Область техники
Настоящее изобретение относится к новым производным гидразона, в которых концевая аминогруппа замещена арильной или гетероарильной группой, и их применению.
Уровень техники
Белок тау (тау (τ) белок), который представляет собой белок, связанный с микротрубочками (MAP), в основном экспрессируемый в аксонах нервных клеток с молекулярной массой от 50000 до 70000, служит для стабилизации микротрубочек и представляет собой молекулярное разнообразие за счет фосфорилирования. У человека тау-белок образуется в шести изоформах путем вставки 29 или 58 аминокислотных остатков на N-конце и альтернативного сплайсинга мРНК из 3 или 4 повторяющихся структур (называемых доменом связывания микротрубочек) на С-конце.
В здоровых нервах тау-белок стабилизирует микротрубочки, способствуя росту аксонов и поляризации нервных клеток. Когда происходит патологическое гиперфосфорилирование, тау-белок отделяется от микротрубочек, что приводит к нерастворимой агрегации. Кроме того, был предложен структурный скелет, вызывающий агрегацию тау-белка, и были представлены доказательства того, что нерастворимые филаменты образуются из 10 растворимых мономеров, и что эти филаменты связаны в высокоразмерные структуры, называемые нейрофибриллярными клубками (NFT). Полноразмерный тау-белок человека содержит связывающий микротрубочки домен, состоящий из четырех повторяющихся консервативных последовательностей. Среди этих повторяющихся последовательностей положительно заряженные остатки играют важную роль в связывании с сильно отрицательно заряженными микротрубочками (от 20 до 30 электронов на димер αβ-тубулина). Сродство связывания с микротрубочками тау также активно регулируется фосфорилированием тау-белка, и это фосфорилирование вызывает динамическую перестройку сетей микротрубочек. Когда тау-белок чрезмерно фосфорилируется, баланс этой динамической перестройки нарушается, и сродство к микротрубочкам быстро снижается.
Гиперфосфорилирование и/или агрегация тау-белков вызывает аномальное накопление этих тау-белков в нервных клетках, что указывается как причина разнообразных нейродегенеративных заболеваний и т.п. Агрегаты тау-белка в основном обнаруживают в телах клеток и дендритах нервных клеток, и эти агрегаты тау-белка называются нейрофибриллярными клубками (NFT) и нейропильными нитями. Исследование микроструктур нейрофибриллярных клубков (NFT) показывает, что такие их микроструктуры состоят из парных спиральных нитей (PHF), в которых тау-белки запутаны, как тонкие нити, и агрегированы и гиперфосфорилированы, в отличие от нормального тау-белка. Феномен аномальной агрегации тау-белка также проявляется при таупатии. В этом случае, хотя точно не известно, какую роль агрегация тау-белка играет в развитии тауопатии, этот феномен агрегации тау-белка похож на феномен агрегации, который является обычным при общих нейродегенеративных заболеваниях.
Таким образом, хотя известно, что гиперфосфорилирование и/или агрегация тау-белка вызывает разнообразные нейродегенеративные заболевания, включая болезнь Альцгеймера и таупатию, конкретный механизм того, как эти аномальные виды тау вызывают изменения в сигнальном пути и вызывают нейротоксичность, еще не подтвержден, и пока нет эффективных методов лечения или терапевтических средств для лечения данных заболеваний.
Описание
Техническая проблема
Авторы настоящего изобретения приложили много усилий для открытия новых низкомолекулярных соединений, способных ингибировать агрегацию и/или гиперфосфорилирование тау-белка. В результате они обнаружили, что ряд производных гидразона, в которых концевая аминогруппа замещена арильной или гетероарильной группой, эффективно ингибирует агрегацию тау-белка и не проявляет цитотоксичности при эффективных концентрациях. На основании этого открытия было завершено настоящее изобретение.
Техническое решение
В первом аспекте настоящего изобретения предложено соединение, представленное приведенной ниже формулой 1, или его фармацевтически приемлемая соль:
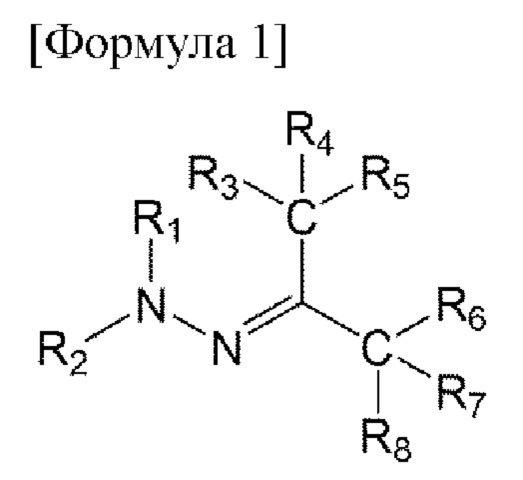
в формуле 1, приведенной выше,
R1 представляет собой водород или C1-6 алкил;
R2 представляет собой незамещенный или замещенный С6-14 и арил, 5-14-членный гетероарил или 5-14-членный гетероциклил;
от R3 до R5 представляют собой i) все атомы водорода, ii) образуют тройную связь вместе с одним N, iii) R3 и R4 образуют двойную связь вместе с NH, О или S с образованием имина, оксо или тиоксо, соответственно, и R5 представляет собой C1-6 алкокси, амино, алкиламино, ди(С1-6 алкил)амино или 5-14-членный гетероциклил, или iv) R3 до R5 связаны с гетероатомом О, N или S одинарной или двойной связью и содержат углерод и гетероатом, связанный с ним, с образованием незамещенного или замещенного 5-14-членного гетероарила или гетероциклила; и
от R6 до R8 представляют собой i) все атомы водорода, ii) образуют тройную связь вместе с одним N, iii) R6 и R7 образуют двойную связь вместе с NH, О или S с образованием имина, оксо или тиоксо, соответственно, и R8 представляет собой C1-6 алкокси, амино, C1-6 алкиламино, ди(С1-6 алкил)амино или 5-14-членный гегероциклил, или iv) от R6 до R8 связаны с гегероатомом О, N или S одинарной или двойной связью и содержат углерод и гетероатом, связанный с ним, с образованием незамещенного или замещенного 5-14-членного гетероарила или гетероциклила;
где гетероарил или гетероциклил содержит по меньшей мере один из О, N и S,
замещенный арил, гетероарил или гетероциклил могут содержать C1-6 алкил; C1-6 алкокси; галоген; С1-6 перфторалкил; С1-6 перфторалкокси; C1-6 галогеналкил; карбамоил; карбоксамидо; С6-10 арилокси; незамещенный С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил; или С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил, замещенный по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, C1-6 алкокси, галогена, нитро, C1-6 алкоксикарбонила, С1-6 перфторалкилa, C1-6 перфторалкокси, C1-6 галогеналкила, амино, C1-6 алкиламино, ди(С1-6 алкил)амино и алкоксикарбонила, в качестве заместителя, и
карбамоил и карбоксамидо могут быть замещены по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, арила и гетероарила, не замещенного или замещенного С1-6 алкокси, гидрокси, циано или C1-6 алкила.
Во втором аспекта настоящего изобретения предложен способ получения соединения первого аспекта, включающий стадию введения в реакцию нитрита натрия, аминопроизводного R2 (R2-NH2) и  в присутствии кислоты с образованием иминосвязи.
в присутствии кислоты с образованием иминосвязи.
В третьем аспекте настоящего изобретения предложен способ получения соединения по первому аспекту, включающий стадии:
a1) получение производного анилина  замещенного арилом или гетероарилом, незамещенным или замещенным R2' или R2"; и
замещенного арилом или гетероарилом, незамещенным или замещенным R2' или R2"; и
а2) введение в реакцию с нитритом натрия, производным анилина  и малононитрилом в присутствии кислоты с образованием иминосвязи.
и малононитрилом в присутствии кислоты с образованием иминосвязи.
В четвертом аспекте настоящего изобретения предложен способ получения соединения по первому аспекту, включающий стадию введения в реакцию нитрита натрия, 5-14-членного гетероарильного или 5-14-членного гетероциклильного производного, содержащего аминогруппу, и малононитрила в присутствии кислоты с образованием иминосвязи.
В пятом аспекте настоящего изобретения предложен способ получения соединения по первому аспекту, включающий стадии:
введение в реакцию карбоновой кислоты R2''' или ее ангидрида 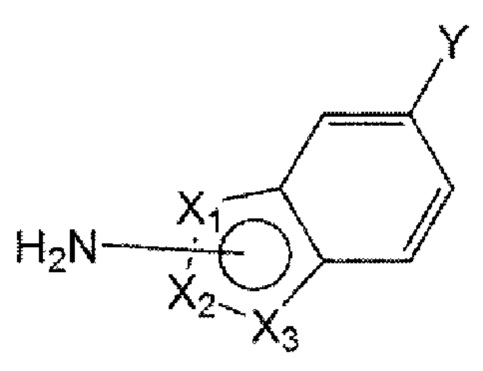 (здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
(здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
введение в реакцию нитрита натрия, гетероциклического производного, содержащего R2''', связанного с карбоксамидогруппой, полученного на предыдущей стадии, и малононитрилом в присутствии кислоты с образованием иминосвязи.
В шестом аспекте настоящего изобретения предложен способ получения соединения по первому аспекту, включающий стадии:
введение в реакцию нитрита натрия, аминопроизводной R2 (R2-NH2) и 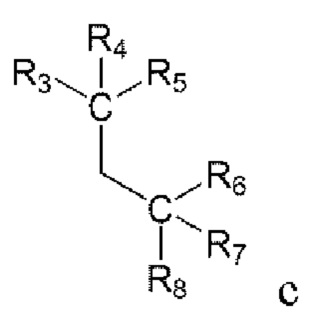 образованием иминосвязи; и
образованием иминосвязи; и
доведение рН реакционного раствора до 5-7 с помощью основания.
В седьмом аспекте настоящего изобретения предложена композиция для ингибирования агрегации тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В восьмом аспекте настоящего изобретения предложена композиция для ингибирования гиперфосфорилирования тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В девятом аспекте настоящего изобретения предложена фармацевтическая композиция для предотвращения или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В десятом аспекте настоящего изобретения предложен способ предотвращения или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка, причем данный способ включает этап введения фармацевтической композиции по настоящему изобретению субъекту, нуждающемуся в этом.
Благоприятные эффекты
В соответствии с настоящим изобретением, поскольку новое производное гидразона, в котором концевая аминогруппа замещена арильной или гетероарильной группой, может эффективно ингибировать агрегацию и/или гиперфосфорилирование тау-белка, данное производное гидразона может эффективно применяться для профилактики или лечения вызванных им заболеваний, таких как болезнь Альцгеймера и различные таупатии.
Лучший способ по данному изобретению
В первом аспекте настоящего изобретения предложено соединение, представленное приведенной ниже формулой 1, или его фармацевтически приемлемая соль:
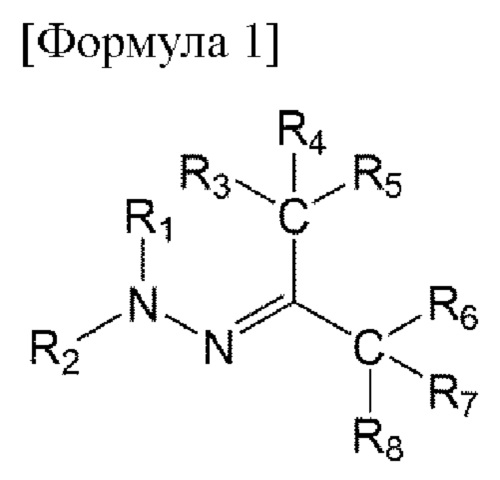
в формуле 1, приведенной выше,
R1 представляет собой водород или C1-6 алкил;
R1 представляет собой незамещенный или замещенный С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил;
от R3 до R5 представляют собой i) все атомы водорода, ii) образуют тройную связь вместе с одним N, iii) R3 и R4 образуют двойную связь вместе с NH, О или S с образованием имина, оксо или тиоксо, соответственно, и R5 представляет собой C1-6 алкокси, амино, алкиламино, ди(С1-6 алкил)амино или 5-14-членный гетероциклил, или iv) R3 до R5 связаны с гетероатомом О, N или S одинарной или двойной связью и содержат углерод и гетероатом, связанный с ним, с образованием незамещенного или замещенного 5-14-членного гетероарила или гетероциклила; и
от R6 до R8 представляют собой i) все атомы водорода, ii) образуют тройную связь вместе с одним N, iii) R6 и R7 образуют двойную связь вместе с NH, О или S с образованием имина, оксо или тиоксо, соответственно, и R8 представляет собой C1-6 алкокси, амино, C1-6 алкиламино, ди(С1-6 алкил)амино или 5-14-членный гетероциклил, или iv) от R6 до R8 связаны с гетероатомом О, N или S одинарной или двойной связью и содержат углерод и гетероатом, связанный с ним, с образованием незамещенного или замещенного 5-14-членного гетероарила или гетероциклила;
где гетероарил или гетероциклил содержит по меньшей мере один из О, N и S,
замещенный арил, гетероарил или гетероциклил могут содержать C1-6 алкил; C1-6 алкокси; галоген; С1-6 перфторалкил; С1-6 перфторалкокси; C1-6 галогеналкил; карбамоил; карбоксамидо; С6-10 арилокси; незамещенный С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил; или С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил, замещенный по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, C1-6 алкокси, галогена, нитро, C1-6 алкоксикарбонила, C1-6 перфторалкилa, C1-6 перфторалкокси, C1-6 галогеналкила, амино, C1-6 алкиламино, ди(С1-6 алкил)амино и С1-6 алкоксикарбонила, в качестве заместителя, и
карбамоил и карбоксамидо могут быть замещены по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, арила и гетероарила, не замещенного или замещенного C1-6 алкокси, гидрокси, циано или C1-6 алкила.
В частности, в соединении по настоящему изобретению,
R1 представляет собой водород или метил;
R2 представляет собой незамещенный или замещенный фенил, бензотиазолил, карбазолил, индазолил, хинолинил, индолил, пирролопиридинил, бензоизоксазолил, дигидробензотиазолил, оксодигидробензотиазолил, бензоксазолил, оксодигидробензоксазолил, бензоимидазолил или оксодигидробензоимидазолил;
от R3 до R5 образуют -ON, -СН3, -CO2Me, -CO2Et, -CONH2, -CSNH2,
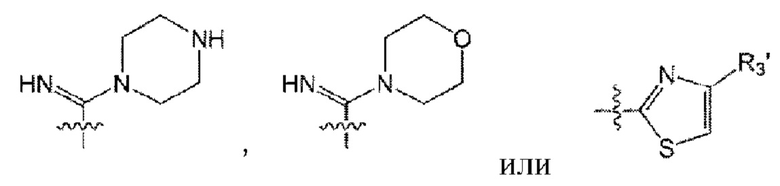 (здесь R3' представляет собой алкил) вместе с С, связанным с ним; а также от R6 до R8 образуют -ON, -СН3, -CO2Me, -CO2Et, -CONH2, -CSNH2,
(здесь R3' представляет собой алкил) вместе с С, связанным с ним; а также от R6 до R8 образуют -ON, -СН3, -CO2Me, -CO2Et, -CONH2, -CSNH2,
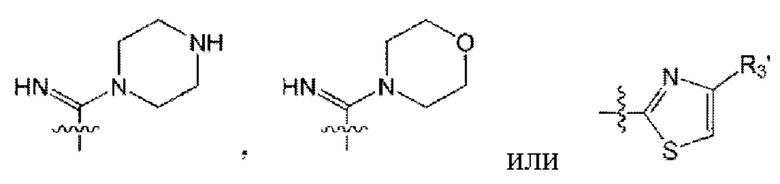 (здесь R3' представляет собой C1-6 алкил),
(здесь R3' представляет собой C1-6 алкил),
где гетероарил и гетероциклил содержит по меньшей мере один О, N или S,
замещенный фенил, бензотиазолил, карбазолил, индазолил, хинолинил, индолил, пирролопиридинил, бензоизоксазолил, дигидробензотиазолил, оксодигидробензотиазолил, бензоксазолил, оксодигидро бензоксазолил, бензоимидазолил или оксодигидробензоимидазолил могут содержать метил, этил, пропил, изопропил, трифторметил, трифторэтил, метокси, этокси, трифторметокси, фтор, хлор, бром, морфолинил, карбамоил, карбоксамидо, фенокси или фенил, пиперидинил, тиазолил, пиридинил, пиридазинил, пирамидинил, пиразил, пиразолил, бензотиазолил, нафтил, пиразолопиримидинил, имидазопиримидинил, хиноксалинил, имидазопиридинил, изохинолинил, индазолил, индолил, фуранил, тиофенил, оксотетрагидропиридазинил или оксодигидропиридазинил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из метила, этила, этинила, метокси, фтора, хлора, брома, нитро, диметиламино, метоксикарбонила, этоксикарбонила и трифторметила, в качестве заместителя, и
карбамоил и карбоксамид могут быть заменены по меньшей мере одним, выбранным из группы, состоящей из метила, этила и фенила, пиридинила, пиримидинила и пиразинила, незамещенных или замещенных метилом, метокси, гидрокси или циано.
Например, соединение по настоящему изобретению может быть представлено формулой 2 ниже:
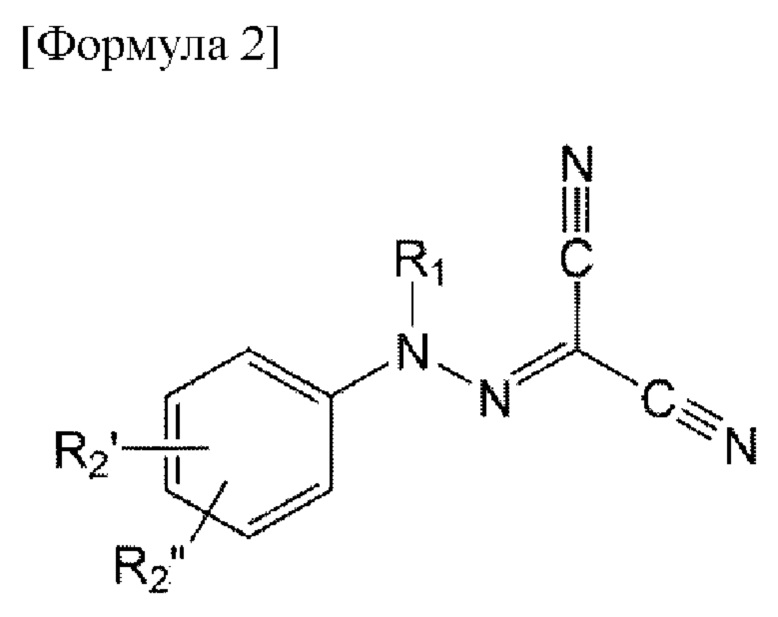
в формуле 2, приведенной выше,
R1 представляет собой водород или C1-6 алкил;
R2' и R2", каждый независимо, представляют собой водород, C1-6 алкил, алкокси, галоген, C1-6 перфторалкил, С1-6 перфторалкокси, C1-6 галогеналкил, карбамоил, незамещенный или замещенный C1-6 алкил или С6-14 арил, 5-14-членный гетероарил или 5-14-членный гетероциклил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, C1-6 алкокси, галогена, нитро, C1-6 алкоксикарбонила, C1-6 перфторалкила, C1-6 перфторалкокси, C1-6 галогеналкила, амино, C1-6 алкиламино, ди(С1-6 алкил)амино, C1-6 алкоксикарбонила и С6-10 арилокси.
Конкретно, в формуле 2, приведенной выше,
R1 представляет собой водород или метил;
R2' представляет собой водород, фтор, метокси или морфолинил; и
R2'’ представляет собой метил, этил, пропил, изопропил, трифторметил, трифторэтил, метокси, этокси, трифторметокси, фтор, хлор, бром, морфолинил, карбамоил, карбоксамидо, фенокси или тиазолил, пиридинил, пиридазинил, пиразинил, пиримидинил, триазолил, пиразолил, бензотиазолил, нафтил, пиразолопиримидинил, имидазопиримидинил, хиноксалинил, имидазопиридинил, изохинолинил, индазолил, индолил, оксотетрагидропиридазинил или оксодигидропиридазинил, незамещенный или замещенный по меньшей мере одним выбранным из группы, состоящей из метила, этила, этинила, метокси, фтора, хлора, брома, нитро, диметиламино, этоксикарбонила и трифторметила.
Например, соединение по настоящему изобретению может быть представлено формулой 3 ниже:
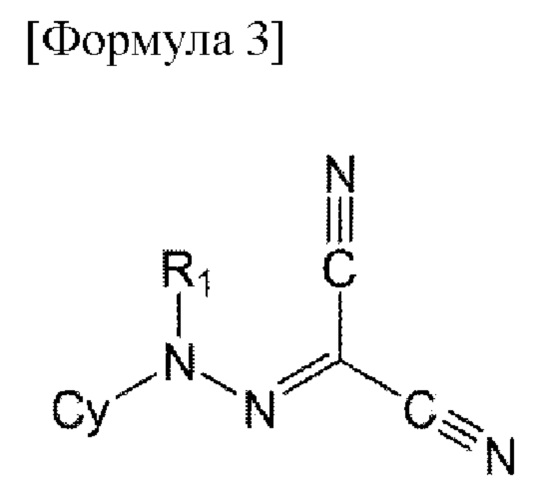
в формуле 3, приведенной выше,
R1 представляет собой водород или C1-6 алкил;
Су представляет собой незамещенный или замещенный 5-14-членный гетероарил или 5-14-членный гетероциклил: и
замещенный 5-14-членный гетероарил или 5-14-членный гетероциклил не замещен или замещен C1-6 алкилом; C1-6 алкокси; галогеном; C1-6 перфторалкилом; перфторалкокси; C1-6 галогеналкилом; карбамоилом, незамещенным или замещенным C1-6 алкилом; С6-10 арилокси; или C6-14 арилом, 5-14-членным гетероарилом или 5-14-членным гетероциклилом, незамещенным или замещенным по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкила, галогена, и ди(С1-6 алкил)амино.
Конкретно, в формуле 3, приведенной выше,
R1 представляет собой водород;
Су представляет собой незамещенный или замещенный бензотиазолил, карбазолил, индазолил, хинолинил, индолил, пирролопиридинил, оксодигидробензотиазолил, оксодигидробензоксазолил или оксодигидробензоимидазолил; и
замещенный 5-14-членный гетероарил или 5-14-членный гетероциклил не замещен или замещен метилом, этилом, пропилом, бутилом, изопропилом, изобутилом, метокси, этокси, фтором, хлором, бромом, трифторэтилом, дифторметилом, трифторметокси, диметилкарбамоилом, фенокси или фенилом, пиридинилом, фуранилом или тиофенилом, не замещенным или замещенным по меньшей мере одним, выбранным из группы, состоящей из метила, фтора и диметиламино.
Например, соединение по настоящему изобретению может быть представлено формулой 4 ниже:
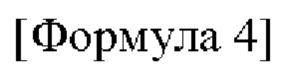
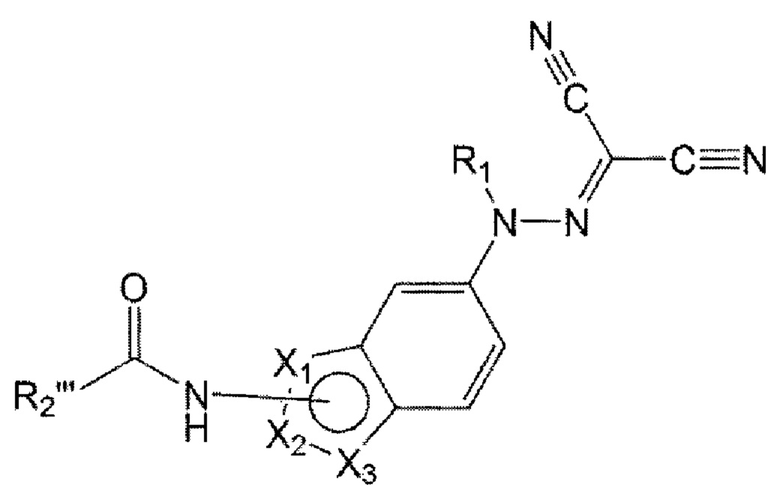
в формуле 4, приведенной выше,
от X1 до Х3 отличаются друг от друга, каждый выбран из С, N, О и S, и каждый связан с карбоксамидогруппой через С-сайт;
R1 представляет собой водород или C1-6 алкил; и
R2''' представляет собой C1-6 алкил или арил, или гетероарил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкокси, гидрокси, циано и С1-6 алкила.
Конкретно, в формуле 4, приведенной выше,
от X1 до Х3 представляют собой последовательно N, С и S или О, N и С и связаны с карбоксамидогруппой через С-сайт;
R1 представляет собой водород; и
R2''' представляет собой метил или фенил, пиридинил, пиразинил или пиримидинил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из метокси, гидрокси, циано и метила.
Например, соединение по настоящему изобретению может быть представлено формулой 5 ниже:
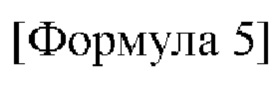
в формуле 5, приведенной выше,
R1 представляет собой водород или C1-6 алкил;
R2"" представляет собой 5-14-членный гетероарил или 5-14-членный гетероциклил, незамещенный или замещенный С1-6 алкилом; и
от R3 до R5 представляют собой i) все атомы водорода, ii) все образуют тройную связь вместе с одним N, iii) R3 и R4 образуют двойную связь вместе с NH, О или S с образованием имина, оксо или тиоксо, соответственно, и R5 представляет собой C1-6 алкокси, амино, С1-6 алкиламино, ди(С1-6 алкил)амино или 5-14-членный гетероциклил, или iv) от R3 до R5 связаны с гетероатомом О, N или S одинарной или двойной связью и содержат углерод и гетероатом, связанный с ним, с образованием незамещенного или замещенного 5-14-членного гетероарила или гетероциклила.
Конкретно, в формуле 5, приведенной выше,
R1 представляет собой водород;
R2"" представляет собой пиримидинил или оксотетрагидропиридазинил, незамещенный или замещенный по меньшей мере одним заместителем, выбранным из группы, состоящей из метила, этила, пропила, бутила, изопропила, изобутила и дифторметила;
от R3 до R5 образуют -ON, -СН3, -CO2Me, -CO2Et, -CONH2, -CSNH2, 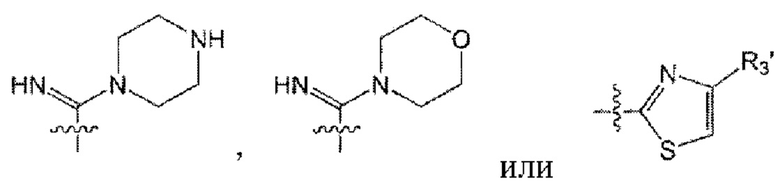 (здесь R3' представляет собой метил) вместе со связанным с ним С.
(здесь R3' представляет собой метил) вместе со связанным с ним С.
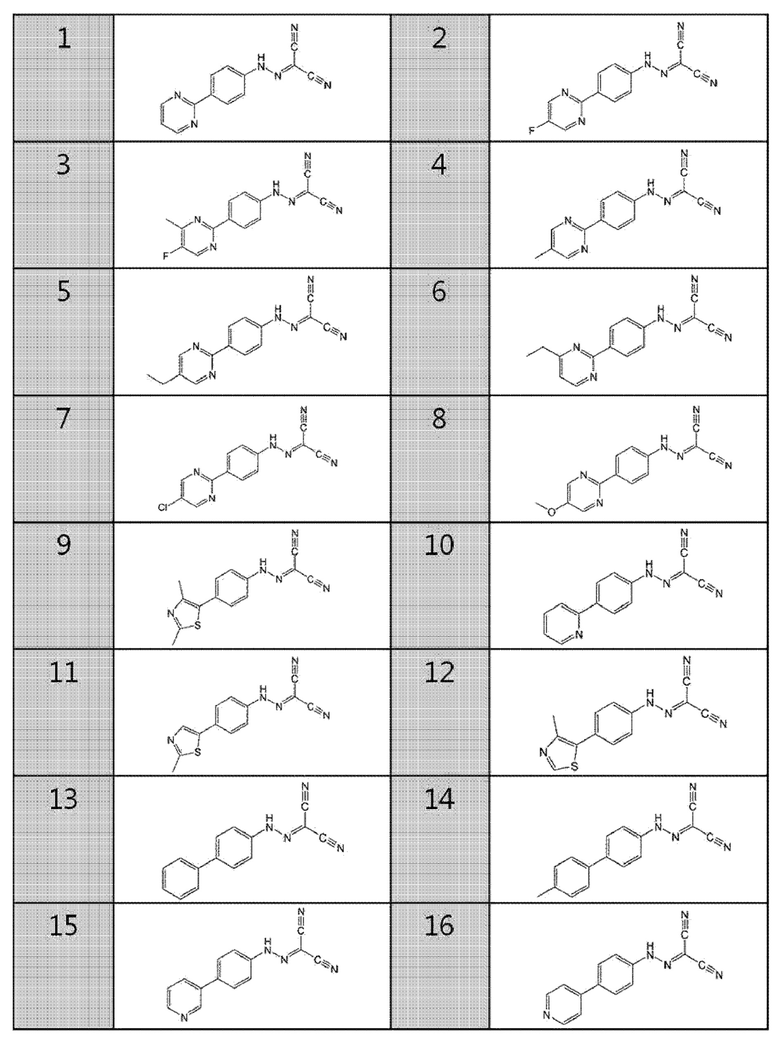
Более конкретно, соединение может представлять собой
1. (4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
2. (4-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
3. (4-(5-фтор-4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
4. (4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
5. (4-(5-этилпиримидин-2-ил)фенил))карбоногидразоноилдицианид,
6. (4-(4-этилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
7. (4-(5-хлорпиримидин-2-ил)карбоногидразоноилдицианид,
8. (4-(5-метоксипиримидин-2-ил)-фенил)карбоногидразоноилдицианид,
9. (4-(2,4-диметилтиазол-5-ил)фенил)карбоногидразоноилдицианид,
10. (4-(пиридин-2-ил)фенил)карбоногидразоноилдицианид,
11. (4-(2-метилтиазол-5-ил)фенил)карбоногидразоноилдицианид,
12. (4-(4-метилтиазол-5-ил)фенил)карбоногидразоноилдицианид,
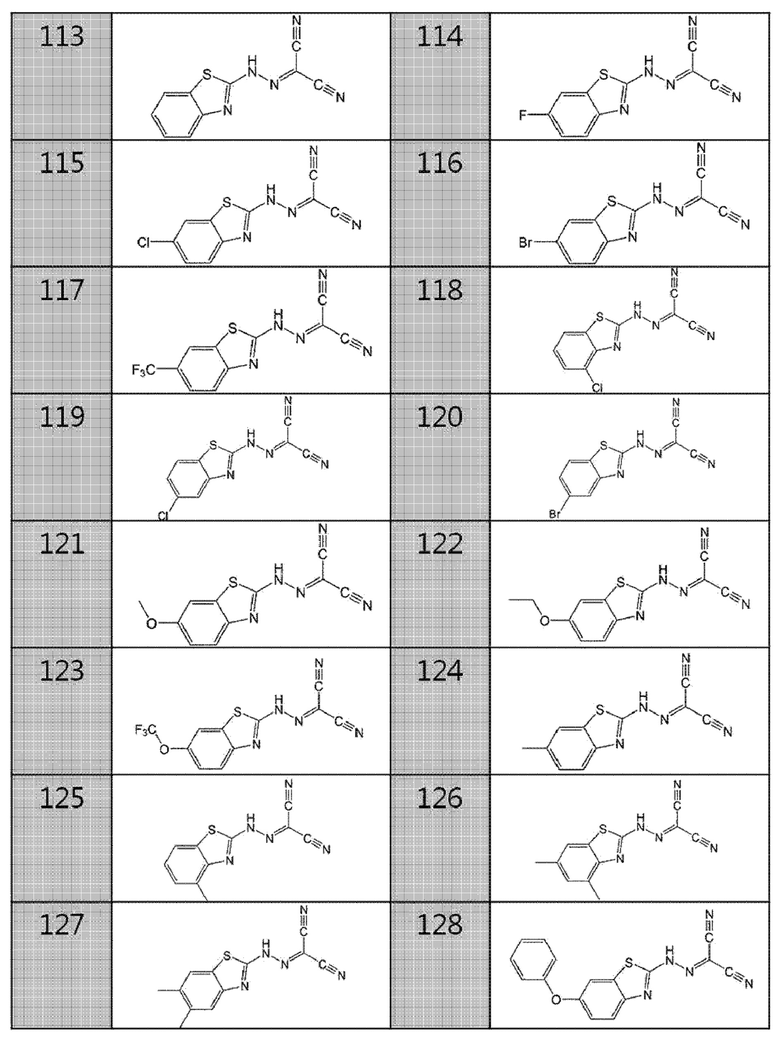
13. бифенил-4-илкарбоногидразоноилдицианид,
14. (4'-метилбифенил-4-ил)карбоногидразоноилдицианид,
15. (4-(пиридин-3-ил)фенил)карбоногидразоноилдицианид,
16. (4-(пиридин-4-ил)фенил)карбоногидразоноилдицианид,
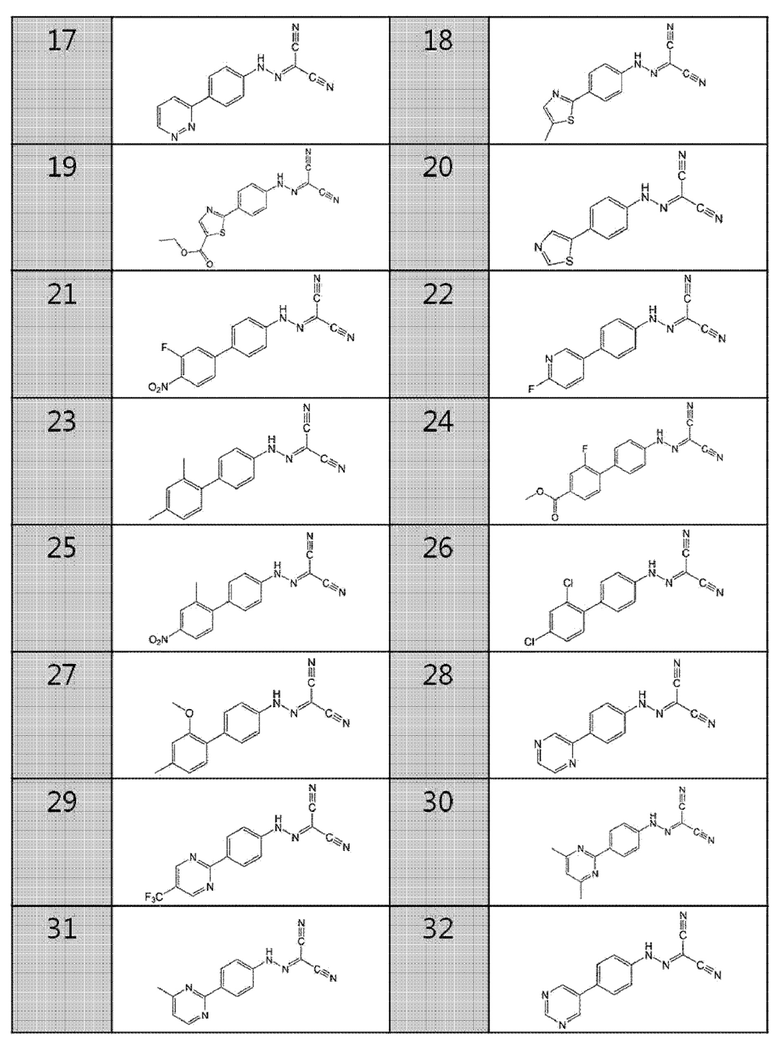
17. (4-(пиридазин-3-ил)фенил)карбоногидразоноилдицианид,
18. (4-(5-метилтиазол-2-ил)фенил)карбоногидразоноилдицианид,
19. этил-2-(4-(2-(дицианометилен)гидразинил)фенил)тиазол-5-карбоксилат,
20. (4-(тиазол-5-ил)фенил)карбоногидразоноилдицианид,
21. (3'-фтор-4'-нитробифенил-4-ил)карбоногидразоноилдицианид,
22. (4-(6-фторпиридин-3-ил)фенил)карбоногидразоноилдицианид,
23. (2',4'-диметилбифенил-4-ил)карбоногидразоноилдицианид,
24. метил-4'-(2-(дицианометилен)гидразинил)-2-фторбифенил-4-карбоксилат,
25. (2'-метил-4'-нитробифенил-4-ил)карбоногидразоноилдицианид,
26. (2',4'-дихлорбифенил-4-ил)карбоногидразоноилдицианид,
27. (2'-метокси-4'-метилбифенил-4-ил)карбоногидразоноилдицианид,
28. (4-(пиразин-2-ил)фенил)карбоногидразоноилдицианид,
29. (4-(5-(трифторметил)пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
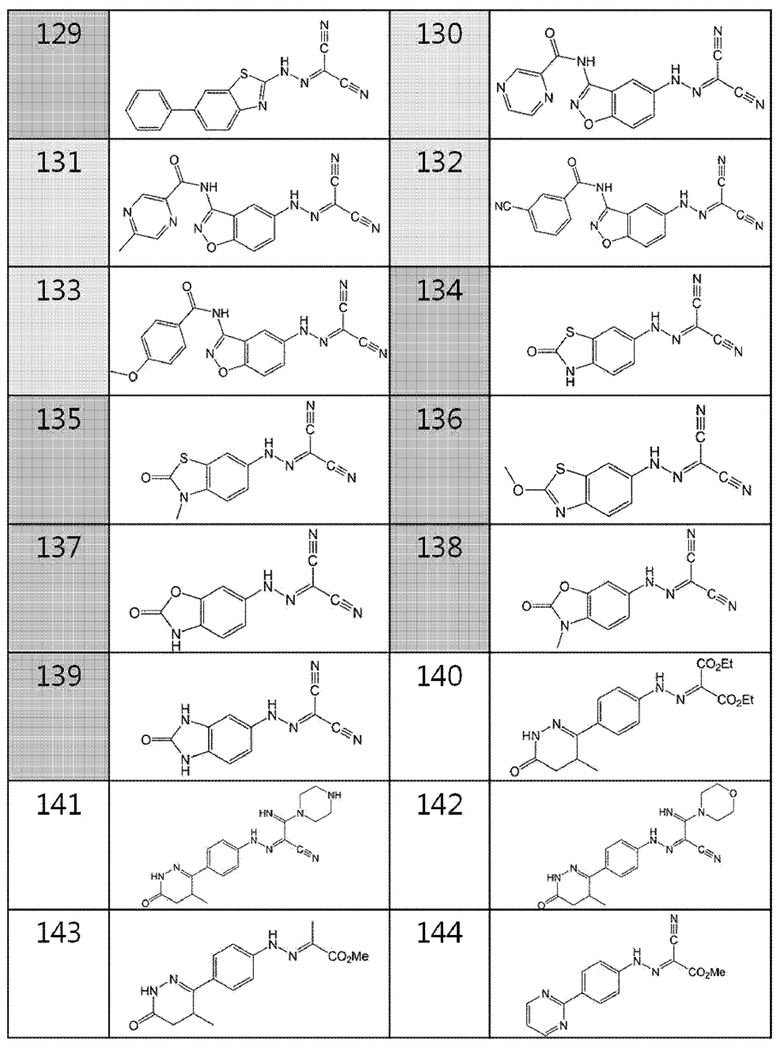
30. (4-(4,6-диметилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
31. (4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
32. (4-(пиримидин-5-ил)фенил)карбоногидразоноилдицианид,
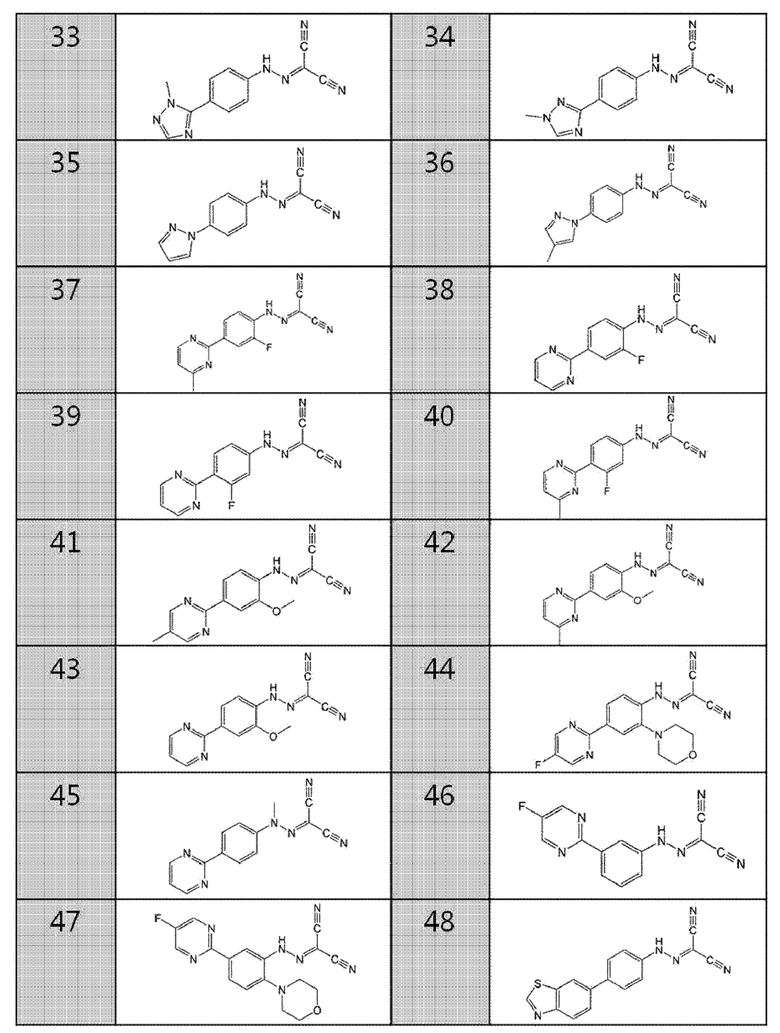
33. (4-(1-метил-1H-1,2,4-триазол-5-ил)фенил)карбоногидразоноилдицианид,
34. (4-(1-метил-1H-1,2,4-триазол-3-ил)фенил)карбоногидразоноилдицианид,
35. (4-(1H-пиразол-1-ил)фенил) карбоногидразоноилдицианид,
36. (4-(4-метил-1H-пиразол-1-ил)фенил)карбоногидразоноилдицианид,
37. (2-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
38. (2-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
39. (3-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
40. (3-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
41. (2-метокси-4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
42. (2-метокси-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
43. (2-метокси-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
44. (4-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианид,
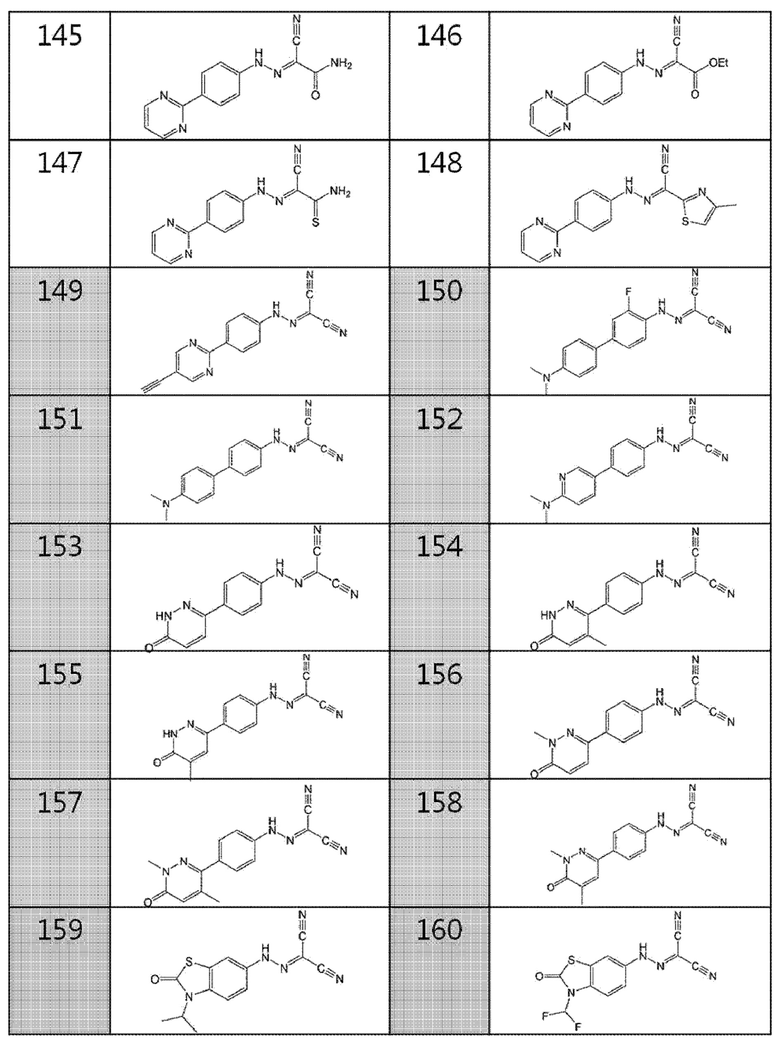
45. метил-(4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианид,
46. (3-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианид,
47. (5-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианид,
48. (4-(бензо[d]тиазол-6-ил)фенил)карбоногидразоноилдицианид,
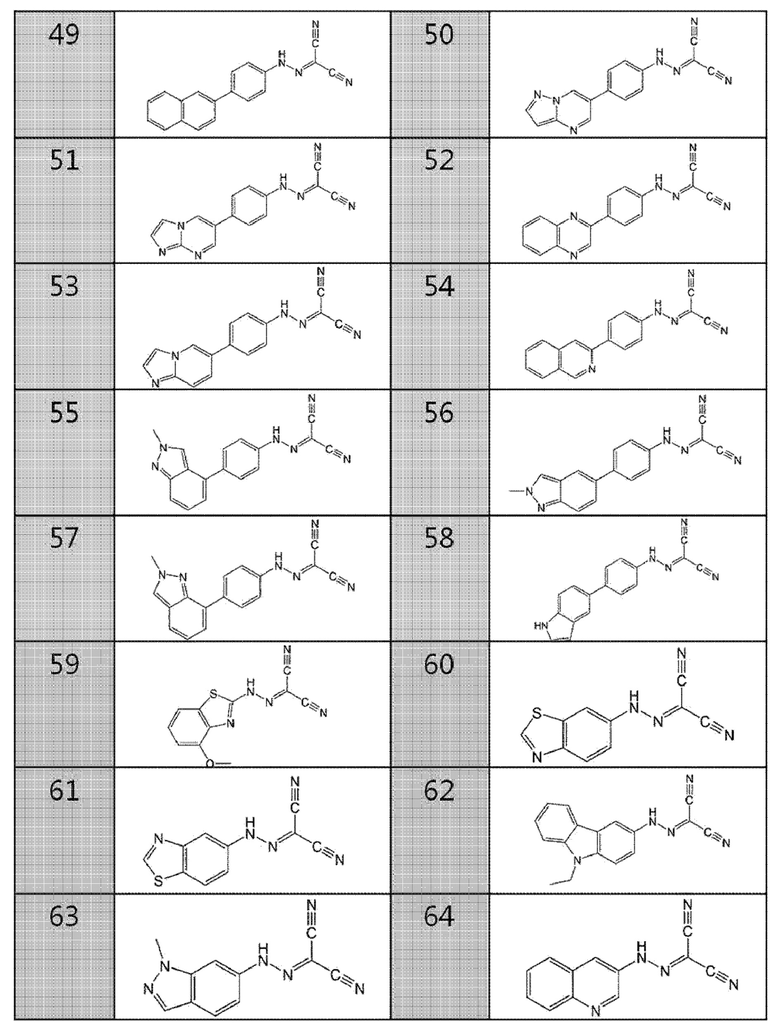
49. (4-(нафталин-2-ил)фенил)карбоногидразоноилдицианид,
50. (4-(пиразоло[1,5-а]пиримидин-6-ил)фенил)карбоногидразоноилдицианид,
51. (4-(имидазо[1,2-а]пиримидин-6-ил)фенил)карбоногидразоноилдицианид,
52. (4-(хиноксалин-2-ил)фенил)карбоногидразоноилдицианид,
53. (4-(имидазо[1,2-а]пиридин-6-ил)фенил)карбоногидразоноилдицианид,
54. (4-(изохинолин-3-ил)фенил)карбоногидразоноилдицианид,
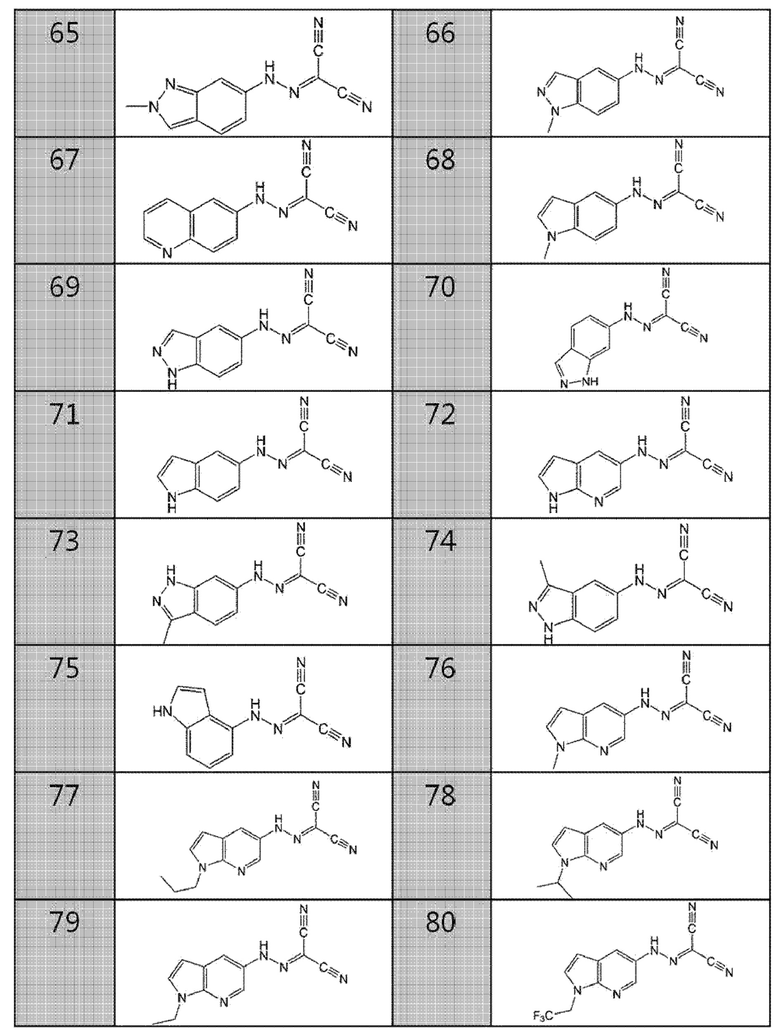
55. (4-(2-метил-2H-индазол-4-ил)фенил)карбоногидразоноилдицианид,
56. (4-(2-метил-2H-индазол-5-ил)фенил)карбоногидразоноилдицианид,
57. (4-(2-метил-2H-индазол-7-ил)фенил)карбоногидразоноилдицианид,
58. (4-(1H-индол-5-ил)фенил)карбоногидразоноилдицианид,
59. (4-метоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
60. бензо[d]тиазол-6-илкарбоногидразоноилдицианид,
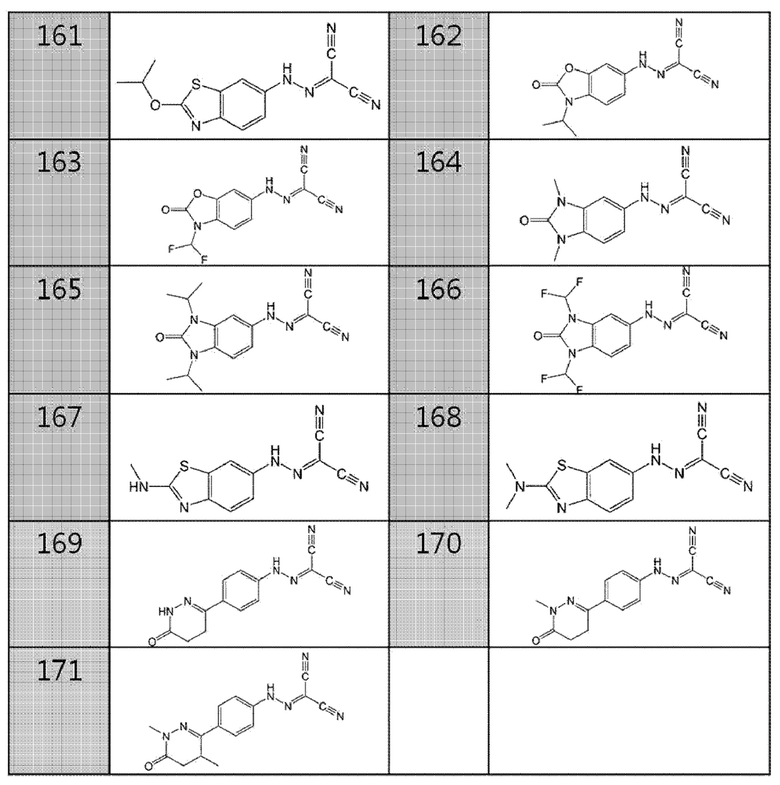
61. бензо[d]тиазол-5-илкарбоногидразоноилдицианид,
62. (9-этил-9H-карбазол-3-ил)карбоногидразоноилдицианид,
63. (1-метил-1H-индазол-6-ил)карбоногидразоноилдицианид,
64. хинолин-3-илкарбоногидразоноилдицианид,
65. (2-метил-2H-индазол-6-ил)карбоногидразоноилдицианид,
66. (1-метил-1H-индазол-5-ил)карбоногидразоноилдицианид,
67. хинолин-6-илкарбоногидразоноилдицианид,
68. (1-метил-1H-индол-5-ил)карбоногидразоноилдицианид,
69. (1H-индазол-5-ил)карбоногидразоноилдицианид,
70. (1H-индазол-6-ил)карбоногидразоноилдицианид,
71. (1H-индол-5-ил)карбоногидразоноилдицианид,
72. (1Н-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
73. (3-метил-1H-индазол-6-ил)карбоногидразоноилдицианид,
74. (3-метил-1H-индазол-5-ил)карбоногидразоноилдицианид,
75. (1H-индол-4-ил)карбоногидразоноилдицианид,
76. (1-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
77. (1-пропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
78. (1-изопропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
79. (1-этил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
80. (1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
81. (2-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
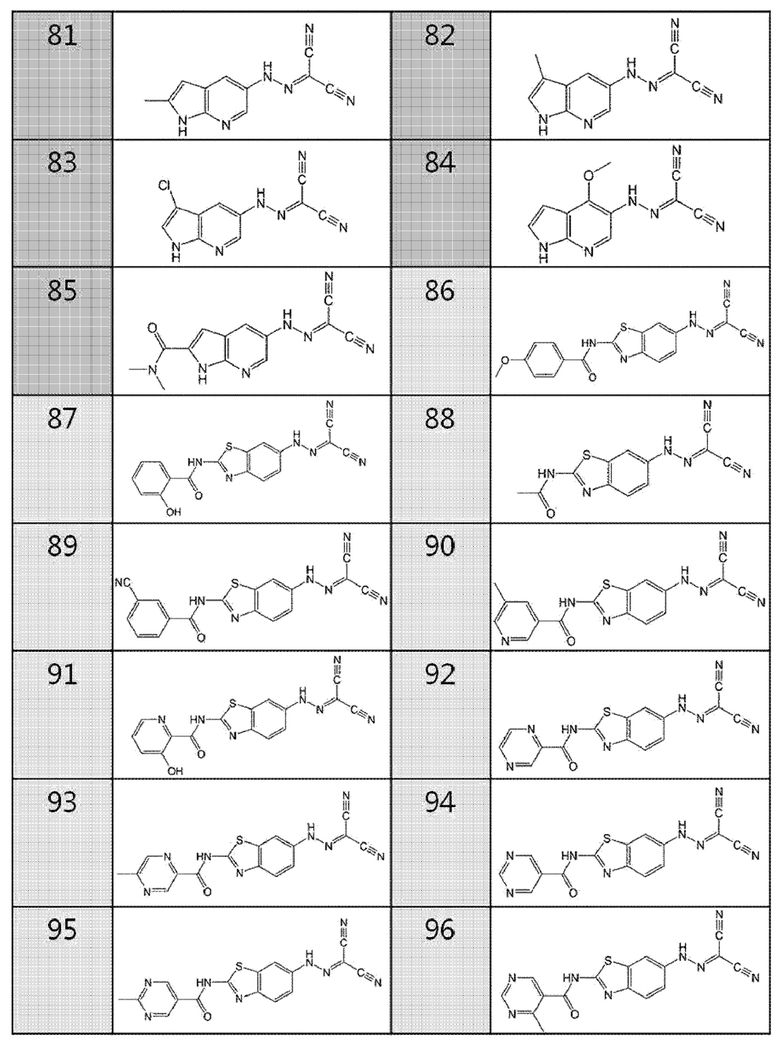
82. (3-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
83. (3-хлор-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
84. (4-метокси-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
85. (2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианид,
86. (2-(4-метоксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
87. (2-(2-гидроксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
88. (2-ацетамидобензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
89. (2-(3-цианобензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
90. (2-(5-метилникотинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
91. (2-(3-гидроксипиколинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
92. (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
93. (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
94. (2-(пиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
95. (2-(2-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
96. (2-(4-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
97. (2-(3-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
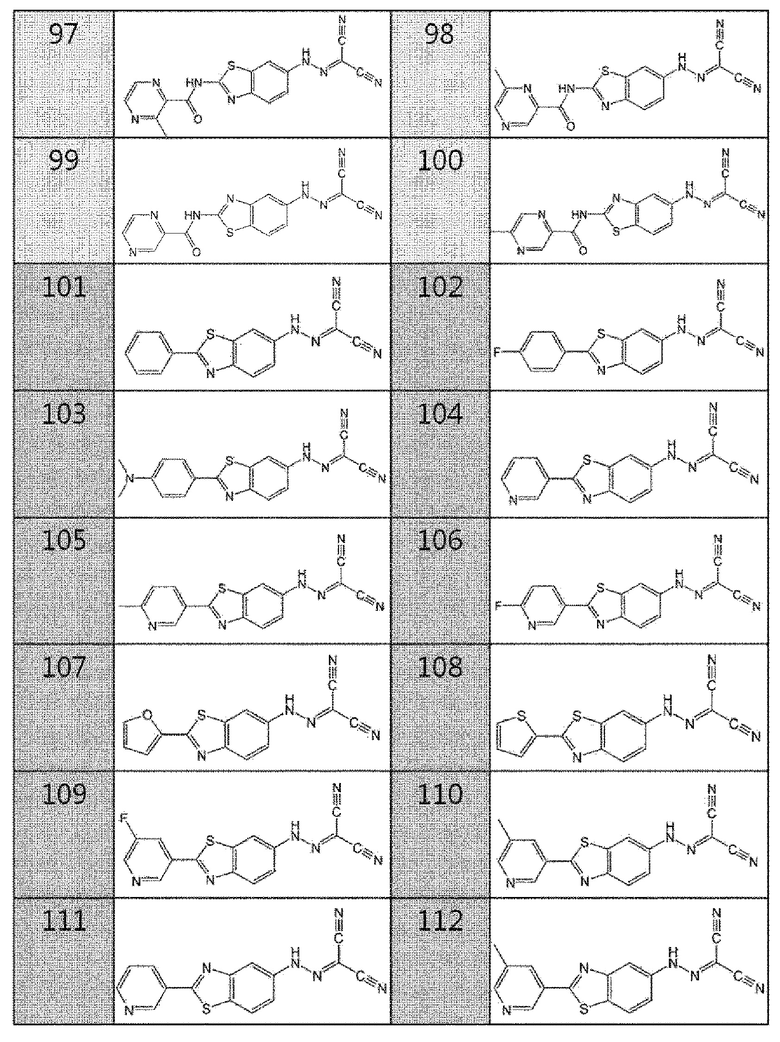
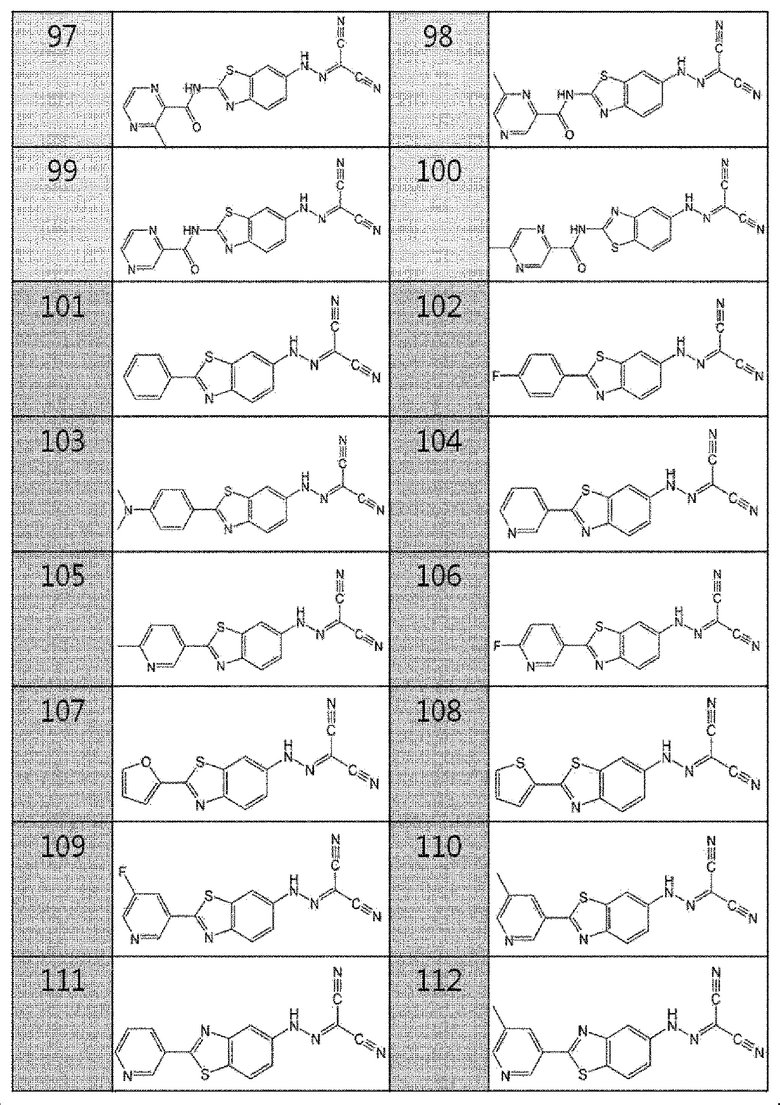
98. (2-(6-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
99. (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
100. (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
101. (2-фенилбензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
102. (2-(4-фторфенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
103. (2-(4-(диметиламино)фенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
104. (2-(пиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
105. (2-(6-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
106. (2-(6-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
107. (2-(фуран-2-ил)бензо [d]тиазол-6-ил)карбоногидразоноилдицианид,
108. (2-(тиофен-2-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
109. (2-(5-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
110. (2-(5-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
111. (2-(пиридин-3-ил)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
112. (2-(5-метилпиридин-3-ил)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
113. бензо[d]тиазол-2-илкарбоногидразоноилдицианид,
114. (6-фторбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
115. (6-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
116. (6-бромбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
117. (6-(трифторметил)бензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
118. (4-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
119. (5-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
120. (5-бромбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
121. (6-метоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
122. (6-этоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
123. (6-(трифторметокси)бензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
124. (6-метилбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
125. (4-метилбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
126. (4,6-диметилбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
127. (5,6-диметилбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
128. (6-феноксибензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
129. (6-фенилбензо[d]тиазол-2-ил)карбоногидразоноилдицианид,
130. (3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
131. (3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
132. (3-(3-цианобензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
133. (3-(4-метоксибензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
134. (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
135. (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
136. (2-метоксибензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
137. (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
138. (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
139. (2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
140. диэтил-2-(2-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)гидразоно)малонат,
141. 2-имино-N'-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)-2-(пиперазин-1-ил)ацетогидразоноилцианид,
142. 2-имино-N'-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)-2-морфолиноацетогидразоноилцианид,
143. метил-2-(2-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)гидразоно)пропаноат,
144. метил-2-циано-2-(2-(4-(пиримидин-2-ил)фенил)гидразоно)ацетат,
145. 2-амино-2-оксо-N'-(4-(пиримидин-2-ил)фенил)ацетогидразоноилцианид,
146. этил-2-циано-2-(2-(4-(пиримидин-2-ил)фенил)гидразоно)ацетат,
147. 2-амино-N'-(4-(пиримидин-2-ил)фенил)-2-тиоксоацетогидразоноил цианид,
148. 4-метил-N'-(4-(пиримидин-2-ил)фенил)тиазол-2-карбогидразоноилцианид,
149. (4-(5-этинилпиримидин-2-ил)фенил)карбогидразоноилдицианид,
150. (4'-(диметиламино)-3-фторбифенил-4-ил)карбогидразоноилдицианид,
151. (4'-(диметиламино)бифенил-4-ил)карбогидразоноилдицианид,
152. (4-(6-(диметиламино)пиридин-3-ил)фенил)карбогидразоноилдицианид,
153. (4-(6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
154. (4-(4-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
155. (4-(5-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
156. (4-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
157. (4-(1,4-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
158. (4-(1,5-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
159. (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбогидразоноилдицианид,
160. (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбогидразоноилдицианид,
161. (2-изопропоксибензо[d]тиазол-6-ил)карбогидразоноилдицианид,
162. (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбо гидразо ноилдицианид,
163. (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбогидразоноилдицианид,
164. (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианид,
165. (1,3-диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианид,
166. (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианид,
167. (2-(метиламино)бензо[d]тиазол-6-ил)карбогидразоноилдицианид,
168. (2-(диметиламино)бензо[d]тиазол-6-ил)карбогидразоноилдицианид,
169. (4-(6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианид,
170. (4-(1-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианид, или
171. (4-(1,4-диметил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианид.
Кроме того, данные соединения могут представлять собой соединения, представленные следующими формулами.
Между тем соединение по настоящему изобретению может существовать в форме фармацевтически приемлемой соли. В качестве соли можно применять соль кислоты, образованную фармацевтически приемлемой свободной формой кислоты. В контексте данного документа термин «фармацевтически приемлемая соль» относится к любой органической или неорганической аддитивной соли соединения, представленного формулой 1, которая относительно нетоксична и безвредна для пациентов, и побочные эффекты, вызванные данной солью, не влияют на благоприятные эффекты данного соединения.
Соль присоединения кислоты получают обычным способом, например, растворением соединения в избыточном количестве водного раствора кислоты и осаждением этого раствора с использованием смешивающегося с водой органического растворителя, такого как метанол, этанол, ацетон или ацетонитрил. Одни и те же молярные количества соединения и кислоты или спирта (например, моноэтилового эфира гликоля) в воде нагревают, а затем смесь можно упарить и высушить, или осажденную соль можно отфильтровать под вакуумом.
В этом случае в качестве свободной формы кислоты можно использовать органическую кислоту или неорганическую кислоту. В качестве неорганической кислоты можно использовать хлористоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, винную кислоту или тому подобное. В качестве органической кислоты может быть использована метансульфоновая кислота, n-толуолсульфоновая кислота, уксусная кислота, трифгоруксусная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, лимонная кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, углекислота, ванилиновая кислота, гидроиодная кислота или тому подобное. Однако настоящее изобретение этим не ограничивается.
Кроме того, фармацевтически приемлемая соль металла может быть получена с использованием основания. Соль щелочного металла или соль щелочноземельного металла получают растворением соединения в избыточном количестве раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, фильтрованием нерастворимой соли соединения, а затем выпариванием и сушкой фильтрата. В этом случае для фармацевтического применения подходит получение соли натрия, калия или кальция в качестве соли металла, но настоящее изобретение этим не ограничивается. Кроме того, соответствующая соль серебра может быть получена путем введения в реакцию соли щелочного или щелочноземельного металла с подходящей солью серебра (например, нитратом серебра).
Фармацевтически приемлемые соли соединений по настоящему изобретению включают в себя соли кислотных или основных групп, которые могут присутствовать в соединениях формул 15, если не указано иное. Например, фармацевтически приемлемые соли могут включать натриевые, кальциевые и калиевые соли гидрокси-групп, а другие фармацевтически приемлемые соли аминогрупп могут включать в себя гидробромид, сульфат, гидросульфат, фосфат, гидрофосфат, дигидрофосфат, ацетат, сукцинат, цитрат, тартрат, лактат, манделат, метансульфонат (мезилат) и n-толуолсульфонат (тозилат). Данные фармацевтически приемлемые соли могут быть получены способами получения солей, известными в данной области.
В качестве солей соединений формул 15 настоящего изобретения можно использовать любую соль в виде фармацевтически приемлемой соли без ограничения при условии, что она проявляет фармакологическую активность, эквивалентную соединению формулы 1, например, она ингибирует агрегацию и/или гиперфосфорилирование тау-белка.
Кроме того, соединения, представленные формулами 1-5 согласно настоящему изобретению, включают, без ограничения, их фармацевтически приемлемые соли, а также сольваты, такие как возможные гидраты, которые могут быть получены из них, и все возможные стереоизомеры. Сольваты и стереоизомеры соединений, представленных формулами 1-5, могут быть получены из соединений, представленных формулами 1-5, с использованием метода, известного в данной области.
Более того, соединения, представленные формулами 15 согласно настоящему изобретению, могут быть получены в кристаллической или аморфной форме и могут быть необязательно гидратированы или сольватированы, если получены в кристаллической форме. В настоящем изобретении могут быть предложены соединения, содержащие разнообразные количества воды, а также стехиометрические гидраты соединений, представленных формулами 1-5. Сольваты соединений, представленных формулами 1-5 согласно настоящему изобретению, включают в себя как стехиометрические сольваты, так и нестехиометрические сольваты.
Во втором аспекте настоящего изобретения предложен способ получения соединения формулы 1, включающий стадию введения в реакцию нитрита натрия, аминопроизводной R2 (R2-NH2) и 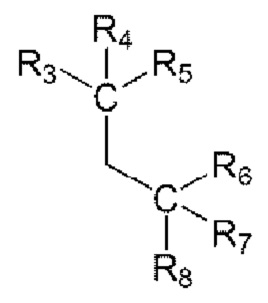 в присутствии кислоты с образованием иминосвязи. В способе получения по настоящему изобретению для используемых в нем реагентов коммерчески доступные соединения могут быть использованы в том виде, в каком они были куплены, или могут быть синтезированы путем точного проведения одной или более реакций, известных в данной области, или путем изменения некоторых условий реакций, известных в данной области. Например, соединения могут быть синтезированы путем последовательного выполнения одной или более реакций с учетом присутствия, вида и положения реакционноспособных функциональных групп и/или гетероэлементов, существующих в структуре скелета, но настоящее изобретение этим не ограничивается. Этот процесс может быть аналогичным образом применен к следующим аспектам с третьего по шестой.
в присутствии кислоты с образованием иминосвязи. В способе получения по настоящему изобретению для используемых в нем реагентов коммерчески доступные соединения могут быть использованы в том виде, в каком они были куплены, или могут быть синтезированы путем точного проведения одной или более реакций, известных в данной области, или путем изменения некоторых условий реакций, известных в данной области. Например, соединения могут быть синтезированы путем последовательного выполнения одной или более реакций с учетом присутствия, вида и положения реакционноспособных функциональных групп и/или гетероэлементов, существующих в структуре скелета, но настоящее изобретение этим не ограничивается. Этот процесс может быть аналогичным образом применен к следующим аспектам с третьего по шестой.
В третьем аспекте настоящего изобретения предложен способ получения соединения формулы 2, включающий стадии:
a1) получение производного анилина 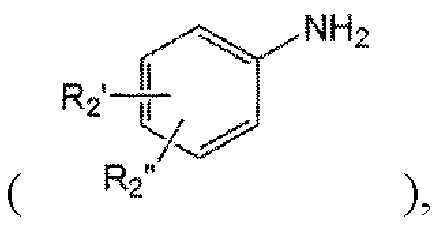 замещенного арилом или гетероарил ом, незамещенным или замещенным R2' или R2"; и
замещенного арилом или гетероарил ом, незамещенным или замещенным R2' или R2"; и
а2) введение в реакцию с нитритом натрия, производным анилина 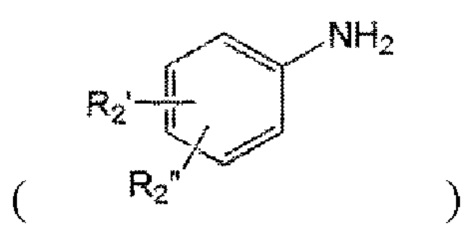 и малононитрилом в присутствии кислоты с образованием иминосвязи.
и малононитрилом в присутствии кислоты с образованием иминосвязи.
Например, когда R1 в приведенной выше формуле представляет собой C1-6 алкил, способ может дополнительно включать стадию выполнения алкилирования реакцией с галогенированным алканом в присутствии трет-бутоксида калия или гидрида натрия в органическом растворителе после стадии образования иминосвязи. Однако настоящее изобретение не ограничивается этим, и реакция алкилирования, известная в данной области, может быть соответствующим образом изменена и проведена с учетом присутствия или отсутствия других реакционноспособных заместителей или гетероэлементов.
В частности, производное анилина, замещенное арилом или гетероарилом, незамещенным или замещенным R2' или R2", может быть синтезировано путем введения в реакцию пинаколового эфира бороновой кислоты, производного анилина, незамещенного или замещенного R2' с галогенированным арильным или гетероарильным производным, незамещенным или замещенным R2" с использованием катализатора Pd (II) в присутствии основания. Однако настоящее изобретение не ограничивается этим, и коммерчески доступные соединения можно использовать в том виде, в котором они были куплены, или их можно синтезировать с помощью метода, известного в данной области.
Кроме того, учитывая потенциальную реакционную способность заместителей и/или гетероэлементов, присутствующих в скелете соединения, которое должно быть синтезировано в реакции, производное анилина, замещенное арилом или гетероарилом, незамещенным или замещенным R2' или R2", может быть получено восстановлением нитрофенильного производного, содержащего нитрогруппу вместо аминогруппы в том же скелете, с использованием палладиевого катализатора, активированного углем в атмосфере газообразного водорода..
В четвертом аспекте настоящего изобретения предложен способ получения соединения формулы 3, включающий стадию введения в реакцию нитрита натрия, 5-14-членного гетероарильного или 5-14-членного гетероциклильного производного, содержащего аминогруппу, и малононитрила в присутствии кислоты с образованием иминосвязи.
Например, когда 5-14-членный гетероарил или 5-14-членный гетероциклил содержит связанный с ним азот, отличный от аминогруппы, он содержит защитную группу в сайте азота. Способ может дополнительно включать стадию удаления защитной группы после стадии образования иминосвязи.
Кроме того, с учетом реакционной способности функциональных групп и/или гетероэлементов, присутствующих в скелете, 5-14-членный гетероарил или 5-14-членный гетероциклил можно получить восстановлением нитрофенильного производного, содержащего нитрогруппу вместо аминогруппы в том же скелете, но настоящее изобретение этим не ограничивается.
В пятом аспекте настоящего изобретения предложен способ получения соединения формулы 4, включающий стадии:
введение в реакцию карбоновой кислоты R2''' или ее ангидрида 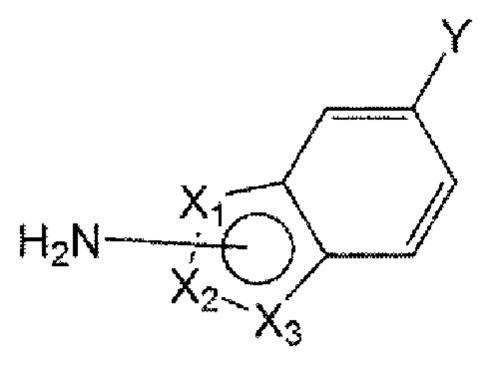 (здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
(здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
введение в реакцию нитрита натрия, гетероциклического производного, содержащего R2''', связанного с карбоксамидогруппой, полученного на предыдущей стадии, и малононитрилом в присутствии кислоты с образованием иминосвязи.
В данном случае, когда Y представляет собой защищенную аминогруппу или нитрогруппу, причем данный способ может дополнительно включать стадию превращения защищенной аминогруппы или нитрогруппы в аминогруппу перед стадией образования иминогруппы.
В шестом аспекте настоящего изобретения предложен способ получения соединения формулы 5, включающий стадии:
введение в реакцию нитрита натрия, аминопроизводной R2 (R2-NH2) и 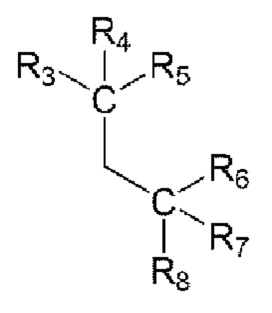 с образованием иминосвязи; и
с образованием иминосвязи; и
доведение рН реакционного раствора до 5-7 с помощью основания.
В седьмом аспекте настоящего изобретения предложена композиция для ингибирования агрегации тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В восьмом аспекте настоящего изобретения предложена композиция для ингибирования гиперфосфорилирования тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В девятом аспекте настоящего изобретения предложена фармацевтическая композиция для предотвращения или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка, содержащая соединение по настоящему изобретению в качестве активного ингредиента.
В конкретных вариантах осуществления настоящего изобретения в общей сложности 171 соединение, пронумерованное от 1 до 171 и представленное формулой 1, было вновь синтезировано, и были подтверждены эффекты ингибирования агрегации и гиперфосфорилирования его тау-белка. Более того, чтобы подтвердить возможность использования в качестве фармацевтической композиции, было подтверждено, что эти соединения не проявляют токсичности для клеток.
В контексте данного документа термин «профилактика» относится к любому действию, которое ингибирует или задерживает возникновение, распространение и рецидив заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка при введении фармацевтической композиции по настоящему изобретению, и термин «лечение» относится к любому действию, при котором симптомы заболевания улучшаются или благоприятно изменяются путем введения фармацевтической композиции по настоящему изобретению.
Как описано выше, поскольку соединение по настоящему изобретению не только ингибирует агрегацию или гиперфосфорилирование тау-белка, но также не проявляет токсичности для клеток, фармацевтическая композиция, содержащая это соединение в качестве активного ингредиента, может использоваться для профилактики или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка. Заболевание, вызванное агрегацией или гиперфосфорилированием тау-белка, к которому может применяться фармацевтическая композиция по настоящему изобретению, может представлять собой болезнь Альцгеймера, болезнь Паркинсона, сосудистую деменцию, острый инсульт, травму, цереброваскулярное заболевание, травму головного мозга, травму спинного мозга, периферическую невропатию, ретинопатию, глаукому или таупатию. Неограничивающие примеры таупатии могут включать в себя хроническую травматическую энцефалопатию (СТЕ), первичную возрастную таупатию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию, болезнь Пика, болезнь аргирофильного зерна (AGD), лобно-височную деменцию (FTD), паркинсонизм, связанный с хромосомой 17, литико-бодиговая болезнь (комплекс паркинсонизма-деменции Гуама), ганглио глиома, ганглиоцитома, менингиоангиоматоз, постэнцефалитный паркинсонизм, подострый склерозирующий панэнцефалит, свинцовая энцефалопатия, туберозный склероз, связанная с пантотенат-киназой нейродегенерация, липофусциноз и травматическое повреждение мозга.
Например, композиция по настоящему изобретению может дополнительно содержать фармацевтически приемлемый носитель, разбавитель или эксципиент, может быть составлена и применяться в разнообразных формах, таких как пероральные композиции, такие как порошки, гранулы, таблетки, капсулы, суспензии, эмульсии, сиропы, аэрозоли и препараты для инъекций стерильных растворов для инъекций в соответствии с общим способом для каждой цели применения, и их можно вводить перорально или можно вводить разнообразными путями, включая внутривенное, внутрибрюшинное, подкожное, ректальное и местное введение. Примеры подходящего носителя, эксципиента или разбавителя, включенных в данную композицию, могут включать в себя лактозу, декстрозу, сахарозу, сорбит, маннит, ксилит, эритрит, мальтит, крахмал, каучук камеди, альгинат, желатин, фосфат кальция, силикат кальция, целлюлозу, метилцеллюлозу, микрокристаллическую целлюлозу, поливинилпирролидон, воду, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральное масло. Композиция по настоящему изобретению может дополнительно содержать наполнитель, антиагрегантный агент, смазывающий агент, смачивающий агент, ароматизатор, эмульгирующий агент, консервант и тому подобное.
Твердые препараты для перорального введения включают в себя таблетки, пилюли, порошки, гранулы, капсулы и т.п., и такой твердый препарат получают путем смешивания одного или более эксципиентов, таких как крахмал, карбонат кальция, сахароза, лактоза и желатин, с композицией. Между тем помимо простого эксципиента можно использовать смазывающее средство, такое как стеарат магния или тальк.
В качестве жидкого препарата для перорального применения можно привести суспензию, агент, препятствующий растворению, эмульсию, сироп и т.п., а жидкий препарат для перорального применения может содержать разнообразные эксципиенты, такие как смачивающий агент, подслащивающий агент, ароматизатор и консервант в дополнение к воде и жидкому парафину, которые обычно используют в качестве простого разбавителя.
Препараты для парентерального введения включают водный растворитель, неводный растворитель, суспендирующий агент, эмульгирующий агент, лиофилизированный препарат и суппозиторий, которые стерилизуют.
В качестве неводного растворителя или суспендирующего агента можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, сложный эфир для инъекций, такой как этилолеат, и т.п. В качестве основы суппозитория можно использовать витепсол, макрогол, твин 61, масло какао, лауриновое масло, глицерогелатин или т.п. Между тем инъекционные препараты могут содержать обычные добавки, такие как солюбилизирующий агент, изотонический агент, суспендирующий агент, эмульгирующий агент, стабилизирующий агент и консервант.
Композиция может быть приготовлена обычным способом смешивания, гранулирования или нанесения покрытия и может содержать активный ингредиент в количестве от около 0,1% масс. до 75% масс. предпочтительно от около 0,1% масс. до 50% масс. Стандартная лекарственная форма для млекопитающего весом от около 50 кг до 70 кг содержит от около 10 мг до 200 мг активного ингредиента.
В этом случае композицию по настоящему изобретению вводят в фармацевтически эффективном количестве. В контексте данного документа термин «фармацевтически эффективное количество» относится к количеству, достаточному для лечения заболевания с разумным соотношением польза/риск, применимым к медикаментозному лечению и не вызывающим побочных эффектов, и уровень эффективного количества может быть определен в зависимости от состояния пациента, состояния здоровья, типа заболевания, тяжести, активности лекарства, чувствительности к лекарству, способа введения, времени введения, пути введения, скорости выведения, периода лечения, факторов, включающих лекарства, используемые в комбинации или одновременно, и других факторов, хорошо известных в области медицины. Композицию по настоящему изобретению можно вводить в виде индивидуального терапевтического агента или вводить в комбинации с другими терапевтическими агентами, можно вводить последовательно или одновременно с обычным терапевтическим агентом и можно вводить в виде одной дозы или нескольких доз. Важно вводить количество, позволяющее получить максимальный эффект в минимальном количестве без побочных эффектов, принимая во внимание все вышеперечисленные факторы, которые могут быть легко определены специалистами в данной области.
Например, поскольку вводимое количество может увеличиваться или уменьшаться в зависимости от пути введения, тяжести заболевания, пола, веса, возраста и т.п., вводимое количество никоим образом не ограничивает объем настоящего изобретения.
Предпочтительное вводимое количество соединения по настоящему изобретению варьируется в зависимости от состояния и веса пациента, степени заболевания, формы лекарственного средства, а также пути и продолжительности введения, но может быть соответствующим образом выбрано специалистами в данной области техники. Однако для желаемого эффекта соединение по настоящему изобретению можно вводить в количестве от 0,0001 мг/кг до 100 мг/кг (вес тела), предпочтительно от 0,001 мг/кг до 100 мг/кг (вес тела) в день. Соединение можно вводить один раз в день или можно разделить и вводить пероральным или парентеральным путем.
В десятом аспекте настоящего изобретения предложен способ предотвращения или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка, причем данный способ включает этап введения фармацевтической композиции по настоящему изобретению субъекту, нуждающемуся в этом.
В контексте данного документа термин «субъект» относится к любому животному, включая обезьян, коров, лошадей, овец, свиней, кур, индеек, перепелов, кошек, собак, мышей, кроликов и морских свинок в дополнение к людям, у которых развились или могут развиваться заболевания, вызванные агрегацией или гиперфосфорилированием тау-белка. Заболевания можно эффективно предотвращать или лечить путем введения субъекту фармацевтической композиции по настоящему изобретению. Кроме того, поскольку фармацевтическая композиция по настоящему изобретению проявляет терапевтический эффект за счет ингибирования агрегации или гиперфосфорилирования тау-белка, синергетический эффект может проявляться при введении этой композиции в комбинации с обычным терапевтическим агентом.
В контексте данного документа термин «введение» относится к введению заранее определенного вещества пациенту любым подходящим способом, и путь введения композиции по настоящему изобретению можно вводить любым общим путем, если он может достигать тканей-мишеней. Композицию можно вводить посредством внутрибрюшинного введения, внутривенного введения, внутримышечного введения, подкожного введения, внутрикожного введения, перорального введения, местного введения, интраназального введения, внутрилегочного введения или ректального введения, но настоящее изобретение не ограничивается этим. Между тем фармацевтическая композиция по настоящему изобретению могут вводить с помощью любого устройства, способного перемещать активное вещество к клетке-мишени. Предпочтительные введения и составы включают лекарства для внутривенной инъекции, лекарства для подкожной инъекции, лекарства для внутрикожной инъекции, лекарства для внутримышечной инъекции и лекарства для капельной инъекции. Лекарственные средства для инъекций могут быть приготовлены с использованием водного растворителя, такого как физиологический раствор или раствор Рингера, или неводного растворителя, такого как растительное масло, сложный эфир высших жирных кислот (например, этилолеат) или спирт (например, этанол, бензиловый спирт, пропиленгликоль или глицерин) и может содержать фармацевтический носитель, такой как стабилизирующий агент для предотвращения денатурирования (например, аскорбиновая кислота, гидросульфит натрия, пиросульфит натрия, ВНА, токоферол или ЭДТА), эмульгирующий агент агент, буферный агент для контроля рН или консервант для подавления роста микроорганизмов (например, фенилртутьнитрат, тимеросал, хлорид бензалкония, фенол, крезол или бензиловый спирт).
Принцип изобретения
В дальнейшем настоящее изобретение будет описано более подробно со ссылкой на примеры и экспериментальные примеры. Однако эти примеры и экспериментальные примеры являются только иллюстрацией настоящего изобретения, и объем настоящего изобретения не ограничивается данными примерами и экспериментальными примерами.
Пример 1: Получение (4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 1)
Стадия 1-1: Получение 4-(пиримидин-2-ил)анилина
2-Бромпиримидин (175 мг, 1,10 ммоль) и пинаколовый эфир 4-аминофенилбороновой кислоты (4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин) (200 мг, 0,91 ммоль) растворяли в 15 мл раствора 1,4-диоксана в присутствии азота, а затем добавляли Pd(PPh3)2Cl2 (63 мг, 0,09 ммоль) и 1,0 М водный раствор карбоната натрия (3,64 мл, 3,64 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 80°С в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 115 мг (74%-й выход) указанного в заголовке соединения.
Стадия 1-2: Получение (4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида
4-(Пиримидин-2-ил)анилин (50 мг, 0,29 ммоль), полученный в соответствии со стадией 1-1, и нитрит натрия (24 мг, 0,35 ммоль) растворяли в этаноле (2,5 мл) в присутствии азота, а затем добавляли 1,0 М водный раствор соляной кислоты (0,73 мл, 0,73 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 0°С в течение 1 часа с образованием диазониевой соли. После образования диазониевой соли добавляли малононитрил (23 мг, 0,35 ммоль) с последующим перемешиванием при комнатной температуре в течение 5 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 57 мг (79%-й выход) указанного в заголовке соединения.
Пример 2: Получение (4-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 2)
Стадия 2-1: Получение 4-(5-фторпиримидин-2-ил)анилина
71 мг (41%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-фторпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 2-2: Получение (4-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианида
30 мг (43%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-фторпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 3: Получение (4-(5-фтор-4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 3)
Стадия 3-1: Получение 4-(5-фтор-4-метилпиримидин-2-ил)анилина
139 мг (75%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-фтор-4-метилпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 3-2: Получение (4-(5-фтор-4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
40 мг (29%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-фтор-4-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 4: Получение (4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 4)
Стадия 4-1: Получение 4-(5-метилпиримидин-2-ил)анилина
65 мг (39%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-метилпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 4-2: Получение (4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
35 мг (50%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 5: Получение 4-(5-этилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 5)
Стадия 5-1: Получение 4-(5-этилпиримидин-2-ил)анилина
169 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-этилпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 5-2: Получение (4-(5-этилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
35 мг (25%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-этилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 6: Получение (4-(4-этилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 6)
Стадия 6-1: Получение 4-(4-этилпиримидин-2-ил)анилина
169 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-4-этилпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 6-2: Получение (4-(4-этилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
80 мг (66%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(4-этилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 7: Получение (4-(5-хлорпиримидин-2-ил)карбоногидразоноилдицианида (Соединение 7)
Стадия 7-1: Получение 4-(5-хлорпиримидин-2-ил)анилина
67 мг (36%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5- хлорпиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 7-2: Получение 4-(5-хлорпиримидин-2-ил)фенил)карбоногидразоноилдицианида
30 мг (16%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-хлорпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 8: Получение (4-(5-метоксипиримидин-2-ил)-фенил)карбоногидразоноилдицианида (Соединение 8)
Стадия 8-1: Получение 4-(5-метоксипиримидин-2-ил)анилина
67 мг (36%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-метоксипиримидин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 8-2: Получение (4-(5-метоксипиримидин-2-ил)фенил)карбоногидразоноилдицианида
26 мг (30%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-метоксипиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 9: Получение (4-(2,4-диметилтиазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 9)
Стадия 9-1: Получение 4-(2,4-диметилтиазол-5-ил)анилина
48 мг (26%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бром-2,4-диметилтиазол (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 9-2: Получение (4-(2,4-диметилтиазол-5-ил)фенил)карбоногидразоноилдицианида
55 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(2,4-диметилтиазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 10: Получение (4-(пиридин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 10)
Стадия 10-1: Получение 4-(пиридин-2-ил)анилина
101 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бромпиридин (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 10-2: Получение (4-(пиридин-2-ил)фенил)карбоногидразоноилдицианида
18 мг (25%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиридин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 11: Получение (4-(2-метилтиазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 11)
Стадия 11-1: Получение 4-(2-метилтиазол-5-ил)анилина
112 мг (57%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бром-2-метилтиазол (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 11-2: Получение (4-(2-метилтиазол-5-ил)фенил)карбоногидразоноилдицианида
11 мг (16%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(2-метилтиазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 12: Получение (4-(4-метилтиазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 12)
Стадия 12-1: Получение 4-(4-метилтиазол-5-ил)анилина
133 мг (76%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бром-4-метилтиазол (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 12-2: Получение (4-(4-метилтиазол-5-ил)фенил)карбоногидразоноилдицианида
62 мг (89%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(4-метилтиазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 13: Получение бифенил-4-илкарбоногидразоноилдицианида (Соединение 13)
Стадия 13-1: Получение бифенил-4-амина
109 мг (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что бромбензол (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 13-2: Получение бифенил-4-илкарбоногидразоноилдицианида
29 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что бифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 14: Получение (4'-метилбифенил-4-ил)карбоногидразоноилдицианида (Соединение 14)
Стадия 14-1: Получение 4'-метилбифенил-4-амина
Указанное в заголовке соединение получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 1-бром-4-метилбензол (1,10 ммоль) использовали вместо 2-бромпиримидина.
Стадия 14-2: Получение (4'-метилбифенил-4-ил)карбоногидразоноилдицианида
48 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4'-метилбифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 15: Получение (4-(пиридин-3-ил)фенил)карбоногидразоноилдицианида (Соединение 15) Стадия 15-1: Получение 4-(пиридин-3-ил)анилина
367 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 3-бромпиридин (10,95 ммоль) использовали вместо 2-бромпиримидина.
Стадия 15-2: Получение (4-(пиридин-3-ил)фенил)карбоногидразоноилдицианида
533 мг (100%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиридин-3-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 16: Получение (4-(пиридин-4-ил)фенил)карбоногидразоноилдицианида (Соединение 16)
Стадия 16-1: Получение 4-(пиридин-4-ил)анилина
70 мг (32%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 4-бромпиридина гидрохлорид (1,53 ммоль) использовали вместо 2-бромпиримидина.
Стадия 16-2: Получение (4-(пиридин-4-ил)фенил)карбоногидразоноилдицианида
9 мг (9%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиридин-4-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 17: Получение (4-(пиридазин-3-ил)фенил)карбоногидразоноилдицианида (Соединение 17)
Стадия 17-1: Получение 4-(пиридазин-3-ил)анилина
76 мг (84%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 3-бромпиридазин (0,64 ммоль) использовали вместо 2-бромпиримидина.
Стадия 17-2: Получение (4-(пиридазин-3-ил)фенил)карбоногидразоноилдицианида
42 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиридазин-3-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 18: Получение (4-(5-метилтиазол-2-ил)фенил)карбоногидразоноилдицианида (Соединение 18)
Стадия 18-1: Получение 4-(5-метилтиазол-2-ил)анилин
88 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бром-5-метилтиазол (0,69 ммоль) использовали вместо 2-бромпиримидина.
Стадия 18-2: Получение (4-(5-метилтиазол-2-ил)фенил)карбоногидразоноилдицианида
30 мг (42%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-метилтиазол-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 19: Получение этил-2-(4-(2-(дицианометилен)гидразинил)фенил)тиазол-5-карбоксилата (Соединение 19)
Стадия 19-1: Получение этил-2-(4-аминофенил)тиазол-5-карбоксилата
12 мг (3%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что этил-2-бромтиазол-5-карбоксилат (1,61 ммоль) использовали вместо 2-бромпиримидина.
Стадия 19-2: Получение этил-2-(4-(2-(дицианометилен)гидразинил)фенил)тиазол-5-карбоксилата
12 мг (75%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что этил-2-(4-аминофенил)тиазол-5-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 20: Получение (4-(тиазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 20)
Стадия 20-1: Получение 4-(тиазол-5-ил)анилина
137 мг (85%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бромтиазол (0,91 ммоль) использовали вместо 2-бромпиримидина.
Стадия 20-2: Получение (4-(тиазол-5-ил)фенил)карбоногидразоноилдицианида
60 мг (83%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(тиазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 21: Получение (3'-фтор-4'-нитробифенил-4-ил)карбоногидразоноилдицианида (Соединение 21)
6 мг (7%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 3'-фтор-4'-нитробифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 22: Получение (4-(6-фторпиридин-3-ил)фенил)карбоногидразоноилдицианида (Соединение 22)
Стадия 22-1: Получение 4-(6-фторпиридин-3-ил)анилина
38 мг (45%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 3-бром-6-фторпиридин (0,55 ммоль) использовали вместо 2-бромпиримидина.
Стадия 22-2: Получение (4-(6-фторпиридин-3-ил)фенил)карбоногидразоноилдицианида
23 мг (53%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(6-фторпиридин-3-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 23: Получение (2',4'-диметилбифенил-4-ил)карбоногидразоноилдицианида (Соединение 23)
8 мг (6%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2',4'-диметилбифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 24: Получение метил-4'-(2-(дицианометилен)гидразинил)-2-фторбифенил-4-карбоксилата (Соединение 24)
Стадия 24-1: Получение метил-4'-амино-2-фторбифенил-4-карбоксилата
93 мг (60%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что метил-4-бром-3-фторбензоат (0,69 ммоль) использовали вместо 2-бромпиримидина.
Стадия 24-2: Получение метил-4'-(2-(дицианометилен)гидразинил)-2-фторбифенил]-4-карбоксилата
16 мг (16%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4'-амино-2-фторбифенил-4-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 25: Получение (2'-метил-4'-нитробифенил-4-ил)карбоногидразоноилдицианида (Соединение 25)
Стадия 25-1: Получение 2'-метил-4'-нитробифенил-4-амина
98 мг (63%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 1-бром-2-метил-4-нитробензол (0,69 ммоль) использовали вместо 2-бромпиримидина.
Стадия 25-2: Получение (2'-метил-4'-нитробифенил-4-ил)карбоногидразоноилдицианида
38 мг (27%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2'-метил-4'-нитробифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 26: Получение (2',4'-дихлорбифенил-4-ил)карбоногидразоноилдицианида (Соединение 26)
Стадия 26-1: Получение 2',4'-дихлорбифенил-4-амина
182 мг (84%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 1-бром-2,4-дихлорбензол (0,91 ммоль) использовали вместо 2-бромпиримидина.
Стадия 26-2: Получение (2',4'-(дихлорбифенил-4-ил)карбоногидразоноилдицианида
47 мг (20%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2',4'-дихлорбифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 27: Получение (2'-метокси-4'-метилбифенил-4-ил)карбоногидразоноилдицианида (Соединение 27)
Стадия 27-1: Получение 2'-метокси-4'-метилбифенил-4-амина
36 мг (24%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 1-бром-2-метокси-4-метилбензол (0,69 ммоль) использовали вместо 2-бромпиримидина.
Стадия 27-2: Получение (2'-метокси-4'-метилбифенил-4-ил)карбоногидразоноилдицианида
3 мг (6%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2'-метокси-4'-метилбифенил-4-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 28: Получение (4-(пиразин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 28) Стадия 28-1: Получение 4-(пиразин-2-ил)анилина
72 мг (33%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бромпиразин (1,26 ммоль) использовали вместо 2-бромпиримидина.
Стадия 28-2: Получение (4-(пиразин-2-ил)фенил)карбоногидразоноилдицианида
12 мг (12%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиразин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 29: Получение (4-(5-(трифторметил)пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 29)
Стадия 29-1: Получение 4-(5-(трифторметил)пиримидин-2-ил)анилин
127 мг (78%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-5-(трифторметил)пиримидин (0,83 ммоль) использовали вместо 2-бромпиримидина.
Стадия 29-2: Получение (4-(5-(трифторметил)пиримидин-2-ил)фенил)карбоногидразоноилдицианида
47 мг (31%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-(трифторметил)пиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 30: Получение (4-(4,6-диметилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 30)
Стадия 30-1: Получение 4-(4,6-диметилпиримидин-2-ил)анилина
118 мг (67%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлор-4,6-диметилпиримидин (1,05 ммоль) использовали вместо 2-бромпиримидина.
Стадия 30-2: Получение (4-(4,6-диметилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
110 мг (77%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(4,6-диметилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 31: Получение (4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 31)
Стадия 31-1: Получение 4-(4-метилпиримидин-2-ил)анилина
77 мг (57%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бром-4-метилпиримидин (0,87 ммоль) использовали вместо 2-бромпиримидина.
Стадия 31-2: Получение (4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
66 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(4-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 32: Получение (4-(пиримидин-5-ил)фенил)карбоногидразоноилдицианида (Соединение 32)
Стадия 32-1: Получение 4 (пиримидин 5-ил)анилина
502 мг (64%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бромпиримидин (0,87 ммоль) использовали вместо 2-бромпиримидина.
Стадия 32-2: Получение (4-(пиримидин-5-ил)фенил)карбоногидразоноилдицианида
100 мг (69%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиримидин-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 33: Получение (4-(1-метил-1H-1,2,4-триазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 33)
Стадия 33-1: Получение 4-(1-метил-1H-1,2,4-триазол-5-ил)анилина
40 мг (20%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бром-1-метил-1H-1,2,4-триазол (0,96 ммоль) использовали вместо 2-бромпиримидина.
Стадия 33-2: Получение (4-(1-метил-1H-1,2,4-триазол-5-ил)фенил)карбоногидразоноилдицианида
18 мг (25%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(1-метил-1H-1,2,4-триазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 34: Получение (4-(1-метил-1H-1,2,4-триазол-3-ил)фенил)карбоногидразоноилдицианида (Соединение 34)
Стадия 34-1: Получение 4-(1-метил-1H-1,2,4-триазол-3-ил)анилина
100 мг (50%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 3-бром-1-метил-1H-1,2,4-триазол (0,96 ммоль) использовали вместо 2-бромпиримидина.
Стадия 34-2: Получение (4-(1-метил-1H-1,2,4-триазол-3-ил)фенил)карбоногидразоноилдицианида
29 мг (40%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(1-метил-1H-1,2,4-триазол-3-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 35: Получение (4-(1H-пиразол-1-ил)фенил)карбоногидразоноилдицианида (Соединение 35)
163 мг (55%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(1H-пиразол-1-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 36: Получение (4-(4-метил-1H-пиразол-1-ил)фенил)карбоногидразоноилдицианида (Соединение 36)
Стадия 36-1: Получение 4-метил-1-(4-нитрофенил)-1H-пиразола
4-Метил-1H-пиразол (100 мг, 1,22 ммоль) растворяли в 1,5 мл ДМСО в присутствии азота, а затем добавляли твердый трет-бутоксид калия (164 мг, 1,46 ммоль) и 1-фтор-4-нитробензол (189 мг, 1,46 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 72°С в течение 20 часов. Когда реакция завершалась, продукт реакции охлаждали, а затем экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали перекристаллизацией, используя этанольный растворитель, с получением 96 мг (39%-й выход) указанного в заголовке соединения.
Стадия 36-2: Получение 4-(4-метил-1H-пиразол-1-ил)анилина
4-Метил-1-(4-нитрофенил)-1H-пиразол, полученный в соответствии со стадией 1-1, растворяли в 2,5 мл этанола, а затем добавляли 10% (32 мг, 0,06 ммоль) палладиевого катализатора, активированного углем, с последующим перемешиванием при комнатной температуре в течение 1 часа в присутствии газообразного водорода. После завершения реакции катализатор в реакционной смеси фильтровали через целит.Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 50 мг (98%-й выход) указанного в заголовке соединения.
Стадия 36-3: Получение (4-(4-метил-1H-пиразол-1-ил)фенил)карбоногидразоноилдицианида
50 мг (69%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(4-метил-1H-пиразол-1-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 37: Получение (2-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 37)
Стадия 37-1: Получение 2-фтор-4-(4-метилпиримидин-2-ил)анилина
100 мг (50%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что пинаколовый эфир 4-амино-3-фторфенилбороновой кислоты (2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин) (0,84 ммоль) использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты, и 2-бром-4-метилпиримидин (1,01 ммоль) использовали вместо 2-бромпиримидина.
Стадия 37-2: Получение (2-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
46 мг (67%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-фтор-4-(4-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 38: Получение (2-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 38)
Стадия 38-1: Получение 2-фтор-4-(пиримидин-2-ил)анилина
41 мг (17%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 37-1 Примера 37, за исключением того, что 2-бромпиримидин (1,26 ммоль) использовали вместо 2-бром-4-метилпиримидина.
Стадия 38-2: Получение (2-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида
11 мг (19%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-фтор-4-(пиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 39: Получение (3-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 39)
Стадия 39-1: Получение 3-фтор-4-(пиримидин-2-ил)анилина
156 мг (66%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что пинаколовый эфир 4-амино-2-фторфенилбороновой кислоты (1,38 ммоль) использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты.
Стадия 39-2: Получение (3-фтор-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида
80 мг (81%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 3-фтор-4-(пиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 40: Получение (3-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 40)
Стадия 40-1: Получение 3-фтор-4-(4-метилпиримидин-2-ил)анилина
82 мг (35%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 39-1 Примера 39, за исключением того, что 2-бром-4-метилпиримидин (1,16 ммоль) использовали вместо 2-бромпиримидина.
Стадия 40-2: Получение (3-фтор-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
56 мг (92%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 3-фтор-4-(4-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 41: Получение (2-метокси-4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 41)
Стадия 41-1: Получение 2-метокси-4-(5-метилпиримидин-2-ил)анилина
99 мг (47%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что пинаколовый эфир 4-амино-3-метоксибороновой кислоты (0,99 ммоль) использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты, и 2-хлор-5-метилпиримидин (1,19 ммоль) использовали вместо 2-бромпиримидина.
Стадия 41-2: Получение (2-метокси-4-(5-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
25 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-метокси-4-(5-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 42: Получение (2-метокси-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 42)
Стадия 42-1: Получение 2-метокси-4-(4-метилпиримидин-2-ил)анилина
80 мг (42%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 41-1 Примера 41, за исключением того, что 2-бром-4-метилпиримидин (1,12 ммоль) использовали вместо 2-хлор-5-метилпиримидина.
Стадия 42-2: Получение (2-метокси-4-(4-метилпиримидин-2-ил)фенил)карбоногидразоноилдицианида
10 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-метокси-4-(4-метилпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 43: Получение (2-метокси-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 43)
Стадия 43-1: Получение 2-метокси-4-(пиримидин-2-ил)анилина
29 мг (18%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 41-1 Примера 41, за исключением того, что 2-бромпиримидин (0,96 ммоль) использовали вместо 2-хлор-5-метилпиримидина.
Стадия 43-2: Получение (2-метокси-4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида
26 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-метокси-4-(пиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 44: Получение (4-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианида (Соединение 44)
Стадия 44-1: Получение 2-(3-фтор-4-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана
4-Бром-2-фтор-1-нитробензол (2 г, 7,88 ммоль) и бис(пинаколато)дибор(4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан); 2,1 г, 9,45 ммоль) растворяли в 60 мл раствора 1,4-диоксана в присутствии азота, а затем добавляли Pd(dppf)2Cl2 (1,3 г, 1,58 ммоль) и ацетат калия (3,1 г, 31,5 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 80°С в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 1,6 г указанного в заголовке соединения.
Стадия 44-2: Получение 4-(2-нитро-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолина
2-(3-Фтор-4-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан, полученный в соответствии со стадией 44-1, и морфолин (0,68 мл, 7,79 ммоль) растворяли в ацетонитриле (20 мл) в присутствии азота с получением реакционной смеси, а затем реакционную смесь перемешивали при 80°С в течение 12 часов.. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 700 г указанного в заголовке соединения.
Стадия 44-3: Получение 4-(5-(5-фторпиримидин-2-ил)-2-нитрофенил)морфолина
164 мг (26%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 4-(2-нитро-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолин, полученный в соответствии со стадией 44-2, использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты, и 2-хлор-5-фторпиримидин (2,51 ммоль) использовали вместо 2-бромпиримидина.
Стадия 44-4: Получение 4-(5-фторпиримидин-2-ил)-2-морфолиноанилина
4-(5-(5-Фторпиримидин-2-ил)-2-нитрофенил)морфолин, полученный в соответствии со стадией 44-3, растворяли в этилацетате, и восстанавливали способом, аналогичным указанному на стадии 36-2 Примера 36 с получением 84 мг (57%-й выход) указанного в заголовке соединения.
Стадия 44-5: Получение (4-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианида
70 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(5-фторпиримидин-2-ил)-2-морфолиноанилин получали в соответствии со стадией 44-4 использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 45: Получение метил-(4-(пиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 45)
(4-(Пиримидин-2-ил)фенил)карбоногидразоноилдид (50 мг, 0,20 ммоль), полученный согласно примеру 1, растворяли в 1,5 мл тетрагидрофурана, а затем добавляли трет-бутоксид калия (29 мг, 0,26 ммоль) при комнатной температуре, и дополнительно добавляли иодметан (25 мкл, 0,40 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 60°С в течение 4 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали перекристаллизацией, используя этанольный растворитель, с получением 3 мг (6%-й выход) указанного в заголовке соединения.
Пример 46: Получение (3-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 46)
Стадия 46-1: Получение 3-(5-фторпиримидин-2-ил)анилина
180 мг указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 41, за исключением того, что пинаколовый эфир 3-аминофенилбороновой кислоты (1,37 ммоль) использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты, и 2-хлор-5-фторпиримидин (1,64 ммоль) использовали вместо 2-бромпиримидина.
Стадия 46-2: Получение (3-(5-фторпиримидин-2-ил)фенил)карбоногидразоноилдицианида
70,5 мг указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 3-(5-фторпиримидин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 47: Получение (5-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианида (Соединение 47)
Стадия 47-1: Получение 4-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолина
480 мг (41%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 44-2 Примера 44, за исключением того, что 2-(4-хлор-3-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан использовали вместо 2-(3-фтор-4-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана.
Стадия 47-2: Получение 4-(4-(5-фторпиримидин-2-ил)-2-нитрофенил)морфолина
300 мг (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 4-(2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолин, полученный в соответствии со стадией 47-1, использовали вместо 4-аминопинаколового эфира фенилбороновой кислоты, и 2-хлор-5-фторпиримидин использовали вместо 2-бромпиримидина.
Стадия 47-3: Получение 5-(5-фторпиримидин-2-ил)-2-морфолиноанилина
4-(5-(5-Фторпиримидин-2-ил)-2-нитрофенил)морфолин, полученный в соответствии со стадией 44-3, растворяли в дихлорметане (ДХМ), и восстанавливали способом, аналогичным указанным на стадии 36-2 Примера 36, с получением 86 мг (48%-й выход) указанного в заголовке соединения.
Стадия 47-4: Получение (5-(5-фторпиримидин-2-ил)-2-морфолинофенил)карбоногидразоноилдицианида
84 мг (77%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 5-(5-фторпиримидин-2-ил)-2-морфолиноанилин, полученный в соответствии со стадией 47-3, использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 48: Получение (4-(бензо[d]тиазол-6-ил)фенил)карбоногидразоноилдицианида (Соединение 48)
Стадия 48-1: Получение 4-(бензо[d]тиазол-6-ил)анилина
58 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 6-бромбензо[d]тиазол (0,55 ммоль) использовали вместо 2-бромпиримидина.
Стадия 48-2: Получение (4-(бензо[d]тиазол-6-ил)фенил)карбоногидразоноилдицианида
10 мг (13%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(бензо[d]тиазол-6-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 49: Получение (4-(нафталин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 49)
Стадия 49-1: Получение 4-(нафталин-2-ил)анилина
40 мг (21%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бромнафталин (0,46 ммоль) использовали вместо 2-бромпиримидина.
Стадия 49-2: Получение (4-(нафталин-2-ил)фенил)карбоногидразоноилдицианида
5 мг (9%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(нафталин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 50: Получение (4-(пиразоло[1,5-a]пиримидин-6-ил)фенил)карбоногидразоноилдицианида (Соединение 50)
Стадия 50-1: Получение 4-(пиразоло[1,5-а]пиримидин-6-ил)анилина
180 мг (62%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 6-бромпиразоло[1,5-а]пиримидин (1,65 ммоль) использовали вместо 2-бромпиримидина.
Стадия 50-2: Получение (4-(пиразоло[1,5-a]пиримидин-6-ил)фенил)карбоногидразоноилдицианида
79 мг (32%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(пиразоло[1,5-а]пиримидин-6-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 51: Получение (4-(имидазо[1,2-a]пиримидин-6-ил)фенил)карбоногидразоноилдицианида (Соединение 51)
Стадия 51-1: Получение 4-(имидазо[1,2-a]пиримидин-6-ил)анилина
76 мг (17%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 6-бромимидазо[1,2-а]пиримидин (2,53 ммоль) использовали вместо 2-бромпиримидина.
Стадия 51-2: Получение (4-(имидазо[1,2-a]пиримидин-6-ил)фенил)карбоногидразоноилдицианида
40 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(имидазо[1,2-a]пиримидин-6-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 52: Получение (4-(хиноксалин-2-ил)фенил)карбоногидразоноилдицианида (Соединение 52)
Стадия 52-1: Получение 4-(хиноксалин-2-ил)анилина
100 мг (33%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-хлорхиноксалин (1,64 ммоль) использовали вместо 2-бромпиримидина.
Стадия 52-2: Получение (4-(хиноксалин-2-ил)фенил)карбоногидразоноилдицианида
30 мг (44%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(хиноксалин-2-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 53: Получение (4-(имидазо[1,2-a]пиридин-6-ил)фенил)карбоногидразоноилдицианида (Соединение 53)
Стадия 53-1: Получение 4-(имидазо[1,2-a]пиридин-6-ил)анилина
100 мг (33%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 6-бромимидазо[1,2-а]пиридин (1,64 ммоль) использовали вместо 2-бромпиримидина.
Стадия 53-2: Получение 4-(имидазо[1,2-a]пиридин-6-ил)фенил)карбоногидразоноилдицианида
22 мг (32%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(имидазо[1,2-a]пиридин-6-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 54: Получение (4-(изохинолин-3-ил)фенил)карбоногидразоноилдицианида (Соединение 54)
Стадия 54-1: Получение 4-(изохинолин-3-ил)анилина
80 мг (16%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 3-хлоризохинолин (2,80 ммоль) использовали вместо 2-бромпиримидина.
Стадия 54-2: Получение (4-(изохинолин-3-ил)фенил)карбоногидразоноилдицианида
23 мг (42%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(изохинолин-3-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 55: Получение (4-(2-метил-2H-индазол-4-ил)фенил)карбоногидразоноилдицианида (Соединение 55)
Стадия 55-1: Получение 4-(2-метил-2H-индазол-4-ил)анилина
331 мг (80%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 4-бром-2-метил-2H-индазол (2,20 ммоль) использовали вместо 2-бромпиримидина.
Стадия 55-2: Получение (4-(2-метил-2H-индазол-4-ил)фенил)карбоногидразоноилдицианида
100 мг (24%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(2-метил-2H-индазол-4-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 56: Получение (4-(2-метил-2H-индазол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 56)
Стадия 56-1: Получение 4-(2-метил-2H-индазол-5-ил)анилина
100 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 5-бром-2-метил-2H-индазол (2,20 ммоль) использовали вместо 2-бромпиримидина.
Стадия 56-2: Получение (4-(2-метил-2H-индазол-5-ил)фенил)карбоногидразоноилдицианида
26 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(2-метил-2H-индазол-5-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 57: Получение (4-(2-метил-2H-индазол-7-ил)фенил)карбоногидразоноилдицианида (Соединение 57)
Стадия 57-1: Получение 4-(2-метил-2H-индазол-7-ил)анилина
550 мг (86%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 7-бром-2-метил-2H-индазол (2,20 ммоль) использовали вместо 2-бромпиримидина.
Стадия 57-2: Получение (4-(2-метил-2H-индазол-7-ил)фенил)карбоногидразоноилдицианида
345 мг (47%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-(2-метил-2H-индазол-7-ил)анилин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 58: Получение (4-(1H-индол-5-ил)фенил)карбоногидразоноилдицианида (Соединение 58)
Стадия 58-1: Получение трет-бутил-5-(4-аминофенил)-1H-индол-1-карбоксилата
596 мг (6%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что трет-бутил-5-бром-1H-индол-1-карбоксилат (3,38 ммоль) использовали вместо 2-бромпиримидина.
Стадия 58-2: Получение трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата
107 мг (14%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-(4-аминофенил)-1H-индол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 58-3: Получение (4-(1H-индол-5-ил)фенил)карбоногидразоноилдицианида
трет-Бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилат растворяли в 8,0 мл дихлорметана в присутствии азота, а затем трифторуксусную кислоту добавляли с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Когда реакция завершилась, продукт реакции концентрировали при пониженном давлении с получением 40 мг (44%-й выход) указанного в заголовке соединения.
Пример 59: Получение (4-метоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 59)
141 мг (51%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 4-метоксибензо[d]тиазол-2-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 60: Получение бензо[d]тиазол-6-илкарбоногидразоноилдицианида (Соединение 60)
290 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что бензо[d]тиазол-6-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 61: Получение бензо[d]тиазол-5-илкарбоногидразоноилдицианида (Соединение 61)
180 мг (40%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что бензо[d]тиазол-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 62: Получение (9-этил-9Н-карбазол-3-ил)карбоногидразоноилдицианида (Соединение 62)
77 мг (11%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 9-этил-9H-карбазол-3-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 63: Получение (1-метил-1H-индазол-6-ил)карбоногидразоноилдицианида (Соединение 63)
246 мг (54%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-метил-1H-индазол-6-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 64: Получение хинолин-3-илкарбоногидразоноилдицианида (Соединение 64)
163 мг (27%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что хинолин-3-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 65: Получение (2-метил-2H-индазол-6-ил)карбоногидразоноилдицианида (Соединение 65)
23 мг (3%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 2-метил-2H-индазол-6-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 66: Получение (1-метил-1H-индазол-5-ил)карбоногидразоноилдицианида (Соединение 66)
128 мг (17%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-метил-1H-индазол-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 67: Получение хинолин-6-илкарбоногидразоноилдицианида (Соединение 67)
56 мг (9%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что хинолин-6-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 68: Получение (1-метил-1H-индол-5-ил)карбоногидразоноилдицианида (Соединение 68)
69 мг (11%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-метил-1H-индол-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 69: Получение (1H-индазол-5-ил)карбоногидразоноилдицианида (Соединение 69)
70 мг (19%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-1H-индазол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 70: Получение (1H-индазол-6-ил)карбоногидразоноилдицианида (Соединение 70)
62 мг (12%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-6-амино-1H-индазол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 71: Получение (1H-индол-5-ил)карбоногидразоноилдицианида (Соединение 71)
Стадия 71-1: Получение трет-бутил-5-(2-(дицианометилен)гидразинил)-1H-индол-1-карбоксилата
25 мг (6%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-1H-индол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 71-2: Получение (1H-индол-5-ил)карбоногидразоноилдицианида
15 мг (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 58-3 Примера 58, за исключением того, что трет-бутил-5-(2-(дицианометилен)гидразинил)-1H-индол-1-илкарбоксилат использовали вместо трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата.
Пример 72: Получение (1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 72)
Стадия 72-1: Получение трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
5-Нитро-1H-пирроло[2,3-b]пиридин (2 г, 12,27 ммоль), ди-трет-бутилдикарбонат (3,2 г, 14,71 ммоль) и 4-диметиламинопиридин (150 мг, 1,23 ммоль) растворяли в 40 мл раствора ацетонитрила в присутствии азота, а затем перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 1,23 г (38%-й выход) указанного в заголовке соединения.
Стадия 72-2: Получение трет-бутил-5-амино-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат (600 мг, 2,28 ммоль), полученный в соответствии со стадией 72-1, растворяли в 5 мл раствора уксусной кислоты в присутствии азота, а затем добавляли железо (1,2 г, 22,80 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали.
Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 261 мг (48%-й выход) указанного в заголовке соединения.
Стадия 72-3: Получение (1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
128 мг (54%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 73: Получение (3-метил-1H-индазол-6-ил)карбоногидразоноилдицианида (Соединение 73)
Стадия 73-1: Получение трет-бутил-3-метил-6-нитро-1H-индазол-1-карбоксилата
1,43 г (92%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 3-метил-6-нитро-1H-индазол использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 73-2: Получение трет-бутил-6-амино-3-метил-1H-индазол-1-карбоксилата
трет-Бутил-3-метил-6-нитро-1H-индазол-1-карбоксилат, полученный в соответствии со стадией 73-1, растворяли в этилацетате, и восстанавливали способом, аналогичным указанному на стадии 36-2 Примера 36 с получением 500 мг (80%-й выход) указанного в заголовке соединения.
Стадия 73-3: Получение (3-метил-1H-индазол-6-ил)карбоногидразоноилдицианида
40 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-6-амино-3-метил-1H-индазол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 74: Получение (3-метил-1H-индазол-5-ил)карбоногидразоноилдицианида (Соединение 74)
Стадия 74-1: Получение трет-бутил-3-метил-5-нитро-1H-индазол-1-карбоксилата
2,83 г (90%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 3-метил-5-нитро-1H-индазол (1,29 ммоль) использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 74-2: Получение трет-бутил-5-амино-3-метил-1H-индазол-1-карбоксилата
трет-Бутил-3-метил-5-нитро-1H-индазол-1-карбоксилат, полученный в соответствии со стадией 74-1, растворяли в этилацетате, и восстанавливали способом, аналогичным указанному на стадии 36-2 Примера 36 с получением 761 мг (85%-й выход) указанного в заголовке соединения.
Стадия 74-3: Получение (3-метил-1H-индазол-5-ил)карбоногидразоноилдицианида
100 мг (15%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-3-метил-1H-индазол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 75: Получение (1H-индол-4-ил)карбоногидразоноилдицианида (Соединение 75)
Стадия 75-1: Получение траря-бутил-4-амино-1H-индол-1-карбоксилата
трет-Бутил-4-нитро-1H-индол-1-карбоксилат растворяли в ацетонитриле (MeCN), и восстанавливали способом, аналогичным указанному на стадии 36-2 Примера 36, с получением 198 мг (75%-й выход) указанного в заголовке соединения.
Стадия 75-2: Получение (1H-индол-4-ил)карбоногидразоноилдицианида
40 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-4-амино-1H-индол-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 76: Получение (1-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 76)
Стадия 76-1: Получение 1-метил-5-нитро-1H-пирроло[2,3-b]пиридина
5-Нитро-1H-пирроло[2,3-b]пиридин (600 мг, 3,72 ммоль) растворяли в ДМФА (6 мл) в присутствии азота, а затем добавляли гидрид натрия (60%, 300 мг, 7,44 ммоль) с последующим добавлением подметана (0,93 мл, 14,88 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 380 мг (57%-й выход) указанного в заголовке соединения.
Стадия 76-2: Получение 1-метил-1H-пирроло[2,3-b]пиридин-5-амина
69 мг (55%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что 1-метил-5-нитро-1H-пирроло[2,3-b]пиридин, полученный на стадии 76-1, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 76-3: Получение (1-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
8 мг (9%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-метил-1H-пирроло[2,3-b]пиридин-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 77: Получение (1-пропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 77)
Стадия 77-1: Получение 5-нитро-1-пропил-1H-пирроло[2,3-b]пиридина
5-Нитро-1H-пирроло[2,3-b]пиридин (600 мг, 3,72 ммоль), карбонат цезия (CsCO3, 2,4 г, 7,44 ммоль) и 1-иодпропан (1,10 мл, 11,01 ммоль) растворяли в ДМФА с получением реакционной смеси, а затем реакционную смесь перемешивали при 60°С в течение 17 часов с получением 523 мг (70%-й выход) указанного в заголовке соединения.
Стадия 77-2: Получение 1-пропил-1H-пирроло[2,3-b]пиридин-5-амина
297 мг (67%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что 5-нитро-1-пропил-1H-пирроло[2,3-b]пиридин, полученный на стадии 77-1, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 77-3: Получение (1-пропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
98 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-пропил-1H-пирроло[2,3-b]пиридин-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 78: Получение (1-изопропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 78)
Стадия 78-1: Получение 5-нитро-1-изопропил-1H-пирроло[2,3-b]пиридина
591 мг (78%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 77-1 Примера 77, за исключением того, что 2-иодпропан (1,10 мл, 11,01 ммоль) использовали вместо 1-иодпропана.
Стадия 78-2: Получение 1-изопропил-1H-пирроло[2,3-b]пиридин-5-амина
270 мг (54%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что 5-нитро-1-изопропил-1H-пирроло[2,3-b]пиридин, полученный на стадии 78-1, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 78-3: Получение (1-изопропил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
81 мг (21%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-изопропил-1H-пирроло[2,3-b]пиридин-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 79: Получение (1-этил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 79)
Стадия 79-1: Получение 1-этил-5-нитро-1H-пирроло[2,3-b]пиридина
363 мг (52%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 77-1 Примера 77, за исключением того, что иодэтан (0,83 мл, 10,14 ммоль) использовали вместо 1-иодпропана.
Стадия 79-2: Получение 1-этил-1H-пирроло[2,3-b]пиридин-5-амина
184 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что 1-этил-5-нитро-1H-пирроло[2,3-b]пиридин, полученный на стадии 79-1, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 79-3: Получение (1-этил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
187 мг (69%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-этил-1H-пирроло[2,3-b]пиридин-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 80: Получение (1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 80)
Стадия 80-1: Получение 5-нитро-1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридина
784 мг (52%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 77-1 Примера 77, за исключением того, что 1,1,1-трифтор-2-иодэтан (1,80 мл, 18,39 ммоль) использовали вместо 1-иодпропана.
Стадия 80-2: Получение 1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-5-амина
235 мг (53%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что 5-нитро-1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин, полученный на стадии 80-1, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 80-3: Получение (1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
150 мг (55%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что 1-(2,2,2-трифторэтил)-1H-пирроло[2,3-b]пиридин-5-амин использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 81: Получение (2-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 81)
Стадия 81-1: Получение трет-бутил-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
1,83 г (98%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 2-метил-1H-пирроло[2,3-b]пиридин использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 81-2: Получение трет-бутил-2-метил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилат и нитрат тетрабутил аммония растворяли в дихлорметане в присутствии азота, а затем перемешивали при 0°С в течение 10 минут. После этого добавляли трифторуксусный ангидрид (TFAA, 1,43 мл, 0,125 ммоль), перемешивание выполняли при 0°С в течение 5 минут, а затем перемешивание осуществляли дополнительно при комнатной температуре в течение 3 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 1,34 г (61%-й выход) указанного в заголовке соединения.
Стадия 81-3: Получение трет-бутил-5-амино-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
311 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-2 Примера 72, за исключением того, что трет-бутил-2-метил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат, полученный на стадии 81-2, использовали вместо трет-бутил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 81-4: Получение трет-бутал-5-(2-(дицианометилен)гидразинил)-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
160 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 81-5: Получение (2-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
29 мг (71%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 58-3 Примера 58, за исключением того, что трет-бутил-5-(2-(дицианометилен)гидразинил)-2-метил-1H-пирроло[2,3-b]пиридин-1-илкарбоксилат использовали вместо трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата.
Пример 82: Получение (3-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 82)
Стадия 82-1: Получение трет-бутил-3-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
3,4 г (97%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 3-метил-1H-пирроло[2,3-b]пиридин использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 82-2: Получение трет-бутил-3-метил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
800 мг (42%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 81-2 Примера 81, за исключением того, что трет-бутил-3-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо трет-бутил-2-метил-1Н-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 82-3: Получение трет-бутил-5-амино-3-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-3-метил-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат растворяли в этилацетате и восстанавливали способом аналогичным, указанному на стадии 36-2 Примера 36 с получением 367 мг (82%-й выход) указанного в заголовке соединения.
Стадия 82-4: Получение трет-бутал-5-(2-(дицианометилен)гидразинил)-3-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата
106 мг (40%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-3-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 82-5: Получение (3-метил-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
30 мг (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 58-3 Примера 58, за исключением того, что трет-бутил-5-(2-(дицианометилен)гидразинил)-3-метил-1H-пирроло[2,3-b]пиридин-1-илкарбоксилат использовали вместо трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата.
Пример 83: Получение (3-хлор-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 83)
Стадия 83-1: Получение 3-хлор-5-нитро-1H-пирроло[2,3-b]пиридина
5-Нитро-1H-пирроло[2,3-b]пиридин и N-хлорсукцинимид растворяли в ДМФА (7 мл) в присутствии азота, а затем перемешивали при комнатной температуре в течение 12 минут. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 664 мг (55%-й выход) указанного в заголовке соединения.
Стадия 83-2: Получение трет-бутил-3-хлор-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
419 г (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 3-хлор-5-нитро-1H-пирроло[2,3-b]пиридин использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 83-3: Получение трет-бутил-5-амино-3-хлор-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-3-хлор-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат растворяли в этилацетате и восстанавливали способом аналогичным, указанному на стадии 36-2 Примера 36 с получением 118 мг (60%-й выход) указанного в заголовке соединения.
Стадия 83-4: Получение трет-бутил-5-(2-(дицианометилен)гидразинил)-3-хлор-1H-пирроло[2,3-b]пиридин-1-карбоксилата
35 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-3-хлор-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 83-5: Получение (3-хлор-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
20 мг (91%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 58-3 Примера 58, за исключением того, что трет-бутил-5-(2-(дицианометилен)гидразинил)-3-хлор-1H-пирроло[2,3-b]пиридин-1-илкарбоксилат использовали вместо трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата.
Пример 84: Получение (4-метокси-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 84)
Стадия 84-1: Получение 4-хлор-5-нитро-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридина
2,0 мг (60%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 81-2 Примера 81, за исключением того, что 4-хлор-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин использовали вместо трет-бутил-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 84-2: Получение 4-метокси-5-нитро-1H-пирроло[2,3-b]пиридина
4-Хлор-5-нитро-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин и твердый карбонат калия растворяли в смешанном растворе метанол:вода=3:1 в присутствии азота и кипятили с обратным холодильником в течение 7 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 235 мг (68%-й выход) указанного в заголовке соединения.
Стадия 84-3: Получение трет-бутил-4-метокси-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
234 мг (77%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что 4-метокси-5-нитро-1H-пирроло[2,3-b]пиридин использовали вместо 5-нитро-1Н-пирроло[2,3-b]пиридина.
Стадия 84-4: Получение трет-бутил-5-амино-4-метокси-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-4-метокси-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат растворяли в этилацетате и восстанавливали способом аналогичным, указанному на стадии 36-2 Примера 36 с получением 107 мг (99%-й выход) указанного в заголовке соединения.
Стадия 84-5: Получение (4-метокси-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
22 мг (17%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-4-метокси-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 85: Получение (2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида (Соединение 85)
Стадия 85-1: Получение N,N-диметил-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбоксамида
N,N-Диизопропиламин (DIPEA, 1,20 мл, 6,62 ммоль) и гексафторфосфат азабензотриазолтетраметилурония (HATU, 2 г, 4,30 ммоль) добавляли к 1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбоновой кислоты (1 г, 3,31 ммоль) в присутствии азота, а затем перемешивали при комнатной температуре в течение 10 минут. После этого добавляли раствор диметиламина (2,0 М в ТГФ, 3,32 мл, 6,62 ммоль) с последующим перемешиванием при комнатной температуре в течение 10 минут. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 800 мг (79%-й выход) указанного в заголовке соединения.
Стадия 85-2: Получение N,N-диметил-5-нитро-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбоксамида
373 мг (41%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 81-2 Примера 81, за исключением того, что N,N-диметил-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбоксамид использовали вместо трет-бутил-2-метил-1H-пирроло[2,3-b]пиридин-1-карбоксилата.
Стадия 85-3: Получение N,N-диметил-5-нитро-1H-пирроло[2,3-b]пиридин-2-карбоксамида
N,N-Диметил-5-нитро-1-(фенилсульфонил)-1H-пирроло[2,3-b]пиридин-2-карбоксамид (0,95 ммоль) и твердый трет-бутоксид калия (0,95 ммоль) растворяли в диоксане (15 мл) в присутствии азота и перемешивали при 80°С в течение 12 часов.
Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 71 мг (48%-й выход) указанного в заголовке соединения.
Стадия 85-4: Получение трет-бутил-2-(диметилкарбамоил)-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилата
100 мг (99%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 72-1 Примера 72, за исключением того, что N,N-диметил-5-нитро-1H-пирроло[2,3-b]пиридин-2-карбоксамид использовали вместо 5-нитро-1H-пирроло[2,3-b]пиридина.
Стадия 85-5: Получение трет-бутил-5-амино-2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-1-карбоксилата
трет-Бутил-2-(диметилкарбамоил)-5-нитро-1H-пирроло[2,3-b]пиридин-1-карбоксилат растворяли в этилацетате и восстанавливали способом аналогичным, указанному на стадии 36-2 Примера 36, с получением 90 мг (97%-й выход) указанного в заголовке соединения.
Стадия 85-6: Получение трет-бутал-5-(2-(дицианометилен)гидразинил)-2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-1-карбоксилата
78 мг (69%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что трет-бутил-5-амино-2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо 4-(пиримидин-2-ил)анилина.
Стадия 85-7: Получение (2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-5-ил)карбоногидразоноилдицианида
41 мг (72%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 58-3 Примера 58, за исключением того, что трет-бутил-5-(2-(дицианометилен)гидразинил)-2-(диметилкарбамоил)-1H-пирроло[2,3-b]пиридин-1-карбоксилат использовали вместо трет-бутил-5-(4-(2-(дицианометилен)гидразинил)фенил)-1H-индол-1-илкарбоксилата.
Пример 86: Получение (2-(4-метоксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 86)
Стадия 86-1: Получение 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида
4-Метоксибензойную кислоту (1 г, 6,57 ммоль) растворяли в MeCN в присутствии азота, а затем HATU (2,75 г, 7,23 ммоль) и триэтиламин (TEA, 1,39 мл, 9,86 ммоль) добавляли с получением реакционной смеси, и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого добавляли 6-нитробензо[d]тиазол-2-амин (1,28 г, 6,57 ммоль), и реакционную смесь кипятили с обратным холодильником в течение 24 часов. Когда реакция была завершена, продукт реакции фильтровали, используя MeCN, с получением 1,6 г (74%-й выход) соединения в качестве твердого фильтрата.
Стадия 86-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида
4-Метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамид (100 мг, 0,30 ммоль) и 10% Pd/C (64 мг, 0,06 ммоль) растворяли в ДМФА с получением реакционной смеси. Реакционную смесь перемешивали при 60°С в течение 2 часов в присутствии водорода. Когда реакция была завершена, продукт реакции фильтровали, используя ДМФА. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с этилацетатом с получением 39 мг (43%-й выход) указанного в заголовке соединения.
Стадия 86-3: Получение (2-(4-метоксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
78 мг (69%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-2 Примера 1, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамид использовали вместо 4-(пиримидин-2-ил)анилина, смешанный раствор этанола и воды 10:3 использовали в качестве растворителя, а 35% водный раствор HCl использовали в качестве кислого раствора.
Пример 87: Получение (2-(2-гидроксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 87)
Стадия 87-1: Получение N-(6-нитробензо[d]тиазол-2-ил)-2-гидроксибензамида
611 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 2-гидроксибензойную кислоту (3,62 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 87-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-2-гидроксибензамида
48 мг (53%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 2-гидрокси-N-(6-нитробензо[d]тиазол-2-ил)бензамид, полученный в соответствии со стадией 87-1, использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 87-3: Получение (2-(2-гидроксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
36 мг (59%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-2-гидроксибензамид, полученный в соответствии со стадией 87-2, использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 88: Получение (2-ацетамидобензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 88)
Стадия 88-1: Получение N-(6-нитробензо[d]тиазол-2-ил)ацетамида
6-Нитробензо[d]тиазол-2-амин растворяли в MeCN в присутствии азота, затем добавляли DIPEA и уксусный ангидрид с получением реакционной смеси, и реакционную смесь перемешивали при 60°С в течение 14 часов. Когда реакция была завершена, продукт реакции фильтровали, используя MeCN, с получением 200 мг (75%-й выход) соединения в качестве твердого фильтрата.
Стадия 88-2: Получение N-(6-аминобензо[d]тиазол-2-ил)ацетамида
66 мг (83%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что N-(6-нитробензо[d]тиазол-2-ил)ацетамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 88-3: Получение (2-ацетамидобензо[d]тиазол-6-ил)карбоногидразоноилдицианида
28 мг (42%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)ацетамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 89: Получение (2-(3-цианобензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 89)
Стадия 89-1: Получение 3-циано-N-(6-нитробензо[d]тиазол-2-ил)бензамида
718 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 3-цианобензойную кислоту (3,62 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 89-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-3-цианобензамида
82 мг (90%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 3-циано-N-(6-нитробензо[d]тиазол-2-ил)бензамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 89-3: Получение (2-(3-цианобензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
33 мг (52%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-3-цианобензамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 90: Получение (2-(5-метилникотинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 90)
Стадия 90-1: Получение 5-метил-N-(6-нитробензо[d]тиазол-2-ил)никотинамида
1,02 г (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 5-метилникотиновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 90-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-5-метилникотинамида
75 мг (83%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 5-метил-N-(6-нитробензо[d]тиазол-2-ил)никотинамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 90-3: Получение (2-(5-метилникотинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
32 мг (49%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-5-метилникотинамид получали в соответствии со стадией 90-2 использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 91: Получение (2-(3-гидроксипиколинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 91)
Стадия 91-1: Получение 3-гидрокси-N-(6-нитробензо[d]тиазол-2-ил)пиколинамида
327 мг (72%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 3-гидроксипиколиновую кислоту (1,44 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 91-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-3-гидроксипиколинамида
46 мг (51%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 3-гидрокси-N-(6-нитробензо[d]тиазол-2-ил)пиколинамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 91-3: Получение (2-(3-гидроксипиколинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
22 мг (35%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-3-гидроксипиколинамид, полученный в соответствии со стадией 91-2, использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 92: Получение (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 92)
Стадия 92-1: Получение N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
870 мг (71%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что пиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 92-2: Получение N-(6-аминобензо[d]тиазол-2-ил)пиразин-2-карбоксамиды
155 мг (87%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 92-3: Получение (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
55 мг (43%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 93: Получение (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 93)
Стадия 93-1: Получение 5-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
766 мг (67%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 5-метилпиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 93-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-5-метилпиразин-2-карбоксамида
165 мг (92%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 5-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 93-3: Получение (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
20 мг (16%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-5-метилпиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 94: Получение (2-(пиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 94)
Стадия 94-1: Получение N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамида
510 мг (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что пиримидин-5-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 94-2: Получение N-(6-аминобензо[d]тиазол-2-ил)пиримидин-5-карбоксамида
113 мг (83%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 94-3: Получение (2-(пиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
15 мг (24%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)пиримидин-5-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 95: Получение (2-(2-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 95)
Стадия 95-1: Получение 2-метил-N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамида
414 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 2-метилпиримидин-5-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 95-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-2-метилпиримидин-5-карбоксамида
119 мг (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 2-метил-N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 95-3: Получение (2-(2-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
21 мг (21%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-2-метилпиримидин-5-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 96: Получение (2-(4-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 96)
Стадия 96-1: Получение 4-метил-N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамида
327 мг (72%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 4-метилпиримидин-5-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 96-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-4-метилпиримидин-5-карбоксамида
131 мг (96%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 4-метил-N-(6-нитробензо[d]тиазол-2-ил)пиримидин-5-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 96-3: Получение (2-(4-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
28 мг (28%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-4-метилпиримидин-5-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 97: Получение (2-(3-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 97)
Стадия 97-1: Получение 3-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
542 мг (79%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 3-метилпиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 97-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-3-метилпиразин-2-карбоксамида
158 мг (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 3-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 97-3: Получение (2-(3-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
86 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-3-метилпиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 98: Получение (2-(6-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 98)
Стадия 98-1: Получение 6-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
542 мг (79%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 6-метилпиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты.
Стадия 98-2: Получение N-(6-аминобензо[d]тиазол-2-ил)-6-метилпиразин-2-карбоксамида
105 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 6-метил-N-(6-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 98-3: Получение (2-(6-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
54 мг (53%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(6-аминобензо[d]тиазол-2-ил)-6-метилпиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 99: Получение (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида (Соединение 99)
Стадия 99-1: Получение N-(5-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
486 мг (67%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что пиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты, и 5-нитробензо[d]тиазол-2-амин использовали вместо 6-нитробензо[d]тиазол-2-амина.
Стадия 99-2: Получение N-(5-аминобензо[d]тиазол-2-ил)пиразин-2-карбоксамиды
110 мг (81%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что N-(5-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 99-3: Получение (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида
22 мг (21%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(5-аминобензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 100: Получение (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида (Соединение 100)
Стадия 100-1: Получение 5-метил-N-(5-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамида
512 мг (75%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-1 Примера 86, за исключением того, что 5-метилпиразин-2-карбоновую кислоту (3,65 ммоль) использовали вместо 4-метоксибензойной кислоты, и 5-нитробензо[d]тиазол-2-амин использовали вместо 6-нитробензо[d]тиазол-2-амина.
Стадия 100-2: Получение N-(5-аминобензо[d]тиазол-2-ил)-5-метилпиразин-2-карбоксамида
83 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-2 Примера 86, за исключением того, что 5-метил-N-(5-нитробензо[d]тиазол-2-ил)пиразин-2-карбоксамид использовали вместо 4-метокси-N-(6-нитробензо[d]тиазол-2-ил)бензамида.
Стадия 100-3: Получение (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида
102 мг (80%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(5-аминобензо[d]тиазол-2-ил)-5-метилпиразин-2-карбоксамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 101: Получение (2-фенилбензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 101)
Стадия 101-1: Получение 2-бром-6-нитробензо[d]тиазола
6-Нитробензо[d]тиазол-2-амин (500 мг, 2,56 ммоль) растворяли в воде, а затем добавляли CuBr (55 мг, 0,38 ммоль) и 48% HBr (5 мл) с получением реакционной смеси. После этого к реакционной смеси добавляли нитрит натрия (1,41 г, 20,48 ммоль) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя насыщенный водный раствор NaCl и этилацетат дважды, и влагу сушили безводным сульфатом магния с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал, его фильтровали метанолом с получением 564 мг (85%-й выход) указанного в заголовке соединения в твердом состоянии.
Стадия 101-2: Получение 2-бром-бензо[d]тиазол-6-амина
2-Бром-6-нитробензо[d]тиазол, полученный на стадии 101-1, и Fe (539 мг, 9,65 ммоль) растворяли в уксусной кислоте с получением реакционной смеси, а затем реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя насыщенный водный раствор NaCl и этилацетат дважды, и влагу сушили безводным сульфатом магния с последующей фильтрацией. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 342 мг (69%-й выход) указанного в заголовке соединения.
Стадия 101-3: Получение 2-фенилбензо[d]тиазол-6-амина
56 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 1-1 Примера 1, за исключением того, что 2-бромбензо[d]тиазол-6-амин использовали вместо 2-бромпиримидина, пинаколовый эфир фенилбороновой кислоты использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты, а водный раствор карбоната калия использовали вместо водного раствора карбоната натрия.
Стадия 101-4: Получение (2-фенилбензо[d]тиазол-6-ил)карбоногидразоноилдицианида
46 мг (76%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-фенилбензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 102: Получение (2-(4-фторфенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 102)
Стадия 102-1: Получение 2-(4-фторфенил)бензо[d]тиазол-6-амина
30 мг (28%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 4-фторфенилбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 102-2: Получение (2-(4-фторфенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
31 мг (79%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(4-фторфенил)бензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 103: Получение 2-(4-(диметиламино)фенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 103)
Стадия 103-1: Получение 2-(4-(диметиламино)фенил)бензо[d]тиазол-6-амина
43 мг (37%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 4-(диметиламино)фенилбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 103-2: Получение (2-(4-(диметиламино)фенил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
31 мг (79%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(4-(диметиламино)фенил)бензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 104: Получение (2-(пиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 104)
Стадия 104-1: Получение 2-(пиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорида
Реакцию проводили таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир пиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты. 48 мг (41%-й выход) указанного в заголовке соединения в форме гидрохлорида получали с помощью раствора 4 М HCl, растворенного в диоксане.
Стадия 104-2: Получение (2-(пиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
15 мг (26%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(пиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 105: Получение (2-(6-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 105)
Стадия 105-1: Получение 2-(6-метилпиридин-3-ил)бензо[d]тиазол-6-амина
68 мг (52%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 6-метилпиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 105-2: Получение (2-(6-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
6 мг (9%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(6-метилпиридин-3-ил)бензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 106: Получение (2-(6-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 106)
Стадия 106-1: Получение 2-(6-фторпиридин-3-ил)бензо[d]тиазол-6-амина
25 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 6-фторпиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 106-2: Получение (2-(6-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
22 мг (63%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(6-фторпиридин-3-ил)бензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 107: Получение (2-(фуран-2-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 107)
Стадия 107-1: Получение 2-(фуран-2-ил)бензо[d]тиазол-6-амина гидрохлорида
Реакцию проводили таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир фуран-2-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты. 68 мг (61%-й выход) указанного в заголовке соединения в форме гидрохлорида получали с помощью раствора 4 М HCl, растворенного в диоксане.
Стадия 107-2: Получение (2-(фуран-2-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
53 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(фуран-2-ил)бензо[d]тиазол-6-амина гидрохлорид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 108: Получение (2-(тиофен-2-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 108)
Стадия 108-1: Получение 2-(тиофен-2-ил)бензо[d]тиазол-6-амина гидрохлорида
Реакцию проводили таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир тиофен-2-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты. 32 мг (27%-й выход) указанного в заголовке соединения в форме гидрохлорида получали с помощью раствора 4 М HCl, растворенного в диоксане.
Стадия 108-2: Получение (2-(тиофен-2-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
13 мг (27%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(тиофен-2-ил)бензо[d]тиазол-6-амина гидрохлорид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 109: Получение (2-(5-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 109)
Стадия 109-1: Получение 2-(5-фторпиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорид
Реакцию проводили таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 5-фторпиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты. 56 мг (45%-й выход) указанного в заголовке соединения в форме гидрохлорида получали с помощью раствора 4 М HCl, растворенного в диоксане.
Стадия 109-2: Получение (2-(5-фторпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
42 мг (57%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(5-фторпиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 110: Получение (2-(5-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 110)
Стадия 110-1: Получение 2-(5-метилпиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорид
Реакцию проводили таким же образом, как на стадии 101-3 Примера 101, за исключением того, что пинаколовый эфир 5-метилпиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты. 56 мг (45%-й выход) указанного в заголовке соединения в форме гидрохлорида получали с помощью раствора 4 М HCl, растворенного в диоксане.
Стадия 110-2: Получение (2-(5-метилпиридин-3-ил)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
9 мг (36%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(5-метилпиридин-3-ил)бензо[d]тиазол-6-амина гидрохлорид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 111: Получение (2-(пиридин-3-ил)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида (Соединение 111)
Стадия 111-1: Получение 2-бром-5-нитробензо[d]тиазола
780 мг (59%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-1 Примера 101, за исключением того, что 5-нитробензо[d]тиазол-2-амин использовали вместо 6-нитробензо[d]тиазол-2-амина.
Стадия 111-2: Получение 2-бромбензо[d]тиазол-5-амина
153 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-2 Примера 101, за исключением того, что 2-бром-5-нитробензо[d]тиазол использовали вместо 2-бром-6-нитробензо[d]тиазола.
Стадия 111-3: Получение 2-(пиридин-3-ил)бензо[d]тиазол-5-амина
48 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что 2-бромбензо[d]тиазол-5-амин использовали вместо 2-бромбензо[d]тиазол-6-амина, и пинаколовый эфир пиридин-3-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 111-4: Получение 2-(гшридин-3-ил)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида
18 мг (23%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(пиридин-3-ил)бензо[d]тиазол-5-амин использовали вместо 2 N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 112: Получение (2-(5-метилпиридин-3-ил)бензо[d]тиазол-5-ил)карбоногидразоноилдицианида (Соединение 112)
26 мг (39%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-(5-метилпиридин-3-ил)бензо[d]тиазол-5-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 113: Получение бензо[d]тиазол-2-илкарбоногидразоноилдицианида (Соединение 113)
29 мг (19%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что бензо[d]тиазол-2-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида, и воду использовали в качестве растворителя.
Пример 114: Получение (6-фторбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 114)
29 мг (19%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-фторбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 115: Получение (6-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 115)
29 мг (21%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-хлорбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 116: Получение (6-бромбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 116)
23 мг (17%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-бромбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 117: Получение (6-(трифторметил)бензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 117)
27 мг (20%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-(трифторметил)бензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 118: Получение (4-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 118)
20 мг (15%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 4-хлорбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 119: Получение (5-хлорбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 119)
11 мг (8%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 5-хлорбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 120: Получение (5-бромбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 120)
18 мг (7%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 5-бромбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 121: Получение (6-метоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 121)
26 мг (18%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-метоксибензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 122: Получение (6-этоксибензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 122)
40 мг (29%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-этоксибензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 123: Получение (6-(трифторметокси)бензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 123)
15 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-(трифторметокси)бензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 124: Получение (6-метилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 124)
36 мг (24%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-метилбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 125: Получение (4-метилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 125)
68 мг (46%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 4-метилбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 126: Получение (4,6-диметилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 126)
41 мг (29%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 4,6-диметилбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 127: Получение (5,6-диметилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 127)
32 мг (22%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 5,6-диметилбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 128: Получение (6-феноксибензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 128)
6 мг (4%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-феноксибензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 129: Получение (6-фенилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида (Соединение 129)
Стадия 129-1: Получение 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[d]тиазол-2-амина
6-Бромбензо[d]тиазол-2-амин (50 мг, 0,22 ммоль) растворяли в диоксане в присутствии азота, а затем добавляли Pd(dppf)Cl2-ДХМ (36 мг, 0,04 ммоль), бис(пинаколато)дибор (84 мг, 0,33 ммоль) и ацетат калия (65 мг, 0,66 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 90°С в течение 3 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 38 мг (62%-й выход) указанного в заголовке соединения.
Стадия 129-2: Получение 6-фенилбензо[d]тиазол-2-амина
81 мг (50%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 101-3 Примера 101, за исключением того, что бромбензол использовали вместо 2-бромбензо[d]тиазол-6-амина, и пинаколовый эфир 2-аминобензо[d]тиазол-6-илбороновой кислоты использовали вместо пинаколового эфира фенилбороновой кислоты.
Стадия 129-3: Получение (6-фенилбензо[d]тиазол-2-ил)карбоногидразоноилдицианида
6 мг (5%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что 6-фенилбензо[d]тиазол-2-амин использовали вместо бензо[d]тиазол-2-амина.
Пример 130: Получение (3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида (Соединение 130)
Стадия 130-1: Получение 5-(трет-бутоксикарбонил)амино-2-фторбензонитрила и 5-ди(трет-бутоксикарбонил)амино-2-фторбензонитрила
5-Амино-2-фторбензонитрил (1 г, 7,35 ммоль) и DMAP (4-диметиламинопиридин, 90 мг, 0,74 ммоль) растворяли в ДХМ в присутствии азота, а затем ди-трет-бутилдикарбонат ((Вос)2О, 2,4 г, 11,02 ммоль) добавляли с получением реакционной смеси. Реакционную смесь кипятили с обратным холодильником в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 1,8 г указанных в заголовке соединений (5-(трет-бутоксикарбонил)амино-2-фторбензонитрил и 5-ди(трет-бутоксикарбонил)амино-2-фторбензонитрил).
Стадия 130-2: Получение трет-бутил-3-аминобензо[d]изоксазол-5-илкарбамата
Карбонат калия (8,34 г, 60,36 ммоль) и ацетогидроксамовую кислоту (2,27 г, 30,18 ммоль) растворяли в ДХМ/H2O в присутствии азота, а затем смесь (1,44 г) 5-(трет-бутоксикарбонил)амино-2-фторбензонитрила и 5-ди(трет-бутоксикарбонил)амино-2-фторбензонитрила, полученного в соответствии со стадией 130-1, добавляли с получением реакционной смеси. Реакционную смесь перемешивали в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с эфиром, а затем фильтровали с получением 743 мг указанного в заголовке соединения.
Стадия 130-3: Получение траря-бутил-3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-илкарбамата
Пиразин-2-карбоновую кислоту (200 мг, 1,61 ммоль) растворяли в ДХМ в присутствии азота, затем добавляли оксалилхлорид (0,25 мл, 2,93 ммоль) и каплю ДМФА с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После этого добавляли трет-бутил-3-аминобензо[d]изоксазол-5-илкарбамат (365 мг, 1,47 ммоль), полученный в соответствии со стадией 130-2, и пиридин, и реакционная смесь перемешивали в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с метанолом, а затем фильтровали с получением 225 мг (43%-й выход) указанного в заголовке соединения.
Стадия 130-4: Получение 3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-аммония 2,2,2-трифторацетата
трет-Бутил-3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-илкарбамат, полученный в соответствии со стадией 130-3 (100 мг, 0,28 ммоль), растворяли в ДХМ в присутствии азота, а затем добавляли трифторуксусную кислоту (0,43 мл, 5,63 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Когда реакция завершалась, продукт реакции концентрировали при пониженном давлении, а затем сушили в течение 4 часов. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с метанолом, а затем фильтровали с получением 225 мг (43%-й выход) указанного в заголовке соединения. После этого концентрат затвердевал с этилацетатом, а затем фильтровали с получением 97 мг (93%-й выход) указанного в заголовке соединения.
Стадия 130-5: Получение (3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида
29 мг (63%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-аммония 2,2,2-трифторацетат использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 131: Получение (3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида (Соединение 131)
Стадия 131-1: Получение трет-бутил-3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-илкарбамата
209 мг (43%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 130-3 Примера 130, за исключением того, что 5-метилпиразин-2-карбоновую кислоту использовали вместо пиразин-2-карбоновой кислоты.
Стадия 131-2: Получение 3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-аммония 2,2,2-трифторацетата
84 мг (81%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 130-4 Примера 130, за исключением того, что трет-бутил-3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-илкарбамат использовали вместо трет-бутил-3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-илкарбамата.
Стадия 131-3: Получение (3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида
34 мг (75%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-аммония 2,2,2-трифторацетат использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 132: Получение (3-(3-цианобензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида (Соединение 132)
Стадия 132-1: Получение трет-бутил-3-(3-цианобензамидо)бензо[d]изоксазол-5-илкарбамата
203 мг (43%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 130-3 Примера 130, за исключением того, что 3-цианобензойную кислоту использовали вместо пиразин-2-карбоновой кислоты.
Стадия 132-2: Получение N-(5-аминобензо[d]изоксазол-3-ил)-3-цианобензамида
трет-Бутил-3-(3-цианобензамидо)бензо[d]изоксазол-5-илкарбамат, полученный в соответствии со стадией 132-1, растворяли в ДХМ в присутствии азота, а затем трифторуксусную кислоту добавляли с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении и сушили в течение 4 часов. После этого концентрат затвердевал с этилацетатом, а затем фильтровали с получением 71 мг (96%-й выход) указанного в заголовке соединения.
Стадия 132-3: Получение (3-(3-цианобензамипирадо)бензо[d]изооксазол-5-ил)карбоногидразоноилдицианида
42 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(5-аминобензо[d]изоксазол-3-ил)-3-цианобензамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 133: Получение (3-(4-метоксибензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида (Соединение 133)
Стадия 133-1: Получение трет-бутил-3-(4-метоксибензамидо)бензо[d]изоксазол-5-илкарбамата
312 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 130-3 Примера 130, за исключением того, что 4-метоксибензойную кислоту использовали вместо пиразин-2-карбоновой кислоты.
Стадия 133-2: Получение N-(5-аминобензо[d]изоксазол-3-ил)-4-метоксибензамида
116 мг (78%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 132-2 Примера 132, за исключением того, что трет-бутил-3-(4-метоксибензамидо)бензо[d]изоксазол-5-илкарбамат использовали вместо трет-бутил-3-(3-цианобензамидо)бензо[d]изоксазол-5-илкарбамата.
Стадия 133-3: Получение (3-(4-метоксибензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианида
38 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что N-(5-аминобензо[d]изоксазол-3-ил)-4-метоксибензамид использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида
Пример 134: Получение (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 134)
Стадия 134-1: Получение 6-аминобензо[d]тиазол-2(3H)-она
6-Нитробензо[d]тиазол-2(3H)-он (200 мг, 1,02 ммоль) растворяли в этаноле/воде в присутствии азота, а затем добавляли Fe (228 мг, 4,08 ммоль) и хлорид аммония (545 мг, 10,19 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при 80°С в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали.
Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 83 мг (49%-й выход) указанного в заголовке соединения.
Стадия 134-2: Получение (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
34 мг (47%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-аминобензо[d]тиазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 135: Получение (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 135)
Стадия 135-1: Получение 3-метил-6-нитробензо[d]тиазол-2(3Н)-она
6-Нитробензо[d]тиазол-2(3H)-он (100 мг, 0,48 ммоль) и 10% Pd/C (101 мг, 0,10 ммоль) растворяли в метаноле в присутствии азота, а затем метилиодид (0,05 мл, 0,76 ммоль) и DBU (1,8-диазабицикло[5.4.0]ундец-7-ен, 0,11 мл, 0,76 ммоль) добавляли с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с метанолом с получением 68 мг (64%-й выход) указанного в заголовке соединения.
Стадия 135-2: Получение 6-амино-3-метилбензо[d]тиазол-2(3H)-она
3-Метил-6-нитробензо[d]тиазол-2(3H)-он (100 мг, 0,48 ммоль) и 10% Pd/C (101 мг, 0,10 ммоль) растворяли в метаноле в присутствии азота с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа в присутствии водорода. Когда реакция завершилась, продукт реакции фильтровали, а затем фильтрат концентрировали при пониженном давлении с получением 79 мг (91%-й выход) указанного в заголовке соединения.
Стадия 135-3: Получение (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
94 мг (83%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-метилбензо[d]тиазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 136: Получение (2-метоксибензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 136)
Стадия 136-1: Получение 2-метокси-6-нитробензо[d]тиазола
60% гидрид натрия (45 мг, 1,12 ммоль) и метанол (0,05 мл, 1,12 ммоль) растворяли в тетрагидрофуране при 0°С в присутствии азота, а затем добавляли 2-хлор-6-нитробензо[d]тиазол (200 мг, 0,93 ммоль) с получением реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 95 мг (49%-й выход) указанного в заголовке соединения.
Стадия 136-2: Получение 2-метоксибензо[d]тиазол-6-амина 69 мг (90%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-метокси-6-нитробензо[d]тиазол использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 136-3: Получение (2-метоксибензо[d]тиазол-6-ил)карбоногидразоноилдицианида
60 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 2-метоксибензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 137: Получение (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 137)
Стадия 137-1: Получение 6-аминобензо[d]оксазол-2(3Н)-она
131 мг (78%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 137-2: Получение (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
115 мг (76%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-аминобензо[d]оксазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 138: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 138)
Стадия 138-1: Получение 3-метил-6-нитробензо[d]оксазол-2(3H)-она
111 мг (88%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-1 Примера 135, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она.
Стадия 138-2: Получение 6-амино-3-метилбензо[d]оксазол-2(3H)-она
282 мг (87%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3-метил-6-нитробензо[d]оксазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 138-3: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
107 мг (73%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-метилбензо[d]оксазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 139: Получение (2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 139)
Стадия 139-1: Получение 5-амино-1H-бензо[d]имидазол-2(3H)-она
122 мг (73%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 139-2: Получение (2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
15 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 5-амино-1H-бензо[d]имидазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 140: Получение диэтил-2-(2-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)гидразоно)малоната (Соединение 140)
6-(4-Аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)-он (50 мг, 0,25 ммоль) и нитрит натрия (40 мг, 0,29 ммоль) растворяли в этаноле в присутствии азота, а затем 1,0 М водный раствор соляной кислоты (0,74 мл, 0,74 ммоль) добавляли с получением реакционной смеси. Реакционную смесь перемешивали при 0°С в течение 10 минут с образованием диазониевой соли. После образования диазониевой соли добавляли диэтилмалонат (80 мкл, 0,50 ммоль) с последующим перемешиванием при комнатной температуре в течение 10 минут. После этого рН реакционной смеси регулировали с помощью водного раствора ацетата натрия, а затем реакционную смесь дополнительно перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 17 мг (18%-й выход) указанного в заголовке соединения.
Пример 141: Получение 2-имино-N'-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)-2-(пиперазин-1-ил)ацетогидразоноилцианида (Соединение 141)
(4-(4-Метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбоногидразоноилдицианид (50 мг, 0,18 ммоль) и пиперазин (23 мг, 0,27 ммоль) растворяли в этаноле, а затем перемешивали при 80°С в течение 17,5 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с этанолом с получением 50 мг (76%-й выход) указанного в заголовке соединения.
Пример 142: Получение 2-амино-N'-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)-2-морфолиноацетогидразоноилцианида (Соединение 142)
61 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 141, за исключением того, что морфолин использовали вместо пиперазина.
Пример 143: Получение метил-2-(2-(4-(4-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)гидразоно)пропаноата (Соединение 143)
6-(4-Аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)-он (200 мг, 0,98 ммоль), нитрит натрия (81,4 мг, 1,18 ммоль) и 1,0 М водный раствор соляной кислоты (2,9 мл, 2,95 ммоль) смешивали с получением реакционной смеси, и реакционную смесь перемешивали при 0°С в течение 10 минут. После этого добавляли гидрат хлорида олова(II) (666 мг, 2,95 ммоль) и 1,0 М водный раствор соляной кислоты (0,68 мл) с последующим перемешиванием при 0°С в течение 10 минут, а затем добавляли метилпируват (0,18 мл, 1,97 ммоль) с последующим перемешиванием при комнатной температуре в течение 30 минут. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с этанолом с получением 177 мг (60%-й выход) указанного в заголовке соединения.
Пример 144: Получение метил-2-циано-2-(2-(4-(пиримидин-2-ил)фенил)гидразоно)ацетата (Соединение 144)
4-(Пиримидин-2-ил)анилин (50 мг, 0,29 ммоль) и нитрит натрия (24 мг, 0,35 ммоль) растворяли в этаноле в присутствии азота, а затем в 1,0 М водном растворе соляной кислоты (0,88 мл, 0,88 ммоль) добавляли с получением реакционной смеси, и реакционную смесь перемешивали при 0°С в течение 10 минут. После образования диазониевой соли добавляли диэтилмалонат (80 мкл, 0,50 ммоль) с последующим перемешиванием при комнатной температуре в течение 10 минут. Добавляли метил-2-цианоацетат (31 мкл, 0,35 ммоль) с последующим перемешиванием при комнатной температуре в течение 10 минут. рН реакционной смеси доводили до 6,0 с помощью водного раствора гидроксида натрия, а затем реакционную смесь дополнительно перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 25 мг (64%-й выход) указанного в заголовке соединения.
Пример 145: Получение 2-амино-2-оксо-N'-(4-(пиримидин-2-ил)фенил)ацетогидразоноилцианида (Соединение 145)
14 мг (18%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 144, за исключением того, что 2-цианоацетамид использовали вместо метил-2-цианоацетата.
Пример 146: Получение этил-2-циано-2-(2-(4-(пиримидин-2-ил)фенил)гидразоно)ацетата (Соединение 146)
24 мг (30%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 144, за исключением того, что 2-цианоацетамид использовали вместо метил-2-цианоацетата.
Пример 147: Получение 2-амино-N'-(4-(пиримидин-2-ил)фенил)-2-тиоксоацетогидразоноилцианида (Соединение 147)
35 мг (43%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 144, за исключением того, что 2-цианоэтантиоамид использовали вместо метил-2-цианоацетата.
Пример 148: Получение 4-метил-N'-(4-(пиримидин-2-ил)фенил)тиазол-2-карбогидразоноилцианида (Соединение 148)
Стадия 148-1: Получение 2-(4-метилтиазол-2-ил)ацетонитрила
2-Цианоэтантиоамид (500 мг, 4,99 ммоль), хлорацетон (0,45 мл, 5,49 ммоль) и триэтиламин (0,70 мл, 4,99 ммоль) растворяли в диметилформамиде с получением реакционной смеси, и реакционную смесь перемешивали при 40°С в течение 1,5 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 163 мг (24%-й выход) указанного в заголовке соединения.
Стадия 148-2: Получение 4-метил-N'-(4-(пиримидин-2-ил)фенил)тиазол-2-карбогидразоноилцианида
18 мг (19%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 144, за исключением того, что 2-(4-метилтиазол-2-ил)ацетонитрил, полученный в соответствии со стадией 148-1, использовали вместо 2-цианоэтантиоамида.
Пример 149: Получение (4-(5-этинилпиримидин-2-ил)фенил)карбогидразоноилдицианида (Соединение 149)
Стадия 149-1: Получение 4-(5-бромпиримидин-2-ил)анилина
2,5-Дибромпиримидин (2 г, 8,41 ммоль) и пинаколовый эфир 4-аминофенилбороновой кислоты (2,3 г, 10,51 ммоль) растворяли в растворе 1,4-диоксана, а затем Pd(PPh3)4 (484 мг, 0,42 ммоль) и 2,0 М водный раствор фосфата калия (12,6 мл, 25,2 ммоль) добавляли с получением реакционной смеси. Реакционную смесь перемешивали при 90°С в течение 2,5 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 1,8 г (68%-й выход) указанного в заголовке соединения.
Стадия 149-2: Получение 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина
4-(5-Бромпиримидин-2-ил)анилин (220 мг, 0,88 ммоль) получали в соответствии со стадией 149-1 и этинилтриметилсилан (1,32 ммоль) растворяли в диметилформамиде, а затем Pd(PPh3)2Cl2 (31 мг, 0,04 ммоль), иодид меди (CuI, 13 мг, 0,07 ммоль) и триэтиламин (0,3 мл, 2,2 ммоль) добавляли с получением реакционной смеси. Реакционную смесь перемешивали при 100°С в течение 2 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 148 мг (63%-й выход) указанного в заголовке соединения.
Стадия 149-3: Получение (4-(5-этинилпиримидин-2-ил)фенил)карбогидразоноилдицианида
92 мг (22%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 144, за исключением того, что малононитрил использовали вместо метил-2-цианоацетата, и 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилин, полученный в соответствии со стадией 149-2, использовали вместо 4-(пиримидин-2-ил)анилина.
Пример 150: Получение (4'-(диметиламино)-3-фторбифенил-4-ил)карбогидразоноилдицианида (Соединение 150)
Стадия 150-1: Получение 3'-фтор-N,N-диметил-4'-нитробифенил-4-амина
134 мг (54%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-1 Примера 149, за исключением того, что 4-бром-2-фтор-1-нитробензол использовали вместо 2,5-дибромпиримидина, и пинаколовый эфир 4-диметиламинофенилбороновой кислоты использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты.
Стадия 150-2: Получение 3-фтор-N4',N4'-диметилбифенил-4,4'-диамина
51 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3'-фтор- N,N-диметил-4'-нитробифенил-4-амин использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 150-3: Получение (4'-(диметиламино)-3-фторбифенил-4-ил)карбогидразоноилдицианида
11 мг (14%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 3-фтор-N4',N4'-диметилбифенил-4,4'-диамин использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 151: Получение (4'-(диметиламино)бифенил-4-ил)карбогидразоноилдицианида (Соединение 151)
Стадия 151-1: Получение N,N-диметил-4'-нитробифенил-4-амина
182 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-1 Примера 149, за исключением того, что 4-бром-1-нитробензол использовали вместо 2,5-дибромпиримидина, и пинаколовый эфир 4-диметиламинофенилбороновой кислоты использовали вместо пинаколового эфира 4-аминофенилбороновой кислоты.
Стадия 151-2: Получение N4',N4'-диметилбифенил-4,4'-диамина
51 мг (48%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что N,N-диметил-4'-нитробифенил-4-амин использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 151-3: Получение (4'-(диметиламино)бифенил-4-ил)карбогидразоноилдицианида
10 мг (6%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что N4',N4'-диметилбифенил-4,4'-диамин использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 152: Получение (4-(6-(диметиламино)пиридин-3-ил)фенил)карбогидразоноилдицианида (Соединение 152)
Стадия 152-1: Получение 5-(4-аминофенил)-N,N-диметилпиридин-2-амина
127 мг (60%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-1 Примера 149, за исключением того, что 5-бром-N,N-диметилпиридин-2-амин использовали вместо 2,5-дибромпиримидина.
Стадия 152-2: Получение (4-(6-(диметиламино)пиридин-3-ил)фенил)карбогидразоноилдицианида
16 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 5-(4-аминофенил)-N,N-диметилпиридин-2-амин использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 153: Получение (4-(6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 153)
Стадия 153-1: Получение 6-хлорпиридазин-3(2H)-она
3,6-Дихлорпиридазин (1 г, 6,71 ммоль) растворяли в уксусной кислоте (26 мл) с получением реакционной смеси, а затем реакционную смесь перемешивали при 110°С в течение 12 часов. Когда реакция завершалась, продукт реакции концентрировали и добавляли водный раствор гидрокарбоната натрия, а затем продукт реакции перемешивали при комнатной температуре до затвердевания и концентрировали при пониженном давлении с получением 420 мг (48%-й выход) указанного в заголовке соединения.
Стадия 153-2: Получение 6-(4-аминофенил)пиридазин-3(2H)-она
6-Хлорпиридазин-3(2H)-он (100 мг, 0,77 ммоль) и пинаколовый эфир 4-аминофенилбороновой кислоты (168 мг, 0,77 ммоль) растворяли в растворе 1,4-диоксана, а затем Pd(PPh3)4 (89 мг, 0,08 ммоль) и водный раствор карбоната калия (423 мг, 3,06 ммоль) добавляли с получением реакционной смеси. Реакционную смесь перемешивали в микроволновой печи при 160°С в течение 1,5 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 36 мг (25%-й выход) указанного в заголовке соединения.
Стадия 153-3: Получение (4-(6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
96 мг (68%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)пиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 154: Получение (4-(4-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 154)
Стадия 154-1: Получение 6-хлор-5-метилпиридазин-3(2H)-она и 6-хлор-4-метилпиридазин-3(2H)-она
3,6-Дихлор-4-метилпиридазин (500 мг, 3,07 ммоль) растворяли в уксусной кислоте (13 мл) с получением реакционной смеси, а затем реакционную смесь перемешивали при 110°С в течение 12 часов. Когда реакция завершалась, продукт реакции концентрировали, затем добавляли водный раствор гидрокарбоната натрия, и продукт реакции перемешивали при комнатной температуре. После этого продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением указанных в заголовке соединений, 184 мг (41%-й выход) 6-хлор-5-метилпиридазин-3(2H)-она и 31 мг (7%-й выход) 6-хлор-4-метилпиридазин-3(2H)-она.
Стадия 154-2: Получение 6-(4-аминофенил)-5-метилпиридазин-3(2H)-она
122 мг (44%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 153-2 Примера 153, за исключением того, что смесь 6-хлор-5-метилпиридазин-3(2H)-она и 6-хлор-4-метилпиридазин-3(2H)-она использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 154-3: Получение (4-(4-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
96 мг (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-5-метилпиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 155: Получение (4-(5-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 155)
Стадия 155-1: Получение 6-(4-аминофенил)-4-метилпиридазин-3(2H)-она
87 мг (62%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 153-2 Примера 153, за исключением того, что смесь 6-хлор-5-метилпиридазин-3(2H)-она и 6-хлор-4-метилпиридазин-3(2H)-она использовали вместо 6-хлорпиридазин-3(2Н)-она.
Стадия 155-2: Получение (4-(5-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
60 мг (87%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-4-метилпиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 156: Получение (4-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 156)
Стадия 156-1: Получение 6-хлор-2-метилпиридазин-3(2H)-она
6-Хлорпиридазин-3(2H)-он (100 мг, 0,77 ммоль), метилиодид (95 мкл, 1,53 ммоль) и карбонат калия (212 мг, 1,53 ммоль) растворяли в диметилформамиде с получением реакционной смеси, а затем реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 82 мг (74%-й выход) указанного в заголовке соединения.
Стадия 156-2: Получение 6-(4-аминофенил)-2-метилпиридазин-3(2H)-она
86 мг (95%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 153-2 Примера 153, за исключением того, что 6-хлор-2-метилпиридазин-3(2H)-он использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 156-3: Получение (4-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
41 мг (59%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-2-метилпиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 157: Получение (4-(1,4-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 157)
Стадия 157-1: Получение 6-хлор-2,5-диметилпиридазин-3(2Н)-она
53 мг (49%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 156-1 Примера 156, за исключением того, что 6-хлор-5-метилпиридазин-3(2H)-он использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 157-2: Получение 6-(4-аминофенил)-2,5-диметилпиридазин-3(2H)-она
51 мг (76%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 153-2 Примера 153, за исключением того, что 6-хлор-2,5-диметилпиридазин-3(2H)-он использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 157-3: Получение (4-(1,4-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
37 мг (77%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-2,5-диметилпиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 158: Получение (4-(1,5-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 158)
Стадия 158-1: Получение 6-хлор-2,4-диметилпиридазин-3(2H)-она
50 мг (65%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 156-1 Примера 156, за исключением того, что 6-хлор-4-метилпиридазин-3(2H)-он использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 158-2: Получение 6-(4-аминофенил)-2,4-диметилпиридазин-3(2H)-она
47 мг (70%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 153-2 Примера 153, за исключением того, что 6-хлор-2,4-диметилпиридазин-3(2H)-он использовали вместо 6-хлорпиридазин-3(2H)-она.
Стадия 158-3: Получение (4-(1,5-диметил-6-оксо-1,6-дигидропиридазин-3-ил)фенил)карбогидразоноилдицианида
11 мг (22%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-2,4-диметилпиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 159: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбогидразоноилдицианида (Соединение 159)
Стадия 159-1: Получение 3-изопропил-6-нитробензо[d]тиазол-2(3H)-она
162 мг (44%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-1 Примера 135, за исключением того, что 2-иодпропан использовали вместо метилиодида.
Стадия 159-2: Получение 6-амино-3-изопропилбензо[d]тиазол-2(3H)-она
112 мг (85%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3-изопропил-6-нитробензо[d]тиазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 159-3: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
109 мг (80%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-изопропилбензо[d]тиазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 160: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбогидразоноилдицианида (Соединение 160)
Стадия 160-1: Получение 3-(дифторметил)-6-нитробензо[d]тиазол-2(3H)-она
6-Нитробензо[d]тиазол-2(3H)-он (300 мг, 1,53 ммоль) растворяли в ДМФА в присутствии азота, а затем добавляли 2-хлор-2,2-дифторацетат натрия (467 мг, 3,06 ммоль) и DBU (0,46 мл, 3,06 ммоль) с получением реакционной смеси, и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал с метанолом с получением 176 мг (47%-й выход) указанного в заголовке соединения.
Стадия 160-2: Получение 6-амино-3-(дифторметил)бензо[d]тиазол-2(3H)-она
125 мг (95%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3-(дифторметил)-6-нитробензо[d]тиазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 160-3: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
85 мг (63%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-(дифторметил)бензо[d]тиазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 161: Получение (2-изопропоксибензо[d]тиазол-6-ил)карбогидразоноилдицианида (Соединение 161)
Стадия 161-1: Получение 2-изопропокси-6-нитробензо[d]тиазола -
60% гидрид натрия (84 мг, 2,10 ммоль) и изопропанол (0,16 мл, 2,10 ммоль) растворяли в ТГФ при 0°С в присутствии азота, азатем 2-хлор-6-нитробензо[d]тиазол (300 мг, 1,40 ммоль) добавляли с получением реакционной смеси, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 184 мг (55%-й выход) указанного в заголовке соединения.
Стадия 161-2: Получение 2-изопропоксибензо[d]тиазол-6-амина
126 мг (96%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 2-изопропокси-6-нитробензо[d]тиазол использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 161-3: Получение (2-изопропоксибензо[d]тиазол-6-ил)карбоногидразоноилдицианида
30 мг (22%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 2-изопропоксибензо[d]тиазол-6-амин использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 162: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбогидразоноилдицианида (Соединение 162)
Стадия 162-1: Получение 3-изопропил-6-нитробензо[d]оксазол-2(3H)-она
201 мг (54%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-1 Примера 135, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она, и 2-иодпропан использовали вместо метилиодида.
Стадия 162-2: Получение 6-амино-3-изопропилбензо[d]оксазол-2(3H)-она
124 мг (85%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3-изопропил-6-нитробензо[d]тиазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 162-3: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
99 мг (59%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-изопропилбензо[d]оксазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 163: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбогидразоноилдицианида (Соединение 163)
Стадия 163-1: Получение 3-(дифторметил)-6-нитробензо[d]оксазол-2(3H)-она
144 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 160-1 Примера 160, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она.
Стадия 163-2: Получение 6-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-она
110 мг (98%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 3-(дифторметил)-6-нитробензо[d]оксазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 163-3: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
85 мг (63%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 6-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 164: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианида (Соединение 164)
Стадия 164-1: Получение 1,3-диметил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
207 мг (64%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-1 Примера 135, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она.
Стадия 164-2: Получение 5-амино-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она
105 мг (82%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 1,3-диметил-5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 164-3: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
11 мг (8%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 5-амино-1,3-диметил-1H-бензо[d]имидазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 165: Получение (1,3-диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианида (Соединение 165)
Стадия 165-1: Получение 1,3-диизопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
204 мг (46%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-1 Примера 135, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она, и 2-иодпропан использовали вместо метилиодида.
Стадия 165-2: Получение 5-амино-1,3-диизопропил-1H-бензо[d]имидазол-2(3H)-она
118 мг (89%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 1,3-диизопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 165-3: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
12 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 5-амино-1,3-диизопропил-1H-бензо[d]имидазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 166: Получение (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбогидразоноилдицианида (Соединение 166)
Стадия 166-1: Получение 1,3-бис(дифторметил)-5-нитро-1H-бензо[d]имидазол-2(3Н)-она
179 мг (38%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 160-1 Примера 160, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3H)-она.
Стадия 166-2: Получение 5-амино-1,3-бис(дифторметил)-1H-бензо[d]имидазол-2(3H)-она
69 мг (96%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что 1,3-бис(дифторметил)-6-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 166-3: Получение (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
45 мг (58%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 86-3 Примера 86, за исключением того, что 5-амино-1,3-бис(дифторметил)-1H-бензо[d]имидазол-2(3H)-он использовали вместо N-(6-аминобензо[d]тиазол-2-ил)-4-метоксибензамида.
Пример 167: Получение (2-(метиламино)бензо[d]тиазол-6-ил)карбогидразоноилдицианида (Соединение 167)
Стадия 167-1: Получение N-метил-6-нитробензо[d]тиазол-2-амина
2-Хлор-6-нитробензо[d]тиазол (500 мг, 2,33 ммоль) растворяли в ТГФ при комнатной температуре в присутствии азота, а затем 2,0 М метиламина (2,56 мл, 5,13 ммоль) растворяли в ТГФ добавляли с получением реакционной смеси, и реакционную смесь перемешивают при комнатной температуре в течение 15 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат затвердевал этилацетатом с получением 442 мг (91%-й выход) указанного в заголовке соединения.
Стадия 167-2: Получение N2-метилбензо[d]тиазол-2,6-диамина
101 мг (78%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что N-метил-6-нитробензо[d]тиазол-2-амин использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 167-3: Получение (2-(метиламино)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
5 мг (4%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что N2-метилбензо[d]тиазол-2,6-диамин использовали вместо бензо[d]тиазол-2-амина.
Пример 168: Получение (2-(диметиламино)бензо[d]тиазол-6-ил)карбогидразоноилдицианида (Соединение 168)
Стадия 168-1: Получение N,N-диметил-6-нитробензо[d]тиазол-2-амина
392 мг (75%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 167-1 Примера 167, за исключением того, что диметиламин использовали вместо метиламина.
Стадия 168-2: Получение N2,N2-диметилбензо[d]тиазол-2,6-диамина
103 мг (80%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 135-2 Примера 135, за исключением того, что N,N-диметил-6-нитробензо[d]тиазол-2-амин использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
Стадия 168-3: Получение (2-(диметиламино)бензо[d]тиазол-6-ил)карбоногидразоноилдицианида
11 мг (10%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как в Примере 113, за исключением того, что N2,N2-диметилбензо[d]тиазол-2,6-диамин использовали вместо бензо[d]тиазол-2-амина.
Пример 169: Получение (4-(6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 169)
Стадия 169-1: Получение N-(4-(6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)ацетамида
4-(4-Ацетамидофенил)-4-оксобутановую кислоту (100 мг, 0,42 ммоль) растворяли в этаноле, а затем добавляли гидразин-гидрат (25 мкл, 0,51 ммоль) с получением реакционной смеси, и реакционную смесь перемешивали при 90°С в течение 3,5 часов. Когда реакция завершилась, продукт реакции отфильтровали с получением 47 мг (48%-й выход) указанного в заголовке соединения.
Стадия 169-2: Получение 6-(4-аминофенил)-4,5-дигидропиридазин-3(2H)-она
1,0 М водный раствор соляной кислоты (21,6 мл, 21,6 ммоль) добавляли в N-(4-(6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)ацетамид, полученный в соответствии со стадией 169-1, а затем вводили в реакцию при 110°С в течение 1,5 часов. После этого продукт реакции нейтрализовали водным раствором гидроксида аммония и фильтровали с получением 118 мг (28%-й выход) указанного в заголовке соединения.
Стадия 169-3: Получение (4-(6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида
66 мг (93%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-4,5-дигидропиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 170: Получение (4-(1-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 170)
Стадия 170-1: Получение 6-(4-аминофенил)-2-метил-4,5-дигидропиридазин-3(2H)-она
6-(4-Аминофенил)-4,5-дигидропиридазин-3(2H)-он (105 мг, 0,55 ммоль) и 60% гидрид натрия (24 мг, 0,61 ммоль) растворяли в смешанном растворителе ДМФА и ТГФ, а затем добавляли метилиодид (34 мкл, 0,55 ммоль) с получением реакционной смеси, и реакционную смесь перемешивали при 0°С в течение 12 часов. Когда реакция завершалась, продукт реакции экстрагировали, используя дистиллированную воду и этилацетатом с получением органического слоя, и органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией с получением 33 мг (29%-й выход) указанного в заголовке соединения.
Стадия 170-2: Получение (4-(1-метил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида
90 мг (95%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-2-метил-4,5-дигидропиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Пример 171: Получение (4-(1,4-диметил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида (Соединение 171)
Стадия 171-1: Получение 6-(4-аминофенил)-2,5-диметил-4,5-дигидропиридазин-3(2Н)-она
131 мг (61%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 170-1 Примера 170, за исключением того, что 6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)-он использовали вместо 6-(4-аминофенил)-4,5-дигидропиридазин-3(2H)-она.
Стадия 171-2: Получение (4-(1,4-диметил-6-оксо-1,4,5,6-тетрагидропиридазин-3-ил)фенил)карбогидразоноилдицианида
82 мг (76%-й выход) указанного в заголовке соединения получали проведением реакции таким же образом, как на стадии 149-3 Примера 149, за исключением того, что 6-(4-аминофенил)-2,5-диметил-4,5-дигидропиридазин-3(2H)-он использовали вместо 4-(5-((триметилсилил)этинил)пиримидин-2-ил)анилина.
Получение примера 1: Получение таблеток прямым прессованием
5,0 мг каждого из активных ингредиентов, полученных в примерах с 1 по 171, просеивали, смешивали с 14,1 мг лактозы, 0,8 мг кросповидона USNF и 0,1 мг стеарата магния, а затем прессовали с получением таблеток.
Получение примера 2: Получение таблеток методом влажной грануляции
Просеивали 5,0 мг каждого из активных ингредиентов, полученных в примерах с 1 по 171, а затем смешивали с 16,0 мг лактозы и 4,0 мг крахмала. 0,3 мг полисорбата 80 растворяли в чистой воде, и этот раствор добавляли к смеси в подходящем количестве с последующим распылением с получением мелких частиц. После сушки мелкие частицы просеивали, смешивали с 2,7 мг коллоидного диоксида кремния и 2,0 мг стеарата магния и прессовали с получением таблеток.
Получение примера 3: Получение порошка и капсул
Просеивали 5,0 мг каждого из активных ингредиентов, полученных в примерах с 1 по 171, а затем смешивали с 14,8 мг лактозы, 10,0 мг поливинилпирролидона и 0,2 мг стеарата магния. Смесью заполняли твердые желатиновые капсулы №5, используя подходящее устройство для получения капсул.
Получение примера 4: Получение инъекционных препаратов
100 мг каждого из активных ингредиентов, полученных в примерах с 1 по 171, смешивали с 180 мг маннита, 26 мг Na2HPO4⋅12Н2О, и 2974 мг дистиллированной воды для получения инъекционных препаратов.
Экспериментальный пример 1: Выбор материалов, ингибирующих агрегацию тау-белка, с использованием клеточных моделей
Для выбора новых материалов, ингибирующих агрегацию тау-белка, использовали клеточные модели тау-BiFC, в которых легко наблюдать образование тау-олигомеров в живых клетках. Клетки Tau-BiFC разливали в 384-луночные планшеты, и на следующий день соединения, полученные в соответствии с примерами с 1 по 171, обрабатывали в концентрации 1 мкМ, 3 мкМ и 10 мкМ соответственно вместе с форсколином (концентрация обработки 30 мкМ), который стимулирует тау-фосфорилирование за счет активации PKA и индуцирует агрегацию тау-белка. Через 48 часов внутриклеточные ядра окрашивали с помощью Hoechst (концентрация обработки 2 мкг/мл), и интенсивность флуоресценции BiFC автоматически определяли количественно с помощью Operetta (PerkinElmer) для подсчета окрашенных ядер в каждой лунке из всех лунок планшетов. Группа, получавшая только форсколин, который индуцирует агглютинацию тау-белка, была установлена в качестве контрольной точки в состоянии агрегации 100% тау-белка, и ее эффект был подтвержден уравнением «Интенсивность флуоресценции BiFC соединением, синтезированным в соответствии с вариантом осуществления настоящего изобретения / (интенсивность флуоресценции контрольной группы, обработанной только форсколином, вызывающим агрегацию тау-белка - интенсивность флуоресценции необработанной контрольной группы) х 100». Кроме того, степень цитотоксичности, вызванной вновь синтезированным соединением, также основана на 100% выживаемости группы, получавшей только форсколин, и значение цитотоксичности соединения рассчитывали по уравнению «количество окрашенных ядер в группе, обработанной соединением / количество окрашенных ядер в группе, получавшей форсколин х 100». По результатам лечения материалы, которые ингибируют внутриклеточную агрегацию тау-белка, выбирали из ряда групп кандидатов на основании 70% или более степени ингибирования агрегации тау-белка и 100% выживаемости клеток при концентрации обработки соединения 10 мкМ или более.
Экспериментальный пример 2: Подтверждение концентрационно-зависимых эффектов ингибирования агрегации тау-белка новыми соединениями
Чтобы оценить дозозависимые эффекты ингибирования агрегации тау-белка соединений, выбранных в соответствии с экспериментальным примером 1, выбранные соединения обрабатывали в клетках тау-BiFC в концентрациях 0,03 мкМ, 0,01 мкМ, 0,3 мкМ, 1 мкМ, 3 мкМ, 10 мкМ и 30 мкМ, соответственно, вместе с форсколином (концентрация обработки 30 мкМ) в качестве материала, индуцирующего агрегацию тау-белка. Через 48 часов реакцию агрегации тау-белка и степень цитотоксичности анализировали путем наблюдения изображений клеток. IC50 и токсичность соединений анализировали с помощью нелинейного регрессионного анализа программного обеспечения призмы (Graph Pad). Результаты, рассчитанные для репрезентативных соединений, проиллюстрированы в Таблице 1 ниже.
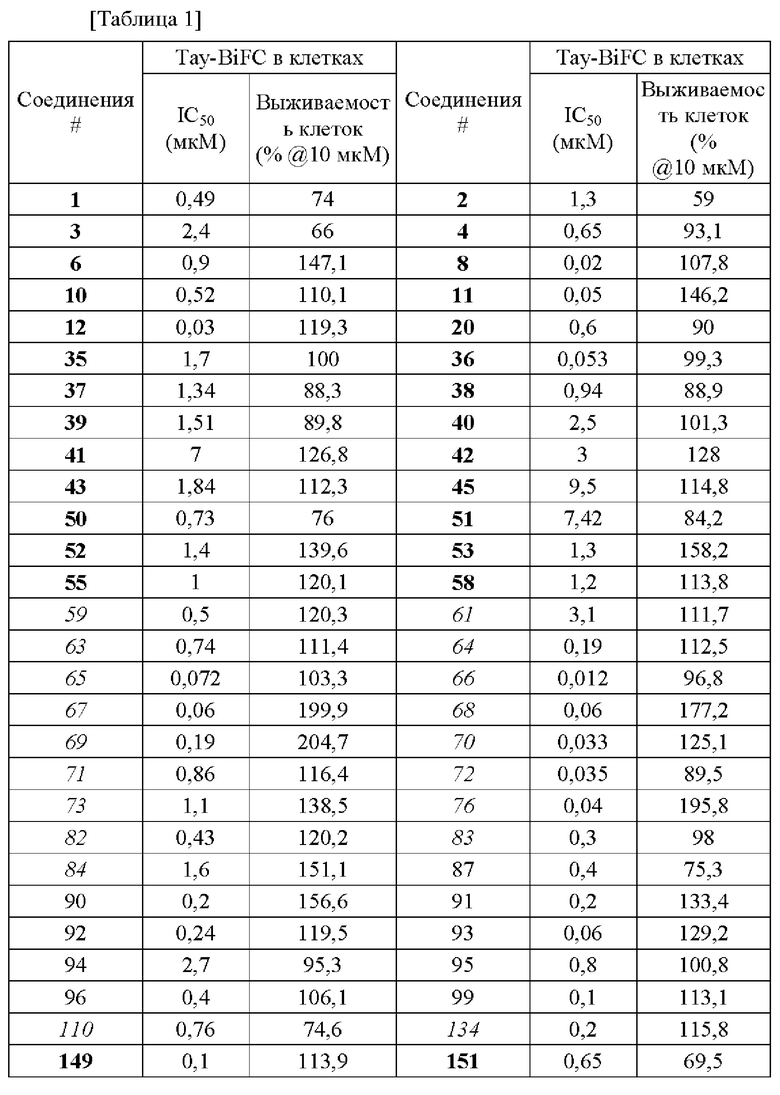
Экспериментальный пример 3: Эффекты ингибирования активности кофермента CYP новыми соединениями
Подтверждены эффекты ингибирования активности кофермента CYP соединениями, полученными в соответствии с примерами 1-171. В частности, микросомы печени человека (0,25 мг/мл), 0,1 М фосфатный буферный раствор (рН 7,4), коктейли субстратных препаратов (фенацетин 50 мкМ, диклофенак 10 мкМ, S-мефенитоин (100 мкМ, декстрометорфан 5 мкМ и мидазолам 2,5 мкМ) пяти видов ферментов метаболизма лекарственных средств, и добавляли соединения с концентрацией 0 мкМ или 10 мкМ, соответственно. Затем смесь предварительно культивировали при 37°С в течение 5 минут, а затем дополнительно культивировали при 37°С в течение 15 минут вместе с добавлением раствора системы генерации НАДФН. После этого реакцию останавливали добавлением раствора ацетонитрила, содержащего материал внутреннего стандарта (терфенадин), разделение центрифугированием (14000 об./мин, 4°С) выполняли в течение 5 минут, а затем супернатант вводили в систему ЖХ-МС/МС.Метаболиты субстратов лекарственных средств одновременно анализировали для оценки способности ингибировать метаболизм лекарственного средства.
Метаболиты каждого препарата-индикатора кофермента CYP, образующегося в ходе реакции, анализировали с использованием системы Shimadzu Nexera XR и TSQ vantage (Thermo). Kinetex С18 (2,1 мм × 100 мм, 2,6 мкм, размер частиц; Phenomenex, США) использовали в колонке для ВЭЖХ, и (А) дистиллированную воду, содержащую 0,1% муравьиной кислоты, и (В) ацетонитрил, содержащий 0 1% муравьиной кислоты, использовали в качестве подвижных фаз и применяли программу градиента, проиллюстрированную в Таблице 2.
Сгенерированные метаболиты были количественно определены с использованием режима количественной оценки MRM (мониторинг множественных реакций), a Xcalibur (версия 1.6.1) использовали для анализа данных. В Таблице 3 проиллюстрированы эффекты ингибирования активности кофермента CYP новых соединений, полученных в соответствии с вариантами осуществления настоящего изобретения.
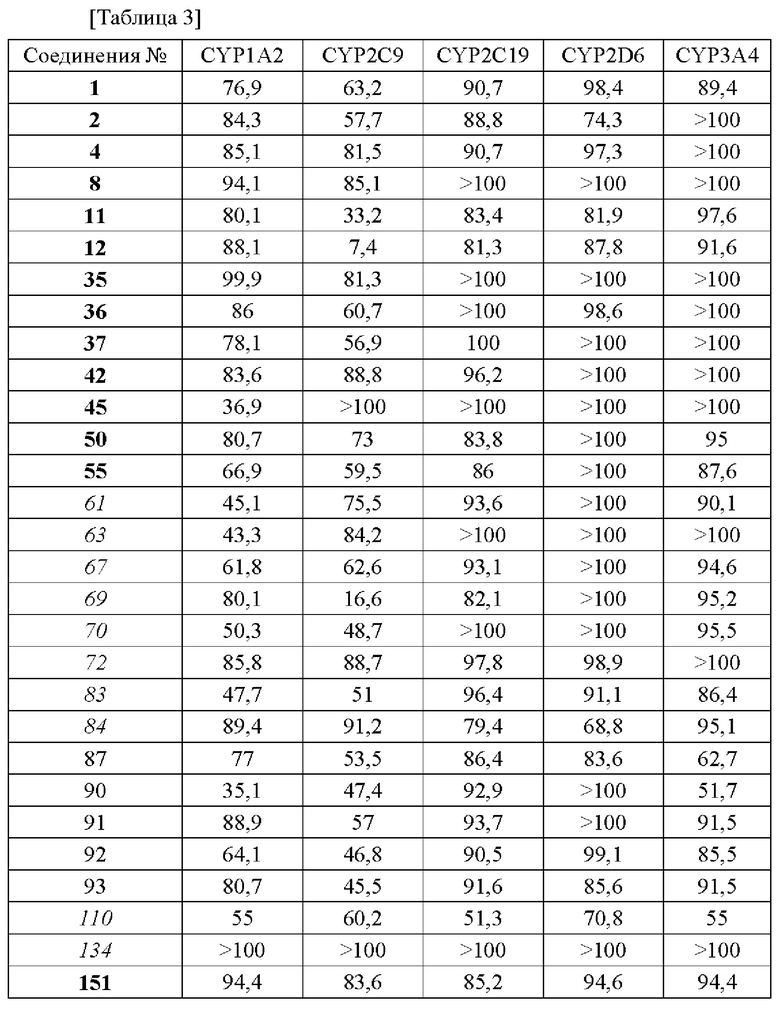
Экспериментальный пример 4: Подтверждение стабильности микросом печени новых соединений
Подтверждали микросомальную стабильность в печени соединений, полученных в соответствии с примерами 1-171. В частности, четыре вида микросом печени (человека, собаки, крысы и мыши, каждая по 0,25 мг/мл), 0,1 М фосфатный буферный раствор (рН 7,4) и соединения с концентрацией 1 мкМ добавляли, соответственно. Затем смесь предварительно культивировали при 37°С в течение 5 минут, а затем дополнительно культивировали при 37°С в течение 30 минут вместе с добавлением раствора системы генерации НАДФН. После этого реакцию останавливали добавлением раствора ацетонитрила, содержащего материал внутреннего стандарта (хлорпропамид), разделение центрифугированием (14000 об./мин, 4°С) выполняли в течение 5 минут, а затем супернатант вводили в систему ЖХ-МС/МС. Субстраты лекарственных средств анализировали для оценки метаболической стабильности 8 видов соединений.
Количество субстрата, оставшегося после реакции, анализировали с использованием системы Shimadzu Nexera XR и TSQ vantage (Thermo). Kinetex XB-C18 (2,1 мм × 100 мм, 2,6 мкм, размер частиц; Phenomenex, США) использовали в колонке для ВЭЖХ, и (А) дистиллированную воду, содержащую 0,1% муравьиной кислоты, и (В) ацетонитрил, содержащий 0 1% муравьиной кислоты, использовали в качестве подвижных фаз. Для анализа данных использовали аналитическое программное обеспечение (версия 1.6.3) и Xcalibur (версия 1.6.1). Расчетные результаты проиллюстрированы в Таблице 4 ниже.
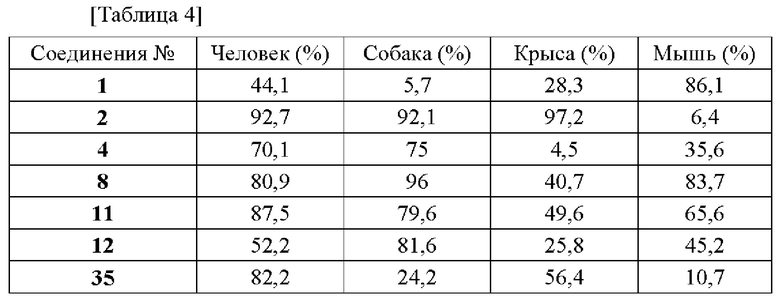
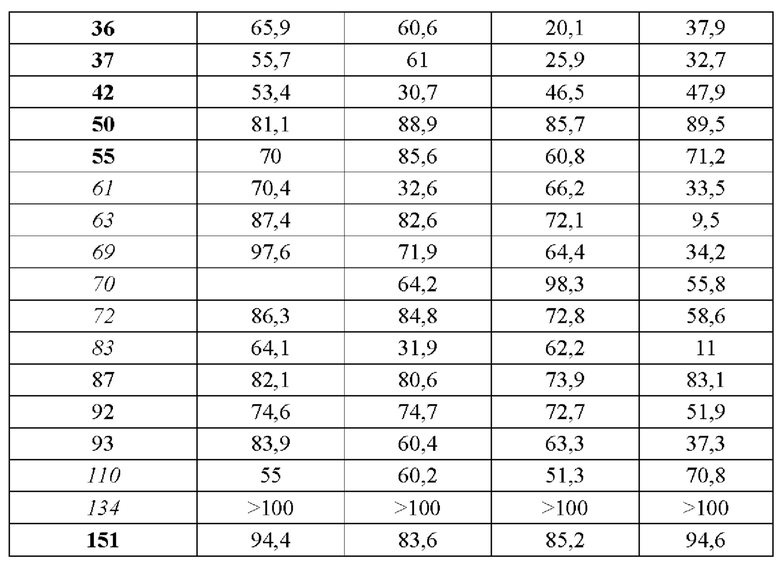
Экспериментальный пример 5: Изучение фармакокинетики новых соединений
Фармакокинетику соединений, полученных в соответствии с примерами 1-171, изучали с использованием экспериментальных моделей на животных. В частности, приобретали крысу SD в возрасте от 7 до 8 недель (Core Tech, Hana Trading Co., Ltd.), имеющую массу тела от около 250 до 300 г, и содержали в простой комнате для разведения мелких животных при температуре 22°С±2°С, относительной влажности 50%±5%, времени освещения 12 часов (загорается в 8:00 утра, выключается в 8:00 вечера) и освещенности от 150 люкс до 300 люкс. течение всего периода испытания обеспечивали свободный доступ к корму, ОС воде, и голодание проводили в течение 16 часов перед пероральным введением лекарственного средства. После приготовления лекарственного средства в условиях, проиллюстрированных в Таблице 5 ниже, лекарственное средство вводили внутривенно (IV) или перорально (РО). 500 мкл перелитой крови собирали в пробирку, содержащую гепарин натрия, и центрифугировали при 2-8°С при 8000 об./мин в течение 6 минут с получением плазмы из крови.
Полученную плазму добавляли к 180 мкл ацетонитрила, содержащего материалы внутреннего стандарта, встряхивали, а затем центрифугировали при 15000 об./мин при 4°С в течение 5 минут с получением супернатанта. Полученный супернатант анализировали с помощью ЖХ-МС/МС. Условия ВЭЖХ и МС, использованные для анализа, проиллюстрированы в Таблицах 6 и 7 ниже.
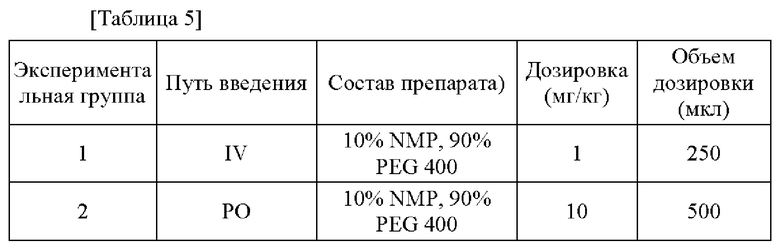
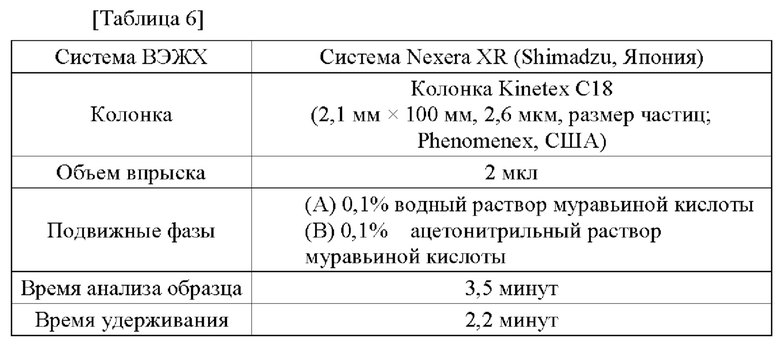
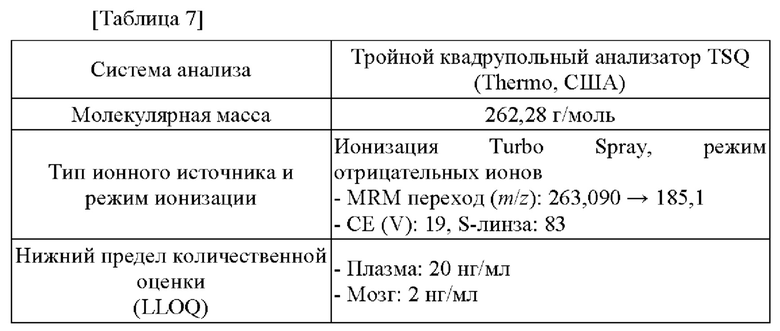

Конкретные параметры ФК получали с помощью программы Phoenix WinNonlin версии 6.4 (Pharsight, США) и рассчитывали с помощью модели некомпартментального анализа. Результаты представлены в Таблице 8 ниже:
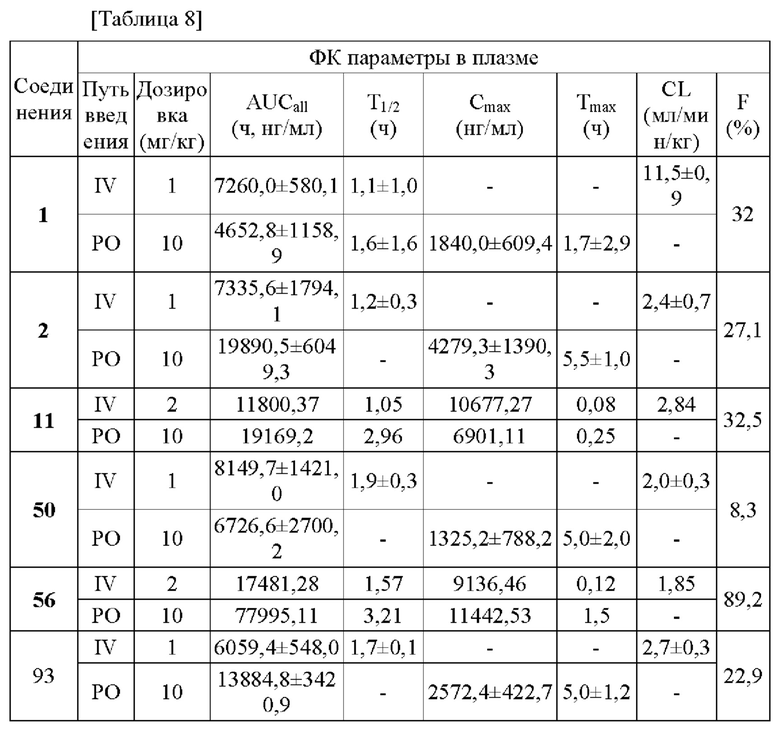
Специалистам в данной области техники будет понятно, что настоящее изобретение, как описано выше, может быть реализовано в других конкретных формах без отступления от технического духа или его основных характеристик. Таким образом, следует принимать во внимание, что варианты осуществления данного изобретения, описанные выше, предназначены для иллюстрации во всех аспектах, а не для ограничения. Объем настоящего изобретения представлен прилагаемой формулой изобретения, которая будет описана ниже, а не подробным описанием, и следует интерпретировать, что смысл и объем прилагаемой формулы изобретения и всех изменений или модифицированных форм, полученных из их эквивалентов, находятся в пределах объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ КОНДЕНСИРОВАННЫХ ГЕТЕРОЦИКЛИЛ-КАРБОНОГИДРАЗОНОИЛДИЦИАНИДОВ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2826002C1 |
| БЛОКАТОРЫ НАТРИЕВЫХ КАНАЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2645711C2 |
| НОВЫЕ ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2636585C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788147C2 |
| БИАРИЛЬНОЕ ПРОИЗВОДНОЕ В КАЧЕСТВЕ АГОНИСТА GPR120 | 2015 |
|
RU2683648C2 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2756505C1 |
| ПРОИЗВОДНЫЕ ОКСИФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2758370C1 |
| БЛОКАТОР НАТРИЕВЫХ КАНАЛОВ | 2016 |
|
RU2705578C1 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
Группа изобретений относится к области медицины, а именно к терапии, и предназначена для профилактики или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка. Представлены новые производные гидразона формулы (4). В другом воплощении обеспечивается способ их получения, включающий введение в реакцию карбоновой кислоты С1-6 алкила, арила или гетероарила или ее ангидрида с 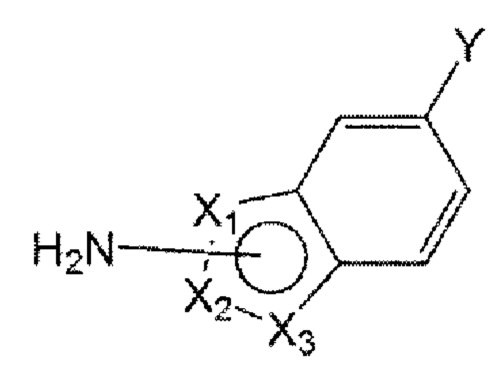 с образованием амидной связи, где Y представляет собой незащищенный или защищенный амин или нитрогруппу; и введение в реакцию нитрита натрия, гетероцикла, связанного с карбоксамидогруппой, полученного на предыдущей стадии, и малононитрила в присутствии кислоты с образованием иминосвязи. Также представлены фармацевтические композиции, содержащие указанные соединения и применяемые для ингибирования агрегации тау-белка, для ингибирования гиперфосфорилирования тау-белка, для профилактики или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка. Группа изобретений обеспечивает новые производные гидразона формулы (4), обладающие ингибирующей активностью по отношению к агрегации тау-белка и гиперфосфорилированию тау-белка. 5 н. и 5 з.п. ф-лы, 8 табл., 176 пр.
с образованием амидной связи, где Y представляет собой незащищенный или защищенный амин или нитрогруппу; и введение в реакцию нитрита натрия, гетероцикла, связанного с карбоксамидогруппой, полученного на предыдущей стадии, и малононитрила в присутствии кислоты с образованием иминосвязи. Также представлены фармацевтические композиции, содержащие указанные соединения и применяемые для ингибирования агрегации тау-белка, для ингибирования гиперфосфорилирования тау-белка, для профилактики или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка. Группа изобретений обеспечивает новые производные гидразона формулы (4), обладающие ингибирующей активностью по отношению к агрегации тау-белка и гиперфосфорилированию тау-белка. 5 н. и 5 з.п. ф-лы, 8 табл., 176 пр.
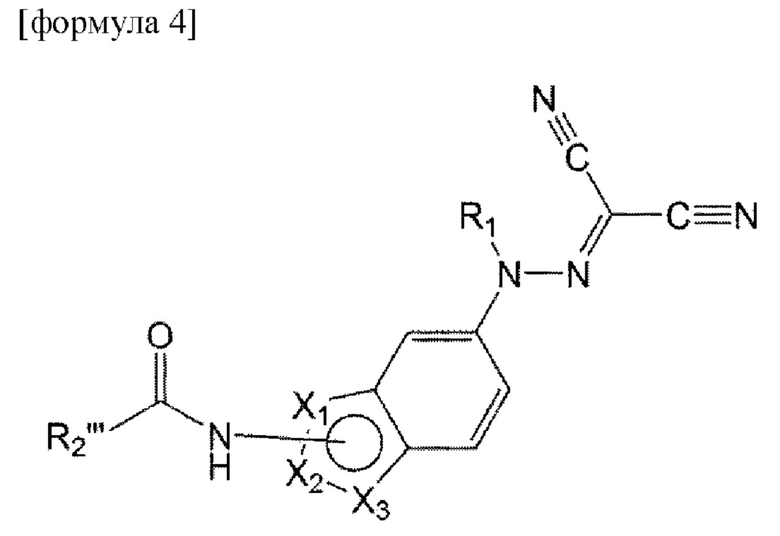
1. Соединение, представленное приведенной ниже формулой 4, или его фармацевтически приемлемая соль,
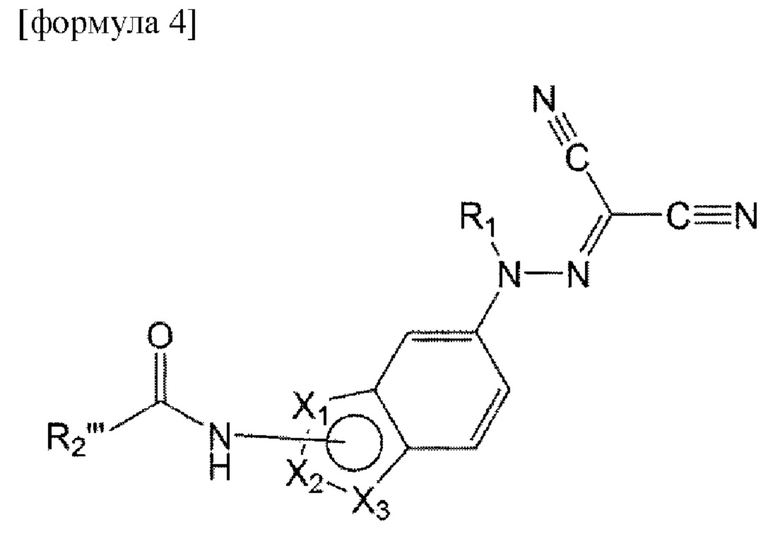
в приведенной выше формуле 4
от X1 до Х3 отличаются друг от друга, каждый выбран из С, N, О и S, и каждый связан с карбоксамидогруппой через С-сайт;
R1 представляет собой водород или C1-6 алкил; и
R2''' представляет собой C1-6 алкил или арил, или гетероарил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из C1-6 алкокси, гидрокси, циано и C1-6 алкила.
2. Соединение по п. 1 или его фармацевтически приемлемая соль,
где от X1 до Х3 представляют собой последовательно N, С и S или О, N и С и связаны с карбоксамидогруппой через С-сайт;
R1 представляет собой водород; и
R2''' представляет собой метил или фенил, пиридинил, пиразинил или пиримидинил, незамещенный или замещенный по меньшей мере одним, выбранным из группы, состоящей из метокси, гидрокси, циано и метила.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение представляет собой
86. (2-(4-метоксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
87. (2-(2-гидроксибензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
88. (2-ацетамидобензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
89. (2-(3-цианобензамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
90. (2-(5-метилникотинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
91. (2-(3-гидроксипиколинамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
92. (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
93. (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
94. (2-(пиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
95. (2-(2-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
96. (2-(4-метилпиримидин-5-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
97. (2-(3-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
98. (2-(6-метилпиразин-2-карбоксамидо)бензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
99. (2-(пиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
100. (2-(5-метилпиразин-2-карбоксамидо)бензо[d]тиазол-5-ил)карбоногидразоноилдицианид,
130. (3-(пиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
131. (3-(5-метилпиразин-2-карбоксамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
132. (3-(3-цианобензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид,
133. (3-(4-метоксибензамидо)бензо[d]изоксазол-5-ил)карбоногидразоноилдицианид.
4. Способ получения соединения по п. 1, включающий стадии:
введение в реакцию карбоновой кислоты R2''' или ее ангидрида с  (здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
(здесь Y представляет собой незащищенный или защищенный амин или нитрогруппу) с образованием амидной связи; и
введение в реакцию нитрита натрия, гетероцикла, содержащего R2''', связанного с карбоксамидогруппой, полученного на предыдущей стадии, и малононитрила в присутствии кислоты с образованием иминосвязи.
5. Способ по п. 4,
где, когда Y представляет собой защищенную аминогруппу или нитрогруппу, данный способ дополнительно включает стадию превращения защищенной аминогруппы или нитрогруппы в аминогруппу перед стадией образования имино-группы.
6. Фармацевтическая композиция для ингибирования агрегации тау-белка, содержащая эффективное количество соединения по любому из пп. 1-3 в качестве активного ингредиента и фармацевтически приемлемый носитель.
7. Фармацевтическая композиция для ингибирования гиперфосфорилирования тау-белка, содержащая эффективное количество соединения по любому из пп. 1-3 в качестве активного ингредиента и фармацевтически приемлемый носитель.
8. Фармацевтическая композиция для профилактики или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка, содержащая эффективное количество соединения по любому из пп. 1-3 в качестве активного ингредиента и фармацевтически приемлемый носитель.
9. Фармацевтическая композиция по п. 8, отличающаяся тем, что заболевание, вызванное агрегацией или гиперфосфорилированием тау-белка, выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, сосудистой деменции, острого инсульта, травмы, цереброваскулярного заболевания, травмы головного мозга, травмы спинного мозга, периферической невропатии, ретинопатии, глаукомы и таупатии.
10. Фармацевтическая композиция по п. 9, отличающаяся тем, что таупатия выбрана из группы, состоящей из неограничивающих примеров таупатии, которые могут включать в себя хроническую травматическую энцефалопатию (СТЕ), первичную возрастную таупатию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию, болезнь Пика, болезнь аргирофильного зерна (AGD), лобно-височную деменцию (FTD), паркинсонизм, связанный с хромосомой 17, литико-бодиговую болезнь (комплекс паркинсонизма-деменции Гуама), ганглиоглиому, ганглиоцитому, менингиоангиоматоз, постэнцефалитный паркинсонизм, подострый склерозирующий панэнцефалит, свинцовую энцефалопатию, туберозный склероз, связанную с пантотенат-киназой нейродегенерацию, липофусциноз и травматическое повреждение мозга.
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| LEVIJOKI J | |||
| et al | |||
| Further evidence for the cardiac troponin C mediated calcium sensitization by levosimendan: structure-response and binding analysis with analogs of levosimendan | |||
| Journal of Molecular and Cellular Cardiology, 2000, Vol.32, No.3, P.479-491 | |||
| TSAI P | |||
| C | |||
| et al | |||
| Synthesis and solvatochromic | |||
