ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет согласно предварительной заявке на патент США с регистрационным № 62/537741, поданной 27 июля 2017 года. Полное содержание вышеупомянутой предварительной заявки на патент включено в данный документ посредством ссылки.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящая заявка содержит перечень последовательностей, который был подан в электронном виде в формате ASCII и включен в данный документ посредством ссылки во всей своей полноте. Указанная копия в формате ASCII, созданная 26 июля 2018 г., называется AXJ-226PC_SL.txt и имеет размер 32987 байтов.
УРОВЕНЬ ТЕХНИКИ
Система комплемента действует в сочетании с другими иммунологическими системами организма для защиты от проникновения клеточных и вирусных патогенов. Известно по меньшей мере 25 белков системы комплемента, которые были обнаружены в виде сложной совокупности белков плазмы крови и мембранных кофакторов. Белки плазмы крови составляют приблизительно 10% от глобулинов в сыворотке крови позвоночных. Компоненты системы комплемента осуществляют свою функцию иммунной защиты путем взаимодействий в ряде сложных, но точных событий ферментативного расщепления и связывания с мембраной. Получаемый в результате каскад системы комплемента приводит к образованию продуктов с опсонической, иммунорегуляторной и литической функцией. Краткое описание форм биологической активности, ассоциированных с активацией системы комплемента, представлено, например, в The Merck Manual, 16th Edition.
Хотя правильно функционирующая система комплемента обеспечивает надежную защиту от заражения микроорганизмами, ненадлежащая регуляция или активация путей системы комплемента вовлечена в патогенез ряда нарушений, в том числе пароксизмальной ночной гемоглобинурии (PNH) и атипичного гемолитико-уремического синдрома (aHUS) (см., например, Socié G, et al., French Society of Haematology Haematet. Lancet. 1996;348(9027):573-577; Brodsky, R., Blood. 2014;124(18):2804-2811); Hillmen, P., et al., Am. J. Hematol. 2010;85(8):553-559; Caprioli et al. (2006) Blood 108:1267-1279; и Kavanagh et al. (2006) British Medical Bulletin 77 и 78:5-22).
Пациенты с нарушениями, ассоциированными с системой комплемента, такими как PNH или aHUS, имели риск значительной заболеваемости и смертности. Соответственно, целью настоящего изобретения является обеспечение улучшенных композиций и способов лечения пациентов с нарушениями, ассоциированными с системой комплемента.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В данном документе предусмотрены стабильные, высококонцентрированные водные растворы антител к C5, а также способы получения и применения составов. В настоящем изобретении предусмотрены, помимо других аспектов, условия состава, подходящие для поддержания физической и функциональной стабильности антитела к C5 (например, равулизумаба, также известного как «антитело BNJ441» и «ALXN1210») в течение значительного времени в высококонцентрированных растворах. Например, в настоящем изобретении предусмотрены условия состава, способные обеспечивать поддержание антитела к C5 в преимущественно мономерной форме в течение периода до 2 лет при 2°C-8°C, даже если антитело содержится в растворах в концентрациях, составляющих примерно 100 мг/мл или выше. Кроме того, как описано в данном документе и проиллюстрировано в демонстрационных примерах, в таких составах также сводятся к минимуму агрегация, фрагментация или разрушение антитела к C5 (например, равулизумаба) в высококонцентрированных растворах. Например, в настоящем изобретении предусмотрены условия состава, способные обеспечивать поддержание в течение двух лет антитела к C5 в высококонцентрированной форме без выявляемых продуктов фрагментации или разрушения антитела (как определяется с помощью методики высокоэффективной жидкостной хроматографии в режиме эксклюзионной хроматографии (SEC-HPLC), такой как гель-проникающая HPLC) и не более чем с 2% агрегатов. Также в данном документе предусмотрены условия, подходящие для составления растворов антитела к C5, такого как равулизумаб, в концентрации более 200 мг/мл.
Преимущества стабильных высококонцентрированных водных растворов антитела к C5 являются многочисленными. Во-первых, для терапевтических путей применения, требующих введения антитела пациенту в небольшом объеме, терапевтическая эффективность часто зависит от количества антитела, которое можно вводить в этом небольшом объеме. При отсутствии возможности составлять антитела к C5 в высоких концентрациях применение, например, подкожных, интравитреальных и/или внутрисуставных путей доставки часто будет исключено. Подобным образом, высококонцентрированные составы антител обеспечивают для пациента больший выбор путей введения. В терапевтических путях применения, которые требуют частых, постоянных введений и/или самостоятельной доставки, введение делается возможным благодаря высокой концентрации составов и может быть более привлекательным для пациентов, чем внутривенная инфузия. Например, высококонцентрированные составы антитела к C5 могут позволять пациенту самостоятельно вводить антитело, например, путем подкожной или внутривенной инъекции. Таким образом, возможность составлять антитела в высоких концентрациях может повышать степень соблюдения режима введения путем обеспечения простой альтернативы введения в домашних условиях для пациентов с нарушениями, ассоциированными с системой комплемента.
Кроме того, способы получения водных растворов, описанных в данном документе, не требуют стадии лиофилизации, также как и представленные высококонцентрированные водные растворы не требуют восстановления из лиофилизированного материала. Представленные в настоящем изобретении высококонцентрированные растворы антител обеспечивают некоторые преимущества по сравнению с восстановленными составами лиофилизированных антител. Во-первых, медицинские работники должны на месте восстанавливать растворы лиофилизированных антител в стерильных условиях, что увеличивает возможность микробного загрязнения раствора перед введением. Кроме того, восстановление требует значительной осторожности, чтобы быть уверенными, что все твердые вещества, содержащиеся в сосуде для восстановления, надлежащим образом растворены в растворе. Высококонцентрированные водные растворы, предусмотренные в данном документе, таким образом, обеспечивают для медицинского работника, лица, осуществляющего уход, и/или пациента быстрые, простые, безопасные и эффективные средства для доставки терапевтического антитела пациенту, нуждающемуся в этом.
Другие преимущества высококонцентрированных составов включают, например, снижение стоимости производства за счет уменьшения места для бестарного хранения и/или количества отходов продукта. Кроме того, возможность производства продукта, характеризующегося более длительным сроком годности, в конечном итоге требует меньше производственных циклов, что в конечном итоге снижает затраты для производителя и потребителя высококонцентрированного терапевтического антитела.
Иллюстративное антитело к C5 представляет собой равулизумаб (также известный как антитело BNJ441 и ALXN1210), содержащий тяжелые и легкие цепи, имеющие последовательности, показанные под SEQ ID NO: 14 и 11 соответственно, или его антигенсвязывающие фрагменты и варианты. В других вариантах осуществления антитело содержит области, определяющие комплементарность (CDR), тяжелой и легкой цепей или вариабельные области (VR) равулизумаба. Соответственно, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 вариабельной области тяжелой цепи (VH) равулизумаба, имеющие последовательность, показанную под SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 вариабельной области легкой цепи (VL) равулизумаба, имеющие последовательность, показанную под SEQ ID NO: 8. В другом варианте осуществления антитело содержит последовательности CDR1, CDR2 и CDR3 тяжелой цепи, приведенные под SEQ ID NO: 19, 18 и 3 соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, приведенные под SEQ ID NO: 4, 5 и 6 соответственно.
В другом варианте осуществления антитело содержит VH- и VL-области, имеющие аминокислотные последовательности, приведенные под SEQ ID NO: 12 и SEQ ID NO: 8 соответственно.
В другом варианте осуществления антитело содержит константную область тяжелой цепи, приведенную под SEQ ID NO: 13.
В другом варианте осуществления антитело содержит вариант константной области Fc человека, который связывается с неонатальным Fc-рецептором человека (FcRn), где вариант константной области CH3 Fc человека содержит замены Met-429-Leu и Asn-435-Ser по остаткам, соответствующим метионину 428 и аспарагину 434 нативной константной области Fc IgG человека, каждый из которых указан согласно нумерации EU.
В другом варианте осуществления антитело содержит последовательности тяжелой цепи CDR1, CDR2 и CDR3, приведенные под SEQ ID NO: 19, 18 и 3 соответственно, и последовательности легкой цепи CDR1, CDR2 и CDR3, приведенные под SEQ ID NO: 4, 5 и 6 соответственно, и вариант константной области Fc человека, который связывается с неонатальным Fc-рецептором человека (FcRn), где вариант константной области Fc CH3 человека содержит замены Met-429-Leu и Asn-435-Ser по остаткам, соответствующим метионину 428 и аспарагину 434 нативной константной области Fc IgG человека, каждый из которых указан согласно нумерации EU.
В другом варианте осуществления антитело конкурирует за связывание и/или связывается с тем же эпитопом на C5, что и вышеупомянутые антитела. В другом варианте осуществления антитело характеризуется по меньшей мере приблизительно 90% идентичностью аминокислотной последовательности вариабельной области с вышеуказанными антителами (например, по меньшей мере приблизительно 90%, 95% или 99% идентичностью вариабельной области с SEQ ID NO: 12 и SEQ ID NO: 8).
В другом варианте осуществления антитело связывается с C5 человека при pH 7,4 и 25°C с аффинной константой диссоциации (KD), которая находится в диапазоне 0,1 нм ≤ KD ≤ 1 нм. В другом варианте осуществления антитело связывается с C5 человека при pH 6,0 и 25°C с KD ≥ 10 нм. В еще одном варианте осуществления [(KD антитела или его антигенсвязывающего фрагмента в отношении C5 человека при pH 6,0 и при 25°C)/(KD антитела или его антигенсвязывающего фрагмента в отношении C5 человека при pH 7,4 и при 25°C)] антитела составляет более 25.
В одном аспекте предусмотрен стабильный водный раствор (например, стерильный раствор), где раствор содержит антитело к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6. В другом варианте осуществления раствор содержит антитело к C5 (например, равулизумаб) в концентрации, составляющей или составляющей приблизительно 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 205, 210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295 или 300 мг/мл.
В другом варианте осуществления стабильный водный раствор содержит одно или более дополнительных средств (например, стабилизирующих средств, буферных средств, поверхностно-активных средств и/или консервантов). Например, в одном варианте осуществления стабильный водный раствор содержит стабилизатор. Иллюстративные стабилизаторы включают без ограничения полиолы, сахара (например, сахарозу или трегалозу), аминокислоты (например, аргинин), амины и высаливающие соли. В одном варианте осуществления раствор содержит по меньшей мере одно стабилизирующее средство в концентрации 2-10% включительно. В одном варианте осуществления раствор содержит 5% сахарозу. В другом варианте осуществления раствор содержит по меньшей мере одно или более стабилизирующих средств в концентрации от 10 мМ до 50 мМ включительно. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 20 мМ. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 25 мМ. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 50 мМ. В другом варианте осуществления раствор содержит 25 мМ аргинина.
В другом варианте осуществления раствор содержит по меньшей мере одно или более буферных средств. Неограничивающие примеры типичных буферов, которые могут содержаться в промывочном (промывочных) растворе(растворах), включают Tris (трис(гидроксиметил)метиламин), бис-Tris, бис-Tris-пропан, гистидин, триэтаноламин, диэтаноламин, формиат, ацетат, MES (2-(N-морфолино)этансульфоновую кислоту), фосфат, HEPES (4-2-гидроксиэтил-1-пиперазинэтансульфоновую кислоту), цитрат, MOPS (3-(N-морфолино)пропансульфоновую кислоту), TAPS (3{[трис(гидроксиметил)метил]амино}пропансульфоновую кислоту), бицин (N, N-бис(2-гидроксиэтил)глицин), трицин (N-трис(гидроксиметил)метилглицин), TES (2-{[трис(гидроксиметил)метил]амино}этансульфоновую кислоту), PIPES (пиперазин-N, N’-бис(2-этансульфоновую кислоту), какодилат (диметиларсиновую кислоту), SSC (солевой раствор с цитратом натрия) и фосфат натрия. В другом варианте осуществления буферное средство представляет собой аминокислоту. Аминокислота может представлять собой, например, аминокислоту, выбранную из группы, состоящей из гистидина (например, L-гистидина), серина (например, L-серина) и глицина (например, L-глицина). В другом варианте осуществления раствор содержит два или более буферных средства. В конкретном варианте осуществления буферное средство представляет собой фосфат натрия.
В другом варианте осуществления раствор содержит по меньшей мере одно или более буферных средств в концентрации от 10 мМ до 300 мМ включительно. В другом варианте осуществления раствор содержит по меньшей мере одно буферное средство в концентрации от 10 мМ до 200 мМ включительно. В другом варианте осуществления раствор содержит по меньшей мере одно буферное средство в концентрации от 10 мМ до 100 мМ включительно. В другом варианте осуществления раствор содержит по меньшей мере одно буферное средство в концентрации от 10 мМ до 50 мМ включительно. В другом варианте осуществления раствор содержит по меньшей мере одно буферное средство в концентрации от 20 мМ до 50 мМ включительно. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 20 мМ. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 25 мМ. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 50 мМ.
В другом варианте осуществления раствор содержит углеводный наполнитель в концентрации от 0,1 до 5%. В одном варианте осуществления углеводный наполнитель присутствует в растворе в концентрации, составляющей по меньшей мере или равной 1,5%. В другом варианте осуществления углеводный наполнитель присутствует в растворе в концентрации, составляющей по меньшей мере или равной 3%. Углеводный наполнитель может быть, например, выбран из группы, состоящей из сорбита и маннита. В другом варианте осуществления раствор содержит два или более углеводных наполнителя.
В другом варианте осуществления раствор содержит поверхностно-активное вещество. Поверхностно-активные вещества, подходящие для применения в составах по настоящему изобретению, включают без ограничения сложные эфиры жирных кислот (например, сорбитанмонокаприлат, сорбитанмонолаурат, сорбитанмонопальмитат), сорбитантриолеат, сложные эфиры глицерина и жирных кислот (например, монокаприлат глицерина, мономиристат глицерина, моностеарат глицерина), сложные эфиры полиглицерина и жирных кислот (например, декаглицерилмоностеарат, декаглицерилдистеарат, декаглицерилмонолинолеат), полиоксиэтиленовые сложные эфиры сорбитана и жирных кислот (например, полиоксиэтиленсорбитанмонолаурат, полиоксиэтиленсорбитанмоноолеат, полиоксиэтиленсорбитанмоностеарат, полиоксиэтиленсорбитанмонопальмитат, полиоксиэтиленсорбитантриолеат, полиоксиэтиленсорбитантристеарат), полиоксиэтиленовые сложные эфиры сорбита и жирных кислот (например, полиоксиэтиленсорбиттетрастеарат, полиоксиэтиленсорбиттетраолеат), полиоксиэтиленовые сложные эфиры глицерина и жирных кислот (например, полиоксиэтиленглицерилмоностеарат), сложные эфиры полиэтиленгликоля и жирных кислот (например, дистеарат полиэтиленгликоля), алкиловые эфиры полиоксиэтилена (например, лауриловый эфир полиоксиэтилена), алкиловые эфиры полиоксиэтилена-полиоксипропилена (например, полиоксиэтилен-полиоксипропиленгликоль, пропиловый эфир полиоксиэтилена-полиоксипропилена, цетиловый эфир полиоксиэтилена-полиоксипропилена), алкилфениловые эфиры полиоксиэтилена (например, нонилфениловый эфир полиоксиэтилена), полиоксиэтиленовые гидрогенизированные касторовые масла (например, полиоксиэтиленовое касторовое масло, полиоксиэтиленовое гидрогенизированное касторовое масло), полиоксиэтиленовые производные восков (например, полиоксиэтиленсорбитовый воск), полиоксиэтиленовые производные ланолина (например, полиоксиэтиленланолин) и полиоксиэтиленовые амиды жирных кислот (например, полиоксиэтиленовый амид стеариновой кислоты); C12-C18-алкилсульфаты (например, цетилсульфат натрия, лаурилсульфат натрия, олеилсульфат натрия), полиоксиэтилен-C10-C18-алкилэфирсульфат с добавлением в среднем от 2 до 4 молей этиленоксидных звеньев (например, полиоксиэтиленлаурилсульфат натрия) и соли C10-C18-алкилсульфосукцинатных сложных эфиров (например, натриевую соль лаурилсульфосукцинатного сложного эфира); а также природные поверхностно-активные вещества, такие как лецитин, глицерофосфолилипид, сфингофосфолипиды (например, сфингомиелин) и сложные эфиры сахарозы и C12-C18-жирных кислот.
В одном варианте осуществления поверхностно-активное вещество в составе представляет собой неионогенное поверхностно-активное вещество. В определенных вариантах осуществления поверхностно-активное вещество в составе представляет собой полиоксиэтиленовый сложный эфир сорбитана и жирной кислоты, например, полисорбат 20, 40, 60, 80, или комбинацию одного или более из них. В одном варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 80 (Tween 80). В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 60. В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 40. В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 20 (Tween 20). Концентрация поверхностно-активного вещества в растворе может составлять, например, от 0,001% до 0,02% включительно. Например, поверхностно-активное вещество может присутствовать в составе в количестве от приблизительно 0,001% до приблизительно 1%, или от приблизительно 0,001% до приблизительно 0,5%, или от приблизительно 0,01% до приблизительно 0,2%. В одном варианте осуществления водные растворы содержат поверхностно-активное вещество в концентрации, составляющей по меньшей мере или примерно 0,001 (например, по меньшей мере или примерно 0,002, 0,003, 0,004, 0,005, 0,006, 0,007, 0,008, 0,009, 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,11, 0,12, 0,13, 0,14, 0,15, 0,16, 0,17, 0,18, 0,19, 0,2, 0,21, 0,22, 0,23, 0,24, 0,25, 0,26, 0,27, 0,28, 0,29, 0,3, 0,31, 0,32, 0,33, 0,34, 0,35, 0,36, 0,37, 0,38, 0,39, 0,4, 0,41, 0,42, 0,43, 0,44, 0,45, 0,46, 0,47, 0,48, 0,49, 0,5 или больше) %. В другом варианте осуществления водный раствор содержит не более 0,2 (например, не более 0,19, 0,18, 0,17, 0,16, 0,15, 0,14, 0,13, 0,12, 0,11, 0,10, 0,09, 0,08, 0,07, 0,06, 0,05, 0,04, 0,03, 0,02, 0,01, 0,009, 0,008, 0,007, 0,006, 0,005, 0,004, 0,003, 0,002 или 0,001) % фармацевтически приемлемого поверхностно-активного вещества. В конкретном варианте осуществления поверхностно-активное вещество представляет собой 0,05% полисорбат 80.
В другом варианте осуществления раствор содержит консервант. Иллюстративные консерванты включают без ограничения бензиловый спирт, м-крезол и фенол.
В одном варианте осуществления стабильный водный раствор содержит не более пяти средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более четырех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более трех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более двух средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более одного средства в дополнение к антителу к C5.
В другом варианте осуществления стабильный водный раствор содержит антитело к C5, содержащее CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу и 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор состоит из антитела к C5, которое содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу и 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор содержит антитело к C5, содержащее CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу; 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; а также 0,05 ± 0,03 (например, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07 и 0,08) % полисорбат 80, где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор состоит из антитела к C5, которое содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу; 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; а также 0,05 ± 0,03 (например, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07 и 0,08) % полисорбат 80, где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления предусмотрен стабильный водный раствор (например, стерильный раствор), где раствор содержит (а) антитело к C5 (например, равулизумаб), (b) приблизительно 50 мМ фосфатного буфера; (c) приблизительно 5% сахарозу и (d) приблизительно 25 мМ аргинина. В другом варианте осуществления предусмотрен стабильный водный раствор (например, стерильный раствор), где раствор содержит (a) антитело к C5 (например, равулизумаб) в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера; (c) приблизительно 5% сахарозу и (d) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит a) антитело к C5 (например, равулизумаб), (b) 50 мМ фосфатного буфера; (c) 5% сахарозу и (d) 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 (например, равулизумаб) в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера; (c) 5% сахарозу и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу, (d) приблизительно 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу, (d) приблизительно 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит не более четырех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более трех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более двух средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более одного средства в дополнение к антителу к C5.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы и (d) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера; (c) 5% сахарозы и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы; (d) приблизительно 0,05% полисорбата 80 и (e) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера, (c) 5% сахарозы, (d) 0,05% полисорбата 80 и (e) 25 мМ аргинина.
В одном варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу и (d) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу, (d) приблизительно 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы, (d) приблизительно 0,05% полисорбата 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозы, (d) 0,05% полисорбата 80 и (e) 25 мМ аргинина.
В одном варианте осуществления pH составляет 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9. В другом варианте осуществления pH раствора составляет от 7,0 до 7,4. В другом варианте осуществления pH раствора составляет от 7,2 до 7,8. В другом варианте осуществления pH раствора составляет от 7,2 до 7,6. В конкретном варианте осуществления pH раствора составляет 7,4.
Растворы, описанные в данном документе, можно составлять для любого подходящего способа введения. В одном варианте осуществления раствор составлен для введения парентеральным способом (например, путем внутривенной, подкожной, внутрибрюшинной или внутримышечной инъекции). В конкретном варианте осуществления раствор составлен для подкожного введения. Например, в одном варианте осуществления стабильный водный раствор содержит антитело к C5 в концентрации 100 мг/мл и составлен для подкожного введения. В другом конкретном варианте осуществления раствор составлен для внутривенного введения. Например, в одном варианте осуществления стабильный водный раствор содержит антитело к C5 в концентрации 100 мг/мл и составлен для внутривенного введения.
В одном варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев при определении с помощью SEC-HPLC (например, гель-проникающей HPLC). В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере одного года при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере 18 месяцев при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере двух лет при определении с помощью SEC-HPLC.
В другом варианте осуществления любого из растворов, описанных в данном документе, менее 5% антител к C5 (например, равулизумаба) в растворе являются агрегированными при определении с помощью SEC-HPLC (например, гель-проникающей HPLC). В другом варианте осуществления менее 4% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 3% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 2% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 1% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC.
В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере одного года по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере восемнадцати месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере двух лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере трех лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения.
В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере одного года по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере 18 месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере двух лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере трех лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения.
В другом аспекте предусмотрены способы получения стабильного концентрированного раствора антитела, содержащего антитело к C5 в концентрации 100 мг/мл, 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина, при этом способ включает:
i) получение первого водного раствора, содержащего антитело к C5, при этом первый водный раствор имеет первый состав и содержит не более 10 мг/мл антитела к C5;
ii) проведение диафильтрации первого водного раствора с получением состава, содержащего 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина, при pH 7,4, вследствие чего обеспечивается получение второго водного раствора, при этом второй водный раствор имеет второй состав в результате диафильтрации; и
iii) концентрирование второго водного раствора с получением стабильного концентрированного раствора антитела, содержащего 100 мг/мл антитела к C5, 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина.
В другом варианте осуществления предусмотрен способ получения стабильного концентрированного раствора антитела, содержащего антитело к C5 в концентрации 100 мг/мл, 50 мМ фосфатного буфера, 5% сахарозу; 25 мМ аргинина и 0,05% полисорбат 80, при этом способ включает:
i) получение первого водного раствора, содержащего антитело к C5, при этом первый водный раствор имеет первый состав и содержит не более 10 мг/мл антитела к C5;
ii) проведение диафильтрации первого водного раствора с получением состава, содержащего 50 мМ фосфатного буфера, 5% сахарозу, 25 мМ аргинина и 0,05% полисорбат 80, при pH 7,4, вследствие чего обеспечивается получение второго водного раствора, при этом второй водный раствор имеет второй состав в результате диафильтрации; и
iii) концентрирование второго водного раствора с получением стабильного концентрированного раствора антитела, содержащего 100 мг/мл антитела к C5, 50 мМ фосфатного буфера, 5% сахарозу, 25 мМ аргинина и 0,05% полисорбат 80.
Также предусмотрены способы лечения пациента-человека, с состоянием, ассоциированным с системой комплемента, включающие введение пациенту стабильного водного раствора (например, подкожно или внутривенно), описанного в данном документе, в количестве, эффективном для лечения состояния, ассоциированного с системой комплемента. Иллюстративные состояния, ассоциированные с системой комплемента, включают без ограничения ревматоидный артрит, синдром антифосфолипидных антител, волчаночный нефрит, ишемическое/реперфузионное повреждение, атипичный гемолитико-уремический синдром (aHUS), типичный гемолитико-уремический синдром, пароксизмальную ночную гемоглобинурию (PNH), болезнь плотного осадка, оптиконевромиелит, мультифокальную моторную нейропатию, рассеянный склероз, макулодистрофию, синдром HELLP, самопроизвольный аборт, тромботическую тромбоцитопеническую пурпуру, малоиммунный васкулит, буллезный эпидермолиз, повторный выкидыш, черепно-мозговую травму, миокардит, цереброваскулярное нарушение, нарушение со стороны периферических сосудов, реноваскулярное нарушение, мезентериальное/энтеральное сосудистое нарушение, васкулит, нефрит при пурпуре Шенлейна-Геноха, васкулит, ассоциированный с системной красной волчанкой, васкулит, ассоциированный с ревматоидным артритом, иммунокомплексный васкулит, болезнь Такаясу, дилатационную кардиомиопатию, диабетическую ангиопатию, болезнь Кавасаки, венозную газовую эмболию, рестеноз после стентирования, ротационную атерэктомию, чрескожную транслюминальную коронарную ангиопластику, тяжелую миастению, болезнь холодовых агглютининов, дерматомиозит, пароксизмальную холодовую гемоглобинурию, антифосфолипидный синдром, болезнь Грейвса, атеросклероз, болезнь Альцгеймера, септический системный воспалительный ответ, септический шок, повреждение спинного мозга, гломерулонефрит, отторжение трансплантата, тиреоидит Хашимото, сахарный диабет I типа, псориаз, пузырчатку, аутоиммунную гемолитическую анемию, идиопатическую тромбоцитопеническую пурпуру, синдром Гудпасчера, болезнь Дегоса и катастрофический антифосфолипидный синдром. В конкретном варианте осуществления состояние, ассоциированное с системой комплемента, представляет собой атипичный гемолитико-уремический синдром (aHUS). В другом варианте осуществления состояние, ассоциированное с системой комплемента, представляет собой пароксизмальную ночную гемоглобинурию (PNH).
Дополнительно предусмотрены наборы, которые содержат стабильный водный раствор, описанный в данном документе, в терапевтически эффективном количестве, приспособленный для применения в способах, описанных в данном документе. В одном варианте осуществления набор содержит: (i) любой из растворов, описанных в данном документе; и (ii) средство для доставки раствора пациенту, нуждающемуся в этом (например, шприц). В одном варианте осуществления средство подходит для подкожной доставки раствора пациенту. В одном варианте осуществления средство подходит для внутривенной доставки раствора пациенту. В другом варианте осуществления наборы дополнительно содержат по меньшей мере одно дополнительное активное средство для применения в лечении нарушения, ассоциированного с системой комплемента, у субъекта.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 представлены результаты динамического рассеяния света для титрования равулизумаба (ALXN1210) при 50 мг/мл солью с заменой буфера на гистидиновый.
На фигуре 2 представлены результаты динамического рассеяния света для титрования равулизумаба (ALXN1210) при 50 мг/мл L-аргинином c заменой буфера.
На фигуре 3 представлены результаты динамического рассеяния света для титрования равулизумаба (ALXN1210) при 50 мг/мл солью c заменой буфера на фосфатный.
На фигуре 4 представлены результаты дифференциальной сканирующей флуориметрии равулизумаба (ALXN1210) при 50 мг/мл c заменой буфера.
На фигуре 5 представлены результаты динамического рассеяния света для равулизумаба (ALXN1210) при 10 мг/мл и 114 мг/мл без L-аргинина и при 114 мг/мл с добавлением L-аргинина.
На фигуре 6 показаны данные о стабильности для равулизумаба (ALXN1210) (с T=0 по T=2 недели включительно при 2-8°C).
На фигуре 7 показаны данные о стабильности для равулизумаба (ALXN1210) (с T=3 недели по T=2 месяца включительно при 2-8°C).
На фигуре 8 показаны данные о стабильности для равулизумаба (ALXN1210) (с T=0 по T=3 недели включительно при 23-27°C).
На фигуре 9 показаны данные о стабильности равулизумаба (ALXN1210) (с T=1 месяц по T=2 месяца включительно при 23-27°C).
На фигуре 10 показаны данные о стабильности равулизумаба (ALXN1210) (с T=1 неделя по T=3 недели включительно при 37°C).
На фигуре 11 показаны данные о стабильности равулизумаба (ALXN1210) (с T=1 месяц по T=2 месяца включительно при 37°C).
На фигуре 12 показаны данные о стабильности, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=2 месяца включительно при 2-8°C).
На фигуре 13 показаны данные о стабильности, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=2 месяца включительно при 23-27°C).
На фигуре 14 показаны данные о стабильности, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=2 месяца включительно при 37°C).
На фигуре 15 показаны данные о стабильности, полученные с помощью динамического рассеяния света, для равулизумаба (ALXN1210) в образцах с гистидином (T=0).
На фигуре 16 показаны данные о стабильности, полученные с помощью динамического рассеяния света, для образцов равулизумаба AS в гистидиновом буфере (ALXN1210) (T=0).
На фигуре 17 показаны данные о стабильности, полученные с помощью динамического рассеяния света, для образцов равулизумаба в фосфатном буфере (ALXN1210) (T=0).
На фигуре 18 показаны данные о стабильности, полученные с помощью динамического рассеяния света, для образцов равулизумаба (ALXN1210) в фосфатном буфере (T=2 месяца, 2-8°C).
На фигуре 19 показаны данные о стабильности равулизумаба, полученные в ходе замораживания-размораживания (ALXN1210) (с T=0 до цикла 2 включительно при T=1 месяц, -20°C).
На фигуре 20 показаны данные о стабильности равулизумаба (ALXN1210), полученные в ходе циклов 3-5 замораживания-размораживания включительно при T=1 месяц, -20°C.
На фигуре 21 показаны данные о стабильности, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (замораживание-размораживание при T=1 месяц, -20°C).
На фигуре 22 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=0 по T=2 месяца включительно при 2-8°C).
На фигуре 23 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (c T=3 месяца по T=6 месяцев включительно при 2-8°C).
На фигуре 24 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=9 месяцев по T=12 месяцев включительно при 2-8°C).
На фигуре 25 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=1 месяц по T=2 месяца включительно при 23-27°C).
На фигуре 26 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=3 месяца по T=6 месяцев включительно при 23-27°C).
На фигуре 27 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=9 месяцев по T=12 месяцев включительно при 23-27°C).
На фигуре 28 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=2 недели по T=2 месяца включительно при 37°C).
На фигуре 29 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=1 месяц по T=3 месяца включительно при -20°C).
На фигуре 30 показаны данные о стабильности опытных образцов для равулизумаба (ALXN1210) (с T=6 месяцев по T=12 месяцев включительно при -20°C).
На фигуре 31 показаны результаты исследования стабильности опытных образцов для равулизумаба (ALXN1210) (с T=3 месяца по T=6 месяцев включительно при -80°C).
На фигуре 32 показаны результаты исследования стабильности опытных образцов для равулизумаба (ALXN1210) (T=12 месяцев при -80°C).
На фигуре 33 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=12 месяцев включительно при 2-8°C).
На фигуре 34 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=12 месяцев включительно при 23-27°C).
На фигуре 35 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=12 месяцев включительно при 37°C).
На фигуре 36 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=12 месяцев включительно при -20°C).
На фигуре 37 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) (с T=0 по T=12 месяцев включительно при -80°C).
На фигуре 38 показаны данные о стабильности опытных образцов, полученные с помощью динамического рассеяния света, для образцов равулизумаба ALXN1210 при 75 мг/мл в фосфатном буфере (T=0).
На фигуре 39 показаны данные о стабильности опытных образцов, полученные с помощью динамического рассеяния света, для образцов равулизумаба ALXN1210 при 75 мг/мл в фосфатном буфере (T=1 месяц при 2-8°C).
На фигуре 40 показаны данные о стабильности опытных образцов, полученные с помощью динамического рассеяния света, для образцов равулизумаба ALXN1210 при 100 мг/мл в фосфатном буфере (T=1 месяц при 2-8°C).
На фигуре 41 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 1-3 замораживания-размораживания при T=1 месяц, -20°C.
На фигуре 42 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 4-5 замораживания-размораживания при T=1 месяц, -20°C.
На фигуре 43 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 1-3 замораживания-размораживания при T=3 месяца, -80°C.
На фигуре 44 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 4-5 замораживания-размораживания при T=3 месяца, -80°C.
На фигуре 45 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 1-5 замораживания-размораживания при T=1 месяц, -20°C.
На фигуре 46 показаны данные о стабильности опытных образцов, полученные с помощью эксклюзионной хроматографии (SEC), в виде % мономеров для равулизумаба (ALXN1210) в циклах 1-5 замораживания-размораживания при T=3 месяца, -80°C.
На фигуре 47 представлена общая схема исследования фазы 1, разработанного для оценки безопасности, переносимости, PK, PD и иммуногенности однократной дозы 400 мг равулизумаба (ALXN1210), вводимого подкожно, по сравнению с однократной дозой 400 мг равулизумаба (ALXN1210), вводимого внутривенно, или плацебо, вводимого подкожно, у 42 здоровых субъектов.
На фигуре 48 приведено общее описание распределения всех субъектов.
Фигура 49 представляет собой график, на котором представлены отдельные концентрации ALXN1210 в сыворотке крови в зависимости от номинального времени с применением линейной шкалы.
Фигура 50 представляет собой график, на котором представлены отдельные концентрации ALXN1210 в сыворотке крови в зависимости от номинального времени с применением линейно-логарифмической шкалы.
Фигура 51 представляет собой график, на котором представлено процентное изменение среднего значения (± SD) концентрации свободного C5 в сыворотке крови относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV.
Фигура 52 представляет собой график, на котором представлено процентное изменение средних значений (± SD) концентраций свободного C5 в сыворотке крови относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV.
Фигура 53 представляет собой график, на котором представлено процентное изменение среднего значения (± SD) степени гемолиза куриных эритроцитов (cRBC) относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении представлены стабильные водные растворы, содержащие высокую концентрацию антитела к C5 (например, равулизумаба). Растворы можно применять в различных терапевтических путях применения, таких как способы лечения или предупреждения нарушений, ассоциированных с системой комплемента. Иллюстративные решения, составы, терапевтические наборы и способы получения и применения любого из вышеприведенного раскрыты ниже и проиллюстрированы в демонстрационных примерах, и при этом они никоим образом не предполагаются как ограничивающие.
I. Определения
Если не определено иное, то все технические и научные термины, используемые в данном документе, имеют такое же значение, какое обычно понятно специалисту в данной области. Хотя при практическом осуществлении или тестировании настоящего изобретения можно применять любые способы и композиции, подобные или эквивалентные описанным в данном документе, в данном документе описаны предпочтительные способы и композиции.
Формы единственного числа включают ссылки на множественное число, если контекст явно не требует иного.
Термин «приблизительно», в частности в отношении заданного количества или числа, подразумевается как охватывающий отклонения в пределах плюс-минус десяти процентов (± 10%), (например, ± 5%).
Термин «фармацевтический состав» относится к препаратам, которые находятся в такой форме, чтобы обеспечить однозначно эффективную биологическую активность активных ингредиентов, и которые не содержат дополнительных компонентов, обладающих значительной токсичностью для субъектов, которым будет вводиться состав.
В контексте данного документа «водная» фармацевтическая композиция представляет собой композицию, подходящую для фармацевтического применения, где водный носитель представляет собой воду. Композиция, подходящая для фармацевтического применения, может быть стерильной, однородной и/или изотонической. Водные фармацевтические композиции можно получать непосредственно в водной форме и/или можно восстанавливать из лиофилизата.
«Изотонический» состав представляет собой состав, который имеет по существу такое же осмотическое давление, как кровь человека. Изотонические составы обычно будут характеризоваться осмотическим давлением, составляющим от приблизительно 275 до 350 мОсм/кг. Термин «гипотонический» описывает состав с осмотическим давлением, более низким, чем в крови человека. Соответственно, термин «гипертонический» используется для описания состава с осмотическим давлением, более высоким, чем в крови человека. Изотоничность может быть измерена, например, с помощью осмометра давления пара или с помощью осмометра по точке замерзания. «Средство, регулирующее тоничность» представляет собой соединение, которое обеспечивает изотоничность состава.
В контексте данного документа «осмоляльность» раствора представляет собой число осмолей растворенного вещества на килограмм растворителя. Осмоляльность представляет собой меру количества частиц, присутствующих в растворе, и не зависит от размера или веса частиц. Ее можно измерять только при использовании свойства раствора, которое зависит только от концентрации частиц. Эти свойства представляют собой понижение давления пара, понижение температуры замерзания, повышение температуры кипения и осмотическое давление и совокупно называются коллигативными свойствами.
«Стерильный» состав является асептическим или не содержит или практически не содержит никаких живых микроорганизмов и их спор.
«Стабильный» состав в контексте данного документа представляет собой состав, в котором антитело по существу сохраняет свою физическую стабильность, и/или химическую стабильность, и/или биологическую активность при хранении. Различные аналитические методики для измерения стабильности белка доступны из уровня техники и рассматриваются в Peptide and Protein Drug Delivery, 247-301, Vincent Lee Ed., Marcel Dekker, Inc., New York, N.Y., Pubs. (1991) и Jones, A. Adv. Drug Delivery Rev. 10: 29-90 (1993). Стабильность составов антитела к C5 можно измерять при выбранных температурах по истечении выбранных периодов времени. Например, увеличение образования агрегатов после хранения является индикатором нестабильности водного состава антитела к C5. В дополнение к образованию агрегатов, сохранение исходной прозрачности, цвета и запаха в течение всего срока годности представляют собой индикаторы, используемые для контроля стабильности водных растворов антител к C5, описанных в данном документе.
Антитело «сохраняет свою физическую стабильность» в фармацевтическом составе, если оно практически не проявляет признаков агрегации, осаждения и/или денатурации при визуальном исследовании цвета и/или прозрачности или при измерении с помощью рассеяния УФ-света или с помощью эксклюзионной хроматографии.
Термин «агрегация» относится к сборке нативных свернутых белков с образованием агрегатов, содержащих ненативные структуры. Агрегация может происходить даже в физиологических, неденатурирующих условиях и часто является необратимой, что приводит к образованию ненативных агрегатов, которые являются неактивными и иногда иммуногенными и токсичными.
Фраза «низкие, вплоть до невыявляемых, уровни агрегации» в контексте данного документа относится к образцам, содержащим не более чем приблизительно 5%, не более чем приблизительно 4%, не более чем приблизительно 3%, не более чем приблизительно 2%, не более чем приблизительно 1% и не более чем приблизительно 0,5% агрегатов по весу белка при измерении с помощью методик высокоэффективной гель-проникающей жидкостной хроматографии (GP-HPLC), высокоэффективной эксклюзионной хроматографии (HPSEC) или статического рассеяния света (SLS).
Антитело «сохраняет свою химическую стабильность» в фармацевтическом составе, если химическая стабильность в данный момент времени является таковой, что считается, что антитело по-прежнему сохраняет свою биологическую активность, как определено ниже. Химическую стабильность можно оценивать путем выявления и количественной оценки химически измененных форм антитела. Химические изменения могут включать модификацию размера (например, усечение), дезамидирование, рацемизацию, гидролиз, окисление, бета-элиминирование и дисульфидный обмен, которые можно оценивать с помощью известных методик, например, эксклюзионной хроматографии, SDS-PAGE, матрично-активированной лазерной десорбции-ионизации/времяпролетной масс-спектрометрии (MALDI/TOF MS) и/или ионообменной хроматографии.
Антитело «сохраняет свою биологическую активность» в фармацевтическом составе, если антитело в фармацевтическом составе является биологически активным для его предполагаемой цели. Например, биологическая активность сохраняется, если биологическая активность антитела в фармацевтическом составе находится в пределах приблизительно 30%, приблизительно 20% или приблизительно 10% (в пределах погрешности анализа) биологической активности, проявляемой на момент получения фармацевтической лекарственной формы (например, как определено в анализе связывания антигена). В данном документе «биологическая активность» моноклонального антитела относится к способности антитела связываться с антигеном. Она может дополнительно включать связывание антитела с антигеном и приводить к измеримому биологическому ответу, который можно измерить in vitro или in vivo.
«Срок годности» фармацевтического продукта, например, водного раствора, содержащего антитело к C5, представляет собой период времени, в течение которого продукт сохраняется перед тем, как произойдет разложение. Например, срок годности может быть определен как время для разложения 0,1%, 0,5%, 1%, 5% или 10% продукта.
В контексте данного документа термин «антитело» описывает полипептиды, содержащие по меньшей мере один антигенсвязывающий участок, полученный из антитела (например, VH/VL-область, или Fv, или CDR). Антитела включают известные формы антител. Например, антитело может представлять собой антитело человека, гуманизированное антитело, биспецифическое антитело или химерное антитело. Антитело также может представлять собой Fab, Fab’2, ScFv, SMIP, Affibody®, нанотело или доменное антитело. Антитело также может относиться к любому из следующих изотипов: IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, секреторному IgA, IgD и IgE. Антитело может представлять собой антитело, встречающееся в природе, или может представлять собой антитело, которое было изменено с помощью технологии белковой инженерии (например, посредством мутации, делеции, замены, конъюгирования с фрагментом, отличным от антитела). Например, антитело может содержать один или более вариантов аминокислот (по сравнению с встречающимся в природе антителом), которые изменяют свойство (например, функциональное свойство) антитела. Например, из уровня техники известны такие многочисленные изменения, которые влияют на, например, время полужизни, эффекторную функцию и/или иммунные ответы на антитело у пациента. Термин «антитело» также включает искусственные или сконструированные полипептидные конструкции, которые содержат по меньшей мере один антигенсвязывающий участок, полученный из антитела.
В контексте данного документа термины «специфичное связывание», «селективное связывание», «селективно связывает» и «специфично связывает» относятся к связыванию антитела с эпитопом в предварительно определенном антигене, но не с другими антигенами. Как правило, антитело (i) связывается с равновесной константой диссоциации (KD), составляющей примерно менее чем 10-7 M, как, например, примерно менее чем 10-8 М, 10-9 M или 10-10 М или даже ниже, при определении, например, с помощью технологии поверхностного плазмонного резонанса (SPR) с использованием прибора BIACORE® 2000, работающего на основе принципа поверхностного плазмонного резонанса, с применением предварительно определенного антигена, например C5, в качестве аналита и антитела в качестве лиганда, или анализа связывания антитела с антиген-положительными клетками по методу Скэтчарда, и (ii) связывается с предварительно определенным антигеном с аффинностью, которая по меньшей мере в два раза превышает его аффинность связывания с неспецифическим антигеном (например, BSA, казеином), отличным от предварительно определенного антигена или близкородственного антигена. Соответственно, если не указано иное, антитело, которое «специфично связывается с C5 человека», относится к антителу, которое связывается с растворимым или связанным с клетками C5 с KD, составляющей 10-7 M или меньше, как, например, примерно менее чем 10-8 М, 10-9 M или 10-10 М или даже ниже.
Термин «поверхностный плазмонный резонанс» в контексте данного документа относится к оптическому явлению, которое обеспечивает анализ биоспецифических взаимодействий в режиме реального времени путем выявления изменений концентраций белков в биосенсорной матрице, например, с применением системы BIAcore (Pharmacia Biosensor AB, Уппсала, Швеция и Пискатауэй, Нью-Джерси). Для получения дополнительного описания см. Jonsson, U., et al. (1993) Ann. Biol. Clin. 51:19-26; Jonsson, U., et al. (1991) Biotechniques 11:620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8:125-131; и Johnnson, B., et al. (1991) Anal. Biochem. 198:268-277.
Термин «Koff» в контексте данного документа предназначен для обозначения константы скорости диссоциации антитела из комплекса антитело/антиген.
Термин "Kd" в контексте данного документа предназначен для обозначения константы диссоциации для конкретного взаимодействия антитело-антиген.
В контексте данного документа термины «субъект» или «пациент» используются в данном документе взаимозаменяемо и относятся к млекопитающему, такому как человек, мышь, крыса, хомяк, морская свинца, кролик, кошка, собака, обезьяна, корова, лошадь, свинья и т. п. В одном варианте осуществления пациент является пациентом-человеком (например, пациентом-человеком с состоянием, ассоциированным с системой комплемента).
Термины «лечить», «осуществление лечения» и «лечение» в контексте данного документа относятся к терапевтическим мерам, описанным в данном документе. В способах «лечения» используется введение субъекту комбинации, раскрытой в данном документе, для излечения, задержки, снижения тяжести или уменьшения интенсивности одного или более симптомов заболевания или нарушения или рецидивирующего заболевания и нарушения или для продления выживания субъекта за пределы ожидаемого при отсутствии такого лечения.
В контексте данного документа термин «эффективное лечение» относится к лечению, которое дает благоприятный эффект, например, уменьшение интенсивности по меньшей мере одного симптома заболевания или нарушения. Благоприятный эффект может принимать форму улучшения по сравнению с исходным уровнем, например, улучшение по сравнению с измерением или наблюдением, осуществляемым перед началом терапии в соответствии со способом. Эффективное лечение может относиться к смягчению по меньшей мере одного симптома заболевания или состояния.
Термин «эффективное количество» относится к количеству средства, которое обеспечивает желаемый биологический, терапевтический и/или профилактический результат. Результатом этого может являться ослабление, уменьшение интенсивности, временное облегчение, снижение тяжести, задержка и/или смягчение одного или более признаков, симптомов или причин заболевания или состояния или любое другое желаемое изменение биологической системы. В одном примере «эффективное количество» представляет собой количество стабильного водного раствора для смягчения по меньшей мере одного симптома заболевания или состояния. Эффективное количество можно вводить за одно или более введений.
В контексте данного документа термины «индукция» и «фаза индукции» используются взаимозаменяемо и относятся к первой фазе лечения.
В контексте данного документа термины «поддержание» и «фаза поддержания» используются взаимозаменяемо и относятся ко второй фазе лечения. В определенных вариантах осуществления лечение продолжается при условии, что наблюдается клиническая польза, или до тех пор, пока не появляется неконтролируемая токсичность или прогрессирование заболевания.
II. Антитела к C5
Антитела к C5, описанные в данном документе, связываются с компонентом С5 системы комплемента (например, C5 человека) и ингибируют расщепление C5 на фрагменты C5a и C5b. Как описано выше, такие антитела также характеризуются, например, улучшенными фармакокинетическими свойствами по сравнению с другими антителами к C5 (например, экулизумабом), используемыми в терапевтических целях.
Антитела к C5 (или домены VH/VL, полученные из них), подходящие для применения в настоящем изобретении, могут быть созданы с применением способов, хорошо известных из уровня техники. В качестве альтернативы, можно применять известные в данной области техники антитела к C5. Также можно применять антитела, которые конкурируют за связывание с C5 с любым из известных в данной области техники антител.
Иллюстративное антитело к C5 представляет собой равулизумаб, содержащий тяжелые и легкие цепи, имеющие последовательности, показанные под SEQ ID NO: 14 и 11 соответственно, или его антигенсвязывающие фрагменты и варианты. Равулизумаб (также известный как BNJ441 и ALXN1210) описан в PCT/US2015/019225 и патенте США № 9079949, идеи которого включены в данный документ посредством ссылки. Во всем данном документе термины равулизумаб, BNJ441 и ALXN1210 могут использоваться взаимозаменяемо. Равулизумаб селективно связывается с белком C5 системы комплемента человека, ингибируя его расщепление на C5a и C5b во время активации системы комплемента. Данное ингибирование предотвращает высвобождение провоспалительного медиатора C5a и образование цитолитического порообразующего мембраноатакующего комплекса (MAC) C5b-9 при сохранении проксимальных или ранних компонентов пути активации системы комплемента (например, C3 и C3b), существенных для опсонизации микроорганизмов и выведения иммунных комплексов.
В других вариантах осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области равулизумаба. Соответственно, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 VH-области равулизумаба, имеющей последовательность, приведенную под SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 VL-области равулизумаба, имеющей последовательность, приведенную под SEQ ID NO: 8. В другом варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, приведенные под SEQ ID NO: 19, 18 и 3 соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, приведенные под SEQ ID NO: 4, 5 и 6 соответственно. В другом варианте осуществления антитело содержит VH- и VL-области, имеющие аминокислотные последовательности, приведенные под SEQ ID NO: 12 и SEQ ID NO: 8 соответственно.
Другим иллюстративным антителом к C5 является антитело BNJ421, содержащее тяжелые и легкие цепи, имеющие последовательности, показанные под SEQ ID NO: 20 и 11 соответственно, или его антигенсвязывающие фрагменты и варианты. BNJ421 (также известный как ALXN1211) описан в PCT/US2015/019225 и патенте США № 9079949, идеи которых включены в данный документ посредством ссылки.
В других вариантах осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области BNJ421. Соответственно, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 VH-области BNJ421, имеющей последовательность, приведенную под SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 VL-области BNJ421, имеющей последовательность, приведенную под SEQ ID NO: 8. В другом варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, приведенные под SEQ ID NO: 19, 18 и 3 соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, приведенные под SEQ ID NO: 4, 5 и 6 соответственно. В другом варианте осуществления антитело содержит VH- и VL-области, имеющие аминокислотные последовательности, приведенные под SEQ ID NO: 12 и SEQ ID NO: 8 соответственно.
Точные границы CDR определялись различным образом в соответствии с различными способами. В некоторых вариантах осуществления положения CDR или каркасных областей в вариабельном домене легкой цепи или тяжелой цепи могут быть определены в соответствии с Kabat et al. [(1991) «Sequences of Proteins of Immunological Interest». NIH Publication No. 91-3242, U.S. Department of Health and Human Services, Bethesda, MD]. В таких случаях CDR могут называться «CDR согласно Kabat» (например, «LCDR2 согласно Kabat» или «HCDR1 согласно Kabat»). В некоторых вариантах осуществления положения CDR вариабельной области легкой или тяжелой цепи могут быть определены в соответствии с Chothia et al. (1989) Nature 342:877-883. Соответственно, данные области могут называться «CDR согласно Chothia» (например, «LCDR2 согласно Chothia» или «HCDR3 согласно Chothia»). В некоторых вариантах осуществления положения CDR вариабельной области легкой и тяжелой цепей могут быть определены с помощью комбинированного определения Kabat и Chothia. В таких вариантах осуществления данные области могут называться «CDR согласно комбинированному определению Kabat и Chothia». В Thomas et al. [(1996) Mol Immuno 33(17/18):1389-1401] проиллюстрирована идентификация границ CDR согласно определению Kabat и Chothia.
В некоторых вариантах осуществления антитело к C5, описанное в данном документе, содержит CDR1 тяжелой цепи, содержащую следующую аминокислотную последовательность или состоящую из нее: GHIFSNYWIQ (SEQ ID NO: 19). В некоторых вариантах осуществления антитело к C5, описанное в данном документе, содержит CDR2 тяжелой цепи, содержащую следующую аминокислотную последовательность или состоящую из нее: EILPGSGHTEYTENFKD (SEQ ID NO: 18). В некоторых вариантах осуществления антитело к C5, описанное в данном документе, содержит вариабельную область тяжелой цепи, содержащую следующую аминокислотную последовательность:
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEWMGEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCARYFFGSSPNWYFDVWGQGTLVTVSS (SEQ ID NO: 12).
В некоторых вариантах осуществления антитело к C5, описанное в данном документе, содержит вариабельную область легкой цепи, содержащую следующую аминокислотную последовательность:
DIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKPGKAPKLLIYGATNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQGTKVEIK (SEQ ID NO: 8).
Антитело к C5, описанное в данном документе, может в некоторых вариантах осуществления содержать вариант константной области Fc человека, который связывается с неонатальным Fc-рецептором человека (FcRn) с большей аффинностью, чем нативная константная область Fc человека, из которой был получен вариант константной области Fc человека. Например, константная область Fc может содержать одну или более (например, две, три, четыре, пять, шесть, семь или восемь или больше) аминокислотных замен по сравнению с нативной константной областью Fc человека, из которой был получен вариант константной области Fc человека. Замены могут повышать аффинность связывания антитела IgG, содержащего вариант константной области Fc, с FcRn при pH 6,0, при сохранении зависимости взаимодействия от pH. Способы тестирования того, увеличивает ли одна или более замен в константной области Fc антитела аффинность константной области Fc к FcRn при pH 6,0 (при сохранении зависимости взаимодействия от pH), известны из уровня техники и проиллюстрированы в демонстрационных примерах. См., например, PCT/US2015/019225 и патент США № 9079949, раскрытие каждого из которых включено в данный документ посредством ссылки во всей своей полноте.
Замены, которые усиливают аффинность связывания константной области Fc антитела с FcRn, известны из уровня техники и включают, например, (1) тройную замену M252Y/S254T/T256E, описанную в Dall’Acqua et al. (2006) J Biol Chem 281: 23514-23524; (2) замены M428L или T250Q/M428L, описанные в Hinton et al. (2004) J Biol Chem 279:6213-6216 и в Hinton et al. (2006) J Immunol 176:346-356; и (3) замены N434A или T307/E380A/N434A, описанные в Petkova et al. (2006) Int Immunol 18(12):1759-69. Дополнительные пары замен P257I/Q311I, P257I/N434H и D376V/N434H описаны, например, в Datta-Mannan et al. (2007) J Biol Chem 282(3):1709-1717, раскрытие которого включено в данный документ посредством ссылки во всей своей полноте.
В некоторых вариантах осуществления вариант константной области имеет замену валина в положении аминокислотного остатка 255 согласно EU. В некоторых вариантах осуществления вариант константной области имеет замену аспарагина в положении аминокислотного остатка 309 согласно EU. В некоторых вариантах осуществления вариант константной области имеет замену изолейцина в положении аминокислотного остатка 312 согласно EU. В некоторых вариантах осуществления вариант константной области имеет замену в положении аминокислотного остатка 386 согласно EU.
В некоторых вариантах осуществления вариант константной области Fc содержит не более 30 (например, не более 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, девяти, восьми, семи, шести, пяти, четырех, трех или двух) аминокислотных замен, вставок или делеций по сравнению с нативной константной областью, из которой он был получен. В некоторых вариантах осуществления вариант константной области Fc содержит одну или более аминокислотных замен, выбранных из группы, состоящей из M252Y, S254T, T256E, N434S, M428L, V259I, T250I и V308F. В некоторых вариантах осуществления вариант константной области Fc человека содержит метионин в положении 428 и аспарагин в положении 434, каждый из которых указан согласно нумерации EU. В некоторых вариантах осуществления вариант константной области Fc содержит двойную замену 428L/434S, как описано, например, в патенте США № 8088376.
В некоторых вариантах осуществления точное местоположение данных мутаций может быть смещено относительно положения нативной константной области Fc человека в связи с конструированием антител. Например, двойная замена 428L/434S при использовании в химерном Fc-фрагменте IgG2/4 может соответствовать 429L и 435S, как в вариантах M429L и N435S, обнаруженных для равулизумаба (BNJ441) и описанных в патенте США № 9079949, раскрытие которого включено в данный документ посредством ссылки во всей своей полноте.
В некоторых вариантах осуществления вариант константной области содержит замену в положении аминокислоты 237, 238, 239, 248, 250, 252, 254, 255, 256, 257, 258, 265, 270, 286, 289, 297, 298, 303, 305, 307, 308, 309, 311, 312, 314, 315, 317, 325, 332, 334, 360, 376, 380, 382, 384, 385, 386, 387, 389, 424, 428, 433, 434 или 436 (нумерация согласно EU) по сравнению с нативной константной областью Fc человека. В некоторых вариантах осуществления замена выбрана из группы, состоящей из метионина вместо глицина в положении 237; аланина вместо пролина в положении 238; лизина вместо серина в положении 239; изолейцина вместо лизина в положении 248; аланина, фенилаланина, изолейцина, метионина, глутамина, серина, валина, триптофана или тирозина вместо треонина в положении 250; фенилаланина, триптофана или тирозина вместо метионина в положении 252; треонина вместо серина в положении 254; глутаминовой кислоты вместо аргинина в положении 255; аспарагиновой кислоты, глутаминовой кислоты или глутамина вместо треонина в положении 256; аланина, глицина, изолейцина, лейцина, метионина, аспарагина серина, треонина или валина вместо пролина в положении 257; гистидина вместо глутаминовой кислоты в положении 258; аланина вместо аспарагиновой кислоты в положении 265; фенилаланина вместо аспарагиновой кислоты в положении 270; аланина или глутаминовой кислоты вместо аспарагина в положении 286; гистидина вместо треонина в положении 289; аланина вместо аспарагина в положении 297; глицина вместо серина в положении 298; аланина вместо валина в положении 303; аланина вместо валина в положении 305; аланина, аспарагиновой кислоты, фенилаланина, глицина, гистидина, изолейцина, лизина, лейцина, метионина, аспарагина, пролина, глутамина, аргинина, серина, валина, триптофана или тирозина вместо треонина в положении 307; аланина, фенилаланина, изолейцина, лейцина, метионина, пролина, глутамина или треонина вместо валина в положении 308; аланина, аспарагиновой кислоты, глутаминовой кислоты, пролина или аргинина вместо лейцина или валина в положении 309; аланина, гистидина или изолейцина вместо глутамина в положении 311; аланина или гистидина вместо аспарагиновой кислоты в положении 312; лизина или аргинина вместо лейцина в положении 314; аланина или гистидина вместо аспарагина в положении 315; аланина вместо лизина в положении 317; глицина вместо аспарагина в положении 325; валина вместо изолейцина в положении 332; лейцина вместо лизина в положении 334; гистидина вместо лизина в положении 360; аланина вместо аспарагиновой кислоты в положении 376; аланина вместо глутаминовой кислоты в положении 380; аланина вместо глутаминовой кислоты в положении 382; аланина вместо аспарагина или серина в положении 384; аспарагиновой кислоты или гистидина вместо глицина в положении 385; пролина вместо глутамина в положении 386; глутаминовой кислоты вместо пролина в положении 387; аланина или серина вместо аспарагина в положении 389; аланина вместо серина в положении 424; аланина, аспарагиновой кислоты, фенилаланина, глицина, гистидина, изолейцина, лизина, лейцина, аспарагина, пролина, глутамина, серина, треонина, валина, триптофана или тирозина вместо метионина в положении 428; лизина вместо гистидина в положении 433; аланина, фенилаланина, гистидина, серина, триптофана или тирозина вместо аспарагина в положении 434 и гистидина вместо тирозина или фенилаланина в положении 436, каждый из которых указан согласно нумерации EU.
Подходящие антитела к C5 для применения в способах, описанных в данном документе, в некоторых вариантах осуществления содержат полипептидную тяжелую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 14, и/или полипептидную легкую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 11. В качестве альтернативы, антитела к C5 для применения в способах, описанных в данном документе, в некоторых вариантах осуществления содержат полипептидную тяжелую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 20, и/или полипептидную легкую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 11.
В одном варианте осуществления антитело связывается с C5 при pH 7,4 и 25°C (и в иных случаях при физиологических условиях) с аффинной константой диссоциации (KD), которая составляет по меньшей мере 0,1 (например, по меньшей мере 0,15, 0,175, 0,2, 0,25, 0,275, 0,3, 0,325, 0,35, 0,375, 0,4, 0,425, 0,45, 0,475, 0,5, 0,525, 0,55, 0,575, 0,6, 0,625, 0,65, 0,675, 0,7, 0,725, 0,75, 0,775, 0,8, 0,825, 0,85, 0,875, 0,9, 0,925, 0,95 или 0,975) нМ. В некоторых вариантах осуществления KD антитела к C5 или его антигенсвязывающего фрагмента составляет не более 1 (например, не более 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3 или 0,2) нМ.
В других вариантах осуществления [(KD антитела к C5 при pH 6,0 при C)/(KD антитела к C5 при pH 7,4 при 25°C)] составляет более 21 (например, более 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500 или 8000).
Способы определения того, связывается ли антитело с белковым антигеном, и/или аффинности антитела к белковому антигену известны из уровня техники. Например, связывание антитела с белковым антигеном можно выявить и/или рассчитать количественно с применением разнообразных методик, таких как без ограничения вестерн-блоттинг, дот-блоттинг, способ поверхностного плазмонного резонанса (SPR) (например, в системе BIAcore; Pharmacia Biosensor AB, Уппсала, Швеция и Пискатауэй, Нью-Джерси) или твердофазный иммуноферментный анализ (ELISA). См., например, Benny K. C. Lo (2004) «Antibody Engineering: Methods and Protocols», Humana Press (ISBN: 1588290921); Johne et al. (1993) J Immunol Meth 160:191-198; Jonsson et al. (1993) Ann Biol Clin 51:19-26; и Jonsson et al. (1991) Biotechniques 11:620-627. Кроме того, способы измерения аффинности (например, констант диссоциации и ассоциации) изложены в демонстрационных примерах.
В контексте данного документа термин «ka» относится к константе скорости ассоциации антитела с антигеном. Термин «kd» относится к константе скорости диссоциации антитела из комплекса антитело/антиген. А термин «KD» относится к равновесной константе диссоциации для взаимодействия антитело-антиген. Равновесная константа диссоциации выводится из соотношений кинетических констант скоростей KD=ka/kd. Такие определения предпочтительно определяют при 25°C или 37°C (см. демонстрационные примеры). Например, кинетику связывания антитела с C5 человека можно определить при pH 8,0, 7,4, 7,0, 6,5 и 6,0 посредством поверхностного плазмонного резонанса (SPR) на приборе BIAcore 3000 с применением способа с использованием антител к Fc для иммобилизации антитела.
В одном варианте осуществления антитело к C5 или его антигенсвязывающий фрагмент блокируют образование или активность активных фрагментов C5a и/или C5b белка C5 (например, белка C5 человека). Благодаря такому эффекту блокирования антитела ингибируют, например, провоспалительные эффекты C5a и образование мембраноатакующего комплекса (MAC) C5b-9 на поверхности клетки.
Способы определения того, ингибирует ли конкретное антитело, описанное в данном документе, расщепление C5, известны из уровня техники. Ингибирование компонента С5 системы комплемента человека может снижать способность системы комплемента к клеточному лизису в биологических жидкостях организма субъекта. Такие снижения способности системы комплемента, присутствующей в биологической(биологических) жидкости(жидкостях) организма, к клеточному лизису можно измерить с помощью способов, хорошо известных из уровня техники, как, например, с помощью традиционного анализа гемолиза, такого как анализ гемолиза, описанный Kabat и Mayer (eds.), «Experimental Immunochemistry, 2nd Edition», 135-240, Springfield, IL, CC Thomas (1961), pages 135-139, или традиционного варианта такого анализа, такого как способ с использованием гемолиза куриных эритроцитов, описанный, например, в Hillmen et al. (2004) N Engl J Med 350(6):552. Способы определения того, ингибирует ли соединение-кандидат расщепление C5 человека на формы C5a и C5b, известны из уровня техники и описаны в Evans et al. (1995) Mol Immunol 32(16):1183-95. Например, концентрацию и/или физиологическую активность C5a и C5b в биологической жидкости можно измерять с помощью способов, хорошо известных из уровня техники. Для C5b можно применять анализы гемолиза или анализы на растворимый C5b-9, как обсуждается в данном документе. Также можно применять другие анализы, известные из уровня техники. С применением анализов этих или других подходящих типов можно подвергать скринингу средства-кандидаты, способные ингибировать компонент С5 системы комплемента человека.
Иммунологические методики, такие как без ограничения ELISA, можно использовать для измерения концентрации белка C5 и/или его продуктов расщепления для определения способности антитела к C5 или его антигенсвязывающего фрагмента ингибировать преобразование C5 в биологически активные продукты. В некоторых вариантах осуществления измеряют образование C5a. В некоторых вариантах осуществления для выявления образования терминальных компонентов системы комплемента применяют антитела, специфичные к неоэпитопу C5b-9.
Анализы гемолиза можно применять для определения ингибирующей активности антитела к C5 или его антигенсвязывающего фрагмента в отношении активации системы комплемента. Для определения эффекта антитела к C5 или его антигенсвязывающего фрагмента в отношении гемолиза, опосредованного классическим путем активации системы комплемента, в тестируемом растворе сыворотки крови in vitro, например, в качестве клеток-мишеней используют эритроциты овцы, покрытые гемолизином, или куриные эритроциты, сенсибилизированные антителами к куриным эритроцитам. Процент лизиса нормализовали, рассматривая 100% лизис как равный лизису, происходящему в отсутствие ингибитора. В некоторых вариантах осуществления система комплемента активируется по классическому пути антителом IgM человека, например, используемым в наборе для активации системы комплемента по классическому пути Wieslab® (Wieslab® COMPL CP310, Euro-Diagnostica, Швеция). Вкратце, тестируемую сыворотку крови инкубируют с антителом к C5 или его антигенсвязывающим фрагментом в присутствии антитела IgM человека. Образующееся количество C5b-9 измеряют путем приведения смеси в контакт с антителом к C5b-9, конъюгированным с ферментом, и флуорогенным субстратом и измерения поглощения при соответствующей длине волны. В качестве контроля тестируемую сыворотку крови инкубируют в отсутствие антитела к C5 или его антигенсвязывающего фрагмента. В некоторых вариантах осуществления тестируемая сыворотка крови представляет собой сыворотку крови с дефицитом С5, восстановленную с помощью полипептида C5.
Для определения эффекта антитела к C5 или его антигенсвязывающего фрагмента в отношении гемолиза, опосредованного альтернативным путем, в качестве клеток-мишеней можно использовать несенсибилизированные эритроциты кролика или морской свинки. В некоторых вариантах осуществления тестируемый раствор сыворотки крови представляет собой сыворотку крови с дефицитом C5, восстановленную с помощью полипептида C5. Процент лизиса нормализовали, рассматривая 100% лизис как равный лизису, происходящему в отсутствие ингибитора. В некоторых вариантах осуществления система комплемента активируется по альтернативному пути молекулами липополисахаридов, например, используемыми в наборе для активации системы комплемента по альтернативному пути Wieslab® (Wieslab® COMPL AP330, Euro-Diagnostica, Швеция). Вкратце, тестируемую сыворотку крови инкубируют с антителом к C5 или его антигенсвязывающим фрагментом в присутствии липополисахарида. Образующееся количество C5b-9 измеряют путем приведения смеси в контакт с антителом к C5b-9, конъюгированным с ферментом, и флуорогенным субстратом и измерения флуоресценции при соответствующей длине волны. В качестве контроля тестируемую сыворотку крови инкубируют в отсутствие антитела к C5 или его антигенсвязывающего фрагмента.
В некоторых вариантах осуществления активность C5 или ее ингибирование оценивают количественно с применением анализа CH50eq. Анализ CH50eq представляет собой способ измерения общей активности системы комплемента, активируемой по классическому пути, в сыворотке крови. Данный тест представляет собой анализ лизиса, в котором используют эритроциты, сенсибилизированные антителами, в качестве активатора системы комплемента по классическому пути и различные разведения тестируемой сыворотки крови для определения количества, необходимого для обеспечения 50% лизиса (CH50). Процентную степень гемолиза можно определить, например, с помощью спектрофотометра. Анализ CH50eq обеспечивает косвенную меру образования терминального комплекса системы комплемента (TCC), поскольку сам TCC непосредственно отвечает за измеряемую степень гемолиза.
Анализ хорошо известен и обычно практикуется специалистами в данной области. Вкратце, для активации системы комплемента по классическому пути неразбавленные образцы сыворотки крови (например, восстановленные образцы сыворотки крови человека) добавляют в ячейки микропланшета, содержащие эритроциты, сенсибилизированные антителами, с получением таким образом TCC. Затем активированные образцы сыворотки крови разбавляют в ячейках микропланшета, которые покрыты реагентом захвата (например, антителом, которое связывается с одним или более компонентами TCC). TCC, присутствующий в активированных образцах, связывается с моноклональными антителами, покрывающими поверхность ячеек микропланшета. Ячейки промывают, и в каждую лунку добавляют детекторный реагент, который имеет детектируемую метку и распознает связанный TCC. Детектируемая метка может представлять собой, например, флуоресцентную метку или ферментативную метку. Результаты анализа выражают в единицах эквивалентов CH50 на миллилитр (ед. экв. CH50/мл).
Ингибирование, например, в той мере, в какой оно относится к активности терминальных компонентов системы комплемента, включает по меньшей мере 5% (например, по меньшей мере 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55 или 60%) снижение активности терминальных компонентов системы комплемента, например, в анализе гемолиза или анализе CH50eq по сравнению с эффектом контрольного антитела (или его антигенсвязывающего фрагмента) в аналогичных условиях и при эквимолярной концентрации. Значительное ингибирование в контексте данного документа относится к ингибированию указанной активности (например, активности терминальных компонентов системы комплемента) на по меньшей мере 40% (например, по меньшей мере 45, 50, 55, 60, 65, 70, 75, 80, 85, 90 или 95% или больше). В некоторых вариантах осуществления антитело к C5, описанное в данном документе, содержит одну или более аминокислотных замен по сравнению с CDR экулизумаба (т.е. SEQ ID NO: 1-6), но при этом сохраняет по меньшей мере 30% (например, по меньшей мере 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90 или 95%) от ингибирующей активности экулизумаба в отношении системы комплемента в анализе гемолиза или анализе CH50eq.
Антитело к C5, описанное в данном документе, имеет период полужизни в сыворотке крови человека, составляющий по меньшей мере 20 (например, по меньшей мере 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54 или 55) дней. В другом варианте осуществления антитело к C5, описанное в данном документе, имеет период полужизни в сыворотке крови человека, составляющий по меньшей мере 40 дней. В другом варианте осуществления антитело к C5, описанное в данном документе, имеет период полужизни в сыворотке крови человека, составляющий примерно 43 дня. В другом варианте осуществления антитело к C5, описанное в данном документе, имеет период полужизни в сыворотке крови человека, составляющий 39-48 дней. Способы измерения периода полужизни антитела в сыворотке крови известны из уровня техники. В некоторых вариантах осуществления, описанных в данном документе, антитело к C5 или его антигенсвязывающий фрагмент имеет период полужизни в сыворотке крови, на по меньшей мере 20% (например, на по меньшей мере 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 250, 300, 400, 500%) превышающий период полужизни экулизумаба в сыворотке крови, например, при измерении в одной из мышиных модельных систем, описанных в демонстрационных примерах (например, в модельной системе на мышах c дефицитом С5/NOD/SCID или мышах, трансгенных по hFcRn).
В одном варианте осуществления антитело конкурирует за связывание и/или связывается с тем же эпитопом на C5, что и антитела, описанные в данном документе. Термин «связывается с тем же эпитопом» в отношении двух или более антител означает, что антитела связываются с одним и тем же сегментом из аминокислотных остатков, определяемым с помощью данного способа. Методики определения того, связываются ли антитела с «тем же эпитопом на C5», что и антитела, описанные в данном документе, включают, например, способы картирования эпитопов, такие как рентгеновские анализы кристаллов комплексов антиген:антитело, которые обеспечивают атомное разрешение эпитопа, и масс-спектрометрия водородно-дейтериевого обмена (HDX-MS). Другие способы обеспечивают контроль связывания антитела с пептидными фрагментами антигена или мутантными вариантами антигена, где утрата связывания вследствие модификации аминокислотного остатка в пределах последовательности антигена часто считается указанием на компонент эпитопа. Кроме того, также можно применять способы вычислительной комбинаторики для картирования эпитопов. В основе этих способов лежит способность антитела, представляющего интерес, к аффинному выделению специфических коротких пептидов из комбинаторных фаг-дисплейных библиотек пептидов. Ожидается, что антитела, имеющие одинаковые последовательности VH и VL или одинаковые последовательности CDR1, 2 и 3, связываются с одним и тем же эпитопом.
Антитела, которые «конкурируют с другим антителом за связывание с мишенью», относятся к антителам, которые ингибируют (частично или полностью) связывание другого антитела с мишенью. То, конкурируют ли два антитела друг с другом за связывание с мишенью, т. e. то, ингибирует ли одно антитело связывание другого антитела с мишенью и в какой степени это происходит, можно определить с применением известных экспериментов по конкуренции. В определенных вариантах осуществления антитело конкурирует с другим антителом и ингибирует его связывание с мишенью на по меньшей мере 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%. Уровень ингибирования или конкуренции может отличаться в зависимости от того, какое антитело представляет собой «блокирующее антитело» (т. е. холодовое антитело, которое инкубируется с мишенью первым). Конкурирующие антитела связываются с тем же эпитопом, перекрывающимся эпитопом или смежными эпитопами (например, как свидетельствует стерическое несоответствие).
Антитела к C5 или их антигенсвязывающие фрагменты, описанные в данном документе, применяемые в способах, описанных в данном документе, могут быть получены с применением разнообразных методик, известных в данной области техники. Моноклональные антитела можно получить с помощью различных методик, известных специалистам в данной области. Вкратце, клетки селезенки животного, иммунизированного желаемым антигеном, иммортализируют, обычно путем слияния с клеткой миеломы (см. Kohler & Milstein, Eur. J. Immunol. 6: 511-519 (1976)). Альтернативные способы иммортализации включают трансформацию с помощью вируса Эпштейна-Барр, онкогенов или ретровирусов или других способов, хорошо известных из уровня техники. Колонии, возникающие из отдельных иммортализованных клеток, подвергают скринингу в отношении продуцирования антител желаемой специфичности и аффинности к антигену, а выход моноклональных антител, продуцируемых такими клетками, можно усилить с помощью различных методик, включая инъекцию в брюшную полость позвоночного хозяина. В качестве альтернативы, можно выделять последовательности ДНК, которые кодируют моноклональное антитело или его связывающий фрагмент, посредством скрининга библиотеки ДНК из B-клеток человека в соответствии с общим протоколом, описанным в Huse, et al.,Science 246: 1275-1281 (1989).
III. Высококонцентрированные растворы антител к C5
В данном документе предусмотрены стабильные водные растворы, содержащие антитело к C5 (например, равулизумаб). Водные растворы, описанные в данном документе, могут представлять собой стерильные фармацевтически чистые композиции, например, для введения субъекту для лечения или предупреждения нарушения, ассоциированного с системой комплемента, такого как PNH или aHUS. Растворы, описанные в данном документе, можно составлять в соответствии со стандартными способами. Получение фармацевтических составов является хорошо разработанной областью техники и дополнительно описано, например, в Gennaro (2000) «Remington: The Science and Practice of Pharmacy», 20th Edition, Lippincott, Williams & Wilkins (ISBN: 0683306472); Ansel et al. (1999) «Pharmaceutical Dosage Forms and Drug Delivery Systems», 7th Edition, Lippincott Williams & Wilkins Publishers (ISBN: 0683305727); и Kibbe (2000) «Handbook of Pharmaceutical Excipients American Pharmaceutical Association», 3rd Edition (ISBN: 091733096X). Подходящие способы составления высококонцентрированных растворов антител, описанных в данном документе, проиллюстрированы в демонстрационных примерах.
Водные растворы, описанные в данном документе, имеют высокую концентрацию антитела, которое связывается с компонентом C5 системы комплемента человека, такого как равулизумаб. Такие растворы в данном документе иногда называют «высококонцентрированными растворами антител». В контексте данного документа «высокая концентрация» антитела к C5 (например, равулизумаба) в водном растворе представляет собой концентрацию антитела, которая составляет по меньшей мере, равна или превышает 40 (например, составляет по меньшей мере, равна или превышает 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 205, 210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295 или 300) мг/мл. В одном варианте осуществления антитело к C5 присутствует в растворе в концентрации, составляющей более 100 (например, более 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190 или 195) мг/мл. В другом варианте осуществления антитело к C5 присутствует в растворе в концентрации более 200 (например, более 200, 205, 210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290 или 295) мг/мл. В другом варианте осуществления антитело к C5 присутствует в растворе в концентрации более 300 мг/мл. В другом варианте осуществления антитело присутствует в растворе в концентрации, составляющей, например, от 40 мг/мл до 200 мг/мл, от 50 мг/мл до 200 мг/мл, от 60 мг/мл до 200 мг/мл, от 70 мг/мл до 200 мг/мл, от 80 мг/мл до 200 мг/мл, от 90 мг/мл до 200 мг/мл, от 100 мг/мл до 200 мг/мл, от 110 мг/мл до 200 мг/мл, от 120 мг/мл до 200 мг/мл, от 130 мг/мл до 200 мг/мл, от 140 мг/мл до 200 мг/мл, от 150 мг/мл до 200 мг/мл, от 40 мг/мл до 100 мг/мл, от 50 мг/мл до 100 мг/мл, от 60 мг/мл до 100 мг/мл, от 70 мг/мл до 100 мг/мл, от 80 мг/мл до 100 мг/мл, от 90 мг/мл до 100 мг/мл, от 40 мг/мл до 150 мг/мл, от 50 мг/мл до 150 мг/мл, от 60 мг/мл до 150 мг/мл, от 70 мг/мл до 150 мг/мл, от 80 мг/мл до 150 мг/мл, от 90 мг/мл до 150 мг/мл, от 100 мг/мл до 150 мг/мл, от 110 мг/мл до 150 мг/мл, от 120 мг/мл до 150 мг/мл, от 40 мг/мл до 50 мг/мл, от 40 мг/мл до 250 мг/мл, от 50 мг/мл до 250 мг/мл, от 60 мг/мл до 250 мг/мл, от 70 мг/мл до 250 мг/мл, от 80 мг/мл до 250 мг/мл, от 90 мг/мл до 250 мг/мл, от 100 мг/мл до 250 мг/мл, от 110 мг/мл до 250 мг/мл, от 120 мг/мл до 250 мг/мл, от 130 мг/мл до 250 мг/мл, от 140 мг/мл до 250 мг/мл, от 150 мг/мл до 250 мг/мл, от 160 мг/мл до 250 мг/мл, от 170 мг/мл до 250 мг/мл, от 180 мг/мл до 250 мг/мл, от 190 мг/мл до 250 мг/мл, от 200 мг/мл до 250 мг/мл, от более 200 мг/мл (например, от по меньшей мере 201 мг/мл) до 250 мг/мл или от более 200 мг/мл (например, от 201 мг/мл или больше) до 300 мг/мл.
Как описано в данном документе и проиллюстрировано в демонстрационных примерах, представленные водные растворы обеспечивают выраженную физическую и химическую стабильность, а также функциональную стабильность антитела к C5, составленного в данном документе. Например, составы, описанные в данном документе, способны поддерживать структурную целостность антитела к C5 (например, равулизумаба), присутствующего в растворе в высоких концентрациях. В одном варианте осуществления раствор подходит для хранения при 2-8°C (например, при 4°C). В другом варианте осуществления раствор составлен для хранения при температуре ниже 0°C (например, при -20°C или -80°C). В другом варианте осуществления раствор составлен для хранения в течение периода до трех лет (например, в течение одного месяца, двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 1 года, 1 ½ лет, 2 лет, 2 ½ лет или 3 лет) при 2-8°C (например, при 4°C). В другом варианте осуществления раствор подходит для хранения в течение периода по меньшей мере 1, 2 или 3 лет при 2-8°C (например, при 4°C).
Как проиллюстрировано в демонстрационных примерах, описанных в данном документе, растворы, описанные в данном документе, подходят для поддержания антитела к C5 при примерно 100 мг/мл в преимущественно мономерной форме в течение периода до двух лет при примерно 2°C-8°C. В контексте данного документа антитело к C5, составленное в высокой концентрации в представленном водном растворе, является «преимущественно мономерным» или находится в «преимущественно мономерной форме», если антитело, присутствующее в растворе, является на по меньшей мере 95% (например, на по меньшей мере 95,1, 95,2, 95,3, 95,4, 95,5, 95,6, 95,7, 95,8, 95,9, 96, 96,1, 96,2, 96,3, 96,4, 96,5, 96,6, 96,7, 96,8, 96,9, 97, 97,1, 97,2, 97,3, 97,4, 97,5, 97,6, 97,7, 97,8, 97,9, 98, 98,1, 98,2, 98,3, 98,4, 98,5, 98,6, 98,7, 98,8, 98,9, 99, 99,1, 99,2, 99,3, 99,4, 99,5, 99,6, 99,7, 99,8 или 99,9% или больше) мономерным, например, при определении с помощью высокоэффективной жидкостной хроматографии в режиме эксклюзионной хроматографии (SEC-HPLC, такой как гель-проникающая HPLC). В одном варианте осуществления антитело к C5 в растворах, описанных в данном документе, может оставаться преимущественно мономерным после хранения в течение по меньшей мере одного месяца (например, в течение по меньшей мере двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 13 месяцев, 14 месяцев, 15 месяцев, 16 месяцев, 17 месяцев, 18 месяцев, 19 месяцев, 20 месяцев, 21 месяца, 22 месяцев, 23 месяцев, 24 месяцев или больше) при примерно от 2°C до 8°C (например, после хранения при, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10°C).
В одном варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев при определении с помощью SEC-HPLC (например, гель-проникающей HPLC). В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере одного года при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере 18 месяцев при определении с помощью SEC-HPLC. В другом варианте осуществления антитело к C5 остается на по меньшей мере 95% (например, на по меньшей мере 96, 97, 98 или 99%) мономерным во время хранения при 2°C-8°C в течение по меньшей мере двух лет при определении с помощью SEC-HPLC.
В другом варианте осуществления менее 5% (например, менее 4,9, 4,8, 4,7, 4,6, 4,5, 4,4, 4,3, 4,2, 4,1, 4,0, 3,9, 3,8, 3,7, 3,6, 3,5, 3,4, 3,3, 3,2, 3,1, 3,0, 2,9, 2,8, 2,7, 2,6, 2,5, 2,4, 2,3, 2,2, 2,1, 2, 1,9, 1,8, 1,7, 1,6, 1,5, 1,4, 1,3, 1,2, 1,1, 1, 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, 0,2 или 0,1%) антител в растворе являются олигомерными, агрегированными и/или фрагментированными. В контексте данного документа фрагментация антител относится к ненадлежащим образом собранным составляющим или продуктам разрушения целого антитела, имеющим более низкую молекулярную массу, чем целое антитело. Такие формы фрагментации включают без ограничения свободную мономерную полипептидную тяжелую цепь, димерную полипептидную тяжелую цепь (например, полипептидную тяжелую цепь, стабилизированную дисульфидными связями), димерную полипептидную тяжелую цепь, связанную с одной полипептидной легкой цепью, мономерную полипептидную тяжелую цепь, связанную с одной полипептидной легкой цепью, или дополнительный (дополнительные) продукт (продукты) разрушения или фрагмент (фрагменты) полипептидной легкой цепи или тяжелой цепи. В некоторых вариантах осуществления менее 2% (например, менее 1,9, 1,8, 1,7, 1,6, 1,5, 1,4, 1,3, 1,2, 1,1, 1, 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, 0,2 или 0,1%) антител являются агрегированными после хранения в течение по меньшей мере одного месяца (например, в течение по меньшей мере двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 13 месяцев, 14 месяцев, 15 месяцев, 16 месяцев, 17 месяцев, 18 месяцев, 19 месяцев, 20 месяцев, 21 месяца, 22 месяцев, 23 месяцев, 24 месяцев или больше) при 2°C-8°C. В некоторых вариантах осуществления менее 1% (например, 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, 0,2 или 0,1%) антител являются фрагментированными после хранения в течение по меньшей мере одного месяца (например, в течение по меньшей мере двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 13 месяцев, 14 месяцев, 15 месяцев, 16 месяцев, 17 месяцев, 18 месяцев, 19 месяцев, 20 месяцев, 21 месяца, 22 месяцев, 23 месяцев, 24 месяцев или больше) при 2°C-8°C. Способы определения количества мономерного антитела, а также количества олигомерных, агрегированных или фрагментированных форм антитела к С5, присутствующего в растворе, описаны в данном документе и проиллюстрированы в демонстрационных примерах. Например, специалист в данной области может определить процентную долю целых, фрагментированных, несвернутых промежуточных продуктов и/или агрегированных молекул антител, присутствующих в данном растворе, с помощью, например, высокоэффективной жидкостной хроматографии в режиме эксклюзионной хроматографии (SEC-HPLC, такой как гель-проникающая HPLC), статического рассеяния света (SLS), инфракрасной спектроскопии с преобразованием Фурье (FTIR), кругового дихроизма (CD), методик разворачивания белка, индуцируемого мочевиной, собственной флуоресценции триптофана, электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия в невосстанавливающих условиях (SDS-PAGE) и дифференциальной сканирующей калориметрии (DSC).
В одном варианте осуществления любого из растворов, описанных в данном документе, менее 5% антител к C5 (например, равулизумаба) в растворе являются агрегированными при определении с помощью SEC-HPLC (например, гель-проникающей HPLC). В другом варианте осуществления менее 4% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 3% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 2% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC. В другом варианте осуществления менее 1% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC.
Как описано в данном документе и проиллюстрировано в демонстрационных примерах, растворы, содержащие антитела к C5, представленные в данном документе, могут сохранять по меньшей мере 90% (например, 91, 92, 93, 94, 95, 96, 97, 98, 99 или даже 100%) от их биологической/функциональной активности (например, способности связываться с C5 человека) после хранения в течение по меньшей мере одного месяца (например, в течение по меньшей мере двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 13 месяцев, 14 месяцев, 15 месяцев, 16 месяцев, 17 месяцев, 18 месяцев, 19 месяцев, 20 месяцев, 21 месяца, 22 месяцев, 23 месяцев, 24 месяцев, 25 месяцев, 26 месяцев, 27 месяцев, 28 месяцев, 29 месяцев, 30 месяцев, 31 месяца, 32 месяцев, 33 месяцев, 34 месяцев, 35 месяцев, 36 месяцев или больше) при 2°C-8°C.
В другом варианте осуществления антитело к C5 (например, равулизумаб), присутствующее в растворе, описанном в данном документе, может сохранять по меньшей мере 90% (например, 91, 92, 93, 94, 95, 96, 97, 98, 99 или даже 100%) от своей активности ингибирования гемолиза после хранения в течение по меньшей мере одного месяца (например, в течение по меньшей мере двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 13 месяцев, 14 месяцев, 15 месяцев, 16 месяцев, 17 месяцев, 18 месяцев, 19 месяцев, 20 месяцев, 21 месяца, 22 месяцев, 23 месяцев, 24 месяцев, 25 месяцев, 26 месяцев, 27 месяцев, 28 месяцев, 29 месяцев, 30 месяцев, 31 месяца, 32 месяцев, 33 месяцев, 34 месяцев, 35 месяцев, 36 месяцев или более) при 2°C-8°C. Подходящие способы анализа гемолиза для определения того, сохраняет ли свою активность антитело в представленном растворе, описаны в данном документе и известны из уровня техники, например, анализ гемолиза с использованием птичьих или свиных эритроцитов in vitro. Подходящие способы оценки способности препарата антител к связыванию с компонентом С5 системы комплемента человека известны из уровня техники и описаны в данном документе.
В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере одного года по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере восемнадцати месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере двух лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей C5-связывающей активности во время хранения при 2°C-8°C в течение по меньшей мере трех лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения.
В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере девяти месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере шести месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере одного года по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере 18 месяцев по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере двух лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения. В другом варианте осуществления любого из растворов, описанных в данном документе, антитело к C5 (например, равулизумаб) сохраняет по меньшей мере 80% (например, по меньшей мере 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%) своей способности ингибировать гемолиз во время хранения при 2°C-8°C в течение по меньшей мере трех лет по сравнению с эталонным антителом к C5, соответствующим антителу к C5 до хранения.
Водные растворы, описанные в данном документе, могут содержать одно или более обычных средств (например, один или более наполнителей и/или добавок, таких как буферные средства, сахара или сахариды, соли, поверхностно-активные вещества, солюбилизаторы, разбавители, связующие вещества, стабилизаторы, соли, липофильные растворители, аминокислоты, хелаторы и/или консерванты).
В одном варианте осуществления водный раствор содержит одно или более буферных средств. В контексте данного документа термин «буферное средство» относится к одному или более компонентам, которые при добавлении в водный раствор способны защищать раствор от изменений pH при добавлении кислоты или щелочи или при разбавлении растворителем. В одном варианте осуществления раствор содержит по меньшей мере одно или более буферных средств. Неограничивающие примеры типичных буферов, которые могут быть включены в промывочный(промывочные) раствор(растворы), включают трис (трис(гидроксиметил)метиламин), бис-трис, бис-трис-пропан, гистидин, триэтаноламин, диэтаноламин, формиат, ацетат, MES (2-(N-морфолинo)этансульфоновую кислоту), фосфат, HEPES (4-2-гидроксиэтил-1-пиперазинэтансульфоновую кислоту), цитрат, MOPS (3-(N-морфолинo)пропансульфоновую кислоту), TAPS (3{[трис(гидроксиметил)метил]амино}пропансульфоновую кислоту), бицин (N, N-бис(2-гидроксиэтил)глицин), трицин (N-трис(гидроксиметил)метилглицин), TES (2-{[трис(гидроксиметил)метил]амино}этансульфоновую кислоту), PIPES (пиперазин-N, N’-бис(2-этансульфоновую кислоту), какодилат (диметиларсиновую кислоту), SSC (солевой раствор с цитратом натрия) и фосфат натрия.
В другом варианте осуществления буферное средство представляет собой аминокислоту. Аминокислота может представлять собой например, аминокислоту, выбранную из группы, состоящей из гистидина (например, L-гистидина), серина (например, L-серина) и глицина (например, L-глицина). В другом варианте осуществления раствор содержит два или более буферных средства. В конкретном варианте осуществления буферное средство представляет собой фосфат натрия. В одном варианте осуществления представленные растворы не содержат свободную аминокислоту в качестве буферного средства. В другом варианте осуществления представленные растворы содержат одну свободную аминокислоту (например, гистидин) в качестве буферного средства. В другом варианте осуществления представленные растворы могут содержать две или более (например, две, три, четыре, пять, шесть или семь или более) различные аминокислоты в качестве буферных средств, например, серин и гистидин.
Концентрация буфера является достаточной для поддержания желаемого pH и также может варьироваться, например, для поддержания изотоничности состава. Типичные концентрации традиционных буферных средств, используемых в составах для парентерального введения, можно найти в: Pharmaceutical Dosage Form: Parenteral Medications, Volume 1, 2nd Edition, Chapter 5, p. 194, De Luca and Boylan, «Formulation of Small Volume Parenterals», Table 5: Commonly used additives in Parenteral Product. В одном варианте осуществления концентрация одного или более буферных средств в составе составляет от приблизительно 10 мМ до 300 мМ включительно. В другом варианте осуществления раствор содержит по меньшей мере одно буферное средство в концентрации от 10 мМ до 200 мМ включительно. В другом варианте осуществления водный раствор, описанный в данном документе, содержит буферное средство в концентрации по меньшей мере 10 (например, по меньшей мере 15, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 или 300 или больше) мМ. В другом варианте осуществления водный раствор содержит буферное средство в концентрации от приблизительно 10 мМ до 50 мМ, от 15 мМ до 50 мМ, от 20 мМ до 50 мМ, от 25 мМ до 50 мМ, от 30 мМ до 50 мМ, от 40 мМ до 50 мМ, от 10 мМ до 100 мМ, от 15 мМ до 100 мМ, от 20 мМ до 100 мМ, от 25 мМ до 100 мМ, от 30 мМ до 100 мМ, от 40 мМ до 100 мМ, от 10 мМ до 150 мМ, от 15 мМ до 150 мМ, от 20 мМ до 150 мМ, от 25 мМ до 150 мМ, от 30 мМ до 150 мМ, от 40 мМ до 150 мМ, от 50 мМ до 100 мМ, от 60 мМ до 100 мМ, от 70 мМ до 100 мМ, от 80 мМ до 100 мМ, от 50 мМ до 150 мМ, от 60 мМ до 150 мМ, от 70 мМ до 150 мМ, от 80 мМ до 150 мМ, от 90 мМ до 150 мМ, от 100 мМ до 150 мМ, от 10 мМ до 200 мМ, от 15 мМ до 200 мМ, от 20 мМ до 200 мМ, от 25 мМ до 200 мМ, от 30 мМ до 200 мМ, от 40 мМ до 200 мМ, от 50 мМ до 200 мМ, от 60 мМ до 200 мМ, от 70 мМ до 200 мМ, от 80 мМ до 200 мМ, от 90 мМ до 200 мМ, от 100 мМ до 200 мМ, от 150 мМ до 200 мМ, от 10 мМ до 250 мМ, от 15 мМ до 250 мМ, от 20 мМ до 250 мМ, от 25 мМ до 250 мМ, от 30 мМ до 250 мМ, от 40 мМ до 250 мМ, от 50 мМ до 250 мМ, от 60 мМ до 250 мМ, от 70 мМ до 250 мМ, от 80 мМ до 250 мМ, от 90 мМ до 250 мМ, от 100 мМ до 250 мМ, от 150 мМ до 250 мМ или от 200 мМ до 250 мМ. В другом варианте осуществления концентрация буфера в составе составляет приблизительно 20 мМ, 25 мМ, 30 мМ, 35 мМ, 40 мМ, 45 мМ, 50 мМ, 55 мМ, 60 мМ, 65 мМ, 70 мМ, 75 мМ, 80 мМ, 90 мМ, 95 мМ или приблизительно 100 мМ. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 20 мМ. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 25 мМ. В другом варианте осуществления буферное средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 50 мМ. В вариантах осуществления, в которых представленный раствор содержит два или более различных буферных средства (например, по меньшей мере два, три, четыре, пять, шесть, семь, восемь, девять или 10 или больше), каждое из двух или более буферных средств может независимо присутствовать, например, в одной из вышеописанных концентраций.
В одном варианте осуществления водный раствор имеет нейтральный pH или может быть доведен до него. В контексте данного документа термин «нейтральный pH» означает pH, составляющий от 7 до 8 включительно. Соответственно, в контексте данного документа нейтральный pH включает конкретные значения pH, такие как 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9 и 8,0. В некоторых вариантах осуществления нейтральный pH составляет по меньшей мере pH 7 (например, по меньшей мере pH 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9), но менее чем pH 8 (например, менее чем pH 7,9, 7,8, 7,7, 7,6, 7,5, 7,4, 7,3, 7,2 или 7,1). Иными словами, в некоторых вариантах осуществления нейтральный pH может составлять, например, по меньшей мере pH 7, но менее чем pH 7,5. В некоторых вариантах осуществления нейтральный pH может составлять от pH 7 до pH 7,5. В некоторых вариантах осуществления нейтральный pH может составлять от pH 7 до pH 7,2. В другом варианте осуществления pH раствора составляет от 7,0 до 7,4. В другом варианте осуществления pH раствора составляет от 7,2 до 7,8. В другом варианте осуществления pH раствора составляет от 7,2 до 7,6. В некоторых вариантах осуществления нейтральный pH может составлять, например, pH 7. Специалисту в данной области также будет понятно, что человеческая кровь (такая как человеческая кровь от здорового пациента) имеет нейтральный pH, определенный в данном документе, например,pH человеческой крови составляет примерно от 7,35 до pH 7,45. См., например, Boron and Boulpaep (2003) «Medical physiology: a cellular and molecular approach», W.B. Saunders, New York (ISBN:0721632564). В некоторых вариантах осуществления pH высококонцентрированного раствора антитела, описанного в данном документе составляет примерно от 6,4 до 7,5 включительно (например, примерно 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6 или 7,7). В одном варианте осуществления pH раствора составляет от 7,2 до 7,6. В конкретном варианте осуществления pH раствора составляет 7,4.
В одном варианте осуществления раствор содержит одно или более поверхностно-активных веществ, таких как анионное, катионное или неионогенное поверхностно-активное вещество. В контексте данного документа термин «поверхностно-активное вещество» относится к поверхностно-активной молекуле, содержащей как гидрофобную часть (например, алкильную цепь), так и гидрофильную часть (например, карбоксильные и карбоксилатные группы). Поверхностно-активные вещества, подходящие для применения в составах по настоящему изобретению, включают без ограничения сложные эфиры жирных кислот (например, сорбитанмонокаприлат, сорбитанмонолаурат, сорбитанмонопальмитат), сорбитантриолеат, сложные эфиры глицерина и жирных кислот (например, монокаприлат глицерина, мономиристат глицерина, моностеарат глицерина), сложные эфиры полиглицерина и жирных кислот (например, декаглицерилмоностеарат, декаглицерилдистеарат, декаглицерилмонолинолеат), полиоксиэтиленовые сложные эфиры сорбитана и жирных кислот (например, полиоксиэтиленсорбитанмонолаурат, полиоксиэтиленсорбитанмоноолеат, полиоксиэтиленсорбитанмоностеарат, полиоксиэтиленсорбитанмонопальмитат, полиоксиэтиленсорбитантриолеат, полиоксиэтиленсорбитантристеарат), полиоксиэтиленовые сложные эфиры сорбита и жирных кислот (например, полиоксиэтиленсорбиттетрастеарат, полиоксиэтиленсорбиттетраолеат), полиоксиэтиленовые сложные эфиры глицерина и жирных кислот (например, полиоксиэтиленглицерилмоностеарат), сложные эфиры полиэтиленгликоля и жирных кислот (например, дистеарат полиэтиленгликоля), алкиловые эфиры полиоксиэтилена (например, лауриловый эфир полиоксиэтилена), алкиловые эфиры полиоксиэтилена-полиоксипропилена (например, полиоксиэтилен-полиоксипропиленгликоль, пропиловый эфир полиоксиэтилена-полиоксипропилена, цетиловый эфир полиоксиэтилена-полиоксипропилена), алкилфениловые эфиры полиоксиэтилена (например, нонилфениловый эфир полиоксиэтилена), полиоксиэтиленовые гидрогенизированные касторовые масла (например, полиоксиэтиленовое касторовое масло, полиоксиэтиленовое гидрогенизированное касторовое масло), полиоксиэтиленовые производные восков (например, полиоксиэтиленсорбитовый воск), полиоксиэтиленовые производные ланолина (например, полиоксиэтиленланолин) и полиоксиэтиленовые амиды жирных кислот (например, полиоксиэтиленовый амид стеариновой кислоты); C12-C18-алкилсульфаты (например, цетилсульфат натрия, лаурилсульфат натрия, олеилсульфат натрия), полиоксиэтилен-C10-C18-алкилэфирсульфат с добавлением в среднем от 2 до 4 молей этиленоксидных звеньев (например, полиоксиэтиленлаурилсульфат натрия) и соли C10-C18-алкилсульфосукцинатных сложных эфиров (например, натриевую соль лаурилсульфосукцинатного сложного эфира); а также природные поверхностно-активные вещества, такие как лецитин, глицерофосфолилипид, сфингофосфолипиды (например, сфингомиелин) и сложные эфиры сахарозы и C12-C18-жирных кислот.
В одном варианте осуществления поверхностно-активное вещество в составе представляет собой неионогенное поверхностно-активное вещество. В определенных вариантах осуществления поверхностно-активное вещество в составе представляет собой полиоксиэтиленовый сложный эфир сорбитана и жирной кислоты, например, полисорбат 20, 40, 60, 80, или комбинацию одного или более из них. В одном варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 80 (Tween 80). В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 60. В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 40. В другом варианте осуществления поверхностно-активное вещество в составе представляет собой полисорбат 20 (Tween 20).
Количество поверхностно-активного вещества, добавленного в состав, является достаточным для уменьшения агрегации составленного антитела и/или минимизации образования частиц в составе. Например, поверхностно-активное вещество может присутствовать в составе в количестве от приблизительно 0,001% до приблизительно 1%, или от приблизительно 0,001% до приблизительно 0,5%, или от приблизительно 0,01% до приблизительно 0,2%. В одном варианте осуществления водные растворы содержат поверхностно-активное вещество в концентрации, составляющей по меньшей мере или примерно 0,001 (например, по меньшей мере или примерно 0,002, 0,003, 0,004, 0,005, 0,006, 0,007, 0,008, 0,009, 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,11, 0,12, 0,13, 0,14, 0,15, 0,16, 0,17, 0,18, 0,19, 0,2, 0,21, 0,22, 0,23, 0,24, 0,25, 0,26, 0,27, 0,28, 0,29, 0,3, 0,31, 0,32, 0,33, 0,34, 0,35, 0,36, 0,37, 0,38, 0,39, 0,4, 0,41, 0,42, 0,43, 0,44, 0,45, 0,46, 0,47, 0,48, 0,49 или 0,5 или больше) %. В другом варианте осуществления водный раствор содержит не более 0,2 (например, не более 0,19, 0,18, 0,17, 0,16, 0,15, 0,14, 0,13, 0,12, 0,11, 0,10, 0,09, 0,08, 0,07, 0,06, 0,05, 0,04, 0,03, 0,02, 0,01, 0,009, 0,008, 0,007, 0,006, 0,005, 0,004, 0,003, 0,002 или 0,001) % фармацевтически приемлемого поверхностно-активного вещества.
В другом варианте осуществления составы содержат полисорбат в концентрации от приблизительно 0,001% до приблизительно 0,5%, от приблизительно 0,005% до приблизительно 0,2%, от приблизительно 0,01% до приблизительно 0,1%, или от приблизительно 0,02% до приблизительно 0,06%, или от приблизительно 0,03% до приблизительно 0,05% (вес/объем). В определенных вариантах осуществления состав содержит полисорбат в концентрации 0,01%, или 0,02%, или 0,03%, или 0,04%, или 0,05%, или 0,06%, или 0,07%, или 0,08%, или 0,09%, или 0,1%, или 0,15%, или 0,2% (вес/объем). В определенных вариантах осуществления поверхностно-активное вещество присутствует в составе в количестве 0,02% или приблизительно 0,04% (вес/объем). В одном варианте осуществления поверхностно-активное вещество присутствует в составе в количестве 0,05% (вес/объем).
В одном варианте осуществления состав содержит по меньшей мере приблизительно 0,01%, по меньшей мере приблизительно 0,02%, по меньшей мере приблизительно 0,05%, по меньшей мере приблизительно 0,1%, по меньшей мере приблизительно 0,2%, по меньшей мере приблизительно 0,3%, по меньшей мере приблизительно 0,4% или по меньшей мере приблизительно 0,5% полисорбат 80. В определенном варианте осуществления состав содержит полисорбат 80 в концентрации от приблизительно 0,01% до приблизительно 0,5%, от приблизительно 0,01% до приблизительно 0,3%, от приблизительно 0,001% до приблизительно 0,2%, от приблизительно 0,02% до приблизительно 0,5%, от приблизительно 0,02% до приблизительно 0,3%, от приблизительно 0,02% до приблизительно 0,2%, от приблизительно 0,05% до приблизительно 0,5%, от приблизительно 0,05% до приблизительно 0,3%, от приблизительно 0,05% до приблизительно 0,2%, от приблизительно 0,075% до приблизительно 0,5%, от приблизительно 0,075% до приблизительно 0,3% или от приблизительно 0,075% до приблизительно 0,2%. В дополнительном варианте осуществления состав содержит приблизительно 0,01%, приблизительно 0,02%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4% или приблизительно 0,5% полисорбат 80. В одном варианте осуществления состав содержит приблизительно 0,05% полисорбат 80. В одном варианте осуществления состав содержит приблизительно 0,04% полисорбат 80. В одном варианте осуществления состав содержит приблизительно 0,03% полисорбат 80. В одном варианте осуществления состав содержит приблизительно 0,02% полисорбат 80. В одном варианте осуществления состав содержит приблизительно 0,01% полисорбат 80.
В одном варианте осуществления водный раствор содержит одну или несколько солей, например, хлорид натрия, хлорид калия или хлорид магния. В некоторых вариантах осуществления водный раствор, описанный в данном документе, содержит соль в концентрации по меньшей мере 10 (например, по меньшей мере 15, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 или 300 или больше) мМ. В некоторых вариантах осуществления водный раствор, описанный в данном документе, может содержать соль в концентрации, составляющей менее чем или примерно 200 (например, менее чем или примерно 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 70, 60, 50, 40, 30, 25, 20, 15 или 10) мМ. В некоторых вариантах осуществления водный раствор, описанный в данном документе, может содержать соль в концентрации от приблизительно 10 мМ до 50 мМ, от 15 мМ до 50 мМ, от 20 мМ до 50 мМ, от 25 мМ до 50 мМ, от 30 мМ до 50 мМ, от 40 мМ до 50 мМ, от 10 мМ до 100 мМ, от 15 мМ до 100 мМ, от 20 мМ до 100 мМ, от 25 мМ до 100 мМ, от 30 мМ до 100 мМ, от 40 мМ до 100 мМ, от 10 мМ до 150 мМ, от 15 мМ до 150 мМ, от 20 мМ до 150 мМ, от 25 мМ до 150 мМ, от 30 мМ до 150 мМ, от 40 мМ до 150 мМ, от 50 мМ до 100 мМ, от 60 мМ до 100 мМ, от 70 мМ до 100 мМ, от 80 мМ до 100 мМ, от 50 мМ до 150 мМ, от 60 мМ до 150 мМ, от 70 мМ до 150 мМ, от 80 мМ до 150 мМ, от 90 мМ до 150 мМ, от 100 мМ до 150 мМ, от 10 мМ до 200 мМ, от 15 мМ до 200 мМ, от 20 мМ до 200 мМ, от 25 мМ до 200 мМ, от 30 мМ до 200 мМ, от 40 мМ до 200 мМ, от 50 мМ до 200 мМ, от 60 мМ до 200 мМ, от 70 мМ до 200 мМ, от 80 мМ до 200 мМ, от 90 мМ до 200 мМ, от 100 мМ до 200 мМ, от 150 мМ до 200 мМ, от 10 мМ до 250 мМ, от 15 мМ до 250 мМ, от 20 мМ до 250 мМ, от 25 мМ до 250 мМ, от 30 мМ до 250 мМ, от 40 мМ до 250 мМ, от 50 мМ до 250 мМ, от 60 мМ до 250 мМ, от 70 мМ до 250 мМ, от 80 мМ до 250 мМ, от 90 мМ до 250 мМ, от 100 мМ до 250 мМ, от 150 мМ до 250 мМ или от 200 мМ до 250 мМ. В вариантах осуществления, в которых представленный раствор содержит две или более различные соли (например, по меньшей мере две, три, четыре, пять, шесть, семь, восемь, девять или 10 или больше), каждая из двух или более солей может независимо присутствовать, например, в одной из вышеописанных концентраций.
В одном варианте осуществления водный раствор содержит один или более углеводных наполнителей. Подходящие углеводные наполнители описаны, например, в Katakam and Banga (1995) J Pharm Pharmacol 47(2):103-107; Andya et al. (2003) AAPS PharmSci 5(2): Article 10; и Shire (2009) «Current Trends in Monoclonal Antibody Development and Manufacturing», Volume 11, Springer, 354 pages. Углеводные наполнители, подходящие для применения в растворах, описанных в данном документе, включают без ограничения моносахариды, такие как фруктоза, мальтоза, галактоза, глюкоза, D-манноза и сорбоза; дисахариды, такие как лактоза, сахароза, трегалоза и целлобиоза; полисахариды, такие как мальтодекстрины, декстраны и крахмалы; и сахарные спирты, такие как маннит, ксилит, мальтит, лактит и сорбит. В одном варианте осуществления углеводный наполнитель присутствует в растворе, представленном в данном документе, в концентрации, составляющей по меньшей мере или примерно 0,5 (например, по меньшей мере или примерно 0,6, 0,7, 0,8, 0,9, 1, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3, 3,25, 3,5, 3,75, 4, 4,25, 4,5, 4,75, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5, 9, 9,5, 10 или больше) %. В вариантах осуществления, в которых представленный раствор содержит два или более различных углеводных наполнителя (например, по меньшей мере два, три, четыре, пять, шесть, семь, восемь, девять или 10 или больше), каждый наполнитель может независимо присутствовать в любой из вышеописанных концентраций.
В другом варианте осуществления стабильный водный раствор содержит одно или более стабилизирующих средств. Иллюстративные стабилизаторы включают без ограничения полиолы, сахара (например, сахарозу или трегалозу), аминокислоты (например, аргинин), амины и высаливающие соли. В одном варианте осуществления раствор содержит по меньшей мере одно стабилизирующее средство в концентрации 2-10% включительно. В одном варианте осуществления раствор содержит 5% сахарозу. В другом варианте осуществления раствор содержит по меньшей мере одно или более стабилизирующих средств в концентрации от 10 мМ до 50 мМ включительно. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 20 мМ. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 25 мМ. В другом варианте осуществления стабилизирующее средство присутствует в растворе в концентрации, составляющей по меньшей мере или равной 50 мМ. В другом варианте осуществления раствор содержит 25 мМ аргинина.
В одном варианте осуществления растворы, описанные в данном документе, содержат один или более консервантов.
В контексте данного документа термин «консервант» относится к средству, которое уменьшает жизнедеятельность бактерий и необязательно может быть добавлено в составы, описанные в данном документе. Добавление консерванта может, например, облегчать получение состава для многократного применения (многодозового состава). Примеры потенциальных консервантов включают хлорид октадецилдиметилбензиламмония, хлорид гексаметония, хлорид бензалкония (смесь хлоридов алкилбензилдиметиламмония, в которых алкильные группы представляют собой длинноцепочечные соединения) и хлорид бензетония. Другие типы консервантов включают ароматические спирты, такие как фенол, бутил и бензиловый спирт, алкилпарабены, такие как метил- или пропилпарабен, катехол, резорцин, циклогексанол, 3-пентанол и м-крезол.
В одном варианте осуществления стабильный водный раствор содержит не более пяти средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более четырех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более трех средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более двух средств в дополнение к антителу к C5. В другом варианте осуществления стабильный водный раствор содержит не более одного средства в дополнение к антителу к C5.
В другом варианте осуществления стабильный водный раствор содержит антитело к C5, которое содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу и 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор состоит из антитела к C5, которое содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу и 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор содержит антитело к C5, содержащее CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу; 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; а также 0,05 ± 0,03 (например, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07 и 0,08) % полисорбат 80, где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления стабильный водный раствор состоит из антитела к C5, которое содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, в концентрации 100 ± 20 (например, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 или 120) мг/мл; 50 ± 15 (например, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 или 65) мМ фосфатного буфера; 5 ± 3 (например, 2, 3, 4, 5, 6, 7 или 8) % сахарозу; 25 ± 10 (например, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35) мM аргинина; а также 0,05 ± 0,03 (например, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07 и 0,08) % полисорбат 80, где раствор имеет pH 7,4 ± 0,5 (например, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8 или 7,9).
В другом варианте осуществления предусмотрен стабильный водный раствор (например, стерильный раствор), где раствор содержит (a) антитело к C5 (например, равулизумаб) в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера; (c) приблизительно 5% сахарозу и (d) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 (например, равулизумаб) в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера; (c) 5% сахарозу и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу, (d) приблизительно 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит не более трех дополнительных средств. В другом варианте осуществления стабильный водный раствор содержит не более двух дополнительных средств. В другом варианте осуществления стабильный водный раствор содержит не более одного дополнительного средства.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы и (d) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера; (c) 5% сахарозы и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы; (d) приблизительно 0,05% полисорбата 80 и (e) приблизительно 25 мМ аргинина. В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, (b) 50 мМ фосфатного буфера, (c) 5% сахарозы, (d) 0,05% полисорбата 80 и (e) 25 мМ аргинина.
В одном варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу и (d) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу и (d) 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозу, (d) приблизительно 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор содержит (a) антитело к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозу, (d) 0,05% полисорбат 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации приблизительно 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) приблизительно 50 мМ фосфатного буфера, (c) приблизительно 5% сахарозы, (d) приблизительно 0,05% полисорбата 80 и (e) приблизительно 25 мМ аргинина.
В другом варианте осуществления стабильный водный раствор состоит из (a) антитела к C5 в концентрации 100 мг/мл, где антитело к C5 содержит CDR1 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 19, CDR2 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 18, CDR3 тяжелой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 3, CDR1 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 4, CDR2 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 5, и CDR3 легкой цепи, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 6, (b) 50 мМ фосфатного буфера, (c) 5% сахарозы, (d) 0,05% полисорбата 80 и (e) 25 мМ аргинина.
IV. Способы получения высококонцентрированных растворов антител
Также в данном документе предусмотрены способы получения высококонцентрированного раствора антитела к C5. В одном варианте осуществления предусмотрены способы получения стабильного концентрированного раствора антитела, содержащего антитело к C5 в концентрации 100 мг/мл, 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина, при этом способ включает:
i) получение первого водного раствора, содержащего антитело к C5, при этом первый водный раствор имеет первый состав и содержит не более 10 мг/мл антитела к C5;
ii) проведение диафильтрации первого водного раствора с получением состава, содержащего 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина, при pH 7,4, вследствие чего обеспечивается получение второго водного раствора, при этом второй водный раствор имеет второй состав в результате диафильтрации; и
iii) концентрирование второго водного раствора с получением стабильного концентрированного раствора антитела, содержащего 100 мг/мл антитела к C5, 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина.
В другом варианте осуществления предусмотрен способ получения стабильного концентрированного раствора антитела, содержащего антитело к C5 в концентрации 100 мг/мл, 50 мМ фосфатного буфера, 5% сахарозу; 25 мМ аргинина и 0,05% полисорбат 80, при этом способ включает:
i) получение первого водного раствора, содержащего антитело к C5, при этом первый водный раствор имеет первый состав и содержит не более 10 мг/мл антитела к C5;
ii) проведение диафильтрации первого водного раствора с получением состава, содержащего 50 мМ фосфатного буфера, 5% сахарозу, 25 мМ аргинина и 0,05% полисорбат 80, при pH 7,4, вследствие чего обеспечивается получение второго водного раствора, при этом второй водный раствор имеет второй состав в результате диафильтрации; и
iii) концентрирование второго водного раствора с получением стабильного концентрированного раствора антитела, содержащего 100 мг/мл антитела к C5, 50 мМ фосфатного буфера, 5% сахарозу, 25 мМ аргинина и 0,05% полисорбат 80.
V. Пути введения
Растворы, описанные в данном документе, можно вводить пациенту с помощью ряда способов, которые отчасти зависят от пути введения. Путь введения может представлять собой парентеральный способ введения, например, внутривенную инъекцию или инфузию (IV), подкожную инъекцию (SC), внутрибрюшинную (IP) инъекцию, внутриглазную инъекцию, внутрисуставную инъекцию или внутримышечную инъекцию (IM). Термины «парентеральное введение», «вводимый парентерально» и другие грамматически эквивалентные фразы в контексте данного документа относятся к способам введения, отличным от энтерального и местного введения, обычно путем инъекции, и включают без ограничения внутривенную, интраназальную, внутриглазную, легочную, внутримышечную, внутриартериальную, интратекальную, внутрикапсулярную, внутриглазничную, внутрисердечную, внутрикожную, внутрилегочную, внутрибрюшинную, транстрахеальную, подкожную, субкутикулярную, внутрисуставную, субкапсулярную, субарахноидальную, интраспинальную, эпидуральную, интрацеребральную, внутричерепную, интракаротидную и интрастернальную инъекцию и инфузию.
В конкретном варианте осуществления раствор вводят посредством подкожной инъекции. Подкожное введение можно осуществлять с помощью устройства. Устройство может представлять собой шприц, предварительно заполненный шприц, одноразовый либо многоразовый автоинъектор, шприц-ручку, пластырный инъектор, нательный инъектор, амбулаторный шприцевой инфузионный насос с набором для подкожной инфузии или другое устройство.
В одном варианте осуществления раствор, описанный в данном документе, доставляется субъекту посредством местного введения. В контексте данного документа термин «местное введение» или «местная доставка» относится к доставке, в основе которой не лежит транспортировка композиции или активного средства (например, антитела к C5) к их предполагаемым ткани- или участку-мишени через сосудистую систему. После местного введения вблизи ткани- или участка-мишени раствор или один или более его компонентов могут диффундировать в предполагаемые ткань- или участок-мишень.
Например, раствор может доставляться путем инъекции или путем имплантации устройства, содержащего раствор. Имплантат может быть изготовлен из пористого, непористого или гелеобразного материала, в том числе из мембран, таких как силастические мембраны, или волокна. Имплантат может быть выполнен с возможностью длительного или периодического высвобождения раствора в организм субъекта. См., например, публикацию заявки на патент США № 20080241223; патенты США №№ 5501856; 4863457 и 3710795; EP488401 и EP 430539, раскрытие каждого из которых включено в данный документ посредством ссылки во всей своей полноте. Раствор, описанный в данном документе, может доставляться субъекту посредством имплантируемого устройства на основе, например, диффузионных, эродируемых или конвекционных систем, например, осмотических насосов, биоразлагаемых имплантатов, электродиффузионных систем, электроосмотических систем, насосов давления пара, электролитических насосов, насосов для газообразующих веществ, пьезоэлектрических насосов, эродируемых систем или электромеханических систем.
В одном варианте осуществления раствор, описанный в данном документе, можно местно вводить в сустав (например, в шарнирный сустав). Например, в вариантах осуществления, где нарушение представляет собой артрит, терапевтически подходящий раствор можно вводить непосредственно в сустав (например, в суставную щель) или вблизи сустава. Примеры суставных соединений, в которые можно местно вводить композицию, описанную в данном документе, включают, например, тазобедренный, коленный, локтевой, лучезапястный, грудинно-ключичный, височно-нижнечелюстной, запястный, предплюсневой, голеностопный и любой другой сустав, подверженный артритным состояниям. Композицию, описанную в данном документе, можно также вводить в синовиальную сумку, такую как, например, ключично-акромиальная, двуглаво-лучевая, локтево-лучевая, дельтовидная, поднадколенная, седалищная и любую другую синовиальную сумку, известную в области медицины.
В другом варианте осуществления раствор, описанный в данном документе, можно местно вводить в глаз. Используемый в данном документе термин «глаз» относится ко всем без исключения анатомическим тканям и структурам, связанным с глазом. В одном варианте осуществления раствор, описанный в данном документе, вводят в заднюю камеру глаза. В другом варианте осуществления раствор, описанный в данном документе, вводят в стекловидное тело. В другом варианте осуществления раствор, описанный в данном документе, вводят через склеру.
В некоторых вариантах осуществления, например, в вариантах осуществления лечения или предупреждения нарушения, такого как COPD или астма, раствор, описанный в данном документе, можно вводить субъекту через легкое. Легочная доставка лекарственного средства может осуществляться путем вдыхания, и введение путем вдыхания в данном документе может быть пероральным и/или интраназальным. В одном варианте осуществления раствор, описанный в данном документе, можно вводить в легкие субъекта посредством ингалятора. В ингаляторах используется сжатый воздух для доставки соединения в виде сжиженного аэрозоля или тумана. Ингалятор может представлять собой, например, струйный ингалятор (например, воздушно-струйный или жидкостно-струйный ингалятор) или ультразвуковой ингалятор. Дополнительные устройства и способы для внутрилегочного введения изложены, например, в публикациях заявок на патент США № 20050271660 и 20090110679, раскрытие каждой из которых включено в данный документ посредством ссылки во всей своей полноте.
В других вариантах осуществления растворы, описанные в данном документе, присутствуют в виде лекарственной формы с однократной дозировкой, которая может быть особенно подходящей для самостоятельного введения. Составленный продукт по настоящему изобретению может быть включен в контейнер, как правило, например, флакон, картридж, предварительно заполненный шприц или одноразовый шприц-ручку. Также можно использовать дозатор, такой как дозирующее устройство, описанное в патенте США № 6302855. Инъекционная система может содержать шприц-ручку для доставки, описанную в патенте США № 5308341. Шприц-ручки, наиболее широко применяемые для самостоятельной доставки инсулина пациентами с сахарным диабетом, хорошо известны из уровня техники. Такие устройства могут содержать по меньшей мере одну инъекционную иглу (например, иглу 31 калибра длиной приблизительно 5-8 мм), как правило, предварительно заполнены одной или более однократными терапевтическими дозами раствора и являются применимыми для быстрой доставки раствора субъекту, причиняя ему как можно меньше боли.
VI. Способы лечения
Описанные растворы можно применять для лечения различных заболеваний и состояний у пациента-человека. В одном варианте осуществления растворы можно применять для лечения нарушения, ассоциированного с системой комплемента, в том числе без ограничения ревматоидного артрита (RA); синдрома антифосфолипидных антител; волчаночного нефрита; ишемического/реперфузионного повреждения; атипичного гемолитико-уремического синдрома (aHUS), типичного или инфекционного гемолитико-уремического синдрома (tHUS); болезни плотного осадка (DDD); пароксизмальной ночной гемоглобинурии (PNH); оптиконевромиелита (NMO); мультифокальной моторной нейропатии (MMN); рассеянного склероза (MS); макулодистрофии (например, возрастной макулодистрофии (AMD)); синдрома гемолиза, повышенной активности печеночных ферментов и тромбоцитопении (HELLP); тромботической тромбоцитопенической пурпуры (TTP); самопроизвольного аборта; малоиммунного васкулита; буллезного эпидермолиза; повторного выкидыша и черепно-мозговой травмы (см., например, Holers (2008), Immunological Reviews 223:300-316, и Holers and Thurman (2004) Molecular Immunology 41:147-152).
В другом варианте осуществления нарушение, ассоциированное с системой комплемента, представляет собой сосудистое нарушение, ассоциированное с системой комплемента, такое как, без ограничения, сосудистое нарушение, ассоциированное с сахарным диабетом (например, в глазу), окклюзия центральной вены сетчатки, сердечно-сосудистое нарушение, миокардит, цереброваскулярное нарушение, нарушение со стороны периферических (например, скелетно-мышечных) сосудов, реноваскулярное нарушение, мезентериальное/энтеральное сосудистое нарушение, реваскуляризация трансплантатов и/или реплантатов, васкулит, нефрит при пурпуре Шенлейна-Геноха, васкулит, ассоциированный с системной красной волчанкой, васкулит, ассоциированный с ревматоидным артритом, иммунокомплексный васкулит, болезнь Такаясу, дилатационная кардиомиопатия, диабетическая ангиопатия, болезнь Кавасаки (артериит), венозная газовая эмболия (VGE) и рестеноз после стентирования, ротационная атерэктомия и чрескожная транслюминальная коронарная ангиопластика (PTCA) (см., например, публикацию заявки на патент США № 20070172483).
Дополнительные нарушения, ассоциированные с системой комплемента, включают без ограничения тяжелую миастению, болезнь холодовых агглютининов, дерматомиозит, болезнь Грейвса, атеросклероз, болезнь Альцгеймера, синдром Гийена-Барре, болезнь Дегоса, отторжение ткани (например, отторжение трансплантата), сепсис, ожог (например, тяжелый ожог), септический системный воспалительный ответ, септический шок, повреждение спинного мозга, гломерулонефрит, тиреоидит Хашимото, сахарный диабет I типа, псориаз, пузырчатку, аутоиммунную гемолитическую анемию (AIHA), идиопатическую тромбоцитопеническую пурпуру (ITP), синдром Гудпасчера, антифосфолипидный синдром (APS), катастрофический APS (CAPS), боковой амиотрофический склероз (ALS), болезнь Альцгеймера и хроническую воспалительную демиелинизирующую нейропатию.
В другом варианте осуществления растворы, описанные в данном документе, можно применять для лечения тромботической микроангиопатии (TMA), например, TMA, ассоциированной с нарушением, ассоциированным с системой комплемента, таким как любое из нарушений, ассоциированных с системой комплемента, описанных в данном документе.
Нарушения, ассоциированные с системой комплемента, также включают легочные нарушения, ассоциированные с системой комплемента, такие как без ограничения астма, бронхит, хроническое обструктивное заболевание легких (COPD), интерстициальное заболевание легких, дефицит α-1-антитрипсина, эмфизема, бронхоэктаз, облитерирующий бронхиолит, альвеолит, саркоидоз, легочный фиброз и коллагенозные сосудистые нарушения.
В другом варианте осуществления раствор, описанный в данном документе, вводят субъекту для лечения, предупреждения или уменьшения интенсивности по меньшей мере одного симптома воспалительного ответа, ассоциированного с системой комплемента (например, аспекта нарушения, ассоциированного с системой комплемента, который представляет собой воспалительный ответ, ассоциированный с системой комплемента), у субъекта. Например, композицию можно применять для лечения, предупреждения и/или уменьшения интенсивности одного или более симптомов, ассоциированных с воспалительным ответом, ассоциированным с системой комплемента, таких как отторжение ткани/реакция «трансплантат против хозяина» (GVHD), реперфузионные повреждения (например, после сердечно-легочного шунтирования или трансплантации ткани) и повреждение тканей после других форм травматических повреждений, таких как ожог (например, тяжелый ожог), тупая травма, повреждение позвоночника или обморожение. См., например, Park et al. (1999) Anesth Analg 99(1):42-48; Tofukuji et al. (1998) J Thorac Cardiovasc Surg 116(6):1060-1068; Schmid et al. (1997) Shock 8(2):119-124 и Bless et al. (1999) Am J Physiol 276(1):L57-L63.
В другом варианте осуществления нарушение, опосредованное системой комплемента, представляет собой сосудистое нарушение, опосредованное системой комплемента, такое как, без ограничения, сердечно-сосудистое нарушение, миокардит, цереброваскулярное нарушение, нарушение со стороны периферических (например, скелетно-мышечных) сосудов, реноваскулярное нарушение, мезентериальное/энтеральное сосудистое нарушение, реваскуляризация трансплантатов и/или реплантатов, васкулит, нефрит при пурпуре Шенлейна-Геноха, васкулит, ассоциированный с системной красной волчанкой, васкулит, ассоциированный с ревматоидным артритом, иммунокомплексный васкулит, трансплантация органа или ткани, болезнь Такаясу, синдром повышенной проницаемости капилляров, дилатационная кардиомиопатия, диабетическая ангиопатия, аневризма грудного и брюшного отдела аорты, болезнь Кавасаки (артериит), венозная газовая эмболия (VGE) и рестеноз после стентирования, ротационная атерэктомия и чрескожная транслюминальная коронарная ангиопластика (PTCA) (см., например, публикацию заявки на патент США № 20070172483).
VII. Виды комбинированного лечения
В одном варианте осуществления растворы, описанные в данном документе, вводят пациенту в качестве монотерапии. В другом варианте осуществления их вводят совместно с одним или более дополнительными средствами и/или другими видами терапии (например, которые являются подходящими для лечения нарушений, ассоциированных с системой комплемента). Например, комбинированная терапия может включать введение пациенту-человеку одного или более дополнительных средств (например, антикоагулянтов, антигипертензивных средств или противовоспалительных лекарственных средств (например, стероидов)), которые обеспечивают терапевтически благоприятное воздействие на пациента. В одном варианте осуществления растворы, описанные в данном документе, вводят в комбинации с противовоспалительным средством (например, NSAID, кортикостероидами, метотрексатом, гидроксихлорохином, средствами на основе антител к TNF, такими как этанерцепт и инфликсимаб, средством, истощающим популяции В-клеток, таким как ритуксимаб, антагонистом интерлейкина-1 или средством, блокирующим костимуляцию T-клеток, таким как абатацепт).
Дополнительные средства для лечения нарушения, ассоциированного с системой комплемента, у субъекта будут отличаться в зависимости от конкретного нарушения, лечение которого осуществляется, но могут включать без ограничения одно или несколько антигипертензивных средств (например, ингибитор ангиотензинпревращающего фермента, лабеталол, гидралазин, нифедипин, антагонисты кальциевых каналов, нитроглицерин или нитропруссид натрия), антикоагулянтов, кортикостероидов (например, преднизон), иммунодепрессантов (например, винкристин или циклоспорин A), антикоагулянтов (например варфарин (кумадин), аспирин, гепарин, фениндион, фондапаринукс, идрапаринукс), ингибиторов тромбина (например, аргатробан, лепирудин, бивалирудин или дабигатран), фибринолитических средств (например, анкрод, α-аминокапроновую кислоту, антиплазмин-a1, простациклин и дефибротид), антигипертензивных средств (например, лабеталол, гидралазин, нифедипин, антагонисты кальциевых каналов, нитроглицерин или нитропруссид натрия), гиполипидемических средств (например, ингибитор гидроксиметилглутарил-CoA-редуктазы), противосудорожных средств (например, сульфат магния), антитромботических средств (например, гепарин, антитромбин, простациклин или низкодозовый аспирин), симпатомиметиков (например, альбутерол), антибиотиков, дезоксирибонуклеаз (например, Pulmozyme®), антихолинергических лекарственных средств, ингибиторов IgE (например, антитела к IgE), кортикостероидов или нестероидных противовоспалительных лекарственных средств (NSAID). Доступно множество различных NSAIDS, некоторые из которых отпускаются без рецепта, включая ибупрофен (Advil®, Motrin®, Nuprin®) и напроксен (Alleve®), и множество других доступно по рецепту, включая мелоксикам (Mobic®), этодолак (Lodine®), набуметон (Relafen®), сулиндак (Clinoril®), толметин (Tolectin®), салицилат холина и магния (Trilasate®), диклофенак (Cataflam®, Voltaren®, Arthrotec®), дифлунисал (Dolobid®), индометацин (Indocin®), кетопрофен (Orudis®, Oruvail®), оксапрозин (Daypro®) и пироксикам (Feldene®) (см., например, Mihu et al. (2007) J Gastrointestin Liver Dis 16(4):419-424). В другом варианте осуществления раствор, описанный в данном документе, может быть составлен для введения пациенту вместе с терапией внутривенными гамма-глобулинами (IVIG), плазмаферезом, замещением плазмы крови или плазмообменом.
В одном варианте осуществления раствор и одно или более дополнительных средств и/или видов терапии вводят одновременно. В другом варианте осуществления раствор вводят перед введением одного или более дополнительных средств и/или видов терапии. В другом варианте осуществления раствор вводят после введения одного или более дополнительных средств и/или видов терапии.
Если раствор антитела, описанный в данном документе, применяют в комбинации со вторым активным средством, то средства (например, антитело к C5 и второе средство) можно составлять по отдельности или вместе. Например, раствор и средство можно смешивать, например, непосредственно перед введением, и вводить вместе или по отдельности, например, в одно и то же или в разное время.
VIII. Наборы и лекарственные формы с однократной дозировкой
Также в данном документе предусмотрены наборы, которые содержат стабильный водный раствор, содержащий антитело к C5, такое как равулизумаб или BNJ421, или его антигенсвязывающий фрагмент в терапевтически эффективном количестве, подходящем для введения пациенту-человеку (например, пациенту с нарушением, ассоциированным с системой комплемента). Наборы необязательно также могут содержать инструкции, например, содержать графики введения, позволяющие практикующему специалисту (например, врачу, медсестре или пациенту) вводить композицию, содержащуюся в них, для введения раствора пациенту.
Наборы могут также содержать подходящее средство для доставки одного или нескольких растворов пациенту, нуждающемуся в этом, например, пациенту, страдающему нарушением, ассоциированным с системой комплемента, предположительно имеющему его или имеющему риск его развития. В одном варианте осуществления средство подходит для инвазивной (например, внутрисосудистой (например, внутривенной), подкожной, внутрисуставной, внутриглазной, интравитреальной или внутримышечной) доставки раствора пациенту. В другом варианте осуществления средство подходит для подкожной доставки раствора пациенту. В другом варианте осуществления средство подходит для внутривенной доставки раствора пациенту. Например, средство может представлять собой шприц или осмотический насос. В другом варианте осуществления раствор может быть составлен в виде глазных капель, при этом средство представляет собой пипетку для глаз.
Наборы необязательно содержат несколько упаковок раствора с однократной дозой, каждая из которых содержит эффективное количество раствора для однократного введения. Приборы или устройства, необходимые для введения раствора, также могут быть включены в наборы. Например, в наборе могут быть представлены один или несколько предварительно заполненных шприцев, содержащих раствор.
Следующие примеры являются лишь иллюстративными и не должны рассматриваться как ограничивающие объем настоящего изобретения каким-либо образом, поскольку многие изменения и эквиваленты станут очевидными специалистам в данной области после прочтения настоящего изобретения.
Содержимое всех литературных источников, записей в Genbank, патентов и опубликованных патентных заявок, цитируемых во всей настоящей заявке, явным образом включено в данный документ посредством ссылки.
ПРИМЕРЫ
Пример 1. Разработка высококонцентрированного состава равулизумаба (AXLN1210) для подкожного введения
В данном примере обобщена разработка высококонцентрированного состава ALXN1210 для подкожного введения (например, 50 мМ фосфатного буфера, 5% сахароза, 25 мМ аргинина, pH 7,4 при 100 мг/мл). Предварительные эксперименты проводили на ранней стадии разработки составов для получения данных скрининга соединений-кандидатов и для оценки снижения степени опалесценции при более высоких концентрациях ALXN1210. Исходный состав ALXN1210 (10 мМ фосфата, 150 мМ хлорида натрия, pH 7,0, 0,02% Tween 80, при 10 мг/мл) являлся бесцветным и слабо опалесцирующим. По мере повышения концентрации ALXN1210 степень опалесценции также повышалась. С учетом результатов скрининга соединений-кандидатов проводили исследование стабильности с получением данных о стабильности соединений-лидеров. После начального исследования стабильности проводили исследование стабильности опытных образцов для получения оптимального состава нерасфасованного лекарственного вещества и лекарственного препарата. Предварительные исследования и исследования стабильности подробно обсуждаются ниже.
1. Способы
Внешний вид
Внешний вид определяли путем визуального наблюдения с применением обычного лабораторного освещения как на белом, так и на черном фоне.
Связывание C5
ELISA для связывания C5 представляет собой анализ активности ALXN1210. Данная процедура тестирования представляет собой иммунологический анализ прямого связывания с колориметрическим выявлением, применяемый для тестирования способности ALXN1210 связываться с его мишенью, представляющей собой белок C5 системы комплемента человека. Титрационный микропланшет PolySorp покрывали белком C5 человека и блокировали бычьим сывороточным альбумином (BSA). Калибровочную кривую получали по эталонному материалу ALXN1210. Эталонный материал и тестовые образцы получали в трех разведениях, предназначенных для попадания в рабочий диапазон анализа. После инкубирования со стандартами и образцами планшет затем промывали и инкубировали с антителом мыши к IgG4 человека, конъюгированным с пероксидазой хрена (HRP). Планшет снова промывали и затем проявляли с применением субстрата, представляющего собой 2,2'-азино-бис(3-этилбензотиазолин-6-сульфоновую кислоту) (ABTS). Количество прореагировавшего субстрата считывали спектрофотометрическим способом на планшет-ридере при 405 нм. Показатель поглощения являлся пропорциональным концентрации ALXN1210, связывающегося с C5 на планшете. В отношении калибровочной кривой применяли четырехпараметрическую аппроксимацию кривой, и результаты для эталонного материала и тестовых образцов интерполировали по кривой. Результаты тестирования образцов сравнивали с результатами для эталонного материала, и регистрировали относительную активность (%).
Плотность
Измерения плотности осуществляли с помощью измерителя плотности DMA 4500, определяя ее посредством метода U-образной трубки. Полую U-образную стеклянную трубку заполняли образцом, а затем переводили в электронно-возбужденное состояние при наиболее низкой возможной амплитуде. Плотность определяли посредством следующего уравнения:
ρ=A(τ2)-B,
ρ=плотность;
τ=период колебаний.
A и B представляют собой постоянные измерительного прибора, определенные посредством калибровки прибора с помощью двух веществ известной плотности.
Дифференциальная сканирующая флуориметрия
Посредством дифференциальной сканирующей флуориметрии измеряют изменение термической стабильности путем получения кривой термической денатурации в присутствии флуоресцентного красителя, такого как Sypro оранжевый. При разворачивании белка доступные гидрофобные поверхности связываются с красителем, повышая уровень флуоресценции и обеспечивая получение кривой стабильности с характеристическим значением срединной точки при температуре воздействия на гидрофобные поверхности Th.
Динамическое рассеяние света
Посредством динамического рассеяния света измеряют размер и взаимодействия белков, наночастиц и других макромолекул in situ в планшетах с микролунками посредством применения осветительной системы, которая обеспечивает возможность визуализации лунок в микропланшете с применением встроенной 3-мегапиксельной камеры. Отклонения рассеяния света вследствие броуновского движения дают коэффициент диффузии, который связан с гидродинамическим радиусом частиц, присутствующих в растворе.
Гель-проникающая HPLC
Гель-проникающую (эксклюзионную) HPLC применяли с целью отделения мономерного IgG от молекул мультимерных антител большего размера, которые могут образовываться в результате агрегации мономеров. Тестовые образцы вводили в колонку с гелем TSK G3000 SWXL, уравновешенную фосфатно-солевым буфером, pH 7,0, с последующим изократическим элюированием. Пики белков контролировали при 214 нм, и процентную чистоту мономерного IgG выражали в виде процентной доли от общей интегрированной площади пиков. Выявление мультимеров с большей массой осуществляли посредством наблюдения пиков, появляющихся при элюировании до пика мономера.
Капиллярный электрофорез с визуализацией (iCE)
В данном способе применяли систему Protein Simple iCE280 или iCE3, которые обеспечивают получение раствора для IEF без примесей в капиллярной колонке, и осуществляли выявление зон скопления белков с применением УФ-детектора для всей колонки. Образцы получали посредством предварительного смешивания ALXN1210, амфолитов-носителей и маркеров pI. Образец загружали в картридж для капиллярного электрофореза, и электролизеры на каждом конце капилляра заполняли кислотой и основанием. Прилагали напряжение, и аналиты образовывали скопления при их pI. CCD-камера осуществляет визуализацию поглощения УФ-излучения всей капиллярной колонки каждые 30 секунд, обеспечивая контроль стадии образования скоплений в реальном времени. Полученный характер разделения регистрировали и анализировали. pI белков, присутствующих в образце, интерполировали по положению маркеров pI, внесенных в образец.
Лаборатория на чипе (LoC)
Посредством данного способа проводили тестирование однородности и чистоты продукта. Денатурацию невосстановленных образцов осуществляли посредством обработки с помощью додецилсульфата лития (LDS). Денатурацию восстановленных образцов осуществляли посредством обработки с помощью додецилсульфата лития (LDS), и дисульфидные связи разрушали с помощью дитиотреитола (DTT). Полипептидные цепи смешивали с флуоресцентным красителем, который связывается с LDS, и разделяли в соответствии с размером молекул посредством микрокапиллярного электрофореза. Белок выявляли и количественно определяли посредством лазерно-индуцированной флуоресценции.
Осмоляльность
Осмоляльность образцов определяли с применением осмометра, измеряющего снижение температуры замерзания. Осмометр калибровали перед применением с помощью коммерчески доступных сертифицированных стандартов осмоляльности при 50 мОсм/кг и 850 мОсм/кг, которые охватывают диапазон для образцов. Эталонный раствор с 290 мОсм/кг применяли для подтверждения успешной калибровки перед тестированием образцов. Образцы тестировали в трех повторностях, и регистрировали среднее значение из определенных для образцов.
pH
Измерение pH проводили с применением устойчивого к белкам насыщенного не содержащего серебра комбинированного электрода c KCl и соответствующего измерительного прибора и устройства контроля температуры. Перед применением измерительный прибор калибровали с помощью коммерчески доступных растворов в соответствующем диапазоне pH (т. е. pH 4,0 - pH 7,0).
Определение концентрации белков с применением SoloVPE
Для определения концентрации белков в тестовых образцах применяли поглощение при 280 нм с применением технологии с различными значениями длины оптического пути с применением теоретически определенного коэффициента экстинкции, составляющего 1,479. В способе показатели поглощения определяли в трех повторностях для каждого образца.
Вязкость
Измеренные значения вязкости определяли с применением вискозиметра AMVn посредством метода падающего шарика. Полую трубку заполняли образцом и твердым шариком известной плотности, а затем наклоняли под известным углом. Определяли время, необходимое для того, чтобы шарик переместился с одного конца трубки на другой, и применяли для вычисления вязкости посредством следующего уравнения:
η=K*(ρb-ρs)*tr,
η=динамическая вязкость (мПа*с);
K=константа пропорциональности;
ρb=плотность шарика (г/мл);
ρs=плотность образца (г/мл);
tr=время падения шарика.
Для вычисления вязкости плотность образца, определенную с применением измерительного прибора DMA 4500 M для определения плотности, использовали в качестве ρs.
Определение невидимых невооруженным глазом частиц посредством микропотоковой визуализации (MGI)
Целью является количественная оценка всех невидимых невооруженным глазом частиц в составе посредством микропотоковой визуализации (MFI). Образец отбирали из хранилища при температуре 2-8°C и непосредственно тестировали посредством MFI с применением автоматического пробоотборника BOT1. Образец переворачивали шесть раз перед загрузкой образца в BOT1 для обеспечения полного перемешивания частиц. Образцы загружали в три находящиеся друг за другом лунки, и осуществляли одно измерение для каждой из них с получением в общей сложности 3 повторностей. В BOT1 проводили три цикла смешивания для дополнительного обеспечения однородности смешивания.
2. Разработка состава
На фигурах 1-5 и в таблицах 1-5 показаны результаты экспериментов, которые проводили на ранней стадии разработки высококонцентрированных составов ALXN1210.
В первом эксперименте наблюдался эффект добавления аминокислоты к ALXN1210 в натрий-фосфатном буфере в отношении опалесценции. Причину возникновения опалесценции определяли как отсутствие заряда для обеспечения отталкивания зарядов между молекулами антител в растворе при высокой концентрации. Проводили ряд экспериментов, описанных ниже, с целью обеспечения оптимального выбора конкретной аминокислоты и концентрации, необходимой для получения стабильного прозрачного раствора. На основании результатов таких экспериментов определяли, что добавление положительно заряженной аминокислоты (L-аргинина) обеспечивает снижение степени опалесценции в образце, содержащем 50 мг/мл ALXN1210 в натрий-фосфатном буфере. К тому же выводу пришли посредством визуального осмотра флаконов (данные не показаны).
Кроме того, последующие эксперименты проводили посредством применения состава ALXN1210 IV при 10 мг/мл и посредством концентрирования антитела и осуществления замены буфера для оценки различных исходных буферных систем для применения при нахождении высококонцентрированного состава ALXN1210. Как показано ниже в таблице 1, для всех объединенных образцов проводили заключительную замену буфера при 1:1000 с получением желаемого pH. Объединенные образцы характеризовались диапазоном концентраций от 35,3 до 54,0 мг/мл. % извлечения после замены буфера находится в диапазоне от 70,6% до 108%. Результаты анализа внешнего вида показали, что содержимое флаконов с заменой буфера на 25 мМ гистидина с pH 7 и на 25 мМ фосфата с pH 7 было прозрачным и бесцветным, что сопоставимо с экулизумабом, и растворы во всех остальных флаконах с заменой буфера были опалесцирующими. Результаты капиллярного электрофореза с визуализацией (iCE) показывают, что pI находится в диапазоне от 5,98 до 6,54, главная pI находится в диапазоне от 6,19 до 6,24, и % площади пиков находится в диапазоне от 63,1% до 65,9%. Результаты эксклюзионной хроматографии (SEC) показывают, что % мономеров (чистоты) составляет от 98,48% до 98,98%.
Таблица 1. ALXN1210 с заменой буфера, от 10 мг/мл до 50 мг/мл (группы объединенных образцов 1-3)
*Сопоставимо с экулизумабом.
Как показано на фигуре 1, результаты, полученные при титровании солью для образцов с заменой буфера на гистидиновый при pH 7 с применением DLS, показывают, что с повышением концентрации соли самоассоциация у ALXN1210 также повышается.
Как показано на фигуре 2, результаты, полученные при титровании L-аргинином с применением динамического рассеяния света (DLS), показывают, что 25 мМ L-аргинина представляет собой минимальное количество, требуемое для снижения степени опалесценции у ALXN1210 при 50 мг/мл.
Как показано на фигуре 3, результаты, полученные при титровании солью для образцов с заменой буфера на фосфатный при pH 7 с применением DLS, показывают, что отсутствие добавления соли и добавление 150 мМ соли также обеспечивают наиболее низкую самоассоциацию у ALXN1210. Для сравнения см. пики, помеченные как 2 и 5.
Как показано на фигуре 4, результаты, полученные для образцов с заменой буфера с применением DSF, показывают, что гидрофобные карманы у ALXN1210 не являются доступными. Цитратный и ацетатный буферы при pH 5 и 6 обладают низкой термической стабильностью с наиболее низкими значениями температуры плавления (Tm), а гистидиновый и фосфатный буферы при pH 7 являются наиболее стабильными с наиболее высоким значением Tm.
Как показывают результаты анализов внешнего вида, представленные в таблице 2, ALXN1210 при приблизительно 100 мг/мл является прозрачным и бесцветным с добавлением 25 мМ L-аргинина в 25 мМ фосфатного буфера при pH 7.
Таблица 2. Внешний вид образцов ALXN1210 при 100 мг/мл
Как показано на фигуре 5, результаты для ALXN1210 при 10 мг/мл и 114 мг/мл с внесением L-аргинина и без него, полученные с применением DLS, показывают, что добавление 25 мМ L-аргинина в образец при по меньшей мере 100 мг/мл обеспечивает результат, наиболее близко сопоставимый с эталонным раствором при 10 мг/мл. В образце с по меньшей мере 100 мг/мл без добавления L-аргинина происходит образование молекул высшего порядка, что указывает на самоассоциацию.
Результаты анализа на осмоляльность, представленные в таблице 3, показывают, что ALXN1210 в 25 мМ гистидина, pH 7,2, с 8% сахарозой или 4,5% сорбитом находятся в пределах диапазона желаемых значений осмоляльности 275-320. Значение осмоляльности ALXN1210 в буфере с 25 мМ фосфата, 25 мМ L-аргинина, pH 7, дополненном 7% сахарозой или 4% сорбитом, также находится в пределах диапазона желаемых значений осмоляльности.
Таблица 3. Осмоляльность ALXN1210 в различных составах
Результаты анализа на вязкость, представленные в таблице 4, показывают, что при повышении концентрации ALXN1210 также повышается вязкость растворов ALXN1210 в гистидиновом и фосфатном буферах. Результаты анализа на плотность, представленные в таблице 4, показывают отсутствие значительного изменения в плотности для гистидинового и фосфатного буферов при изменениях концентрации.
Таблица 4. Вязкость и плотность образцов ALXN1210 при различных концентрациях
Как показано в таблице 5, добавление L-аргинина в форме основания в значительной степени повышает pH образца. L-аргинин в достаточном количестве с одноосновным фосфатом натрия, внесенный в образец, обеспечивал повышение pH на 1 единицу pH. L-аргинин⋅HCl, внесенный в образец, обеспечивал снижение pH на приблизительно 0,25 единицы pH. Однако что касается внешнего вида, степень опалесценции снижалась от внесения L-аргинина⋅HCl к внесению L-аргинина в достаточном количестве с одноосновным фосфатом натрия и к внесению L-аргинина в форме основания.
Таблица 5. Эффекты внесения в L-аргининовые буферы 25 мМ L-аргинина в отношении pH и внешнего вида для ALXN1210
3. Стабильность при хранении
На фигурах 6-21 показаны результаты первоначального исследования стабильности. Эти результаты показывают, что составы, содержащие гистидин, являются наименее стабильными, и составы, содержащие фосфат, являются наиболее стабильными спустя 2 месяца при 2-8°C, 23-27°C и 37°C. Также, как свидетельствуют результаты эксклюзионной хроматографии, составы, содержащие фосфат, с сорбитом и сахарозой были сопоставимыми спустя 2 месяца при 2-8°C и 23-27°C. Однако составы с сорбитом были немного более стабильными при 37°C по сравнению с составами с сахарозой спустя 2 месяца. Результаты динамического рассеяния света показывают отсутствие значительного изменения в образцах в фосфатном буфере с 25 мМ L-аргинина спустя 2 месяца при 2-8°C с добавлением сахарозы либо сорбита. Результаты динамического рассеяния света для образцов, содержащих гистидин, при T=2 месяца могут не перекрываться из-за высокой полидисперсности между сеансами сбора данных, что указывает на менее стабильный состав по сравнению с составами, содержащими фосфат. Результаты 5-дневного цикла замораживания-размораживания не показали какое-либо значительное изменение между T=0 и 5 циклами замораживания-размораживания.
4. Состав опытных образцов
На фигурах 22-46 показаны результаты исследования стабильности опытных образцов.
Эти результаты показывают, что все составы, содержащие фосфат, при 75 мг/мл и 100 мг/мл (нерасфасованное лекарственное вещество (BDS) и лекарственный препарат (DP)) являются стабильными в течение всего исследования стабильности при 2-8°C, -20°C и -80°C. Все составы, содержащие нерасфасованное лекарственное вещество, с концентрацией 100 мг/мл после 5 циклов замораживания-размораживания при -20°C и -80°C являлись стабильными и не демонстрировали значительное изменение.
5. Промежуточные заключения перед краткосрочными тестированиями разрушения
На основании результатов данных исследований определяли оптимальный состав для высококонцентрированного материала ALXN1210. В предварительных экспериментах предлагали добавление L-аргинина для снижения степени опалесценции для ALXN1210 при 100 мг/мл. Первоначальное исследование стабильности приводило к выбору состава-лидера с фосфатным буфером и L-аргинином при >50 мг/мл. По результатам исследования стабильности опытных образцов определяли исходный оптимальный состав для ALXN1210, содержащий 50 мМ фосфатного буфера, 5% сахарозу, 25 мМ аргинина, pH 7,4 при 100 мг/мл.
6. Разработка конечного оптимального состава
С целью оценки пригодности исходного оптимального состава ALXN1210 с концентрацией 100 мг/мл в буфере для состава (50 мМ фосфата натрия, 25 мМ аргинина и 5% сахароза при pH 7,4) его подвергали краткосрочным исследованиям разрушения, чтобы установить, является ли необходимым полисорбат 80 (PS80) либо другое поверхностно-активное вещество для предупреждения разрушения. Две марки PS 80 представляли собой 0,05% (вес/объем) POLYSORBATE 80 (HX2)™ от NOF America Corporation, о котором сообщается, что он содержит олеиновую кислоту >99% чистоты, и полисорбат 80 AVANTOR™ 4117 от J.T. Baker®, который является широко используемым поверхностно-активным веществом, которое состоит из смеси жирных кислот, содержащей олеиновую кислоту и пальмитиновую кислоту. Оба продукта часто называются TWEEN 80® и представляют собой неионогенное поверхностно-активное вещество, полученное из полиэтоксилированного сорбитана и олеиновой кислоты с гидрофильными группами, происходящими из полимеров этиленоксида.
Способы тестирования, применяемые для оценки потенциального применения PS80 Avantor J.T. Baker 4117 в составе ALXN1210 с концентрацией 100 мг/мл, перечислены ниже в таблице 6. См. способ индивидуального тестирования для получения дополнительных подробностей и описания способа.
Таблица 6. Способы тестирования для определения потенциального применения PS80 Avantor J.T. Baker 4117
Флаконы, содержащие 100 мг/мл ALXN1210 с PS80 Avantor J.T. Baker 4117 либо с PS80 HX2 NOF, подвергали визуальному осмотру. Для всех образцов было показано отсутствие видимых частиц или явных изменений цвета во всех образцах, подвергнутых разрушению при хранении при 45°C и дополнительному взбалтыванию в течение 5 дней при 2-8°C. Результаты показаны в таблице 7.
Таблица 7. Визуальный осмотр 100 мг/мл ALXN1210 во флаконах объемом 5 см3, содержащих PS80 Avantor J.T. Baker 4117 либо PS80 HX2 NOF
Для концентрации ALXN1210 в мг/мл было показано незначительное снижение, обусловленное условием разрушения. Все измерения концентрации приведены в таблице 8.
Составы ALXN1210 с концентрацией 100 мг/мл, содержащие 0,05% PS80 Avantor либо PS80 HX2 NOF, не имели значительного изменения в концентрации, будучи подвергнутыми такому же условию разрушения, как показано на фигуре 2.
Таблица 8. Измерения концентрации
Как показано выше в таблице 8, составы ALXN1210 с концентрацией 100 мг/мл, содержащие 0,05% PS80 Avantor либо PS80 HX2 NOF, не имели значительного изменения в концентрации, будучи подвергнутыми такому же условию разрушения. Концентрация оставалась сопоставимой в составах ALXN1210 с концентрацией 100 мг/мл, содержащих 0,05% PS80 Avantor либо PS80 HX2 NOF.
Мутность измеряли путем контроля поглощения при 650 нм. Измерения показаны в таблице 9.
Составы ALXN1210 с концентрацией 100 мг/мл, содержащие 0,05% PS80 Avantor либо 0,05% PS80 HX2 NOF, не имеют существенных изменений мутности. Показатели мутности остаются стабильными во все моменты времени и при всех условиях разрушения в данном исследовании.
Таблица 9. Мутность, измеряемая путем контроля поглощения при 650 нм
Наблюдалось снижение процентной доли мономеров для образцов, которые инкубировали при 45°C в течение 7 дней или 14 дней с дополнительным встряхиванием (200 об/мин) при 2-8°C, которое ожидалось в связи с условиями разрушения. Однако для процентной доли мономеров не было показано значительного отличия между обоими составами ALXN1210 с концентрацией 100 мг/мл, содержащими 0,05% PS80 Avantor либо 0,05% PS80 HX2 NOF, в одни и те же моменты времени и при воздействии одного и того же условия.
Данные о процентной доле мономеров показаны в таблице 10. На фигуре 4 показано снижение количества % мономеров, которое ожидалось после воздействия условий разрушения, и отсутствие значительного отличия между обоими составами ALXN1210 с концентрацией 100 мг/мл, содержащими 0,05% PS80 Avantor либо 0,05% PS80 HX2 NOF, в одни и те же моменты времени и при воздействии одного и того же условия.
Таблица 10. Процентная доля мономеров для образцов, которые инкубировали при 45°C в течение 7 дней или 14 дней с дополнительным встряхиванием (200 об/мин) при 2-8°C
Сдвиг в направлении кислых молекул выявляли посредством изоэлектрического фокусирования после инкубирования ALXN1210 с 0,05% полисорбатом 80 Avantor J.T. Baker либо с 0,05% полисорбатом 80 HX2 NOF при 45°C в течение 7 дней и 14 дней, как показано в таблице 11. Дополнительное встряхивание образцов не оказывало значительное влияние на дополнительный сдвиг главного пика в направлении кислых молекул.
Таблица 11. Изоэлектрическое фокусирование посредством CE-SDS
Тестирование с нагрузкой при встряхивании проводили в отношении исходного оптимального состава ALXN1210 с концентрацией 100 мг/мл в буфере для состава, содержащем 50 мМ фосфата натрия, 25 мМ аргинина и 5% сахарозу, при pH 7,4 в присутствии двух марок PS 80, включенных в состав при концентрации 0,05%, и в их отсутствие. Степень образования невидимых невооруженным глазом частиц применяли для оценки разрушения. Образцы встряхивали при 200 об./мин при температуре 2-8°C. Моменты времени для проведения измерений в отношении того, происходит ли образование невидимых невооруженным глазом частиц, представляли собой дни 0, 1, 3 и 5. Результаты, показанные в таблице 12, указывают на то, что добавление 0,05% PS80 в состав значительно снижает степень образования невидимых невооруженным глазом частиц, если высококонцентрированный состав подвергали краткосрочной нагрузке, такой как встряхивание при 200 об/мин.
Таблица 12. Снижение образования невидимых невооруженным глазом частиц посредством добавления PS80 и фильтрации
7. Заключение
В заключение, оптимальный с точки зрения количества невидимых невооруженным глазом частиц состав ALXN1210 при концентрации 100 мг/мл содержит буфер, содержащий 50 мМ фосфата натрия, 25 мМ аргинина и 5% сахарозу, и 0,05% PS80 при pH 7,4.
Пример 2. Исследование фазы 1 для оценки однократной дозы ALXN1210, вводимого подкожно в сравнении с вводимым внутривенно, у здоровых субъектов
Исследование фазы 1 проводили для оценки безопасности, переносимости, эффективности, фармакокинетических (PK)/фармакодинамических (PD) характеристик и иммуногенности антитела BNJ441 (также известного как ALXN1210), вводимого подкожно (SC) в сравнении с вводимым внутривенно (IV), у здорового субъекта.
1. Цели
Первичные цели данного исследования представляли собой (1) оценку безопасности и переносимости однократной дозы ALXN1210, вводимого подкожно, по сравнению с ALXN1210, вводимым внутривенно, у здоровых субъектов, что оценивали по данным физикального обследования, измерениям основных показателей жизнедеятельности, данным об иммуногенности, данным лабораторного анализа и оценкам нежелательных явлений (AE), и (2) определение абсолютной биологической доступности ALXN1210, вводимого подкожно.
Вторичная цель представляла собой оценку PD-эффектов ALXN1210, вводимого подкожно, по сравнению с ALXN1210, вводимым внутривенно, которые оценивали по уровню свободного C5 и степени гемолиза куриных эритроцитов (cRBC).
2. План исследования
Общий план исследования выполняли, как показано на изображении на фигуре 47. Это было исследование фазы 1, разработанное для оценки безопасности, переносимости, PK, PD и иммуногенности однократной дозы 400 мг ALXN1210, вводимого подкожно, по сравнению с однократной дозой 400 мг ALXN1210, вводимого внутривенно, или плацебо, вводимого подкожно, у 42 здоровых субъектов. Всех субъектов подвергали скринингу на предмет соответствия требованиям к включению в исследование. Субъектов, которые не соответствуют критериям включения в исследование, не подвергали повторному скринингу в отношении участия в исследовании, если только условие, которое ведет к несоответствию требованиям к включению в исследование, не было временным, самоограничивающимся и легко поддающимся лечению, и, как ожидается, будет устранено к моменту введения доз.
Шестерых субъектов изначально произвольным образом распределяли в соотношении 2:1 на когорту 1a слепым способом для получения однократной дозы 400 мг ALXN1210, вводимого подкожно, либо однократной дозы плацебо, вводимого подкожно. Данные о клинической безопасности, полученные в течение первых 48 часов после введения дозы, оценивали для субъектов в когорте 1a перед началом включения для исследования в когорты 1b или 2. Затем тридцать шесть субъектов произвольным образом распределяли в соотношении 2:1 в когорту 1b (N=24) либо в когорту 2 (N=12). В когорте 1b 24 субъекта дополнительно произвольным образом распределяли в соотношении 5:1 слепым способом для получения соответственно однократной дозы 400 мг ALXN1210, вводимого подкожно (20 субъектов), либо однократной дозы плацебо, вводимого подкожно (4 субъекта). 12 субъектов в когорте 2 получали однократную дозу 400 мг ALXN1210, вводимого внутривенно, в немаскированном режиме.
Всех субъектов, включенных в исследование, включали в анализы соответствующим образом. Субъектов в когортах 1a и 1b объединяли для проведения анализов. Субъекты принимали участие в исследовании в течение периода до 39 недель, включая период проведения скрининга, составляющий до 70 дней, с последующим 200-дневным периодом последующего наблюдения для осуществления оценки безопасности, PK, PD и иммуногенности после введения исследуемого лекарственного средства.
Сорок два субъекта оценивали в отношении первичных и вторичных целей в данном исследовании: 6 (4 получали ALXN1210 подкожно, 2 получали плацебо подкожно) субъектов в когорте 1a, 24 (20 получали ALXN1210 подкожно, 4 получали плацебо подкожно) субъекта в когорте 1b и 12 (ALXN1210 IV) субъектов в когорте 2.
3. Обоснование выбора дозы
Однократную дозу 400 мг, эквивалентную 4 мл, вводили подкожно посредством 4 инъекций по 1 мл в область живота. Ожидалось, что введение SC однократной дозы 400 мг ALXN1210 будет характеризоваться приемлемым профилем безопасности. Предполагалось, что однократные дозы 400 мг ALXN1210 SC и плацебо SC, вводимые, как описано в данном протоколе, обеспечат получение данных, на основании которых можно осуществить моделирование введения многократных доз с целью планирования режимов дозирования, необходимых для достижения терапевтических концентраций в сыворотке крови (>50 мкг/мл) у пациентов.
Параллельную рандомизацию 36 субъектов в когорте 1b и когорте 2 проводили на основании оценки данных о клинической безопасности, полученных в течение первых 48 часов после введения дозы от 6 субъектов в когорте 1a. Включение для исследования в когорту 1b и когорту 2 осуществляли, как описано в таблице 13.
Правила определения токсичности в группах были следующими. Токсичность относится к клинически значимой (значимым) нежелательной (нежелательным) реакции (реакциям), связанной (связанным) с лекарственным средством. «Продолжение участия когорты в исследовании» относится к продолжению участия в исследовании с последовательным введением доз/режимом дозирования с соблюдением правил продолжения введения доз в ходе исследования и при минимальных требованиях к данным. «Приостановка клинического исследования» означает, что дополнительный IMP не вводили при соответствующем уровне дозы/режиме дозирования, и что дальнейшее продолжение участия когорты в исследовании было приостановлено.
Таблица 13. Правила определения токсичности
серьезность
Сокращения: CTCAE=общие терминологические критерии нежелательных явлений; SAE=серьезное нежелательное явление; SOC=системно-органный класс.
4. График проведения оценок
Временные рамки применяемых процедур исследования представлены в таблицах 14-15.
Таблица 14. График проведения оценок: от скрининга до визита 1
1 Допустимые отклонения для оценок в рамках исследования описаны в руководстве по проведению процедур в ходе исследования.
2 Завершение инфузии (EOI) происходило примерно через 15 минут после начала инфузии (SOI).
3 Субъекта исключали из отделения клинического исследования после завершения всех оценок в день 5. Для субъектов заводили «личную карточку пациента, участвующего в исследовании», с информацией для медицинских работников и участника исследования о симптомах инфекционного менингита.
4 Перед осуществлением каких-либо связанных с исследованием процедур скрининга получали подписанные и датированные формы информированного согласия.
5 Для субъектов, у которых нет соответствующей документации о предшествующей иммунизации MCV4 или вакцинации против серологической группы B, иммунизацию MCV4 проводили за по меньшей мере 56 дней до введения первой дозы в день 1, а вакцину против менингококковых инфекций из серологической группы B вводили за по меньшей мере 56 дней до введения дозы в день 1 с введением бустерной инъекции за по меньшей мере 28 дней до введения дозы в день 1.
6 Определение активности системы комплемента, подтвержденной посредством подходящего анализа, такого как ELISA для альтернативного пути активации системы комплемента (CAP)/анализ ингибирования C5 (гемолиза), проводили при скрининге для подтверждения того, что у субъектов нет недостаточности комплемента.
7 Образец, отобранный в день -1, хранили для проведения анализа в будущем, который нужно будет провести в случае, если для образца, отобранного после введения дозы, будет показано, что функционирование системы комплемента не нормализовано.
8 Тестирование сыворотки крови на беременность проводили для всех субъектов-женщин для подтверждения того, что субъекты-женщины не находились в состоянии беременности.
9 В день 1 оценивали измерения основных показателей жизнедеятельности до введения дозы (в течение 15 минут до SOI) и по завершении инфузии, через 30 минут после завершения инфузии, через 2 часа после начала инфузии, через 4 часа после начала инфузии и через 8 часов после начала инфузии.
10 В день 1 проводили ECG в трех повторностях в 12 отведениях до введения дозы и примерно через 15 минут после завершения инфузии.
11 Проводили непрерывную регистрацию сердечной деятельности с момента до введения дозы в течение периода IV инфузии (когорта 2) и до момента через 3 часа после SC инъекции (когорты 1a и 1b).
12 Оценку в месте инфузии или инъекции осуществляли в течение 15 минут от начала инфузии/инъекции и ±15 минут от другого запланированного времени в день 1. Уплотнения или реакции размером <1 см не указывали в качестве нежелательного явления, если они не сохранялись в течение более 24 ч. Боль в месте инфузии или инъекции оценивали с помощью визуально-аналоговой шкалы (0-10). Боль не оценивали до введения дозы.
13 Исследователь или уполномоченное лицо встречались с субъектом при каждом визите для обсуждения потенциальных рисков, связанных с безопасностью ALXN1210, и для того, чтобы отреагировать на любые проблемы безопасности со стороны субъекта.
14 Сбор данных о нежелательных явлениях (AE) и серьезных нежелательных явлениях (SAE) начинали с момента подписания формы информированного согласия.
15 Для осуществления профилактического лечения антибиотиками субъектам перорально вводили пенициллин V в количестве 500 мг два раза в день (что эквивалентно 1×106 единицам), начиная с вечера дня -1, до тех пор, пока активность системы комплемента не нормализовалась, что определяли посредством анализа CH50.
Сокращения: ADA=антитело к лекарственному средству; BMI=индекс массы тела; cRBC=куриный эритроцит; CRU=отделение клинического исследования; ECG=электрокардиограмма; EOI=завершение инфузии/инъекции; HIV=вирус иммунодефицита человека; ICF=форма информированного согласия; MCV4=тетравалентная менингококковая конъюгатная вакцина; OP=амбулаторный пациент; SOI=начало инфузии/инъекции; TB=туберкулез.
Таблица 15. График проведения оценок: от визита 2 до визита 14
1 Дополнительные образцы отбирали после дня 57.
2 Исследователь или уполномоченное лицо встречались с субъектом при каждом визите для обсуждения потенциальных рисков, связанных с безопасностью ALXN1210, и для того, чтобы отреагировать на любые проблемы безопасности со стороны субъекта.
3 Сбор данных о нежелательных явлениях начинали с момента подписания формы информированного согласия.
4 Для осуществления профилактического лечения антибиотиками субъектам перорально вводили пенициллин V в количестве 500 мг два раза в день (что эквивалентно 1×106 единицам) до тех пор, пока активность системы комплемента не нормализовалась, что определяли посредством анализа CH50.
Сокращения: ADA=антитело к лекарственному средству; cRBC=куриный эритроцит; CRU=отделение клинического исследования; ECG=электрокардиограмма; ICF=форма информированного согласия; OP=амбулаторный пациент.
5. Отбор и прекращение участия субъектов
Для соответствия требованиям к включению в исследование субъекты должны соответствовать всем следующим критериям.
1. Здоровые субъекты возрастом от 25 до 55 лет включительно на момент введения доз.
2. Индекс массы тела (BMI), составляющий от 18 до 29,9 кг/м2 включительно, и вес, составляющий от 50 до 100 кг включительно.
3. Интервал QT, скорректированный с применением формулы Фридеричиа (QTcF), составляющий ≤450 мс для субъектов-мужчин и ≤470 мс для субъектов-женщин при скрининге и до введения доз в день 1.
4. Согласие и способность предоставить письменное информированное согласие и соблюдать график визитов в ходе исследования.
5. Документально подтвержденная вакцинация с помощью MCV4 за по меньшей мере 56 дня и не более чем за 3 года до введения доз. Документальное подтверждение должно включать положительный титр антител для подтверждения иммунного ответа до введения исследуемого лекарственного средства.
6. Вакцинация с помощью вакцины против менингококковых инфекций из серологической группы B за по меньшей мере 56 дней до введения дозы в день 1 с введением бустерной инъекции за по меньшей мере 28 дней до введения дозы в день 1 с интервалом, составляющим по меньшей мере 28 дней, между первой и второй инъекциями.
7. Субъекты-женщины, способные к деторождению, если они проявляют гетеросексуальную активность, должны применять высокоэффективные или приемлемые противозачаточные средства, как определено ниже, начиная со скрининга и продолжая в течение по меньшей мере 6 месяцев после введения исследуемого лекарственного средства. В ходе данного исследования требовалась профилактика антибиотиками, которая может негативно влиять на эффективность гормональных противозачаточных средств. Следовательно, рекомендуется, чтобы субъекты, применяющие гормональные противозачаточные средства, также применяли барьерные противозачаточные средства (например, презерватив или диафрагму со спермицидом) в течение всего периода профилактики антибиотиками. Субъекты-мужчины, если они проявляют гетеросексуальную активность и имеют супругу- или партнера-женщину, способную к деторождению, или супругу или партнера, находящихся в состоянии беременности или осуществляющих грудное вскармливание, должны быть согласны применять барьерные противозачаточные средства (мужской презерватив) во время периода лечения и в течение по меньшей мере 6 месяцев после введения исследуемого лекарственного средства. Барьерные противозачаточные средства были необходимы даже в случае предоставления документального медицинского заключения об успешной хирургической вазэктомии. Супруги- или партнеры-женщины субъектов-мужчин, способные к деторождению, должны применять высокоэффективные противозачаточные средства, определенные выше, или приемлемые противозачаточные средства, определенные ниже, начиная со скрининга и продолжая в течение по меньшей мере 6 месяцев после введения исследуемого лекарственного средства. Субъекты-мужчины не должны быть донорами спермы в течение периодов скрининга и лечения и в течение по меньшей мере 6 месяцев после введения исследуемого лекарственного средства.
Субъекты, удовлетворяющие любому из следующих критериев исключения, не соответствовали требованиям к участию в исследовании.
1. Субъекты, которые находились в тесном и длительном контакте (определенном как проживание под одной крышей или обеспечение персонального ухода) с людьми в возрасте моложе 2 лет или в возрасте старше 65 лет или которые имели ослабленный иммунитет либо одно из следующих первичных медицинских состояний: анатомическую или функциональную асплению (включая серповидноклеточную анемию); врожденные дефициты системы комплемента, пропердина, фактора D или первичных антител; приобретенные дефициты системы комплемента (например, пациенты, получающие экулизумаб) или вирус иммунодефицита человека (HIV).
2. Субъекты, которые относились к одной из следующих групп: специалисты, которые были подвержены воздействию условий окружающей среды с повышенным риском возникновения менингококкового заболевания; научный, производственный и клинико-лабораторный персонал, который регулярно подвергался воздействию N. meningitidis; военный персонал во время обучения новобранцев (военный персонал может подвергаться повышенному риску возникновения менингококковой инфекции при размещении в тесных условиях); сотрудники детских дошкольных учреждений; субъекты, проживающие в общежитии колледжей или университетов; и субъекты, которые планируют путешествие в течение исследования или путешествовали в области, эндемичные для менингококкового менингита (например, в Индию, страны Африки, расположенные к югу от Сахары, паломничество в Саудовскую Аравию для осуществления хаджа) в течение 6 месяцев до введения дозы.
3. Любая инфекция, вызванная Neisseria, в анамнезе.
4. Рецидивирующая инфекция неизвестной этиологии или инфекция, требующая лечения с помощью системных антибиотиков, в течение 90 дней до введения дозы в анамнезе.
5. HIV-инфекция (о которой свидетельствует титр антител к HIV-1 или HIV-2).
6. Острая или хроническая инфекция, вызванная вирусом гепатита B (HBV). Для всех субъектов перед включением в исследование требовалось тестирование на поверхностный антиген вируса гепатита В (HBsAg). Субъектов с положительными результатами анализа на HBsAg не включали в исследование. Для субъектов с отрицательным результатом анализа на HBsAg был необходим следующий алгоритм тестирования. Если результат анализа на антитела к сердцевинному антигену вируса гепатита B (HBcAb) был отрицательным, то субъект соответствовал требованиям к включению в исследование. Если результат анализа на HBcAb был положительным, то проводили тестирование на антитела к поверхностному антигену вируса гепатита B (HBsAb). Если результаты анализа как на HBcAb, так и на HBsAb были положительными, то субъект соответствовал требованиям к включению в исследование. Если результат анализа на HBcAb был положительным, а результат анализа на HBsAb был отрицательным, то субъекта не включали в исследование.
7. Острая или хроническая инфекция, вызванная вирусом гепатита C (о которой свидетельствует титр антител).
8. Активная системная вирусная или грибковая инфекция в течение 14 дней до введения дозы.
9. Положительный или неясный результат теста QuantiFERON®-TB, указывающий на возможное наличие туберкулезной инфекции (TB).
10. Латентная или активная TB или нахождение в эндемичных областях в течение 8 недель до скринингового визита в анамнезе.
11. Субъекты-женщины, которые осуществляют грудное вскармливание или проявляют гетеросексуальную активность и не согласны применять противозачаточные средства и не находятся в периоде постменопаузы. Период постменопаузы определяли как отсутствие менструаций в течение ≥12 последовательных месяцев без другой причины и документально подтвержденные уровень фолликулостимулирующего гормона в сыворотке крови, составляющий ≥40 мМЕ/мл, и концентрация эстрадиола, составляющая ≤110 пмоль/л, в течение 6 месяцев до введения исследуемого лекарственного средства.
12. Положительный результат тестирования сыворотки крови на беременность при скрининге или в день -1.
13. Уровень креатинина в сыворотке крови, превышающий верхний предел нормы (ULN) в диапазоне эталонных значений в лабораторных тестах при скрининге или в день -1.
14. Уровень аланинаминотрансферазы (ALT) или аспартатаминотрансферазы (AST) > ULN в диапазоне эталонных значений в лабораторных тестах при скрининге или > 1,5 × ULN в диапазоне эталонных значений в лабораторных тестах в день -1.
15. Любой из следующих результатов гематологического анализа: уровень гемоглобина <130 г/л для субъектов-мужчин и < 115 г/л для субъектов-женщин, гематокритное число <0,37 л/л для субъектов-мужчин и <0,33 л/л для субъектов-женщин, количество белых кровяных телец (WBC) <3,0×103/мкл, абсолютное количество нейтрофилов <2,0×103/мкл и количество тромбоцитов <150 или >400×103/мкл при скрининге или в день -1. Результаты клинико-лабораторного развернутого анализа крови (CBC), которые исследователь считает важными с клинической точки зрения и неприемлемыми, в день -1.
16. Недостаточность комплемента или активность системы комплемента ниже диапазона нормальных эталонных значений, оцениваемые посредством ELISA для СAP при скрининге, в анамнезе.
17. Злокачественные новообразования за исключением немеланомного рака кожи или карциномы шейки матки in situ, которые были подвергнуты лечению без признаков рецидива, в анамнезе.
18. Участие в клиническом исследовании в течение 30 дней до начала введения дозы в день 1 или применение любого экспериментального низкомолекулярного терапевтического средства в течение 30 дней до введения дозы в день 1.
19. Участие в более чем одном клиническом исследовании mAb или участие в клиническом исследовании mAb в течение 12 месяцев до проведения скрининга, во время которого субъект подвергался воздействию активного исследуемого лекарственного средства. Субъекты, которые принимали участие только в одном исследовании mAb, могли считаться подходящими для включения в данное исследование, если они завершили свое участие в предыдущем исследовании более чем за 12 месяцев до проведения скрининга.
20. Предшествующее воздействие ALXN1210.
21. Обширное хирургическое вмешательство или госпитализация в течение 90 дней до введения дозы.
22. Аллергия на наполнители для ALXN1210 (например, полисорбат 80) в анамнезе.
23. Документально подтвержденная аллергия на пенициллин или цефалоспорин в анамнезе.
24. Значительная аллергическая реакция (например, анафилаксия или ангионевротический отек) на любой продукт (пищевой, фармацевтический и т.д.) в анамнезе.
25. Курящие в настоящее время >10 сигарет в день (может быть позволено включить в исследование бывших курильщиков по усмотрению исследователя).
26. Злоупотребление запрещенными наркотическими средствами в анамнезе, выраженное злоупотребление алкоголем в течение 1 года до скринингового визита или регулярный прием алкоголя в течение 6 месяцев до скринингового визита (более 14 питейных доз алкоголя в неделю [1 единица=150 мл вина, 360 мл пива или 45 мл 40% спирта]) в анамнезе.
27. Положительный результат токсикологического скрининга мочи на наркотические вещества при скрининге или в день -1.
28. Употребление алкоголя в течение 48 часов до введения исследуемого лекарственного средства или положительный результат тестирования на алкоголь в выдыхаемом воздухе в день -1.
29. Донорство плазмы крови в течение 7 дней до введения дозы. Донорство или потеря (за исключением объема, отбираемого при скрининге) более 50 мл крови в течение 30 дней до введения дозы или более 499 мл крови в течение 56 дней до введения дозы.
30. Непрерывное местное, ингаляционное или системное применение стероидов в течение >28 дней в анамнезе или ингаляционное или местное применение любого иммунодепрессивного терапевтического средства в течение 90 дней до введения исследуемого лекарственного средства в анамнезе.
31. Применение рецептурных лекарственных препаратов (за исключением пероральных противозачаточных средств) в течение 14 дней до введения исследуемого лекарственного средства, кроме случаев предварительного одобрения спонсором клинического исследования.
32. Регулярное применение продаваемых без рецепта, безрецептурных лекарственных препаратов, в том числе препаратов растительного происхождения и пищевых добавок, в течение 14 дней до введения исследуемого лекарственного средства. Допускается применение поливитаминов, ацетаминофена ≤2 г в день и препаратов для кожи местного действия без значительной системной абсорбции.
33. Любое клинически диагностированное аутоиммунное или ревматологическое заболевание (например, системная красная волчанка, ревматоидный артрит).
34. Иммунизация с помощью живой аттенуированной вакцины за 28 дней до введения дозы или плановая вакцинация в течение исследования (за исключением вакцинации, запланированной согласно протоколу исследования). Допускалась иммунизация инактивированной или рекомбинантной вакциной против гриппа.
35. Наличие лихорадки (подтвержденной температуры тела >37,6°C) (например, лихорадки, ассоциированной с симптоматической вирусной или бактериальной инфекцией) в течение 14 дней до введения дозы.
36. Субъекты с любым анамнезом, состояниями или рисками, которые по мнению исследователя могли бы воспрепятствовать полному участию субъекта в исследовании или соблюдению протокола или могли бы подвергать субъекта каким-либо дополнительным рискам или искажать оценку субъекта или исхода исследования.
6. Инфекция
Для снижения риска инфекции, ассоциированной с ингибированием терминальных компонентов системы комплемента, субъектам в данном исследовании вводили следующее.
1. Вакцинация с помощью MCV4 за по меньшей мере 56 дней до введения дозы ALXN1210 в день 1 (если субъекты не были вакцинированы с помощью MCV4 в течение последних 3 лет или если субъекты были вакцинированы ранее, но отсутствовала соответствующая документация для подтверждения предшествующей вакцинации).
2. Две инъекции вакцины против менингококковых инфекций из серологической группы B. Первую инъекцию нужно вводить за по меньшей мере 56 дней до введения дозы в день 1 с введением бустерной инъекции за по меньшей мере 28 дней до введения дозы в день 1 с интервалом, составляющим по меньшей мере 28 дней, между первой и второй инъекциями.
3. Для осуществления профилактического лечения антибиотиками перорально применяли пенициллин V в количестве 500 мг два раза в день (что эквивалентно 1×106 единицам) до тех пор, пока активность системы комплемента не нормализовалась (что определяли посредством анализа CH50).
Первую дозу антибиотика вводили перорально в день -1 вечером перед днем 1 введения дозы исследуемого лекарственного средства. Для амбулаторной части исследования субъектов инструктировали в отношении того, чтобы они принимали антибиотик примерно в одно и то же время (два раза в день) в каждый запланированный день. Для ежедневного контроля соблюдения субъектами режима профилактики антибиотиками применяли подходящую систему (такую как обмен текстовыми сообщениями).
Следующие наблюдения подтверждают обоснованность введения средств для профилактики антибиотиками в данном исследовании с однократной дозой.
1. Пенициллин представлял собой предпочтительное лекарственное средство для уничтожения N. meningitidis у носителей.
2. Пациенты с недостаточностью комплемента, которые один раз в месяц получали инъекции бензатинпенициллина G в качестве средства профилактики рецидивирующего менингококкового заболевания в течение 2-4-летнего периода, испытывали значительно меньшее количество эпизодов инфекции, вызванной Neisseria, чем индивидуумы с недостаточностью, не получающие профилактику (Figueroa JE, et al., Clin. Microbiol. Rev. 1991 Jul;4(3):359-95).
3. Высокие уровни устойчивости к пенициллину, обусловленные β-лактамазами, кодируемыми плазмидами, редко встречаются у штаммов менингококков (Yazdankhah SP, et al., J. Med. Microbiol. 2004 Sep;53(Pt 9):821-32).
4. Некоторые врачи предлагали профилактику антибиотиками с помощью перорального введения пенициллина V в количестве 500 мг два раза в день при лечении пациентов с PNH и aHUS с помощью экулизумаба (Kelly RJ, et al., Blood 2011 Jun 23;117(25):6786-92 and Leeds Teaching Hospitals NHS Trust, Kings College Hospital NHS Foundation Trust, National Specialised Commissioning Team (NSCT) Service Specification Paroxysmal Nocturnal Haemoglobinuria (PNH) 2013).
5. Неопределенность в отношении эффективности вакцин у пациентов с ослабленным иммунитетом вынудила несколько стран, таких как Франция, рекомендовать непрерывную профилактику антибиотиками в течение всего периода лечения экулизумабом у пациентов с PNH и aHUS (Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat.Rev.Nephrol. 2012 Nov;8(11):643-57).
7. Ранее назначенные и сопутствующие лекарственные препараты и процедуры
Ранее назначенные лекарственные препараты (любое лекарственное средство или вещество, принимаемое субъектом в течение 14 дней до момента времени подписания субъектом ICF перед введением исследуемого лекарственного средства) и сопутствующие лекарственные препараты (любое лекарственное средство или вещество, принимаемое субъектом после введения исследуемого лекарственного средства до завершения последнего визита в ходе исследования) записывали в электронную индивидуальную регистрационную карту субъекта (eCRF). Ранее назначенные процедуры (любое терапевтическое вмешательство [например, хирургическая операция/биопсия, физиотерапия], проводимые в течение 14 дней до момента времени подписания субъектом информированного согласия перед введением исследуемого лекарственного средства) и сопутствующие процедуры (любое терапевтическое вмешательство [например, хирургическая операция/биопсия, физиотерапия], проводимое после введения исследуемого лекарственного средства до завершения последнего визита в ходе исследования) записывали в eCRF субъекта.
Сопутствующее терапевтическое средство представляло собой любое лекарственное средство или вещество, вводимое с момента времени скрининга субъекта для исследования до завершения последнего визита в ходе исследования. Субъектов инструктировали в отношении того, чтобы они не начинали принимать какие-либо новые лекарственные препараты, включая продаваемые без рецепта лекарственные средства и препараты растительного происхождения, если они не получили разрешения от исследователя, в течение всего периода исследования. Во время исследования было разрешено эпизодическое применение безрецептурных жаропонижающих или болеутоляющих средств (например, ацетаминофена).
Сопутствующая процедура представляла собой любое терапевтическое вмешательство (например, хирургическую операцию/биопсию, физиотерапию) или диагностическую оценку за рамками исследования (например, измерение газов крови, анализ на микрофлору), выполняемые с момента подписания субъектом информированного согласия до последнего визита в ходе исследования. Сопутствующие процедуры не были разрешены, если они не были назначены по медицинским показаниям.
8. Рандомизация и маскирование
Субъектам, соответствующим требованиям к участию в исследовании, которые соответствовали критериям включения и исключения, присваивали индивидуальные номера для включения в исследование и рандомизации.
Данное исследование представляло собой следующее частично слепое исследование.
Когорта 1a. Введение дозы (однократной дозы 400 мг ALXN1210 или плацебо SC) происходило в двойном слепом режиме. Субъектов в когорте 1a произвольным образом распределяли в соотношении 2:1 (4 ALXN1210 SC, 2 плацебо SC; N=6).
Когорта 1b. Введение дозы (однократной дозы 400 мг ALXN1210 или плацебо SC) происходило в двойном слепом режиме. Субъектов в когорте 1b произвольным образом распределяли в соотношении 5:1 (20 ALXN1210 SC, 4 плацебо SC; N=24).
Когорта 2 . Введение дозы (однократной дозы 400 мг ALXN1210 IV) происходило в немаскированном режиме (N=12).
Во время введения доз когорте 2 как субъектам, так и присутствующему на месте врачебному/сестринскому персоналу были известны лекарственное средство/доза, предназначенные для введения.
Во время введения доз когортам 1a и 1b как для субъектов, так и для присутствующего на месте врачебного/сестринского персонала в исследовательском центре были замаскированы данные о назначении исследуемого лекарственного средства. Для фармацевтического персонала, который готовил SC инъекции, и для персонала, осуществляющего введение исследуемого лекарственного средства, данные не были замаскированы, тогда как для всего остального персонала исследовательского центра, участвующего в оценивании безопасности, данные о назначении исследуемого лекарственного средства оставались замаскированными. Для персонала спонсора клинического исследования при необходимости проводили демаскирование данных (например, для контроля приготовления SC инъекций соответствующим образом, для определения возможности сообщения данных о SAE), и его удерживали от предоставления любой информации о назначении исследуемого лекарственного средства персоналу исследовательского центра.
9. Описание исследуемого лекарственного средства
Исследуемый препарат описан в таблице 16.
Таблица 16. Исследуемый препарат
1 Каждый флакон с лекарственным препаратом ALXN1210 IV имел незначительное переполнение для обеспечения возможности извлечения 15 мл (150 мг ALXN1210) для введения.
2 Каждый флакон с лекарственным препаратом ALXN1210 SC имел незначительное переполнение для обеспечения возможности извлечения 1 мл (100 мг ALXN1210) для введения.
Сокращения: BP=Британская фармакопея; IV=внутривенный; н.д.=нет данных; Ph Eur=Европейская фармакопея; SC=подкожный.
10. ALXN1210 и плацебо
Каждый флакон с ALXN1210 SC содержал 100 мг ALXN1210 (100 мг/мл) в 50 мM фосфата натрия, 25 мM аргинина, 5% сахарозе и 0,05% полисорбате 80. ALXN1210 SC составляли при pH 7,4 и предоставляли в виде полностью составленного, стерильного, не содержащего консервантов водного раствора ALXN1210 в концентрации 100 мг/мл, поставляемого в одноразовых флаконах на 2 мл. Каждый флакон с ALXN1210 SC имел незначительное переполнение для обеспечения возможности извлечения 1 мл (100 мг ALXN1210) для введения.
Каждая доза плацебо SC содержала 0,9% хлорид натрия для инъекций согласно Европейской или Британской фармакопее в таком же объеме, который указан для когорт 1a и 1b.
Каждый флакон с ALXN1210 IV содержит 150 мг ALXN1210 в 10 мM фосфата натрия, 150 мM хлорида натрия, 0,02% полисорбате 80 и воде для инъекций. ALXN1210 IV составляли при pH 7,0 и предоставляли в виде полностью составленного, стерильного, не содержащего консервантов водного раствора ALXN1210 в концентрации 10 мг/мл, поставляемого в одноразовых флаконах на 20 мл. ALXN1210 IV разбавляли в 0,9% хлориде натрия для инъекций согласно Европейской или Британской фармакопее и вводили путем IV инфузии при максимальной скорости 333 мл/ч, не считая прерывания в целях безопасности или по техническим причинам.
Флаконы с ALXN1210 хранили в охлажденном состоянии при 2°C-8°C (36°F-46°F) и защищали от света. Флаконы с ALXN1210 не замораживали и не встряхивали.
ALXN1210 SC и плацебо SC получали слепым способом в шприце для SC введения. Разбавление ALXN1210 SC или плацебо SC отсутствовало. ALXN1210 SC и плацебо SC помещали непосредственно в шприц.
ALXN1210 IV разрабатывали для инфузии путем разбавления в коммерчески доступном солевом растворе (0,9% хлориде натрия для инъекций согласно Европейской или Британской фармакопее) для IV инфузии при максимальной скорости 333 мл/ч, не считая прерывания в целях безопасности или по техническим причинам.
ALXN1210 IV разбавляли с помощью 0,9% хлорида натрия для инъекций согласно Европейской или Британской фармакопее перед введением (раствор для дозирования). Срок годности раствора для дозирования после вскрытия упаковки составлял 4 часа при комнатной температуре от 15°C до 25°C (от 59°F до 77°F). Дату и время истечения срока годности раствора для дозирования рассчитывали от момента вскрывания первого флакона. Дозу вводили до даты и времени истечения срока годности. Набранное содержимое каждого шприца с ALXN1210 SC или плацебо SC на 1 мл (4 шприца на субъекта) вводили в течение 1 часа после набора содержимого флакона в шприц.
11. Введение
Все дозы ALXN1210 SC или плацебо SC вводили посредством четырех инъекций 100 мг SC в объеме 1 мл каждая (таблица 17) в область живота. Все четыре инъекции объемом 1 мл вводили в течение 15-минутного периода, и между завершением инъекции у одного субъекта и началом инъекции у следующего субъекта должно было пройти по меньшей мере 15 минут.
Таблица 17. Справочная схема дозирования для препаратов ALXN1210 SC и плацебо SC
Все дозы ALXN1210 IV вводили путем IV инфузии с применением наборов для IV введения со встроенными фильтрами при максимальной скорости 333 мл/ч, не считая прерывания в целях безопасности или по техническим причинам. Между завершением инъекции у одного субъекта и началом инъекции у следующего субъекта должно было пройти по меньшей мере 15 минут.
Таблица 18. Справочная схема дозирования для препарата ALXN1210 IV
1 Продолжительность инфузии была примерной.
12. Контроль потенциальных нежелательных явлений во время введения исследуемого лекарственного средства
Некоторые субъекты, которые получали лечение путем IV инфузий моноклональных антител, испытывали сопутствующие инфузионные реакции с признаками или симптомами, которые можно классифицировать как острые аллергические реакции/реакции гиперчувствительности или синдром высвобождения цитокинов.
Субъектов тщательно контролировали во время и после введения исследуемого лекарственного средства в отношении любых симптомов анафилаксии и других реакций гиперчувствительности, включая изменения и остановку кровообращения и/или дыхания или крапивницу, артралгию, миалгию или другие признаки связанных реакций. Соответствующие средства лечения были доступны для немедленного применения. Могли возникнуть нежелательные явления, ассоциированные с инфузией, и в зависимости от их типа и степени тяжести могло потребоваться прекращение инфузии. Субъекты были проинформированы о ранних симптомах и признаках реакций гиперчувствительности, включающих крапивницу, отек лица, век, губ или языка или проблемы с дыханием. Для контроля инфузионных реакций использовали алгоритм действий при острой реакции на инфузию. В данном исследовании проводили регулярные оценки для контроля инфузионных реакций и реакций в месте инфузии. Для обеспечения безотлагательного решения проблем с этими реакциями в распоряжении имелось по меньшей мере 15 минут между завершением инфузии/инъекции у одного субъекта и началом инфузии/инъекции у следующего субъекта. Введение доз проводили не более чем 6 субъектам в день. Любые реакции подвергали лечению и учитывали в правилах продолжения приема/увеличения дозы и определения токсичности. В случае возникновения анафилактических реакций следовали действующему руководству «UK Treatment Guideline for Anaphylactic Reactions» Реанимационного совета Великобритании.
13. Оценки фармакокинетических (PK) и фармакодинамических (PD) характеристик
После исследования введения лекарственного средства образцы сыворотки крови для определения концентраций ALXN1210 в сыворотке крови и для анализов концентраций общего и свободного C5, степени гемолиза cRBC и других возможных показателей активации C5 собирали в следующие моменты времени, регистрируя фактические даты и моменты времени отбора образцов сыворотки крови и используя их в расчетах PK и PD.
Концентрации ALXN1210 в сыворотке крови анализировали в следующие моменты времени отбора образцов: до введения дозы (в течение 15 минут до начала инфузии/инъекции [SOI]); в день 1 по завершении инфузии/инфекции (EOI), через 30 минут после EOI и в следующие моменты времени после SOI: 2 ч, 4 ч и 8 ч; день 2 (24 ч); день 3 (48 ч); день 5 (96 ч); день 8 (168 ч); день 15 (336 ч); день 22 (504 ч); день 29 (672 ч); день 36 (840 ч); день 43 (1008 ч; день 50 (1176 ч); день 57 (1344 ч); день 71 (1680 ч); день 90 (2136 ч); день 120 (2856 ч); день 150 (3576 ч); и день 200 (4776 ч).
Всех субъектов, которые предоставили достаточное количество образцов сыворотки крови для анализа PK для определения характеристик профиля зависимости концентрации от времени, включали в популяцию для анализа PK. Всех субъектов, которые предоставили образцы для анализа PD, включали в популяцию для анализа PD.
14. Оценки иммуногенности
Образцы сыворотки крови собирали в следующие моменты времени: перед введением дозы (в течение 15 минут до SOI) и в дни 15 (336 ч), 29 (672 ч), 57 (1344 ч), 90 (2136 ч), 120 (2856 ч), 150 (3576 ч) и 200 (4776 ч) и анализировали на выработку ADA в отношении ALXN1210. Определение дополнительных характеристик гуморального ответа проводили соответствующим образом на основании данных о PK/PD и безопасности ALXN1210.
Всех субъектов, которые предоставили образец перед введением дозы и после введения дозы для анализа на ADA, включали в популяцию для анализа иммуногенности.
С помощью анализа иммуногенности оценивали выработку антитела к лекарственному средству (ADA) в отношении ALXN1210. Подробные инструкции по процедуре сбора, обработки, хранения и транспортировки образцов сыворотки крови для анализа иммуногенности представлены в лабораторном руководстве.
15. Оценка безопасности
Оценки безопасности включали тестирование на TB, данные физикального обследования, измерения основных показателей жизнедеятельности, тестирование иммуногенности (ADA), лабораторные оценивания, ECG, оценивания в местах инфузии и местах инъекции (например, кровотечения, кровоподтеков, покраснения, опухания, уплотнения и боли) и контроль нежелательных явлений. Нежелательные явления классифицировали по степеням в соответствии с общими терминологическими критериями нежелательных явлений Национального института онкологии версии 4.03 (CTCAE v4.03), опубликованными 14 июня 2010 г. Лабораторные оценивания включают гематологическую, биохимическую и коагуляционную панели; CBC с подсчетом лейкоцитарной формулы; общий анализ мочи и тестирование сыворотки крови на беременность для субъектов-женщин.
Для оценки безопасности ALXN1210 проводили клинические и лабораторные оценки. Временные рамки проведения оценок описаны в графике проведения оценок. За аномальными результатами следили до их устранения или стабилизации.
Рассмотрение демографических параметров, включая возраст, пол, расу и этническую принадлежность, проводили согласно описанному в графике проведения оценок. Собирали полный анамнез.
Измерения основных показателей жизнедеятельности проводили после того, как субъект находился в положении лежа на спине или в полулежачем положении в течение по меньшей мере 5 минут, и они включали температуру тела (°C; перорально), частоту дыхательных движений, кровяное давление в положении лежа на спине и частоту пульса. Временные рамки проведения измерений основных показателей жизнедеятельности описаны в графике проведения оценок. Измерения кровяного давления или частоты пульса, которые выходили за пределы допустимого диапазона, повторяли по усмотрению исследователя. Любые подтвержденные клинически значимые измерения основных показателей жизнедеятельности регистрировали в качестве нежелательных явлений.
Вес, рост и BMI регистрировали согласно описанному в графике проведения оценок. Каждое обследование включало следующие оценки: общий внешний вид; кожа; голова, уши, глаза, нос и горло; шея; лимфоузлы; грудная клетка; сердце; брюшная полость; конечности; центральная нервная система и скелетно-мышечная система.
ECG в трех повторностях в 12 отведениях получали после того, как субъект находился в состоянии покоя в течение по меньшей мере 5 минут. Временные рамки проведения ECG описаны в графике проведения оценок. Кроме того, проводили непрерывную регистрацию сердечной деятельности при каждом введении дозы с момента до введения дозы до момента завершения IV инфузии в когорте 2 и с момента до введения дозы до момента через 3 часа после завершения SC инъекции в когортах 1a и 1b. Измеряли частоту сердечных сокращений, PR, QRS, RR и QT и рассчитывали скорректированные интервалы QTcF.
Образцы крови для гематологического, клинико-биохимического анализа, анализа коагуляции и серологического анализа на вирусы, а также образцы мочи для общего анализа мочи, биохимического анализа мочи и скрининга на наркотические вещества и алкоголь собирали согласно описанному в графике проведения оценок.
Образцы крови анализировали в отношении следующих гематологических параметров: число тромбоцитов, число красных кровяных телец (RBC) и число WBC; лейкоцитарная формула при автоматизированном подсчете (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы); уровень гемоглобина; гематокритное число и индексы RBC (средний объем эритроцита, среднее содержание гемоглобина в эритроците и средняя концентрация гемоглобина в эритроците). Временные рамки проведения гематологических оценок описаны в графике проведения оценок.
Образцы крови анализировали в отношении следующих клинико-биохимических параметров: уровень азота мочевины крови; креатинина; глюкозы; натрия; фосфора; калия; хлорида; общего диоксида углерода; общего кальция; магния; AST; ALT; гамма-глутамилтранспептидазы; щелочной фосфатазы; лактата; общего, прямого и непрямого билирубина; мочевой кислоты; альбумина и общего белка. С учетом того, что уровень непрямого билирубина рассчитывали на основании значений для общего и прямого билирубина, результаты для непрямого билирубина были не были доступны, если уровень прямого билирубина был ниже предела количественного определения.
Уровень фолликулостимулирующего гормона и концентрации эстрадиола в сыворотке крови измеряли при скрининге для субъектов-женщин в период постменопаузы для подтверждения их постменопаузального статуса.
Временные рамки проведения биохимических оценок описаны в графике проведения оценок.
Образцы крови анализировали в отношении протромбинового времени, международного нормализационного индекса и частичного тромбопластинового времени. Временные рамки проведения оценок коагуляции описаны в графике проведения оценок.
Общий анализ мочи включает исследования в отношении удельного веса, pH, содержания глюкозы, белка, крови и кетонов. Микроскопическое исследование образцов мочи проводили только в случае обнаружения аномалий. Образцы мочи также отправляли в патологическую лабораторию для измерения содержания белка и креатинина с целью расчета соотношения белок:креатинин в моче. Временные рамки проведения оценок в общем анализе мочи и биохимическом анализе мочи описаны в графике проведения оценок.
Образцы крови, собранные при скрининге, анализировали в отношении титров антител к HIV-1, HIV-2, HBsAg и HCV. Для всех субъектов перед включением в исследование требовалось тестирование на поверхностный антиген вируса гепатита В. Субъекты с положительным результатом анализа на HBsAg не были включены в исследование. Для субъектов с отрицательным результатом анализа на HBsAg был необходим следующий алгоритм тестирования.
1. Если результат анализа на HBcAb был отрицательным, то субъект соответствовал требованиям к включению в исследование.
2. Если результат анализа на HBcAb был положительным, то проводили тестирование на антитела к поверхностному антигену вируса гепатита B (HBsAb).
Если результаты анализа как на HBcAb, так и на HBsAb были положительными, то субъект соответствовал требованиям к включению в исследование.
Если результат анализа на HBcAb был положительным, а результат анализа на HBsAb был отрицательным, то субъекта не включали в исследование.
Образец мочи для скрининга на наркотические вещества анализировали в отношении следующих соединений: амфетамины, барбитураты, бензодиазепины, кокаин, метадон, опиаты, фенциклидин, метамфетамин, 3,4-метилендиоксиметамфетамин и тетрагидроканнабинол (каннабиноиды). Проводили тестирование на алкоголь в выдыхаемом воздухе. Если до введения доз наблюдался положительный результат, то введение доз не продолжали. Временные рамки проведения тестирования мочи на наркотические вещества и тестирования на алкоголь в выдыхаемом воздухе описаны в графике проведения оценок.
Тестирование на беременность (бета-хорионический гонадотропин человека) проводили у всех субъектов-женщин. Временные рамки проведения тестирования на беременность описаны в графике проведения оценок.
Образцы сыворотки крови для тестирования QuantiFERON-TB собирали согласно описанному в графике проведения оценок.
Подходящий анализ для определения активности системы комплемента, такой как ELISA для CAP/анализ ингибирования C5 (гемолиза), проводили при скрининге для подтверждения того, что у субъектов нет недостаточности комплемента. Субъектов, у которых обнаружили недостаточность комплемента, исключали из участия в исследовании.
Образцы сыворотки крови собирали на исходном уровне и в ходе последующего наблюдения для измерения активности CH50 с применением LIA in vitro для подтверждения нормализации активности системы комплемента. Если нормальный результат CH50 получали для первого образца для CH50 от субъекта, собранного в ходе последующего наблюдения, то профилактику антибиотиками могли остановить, а второй запланированный образец для CH50 не требовался. Если первый и второй образцы для CH50 не были нормальными, образец на исходном уровне могли проанализировать, и дополнительные образцы для CH50 собирали до тех пор, пока активность системы комплемента не восстанавливалась.
Титр сывороточных бактерицидных антител (SBA) к менингококкам из серологических групп A, C, W135 и Y проводили при скрининге. Измерения титра использовали для исключения субъектов без иммунного ответа из введения доз.
Проводили оценки в месте подкожной инъекции или в месте IV инфузии. Боль в месте SC инъекции или IV инфузии оценивали с помощью визуально-аналоговой шкалы (0-10). Боль не оценивали до введения дозы. Уплотнения или реакции размером≤1 см не указывали в качестве нежелательного явления, если они не сохранялись в течение более 24 часов.
Образцы сыворотки крови анализировали в отношении антител к лекарственному средству (ADA). Временные рамки сбора образцов сыворотки крови для анализа на ADA описаны в графике проведения оценок.
16. Контроль нежелательных явлений
Исследователь отвечал за выявление, оценку, документирование всех нежелательных явлений (AE) и сообщение о них. Все AE регистрировали с момента подписания формы информированного согласия до завершения исследования. Для SAE, которые считались имеющими причинно-следственную связь, не было ограничения по времени.
Обо всех наблюдаемых или предполагаемых AE, независимо от причинно-следственной связи, сообщалось и все они регистрировались в системе сбора данных. Нежелательные явления, о которых сообщали субъект и/или его родитель или законный опекун, и/или выявленные в ответ на открытый вопрос от исследовательской группы, или выявленные в результате наблюдения, физикального обследования или других процедур исследования, собирали и регистрировали.
AE определяли как любые неблагоприятные и непредусмотренные признак (например, в том числе аномальные данные лабораторного исследования), симптом или заболевание, соответствующие по времени применению лекарственного препарата или процедуры, независимо от того, считаются ли они связанными с применением лекарственного препарата или процедуры, которое происходит в ходе клинического исследования.
Все обострения хронического или перемежающегося уже имеющегося состояния, включая также увеличение частоты и/или интенсивности состояния, считались AE.
Аномальные результаты тестирования считались AE. При выявлении аномальных показателей лабораторных анализов исследователям настоятельно рекомендовалось сообщать о диагнозе или признаке или симптоме, а не об отдельном аномальном показателе тестирования. Аномальный результат тестирования документировали как AE, если он соответствовал любому из следующих условий: был ассоциирован с признаком или симптомом; требовал дополнительного диагностического тестирования (повторные тестирования не считались дополнительным тестированием); требовал медицинского или хирургического вмешательства; приводил к изменению дозы исследуемого препарата за пределами доз, определенных протоколом, или приводил к прекращению участия в исследовании; требовал значительного дополнительного лечения; не соответствовал ни одному из условий, перечисленных выше.
Данное определение также включает признаки или симптомы, обусловленные следующим: передозировкой лекарственного средства, отменой лекарственного средства, злоупотреблением лекарственным средством, взаимодействиями лекарственных средств, экстравазацией, воздействием во время беременности, воздействием посредством грудного вскармливания, ошибкой применения лекарственного препарата и воздействием, связанным с родом занятий.
AE не обязательно включает следующее.
Медицинские или хирургические процедуры ( например, хирургическую операцию, эндоскопию, удаление зуба, переливание крови); состояние, которое приводит к такой процедуре, представляло собой AE ( например, лапароскопическая холецистэктомия представляла собой процедуру или лечение SAE в виде некротического процесса в желчном пузыре)
Существовавшие ранее заболевания или состояния, присутствующие на момент скринингового оценивания или выявленные до него, которые не усугубляются
Ситуации, когда неблагоприятное медицинское событие не произошло (например, госпитализацию для неэкстренного хирургического вмешательства, если оно запланировано до начала исследования; поступление в стационар по социальным причинам и/или для целей удобства)
Любое AE, которое соответствует любому 1 из критериев, перечисленных ниже, должно быть зарегистрировано в качестве SAE.
SAE описывали как любое неблагоприятное медицинское событие, которое при любой дозе:
1. Приводит к смерти
2. Угрожает жизниa
3. Требует госпитализации или продления госпитализацииb. Госпитализация не обязательно включает следующее:
Медицинскую реабилитацию/помещение в учреждение паллиативной помощи/помещение в учреждение сестринского ухода
Визит в отделение неотложной помощи в течение периода менее 24 часов
Неэкстренное или предварительно запланированное поступление в стационар/хирургическое вмешательство/поступление в дневной хирургический стационар
Указанное в протоколе поступление в стационар
Поступление в стационар в случае ранее существовавшего состояния, не ассоциированного ни с новым AE, ни с усугублением ранее существовавшего AE
4. Приводит к постоянной или значительной инвалидизации/недееспособности
5. Представляет собой врожденную аномалию/врожденный дефект
6. Является важным медицинским явлениемc
a Термин «угрожающий жизни» в значении «серьезный» относится к явлению, при котором субъект имел риск смерти во время этого явления; оно не относится к явлению, которое гипотетически могло бы вызвать смерть, если бы оно было более сильным.
b Госпитализация требует помещения пациента в стационар или продления текущей госпитализации. AE, которые были ассоциированы с госпитализацией или продлением госпитализации, считались SAE.
c Важное медицинское явление. Следует руководствоваться медицинским и научным суждением при принятии решения о необходимости срочного сообщения в других ситуациях, таких как важные медицинские явления, которые не могут непосредственно угрожать жизни или приводить к смерти или госпитализации, но могут подвергать субъекта опасности или могут потребовать вмешательства для предотвращения 1 из других исходов, перечисленных в определении выше. Также обычно их следует считать серьезными. Примерами таких явлений были интенсивное лечение в отделении неотложной помощи или дома при аллергическом бронхоспазме; гиперчувствительность немедленного типа или конвульсии, которые не приводят к госпитализации; или развитие зависимости от лекарственного средства или злоупотребление лекарственным средством.
Необходимо дифференцировать тяжесть и серьезность. Тяжесть описывает интенсивность AE, тогда как термин «серьезность» относится к AE, которое соответствует критериям для SAE, описанным выше.
Все AE классифицировали по степеням в соответствии со следующими критериями из CTCAE v4.03, опубликованными 14 июня 2010 г.
Степень 1: легкая (осведомленность о признаке или симптоме, но легкая его переносимость)
Степень 2: умеренная (дискомфорт, достаточный для того, чтобы воспрепятствовать выполнению обычных видов деятельности)
Степень 3: сильная (ограничение способностей, с неспособностью выполнять обычные виды деятельности)
Степень 4: угрожающая жизни
Степень 5: смертельная.
Изменения тяжести AE задокументировали для обеспечения возможности оценки продолжительности AE на каждом уровне интенсивности, подлежащем оцениванию. Нежелательные явления, характеризующиеся как перемежающиеся, требуют документирования начала и продолжительности каждого эпизода, если тяжесть перемежающегося эпизода изменялась.
Оценка исследователем причинно-следственной связи предоставлялась для всех AE (как несерьезных, так и серьезных). Эту оценку регистрировали в системе сбора данных и в любых дополнительных формах соответствующим образом. Определения для исследований причинно-следственных связей были следующими:
Не связано (несвязанное). Эта взаимосвязь позволяет предположить отсутствие связи между исследуемым препаратом и явлением, о котором сообщается.
Маловероятно связанное. Эта взаимосвязь позволяет предположить, что клиническая картина в высокой степени соответствовала причине, отличной от применения исследуемого препарата, но такое отнесение не может быть сделано с абсолютной определенностью, и взаимосвязь между исследуемым препаратом и AE не может быть исключена с полной уверенностью.
Возможно связанное. Эта взаимосвязь позволяет предположить, что лечение с помощью исследуемого препарата может стать причиной AE или способствовать его развитию; например, явление соответствует разумно обоснованной временной последовательности от момента введения исследуемого лекарственного средства и/или соответствует известной картине ответа на исследуемый препарат, но также могло быть вызвано другими факторами.
Вероятно связанное. Эта взаимосвязь позволяет предположить, что существует разумно обоснованная временная последовательность явления и введения исследуемого препарата, а также вероятная связь явления с исследуемым препаратом. В основе этого лежат известное фармакологическое действие исследуемого препарата, известные или те, о которых сообщалось ранее, нежелательные реакции на исследуемый препарат или класс лекарственных средств или суждение, основанное на клиническом опыте исследователя.
Определенно связанное. Связь по времени с исследуемым препаратом. Другие условия (сопутствующее заболевание, сопутствующая реакция на лекарственный препарат или прогрессирование/проявление болезненного состояния), по-видимому, не являются объяснением явления; явление соответствует известному фармацевтическому профилю; улучшение по завершении лечения; повторное появление при повторном назначении препарата.
Если субъект подвергся SAE со смертельным исходом, проводили следующие процедуры. SAE, которое привело к смерти, имеет исход, задокументированный как смерть/летальный исход, с датой окончания, являющейся датой смерти. Если у субъекта были дополнительные AE/SAE, которые продолжались на момент смерти, эти явления документировали как продолжающиеся без даты окончания. Только 1 явление имеет смертельный/летальный исход, если в отчете о вскрытии или в сообщении исследователя не указано иное.
17. Статистический анализ
Официальный план статистического анализа (SAP) был разработан и окончательно утвержден перед блокированием базы данных.
Популяция для анализа безопасности состоит из всех субъектов, которые получили по меньшей мере 1 дозу исследуемого лекарственного средства. Субъекты этой популяции использовались для анализа безопасности.
Популяция для PK состоит из всех субъектов, для которых имелись достаточные данные о концентрации в сыворотке крови для обеспечения возможности расчета PK-параметров. Для получения сводных данных PK-анализа использовали популяцию для PK.
Популяция для PD состоит из всех субъектов, для которых имелись достаточные данные о концентрации общего и свободного C5 и данные о степени гемолиза cRBC. Для получения сводных данных PD-анализа использовали популяцию для PD.
Популяция для анализа иммуногенности состояла из всех субъектов, от которых были собраны образцы для анализа ADA до введения дозы и после введения дозы.
Общий оцениваемый размер выборки, составляющий 36 субъектов - 24 субъекта с ALXN1210 SC из когорты 1 и 12 субъектов с ALXN1210 IV из когорты 2, обеспечивал > 80% мощность, позволяющую сделать вывод, что нижняя граница 90% доверительного интервала для соотношения биологической доступности ALXN1210 SC и IV составляла > 0,4 при предположении, что абсолютная биологическая доступность составляет 0,6, и коэффициент изменчивости составляет 0,35. Кроме того, 6 субъектов получали плацебо SC: 2 в когорте 1a и 4 в когорте 1b. Рандомизацию в когорту 1a проводили в соотношении 2:1, а в когорту 1b в соотношении 5:1 для получения ALXN1210 SC либо плацебо SC. Это довело общее запланированное количество субъектов до N=42.
В целом, параметры описательной статистики для непрерывных переменных включает количество непропущенных значений, среднее арифметическое, стандартное отклонение, медианное значение, минимальное значение и максимальное значение. Параметры описательной статистики для PK-параметров включала количество наблюдений, среднее арифметическое, стандартное отклонение, арифметический коэффициент изменчивости (% CV), медианное значение, минимальное значение, максимальное значение, среднее геометрическое и геометрический % CV. Категориальные переменные были обобщены с использованием процентных долей и подсчета частотности по когортам и моментам времени.
Все субъекты были включены в краткое описание распределения субъектов, в котором обобщалась частота и процентная доля субъектов, подвергнутых скринингу и лечению, которые завершили или прекратили участие в исследовании, а также причина прекращения по когортам. Демографические данные и исходные характеристики были обобщены для всех субъектов по каждой когорте и в целом.
Анализы безопасности проводили на популяции для анализа безопасности, и результаты сообщали по каждой группе. Анализы безопасности включали анализ всех AE, ECG, клинико-лабораторных данных, физикальных обследований и измерений основных показателей жизнедеятельности и были представлены с применением параметров описательной статистики. Никакие инференциальные статистические анализы не были запланированы в отношении параметров безопасности этого исследования. Частоту возникновения AE и SAE, возникших в ходе лечения, обобщали по системно-органным классам и предпочтительным терминам для каждой когорты и в целом, по взаимосвязи с исследуемым лекарственным средством. AE, возникшие в ходе лечения, также обобщали по когортам и в целом по степени тяжести. Перечисляли серьезные AE и AE, приводящие к прекращению участия в исследовании. Субъектов с несколькими AE в пределах одной категории ( например, в целом, в системно-органном классе, под предпочтительным термином) учитывали один раз в этой категории. Для таблиц степени тяжести учитывали наиболее тяжелое явление у субъекта в пределах категории.
Изменения измерений основных показателей жизнедеятельности и лабораторных оценок (например, в биохимическом анализе, CBC с подсчетом лейкоцитарной формулы и общем анализе мочи) по сравнению с исходным уровнем обобщали по каждой когорте. Значения лабораторных параметров классифицировали по степеням согласно CTCAE. Для данных лабораторных параметров получали таблицы сдвигов по когортам. В этих таблицах обобщены количество субъектов с каждой степенью на исходном уровне относительно диапазона эталонных значений и изменения до наихудшей самой высокой степени, оцененной после введения дозы во время исследования.
Параметры ECG измеряли в указанные моменты времени, включая частоту сердечных сокращений, интервалы PR, RR, QRS, QT и скорректированный QTcF. Рассчитывали среднее значение показателей ECG в трех повторностях в моменты времени сбора и оценивали изменения относительно исходных значений до лечения в каждой когорте.
Проводили анализ выпадающих значений, в котором обобщалась частота и процентная доля субъектов, которые соответствовали любому из следующих критериев выпадающих значений во время каждого визита по когортам:
QT, интервал QTcF >450 мс
QT, интервал QTcF >480 мс
QT, интервал QTcF >500 мс
QT, интервал QTcF увеличивается относительно исходного уровня >30 мс
QT, интервал QTcF увеличивается относительно исходного уровня > 0 мс.
Все сопутствующие лекарственные препараты кодировали с использованием словаря лекарственных средств WHO, а также обобщали частоту и процентную долю применения сопутствующих лекарственных препаратов.
Индивидуальные данные о концентрации в сыворотке крови для субъектов, получавших лечение с помощью ALXN1210, с фактическими датами и временем отбора образцов использовали для получения PK-параметров с помощью некомпартментных способов анализа с применением Phoenix WinNonlin 6.3 или более поздней версии.
Получали следующие PK-параметры: максимальная наблюдаемая концентрация в сыворотке крови (Cmax), время достижения максимальной наблюдаемой концентрации в сыворотке крови (Tmax), площадь под кривой зависимости концентрации в сыворотке крови от времени от нуля до последней поддающейся количественному определению концентрации (AUCt), площадь под кривой от нулевого момента времени до бесконечности (AUC∞), константа скорости терминальной элиминации (λz), терминальный период полувыведения (T½), общий клиренс (CL или CL/F) и объем распределения (Vd или Vd/F).
Соотношение средних геометрических (ALXN1210 SC/ALXN1210 IV) и его 90% CI рассчитывали для Cmax, AUCt и AUC∞ и представляли в табличной форме. CI рассчитывали с использованием межсубъектной вариабельности. Были представлены оценки изменения концентрации с течением времени.
PD-эффекты ALXN1210 SC и IV оценивали путем оценки изменений концентраций общего и свободного C5 в сыворотке крови, степени гемолиза cRBC и других показателей активации C5 с течением времени. Анализы проводили на образцах, собранных согласно описанному в графике проведения оценок.
Иммуногенность, измеренную по ADA, обобщали в табличной форме по когортам и по перечням субъектов.
Пример 3. Результаты исследования фазы 1 для оценивания однократной дозы ALXN1210, вводимого подкожно, по сравнению с внутривенным введением у здоровых субъектов
Ниже приведено краткое описание данных из исследования фазы 1 с однократной дозой, которое проводили по существу так, как описано выше в примере 2. В частности, исследование было разработано для оценивания безопасности, переносимости, фармакокинетических характеристик (PK), фармакодинамических характеристик (PD) и иммуногенности однократной дозы 400 мг ALXN1210, вводимого подкожно, по сравнению с однократной дозой 400 мг ALXN1210, вводимого внутривенно, или плацебо, вводимого подкожно, у 42 здоровых субъектов.
1. Распределение субъектов
Из 161 субъекта, подвергнутых скринингу, 42 (26,09%) субъекта были случайным образом распределены для получения исследуемого лекарственного средства: плацебо SC (n=6), ALXN1210 SC (n=24) и ALXN1210 IV (n=12) (фигура 48). Ни один из рандомизированных субъектов досрочно не прекратил участие в исследовании.
2. Отклонения от протокола
Сообщалось по меньшей мере об одном отклонении от протокола у 36 субъектов (плацебо SC: n=6; ALXN1210 SC: n=20 и ALXN1210 IV: n=10). Категории отклонений от протокола включали: отклонение временного окна, соблюдение субъектом режима лечения, невыполнение оценки, критерии исключения и введение дозы.
У двух субъектов в группе ALXN1210 IV отклонения от протокола были оценены как значительные. У одного субъекта в день 29 оценки ADA, PK, PD и лабораторные оценки не проводили, поскольку субъект не совершил визит последующего наблюдения. У другого субъекта образцы для анализа PK и PD в день 71 не собирали, поскольку субъект не совершил визит последующего наблюдения. Несмотря на то, что данные отклонения были оценены как значительные из-за природы плана клинического исследования (первичной конечной точки, связанной с PK), они не считались оказывающими влияние на интерпретацию результатов. Ни одно из других отклонений от протокола не считалось влияющим на интерпретацию результатов или безопасность субъектов. Результаты тестирования сыворотки крови на беременность у всех субъектов во время исследования были отрицательными.
3. Оценивание фармакокинетических характеристик, фармакодинамических характеристик и иммуногенности
Все 42 рандомизированных субъекта получали исследуемое лекарственное средство и были включены в группу для анализа безопасности (таблица 19). Все эти субъекты также были включены в группу для анализа PD и группу для анализа иммуногенности на основании определений. Для 36 субъектов в группе для анализа безопасности, которые получали ALXN1210 SC либо ALXN1210 IV, имелись достаточные данные о концентрации в сыворотке крови для обеспечения возможности расчета PK-параметров, и они были включены в группу для анализа PK (таблица 19).
Таблица 19. Анализ популяций (все рандомизированные субъекты)
Примечание: процентная доля (%) равняется n/N × 100.
Сокращения: IV=внутривенный; N=общее количество субъектов; n=количество субъектов; SC=подкожный.
4. Демографические и другие исходные характеристики
Среди групп лечения большинство субъектов являлись мужчинами (66,7%) европеоидной расы (69,0%) со средним возрастом (± SD) 35,0 (± 7,65) лет. Среднее значение (± SD) BMI для общей популяции составляло 24,035 (± 3,1582). В целом, демографические данные были хорошо сбалансированы среди групп лечения (таблица 20).
Таблица 20. Демографические данные - параметры описательной статистики по видам лечения (группа для анализа безопасности)
Примечание: процентная доля (%) равняется n/N × 100.
Сокращения: BMI=индекс массы тела; IV=внутривенный; макс. = максимум; мин. = минимум; N=общее количество субъектов; n=количество субъектов; SC=подкожный; SD=стандартное отклонение.
О применении ранее назначенных лекарственных препаратов сообщали 5 (20,8%) субъектов в группе ALXN1210 SC. Сообщения о применении ранее назначенных препаратов отсутствовали в группах плацебо SC и ALXN1210 IV. О применении сопутствующих лекарственных препаратов сообщали 3 (50,0%), 13 (54,2%) и 8 (66,7%) субъектов в группах плацебо SC, ALXN1210 SC и ALXN1210 IV соответственно. Наиболее часто применяемыми сопутствующими лекарственными средствами были анилиды, такие как ацетаминофен/парацетамол (15 субъектов), для лечения AE, затем прогестогены и эстрогены, комбинированный препарат с фиксированной дозой (7 субъектов) для предупреждения зачатия. Ни один из сопутствующих лекарственных препаратов, о которых сообщалось, не оказывал влияние на результаты исследования.
Ни один субъект не получал каких-либо нефармакологических видов терапии и процедур. Все дозы ALXN1210 SC или плацебо SC вводили посредством четырех SC инъекций по 100 мг в объеме 1 мл каждая в область живота. Все дозы ALXN1210 IV вводили путем IV инфузии с применением наборов для IV введения со встроенными фильтрами. Все субъекты получали назначенные им дозы.
5. Результаты анализа фармакокинетических характеристик, фармакодинамических характеристик и иммуногенности и таблицы данных по отдельным субъектам
Анализы PK проводили в группе для анализа PK, которая состояла из всех субъектов из группы для анализа безопасности, которые получали ALXN1210 SC либо ALXN1210 IV и для которых имелись достаточные данные о концентрации в сыворотке крови для обеспечения возможности расчета PK-параметров.
На фигурах 49-50 проиллюстрированы профили зависимости среднего значения (± SD) концентрации в сыворотке крови от времени для здоровых субъектов после SC и IV введения ALXN1210 (линейные и линейно-логарифмические шкалы). Графики отдельных концентраций ALXN1210 в сыворотке крови в зависимости от номинального времени представлены с применением линейной шкалы (фигура 49) и линейно-логарифмической шкалы (фигура 50) соответственно.
Фармакокинетические параметры ALXN1210 после SC и IV введения обобщены в Таблица 21. В общей сложности 24 субъекта получили введение ALXN1210 SC; медианное значение (диапазон) tmax составляло 169,8 (96,0-508,1) часа после SC инъекции. Среднее геометрическое (CV%) t½ было сходным, составляя 31,3 (13,6) дня и 29,9 (15,4) дня для введения ALXN1210 SC и IV соответственно. Выведение ALXN1210 было сходным для путей IV и SC (фигура 49).
Таблица 21. Краткое описание фармакокинетических параметров ALXN1210 (группа для фармакокинетического анализа)
aМедианное значение представлено для tmax.
Примечание: для лечения ALXN1210 SC столбцы CL и Vd представляют CL/F и Vd/F соответственно.
Сокращения: AUCt=площадь под кривой зависимости концентрации в сыворотке крови от времени от момента времени 0 до последней поддающейся количественному определению концентрации; AUC∞ = площадь под кривой зависимости концентрации в сыворотке крови от времени от момента времени 0 с экстраполяцией до бесконечности; CL или CL/F=общий клиренс лекарственного средства из сыворотки крови; Cmax=максимальная наблюдаемая концентрация в сыворотке крови; CV=коэффициент вариации; ч=час; IV=внутривенный; л=литр; макс.=максимум; мин.=минимум; n=количество субъектов; н.д.=нет данных; SC=подкожный; t½=терминальный период полувыведения; tmax=время достижения максимальной наблюдаемой концентрации в сыворотке крови; Vd или Vd/F=объем распределения; λz=константа скорости терминальной элиминации.
В таблице 22 обобщена абсолютная биологическая доступность ALXN1210 SC. PK-параметры для ALXN1210 SC (Cmax, AUCt, и AUC∞) сравнивали с эталоном (ALXN1210 IV) с помощью статистического анализа с применением смешанной модели после логарифмического преобразования данных. GMR Cmax для ALXN1210 (SC/IV) составляло 26,1% (95% CI: 21,3, 32,0). Абсолютная биологическая доступность ALXN1210 SC, определенная на основании GMR оценочных показателей AUC∞ (SC/IV), составляла 60,4% (95% CI: 49,7, 73,3).
Таблица 22. Статистический анализ абсолютной биологической доступности ALXN1210 при подкожном введении (группа для фармакокинетического анализа)
Примечание: соотношение определяли как среднее геометрическое для группы ALXN1210 SC, деленное на среднее геометрическое для группы ALXN1210 IV × 100. Использовали линейную смешанную модель с фиксированными и случайными эффектами для субъекта.
Сокращения: AUCt=площадь под кривой зависимости концентрации в сыворотке крови от времени от момента времени 0 до последней поддающейся количественному определению концентрации; AUC∞ = площадь под кривой зависимости концентрации в сыворотке крови от времени от момента времени 0 с экстраполяцией до бесконечности; CI=доверительный интервал; Cmax=максимальная наблюдаемая концентрация в сыворотке крови; GMR=соотношение средних геометрических; ч=час; IV=внутривенный; n=количество субъектов; SC=подкожный.
Анализы PD проводили в группе для анализа PD, которая состояла из всех субъектов из группы для анализа безопасности, для которых имелись достаточные данные о концентрации свободного и общего C5 и данные о степени гемолиза cRBC.
На фигуре 51 изображено процентное изменение среднего значения (± SD) концентрации свободного C5 в сыворотке крови относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV. Среднее значение концентрации свободного C5 оставалось относительно постоянным после SC введения плацебо. Введение однократной дозы 400 мг ALXN1210 IV приводило к немедленному и практически полному ингибированию свободного C5 (≥99%) в течение периода до дня 8 включительно после IV введения. Введение однократной дозы 400 мг ALXN1210 SC также приводило к снижению количества свободного C5, но не в той же степени или не с таким же быстрым началом, как это наблюдалось после IV введения. После введения ALXN1210 SC максимальная средняя степень ингибирования свободного C5 (77%) наблюдалась через 1 неделю после введения дозы.
Продолжительность и степень снижения средней концентрации свободного C5 зависели от воздействия. На фигуре 52 изображено процентное изменение средних значений (± SD) концентраций общего C5 в сыворотке крови относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV. Средние значения концентрации общего C5 концентраций оставались относительно постоянными после SC введения плацебо. Однако введение однократной дозы 400 мг ALXN1210 приводило к максимальному среднему увеличению концентрации общего C5 на 82% и 107% относительно исходного уровня после SC и IV введения дозы соответственно.
На фигуре 53 изображено процентное изменение среднего значения (± SD) степени гемолиза куриных эритроцитов (cRBC) относительно исходного уровня с течением времени для субъектов, которым вводили плацебо SC, ALXN1210 SC и ALXN1210 IV. Средняя степень гемолиза cRBC оставалась относительно постоянной после SC введения плацебо. Введение однократной дозы 400 мг ALXN1210 IV приводило к немедленному ингибированию средней степени гемолиза cRBC с максимальным средним снижением на 88%. Введение однократной дозы 400 мг ALXN1210 SC также приводило к ингибированию гемолиза cRBC, но не в той же степени или не с таким же быстрым началом по сравнению с IV введением. Максимальная средняя степень ингибирования гемолиза cRBC, составляющая 29%, наблюдалась через примерно 1 неделю после введения дозы ALXN1210 SC. Продолжительность и степень ингибирования cRBC зависели от воздействия.
Анализ иммуногенности проводили в группе для анализа иммуногенности, которая состояла из всех субъектов из группы для анализа безопасности, от которых были собраны образцы для анализа ADA до введения дозы и после введения дозы. Тестирование на антитела к лекарственному средству проводили до введения дозы и после введения дозы в дни 15, 29, 57, 90, 120, 150 и 200.
У одного субъекта (субъекта 0344-185) в группе лечения ALXN1210 SC был подтвержденный ADA-положительный образец на исходном уровне (до введения дозы) и во всех образцах после введения дозы. Все значения титра антител после введения дозы у данного субъекта были ниже значения титра до введения дозы. Ответ, положительный по выработке антител к лекарственному средству, у данного субъекта не считался клинически значимым или связанным с ALXN1210. Поэтому данного субъекта не включали в сводные данные по иммуногенности, приведенные ниже.
В общей сложности у 4 субъектов (группа ALXN1210 SC: 3/23 [13%] субъектов и группа ALXN1210 IV: 1/12 [8,3%] субъектов) вырабатывались ADA в ходе лечения. В группе ALXN1210 SC первый субъект имел положительный результат в отношении ADA в дни 57, 90, 120, 150 и 200. Все положительные значения ADA были положительными по перекрестной реактивности в отношении экулизумаба. Второй субъект имел положительный результат в отношении ADA в дни 29, 57, 90, 120, 150 и 200. Все положительные значения ADA были положительными по перекрестной реактивности в отношении экулизумаба, за исключением дня 90, когда они были отрицательными. Третий субъект имел положительный результат в отношении ADA в дни 90, 120, 150 и 200. Все положительные значения ADA были положительными по перекрестной реактивности в отношении экулизумаба.
В группе ALXN1210 IV один субъект имел положительный результат в отношении ADA в дни 15, 29, 90, 120, 150 и 200. Все положительные значения ADA были отрицательными по перекрестной реактивности в отношении экулизумаба.
Наиболее ранние ADA-положительные ответы после введения дозы наблюдались в день 29 и день 15 для SC и IV введения дозы соответственно. Титры ADA для ADA-положительных образцов были низкими и находились в диапазоне от < 1,0 до 27. В большинстве ADA-положительных образцов после SC введения ADA характеризовались перекрестной реактивностью в отношении экулизумаба. После IV введения ADA не характеризовались перекрестной реактивностью в отношении экулизумаба. Все субъекты с положительным результатом в отношении ADA оставались с положительным результатом до завершения периода последующего наблюдения. Формальную оценку влияния ADA на PK и PD было нельзя провести из-за небольшого количества субъектов с положительным результатом в отношении ADA. Изучение ограниченных отдельных результатов анализа PK и PD у этих субъектов позволяет предположить, что явное влияние иммуногенности на PK или PD ALXN1210 отсутствует.
6. Заключения в отношении фармакокинетических характеристик, фармакодинамических характеристик и иммуногенности
Медианное значение (диапазон) tmax составляло 169,8 (96,0-508,1 часа) после SC инъекции. Среднее геометрическое значение терминального периода полувыведения было сходным, составляя 31,3 дня и 29,9 дня после введения ALXN1210 SC и IV соответственно.
GMR оценочных показателей Cmax (SC/IV) составляло 26,1% (95% CI: 21,3, 32,0). Абсолютная биологическая доступность ALXN1210 SC на основании GMR оценочных показателей AUC∞ (SC/IV) составляла 60,4% (95% CI: 49,7, 73,3).
Степень и продолжительность PD-ответа, оцениваемые по концентрации свободного и общего C5 в сыворотке крови и степени гемолиза cRBC, зависели от воздействия. Введение однократной дозы 400 мг ALXN1210 IV приводило к немедленному и практически полному ингибированию свободного C5 (≥99%) в течение периода до дня 8 включительно после введения исследуемого лекарственного средства. Введение однократной дозы 400 мг ALXN1210 SC, вводимой в виде четырех SC инъекций по 100 мг, также приводило к снижению количества свободного C5, но не в той же степени или не с таким же быстрым началом, как при IV введении. Максимальная средняя степень ингибирования свободного C5 составляла 77%, что происходило через примерно 1 неделю после SC введения. Введение 400 мг ALXN1210 приводило к максимальному среднему увеличению концентрации общего C5 на 82% и 107% относительно исходного уровня после SC и IV введения дозы соответственно. Введение однократной дозы 400 мг ALXN1210 IV приводило к немедленному ингибированию средней степени гемолиза cRBC с максимальным средним снижением на 87%. Введение однократной дозы 400 мг ALXN1210 SC также приводило к ингибированию гемолиза cRBC, но не в той же степени или не с таким же быстрым началом по сравнению с IV введением. Максимальная средняя степень ингибирования гемолиза cRBC, составляющая 29%, наблюдалась примерно в день 8 после SC введения.
Об ADA, вырабатывающихся в ходе лечения, сообщалось для 3/23 (13%) субъектов и 1/12 (8,3%) субъектов в группах ALXN1210 SC и ALXN1210 IV соответственно с низкими значениями титра ADA в диапазоне от <1,0 до 27. Наиболее ранний ответ с выработкой ADA после введения дозы наблюдался в день 29 и день 15 для SC и IV введения дозы соответственно. После SC введения ADA характеризовались перекрестной реактивностью в отношении экулизумаба в большинстве ADA-положительных образцов. После IV введения ADA не характеризовались перекрестной реактивностью в отношении экулизумаба. Все субъекты с положительным результатом в отношении ADA оставались с положительным результатом до завершения периода последующего наблюдения. Явное влияние иммуногенности на PK или PD ALXN1210 не наблюдалось.
У одного дополнительного субъекта в группе лечения ALXN1210 SC был подтвержденный ADA-положительный образец на исходном уровне (до введения дозы) и во всех образцах после введения дозы. Все значения титра антител после введения дозы у данного субъекта были ниже значения титра до введения дозы. Ответ, положительный по выработке антител к лекарственному средству, у данного субъекта не был связан с ALXN1210.
7. Степень воздействия
Всех субъектов, которые получили одну дозу исследуемого лекарственного средства, включали в группу для анализа безопасности (N=42): плацебо SC (n=6); ALXN1210 SC (n=24) и ALXN1210 IV (n=12). Общий объем инфузии (80 мл) исследуемого лекарственного средства вводили каждому субъекту, которому назначали получение ALXN1210 IV. У одного субъекта инфузию прерывали на минуту, так как в насосе было запрограммировано недостаточно времени для полной инфузии. Каждому субъекту, который получал ALXN1210 SC либо плацебо SC, вводили общий объем исследуемого лекарственного средства (4 мл).
8. Нежелательные явления
Во всех трех группах лечения 35/42 (83,3%) субъектов испытывали 75 TEAE (все 1 степени). Доля субъектов, у которых наблюдалось по меньшей мере 1 TEAE, составляла 91,7%, 83,3% и 79,2% соответственно в группе ALXN1210 IV, группе плацебо SC и группе ALXN1210 SC. О случаях смерти или SAE в ходе исследования не сообщалось. Ни один из субъектов не прекратил прием исследуемого лекарственного средства и не прекратил участие в исследовании вследствие TEAE (таблица 23).Все TEAE были устранены в ходе исследования. Большинство TEAE не требовали приема какого-либо лекарственного препарата, и ни один из субъектов не нуждался в нефармакологических вмешательствах ни в один момент времени.
Таблица 22. Нежелательные явления, возникшие в ходе лечения (TEAE) - общее краткое описание (группа для анализа безопасности)
Примечание: процентная доля (%) равняется n/N × 100.
1 степень=легкая; 2 степень=умеренная; 3 степень=сильная; 4 степень=угрожающая жизни или инвалидизирующая; 5 степень=смерть, связанная с TEAE.
Связанное TEAE=возможно связанное, вероятно связанное или определенно связанное TEAE; несвязанное TEAE=несвязанное или маловероятно связанное TEAE.
Для ALXN1210 IV считалось, что TEAE имело место во время введения исследуемого лекарственного средства, если TEAE возникло во время инфузии; для плацебо SC и ALXN1210 SC считалось, что нежелательное явление имело место во время введения исследуемого лекарственного средства, если нежелательное явление возникло между первой и последней инъекциями.
Сокращения: E=количество явлений; IV=внутривенный; N=общее количество субъектов, имеющих риск; n=количество субъектов, имеющих AE; SC=подкожный; SAE=серьезное нежелательное явление; TEAE=нежелательное явление, возникшее в ходе лечения.
В общей сложности сообщалось о 75 TEAE у 35 субъектов. Среди групп лечения TEAE, о которых сообщалось наиболее часто, представляли собой назофарингит (23/42 субъектов, 54,8%) и головную боль (7/42 субъектов, 16,7%). Все TEAE обобщали по системно-органным классам (SOC) и предпочтительным терминам по видам лечения в таблице 23.
Таблица 23. Нежелательные явления, возникшие в ходе лечения - таблица частот по системно-органным классам и предпочтительным терминам (группа для анализа безопасности)
Предпочтительный термин
Примечание: процентная доля (%) равняется n/N × 100.
Каждый субъект учитывался только один раз для данного SOC и предпочтительного термина независимо от фактического количества возникших нежелательных явлений.
Классификация SOC и предпочтительный термин соответствуют MedDRA v20.0.
Сокращения: E=количество явлений; IV=внутривенный; MedDRA=Медицинский словарь для регуляторной деятельности; N=общее количество субъектов, имеющих риск; n=количество субъектов, имеющих нежелательное явление; SC=подкожный; SOC=системно-органный класс; TEAE=нежелательное явление, возникшее в ходе лечения.
Большинство TEAE (72/75 TEAE, 96%) считались не связанными с лечением с помощью ALXN1210. Среди групп лечения 3/42 (7,1%) субъектов сообщали о 3 TEAE, которые исследователь оценивал как связанные («возможно связанные») с лечением с помощью ALXN1210 и имеющие 1 степень (легкую): (1) инфекции верхних дыхательных путей у одного субъекта из группы ALXN1210 SC (2), мигрени у одного субъекта в группе ALXN1210 SC и (3) головной боли у одного субъекта в группе ALXN1210 IV. Все 75 TEAE классифицировали как имеющие 1 степень (легкую). Ни один субъект не умер, не испытывал SAE и не прекратил прием исследуемого лекарственного средства или участие в исследовании вследствие TEAE.
В целом средние значения в гематологическом анализе, анализе коагуляции, биохимическом анализе крови, общем анализе мочи и биохимическом анализе мочи находились в пределах диапазона эталонных значений, и не наблюдалось явных тенденций к изменению среднего значения относительно исходного уровня.
Большинство субъектов включались в исследование с нормальными значениями (т.е. в пределах соответствующих диапазонов эталонных значений) параметров гематологического анализа, общего анализа мочи, анализа коагуляции, биохимического анализа крови и биохимического анализа мочи. Среди групп лечения не наблюдались никакие очевидные тенденции к сдвигам. Для некоторых лабораторных параметров во время исследования наблюдали сдвиг от нормальных значений на исходном уровне до аномальных значений (1 степень [легкая] или 2 степень [умеренная]), которые, однако, не считались клинически значимыми. Большинство сдвигов были временными и устранялись во время исследования.
Во время исследования для 3 субъектов в группе ALXN1210 SC сообщалось о сдвиге до аномальных значений 3 степени. Ни об одном из сдвигов до аномальных значений 3 степени не сообщалось в качестве AE.
Во-первых, сообщалось о снижении количества нейтрофилов у одного субъекта. Количество нейтрофилов у данного субъекта на исходном уровне составляло 3,77×109/л. Оцененное количество нейтрофилов в день 43 составляло 0,95×109/л (диапазон нормальных значений: 2,0-7,5×109/л). Количество нейтрофилов находилось в диапазоне нормальных значений в день 57.
Для двух субъектов сообщалось о повышении уровней калия (диапазон нормальных значений: 3,5-5,1 ммоль/л). У 1 субъекта с исходным уровнем калия, составляющим 4,5 ммоль/л, оцениваемый уровень калия в день 150 составлял 6,1 ммоль/л. Повторно измеренный уровень калия находился в нормальном диапазоне в тот же день (внеплановый визит). У другого субъекта с исходным уровнем калия, составляющим 4,6 ммоль/л, оцениваемый уровень калия в день 90 составлял 6,4 ммоль/л. У данного субъекта были выявлены аномальные значения уровня калия при скрининге (находящиеся в диапазоне 5,2-6,2 ммоль/л во время различных скрининговых визитов) и в ходе большинства визитов в рамках исследования. Повышение уровней калия было временным; зарегистрированные значения находились в пределах диапазона нормальных значений в день 150 и день 200.
Никаких заметных изменений измерений основных показателей жизнедеятельности относительно исходного уровня и каких-либо клинически значимых аномалий основных показателей жизнедеятельности, стабильно наблюдаемых для отдельных субъектов, не выявляли.
Ни один субъект не имел клинически значимых результатов физикального обследования, отличных от тех результатов, о которых сообщалось в качестве AE. Значимые изменения средних значений относительно исходного уровня в результатах ECG или телеметрического контроля не наблюдались.
Изменения интервалов QT корректировали с применением формулы Фридеричиа (QTcF). У одного субъекта в группе плацебо SC средний интервал QT >500 мс наблюдался при скрининге (510,0 мс) и в день 2 (508,7 мс), день 150 (516,6 мс) и день 200 (612,9 мс). Средний интервал QTcF у одного и того же субъекта составлял 449,7 мс, 443,7 мс, 451,9 мс и 501,3 мс при скрининге и в дни 2, 150 и 200 соответственно. Увеличение интервала QT и QTcF не считалось клинически значимым у данного субъекта-женщины в группе плацебо. О таких изменениях также не сообщалось в качестве AE. Во время исследования не наблюдались значимые изменения средних интервалов QT и QTcF относительно исходного уровня.
Оценивания в месте инфузии или инъекции проводили в течение 15 минут после SOI и ± 15 минут в моменты времени 30 минут, 2 ч, 4 ч, 8 ч и в день 2 (48 ч.), день 3 (72 ч, в общей сложности 6 оценок). Уплотнения или реакции размером <1 см не считались AE, если они не сохранялись в течение более 24 часов. Через 30 минут после EOI у 5/24 субъектов в группе ALXN1210 SC наблюдалось покраснение, а у 1 субъекта в 2/4 мест инъекции наблюдалось минимальное (3 мм) покраснение через 2 часа после инъекции и ни одной в последние моменты времени. Минимальное уплотнение или набухание (10 мм) регистрировали через 30 минут после EOI у 1/24 субъектов в группе ALXN1210 SC, что не наблюдалось при последних оценках. Однако ни одно из них не соответствовало критериям, определенным в протоколе, чтобы считаться AE. Боль в месте инфузии или инъекции оценивалась субъектами с применением VAS (0-100 мм). Для большинства инфузий и инъекций боль в месте инфузии была оценена как 0 мм при всех оценках. Два субъекта в группе SC сообщали о временной боли, соответствующей 3-5 мм, в день 1, а три субъекта в группе IV сообщали о минимальной (1-5 мм) боли после инфузии.
9. Заключения в отношении безопасности
Всех субъектов, которые получили одну дозу исследуемого лекарственного средства (плацебо SC, ALXN1210 SC или ALXN1210 IV), включали в группу для анализа безопасности (N=42). Среди 3 групп лечения 35/42 субъектов (83,3%) испытывали 75 TEAE. Только 3/75 TEAE (4%) считались связанными с ALXN1210, тогда как 72/75 (96%) считались не связанными с лечением ALXN1210. Все TEAE были легкими (1 степени) и были устранены в ходе исследования. Большинство TEAE не требовали приема какого-либо лекарственного препарата, и ни один из субъектов не нуждался в нефармакологических вмешательствах ни в один момент времени. TEAE, о которых сообщалось наиболее часто, представляли собой назофарингит (23/42 субъектов, 54,8%) и головную боль (7/42 субъектов, 16,7%).
Случаи смерти или SAE во время исследования не наблюдались. Ни одно из TEAE, о которых сообщалось, не приводило к прекращению приема исследуемого лекарственного средства или к прекращению участия субъекта в исследовании. В целом во время исследования или последующего наблюдения не наблюдались клинически значимые изменения лабораторных параметров, основных показателей жизнедеятельности, параметров физикального обследования, ECG или телеметрии. Никакие клинические проявления гиперчувствительности во время или после введения какой-либо однократной дозы путем SC инъекции или IV инфузии не наблюдались. У субъектов с положительными результатами в отношении ADA отсутствуют клинические признаки или симптомы, ассоциированные с аллергической реакцией или гиперчувствительностью.
10. Обсуждение и общие заключения
Целью данного исследования фазы 1 было оценивание безопасности, переносимости, PK, PD и иммуногенности однократной дозы 400 мг ALXN1210 SC по сравнению с однократной дозой 400 мг ALXN1210 IV или плацебо SC, вводимой путем инъекции, у здоровых субъектов. В общей сложности 42 субъекта были рандомизированы и получали исследуемое лекарственное средство: плацебо SC (n=6); ALXN1210 SC (n=24) и ALXN1210 IV (n=12).
Введение ALXN1210 в дозе 400 мг хорошо переносилось при SC пути введения у здоровых субъектов. Абсолютная биологическая доступность ALXN1210 SC на основании GMR оценочных показателей AUC∞ (SC/IV) составляла 60,4% (95% CI: 49,7, 73,3). Оценочные показатели среднего геометрического значения t½ составляли 31,3 дня и 29,9 дня после введения ALXN1210 SC и IV. Степень и продолжительность PD-ответа, оцениваемые по концентрации свободного и общего C5 в сыворотке крови и степени гемолиза cRBC, зависели от воздействия.
Об антителах к лекарственному средству сообщалось для 3/23 (13%) субъектов и 1/12 (8,3%) субъектов в группах ALXN1210 SC и ALXN1210 IV соответственно с положительными значениями титра ADA в диапазоне от <1,0 до 27. Наиболее ранний ответ после введения дозы наблюдался в день 29 и день 15 для SC и IV введения дозы соответственно. В большинстве ADA-положительных образцов после SC введения ADA характеризовались перекрестной реактивностью в отношении экулизумаба. У субъектов, у которых наблюдался ADA-положительный ответ, отсутствовали клинические признаки или симптомы, соответствующие аллергической реакции или гиперчувствительности (включая анафилаксию). Кроме того, нельзя было идентифицировать явное влияние ALXN1210 на PK или PD.
Никаких неожиданных проблем безопасности не наблюдалось ни в одной из групп лечения во время исследования. В ходе исследования не происходили случаи смерти или SAE, и ни один субъект не испытывал каких-либо TEAE, приводящих к прекращению приема исследуемого лекарственного средства или к прекращению участия в исследовании.
КРАТКОЕ ОПИСАНИЕ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
аминокислотная последовательность CDR1 тяжелой цепи экулизумаба (как определено согласно комбинированному определению Kabat и Chothia)
GYIFSNYWIQ
аминокислотная последовательность CDR2 тяжелой цепи экулизумаба (как определено согласно определению Kabat)
EILPGSGSTEYTENFKD
аминокислотная последовательность CDR3 тяжелой цепи экулизумаба (как определено согласно комбинированному определению Kabat)
YFFGSSPNWYFDV
аминокислотная последовательность легкой цепи CDR1 экулизумаба (как определено согласно определению Kabat)
GASENIYGALN
аминокислотная последовательность CDR2 легкой цепи экулизумаба
(как определено согласно определению Kabat)
GATNLAD
аминокислотная последовательность CDR3 легкой цепи экулизумаба (как определено согласно определению Kabat)
QNVLNTPLT
аминокислотная последовательность вариабельной области тяжелой цепи экулизумаба
QVQLVQSGAEVKKPGASVKVSCKASGYIFSNYWIQWVRQAPGQGLEWM
GEILPGSGSTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCARY
FFGSSPNWYFDVWGQGTLVTVSS
аминокислотная последовательность вариабельной области легкой цепи экулизумаба, равулизумаба и антитела BNJ421
DIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKPGKAPKLLIYGA
TNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQGTK
VEIK
аминокислотная последовательность константной области тяжелой цепи экулизумаба и антитела BNJ421
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVH
TFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKC
CVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQF
NWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKV
SNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPS
DIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCS
VMHEALHNHYTQKSLSLSLGK
аминокислотная последовательность всей тяжелой цепи экулизумаба
QVQLVQSGAEVKKPGASVKVSCKASGYIFSNYWIQWVRQAPGQGLEWM
GEILPGSGSTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCAR
YFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCL
VKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYT
CNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISR
TPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLT
VLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMT
KNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV
DKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
аминокислотная последовательность всей легкой цепи экулизумаба, равулизумаба и антитела BNJ421
DIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKPGKAPKLLIYG
ATNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATFATCQNVLNTPLTFGQ
GTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDN
ALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPV
TKSFNRGEC
аминокислотная последовательность вариабельной области тяжелой цепи равулизумаба и антитела BNJ421
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEW
MGEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYC
ARYFFGSSPNWYFDVWGQGTLVTVSS
аминокислотная последовательность константной области тяжелой цепи равулизумаба
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGV
HTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVER
KCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPE
VQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYK
CKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKG
FYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGN
VFSCSVLHEALHSHYTQKSLSLSLGK
аминокислотная последовательность всей тяжелой цепи равулизумаба
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEWM
GEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCAR
YFFGSSPNWYFDVWGQGTLVTVSS ASTKGPSVFPLAPCSRSTSESTAALGCL
VKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYT
CNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISR
TPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLT
VLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMT
KNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV
DKSRWQEGNVFSCSVLHEALHSHYTQKSLSLSLGK
аминокислотная последовательность варианта константной области тяжелой цепи IgG2, содержащего замены YTE
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVH
TFPAVLQSSGLYSLSSVVTVTSSNFGTQTYTCNVDHKPSNTKVDKTVERKC
CVECPPCPAPPVAGPSVFLFPPKPKDTLYITREPEVTCVVVDVSHEDPEVQF
NWYVDGMEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKV
SNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYP
SDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVF
SCSVMHEALHNHYTQKSLSLSPGK
аминокислотная последовательность всей тяжелой цепи варианта экулизумаба, содержащей константную область тяжелой цепи, приведенную под SEQ ID NO: 15 (выше)
QVQLVQSGAEVKKPGASVKVSCKASGYIFSNYWIQWVRQAPGQGLEWM
GEILPGSGSTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCAR
YFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALG
CLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVTSSNF
GTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKP
KDTLYITREPEVTCVVVDVSHEDPEVQFNWYVDGMEVHNAKTKPREEQ
FNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPRE
PQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTP
PMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLS
PGK
аминокислотная последовательность CDR1 легкой цепи экулизумаба (как определено согласно определению Kabat) с заменой глицина на гистидин в положении 8 по сравнению с SEQ ID NO: 4
GASENIYHALN
приведена аминокислотная последовательность CDR2 тяжелой цепи экулизумаба, в которой серин в положении 8 по сравнению с SEQ ID NO: 2 заменен гистидином.
EILPGSGHTEYTENFKD
аминокислотная последовательность CDR1 тяжелой цепи экулизумаба, в которой тирозин в положении 2 (по сравнению с SEQ ID NO: 1) заменен гистидином
GHIFSNYWIQ
аминокислотная последовательность всей тяжелой цепи антитела BNJ421
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEW
MGEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYC
ARYFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALG
CLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQT
YTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMIS
RTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVL
TVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMT
KNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV
DKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> АЛЕКСИОН ФАРМАСЬЮТИКАЛС, ИНК.
<120> ВЫСОКОКОНЦЕНТРИРОВАННЫЕ СОСТАВЫ АНТИТЕЛ К C5
<130> AXJ-226PC
<140>
<141>
<150> 62/537741
<151> 2017-07-27
<160> 20
<170> PatentIn, версия 3.5
<210> 1
<211> 10
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 1
Gly Tyr Ile Phe Ser Asn Tyr Trp Ile Gln
1 5 10
<210> 2
<211> 17
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 2
Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe Lys
1 5 10 15
Asp
<210> 3
<211> 13
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 3
Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val
1 5 10
<210> 4
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /=примечание"Описание искусственной последовательности: синтетический
пептид"
<400> 4
Gly Ala Ser Glu Asn Ile Tyr Gly Ala Leu Asn
1 5 10
<210> 5
<211> 7
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 5
Gly Ala Thr Asn Leu Ala Asp
1 5
<210> 6
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 6
Gln Asn Val Leu Asn Thr Pro Leu Thr
1 5
<210> 7
<211> 122
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 7
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 8
<211> 107
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 8
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Gly Ala Ser Glu Asn Ile Tyr Gly Ala
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Thr Asn Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Asn Val Leu Asn Thr Pro Leu
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105
<210> 9
<211> 326
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 9
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Pro Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser Gln Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ser Ser Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Glu Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Leu Gly Lys
325
<210> 10
<211> 448
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 10
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 11
<211> 214
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 11
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Gly Ala Ser Glu Asn Ile Tyr Gly Ala
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Thr Asn Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Asn Val Leu Asn Thr Pro Leu
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 12
<211> 122
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 12
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 13
<211> 326
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 13
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Pro Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser Gln Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ser Ser Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Glu Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Leu His Glu Ala Leu His Ser His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Leu Gly Lys
325
<210> 14
<211> 448
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 14
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Leu His Glu Ala
420 425 430
Leu His Ser His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 15
<211> 326
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 15
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Thr Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Tyr Ile Thr Arg Glu Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser His Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Met Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Phe Arg Val Val Ser Val Leu Thr Val Val His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ala Pro Ile Glu Lys Thr Ile Ser Lys Thr Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Met Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Pro Gly Lys
325
<210> 16
<211> 448
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 16
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Thr Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Tyr Ile Thr Arg
245 250 255
Glu Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Met Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Phe Arg Val Val
290 295 300
Ser Val Leu Thr Val Val His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ala Pro Ile Glu Lys Thr
325 330 335
Ile Ser Lys Thr Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Met Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
435 440 445
<210> 17
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 17
Gly Ala Ser Glu Asn Ile Tyr His Ala Leu Asn
1 5 10
<210> 18
<211> 17
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 18
Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe Lys
1 5 10 15
Asp
<210> 19
<211> 10
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
пептид"
<400> 19
Gly His Ile Phe Ser Asn Tyr Trp Ile Gln
1 5 10
<210> 20
<211> 448
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="Описание искусственной последовательности: синтетический
полипептид"
<400> 20
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ДОЗЫ И ВВЕДЕНИЕ АНТИТЕЛ ПРОТИВ C5 ДЛЯ ЛЕЧЕНИЯ АТИПИЧНОГО ГЕМОЛИТИКО-УРЕМИЧЕСКОГО СИНДРОМА (аГУС) | 2020 |
|
RU2822664C2 |
| ДОЗИРОВКА И СХЕМА ВВЕДЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ СВЯЗАННЫХ С С5 ЗАБОЛЕВАНИЙ ПОСРЕДСТВОМ ПРИМЕНЕНИЯ АНТИТЕЛА ПРОТИВ С5 КРОВАЛИМАБА | 2020 |
|
RU2832188C2 |
| АНТИТЕЛА ПРОТИВ E-СЕЛЕКТИНА, КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2021 |
|
RU2805176C1 |
| АНТИТЕЛА, НЕЙТРАЛИЗУЮЩИЕ GM-CSF, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РЕВМАТОИДНОГО АРТРИТА ИЛИ В КАЧЕСТВЕ АНАЛЬГЕТИКОВ | 2014 |
|
RU2714919C2 |
| АНТИТЕЛА К ПАРВОВИРУСУ ДЛЯ ПРИМЕНЕНИЯ В ВЕТЕРИНАРИИ | 2020 |
|
RU2830441C2 |
| АНТАГОНИСТЫ | 2019 |
|
RU2839429C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АНТИТЕЛ К CD40 И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2778572C1 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТИТЕЛО ПРОТИВ ПРОТОФИБРИЛЛ АБЕТА И ИНГИБИТОР БЕТА-СЕКРЕТАЗЫ ВАСЕ1 ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2017 |
|
RU2786476C2 |
| КОМБИНИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ | 2020 |
|
RU2829637C2 |
| СТАБИЛЬНЫЕ СОСТАВЫ АНТИТЕЛ ПРОТИВ TIGIT, ОТДЕЛЬНО И В КОМБИНАЦИИ С АНТИТЕЛАМИ ПРОТИВ РЕЦЕПТОРА 1 ПРОГРАММИРУЕМОЙ СМЕРТИ (PD-1), И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2820576C2 |
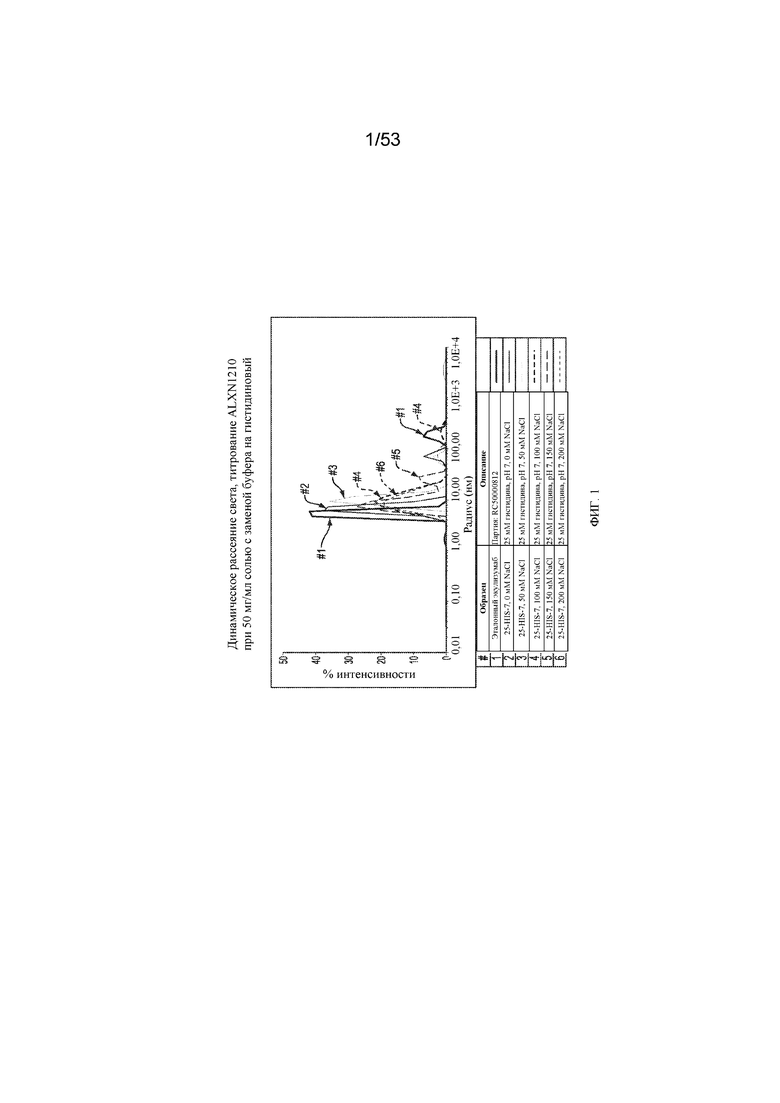

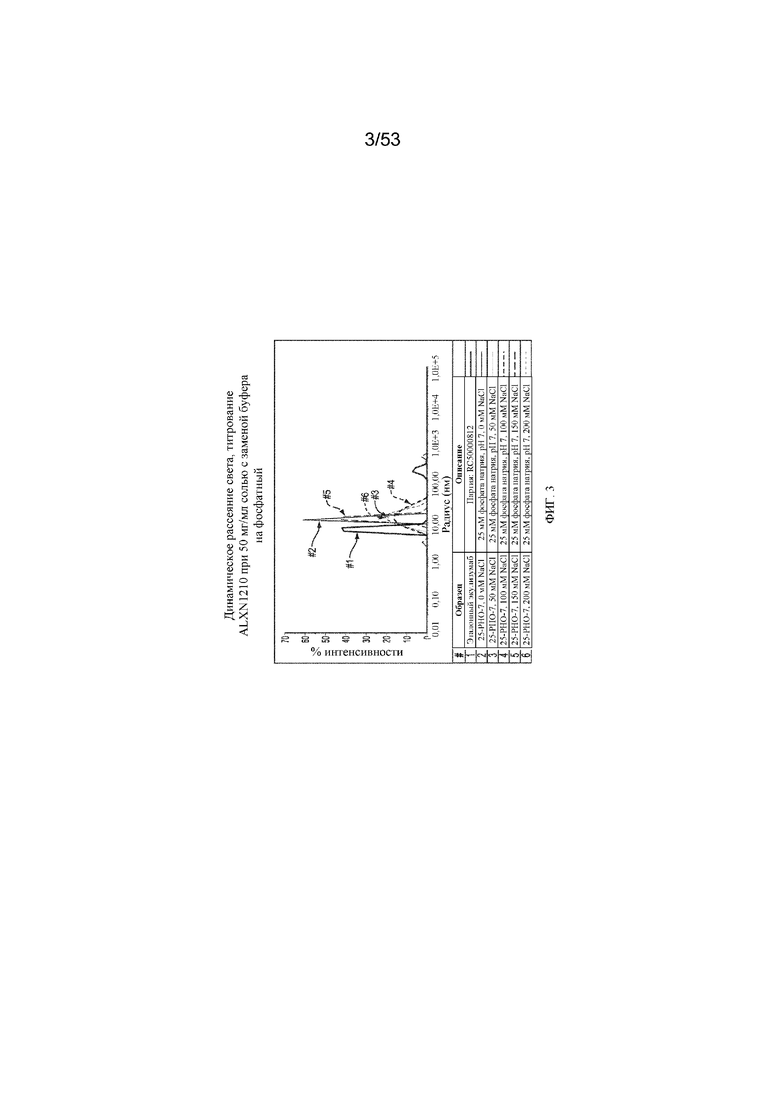
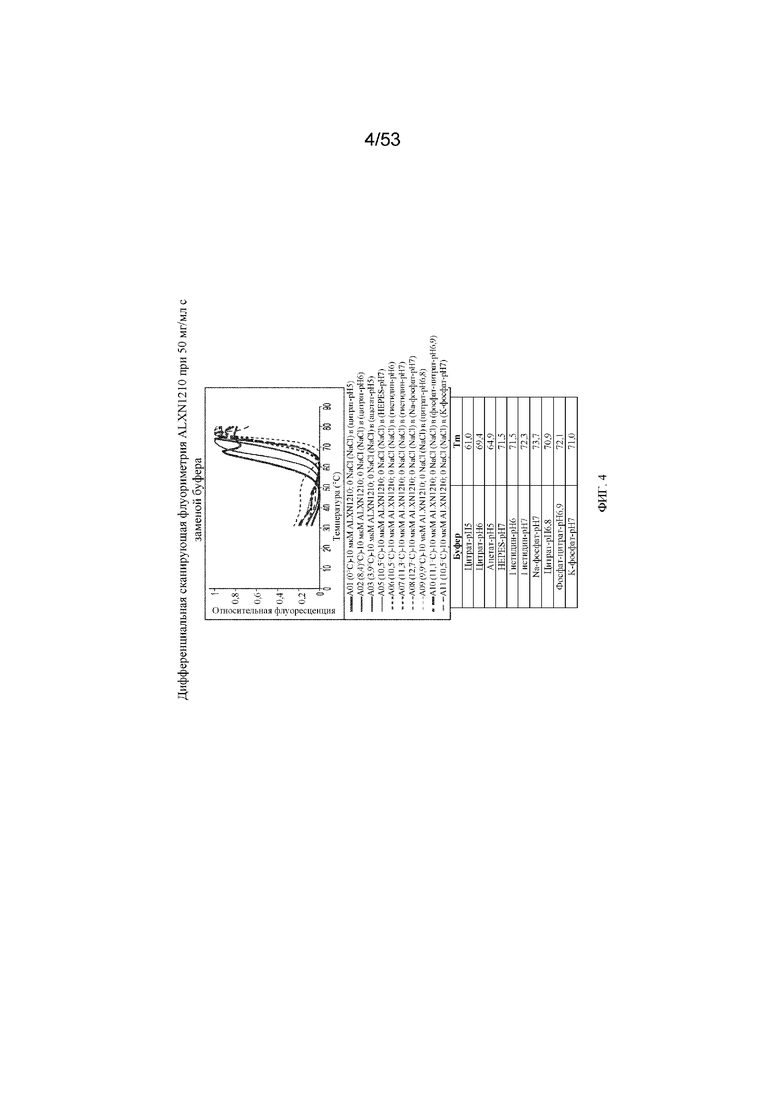
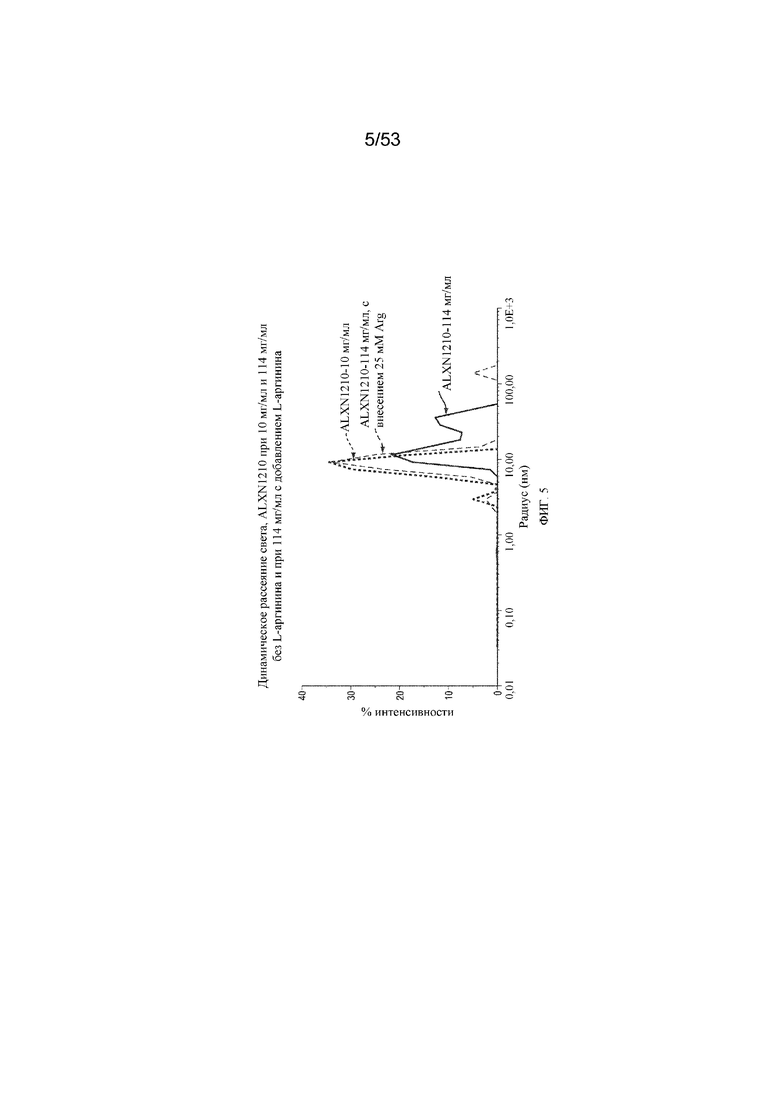
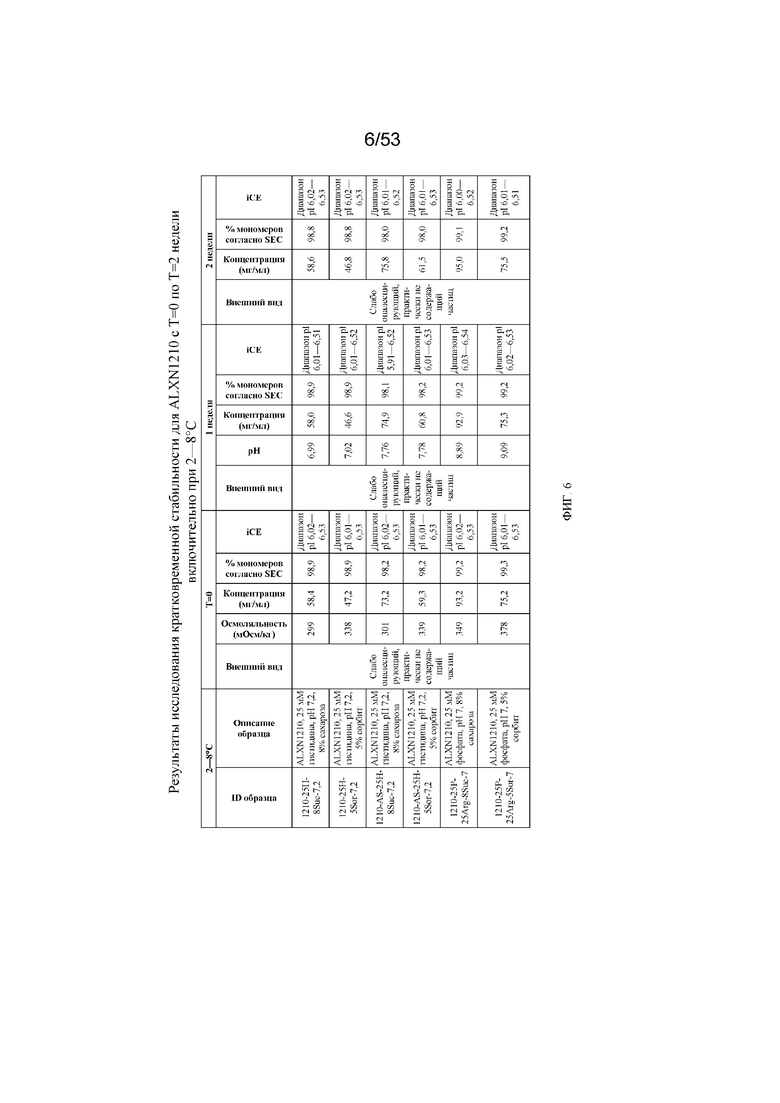
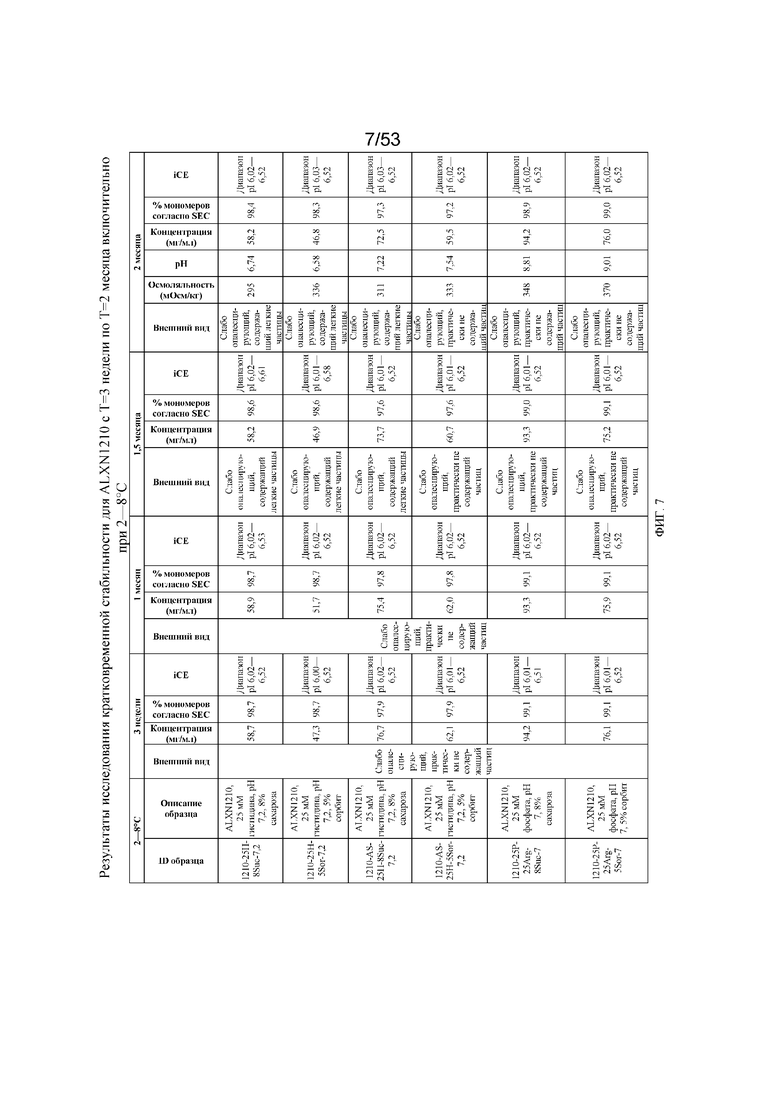
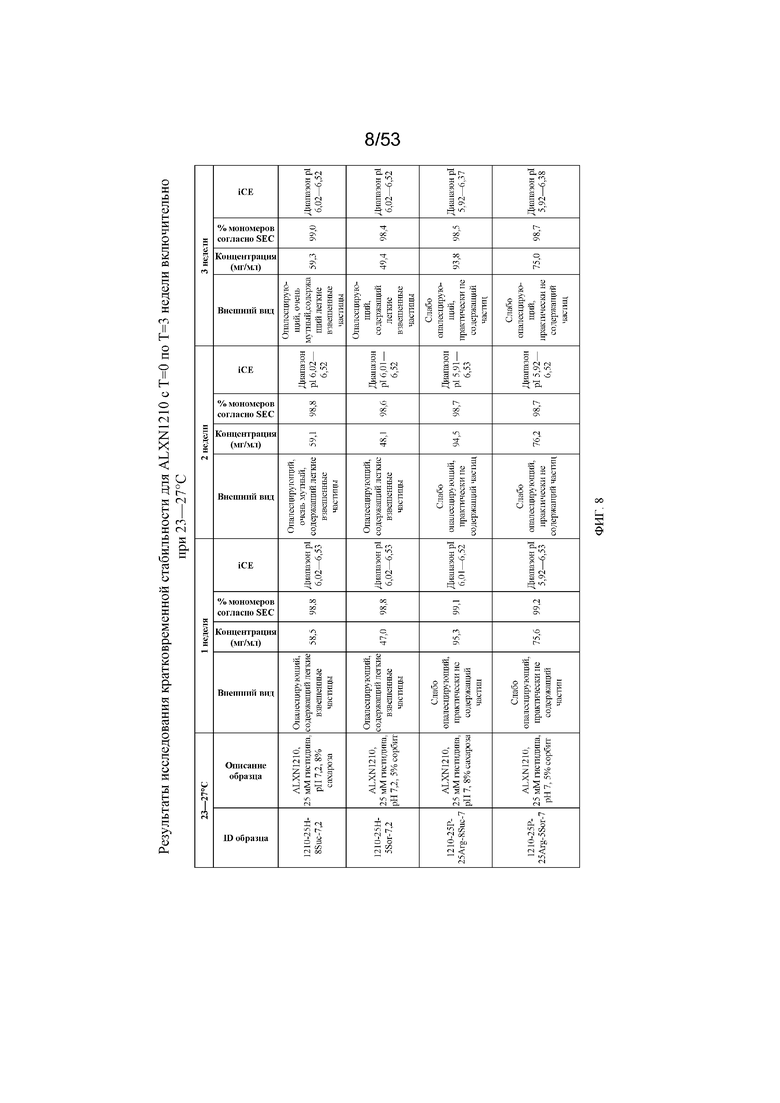
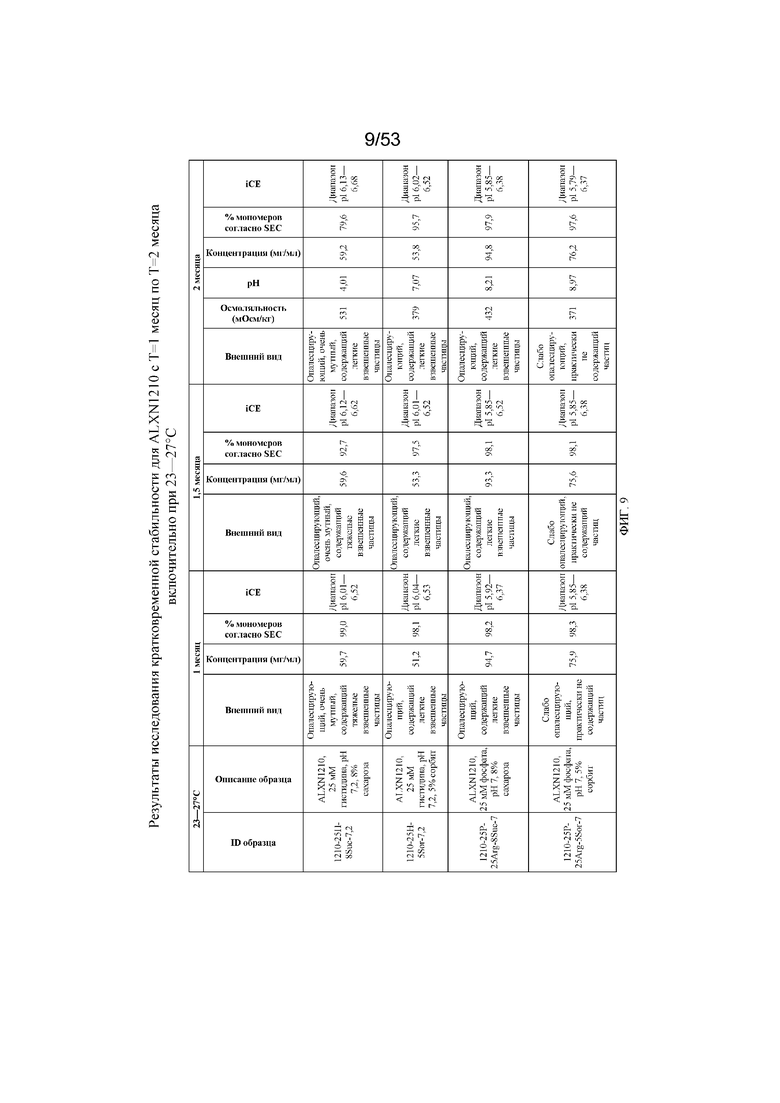
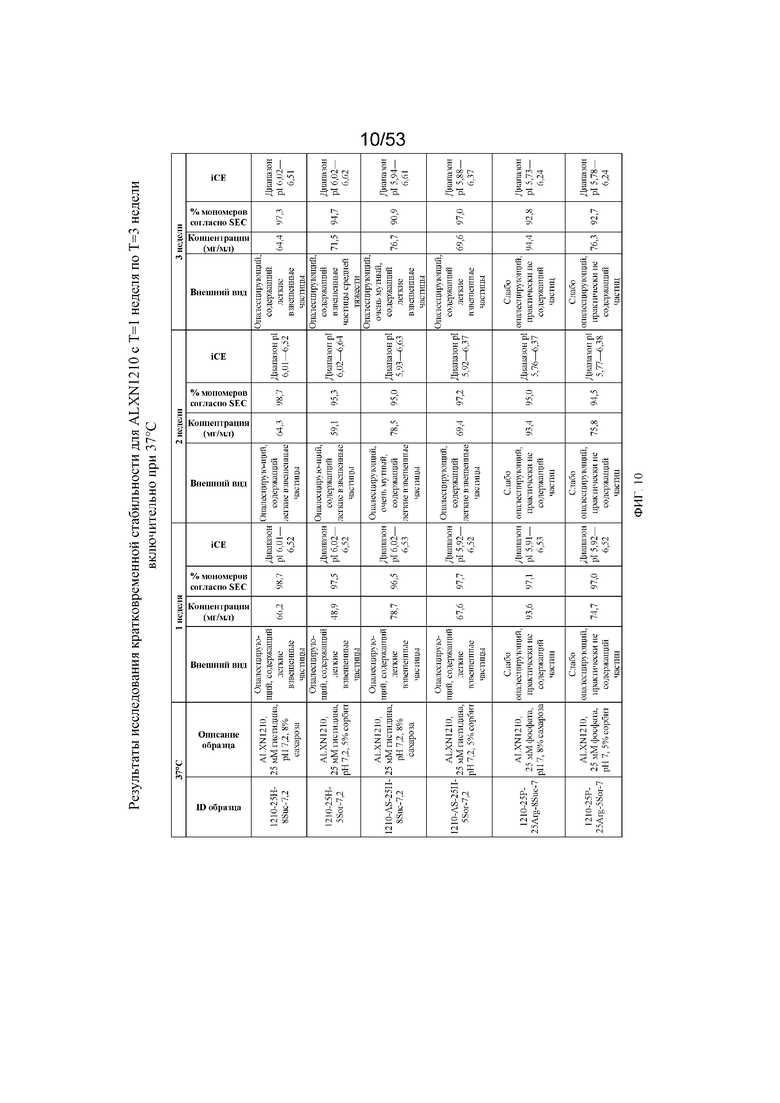
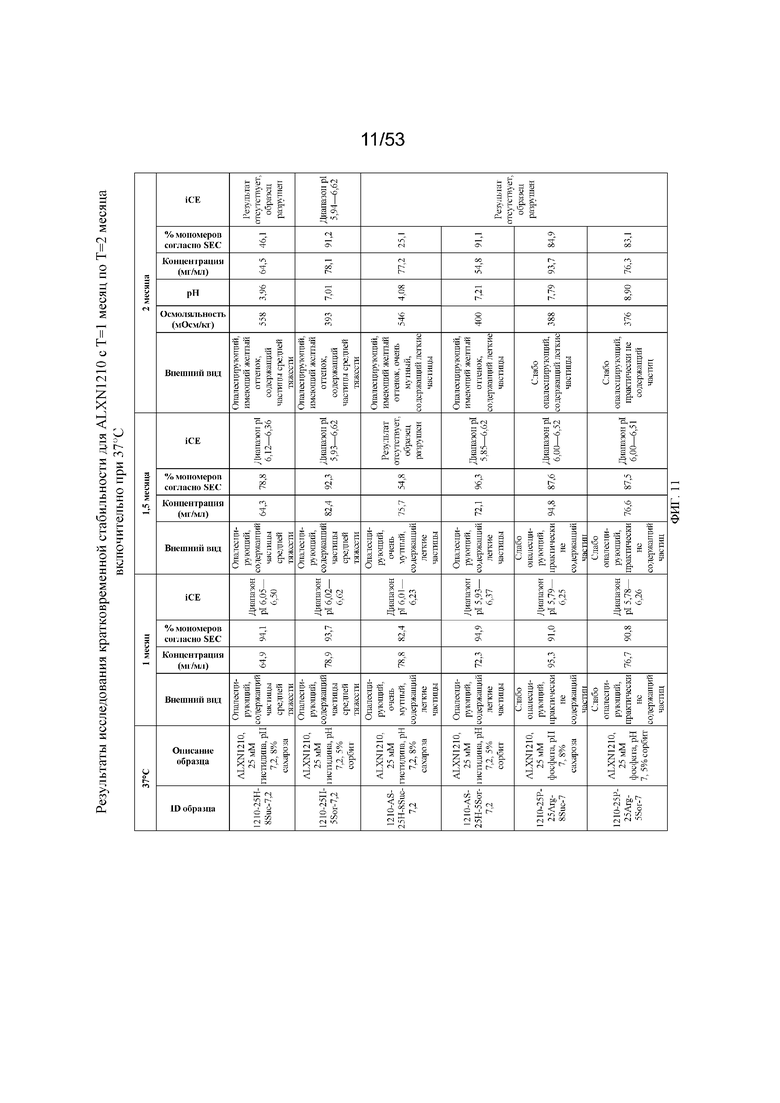

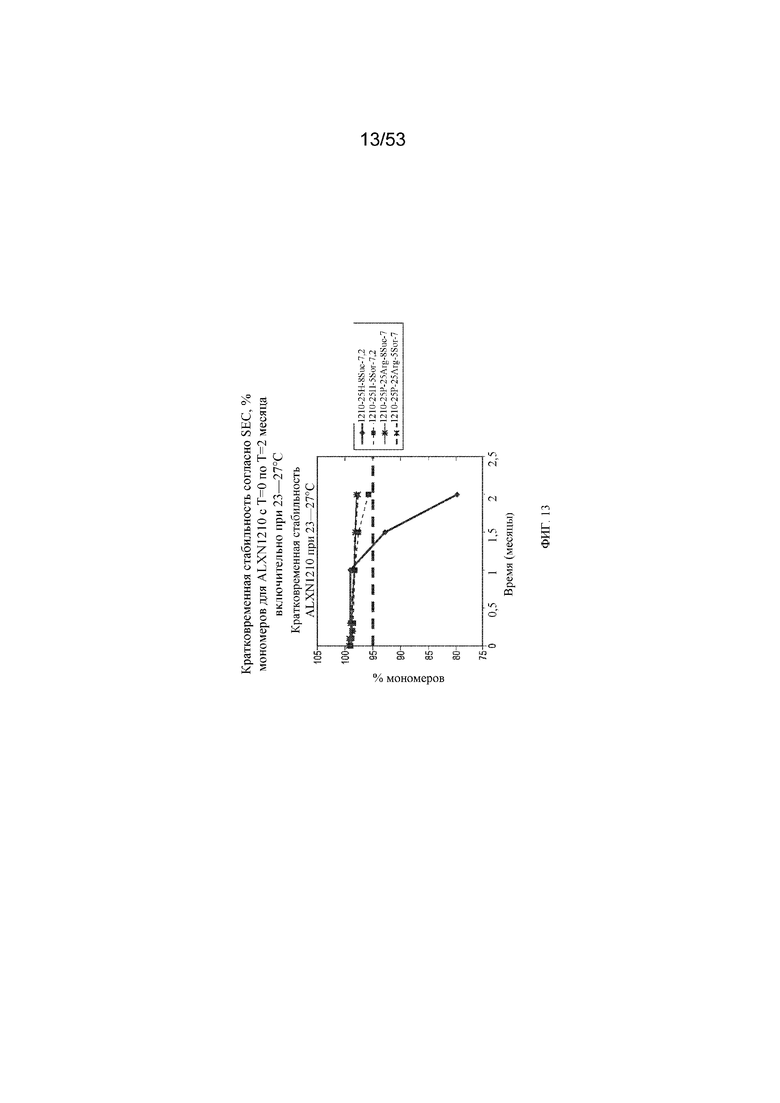
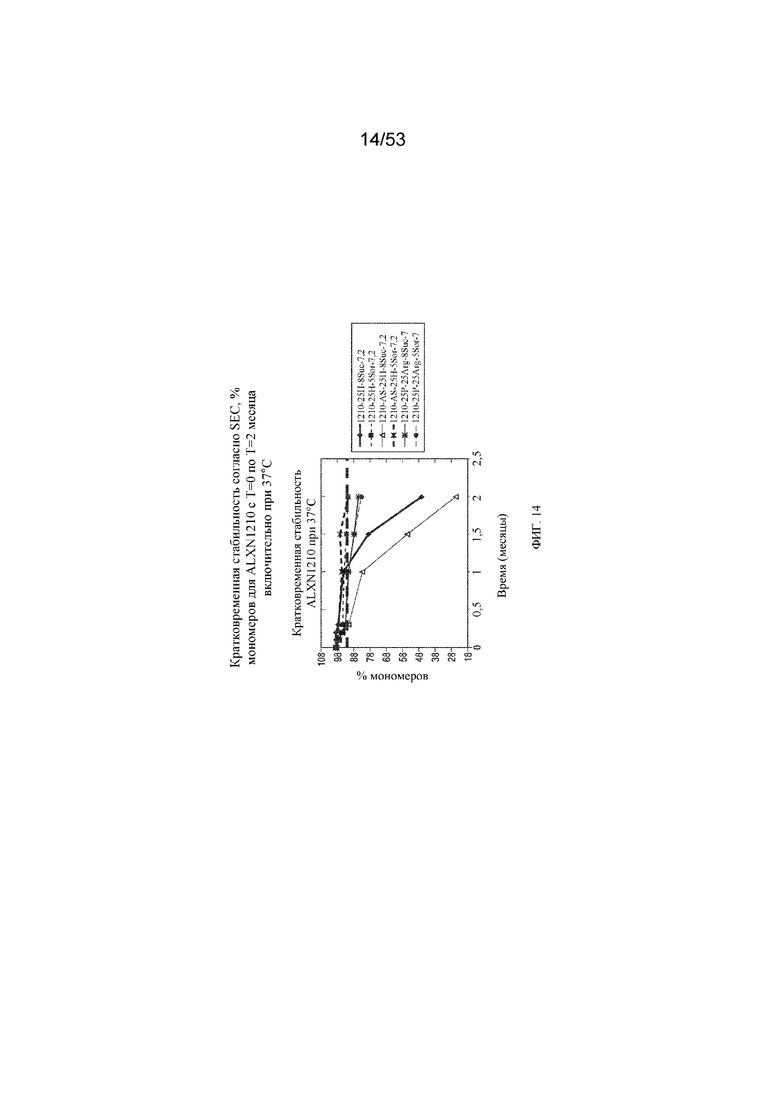
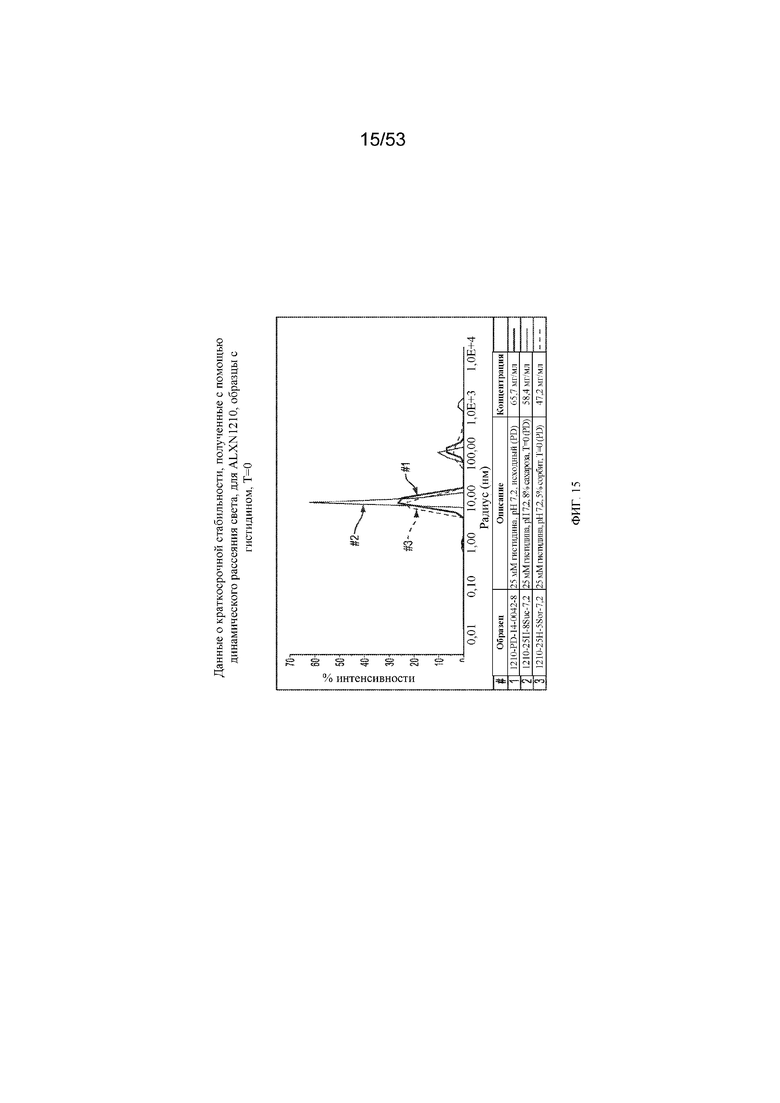




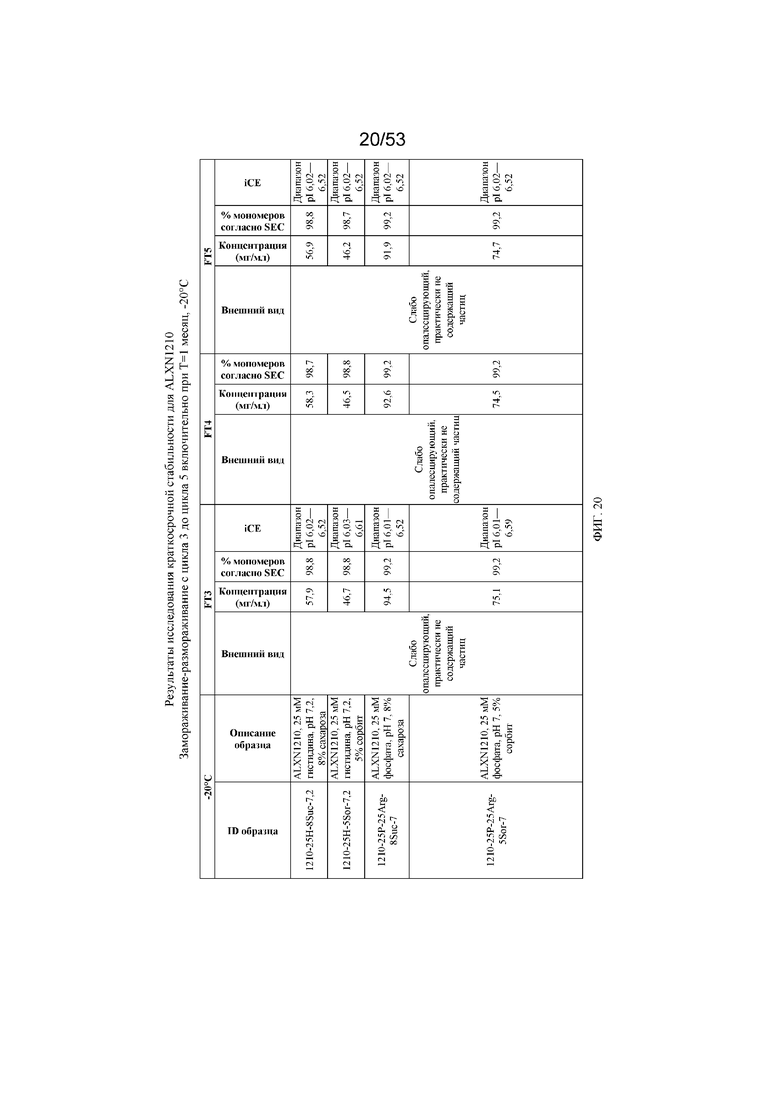

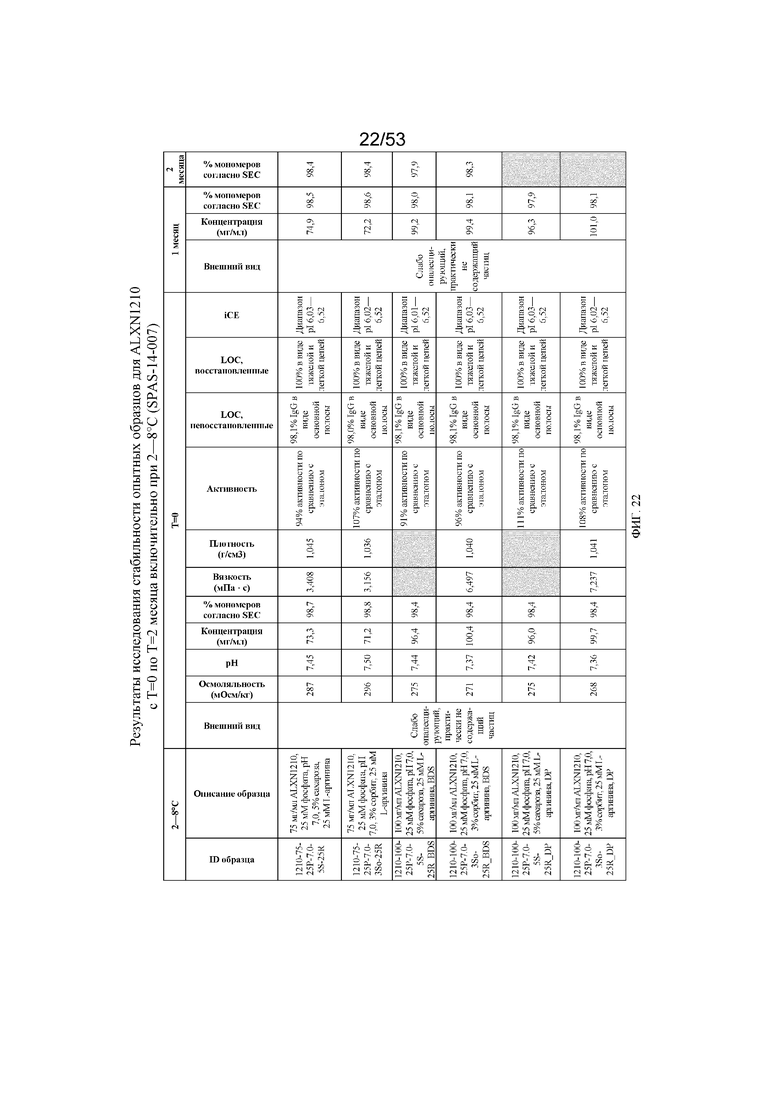
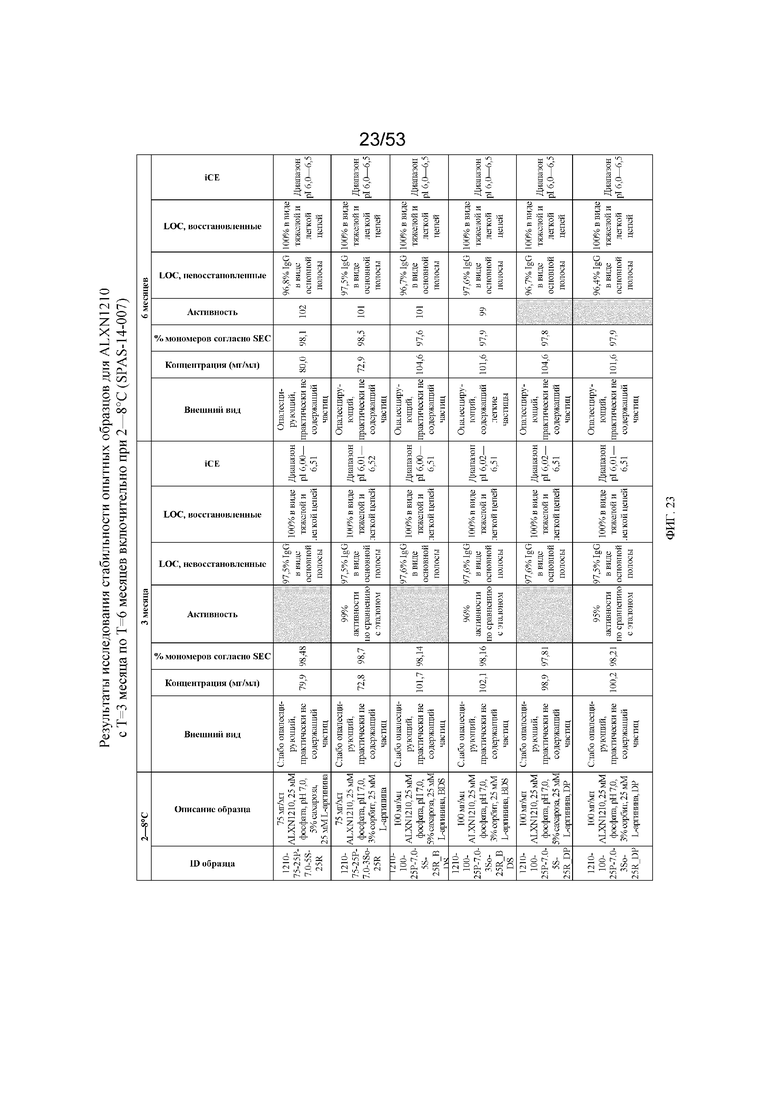

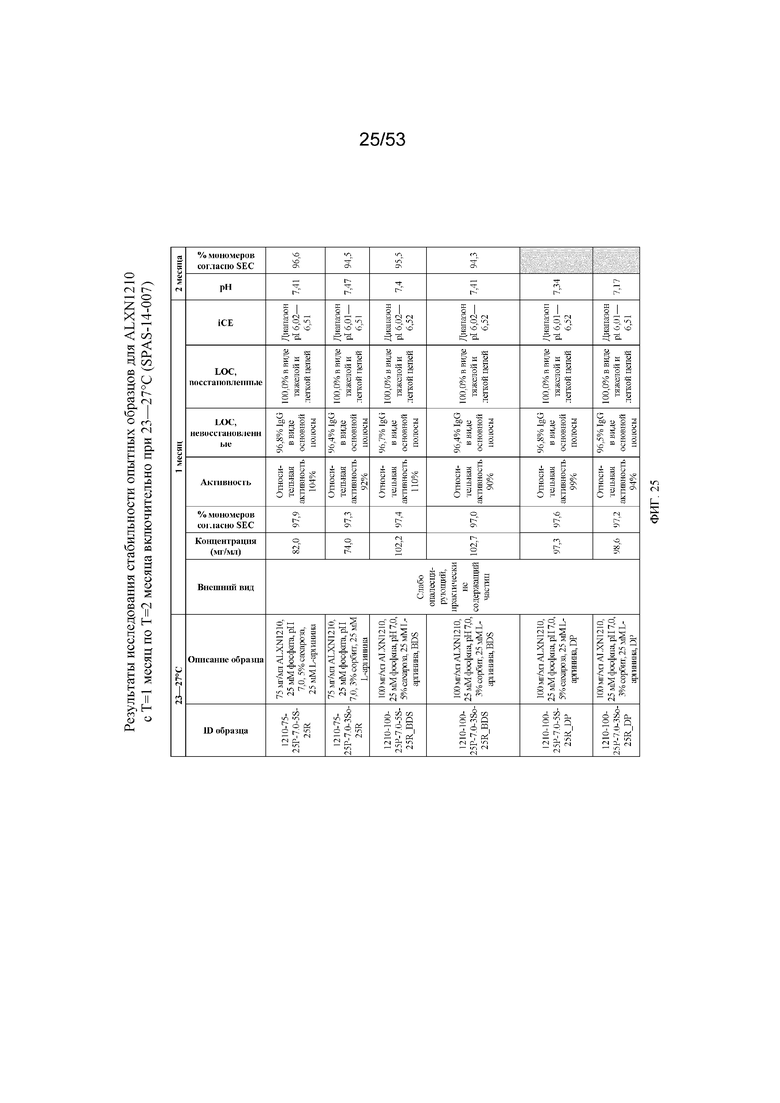
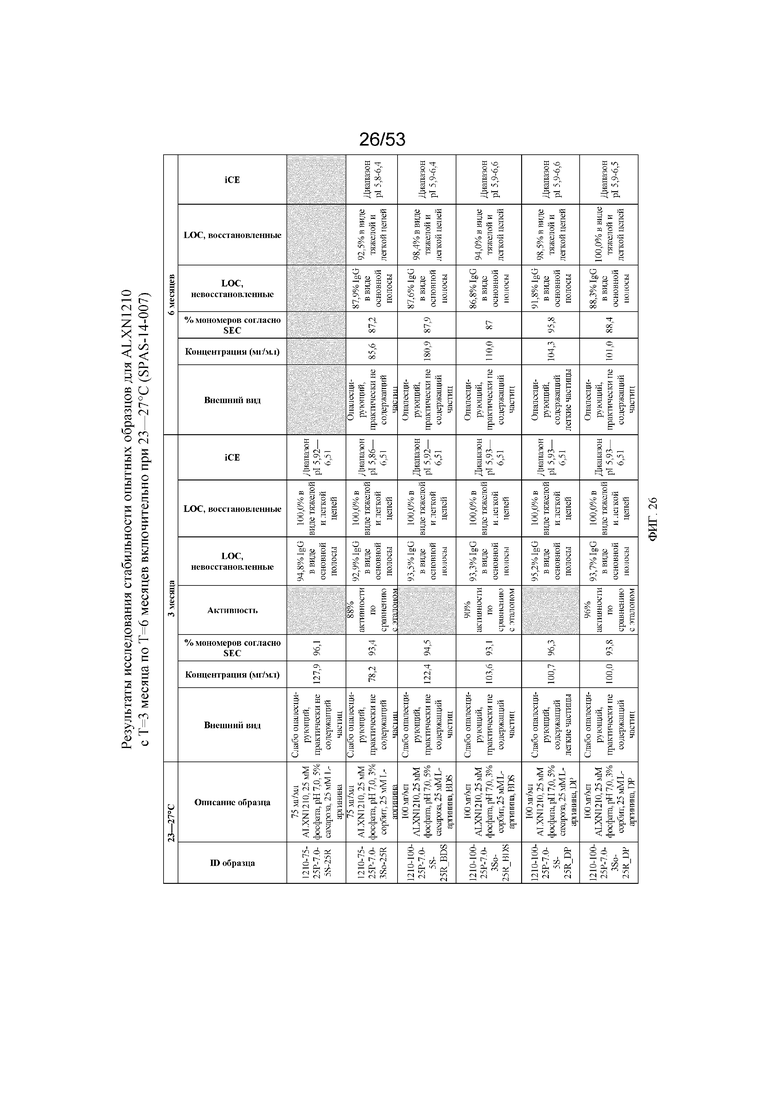
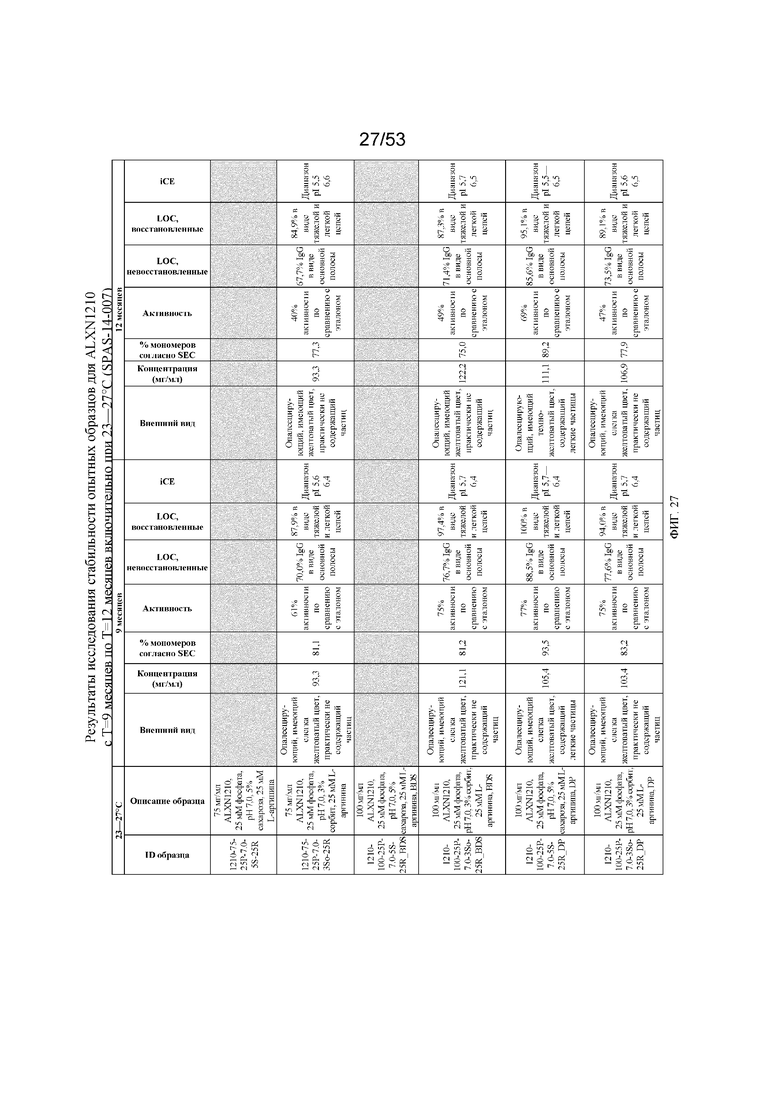
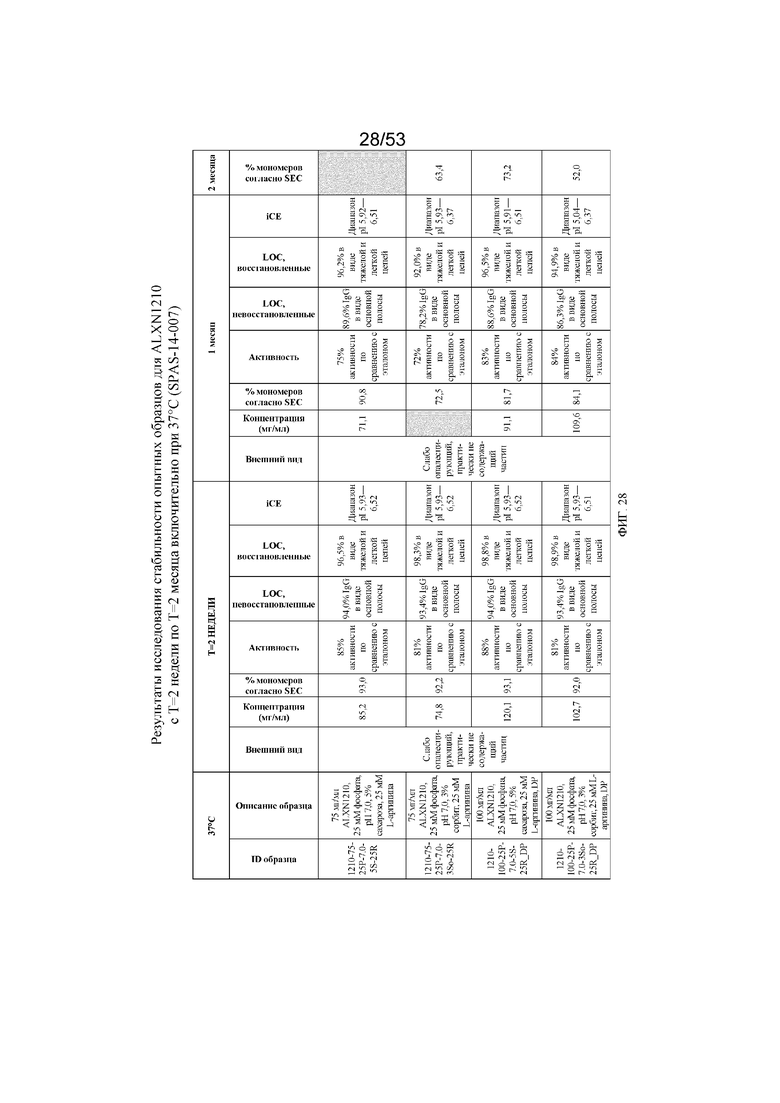

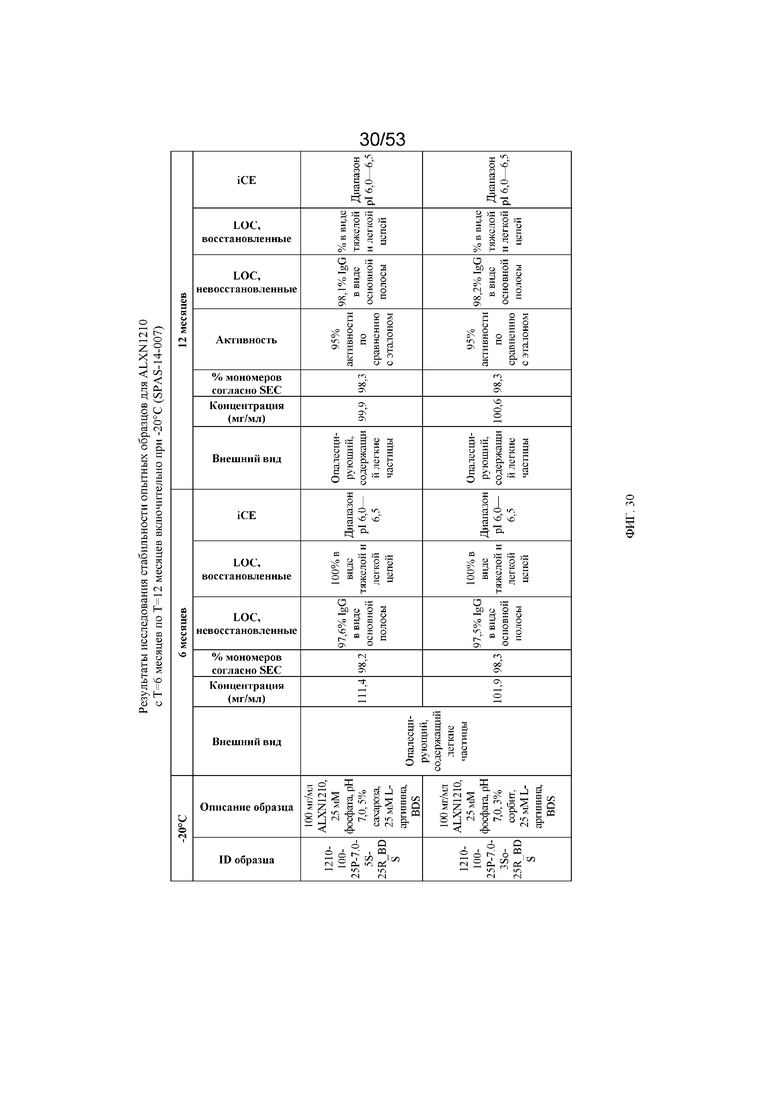
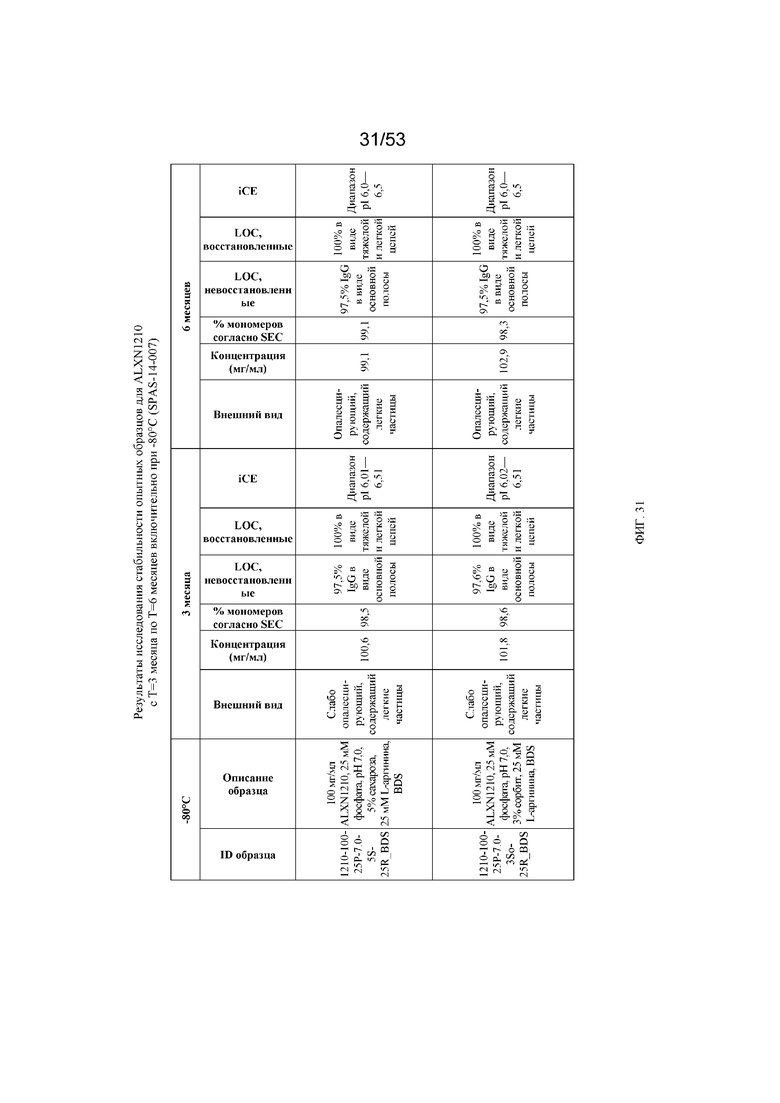
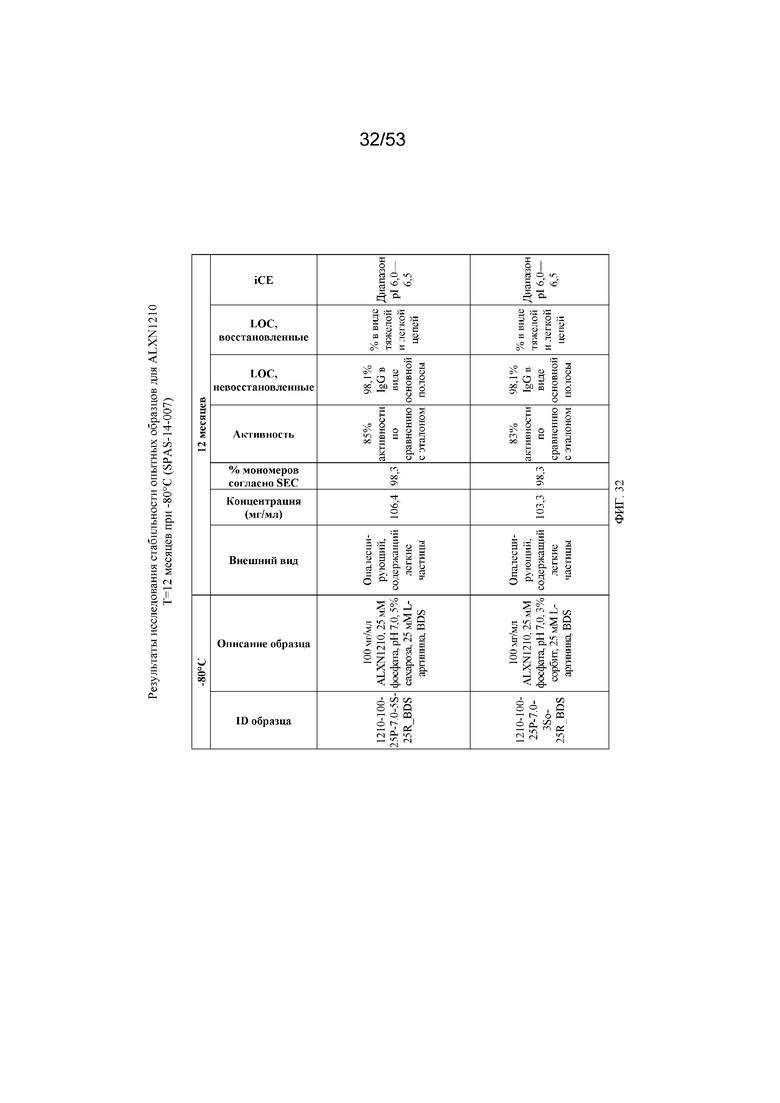
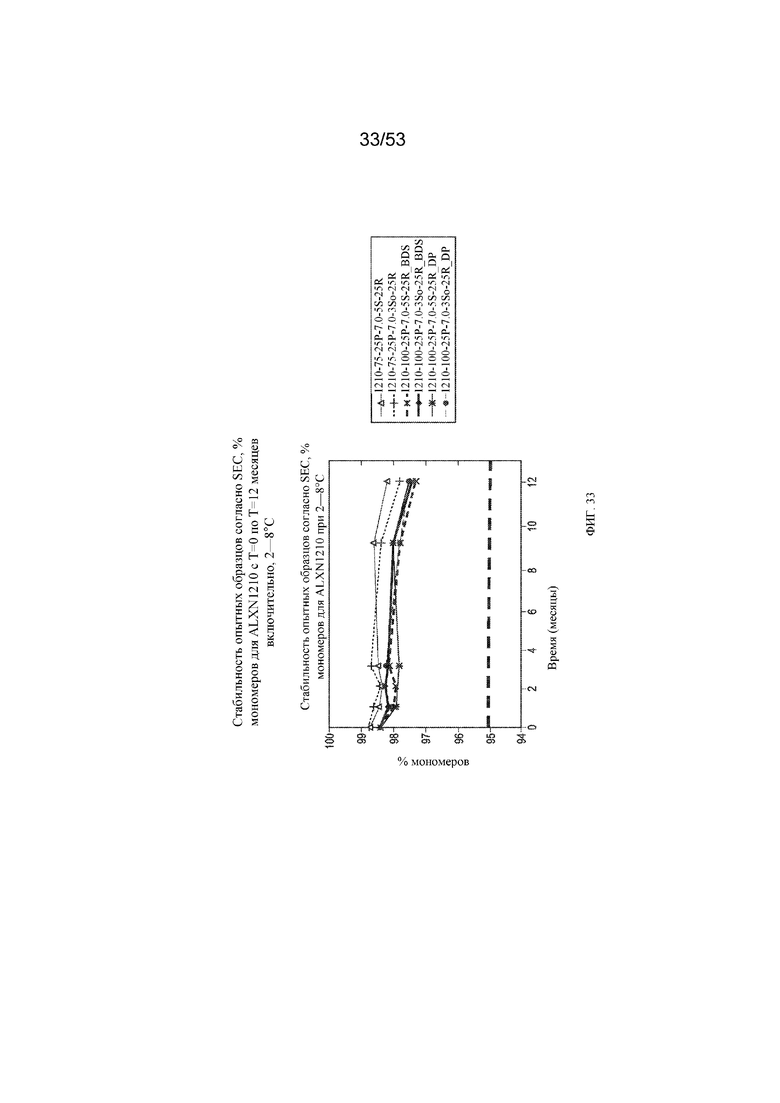
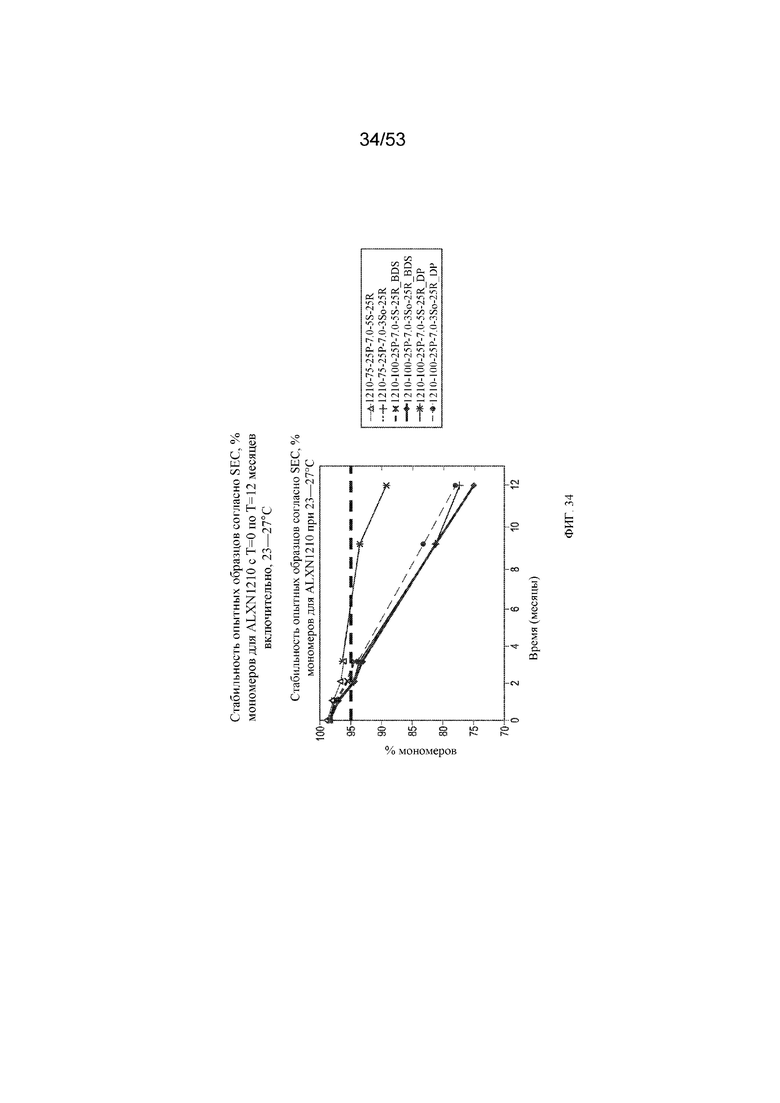
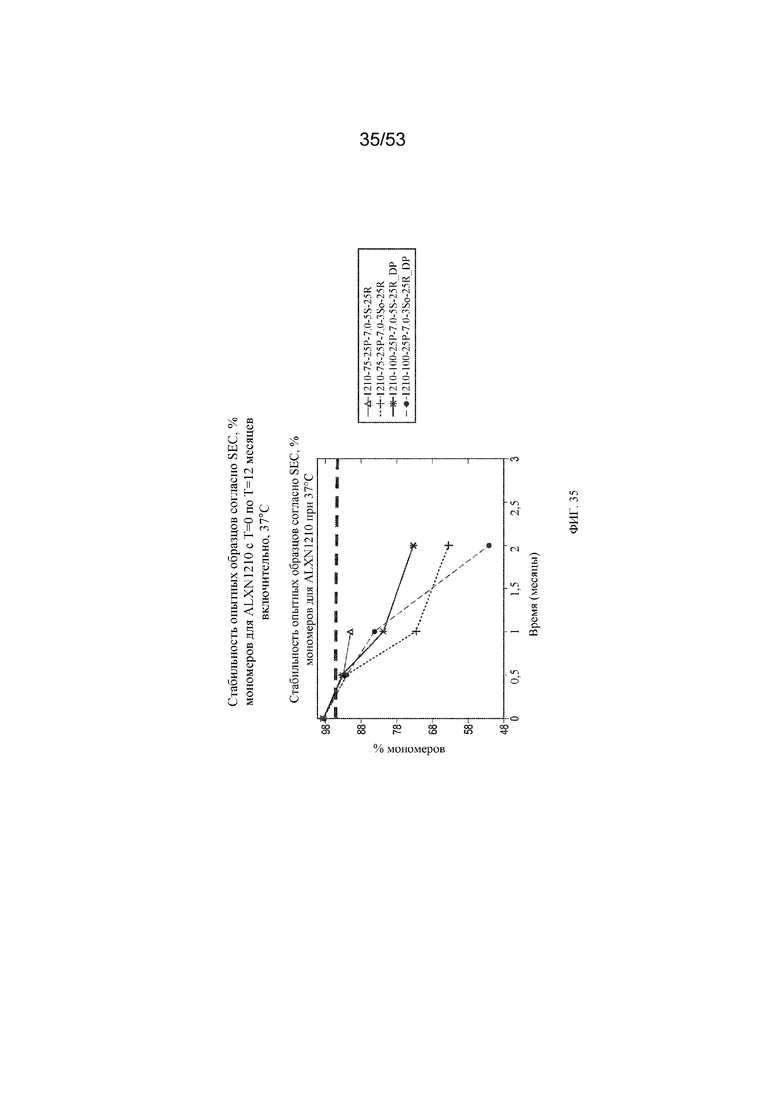

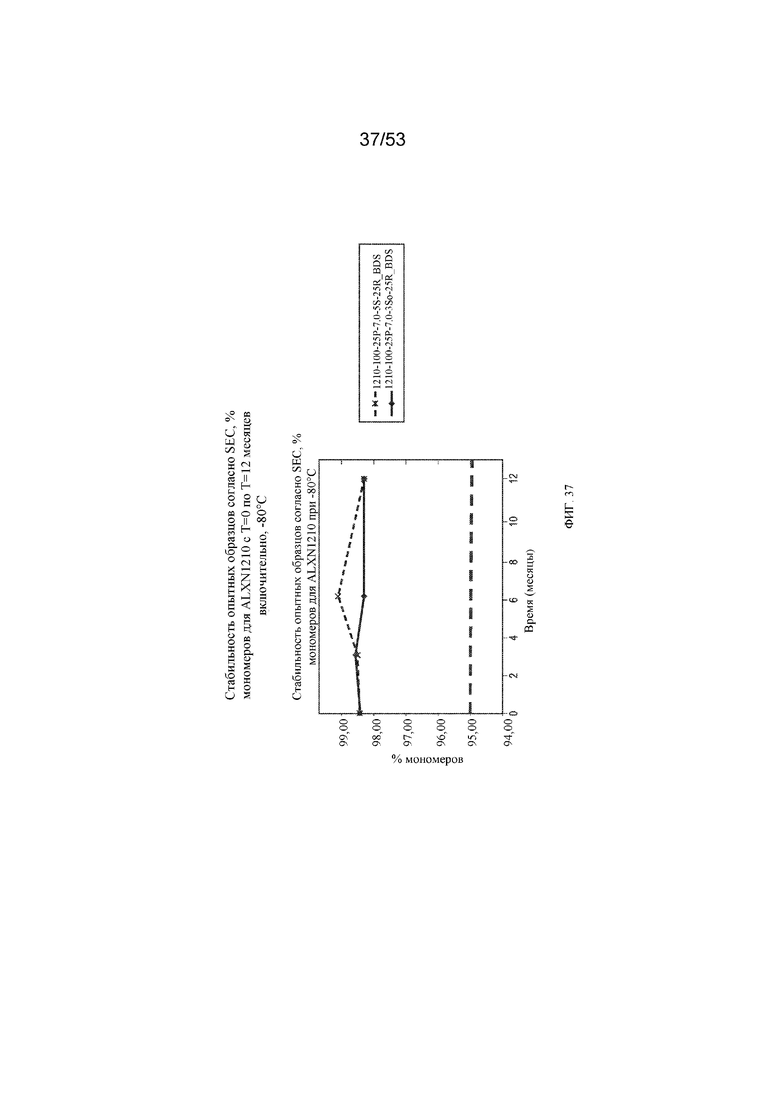
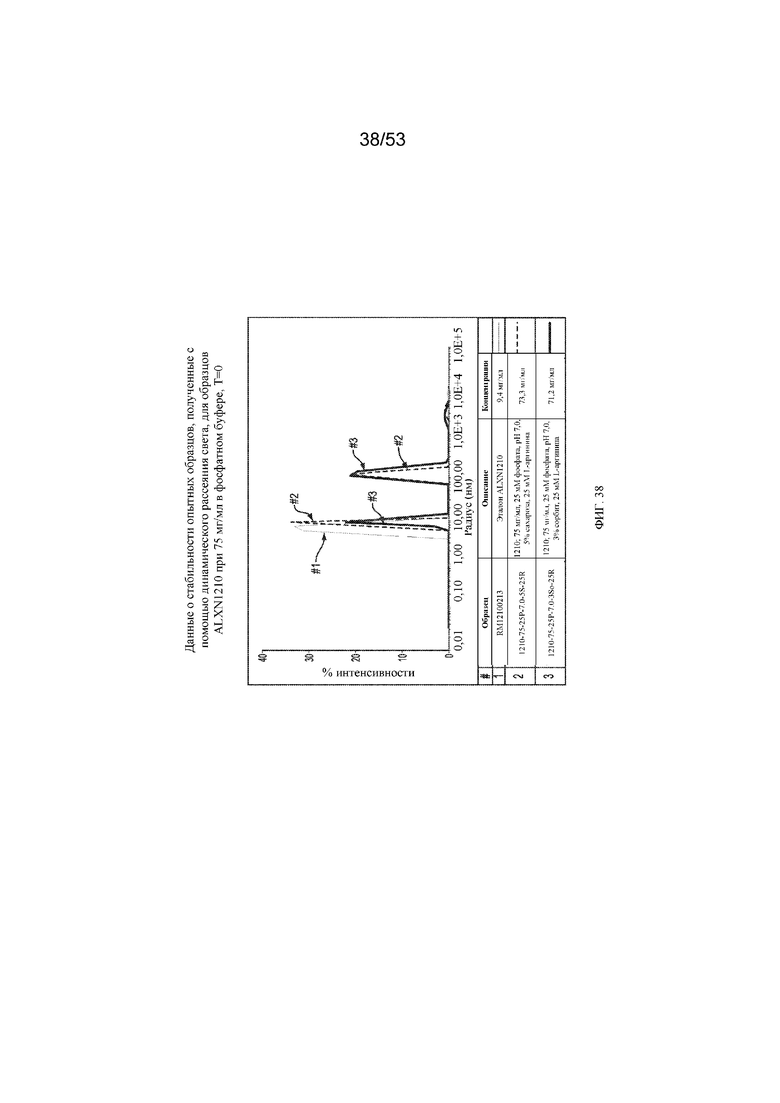




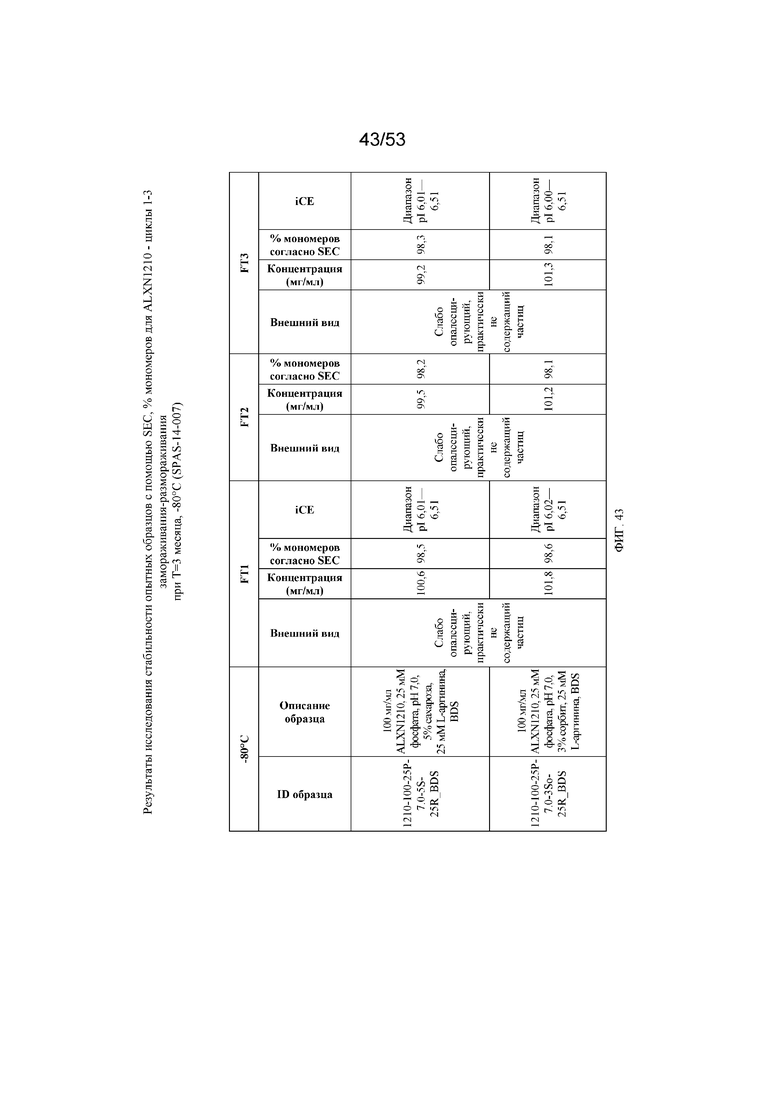


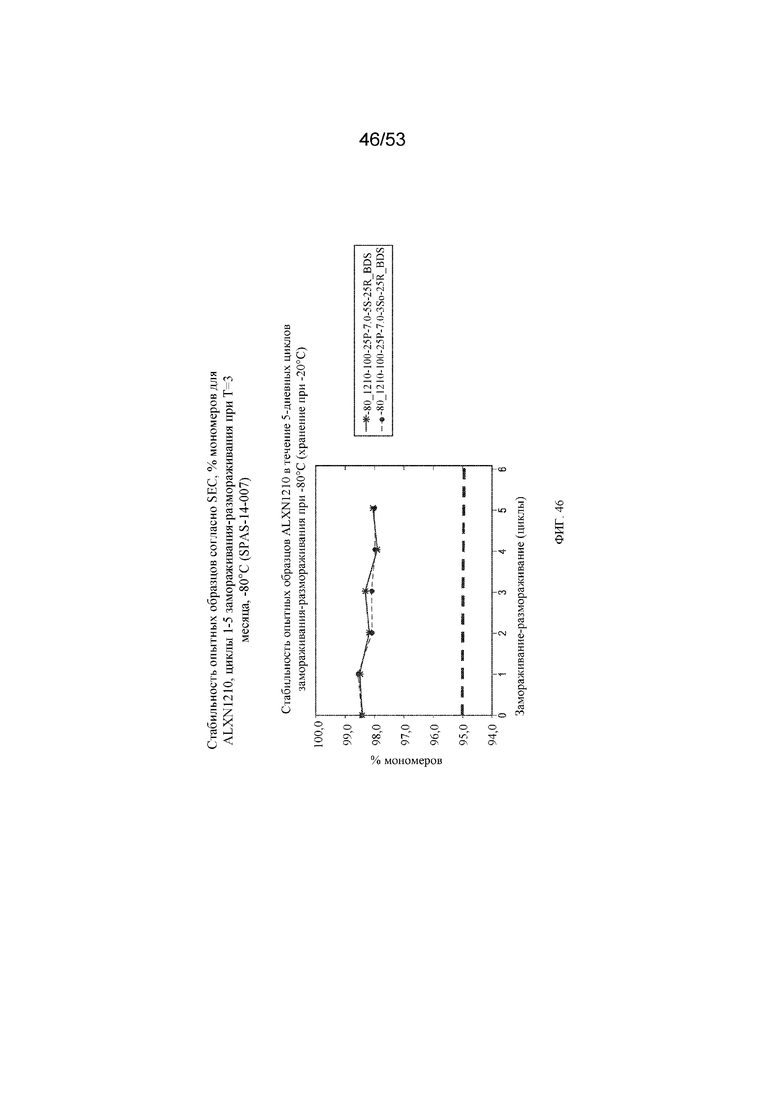


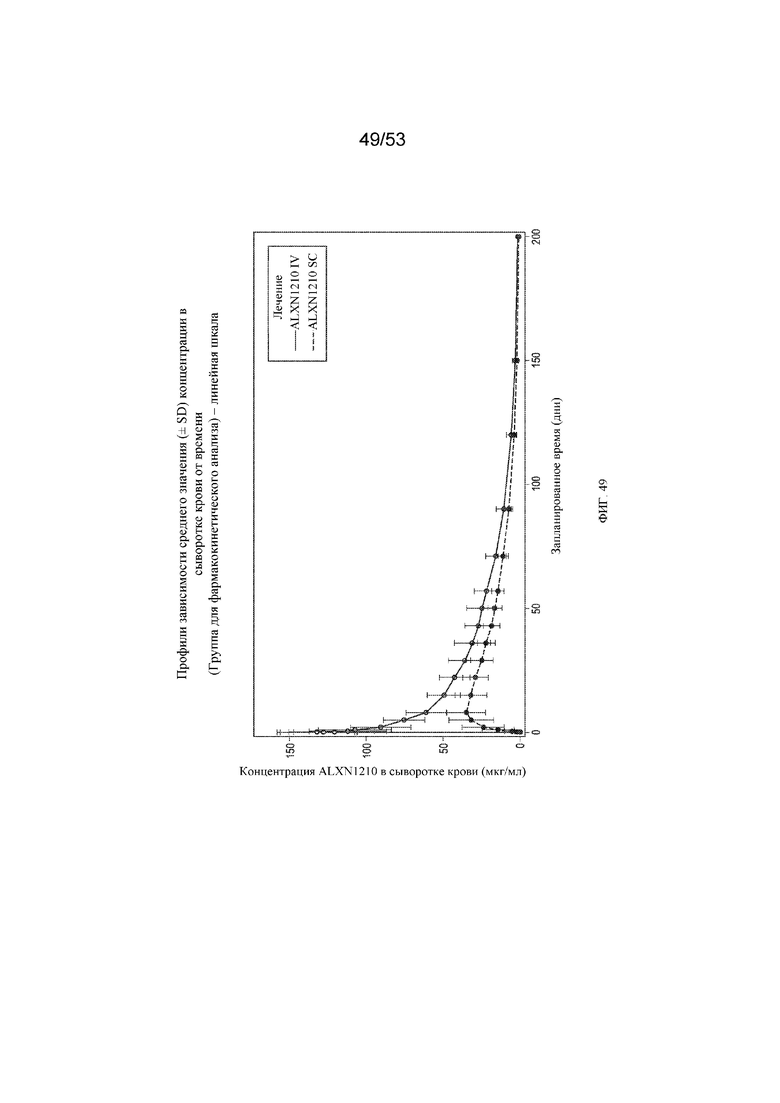
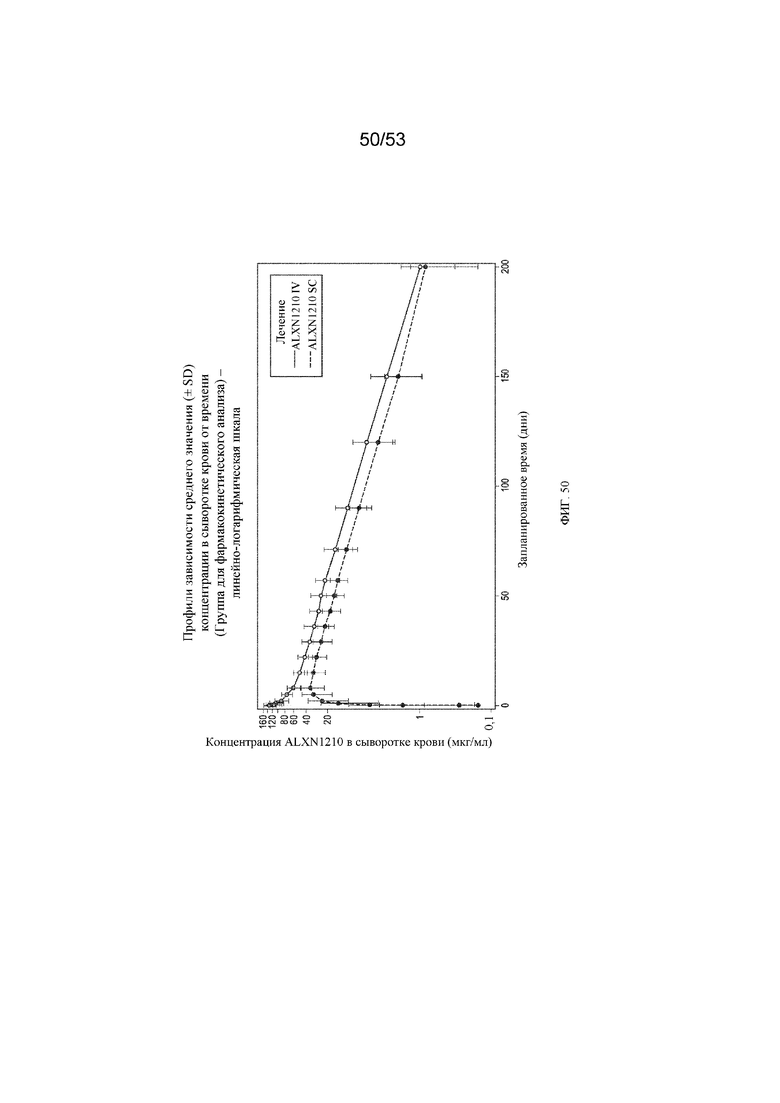
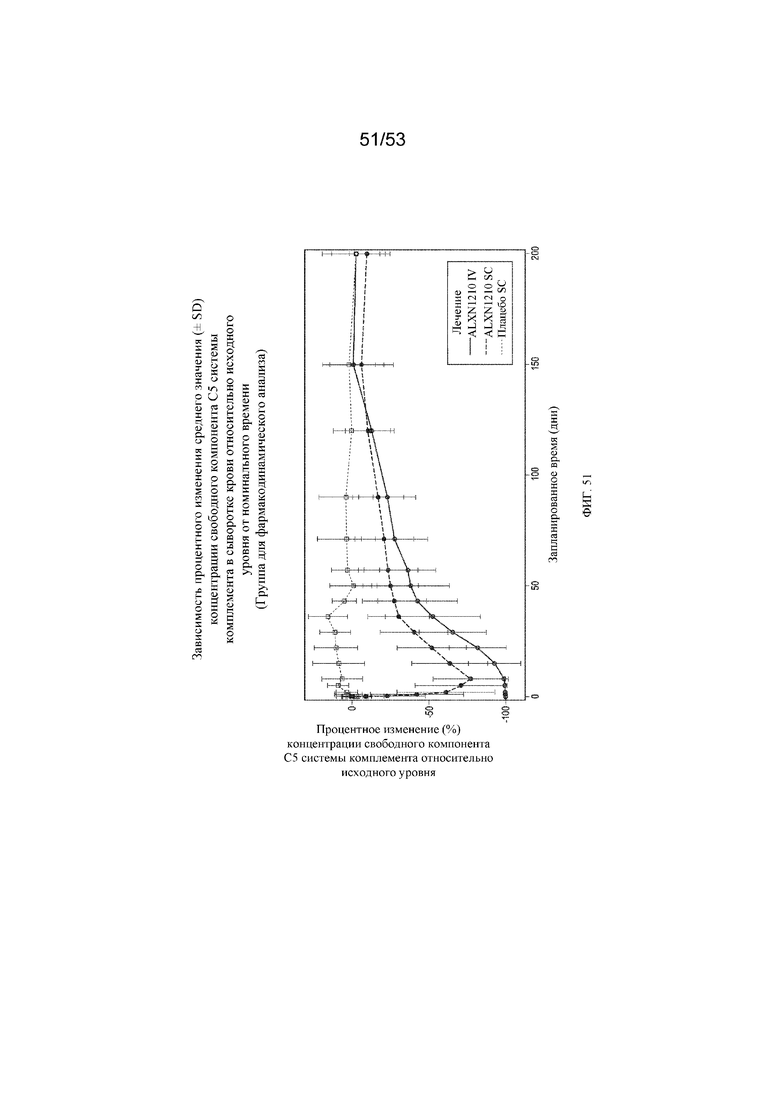
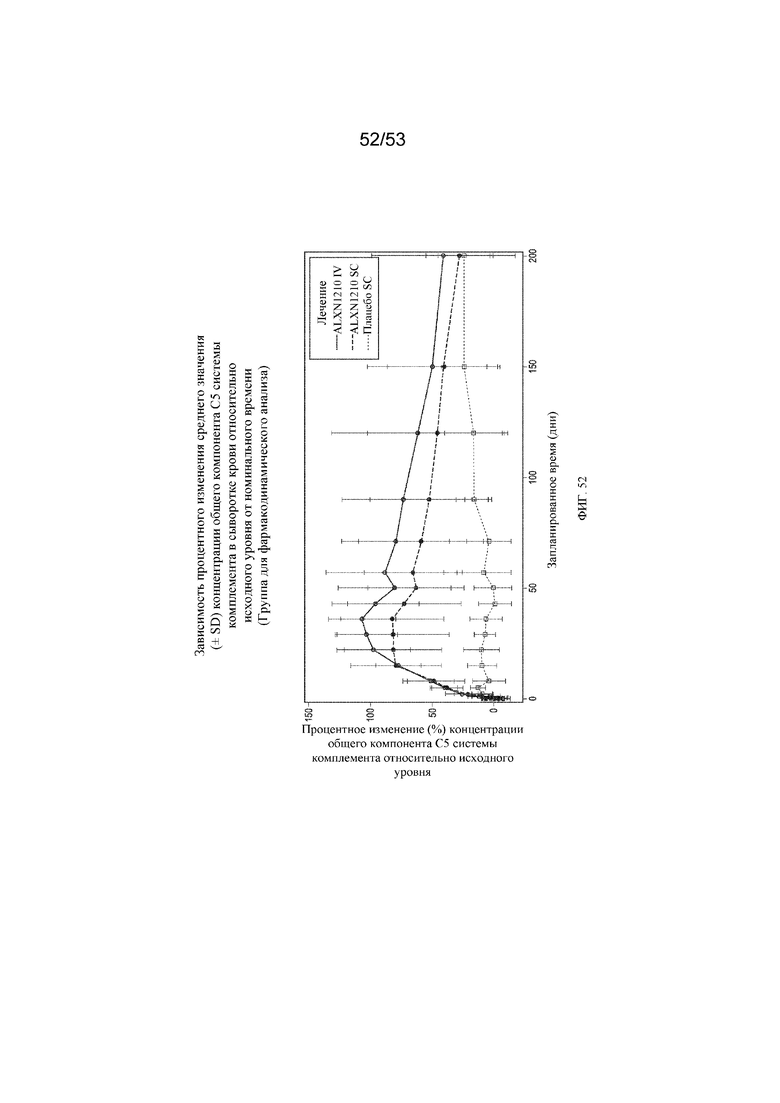

Группа изобретений относится к стабильным водным растворам, содержащим антитело к C5 в высокой концентрации, и способам получения данных растворов. Стабильный водный раствор содержит антитело к C5 в концентрации 100 мг/мл ± 5%, 50 мМ ± 5% фосфатного буфера, 5% ± 5% сахарозы. В другом варианте раствор также содержит 0,05% ± 5% полисорбата 80. Антитело к C5 содержит тяжелую цепь с SEQ ID NO: 14 и легкую цепь с SEQ ID NO: 11. Также предложены способы лечения или предупреждения нарушений, ассоциированных с системой комплемента C5, таких как пароксизмальная ночная гемоглобинурия (PNH) и атипичный гемолитико-уремический синдром (aHUS), с помощью данных растворов. Также представлены терапевтические наборы, содержащие указанный раствор и средство для введения раствора пациенту, нуждающемуся в лечении. Изобретение обеспечивает стабильный высококонцентрированный водный раствор антитела к C5, которое остается на по меньшей мере 97% мономерным и сохраняет по меньшей мере 90% своей C5-связывающей активности во время хранения при 2-8°C в течение по меньшей мере шести месяцев. 5 н. и 22 з.п. ф-лы, 53 ил., 23 табл., 3 пр.
1. Стабильный водный раствор для лечения пациента-человека с состоянием, ассоциированным с белком C5 системы комплемента, содержащий:
(a) антитело к C5 в концентрации 100 мг/мл ± 5%, где антитело к C5 содержит полипептидную тяжелую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 14, и полипептидную легкую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 11;
(b) 50 мМ ± 5% фосфатного буфера;
(c) 5% ± 5% сахарозы (вес/объем); и
(d) 25 мМ ± 5% аргинина.
2. Стабильный водный раствор для лечения пациента-человека с состоянием, ассоциированным с белком C5 системы комплемента, содержащий:
(a) антитело к C5, где антитело к C5 содержит полипептидную тяжелую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 14, и полипептидную легкую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 11;
(b) 50 мМ ± 5% фосфатного буфера;
(c) 5% ± 5% сахарозы (вес/объем);
(d) 0,05% ± 5% полисорбата 80 и
(е) 25 мМ ± 5% аргинина.
3. Стабильный водный раствор по п.1, дополнительно содержащий поверхностно-активное вещество.
4. Стабильный водный раствор по п.3, где поверхностно-активное вещество представляет собой 0,05% ± 5% полисорбата 80.
5. Стабильный водный раствор по п.2, где антитело к C5 представлено в концентрации 100 мг/мл ± 5%.
6. Стабильный водный раствор по любому из пп. 1-5, где антитело к C5 представляет собой ALXN1210 (равулизумаб).
7. Стабильный водный раствор по любому из пп. 1-6, где pH раствора составляет от 7,2 до 7,6.
8. Стабильный водный раствор по п.7, где pH раствора составляет 7,4.
9. Стабильный водный раствор по любому из пп. 1-8, где раствор является стерильным.
10. Стабильный водный раствор по любому из пп. 1-9, где антитело к C5 остается на по меньшей мере 97% мономерным во время хранения при 2-8°C в течение по меньшей мере шести месяцев при определении с помощью SEC-HPLC.
11. Стабильный водный раствор по любому из пп. 1-10, где антитело к C5 остается на по меньшей мере 97% мономерным во время хранения при 2-8°C в течение по меньшей мере одного года при определении с помощью SEC-HPLC.
12. Стабильный водный раствор по любому из пп. 1-11, где менее 3% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC.
13. Стабильный водный раствор по любому из пп. 1-12, где менее 2% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC.
14. Стабильный водный раствор по любому из пп. 1-13, где менее 1% антител к C5 в растворе являются агрегированными при определении с помощью SEC-HPLC.
15. Стабильный водный раствор по любому из пп. 1-14, где во время хранения при 2-8°C в течение по меньшей мере шести месяцев антитело к C5 сохраняет по меньшей мере 90% своей C5-связывающей активности по сравнению с антителом к C5 до хранения.
16. Стабильный водный раствор по любому из пп. 1-15, где во время хранения при 2-8°C в течение по меньшей мере шести месяцев антитело к C5 сохраняет по меньшей мере 95% своей способности ингибировать гемолиз по сравнению с антителом к C5 до хранения.
17. Стабильный водный раствор по любому из пп. 1-16, где раствор подходит для подкожного введения.
18. Стабильный водный раствор по любому из пп. 1-17, где раствор подходит для внутривенного введения.
19. Способ получения стабильного концентрированного раствора антитела, содержащего антитело к C5, содержащее полипептидную тяжелую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 14, и полипептидную легкую цепь, содержащую аминокислотную последовательность, приведенную под SEQ ID NO: 11, в концентрации 100 мг/мл, 50 мМ фосфатного буфера, 5% сахарозы (вес/объем) и 25 мМ аргинина, включающий:
i) получение первого водного раствора, содержащего не более 10 мг/мл антитела к C5;
ii) проведение диафильтрации первого водного раствора с получением состава, содержащего 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина, при pH 7,4, вследствие чего обеспечивается получение второго водного раствора; и
iii) концентрацию второго водного раствора с получением стабильного концентрированного раствора антитела, содержащего 100 мг/мл антитела к C5, 50 мМ фосфатного буфера, 5% сахарозу и 25 мМ аргинина.
20. Способ лечения пациента-человека с состоянием, ассоциированным с белком C5 системы комплемента, включающий введение пациенту стабильного водного раствора по любому из пп. 1-17 в количестве, эффективном для лечения состояния.
21. Способ по п.20, где состояние, ассоциированное с белком C5 системы комплемента, выбрано из группы, состоящей из ревматоидного артрита, синдрома антифосфолипидных антител, волчаночного нефрита, ишемического/реперфузионного повреждения, атипичного гемолитико-уремического синдрома (aHUS), типичного гемолитико-уремического синдрома, пароксизмальной ночной гемоглобинурии (PNH), болезни плотного осадка, оптиконевромиелита, мультифокальной моторной нейропатии, рассеянного склероза, макулодистрофии, синдрома HELLP, самопроизвольного аборта, тромботической тромбоцитопенической пурпуры, малоиммунного васкулита, буллезного эпидермолиза, повторного выкидыша, черепно-мозговой травмы, миокардита, цереброваскулярного нарушения, нарушения со стороны периферических сосудов, реноваскулярного нарушения, мезентериального/энтерального сосудистого нарушения, васкулита, нефрита при пурпуре Шенлейна-Геноха, васкулита, ассоциированного с системной красной волчанкой, васкулита, ассоциированного с ревматоидным артритом, иммунокомплексного васкулита, болезни Такаясу, дилатационной кардиомиопатии, диабетической ангиопатии, болезни Кавасаки, венозной газовой эмболии, рестеноза после стентирования, ротационной атерэктомии, чрескожной транслюминальной коронарной ангиопластики, тяжелой миастении, болезни холодовых агглютининов, дерматомиозита, пароксизмальной холодовой гемоглобинурии, антифосфолипидного синдрома, болезни Грейвса, атеросклероза, болезни Альцгеймера, септического системного воспалительного ответа, септического шока, повреждения спинного мозга, гломерулонефрита, отторжения трансплантата, тиреоидита Хашимото, сахарного диабета I типа, псориаза, пузырчатки, аутоиммунной гемолитической анемии, идиопатической тромбоцитопенической пурпуры, синдрома Гудпасчера, болезни Дегоса и катастрофического антифосфолипидного синдрома.
22. Способ по п.20, где состояние, ассоциированное с белком C5 системы комплемента, представляет собой атипичный гемолитико-уремический синдром (aHUS).
23. Способ по п.20, где состояние, ассоциированное с белком C5 системы комплемента, представляет собой пароксизмальную ночную гемоглобинурию (PNH).
24. Способ по любому из пп. 20-22, где стабильный водный раствор вводят пациенту подкожно.
25. Способ по любому из пп. 20-22, где стабильный водный раствор вводят пациенту внутривенно.
26. Терапевтический набор для лечения пациента-человека с состоянием, ассоциированным с белком C5 системы комплемента, содержащий (i) стабильный водный раствор по любому из пп. 1-17 и (ii) средство для доставки стабильного водного раствора человеку.
27. Терапевтический набор по п.26, где средство представляет собой шприц.
| US 9371377 B2, 21.06.2016 | |||
| US 9556263 B2, 31.01.2017 | |||
| US 2014056888 A1, 27.02.2014 | |||
| US 2013344088 A1, 26.12.2013 | |||
| US 9556263 B2, 31.01.2017 | |||
| EA 201691764 A1, 30.12.2016 | |||
| WANG, W | |||
| Instability, stabilization, and formulation of liquid protein pharmaceuticals | |||
| International Journal of Pharmaceutics, 1999, 185(2), p.129-188, |

