ОБЛАСТЬ ТЕХНИКИ
[0001]
Настоящее изобретение относится к способу получения ароматического нитрильного соединения, такого как цианопиридин, и к способу получения карбонатного эфира.
УРОВЕНЬ ТЕХНИКИ
[0002]
"Карбонатный эфир" является общим названием для соединения, получаемого путем замещения одного или обоих из двух атомов водорода в угольной кислоте CO(OH)2 на алкильную группу или арильную группу, и он имеет структуру RO-C(=O)-OR' (R и R' представляют насыщенную углеводородную группу или ненасыщенную углеводородную группу).
[0003]
Карбонатный эфир используется в качестве присадки, например, присадки к бензину для улучшения октанового числа и присадки к дизельному топливу для уменьшения количества частиц в выхлопном газе. Карбонатный эфир также используется, например, в качестве алкилирующего агента, карбонилирующего агента, растворителя или т.п. для синтеза смол или органических соединений, таких как поликарбонат, уретан, фармацевтические препараты, сельскохозяйственные химикаты или т.п., материала электролитического раствора литий-ионных элементов, материала смазочного масла или материала поглотителя кислорода для ингибирования ржавления бойлерных труб. Таким образом, карбонатный эфир является очень полезным соединением.
[0004]
В качестве обычного способа получения карбонатного эфира в основном применяется способ прямого проведения реакции фосгена, используемого в качестве источника карбонила, со спиртом. Фосген, используемый в этом способе, очень вреден и имеет высокую коррозионную активность, и поэтому требует крайней осторожности при обращении, например, при транспортировке или хранении. Контроль и управление оборудованием для получения фосгена, а также обеспечение безопасности требует больших затрат. Кроме того, в случае получения с использованием данного способа исходные материалы и катализаторы содержат галоген, такой как хлор, и полученный карбонатный эфир содержит следовое количество галогена, которое не может быть удалено с помощью простого этапа очистки. При использовании карбонатного эфира для присадки к бензину, легкой присадки к маслу или материала для электронного оборудования, такой галоген может нежелательным образом вызывать коррозию. По этой причине необходим этап тщательной очистки для уменьшения следового количества галогена, присутствующего в карбонатном эфире, до уровня чрезвычайно малого следового количества. Помимо этого, в последнее время административные учреждения вводят строгие административные указания и не разрешают создавать новые производственные сооружения с использованием данного способа, поскольку в этом способе применяется фосген, который очень вреден для человеческого организма. Соответственно, крайне необходим новый способ получения карбонатного эфира, в котором фосген не используется.
[0005]
Также известен способ прямого синтеза карбонатного эфира из спирта и диоксида углерода с использованием гетерогенного катализатора. В отношении этого способа были проведены исследования по использованию 2-цианопиридина или бензонитрила в качестве смачиваемого порошка для значительного увеличения получаемого количества и производительности при получении карбонатного эфира, для обеспечения легкого протекания реакция при давлении, близком к нормальному давлению, и для увеличения скорости реакции (см. Патентные документы 1 и 2). Однако существует проблема, касающаяся способа обработки или использования бензамида или т.п., образующегося в виде побочного продукта.
Например, использование бензамида, образующегося в результате реакции между бензонитрилом и водой, ограничено некоторыми фармацевтическими и агрохимическими промежуточными соединениями. Таким образом, использование бензамида до некоторой степени ограничено. Поэтому при получении карбонатного эфира с использованием бензонитрила в качестве смачиваемого порошка желательно регенерировать получаемый побочно бензамид с получением бензонитрила и повторно использовать его. В настоящее время существует проблема осуществления реакции регенерации с высокой селективностью (поскольку считается, что если образуется побочный продукт, то повторное использование бензонитрила в качестве смачиваемого порошка является затруднительным) и с высоким выходом (поскольку, если выход низок, бензамид остается в большом количестве, что увеличивает объем работы, а именно загрузку, по отделению бензамида и бензонитрила друг от друга).
[0006]
Ввиду вышеописанных проблем, относящихся к регенерации бензамида или т.п. с получением бензонитрила или т.п., существует известный способ выполнения вышеописанной регенерации без использования сильного реагента при подавлении образования побочного продукта (Патентный документ 3).
Однако согласно этому способу для образования нитрила посредством дегидратации амидного соединения требуется 400 часов, и поэтому он не может быть сбалансирован, а именно, не может быть использован в сочетании с реакцией синтеза карбонатного эфира, для которой требуется только 24 часа. С этим способом также связана проблема, заключающаяся в том, что для разделения катализатора на твердую и жидкую фазу необходимы этапы экстракции, инфильтрации и т.п., что приводит к сложному процессу, имеющему множество этапов.
ДОКУМЕНТЫ, ИЗВЕСТНЫЕ ИЗ УРОВНЯ ТЕХНИКИ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
[0007]
Патентный документ 1: открытая публикация патента Японии №2010-77113
Патентный документ 2: открытая публикация патента Японии №2012-162523
Патентный документ 3: WO 2015/099053
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ В ИЗОБРЕТЕНИИ
[0008]
Ввиду вышеописанных проблем предшествующего уровня техники, задачей настоящего изобретения является создание способа, обеспечивающего возможность реакции дегидратации, в котором может быть селективно получено требуемое соединение с высоким выходом при подавлении образования побочных продуктов в течение регенерации ароматического амидного соединения, такого как пиридинкарбоамид, с получением цианопиридина, представляющего собой соответствующее ароматическое нитрильное соединение. Другой задачей настоящего изобретения является создание способа получения ароматического нитрильного соединения, в котором может быть уменьшено количество этапов реакции дегидратации и значительно повышена скорость реакции для уменьшения продолжительности реакции.
Еще одной задачей настоящего изобретения является применение вышеописанного способа получения ароматического нитрильного соединения к способу получения карбонатного эфира для реализации способа эффективного получения карбонатного эфира.
СРЕДСТВА РЕШЕНИЯ ЗАДАЧ
[0009]
Для решения вышеописанных проблем авторами настоящего изобретения были проведены исследования касательно способа получения ароматического нитрильного соединения, такого как цианопиридин, посредством дегидратации ароматического амидного соединения. Авторами настоящего изобретения были исследованы условия реакции дегидратации ароматического амидного соединения и реализован процесс реакции дегидратации, в котором путем приведения ароматического амидного соединения в виде газа или тумана в контакт с катализатором в газовой фазе в течение короткого времени может быть значительно повышена скорость реакции для уменьшения продолжительности реакции, и, кроме того, может быть избирательно получено требуемое соединение с высоким выходом при подавлении образования побочных продуктов.
[0010]
Согласно настоящему изобретению, описанному выше, может быть увеличена скорость регенерации ароматического амидного соединения с получением ароматического нитрильного соединения посредством реакции дегидратации, причем реакция дегидратации и скорость синтеза карбонатного эфира из CO2 и спирта с использованием ароматического нитрильного соединения может быть реализована в виде группы эффективных промышленных процессов. Авторами настоящего изобретения также были проведены исследования по применению вышеописанного решения к способу получения карбонатного эфира. Как следствие, также и в способе получения карбонатного эфира, в котором карбонатный эфир синтезируют непосредственно из спирта и диоксида углерода, было подтверждено, что преимущества достигаются, если ароматическое нитрильное соединение эффективно регенерируют на этапе контакта, на котором ароматическое амидное соединение в виде газа или тумана приводят в контакт с катализатором в газовой фазе в течение короткого времени. Ниже описывается сущность настоящего изобретения.
[0011]
(1) Способ получения ароматического нитрильного соединения, включающий реакцию дегидратации, в которой дегидратируют ароматическое амидное соединение,
при этом способ включает этап контакта для приведения ароматического амидного соединения в контакт с катализатором в газовой фазе в ходе реакции дегидратации.
(2) Способ получения ароматического нитрильного соединения по пункту (1), в котором катализатор включает щелочной металл.
(3) Способ получения ароматического нитрильного соединения по пункту (1) или (2), в котором ароматическое амидное соединение включает по меньшей мере гетероариламидное соединение, при этом ароматическое нитрильное соединение включает по меньшей мере гетероарилнитрильное соединение.
(4) Способ получения ароматического нитрильного соединения по пункту (3), в котором гетероариламидное соединение включает 2-пиколинамид, при этом гетероарилнитрильное соединение включает 2-цианопиридин.
(5) Способ получения ароматического нитрильного соединения по любому из пунктов (1)-(4), в котором на этапе контакта дополнительно приводят в контакт с катализатором инертный газ и/или растворитель в парообразном состоянии.
(6) Способ получения ароматического нитрильного соединения по пункту (5), в котором инертный газ включает по меньшей мере газообразный азот.
(7) Способ получения ароматического нитрильного соединения по пункту (5) или (6), в котором температура кипения растворителя при нормальном давлении составляет от 20 до 300°С.
(8) Способ получения ароматического нитрильного соединения по любому из пунктов (5)-(7), в котором растворитель совместим с ароматическим амидным соединением.
(9) Способ получения ароматического нитрильного соединения по любому из пунктов (5)-(8), в котором растворитель включает пиридиновое соединение и/или циклический кетон.
(10) Способ получения ароматического нитрильного соединения по любому из пунктов (1)-(9), в котором на этапе контакта температура, при которой ароматическое амидное соединение приводят в контакт с катализатором в газовой фазе, составляет 170°С или выше, но ниже 300°С.
(11) Способ получения ароматического нитрильного соединения по любому из пунктов (1)-(10), в котором продолжительность приведения ароматического амидного соединения в контакт с катализатором в газовой фазе составляет 0,001 сек или больше, но меньше 10 сек.
(12) Способ получения карбонатного эфира, содержащий:
первый этап реакции, включающий: реакцию получения карбонатного эфира, в которой проводят реакцию спирта с диоксидом углерода в присутствии ароматического нитрильного соединения для получения карбонатного эфира и воды, и реакцию гидратации, в которой гидратируют ароматическое нитрильное соединение с полученной водой для получения ароматического амидного соединения; и
второй этап реакции, на котором отделяют ароматическое амидное соединение от реакционной системы первого этапа реакции и затем регенерируют ароматическое амидное соединение с получением ароматического нитрильного соединения посредством реакции дегидратации для дегидратации ароматического амидного соединения, причем реакция дегидратации имеет этап контакта для приведения ароматического амидного соединения в контакт с катализатором в газовой фазе,
при этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на втором этапе реакции, используют на первом этапе реакции.
(13) Способ получения карбонатного эфира по пункту (12), в котором на втором этапе реакции получают ароматическое нитрильное соединение из ароматического амидного соединения согласно способу получения ароматического нитрильного соединения по любому из пунктов (2)-(11) для регенерации ароматического нитрильного соединения.
(14) Способ получения карбонатного эфира по пункту (12) или (13), в котором в реакции получения карбонатного эфира используют катализатор, включающий оксид церия.
(15) Способ получения карбонатного эфира по любому из пунктов (12)-(14), в котором спирт включает спирт, содержащий от 1 до 6 атомов углерода.
ПРЕИМУЩЕСТВА ИЗОБРЕТЕНИЯ
[0012]
Согласно настоящему изобретению может быть эффективно осуществлено получение (регенерация) ароматического нитрильного соединения, такого как цианопиридин, из ароматического амидного соединения, такого как пиридинкарбоамид (пиколинамид, никотинамид и т.д.) и бензамид. В частности, в реакции дегидратации ароматического амидного соединения для вышеописанной регенерации может быть подавлено образование побочных продуктов, может быть избирательно получено требуемое соединение с высоким выходом и может быть повышена скорость реакции. Таким образом, согласно настоящему изобретению может быть значительно уменьшена продолжительность реакции дегидратации для регенерации ароматического нитрильного соединения по сравнению с обычными способами. Кроме того, за счет применения газофазной реакции размер реакционной емкости может быть уменьшен в большей степени по сравнению с этапом дегидратации амидного соединения с использованием обычной жидкофазной реакции. Таким образом, согласно настоящему изобретению этап регенерации соответствующего ароматического нитрильного соединения из ароматического амидного соединения может быть легко реализован в промышленности.
Кроме того, согласно настоящему изобретению за счет вышеописанного получения ароматического нитрильного соединения также может быть реализован способ эффективного получения карбонатного эфира.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0013]
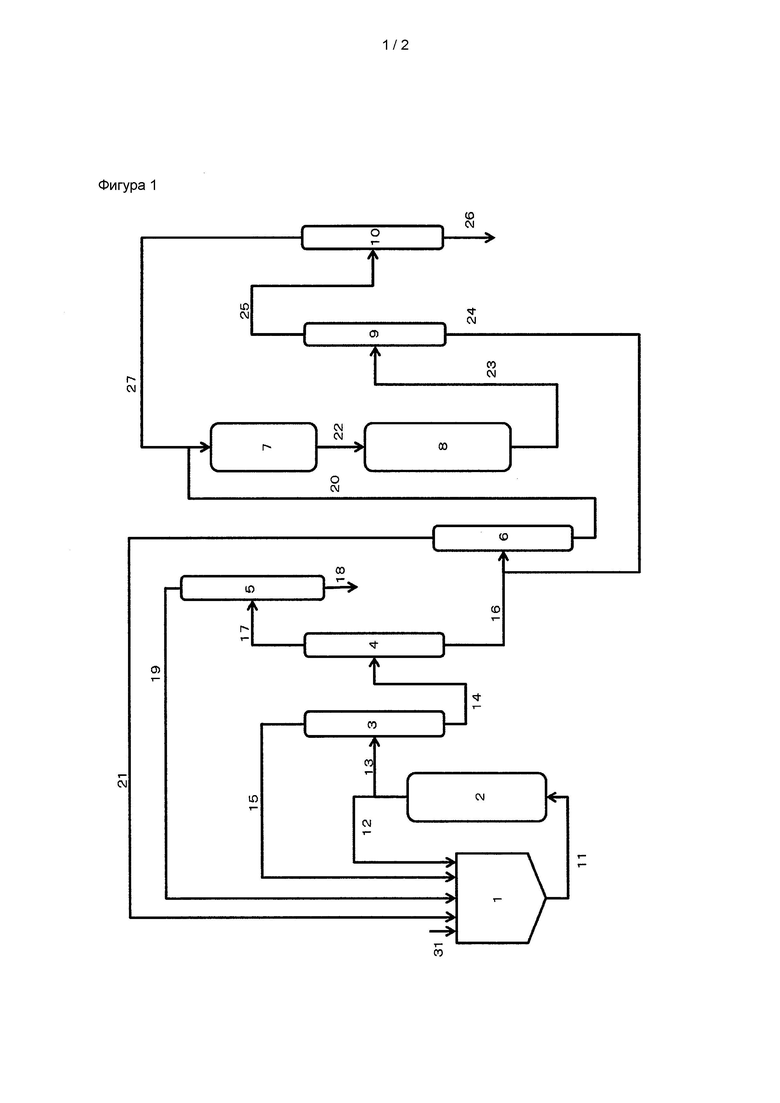
На фиг. 1 показан пример устройства для получения карбонатного эфира.
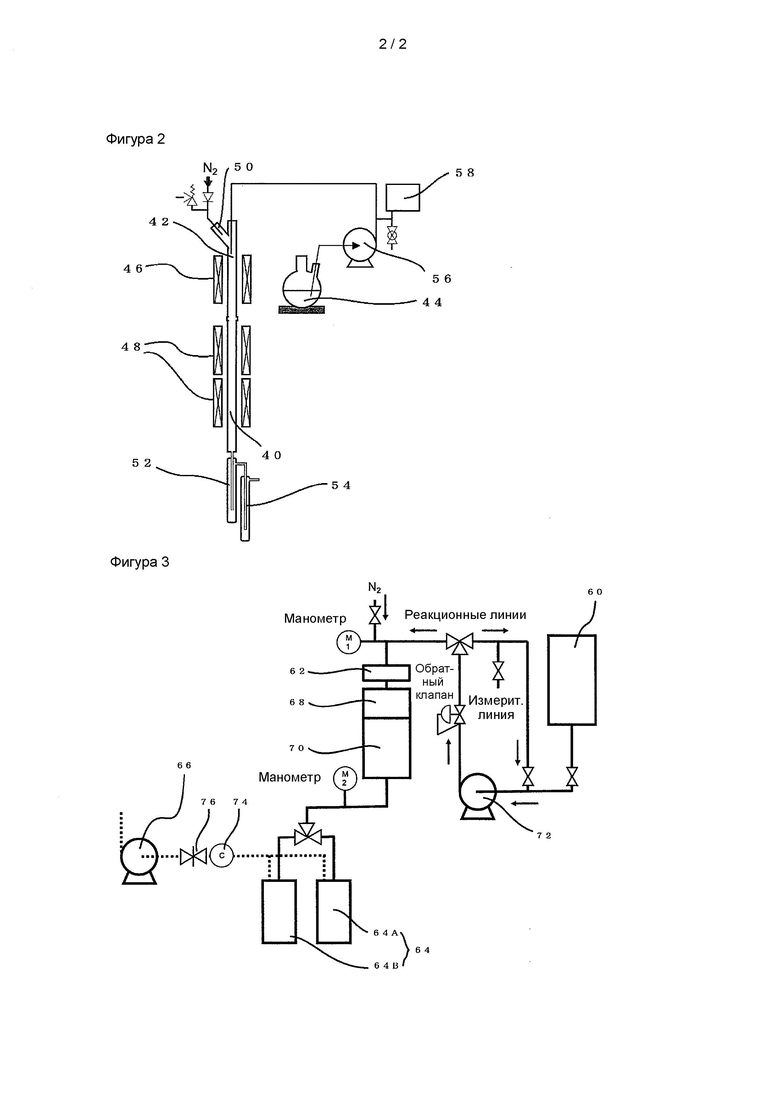
На фиг. 2 схематично показано устройство для получения цианопиридина, которое использовалось в рабочих примерах, в которых цианопиридин получали при повышенном или нормальном давлении.
На фиг. 3 схематично показано устройство для получения цианопиридина, которое использовалось в рабочих примерах, в которых цианопиридин получали при пониженном давлении.
ВАРИАНТЫ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[0014]
Ниже подробно описываются предпочтительные варианты реализации настоящего изобретения.
[0015]
<1. Способ получения ароматического нитрильного соединения>
В способе получения ароматического нитрильного соединения согласно настоящему изобретению ароматическое амидное соединение, такое как пиридинкарбоамид (2-пиридинкарбоамид, 3-пиридинкарбоамид или 4-пиридинкарбоамид), дегидратируют в газовой фазе и превращают в ароматическое нитрильное соединение, такое как цианопиридин. В частности, в способе получения ароматического нитрильного соединения согласно настоящему изобретению, например, реакцию дегидратации вызывают с помощью этапа контакта, на котором ароматическое амидное соединение приводят в контакт с катализатором, несущим оксид основного металла в газовой фазе, тем самым получая ароматическое нитрильное соединение.
[0016]
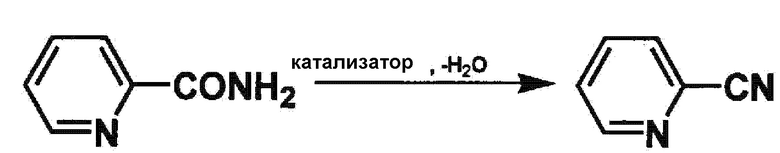
[0017]
(Реакционная подложка)
Примеры ароматического амидного соединения, используемого в способе получения ароматического нитрильного соединения, включают в себя амидные соединения, имеющие ароматическое углеводородное кольцо, такое как бензольное кольцо, нафталиновое кольцо и антраценовое кольцо или гетероарильное кольцо. Среди этих ароматических амидных соединений предпочтительно используется ароматическое амидное соединение, имеющее гетероарильное кольцо, т.е. гетероариламидное соединение, примеры гетероариламидного соединения включают в себя амидные соединения, имеющие пиридиновое кольцо, пиримидиновое кольцо, пиридазиновое кольцо, пиразиновое кольцо, триазиновое кольцо, пиррольное кольцо, фурановое кольцо, тиофеновое кольцо, имидазольное кольцо, пиразольное кольцо, оксазольное кольцо или т.п.
Предпочтительные частные примеры гетероариламидного соединения включают в себя соединения, имеющие пиридиновое кольцо, такие как вышеописанный пиридинкарбоамид (2-пиридинкарбоамид, 3-пиридинкарбоамид и 4-пиридинкарбоамид).
Ароматическое нитрильное соединение, получаемое способом получения согласно настоящему изобретению, является продуктом реакции дегидратации, соответствующим вышеописанному ароматическому амидному соединению, как ясно из описанной выше формулы реакции. Соответственно, частные примеры ароматического нитрильного соединения, получаемого в способе получения согласно настоящему изобретению, включают в себя гетероарилнитрильное соединение. Предпочтительные частные примеры гетероарилнитрильного соединения включают в себя соединения, имеющие пиридиновое кольцо, такие как цианопиридин (2-цианопиридин, 3-цианопиридин и 4-цианопиридин).
[0018]
(Катализатор)
В связи этим катализатор, используемый в вышеописанной реакции дегидратации согласно настоящему изобретению, предпочтительно включает оксид щелочного металла (K, Li, Na, Rb, Cs). В частности, в качестве катализатора, используемого в вышеописанной реакции, предпочтительно используется катализатор, включающий оксид по меньшей мере одного из Na, K, Rb и Cs (цезий). Кроме того, в качестве носителя вышеописанного катализатора может использоваться вещество, обычно служащее носителем катализатора, однако в результате исследования различных носителей предпочтительно включать SiO2 или ZrO2.
[0019]
Ниже описывается пример способа получения катализатора, используемого в реакции дегидратации согласно настоящему изобретению. В случае, когда носитель представляет собой SiO2, может быть использован коммерчески доступный порошковый или сферический SiO2, при этом предпочтительно подогнать размер частиц, например, до 4,0 мм или меньше, так что активный металл может переноситься равномерно, и выполнять предварительную термообработку на воздухе при 700°С в течение 1 часа для удаления влаги. Существуют продукты SiO2 с различными характеристиками, однако чем больше удельная площадь поверхности, тем лучше, поскольку активный металл может быть высокодисперсным, и количество получаемого ароматического нитрильного соединения увеличивается. В частности, удельная площадь поверхности предпочтительно составляет 300 м2/г или более. Однако удельная площадь поверхности катализатора после приготовления может быть уменьшена так, чтобы быть меньше удельной площади поверхности только SiO2 из-за взаимодействия между SiO2 и активным металлом или т.п. В этом случае удельная площадь поверхности катализатора после получения предпочтительно составляет 150 м2/г или более. Оксид металла, служащий в качестве активного вещества, может быть перенесен в соответствии со способом пропитки, таким как способ пропитки по влагоемкости и способ сушки выпариванием.
[0020]
Достаточно, если соль металла, являющаяся предшественником катализатора, является водорастворимой, и если это щелочной металл, то могут быть использованы различные соединения, такие как карбонат, гидрокарбонат, хлористая соль, нитрат и силикат. Носитель пропитывают водным раствором предшественника основного металла с последующей сушкой и термообработкой, затем полученный продукт может быть использован в качестве катализатора. Температура термообработки варьируется в зависимости от используемого предшественника, но предпочтительно составляет от 400 до 600°С.
[0021]
Кроме того, в качестве катализатора, используемого в настоящем изобретении, предпочтителен катализатор, в котором только один или более оксидов щелочных металлов переносятся на носителе, состоящем из одного или обоих из SiO2 и ZrO2, однако кроме вышеописанных элементов, катализатор может содержать неизбежную примесь, примешанную в течение процесса получения катализатора или т.п. Однако целесообразно подавлять примешивание примеси, насколько это возможно.
[0022]
В этой связи используемый в настоящем изобретении катализатор, в котором оксид металла, служащий активным веществом, переносится на носителе, может быть либо в виде порошка или формованного тела, при этом в случае формованного тела он может быть любым из сферического типа, типа дробинки, цилиндрического типа, кольцевого типа, дискового типа, гранулированного типа и т.д. Кроме того, катализатор может быть фиксирован на несущей конструкции, такой как сотовый носитель, и использован.
[0023]
Размер носителя не ограничен, однако при использовании сферического носителя средний диаметр частиц катализатора (диаметр носителя) предпочтительно составляет от 0,01 до 8,0 мм, более предпочтительно от 0,03 до 6,0 мм и еще более предпочтительно от 0,05 до 5,0 мм. За счет использования катализатора, диаметр носителя которого относительно больше среднего диаметра (диаметр носителя) частиц катализатора, используемого в жидкофазной реакции, как описано выше, обеспечивается проточный канал реакционной подложки в газофазной реакции, и может быть обеспечена высокая объемная скорость. Следует отметить, что при использовании псевдоожиженного слоя в газофазной реакции предпочтительно используют катализатор с относительно небольшим диаметром частиц, так что катализатор может перемешиваться с небольшим количеством газового потока, и что при использовании неподвижного слоя в газофазной реакции предпочтительны относительно большой диаметр или форма частиц, так что возникновение потерь давления может быть предотвращено, насколько это возможно.
Диаметр носителя, т.е. средний диаметр частиц катализатора, относится к среднему диаметру частиц всего катализатора, включая каталитический компонент и носитель. Значение диаметра носителя катализатора измеряется на основе способа просеивания: Тестовое просеивание - Общие требования, определенные в JISZ8815 или т.п.
[0024]
Кроме того, переносимое количество катализатора может быть задано подходящим образом, однако, исходя из общей массы катализатора, переносимое количество при превращении металла активных частиц, таких как оксид щелочного металла, предпочтительно составляет от 0,05 до 2,0 ммоль/г, более предпочтительно от 0,10 до 1,5 ммоль/г и еще более предпочтительно от 0,30 до 1,0 ммоль/г. Кроме того, количество катализатора, используемого в течение реакции, может быть также задано подходящим образом.
[0025]
(Реакционная система и реакционная емкость)
В способе получения ароматического нитрильного соединения согласно настоящему изобретению предпочтительно использовать газофазную реакцию, в которой амидное соединение в виде газа или тумана пропускают через слой катализатора вместе с инертным газом, и к нему может быть применено устройство газофазной реакции с неподвижным слоем или псевдоожиженным слоем. Таким образом, на этапе контакта, на котором ароматическое амидное соединение приводят в контакт с катализатором в газовой фазе, предпочтительно используют ароматическое амидное соединение в полностью испаренном состоянии, однако также может быть использовано ароматическое амидное соединение в состоянии, в котором туман частично включен в газ.
[0026]
Целесообразно выполнять способ получения ароматического нитрильного соединения при удалении получаемой побочно воды посредством реакции дегидратации. Авторы настоящего изобретения провели тщательные исследования и обнаружили, что, например, когда испарительная камера присоединена к верхней части реакционной трубы, в которую помещен катализатор, ароматическое амидное соединение вводят в испарительную камеру, и амидное соединение проходит через слой катализатора в газовой фазе в течение короткого времени, может быть увеличено количество получаемого ароматического нитрильного соединения и может быть подавлено образование побочного продукта.
[0027]
(Инертный газ)
В вышеописанной реакции дегидратации предпочтительно приводить инертный газ в контакт с катализатором вместе с ароматическим амидным соединением в газовой фазе. Частные примеры инертного газа, используемого в реакции дегидратации, включают в себя газообразный азот и инертные газы, такие как гелий и аргон, и предпочтительно используется газообразный азот. Скорость потока и т.д. инертного газа описывается ниже.
[0028]
(Растворитель)
В вышеописанной реакции дегидратации может быть использован растворитель. В частности, на этапе испарения ароматического амидного соединения с высокой температурой плавления путем предварительного смешивания ароматического амидного соединения с совместимым с ним растворителем и подачи этой смеси может быть предотвращена проблема закупоривания из-за осаждения ароматического амидного соединения. То есть предпочтительно использовать растворитель, совместимый с ароматическим амидным соединением в качестве мишени реакции дегидратации. Растворитель, совместимый с ароматическим амидным соединением, включает не только растворитель, который может быть смешан с ароматическим амидным соединением в любом соотношении, но также растворитель, который может растворять ароматическое амидное соединение в качестве мишени только до заданной степени растворимости, в количестве, при котором ароматическое амидное соединение может быть растворено до концентрации, равной или меньшей верхнего предела степени растворимости при заданной температуре. Кроме того, предпочтительно также испарять и использовать растворитель в реакции дегидратации (этап контакта). Температура кипения растворителя, испаряемого и используемого в вышеописанной реакции дегидратации, предпочтительно составляет от 20 до 300°С, более предпочтительно от 80 до 250°С, и еще более предпочтительно от 110 до 200°С при нормальном давлении.
[0029]
Частные примеры растворителя, используемого в реакции дегидратации, включают в себя пиридиновое соединение, кетоновое соединение, соединение простого эфира, сложноэфирное соединение и спирт, предпочтительно используют пиридиновое соединение, кетоновое соединение и т.д.
Примеры пиридинового соединения, т.е. соединения, имеющего пиридиновый скелет, служащего в качестве растворителя, включают в себя 2-алкилпиридин, 3-алкилпиридин и 4-алкилпиридин, такой как 4-метилпиридин и пиридин. В частности, в реакции дегидратации, в которой пиридин может образовываться в качестве побочного продукта, при использовании пиридинового соединения в качестве растворителя с предварительно заданными предпочтительными условиями не требуется этап удаления пиридина в качестве основного побочного продукта.
Кроме того, примеры кетонового соединения, служащего в качестве растворителя, включают в себя соединение циклического кетона, такое как циклопентанон, циклогексан и ацетон.
[0030]
В качестве растворителя для реакции дегидратации предпочтительно использовать растворитель, состоящий только из одного или более веществ, выбранных из вышеописанных соединений, однако также может быть использован смешанный растворитель, дополнительно содержащий другое соединение.
[0031]
(Условия реакции дегидратации, включая этап контакта)
Как описано выше, способ получения ароматического нитрильного соединения содержит этап контакта для приведения ароматического амидного соединения в контакт с катализатором в газовой фазе в течение реакции дегидратации.
На этапе контакта температура, при которой ароматическое амидное соединение и т.д. приводят в контакт с катализатором, предпочтительно составляет 170°С или выше, но ниже 300°С. Температура составляет, например, 180°С или выше, но ниже 290°С, или 190°С или выше, но ниже 280°С, более предпочтительно 210°С или выше, но ниже 280°С и еще более предпочтительно 220°С или выше, но ниже 260°С. Считается, что температура, при которой ароматическое амидное соединение и т.д. приводят в контакт с катализатором, равна, например, температуре внутри реакционной трубы (реакционной емкости), к которой прикреплен катализатор.
Кроме того, продолжительность приведения ароматического амидного соединения в контакт с катализатором на этапе контакта и в случае, когда инертный газ, растворитель и т.д. включены вместе с ароматическим амидным соединением, продолжительность приведения смешанного газа этих газовых композиций в контакт с катализатором предпочтительно составляют 0,001 сек или более, или 0,005 сек или более, например 0,01 сек или больше, но меньше 10 сек. Вышеописанная продолжительность контакта с катализатором более предпочтительно составляет 0,1 сек или больше, но меньше 5 сек, и еще более предпочтительно 0,5 сек или больше, но меньше 2 сек.
Следует отметить, что продолжительность контакта с катализатором представляет собой среднюю продолжительность, в течение которой газ ароматического амидного соединения или вышеописанный смешанный газ проходит через слой катализатора, и она рассчитывается по формуле: Продолжительность контакта с катализатором (сек) = Высота слоя катализатора в реакторе (см) ÷ Линейная скорость газа в реакторе (см/сек).
[0032]
На этапе контакта молярное отношение между скоростью потока ароматического амидного соединения и скоростью потока газовой композиции предпочтительно составляет от 1:0 до 1:200, более предпочтительно от 1:1 до 1:100 и еще более предпочтительно от 1:1 до 1:20.
Объемная скорость (SV) всего газового компонента на этапе контакта предпочтительно составляет от 1000 до 50000 (час-1), более предпочтительно от 1500 до 30000 (час-1) и еще более предпочтительно от 2000 до 25000 (час-1). Кроме того, при выполнении этапа контакта при пониженном давлении объемная скорость (SV) всего газового компонента предпочтительно составляет от 1000 до 1000000 (час-1), более предпочтительно от 1500 до 700000 (час-1) и еще более предпочтительно от 1900 до 504000 (час-1).
[0033]
Касательно условий реакции дегидратации, давление может находиться в диапазоне от повышенного давления (например, 506,5 (кПа)) до пониженного давления (например, 0,1 (кПа)), но без конкретного ограничения.
Например, давление реакции составляет от 303,9 до 0,7 (кПа), предпочтительно от 202,6 до 0,9 (кПа) и более предпочтительно от 101,3 до 1,0 (кПа).
[0034]
Кроме того, предпочтительно дегидратировать ароматическое амидное соединение, когда оно находится в виде жидкости перед испарением. При использовании молекулярного сита в качестве дегидратирующего средства его тип и форма конкретно не ограничиваются, но, например, могут использоваться сферические или гранулированные молекулярные сита 3A, 4A, 5A и т.д., как правило имеющие высокую водопоглотительную способность. Например, подходящим образом может быть использовано ZEOLUM, производимое Tosoh Corporation.
[0035]
(Пример побочного продукта в реакции дегидратации)
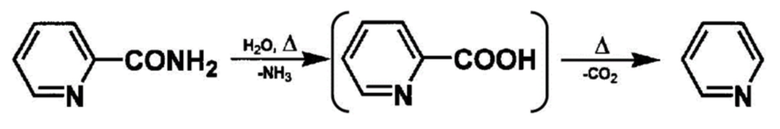
[0036]
Считается, что в реакции дегидратации ароматического амидного соединения пиридин получают побочно через ароматическую карбоновую кислоту из-за разложения вышеописанного ароматического амидного соединения. Однако в реакционном растворе почти не образуется побочный продукт, такой как пиридин, показанный в вышеописанной формуле, после реакции дегидратации с использованием этапа контакта согласно настоящему изобретению.
[0037]
<2. Способ получения карбонатного эфира с использованием ароматического нитрильного соединения>
За счет использования этапа контакта в газовой фазе при регенерации ароматического амидного соединения с получением ароматического нитрильного соединения посредством реакции дегидратации, была успешно увеличена скорость реакции для значительного уменьшения продолжительности реакции. Таким образом, стало возможным достижение баланса между скоростью регенерации ароматического амидного соединения с получением ароматического нитрильного соединения посредством реакции дегидратации и скоростью синтеза карбонатного эфира из CO2 и спирта с использованием ароматического нитрильного соединения, и стала возможной реализация этих реакций в виде серии промышленных процессов. Соответственно, за счет применения этого открытия к способу получения карбонатного эфира авторами настоящего изобретения был успешно разработан описанный ниже способ получения карбонатного эфира.
[0038]
(Первый этап реакции)
Первый этап реакции в способе получения карбонатного эфира согласно настоящему изобретению включает реакцию, в которой непосредственно проводят реакцию спирта с диоксидом углерода в присутствии твердого катализатора, содержащего, например, CeO2 (оксид церия) и т.д., и ароматического нитрильного соединения для получения карбонатного эфира (реакция получения карбонатного эфира).
[0039]
На этом этапе, когда проводят реакцию спирта с диоксидом углерода, также получают воду в дополнение к карбонатному эфиру и за счет реакции гидратации ароматического нитрильного соединения, присутствующего в реакционной системе, и полученной воды получают ароматическое амидное соединение, причем полученная вода может быть удалена из реакционной системы или восстановлена. За счет эффективного удаления воды из реакционной системы таким способом можно способствовать получению карбонатного эфира. Например, как показано в формуле ниже.
[0040]
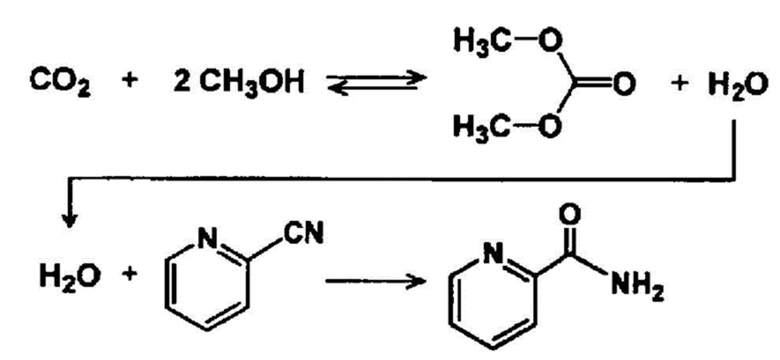
[0041]
(Спирт)
В этой связи в качестве спирта может быть использован любой спирт, выбранный из одного или более из первичного спирта, вторичного спирта и третичного спирта. Например, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, 1-пентанол, 1-гексанол, 1-гептанол, 1-октанол, 1-нонанол, аллиловый спирт, 2-метил-1-пропанол, циклогексанметанол, бензиловый спирт, этиленгликоль, 1,2-пропандиол и 1,3-пропандиол предпочтительно используют ввиду получения высокого выхода продукта и высокой скорости реакции. В этих случаях получаемые карбонатные эфиры представляют собой соответственно диметилкарбонат, диэтилкарбонат, дипропилкарбонат, диизопропилкарбонат, дибутилкарбонат, дипентилкарбонат, дигексилкарбонат, дигептилкарбонат, диоктилкарбонат, динонанкарбонат, диаллилкарбонат, ди-2-метилпропилкарбонат, дициклогексанметилкарбонат, дибензилкарбонат, этиленкарбонат, 1,2-пропиленкарбонат и 1,3-пропиленкарбонат.
Кроме того, на первом этапе реакции предпочтительно используют спирт, содержащий от 1 до 6 атомов углерода, и более предпочтительно используют спирт, содержащий от 2 до 4 атомов углерода. В частности, при использовании карбонатного эфира, полученного в качестве исходного материала для диарилкарбоната, число атомов углерода в спирте предпочтительно устанавливают в вышеописанном диапазоне.
Кроме того, на первом этапе реакции предпочтительно используют одноатомный спирт или двухатомный спирт.
[0042]
(Катализатор для получения карбонатного эфира)
Кроме того, на первом этапе реакции при получении карбонатного эфира предпочтительно использовать один или оба из CeO2 и ZrO2 в качестве твердого катализатора. Например, предпочтительно использовать только CeO2, только ZrO2, смесь CeO2 и ZrO2, твердый раствор или сложный оксид CeO2 и ZrO2 или т.п., и в частности предпочтительно использовать только CeO2. Коэффициент смешивания CeO2 и ZrO2 в твердом растворе или его сложном оксиде как правило составляет 50:50, но может быть соответствующим образом изменен.
[0043]
В этой связи катализатор, используемый на первом этапе реакции, может быть либо в виде порошка или формованного тела, и в случае формованного тела он может быть любым из сферического типа, типа дробинки, цилиндрического типа, кольцевого типа, дискового типа, гранулированного типа и т.д.
[0044]
(Диоксид углерода)
В качестве диоксида углерода, используемого в настоящем изобретении, может быть использован не только диоксид углерода, полученный в виде промышленного газа, но также диоксид углерода, отделенный и уловленный из выхлопных газов предприятий, производящих различные продукты, металлургических заводов, электростанций и т.д.
[0045]
(Растворитель для реакции получения карбонатного эфира)
В реакции получения карбонатного эфира предпочтительно использовать растворитель, имеющий более высокую точку кипения, чем точка кипения получаемого амидного соединения. Более предпочтительно растворитель в реакции получения карбонатного эфира содержит по меньшей мере один из диалкилбензола, алкилнафталина и дифенилбензола. Частные примеры включают в себя Barrel Process Oil B28AN и Barrel Process Oil B30 (изготавливаемые Matsumura Oil Co., Ltd.), каждый из которых содержит компоненты, включающие в себя диалкилбензол, алкилнафталин, дифенилбензол и т.д.
[0046]
(Отделение перегонкой)
После этой реакции карбонатный эфир в качестве основного продукта, ароматическое амидное соединение в качестве побочного продукта, ароматическое нитрильное соединение, не вступившее в реакцию, и твердый катализатор, такой как CeO2, отделяют перегонкой, тем самым улавливая эти продукты.
[0047]
(Второй этап реакции)
Затем, на втором этапе реакции в настоящем изобретении, ароматическое амидное соединение, получаемое побочно на первом этапе реакции, предпочтительно отделяют от системы, полученной после реакции получения карбонатного эфира, и затем получают ароматическое нитрильное соединение посредством реакции дегидратации. Второй этап реакции соответствует вышеописанному способу получения ароматического нитрильного соединения. В частности, на втором этапе реакции получения карбонатного эфира ароматическое нитрильное соединение получают из ароматического амидного соединения в соответствии со способом, описанном выше в разделе, относящемся к способу получения ароматического нитрильного соединения, тем самым регенерируя ароматическое нитрильное соединение. Соответственно, подробное описание второго этапа реакции опускается.
[0048]
(Повторное использование ароматического нитрильного соединения)
Ароматическое нитрильное соединение, регенерированное на втором этапе реакции, может быть повторно использовано на первом этапе реакции (реакция гидратации).
[0049]
Согласно настоящему изобретению, как описано выше, путем проведения реакции дегидратации ароматического амидного соединения в газовой фазе ароматическое нитрильное соединение может быть эффективно регенерировано из ароматического амидного соединения при подавлении образования побочного продукта. Кроме того, за счет фиксации катализатора в реакционной трубе отпадает необходимость в этапе разделения катализатора на твердую и жидкую фазы, и ароматическое нитрильное соединение может быть легко уловлено. Таким образом, в настоящем изобретении можно избирательно регенерировать ароматическое нитрильное соединение из ароматического амидного соединения и способствовать серии реакций, отделяя соответствующие компоненты только путем дистилляции без разделения катализатора на твердую и жидкую фазы. Соответственно, может быть реализован эффективный процесс, описываемый подробно ниже.
[0050]
<3. Устройство для получения карбонатного эфира>
Ниже посредством частного примера подробно описывается устройство для получения карбонатного эфира, используемого в настоящем изобретении. На фиг. 1 показан пример предпочтительного устройства.
[0051]
(Первый этап реакции)
На первом этапе реакции исходные вещества, т.е. спирт (1-пропанол (PrOH); жидкая фаза), 2-цианопиридин (2-CP; жидкая фаза) и диоксид углерода (CO2; жидкая фаза, может подаваться через вспомогательный насос) непрерывно подают в промежуточную емкость 1 с использованием трубопровода 31 для подачи исходных материалов, трубопровода 15 перекачки СО2 из верхней части колонны для улавливания CO2 (трубопровод перекачки CO2 на верхней стороне колонны для улавливания CO2), трубопровода 19 перекачки жидкости из верхней части колонны для улавливания карбонатного эфира (трубопровод перекачки жидкости на верхней стороне колонны для улавливания карбонатного эфира) и трубопровода 21 перекачки жидкости из верхней части колонны отделения амидов (трубопровод перекачки жидкости на верхней стороне колонны отделения амидов). Этот смешанный раствор исходных материалов циркулирует между реактором 2 для получения карбонатного эфира проточного типа с неподвижным слоем, в котором твердый катализатор (твердая фаза), состоящий из одного или обоих из CeO2 и ZrO2, прикреплен к материалу-подложке (первая реакционная часть), и промежуточной емкостью 1 через трубопровод 11 циркуляции первого реакционного раствора и трубопровод 12 циркуляции второго реакционного раствора с использованием насоса (не показан), обеспечивая тем самым синтез дипропилкарбоната (DPrC). Реакционный раствор, содержащий DPrC, непрерывно отводят из трубопровода 12 циркуляции второго реакционного раствора в том же количестве, что и количество исходных материалов, подаваемых в промежуточную емкость 1, улавливают с использованием трубопровода 13 и подают на этап улавливания DPrC. PrOH и CO2 улавливают из реакционного раствора и перемещают в буферный резервуар 1 соответственно через трубопровод 19 перекачки жидкости из верхней части колонны для улавливания карбонатного эфира и трубопровод 15 перекачки СО2 из верхней части колонны для улавливания СО2, тем самым повторно используя их. В начале этой реакции используют новый 2-цианопиридин, однако 2-цианопиридин, регенерируемый из 2-пиколинамида, отделяют и очищают в колонне 6 отделения амидов и перемещают в промежуточную емкость 1 через верхний трубопровод 21 перекачки жидкости из верхней части колонны отделения амидов, тем самым повторно используя его.
[0052]
В качестве устройства прямого синтеза для карбонатного эфира с использованием CeO2, ZrO2 и т.д. в качестве твердого катализатора (реактор 2 для получения карбонатного эфира), может быть использован любой из проточных реакторов, такой как реактор периодического действия, реактор полупериодического действия, емкостный реактор непрерывного действия и трубчатый реактор. Если катализатор прикреплен к реактору, нет необходимости фильтровать и отделять катализатор. По этой причине предпочтителен реактор с неподвижным слоем.
[0053]
(Температура реакционного раствора)
Температура реакционного раствора в реакторе 2 для получения карбонатного эфира предпочтительно составляет от 50 до 300°С. Если температура реакционного раствора ниже 50°С, то скорость реакции является низкой, реакция синтеза карбонатного эфира и реакция гидратации с 2-цианопиридином почти не протекают, и эффективность получения карбонатного эфира, как правило, является низкой. Если температура реакционного раствора выше 300°С, то скорость каждой реакции является высокой, однако карбонатный эфир легко разлагается или денатурируется, и 2-пиколинамид легко реагирует со спиртом. По этой причине выход карбонатного эфира, как правило, является низким. Температура реакционного раствора более предпочтительно составляет от 100 до 150°С. Однако, поскольку считается, что идеальная температура реакционного раствора варьируется в зависимости от типа и количества твердого катализатора, а также количества и соотношения исходных материалов (спирт и 2-цианопиридин), целесообразно установить оптимальные условия подходящим образом. Поскольку предпочтительная температура реакционного раствора составляет от 100 до 150°С, целесообразно предварительно нагреть исходные материалы (спирт и 2-цианопиридин) водяным паром или т.п. на стадии перед реактором для получения карбонатного эфира.
[0054]
(Давление реакции)
Давление реакции в реакторе 2 для получения карбонатного эфира предпочтительно составляет от 0,1 до 20 МПа (абсолютное давление). Если давление реакции ниже 0,1 МПа (абсолютное давление), то требуется устройство уменьшения давления. Как следствие, оборудование усложняется, а стоимость увеличивается, и, кроме того, требуется энергия для уменьшения давления, что приводит к снижению энергетической эффективности. Если давление реакции выше 20 МПа, то реакция гидратации с 2-цианопиридином не протекает легко, что приводит к уменьшению выхода карбонатного эфира. Кроме того, требуется энергия для увеличения давления, что приводит к снижению энергетической эффективности. С точки зрения увеличения выхода карбонатного эфира давление реакции более предпочтительно составляет от 0,5 до 15 МПа (абсолютное давление) и еще более предпочтительно от 1,0 до 10 МПа (абсолютное давление).
[0055]
(Количество 2-цианопиридина)
2-цианопиридин, используемый для реакции гидратации, предпочтительно используют в молярном количестве, в 0,1-5 раз большем теоретического молярного количества воды, получаемой побочно при реакции спирта и CO2 в качестве исходных материалов, и предпочтительно его вводят в реактор перед реакцией. Молярное количество 2-цианопиридина более предпочтительно в 0,2-3 раза и в частности предпочтительно в 0,3-1,5 раза больше теоретического молярного количества воды, получаемой побочно при реакции спирта и СО2 в качестве исходных материалов. Если молярное количество 2-цианопиридина слишком мало, поскольку количество 2-цианопиридина, участвующего в реакции гидратации, мало, то выход карбонатного эфира может быть уменьшен. Вместе с тем, если 2-цианопиридин вводят в избыточном молярном количестве относительно спирта в качестве исходного материала, то побочная реакция 2-цианопиридина увеличивается, и, таким образом, это нежелательно. Поскольку считается, что идеальные количества спирта и 2-цианопиридина относительно твердого катализатора варьируются в зависимости от типа и количества твердого катализатора, типа спирта и соотношения между спиртом и 2-цианопиридином, целесообразно установить оптимальные условия подходящим образом.
[0056]
(Отделение продуктов реакции)
Предпочтительно отделение продуктов реакции полностью выполняют путем перегонки. После реакции в реакторе 2 для получения карбонатного эфира реакционный раствор 13 перемещают в колонну 3 для улавливания CO2. Смесь PrOH, DPrC, 2-цианопиридина и 2-пиколинамида улавливают из нижней части колонны 3 для улавливания CO2 через трубопровод 14 перекачки жидкости из нижней части колонны для улавливания CO2, и CO2 улавливают из верхней части колонны 3 для улавливания CO2 через трубопровод 15 перекачки CO2 из верхней части колонны для улавливания CO2. Улавливаемый CO2 перемещают в промежуточную емкость 1 и повторно используют в реакции в реакторе 2 для получения карбонатного эфира.
[0057]
Смесь, улавливаемую из колонны 3 для улавливания CO2, подают в колонну 4 отделения дегидратирующего средства через трубопровод 14 перекачки жидкости из нижней части колонны для улавливания CO2. Смесь 2-цианопиридина и 2-пиколинамида улавливают из нижней части колонны 4 отделения дегидратирующего средства через трубопровод 16 перекачки жидкости из нижней части колонны отделения дегидратирующего средства, а PrOH и DPrC улавливают из верхней части колонны 4 отделения дегидратирующего средства через трубопровод 17 перекачки жидкости из верхней части колонны отделения дегидратирующего средства.
[0058]
Смесь, улавливаемую из нижней части колонны 4 отделения дегидратирующего средства, перемещают в колонну 6 отделения амидов через трубопровод 16 перекачки жидкости из нижней части колонны отделения дегидратирующего средства. 2-пиколинамид (20) улавливают из нижней части колонны 6 отделения амидов, и 2-цианопиридин улавливают из верхней части колонны 6 отделения амидов. Улавливаемый 2-цианопиридин перемещают в промежуточную емкость 1 через трубопровод 21 перекачки жидкости из верхней части колонны отделения амидов и повторно используют в реакции в реакторе 2 для получения карбонатного эфира.
[0059]
PrOH и DPrC, улавливаемые из верхней части колонны 4 отделения дегидратирующего средства, перемещают в колонну 5 для улавливания карбонатного эфира через трубопровод 17 перекачки жидкости из верхней части колонны отделения дегидратирующего средства. DPrC улавливают из нижней части колонны 5 для улавливания карбоната эфира через трубопровод 18 перекачки жидкости из нижней части колонны для улавливания карбонатного эфира, и PrOH улавливают из верхней части колонны 5 для улавливания карбонатного эфира через трубопровод 19 перекачки жидкости из верхней части колонны для улавливания карбонатного эфира. Улавливаемый PrOH перемещают в промежуточную емкость 1 и повторно используют в реакции в реакторе 2 для получения карбонатного эфира.
[0060]
(Второй этап реакции)
На втором этапе реакции получают 2-цианопиридин за счет реакции дегидратации 2-пиколинамида в газофазном реакторе 8 для регенерации нитрила.
2-пиколинамид, улавливаемый из колонны 6 отделения амидов, перемещают в испаритель 7 через трубопровод 20 перекачки жидкости из нижней части колонны отделения амидов. Предпочтительно его смешивают с инертным газом, которым является азот или т.п., полученную смесь нагревают до температуры вблизи точки кипения амида для получения газа или смешанного газа, состоящего из газа и капель, и перемещают в газофазный реактор 8 для регенерации нитрила через трубопровод 22, соединяющий газофазный реактор регенерации нитрила и испаритель. Форма испарителя 7 для испарения амидного соединения и т.д. конкретно не ограничивается, и может быть использован любой испаритель эжекторного типа, испаритель контактного типа, барботажное устройство и т.д.
[0061]
В устройстве для получения нитрильного соединения, используемого в настоящем изобретении (газофазный реактор 8 для регенерации нитрила), 2-пиколинамид и предпочтительно инертный газ, который представляет собой азот или т.п., и т.д. вводят в контакт с катализатором, содержащим переносимый оксид основного металла в газовой фазе, чтобы вызвать реакцию дегидратации 2-пиколинамида. В результате этой реакции дегидратации получают 2-цианопиридин. При перемещении амидного соединения с целью предотвращения проблемы закупоривания амидное соединение может быть растворено в переносящем растворителе для перемещения жидкости. При использовании переносящего растворителя для амида предпочтительно, чтобы растворитель также испарялся вместе с амидом для проведения газофазной реакции регенерации нитрила. В этом случае вместо инертного газа может быть использован пар растворителя.
[0062]
Форма газофазного реактора 8 для регенерации нитрила конкретно не ограничивается. Предпочтительно использовать газофазную реакцию, в которой амидное соединение в виде газа или тумана пропускают через слой катализатора вместе с инертным газом или т.п., и к нему может быть применено устройство газофазной реакции с неподвижным слоем, псевдоожиженным слоем или т.п.
Таким образом, для эффективного способствования реакции дегидратации путем приведения 2-пиколинамида в контакт с катализатором и т.д. в газовой фазе соответственно используют вышеописанные различные условия реакции, относящиеся к реакции дегидратации, или условия реакции, описанные в примерах ниже.
[0063]
Смешанный газ, содержащий 2-цианопиридин, перемещают из газофазного реактора 8 в устройство 9 отделения H2O через трубопровод 23 перекачки продуктов газофазной реакции. Воду и газообразный азот отделяют от смешанного газа в устройстве 9 отделения H2O, и улавливаемые 2-цианопиридин и 2-пиколинамид перемещают из устройства 9 отделения H2O в колонну 6 отделения амидов через трубопровод 24 перекачки сред с высокой температурой кипения. Затем 2-пиколинамид улавливают из нижней части колонны 6 отделения амидов через трубопровод 20 перекачки жидкости из нижней части колонны отделения амидов и перемещают в испаритель 7, и 2-цианопиридин улавливают из верхней части колонны 6 отделения амидов. Улавливаемый 2-цианопиридин перемещают в промежуточную емкость 1 через трубопровод 21 перекачки жидкости из верхней части колонны отделения амидов и повторно используют в реакции в реакторе 2 для получения карбонатного эфира.
[0064]
Затем газообразный азот и воду, отделенные в устройстве 9 отделения H2O, перемещают в устройство 10 улавливания N2 через трубопровод 25 перекачки сред с низкой температурой кипения, и воду отделяют в устройстве 10 улавливания N2 для улавливания газообразного азота. Воду, отделенную в устройстве 10 улавливания N2, перемещают наружу устройства получения карбонатного эфира через трубопровод 26 перекачки воды из устройства улавливания N2. Газообразный азот, улавливаемый в устройстве 10 улавливания N2, перекачивают в испаритель 7 через трубопровод 27 перекачки из устройства улавливания N2, и может быть использован в газофазной реакции. В случае использования переносящего растворителя для амидного соединения этап улавливания растворителя применяют отдельно, тем самым повторно используя растворитель для перемещения амидного соединения. Форма каждого из устройства 9 отделения H2O и устройства 10 улавливания N2 конкретно не ограничивается, и может быть использовано любое из охлаждающей системы, устройства мембранного отделения и т.д.
[0065]
Как описано выше, в настоящем изобретении дегидратации амидного соединения может способствовать этап контакта в газовой фазе, и, кроме того, продукт реакции и повторно используемые соединения могут быть отделены путем дистилляции или т.п. без разделения твердой и жидкой фаз. По этой причине, согласно настоящему изобретению, карбонатный эфир может быть эффективно получен за меньшее количество этапов получения с одновременным упрощением установки.
ПРИМЕРЫ
[0066]
Настоящее изобретение описывается более подробно ниже посредством примеров, однако настоящее изобретение не ограничивается ими. Ниже сначала описываются примеры и сравнительные примеры способа получения цианопиридина.
[0067]
(Пример 1)
SiO2 в качестве носителя (изготовлен Fuji Silysia Chemical Ltd., CARiACT, Q-6 (диаметр носителя: от 0,075 до 0,15 мм)) предварительно термообрабатывали при 700°С в течение примерно 1 часа. Затем для переноса Cs в качестве щелочного металла приготовляли водный раствор с использованием Cs2CO3 (изготовлен Wako Pure Chemical Industries, Ltd.) так, чтобы конечное количество переносимого металлического Cs составляло 0,5 ммоль/г, и пропитывали SiO2 водным раствором. Затем полученный материал сушили при 110°С в течение примерно 6 часов и затем термообрабатывали при 500°С в течение примерно 3 часов с получением при этом катализатора Cs2O/ SiO2.
Реакционную трубу 40, выполненную из SUS 304, с внутренним диаметром 10,7 мм и длиной 30 см заполняли катализатором, полученным вышеописанным способом получения (см. фиг. 2). Кроме того, непосредственно над реакционной трубой 40 размещали испарительную камеру 42, выполненную из SUS 304, с внутренним диаметром 10,7 мм и длиной 30 см, и заполненную кольцом Рашига.
[0068]
10 г 2-пиколинамида (2-PA) растворяли в 4-метилпиридине, служащем переносящим растворителем (90 г), и помещали полученную смесь в емкость 44 для исходного материала. Емкость 44 для исходного материала помещали на высокоточные весы, в раствор исходного материала опускали всасывающую трубу с фильтром, и уравновешивали весы.
Через испарительную камеру 42, соединенную с емкостью 44 для исходного материала, и реакционную трубу 40 пропускали азот со скоростью потока 1000 мл/мин, и нагревали испарительную камеру 42 и реакционную трубу 40 до 230°С соответственно с помощью колбовых нагревателей 46 и 48. Кроме того, трубопровод 50 для подачи азота и трубопровод на выходной стороне реакционной трубы 40 поддерживали при температуре 150°С. Для улавливания продукта реакции улавливающую емкость 52 с охлаждением водой расположили на первой стадии, улавливающую емкость 54 с охлаждением сухим льдом/метанолом расположили на второй стадии, и улавливающую емкость с охлаждением жидким азотом (не показана) расположили на последней стадии.
[0069]
После того, как температура слоя катализатора стала стабильной, включали плунжерный насос 56 и проводили реакцию в течение времени, указанного в столбце продолжительность контакта с катализатором в таблице ниже. В течение реакции предотвращали чрезмерное повышение давления с помощью ограничителя 58 давления, так что давление в реакционной системе устройства для получения цианопиридина, показанного на фиг. 2, не превышало 506,5 (кПа).
[0070]
Условия для вышеописанной контактной реакции (реакция дегидратации) были такими, как представлено в Таблице 1. После завершения реакции полученный продукт реакции улавливали и анализировали с помощью GC-FID.
[0071]
Условия анализа результатов контактной реакции (реакция дегидратации) были такими, как показано ниже.
[Условия анализа]
(GC-FID)
Shimadzu GC-2014, колонна: TC-17 (длина: 30 м, внутренний диаметр: 0,25 мм ID, толщина жидкой фазы: 0,25 мкм)
Температура испарительной камеры: 250°С, детектор: 260°С, носитель He: 175 кПа, скорость потока колонны: 2,5 мл/мин, деление потока: 50
Температурная программа: [выдерживали при 70°С в течение 5 мин] → [повышали до 190°С, 12°/мин] → [выдерживали при 190°С в течение 5 минут] → [повышали до 250°С, 12°С/мин] → [выдерживали при 250°С в течение 10 мин]
[0072]
[24-часовой выход на 1 г катализатора (ммоль/24 ч⋅г)] =
(Количество улавливаемого продукта после 24-часовой реакции (ммоль))/( Количество катализатора (г))
[Объемная скорость: SV (час-1)] =
(Количество газа, прошедшего через слой катализатора (л⋅час-1))/(Количество катализатора (л))
Количество газа: сумма объемов газов азота, 2-PA и растворителя (л⋅час-1)
[Высота заполнения катализатором (высота слоя катализатора в реакторе)]: высота катализатора, заполненного в реакционной трубе (см)
[Продолжительность контакта с катализатором (сек)] =
[Высота заполнения катализатором (см)]/[ Линейная скорость (линейная скорость газа в реакторе) (см/сек)]
[0073]
[Диаметр носителя катализатора]
Значение диаметра носителя катализатора измерялось на основе способа просеивания: Тестовое просеивание - Общие требования, определенные в JIS Z8815.
[0074]
(Примеры 2-15)
В Примере 2 или далее реакцию проводили так же, как в Примере 1, за исключением того, что условия были изменены, как представлено в Таблице 1. Результаты представлены в Таблице 1.
В каждом из примеров и сравнительных примеров активным компонентом катализатора был Cs2O; в качестве носителя использовали CARiACT Q-6 (основным компонентом является SiO2), изготовленный Fuji Silysia Chemical Ltd.; и количество активного металла, переносимого катализатором (переносимое количество при превращении металла активных веществ в расчете на общую массу катализатора), составляло 0,5 ммоль/г. Кроме того, в каждом из примеров и сравнительных примеров использовали 2-пиколинамид (2-PA) в качестве ароматического амидного соединения.
[0075]
(Пример 16)
В отличие от примеров 1-15, в примере 16 контактную реакцию (реакция дегидратации) ароматического амидного соединения проводили при пониженном давлении. В частности, ее выполняли так, как описано ниже.
100 г 2-пиколинамида (2-PA) растворяли в циклопентаноне, служащем переносящим растворителем (900 г), и размещали эту смесь в емкость для исходного материала.
Пропускали азот через реакционную линию (от эжектора 62 к ловушке 64, показанной на фиг. 3), соединенную с емкостью 60 для исходного материала, со скоростью потока 1000 мл/мин. После прекращения подачи азота, к ловушке 64 присоединяли вакуумный насос 66 и снижали давление в реакционной системе до 0,5 кПа. Испаритель 68 и реактор 70 нагревали до 220°С соответственно с помощью электрических печей (не показаны). Затем нагревали трубопровод на выходной стороне реакционной линии до 150°С с использованием ленточного нагревателя (не показан). Для улавливания продукта реакции расположили улавливающую емкость 64 с охлаждением сухим льдом/метанолом (ловушка 64A для первой фракции и улавливающая ловушка 64B на фиг. 3), и рядом с ней расположили улавливающую емкость с охлаждением жидким азотом (не показана).
После того, как температура слоя катализатора (не показан), помещенного в реакторе 70, стала стабильной, запускали плунжерный насос 72 и проводили реакцию. В течение 90 минут с момента, когда внутренняя часть слоя катализатора достигла стационарного состояния, продукт реакции улавливали с помощью улавливающей ловушки 64B. В течение реакции давление в реакционной системе устройства для получения цианопиридина, показанного на фиг. 3, контролировали с помощью регулятора 74 понижения давления и игольчатого клапана 76. Следует отметить, что до достижения внутренней частью слоя катализатора стационарного состояния и после улавливания продукта реакции с помощью улавливающей ловушки 64B после того, как внутренняя часть слоя катализатора достигла стационарного состояния, продукт реакции улавливали с помощью ловушки 64A для первой фракции.
Условия для вышеописанной контактной реакции (реакция дегидратации) были такими, как представлено в Таблице 1. После завершения реакции полученный продукт реакции улавливали и анализировали с помощью GC-FID.
[0076]
(Примеры 17-21)
В Примере 17 или далее реакцию проводили так же, как в Примере 16, за исключением того, что условия были изменены, как представлено в Таблице 1.
[0077]
Результаты примеров 1-21 представлены ниже в Таблице 1.
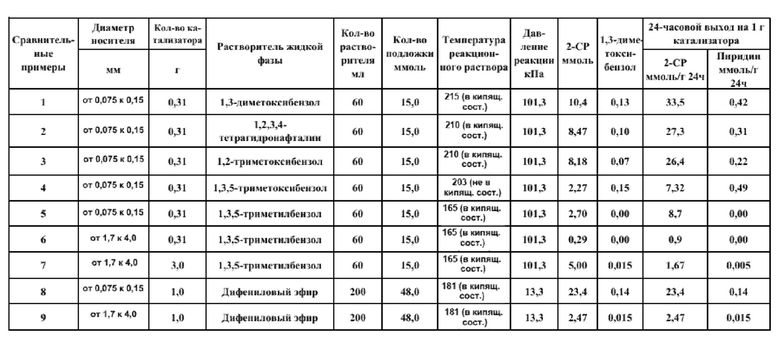
Таблица 1
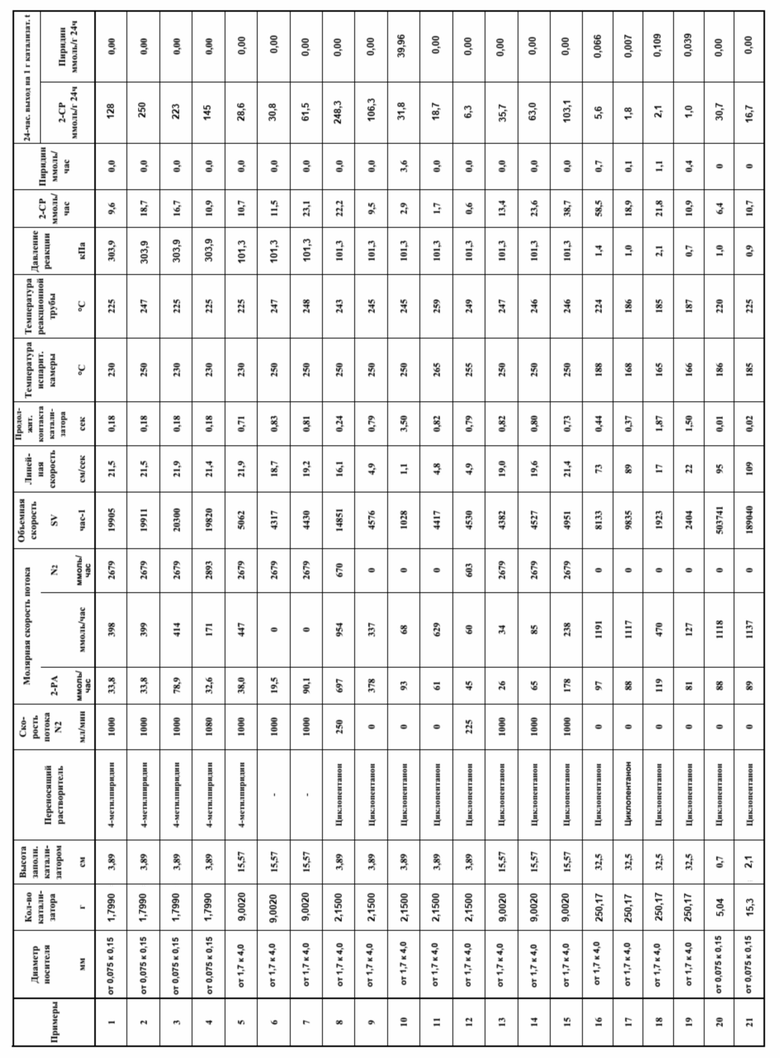
[0078]
(Сравнительные примеры 1-9)
В сравнительных примерах 1-9 использовали растворитель для удаления воды, получаемой в результате реакции, наружу реакционной системы, и проводили реакцию дегидратации в жидкой фазе.
[0079]
(Сравнительный пример 1)
SiO2 в качестве носителя (изготовлен Fuji Silysia Chemical Ltd., CARiACT, Q-6 (диаметр носителя: от 0,075 до 0,15 мм)) предварительно термообрабатывали при 700°С в течение примерно 1 часа. Затем для переноса Cs в качестве щелочного металла приготовляли водный раствор с использованием Cs2CO3 (изготовлен Wako Pure Chemical Industries, Ltd.) так, чтобы конечное количество переносимого металлического Cs составляло 0,5 ммоль/г, и пропитывали SiO2 водным раствором. Затем полученный материал сушили при 110°С в течение примерно 6 часов и затем термообрабатывали при 500°C в течение примерно 3 часов с получением при этом катализатора Cs2O/SiO2.
[0080]
Затем в качестве реактора использовали трехгорлую круглодонную колбу и в реактор вводили магнитную мешалку, вышеописанный катализатор Cs2O/SiO2, 2-пиколинамид (изготовленный Tokyo Chemical Industry Co., Ltd.) и 1,3-диметоксибензол (изготовленный Tokyo Chemical Industry Co., Ltd.).
Затем к реактору прикрепляли термометр и трубу воздушного охлаждения, содержащую 10 г молекулярного сита 4А, и к верхнему концу трубы воздушного охлаждения прикрепляли конденсатор Либиха для создания реакционного устройства.
Затем нагревали реакционный раствор при нормальном давлении и поддерживали в кипящем состоянии, адсорбировали побочно получаемую воду на молекулярном сите без возврата в реактор для проведения дегидратации, тем самым проводя реакцию.
Реакцию начинали, когда реакционный раствор начинал кипеть, и проводили реакцию в течение 24 часов.
После реакции температуру понижали до комнатной температуры. Из реакционного раствора отбирали пробу и разбавляли в два раза этанолом, к нему добавляли 1-гексанол в качестве вещества внутреннего стандарта. Полученное вещество подвергали качественному анализу с помощью GC-MS (газовый хроматограф - масс-спектрометр) и количественному анализу с помощью GC-FID.
[0081]
(Сравнительные примеры 2-7)
В сравнительных примерах 2-7 реакцию проводили так же, как в сравнительном примере 1, за исключением того, что условия были изменены, как представлено в Таблице 2.
[0082]
(Сравнительные примеры 8-9)
В сравнительных примерах 8-9 условия были изменены, как представлено в Таблице 2, и, кроме того, были изменены конфигурация устройства и условия реакции, как описано ниже, тем самым проводя реакцию.
[0083]
SiO2 в качестве носителя (изготовлен Fuji Silysia Chemical Ltd., CARiACT, Q-6 (диаметр носителя: от 0,075 до 0,15 мм)) измельчали до 100 меш или меньше и предварительно термообрабатывали при 700°С в течение примерно 1 часа. Затем для переноса Cs в качестве щелочного металла приготовляли водный раствор с использованием Cs2CO3 (изготовлен Wako Pure Chemical Industries, Ltd.) так, чтобы конечное количество переносимого металлического Cs составляло 0,5 ммоль/г, и пропитывали SiO2 водным раствором. Затем полученный материал сушили при 110°С в течение примерно 6 часов и затем термообрабатывали при 500°С в течение примерно 3 часов с получением при этом катализатора Cs2O/SiO2.
[0084]
Затем в качестве реактора использовали трехгорлую круглодонную колбу и в реактор вводили магнитную мешалку, вышеописанный катализатор Cs2O/SiO2, 2-пиколинамид (изготовленный Tokyo Chemical Industry Co., Ltd.) и дифениловый эфир (изготовленный Tokyo Chemical Industry Co., Ltd.).
Затем к реактору прикрепляли термометр и первую трубу воздушного охлаждения в качестве дистилляционной колонны, к верхнему концу первой трубы воздушного охлаждения прикрепляли дистилляционную головку, оснащенную термометром, и к дистилляционной головке присоединяли вторую трубу воздушного охлаждения, приемник и вакуумный насос для создания устройства для реакционной дистилляции. Следует отметить, что вокруг первой трубы воздушного охлаждения намотали ленточный нагреватель для регулировки температуры. Затем охлаждали охлаждающую ловушку жидким азотом для улавливания испаренного пиридина.
Затем уменьшали давление в устройстве для реакционной дистилляции до 13,3 кПа (100 торр) с использованием вакуумного насоса. Первую трубу воздушного охлаждения нагревали до 60°С, что было выше точки кипения воды и ниже точки кипения дифенилового эфира при давлении реакции. Реакционный раствор поддерживали в кипящем состоянии при 184°С, что было равно или выше точки кипения дифенилового эфира и ниже точки кипения 2-пиколинамида при давлении реакции. Путем регулировки температуры данным способом частично испаренный дифениловый эфир в реакционной системе охлаждали в первой трубе воздушного охлаждения и возвращали в реактор, тогда как получаемую побочно воду перегоняли наружу системы без возврата в реактор, тем самым проводя реакцию.
Реакцию начинали, когда реакционный раствор начинал кипеть, и проводили реакцию в течение 24 часов.
После реакции температуру понижали до комнатной температуры. Из реакционного раствора отбирали пробу и разбавляли в два раза этанолом, к нему добавляли 1-гексанол в качестве вещества внутреннего стандарта. Полученное вещество подвергали качественному анализу с помощью GC-MS (газовый хроматограф - масс-спектрометр) и количественному анализу с помощью GC-FID.
[0085]
Результаты сравнительных примеров 1-9 представлены ниже в Таблице 2.
Таблица 2
[0086]
Выше со ссылкой на прилагаемые чертежи подробно описаны предпочтительные варианты реализации настоящего изобретения, однако настоящее изобретение не ограничивается этими примерами. Очевидно, что специалисты в области техники, относящейся к настоящему изобретению, могут предложить любой из различных измененных или модифицированных примеров в пределах объема технической идеи, изложенной в формуле изобретения, понятно, что такие измененные или модифицированные примеры должным образом включены в объем правовой охраны настоящего изобретения.
СПИСОК НОМЕРОВ ПОЗИЦИЙ
[0087]
1 промежуточная емкость (CO2, PrOH, DPrC, 2-CP, 2-PA)
2 реактор для получения карбонатного эфира (CO2, PrOH, DPrC, 2-CP, 2-PA, фиксированный катализатор)
3 колонна для улавливания CO2 (CO2, PrOH, DPrC, 2-CP, 2-PA)
4 колонна отделения дегидратирующего средства (PrOH, DPrC, 2-CP, 2-PA)
5 колонна для улавливания карбонатного эфира (PrOH, DPrC)
6 колонна отделения амидов (2-CP, 2-PA)
7 испаритель (N2, 2-PA)
8 газофазный реактор для регенерации нитрила (N2, H2O, 2-CP, 2-PA, катализатор)
9 устройство отделения H2O (N2, H2O, 2-CP, 2-PA)
10 устройство для улавливания N2 (N2, H2O)
11 трубопровод циркуляции первого реакционного раствора (CO2, PrOH, DPrC, 2-CP, 2-PA)
12 трубопровод циркуляции второго реакционного раствора (CO2, PrOH, DPrC, 2-CP, 2-PA)
13 трубопровод для отвода реакционного раствора (CO2, PrOH, DPrC, 2-CP, 2-PA)
14 трубопровод перекачки жидкости из нижней части колонны для улавливания CO2 (PrOH, DPrC, 2-CP, 2-PA)
15 трубопровод перекачки СО2 из верхней части колонны для улавливания CO2 (CO2)
16 трубопровод перекачки жидкости из нижней части колонны отделения дегидратирующего средства (2-CP, 2-PA)
17 трубопровод перекачки жидкости из верхней части колонны отделения дегидратирующего средства (PrOH, DPrC)
18 трубопровод перекачки жидкости из нижней части колонны для улавливания карбонатного эфира (DPrC)
19 трубопровод перекачки жидкости из верхней части колонны для улавливания карбонатного эфира (PrOH)
20 трубопровод перекачки жидкости из нижней части колонны отделения амидов (2-PA)
21 трубопровод перекачки жидкости из верхней части колонны отделения амидов (2-CP)
22 трубопровод, соединяющий газофазный реактор для регенерации нитрила и испаритель (N2, 2-PA)
23 трубопровод перекачки продуктов газофазной реакции (N2, H2O, 2-CP, 2-PA)
24 трубопровод перекачки сред с высокой температурой кипения из устройства отделения H2O (2-CP, 2-PA)
25 трубопровод перекачки сред с низкой температурой кипения из устройства отделения H2O (N2, H2O)
26 трубопровод перекачки воды из устройства улавливания N2 (H2O)
27 трубопровод перекачки N2 из устройства улавливания N2 (N2)
31 трубопровод для подачи исходных материалов (CO2, PrOH)
40 реакционная труба
42 испарительная камера
44 емкость для исходного материала
46, 48 колбовый нагреватель
50 трубопровод для подачи газообразного азота
52 улавливающая емкость с охлаждением водой
54 улавливающая емкость с охлаждением сухим льдом/метанолом
56 плунжерный насос
58 ограничитель давления
60 емкость для исходного материала
62 эжектор
64 ловушка
64A ловушка для первой фракции
64B улавливающая ловушка
66 вакуумный насос
68 испаритель
70 реактор
72 плунжерный насос
74 регулятор понижения давления
76 игольчатый клапан
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРА УГОЛЬНОЙ КИСЛОТЫ | 2019 |
|
RU2783523C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО НИТРИЛЬНОГО СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СЛОЖНОГО ЭФИРА | 2017 |
|
RU2739444C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНЫХ СЛОЖНЫХ ЭФИРОВ И КАТАЛИТИЧЕСКАЯ КОНСТРУКЦИЯ ДЛЯ ПОЛУЧЕНИЯ КАРБОНАТНЫХ СЛОЖНЫХ ЭФИРОВ | 2019 |
|
RU2796601C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДНОГО СОЕДИНЕНИЯ И АКРИЛАМИДНЫЙ ПОЛИМЕР | 2005 |
|
RU2402527C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2012 |
|
RU2602080C2 |
| СПОСОБ ОЧИСТКИ И УСТАНОВКА ДЛЯ ОЧИСТКИ СТОЧНЫХ ВОД | 2009 |
|
RU2516746C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2454399C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛЕГКИХ ОЛЕФИНОВ | 2015 |
|
RU2698107C2 |
| СПОСОБ ПОЛУЧЕНИЯ АДИПОНИТРИЛА | 2005 |
|
RU2373191C2 |
| НЕПРЕРЫВНЫЕ СПОСОБЫ ГИДРОЛИЗА ЦИАНОПИРИДИНОВ В АДИАБАТИЧЕСКИХ УСЛОВИЯХ | 1997 |
|
RU2175968C2 |
Группа изобретений относится к органической химии, а именно к способу получения ароматического нитрильного соединения, представляющего собой 2-цианопиридин, 3-цианопиридин или 4-цианопиридин. Способ включает приведение в контакт амидного соединения, представляющего собой 2-пиколинамид, 3-пиколинамид или 4-пиколинамид, с катализатором, включающим щелочной металл, в газовой фазе в ходе реакции дегидратации. Также группа изобретений относится к способу получения карбонатного эфира, содержащему первый этап, включающий получение карбонатного эфира и воды реакцией спирта с диоксидом углерода в присутствии ароматического нитрильного соединения, и гидратацию ароматического нитрильного соединения с полученной водой с образованием ароматического амидного соединения; и второй этап, на котором отделяют ароматическое амидное соединение от реакционной системы первого этапа и затем регенерируют ароматическое амидное соединение с получением ароматического нитрильного соединения посредством процесса дегидратации, указанного выше. При этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на втором этапе реакции, используют на первом этапе реакции. Технический результат - усовершенствование способа получения ароматического нитрильного соединения за счет уменьшения этапов реакции и повышения скорости дегидратации для уменьшения продолжительности процесса. Дополнительно настоящее изобретение реализует способ эффективного получения карбонатного эфира путем применения вышеупомянутого способа получения ароматического нитрильного соединения. 2 н. и 12 з.п. ф-лы, 3 ил., 2 табл., 7 пр.
1. Способ получения ароматического нитрильного соединения, включающий реакцию дегидратации, в которой дегидратируют ароматическое амидное соединение,
при этом способ включает этап контакта для приведения ароматического амидного соединения в контакт с катализатором в газовой фазе в ходе реакции дегидратации,
при этом катализатор включает щелочной металл и
при этом ароматическое амидное соединение представляет собой 2-пиколинамид, 3-пиколинамид или 4-пиколинамид; и ароматическое нитрильное соединение представляет собой 2-цианопиридин, 3-цианопиридин или 4-цианопиридин.
2. Способ получения ароматического нитрильного соединения по п.1, в котором катализатор включает оксид щелочного металла.
3. Способ получения ароматического нитрильного соединения по п.1, в котором ароматическое амидное соединение представляет собой 2-пиколинамид, при этом ароматическое нитрильное соединение представляет собой 2-цианопиридин.
4. Способ получения ароматического нитрильного соединения по любому из пп.1-3, в котором на этапе контакта дополнительно приводят в контакт с катализатором инертный газ и/или растворитель в парообразном состоянии.
5. Способ получения ароматического нитрильного соединения по п.4, в котором инертный газ включает по меньшей мере газообразный азот.
6. Способ получения ароматического нитрильного соединения по п.4 или 5, в котором температура кипения растворителя при нормальном давлении составляет от 20 до 300 °C.
7. Способ получения ароматического нитрильного соединения по любому из пп.4-6, в котором растворитель совместим с ароматическим амидным соединением.
8. Способ получения ароматического нитрильного соединения по любому из пп.4-7, в котором растворитель представляет собой пиридиновое соединение и/или кетоновое соединение.
9. Способ получения ароматического нитрильного соединения по любому из пп.1-8, в котором на этапе контакта температура, при которой ароматическое амидное соединение приводят в контакт с катализатором в газовой фазе, составляет 170 °C или выше, но ниже 300 °C.
10. Способ получения ароматического нитрильного соединения по любому из пп.1-9, в котором продолжительность приведения ароматического амидного соединения в контакт с катализатором в газовой фазе составляет 0,001 с или больше, но меньше 10 с.
11. Способ получения карбонатного эфира, включающий:
первый этап реакции, включающий: реакцию получения карбонатного эфира, в которой проводят реакцию спирта с диоксидом углерода в присутствии ароматического нитрильного соединения с получением карбонатного эфира и воды, и реакцию гидратации, в которой гидратируют ароматическое нитрильное соединение с полученной водой с получением ароматического амидного соединения; и
второй этап реакции, на котором отделяют ароматическое амидное соединение от реакционной системы первого этапа реакции и затем регенерируют ароматическое амидное соединение с получением ароматического нитрильного соединения посредством реакции дегидратации для дегидратации ароматического амидного соединения согласно способу по любому по любому из пп.1-10,
при этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на втором этапе реакции, используют на первом этапе реакции.
12. Способ получения карбонатного эфира по п.11, в котором на втором этапе реакции получают ароматическое нитрильное соединение из ароматического амидного соединения согласно способу получения ароматического нитрильного соединения по любому из пп.2-10 для регенерации ароматического нитрильного соединения.
13. Способ получения карбонатного эфира по п.11 или 12, в котором в реакции получения карбонатного эфира используют катализатор, включающий оксид церия.
14. Способ получения карбонатного эфира по любому из пп.11-13, в котором спирт включает спирт, содержащий от 1 до 6 атомов углерода.
| EP 0492233 A1, 01.07.1992 | |||
| Одинарный кулирный трикотаж и способ его изготовления | 1978 |
|
SU749956A1 |
| J | |||
| Campbell et al., Laboratory-scale synthesis of nitriles by catalysed dehydration of amides and oximes under flash vacuum pyrolysis (FVP) conditions | |||
| Synthesis, 2007, no.20, pp.3179-3184 | |||
| WO 2015099053 A1, 02.07.2015 | |||
| JP 2012162523 A, 30.08.2012 | |||
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |

