Область техники
Настоящая заявка испрашивает приоритет на основании корейской заявки № 10-2018-0156031, поданной 6 декабря 2018 года, и полное содержание описания и чертежей указанной заявки включено в настоящий документ посредством ссылки.
Настоящее изобретение относится к группе соединений, характеризующихся конкретной структурой и превосходной ингибирующей активностью по отношению к PDE9A. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения. Настоящее изобретение относится к способу, который можно применять для лечения заболеваний, связанных с PDE9A, с использованием указанных соединений. Таким образом, настоящее изобретение относится к медицинскому применению соединений согласно настоящему изобретению для лечения или предотвращения заболеваний, связанных с PDE9A.
Уровень техники
Фосфодиэстераза 9A (PDE9A) в основном экспрессируется в головном мозге, в частности, в неокортексе, мозжечке и гиппокампе; известно, что она ассоциирована с регулированием концентрации цГМФ, т.е. сигналами, связанными с глутаматом и вовлеченными в механизмы памяти и обучения. Таким образом, известно, что ингибирование фосфодиэстеразы 9А можно применять при лечении болезни Альцгеймера, расстройств ЦНС или когнитивных расстройств, вызванных различными нейродегенеративными процессами. Поэтому она считается полезной фармакологической мишенью для лечения или облегчения когнитивных расстройств, например, деменции с дегенерацией лобной доли, включая деменцию с тельцами Леви, синдром Пика, болезнь Паркинсона и болезнь Альцгеймера с проблемами обучения и памяти.
Кроме того, в дополнение к заболеваниям черепно-мозговых нервов, сверхэкспрессия фосфодиэстеразы 9А недавно была обнаружена у пациентов с заболеваниями сердца, особенно у пациентов с сердечной недостаточностью с сохранением сердечного выброса; кроме того, сверхэкспрессию фосфодиэстеразы 9А наблюдали на животных моделях, индуцирующих патологические состояния сердца. Кроме того, зарегистрированы эффекты улучшения функции сердца и облегчения гипертрофии миокарда за счет ингибирования фосфодиэстеразы 9А; таким образом, в последнее время она привлекает внимание в качестве фармакологической мишени, которую можно использовать у пациентов с сердечно-сосудистыми заболеваниями, особенно с сердечной недостаточностью с сохранением сердечного выброса.
Описание
Техническая задача
Соответственно, задача, которую должно решить настоящее изобретение, заключается в обеспечении соединения, характеризующегося ингибирующей активностью по отношению к PDE9A, фармацевтической композиции, содержащей указанное соединение в качестве активного ингредиента, и медицинского применения для лечения или предотвращения заболеваний, связанных с PDE9A.
Еще одна задача, которую должно решить настоящее изобретение, заключается в обеспечении способа лечения или облегчения заболеваний, связанных с PDE9A, характеризующегося тем, что он ингибирует активность PDE9A и включает введение соединения согласно настоящему изобретению пациенту, нуждающемуся в лечении, улучшении или предотвращении заболеваний, связанных с PDE9A.
Техническое решение
Для решения вышеуказанной задачи в одном варианте реализации настоящего изобретения предложено соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль,
[Химическая формула 1]

при этом в химической формуле 1
A представляет собой фенил, незамещенный или замещенный одним или более атомами галогена, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или более атомами галогена, 3-6-членный циклоалкил, незамещенный или замещенный одним или более атомами галогена, или 3-6-членный гетероциклоалкил, незамещенный или замещенный одним или более атомами галогена,
каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, фенил, незамещенный или замещенный одним или более атомами галогена, C1-5 линейный или разветвленный алкил, незамещенный или замещенный одним или более атомами галогена, или C1-5 линейную или разветвленную алкокси-группу, незамещенную или замещенную одним или более атомами галогена.
В настоящем документе термины «галоген» и «гало» означают фтор, хлор, бром или иод.
В настоящем документе термин "циклоалкил" означает моноциклическое или полициклическое насыщенное кольцо, содержащее атомы углерода и водорода и не содержащее углерод-углеродных кратных связей. Примеры циклоалкильных групп включают (C3-C6)-циклоалкильные группы, включая циклопропильные, циклобутильные, циклопентильные, циклогексильные и циклогептильные группы, но не ограничиваются ими. Циклоалкильная группа может быть незамещенной или необязательно замещенной. В одном варианте реализации циклоалкильная группа представляет собой моноциклическое кольцо или бициклическое кольцо.
В настоящем документе термин "гетероциклоалкил" означает кольцо, в котором по меньшей мере один из атомов углерода циклоалкильной группы независимо замещен атомом азота, кислорода и серы.
В настоящем документе термин "фармацевтически приемлемая соль (соли)" относится к соли, полученной из активных соединений согласно настоящему изобретению с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей этих соединений. Если соединения содержат относительно кислую группу, соли присоединения основания можно получать путем приведения нейтральных соединений в контакт с достаточным количеством желательного основания и чистым или инертным растворителем. Подходящие фармацевтически приемлемые соли присоединения оснований включают соли натрия, калия, кальция, алюминия, органических аминов, магния и т.п., но не ограничиваются ими. Если соединения содержат относительно основную группу, соли присоединения кислоты можно получать путем приведения нейтральных соединений в контакт с достаточным количеством желательной кислоты и чистого или инертного растворителя. Подходящие фармацевтически приемлемые соли присоединения кислоты включают соли - производные нетоксичных органических кислот, включая уксусную кислоту, пропионовую кислоту, изобутиловую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т.п., и нетоксичных неорганических кислот, включая соляную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, моногидрокарбоновую кислоту, фосфорную кислоту, моногидрофосфорную кислоту, дигидрофосфорную кислоту, серную кислоту, моногидросерную кислоту, иодид водорода, фосфорную кислоту и т.п., но не ограничиваются ими. Также они включают соли аминокислот, например, аргинат или его аналоги, а также аналоги органических кислот, например, глюкуроновую или галактуроновую кислоту. Некоторые конкретные соединения согласно настоящему изобретению содержат как основные, так и кислые функциональные группы для преобразования соединений в соли с основным или кислым фрагментом (присоединения основания или кислоты).
В настоящем документе фраза «соединение(я) согласно настоящему изобретению» включает любое соединение(я) согласно химической формуле 1, а также его клатраты, гидраты, сольваты или полиморфные модификации. Даже если в термине “соединение(я) согласно настоящему изобретению” не упоминается его фармацевтически приемлемая соль, этот термин включает соли указанного соединения. В одном варианте реализации соединения согласно настоящему изобретению включают стереохимически чистые соединения, например, практически не содержащие других стереоизомеров (например, характеризующиеся более чем 85% э.и., более чем 90% э.и., более чем 95% э.и., более чем 97% э.и. или более чем 99% э.и.). Таким образом, если соединения согласно химической формуле 1 в соответствии с настоящим изобретением или их соли представляют собой таутомерные изомеры и/или стереоизомеры (например, геометрические изомеры и конформационные изомеры), такие выделенные изомеры и их смеси также включены в настоящее изобретение. Если соединения согласно настоящему изобретению или их соли содержат асимметричный атом углерода, их активные оптические изомеры и их рацемические смеси также включены в настоящее изобретение.
В настоящем документе термин «полиморфный» относится к твердым кристаллическим формам соединения, описываемого в настоящем изобретении, или его комплексам. Различные полиморфные модификации одного и того же соединения могут проявлять различные физические, химические и/или спектроскопические свойства. Различные физические свойства включают стабильность (например, к действию теплоты или света), способность к сжатию и плотность (важно при получении лекарственной формы и производстве продукта), а также скорости растворения (которые могут повлиять на биодоступность), но не ограничиваются ими. Различия в стабильности могут привести к изменениям реакционной способности (например, разница в окислении, при которой состоящая из одной полиморфной модификации лекарственная форма выцветает быстрее, чем лекарственная форма, состоящая из другой) или механических свойств (например, таблетки рассыпаются при хранении, поскольку кинетически более подходящая полиморфная модификация переходит в более термодинамически стабильную) или обеих вышеуказанных характеристик (например, таблетки одной полиморфной модификации более склонны к разрушению при высоких значениях влажности). Различные физические свойства полиморфных модификаций могут повлиять на их переработку. Например, одна полиморфная модификация может с большей вероятностью образовывать сольваты или может труднее отфильтровываться или промываться от примесей, чем другая, вследствие, например, формы частиц или распределения частиц по размерам.
Используемый в настоящей заявке термин «сольват» означает соединение или его соль, описываемое в настоящем документе, которое дополнительно включает стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными силами межмолекулярного взаимодействия. Предпочительные растворители летучи, нетоксичны и приемлемы для употребления человеком в следовых количествах.
В настоящем документе термин «гидрат» означает соединение или его соль, описываемое в настоящем документе, которое дополнительно содержит стехиометрическое или нестехиометрическое количество воды, связанной за счет нековалентных межмолекулярных сил.
В настоящем документе термин «клатрат» означает соединение или его соль в форме кристаллической решетки, содержащей полости (например, каналы), внутри которых присутствуют примесные молекулы (например, растворителя или воды).
В одном варианте реализации в соответствии с настоящим изобретением заместитель A химической формулы 1 предпочтительно представляет собой фенил, линейный или разветвленный C1-5-алкил, незамещенный или замещенный одним или более атомами галогена, или 3-или 6-членный гетероциклоалкил, содержащий гетероатом O или S, незамещенный или замещенный одним или более атомами галогена, причем каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, фенил, линейный или разветвленный C1-5-алкил, незамещенный или замещенный одним или более атомами галогена, или линейную или разветвленную C1-5-алкокси-группу, незамещенную или замещенную одним или более атомами галогена.
В одном варианте реализации в соответствии с настоящим изобретением заместитель A химической формулы 1 более предпочтительно представляет собой фенил,

и каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, -CH3, -C(CH3)3 или фенил.
Для достижения вышеупомянутой цели авторы настоящего изобретения выполнили различные оценочные эксперименты после синтеза различных соединений с целью обеспечения надежности соединений, обладающих высокой ингибирующей активностью по отношению к PDE9A и высокой селективностью в отношении них и их применения, и, наконец, настоящее изобретение было завершено подтверждением того, что соединения согласно настоящему изобретению подходят для целей настоящего изобретения.
Неограничивающие примеры предпочтительных соединений согласно настоящему изобретению включают соединения, приведенные ниже в Таблице 1, и их фармацевтически приемлемые соли.
Кроме того, в настоящем изобретении, как показано ниже на схеме 1, предложен
способ A для получения соединения, представленного химической формулой 1, путем приведения во взаимодействие соединения, представленного химической формулой 2, с соединением, представленным химической формулой 3; и
способ B получения соединения, представленного химической формулой 1, путем приведения во взаимодействие соединения, представленного химической формулой 4, с соединением, представленным химической формулой 5.
[Схема 1]
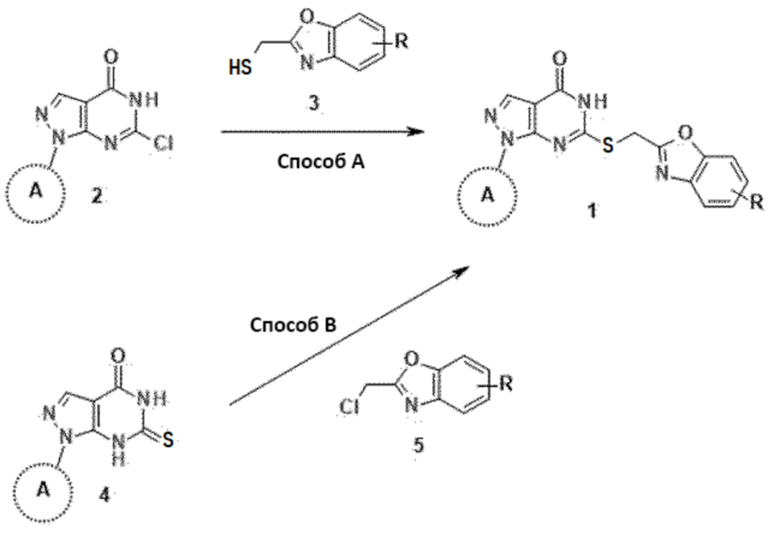
На схеме 1A и R соответствуют определениям, приведенным в химической формуле 1.
Далее будет подробно описан способ получения, представленный схемой 1 согласно настоящему изобретению.
Соединение, представленное химической формулой 1 согласно настоящему изобретению, можно получить путем присоединения пиразолпиримидинонового соединения, представленного химической формулой 2 в качестве исходного материала, к бензоксазольному соединению, представленному химической формулой 3, в присутствии основания (способ A), а также путем присоединения тиоксо-соединения, представленного химической формулой 4 в качестве исходного материала, к соединению галогена, представленному химической формулой 5, в присутствии основания (способ B), как показано выше на схеме 1. Ниже представлены конкретные примеры.
Способ получения
Как показано на схеме 1, соединение, представленное химической формулой 1, можно получить путем приведения во взаимодействие соединения, представленного химической формулой 2, и соединения, представленного химической формулой 3 (способ A), или приведения во взаимодействие соединения, представленного химической формулой 4, и соединения, представленного химической формулой 5 (способ B), в присутствии основания.
При этом основание используют для ускорения реакции и увеличения выхода, и доступные основания включают органические основания, например, N,N-диметиламинопиридин (ДМАП), пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (ДБУ) или неорганические основания, например, бикарбонат натрия, карбонат цезия, карбонат калия, гидроксид натрия и гидроксид калия. Их можно применять по отдельности или в комбинации и в эквивалентном количестве или в избытке.
Кроме того, растворители, которые можно применять в указанной реакции, включают эфирные растворители, например, тетрагидрофуран, диоксан, дихлорметан и 1,2-диметоксиэтан; ароматические углеводородные растворители, например, бензол, толуол и ксилол; диметилформамид (ДМФА); диметилсульфоксид; ацетонитрил и т.п. Их можно применять по отдельности или в комбинации. Температура реакции составляет от 0°C до температуры кипения растворителя.
Получение 1 исходного материала (соединения, представленного химической формулой 2)
Соединение согласно химической формуле 2, которое является исходным материалом схемы 1, можно получить в соответствии со способом производства, включающим этап получения соединения, представленного химической формулой 8, за счет приведения во взаимодействие соединения, представленного химической формулой 6, с соединением, представленным химической формулой 7 (этап 1); и
этап приведения во взаимодействие соединения, представленного химической формулой 8, полученного на указанном этапе 1, с получением соединения, представленного химической формулой 2 (этап 2), как показано ниже на схеме 2.
[Схема 2]

На схеме 2 A соответствует определению, приведенному в химической формуле 1.
Далее подробно описан способ получения, представленный на схеме 2.
В способе получения, представленном на схеме 2, этап 1 представляет собой этап получения соединения, представленного химической формулой 8, путем приведения во взаимодействие соединения, представленного химической формулой 6, с соединением, представленным химической формулой 7. В частности, это этап получения соединения, представленного химической формулой 8, за счет реакции конденсации галогенида, представленного химической формулой 6, в основных условиях.
В указанной выше реакции соединение, представленное химической формулой 6, представляло собой доступное для приобретения соединение.
Кроме того, основания, применяемые в указанной реакции, включают органические основания, например, пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), и неорганические основания, например, гидроксид натрия, карбонат натрия, карбонат калия, карбонат цезия и т.п., и их можно использовать в эквивалентном количестве или в избытке.
Кроме того, растворитель для реакции представляет собой эфирный растворитель, например, тетрагидрофуран, диоксан и 1,2-диметоксиэтан, ароматический углеводородный растворитель, например, бензол, толуол и ксилол, низший спирт, например, метанол, этанол, пропанол и бутанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.д., который можно применять по отдельности или в комбинации, а температура реакции составляет от 0°С до температуры кипения растворителя.
В способе получения, представленном на схеме 2, этап 2 представляет собой этап получения соединения, представленного химической формулой 2, за счет приведения во взаимодействие соединения, представленного химической формулой 8, полученного на этапе 1. В частности, на этом этапе пиразолпиримидиноновое соединение, представленное химической формулой 2, получают путем гидролиза соединения галогена, представленного химической формулой 8, которое получают на этапе 1, в основных условиях.
В этой реакции основание включает неорганические основания, например, карбонат натрия, карбонат калия, гидроксид калия, гидроксид натрия, карбонат цезия, гидроксид бария и т.п., и его можно применять по отдельности или в комбинации в эквивалентном количестве или в избытке.
Кроме того, растворители, которые можно применять в реакции, включают эфирные растворители, например, тетрагидрофуран, диоксан, дихлорметан и 1,2-диметоксиэтан, ароматические углеводородные растворители, например, бензол, толуол и ксилол, растворители на спиртовой основе, например, метанол и этанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.п. Их можно применять по отдельности или в комбинации. Температура реакции составляет от 0°C до температуры кипения растворителя.
Получение 2 исходного материала (соединения, представленного химической формулой 3 или 5)
Соединение согласно химической формуле 3, которое является исходным материалом схемы 1, можно получить в соответствии со способом производства, включающим этап получения соединения, представленного химической формулой 5, за счет приведения во взаимодействие соединения, представленного химической формулой 9 (этап 1); этап получения соединения, представленного химической формулой 10, путем приведения во взаимодействие соединения, представленного химической формулой 5, полученного на этапе 1 (этап 2); и этап получения соединения, представленного химической формулой 3, путем приведения во взаимодействие соединения, представленного химической формулой 10, полученного на этапе 2 (этап 3), как показано ниже на схеме 3.
[Схема 3]

На схеме реакции 3 R соответствует определению в химической формуле 1.
В настоящем документе подробно описан способ получения, представленный на схеме 3.
В способе получения, представленном на схеме 3, этап 1 представляет собой этап, на котором соединение, представленное химической формулой 5, получают из соединения, представленного химической формулой 9. В частности, это этап получения соединения, представленного химической формулой 5, путем реакции конденсации аминофенола, представленного химической формулой 9, в основных условиях.
В вышеуказанной реакции соединение, представленное химической формулой 9, представляло собой доступное для приобретения соединение.
Кроме того, основания, применяемые в указанной реакции, включают органические основания, например, пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), и неорганические основания, например, гидроксид натрия, карбонат натрия, карбонат калия, карбонат цезия и т.п., и их можно использовать в эквивалентном количестве или в избытке.
Кроме того, растворитель для реакции представляет собой эфирный растворитель, например, тетрагидрофуран, диоксан и 1,2-диметоксиэтан, ароматический углеводородный растворитель, например, бензол, толуол и ксилол, низший спирт, например, метанол, этанол, пропанол и бутанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.д., который можно применять по отдельности или в комбинации, а температура реакции составляет от 0 °С до температуры кипения растворителя.
В способе производства, представленном на схеме 3, этап 2 представляет собой этап получения соединения, представленного химической формулой 10, за счет приведения во взаимодействие соединения, представленного химической формулой 5, полученного на этапе 1. В частности, это этап получения тиоацетилового соединения, представленного химической формулой 10, путем замены соединения галогена, представленного химической формулой 5, полученного на этапе 1.
При этом растворители, которые можно применять в данной реакции, включают эфирные растворители, например, тетрагидрофуран, диоксан, дихлорметан и 1,2-диметоксиэтан, ароматические углеводородные растворители, например, бензол, толуол и ксилол, растворители на спиртовой основе, например, метанол и этанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, ацетон, воду и т.п. Их можно применять по отдельности или в комбинации. Температура реакции составляет от 0°C до температуры кипения растворителя.
В способе производства, представленном на схеме 3, этап 3 представляет собой этап получения соединения, представленного химической формулой 3, за счет приведения во взаимодействие соединения, представленного химической формулой 10, полученного на этапе 2. В частности, это этап получения тиолового соединения, представленного химической формулой 3, путем снятия защиты с тиоацетилового соединения, представленного химической формулой 10, полученного на этапе 2.
В данной реакции основания, применяемые в указанной реакции, включают органические основания, например, пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), и неорганические основания, например, гидроксид натрия, карбонат натрия, карбонат калия, карбонат цезия и т.п., и их можно использовать в эквивалентном количестве или в избытке.
Кроме того, растворитель для реакции включает эфирный растворитель, например, тетрагидрофуран, диоксан и 1,2-диметоксиэтан, ароматический углеводородный растворитель, например, бензол, толуол и ксилол, низший спирт, например, метанол, этанол, пропанол и бутанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.д. Их можно применять по отдельности или в комбинации, а температура реакции составляет от 0°C до температуры кипения растворителя.
Получение 3 исходного материала (соединения, представленного химической формулой 4)
Соединение согласно химической формуле 4, которое является исходным материалом схемы 1, можно получить в соответствии со способом получения, включающим этап получения соединения, представленного химической формулой 12, за счет приведения во взаимодействие соединения, представленного химической формулой 11, с соединением, представленным химической формулой 7 (этап 1); этап получения соединения, представленного химической формулой 13, путем приведения во взаимодействие соединения, представленного химической формулой 12, полученного на этапе 1 (этап 2); и этап получения соединения, представленного химической формулой 4, путем приведения во взаимодействие соединения, представленного химической формулой 13, полученного на этапе 2 (этап 3), как показано ниже на схеме 4.
[Схема 4]

На схеме реакции 4 A соответствует определению в химической формуле 1.
В настоящем документе подробно описан способ получения, представленный на схеме 4.
В способе получения, представленном на схеме 4, этап 1 представляет собой этап получения соединения, представленного химической формулой 12, путем приведения во взаимодействие соединения, представленного химической формулой 11, с соединением, представленным химической формулой 7. В частности, это этап получения соединения, представленного химической формулой 12, путем реакции конденсации акрилатного соединения, представленного химической формулой 11, с гидразиновым соединением в основных условиях.
В вышеуказанной реакции соединение, представленное химической формулой 11, представляло собой доступное для приобретения соединение.
Кроме того, основания, применяемые в указанной реакции, включают органические основания, например, пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), и неорганические основания, например, гидроксид натрия, карбонат натрия, карбонат калия, карбонат цезия и т.п. Их можно применять в эквивалентном количестве или в избытке.
Кроме того, растворитель для реакции включает эфирный растворитель, например, тетрагидрофуран, диоксан и 1,2-диметоксиэтан, ароматический углеводородный растворитель, например, бензол, толуол и ксилол, низший спирт, например, метанол, этанол, пропанол и бутанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.д. Их можно применять по отдельности или в комбинации, а температура реакции составляет от 0°C до температуры кипения растворителя.
В способе производства, представленном на схеме 4, этап 2 представляет собой этап получения соединения, представленного химической формулой 13, за счет приведения во взаимодействие соединения, представленного химической формулой 12, полученного на этапе 1. В частности, это этап получения соединения - производного тиомочевины, представленного химической формулой 13, путем реакции присоединения аминного соединения, представленного химической формулой 12, полученного на этапе 1, к тиоизоцианатному соединению.
При этом растворители, которые можно применять в данной реакции, включают эфирные растворители, например, тетрагидрофуран, диоксан, дихлорметан и 1,2-диметоксиэтан, ароматические углеводородные растворители, например, бензол, толуол и ксилол, растворители на спиртовой основе - метанол и этанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.п. Их можно применять по отдельности или в комбинации. Температура реакции составляет от 0°C до температуры кипения растворителя.
В способе производства, представленном на схеме 4, этап 3 представляет собой этап получения соединения, представленного химической формулой 4, за счет приведения во взаимодействие соединения, представленного химической формулой 13, полученного на этапе 2. В частности, это этап получения тиоксо-соединения, представленного химической формулой 4, путем циклизации соединения - производного тиомочевины, представленного химической формулой 13, полученного на этапе 2.
В данном случае основания, применяемые в указанной реакции, включают органические основания, например, пиридин, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), и неорганические основания, например, гидроксид натрия, карбонат натрия, карбонат калия, карбонат цезия и т.п. Их можно применять в эквивалентном количестве или в избытке.
Кроме того, растворитель для реакции представляет собой эфирный растворитель, например, тетрагидрофуран, диоксан и 1,2-диметоксиэтан, ароматический углеводородный растворитель, например, бензол, толуол и ксилол, низший спирт, например, метанол, этанол, пропанол и бутанол, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), ацетонитрил, воду и т.д. Их можно применять по отдельности или в комбинации, а температура реакции составляет от 0°C до температуры кипения растворителя.
В еще одном варианте реализации настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения согласно химической формуле 1 в соответствии с настоящим изобретением или ее фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
В еще одном варианте реализации настоящего изобретения предложен способ лечения заболевания или состояния, включающий введение терапевтически эффективного количества соединения согласно химической формуле 1 или его фармацевтически приемлемой соли индивиду, нуждающемуся в этом, причем указанное заболевание или патологическое состояние представляет собой заболевание, связанное с фосфодиэстеразой 9A.
Таким образом, в настоящем изобретении предложено медицинское применение, характеризующееся тем, что соединение согласно химической формуле 1 в соответствии с настоящим изобретением или его фармацевтически приемлемую соль применяют в качестве активного ингредиента. В одном варианте реализации медицинское применение согласно настоящему изобретению представляет собой заболевание, связанное с фосфодиэстеразой 9А.
В одном варианте реализации связанное заболевание, связанное с фосфодиэстеразой 9А, представляет собой неврологическое заболевание или психическое заболевание. В одном варианте реализации неврологическое или психическое расстройство представляет собой болезнь Альцгеймера, болезнь Хантингтона, деменцию с тельцами Леви или синдром Пика.
В еще одном варианте реализации заболевание, связанное с фосфодиэстеразой 9А, представляет собой сердечную недостаточность, в частности, сердечную недостаточность с сохранением сердечного выброса или серповидноклеточную анемию.
Соединения согласно настоящему изобретению обычно вводят в терапевтически эффективном количестве. Соединения согласно настоящему изобретению можно вводить любым подходящим путем в форме фармацевтической композиции, подходящей для такого пути введения, и в эффективной дозировке для предполагаемого лечения. Обычно эффективные дозы составляют от приблизительно 0,001 до приблизительно 100 мг/кг массы тела/сутки, предпочтительно от приблизительно 0,01 до приблизительно 50 мг/кг/сутки, и их вводят в виде разовой или нескольких доз. Подходящие уровни дозировки могут быть меньше нижнего предела этого диапазона в зависимости от возраста, вида животного и заболевания или патологического состояния, подлежащего лечению. В прочих случаях могут использоваться еще более высокие дозировки без негативных побочных эффектов. Более высокие дозировки можно разделять на несколько меньших дозировок для введения в течение суток. Способы определения подходящей дозировки хорошо известны в данной области техники, к которой относится настоящее изобретение, и, например, можно использовать Remington: The Science and Practice°f Pharmacy, Mack Publishing Co., 20th ed., 2000.
Для лечения вышеуказанных заболеваний или состояний описанные в настоящем документе соединения или их фармацевтически приемлемые соли можно вводить следующими способами.
Пероральное введение
Соединения, описываемые в настоящем документе, можно вводить перорально, в том числе путем проглатывания, чтобы они попадали в желудочно-кишечный тракт или всасывались в кровоток непосредственно из ротовой полости (например, буккальное или сублингвальное введение).
Подходящие для перорального введения композиции включают твердые, жидкие, гелевые или порошкообразные составы и представлены в таких лекарственных формах, как таблетка, пастилка, капсула, гранула или порошок.
Композиции для перорального введения можно изготавливать как с немедленным, так и модифицированным высвобождением, включая отсроченное или замедленное высвобождение, необязательно с кишечнорастворимым покрытием.
Жидкие составы могут включать растворы, сиропы и суспензии, которые можно использовать в мягких или жестких капсулах. Такие составы могут включать фармацеветически приемлемый носитель, например, воду, этанол, полиэтиленгликоль, целлюлозу или масло. Состав также может содержать один или более из эмульгирующих и/или суспендирующих агентов.
В таблетированной лекарственной форме количество присутствующего лекарственного средства может составлять от приблизительно 0,05 до приблизительно 95% по массе, в более типичном случае - от приблизительно 2 до приблизительно 50% от массы лекарственной формы. Кроме того, в таблетках может содержаться разрыхлитель, содержание которого составляет от 0,5 до 35% по массе, в более типичном случае - от приблизительно 2 до приблизительно 25% от массы лекарственной формы. Примеры разрыхлителей включают лактозу, крахмал, гликолят крахмала натрия, кросповидон, кроскармелозу натрия, мальтодекстрин или их смеси, но не ограничиваются ими.
Подходящие смазывающие вещества в таблетке могут присутствовать в количествах от приблизительно 0,1 до приблизительно 5% по массе и включают тальк, диоксид кремния, стеариновую кислоту, стеарат кальция, цинка или магния, стеарилфумарат натрия и т.п., но не ограничиваются ими.
Подходящие связующие для применения в таблетке включают желатин, полиэтиленгликоль, углеводы, камеди, крахмал, поливинилпироллидон, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и т.п., но не ограничиваются ими. Подходящие разбавители для применения в таблетке включают маннит, ксилит, лактозу, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу и крахмал, но не ограничиваются ими.
Подходящие солюбилизаторы для применения в таблетке могут присутствовать в количествах от приблизительно 0,1 до приблизительно 3% по массе и включают полисорбаты, лаурилсульфат натрия, додецилсульфат натрия, пропиленкарбонат, моноэтиловый эфир диэтиленгликоля, диметилизосорбид, полиэтиленгликоль, касторовое масло (естественное или гидрогенизированное), HCOR™ (Nikkol), олеиловый эфир, Gelucire™, моно/диглицерид каприловой/каприловой кислоты, эфиры жирных кислот и сорбитана и Solutol HS™, но не ограничиваются ими.
Парентеральное введение
Соединения согласно настоящему изобретению можно вводить непосредственно в кровоток, мышцы или внутренние органы. Подходящие способы парентерального введения включают внутривенное, внутримышечное, подкожное, внутриартериальное, интраперитонеальное, интратекальное, интракраниальное и им подобные способы введения. Подходящие устройства для парентерального введения включают инъекционные (включая шприцы с иглой и без) и инфузионные методы.
Композиции для парентерального введения можно изготавливать как с немедленным, так и модифицированным высвобождением, включая отсроченное или замедленное.
Большинство парентеральных составов являются водными растворами, содержащими вспомогательные вещества, в том числе соли, буферные агенты и изотонические агенты.
Парентеральные составы также могут быть изготовлены в безводной форме (например, путем лиофилизации) или в форме стерильных неводных растворов. Такие формы можно применять с подходящей средой-носителем, например, стерильной водой. При получении парентеральных растворов можно также использовать усилители растворимости.
Местное применение
Соединения согласно настоящему изобретению можно наносить местно на кожу или вводить трансдермально. Составы для местного применения могут включать лосьоны, растворы, крема, гели, гидрогели, мази, пены, имплантаты, пластыри и т. п. Фармацевтически приемлемые носители для составов для местного применения могут включать воду, спирт, минеральное масло, глицерин, полиэтиленгликоль и т.п. Местное применение также можно осуществлять с помощью электропорации, ионофореза, фонофореза и т.п.
Композиции для местного применения можно изготавливать как с немедленным, так и модифицированным высвобождением, включая отсроченное или замедленное высвобождение.
Положительные эффекты
В настоящем изобретении предложено соединение, способное проявлять различную фармакологическую активность путем ингибирования активности PDE 9A, фармацевтическая композиция, содержащая указанное соединение в качестве активного ингредиента, их медицинское применение (особенно для лечения или облегчения нейропатических заболеваний и психических заболеваний), и способ лечения или предотвращения, включающий введение индивиду, нуждающемуся в этом.
Принцип изобретения
Настоящее изобретение будет подробнее описано на основании нижеследующих примеров, однако они не предназначены для ограничения сущности настоящего изобретения. Кроме того, специалисты в данной области техники смогут добавлять различные модификации и изменения в настоящее изобретению в объеме, не изменяющем сущность настоящего изобретения.
<Пример получения 1> Получение 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

Этап 1: Получение трет-бутил-2-(тетрагидро-4H-пиран-4-илиден)гидразин-1-карбоксилата

3 г тетрагидро-4H-пиран-4-она (29,96 ммоль) растворяли в 60 мл гексана, добавляли 3,96 г трет-бутилкарбазита (29,96 ммоль) с последующим перемешиванием с обратным холодильником в течение 3 часов. После завершения реакции смесь концентрировали при пониженном давлении, получая 6,4 г соединения, указанного в заголовке (29,95 ммоль) со 100% выходом.
Rf = 0,19 (гексан:этилацетат = 1:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 9,65 (br s, 1H), 3,69 (t, J = 5,7 Гц, 2H), 3,60 (t, J = 5,7 Гц, 2H), 2,41 (t, J = 5,7 Гц, 2H), 2,27 (t, J = 5,7 Гц, 2H), 1,43 (s, 9H)
Этап 2: Получение трет-бутил-2-(тетрагидро-2H-пиран-4-ил)гидразин-1-карбоксилата
6,4 г трет-бутил-2-(тетрагидро-4H-пиран-4-илиден)гидразин-1-карбоксилата (29,95 ммоль), полученного на этапе 1, растворяли в 80 мл ТГФ, добавляли 12,7 г триацетоксиборгидрида натрия (59,92 ммоль) и перемешивали при комнатной температуре в течение 15 часов. После завершения реакции реакционный раствор экстрагировали 30 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 1:2, об./об.), получая 6 г указанного в заголовке соединения (27,74 ммоль) с 92% выходом.
Rf = 0,38 (гексан:этилацетат = 1:2, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 6,24 (br s, 1H), 3,95-3,99 (m, 2H), 3,38-3,43 (m, 2H), 3,05-3,11 (m, 1H), 1,75-1,81 (m, 2H), 1,40-1,52 (m, 11H)
Этап 3: Получение гидрохлорида (тетрагидро-2H-пиран-4-ил)гидразина
6 г трет-бутил-2-(тетрагидро-2H-пиран-4-ил)гидразин-1-карбоксилата (27,74 ммоль), полученного на этапе 2, растворяли в 60 мл метанола и добавляли соляную кислоту, а затем перемешивали при 50 °C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, получая 4 г указанного в заголовке соединения (26,21 ммоль) с 94% выходом.
1H ЯМР (500 МГц, ДМСО-d6) δ 7,25 (br s, 1H), 3,83-3,92(m, 2H), 3,23-3,31 (m, 2H), 3,08-3,18 (m, 1H), 1,84-1,95 (m, 2H), 1,41-1,54 (m, 2H)
Этап 4: Получение 4,6-дихлор-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[3,4-d]пиримидина

5,55 г 2,4,6-трихлорпиримидин-5-карбальдегида (26,21 ммоль) растворяли в 60 мл этанола и добавляли 4 г гидрохлорида (тетрагидро-2H-пиран-4-ил)гидразина (26,21 ммоль), полученного на вышеописанном этапе 3, и 14 мл N,N-диизопропилэтиламина (78,63 ммоль) при -78 °C и перемешивали в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 6,5 г указанного в заголовке соединения (23,80 ммоль) с 91% выходом.
Rf = 0,20 (гексан:этилацетат = 9:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,57 (s, 1H), 4,96-5,03 (m, 1H), 3,98-4,02 (m, 2H), 3,56-3,61 (m, 2H), 2,10-2,19 (m, 2H), 1,90-1,95 (m, 2H)
Этап 5: Получение 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
6,5 г 4,6-дихлор-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[3,4-d]пиримидина (23,80 ммоль), полученного на вышеописанном этапе 4, растворяли в 50 мл ТГФ и добавляли 25 мл 2 н. гидроксида натрия (47,60 ммоль), и смесь перемешивали с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 5,4 г указанного в заголовке соединения (21,20 ммоль) с 89% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,21 (br s, 1H), 8,11 (s, 1H), 4,75-4,81 (m, 1H), 3,95-3,99 (m, 2H), 3,51-3,56 (m, 2H), 2,05-2,12 (m, 2H), 1,81-1,86 (m, 2H)
<Пример получения 2> Получение бензо[d]оксазол-2-илметантиола

Этап 1: Получение 2-(хлорметил)бензо[d]оксазола

500 мг 2-аминофенола (4,58 ммоль) растворяли в 15 мл ксилола и добавляли 0,55 мл хлорацетилхлорида (6,87 ммоль) при 0 °C с последующим перемешиванием. Через 30 минут добавляли 0,7 мл триэтиламина (5,04 ммоль) и перемешивали смесь с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 7:1, об./об.), получая 600 г указанного в заголовке соединения (3,58 ммоль) с 78% выходом.
Rf = 0,47 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,76-7,80 (m, 2H), 7,41-7,48 (m, 2H), 5,07 (s, 2H)
Этап 2: Получение S-(бензо[d]оксазол-2-илметил)этантиоата
280 мг 2-(хлорметил)бензо[d]оксазола (1,67 ммоль), полученного на этапе 1, растворяли в 8 мл ацетона и добавляли 286 мг тиоацетата калия (2,50 ммоль) с последующим перемешиванием в течение 1 часа при 60 °C. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 290 мг указанного в заголовке соединения (1,39 ммоль) с 84% выходом.
Rf = 0,28 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,68-7,73 (m, 1H), 7,49-7,54 (m, 1H), 7,30-7,37 (m, 2H), 4,41 (s, 2H), 2,45 (s, 3H)
Этап 3: Получение бензо[d]оксазол-2-илметантиола
290 мг S-(бензо[d]оксазол-2-илметил)этантиоата (1,39 ммоль), полученного на этапе 1, растворяли в смеси 4 мл метанола и 1 мл воды и добавляли 386 мг карбоната калия (2,79 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 30 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении, получая 170 мг указанного в заголовке соединения (1,03 ммоль) с 74% выходом.
Rf = 0,40 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,70-7,73 (m, 1H), 7,49-7,55 (m, 1H), 7,32-7,39 (m, 2H), 4,17 (s, 1,3H), 3,99 (d, J = 8,3 Гц, 0,7H), 2,24 (t, J = 8,3 Гц, 0,4H), 1,65 (s, 0,6H)
<Пример 1> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в вышеописанном примере получения 1, растворяли в 3 мл ДМФА и добавляли 65 мг бензо[d]оксазол-2-илметантиола (0,39 ммоль), полученного в примере получения 2, и 83 мг карбоната калия (0,60 ммоль). Смесь перемешивали при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 55 мг указанного в заголовке соединения (0,14 ммоль) с 72% выходом.
Rf = 0,21 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,65 (br s, 1H), 7,95 (s, 1H), 7,68-7,71 (m, 2H), 7,33-7,39 (m, 2H), 4,80 (s, 2H), 4,50-4,57 (m, 1H), 3,84-3,89 (m, 2H), 3,35-3,40 (m, 2H), 1,89-1,97 (m, 2H), 1,51-1,56 (m, 2H)
<Пример получения 3> Получение 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
Этап 1: Получение 4,6-дихлор-1-фенил-1H-пиразоло[3,4-d]пиримидина

2 г 2,4,6-трихлорпиримидин-5-карбальдегида (9,44 ммоль) растворяли в 30 мл этанола и добавляли 0,9 мл гидрохлорида фенилгидразина (9,44 ммоль) и 4,8 мл N,N-диизопропилэтиламина (28,32 ммоль) при -78°C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 300 мл этилацетата, промывали 200 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 20:1, об./об.), получая 1,7 г указанного в заголовке соединения (6,41 ммоль) с 68% выходом.
Rf = 0,50 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (300 МГц, CD3OD) δ 8,35 (s, 1H), 8,17 (d, J = 8,5 Гц, 2H), 7,59 (dd, J = 8,5, 7,6 Гц, 2H), 7,44 (dd, J = 7,6, 7,6 Гц, 1H)
Этап 2: Получение 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
1,9 г 4,6-дихлор-1-фенил-1H-пиразоло[3,4-d]пиримидина (8,98 ммоль), полученного на этапе 1, растворяли в 20 мл ТГФ, добавляли 10 мл 2 н. гидроксида натрия (17,96 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 2,2 г указанного в заголовке соединения (8,98 ммоль) с 99% выходом.
Rf = 0,23 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,47 (br s, 1H), 8,35 (s, 1H), 7,94 (d, J = 8,0 Гц, 2H), 7,58 (dd, J = 8,0, 7,4 Гц, 2H), 7,43 (dd, J = 7,4, 7,4 Гц, 1H)
<Пример 2> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
60 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,24 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 150 мг бензо[d]оксазол-2-илметантиола (0,91 ммоль), полученного в примере получения 2, и 186 мг карбоната калия (1,35 ммоль), с последующим перемешиванием при 60°C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 70 мг указанного в заголовке соединения (0,19 ммоль) с 78% выходом.
Rf = 0,27 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,96 (br s, 1H), 8,22 (s, 1H), 7,93 (d, J = 7,8 Гц, 2H), 7,69 (dd, J = 8,8, 8,3 Гц, 2H), 7,45 (dd, J = 7,8, 7,5 Гц, 2H), 7,32-7,38 (m, 3H), 5,75 (s, 2H)
<Пример получения 4> Получение (5-метилбензо[d]оксазол-2-ил)метантиола
Этап 1: Получение 2-(хлорметил)-5-метилбензо[d]оксазола
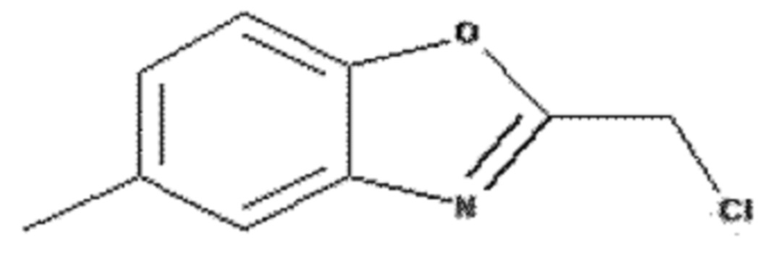
500 мг 2-амино-4-метилфенола (4,06 ммоль) растворяли в 15 мл ксилола и добавляли 0,48 мл хлорацетилхлорида (6,09 ммоль) при 0°C с последующим перемешиванием. Через 30 минут добавляли 0,62 мл триэтиламина (4,47 ммоль) и перемешивали смесь с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 240 мг указанного в заголовке соединения (1,32 ммоль) с 33% выходом.
Rf = 0,54 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,54 (s, 1H), 7,45 (d, J = 8,3 Гц, 1H), 7,22 (d, J = 8,3 Гц, 1H), 4,76 (s, 2H), 2,49 (s, 3H)
Этап 2: Получение S-((-5-метилбензо[d]оксазол-2-ил)метил)этантиоата
220 мг 2-(хлорметил)-5-метилбензо[d]оксазола (1,21 ммоль), полученного на этапе 1, растворяли в 6 мл ацетона и добавляли 208 мг тиоацетата калия (1,82 ммоль) с последующим перемешиванием при 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 250 мг указанного в заголовке соединения (1,13 ммоль) с 93% выходом.
Rf = 0,40 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,49 (s, 1H), 7,39 (d, J = 8,3 Гц, 1H), 7,16 (d, J = 8,3 Гц, 1H), 4,40 (s, 2H), 2,47 (s, 3H), 2,45 (s, 3H)
Этап 3: Получение (5-метилбензо[d]оксазол-2-ил)метантиола
230 мг S-((-5-метилбензо[d]оксазол-2-ил)метил)этантиоата (1,04 ммоль), полученного на этапе 2, растворяли в смеси 4 мл метанола и 1 мл воды и добавляли 287 мг карбоната калия (2,08 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 170 мг указанного в заголовке соединения (0,95 ммоль) с 91% выходом.
Rf = 0,17 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,49 (s, 1H), 7,41 (d, J = 8,3 Гц, 1H), 7,17 (d, J = 8,3 Гц, 1H), 3,96 (d, J = 8,4 Гц, 2H), 2,48 (s, 3H), 2,23 (t, J = 8,4 Гц, 1H)
<Пример 3> Получение 6-(((5-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
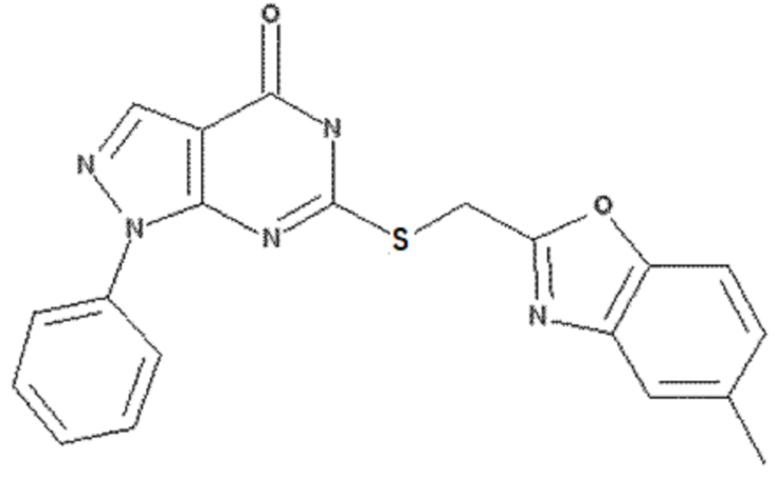
50 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d] пиримидин-4-она (0,20 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 100 мг (5-метилбензо[d]оксазол-2-ил)метантиола (0,55 ммоль), полученного в вышеописанном примере получения 4, и 83 мг карбоната калия (0,60 ммоль) с последующим перемешиванием при 60°C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 60 мг указанного в заголовке соединения (0,15 ммоль) с 77% выходом.
Rf = 0,27 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,91 (br s, 1H), 7,94 (d, J = 7,8 Гц, 2H), 7,56 (d, J = 8,5 Гц, 1H), 7,49 (s, 1H), 7,47 (dd, J = 7,8, 7,4 Гц, 2H), 7,36 (dd, J = 7,4, 7,4 Гц, 1H), 7,18 (d, J = 8,5 Гц, 1H), 4,79 (s, 2H), 2,38 (s, 3H)
<Пример получения 5> Получение (5-фторбензо[d]оксазол-2-ил)метантиола
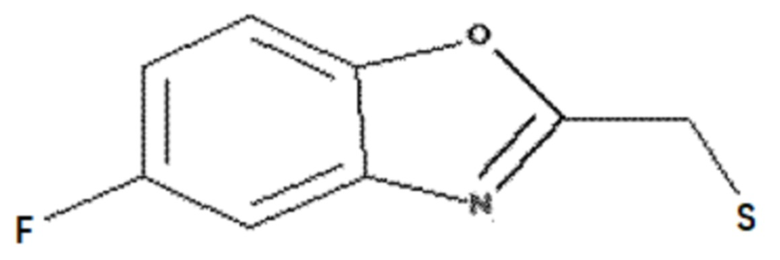
Этап 1: Получение 2-(хлорметил)-5-метилбензо[d]оксазола

500 мг 2-амино-4-фторфенола (3,93 ммоль) растворяли в 15 мл ксилола и добавляли 0,50 мл хлорацетилхлорида (5,90 ммоль) при 0°C с последующим перемешиванием. Через 30 минут добавляли 0,60 мл триэтиламина (4,32 ммоль) и перемешивали смесь с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 200 мг указанного в заголовке соединения (1,08 ммоль) с 27% выходом.
Rf = 0,56 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3)δ 7,52 (dd, J = 8,9, 4,8 Гц, 1H), 7,45 (dd, J = 8,1, 2,5 Гц, 1H), 7,16 (ddd, J = 9,1, 8,9, 2,5 Гц, 1H), 4,76 (s, 2H)
Этап 2: Получение S-((-5-фторбензо[d]оксазол-2-ил)метил)этантиоата
180 мг 2-(хлорметил)-5-фторбензо[d]оксазола (0,97 ммоль), полученного на этапе 1, растворяли в 6 мл ацетона и добавляли 166 мг тиоацетата калия (1,45 ммоль) с последующим перемешиванием при 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 210 мг указанного в заголовке соединения (0,93 ммоль) с 96% выходом.
Rf = 0,44 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,45 (dd, J = 8,8, 4,2 Гц, 1H), 7,39 (dd, J = 8,3, 2,7 Гц, 1H), 7,09 (ddd, J = 9,0, 8,8, 2,7 Гц, 1H), 4,40 (s, 2H), 2,46 (s, 3H)
Этап 3: Получение (5-фторбензо[d]оксазол-2-ил)метантиола
190 мг S-((-5-фторбензо[d]оксазол-2-ил)метил)этантиоата (0,84 ммоль), полученного на этапе 2, растворяли в смеси 4 мл метанола и 1 мл воды и добавляли 233 мг карбоната калия (1,69 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 115 мг указанного в заголовке соединения (0,63 ммоль) с 75% выходом.
Rf = 0,22 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,47 (dd, J = 8,8, 4,1 Гц, 1H), 7,40 (dd, J = 9,2, 2,5 Гц, 1H), 7,10 (ddd, J = 9,2, 8,8, 2,5 Гц, 1H), 3,97 (d, J = 8,2 Гц, 2H), 2,25 (t, J = 8,2 Гц, 1H)
<Пример 4> Получение 6-(((5-фторбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 90 мг (5-фторбензо[d]оксазол-2-ил)метантиола (0,49 ммоль), полученного в примере получения 5 и 83 мг карбоната калия (0,60 ммоль), с последующим перемешиванием при 60°C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 54 мг указанного в заголовке соединения (0,14 ммоль) с 69% выходом.
Rf = 0,27 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,91 (br s, 1H), 8,22 (s, 1H), 7,90 (d, J = 8,3 Гц, 2H), 7,72 (dd, J = 8,3, 4,7 Гц, 1H), 7,60 (dd, J = 9,2, 2,6 Гц, 1H), 7,46 (dd, J = 8,3, 7,4 Гц, 2H), 7,36 (dd, J = 7,4, 7,4 Гц, 1H), 7,22 (ddd, J = 9,1, 8,3, 2,6 Гц, 1H), 4,82 (s, 2H)
<Пример получения 6> Получение (6-метилбензо[d]оксазол-2-ил)метантиола
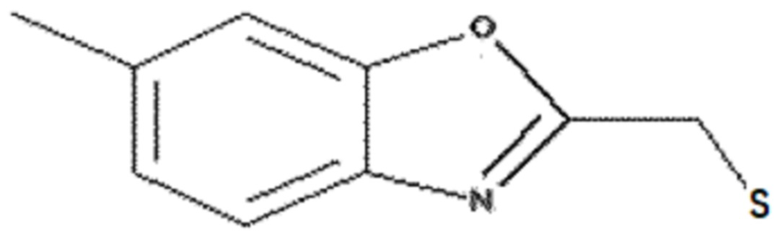
Этап 1: Получение 2-(хлорметил)-6-метилбензо[d]оксазола

500 мг 6-амино-м-крезола (4,00 ммоль) растворяли в 15 мл ксилола и добавляли 0,48 мл хлорацетилхлорида (6,09 ммоль) при 0 °C с последующим перемешиванием. Через 30 минут добавляли 0,62 мл триэтиламина (4,47 ммоль) и перемешивали смесь с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 20:1, об./об.), получая 640 г указанного в заголовке соединения (3,52 ммоль) с 88% выходом.
Rf = 0,64 (гексан:этилацетат = 9:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,52 (dd, J = 8,9, 4,8 Гц, 1H), 7,45 (dd, J = 8,1, 2,5 Гц, 1H), 7,16 (ddd, J = 9,1, 8,9, 2,5 Гц, 1H), 4,76 (s, 2H)
Этап 2: Получение S-((-6-метилбензо[d]оксазол-2-ил)метил)этантиоата
600 мг 2-(хлорметил)-6-метилбензо [d]оксазола (3,30 ммоль), полученного на этапе 1, растворяли в 10 мл ацетона и добавляли 566 мг тиоацетата калия (4,96 ммоль) с последующим перемешиванием при 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 690 мг указанного в заголовке соединения (3,12 ммоль) с 94% выходом.
Rf = 0,18 (гексан:этилацетат = 9:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,57 (d, J = 8,0 Гц, 1H), 7,31 (s, 1H), 7,15 (d, J = 8,0 Гц, 1H), 4,39 (s, 2H), 2,48 (s, 3H), 2,44 (s, 3H)
Этап 3: Получение (6-метилбензо [d]оксазол-2-ил)метантиола
500 мг S-((-6-метилбензо[d]оксазол-2-ил)метил)этантиоата (2,26 ммоль), полученного на этапе 2, растворяли в смеси 6 мл метанола и 1 мл воды и добавляли 624 мг карбоната калия (4,52 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 50 мл этилацетата, промывали 50 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 370 мг указанного в заголовке соединения (2,06 ммоль) с 91% выходом.
Rf = 0,36 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,58 (d, J = 8,0 Гц, 1H), 7,33 (s, 1H), 7,17 (d, J = 8,0 Гц, 1H), 3,96 (d, J = 8,3 Гц, 2H), 2,50 (s, 3H), 2,22 (t, J = 8,3 Гц, 1H)
<Пример 5> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 108 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,60 ммоль), полученного в примере получения 6, и 83 мг карбоната калия (0,60 ммоль), с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 67 мг указанного в заголовке соединения (0,17 ммоль) с 86% выходом.
Rf = 0,27 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,94 (br s, 1H), 8,23 (s, 1H), 7,94 (d, J = 7,8 Гц, 1H), 7,57 (d, J = 8,2 Гц, 1H), 7,45-7,49 (m, 3H), 7,37 (dd, J = 7,5, 7,5 Гц, 1H), 7,16 (d, J = 8,2 Гц, 1H), 4,79 (s, 2H), 2,41 (s, 3H)
<Пример получения 7> Получение (5-хлорбензо[d]оксазол-2-ил)метантиола
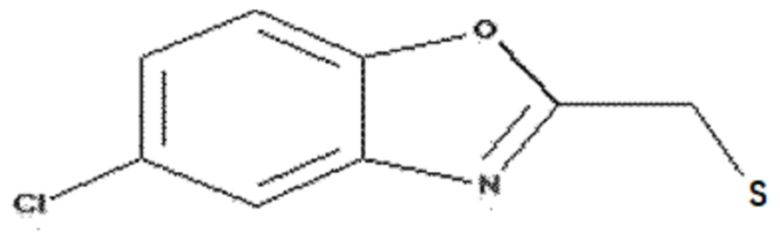
Этап 1: Получение 2-хлор-N-(5-хлор-2-гидроксифенил)ацетамида

1 г 2-амино-4-хлорфенола (6,96 ммоль) растворяли в 20 мл MC и добавляли 0,83 мл хлорацетилхлорида (10,44 ммоль) при 0°C с последующим перемешиванием. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 3:1, об./об.), получая 1,3 г указанного в заголовке соединения (5,91 ммоль) с 85% выходом.
Rf = 0,20 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) 10,81 (br s, 1H), 6,94-6,98 (m, 2H), 6,90 (d, J = 2,0 Гц, 1H), 4,59 (s, 2H)
Этап 2: Получение 5-хлор-2-(хлорметил)бензо[d]оксазола
200 мг 2-хлор-N-(5-хлор-2-гидроксифенил)ацетамида (0,91 ммоль), полученного на этапе 1, растворяли в 5 мл фенола и добавляли 78 мг п-толуолсульфониловой кислоты (0,45 ммоль)) с последующим перемешиванием с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 180 мг указанного в заголовке соединения (0,89 ммоль) с 98% выходом.
Rf = 0,48 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,93 (d, J = 2,0 Гц, 1H), 7,85 (d, J = 8,6 Гц, 1H), 7,53 (dd, J = 8,6, 2,0 Гц, 1H), 5,09 (s, 2H)
Этап 3: Получение S-((5-хлорбензо[d]оксазол-2-ил)метил)этантиоата
180 мг 5-хлор-2-(хлорметил)бензо[d]оксазола (0,89 ммоль), полученного на этапе 2, растворяли в 6 мл ацетона и добавляли 152 мг тиоацетата калия (1,33 ммоль) с последующим перемешиванием при 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 200 мг указанного в заголовке соединения (0,83 ммоль) с 93% выходом.
Rf = 0,38 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,84 (d, J = 1,9 Гц, 1H), 7,76 (d, J = 8,5 Гц, 1H), 7,45 (dd, J = 8,5, 1,9 Гц, 1H), 4,47 (s, 2H), 2,43 (s, 3H)
Этап 4: Получение (5-хлорбензо[d]оксазол-2-ил)метантиола
200 мг S-((5-хлорбензо[d]оксазол-2-ил)метил)этантиоата (0,83 ммоль), полученного на этапе 3, растворяли в смеси 5 мл метанола и 1 мл воды и добавляли 229 мг карбоната калия (1,66 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 30 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 160 мг указанного в заголовке соединения (0,80 ммоль) с 96% выходом.
Rf = 0,28 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,70 (d, J = 2,0 Гц, 1H), 7,46 (d, J = 8,4 Гц, 1H), 7,35 (dd, J = 8,4, 2,0 Гц, 1H), 3,97 (d, J = 8,2 Гц, 2H), 2,25 (t, J = 8,2 Гц, 1H)
<Пример 6> Получение 6-(((5-хлорбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

60 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d] пиримидин-4-она (0,24 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 150 мг (5-хлорбензо[d]оксазол-2-ил)метантиола (0,75 ммоль), полученного в примере получения 7, и 100 мг карбоната калия (0,72 ммоль) с последующим перемешиванием при 60°C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 95 мг указанного в заголовке соединения (0,23 ммоль) с 96% выходом.
Rf = 0,17 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,95 (br s, 1H), 8,23 (s, 1H), 7,91 (d, J = 8,3 Гц, 2H), 7,83 (d, J = 2,1 Гц, 1H), 7,75 (d, J = 8,6 Гц, 1H), 7,47 (dd, J = 8,3, 7,5 Гц, 2H), 7,42 (dd, J = 8,6, 2,1 Гц, 1H), 7,36 (dd, J = 7,5, 7,5 Гц, 1H), 4,84 (s, 2H)
<Пример получения 8> Получение (5-бромбензо[d]оксазол-2-ил)метантиола

Этап 1: Получение 2-хлор-N-(5-бром-2-гидроксифенил)ацетамида

500 мг 2-амино-4-бромфенола (2,66 ммоль) растворяли в 10 мл MC и добавляли 0,32 мл хлорацетилхлорида (3,99 ммоль) при 0 °C с последующим перемешиванием. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 3:1, об./об.), получая 600 мг указанного в заголовке соединения (2,27 ммоль) с 85% выходом.
Rf = 0,23 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,37 (br s, 1H), 7,12 (dd, J = 8,5, 2,3 Гц, 1H), 6,98 (d, J = 2,3 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,65 (s, 2H)
Этап 2: Получение 5-бром-2-(хлорметил)бензо[d]оксазола
600 мг 2-хлор-N-(5-бром-2-гидроксифенил)ацетамида (2,27 ммоль), полученного на этапе 1, растворяли в 8 мл фенола и добавляли 195 мг п-толуолсульфониловой кислоты (1,13 ммоль) с последующим перемешиванием с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 340 мг указанного в заголовке соединения (1,38 ммоль) с 61% выходом.
Rf = 0,50 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,06 (d, J = 2,0 Гц, 1H), 7,79 (d, J = 8,6 Гц, 1H), 7,65 (dd, J = 8,6, 2,0 Гц, 1H), 5,09 (s, 2H)
Этап 3: Получение S-((5-бромбензо[d]оксазол-2-ил)метил)этантиоата
320 мг 5-бром-2-(хлорметил)бензо[d]оксазола (1,30 ммоль), полученного на этапе 2, растворяли в 6 мл ацетона и добавляли 222 мг тиоацетата калия (1,95 ммоль) с последующим перемешиванием при 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 100 мл этилацетата, промывали 100 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 360 мг указанного в заголовке соединения (1,26 ммоль) с 97% выходом.
Rf = 0,39 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,97 (d, J = 1,7 Гц, 1H), 7,72 (d, J = 8,5 Гц, 1H), 7,57 (dd, J = 8,5, 1,7 Гц, 1H), 4,48 (s, 2H), 2,43 (s, 3H)
Этап 4: Получение (5-бромбензо[d]оксазол-2-ил)метантиола
350 мг S-((5-бромбензо[d]оксазол-2-ил)метил)этантиоата (1,22 ммоль), полученного на этапе 3, растворяли в смеси 5 мл метанола и 1 мл воды и добавляли 338 мг карбоната калия (2,45 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После завершения реакции полученный продукт экстрагировали 30 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 290 мг указанного в заголовке соединения (1,19 ммоль) с 97% выходом.
Rf = 0,30 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,85 (d, J = 1,9 Гц, 1H), 7,49 (dd, J = 8,5, 1,9 Гц, 1H), 7,42 (d, J = 8,5 Гц, 1H), 3,97 (d, J = 8,3 Гц, 2H), 2,25 (t, J = 8,3 Гц, 1H)
<Пример 7> Получение 6-(((5-бромбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
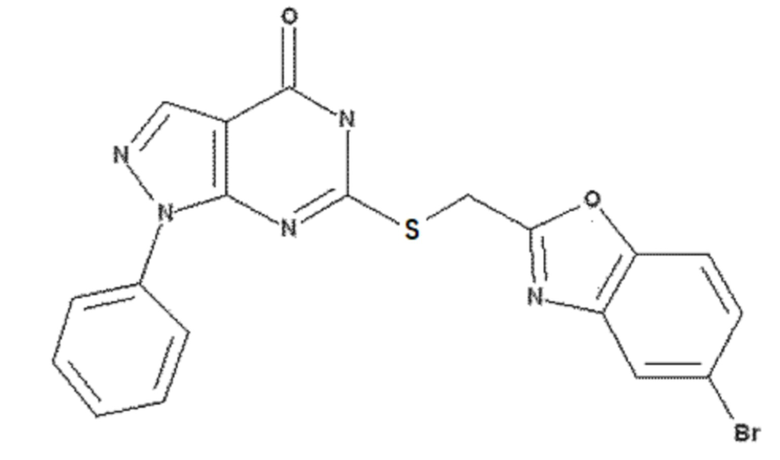
60 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,24 ммоль), полученного в примере получения 2, растворяли в 5 мл ДМФА и добавляли 180 мг (5-бромбензо[d]оксазол-2-ил)метантиола (0,75 ммоль), полученного в примере получения 8, и 100 мг карбоната калия (0,72 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции выполняли экстракцию 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали реакционный раствор при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 88 мг указанного в заголовке соединения (0,19 ммоль) с 81% выходом.
Rf = 0,28 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,95 (br s, 1H), 8,23 (s, 1H), 7,96 (d, J = 1,9 Гц, 1H), 7,91 (d, J = 8,5 Гц, 2H), 7,70 (d, J = 8,5 Гц, 1H), 7,54 (dd, J = 8,5, 1,9 Гц, 1H), 7,47 (dd, J = 8,5, 7,2 Гц, 2H), 7,36 (dd, J = 7,2, 7,2 Гц, 1H), 4,84 (s, 2H)
<Пример получения 9> Получение 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
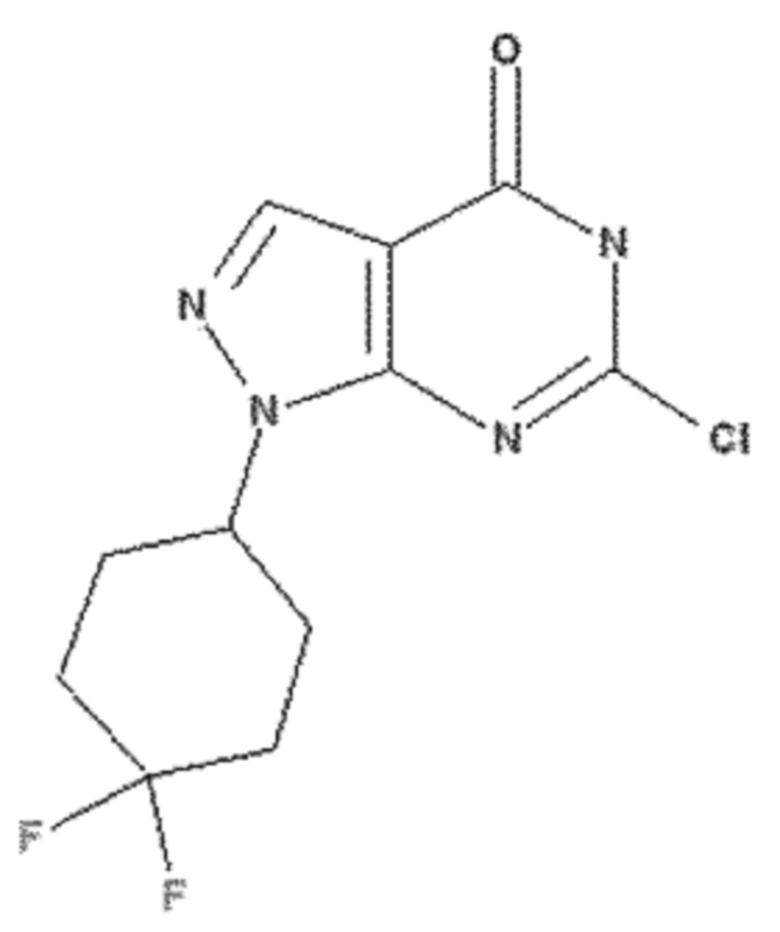
Этап 1: Получение трет-бутил-2-(4,4-дифторциклогексилиден)гидразин-1-карбоксилата

300 мг 4,4-дифторциклогексанона (2,24 ммоль) растворяли в 6 мл гексана, добавляли 296 мг трет-бутилкарбазита (2,24 ммоль) с последующим перемешиванием с обратным холодильником в течение 3 часов. После завершения реакции полученный продукт концентрировали при пониженном давлении, получая 550 г соединения, указанного в заголовке (2,24 ммоль) со 100% выходом.
Rf = 0,30 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (300 МГц, ДМСО-d6) δ 9,72 (br s, 1H), 2,45 (t, J = 6,4 Гц, 2H), 2,37 (t, J = 6,4 Гц, 2H), 1,99-2,12 (m, 4H), 1,43 (s, 9H)
Этап 2: Получение трет-бутил-2-(4,4-дифторциклогексил)гидразин-1-карбоксилата
550 мг трет-бутил-2-(4,4-дифторциклогексилиден)гидразин-1-карбоксилата (2,21 ммоль), полученного на этапе 1, растворяли в 5 мл ТГФ и добавляли 950 мг триацетоксиборгидрида натрия (4,48 ммоль) с последующим перемешиванием при комнатной температуре в течение 15 часов. После завершения реакции полученный продукт экстрагировали 30 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 3:1, об./об.), получая 465 мг указанного в заголовке соединения (1,86 ммоль) с 84% выходом.
Rf = 0,45 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (300 МГц, ДМСО-d6) δ 8,20 (br s, 1H), 4,38 (br s, 1H), 2,86-2,95 (m, 1H), 1,93-2,11 (m, 2H), 1,59-1,83 (m, 4H), 1,41-1,49 (m, 2H), 1,38 (s, 9H)
Этап 3: Получение гидрохлорида (4,4-дифторциклогексил)гидразина
465 мг трет-бутил-2-(4,4-дифторциклогексил)гидразин-1-карбоксилата (1,86 ммоль), полученного на этапе 2, растворяли в 5 мл метанола и добавляли к нему соляную кислоту с последующим перемешиванием при 50 °C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, получая 335 мг указанного в заголовке соединения (1,79 ммоль) с 96% выходом.
Rf = 0,00 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (300 МГц, ДМСО-d6) δ 3,05-3,12 (m, 1H), 1,96-2,09 (m, 4H), 1,79-1,91 (m, 2H), 1,52-1,59 (m, 2H)
Этап 4: Получение 4,6-дихлор-1-(4,4-дифторциклогексил)-1H-пиразоло[3,4-d]пиримидина
340 мг 2,4,6-трихлорпиримидин-5-карбальдегида (1,61 ммоль) растворяли в 5 мл этанола и 300 мг гидрохлорида (4,4-дифторциклогексил)гидразина (1,61 ммоль), полученного на вышеописанном этапе 3, и добавляли 0,86 мл N,N-диизопропилэтиламина (4,82 ммоль) при -78°C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 360 мг указанного в заголовке соединения (1,17 ммоль) с 73% выходом.
Rf = 0,35 (гексан:этилацетат = 9:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,57 (s, 1H), 4,97-5,04 (m, 1H), 2,13-2,23 (m, 6H), 2,03-2,07 (m, 2H)
Этап 5: Получение 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
340 мг 4,6-дихлор-1-(4,4-дифторциклогексил)-1H-пиразоло[3,4-d]пиримидина (1,11 ммоль), полученного на этапе 4, растворяли в 5 мл ТГФ и добавляли 1,2 мл 2 н. гидроксида натрия (2,21 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 320 мг указанного в заголовке соединения (1,11 ммоль) с 99% выходом.
Rf = 0,38 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,11 (s, 1H), 4,77-4,81 (m, 1H), 2,08-2,17 (m, 6H), 1,94-2,00 (m, 2H)
<Пример 8> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
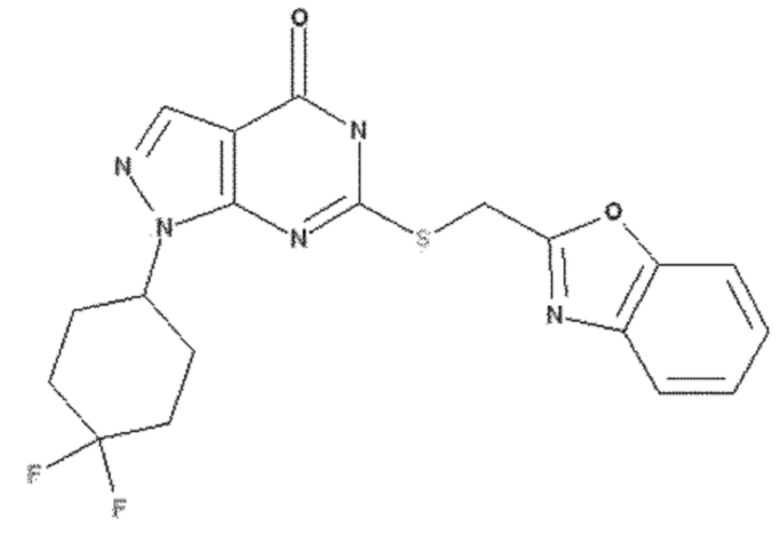
50 мг 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,17 ммоль), полученного в примере получения 9, растворяли в 3 мл ДМФА и добавляли 43 мг бензо[d]оксазол-2-илметантиола (0,26 ммоль), полученного в примере получения 2, и 70 мг карбоната калия (0,51 ммоль) с последующим перемешиванием при 60°C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 57 мг указанного в заголовке соединения (0,13 ммоль) с 80% выходом.
Rf = 0,20 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (br s, 1H), 7,96 (s, 1H), 7,68-7,71 (m, 2H), 7,34-7,40 (m, 2H), 4,83 (s, 2H), 3,54-3,60 (m, 1H), 1,90-2,08 (m, 6H), 1,74-1,78 (m, 2H)
<Пример получения 10> Получение 6-хлор-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

Этап 1: Получение трет-бутил-2-(тетрагидро-4H-тиопиран-4-илиден)гидразин-1-карбоксилата
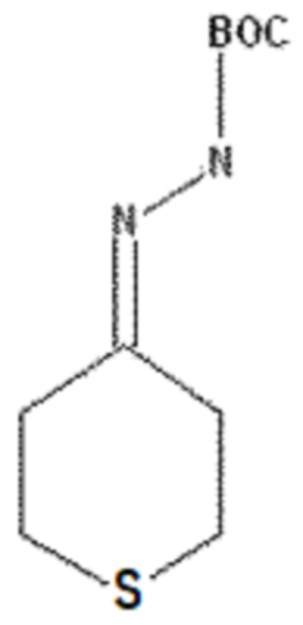
3,3 г тетрагидро-4H-тиопиран-4-она (28,40 ммоль) растворяли в 60 мл гексана и добавляли 4,13 г трет-бутилкарбазита (31,24 ммоль) с последующим перемешиванием с обратным холодильником в течение 3 часов. После завершения реакции смесь концентрировали при пониженном давлении, получая 6,5 г соединения, указанного в заголовке (28,40 ммоль) со 100% выходом.
Rf = 0,15 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 9,67 (br s, 1H), 2,73-2,76 (m, 2H), 2,65-2,68 (m, 2H), 2,60-2,63 (m, 2H), 2,47-2,50 (m, 2H), 1,43 (s, 9H)
Этап 2: Получение трет-бутил-2-(тетрагидро-2H-тиопиран-4-ил)гидразин-1-карбоксилата
6,4 г трет-бутил-2-(тетрагидро-4H-тиопиран-4-илиден)гидразин-1-карбоксилата (28,40 ммоль), полученного на этапе 1, растворяли в 80 мл ТГФ и добавляли 12 г триацетоксиборгидрида натрия (56,80 ммоль) с последующим перемешиванием при комнатной температуре в течение 15 часов. После завершения реакции полученный продукт экстрагировали 300 мл этилацетата, промывали 300 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 2:1, об./об.), получая 6,5 г указанного в заголовке соединения (27,97 ммоль) с 98% выходом.
Rf = 0,34 (гексан:этилацетат = 2:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,20 (br s, 1H), 4,28 (br s, 1H), 2,65-2,73 (m, 3H), 2,05-2,49 (m, 2H), 1,93-1,98 (m, 2H), 1,34-1,43 (m, 1H)
Этап 3: Получение гидрохлорида (тетрагидро-2H-тиопиран-4-ил)гидразина
6,5 г трет-бутил-2-(тетрагидро-2H-тиопиран-4-ил)гидразин-1-карбоксилата (27,97 ммоль), полученного на этапе 2, растворяли в 60 мл метанола и добавляли соляную кислоту с последующим перемешиванием при 50°C в течение 3 часов. После завершения реакции смесь концентрировали при пониженном давлении, получая 4,7 г соединения, указанного в заголовке (27,97 ммоль) со 94% выходом.
1H ЯМР (500 МГц, ДМСО-d6) δ 2,91-2,96 (m, 1H), 2,66-2,72 (m, 2H), 2,57-2,62 (m, 2H), 2,25-2,29 (m, 2H), 1,53-1,60 (m, 2H)
Этап 4: Получение 4,6-дихлор-1-(тетрагидро-2H-тиопиран-4-ил)-1H-пиразоло[3,4-d]пиримидина
5,9 г 2,4,6-трихлорпиримидин-5-карбальдегида (27,97 ммоль) растворяли в 60 мл этанола и добавляли 4,7 г гидрохлорида (тетрагидро-2H-тиопиран-4-ил)гидразина (27,97 ммоль), полученного на этапе 3, и 15 мл N,N-диизопропилэтиламина (83,91 ммоль) при -78 °C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 200 мл этилацетата, промывали 200 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 7 г указанного в заголовке соединения (24,20 ммоль) с 86% выходом.
Rf = 0,41 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,57 (s, 1H), 4,78-4,85 (m, 1H), 2,93-2,98 (m, 2H), 2,73-2,78 (m, 2H), 2,15-2,25 (m, 4H)
Этап 5: Получение 6-хлор-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
7 г 4,6-дихлор-1-(тетрагидро-2H-тиопиран-4-ил)-1H-пиразоло[3,4-d]пиримидина (24,20 ммоль), полученного на вышеописанном этапе 4, растворяли в 50 мл ТГФ и добавляли 25 мл 2 н. гидроксида натрия (47,60 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 5,7 г указанного в заголовке соединения (21,05 ммоль) с 87% выходом.
Rf = 0,31 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,20 (br s, 1H), 8,11 (s, 1H), 4,56-4,63 (m, 1H), 2,88-2,94 (m, 2H), 2,72-2,76 (m, 2H), 2,10-2,20 (m, 4H)
<Пример 9> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,18 ммоль), полученного в вышеописанном примере получения 10, растворяли в 3 мл ДМФА и добавляли 46 мг бензо[d]оксазол-2-илметантиола (0,28 ммоль), полученного в примере получения 2, и 75 мг карбоната калия (0,54 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 62 мг (0,15 ммоль) указанного в заголовке соединения с 86% выходом.
Rf = 0,36 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,65 (br s, 1H), 7,94 (s, 1H), 7,69-7,72 (m, 2H), 7,34-7,40 (m, 2H), 4,81 (s, 2H), 4,33-4,39 (m, 1H), 2,68-2,74 (m, 2H), 2,59-2,64 (m, 2H), 1,97-2,04 (m, 2H), 1,88-1,93 (m, 2H)
<Пример получения 11> Получение 6-хлор-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

Этап 1: Получение 4,6-дихлор-1-циклопропил-1H-пиразоло[3,4-d]пиримидина

1,95 г 2,4,6-трихлорпиримидин-5-карбальдегида (9,21 ммоль) растворяли в 30 мл этанола и добавляли 1 г гидрохлорида циклопропилгидразина (9,21 ммоль) и 4,8 мл N,N-диизопропилэтиламина (27,63 ммоль) при -78°C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 200 мл этилацетата, промывали 200 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 950 мг указанного в заголовке соединения (4,15 ммоль) с 45% выходом.
Rf = 0,45 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, CDCl3)δ 8,10 (s, 1H), 3,88-3,92 (m, 1H), 1,35-1,38 (m, 2H), 1,21-1,25 (m, 2H)
Этап 2: Получение 6-хлор-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
930 мг 4,6-дихлор-1-циклопропил-1H-пиразоло[3,4-d]пиримидина (4,06 ммоль), полученного на этапе 1, растворяли в 12 мл ТГФ и добавляли 4 мл 2 н. гидроксида натрия (8,12 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 680 мг указанного в заголовке соединения (3,23 ммоль) с 80% выходом.
Rf = 0,28 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,20 (br s, 1H), 8,02 (s, 1H), 3,78-3,82 (m, 1H), 1,05-1,15 (m, 4H)
<Пример 10> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,24 ммоль), полученного в примере получения 11, растворяли в 3 мл ДМФА и добавляли 59 мг бензо[d]оксазол-2-илметантиола (0,36 ммоль), полученного в примере получения 2, и 100 мг карбоната калия (0,72 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 48 мг указанного в заголовке соединения (0,14 ммоль) с 59% выходом.
Rf = 0,28 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,64 (br s, 1H), 7,89 (s, 1H), 7,69-7,72 (m, 2H), 7,34-7,40 (m, 2H), 4,83 (s, 2H), 3,65-3,69 (m, 1H), 0,95-0,99 (m, 2H), 0,90-0,94 (m, 2H)
<Пример получения 12> Получение 6-хлор-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

Этап 1: Получение 4,6-дихлор-1-изопропил-1H-пиразоло[3,4-d]пиримидина

2,22 г 2,4,6-трихлорпиримидин-5-карбальдегида (10,49 ммоль) растворяли в 30 мл этанола и добавляли 1,16 г гидрохлорида изопропилгидразина (10,49 ммоль) и 5,5 мл N,N-диизопропилэтиламина (31,47 ммоль) при -78°C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 200 мл этилацетата, промывали 200 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 2,1 г указанного в заголовке соединения (9,08 ммоль) с 87% выходом.
Rf = 0,48 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,16 (s, 1H), 5,16-5,22 (m, 1H), 1,60 (d, J = 6,7 Гц, 6H)
Этап 2: Получение 6-хлор-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
2 г 4,6-дихлор-1-изопропил-1H-пиразоло[3,4-d]пиримидина (8,65 ммоль), полученного на этапе 1, растворяли в 12 мл ТГФ и добавляли 8,6 мл 2 н. гидроксида натрия (17,13 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 1,7 г указанного в заголовке соединения (7,99 ммоль) с 92% выходом.
Rf = 0,29 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,16 (br s, 1H), 8,08 (s, 1H), 4,85-4,91 (m, 1H), 1,44 (d, J = 7,0 Гц, 6H)
<Пример 11> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,23 ммоль), полученного в примере получения 12, растворяли в 3 мл ДМФА и добавляли 58 мг бензо[d]оксазол-2-илметантиола (0,35 ммоль), полученного в примере получения 2, и 100 мг карбоната калия (0,72 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 44 мг указанного в заголовке соединения (0,13 ммоль) с 56% выходом.
Rf = 0,44 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,60 (br s, 1H), 7,91 (s, 1H), 7,68-7,72 (m, 2H), 7,33-7,39 (m, 2H), 4,79 (s, 2H), 4,70-4,75 (m, 1H), 1,22 (d, J = 6,5 Гц, 6H)
<Пример получения 13> Получение 6-хлор-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

Этап 1: Получение 4,6-дихлор-1-циклогексил-1H-пиразоло[3,4-d]пиримидина

4,39 г 2,4,6-трихлорпиримидин-5-карбальдегида (20,71 ммоль) растворяли в 50 мл этанола и добавляли 3,12 г гидрохлорида циклогексилгидразина (20,71 ммоль) и 11 мл N,N-диизопропилэтиламина (62,13 ммоль) при -78°C с последующим перемешиванием в течение 3 часов. После завершения реакции полученный продукт экстрагировали 200 мл этилацетата, промывали 200 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 5,2 г указанного в заголовке соединения (19,18 ммоль) с 92% выходом.
Rf = 0,39 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,15 (s, 1H), 4,75-4,82 (m, 1H), 2,01-2,08 (m, 2H), 1,93-1,99 (m, 2H), 1,77-1,81 (m, 1H), 1,48-1,57 (m, 2H), 1,30-1,39 (m, 1H), 1,01-1,04 (m, 2H)
Этап 2: Получение 6-хлор-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
5 г 4,6-дихлор-1-циклогексил-1H-пиразоло[3,4-d]пиримидина (18?44 ммоль), полученного на этапе 1, растворяли в 12 мл ТГФ и добавляли 19 мл 2 н. гидроксида натрия (36,88 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции смесь подкисляли 6 н. соляной кислотой, а осажденное твердое соединение фильтровали с получением 4,6 г указанного в заголовке соединения (18,20 ммоль) с 98% выходом.
Rf = 0,35 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,18 (br s, 1H0, 8,07 (s, 1H), 4,46-4,52 (m, 1H), 1,80-1,87 (m, 4H), 1,66-1,69 (m, 1H), 1,40-1,48 (m, 2H), 1,18-1,25 (m, 1H)
<Пример 12> Получение 6-((бензо[d]оксазол-2-илметил)тио)-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в примере получения 13, растворяли в 3 мл ДМФА и добавляли 49 мг бензо[d]оксазол-2-илметантиола (0,30 ммоль), полученного в примере получения 2, и 83 мг карбоната калия (0,60 ммоль) с последующим перемешиванием при 60 ° С в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 55 мг указанного в заголовке соединения (0,14 ммоль) с 72% выходом.
Rf = 0,27 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,61 (br s, 1H), 7,90 (s, 1H), 7,67-7,69 (m, 2H), 7,33-7,39 (m, 2H), 4,79 (s, 2H), 4,25-4,31 (m, 1H), 1,56-1,74 (m, 7H), 1,26-1,34 (m, 2H), 1,16-1,21 (m, 1H)
<Пример 13> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
50 мг 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,17 ммоль), полученного в вышеописанном примере получения 9, растворяли в 3 мл ДМФА и добавляли 61 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,34 ммоль), полученного в примере получения 6, и 70 мг карбоната калия (0,51 ммоль) с последующим перемешиванием при 60 °С в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 60 мг указанного в заголовке соединения (0,14 ммоль) с 82% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (br s, 1H), 7,96 (s, 1H), 7,55 (d, J = 8,0 Гц, 1H), 7,52 (s, 1H), 7,18 (d, J = 8,0 Гц, 1H), 4,80 (s, 2H), 4,52-4,58 (m, 1H), 2,42 (s, 3H), 1,89-2,10 (m, 6H), 1,74-1,78 (m, 2H)
<Пример 14> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в вышеописанном примере получения 1, растворяли в 3 мл ДМФА и добавляли 72 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,40 ммоль), полученного в вышеописанном примере получения 6, и 83 мг карбоната калия (0,60 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 51 мг указанного в заголовке соединения (0,13 ммоль) с 64% выходом.
Rf = 0,40 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,65 (br s, 1H), 7,95 (s, 1H), 7,54 (d, J = 8,2 Гц, 1H), 7,52 (s, 1H), 7,17 (d, J = 8,2 Гц, 1H), 4,77 (s, 2H), 4,51-4,57 (m, 1H), 3,86-3,89 (m, 2H), 3,37-3,41 (m, 2H), 2,42 (s, 3H), 1,90-1,98 (m, 2H), 1,52-1,55 (m, 2H)
<Пример 15> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,18 ммоль), полученного в вышеописанном примере получения 10, растворяли в 3 мл ДМФА и добавляли 64 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,36 ммоль), полученного в вышеописанном примере получения 6, и 75 мг карбоната калия (0,54 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 59 мг указанного в заголовке соединения (0,14 ммоль) с 78% выходом.
Rf = 0,33 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,64 (br s, 1H), 7,95 (s, 1H), 7,56 (d, J = 7,9 Гц, 1H), 7,52 (s, 1H), 7,18 (d, J = 7,9 Гц, 1H), 4,78 (s, 2H), 4,33-4,39 (m, 1H), 2,70-2,75 (m, 2H), 2,62-2,66 (m, 2H), 2,42 (s, 3H), 1,97-2,05 (m, 2H), 1,89-1,93 (m, 2H)
<Пример 16> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в примере получения 13, растворяли в 3 мл ДМФА и добавляли 72 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,40 ммоль), полученного в примере получения 6, и 83 мг карбоната калия (0,60 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 55 мг указанного в заголовке соединения (0,14 ммоль) с 70% выходом.
Rf = 0,45 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,62 (br s, 1H), 7,90 (s, 1H), 7,54 (d, J = 8,1 Гц, 1H), 7,50 (s, 1H), 7,17 (d, J = 8,1 Гц, 1H), 4,76 (s, 2H), 4,25-4,32 (m, 1H), 2,41 (s, 3H), 1,56-1,75 (m, 7H), 1,27-1,36 (m, 2H), 1,15-1,23 (m, 1H)
<Пример 17> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,24 ммоль), полученного в примере получения 11, растворяли в 3 мл ДМФА и добавляли 86 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,48 ммоль), полученного в примере получения 6, и 100 мг карбоната калия (0,72 ммоль) с последующим перемешиванием при 60 °C в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 50 мг указанного в заголовке соединения (0,14 ммоль) с 59% выходом.
Rf = 0,29 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,64 (br s, 1H), 7,89 (s, 1H), 7,57 (d, J = 8,2 Гц, 1H), 7,53 (s, 1H), 7,17 (d, J = 8,2 Гц, 1H), 4,81 (s, 2H), 3,66-3,71 (m, 1H), 2,43 (s, 3H), 0,91-1,01 (m, 4H)
<Пример 18> Получение 6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,24 ммоль), полученного в примере получения 12, растворяли в 3 мл ДМФА и добавляли 82 мг (6-метилбензо[d]оксазол-2-ил)метантиола (0,46 ммоль), полученного в вышеописанном примере получения 6, и 95 мг карбоната калия (0,69 ммоль) с последующим перемешиванием при 60 °С в течение 3 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 56 мг указанного в заголовке соединения (0,16 ммоль) с 68% выходом.
Rf = 0,21 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,60 (br s, 1H), 7,92 (s, 1H), 7,55 (d, J = 8,2 Гц, 1H), 7,52 (s, 1H), 7,17 (d, J = 8,2 Гц, 1H), 4,76 (s, 2H), 4,71-4,75 (m, 1H), 2,42 (s, 3H), 1,23 (d, J = 6,5 Гц, 6H)
<Пример получения 14> Получение 1-(бензо[d]оксазол-2-ил)этан-1-тиола

1 г 2-аминофенола (9,16 ммоль) растворяли в 20 мл толуола и добавляли 1 мл тиомолочной кислоты (9,16 ммоль) с последующим перемешиванием с обратным холодильником в течение 5 часов. Затем добавляли 1,6 г п-толуолсульфоновой кислоты (9,16 ммоль) и перемешивали с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 50 мл этилацетата, промывали 50 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 2:1, об./об.), получая 240 мг указанного в заголовке соединения (1,33 ммоль) с 15% выходом.
Rf = 0,50 (гексан:этилацетат = 2:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,71-7,73 (m, 1H), 7,53-7,55 (m, 1H), 7,35-7,37 (m, 2H), 4,34-4,39 (m, 1H), 2,42 (d, J = 7,4 Гц, 1H), 1,90 (d, J = 6,9 Гц, 2H)
<Пример 19> Получение 6-((1-(бензо[d]оксазол-2-ил)этил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
50 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в примере получения 3, растворяли в 3 мл ДМФА и добавляли 55 мг 1-(бензо[d]оксазол-2-ил)этан-1-тиола (0,30 ммоль), полученного в примере получения 14, и 83 мг карбоната калия (0,61 ммоль) с последующим перемешиванием 60°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 49 мг указанного в заголовке соединения (0,13 ммоль) с 63% выходом.
Rf = 0,37 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,90 (br s, 1H), 8,25 (s, 1H), 7,96-7,99 (m, 2H), 7,74 (d, J = 7,1 Гц, 1H), 7,70 (d, J = 7,1 Гц, 1H), 7,51-7,54 (m, 2H), 7,34-7,40 (m, 3H), 5,38-5,42 (m, 1H), 1,89 (d, J = 5,1 Гц, 3H)
<Пример 20> Получение 6-((1-бензо[d]оксазол-2-ил)этил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

25 мг 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,08 ммоль), полученного в вышеописанном примере получения 9, растворяли в 3 мл ДМФА и добавляли 55 мг 1-(бензо[d]оксазол-2-ил)этан-1-тиола (0,30 ммоль), полученного в вышеописанном примере получения 14, и 36 мг карбоната калия (0,24 ммоль) с последующим перемешиванием при 60 °C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 30 мг указанного в заголовке соединения (0,07 ммоль) с 80% выходом.
Rf = 0,33 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,65 (br s, 1H), 7,97 (s, 1H), 7,71-7,74 (m, 2H), 7,36-7,42 (m, 2H), 5,43 (q, J = 7,4 Гц, 1H), 4,55-4,61 (m, 1H), 2,07-2,17 (m, 4H), 1,96-2,01 (m, 2H), 1,89 (d, J = 7,4 Гц, 3H), 1,83-1,90 (m, 1H), 1,74-1,76 (m, 1H)
<Пример 21> Получение 6-((1-бензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

30 мг 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,12 ммоль), полученного в вышеописанном примере получения 1, растворяли в 3 мл ДМФА и 42 мг 1-(бензо[d]оксазол-2-ил)этан-1-тиола (0,23 ммоль), полученного в вышеописанном примере получения 14, и добавляли 50 мг карбоната калия (0,37 ммоль) с последующим перемешиванием при 60 °С в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 17 мг указанного в заголовке соединения (0,04 ммоль) с 36% выходом.
Rf = 0,33 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,63 (br s, 1H), 7,96 (s, 1H), 7,71-7,73 (m, 2H), 7,35-7,40 (m, 2H), 5,37 (q, J = 7,3 Гц, 1H), 4,50-4,56 (m, 1H), 3,87-3,93 (m, 2H), 3,36-3,45 (m, 2H), 1,90-2,04 (m, 2H), 1,87 (d, J = 7,3 Гц, 3H), 1,63-1,66 (m, 1H), 1,47-1,51 (m, 1H)
<Пример получения 15> Получение 1-(6-метилбензо[d]оксазол-2-ил)этан-1-тиола
1 г 2-амино-5-метилфенола (8,12 ммоль) растворяли в 10 мл толуола и добавляли 0,72 мл тиомолочной кислоты (8,13 ммоль) с последующим перемешиванием при 100 °C в течение 24 часов. После завершения реакции остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 384 мг указанного в заголовке соединения (1,83 ммоль) с 22% выходом.
Rf = 0,48 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,59 (d, J = 8,2 Гц, 1H), 7,33 (s, 1H), 7,17 (d, J = 8,2 Гц, 1H), 4,31-4,37 (m, 1H), 2,50 (s, 3H), 2,40 (d, J = 7,5 Гц, 1H), 1,88 (d, J = 7,1 Гц, 3H)
<Пример 22> Получение 1-(4,4-дифторциклогексил)-6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,17 ммоль), полученного в вышеописанном примере получения 9, растворяли в 3 мл ДМФА и добавляли 72 мг 1-(6-метилбензо[d]оксазол-2-ил)этан-1-тиола (0,35 ммоль), полученного в вышеописанном примере получения 15, и 72 мг карбоната калия (0,52 ммоль) с последующим перемешиванием при 60 °C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 60 мг указанного в заголовке соединения (0,13 ммоль) с 75% выходом.
Rf = 0,18 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (br s, 1H), 7,98 (s, 1H), 7,59 (d, J = 7,9 Гц, 1H), 7,53 (s, 1H), 7,20 (d, J = 7,9 Гц, 1H), 5,40 (q, J = 7,0 Гц, 1H), 4,54-4,59 (m, 1H), 2,42 (s, 3H), 1,91-2,19 (m, 7H), 1,87 (d, J = 7,0 Гц, 3H), 1,73-1,76 (m, 1H)
<Пример 23> Получение 6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,20 ммоль), полученного в вышеописанном примере получения 1, растворяли в 3 мл ДМФА и добавляли 84 мг 1-(6-метилбензо[d]оксазол-2-ил)этан-1-тиола (0,40 ммоль), полученного в вышеописанном примере получения 15, и 83 мг карбоната калия (0,60 ммоль) с последующим перемешиванием при 60 °C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 50 мг указанного в заголовке соединения (0,12 ммоль) с 66% выходом.
Rf = 0,11 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,62 (br s, 1H), 7,96 (s, 1H), 7,59 (d, J = 8,2 Гц, 1H), 7,53 (s, 1H), 7,19 (d, J = 8,2 Гц, 1H), 5,35 (q, J = 7,1 Гц, 1H), 4,51-4,57 (m, 1H), 3,98-3,95 (m, 2H), 3,38-3,47 (m, 2H), 2,42 (s, 3H), 1,87-2,06 (m, 2H), 1,86 (d, J = 7,1 Гц, 3H), 1,65-1,68 (m, 1H), 1,48-1,51 (m, 1H)
<Пример 24> Получение 6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

50 мг 6-хлор-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,18 ммоль), полученного в вышеописанном примере получения 10, растворяли в 3 мл ДМФА и добавляли 77 мг 1-(6-метилбензо[d]оксазол-2-ил)этан-1-тиола (0,37 ммоль), полученного в вышеописанном примере получения 15, и 75 мг карбоната калия (0,54 ммоль) с последующим перемешиванием при 60 °C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 62 мг указанного в заголовке соединения (0,14 ммоль) с 77% выходом.
Rf = 0,19 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,61 (br s, 1H), 7,96 (s, 1H), 7,60 (d, J = 7,9 Гц, 1H), 7,53 (s, 1H), 7,20 (d, J = 7,9 Гц, 1H), 5,37 (q, J = 7,0 Гц, 1H), 4,34-4,40 (m, 1H), 2,66-2,82 (m, 4H), 2,42 (s, 3H), 1,96-2,12 (m, 3H), 1,88-1,91 (m, 1H), 1,86 (d, J = 7,0 Гц, 3H)
<Пример получения 16> Получение (5-фенилбензо[d]оксазол-2-ил)метантиола

Этап 1: Получение 5-бром-2-метилбензо[d]оксазола

220 мг 2-амино-4-бромфенола (1,17 ммоль) растворяли в 0,43 мл триэтилзоацетата (2,34 ммоль) и 7 мкл уксусной кислоты (0,12 ммоль) с последующим перемешиванием при 100 °C в течение 0,5 часов. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 10 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 5:1, об./об.), получая 228 мг указанного в заголовке соединения (1,07 ммоль) с 92% выходом.
Rf = 0,36 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,81 (d, J = 1,8 Гц, 1H), 7,44 (dd, J = 8,6, 1,4 Гц, 1H), 7,37 (d, J = 8,6 Гц, 1H), 2,67 (s, 3H)
Этап 2: Получение 2-метил-5-фенилбензо[d]оксазола
210 мг 5-бром-2-метилбензо[d]оксазола (0,99 ммоль), полученного на этапе 1, растворяли в смеси диоксана и воды и добавляли 181 мг фенилборной кислоты (1,48 ммоль), 40 мг Pd(dppf))Cl2 (0,05 ммоль) и 410 мг карбоната калия (2,97 ммоль) с последующим перемешиванием с обратным холодильником в течение 1 часа. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 4:1, об./об.), получая 199 мг указанного в заголовке соединения (0,95 ммоль) с 96% выходом.
Rf = 0,25 (гексан:этилацетат = 5:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,87 (s, 1H), 7,64 (d, J = 7,3 Гц, 2H), 7,54-7,55 (m, 2H), 7,48 (dd, J = 7,5, 7,3 Гц, 2H), 7,38 (dd, J = 7,3, 7,3 Гц, 1H), 2,69 (s, 3H)
Этап 3: Получение 2-(бромметил)-5-фенилбензо[d]оксазола
199 мг 2-метил-5-фенилбензо[d]оксазола (0,95 ммоль), полученного на этапе 2, растворяли в хлорбензоле и добавляли 169 мг N-бромсукцинимида (0,95 ммоль), 31 мг AIBN (азобизизобутилонитрила) (0,19 ммоль) с последующим перемешиванием с обратным холодильником в течение 2 часов. После завершения реакции остаток очищали колоночной хроматографией (гексан:этилацетат = 8:1, об./об.), получая 110 мг указанного в заголовке соединения (0,38 ммоль) с 40% выходом.
Rf = 0,30 (гексан:этилацетат = 8:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,94 (s, 1H), 7,61-7,66 (m, 4H), 7,49 (dd, J = 7,7, 7,4 Гц, 2H), 7,40 (dd, J = 7,4, 7,4 Гц, 1H), 4,64 (s, 2H)
Этап 4: Получение S-((5-фенилбензо[d]оксазол-2-ил)метил)этантиоата
100 мг 2-(бромметил)-5-фенилбензо[d]оксазола (0,34 ммоль), полученного на этапе 3, растворяли в ацетоне и добавляли 59 мг тиоацетата калия (0,52 ммоль) с последующим перемешиванием при 50°C в течение 1 часа. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли и сушили над безводным сульфатом натрия (Na2SO4), получая 95 мг указанного в заголовке соединения (0,33 ммоль) с 96% выходом.
Rf = 0,23 (гексан:этилацетат = 6:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,90 (s, 1H), 7,58-7,62 (m, 4H), 7,48 (dd, J = 7,7, 7,4 Гц, 2H), 7,39 (dd, J = 7,4, 7,4 Гц, 1H), 4,44 (s, 2H), 2,47 (s, 3H)
Этап 5: Получение (5-фенилбензо[d]оксазол-2-ил)метантиола
90 мг S-((5-фенилбензо[d]оксазол-2-ил)метил)этаноата (0,32 ммоль), полученного на этапе 4, растворяли в 6 мл смеси метанола и воды и добавляли 131 мг карбоната калия (0,95 ммоль) с последующим перемешиванием при комнатной температуре в течение 1 часа. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли и сушили над безводным сульфатом натрия (Na2SO4), получая 75 мг указанного в заголовке соединения (0,31 ммоль) с 99% выходом.
Rf = 0,14 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,90 (s, 1H), 7,58-7,63 (m, 4H), 7,49 (dd, J = 7,8, 7,5 Гц, 2H), 7,39 (dd, J = 7,5, 7,5 Гц, 1H), 4,01 (d, J = 8,1 Гц, 2H), 2,27 (t, J = 8,1 Гц, 1H)
<Пример 25> Получение 1-фенил-6-(((5-фенилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

38 мг 6-хлор-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,15 ммоль), полученного в примере получения 3, растворяли в 3 мл ДМФА и добавляли 75 мг (5-фенилбензо[d]оксазол-2-ил)метантиола (0,31 ммоль), полученного в примере получения 16, и 128 мг карбоната калия (0,93 ммоль) с последующим перемешиванием при 60 °С в течение 1 часа. После завершения реакции полученный продукт экстрагировали 10 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (дихлорметан:метанол = 20:1, об./об.), получая 50 мг указанного в заголовке соединения (0,11 ммоль) с 71% выходом.
Rf = 0,35 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,96 (br s, 1H), 8,24 (s, 1H), 7,95-7,97 (m, 3H), 7,78 (d, J = 8,7 Гц, 1H), 7,65-7,69 (m, 3H), 7,45-7,51 (m, 4H), 7,35-7,39 (m, 2H), 4,86 (s, 2H)
<Пример получения 17> Получение 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она

Этап 1: Получение этил-5-амино-1-фенил-1H-пиразол-4-карбоксилата

5 г этил (E)-2-циан-3-этоксиакрилата (30 ммоль) растворяли в 100 мл этанола и добавляли 3,9 г фенилгидразина (36 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, а остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 7 г указанного в заголовке соединения (30 ммоль) с 100% выходом.
Rf = 0,30 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,75 (s, 1H), 7,55-7,45 (m, 4H), 7,41-7,34 (m, 1H), 5,36 (br s, 2H), 4,28 (q, J = 7,1 Гц, 2H), 1,35 (t, J = 7,1 Гц, 3H)
Этап 2: Получение 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиримидин-4-она
7 г этил-5-амино-1-фенил-1H-пиразол-4-карбоксилата (30 ммоль), полученного на этапе 1, растворяли в 60 мл тетрагидрофурана и добавляли 4,95 мл бензоилизотиоцианата (39 ммоль), после чего перемешивали с обратным холодильником в течение 13 часов. Затем добавляли 92 мл 2 н. гидроксида натрия и перемешивали смесь с обратным холодильником в течение 30 минут. После завершения реакции ее нейтрализовали 2 н. соляной кислотой и добавляли уксусную кислоту для осаждения твердого вещества. Остаток очищали водой, гексаном и дихлорметаном, получая 4,48 г указанного в заголовке соединения (18,34 ммоль) с 60% выходом.
Rf = 0,15 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 11,27 (br s, 1H), 10,46 (s, 1H), 8,13 (s, 1H), 7,87 (d, J = 7,6 Гц, 2H), 7,68-7,47 (m, 3H)
<Пример получения 18> Получение 2-(хлорметил)-4-метилбензо[d]оксазола

300 мг 2-амино-3-метилфенола (2,44 ммоль) растворяли в 6 мл ксилола и медленно добавляли 0,29 мл хлорацетилхлорида (3,66 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,37 мл триэтиламина (2,68 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 30 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 363 мг указанного в заголовке соединения (2,0 ммоль) с 82% выходом.
Rf = 0,5 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,36 (d, J = 8,2 Гц, 1H), 7,28-7,24 (m, 1H), 7,15 (d, J = 7,4 Гц, 1H), 4,77 (s, 2H), 2,62 (s, 3H).
<Пример 26> Получение 6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразола[3,4-d]пиримидин-4-она
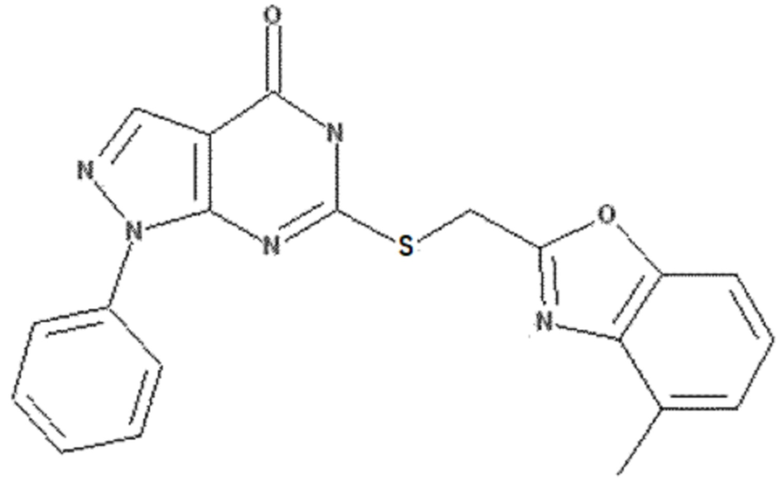
40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в метаноле и добавляли 33 мг 2-(хлорметил)-4-метилбензо[d]оксазола (0,18 ммоль), полученного в примере получения 18, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции полученный продукт концентрировали при пониженном давлении и очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 55 мг указанного в заголовке соединения (0,14 ммоль) с 86% выходом.
Rf = 0,3 (гексан:этилацетат = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,22 (s, 1H), 8,06 (d, J = 8,1 Гц, 2H), 7,53-7,44 (m, 3H), 7,40-7,38 (m, 1H), 7,26-7,23 (m, 1H), 7,15 (d, J = 7,3 Гц, 1H), 4,82 (s, 2H), 2,48 (s, 3H)
<Пример получения 19> Получение 5-(трет-бутил)-2-(хлорметил)бензо[d]оксазола

200 мг 2-амино-4-(трет-бутил)фенола (1,21 ммоль) растворяли в 5 мл ксилола и медленно добавляли 0,14 мл хлорацетилхлорида (1,81 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,19 мл триэтиламина (1,33 ммоль) и перемешивали с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 20:1, об./об.), получая 154 мг указанного в заголовке соединения (0,69 ммоль) с 57% выходом.
Rf = 0,55 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,77 (s, 1H), 7,50-7,46 (m, 2H), 4,76 (s, 2H), 1,40 (s, 9H)
<Пример 27> Получение 6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
20 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,08 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 20 мг 5-(трет-бутил)-2-(хлорметил)бензо[d]оксазола (0,09 ммоль), полученного в вышеописанном примере получения 19, и 0,05 мл 2 н. гидроксида натрия, после чего перемешивали с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 36 мг указанного в заголовке соединения (0,08 ммоль) со 100% выходом.
Rf = 0,3 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,17 (s, 1H), 7,98 (d, J = 8,4 Гц, 2H), 7,66 (s, 1H), 7,57 (d, J = 8,6 Гц, 1H), 7,49-7,45 (m, 2H), 7,43-7,41 (d, J = 8,6 Гц, 1H), 7,36-7,34 (m, 1H), 4,78 (s, 2H), 1,30 (s, 9H)
<Пример получения 20> Получение 2-(хлорметил)-6-нитробензо[d]оксазола

200 мг 2-амино-5-нитрофенола (1,30 ммоль) растворяли в 5 мл ксилола и медленно добавляли 0,16 мл хлорацетилхлорида (1,95 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,2 мл триэтиламина (1,43 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 190 мг указанного в заголовке соединения (0,89 ммоль) с 67% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,47 (s, 1H), 8,33 (d, J = 8,8, 1H), 7,87 (d, J = 8,8, 1H), 4,82 (s, 2H)
<Пример 28> Получение 6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 38 мг 2-(хлорметил)-6-нитробензо[d]оксазола (0,18 ммоль), полученного в примере получения 20, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 22 мг указанного в заголовке соединения (0,05 ммоль) с 32% выходом.
Rf = 0,3 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,59 (d, J = 2,3 Гц, 1H), 8,24 (dd, J = 8,8, 2,3 Гц, 1H), 8,20 (s, 1H), 7,91 (d, J = 8,8 Гц, 1H), 7,87 (d, J = 7,7 Гц, 2H), 7,52-7,45 (m, 2H), 7,39-7,36 (m, 1H), 4,90 (s, 2H)
<Пример получения 21> Получение 2-(хлорметил)-5-нитробензо[d]оксазола

400 мг 2-амино-4-нитрофенола (2,60 ммоль) растворяли в 5 мл ксилола и медленно добавляли 0,32 мл хлорацетилхлорида (3,90 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,4 мл триэтиламина (2,86 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 20 мл этилацетата, промывали 20 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 42 мг указанного в заголовке соединения (0,19 ммоль) с 8% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,66 (d, J = 2,2 Гц, 1H), 8,38 (dd, J = 9,0, 2,2 Гц, 1H), 7,69 (d, J = 9,0 Гц, 1H), 4,80 (s, 2H)
<Пример 29> Получение 6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0?16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 37 мг 2-(хлорметил)-5-нитробензо[d]оксазола (0,17 ммоль), полученного в примере получения 21, и 0,05 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 28 мг указанного в заголовке соединения (0,06 ммоль) с 41% выходом.
Rf = 0,3 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,96 (s, 1H), 8,58 (d, J = 2,3 Гц, 1H), 8,28 (dd, J = 9,0, 2,3 Гц, 1H), 8,21 (s, 1H), 7,94 (d, J = 9,0 Гц, 1H), 7,90 (d, J = 8,1 Гц, 2H), 7,48 (dd, J = 7,7, 8,1 Гц, 2H), 7,36 (d, J = 7,7 Гц, 1H), 4,90 (s, 2H)
<Пример получения 22> Получение 2-(хлорметил)-4-нитробензо[d]оксазола

600 мг 2-амино-3-нитрофенола (3,89 ммоль) растворяли в 10 мл ксилола, медленно добавляли 0,66 мл хлорацетилхлорида (5,83 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,6 мл триэтиламина (4,27 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 508 мг указанного в заголовке соединения (2,39 ммоль) с 61% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,27 (d, J = 8,2 Гц, 1H), 7,96 (d, J = 8,2 Гц, 1H), 7,61 (dd, J = 8,2, 8,2 Гц, 1H), 4,90 (s, 2H)
<Пример 30> Получение 6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 38 мг 2-(хлорметил)-4-нитробензо[d]оксазола (0,17 ммоль), полученного в примере получения 22, и 0,05 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 34 мг указанного в заголовке соединения (0,08 ммоль) с 50% выходом.
Rf = 0,3 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,88 (s, 1H), 8,24 (s, 1H), 8,17-8,14 (m, 1H), 8,01-7,96 (m, 3H), 7,60 (d, J = 8,2 Гц, 1H), 7,55-7,52 (m, 2H), 7,47 (d, J = 8,0 Гц, 1H), 4,13 (s, 2H)
<Пример получения 23> Получение 5-хлор-2-(хлорметил)-6-нитробензо[d]оксазола

500 мг 2-амино-4-хлор-5-нитрофенола (2,65 ммоль) растворяли в 8 мл ксилола и медленно добавляли 0,32 мл хлорацетилхлорида (3,97 ммоль) с последующим перемешиванием при 0°C в течение 2 часов. Затем добавляли 0,41 мл триэтиламина (2,91 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 329 мг указанного в заголовке соединения (1,33 ммоль) с 50% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,15 (s, 1H), 7,94 (s, 1H), 4,80 (s, 2H)
<Пример 31> Получение 6-(((5-нитро-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 45 мг 5-хлор-2-(хлорметил)-6-нитробензо[d]оксазола (0,17 ммоль), полученного в примере получения 23, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 22 мг указанного в заголовке соединения (0,05 ммоль) с 29% выходом.
Rf = 0,2 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 10,63 (s, 1H), 8,59 (s, 1H), 8,22 (s, 1H), 8,18 (s, 1H), 7,96-7,93 (m, 1H), 7,86-7,84 (m, 1H), 7,51-7,47 (m, 2H), 7,37 (d, J = 7,5 Гц, 1H), 4,90 (s, 2H)
<Пример получения 24> Получение 5,7-дихлор-2-(хлорметил)-бензо[d]оксазола

500 мг 2-амино-4,6-дихлорфенола (2,81 ммоль) растворяли в 10 мл ксилола и медленно добавляли 0,34 мл хлорацетилхлорида (4,21 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,43 мл триэтиламина (3,09 ммоль) и перемешивали с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 470 мг указанного в заголовке соединения (1,99 ммоль) с 71% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,62 (d, J = 1,9 Гц, 1H), 7,38 (d, J = 1,9 Гц, 1H), 4,77 (s, 2H)
<Пример 32> Получение 6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 43 мг 5,7-дихлор-2-(хлорметил)-бензо[d]оксазола (0,17 ммоль), полученного в примере получения 24, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 11 мг указанного в заголовке соединения (0,02 ммоль) с 15% выходом.
Rf = 0,3 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,20 (s, 1H), 7,92 (d, J = 7,9 Гц, 2H), 7,86 (d, J = 1,9 Гц, 1H), 7,68 (d, J = 1,9 Гц, 1H), 7,45 (t, J = 7,8 Гц, 2H), 7,34 (t, J = 7,3 Гц, 1H), 4,86 (s, 2H)
<Пример получения 25> Получение 2-(хлорметил)-5,7-диметилбензо[d]оксазола

500 мг 2-амино-4,6-диметилфенола (3,64 ммоль) растворяли в 10 мл ксилола и медленно добавляли 0,44 мл хлорацетилхлорида (5,46 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,56 мл триэтиламина (4,00 ммоль) и перемешивали с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 479 мг указанного в заголовке соединения (2,45 ммоль) с 67% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,31 (s, 1H), 6,95 (s, 1H), 4,71 (s, 2H), 2,46 (s, 3H), 2,38 (s, 3H)
<Пример 33> Получение 6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d] пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 35 мг 2-(хлорметил)-5,7-диметилбензо[d]оксазола (0,17 ммоль), полученного в примере получения 24, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 17 мг указанного в заголовке соединения (0,04 ммоль) с 26% выходом.
Rf = 0,3 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,24 (s, 1H), 7,99 (d, J = 7,9 Гц, 2H), 7,45 (dd, J = 7,9, 7,3 Гц, 2H), 7,37 (d, J = 7,3 Гц, 1H), 7,30 (s, 1H), 7,00 (s, 1H), 4,80 (s, 2H), 2,38 (s, 3H), 2,34 (s, 3H)
<Пример получения 26> Получение 2-(хлорметил)-5-метоксибензо[d]оксазола

412 мг 2-амино-4-метоксифенола (2,96 ммоль) растворяли в 10 мл ксилола и медленно добавляли 0,35 мл хлорацетилхлорида (4,44 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,45 мл триэтиламина (3,25 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 20:1, об./об.), получая 254 мг указанного в заголовке соединения (1,29 ммоль) с 43% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,48-7,38 (m, 1H), 7,21 (d, J = 2,6 Гц, 1H), 6,99 (dd, J = 9,0, 2,6 Гц, 1H), 4,74 (s, 2H), 3,86 (s, 3H)
<Пример 34> Получение 6-(((5-метоксибензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 36 мг 2-(хлорметил)-5метоксибензо[d]оксазола (0,17 ммоль), полученного в примере получения 25, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 15 мг указанного в заголовке соединения (0,04 ммоль) с 23% выходом.
Rf = 0,2 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,94 (s, 1H), 8,24 (s, 1H), 7,94 (d, J = 7,9 Гц, 2H), 7,59 (d, J = 8,9 Гц, 1H), 7,48 (t, J = 7,7 Гц, 2H), 7,37 (t, J = 7,5 Гц, 1H), 7,25 (d, J = 2,5 Гц, 1H), 6,94 (dd, J = 8,9, 2,6 Гц, 1H), 4,80 (s, 2H), 3,77 (s, 3H)
<Пример получения 27> Получение 2-(хлорметил)-6-метоксибензо[d]оксазола

412 мг 2-амино-5-метоксифенола (2,96 ммоль) растворяли в 10 мл ксилола и медленно добавляли 0,35 мл хлорацетилхлорида (4,44 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,45 мл триэтиламина (3,25 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 20:1, об./об.), получая 236 мг указанного в заголовке соединения (1,19 ммоль) с 40% выходом.
Rf = 0,4 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,58 (d, J = 8,8 Гц, 1H), 7,03 (d, J = 2,4 Гц, 1H), 6,94 (dd, J = 8,8, 2,4 Гц, 1H), 4,72 (s, 2H), 3,83 (s, 3H)
<Пример 35> Получение 6-(((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 36 мг 2-(хлорметил)-6-метоксибензо[d]оксазола (0,17 ммоль), полученного в примере получения 26, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 25 мг указанного в заголовке соединения (0,06 ммоль) с 38% выходом.
Rf = 0,2 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,93 (s, 1H), 8,24 (s, 1H), 7,97 (d, J = 8,0 Гц, 2H), 7,58 (d, J = 8,8 Гц, 1H), 7,50 (t, J = 7,7 Гц, 2H), 7,38 (t, J = 7,4 Гц, 1H), 7,30 (d, J = 2,4 Гц, 1H), 6,93 (dd, J = 8,8, 2,4 Гц, 1H), 4,79 (s, 2H), 3,79 (s, 3H)
<Пример получения 28> Получение 2-(хлорметил)-5-(трифторметил)бензо[d]оксазола

776 мг 2-амино-4-(трифторметил)фенола (4,38 ммоль) растворяли в 12 мл ксилола, медленно добавляли 0,52 мл хлорацетилхлорида (6,57 ммоль) с последующим перемешиванием при 0 °C в течение 2 часов. Затем добавляли 0,67 мл триэтиламина (4,82 ммоль) и перемешивали смесь с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 40 мл этилацетата, промывали 40 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 10:1, об./об.), получая 275 мг указанного в заголовке соединения (1,17 ммоль) с 27% выходом.
Rf = 0,5 (гексан:этилацетат = 4:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 8,09-8,02 (m, 1H), 7,73-7,64 (m, 2H), 4,81 (s, 2H)
<Пример 36> Получение 1-фенил-6-(((5-(трифторметил)бензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-фенил-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,16 ммоль), полученного в примере получения 17, растворяли в 3 мл метанола и добавляли 41 мг 2-(хлорметил)-5-(трифторметил)бензо[d]оксазола (0,17 ммоль), полученного в примере получения 27, и 0,1 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 25 мг указанного в заголовке соединения (0,06 ммоль) с 34% выходом.
Rf = 0,25 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,97 (s, 1H), 8,20 (s, 1H), 8,16-8,12 (m, 1H), 7,96-7,90 (m, 3H), 7,75 (d, J = 8,5 Гц, 1H), 7,47 (dd, J = 7,7, 8,1 Гц, 2H), 7,35 (dd, J = 7,5, 7,7 Гц, 1H), 4,88 (s, 2H)
<Пример получения 29> Получение 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она

Этап 1: Получение этил-5-амино-1-(4,4-дифторциклогексил)-1H-пиразол-4-карбоксилата

1 г этил (E)-2-циан-3-этоксиакрилата (5,91 ммоль) растворяли в 30 мл этанола и добавляли 1,32 г (4,4-дифторциклогексил)гидразина (7,09 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 50 мл этилацетата, промывали 50 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 3:1, об./об.), получая 1,16 г указанного в заголовке соединения (4,31 ммоль) с 73% выходом.
Rf = 0,40 (гексан:этилацетат = 3:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,65 (s, 1H), 5,03 (br s, 2H), 429 (q, J = 8,0 Гц, 2H), 3,95-3,90 (m, 1H), 2,34-2,27 (m, 4H), 2,08-1,84 (m, 4H), 1,36 (t, J = 8,0 Гц, 4H)
Этап 2: Получение этил 5-(3-бензоилтиоуреидо)-1-(4,4-дифторциклогексил)-1H-пиразол-4-карбоксилата
200 мг этил-5-амино-1-(4,4-дифторциклогексил)-1H-пиразол-4-карбоксилата (0,73 ммоль), полученного на этапе 1, растворяли в 5 мл тетрагидрофурана и добавляли 0,12 мл бензоилизотиоцианата (0,87 ммоль) с последующим перемешиванием с обратным холодильником в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией (гексан:этилацетат = 2:1, об./об.), получая 312 мг указанного в заголовке соединения (0,72 ммоль) с 98% выходом.
Rf = 0,55 (гексан:этилацетат = 2:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 12,17 (s, 1H), 9,44 (s, 1H), 8,01 (s, 1H), 7,97 (d, J = 7,8 Гц, 2H), 7,73 (dd, J = 7,8 Гц, 7,8 Гц, 1H), 7,61 (dd, J = 7,8 Гц, 7,8 Гц, 2H), 4,29 (q, J = 7,3 Гц, 2H), 2,42-2,26 (m, 4H), 2,16-2,10 (m, 2H), 1,91-1,80 (m, 2H), 1,30 (t, J = 7,3 Гц, 3H)
Этап 3: Получение 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она
100 мг этил 5-(3-бензоилтиоуреидо)-1-(4,4-дифторциклогексил)-1H-пиразол-4-карбоксилата (0,23 ммоль), полученного на этапе 2, растворяли в 3 мл этанола и добавляли 44 мг трет-бутоксида натрия (0,46 ммоль) с последующим перемешиванием при комнатной температуре в течение 3 часов. После завершения реакции смесь нейтрализовали 1 н. соляной кислотой для осаждения твердого вещества, которое очищали водой, простым эфиром и этилацетатом с получением 50 мг указанного в заголовке соединения (0,17 ммоль) с 76% выходом.
1H ЯМР (500 МГц, ДМСО-d6) δ 13,44 (br s, 1H), 12,19 (br s, 1H), 7,96 (s, 1H), 4,77 (s, 1H), 2,37-1,81 (m, 8H)
<Пример 37> Получение 1-(4,4-дифторциклогексил)-6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
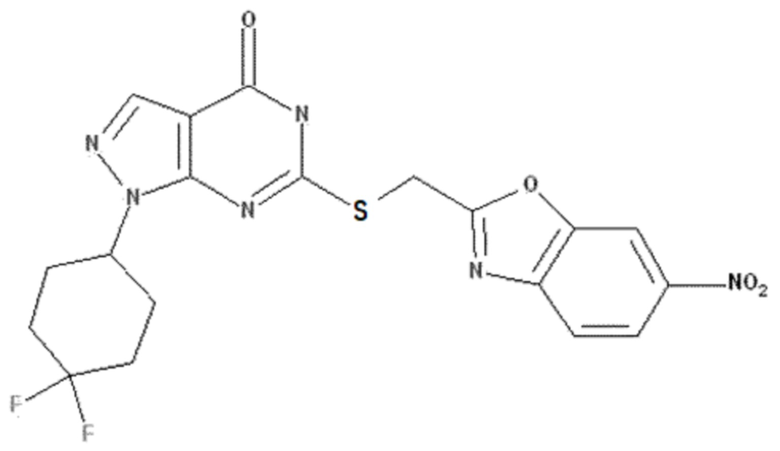
40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 33 мг 2-(хлорметил)-6-нитробензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 20, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 48 мг указанного в заголовке соединения (0,10 ммоль) с 75% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,71 (d, J = 2,3 Гц, 1H), 8,29 (dd, J = 8,8, 2,3 Гц, 1H), 7,95 (s, 1H), 7,93 (d, J = 8,8 Гц, 1H), 4,90 (s, 2H), 4,54-4,49 (m, 1H), 2,09-1,89 (m, 6H), 1,78-1,68 (m, 2H)
<Пример 38> Получение 1-(4,4-дифторциклогексил)-6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

24 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,08 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 20 мг 2-(хлорметил)-5-нитробензо[d]оксазола (0,09 ммоль), полученного в вышеописанном примере получения 21, и 0,05 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 41 мг указанного в заголовке соединения (0,09 ммоль) с 63% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,69 (s, 1H), 8,60 (d, J = 2,3 Гц, 1H), 8,32 (dd, J = 8,9, 2,3 Гц, 1H), 8,00 (d, J = 9,0 Гц, 1H), 7,95 (s, 1H), 4,90 (s, 2H), 4,55 (s, 1H), 2,10-1,90 (m, 6H), 1,79-1,71 (m, 2H)
<Пример 39> Получение 1-(4,4-дифторциклогексил)-6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она, полученного в вышеописанном примере получения 29 (0,14 ммоль), растворяли в 3 мл метанола и добавляли 33 мг 2-(хлорметил)-4-нитробензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 22, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 13 мг указанного в заголовке соединения (0,03 ммоль) с 21% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,54 (s, 1H), 7,98 (s, 1H), 7,35-7,16 (m, 3H), 4,93-4,81 (m, 1H), 4,18 (s, 2H), 2,12-1,96 (m, 5H), 1,95-1,83 (m, 3H)
<Пример 40> Получение 6-(((5-хлор-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 38 мг 5-хлор-2-(хлорметил)-6-нитробензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 23, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 70 мг указанного в заголовке соединения (0,14 ммоль) с 100% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,66 (s, 1H), 8,82 (s, 1H), 7,95 (s, 1H), 4,89 (s, 1H), 4,55-4,47 (m, 1H), 2,29-1,70 (m, 8H)
<Пример 41> Получение 1-(4,4-дифторциклогексил)-6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 28 мг 2-(хлорметил)-4-метилбензо[d]оксазола (0,15 ммоль), полученного в примере получения 18, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 39 мг указанного в заголовке соединения (0,09 ммоль) с 66% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,97 (s, 1H), 7,48 (d, J = 8,0 Гц, 1H), 7,27 (dd, J = 8,0, 7,6 Гц, 1H), 7,18 (d, J = 7,6 Гц, 1H), 4,83 (s, 2H), 4,67-4,54 (m, 1H), 2,49 (s, 3H), 2,18-1,96 (m, 5H), 1,95-1,77 (m, 3H)
<Пример 42> Получение 6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
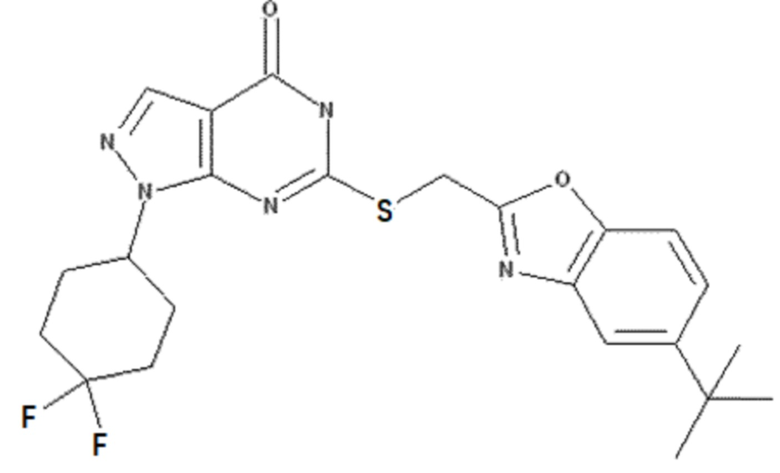
40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 34 мг 5-(трет-бутил)-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 19, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции полученный продукт концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 64 мг указанного в заголовке соединения (0,13 ммоль) с 97% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (s, 1H), 7,95 (s, 1H), 7,64 (d, J = 2,9 Гц, 1H), 7,59 (d, J = 8,6 Гц, 1H), 7,44 (dd, J = 8,6, 2,9 Гц, 1H), 4,79 (s, 2H), 4,55-4,50 (m, 1H), 2,09-1,86 (m, 6H), 1,76-1,71 (m, 2H), 1,32 (s, 9H)
<Пример 43> Получение 6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
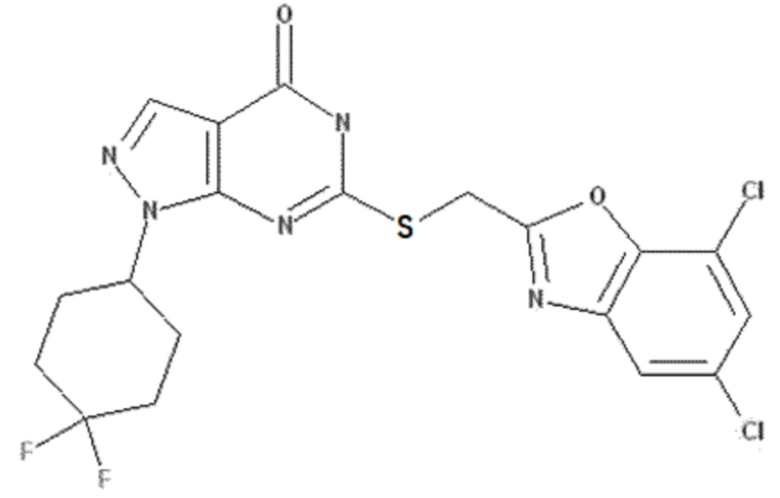
40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 36 мг 5,7-дихлор-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 24, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 17 мг указанного в заголовке соединения (0,03 ммоль) с 25% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,94 (s, 1H), 7,85 (d, J = 1,9 Гц, 1H), 7,69 (d, J = 1,9 Гц, 1H), 4,85 (s, 2H), 4,55-4,50 (m, 1H), 2,12-1,74 (m, 8H)
<Пример 44> Получение 1-(4,4-дифторциклогексил)-6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 30 мг 5,7-диметил-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 25, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 22 мг указанного в заголовке соединения (0,05 ммоль) с 36% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,95 (s, 1H), 7,27 (s, 1H), 7,02 (s, 1H), 4,79 (s, 2H), 4,59 (s, 1H), 2,38 (d, J = 19,4 Гц, 5H), 2,04 (q, J = 10,9, 9,9 Гц, 4H), 1,91 (d, J = 8,3 Гц, 2H), 1,79 (d, J = 11,3 Гц, 2H)
<Пример 45> Получение 1-(4,4-дифторциклогексил)-6-(((5-метоксибензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она
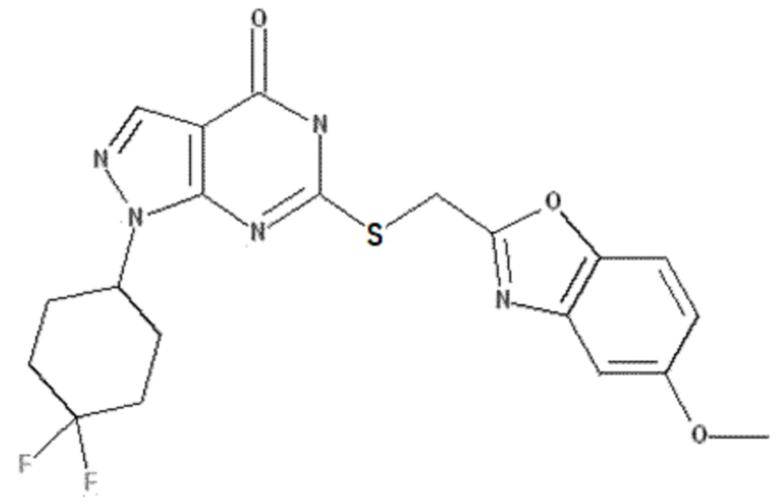
40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 30 мг 5-метокси-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 26, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 43 мг указанного в заголовке соединения (0,09 ммоль) с 70% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (s, 1H), 7,96 (s, 1H), 7,59 (d, J = 8,9 Гц, 1H), 7,23 (d, J = 2,5 Гц, 1H), 6,96 (dd, J = 8,9, 2,6 Гц, 1H), 4,80 (s, 2H), 4,63-4,48 (m, 1H), 3,78 (s, 3H), 2,14-1,87 (m, 6H), 1,81-1,74 (m, 2H)
<Пример 46> Получение 1-(4,4-дифторциклогексил)-6-(((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 30 мг 6-метокси-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в примере получения 27, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 23 мг указанного в заголовке соединения (0,05 ммоль) с 37% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,66 (s, 1H), 7,97 (s, 1H), 7,56 (d, J = 8,7 Гц, 1H), 7,34 (d, J = 2,4 Гц, 1H), 6,95 (dd, J = 8,7, 2,4 Гц, 1H), 4,79 (s, 2H), 4,60 (s, 1H), 3,80 (s, 3H), δ 2,16-1,88 (m, 6H), 1,86-1,73 (m, 2H)
<Пример 47> Получение 1-(4,4-дифторциклогексил)-6-(((5-(трифторметил)бензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(4,4-дифторциклогексил) -6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло [3,4-d] пиридин-4-она (0,14 ммоль), полученного в вышеописанном примере получения 29, растворяли в 3 мл метанола и добавляли 36 мг 5-(трифторметил)-2-(хлорметил)бензо[d]оксазола (0,15 ммоль), полученного в вышеописанном примере получения 28, и 0,08 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 53 мг указанного в заголовке соединения (0,11 ммоль) с 78% выходом.
Rf = 0,25 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,71 (s, 1H), 8,15 (s, 1H), 7,97 (d, J = 8,7 Гц, 1H), 7,90 (s, 1H), 7,77 (dd, J = 8,7, 1,8 Гц, 1H), 4,86 (s, 2H), 4,51 (s, 1H), 2,10-1,93 (m, 6H), 1,76-1,70 (m, 2H)
<Пример получения 30> Получение 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она
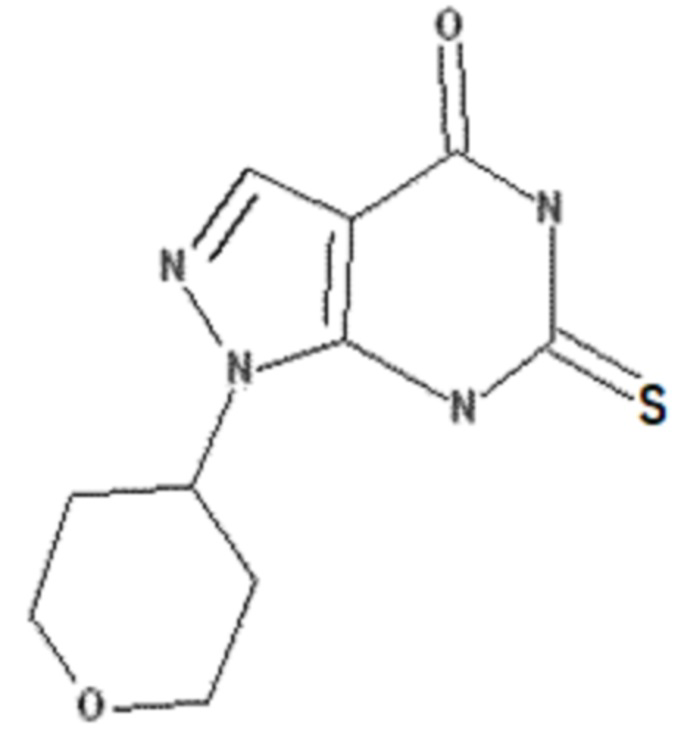
Этап 1: Получение этил 5-амино-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата

2,08 г этил (E)-2-циан-3-этоксиакрилата (12,3 ммоль) растворяли в 30 мл этанола и добавляли 2,25 г (тетрагидро-2H-пиран-4-ил)гидразина (14,76 ммоль) с последующим перемешиванием с обратным холодильником в течение 15 часов. После завершения реакции полученный продукт экстрагировали 50 мл этилацетата, промывали 50 мл раствора неорганической соли, сушили над безводным сульфатом натрия (Na2SO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (гексан:этилацетат = 1:1, об./об.), получая 2,2 г указанного в заголовке соединения (9,22 ммоль) с 75% выходом.
Rf = 0,50 (гексан:этилацетат = 1:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 7,66 (s, 1H), 5,04 (br s, 2H), 4,29 (q, J = 7,1 Гц, 2H), 4,19-4,10 (m, 2H), 4,08-3,94 (m, 1H), 3,60-3,45 (m, 2H), 2,40-2,20 (m, 2H), 1,92-1,80 (m, 2H), 1,36 (t, J = 7,1 Гц, 3H)
Этап 2: Получение этил-5-(3-бензоилтиоуреидо)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
2,20 г этил-5-амино-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата (9,22 ммоль), полученного на этапе 1, растворяли в 30 мл тетрагидрофурана и добавляли 1,48 мл бензоилизотиоцианата (11,06 ммоль) с последующим перемешиванием с обратным холодильником в течение 2 часов. После завершения реакции полученный продукт концентрировали при пониженном давлении и очищали гексаном, получая 3,54 г указанного в заголовке соединения (8,74 ммоль) с 95% выходом.
Rf = 0,60 (гексан:этилацетат = 1:1, об./об.)
1H ЯМР (500 МГц, CDCl3) δ 12,16 (br s, 1H), 9,53 (br s, 1H), 8,02 (s, 1H), 7,98 (d, J = 7,4 Гц, 2H), 7,71 (dd, J = 7,4 Гц, 7,4 Гц, 1H), 7,60 (dd, J = 7,4 Гц, 7,4 Гц, 2H), 4,44-4,34 (m, 1H), 4,28 (q, J = 7,1 Гц, 2H), 4,17-4,09 (m, 2H), 3,58-3,50 (m, 2H), 2,37-2,30 (m, 2H), 2,10-2,00 (m, 2H), 1,31 (t, J = 7,1 Гц, 3H)
Этап 3: Получение 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она
100 мг этил-5- 3-бензоилтиоуреидо)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата (0,25 ммоль), полученного на этапе 2, растворяли в 3 мл этанола и добавляли 0,75 мл 2 н. гидроксида натрия (1,50 ммоль) с последующим перемешиванием с обратным холодильником в течение 30 минут. После завершения реакции его нейтрализовали 1 н. соляной кислотой с осаждением твердого вещества, которое очищали водой и гексаном, получая 30 мг указанного в заголовке соединения (0,12 ммоль) с 48% выходом.
Rf=0,00 (гексан:этилацетат = 1:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 13,48 (br s, 1H), 12,17 (br s, 1H), 7,95 (s, 1H), 4,91-4,82 (m, 1H), 4,08-3,91 (m, 2H), 3,55-3,45 (m, 2H), 2,10-1,90 (m, 2H), 1,90-1,76 (m, 2H)
<Пример 48> Получение 6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 37 мг 2-(хлорметил)-6-нитробензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 20, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 24 мг указанного в заголовке соединения (0,06 ммоль) с 36% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,74 (d, J = 2,2 Гц, 1H), 8,28 (dd, J = 8,8, 2,2 Гц, 1H), 7,95 (s, 1H), 7,92 (s, 1H), 4,88 (s, 2H), 4,52-4,43 (m, 1H), 3,87-3,82 (m, 2H), 3,45-3,36 (m, 2H), 1,95-1,87 (m, 2H), 1,52-1,48 (m, 2H)
<Пример 49> Получение 6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 37 мг 2-(хлорметил)-5-нитробензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 21, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 41 мг указанного в заголовке соединения (0,09 ммоль) с 60% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,68 (s, 1H), 8,60 (d, J = 2,3 Гц, 1H), 8,31 (dd, J = 9,0, 2,3 Гц, 1H), 8,00 (d, J = 9,0 Гц, 1H), 7,94 (s, 1H), 4,87 (s, 2H), 4,53-4,46 (m, 1H), 3,89-3,82 (m, 2H), 3,42-3,36 (m, 2H), 1,96-1,88 (m, 2H), 1,54-1,50 (m, 2H)
<Пример 50> Получение 6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 37 мг 2-(хлорметил)-4-нитробензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 22, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 26 мг указанного в заголовке соединения (0,06 ммоль) с 39% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,97 (s, 1H), 7,29 (dd, J = 7,5, 2,2 Гц, 1H), 7,25-7,18 (m, 2H), 4,90-4,84 (m, 1H), 4,18 (s, 2H), 3,90-3,86 (m, 2H), 3,39-3,35 (m, 2H), 2,04-1,96 (m, 2H), 1,77-1,70 (m, 2H)
<Пример 51> Получение 6-(((5-хлор-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 43 мг 5-хлор-2-(хлорметил)-6-нитробензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 23, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой, простым эфиром и этилацетатом, получая 20 мг указанного в заголовке соединения (0,04 ммоль) с 27% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 8,70 (s, 1H), 8,21 (s, 1H), 7,96 (s, 1H), 4,87 (s, 2H), 4,47-4,40 (m, 1H), 3,89-3,81 (m, 2H), 3,40-3,37 (m, 2H), 1,93-1,86 (m, 2H), 1,53-1,48 (m, 2H)
<Пример 52> Получение 6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 32 мг 2-(хлорметил)-4-метилбензо[d]оксазола (0,16 ммоль), полученного в примере получения 18, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 14 мг указанного в заголовке соединения (0,03 ммоль) с 22% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,58 (br s, 1H), 7,95 (s, 1H), 7,49 (d, J = 8,1 Гц, 1H), 7,25 (dd, J = 8,1, 7,5 Гц, 1H), 7,16 (d, J = 7,5 Гц, 1H), 4,81 (s, 2H), 4,62-4,55 (m, 1H), 3,89 (dd, J = 11,0, 4,4 Гц, 2H), 3,41-3,36 (m, 2H), 2,49 (s, 3H), 2,04-1,91 (m, 2H), 1,65-1,61 (m, 2H)
<Пример 53> Получение 6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 39 мг 5-(трет-бутил)-2-(хлорметил) бензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 19, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции полученный продукт концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол = 100:1, об./об.), получая 23 мг указанного в заголовке соединения (0,05 ммоль) с 33% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 7,94 (s, 1H), 7,64 (d, J = 1,9 Гц, 1H), 7,59 (d, J = 8,6 Гц, 1H), 7,42 (dd, J = 8,7, 2,0 Гц, 1H), 4,77 (s, 2H), 4,55-4,49 (m, 1H), 3,87-3,84 (m, 2H), 3,39-3,35 (m, 2H), 1,99-1,90 (m, 2H), 1,56-1,50 (m, 2H), 1,31 (s, 9H)
<Пример 54> Получение 6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 41 мг 5,7-дихлор-2-(хлорметил)бензо[d]оксазола (0,16 ммоль), полученного в примере получения 24, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 41 мг указанного в заголовке соединения (0,09 ммоль) с 57% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,67 (br s, 1H), 7,95 (s, 1H), 7,86 (d, J = 1,9 Гц, 1H), 7,69 (d, J = 1,9 Гц, 1H), 4,85 (s, 2H), 4,54-4,45 (m, 1H), 3,88-3,84 (m, 2H), 3,39-3,34 (m, 2H), 1,98-1,90 (m, 2H), 1,58-1,54 (m, 2H)
<Пример 55> Получение 6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 34 мг 5,7-диметил-2-(хлорметил)бензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 25, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 22 мг указанного в заголовке соединения (0,05 ммоль) с 36% выходом.
Rf = 0,30 (дихлорметан:метанол = 20:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,62 (br s, 1H), 7,94 (s, 1H), 7,27 (d, J = 1,6 Гц, 1H), 7,00 (s, 1H), 4,78 (s, 2H), 4,61-4,55 (m, 1H), 3,88-3,84 (m, 2H), 3,41-3,35 (m, 2H), 2,41 (s, 3H), 2,35 (s, 3H), 2,00-1,90 (m, 2H), 1,62-1,58 (m, 2H)
<Пример 56> Получение 6-((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она

40 мг 1-(тетрагидро-2H-пиран-4-ил)-6-тиоксо-1,5,6,7-тетрагидро-4H-пиразоло[3,4-d]пиридин-4-она (0,15 ммоль), полученного в вышеописанном примере получения 30, растворяли в 3 мл метанола и добавляли 35 мг 6-метокси-2-(хлорметил)бензо[d]оксазола (0,16 ммоль), полученного в вышеописанном примере получения 27, и 0,09 мл 2 н. гидроксида натрия с последующим перемешиванием с обратным холодильником в течение 12 часов. После завершения реакции осажденное твердое вещество очищали водой и этилацетатом, получая 48 мг указанного в заголовке соединения (0,12 ммоль) с 74% выходом.
Rf = 0,20 (дихлорметан:метанол = 10:1, об./об.)
1H ЯМР (500 МГц, ДМСО-d6) δ 12,64 (s, 1H), 7,95 (s, 1H), 7,55 (d, J = 8,7 Гц, 1H), 7,33 (d, J = 2,4 Гц, 1H), 6,94 (dd, J = 8,7, 2,4 Гц, 1H), 4,76 (s, 2H), 4,64-4,53 (m, 1H), 3,94-3,86 (m, 2H), 3,79 (s, 3H), 3,46-3,38 (m, 2H), 2,03-1,90 (m, 2H), 1,62-1,55 (m, 2H)
Химические структурные формулы соединений, полученных в примерах 1-56, обобщены и представлены в следующей таблице.
Таблица 1
При
мер
Химическая структура


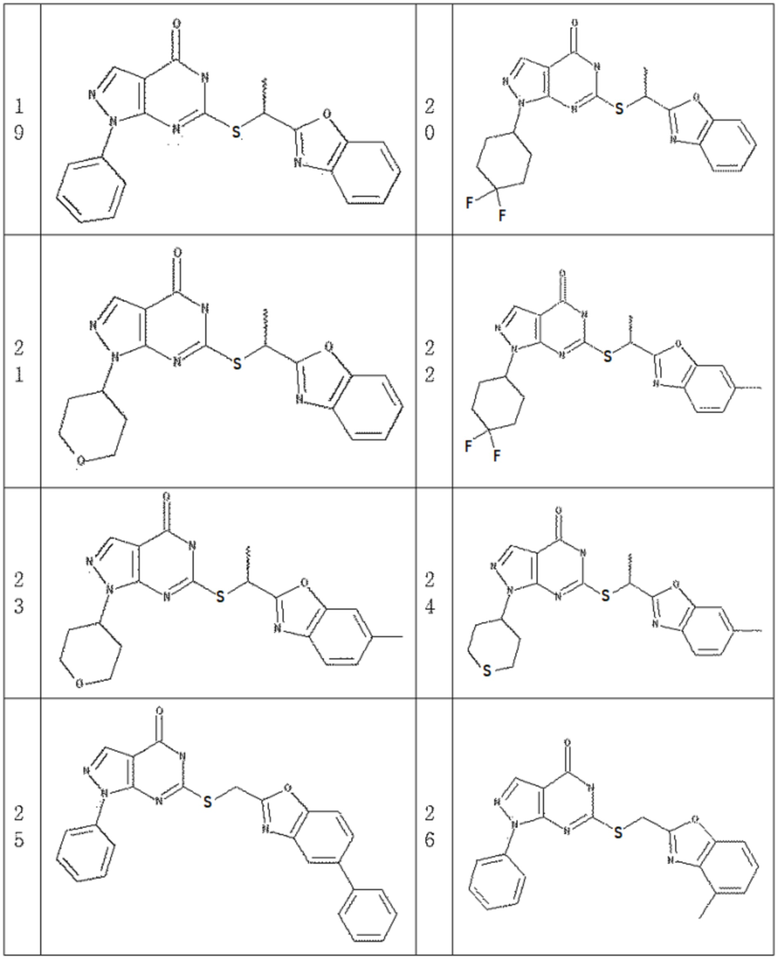



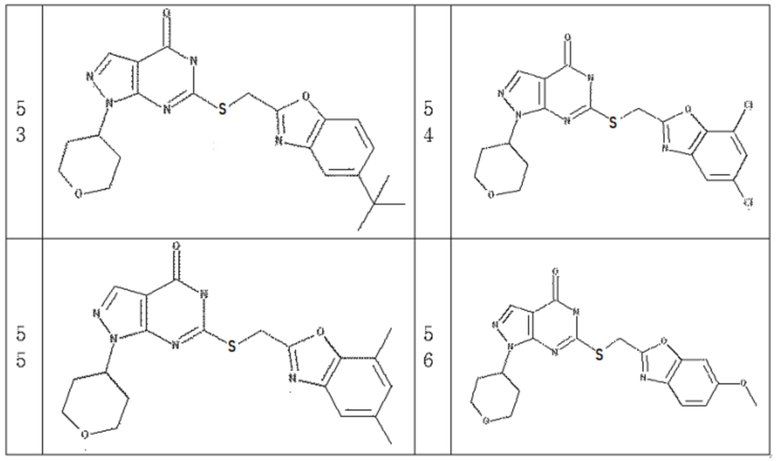
Ингибирующую активность типичных соединений в соответствии с настоящим изобретением в отношении фосфодиэстеразы 9A тестировали на основе анализа поляризации (набор для экспресс-скрининга IMAP-FP), предоставленный MDS (MDS Analytical Technologies, Сунивейл, Калифорния, США). В качестве буферного раствора готовили реакционный раствор (реакционный раствор, содержащий 0,01% Tween 20, предоставленный MDS, разбавляли в 5 раз, затем добавляли 5 мМ DTT) и раствор для обнаружения поляризации (раствор для связывания A и B, предоставленный MDS, смешивали в соотношении 3:1 и добавляли реагент, связывающий IMAP, в соотношении 1/600). Получали 100 мкМ субстрата (субстрат Fl-cGMP; MDS) и 3,6 мкг/мл фермента PDE9A (ab54113; abcam). 3,6 мкг/мл субстрата PDE9A и 100 мкМ разбавляли до 20 нг/мл (конечная концентрация реакции: 5 нг/мл) и 400 нМ (конечная концентрация реакции: 100 нМ), соответственно. Буферный раствор, используемый для всех процессов разбавления и приготовления, представлял собой 1× реакционный раствор с добавлением 1 мМ DTT, а раствор для обнаружения поляризации использовали для индукции поляризации на конечном этапе.
Полученные образцы распределяли в черные микропланшеты (384-луночные планшеты Multiwell, # 3573, Corning Life Sciences, Lowell, MA, USA) с использованием 16-канальной пипетки (multi 16-channel, Finnpipette, Thermo Scientific, Эссекс, Великобритания). Общий реакционный объем на лунку составлял 20 мкл. Вместе с тем, в качестве отрицательного контроля использовали 10 мкл 2% ДМСО, 5 мкл раствора субстрата и 5 мкл реакционного раствора, а в качестве положительного контроля использовали 10 мкл 2% ДМСО, 5 мкл раствора субстрата и 5 мкл раствора PDE9A. В качестве экспериментальной группы использовали 10 мкл соединения, полученного в примере, 5 мкл раствора субстрата и 5 мкл раствора PDE9A. После предварительной обработки соединения и фермента в течение приблизительно 10 минут перед реакцией фермента и субстрата добавляли 5 мкл цГМФ для индукции ферментативной реакции. Во время реакции каждое соединение, фермент и субстрат занимали 50%, 25% и 25% от общего объема, соответственно, и их получали в высоких концентрациях (2-, 4- и 4-кратных, соответственно) непосредственно перед добавлением. После ферментативной реакции смесь слегка встряхивали в течение 1 минуты и индуцировали ферментативную реакцию при комнатной температуре в течение 1 часа. Затем для индукции поляризации добавляли 60 мкл заранее приготовленного раствора для обнаружения поляризации.
Наночастицы, состоящие их трехвалентных ионов металла, смешивали в растворе для обнаружения флуоресценции, который объединяли с фосфорной кислотой при воздействии ферментативной реакции для увеличения молекулярной массы и индукции поляризации. После инкубирования в течение 2 часов при комнатной температуре измеряли значение поляризации (поляризация флуоресценции, FP) с помощью многометочного счетчика (Envision, PerkinElmer, Турку, Финляндия) (длина волны излучения: P-535 нм, S-535 нм, длина волны возбуждения: 480 нм), выражая результат в виде значения IC50, которое представляет собой концентрацию соединения, ингибирующего PDE9A in vitro на 50% (таблица 2).
Таблица 2
В таблице 2 значения, выраженные в %, представляют собой средний % ингибирования PDE9A при концентрации соединения 10 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788148C2 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА, ЗАМЕЩЕННЫЕ ВТОРИЧНЫМИ АМИНАМИ, ВКЛЮЧАЮЩИМИ В СЕБЯ ТЕТРАЗОЛ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2003 |
|
RU2283312C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2792163C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2756505C1 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
Группа изобретений относится к области фармацевтической химии и включает соединение формулы 1 или его фармацевтически приемлемую соль и фармацевтическую композицию на их основе. В формуле 1 A представляет собой фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, 3-6-членный циклоалкил, незамещенный или замещенный одним или двумя атомами галогена, или 3-6-членный гетероциклоалкил, содержащий азот, кислород или серу в качестве гетероатома, каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, или линейную или разветвленную C1-5 алкоксигруппу, незамещенную или замещенную одним или двумя атомами галогена. Технический результат – соединение формулы 1, обладающее ингибирующей активностью в отношении PDE9A. 2 н. и 6 з.п. ф-лы, 2 табл., 56 пр.

1. Соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль,
[Химическая формула 1]
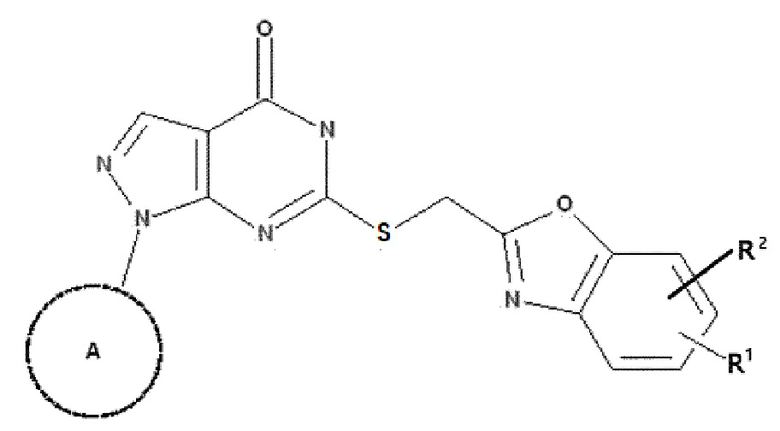
при этом в химической формуле 1
A представляет собой фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, 3-6-членный циклоалкил, незамещенный или замещенный одним или двумя атомами галогена, или 3-6-членный гетероциклоалкил, содержащий азот, кислород или серу в качестве гетероатома, и
каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, или линейную или разветвленную C1-5 алкоксигруппу, незамещенную или замещенную одним или двумя атомами галогена,
при условии, что, когда А представляет собой фенил, любой из R1 и R2 не является водородом.
2. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что A представляет собой фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, 3-6-членный циклоалкил, незамещенный или замещенный одним или двумя атомами галогена, или 3-6-членный гетероциклоалкил, содержащий азот, кислород или серу в качестве гетероатома, и
каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, фенил, линейный или разветвленный C1-5 алкил, незамещенный или замещенный одним или двумя атомами галогена, или линейную или разветвленную C1-5 алкоксигруппу, незамещенную или замещенную одним или двумя атомами галогена.
3. Соединение или его фармацевтически приемлемая соль по п. 2, отличающееся тем, что
A представляет собой фенил,  или
или  и
и
каждый из R1 и R2 независимо представляет собой -H, атом галогена, -NO2, -CF3, -CH3, -C(CH3)3 или фенил.
4. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что указанное соединение представляет собой
6-((бензо[d]оксазол-2-илметил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-фторбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-хлорбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-бромбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((бензо[d]оксазол-2-илметил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((бензо[d]оксазол-2-илметил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((бензо[d]оксазол-2-илметил)тио)-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((бензо[d]оксазол-2-илметил)тио)-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((бензо[d]оксазол-2-илметил)тио)-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-циклогексил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-циклопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метилбензо[d]оксазол-2-ил)метил)тио)-1-изопропил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((1-(бензо[d]оксазол-2-ил)этил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((1-бензо[d]оксазол-2-ил)этил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((1-бензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-((1-(6-метилбензо[d]оксазол-2-ил)этил)тио)-1-(тетрагидро-2H-тиопиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-фенил-6-(((5-фенилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразол[3,4-d]пиримидин-4-он,
6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-нитро-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-метоксибензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1-фенил-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-фенил-6-(((5-(трифторметил)бензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-хлор-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-(4,4-дифторциклогексил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((5-метоксибензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
1-(4,4-дифторциклогексил)-6-(((5-(трифторметил)бензо[d]оксазол-2-ил)метил)тио)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((4-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-хлор-6-нитробензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((4-метилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5-(трет-бутил)бензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5,7-дихлорбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он,
6-(((5,7-диметилбензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он или
6-(((6-метоксибензо[d]оксазол-2-ил)метил)тио)-1-(тетрагидро-2H-пиран-4-ил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-он.
5. Фармацевтическая композиция для лечения или предотвращения заболевания, связанного с фосфодиэстеразой 9A, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-4 в качестве активного ингредиента и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п. 5, отличающаяся тем, что указанное заболевание представляет собой неврологическое заболевание или психическое заболевание.
7. Фармацевтическая композиция по п. 6, отличающаяся тем, что указанное неврологическое или психическое расстройство представляет собой болезнь Альцгеймера, болезнь Хантингтона, деменцию с тельцами Леви или синдром Пика.
8. Фармацевтическая композиция по п. 5, отличающаяся тем, что указанное заболевание представляет собой сердечную недостаточность, в частности сердечную недостаточность с сохранением сердечного выброса, или серповидно-клеточную анемию.
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| База данных REGISTRY [онлайн], RN 2183345-16-4, 02.03.2018, найдено в STN | |||
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| VALENCIA A.et al., Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington's disease | |||
| Human molecular | |||

