ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к соединениям и их фармацевтически приемлемым солям, а также способам их применения для лечения онкологических заболеваний в качестве монотерапии или в комбинации с лучевой терапией, химиотерапией и/или иммунотерапией.
УРОВЕНЬ ТЕХНИКИ
Несколько представителей семейства PIKK (PI-3K-подобных киназ) серин-треониновых киназ известны как медиаторы сигнализации, связанной с повреждением ДНК.
Лучевую терапию (ЛТ) применяют для лечения > 50% всех онкологических пациентов в определенный момент времени на протяжении их болезни. Несмотря на затраченные значительные усилия предыдущие подходы к разработке клинических радиосенсибилизаторов не были особо эффективны, главным образом вследствие нацеливания на неспецифические пути, которые не являются непосредственными регуляторами клеточного ответа на облучение.
Существует потребность в новых вариантах терапии для онкологических заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В целом, в данном изобретении предложено соединение формулы (I):
 ,
,
(I)
или его фармацевтически приемлемая соль,
где
Z представляет собой СH, CR3 или N;
Y представляет собой СHR5 или NR6;
n равно 0, 1, 2 или 3;
R1 представляет собой -O-L-N(R7)2 или необязательно замещенный четырехчленный насыщенный N-гетероциклил;
R2 представляет собой С1-3 aлкил;
каждый R3 независимо представляет собой галоген;
R4 представляет собой необязательно замещенный aлкил;
R5 представляет собой водород, необязательно замещенный C1-3 aлкил или бензилокси;
R6 представляет собой необязательно замещенный C1-3 алкил;
каждый R7 независимо представляет собой H или необязательно замещенный C1-3 aлкил; и
L представляет собой необязательно замещенный этилен.
В некоторых вариантах реализации n равно 1. В определенных вариантах реализации, соединение представляет собой соединение формулы (IA):
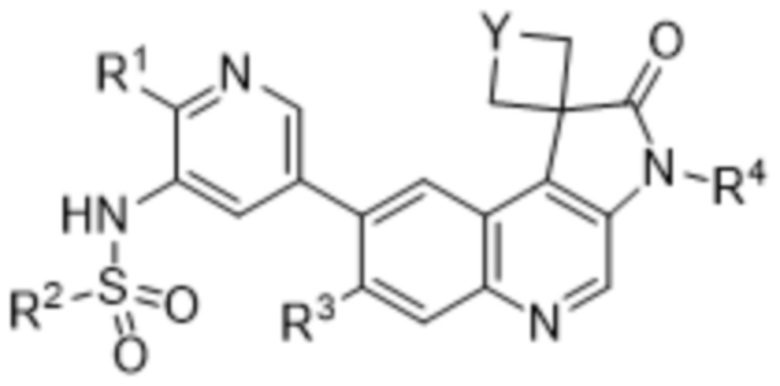 ,
,
(IA)
или его фармацевтически приемлемую соль.
В конкретных вариантах реализации, R3 представляет собой галоген (например, фтор). В дополнительных вариантах реализации n равно 0.
В еще дополнительных вариантах реализации соединение представляет собой соединение формулы (IB):
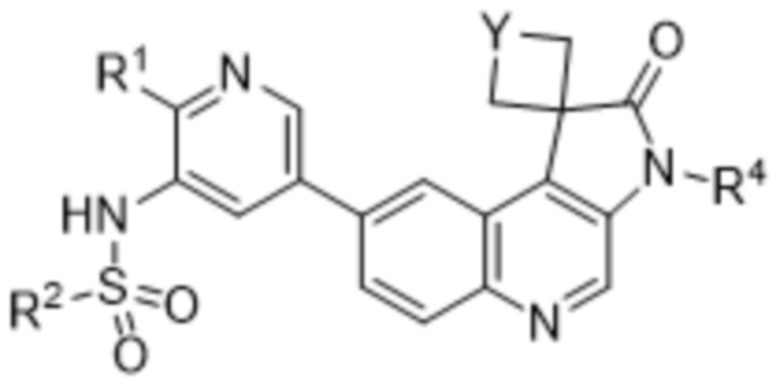 ,
,
(IB)
или его фармацевтически приемлемую соль.
В еще дополнительных вариантах реализации, R1 представляет собой -O-L-N(R7)2. В некоторых вариантах реализации один R7 представляет собой H, а оставшийся R7 представляет собой необязательно C1-3 aлкил. В определенных вариантах реализации по меньшей мере один R7 представляет собой изопропил. В конкретных вариантах реализации, R2 представляет собой метил, этил или изопропил. В дополнительных вариантах реализации R2 представляет собой метил. В еще дополнительных вариантах реализации, R4 представляет собой метил. В еще дополнительных вариантах реализации, Y представляет собой СHR5. В как и предыдущих дополнительных вариантах реализации, R5 представляет собой водород. В других вариантах реализации, R5 представляет собой необязательно замещенный C1-3 aлкил. В еще других вариантах реализации, R5 представляет собой бензилокси. В как и предыдущих других вариантах реализации, Y представляет собой NR6. В некоторых вариантах реализации, R6 представляет собой необязательно замещенный C3 aлкил. В определенных вариантах реализации, R6 представляет собой изопропил.
В конкретных вариантах реализации, соединение выбрано из группы, состоящей из:
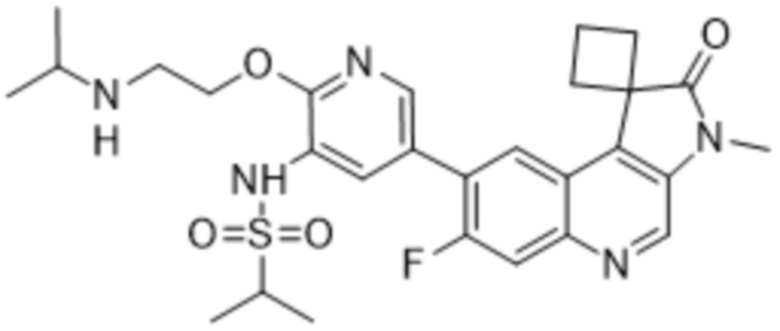 ,
,  ,
, 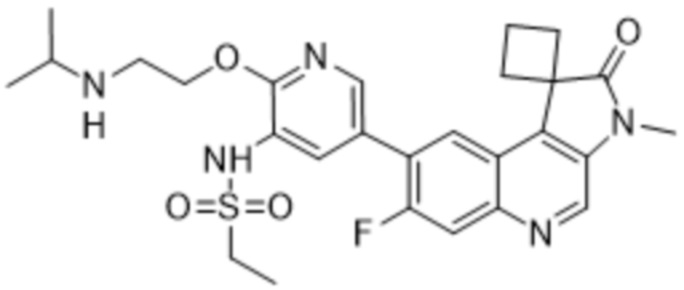 ,
,  ,
, 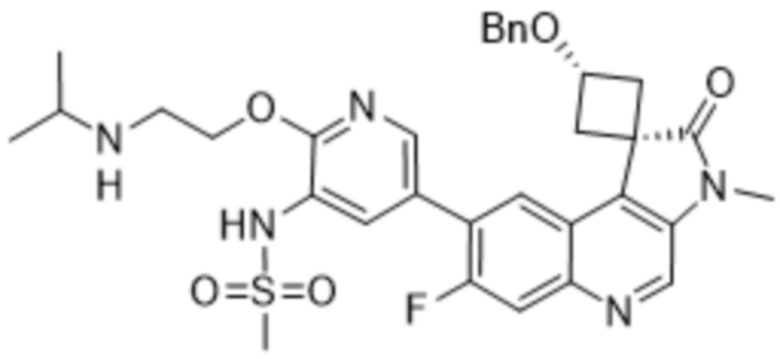 ,
, 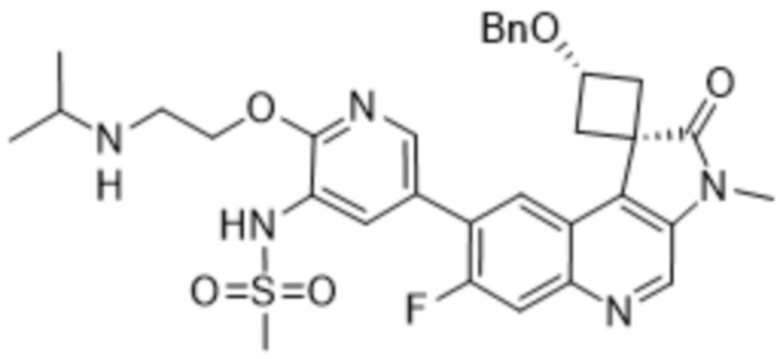 ,
,  ,
, 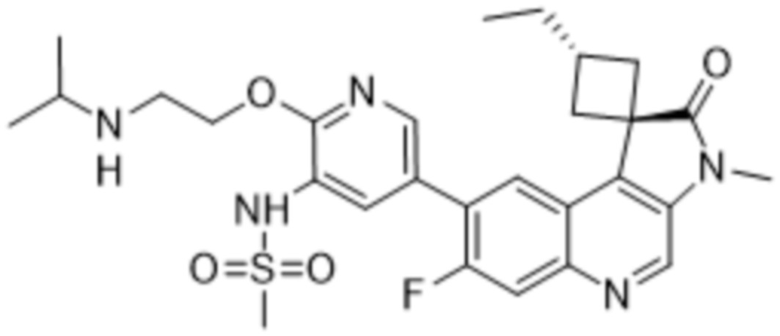 ,
, 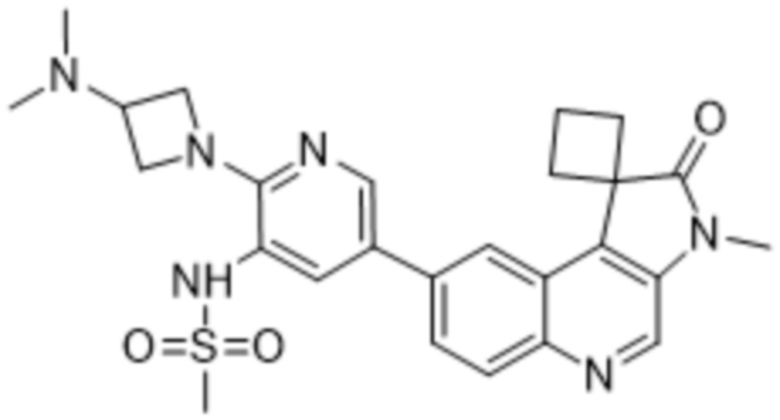 ,
,
и их фармацевтически приемлемых солей.
В дополнительных вариантах реализации соединение представляет собой следующую структуру:
,
или его фармацевтически приемлемую соль.
В еще дополнительных вариантах реализации соединение представляет собой следующую структуру:
,
или его фармацевтически приемлемую соль.
В как и предыдущих дополнительных вариантах реализации, соединение представляет собой следующую структуру:
,
или его фармацевтически приемлемую соль.
В другом аспекте изобретение обеспечивает фармацевтическую композицию, включающую соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество.
В еще другом аспекте изобретение обеспечивает способ лечения онкологического заболевания (например, рака, например, тех видов рака, которые описаны в данном документе) путем введения терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли, или фармацевтической композиции изобретения пациенту, нуждающемуся в этом. В как и предыдущем другом аспекте изобретение относится к фармацевтическим композициям для применения при лечении онкологического заболевания (например, рака, например, рака, описанного в данном документе). Фармацевтические композиции включают соединение изобретения. В дополнительном аспекте изобретение обеспечивает применение соединения по изобретению в производстве лекарственного средства для лечения онкологического заболевания (например, рака, например, рака, описанного в данном документе). В некоторых вариантах реализации онкологическое заболевание представляет собой рак головного мозга, рак мочевого пузыря, рак груди, рак центральной нервной системы, рак шейки матки, рак толстой кишки, рак эндометрия, рак пищевода, стромальную опухоль желудочно-кишечного тракта, рак желудка, рак головы и шеи, буккальный рак, рак ротовой полости, гепатоцеллюлярный рак, рак легких, меланома, карцинома из клеток Меркеля, мезотелиома, рак носоглотки, нейробластома, остеосаркома, рак яичников, рак поджелудочной железы, рак предстательной железы, рак почек, рак слюнной железы, саркомы, рак яичек, рак уротелия, рак вульвы или опухоль Вильма. В дополнительных вариантах реализации онкологическое заболевание представляет собой рак груди, рак легких, рак головы и шеи, рак поджелудочной железы, рак прямой кишки, глиобластому, гепатоцеллюлярную карциному, холангиокарциному, метастатические поражения печени, меланому, саркому кости, саркому мягких тканей, рак эндометрия, рак шейки матки, рак предстательной железы или карциному из клеток Меркеля.
В некоторых вариантах реализации пациент получает лучевую терапию. В определенных вариантах реализации соединение или фармацевтическую композицию вводят пациенту одновременно с лучевой терапией. В конкретных вариантах реализации соединение или фармацевтическую композицию вводят пациенту перед лучевой терапией. В дополнительных вариантах реализации соединение или фармацевтическую композицию вводят пациенту после лучевой терапии. В еще дополнительных вариантах реализации лучевая терапия включает внешнее, внутреннее, брахитерапевтическое или системное облучение, например, с помощью радионуклида (например, β-излучающего радионуклида (например, 32Фосфора, 67Меди, 77Брома, 89Стронция, 90Иттрия, 105Родия, 131Иода, 137Цезия, 149Прометея, 153Самария, 166Гольмия, 177Лютеция, 186Рения, 188Рения, или 199Золота), α-излучающего радионуклида (например, 211Aстатина, 213Висмута, 223Радия, 225Aктиния, или 227Tория), радионуклида, излучающего гамма-лучи (например, 192Иридия), или радионуклидов, улавливающих электроны (например, 67Галлия, 103Палладия, или 125Иода)), радионуклидного конъюгата антитела (например, 90Y-ибритумомаб тиуксетана, 131I-тозитумомаба, 225Ac-линтузумаб сатетраксетана, 227Th-анетумаб кориксетана, 90Y-эпитумомаб цитуксетана, 90Y-кливатузумаб тетраксетана, 177Lu-лилотомаб сатетраксетана, 90Y-розопатамаб тетраксетана, 90Y-табитуксимаб барзуксетана, или 90Y-такатузумаб тетраксетана), или другого целевого радионуклидного конъюгата (например, 131I-PSMA, 90Y-PSMA, 177Lu-PSMA или 177Lu-сатореотид тетраксетана). Предпочтительно лучевая терапия включает введение радионуклидного конъюгата антитела. В как и предыдущих дополнительных вариантах реализации пациент получает противоопухолевое средство. В других вариантах реализации противоопухолевый агент представляет собой один или более из цисплатин, оксалиплатин, карбоплатин, валрубицин, идарубицин, калихеамицин или ингибитор PARP. В еще других вариантах реализации противоопухолевый агент представляет собой противоопухолевый биологический агент и/или противоопухолевый иммунотерапевтический агент. В как и предыдущих других вариантах реализации соединение или фармацевтическую композицию вводят пациенту одновременно с противоопухолевым агентом. В некоторых вариантах реализации соединение или фармацевтическая композиция вводится пациенту перед противоопухолевым агентом. В определенных вариантах реализации соединение или фармацевтическая композиция вводится пациенту после противоопухолевого агента.
ПОДРОБНОЕ ОПИСАНИЕ
Определения
Следует понимать, что используемая в данном документе терминология предназначена для описания конкретных вариантов осуществления и не является ограничительной. Кроме того, хотя любые способы, устройства и материалы, аналогичные или эквивалентные описанным в данном документе, можно использовать при практической реализации или тестировании данного изобретения, далее будут описаны предпочтительные способы, устройства и материалы. В дополнение к вышесказанному, в контексте описания и прилагаемой формулы изобретения, если не указано иное, следующие термины имеют такие значения:
«Амино» относится к радикалу -NH2.
«Циано» относится к радикалу -CN.
«Гидроксил» относится к радикалу -OH.
«Имино» относится к заместителю=NH.
«Нитро» относится к радикалу -NO2.
«Оксо» относится к заместителю=O.
«Тиоксо» относится к заместителю=S.
«Трифторметил» относится к радикалу -CF3.
«Алкил» относится к линейному, насыщенному, ациклическому, одновалентному углеводородному радикалу или разветвленному, насыщенному, ациклическому, одновалентному углеводородному радикалу, имеющему от одного до двенадцати атомов углерода, предпочтительно от одного до восьми атомов углерода или от одного до шести атомов углерода, и который является присоединенным к остальной части молекулы одинарной связью, например, метил, этил, н-пропил, 1-метилэтил (изо-пропил), н-бутил, н-пентил, 1,1-диметилэтил (трет-бутил), 3-метилгексил, 2-метилгексил и т.п. Необязательно замещенный алкильный радикал представляет собой алкильный радикал, который необязательно замещен, если позволяет валентность, одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила, оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1 или 2) и -S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил.
«Алкенил» относится к линейному, ациклическому, одновалентному углеводородному радикалу или разветвленному, ациклическому, одновалентному углеводородному радикалу, содержащему одну, две или три двойные связи углерод-углерод, имеющему от двух до двенадцати атомов углерода, предпочтительно от двух до восьми атомов углерода и который присоединен к остальной части молекулы одинарной связью, например этенил, проп-1-енил, бут-1-енил, пент-1-енил, пента-1,4--диенил и т. п. Необязательно замещенный алкенильный радикал представляет собой алкенильный радикал, который необязательно замещен, если позволяет валентность, одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из: галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила, оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1 или 2) и -S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, гетероциклил или гетероарил.
«Алкинил» относится к линейному, ациклическому, одновалентному углеводородному радикалу или разветвленному, ациклическому, одновалентному углеводородному радикалу, содержащему одну или две тройные связи углерод-углерод и, необязательно, одну, две или три двойные связи углерод-углерод, и имеющий от двух до двенадцати атомов углерода, предпочтительно от двух до восьми атомов углерода, который присоединен к остальной части молекулы одинарной связью, например, этинил, проп-1-инил, бут-1-инил, пент-1-инил, пента-1-ен-4-инил и подобные.. Необязательно замещенный алкинильный радикал представляет собой алкинильный радикал, который необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из: галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила, оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1, или 2) и -S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил.
«Алкилен» или «алкиленовая цепь» относится к линейной, ациклической, насыщенной, двухвалентной углеводородной цепи или разветвленной, ациклической, насыщенной, двухвалентной углеводородной цепи, имеющей от одного до двенадцати атомов углерода, например, метилен, этилен, пропилен, н-бутилен, и подобные. Алкиленовая цепь присоединена посредством одинарных связей. Точки присоединения алкиленовой цепи могут находиться на одном атоме углерода или на разных атомах углерода в алкиленовой цепи. Необязательно замещенная алкиленовая цепь представляет собой алкиленовую цепь, которая необязательно замещена, если позволяет валентность, одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из: галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила, оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1, или 2) и -S(O)tN(R14)2 (где t равно 1 или 2) где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил. В некоторых вариантах реализации алкилен представляет собой этилен.
«Алкенилен» или «алкениленовая цепь» относится к линейной, ациклической, двухвалентной углеводородной цепи или разветвленной, ациклической, двухвалентной углеводородной цепи, содержащей одну, две или три двойные углерод-углеродные связи и имеющей от двух до двенадцати атомов углерода, например, этенилен, пропенилен, н-бутенилен и тому подобное. Алкениленовая цепь присоединена посредством одинарных связей. Точки присоединения алкениленовой цепи могут находиться на одном атоме углерода или на разных атомах углерода в алкениленовой цепи. Необязательно замещенная алкениленовая цепь представляет собой алкениленовую цепь, которая необязательно замещена, если позволяет валентность, одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из: галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила , оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1, или 2) и -S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил.
«Алкинилен» или «алкиниленовая цепь» относится к линейной, ациклической, двухвалентной углеводородной цепи или разветвленной, ациклической, двухвалентной углеводородной цепи, содержащей одну или две тройные связи углерод-углерод и, необязательно, одну, две или три углерод-углерод двойные связи и имеющие от двух до двенадцати атомов углерода, например пропинилен, н-бутинилен и т.п. Алкиниленовая цепь присоединена посредством одинарных связей. Точки присоединения алкиниленовой цепи могут находиться на одном атоме углерода или на разных атомах углерода в алкиниленовой цепи. Необязательно замещенная алкиниленовая цепь представляет собой алкинеленовую цепь, которая необязательно замещена одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из: галогена, циано, нитро, арила, циклоалкила, гетероциклила, гетероарила, оксо, триметилсиланила, -OR14, -OC(O)-R14, -N(R14)2, -C(O)R15, -C(O)OR14, -C(O)N(R14)2, -N(R14)C(O)OR16, -N(R14)C(O)R16, -N(R14)S(O)tR16 (где t равно 1 или 2), -S(O)tOR16 (где t равно 1 или 2), -S(O)pR16 (где p равно 0, 1, или 2) и -S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой водород, циклоалкил, арил, гетероциклил или гетероарил; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил.
«Алкокси» относится к радикалу формулы -ORa, где Ra представляет собой алкильный радикал по определению выше, содержащий от одного до двенадцати атомов углерода. Алкильная часть необязательно замещенного алкокси-радикала необязательно замещена по определению выше для алкильного радикала.
«Алкоксиалкил» относится к радикалу формулы -Ra-O-Rb, где Ra представляет собой алкилен, а Rb представляет собой алкил, как определено выше. Алкильная и алкиленовая части необязательно замещенного алкоксиалкильного радикала необязательно замещены по определению выше для алкильного радикала и алкиленовой цепи, соответственно.
«Аралкил» относится к радикалу формулы -Ra-Rb, где Ra представляет собой алкилен, а Rb представляет собой арил по описанию в данном документе. Алкиленовая и арильная части необязательно замещенного аралкила необязательно замещены, как описано в данном документе для алкилена и арила, соответственно.
«Арил» относится к радикалу ароматической моноциклической или мультициклической углеводородной кольцевой системы, содержащему от 6 до 18 атомов углерода, причем мультициклическая арильная кольцевая система является бициклической, трициклической или тетрациклической кольцевой системой. Арильные радикалы включают, но не ограничиваются этим, такие группы как флуоренил, фенил и нафтил. Необязательно замещенный арил представляет собой арильный радикал, который необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из алкила, акенила, галогена, галогеналкила, галогеналкенила, циано, нитро, арила, гетероарила, гетероарилалкила, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)pR16 (где p равно 0, 1, или 2), и -R15-S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой прямую связь или линейную или разветвленную алкиленовую или алкениленовую цепь; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, гетероциклил или гетероарил.
«Арилалкокси» относится к группе формулы -O-R, где R представляет собой аралкил. Необязательно замещенный арилалкокси представляет собой арилалкокси, который необязательно замещен, как описано в данном документе для аралкила. В некоторых вариантах реализации арилалкокси представляет собой бензилокси.
«Циклоалкил» относится к стабильному не-ароматическому моноциклическому или полициклическому углеводородному радикалу, имеющему от трех до пятнадцати атомов углерода, предпочтительно имеющему от трех до десяти атомов углерода, который является насыщенным или ненасыщенным и который присоединяется к остальной части молекулы посредством одинарная связи. Полициклический углеводородный радикал является бициклической, трициклической или тетрациклической кольцевой системой. Ненасыщенный циклоалкил содержит одну, две или три углерод-углеродные двойные связи и/или одну углерод-углеродную тройную связь. Моноциклические циклоалкильные радикалы включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Полициклические циклоалкильные радикалы включают, например, адамантил, норборнил, декалинил и т. п. Необязательно замещенный циклоалкил представляет собой циклоалкильный радикал, который необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, нитро, оксо, арила, аралкила, циклоалкила, гетероциклила, гетероарила, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)pR16 (где p равно 0, 1, или 2) и -R15-S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой прямую связь или линейную или разветвленную алкиленовую или алкениленовую цепь; и каждый R16 независимо представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил или гетероарил.
«Конденсированный» относится к любой описанной в данном документе кольцевой системе, которая конденсирована с существующей кольцевой структурой в соединениях по изобретению. Если конденсированная кольцевая система представляет собой гетероциклил или гетероарил, любой атом углерода на существующей кольцевой структуре, который становится частью конденсированной кольцевой системы, может быть замещен атомом азота.
«Галоген» относится к заместителям-галогенам: брому, хлору, фтору и йоду.
«Галогеналкил» относится к алкильному радикалу по определению выше, который дополнительно замещен одним или более заместителями-галогенами. Число заместителей-галогенов, включенных в галогеналкил, составляет от одного и до общего числа атомов водорода, доступных для замещения заместителями-галогенами (например, перфторалкил). Неограничивающие примеры галогеналкила включают трифторметил, дифторметил, трихлорметил, 2,2,2-трифторэтил, 1-фторметил-2-фторэтил, 3-бром-2- фторпропил, 1-бромметил-2-бромэтил и тому подобное. В случае необязательно замещенного галогеналкила атомы водорода, связанные с атомами углерода алкильной части галогеналкильного радикала, могут быть необязательно замещены заместителями, определенными выше для необязательно замещенного алкила.
«Галогеналкенил» относится к алкенильному радикалу по определению выше, который дополнительно замещен одним или более заместителями-галогенами. Число заместителей-галогенов, включенных в галогеналкенил, составляет от одного и до общего числа атомов водорода, доступных для замещения заместителями-галогенами (например, перфторалкенил). Неограничивающие примеры галогеналкенила включают 2,2-дифторэтенил, 3-хлорпроп-1-енил и тому подобное. В случае необязательно замещенного галогеналкенила атомы водорода, связанные с атомами углерода алкенильной части галогеналкенильного радикала, могут быть необязательно замещены заместителями, определенными выше для необязательно замещенной алкенильной группы.
«Галогеналкинил» относится к алкинильному радикалу по определению выше, который дополнительно замещен одним или более заместителями-галогенами. Число заместителей-галогенов, включенных в галогеналкинил, составляет от одного и до общего числа атомов водорода, доступных для замещения заместителями-галогенами (например, перфторалкинил). Неограничивающие примеры галогеналкинила включают 3-хлорпроп-1-инил и тому подобное. Алкинильная часть галогеналкинильного радикала может быть дополнительно необязательно замещена по определению выше для алкинильной группы.
«Гетероарилалкил» относится к радикалу формулы -Ra-Rb, где Ra представляет собой алкилен, а Rb представляет собой гетероарил по описанию в данном документе. Алкиленовая и гетероарильная части необязательно замещенного гетероарилалкила необязательно замещены, как описано в данном документе для алкилена и гетероарила, соответственно.
«Гетероциклил» относится к стабильному 3-18-членному радикалу не-ароматической кольцевой системы, имеющему число атомов углерода от двух до двенадцати и содержащему в общей сложности от одного до шести гетероатомов, независимо выбранных из группы, состоящей из азота, кислорода, фосфора и серы. Гетероциклильный радикал является моноциклической, бициклической, трициклической или тетрациклической кольцевой системой. Бициклический, трициклический или тетрациклический гетероциклил является конденсированной спиро- и/или мостиковой кольцевой системой. Гетероциклильный радикал может быть насыщенным или ненасыщенным. Ненасыщенный гетероциклил содержит одну, две или три углерод-углеродные двойные связи и/или одну углерод-углеродную тройную связь. Необязательно замещенный гетероциклил представляет собой гетероциклильный радикал, который необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, оксо, тиоксо, нитро, арила, аралкила, циклоалкила, гетероциклила, гетероарила, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)pR16 (где p равно 0, 1, или 2), и -R15-S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, алкенил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой прямую связь или линейную или разветвленную алкиленовую или алкениленовую цепь; и каждый R16 независимо представляет собой алкил, алкенил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил. Атомы азота, углерода или серы в гетероциклильном радикале могут быть необязательно окисленными (когда заместитель представляет собой оксо и присутствует на гетероатоме); атом азота может быть необязательно кватернизован (когда заместитель представляет собой алкил, алкенил, арил, аралкил, циклоалкил, гетероциклил, гетероарил, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)pR16 (где p равно 0, 1, или 2), и -R15-S(O)tN(R14)2 (где t равно 1 или 2), где R15 представляет собой линейную или разветвленную алкиленовую или алкениленовую цепь, а R14 и R16 имеют указанные выше значения). Примеры необязательно замещенных гетероциклильных радикалов включают, но не ограничиваются ими, азетидинил, диоксоланил, тиенил[1,3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4-пиперидонил, пирролидинил, пиразолидинил, тиазолидинил, тетрагидрофурил, тритианил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1-оксо-тиоморфолинил и 1,1-диоксо-тиоморфолинил.
«Гетероциклилен» относится к гетероциклилу, в котором один атом водорода замещен валентностью. Необязательно замещенный гетероциклилен необязательно замещен, как описано в данном документе для гетероциклила.
«Гетероарил» относится к 5-18-членному радикалу кольцевой системы, содержащему по крайней мере одно ароматическое кольцо, имеющему количество атомов углерода от одного до семнадцати атомов углерода и содержащему в общей сложности от одного до десяти гетероатомов, независимо выбранных из группы, состоящей из азота., кислорода и серы. Гетероарильный радикал является моноциклической, бициклической, трициклической или тетрациклической кольцевой системой. Бициклический, трициклический или тетрациклический гетероарильный радикал является конденсированной и/или мостиковой кольцевой системой. Необязательно замещенный гетероарил представляет собой гетероарильный радикал, который необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкокси, галогена, галогеналкила, галогеналкенила, циано, оксо, тиоксо, нитро, оксо, арила, аралкила, циклоалкила, гетероциклила, гетероарила или гетероарилалкила, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)tR16 (где p равно 0, 1, или 2), and -R15-S(O)tN(R14)2 (где t равно 1 или 2), где каждый R14 независимо представляет собой водород, алкил, алкенил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил; каждый R15 независимо представляет собой прямую связь или линейную или разветвленную алкиленовую или алкениленовую цепь; и каждый R16 представляет собой алкил, алкенил, галогеналкил, циклоалкил, арил, гетероциклил или гетероарил. Атомы азота, углерода или серы в гетероциклильном радикале могут быть необязательно окисленными (когда заместитель представляет собой оксо и присутствует на гетероатоме) при условии, что по крайней мере одно кольцо в гетероариле остается ароматическим; атом азота может быть необязательно кватернизован (когда заместитель представляет собой алкил, алкенил, арил, аралкил, циклоалкил, гетероциклил, гетероарил, -R15-OR14, -R15-OC(O)-R14, -R15-N(R14)2, -R15-C(O)R14, -R15-C(O)OR14, -R15-C(O)N(R14)2, -R15-N(R14)C(O)OR16, -R15-N(R14)C(O)R16, -R15-N(R14)S(O)tR16 (где t равно 1 или 2), -R15-S(O)tOR16 (где t равно 1 или 2), -R15-S(O)pR16 (где p равно 0, 1, или 2), и -R15-S(O)tN(R14)2 (где t равно 1 или 2), где R15 представляет собой линейную или разветвленную алкиленовую или алкениленовую цепь, а R14 и R16 имеют указанные выше значения) при условии, что по крайней мере одно кольцо в гетероариле остается ароматическим. Примеры необязательно замещенных гетероарильных радикалов включают, но не ограничиваются ими, азепинил, акридинил, бензимидазолил, бензтиазолил, бензиндолил, бензодиоксолил, бензофуранил, бензооксазолил, бензотиазолил, бензотиадиазолил, бензо[b][1,4]диоксепин, 1,4-бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензопиранил, бензопиранонил, бензофуранил, бензофуранонил, бензотиенил (бензотиофенил), бензотриазолил, бензо [4,6]имидазо[1,2-a]пиридинил, карбазолил, циннолинил, дибензофуранил, дибензотиофенил, фуранил, фуранонил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолил, индолизинил, изоксазолил, нафтил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, оксиранил, 1-фенил-1H-пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пирролил, пиразолил, пиридинил, пиразинил, пиримидинил, пиридазинил, пирролил, хиназолинил, хиноксалинил, хинолинил, хинуклидинил, изохинолинил, тетрагидрохинолинил, тиазолил, тиадиазолил, триазолил, тетразолил, триазинил и тиофенил (т.е. тиенил).
Также подразумевается, что описанное в данном документе изобретение охватывает все фармацевтически приемлемые соединения формулы (I), меченные изотопами посредством замещения одного или более атомов на атом, имеющий отличные атомную массу или массовое число. Примеры изотопов, которые могут быть включены в описанные соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl, 123I и 125I, соответственно. Эти радиоактивно меченные соединения могут быть полезны при определении или измерении эффективности соединений, характеризуя, например, сайт или способ действия ферментов ATM и DNA-PK, или являясь аффинными в отношении фармакологически важного сайта действия на ферментах ATM и DNA-PK. Некоторые меченные изотопами соединения формулы (I), например, содержащие радиоактивный изотоп, полезны при исследованиях распределения лекарственного препарата и/или субстрата в тканях. В частности, ввиду легкости включения и наличия готовых средств обнаружения в этих целях применимы такие радиоактивные изотопы, как тритий, т. е. 3H, и углерод-14, т. е. 14C.
Замена на более тяжелые изотопы, такие как дейтерий, т. е. 2H, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, повышенным временем полужизни in vivo или необходимостью меньших дозировок, и, следовательно, может быть предпочтительной в некоторых обстоятельствах.
Замена на испускающие позитроны изотопы, такие как 11C, 18F, 15O и 13N, может быть полезна в исследованиях методом позитронной эмиссионной топографии (ПЭТ) для изучения занятости рецепторов субстрата. Меченные изотопами соединения формулы (I) в общем случае можно получать традиционными методами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в разделе примеров и способов получения, с использованием подходящих меченных изотопами реагентов вместо не меченных изотопами реагентов, которые использовали до этого.
Также подразумевается, что описанное в данном документе изобретение охватывает продукты in vivo метаболизма описанных соединений. Такие продукты могут образовываться в результате, например, окисления, восстановления, гидролиза, амидирования, эстерификации и т. п. вводимого соединения, главным образом в результате ферментативного процесса. Соответственно, данное изобретение включает соединения, полученные способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения его метаболического продукта. Такие продукты, как правило, идентифицируют путем введения радиоактивно меченного соединения по изобретению в выявляемой дозе млекопитающему, такому как крыса, мышь, морская свинка, собака, обезьяна или человек, ожидания в течение времени, достаточного для метаболизма, и выделения продуктов его превращения из мочи, крови или других биологических образцов.
Подразумевается, что «стабильное соединение» или «стабильная структура» обозначают соединение, которое является достаточно стойким, чтобы перенести выделение в достаточной степени чистоты из реакционной смеси и включение в эффективный терапевтический агент.
«Млекопитающее» включает людей и как одомашненных животных, таких как лабораторные животные или домашние животные (например, кошки, собаки, свиньи, крупный рогатый скот, овцы, козы, лошади, кролики), так и не одомашненных животных, таких как дикие животные и т. п.
Термин «необязательный» или «необязательно» означает, что описанное после него событие или обстоятельство может произойти или может не произойти, и что описание включает случаи, когда указанное событие или обстоятельство происходит, и случаи, когда оно не происходит. Например, «необязательно замещенный арил» означает, что арильный радикал может быть или не быть замещенным и что описание включает как замещенные арильные радикалы, так и арильные радикалы без замещений.
«Пациент» означает человека или животное, не относящееся к человеку (например, млекопитающее), которое страдает заболеванием или состоянием, как определено квалифицированным специалистом (например, врачом, практикующей медсестрой или ветеринаром), при наличии или отсутствии известных сведений лабораторных тест (ов) образца (ов) от пациента.
Термин «фармацевтически приемлемые носитель, разбавитель или вспомогательное вещество» включает, без ограничения, любые адъювант, носитель, вспомогательное вещество, вещество, способствующее скольжению, подсластитель, разбавитель, консервант, краситель/пигмент, усилитель вкуса, поверхностно-активное вещество, смачивающий агент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, который был одобрен Управлением по контролю за продуктами питания и лекарственными препаратами США как приемлемый для использования для людей или одомашненных животных.
«Фармацевтически приемлемая соль», как используется в данном документе, представляет те соли, которые, в рамках здравого медицинского заключения, подходят для использования в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного и являются соразмерными с разумным соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Например, фармацевтически приемлемые соли описаны в: Berge et al., J. Pharmaceutical Sciences 66:1-19, 1977 and in Pharmaceutical Salts: Properties, Selection, and Use, (Eds. P.H. Stahl и C.G. Wermuth), Wiley-VCH, 2008. Фармацевтически приемлемые соли включают соли присоединения кислот и оснований.
«Фармацевтически приемлемая соль присоединения кислоты» относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются в биологическом или ином смысле нежелательными, и которые образованы с неорганическими кислотами, такими как, без ограничения, хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т. п., и органическими кислотами, такими как, без ограничения, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфора-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламиновая кислота, додецилсерная кислота, этан-1,2 -дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксоглутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муциновая кислота, нафталин-1,5-дисульфоновая кислота, нафталин- 2-сульфоновая кислота, 1-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и т. п.
«Фармацевтически приемлемая соль присоединения основания» относится к солям, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются в биологическом или ином смысле нежелательными. Эти соли получают при добавлении неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, но не ограничиваются этим, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т. п. Предпочтительными неорганическими солями являются соли аммония, натрия, калия, кальция и магния. Соли, полученные из органических оснований, включают, но не ограничиваются ими, соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2-диметиламиноэтанол, 2 - диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N - этилпиперидин, полиаминовые смолы и тому подобное. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто соединение по изобретению получают при кристаллизации. В контексте данного документа термин «сольват» относится к агрегату, который содержит одну или более молекул соединения по изобретению с одной или более молекулами растворителя. Растворителем может быть вода, в случае чего сольват может представлять собой гидрат. В альтернативном варианте растворителем может быть органический растворитель. Таким образом, соединения по настоящему изобретению могут существовать в виде гидрата, включая моногидрат, дигидрат, гемигидрат, сесквигидрат, тригидрат, тетрагидрат и т. п., а также в виде соответствующих сольватированных форм. Соединения по изобретению могут представлять собой истинные сольваты, тогда как в других случаях соединения по изобретению могут просто сохранять занесенную воду или представлять собой смесь воды и некоторого занесенного растворителя.
«Фармацевтическая композиция» относится к составу соединения по изобретению и среды, в общем случае приемлемой в данной области техники для введения биологически активного соединения млекопитающим, например, людям. Такая среда включает фармацевтически приемлемые носители, разбавители или вспомогательные вещества.
«Терапевтически эффективное количество» относится к такому количеству соединения по изобретению, которое при введении млекопитающему, предпочтительно человеку, является достаточным для осуществления лечения, по определению ниже, млекопитающего, предпочтительно человека или представителя собачьих. Количество соединения по изобретению или другого фармацевтического агента (например, противоопухолевого агента), которое составляет «терапевтически эффективное количество», будет варьироваться в зависимости от соединения, состояния и его тяжести, способа введения и возраста млекопитающего, подлежащего лечению, но может быть определено обычным образом специалистом в данной области техники с учетом его собственных знаний и данного раскрытия.
Термин «лечение» или «терапия» в контексте данного описания охватывает лечение представляющего интерес заболевания или состояния у млекопитающего, предпочтительно человека, имеющего интересующее заболевание или состояние, и включает: (i) предотвращение возникновения заболевания или состояния у млекопитающего, в частности, когда такое млекопитающее предрасположено к состоянию, но еще не диагностировано как имеющее его; (ii) подавление заболевания или состояния, то есть остановку его развития; (iii) облегчение заболевания или состояния, т.е. вызывание регресса заболевания или состояния; или (iv) облегчение симптомов, вызванных заболеванием или состоянием, т.е. снятие боли без устранения основного заболевания или состояния. В контексте данного документа термины «заболевание» и «патологическое состояние» могут использоваться взаимозаменяемо или могут отличаться в том, что конкретное недомогание или патологическое состояние может не иметь известной причины (т. е. его этиология еще не была выяснена) и, следовательно, не считается заболеванием, но только нежелательным состоянием или синдромом, более или менее конкретный набор симптомов которого был определен клиницистами.
Соединения по изобретению или их фармацевтически приемлемые соли могут содержать один или более асимметричных центров и, таким образом, могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены с точки зрения абсолютной стереохимии как (R)- или (S)-, или же, как (D)- или (L)- в случае аминокислот. Подразумевается, что настоящее изобретение включает все такие возможные изомеры, а также их рацемические и оптически чистые формы. Оптически активные (+) и (-), (R) - и (S)- или (D) - и (L) -изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или разделены с использованием обычных методик, например хроматографии и фракционной кристаллизации. Традиционные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разрешение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Если описанные в данном документе соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иное, подразумевается, что такие соединения включают как E-, так и Z-геометрические изомеры. Также подразумевается, что включены все таутомерные формы.
«Стереоизомер» относится к соединению, состоящему из тех же атомов, связанных такими же связями, но имеющими отличные трехмерные структуры, которые не являются взаимозаменяемыми. В настоящем изобретении предусмотрены различные стереоизомеры и их смеси и включены «энантиомеры», которые относятся к двум стереоизомерам, молекулы которых являются несовпадающими зеркальными изображениями друг друга.
«Таутомер» относится к протонному сдвигу от одного атома молекулы к другому атому той же молекулы. Настоящее изобретение включает таутомеры любых указанных соединений.
Также в объем данного изобретения входят промежуточные соединения формулы (I) и все полиморфные формы вышеуказанных молекул и их кристаллические формы.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
ФИГ. 1 представляет собой изображение иммуноблота из репрезентативного эксперимента по оценке воздействия соединения 569 на клетки MCF7 с облучением или без него.
ФИГ. 2 представляет собой график, показывающий результаты анализа клоногенной выживаемости, исследующего жизнеспособность клеток MCF7 после воздействия несущей среды (ДМСО) или соединения 569 с облучением или без него.
ФИГ. 3 представляет собой изображение иммуноблота, показывающее индукцию фосфорилирования TBK1 соединением 569, селективным ингибитором ATM (ATMi), селективным ингибитором DNA-PK (DNA-PKi), и комбинацией селективного ингибитора ATM и селективного ингтибитора DNA-PK (Ai+Di) в клетках HCT116, экспрессирующих p53 дикого типа, или клетках HCT116, которые были отрицательными по экспрессии p53. На ФИГ. 3, p.ATM, p.DNA-PK, p.TBK1 и p.STING указывают фосфорилированные формы ATM, DNA-PK, TBK1 и STING соответственно.
ФИГ. 4 представляет собой иммуноблот, показывающий ингибирование индуцированного излучением аутофосфорилирования киназы DNA-PK индуцированного излучением фосфорилирования KAP1, субстрата ATM, соединением 569 в ксенотрансплантатах опухоли человека плоскоклеточной карциномы головы и шеи FADU (HNSCC). На ФИГ. 4 pDNA-PK и pKAP1 указывают фосфорилированные формы DNA-PK и KAP1 соответственно.
ФИГ. 5 представляет собой иммуноблот, показывающий ингибирование радиационно-индуцированного аутофосфорилирования киназы DNA-PK и радиационно-индуцированного фосфорилирования KAP1, субстрата АТМ, соединением 569 в ксенотрансплантатах опухоли человека карциномы молочной железы MDA-MB-231. На ФИГ. 5 pDNA-PK и pKAP1 указывают фосфорилированные формы DNA-PK и KAP1 соответственно.
ФИГ. 6 иллюстрирует дозирование модели мыши с подкожным ксенотрансплантатом человека FADU с соединением 569 и/или IR, qd × 3. Показан средний относительный объем опухоли с течением времени для каждой группы в исследовании. На ФИГ. 6, “569” означает соединение 569, “Veh.” означает несущую среду и "Rad." означает облучение.
ФИГ. 7 представляет пятикратную выживаемость без пробы Каплана-Мейера для каждой группы, получавшей в модели мыши с подкожным ксенотрансплантатом человека FADU соединение 569 и/или IR, qd × 3. На ФИГ. 7, “569” означает соединение 569, “Veh.” означает несущую среду и "Rad." означает облучение.
Фигура 8 иллюстрирует дозирование модели мыши с подкожным ксенотрансплантатом человека MDA-MB-231 с соединением 569 и/или IR, qd × 3. Показан средний относительный объем опухоли с течением времени для каждой группы в исследовании. На ФИГ. 8, “569” означает соединение 569, “Veh.” означает несущую среду и "Rad." означает облучение.
ФИГ. 9 представляет данные о выживании без пяти выборок Каплана-Мейера для каждой группы, получавшей в модели мыши с подкожным ксенотрансплантатом человека MDA-MB-231 соединение 569 и/или IR, qd × 3. На ФИГ. 9, “569” означает соединение 569, “Veh.” означает несущую среду и "Rad." означает облучение.
СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ
В изобретении предложены соединения и композиции, которые могут быть полезны при лечении онкологических заболеваний (например, рака, например, рака, описанного в данном документе), например, отдельно или в сочетании с лучевой терапией и/или противоопухолевой терапией. Соединение может быть соединением формулы (I):
,
(I)
или его фармацевтически приемлемая соль,
где
Z представляет собой СH, CR3 или N;
Y представляет собой СHR5 или NR6;
n равно 0, 1, 2 или 3;
R1 представляет собой -O-L-N(R7)2 или необязательно замещенный четырехчленный насыщенный N-гетероциклил;
R2 представляет собой С1-3 aлкил;
каждый R3 независимо представляет собой галоген или необязательно замещенный C1-3 aлкил;
R4 представляет собой необязательно замещенный aлкил;
R5 представляет собой водород, необязательно замещенный C1-3 aлкил или бензилокси;
R6 представляет собой необязательно замещенный C1-3 aлкил;
каждый R7 независимо представляет собой H или необязательно замещенный C1-3 aлкил; и
L представляет собой необязательно замещенный этилен.
Преимущественно соединения по изобретению (например, соединение 568, 569 или 570) могут проявлять превосходную ингибирующую активность в отношении ATM и DNA-PK. Преимущественно соединения по данному изобретению (например, соединение 568, 569 или 570) могут демонстрировать превосходную селективность, измеряемую по уменьшенной нецелевой активности (например, ингибирование mTOR, ингибирование PI3K α/δ и/или ингибирование hERG). Например, соединение по данному изобретению (например, соединение 568, 569 или 570) может иметь mTOR IC50 по меньшей мере в 10 раз (например, по меньшей мере в 20 раз) выше, чем IC50 ATM или IC50 DNA-PK. Соединение по изобретению (например, соединение 568, 569 или 570) может иметь mTOR IC50 10 нМ или больше (например,> 100 нМ). Дополнительно или альтернативно, соединение по изобретению (например, соединение 568, 569 или 570) может иметь hERG IC50 по меньшей мере в 100 раз (например, по меньшей мере в 500 раз, по меньшей мере в 1000 раз или по меньшей мере в 3000 раз) больше чем IC50 ATM или IC50 DNA-PK при измерении при той же концентрации соединения. Соединение по изобретению (например, соединение 568, 569 или 570) может иметь IC50 hERG 3 мкМ или больше (например, 10 мкМ или больше).
Преимущественно соединения по изобретению (например, соединение 568, 569 или 570) могут проявлять превосходные фармакокинетические свойства (например, Cmax, AUC и/или t1/2).
В некоторых вариантах реализации соединение выбрано из группы, состоящей из:
Соединения по настоящему изобретению имеют то преимущество, что они могут ингибировать ATM (атаксия-телеангиэктазия, мутированные) и DNA-PK киназы. ATM (мутированная при атаксии-телеангиэктазии) и DNA-PK-киназы, в частности, являются важными модуляторами клеточных ответов на разрывы в ДНК, а ингибирование любой из этих молекул заметно повышает чувствительность клеток к ионизирующему излучению. Таким образом, соединения по данному изобретению могут быть эффективными ингибиторами действия ATM и DNA-PK с или без излучения и с химиотерапией или иммунотерапией или без нее, чтобы обеспечить эффективную терапию для лечения онкологических заболеваний (например, рака, например, этих видов рака, что описаны в данном документе). Лечение пациента соединением по изобретению может отсрочить или устранить восстановление повреждений ДНК с помощью лучевой терапии. В результате пациенты, получающие соединение по данному изобретению, могут лучше реагировать на противоопухолевую терапию. Преимущественно пациенты, получающие соединение по изобретению, могут получить терапевтический эффект за счет увеличения контроля над опухолью от стандартных доз лучевой терапии или за счет достижения аналогичных уровней контроля над опухолью от более низких доз ионизирующего излучения, чем обычно используются у пациентов, не получающих соединение по изобретению. Преимущественно более низкие дозы ионизирующего излучения могут быть менее повреждающими для незлокачественных тканей, чем дозы, необходимые для пациентов, не получающих соединение по данному изобретению.
Люди и мыши, имеющие мутации с потерей функции в генах ATM или PRKDC, которые кодируют киназу, мутированную при атаксии-телеангиэктазии (ATM), и ДНК-зависимую протеинкиназу (DNA-PK), соответственно, являются гиперчувствительными к ионизирующему излучению. Совместное ингибирование киназ ATM и DNA-PK может быть эффективным для сенсибилизации опухолевых клеток к радиации или повреждающим ДНК агентам (например, противоопухолевым агентам). Эффективность двойного ингибирования киназ ATM и DNA-PK может превосходить ингибирование любой киназы отдельно.
Кроме того, соединения по данному изобретению могут преимущественно проявлять пониженное ингибирование других киназ (ATR и mTOR) и, таким образом, могут проявлять пониженную токсичность.
Соединения по данному изобретению могут повышать чувствительность опухолевых клеток к радиации и/или противоопухолевым агентам.
Способы
В другом аспекте изобретение обеспечивает способы лечения онкологического заболевания (например, рака) у млекопитающего, предпочтительно человека или собаки, где способы включают введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению. . В некоторых вариантах осуществления соединение вводят млекопитающему, проходящему лучевую терапию.
В другом аспекте изобретение обеспечивает способы лечения онкологического заболевания (например, рака) у млекопитающего, при этом способы включают введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению. В некоторых вариантах осуществления соединение вводят млекопитающему в комбинации с повреждающим ДНК агентом. Неограничивающие примеры повреждающих ДНК агентов включают цисплатин, оксалиплатин, карбоплатин, валрубицин, идарубицин, калихеамицин, ингибиторы PARP.
В другом аспекте в данном изобретении предложены фармацевтические композиции, содержащие соединения по изобретению и фармацевтически приемлемые вспомогательные вещества. В одном варианте реализации фармацевтическая композиция содержит соединение по изобретению в фармацевтически приемлемом носителе и в количестве, эффективном для лечения онколигитального заболевания у животного, предпочтительно млекопитающего.
Соединение по данному изобретению, при использовании в комбинированной терапии, может увеличивать эффективность другой лучевой или лекарственной терапии, если оно позволяет снизить дозу другого лечения, что может снизить частоту и/или тяжесть побочных явлений, связанных с другой лекарственной терапией. Например, побочные эффекты лучевой терапии (например, мукозит полости рта или желудочно-кишечного тракта, дерматит, пневмонит или утомляемость) могут быть уменьшены у пациентов, получающих комбинированную терапию, включающую соединение по данному изобретению, и лучевую терапию с уменьшенной дозой (например, частота нежелательных явлений может быть сниженным по крайней мере на 1%, 5%, 10% или 20%) по сравнению с пациентами, получающими стандартную полную дозу лучевой терапии без соединения по данному изобретению. Кроме того, другие нежелательные явления, которые могут быть уменьшены у пациентов, получающих комбинированную терапию, включающую соединение по данному изобретению и лучевую терапию с уменьшенной дозой (например, частота нежелательных явлений может быть снижена по крайней мере на 1%, 5%, 10% или 20%) по сравнению с пациентами, получающими стандартную полную дозу лучевой терапии без соединения по данному изобретению, могут быть поздние эффекты излучения, например, радиационно-индуцированный фиброз легких, сердечное повреждение, непроходимость кишечника, повреждение нервов, сосудистое повреждение, лимфедема, некроз мозга или индуцированный облучением рак. Аналогично, когда соединение вводят в комбинации с другим противораковым лекарственным препаратом (например, одним из описанных в данном документе), комбинированная терапия может приводить к такой же или даже повышенной гибели опухолевых клеток даже при снижении дозы другого противоракового лекарственного препарата. Таким образом, снижение дозировок других противораковых лекарственных препаратов может снизить тяжесть нежелательных явлений, вызванных другими противораковыми лекарственными препаратами.
В другом аспекте данное изобретение относится к применению соединений по изобретению, описанных выше, их стереоизомеров, энантиомеров, таутомеров или их смесей, или их фармацевтически приемлемых солей или сольватов, или к применению фармацевтической композиции, содержащей фармацевтически приемлемое вспомогательное вещество и соединение по изобретению, описанное выше, его стереоизомер, энантиомер, таутомер или их смеси, или его фармацевтически приемлемые соль или сольват, при изготовлении лекарственного средства для применения при лечении заболевания. В некоторых вариантах осуществления соединение по изобретению вводят в комбинации с лучевой терапией. В других вариантах осуществления соединение по изобретению вводят в комбинации с повреждающим ДНК агентом. В дополнительных вариантах реализации соединение по изобретению вводят в комбинации с противоопухолевым иммунотерапевтическим агентом (например, ипилимумаб, офатумумаб, ниволумаб, пембролизумаб, атезолизумаб, авелумаб, дурвалумаб, цемиплимаб, обинутузумаб, окаратузумаб, тремелимумаб, спартализумаб, камрелизумаб, синтилимаб, тислелизумаб, торипалимаб, достарлимаб, велтузумаб, INCMGA00012, AMP-224, AMP-514, KN035, CK-301, AUNP12, CA-170 или BMS-986189). В других вариантах реализации противоопухолевый иммунотерапевтический агент представляет собой офатумумаб, обинутузумаб, окаратузумаб или велтузумаб. В еще других вариантах реализации противоопухолевый иммунотерапевтический агент представляет собой ниволумаб, пембролизумаб, цемиплимаб, спартализумаб, камрелизумаб, синтилимаб, тислелизумаб, торипалимаб, достарлимаб, INCMGA00012, AMP-224 или AMP-514. Как и в предыдущих других вариантах реализации противоопухолевый иммунотерапевтический агент представляет собой атезолизумаб, авелумаб, дурвалумаб, KN035, CK-301, AUNP12, CA-170 или BMS-986189. В определенных предпочтительных вариантах реализации комбинированной терапии соединение по данному изобретению вводят в сочетании с противоопухолевым иммунотерапевтическим средством.Способы по изобретению можно использовать при лечении онкологического заболевания, как описано в данном документе. Онкологическим заболеванием может быть, например, предраковая опухоль или злокачественная опухоль (например, солидная опухоль или жидкая опухоль). Злокачественные опухоли обычно называют раком. В определенных вариантах реализации онкологическое заболевание представляет собой Рак.
В дополнительных вариантах реализации примеры рака, который необходимо лечить с использованием раскрытых в данном документе способов и применений, включают, но не ограничиваются ими, гематологические раковые заболевания, например лейкемии и лимфомы. Неограничивающие примеры рака включают острый миелогенный лейкоз, острый лимфобластный лейкоз, острый мегакариоцитарный лейкоз, промиелоцитарный лейкоз, эритролейкоз, лимфобластный Т-клеточный лейкоз, хронический миелоидный лейкоз, хронический лимфоцитарный лейкоз, лейкоз ворсистых клеток, хронический нейрофильный лейкоз. плазмацитома, иммунобластный крупноклеточный лейкоз, лейкоз из мантийных клеток, множественные миеломы, злокачественная лимфома, диффузная крупноклеточная лимфома, лимфома Ходжкина, неходжкинская лимфома, лимфобластная Т-клеточная лимфома, лимфома Беркитта и фолликулярная лимфома.
В еще дополнительных вариантах реализации примеры рака, который необходимо лечить с использованием раскрытых в данном документе способов и применений, включают, но не ограничиваются ими, солидные опухоли. Неограничивающие примеры солидных опухолей включают рак головного мозга (например, астроцитому, глиому, глиобластому, медуллобластому или эпендимому), рак мочевого пузыря, рак груди, рак центральной нервной системы, рак шейки матки, рак толстой кишки, рак эндометрия, рак пищевода, гастроинтестинальную стромальную опухоль, рак желудка, рак головы и шеи, буккальный рак, рак ротовой полости, гепатоцеллюлярный рак, рак легких, меланому, карциному из клеток Меркеля, мезотелиому, рак носоглотки, нейробластому, остеосаркому, рак яичников, рак поджелудочной железы, рак предстательной железы, рак почек, рак слюнных желез, саркомы, рак яичек, уротелиальный рак, рак вульвы и опухоль Вильма. Предпочтительно способы по настоящему изобретению используются при лечении рака легких, рака головы и шеи, рака поджелудочной железы, рака прямой кишки, глиобластомы, гепатоцеллюлярной карциномы, холангиокарциномы, метастических поражений печени, меланомы, саркомы кости, саркомы мягких тканей, рака эндометрия, рака шейки матки, рака простаты или карциномы из клеток Меркеля.
В других дополнительных вариантах осуществления примеры онкологических заболеваний, подлежащих лечению с помощью способов и применений, описанных в данном документе, включают, но не ограничиваются этим, метастазы и метатстатический рак. Например, описанные в данном документе способы и применения для лечения онкологического заболевания могут включать лечение как первичных опухолей, так и метастазов.
В некоторых вариантах реализации способы по данному изобретению могут уменьшить размер опухоли у субъекта, например, по меньшей мере, на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или 90%, или могут устранить опухоль (например, относительно размера опухоли на момент начала терапии или относительно контрольного субъекта, который получает плацебо вместо соединения изобретения). В некоторых вариантах реализации способы по данному изобретению могут снизить опухолевую нагрузку у субъекта, например, по меньшей мере, на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или 90%, или могут устранить опухоль (например, относительно опухолевой нагрузки на момент начала терапии или относительно контрольного субъекта, который получает плацебо вместо соединения изобретения). В некоторых вариантах реализации способы по данному изобретению могут увеличивать среднее время выживания субъекта, например, по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%. %, 150% или 200% (например, относительно контрольного субъекта, который получает плацебо вместо соединения изобретения).. В некоторых вариантах реализации способы по данному изобретению могут увеличивать способность лучевой терапии или лекарственной терапии ослаблять боль или другие симптомы в течение более длительного времени для субъекта, например, по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150% или 200% (например, относительно контрольного субъекта, который получает плацебо вместо соединения изобретения).
В некоторых вариантах реализации описанные в данном документе способы и применения включают предварительное лечение субъекта двойным ингибитором ATM и DNA-PK перед применением лучевой терапии или повреждающего ДНК агента. Предварительное лечение пациента двойным ингибитором ATM и DNA-PK может отсрочить или устранить восстановление повреждений ДНК после лучевой терапии.
Лучевая терапия включает, но не ограничивается этим, наружную дистанционную лучевую терапию рентгеновским излучением (фотонами), гамма-излучением от 60кобальта или других радиоактивных изотопов, нейтронами, электронами, протонами, ионами углерода, ионами гелия и другими заряженными частицами. Лучевая терапия также включает брахитерапию и радиофармпрепараты, которые испускают гамма-лучи, альфа-частицы, бета-частицы, электроны Оже или другие типы радиоактивных частиц из изотопов, включая 32Фосфор, 67Медь, 77Бром, 89Стронций, 90Иттрий, 105Родий, 131Иод, 137Цезий, 149Прометей, 153Самарий, 166Гольмий, 177Лютеций, 186Рений, 188Рений, 199Золото, 211Aстатин, 213Bисмут, 223Радий, 225Aктиний, или 227Tорий, 192Иридий, 67Галлий, 103Палладий, 125Иод, и другие радиоактивные изотопы (например, 192Иридий, 125Иод, 137Цезий, 103Палладий, 32Фосфор, 90Иттрий, 67Галлий, 211Aстатин, или 223Радий). Лучевая терапия также включает радиоиммунотерапию (РИТ) антителами или небольшими молекулами, которые конъюгированы с радиоактивными изотопами, включая 131Иод, 90Иттрий, 225Aктиний, 211Aстатин, 67Галлий, 177Лютеций, 227Торий, и другие радиоактивные изотопы.
В некоторых вариантах реализации комбинированная терапия включает введение субъекту ингибитора ATM и DNA-PK и противоопухолевого агента, например, цисплатина, оксалиплатина, карбоплатина, ингибиторов топоизомеразы I, ингибиторов топоизомеразы II, антрациклинов, валрубицина, идарубицина, калихеамицина, ингибиторов PARP (например, олапариба, рукапариба, нирапариба, велипариба, талазопариба), а также других противораковых агентов, известных специалистам в данной области техники.
В определенных вариантах реализации комбинированная терапия включает введение субъекту ингибитора ATM и DNA-PK и противоопухолевых иммунотерапевтических агентов, включая, но не ограничиваясь ими, ипилимумаб, офатумумаб, ниволумаб, пембролизумаб, атезолизумаб, авелумаб, дурвалумаб и т. д.
В описанных в данном документе комбинированных терапиях ингибитор ATM и DNA-PK можно вводить пациенту одновременно или последовательно (например, до или после) другого лекарственного средства.
Получение соединений по изобретению
Соединения по данному изобретению можно получить с использованием способов и методик, известных в данной области. Подходящие способы синтеза этих соединений представлены в Примерах. В общем случае соединения формулы (I) можно получать в соответствии с описанными ниже схемами. Также описаны источники исходных материалов для этих реакций.
При получении соединений по изобретению можно добавлять или удалять защитные группы в соответствии со стандартными методиками, которые известны специалисту в данной области техники, и как описано в данном документе. Использование защитных групп подробно описано в Greene, T.W.и P.G.M. Wuts, Greene's Protective Groups in Organic Synthesis (2006), 4th Ed., Wiley. Защитная группа также может представлять собой полимерную смолу, такую как смола Ванга или 2-хлоротритил-хлоридная смола.
Также специалистам в данной области техники понятно, что хотя такие защищенные производные соединений по данному изобретению могут не обладать фармакологической активностью как таковой, их можно вводить млекопитающему, после чего они будут метаболизироваться в организме с образованием соединений по изобретению, которые являются фармакологически активными.
Все соединения, описанные ниже как полученные, которые могут существовать в форме свободных основания или кислоты, можно превращать в их фармацевтически приемлемые соли путем обработки соответствующими неорганическими или органическими основанием или кислотой. Соли соединений, полученных ниже, можно превращать в форму свободных основания или кислоты с помощью стандартных методик. Следует понимать, что все полиморфные формы, аморфные формы, ангидраты, гидраты, сольваты и соли соединений по изобретению, включены в объем данного изобретения. Кроме того, все соединения по изобретению, которые содержат кислотную или сложноэфирную группу, можно превращать в соответствующие сложный эфир или кислоту, соответственно, способами, известными специалисту в данной области техники, или способами, описанными в данном документе.
Общая схема получения многих из этих соединений приведена ниже на схеме 1. Соединения получают путем сочетания различных компонентов молекулы: Сочетание Сузуки галогензамещенного соединения 3 (или 2’) с бороновой кислотой или боратным соединением 2 (3’). Для завершения синтеза соединений по данному изобретению могут быть нужны дополнительные реакции или нет. Получение конкретных соединений по этому изобретению проиллюстрировано на нижеприведенных схемах.
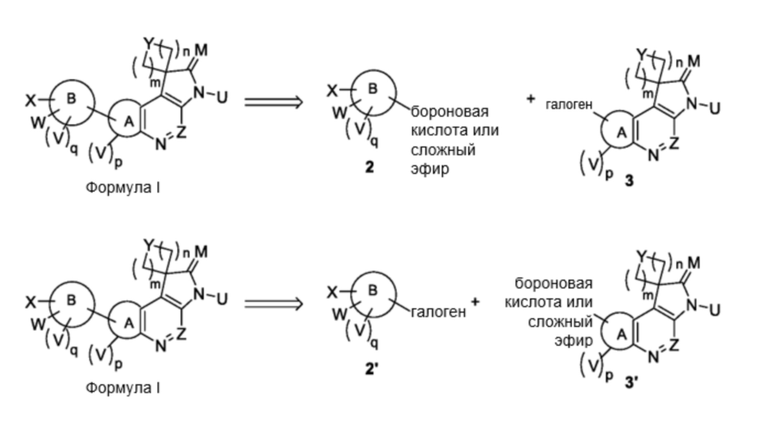
Схема 1
В арил-арильных реакциях сочетания галоген может представлять собой йод, бром или хлор, предпочтительно бром или йод. В этом способе замещения галогенами можно преобразовывать в замещения арилами, используя условия для реакции сочетания Сузуки. Условия этого способа описаны во многих публикациях, обзор которых был сделан A. Suzuki в статье под названием “The Suzuki reaction with arylboron compounds in arene chemistry” в Modern Arene Chemistry 2002, 53-106. При проведении этой реакции можно использовать любые подходящие условия, общепринятые для реакции Сузуки. В общем случае реакции сочетания Сузуки проводят в присутствии катализатора из переходного металла, такого как палладиевый катализатор, используя любой традиционный для этой реакции органический растворитель и слабое неорганическое или органическое основание. К предпочтительным органическим растворителям относятся полярные апротонные растворители. При получении соединений по изобретению можно использовать любые традиционные полярные апротонные растворители. Подходящие растворители являются общепринятыми, в особенности высококипящие растворители, например, диметоксиэтан. Слабое неорганическое основание может представлять собой карбонат или бикарбонат, например, карбонат калия или карбонат цезия. Органическое основание может представлять собой амин, такой как триэтиламин.
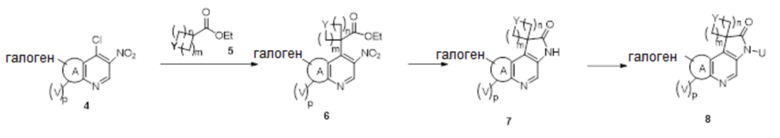
Схема 2
В частности, другое спирооксиндольное промежуточное соединение 7 синтезируют, как проиллюстрировано на схеме 2. Циклический или гетероциклический замещенный сложный эфир 5 обрабатывают сильным основанием, таким как, но не ограничиваясь этим, диизопропиламид лития, при низкой температуре в безводном растворителе, таком как, но не ограничиваясь этим, тетрагидрофуран, для проведения реакции с исходным материалом 4, который может быть приобретен на коммерческой основе или получен специалистами в данной области техники по описанным в литературе способам, с получением промежуточного соединения 6. Промежуточное соединение 6 восстанавливают восстанавливающим агентом, таким как, но не ограничиваясь этим, железо, с получением соответствующего промежуточного амино-соединения, которое циклизуется с получением оксиндольного соединения 7 in situ. Таким образом, соединение 7 затем N-алкилируют алкилирующим реагентом в присутствии основания, такого как, но не ограничиваясь ими, карбонат калия или гидрид натрия, в полярном растворителе, таком как, но не ограничиваясь ими, N, N-диметилформамид или тетрагидрофуран с образованием промежуточного спирооксиндола 8.
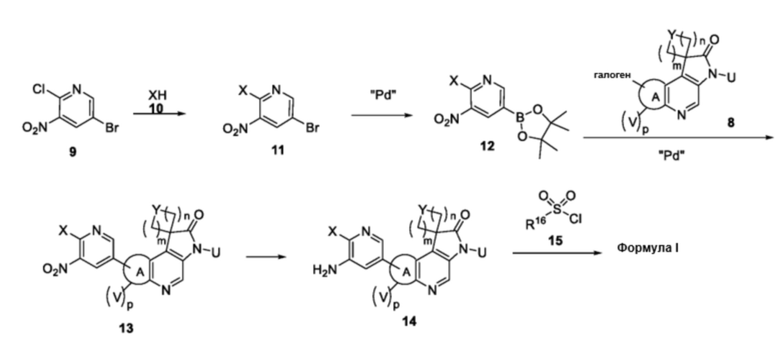
Схема 3
В частности, соединения формулы (I) в данном изобретении можно синтезировать, как проиллюстрировано на схеме 3. Коммерчески доступный 5-бром-2-хлор-3-нитропиридин (9) вступает в реакцию с нуклеофилом XH (10) в присутствии сильного основания, такого как, но не ограничиваясь этим, гидрид натрия, с получением промежуточного соединения 11. В условиях применения палладиевого катализатора можно получать борат 12 , который впоследствии приводят в реакцию с промежуточным спиро-соединением 8 с получением продукта перекрестного сочетания 13. Нитро-группу в соединении 13 восстанавливают до амино-группы, используя восстанавливающий реагент, такой как, но не ограничиваясь этим, железо, с получением промежуточного соединения 14. Реакция 14 с разными сульфонилхлоридами (15) завершает синтез соединений формулы (I).

Схема 4
В частности, соединения формулы (I) в данном изобретении также можно синтезировать, как проиллюстрировано на схеме 4. Нитро-группу в соединении 11 восстанавливают до амино-группы, используя восстанавливающий реагент, такой как, но не ограничиваясь этим, железо, с получением промежуточного соединения 16. Реакция 16 с разными сульфонилхлоридами (15) обеспечивает получение сульфонамидного промежуточного соединения 17, которое превращается в соответствующий борат 18 в условиях применения палладиевого катализатора. Борат 18 можно сочетать с галоген-содержащим соединением 8 в условиях реакции Сузуки с получением соединений формулы (I).
На схеме 3 и схеме 4перекрестно-связанные соединения синтезируют также, используя химическую реакцию сочетания Сузуки с компонентами, имеющими обратный порядок замещения галогена и бороната/бороновой кислоты, например, как проиллюстрировано на схеме 5
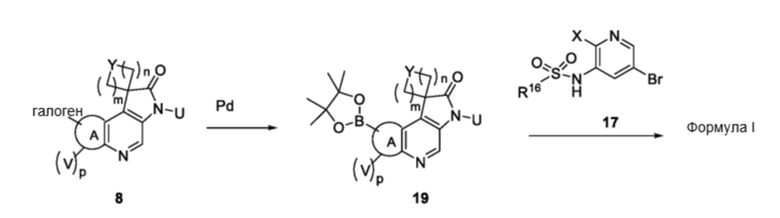 Схема 5
Схема 5
В частности, соединения формулы (I) в данном изобретении также можно синтезировать, как проиллюстрировано на схеме 5. Галоген-содержащее соединение 8 можно превращать в соответствующий борат 19 в условиях применения палладиевого катализатора. Борат 19 можно сочетать с галоген-содержащим 17 в условиях реакции Сузуки с получением соединений формулы (I).
При практической реализации способов по настоящему изобретению эффективное количество любого из соединений по данному изобретению или комбинации любых соединений по данному изобретению или их фармацевтически приемлемых солей вводят с помощью любого из обычных и приемлемых способов, известных в данной области техники, по отдельности или в комбинации. Таким образом, соединения или композиции можно вводить перорально (например, в полость рта), подъязычно, парентерально (например, внутримышечно, внутривенно или подкожно), ректально (например, посредством суппозиториев или промываний), трансдермально (например, посредством электропорации кожи) или путем ингаляции (например, посредством аэрозоля), и в виде твердых, жидких или газообразных дозированных форм, включая таблетки и суспензии. Введение можно проводить в единичной дозированной форме посредством непрерывной терапии или в виде терапии с применением единичных доз ad libitum. Терапевтическая композиция также может иметь форму масляной эмульсии или дисперсии в сочетании с лиофильной солью, такой как соль памовой кислоты, или форму биоразлагаемой композиции с замедленным высвобождением для подкожного или внутримышечного введения.
Применимые фармацевтические носители для получения композиций могут представлять собой твердые вещества, жидкости или газы; таким образом, композиции могут иметь форму таблеток, пилюль, капсул, суппозиториев, порошков, покрытых энтеросолюбильным слоем или иным образом защищенных лекарственных форм (например, с применением связывания на ионообменных смолах или упаковки в липидно-белковые везикулы), лекарственных форм с замедленным высвобождением, растворов, суспензий, эликсиров, аэрозолей и т. п. Носитель может быть выбран из различных масел, включая нефтяные масла, масла животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т. п. Вода, солевой раствор, водный раствор декстрозы и гликоли являются предпочтительными жидкими носителями, в частности (при изотоничности с кровью) для инъекционных растворов. Например, составы для внутривенного введения содержат стерильные водные растворы активного(ых) ингредиента(ов), которые готовят путем растворения твердого(ых) активного(ых) ингредиента(ов) в воде с получением водного раствора и придания раствору стерильности. Подходящие фармацевтические вспомогательные вещества включают крахмал, целлюлозу, тальк, глюкозу, лактозу, желатин, солод, рис, муку, мел, диоксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и т. п. В композиции можно добавлять традиционные фармацевтические добавки, такие как консерванты, стабилизирующие агенты, смачивающие или эмульгирующие агенты, соли для регуляции осмотического давления, буферы и т. п. Подходящие фармацевтические носители и их составление описаны в Remington's Pharmaceutical Sciences авторства E. W. Martin. В любом случае такие композиции будут содержать эффективное количество активного соединения вместе с подходящим носителем так, чтобы приготовить надлежащую дозированную форму для надлежащего введения реципиенту.
Доза соединения по данному изобретению зависит от ряда факторов, таких как, например, способ введения, возраст и масса тела пациента, а также состояния пациента, подлежащего лечению, и в конечном итоге будет решаться лечащим врачом или ветеринаром. Такое количество активного соединения, определенное лечащим врачом или ветеринаром, называется в данном документе и в формуле изобретения «эффективным количеством».
Данное изобретение будет дополнительно описано в нижеприведенных примерах, которые предназначены исключительно в качестве иллюстрации и не ограничивают объем изобретения.
ПРИМЕРЫ
Реагенты были приобретены у Aldrich, Sigma, TCI (Shanghai) Development, Chembon Pharmaceutical Co., Ltd, Zhangjiagang Aimate Huaxue Youxiangongsi, Changzhou Qinuo BioTech Co. Ltd, and Shanghai Weiyuan Fine Fluorine Technology Development Co., Ltd или других поставщиков, как указано ниже, и используемых без дополнительной очистки. Реакции с применением микроволнового излучения для нагревания проводили, используя Biotage Initiator+. Очистку в масштабе от миллиграмов до граммов проводили способами, известными специалистам в данной области техники, такими как элюирование методом колоночной флэш-хроматографии на силикагеле; очистку методом препаративной колоночной флэш-хроматографии также проводили в некоторых случаях, используя одноразовые предварительно упакованные силикагелевые колонки (Welch/Agela) с элюированием с помощью системы Biotage CombiFlash.
В целях определения идентичности и чистоты соединения обычно использовали систему для аналитической ЖХ-МС (жидкостной хроматографии/масс-спектроскопии) на основе платформы Waters ZQTM с ионизацией электрораспылением в режиме детектирования положительных ионов с помощью ВЭЖХ с автодозатором Agilent 1100. Колонка обычно представляла собой Water Xterra MS C18, 3,0 × 50 мм, 5 мкм. Скорость потока составляла 1 мл/мин, а объем вводимой пробы составлял 10 мкл. УФ-детектирование проводили в диапазоне 210-400 нм. Подвижная фаза состояла из растворителя A (вода плюс 0,06% TФУ) и растворителя B (ацетонитрил плюс 0,05% TФУ) с градиентом 100% растворителя A в течение 0,7 мин, с заменой на 100% растворитель B в течение 3,75 мин, поддержанием в течение 1,1 мин, с последующей обратной заменой на 100% растворитель A в течение 0,2 мин..
В некоторых случаях разделения также может быть полезно использовать сверхкритическую жидкостную хроматографию. Разделение с помощью сверхкритической жидкостной хроматографии проводили с использованием системы Mettler-Toledo Minigram со следующими типичными условиями: 100 бар, 30 °C, 2,0 мл/мин, элюируя колонку AD 12 мм 40% MeOH в сверхкритическом флюиде CO2. В случае аналитов с кислотными амино-группами в метаноловый модификатор добавляли 0,2% изопропиламин.
Многие соединения Формулы (I) также очищали обращенно-фазовой ВЭЖХ с использованием методов, хорошо известных специалистам в данной области. В некоторых случаях очистку препаративной ВЭЖХ проводили с использованием масс-спектрометра PE Sciex 150 EX, контролирующим коллектор Gilson 215, присоединенный к системе препаративной ВЭЖХ Shimadzu, и автоинжектор Leap. Соединения собирали из потока элюирования с использованием МС-детекции при детектировании положительных ионов: На элюцию соединений из колонок C-18 (2,0 × 10 см, элюирование при 20 мл/мин) влияли соответствующие режимы линейной градации в течение 10 минут: растворитель (A) 0,05% TФУ/H2O и растворитель (B) 0,035% TФУ/ацетонитрил. Для ввода в ВЭЖХ-системы неочищенные образцы растворяли в смеси метанола, ацетилнитрила и ДМСО.
Характеристики соединений получали с помощью 1H-ЯМР, используя спектрометр Bruker ADVANCE III HD (400 МГц) или спектрометр AVANCE (300 МГц).
ПЕРЕЧЕНЬ СОКРАЩЕНИЙ
DCE 1,2-дихлорэтан
DCM дихлорметан
DIPEA диизопропилэтиламин
DMF N, N-диметилформамид
DMSO диметилсульфоксид
EtOAc этилацетат
HOAc Уксусная кислота
HPLC жидкостная хроматография высокого давления
MeI йодистый метил
MeOH метиловый спирт
MW микроволновая печь
NMP 1-метил-2-пирролидинон
rt температура окружающей среды
TBDMS трет-бутил-диметилсилил
TEA триэтиламин
TFA трифторуксусная кислота
TEMPO 2,2,6,6-тетраметил-1-пиперидинилокси
THF тетрагидрофуран
ПРИГОТОВЛЕНИЕ СОЕДИНЕНИЙ
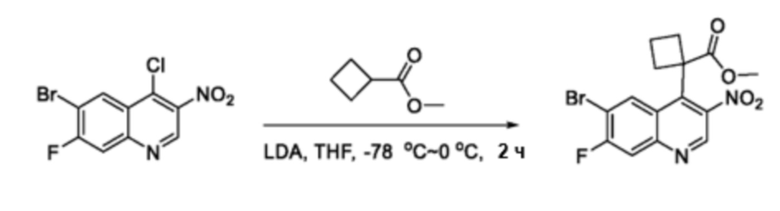
Метил 1-(6-бром-7-фтор-3-нитрохинолин-4-ил)циклобутан-1-карбоксилат: Раствор метилциклобутанкарбоксилата (0,73 г, 6,38 ммоль) в тетрагидрофуране (5,00 мл) обрабатывали свежеприготовленным диизопропиламидом лития (6,38 ммоль) в тетрагидрофуране (45,0 мл) в течение 1 часа при -78°C в атмосфере азота с последующим добавлением 6-бром-4-хлор-7-фтор-3-нитрохинолина (1,50 г, 4,91 ммоль) порциями в течение 2 мин. После перемешивания в течение дополнительного 1 часа при температуре окружающей среды реакцию гасили насыщенным водным раствором хлорида аммония (60,0 мл) и разбавляли водой (120 мл). Полученную в результате смесь экстрагировали этилацетатом (3×60,0 мл). Объединенные органические слои промывали солевым раствором (2×50,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 1% ~ 2% этилацетатом в петролейном эфире, с получением указанного в заголовке соединения в виде бесцветного твердого вещества (240 мг, 13%): 1H ЯМР (400 МГц, CDCl3) δ 9,14 (с, 1H), 8,20 (д, J=7,2 Гц, 1H), 7,90 (д, J=8,8 Гц, 1H), 3,84 (с, 3H), 3,12-2,99 (м, 1H), 2,58-2,48 (м, 3H), 1,91-1,83 (м, 1H), 1,45-1,27 (м, 1H); МС: [(M+1)]+=383.17, 385.17.

8'-бром-7'-фторспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: Смесь метилового эфира 1-(6-бром-7-фтор-3-нитрохинолин-4-ил) циклобутан-1-карбоксилата (240 мг, 0,63 ммоль) и порошка железа (350 мг, 6,26 ммоль) в уксусной кислоте (10,0 мл) перемешивали в течение 18 часов при температуре окружающей среды. Полученную смесь фильтровали, и осадок на фильтре промывали этилацетатом (5 х 100 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 1% ~ 2% метанолом в дихлорметане, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (100 мг, 50%): 1H ЯМР (400 МГц, ДМСО-d6) δ 10,75 (с, 1H), 8,68 (с, 1H), 8,52 (д, J=7,5 Гц, 1H), 7,98 (д, J=10,1 Гц, 1H), 2,90 -2,75 (м, 2H), 2,50-2,37 (м, 4H); MС: [(M+1)]+=321,15, 323,15.
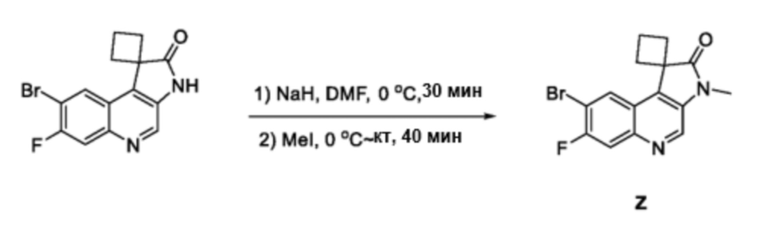
8'-бром-7'-фтор-3-метилспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: Раствор 8-бром-7-фтор-2,3-дигидроспиро[циклобутан-1,1-пирроло[2,3-c]хинолин]-2-она (100 мг, 0,31 ммоль) в N, N-диметилформамиде (10,0 мл) обрабатывали гидридом натрия (19,9 мг, 0,50 ммоль, 60% диспергировано в минеральном масле) при 0°C в течение 30 минут в атмосфере азота с последующим добавлением йодметана (66,3 мг, 0,47 ммоль). После перемешивания в течение дополнительных 40 мин при температуре окружающей среды реакцию гасили насыщенным водным раствором хлорида аммония (10,0 мл). Полученную в результате смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×30,0 мл). Объединенные органические слои промывали солевым раствором (2×20,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью преп-TСХ (ДХM/MeOH=20/1, об:об) с получением указанного в заголовке соединения в виде бесцветного твердого вещества (102 мг, 98%): 1H МР (400 МГц, CD3OD) δ 8,78 (с, 1H), 8,61 (д, J=7,4 Гц, 1H), 7,85 (д, J=9,8 Гц, 1H), 3,36 (с, 3H), 2,94-2,85 (м, 2H), 2,72-2,61 (м, 3H), 2,56-2,48 (м, 1H); MС: [(M+1)]+=335,00, 337,00.
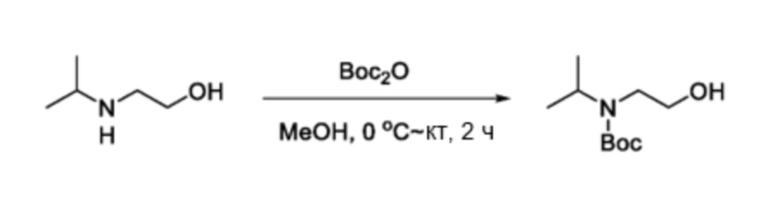
трет-Бутил N-(2-гидроксиэтил)-N-(пропан-2-ил)карбамат: К раствору 2-[(пропан-2-ил)амино]этан-1-ола (40,0 г, 388 ммоль) в метаноле (300 мл) по каплям добавляли ди-трет-бутилдикарбонат (127 г, 586 ммоль) при 0 0С. Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды и упаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0% ~ 4% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (65,0 г, 82%): 1H ЯМР (400 МГц, CDCl3) δ 4,17 (с, 1H), 3,71 (т, J=5,4 Гц, 2H), 3,30 (т, J=5,4 Гц, 2H), 1,47 (с, 9H), 1,12 (д , J=6,8 Гц, 6H).

трет-Бутил N-[2-[(5-бром-3-нитропиридин-2-ил)окси]этил]-N-(пропан-2-ил)карбамат: Раствор трет-бутил-N-(2-гидроксиэтил)-N-(пропан-2-ил)карбамата (15,4 г, 75,8 ммоль) в безводном тетрагидрофуране (250 мл) обрабатывали гидридом натрия (3,30 г, 82,1 ммоль, 60% масс. диспергировано в минеральном масле) в течение 1 часа при 0°C в атмосфере азота с последующим добавлением 5-бром-2-хлор-3-нитропиридина (15,0 г, 63,2 ммоль) в течение 2 минут при 0°C. Еще через 2 часа при 25°C реакцию гасили насыщенным водным хлоридом аммония (50,0 мл) и разбавляли водой (500 мл). Водный слой экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 18% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого масла (18,0 г, 71%): 1H ЯМР (400 МГц, CDCl3) δ 8,42 (д, J=2,4 Гц, 1H), 8,37 (д, J=2,4 Гц, 1H), 4,57 (т, J=6,3 Гц, 2H), 4,32 (с, 1H ), 3,51 (т, J=6,3 Гц, 2H), 1,47 (с, 9H), 1,15 (д, J=6,9 Гц, 6H); MС: [(M+1)]+=404,00, 406,00.
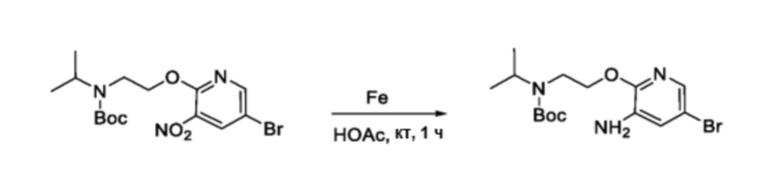
трет-Бутил N-[2-[(3-амино-5-бромпиридин-2-ил)окси]этил]-N-(пропан-2-ил)карбамат: К раствору трет-бутил-N-[2-[(5-бром-3-нитропиридин-2-ил)окси]этил]-N-(пропан-2-ил) карбамата (15,0 г, 37,1 ммоль) в уксусной кислоте. кислоты (150 мл) добавляли порошок железа (20,7 г, 371 ммоль) при температуре окружающей среды. После перемешивания в течение дополнительного 1 часа при температуре окружающей среды полученную в результате смесь фильтровали, а фильтрационный осадок промывали тетрагидрофураном (4×100 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 20% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (12,0 г, 86%): 1H ЯМР (400 МГц, CD3OD) δ 7,40 (д, J=2,2 Гц, 1H), 7,04 (д, J=2,2 Гц, 1H), 4,40 (т, J=6,3 Гц, 2H), 4,25-3,99 (м , 1H), 3,52 (т, J=6,3 Гц, 2H), 1,46 (с, 9H), 1,17 (д, J=6,8 Гц, 6H); MС: [(M+1)]+=374,10, 376,10.

трет-Бутил (2-((5-бром-3-(метилсульфонамидо)пиридин-2-ил)окси)этил)(изопропил) карбамат: К раствору трет-бутил N-[2-[(3-амино-5-бромпиридин-2-ил)окси]этил]-N-(пропан-2-ил) карбамата (18,8 г, 50,3 ммоль) в пиридине (400 мл) добавляли по каплям метансульфонилхлорид (8,63 г, 75,4 ммоль) при температуре окружающей среды. После перемешивания в течение 6 часов при температуре окружающей среды полученную в результате смесь упаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 3% ~ 25% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (16,3 г, 72%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,41 (с, 1H), 8,05 (д, J=2,2 Гц, 1H), 7,79 (д, J=2,2 Гц, 1H), 4,35 (т, J=6,3 Гц, 2H), 4,15 (с, 1H), 3,43 (т, J=6,3 Гц, 2H), 3,11 (с, 3H), 1,39 (с, 9H), 1,09 (д, J=6,8 Гц, 6H); MС: [(M+1)]+=452,00, 454,00.
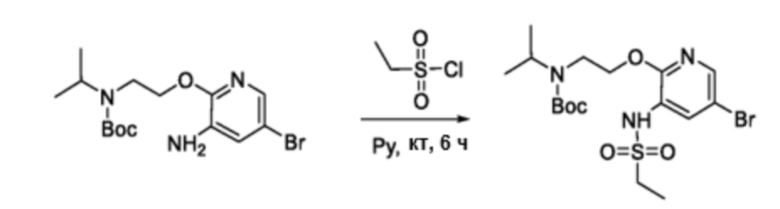
трет-Бутил (2-((5-бром-3-(этилсульфонамидо)пиридин-2-ил)окси)этил)(изопропил) карбамат: К перемешиваемому раствору трет-бутил (2-((3-амино-5-бромпиридин-2-ил) окси)этил)(изопропил)карбамата (5,00 г, 13,4 ммоль) в пиридине (120 мл) добавляли этансульфонилхлорид. (5,15 г, 40,1 ммоль) по каплям при температуре окружающей среды в атмосфере азота. После перемешивания в течение 6 часов при температуре окружающей среды в атмосфере азота полученную смесь упаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 3% ~ 25% этилацетатом в петролейном эфире. Желаемые фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (3.78 g, 61%): 1H ЯМР (400 МГц, CD3OD) δ 7,92 (с, 1H), 7,87 (с, 1H), 4,48 (т, J=6,6 Гц, 2H), 4,25-4,20 (м, 1H), 3,54 (т, J=6,6 Гц, 2H), 3,13 (т, J=7,3 Гц, 2H), 1,49 (с, 9H), 1,35 (т, J=7,4 Гц, 3H), 1,19 (д, J=6,8 Гц, 6H); MС: [(M+1)]+=466,10, 468,10.

трет-Бутил N-[2-[(5-бром-3-иодпиридин-2-ил)окси]этил]-N-изопропилкарбамат: К раствору 5-бром-3-иодпиридин-2-ола (10,0 г, 33,3 ммоль) в безводном тетрагидрофуране (300 мл) добавляли трифенилфосфин (11,4 г, 43,3 ммоль), трет-бутил N-(2-гидроксиэтил)-N-изопропилкарбамат (8,80 г, 43,3 ммоль) и диизопропилазодиформат (8,80 г, 43,4 ммоль) по каплям при 0 oC. Полученную смесь перемешивали еще 16 часов при температуре окружающей среды в атмосфере азота. Полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 2% ~ 10% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (14,0 г, 87%): 1H ЯМР (400 МГц, CDCl3) δ 8,14 (с, 2H), 4,43 (т, J=6,4 Гц, 2H), 4,38-3,96 (м, 1H), 3,50 (т, J=6,4 Гц, 2H), 1,49 (с, 9H), 1,19 (д, J=6,8 Гц, 6H); MС: [(M+1)]+=484,95, 486,95.
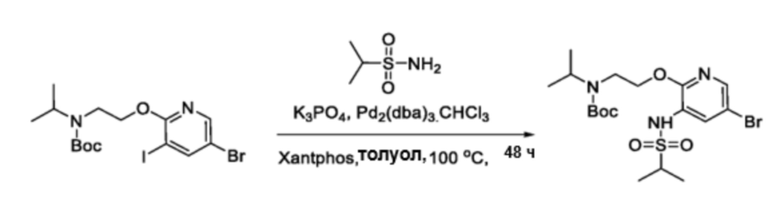
трет-Бутил (2-((5-бром-3-((1-метилэтил)сульфонамидо)пиридин-2-ил)окси)этил) (изопропил)карбамат: К смеси трет-бутил (2-((5-бром-3-иодпиридин-2-ил)окси)этил)(изопропил)карбамата (5,00 г, 10,3 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (1,80 г, 3,10 ммоль) и пропан-2-сульфонамида (1,50 г, 12,4 ммоль) в толуоле (125 мл) добавляли трикалия фосфат (10,9 г, 51,5 ммоль) и аддукт трис(дибензилиденацетон) дипалладий-хлороформ (1,10 г, 1,10 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 48 часов при 100°C в атмосфере аргона. После охлаждения до температуры окружающей среды полученную в результате смесь фильтровали. Отфильтрованный осадок промывали этилацетатом (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 20% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,90 г, 39%): 1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=2,1 Гц, 1H), 7,89 (д, J=2,2 Гц, 1H), 4,44 (т, J=6,3 Гц, 2H), 4,11 (с, 1H), 3,48 (т, J=6,3 Гц, 2H), 3,24-3,30 (м, 1H), 1,48 (с, 9H), 1,41 (д, J=6,8 Гц, 6H), 1,14 (д, J=6,8 Гц , 6H); MС: [(M+1)]+=480,20, 482,20.

N-(5-Бром-2-[2-[(пропан-2-ил)амино]этокси]пиридин-3-ил)метансульфонамид: К раствору трет-бутил N-[2-[(5-бром-3-метансульфонамидопиридин-2-ил)окси]этил]-N- (пропан-2-ил)карбамата (3,00 г, 6,63 ммоль) в дихлорметане. (5,00 мл) обрабатывали хлористым водородом (20,0 мл, 4 M в 1,4-диоксане) в течение 40 минут при температуре окружающей среды. Полученную в результате смесь концентрировали при пониженном давлении. Остаток подщелачивали до pH=8 насыщенным водным раствором бикарбоната натрия (30,0 мл). Полученную в результате смесь экстрагировали этилацетатом (6×200 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 10% метанолом в дихлорметане. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,20 г, 50%): 1H ЯМР (400 МГц, ДМСО-d6) δ 7,68 (д, J=2,3 Гц, 1H), 7,61 (д, J=2,3 Гц, 1H), 5,75 (с, 1H), 4,36 (т, J=5,2 Гц , 2H), 3,19-3,12 (м, 1H), 3,07 (т, J=5,1 Гц, 2H), 2,84 (с, 3H), 1,15 (д, J=6,4 Гц, 6H); MС: [(M+1)]+=352,10, 354,10.
Следующие промежуточные соединения получали в соответствии с описанной выше процедурой:
[(M+1)]+
368,10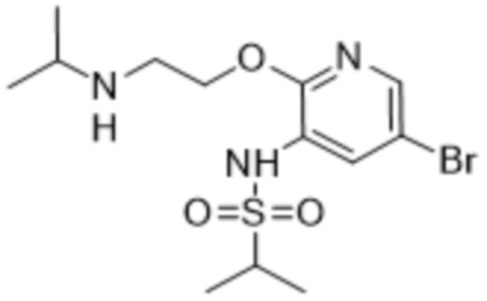
382,15
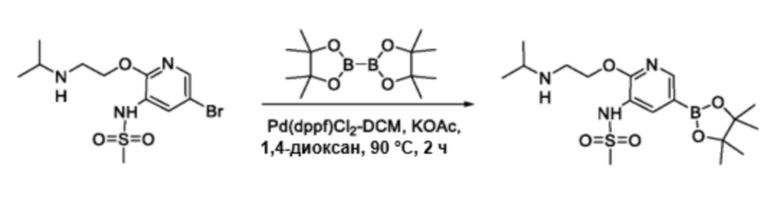
N-(2-(2-(Изопропиламино)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) пиридин-3-ил)метансульфонамид: К раствору трет-бутил N-[2-[(5-бром-3-метансульфонамидопиридин-2-ил)окси]этил]-N- (пропан-2-ил)карбамата (2,00 г, 5,68 ммоль) и бис(пинаколато)диборона (2,88 г, 11,4 ммоль) в 1,4-диоксане (50,0 мл) добавляли ацетат калия (2,23 г, 22,7 ммоль) и аддукт бис(дифенилфосфино)ферроцен]дихлорпалладий (II) дихлорметана (0,46 г, 0,57 ммоль) при температуре окружающей среды. После перемешивания в течение 2 часов при 90°C в атмосфере азота полученную в результате смесь концентрировали при пониженном давлении. Полученную смесь разбавляли дихлорметаном (100 мл). После фильтрации отфильтрованный осадок промывали дихлорметаном (3 × 10,0 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 330 г; Подвижная фаза А: Вода (плюс 10 мМ NH4HCO3), подвижная фаза B: ацетонитрил; Скорость потока: 65 мл/мин; Градиент (B%): 5%~22%, 6 мин; 22%~40%, 20 мин; 40%~95%; 2 мин; 95%, 5 мин; Детектор: УФ 254 нм; Rt: 15 мин. Фракции, содержащие желаемый продукт, собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1,57 г, 87%): 1HЯМР (400 МГц, CDCl3) δ 8,27 (д, J=1,7 Гц, 1H), 8,06 (д, J=1,7 Гц, 1H), 4,51 (т, J=5,2 Гц, 2H), 3,05-2,99 (м , 5H), 2,95-2,84 (м, 1H), 1,33 (с, 12H), 1,11 (д, J=6,3 Гц, 6H); MС: [(M+1)]+=400,30.
COЕДИНЕНИЕ 568
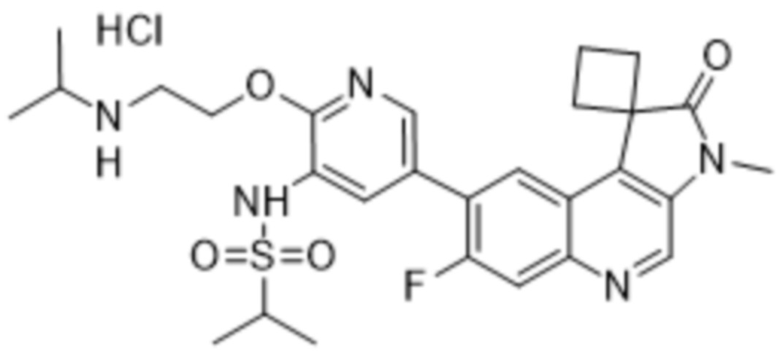
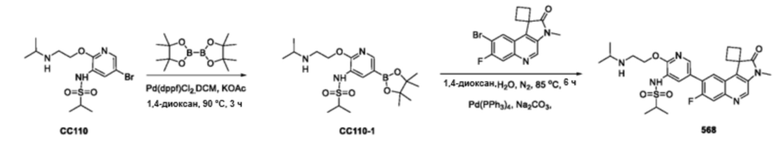
N-(5-(7'-Фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)пропан-2-сульфонамид: К раствору N-[5-бром-2-[2-(изопропиламино)этокси]пиридин-3-ил]пропан-2-сульфонамида (2,00 г, 5,26 ммоль) в 1,4-диоксане (50,0 мл) добавляли бис(пинаколато)диборон (4,01 г, 15,8 ммоль), ацетат калия (2,06 г, 21,1 ммоль) и аддукт бис(дифенилфосфино)ферроцен]дихлор палладий (II) дихлорметана (0,34 г, 0,42 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 3 часов при 90°C в атмосфере азота. Полученную смесь охлаждали до температуры окружающей среды с последующим добавлением 8-бром-7-фтор-3-метилспиро[циклобутан-1,1-пирроло[2,3-c]хинолин]-2-она (1,26 г, 3,76 ммоль), воды (12,5 мл), карбоната натрия (0,80 г, 7,55 ммоль) и тетракис (трифенилфосфин)палладий (0) (0,43 г, 0,38 ммоль). Полученную в результате смесь перемешивали в течение 6 часов при 85°C в атмосфере азота. После охлаждения до температуры окружающей среды полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 120 г; Подвижная фаза А: вода (плюс 10 мМ NH4HCO3); Подвижная фаза B: ацетонитрил; Скорость потока: 45 мл/мин; Градиент (B%): 5%, 2 мин; 5%~25%, 8 мин; 25%~39%, 9 мин; 39%, 10 мин; 39%~95%; 3 мин; 95%, 2 мин; Детектор: УФ 254 нм; Rt: 21 мин. Фракции, содержащие желаемый продукт, собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,66 г, 80%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (с, 1H), 8,35-8,31 (м, 2H), 8,07 (с, 1H), 7,99 (с, 1H), 7,96 (с, 1H), 4,55 ( уш, 1H), 4,44 (т, J=5,2 Гц, 2H), 3,35-3,30 (м, 4H), 2,93-2,79 (м, 5H), 2,55-2,38 (м, 4H), 1,29 (д, J=7,2 Гц, 6H), 1,02 (д, J=6,0 Гц, 6H); MС: [(M+1)]+=556,30.

N-(5-(7'-Фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)пропан-2-сульфонамид гидрохлорид. Раствор N-(5-[7-фтор-3-метил-2-оксоспиро[циклобутан-1,1-пирроло[2,3-c]хинолин]-8-ил]-2-[2-(изопропиламино)этокси]пиридин-3-ил)пропан-2-сульфонамида (1,66 г, 2,99 ммоль) в разбавленном водном растворе соляной кислоты (392 мл, 3,14 ммоль, 0,008 M) и ацетонитриле (79,0 мл) лиофилизировали с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (1,76 г, 100%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,45 (с, 1H), 9,02 (с, 2H), 8,90 (с, 1H), 8,40 (с, 1H), 8,32 (д, J=8,4 Гц, 1H) , 8,16 (с, 1H), 8,00 (д, J=8,4 Гц, 1H), 4,65 (т, J=4,4 Гц, 2H), 3,54-3,40 (м, 3H), 3,31 (с, 3H), 2,91 ( т, J=11,2 Гц, 2H), 2,55-2,45 (м, 5H), 1,34-1,30 (м, 12H); MС: [(M+1)]+=556,30.
Аналогичным образом на пары палладия (II) воздействовали промежуточными соединениями CC108 и CC110 с получением следующих соединений примера 569 и примера 570.
[(M+1)]+
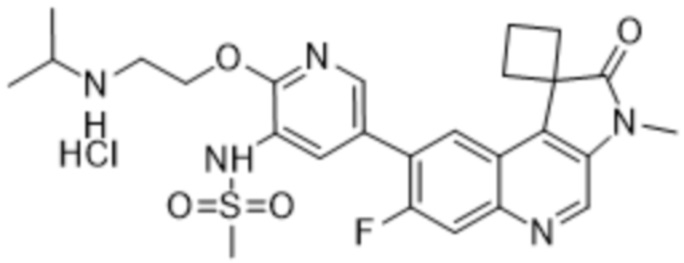
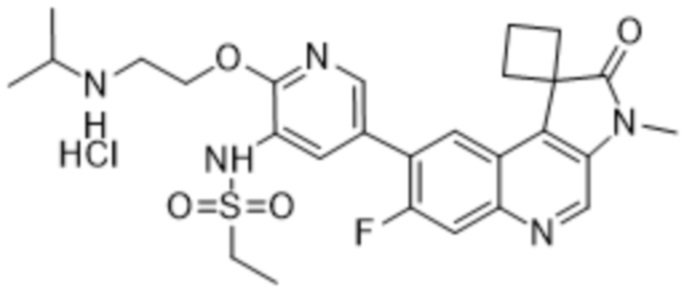
COЕДИНЕНИЕ 571

1- (трет-Бутил) 3-метил 3-(6-бром-7-фтор-3-нитрохинолин-4-ил)азетидин-1,3-дикарбоксилат: К раствору свежеприготовленного диизопропиламида лития (137 ммоль) в безводном тетрагидрофуране (110 мл) добавляли раствор 1-трет-бутил 3-метилазетидин-1,3-дикарбоксилата (29,3 г, 137 ммоль) в тетрагидрофуране (100 мл) при -78 oC. После перемешивания в течение 1 часа к реакционной смеси в течение 20 минут добавляли раствор 6-бром-4-хлор-7-фтор-3-нитрохинолина (32,0 г, 105 ммоль) в тетрагидрофуране (100 мл). Полученную смесь медленно нагревали до 0°C в течение 2 часов. Реакцию гасили насыщенным водным раствором хлорида аммония (20,0 мл) и разбавляли водой (800 мл). Полученную в результате смесь экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 5-20% этилацетатом в петролейном эфире, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (25,0 г, 49%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,35 (с, 1H), 8,21 (д, J=9,3 Гц, 1H), 8,14 (д, J=7,1 Гц, 1H), 4,18-4,11 (м, 2H), 3,87-3,74 (м, 2H), 3,66 (с, 3H), 1,37 (с, 9H); MС: [(M+1)]+=484,20, 486,20.

трет-Бутил 8'-бром-7'-фтор-2'-оксо-2',3'-дигидроспиро[азетидин-3,1'-пирроло[2,3-c] хинолин]-1-карбоксилат: К раствору 1-трет-бутил 3-метил-3-(6-бром-7-фтор-3-нитрохинолин-4-ил) азетидин-1,3-дикарбоксилата (12,0 г, 24,8 ммоль) в уксусной кислоте (300 мл) добавляли порошок железа (9,69 г, 174 ммоль) при температуре окружающей среды. После перемешивания в течение 3 часов при температуре окружающей среды полученную в результате смесь концентрировали при пониженном давлении. Остаток разводили водой (200 мл) и экстрагировали этилацетатом (4×100 мл). Объединенные органические слои промывали солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 1%~5% метанолом в дихлорметане, с получением указанного в заголовке соединения в виде бесцветного твердого вещества (10,4 г, 99%): 1H ЯМР (400 МГц, ДМСО-d6) δ 11,02 (широкий, 1H), 8,71 (с, 1H), 8,24 (д, J=7,3 Гц, 1H), 8,03 (д, J=10,1 Гц, 1H), 4,29 (д, J=9,0 Гц, 2H), 4,21 (д, J=9,0 Гц, 2H), 1,49 (с, 9H); MС: [(M+1)]+=422,20, 424,20.
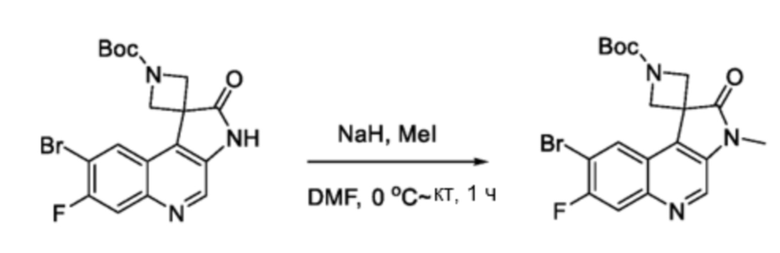
трет-Бутил 8'-бром-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[азетидин-3,1'-пирроло [2,3-c]хинолин]-1-карбоксилат: К перемешиваемому раствору трет-бутил 8-бром-7-фтор-2-оксо-2,3-дигидроспиро [азетидин-3,1-пирроло[2,3-c]хинолин]-1-карбоксилата (4,22 г, 9,99 ммоль) в N, N-диметилформамиде (100 мл) добавляли гидрид натрия (0,52 г, 13,0 ммоль, 60% диспергированный в минеральном масле) при 0°C в атмосфере азота. Полученную в результате смесь перемешивали в течение 1 часа при температуре окружающей среды с последующим добавлением йодометана (1,70 г, 12,0 ммоль). После перемешивания в течение дополнительного 1 часа при температуре окружающей среды реакцию гасили насыщенным водным раствором хлорида аммония (20,0 мл) и разводили водой (1,00 л). Осажденное твердое вещество собирали путем фильтрации, промывали водой (3×30,0 мл) и гексаном (2×30,0 мл). Полученное твердое вещество сушили под инфракрасным светом с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (3,93 г, 90%): 1H ЯМР (400 МГц, CDCl3) δ 8,71 (с, 1H), 8,47 (д, J=7,0 Гц, 1H), 7,94 (д, J=9,1 Гц, 1H), 4,54 (д, J=9,1 Гц, 2H ), 4,31 (д, J=9,0 Гц, 2H), 3,40 (с, 3H), 1,56 (с, 9H); MС: [(M+1)]+=436,15, 438,15.
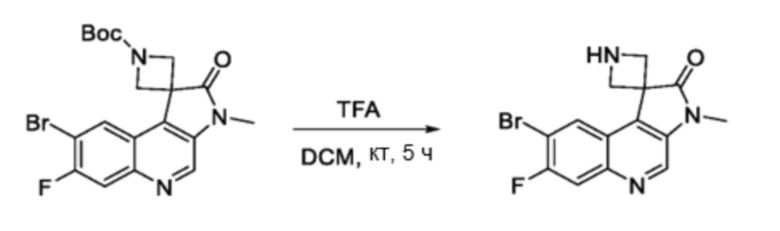
8-Бром-7-фтор-3-метил-2,3-дигидроспиро[азетидин-3,1-пирроло[2,3-c]хинолин]-2-он: Раствор трет-бутил-8-бром-7-фтор-3-метил-2-оксо-2,3-дигидроспиро [азетидин-3,1-пирроло[2,3-c]хинолин]-1-карбоксилата (3.93 г, 9,01 ммоль) и трифторуксусную кислоту (20,0 мл) в дихлорметане (100 мл) перемешивали в течение 5 часов при температуре окружающей среды. Полученную в результате смесь концентрировали при пониженном давлении. Остаток подщелачивали до pH 8 насыщенным водным раствором бикарбоната натрия. Полученную в результате смесь экстрагировали этилацетатом (6×300 мл). Объединенные органические слои промывали солевым раствором (2×300 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат упаривали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (3,00 г, 99%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,56 (д, J=7,7 Гц, 1H), 8,92 (с, 1H), 8,02 (д, J=10,2 Гц, 1H), 4,18 (д, J=7,5 Гц , 2H), 3,59 (д, J=7,5 Гц, 2H), 3,29 (с, 3H); MС: [(M+1)]+=335,95, 337,95.

8'-бром-7'-фтор-1-изопропил-3'-метилспиро[азетидин-3,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: К перемешиваемому раствору 8'-бром-7'-фтор-3'-метилспиро[азетидин-3,1'-пирроло [2,3-c]хинолин]-2'(3'H)-она (100 мг , 0,30 ммоль) и ацетона (4,00 мл) в этаноле (8,00 мл) добавляли цианоборгидрид натрия (94,0 мг, 1,50 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 3 часов при 50°C в атмосфере азота. Полученную смесь очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 120 г; Подвижная фаза А: вода (плюс 10 мМ NH4HCO3); Подвижная фаза B: ацетонитрил; Скорость потока: 50 мл/мин; Градиент (B): 5%~20%, 6 мин; 20%~50%, 30 мин; 50%~95%, 5 мин; 95%, 5мин; Детектор: УФ 254 нм. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (85,0 мг, 77%): 1H ЯМР: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,84 (д, J=8,0 Гц, 1H), 8,93 (с, 1H), 8,00 (д, J=10,2 Гц, 1H), 3,69 (д, J=7,6 Гц, 2H), 3,46 (д, J=7,3 Гц, 2H), 3,30 (с, 3H), 2,63-2,56 (м, 1H), 1,01 (д, J=6,1 Гц, 6H); MС: [(M+1)]+=378,10, 380,10.
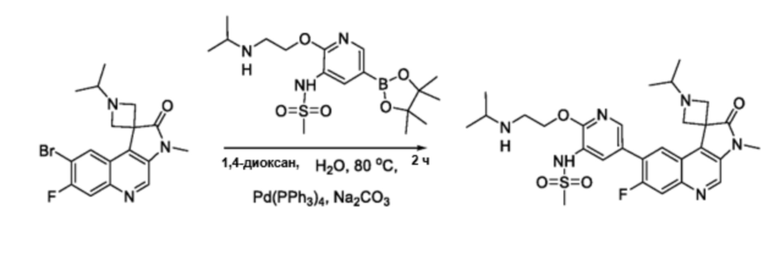
N-[5-[7-фтор-3-метил-2-оксо-1-(пропан-2-ил)-2,3-дигидроспиро[азетидин-3,1-пирроло [2,3-c]хинолин]-8-ил]-2-[2 [(пропан-2-ил)амино]этокси]пиридин-3-ил]метансульфонамид: К раствору 8-бром-7-фтор-3-метил-1-(пропан-2-ил)-2,3-дигидроспиро[азетидин-3,1-пирроло[2,3-c]хинолин]-2-она (80,0 мг, 0,21 ммоль) и N-(2 [2-[(пропан-2-ил)амино]этокси]-5- (4,4,5,5-тетраметил-1,3,2- диоксаборолан-2-ил)пиридин-3-ил)метансульфонамида (169 мг, 0,42 ммоль) в воде (2,00 мл) и 1,4-диоксане (10,00 мл) добавляли карбонат натрия (33,6 мг, 0,32 ммоль) и тетракис(трифенилфосфин) палладий (0) (24,3 мг, 0,021 ммоль). После перемешивания в течение 2 часов при 80°C в атмосфере азота полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=10/1, об./об.) с получением неочищенного продукта, который дополнительно очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 120 г; Подвижная фаза А: Вода (плюс 10 мМ NH4HCO3), подвижная фаза B: ацетонитрил; Скорость потока: 45 мл/мин; Градиент (B%): 5%, 2 мин; 5%~24%, 5 мин; 24%~34%, 9 мин; 34%, 8 мин; 34%~95%; 3 мин; 95%, 2 мин; Детектор: УФ 254 нм; Rt: 21 мин. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (65,0 мг, 54%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,66 (д, J=8,9 Гц, 1H), 8,89 (с, 1H), 8,25 (т, J=1,9 Гц, 1H), 7,99-7,91 (м, 2H), 4,42 (т, J=5,4 Гц, 2H), 3,76 (д, J=7,2 Гц, 2H), 3,47 (д, J=7,3 Гц, 2H), 3,31 (с, 3H), 3,02 (с, 3H), 2,96 (т, J=5,4 Гц, 2H), 2,92-2,84 (м, 1H), 2,62-2,53 (м, 1H), 1,05 (д, J=6,3 Гц, 6H), 0,97 (д, J=6,1 Гц , 6H); MС: [(M+1)]+=571,25.
COЕДИНЕНИЕ 572

Метил 1-(6-бром-7-фтор-3-нитрохинолин-4-ил)-3-метоксициклобутан-1-карбоксилат: К раствору свежеприготовленного диизопропиламида лития (10,6 ммоль) в безводном тетрагидрофуране (100 мл) добавляли метил-3-метоксициклобутан-1-карбоксилат (1,53 г, 10,6 ммоль) при -78 °C. После перемешивания в течение дополнительного 1 часа в течение 5 минут добавляли раствор 6-бром-4-хлор-7-фтор-3-нитрохинолина (2,50 г, 8,18 ммоль) в тетрагидрофуране (5,00 мл). Полученную смесь медленно нагревали до 0°C и затем гасили насыщенным водным хлоридом аммония (100 мл). Полученную в результате смесь разводили водой (1,00 л) и отделяли органический слой. Водный слой экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (2×200 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 5%-50% этилацетатом в петролейном эфире, с получением указанного в заголовке соединения в виде коричневого сиропа (720 мг, 21%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,25 (с, 1H), 8,31 (д, J=7,3 Гц, 1H), 8,17 (д, J=9,3 Гц, 1H), 4,16-4,13 (м, 1H) , 3,71 (с, 3 H), 3,11 (с, 3H), 2,47-2,38 (м, 2H), 2,00-1,89 (м, 2H); MС: [(M+1)]+=413,20, 415,20.
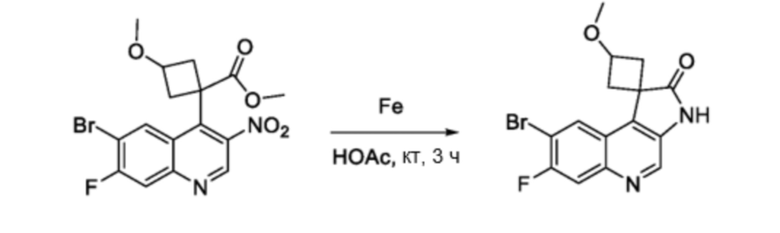
8'-Бром-7'-фтор-3-метоксиспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: К раствору метил 1-(6-бром-7-фтор-3-нитрохинолин-4-ил)-3-метоксициклобутан-1-карбоксилата (720 мг, 1,74 ммоль) в уксусной кислоте (10,0 мл) добавляли порошок железа (681 мг, 12,2 ммоль). Полученную в результате смесь перемешивали в течение 3 часов при температуре окружающей среды. Полученную в результате смесь разводили водой (150 мл) и экстрагировали этилацетатом (4×50,0 мл). Объединенные органические слои промывали солевым раствором (2×50,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 5-30% этилацетатом в петролейном эфире, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (550 мг, 89%): 1H ЯМР (400 МГц, CDCl3) δ 8,93 (с, 0,45 H), 8,88 (д, J=7,2 Гц, 0,55H), 8,84 (с, 1H), 8,78 (с, 0,55H), 8,27 (д, J=7,0 Гц, 0,45H) 7,92 (д, J=9,3 Гц, 1H), 4,71-4,63 (м, 0,45H), 4,57-4,49 (м, 0,55H), 3,48 (д, J=6,6 Гц, 3H) 3,07-2,83 (м, 4H); MС: [(M+1)]+=351,00, 353,00.

8'-Бром-7'-фтор-3-метокси-3'-метилспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H) -он: Раствор 8-бром-7-фтор-3-метокси-2,3-дигидроспиро[циклобутан-1,1-пирроло[2,3-c] хинолин]-2-она (550 мг, 1,56 ммоль) в N, N-диметилформамиде (10,0 мл) обрабатывали гидридом натрия (81,4 мг, 2,04 ммоль, 60% диспергированный в минеральном масле) в течение 0,5 часа при 0°C с последующим добавлением йодметана (265 мг, 1,87 ммоль) по каплям в течение 2 минут. при 0 oC. После дополнительного перемешивания в течение 1 часа при температуре окружающей среды, реакцию гасили насыщенным водным раствором хлорида аммония (20,0 мл) и разводили водой (150 мл). Полученную в результате смесь экстрагировали этилацетатом (3×50,0 мл). Объединенные органические слои промывали солевым раствором (2×30,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 5-10% этилацетатом в петролейном эфире, с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (500 мг, 87%): MС: [(M+1)]+=365,10, 367,10.
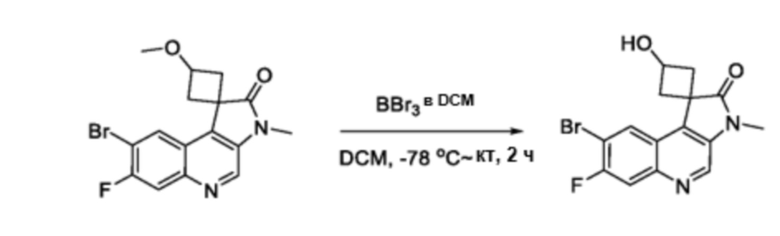
8'-Бром-7'-фтор-3-гидрокси-3'-метилспир [циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H) -он: К перемешиваемому раствору 8'-бром-7'-фтор-3-метокси-3'-метилспиро[циклобутан-1,1'-пирроло [2,3-c] хинолин]-2'(3'H) -она (900 мг, 2,46 ммоль) в дихлорметане (20,0 мл) по каплям при -78°C в атмосфере азота добавляли трибромид бора (24,6 мл, 24,6 ммоль, 1M в дихлорметане). Полученную смесь медленно нагревали до температуры окружающей среды. Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды в атмосфере азота.. Смесь нейтрализовали насыщенным водным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 5% метанолом в дихлорметане. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (530 мг, 62%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,96-8,89 (м, 1,6H), 8,32 (д, J=7,4 Гц, 0,4H), 8,05-7,98 (м, 1H), 5,99 (д, J=6,5 Гц, 0,6H), 5,73 (д, J=5,7 Гц, 0,4H), 4,97-4,87 (м, 0,4H), 4,75-4,65 (м, 0,6H), 3,31 (с, 1,2 H), 3,29 (с , 1,8H), 2,98-2,87 (м, 0,8H), 2,81-2,67 (м, 2,4H), 2,65-2,55 (м, 0,8H); MС: [(M+1)]+=351,00, 353,00.

3-(бензилокси)-8'-бром-7'-фтор-3'-метилспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: К раствору 8'-бром-7'-фтор-3-гидрокси-3'-метилспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-2'(3'H)-она (100 мг, 0,29 ммоль) в N, N-диметилформамиде (5,00 мл) добавляли гидрид натрия (13,7 мг, 0,35 ммоль, 60% диспергированный в минеральном масле) при 0°C в атмосфере азота. Полученную смесь перемешивали в течение 1 часа при 25°C с последующим добавлением бензилбромида (97,4 мг, 0,57 ммоль) при 0°C. После перемешивания еще в течение 1 часа при 25°C реакцию гасили насыщенным водным раствором хлорида аммония (10,0 мл). Полученную в результате смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×50,0 мл). Объединенные органические слои промывали солевым раствором (2 × 50,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=25/1, об./об.) с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (64,0 мг, 51%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,92 (д, J=3,6 Гц, 1H), 8,79 (д, J=7,6 Гц, 0,5H), 8,25 (д, J=7,2 Гц, 0,5H), 8,03 (д, J=10,1 Гц, 1H), 7,50 (д, J=7,0 Гц, 1H), 7,46-7,37 (м, 3H), 7,37-7,30 (м, 1H), 4,87-4,79 (м, 0,5H) , 4,62-4,50 (м, 2,5H), 3,30 (д, J=5,7 Гц, 3H), 2,98-2,90 (м, 1H), 2,80 (д, J=6,7 Гц, 2H), 2,72-2,64 (м, 1H); MС: [(M+1)]+=441,00, 443,00.

N-(5-(3-(Бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид: К раствору 3-(бензилокси)-8'-бром-7'-фтор-3'-метилспиро[циклобутан-1,1'-пирроло [2,3-c]хинолин]-2'(3'H)-она (64,0 мг, 0,15 ммоль) и N-[2-[2-(изопропиламино)этокси]-5- (4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил]метансульфонамида (69,5 мг, 0,17 ммоль) в воде (1,00 мл) и 1,4-диоксане (4,00 мл), добавляли карбонат натрия (15,4 мг, 0,15 ммоль) и тетракис (трифенилфосфин) палладий (0) ( 33,5 мг, 0,029 ммоль). После перемешивания в течение 2 часов при 80°C в атмосфере азота полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=10/1, об./об.) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (55,0 мг, 60%): MС: [(M+1)]+=634,55.
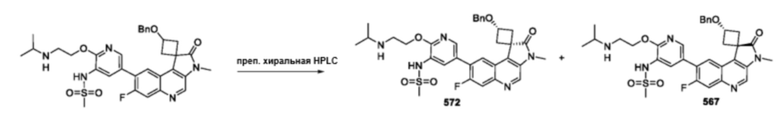
цис-N-(5-(3-(Бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 572) и транс-N-(5-(3-(Бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро [циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 567). Вышеупомянутый N-(5-(3-(бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро [циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)-2-(2-(изопропиламино)этокси) пиридин-3-ил)метансульфонамид (55,0 мг, 0,087 ммоль) разделяли с помощью Преп-Chiral-ВЭЖХ в следующих условиях: (Колонка: CHIRALPAK ID, 2×25 см (5 мкм); Подвижная фаза A: метил трет-бутиловый эфир (плюс 0,2% изопропиламина), подвижная фаза B: EtOH; Скорость потока: 17 мл/мин; Градиент: 10% B за 13 мин; Детектор: УФ 220/254 нм) с получением цис-N-(5-(3-(бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамида (ПРИМЕР 572, RT1: 8,70 мин) в виде светло-желтого твердого вещества (15,3 мг, 28%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,90 (с, 1H), 8,29 (с, 1H), 8,09 (д, J=8,3 Гц, 1H), 8,02 (д, J=1,9 Гц, 1H), 7,98 (д, J=12,1 Гц, 1H), 7,42-7,34 (м, 4H), 7,32-7,27 (м, 1H), 4,90-4,82 (м, 1H), 4,53 (с, 2H), 4,47 (т, Дж=5,4 Гц, 2H), 3,32 (с, 3H), 3,06 (с, 3H), 3,05-2,88 (м, 5H), 2,68 (дд, J=13,9, 6,2 Гц, 2H), 1,08 (д, J=6,2 Гц, 6H); MС: [(M+1)]+=634,50 и транс-N-(5-(3-(бензилокси)-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло [2,3-c]хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамида (ПРИМЕР 567, RT2: 11,2 мин) в виде бесцветное твердое вещество (12,6 мг, 23%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (с, 1H), 8,59 (д, J=8,3 Гц, 1H), 8,33 (с, 1H), 8,02-7,94 (м, 2H), 7,36-7,21 ( м, 5H), 4,58 (кв, J=6,3 Гц, 1H), 4,44 (т, J=5,4 Гц, 2H), 4,41-4,33 (м, 1H), 3,28 (с, 3H), 3,05 (с, 3H ), 2,00-3,94 (м, 3H), 2,92-2,84 (м, 1H), 2,83-2,74 (м, 1H), 2,69 (дд, J=13,1, 5,8 Гц, 2H), 2,59-2,54 (м, 1H), 1,35 (д, J=6,4 Гц, 3H), 1,05 (д, J=6,3 Гц, 6H); MС: [(M+1)]+=634,55.
COЕДИНЕНИЕ 573
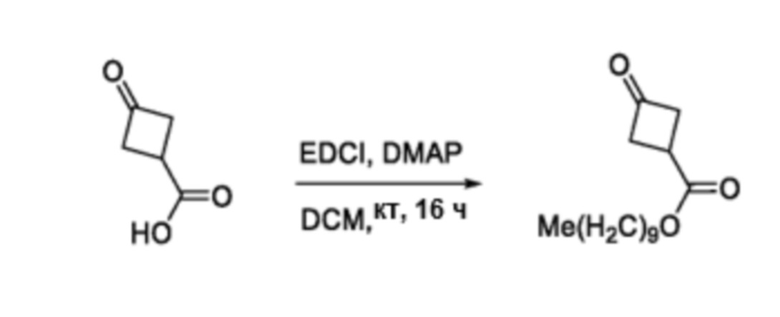
Децил-3-оксоциклобутан-1-карбоксилат: К перемешиваемому раствору 3-метилциклобутан-1-карбоновой кислоты (3,00 г, 26,3 ммоль) и 1-деканола (4,16 г, 26,3 ммоль) в дихлорметане (90,0 мл) добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (7,56 г, 39,4 ммоль) и 4-диметиламинопиридин (0,32 г, 2,62 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 16 часов при 25°C и гасили водой (30,0 мл). Полученную в результате смесь экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором (2×50,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 8% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого масла (3,10 г, 47%): 1H ЯМР (400 МГц, CDCl3) δ 4,15 (т, J=6,7 Гц, 2H), 3,47-3,35 (м, 2H), 3,35-3,15 (м, 3H), 1,69-1,61 (м, 2H), 1,39- 1,22 (м, 14H), 0,88 (т, J=6,7 Гц, 3H); MС: [(M+1)]+=252,20.
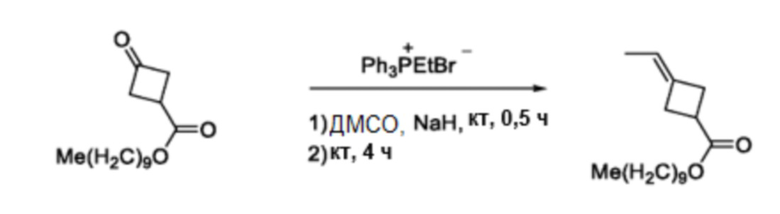
Децил-3-этилиденциклобутан-1-карбоксилат: К раствору бромида этилтрифенилфосфония (17,3 г, 46,5 ммоль) в диметилсульфоксиде (300 мл) добавляли трет-бутоксид калия (4,94 г, 44,1 ммоль) порциями при 14 °C. Полученную смесь перемешивали в течение 0,5 часа при 25°C в атмосфере азота с последующим добавлением децил 3-оксоциклобутан-1-карбоксилата (8,00 г, 31,5 ммоль) по каплям в течение 2 минут при 14 °C. После перемешивания в течение 4 часов при 25°C реакцию гасили насыщенным водным раствором хлорида аммония (20,0 мл) при 0 oC. Полученную смесь разбавляли водой (1,00 л) и экстрагировали этилацетатом (3 х 200 мл). Объединенные органические слои промывали солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% ~ 5% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (1,70 г, 21%): 1H ЯМР (400 МГц, CDCl3) δ 5,22-5,14 (м, 1H), 4,09 (т, J=6,7 Гц, 2H), 3,10 (тт, J=9,2, 7,2 Гц, 1H), 2,96-2,79 (м, 4H), 1,67-1,58 (м, 2H), 1,49 (дкв, J=6,0, 2,0 Гц, 3H), 1,39-1,20 (м, 14H), 0,88 (т, J=6,7 Гц, 3H).

Децил-3-этилциклобутан-1-карбоксилат: К перемешиваемому раствору децил 3-этилиденциклобутан-1-карбоксилата (700 мг, 4,94 ммоль) в метаноле (10,0 мл) добавляли безводный Pd/C (70,0 мг, 10% палладий на угле) при температуре окружающей среды. После перемешивания в течение 16 часов при температуре окружающей среды в атмосфере водорода (2 атм) полученную смесь фильтровали. Фильтрационный осадок промывали метанолом (3×20,0 мл). Фильтрат упаривали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого масла (666 мг, 95%): 1H ЯМР (400 МГц, CDCl3) δ 4,10-4,02 (м, 2H), 3,10-2,87 (м, 1H), 2,40-2,22 (м, 2H), 2,16-2,03 (м, 1H), 1,91-1,76 (м , 2H), 1,67-1,56 (м, 2H), 1,48-1,21 (м, 16H), 0,92-0,75 (м, 6H).

децил 1-(6-бром-7-фтор-3-нитрохинолин-4-ил)-3-этилциклобутан-1-карбоксилат: К раствору свежеприготовленного диизопропиламида лития (2,45 ммоль) в безводном тетрагидрофуране (20,0 мл) добавляли децил 3-этилциклобутан-1-карбоксилат (657 мг, 2,45 ммоль) при -78°C в атмосфере азота. Полученную смесь перемешивали в течение 1 часа с последующим добавлением 6-бром-4-хлор-7-фтор-3-нитрохинолина (575 мг, 1,88 ммоль) при -78 °C. После перемешивания еще в течение 1 часа при 25°C реакцию гасили насыщенным водным раствором хлорида аммония (10,0 мл). Полученную смесь разбавляли водой (20,0 мл) и разделяли. Водный слой экстрагировали этилацетатом (5 х 30 мл). Объединенные органические слои промывали солевым раствором (4×30,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 3% ~ 9% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла (450 мг, неочищенное): MС: [(M+1)]+=537,25, 539,25.

8'-бром-3-этил-7'-фторспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: Смесь неочищенного децил 1-(6-бром-7-фтор-3-нитрохинолин-4-ил)-3-этилциклобутан-1-карбоксилата (450 мг, 0,84 ммоль) и железа (327 мг, 5,86 ммоль) в уксусную кислоту (8,00 мл) перемешивали в течение 16 ч при температуре окружающей среды.. Полученную смесь фильтровали, отфильтрованный осадок промывали тетрагидрофураном (5 х 300 мл). Фильтрат концентрировали при пониженном давлении. Остаток подщелачивали до pH 8 насыщенным водным раствором бикарбоната натрия. Полученную в результате смесь экстрагировали этилацетатом (5×100 мл). Объединенные органические слои промывали солевым раствором (50,0 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=15/1, об./об.) с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (40,0 мг, 14%): 1H ЯМР (400 МГц, ДМСО-d6) δ 10,76 (д, J=5,3 Гц, 1H), 8,67 (д, J=1,7 Гц, 1H), 8,45 (д, J=7,2 Гц, 0,4H), 8,39 (д, J=7,4 Гц, 0,6H), 7,98 (дд, J=10,1, 3,4 Гц, 1H), 2,88-2,73 (м, 2H), 2,55-2,51 (м, 2H), 2,29-2,19 (м, 1H), 1,78-1,64 (м, 2H), 0,99-0,88 (м, 3H); MС: [(M+1)]+=349,05, 351,05.

8'-бром-3-этил-7'-фтор-3'-метилспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H)-он: К раствору 8'-бром-3-этил-7'-фтороспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-2'(3'H) -она (35,0 мг, 0,10 ммоль) в N, N-диметилформамиде (3,00 мл) добавляли гидрид натрия (5,21 мг, 0,13 ммоль, 60% диспергированный в минеральном масле) при 0°C в атмосфере азота. Полученную смесь перемешивали в течение 30 минут при 25°C с последующим добавлением йодметана (21,4 мг, 0,15 ммоль) при 0 °C. После перемешивания в течение дополнительных 40 минут при 25°C реакцию гасили насыщенным водным раствором хлорида аммония (20,0 мл). Полученную в результате смесь разводили водой (100 мл) и экстрагировали этилацетатом (4×100 мл). Объединенные органические слои промывали солевым раствором (4×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ(ДХM/MeOH=20/1, об./об.) с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (30,0 мг, 83%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,90 (д, J=2,9 Гц, 1H), 8,49 (д, J=7,4 Гц, 0,4H), 8,42 (д, J=7,4 Гц, 0,6H), 8,02 (дд, J=10,1, 3,1 Гц, 1H), 3,29 (д, J=4,5 Гц, 3H), 2,93-2,75 (м, 2H), 2,61-2,52 (м, 2H), 2,29-2,21 (м, 1H), 1,82-1,67 (м, 2H), 0,98-0,89 (м, 3H); MС: [(M+1)]+=363,05, 365,05.
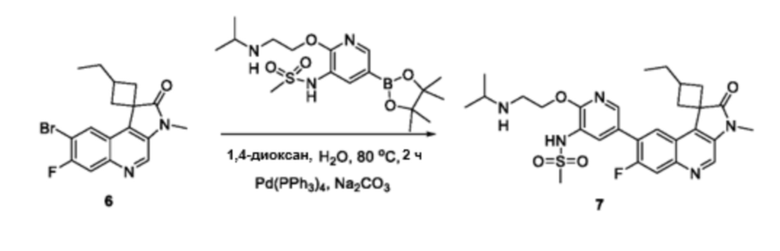
N-(5-(3-Этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид: К раствору 8'-бром-3-этил-7'-фтор-3'-метилспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-2'(3'H)-она (50,0 мг, 0,14 ммоль) и N-[2 [2-(изопропиламино)этокси]-5- (4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил]метансульфонамида(165 мг, 0,41 ммоль) в воде (0,75 мл) и 1,4-диоксане (3,00 мл) добавляли карбонат натрия (17,5 мг, 0,17 ммоль) и тетракис(трифенилфосфин) палладий (0) (31,8 мг 0,028 ммоль). После перемешивания в течение 2 часов при 80°C в атмосфере азота полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=10/1, об./об.) с получением неочищенного продукта, который дополнительно очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 120 г; Подвижная фаза А: Вода (плюс 10 мМ NH4HCO3), подвижная фаза B: ацетонитрил; Скорость потока: 45 мл/мин; Градиент (B%): 5%~25%, 7 мин; 25%~45%, 25 мин; 45%~65%, 8 мин; 95%, 5 мин; Детектор: УФ 254 нм. Фракции, содержащие желаемый продукт, собирали через 17 мин и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (30,0 мг, 40%): MС: [(M+1)]+=556,20.
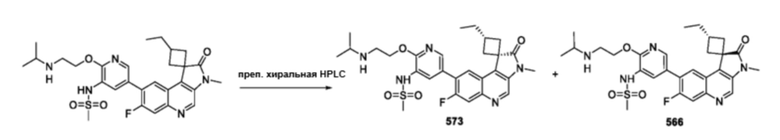 цис-N-(5-(3-Этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 573) и транс-N-(5-(3-Этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 566): Указанный выше N-(5-(3-этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (30,0 мг) разделяли препаративной хиральной ВЭЖХ в следующих условиях: Колонка: Chiralpak IC, 2×25 см, 5 мкм; Подвижная фаза A: гексан/ДХM=3/1 (плюс 0,2% изопропиламина), подвижная фаза B: EtOH; Скорость потока: 20 мл/мин; Градиент: 30% B за 15 мин; Детектор: УФ 220/254 нм. Желаемые фракции собирали и упаривали при пониженном давлении с получением цис-N-(5-(3-этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамида (ПРИМЕР 573, RT1: 10,94 мин) в виде бесцветного твердого вещества (12,8 мг, 43%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (с, 1H), 8,32-8,22 (м, 2H), 7,98 (д, J=12,2 Гц, 2H), 4,44 (т, J=5,3 Гц, 2H) , 3,30 (с, 3H), 3,04 (с, 3H), 2,99 (т, J=5,3 Гц, 2H), 2,94-2,78 (м, 4H), 2,26 (дд, J=12,2, 5,7 Гц, 2H), 1,75 (т, J=7,2 Гц, 2H), 1,07 (д, J=6,3 Гц, 6H), 0,89 (т, J=7,3 Гц, 3H); MС: [(M+1)]+=556,25
цис-N-(5-(3-Этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 573) и транс-N-(5-(3-Этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (ПРИМЕР 566): Указанный выше N-(5-(3-этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамид (30,0 мг) разделяли препаративной хиральной ВЭЖХ в следующих условиях: Колонка: Chiralpak IC, 2×25 см, 5 мкм; Подвижная фаза A: гексан/ДХM=3/1 (плюс 0,2% изопропиламина), подвижная фаза B: EtOH; Скорость потока: 20 мл/мин; Градиент: 30% B за 15 мин; Детектор: УФ 220/254 нм. Желаемые фракции собирали и упаривали при пониженном давлении с получением цис-N-(5-(3-этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил)метансульфонамида (ПРИМЕР 573, RT1: 10,94 мин) в виде бесцветного твердого вещества (12,8 мг, 43%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (с, 1H), 8,32-8,22 (м, 2H), 7,98 (д, J=12,2 Гц, 2H), 4,44 (т, J=5,3 Гц, 2H) , 3,30 (с, 3H), 3,04 (с, 3H), 2,99 (т, J=5,3 Гц, 2H), 2,94-2,78 (м, 4H), 2,26 (дд, J=12,2, 5,7 Гц, 2H), 1,75 (т, J=7,2 Гц, 2H), 1,07 (д, J=6,3 Гц, 6H), 0,89 (т, J=7,3 Гц, 3H); MС: [(M+1)]+=556,25
и транс-N-(5-(3-этил-7'-фтор-3'-метил-2'-оксо-2',3'-дигидроспиро[циклобутан-1,1'-пирроло[2,3-c] хинолин]-8'-ил)-2-(2-(изопропиламино)этокси)пиридин-3-ил) метансульфонамида (ПРИМЕР 566, RT2: 13,63 мин) в виде бесцветного твердого вещества (8,6 мг, 29%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (с, 1H), 8,34-8,26 (м, 2H), 8,01-7,93 (м, 2H), 4,44 (т, J=5,4 Гц, 2H), 3,31 ( с, 3H), 3,05 (с, 3H), 3,00 (д, J=5,4 Гц, 2H), 2,96-2,83 (м, 2H), 2,70-2,60 (м, 2H), 1,66 (кв, J=7,2, 6,7 Гц, 2H), 2,56-2,52 (м, 2H), 1,07 (д, J=6,2 Гц, 6H), 0,89 (т, J=7,3 Гц, 3H); MС: [(M+1)]+=556,20.
COЕДИНЕНИЕ 574
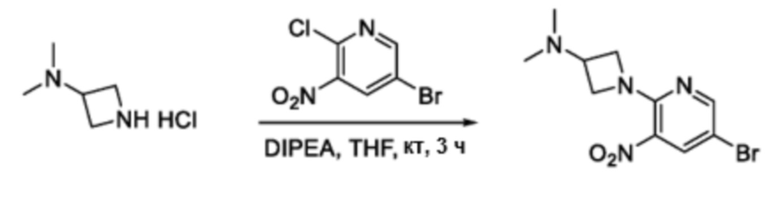
1-(5-бром-3-нитропиридин-2-ил)-N, N-диметилазетидин-3-амин: К перемешиваемому раствору гидрохлорида N, N-диметилазетидин-3-амина (0,27 г, 2,08 ммоль) и 5-бром-2-хлор-3-нитропиридина (0,49 г, 2,08 ммоль) в тетрагидрофуране (40,0 мл) добавляли диизопропилэтиламин. (0,67 г, 5,19 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 3 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 3% ~ 9% этилацетатом в петролейном эфире. Желаемые фракции собирали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (0,50 г, 59%): 1H ЯМР (400 МГц, CDCl3) δ 8,37 (д, J=2,2 Гц, 1H), 8,29 (д, J=2,2 Гц, 1H), 4,20 (ддд, J=10,0, 7,0, 1,2 Гц, 2H), 3,93 (ддд, J=9,9, 5,1, 1,2 Гц, 2H), 3,16 (тт, J=7,0, 5,1 Гц, 1H), 2,20 (с, 6H); MС: [(M+1)]+=301,00, 303,00.

1-(5-бром-3-нитропиридин-2-ил)-N, N-диметилазетидин-3-амин. К раствору 1-(5-бром-3-нитропиридин-2-ил)-N, N-диметилпиперидин-4-амина (6,20 г, 20,7 ммоль) в уксусной кислоте (90,0 мл) добавляли порошок железа (11,5 г , 206 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 2 часов при температуре окружающей среды. Полученную в результате смесь фильтровали, а осадок после фильтрования промывали тетрагидрофураном (3×100 мл). Фильтрат концентрировали при пониженном давлении. Остаток разводили насыщенным водным раствором карбоната натрия (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем, элюируя 2%~4% метанолом в дихлорметане, с получением указанного в заголовке соединения в виде серого твердого вещества (5,20 г, 92%): 1H ЯМР (400 МГц, CDCl3) δ 7,74 (д, J=2,0 Гц, 1H), 6,93 (д, J=2,0 Гц, 1H), 4,19-4,04 (м, 2H), 3,93-3,88 (м, 2H) , 3,21-3,14 (м, 1H), 2,24 (с, 6H); MС: [(M+1)]+=271,00, 273,00.
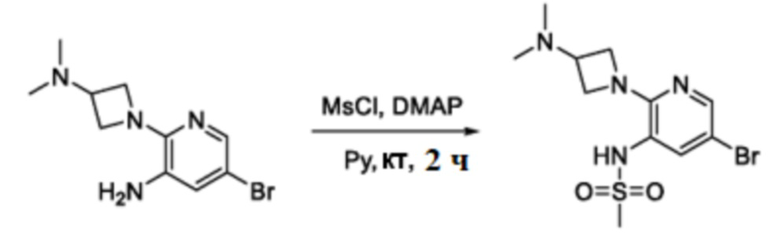
N-(5-Бром-2-(3-(диметиламино)азетидин-1-ил)пиридин-3-ил)метансульфонамид: К перемешиваемому раствору 5-бром-2-[3-(диметиламино)азетидин-1-ил] пиридин-3-амина (2,10 г, 7,77 ммоль) и N, N-4-диметиламинопиридина (77,0 мг, 0,63 ммоль) в пиридине (70,0 мл) по каплям добавляли метансульфонилхлорид (1,77 г, 15,5 ммоль) при температуре окружающей среды. После перемешивания в течение 3 часов при температуре окружающей среды в атмосфере азота полученную смесь упаривали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии с обращенной фазой в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 330 г; Подвижная фаза А: Вода (плюс 10 мМ NH4HCO3), подвижная фаза B: ацетонитр; скорость потока: 65 мл/мин; Градиент (B%): 5%~20%, 8 мин; 20%~40%, 20 мин; 40%~95%, 2 мин; 95%, 5 мин; Детектор: УФ 254 нм. Желаемые фракции собирали через 19 мин и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного твердого вещества (1,40 г, 52%): 1H ЯМР (400 МГц, CD3OD) δ 7,92 (д, J=2,2 Гц, 1H), 7,61 (д, J=2,2 Гц, 1H), 4,27 (дд, J=8,8, 7,2 Гц, 2H), 3,96 (дд , J=9,0, 5,6 Гц, 2H), 3,24-3,16 (м, 1H), 3,00 (с, 3H), 2,20 (с, 6H); MС: [(M+1)]+=349,00, 351,00.
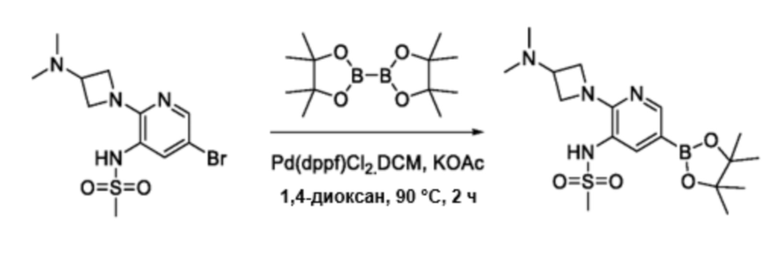
N-(2-(3-(Диметиламино)азетидин-1-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) пиридин-3-ил)метансульфонамид : К раствору N-[5-бром-2-[3-(диметиламино)азетидин-1-ил]пиридин-3-ил] метансульфонамида (1,00 г, 2,86 ммоль) и 4-бис(пинаколато)диборона (2,18 г, 8,59 ммоль) в 1,4-диоксане (30,0 мл) добавляли ацетат калия (1,12 г, 11,5 ммоль) и аддукт бис(дифенилфосфино)ферроцен]дихлорпалладий (II) дихлорметана (351 мг, 0,43 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 2 часов при 85°C в атмосфере азота. Полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали обращенной флэш-хроматографией в следующих условиях: Колонка: Сферический C18, 20~40 мкм, 330 г; Подвижная фаза А: Вода (плюс 10 мМ NH4HCO3), подвижная фаза B: ацетонитрил; Скорость потока: 65 мл/мин; Градиент (B%): 5%~20%, 7 мин; 20%~40%, 12 мин; 40%~95%; 2 мин; 95%, 5 мин; Детектор: УФ 254 нм. Желаемые фракции собирали через 20 мин и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (800 мг, 71%): 1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (с, 1H), 8,18 (д, J=1,5 Гц, 1H), 7,50 (д, J=1,6 Гц, 1H), 4,19 (т, J=8,0 Гц , 2H), 3,93 (дд, J=8,9, 5,0 Гц, 2H), 3,12-3,04 (м, 1H), 2,99 (с, 3H), 2,09 (с, 6H), 1,27 (с, 12H); MС: [(M+1)]+=397,20.
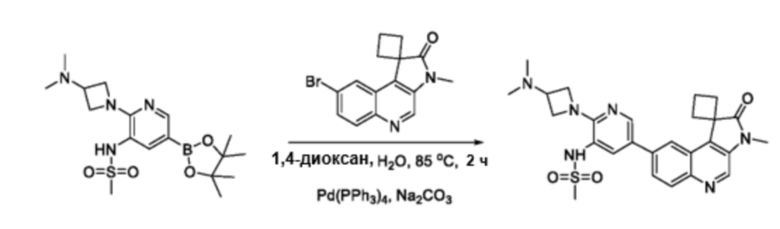
N-(2-(3-(Диметиламино)азетидин-1-ил)-5-(3'-метил-2'-оксо-2 ',3'-дигидроспиро [циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)пиридин-3-ил)метансульфонамид: К раствору 8-бром-3-метил-2,3-дигидроспиро[циклобутан-1,1-пирроло[2,3-c]хинолин] -2-она (320 мг, 1,01 ммоль) и N-[2-[3-(диметиламино)азетидин-1-ил]-5- (4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил]метансульфонамида (600 мг, 1,51 ммоль) в 1,4-диосане (13,0 мл) и воде (2,00 мл) добавляли карбонат натрия (160 мг, 1,51 ммоль) и теткракис(трифенилфосфин) палладий (0) (175 мг, 0,15 ммоль). После перемешивания в течение 2 часов при 85°C в атмосфере азота полученную в результате смесь концентрировали при пониженном давлении. Остаток очищали с помощью преп.-TСХ (ДХM/MeOH=8/1, об./об.) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (350 мг, 69%): 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (с, 1H), 8,80 (с, 1H), 8,58 (с, 1H), 8,33 (с, 1H), 8,13 (д, J=8,9 Гц, 1H) , 7,95 (д, J=9,2 Гц, 1H), 7,91 (д, J=2,1 Гц, 1H), 4,28-4,21 (м, 2H), 4,02-3,95 (м, 2H), 3,30 (с, 3H), 3,14 (с, 4H), 2,98-2,90 (м, 2H), 2,60-2,52 (м, 4H), 2,13 (с, 6H); MС: [(M+1)]+=507,20.
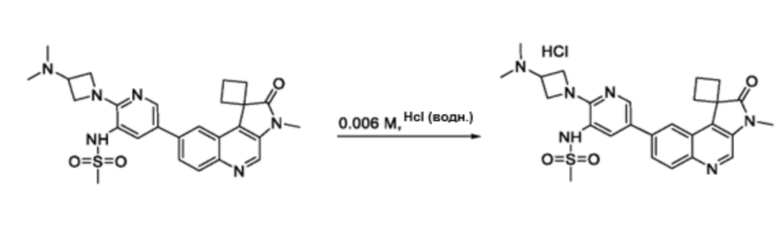
N-(2-(3-(Диметиламино)азетидин-1-ил)-5-(3'-метил-2'-оксо-2 ',3'-дигидроспиро [циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)пиридин-3-ил)метансульфонамид гидрохлорид: Раствор N-(2-(3-(диметиламино)азетидин-1-ил)-5-(3'-метил-2'-оксо-2 ',3'-дигидроспиро [циклобутан-1,1'-пирроло[2,3-c]хинолин]-8'-ил)пиридин-3-ил)метансульфонамида (350 мг, 0,69 ммоль) в разбавленном водном растворе соляной кислоты (115 мл, 0,69 ммоль, 0,006 M) и ацетонитриле (20,0 мл. ) лиофилизировали непосредственно с получением указанного в заголовке соединения в виде оранжевого твердого вещества (375 мг, 100%): 1H ЯМР (400 МГц, ДМСО-d6) δ 10,77 (с, 1H), 9,24 (с, 1H), 8,83 (с, 1H), 8,64 (д, J=2,1 Гц, 1H), 8,35 (с, 1H) , 8,17 (д, J=8,9 Гц, 1H), 8,02-7,94 (м, 2H), 4,50-4,41 (м, 2H), 4,39-4,32 (м, 2H), 4,27-4,16 (м, 1H), 3,31 (с, 3H), 3,18 (с, 3H), 2,99-2,88 (м, 2H), 2,82 (д, J=4,1 Гц, 6H), 2,63-2,52 (м, 4H); MС: [(M+1)]+=507,20.
ЦЕЛЕВАЯ И НЕЦЕЛЕВАЯ ИНГИБИРУЮЩАЯ АКТИВНОСТЬ
ATM в клеточном вестерн-блот анализе:
Утром клетки рака молочной железы MCF-7 помещали в 384-луночный планшет с плотностью 10 000 клеток/лунку (Corning, # 356663), 25 мкл клеток на лунку. На следующий день соединения добавляли в планшет с помощью булавочного инструмента (Echo 550) до конечной концентрации 1 мкМ посредством 3-кратного серийного разведения (всего 10 доз). Затем добавляли этопозид (Sigma, # E1383) до конечной концентрации 100 мкМ. Планшет инкубировали при 37°C в течение 1 ч, и клетки фиксировали добавлением 25 мкл фиксирующего раствора (8% параформальдегид) в течение 20 минут при температуре окружающей среды. Клетки были проницаемы с помощью 5 промывок 1X PBS (забуференный фосфатом физиологический раствор), содержащим 0,1% Triton X-100; каждая стирка длилась 5 минут. Клетки блокировали добавлением 50 мкл блокирующего буфера Odyssey (LI-COR, # 927-40000) в 384-луночные планшеты на 1,5 часа при встряхивании при температуре окружающей среды. Затем удаляли блокирующий буфер и добавляли 20 мкл раствора антитела против pKAP1 (Bethyl Laboratories, # A300-767A) (1/2000) в каждую лунку 384-луночного планшета. Планшет инкубировали в течение ночи при 4 °C, а затем промывали 5 раз 1X PBST (1X PBS, содержащий 0,1% Tween-20). К каждой лунке в планшете добавляли раствор вторичных антител (IRDye 800CW Goat anti-Rabbit IgG, LI-COR, # 926-32211) (1/5 000), содержащий краситель ДНК DRAQ5 (CST, # 4084L) (1/5 000) (20 мкл) и планшет инкубировали 1 час при осторожном встряхивании в темноте. Клетки промывали 5 раз 1X PBST (1X PBS, содержащий 0,1% Tween-20) при температуре окружающей среды с осторожным встряхиванием в темноте. После последней промывки промывочный раствор удаляли, планшет переворачивали вверх дном на тонкое бумажное полотенце и центрифугировали при 1000 об/мин в течение 1 мин для поглощения всего промывочного буфера. Дно планшета очищали влажной безворсовой бумагой. Планшет немедленно сканировали с использованием ODYSSEY CLx (LI-COR).
Ферментный иммуносорбентный анализ DNA-PK
В первый день 96-луночный планшет (ThermoFisher, № по каталогу: 442404) покрывали пептидом GST-p53 (1-101) (очищенным Pharmaron, отдел BCS) путем разбавления 3 мкг GST-p53 в каждой лунке 0,1 М Na2CO3/NaHCO3 (pH 9,6). Планшет инкубировали в течение ночи при 4 ° C. На второй день буфер для покрытия удаляли, и планшет дважды промывали PBST (1X PBS, содержащий 0,1% Tween-20). Раствор фермента DNA-PK (Invitrogen, # PR9107A; конечная концентрация DNA-PK: 0,1 мкг/мл) затем был добавлен. Соединения были серийно разбавлены до конечной максимальной концентрации 100 нМ (3-кратное серийное разведение, всего 10 доз) и раствор АТФ (конечная концентрация АТФ: 20 мкМ) добавляли в планшет. Инкубировать планшет при 25°C в течение 1 часа. Планшет промывали трижды PBST (1X PBS, содержащий 0,1% Tween-20) и блокировали раствором PBST и 1% BSA при 4°C в течение ночи. На третий день планшет промывали четыре раза PBST (1X PBS, содержащий 0,1% Tween-20). В каждую лунку добавляли первичное антитело против фосфо-p53 (Cell signaling Technology, # 9286, фосфо-p53 (Ser15) (16G8) Mouse mAb) (1/1000). Планшет герметично закрывали, инкубировали 1 час при 37°C и четыре раза промывали PBST (1X PBS, содержащий 0,1% Tween-20). Вторичное антитело, связанное с HRP (Cell signaling Technology, # 7076, против IgG мыши, антитело, связанное с HRP) (1/1000) (100 мкл), добавляли в каждую лунку. Планшет заклеивали лентой, инкубировали 30 мин при 37°C и четыре раза промывали PBST (1X PBS, содержащий 0,1% Tween-20). В это время в каждую лунку добавляли 100 мкл субстрата TMB (Cell signaling Technology, # 7004). Планшет заклеивали лентой и инкубировали планшет 10 мин при 37 °C. Стоп-раствор (Cell signaling Technology, # 7002) (100 мкл) добавляли в каждую лунку, и планшет подвергали детекции поглощения при 450 нм.
Биохимический анализ mTOR:
Реакции с mTOR-киназой проводили в объеме 10 мкл в 384-луночных планшетах малого объема. Обычно использовались пластины PerkinElmer модели 6008260. Состав буфера для реакции 1x киназы был: 50 мМ HEPES pH 7,5, 0,01% Твин 20, 1 мМ EGTA, 10 мМ MnCl2 и 2 мМ DTT. Раствор фермента mTOR (ThermoFisher, # PR8683B; конечная концентрация mTOR: 0,5 мкг/мл) добавляли, и соединения последовательно разбавляли до конечной максимальной концентрации 100 нМ (3-кратное серийное разведение, всего 10 доз). GFP-4E-BP1 (конечная концентрация: 0,4 мкМ) и раствора АТФ (конечная концентрация АТФ: 3 мкМ) добавляли в 384-луночный планшет. Планшет инкубировали при 25°C в течение 1 часа, и в каждую лунку добавляли 10 мкл раствора EDTA (20 мМ) и меченого Tb антитела против p4E-BP1 (4 нМ) в буфере для разведения TR-FRET. Планшет герметично закрывали, инкубировали 30 мин при 25 ° C и считывали на ридере для планшетов, сконфигурированном для LanthaScreen ™ TRFRET.
Биохимический анализ PI3Kα и PI3Kδ:
Реакции с киназой PI3Kα и PI3Kδ проводили в объеме 5 мкл в 384-луночных планшетах малого объема. Обычно использовались планшеты PerkinElmer модели 6008280. Буфер для реакции 1x киназы состоял из 50 мМ HEPES pH 7,5, 3 мМ MgCl2, 0,03% CHAPS, 1 мМ EGTA, 100 мМ NaCl и 2 мМ DTT. PI3Kα (ThermoFisher, # PV4788; конечная концентрация PI3K: 120 нг мл) или PI3Kδ раствор фермента (ThermoFisher, # PV6451; конечная концентрация PI3Kδ: 250 нг/мл) был добавлен в планшет, соединения последовательно разбавляли до конечной максимальной концентрации 100 нМ (3-кратное серийное разведение, всего 10 доз) и PIP2:3PS (конечная концентрация: 10 мкг/мл) и раствор АТФ (конечная концентрация АТФ: 10 мкМ) добавляли в 384-луночный планшет. Планшет инкубировали при 25°C в течение 1 часа. Буфер для реагента ADP-Glo (5 мкл) добавляли в каждую лунку. Планшет закрывали и инкубировали 40 мин при 25 ° C. Буфер для обнаружения ADP-Glo (10 мкл) добавляли в каждую лунку, и планшет инкубировали в течение 40 минут при 25°C и считывали на считывающем устройстве для планшетов, сконфигурированном для люминесценции.
Анализ ингибирования hERG in vitro:
Клетки hERG-T-RExTM HEK 293 (Invitrogen, K1236) были получены путем трансфекции последовательности, кодирующей hERG в Tet-регулируемом векторе экспрессии pT-Rex-DEST30, в клетки, экспрессирующие Tet-репрессор (T-RexTM HEK293), тем самым продуцируя клетки, которые можно индуцировать для экспрессии высокого уровня каналов hERG. Клетки культивировали в среде, содержащей 85% DMEM, 10% диализованного FBS, 0,1 мМ NEAA, 25 мМ HEPES, 100 Ед/мл пенициллин-стрептомицина, 5 мкг/мл бластицидина и 400 мкг/мл генетицина. Клетки разделяли с использованием TrypLE ™ Express (Gibco, 12604) примерно три раза в неделю и поддерживали конфлюэнтность от ~ 40% до ~ 80%. Перед анализом клетки индуцировали доксициклином (Sigma, D9891) в концентрации 1 мкг/мл в течение 48 часов. В день эксперимента индуцированные клетки ресуспендировали и высевали на покровные стекла из расчета 5 × 105 клеток/на 6 см чашку для культивирования клеток перед использованием. Ток, опосредованный каналом hERG, регистрировали с помощью записывающих систем с ручным зажимом, оснащенных усилителями (HEKA, EPC10 и Molecular Devices, multiclamp 700B) и инвертированным фазово-контрастным микроскопом (Olympus, IX51/71/73). Стеклянные пипетки готовили с помощью съемника для микропипеток (Sutter, P97 и Narishige, PC-10) и оценивали по сопротивлению пипеток в диапазоне 2-4 МОм. Раствор внутренней пипетки представлял собой 140 мМ KCl, 2 мМ MgCl2, 10 мМ EGTA, 5 мМ MgATP и 10 мМ HEPES (pH доведен до 7,35 с помощью КОН), а внешний буфер - 132 мМ NaCl, 4 мМ KCl, 3 мМ CaCl2., 0,5 мМ MgCl2, 11,1 мМ глюкозы и 10 мМ HEPES (pH доведен до 7,35 с помощью NaOH). Конфигурация целых клеток поддерживалась с постоянным контролем сопротивления доступа (<15 МОм). Ток hERG вызывали деполяризацией мембраны до +30 мВ в течение 4,8 с, а напряжение снова устанавливали на -50 мВ на 5,2 с, чтобы удалить инактивацию и измерить деактивирующий хвостовой ток. Максимальный размер хвостового тока использовался для определения амплитуды тока hERG. Для оценки ингибирования hERG пустой носитель и испытуемые образцы перфузировали в клетки в конфигурации записи целых клеток через систему перфузии жидкости (ALA, система доставки гравитационного потока VM8). Для анализа реакции на дозу испытуемое вещество наносили на клетки накопительно от низких до высоких концентраций. Положительный контроль (дофетилид) использовался в экспериментах для обеспечения работоспособности клеток и операций в качестве основной части валидации метода. Процент подавления тока hERG сравнивали с концентрациями доз для построения кривой доза-ответ и определения IC50.
В некоторых вариантах осуществления соединение по изобретению выбрано из группы, состоящей из соединений, перечисленных в таблице ниже.
ТАБЛИЦА 1 Результаты анализа
IC50 (нM)
IC50 (нM)
IC50 (мкM)
14
КЛОНОГЕННЫЕ И АНАЛИЗЫ ЭФФЕКТИВНОСТИ IN VIVO
Способы
Клеточная культура Клеточная линия карциномы молочной железы человека MCF-7 и клеточные линии HCT116 p53 дикого типа и HCT116 p53 - / - были получены от ATCC. Клетки MCF7 и HCT116 культивировали в среде Игла, модифицированной Дульбекко (DMEM (Gibco # 11995-065). Линия клеток тройного отрицательного рака груди человека MDA-MB-231 и линия плоскоклеточной карциномы головы и шеи человека FADU были получены от Charles River Laboratories (Моррисвилл, Северная Каролина). Клетки MDA-MB-231 культивировали в среде RPMI 1640 (Gibco, 11875-093). Клетки FADU культивировали в минимальной необходимой среде α (MEM α) (Gibco, 12571-063) с добавлением 1 мМ пирувата натрия (Gibco, 11360-070), незаменимых аминокислот 1X MEM (Gibco, 11140-050). Во все клеточные линии добавляли 10% фетальную бычью сыворотку (FBS) (Corning, 35-010-CV) и 1X антибиотик-антимикотик (Gibco # 15240-062) и выращивали при 37°C в 5% CO2. Все клеточные линии были аутентифицированы с помощью профилирования коротких тандемных повторов и дали отрицательный результат на микоплазму.
Антитела. Антитела, распознающие фосфорилированный/активированный ATM (S1981, # ab81292) и DNA-PKcs (S2056, # ab18192), были приобретены в Abcam Biotechnology (Кембридж, Массачусетс). Антитела к ATM (# A1106) и DNA-PKcs (# ab1832) были от Sigma и Abcam, соответственно. Антитела Phospho-KAP1 (S824, # A300-767A) и KAP1 (A700-014) были от Bethyl Labs (Montgomery, TX). Антитела Phospho-TBK1 (# 5483), cGAS (# 15102) и pSTING (S366, # 19781S) были от Cell Signaling Technology (CST) (Danvers, MA). Антитела к STING (PA5-23381) были от ThermoFisher Scientific (Уолтем, Массачусетс), а антитела к актину (# A5316) были от Sigma. Все антитела были разведены 1: 1000, за исключением актина, который был разведен 1: 3000 в 1X TBST (1X TBS -10X TBS, (Corning, 46-012-CM), 0,05% Tween-20 (P7949) Sigma и 1% BSA. В качестве вторичных антител использовали (а) метод Li-Cor - козий анти-кроличий (926-3221) и козий анти-мышиный (926-68020) Li-Cor (b) метод ECL: Анти-кроличий IgG, связанный с HRP (# 7074S), CST.
Иммуноблоттинг. Клетки предварительно обрабатывали соединением 569 при 20, 100, 500 или 1000 нМ или ДМСО в течение 30 минут, а затем облучали 0 или 10 Гр (XRAD 160, Precision X Ray), как показано на ФИГ. 1. На ФИГ. 3, клетки предварительно обрабатывали Соединением 569 в концентрации 1 мкМ, ATMi (AZD0156) в концентрации 1 мкМ, DNA-PKi (пепосертиб) в концентрации 1 мкМ в виде отдельных агентов или ДМСО, или ATMi и DNA-PKi в комбинации по 0,5 мкмоль каждый или 1 мкМ каждый в течение 30 мин, а затем облучение 0 или 10 Гр (XRAD 160, Precision X Ray), как показано на ФИГ. 1. Через час после IR клетки лизировали в буфере RIPA (50 нМ Трис, pH 8,0, 150 мМ NaCl, 0,1% SDS, 0,5% дезоксихолат натрия, 1% IGEPAL® CA-630 (Sigma, I8896) в течение 20 минут при 4°C в присутствии коктейлей с ингибиторами протеаз и фосфатаз (Roche, # 04693159001 & # 04906837001). Лизаты клеток центрифугировали при 13 200 g в течение 10 минут при 4ºC, и супернатанты анализировали на содержание белка с помощью анализа BCA (Pierce ™ BCA Protein Assay Kit, каталожный № 23227, ThermoFisher Waltham, MA 02451) и использовали равные количества белка на дорожку для последующих иммуноблот-анализов. Белки денатурировали добавлением буфера для образцов NuPAGE LSD [4X] (Invitrogen, NP0007), содержащего β-меркаптоэтанол в конечной концентрации 2,5%, и образцы кипятили в течение 5 минут. Образцы загружали в гели NuPAGE 3-8% протеина трис-ацетата (Invitrogen, EA03785BOX) и подвергали электрофорезу с 1X трис-ацетатным SDS-буфером (Invitrogen, LA0041) или загружали в гели NuPAGE 4-12% протеина Bis-Tris ( Invitrogen, NP0336BOX) и подвергали электрофорезу с использованием 1X MOPS SDS рабочего буфера (Invitrogen, NP0001).
Белки переносили на ночь на нитроцеллюлозные мембраны (Amersham, 10600003). Мембраны блокировали 5% обезжиренным сухим молоком в 1X TBST в течение 1 часа и инкубировали с первичными антителами в течение ночи при 4ºC. Мембраны промывали 4 × 10 мин с помощью 1x TBST при комнатной температуре, а затем инкубировали со вторичными антителами в течение 1 часа при комнатной температуре с последующей отмывкой 3 × 10 минут с помощью 1x TBST при комнатной температуре. Мембраны визуализировали с помощью системы ODYSSEY CLX или с помощью ECL (субстрат для блоттинга ECL, Pierce, 32209, 34075, 34095).
Анализ клоногенной выживаемости. Этот анализ был выполнен с соединением 569. Клетки высевали в 6-луночные планшеты различной плотности: 250 клеток без IR контроля, 5000 клеток для 2 Гр IR и 10000 клеток для 4 Гр IR и культивировали в течение ночи. На следующий день клетки предварительно инкубировали с соединением 569 при 100, 250, 500 или 1000 нМ или с ДМСО в течение 30 минут перед воздействием возрастающих доз IR (0, 2 или 4 Гр). После IR клетки непрерывно инкубировали с ДМСО или соединением 569 в течение 5 ч перед удалением культуральной среды и промыванием клеток PBS. Клетки культивировали в полной среде в отсутствие ингибитора в течение 9 дней. Затем клетки окрашивали (PBS, 0,0037% об./об. формальдегида, 0,1% кристаллического фиолетового), промывали водой и сушили.
Исследования опухолей. Самок мышей NCr nu/nu (Crl: NU (NCr) -Foxn1nu, штамм 490) получали из Charles River в возрасте 5-6 недель. Все процедуры на животных, использованные в этих исследованиях, были одобрены Комитетом по уходу и использованию животных (IACUC) в Университете Дьюка.
Клетки FADU или MDA-MB-231 собирали во время логарифмической фазы роста для имплантации in vivo. Ресуспендированные клетки трижды промывали фосфатно-солевым буфером (PBS) перед приготовлением рабочего разведения в PBS. Клетки FADU разбавляли до 1 × 107 клеток/мл, а клетки MDA-MB-231 разбавляли до 5 × 107 клеток/мл. Каждой мыши вводили подкожно в правый бок 100 мкл клеточной суспензии. За ростом опухоли следили до тех пор, пока опухоли не достигли целевого объема не менее 100 мм3, после чего мышей рандомно разделили на четыре группы лечения.
После имплантации линий опухолевых клеток мышей еженедельно контролировали на предмет развития опухоли. После обнаружения опухоли были измерены в двух измерениях с помощью цифровых штангенциркулей. Опухоли лечили после достижения по крайней мере 100 мм3 и измеряли два-три раза в неделю после включения в одну из групп лечения. Конечная точка исследования была определена как пятикратное увеличение опухоли от объема во время лечения.
Несущая среда, использованная в этом исследовании, представляла собой 0,5% (гидроксипропил)метилцеллюлозу (HPMC) и 0,2% Tween80, растворенные в деионизированной воде. PH несущей среды доводили до 7,0-8,0, и несущую среду хранили при 4 °C. В каждый день обработки in vivo к несущей среде добавляли подходящую массу тестируемого соединения при комнатной температуре. Смесь встряхивали по мере необходимости до достижения конечной концентрации 1,6 мг/мл.
Фармакодинамические (PD) анализы. Опухолевые исследования проводили следующим образом. Мышам, несущим опухоли FADU или MDA-231, предварительно вводили только несущую среду или только соединение 569 в дозах 3, 6 и 10 мг/кг. Затем опухоли облучали дозой 10 Гр через 45 минут после введения несущей среды или соединения 569 или оставляли необлученными (несущая среда и соединение 569 в дозе 10 мг/кг). Опухоли собирали через 1 час после облучения.
Гомогенаты тканей для фармакодинамического анализа собирали и обрабатывали следующим образом. Опухолевые ткани из опухолей FADU или MDA-MB-231 мгновенно замораживали в жидком азоте и измельчали на сухом льду с использованием измельчителя тканей. Часть опухолевого порошка переносили в микропробирку с последующим добавлением 500 мкл буфера RIPA [50 нМ Трис, pH 8,0, 150 мМ NaCl, 0,1% SDS, 0,5% дезоксихолат натрия, 1% IGEPAL® CA-630 ( Sigma, I8896)]. К 10 мл буфера RIPA добавляли по 2 таблетки ингибитора протеазы и ингибитора фосфатазы (Roche, # 04693159001 и # 04906837001), 200 мкл 0,1 M PMSF и 160 мкл апротинина (Sigma, A6279). Суспензию гомогенизировали на льду с помощью пестика для гранул (KONTES) и инкубировали на льду в течение 30 минут. Лизаты клеток центрифугировали при 13 200 об/мин в течение 15 минут при 4ºC, супернатанты переносили в новую пробирку и анализировали на концентрацию белка с помощью набора BCA (Pierce, 23227) с последующим иммуноблоттингом.
Исследование задержки роста опухоли in vivo проводили следующим образом. После имплантации линий опухолевых клеток мышей еженедельно контролировали на предмет развития опухоли. При развитии опухоли размером не менее 100 мм3 мышей с опухолью разделяли на одну из 4 групп, используя по 5 мышей на группу. Объем опухоли составлял от 100 до 500 мм3. Несущую среду или соединение 569 (10 мг/кг) вводили через желудочный зонд один раз в день в течение трех последовательных дней (qd × 3), отдельно или в комбинации с очаговым излучением. Доставленный объем несущей среды или соединения 569 регулировали в зависимости от массы тела отдельных животных. Мыши, разделенные на группы лучевой терапии, получали 3 Гр в день в течение трех последовательных дней (qd × 3) через 45 минут после введения несущей среды или соединения 569. Мышей анестезировали изофлураном во время лучевой терапии, которую проводили с помощью облучателя с визуальным контролем для небольших животных X-RAD 225Cx (Precision X-Ray). Поле облучения 40 мм x 40 мм центрировали на ноге, несущей опухоль, с помощью рентгеноскопии. Мышей облучали параллельно-противоположными передним и задним полями рентгеновскими лучами 225 кВп, 13 мА с использованием 0,3-миллиметрового Cu-фильтра при средней мощности дозы 300 сГр/мин.
Мыши Группы 1 получали несущую среду qd × 3.
Мыши Группы 2 получали 10 мг/кг соединения 569 qd × 3.
Мыши Группы 3 получали несущую среду за 45 минут до фокусного облучения 3 Гр qd × 3.
Мыши Группы 4 получали соединение 569 в дозе 10 мг/кг за 45 минут до фокусного облучения 3 Гр qd × 3.
Статистический и графический анализ данных in vivo. Призма (GraphPad) использовалась для графических презентаций. Для статистического анализа использовались SAS 9.4 и R 3.6.1. Сравнивали темпы роста опухолей в четырех группах лечения. Для того чтобы приспособиться к разным размерам опухолей в начале лечения, относительный объем опухоли был рассчитан путем деления объема опухоли в каждый момент времени на объем на исходном уровне. Линейные графики использовались для отображения среднего относительного объема опухоли с течением времени (ФИГУРЫ 6 и 8). Когда животное выходило из исследования из-за пятикратного увеличения опухоли, окончательный объем опухоли, зарегистрированный для животного, был включен в данные, используемые для расчета среднего объема в последующие моменты времени. Графики Каплана-Мейера использовались, чтобы показать процент мышей в каждой экспериментальной группе, оставшихся в исследовании с течением времени (ФИГУРЫ 7 и 9). Мышей, которые были найдены мертвыми до достижения конечной точки пятикратной выборки, подвергали цензурированию справа. Различия в выживаемости без пяти выборок между четырьмя группами лечения оценивали с помощью лог-рангового теста. Значение P менее 0,05 считалось статистически значимым.
Результаты
ФИГ. 1 показывает изображение иммуноблота из эксперимента in vitro по оценке действия соединения 569 на клетки MCF7 с облучением или без него. Иммуноблоттинг показывает ингибирование радиационно-индуцированного аутофосфорилирования киназ ATM и DNA-PK и радиационно-индуцированного фосфорилирования KAP1, субстрата ATM, соединением 569 в опухолевых клетках. MCF7, линию клеток карциномы груди человека, предварительно обрабатывали в течение 30 мин соединением 569 в концентрациях 20, 100, 500 и 1000 нМ. ДМСО использовали в качестве отрицательного контроля. Затем клетки подвергали воздействию 0 или 10 Гр ионизирующего излучения (ИК), и через час клетки собирали для анализа иммуноблоттинга. Соединение 569 показало сильное дозозависимое ингибирование радиационно-индуцированного фосфорилирования DNA-PKcs на Ser2056 и ATM на Ser1981 (оба события аутофосфорилирования) и субстрата ATM KAP1 (ser824), демонстрируя, что соединение 569 ингибирует как DNA-PK, так и киназы ATM в клетке. .
ФИГ. 2 демонстрирует радиосенсибилизирующие свойства соединения 569 в анализе клоногенной выживаемости in vitro. Клетки MCF7 предварительно обрабатывали несущей средой или соединением 569 при 100, 250, 500 и 1000 нМ в течение 30 минут перед облучением возрастающими дозами IR (0, 2 и 4 Гр). Через 5 часов среду удаляли и к клеткам добавляли свежую среду без ингибиторов, и они культивировали в течение 9 дней перед фиксацией, окрашиванием и подсчетом колоний. 5-часовое воздействие на клетки соединения 569 индуцировало значительную степень радиосенсибилизации без доказательств клеточной токсичности в отсутствие IR в клетках MCF7 (ФИГ. 2, Таблица 1).
ФИГ. 3 представляет собой иммуноблот, показывающий индукцию фосфорилирования TBK1 соединением 569, AZD0156 (селективный ингибитор ATM; ATMi), пепосертибом (селективным ингибитором DNA-PK; DNA-PKi) и комбинацией AZD0156 и пепосертиба (Ai+Di ) в клетках HCT116, экспрессирующих p53 дикого типа или клетках HCT116, которые были отрицательными по экспрессии p53. Фосфорилирование TBK1 является маркером активации ответа интерферона типа I и связано с усилением ответа опухоли на терапию блокадой иммунных контрольных точек. Двойное ингибирование ATM и DNA-PKcs путем объединения двух селективных химических ингибиторов (AZD0156 и пепосертиб) привело к более глубокой активации TBK1, чем любой из селективных ингибиторов по отдельности. Точно так же воздействие на клетки соединения 569, двойного ингибитора ATM и DNA-PKcs, было более сильным активатором фосфорилирования TBK1, чем любой из ингибиторов одиночной киназы-мишени, и не зависело от статуса p53. Активация TBK1 с помощью 569 и двойное ингибирование ATM и DNA-PKcs оказались независимыми от индукции cGAS и фосфо-STING.

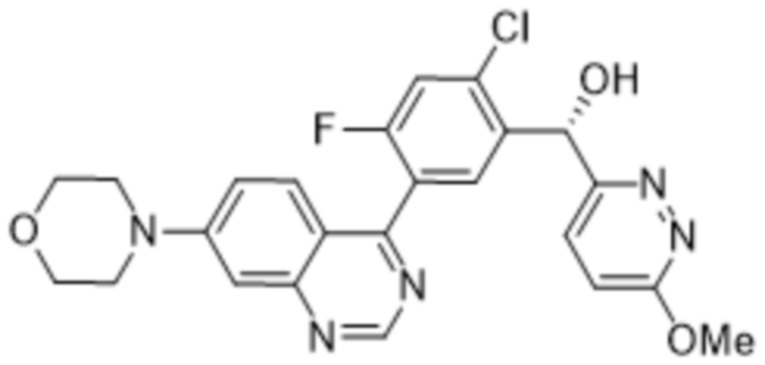
AZD0156 пепосертиб
Для оценки фармакодинамической активности соединения 569 in vivo клетки FADU и MDA-MB-231 выращивали в виде ксенотрансплантатов опухоли на голых мышах. Несущую среду или соединение 569 вводили через желудочный зонд отдельно или в сочетании с очаговым облучением.. Мышам вводили несущую среду или соединение 569 в дозах 3, 6 и 10 мг/кг. Затем опухоли получали 10 Гр очагового облучения через 45 минут или оставались необлученными (0 Гр, несущая среда и 0 Гр+соединение 569 в дозе 10 мг/кг). Опухоли собирали через 1 час после облучения. Иммуноблоттинг лизатов этих опухолей показывает индуцированное излучением аутофосфорилирование pDNA-PK и индуцированное излучением фосфорилирование KAP1, нижестоящей мишени ATM, и дозозависимое ингибирование этих событий фосфорилирования в опухолях FADU и MDA231, обработанных облучением плюс соединение 569. Опухолевые уровни радиационно-индуцированного аутофосфорилирования DNA-PK и радиационно-индуцированного фосфорилирования KAP1 значительно ингибировались на ~ 55% и > 80%, соответственно, при 10 мг/кг (ФИГУРЫ 4 и 5, Таблицы 2-5). Базальное фосфорилирование DNA-PK также ингибировалось соединением 569 в опухолях.
ТАБЛИЦА 1 Клоногенный анализ соединения 569 в клетках MCF7 (см. ФИГ. 2)
ТАБЛИЦА 2 Количественное определение pDNA-PK/DNA-PK в опухолях FADU
ТАБЛИЦА 3 Количественная оценка pKAP1/KAP1 в опухолях FADU
ТАБЛИЦА 4 Количественное определение pDNA-PK/DNA-PK в опухолях MDA-231
ТАБЛИЦА 5 Количественная оценка pKAP1/KAP1 в опухолях MDA-231
Опухоли FADU. Мыши в Группе 1 получали несущую среду, qd × 3. Совокупный рост опухоли в этой группе был прогрессивным. Среднее время до конечной точки составляло 14 дней с диапазоном 11-14 дней (ФИГУРЫ 6 и 7). Мыши в Группе 2 получали соединение 569 в дозе 10 мг/кг qd × 3. Среднее время до конечной точки составляло 14 дней с диапазоном от 11 до 17 дней. Кривые роста опухоли и выживаемость без пяти выборок для животных Группы 2 почти идентичны таковым для Группы 1 (ФИГУРЫ 6 и 7). Мыши в Группе 3 получили несущую среду и очаговую дозу облучения 3 Гр, которая была доставлена через 45 минут после введения несущей среды. И несущая среда и облучение были qd × 3. Одно животное было найдено мертвым на 16-й день по неизвестным причинам. Среднее время до конечной точки составило 21 день с диапазоном от 19 до 25 дней. Общая выживаемость была значительно улучшена по сравнению с Группой 1 и Группой 2 (P <0,01, логранг). Задержка совокупного роста опухоли и увеличение выживаемости без пяти выборок для Группы 3 по сравнению с Группами 1 и 2 показаны на ФИГУРАХ 6 и 7. Мыши в Группе 4 получали соединение 569 и очаговую дозу облучения 3 Гр, которая была доставлена через 45 минут после введения соединения. И соединение 569, и облучение вводили qd × 3. Среднее время до конечной точки составило 42 дня с диапазоном от 31 до 51 дня. Общая выживаемость была значительно улучшена по сравнению с Группой 1, Группой 2 и Группой 3 (P <0,01, логранг). Задержка совокупного роста опухоли и увеличение выживаемости без пятикратной выборки для Группы 4 по сравнению со всеми другими группами показано на ФИГУРАХ 6 и 7. Все виды лечения переносились хорошо.
MDA-MB-231 Опухоли. Мыши в Группе 1 получали несущую среду, qd × 3. Среднее время до конечной точки составило 12 дней с диапазоном от 12 до 17 дней. Мыши в Группе 2 получали соединение 569 в дозе 10 мг/кг qd × 3. Среднее время до конечной точки составило 17 дней с диапазоном от 12 до 22 дней. Кривые роста опухоли и выживаемость без пяти выборок для животных Группы 2 почти идентичны таковым для Группы 1 (ФИГУРЫ 8 и 9). Мыши в Группе 3 получили несущую среду и очаговую дозу облучения 3 Гр, которая была доставлена через 45 минут после введения несущей среды. И несущая среда, и облучение были qd × 3. Среднее время до конечной точки составило 22 дня с диапазоном от 16 до 22 дней. Общая выживаемость была значительно улучшена по сравнению с животными Группы 1, получавшими только несущую среду (P <0,01, логранг). Увеличение выживаемости без пятикратной выборки для Группы 3 по сравнению с Группами 1 и 2 показано на ФИГ. 9. Мыши в Группе 4 получили 10 мг/кг соединения 569 и дозу очагового излучения 3 Гр, которая была доставлена через 45 минут после введения соединения. И соединение 569, и облучение были qd × 3. Одно животное было найдено мертвым на 35-й день по неизвестным причинам. Среднее время до конечной точки составило 43 дня с диапазоном от 37 до 58 дней. Общая выживаемость была значительно улучшена по сравнению с Группой 1, Группой 2 и Группой 3 (P <0,01, логранг). Задержка совокупного роста опухоли и увеличение выживаемости без пятикратной выборки для Группы 4 по сравнению со всеми другими группами показано на ФИГУРАХ 8 и 9. Все виды лечения переносились хорошо.
Другие варианты реализации
Различные модификации и вариации описанного изобретения будут очевидны специалистам в данной области без отклонения от объема и сущности изобретения. Хотя изобретение было описано в связи с конкретными вариантами реализации, следует понимать, что заявленное изобретение не должно чрезмерно ограничиваться такими конкретными вариантами реализации. Действительно, предполагается, что различные модификации описанных способов реализации изобретения, которые очевидны для специалистов в данной области техники, находятся в пределах объема изобретения.
Другие варианты реализации указаны в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛА КАК ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА | 2005 |
|
RU2470916C2 |
| ПРОИЗВОДНОЕ БИАРИЛА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2768830C2 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2773444C2 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| ИНГИБИТОР CD73, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2787428C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
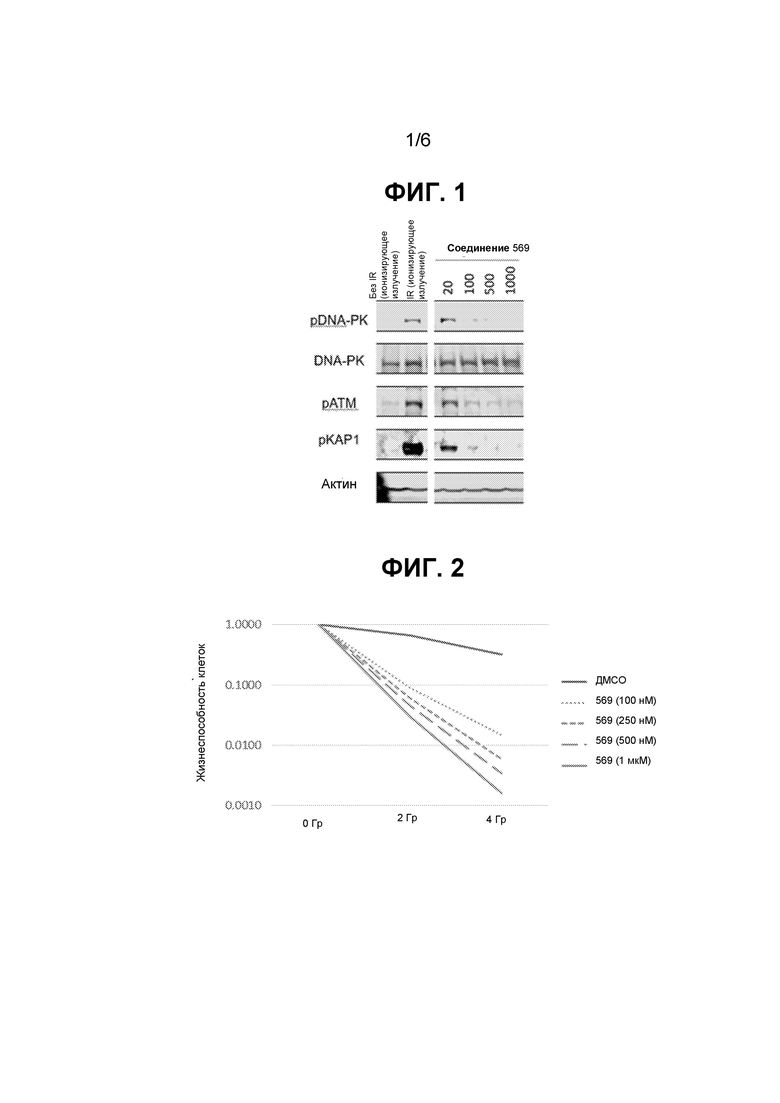



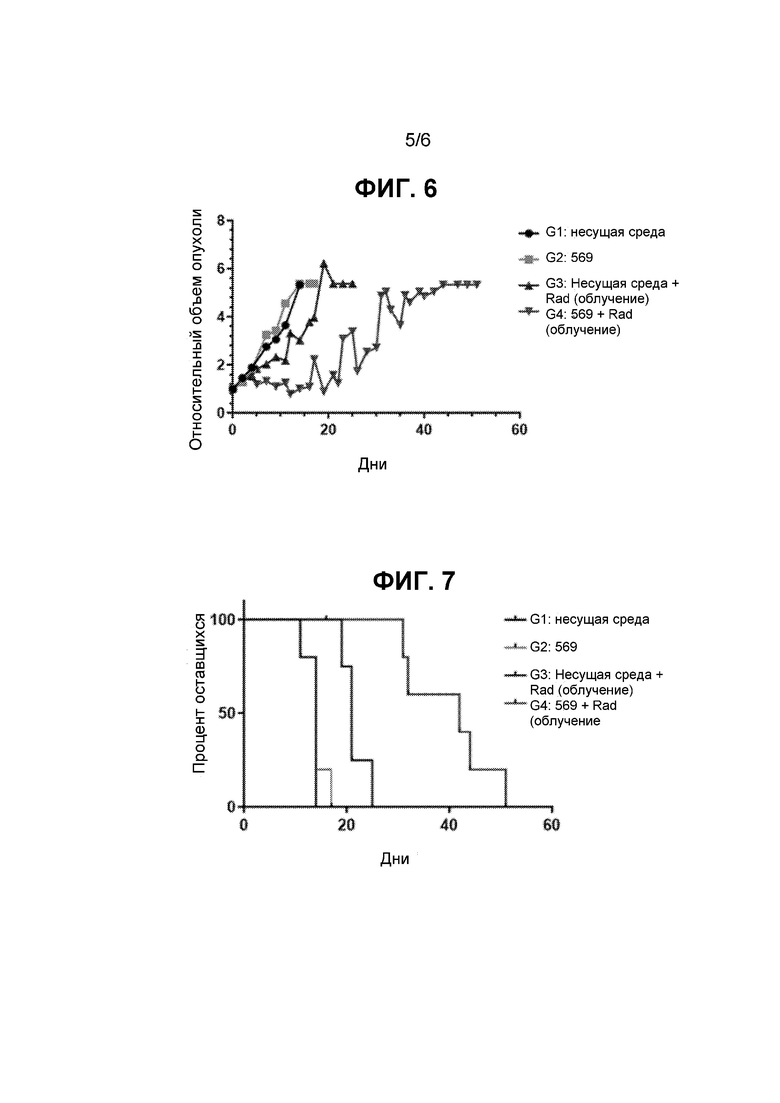

Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его фармацевтически приемлемой соли, где Y представляет собой СHR5 или NR6; Z представляет собой СH; n равно 0 или 1; R1 представляет собой -O-L-N(R7)2 или четырехчленный насыщенный N-гетероциклил, замещённый NMe2; R2 представляет собой С1-3 aлкил; каждый R3 независимо представляет собой галоген; R4 представляет собой С1-6 aлкил; R5 представляет собой водород, C1-3 aлкил или бензилокси; R6 представляет собой C1-3 aлкил; каждый R7 независимо представляет собой H или C1-3 aлкил; и L представляет собой этилен. Также изобретение относится к конкретным соединениям, фармацевтичекой композиции на основе соединения формулы (I) или конкретных указанных соединений, способу лечения онкологического заболевания указанными соединениями. Изобретение обеспечивает эффективное лечение онкологического заболевания двойным ингибитором ATM и DNA-PK. 7 н. и 32 з.п. ф-лы. 9 ил., 6 табл., 6 пр.
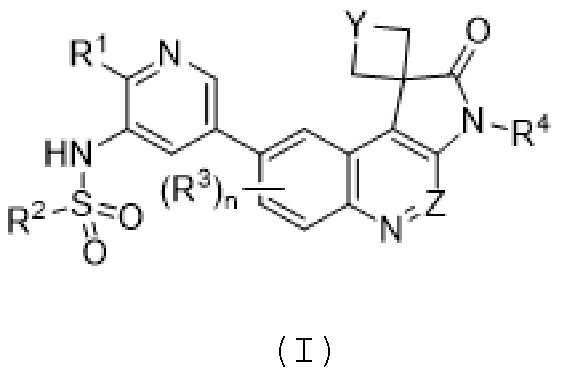
1. Соединение формулы (I):

или его фармацевтически приемлемая соль,
где
Y представляет собой СHR5 или NR6;
Z представляет собой СH;
n равно 0 или 1;
R1 представляет собой -O-L-N(R7)2 или четырехчленный насыщенный N-гетероциклил, замещённый NMe2;
R2 представляет собой С1-3 aлкил;
каждый R3 независимо представляет собой галоген;
R4 представляет собой С1-6 aлкил;
R5 представляет собой водород, C1-3 aлкил или бензилокси;
R6 представляет собой C1-3 aлкил;
каждый R7 независимо представляет собой H или C1-3 aлкил; и
L представляет собой этилен.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где n равно 1.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (IA):
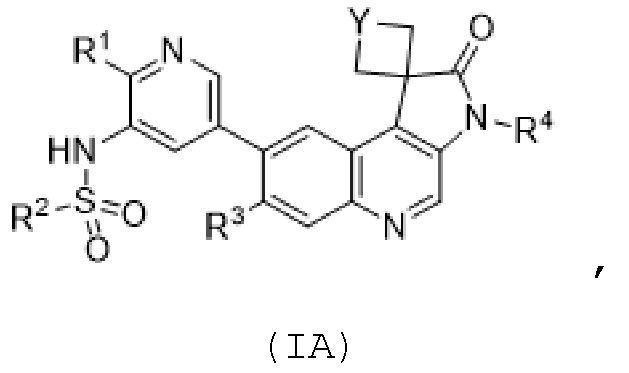
или его фармацевтически приемлемую соль.
4. Соединение по любому одному из пп. 1-3 или его фармацевтически приемлемая соль, где R3 представляет собой фтор.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где n равно 0.
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (IB):

или его фармацевтически приемлемую соль.
7. Соединение по любому из пп. 1-6 или его фармацевтически приемлемая соль, где R1 представляет собой -O-L-N(R7)2.
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, где один R7 представляет собой H, а оставшийся R7 необязательно представляет собой C1-3 алкил.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль, где по меньшей мере один R7 представляет собой изопропил.
10. Соединение по любому из пп. 1-9 или его фармацевтически приемлемая соль, где R2 представляет собой метил, этил или изопропил.
11. Соединение по любому из пп. 1-10 или его фармацевтически приемлемая соль, где R2 представляет собой метил.
12. Соединение по любому из пп. 1-11 или его фармацевтически приемлемая соль, где R4 представляет собой метил.
13. Соединение по любому из пп. 1-12 или его фармацевтически приемлемая соль, где Y представляет собой СHR5.
14. Соединение по любому из пп. 1-13 или его фармацевтически приемлемая соль, где R5 представляет собой водород.
15. Соединение по любому из пп. 1-13 или его фармацевтически приемлемая соль, где R5 представляет собой C1-3 алкил.
16. Соединение по любому из пп. 1-13 или его фармацевтически приемлемая соль, где R5 представляет собой бензилокси.
17. Соединение по любому из пп. 1-12 или его фармацевтически приемлемая соль, где Y представляет собой NR6.
18. Соединение по любому из пп. 1-17 или его фармацевтически приемлемая соль, где R6 представляет собой C3 алкил.
19. Соединение по любому из пп. 1-17 или его фармацевтически приемлемая соль, где R6 представляет собой изопропил.
20. Соединение, выбранное из группы, состоящей из:



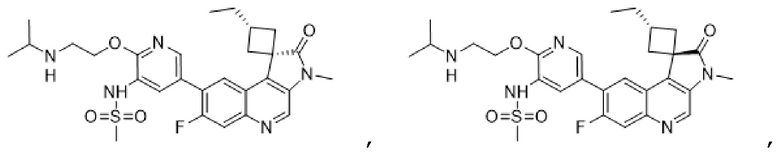

и их фармацевтически приемлемых солей.
21. Соединение, имеющее следующую структуру:

или его фармацевтически приемлемая соль.
22. Соединение, имеющее следующую структуру:
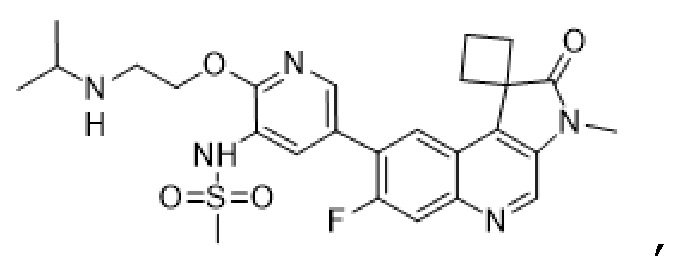
или его фармацевтически приемлемая соль.
23. Соединение, имеющее следующую структуру:
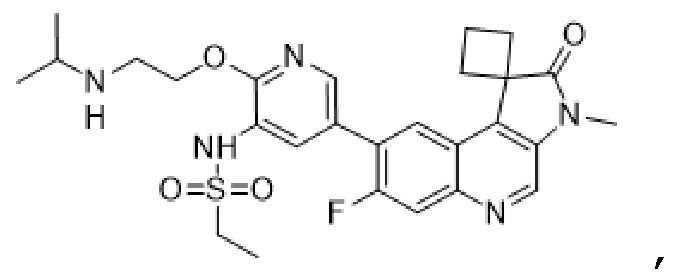
или его фармацевтически приемлемая соль.
24. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ATM и DNA-PK, содержащая терапевтически эффективное количество соединения по любому из пп. 1-23 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество.
25. Способ лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-23 или его фармацевтически приемлемой соли, или фармацевтической композиции по п. 24 пациенту, нуждающемуся в этом.
26. Способ по п. 25, в котором пациент получает лучевую терапию.
27. Способ по п. 26, в котором соединение или фармацевтическую композицию вводят пациенту одновременно с проведением лучевой терапией.
28. Способ по п. 26, в котором соединение или фармацевтическую композицию вводят пациенту перед проведением лучевой терапии.
29. Способ по п. 26, в котором соединение или фармацевтическую композицию вводят пациенту после проведения лучевой терапии.
30. Способ по любому из пп. 26-29, в котором лучевая терапия включает внешнее, внутреннее, брахитерапевтическое или системное облучение.
31. Способ по любому из пп. 26-30, в котором лучевая терапия включает введение радионуклидного конъюгата антитела.
32. Способ по любому из пп. 25-31, в котором пациент получает противоопухолевый агент.
33. Способ по п. 32, в котором противоопухолевый агент представляет собой цисплатин, оксалиплатин, карбоплатин, валрубицин, идарубицин, калихеамицин или ингибитор PARP.
34. Способ по п. 33, в котором противоопухолевый агент представляет собой противоопухолевый биологический агент или противоопухолевый иммунотерапевтический агент.
35. Способ по любому из пп. 32-34, в котором соединение или фармацевтическую композицию вводят пациенту одновременно с противоопухолевым агентом.
36. Способ по любому из пп. 32-34, в котором соединение или фармацевтическую композицию вводят пациенту перед противоопухолевым агентом.
37. Способ по любому из пп. 32-34, в котором соединение или фармацевтическую композицию вводят пациенту после противоопухолевого агента.
38. Способ по любому из пп. 25-37, в котором онкологическое заболевание представляет собой рак головного мозга, рак мочевого пузыря, рак груди, рак центральной нервной системы, рак шейки матки, рак толстой кишки, рак эндометрия, рак пищевода, стромальную опухоль желудочно-кишечного тракта, рак желудка, рак головы и шеи, буккальный рак, рак ротовой полости, гепатоцеллюлярный рак, рак легких, меланому, карциному из клеток Меркеля, мезотелиому, рак носоглотки, нейробластому, остеосаркому, рак яичников, рак поджелудочной железы, рак предстательной железы, рак почек, рак слюнной железы, саркомы, рак яичек, рак уротелия, рак вульвы или опухоль Вильма.
39. Способ по любому из пп. 25-37, в котором онкологическое заболевание представляет собой рак груди, рак легких, рак головы и шеи, рак поджелудочной железы, рак прямой кишки, глиобластому, гепатоцеллюлярную карциному, холангиокарциному, метастатические поражения печени, меланому, саркому кости, саркому мягких тканей, рак эндометрия, рак шейки матки, рак предстательной железы или карциному из клеток Меркеля.
| WO 2017076898 A1, 11.05.2017 | |||
| WO 2016155884 A1, 06.10.2016 | |||
| EP 3159342 B1, 05.08.2020 | |||
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |

