Область, к которой относится изобретение
Настоящее изобретение относится к способу очистки сточных вод в процессе получения ароматической карбоновой кислоты и к установке для очистки данных сточных вод.
Уровень техники
Сточные воды, содержащие соединения тяжелых металлов, сбрасываются из целого ряда промышленных установок, таких как различные химические установки, оборудование по производству компонентов электроники, оборудование по переработке пищевых продуктов, металлообрабатывающее оборудование, оборудование для нанесения гальванических покрытий, оборудование для изготовления печатных форм, фотографическое оборудование и дальнейшее вырабатывающее электроэнергию оборудование для получения тепловой энергии, генерирования ядерной энергии и аналогичное. Примеры химических установок включают технологическое оборудование и аналогичное для получения этиленоксида, этиленгликоля, метанола, этанола, высших спиртов, акриловой кислоты, сложных эфиров акриловой кислоты и ароматических карбоновых кислот или их сложных эфиров, например, терефталевой кислоты, сложных эфиров терефталевой кислоты, изофталевой кислоты и сложных эфиров изофталевой кислоты.
Например, когда в качестве примера указывается производство терефталевой кислоты, терефталевую кислоту получают жидкофазным окислением п-ксилола молекулярным кислородом под давлением в присутствии катализатора, состоящего из соединения тяжелого металла, такого как кобальт или марганец, и соединения брома в растворителе, представляющем собой уксусную кислоту. Суспензию, содержащую терефталевую кислоту, после реакции подвергают разделению на твердую и жидкую фазы и промывке, и полученный в результате отфильтрованный осадок терефталевой кислоты сушат, получая неочищенную терефталевую кислоту, которую затем перемещают в стадию гидрирования для очистки с получением терефталевой кислоты высокой чистоты. С другой стороны, поскольку отделенная маточная жидкость и промывающая жидкость (в дальнейшем называемые маточной жидкостью и аналогичным), образующиеся при разделении на твердую и жидкую фазы, содержат, кроме растворителя, представляющего собой уксусную кислоту, органические вещества, такие как терефталевая кислота, и компоненты, такие как металлический компонент катализатора (в дальнейшем называемые ценными компонентами), маточную жидкость и аналогичное вновь направляют в стадию реакции окисления и повторно там используют. Однако, поскольку органические вещества в маточной жидкости и аналогичном содержат примеси, такие как п-толуиловая кислота, 4-карбоксибензальдегид и бензойная кислота, часть маточной жидкости и аналогичного удаляют за пределы производственных стадий и остающуюся часть повторно используют, чтобы избежать ухудшения качества терефталевой кислоты из-за накопления таких примесей. Однако, поскольку маточная жидкость и аналогичное, которая должна быть удалена за пределы производственных стадий, содержит значительное количество ценных компонентов, как указано выше, с экономической и экологической точки зрения необходимо собрать и повторно использовать ценные компоненты. В качестве метода извлечения ценных компонентов, как правило, уксусную кислоту выпаривают, затем воду отделяют дистилляцией для повторного использования и компоненты катализатора, представляющие собой тяжелый металл, извлекают из остатка при выпаривании.
Патентный документ 1 предлагает общепринятый способ, который нацелен на улучшение величины извлечения металлических компонентов при получении терефталевой кислоты. Согласно данному документу к остающемуся концентрату реакционной маточной жидкости добавляют воду для растворения компонентов катализатора, представляющих собой тяжелый металл, с образованием водного раствора и, в случае, когда незначительное количество терефталевой кислоты и других побочных продуктов реакции осаждается в виде твердого вещества, проводят разделение на твердую и жидкую фазы. Сообщенный способ далее включает следующее: к полученному в результате водному раствору добавляют карбонат щелочного металла для получения карбонатов кобальта и марганца в качестве компонентов, представляющих собой тяжелый металл, из которых получают суспензию в аппарате концентрирования посредством непрерывного осаждения; сконцентрированную суспензию отбирают из нижней части аппарата, и после того как суспензия взаимодействует с уксусной кислотой, суспензию повторно используют в реакции окисления.
Патентный документ 2 сообщает способ предотвращения уменьшения величины извлечения катализатора в результате недостаточного разделения на твердую и жидкую фазы, вызванного микрочастицами в суспензии, причем данный способ включает, при получении терефталевой кислоты, добавление воды к концентрату реакционной маточной жидкости с получением суспензии и регулирование концентрации ароматического альдегида в суспензии, концентрации в ней уксусной кислоты, и понижение температуры для осуществления разделения на твердую и жидкую фазы.
Патентный документ 3 сообщает способ улавливания водного раствора катализатора, который включает, при получении терефталевой кислоты, обработку остающегося концентрата реакционной маточной жидкости горячей водой при перемешивании, затем гранулирование полученной в результате суспензии в горячей воде при перемешивании для получения гранулированной суспензии, осуществление обработки горячей водой и гранулирующей обработки в отдельных резервуарах с перемешиванием в данное время и, наконец, разделение полученной в результате гранулированной суспензии на твердую и жидкую фазы.
Кроме того, патентный документ 4 предлагает способ очистки отработанной промывающей жидкости, источником которой является получение ароматической кислоты, который включает отделение нерастворимой ароматической кислоты с помощью фильтрационной установки, затем адсорбцию металлических компонентов, таких как железо, никель и хром, из фильтрата на сильнокислой катионообменной смоле, в дальнейшем адсорбцию кобальта и марганца, являющихся каталитическими металлами, на хелатообразующей смоле, далее удаление растворенных органических веществ, пропусканием фильтрата через мембранную систему обратного осмоса, и повторное использование полученной в результате жидкости в технологических стадиях.
Перечень документов
Патентный документ
[Патентный документ 1] JP-A-5-15788
[Патентный документ 2] JP-A-2004-321889
[Патентный документ 3] JP-A-2006-312166
[Патентный документ 4] JP-A-2003-507156
Сущность изобретения
Проблемы, которые необходимо решить изобретением
Однако способ, описанный в вышеуказанном патентном документе 4, обнаруживает недостатки, состоящие в том, что на ранней стадии происходит снижение адсорбционной способности, срок службы хелатообразующей системы является недостаточным и т.д., так что способ не достигает промышленного практического уровня с технической и экономической точек зрения.
Согласно технологиям, показанным выше, величина извлечения соединений тяжелых металлов, по вышесказанному, улучшается до некоторой степени, но не является достаточным уровнем для промышленного использования. В частности, на промышленных установках крупного масштаба, например, на установке для получения терефталевой кислоты, было желательно дальнейшее решение по охране окружающей среды.
Части, в которых соединения тяжелых металлов остаются и сбрасываются за пределы технологического процесса, представляют собой твердое вещество, получаемое при разделении на твердую и жидкую фазы из остающегося концентрата, и фильтрат, полученный при разделении на твердую и жидкую фазы после образования карбонатных солей в патентном документе 1. Кроме того, соединения тяжелых металлов также остаются в фильтрате, полученном при разделении на твердую и жидкую фазы суспензии терефталевой кислоты высокой чистоты. Обычно твердое вещество подвергают обработке прокаливанием или утилизации, и фильтрат обычно сбрасывают в виде сточных вод после биологической очистки. Соединения тяжелых металлов в фильтрате, образующемся в таком способе получения терефталевой кислоты, присутствуют в незначительной концентрации, и способ эффективного улавливания соединений тяжелых металлов, присутствующих в незначительной концентрации, до настоящего времени не был известен. Поэтому сточные воды, содержащие низкие концентрации соединений тяжелых металлов, часто сбрасывают в реку или аналогичный водоем после биологической очистки. Таким образом, эффективный способ улавливания соединений тяжелых металлов еще не создан.
Реальным условием является, когда количество соединений тяжелых металлов, сбрасываемых за пределы технологического процесса, достигает, в общем, от 5 до 10% от их общего используемого количества. При такой ситуации, когда нормативы по сбросу соединений тяжелых металлов в виде сточных вод ужесточаются, эффективное улавливание соединений тяжелых металлов из сточных вод становится крайне важным фактором не только с экономической точки зрения, но также и с экологической точки зрения. Кроме того, с данных точек зрения, сброс соединений тяжелых металлов во внешнюю среду следует снижать, насколько это возможно. Однако способ очистки сточных вод, который удовлетворяет вышеуказанным требованиям, до настоящего времени не известен.
Настоящее изобретение осуществлено, принимая во внимание вышеуказанные проблемы, и его цель состоит в предложении способа очистки сточных вод, который может эффективно улавливать соединение тяжелого металла из сточных вод, содержащих соединение тяжелого металла, образующихся в способе получения ароматической карбоновой кислоты.
В результате обширных исследований, принимая во внимание вышеуказанные действительные ситуации, в способе, включающем приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, авторы настоящего изобретения обнаружили, что можно эффективно улавливать соединение тяжелого металла, используя хелатообразующую смолу, имеющую особую форму. Таким образом, авторы осуществили следующее первое изобретение.
Более того, в результате обширных исследований, принимая во внимание вышеуказанные действительные ситуации, в способе, включающем приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, авторы настоящего изобретения обнаружили, что можно эффективно улавливать соединение тяжелого металла, используя хелатообразующую смолу, показывающую особую величину снижения адсорбционной емкости по Cu. Таким образом, авторы осуществили следующее второе изобретение.
Кроме того, в результате обширных исследований, принимая во внимание вышеуказанные действительные ситуации, в способе, включающем приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, и затем регенерацию хелатообразующей смолы для получения жидкости регенерации, содержащей соединение тяжелого металла, авторы настоящего изобретения обнаружили, что можно эффективно улавливать соединение тяжелого металла, приводя сточные воды в контакт с хелатообразующей смолой при особых условиях и регенерируя хелатообразующую смолу после контакта со сточными водами при особых условиях. Таким образом, авторы осуществили следующее третье изобретение.
А именно, первое изобретение представляет собой способ очистки сточных вод, включающий приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, где хелатообразующая смола представляет собой частицы, имеющие коэффициент однородности 1,4 или менее. Метод измерения коэффициента однородности в изобретении будет подробно описан далее.
Кроме того, второе изобретение представляет собой способ очистки сточных вод, включающий приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, где величина снижения адсорбционной емкости по Cu для хелатообразующей смолы составляет 11%/месяц или менее.
В способах очистки сточных вод согласно первому и второму изобретению pH сточных вод, которые следует привести в контакт с хелатообразующей смолой, предпочтительно, составляет от 5,1 или более до 5,9 или менее. Когда pH сточных вод превышает вышеуказанный диапазон, соединение тяжелого металла может выпасть в осадок на поверхности хелатообразующей смолы и, таким образом, может оказаться невозможным извлечь данное соединение. С другой стороны, когда pH сточных вод меньше вышеуказанного диапазона, терефталевая кислота и органические вещества, такие как п-толуиловая кислота, содержащиеся в сточных водах, могут выпасть в осадок, или может уменьшиться величина извлечения соединения тяжелого металла.
Кроме того, в способах очистки сточных вод согласно первому и второму изобретению скорость потока сточных вод, которые следует привести в контакт с хелатообразующей смолой, предпочтительно, составляет от 5 м/час или более до 14 м/час или менее. Когда скорость потока сточных вод превышает вышеуказанный диапазон, существует тенденция, что соединение тяжелого металла, содержащееся в сточных водах, не может эффективно адсорбироваться хелатообразующей смолой вследствие влияния уноса и образования сквозных протоков. Когда скорость потока сточных вод меньше вышеуказанного диапазона, необходимо увеличить сосуд для помещения хелатообразующей смолы, так что данный случай невыгоден с экономической точки зрения.
Третье изобретение представляет собой способ очистки сточных вод, включающий приведение сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы; и затем регенерацию хелатообразующей смолы для получения жидкости регенерации, содержащей соединение тяжелого металла, где pH сточных вод, которые следует привести в контакт с хелатообразующей смолой, составляет от 5,1 или более до 5,9 или менее, скорость потока сточных вод, которые следует привести в контакт с хелатообразующей смолой, составляет от 5 м/час или более до 14 м/час или менее, и хелатообразующую смолу регенерируют, используя водный раствор бромистого водорода с концентрацией от 7,1% по массе или более до 19% по массе или менее.
В способе очистки сточных вод согласно третьему изобретению хелатообразующая смола, предпочтительно, представляет собой частицы, имеющие коэффициент однородности 1,4 или менее. В случае, когда коэффициент однородности превышает вышеуказанный диапазон, когда сточные воды адсорбируют и регенерируют методом потока через неподвижный слой, вероятным является потеря давления, что может являться одной из причин снижения величины ухудшения, что будет подробно обсуждено далее.
Кроме того, в способах очистки сточных вод согласно первому, второму и третьему изобретениям температура сточных вод, которые следует привести в контакт с хелатообразующей смолой, предпочтительно, составляет от 51°C или выше до 59°C или ниже. Когда температура сточных вод ниже, чем вышеуказанный диапазон, в случае, когда в сточных водах содержится терефталевая кислота, она стремится легко выпасть в осадок, и величина улавливания соединения тяжелого металла снижается. С другой стороны, когда температура сточных вод превышает вышеуказанный диапазон, хелатообразующая смола имеет тенденцию легко портиться.
Более того, в способах очистки сточных вод согласно первому, второму и третьему изобретениям адсорбционная емкость хелатообразующей смолы по Cu, предпочтительно, составляет 0,5 ммоль/мл или более. Верхний предел адсорбционной емкости по Cu для хелатообразующей смолы не ограничивается и более значительная хелатообразующая способность перед очисткой сточных вод является более предпочтительной. Метод измерения адсорбционной емкости по Cu будет подробно описан далее.
Кроме того, в способе очистки сточных вод согласно третьему изобретению жидкость регенерации можно повторно использовать в системе реакции окисления в способе получения ароматической карбоновой кислоты.
Более того, в качестве четвертого изобретения предлагается установка очистки сточных вод для приведения сточных вод, образующихся в процессе получения ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, хелатообразующей смолой, где хелатообразующая смола представляет собой частицы, имеющие коэффициент однородности 1,4 или менее.
Преимущество изобретения
Согласно первому, второму и четвертому изобретениям можно весьма эффективно извлечь соединения тяжелого металла, такого как кобальт и марганец, содержащиеся в сточных водах, образующихся в процессе получения ароматической карбоновой кислоты. В случае, когда коэффициент однородности превышает заранее определенный диапазон, вероятным является потеря давления в слое смолы, когда сточные воды адсорбируют и регенерируют методом потока через неподвижный слой.
Согласно третьему изобретению, становится возможным извлечь соединения тяжелого металла, такого как кобальт и марганец, содержащиеся в сточных водах, образующихся в процессе получения ароматической карбоновой кислоты, с высокой эффективностью и повторно их использовать.
Согласно изобретению, благодаря получению вышеуказанных эффектов, количество вышеуказанных металлических компонентов, сбрасываемых за пределы способа получения, можно подавить до минимума относительно общего количества вышеуказанных используемых металлических компонентов, и, таким образом, становится возможным обеспечить благоприятный для окружающей среды способ получения ароматической карбоновой кислоты, который является беспрецедентным. Способ далее дает существенный экономический эффект, который получают посредством улавливания и повторного использования соединений тяжелых металлов. В общем, нефтехимические заводы часто состоят из химических установок различного вида на одной площади или в одном промышленном районе в виде комплекса. Поэтому виды промышленных сточных вод, сбрасываемых из них, отличаются друг от друга, но имеется случай, кода сточные воды от химических установок различного типа, сбрасывающие соединения тяжелых металлов, как описано выше, смешивают и сбрасывают наружу, в зависимости от ситуации. Поскольку способ очистки сточных вод по изобретению может обеспечить улавливание и повторное использование соединений тяжелых металлов, таких как кобальт и марганец, содержащихся в сточных водах, с высокой эффективностью, так как сточные воды содержат сточные воды, образующиеся в процессе получения ароматической карбоновой кислоты, представленной терефталевой кислотой, даже когда присутствуют сточные воды из химических установок различного типа, преимущества имеются не только с экономической точки зрения, но также и с экологической точки зрения.
Краткое описание чертежей
[Фиг.1] Фиг.1 представляет собой блок-схему, схематически показывающую один пример стадий получения терефталевой кислоты высокой чистоты по изобретению.
[Фиг.2] Фиг.2 представляет собой пример технологической схемы стадий получения терефталевой кислоты высокой чистоты по изобретению.
[Фиг.3] Фиг.3 представляет собой пример технологической схемы стадий улавливания соединений тяжелых металлов по изобретению.
Режим осуществления изобретения
Следующее ниже будет описывать изобретение более подробно. Изобретение не ограничивается следующими ниже режимами осуществления изобретения, и оно может быть осуществлено с различными изменениями внутри его сущности.
<Способ получения ароматической карбоновой кислоты>
Следующее ниже будет описывать способ получения ароматической карбоновой кислоты. Исходное вещество для получения ароматической карбоновой кислоты не ограничивается, но обычно используют ароматическое соединение, имеющее алкильную группу. Ароматическое кольцо, входящее в состав ароматического соединения, может представлять собой моноциклическое кольцо или полициклическое кольцо.
Примеры вышеуказанной алкильной группы включают метильную группу, этильную группу, н-пропильную группу и изопропильную группу.
Конкретные примеры ароматического соединения, содержащего алкильную группу, включают ди- и триалкилбензолы, ди- и триалкилнафталины и ди- и триалкилдифенилы. Их предпочтительные примеры включают м-диизопропилбензол, п-диизопропилбензол, м-цимол, п-цимол, о-ксилол, м-ксилол, п-ксилол, триметилбензолы, 2,6- или 2,7-диметилнафталин, 2,6-диизопропилнафталин и 4,4′-диметилдифенил. Среди данных соединений предпочтительные соединения, благодаря высокой реакционной способности, включают алкилбензолы, алкилнафталины и алкилдифенилы, имеющие от 2 до 4 алкильных групп, содержащих от 1 до 4 атомов углерода, таких как метильная группа, этильная группа, н-пропильная группа и изопропильная группа.
Кроме того, ароматическое соединение, имеющее алкильную группу, может быть частично окисленным. Оно представляет собой соединение, в котором алкильная группа в вышеуказанном ароматическом соединении, имеющем алкильную группу, окислена до альдегидной группы, ацильной группы, карбоксильной группы или гидроксиалкильной группы, но не окислена, так чтобы представлять собой целевую ароматическую карбоновую кислоту. Конкретно, его примеры включают 3-метилбензальдегид, 4-метилбензальдегид, м-толуиловую кислоту, п-толуиловую кислоту, 3-формилбензойную кислоту, 4-формилбензойную кислоту и 2-метил-6-формилнафталины.
Данные исходные материалы можно использовать по отдельности или в сочетании двух или более соединений.
Рассматривая все вышеуказанное, в качестве исходного материала предпочтительными являются ксилолы (о-ксилол, м-ксилол, п-ксилол) и, в особенности, предпочтительным является п-ксилол. В случае использования п-ксилола в качестве исходного материала, примеры ароматического соединения, имеющего частично окисленную алкильную группу, включают 4-карбоксибензальдегид (в дальнейшем иногда называемый 4CBA), п-толуальдегид и п-толуиловую кислоту, и терефталевую кислоту получают в качестве ароматической карбоновой кислоты.
Данные исходные материалы обычно окисляют в жидкой фазе при высокой температуре и высоком давлении в присутствии молекулярного кислорода в реакционном растворителе, содержащем низшую алифатическую карбоновую кислоту, используя соединение тяжелого металла, и, если необходимо, соединение брома в качестве катализаторов окисления, посредством чего получают ароматические карбоновые кислоты. Полученные в результате ароматические карбоновые кислоты превращают в продукты посредством стадий очистки/разделения в зависимости от намеченного качества.
Вид ароматической карбоновой кислоты, на которую направлено изобретение, конкретно не ограничивается, и ее примеры включают о-фталевую кислоту, изофталевую кислоту, терефталевую кислоту, тримеллитовую кислоту (бензолтрикарбоновую кислоту), 2,6- или 2,7-нафталиндикарбоновую кислоту и 4,4′-дифенилдикарбоновую кислоту. Из данных соединений изобретение предпочтительно применять для получения фталевых кислот (о-фталевой кислоты, изофталевой кислоты, терефталевой кислоты), и особенно предпочтительно применять его для получения терефталевой кислоты.
<Способ получения терефталевой кислоты высокой чистоты>
В качестве типичной ароматической карбоновой кислоты по изобретению можно указать терефталевую кислоту высокой чистоты. Следующее ниже будет описывать способ получения терефталевой кислоты высокой чистоты с использованием п-кислола в качестве исходного материала, но, при получении других ароматических карбоновых кислот, их также можно получать аналогичным образом с подходящими изменениями способа. Способ получения высокочистой терефталевой кислоты конкретно включает, например, следующие стадии.
Фиг.1 представляет собой блок-схему, схематически показывающую один пример стадий получения терефталевой кислоты высокой чистоты. Как показано на Фиг.1, способ получения терефталевой кислоты высокой чистоты включает 11 стадий, показанных ниже.
(S1) Стадия окисления: стадия окисления п-ксилола в присутствии катализатора и, если необходимо, проведения кристаллизации для получения суспензии неочищенной терефталевой кислоты 11′.
(S2) Первая стадия разделения на твердую и жидкую фазы: стадия разделения вышеуказанной суспензии неочищенной терефталевой кислоты 11′ на твердую и жидкую фазы, промывки и сушки полученного в результате отфильтрованного осадка терефталевой кислоты с получением отделенной маточной жидкости/промывающей жидкости 17′ и неочищенной терефталевой кислоты 12′.
(S3) Стадия гидрирования: стадия растворения вышеуказанной неочищенной терефталевой кислоты 12′ в воде и гидрирования раствора с получением жидкости обработки гидрированием 13′.
(S4) Стадия кристаллизации: стадия кристаллизации вышеуказанной жидкости обработки гидрированием 13′ с получением суспензии терефталевой кислоты высокой чистоты 14′.
(S5) Вторая стадия разделения на твердую и жидкую фазы: стадия разделения вышеуказанной суспензии терефталевой кислоты высокой чистоты 14′ на твердую и жидкую фазы и промывки полученного в результате продукта с получением отфильтрованного осадка терефталевой кислоты высокой чистоты 14″ и маточной жидкости, отделенной от терефталевой кислоты высокой чистоты, и аналогичного 19′.
(S6) Стадия сушки: стадия сушки вышеуказанного отфильтрованного осадка терефталевой кислоты высокой чистоты 14″ для получения терефталевой кислоты высокой чистоты 15′ и сухой конденсированной жидкости 15″.
(S7) Стадия улавливания органического соединения: стадия улавливания органических соединений (органических веществ, таких как п-толуиловая кислота 27′) из всего количества или части маточной жидкости, отделенной от терефталевой кислоты высокой чистоты, и аналогичного 19′ и сухой конденсированной жидкости 15”, отводимых из вышеуказанных стадий S5 и S6.
(S8) Стадия рециркуляции: стадия повторного использования в стадии S1 части или всего количества отделенной маточной жидкости/промывающей жидкости 17′, полученной на вышеуказанной стадии S2.
(S9) Стадия первичного улавливания, стадия улавливания уксусной кислоты 22′ из отделенной маточной жидкости/промывающей жидкости 17′, повторно не используемой в вышеуказанной стадии S8, и затем улавливания жидкости первичного улавливания стадии регенерации катализатора 25′, содержащей каталитический металлический компонент.
(S10) Стадия вторичного улавливания: стадия улавливания и повторного использования жидкости вторичного улавливания катализатора 29′, содержащей остающееся соединение тяжелого металла, из сточных вод 26′ и 28′, образующихся на вышеуказанных стадиях S7 и S9.
(S11) Стадия биологической очистки: стадия биологической очистки всего количества или части сточных вод, сбрасываемых из вышеуказанных стадий S1-S10.
Данные стадии будут более подробно описаны на основе Фиг.2. Фиг.2 представляет собой чертеж, показывающий пример технологической схемы стадий получения терефталевой кислоты высокой чистоты согласно изобретению.
1. Стадия окисления (S1)
Стадия S1 представляет собой стадию реакции окисления п-ксилола в уксусной кислоте, содержащей воду, с получением суспензии неочищенной терефталевой кислоты. А именно, п-ксилол 1′ и растворитель 3′, включающий уксусную кислоту и аналогичное, сначала смешивают и перемещают в аппарат реакции окисления 11, и п-ксилол 1′ окисляют в растворителе 3′ в присутствии катализатора 31′, состоящего из тяжелого металла и соединения брома, используя молекулярный кислород 2′. Посредством этого получают суспензию неочищенной терефталевой кислоты 11′ и затем ее перемещают на стадию S2. Кроме того, воду, промывающую сбрасываемый из стадии окисления газ (сточные воды), 16′ обрабатывают на стадии S11, что будет подробно описано далее.
Катализатор, используемый при окислении п-ксилола 1′, конкретно не ограничивается при условии, что он обладает способностью окислять органическое соединение, имеющее алкильную группу, чтобы превратить его в ароматическую карбоновую кислоту, но обычно используют соединение тяжелого металла. Примеры тяжелых металлов в соединении тяжелого металла включают кобальт, марганец, никель, хром, цирконий, медь, свинец, гафний и церий. Их можно использовать по одиночке или в комбинации, и предпочтительным является комбинированное использование кобальта и марганца. Примеры такого соединения тяжелого металла включают ацетатные соли, нитратные соли, ацетилацетонатные соли, нафтенатные соли, стеаратные соли и бромиды, и предпочтительными являются ацетатные соли и бромиды.
Более того, катализатор может содержать, при необходимости, ускоритель, и в качестве ускорителя обычно используют соединение брома. Примеры соединения брома включают неорганические соединения брома, такие как молекулярный бром, бромистый водород, бромид натрия, бромид калия, бромид кобальта и бромид марганца, и органические соединения брома, такие как метилбромид, метиленбромид, бромоформ, бензилбромид, бромксилол, дибромэтан, трибромэтан и тетрабромэтан. Данные соединения брома также можно использовать по отдельности или в комбинации двух или более данных соединений.
А именно, катализатор, который следует использовать для окисления п-ксилола 1′ в изобретении, особенно предпочтительно представляет собой комбинацию соединения кобальта, соединения марганца и соединения брома. В частности, более предпочтительной является комбинация ацетата кобальта, ацетата марганца и бромистого водорода.
В данном изобретении катализатор, состоящий из комбинации вышеуказанных соединений тяжелых металлов и соединения брома, содержит атом брома в количестве от 0,05 моль или более до 10 моль или менее, более предпочтительно, от 0,1 моль или более до 5 моль или менее, исходя из 1 моль тяжелого(ых) металла(ов).
Такой катализатор используют в диапазоне от 10 ч/млн по массе или более до 10000 ч/млн по массе или менее, более предпочтительно, от 100 ч/млн по массе или более до 5000 ч/млн по массе или менее, еще более предпочтительно, от 200 ч/млн по массе или более до 3000 ч/млн по массе или менее. Когда количество катализатора составляет вышеуказанный нижний предел или более, скорость реакции имеет тенденцию увеличиваться, а когда количество составляет вышеуказанный верхний предел или менее, стоимость имеет тенденцию снижаться.
Температура реакции окисления п-ксилола 1′ в аппарате реакции окисления 11, предпочтительно, составляет от 140°C или выше до 230°C или ниже, более предпочтительно, от 150°C или выше до 210°C или ниже и, еще более предпочтительно, от 170°C или выше до 200°C или ниже. Когда температура реакции ниже вышеуказанного диапазона, скорость реакции имеет тенденцию снижаться, а когда температура превышает вышеуказанный диапазон, потеря растворителя, представляющего собой уксусную кислоту, вследствие окисления имеет тенденцию увеличиваться. Давление реакции обязательно представляет собой, по меньшей мере, давление, при котором смесь может сохранять свою жидкую фазу при температуре реакции, и должно быть выше, чем нормальное давление. Конкретно, давление, предпочтительно, составляет от 0,2 МПа или более до 6 МПа или менее (абсолютное давление) и, более предпочтительно, от 0,4 МПа или более до 3 МПа или менее (абсолютное давление).
В качестве аппарата реакции окисления 11 обычно применяют резервуар с перемешиванием, барботажную колонну или аналогичное устройство. В случае, когда в качестве аппарата реакции окисления 11 используют барботажную колонну, температура реакции слегка ниже, чем в случае, когда используют резервуар с перемешиванием и, предпочтительно, составляет от 140°C или выше до 180°C или ниже и, более предпочтительно, от 150°C или выше до 170°C или ниже.
2. Стадия первого разделения на твердую и жидкую фазы (S2)
Стадия S2 представляет собой стадию разделения вышеуказанной суспензии неочищенной терефталевой кислоты 11′ на твердую и жидкую фазы, промывки и сушки полученного в результате отфильтрованного осадка терефталевой кислоты, чтобы посредством этого получить отделенную маточную жидкость/промывающую жидкость 17′ и неочищенную терефталевую кислоту 12′ в аппарате разделения на твердую и жидкую фазы/промывки/сушки 12.
В качестве метода разделения на твердую и жидкую фазы используют метод обработки суспензии в сепараторе жидкой и твердой фаз, в то же время поддерживая суспензию при высокой температуре и высоком давлении. Более того, поскольку суспензия неочищенной терефталевой кислоты 11′ находится под давлением, растворенную неочищенную терефталевую кислоту можно осадить, когда давление понижают. Поэтому имеется способ очистки суспензии неочищенной терефталевой кислоты 11′ в сепараторе жидкой и твердой фаз, после того как суспензию перемещают в резервуар кристаллизации (не показан на фигуре) и осуществляют охлаждение посредством сброса давления, чтобы осадить растворенную неочищенную терефталевую кислоту. В этой связи, вышеуказанное охлаждение посредством сброса давления обозначает операцию, в которой заданной жидкости дают возможность охладиться посредством расширения и испарения компонента, представляющего собой растворитель, посредством сброса давления (падения давления) до условия более низкого давления, чем давление заданной жидкости.
Неочищенную терефталевую кислоту 12′ получают промывкой и сушкой отфильтрованного осадка терефталевой кислоты, полученного таким способом. В качестве промывающей жидкости обычно применяют уксусную кислоту, и можно использовать уксусную кислоту 22′, собранную на указанной далее стадии S9, или свежую уксусную кислоту.
Полученная в результате неочищенная терефталевая кислота 12′ содержит 4-карбоксибензальдегид (в дальнейшем иногда называемый 4CBA) и аналогичные соединения, которые представляют собой промежуточные продукты окисления, в качестве примесей. Для их удаления неочищенную терефталевую кислоту 12′ передают на стадию S3.
3. Стадия гидрирования (S3)
Стадия S3 представляет собой стадию растворения вышеуказанной неочищенной терефталевой кислоты 12′ в воде и гидрирование раствора для осуществления восстановительной обработки добавлением водорода 4′ в аппарате реакции гидрирования 13. А именно, данная стадия представляет собой стадию восстановления 4CBA в качестве примеси с образованием п-толуиловой кислоты. Поскольку п-толуиловая кислота является более водорастворимой по сравнению с терефталевой кислотой, п-толуиловую кислоту можно отделить от жидкости обработки гидрированием 13′ на стадии S5, более подробно описанной далее. Возвращением п-толуиловой кислоты в вышеуказанную стадию S1, ее используют в качестве исходного материала для терефталевой кислоты. Жидкость обработки гидрированием 13′ затем перемещают на стадию S4.
4. Стадия кристаллизации (S4)
Стадия S4 представляет собой стадию кристаллизации вышеуказанной жидкости обработки гидрированием 13′ для получения суспензии терефталевой кислоты высокой чистоты 14′ в аппарате кристаллизации 14. В качестве методов кристаллизации можно указать метод, включающий выпаривание и удаление воды в качестве растворителя и последующее охлаждение, метод, включающий охлаждение посредством сброса давления, и аналогичные методы. На данной стадии, как указано выше, поскольку п-толуиловая кислота хорошо растворима в воде, основная часть кислоты остается растворенной, так что п-толуиловую кислоту и терефталевую кислоту можно отделить на следующей стадии S5. Суспензию терефталевой кислоты высокой чистоты 14′ перемещают на следующую стадию S5, а конденсированную воду (сточные воды) 18′, образующуюся при кристаллизации на стадии S4, обрабатывают на стадии S11, более подробно описанной далее.
5. Стадия второго разделения на твердую и жидкую фазы (S5)
Стадия S5 представляет собой стадию разделения вышеуказанной суспензии терефталевой кислоты высокой чистоты 14′ на твердую и жидкую фазы, промывки полученного в результате продукта и его разделения на отфильтрованный осадок терефталевой кислоты высокой чистоты 14′ и отделенную маточную жидкость и промывающую жидкость (маточную жидкость, отделенную от терефталевой кислоты высокой чистоты, и аналогичное 19′) в аппарате разделения на твердую и жидкую фазы/промывки 15a. В качестве сепаратора можно применить известный сепаратор, такой как аппарат фильтрации или центробежный сепаратор.
6. Стадия сушки (S6)
Стадия S6 представляет собой стадию сушки отфильтрованного осадка терефталевой кислоты высокой чистоты для получения терефталевой кислоты высокой чистоты 15′ и сухой конденсированной жидкости 15″ в сушильной установке 15b. В качестве сушильной установки используют сушилку, работающую посредством сброса давления, обычную сушилку с кипящим слоем или аналогичное устройство. Сухая конденсированная жидкость 15″, которую получают в результате конденсации паровой фазы, образующейся при выпаривании посредством сброса давления, содержит компоненты, аналогичные компонентам в маточной жидкости, отделенной от терефталевой кислоты высокой чистоты, 19′.
7. Стадия улавливания органического соединения (S7)
Стадия S7 представляет собой стадию улавливания органических соединений из всего количества или из части маточной жидкости, отделенной от терефталевой кислоты высокой чистоты, и аналогичного 19′, выгружаемой из вышеуказанной стадии S5, и из сухой конденсированной жидкости 15″, выгружаемой из вышеуказанной стадии S6. Маточная жидкость, отделенная от терефталевой кислоты высокой чистоты, и аналогичное 19′ и сухая конденсированная жидкость 15″, выгружаемая из вышеуказанной стадии S6, содержат примеси, например, п-толуиловую кислоту, катализатор, терефталевую кислоту и аналогичные соединения и, следовательно, п-толуиловую кислоту, терефталевую кислоту и аналогичные соединения улавливают осаждением посредством охлаждения в аппарате улавливания п-толуиловой кислоты и аналогичного 19 и органические вещества, например, п-толуиловую кислоту 27′, возвращают на стадию S1. Поскольку соединения тяжелых металлов и аналогичные остаются в маточной жидкости, отделенной от п-толуиловой кислоты и аналогичного 28′, маточную жидкость перемещают на стадию S10, подробно описанную далее, и соединения тяжелых металлов и аналогичные будут улавливаться и повторно там использоваться.
8. Стадия рециркуляции (S8)
Стадия S8 представляет собой стадию рециркуляции в стадию S1 части или всего количества отделенной маточной жидкости/промывающей жидкости 17′, полученной на вышеуказанной стадии S2. Из отделенной маточной жидкости/промывающей жидкости 17′ отделенную маточную жидкость, предпочтительно, разветвляют на рециркулирующую маточную жидкость и промывающую маточную жидкость, причем давление поддерживают выше нормального давления. Более того, давление, предпочтительно, равно давлению, которое по существу поддерживает рабочее давление на стадии S2. Отношение разветвления рециркулирующей маточной жидкости к промывающей маточной жидкости можно произвольно контролировать в зависимости от технологического процесса, но нижний предел коэффициента рециркуляции отделенной маточной жидкости (масса рециркулирующей маточной жидкости × 100/(масса рециркулирующей маточной жидкости + масса промывающей маточной жидкости), предпочтительно, составляет 50% или более, более предпочтительно, 60% или более и, еще более предпочтительно, 70% или более. Контролируя коэффициент рециркуляции в вышеуказанном диапазоне, можно повторно использовать ценные компоненты отделенной маточной жидкости, снизить нагрузку на стадию очистки сточных вод, а также можно снизить количество отходов так, что вышеуказанный коэффициент рециркуляции является предпочтительным. Более того, можно повторно использовать все количество отделенной маточной жидкости, но верхний предел коэффициента рециркуляции, предпочтительно, составляет 90% или менее, более предпочтительно, 80% или менее. Контролируя коэффициент рециркуляции в вышеуказанном диапазоне, можно подавить накопление примесей в системе, и достигается улучшение качества продукта, представляющего собой терефталевую кислоту, так, что вышеуказанный коэффициент рециркуляции является предпочтительным.
Для отделенной маточной жидкости/промывающей жидкости 17′ рециркуляция промывающей воды не всегда является необходимой, но, предпочтительно, промывающая вода направляется на рециркуляцию. Коэффициент рециркуляции промывающей жидкости (масса рециркулирующей промывающей жидкости × 100/(масса рециркулирующей промывающей жидкости + масса очищающей промывающей жидкости) находится в диапазоне, обычно, 60% или более, более предпочтительно, 75% или более и 100% или менее. Посредством рециркуляции промывающей жидкости можно повторно использовать ценные компоненты в промывающей жидкости, снижается нагрузка на стадию очистки сточных вод, а также можно снизить количество отходов так, что рециркуляция является предпочтительной.
9. Стадия первичного улавливания (S9)
Стадия S9 представляет собой стадию улавливания уксусной кислоты 22′ из отделенной маточной жидкости (промывающей маточной жидкости) и промывающей жидкости (очищающей промывающей жидкости), не используемой повторно на вышеуказанной стадии S8, причем отделенную маточную жидкость и промывающую жидкость 17′ получают на стадии 2, и затем улавливания ценных компонентов, таких как каталитический металлический компонент. Конкретно, испаренный растворитель 20′, полученный испарением отделенной маточной жидкости/промывающей жидкости 17′ в аппарате испарения растворителя 16, подвергают дегидратации в аппарате дегидратации/дистилляции 17, посредством чего улавливают уксусную кислоту 22′, содержащуюся в отделенной маточной жидкости/промывающей жидкости 17′ в наибольшем количестве. В данной ситуации, жидкость кубового остатка (сточные воды) 23′, образующуюся в аппарате дегидратации/дистилляции 17, будут перерабатывать на стадии S11, подробно описанной далее. Затем остаток концентрирования 21′, образовавшийся в аппарате испарения растворителя 16, снова превращают в суспензию, добавляя воду в аппарат улавливания/регенерации катализатора 18, и разделяют на твердое вещество и отделенную маточную жидкость с помощью фильтрационной установки. После того как осуществляют эффективную промывку для минимизации маточной жидкости, связанной в твердом веществе, остаток фильтрации 24′ сжигают вместе с активированным илом 32′, образующимся на стадии S11, с помощью установки для сжигания 22. Образовавшийся на данной стадии фильтрат будут перерабатывать в виде сточных вод стадии регенерации катализатора 26′ на стадии S10, указанной далее.
Затем, после того как отделенную маточную жидкость и промывающую жидкость, полученные фильтрацией после повторного образования вышеуказанной суспензии, нейтрализуют водным раствором гидроксида натрия, осуществляют их взаимодействие с водным раствором карбоната натрия 5′ с образованием карбоната кобальта и карбоната марганца, и суспензию, содержащую карбонатные соли, подвергают разделению на твердую и жидкую фазы, отделяя отфильтрованный осадок карбонатных солей. После этого снова получают суспензию, добавляя воду, и затем осуществляют взаимодействие с уксусной кислотой, получая ацетат кобальта и ацетат марганца. Полученную в результате суспензию называют жидкостью первичного улавливания стадии регенерации катализатора 25′. Жидкость первичного улавливания стадии регенерации катализатора 25′ смешивают с жидкостью вторичного улавливания катализатора 29′, указанной далее, в резервуаре смешивания жидкостей катализатора 21, и полученную в результате смесь пополняют пополняющим катализатором 6′ в количестве, которое пополняет потерю катализатора на технологических стадиях, причем пополненную смесь возвращают на стадию S1 в качестве катализатора 31′.
10. Стадия вторичного улавливания (S10)
Стадия S10 представляет собой стадию получения жидкости вторичного улавливания катализатора 29′, содержащую остающееся соединение тяжелого металла, из маточной жидкости отделенной от п-толуиловой кислоты и аналогичного 28′, образующейся на стадии S7, и/или сточных вод стадии регенерации катализатора 26′, образующихся на стадии S9 (в дальнейшем иногда обобщенно называемых очищаемой сточной водой) с помощью установки улавливания/очистки катализатора 20. А именно, стадия S10 представляет собой стадию улавливания компонентов, представляющих собой тяжелый металл, таких как кобальт и марганец, получаемых из катализатора, остающегося в перерабатываемых сточных водах. Конкретно, сточные воды 28′ с вышеуказанной стадии S7 и/или сточные воды 26′ со стадии S9 пропускают через стадию адсорбции для поглощения соединений тяжелых металлов хелатообразующей смолой и, после адсорбции, их десорбируют и регенерируют водным раствором бромистого водорода.
В частности, очищаемые сточные воды, получаемые в изобретении, могут содержать одно или несколько соединений, выбранных из терефталевой кислоты и п-толуиловой кислоты, 4-карбоксибензальдегида, 3-карбоксибензальдегида, бензойной кислоты, изофталевой кислоты и 2,6-дикарбоксифлуоренона, которые образуются в качестве примесей.
Нижеследующее будет более подробно описывать стадию S10, которая является характерной особенностью изобретения. Хелатообразующая смола для использования в изобретении представляет собой смолу, способную селективно адсорбировать конкретный металл посредством введения хелатообразующей группы (в дальнейшем, хелатной группы), которая легко образует хелатную связь с металлом, вместо ионообменной группы ионообменной смолы. Хелатная группа содержит комбинацию одинаковых или различных двух или более элементов, таких как N, S, O и P в качестве электронодонорных элементов, как в случае обычных хелатных агентов, и примеры их типов включают соединения на основе N-O, S-N, N-N, O-O. Когда любая из данных хелатных групп связана с трехмерной полимерной подложкой, получают хелатообразующую смолу, обладающую селективностью по отношению к конкретному металлу, которая характерна для хелатной группы. Хотя удаление тяжелых металлов также возможно с помощью катионообменных смол, в сточных водах присутствует огромное количество металлов, которые нет необходимости удалять, таких как щелочные металлы и щелочноземельные металлы, и, таким образом, они одновременно адсорбируются, приводя к истощению регенерирующего агента так, что необходимо использовать хелатообразующую смолу. Хелатная группа конкретно не ограничивается при условии, что она обладает селективностью по отношению к металлу, и ее примеры включают группу иминодиуксусной кислоты, полиаминовую группу, группу тиомочевины, группу аминофосфоновой кислоты, группу полиакриловой кислоты и N-метилглюкаминовую группу. Из данных групп более предпочтительной является группа иминодиуксусной кислоты.
Данные хелатные группы, предпочтительно, ионизируют щелочным металлом полностью или частично перед использованием. В качестве щелочного металла, который следует использовать в данное время, можно указать литий, калий или натрий. Из данных металлов натрий является наиболее обычным и предпочтительным.
Для ионизации такой хелатообразующей смолы натрием предпочтительно проводить ионизацию, используя водный раствор гидроксида натрия методом течения в неподвижном слое, в котором в качестве насадки используют хелатообразующую смолу, имеющую водородную форму в качестве функциональной концевой группы. В качестве гидроксида натрия, который следует использовать в данный момент, выбирают гидроксид натрия, имеющий концентрацию в водном растворе, предпочтительно, от 0,5% по массе или более до 20% по массе или менее, более предпочтительно, от 1% по массе или более до 10% по массе или менее. Кроме того, линейная скорость, принятая для ионизации при методе течения в неподвижном слое, предпочтительно, составляет от 1 м/ч или более до 20 м/ч или менее, более предпочтительно, от 2 м/ч или более до 15 м/ч или менее. Для режима ионизации в неподвижном слое режим восходящего потока является более предпочтительным, чем режим нисходящего потока, и предпочтительным является условие, когда хелатообразующая смола погружена в водный раствор гидроксида натрия в течение 2 часов или более. Выбранная температура при ионизации, предпочтительно, составляет от 10°C или выше до 80°C или ниже, более предпочтительно, представляет собой примерно обычную температуру (от 15°C или выше до 40°C или ниже). В изобретении, в случае, когда функциональная группа хелатообразующей смолы уже была ионизирована полностью или частично щелочным металлом, как указано выше, в момент времени поставки, вышеуказанная операция не всегда необходима.
В качестве подложки смолы, к которой присоединена хелатная группа, необходимая для использования, можно указать сшитый полистирол, сополимер стирола и дивинилбензола, сшитую полиакриловую кислоту и аналогичные полимеры, но сшитый полистирол и сополимер стирола и дивинилбензола являются особенно предпочтительными.
Структуры подложки более высокого порядка, составляющей хелатообразующую смолу, как правило, разделяют на гелевый тип, пористый тип и высокопористый тип, и в изобретении можно использовать любой тип. Из данных подложек пористый тип или высокопористый тип являются предпочтительными, и наиболее предпочтительным является пористый тип.
(Коэффициент однородности)
Изобретение относится к способу очистки сточных вод, включающему приведение перерабатываемых сточных вод в контакт с хелатообразующей смолой, имеющей особую форму, для улавливания соединения тяжелого металла, содержащегося в сточных водах, данной хелатообразующей смолой. А именно, в первом и четвертом изобретениях коэффициент однородности, показывающий однородность размера частиц хелатообразующей смолы, составляет 1,4 или менее, предпочтительно, 1,2 или менее, более предпочтительно, 1,1 или менее и, особенно предпочтительно, 1,05 или менее. Когда коэффициент однородности превышает вышеуказанный диапазон, имеется тенденция к падению давления в слое смолы, когда на ней адсорбируют очищаемые сточные воды, и затем смолу регенерируют методом течения в неподвижном слое, и потеря давления может являться одной причиной снижения величины ухудшения, о чем будет указано далее.
Коэффициент однородности можно измерить следующим методом. Во-первых, примерно 3 г образца (хелатообразующую смолу, набухшую в воде) взвешивают и связанную воду удаляют, смешивая образец примерно с 0,06 г частиц мелкодисперсного кремнезема. Смесь загружают в верхнюю часть работающего с использованием звуковых колебаний автоматического просеивающего анализатора распределения частиц по размерам (изготовленного Seishin Enterprise Co., Ltd., Robot Sifter RPS-85), на котором были установлены сита стандарта JIS (размер ячейки: 1180 мкм, 850 мкм, 710 мкм, 600 мкм, 425 мкм, 355 мкм). В наиболее нижней части просеивателя используют сито, имеющее размер ячейки 106 мкм для удаления только частиц мелкодисперсного кремнезема (белой сажи). Рабочие условия анализатора распределения частиц по размерам являются следующими: УРОВЕНЬ (интенсивность) от 7 до 9, ВРЕМЯ (период) 5 минут, ИНТЕРВАЛ (интервал) 1 секунда. Автоматически измеряют массу отсеянного образца на каждом сите. Из полученных значений рассчитывают объемное отношение каждого размера частиц в соответствии со следующими ниже уравнениями. В данной связи, белая сажа не содержится в расчетах, поскольку она проходит через сито, имеющее размер ячейки 106 мкм.
V (г)=a+b+c+d+e+f+g (от a до g: масса образца на каждом сите)
a′ (%)=a/V×100 a′: объемное отношение частиц, имеющих размер частиц 1180 мкм или более
b′ (%)=b/V×100 b′: объемное отношение частиц, имеющих размер частиц 850 мкм или более и менее чем 1180 мкм
c′ (%)=c/V×100 c′: объемное отношение частиц, имеющих размер частиц 710 мкм или более и менее чем 850 мкм
d′ (%)=d/V×100 d′: объемное отношение частиц, имеющих размер частиц 600 мкм или более и менее чем 710 мкм
e′ (%)=e/V×100 e′: объемное отношение частиц, имеющих размер частиц 425 мкм или более и менее чем 600 мкм
f′ (%)=f/V×100 f′: объемное отношение частиц, имеющих размер частиц 355 мкм или более и менее чем 425 мкм
g′ (%)=g/V×100 g′: объемное отношение частиц, имеющих размер частиц менее чем 355 мкм
Из значений от a′ до g′ строят график координаты логарифмической вероятности, где абсцисса показывает нарастающую сумму (%) частиц, остающихся на каждом сите, и ордината показывает размер ячейки (мкм) на каждом сите. Например, поскольку зависимость на ординате 425 мкм представляет собой нарастающую сумму частиц, имеющих размер 425 мкм или более, на абсциссу наносят общее значение от a′ до e′. Из графических результатов выбирают три зависимости в порядке более крупных масс частиц (в порядке более высокого значения на абсциссе) и проводят линию настолько близко к точкам, насколько это возможно (если необходимо, используя метод наименьших квадратов или аналогичный). Из данной линии определяют размер ячейки (мкм), когда нарастающая сумма остающихся частиц (абсцисса) составляет 90%, и размер ячейки (мкм), когда нарастающая сумма составляет 40%, и затем определяют коэффициент однородности согласно следующему уравнению.
[Suu 1]
Коэффициент однородности = размер ячейки (мкм), соответствующий нарастающей сумме 40%/размер ячейки (мкм), соответствующий нарастающей сумме 90% (эффективный размер)
(Средний размер частиц)
Кроме того, средний размер частиц хелатообразующей смолы для использования в настоящем изобретении составляет, предпочтительно, 0,2 мм или более, более предпочтительно, 0,4 мм или более, еще более предпочтительно, 0,5 мм или более и, предпочтительно, 2 мм или менее, более предпочтительно, 1 мм или менее, еще более предпочтительно, 0,7 мм или менее и, особенно предпочтительно, 0,65 мм или менее. Когда средний размер частиц хелатообразующей смолы меньше вышеуказанного диапазона, частицы, имеющие небольшой размер, имеют тенденцию осаждаться в нижней части колонны с хелатообразующей смолой, и это приводит к закупориванию колонны с хелатообразующей смолой или к снижению эффективности адсорбции из-за плотного заполнения, далее происходит вытекание хелатообразующей смолы из верхней части колонны с хелатообразующей смолой, закупоривание фильтра или аналогичного, в результате чего стабильная операция адсорбции становится затруднительной. Когда средний размер частиц хелатообразующей смолы превышает вышеуказанный диапазон, поток в колонне с хелатообразующей смолой становится нестабильным/неоднородным, в результате чего эффективность адсорбции снижается.
В изобретении для измерения среднего размера частиц осуществляют те же операции, что и при вышеуказанном измерении коэффициента однородности, и используют размер ячейки (мкм), соответствующий нарастающей сумме остающихся частиц, составляющей 50%.
(Адсорбционная емкость по Cu)
Хелатообразующая смола для использования в изобретении, предпочтительно, имеет более существенную хелатообразующую способность перед обработкой сточных вод, и адсорбционная емкость по Cu перед использованием, предпочтительно, составляет 0,5 ммоль/мл или более, более предпочтительно, 0,7 ммоль/мл или более. Верхний предел адсорбционной емкости по Cu не ограничивается, но обычно он составляет 2 ммоль/мл или менее. Адсорбционную емкость по Cu измеряют в виде общей обменной емкости, используя Cu в качестве металла, который следует адсорбировать, согласно следующему методу.
Аккуратно взвешивают примерно 6 г образца хелатообразующей смолы. Затем ее помещают в 1 л колбу Эрленмейера, снабженную запорным краном, и в нее добавляют 200 мл 0,05 М CuCl2, используя целую пипетку. Колбу Эрленмейера, содержащую образец и 0,05 М CuCl2, встряхивают в термостатируемом встряхивающем устройстве при 30±2°C в течение 6 часов. После встряхивания 10 мл надосадочной жидкости отбирают в 300 мл коническую мензурку, используя целую пипетку.
Затем туда добавляют 90 мл дистиллированной воды, 1 мл буферного раствора, раскрытого ниже, и подходящее количество индикатора, который указан далее, и смесь титруют 0,01М раствором ЭДТУ (a мл). В качестве холостого опыта титрование проводят аналогичным образом за исключением того, что используют 10 мл 0,05 М CuCl2 вместо 10 мл вышеуказанной надосадочной жидкости (b мл).
Буферный раствор: раствор, полученный, растворением 5,8 мл ледяной уксусной кислоты в 100 мл воды, целиком смешивают с раствором, полученным растворением 13,6 г ацетата натрия в 100 мл воды, получая буферный раствор.
Индикатор: раствор, полученный растворением 0,1 г PAN (1-пиридилазо-2-нафтола) в 100 мл метанола.
Обменную емкость рассчитывают в соответствии со следующим ниже уравнением.
[Suu 2]
Адсорбционная емкость по Cu (ммоль/мл)={(b-a)×F×0,01×200/10}/образец (г) x Объемная плотность/1000
F: титр 0,01М ЭДТУ
Во втором изобретении величина снижения адсорбционной емкости хелатообразующей смолы при условиях адсорбции сточных вод и регенерации, о чем будет указано далее, составляет 11%/месяц или менее, предпочтительно, 10%/месяц или менее, более предпочтительно, 7%/месяц или менее, и еще более предпочтительно, 6%/месяц или менее. Нижний предел величины снижения адсорбционной емкости по Cu не ограничивается, но обычно составляет 0,3%/месяц или более. Кроме того, желательно выбрать и использовать хелатообразующую смолу, для которой величина снижения адсорбционной емкости по Cu попадает внутрь вышеуказанного диапазона. Когда величина снижения адсорбционной емкости по Cu превышает вышеуказанный диапазон, адсорбция тяжелых металлов и регенерация имеют тенденцию требовать существенных усилий и, таким образом, может снижаться очистная способность и увеличиваться производственные затраты, так что данная ситуация является невыгодной с промышленной точки зрения.
Следующее ниже будет описывать метод измерения адсорбционной емкости хелатообразующей смолы по Cu для использования в изобретении.
Колонна с хелатообразующей смолой, которую следует использовать, представляет собой проточный реактор с неподвижным слоем, имеющий почти цилиндрическую форму (тип с неподвижным слоем описан на Фиг.11·67 на странице 901 Kagaku Kogyo Binran, исправленное 4 издание, Maruzen Co., Ltd., опубликованное 24 октября 1978 года). Отношение внутреннего диаметра к высоте части, которая заполнена хелатообразующей смолой, составляет 1,0:0,7, и частицами хелатообразующей смолы заполняют до 50% высоты в неподвижном состоянии. А именно, ее верхние 50% заполняют жидкостью, и частицы хелатообразующей смолы способны течь в верхнюю часть с потоком жидкости. Объем части, заполненной хелатообразующей смолой, составляет 1 м3 или более.
(1) Операция адсорбции: Водный раствор (приготовленный растворением тетрагидрата ацетата Co), имеющий концентрацию Co 5,0±0,5 ч/млн по массе, pH которого равна 5,5±0,2 и температура составляет 55°C±2°C, пропускают через колонну с хелатообразующей смолой при линейной скорости 0,15±0,02 м/мин, времени пребывания 13±2,0 мин и восходящем режиме течения. Момент времени, когда концентрация Co на выходе из колонны с хелатообразующей смолой достигает 1,0 ч/млн по массе, считают областью проскока, и, таким образом, операцию адсорбции заканчивают. В данной связи, для измерения концентрация Co используют JIS K 0102 60 (60. 3: эмиссионный спектральный анализ с индуктивно связанной плазмой).
(2) Операция регенерации: Водный раствор бромистого водорода с концентрацией 10±1% по массе в качестве регенерирующего агента пропускают через колонну со смолой, которая достигла области проскока, при комнатной температуре и линейной скорости 0,083±0,008 м/мин, и момент времени, когда 55±2% или более адсорбированного Co десорбировалось в результате операции десорбции, считают концом десорбции. Затем, используя чистую воду в объеме, таком же, как и объем колонны с хелатообразующей смолой или более, промывают колонну со смолой, пропуская воду с линейной скоростью 0,15±0,02 м/мин.
После промывки чистой водой хелатообразующую смолу конвертируют из H-формы (водородной) в Na-форму, пропуская 5,0±0,5 по массе водный раствор гидроксида натрия через колонну при температуре 35±2°C и линейной скорости 0,083±0,008 м/мин в восходящем режиме течения в течение 1 часа. Посредством данной операции 90% смолы или более конвертируют в Na-форму и затем начинают операцию адсорбции (1).
После того, как вышеуказанную операцию адсорбции и операцию регенерации повторяют в течение 6 месяцев, рассчитывают величину снижения адсорбционной емкости по Cu в соответствии со следующим уравнением:
Величина снижения адсорбционной емкости по Cu [%/месяц]=(a-b)/(a×6)
a: Адсорбционная емкость хелатообразующей смолы по Cu до тестирования [ммоль/мл]
b: Адсорбционная емкость хелатообразующей смолы по Cu через 6 месяцев тестирования [ммоль/мл]
Хелатообразующая смола, используемая в настоящем изобретении, предпочтительно обладает эластичностью в состоянии погружения в воду.
В качестве одного конкретного примера подходящей хелатообразующей смолы, обладающей вышеуказанными свойствами, не ограничиваясь этим, можно, например, указать Lewatit (R) MonoPlus TP 207, продаваемую Sybron Chemicals Inc. A LANXESS COMPANY.
Метод регулирования величины снижения адсорбционной емкости по Cu хелатообразующей смолы внутри вышеуказанного диапазона можно осуществить, выбирая хелатообразующую смолу, как указано выше, а также с помощью метода заполнения хелатообразующей смолой колонны с хелатообразующей смолой или оптимизируя конструкцию колонны с хелатообразующей смолой. Конкретно, предпочтительно разработать конструкцию таким образом, чтобы очищаемые сточные воды могли равномерно протекать через пространство между частицами хелатообразующей смолы в течение прохождения очищаемых сточных вод через область, которая заполнена хелатообразующей смолой. Другими словами, предпочтительно разработать конструкцию так, чтобы отсутствовала зона задержки, через которую не проходят очищаемые сточные воды, или очищаемые сточные воды не проходят только через определенную область. Кроме того, предпочтительно разработать конструкцию так, чтобы части, такие как ячейки сита, перфорированный металл или фильтр, обеспечивались в области, которую заполняют хелатообразующей смолой, и, таким образом, частицы хелатообразующей смолы, имеющие маленький размер, не рассеиваются вместе с очищаемыми сточными водами. В данной связи, когда используется хелатообразующая смола, имеющая особый коэффициент однородности, определенный изобретением, можно подавить снижение эксплуатационных характеристик из-за рассеивания частиц хелатообразующей смолы, имеющих малый размер. Кроме того, во внутренней части колонны с хелатообразующей смолой в течение выгрузки в нижней части образуется слой хелатообразующей смолы, и в верхней части образуется слой только сточных вод. Таким образом, частицы хелатообразующей смолы могут суспендироваться и переноситься в слое только сточных вод в верхней части, и можно увеличить снижающуюся величину адсорбционной емкости по Cu оптимизацией объема текучей среды слоя только сточной воды в верхней части или количественного баланса между верхним слоем и слоем хелатообразующей смолы.
Третье изобретение относится к способу очистки сточных вод, включающему приведение очищаемых сточных вод в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в очищаемых сточных водах, посредством хелатообразующей смолы, и затем регенерацию хелатообразующей смолы для получения жидкости регенерации, содержащей соединение тяжелого металла, где сточные воды приводят в контакт с хелатообразующей смолой при определенных условиях, и далее хелатообразующую смолу после контакта со сточными водами регенерируют при определенных условиях.
Нижеследующее будет описывать способ и условия адсорбции сточных вод, содержащих соединения тяжелых металлов, таких как кобальт и марганец, с использованием хелатообразующей смолы со ссылкой на Фиг.3. Фиг.3 представляет собой чертеж, показывающий пример технологических операций для стадии улавливания соединения тяжелого металла согласно изобретению.
Маточную жидкость, отделенную от п-толуиловой кислоты и аналогичного, 28′, образовавшуюся на стадии S7, и сточные воды стадии регенерации катализатора 26′, образовавшиеся на стадии S9, смешивают в резервуаре корректировки pH 101, чтобы скорректировать pH, и смесь сначала пропускают через любую из колонн с хелатообразующей смолой 102 или 103, которые заполнены вышеуказанной хелатообразующей смолой. Колонны с хелатообразующей смолой 102 и 103 обычно представляют собой аппараты с неподвижным слоем, и поточный способ принят для прохождения жидкости. Жидкость, прошедшую через любую из колонн с хелатообразующей смолой 102 или 103, перемещают на стадию S11 через резервуар сточных вод 104. Когда колонна с хелатообразующей смолой 102 достигает области проскока, пропускание очищаемых сточных вод переключают на колонну с хелатообразующей смолой 103 и прошедшую жидкость перемещают в резервуар сточных вод 104, как описано выше. Толстая сплошная линия на Фиг.3 показывает поток сточных вод, которые следует очищать.
Величину pH сточных вод, которые следует пропускать через колонну с хелатообразующей смолой 102 или 103 корректируют в резервуаре корректировки pH 101, и pH очищаемых сточных вод, предпочтительно, составляет 5,1 или более, более предпочтительно, 5,2 или более, еще более предпочтительно, 5,3 или более и, предпочтительно, 5,9 или менее, более предпочтительно, 5,8 или менее, еще более предпочтительно, 5,7 или менее. Когда pH сточных вод превышает вышеуказанный диапазон, соединения тяжелых металлов могут осаждаться, накапливаясь в виде осадка на поверхности хелатообразующей смолы, так что их улавливание становится невозможным. Когда pH сточных вод меньше вышеуказанного диапазона, могут осаждаться терефталевая кислота и органические вещества, такие как п-толуиловая кислота, содержащиеся в сточных водах, или величина улавливания соединений тяжелых металлов может снизиться.
Температура очищаемых сточных вод, которые следует пропускать через колонну с хелатообразующей смолой 102 или 103, предпочтительно, составляет 51°C или выше, более предпочтительно, 53°C или выше, еще более предпочтительно, 55°C или выше и, предпочтительно, 59°C или ниже, более предпочтительно, 57°C или ниже. Когда температура сточных вод ниже, чем вышеуказанный диапазон, в случае, когда в сточных водах содержится терефталевая кислота, она стремится легко выпасть в осадок, и величина извлечения соединений тяжелых металлов снижается. Когда температура сточных вод превышает вышеуказанный диапазон, хелатообразующая смола имеет тенденцию легко деградировать.
Скорость протекания очищаемых сточных вод, которые следует пропускать через колонну с хелатообразующей смолой 102 или 103, предпочтительно, составляет 5 м/ч или более, более предпочтительно, 7 м/ч или более и, предпочтительно, 14 м/ч или менее, более предпочтительно, 10 м/ч или менее, в виде линейной скорости. Когда скорость потока превышает вышеуказанный диапазон, внутри колонны с хелатообразующей смолой 102 или 103 может иметь место унос и каналообразование, и эффективная адсорбция, таким образом, становится невозможной. Когда скорость потока меньше вышеуказанного диапазона, колонну с хелатообразующей смолой 102 или 103 надо увеличивать, и, следовательно, данный случай имеет тенденцию быть экономически невыгодным.
В качестве способа адсорбции соединений тяжелых металлов существует периодический способ и непрерывный способ, но непрерывный способ является общепринятым, и желательно осуществлять адсорбцию непрерывным проточным способом с неподвижным слоем. Параллельно размещают два или более резервуаров, которые действуют посредством метода одновременного пропускания жидкости или метода переключения. В качестве материала для резервуара очистки обычно используют углеродистую сталь, футерованную волокнистонаполненным пластиком, или аналогичное.
Величина адсорбции кобальта и марганца при вышеуказанных условиях адсорбции составляет 80% или более, исходя из содержания металла на входе, и 90% или более является более предпочтительным.
После того, как стадия адсорбция закончена и колонну переключают на другую колонну с хелатообразующей смолой 102 или 103, колонну с хелатообразующей смолой 102 или 103, на которой адсорбция была закончена, затем промывают водой восходящим потоком и нисходящим потоком, чтобы вымыть адсорбированные или осажденные металлы и органические соединения. Что касается температуры и давления, в данной ситуации используются примерно такие же условия, как для вышеуказанной адсорбции тяжелых металлов.
Нижеследующее будет описывать способ и условия десорбции/регенерации колонны с хелатообразующей смолой 102 или 103.
В качестве регенерирующего агента для адсорбированных соединений тяжелых металлов используют раствор бромистого водорода. В общем, для регенерации хелатообразующих смол используют серную кислоту, хлористоводородную кислоту или аналогичную, но в случае данного изобретения использование бромистого водорода является наиболее предпочтительным по причине, что кобальт, марганец и соединение брома используются в качестве катализаторов для реакции окисления.
Концентрация водного раствора бромистого водорода для использования в изобретении составляет 7,1% по массе или более, предпочтительно, 7,5% по массе или более, более предпочтительно, 8% по массе или более и 19% по массе или менее, предпочтительно, 15% по массе или менее, более предпочтительно, 12% по массе или менее. В изобретении было обнаружено, что извлеченное количество соединений тяжелых металлов зависит от концентрации водного раствора бромистого водорода, и существует оптимальная концентрация. Когда концентрация водного раствора бромистого водорода меньше чем вышеуказанный диапазон или превышает вышеуказанный диапазон, нельзя добиться хорошего улавливания соединений тяжелых металлов.
Скорость протекания водного раствора бромистого водорода 7′ в колонне с хелатообразующей смолой 102 или 103, предпочтительно, находится в диапазоне от 1 м/ч или более до 10 м/ч или менее в виде линейной скорости. Когда скорость попадает внутрь данного диапазона, никакого влияния на извлекаемое количество не наблюдается. Температура водного раствора бромистого водорода в течение протекания жидкости, желательно, находится в диапазоне примерно обычной температуры (от 15°C или выше до 40°C или ниже). Величина десорбции соединений тяжелых металлов водным раствором бромистого водорода, предпочтительно, составляет 90% или более, более предпочтительно, 95% или более, и еще более предпочтительным является отношение, близкое к 100%.
На Фиг.3 водный раствор бромистого водорода 7′ хранят в резервуаре регенерирующего агента 106 и пропускают через колонну с хелатообразующей смолой 102 или 103, на которой была закончена операция адсорбции (102 в случае Фиг.3) вдоль пунктирной линии, чтобы осуществить десорбцию соединений тяжелых металлов. Затем, после того как башню промывают водой 9′ для удаления остающихся в смоле металлов и аналогичного, водный раствор гидроксида натрия 8′, хранящийся в резервуаре гидроксида натрия 108 или запасном резервуаре гидроксида натрия 109, пропускают через колонну с хелатообразующей смолой 102 или 103, на которой была закончена операция регенерации, для осуществления регенерации хелатообразующей смолы, ионизированной натрием, или хелатообразующей смолы, частично ионизированной натрием.
Концентрация гидроксида натрия в водном растворе гидроксида натрия 8′, предпочтительно, составляет от 1% по массе или более до 10% по массе или менее, более предпочтительно, от 3% по массе или более до 7% по массе или менее.
В качестве линейной скорости при прохождении водного раствора гидроксида натрия 8′ в колонне с хелатообразующей смолой 102 или 103, предпочтительно, выбирают от 1 м/ч или более до 20 м/ч или менее, более предпочтительно, от 2 м/ч или более до 15 м/ч или менее. Метод ионизации в случае, когда колонна с хелатообразующей смолой 102 или 103 является аппаратом с неподвижным слоем, предпочтительно, представляет собой режим восходящего потока, а не нисходящего потока. В данной ситуации предпочтительным является условие, когда хелатообразующая смола погружена в водный раствор гидроксида натрия в течение 2 часов или более. Температура ионизации, предпочтительно, составляет от 10°C или выше до 80°C или ниже, более предпочтительно, представляет собой примерно обычную температуру (от 15°C или выше до 40°C или ниже).
Соединения тяжелых металлов, извлеченные со стадии S10, хранят в резервуаре жидкости вторичного улавливания катализатора 105 вместе с бромистым водородом. Затем, в качестве жидкости вторичного улавливания катализатора 29′, хранимую жидкость смешивают с жидкостью первичного улавливания регенерации катализатора 25′ в резервуаре смешивания каталитической жидкости 21 на Фиг.1 и, после пополнения утраченного количества катализатора, смесь направляют на стадию S1 для повторного использования.
Сточные воды стадии вторичного улавливания катализатора 30′, используемые для промывки, перемещают в установку биологической очистки 23 через резервуар сточных вод 104, и там очищают.
Вблизи конечной точки операции десорбции поток проходящей жидкости переключают, и жидкость хранят в резервуаре конечной жидкости операции десорбции 107. Жидкость, хранимую в резервуаре конечной жидкости операции десорбции 107, используют для пропускания через колонну с хелатообразующей смолой 102 или 103 при следующей десорбции.
Кроме того, колонна с хелатообразующей смолой не ограничивается случаем поочередной работы двух колонн, а включает случай одновременной работы двух колонн последовательно и случай, должным образом, объединения чередующейся работы и работы при последовательном расположении с использованием трех и более колонн.
11. Стадия биологической очистки (S11)
Стадия S11 представляет собой стадию биологической очистки всего количества или части сточных вод, выгружаемых из вышеуказанных стадий S1-S10. Примеси в данных конечных сточных водах разлагаются с помощью установки биологической очистки 23. Установка биологической очистки 23 обычно состоит из комбинации резервуара общего осаждения и резервуара аэрации.
Сточные воды 16′, 17′, 18′, 19′, 23′ и 30′, выгружаемые со стадий S1-S10, не всегда передают на стадию биологической очистки непосредственно без какой-либо обработки и очищают, а в некоторых случаях каждую из сточных вод подвергают обработке, соответствующей составу сточных вод, характерному для каждой стадии, и используют без дополнительно очистки для рециркуляции без их передачи на стадию биологической очистки.
Очищаемые сточные воды 33′ после биологической очистки обычно сбрасывают в общую акваторию, такую как река. Избыточный активный ил 32′ сжигают в установке для сжигания 22, а золу 34′ удаляют в виде промышленных отходов.
Согласно способам изобретения, количество соединения тяжелого металла, выгружаемое за пределы системы, можно понизить до минимального значения, составляющего примерно 1%, исходя из общего количества, используемого на стадии S1. Соответственно, можно добиться существенного снижения относительно обычного выгружаемого количества, составляющего от 5 до 10%.
В указанном выше, изобретение описывается в части способа получения терефталевой кислоты высокой чистоты в качестве типичного примера ароматической карбоновой кислоты, но изобретение можно использовать, например, для случая терефталевой кислоты средней чистоты.
В случае получения терефталевой кислоты средней чистоты в способе содержатся стадии разделения на твердую и жидкую фазы, промывки и сушки после дополнительного окисления при высокой температуре и высоком давлении суспензии неочищенной терефталевой кислоты, полученной реакцией окисления без гидрирования. Отделенную маточную жидкость, промывающую воду и аналогичное после дополнительного окисления частично повторно используют для рециркуляции в реакционной стадии и остающуюся отделенную маточную жидкость и промывающую воду обрабатывают для улавливания и повторного использования ценных компонентов аналогично случаю терефталевой кислоты высокой чистоты. Более того, содержащиеся соединения тяжелых металлов также можно извлечь и повторно использовать посредством аналогичных стадий.
Кроме того, изобретение также применимо к способам получения других ароматических карбоновых кислот, таких как изофталевая кислота, которые содержат аналогичные технологические стадии, где в реакции окисления используют катализатор на основе тяжелого металла, и преимущества с экологической и экономической точки зрения являются заметными.
Способ очистки сточных вод по изобретению можно применить не только к сточным водам, образующимся в процессе получения ароматической карбоновой кислоты, но также к сточным водам, содержащим сточные воды, полученные из других химических установок. Даже в таком случае соединение тяжелого металла эффективно улавливается, и регенерация хелатообразующей смолы и дальнейшее повторное использование жидкости для регенерации осуществляется без каких-либо проблем.
Изобретение может обеспечить технологию, которая не только приводит к экономическому эффекту, получаемому от очистки сточных вод, но также не оказывает вредного воздействия на окружающую среду с практической применимостью и, таким образом, изобретение является крайне важным и высокоэффективным также с экологической точки зрения.
[Примеры]
Нижеследующее будет описывать изобретение более конкретно со ссылкой к примерам, но изобретение не ограничивается следующими ниже примерами.
Пример 1
В оборудовании для получения терефталевой кислоты высокой чистоты (производительность 75 тонн/час) в реактор жидкофазного окисления непрерывно подавали п-ксилол и примерно 3 массовых эквивалента уксусной кислоты, исходя из п-ксилола, ацетат кобальта, ацетат марганца и бромистый водород в качестве катализаторов, и реакцию окисления осуществляли при температуре от 185°C или выше до 195°C или ниже и давлении от 1,0 МПа или более до 1,7 МПа или менее в течение времени реакции (среднее время пребывания) 90 минут. Количества использованных катализаторов были следующие: кобальтовый компонент и марганцевый компонент, каждый, составляли 300 ч/млн по массе в пересчете на металл; бромистый компонент составлял 700 ч/млн, исходя из растворителя. В качестве газа для осуществления реакции окисления молекулярным кислородом использовали воздух. В данной ситуации содержание кислорода в воздухе составляло 21% и, таким образом, сжатый воздух подавали в реактор так, чтобы концентрация кислорода в газе, сбрасываемом из реактора, составляла от 3% по объему или более до 7% по объему или менее.
Затем полученную в результате окисления суспензию непрерывно подавали в реактор низкотемпературного окисления и дополнительную реакцию окисления осуществляли при температуре от 180°C или выше до 195°C или ниже и давлении от 0,9 МПа или более до 1,7 МПа или менее в течение времени реакции от 40 минут или более до 60 минут или менее. В качестве газа для осуществления реакции окисления подавали сжатый воздух (содержание кислорода 21%) так, чтобы концентрация кислорода в сбрасываемом газе составляла от 3% по объему или более до 7% по объему или менее. Поддерживая температуру и давление полученной в результате суспензии неочищенной терефталевой кислоты после завершения реакции, суспензию подвергали разделению на твердую и жидкую фазы посредством декантерной центрифуги, и затем полученный в результате отфильтрованный осадок неочищенной терефталевой кислоты промывали водным раствором уксусной кислоты. Отфильтрованный осадок неочищенной терефталевой кислоты сушили выпариванием связанной маточной жидкости посредством сброса давления в сушильной установке для работы под давлением и перемещали на следующую стадию очистки.
Все количество промывающей жидкости и 80% отделенной маточной жидкости, полученной разделением суспензии неочищенной терефталевой кислоты на твердую и жидкую фазы, повторно использовали в реакторе жидкофазного окисления. Что касается остающихся 20% отделенной маточной жидкости, низкокипящие компоненты, такие как уксусная кислота, сначала выпаривали на двух стадиях на выпарной установке и полученную уксусную кислоту повторно использовали в реакционной стадии после дегидратации. Концентрат на выпарной установке снова превращали в суспензию, добавляя воду, и затем подвергали разделению на твердую и жидкую фазы на горизонтальном ленточном фильтре, после чего следовала промывка. Твердое вещество рассматривали в качестве отходов, а компоненты катализатора извлекали из отделенной жидкости и повторно использовали на реакционной стадии. Остающуюся жидкость (сточные воды 1), из которой были извлечены компоненты катализатора, подвергали очистке хелатообразующей смолой, как указано далее.
Отфильтрованный осадок неочищенной терефталевой кислоты смешивали с водой в смесительном резервуаре и затем нагревали и повышали давление от 280°C или выше до 300°C или ниже и от 7 МПа или более до 10 МПа или менее паром и переносящим тепло маслом для осуществления растворения. После этого раствор вводили в колонну реакции гидрирования, содержащую неподвижный слой активированного палладием угля, вместе с водородом для осуществления реакции гидрирования. Водный раствор терефталевой кислоты после реакции непрерывно перемещали в резервуар кристаллизации и затем охлаждали посредством сброса давления и кристаллизовали в четырехстадийной операции кристаллизации. Затем полученную в результате смесь подвергали разделению на твердую и жидкую фазы при 100°C, и отфильтрованный осадок терефталевой кислоты высокой чистоты промывали водой и сушили. В качестве сушильных установок использовали сушильную установку для работы под давлением для выпаривания посредством сброса давления и сушилку с псевдоожиженным слоем, и получали отфильтрованный осадок терефталевой кислоты высокой чистоты с выходом 75 тонн/ч. Поскольку отделенная маточная жидкость содержала промежуточные продукты окисления, такие как п-толуиловая кислота, маточную жидкость дополнительно охлаждали, чтобы осадить кристаллы, и после разделения на твердую и жидкую фазы промежуточные продукты окисления собирали в виде твердого вещества и повторно использовали на стадии жидкофазной реакции окисления. Маточную жидкость (сточные воды 2) после того, как осадок промежуточных продуктов окисления был отделен, объединяли с вышеуказанными сточными водами 1 (в дальнейшем называемыми исходными сточными водами), которые подвергали следующему ниже методу очистки сточных вод.
Во-первых, pH вышеуказанных исходных сточных вод корректировали до 5,5 в резервуаре 101 корректировки pH и полученные в результате сточные воды пропускали через колонны с хелатообразующей смолой 102 и 103, заполненные хелатообразующей смолой (Lewatit (R) MonoPlus TP 207, изготовленной Sybron Chemicals Inc. A LANXESS COMPANY), показанной в таблице 1. В этой связи, ион хелатообразующей смолы был выражен теоретической величиной, и почти все его количество было конвертировано в натриевую форму. В исходных сточных водах Co, в качестве тяжелого металла, содержался в количестве 5 ч/млн, а также содержались незначительные количества Mn и, в качестве органических соединений, терефталевая кислота и п-толуиловая кислота, таким образом, величина CODCr (химическая потребность в кислороде) была равна 4000 ч/млн. В этой связи, измерение концентрации Co осуществляли согласно JIS K 0102 60 (60.3: эмиссионный спектральный анализ с индуктивно связанной плазмой) и CODCr измеряли согласно JIS K 0102 20.
(ммоль/г)
Колонны с хелатообразующей смолой 102 и 103 представляли собой проточные реакторы с неподвижным слоем почти цилиндрической формы, отношение внутреннего диаметра к высоте у части, которая была заполнена хелатообразующей смолой, составляло 1,0:0,7, и частицами хелатообразующей смолы заполняли до 50% высоты в неподвижном состоянии. Объем части, которую заполняли хелатообразующей смолой, составлял 30 м3 или более. Жидкость пропускали в режиме потока и две колонны (102 и 103) помещали параллельно, причем их можно было переключать. Колонну с хелатообразующей смолой 102 или 103, которая достигала области проскока, подвергали таким операциям, как регенерация, промывка и Na ионный обмен, как будет указано далее.
Температуру исходных сточных вод, величина pH которых была скорректирована до 5,5, повышали до 55°C, и их пропускали через колонну с хелатообразующей смолой 102 при скорости потока 67,5 м3/ч и линейной скорости 8,9 м/ч. Сточные воды после обработки, осуществляемой пропусканием жидкости, перемещали в резервуар сточных вод 104 и хранили в нем, и затем сточные воды перемещали в установку для очистки сточных вод.
Момент времени, когда концентрация Co на выходе из колонны с хелатообразующей смолой 102 достигала 1 ч/млн или более, считался областью проскока, и пропускание жидкости проводили непрерывно, переключая поток жидкости в колонну с хелатообразующей смолой 103.
Концентрация Co в сточных водах после пропускания жидкости составляла примерно от 0,01 до 0,04 ч/млн на ранней стадии операции. Затем концентрация Co в сточных водах после пропускания жидкости постепенно повышалась, но ее можно было поддерживать на уровне менее чем 1 ч/млн, как указано выше. Из данных результатов, хотя концентрация Co в сточных водах, сбрасываемых за пределы системы, обычно составляла 5 ч/млн, концентрацию можно было подавить до менее чем 1 ч/млн, осуществляя обработку по изобретению. В данной связи, в настоящем примере момент времени, когда концентрация Co достигала 1 ч/млн или более, считался областью проскока, но область проскока можно, соответственно, установить при любом значении при условии, что данное значение больше чем концентрация Co на ранней стадии операции, и может быть, соответственно, установлена в зависимости от нормативов или аналогичного для сточных вод.
Затем, для того чтобы регенерировать (десорбировать) адсорбированный Co и другие тяжелые металлы, пропускание жидкости осуществляли через колонну с хелатообразующей смолой 102, которая достигла области проскока, используя 10% по массе водный раствор бромистого водорода в качестве регенерирующего агента из резервуара регенерирующего агента 106. Температура регенерирующего агента в данной ситуации представляла собой обычную температуру, и скорость пропускания жидкости составляла 5 м/ч. Осуществляя пропускание жидкости в течение 1 часа или более, десорбировали 90% Co или более. Извлеченные соединения тяжелых металлов, таких как Co, хранили в резервуаре вторичного улавливания катализатора 105 вместе с водным раствором бромистого водорода в качестве регенерирующего агента и затем смешивали, в качестве жидкости вторичного улавливания 29′, с жидкостью первичного улавливания 25′ в резервуаре смешивания катализатора 21 на Фиг.1, возвращали на стадию реакции окисления и повторно использовали.
После того, как операция регенерации водным раствором бромистого водорода закончена, воду 9′ пропускали через колонну с хелатообразующей смолой 102 для промывки, чтобы удалить незначительное количество металлов, остающихся в смоле. После этого, чтобы приготовиться к следующей адсорбции, 5% по массе водный раствор гидроксида натрия пропускали из резервуара гидроксида натрия 108 и резервного резервуара 109 через колонну с хелатообразующей смолой 102, которая была переведена в водородную форму регенерирующей жидкостью. Пропускание жидкости в данной ситуации осуществляли в режиме восходящего потока при линейной скорости 5 м/ч и температуре 35°C.
Осуществляя ряд непрерывных повторяющихся операций адсорбции, промывки, регенерации (десорбции), промывки и обмена натриевой формы, как указано выше, с переключением между колоннами с хелатообразующей смолой 102 и 103, операции улавливания соединений тяжелого металла многократно осуществляли в течение 6 месяцев.
В результате, адсорбционная емкость по Cu для хелатообразующей смолы, использованной в настоящем примере, понизилась от 0,71 ммоль/см3 в начале ее использования (новая смола) до 0,47 ммоль/см3, и величина снижения адсорбционной емкости по Cu составляла 5,6%/месяц. Данный факт означает, что в течение периода 6 месяцев было потеряно 34% ионообменных групп по сравнению с неиспользованной смолой.
Пример сравнения 1
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что хелатообразующую смолу меняли на смолу, имеющую коэффициент однородности 1,41, как показано в таблице 1.
В результате на четвертый месяц после начала использования ряд непрерывных повторяющихся операций адсорбции, промывки, регенерации (десорбции), промывки и обмена натриевой формы перешел на промышленно непригодный уровень и, таким образом, смолу следовало заменить новой смолой.
В результате измерения адсорбционной емкости хелатообразующей смолы по Cu таким же способом, как в примере 1, после использования в настоящем примере сравнения, адсорбционная емкость по Cu понизилась от 0,71 ммоль/см3 в начале ее использования (новая смола) до 0,40 ммоль/см3 после 4 месяцев использования. Данный факт означает, что было потеряно 46% ионообменных групп в течение периода только 4 месяцев по сравнению с неиспользованной смолой, и величина снижения адсорбционной емкости по Cu составляла примерно 11,5%/месяц.
Справочный пример 1
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что pH исходных сточных вод корректировали до 6,0. В результате было обнаружено, что тяжелые металлы в исходных сточных водах выпали в осадок и, таким образом, не только протекание жидкости, но также ряд непрерывных повторяющихся операций адсорбции, промывки, регенерации (десорбции), промывки и обмена натриевой формы нельзя было осуществить.
Справочный пример 2
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что pH исходных сточных вод корректировали до 5,0. В результате было обнаружено, что незначительное количество органических соединений в исходных сточных водах выпало в осадок и, таким образом, не только протекание жидкости, но также ряд непрерывных повторяющихся операций адсорбции, промывки, регенерации (десорбции), промывки и обмена натриевой формы нельзя было осуществить.
Справочный пример 3
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что линейную скорость протекания исходных сточных вод через колонны с хелатообразующей смолой 102 и 103 меняли до 15 м/ч. В результате проскок для смолы ускорился из-за быстрой линейной скорости, и величина снижения адсорбционной емкости по Cu составляла 11,0%/месяц.
Пример сравнения 2
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что 20% по массе водный раствор бромистого водорода использовали в регенерационной обработке. В результате было обнаружено, что для адсорбированных тяжелых металлов величина улавливания Co составляла только 80% по сравнению с величиной в примере 1 и, таким образом, было обнаружено, что величина улавливания тяжелого металла снизилась.
Пример сравнения 3
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что 5% по массе водный раствор бромистого водорода использовали в регенерационной обработке. В результате для адсорбированных тяжелых металлов величина улавливания Co составляла только 75% по сравнению с величиной в примере 1 и, таким образом, было обнаружено, что величина улавливания тяжелого металла снизилась.
Пример сравнения 4
Исходные сточные воды очищали таким же способом и при тех же условиях, как в примере 1, за исключением того, что 30% по массе водный раствор бромистого водорода использовали в регенерационной обработке. В результате для адсорбированных тяжелых металлов величина улавливания Co составляла только 25% по сравнению с величиной в примере 1 и, таким образом, было обнаружено, что величина улавливания тяжелого металла снизилась в большей степени. В результате непрерывная работа посредством повторения адсорбции и регенерации хелатообразующей смолы была затруднительна.
В этой связи, все содержание описания, формулы изобретения, чертежей и реферата заявки на патент Японии № 2008-186235, поданной 17 июля 2008 года, цитируется здесь и включается в качестве раскрытия описания настоящего изобретения.
Применимость в промышленности
Согласно изобретению становится возможным уловить соединения тяжелых металлов, таких как кобальт и марганец, содержащиеся в сточных водах, образующихся в процессе производства ароматической карбоновой кислоты, с высокой эффективностью, и повторно их использовать. Согласно изобретению, поскольку получаются вышеуказанные эффекты, количество вышеуказанных металлических компонентов, сбрасываемых за пределы технологического процесса, можно снизить до минимума относительно общего количества используемых вышеуказанных металлических компонентов и, таким образом, становится возможным обеспечить безопасный для окружающей среды способ получения ароматической карбоновой кислоты, который является беспрецедентным. Способ дополнительно дает существенный экономический эффект, который получают улавливанием и повторным использованием соединений тяжелых металлов. Как правило, нефтехимические заводы часто включают в себя различные виды химических установок на одной площади или в одном промышленном районе в виде комплекса. Поэтому виды промышленных сточных вод, сбрасываемые из них, отличаются друг от друга, но имеется случай, когда технологические сточные воды из химических установок различного типа, сбрасывающих соединения тяжелых металлов, как указано выше, объединяют и сбрасывают в окружающую среду в зависимости от ситуации. Поскольку способ очистки сточных вод по изобретению может выполнить улавливание и повторное использование соединений тяжелых металлов, таких как кобальт и марганец, содержащихся в сточных водах, с высокой эффективностью, так как сточные воды содержат сточные воды, образующиеся в процессе получения ароматической карбоновой кислоты, представленной терефталевой кислотой, даже когда присутствуют сточные воды из химических установок различного типа, преимущества являются существенными не только с экономической точки зрения, но также с экологической точки зрения.
Описание номеров позиций и обозначений

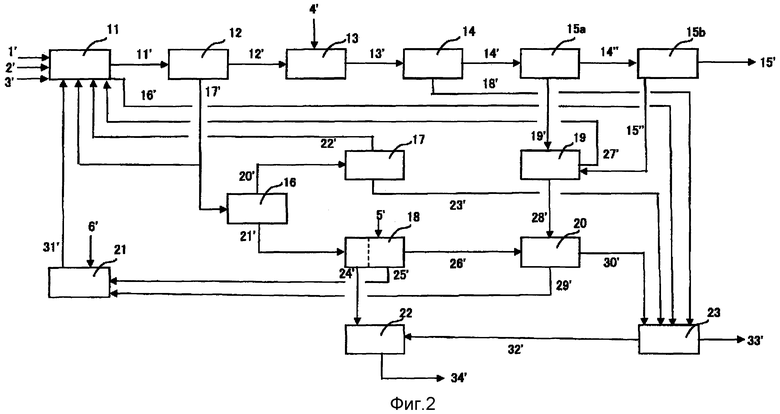
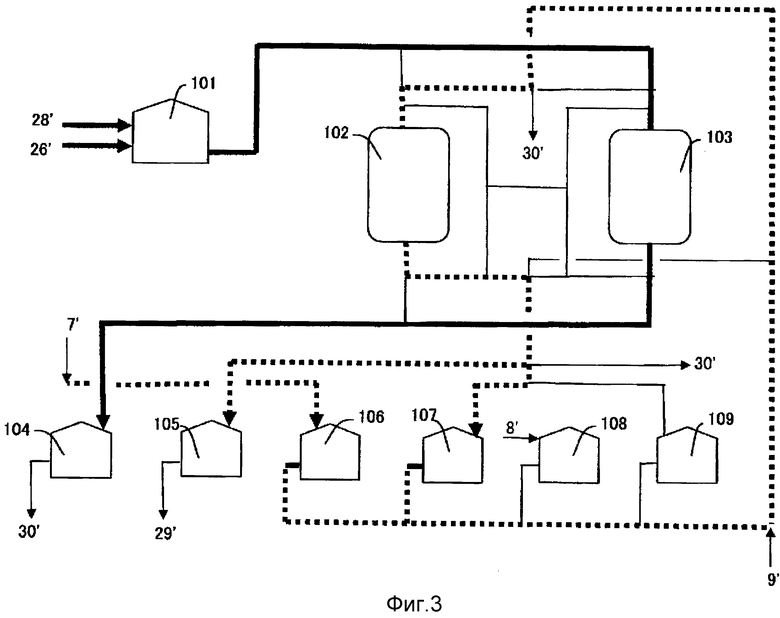
Изобретения могут быть использованы для очистки сточных вод, образующихся в процессе получения ароматических карбоновых кислот, от соединений тяжелых металлов. Для осуществления способа сточные воды приводят в контакт с частицами хелатообразующей смолы, имеющими коэффициент однородности 1,4 или менее, при этом pH сточных вод составляет 5,1-5,9 и скорость потока сточных вод составляет 5-14 м/час. Величина снижения адсорбционной емкости хелатообразующей смолы по Cu составляет 11% в месяц или менее. Регенерацию хелатообразующей смолы проводят водным раствором бромистого водорода с концентрацией от 7,1% до 19% по массе. В предпочтительных вариантах осуществления способа температура очищаемых сточных вод составляет от 51°C до 59°C, адсорбционная емкость хелатообразующей смолы по Cu составляет 0,5 ммоль/мл или более, а жидкость регенерации повторно направляют в систему реакции окисления при получении ароматических карбоновых кислот. Изобретения обеспечивают эффективное извлечение ионов тяжелых металлов при их низких концентрациях в очищаемых сточных водах. 3 н. и 8 з.п. ф-лы, 3 ил., 1 табл., 4 пр.
1. Способ очистки сточных вод, включающий приведение сточных вод, образующихся при получении ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, где хелатообразующая смола представляет собой частицы, имеющие коэффициент однородности 1,4 или менее, причем pH сточных вод, которые контактируют с хелатообразующей смолой, составляет от 5,1 или более до 5,9 или менее и скорость потока сточных вод, которые контактируют с хелатообразующей смолой, составляет от 5 м/час или более до 14 м/час или менее.
2. Способ очистки сточных вод, включающий приведение сточных вод, образующихся при получении ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, где величина снижения адсорбционной емкости по Cu для хелатообразующей смолы составляет 11%/месяц или менее, причем pH сточных вод, которые контактируют с хелатообразующей смолой, составляет от 5,1 или более до 5,9 или менее и скорость потока сточных вод, которые контактируют с хелатообразующей смолой, составляет от 5 м/час или более до 14 м/час или менее.
3. Способ очистки сточных вод, включающий приведение сточных вод, образующихся при получении ароматической карбоновой кислоты, в контакт с хелатообразующей смолой для улавливания соединения тяжелого металла, содержащегося в сточных водах, посредством хелатообразующей смолы, и затем регенерацию хелатообразующей смолы для получения жидкости регенерации, содержащей соединение тяжелого металла, где pH сточных вод, которые следует привести в контакт с хелатообразующей смолой, составляет от 5,1 или более до 5,9 или менее, скорость потока сточных вод, которые следует привести в контакт с хелатообразующей смолой, составляет от 5 м/час или более до 14 м/час или менее, и хелатообразующую смолу регенерируют, используя водный раствор бромистого водорода с концентрацией от 7,1% по массе или более до 19% по массе или менее.
4. Способ очистки сточных вод по п.3, где хелатообразующая смола представляет собой частицы, имеющие коэффициент однородности 1,4 или менее.
5. Способ очистки сточных вод по любому одному из пп.1, 2, 3 или 4, где температура сточных вод, которые следует привести в контакт с хелатообразующей смолой, составляет от 51°C или выше до 59°C или ниже.
6. Способ очистки сточных вод по любому одному из пп.1, 2, 3 или 4, где адсорбционная емкость хелатообразующей смолы по Cu составляет 0,5 ммоль/мл или более.
7. Способ очистки сточных вод по п.5, где адсорбционная емкость хелатообразующей смолы по Cu составляет 0,5 ммоль/мл или более.
8. Способ очистки сточных вод по п.6, где адсорбционная емкость хелатообразующей смолы по Cu составляет 0,5 ммоль/мл или более.
9. Способ очистки сточных вод по любому одному из пп.3, 4, 7 или 8, где жидкость регенерации повторно направляют в систему реакции окисления при получении ароматической карбоновой кислоты.
10. Способ очистки сточных вод по п.5, где жидкость регенерации повторно направляют в систему реакции окисления при получении ароматической карбоновой кислоты.
11. Способ очистки сточных вод по п.6, где жидкость регенерации повторно направляют в систему реакции окисления при получении ароматической карбоновой кислоты.
| Бандажированный прокатный валок | 1984 |
|
SU1224026A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| БУРКХАРД БРИНГС, Удаление радионуклидов современными ионообменными смолами, Водоочистка | |||
| Водоподготовка | |||
| Водоснабжение, 2010, N2, с.с | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| АШИРОВ А., Ионообменная очистка сточных вод, растворов и газов, Ленинград, "Химия", 1983, с.с | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| EP | |||

