 59,1%, чистота 95%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 277,2 (M+H)+ через 1,63 мин.
Стадия 2: 5-хлор-2-(3,3-дифторпиперидин-1-ил)анилин: Порошок железа (1,62 г, 28,9 ммоль) добавляли к суспензии продукта из стадии 1 выше (0,490 г, 1,77 ммоль) и хлорида аммония (0,093 г, 1,74 ммоль) в ИПС (10 мл) и воде (5 мл) при КТ. Полученную суспензию нагревали и перемешивали при 90°C в течение 2 ч. Реакционную смесь фильтровали через Celite®, промывали MeOH (100 мл) и выпаривали в вакууме. Остаток растворяли в ДХМ (25 мл) и последовательно промывали водой (10 мл) и солевым раствором (10 мл), сушили (MgSO4) и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,252 г, 1,02 ммоль, выход 70,6%, чистота 100%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 247,2 (M+H)+ через 1,59 мин.
Стадия 3: метил-3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Пиридин (0,123 мл, 1,52 ммоль) добавляли к раствору продукта из стадии 2 выше (0,125 г, 0,507 ммоль) и метил-3-(хлорсульфонил)-4-метоксибензоата (0,168 г, 0,633 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,241 г, 0,502 ммоль, выход 99%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 472,8 (M-H)- через 1,73 мин.
Стадия 4: 3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: 1М LiOH (водн.) (3,04 мл, 3,04 ммоль) добавляли к раствору продукта из стадии 3 выше (0,241 г, 0,507 ммоль) в ТГФ (12 мл) и MeOH (3 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,211 г, 0,458 ммоль, выход 90%, чистота 100%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 461,3 (M+H)+, 459,1 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,25 (с, 1H), 8,46 (с, 1H), 8,40 (д, J=2,2 Гц, 1H), 8,19 (дд, J=8,7, 2,2 Гц, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,31 (д, J=8,6 Гц, 1H), 7,22 (д, J=2,4 Гц, 1H), 7,09 (дд, J=8,5, 2,4 Гц, 1H), 3,94 (с, 3H), 3,04 (т, J=11,1 Гц, 2H), 2,85-2,76 (м, 2H), 2,11-1,98 (м, 2H), 1,90-1,79 (м, 2H).
Пример 220: 3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)-фенил)сульфамоил)-4-этилбензойная кислота
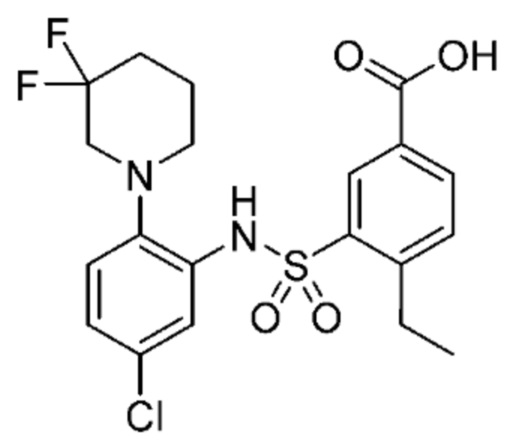
Стадия 1: метил-3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Пиридин (0,123 мл, 1,52 ммоль) добавляли к раствору продукта из Примера 219, стадии 2 (0,125 г, 0,507 ммоль) и продукта из Примера 203, стадии 2 (0,166 г, 0,633 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,166 г, 0,351 ммоль, выход 69,3%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,2 (M-H)- через 1,89 мин.
Стадия 2: 3-(N-(5-хлор-2-(3,3-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (2,11 мл, 2,11 ммоль) добавляли к раствору продукта из стадии 1 выше (0,166 г, 0,351 ммоль) в ТГФ (8,5 мл) и MeOH (2,1 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,143 г, 0,308 ммоль, выход 88%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,1 (M+H)+, 457,0 (M-H)- через 1,75 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,36 (с, 1H), 8,98 (с, 1H), 8,37 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,26 (д, J=8,6 Гц, 1H), 7,15 (дд, J=8,6, 2,5 Гц, 1H), 7,07 (д, J=2,4 Гц, 1H), 3,06-2,96 (м, 4H), 2,81-2,72 (м, 2H), 2,06-1,93 (м, 2H), 1,80-1,71 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 221: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
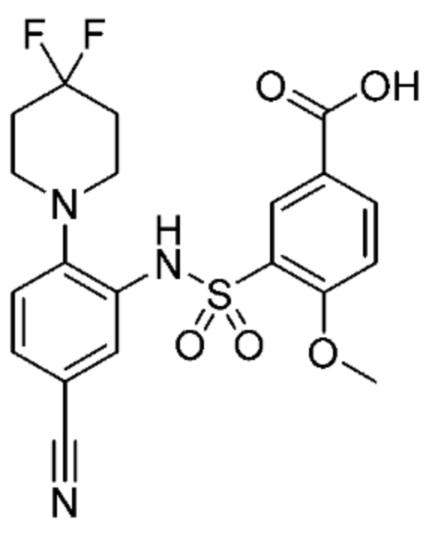
Стадия 1: 4-(4,4-дифторпиперидин-1-ил)-3-нитробензонитрил: Смесь 4-фтор-3-нитробензонитрила (500 мг, 3,01 ммоль), 4,4-дифторпиперидина (400 мг, 3,30 ммоль) и Et3N (0,65 мл, 4,66 ммоль) в ДМФА (5 мл) перемешивали при 90°C в течение ночи. Смесь разбавляли водой (20 мл) и экстрагировали EtOAc (3×35 мл). Объединенные органические экстракты промывали солевым раствором (2×30 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-10% EtOAc/изогексан) с получением указанного в заголовке соединения (541 мг, 2,02 ммоль, выход 67,3%, чистота 100%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 1,39 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,37 (д, J=2,1 Гц, 1H), 7,94 (дд, J=8,7, 2,1 Гц, 1H), 7,46 (д, J=8,7 Гц, 1H), 3,31-3,25 (м, 4H), 2,15-2,04 (м, 4H).
Стадия 2: 3-амино-4-(4,4-дифторпиперидин-1-ил)бензонитрил: Смесь продукта из стадии 1 выше (541 мг, 2,02 ммоль), порошка железа (2,5 г, 44,8 ммоль), хлорида аммония (130 мг, 2,43 ммоль), ИПС (16 мл) и воды (8 мл) нагревали при 90°C в течение ночи. Смесь фильтровали через Celite®, промывали MeOH и удаляли растворитель в вакууме. Остаток разбавляли ДХМ (20 мл), сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 100% ДХМ) с получением указанного в заголовке соединения (260 мг, 1,10 ммоль, выход 54,1%, чистота 100%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 238,2 (M+H)+, 236,0 (M-H)-, через 1,34 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,04-6,90 (м, 3H), 5,29 (с, 2H), 2,94 (т, J=5,5 Гц, 4H), 2,24-2,08 (м, 4H).
Стадия 3: метил-3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Смесь продукта из стадии 2 выше (130 мг, 0,548 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (0,160 г, 0,603 ммоль), пиридина (140 мкл, 1,73 ммоль) и ДХМ (3,5 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (148 мг, 0,312 ммоль, выход 56,9%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 466,3 (M+H)+, 464,1 (M-H)-, через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,32 (д, J=2,3 Гц, 1H), 8,21 (дд, J=8,7, 2,3 Гц, 1H), 7,54 (дд, J=8,3, 1,9 Гц, 1H), 7,42-7,35 (м, 2H), 7,28 (д, J=8,3 Гц, 1H), 3,90 (с, 3H), 3,85 (с, 3H), 2,98 (т, J=5,6 Гц, 4H), 2,13-2,01 (м, 4H).
Стадия 4: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: Смесь продукта из стадии 3 выше (0,148 г, 0,312 ммоль) и LiOH (30 мг, 1,253 ммоль) в ТГФ/MeOH/воде (4:1:1, 3,6 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл) и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (85,6 мг, 0,185 ммоль, выход 59,3%, чистота 97%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 452,2 (M+H)+, 450,2 (M-H)-, через 1,40 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,17 (с, 1H), 9,40 (с, 1H), 8,32 (д, J=2,3 Гц, 1H), 8,18 (дд, J=8,8, 2,3 Гц, 1H), 7,53 (дд, J=8,3, 2,0 Гц, 1H), 7,41 (д, J=2,0 Гц, 1H), 7,35 (д, J=8,8 Гц, 1H), 7,28 (д, J=8,3 Гц, 1H), 3,89 (с, 3H), 3,04-2,92 (м, 4H), 2,15-2,01 (м, 4H).
Пример 222: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)-фенил)сульфамоил)-4-этилбензойная кислота
Стадия 1: метил-3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Смесь продукта из Примера 221, стадии 2 (130 мг, 0,548 ммоль), продукта из Примера 203, стадии 2 (0,244 г, 0,603 ммоль), пиридина (140 мкл, 1,73 ммоль) и ДХМ (3,5 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (102 мг, 0,211 ммоль, выход 38,6%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 464,3 (M+H)+, 462,1 (M-H)- через 1,68 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,91 (с, 1H), 8,33 (д, J=1,9 Гц, 1H), 8,13 (дд, J=8,0, 1,9 Гц, 1H), 7,66 (д, J=8,0 Гц, 1H), 7,59 (дд, J=8,4, 1,9 Гц, 1H), 7,33 (д, J=1,9 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 3,86 (с, 3H), 3,01 (к, J=7,4 Гц, 2H), 2,91 (т, J=5,6 Гц, 4H), 2,05-1,94 (м, 4H), 1,20 (т, J=7,4 Гц, 3H).
Стадия 2: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 3 выше (0,102 г, 0,211 ммоль) и LiOH (30 мг, 1,253 ммоль) в ТГФ/MeOH/воде (4:1:1, 3,6 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl. Смесь экстрагировали EtOAc (3×20 мл) и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (32,6 мг, 0,070 ммоль, выход 33,2%, чистота 97%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 450,2 (M+H)+, 448,2 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (с, 1H), 9,88 (с, 1H), 8,33 (д, J=1,9 Гц, 1H), 8,11 (дд, J=8,1, 1,9 Гц, 1H), 7,63 (д, J=8,1 Гц, 1H), 7,58 (дд, J=8,4, 2,0 Гц, 1H), 7,33 (д, J=2,0 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 3,01 (к, J=7,4 Гц, 2H), 2,94-2,88 (м, 4H), 2,06-1,97 (м, 4H), 1,20 (т, J=7,3 Гц, 3H).
Пример 223: 3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
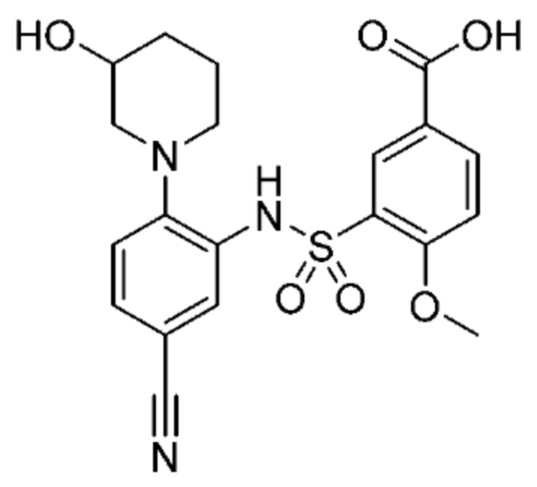
Стадия 1: 4-(3-гидроксипиперидин-1-ил)-3-нитробензонитрил: Смесь 4-фтор-3-нитробензонитрила (500 мг, 3,01 ммоль), пиперидин-3-ола (350 мг, 3,46 ммоль) и Et3N (0,65 мл, 4,66 ммоль) в ДХМ (10 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (698 мг, 2,63 ммоль, выход 87%, чистота 93%) в виде густого оранжевого масла. СВЭЖХ-МС (Метод 2) m/z 248,2 (M+H)+, 246,4 (M-H)- через 1,02 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,28 (д, J=2,1 Гц, 1H), 7,83 (дд, J=8,9, 2,1 Гц, 1H), 7,35 (д, J=8,9 Гц, 1H), 4,92 (д, J=4,2 Гц, 1H), 3,67-3,57 (м, 1H), 3,29-3,21 (м, 2H), 3,01 (ддд, J=12,8, 9,6, 3,1 Гц, 1H), 2,81 (дд, J=12,8, 8,2 Гц, 1H), 1,91-1,84 (м, 1H), 1,82-1,73 (м, 1H), 1,54-1,44 (м, 1H), 1,43-1,33 (м, 1H).
Стадия 2: 3-амино-4-(3-гидроксипиперидин-1-ил)бензонитрил: Смесь продукта из стадии 1 выше (698 мг, 2,65 ммоль), порошка железа (3 г, 53,7 ммоль), хлорида аммония (170 мг, 3,18 ммоль), ИПС (20 мл) и вода (10 мл) нагревали до 90°C в течение ночи. Смесь фильтровали через Celite®, промывали MeOH и выпаривали фильтрат в вакууме. Остаток разбавляли ДХМ (20 мл), сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (381 мг, 1,72 ммоль, выход 64,8%, чистота 98%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 218,2 (M+H)+, 216,2 (M-H)- через 0,94 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,98-6,90 (м, 3H), 5,15 (с, 2H), 4,80 (д, J=5,4 Гц, 1H), 3,75-3,66 (м, 1H), 3,06-2,97 (м, 1H), 2,96-2,87 (м, 1H), 2,56 (т, J=10,3 Гц, 1H), 2,49-2,42 (м, 1H), 1,89-1,74 (м, 2H), 1,67-1,54 (м, 1H), 1,41-1,27 (м, 1H).
Стадия 3: метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Смесь продукта из стадии 2 выше (100 мг, 0,451 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (131 мг, 0,496 ммоль), пиридина (110 мкл, 1,36 ммоль) и ДХМ (3 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (111 мг, 0,219 ммоль, выход 48,6%, чистота 88%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 446,3 (M+H)+, 444,1 (M-H)- через 1,31 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,28 (с, 1H), 8,36 (д, J=2,3 Гц, 1H), 8,23-8,17 (м, 1H), 7,49-7,43 (м, 1H), 7,43-7,39 (м, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 5,09 (д, J=5,2 Гц, 1H), 3,90 (с, 3H), 3,86 (с, 3H), 3,75-3,67 (м, 1H), 2,96-2,86 (м, 2H), 2,78-2,70 (м, 1H), 2,70-2,61 (м, 1H), 1,90-1,80 (м, 1H), 1,76-1,66 (м, 1H), 1,57-1,40 (м, 2H).
Стадия 4: 3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: Смесь продукта из стадии 3 выше (111 мг, 0,219 ммоль) и LiOH (21,0 мг, 0,877 ммоль) в ТГФ/MeOH/воде (4:1:1, 3 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, MeOH/ДХМ на 0-5%) с получением указанного в заголовке соединения (69,3 мг, 0,157 ммоль, выход 71,8%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 432,2 (M+H)+, 430,2 (M-H)- через 1,14 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (с, 1H), 9,24 (с, 1H), 8,36 (д, J=2,3 Гц, 1H), 8,16 (дд, J=8,7, 2,3 Гц, 1H), 7,45 (дд, J=8,3, 2,0 Гц, 1H), 7,41 (д, J=2,0 Гц, 1H), 7,32 (д, J=8,7 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 5,10 (шс, 1H), 3,89 (с, 3H), 3,72 (шс, 1H), 2,96-2,84 (м, 2H), 2,78-2,71 (м, 1H), 2,67 (дд, J=11,6, 6,3 Гц, 1H), 1,91-1,80 (м, 1H), 1,75-1,67 (м, 1H), 1,56-1,44 (м, 2H).
Пример 224: 3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)-фенил)сульфамоил)-4-этилбензойная кислота
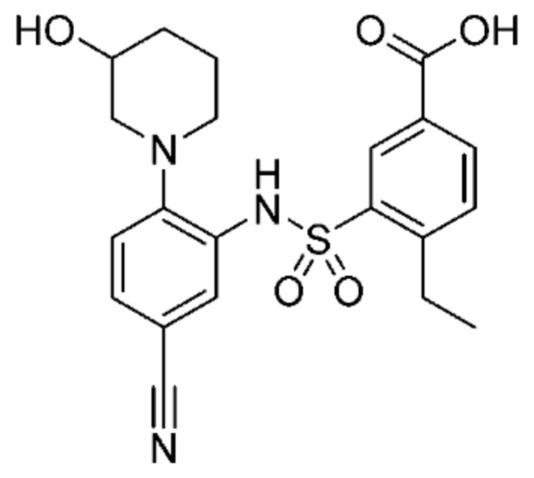
Стадия 1: метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Смесь продукта из Примера 223, стадии 2 (100 мг, 0,451 ммоль), продукта из Примера 203, стадии 2 (201 мг, 0,496 ммоль), пиридина (110 мкл, 1,36 ммоль) и ДХМ (3 мл) перемешивали при КТ в течение ночи. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (108 мг, 0,219 ммоль, выход 48,6%, чистота 90%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 444,2 (M+H)+, 442,1 (M-H)- через 1,49 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,79 (с, 1H), 8,35 (д, J=1,9 Гц, 1H), 8,15-8,09 (м, 1H), 7,64 (д, J=8,2 Гц, 1H), 7,50 (д, J=8,4 Гц, 1H), 7,32 (д, J=1,9 Гц, 1H), 7,15 (д, J=8,2 Гц, 1H), 5,15 (д, J=5,5 Гц, 1H), 3,87 (с, 3H), 3,71-3,66 (м, 1H), 3,11-3,00 (м, 2H), 2,98-2,93 (м, 1H), 2,93-2,87 (м, 1H), 2,67-2,59 (м, 2H), 1,84-1,74 (м, 1H), 1,71-1,62 (м, 1H), 1,52-1,40 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Стадия 2: 3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 2 выше (108 мг, 0,219 ммоль) и LiOH (21,0 мг, 0,877 ммоль) в ТГФ/MeOH/воде (4:1:1, 3 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (75 мг, 0,169 ммоль, выход 77%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 430,3 (M+H)+, 428,2 (M-H)- через 1,33 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (с, 1H), 9,76 (с, 1H), 8,35 (д, J=1,9 Гц, 1H), 8,10 (дд, J=8,0, 1,9 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,48 (дд, J=8,4, 2,0 Гц, 1H), 7,31 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 5,16 (шс, 1H), 3,70 (шс, 1H), 3,13-2,98 (м, 2H), 2,98-2,93 (м, 1H), 2,93-2,85 (м, 1H), 2,70-2,61 (м, 2H), 1,85-1,75 (м, 1H), 1,72-1,62 (м, 1H), 1,52-1,41 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 225: 4-(метилсульфонил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
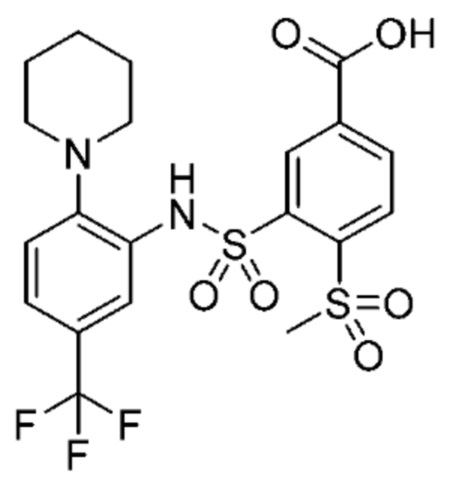
Стадия 1: метил-4-фтор-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Пиридин (0,199 мл, 2,46 ммоль) добавляли к раствору 2-(пиперидин-1-ил)-5-(трифторметил)анилина (200 мг, 0,819 ммоль) и метил-3-(хлорсульфонил)-4-фторбензоата (259 мг, 1,02 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,183 г, 0,389 ммоль, выход 47,6%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 461,3 (M+H)+, 459,2 (M-H)- через 1,91 мин.
Стадия 2: метил-4-(метилтио)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Тиометилат натрия (0,084 г, 1,19 ммоль) добавляли к раствору продукта из стадии 1 выше (0,183 г, 0,397 ммоль) в ДМФА (4 мл) и перемешивали при КТ в течение 18 ч. Смесь делили между ДХМ (10 мл) и водой (10 мл) и отделяли органическую фазу. Водную фазу экстрагировали ДХМ (2×10 мл) и объединенные органические фазы сушили при пропускании через фазовый сепаратор. Растворитель удаляли в вакууме с получением указанного в заголовке соединения (0,095 г, 0,193 ммоль, выход 48,4%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 486,8 (M-H)- через 1,95 мин.
Стадия 3: метил-4-(метилсульфонил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат и метил-4-(метилсульфинил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензоат: 3-Хлорпероксибензойную кислоту (0,044 г, 0,194 ммоль, 77% об/об) добавляли к продукту из стадии 2 выше (0,095 г, 0,194 ммоль) в ДХМ (4 мл) и перемешивали при КТ в течение 72 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением метил-4-(метилсульфонил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензоата (0,060 г, 0,112 ммоль, выход 57,5%, чистота 97%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 521,3 (M+H)+, 518,9 (M-H)- через 1,28 мин.
Метил-4-(метилсульфинил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат (0,032 г, 0,058 ммоль, выход на 30,0%, чистота 92%) также выделяли в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 505,3 (M+H)+, 503,1 (M-H)- через 1,69 мин.
Стадия 4: 4-(метилсульфонил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,692 мл, 0,692 ммоль) добавляли к раствору метил-4-(метилсульфонил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)бензоата из стадии 3 выше (0,06 г, 0,115 ммоль) в ТГФ (3 мл) и MeOH (0,7 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли конц. HCl и экстрагировали МТБЭ (3×10 мл). Органические фазы объединяли и сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (4 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (0,049 г, 0,096 ммоль, выход 83%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 507,3 (M+H)+, 504,8 (M-H)- через 1,13 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 15,98 (с, 1H), 13,59 (с, 1H), 8,43 (д, J=1,7 Гц, 1H), 8,30 (дд, J=8,1, 1,7 Гц, 1H), 8,24 (д, J=8,1 Гц, 1H), 7,87 (д, J=8,7 Гц, 1H), 7,76 (д, J=2,0 Гц, 1H), 7,23 (дд, J=8,9, 2,1 Гц, 1H), 4,37-4,25 (м, 2H), 3,92 (д, J=11,8 Гц, 1H), 3,72 (д, J=11,8 Гц, 1H), 2,85 (с, 3H), 2,28-2,15 (м, 2H), 1,87-1,72 (м, 3H), 1,59-1,44 (м, 1H).
Пример 227: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(2,2,2-трифторэтокси)бензойная кислота
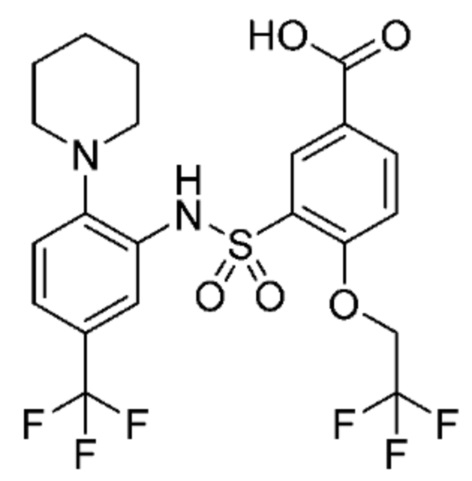
Стадия 1: 3-(хлорсульфонил)-4-(2,2,2-трифторэтокси)-бензойная кислота: 4-(2,2,2-трифторэтокси)бензойную кислоту (1 г, 4,54 ммоль) в хлоросульфоновой кислоте (5 мл, 74,7 ммоль) нагревали при 80°C в течение 2 ч. Смесь охлаждали и осторожно добавляли в перемешиваемую воду со льдом (100 мл). Образовавшийся осадок собирали с помощью фильтрации, промывали водой (100 мл) и сушили в вакууме с получением указанного в заголовке соединения (1,20 г, 3,58 ммоль, выход 79%, чистота 95%) в виде кремового твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,34 (д, J=2,3 Гц, 1H), 7,89 (дд, J=8,5, 2,4 Гц, 1H), 7,16 (д, J=8,5 Гц, 1H), 4,82 (к, J=8,9 Гц, 2H). Один обмениваемый протон не наблюдали.
Стадия 2: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-сульфамоил)-4-(2,2,2-трифторэтокси)бензойная кислота (2393-12): Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0.100 г, 0,409 ммоль) в ДХМ (5 мл) и пиридине (0,199 мл, 2,46 ммоль) добавляли к раствору продукта из стадии 1 выше (0,130 г, 0,409 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 24 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (42,8 мг, 0,077 ммоль, выход 18,9%, чистота 95%) в виде кремового воскообразного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 527,4 (M+H)+, 525,1 (M-H)- через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,36 (шс, 1H), 8,50 (шс, 1H), 8,44 (д, J=2,2 Гц, 1H), 8,22 (дд, J=8,7, 2,2 Гц, 1H), 7,47 (д, J=8,8 Гц, 1H), 7,40-7,35 (м, 2H), 7,31 (д, J=8,6 Гц, 1H), 5,02 (к, J=8,6 Гц, 2H), 2,77 (т, J=5,2 Гц, 4H), 1,61 (п, J=5,7 Гц, 4H), 1,52 (п, J=6,2 Гц, 2H).
Пример 228: 4-(2-гидроксипропан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
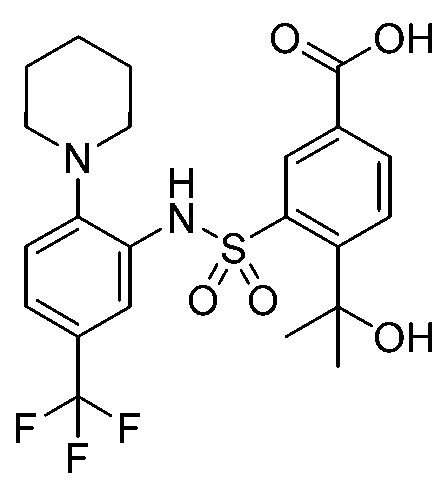
Стадия 1: метил-4-бром-2-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор 2-(пиперидин-1-ил)-5-(трифторметил)анилина (0,200 г, 0,819 ммоль) в ДХМ (1 мл) и пиридине (0,397 мл, 4,91 ммоль) добавляли к раствору метил-4-бром-2-(хлорсульфонил)бензоата (0,257 г, 0,819 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 24 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,33 г, 0,601 ммоль, выход 73,4%, чистота 95%) в виде кремового воскообразного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 521,2 (M+H)+, 518,7 (M-H)- через 2,07 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,06 (д, J=2,0 Гц, 1H), 8,01 (дд, J=8,2, 2,0 Гц, 1H), 7,74 (д, J=8,2 Гц, 1H), 7,54 (д, J=2,1 Гц, 1H), 7,50-7,45 (м, 1H), 7,33 (д, J=8,4 Гц, 1H), 3,81 (с, 3H), 2,69 (т, J=5,2 Гц, 4H), 1,56 (п, J=5,5 Гц, 4H), 1,51-1,42 (м, 2H).
Стадия 2: 5-бром-2-(2-гидроксипропан-2-ил)-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)бензолсульфонамид: Раствор продукта из стадии 1 выше (0,150 г, 0,288 ммоль) в сухом ТГФ (10 мл) обрабатывали 3,0 М метилмагнийбромида в Et2O (0,384 мл, 1,15 ммоль) и перемешивали раствор при КТ в течение 16 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (91 мг, 0,150 ммоль, выход 52,2%, чистота 87%) кав виде бесцветного воскообразного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 521,2 (M+H)+, 519,1 (M-H)- через 2,11 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,76 (шс, 1H), 8,15 (д, J=2,2 Гц, 1H), 7,79 (дд, J=8,5, 2,2 Гц, 1H), 7,74 (д, J=2,0 Гц, 1H), 7,53 (д, J=8,6 Гц, 1H), 7,38-7,23 (м, 2H), 6,15 (с, 1H), 2,71 (т, J=5,3 Гц, 4H), 1,67 (п, J=5,3 Гц, 4H), 1,58 (с, 6H), 1,54-1,49 (м, 2H).
Стадия 3: метил-4-(2-гидроксипропан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 2 выше (0,091 г, 0,175 ммоль), Et3N (0,049 мл, 0,349 ммоль) и PdCl2(dppf)∙ДХМ (0,029 г, 0,035 ммоль) в MeOH (10 мл) перемешивали под атмосферой CO (4 бара) в течение ночи при 100°C. Через 24 ч реакцию охлаждали, фильтровали через Celite® и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,090 г, 0,171 ммоль, выход 98%, чистота 95%) в виде голубого масла. СВЭЖХ-МС (Метод 1) m/z 501,4 (M+H)+, 499,3 (M-H)- через 1,97 мин.
Стадия 4: 4-(2-гидроксипропан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,013 г, 0,539 ммоль) добавляли к раствору продукта из стадии 3 выше (0,090 г, 0,180 ммоль) в ТГФ (5 мл) и перемешивали раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме и полученную водную фазу доводили до pH 6 1M HCl. Осадок отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл) с получением указанного в заголовке соединения (54,3 мг, 0,106 ммоль, выход 59,0%, чистота 95%) в виде светло-серого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,3 (M-H)- через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,38 (шс, 1H), 9,74 (шс, 1H), 8,66 (д, J=1,9 Гц, 1H), 8,06 (дд, J=8,3, 1,9 Гц, 1H), 7,78 (с, 1H), 7,71 (д, J=8,3 Гц, 1H), 7,33-7,28 (м, 2H), 6,20 (шс, 1H), 2,70 (т, J=5,3 Гц, 4H), 1,67 (п, J=5,2 Гц, 4H), 1,62 (с, 6H), 1,55-1,48 (м, 2H).
Пример 229: 4-(гидроксиметил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
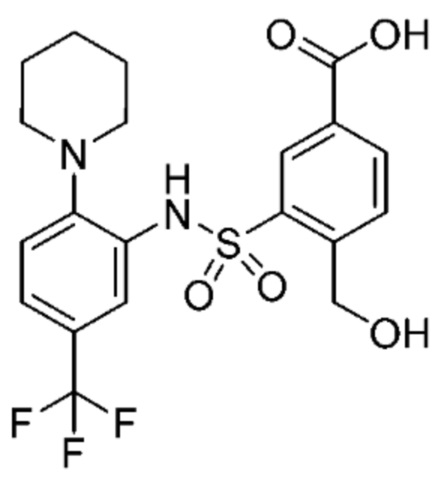
Стадия 1: 5-бром-2-(гидроксиметил)-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)бензолсульфонамид: Раствор продукта из Примера 228, стадии 1 (0,285 г, 0,547 ммоль) в ТГФ (5 мл) охлаждали до 0°C, затем обрабатывали 2,0М LiBH4 в ТГФ (0,273 мл, 0,547 ммоль). Смесь перемешивали при КТ в течение 16 ч, затем смесь разбавляли водой (100 мл), экстрагировали EtOAc (100 мл), сушили (MgSO4) и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,212 г, 0,408 ммоль, выход 74,7%, чистота 95%) в виде бесцветного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 493,2 (M+H)+, 491,1 (M-H)- через 1,86 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,53 (шс, 1H), 7,87 (дд, J=8,3, 2,1 Гц, 1H), 7,83 (д, J=2,1 Гц, 1H), 7,71 (д, J=8,3 Гц, 1H), 7,45 (дд, J=8,5, 2,2 Гц, 1H), 7,38 (д, J=2,2 Гц, 1H), 7,26 (д, J=8,4 Гц, 1H), 5,67 (шс, 1H), 4,82 (с, 2H), 2,71 (т, J=5,3 Гц, 4H), 1,58-1,54 (м, 4H), 1,48-1,45 (м, 2H).
Стадия 2: метил-4-(гидроксиметил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 1 выше (0,210 г, 0,426 ммоль), Et3N (0,119 мл, 0,851 ммоль) и PdCl2(dppf)∙ДХМ (0,070 г, 0,085 ммоль) в MeOH (10 мл) перемешивали под атмосферой CO (4 бара) в течение ночи при 100°C. Через 24 ч реакцию охлаждали, фильтровали через Celite® и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,180 г, 0,376 ммоль, выход 88%, чистота 99%) в виде кремового воскообразного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+, 471,3 (M-H)- через 1,73 мин.
Стадия 3: 4-(гидроксиметил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,027 г, 1,14 ммоль) добавляли к раствору продукта из стадии 2 выше (0,180 г, 0,381 ммоль) в ТГФ (5 мл) и перемешивали раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме и экстрагировали полученную водную фазу EtOAc (50 мл). Затем водную фазу доводили до pH 6 1М HCl (водн.) с образованием осадка, который отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл) с получением указанного в заголовке соединения (134 мг, 0,277 ммоль, выход 72,8%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,3 (M+H)+, 457,3 (M-H)- через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (шс, 1H), 9,49 (шс, 1H), 8,30 (д, J=1,8 Гц, 1H), 8,17 (дд, J=8,0, 1,8 Гц, 1H), 7,90 (д, J=8,1 Гц, 1H), 7,42 (дд, J=8,4, 2,2 Гц, 1H), 7,37 (д, J=2,2 Гц, 1H), 7,24 (д, J=8,4 Гц, 1H), 5,74-5,30 (м, 1H), 4,94 (с, 2H), 2,69 (т, J=5,2 Гц, 4H), 1,55 (п, J=5,4 Гц, 4H), 1,46 (п, J=6,0 Гц, 2H).
Пример 230: 4-этил-3-(N-(2-(пиперидин-1-ил)-4-(трифтор-метил)фенил)сульфамоил)бензойная кислота
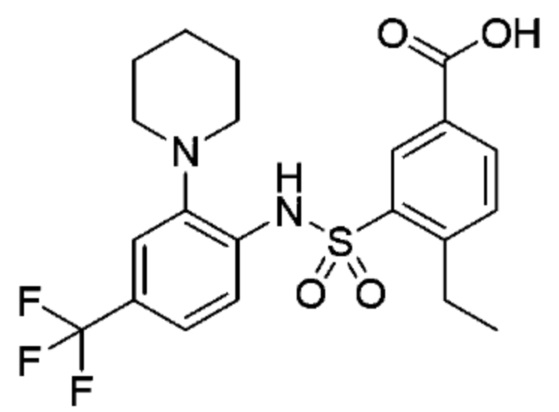
Стадия 1: 1-(2-нитро-5-(трифторметил)фенил)пиперидин: 2-фтор-1-нитро-4-(трифторметил)бензол (1,1 г, 5,26 ммоль) растворяли в ДМСО (10 мл) и обрабатывали K2CO3 (0,872 г, 6,31 ммоль), затем пиперидином (0,779 мл, 7,89 ммоль) и нагревали смесь при 100°C в течение 2 ч. Реакционную смесь добавляли в воду со льдом (100 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали водой (100 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (1,32 г, 4,81 ммоль, выход 91%, чистота 95%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 275,2 (M+H)+ через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,97 (д, J=8,4 Гц, 1H), 7,53 (д, J=1,8 Гц, 1H), 7,36 (дд, J=8,5, 1,8 Гц, 1H), 3,05-3,03 (м, 4H), 1,62-1,52 (м, 6H).
Стадия 2: 2-(пиперидин-1-ил)-4-(трифторметил)анилин: Продукт из стадии 1 выше (1,32 г, 4,81 ммоль) добавляли к суспензии 10% Pd/C (0,051 г, 0,481 ммоль) в EtOH (40 мл, 685 ммоль) и перемешивали смесь при КТ под H2 (давление 3 бара) в течение 2 ч. Реакционную смесь фильтровали через Celite® и выпаривали фильтрат в вакууме с получением указанного в заголовке соединения (1,15 г, 4,61 ммоль, выход 96%, чистота 99%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 245,3 (M+H)+ через 1,69 мин.
Стадия 3: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-4-(трифторметил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 2 выше (0,100 г, 0,409 ммоль) в ДХМ (1 мл) и пиридине (0,199 мл, 2,46 ммоль) добавляли к раствору продукта из Примера 203, стадии 2 (0,108 г, 0,409 ммоль) в ДХМ (10 мл) и перемешивали полученную смесь при КТ в течение 24 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (86 мг, 0,174 ммоль, выход 42,4%, чистота 96%) в виде бледно-желтого масла, которое кристаллизовалось при стоянии. СВЭЖХ-МС (Метод 2) m/z 471,4 (M+H)+, 469,2 (M-H)- через 2,02 мин.
Стадия 4: 4-этил-3-(N-(2-(пиперидин-1-ил)-4-(трифторметил)-фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,555 мл, 0,555 ммоль) добавляли к раствору продукта из стадии 3 выше (0,087 г, 0,185 ммоль) в ТГФ (5 мл, 61,0 ммоль) и перемешивали раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме и полученный водный раствор подокисляли до pH 6 1М HCl (водн.). Осадок отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл) с получением указанного в заголовке соединения (17,1 мг, 0,036 ммоль, выход 19,3%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 457,4 (M+H)+, 455,2 (M-H)- через 1,91 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,34 (шс, 1H), 9,38 (шс, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,43 (с, 1H), 7,40 (д, J=8,6 Гц, 1H), 7,32 (д, J=8,6 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,70 (т, J=5,2 Гц, 4H), 1,62 (п, J=5,7 Гц, 4H), 1,49 (п, J=5,6 Гц, 2H), 1,23 (т, J=7,4 Гц, 3H).
Пример 231: 4-метокси-3-(N-(2-(пиперидин-1-ил)-4-(трифтор-метил)фенил)сульфамоил)бензойная кислота
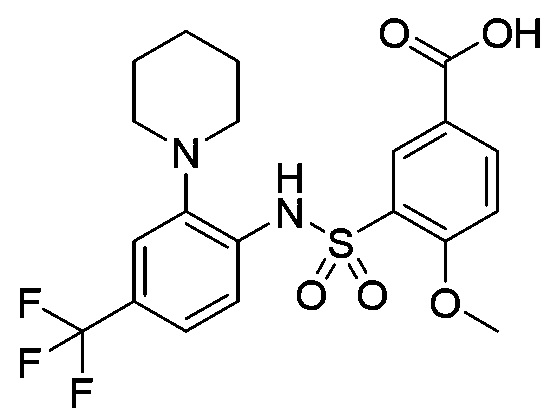
Стадия 1: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-4-(трифторметил)фенил)сульфамоил)бензоат: Раствор продукта из Примера 230, стадии 2 выше (0.100 г, 0,409 ммоль) в ДХМ (1 мл) и пиридине (0,199 мл, 2,46 ммоль) добавляли к раствору метил-3-(хлорсульфонил)-4-метоксибензоата (0,108 г, 0,409 ммоль) в ДХМ (10 мл) и перемешивали полученный раствор при КТ в течение 24 ч. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,150 г, 0,317 ммоль, выход 78%, чистота 100%) в виде бледно-желтого медленно кристаллизующегося масла. СВЭЖХ-МС (Метод 2) m/z 473,4 (M+H)+, 471,2 (M-H)- через 1,85 мин.
Стадия 2: 4-метокси-3-(N-(2-(пиперидин-1-ил)-4-(трифторметил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,952 мл, 0,952 ммоль) добавляли к раствору продукта из стадии 1 выше (0,150 г, 0,317 ммоль) в ТГФ (5 мл) и перемешивали полученный раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме и полученный водный раствор подкисляли до pH 6 1М HCl (водн.). Осадок отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл) с получением указанного в заголовке соединения (41,4 мг, 0,086 ммоль, выход 27,0%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,4 (M+H)+, 457,0 (M-H)- через 1,73 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 8,81 (шс, 1H), 8,43 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,52 (д, J=1,9 Гц, 1H), 7,47-7,39 (м, 2H), 7,33 (д, J=8,8 Гц, 1H), 3,97 (с, 3H), 2,75 (т, J=5,3 Гц, 4H), 1,71 (п, J=5,2 Гц, 4H), 1,57 (п, J=5,4 Гц, 2H).
Пример 232: 4-(метилтио)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
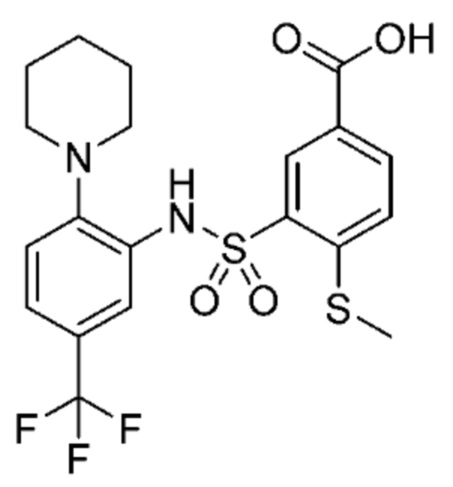
Водную фазу из реакции в Примере 225, стадии 2 подкисляли конц. HCl и экстрагировали ДХМ (3×15 мл). Органические фазы объединяли и экстрагировали 0,5 М раствором NaOH (3×20 мл). Водные экстракты объединяли, подкисляли конц. HCl и экстрагировали МТБЭ (3×30 мл). Все органические фазы после этого объединяли, сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,075 г, 0,156 ммоль, выход 39,4%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 472,9 (M-H)- через 1,79 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (с, 1H), 9,26 (с, 1H), 8,39 (д, J=1,9 Гц, 1H), 8,04 (дд, J=8,3, 1,9 Гц, 1H), 7,60 (д, J=8,5 Гц, 1H), 7,46 (д, J=2,0 Гц, 1H), 7,39-7,32 (м, 2H), 2,72 (т, J=5,2 Гц, 4H), 2,57 (с, 3H), 1,64 (п, J=5,5 Гц, 4H), 1,54-1,51 (м, 2H).
Пример 233: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
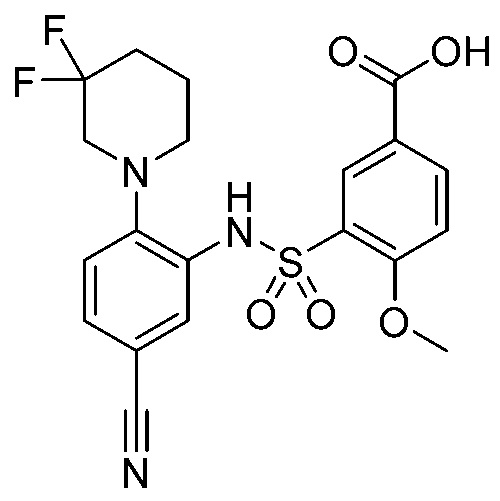
Стадия 1: 4-(3,3-дифторпиперидин-1-ил)-3-нитробензонитрил: Смесь 4-фтор-3-нитробензонитрила (500 мг, 3,01 ммоль), гидрохлорида 3,3-дифторпиперидина (569 мг, 3,61 ммоль) и Et3N (1,6 мл, 11,5 ммоль) в ДМФА (5 мл) оставляли с перемешиванием при 90°C на выходные. Смесь разбавляли водой (20 мл) и экстрагировали EtOAc (3×35 мл). Органические экстракты объединяли, промывали солевым раствором (2×30 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (635 мг, 2,35 ммоль, выход 78%, чистота 99%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,34 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,36 (д, J=2,1 Гц, 1H), 7,93 (дд, J=8,8, 2,1 Гц, 1H), 7,42 (д, J=8,8 Гц, 1H), 3,55 (т, J=11,7 Гц, 2H), 3,18 (т, J=5,4 Гц, 2H), 2,15-2,03 (м, 2H), 1,81-1,72 (м, 2H).
Стадия 2: 3-амино-4-(3,3-дифторпиперидин-1-ил)бензонитрил: Смесь продукта из стадии 1 выше (635 мг, 2,35 ммоль), порошка железа (2,6 г, 46,6 ммоль), хлорида аммония (151 мг, 2,82 ммоль), ИПС (18 мл) и воды (9 мл) перемешивали при 90°C в течение ночи. Смесью фильтровали через Celite®, промывали MeOH и выпаривали фильтрат в вакууме. Остаток разбавляли ДХМ (20 мл), сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (334 мг, 1,41 ммоль, выход 60%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,34 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,05-6,96 (м, 3H), 5,13-5,01 (м, 2H), 3,14 (т, J=11,3 Гц, 2H), 2,88 (т, J=5,4 Гц, 2H), 2,09-1,97 (м, 2H), 1,88-1,81 (м, 2H).
Стадия 3: метил-3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Смесь продукта из стадии 2 выше (80 мг, 0,337 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (100 мг, 0,378 ммоль), пиридина (0,1 мл, 1,24 ммоль) и ДХМ (2,2 мл) перемешивали при КТ в течение 4 ч, а затем при 35°C в течение 5 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (105 мг, 0,219 ммоль, выход 64,9%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 466,2 (M+H)+, 464,1 (M-H)- через 1,53 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,85 (с, 1H), 8,32 (д, J=2,2 Гц, 1H), 8,21 (дд, J=8,8, 2,2 Гц, 1H), 7,56-7,51 (м, 1H), 7,39 (д, J=8,8 Гц, 1H), 7,34-7,28 (м, 2H), 3,93 (с, 3H), 3,86 (с, 3H), 3,26 (т, J=11,2 Гц, 2H), 3,04-3,01 (м, 2H), 2,09-1,99 (м, 2H), 1,84-1,77 (м, 2H).
Стадия 4: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)-сульфамоил)-4-метоксибензойная кислота: Смесь продукта из стадии 3 выше (105 мг, 0,219 ммоль) и LiOH (21,0 мг, 0,875 ммоль) в ТГФ/воде/MeOH (4:1:1, 2,7 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (36,1 мг, 0,078 ммоль, выход 36%, чистота 98%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 452,2 (M+H)+, 450,1 (M-H)- через 1,37 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (с, 1H), 8,79 (с, 1H), 8,32 (д, J=2,2 Гц, 1H), 8,18 (дд, J=8,7, 2,2 Гц, 1H), 7,52 (дд, J=8,3, 2,0 Гц, 1H), 7,38-7,28 (м, 3H), 3,92 (с, 3H), 3,25 (т, J=11,3 Гц, 2H), 3,02 (т, J=5,5 Гц, 2H), 2,11-2,00 (м, 2H), 1,86-1,77 (м, 2H).
Пример 234: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
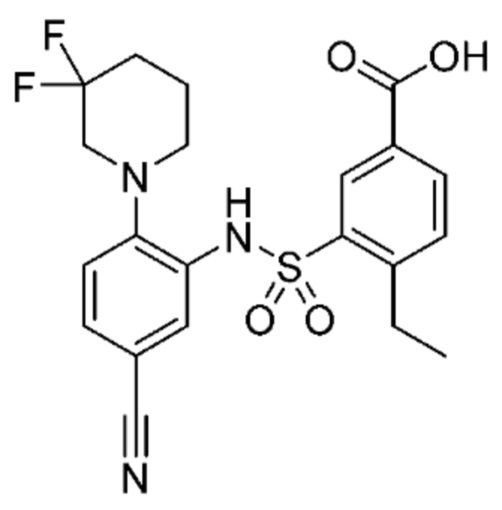
Стадия 1: метил-3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Смесь продукта из Примера 233, стадии 2 (80 мг, 0,337 ммоль), продукта из Примера 203, стадии 2 (99 мг, 0,378 ммоль), пиридина (0,1 мл, 1,24 ммоль) и ДХМ (2,2 мл) перемешивали при КТ в течение 4 ч, а затем при 35°C в течение 5 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (59 мг, 0,120 ммоль, выход 36%, чистота 94%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 464,2 (M+H)+, 462,2 (M-H)- через 1,68 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,59 (с, 1H), 8,29 (д, J=1,9 Гц, 1H), 8,14 (дд, J=8,0, 1,9 Гц, 1H), 7,66 (д, J=8,0 Гц, 1H), 7,59 (дд, J=8,5, 2,1 Гц, 1H), 7,23 (д, J=8,5 Гц, 1H), 7,13 (д, J=2,1 Гц, 1H), 3,86 (с, 3H), 3,28-3,24 (м, 2H), 3,05-2,95 (м, 4H), 2,04-1,95 (м, 2H), 1,77-1,69 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Стадия 2: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 1 выше (59 мг, 0,120 ммоль) и LiOH (21,0 мг, 0,875 ммоль) в ТГФ/воде/MeOH (4:1:1, 2,7 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (12,4 мг, 0,028 ммоль, выход 22%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 450,2 (M+H)+, 448,1 (M-H)- через 1,53 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (с, 1H), 9,53 (с, 1H), 8,30 (с, 1H), 8,11 (д, J=8,6 Гц, 1H), 7,65-7,54 (м, 2H), 7,29-7,19 (м, 1H), 7,13 (с, 1H), 3,29-3,23 (м, 2H), 3,05-2,96 (м, 4H), 2,05-1,95 (м, 2H), 1,78-1,72 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 235: (R)-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
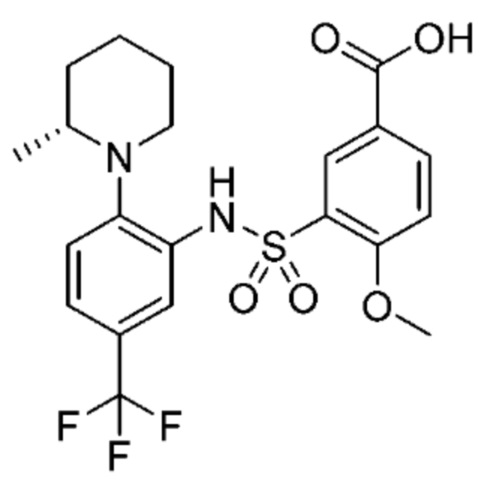
Стадия 1: (R)-2-метил-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Смесь 1-фтор-2-нитро-4-(трифторметил)бензола (220 мкл, 1,57 ммоль), (R)-2-метилпиперидина (220 мкл, 1,87 ммоль) и Et3N (0,6 мл, 4,30 ммоль) в ДХМ (8 мл) перемешивали при КТ в течение 2 ч, а затем при 35°C в течение ночи. Смесь промывали 1М HCl (водн.) (10 мл), сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (439 мг, 1,48 ммоль, выход 94%, чистота 97%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 289,2 (M+H)+ через 1,87 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,14 (д, J=2,3 Гц, 1H), 7,85 (дд, J=8,8, 2,3 Гц, 1H), 7,54 (д, J=8,8 Гц, 1H), 3,58-3,51 (м, 1H), 3,16 (ддд, J=12,5, 8,5, 4,0 Гц, 1H), 2,81 (дт, J=12,5, 4,6 Гц, 1H), 1,80-1,63 (м, 2H), 1,62-1,47 (м, 3H), 1,45-1,37 (м, 1H), 0,99 (д, J=6,5 Гц, 3H).
Стадия 2: (R)-2-(2-метилпиперидин-1-ил)-5-(трифторметил)-анилин: Раствор продукта из стадии 1 выше (438 мг, 1,47 ммоль) в EtOH (35 мл) гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Полученный раствор выпаривали в вакууме с получением указанного в заголовке соединения (361 мг, 1,34 ммоль, выход 91%, чистота 96%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 2) m/z 259,2 (M+H)+ через 1,88 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,09 (д, J=8,1 Гц, 1H), 6,95 (д, J=2,2 Гц, 1H), 6,81 (дд, J=8,1, 2,2 Гц, 1H), 5,24 (с, 2H), 3,09-2,96 (м, 1H), 2,91-2,84 (м, 1H), 2,44-2,35 (м, 1H), 1,81-1,69 (м, 2H), 1,66-1,57 (м, 2H), 1,49-1,28 (м, 2H), 0,78 (д, J=6,1 Гц, 3H).
Стадия 3: (R)-метил-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь продукта из стадии 3 выше (100 мг, 0,372 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (113 мг, 0,427 ммоль) и пиридина (0,1 мл, 1,24 ммоль) в ДХМ (2,5 мл) оставляли с перемешиванием при 35°C на выходные. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (181 мг, 0,337 ммоль, выход 91%, чистота 91%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,2 (M-H)- через 1,89 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,92 (с, 1H), 8,39 (д, J=2,3 Гц, 1H), 8,19 (дд, J=8,8, 2,3 Гц, 1H), 7,65 (д, J=2,1 Гц, 1H), 7,48 (д, J=8,3 Гц, 1H), 7,41-7,34 (м, 2H), 3,96 (с, 3H), 3,85 (с, 3H), 3,01-2,93 (м, 1H), 2,63-2,52 (м, 2H), 1,79-1,74 (м, 2H), 1,69-1,55 (м, 2H), 1,45-1,33 (м, 2H), 0,59 (д, J=6,1 Гц, 3H).
Стадия 4: (R)-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 3 выше (181 мг, 0,337 ммоль) и LiOH (32,3 мг, 1,35 ммоль) в ТГФ/MeOH/воде (4:1:1, 4,5 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (15,9 мг, 0,033 ммоль, выход 10%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+, 471,2 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (с, 1H), 8,89 (с, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,64 (д, J=2,1 Гц, 1H), 7,48 (д, J=8,3 Гц, 1H), 7,37 (дд, J=8,3, 2,1 Гц, 1H), 7,34 (д, J=8,7 Гц, 1H), 3,95 (с, 3H), 3,00-2,93 (м, 1H), 2,63-2,57 (м, 1H), 2,55-2,51 (м, 1H), 1,80-1,74 (м, 2H), 1,70-1,56 (м, 2H), 1,48-1,34 (м, 2H), 0,60 (д, J=6,1 Гц, 3H).
Пример 236: (R)-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
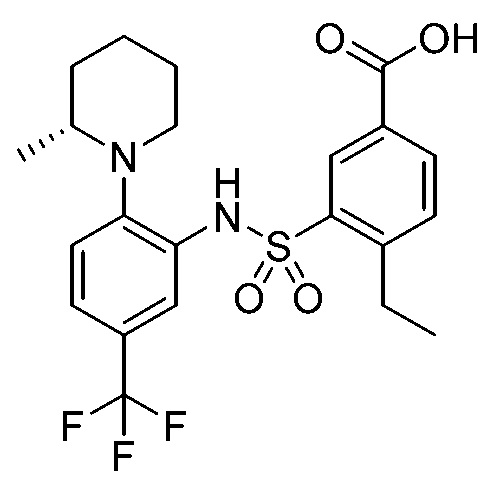
Стадия 1: (R)-метил-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 235, стадии 2 (100 мг, 0,372 ммоль), продукта из Примера 203, стадии 2 (112 мг, 0,427 ммоль) и пиридина (0,1 мл, 1,24 ммоль) в ДХМ (2,5 мл) оставляли с перемешиванием при 35°C на выходные. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (120 мг, 0,238 ммоль, выход 64%, чистота 96%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,2 (M-H)- через 2,06 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,42 (с, 1H), 8,38 (д, J=1,9 Гц, 1H), 8,10 (дд, J=8,0, 1,9 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,57 (шс, 1H), 7,42 (шс, 2H), 3,84 (с, 3H), 3,16-2,99 (м, 2H), 2,98-2,91 (м, 1H), 2,46-2,38 (м, 2H), 1,72-1,65 (м, 2H), 1,63-1,48 (м, 2H), 1,41-1,32 (м, 2H), 1,23 (т, J=7,4 Гц, 3H), 0,57 (д, J=6,1 Гц, 3H).
Стадия 2: (R)-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 1 выше (120 мг, 0,238 ммоль) и LiOH (32,3 мг, 1,35 ммоль) в ТГФ/MeOH/воде (4:1:1, 4,5 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (28,6 мг, 0,060 ммоль, выход 27%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,2 (M+H)+, 469,2 (M-H)- через 1,91 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (с, 1H), 9,36 (с, 1H), 8,39 (д, J=1,9 Гц, 1H), 8,08 (дд, J=8,0, 1,9 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,45-7,38 (м, 2H), 3,15-2,99 (м, 2H), 2,97-2,91 (м, 1H), 2,47-2,38 (м, 2H), 1,73-1,66 (м, 2H), 1,63-1,48 (м, 2H), 1,43-1,33 (м, 2H), 1,22 (т, J=7,4 Гц, 3H), 0,58 (д, J=6,2 Гц, 3H).
Пример 237: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
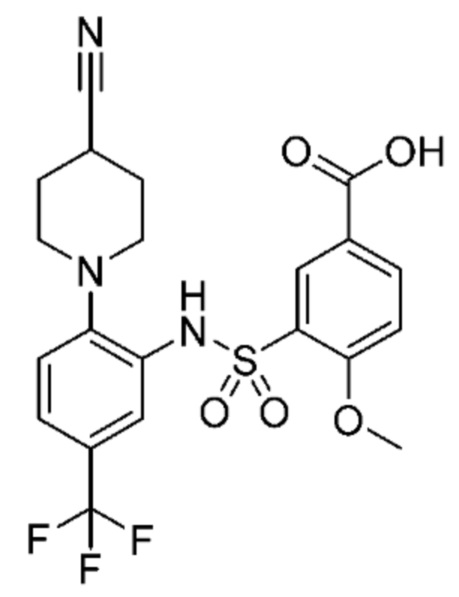
Стадия 1: 1-(2-нитро-4-(трифторметил)фенил)пиперидин-4-карбонитрил: Раствор 1-фтор-2-нитро-4-(трифторметил)бензола (200 мкл, 1,43 ммоль), пиперидин-4-карбонитрила (250 мкл, 2,24 ммоль) и Et3N (500 мкл, 3,59 ммоль) в ДХМ (6 мл) оставляли при КТ на 4 ч. Реакционную смесь промывали 1М HCl (водн.) (2×2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (470 мг, 1,54 ммоль, колич. выход, чистота 98%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 299,7 (M+H)+ через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,17 (д, J=2,2 Гц, 1H), 7,87 (дд, J=8,9, 2,3 Гц, 1H), 7,47 (д, J=8,8 Гц, 1H), 3,29-3,20 (м, 2H), 3,20-3,02 (м, 3H), 2,05-1,94 (м, 2H), 1,91-1,75 (м, 2H).
Стадия 2: 1-(2-амино-4-(трифторметил)фенил)пиперидин-4-карбонитрил: Продукт из стадии 1 выше (465 мг, 1,52 ммоль) растворяли в EtOH (40 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Полученный бесцветный раствор выпаривали в вакууме с получением указанного в заголовке соединения (407 мг, 1,50 ммоль, выход 98%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 270,4 (M+H)+ через 1,48 мин.
Стадия 3: метил-3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (100 мг, 0,371 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,1 мл, 1,24 ммоль) и обрабатывали метил-3-(хлорсульфонил)-4-метоксибензоатом (140 мг, 0,529 ммоль). Полученный раствор оставляли при КТ на 18 ч. Смесь выпаривали в вакууме, а остаток растворяли в EtOAc (4 мл) и последовательно промывали насыщенным NaHCO3 (водн.) (3 мл) и солевым раствором (2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (157 мг, 0,309 ммоль, выход 83%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 498,3 (M+H)+, 496,2 (M-H)- через 1,64 мин.
Стадия 4: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (50 мг, 0,098 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (400 мкл, 0.400 ммоль). Добавляли MeOH с получением прозрачного раствора и перемешивали полученную смесь при КТ в течение 3 дней. Раствор разбавляли водой (4 мл) и выпаривали в вакууме при 22°C. Полученный водный раствор подкисляли 1М HCl (водн.). Осадок собирали с помощью фильтрации, промывали водой и сушили в вакууме с получением указанного в заголовке соединения (40 мг, 0,079 ммоль, выход 80%, чистота 98%) в виде белого порошка. СВЭЖХ-МС (Метод 2) m/z 484,3 (M+H)+, 482,3 (M-H)- через 0,98 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,06 (с, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,3 Гц, 1H), 7,46 (д, J=2,2 Гц, 1H), 7,39 (дд, J=8,6, 2,2 Гц, 1H), 7,35-7,29 (м, 2H), 3,90 (с, 3H), 3,02 (тт, J=8,4, 4,1 Гц, 1H), 2,94-2,86 (м, 2H), 2,82-2,71 (м, 2H), 2,08-1,95 (м, 2H), 1,95-1,80 (м, 2H).
Пример 238: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифтор-метил)фенил)сульфамоил)-4-этилбензойная кислота
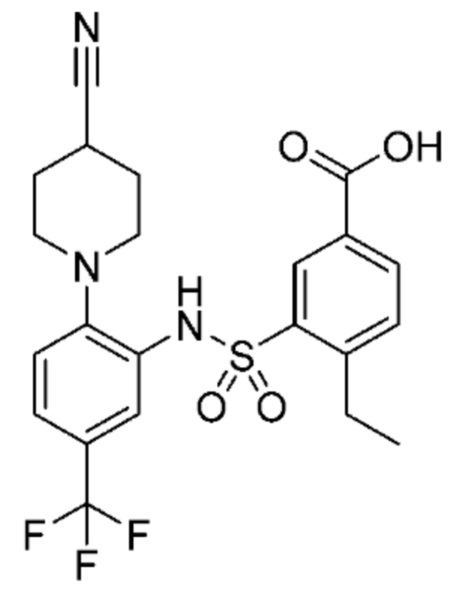
Стадия 1: метил-3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензоат: Продукт из Примера 237, стадии 2 (100 мг, 0,371 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (0,1 мл, 1,24 ммоль) и обрабатывали продуктом из Примера 203, стадии 2 (140 мг, 0,533 ммоль). Полученный раствор оставляли при КТ на 18 ч. Смесь выпаривали в вакууме, остаток растворяли в EtOAc (4 мл) и последовательно промывали насыщенным NaHCO3 (водн.) (3 мл) и солевым раствором (2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (105 мг, 0,208 ммоль, выход 56%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 496,3 (M+H)+, 494,3 (M-H)- через 1,78 мин.
Стадия 2: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-этилбензойная кислота: Продукт из стадии 1 выше (50 мг, 0,099 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (400 мкл, 0.400 ммоль). Добавляли MeOH с получением прозрачного раствора и перемешивали полученную смесь при КТ в течение 3 дней. Раствор разбавляли водой (4 мл) и выпаривали в вакууме при 22°C. Полученный водный раствор подкисляли 1М HCl (водн.). Осадок собирали с помощью фильтрации, промывали водой и сушили в вакууме с получением указанного в заголовке соединения (44 мг, 0,090 ммоль, выход 91%, чистота 98%) в виде белого порошка. СВЭЖХ-МС (Метод 2) m/z 482,3 (M+H)+, 480,2 (M-H)- через 1,09 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (шс, 1H), 9,70 (шс, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,9 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,44 (дд, J=8,5, 2,2 Гц, 1H), 7,32 (д, J=2,2 Гц, 1H), 7,28 (д, J=8,4 Гц, 1H), 3,04 (к, J=7,4 Гц, 2H), 2,96 (тт, J=8,7, 4,2 Гц, 1H), 2,89-2,78 (м, 2H), 2,74-2,64 (м, 2H), 1,99-1,88 (м, 2H), 1,88-1,76 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 239: (S)-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
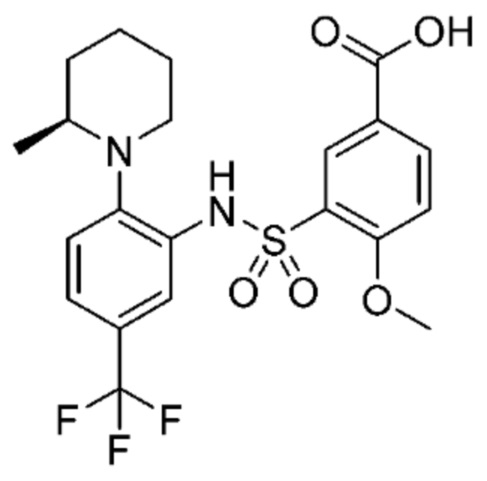
Стадия 1: (S)-2-метил-1-(2-нитро-4-(трифторметил)фенил)-пиперидин: Смесь 1-фтор-2-нитро-4-(трифторметил)бензола (250 мкл, 1,79 ммоль), (S)-2-метилпиперидина (250 мкл, 2,13 ммоль) и Et3N (0,6 мл, 4,30 ммоль) в ДХМ (8 мл) перемешивали при КТ в течение 2 ч, а затем при 35°C в течение ночи. Смесь промывали 1М HCl (водн.) (10 мл), сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% ДХМ/изогексан) с получением указанного в заголовке соединения (503 мг, 1,68 ммоль, выход 94%, чистота 96%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 289,2 (M+H)+ через 1,87 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,14 (д, J=2,3 Гц, 1H), 7,85 (дд, J=8,8, 2,3 Гц, 1H), 7,54 (д, J=8,8 Гц, 1H), 3,58-3,50 (м, 1H), 3,16 (ддд, J=12,5, 8,5, 4,0 Гц, 1H), 2,82 (дт, J=12,5, 4,6 Гц, 1H), 1,80-1,63 (м, 2H), 1,63-1,46 (м, 3H), 1,45-1,37 (м, 1H), 0,99 (д, J=6,5 Гц, 3H).
Стадия 2: (S)-2-(2-метилпиперидин-1-ил)-5-(трифторметил)-анилин: Раствор продукта из стадии 1 выше (503 мг, 1,68 ммоль) в EtOH (35 мл) гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, картридж 30×4 мм, режим полной подачи водорода, КТ, скорость потока 1 мл/мин, 1 цикл). Растворитель выпаривали с получением указанного в заголовке соединения (410 мг, 1,38 ммоль, выход 82%, 87% чистота) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 2) m/z 259,2 (M+H)+, 257,0 (M-H)- через 1,86 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,10 (д, J=8,2 Гц, 1H), 6,96 (д, J=2,2 Гц, 1H), 6,82 (дд, J=8,2, 2,2 Гц, 1H), 5,25 (с, 2H), 3,09-2,98 (м, 1H), 2,91-2,85 (м, 1H), 2,45-2,36 (м, 1H), 1,84-1,70 (м, 2H), 1,67-1,58 (м, 2H), 1,50-1,29 (м, 2H), 0,79 (д, J=6,2 Гц, 3H).
Стадия 3: (S)-метил-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь продукта из стадии 2 выше (100 мг, 0,337 ммоль), метил-3-(хлорсульфонил)-4-метоксибензоата (103 мг, 0,387 ммоль) и пиридина (0,1 мл, 1,24 ммоль) в ДХМ (2,5 мл) перемешивали при 35°C в течение 4 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (159 мг, 0,321 ммоль, выход 95%, чистота 98%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 487,2 (M+H)+, 485,1 (M-H)- через 1,90 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,92 (с, 1H), 8,39 (д, J=2,3 Гц, 1H), 8,19 (дд, J=8,8, 2,3 Гц, 1H), 7,65 (д, J=2,1 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,41-7,34 (м, 2H), 3,96 (с, 3H), 3,85 (с, 3H), 2,98-2,95 (м, 1H), 2,62-2,51 (м, 2H), 1,76 (шд, J=10,8 Гц, 2H), 1,70-1,55 (м, 2H), 1,45-1,33 (м, 2H), 0,59 (д, J=6,1 Гц, 3H).
Стадия 4: (S)-4-метокси-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 3 выше (159 мг, 0,321 ммоль) и LiOH∙H2O (55 мг, 1,31 ммоль) в ТГФ/MeOH/воде (4:1:1, 4,5 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением продукта (29,6 мг, 0,062 ммоль, выход 19%, чистота 99%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 473,2 (M+H)+, 471,1 (M-H)- через 1,76 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (с, 1H), 8,89 (с, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,64 (д, J=2,1 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,37 (дд, J=8,2, 2,1 Гц, 1H), 7,34 (д, J=8,7 Гц, 1H), 3,95 (с, 3H), 3,01-2,92 (м, 1H), 2,63-2,51 (м, 2H), 1,80-1,74 (м, 2H), 1,70-1,58 (м, 2H), 1,48-1,31 (м, 2H), 0,60 (д, J=6,1 Гц, 3H).
Пример 240: (S)-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
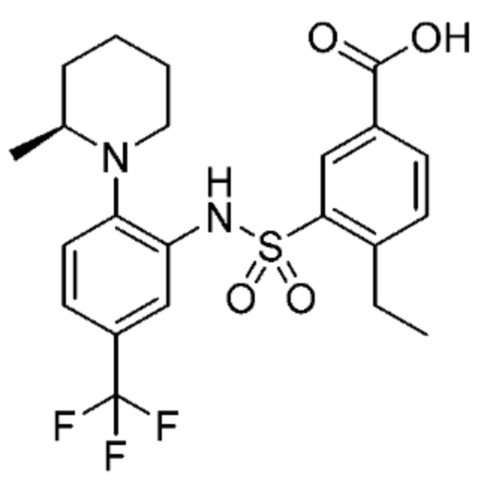
Стадия 1: (S)-метил-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 239, стадии 2 (100 мг, 0,337 ммоль), продукта из Примера 203, стадии 2 (102 мг, 0,387 ммоль) и пиридина (0,1 мл, 1,24 ммоль) в ДХМ (2,5 мл) перемешивали при 35°C в течение 4 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (159 мг, 0,320 ммоль, выход 95%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,1 (M-H)- через 2,06 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,42 (с, 1H), 8,38 (д, J=1,9 Гц, 1H), 8,10 (дд, J=8,1, 1,9 Гц, 1H), 7,63 (д, J=8,1 Гц, 1H), 7,59-7,55 (м, 1H), 7,43-7,40 (м, 2H), 3,84 (с, 3H), 3,15-2,99 (м, 2H), 2,98-2,91 (м, 1H), 2,45-2,38 (м, 2H), 1,72-1,66 (м, 2H), 1,63-1,47 (м, 2H), 1,41-1,32 (м, 2H), 1,23 (т, J=7,4 Гц, 3H), 0,57 (д, J=6,2 Гц, 3H).
Стадия 2: (S)-4-этил-3-(N-(2-(2-метилпиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 1 выше (159 мг, 0,320 ммоль) и LiOH⋅H2O (55 мг, 1,31 ммоль) в ТГФ/MeOH/воде (4:1:1, 4,5 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Смесь экстрагировали EtOAc (3×20 мл), и объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (26,1 мг, 0,055 ммоль, выход 17%, чистота 99%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 471,3 (M+H)+, 469,1 (M-H)- через 1,94 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,36 (с, 1H), 8,39 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,55 (с, 1H), 7,45-7,38 (м, 2H), 3,15-2,99 (м, 2H), 2,99-2,91 (м, 1H), 2,48-2,38 (м, 2H), 1,73-1,64 (м, 2H), 1,62-1,48 (м, 2H), 1,42-1,32 (м, 2H), 1,22 (т, J=7,4 Гц, 3H), 0,58 (д, J=6,2 Гц, 3H).
Пример 241: 4-этил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
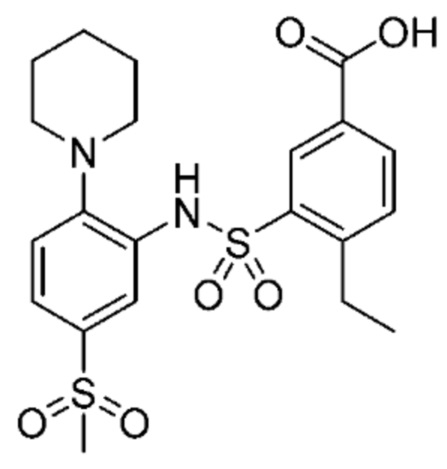
Стадия 1: метил-4-этил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из Примера 207, стадии 2 (0,120 г, 0,472 ммоль) и продукта из Примера 203, стадии 2 (0,124 г, 0,472 ммоль) в ДХМ (10 мл) обрабатывали пиридином (0,229 мл, 2,83 ммоль) и перемешивали раствор при КТ в течение 24 ч, а затем нагревали с обратным холодильником в течение 20 ч. Реакционную смесь выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-70% EtOAc/изогексан) с получением указанного в заголовке соединения (0,180 г, 0,375 ммоль, выход 79%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 481,3 (M+H)+, 479,3 (M-H)- через 1,66 мин.
Стадия 2: 4-этил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,12 мл, 1,12 ммоль) добавляли к раствору продукта из стадии 1 выше (0,180 г, 0,375 ммоль) в ТГФ (5 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а полученный водный раствор промывали EtOAc (50 мл). Водную фазу доводили до pH 6 1M HCl (водн.) с образованием осадка, который отфильтровывали и промывали водой (10 мл) и изогексаном (20 мл), с получением указанного в заголовке соединения (142 мг, 0,289 ммоль, выход 77%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 467,3 (M+H)+, 465,3 (M-H)- через 1,52 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,23 (шс, 1H), 9,58 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,06 (д, J=7,9 Гц, 1H), 7,58-7,51 (м, 3H), 7,20 (с, 1H), 3,06 (к, J=7,4 Гц, 2H), 3,02 (с, 3H), 2,81-2,80 (м, 4H), 1,58-1,54 (м, 4H), 1,49-1,48 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 242: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
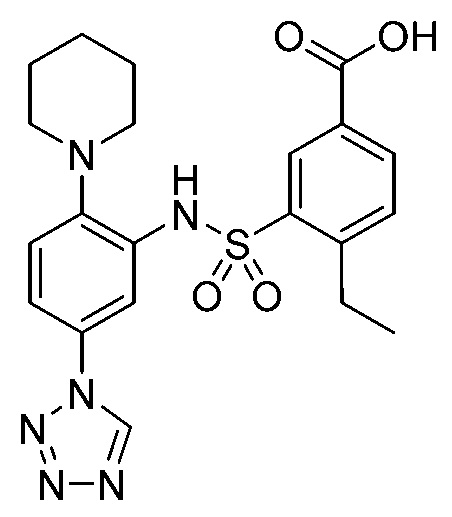
Стадия 1: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Пиридин (0,099 мл, 1,23 ммоль) добавляли к раствору продукта из Примера 214, стадии 3 (100 мг, 0,409 ммоль) и продукта из Примера 203, стадии 2 (129 мг, 0,491 ммоль) в ДХМ (10 мл) и перемешивали раствор при КТ в течение 18 ч. Раствор выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,120 г, 0,245 ммоль, выход 60%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,3 (M+H)+, 469,4 (M-H)- через 1,72 мин.
Стадия 2: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,5 мл, 1,50 ммоль) добавляли к раствору продукта из стадии 1 выше (0,120 г, 0,245 ммоль) в ТГФ (6 мл) и метаноле (1,5 мл) и перемешивали раствор при КТ в течение ночи. Растворитель удаляли в вакууме, а остаток растворяли в воде (5 мл) и промывали МТБЭ (3×5 мл). Водную фазу подкисляли до ~pH 2 конц. HCl и экстрагировали продукт МТБЭ (3×10 мл). Органические фазы объединяли, сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (0,103 г, 0,220 ммоль, выход 90%, чистота 98%). СВЭЖХ-МС (Метод 1) m/z 457,3 (M+H)+, 455,3 (M-H)- через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,27 (с, 1H), 9,97 (с, 1H), 9,44 (с, 1H), 8,38 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,9 Гц, 1H), 7,70 (д, J=2,5 Гц, 1H), 7,66-7,57 (м, 2H), 7,37 (д, J=8,6 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,71-2,58 (м, 4H), 1,62-1,49 (м, 4H), 1,49-1,40 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 243: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
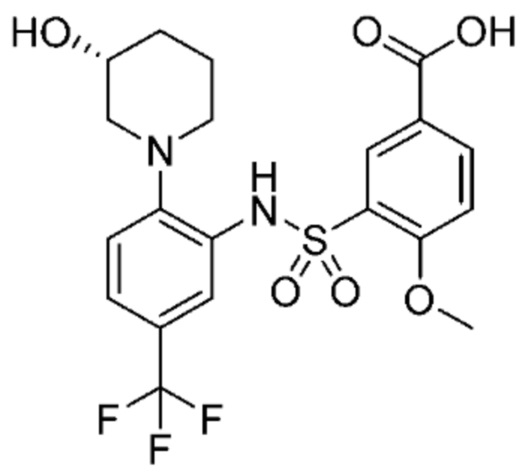
Стадия 1: (R)-1-(2-нитро-4-(трифторметил)фенил)пиперидин-3-ол: Et3N (720 мкл, 5,17 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и гидрохлорида (R)-пиперидин-3-ола (257 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (421 мг, 1,44 ммоль, выход 100%, чистота 99%) в виде темно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 291,2 (M+H)+ через 1,35 мин.
Стадия 2: (R)-1-(2-амино-4-(трифторметил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к раствору продукта из стадии 1 выше (416 мг, 1,44 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (384 мг, 1,48 ммоль, колич. выход) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 261,1 (M+H)+, 259,1 (M-H)- через 1,28 мин.
Стадия 3: (R)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 3 выше (65,6 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (102 мг, 0,207 ммоль, выход 82%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 489,2 (M+H)+, 487,1 (M-H)- через 1,54 мин.
Стадия 4: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (100 мг, 0,205 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (744 мкл, 0,819 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (47,8 мг, 0,096 ммоль, выход 47%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 473,2 (M-H)- через 0,93 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,16 (с, 1H), 8,40 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,47 (д, J=2,1 Гц, 1H), 7,38-7,16 (м, 3H), 5,11 (с, 1H), 3,90 (с, 3H), 3,82-3,70 (м, 1H), 2,89-2,78 (м, 2H), 2,77-2,55 (м, 2H), 1,96-1,85 (м, 1H), 1,79-1,68 (м, 1H), 1,63-1,37 (м, 2H).
Пример 244: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота
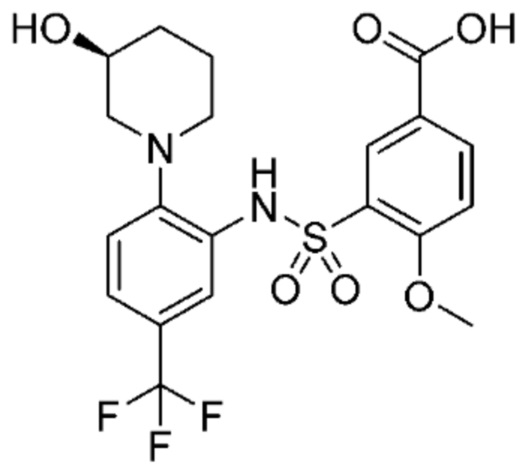
Стадия 1: (S)-1-(2-нитро-4-(трифторметил)фенил)пиперидин-3-ол: Et3N (720 мкл, 5,17 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и гидрохлорида (S)-пиперидин-3-ола (257 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (416 мг, 1,44 ммоль, выход 100%) в виде темно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 291,3 (M+H)+, 289,1 (M-H)- через 1,35 мин.
Стадия 2: (S)-1-(2-амино-4-(трифторметил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к раствору продукта из стадии 1 выше (416 мг, 1,44 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, и остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (376 мг, 1,43 ммоль, выход 100%, чистота 99%) в виде светло-желтого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 261,1 (M+H)+, 259,0 (M-H)- через 1,28 мин.
Стадия 3: (S)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (65,6 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (93,1 мг, 0,191 ммоль, выход 76%) в виде светло-желтого клейкого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 489,3 (M+H)+, 487,1 (M-H)- через 1,55 мин.
Стадия 4: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (91 мг, 0,186 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (677 мкл, 0,745 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации и промывали водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (79,7 мг, 0,163 ммоль, выход 87%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 473,2 (M-H)- через 0,94 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,15 (с, 1H), 8,39 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,47 (д, J=2,1 Гц, 1H), 7,38-7,10 (м, 3H), 5,10 (с, 1H), 3,90 (с, 3H), 3,80-3,74 (м, 1H), 2,90-2,78 (м, 2H), 2,76-2,58 (м, 2H), 1,96-1,84 (м, 1H), 1,80-1,69 (м, 1H), 1,65-1,37 (м, 2H).
Пример 245: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота
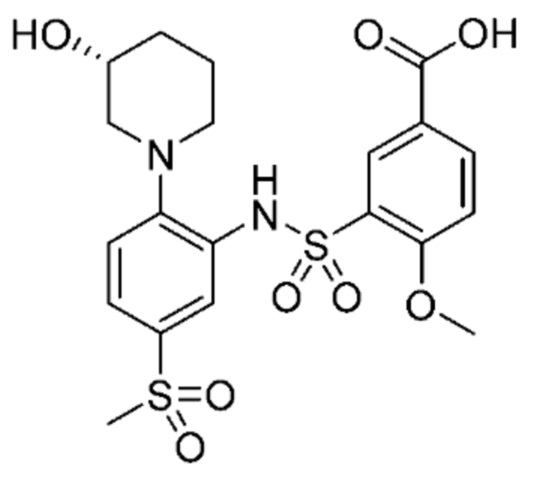
Стадия 1: (R)-1-(4-(метилсульфонил)-2-нитрофенил)пиперидин-3-ол: Et3N (687 мкл, 4,93 ммоль) добавляли к раствору 1-фтор-4-(метилсульфонил)-2-нитробензола (300 мг, 1,37 ммоль) и гидрохлорида (R)-пиперидин-3-ола (245 мг, 1,78 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (411 мг, 1,37 ммоль, выход 100%) в виде темно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 301,2 (M+H)+, 299,1 (M-H)- через 0,87 мин.
Стадия 2: (R)-1-(2-амино-4-(метилсульфонил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к раствору продукта из стадии 1 выше (411 мг, 1,37 ммоль) в EtOH (15 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (368 мг, 1,36 ммоль, выход 99%) в виде светло-желтого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 271,1 (M+H)+, 269,2 (M-H)- через 0,78 мин.
Стадия 3: (R)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (68,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (96 мг, 0,189 ммоль, выход 75%, чистота 98%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 499,3 (M+H)+, 497,2 (M-H)- через 1,16 мин.
Стадия 4: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (94 мг, 0,185 ммоль, чистота 98%) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (686 мкл, 0,754 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (32,4 мг, 0,062 ммоль, выход 34%, чистота 93%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,2 (M+H)+, 483,2 (M-H)- через 0,68 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (шс, 1H), 9,19 (шс, 1H), 8,38 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,69 (д, J=2,2 Гц, 1H), 7,50 (дд, J=8,4, 2,2 Гц, 1H), 7,29 (д, J=8,8 Гц, 1H), 7,27 (д, J=8,5 Гц, 1H), 5,55-4,80 (м, 1H), 3,91 (с, 3H), 3,80-3,74 (м, 1H), 2,98 (с, 3H), 2,94-2,86 (м, 2H), 2,82-2,61 (м, 2H), 1,95-1,83 (м, 1H), 1,79-1,68 (м, 1H), 1,63-1,38 (м, 2H).
Пример 246: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота
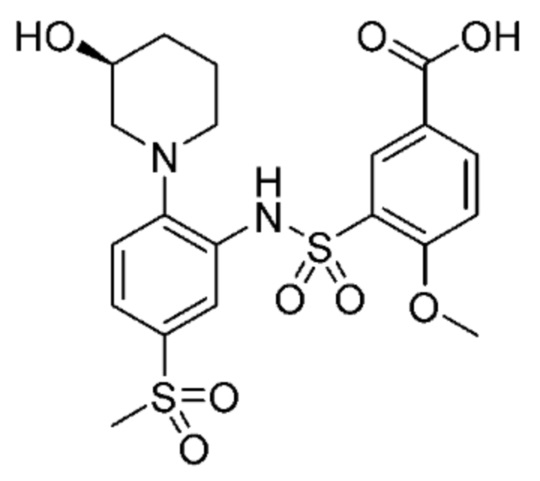
Стадия 1: (S)-1-(4-(метилсульфонил)-2-нитрофенил)пиперидин-3-ол: Et3N (687 мкл, 4,93 ммоль) добавляли к раствору 1-фтор-4-(метилсульфонил)-2-нитробензола (300 мг, 1,37 ммоль) и гидрохлорида (S)-пиперидин-3-ола (245 мг, 1,78 ммоль) в ДХМ (6 мл) и перемешивали раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и сушили фильтрат при пропускании через фазовый сепаратор. Органическую фазу выпаривали в вакууме с получением указанного в заголовке соединения (415 мг, 1,37 ммоль, выход 100%, чистота 99%) в виде темно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 301,1 (M+H)+, 299,1 (M-H)- через 0,88 мин.
Стадия 2: (S)-1-(2-амино-4-(метилсульфонил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к раствору продукта из стадии 1 выше (415 мг, 1,37 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 3 дней. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (375 мг, 1,37 ммоль, выход 100%, чистота 99%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 269,0 (M-H)- через 0,76 мин.
Стадия 3: (S)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 3 выше (68,1 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (68,4 мг, 0,136 ммоль, выход 54%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 499,3 (M+H)+, 497,2 (M-H)- через 1,16 мин.
Стадия 4: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (66 мг, 0,132 ммоль) растворяли в ТГФ (5 мл) и обрабатывали 1,1 М LiOH (водн.) (481 мкл, 0,530 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (31,3 мг, 0,063 ммоль, выход 48%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,2 (M-H)- через 0,69 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (с, 1H), 9,21 (с, 1H), 8,38 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,69 (д, J=2,2 Гц, 1H), 7,51 (дд, J=8,4, 2,2 Гц, 1H), 7,31 (д, J=8,7 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 5,11 (с, 1H), 3,91 (с, 3H), 3,80-3,73 (м, 1H), 2,98 (с, 3H), 2,95-2,82 (м, 2H), 2,82-2,63 (м, 2H), 1,98-1,82 (м, 1H), 1,79-1,69 (м, 1H), 1,64-1,41 (м, 2H).
Пример 247: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
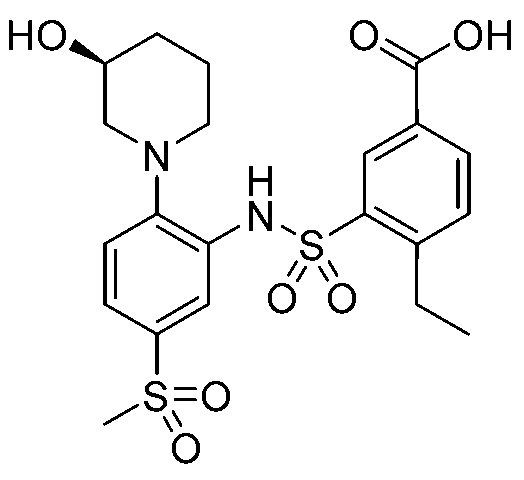
Стадия 1: (S)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 246, стадии 2 (68,6 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (41,9 мг, 0,084 ммоль, выход 33%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 497,3 (M+H)+, 495,3 (M-H)- через 1,34 мин.
Стадия 2: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (40 мг, 0,081 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (293 мкл, 0,322 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (30,5 мг, 0,058 ммоль, выход 72%, чистота 92%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,3 (M+H)+, 481,2 (M-H)- через 0,77 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (шс, 1H), 9,76 (шс, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,63-7,58 (м, 2H), 7,54 (дд, J=8,4, 2,2 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 5,21 (с, 1H), 3,87-3,69 (м, 1H), 2,98-3,15 (м, 7H), 2,77-2,59 (м, 2H), 1,90-1,79 (м, 1H), 1,73-1,64 (м, 1H), 1,56-1,45 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 248: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
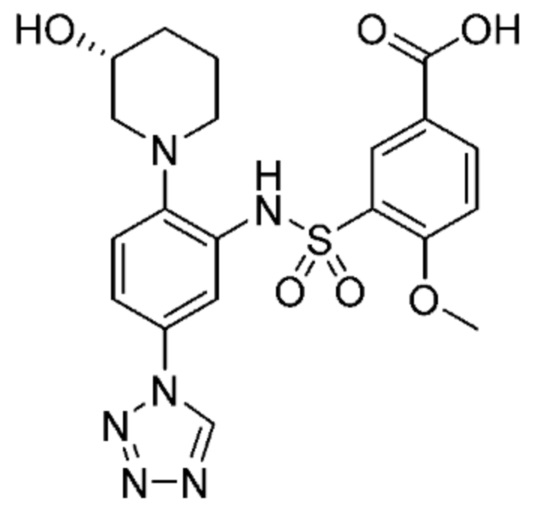
Стадия 1: (R)-1-(2-нитро-4-(тетразол-1-ил)фенил)пиперидин-3-ол: Et3N (720 мкл, 5,16 ммоль) добавляли к раствору продукта из Примера 214, стадии 1 (300 мг, 1,43 ммоль) и гидрохлорида (R)-пиперидин-3-ола (257 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл), и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (421 мг, 1,43 ммоль, выход 100%, чистота 99%) в виде темно-красного вязкого масла. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,92 мин.
Стадия 2: (R)-1-(2-амино-4-(тетразол-1-ил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к раствору продукта из стадии 1 выше (421 мг, 1,43 ммоль, чистота 99%) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 3 дней. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (369 мг, 1,42 ммоль, выход 99%, чистота 100%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 259,1 (M-H)- через 0,82 мин.
Стадия 3: (R)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (65,6 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (83,9 мг, 0,170 ммоль, выход 68%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,3 (M-H)- через 1,20 мин.
Стадия 4: (R)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (81 мг, 0,166 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (603 мкл, 0,663 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (30,2 мг, 0,059 ммоль, выход 35%, чистота 92%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 473,2 (M-H)- через 0,73 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,15 (шс, 1H), 9,94 (с, 1H), 9,18 (с, 1H), 8,44 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,80 (д, J=2,5 Гц, 1H), 7,50 (дд, J=8,5, 2,5 Гц, 1H), 7,38 (д, J=8,7 Гц, 1H), 7,31 (д, J=8,6 Гц, 1H), 5,06 (с, 1H), 3,93 (с, 3H), 3,82-3,73 (м, 1H), 2,92-2,56 (м, 4H), 1,99-1,80 (м, 1H), 1,82-1,70 (м, 1H), 1,65-1,51 (м, 1H), 1,52-1,41 (м, 1H).
Пример 249: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
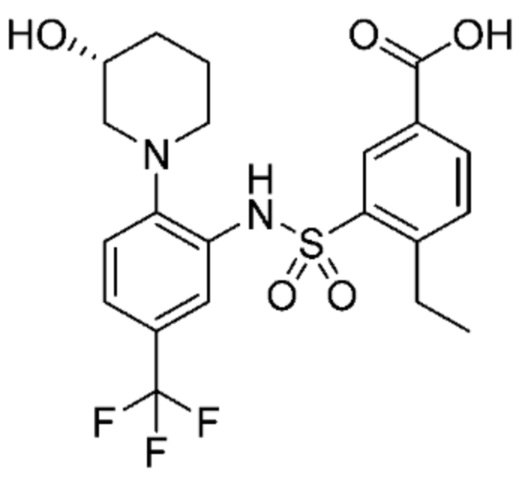
Стадия 1: (R)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 243, стадии 2 (66,0 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (93,1 мг, 0,180 ммоль, выход 71%, чистота 94%) в виде темно-желтого клейкого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+ через 1,72 мин.
Стадия 2: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (91 мг, 0,187 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (680 мкл, 0,748 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (30,2 мг, 0,062 ммоль, выход 33%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,2 (M-H)- через 1,06 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,22 (шс, 1H), 9,71 (шс, 1H), 8,42 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,59 (д, J=8,1 Гц, 1H), 7,47-7,27 (м, 2H), 7,20 (д, J=8,2 Гц, 1H), 5,31 (с, 1H), 3,84-3,71 (м, 1H), 3,15-3,00 (м, 2H), 2,95-2,75 (м, 2H), 2,71-2,63 (м, 2H), 1,95-1,77 (м, 1H), 1,77-1,59 (м, 1H), 1,57-1,45 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 250: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
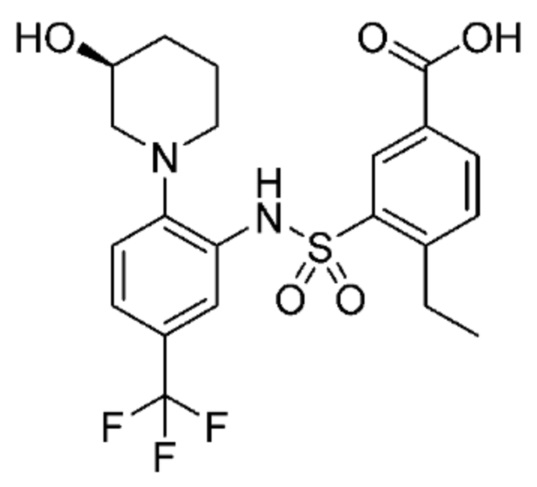
Стадия 1: (S)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 244, стадии 2 (66,0 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (86,6 мг, 0,174 ммоль, выход 69%, чистота 98%) в виде клейкого кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,2 (M-H)- через 1,71 мин.
Стадия 2: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (84 мг, 0,173 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (628 мкл, 0,691 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (66,6 мг, 0,133 ммоль, выход 77%, чистота 94%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+, 471,2 (M-H)- через 1,05 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (шс, 1H), 9,71 (шс, 1H), 8,41 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,42-7,27 (м, 2H), 7,21 (д, J=8,1 Гц, 1H), 5,20 (с, 1H), 3,84-3,68 (м, 1H), 3,15-2,99 (м, 2H), 2,94-2,75 (м, 2H), 2,72-2,61 (м, 2H), 1,94-1,74 (м, 1H), 1,73-1,61 (м, 1H), 1,58-1,41 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 251: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
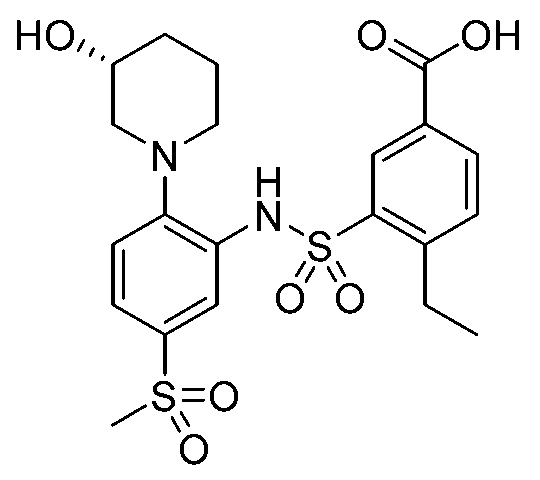
Стадия 1: (R)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 245, стадии 2 (68,6 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (75,4 мг, 0,152 ммоль, выход 60%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 497,3 (M+H)+, 495,2 (M-H)- через 1,32 мин.
Стадия 2: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (73 мг, 0,147 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (535 мкл, 0,588 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (28,4 мг, 0,056 ммоль, выход 38%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,3 (M+H)+, 481,2 (M-H)- через 0,78 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (шс, 1H), 9,77 (с, 1H), 8,41 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,65-7,58 (м, 2H), 7,55 (дд, J=8,4, 2,2 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 5,23 (с, 1H), 3,84-3,69 (м, 1H), 3,17-2,96 (м, 5H), 2,96-2,82 (м, 2H), 2,76-2,65 (м, 2H), 1,97-1,78 (м, 1H), 1,73-1,62 (м, 1H), 1,54-1,47 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 252: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
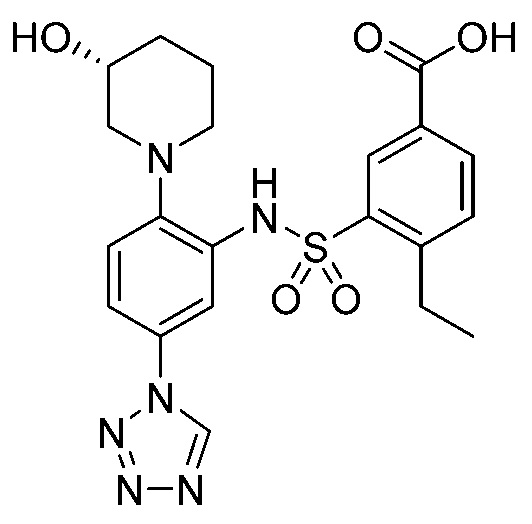
Стадия 1: (R)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 248, стадии 2 (66,1 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (96,0 мг, 0,195 ммоль, выход 77%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,3 (M-H)- через 1,38 мин.
Стадия 2: (R)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (94 мг, 0,193 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (703 мкл, 0,773 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (58,4 мг, 0,120 ммоль, выход 62%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+, 471,3 (M-H)- через 0,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (шс, 1H), 9,93 (с, 1H), 9,71 (шс, 1H), 8,44 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,73 (д, J=2,5 Гц, 1H), 7,60 (д, J=8,1 Гц, 1H), 7,54 (дд, J=8,6, 2,5 Гц, 1H), 7,32 (д, J=8,6 Гц, 1H), 5,17 (с, 1H), 3,87-3,66 (м, 1H), 3,18-3,03 (м, 2H), 2,80-2,72 (м, 1H), 2,69-2,59 (м, 1H), 2,68-2,60 (м, 2H), 1,92-1,80 (м, 1H), 1,74-1,62 (м, 1H), 1,59-1,38 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 253: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
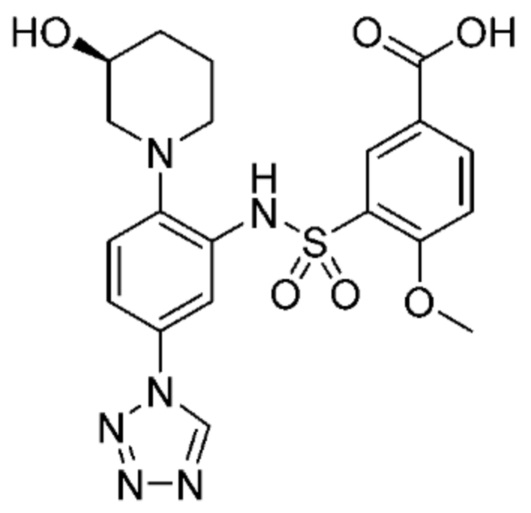
Стадия 1: (S)-1-(2-нитро-4-(тетразол-1-ил)фенил)пиперидин-3-ол: Et3N (720 мкл, 5,16 ммоль) добавляли к раствору продукта из Примера 214, стадии 1 (300 мг, 1,43 ммоль) и гидрохлорида (S)-пиперидин-3-ола (257 мг, 1,87 ммоль) в ДХМ (6 мл) и полученный раствор перемешивали при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (416 мг, 1,43 ммоль, выход 100%) в виде темно-красного вязкого масла. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,92 мин.
Стадия 2: (S)-1-(2-амино-4-(тетразол-1-ил)фенил)пиперидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к фильтрованному раствору продукта из стадии 1 выше (416 мг, 1,43 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (275 мг, 1,05 ммоль, выход 73%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 258,8 (M-H)- через 0,82 мин.
Стадия 3: (S)-метил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (65,6 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (57,8 мг, 0,115 ммоль, выход 46%, чистота 97%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,2 (M-H)- через 1,20 мин.
Стадия 4: (S)-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 1 выше (56 мг, 0,115 ммоль) растворяли в ТГФ (5 мл) и обрабатывали 1,1 М LiOH (водн.) (417 мкл, 0,459 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Добавляли дополнительное количество 1,1 М LiOH (водн.) (417 мкл, 0,459 ммоль) и нагревали реакционную смесь при 40°C в течение 4 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (27,3 мг, 0,052 ммоль, выход 45%, чистота 90%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 475,3 (M+H)+, 473,3 (M-H)- через 0,72 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,14 (шс, 1H), 9,94 (с, 1H), 9,18 (шс, 1H), 8,43 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,80 (д, J=2,5 Гц, 1H), 7,50 (дд, J=8,5, 2,5 Гц, 1H), 7,38 (д, J=8,7 Гц, 1H), 7,31 (д, J=8,7 Гц, 1H), 5,08 (с, 1H), 3,93 (с, 3H), 3,83-3,73 (м, 1H), 2,95-2,56 (м, 4H), 1,96-1,85 (м, 1H), 1,82-1,71 (м, 1H), 1,66-1,37 (м, 2H).
Пример 254: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
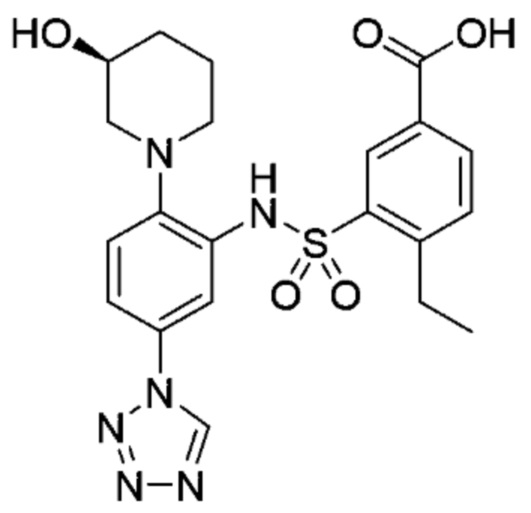
Стадия 1: (S)-метил-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 253, стадии 2 (66,1 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (99,6 мг, 0,205 ммоль, выход 81%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+, 485,3 (M-H)- через 1,37 мин.
Стадия 2: (S)-4-этил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (97 мг, 0,199 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (725 мкл, 0,797 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 24 ч. Добавляли дополнительное количество 1,1 М LiOH (водн.) (725 мкл, 0,797 ммоль) и перемешивали реакцию при 40°C в течение 24 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (39,9 мг, 0,080 ммоль, выход 40%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,3 (M-H)- через 0,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,22 (шс, 1H), 9,93 (с, 1H), 9,71 (шс, 1H), 8,44 (д, J=1,8 Гц, 1H), 8,07 (дд, J=7,9, 1,8 Гц, 1H), 7,72 (д, J=2,5 Гц, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,52 (дд, J=8,6, 2,5 Гц, 1H), 7,31 (д, J=8,6 Гц, 1H), 5,17 (с, 1H), 3,82-3,68 (м, 1H), 3,18-3,04 (м, 2H), 2,86 (дд, J=11,5, 2,7 Гц, 1H), 2,81-2,69 (м, 1H), 2,69-2,56 (м, 2H), 1,91-1,80 (м, 1H), 1,73-1,62 (м, 1H), 1,56-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 255: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
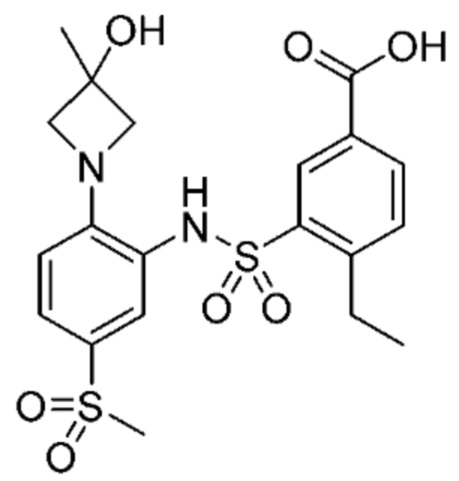
Стадия 1: 3-метил-1-(4-(метилсульфонил)-2-нитрофенил) азетидин-3-ол: Et3N (687 мкл, 4,93 ммоль) добавляли к раствору 1-фтор-4-(метилсульфонил)-2-нитробензола (300 мг, 1,37 ммоль) и гидрохлорида 3-метилазетидин-3-ола (220 мг, 1,78 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и сушили органическую фазу при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (392 мг, 1,37 ммоль, выход 100%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,83 мин.
Стадия 2: 1-(2-амино-4-(метилсульфонил)фенил)-3-метилазетидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к тонкодисперсной суспензии продукта из стадии 1 выше (392 мг, 1,37 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в EtOAc (10 мл). Органическую фазу промывали водой (5 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением темно-коричневого масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (270 мг, 1,03 ммоль, выход 74%, чистота 98%) в виде темно-розового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 257,2 (M+H)+ через 0,61 мин.
Стадия 3: метил-4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (65,0 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (74,7 мг, 0,155 ммоль, выход 61%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,2 (M+H)+, 481,1 (M-H)- через 1,16 мин.
Стадия 4: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (72 мг, 0,149 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1,1 М LiOH (водн.) (543 мкл, 0,597 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 45°C с получением указанного в заголовке соединения (66,2 мг, 0,137 ммоль, выход 92%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 467,1 (M-H)- через 1,02 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (шс, 1H), 9,60 (шс, 1H), 8,25 (д, J=1,8 Гц, 1H), 8,12 (дд, J=8,0, 1,9 Гц, 1H), 7,65 (д, J=8,0 Гц, 1H), 7,51 (дд, J=8,7, 2,2 Гц, 1H), 6,58-6,50 (м, 2H), 5,58 (с, 1H), 4,07 (д, J=8,5 Гц, 2H), 3,98 (д, J=8,5 Гц, 2H), 2,95 (к, J=7,4 Гц, 2H), 2,80 (с, 3H), 1,43 (с, 3H), 1,20 (т, J=7,4 Гц, 3H).
Пример 257: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
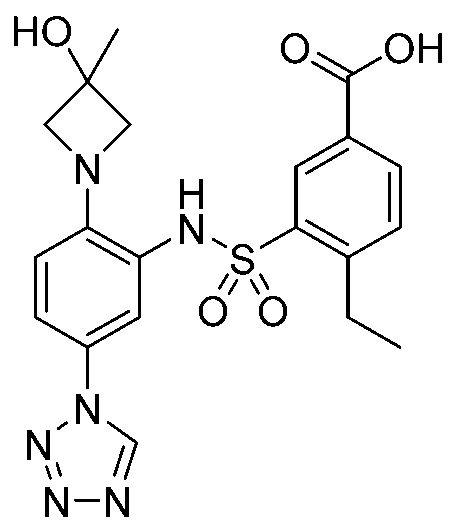
Стадия 1: 3-метил-1-(2-нитро-4-(тетразол-1-ил)фенил)-азетидин-3-ол: Et3N (720 мкл, 5,16 ммоль) добавляли к раствору продукта из Примера 214, стадии 1 (300 мг, 1,43 ммоль) и гидрохлорида 3-метилазетидин-3-ола (230 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл), затем 10% MeOH в ДХМ (200 мл) и сушили органическую фазу при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (413 мг, 1,43 ммоль, выход 100%) в виде темно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,87 мин.
Стадия 2: 1-(2-амино-4-(тетразол-1-ил)фенил)-3-метилазетидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (214 мг, 0,050 ммоль) в EtOH (1 мл) добавляли к фильтрованному раствору продукта из стадии 1 выше (396 мг, 1,44 ммоль) в EtOH (150 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (117 мг, 0,474 ммоль, выход 33%, чистота 100%) в виде светло-фиолетового твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,65 мин.
Стадия 3: метил-4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (62,5 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (31,0 мг, 0,064 ммоль, выход 25%, чистота 98%) в виде очень бледного розового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,3 (M-H)- через 1,20 мин.
Стадия 4: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (29 мг, 0,061 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (245 мкл, 0,245 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (27,5 мг, 0,058 ммоль, выход 94%, чистота 96%) в виде светло-розового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,2 (M+H)+, 457,2 (M-H)- через 1,05 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,23 (шс, 1H), 9,74 (с, 1H), 9,70 (шс, 1H), 8,32 (д, J=1,9 Гц, 1H), 8,10 (дд, J=7,9, 1,9 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,59-7,52 (м, 1H), 6,82 (д, J=2,6 Гц, 1H), 6,62 (д, J=8,8 Гц, 1H), 5,48 (с, 1H), 3,92 (д, J=8,0 Гц, 2H), 3,83 (д, J=7,9 Гц, 2H), 2,96 (к, J=7,4 Гц, 2H), 1,38 (с, 3H), 1,18 (т, J=7,4 Гц, 3H).
Пример 258: (R)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
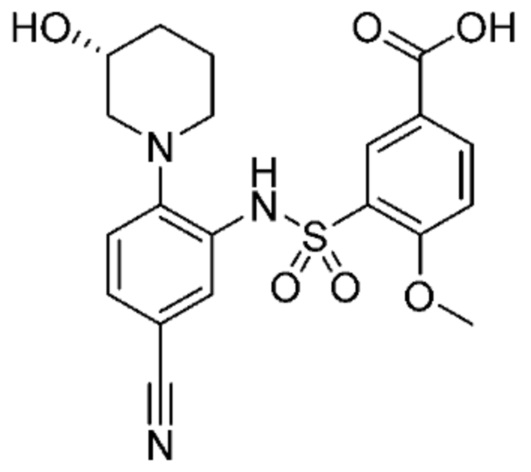
Стадия 1: (R)-4-(3-гидроксипиперидин-1-ил)-3-нитробензонитрил: Et3N (0,252 мл, 1,81 ммоль) добавляли к раствору 4-фтор-3 нитробензонитрила (300 мг, 1,81 ммоль) и гидрохлорида (R)-пиперидин-3-ола (249 мг, 1,81 ммоль) в ДХМ (20 мл) и перемешивали полученный раствор при КТ в течение ночи. Добавляли 1М HCl (водн.) (10 мл) и сушили органическую фазу при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (447 мг, 1,81 ммоль, выход 100%) в виде темно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 248,3 (M+H)+, 246,2 (M-H)- через 1,01 мин.
Стадия 2: (R)-3-амино-4-(3-гидроксипиперидин-1-ил)бензонитрил: Смесь продукта из стадии 1 выше (447 мг, 1,81 ммоль), порошка железа (2,48 г, 44,4 ммоль), хлорида аммония (116 мг, 2,17 ммоль), ИПС (15 мл) и воды (7,6 мл) нагревали при 90°C в течение 20 ч. Реакционную смесь фильтровали через Celite®, промывали MeOH (25 мл) и выпаривали фильтрат в вакууме. Остаток разбавляли ДХМ (20 мл), сушили при пропускании через фазовый сепаратор, и неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (191 мг, 0,870 ммоль, выход 48%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 218,3 (M+H)+ через 0,95 мин.
Стадия 3: (R)-метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (54,7 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (89,5 мг, 0,201 ммоль, выход 80%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 446,3 (M+H)+, 444,3 (M-H)- через 1,32 мин.
Стадия 4: (R)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (87 мг, 0,195 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (781 мкл, 0,781 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (74 мг, 0,166 ммоль, выход 85%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 432,2 (M+H)+, 430,3 (M-H)- через 1,15 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 9,25 (шс, 1H), 8,37 (д, J=2,2 Гц, 1H), 8,17 (дд, J=8,7, 2,2 Гц, 1H), 7,45 (дд, J=8,3, 2,0 Гц, 1H), 7,42 (д, J=1,9 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 5,11 (шс, 1H), 3,90 (с, 3H), 3,77-3,70 (м, 1H), 2,95-2,86 (м, 2H), 2,79-2,72 (м, 1H), 2,71-2,63 (м, 1H), 1,91-1,80 (м, 1H), 1,77-1,67 (м, 1H), 1,58-1,41 (м, 2H).
Пример 259: 4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензойная кислота
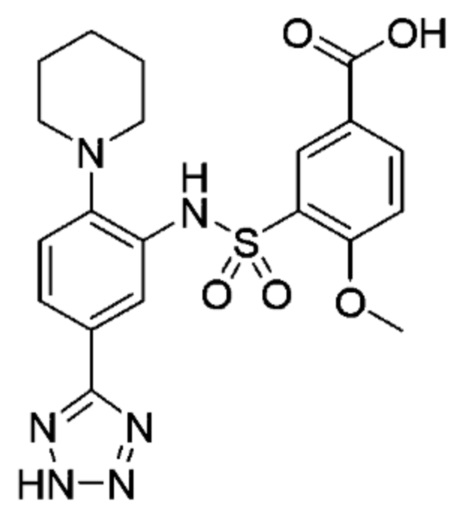
Стадия 1: 1-(2-нитро-4-(тетразол-5-ил)фенил)пиперидин: Et3N (720 мкл, 5,16 ммоль) добавляли к раствору 5-(4-фтор-3-нитрофенил)тетразола (300 мг, 1,43 ммоль) и пиперидина (185 мкл, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и сушили органическую фазу при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (406 мг, 1,43 ммоль, выход 100%, чистота 97%) в виде темно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 275,2 (M+H)+, 273,1 (M-H)- через 0,92 мин.
Стадия 2: 2-(пиперидин-1-ил)-5-(тетразол-5-ил)анилин: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к фильтрованному раствору продукта из стадии 1 выше (394 мг, 1,44 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток растворяли в ДХМ (5 мл) и сушили при пропускании через фазовый сепаратор. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (206 мг, 0,801 ммоль, выход 56%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 243,1 (M-H)- через 0,78 мин.
Стадия 3: метил-4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (61,5 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (37,4 мг, 0,078 ммоль, выход 31%, чистота 99%) в виде клейкого кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,3 (M-H)- через 1,45 мин.
Стадия 4: 4-метокси-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (35 мг, 0,074 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (296 мкл, 0,296 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (29,1 мг, 0,060 ммоль, выход 81%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,2 (M+H)+, 457,2 (M-H)- через 1,31 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 16,79 (шс, 1H), 13,10 (шс, 1H), 8,74 (с, 1H), 8,42 (д, J=2,2 Гц, 1H), 8,12 (дд, J=8,7, 2,2 Гц, 1H), 7,98 (д, J=2,0 Гц, 1H), 7,67 (дд, J=8,3, 2,0 Гц, 1H), 7,37 (д, J=8,3 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 3,95 (с, 3H), 2,83-2,71 (м, 4H), 1,71-1,63 (м, 4H), 1,58-1,50 (м, 2H).
Пример 260: (R)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
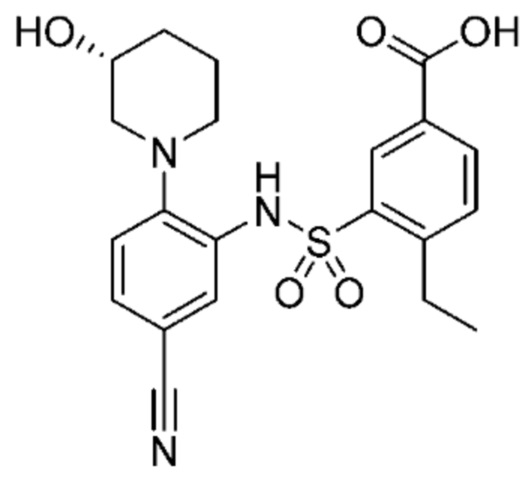
Стадия 1: (R)-метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Продукт из Примера 258, стадии 2 (55,1 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (62,7 мг, 0,141 ммоль, выход 56%, чистота 100%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 444,4 (M+H)+, 442,3 (M-H)- через 1,50 мин.
Стадия 2: (R)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: Продукт из стадии 1 выше (60 мг, 0,135 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (541 мкл, 0,541 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (57,8 мг, 0,125 ммоль, выход 93%, чистота 93%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 430,2 (M+H)+, 428,2 (M-H)- через 1,33 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,34 (шс, 1H), 9,77 (шс, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,48 (шд, J=8,2 Гц, 1H), 7,32 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,3 Гц, 1H), 5,16 (шс, 1H), 3,71 (шс, 1H), 3,15-2,85 (м, 4H), 2,72-2,62 (м, 2H), 1,86-1,76 (м, 1H), 1,73-1,63 (м, 1H), 1,51-1,41 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 261: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензойная кислота
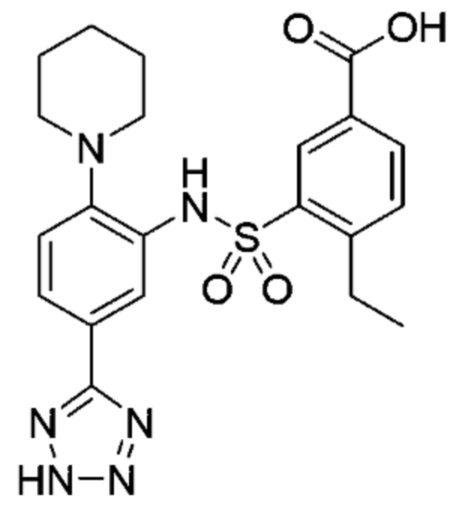
Стадия 1: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензоат: Продукт из Примера 259 стадии 2 (62,0 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (37,6 мг, 0,076 ммоль, выход 30%, чистота 95%) в виде светло-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,3 (M+H)+, 469,3 (M-H)- через 1,65 мин.
Стадия 2: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-5-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 1 выше (35 мг, 0,074 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (298 мкл, 0,298 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (30,9 мг, 0,068 ммоль, выход 91%, чистота 95%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 457,3 (M+H)+, 455,2 (M-H)- через 1,51 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 16,75 (шс, 1H), 13,22 (шс, 1H), 9,33 (шс, 1H), 8,36 (д, J=1,9 Гц, 1H), 8,07 (дд, J=8,0, 1,9 Гц, 1H), 7,86 (д, J=2,1 Гц, 1H), 7,75 (дд, J=8,4, 2,1 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,29 (д, J=8,4 Гц, 1H), 3,05 (к, J=7,4 Гц, 2H), 2,77-2,67 (м, 4H), 1,56-1,49 (м, 4H), 1,48-1,41 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 262: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
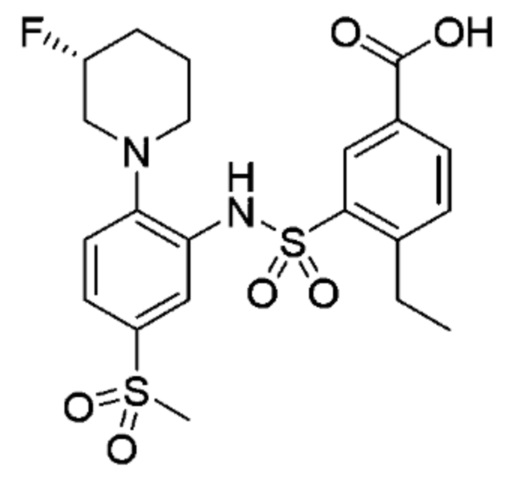
Стадия 1: (R)-3-фтор-1-(4-(метилсульфонил)-2-нитрофенил)-пиперидин: Et3N (449 мкл, 3,22 ммоль) добавляли к раствору 1-фтор-4-(метилсульфонил)-2-нитробензола (196 мг, 0,895 ммоль) и гидрохлорида (R)-3-фторпиперидина (125 мг, 0,895 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и сушили органическую фазу при пропускании через фазовый сепаратор. Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (273 мг, 0,895 ммоль, выход 100%, чистота 99%) в виде темно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,15 мин.
Стадия 2: (R)-2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)-анилин: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к суспензии продукта из стадии 1 выше (273 мг, 0,895 ммоль, чистота 99%) в EtOH (19 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 3 дней. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (239 мг, 0,867 ммоль, выход 96%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 273,1 (M+H)+ через 1,37 мин.
Стадия 3: (R)-метил-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (69,1 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 2 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (55,7 мг, 0,112 ммоль, выход 44%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 499,3 (M+H)+, 497,2 (M-H)- через 1,54 мин.
Стадия 4: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (53 мг, 0,106 ммоль) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (425 мкл, 0,425 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Добавляли еще 1М LiOH (водн.) (425 мкл, 0,425 ммоль) и перемешивали реакционную смесь при 40°C в течение 1 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (21,3 мг, 0,043 ммоль, выход 40%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,1 (M-H)- через 1,39 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,32 (шс, 1H), 9,45 (шс, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,64-7,57 (м, 2H), 7,49 (д, J=2,2 Гц, 1H), 7,31 (д, J=8,5 Гц, 1H), 4,86-4,64 (м, 1H), 3,21-3,11 (м, 1H), 3,07-2,98 (м, 5H), 2,95-2,85 (м, 2H), 2,81-2,74 (м, 1H), 2,00-1,86 (м, 1H), 1,82-1,72 (м, 1H), 1,71-1,53 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 263: (S)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
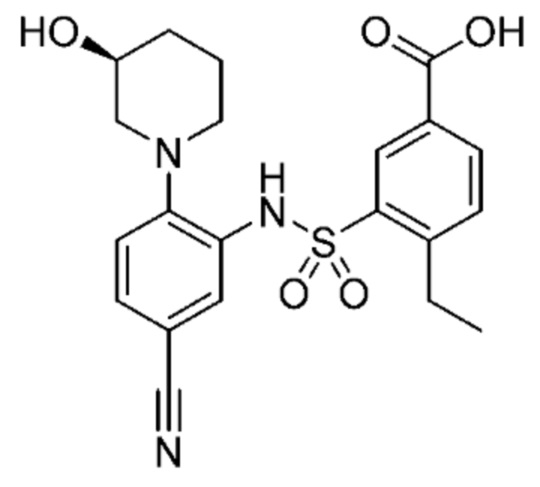
Стадия 1: (S)-4-(3-гидроксипиперидин-1-ил)-3-нитробензонитрил: Et3N (906 мкл, 6,50 ммоль) добавляли к раствору 4-фтор-3 нитробензонитрила (300 мг, 1,81 ммоль) и гидрохлорида (S)-пиперидин-3-ола (249 мг, 1,81 ммоль) в ДХМ (20 мл) и перемешивали полученный раствор при КТ в течение ночи. Добавляли 1М HCl (водн.) (2 мл) и выпаривали органическую фазу в вакууме с получением указанного в заголовке соединения (465 мг, 1,81 ммоль, выход 100%, чистота 96%) в виде темно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z 248,3 (M+H)+, 246,2 (M-H)- через 1,02 мин.
Стадия 2: (S)-3-амино-4-(3-гидроксипиперидин-1-ил)бензонитрил: Смесь продукта из стадии 1 выше (447 мг, 1,81 ммоль), порошка железа (2,48 г, 44,4 ммоль), хлорида аммония (116 мг, 2,17 ммоль), ИПС (15 мл) и воды (7,6 мл) перемешивали при 90°C в течение 20 ч. Смесь фильтровали через Celite®, промывали MeOH (25 мл) и выпаривали фильтрат в вакууме. Остаток разбавляли ДХМ (20 мл), сушили при пропускании через фазовый сепаратор, и неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (190 мг, 0,875 ммоль, выход 48%, чистота 100%) в виде темно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 218,3 (M+H)+ через 0,95 мин.
Стадия 3: (S)-метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Продукт из стадии 2 выше (55,1 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (42,7 мг, 0,094 ммоль, выход 37%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 444,4 (M+H)+, 442,3 (M-H)- через 1,50 мин.
Стадия 4: (S)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: Продукт из стадии 3 выше (40 мг, 0,090 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (361 мкл, 0,361 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (34,7 мг, 0,078 ммоль, выход 87%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 430,3 (M+H)+, 428,2 (M-H)- через 1,33 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,34 (шс, 1H), 9,76 (шс, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,48 (дд, J=8,3, 1,9 Гц, 1H), 7,31 (д, J=1,9 Гц, 1H), 7,15 (д, J=8,3 Гц, 1H), 5,16 (с, 1H), 3,70 (шс, 1H), 3,13-2,82 (м, 4H), 2,71-2,61 (м, 2H), 1,85-1,75 (м, 1H), 1,73-1,62 (м, 1H), 1,51-1,41 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 264: (S)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
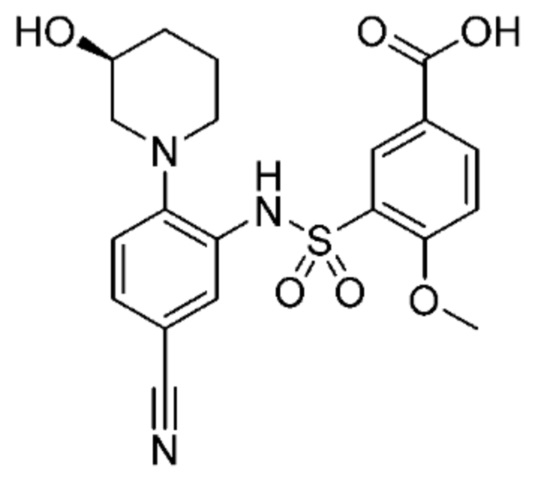
Стадия 1: (S)-метил-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензоат: Продукт из Примера 263, стадии 2 (54,7 мг, 0,252 ммоль) суспендировали в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (91,9 мг, 0,206 ммоль, выход 82%, чистота 100%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 446,3 (M+H)+, 444,3 (M-H)- через 1,31 мин.
Стадия 2: (S)-3-(N-(5-циано-2-(3-гидроксипиперидин-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 2 выше (89 мг, 0.200 ммоль) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (799 мкл, 0,799 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (79,6 мг, 0,179 ммоль, выход 90%, чистота 97%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 432,2 (M+H)+, 430,1 (M-H)- через 1,14 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,19 (шс, 1H), 9,24 (шс, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,16 (дд, J=8,7, 2,2 Гц, 1H), 7,45 (дд, J=8,3, 1,9 Гц, 1H), 7,41 (д, J=1,9 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 5,10 (с, 1H), 3,89 (с, 3H), 3,77-3,69 (м, 1H), 2,95-2,85 (м, 2H), 2,79-2,71 (м, 1H), 2,70-2,62 (м, 1H), 1,90-1,81 (м, 1H), 1,76-1,67 (м, 1H), 1,58-1,37 (м, 2H).
Пример 265: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота
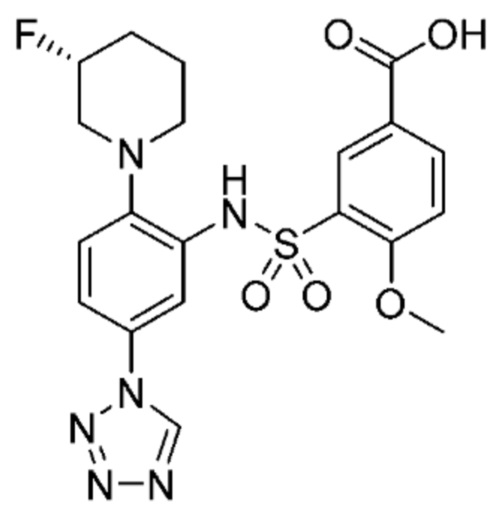
Стадия 1: (R)-3-фтор-1-(2-нитро-4-(тетразол-1-ил)фенил)пиперидин: Et3N (449 мкл, 3,22 ммоль) добавляли к раствору продукта из Примера 214, стадии 1 (187 мг, 0,895 ммоль) и гидрохлорида (R)-3-фторпиперидина (125 мг, 0,895 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и сушили органическую фазу при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (281 мг, 0,895 ммоль, выход 100%, чистота 93%) в виде темно-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,20 мин.
Стадия 2: (R)-2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)анилин: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к суспензии продукта из стадии 1 выше (281 мг, 0,895 ммоль, чистота 93%) в EtOH (19 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 3 дней. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (241 мг, 0,863 ммоль, выход 96%, чистота 96%) в виде светло-желтого вязкого масла. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,14 мин.
Стадия 3: (R)-метил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензоат: Продукт из стадии 2 выше (66,1 мг, 0,237 ммоль, чистота 96%) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (73,0 мг, 0,144 ммоль, выход 61%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 491,3 (M+H)+, 489,2 (M-H)- через 1,31 мин.
Стадия 4: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-метоксибензойная кислота: Продукт из стадии 3 выше (71 мг, 0,141 ммоль, чистота 97%) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (579 мкл, 0,579 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (55,8 мг, 0,111 ммоль, выход 77%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 477,2 (M+H)+, 475,2 (M-H)- через 1,25 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,16 (шс, 1H), 9,95 (с, 1H), 8,81 (шс, 1H), 8,42 (д, J=2,2 Гц, 1H), 8,14 (дд, J=8,7, 2,2 Гц, 1H), 7,79 (д, J=2,4 Гц, 1H), 7,54 (дд, J=8,6, 2,5 Гц, 1H), 7,47 (д, J=8,6 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 4,95-4,79 (м, 1H), 3,93 (с, 3H), 3,07-2,70 (м, 4H), 1,97-1,74 (м, 3H), 1,74-1,64 (м, 1H).
Пример 266: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
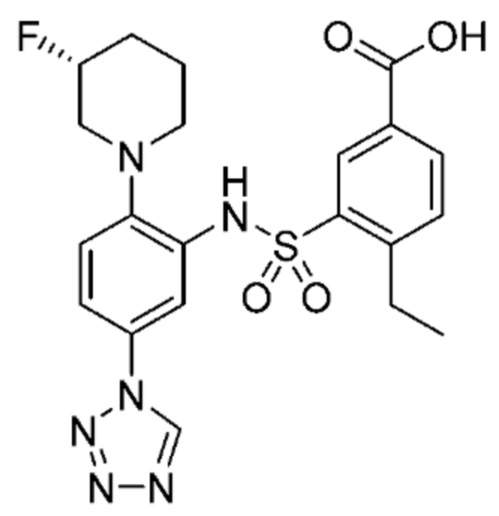
Стадия 1: (R)-метил-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 265, стадии 2 (66,6 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (35,6 мг, 0,071 ммоль, выход 28%, чистота 97%) в виде клейкого кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 489,3 (M+H)+, 487,3 (M-H)- через 1,58 мин.
Стадия 2: (R)-4-этил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 2 выше (33 мг, 0,068 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (270 мкл, 0,270 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (29,4 мг, 0,061 ммоль, выход 90%, чистота 98%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,2 (M-H)- через 1,43 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,31 (шс, 1H), 9,97 (с, 1H), 9,38 (шс, 1H), 8,41 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,72 (д, J=2,5 Гц, 1H), 7,62 (кажущ, d, J=8,0 Гц, 2H), 7,44 (д, J=8,6 Гц, 1H), 4,84-4,67 (м, 1H), 3,13-2,94 (м, 3H), 2,83-2,74 (м, 2H), 2,69-2,61 (м, 1H), 2,00-1,85 (м, 1H), 1,82-1,70 (м, 1H), 1,70-1,55 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 267: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
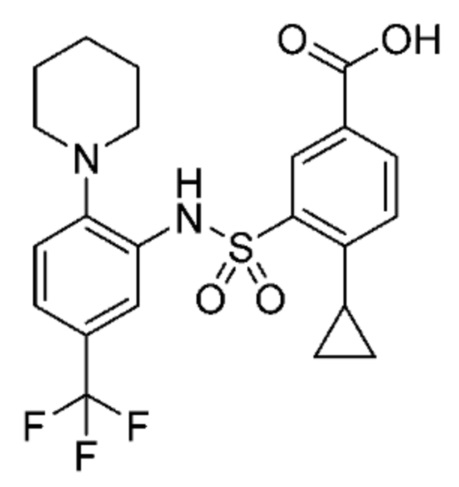
Стадия 1: метил-4-бром-3-(хлорсульфонил)бензоат: Смесь 4-бром-3-(хлорсульфонил)бензойной кислоты (500 мг, 1,67 ммоль) и SOCl2 (5 мл) нагревали с обратным холодильником в течение 4 ч. После охлаждения до КТ смесь выпаривали в вакууме, а остаток медленно добавляли к MeOH (10 мл) при 0°C. Смесь выпаривали в вакууме с получением указанного в заголовке соединения (758 мг, 1,45 ммоль, выход 87%, чистота 60%), содержащего примесь 4-бром-3-(хлорсульфонил)бензойной кислоты, в виде бежевого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,50-8,46 (м, 1H), 7,78-7,68 (м, 2H), 3,86 (с, 3H).
Стадия 2: метил-4-бром-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь 2-(пиперидин-1-ил)-5-(трифторметил)анилина (240 мг, 0,653 ммоль, чистота 60%), продукта из стадии 1 выше (341 мг, 1,09 ммоль) и пиридина (0,25 мл, 3,09 ммоль) в ДХМ (6,5 мл) перемешивали при КТ в течение 2 дней. Смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-30% EtOAc/изогексан) с получением указанного в заголовке соединения (325 мг, 0,605 ммоль, выход 93%, чистота 97%) в виде бежевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 521,1 (M+H)+, 519,0 (M-H)- через 2,00 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,49 (с, 1H), 8,46 (шс, 1H), 8,04 (шс, 2H), 7,47-7,41 (м, 1H), 7,38-7,34 (м, 1H), 7,34-7,28 (м, 1H), 3,88 (с, 3H), 2,77 (т, J=5,2 Гц, 4H), 1,62-1,54 (м, 4H), 1,51-1,44 (м, 2H).
Стадия 3: метил-4-бром-3-(N-(2-(пиперидин-1-ил)-5-(трифтор-метил)фенил)-N-((2-(триметилсилил)этокси)метил)сульфамоил)-бензоат: Суспензию NaH (36 мг, 0,900 ммоль, 60% об/об в минеральном масле) в ТГФ (5 мл) охлаждали до 0°C и медленно обрабатывали продуктом из стадии 2 выше (325 мг, 0,605 ммоль) в ТГФ (5 мл). Смесь нагревали до КТ и перемешивали в течение 1 ч, затем обрабатывали SEM-Cl (0,150 мл, 0,847 ммоль). Полученную смесь перемешивали при КТ в течение ночи. В смесь осторожно вливали воду (15 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические фазы промывали солевым раствором (15 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (275 мг, 0,418 ммоль, выход 69%, чистота 99%) в виде прозрачного бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 651,7 (M+H)+ через 2,30 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,44-8,40 (м, 1H), 8,11-8,04 (м, 2H), 7,69-7,63 (м, 1H), 7,34-7,28 (м, 2H), 5,45 (шс, 1H), 5,00 (шс, 1H), 3,86 (с, 3H), 3,38 (шс, 2H), 2,90-2,79 (м, 4H), 1,55 (шс, 4H), 1,53-1,48 (м, 2H), 0,67 (т, J=8,0 Гц, 2H), -0,16 (с, 9H).
Стадия 4: метил-4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Были подготовлены две реакции. Смесь продукта из стадии 3 выше (20 мг, 0,030 ммоль) и трибутил(циклопропил)станнана (20 мг, 0,060 ммоль) в диоксане (0,5 мл) барботировали N2 в течение 10 мин, после чего добавляли tBuXPhos Pd G3 (2,5 мг, 3,15 мкмоль). Смесь барботировали N2 в течение 5 мин, а затем нагревали с обратным холодильником и перемешивали в течение ночи. Такую же реакцию проводили с использованием продукта из стадии 3 выше (80 мг, 0,122 ммоль). Две реакционных смеси объединяли, концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (80 мг, 0,078 ммоль, выход 51%, чистота 47%) в виде смеси с метил-4-бутил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоатом (29% примеси). СВЭЖХ-МС (Метод 1) m/z 483,3 (M+H)+, 481,3 (M-H)- через 2,03 мин.
Стадия 5: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 4 выше (80 мг, 0,078 ммоль, чистота 47%) и LiOH (30 мг, 0,702 ммоль) в ТГФ/MeOH/воде (4:1:1, 2,4 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл) и pH доводили до ~pH 4 1М HCl (водн.). Водную фазу экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 60-90% MeCN в воде) с получением указанного в заголовке соединения (17,4 мг, 0,037 ммоль, выход 47%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,3 (M+H)+, 467,2 (M-H)- через 1,88 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,44 (д, J=1,9 Гц, 1H), 7,98 (д, J=7,8 Гц, 1H), 7,35-7,26 (м, 2H), 7,25-7,17 (м, 1H), 7,12 (д, J=8,2 Гц, 1H), 2,88-2,77 (м, 5H), 1,63-1,55 (м, 4H), 1,52-1,45 (м, 2H), 1,11-1,04 (м, 2H), 0,86-0,79 (м, 2H). Два обмениваемых протона не наблюдали.
Пример 268: 4-бутил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
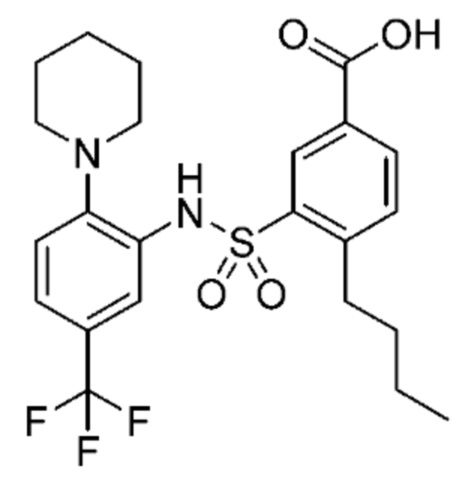
Указанное в заголовке соединение (11,7 мг, 0,023 ммоль, выход 53%, чистота 95%) получали в виде белого твердого вещества с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 60-90% MeCN в воде) в качестве побочного продукта реакции в Примере 267, стадии 5. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,2 (M-H)- через 2,05 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,39 (д, J=1,8 Гц, 1H), 8,03 (д, J=7,5 Гц, 1H), 7,53 (д, J=7,9 Гц, 1H), 7,43-7,28 (м, 2H), 7,26-7,15 (м, 1H), 2,92 (т, J=7,9 Гц, 2H), 2,81-2,66 (м, 4H), 1,61-1,41 (м, 8H), 1,39-1,29 (м, 2H), 0,87 (т, J=7,3 Гц, 3H). Два обмениваемых протона не наблюдали.
Пример 269: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота
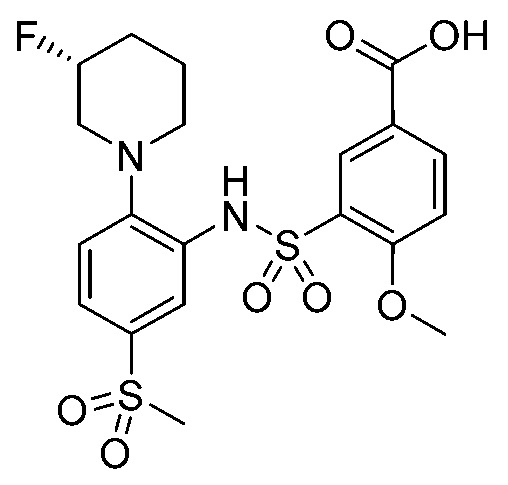
Стадия 1: (R)-метил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензоат: Продукт из Примера 262, стадии 2 (68,6 мг, 0,252 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (81 мкл, 1,01 ммоль) и обрабатывали раствором метил-3-(хлорсульфонил)-4-метоксибензоата (80 мг, 0,302 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 2 дней. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (95,6 мг, 0,191 ммоль, выход 76%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 501,3 (M+H)+, 499,2 (M-H)- через 1,37 мин.
Стадия 2: (R)-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-метоксибензойная кислота: продукт из стадии 2 выше (93 мг, 0,186 ммоль) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (743 мкл, 0,743 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 20 ч. Добавляли дополнительное количество 1М LiOH (водн.) (743 мкл, 0,743 ммоль) и перемешивали реакцию при 40°C еще в течение 3 дней. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C с получением указанного в заголовке соединения (69,4 мг, 0,134 ммоль, выход 72%, чистота 94%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 487,2 (M+H)+, 485,1 (M-H)- через 1,22 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (шс, 1H), 8,82 (шс, 1H), 8,36 (д, J=2,2 Гц, 1H), 8,15 (дд, J=8,7, 2,2 Гц, 1H), 7,65 (д, J=2,2 Гц, 1H), 7,54 (дд, J=8,4, 2,2 Гц, 1H), 7,41-7,27 (м, 2H), 4,94-4,78 (м, 1H), 3,92 (с, 3H), 3,15-2,91 (м, 3H), 2,89-2,81 (м, 1H), 1,98-1,74 (м, 3H), 1,73-1,61 (м, 1H). Три протона были скрыты растворителем.
Пример 270: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
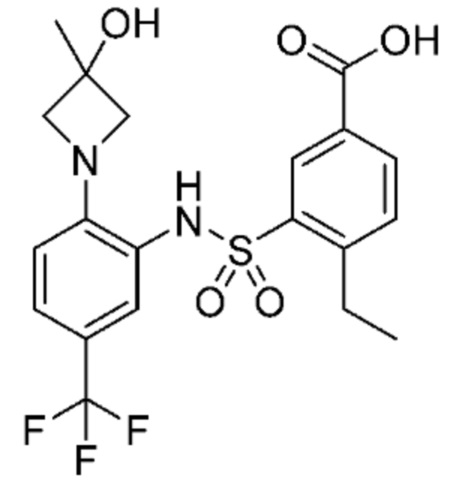
Стадия 1: 3-метил-1-(2-нитро-4-(трифторметил)фенил)-азетидин-3-ол: Et3N (720 мкл, 5,18 ммоль) добавляли к раствору 1-фтор-2-нитро-4-(трифторметил)бензола (201 мкл, 1,44 ммоль) и гидрохлорида 3-метилазетидин-3-ола (230 мг, 1,87 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 20 ч. Добавляли 1М HCl (водн.) (2 мл) и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (396 мг, 1,44 ммоль, выход 100%) в виде светло-оранжевого вязкого масла. СВЭЖХ-МС (Метод 2) m/z без ионизации через 1,34 мин.
Стадия 2: 1-(2-амино-4-(трифторметил)фенил)-3-метилазетидин-3-ол: 5% Pd/C (50% об/об вода) Тип 87L (107 мг, 0,025 ммоль) в EtOH (1 мл) добавляли к суспензии продукта из стадии 1 выше (396 мг, 1,44 ммоль) в EtOH (6,4 мл) при КТ. Реакционную смесь перемешивали при КТ под H2 (давление 4 бара) в течение 19 ч. Катализатор удаляли с помощью фильтрации через Celite® и промывали MeOH (20 мл). Фильтрат выпаривали в вакууме, а остаток суспендировали в ДХМ (5 мл) и сушили при пропускании через фазовый сепаратор. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (326 мг, 1,32 ммоль, выход 92%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 247,3 (M+H)+ через 1,11 мин.
Стадия 3: метил-4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (62,5 мг, 0,254 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль) и обрабатывали раствором продукта из Примера 203, стадии 2 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 20 ч. Неочищенный продукт непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (12,7 мг, 0,026 ммоль, выход 10%, чистота 95%) в виде светло-розового вязкого масла. СВЭЖХ-МС (Метод 1) m/z 473,4 (M+H)+, 471,3 (M-H)- через 1,52 мин.
Стадия 4: 4-этил-3-(N-(2-(3-гидрокси-3-метилазетидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (12,7 мг, 0,027 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (108 мкл, 0,108 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором, и перемешивали реакцию при 30°C в течение 2 дней. Добавляли дополнительное количество 1М LiOH (водн.) (108 мкл, 0,108 ммоль) и перемешивали реакцию при 40°C в течение 5 ч. Добавляли дополнительное количество 1М LiOH (водн.) (108 мкл, 0,108 ммоль) и перемешивали реакцию при 40°C в течение 3 дней. Добавляли дополнительное количество 1М LiOH (водн.) (108 мкл, 0,108 ммоль) и перемешивали реакцию при 40°C в течение 24 ч. Добавляли дополнительное количество 1М LiOH (водн.) (500 мкл, 0.500 ммоль) и перемешивали реакцию при 40°C в течение 24 ч. Реакционную смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и нейтрализовали до ~pH 6 1М HCl (водн.). Полученную комковатую суспензию обрабатывали ультразвуком с получением мутной суспензии, которую выпаривали в вакууме до ~2 мл. Осадок собирали с помощью фильтрации, промывая водой (2×2 мл). Твердое вещество суспендировали в MeCN (4 мл), выпаривали в вакууме и сушили при 50°C. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (4 мг, 8,46 мкмоль, выход 32%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 459,3 (M+H)+, 457,2 (M-H)- через 1,37 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,03 (шс, 1H), 9,60 (шс, 1H), 8,26 (д, J=1,8 Гц, 1H), 8,09 (д, J=7,7 Гц, 1H), 7,60 (д, J=7,5 Гц, 1H), 7,28 (шс, 1H), 6,49 (д, J=8,6 Гц, 1H), 6,33 (шс, 1H), 5,50 (с, 1H), 4,00 (д, J=8,1 Гц, 2H), 3,89 (д, J=8,1 Гц, 2H), 2,94 (к, J=7,4 Гц, 2H), 1,42 (с, 3H), 1,17 (т, J=7,4 Гц, 3H).
Пример 271: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(пиразол-1-ил)бензойная кислота
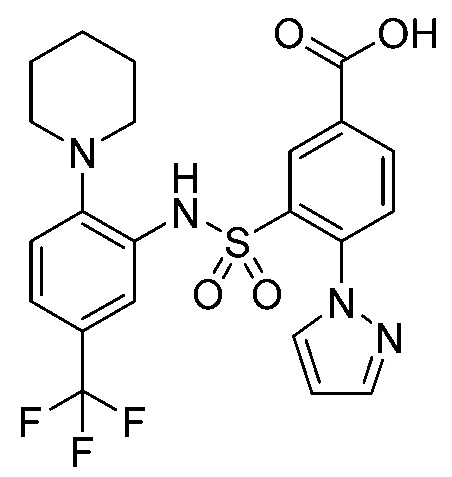
Стадия 1: метил-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(пиразол-1-ил)бензоат: К дегазированному раствору Примера 267, Стадии 3 (100 мг, 0,152 ммоль) в ДМСО (0,75 мл) добавляли пиразол (25,0 мг, 0,367 ммоль), CuI (10 мг, 0,053 ммоль), L-пролин (8,0 мг, 0,069 ммоль) и K2CO3 (75,0 мг, 0,543 ммоль). Смесь нагревали до 90°C в течение ночи. Смесь разбавляли водой (10 мл) и EtOAc (10 мл) и удаляли твердую фазу с помощью фильтрации. Фильтрат экстрагировали EtOAc (3×15 мл), затем органические фазы объединяли, промывали солевым раствором (10 мл) и сушили при пропускании через фазовый сепаратор. Растворитель удаляли в вакууме, а неочищенный продукт очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,30 г, 0,047 ммоль, выход 31%, чистота 80%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 509,2 (M+H)+, 507,2 (M-H)- через 1,98 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,68 (с, 1H), 10,00 (с, 1H), 8,51 (д, J=2,0 Гц, 1H), 8,45-8,35 (м, 1H), 8,31-8,25 (м, 1H), 8,02-7,96 (м, 1H), 7,85 (д, J=8,3 Гц, 1H), 7,81-7,77 (м, 1H), 7,46-7,40 (м, 1H), 7,38-7,33 (м, 1H), 6,69 (с, 1H), 2,74-2,66 (м, 4H), 1,63-1,55 (м, 4H), 1,52-1,46 (м, 2H).
Стадия 2: 3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-(пиразол-1-ил)бензойная кислота: Смесь продукта из стадии 1 выше (30 мг, 0,047 ммоль) и LiOH (5,6 мг, 0,23 ммоль) в ТГФ/MeOH/воде (4:1:1, 1 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (9,0 мг, 0,018 ммоль, выход 37%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 495,2 (M+H)+, 493,1 (M-H)- через 1,33 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,68 (с, 1H), 10,00 (с, 1H), 8,51 (д, J=2,0 Гц, 1H), 8,45-8,35 (м, 1H), 8,31-8,25 (м, 1H), 8,02-7,96 (м, 1H), 7,85 (д, J=8,3 Гц, 1H), 7,81-7,77 (м, 1H), 7,46-7,40 (м, 1H), 7,38-7,33 (м, 1H), 6,69 (с, 1H), 2,74-2,66 (м, 4H), 1,63-1,55 (м, 4H), 1,52-1,46 (м, 2H).
Пример 272: 4-(оксетан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
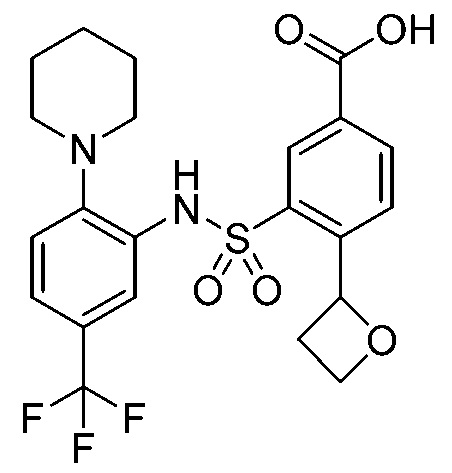
Стадия 1: метил-4-бром-2-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-N-((2-(триметилсилил)этокси)метил)-сульфамоил)бензоат: Раствор продукта из Примера 228, Стадии 1 (1,20 г, 2,30 ммоль) в ТГФ (10 мл, 122 ммоль) охлаждали до 0°C, затем обрабатывали гидридом натрия (0,138 г, 3,45 ммоль, 60% об/об в минеральном масле). Смесь нагревали до КТ и перемешивали в течение 1 ч, затем обрабатывали SEM-Cl (0,572 мл, 3,22 ммоль). Полученную смесь перемешивали при КТ в течение ночи, затем разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% МТБЭ/изогексан) с получением указанного в заголовке соединения (1,33 г, 1,8 ммоль, выход 80%, чистота 90%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 651,3 (M+H)+ через 2,30 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,11 (д, J=1,9 Гц, 1H), 8,04 (дд, J=8,2, 1,9 Гц, 1H), 7,71-7,64 (м, 1H), 7,62 (дд, J=8,3, 1,8 Гц, 1H), 7,30 (д, J=8,6 Гц, 1H), 7,27-7,21 (м, 1H), 5,76 (с, 2H), 3,61 (с, 3H), 3,52-3,42 (м, 2H), 3,00-2,75 (м, 4H), 1,54-1,48 (м, 6H), 0,84-0,76 (м, 2H), -0,13 (с, 9H).
Стадия 2: 5-бром-2-(гидроксиметил)-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-N-((2-(триметилсилил)этокси)метил)-бензолсульфонамид: Раствор продукта из стадии 1 выше (1,20 г, 1,66 ммоль, чистота 90%) в ТГФ (20 мл, 244 ммоль) охлаждали до 0°C, затем обрабатывали 2,0М LiAlH4 в ТГФ (0,921 мл, 1,8 ммоль, чистота 90%). Смесь перемешивали при 0°C в течение 1 ч. В смесь осторожно вливали воду (20 мл) и экстрагировали EtOAc (100 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-20% МТБЭ/изогексан) с получением указанного в заголовке соединения (0,685 г, 1,00 ммоль, выход 60%, чистота 90%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 622,9 (M+H)+ через 2,25 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,98-7,88 (м, 2H), 7,80 (д, J=8,3 Гц, 1H), 7,70-7,61 (м, 1H), 7,29 (д, J=8,6 Гц, 1H), 7,19 (д, J=2,3 Гц, 1H), 5,48 (т, J=5,3 Гц, 1H), 4,64 (д, J=45,8 Гц, 2H), 3,32 (с, 2H), 2,97-2,92 (м, 4H), 1,62-1,45 (м, 6H), 0,90-0,80 (м, 2H), 0,77-0,68 (м, 2H), -0,15 (с, 9H).
Стадия 3: 5-бром-2-формил-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-N-((2-(триметилсилил)этокси)метил)-бензолсульфонамид: Продукт из Стадии 2 выше (0,685 г, 1,00 ммоль, чистота 90%) в ДХМ (20 мл, 311 ммоль) обрабатывали MnO2 (0,955 г, 10,9 ммоль). Смесь перемешивали при КТ в течение 2 ч, затем фильтровали через Celite® и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-20% МТБЭ/изогексан) с получением указанного в заголовке соединения (0,410 г, 0,60 ммоль, выход 61%, чистота 92%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 621,3 (M+H)+ через 2,35 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 10,10 (д, J=0,8 Гц, 1H), 8,23-8,07 (м, 1H), 8,02 (д, J=2,0 Гц, 1H), 7,90 (д, J=8,3 Гц, 1H), 7,65 (дд, J=8,8, 2,3 Гц, 1H), 7,28 (д, J=8,6 Гц, 1H), 7,08 (д, J=2,3 Гц, 1H), 5,48 (д, J=10,7 Гц, 1H), 4,87 (д, J=10,8 Гц, 1H), 3,08-3,02 (шм, 4H), 1,65-1,52 (м, 6H), 0,93-0,78 (м, 2H), 0,73-0,62 (м, 2H), -0,15 (с, 9H).
Стадия 4: 5-бром-2-(оксетан-2-ил)-N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-N-((2-(триметилсилил)этокси)метил)-бензолсульфонамид: Иодид триметилсульфоксония (0,581 г, 2,64 ммоль) в трет-бутаноле (10 мл, 105 ммоль) обрабатывали KOtBu (0,296 г, 2,64 ммоль). Смесь перемешивали при 50°C в течение 30 мин, после чего обрабатывали продуктом из Стадии 3 выше (0,410 г, 0,607 ммоль, чистота 92%). Полученную смесь перемешивали при 50°C в течение ночи, затем фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-20% МТБЭ/изогексан) с получением указанного в заголовке соединения (0,165 г, 0,24 ммоль, выход 40%, чистота 95%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 649,3 в 2,40 мин.
Стадия 5: метил-4-(оксетан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)-N-((2-(триметилсилил)этокси)метил)-сульфамоил)бензоат: Раствор продукта из стадии 4 выше (0,160 г, 0,234 ммоль), триэтиламина (0,069 мл, 0,494 ммоль) и PdCl2(dppf)∙ДХМ (0,040 г, 0,049 ммоль) в MeOH (10 мл) перемешивали при 100°C под атмосферой CO (4 бара). Через 24 ч реакцию охлаждали, фильтровали через Celite® и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (0,085 г, 0,134 ммоль, выход 57%, чистота 99%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 629,4 (M+H)+ через 2,28 мин.
Стадия 6: 4-(оксетан-2-ил)-3-(N-(2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Раствор продукта из стадии 5 выше (0,085 г, 0,134 ммоль, чистота 99%) в ТГФ (5 мл, 0,13 ммоль) обрабатывали 1,0 М TBAF в ТГФ (0,135 мл, 0,135 ммоль) и перемешивали при КТ в течение 16 ч. Добавляли дополнительную порцию 1,0 М TBAF в ТГФ (0,135 мл, 0,135 ммоль) и нагревали полученную смесь при 40°C в течение 80 ч. Затем смесь обрабатывали раствором LiOH (9,71 мг, 0,40 ммоль) в воде (2 мл) и перемешивали при КТ в течение 3 ч. Смесь подкисляли 1,0 М лимонной кислотой (водн.) и экстрагировали EtOAc (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан, затем 0-10% MeOH/EtOAc) с получением указанного в заголовке соединения (10,3 мг, 0,020 ммоль, выход 15%, чистота 98%) в виде бесцветного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+ через 1,73 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,37 (с, 1H), 9,61 (с, 1H), 8,31-8,21 (м, 2H), 8,16 (д, J=8,0 Гц, 1H), 7,45 (д, J=8,5 Гц, 1H), 7,31 (д, J=2,2 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 6,34 (т, J=7,5 Гц, 1H), 4,69 (тд, J=7,9, 5,9 Гц, 1H), 4,57 (дт, J=9,1, 6,1 Гц, 1H), 3,19-3,09 (м, 1H), 2,79-2,67 (м, 2H), 2,60-2,53 (м, 2H), 2,42-2,32 (м, 1H), 1,54-1,46 (м, 4H), 1,45-1,38 (м, 2H).
Пример 273: 4-этил-3-(N-(5-(1-метил-1,2,3-триазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
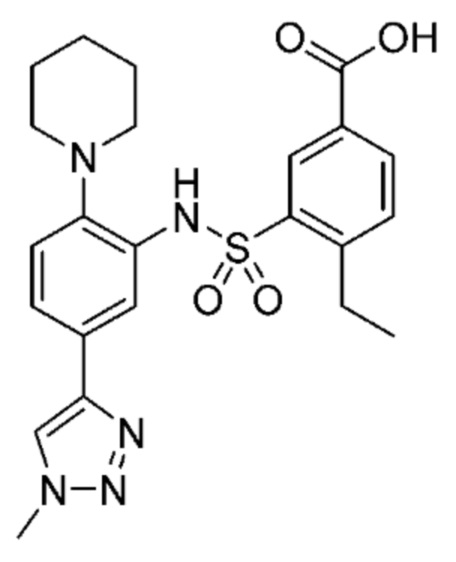
Стадия 1: 1-(4-бром-2-нитрофенил)пиперидин: Пиперидин (9,88 мл, 45,5 ммоль) добавляли к раствору 4-бром-1-фтор-2-нитробензола (5,60 мл, 45,5 ммоль) в MeCN (50 мл), а затем полученный раствор перемешивали при КТ в течение 2 ч. Раствор выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (13,4 г, 44,7 ммоль, выход 97%, чистота 94%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 285,1 (M+H)+ через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) 7,99 (д, J=2,4 Гц, 1H), 7,71 (дд, J=8,9, 2,5 Гц, 1H), 7,24 (д, J=8,9 Гц, 1H), 3,01-2,86 (м, 4H), 1,63-1,55 (м, 4H), 1,55-1,49 (м, 2H).
Стадия 2: 1-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (12,30 г, 43,1 ммоль, чистота 94%), бис(пинаколато)дибора (13,2 г, 51,8 ммоль), KOAc (12,7 г, 129 ммоль) и PdCl2(dppf)∙ДХМ (3,16 г, 4,31 ммоль) в диоксане (10 мл) дегазировали N2 в течение 15 мин и затем нагревали при 80°C в течение 16 ч. Смесь разбавляли водой (250 мл) и экстрагировали EtOAc (250 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (15,6 г, 44,7 ммоль, выход 97%, чистота 85%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 333,3 (M+H)+ через 1,99 мин.
1H-ЯМР (500 МГц, ДМСО-d6) δ 7,95 (д, J=1,6 Гц, 1H), 7,73 (дд, J=8,4, 1,6 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 3,10-2,99 (м, 4H), 1,67-1,49 (м, 6H), 1,29 (с, 12H).
Стадия 3: 2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Раствор продукта из стадии 2 выше (15,6 г, 35,2 ммоль, чистота 85%) в MeOH (20 мл, 494 ммоль) обрабатывали 10% Pd/C (3,75 г, 3,52 ммоль). Полученную смесь гидрировали (2 бара) в течение 16 ч, а затем фильтровали через Celite® и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (5,20 г, 12,0 ммоль, выход 41%, чистота 95%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 303,3 (M+H)+ через 1,41 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,04 (д, J=1,4 Гц, 1H), 6,90 (дд, J=7,7, 1,5 Гц, 1H), 6,84 (д, J=7,7 Гц, 1H), 4,63 (с, 2H), 2,80-2,70 (м, 4H), 1,65 (п, J=5,6 Гц, 4H), 1,55-1,49 (м, 2H), 1,26 (с, 12H).
Стадия 4: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 3 выше (3,53 г, 11,1 ммоль, чистота 95%), метил-3-(хлорсульфонил)-4-этилбензоата (3,20 г, 12,18 ммоль) и пиридина (3,60 мл, 44,5 ммоль) в ДХМ (20 мл) энергично перемешивали при КТ в течение 41 ч. Реакционную смесь наносили на Celite® и очищали с помощью хроматографии на силикагеле (80 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (4,89 г, 9,16 ммоль, выход 83%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 529,4 (M+H)+ через 2,12 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,04 (шс, 1H), 8,38 (д, J=1,9 Гц, 1H), 8,11 (дд, J=8,0, 1,9 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,34 (дд, J=7,9, 1,5 Гц, 1H), 7,31 (д, J=1,4 Гц, 1H), 7,07 (д, J=7,9 Гц, 1H), 3,85 (с, 3H), 3,05 (к, J=7,4 Гц, 2H), 2,68 (кажущ. т, J=5,2 Гц, 4H), 1,59-1,49 (м, 4H), 1,49-1,41 (м, 2H), 1,24-1,18 (м, 15H).
Стадия 5: метил-4-этил-3-(N-(5-(1-метил-1,2,3-триазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из стадии 4 выше (0,150 г, 0,28 ммоль, чистота 99%), 4-бром-1-метил-1,2,3-триазол (0,055 г, 0,34 ммоль), K3PO4 (0,078 г, 0,37 ммоль) в диоксане (5 мл, 0,28 ммоль) и воду (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь барботировали N2 в течение 15 минут, а затем нагревали при 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,106 г, 0,22 ммоль, 77% выход) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 484,4 (M+H)+ через 1,67 мин.
Стадия 6: 4-этил-3-(N-(5-(1-метил-1,2,3-триазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,658 мл, 0,658 ммоль) добавляли к раствору продукта из стадии 5 выше (0,106 г, 0,219 ммоль) в ТГФ (5 мл, 61,0 ммоль) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.), а образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (67,2 мг, 0,136 ммоль, выход 65%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 470,4 (M+H)+ через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,37 (шс, 1H), 9,26 (шс, 1H), 8,38 (д, J=1,9 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,71 (с, 1H), 7,61 (д, J=8,1 Гц, 1H), 7,32 (дд, J=8,2, 2,1 Гц, 1H), 7,29-7,24 (м, 2H), 3,95 (с, 3H), 3,07 (к, J=7,4 Гц, 2H), 2,66 (т, J=5,3 Гц, 4H), 1,62-1,55 (м, 4H), 1,51-1,44 (м, 2H), 1,23 (т, J=7,4 Гц, 3H).
Пример 274: 4-этил-3-(N-(5-(1-метил-1,2,3-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
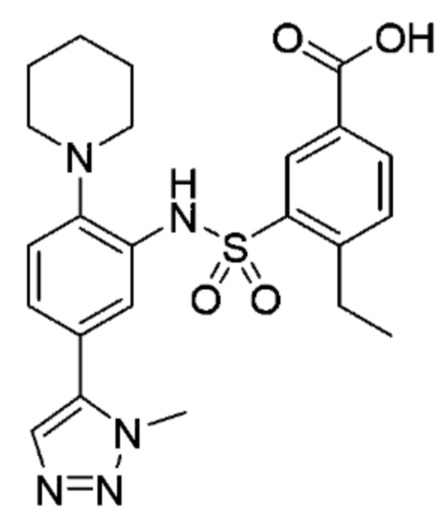
Стадия 1: метил-4-этил-3-(N-(5-(1-метил-1,2,3-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 5-бром-1-метил-1,2,3-триазол (0,055 г, 0,34 ммоль), K3PO4 (0,078 г, 0,36 ммоль) в диоксане (5 мл, 0,28 ммоль) и воду (1 мл), добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,115 г, 0,238 ммоль, выход 85%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 484,4 (M+H)+ через 1,65 мин.
Стадия 2: 4-этил-3-(N-(5-(1-метил-1,2,3-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,713 мл, 0,71 ммоль) добавляли в раствор продукта из стадии 1 выше (0,115 г, 0,238 ммоль) в ТГФ (5 мл) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (76,1 мг, 0,154 ммоль, выход 67%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 470.4 (M+H)+ через 1,55 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,99 (шс, 1H), 8,42 (д, J=1,8 Гц, 1H), 8,36 (с, 1H), 8,05 (дд, J=7,9, 1,8 Гц, 1H), 7,70 (д, J=2,0 Гц, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,49 (дд, J=8,2, 2,0 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 4,06 (с, 3H), 3,06 (к, J=7,4 Гц, 2H), 2,61 (т, J=5,2 Гц, 4H), 1,57 (п, J=5,4 Гц, 4H), 1,50-1,45 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 275: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тиазол-5-ил)фенил)сульфамоил)бензойная кислота
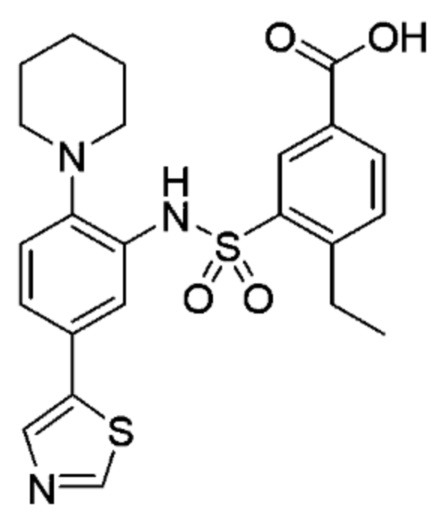
Стадия 1: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тиазол-5-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 5-бромтиазол (0,056 г, 0,34 ммоль), K3PO4 (0,078 г, 0,37 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 6 ч. Реакционной смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0.100 г, 0,19 ммоль, выход 69%, чистота 95%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 486,3 (M+H)+ через 1,80 мин.
Стадия 2: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(тиазол-5-ил)фенил)сульфамоил)бензойная кислота: 1,0 M LiOH (водн.) (0,587 мл, 0,587 ммоль) добавляли к раствору продукта из стадии 1 выше (0,100 г, 0,196 ммоль, чистота 95%) в ТГФ (5 мл) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.) и собирали образовавшийся осадок с помощью фильтрации с получением указанного в заголовке соединения (71,2 мг, 0,143 ммоль, выход 75%, чистота 97%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 472,3 (M+H)+ через 1,75 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,36 (шс, 1H), 9,16 (шс, 1H), 9,03 (с, 1H), 8,44 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 8,05 (с, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,42 (дд, J=8,3, 2,2 Гц, 1H), 7,28 (д, J=2,2 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,67 (т, J=5,2 Гц, 4H), 1,59 (п, J=5,7 Гц, 4H), 1,48 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 276: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразол-3-ил)фенил)сульфамоил)бензойная кислота
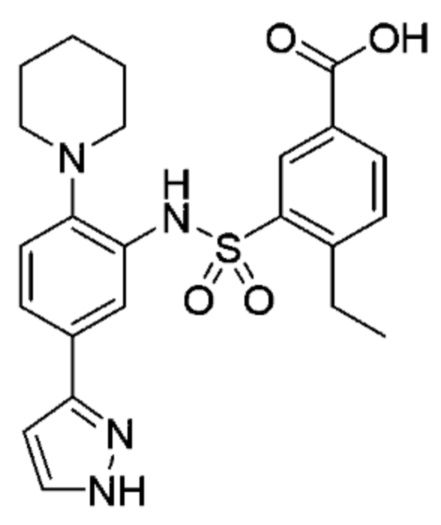
В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 3-бромпиразол (0,050 г, 0,34 ммоль), K3PO4 (0,078 г, 0,37 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 10 ч. Смесь обрабатывали 1М NaOH (водн.) (2 мл) и перемешивали в течение 1 ч при КТ, а затем экстрагировали МТБЭ (50 мл). Водную фазу нейтрализовали насыщенным NH4Cl (водн.) (1 мл) и экстрагировали EtOAc (20 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 10-80% MeCN в воде) с получением указанного в заголовке соединения (18,2 мг, 0,04 ммоль, выход 14%, чистота 99%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 455,4 (M+H)+ через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,13 (шс, 1H), 8,99 (с, 1H), 8,42 (д, J=1,8 Гц, 1H), 8,07 (дд, J=8,0, 1,8 Гц, 1H), 7,67 (с, 1H), 7,62-7,52 (м, 2H), 7,48 (дд, J=8,2, 2,0 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,43 (д, J=2,2 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,68-2,58 (м, 4H), 1,57 (п, J=5,4 Гц, 4H), 1,50-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 277: 4-этил-3-(N-(5-(1-метилпиразол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
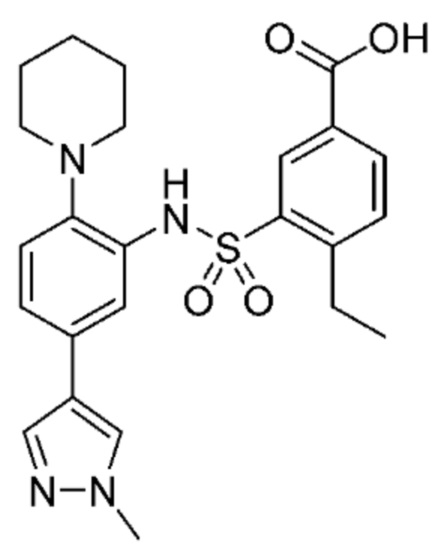
Стадия 1: метил-4-этил-3-(N-(5-(1-метилпиразол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 4-бром-1-метилпиразол (0,054 г, 0,34 ммоль), K3PO4 (0,078 г, 0,37 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (0,105 г, 0,214 ммоль, выход 76%, чистота 98%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 483,4 (M+H)+ через 1,76 мин.
Стадия 2: 4-этил-3-(N-(5-(1-метилпиразол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1,0 M LiOH (водн.) (0,640 мл, 0,640 ммоль) добавляли к раствору продукта из стадии 1 выше (0,105 г, 0,21 ммоль, чистота 98%) в ТГФ (5 мл) и перемешивали полученную смесь при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (41,9 мг, 0,085 ммоль, выход 41%, чистота 98%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,4 (M+H)+ через 1,64 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,40 (с, 1H), 8,92 (с, 1H), 8,48 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,91 (с, 1H), 7,59 (д, J=8,1 Гц, 1H), 7,58 (д, J=0,8 Гц, 1H), 7,28 (д, J=2,0 Гц, 1H), 7,21 (дд, J=8,2, 2,1 Гц, 1H), 7,14 (д, J=8,3 Гц, 1H), 3,84 (с, 3H), 3,06 (к, J=7,4 Гц, 2H), 2,59 (т, J=5,2 Гц, 4H), 1,59 (п, J=5,5 Гц, 4H), 1,52-1,45 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 278: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,3,4-тиадиазол-2-ил)фенил)сульфамоил)бензойная кислота
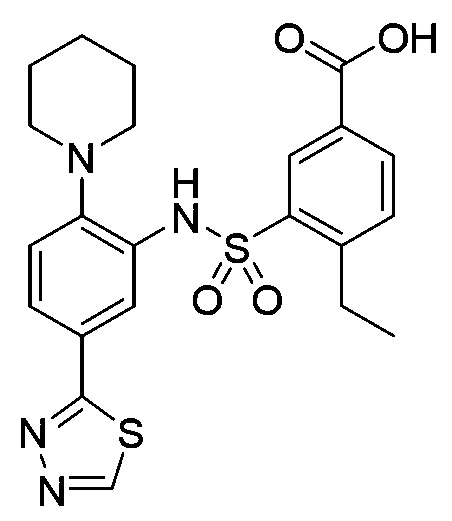
Стадия 1: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,3,4-тиадиазол-2-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 2-бром-1,3,4-тиадиазол (0,056 г, 0,34 ммоль), K3PO4 (0,078 г, 0,36 ммоль) в диоксане (4 мл, 0,28 ммоль) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Реакционной смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,021 г, 0,04 ммоль, выход 13%, 86% чистота) в виде бесцветного масла. СВЭЖХ-МС (Метод 1) m/z 487,3 (M+H)+ через 1,78 мин.
Стадия 2: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,3,4-тиадиазол-2-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,111 мл, 0,11 ммоль) добавляли к раствору продукта из стадии 1 выше (0,021 г, 0,04 ммоль, чистота 86%) в ТГФ (5 мл) и перемешивали полученную смесь при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (1,8 мг, 3,62 мкмоль, выход 10%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 473,3 (M+H)+ через 1,63 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,55 (шс, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,07 (д, J=8,1 Гц, 1H), 7,66 (д, J=2,0 Гц, 2H), 7,59 (д, J=8,0 Гц, 1H), 7,21 (д, J=8,1 Гц, 1H), 3,08 (к, J=7,4 Гц, 2H), 2,77 (т, J=5,8 Гц, 4H), 1,60-1,54 (м, 4H), 1,51-1,45 (м, 2H), 1,20 (т, J=7,4 Гц, 3H).
Пример 279: 4-этил-3-(N-(5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
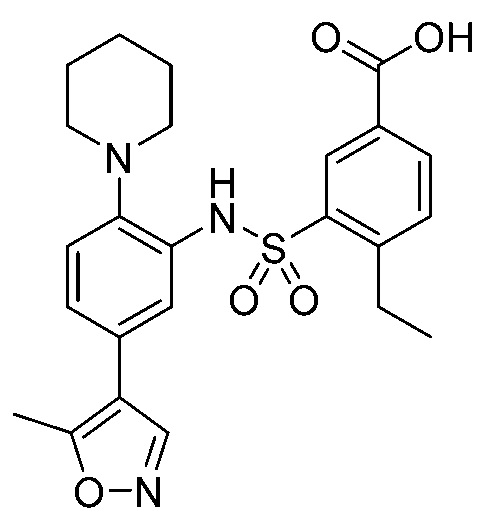
Стадия 1: метил-4-этил-3-(N-(5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 4-бром-5-метилизоксазол (0,055 г, 0,34 ммоль), K3PO4 (0,078 г, 0,36 ммоль) в диоксане (4 мл, 0,28 ммоль) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Реакционной смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-30% EtOAc/изогексан) с получением указанного в заголовке соединения (0,110 г, 0,22 ммоль, выход 76%, чистота 95%) в виде желтого масла. СВЭЖХ-МС (Метод 1) m/z 484,4 (M+H)+ через 1,90 мин.
Стадия 2: 4-этил-3-(N-(5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,682 мл, 0,682 ммоль) добавляли к раствору продукта из стадии 1 выше (0,110 г, 0,23 ммоль, чистота 95%) в ТГФ (5 мл) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением неочищенной смеси продуктов. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 10-80% MeCN в воде) с получением указанного в заголовке соединения (13,3 мг, 0,03 ммоль, выход 12%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 470,4 (M+H)+ через 1,79 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,69 (с, 1H), 8,43 (д, J=1,8 Гц, 1H), 8,08 (дд, J=7,9, 1,8 Гц, 1H), 7,64 (дд, J=7,8 Гц, 1H), 7,34-7,13 (м, 3H), 3,07 (к, J=7,4 Гц, 2H), 2,63 (т, J=5,2 Гц, 4H), 2,42 (с, 3H), 1,59 (п, J=5,4 Гц, 4H), 1,48 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 280: 4-этил-3-(N-(5-(1-метилпиразол-3-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
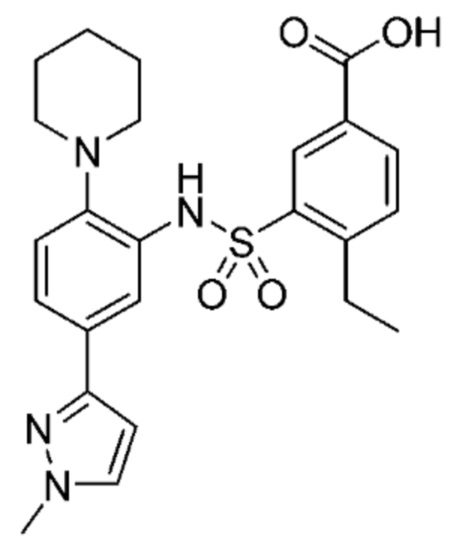
Стадия 1: метил-4-этил-3-(N-(5-(1-метилпиразол-3-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,150 г, 0,28 ммоль, чистота 99%), 3-бром-1-метилпиразол (0,035 мл, 0,34 ммоль), K3PO4 (0,078 г, 0,34 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,024 г, 0,04 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ, фильтровали и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,085 г, 0,16 ммоль, выход 57%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 483,3 (M+H)+ через 1,82 мин.
Стадия 2: 4-этил-3-(N-(5-(1-метилпиразол-3-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (2,82 мл, 0,582 ммоль) добавляли к раствору продукта из стадии 1 выше (0,085 г, 0,176 ммоль, чистота 92%) в ТГФ (3,5 мл) и перемешивали полученную смесь при КТ в течение 72 ч. Затем смесь подкисляли до pH 6 1М лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (44 мг, 0,089 ммоль, выход 51%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,2 (M+H)+ через 1,66 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,4 (шс, 1H), 8,91 (с, 1H), 8,45 (д, J=1,8 Гц, 1H), 8,07 (дд, J=8,0, 1,8 Гц, 1H), 7,66 (д, J=2,2 Гц, 1H), 7,60-7,53 (м, 2H), 7,43 (дд, J=8,2, 2,0 Гц, 1H), 7,15 (д, J=8,3 Гц, 1H), 6,43 (д, J=2,2 Гц, 1H), 3,84 (с, 3H), 3,06 (к, J=7,4 Гц, 2H), 2,63 (т, J=5,2 Гц, 4H), 1,63-1,55 (м, 4H), 1,51-1,44 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 281: 4-этил-3-(N-(5-(1-метилпиразол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
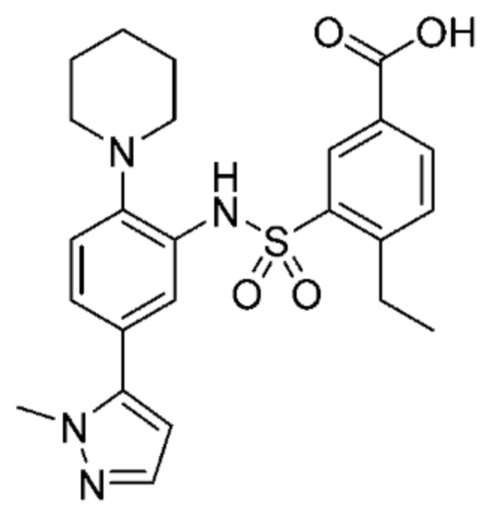
Стадия 1: метил-4-этил-3-(N-(5-(1-метилпиразол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,140 г, 0,26 ммоль, чистота 99%), 5-бром-1-метилпиразол (51,2 мг, 0,32 ммоль), K3PO4 (0,073 г, 0,34 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,022 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ, фильтровали и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,105 г, 0,22 ммоль, выход 82%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 483,6 (M+H)+ через 1,79 мин.
Стадия 2: 4-этил-3-(N-(5-(1-метилпиразол-3-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (3,45 мл, 3,49 ммоль) добавляли к раствору продукта из стадии 1 выше (0,105 г, 0,22 ммоль) в ТГФ (4 мл) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 1М лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (71 мг, 0,144 ммоль, выход 66%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,6 (M+H)+ через 1,60 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,37 (шс, 1H), 9,18 (с, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (д, J=8,1 Гц, 1H), 7,42 (д, J=1,9 Гц, 1H), 7,26-7,19 (м, 3H), 6,22 (д, J=1,9 Гц, 1H), 3,71 (с, 3H), 3,07 (к, J=7,4 Гц, 2H), 2,66 (т, J=5,3 Гц, 4H), 1,65-1,55 (м, 4H), 1,53-1,44 (м, 2H), 1,23 (т, J=7,4 Гц, 3H).
Пример 282: 4-этил-3-(N-(5-(2-метилтиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
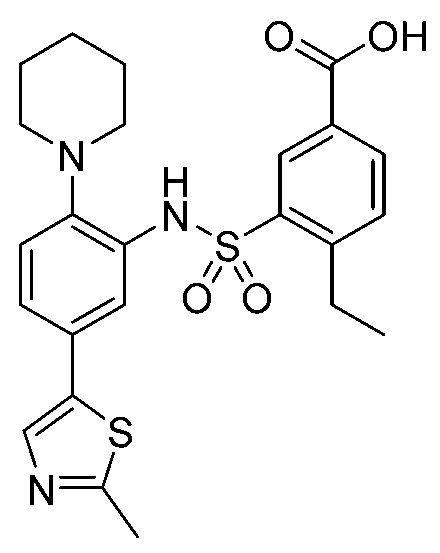
Стадия 1: метил-4-этил-3-(N-(5-(2-метилтиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из Примера 273, Стадии 4 (0,140 г, 0,27 ммоль, чистота 99%), 5-бром-2-метилтиазол (56,6 мг, 0,318 ммоль), K3PO4 (0,073 г, 0,36 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли XPhos Pd G3 (0,023 г, 0,03 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 3 ч. Реакционной смеси позволяли охладиться до КТ, фильтровали и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,096 г, 0,19 ммоль, выход 71%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 500,2 (M+H)+ через 1,92 мин.
Стадия 2: 4-этил-3-(N-(5-(2-метилтиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (2,88 мл, 2,88 ммоль) добавляли к раствору продукта из стадии 1 выше (0,096 г, 0,19 ммоль) в ТГФ (4 мл) и перемешивали при КТ в течение ночи. Затем смесь подкисляли до pH 6 1М лимонной кислотой (водн.), и образовавшийся осадок собирали с помощью фильтрации с получением материала, который подвергали азеотропной перегонке с MeCN (5 мл), с получением указанного в заголовке соединения (68 мг, 0,133 ммоль, выход 69%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 486,5 (M+H)+ через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,37 (шс, 1H), 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,36 (шс, 1H), 9,12 (с, 1H), 8,45 (д, J=1,8 Гц, 1H), 8,10 (дд, J=7,9, 1,9 Гц, 1H), 7,75 (с, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,34 (дд, J=8,3, 2,2 Гц, 1H), 7,21 (д, J=2,2 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,69-2,62 (м, 7H), 1,64-1,55 (м, 4H), 1,51-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 283: 4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
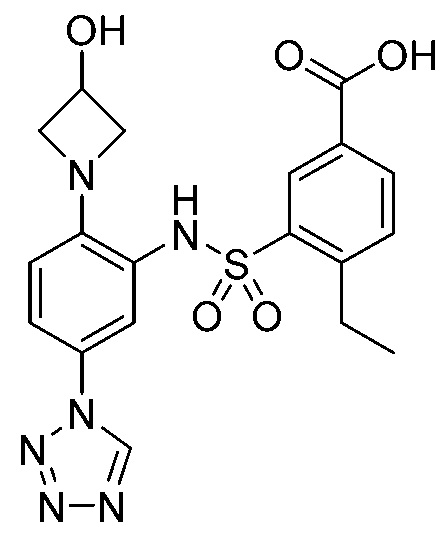
Стадия 1: 1-(2-нитро-4-(тетразол-1-ил)фенил)азетидин-3-ол: Et3N (720 мкл, 5,16 ммоль) добавляли к раствору продукта из Примера 214, Стадии 1 (300 мг, 1,434 ммоль) и гидрохлорида азетидин-3-ола (204 мг, 1,86 ммоль) в ДХМ (6 мл) и перемешивали полученный раствор при КТ в течение 3 дней. Реакционную смесь выпаривали в вакууме, а остаток растирали с водой (10 мл), фильтровали и промывали водой (2×10 мл). Твердое вещество сушили при 50°C в течение 4 ч с получением указанного в заголовке соединения (368 мг, 1,40 ммоль, выход 98%) в виде темно-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z без ионизации через 0,77 мин.
Стадия 2: 1-(2-амино-4-(тетразол-1-ил)фенил)азетидин-3-ол: Продукт из стадии 1 выше (368 мг, 1,40 ммоль) растворяли в MeOH (500 мл) и делили на три порции. К каждой порции добавляли 5% Pd/C (50% об/об воды) Тип 87L (140 мг, 0,033 ммоль) в MeOH (1 мл). Каждую реакционную смесь гидрировали под давлением 4 бара при 40°C в течение 6-20 ч. Реакционные смеси объединяли, фильтровали через Celite® и выпаривали в вакууме с получением указанного в заголовке соединения (316 мг, 1,32 ммоль, выход 94%, чистота 97%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 233,3 (M+H)+ через 0,56 мин.
Стадия 3: метил-4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из стадии 2 выше (58,9 мг, 0,246 ммоль, чистота 97%) суспендировали в смеси ДХМ (1 мл) и пиридина (82 мкл, 1,02 ммоль), затем обрабатывали раствором продукта из Примера 183, Стадии 1 (80 мг, 0,305 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 4 дней. Реакционную смесь очищали непосредственно с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (37 мг, 0,081 ммоль, выход 33%) в виде почти белого твердого вещества.
Стадия 4: 4-этил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (37 мг, 0,081 ммоль) растворяли в ТГФ (2 мл) и обрабатывали 1М LiOH (водн.) (323 мкл, 0,323 ммоль). По каплям добавляли MeOH, пока смесь не становилась раствором. Реакционную смесь перемешивали при 30°C в течение 20 ч. Смесь разбавляли водой (3 мл), выпаривали в вакууме и полученный водный раствор разбавляли водой (до ~5 мл). Водную фазу промывали EtOAc (2×5 мл) и затем нейтрализовали до ~pH 6 10% в/об лимонной кислотой (водн.). Образовавшийся осадок отфильтровывали и промывали водой (2×2 мл), затем сушили в вакууме при 50°C с получением указанного в заголовке соединения (7,1 мг, 0,015 ммоль, выход 19%, чистота 97%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 443,2 (M-H)- через 0,98 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,18 (шс, 1H), 9,72 (с, 1H), 8,32 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,9 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,59-7,49 (м, 1H), 6,76 (д, J=2,5 Гц, 1H), 6,63 (д, J=8,9 Гц, 1H), 5,61 (д, J=6,0 Гц, 1H), 4,53-4,46 (м, 1H), 4,33-4,28 (м, 2H), 3,75 (дд, J=8,6, 5,0 Гц, 2H), 2,98 (к, J=7,4 Гц, 2H), 1,19 (т, J=7,4 Гц, 3H). Один обмениваемый протон не наблюдали.
Пример 286: 4-этил-3-(N-(5-(изотиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
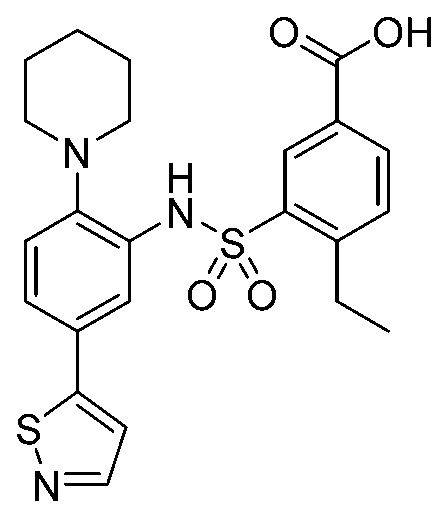
Стадия 1: Метил-4-этил-3-(N-(5-(изотиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 273, Стадии 4 (140 мг, 0,265 ммоль), 5-бромизотиазола (52,1 мг, 0,318 ммоль) и K3PO4 (73,1 мг, 0,344 ммоль) в диоксане (5 мл) и воде (1 мл) обрабатывали XPhos Pd G3 (22,4 мг, 0,026 ммоль). Реакционную смесь дегазировали N2 в течение 15 мин, затем нагревали при 80°C в течение 3 ч. Смеси позволяли охладиться до КТ, затем фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (121 мг, 0,239 ммоль, выход 90%, чистота 96%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 486,3 (M+H)+ через 1,94 мин.
Стадия 2: 4-этил-3-(N-(5-(изотиазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (2,5 мл, 2,50 ммоль) добавляли к раствору продукта из стадии 1 выше (121 мг, 0,239 ммоль, чистота 96%) в ТГФ (5 мл) и перемешивали раствор при КТ в течение ночи. Добавляли дополнительное количество 1М LiOH (водн.) (1,75 мл, 1,75 ммоль) и перемешивали раствор в течение ночи. Смесь доводили до pH 6 1М лимонной кислотой (водн.), осадок собирали с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (37,8 мг, 0,076 ммоль, выход 32%, чистота 95%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 472,5 (M+H)+, 470,2 (M-H)- через 1,85 мин. 1H-ЯМР (500 МГц, ДМСО-d6) 13,3 (шс, 1H), 9,26 (шс, 1H), 8,53 (д, J=1,8 Гц, 1H), 8,45 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,55 (д, J=1,8 Гц, 1H), 7,48 (дд, J=8,3, 2,2 Гц, 1H), 7,29 (д, J=2,2 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 3,08 (к, J=7,4 Гц, 2H), 2,71 (т, J=5,3 Гц, 4H), 1,64-1,55 (м, 4H), 1,53-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 287: 4-этил-3-(N-(5-(1-метилимидазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
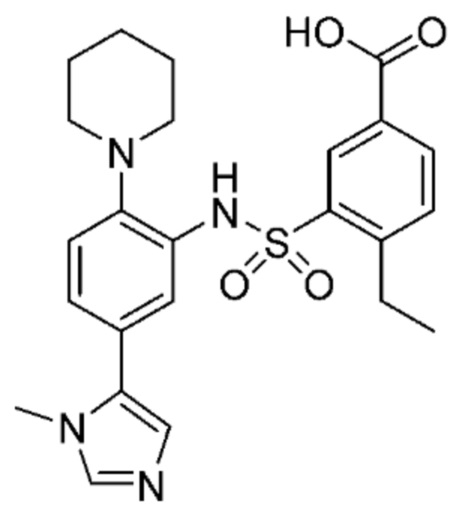
Стадия 1: 1-(4-бром-2-нитрофенил)пиперидин: Пиперидин (9,88 мл, 100 ммоль) добавляли к раствору 4-бром-1-фтор-2-нитробензола (5,60 мл, 45,5 ммоль) в MeCN (50 мл) и перемешивали полученный раствор при КТ в течение 2 ч. Реакционную смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (13,4 г, 44,2 ммоль, выход 97%, чистота 94%) в виде красного масла. СВЭЖХ-МС (Метод 1): m/z 285,1 (M+H)+, в 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,99 (д, J=2,4 Гц, 1H), 7,71 (дд, J=8,9, 2,5 Гц, 1H), 7,24 (д, J=8,9 Гц, 1H), 3,01-2,86 (м, 4H), 1,63-1,55 (м, 4H), 1,55-1,49 (м, 2H).
Стадия 2: 1-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (1 г, 3,33 ммоль, чистота 94%), бис(пинаколато)дибора (1,27 г, 5,00 ммоль), KOAc (0,981 г, 10,0 ммоль), PdCl2(dppf) (0,244 г, 0,333 ммоль) в диоксане (10 мл) дегазировали N2 в течение 5 мин. Реакцию нагревали при 80°C в течение 12 ч. Затем смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (1,40 г, 3,16 ммоль, выход 95%, чистота 75%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 333,3 (M+H)+, в 1,99 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,95 (д, J=1,6 Гц, 1H), 7,73 (дд, J=8,4, 1,6 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 3,04 (т, J=5,1 Гц, 4H), 1,67-1,52 (м, 6H), 1,29 (с, 12H).
Стадия 3: 2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Раствор продукта из стадии 2 выше (15,6 г, 39,9 ммоль, чистота 75%) в MeOH (20 мл, 494 ммоль) обрабатывали 10% Pd/C (4,25 г, 3,99 ммоль). Раствор гидрировали под давлением 2 бара в течение 16 ч. Смесь фильтровали через Celite® и выпаривали фильтрат в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (5,20 г, 16,4 ммоль, выход 41%, чистота 95%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 303,3 (M+H)+, через 1,41 мин, чистота 95% (254 нм). 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,04 (д, J=1,4 Гц, 1H), 6,90 (дд, J=7,7, 1,5 Гц, 1H), 6,84 (д, J=7,7 Гц, 1H), 4,63 (с, 2H), 2,80-2,70 (м, 4H), 1,65 (п, J=5,6 Гц, 4H), 1,55-1,49 (м, 2H), 1,26 (с, 12H).
Стадия 4: Метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 3 выше (3,53 г, 11,1 ммоль, чистота 95%), продукта из Примера 203, Стадии 2 (3,20 г, 12,2 ммоль) и пиридина (3,60 мл, 44,5 ммоль) в ДХМ (20 мл) энергично перемешивали при КТ в течение 41 ч. Реакционную смесь выпаривали в вакууме на Celite®. Неочищенный продукт очищали с помощью хроматографии на силикагеле (80 г картридж, 0-30%, затем 100% EtOAc/изогексан) с получением указанного в заголовке соединения (4,89 г, 9,16 ммоль, выход 83%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 529,4 (M+H)+, 527,3 (M-H)- через 2,12 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,04 (шс, 1H), 8,38 (д, J=1,9 Гц, 1H), 8,11 (дд, J=8,0, 1,9 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,34 (дд, J=7,9, 1,5 Гц, 1H), 7,31 (д, J=1,4 Гц, 1H), 7,07 (д, J=7,9 Гц, 1H), 3,85 (с, 3H), 3,05 (к, J=7,4 Гц, 2H), 2,68 (т, J=5,2 Гц, 4H), 1,59-1,49 (м, 4H), 1,49-1,41 (м, 2H), 1,24-1,18 (м, 15H).
Стадия 5: метил-4-этил-3-(N-(5-(1-метилимидазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: В реакционный сосуд, содержащий продукт из стадии 4 выше (0,150 г, 0,284 ммоль, чистота 99%), 5-бром-1-метилимидазол (0,055 г, 0,341 ммоль), K3PO4 (0,078 г, 0,369 ммоль) и диоксан (5 мл, 0,284 ммоль) и воду (1 мл), добавляли XPhos Pd G3 (0,024 г, 0,028 ммоль). Полученную реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали до 80°C в течение 6 ч. Реакционной смеси позволяли охладиться до КТ, фильтровали и затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% 10% (MeOH/ДХМ) в ДХМ) с получением указанного в заголовке соединения (0,100 г, 0,204 ммоль, выход 72%, чистота 99%) в виде желтого масла. СВЭЖХ-МС (Метод 1): m/z 483,4 (M+H)+, 481,3 (M-H)- через 1,15 мин.
Стадия 6: 4-этил-3-(N-(5-(1-метилимидазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,622 мл, 0,622 ммоль) добавляли к раствору продукта из стадии 5 выше (0.100 г, 0,207 ммоль, чистота 99%) в ТГФ (5 мл, 61,0 ммоль) и перемешивали раствор при КТ в течение ночи. Затем смесь доводили до pH 6 лимонной кислотой с образованием осадка, который отфильтровывали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Основный (0,1% бикарбонат аммония), Основный, Waters X-Bridge Prep-C18, 5 мкм, колонка 19×50 мм, 20-50% MeCN в воде) с получением указанного в заголовке соединения (4,4 мг, 9,30 мкмоль, выход 5%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 469,4 (M+H)+, 467,3 (M-H)- через 1,08 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,08 (шс, 1H), 8,41 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,65 (с, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,26-7,13 (м, 3H), 6,86 (с, 1H), 3,54 (с, 3H), 3,06 (к, J=7,4 Гц, 2H), 2,71-2,59 (м, 4H), 1,60 (п, J=5,6 Гц, 4H), 1,52-1,45 (м, 2H), 1,23 (т, J=7,4 Гц, 3H). 1 обмениваемый протон не наблюдали.
Пример 288: 4-этил-3-(N-(5-(1-метил-1,2,4-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
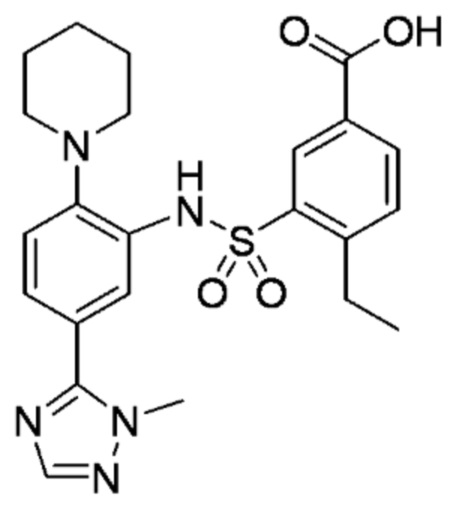
Стадия 1: 1-(4-бром-2-нитрофенил)пиперидин: Пиперидин (4,95 мл, 50,0 ммоль) добавляли к раствору 4-бром-1-фтор-2-нитробензола (2,79 мл, 22,7 ммоль) в MeCN (25 мл) и перемешивали полученный раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (120 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (6,8 г, 22,7 ммоль, выход 100%, чистота >95%) в виде красной жидкости. СВЭЖХ-МС (Метод 2): m/z 285,1 (M+H)+, через 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,00 (д, J=2,5 Гц, 1H), 7,71 (дд, J=8,9, 2,5 Гц, 1H), 7,25 (д, J=8,8 Гц, 1H), 2,98-2,92 (м, 4H), 1,63-1,50 (м, 6H).
Стадия 2: 1-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (6,8 г, 22,7 ммоль, чистота >95%), бис(пинаколато)дибора (9,08 г, 35,8 ммоль), KOAc (7,02 г, 71,5 ммоль), Pd(dppf)Cl2∙ДХМ (1,95 г, 2,39 ммоль) в 1,4-диоксане (70 мл) дегазировали азотом в течение 15 мин. Реакцию нагревали при 80°C в течение 12 ч. Смесь разбавляли солевым раствором (350 мл) и экстрагировали EtOAc (350 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (7,7 г, 16,2 ммоль, выход 72%, чистота 70%) в виде оранжевого масла. СВЭЖХ-МС (Метод 2): m/z 333,6 (M+H)+, через 1,98 мин.
Стадия 3: 1-(4-(1-метил-1,2,4-триазол-5-ил)-2-нитрофенил)-пиперидин: В реакционный сосуд, содержащий 5-бром-1-метил-1,2,4-триазол (0,102 г, 0,632 ммоль), продукт из стадии 2 выше (0,250 г, 0,527 ммоль, чистота 70%), K3PO4 (0,145 г, 0,685 ммоль) и диоксан:воду 5:1 (12 мл), добавляли XPhos Pd G3 (0,045 г, 0,053 ммоль). Полученную реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали до 80°C в течение 2 ч. Смесь разбавляли EtOAc (50 мл) и промывали водой (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,150 г, 0,517 ммоль, выход 98%, чистота 99%) в виде оранжевого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 288,2 (M+H)+, в 1,29 мин.
Стадия 4: 5-(1-метил-1,2,4-триазол-5-ил)-2-(пиперидин-1-ил)анилин: Продукт из стадии 3 выше (0,150 г, 0,522 ммоль) растворяли в MeOH, добавляли 10% Pd/C (на 10 мл) (50% об/об вода) Тип 39 (0,013 г, 6,11 мкмоль) и реакцию помещали под атмосферу водорода (2 бара) и перемешивали при комнатной температуре в течение 16 ч. Смесь фильтровали через Celite®, промывали MeOH (20 мл) и выпаривали фильтрат в вакууме с получением указанного в заголовке соединения (0,121 г, 0,353 ммоль, выход 68%, чистота 75%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 258,3 (M+H)+ через 0,88 мин.
Стадия 5: Метил-4-этил-3-(N-(5-(1-метил-1,2,4-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (0,121 г, 0,353 ммоль, чистота 75%) в ДХМ (3 мл) и пиридине (0,171 мл, 2,12 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (0,093 г, 0,353 ммоль) в ДХМ (3 мл) и перемешивали раствор при КТ в течение 48 ч. Смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,095 г, 0,187 ммоль, выход 53%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 484,4 (M+H)+, 482,3 (M-H)- через 1,64 мин.
Стадия 6: 4-этил-3-(N-(5-(1-метил-1,2,4-триазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,560 мл, 0,560 ммоль) добавляли к раствору продукта из стадии 5 выше (0,095 г, 0,187 ммоль, чистота 95%) в ТГФ (5 мл, 61,0 ммоль) и перемешивали раствор при КТ в течение 16 ч. Затем смесь доводили до pH 6 лимонной кислотой с образованием осадка, который отфильтровывали под вакуумом с получением указанного в заголовке соединения (46,8 мг, 0,098 ммоль, выход 52%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 470,3 (M+H)+, 468,3 (M-H)- через 1,49 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,37 (д, J=1,8 Гц, 1H), 8,06 (дд, J=8,0, 1,8 Гц, 1H), 7,92 (с, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,53-7,43 (м, 2H), 7,24 (д, J=8,2 Гц, 1H), 3,84 (с, 3H), 3,07 (к, J=7,4 Гц, 2H), 2,71 (т, J=5,2 Гц, 4H), 1,58 (п, J=5,5 Гц, 4H), 1,52-1,45 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). 2 обмениваемых протона не наблюдали.
Пример 289: 4-этил-3-(N-(5-(оксазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
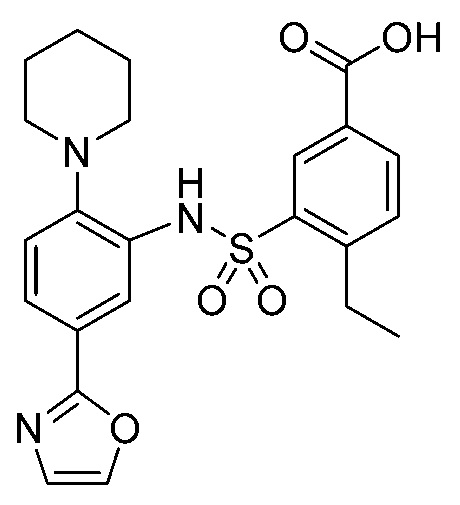
Стадия 1: 2-(3-нитро-4-(пиперидин-1-ил)фенил)оксазол: В реакционный сосуд, содержащий 2-бромоксазол (0,094 г, 0,632 ммоль), продукт из Примера 287, Стадии 2 (0,250 г, 0,527 ммоль), K3PO4 (0,145 г, 0,685 ммоль) и диоксан:воду 5:1 (12 мл), добавляли XPhos Pd G3 (0,045 г, 0,053 ммоль). Полученную реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали до 80°C в течение 2 ч. Смесь разбавляли EtOAc (50 мл) и промывали с водой (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,137 г, 0,481 ммоль, выход 91%, чистота 96%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1): m/z 274,2 (M+H)+, в 1,60 мин.
Стадия 2: 5-(оксазол-2-ил)-2-(пиперидин-1-ил)анилин: Продукт из стадии 1 выше (0,137 г, 0,501 ммоль) растворяли в MeOH (10 мл), добавляли 10% Pd/C (50% об/об вода) Тип 39 (0,013 г, 5,64 мкмоль) и перемешивали реакцию под водородом (2 бара) при комнатной температуре в течение 16 ч. Смесь фильтровали через Celite®, промывали MeOH (20 мл) и выпаривали в вакууме с получением указанного в заголовке соединения (0,119 г, 0,489 ммоль, выход 98%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 244?3 (M+H)+ через 1,18 мин.
Стадия 3: Метил-4-этил-3-(N-(5-(оксазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 2 выше (0,119 г, 0,489 ммоль) в ДХМ (3 мл) и пиридине (0,237 мл, 2,93 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (0,128 г, 0,489 ммоль) в ДХМ (3 мл, 46,6 ммоль) и перемешивали полученный раствор при КТ в течение 48 ч. Смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,140 г, 0,298 ммоль, выход 61%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 470,4 (M+H)+, 468,3 (M-H)- через 1,86 мин.
Стадия 4: 4-этил-3-(N-(5-(оксазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,894 мл, 0,894 ммоль) добавляли к раствору продукта из стадии 3 выше (0,140 г, 0,298 ммоль) в ТГФ (5 мл) и перемешивали раствор при КТ в течение 16 ч. Затем смесь доводили до pH 6 лимонной кислотой с образованием осадка, который отфильтровывали под вакуумом с получением указанного в заголовке соединения (95 мг, 0,207 ммоль, выход 70%, чистота 99%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 456,4 (M+H)+, 454,3 (M-H)- через 1,72 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,28 (шс, 1H), 9,27 (шс, 1H), 8,38 (д, J=1,8 Гц, 1H), 8,13 (с, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,75 (д, J=2,0 Гц, 1H), 7,69 (дд, J=8,3, 2,1 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,31 (с, 1H), 7,23 (д, J=8,4 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,70 (т, J=5,2 Гц, 4H), 1,56 (п, J=5,6 Гц, 4H), 1,50-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 290: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразин-2-ил)фенил)сульфамоил)бензойная кислота
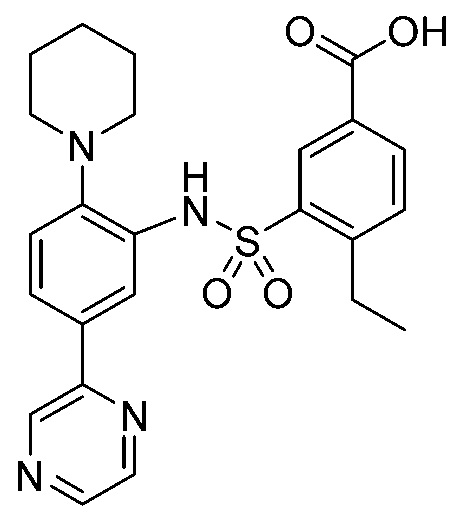
Стадия 1: 2-(3-нитро-4-(пиперидин-1-ил)фенил)пиразин: В реакционный сосуд с продуктом из Примера 287, Стадии 2 (0,250 г, 0,527 ммоль), 2-бромпиразином (0,084 г, 0,527 ммоль), K3PO4 (0,145 г, 0,685 ммоль) и диоксан:водой 5:1 (12 мл) добавляли XPhos Pd G3 (0,045 г, 0,053 ммоль). Полученную реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали до 80°C в течение 16 ч. Смесь разбавляли EtOAc (50 мл) и промывали с водой (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (0,145 г, 0,500 ммоль, выход 95%, чистота 98%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1): m/z 285,2 (M+H)+, в 1,57 мин.
Стадия 2: 2-(пиперидин-1-ил)-5-(пиразин-2-ил)анилин: Продукт из стадии 1 выше (0,145 г, 0,510 ммоль, чистота 98%) растворяли в MeOH и добавляли 10% Pd/C (на 10 мл) (50% об/об вода), Тип 39 (0,013 г, 6,11 мкмоль), и перемешивали реакцию под водородом (3 бара) при комнатной температуре в течение 1 ч. Смесь фильтровали через Celite®, промывали MeOH (20 мл) и выпаривали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,051 г, 0,201 ммоль, выход 39%) в виде желтого масла. СВЭЖХ-МС (Метод 1): m/z 255,3 (M+H)+ через 0,95 мин.
Стадия 3: Метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразин-2-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 2 выше (0,051 г, 0,201 ммоль) в ДХМ (3 мл) и пиридина (0,097 мл, 1,20 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (0,063 г, 0,241 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 72 ч. Смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,090 г, 0,176 ммоль, выход 88%, чистота 94%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 481,4 (M+H)+, 479,3 (M-H)- через 1,86 мин.
Стадия 4: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразин-2-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (0,562 мл, 0,562 ммоль) добавляли к раствору продукта из стадии 3 выше (0,090 г, 0,187 ммоль, чистота 94%) в ТГФ (5 мл) и перемешивали раствор при КТ в течение 16 ч. Затем смесь доводили до pH 6 лимонной кислотой с образованием осадка, который отфильтровывали под вакуумом и промывали водой (10 мл) с получением указанного в заголовке соединения (67 мг, 0,141 ммоль, выход 75%, чистота 98%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 467,3 (M+H)+, 465,3 (M-H)- через 1,70 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,28 (шс, 1H), 9,18 (шс, 1H), 9,04 (д, J=1,5 Гц, 1H), 8,62 (дд, J=2,5, 1,5 Гц, 1H), 8,55 (д, J=2,5 Гц, 1H), 8,44 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,91 (д, J=2,1 Гц, 1H), 7,85 (дд, J=8,4, 2,1 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,24 (д, J=8,4 Гц, 1H), 3,08 (к, J=7,4 Гц, 2H), 2,71 (т, J=5,2 Гц, 4H), 1,58 (к, J=5,6 Гц, 4H), 1,52-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 291: 3-(N-(5-(3,5-диметилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
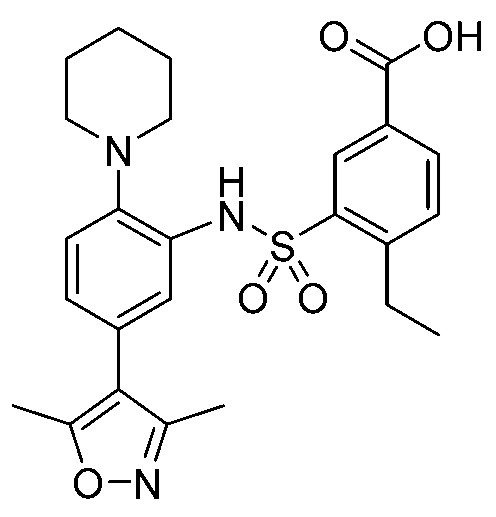
Стадия 1: 1-(4-бром-2-нитрофенил)пиперидин: Пиперидин (9,88 мл, 100 ммоль) добавляли к раствору 4-бром-1-фтор-2-нитробензола (5,60 мл, 45,5 ммоль) в MeCN (50 мл) и перемешивали полученный раствор при КТ в течение 2 ч. Реакционную смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (13,4 г, 44,2 ммоль, выход 97%, чистота 94%) в виде красного масла. СВЭЖХ-МС (Метод 1): m/z 285,1 (M+H)+, в 1,82 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,99 (д, J=2,4 Гц, 1H), 7,71 (дд, J=8,9, 2,5 Гц, 1H), 7,24 (д, J=8,9 Гц, 1H), 3,01-2,86 (м, 4H), 1,63-1,55 (м, 4H), 1,55-1,49 (м, 2H).
Стадия 2: 1-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (1 г, 3,33 ммоль, чистота 94%), бис(пинаколато)дибора (1,27 г, 5,00 ммоль), KOAc (0,981 г, 10,0 ммоль) и PdCl2(dppf) (0,244 г, 0,333 ммоль) в диоксане (10 мл) дегазировали N2 в течение 5 мин. Реакцию нагревали при 80°C в течение 12 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (1,40 г, 3,16 ммоль, выход 95%, чистота 75%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 333,3 (M+H)+ через 1,99 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,95 (д, J=1,6 Гц, 1H), 7,73 (дд, J=8,4, 1,6 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 3,04 (т, J=5,1 Гц, 4H), 1,67-1,52 (м, 6H), 1,29 (с, 12H).
Стадия 3: 2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Раствор продукта из стадии 2 выше (1,40 г, 3,16 ммоль, чистота 75%) в MeOH (20 мл) обрабатывали 10% Pd/C (0,336 г, 0,316 ммоль). Раствор гидрировали (2 бара) в течение 2 ч. Смесь фильтровали через Celite® и выпаривали фильтрат в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (80 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,695 г, 2,21 ммоль, выход 70%, чистота 96%) в виде воскообразного белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 303,3 (M+H)+, в 1,41 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,04 (д, J=1,4 Гц, 1H), 6,90 (дд, J=7,7, 1,5 Гц, 1H), 6,84 (д, J=7,7 Гц, 1H), 4,63 (с, 2H), 2,80-2,70 (м, 4H), 1,55-1,49 (м, 2H), 1,55-1,49 (м, 2H), 1,26 (с, 12H).
Стадия 4: 5-(3,5-диметилизоксазол-4-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (0,150 г, 0,496 ммоль, 96% чистота), 4-бром-3,5-диметилизоксазола (0,105 г, 0,596 ммоль) и K3PO4 (0,137 г, 0,645 ммоль) в диоксане (5 мл) и воде (1 мл) обрабатывали XPhos Pd G3 (0,042 г, 0,050 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали до 80°C в течение 2 ч. Реакционной смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0.100 г, 0,276 ммоль, выход 56%, чистота 75%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 272,3 (M+H)+, через 1,11 мин.
Стадия 5: Метил-3-(N-(5-(3,5-диметилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Раствор продукта из стадии 4 выше (0,10 г, 0,276 ммоль, чистота 75%) в ДХМ (3 мл) и пиридине (0,134 мл, 1,66 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (0,087 г, 0,332 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 72 ч. Смесь выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-40% EtOAc/изогексан) с получением указанного в заголовке соединения (0,110 г, 0,206 ммоль, выход 74%, чистота 93%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 498,4 (M+H)+, 496,3 (M-H)- через 1,94 мин.
Стадия 6: 3-(N-(5-(3,5-диметилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: 4М HCl (водн.) (0,276 мл, 1,11 ммоль) добавляли в раствор продукта из стадии 5 выше (0,110 г, 0,221 ммоль, 93%) в диоксане (5 мл) и перемешивали раствор при 60°C в течение 16 ч. Добавляли конц. HCl (водн.) (2 мл) и нагревали смесь до 70°C в течение 16 ч. Смесь выпаривали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии (40 г обращенно-фазовый картридж C18, 15-70% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (23 мг, 0,045 ммоль, выход 20%, чистота 97%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1): m/z 484,4 (M+H)+, 482,3 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,39 (д, J=1,8 Гц, 1H), 8,09 (дд, J=7,9, 1,9 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,23 (д, J=8,1 Гц, 1H), 7,09 (дд, J=8,1, 2,1 Гц, 1H), 7,06 (д, J=2,0 Гц, 1H), 3,10-3,02 (к, J=7,4 Гц, 2H), 2,64 (т, J=5,1 Гц, 4H), 2,27 (с, 3H), 2,09 (с, 3H), 1,60 (п, J=5,4 Гц, 4H), 1,53-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). 2 обмениваемых протона не наблюдали.
Пример 292: 4-этил-3-(N-(5-(5-метил-1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
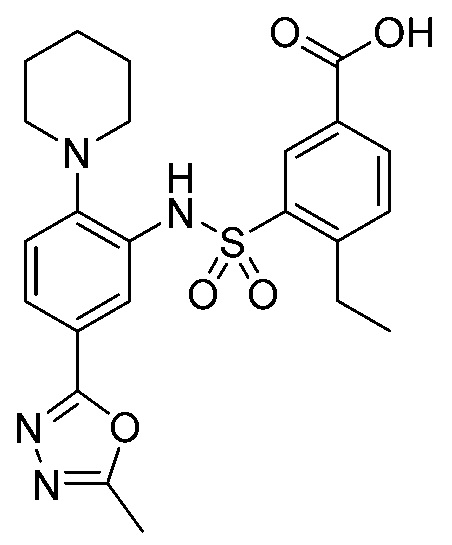
Стадия 1: 1-(4-бром-2-нитрофенил)пиперидин: Пиперидин (4,95 мл, 50 ммоль) добавляли к раствору 4-бром-1-фтор-2-нитробензола (2,79 мл, 22,7 ммоль) в MeCN (25 мл). Реакционную смесь перемешивали при КТ в течение ночи, после чего выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (120 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (6,8 г, 22,7 ммоль, выход 100%, чистота 95%) в виде красной жидкости. СВЭЖХ-МС (Метод 2) m/z 285,1 (M+H)+ через 1,82 мин.
Стадия 2: 1-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (6,8 г, 22,7 ммоль, чистота 95%), бис(пинаколато)дибора (9,08 г, 35,8 ммоль), KOAc (7,02 г, 71,5 ммоль) и PdCl2(dppf)∙ДХМ (1,95 г, 2,39 ммоль) в диоксане (70 мл) дегазировали N2 в течение 15 мин. Полученную смесь нагревали при 80°C в течение 12 ч. Смеси позволяли охладиться до КТ, а затем разбавляли солевым раствором (350 мл) и экстрагировали EtOAc (350 мл). Фазы разделяли и органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (220 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (7,7 г, 16,2 ммоль, выход 72%, чистота 70%) в виде оранжевого масла. СВЭЖХ-МС (Метод 2) m/z 333,6 (M+H)+ через 1,98 мин.
Стадия 3: 2-метил-5-(3-нитро-4-(пиперидин-1-ил)фенил)-1,3,4-оксадиазол: Смесь продукта из стадии 2 выше (360 мг, 0,759 ммоль, 70% чистота), 2-бром-5-метил-1,3,4-оксадиазола (212 мг, 1,3 ммоль) и K3PO4 (299 мг, 1,41 ммоль) в диоксане (10 мл) и воде (2 мл) обрабатывали XPhos Pd G3 (92 мг, 0,108 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ, затем фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (115 мг, 0,279 ммоль, выход 37%, чистота 70%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 289,5 (M+H)+ через 1,39 мин.
Стадия 4: 5-(5-метил-1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)анилин: Раствор продукта из стадии 3 выше (115 мг, 0,279 ммоль, чистота 70%) в EtOH (3 мл) обрабатывали 5% Pd/C (50% об/об вода) Тип 87L (42,4 мг, 0,029 ммоль). Полученную смесь гидрировали (1 бар) в течение 1 ч, фильтровали через Celite® и выпаривали фильтрат в вакууме с получением неочищенного продукта (75 мг). СВЭЖХ-МС (Метод 2) m/z 259,6 (M+H)+ через 1,33 мин.
Стадия 5: метил-4-этил-3-(N-(5-(5-метил-1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (75 мг) в ДХМ (1 мл) и пиридине (73,3 мкл, 0,906 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (43,6 мг, 0,166 ммоль, чистота 95%) в ДХМ (1 мл) и перемешивали полученный раствор при КТ в течение 2 дней. Растворитель удаляли в вакууме, а остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (20 мг, 0,039 ммоль, выход 14% в двух стадиях, чистота 95%) в виде бесцветного масла. СВЭЖХ-МС (Метод 2) m/z 485,3 (M+H)+ через 1,69 мин.
Стадия 6: 4-этил-3-(N-(5-(5-метил-1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (413 мкл, 0,413 ммоль) добавляли к раствору продукта из стадии 5 выше (20 мг, 0,039 ммоль, чистота 95%) в ТГФ (1 мл) и перемешивали полученную смесь при КТ в течение ночи. Реакционную смесь выпаривали в вакууме для удаления ТГФ, подкисляли до ~pH 6 1М лимонной кислотой (водн.) и собирали образовавшийся осадок с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (6 мг, 0,012 ммоль, выход 31%, чистота 95%) в виде бледно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,4 (M+H)+, 469,3 (M-H)- через 1,61 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,37 (шс, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,68 (дд, J=8,4, 2,1 Гц, 1H), 7,64-7,61 (м, 2H), 7,26 (д, J=8,4 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,75 (т, J=5,2 Гц, 4H), 2,53 (с, 3H), 1,57 (м, J=6,5 Гц, 4H), 1,52-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 293: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-3-ил)фенил)сульфамоил)бензойная кислота
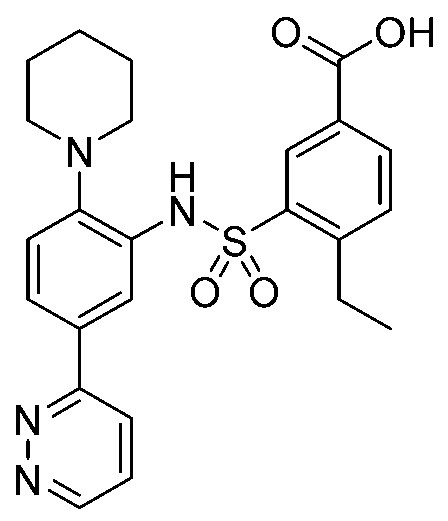
Стадия 1: 3-(3-нитро-4-(пиперидин-1-ил)фенил)пиридазин: Смесь продукта из Примера 287, Стадии 2 (360 мг, 0,759 ммоль, чистота 70%), 3-бромпиридазина (258 мг, 1,63 ммоль) и K3PO4 (299 мг, 1,41 ммоль) в диоксане (10 мл) и воде (2 мл) обрабатывали XPhos Pd G3 (92 мг, 0,108 ммоль). Реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение ночи. Добавляли дополнительную порцию 3-бромпиридазина (129 мг, 0,813 ммоль), XPhos Pd G3 (56 мг, 0,054 ммоль) и K3PO4 (150 мг, 0,75 ммоль) и нагревали смесь при 80°C в течение 4 дней. Смеси позволяли охладиться до КТ, затем фильтровали через Celite® и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (53 мг, 0,186 ммоль, выход 19%, чистота 80%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 285,2 (M+H)+ через 1,33 мин. Две партии продукта объединяли с получением указанного в заголовке соединения (95 мг, 0,294 ммоль, выход 19%, чистота 88%).
Стадия 2: 2-(пиперидин-1-ил)-5-(пиридазин-3-ил)анилин: Раствор продукта из стадии 1 выше (95 мг, 0,294 ммоль, чистота 88%) в EtOH (3 мл) обрабатывали 10% Pd/C (50% об/об воды) Тип 87L (35,6 мг, 0,023 ммоль). Полученную смесь гидрировали (1 бар) в течение 1,5 ч, фильтровали через Celite® и выпаривали фильтрат в вакууме с получением неочищенного продукта (83 мг). СВЭЖХ-МС (Метод 2) m/z 255,6 (M+H)+ через 1,21 мин.
Стадия 3: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-3-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 2 выше (83 мг) в ДХМ (1 мл) и пиридине (60,2 мкл, 0,744 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (35,8 мг, 0,129 ммоль, чистота 95%) в ДХМ (1 мл) и перемешивали полученный раствор при КТ в течение 72 ч. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (4 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (22 мг, 0,043 ммоль, выход 15% в двух стадиях, чистота 95%) в виде бесцветного масла. СВЭЖХ-МС (Метод 2) m/z 481,1 (M+H)+ через 2,59 мин.
Стадия 4: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-3-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (458 мкл, 0,458 ммоль) добавляли к раствору продукта из стадии 3 выше (22 мг, 0,043 ммоль, чистота 95%) в ТГФ (1 мл) и перемешивали полученную смесь при КТ в течение ночи. Реакционную смесь выпаривали в вакууме для удаления ТГФ, подкисляли до ~pH 6 1М лимонной кислотой (водн.), собирали осадок с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (11 мг, 0,022 ммоль, выход 52%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 467,3 (M+H)+, 465,3 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,16 (дд, J=4,9, 1,5 Гц, 2H), 8,40 (д, J=1,8 Гц, 1H), 8,07 (дд, J=8,0, 1,8 Гц, 1H), 8,03-7,98 (м, 2H), 7,85 (дд, J=8,4, 2,2 Гц, 1H), 7,72 (дд, J=8,7, 4,9 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 3,08 (к, J=7,5 Гц, 2H), 2,70 (т, J=5,2 Гц, 4H), 1,60-1,53 (м, 4H), 1,51-1,44 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 294: 3-(N-(4-бром-2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензойная кислота
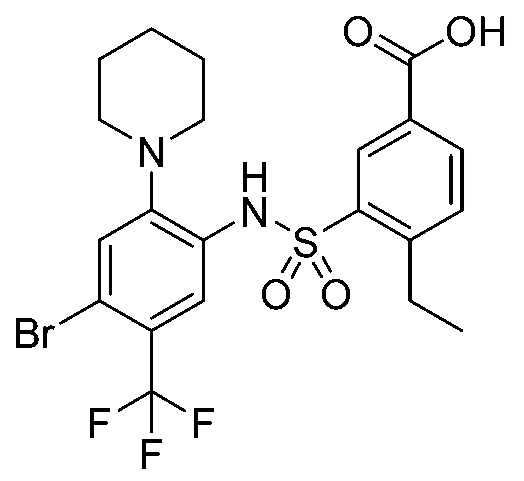
Стадия 1: 1-(5-бром-2-нитро-4-(трифторметил)фенил)-пиперидин: Раствор 1-бром-5-фтор-4-нитро-2-(трифторметил)бензола (300 мг, 1,04 ммоль) и пиперидина (250 мкл, 2,53 ммоль) в ДХМ (6 мл) оставляли при КТ на 1 ч. Реакционную смесь промывали 1М HCl (водн.) (2×2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме, подвергая азеотропной перегонке с толуолом (3 мл), с получением указанного в заголовке соединения (353 мг, 1,00 ммоль, 96% выход) в виде ярко-оранжевого масла, которое кристаллизовалось при стоянии. СВЭЖХ-МС (Метод 1) m/z 352,9 (M+H)+ через 1,96 мин.
Стадия 2: 4-бром-2-(пиперидин-1-ил)-5-(трифторметил)анилин: Продукт из стадии 1 выше (353 мг, 1,00 ммоль) объединяли с цинковой пылью (500 мг, 7,65 ммоль) и NH4Cl (тв) (410 мг, 7,66 ммоль) в ТГФ (9 мл) и воде (3 мл). Полученную смесь перемешивали при КТ в течение 4 дней, затем оставляли на 1 день. Смесь фильтровали через Celite®, промывали EtOAc (3×5 мл). Фазы разделяли, органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (315 мг, 0,799 ммоль, выход 80%, чистота 82%) в виде темно-оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 323,2 (M+H)+ через 1,96 мин.
Стадия 3: Метил-3-(N-(4-бром-2-(пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-этилбензоат: Продукт из стадии 2 выше (315 мг, 0,799 ммоль, чистота 82%) растворяли в смеси ДХМ (1 мл) и пиридина (150 мкл, 1,86 ммоль) и обрабатывали продуктом из Примера 203, Стадии 2 (250 мг, 0,952 ммоль). Полученный раствор оставляли при КТ на 18 ч, затем нагревали при 35°C в течение 4 дней. Смесь выпаривали в вакууме, а остаток растворяли в EtOAc (4 мл) и последовательно промывали водой (3 мл), насыщенным NaHCO3 (водн.) (3 мл) и солевым раствором (2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (363 мг, 0,614 ммоль, выход 77%, чистота 93%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 549,2 (M+H)+, 547,1 (M-H)- через 2,13 мин.
Стадия 4: 3-(N-(4-бром-2-(пиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-этилбензойная кислота: Продукт из стадии 3 выше (63 мг, 0,107 ммоль, чистота 93%) растворяли в ТГФ (1 мл) и обрабатывали 1М LiOH (водн.) (427 мкл, 0,427 ммоль). Полученный раствор оставляли при КТ на 18 ч. Смесь разбавляли водой (2 мл) и выпаривали в вакууме. Полученный водный раствор разбавляли водой (1 мл) и подкисляли до pH ~5 1М HCl (водн.). Образовавшийся осадок отфильтровывали, промывали водой (3×1 мл) и сушили в вакууме с получением рыжевато-коричневого твердого вещества (50 мг). Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (32 мг, 0,057 ммоль, выход 53%, чистота 95%) в виде рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 535,2 (M+H)+, 533,1 (M-H)- через 1,34 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,27 (шс, 1H), 9,68 (шс, 1H), 8,32 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,62 (д, J=8,0 Гц, 1H), 7,36 (с, 1H), 7,24 (с, 1H), 3,04 (к, J=7,4 Гц, 2H), 2,88-2,77 (м, 4H), 1,59-1,40 (м, 6H), 1,21 (т, J=7,4 Гц, 3H).
Пример 295: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
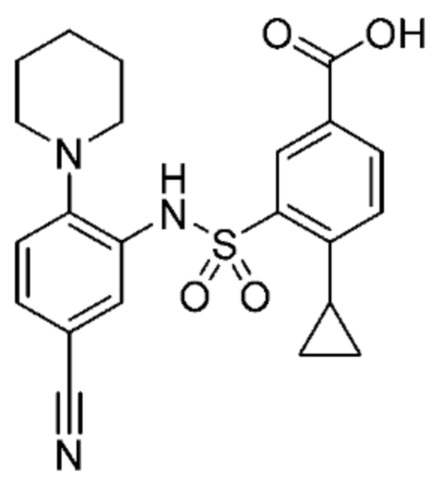
Стадия 1: Метил-4-бром-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 182, Стадии 2 (250 мг, 1,24 ммоль), продукта из Примера 316, Стадии 1 (433 мг, 1,37 ммоль) и пиридина (300 мкл, 3,71 ммоль) в ДХМ (7 мл) перемешивали при 35°C в течение 3 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (403 мг, 0,834 ммоль, выход 67%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 478,2 (M+H)+, 476,0 (M-H)- через 1,79 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,65 (с, 1H), 8,45-8,41 (м, 1H), 8,07-8,01 (м, 2H), 7,57 (дд, J=8,4, 2,0 Гц, 1H), 7,37 (д, J=2,0 Гц, 1H), 7,22 (д, J=8,4 Гц, 1H), 3,88 (с, 3H), 2,85-2,79 (м, 4H), 1,54-1,47 (м, 4H), 1,47-1,42 (м, 2H).
Стадия 2: Метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензоат: Продукт из Стадии 1 выше (403 мг, 0,834 ммоль, чистота 99%) и Pd-174 (61 мг, 85,0 мкмоль) в ТГФ (17 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (6,7 мл, 3,35 ммоль) и перемешивали смесь при КТ в течение 2 ч, а затем при 55°C в течение 3 ч. После охлаждения до КТ в смесь вливали MeOH (5 мл). Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (267 мг, 0,565 ммоль, выход 67%, чистота 93%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 440,3 (M+H)+, 438,2 (M-H)- через 1,81 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,53 (с, 1H), 8,37 (д, J=1,9 Гц, 1H), 8,04 (дд, J=8,3, 1,9 Гц, 1H), 7,54 (д, J=8,2 Гц, 1H), 7,24 (д, J=2,0 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 7,17 (д, J=8,5 Гц, 1H), 3,86 (с, 3H), 2,86-2,81 (м, 4H), 2,75-2,70 (м, 1H), 1,53-1,48 (м, 4H), 1,47-1,42 (м, 2H), 1,13-1,06 (м, 2H), 0,92-0,85 (м, 2H).
Стадия 3: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: смесь продукта из стадии 2 выше (267 мг, 0,565 ммоль, чистота 93%) и LiOH (97 мг, 2,26 ммоль) в ТГФ/MeOH/воде (4:1:1, 10,8 мл) перемешивали при 40°C в течение 4 дней. Смесь разбавляли водой (10 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток растворяли в ДХМ, концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ). Остаток растирали с МТБЭ с получением указанного в заголовке соединения (138 мг, 0,311 ммоль, выход 55%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 426,3 (M+H)+, 424,3 (M-H)- через 1,65 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,27 (с, 1H), 9,48 (с, 1H), 8,37 (д, J=1,9 Гц, 1H), 8,02 (дд, J=8,2, 1,9 Гц, 1H), 7,54 (дд, J=8,4, 2,0 Гц, 1H), 7,24 (д, J=2,0 Гц, 1H), 7,17 (дд, J=8,4, 1,9 Гц, 2H), 2,86-2,80 (м, 4H), 2,77-2,68 (м, 1H), 1,55-1,41 (м, 6H), 1,12-1,05 (м, 2H), 0,90-0,82 (м, 2H).
Пример 296: 4-циклопропил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
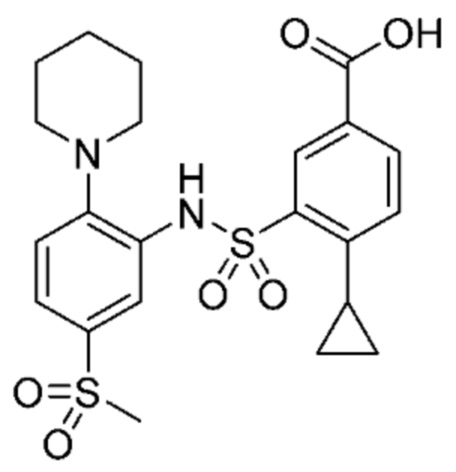
Стадия 1: Метил-4-бром-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 207, Стадии 2 (250 мг, 0,983 ммоль), продукта Примера 316, Стадии 1 (342 мг, 1,08 ммоль) и пиридина (240 мкл, 2,97 ммоль) в ДХМ (6 мл) перемешивали при 35°C в течение 4 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (326 мг, 0,583 ммоль, выход 59%, чистота 95%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 531,1 (M+H)+, 529,0 (M-H)- через 1,63 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,57 (с, 1H), 8,48-8,44 (м, 1H), 8,08-8,01 (м, 2H), 7,62 (дд, J=8,6, 2,2 Гц, 1H), 7,53 (д, J=2,2 Гц, 1H), 7,32 (д, J=8,6 Гц, 1H), 3,87 (с, 3H), 3,04 (с, 3H), 2,86-2,81 (м, 4H), 1,60-1,53 (м, 4H), 1,51-1,46 (м, 2H).
Стадия 2: Метил-4-циклопропил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Продукт из Стадии 1 выше (326 мг, 0,583 ммоль, чистота 95%) и Pd-174 (42 мг, 58,0 мкмоль) в ТГФ (12 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (4,7 мл, 2,35 ммоль) и перемешивали смесь при КТ в течение 2 ч, а затем при 55°C в течение 3 ч. После охлаждения до КТ в смесь вливали MeOH (5 мл). Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (284 мг, 0,461 ммоль, выход 79%, чистота 80%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 493,3 (M+H)+, 491,2 (M-H)- через 1,66 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,46 (с, 1H), 8,39 (д, J=1,9 Гц, 1H), 8,03 (дд, J=8,3, 1,9 Гц, 1H), 7,62-7,57 (м, 1H), 7,46 (д, J=2,2 Гц, 1H), 7,26 (д, J=8,5 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 3,85 (с, 3H), 3,00 (с, 3H), 2,87-2,82 (м, 4H), 2,80-2,72 (м, 1H), 1,58-1,52 (м, 4H), 1,50-1,45 (м, 2H), 1,12-1,08 (м, 2H), 0,91-0,85 (м, 2H).
Стадия 3: 4-циклопропил-3-(N-(5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 2 выше (284 мг, 0,461 ммоль, чистота 80%) и LiOH (79 мг, 1,85 ммоль) в ТГФ/MeOH/воде (4:1:1, 9 мл) перемешивали при 40°C в течение 4 дней. Смесь разбавляли водой (10 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (121 мг, 0,245 ммоль, выход 53%, чистота 97%) в виде белого твердого вещества после растирания с МТБЭ. СВЭЖХ-МС (Метод 1) m/z 479,2 (M+H)+, 477,2 (M-H)- через 1,50 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,26 (с, 1H), 9,41 (с, 1H), 8,39 (д, J=1,9 Гц, 1H), 8,01 (дд, J=8,3, 1,9 Гц, 1H), 7,59 (дд, J=8,4, 2,2 Гц, 1H), 7,47 (д, J=2,2 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 2,99 (с, 3H), 2,88-2,81 (м, 4H), 2,79-2,71 (м, 1H), 1,60-1,52 (м, 4H), 1,50-1,44 (м, 2H), 1,14-1,06 (м, 2H), 0,90-0,83 (м, 2H).
Пример 297: 4-этил-3-(N-(5-(изоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
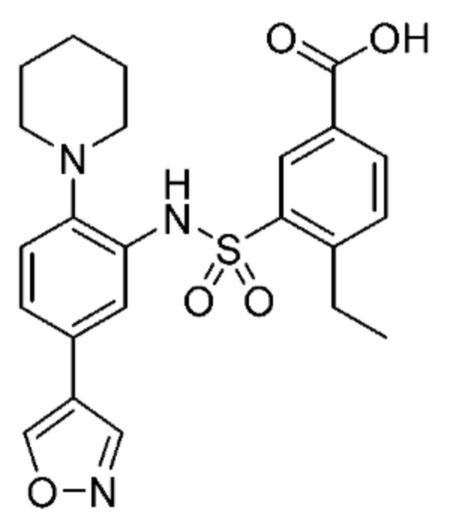
Стадия 1: 5-(изоксазол-4-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из Примера 273, Стадии 3 (250 мг, 0,827 ммоль), 4-бромизоксазола (147 мг, 0,993 ммоль), K3PO4 (228 мг, 1,08 ммоль) в диоксане (10 мл) и воде (2 мл) обрабатывали XPhos Pd G3 (70 мг, 0,083 ммоль). Реакционную смесь дегазировали N2 в течение 15 мин, затем нагревали при 80°C в течение ночи. Добавляли дополнительную порцию 4-бромизоксазола (74 мг, 0,50 ммоль), K3PO4 (114 мг, 0,540 ммоль) и XPhos Pd G3 (35 мг, 0,042 ммоль). Смесь нагревали при 80°C в течение ночи. Смеси позволяли охладиться до КТ, затем фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (90 мг, 0,277 ммоль, выход 34%, чистота 75%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 244,2 (M+H)+ через 1,51 мин.
Стадия 2: Метил-4-этил-3-(N-(5-(изоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 1 выше (90 мг, 0,277 ммоль, чистота 75%) в ДХМ (1 мл) и пиридине (180 мкл, 2,22 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (107 мг, 0,407 ммоль, чистота 95%) в ДХМ (1 мл) и перемешивали полученный раствор при КТ в течение 2 дней. Смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (139 мг, 0,237 ммоль, выход 85%, чистота 80%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 470,6 (M+H)+ через 1,85 мин.
Стадия 3: 4-этил-3-(N-(5-(изоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 4М HCl в диоксане (370 мкл, 1,48 ммоль) добавляли к раствору продукта из стадии 2 выше (139 мг, 0,237 ммоль, чистота 80%) в диоксане (3 мл) и перемешивали полученный раствор при 60°C в течение ночи. Добавляли воду (1 мл) и нагревали раствор при 60°C в течение ночи. Добавляли дополнительное количество 4М HCl в диоксане (370 μL, 1,48 ммоль) и воду (1 мл) и нагревали раствор при 60°C в течение 2 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии (13 г обращенно-фазовый картридж C18, 5-90% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (33 мг, 0,069 ммоль, выход 29%, чистота 95%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 456,3 (M+H)+, 454,2 (M-H)- через 1,76 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,27 (с, 1H), 9,08 (шс, 1H), 8,93 (с, 1H), 8,41 (д, J=1,8 Гц, 1H), 8,06 (дд, J=8,0, 1,9 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,44 (д, J=2,1 Гц, 1H), 7,39 (дд, J=8,2, 2,1 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 3,05 (к, J=7,4 Гц, 2H), 2,57 (т, J=5,2 Гц, 4H), 1,57-1,49 (м, 4H), 1,48-1,40 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 298: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,2,3-триазол-4-ил)фенил)сульфамоил)бензойная кислота
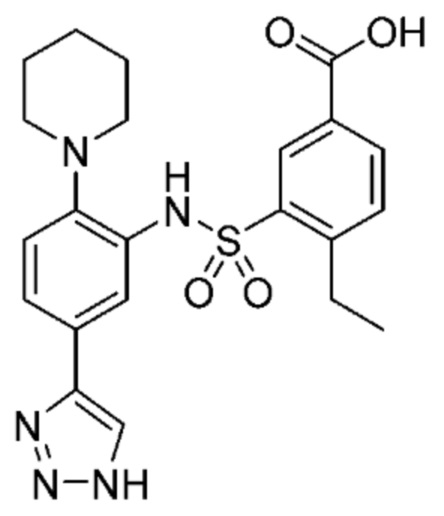
Стадия 1: 1-(2-нитро-4-((триметилсилил)этинил)фенил)-пиперидин: продукт из Примера 273, Стадии 1 (0,680 г, 2,39 ммоль) в сухом ТГФ (5 мл) обрабатывали Et3N (0,499 мл, 3,58 ммоль), Pd(PPh3)4 (0,055 г, 0,048 ммоль) и CuI (тв) (0,018 г, 0,095 ммоль), а затем этинилтриметилсиланом (0,219 мл, 3,10 ммоль). Полученную темную смесь перемешивали при КТ в течение 16 ч, затем нагревали при 70°C в течение 16 ч, а затем при 100°C в течение 16 ч. Смесь охлаждали, разбавляли водой (50 мл) и экстрагировали ДХМ (2×50 мл). Органические фазы объединяли и сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-10% МТБЭ в изогексане) с получением указанного в заголовке соединения (0,520 г, 1,31 ммоль, выход 55%, чистота 76%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 303,3 (M+H)+, в 2,17 мин.
Стадия 2: 1-(4-этинил-2-нитрофенил)пиперидин: Продукт из стадии 1 выше (0,520 г, 1,31 ммоль, чистота 76%) в сухом ТГФ (5 мл) обрабатывали 1,0 М TBAF (1,57 мл, 1,57 ммоль). Полученную смесь перемешивали при КТ в течение 16 ч, затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,240 г, 0,907 ммоль, выход 69%, чистота 87%) в виде коричневого масла. СВЭЖХ-МС (Метод 1): m/z 231,3 (M+H)+ через 1,72 мин.
Стадия 3: 1-(2-нитро-4-(1,2,3-триазол-4-ил)фенил)пиперидин: Продукт из Стадии 2 выше (0,240 г, 0,907 ммоль, чистота 87%) в сухом MeOH (1 мл) и ДМФА (9 мл) обрабатывали CuI (тв) (8,6 мг, 0,045 ммоль), а затем триметилсилилазидом (0,169 мл, 1,27 ммоль). Полученную смесь нагревали при 100°C в течение 16 ч, затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,090 г, 0,319 ммоль, выход 35%, чистота 97%) в виде красного масла. СВЭЖХ-МС (Метод 1): m/z 274,3 (M+H)+, 272,2 (M-H)- через 1,39 мин.
Стадия 4: 2-(пиперидин-1-ил)-5-(1,2,3-триазол-4-ил)анилин: Продукт из стадии 3 выше (0,090 г, 0,319 ммоль, чистота 97%) растворяли в MeOH (10 мл) и обрабатывали 10% Pd/C (50% об/об вода) Тип 39 (8,1 мг, 3,80 мкмоль). Полученную смесь гидрировали (2 бара) при КТ в течение 16 ч. Смесь фильтровали через Celite®, промывая MeOH (20 мл). Фильтрат выпаривали в вакууме с получением указанного в заголовке соединения (70 мг, 0,282 ммоль, выход 88%, чистота 98%) в виде бесцветного масла. СВЭЖХ-МС (Метод 1): m/z 244,3 (M+H)+ через 0,72 мин.
Стадия 5: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,2,3-триазол-4-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (70 мг, 0,282 ммоль, чистота 98%) в ДХМ (3 мл) и пиридине (0,047 мл, 0,575 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (0,076 г, 0,288 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 16 ч, затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (75 мг, 0,152 ммоль, выход 54%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 470,4 (M+H)+, 468,3 (M-H)- через 1,69 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,12 (шс, 1H), 8,41 (д, J=1,9 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,75-7,58 (м, 2H), 7,54 (д, J=8,1 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 3,83 (с, 3H), 3,13-2,97 (к, J=7,4 Гц, 2H), 2,66-2,58 (м, 4H), 1,60-1,53 (м, 4H), 1,50-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). Два обмениваемых протона не наблюдали.
Стадия 6: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(1,2,3-триазол-4-ил)фенил)сульфамоил)бензойная кислота: Раствор продукта из стадии 5 выше (75 мг, 0,152 ммоль, чистота 95%) в ТГФ (5 мл) обрабатывали 1М LiOH (водн.) (0,479 мл, 0,479 ммоль) и оставляли полученную смесь с перемешиванием при КТ на выходные. Затем смесь подкисляли до pH 7 10% в/об лимонной кислотой (водн.) и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии (12 г обращенно-фазовый картридж C18, 10-45% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (40,2 мг, 0,086 ммоль, выход 57%, чистота 98%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 456,4 (M+H)+, 454,3 (M-H)- через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,02 (шс, 1H), 8,42 (д, J=1,8 Гц, 1H), 8,12 (шс, 1H), 8,05 (дд, J=7,9, 1,8 Гц, 1H), 7,67 (с, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,53 (дд, J=8,3, 2,0 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 3,06 (к, J=7,4 Гц, 2H), 2,66-2,60 (м, 4H), 1,60-1,53 (м, 4H), 1,50-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 299: 3-(N-(5-(1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил) фенилсульфамоил)-4-этилбензойная кислота
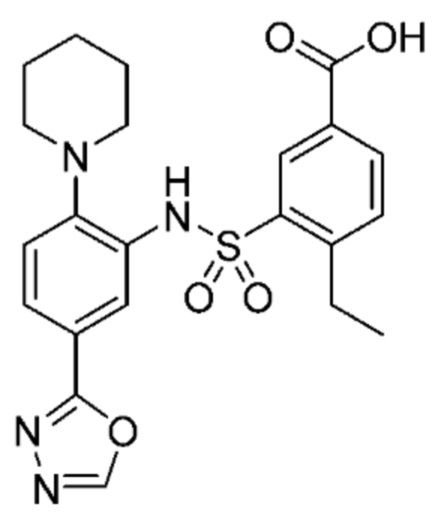
Стадия 1: метил-3-нитро-4-(пиперидин-1-ил)бензоат: метил-4-фтор-3-нитробензоат (500 мг, 2,51 ммоль) в сухом ДМФА (5 мл) обрабатывали пиперидином (744 мкл, 7,53 ммоль) и перемешивали при КТ в течение 2 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили (MgSO4), фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (650 мг, 2,31 ммоль, выход 92%, чистота 94%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 265,2 (M+H)+ через 1,62 мин.
Стадия 2: 3-нитро-4-(пиперид-1-ил)бензогидразид: Продукт из стадии 1 выше (650 мг, 2,31 ммоль, чистота 94%) в сухом EtOH (5 мл) обрабатывали гидразин-гидратом (1,04 мл, 11,5 ммоль) и нагревали при 80°C в течение 16 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-5% MeOH/ДХМ) с получением указанного в заголовке соединения (530 мг, 2,00 ммоль, выход 86%, чистота 99%) в виде оранжевого масла. СВЭЖХ-МС (Метод 1) m/z 265,2 (M+H)+ через 0,99 мин.
Стадия 3: 2-(3-нитро-4-(пиперидин-1-ил)фенил)-1,3,4-оксадиазол: Продукт из Стадии 2 выше (530 мг, 2,00 ммоль, чистота 99%) обрабатывали триэтилортоформиатом (5 мл) и нагревали смесь при 100°C в течение 16 ч. Смеси позволяли охладиться до КТ, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, MeOH/ДХМ на 0-5%) с получением указанного в заголовке соединения (486 мг, 1,75 ммоль, выход 87%, чистота 99%) в виде красного твердого вещества. СВЭЖХ-МС (Метод 1) m/z 275,3 (M+H)+ через 1,38 мин.
Стадия 4: 5-(1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)анилин: Продукт из стадии 3 выше (486 мг, 1,75 ммоль, чистота 99%) в MeOH (10 мл) обрабатывали 10% Pd/C (50% об/об воды) Тип 39 (45,0 мг, 0,180 ммоль). Реакционную смесь гидрировали (2 бара) при КТ в течение 16 ч. Смесь фильтровали через Celite®, промывая MeOH (20 мл), а затем выпаривали в вакууме с получением указанного в заголовке соединения (302 мг, 1,17 ммоль, выход 66%, чистота 95%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 245,3 (M+H)+ через 1,15 мин.
Стадия 5: метил-3-(N-(5-(1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Раствор продукта из стадии 4 выше (100 мг, 0,409 ммоль, чистота 95%) в ДХМ (3 мл) и пиридине (199 мкл, 2,45 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (108 мг, 0,409 ммоль) в ДХМ (1 мл) и перемешивали раствор при КТ в течение 4 дней. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (133 мг, 0,257 ммоль, выход 63%, чистота 91%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 471,4 (M+H)+, 469,3 (M-H)- через 1,72 мин.
Стадия 6: 3-(N-(5-(1,3,4-оксадиазол-2-ил)-2-(пиперидин-1-ил)фенилсульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (772 мкл, 0,772 ммоль) добавляли к раствору продукта из стадии 5 выше (133 мг, 0,257 ммоль, 91% чистота) в ТГФ (5 мл) и перемешивали смесь при КТ в течение ночи. Смесь подкисляли до pH 6 10% в/об лимонной кислотой (водн.) и выпаривали полученную смесь в вакууме. Остаток очищали с помощью хроматографии (12 г обращенно-фазовый картридж C18, 10-45% MeCN/0,1% муравьиная кислота (водн.)) с получением указанного в заголовке соединения (55,8 мг, 0,12 ммоль, выход 47%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 457,3 (M+H)+, 455,3 (M-H)- через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,24 (шс, 1H), 9,43 (шс, 1H), 9,24 (с, 1H), 8,37 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,80-7,68 (м, 2H), 7,62 (д, J=8,0 Гц, 1H), 7,27 (д, J=8,3 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,76 (т, J=5,2 Гц, 4H), 1,58-1,52 (м, 4H), 1,47 (м, 2H), 1,23 (т, J=7,4 Гц, 3H).
Пример 300: 4-этил-3-(N-(5-(изоксазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
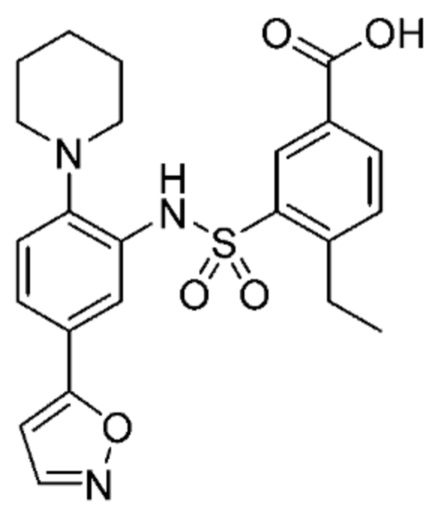
Стадия 1: 1-(3-нитро-4-(пиперидин-1-ил)фенил)этанон: Пиперидин (539 мкл, 5,46 ммоль) добавляли к раствору 1-(4-фтор-3-нитрофенил)этанона (1 г, 5,46 ммоль) и Et3N (2,28 мл, 16,4 ммоль) в MeCN (10 мл) и перемешивали полученный раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме с получением указанного в заголовке соединения (1,2 г, 4,64 ммоль, выход 85%, чистота 96%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 249,3 (M+H)+ через 1,48 мин.
Стадия 2: (E)-3-(диметиламино)-1-(3-нитро-4-(пиперидин-1-ил)фенил)проп-2-ен-1-он: Продукт из Стадии 1 выше (1,2 г, 4,64 ммоль, чистота 96%) обрабатывали диметилацеталем N, N-диметилформамида (10,0 мл, 4,64 ммоль) и нагревали смесь при 120°C в течение 16 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме с получением указанного в заголовке соединения (1,3 г, 4,07 ммоль, выход 88%, чистота 95%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 304,3 (M+H)+ через 1,39 мин.
Стадия 3: 5-(3-нитро-4-(пиперидин-1-ил)фенил)изоксазол: Продукт из стадии 2 выше (1,3 г, 4,07 ммоль, чистота 95%) объединяли с гидрохлоридом гидроксиламина (339 мг, 4,89 ммоль) в MeOH (10 мл) и нагревали смесь при 70°C в течение 3 ч. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (937 мг, 3,26 ммоль, выход 80%, чистота 95%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 274,2 (M+H)+ через 1,62 мин.
Стадия 4: 5-(изоксазол-5-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (200 мг, 0,730 ммоль, чистота 95%) в ТГФ (9 мл) и воде (3 мл) обрабатывали цинковой пылью (287 мг, 4,39 ммоль) и NH4Cl (тв) (235 мг, 4,39 ммоль). Полученную смесь перемешивали при КТ в течение 1 ч. Смесь фильтровали через Celite®, промывая EtOAc, а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% МТБЭ/изогексан) с получением указанного в заголовке соединения (102 мг, 0,419 ммоль, выход 57%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 244,3 (M+H)+ через 1,27 мин.
Стадия 5: метил-4-этил-3-(N-(5-(изоксазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (102 мг, 0,419 ммоль) в ДХМ (3 мл) и пиридине (203 мкл, 2,52 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (110 мг, 0,419 ммоль) в ДХМ (5 мл) и перемешивали раствор при КТ в течение 2 дней. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (150 мг, 0,319 ммоль, выход 76%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 470,4 (M+H)+, 468,3 (M-H)- через 1,85 мин.
Стадия 6: 4-этил-3-(N-(5-(изоксазол-5-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 4М HCl в диоксане (399 мкл, 1,59 ммоль) добавляли к раствору продукта из стадии 5 выше (150 мг, 0,319 ммоль) в диоксане (5 мл) и нагревали смесь при 60°C в течение 16 ч. Добавляли концентрированную HCl (водн.) (2 мл) и нагревали смесь при 70°C еще в течение 16 ч. Смесь выпаривали в вакууме и суспендировали твердое вещество в МТБЭ (20 мл) в течение 30 мин. Твердое вещество собирали с получением указанного в заголовке соединения (145 мг, 0,309 ммоль, выход 97%, чистота 97%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 456,4 (M+H)+, 454,3 (M-H)- через 1,74 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,32 (шс, 1H), 8,59 (д, J=1,9 Гц, 1H), 8,39 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,8 Гц, 1H), 7,61 (дд, J=8,2, 1,9 Гц, 2H), 7,54 (д, J=2,1 Гц, 1H), 7,25 (д, J=8,4 Гц, 1H), 6,79 (д, J=1,9 Гц, 1H), 3,06 (к, J=7,4 Гц, 2H), 2,75-2,68 (м, 4H), 1,60-1,53 (м, 4H), 1,50-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). Один обмениваемый протон не наблюдали.
Пример 301: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразол-4-ил)фенил)сульфамоил)бензойная кислота
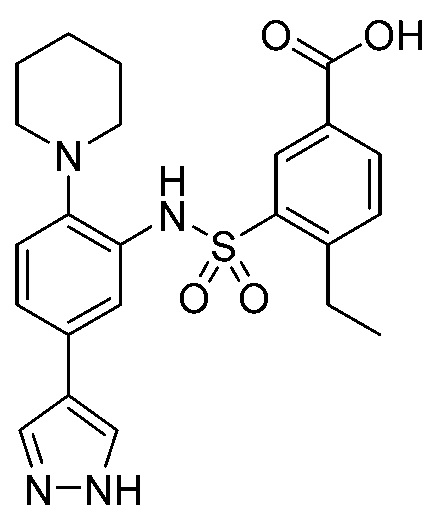
Стадия 1: трет-бутил-4-(3-нитро-4-(пиперидин-1-ил)фенил)-пиразол-1-карбоксилат: Смесь продукта из Примера 287, Стадии 2 (291 мг, 0,714 ммоль, чистота 70%), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-пиразол-1-карбоксилата (250 мг, 0,85 ммоль) и K3PO4 (235 мг, 1,11 ммоль) в диоксане (10 мл) и воде (2 мл) обрабатывали XPhos Pd G3 (71,9 мг, 0,085 ммоль). Реакционную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 1 ч. Смеси позволяли охладиться до КТ, затем фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (201 мг, 0,497 ммоль, выход 83%, чистота 92%) в виде красного масла. СВЭЖХ-МС (Метод 2) m/z 373,3 (M+H)+ через 1,85 мин.
Стадия 2: трет-бутил-4-(3-амино-4-(пиперидин-1-ил)фенил)-пиразол-1-карбоксилат: Раствор продукта из стадии 1 выше (201 мг, 0,497 ммоль, чистота 92%) в MeOH (10 мл) обрабатывали 10% Pd/C (57,4 мг, 0,027 ммоль). Раствор гидрировали под давлением 1 бар в течение 1 ч. Реакционную смесь фильтровали через Celite® и выпаривали фильтрат в вакууме с получением указанного в заголовке соединения (160 мг, 0,439 ммоль, выход 88%, чистота 94%) в виде прозрачного масла. СВЭЖХ-МС (Метод 2) m/z 343,3 (M+H)+ через 1,77 мин.
Стадия 3: трет-бутил-4-(3-(2-этил-5-(метоксикарбонил) фенилсульфонамидо)-4-(пиперидин-1-ил)фенил)-пиразол-1-карбоксилат: Раствор продукта из стадии 2 выше (160 мг, 0,439 ммоль, чистота 94%) в ДХМ (5 мл) и пиридине (227 мкл, 2,80 ммоль) обрабатывали продуктом из Примера 203, Стадии 2 (135 мг, 0,488 ммоль, чистота 95%) и перемешивали раствор при КТ в течение 2 дней. Растворитель удаляли в вакууме, а остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (112 мг, 0,158 ммоль, выход 36%, чистота 80%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 569,4 (M+H)+ через 2,06 мин.
Стадия 4: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиразол-4-ил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 3 выше (112 мг, 0,158 ммоль, чистота 80%) обрабатывали 4М HCl в диоксане (59,8 мкл, 1,97 ммоль) и водой (0,5 мл). Раствор нагревали при 60°C в течение 6 ч. Добавляли концентрированную HCl (водн.) (1 мл) и воду (1 мл) и нагревали реакционную смесь при 60°C в течение ночи. Реакционную смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии (13 г обращенно-фазовый картридж C18, 15-80% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (35,5 мг, 0,074 ммоль, выход 47%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 455,4 (M+H)+, 453,3 (M-H)- через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,1 (шс, 1H), 8,94 (шс, 1H), 8,47 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,9 Гц, 1H), 7,79 (с, 2H), 7,59 (д, J=8,0 Гц, 1H), 7,33-7,22 (м, 2H), 7,14 (д, J=8,2 Гц, 1H), 3,06 (к, J=7,4 Гц, 2H), 2,59 (т, J=5,2 Гц, 4H), 1,62-1,54 (м, 4H), 1,51-1,43 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). Один обмениваемый протон не наблюдали.
Пример 302: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-4-ил)фенил)сульфамоил)бензойная кислота
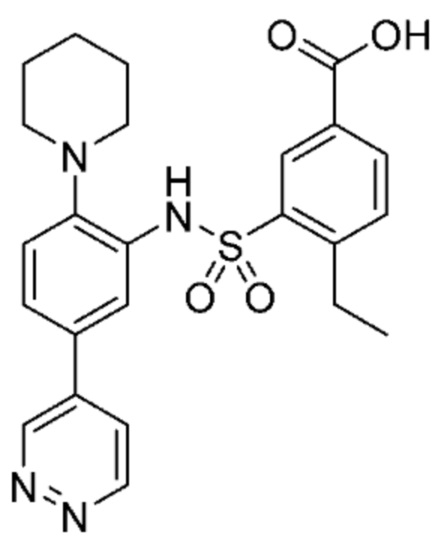
Стадия 1: метил-4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-4-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 273, Стадии 4 (150 мг, 0,281 ммоль, чистота 99%), гидрохлорида 4-хлорпиридазина (51,4 мг, 0,341 ммоль) и K3PO4 (139 мг, 0,653 ммоль) в диоксане (5 мл) и воде (1 мл) обрабатывали XPhos Pd G3 (24 мг, 0,028 ммоль). Реакционную смесь дегазировали N2 в течение 15 мин, затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ и затем разбавляли водой (50 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (82 мг, 0,171 ммоль, выход 61%) в виде желтого масла. СВЭЖХ-МС (Метод 2) m/z 481,4 (M+H)+ через 1,64 мин.
Стадия 2: 4-этил-3-(N-(2-(пиперидин-1-ил)-5-(пиридазин-4-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,71 мл, 1,71 ммоль) добавляли к раствору продукта из стадии 1 выше (82 мг, 0,171 ммоль) в ТГФ (2 мл) и перемешивали раствор при КТ в течение ночи. Реакционную смесь выпаривали в вакууме для удаления ТГФ, подкисляли до ~pH 6 1М HCl (водн.) и образовавшийся в результате осадок собирали с помощью фильтрации и сушили в вакууме с получением указанного в заголовке соединения (60 мг, 0,122 ммоль, выход 72%, чистота 95%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 467,4 (M+H)+, 465,3 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,4 (шс, 1H), 9,38 (дд, J=2,5, 1,2 Гц, 1H), 9,30 (шс, 1H), 9,21 (дд, J=5,5, 1,2 Гц, 1H), 8,43 (д, J=1,8 Гц, 1H), 8,09 (дд, J=8,0, 1,9 Гц, 1H), 7,74 (дд, J=5,5, 2,6 Гц, 1H), 7,65 (дд, J=8,3, 2,2 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,53 (д, J=2,2 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 3,07 (к, J=7,4 Гц, 2H), 2,70 (т, J=5,2 Гц, 4H), 1,63-1,53 (м, 4H), 1,51-1,42 (м, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 303: 4-этил-3-(N-(4-метил-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
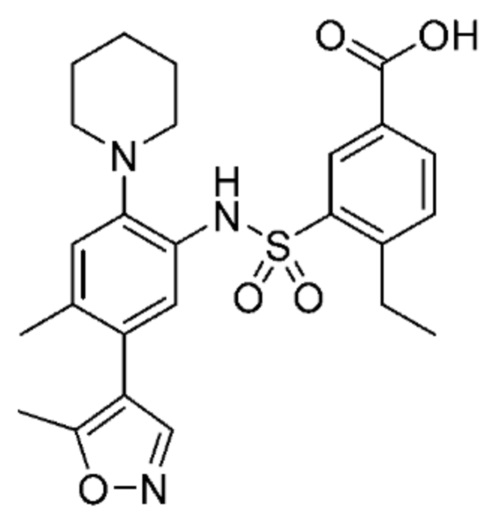
Стадия 1: 1-(4-бром-5-метил-2-нитрофенил)пиперидин: 1-бром-4-фтор-2-метил-5-нитробензол (1,1 г, 4,70 ммоль) в сухом ДМФА (5 мл) обрабатывали пиперидином (1,39 мл, 14,1 ммоль) и перемешивали полученную смесь при КТ в течение 72 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (1,4 г, 4,63 ммоль, выход 99%, чистота 99%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 299,1 (M+H)+ через 1,95 мин.
Стадия 2: 1-(5-метил-2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (1,4 г, 4,63 ммоль, чистота 99%), бис(пинаколато)дибора (1,76 г, 6,95 ммоль), KOAc (1,36 г, 13,9 ммоль) и PdCl2(dppf)∙ДХМ (339 мг, 0,463 ммоль) в диоксане (10 мл) дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (1,6 г, 3,23 ммоль, выход 70%, чистота 70%) в виде оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 347,4 (M+H)+ через 2,15 мин.
1H-ЯМР (500 МГц, ДМСО-d6) δ 8,01 (с, 1H), 7,02 (с, 1H), 3,02 (т, J=5,1 Гц, 4H), 2,48 (с, 3H), 1,67-1,50 (м, 6H), 1,29 (с, 12H).
Стадия 3: 4-метил-2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Продукт из стадии 2 выше (500 мг, 1,01 ммоль, чистота 70%) в MeOH (5 мл) обрабатывали 10% Pd/C (108 мг, 0,101 ммоль). Реакционную смесь гидрировали (2 бара) при КТ в течение 2 ч. Смесь фильтровали через Celite®, промывая MeOH (20 мл), а затем выпаривали в вакууме с получением указанного в заголовке соединения (340 мг, 1,00 ммоль, выход 99%, чистота 93%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 317,7 (M+H)+ через 1,44 мин.
Стадия 4: 4-метил-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (200 мг, 0,588 ммоль, чистота 93%), 4-иод-5-метилизоксазола (135 мг, 0,647 ммоль), K3PO4 (162 мг, 0,765 ммоль), диоксана (4 мл) и воды (1 мл) обрабатывали XPhos Pd G3 (50 мг, 0,059 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-30% EtOAc/изогексан) с получением указанного в заголовке соединения (150 мг, 0,531 ммоль, выход 90%, чистота 96%) в виде воскообразного рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 272,3 (M+H)+ через 1,07 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,53 (с, 1H), 6,79 (с, 1H), 6,52 (с, 1H), 4,58 (с, 2H), 2,82-2,70 (м, 4H), 2,33 (с, 3H), 2,04 (с, 3H), 1,70-1,63 (м, 4H), 1,57-1,47 (м, 2H).
Стадия 5: метил-4-этил-3-(N-(4-метил-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (150 мг, 0,531 ммоль, чистота 96%) в ДХМ (3 мл) и пиридине (258 мкл, 3,18 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (139 мг, 0,531 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 2 дней. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-40% EtOAc/изогексан) с получением указанного в заголовке соединения (220 мг, 0,438 ммоль, выход 82%, чистота 99%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 498,4 (M+H)+, 496,2 (M-H)- через 1,95 мин.
Стадия 6: 4-этил-3-(N-(4-метил-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 4М HCl в диоксане (553 мкл, 2,21 ммоль) добавляли к раствору продукта из стадии 5 выше (220 мг, 0,438 ммоль, чистота 99%) в диоксане (5 мл) и нагревали смесь при 60°C в течение 16 ч. Смесь выпаривали в вакууме и твердое вещество суспедировали в MeCN (5 мл) в течение 30 мин. Твердое вещество собирали с помощью фильтрации с получением указанного в заголовке соединения (195 мг, 0,390 ммоль, выход 89%, чистота 98%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 484,4 (M+H)+, 482,3 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,89 (шс, 1H), 8,57 (с, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,08 (дд, J=7,9, 1,8 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,13 (шс, 1H), 6,92 (шс, 1H), 3,06 (к, J=7,4 Гц, 2H), 2,82-2,55 (м, 4H), 2,20 (с, 3H), 2,12 (с, 3H), 1,73-1,55 (м, 4H), 1,54-1,40 (м, 2H), 1,22 (т, J=7,4 Гц, 3H). Один обмениваемый протон не наблюдали.
Пример 304: 4-этил-3-(N-(4-фтор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
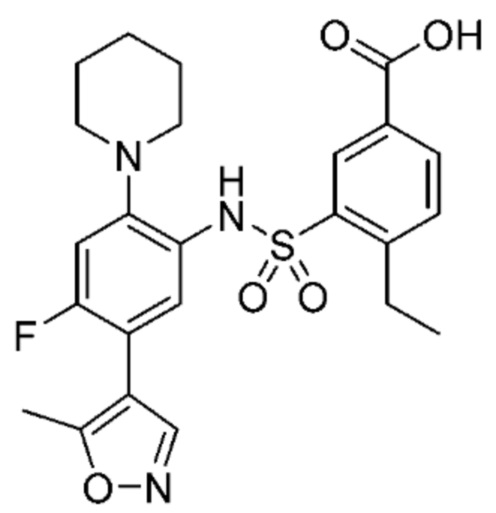
Стадия 1: 1-(4-бром-5-фтор-2-нитрофенил)пиперидин: 1-бром-2,4-дифтор-2-метил-5-нитробензол (1 г, 4,20 ммоль) в сухом ДМФА (5 мл) обрабатывали Et3N (586 мкл, 4,20 ммоль), затем пиперидином (415 мкл, 4,20 ммоль), и перемешивали смесь при КТ в течение 3 дней. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (1,27 г, 3,02 ммоль, выход 72%, 72% чистота) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 303,2 (M+H)+ через 1,84 мин.
Стадия 2: 1-(5-фтор-2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин: Смесь продукта из стадии 1 выше (1,27 г, 3,02 ммоль, чистота 72%), бис(пинаколато)дибора (1,15 г, 4,52 ммоль), KOAc (888 мг, 9,05 ммоль) и PdCl2(dppf)∙ДХМ (221 мг, 0,302 ммоль) в диоксане (10 мл) дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% МТБЭ/изогексан) с получением указанного в заголовке соединения (1,05 г, 1,80 ммоль, выход 59%, чистота 60%) в виде оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 347,4 (M+H)+ через 2,15 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,03 (д, J=6,4 Гц, 1H), 6,97 (д, J=12,1 Гц, 1H), 3,07 (т, J=5,0 Гц, 4H), 1,68-1,49 (м, 6H), 1,29 (с, 12H).
Стадия 3: 4-фтор-2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Продукт из стадии 2 выше (500 мг, 0,857 ммоль, чистота 60%) в MeOH (5 мл) обрабатывали 10% Pd/C (50% об/об воды) Тип 39 (91 мг, 0,086 ммоль). Реакционную смесь гидрировали при 2 барах при КТ в течение 2 ч. Смесь фильтровали через Celite®, промывая MeOH (20 мл), а затем выпаривали в вакууме с получением указанного в заголовке соединения (373 мг, 0,559 ммоль, выход 65%, чистота 48%) в виде темно-голубого масла. СВЭЖХ-МС (Метод 1) m/z 321,4 (M+H)+ через 1,62 мин.
Стадия 4: 4-фтор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (373 мг, 0,559 ммоль, чистота 48%), 4-иод-5-метилизоксазола (128 мг, 0,610 ммоль), K3PO4 (153 мг, 0,721 ммоль), диоксана (4 мл) и воды (1 мл) обрабатывали XPhos Pd G3 (47 мг, 0,060 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-30% EtOAc/изогексан) с получением указанного в заголовке соединения (180 мг, 0,458 ммоль, выход 83%, чистота 70%) в виде темно-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 276,3 (M+H)+ через 1,50 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,60 (д, J=1,6 Гц, 1H), 6,81 (д, J=12,0 Гц, 1H), 6,72 (д, J=7,8 Гц, 1H), 4,66 (шс, 2H), 2,82-2,75 (м, 4H), 2,46 (с, 3H), 1,70-1,57 (м, 6H).
Стадия 5: метил-4-этил-3-(N-(4-фтор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (180 мг, 0,458 ммоль, чистота 70%) в ДХМ (3 мл) и пиридине (222 мкл, 2,75 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (120 мг, 0,458 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 72 ч. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-30% EtOAc/изогексан) с получением указанного в заголовке соединения (120 мг, 0,167 ммоль, выход 37%, чистота 70%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 502,4 (M+H)+, 500,3 (M-H)- через 1,96 мин.
Стадия 6: 4-этил-3-(N-(4-фтор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 4М HCl в диоксане (210 мкл, 0,840 ммоль) добавляли к раствору продукта из стадии 5 выше (120 мг, 0,167 ммоль, чистота 70%) в диоксане (5 мл) и нагревали смесь при 60°C в течение 16 ч. Смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии (12 г обращенно-фазовый картридж C18, 15-85% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (30,5 мг, 0,061 ммоль, выход 36%, чистота 98%) в виде почти белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 488,3 (M+H)+, 486,2 (M-H)- через 1,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (шс, 1H), 9,48-9,07 (с, 1H), 8,59 (д, J=1,8 Гц, 1H), 8,32 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,8 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,12 (д, J=8,0 Гц, 1H), 7,06 (д, J=12,0 Гц, 1H), 3,04 (к, J=7,4 Гц, 2H), 2,64 (т, J=5,1 Гц, 4H), 2,35 (с, 3H), 1,48 (д, J=9,7 Гц, 4H), 1,43 (д, J=8,6 Гц, 2H), 1,22 (т, J=7,4 Гц, 3H).
Пример 305: 4-этил-3-(N-(2-фтор-3-(5-метилизоксазол-4-ил)-6-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
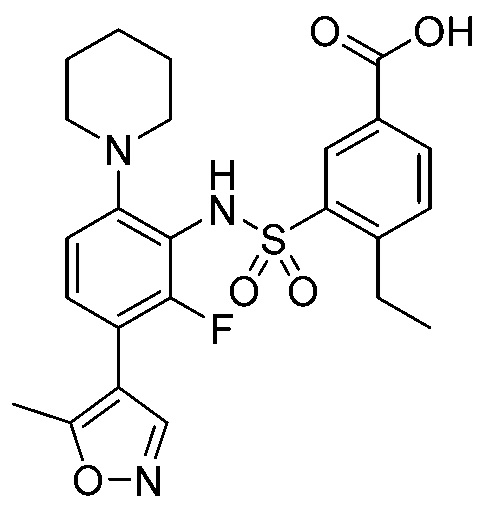
Стадия 1: 1-(4-бром-3 фтор 2-нитрофенил)пиперидин: 1-бром-2,4-фтор-3-нитробензол (1 г, 4,20 ммоль) в сухом ДМФА (5 мл) охлаждали до 0°C. Добавляли Et3N (1,17 мл, 8,40 ммоль), затем пиперидин (415 мкл, 4,20 ммоль) и перемешивали смесь при КТ в течение 24 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (1,2 г, 3,76 ммоль, выход 90%, чистота 95%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 303,4 (M+H)+ через 1,88 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,82 (т, J=8,5 Гц, 1H), 7,13 (дд, J=9,1 Гц, 1H), 2,97 (т, J=5,1 Гц, 4H), 1,62-1,48 (м, 6H).
Стадия 2: 3-бром-2-фтор-6-(пиперидин-1-ил)анилин: Раствор продукта из стадии 1 выше (500 мг, 1,56 ммоль, чистота 95%) в ТГФ (9 мл) и воде (3 мл) обрабатывали цинковой пылью (615 мг, 9,40 ммоль) и NH4Cl (тв) (503 мг, 9,40 ммоль). Полученную смесь перемешивали при КТ в течение 2 ч. Смесь фильтровали через Celite®, промывая EtOAc (3×5 мл), а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (320 мг, 1,14 ммоль, выход 72%, чистота 97%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 273,1 (M+H)+ через 1,79 мин.
Стадия 3: 2-фтор-6-(пиперидин-1-ил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Смесь продукта из стадии 2 выше (320 мг, 1,14 ммоль, чистота 97%), бис(пинаколато)дибора (433 мг, 1,70 ммоль), KOAc (335 мг, 3,41 ммоль) и PdCl2(dppf)∙ДХМ (83 мг, 0,114 ммоль) в диоксане (10 мл) дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (379 мг, 1,11 ммоль, выход 98%, чистота 94%) в виде светло-зеленого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 321,4 (M+H)+ через 1,83 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,01 (с, 1H), 7,02 (с, 1H), 3,02 (т, J=5,1 Гц, 4H), 2,48 (с, 3H), 1,67-1,50 (м, 6H), 1,29 (с, 12H).
Стадия 4: 2-фтор-3-(5-метилизоксазол-4-ил)-6-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (200 мг, 0,587 ммоль, чистота 94%), 4-иод-5-метилизоксазола (135 мг, 0,646 ммоль), K3PO4 (162 мг, 0,763 ммоль), диоксана (4 мл) и воды (1 мл) обрабатывали XPhos Pd G3 (50,0 мг, 0,059 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (140 мг, 0,386 ммоль, выход 66%, чистота 76%) в виде воскообразного рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 276,3 (M+H)+ через 1,58 мин.
Стадия 5: Метил-4-этил-3-(N-(2-фтор-3-(5-метилизоксазол-4-ил)-6-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор продукта из стадии 4 выше (140 мг, 0,386 ммоль, чистота 76%) в ДХМ (3 мл) и пиридине (188 мкл, 2,32 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (102 мг, 0,386 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 48 ч. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-40% EtOAc/изогексан) с получением указанного в заголовке соединения (64 мг, 0,089 ммоль, выход 23%, чистота 70%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 502,4 (M+H)+, 500,3 (M-H)- через 1,81 мин.
Стадия 6: 4-этил-3-(N-(2-фтор-3-(5-метилизоксазол-4-ил)-6-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: 4М HCl в диоксане (110 мкл, 0,440 ммоль) добавляли к раствору продукта из стадии 5 выше (64 мг, 0,089 ммоль, чистота 70%) в диоксане (5 мл) и нагревали смесь при 60°C в течение 72 ч. Смесь выпаривали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (4 мг, 7,88 мкмоль, выход 9%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 488,4 (M+H)+, 486,3 (M-H)- через 1,68 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,21 (шс, 1H), 9,52 (с, 1H), 8,58 (с, 1H), 8,30 (д, J=1,8 Гц, 1H), 8,08 (дд, J=8,0, 1,9 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,32 (т, J=8,4 Гц, 1H), 6,90 (д, J=8,6 Гц, 1H), 3,08 (д, J=7,1 Гц, 2H), 2,80-2,69 (м, 4H), 2,38 (с, 3H), 1,35-1,18 (м, 9H).
Пример 306: 4-этил-3-(N-(4-хлор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
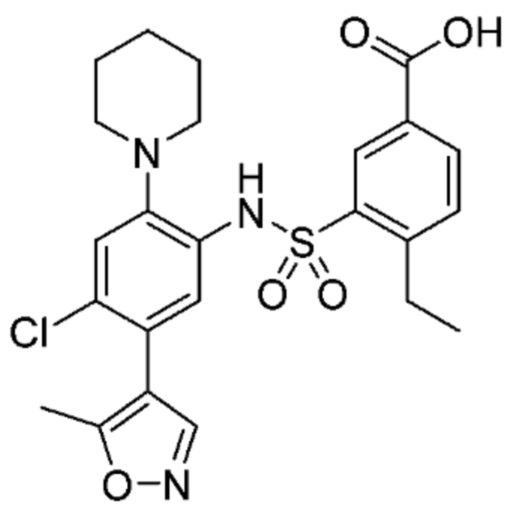
Стадия 1: 1-(4-бром-5-хлор-2-нитрофенил)пиперидин: 1-бром-2-хлор-4-фтор-5-нитробензол (1 г, 3,93 ммоль) в сухом ДМФА (5 мл) охлаждали до 0°C. Последовательно добавляли Et3N (1,09 мл, 7,86 ммоль) и пиперидин (388 мкл, 3,93 ммоль) и перемешивали смесь при КТ в течение 24 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (1,2 г, 3,68 ммоль, выход 94%, чистота 98%) в виде красного масла. СВЭЖХ-МС (Метод 1) m/z 319,2 (M+H)+ через 1,97 мин.
Стадия 2: 5-бром-4-хлор-2-(пиперидин-1-ил)анилин: Раствор продукта из стадии 1 выше (1,20 г, 3,68 ммоль, чистота 98%) в ТГФ (9 мл) и воде (3 мл) обрабатывали цинковой пылью (1,44 г, 22,1 ммоль) и NH4Cl (тв) (1,18 г, 22,1 ммоль). Полученную смесь перемешивали при КТ в течение 2 ч. Смесь фильтровали через Celite®, промывая EtOAc (3×5 мл), а затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (880 мг, 2,92 ммоль, выход 79%, чистота 96%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 289,1 (M+H)+ через 1,87 мин.
Стадия 3: 4-хлор-2-(пиперидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин: Смесь продукта из стадии 2 выше (880 мг, 2,92 ммоль, чистота 96%), бис(пинаколато)дибора (1,11 г, 4,38 ммоль), KOAc (859 мг, 8,75 ммоль) и PdCl2(dppf)∙ДХМ (213 мг, 0,292 ммоль) в диоксане (10 мл) дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 16 ч. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (980 мг, 2,38 ммоль, выход 82%, чистота 82%) в виде коричневого масла. СВЭЖХ-МС (Метод 1) m/z 337,3 (M+H)+ через 1,89 мин.
Стадия 4: 4-хлор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)анилин: Смесь продукта из стадии 3 выше (300 мг, 0,731 ммоль, чистота 82%), 4-иод-5-метилизоксазола (168 мг, 0,804 ммоль), K3PO4 (202 мг, 0,950 ммоль), диоксана (4 мл) и воды (1 мл) обрабатывали XPhos Pd G3 (62,0 мг, 0,073 ммоль). Полученную смесь дегазировали N2 в течение 15 мин, а затем нагревали при 80°C в течение 2 ч. Смеси позволяли охладиться до КТ и затем выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (225 мг, 0,717 ммоль, выход 98%, чистота 93%) в виде воскообразного рыжевато-коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 292,2 (M+H)+ через 1,69 мин.
Стадия 5: метил-3-(N-(4-хлор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Раствор продукта из стадии 4 выше (225 мг, 0,717 ммоль, чистота 93%) в ДХМ (3 мл) и пиридине (348 мкл, 4,30 ммоль) добавляли к раствору продукта из Примера 203, Стадии 2 (188 мг, 0,717 ммоль) в ДХМ (3 мл) и перемешивали полученный раствор при КТ в течение 3 дней. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (40 г картридж, 0-40% EtOAc/изогексан) с получением указанного в заголовке соединения (320 мг, 0,525 ммоль, выход 73%, чистота 85%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 518,3 (M+H)+, 516,1 (M-H)- через 2,02 мин.
Стадия 6: 3-(N-(4-хлор-5-(5-метилизоксазол-4-ил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота: 4М HCl в диоксане (656 мкл, 2,63 ммоль) добавляли к раствору продукта из стадии 5 выше (320 мг, 0,525 ммоль, чистота 85%) в диоксане (5 мл) и нагревали смесь при 60°C в течение 24 ч. Смесь выпаривали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (42,6 мг, 0,083 ммоль, выход 16%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 504,3 (M+H)+, 502,2 (M-H)- через 1,90 мин. 1H-ЯМР (500 МГц, ДМСО-d6) 8,59 (шс, 1H), 8,35 (д, J=1,8 Гц, 1H), 8,08 (дд, J=7,9, 1,8 Гц, 1H), 7,59 (д, J=8,0 Гц, 1H), 7,24 (с, 1H), 7,09 (с, 1H), 3,12-2,97 (м, 2H), 2,67 (т, J=7,4 Гц, 4H), 2,26 (с, 3H), 1,55 (к, J=5,6 Гц, 4H), 1,45 (к, J=5,8 Гц, 2H), 1,22 (т, J=7,4 Гц, 3H). Два обмениваемых протона не наблюдали.
Пример 308: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-этилбензойная кислота
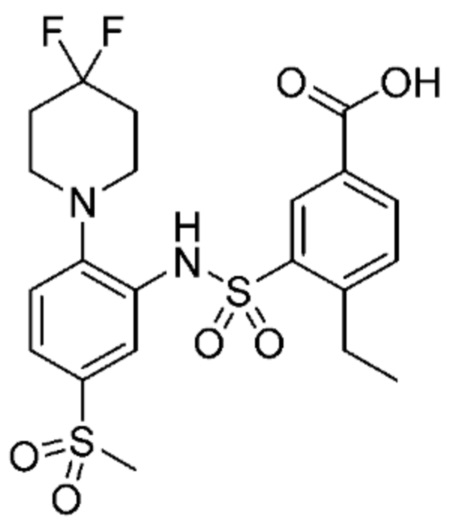
Стадия 1: 4,4-дифтор-1-(4-(метилсульфонил)-2-нитрофенил)-пиперидин: Смесь 1-фтор-4-(метилсульфонил)-2-нитробензола (500 мг, 2,28 ммоль) и 4,4-дифторпиперидина (276 мг, 2,28 ммоль) и Et3N (400 мкл, 2,87 ммоль) обрабатывали ультразвуком в ДХМ (6 мл) до образования прозрачного раствора. Полученный раствор оставляли при КТ на 2 ч. Реакционную смесь последовательно промывали 1М HCl (водн.) (4 мл), водой (4 мл) и солевым раствором (2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (717 мг, 1,90 ммоль, выход 83%, чистота 85%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 321,3 (M+H)+ через 1,28 мин.
Стадия 2: 2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)-анилин: Продукт из стадии 1 выше (717 мг, 2,24 ммоль, чистота 85%) растворяли в EtOH/ТГФ 1:1 (100 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, режим полной подачи водорода, КТ, 1 мл/мин, затем 20 бар, 40°C, рециркуляция 2 ч, затем 40 бар, 50°C рециркуляция 2 ч, затем 40 бар, 50°C). Смесь выпаривали в вакууме. Остаток растворяли в смеси AcOH (50 мл), ТГФ (50 мл) и воды (10 мл) и отфильтровывали. Фильтрат выпаривали в вакууме, растворяли в AcOH (50 мл) и гидрировали в проточном реакторе ThalesNano H-cube® (10% Pd/C, 30×4 мм, Полный водород, КТ, 1 мл/мин, рециркуляция 1,5 ч). Смесь выпаривали в вакууме, подвергая азеотропной перегонке с толуолом (50 мл). Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (162 мг, 0,469 ммоль, выход 21%, чистота 84%) в виде бледно-желтой пены. СВЭЖХ-МС (Метод 1) m/z 291,2 (M+H)+ через 1,18 мин.
Стадия 3: Метил-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-этилбензоат: Продукт из стадии 2 выше (62 мг, 0,179 ммоль, чистота 84%) растворяли в смеси ДХМ (1 мл) и пиридина (50 мкл, 0,618 ммоль) и обрабатывали продуктом из Примера 203, Стадии 2 (50 мг, 0,188 ммоль). Полученный раствор оставляли при КТ на 3 дня. Добавляли дополнительную порцию продукта из Примера 203, Стадии 2 (53 мг, 0,200 ммоль) и ставляли полученный раствор при КТ на 4 дня. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (104 мг, 0,167 ммоль, выход 93%, чистота 83%) в виде светло-бежевой пены. СВЭЖХ-МС (Метод 1) m/z 517,2 (M+H)+, 515,3 (M-H)- через 1,55 мин.
Стадия 4: 3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метил-сульфонил)фенил)сульфамоил)-4-этилбензойная кислота: Смесь продукта из стадии 3 выше (104 мг, 0,167 ммоль, чистота 83%) и LiOH (28 мг, 0,655 ммоль) в ТГФ/MeOH/воде (4:1:1, 6 мл) перемешивали при 40°C в течение 18 ч. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×10 мл). Органические экстракты объединяли и промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (36,3 мг, 72 мкмоль, выход 42%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 503,2 (M+H)+, 501,1 (M-H)- через 1,41 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (с, 1H), 9,89 (с, 1H), 8,36 (д, J=1,8 Гц, 1H), 8,10 (дд, J=8,0, 1,8 Гц, 1H), 7,65-7,59 (м, 2H), 7,55 (д, J=2,2 Гц, 1H), 7,35 (д, J=8,5 Гц, 1H), 3,05 (с, 3H), 3,02 (к, J=7,4 Гц, 2H), 2,94-2,88 (м, 4H), 2,12-2,01 (м, 4H), 1,20 (т, J=7,4 Гц, 3H).
Пример 309: 4-циклопропил-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
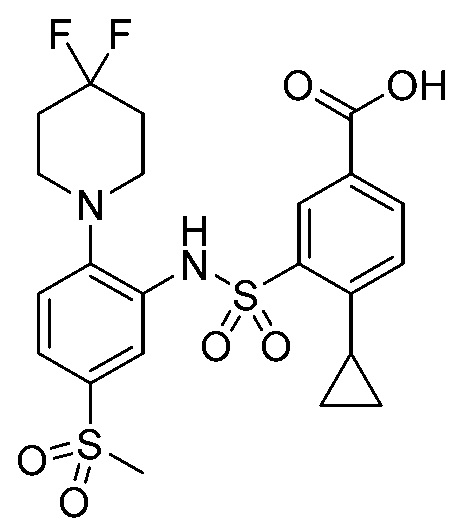
Стадия 1: Метил-4-бром-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 308, Стадии 2 (102 мг, 0,313 ммоль, чистота 84%) растворяли в смеси ДХМ (1 мл) и пиридина (50 мкл, 0,618 ммоль) и обрабатывали продуктом из Примера 316, Стадии 1 (103 мг, 0,325 ммоль). Полученную суспензию обрабатывали ультразвуком, а затем разбавляли ДХМ (1 мл) с получением прозрачного раствора. Смесь оставляли при КТ на 4 дня. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (108 мг, 0,101 ммоль, выход 32%, чистота 53%) в виде бежевой пены. СВЭЖХ-МС (Метод 1) m/z 567,2 (M+H)+, 565,0 (M-H)- через 1,52 мин.
Стадия 2: Метил-4-бром-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Дегазированную смесь продукта из стадии 1 выше (108 мг, 0,101 ммоль, чистота 53%) и Pd-174 (7,00 мг, 9,71 мкмоль) в ТГФ (2 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (800 мкл, 0,400 ммоль). Смесь нагревали при 60°C в течение 18 ч. После охлаждения до КТ смесь концентрировали на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (54 мг, 62,0 мкмоль, выход 61%, чистота 61%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1) m/z 529,2 (M+H)+, 527,1 (M-H)- через 1,55 мин.
Стадия 3: 4-циклопропил-3-(N-(2-(4,4-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 2 выше (54 мг, 62,0 мкмоль, чистота 61%) и LiOH (11 мг, 0,257 ммоль) в ТГФ/MeOH/воде (4:1:1, 2,4 мл) перемешивали при 40°C в течение 18 ч. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×10 мл). Органические экстракты объединяли и промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (13,8 мг, 27,0 мкмоль, выход 42%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 515,2 (M+H)+, 513,0 (M-H)- через 1,40 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,27 (с, 1H), 9,80 (с, 1H), 8,40 (д, J=1,8 Гц, 1H), 8,01 (дд, J=8,2, 1,8 Гц, 1H), 7,65-7,58 (м, 1H), 7,55 (д, J=2,2 Гц, 1H), 7,36 (д, J=8,4 Гц, 1H), 7,15 (д, J=8,2 Гц, 1H), 3,02 (с, 3H), 2,98-2,93 (м, 4H), 2,84-2,75 (м, 1H), 2,10-2,01 (м, 4H), 1,11-1,03 (м, 2H), 0,90-0,83 (м, 2H).
Пример 310: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
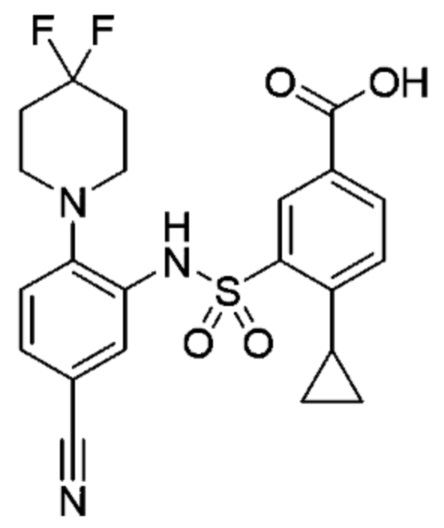
Стадия 1: метил-4-бром-3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 221, Стадия 2 (100 мг, 0,400 ммоль) растворяли в смеси ДХМ (1 мл) и пиридина (100 мкл, 1,23 ммоль) и обрабатывали продуктом из Примера 316, Стадии 1 (130 мг, 0,410 ммоль). Полученный раствор оставляли при КТ на 4 дня. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (123 мг, 0,234 ммоль, выход 58%, чистота 98%) в виде светло-оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 514,2 (M+H)+, 512,0 (M-H)- через 1,65 мин.
Стадия 2: метил-4-бром-3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)бензоат: Дегазированную смесь продукта из стадии 1 выше (123 мг, 0,234 ммоль, чистота 98%) и Pd-174 (17 мг, 24 мкмоль) в ТГФ (4,5 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (1,8 мл, 0,900 ммоль). Смесь нагревали при 60°C в течение 18 ч. После охлаждения до КТ смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (99,4 мг, 0,192 ммоль, выход 82%, чистота 92%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 476,3 (M+H)+, 474,2 (M-H)- через 1,69 мин.
Стадия 3: 3-(N-(5-циано-2-(4,4-дифторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота: Смесь продукта из стадии 2 выше (99,4 мг, 0,192 ммоль, чистота 92%) и LiOH (33 мг, 0,772 ммоль) в ТГФ/MeOH/воде (4:1:1, 6,6 мл) перемешивали при 40°C в течение 18 ч. Смесь разбавляли водой (10 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (55,8 мг, 0,120 ммоль, выход 62%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 462,2 (M+H)+, 460,2 (M-H)- через 1,54 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (с, 1H), 9,81 (с, 1H), 8,37 (д, J=1,8 Гц, 1H), 8,02 (дд, J=8,2, 2,0 Гц, 1H), 7,56 (дд, J=8,4, 1,8 Гц, 1H), 7,33 (д, J=2,0 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,16 (д, J=8,2 Гц, 1H), 2,97-2,91 (м, 4H), 2,80-2,71 (м, 1H), 2,07-1,95 (м, 4H), 1,10-1,03 (м, 2H), 0,90-0,83 (м, 2H).
Пример 311: (R)-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
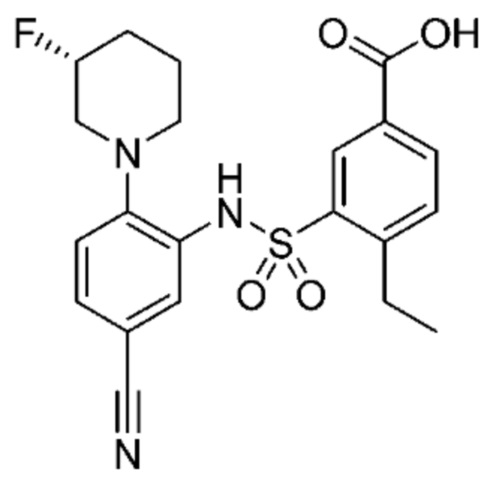
Стадия 1: (R)-4-(3-фторпиперидин-1-ил)-3-нитробензонитрил: Смесь 4-фтор-3-нитробензонитрила (400 мг, 2,41 ммоль) и гидрохлорида (R)-3-фторпиперидина (336 мг, 2,41 ммоль) и Et3N (700 мкл, 5,02 ммоль) обрабатывали ультразвуком в ДХМ (6 мл) в течение 5 мин. Полученную суспензию перемешивали при КТ в течение 18 ч. Реакционную смесь последовательно промывали 1М HCl (водн.) (4 мл), водой (4 мл) и солевым раствором (2 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (596 мг, 2,37 ммоль, выход 98%, чистота 99%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 250,3 (M+H)+ через 1,35 мин.
Стадия 2: (R)-3-амино-4-(3-фторпиперидин-1-ил)бензонитрил: Продукт из стадии 1 выше (596 мг, 2,37 ммоль, чистота 99%) объединяли с порошком железа (2,5 г, 44,8 ммоль) и NH4Cl (тв) (150 мг, 2,80 ммоль) в ИПС (20 мл) и воде (10 мл), затем нагревали при 80°C и перемешивали в течение ночи. Смесь охлаждали и оставляли на 24 ч. Смесь фильтровали через Celite®, промывали EtOAc (2×10 мл) и выпаривали фильтрат в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (304 мг, 1,36 ммоль, выход 62%, чистота 98%). СВЭЖХ-МС (Метод 1): m/z 220,3 (M+H)+ через 1,32 мин.
Стадия 3: (R)-метил-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Продукт из Примера 203, Стадии 2 (300 мг, 1,13 ммоль) добавляли к раствору пиридина (0,1 мл, 1,24 ммоль) и продукта из стадии 2 выше (152 мг, 0,679 ммоль, чистота 98%) в ДХМ (1 мл). Полученный раствор оставляли при КТ на 3 дня. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (254 мг, 0,570 ммоль, выход 84%) в виде белого пенистого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 446,4 (M+H)+, 444,3 (M-H)- через 1,70 мин.
Стадия 4: (R)-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: Продукт из стадии 3 выше (254 мг, 0,570 ммоль) растворяли в ТГФ (10 мл) и обрабатывали 1М LiOH (водн.) (2 мл, 2,00 ммоль). Добавляли MeOH с получением прозрачного раствора. Полученный раствор оставляли при КТ на 1 неделю. Смесь разбавляли водой (4 мл) и выпаривали в вакууме. Полученный водный раствор разбавляли водой (2 мл) и промывали МТБЭ (8 мл), затем выпаривали в вакууме для удаления остаточного МТБЭ. Раствор разбавляли водой (4 мл) и подкисляли 1М HCl (водн.) до ~pH 4 и собирали образовавшийся белый осадок с помощью фильтрации, промывая водой (2×4 мл). Полученное твердое вещество сушили в вакууме с получением указанного в заголовке соединения (214 мг, 0,471 ммоль, выход 83%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 432,4 (M+H)+, 430,3 (M-H)- через 0,99 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,33 (с, 1H), 9,52 (с, 1H), 8,32 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,55 (д, J=8,4 Гц, 1H), 7,31-7,14 (м, 2H), 4,69 (dtt, J=48,2, 7,3, 3,6 Гц, 1H), 3,25-3,09 (м, 1H), 3,02 (к, J=7,4 Гц, 2H), 2,95-2,84 (м, 2H), 2,83-2,70 (м, 1H), 2,05-1,84 (м, 1H), 1,82-1,69 (м, 1H), 1,69-1,45 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 312: (R)-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
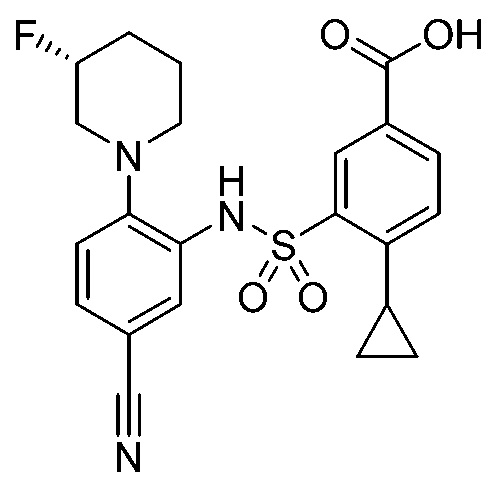
Стадия 1: (R)-метил-4-бром-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (350 мг, 1,11 ммоль) добавляли к раствору пиридина (100 мкл, 1,24 ммоль) и продукта из Примера 311, Стадии 2 (152 мг, 0,679 ммоль, чистота 98%) в ДХМ (1 мл). Полученный раствор оставляли при КТ на 3 дня. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (313 мг, 0,618 ммоль, выход 91%, чистота 98%) в виде белого пенистого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 496,2 (M+H)+, 494,1 (M-H)- через 1,66 мин.
Стадия 2: (R)-метил-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Смесь продукта из стадии 1 выше (313 мг, 0,618 ммоль, чистота 98%) и Pd-174 (45 мг, 0,062 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (5 мл, 2,50 ммоль) и затем нагревали при 60°C в течение 45 мин. Смесь охлаждали в бане со льдом, приливали насыщенный NH4Cl (водн.) (1 мл) и отгоняли ТГФ в вакууме. Остаток экстрагировали ДХМ (4 мл, затем 2×1 мл). Объединенные органические экстракты непосредственно очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением двух партий указанного в заголовке соединения:
Партия 1 (50 мг, 0,105 ммоль, выход 17%, чистота 96%) в виде прозрачного бесцветного масла, которое частично кристаллизовалось при стоянии. СВЭЖХ-МС (Метод 1): m/z 458,4 (M+H)+, 456,3 (M-H)- через 1,70 мин.
Партия 2 (192 мг, 0,386 ммоль, выход 63%, чистота 92%) в виде прозрачного бесцветного масла, которое частично кристаллизовалось при стоянии.
Стадия 3: (R)-3-(N-(5-циано-2-(3-фторпиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: Продукты из Стадии 2 выше: Партию 1 (50 мг, 0,105 ммоль, выход 17%, 96% чистота) и Партию 2 (192 мг, 0,386 ммоль, выход 63%, чистота 92%) растворяли в ТГФ (2 мл или 8 мл соответственно) и обрабатывали 1М LiOH (водн.) (400 мкл, 0,400 ммоль; или 1,6 мл, 1,60 ммоль соответственно). К реакционным смесям добавляли MeOH с образованием прозрачных растворов, которые оставляли при КТ на 30 ч. Смеси нейтрализовали AcOH и выпаривали в вакууме. Остатки объединяли и очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-85% MeCN в воде) с получением указанного в заголовке соединения (127 мг, 0,281 ммоль, выход 57%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 444,4 (M+H)+, 442,3 (M-H)- через 1,01 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,43 (с, 1H), 8,38 (д, J=1,8 Гц, 1H), 8,03 (дд, J=8,2, 1,9 Гц, 1H), 7,54 (д, J=8,3 Гц, 1H), 7,32-7,09 (м, 3H), 4,71 (dtt, J=48,0, 7,3, 3,5 Гц, 1H), 3,26-3,13 (м, 1H), 3,01-2,88 (м, 2H), 2,86-2,77 (м, 1H), 2,76-2,66 (м, 1H), 1,97-1,84 (м, 1H), 1,79-1,70 (м, 1H), 1,69-1,50 (м, 2H), 1,21-1,03 (м, 2H), 0,97-0,73 (м, 2H).
Пример 313: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-этилбензойная кислота
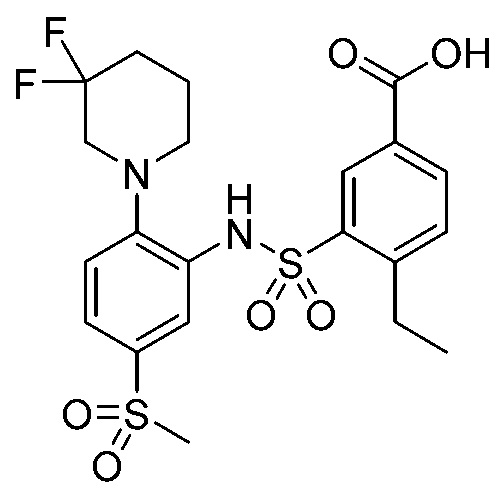
Стадия 1: 3,3-дифтор-1-(4-(метилсульфонил)-2-нитрофенил)-пиперидин: Смесь 1-фтор-4-(метилсульфонил)-2-нитробензола (500 мг, 2,28 ммоль) и гидрохлорида 3,3-дифторпиперидина (359 мг, 2,28 ммоль) и триэтиламина (700 мкл, 5,02 ммоль) обрабатывали ультразвуком в ДХМ (6 мл) в течение 5 мин. Полученную суспензию перемешивали при КТ в течение 18 ч. Реакционную смесь последовательно промывали 1М HCl (4 мл), водой (4 мл) и солевым раствором (2 мл), разбавляли EtOAc (175 мл), промывали солевым раствором (10 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (709 мг, 2,19 ммоль, выход 96%, чистота 99%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 321,1 (M+H)+, в 1,24 мин.
Стадия 2: 2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)-анилин: Продукт из стадии 1 выше (709 мг, 2,19 ммоль, чистота 99%) объединяли с порошком железа (2,5 г, 44,8 ммоль) и NH4Cl (тв) (150 мг, 2,80 ммоль) в ИПС (20 мл) и воде (10 мл), нагревали при 80°C и перемешивали в течение ночи. Смесь охлаждали и оставляли на 24 ч. Смесь фильтровали через Celite®, промывали EtOAc (2×10 мл) и выпаривали фильтрат в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (505 мг, 1,74 ммоль, выход 79%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 291,1 (M+H)+ через 1,16 мин.
Стадия 3: метил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-этилбензоат: Продукт из Примера 203, Стадии 2 (300 мг, 1,13 ммоль) добавляли к раствору пиридина (100 мкл, 1,24 ммоль) и продукта из стадии 2 выше (252 мг, 0,868 ммоль) в ДХМ (2 мл). Полученный раствор оставляли при КТ на 11 дней. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (319 мг, 0,618 ммоль, выход 71%) в виде белого пенистого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 517,4 (M+H)+, 515,2 (M-H)- через 1,57 мин.
Стадия 4: 3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)-4-этилбензойная кислота: Продукт из стадии 3 выше (319 мг, 0,618 ммоль) растворяли в ТГФ (9 мл) и обрабатывали 1М LiOH (водн.) (3 мл, 3,00 ммоль). Полученную двухфазную смесь перемешивали при КТ в течение 18 ч. Смесь выпаривали в вакууме. Полученный водный раствор разбавляли водой (6 мл) и подкисляли 1М HCl (водн.) до ~pH 4 и собирали образовавшийся белый осадок с помощью фильтрации, промывая водой (2×4 мл). Полученное твердое вещество суспендировали в MeCN (5 мл), суспензию выпаривали и сушили в вакууме с получением указанного в заголовке соединения (292 мг, 0,569 ммоль, выход 92%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 503,2 (M+H)+, 501,2 (M-H)- через 0,99 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 12,81 (с, 1H), 8,60-8,35 (м, 1H), 7,96-7,74 (м, 1H), 7,73-7,51 (м, 1H), 7,42-7,20 (м, 1H), 7,12-6,51 (м, 2H), 3,54-3,41 (м, 2H), 3,24-3,11 (м, 4H), 2,84 (с, 3H), 2,05-1,92 (м, 2H), 1,82-1,71 (м, 2H), 1,15 (т, J=7,4 Гц, 3H).
Пример 314: (R)-4-циклопропил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота
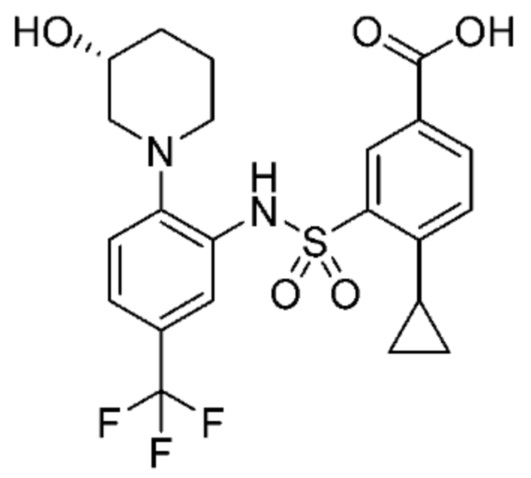
Стадия 1: (R)-метил-4-бром-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (300 мг, 0,947 ммоль) добавляли к раствору пиридина (0,1 мл, 1,24 ммоль) и продукта из Примера 243, Стадии 2 (191 мг, 0,735 ммоль) в ДХМ (2 мл). Полученный раствор оставляли при КТ на 24 ч. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (379 мг, 0,698 ммоль, выход 95%, чистота 99%) в виде белого пенистого твердого вещества. СВЭЖХ (Метод 1): m/z 537,2 (M+H)+, 535,1 (M-H)- через 1,67 мин.
Стадия 2: (R)-метил-4-циклопропил-3-(N-(2-(3-гидрокси-пиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Смесь продукта из стадии 1 выше (379 мг, 0,698 ммоль, чистота 99%) и Pd-174 (50 мг, 0,069 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (5 мл, 2,50 ммоль) и затем нагревали до 60°C и перемешивали в течение 18 ч. Добавляли дополнительную порцию бромида циклопропилцинка (II) (0,5 М в ТГФ) (1 мл, 0,500 ммоль) и продолжали нагревание в течение 1 ч. Смесь нейтрализовали насыщенным NH4Cl (водн.) (1 мл) и выпаривали в вакууме. Остаток экстрагировали ДХМ (2×4 мл). Экстракты объединяли и выпаривали в вакууме. Остаток частично очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (184 мг, 0,343 ммоль, выход 49%, чистота 93%) в виде прозрачной белой пены. СВЭЖХ (Метод 1): m/z 499,3 (M+H)+, 497,3 (M-H)- через 1,73 мин.
Стадия 3: (R)-4-циклопропил-3-(N-(2-(3-гидроксипиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 2 выше (184 мг, 0,343 ммоль, чистота 93%) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (2,5 мл, 2,50 ммоль). Полученную двухфазную смесь перемешивали при КТ в течение 3 дней. Смесь выпаривали в вакууме для удаления ТГФ, затем разбавляли водой (5 мл) и подкисляли до ~pH 4 1М HCl (водн.). Полученный белый осадок собирали с помощью фильтрации, промывали водой и сушили в вакууме с получением белого твердого вещества. Твердое вещество очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (106 мг, 0,214 ммоль, выход 63%, чистота 98%). СВЭЖХ (Метод 2): m/z 485,3 (M+H)+, 483,3 (M-H)- через 1,09 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,29 (с, 1H), 9,61 (с, 1H), 8,49 (д, J=1,9 Гц, 1H), 8,02 (дд, J=8,2, 1,9 Гц, 1H), 7,38-7,29 (м, 2H), 7,22 (д, J=8,7 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 5,42-4,92 (м, 1H), 3,84-3,71 (м, 1H), 3,37-3,22 (м, 1H), 2,94-2,82 (м, 2H), 2,82-2,65 (м, 2H), 1,91-1,74 (м, 1H), 1,74-1,61 (м, 1H), 1,61-1,40 (м, 2H), 1,23-1,03 (м, 2H), 0,92-0,83 (м, 1H), 0,83-0,73 (м, 1H).
Пример 315: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-циклопропилбензойная кислота
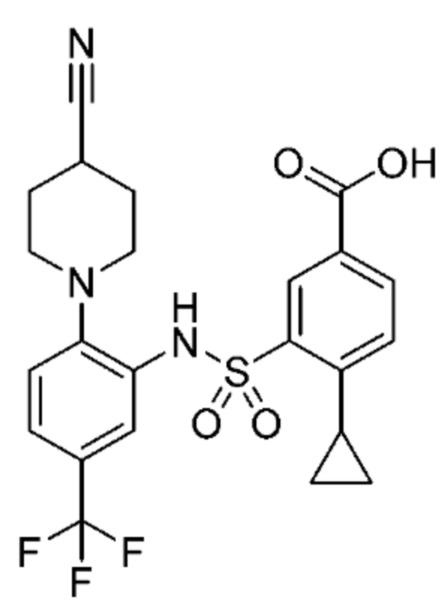
Стадия 1: Метил-4-бром-3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (300 мг, 0,947 ммоль) добавляли к раствору пиридина (100 мкл, 1,24 ммоль) и продукта из Примера 237, Стадии 2 (200 мг, 0,735 ммоль) в ДХМ (1 мл). Полученный раствор оставляли при КТ на 24 ч. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (388 мг, 0,710 ммоль, выход 97%) в виде бледно-желтого пенистого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 546,2 (M+H)+, 544,1 (M-H)- через 1,74 мин.
Стадия 2: Метил-3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)фенил)сульфамоил)-4-циклопропилбензоат: Смесь продукта из стадии 1 выше (388 мг, 0,710 ммоль) и Pd-174 (50 мг, 0,069 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (5 мл, 2,50 ммоль) и затем нагревали при 60°C в течение 1,5 ч. Смесь охлаждали и оставляли при КТ на ночь. Смесь нейтрализовали насыщенным NH4Cl (водн.) (1 мл) и выпаривали в вакууме. Остаток экстрагировали ДХМ (2×4 мл). Экстракты объединяли и выпаривали в вакууме. Остаток частично очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (263 мг, 0,430 ммоль, выход 61%, чистота 83%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 508,3 (M+H)+, 506,2 (M-H)- через 1,77 мин.
Стадия 3: 3-(N-(2-(4-цианопиперидин-1-ил)-5-(трифторметил)-фенил)сульфамоил)-4-циклопропилбензойная кислота: Продукт из стадии 2 выше (263 мг, 0,430 ммоль, чистота 83%) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (2,5 мл, 2,50 ммоль). Полученную двухфазную смесь перемешивали при КТ в течение 3 дней. Смесь выпаривали в вакууме для удаления ТГФ. Полученную водную смесь подкисляли AcOH и выпаривали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (125 мг, 0,248 ммоль, выход 58%, чистота 98%). СВЭЖХ-МС (Метод 2): m/z 494,3 (M+H)+, 492,2 (M-H)- через 1,13 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,26 (с, 1H), 9,59 (с, 1H), 8,42 (д, J=1,9 Гц, 1H), 8,01 (дд, J=8,2, 1,9 Гц, 1H), 7,45-7,35 (м, 1H), 7,35-7,22 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 3,03-2,85 (м, 3H), 2,85-2,78 (м, 1H), 2,78-2,67 (м, 2H), 2,01-1,89 (м, 2H), 1,89-1,78 (м, 2H), 1,13-0,99 (м, 2H), 0,92-0,75 (м, 2H).
Пример 316: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
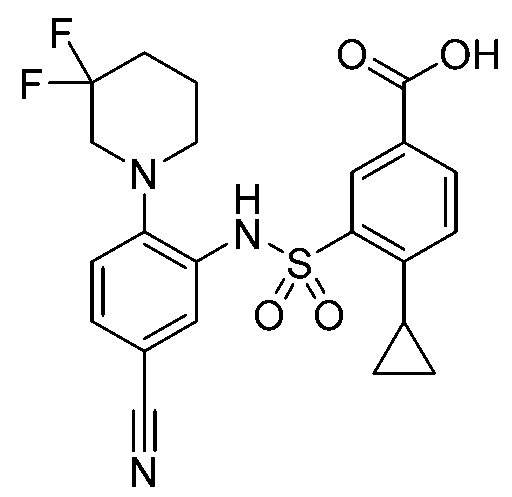
Стадия 1: Метил-4-бром-3-(хлорсульфонил)бензоат: Смесь 4-бром-3-(хлорсульфонил)бензойной кислоты (23,9 г, 76 ммоль) и SOCl2 (160 мл) нагревали с обратным холодильником в течение 4 ч. После охлаждения до КТ летучие вещества удаляли в вакууме, а остаток медленно добавляли в MeOH (500 мл) при 0°C. Осадок собирали и промывали небольшими количествами холодного MeOH с получением указанного в заголовке соединения (17,3 г, 54,7 ммоль, выход 72%, чистота 99%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,47 (д, J=2,0 Гц, 1H), 7,78-7,71 (м, 2H), 3,86 (с, 3H).
Стадия 2: Метил-4-бром-3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 233, Стадии 2 (150 мг, 0,632 ммоль, чистота 99%), продукта из Стадии 1 выше (218 мг, 0,695 ммоль, чистота 99%) и пиридина (153 мкл, 1,9 ммоль) в ДХМ (4 мл) перемешивали при 35°C в течение 6 дней. Реакционную смесь выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (327 мг, 0,547 ммоль, выход 86%, 86% чистота) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 514,1 (M+H)+ через 1,50 мин.
Стадия 3: метил-3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Дегазированную смесь продукта из стадии 2 выше (180 мг, 0,301 ммоль, чистота 86%) и Pd-174 (25,2 мг, 0,035 ммоль) в ТГФ (4 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (2,80 мл, 1,40 ммоль). Реакционную смесь перемешивали при 40°C в течение 1 ч, после чего выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (84 мг, 0,139 ммоль, выход 46%, чистота 79%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 476,3 (M+H)+ через 1,56 мин.
Стадия 4: 3-(N-(5-циано-2-(3,3-дифторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота: 1М LiOH (водн.) (1,39 мл, 1,39 ммоль) добавляли к раствору продукта из стадии 3 выше (84 мг, 0,139 ммоль, чистота 79%) в ТГФ (2 мл) и перемешивали полученную смесь при КТ в течение ночи. Реакционную смесь разбавляли EtOAc (50 мл) и промывали водой (2×50 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (5 мг, 10,3 мкмоль, выход 7%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 462,1 (M+H)+, 460,2 (M-H)- через 1,56 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,63 (д, J=1,8 Гц, 1H), 8,13 (дд, J=8,2, 1,9 Гц, 1H), 7,49 (д, J=1,9 Гц, 1H), 7,40 (дд, J=8,3, 1,9 Гц, 1H), 7,32 (д, J=8,3 Гц, 1H), 7,19 (д, J=8,2 Гц, 1H), 3,13 (т, J=10,9 Гц, 2H), 3,00 (т, J=5,4 Гц, 2H), 2,71 (тт, J=8,5, 5,2 Гц, 1H), 2,06 (тт, J=13,6, 6,4 Гц, 2H), 1,90 (h, J=6,0, 5,4 Гц, 2H), 1,19-1,14 (м, 2H), 0,87 (дт, J=6,8, 4,7 Гц, 2H). Два обмениваемых протона не наблюдали.
Пример 317: 4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
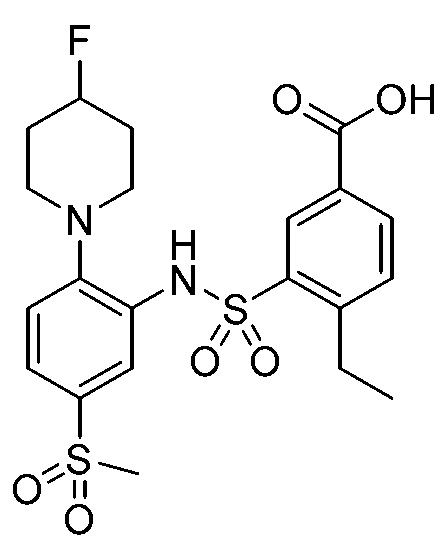
Стадия 1: 4-фтор-1-(4-(метилсульфонил)-2-нитрофенил)-пиперидин: Смесь 1-фтор-4-(метилсульфонил)-2-нитробензола (500 мг, 2,28 ммоль), гидрохлорида 4-фторпиперидина (318 мг, 2,28 ммоль) и триэтиламина (699 мкл, 5,02 ммоль) обрабатывали ультразвуком в ДХМ (6 мл) до образования прозрачного раствора. Реакционный раствор перемешивали при КТ в течение 2 ч, после чего разбавляли ДХМ (10 мл) и последовательно промывали 1М HCl (водн.) (15 мл), водой (15 мл) и солевым раствором (15 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (651 мг, 2,09 ммоль, выход 92%, чистота 97%) в виде ярко-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 303,2 (M+H)+ через 1,15 мин.
Стадия 2: 2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)-анилин: Продукт из стадии 1 выше (651 мг, 2,09 ммоль, чистота 97%) растворяли в уксусной кислоте и добавляли 5% Pd/C (20 мл) (50% об/об воды) Тип 87L (130 мг, 2,09 ммоль). Раствор гидрировали (5 бар) в течение 3 дней. Реакционную смесь фильтровали через Celite® и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (482 мг, 1,65 ммоль, выход 79%, чистота 93%) в виде красного масла, которое затвердевало при стоянии. СВЭЖХ-МС (Метод 2) m/z 273,2 (M+H)+ через 1,04 мин.
Стадия 3: метил-4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 203, Стадии 2 (159 мг, 0,575 ммоль, чистота 95%) добавляли к раствору пиридина (134 мкл, 1,65 ммоль) и продукта из Стадии 2 выше (150 мг, 0,512 ммоль, чистота 93%) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение ночи. Реакционную смесь выпаривали в вакууме, затем частично очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) и после этого очищали с помощью хроматографии (24 г обращенно-фазовый картридж C18, 15-65% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (175 мг, 0,351 ммоль, выход 69%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 499,3 (M+H)+ через 1,40 мин.
Стадия 4: 4-этил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,4 мл, 1,4 ммоль) добавляли к раствору продукта из Стадии 3 выше (175 мг, 0,351 ммоль) в ТГФ (3 мл). Реакционную смесь перемешивали при КТ в течение ночи, затем выпаривали в вакууме. Остаток растворяли в воде (12 мл) и промывали EtOAc (12 мл). Водную фазу подкисляли 1М HCl до pH 4-5 и продукт экстрагировали EtOAc (2×12 мл). Объединенные органические экстракты сушили над MgSO4 и выпаривали в вакууме с получением указанного в заголовке соединения (106 мг, 0,208 ммоль, выход 59%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 485,3 (M+H)+, 483,3 (M-H)- через 1,39 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,77 (шс, 1H), 8,31 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,9 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,57 (д, J=8,3 Гц, 1H), 7,29 (д, J=2,0 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 4,88-4,67 (м, 1H), 3,32 (с, 3H), 3,02 (к, J=7,4 Гц, 2H), 2,99-2,92 (м, 2H), 2,83-2,73 (м, 2H), 1,98-1,79 (м, 2H), 1,80-1,68 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 318: 3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензойная кислота
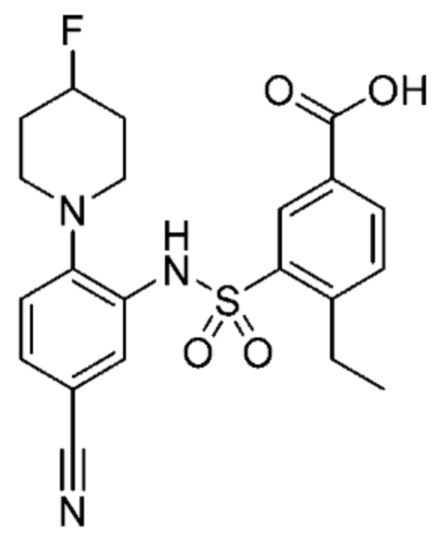
Стадия 1: 4-(4-фторпиперидин-1-ил)-3-нитробензонитрил: Смесь 4-фтор-3 нитробензонитрила (500 мг, 3,01 ммоль), гидрохлорида 4-фторпиперидина (420 мг, 3,01 ммоль) и триэтиламина (923 мкл, 6,62 ммоль) обрабатывали ультразвуком в ДХМ (6 мл) до образования прозрачного раствора. Реакционную смесь перемешивали при КТ в течение ночи, разбавляли ДХМ (40 мл) и последовательно промывали водой (50 мл) и солевым раствором (50 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (735 мг, 2,80 ммоль, выход 93%, чистота 95%) в виде ярко-оранжевого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 250,5 (M+H)+ через 1,31 мин.
Стадия 2: 3-амино-4-(4-фторпиперидин-1-ил)бензонитрил: Продукт из Стадии 1 выше (735 мг, 2,80 ммоль, чистота 95%) объединяли с порошком железа (3,29 г, 59 ммоль) и NH4Cl (тв) (205 мг, 3,83 ммоль) в ИПС/воде 2:1 (30 мл), нагревали при 70°C в течение ночи. Реакционной смеси позволяли охладиться, после чего фильтровали через Celite®, промывали EtOAc (200 мл) и выпаривали фильтрат в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (350 мг, 1,56 ммоль, выход 56%, чистота 98%) в виде красного масла. СВЭЖХ-МС (Метод 2) m/z 220,6 (M+H)+ через 1,27 мин.
Стадия 3: метил-3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)сульфамоил)-4-этилбензоат: Продукт из Примера 203, Стадии 2 (198 мг, 0,753 ммоль) добавляли к раствору пиридина (166 мкл, 2,05 ммоль) и продукта из Стадии 3 выше (150 мг, 0,670 ммоль, чистота 98%) в ДХМ (1 мл). Реакционную смесь перемешивали при КТ в течение 4 дней, после чего выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (322 мг) в виде красного масла. СВЭЖХ-МС (Метод 2) m/z 446,3 (M+H)+ через 1,57 мин.
Стадия 4: 3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)-сульфамоил)-4-этилбензойная кислота: 1М LiOH (водн.) (2,89 мл, 2,89 ммоль) добавляли к раствору продукта из Стадии 3 выше (322 мг) в ТГФ (6 мл). Реакционную смесь перемешивали при КТ в течение ночи, после чего выпаривали в вакууме. Остаток растворяли в воде (12 мл) и промывали EtOAc (12 мл). Водную фазу подкисляли помощью 1М HCl до pH 4-5 и продукт экстрагировали EtOAc (2×15 мл). Объединенные органические экстракты сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (234 мг, 0,515 ммоль, выход 77% в 2 стадиях, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 432,4 (M+H)+, 430,3 (M-H)- через 1,53 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,77 (шс, 1H), 8,31 (д, J=1,8 Гц, 1H), 8,11 (дд, J=8,0, 1,8 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,57 (д, J=8,3 Гц, 1H), 7,29 (д, J=2,0 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 4,77 (дт, J=48,6, 7,0, 3,5 Гц, 1H), 3,02 (к, J=7,4 Гц, 2H), 2,96 (т, J=9,9 Гц, 2H), 2,83-2,71 (м, 2H), 1,95-1,83 (м, 2H), 1,80-1,67 (м, 2H), 1,21 (т, J=7,4 Гц, 3H).
Пример 319: 4-циклопропил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
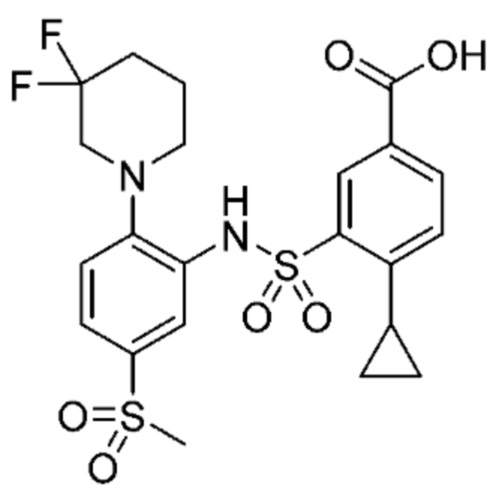
Стадия 1: метил-4-бром-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (350 мг, 1,11 ммоль) добавляли к раствору пиридина (100 мкл, 1,24 ммоль) и продукта из Примера 313, Стадии 2 (252 мг, 0,868 ммоль) в ДХМ (2 мл). Полученный раствор оставляли при КТ на 11 дней. Смесь обрабатывали PhMe (1 мл) и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (276 мг, 0,482 ммоль, выход 56%, чистота 99%) в виде белого пенистого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 567,3 (M+H)+, 565,5 (M-H)- через 1,54 мин.
Стадия 2: метил-4-циклопропил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Смесь продукта из стадии 1 выше (276 мг, 0,482 ммоль, чистота 99%) и Pd-174 (50 мг, 0,069 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (5 мл, 2,50 ммоль) и затем нагревали при 60°C в течение 1,5 ч. Смесь охлаждали и оставляли при КТ на ночь. Смесь нейтрализовали насыщенным NH4Cl (водн.) (1 мл) и выпаривали в вакууме. Остаток экстрагировали ДХМ (2×4 мл). Экстракты объединяли и выпаривали в вакууме. Остаток частично очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/ДХМ), затем хроматографии на силикагеле (24 г картридж, 10-60% EtOAc/изогексан) с получением указанного в заголовке соединения (160 мг, 0,275 ммоль, выход 57%, 91% чистота) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 529,3 (M+H)+, 527,3 (M-H)- через 1,58 мин.
Стадия 3: 4-циклопропил-3-(N-(2-(3,3-дифторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: Продукт из стадии 2 выше (160 мг, 0,275 ммоль, чистота 91%) растворяли в ТГФ (5 мл) и обрабатывали 1М LiOH (водн.) (2,5 мл, 2,50 ммоль). Полученную двухфазную смесь перемешивали при КТ в течение 3 дней. Смесь выпаривали в вакууме для удаления ТГФ. Полученную водную смесь подкисляли AcOH и выпаривали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (86 мг, 0,164 ммоль, выход 60%, чистота 98%). СВЭЖХ-МС (Метод 2): m/z 515,3 (M+H)+, 513,2 (M-H)- через 1,00 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,27 (с, 1H), 9,38 (с, 1H), 8,37 (д, J=1,8 Гц, 1H), 8,02 (д, J=8,2 Гц, 1H), 7,61 (с, 1H), 7,42-7,27 (м, 2H), 7,19 (д, J=8,2 Гц, 1H), 3,40-3,24 (м, 2H), 3,17-3,04 (м, 2H), 2,97 (с, 3H), 2,81-2,66 (м, 1H), 2,09-1,94 (м, 2H), 1,87-1,68 (м, 2H), 1,20-1,00 (м, 2H), 0,96-0,79 (м, 2H). Два протона частично перекрывались водой.
Пример 320: 4-циклопропил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
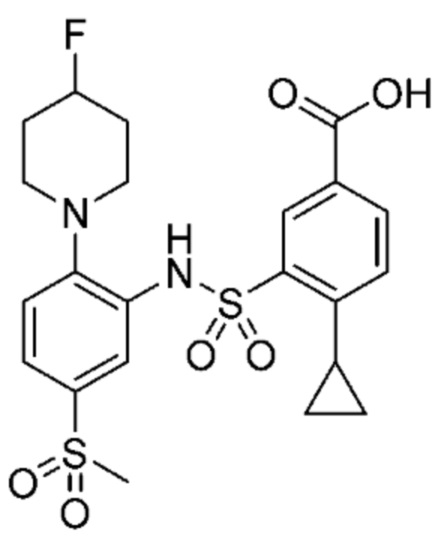
Стадия 1: Метил-4-бром-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (420 мг, 1,33 ммоль, чистота 99%) добавляли к раствору пиридина (296 мкл, 3,66 ммоль) и продукта из Примера 317, Стадии 2 (332 мг, 1,13 ммоль, чистота 93%) в ДХМ (2 мл). Реакционную смесь перемешивали при КТ в течение 6 дней, после чего выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (395 мг, 0,623 ммоль, выход 55%, чистота 87%) в виде бледно-красного твердого вещества. СВЭЖХ-МС (Метод 2) m/z 549,1 (M+H)+ через 1,40 мин.
Стадия 2: метил-4-циклопропил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Дегазированную смесь продукта из Стадии 1 выше (395 мг, 0,623 ммоль, чистота 87%) и Pd-174 (51,8 мг, 0,072 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (5,75 мл, 2,88 ммоль). Реакционную смесь нагревали при 70°C в течение 1 ч. Добавляли дополнительное количество Pd-174 (51,8 мг, 0,072 ммоль) и бромида циклопропилцинка (II) (0,5 М в ТГФ) (5,75 мл, 2,88 ммоль) и перемешивали смесь при 70°C в течение 3 ч. Смеси позволяли охладиться до КТ и выпаривали в вакууме. Остаток делили между солевым раствором (75 мл) и ДХМ (75 мл) и разделяли фазы. Водную фазу экстрагировали ДХМ (75 мл), а органические фазы объединяли и фильтровали через Celite®. Фильтрат сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (226 мг, 0,398 ммоль, выход 64%, чистота 90%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 511,3 (M+H)+ через 1,48 мин.
Стадия 3: 4-циклопропил-3-(N-(2-(4-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1,77 мл, 1,77 ммоль) добавляли к раствору продукта из Стадии 2 выше (226 мг, 0,398 ммоль, чистота 90%) в ТГФ (3,5 мл). Реакционную смесь перемешивали при КТ в течение ночи, после чего выпаривали в вакууме. Остаток растворяли в воде (12 мл) и промывали EtOAc (12 мл). Водную фазу подкисляли до pH 4-5 1М HCl (водн.), собирали осадок с помощью фильтрации и сушили в вакууме. Неочищенный продукт очищали с помощью хроматографии на 24 г обращенно-фазовом картридже (0-100% MeCN/вода с 0,1% муравьиной кислоты) с получением указанного в заголовке соединения (93 мг, 0,178 ммоль, выход 45%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 497.3 (M+H)+, 495.2 (M-H)- через 1,38 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,2 (шс, 1H), 9,65 (шс, 1H), 8,40 (д, J=1,9 Гц, 1H), 8,01 (д, J=8,1 Гц, 1H), 7,59 (с, 1H), 7,51 (д, J=2,2 Гц, 1H), 7,30 (д, J=8,6 Гц, 1H), 7,16 (д, J=8,2 Гц, 1H), 4,87-4,71 (м, 1H), 3,02-2,99 (м, 5H), 2,87-2,74 (м, 3H), 2,01-1,88 (м, 2H), 1,85-1,74 (м, 2H), 1,13-1,05 (м, 2H), 0,89-0,85 (м, 2H).
Пример 321: (R)-4-циклопропил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота
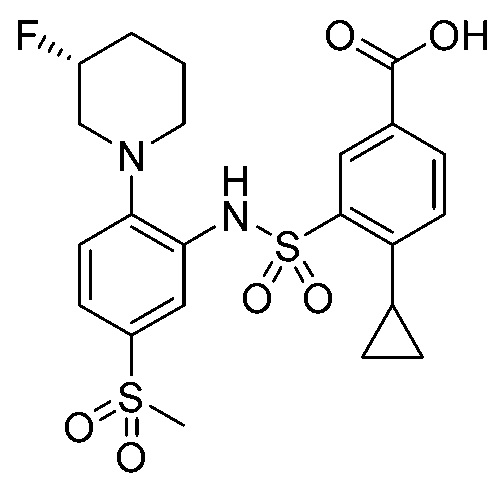
Стадия 1: (R)-метил-4-бром-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (198 мг, 0,63 ммоль, чистота 99%) добавляли к раствору продукта из Примера 262, Стадии 2 (156 мг, 0,567 ммоль, чистота 99%) и пиридина (139 мкл, 1,72 ммоль) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 6 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (207 мг, 0,377 ммоль, выход 66%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 550,1 (M+H)+ через 1,46 мин.
Стадия 2: (R)-метил-4-циклопропил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензоат: Дегазированную смесь продукта из Стадии 1 выше (207 мг, 0,377 ммоль) и Pd-174 (27,2 мг, 0,038 ммоль) в ТГФ (6 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (3,01 мл, 1,51 ммоль). Полученную смесь нагревали при 70°C в течение 1 ч. Добавляли дополнительное количество Pd-174 (27,2 мг, 0,038 ммоль) и бромида циклопропилцинка (II) (0,5 М в ТГФ) (3,01 мл, 1,51 ммоль) и перемешивали реакцию при 70°C в течение 3 ч. Смеси позволяли охладиться до КТ, затем фильтровали через Celite® и выпаривали в вакууме. Остаток делили между солевым раствором (75 мл) и ДХМ (75 мл) и разделяли фазы. Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (128 мг, 0,248 ммоль, выход 66%, чистота 99%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 511,3 (M+H)+ через 1,49 мин.
Стадия 3: (R)-4-циклопропил-3-(N-(2-(3-фторпиперидин-1-ил)-5-(метилсульфонил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (1 мл, 1,00 ммоль) добавляли к раствору продукта из Стадии 2 выше (128 мг, 0,248 ммоль, чистота 99%) в ТГФ (2 мл). Полученную смесь перемешивали при КТ в течение ночи. Реакционную смесь выпаривали в вакууме, растворяли в воде (12 мл) и промывали EtOAc (12 мл). Водную фазу подкисляли до pH 4-5 1М HCl (водн.) и экстрагировали EtOAc (2×12 мл). Продукт осаждался в виде белого твердого вещества из органической фазы, и его растворяли в ТГФ (20 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Для удаления остаточного ТГФ, остаток растворяли в 1М LiOH (водн.) (5 мл) и подкисляли с использованием 1М HCl (водн.) до pH 4-5. Продукт осаждался в виде белого твердого вещества, и его собирали с помощью фильтрации, с получением указанного в заголовке соединения (63 мг, 0,121 ммоль, выход 49%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 497,6 (M+H)+, 495,2 (M-H)- через 1,40 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,37 (шс, 1H), 8,41 (д, J=1,9 Гц, 1H), 8,01 (дд, J=8,2, 1,9 Гц, 1H), 7,58 (д, J=8,4 Гц, 1H), 7,49 (д, J=2,2 Гц, 1H), 7,32 (д, J=8,5 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 4,84-4,68 (м, 1H), 3,26-3,13 (м, 1H), 3,02-2,88 (м, 5H), 2,89-2,79 (м, 1H), 2,79-2,67 (м, 1H), 2,01-1,85 (м, 1H), 1,77 (д, J=6,7 Гц, 1H), 1,71-1,51 (м, 2H), 1,14-1,04 (м, 2H), 0,95-0,88 (м, 1H), 0,87-0,80 (м, 1H).
Пример 322: 3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
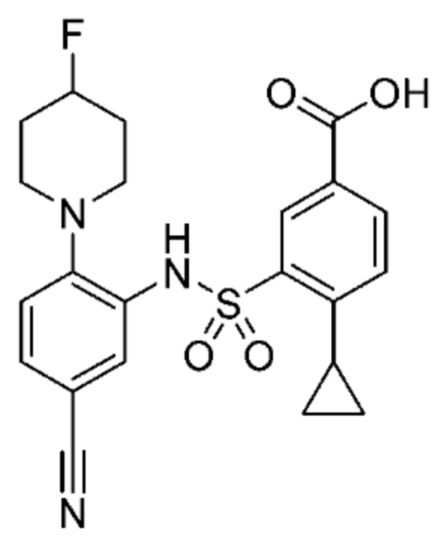
Стадия 1: Метил-4-бром-3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (393 мг, 1,24 ммоль, чистота 99%) добавляли к раствору пиридина (277 мкл, 3,42 ммоль) и продукта из Примера 318, Стадии 2 (250 мг, 1,12 ммоль, чистота 98%) в ДХМ (2 мл). Полученный раствор перемешивали при КТ в течение 6 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (372 мг, 0,749 ммоль, выход 67%) в виде розового твердого вещества. СВЭЖХ-МС (Метод 2) m/z 497,2 (M+H)+ через 1,46 мин.
Стадия 2: Метил-3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Дегазированную смесь продукта из Стадии 1 выше (372 мг, 0,749 ммоль) и Pd-174 (54,1 мг, 0,075 ммоль) в ТГФ (10 мл) обрабатывали бромидом циклопропилцинка (II) (0,5 М в ТГФ) (6 мл, 3,00 ммоль). Полученную смесь нагревали при 70°C в течение 1 ч. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (276 мг, 0,57 ммоль, выход 76%, чистота 95%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 458,4 (M+H)+ через 1,53 мин.
Стадия 3: 3-(N-(5-циано-2-(4-фторпиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: 1М LiOH (водн.) (2,41 мл, 2,41 ммоль) добавляли к раствору продукта из Стадии 2 выше (276 мг, 0,573 ммоль, чистота 95%) в ТГФ (5 мл). Реакционную смесь перемешивали при КТ в течение ночи, после чего выпаривали в вакууме. Реакционную смесь подкисляли до pH 4-5 1М HCl (водн.). Осадок собирали с помощью фильтрации и сушили в вакууме. Неочищенный продукт очищали с помощью хроматографии (24 г обращенно-фазовый картридж C18, MeCN/0,1% муравьиной кислоты 0-100% (водн.)) с получением указанного в заголовке соединения (169 мг, 0,362 ммоль, выход 63%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 444,3 (M+H)+, 442,3 (M-H)- через 1,53 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,69 (шс, 1H), 8,37 (д, J=1,9 Гц, 1H), 8,03 (дд, J=8,2, 1,9 Гц, 1H), 7,55 (д, J=8,5 Гц, 1H), 7,28 (д, J=2,0 Гц, 1H), 7,22 (д, J=8,4 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 4,86-4,69 (м, 1H), 3,04-2,96 (м, 2H), 2,87-2,71 (м, 3H), 1,97-1,83 (м, 2H), 1,82-1,69 (м, 2H), 1,13-1,04 (м, 2H), 0,91-0,84 (м, 2H).
Пример 323: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
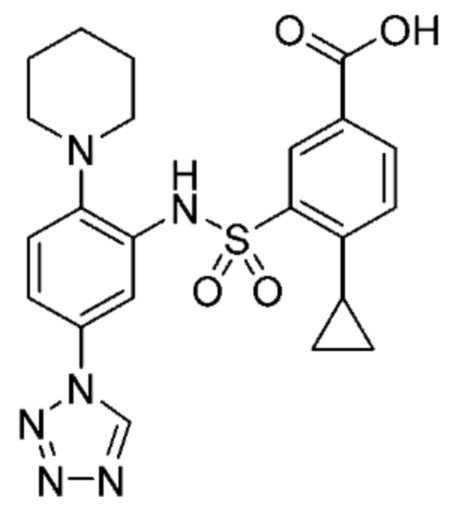
Стадия 1: Метил-3-(бензилтио)-4-циклопропилбензоат: К дегазированной смеси метил-3-бром-4-циклопропилбензоата (850 мг, 3,33 ммоль), DIPEA (1,2 мл, 6,87 ммоль) и XantPhos Pd G3 (300 мг, 0,316 ммоль) в диоксане (13 мл) добавляли фенилметантиол (425 мкл, 3,62 ммоль) и перемешивали смесь при 100°C в течение ночи. Смесь охлаждали до КТ, концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (40 г картридж, 20-70% ДХМ/изогексан) с получением указанного в заголовке соединения (600 мг, 1,91 ммоль, выход 58%, чистота 95%) в виде оранжевого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,86 (д, J=1,8 Гц, 1H), 7,67 (дд, J=8,1, 1,8 Гц, 1H), 7,40-7,35 (м, 2H), 7,35-7,29 (м, 2H), 7,28-7,22 (м, 1H), 7,04 (д, J=8,1 Гц, 1H), 4,27 (с, 2H), 3,83 (с, 3H), 2,21-2,12 (м, 1H), 1,06-0,99 (м, 2H), 0,75-0,68 (м, 2H).
Стадия 2: Метил-3-(хлорсульфонил)-4-циклопропилбензоат: Смесь продукта из Стадии 1 выше (600 мг, 1,91 ммоль, чистота 95%), AcOH (110 мкл, 1,92 ммоль) и воды (250 мкл) в MeCN (9 мл) при -10°C обрабатывали 1,3-дихлор-5,5-диметилимидазолидин-2,4-дионом (565 мг, 2,87 ммоль). Смесь перемешивали при -10°C в течение 3 ч. Смесь разбавляли водой (50 мл) и экстрагировали ДХМ (2×50 мл). Органические фазы объединяли, сушили над MgSO4, фильтровали и концентрировали в вакууме на силикагеле, после чего очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% ДХМ/изогексан) с получением указанного в заголовке соединения (440 мг, 1,52 ммоль, выход 80%, чистота 95%) в виде бледно-желтого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,36 (д, J=2,0 Гц, 1H), 7,77 (дд, J=8,2, 2,1 Гц, 1H), 6,84 (д, J=8,2 Гц, 1H), 3,84 (с, 3H), 3,22-3,01 (м, 1H), 1,08-0,98 (м, 2H), 0,79-0,70 (м, 2H).
Стадия 3: Метил-4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Продукт из Стадии 2 выше (50 мг, 0,182 ммоль) добавляли к раствору пиридина (40 мкл, 0,495 ммоль) и продукта из Примера 214, Стадии 3 (40 мг, 0,164 ммоль, чистота 99%) в ДХМ (300 мкл). Полученный раствор перемешивали при КТ в течение ночи. Добавляли дополнительную порцию пиридина (40 мкл, 0,495 ммоль) и перемешивали реакцию в течение 3 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (76 мг, 0,157 ммоль, выход 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 483,4 (M+H)+ через 1,66 мин.
Стадия 4: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота: 1М LiOH (водн.) (630 мкл, 0,63 ммоль) добавляли к раствору продукта из Стадии 3 выше (76 мг, 0,157 ммоль) в ТГФ (1,3 мл). Реакционную смесь перемешивали при КТ в течение ночи, после чего выпаривали в вакууме. Реакционную смесь доводили до pH 6 1М HCl (водн.). Осадок собирали с помощью фильтрации и сушили в вакууме. Неочищенный продукт очищали с помощью хроматографии на 24 г обращенно-фазовом картридже (0-100% MeCN/вода с 0,1% муравьиной кислоты) с получением указанного в заголовке соединения (52 мг, 0,105 ммоль, выход 67%, чистота 95%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 469,4 (M+H)+, 467,3 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,94 (с, 1H), 8,48 (д, J=1,9 Гц, 1H), 8,00 (дд, J=8,2, 1,9 Гц, 1H), 7,67 (д, J=2,5 Гц, 1H), 7,56 (д, J=8,6 Гц, 1H), 7,37 (д, J=8,6 Гц, 1H), 7,16 (д, J=8,2 Гц, 1H), 2,86-2,76 (м, 1H), 2,73 (т, J=5,2 Гц, 4H), 1,62-1,53 (м, 4H), 1,52-1,43 (м, 2H), 1,13-1,06 (м, 2H), 0,88-0,79 (м, 2H). Два обмениваемых протона не наблюдали.
Пример 324: 3-(N-(5-циано-4-фтор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
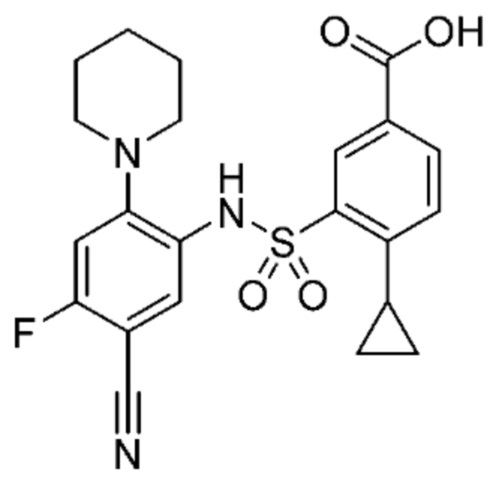
Стадия 1: 2-фтор-5-нитро-4-(пиперидин-1-ил)бензонитрил: Пиперидин (269 мкл, 2,72 ммоль) добавляли к суспензии 2,4-дифтор-5-нитробензонитрила (500 мг, 2,72 ммоль) в ДХМ (5 мл) при 0°C. Полученному раствору позволяли нагреться до КТ и перемешивали в течение 2 ч. Добавляли дополнительную порцию ДХМ (50 мл) и промывали реакционную смесь водой (2×60 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (660 мг, 2,44 ммоль, выход 90%, чистота 92%) в виде желтого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 250,6 (M+H)+ через 1,50 мин.
Стадия 2: 5-амино-2-фтор-4-(пиперидин-1-ил)бензонитрил: Продукт из Стадии 1 выше (660 мг, 2,44 ммоль, чистота 92%) объединяли с цинковой пылью (1,04 г, 15,9 ммоль) и NH4Cl (тв) (850 мг, 15,9 ммоль) в ТГФ (11 мл) и воде (4 мл). Полученный раствор перемешивали при КТ в течение ночи. Реакционную смесь фильтровали через Celite® и выпаривали в вакууме. Остаток растворяли в EtOAc (60 мл) и промывали водой (60 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (533 мг, 2,19 ммоль, выход 90%, чистота 90%) в виде темно-коричневого твердого вещества. СВЭЖХ-МС (Метод 2) m/z 220,3 (M+H)+ через 1,51 мин.
Стадия 3: метил-4-бром-3-(N-(5-циано-4-фтор-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Продукт из Примера 316, Стадии 1 (393 мг, 1,21 ммоль, чистота 99%) добавляли к раствору пиридина (277 мкл, 3,42 ммоль) и продукта из Стадии 2 выше (250 мг, 1,03 ммоль, чистота 90%) в ДХМ (2 мл). Полученный раствор перемешивали при КТ в течение 5 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (357 мг, 0,712 ммоль, выход 69%, чистота 99%) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 497,3 (M+H)+ через 1,63 мин.
Стадия 4: метил-3-(N-(5-циано-4-фтор-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: К дегазированной смеси продукта из Стадии 3 выше (357 мг, 0,712 ммоль, чистота 99%) и Pd-174 (52 мг, 0,072 ммоль) в ТГФ (10 мл) добавляли бромид циклопропилцинка (II) (0,5 М в ТГФ) (5,8 мл, 2,9 ммоль). Полученный раствор нагревали при 70°C в течение 1 ч. Смеси позволяли охладиться до КТ, выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (273 мг, 0,489 ммоль, выход 68%, 82% чистота) в виде коричневого масла. СВЭЖХ-МС (Метод 2) m/z 458,4 (M+H)+ через 1,69 мин.
Стадия 5: 3-(N-(5-циано-4-фтор-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: 1М LiOH (водн.) (2,4 мл, 2,4 ммоль) добавляли к раствору продукта из стадии 4 выше (273 мг, 0,489 ммоль, чистота 82%) в ТГФ (5 мл). Реакционную смесь перемешивали при КТ в течение ночи, выпаривали в вакууме для удаления ТГФ и доводили до pH 6 1М HCl (водн.). Осадок собирали с помощью фильтрации и сушили в вакууме. Неочищенный продукт очищали с помощью хроматографии на 24 г обращенно-фазовом картридже (0-100% MeCN/вода с 0,1% муравьиной кислоты) с получением указанного в заголовке соединения (80 мг, 0,177 ммоль, 36% выход, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 444,4 (M+H)+, 442,4 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,3 (шс, 1H), 9,65 (шс, 1H), 8,30 (д, J=1,9 Гц, 1H), 8,02 (дд, J=8,2, 1,9 Гц, 1H), 7,16 (д, J=8,2 Гц, 1H), 7,13 (д, J=7,2 Гц, 1H), 7,03 (д, J=12,0 Гц, 1H), 3,02-2,92 (м, 4H), 2,74-2,65 (м, 1H), 1,49-1,39 (м, 6H), 1,13-1,04 (м, 2H), 0,95-0,84 (м, 2H).
Пример 325: 4-циклопропил-3-(N-(2-(3-гидроксиазетидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
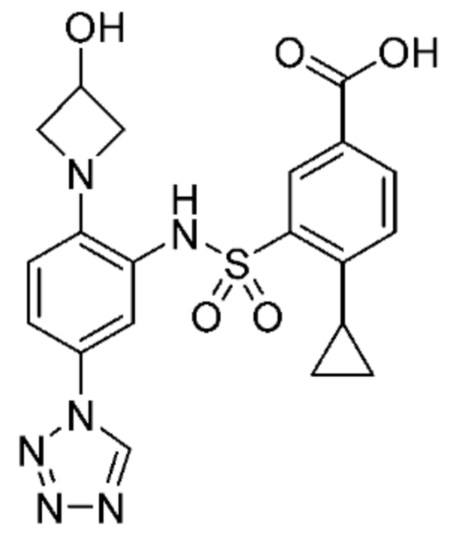
Продукт из Примера 323, Стадии 2 (42,9 мг, 0,148 ммоль) добавляли к раствору пиридина (34,5 мкл, 0,426 ммоль) и продукта из Примера 283, Стадии 2 (33 мг, 0,134 ммоль) в ДХМ (0,2 мл). Полученный раствор перемешивали при КТ в течение 3 дней, затем выпаривали в вакууме. Остаток растворяли в ТГФ (0,5 мл) и обрабатывали 1М LiOH (водн.) (240 мкл, 0,240 ммоль). Полученную смесь перемешивали при КТ в течение ночи. Добавляли дополнительное количество 1М LiOH (водн.) (240 мкл, 0,240 ммоль) и перемешивали смесь в течение 24 ч. Реакционную смесь выпаривали в вакууме, а остаток растворяли в воде (12 мл) и промывали МТБЭ (12 мл). Водную фазу подкисляли 1М HCl (водн.) до pH 4-5 и экстрагировали продукт EtOAc (2×12 мл). Органические фазы объединяли и пропускали через фазовый сепаратор, после чего выпаривали в вакууме. Остаток очищали с помощью хроматографии (24 г обращенно-фазовый картридж C18, 15-40% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (6 мг, 0,013 ммоль, выход 10%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 457,4 (M+H)+, 455,3 (M-H)- через 0,61 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 9,33 (с, 1H), 8,51 (д, J=1,8 Гц, 1H), 8,35 (с, 1H), 8,11 (дд, J=8,2, 1,8 Гц, 1H), 7,50 (дд, J=8,8, 2,5 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,97 (д, J=2,5 Гц, 1H), 6,68 (д, J=8,8 Гц, 1H), 4,69-4,60 (м, 1H), 4,47-4,41 (м, 2H), 3,87 (дд, J=8,6, 4,9 Гц, 2H), 2,86-2,77 (м, 1H), 1,18-1,10 (м, 2H), 0,96-0,89 (м, 2H).
Пример 326: 3-(N-(5-циано-2-(4-цианопиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
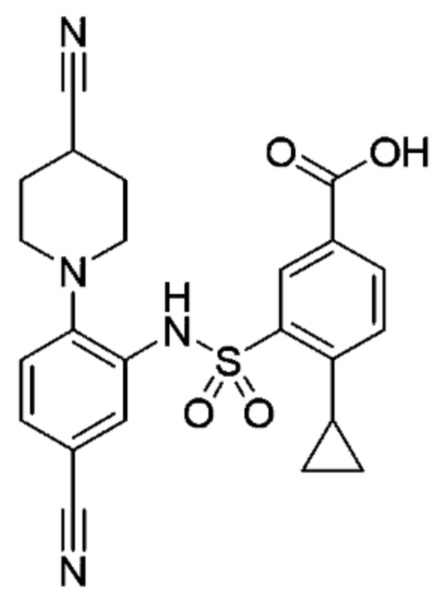
Стадия 1: 1-(4 циано 2-нитрофенил)пиперидин-4-карбонитрил: Смесь 4-фтор-3 нитробензонитрила (300 мг, 1,81 ммоль), пиперидин-4-карбонитрила (220 мкл, 1,97 ммоль) и Et3N (800 мкл, 5,74 ммоль) в ДХМ (9 мл) перемешивали при КТ в течение ночи. Смесь разбавляли ДХМ (15 мл), промывали насыщенным NH4Cl (водн.) (15 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме с получением указанного в заголовке соединения (449 мг, 1,73 ммоль, выход 96%, чистота 99%) в виде желтого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,33 (д, J=2,1 Гц, 1H), 7,90 (дд, J=8,8, 2,1 Гц, 1H), 7,40 (д, J=8,8 Гц, 1H), 3,31-3,23 (м, 2H), 3,18-3,09 (м, 3H), 2,03-1,93 (м, 2H), 1,86-1,76 (м, 2H).
Стадия 2: 1-(2 амина 4 цианофенил) пиперидин-4-карбонитрил: Смесь продукта из Стадии 1 выше (449 мг, 1,73 ммоль, чистота 99%), порошка железа (2 г, 35,8 ммоль) и NH4Cl (тв) (111 мг, 2,08 ммоль) в ИПС (15 мл) и воде (7,5 мл) нагревали при 90°C в течение ночи. После охлаждения до КТ смесь фильтровали через Celite®, промывая EtOAc, а затем выпаривали в вакууме. Остаток экстрагировали ДХМ (2×30 мл), и объединенные органические фазы сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (196 мг, 0,840 ммоль, выход 48%, чистота 97%) в виде светло-коричневого твердого вещества после растирания с МТБЭ. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,00-6,92 (м, 3H), 5,19 (с, 2H), 3,07-2,92 (м, 3H), 2,80-2,70 (м, 2H), 2,07-1,97 (м, 2H), 1,97-1,86 (м, 2H).
Стадия 3: метил-3-(N-(5-циано-2-(4-цианопиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Смесь продукта из Стадии 2 выше (50 мг, 0,214 ммоль, чистота 97%), продукта из Примера 323, Стадии 2 (62 мг, 0,214 ммоль) и пиридина (52,0 мкл, 0,643 ммоль) в ДХМ (1 мл) перемешивали при 35°C в течение 3 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (77 мг, 0,157 ммоль, выход 73%, чистота 95%) в виде светло-фиолетового твердого вещества. СВЭЖХ-МС (Метод 1) m/z 465,4 (M+H)+, 463,3 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,72 (с, 1H), 8,37 (д, J=1,9 Гц, 1H), 8,04 (д, J=8,1 Гц, 1H), 7,62-7,52 (м, 1H), 7,31 (д, J=2,0 Гц, 1H), 7,23-7,16 (м, 2H), 3,86 (с, 3H), 2,99-2,89 (м, 3H), 2,79-2,71 (м, 3H), 1,90-1,84 (м, 2H), 1,76-1,66 (м, 2H), 1,11-1,04 (м, 2H), 0,91-0,84 (м, 2H).
Стадия 4: 3-(N-(5-циано-2-(4-цианопиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: Смесь продукта из стадии 3 выше (77 мг, 0,157 ммоль, чистота 95%) и LiOH (27 мг, 0,631 ммоль) в ТГФ/MeOH/воде (4:1:1, 2,7 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×10 мл). Органические фазы объединяли и промывали солевым раствором (5 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (12,1 мг, 0,026 ммоль, выход 16%, чистота 98%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 451,8 (M+H)+, 449,3 (M-H)- через 1,39 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,28 (с, 1H), 9,68 (с, 1H), 8,36 (д, J=1,9 Гц, 1H), 8,02 (дд, J=8,2, 1,9 Гц, 1H), 7,55 (д, J=8,2 Гц, 1H), 7,30 (д, J=2,0 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 3,01-2,89 (м, 3H), 2,81-2,71 (м, 3H), 1,93-1,85 (м, 2H), 1,80-1,70 (м, 2H), 1,12-1,04 (м, 2H), 0,91-0,84 (м, 2H).
Пример 327: 3-(N-(4-хлор-5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
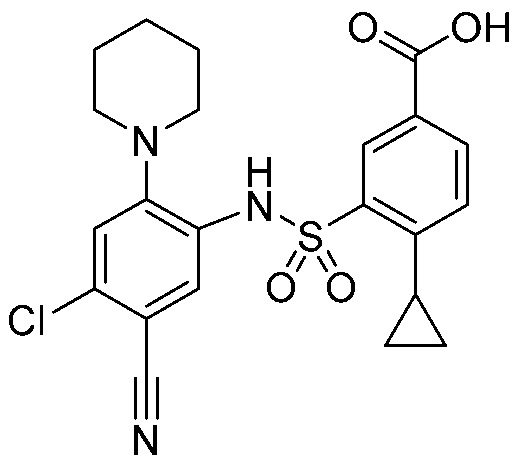
Стадия 1: 2-хлор-4-фтор-5 нитробензонитрил: 2-хлор-4-фтор-бензонитрил (1,00 г, 6,43 ммоль) растворяли в конц. H2SO4 (водн.) (6,85 мл, 129 ммоль) и охлаждали до 0°C, после чего добавляли азотную кислоту (8,45 мл, 129 ммоль). Смесь выдерживали при 0°C в течение 30 мин, после чего перемешивали при КТ в течение ночи. Реакционную смесь разбавляли водой (50 мл) и экстрагировали ДХМ (50 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-50% МТБЭ/изогексан) с получением указанного в заголовке соединения (0,240 г, 1,15 ммоль, выход 18%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1) m/z без ионизации через 1,17 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,97 (д, J=7,7 Гц, 1H), 8,30 (д, J=10,9 Гц, 1H).
Стадия 2: 2-хлор-5-нитро-4-(пиперидин-1-ил)бензонитрил: Продукт из Стадии 1 выше (0,240 г, 1,15 ммоль, чистота 96%) в сухом ДХМ (10 мл) обрабатывали триэтиламином (0,160 мл, 1,15 ммоль) и пиперидином (0,113 мл, 1,15 ммоль) и перемешивали смесь при КТ в течение 24 ч. Реакционную смесь разбавляли водой (50 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили (MgSO4), фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (0,298 г, 1,10 ммоль, выход 96%, чистота 98%) в виде ярко-оранжевого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 266,5 (M+H)+, в 1,63 мин.
Стадия 3: 5-амино-2-хлор-4-(пиперидин-1-ил)бензонитрил: Продукт из Стадии 2 выше (0,298 г, 1,10 ммоль, чистота 98%) объединяли с цинковой пылью (0,431 г, 6,59 ммоль) и NH4Cl (тв) (0,353 г, 6,59 ммоль) в ТГФ (9 мл) и воде (3 мл). Полученную смесь перемешивали при КТ в течение 16 ч, фильтровали через Celite® и затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (0,243 г, 0,897 ммоль, выход 82%, 87% чистота) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 236,3 (M+H)+, в 1,66 мин.
Стадия 4: Метил-3-(N-(4-хлор-5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: К раствору продукта из Стадии 3 выше (0,050 г, 0,212 ммоль, чистота 87%) в ДХМ (3 мл) и пиридине (0,103 мл, 1,27 ммоль) добавляли продукт из Примера 323, Стадии 2 (0,058 г, 0,212 ммоль) и перемешивали реакционную смесь при КТ в течение 72 ч, а затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (0,039 г, 0,081 ммоль, выход 38%, чистота 98%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1) m/z 474,3 (M+H)+, 472,2 (M-H)- через 1,91 мин.
Стадия 5: 3-(N-(4-хлор-5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: 1М LiOH (водн.) (0,242 мл, 0,242 ммоль) добавляли к раствору продукта из стадии 4 выше (39 мг, 0,081 ммоль, чистота 98%) в ТГФ (5 мл) и перемешивали полученный раствор при КТ в течение 16 ч. Затем реакционную смесь доводили до pH 6 10% в/об лимонной кислотой (водн.) и собирали образовавшийся осадок под вакуумом и промывали водой (5 мл) с получением указанного в заголовке соединения (21,6 мг, 0,046 ммоль, выход 57%, чистота 98%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 460,3 (M+H)+, 458,3 (M-H)- через 1,77 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 9,70 (шс, 1H), 8,35 (д, J=1,9 Гц, 1H), 8,00 (д, J=8,2 Гц, 1H), 7,24 (с, 1H), 7,13 (д, J=8,7 Гц, 2H), 3,00-2,87 (м, 4H), 2,85-2,70 (м, 1H), 1,55-1,40 (м, 6H), 1,07 (дд, J=8,2, 2,5 Гц, 2H), 0,87 (д, J=5,5 Гц, 2H).
Пример 328: 3-(N-(5-циано-2-(цис-3,5-диметилпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
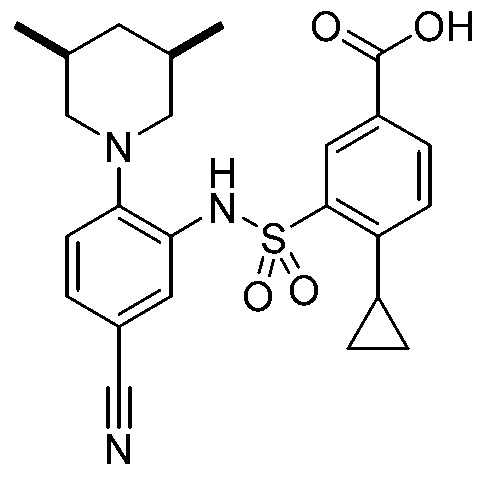
Стадия 1: 4-(цис-3,5-диметилпиперидин-1-ил)-3-нитробензонитрил: Триэтиламин (520 мкл, 3,73 ммоль) добавляли к раствору 4-фтор-3 нитробензонитрила (250 мг, 1,51 ммоль) и цис-3,5-диметилпиперидина (190 мг, 1,68 ммоль) в ДХМ (2 мл) и перемешивали полученный раствор при КТ в течение ночи. Реакционную смесь разбавляли ДХМ (10 мл), промывали водой (2×12 мл) и солевым раствором (12 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (370 мг, 1,41 ммоль, выход 94%, чистота 99%) в виде ярко-желтого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 260,3 (M+H)+ через 1,71 мин.
Стадия 2: 3-амино-4-(цис-3,5-диметилпиперидин-1-ил)бензонитрил: Продукт из Стадии 1 выше (370 мг, 1,43 ммоль) объединяли с цинковой пылью (560 мг, 8,56 ммоль) и хлоридом аммония (458 мг, 8,56 ммоль) в ТГФ (6 мл) и воде (2 мл). Полученную смесь перемешивали при КТ в течение ночи. Реакционную смесь фильтровали через Celite® и выпаривали в вакууме. Остаток растворяли в EtOAc (20 мл) и промывали водой (20 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (300 мг, 1,24 ммоль, выход 87%, чистота 95%) в виде темно-красного твердого вещества. СВЭЖХ-МС (Метод 2): m/z 230,4 (M+H)+ через 1,72 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 6,99-6,93 (м, 3H), 5,07 (с, 2H), 3,11-3,05 (м, 2H), 2,03 (т, J=11,1 Гц, 2H), 1,88-1,75 (м, 3H), 0,87 (д, J=6,5 Гц, 6H), 0,66 (к, J=11,8 Гц, 1H).
Стадия 3: Метил-3-(N-(5-циано-2-(цис-3,5-диметилпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: К смеси продукта из Стадии 2 выше (70 мг, 0,290 ммоль) и продукта из Примера 323, Стадии 2 (88 мг, 0,319 ммоль) в ДХМ (600 μl) при КТ добавляли пиридин (152 мкл, 1,89 ммоль). Полученный раствор перемешивали при КТ в течение 72 ч, а затем выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (108 мг, 0,224 ммоль, выход 77%, чистота 97%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 468,5 (M+H)+, в 1,94 мин.
Стадия 4: 3-(N-(5-циано-2-(цис-3,5-диметилпиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота: Смесь метилового эфира продукта из стадии 3 выше (108 мг, 0,231 ммоль) и 1М LiOH (водн.) (23 мг, 0,924 ммоль) в ТГФ/MeOH/воде (4:1:1, 4 мл) нагревали до 40°C и перемешивали в течение 3 дней. Реакционную смесь выпаривали в вакууме, а остаток доводили до ~pH 4 1М HCl (водн.). Образовавшийся осадок отфильтровывали, промывали водой и сушили с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (37 мг, 0,080 ммоль, выход 35%, чистота 98%) в виде прозрачного бесцветного масла. СВЭЖХ-МС (Метод 1): m/z 454,4 (M+H)+, 452,3 (M-H)- через 1,82 мин. 1H-ЯМР (500 МГц, Метанол-d4) δ 8,60 (д, J=1,9 Гц, 1H), 8,13 (дд, J=8,2, 1,8 Гц, 1H), 7,52 (д, J=1,9 Гц, 1H), 7,41 (дд, J=8,3, 1,9 Гц, 1H), 7,27 (д, J=8,3 Гц, 1H), 7,19 (д, J=8,2 Гц, 1H), 2,99-2,93 (м, 2H), 2,80-2,71 (м, 1H), 2,18 (т, J=11,2 Гц, 2H), 1,86-1,74 (м, 3H), 1,17-1,08 (м, 2H), 0,93-0,83 (м, 8H), 0,69 (к, J=12,2 Гц, 1H).
Пример 329: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-сульфамоилфенил)сульфамоил)бензойная кислота:
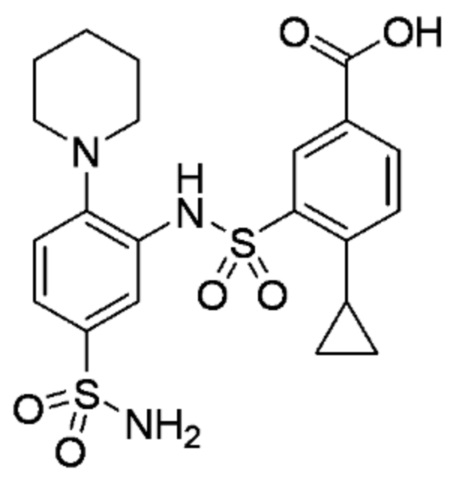
Стадия 1: Метил-4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-сульфамоилфенил)сульфамоил)бензоат: Смесь 3-амино-4-(пиперидин-1-ил)бензолсульфонамида (100 мг, 0,392 ммоль), продукта из Примера 323, Стадии 2 (129 мг, 0,470 ммоль) и пиридина (100 мкл, 1,24 ммоль) в ДХМ (2 мл) перемешивали при КТ в течение 2 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-10% MeOH/ДХМ) с получением указанного в заголовке соединения (174 мг, 0,296 ммоль, выход 76%, 84% чистота) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 494,4 (M+H)+, 492,1 (M-H)- через 1,58 мин.
Стадия 2: 4-циклопропил-3-(N-(2-(пиперидин-1-ил)-5-сульфамоилфенил)сульфамоил)бензойная кислота: Смесь продукта из Стадии 1 выше (174 мг, 0,296 ммоль, чистота 84%) и LiOH (50 мг, 1,17 ммоль) в ТГФ/MeOH/воде (4:1:1, 5,4 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (15 мл), сушили при пропускании через фазовый сепаратор, а затем выпаривали в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 35-65% MeCN в воде) с получением указанного в заголовке соединения (93,9 мг, 0,194 ммоль, выход 66%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 480,4 (M+H)+, 478,3 (M-H)- через 1,44 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,22 (с, 1H), 9,23 (с, 1H), 8,44 (д, J=1,9 Гц, 1H), 8,00 (дд, J=8,2, 1,9 Гц, 1H), 7,59 (д, J=2,2 Гц, 1H), 7,52 (дд, J=8,4, 2,2 Гц, 1H), 7,29-7,23 (м, 3H), 7,15 (д, J=8,4 Гц, 1H), 2,77-2,67 (м, 5H), 1,54-1,46 (м, 4H), 1,46-1,38 (м, 2H), 1,10-1,02 (м, 2H), 0,86-0,79 (м, 2H).
Пример 330: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклобутилбензойная кислота
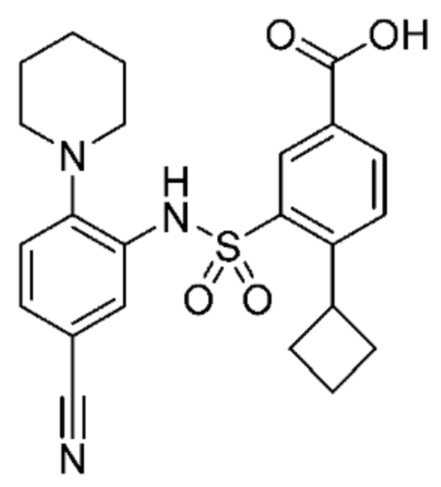
Стадия 1: Метил-4-бром-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 182, Стадии 2 (250 мг, 1,23 ммоль), продукта из Примера 316, Стадии 1 (409 мг, 1,29 ммоль) и пиридина (300 мкл, 3,71 ммоль) в ДХМ (6 мл) перемешивали при КТ в течение 5 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (24 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (360 мг, 0,753 ммоль, выход 61%) в виде светло-коричневого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 478,3 (M+H)+, 476,1 (M-H)- через 1,80 мин.
Стадия 2: Метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклобутилбензоат: В высушенную над пламенем колбу добавляли стружку Mg (137 мг, 5,64 ммоль) и иод (10 мг, 0,039 ммоль). Добавляли небольшую аликвоту бромциклобутана (0,35 мл, 3,72 ммоль) в ТГФ (4 мл) и нагревали смесь с обратным холодильником при помощи технического фена. После исчезновения коричневого цвета добавляли оставшийся раствор с такой скоростью, чтобы сохранялось кипение. После завершения добавления смесь перемешивали при КТ в течение 2 ч. Смесь медленно добавляли к 2 М хлориду цинка в 2-метилтетрагидрофуране (2,8 мл, 5,60 ммоль) при 0°C и затем нагревали до КТ и перемешивали в течение 1 ч. Добавляли раствор продукта из Стадии 1 выше (180 мг, 0,376 ммоль) в ТГФ (2 мл) и PdCl2(dppf)∙ДХМ (62 мг, 0,076 ммоль) и нагревали смесь при 70°C в течение 4 ч, а затем перемешивали при КТ в течение ночи. В смесь вливали насыщенный NH4Cl (водн.) (20 мл) и экстрагировали EtOAc (3×20 мл). Органические экстракты объединяли, промывали солевым раствором (20 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-75% EtOAc/изогексан) с получением указанного в заголовке соединения (118 мг, 0,250 ммоль, выход 66%, чистота 96%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1): m/z 454,4 (M+H)+, 452,4 (M-H)- через 1,95 мин.
Стадия 3: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклобутилбензойная кислота: Смесь продукта из стадии 2 выше (118 мг, 0,250 ммоль, 96% чистота) и LiOH (43 мг, 1,01 ммоль) в ТГФ/MeOH/воде (4:1:1, 4 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (15 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (Waters, Кислотный (0,1% муравьиная кислота), Кислотный, Waters X-Select Prep-C18, 5 мкм, колонка 19×50 мм, 50-80% MeCN в воде) с получением указанного в заголовке соединения (60 мг, 0,134 ммоль, выход 54%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 440,4 (M+H)+, 438,3 (M-H)- через 1,80 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,57 (с, 1H), 8,31 (д, J=2,0 Гц, 1H), 8,16 (д, J=8,2 Гц, 1H), 7,93 (д, J=8,2 Гц, 1H), 7,59-7,48 (м, 1H), 7,21 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,6 Гц, 1H), 4,34-4,23 (м, 1H), 2,84-2,73 (м, 4H), 2,28-2,12 (м, 4H), 1,98-1,82 (м, 2H), 1,51-1,41 (м, 6H).
Пример 331: 4-циклопропил-3-(N-(4-фтор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
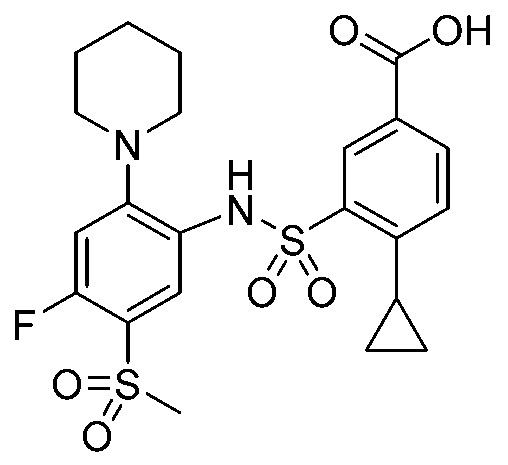
ЧАСТЬ A; получение ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 1; 4-фтор-5-(метилсульфонил)-2-(пиридин-2-ил)анилин.
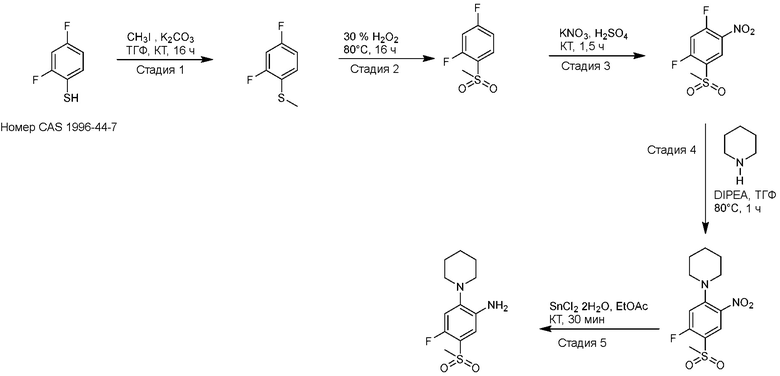
Стадия 1: Синтез (2,4-дифторфенил)(метил)сульфана.
К перемешиваемому раствору 2,4-дифторбензолтиола (номер CAS 1996-44-7; 5 г, 0,03421 моль, 1 экв.) в ТГФ (50 мл, 10 об) добавляли K2CO3 (23,60 г, 0,17105 моль, 5 экв.) при 0°C, затем метилиодид (14,5 г, 0,102 моль, 3 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, затем вливали в воду (500 мл) и экстрагировали этилацетатом (2×200 мл). Объединенный органический слой промывали раствором соли (200 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении с получением указанного в заголовке сульфана (4,3 г, 78,47%).
Стадия 2: Синтез 2,4-дифтор-1-(метилсульфонил)бензола
Смесь сульфана из Стадии 1 (2,5 г, 0,0141 моль, 1 экв.) в 30% H2O2 (1 г (3,5 мл), 0,0312 моль, 2,2 экв.) перемешивали при 80°C в течение 16 ч, охлаждали и разбавляли водой (250 мл), затем экстрагировали EtOAc (2×200 мл). Органический слой сушили над безводным Na2SO4 и выпаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной флеш-хроматографии (силикагель 230-400) при использовании 13% EtOAc в гексане, с получением указанного в заголовке метилсульфона в виде коричневой жидкости (1,9 г, 63,35%). СВЭЖХ-МС (Метод 1) m/z 193,1 (M+H)+ через 1,54 мин.
Стадия 3: Синтез 1,5-дифтор-2-(метилсульфонил)-4-нитробензола.
К перемешиваемому раствору метилсульфона из Стадии 2 (1,4 г, 0,00728 моль, 1 экв.) в конц. H2SO4 (14 мл, 10 об) порциями добавляли KNO3 (2,2 г, 0,0218 моль, 3 экв.). После завершения реакции согласно ТСХ (30% EtOAc в гексане), реакционную смесь вливали в воду со льдом (250 мл) и экстрагировали ДХМ (2×150 мл). Объединенный органический слой промывали нас. раствором NaHCO3 (250 мл), сушили над безводным Na2SO4, выпаривали при пониженном давлении с получением указанного в заголовке нитробензола в виде желтого твердого вещества (1,5 г, 86,82%).
Стадия 4: Синтез 1-(5-фтор-4-(метилсульфонил)-2-нитрофенил)пиперидин.
К перемешиваемому раствору нитробензола из Стадии 3 (1,5 г, 0,00632 моль, 1 экв.) в ТГФ (30 мл) добавляли DIPEA (2,447 г, 0,01897 моль, 3 экв.) и пиперидин (0,538 г, 0,00632 моль, 1 экв.). Реакционную смесь перемешивали при 80°C в течение 1 ч, охлаждали и разбавляли водой (250 мл), затем экстрагировали EtOAc (2×100 мл). Объединенный органический слой сушили над безводным Na2SO4, выпаривали при пониженном давлении с получением указанного в заголовке пиперидина в виде желтого твердого вещества (1,2 г, 62,76%). СВЭЖХ-МС (Метод 1) m/z 303,3 (M+H)+ через 2,14 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,17 (д, 1H), 7,34 (д, 1H), 3,34 (с, 3H), 3,10 (шс, 4H), 1,66 (м, 6H).
Стадия 5: Синтез 4-фтор-5-(метилсульфонил)-2-(пиперидин-1-ил)анилин.
К раствору пиперидина из Стадии 4 (1,2 г, 0,00396 моль, 1 экв.) в EtOAc (10 мл) добавляли SnCl2 2H2O (4,48 г, 0,01984 моль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли водой (200 мл) и доводили pH до ~12 при использвании 1Н раствора NaOH. Водный слой экстрагировали этилацетатом (2×100 мл). Объединенный органический слой сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный неочищенный продукт растворяли в ДХМ (10 мл) с последующим добавлением н-пентана (20 мл). Образовавшийся осадок отфильтровывали с получением указанного в заголовке анилина в виде почти белого твердого вещества (0,9 г, 92,51%). СВЭЖХ-МС (Метод 1) m/z 273,3 (M+H)+ через 2,08 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,12 (д, 1H), 6,89 (д, 1H), 4,99 (с, 2H), 3,20 (с, 3H), 2,83 (шс, 4H), 1,67 (шс, 4H), 1,54 (шс, 2H).
ЧАСТЬ B; Стадия 1: Метил-4-циклопропил-3-(N-(4-фтор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Раствор 4-фтор-5-(метилсульфонил)-2-(пиперидин-1-ил)анилина (ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 1) (0,10 г, 0,367 ммоль) в пиридине (1,04 мл, 12,9 ммоль) обрабатывали продуктом из Примера 323, Стадии 2 (0,131 г, 0,477 ммоль) и перемешивали полученный раствор при КТ в течение 24 ч, затем при 50°C в течение 96 ч. Смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-60% EtOAc/изогексан) с получением указанного в заголовке соединения (73 мг, 0,143 ммоль, выход 39%) в виде коричневого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 511,3 (M+H)+, 509,2 (M-H)- через 1,64 мин.
Стадия 2: 4-циклопропил-3-(N-(4-фтор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: LiOH (10,3 мг, 0,429 ммоль) добавляли к смеси продукта из Стадии 1 выше (0,073 г, 0,143 ммоль) в ТГФ (3 мл) и воде (1 мл) при КТ. Полученную смесь перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме. Остаток подкисляли 10% в/об лимонной кислотой (водн.) и собирали осадок с помощью фильтрации с получением указанного в заголовке соединения (32 мг, 0,064 ммоль, выход 45%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 497,3 (M+H)+, 495,2 (M-H)- через 1,50 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,17 (шс, 1H), 9,60 (шс, 1H), 8,30 (д, J=1,9 Гц, 1H), 8,01 (д, J=8,2 Гц, 1H), 7,21 (д, J=7,7 Гц, 1H), 7,15 (д, J=8,3 Гц, 1H), 7,02 (д, J=12,6 Гц, 1H), 3,11 (с, 3H), 3,06-2,98 (м, 4H), 2,80-2,72 (м, 1H), 1,55-1,45 (м, 6H), 1,17-1,04 (м, 2H), 0,93-0,87 (м, 2H).
Пример 332: 3-(N-(4-хлор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
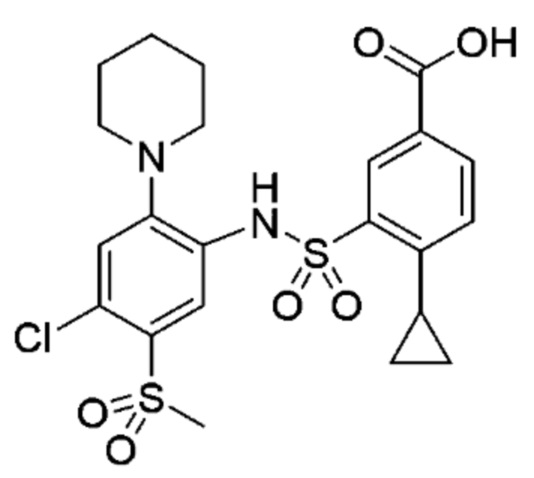
ЧАСТЬ A; Получение ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 4; 4-хлор-5-(метилсульфонил)-2-(пиридин-2-ил)анилин.
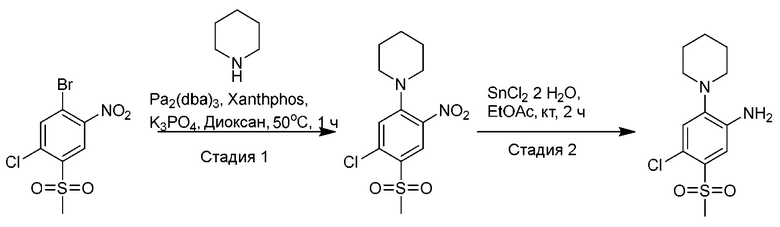
Стадия 1: Синтез 1-(5-хлор-4-(метилсульфонил)-2-нитрофенил)пиперидина.
К смеси 1-бром-5-хлор-4-(метилсульфонил)-2-нитробензола (1 г, 0,00319 моль, 1 экв.) и пиперидина (0,273 г, 0,00319 моль, 1 экв.) в 1,4-диоксане (10 мл) добавляли K3PO4 (1,01 г, 0,00478 моль, 1,5 экв.). Реакционную смесь барботировали N2 в течение 30 мин при комнатной температуре, после чего добавляли Pd2(dba)3 (0,145 г, 0,00015 моль, 0,05 экв.) и Xanthphos (0,184 г, 0,000319). Полученную реакционную смесь перемешивали при 50°C в течение 1 ч, охлаждали, разбавляли водой (100 мл) и экстрагировали этилацетатом (2×50 мл). Объединенный органический слой сушили над безводным Na2SO4 и упаривали в вакууме с получением указанного в заголовке пиперидина в виде коричневой жидкости (1 г, 73,78%). Этот неочищенный материал использовали непосредственно в следующей стадии.
Стадия 2: Синтез 4-хлор-5-(метилсульфонил)-2-(пиперидин-1-ил)анилина.
К раствору пиперидина из Стадии 1 (1,5 г, 0,0047 моль, 1 экв.) в EtOAc (50 мл) добавляли SnCl2 2H2O (5,32 г, 0,0235 моль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, разбавляли водой (100 мл) и доводили pH до ~12 при использовании 1Н раствора NaOH. Водный слой экстрагировали этилацетатом (2×100 мл). Объединенный органический слой сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии при использовании нейтрального оксида алюминия и 20% EtOAc в гексане в качестве элюента. Полученный в результате материал растворяли в ДХМ (10 мл) с последующим добавлением н-пентана (20 мл). Образовавшийся осадок отфильтровывали с получением указанного в заголовке анилина в виде почти белого твердого вещества (0,41 г, 30,18%). СВЭЖХ-МС (Метод 1) m/z 289,2/291,2 (M+H)+ через 2,27 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,37 (д, 1H), 6,99 (д, 1H), 5,31 (с, 2H), 3,24 (с, 3H), 2,82 (шс, 4H), 1,67 (шс, 4H), 1,52 (шс, 2H).
ЧАСТЬ B; Стадия 1: Метил-3-(N-(4-хлор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Раствор 4-хлор-5-(метилсульфонил)-2-(пиперидин-1-ил)анилина (0,10 г, 0,346 ммоль) в пиридине (0,980 мл, 12,1 ммоль) обрабатывали продуктом из Примера 323, Стадии 2 (0,124 г, 0,450 ммоль) и перемешивали полученный раствор при КТ в течение 24 ч, затем при 50°C в течение 96 ч. Смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-10% EtOAc/ДХМ, затем 0-50% EtOAc/изогексан) с получением указанного в заголовке соединения (84 мг, 0,159 ммоль, выход 46%, чистота 100%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1): m/z 527,3 (M+H)+, 525,1 (M-H)- через 1,72 мин.
Стадия 2: 3-(N-(4-хлор-5-(метилсульфонил)-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота: LiOH (0,011 г, 0,478 ммоль) добавляли к смеси продукта из Стадии 1 выше (0,084 г, 0,159 ммоль, чистота 100%) в ТГФ (3 мл) и воде (1 мл) при КТ. Полученную смесь перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме. Остаток подкисляли 10% в/об лимонной кислотой (водн.) и собирали осадок с помощью фильтрации, с получением указанного в заголовке соединения (65 мг, 0,126 ммоль, выход 79%, чистота 99%) в виде бледно-желтого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 513,3 (M+H)+, 511,2 (M-H)- через 1,58 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 9,50 (шс, 1H), 8,34 (д, J=1,9 Гц, 1H), 8,01 (д, J=8,1 Гц, 1H), 7,51 (с, 1H), 7,20 (с, 1H), 7,16 (д, J=8,3 Гц, 1H), 3,16 (с, 3H), 3,02-2,96 (м, 4H), 2,80-2,72 (м, 1H), 1,54-1,50 (м, 4H), 1,49-1,43 (м, 2H), 1,13-1,04 (м, 2H), 0,92-0,86 (м, 2H).
Пример 333: 4-циклопропил-3-(N-(4-фтор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензойная кислота
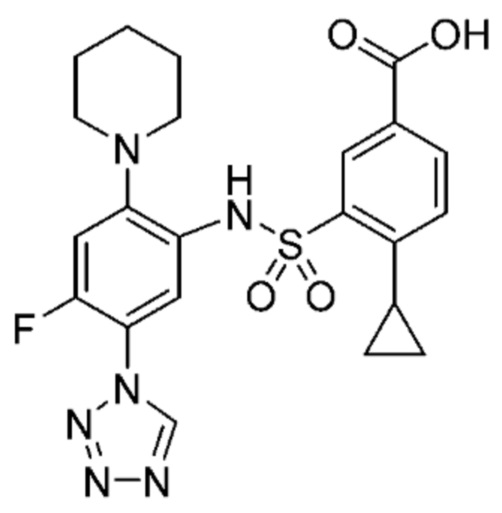
ЧАСТЬ A; Получение ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 3; 4-фтор-2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)анилин.
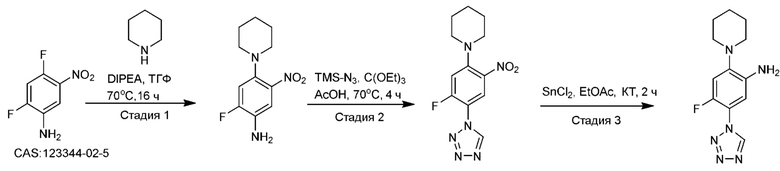
Стадия 1: Синтез 2-фтор-5-нитро-4-(пиперидин-1-ил)анилина.
К перемешиваемому раствору 2,4-дифтор-5-нитроанилина (7 г, 0,0287 моль, 1 экв.) в ТГФ (70 мл, 10 об) добавляли DIPEA (11,1 г, 0,0861 моль, 3 экв.) при комнатной температуре под N2, затем пиперидин (2,44 г, 0,0287 моль, 1 экв.) при 0°C. Затем реакционную смесь перемешивали при 70°C в течение 16 ч, охлаждали и вливали в воду (500 мл), после чего экстрагировали этилацетатом (3×250 мл). Объединенный органический слой сушили над сульфатом натрия и выпаривали при пониженном давлении с получением указанного в заголовке анилина в виде коричневого масла и смеси региоизомеров (10 г, количественный). СВЭЖХ-МС (Метод 1) m/z 240,3 (M+H)+ через 2,14 и 2,44 мин. Этот материал использовали непосредственно в следующей стадии.
Стадия 2: Синтез 1-(5-фтор-2-нитро-4-(1H-тетразол-1-ил)фенил)пиперидина.
Раствор анилина из Стадии 1 (10 г, 0,042 моль, 1 экв.) в уксусной кислоте (200 мл, 20 об) перемешивали в течение 5 мин, после чего добавляли триэтилортоформиат (31,08 г, 0,21 моль, 5 экв.), а затем TMS-N3 (24,15 г, 0,21 моль, 5 экв.) при 0°C. Полученную реакционную смесь перемешивали при 70°C в течение 4 ч, охлаждали и вливали в воду (500 мл), после чего экстрагировали этилацетатом (3×250 мл). Объединенный органический слой промывали раствором NaHCO3 (50 мл), сушили над сульфатом натрия и выпаривали при пониженном давлении с получением указанного в заголовке тетразола в виде коричневого масла и смеси региоизомеров (15 г, количественный). СВЭЖХ-МС (Метод 1) m/z 293,3 (M+H)+ через 2,19 и 2,24 мин. Этот материал использовали непосредственно в следующей стадии.
Стадия 3: Синтез 4-фтор-2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)анилина.
К перемешиваемому раствору тетразола из Стадии 2 (15 г, 0,051 моль, 1 экв.) в этилацетате (100 мл) добавляли SnCl2∙2H2O (38,9 г, 0,205 моль, 4 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем вливали в воду (700 мл) и этилацетат (250 мл). Осадок гидроксида олова отфильтровывали через слой целита, и органический слой сушили над сульфатом натрия и выпаривали при пониженном давлении. Неочищенный продукт очищали с помощью флеш-хроматографии при использовании нейтрального оксида алюминия и 20-100% EtOAc в гексане в качестве элюента с получением смеси региоизомеров. Требуемый региоизомер выделяли с помощью обращенно-фазовой колоночной хроматографии (A) 0,1% FA в воде, (B) ацетонитрил (40:60) и идентифицировали с помощью NOE-экспериментов 1H-ЯМР с получением указанного в заголовке анилина в виде бледно-желтого твердого вещества (0,560 г, 14,18%). СВЭЖХ-МС (Метод 1) m/z 263,3 (M+H)+ через 2,19 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,93 (с, 1H), 7,06 (д, 1H), 7,02 (д, 1H), 5,04 (с, 2H), 2,82 (шс, 4H), 1,69 (м, 4H), 1,54 (м, 2H).
ЧАСТЬ B; Стадия 1: Метил-4-циклопропил-3-(N-(4-фтор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)бензоат: Раствор 4-фтор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)анилина (ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 3) (0,30 г, 1,14 ммоль) в пиридине (3,24 мл, 40,0 ммоль) обрабатывали продуктом из Примера 323, Стадии 2 (0,408 г, 1,49 ммоль) и перемешивали полученный раствор при КТ в течение 24 ч, затем при 50°C в течение 96 ч. Смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан, затем 0-10% EtOAc/ДХМ) с получением продукта (0,277 г, 0,548 ммоль, выход 48%, чистота 99%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 523,4 (M+Na)+, 499,2 (M-H)- через 1,72 мин.
Стадия 2: 4-циклопропил-3-(N-(4-фтор-2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)фенил)сульфамоил)бензойная кислота: LiOH (0,039 г, 1,644 ммоль) добавляли к раствору продукта из Стадии 1 выше (0,277 г, 0,548 ммоль, чистота 99%) в ТГФ (3 мл, 0,548 ммоль) и воде (1 мл) при КТ. Полученную смесь перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме. Остаток подкисляли 10% в/об лимонной кислотой (водн.) и собирали осадок с помощью фильтрации с получением продукта (220 мг, 0,447 ммоль, выход 82%, чистота 99%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1): m/z 509,3 (M+Na)+, 485,2 (M-H)- через 1,57 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,21 (шс, 1H), 9,80 (д, J=1,6 Гц, 1H), 9,55 (шс, 1H), 8,39 (д, J=1,9 Гц, 1H), 8,01 (дд, J=8,2, 1,9 Гц, 1H), 7,46 (д, J=7,8 Гц, 1H), 7,29 (д, J=12,3 Гц, 1H), 7,15 (д, J=8,3 Гц, 1H), 2,79 (м, 5H), 1,48 (д, J=6,4 Гц, 4H), 1,43 (к, J=9,4, 7,3 Гц, 2H), 1,13-1,04 (м, 2H), 0,90-0,81 (м, 2H).
Пример 334: 3-(N-(4-хлор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота
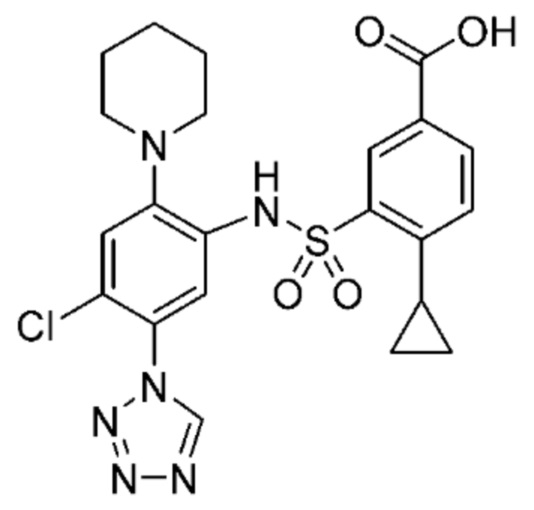
ЧАСТЬ A; Получение ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 4; 4-хлор-2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)анилин.
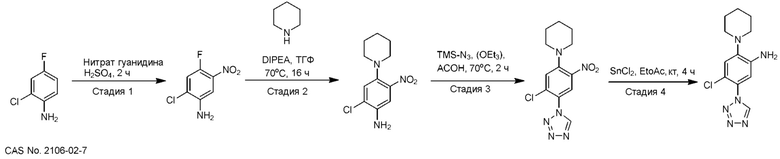
Стадия 1: Синтез 2-хлор-4-фтор-5-нитроанилина.
К раствору 2-хлор-4-фторанилина (5,00 г, 0,0343 моль, 1 экв.) в H2SO4 (15 мл) добавляли нитрат гуанидина (4,19 г, 0,0343 моль, 1 экв.) при 0°C. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, затем подщелачивали 20% раствором NaOH (100 мл) и вливали холодную воду (500 мл). Образовавшийся осадок отфильтровывали и сушили в вакууме с получением указанного в заголовке нитроанилина в виде коричневого твердого вещества (3,7 г, 56,92%).
Стадия 2: Синтез 2-хлор-5-нитро-4-(пиперидин-1-ил)анилина.
К раствору нитроанилина из Стадии 1 (3,56 г, 0,0191 моль, 1 экв.) и пиперидин (3,56 г, 0,038 моль, 2 экв.) в ТГФ (35 мл) добавляли DIPEA (7,5 мл, 0,057 моль, 2 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 16 ч, охлаждали и разбавляли водой (100 мл), после чего экстрагировали этилацетатом (3×100 мл). Объединенный органический слой промывали солевым раствором (50 мл), сушили над безводным Na2SO4, выпаривали при пониженном давлении с получением указанного в заголовке анилина в виде оранжевого твердого вещества (4,20 г, 89,36%). СВЭЖХ-МС (Метод 1) m/z 256,3/258,3 (M+H)+ через 2,43 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,28 (с, 1H), 7,17 (с, 1H), 5,65 (с, 2H), 2,77 (м, 4H), 1,36 (м, 4H), 1,54 (м, 2H).
Стадия 3: Синтез 1-(5-хлор-2-нитро-4-(1H-тетразол-1-ил)фенил)пиперидина.
К раствору анилина из Стадии 2 (4,2 г, 0,0164 моль, 1 экв.) в уксусной кислоте (40 мл) добавляли триэтилортоформиат (12,15 г, 0,0821 моль, 5 экв.) и TMS-азид (9,4 г, 0,0821 моль, 5 экв.) при 0°C. Реакционную смесь перемешивали при 70°C в течение 2 ч, охлаждали и подщелачивали нас. раствором NaHCO3 (400 мл) и экстрагировали этилацетатом (3×100 мл). Объединенный органический слой промывали солевым раствором (100 мл), сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении и полученный неочищенный материал очищали при растирании в пентане (50 мл) и диэтиловом эфире (20 мл) с получением указанного в заголовке пиперидина в виде коричневого твердого вещества (3,20 г, 63,11%). СВЭЖХ-МС (Метод 1) m/z 309,3/311,3 (M+H)+ через 2,31 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,84 (с, 1H), 8,39 (с, 1H), 7,63 (с, 1H), 3,14 (шс, 4H), 1,62 (шс, 6H).
Стадия 4: Синтез 4-хлор-2-(пиперидин-1-ил)-5-(1H-тетразол-1-ил)анилина.
К раствору пиперидина из Стадии 3 (1,0 г, 0,0032 моль, 1.0 экв.) в EtOAC при перемешивании добавляли SnCl2 (3,06 г, 0,0161 моль, 3 eq.) Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, фильтровали через слой целита, разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенный органический слой сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении и неочищенный продукт очищали с помощью комбифлеш-хроматографии на нейтральном силикагеле (5% этилацетата в гексане) с получением указанного в заголовке анилина в виде светло-коричневого твердого вещества (0,587 г, 56,72%). СВЭЖХ-МС (Метод 1) m/z 279,3/281,3 (M+H)+ через 2,36 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,84 (с, 1H), 7,11 (с, 1H), 6,91 (с, 1H), 5,33 (с, 2H), 2,82 (шс, 4H), 1,69 (м, 4H), 1,54 (м, 2H).
ЧАСТЬ B; Стадия 1: Метил-3-(N-(4-хлор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: Раствор 4-хлор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)анилина (ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 4) (0,30 г, 1,08 ммоль) в пиридине (3,05 мл, 37,7 ммоль) обрабатывали продуктом из Примера 323, Стадии 2 (0,384 г, 1,40 ммоль) и перемешивали раствор при КТ в течение 24 ч, затем при 70°C в течение 96 ч. Смесь выпаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (24 г картридж, 0-50% EtOAc/изогексан, затем 0-10% EtOAc/ДХМ) с получением указанного в заголовке соединения (52 мг, 0,099 ммоль, выход 9%, чистота 99%) в виде бесцветного твердого вещества. СВЭЖХ-МС (Метод 1): m/z 539,3 (M+Na)+, 515,2 (M-H)- через 1,74 мин.
Стадия 2: 3-(N-(4-хлор-2-(пиперидин-1-ил)-5-(тетразол-1-ил)фенил)сульфамоил)-4-циклопропилбензойная кислота: LiOH (7,08 мг, 0,296 ммоль) добавляли к смеси продукта из Стадии 1 выше (0,052 г, 0,099 ммоль, чистота 99%) в ТГФ (3 мл) и воде (1 мл) при КТ. Полученную смесь перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме, а остаток подкисляли 10% в/об лимонной кислотой (водн.). Образовавшийся осадок собирали с помощью фильтрации с получением указанного в заголовке соединения (33 мг, 0,062 ммоль, выход 63%, чистота 95%) в виде кремового твердого вещества. СВЭЖХ-МС (Метод 1): m/z 525,3 (M+Na)+, 501,2 (M-H)- через 1,63 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,20 (шс, 1H), 9,80 (с, 1H), 8,40 (д, J=1,9 Гц, 1H), 8,00 (дд, J=8,2, 1,8 Гц, 1H), 7,40 (д, J=7,2 Гц, 2H), 7,17-7,11 (м, 1H), 2,80 (т, J=5,2 Гц, 5H), 1,52 (к, J=5,6 Гц, 4H), 1,48-1,42 (м, 2H), 1,13-0,99 (м, 2H), 0,89-0,77 (м, 2H). 1 обмениваемый протон не наблюдали.
Пример 335: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-(циклопропил-d5)бензойная кислота
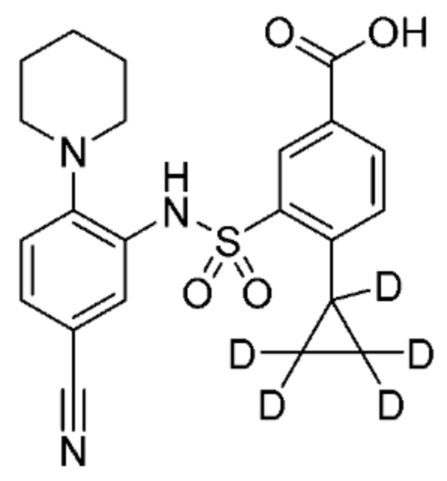
Стадия 1: Метил-4-бром-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 182, стадии 2 (1,00 г, 4,92 ммоль), продукта из Примера 316, Стадии 1 (1,64 г, 5,18 ммоль) и пиридина (1,2 мл, 14,8 ммоль) в ДХМ (25 мл) перемешивали при КТ в течение ночи. Смесь наносили на силикагель и очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% EtOAc/изогексан), а затем растирали с МТБЭ с получением указанного в заголовке соединения (1,44 г, 3,01 ммоль, выход 61%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 478,3 (M+H)+, 476,3 (M-H)- через 1,80 мин.
Стадия 2: Метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-(циклопропил-d5)бензоат: В высушенную на пламенем колбу вносили стружку Mg (72 мг, 2,96 ммоль) и иод (5 мг, 0,020 ммоль). Добавляли небольшую часть (~0,25 мл) раствора циклопропил-d5 бромида (250 мг, 1,98 ммоль) в ТГФ (2 мл) и нагревали смесь с обратным холодильником с помощью технического фена. Когда исчезал коричневый цвет, добавляли оставшийся раствор с такой скоростью, чтобы сохранялось кипение. После завершения добавления смесь перемешивали при КТ в течение 30 мин. Смесь медленно добавляли к 2М ZnCl в 2-метилтетрагидрофуране (1,5 мл, 3,00 ммоль) при 0°C, а затем нагревали до КТ и перемешивали в течение 20 мин. Добавляли раствор продукта из Стадии 1 выше (95 мг, 0,198 ммоль) в ТГФ (1 мл) и PdCl2(dppf)∙ДХМ (30 мг, 0,037 ммоль) и нагревали смесь до 70°C в течение 2 ч. В смесь вливали насыщенный NH4Cl (водн.) (10 мл) и экстрагировали EtOAc (3×20 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор, а затем выпаривали в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (82 мг, 0,179 ммоль, выход 90%, чистота 97%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1): m/z 445,5 (M+H)+, 443,4 (M-H)- через 1,82 мин.
Стадия 3: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-(циклопропил-d5)бензойная кислота: Смесь продукта из стадии 2 выше (82 мг, 0,179 ммоль, чистота 97%) и LiOH∙H2O (30,0 мг, 0,716 ммоль) в ТГФ/MeOH/воде (4:1:1, 3 мл) оставляли с перемешиванием при 40°C на выходные. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×15 мл). Органические фазы объединяли и промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (4 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (27 мг, 0,060 ммоль, выход 33%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 431,6 (M+H)+, 429,4 (M-H)- через 1,66 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,25 (с, 1H), 9,48 (с, 1H), 8,37 (д, J=1,9 Гц, 1H), 8,02 (дд, J=8,3, 1,9 Гц, 1H), 7,54 (дд, J=8,4, 2,0 Гц, 1H), 7,24 (д, J=2,0 Гц, 1H), 7,20-7,14 (м, 2H), 2,87-2,80 (м, 4H), 1,53-1,47 (м, 4H), 1,47-1,43 (м, 2H).
Пример 336: 3-(N-(5-циано-3-метил-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота
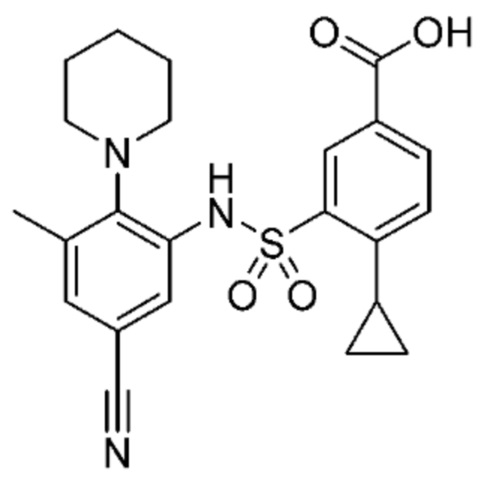
Стадия 1: 1-(4-бром-2-метил-6-нитрофенил)пиперидин: К раствору 5-бром-2-фтор-1-метил-3-нитробензола (1,01 г, 4,32 ммоль) в ДХМ (10 мл) при КТ добавли пиперидин (0,64 мл, 6,4 ммоль), а затем триэтиламин (1,2 мл, 8,6 ммоль). Полученный раствор перемешивали при КТ в течение 17 ч. Добавляли дополнительную порцию пиперидина (1,3 мл, 13 ммоль) и нагревали смесь при 40°C в течение 6 ч. Добавляли дополнительную порцию пиперидина (3,5 мл, 35 ммоль) и нагревали смесь при 40°C в течение 17 ч, а затем при 50°C в течение 3 ч. Добавляли дополнительную порцию пиперидина (7,0 мл, 70 ммоль) и нагревали смесь при 50°C в течение 4 ч. Реакционной смеси позволяли охладиться до КТ и затем промывали водой (3×20 мл). Водную фазу экстрагировали ДХМ (10 мл), и органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток разбавляли ДХМ (100 мл) и промывали 0,5М HCl (водн.) (3×50 мл). Органическую фазу сушили над MgSO4 и выпаривали в вакууме с получением указанного в заголовке соединения (1,17 г, 3,84 ммоль, выход 89%, чистота 98%) в виде оранжевого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,80 (с, 1H), 7,71 (с, 1H), 2,90-2,77 (м, 4H), 2,33 (с, 3H), 1,61-1,51 (м, 6H).
Стадия 2: 3-метил-5-нитро-4-(пиперидин-1-ил)бензонитрил: Раствор продукта из стадии 1 выше (1,17 г, 3,84 ммоль, чистота 98%) и дицианоцинка (0,483 г, 4,12 ммоль) в DMA (5 мл) дегазировали N2 в течение 10 мин, затем добавляли Pd(PPh3)4 (0,453 г, 0,392 ммоль) 4 порциями. Реакционную смесь нагревали до 100°C под N2 в течение 2 ч. Реакционной смеси позволяли охладиться до КТ и затем фильтровали через Celite® с EtOAc (50 мл). Фильтрат промывали насыщенным NaHCO3 (водн.) (2×20 мл), водой (2×20 мл) и солевым раствором (2×20 мл). Органическую фазу сушили над Na2SO4, фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (12 г колонка, 0-40% EtOAc/изогексан) с получением указанного в заголовке соединения (720 мг, 2,88 ммоль, выход 73%, чистота 98%) в виде ярко-желтого бесцветного твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,17 (д, J=2,0 Гц, 1H), 7,93 (д, J=2,1 Гц, 1H), 2,95-2,85 (м, 4H), 2,36 (с, 3H), 1,65-1,50 (м, 6H).
Стадия 3: 3-амино-5-метил-4-(пиперидин-1-ил)бензонитрил: К смеси продукта из стадии 2 выше (720 мг, 2,88 ммоль, чистота 98%) и NH4Cl (188 мг, 3,52 ммоль) в ИПС (10 мл) и воде (3 мл) добавляли порошок железа (1,64 г, 29,4 ммоль) и нагревали реакционную смесь при 90°C в течение 4 ч. Суспензию охлаждали до КТ и фильтровали через Celite®, промывая MeOH (10 мл), и удаляли растворитель в вакууме. Добавляли 0,1M HCl (водн.) (100 мл) и раствор промывали EtOAc (50 мл). Водный раствор нейтрализовали нас. NaHCO3 (водн.) (50 мл) и затем экстрагировали этилацетатом (5×50 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и выпаривали в вакууме с получением указанного в заголовке соединения (220 мг) в виде светло-коричневого масла, и использовали без дополнительной очистки.
Стадия 4: Метил-3-(N-(5-циано-3-метил-2-(пиперидин-1-ил)фенил)сульфамоил)-4-циклопропилбензоат: К раствору продукта из стадии 3 выше (110 мг) и пиридина (0,204 мл, 2,52 ммоль) в ДХМ (5 мл) добавляли продукт из Примера 323, стадии 2 (177 мг, 0,613 ммоль, чистота 95%) при 0°C и перемешивали смесь при КТ в течение 19 ч. Реакционную смесь разбавляли ДХМ (50 мл) и последовательно промывали 0,5М HCl (водн.) (2×50 мл), водой (50 мл) и солевым раствором (50 мл). Объединенные органические фазы сушили над MgSO4 и выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения (104 мг, 0,225 ммоль, выход 16% в 2 стадиях, чистота 98%) в виде кремового твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,52 (с, 1H), 8,33 (с, 1H), 8,06 (д, J=8,3 Гц, 1H), 7,45 (с, 1H), 7,21 (д, J=8,4 Гц, 1H), 6,88 (с, 1H), 3,86 (с, 3H), 3,07-2,96 (м, 4H), 2,77-2,66 (м, 1H), 2,29 (с, 3H), 1,65-1,52 (м, 6H), 1,16-1,10 (м, 2H), 0,98-0,87 (м, 2H).
Стадия 5: 3-(N-(5-циано-3-метил-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-циклопропилбензойная кислота: LiOH (16,5 мг, 0,688 ммоль) добавляли к раствору продукта из стадии 4 выше (104 мг, 0,225 ммоль, чистота 98%) в ТГФ (1 мл) и воде (0,5 мл) при КТ. Полученную смесь перемешивали при КТ в течение 24 ч. Реакционную смесь выпаривали в вакууме, а остаток подкисляли 10% в/об лимонной кислотой (водн.). Образовавшийся осадок собирали с помощью фильтрации и промывали водой (50 мл) с получением указанного в заголовке соединения (57,5 мг, 0,125 ммоль, выход 55%, чистота 96%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 2): m/z 440,7 (M+H)+, 438,3 (M-H)- через 1,29 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,30 (с, 1H), 9,46 (с, 1H), 8,33 (д, J=1,8 Гц, 1H), 8,04 (дд, J=8,2, 1,8 Гц, 1H), 7,44 (с, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,90 (д, J=2,1 Гц, 1H), 3,03-2,95 (м, 4H), 2,75-2,67 (м, 1H), 2,29 (с, 3H), 1,66-1,49 (м, 6H), 1,14-1,09 (м, 2H), 0,94-0,89 (м, 2H).
Пример 337: 4-(трет-бутил)-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота
Стадия 1: 3-бром-4-(трет-бутил)бензойная кислота: К смеси 4-(трет-бутил)бензойной кислоты (10 г, 56,1 ммоль), азотной кислоты (37 мл), воды (28 мл), AcOH (170 мл) и Br2 (5,20 мл, 101 ммоль) добавляли нитрат серебра (9,63 г, 56,7 ммоль) в воде (28,5 мл) через капельную воронку в течение 30 мин. После завершения добавления смесь перемешивали при КТ в течение ночи. Смесь вливали в лед/воду и перемешивали, пока не растаял весь лед. Осадок собирали с помощью фильтрации, растворяли в EtOAc (600 мл) и последовательно промывали водой (200 мл) и солевым раствором (200 мл). Органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме с получением указанного в заголовке соединения (13,4 г, 21,9 ммоль, выход 39%, 42% чистота) в виде желтого твердого вещества, и использовали без дополнительной очистки. СВЭЖХ-МС (Метод 1): 255,1 (M-H)- через 1,73 мин.
Стадия 2: Метил-3-бром-4-(трет-бутил)бензоат: Смесь продукта из стадии 1 выше (13,4 г, 21,9 ммоль, чистота 42%), иодметана (2,7 мл, 43,4 ммоль) и K2CO3 (6,06 г, 43,8 ммоль) в ДМФА (20 мл) перемешивали при КТ в течение 2 ч. Смесь фильтровали и выпаривали фильтрат в вакууме. Остаток растворяли в ДХМ (100 мл), промывали 1М HCl (водн.) (100 мл) и солевым раствором (3×100 мл). Органическую фазу сушили при пропускании через фазовый сепаратор, концентрировали растворитель в вакууме на силикагеле и частично очищали с помощью хроматографии на силикагеле (80 г картридж, 0-10% EtOAc/изогексан), а затем дополнительно очищали с помощью хроматографии (40 г обращенно-фазовый картридж C18, 35-95% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (4,19 г, 15,0 ммоль, выход 68%, чистота 97%) в виде бледно-желтого масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,09 (д, J=1,9 Гц, 1H), 7,88 (дд, J=8,3, 1,9 Гц, 1H), 7,63 (д, J=8,3 Гц, 1H), 3,85 (с, 3H), 1,48 (с, 9H).
Стадия 3: Метил-3-(бензилтио)-4-(трет-бутил)бензоат: В смесь продукта из стадии 2 выше (4,19 г, 15,0 ммоль, чистота 97%), DIPEA (5,50 мл, 31,5 ммоль), Pd2(dba)3 (1,38 г, 1,51 ммоль) и Xantphos (1,30 г, 2,25 ммоль) в диоксане (70 мл) пропускали N2 в течение 15 мин. Добавляли бензилмеркаптан (1,90 мл, 16,1 ммоль) и перемешивали смесь при 100°C в течение 18 ч, а затем при КТ в течение 3 дней. Добавляли дополнительную порцию бензилмеркаптана (1,90 мл, 16,1 ммоль) и перемешивали смесь при 100°C в течение 5 ч. Добавляли дополнительную порцию Pd2(dba)3 (1,38 г, 1,51 ммоль) и xantphos (1,30 г, 2,25 ммоль) и продолжали перемешивание при 100°C в течение ночи. Добавляли дополнительную порцию DIPEA (5,50 мл, 31,5 ммоль) и продолжали перемешивание при 100°C в течение ночи. После охлаждения до КТ смесь фильтровали через Celite® и выпаривали фильтрат в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (120 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (1,18 г, 2,85 ммоль, выход 19%, чистота 76%) в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1): m/z 315,2 (M+H)+, 313,2 (M-H)- (ЭР-) через 2,06 мин.
Стадия 4: Метил-4-(трет-бутил)-3-(хлорсульфонил)бензоат: К раствору продукта из стадии 3 выше (1,18 г, 2,85 ммоль, чистота 76%) в AcOH (0,21 мл), воде (1,5 мл) и MeCN (20 мл) при -10°C добавляли 1,3-дихлор-5,5-диметилимидазолидин-2,4-дион (843 мг, 4,28 ммоль) и перемешивали смесь при -10°C в течение 2 ч. Смесь упаривали в вакууме до ~5 мл, экстрагировали ДХМ (2×40 мл) и объединенную органическую фазу сушили при пропускании через фазовый сепаратор и выпаривали в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (697 мг, 2,28 ммоль, выход 80%, чистота 95%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,76 (д, J=2,2 Гц, 1H), 7,79 (дд, J=8,3, 2,2 Гц, 1H), 7,55 (д, J=8,3 Гц, 1H), 3,84 (с, 3H), 1,53 (с, 9H).
Стадия 5: Метил-4-(трет-бутил)-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензоат: Смесь продукта из Примера 182, стадии 2 (100 мг, 492 мкмоль, чистота 93%), продукта из стадии 4 выше (226 мг, 738 мкмоль, чистота 95%) и пиридина (0,12 мл, 1,52 ммоль) в ДХМ (2 мл) перемешивали при 35°C в течение 3 дней. Смесь концентрировали в вакууме на силикагеле и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (111 мг, 135 мкмоль, выход 27%, чистота 55%) в виде светло-коричневого масла. СВЭЖХ-МС (Метод 1): m/z 456,6 (M+H)+, 454,3 (M-H)- через 1,98 мин.
Стадия 6: 4-(трет-бутил)-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)сульфамоил)бензойная кислота: Смесь продукта из стадии 5 выше (111 мг, 135 мкмоль, чистота 55%) и LiOH⋅H2O (23,0 мг, 548 мкмоль) в ТГФ/MeOH/воде (4:1:1, 2,1 мл) перемешивали при 40°C в течение ночи. Смесь разбавляли водой (5 мл), подкисляли до ~pH 4 1М HCl (водн.) и экстрагировали EtOAc (3×15 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили при пропускании через фазовый сепаратор и удаляли растворитель в вакууме. Остаток наносили на силикагель и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) с получением указанного в заголовке соединения в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 442,6 (M+H)+, 440,3 (M-H)- через 1,98 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,09 (д, J=1,9 Гц, 1H), 7,88 (дд, J=8,3, 1,9 Гц, 1H), 7,63 (д, J=8,3 Гц, 1H), 3,85 (с, 3H), 1,48 (с, 9H).
Пример 338: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этинилбензойная кислота
Стадия 1: метил-3-бром-4-((триметилсилил)этинил)бензоат: Смесь метил-3-бром-4-иодбензоата (1,00 г, 2,93 ммоль), PdCl2(PPh3)2 (51 мг, 0,073 ммоль) и CuI (тв) (14 мг, 0,073 ммоль) в ТГФ (10 мл) подготавливали под атмосферой N2. Добавляли Et3N (2,04 мл, 14,7 ммоль) и этинилтриметилсилан (488 мкл, 3,52 ммоль) и перемешивали смесь при КТ в течение ночи. Реакционную смесь фильтровали через Celite® и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (80 г картридж, 0-20% EtOAc/изогексан) с получением указанного в заголовке соединения (870 мг, 2,52 ммоль, выход 86%, чистота 90%) в виде желтой жидкости. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,17-8,14 (м, 1H), 7,94-7,90 (м, 1H), 7,70 (д, J=8,1 Гц, 1H), 3,87 (с, 3H), 0,27 (с, 9H).
Стадия 2: метил-3-(бензилтио)-4-((триметилсилил)этинил)-бензоат: В смесь продукта из стадии 1 выше (870 мг, 2,52 ммоль, чистота 90%), Pd2(dba)3 (140 мг, 0,598 ммоль), DIPEA (780 мкл, 4,47 ммоль) и диоксана (6,5 мл) пропускали N2 в течение 15 мин, после чего добавляли бензилмеркаптан (280 мкл, 2,37 ммоль). Смесь нагревали до 100°C и перемешивали в течение ночи. После охлаждения до КТ смесь фильтровали через Celite®. Фильтрат наносили на силикагель и очищали с помощью хроматографии на силикагеле (330 г картридж, 0-50% ДХМ/изогексан) с получением указанного в заголовке соединения (880 мг, 2,36 ммоль, выход 94%, чистота 95%) в виде оранжевого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,91-7,86 (м, 1H), 7,70-7,66 (м, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,46-7,42 (м, 2H), 7,37-7,31 (м, 2H), 7,29-7,24 (м, 1H), 4,36 (с, 2H), 3,85 (с, 3H), 0,25 (с, 9H).
Стадия 3: метил-3-(хлорсульфонил)-4-((триметилсилил)-этинил)бензоат: К раствору продукта из стадии 2 выше (880 мг, 2,36 ммоль, чистота 95%), AcOH (150 мкл, 2,62 ммоль) и воды (300 мкл) в MeCN (12 мл) при -10°C добавляли 1,3-дихлор-5,5-диметилимидазолидин-2,4-дион (700 мг, 3,55 ммоль) 4 порциями и перемешивали смесь при -10°C в течение 2 ч. Смесь выпаривали в вакууме, растворяли в воде (30 мл) и экстрагировали ДХМ (3×30 мл). Объединенные органические фазы сушили при пропускании через фазовый сепаратор и концентрировали на силикагеле в вакууме. Неочищенный продукт очищали с помощью хроматографии на силикагеле (40 г картридж, 0-100% ДХМ/изогексан) с получением указанного в заголовке соединения (637 мг, 1,35 ммоль, выход 57%, чистота 70%) в виде прозрачного бесцветного масла. 1H-ЯМР (500 МГц, ДМСО-d6) δ 8,38 (д, J=1,9 Гц, 1H), 7,84 (дд, J=8,0, 1,9 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 3,87 (с, 3H), 0,22 (с, 9H).
Стадия 4: метил-3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-((триметилсилил)этинил)бензоат: Продукт из стадии 3 выше (256 мг, 0,541 ммоль, чистота 70%) добавляли к раствору пиридина (0,12 мл, 1,5 ммоль) и продукта из Примера 182, стадии 2 (100 мг, 0,492 ммоль, чистота 93%) в ДХМ (1 мл). Полученный раствор перемешивали при КТ в течение 3 дней. Реакционную смесь выпаривали в вакууме и очищали с помощью хроматографии на силикагеле (12 г картридж, 0-100% EtOAc/изогексан) до соединения, указанного в заголовке (285 мг, 0,492 ммоль, выход 100%, чистота 85%), в виде бледно-желтого масла. СВЭЖХ-МС (Метод 1): m/z 496,3 (M+H)+, 494,2 (M-H)- через 2,08 мин.
Стадия 5: 3-(N-(5-циано-2-(пиперидин-1-ил)фенил)-сульфамоил)-4-этинилбензойная кислота: К раствору продукта из стадии 4 выше (285 мг, 492 мкмоль, чистота 85%) в ТГФ (3 мл) добавляли 1М LiOH (водн.) (1,50 мл, 1,50 ммоль). Реакционную смесь нагревали до 40°C и перемешивали в течение 3 ч. Реакционную смесь охлаждали до КТ и выпаривали в вакууме. Остаток подкисляли до ~pH 4 1М HCl (водн.). Осадок собирали с помощью фильтрации и очищали с помощью хроматографии (24 г обращенно-фазовый картридж C18, 15-80% MeCN/0,1% муравьиной кислоты (водн.)) с получением указанного в заголовке соединения (100 мг, 0,230 ммоль, выход 47%, чистота 94%) в виде белого твердого вещества. СВЭЖХ-МС (Метод 1): m/z 410,5 (M+H)+, 408,3 (M-H)- через 1,66 мин. 1H-ЯМР (500 МГц, ДМСО-d6) δ 13,75 (шс, 1H), 8,47 (с, 1H), 8,37-8,31 (м, 2H), 7,83 (дд, J=8,7, 2,1 Гц, 1H), 7,71 (д, J=2,1 Гц, 1H), 7,27 (д, J=8,7 Гц, 1H), 5,65 (д, J=2,9 Гц, 1H), 4,41 (д, J=2,9 Гц, 1H), 3,23-3,10 (м, 4H), 1,50-1,38 (м, 6H).
Биологические исследования
Следующие анализы могут использоваться для иллюстрации промышленной применимости соединений согласно настоящему изобретению.
Биологический анализ 1: ERAP1-опосредованный гидролиз амидного субстрата, измеряемый в биохимической системе
Материалы и растворы
1× Аналитический буфер (AB): 25 мМ Бис-трис пропан, 0,05% в/об Гидроксипропилметилцеллюлозы, pH 7,75, приготовленный с использованием воды Optima
Декапептид WRVYEKC(Dnp)ALK-кислота (где Dnp представляет собой динитрофенилмалеимид) (10-мер)
L-лейцин 7-амидо-4-метилкумарин (L-AMC)
Очищенный ERAP1 (37-941)-10His (ERAP1)
Процедура анализа:
12,5 мкл фермента ERAP1 в 1× AB объединяли с 250 нл тестируемого соединения в ДМСО. Либо 12,5 мкл 240 мкм L-AMC в 1× AB, либо 100 мкм 10-мера в 1× AB добавляли к реакции и инкубировали при 23°C в течение 1 ч. Для обнаружения планшеты считывали при возбуждении 365 нм и эмиссии 442 нм (L-AMC) или возбуждении 279 нм и эмиссии (10-мер) 355 нм. Значения IC50 соединений определяли с использованием 4-параметрического уравнения. Результаты для выбранных соединений согласно изобретению показаны в Таблице 1.
Анализ презентации OVA антигена
Клеточный эффект репрезентативных соединений согласно изобретению в отношении презентации антигена измеряли путем оценки их влияния на презентацию овальбумин-специфического пептида (SIINFEKL) T-клеткам, как описано ранее [Reeves et al, (2014) Proc. Natl. Acad. Sci. USA 111; 17594-17599]. Вкратце, клетки SiHa транзиентно трансфицировали плазмидами, кодирующими мышиный H2Kb и ЭР-направленный пептид-предшественник с удлиненным N-концом, полученный из овальбумина (MRYMILGLLALAAVCSAAIVMKSIINFEHL), с использованием Lipofectamine 3000. Клетки собирали через 6 часов после трансфекции и трансфицированные клетки SiHa наносили на кривую зависимости ответа от концентрации, построенную по 12 точкам, для количественного определения IC50 ингибитора ERAP1. Клетки SiHa культивировали в присутствии соединения в течение 48 ч. Затем клетки B3Z [Karttunen et al, (1992) Proc. Natl. Acad. Sci. USA 89; 6020-6024] добавляли к культуре клеток на 4 ч; B3Z T-клеточная гибридома кодирует TCR, специфично распознающий комплекс SIINFEHL/H2Kb на поверхности клеток, который после активации запускает сигнальный каскад, приводящий к транскрипции гена LacZ, который находится под контролем промотора ИЛ-2. Внутриклеточную активность β-галактозидазы в качестве показателя активации T-клеток измеряли путем количественного определения превращения хлорфенолового красного-β-D-галактопиранозида (CPRG) в хлорфеноловый красный при измерении оптической плотности при 570 нм.
Репрезентативная кривая IC50 для примерных соединений согласно изобретению показана на Фигуре 1. Данные нормализовали по сигналу, полученному в отсутствие соединения (высокий) и в отсутствие антигена (низкий), и представлены как среднее значение±стандартное отклонение (n=2). На Фигуре 2 представлены обобщенные данные IC50, полученные для примерных соединений согласно изобретению. Данные представлены как среднее значение±стандартная ошибка среднего (n=6).
Иммунопептидомика
Эффект репрезентативных соединений согласно изобретению в отношении глобального процессинга антигенов определяли с использованием объективного протеомного ассортимента, как описано Перселлом с соавторами [Purcell et al, (2019) Nat Protoc. 14; 1687-1707]. Вкратце, 500 миллионов клеток SiHa обрабатывали соединением в течение 24 часов или миРНК в течение 72 часов, а затем собирали, лизировали и выделяли MHC-связанные пептиды с помощью иммуноаффинного захватом. Пептиды элюировали 10% (об/об) уксусной кислотой и отделяли от белков MHC-1 и β2-микроглобулина с помощью ВЭЖХ перед анализом методом ЖХ-МС/МС. Репрезентативные результаты показаны на Фигуре 3, которая демонстрирует сопоставимые эффекты ингибирования миРНК ERAP1 и соединения в отношении иммунопептидома (и, следовательно, презентации антигенов) клеток SiHa. Нокдаун ERAP1 влияет на длину антигенов, презентируемых на MHC класса I, уменьшая общую долю пептидов из 8 и 9 аминокислот, но увеличивая количество пептидов из 10, 11, 12 и 13 аминокислот. 10 мкМ соединения приводило к большему влиянию на длину антигенного пептида по сравнению с 1 мкМ.
Различные модификации и вариации описанных аспектов изобретения будут очевидны специалистам в данной области без отступления от объема и сущности изобретения. Хотя изобретение было описано в отношении конкретных предпочтительных вариантов осуществления, следует понимать, что заявленное изобретение не должно быть чрезмерно ограничено такими конкретными вариантами осуществления. Действительно, предполагается, что различные модификации описанных способов осуществления изобретения, которые очевидны для специалистов в соответствующих областях, включены в объем следующей формулы изобретения.
IC50 в сравнении с Декапептидом WRVYEKC(Dnp)ALK-кислота (где Dnp представляет собой Динитрофенилмалеимид) (10-мер); Высокий (<500 нМ), Средний (<5 мкМ), Низкий (>5 мкМ).
Таблица 2: Структуры соединений согласно изобретению
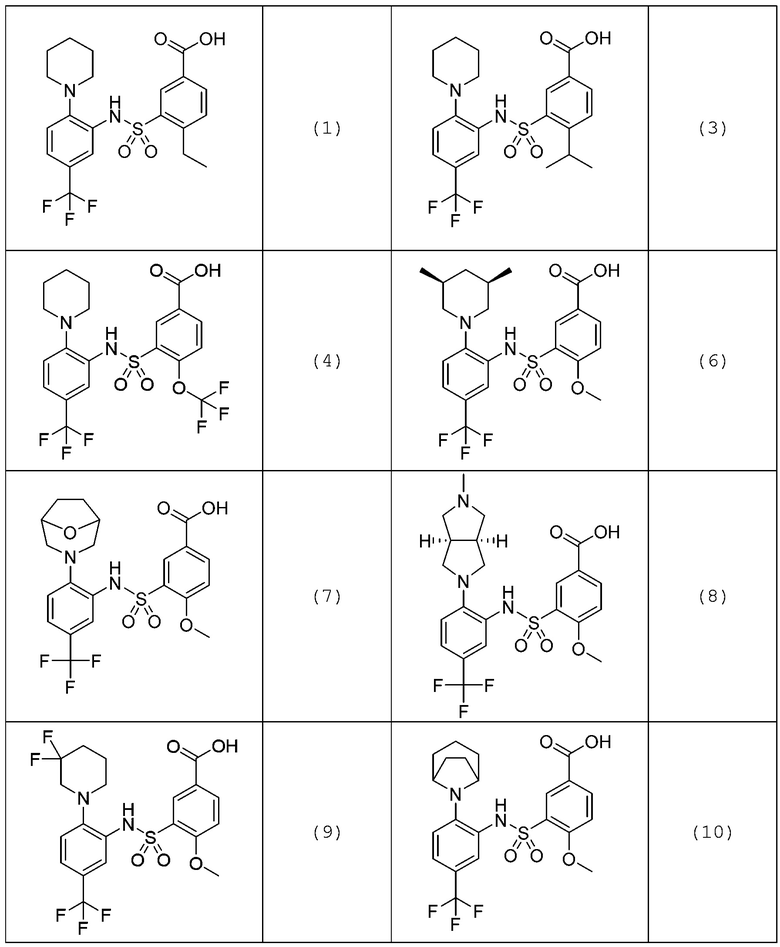
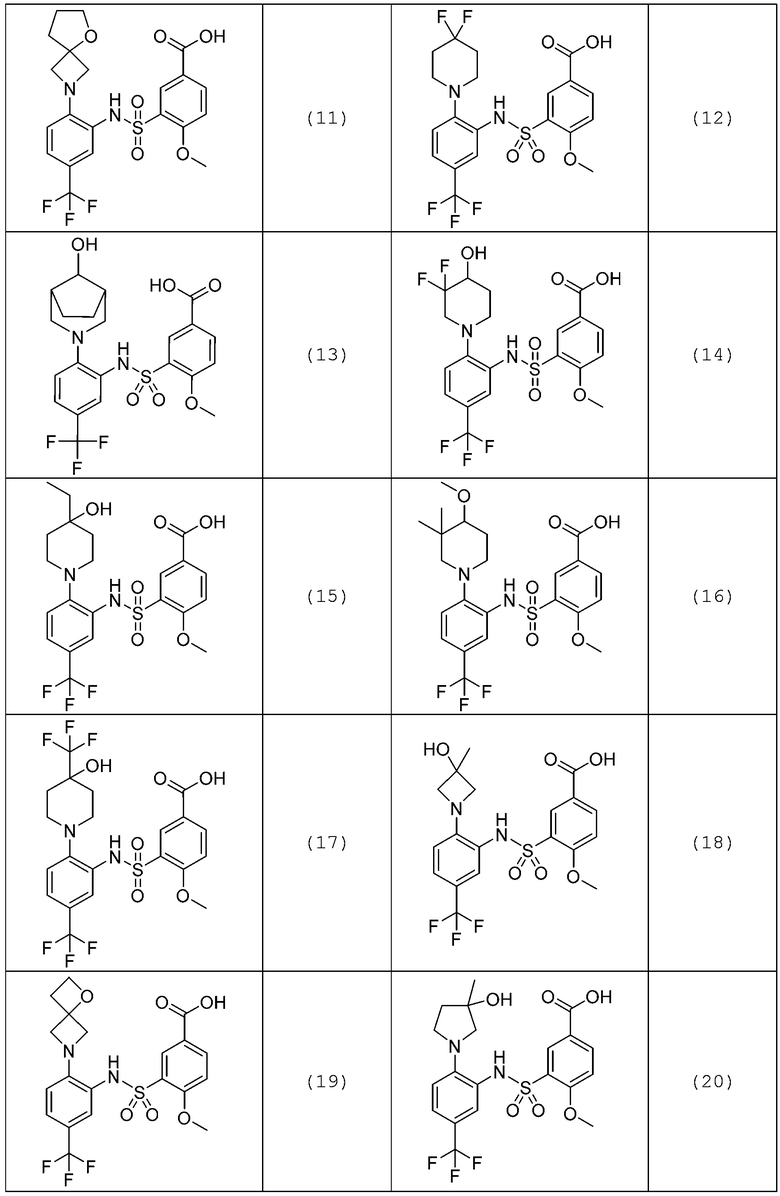
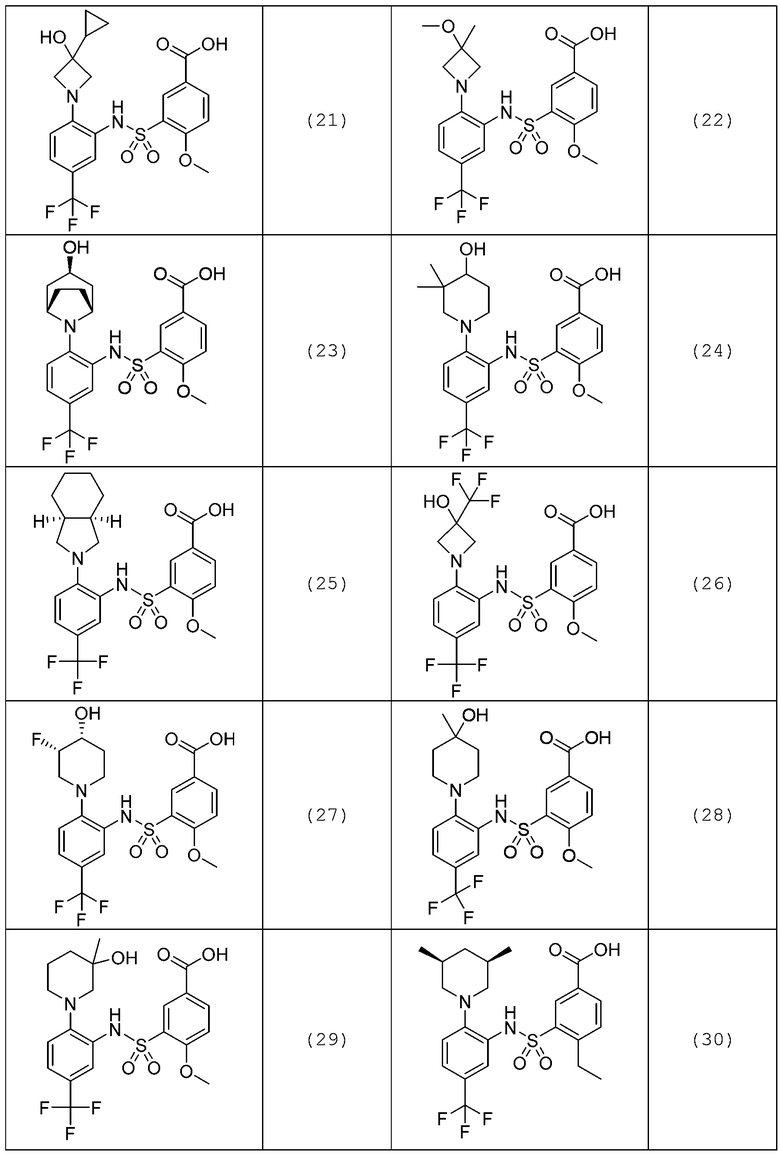
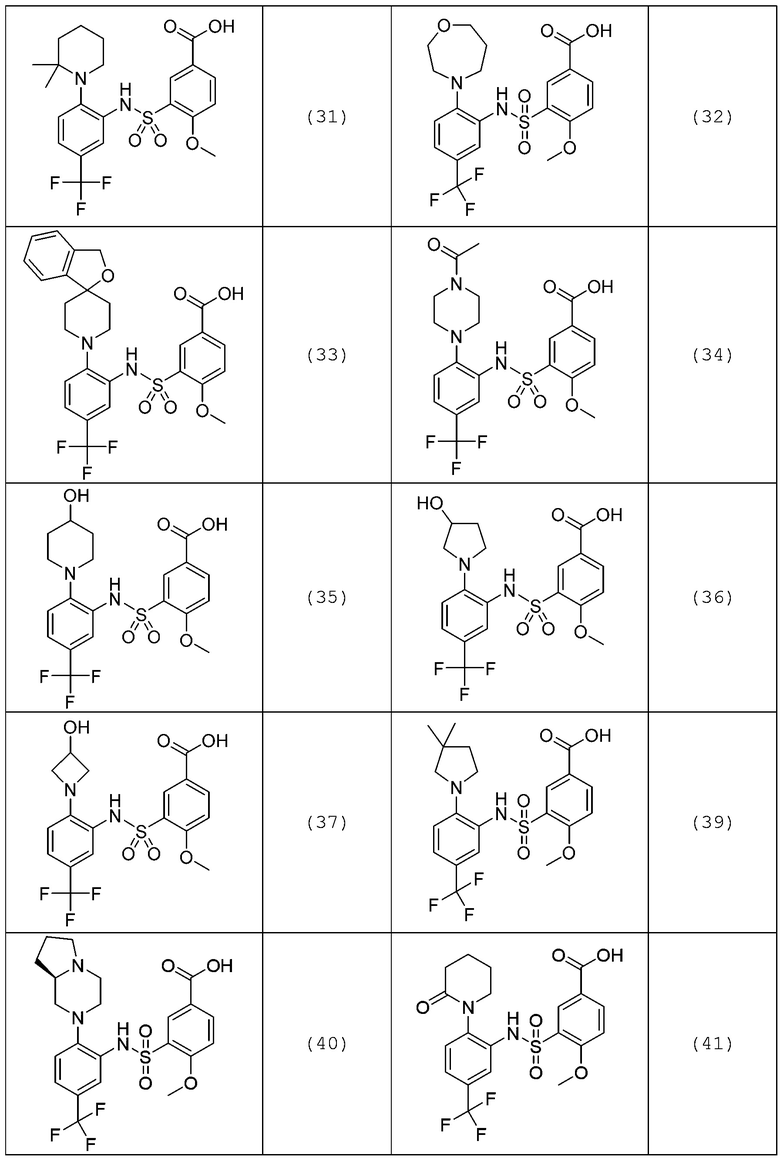
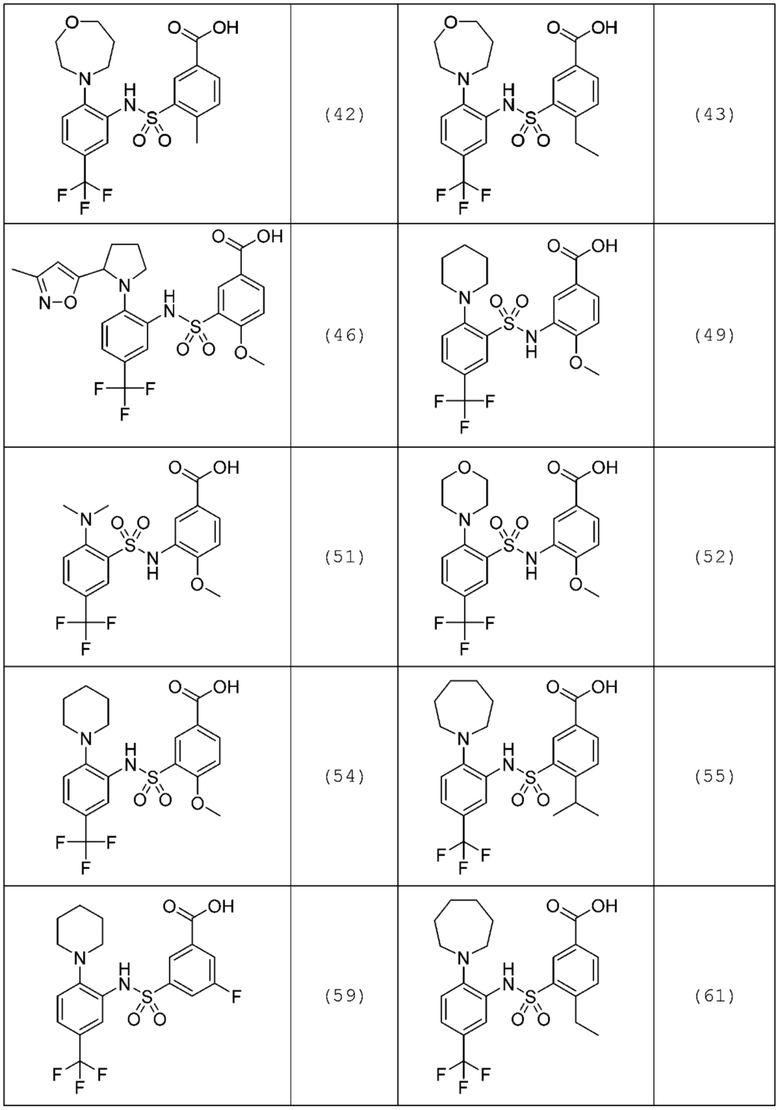
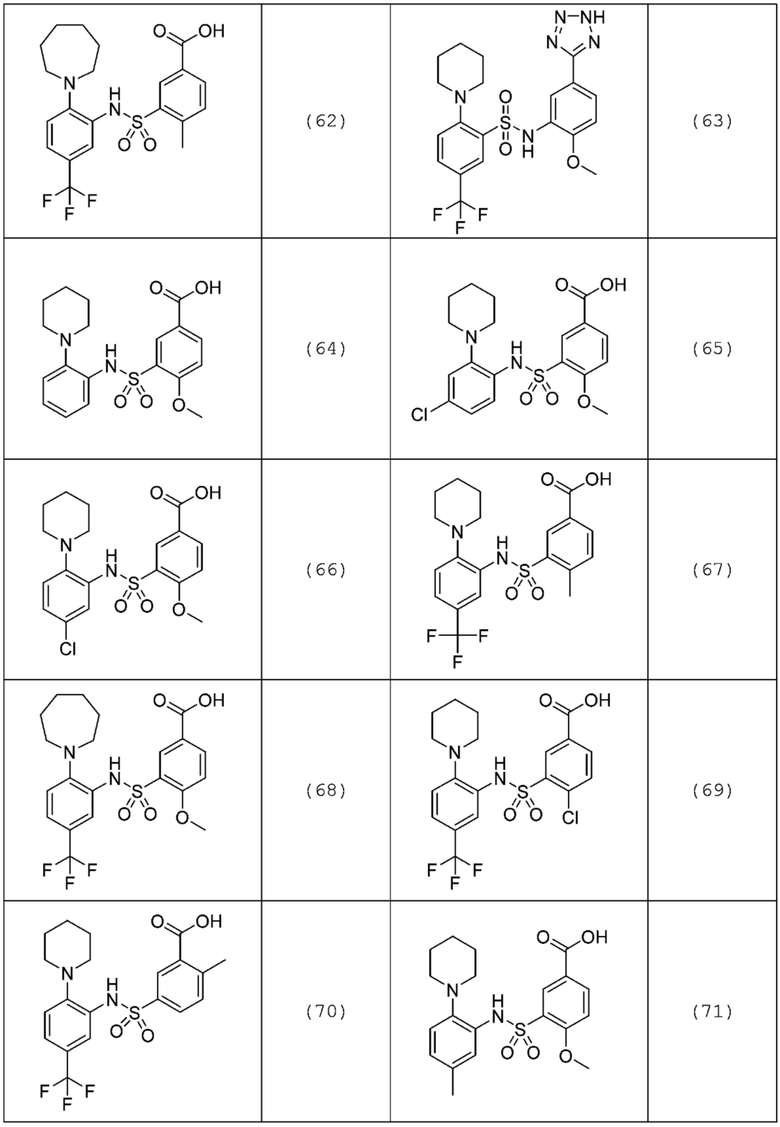
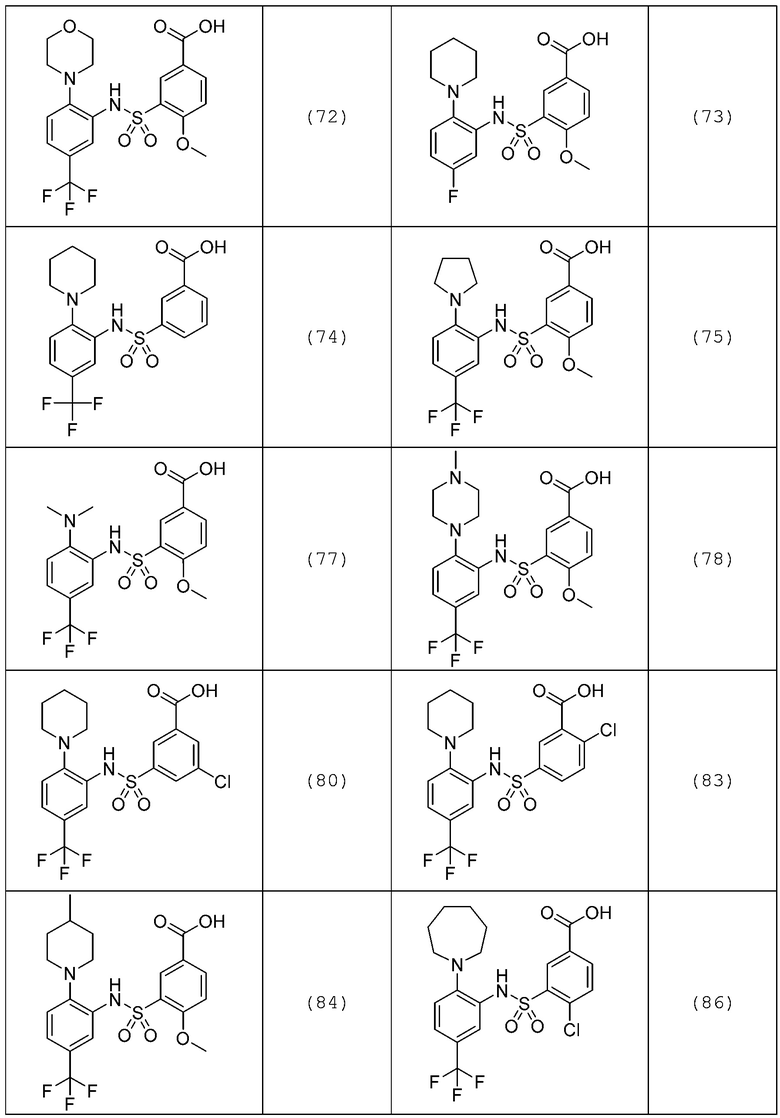
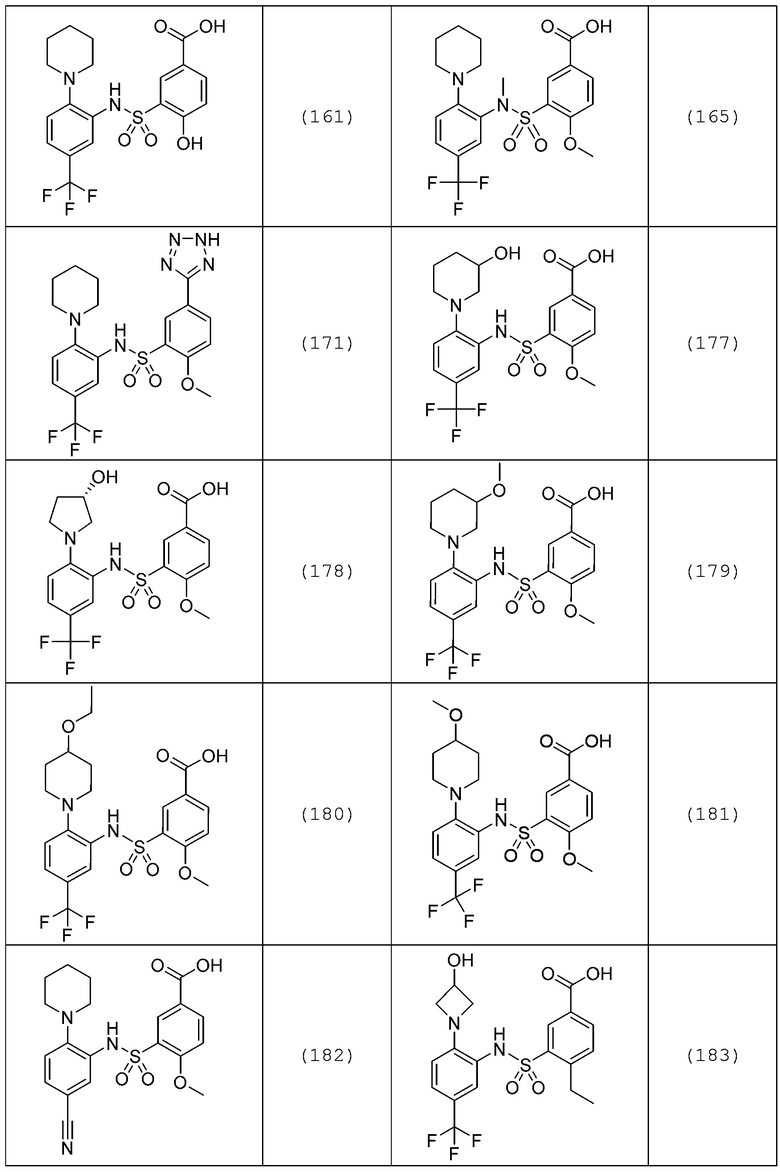
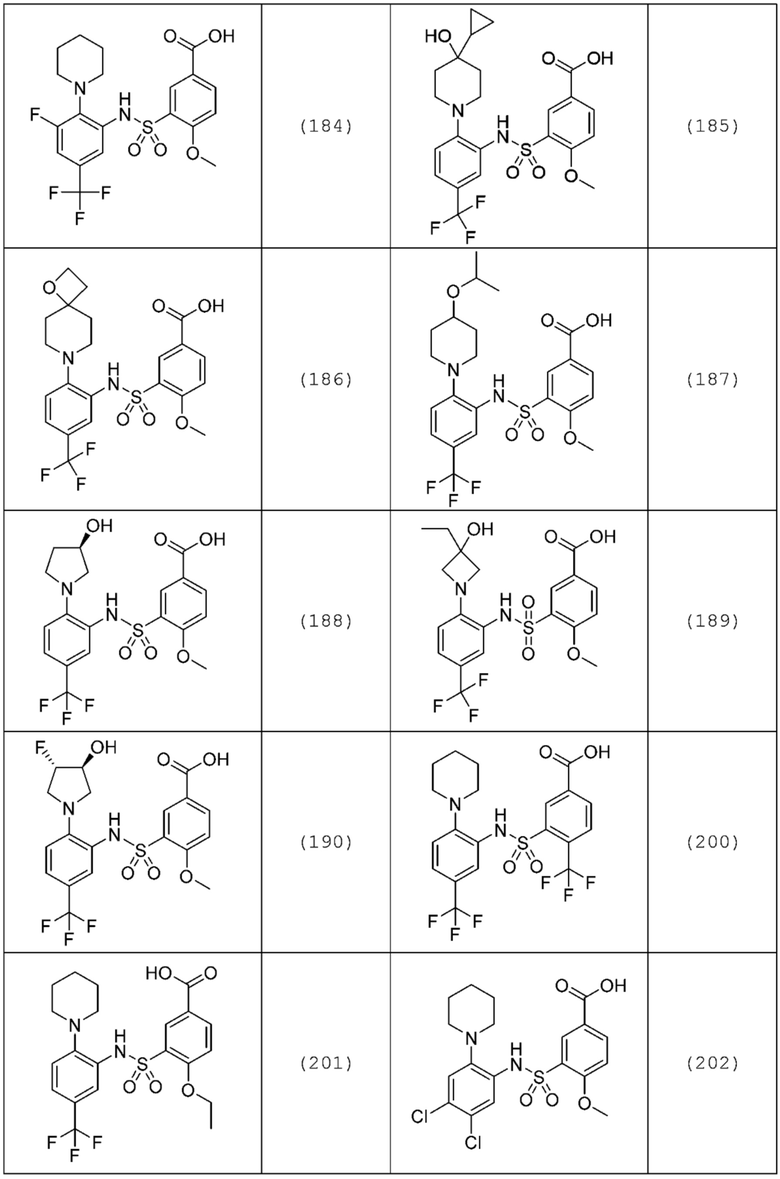
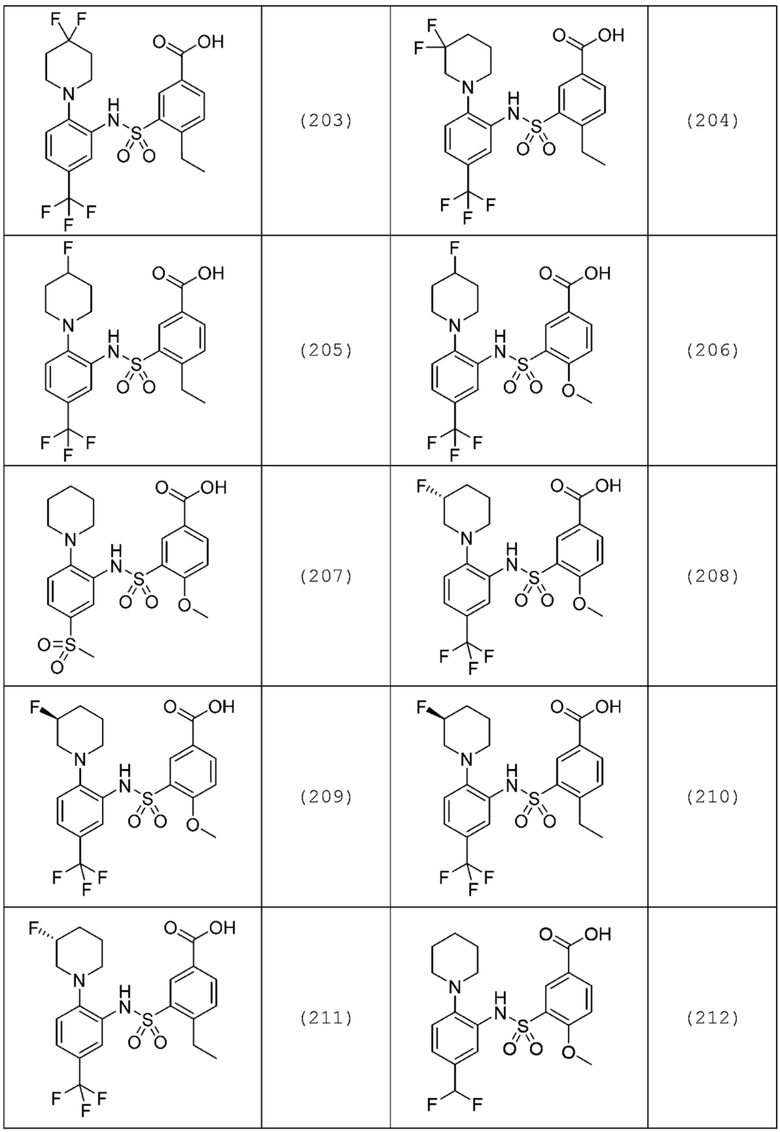
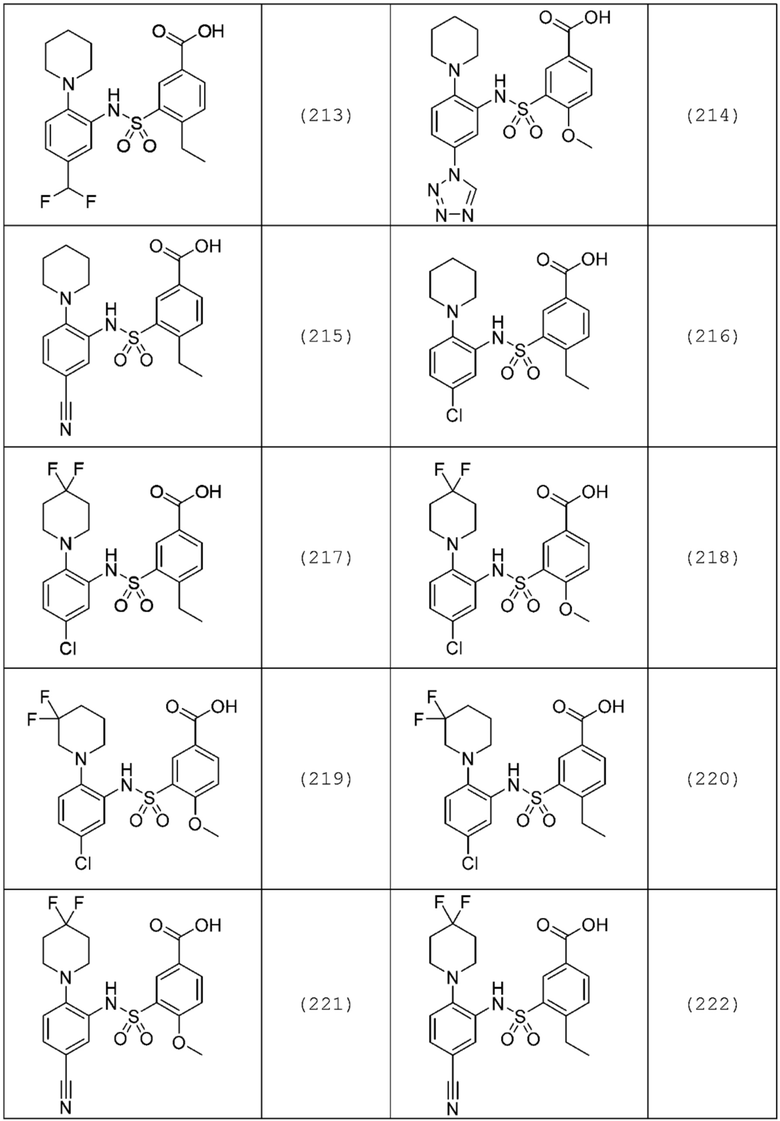
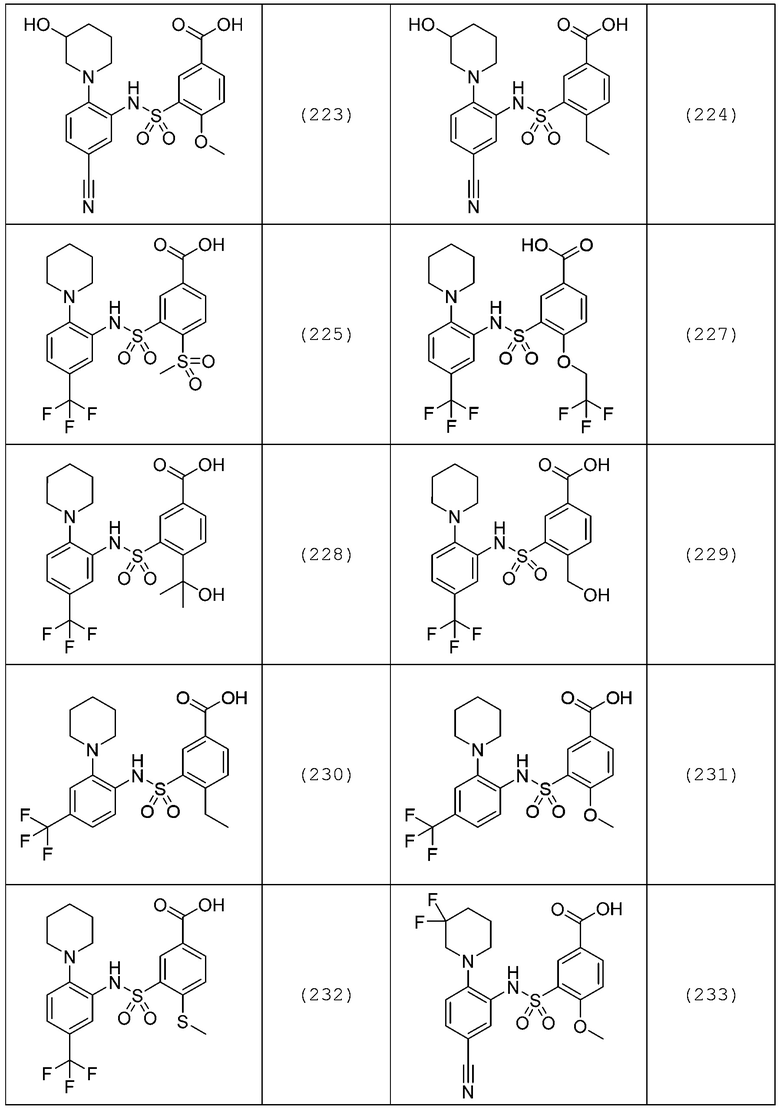
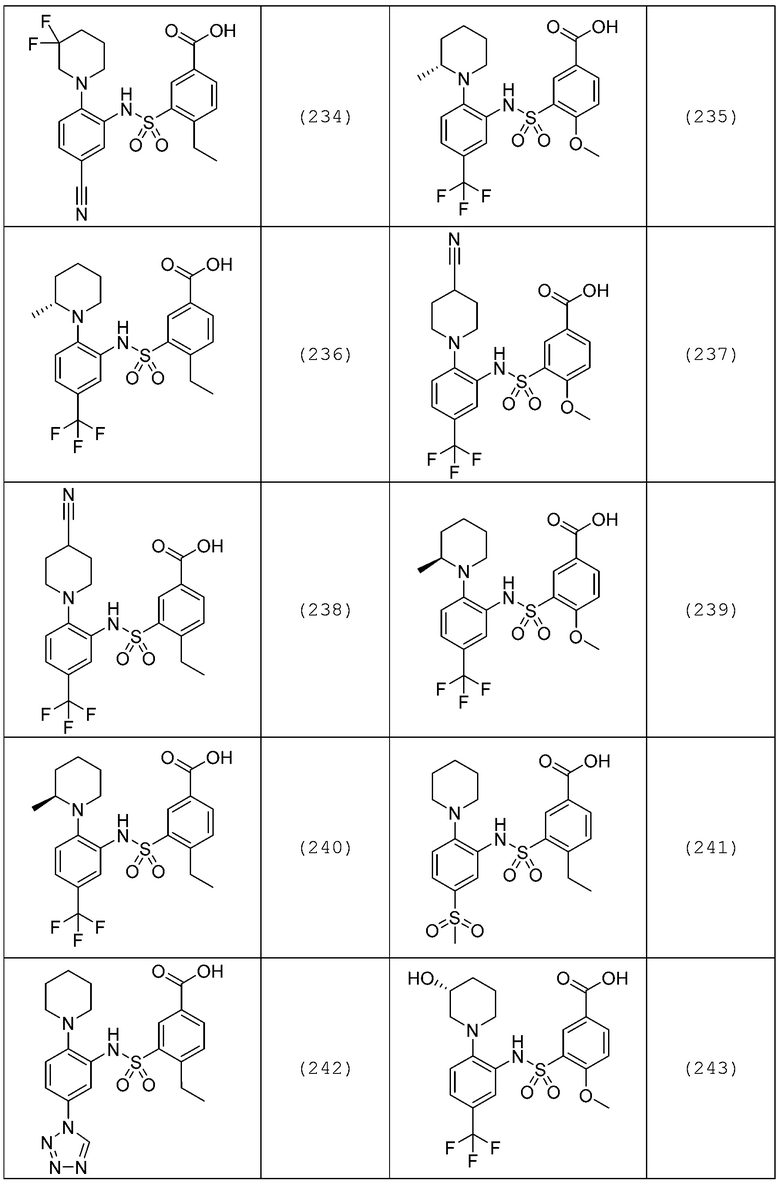
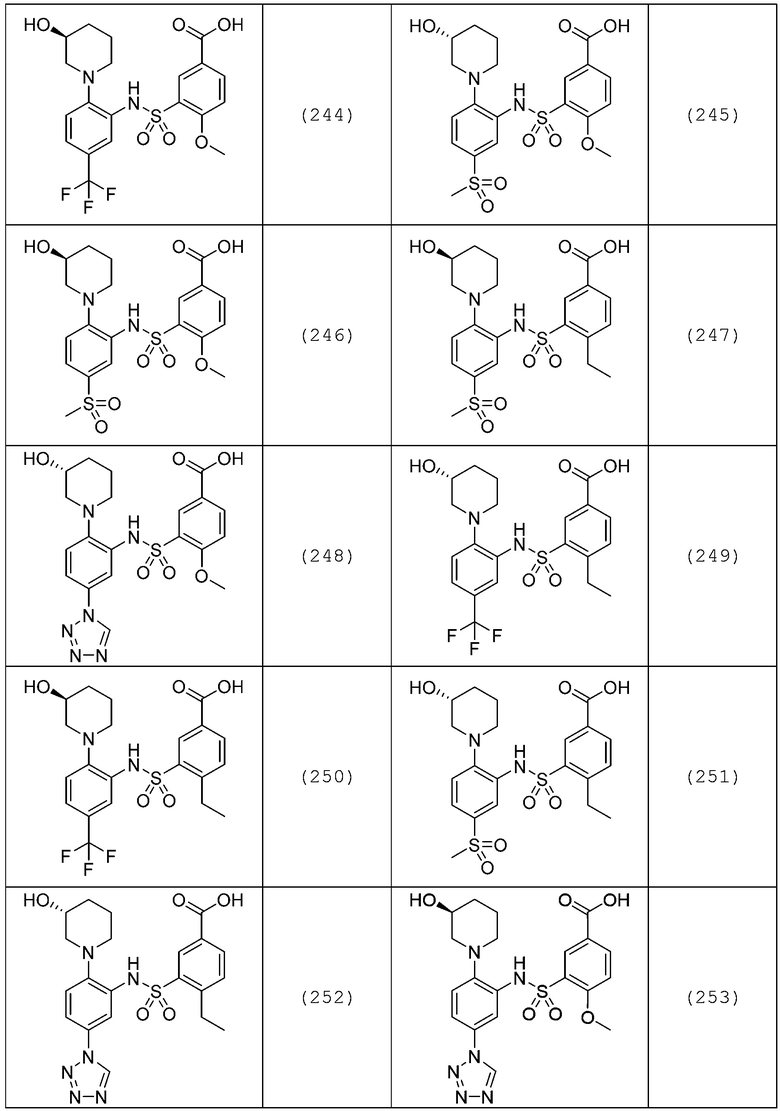
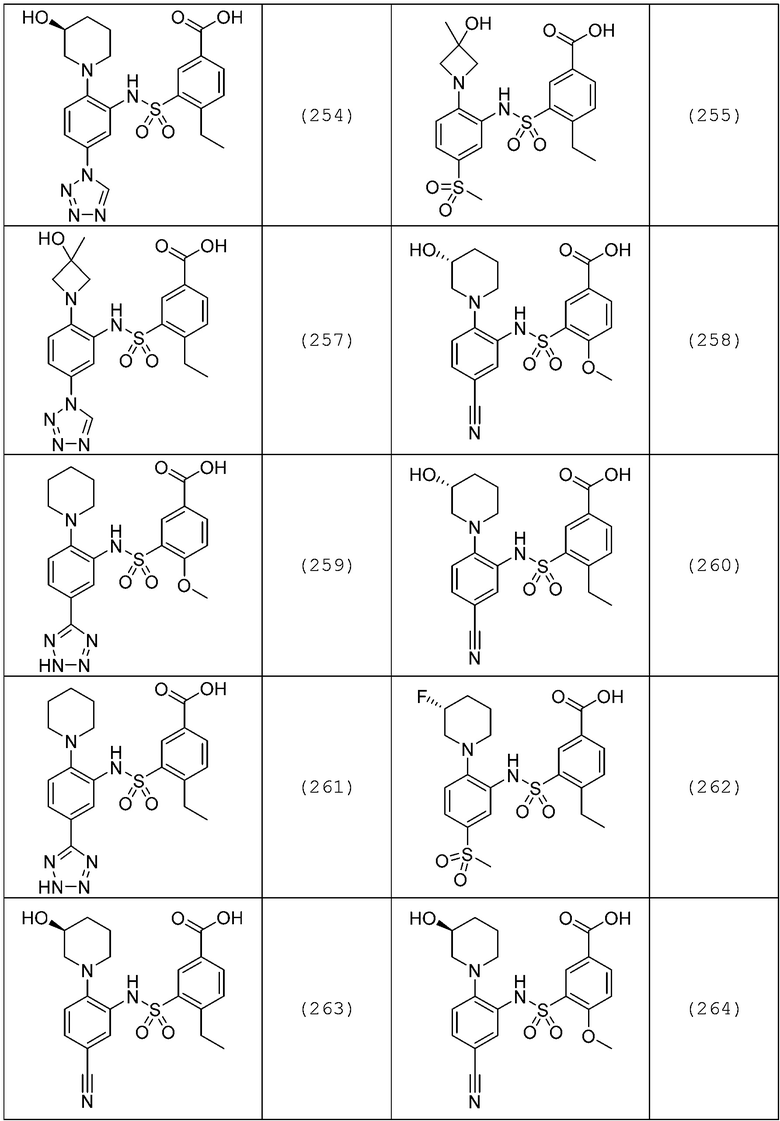
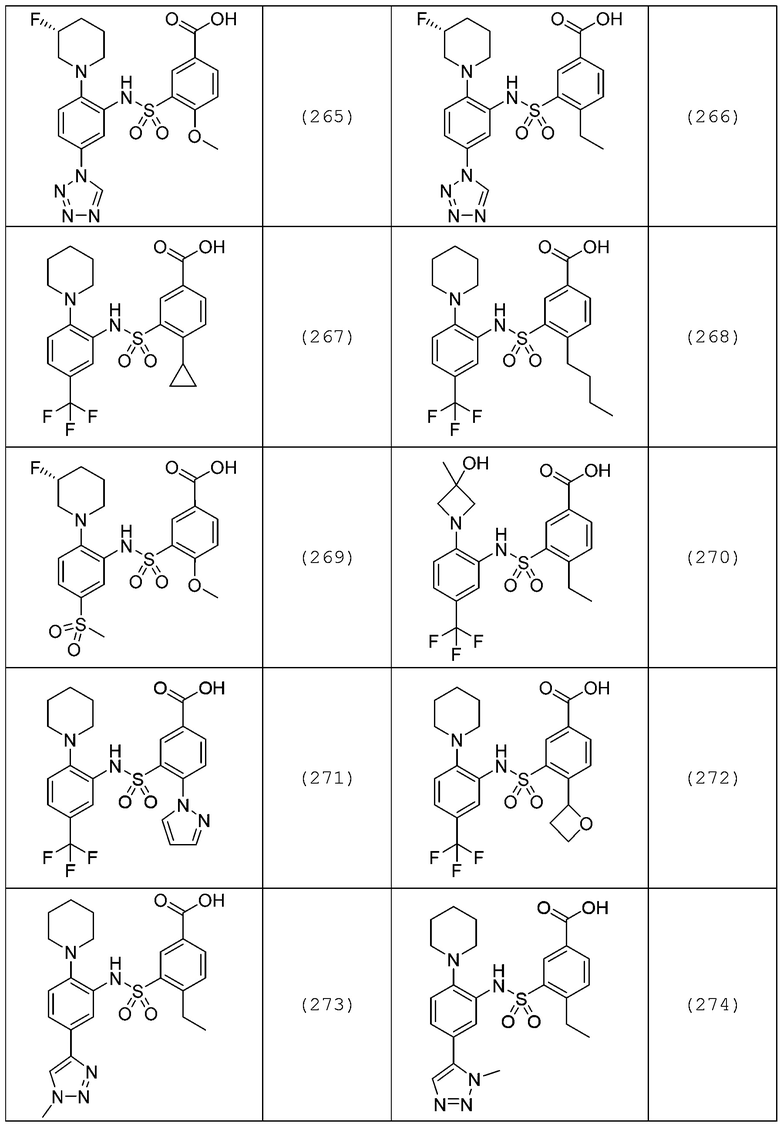
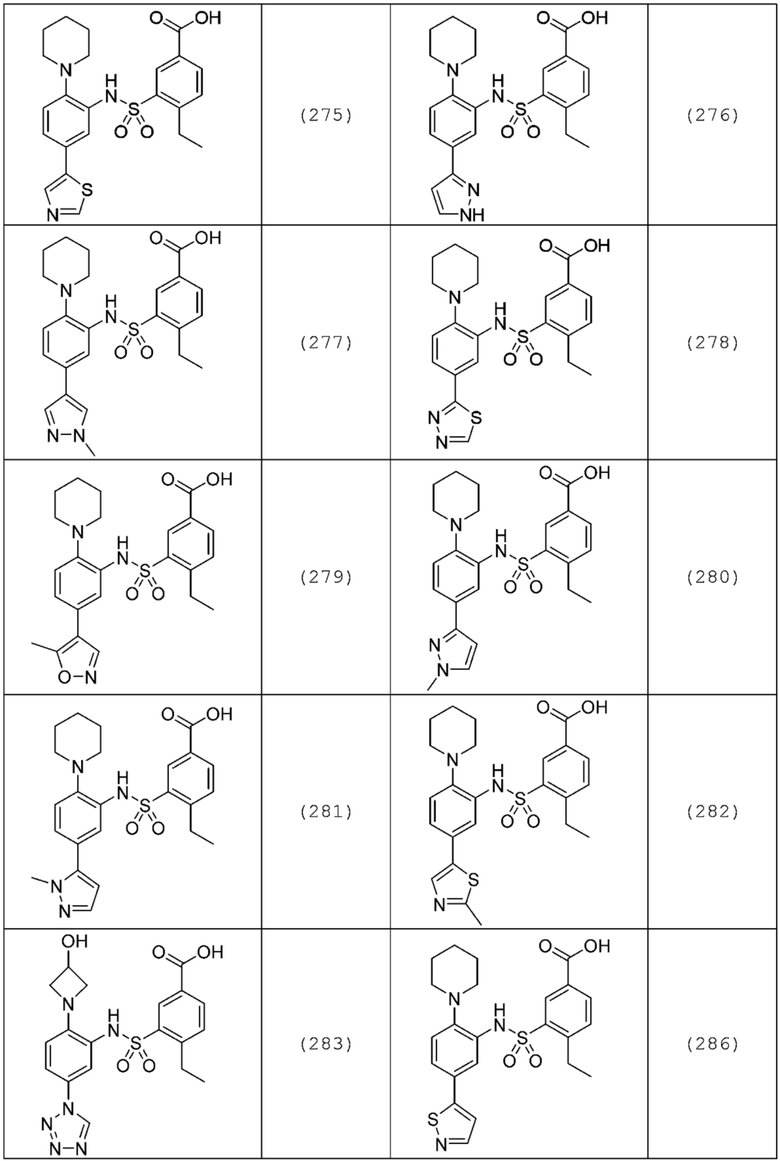
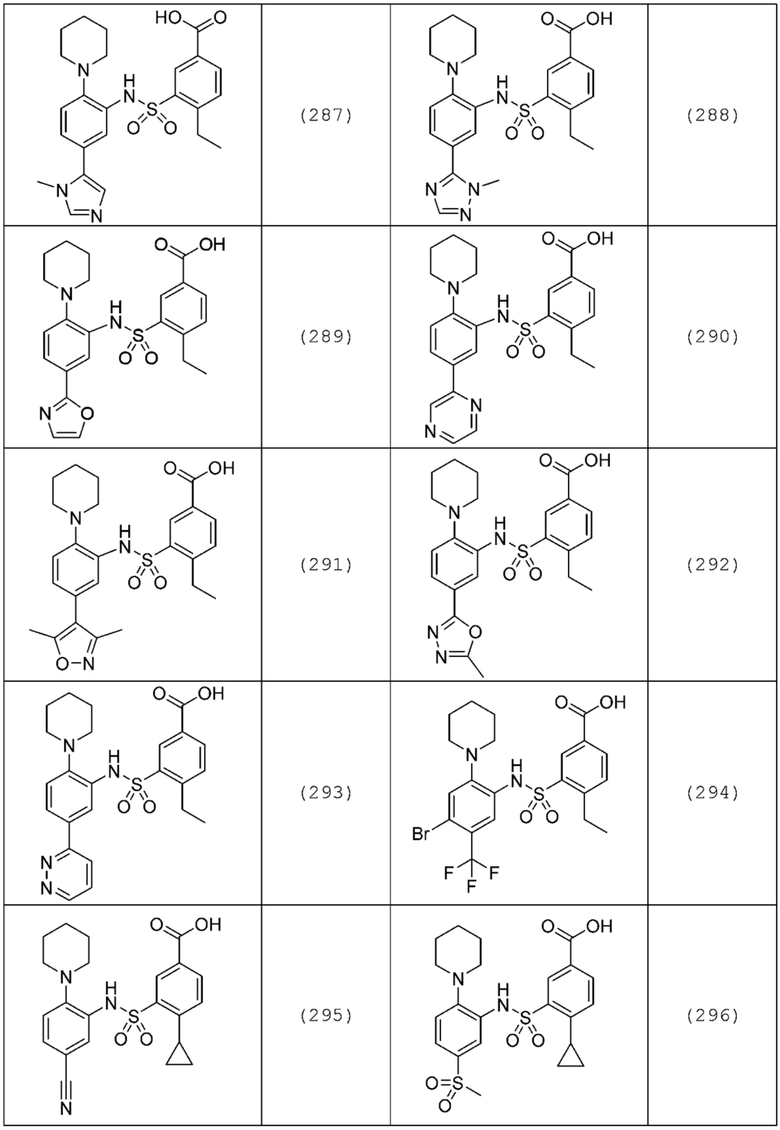
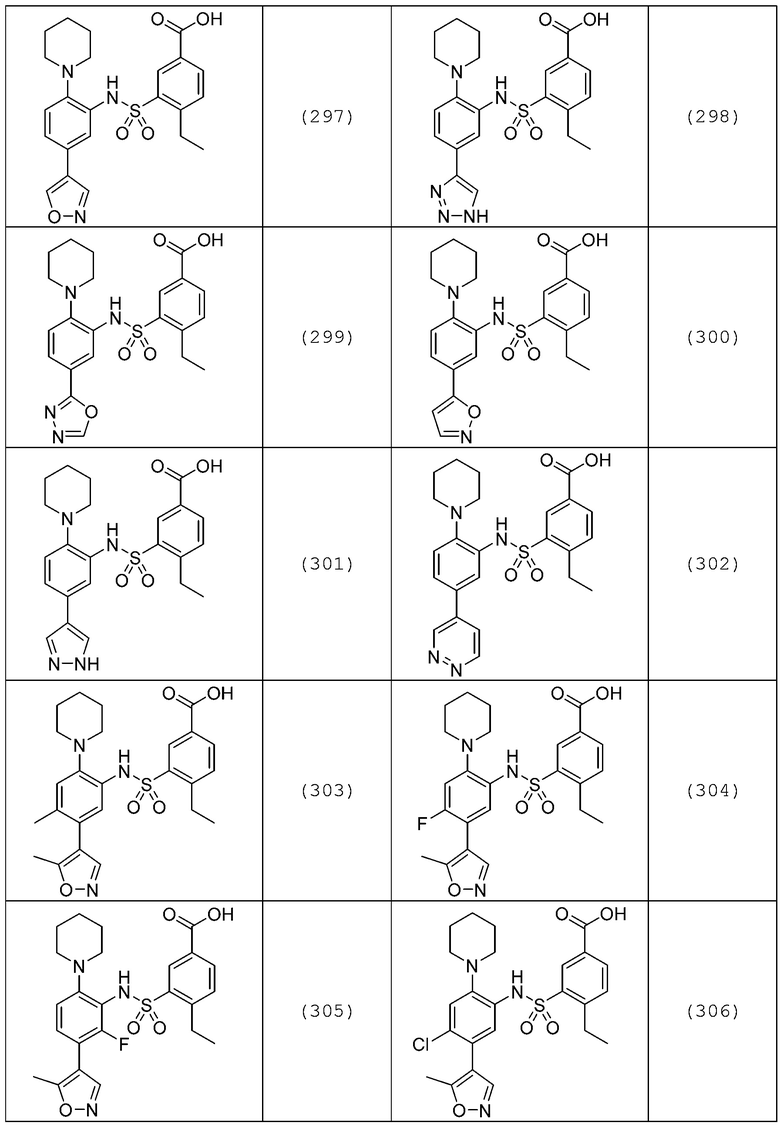
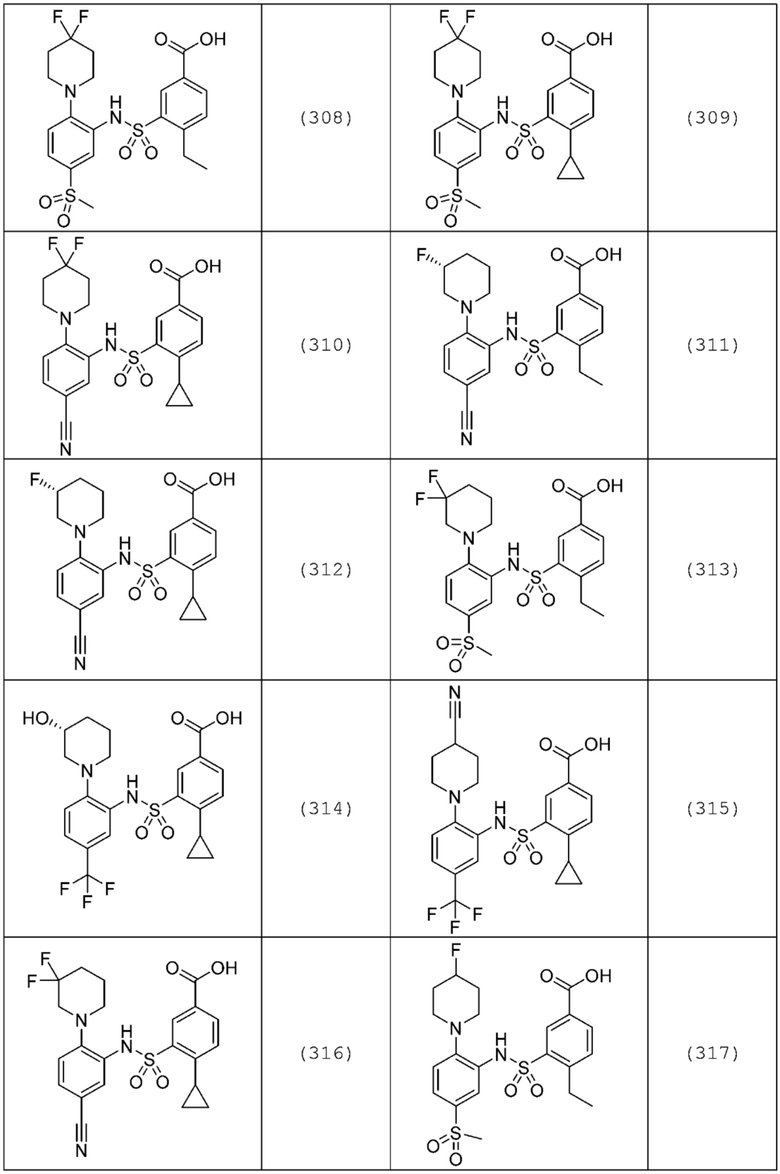
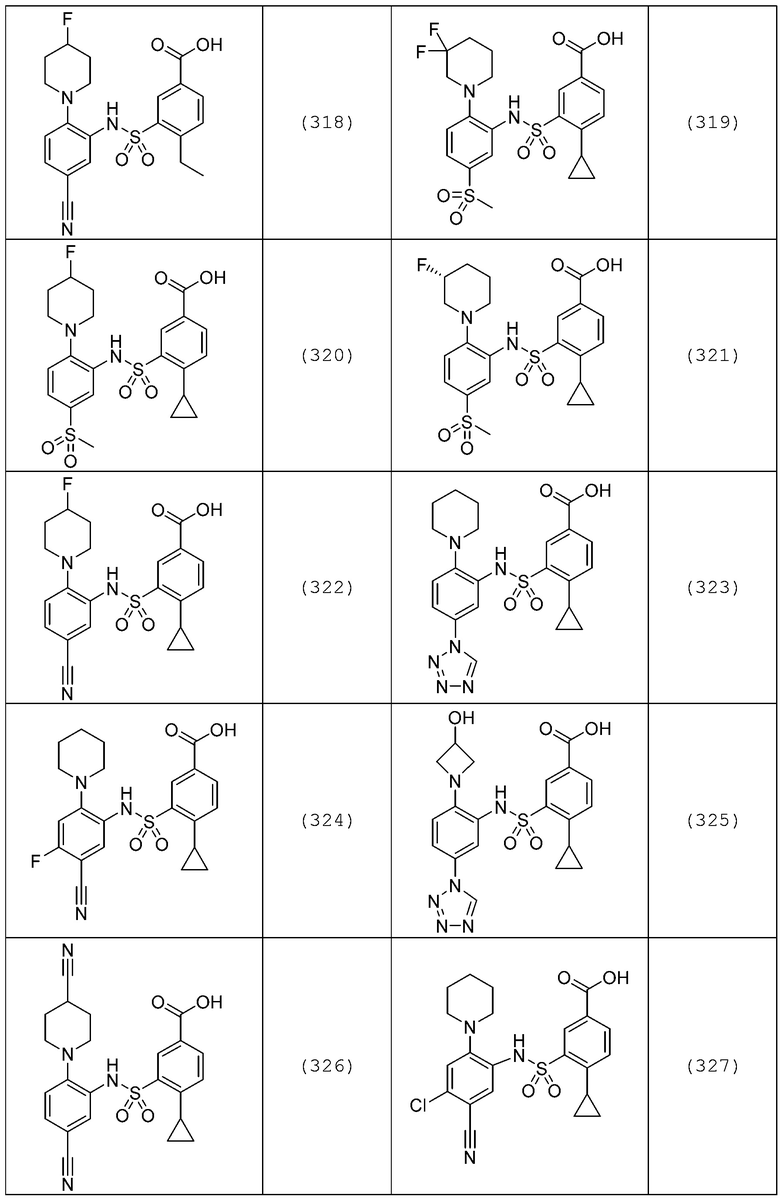
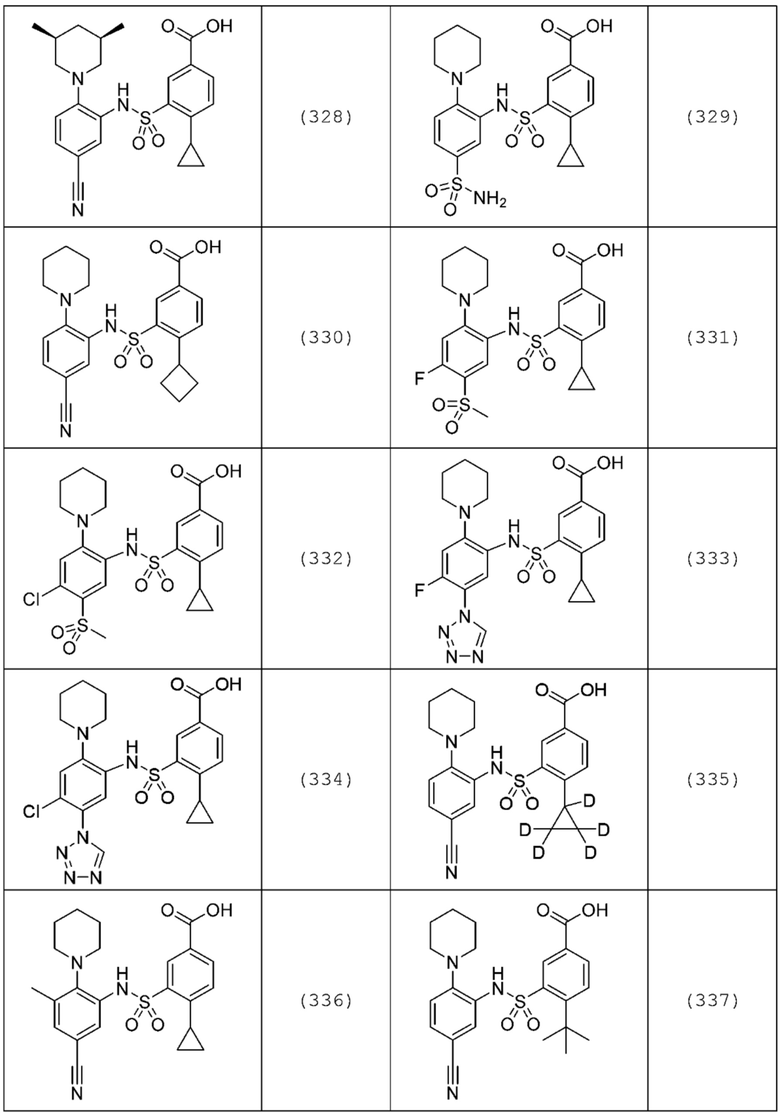

ИСПОЛЬЗОВАННЫЕ ИСТОЧНИКИ
1. Serwold et al., (2002), ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum; Nature: 419, p. 480.
2. Snyder et al., (2014), Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma; NEJM: 371, p. 2189.
3. Van Allen et al., (2015), Genomic correlates of response to CTLA-4 blockade in metastatic melanoma; Science: 348, p. 124.
4. James et al., (2013), Induction of Protective Antitumor Immunity through Attenuation of ERAAP Function; J Immunol: 190, p. 5839.
5. Niranjana et al., (2016), ERAAP Shapes the Peptidome Associated with Classical and Nonclassical MHC Class I Molecules; J Immunol: 197, p. 1035.
6. Pepelyayeva et al., (2018), ERAP1 deficient mice have reduced Type 1 regulatory T cells and develop skeletal and intestinal features of Ankylosing Spondylitis; Sci. Reports: 8: p12464.
7. Cifaldi et al., (2015), ERAP1 Regulates Natural Killer Cell Function by Controlling the Engagement of Inhibitory Receptors, Cancer Res.: 75, p. 824.
8. Steinbach et al., (2017), ERAP1 overexpression in HPV-induced malignancies: A possible novel immune evasion mechanism, Oncoimmunol: 6, e1336594.
9. Kim et al., (2011), Human cytomegalovirus microRNA miR-US4-1 inhibits CD8+ T cell responses by targeting the aminopeptidase ERAP1, Nat. Immunol.: 12, p. 984.
10. Tenzer et al., (2009), Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance, Nat. Immunol.: 10, p. 636.
11. Reeves et al., (2018), The role of polymorphic ERAP1 in autoinflammatory disease, Biosci. Rep.: 29, p. 38.
12. Chen et al., (2014), Silencing or inhibition of endoplasmic reticulum aminopeptidase 1 (ERAP1) suppresses free heavy chain expression and Th17 responses in ankylosing spondylitis, Ann Rheum Dis: 75, p. 916.
13. Sheehan, NJ (January 2004). "The ramifications of HLA-B27". Journal of the Royal Society of Medicine. 97 (1): 10-4.
14. Smith, JA (January 2015). "Update on ankylosing spondylitis: current concepts in pathogenesis". Current allergy and asthma reports. 15 (1): 489.
15. Kuiper JJW, Mutis T, de Jager W, de Groot-Mijnes JD, Rothova A (2011). "Intraocular interleukin-17 and proinflammatory cytokines in HLA-A29-associated birdshot chorioretinopathy". Am J Ophthalmol. 152 (2): 177-182.
16. Kuiper JJW, Emmelot ME, Rothova A, Mutis T (2013). "Interleukin-17 production and T helper 17 cells in peripheral blood mononuclear cells in response to ocular lysate in patients with birdshot chorioretinopathy". Mol Vis. 19: 2606-14.
17. Kuiper JJW, van Setten J, Ripke S, Van't Slot R, Mulder F, Missotten T, Baarsma GS, Francioli LC, Pulit SL, de Kovel CG, Ten Dam-van Loon N, den Hollander AI, Huis In Het Veld P, Hoyng CB, Cordero-Coma M,  Arya B, Thomas D, Bakker SC, Ophoff RA, Rothova A, de Bakker PI, Mutis T, Koeleman BP (2014). "A genome-wide association study identifies a functional ERAP2 haplotype associated with birdshot chorioretinopathy". Hum Mol Genet. 23 (22): 6081-6087.
Arya B, Thomas D, Bakker SC, Ophoff RA, Rothova A, de Bakker PI, Mutis T, Koeleman BP (2014). "A genome-wide association study identifies a functional ERAP2 haplotype associated with birdshot chorioretinopathy". Hum Mol Genet. 23 (22): 6081-6087.
18. Evans et al. (2011), Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 10; 43(8): 761-7.
19. Conde-Jaldon et al. (2014), Epistatic interaction of ERAP1 and HLA-B in  a replication study in the Spanish population. PLoS One. 14; 9(7).
a replication study in the Spanish population. PLoS One. 14; 9(7).
20. Kuiper et al (2018), Functionally distinct ERAP1 and ERAP2 are a hallmark of HLA-A29-(Birdshot) Uveitis. Hum Mol Genet. doi: 10.1093/hmg/ddy319.
21. Strange et al. (2010), A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet.; 42(11): 985-90.
| название | год | авторы | номер документа |
|---|---|---|---|
| РАПП-ПОДДЕРЖИВАЮЩИЕ ТИАЗОЛИДИНДИОНЫ И ИХ КОМБИНАЦИИ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2570424C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| НОВЫЕ СУЛЬФОНАМИДКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2808572C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2667788C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ BTK И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2703301C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
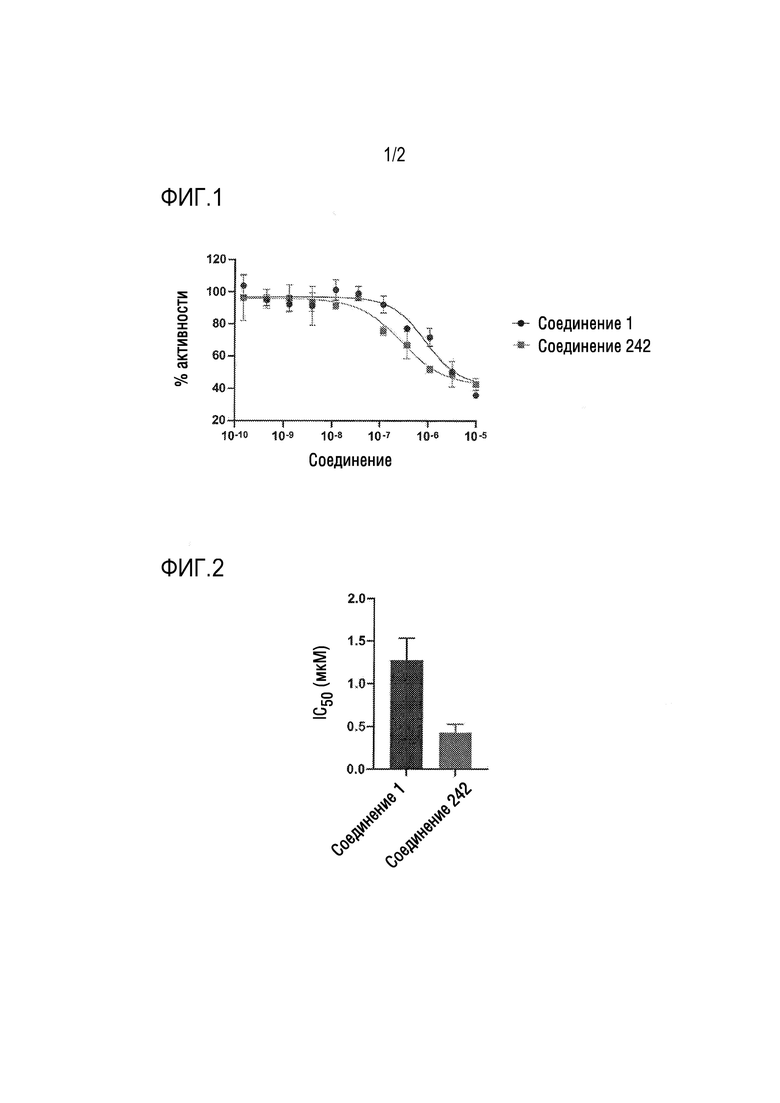

Изобретение относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где группа X-Y представляет собой -NHSO2- или -SO2NH-; R1 представляет собой H; R2 выбран из COOH и тетразолильной группы; R3 выбран из H, Cl и C1-C6-алкила; R4 выбран из H, Cl и F; R5 выбран из H, C1-C6-алкила, C≡CH, C1-C6-галогеналкила, SO2-C1-C6-алкила, Cl, C1-C6-алкокси, OH, C1-C6-гидроксиалкила, C1-C6-алкилтио, пиразолила, C3-C7-циклоалкила, C3-C7-гетероциклоалкила, содержащего один гетероатом, выбранный из кислорода, и C1-C6-галогеналкокси; R6 представляет собой H; R7 выбран из H, CN, C1-C6-галогеналкила, Cl, F, SO2-C1-C6-алкила, SO2NR13R14, 5-6-членного моноциклического гетероарила и C1-C6-алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из C1-C6-алкила; R8 выбран из H, C1-C6-алкила, C1-C6-галогеналкила и галогена; R9 представляет собой H, C1-C3-алкил или галоген; R10 и R11 вместе с азотом, к которому они присоединены, образуют азепанильную группу, где один углерод в указанной азепанильной группе заменен группой, выбранной из O; или R10 и R11 вместе с азотом, к которому они присоединены, образуют азетидинильную, пирролидинильную или пиперидинильную группу, где: (a) указанная азетидинильная, пирролидинильная или пиперидинильная группа замещена одной или более группами, выбранными из C1-C6-алкила, CN, C3-C7-циклоалкила, OH, C1-C6-алкокси, галогена, C1-C6-галогеналкила и оксазолила, где указанная оксазолильная группа в свою очередь необязательно дополнительно замещена одной группой, выбранной из C1-C6-алкила, или (b) один углерод в указанной пиперидинильной группе заменен группой, выбранной из CO; или R10 и R11 вместе с азотом, к которому они присоединены, образуют 8-членную бициклическую гетероциклоалкильную группу, где один углерод в бициклическом гетероциклоалкильном кольце необязательно заменен группой, выбранной из O и NH, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила и OH; или R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен группой, выбранной из O и NH, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила; и каждый R13 и R14 независимо представляет собой H. Изобретение также относится к фармацевтической композиции, обладающей модулирующей ERAP1 активностью, на основе указанного соединения. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения пролиферативного, иммунного, вирусного или воспалительного нарушений. 7 н. и 51 з.п. ф-лы, 3 ил., 2 табл., 338 пр.
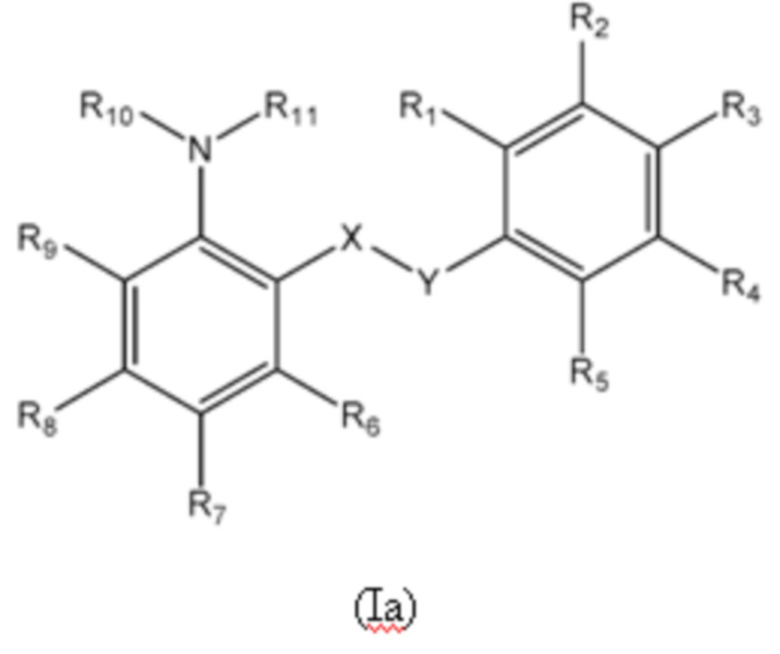
1. Соединение формулы (Ia) или его фармацевтически приемлемая соль,
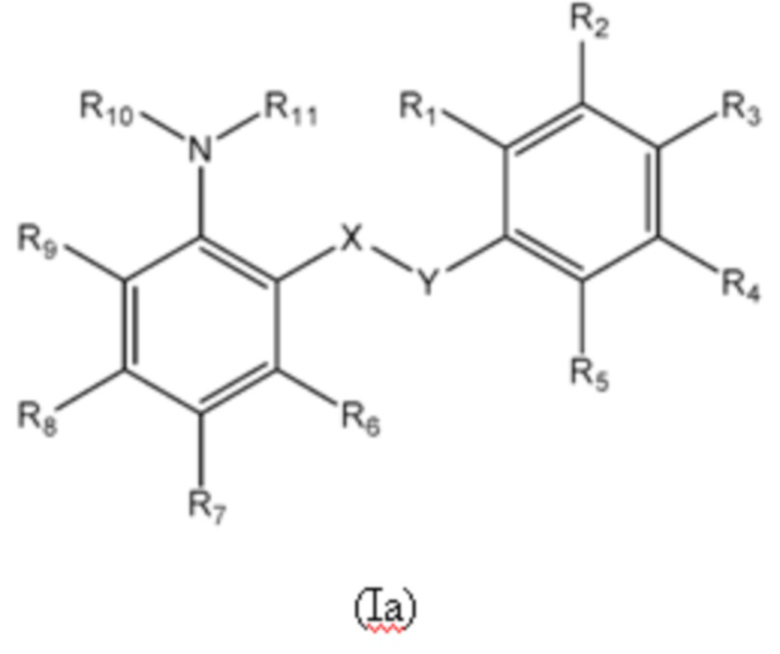 ,
,
где группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и C1-C6-алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, C1-C6-алкила, C≡CH, C1-C6-галогеналкила, SO2-C1-C6-алкила, Cl, C1-C6-алкокси, OH, C1-C6-гидроксиалкила, C1-C6-алкилтио, пиразолила, C3-C7-циклоалкила, C3-C7-гетероциклоалкила, содержащего один гетероатом, выбранный из кислорода, и C1-C6-галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, C1-C6-галогеналкила, Cl, F, SO2-C1-C6-алкила, SO2NR13R14, 5-6-членного моноциклического гетероарила и C1-C6-алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из C1-C6-алкила;
R8 выбран из H, C1-C6-алкила, C1-C6-галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 и R11 вместе с азотом, к которому они присоединены, образуют азепанильную группу, где один углерод в указанной азепанильной группе заменен группой, выбранной из O; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют азетидинильную, пирролидинильную или пиперидинильную группу, где: (a) указанная азетидинильная, пирролидинильная или пиперидинильная группа замещена одной или более группами, выбранными из C1-C6-алкила, CN, C3-C7-циклоалкила, OH, C1-C6-алкокси, галогена, C1-C6-галогеналкила и оксазолила, где указанная оксазолильная группа в свою очередь необязательно дополнительно замещена одной группой, выбранной из C1-C6-алкила, или (b) один углерод в указанной пиперидинильной группе заменен группой, выбранной из CO; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 8-членную бициклическую гетероциклоалкильную группу, где один углерод в бициклическом гетероциклоалкильном кольце необязательно заменен группой, выбранной из O и NH, и указанная бициклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила и OH; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен группой, выбранной из O и NH, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила; и
каждый R13 и R14 независимо представляет собой H.
2. Соединение формулы (Ia) по п. 1, где R2 представляет собой COOH.
3. Соединение формулы (Ia) по п. 1 или 2, где X-Y представляет собой -NH-SO2-.
4. Соединение формулы (Ia) по любому предыдущему пункту, где R5 выбран из C1-C6-алкила, C≡CH, C1-C6-галогеналкила, SO2-C1-C6-алкила, Cl, C1-C6-алкокси, OH, C1-C6-гидроксиалкила, C1-C6-алкилтио, пиразолила, C3-C7-циклоалкила, C3-C7-гетероциклоалкила, содержащего один гетероатом, выбранный из кислорода, и C1-C6-галогеналкокси, и более предпочтительно выбран из OMe, Me, Et, Pr, SMe, CH2OH, этинила, циклопропила, оксетанила и Cl, и еще более предпочтительно представляет собой OMe.
5. Соединение формулы (Ia) по любому предыдущему пункту, где все R1, R3, R4, R6, R8 и R9 представляют собой H.
6. Соединение формулы (Ia) по любому предыдущему пункту, где R7 представляет собой C1-C6-галогеналкил, более предпочтительно CF3.
7. Соединение формулы (Ia) по любому предыдущему пункту, где R10 и R11 вместе с азотом, к которому они присоединены, образуют азетидинильную группу, которая замещена одной или более группами, выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
8. Соединение формулы (Ia) по любому одному из пп. 1-6, где R10 и R11 вместе с азотом, к которому они присоединены, образуют пирролидинильную группу, которая замещена одной или более группами, выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
9. Соединение формулы (Ia) по любому одному из пп. 1-6, где R10 и R11 вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, которая замещена одной или более группами, выбранными из C1-3-алкила, CN, C3-6-циклоалкила, OH, C1-3-алкокси, галогена и CF3.
10. Соединение формулы (Ia) по п. 9, где R10 и R11 вместе с азотом, к которому они присоединены, образуют пиперидинильную группу, которая необязательно замещена одной или более группами, выбранными из C1-C6-алкила, CN, OH и галогена, и где два несмежных углерода в кольце указанной пиперидинильной группы связаны друг с другом через алкиленовый мостик из 2 углеродов или 3 углеродов.
11. Соединение формулы (Ia) по любому одному из пп. 1-6, где R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен O и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила.
12. Соединение формулы (Ia) по п. 11, где R10 и R11 вместе с азотом, к которому они присоединены, образуют 7-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен O и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила.
13. Соединение формулы (Ia) по п. 11, где R10 и R11 вместе с азотом, к которому они присоединены, образуют 8-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен O и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила.
14. Соединение формулы (Ia) по п. 11, где R10 и R11 вместе с азотом, к которому они присоединены, образуют 9-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе заменен O и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила.
15. Соединение формулы (Ia) по любому одному из пп. 1-6, где NR10R11 выбран из следующих групп:
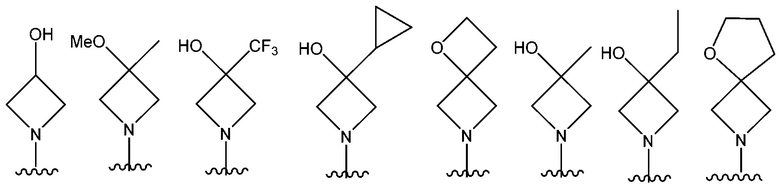 .
.
16. Соединение формулы (Ia) по любому одному из пп. 1-6, где NR10R11 выбран из следующих групп:
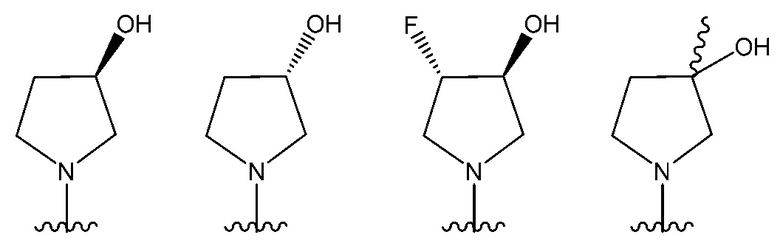 .
.
17. Соединение формулы (Ia) по любому одному из пп. 1-6, где NR10R11 выбран из следующих групп:
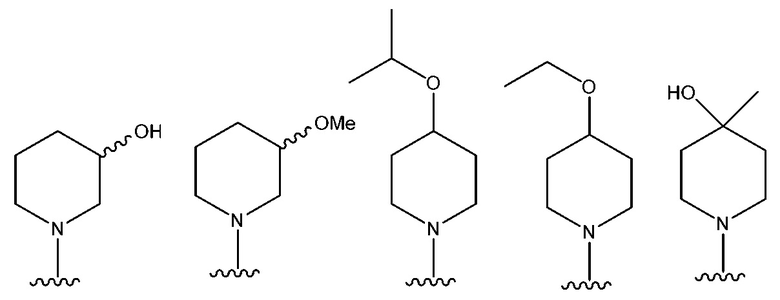
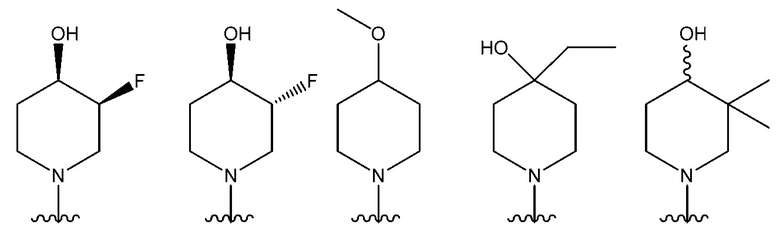
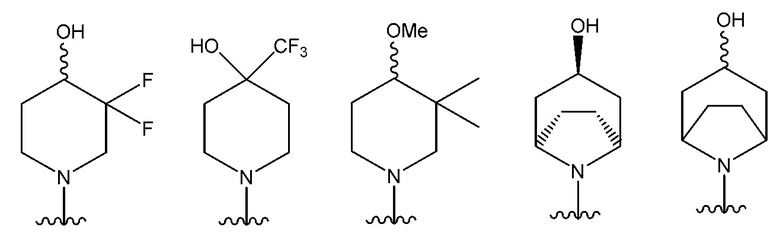
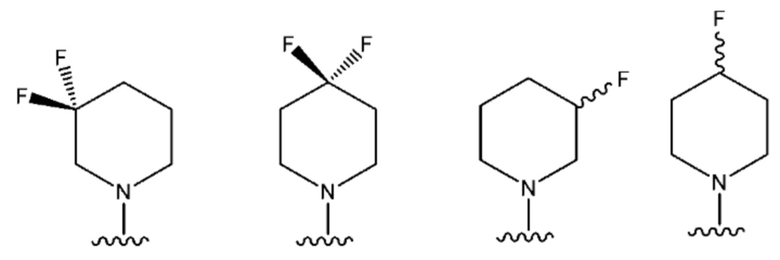
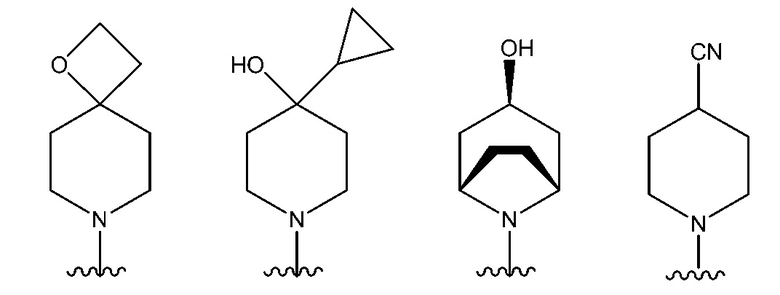 .
.
18. Соединение по п. 1, выбранное из следующего:
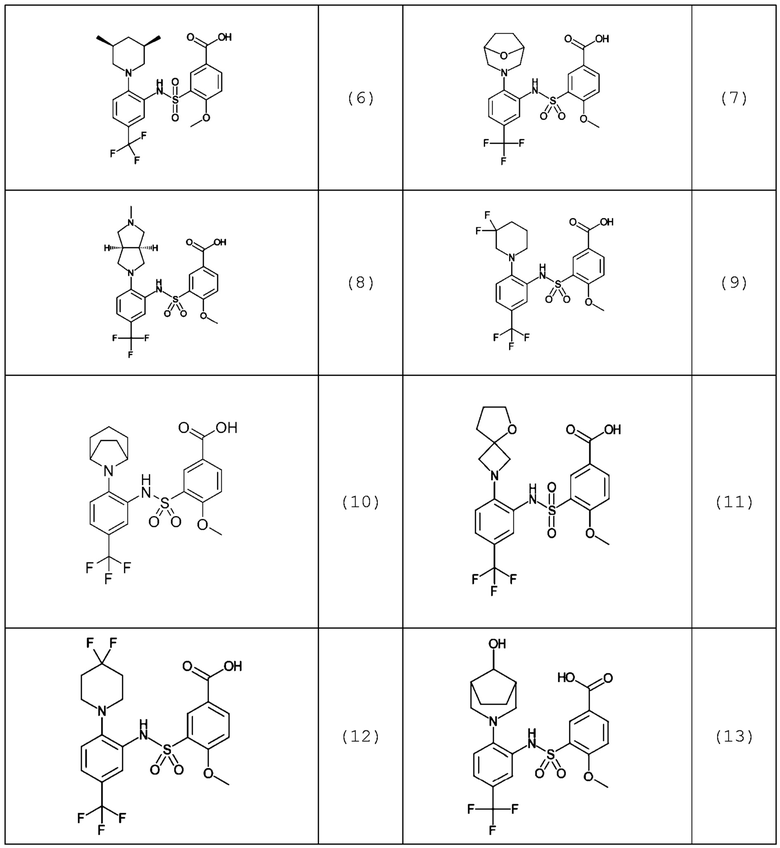
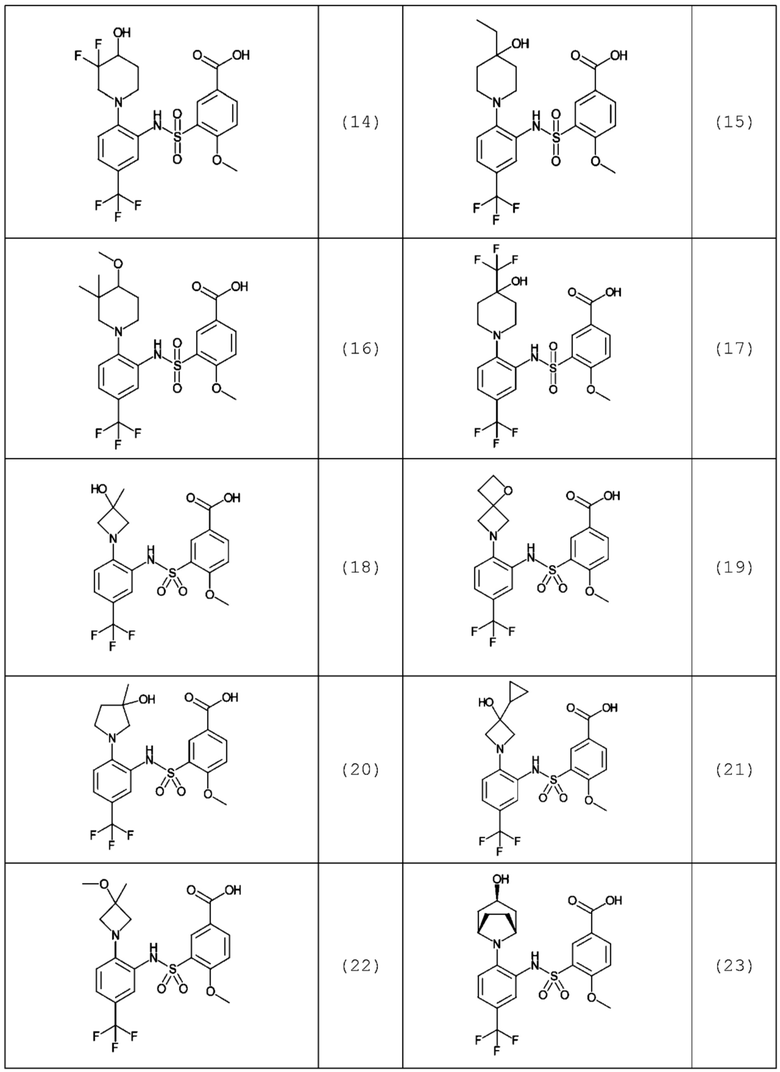
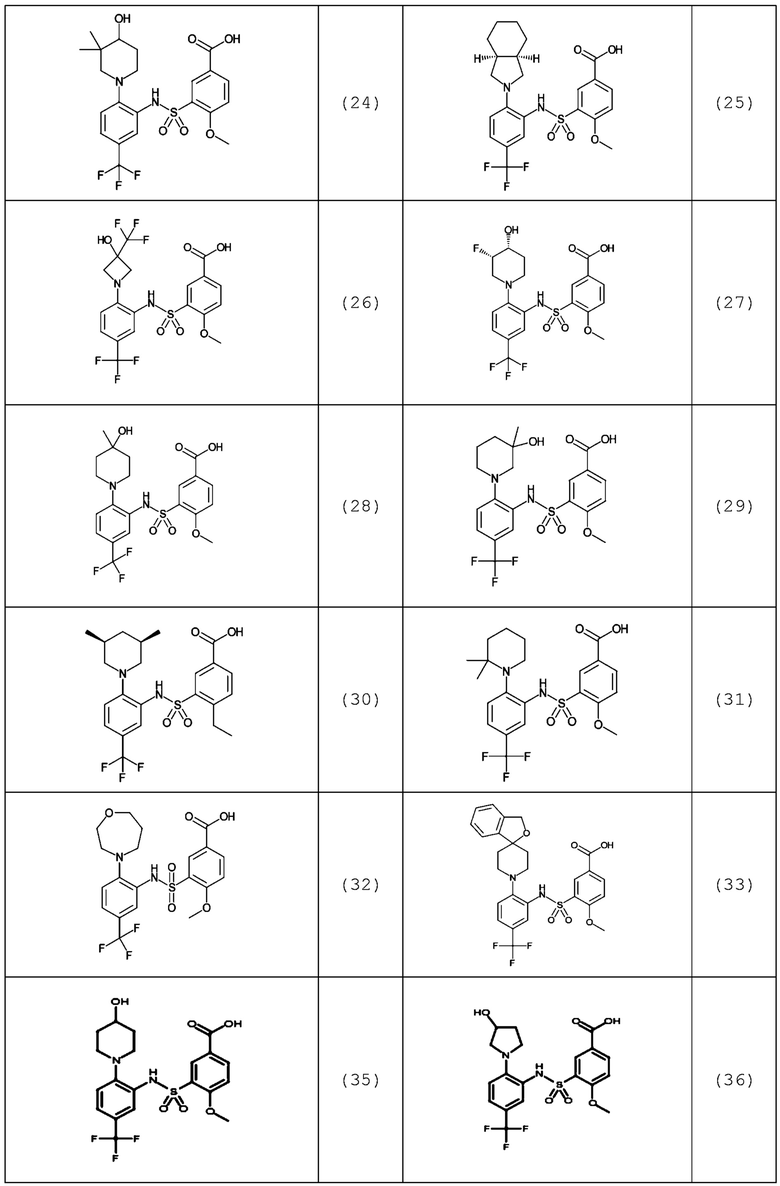
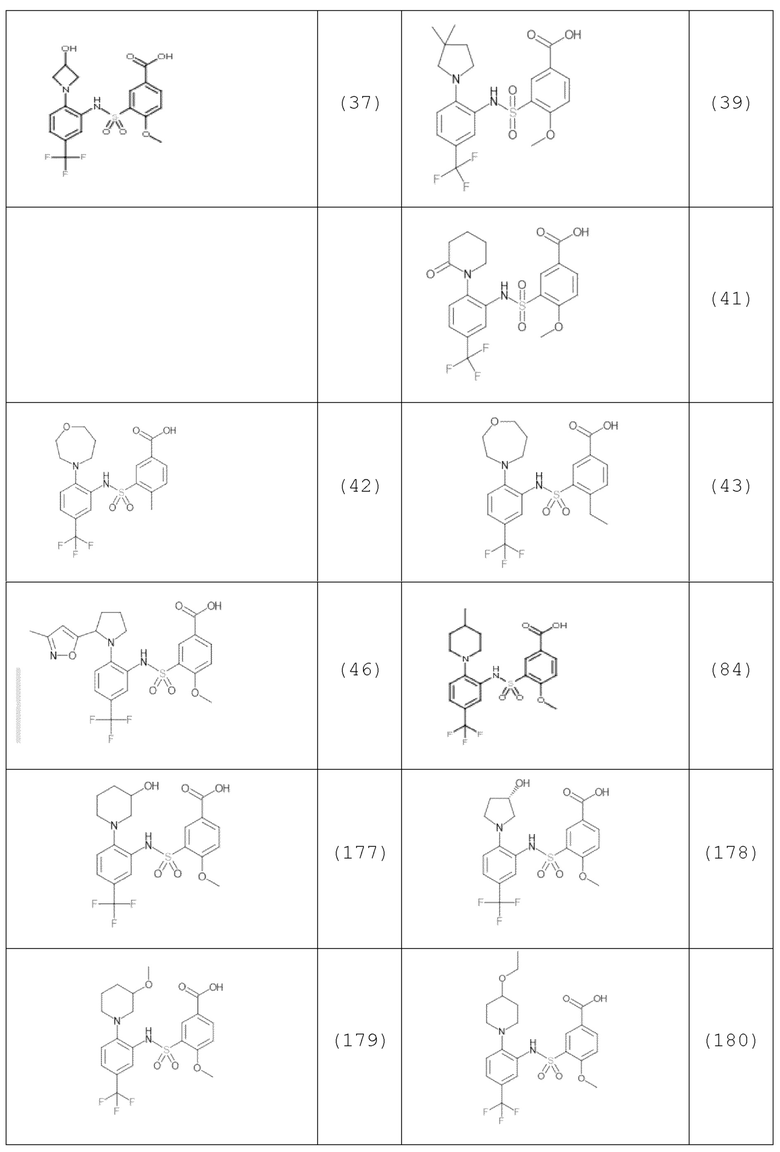
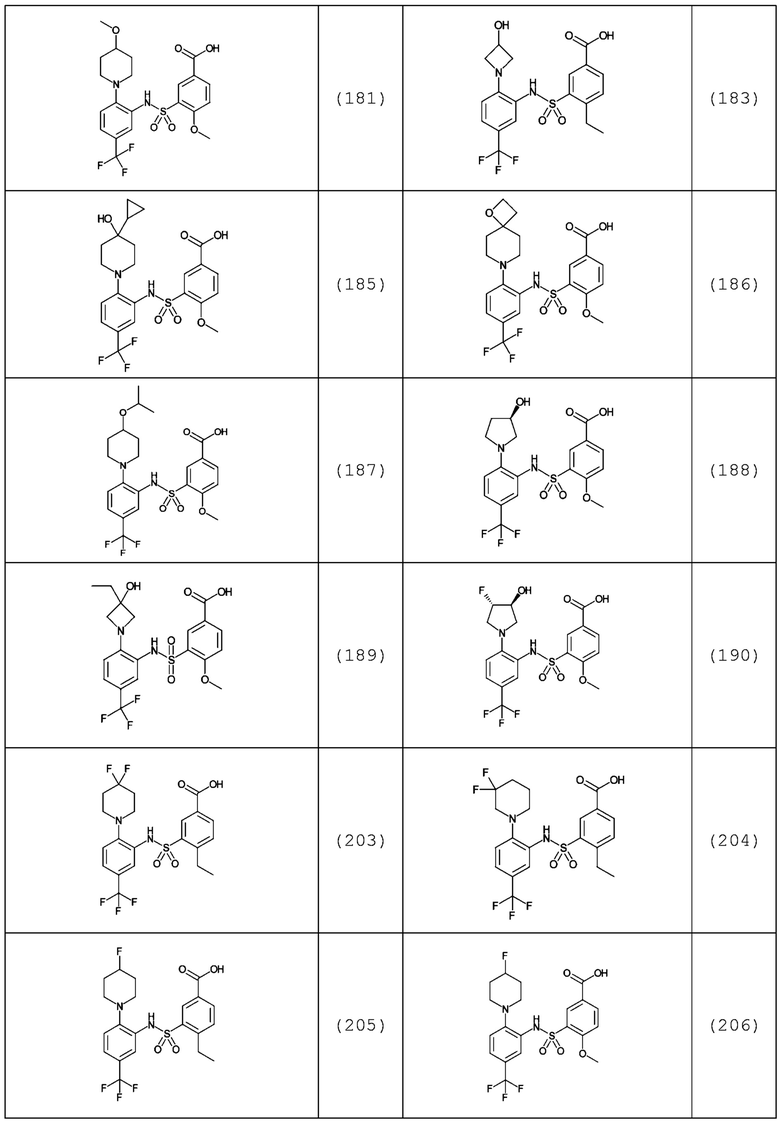
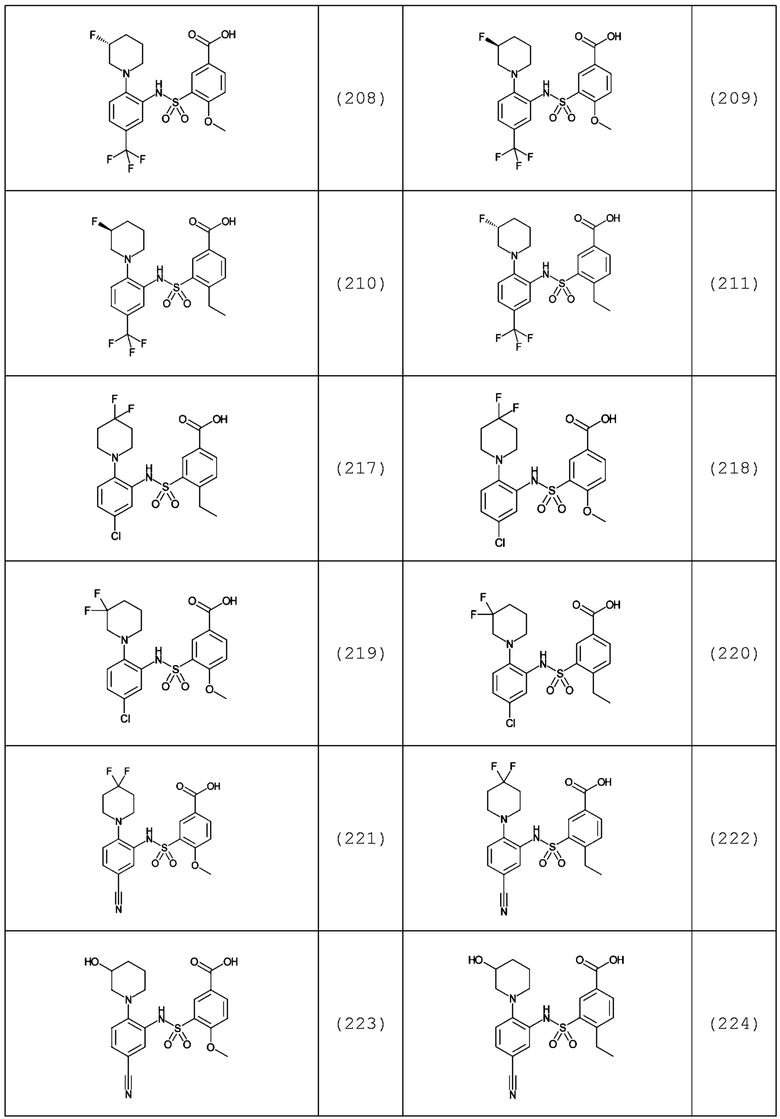
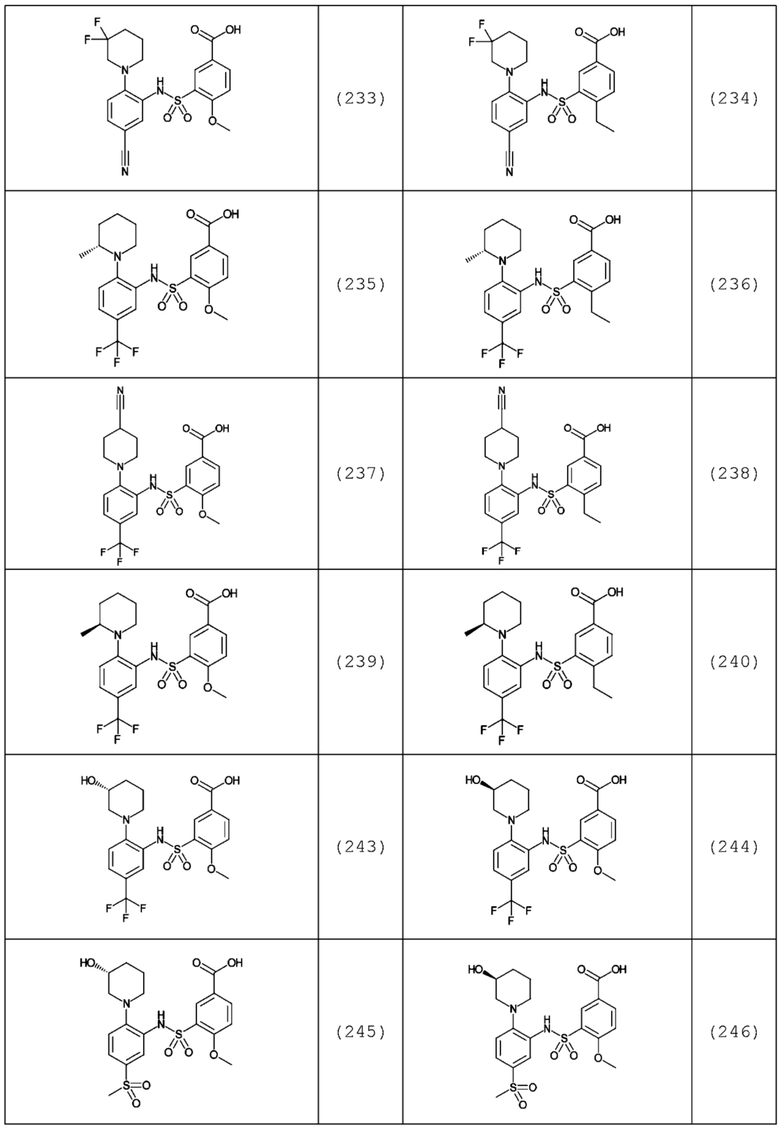
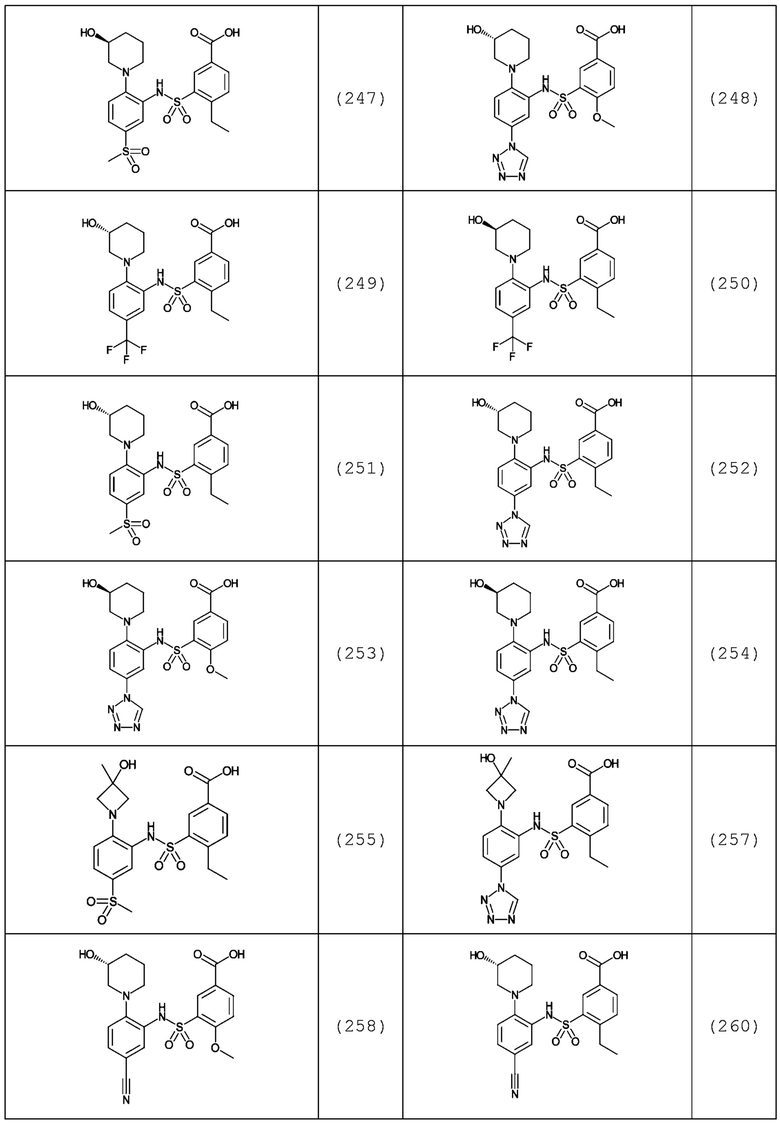
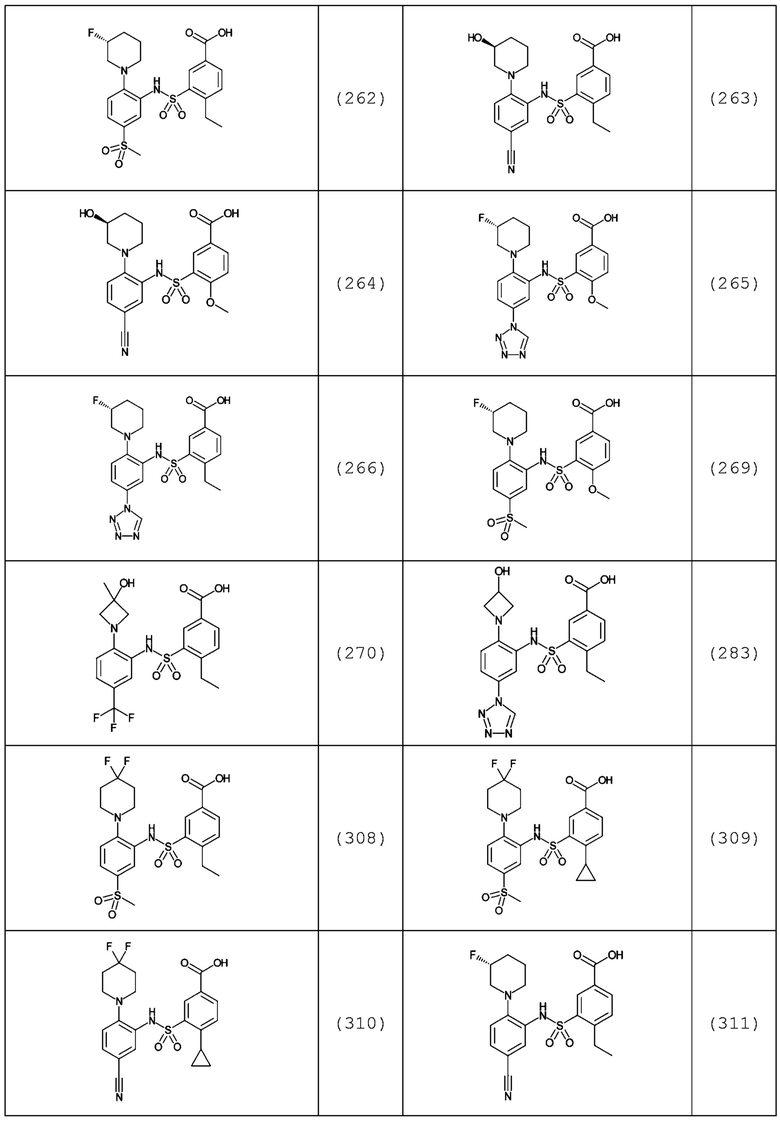
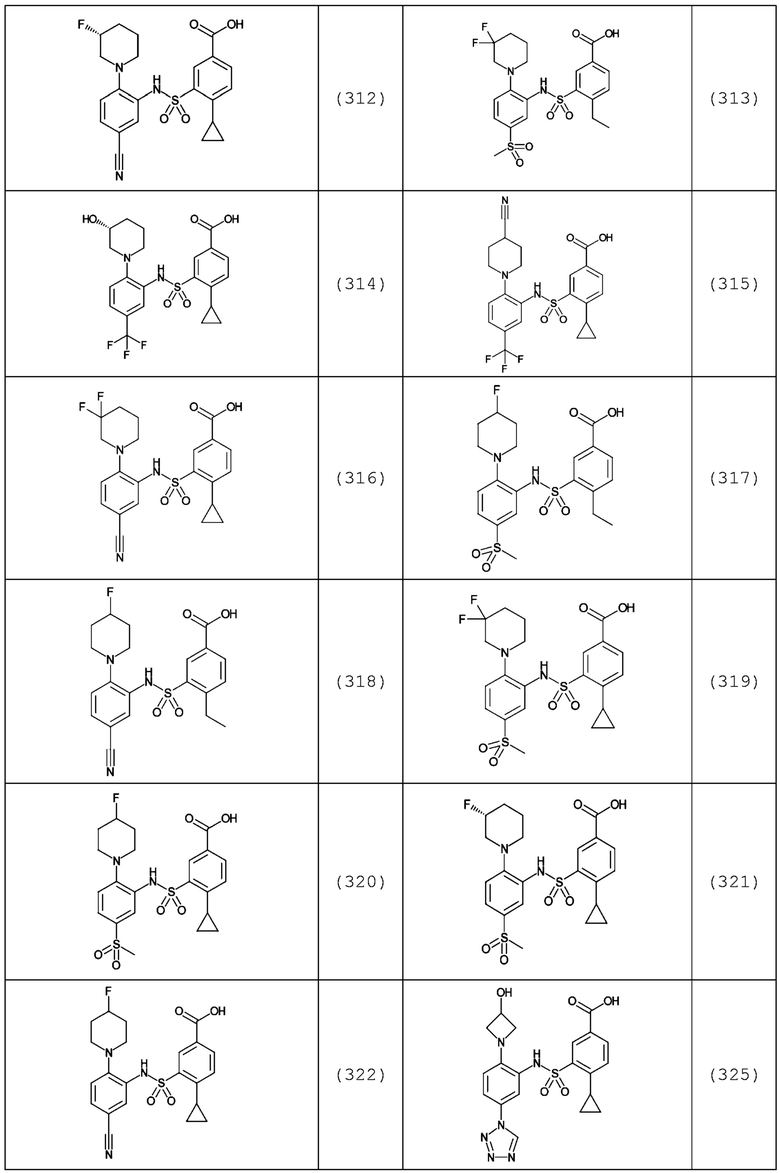
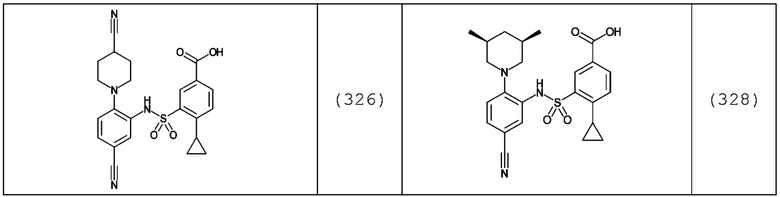
и их фармацевтически приемлемых солей.
19 Соединение формулы (Id) или его фармацевтически приемлемая соль,
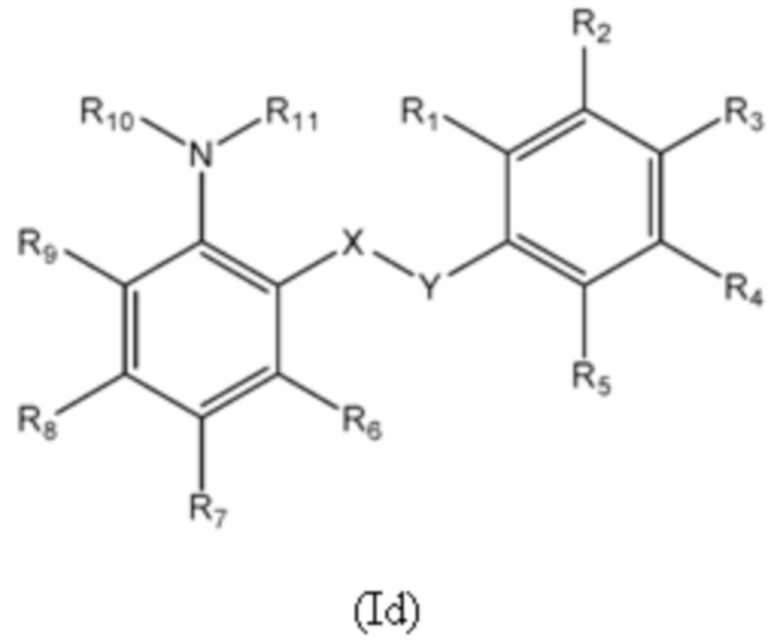 ,
,
где группа X-Y представляет собой -NHSO2-;
R1 представляет собой H;
R2 представляет собой COOH;
R3 представляет собой H;
R4 представляет собой H;
R5 выбран из C1-C6-алкила, C≡CH, C1-C6-алкокси, C3-C7-циклоалкила;
R6 представляет собой H;
R7 представляет собой CN, SO2-C1-C6-алкил, SO2NR13R14 или 5-6-членную моноциклическую гетероарильную группу, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из C1-C3-алкила;
R8 выбран из H и галогена;
R9 представляет собой H или C1-C3-алкил;
R10 и R11 вместе с азотом, к которому они присоединены, образуют 4 или 6-членную моноциклическую гетероциклоалкильную группу, которая необязательно замещена одной или более группами, выбранными из C1-C6-алкила, CN, OH и галогена; и
каждый R13 и R14 независимо представляет собой H или C1-C6-алкил.
20. Соединение по п. 19, где R7 представляет собой гетероарильную группу, выбранную из имидазолила, пиразолила, пиразинила, пиридазинила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, тетразолила и триазолила, каждый из которых необязательно замещен одним или более заместителями, выбранными из C1-C3-алкила.
21. Соединение по п. 19 или 20, где R7 представляет собой гетероарильную группу, выбранную из 1H-пиразола-5-ила, 1H-пиразол-3-ила, 1H-пиразол-4-ила, оксазол-2-ила, 1H-1,2,3-триазол-4-ила, 1H-1,2,3-триазол-5-ила, тиазол-5-ила, 1H-1,2,3,4-тетразол-4-ила, 2H-1,2,3,4-тетразол-5-ила, изоксазол-4-ила, изоксазол-5-ила, изотиазол-5-ила, пиридазин-3-ила, пиридазин-4-ила, пиразинила и 1,3,4-оксадиазол-2-ила, каждый из которых необязательно замещен одним или более заместителями, выбранными из Me.
22. Соединение по любому одному из пп. 19-21, где соединение формулы (Id) выбрано из следующего:
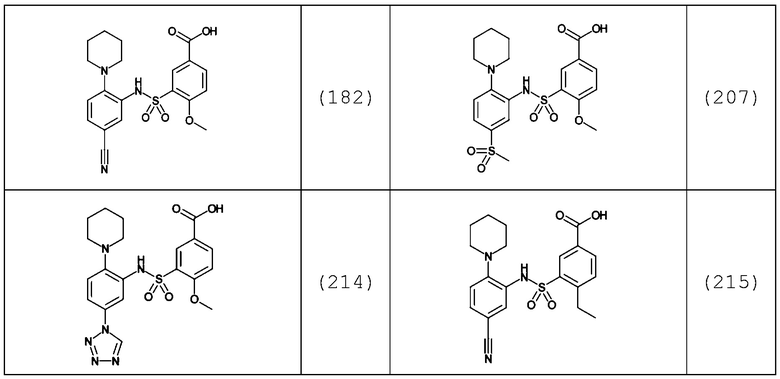
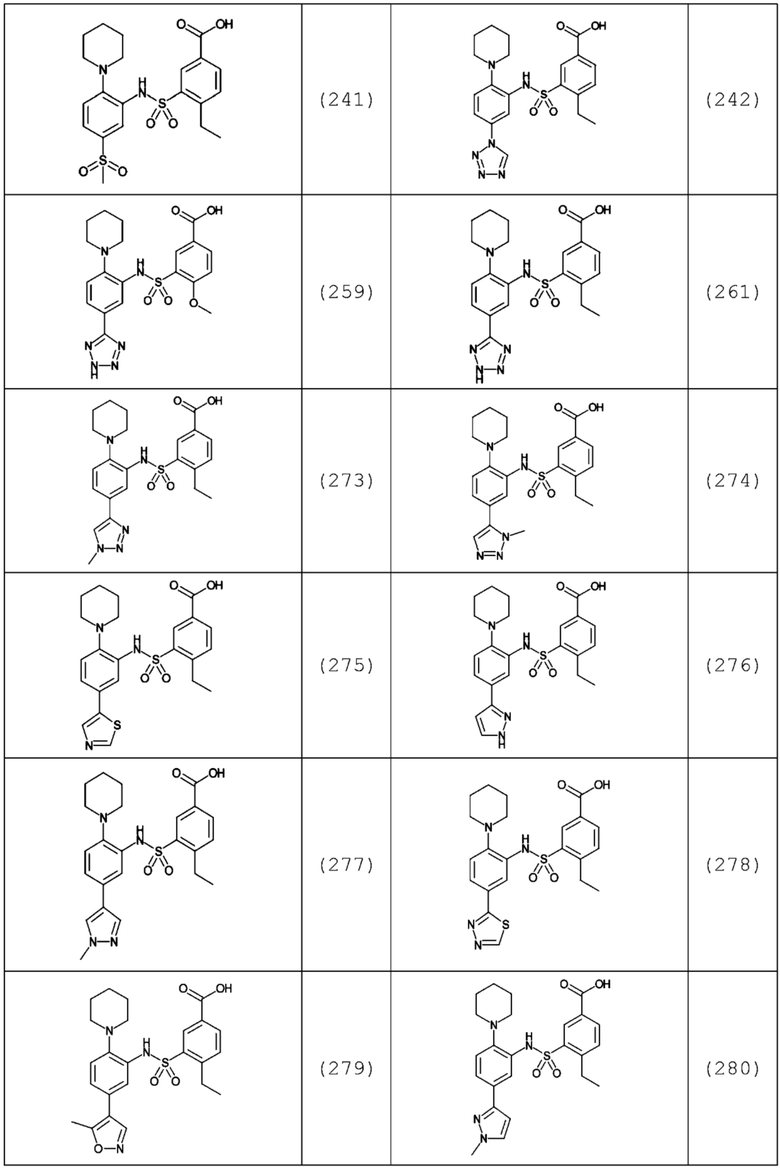
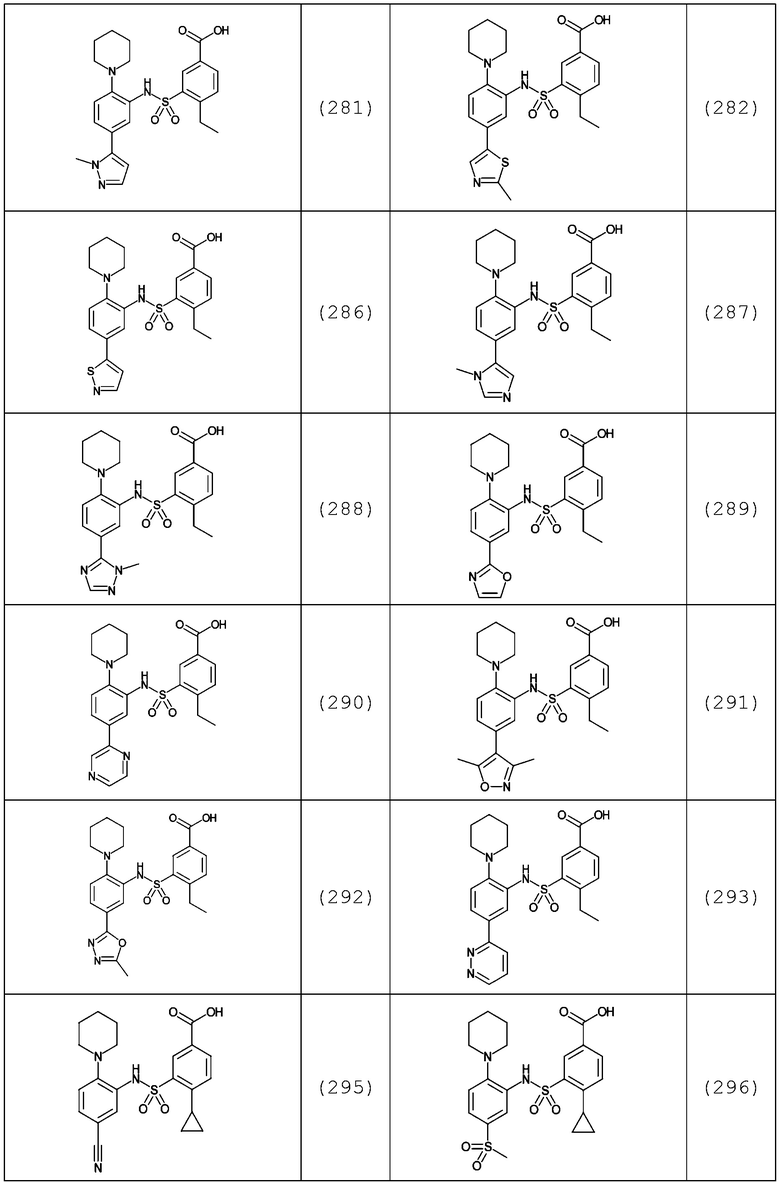
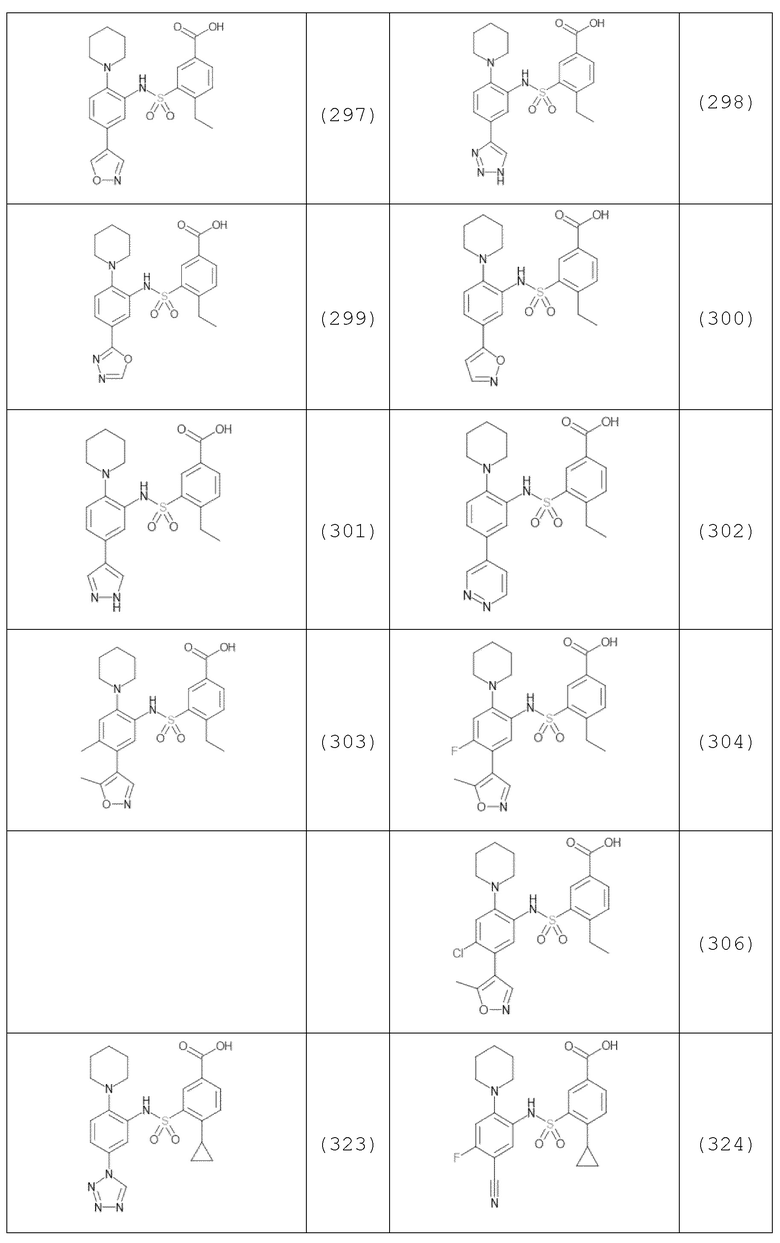
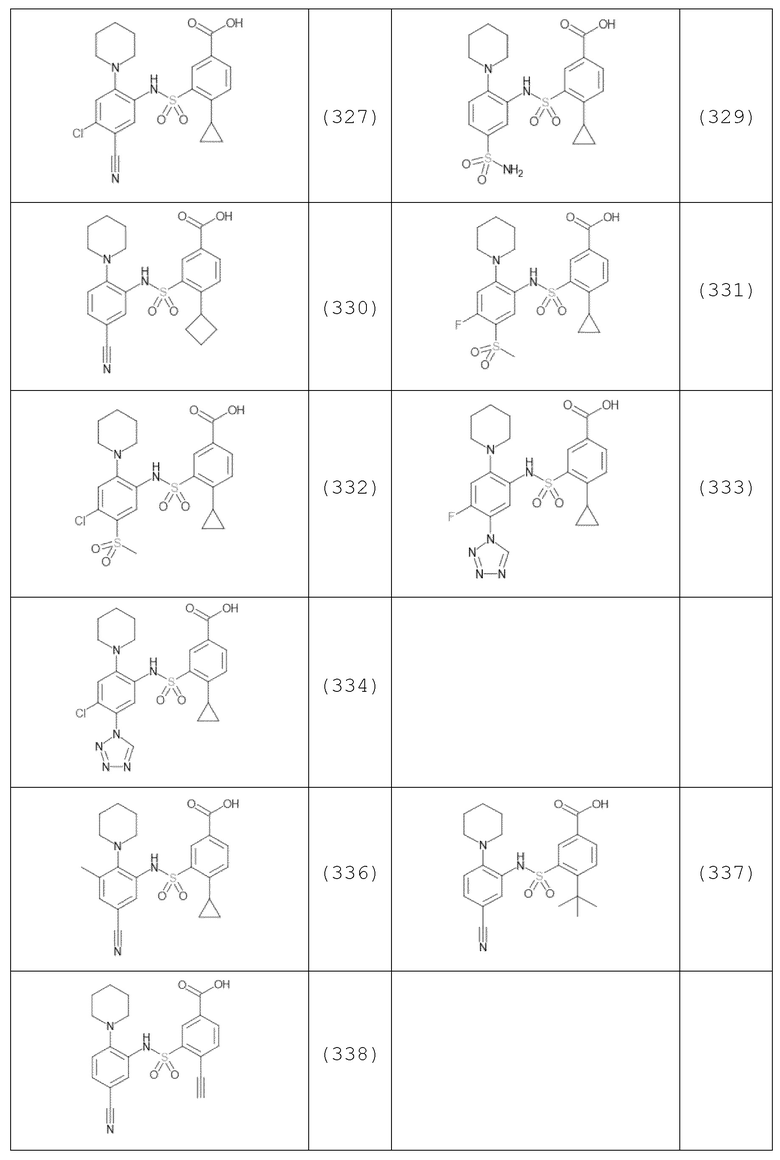
и их фармацевтически приемлемых солей.
23. Соединение формулы (Ib) или его фармацевтически приемлемая соль,
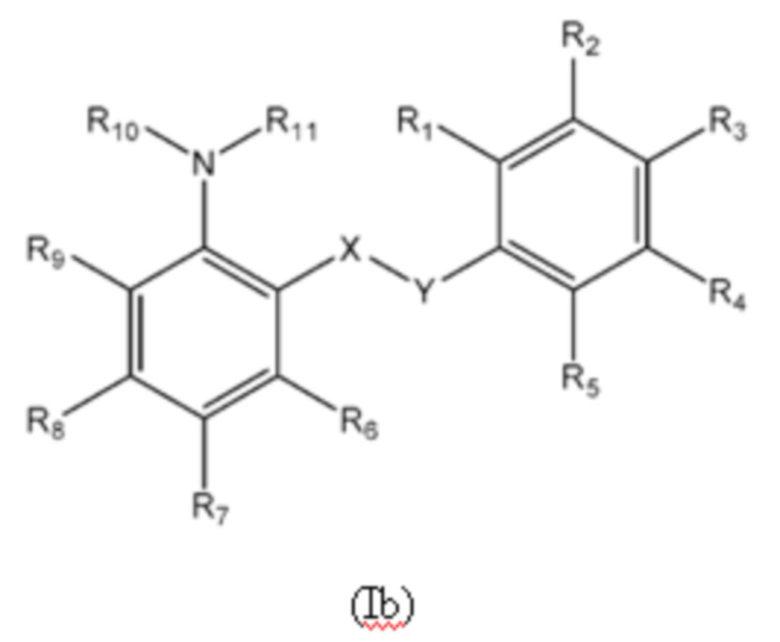 ,
,
где группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H;
R2 представляет собой тетразолильную группу;
R3 представляет собой H;
R4 представляет собой H;
R5 представляет собой C1-C6-алкокси;
R6 представляет собой H;
R7 представляет собой C1-C6-галогеналкил;
R8 представляет собой H;
R9 представляет собой H;
R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу.
24. Соединение по п. 23, которое представляет собой:
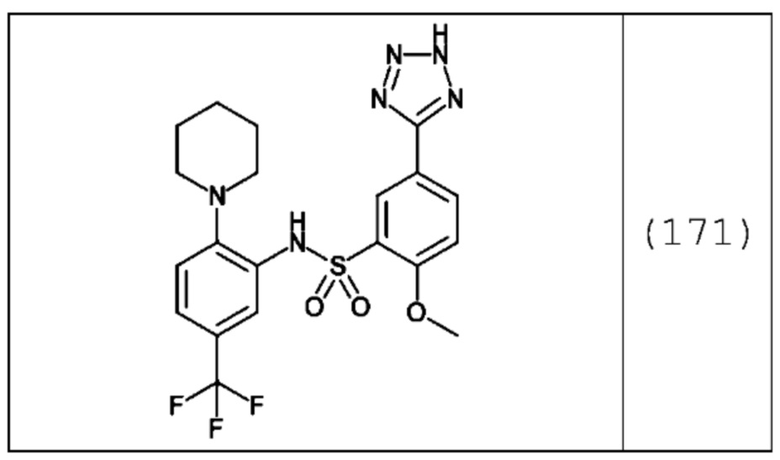
или его фармацевтически приемлемую соль.
25. Соединение формулы (Ic) или его фармацевтически приемлемая соль,
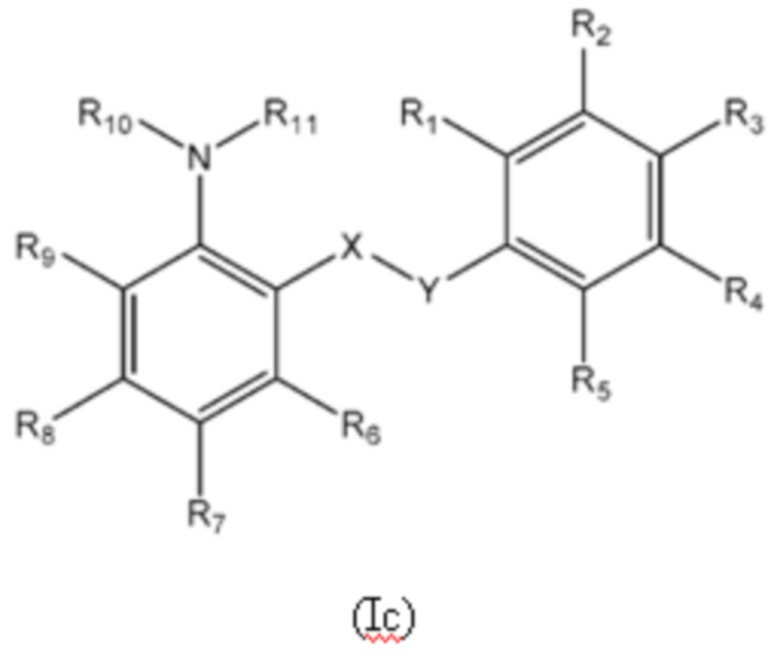 ,
,
где X представляет собой SO2;
Y представляет собой NH;
R1 представляет собой H;
R2 выбран из COOH и тетразолильной группы;
R3 представляет собой H;
R4 представляет собой H;
R5 представляет собой C1-C6-алкокси;
R6 представляет собой H;
R7 представляет собой C1-C6-галогеналкил;
R8 представляет собой H;
R9 представляет собой H;
R10 представляет собой C1-C6-алкил;
R11 представляет собой C1-C6-алкил; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-членную моноциклическую гетероциклоалкильную группу, где один углерод в моноциклической гетероциклоалкильной группе необязательно заменен группой O.
26. Соединение по п. 25, выбранное из следующего:
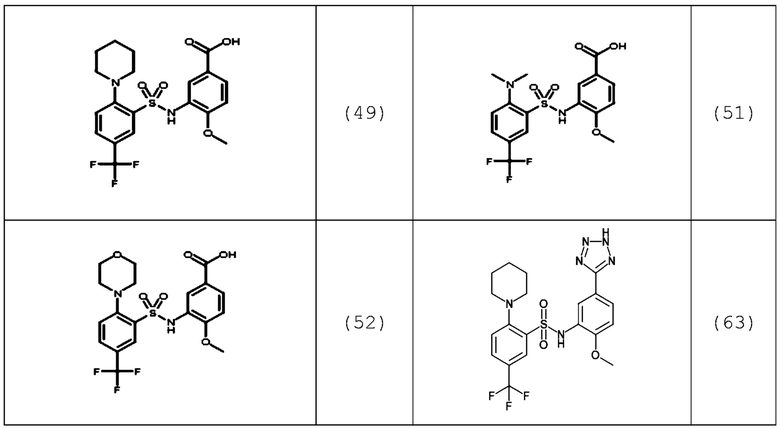
и их фармацевтически приемлемых солей.
27. Соединение, выбранное из следующего:
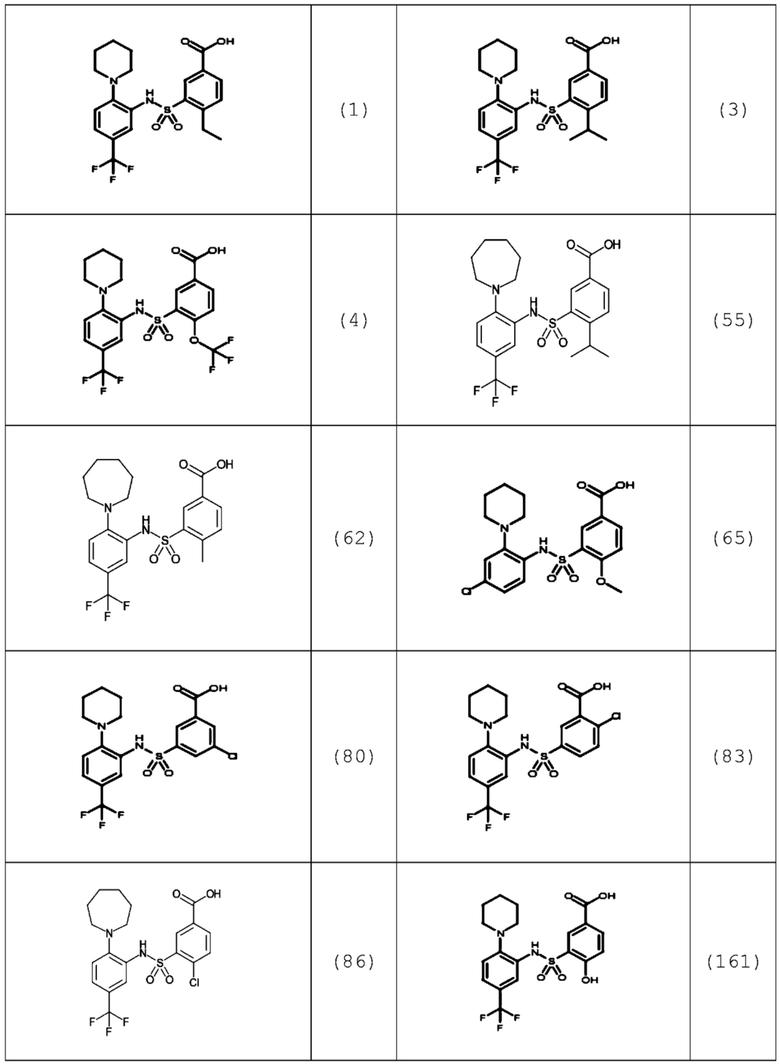
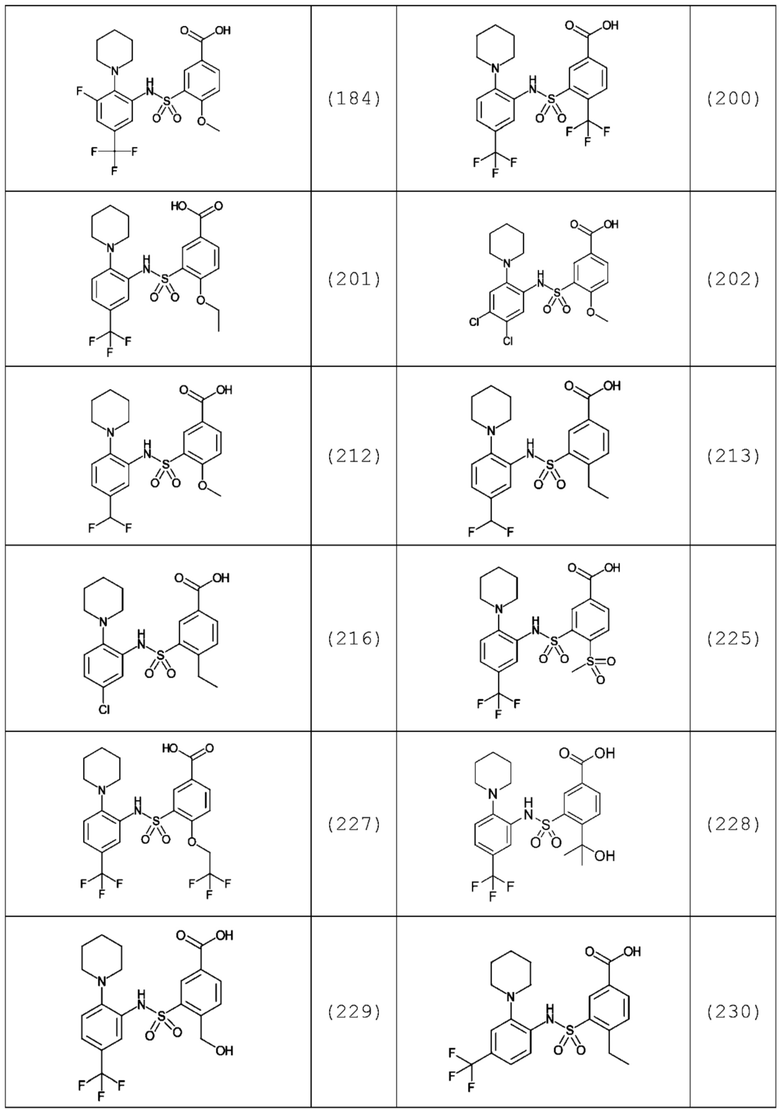
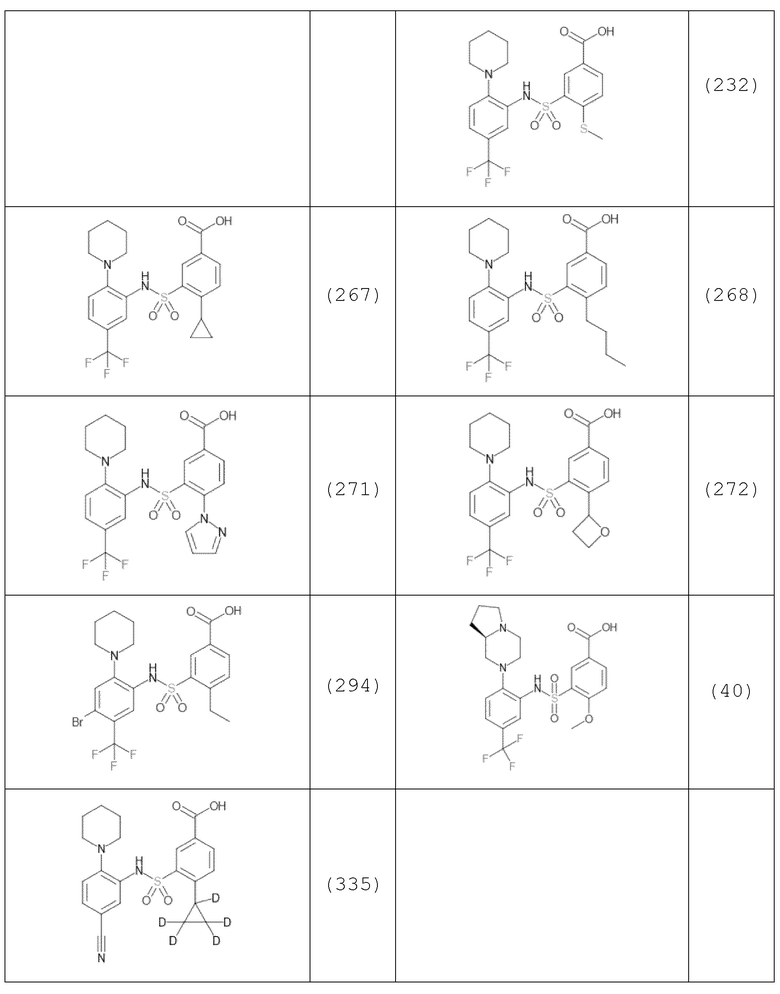
и их фармацевтически приемлемых солей.
28. Фармацевтическая композиция, обладающая модулирующей ERAP1 активностью, включающая терапевтически эффективное количество соединения по любому одному из пп. 1-27, смешанное с фармацевтически приемлемым разбавителем, вспомогательным веществом или носителем.
29. Соединение по любому одному из пп. 1-27 для применения в медицине для лечения заболевания, опосредованного активностью ERAP1.
30. Соединение по любому одному из пп. 1-27 для применения в лечении или предупреждении нарушения, опосредованного активностью ERAP1, выбранного из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
31. Соединение для применения по п. 30, где соединение модулирует ERAP1.
32. Соединение для применения по п. 30 или 31, где нарушение является пролиферативным нарушением и предпочтительно является раком или лейкозом.
33. Соединение для применения по п. 32, где соединение убивает раковые клетки, снижает количество пролиферирующих клеток в злокачественной опухоли, уменьшает объем или размер опухоли, включающей раковые клетки, и/или снижает количество метастазирующих раковых клеток.
34. Соединение для применения по п. 32 или 33, где соединение применяется для предупреждения рака, где соединение предпочтительно индуцирует неоантиген, на который субъект имеет существующий иммунный ответ.
35. Соединение для применения по любому одному из пп. 32-34, где субъект ранее имел рак, имел случай рака в семье, имеет высокий риск развития рака, имеет генетическую предрасположенность к развитию рака, подвергся воздействию канцерогенного агента и/или находится в ремиссии после рака.
36. Соединение для применения по любому одному из пп. 30-35, где указанное соединение применяют в комбинации с иммунотерапией, где субъект предпочтительно имеет рак, и соединение повышает чувствительность раковых клеток к иммунотерапии.
37. Соединение для применения по п. 36, где указанная иммунотерапия является вмешательством в иммунную контрольную точку, предпочтительно антителом-ингибитором контрольной точки.
38. Соединение для применения по п. 37, где указанное антитело-ингибитор контрольной точки является антителом против PD-1, антителом против PD-L1 или антителом против CTLA4.
39. Соединение для применения по п. 30 или 31, где нарушение является иммунологическим нарушением и предпочтительно выбрано из анкилозирующего спондилита, болезни Бехчета, псориаза и birdshot хориоретинопатии.
40. Соединение для применения по п. 30 или 31, где нарушение является воспалительным заболеванием, более предпочтительно аутовоспалительным заболеванием.
41. Соединение для применения по п. 30 или 31, где вирусное нарушение является инфекционным вирусным заболеванием, выбранным из ВИЧ, ВПЧ, ЦМВ и ВГС.
42. Соединение для применения по любому одному из пп. 30-35 и 36-38, где нарушением является рак и где соединение повышает заметность раковых клеток для иммунной системы путем изменения репертуара антигенов и неоантигенов, презентируемых иммунной системе.
43. Соединение для применения по п. 42, где соединение усиливает ответ CD8+T-клеток против раковой клетки.
44. Применение соединения формулы (I) или его фармацевтически приемлемой соли,
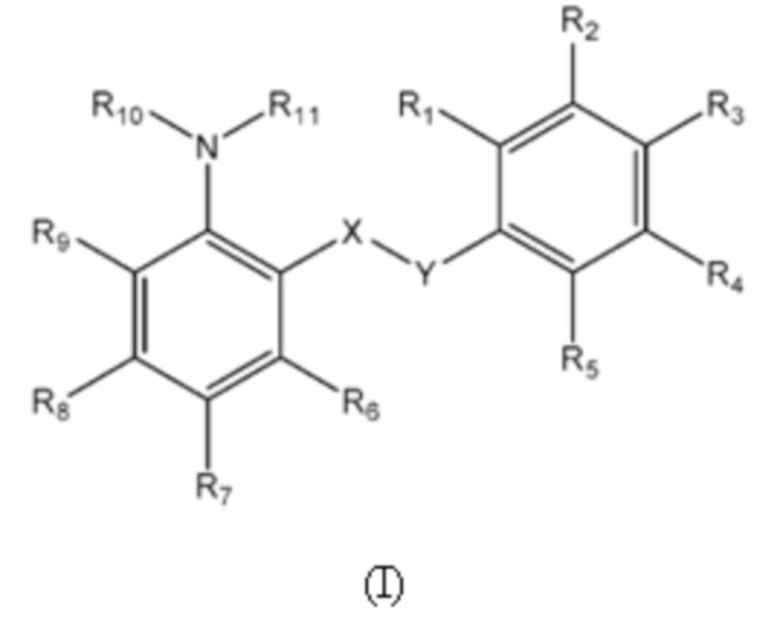 ,
,
где группа X-Y представляет собой -NHSO2- или -SO2NH-;
R1 представляет собой H;
R2 выбран из COOH и тетразолильной группы;
R3 выбран из H, Cl и C1-C6-алкила;
R4 выбран из H, Cl и F;
R5 выбран из H, C1-C6-алкила, C≡CH, C1-C6-галогеналкила, SO2-C1-C6-алкила, Cl, C1-C6-алкокси, OH, C1-C6-гидроксиалкила, C1-C6-алкилтио, гетероарила, C3-C7-циклоалкила, C3-C7-гетероциклоалкила, содержащего один гетероатом, выбранный из кислорода, и C1-C6-галогеналкокси;
R6 представляет собой H;
R7 выбран из H, CN, C1-C6-галогеналкила, Cl, F, SO2-C1-C6-алкила, SO2NR13R14, 5-6-членного моноциклического гетероарила и C1-C6-алкила, где указанная гетероарильная группа необязательно замещена одним или более заместителями, выбранными из C1-C6-алкила;
R8 выбран из H, C1-C6-алкила, C1-C6-галогеналкила и галогена;
R9 представляет собой H, C1-C3-алкил или галоген;
R10 представляет собой C1-C6-алкил;
R11 представляет собой C1-C6-алкил; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 4, 5, 6 или 7-членную моноциклическую гетероциклоалкильную группу, где один углерод в моноциклической гетероциклоалкильной группе необязательно заменен группой, выбранной из O и CO, и указанная моноциклическая гетероциклоалкильная группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила, CN, C3-C7-циклоалкила, OH, C1-C6-алкокси, галогена, C1-C6-галогеналкила и оксазолила, где указанная оксазолильная группа в свою очередь необязательно дополнительно замещена одной группой, выбранной из C1-C6-алкила; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 8 или 9-членную бициклическую гетероциклоалкильную группу; или
R10 и R11 вместе с азотом, к которому они присоединены, образуют 6-12-членную бициклическую группу, содержащую спироциклический атом углерода, где один углерод в бициклической группе замещен группой, выбранной из O и NH, и указанная бициклическая группа необязательно замещена одной или более группами, выбранными из C1-C6-алкила; и
каждый R13 и R14 независимо представляет собой H;
при лечении или предупреждении у субъекта нарушения, опосредованного активностью ERAP1, где указанное расстройство выбрано из пролиферативного нарушения, иммунного нарушения, вирусного нарушения и воспалительного нарушения.
45. Применение соединения формулы (I) по п. 44, которое выбрано из следующего:
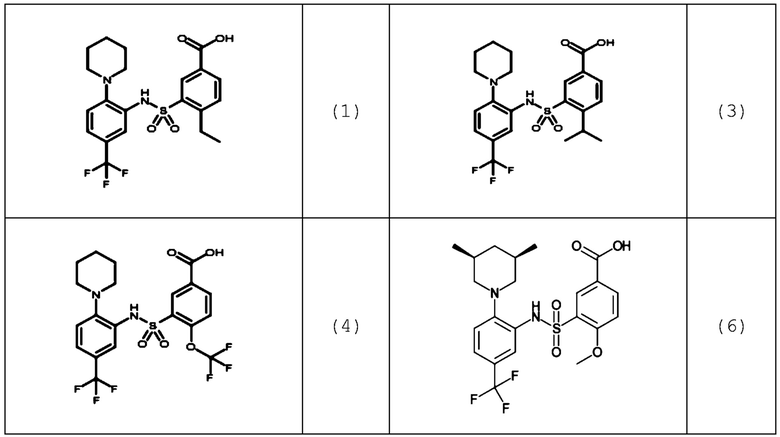
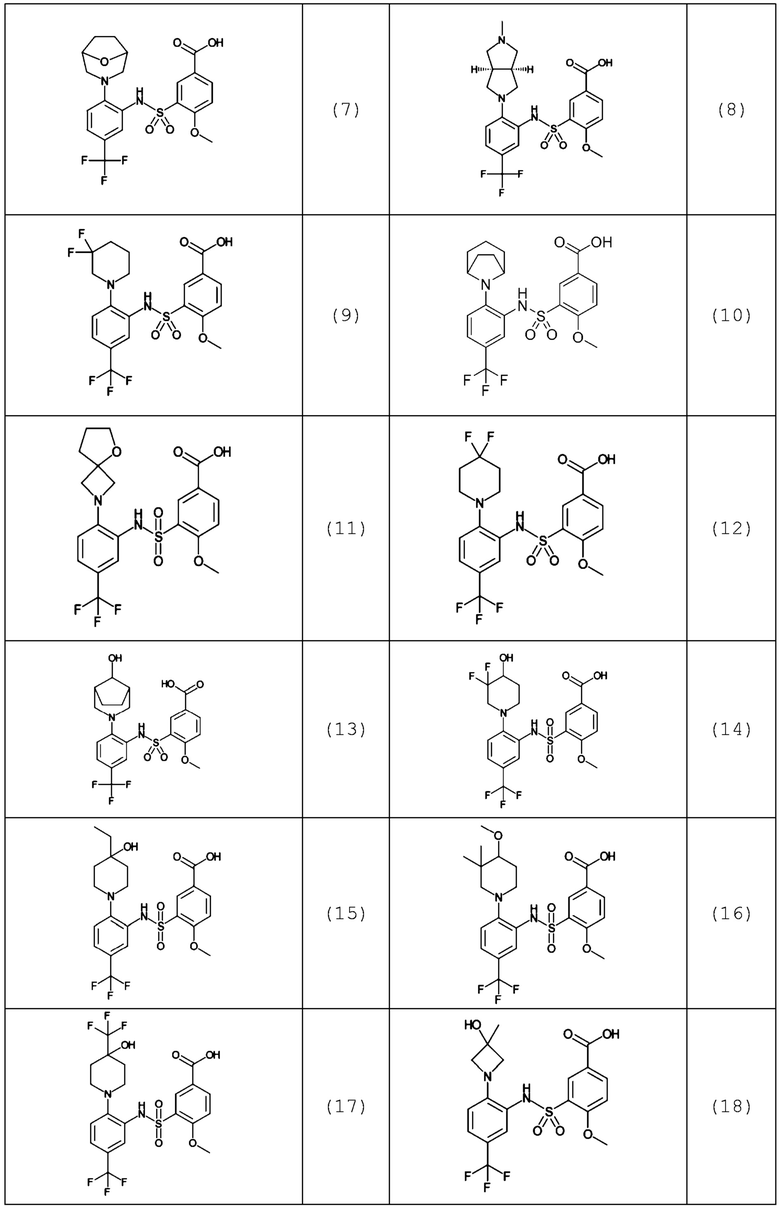
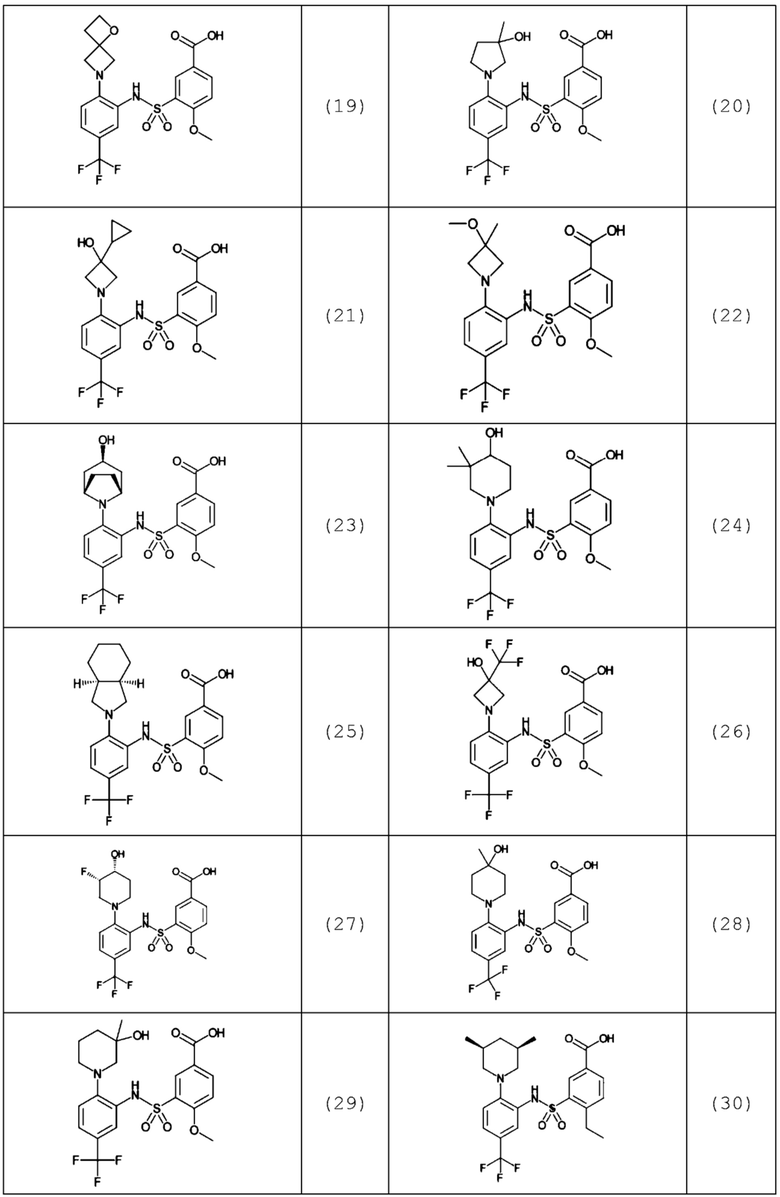
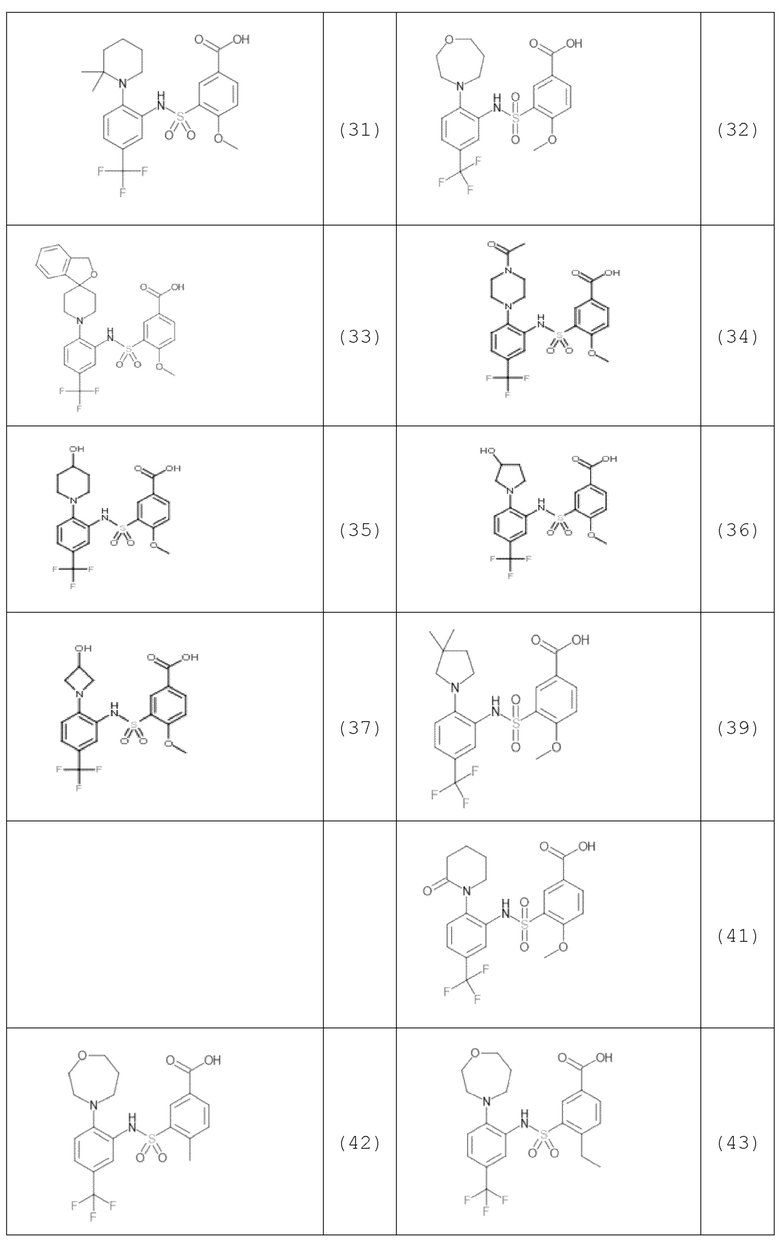
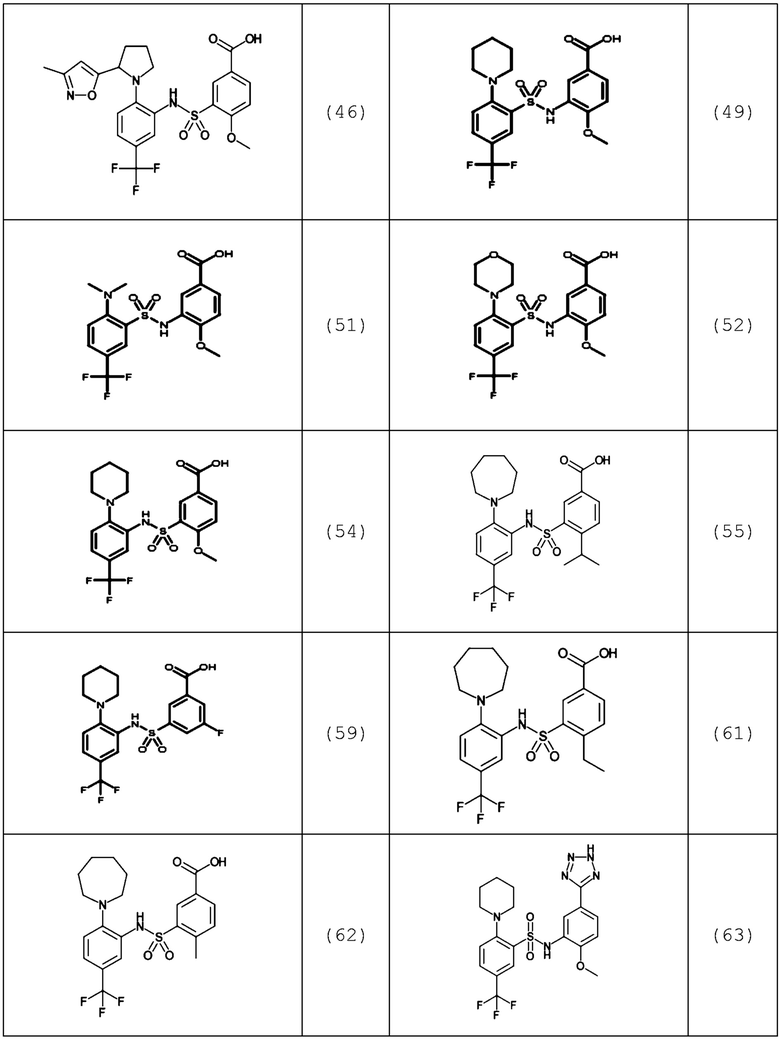
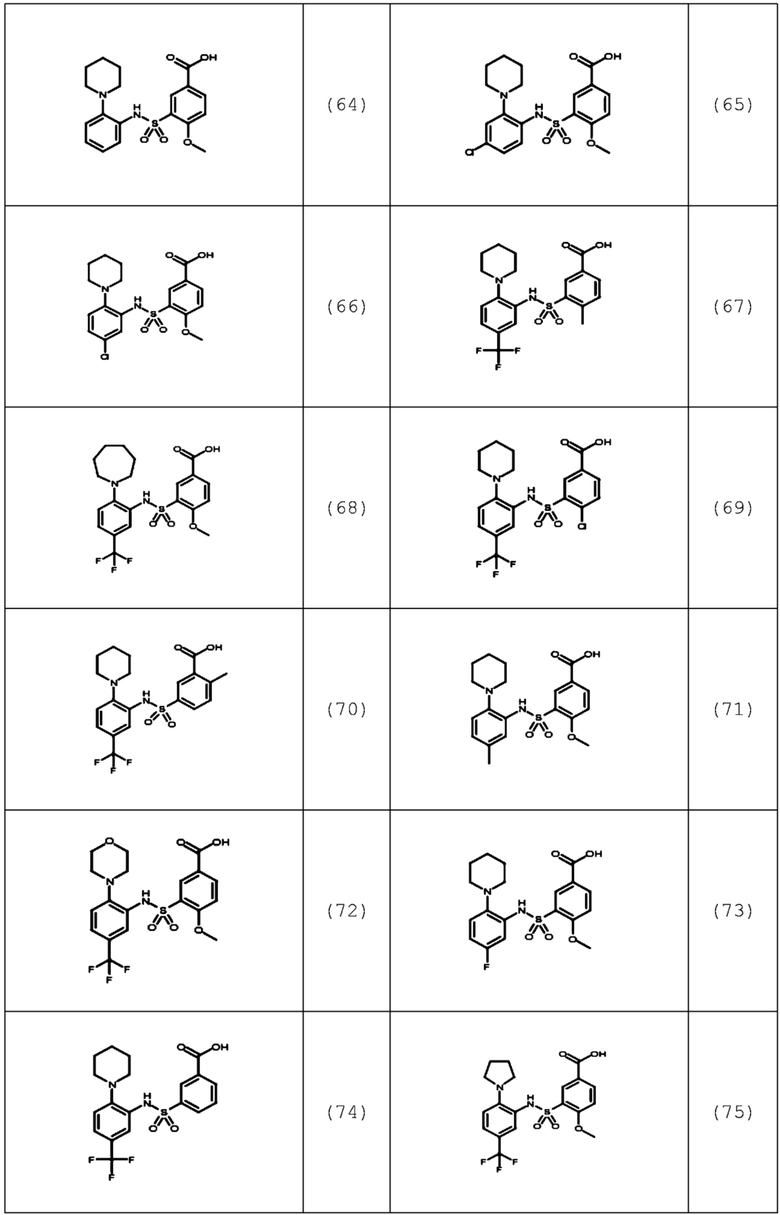
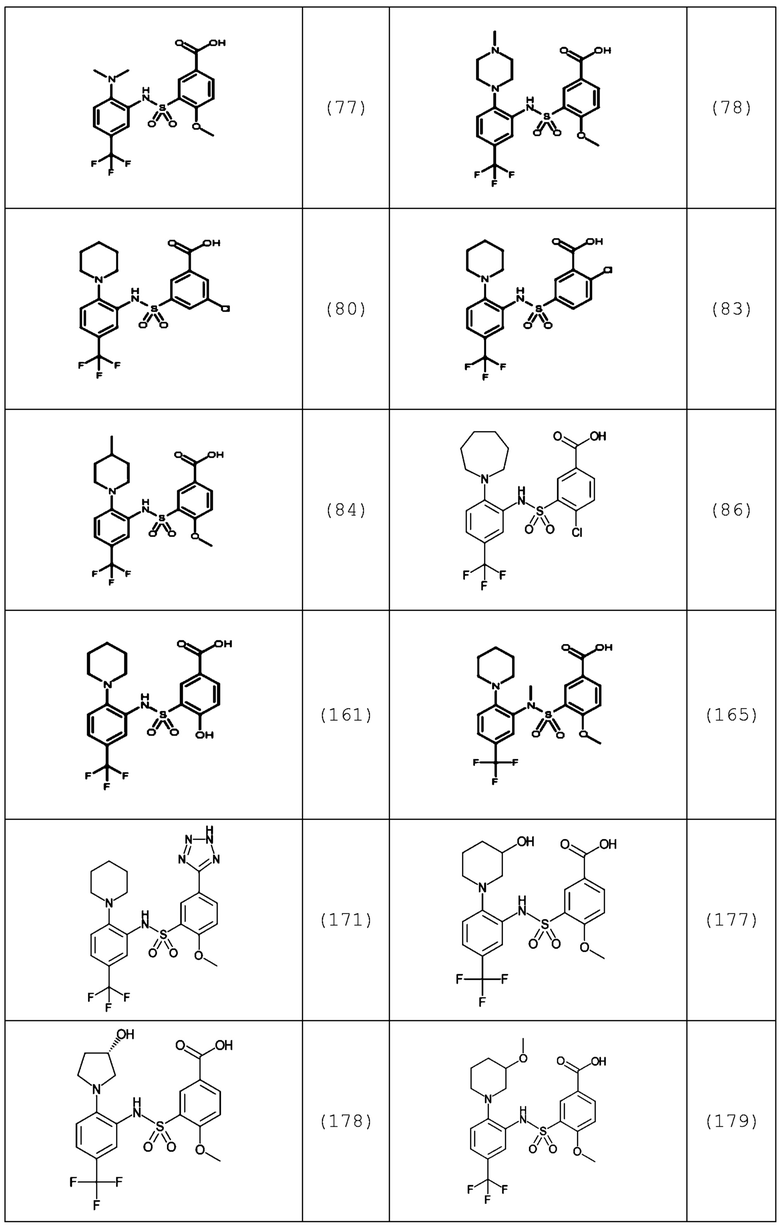
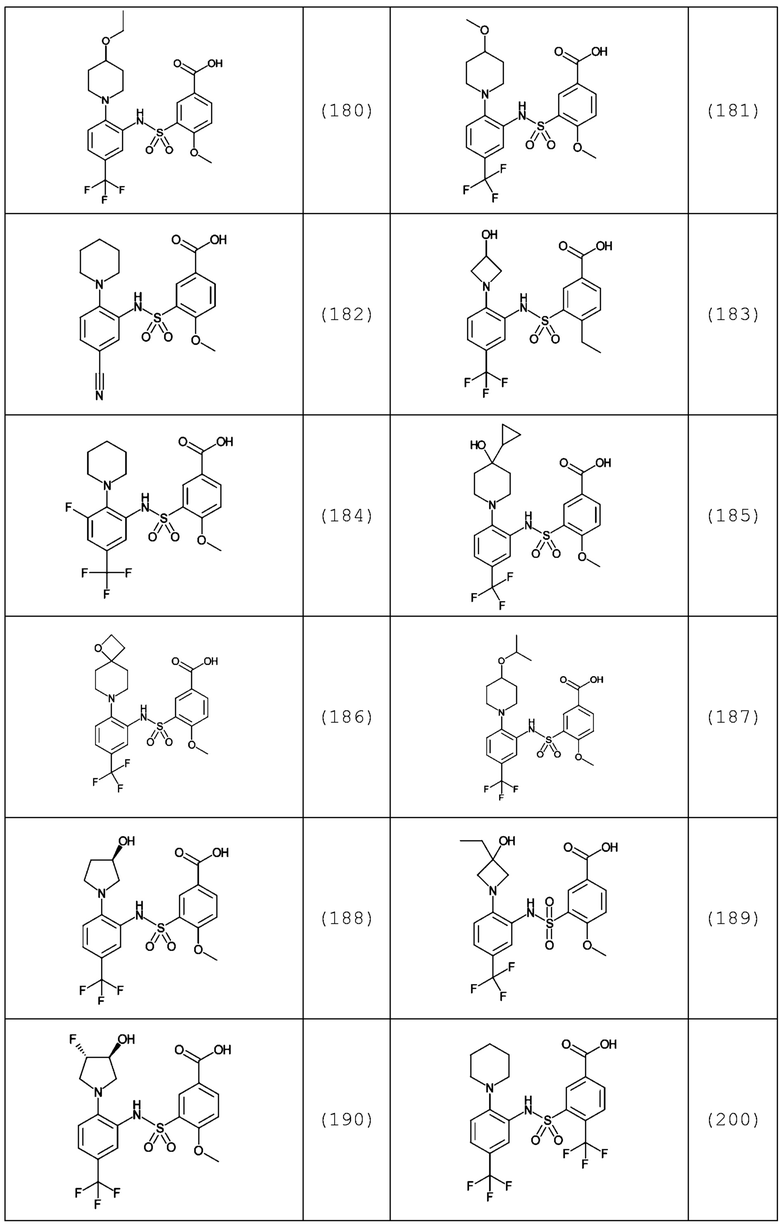
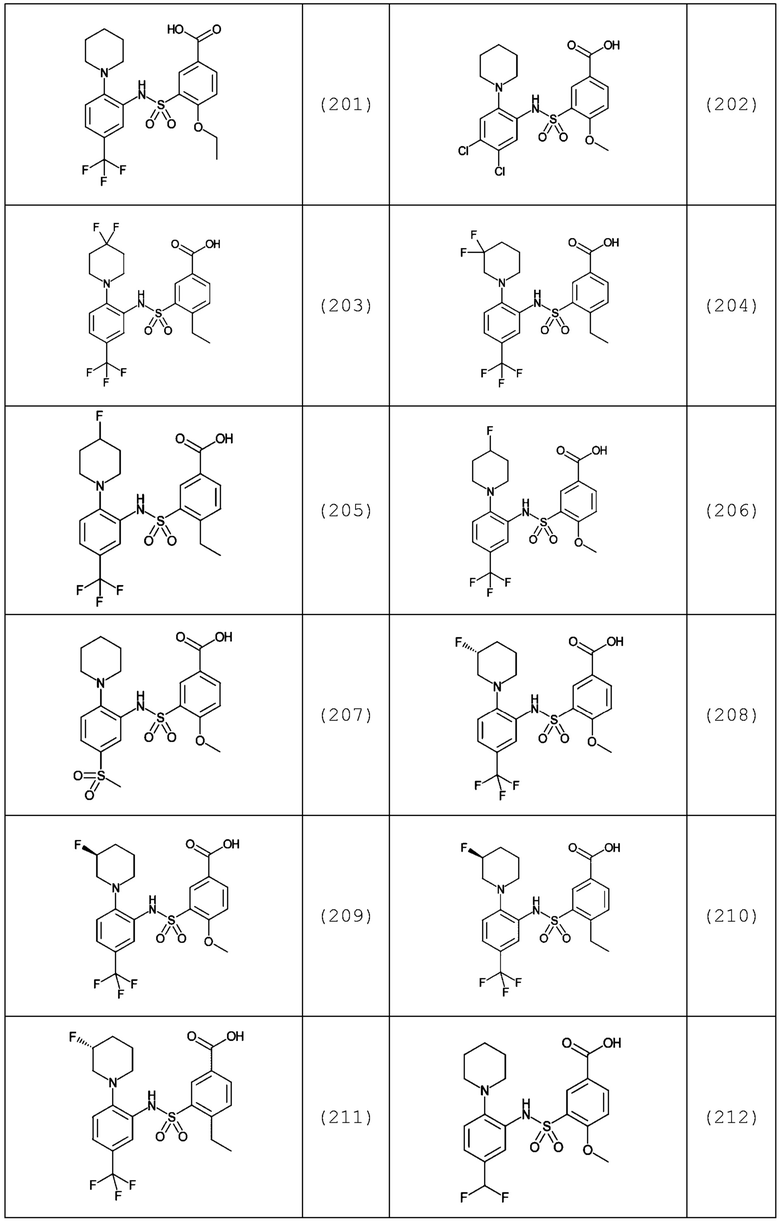
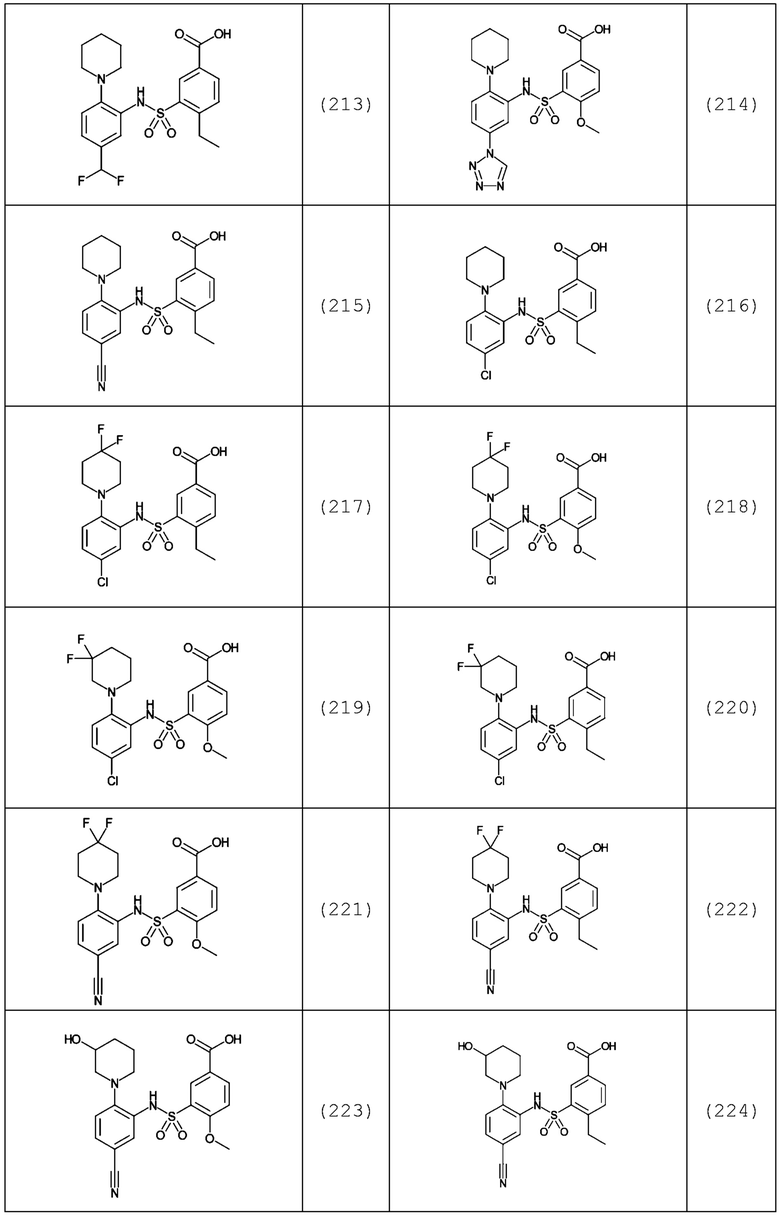
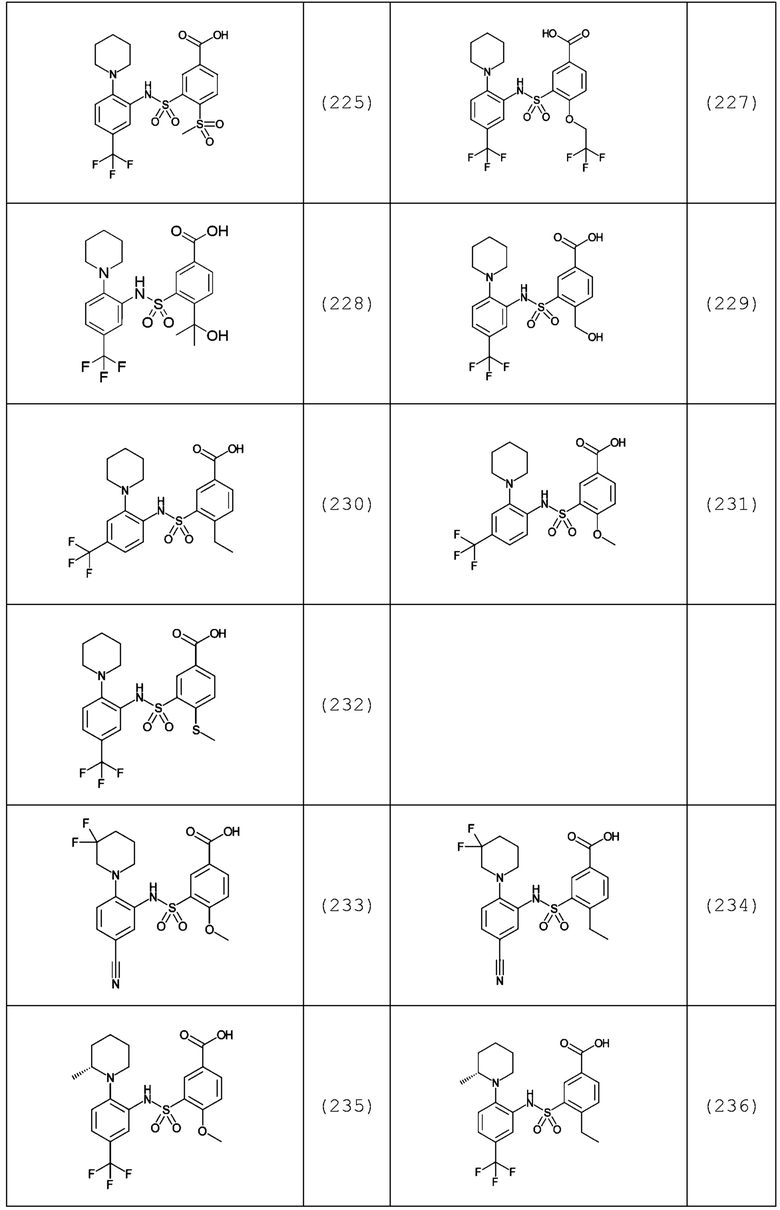
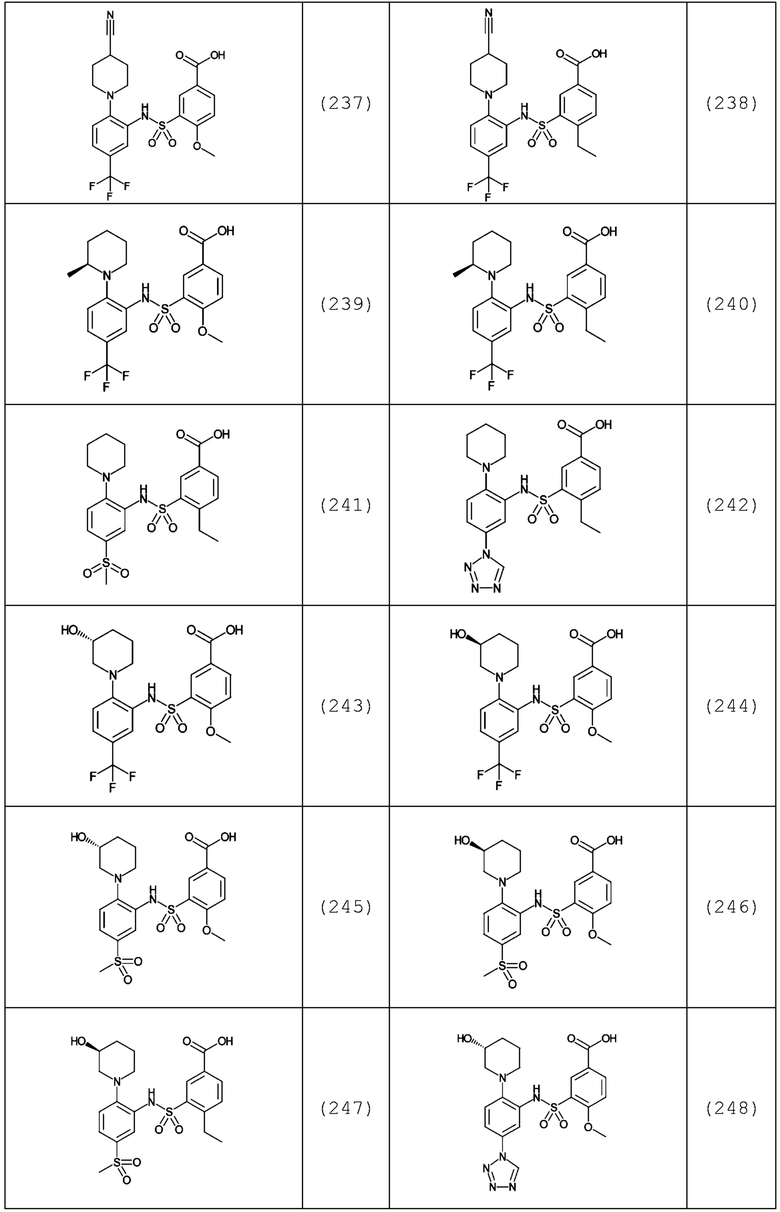
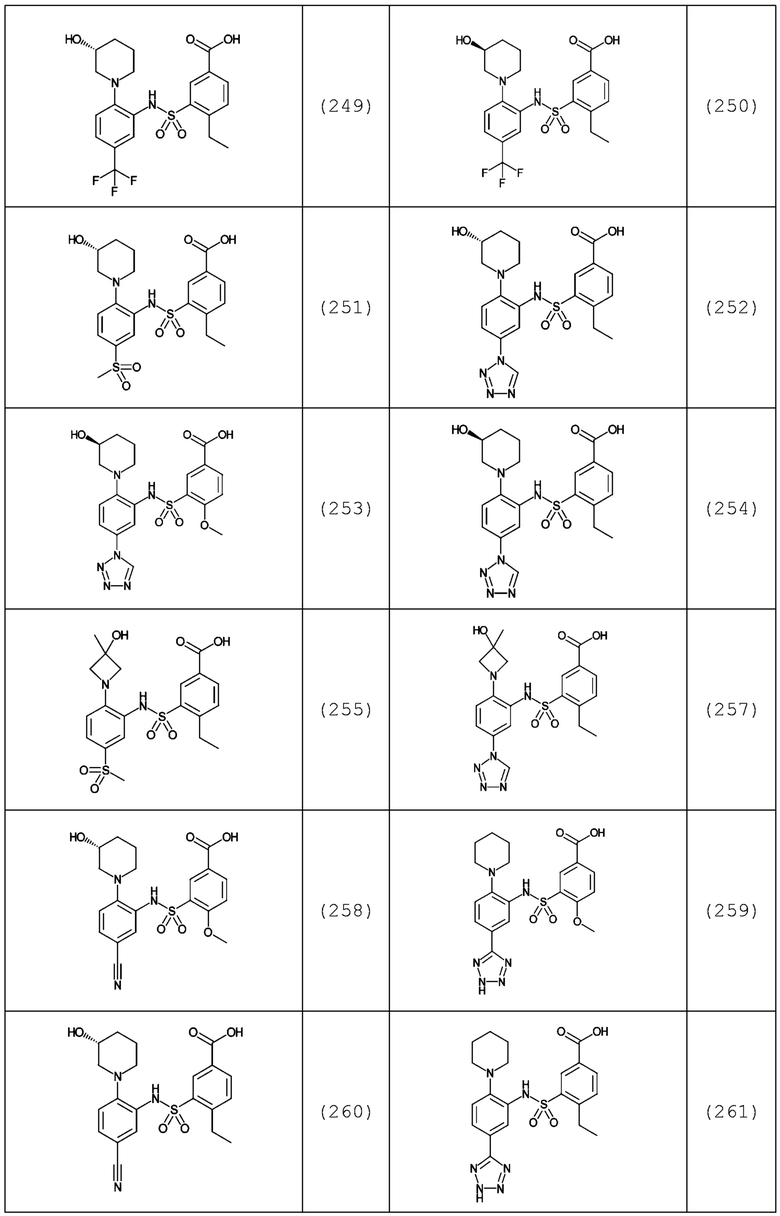
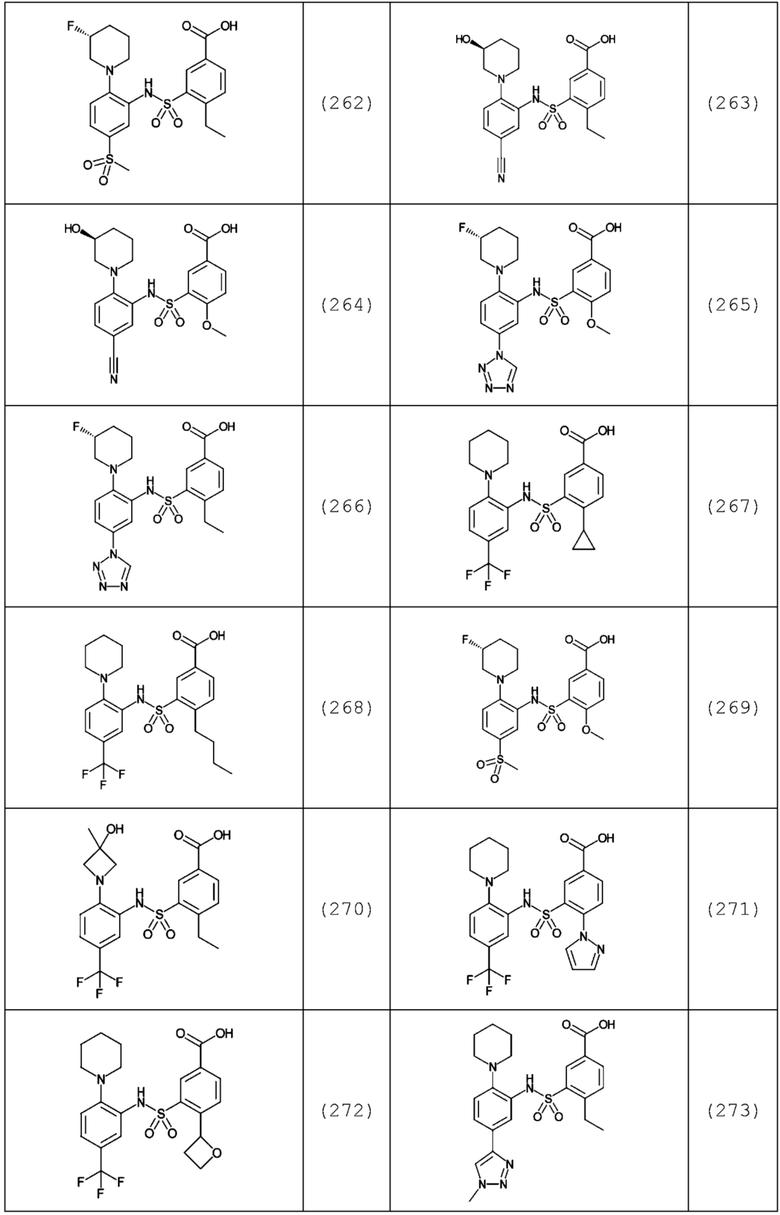
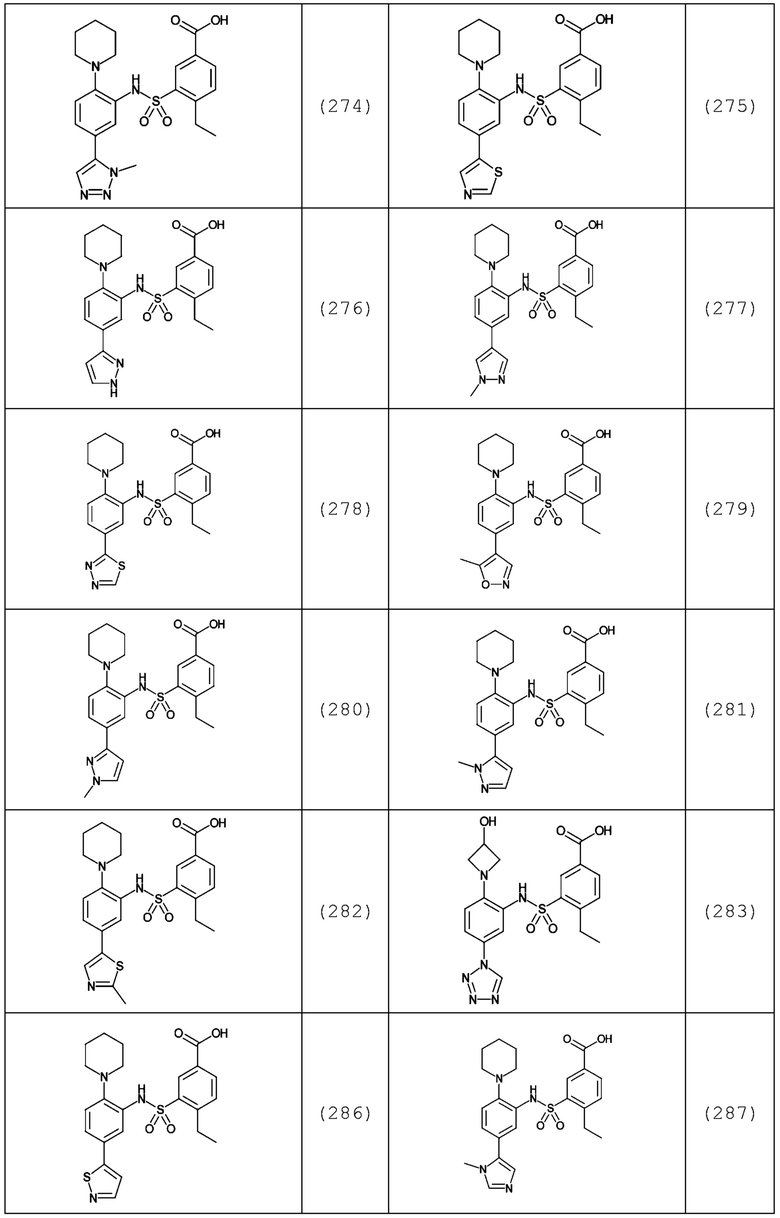
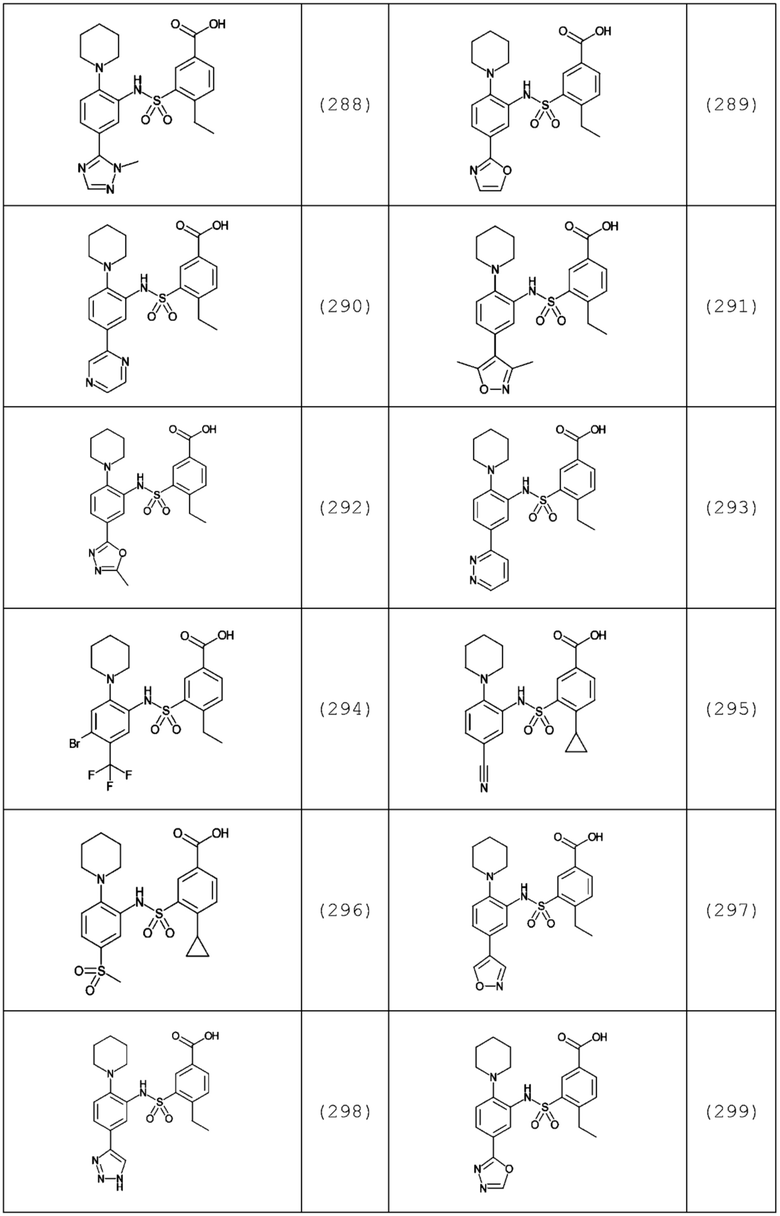
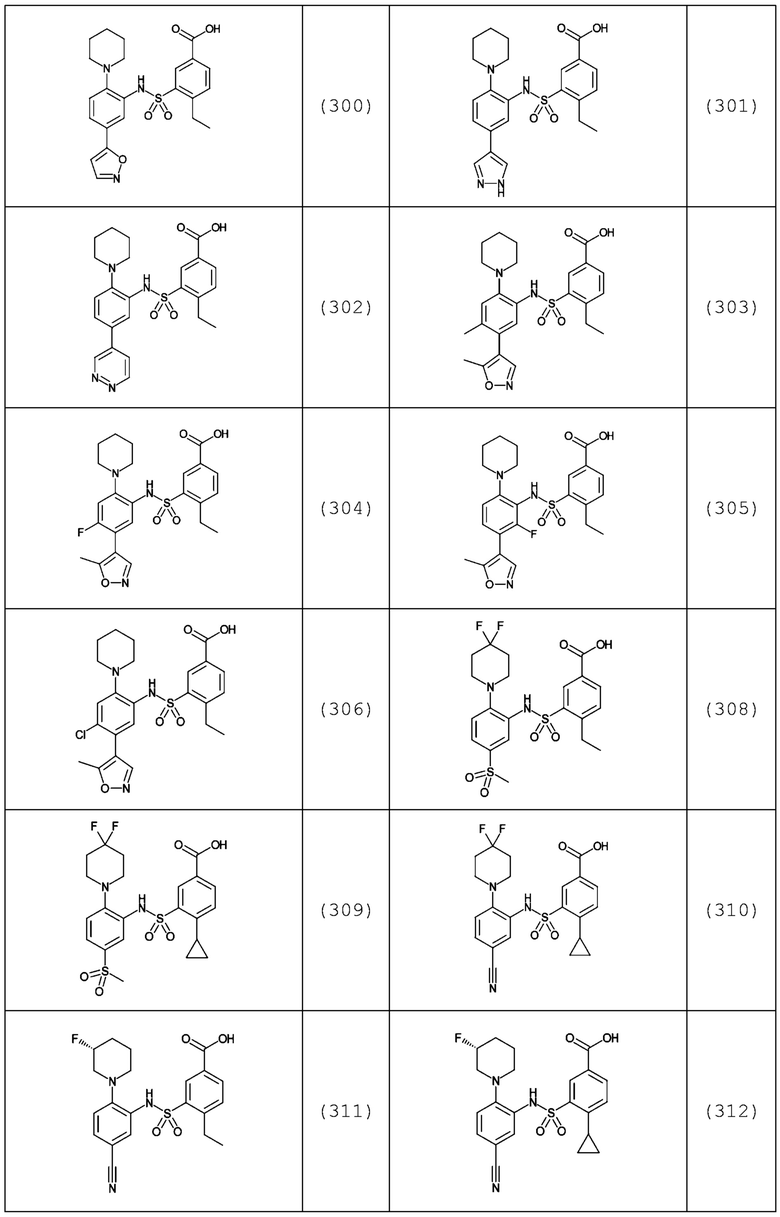
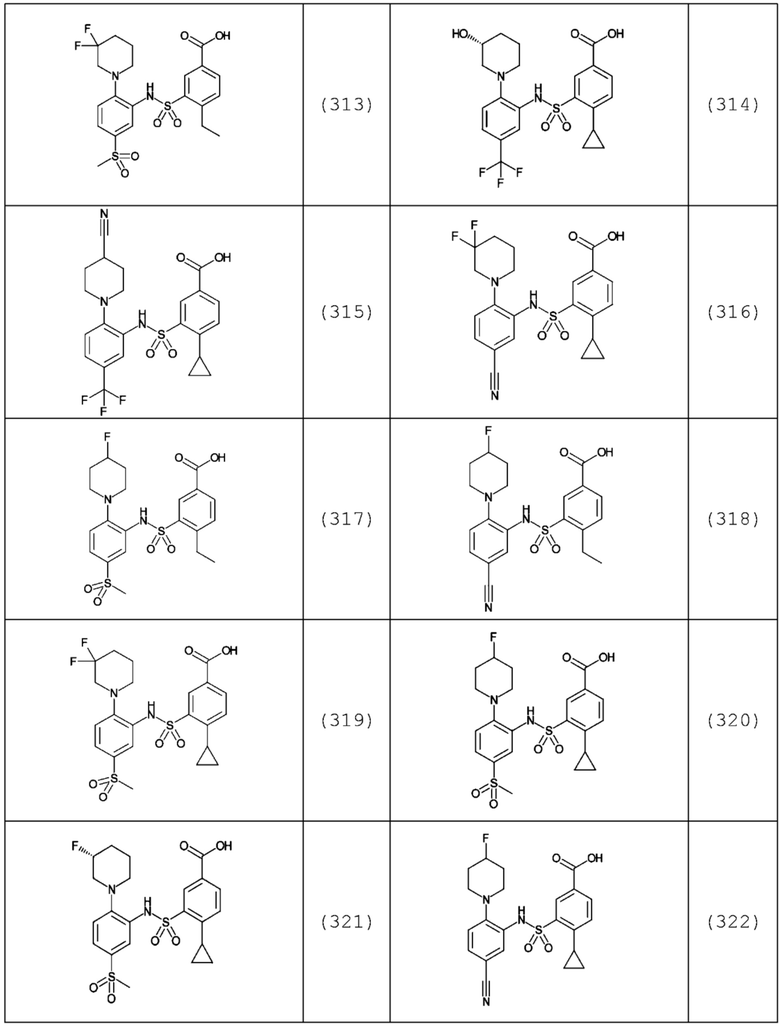
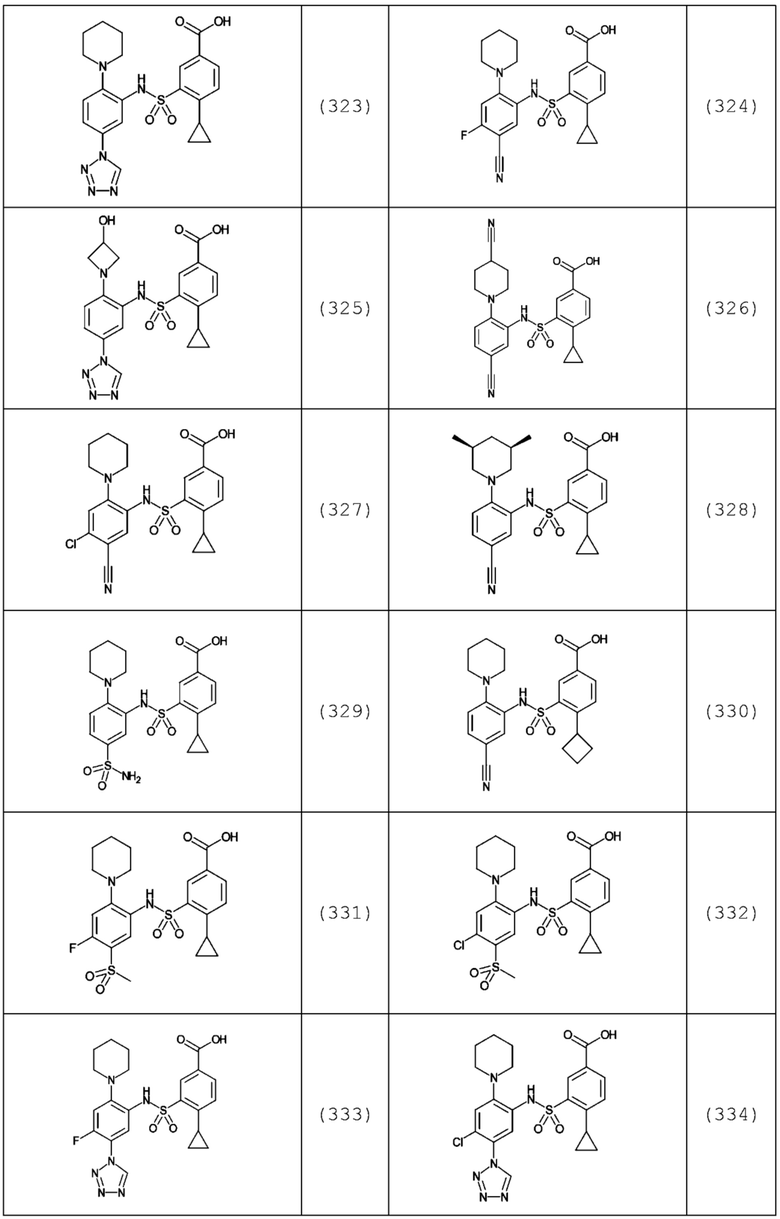
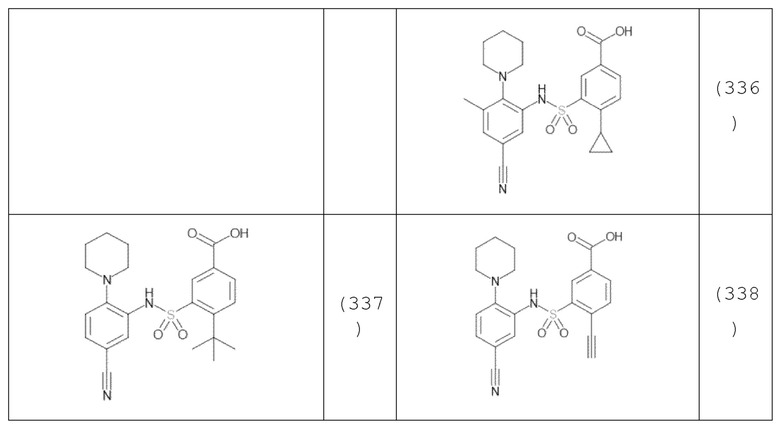
и их фармацевтически приемлемых солей.
46. Применение соединения по п. 44 или 45, где соединение модулирует ERAP1.
47. Применение соединения по любому одному из пп. 44-46, где нарушение является пролиферативным нарушением и предпочтительно является раком или лейкозом.
48. Применение соединения по п. 47, где соединение убивает раковые клетки, снижает количество пролиферирующих клеток в злокачественной опухоли, уменьшает объем или размер опухоли, включающей раковые клетки, и/или снижает количество метастазирующих раковых клеток.
49. Применение соединения по п. 47 или 48, где соединение применяется для предупреждения рака, где соединение предпочтительно индуцирует неоантиген, к которому субъект имеет существующий иммунный ответ.
50. Применение соединения по любому одному из пп. 44-49, где субъект ранее имел рак, имел случай рака в семье, имеет высокий риск развития рака, имеет генетическую предрасположенность к развитию рака, подвергся воздействию канцерогенного агента и/или находится в ремиссии после рака.
51. Применение соединения по любому одному из пп. 47-50, где указанное соединение применяют в комбинации с иммунотерапией, где субъект предпочтительно имеет рак, и соединение повышает чувствительность раковых клеток к иммунотерапии.
52. Применение соединения по п. 51, где указанная иммунотерапия является вмешательством в иммунную контрольную точку, предпочтительно является антителом-ингибитором контрольной точки.
53. Применение соединения по п. 52, где указанный ингибитор контрольной точки антитела является антителом против PD-1, антителом против PD-L1 или антителом против CTLA4.
54. Применение соединения по любому одному из пп. 44-46, где нарушение является иммунологическим нарушением и предпочтительно выбрано из анкилозирующего спондилита, болезни Бехчета, псориаза и birdshot хориоретинопатии.
55. Применение соединения по любому одному из пп. 44-46, где нарушение является воспалительным заболеванием, более предпочтительно аутовоспалительным заболеванием.
56. Применение соединения по любому одному из пп. 44-46, где вирусное нарушение является инфекционным вирусным заболеванием, выбранным из ВИЧ, ВПЧ, ЦМВ и ВГС.
57. Применение соединения по любому одному из пп. 44-46, где нарушением является рак и где соединение повышает заметность раковых клеток для иммунной системы путем изменения репертуара антигенов и неоантигенов, презентируемых иммунной системе.
58. Применение соединения по любому одному из пп. 44-46 или 57, где соединение усиливает ответ CD8+T-клеток против раковой клетки.
| E | |||
| ZERVOUDI ET AL, Proceedings of the National Academy of Sciences, vol | |||
| Облицовка комнатных печей | 1918 |
|
SU100A1 |
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |
| Двух шпиндельный станок для нарезания скоб | 1929 |
|
SU19890A1 |
| WO 2009064388 A2, 22.05.2009 | |||
| WO 2016097389 A1, 23.06.2016 | |||
| Электромагнитный бесклапанный насос | 1931 |
|
SU28614A1 |
| Прозрачные каше для киносъемки | 1928 |
|
SU18032A1 |

