Настоящее изобретение относится к новым фенил-2-гидрокси-ацетиламино-2-метил-фениловым соединениям, к фармацевтическим композициям, включающим соединения, к способам применения соединений для лечения физиологических расстройств, а также к промежуточным соединениям и способам, которые могут использоваться для синтеза соединений.
Настоящее изобретение относится к области лечения рака и других заболеваний и нарушений, связанных с протеинкиназа R (PKR)-подобной протеинкиназой эндоплазматического ретикулума (PERK). PERK, киназа eIF2, участвующая в реакции несвернутых белков (UPR), регулирует синтез белка, помогает клеткам смягчать влияние стресса эндоплазматического ретикулума и участвует в опухолеобразование и выживании раковых клеток.
Опухолевые клетки процветают в агрессивной микроокружающей среде, вызванной главным образом ограничением питательных веществ и кислорода, высокой метаболической потребностью и окислительным стрессом. Известно, что эти стрессы нарушают способность эндоплазматического ретикулума (ER) к сворачиванию белков, вызывая клеточный ответ на ремедиацию, известный как реакция несвернутых белков (UPR). Стресс ER способствует увеличению онкогенного потенциала раковых клеток, метастаз опухоли, устойчивости опухоли к лекарственным средствам и способности раковых клеток избегать эффективных иммунных реакций.
Существует три основных ER трансмембранных сенсора UPR: 1) инозитол-требующий фермент (IRE1α/IRE1β, кодируемый ERN1 и ERN2, соответственно); 2) PKR-подобная ER-киназа (PERK, также известная как PEK, кодируемая EIF2AK3); и 3) активирующий фактор транскрипции 6α (кодируемый ATF6). Каждый из этих трех сенсоров регулируется аналогичным образом посредством связывания люминального белка-шаперона ER GRP78 или BiP (кодируемого HSPA5). Когда потребности ER в сворачивании белка превышают емкость, сниженное связывание BiP приводит к активации этих белков-сенсоров ER, что приводит к индукции скоординированных сигнальных путей для увеличения способности ER к сворачиванию и ослаблению основного стресса. Эффективные ответы приводят к адаптации и выживанию клеток, а непоправимый стресс ER вызывает гибель клеток и апоптоз.
PERK является трансмембранной серин/треонин киназой типа I и членом семейства киназ, которые фосфорилируют эукариотический фактор инициации трансляции 2α (eIF2-α) и регулируют инициацию трансляции. Другие члены семьи включают HRI (EIF2AK1), PKR (EIF2AK2) и GCN2 (EIF2AK4). Каждая киназа eIF2 реагирует на различные сигналы клеточного стресса, чтобы регулировать общую трансляцию и ген-специфичный контроль трансляции. Фосфорилирование eIF2 приводит к снижению инициации общей трансляции из-за снижения активности фактора обмена eIF2B, уменьшая количество белка, поступающего в ER (и, следовательно, нагрузку сворачивания белка) и потребности в трансляции ATP. Фосфорилирование eIF2 также увеличивает трансляцию некоторых мРНК ген-специфическим образом, включая транскрипционный фактор ATF4. Транскрипционные мишени ATF4 включают многочисленные гены, участвующие в адаптации и выживании клеток, в том числе несколько генов, участвующих в фолдинге белка, поглощении питательных веществ, метаболизме аминокислот, окислительно-восстановительном гомеостазе и аутофагии (4). Предполагается, что селективное ингибирование ветви PERK UPR сильно повлияет на рост и выживание опухолевых клеток. Таким образом, считается, что соединения, которые ингибируют PERK, являются полезными при лечении рака.
При современном состоянии медицинского лечения у больных раком поджелудочной железы часто бывает плохой прогноз, даже если заболевание выявляется на ранних стадиях. Таким образом, сохраняется значительная потребность в новых и эффективных способах лечения рака поджелудочной железы. Соединения по настоящему изобретению являются ингибиторами PERK и, как полагают, являются полезными при лечении рака, в частности, рака поджелудочной железы.
В WO 2015/136463 раскрыты некоторые производные пирролидинона, которые обладают ингибирующей активностью в отношении PERK, и, кроме того, раскрыты соединения, полезные для лечения рака и заболеваний, связанных с активированной реакцией несвернутых белков, включая рак поджелудочной железы.
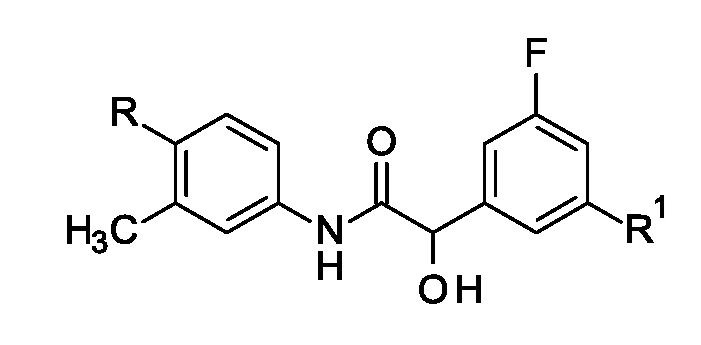
Соответственно, настоящее изобретение относится к соединению формулы I:
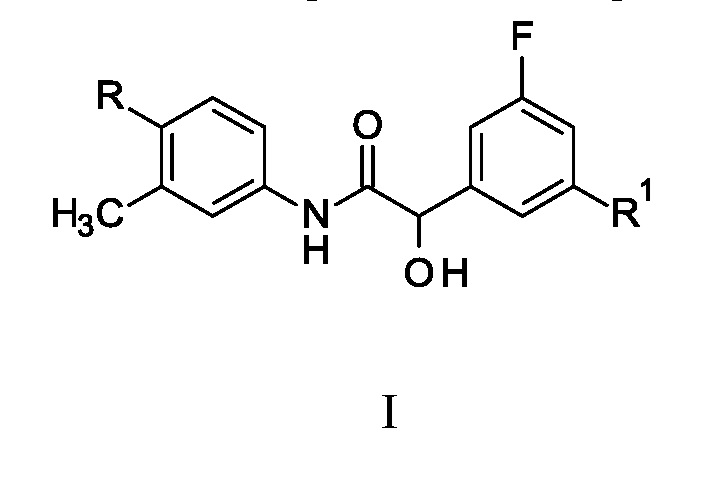
где
R выбран из группы, состоящей из
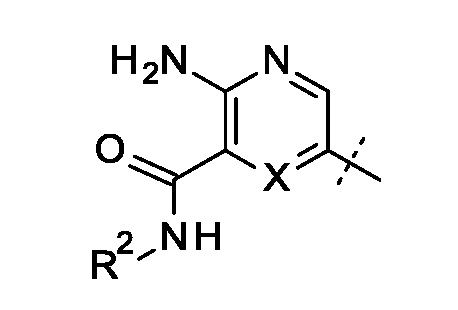 и
и 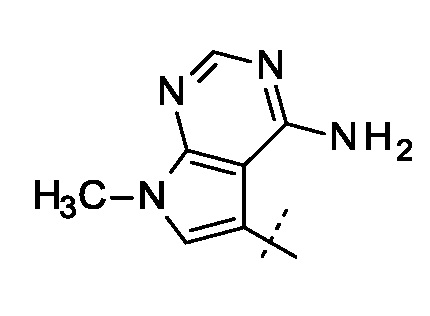 ;
;
X представляет собой CH или N;
R1 представляет собой водород или фтор; и
R2 представляет собой С1-С3 алкил;
или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению формулы Ia:
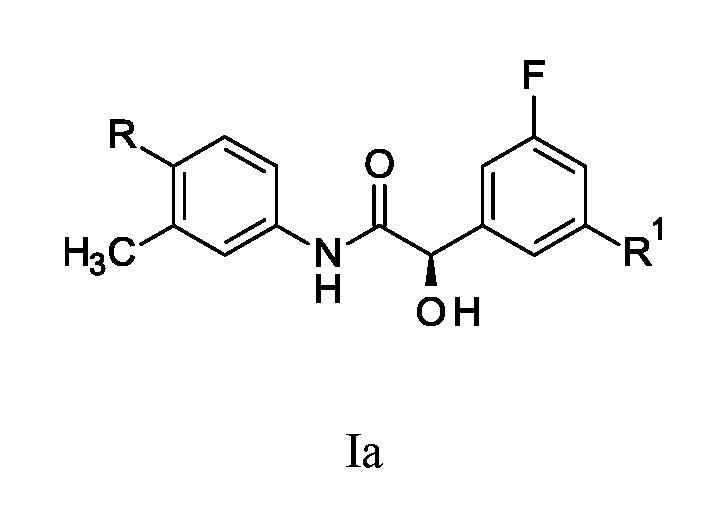
где
R выбран из группы, состоящей из
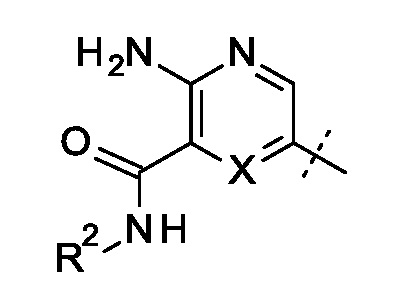 и
и 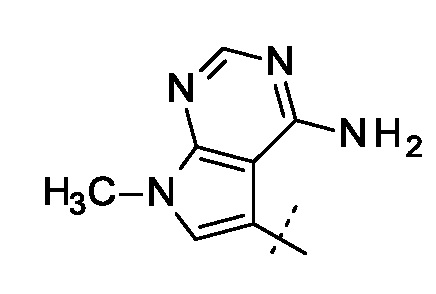 ;
;
X представляет собой CH или N;
R1 представляет собой водород или фтор; и
R2 представляет собой С1-С3 алкил;
или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению формулы I или Ia:
где R представляет собой
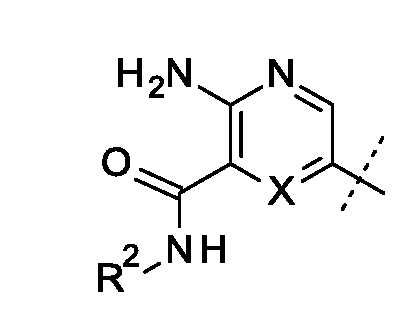 .
.
Кроме того, настоящее изобретение относится к соединению формулы I или Ia: где X представляет собой CH или N; R1 представляет собой водород или фтор; и R2 представляет собой метил или изопропил; или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению формулы I или Ia: где R представляет собой
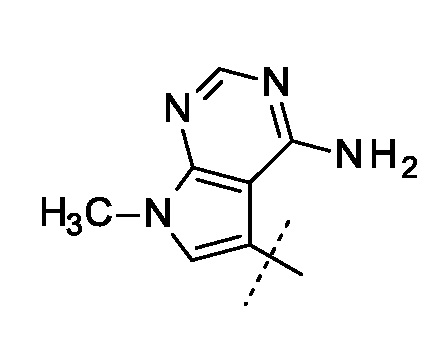
или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению 3-амино-6-[4-[[(2R)-2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамид, которое может быть представлено формулой

или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению 2-амино-5-[4-[[(2R)-2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамид, которое может быть представлено формулой

или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению (2R)-N-[4-(4-амино-7-метил-пирроло[2,3-d]пиримидин-5-ил)-3-метил-фенил]-2-(3-фторфенил)-2-гидрокси-ацетамид, которое может быть представлено формулой
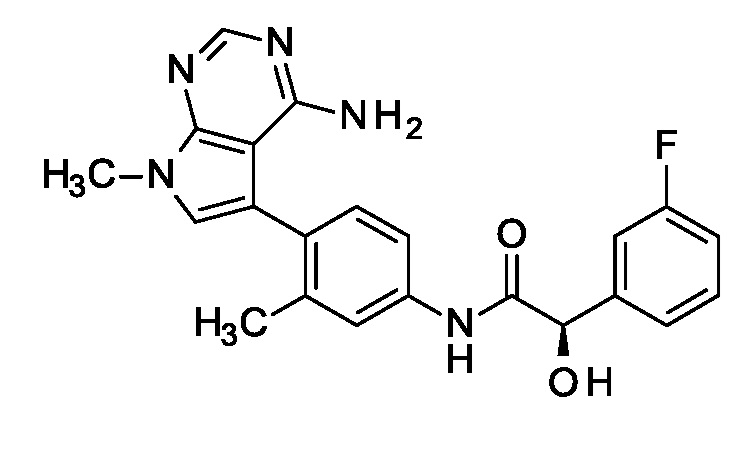
или его фармацевтически приемлемой соли.
Настоящее изобретение относится к способу лечения рака у пациента, нуждающегося в таком лечении, включающему введение пациенту эффективного количества соединения формулы I или Ia, или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу ингибирования активности PERK, приводящей к противоопухолевой активности у пациента, нуждающегося в таком лечении, включающему введение пациенту эффективного количества соединения формулы I или Ia, или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к способу лечения рака поджелудочной железы у пациента, нуждающегося в таком лечении, включающему введение пациенту эффективного количества соединения формулы I или Ia, или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к соединению формулы I или Ia, или его фармацевтически приемлемой соли для применения в терапии, в частности, для лечения рака поджелудочной железы. Кроме того, настоящее изобретение относится к соединению формулы I или Ia или его фармацевтически приемлемой соли для применения при лечении рака поджелудочной железы. В дополнительном варианте осуществления настоящее изобретение относится к применению соединения по изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения рака поджелудочной железы.
Изобретение, кроме того, относится к фармацевтической композиции, содержащей соединение по изобретению или его фармацевтически приемлемую соль с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами. В дополнительном варианте осуществления композиция дополнительно содержит одно или несколько других терапевтических средств. Изобретение также относится к способу получения фармацевтической композиции, включающему смешивание соединения формулы I или Ia или его фармацевтически приемлемой соли с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Настоящее изобретение также охватывает новые промежуточные соединения и способы синтеза соединений формулы I и Ia.
Используемые в настоящем описании термины «лечение» или «лечить» включают ограничение, замедление, остановку или обращение прогрессирования или серьезности существующего симптома или расстройства.
Используемый в настоящем описании термин «эффективное количество» относится к количеству или дозе соединения по изобретению или его фармацевтически приемлемой соли, которое при однократном или многократном введении дозы пациенту обеспечивает желаемый эффект у пациента, подвергаемого диагностике или лечению.
Эффективное количество может быть легко определено специалистом в данной области техники с использованием известных методик и путем наблюдения результатов, полученных при аналогичных обстоятельствах. При определении эффективного количества для пациента учитывается ряд факторов, включая, но не ограничиваясь: род пациента; его размер, возраст и общее состояние здоровья; конкретное заболевание или расстройство; степень или поражение или тяжесть заболевания или расстройства; ответ отдельного пациента; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого препарата; выбранный режим дозирования; использование сопутствующих лекарственных средств; и другие соответствующие обстоятельства.
Соединения по настоящему изобретению обычно эффективны в широком диапазоне доз. Например, дозировки в день обычно находятся в диапазоне от около 0,1 до около 50 мг/кг массы тела. В некоторых случаях уровни дозировки ниже нижнего предела вышеупомянутого диапазона могут быть более чем адекватными, в то время как в других случаях могут использоваться еще большие дозы с приемлемыми побочными эффектами, и, следовательно, указанный выше диапазон дозировок никоим образом не предназначен для ограничения объема изобретения. Понятно, что количество фактически вводимого соединения будет определяться врачом с учетом соответствующих обстоятельств, включая состояние, подлежащее лечению, выбранный способ введения, фактическое вводимое соединение или соединения, возраст, массу и реакцию отдельного пациента, а также тяжесть симптомов пациента.
Соединения по настоящему изобретению предпочтительно получают в виде фармацевтических композиций, вводимых любым путем, который делает соединение биодоступным, включая пероральный, внутривенный и трансдермальный пути введения. Наиболее предпочтительно такие композиции предназначены для перорального введения. Такие фармацевтические композиции и способы их получения хорошо известны в данной области (см., например, Remington: The Science and Practice of Pharmacy, L.V. Allen, Editor, 22nd Edition, Pharmaceutical Press, 2012).
Понятно, что соединения формулы I могут существовать в виде стереоизомеров. Варианты осуществления настоящего изобретения включают все энантиомеры, диастереомеры и их смеси. Конкретный энантиомер соединения формулы I представлен соединением формулы Ia
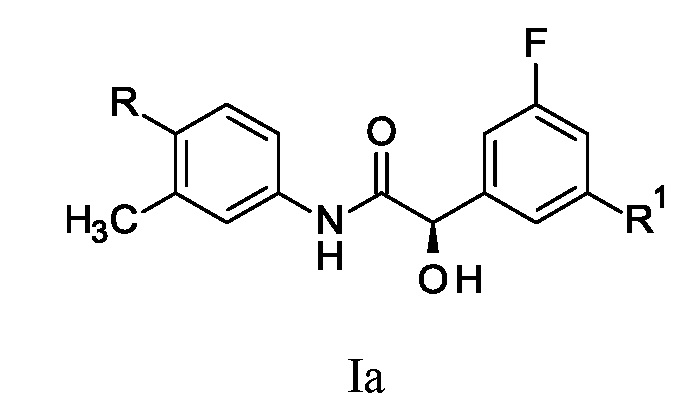
где R и R1 являются такими, как определено ранее.
Специалисту в данной области также понятно, что обозначения Кана-Ингольда-Прелога (R) или (S) для всех хиральных центров будут варьироваться в зависимости от схемы замещения конкретного соединения. Отдельные энантиомеры или диастереомеры могут быть получены, исходя из хиральных реагентов или с помощью стереоселективных или стереоспецифических методов синтеза. Альтернативно, отдельные энантиомеры или диастереомеры могут быть выделены из смесей стандартными методами хиральной хроматографии или кристаллизации в любой удобной точке синтеза соединений по изобретению. См., например, J. Jacques, et al., “Enantiomers, Racemates, and Resolutions”, John Wiley and Sons, Inc., 1981, and E.L. Eliel and S.H. Wilen,” Stereochemistry of Organic Compounds”, Wiley-Interscience, 1994. Отдельные энантиомеры соединений по изобретению являются предпочтительным вариантом осуществления изобретения.
Фармацевтически приемлемая соль соединений по изобретению может быть образована, например, путем взаимдействия подходящего свободного основания соединения по изобретению и подходящей фармацевтически приемлемой кислоты в подходящем растворителе в стандартных условиях, хорошо известных в данной области техники. Образование таких солей хорошо известно и оценено в данной области. См., например, Gould, P.L., “Salt selection for basic drugs,” International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., et al. “Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities,” Organic Process Research and Development, 4: 427-435 (2000); and Berge, S.M., et al., “Pharmaceutical Salts,” Journal of Pharmaceutical Sciences, 66: 1-19, (1977).
In vitro ингибирование ферментативной активности PERK (выделенный)
Получали рекомбинантный каталитический домен EIF2AK2 (PKR) человека (аминокислоты 252-551), каталитический домен EIF2AK3 (PERK) (аминокислоты 536-1116), субстрат GFP-eIF2α и меченое тербием фосфо-eIF2α антитело (Invitrogen, Carlsbad, CA). Каталитический домен HIS-SUMO-GCN2 (аминокислоты 584-1019) экспрессировали и очищали из E. coli. Осуществляли анализы киназы TR-FRET в отсутствие или в присутствии ингибиторов в реакционном буфере, состоящем из 50 мМ HEPES, pH 7,5, 10 мМ MgCl2, 1,0 мМ EGTA и 0,01% Brij-35 и 100-200 нМ GFP-eIF2α субстрата. Анализы PKR содержат 14 нг/мл фермента и 2,5 мкМ ATP (Km, приблизительно ~2,5 мкМ), анализы PERK содержат 62,5 нг/мл фермента и 1,5 мкМ ATP (Km, приблизительно ~1,5 мкМ), и анализы GCN2 содержат 3 нМ фермента и 90 мкМ ATP (Km, приблизительно ~200 мкМ). Добавляли исследуемое соединение, инициировали реакцию путем добавления фермента и инкубировали при комнатной температуре в течение 45 минут. Останавливали реакцию добавлением EDTA до конечной концентрации 10 мМ, добавляли меченное тербием фосфо-eIF2α антитело в конечной концентрации 2 нМ и инкубировали в течение 90 минут. Мониторинг получаемой флуоресценции осуществляли с помощью ридера EnVison® Multilabel reader (PerkinElmer, Waltham, MA). Определяли отношения TR-FRET и полученные значения IC50, используя 4-параметрическое нелинейное логистическое уравнение, как показано(A+((B-A)/(1+((C/x)∧D)))), где Y=% специфическое ингибирование, A=нижняя часть кривой, B=верхняя часть кривой, C=абсолютная IC50 (концентрация, вызывающая 50% ингибирования) и D=угловой коэффициент Хилла.
Соединения примеров 1, 5 и 9 тестировали, по существу, как описано выше, и показали значения IC50, показанные в таблице 1. Эти данные демонстрируют, что соединения примеров 1, 5 и 9 ингибируют активность выделенного фермента PERK in vitro.
Таблица 1
(N=3)
(N=2)
(N=1)
(Ν=3)
(N=4)
(N=1)
(N=4)
(N=4)
In vitro ингибирование активности фермента PERK (целая клетка)
Высевали клетки GripTite™ 293 (Invitrogen, Carlsbad, CA), экспрессирующие GFP-eIF2α, при 10000 клеток на лунку в 384-луночные планшеты и оставляли на ночь для прикрепления. Клетки предварительно обрабатывали исследуемыми соединениями в течение 1 часа. Добавляли туникамицин (1 мкМ), чтобы вызвать активность PERK, и планшеты инкубировали при 37°C в течение 2 часов. Удаляли культуральную среду и лизировали клетки в буфере, состоящем из 20 мМ Tris-HCl, рН 7,5, 150 мМ NaCl, 5 мМ EDTA, 1% NP-40, 5 мМ NaF, ингибиторов протеаз (Sigma, St. Louis, MO), ингибиторов фосфатазы (Sigma, St. Louis, MO) и 2 нМ тербий-меченого антитела против фосфо-eIF2 (Invitrogen, Carlsbad, CA). Инкубировали лизаты клеток в течение 2 часов в темноте при комнатной температуре и контролировали флуоресценцию в ридере EnVison® Multilabel reader (PerkinElmer, Waltham, MA). Определяли соотношения TR-FRET и полученные значения IC50 по соответствующим кривым ингибирования, используя неиндуцированные (100% ингибирование) и индуцированные (0% ингибирование) лунки в качестве контролей.
Соединения примеров 1, 5 и 9 тестировали по существу так же, как описано выше, и показали значения IC50, показанные в таблице 2. Эти данные демонстрируют, что соединения примеров 1, 5 и 9 ингибируют активность фермента PERK целой клетки in vitro.
Таблица 2
(N=9)
(N=14)
(N=12)
In vivo Ингибирование рака поджелудочной железы (модель ксенотрансплантата у мышей)
Имплантировали самкам бестимусных мышей (Harlan Laboratories) подкожно 5×106 клеток BxPC-3 с матригелем в правый бок, и контролировали рост опухоли с помощью калипера. Начинали введение соединения, когда опухоли достигали ~250 мм3, и введение мышам осуществляли два раза в день перорально с помощью желудочного зонда (8 животных на группу) в течение 28 дней. Соединения получают в 10% аравийской камеди, содержащей 0,05% антивспенивателя или 20% каптизола в 25 мМ буфере NaPO4, рН 2, для 30 соединений примеров 5 и 9, соответственно. Контрольным животным вводили только носитель аравийскую камедь. Оценивали объемы опухоли, используя формулу l×w2×(π/6), где l представляет собой больший измеренный диаметр, а w представляет собой меньший перпендикулярный диаметр. Рассчитывали процент дельта T/C, используя формулу 100×[(T-T0)/(C-C0)], и процент регрессии, используя формулу 100×[(T-T0)/T0], где T и C представляют собой средние объемы опухоли в обработанной и контрольной группах, соответственно. T0 и C0 представляют собой соответствующие исходные средние объемы опухоли. Преобразованиее процент дельта Т/С в процент дельта ингибирования роста опухоли (TGI), используя уравнение, 100 - процент дельта Т/С. Для статистического анализа преобразование данных об объеме опухоли в шкалу log10 для выравнивания дисперсии по времени и группам лечения. Анализ данных объема log10 с помощью двухстороннего дисперсионного анализа повторных измерений (корреляционная модель пространственной статистической мощности) по времени и лечению с использованием смешанных процедур в программном пакете SAS (версия 9.3). Сравнение группы, получающей лечения, с контрольной группой в каждый момент времени.
Соединения Примеров 5 и 9 были протестированы, по существу, как описано выше, и показали значения ингибирования роста опухоли, показанные в Таблицах 3 и 4, соответственно. Эти данные демонстрируют, что соединения примеров 5 и 9 ингибируют рост опухоли поджелудочной железы in vivo.
Таблица 3
30 мг/кг п/о
2 р/сут
а) Стандартная ошибка среднего геометрического объема опухоли
b) Рассчитывали с использованием 100×[(T-T0)/(C-C0)], где T и C представляют собой средние объемы опухолей в получающей лечение и контрольной группах, соответственно, T0 и C0 представляют собой соответствующие исходные средние объемы опухолей.
c) TCI представляет собой ингибирование роста опухоли, рассчитанное с использованием 100 - %T/C
d) День рандомизации и начало лечения
*Значимый, p < 0,05
Таблица 4
30 мг/кг п/о
2 р/сут
а) Стандартная ошибка среднего геометрического объема опухоли
b) Рассчитывали с использованием 100×[(T-T0)/(C-C0)], где T и C представляют собой средние объемы опухолей в получающей лечение и контрольной группах, соответственно, T0 и C0 представляют собой соответствующие исходные средние объемы опухолей.
c) TCI представляет собой ингибирование роста опухоли, рассчитанное с использованием 100 - %T/C
d) День рандомизации и начало лечения
*Значимый, p < 0,05
Соединения по настоящему изобретению или их соли могут быть получены различными способами, известными специалисту в данной области, некоторые из которых проиллюстрированы на схемах, препаратах и примерах ниже. Специалист в данной области признает, что конкретные стадии синтеза для каждого из описанных способов могут комбинироваться различными способами или в сочетании со стадиями из различных схем для получения соединений по изобретению или их солей. Продукты каждой стадии в схемах ниже могут быть извлечены обычными способами, хорошо известными в данной области, включая экстракцию, выпаривание, осаждение, хроматографию, фильтрацию, измельчение и кристаллизацию. На схемах ниже все заместители, если не указано иное, являются такими, как определено ранее. Реагенты и исходные материалы легко доступны для специалиста в данной области. Без ограничения объема изобретения для дальнейшей иллюстрации изобретения приводятся следующие схемы, препараты и примеры. Кроме того, специалист в данной области понимает, что соединения формулы Ia могут быть получены с использованием исходного вещества с соответствующей стереохимической конфигурацией, которое может быть получено специалистом в данной области. Например, на приведенных ниже схемах используются исходные вещества с конфигурацией, по существу, соответствующей формуле Ia.
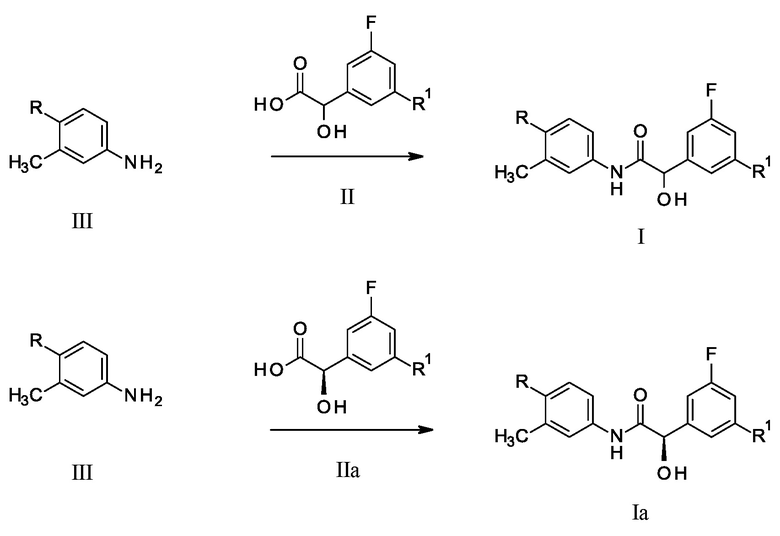
Обычно соединение формулы I может быть получено из соединения формулы III (схема 1). Более конкретно, соединение формулы III взаимодействует с соединением формулы II и подходящим реагентом для реакций сочетания, таким как HATU (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат) в присутствии подходящего аминного основания, такого как N, N-диизопропилэтиламин или триметиламина. Соединение формулы I может быть разделено на его изомеры с помощью хиральной хроматографии.
Соответственно, соединение формулы Ia может быть получено из соединения формулы IIa. Более конкретно, соединение формулы III взаимодействует с соединением формулы IIa и подходящим реагентом для реакций сочетания, таким как HATU (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат) в присутствии подходящего аминного основания, такого как N, N-диизопропилэтиламин или триметиламина. Соединение формулы IIa может быть получено из соединения формулы II с липолитическим ферментом, таким как Lipase PS Amano SD. Дополнительную информацию относительно этого метода оптического разрешения можно найти у Mendiola, J. et al, Org. Process Res. Dev. 2012, 16, 1312−1316.
Схема 1
Обычно соединение формулы III может быть получено из соединения формулы IV. Соединение формулы III может быть получено путем обработки соединения формулы R-Br 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилином в присутствии основания, такого как K2CO3, и палладиевого катализатора, такого как Pd(dppf)2Cl2.
Схема 2
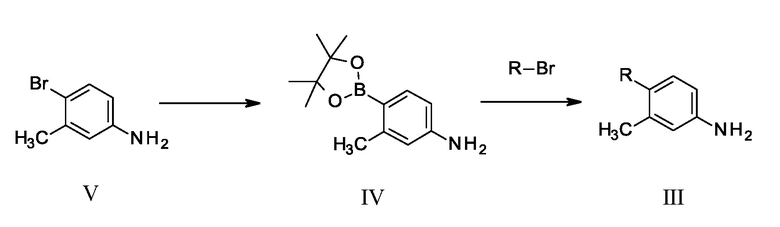
R-Br представляет собой 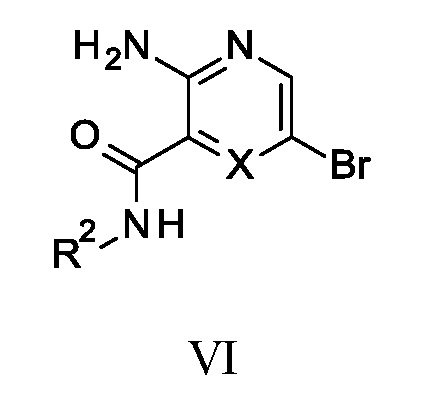 или
или 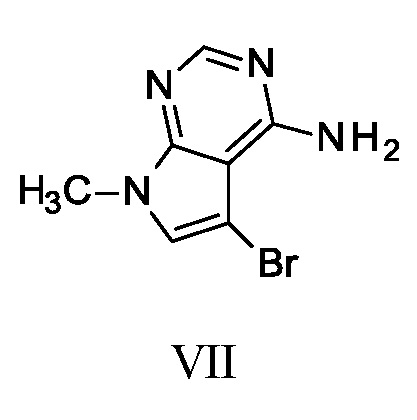
Соединение формулы R-Br, представленное соединением формулы VI или VII, может быть получено способами, известными в области химии, а также способами, описанными в приведенных ниже Получениях и Примерах.
Получение 1
Синтез 4-хлор-7-метил-пирроло[2,3-d]пиримидин.
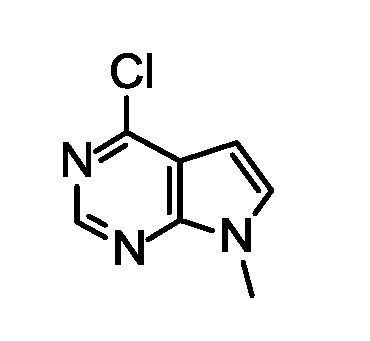
Добавляли Cs2CO3 (845 г, 2,60 моль) при 15°C к раствору 4-хлор-7H-пирроло[2,3-d]пиримидина (200 г, 1,29 моль) в N-метил-2-пирролидоне (1,20 л). Нагревали до 23°C, добавляли MeI (202 г, 1,43 моль) по каплям в течение 30 минут, и перемешивали в течение 4 ч. По истечении этого времени выливали на ледяную воду (2,00 л) и перемешивали в течение 30 мин. Фильтровали, затем суспендировали вещество в H2O (1,00 л). Фильтровали и сушили с получением указаннго в заголовке соединения (180 г, 81%). ES/MS m/z (35Cl) 168,0 (M+H).
Получение 2
Синтез 5-бром-4-хлор-7-метил-пирроло[2,3-d]пиримидина.
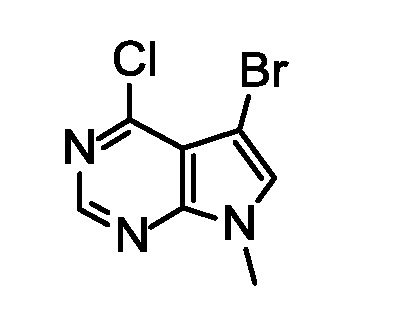
Добавляли N-бромсукцинимид (418 г, 2,35 моль) порциями в течение 20 мин при 15°C к раствору 4-хлор-7-метил-пирроло[2,3-d]пиримидина (355 г, 2,12 моль) в дихлорметане (3,19 л), и перемешивали при 23°C в течение 3 ч. По прошествии этого времени, фильтровали, промывали H2O (5,32 л) и сушили с получением указанного в заголовке соединения (448 г, 86%) в виде белого твердого вещества. ES/MS m/z (35Cl, 79Br) 245,9 (M+H).
Получение 3
Синтез 5-бром-7-метил-пирроло[2,3-d]пиримидин-4-амина.
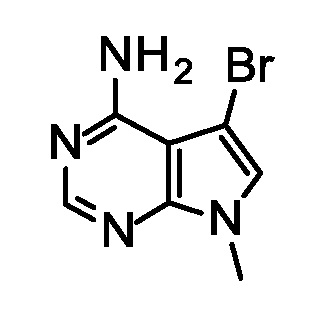
Перемешивали суспензию 5-бром-4-хлор-7-метил-пирроло[2,3-d]пиримидина (454 г 1,84 моль) в аммиаке (30% в H2O, 3,63 л) при 120°C в сосуде под давлением Hastelloy™ в течение 18 ч. Охлаждали до 20°C, фильтровали, промывали H2O (1,80 л) и метанолом (900 мл), и сушили с получением указанного в заголовке соединения (351 г, 82%) в виде белого твердого вещества. ES/MS m/z (79Br) 227,2 (M+H).
Получение 4
Синтез 3-амино-6-бром-пиразин-2-карбоновой кислоты.
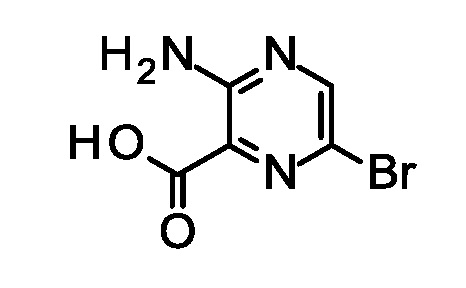
Добавляли 3-аминопиразин-2-карбоновую кислоту (50,0 г, 369,4 ммоль) к раствору N-бромсукцинимида (61,2 г, 377,3 ммоль) и диметилформамида (236,3 г, 3,2 моль) при 0°C. Через 1 час при комнатной температуре образовалось оранжевое твердое вещество. Твердый остаток промывали этилацетатом (500 мл) и отбрасывали его. Сушили органическую фазу сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (32,0 г, 146,7 ммоль, 41%). ES/MS m/z (79Br/81Br) 217,1/219,0 (M+H).
Получение 5
Синтез 3-амино-6-бром-N-метил-пиразин-2-карбоксамида.
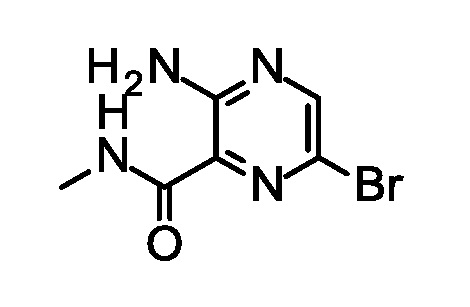
Обрабатывали раствор 3-амино-6-бром-пиразин-2-карбоновой кислоты (214 г, 983 ммоль) в диметилформамиде (1,07 л) метиламин гидрохлоридом (79,7 г, 1,18 моль) и N, N-диизопропилэтиламином (445 г, 3,44 моль) при 23°C. К полученной суспензии, добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат (449 г, 1,18 моль) в течение 30 минут. Через 30 мин, добавляли H2O (4,29 л) в течение 2 ч. Перемешивали при 23°C в течение 30 мин и затем 1 ч при 10°C. Фильтровали, промывали твердое вещество H2O (2×428 мл), и сушили с получением указанного в заголовке соединения (227 г, 82%). ES/MS m/z (79Br) 231,0 (M+H).
Получение 6
Синтез 2-амино-5-бром-N-изопропил-пиридин-3-карбоксамида.
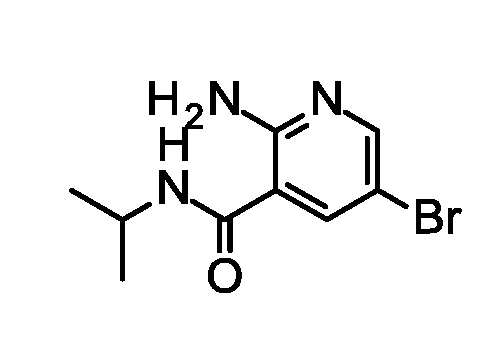
Добавляли пропан-2-амин (42,5 г, 0,719 моль), 1-этил-3-(3-диметиламинопропил)карбодиимид (127 г, 0,664 моль) и гидроксибензотриазол (89,7 г, 0,660 моль) к суспензии 2-амино-5-бром-пиридин-3-карбоновой кислоты (120 г, 0,553 моль) в тетрагидрофуране (1,2 л) при 12°C. Перемешивали смесь при 23°C в течение ночи. Добавляли этилацетат (250 мл) и насыщенный водный NaHCO3 (250 мл), отделяли фазы, и экстрагировали водный слой этилацетатом (2×150 мл). Объединенные органические фазы промывали H2O (300 мл) и насыщенным водным NaCl (300 мл), и концентрировали при пониженном давлении с получением указанного в заголовке соединения (125 г, 88%). ES/MS m/z (79Br) 258,0 (M+H).
Получение 7
Выделение (2R)-2-(3,5-дифторфенил)-2-гидрокси-уксусной кислоты.
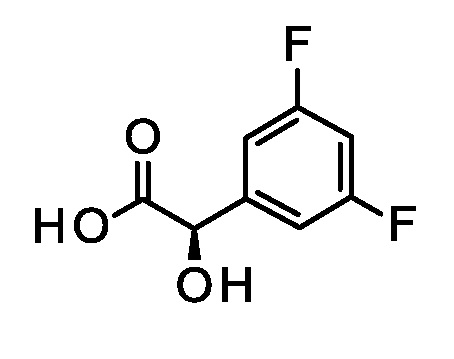
Поддерживали липазу PS Amano (см. Mendiola, J. et al, Org. Process Res. Dev. 2012, 16, 1312-1316) в диатомовой земле перед использованием, смешивая 200 г диатомовой земли и 200 г липазы PS Amano SD. Добавляли H2O для покрытия твердого вещества, и смесь перемешивали. Удаляли H2O в духовом шкафу при 4 мбар и 40°C в течение 16 ч. Контролировали H2O ниже 1% путем титрования по Карлу Фишера для определения воды.
Добавляли нанесенную липазу PS amano SD (250 г) и винилацетат (312 мл; 3,36 моль к суспензии рацемической 2-(3,5-дифторфенил)-2-гидроксиуксусной кислоты (125 г, 664 ммоль) в метил-трет-бутиловом эфире (2,50 л), и смесь перемешивали при 26°C в течение 72 ч. По прошествии этого времени, фильтровали, промывали твердое вещество метил-трет-бутиловым эфиром (1,50 л) и концентрировали объединенные фильтраты при пониженном давлении. Суспендировали остаток в дихлорметане (160 мл) при 23°C в течение 4 ч. Фильтровали, промывали твердое вещество петролейным эфиром (150 мл), и сушили с получением указанного в заголовке соединения (47,0 г, 36%). 1H ЯМР (d6-DMSO) δ 5,11 (с, 1H), 6,20 (шир.с, 1H), 7,11-7,21 (м, 3H), 12,8 (шир.с, 1H). Абсолютную конфигурацию определяли с помощью вибрационного кругового дихроизма (см. Freedman T.B et al, Chirality, 2003 Nov., 15(9), 743-758). Хиральная ВЭЖХ: Rt=7,39 мин (УФ); Колонка: Chiralpak® AD 4,6×150 мм 5 мкм; 5% EtOH в н-гексане (0,05% TFA) изократический; Скорость потока: 1,5 мл/мин, ee >98%.
Получение 8
Выделение (2R)-2-(3-фторфенил)-2-гидрокси-уксусной кислоты.
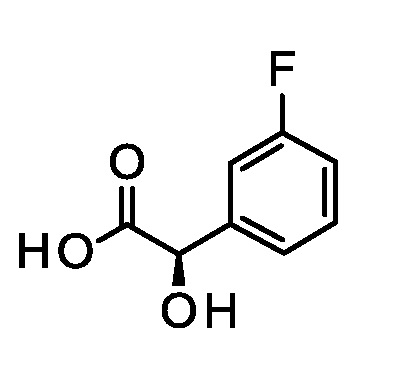
Поддерживали липазу PS Amano SD (см. Mendiola, J. et al, Org. Process Res. Dev. 2012, 16, 1312-1316) в диатомовой земле перед использованием, смешивая 100 г диатомовой земли и 100 г липазы PS Amano SD. Добавляли H2O для покрытия твердого вещества, и смесь перемешивали. Удаляли H2O в духовом шкафу при 4 мбар и 40°C в течение 16 ч. Контролировали H2O ниже 1% путем титрования по Карлу Фишера для определения воды.
Добавляли нанесенную липазу PS amano SD (200 г) и винилацетат (269 мл; 2,90 моль к суспензии рацемической 2-(3-фторфенил)-2-гидроксиуксусной кислоты (96 г, 560 ммоль) в метил-трет-бутиловом эфире (2,00 л), и смесь перемешивали при 26°C в течение 90 ч. По прошествии этого времени, фильтровали, промывали твердое вещество метил-трет-бутиловым эфиром (1,50 л) и концентрировали объединенные фильтраты при пониженном давлении. Суспендировали остаток в дихлорметане (160 мл) при 23°C в течение 4 ч. Фильтровали, промывали твердое вещество петролейным эфиром (150 мл), и сушили с получением указанного в заголовке соединения (31,0 г, 32%). 1H ЯМР (d6-DMSO) δ 5,07 (с, 1H), 6,17 (шир.с, 1H), 7,12 (м, 1H), 7,23 (м, 1H), 7,39 (м, 1H), 12,8 (шир.с, 1H). [α]D20 = -119° (C=2,83, ацетон). Хиральная ВЭЖХ: Rt=10,22 мин (УФ); Колонка: Chiralpak® AD 4,6×150 мм 5 мкм; 5% EtOH в н-гексане (0,05% TFA) изократический; Скорость потока: 1,5 мл/мин, ee >98%.
Получение 9
Синтез 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина.
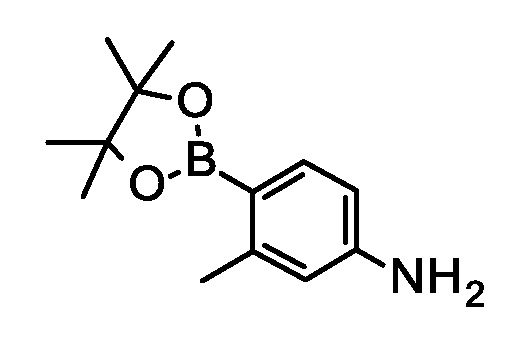
Нагревали суспензию трициклогексилфосфина (59,85 г, 213 ммоль) в 1,4-диоксане (2,98 л) при 95°C в течение 10 мин, пока не получали раствор. Затем, добавляли 4-бром-3-метиланилин (752 г, 2,67 моль), бис(пинаколато)диборон (745,17 г, 2,93 моль), ацетат калия (524 г, 5,34 моль), и ацетат палладия(II) (23,96 г, 107 ммоль), и продолжали нагревать смесь при 95°C в течение 4 ч. По прошествии этого времени, охлаждали до 23°C, разбавляли метил трет-бутиловым эфиром (2,5 л), фильтровали через кизельгур, и промывали твердое вещество метил-трет-бутиловым эфиром (1 л). Объединяли фильтраты, промывали H2O (1,5 л) и насыщенным водным NaCl (1,2 л), и концентрировали при пониженном давлении с получением указанного в заголовке соединения (593 г, 95%). Для получения аналитического образца, суспендировали с гексаном (1,6 мл/г) при 40°C в течение 2 ч, затем охлаждали до 23°C, фильтровали и твердое вещество промывали гексаном (2×0,5 мл/г). ES/MS m/z 234,1 (M+H).
Получение 10
Синтез 2-амино-5-(4-амино-2-метил-фенил)-N-изопропил-пиридин-3-карбоксамида.
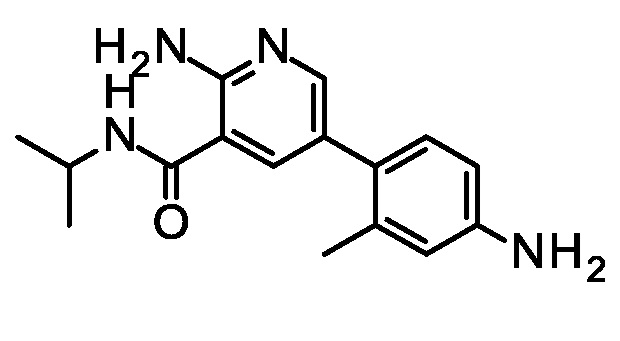
Добавляли 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (93,6 г, 0,401 моль), K2CO3 (119g, 0,860 моль) и Pd(dppf)2Cl2 (10,6 г, 140 ммоль) к раствору 2-амино-5-бром-N-изопропил-пиридин-3-карбоксамида (74,0 г, 0,287 моль) в диоксане (888 мл) и H2O (296 мл), и нагревали смесь до 55°C в течение ночи. Охлаждали до 23°C, добавляли этилацетат (150 мл), фильтровали полученную суспензию через кизельгур, и промывали твердое вещество этилацетатом (50 мл). Промывали объединенные фильтраты H2O (30 мл) и насыщенным водным NaCl (300 мл), и концентрировали при пониженном давлении с получением указанного в заголовке соединения (78,0 г, 96%). ES/MS m/z 285,1 (M+H).
Получение 11
Синтез 3-амино-6-(4-амино-2-метилфенил)-N-метилпиразин-2-карбоксамида.
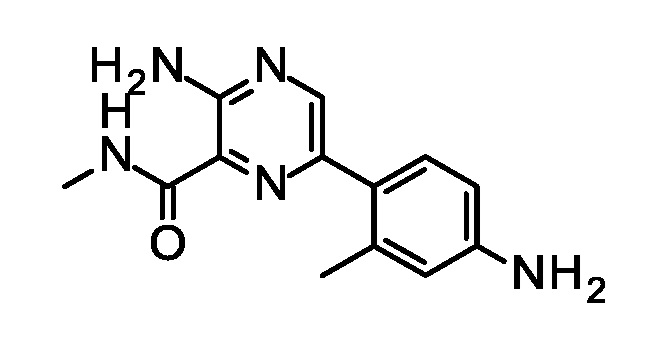
Добавляли 3-амино-6-бром-N-метилпиразин-2-карбоксамид (99,1 г, 429 ммоль), Na2CO3, (2 M в H2O, 500 мл, 1,00 моль), и 1,1'-бис(дифенилфосфино)ферроцен)палладий (II) хлорид (19 г, 22,8 ммоль) к раствору 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (122 г, 450 ммоль) в 1,4-диоксане (3,00 л), и нагревали смесь до 85°C в течение 32 ч. Охлаждали до 30°C, добавляли этилацетат (4,00 л), фильтровали через слой силикагеля, и промывали твердое вещество этилацетатом (3×1,00 л). Промывали объединенные фильтраты H2O (2×1,50 л), и концентрировали при пониженном давлении. Очищали остаток с помощью хроматографии (элюент: петролейный эфир/этилацетат 5:1-1:1) с получением указанного в заголовке соединения (80 г, 72%) в виде желтого твердого вещества. ES/MS m/z 258,1 (M+H).
Получение 12
Синтез 5-(4-амино-2-метил-фенил)-7-метил-пирроло[2,3-d]пиримидин-4-амина.
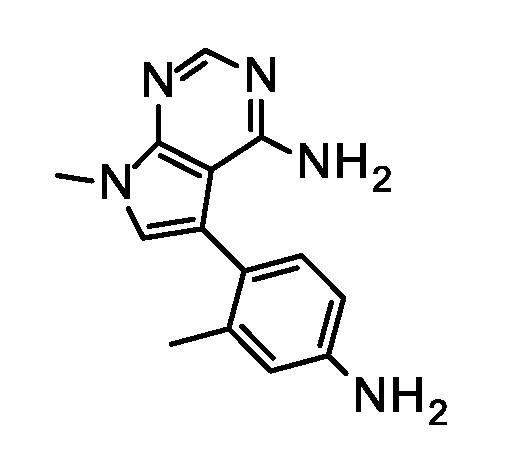
Добавляли Pd(II) ацетат (635 мг, 2,83 ммоль), cataCXium A™ (2,03 г, 5,65 ммоль) и насыщенный водный NaHCO3 (186 мл, 188 ммоль) к суспензии 5-бром-7-метил-пирроло[2,3-d]пиримидин-4-амина (21,4 г, 94,3 ммоль) и 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (28,6 г, 123 ммоль) в 2-метил-тетрагидрофуране (214 мл) при 23°C, и смесь перемешивали в запаянной трубке при 100°C в течение 3 ч. Охлаждали до 23°C, фильтровали через слой кизельгура, и промывали твердое вещество H2O (50 мл) и этилацетатом (100 мл). Отделяли органический слой, промывали его насыщенным водным NaCl (50 мл) и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии (элюент: гексан/ацетон 0-100%) с получением указанного в заголовке соединения (12,1 г, 51%) в виде желтого твердого вещества. ES/MS m/z 254,1 (M+H).
Пример 1
Синтез 2-амино-5-[4-[[(2R)-2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамида.
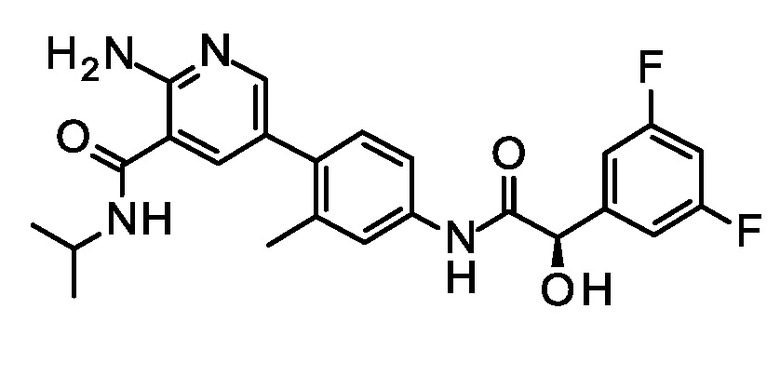
Обрабатывали смесь (2R)-2-(3,5-дифторфенил)-2-гидроксиуксусной кислоты (29,0 г, 0,154 моль), 2-амино-5-(4-амино-2-метил-фенил)-N-изопропил-пиридин-3-карбоксамида (43,83 г, 0,154 моль), и N, N-диизопропилэтиламина (39,8 г, 0,308 моль) в тетрагидрофуране (960 мл), (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфатом) (87,9 г, 0,231 моль) при 0°C в течение 30 мин, и затем нагревали до 20°C и перемешивали в течение 2 ч. Добавляли этилацетат (50 мл), и смесь фильтровали. Концентрировали фильтрата при пониженном давлении, и остаток очищали с помощью хроматографии (элюент: 2:1 петролейный эфир/этилацетат) и затем с помощью сверхкритической жидкостной хроматографии, SFC (Колонка: Chiralpak® IC 30×250 мм 5 мкм (Daicel); MeOH/CO2= 30:70 изократический; Скорость потока: 80 г/мин; Обратное давление: 100 Бар; Температура колонки: 40°C) с получением указанного в заголовке соединения (27,5 г, 39%) в виде белого твердого вещества. ES/MS m/z 455,2 (M+H).
Пример 2
Синтез 2-амино-5-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамида.
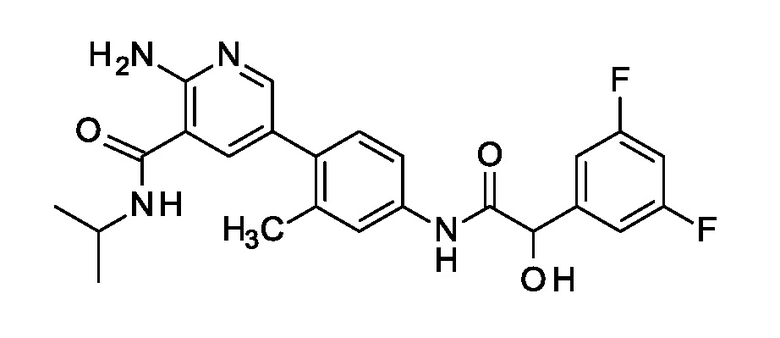
Добавляли 2-амино-5-(4-амино-2-метил-фенил)-N-изопропил-пиридин-3-карбоксамид (1000,5 мг, 3,5 ммоль) к раствору 2-(3,5-дифторфенил)-2-гидрокси-уксусной кислоты (793 мг, 4,2 ммоль), (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфата) (1,7 г, 4,6 ммоль), N, N-диизопропилэтиламина (909,5 мг, 7,0 ммоль) в тетрагидрофуране (7,9 г, 93,6 моль). Через 2 часа при комнатной температуре добавляли 3 мл этилацетата и реакционную смесь перемешивали в течение 10 минут. Отфильтровывали твердое вещество и восстанавливали органическую фазу при пониженном давлении. Промывали остаток насыщенным водным NaHCO3 (10 мл) и экстрагировали DCM (2×10 мл). Сушили органическую фазу сульфатом натрия, фильтровали и концентрировали при пониженном давлении.
Остаток очищают с помощью ВЭЖХ, Rt (время удерживания) = 2,036 минут (УФ), ЖХ-колонка: XTerra MS C18 (2,1×50 мм, 3,5 мкм; H2O:ацетонитрил; градиент 0,25 мин при 5%B; от 5%B до 100%B в 3 мин; выдержка 0,25 мин при 100%B; Температура колонки: 50°C; Скорость потока 1,1 мл/мин с получением указанного в заголовке соединения в виде смеси изомера 1 и изомера 2 в виде твердого вещества белого цвета (0,97 г, 60%). ES/MS (m/z): 455,4 (M+H).
Пример 3 и 4
Разделение 2-амино-5-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамида на изомер 1 и изомер 2.
Смесь изомера 1 и изомера 2 разделяли, используя Chiralcel® OD-H (4,6×100 мм, 5 мкм), 20% MeOH-DMEA (0,2%) в CO2), 2,5 мл/мин, 100 бар давление на выходе, 35°C температура для получения индивидуального изомера 1 и изомера 2 в виде белого твердого вещества.
Пример 3: 2-амино-5-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамид изомер 1. Rt (время удерживания) = 1,131 минут (430 мг, ee > 98%), ES/MS m/z 455,4 (M+H).
Пример 4: 2-амино-5-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-изопропил-пиридин-3-карбоксамид изомер 2. Rt (время удерживания) = 1,823 минут (404 мг, ee > 98%), ES/MS m/z 455,4 (M+H).
Пример 5
Синтез 3-амино-6-[4-[[(2R)-2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамида.
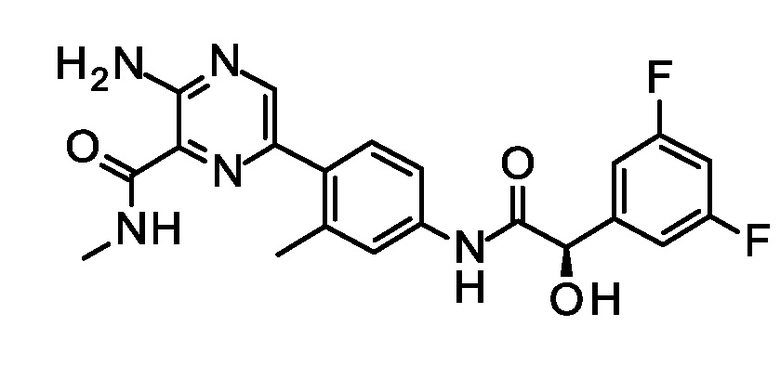
Добавляли N, N- (15,3 мл 87,5 ммоль) и 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат (33,2 г, 87,5 ммоль) к раствору 3-амино-6-(4-амино-2-метилфенил)-N-метилпиразин-2-карбоксамида (18,0 г, 70,0 ммоль) и (2R)-2-(3,5-дифторфенил)-2-гидрокси-уксусной кислоты (13,2 г, 70,0 ммоль) в тетрагидрофуране (90,0 мл), и смесь перемешивали при 23°C в течение 5 ч. По прошествии этого времени, концентрировали смесь при пониженном давлении, суспендировали остаток в этилацетате (100 мл) в течение 15 мин, фильтровали, и промывали твердое вещество этилацетатом (2×25 мл). Концентрировали объединенные фильтраты при пониженном давлении, и остаток очищали с помощью хроматографии (элюент: гексан/ацетон 2:1, затем гексан/этанол 4:1). Растворяли вещество в метаноле (115 мл), добавляли диоксид кремния-тиоловую смолу (0,4 г/г), и полученную суспензию перемешивали при 23°C в течение 8 ч. По прошествии этого времени, фильтровали, и промывали твердое вещество метанолом (2×12 мл). Концентрировали объединенные фильтраты при пониженном давлении. Очищали с помщью SFC (Колонка: Chiralpak® IC 4,6×100 мм 5 мкм; 35% метанол (0,2% N, N-диметилэтиламин) в CO2 изократический; Скорость потока: 2,5 мл/мин; Обратное давление: 100 Бар; Температура колонки: 40°C) с получением указанного в заголовке соединения (19,7 г, 62%). ES/MS m/z 428,1 (M+H).
Пример 6
Синтез 3-амино-6-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамида.
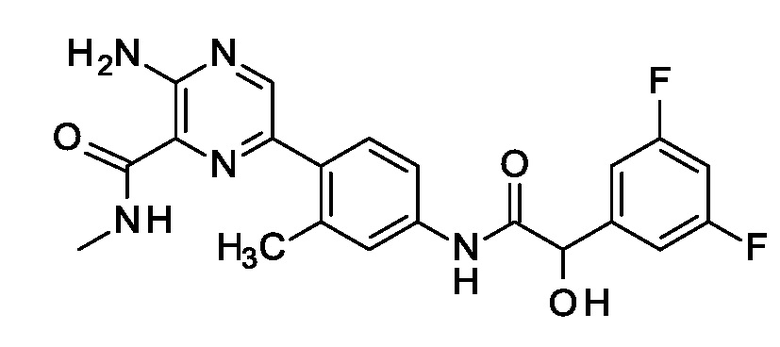
Добавляли 3-амино-6-(4-амино-2-метил-фенил)-N-метил-пиразин-2-карбоксамид (800,0 мг, 3,2 ммоль) к раствору 2-(3,5-дифторфенил)-2-гидрокси-уксусной кислоты (701,9 мг, 3,4 ммоль), (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфата) (1,7 г, 4,6 ммоль), N, N-диизопропилэтиламина (803,2 мг, 6,3 ммоль) в тетрагидрофуране (7,9 г, 93,6 моль). Через 2 часа при комнатной температуре, добавляли 3 мл этилацетата и реакционную смесь перемешивали в течение 10 минут. Отфильтровывали твердое вещество и восстанавливали органическую фазу при пониженном давлении. Промывали остаток насыщенным водным NaHCO3 (10 мл) и экстрагировали дихлорметаном (2×10 мл). Сушили органическую фазу сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии на силикагеле, элюируя этилацетат:гексан (30:70) с получением указанного в заголовке соединения в виде смеси изомера 1 и изомера 2 в виде коричневого твердого вещества (0,72 г, 1,6 ммоль). ES/MS (m/z): 428,3 (M+H).
Примеры 7 и 8
Разделение 3-амино-6-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамид на изомер 1 и изомер 2.
Смесь изомера 1 и изомера 2 разделяли, используя Chiralpak® OD (4,6×50 мм, 5 мкм), 20% MeOH-DMEA (0,2%) в CO2), 5 мл/мин, 100 бар давление на выходе, 35°C температура для получения индивидуального изомера 1 и изомера 2.
Пример 7. 3-амино-6-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамид изомер 1. Rt (время удерживания) = 1,610 минут (258 мг, ee > 98%), ES/MS m/z 428,3 (M+H).
Пример 8. 3-амино-6-[4-[[2-(3,5-дифторфенил)-2-гидрокси-ацетил]амино]-2-метил-фенил]-N-метил-пиразин-2-карбоксамид изомер 2. Rt (время удерживания) = 2,410 минут (278 мг, ee > 98%), ES/MS m/z 428,3 (M+H).
Пример 9
Синтез (2R)-N-[4-(4-амино-7-метил-пирроло[2,3-d]пиримидин-5-ил)-3-метил-фенил]-2-(3-фторфенил)-2-гидрокси-ацетамида.
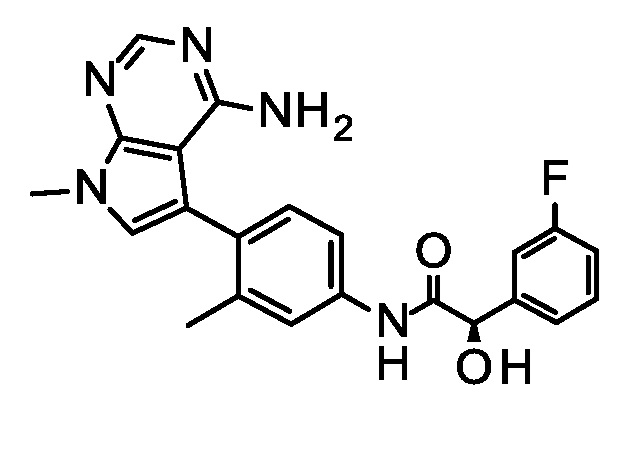
Обрабатывали раствор 5-(4-амино-2-метил-фенил)-7-метил-пирроло[2,3-d]пиримидин-4-амина (15,5 г, 44,1 ммоль) и (2R)-2-(3-фторфенил)-2-гидрокси-уксусной кислоты (8,25 г, 48,5 ммоль) в тетрагидрофуране (56 мл) N, N-диизопропилэтиламином (9,22 мл, 52,9 ммоль) и 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфатом (20,1 г, 52,9 ммоль) при 23°C в течение 3,5 ч. По прошествии этого времени, концентрировали смесь при пониженном давлении, и суспендировали в этилацетате (100 мл) в течение 15 мин. Фильтровали, промывали твердое вещество этилацетатом (2×15 мл), и концентрировали объединенные фильтраты при пониженном давлении. Остаток очищали с помощью хроматографии (элюент: дихлорметан/метанол 0-10%) и затем с помощью SFC (размер колонки: 5 мкм, 2×25 см; неподвижная фаза: 2-этилпиридин; подвижная фаза: CO2 (A)/метанол-N, N-диметилэтиламин (0,2%) (B); состав подвижной фазы (то есть соотношение A/B): изократический 72/25 A/B; Скорость потока: 65 мл/мин; загрузка: 70 мг/4,35 мин) с получением указанного в заголовке соединения (11,7 г, 65%). ES/MS m/z 406,1 (M+H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2017 |
|
RU2744988C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ УВЕИТА | 2019 |
|
RU2757273C1 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ПРОИЗВОДНЫЕ АРИЛСУЛЬФОНИЛСТИЛЬБЕНА ДЛЯ ЛЕЧЕНИЯ БЕССОННИЦЫ И СВЯЗАННЫХ С НЕЙ РАССТРОЙСТВ | 2005 |
|
RU2382766C2 |
Изобретение относится к соединению формулы
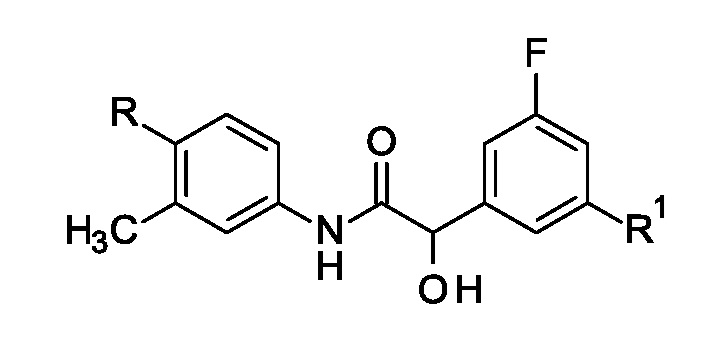 , где R выбран из группы, состоящей из
, где R выбран из группы, состоящей из и
и 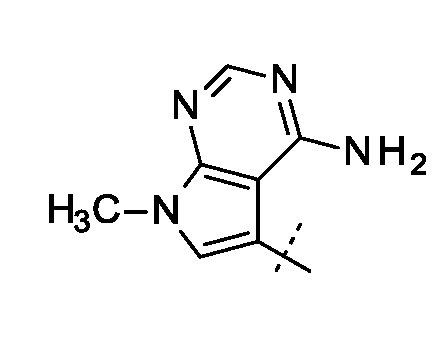 ; X представляет собой CH или N; R1 представляет собой водород или фтор и R2 представляет собой С1–С3 алкил; или к его фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции, обладающей активностью ингибитора PERK, на основе указанного соединения. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине в качестве эффективных лекарственных средств для лечения рака поджелудочной железы. 2 н. и 9 з.п. ф-лы, 4 табл., 9 пр.
; X представляет собой CH или N; R1 представляет собой водород или фтор и R2 представляет собой С1–С3 алкил; или к его фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции, обладающей активностью ингибитора PERK, на основе указанного соединения. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине в качестве эффективных лекарственных средств для лечения рака поджелудочной железы. 2 н. и 9 з.п. ф-лы, 4 табл., 9 пр.
1. Соединение формулы
 ,
,
где R выбран из группы, состоящей из
 и
и 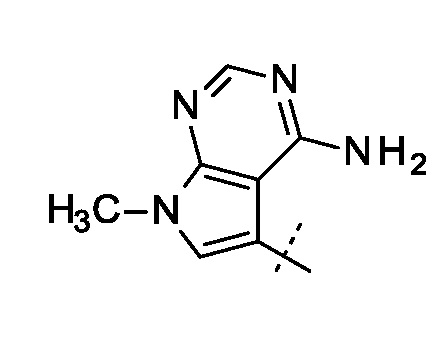 ;
;
X представляет собой CH или N;
R1 представляет собой водород или фтор; и
R2 представляет собой С1-С3 алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 формулы
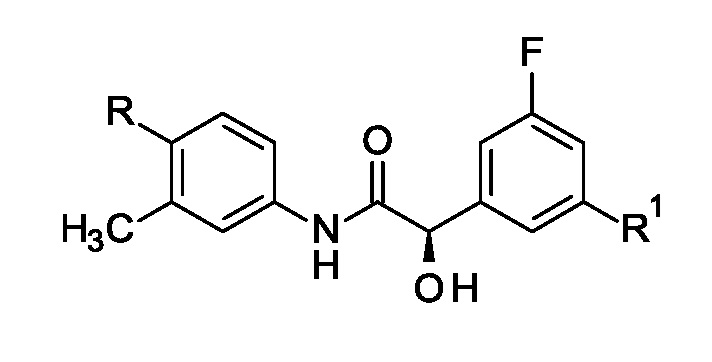 ,
,
где R выбран из группы, состоящей из
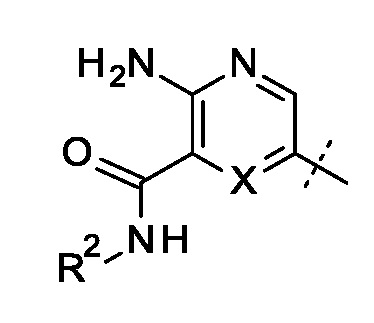 и
и 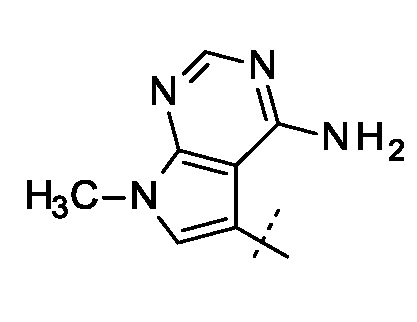 ;
;
X представляет собой CH или N;
R1 представляет собой водород или фтор; и
R2 представляет собой С1-С3 алкил;
или его фармацевтически приемлемая соль.
3. Соединение или соль по п.1 или 2, где R представляет собой
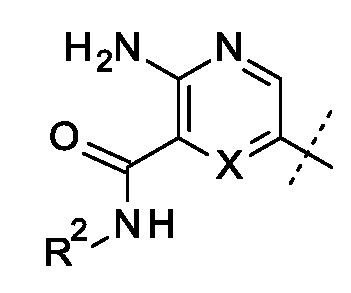 .
.
4. Соединение или соль по п.1 или 2,
где X представляет собой CH или N;
R1 представляет собой водород или фтор; и
R2 представляет собой метил или изопропил.
5. Соединение или соль по п.1 или 2, где R представляет собой
 .
.
6. Соединение по п.1 или 2, которое представляет собой 3–амино–6–[4–[[(2R)–2–(3,5–дифторфенил)–2–гидрокси–ацетил]амино]–2–метил–фенил]–N–метил–пиразин–2–карбоксамид, которое представлено формулой
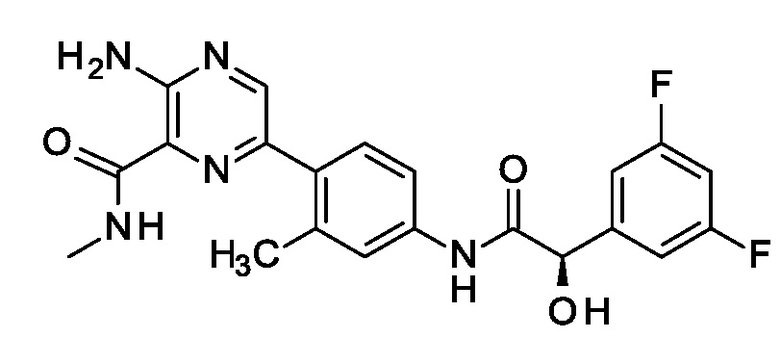 ,
,
или его фармацевтически приемлемая соль.
7. Соединение по п.1 или 2, которое представляет собой 2–амино–5–[4–[[(2R)–2–(3,5–дифторфенил)–2–гидрокси–ацетил]амино]–2–метил–фенил]–N–изопропил–пиридин–3–карбоксамид, которое представлено формулой
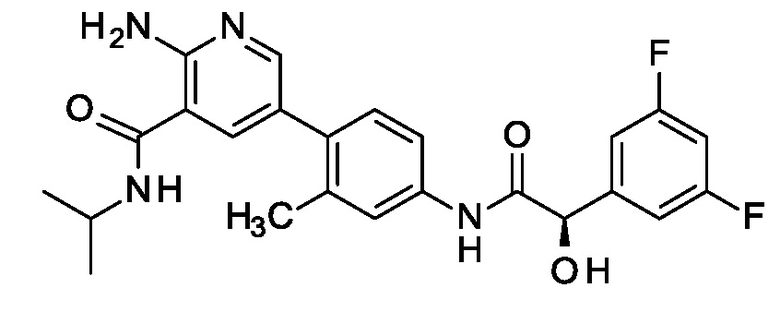 ,
,
или его фармацевтически приемлемая соль.
8. Соединение по п.1 или 2, которое представляет собой (2R)–N–[4–(4–амино–7–метил–пирроло[2,3–d]пиримидин–5–ил)–3–метил–фенил]–2–(3–фторфенил)–2–гидрокси–ацетамид, которое представлено формулой
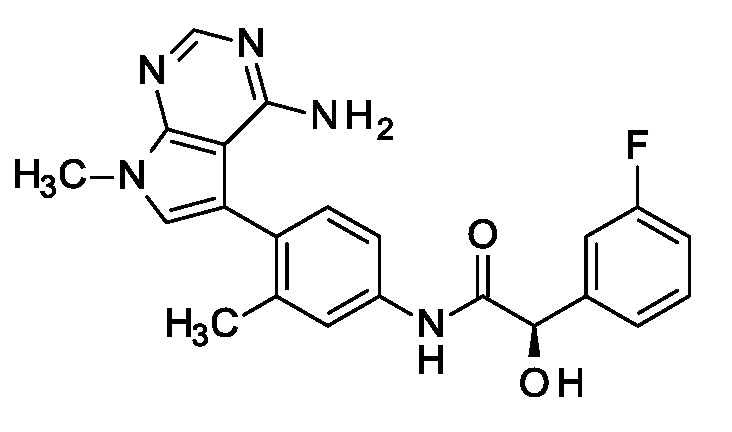 ,
,
или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция, обладающая активностью ингибитора протеинкиназа R (PKR)–подобной протеинкиназы эндоплазматического ретикулума (PERK), содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-8 с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
10. Соединение или его фармацевтически приемлемая соль по любому из пп.1-8 для применения в производстве лекарственного средства для лечения рака поджелудочной железы.
11. Соединение или его фармацевтически приемлемая соль по любому из пп.1-8 для применения при лечении рака поджелудочной железы.
| WO 2007026920 A2, 08.03.2007 | |||
| WO 2003064397 A1, 07.08.2003 | |||
| US 20170022206 A1, 26.01.2017 | |||
| Переходная камера для поверки манометров | 1929 |
|
SU14230A1 |
| Буровой инструмент | 1927 |
|
SU10485A1 |
