ОБЛАСТЬ ТЕХНИКИ
[0001]
Настоящее изобретение относится к способу получения ароматического соединения, такого как цианопиридин, цианопиразин или т.п., и к способу получения карбонатного сложного эфира.
УРОВЕНЬ ТЕХНИКИ
[0002]
«Карбонатный сложный эфир» представляет собой общее название соединений, получаемых в результате замещения одного атома или двух атомов из двух атомов водорода карбоновой кислоты, СО(ОН)2, на алкильную группу или арильную группу, и имеющих структуру RO-C(=O)-OR' (каждый R и R' представляет собой насыщенную углеводородную группу или ненасыщенную углеводородную группу).
[0003]
Карбонатный сложный эфир используют в качестве присадки, например, бензиновой присадки для повышения октанового числа, а также в качестве присадки для дизельного топлива для уменьшения количества частиц в выхлопном газе. Карбонатный сложный эфир также используют, например, в качестве алкилирующего агента, карбонилирующего агента, растворителя или т.п., используемых для синтеза смол или органических соединений, таких как поликарбонат, уретан, фармацевтические препараты, сельскохозяйственные химикаты или т.п., вещества электролитических растворов для литиево-ионных элементов, вещества для смазочных масел или вещества-абсорбенты кислорода для ингибирования коррозии в трубах паровых котлов. Как можно видеть, карбонатный сложный эфир представляет собой весьма ценное соединение.
[0004]
В соответствии с традиционным главным способом получения карбонатного сложного эфира, фосген, который используют в качестве источника карбонила, непосредственно приводят в контакт со спиртом. Фосген, используемый в указанном способе, является чрезвычайно вредным и коррозионно-активным и, следовательно, необходимы исключительные меры предосторожности для работы с ним, например, при транспортировке или хранении. Большие затраты связаны с регулированием и контролированием, а также с обеспечением безопасности на производственных мощностях по выпуску фосгена. В соответствии с указанным способом, материалы и катализаторы, используемые для получения карбонатного сложного эфира, содержат галоген, такой как хлор или т.п., и получаемый карбонатный сложный эфир содержит следовые количества галогена, которые не удалены на стадии простой очистки. Если карбонатный сложный эфир используют для бензиновых присадок, присадок для легкого масла или электронного материала, то указанный галоген может вызывать нежелательную коррозию. Таким образом, неизбежно необходимо использовать стадию тонкой очистки для уменьшения следового количества галогена в карбонатном сложном эфире до крайне низкого следового содержания. Кроме того, недавно административные учреждения установили жесткие административные правила и не допускают создание новых производственных мощностей с применением данного способа, поскольку фосген чрезвычайно опасен для организма человека. В такой ситуации остро необходим новый способ получения карбонатного сложного эфира без применения фосгена.
[0005]
Существует также другой известный способ получения карбонатного сложного эфира. В соответствии с указанным способом, карбонатный сложный эфир напрямую синтезируют из спирта и диоксида углерода, используя гетерогенный катализатор. В отношении указанного способа проведены исследования по использованию 2-цианопиридина или бензонитрила в качестве смачиваемого порошка для существенного улучшения производственного выхода и скорости производства карбонатного сложного эфира, для обеспечения возможности быстрого протекания реакции при давлении, близком к нормальному давлению, а также для увеличения скорости реакции (см. патентные документы 1 и 2). Однако существует проблема, связанная со способом очистки или применением бензамида или подобного агента, образующегося в качестве побочного продукта.
Например, применение бензамида, образующегося в результате взаимодействия бензонитрила и воды, ограничено до небольшого количества фармацевтических и сельскохозяйственных промежуточных соединений. Таким образом, в отношении способа получения карбонатного сложного эфира с применением бензонитрила в качестве смачиваемого порошка, бензамид, образующийся в качестве побочного продукта, необходимо регенерировать в бензонитрил и использовать повторно. Теперь задачей является осуществление реакции регенерации с высокой степенью селективности (поскольку считают, что при образовании побочного продукта бензонитрил не может быть легко использован в качестве смачиваемого порошка) и с высоким выходом (поскольку при низком выходе остается большое количество бензамида, что увеличивает трудозатраты, а именно производственные расходы на отделение бензамида и бензонитрила друг от друга).
[0006]
В свете вышеописанной ситуации, когда регенерация бензамида или подобного вещества в бензонитрил или подобное вещество связана с проблемами, существует известный способ проведения регенерации без использования сильного реагента и с подавлением образования побочного продукта (патентный документ 3).
Однако в соответствии с указанным способом, для получения нитрила посредством дегидратации амидного соединения необходимо 400 часов и, следовательно, такой способ трудно адаптировать к реакции синтеза карбонатного сложного эфира, обычно занимающей лишь 24 часа. Указанный способ также связан с проблемой, которая обусловлена необходимостью использования стадий экстракции, инфильтрации и т.п. для жидкостно-твердофазного выделения катализатора, что увеличивает количество технологических стадий и усложняет производственный процесс.
СПИСОК ЦИТИРОВАННОЙ ЛИТЕРАТУРЫ
ПАТЕНТНАЯ ЛИТЕРАТУРА
[0007]
Патентный документ 1: выложенная для всеобщего ознакомления публикация патента Японии №2010-77113
Патентный документ 2: выложенная для всеобщего ознакомления публикация патента Японии №2012-162523
Патентный документ 3: WO2015/099053
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
[0008]
В свете вышеописанных технологических проблем, задача настоящего изобретения заключается в обеспечении способа регенерации ароматического амидного соединения, например, пиридинкарбоксамида или пиразинамида, в соответствующее ароматическое нитрильное соединение, а именно цианопиридин или цианопиразин, и в указанном способе реализована реакция дегидратации с получением требуемого соединения, селективно и с высоким выходом, с одновременным подавлением образования побочного продукта. Другая задача настоящего изобретения заключается в обеспечении способа получения ароматического нитрильного соединения, который уменьшает количество стадий реакции дегидратации и существенно повышает скорость реакции для сокращения времени реакции даже при давлении, близком к нормальному давлению.
Еще одна задача настоящего изобретения заключается в применении вышеописанного способа получения ароматических нитрильных соединений к способу получения карбонатного сложного эфира с целью обеспечения способа эффективного получения карбонатного сложного эфира.
РЕШЕНИЕ ПРОБЛЕМЫ
[0009]
Для достижения вышеописанных задач, авторами настоящего изобретения проведены исследования способа получения ароматического нитрильного соединения, такого как цианопиридин, цианопиразин или т.п., посредством дегидратации ароматического амидного соединения. Более конкретно, авторами настоящего изобретения изучены условия реакций дегидратации ароматического амидного соединения и в результате обнаружено следующее. В случае использования дифенилового эфира, имеющего температуру кипения выше температуры кипения образующегося ароматического нитрильного соединения и ниже температуры кипения ароматического амидного соединения, используемого в качестве сырья, а также при подборе температуры реакции, протекает процесс реакции дегидратации, благодаря которому существенно улучшается скорость реакции для сокращения времени реакции, требуемое соединение образуется селективно и с высоким выходом, при этом образование побочного продукта подавлено, а ароматическое нитрильное соединение можно легко выделить. Авторами настоящего изобретения также установлено следующее: Поскольку процесс реакции дегидратации, предложенный авторами настоящего изобретения, не требует осуществления жидкостно-твердофазного выделения катализатора, то количество стадий указанной реакции дегидратации уменьшено. Предпочтительно, реакцию дегидратации осуществляют при кипении дифенилового эфира.
[0010]
В результате вышесказанного, скорость регенерации ароматического нитрильного соединения посредством реакции дегидратации ароматического амидного соединения, а также скорость синтеза карбонатного сложного эфира из СО2 и спирта с применением указанного ароматического нитрильного соединения, становятся хорошо сбалансированными. А именно, реакция дегидратации и реакция синтеза карбонатного сложного эфира теперь являются последовательностью промышленных процессов. На основании этого авторами данного изобретения проведены также исследования по применению вышеописанных сведений для способа получения карбонатного сложного эфира. А именно, авторами настоящего изобретения в отношении способа получения карбонатного сложного эфира непосредственным синтезом карбонатного сложного эфира из спирта и диоксида углерода обнаружено следующее. В случае использования растворителя, имеющего температуру кипения выше температуры кипения ароматического амидного соединения, количество стадий реакции сокращается, а сам способ упрощается благодаря отсутствию необходимости жидкостно-твердофазного выделения катализатора. Авторы настоящего изобретения подтвердили, что такой способ синтеза карбонатного сложного эфира можно комбинировать с реакцией дегидратации ароматического амидного соединения с получением ароматического нитрильного соединения, обеспечивая блестящий результат. Ниже представлена сущность настоящего изобретения.
[0011]
(1) Способ получения ароматического нитрильного соединения, включающий:
реакцию дегидратации для дегидратации ароматического амидного соединения;
где в указанной реакции дегидратации используют дифениловый эфир.
(2) Способ получения ароматического нитрильного соединения по п. (1), отличающийся тем, что реакцию дегидратации проводят при кипении дифенилового эфира.
(3) Способ получения ароматического нитрильного соединения по п. (2), отличающийся тем, что дифениловый эфир имеет температуру кипения выше температуры кипения ароматического нитрильного соединения и температуры кипения воды, и ниже температуры кипения ароматического амидного соединения.
(4) Способ получения ароматического нитрильного соединения по любому из пп. (1) - (3), отличающийся тем, что реакцию дегидратации осуществляют при пониженном давлении.
(5) Способ получения ароматического нитрильного соединения по любому из пп. (1) - (4), отличающийся тем, что реакционный раствор реакции дегидратации имеет температуру, составляющую 170°С или выше, но ниже 230°С.
(6) Способ получения ароматического нитрильного соединения по любому из пп. (1) - (5), отличающийся тем, что ароматическое амидное соединение включает пиридинкарбоксамид или пиразинамид, и ароматическое нитрильное соединение включает цианопиридин или цианопиразин.
(7) Способ получения ароматического нитрильного соединения по любому из пп. (1) - (6), отличающийся тем, что в реакции дегидратации используют катализатор, содержащий цезий.
(8) Способ получения карбонатного сложного эфира, включающий:
первую стадию реакции, включающую реакцию получения карбонатного
сложного эфира посредством приведения во взаимодействие спирта и диоксида углерода в присутствии ароматического нитрильного соединения с образованием карбонатного сложного эфира и воды, и реакцию гидратации для гидратации полученной водой ароматического нитрильного соединения с образованием ароматического амидного соединения; и
вторую стадию реакции, осуществляемую после выделения ароматического амидного соединения из реакционной системы после первой стадии реакции, включающую регенерацию ароматического амидного соединения в ароматическое нитрильное соединение посредством реакции дегидратации для дегидратации ароматического амидного соединения при температуре реакционного раствора 170°С или выше и ниже 230°С;
при этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на второй стадии реакции, используют на первой стадии реакции.
(9) Способ получения карбонатного сложного эфира, включающий:
первую стадию реакции, включающую реакцию получения карбонатного сложного эфира посредством приведения во взаимодействие спирта и диоксида углерода в присутствии ароматического нитрильного соединения с образованием карбонатного сложного эфира и воды, и реакцию гидратации для гидратации полученной водой ароматического нитрильного соединения с образованием ароматического амидного соединения; и
вторую стадию реакции, осуществляемую после выделения ароматического амидного соединения из реакционной системы после первой стадии реакции, включающую регенерацию ароматического амидного соединения в ароматическое нитрильное соединение посредством реакции дегидратации для дегидратации ароматического амидного соединения в присутствии дифенилового эфира;
при этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на второй стадии реакции, используют на первой стадии реакции.
(10) Способ получения карбонатного сложного эфира по любому из пп. (8) или (9), отличающийся тем, что ароматическое амидное соединение включает пиридинкарбоксамид или пиразинамид, и ароматическое нитрильное соединение включает цианопиридин или цианопиразин.
(11) Способ получения карбонатного сложного эфира по любому из пп. (8) -(10), отличающийся тем, что в реакции дегидратации используют катализатор, содержащий цезий.
(12) Способ получения карбонатного сложного эфира по любому из пп. (8) - (11), отличающийся тем, что в реакции получения карбонатного сложного эфира используют катализатор, содержащий цезий.
(13) Способ получения карбонатного сложного эфира по любому из пп. (8) - (12), отличающийся тем, что указанный спирт включает спирт, содержащий от 1 до 6 атомов углерода.
(14) Способ получения карбонатного сложного эфира по любому из пп. (8) - (13), отличающийся тем, что на первой стадии реакции используют растворитель, имеющий температуру кипения выше температуры кипения получаемого ароматического амидного соединения.
(15) Способ получения карбонатного сложного эфира по п. (14), отличающийся тем, что указанный растворитель содержит по меньшей мере один из диалкилбензола, алкилнафталина и дифенилбензола.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0012]
В соответствии с настоящим изобретением, описанным выше, ароматическое нитрильное соединение, такое как цианопиридин, цианопиразин или т.п., эффективно получают (регенерируют) из ароматического амидного соединения, такого как пиридинкарбоксамид (пиколинамид, никотинамид или т.п.), бензамид или т.п. Более конкретно, реакцию дегидратации ароматического амидного соединения для указанной регенерации осуществляют для получения требуемого соединения, селективно и с высоким выходом, с одновременным подавлением образования побочного продукта. Даже при мягких условиях реакции, например, при давлении, близком к нормальному давлению, скорость реакции увеличивается. Таким образом, в соответствии с настоящим изобретением, время реакции дегидратации для регенерации ароматического нитрильного соединения существенно сокращено, по сравнению со временем реакции, необходимым при традиционном способе.
Также, в соответствии с настоящим изобретением, ароматическое нитрильное соединение получают так, как описано выше, и, таким образом, эффективно реализуют способ получения карбонатного сложного эфира.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0013]
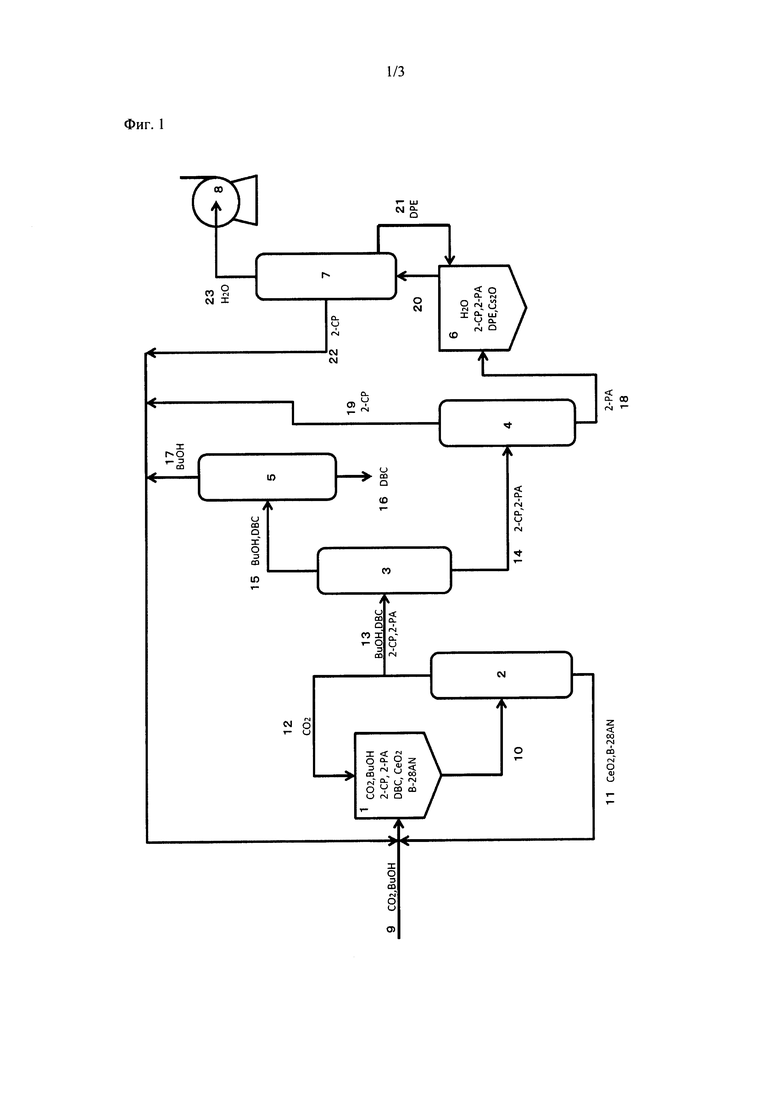
На фиг. 1 представлен пример устройства для получения карбонатного сложного эфира.
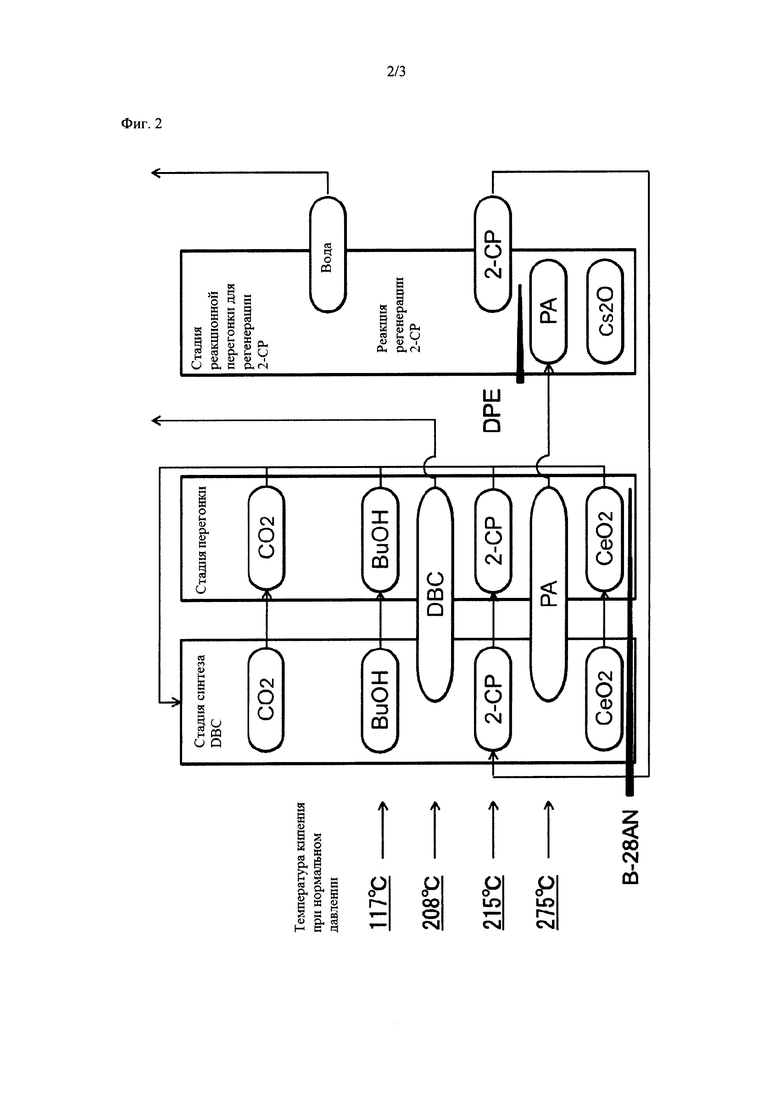
На фиг. 2 представлена диаграмма, демонстрирующая состояние каждого вещества на каждой стадии указанного способа получения, осуществляемого с помощью технологического устройства, представленного на фиг. 1.
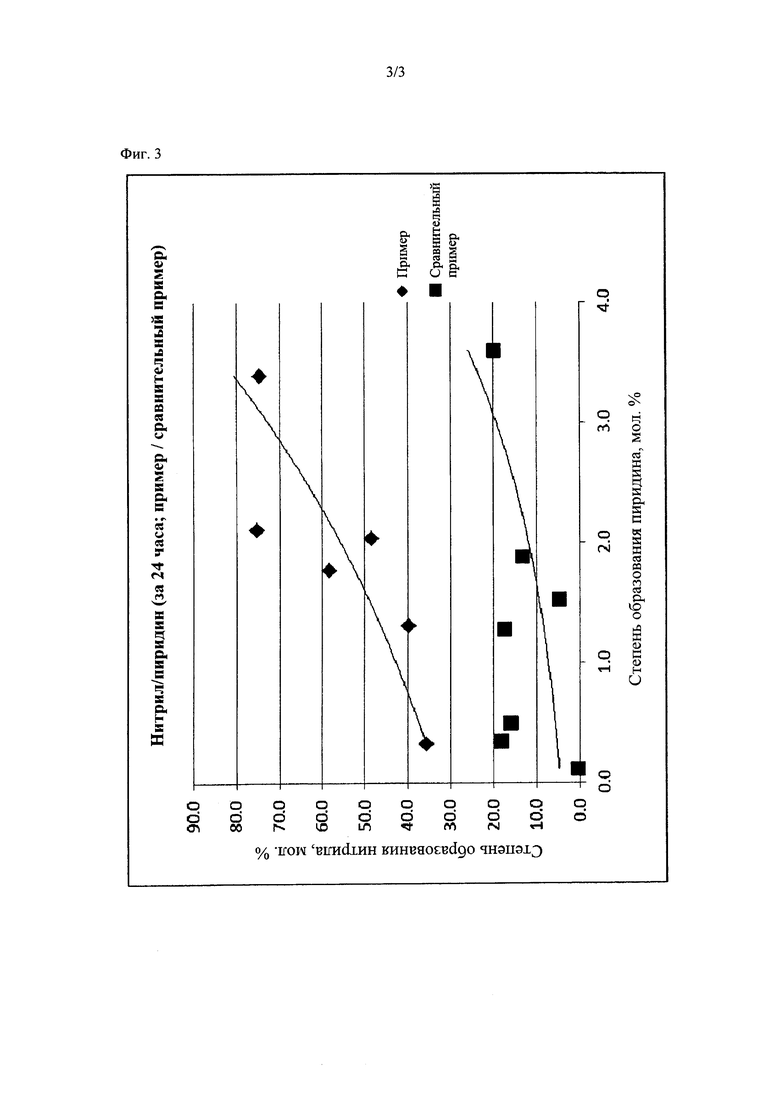
На фиг. 3 представлена диаграмма, демонстрирующая отношение выходов (выходов реакции получения) нитрила и пиридина в примере по данному изобретению и в сравнительном примере.
ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[0014]
Далее подробно описаны предпочтительные варианты реализации настоящего изобретения со ссылкой на прилагаемые графические материалы. В тексте настоящего описания и на чертежах компоненты, имеющие по существу одинаковые функции или структуры, имеют одинаковые ссылочные позиции, и одинаковые описания не повторяются.
[0015]
<1. Способ получения ароматического нитрильного соединения>
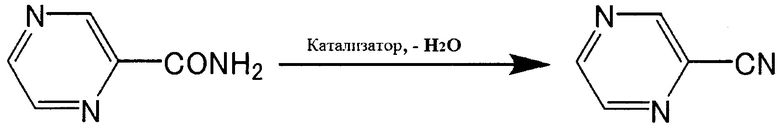
В соответствии со способом получения ароматического нитрильного соединения согласно настоящему изобретению, ароматическое нитрильное соединение, такое как цианопиридин, цианопиразин или т.п. получают дегидратацией ароматического амидного соединения, такого как пиридинкарбоксамид (2-пиридинкарбоксамид, 3-пиридинкарбоксамид или 4-пиридинкарбоксамид), пиразинамид или т.п. В соответствии с указанным способом, ароматическое амидное соединение подвергают реакции дегидратации в присутствии, например, катализатора, содержащего основной оксид металла и дифениловый эфир, с получением ароматического нитрильного соединения.
[0016]
[Химическая формула 1]
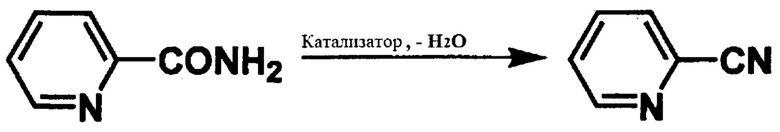
[Химическая формула 2]
[0017]
Катализатор, подходящий для вышеописанной реакции дегидратации согласно настоящему изобретению, содержит оксид щелочного металла (K, Li, Na, Rb, Cs), который является основным. Предпочтительно, катализатор, подходящий для вышеописанной реакции содержит оксид по меньшей мере одного из Na, K, Rb и Cs. Носителем катализатора может быть вещество, которое обычно действует как носитель катализатора. В результате исследований, проведенных на различных носителях, было обнаружено, что указанный катализатор демонстрирует особенно высокие характеристики при использовании в качестве носителя одного или двух из SiO2 и ZrO2.
[0018]
Далее описаны примеры способов получения катализатора, подходящего для вышеописанной реакции дегидратации. В случае если носителем является SiO2, можно использовать имеющийся в продаже SiO2 в форме порошка или сферических частиц. Предпочтительно, размер частиц SiO2 составляет 100 меш (0,15 мм) или менее, так что активный металл равномерно распределен на подложке, и его предварительно прокаливают при 700°С в течение 1 часа на воздухе для удаления влаги. Существуют различные типы SiO2 с разными свойствами. SiO2, имеющий более высокую площадь поверхности, является более предпочтительным, поскольку чем больше площадь поверхности, тем более диспергирован активный металл, что приводит к увеличению количества получаемого ароматического нитрильного соединения. В частности, предпочтительна площадь поверхности 300 м2 или более. Следует отметить, что площадь поверхности полученного катализатора может быть меньше, чем площадь поверхности чистого SiO2, в результате, например, взаимного влияния SiO2 и активного металла. В таком случае площадь поверхности полученного катализатора предпочтительно составляет 150 м2 или более. Оксид металла, действующий в качестве активных частиц, можно наносить методом пропитки, таким как метод пропитки по влагоемкости, методом выпаривания досуха или т.п.
[0019]
Соль металла, используемая в качестве предшественника катализатора, должна быть лишь растворимой в воде. Примеры подходящих солей щелочных металлов включают различные соединения, такие как карбонаты, гидрокарбонаты, хлориды, нитраты, силикаты и т.п. Водным раствором предшественника, содержащего основной металла, пропитывают подложку, затем ее сушат и прокаливают. Полученное вещество можно использовать в качестве катализатора. Температура прокаливания, которая зависит от используемого предшественника, предпочтительно составляет от 400 до 600°С.
[0020]
Количество катализатора на подложке можно задавать в соответствии с требованиями. Например, количество оксида щелочного металла на подложке, в пересчете на металл, предпочтительно составляет от примерно 0,1 до 1,5 ммоль/г, особенно предпочтительно от примерно 0,1 до 1 ммоль/г относительно общей массы катализатора. Если количество, наносимое на подложку, больше указанного значения, то активность может быть снижена. Количество катализатора, используемого для реакции, можно задавать в соответствии с требованиями.
[0021]
Катализатор, предпочтительно используемый согласно настоящему изобретению, содержит подложку, состоящую из одного или двух из SiO2 и ZrO2, и один или по меньшей мере два типа оксидов щелочных металлов на указанной подложке. Катализатор, помимо вышеописанных элементов, может содержать нежелательные примеси, внесенные при получении катализатора. Необходимо избегать включения примесей, насколько это возможно.
[0022]
Катализатор, пригодный согласно настоящему изобретению, содержащий оксид металла, действующий в качестве активных частиц, на подложке катализатора может быть в форме порошка или формованного изделия. В случае формованного изделия катализатор может быть в сферической форме, в форме пеллет, цилиндров, колец, колес, гранул или т.п.
[0023]
Для способа согласно настоящему изобретению для получения ароматического нитрильного соединения с применением катализатора нет никаких специальных ограничений в отношении конструкции реактора. Можно использовать проточный реактор, такой как реактор периодического действия, реактор полунепрерывного действия, реактор непрерывного действия, трубчатый реактор или т.п. Для катализатора можно использовать неподвижный слой, взвешенный слой или т.п.
[0024]
В способе получения ароматических нитрильных соединений согласно настоящему изобретению желательно осуществлять реакцию получения ароматического нитрильного соединения с одновременным удалением побочного продукта, воды, образующейся в реакции дегидратации. Например, желательно осуществлять кипячение с обратным холодильником или перегонку, или подавать в систему агент дегидратации, такой как цеолит или т.п., для осуществления реакции с одновременным удалением побочного продукта, воды. В результате активных исследований, предпринятых авторами настоящего изобретения, было обнаружено, что количество получаемого ароматического нитрильного соединения увеличивается при использовании перегонного реакционного устройства с присоединенным к нему устройством для снижения давления. Катализатор, ароматическое амидное соединение и дифениловый эфир помещают в реакционную пробирку и снижают давление для регулирования температуры реакционного раствора, и кипятят дифениловый эфир с обратным холодильником для перегонки реакционной жидкости с целью выделения и удаления из системы побочного продукта, воды.
[0025]
Дифениловый эфир имеет высокую температуру кипения, составляющую примерно 259°С, и является предпочтительным для реакции дегидратации.
[0026]
Желательно выбирать условия реакции в соответствии со скоростью реакции дегидратации, температурой кипения дифенилового эфира, образованием пиридина в качестве побочного продукта указанной реакции, а также с экономическими показателями.
[0027]
Обычные условия реакции для способа получения ароматического нитрильного соединения согласно настоящему изобретению могут быть следующими. Температура реакционного раствора составляет от 170 до 230°; давление представляет собой нормальное давление (от 101,3 (кПа) (760 торр) до пониженного давления (13,3 (кПа) (100 торр)); и время составляет от нескольких часов до примерно 100 часов. Условия реакции не ограничены вышеуказанными условиями.
Например, температура реакционного раствора предпочтительно составляет от 180 до 228°С, и более предпочтительно от 190 до 210°С. Давление реакции предпочтительно составляет от 1,33 до 60 (кПа) (от 10 до 450 торр), и более предпочтительно от 13,3 до 53,3 (кПа) (от 100 до 400 торр). Предпочтительно, время реакции составляет от 4 до 24 часов, и более предпочтительно от 8 до 24 часов.
[0028]
В случае использования молекулярных сит в качестве агента дегидратации, нет никакого специального ограничения в отношении типа или формы молекулярных сит. Например, можно использовать обычные молекулярные сита, имеющие высокую степень абсорбции воды, такие как 3А, 4А, 5А или т.п., и которые имеют сферическую форму или форму пеллет. Например, можно использовать Zeolum производства компании Tosoh Corporation. Предпочтительно, молекулярные сита предварительно сушат, например, при 300-500°С в течение примерно 1 часа.
[0029]
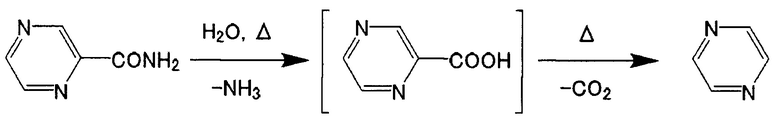
[Химическая формула 3]
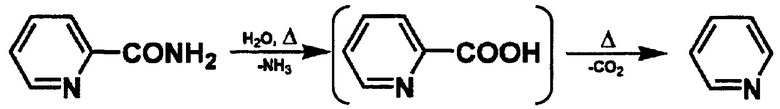
[Химическая формула 4]
[0030]
В реакции дегидратации ароматического амидного соединения считают, что в результате вышеописанного разложения ароматического амидного соединения образуется ароматическая карбоновая кислота, из которой в качестве побочного продукта образуется пиридин или пиразин. Однако реакционный раствор, получаемый в результате реакции дегидратации, осуществляемой в условиях реакции согласно настоящему изобретению, содержит не прореагировавшее ароматическое амидное соединение, ароматическое нитрильное соединение в качестве продукта реакции и дифениловый эфир, но не содержит побочный продукт, представленный каждой из приведенных выше формул, в максимальной степени -совершенно не содержит его.
[0031]
Ниже приведены температуры плавления соединений: 110°С (2-пиколинамид), 24°С (2-цианопиридин), 190°С (пиразинамид), 19°С (цианопиразин) и 28°С (дифениловый эфир). Ниже приведены температуры кипения соединений: 275°С (2-пиколинамид), 215°С (2-цианопиридин), 357°С (пиразинамид), 87°С/6 мм рт.ст. (цианопиразин), 100°С (вода) и 259°С (дифениловый эфир). Таким образом, все реакционные фазы являются жидкими, за исключением твердого катализатора. Используют устройство для перегонки реакционной смеси с присоединенным к нему устройством для снижения давления. Дистилляционную колонну нагревают до температуры выше температуры кипения воды при давлении реакции и ниже температуры кипения дифенилового эфира. Реакционный раствор нагревают до температуры, которая выше или равна температуре кипения дифенилового эфира при давлении реакции и ниже температуры кипения 2-пиколинамида. При таком условии дифениловый эфир, частично испаряющийся в реакционной системе, охлаждают с помощью охлаждающего устройства и возвращают в реакционную пробирку. Побочный продукт, воду, эффективно отделяют от реакционного раствора посредством перегонки и выводят за пределы системы. Таким образом, реакция регенерации нитрила протекает с высокой скоростью и, следовательно, время реакции дегидратации существенно сокращено.
Считают, что температура кипения дифенилового эфира выше, чем температура кипения ароматического нитрильного соединения и температура кипения воды, но ниже температуры кипения ароматического амидного соединения. Используют дифениловый эфир, удовлетворяющий таким взаимным требованиям между температурами кипения реагентов, поэтому реакция дегидратации протекает с высокой эффективностью, и ароматическое нитрильное соединение можно легко выделить.
[0032]
Температуры кипения веществ, присутствующих в реакционной системе, отличаются друг от друга, как описано выше. Таким образом, указанные вещества можно легко разделить перегонкой.
[0033]
<2. Способ получения карбонатного сложного эфира с использованием ароматического нитрильного соединения>
Как описано выше, осуществляют реакцию дегидратации для регенерации ароматического амидного соединения в ароматическое нитрильное соединение, и ароматическое амидное соединение, которое является целевым соединением, получают селективно и с высоким выходом, без использования сильного реагента и с подавлением образования побочного продукта. Скорость реакции существенно улучшена, обеспечивая существенное сокращение времени реакции. Таким образом, скорость регенерации посредством реакции дегидратации из ароматического амидного соединения в ароматическое нитрильное соединение, а также скорость синтеза карбонатного сложного эфира из СО2 и спирта с использованием ароматического нитрильного соединения, становятся хорошо сбалансированными, так что указанные реакции можно осуществлять одновременно. Указанные реакции можно осуществлять в виде последовательности промышленных процессов. Авторы настоящего изобретения использовали полученные сведения в отношении способа получения карбонатного сложного эфира и предложили следующий способ получения карбонатного сложного эфира.
[0034]
(Первая реакционная стадия)
Первая реакционная стадия способа получения карбонатного сложного эфира согласно настоящему изобретению включает, например, непосредственное приведение во взаимодействие спирта и диоксида углерода в присутствии твердого катализатора, такого как СеО2 или т.п., и ароматического нитрильного соединения с получением карбонатного сложного эфира (реакция получения карбонатного сложного эфира).
[0035]
На данной стадии спирт и диоксид углерода взаимодействуют друг с другом. В результате образуется карбонатный сложный эфир, а также вода. Ароматическое нитрильное соединение, присутствующее в системе, и образовавшаяся вода вступают в реакцию гидратации с образованием ароматического амидного соединения. Таким образом, вода, образовавшаяся в результате реакции спирта и диоксида углерода, выводится из реакционной системе или уменьшается ее количество, что приводит к ускорению образования карбонатного сложного эфира. Например, реакцию можно записать следующим образом.
[0036]
[Химическая формула 5]
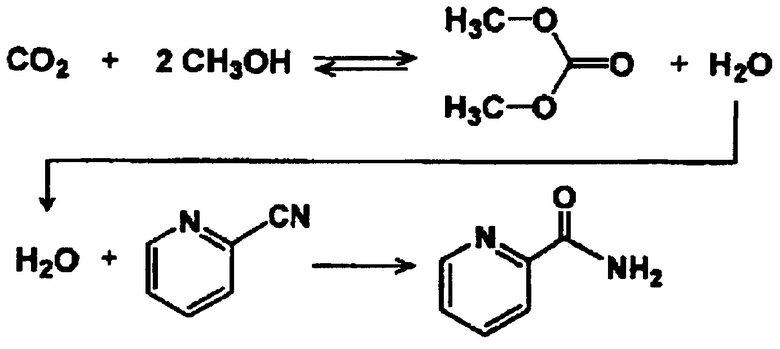
[0037]
(Спирт)
В качестве спирта можно использовать любой один, или два, или более спиртов, выбранных из первичного спирта, вторичного спирта и третичного спирта. Примеры предпочтительных спиртов включают метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, 1-пентанол, 1-гексанол, 1-гептанол, 1-октанол, 1-нонанол, аллиловый спирт, 2-метил-1-пропанол, циклогексанметанол, бензиловый спирт, этиленгликоль, 1,2-пропандиол и 1,3-пропандиол. Указанные спирты увеличивают выход целевого продукта, а также повышают скорость реакции. Карбонатные сложные эфиры, получаемые с применением вышеперечисленных спиртов, соответственно, представляют собой диметилкарбонат, диэтилкарбонат, дипропилкарбонат, диизопропилкарбонат, дибутилкарбонат, дипентилкарбонат, дигексилкарбонат, дигептилкарбонат, диоктилкарбонат, динонилкарбонат, диаллилкарбонат, ди-2-метилпропилкарбонат, дициклогексанметилкарбонат, дибензилкарбонат, этиленкарбонат, 1,2-пропиленкарбонат и 1,3-пропиленкарбонат.
Если полученный карбонатный сложный эфир представляет собой диаллилкарбонат, предпочтительно использовать спирт, содержащий от 1 до 6 атомов углерода, и более предпочтительно использовать спирт, содержащий от 2 до 4 атомов углерода.
Предпочтительно использовать одноатомный спирт или двухатомный спирт.
[0038]
(Катализатор, подходящий для получения карбонатного сложного эфира)
На первой реакционной стадии способа получения карбонатного сложного эфира предпочтительно использовать один или оба из СеО2 и ZrO2 в качестве твердого катализатора. Например, предпочтительно использовать только СеО2, только ZrO2, смесь СеО2 и ZrO2, твердый раствор CeO2 и ZrO2 или сложный оксид СеО2 и ZrO2. Особенно предпочтительно использовать только СеО2. Соотношение смешивания СеО2 и ZrO2 в твердом растворе или сложном оксиде составляет, в основном, 50:50, но может необязательно варьироваться.
[0039]
Катализатор, используемый на первой реакционной стадии, может быть в форме порошка или формованного изделия. В случае формованного изделия катализатор может быть в сферической форме, в форме пеллет, цилиндров, колец, колес, гранул или т.п.
[0040]
(Диоксид углерода)
Согласно настоящему изобретению, можно использовать диоксид углерода, получаемый в качестве промышленного газа, или диоксид углерода, выделяемый и извлекаемый из отработанных газов заводов по производству различных продуктов, металлургических предприятий, электростанций или т.п.
[0041]
(Растворитель для реакции получения карбонатного сложного эфира)
Для реакции получения карбонатного сложного эфира предпочтительно использовать растворитель, имеющий температуру кипения выше температуры кипения получаемого амидного соединения. Более предпочтительно, растворитель в реакции получения карбонатного сложного эфира содержит по меньшей мере один из диалкилбензола, алкилнафталина и дифенилбензола. Конкретные примеры предпочтительных растворителей включают, например, бочковое технологическое масло B-28AN и бочковое технологическое масло В-30 (производства компании Matsumura Oil Co., Ltd.), каждое из которых содержит такой компонент, как диалкилбензол, алкилнафталин, дифенилбензол или т.п.
[0042]
(Разделение перегонкой)
После проведения реакции полученное вещество перегоняют для разделения на карбонатный сложный эфир в качестве основного продукта, ароматическое амидное соединение в качестве побочного продукта, не прореагировавшее ароматическое нитрильное соединение и твердый катализатор, такой как СеО2 или т.п. В результате выделяют продукты.
[0043]
(Вторая реакционная стадия)
На второй реакционной стадии согласно настоящему изобретению ароматическое амидное соединение, полученное в качестве побочного продукта на первой реакционной стадии, выделяют из системы после реакции получения карбонатного сложного эфира и получают ароматическое нитрильное соединение по реакции дегидратации. Вторая реакционная стадия соответствует вышеописанному способу получения ароматического нитрильного соединения и, следовательно, не будет подробно описана.
[0044]
(Повторное использование ароматического нитрильного соединения)
Ароматическое нитрильное соединение, регенерированное на второй реакционной стадии, можно снова использовать для первой реакционной стадии (реакции гидратации).
[0045]
В соответствии с настоящим изобретением, как описано выше, для реакции дегидратации ароматического амидного соединения используют дифениловый эфир, который имеет температуру кипения выше, чем температура кипения получаемого ароматического нитрильного соединения, и ниже температуры кипения ароматического амидного соединения, используемого в качестве реагента, и подбирают температуру реакционного раствора. При таком условии стадия жидкостно-твердофазного выделения катализатора становится необязательной, и ароматическое нитрильное соединение можно легко выделить. В реакции получения карбонатного сложного эфира используют катализатор, имеющий температуру кипения выше, чем температура ароматического карбоксамида, поэтому стадия жидкостно-твердофазного выделения катализатора становится необязательной. Как можно видеть, в соответствии с настоящим изобретением осуществляют серию реакций, при этом компоненты отделяют друг от друга лишь с помощью перегонки, без необходимости проведения стадии жидкостно-твердофазного выделения катализатора. Таким образом, реализован эффективный способ, подробно описанный ниже.
[0046]
<3. Устройство для получения карбонатного сложного эфира>
Далее на конкретном примере приведено подробное описание производственного устройства, пригодного согласно настоящему изобретению. На фиг. 1 представлен пример предпочтительного производственного устройства. На фиг. 2 схематически представлено состояние каждого из веществ на каждой стадии, осуществляемой в производственном устройстве.
[0047]
(Первая реакционная стадия)
На первой реакционной стадии реактор 1 получения карбонатного сложного эфира (первая реакционная часть) наполняют одним или обоими из СеО2 и ZrO2 в качестве твердого катализатора (твердая фаза), спиртом (1-бутанолом (BuOH); жидкая фаза), 2-цианопиридином (2-СР; жидкость), бочковым технологическим маслом (B-28AN; жидкая фаза) в качестве растворителя и диоксидом углерода (CO2; газообразная фаза), который подают через воздуходувку с повышением давления (не показана). Перед началом реакции можно вносить свежий твердый катализатор (CeO2; твердая фаза) или можно регенерировать катализатор из колонны 2 выделения катализатора. В начале реакции можно использовать свежий 2-цианопиридин. Альтернативно, можно повторно использовать 2-цианопиридин 22 (жидкая фаза), выделенный из непрореагировавшего 2-цианопиридина 19 (газообразная фаза), отделенного и очищенного в колонне 3 для выделения агента дегидратации и в колонне 4 выделения амида, а также 2-пиколинамид, очищенный в колонне 7 для отделения воды.
[0048]
В устройстве прямого синтеза карбонатного сложного эфира, пригодном согласно настоящему изобретению, в качестве твердого катализатора используют один или оба из СеО2 и ZrO2. Устройство для синтеза может представлять собой проточный реактор, такой как реактор периодического действия, реактор полунепрерывного действия, реактор непрерывного действия, трубчатый реактор или т.п.
[0049]
(Температура реакционного раствора)
Температура реакционного раствора в реакторе 1 получения карбонатного сложного эфира предпочтительно составляет от 50 до 300°С. Если температура реакционного раствора составляет менее 50°С, то скорость реакции мала, и реакция синтеза карбонатного сложного эфира или реакция гидратации с 2-цианопиридином почти не идет. В таком случае производительность по карбонатному сложному эфиру является низкой. Если температура реакционного раствора выше 300°С, то скорость каждой реакции высока, но карбонатный сложный эфир легко разлагается или денатурирует, и 2-пиколинамид легко взаимодействует со спиртом. Таким образом, выход карбонатного эфира является низким. Более предпочтительно, температура реакционного раствора в реакторе 1 получения карбонатного сложного эфира составляет от 100 до 150°С. Следует учитывать, что предпочтительная температура варьируется в соответствии с типом или количеством твердого катализатора, или с количеством, или с соотношением реагентов (спирта и 2-цианопиридина). Таким образом, предпочтительно опционально настраивать оптимальную температуру. Поскольку предпочтительная температура реакционного раствора составляет от 100 до 150°С, желательно предварительно нагревать реагенты (спирт и 2-цианопиридин) паром или подобным способом на стадии, предшествующей реактору 1 получения карбонатного сложного эфира.
[0050]
(Давление реакции)
Давление реакции в реакторе 1 получения карбонатного сложного эфира предпочтительно составляет от 0,1 до 20 МПа (абсолютное давление). Если давление реакции составляет менее 0,1 МПа (абсолютное давление), то необходимо устройство для сброса давления, в результате чего производственное оборудование становится сложным и дорогостоящим. Кроме того, необходима энергия движущей силы для снижения давления, что уменьшает эффективность использования энергии. Если давление реакции превышает 20 МПа (абсолютное давление), то реакция гидратации с 2-цианопиридином протекает с трудом, что уменьшает выход карбонатного сложного эфира. Кроме того, необходима энергия движущей силы для повышения давления, что уменьшает эффективность использования энергии. С точки зрения увеличения выхода карбонатного сложного эфира, давление реакции более предпочтительно составляет от 0,5 до 15 МПа (абсолютное давление), и еще более предпочтительно от 1,0 до 10 МПа (абсолютное давление).
[0051]
(Количество 2-цианопиридина)
2-Цианопиридин, используемый для реакции гидратации, обычно вводят в реактор до начала реакции в таком молярном количестве, которое составляет от 0,2 раза или более до 5 раз или менее от теоретического молярного количества воды, образующейся в качестве побочного продукта при взаимодействии спирта и CO2 в качестве реагентов. Более предпочтительно, молярное количество 2-цианопиридина составляет от 0,5 раза или более до 3 раз или менее, и особенно предпочтительно от 0,8 раза или более до 1,5 раза или менее от теоретического молярного количества воды, образующейся в качестве побочного продукта при взаимодействии спирта и СО2 в качестве реагентов. Если молярное количество 2-цианопиридина слишком мало, то количество 2-цианопиридина, способствующего реакции гидратации, невелико, что может уменьшать выход карбонатного сложного эфира. Напротив, если молярное количество 2-цианопиридина слишком велико относительно спирта, то происходит нежелательное усиление побочной реакции 2-цианопиридина. Следует учитывать, что количество спирта и 2-цианопиридина относительно твердого катализатора варьируется в соответствии с типом или количеством твердого катализатора, с типом или количеством спирта, или с соотношением спирта и 2-цианопиридина. Таким образом, предпочтительно правильно подбирать оптимальные количества.
[0052]
(Разделение продуктов реакции)
Разделение продуктов реакции полностью осуществляют перегонкой. После реакции в реакторе 1 получения карбонатного сложного эфира, реакционный раствор 10 подают в колонну 2 выделения катализатора. Из нижней части колонны 2 выделения катализатора выделяют катализатор и растворитель (в данном примере бочковое технологическое масло (B-28AN) (жидкая фаза; 11)). Из верхней части колонны 2 выделения катализатора выделяют СО2 (12) и смесь (13) BuOH, дибутилкарбоната (DBC), 2-цианопиридина и 2-пиколинамида. Выделенный катализатор, растворитель и СО2 возвращают в цикл в реактор 1 получения карбонатного сложного эфира.
[0053]
Смесь (13), выделенную из колонны 2 выделения катализатора, подают в колонну 3 выделения агента дегидратации. Из нижней части колонны 3 выделения агента дегидратации выделяют смесь (14) 2-цианопиридина и 2-пиколинамида. Из верхней части колонны 3 выделения агента дегидратации выделяют BuOH и DBC (15).
[0054]
Смесь (14), выделенную из нижней части колонны 3 выделения агента дегидратации, подают в колонну 4 выделения амида. Из нижней части колоны 4 выделения амида выделяют 2-пиколинамид (18). Из верхней части колонны 4 выделения амида выделяют 2-цианопиридин (19). Выделенный 2-цианопиридин возвращают в цикл в реактор 1 получения карбонатного сложного эфира. 2-пиколинамид (18), выделенный из нижней части колонны 4 выделения амида, подают в реактор 6 регенерации нитрила.
[0055]
BuOH и DBC (15), выделенные из верхней части колонны 3 выделения агента дегидратации, подают в колонну 5 выделения карбонатного сложного эфира. Из нижней части колонны 5 выделения карбонатного сложного эфира выделяют DBC (16). Из верхней части колонны 5 выделения карбонатного сложного эфира выделяют BuOH (17). Выделенный BuOH возвращают в цикл в реактор 1 получения карбонатного сложного эфира.
[0056]
2-Пиколинамид (2-РА; 18), выделенный из колонны 4 выделения амида, подают в реактор 6 регенерации нитрила (вторая реакционная часть) для регенерации в 2-цианопиридин.
[0057]
(Вторая реакционная стадия)
На второй реакционной стадии получают 2-цианопиридин (2-СР) посредством реакции дегидратации 2-пиколинамида в реакторе 6 регенерации нитрила. В производственном устройстве, используемом согласно настоящему изобретению (реактор 6 регенерации нитрила), осуществляют реакцию дегидратации 2-пиколинамида в присутствии катализатора, содержащего основной оксид металла и дифениловый эфир, с получением 2-цианопиридина. Не существует никаких специальных ограничений в отношении формы указанного реактора. Можно использовать проточный реактор, такой как реактор периодического действия, реактор полунепрерывного действия, реактор непрерывного действия, трубчатый реактор или т.п. Для катализатора можно использовать неподвижный слой, взвешенный слой или т.п. Температура реактора 6 регенерации нитрила варьируется в соответствии с формой реакции. Используют устройство для перегонки реакционной смеси с присоединенным к нему устройством для снижения давления. Дистилляционную колонну нагревают до температуры выше температуры кипения воды при давлении реакции и ниже температуры кипения дифенилового эфира. Реакционный раствор нагревают до температуры, которая выше или равна температуре кипения дифенилового эфира при давлении реакции и ниже температуры кипения 2-пиколинамида. При таком условии дифениловый эфир, частично испаряющийся в реакционной системе, охлаждают с помощью охлаждающего устройства и возвращают в реакционную пробирку. Побочный продукт, воду, эффективно отделяют от реакционного раствора посредством перегонки и выводят за пределы системы. Таким образом, реакция регенерации нитрила протекает с высокой скоростью.
[0058]
2-Цианопиридин (22) можно выделять из колонны 7 выделения воды в процессе реакции или можно отделять посредством перегонки и выделения после реакции. Выделенный 2-цианопиридин (22) подают в реактор 1 получения карбонатного сложного эфира и повторно используют для получения карбонатного сложного эфира.
[0059]
Как описано выше, согласно настоящему изобретению, продукт реакции и соединение, подлежащее повторному и использованию, отделяют друг от друга лишь с помощью перегонки, без необходимости в жидкостно-твердофазном разделении. Таким образом, в соответствии с настоящим изобретением, карбонатный сложный эфир эффективно получают в более простом производственном устройстве и с меньшим количеством технологических стадий.
[0060]
Далее настоящее изобретение более подробно описано на примерах. Настоящее изобретение не ограничено ни одним из следующих примеров. Сначала будут описаны примеры и сравнительные примеры способа получения цианопиридина.
[0061
(Пример 1)
SiO2 (CARiACT, G-6, площадь поверхности: 535 м2/г; производства компании Fuji Silysia Chemical Ltd.) в качестве носителя измельчали до 100 меш или менее и предварительно прокаливали при 700°С в течение примерно 1 часа. Затем, для нанесения на подложку Cs в качестве щелочного металла, получали водный раствор с использованием CS2CO3 (производства компании Wako Pure Chemical Industries, Ltd.), так чтобы конечное содержание металлического Cs на подложке составляло 0,5 ммоль/г, и пропитывали полученным водным раствором SiO2. Затем полученное вещество сушили при 110°С в течение примерно 6 часов и прокаливали при 500°С в течение примерно 3 часов. В результате получали катализатор Cs2O/SiO2. Катализатор Na2O/SiO2 получали по существу таким же способом, как катализатор Cs2O/SiO2.
[0062]
Затем 3-горлую круглодонную колбу, используемую в качестве реактора, оснащали магнитной мешалкой, добавляли катализатор Cs2O/SiO2 (1,0 г (Cs: 0,5 ммоль)), 2-пиколинамид (2-РА; 6,1 г (50 ммоль); производства компании Tokyo Chemical Industry Co., Ltd.) и дифениловый эфир (212,5 г (1,25 моль); производства компании Tokyo Chemical Industry Co., Ltd.).
К реактору присоединяли термометр и первую трубку воздушного охлаждения в качестве дистилляционной колонны. Дистилляционную головку с прикрепленным к ней термометром устанавливали на верхнем конце первой трубки воздушного охлаждения. С дистилляционной головкой соединяли вторую трубку воздушного охлаждения, приемник, и вакуумный насос. Полученное устройство использовали в качестве реакционного дистилляционного устройства. Первую трубку воздушного охлаждения оборачивали ленточным нагревателем, чтобы можно было регулировать температуру первой трубки воздушного охлаждения. Охлаждающую ловушку охлаждали жидким азотом для выделения газообразного пиридина.
Затем понижали давление в реакционном дистилляционном устройстве с помощью вакуумного насоса до 13,3 кПа (100 торр). Первую трубку воздушного охлаждения нагревали до 60°С, что выше температуры кипения воды при давлении реакции и ниже температуры кипения дифенилового эфира. Реакционный раствор поддерживали в кипящем состоянии при 184°С, что выше или равно температуре кипения дифенилового эфира при давлении реакции и ниже температуры кипения 2-пиколинамида. Температуры регулировали так, чтобы осуществлять реакцию с одновременным охлаждением дифенилового эфира, частично переходящего в реакционной системе в газообразное состояние, в первой трубке воздушного охлаждения и его возвратом в реактор, и выделяли побочный продукт, воду, посредством ее перегонки и вывода за пределы системы без возврата в реактор.
Начало реакции определяли как момент начала кипения реакционного раствора, и продолжали реакцию в течение 24 часов.
По окончании реакции температуру реакционной системы понижали до комнатной. Брали образец реакционного раствора и разбавляли в два раза этанолом, и добавляли к нему 1-гексанол в качестве вещества внутреннего стандарта. Полученное вещество анализировали количественным анализом с помощью ГХ-МС (на газовом хроматомасс-спектрометре) и количественным анализом на ПИД-ГХ. В результате получали 2-цианопиридин, как показано в таблице 1. Выход 2-цианопиридина составлял 35,7 мол. %, и степень получения пиридина в качестве побочного продукта была снижена до 0,3 мол. %.
[0063]
(Примеры 2-5, 7 и 8)
В примерах 2-5, 7 и 8 получали 2-цианопиридин из 2-пиколинамида в условиях, в которых по меньшей мере один из следующих параметров: концентрация 2-пиколинамида в реакционном растворе, тип катализатора, тип добавки для реакционного раствора, температура реакционного раствора, давление реакции, температура первой трубки воздушного охлаждения и время реакции, - был отличным от данного параметра в примере 1 (см. таблицу 1). Выход 2-цианопиридина и степень образования пиридина в качестве побочного продукта представлены в таблице 1.
[0064]
(Пример 6)
В примере 6 использовали пиразинамид (производства компании Sigma-Aldrich) вместо 2-пиколинамида. Цианопиразин получали из пиразинамида в условиях, в которых температура реакционного раствора, давление реакции, температура первой трубки воздушного охлаждения и время реакции были отличными от указанных в примере 1 (см. таблицу 1). Выход цианопиразина и степень образования пиразина в качестве побочного продукта представлены в таблице 1.
[0065]
(Сравнительные примеры 1-17)
В сравнительных примерах 1-6 получали 2-цианопиридин из 2-пиколинамида, или получали цианопиразин из пиразинамида в условиях, в которых по меньшей мере один из следующих параметров: тип добавки для реакционного раствора, температура реакционного раствора, давление реакции, температура первой трубки воздушного охлаждения, время реакции и способ дегидратации, - был отличным от данного параметра в примерах 1-8 (см. таблицу 1). В сравнительных примерах, за исключением сравнительного примера 4, к реакционной трубке присоединяли экстрактор Soxhlet, заполненный молекулярными ситами 4А (предварительно высушенными при 300°С в течение 1 часа), и холодильник Liebig, и использовали полученное устройство в качестве реакционного устройства. Температуру холодильника устанавливали на 10°С, и скорость магнитной мешалки устанавливали на 600 об./мин. Реакцию проводили после продувания холодильника, экстрактора Soxhlet и тестовой пробирки газообразным Ar. Выход 2-цианопиридина или аналогичного продукта и степень образования пиридина или аналогичного соединения в качестве побочного продукта представлены в таблице 1.
[0066]
В таблице 1 представлены результаты примеров 1-8 и сравнительных примеров 1-17.
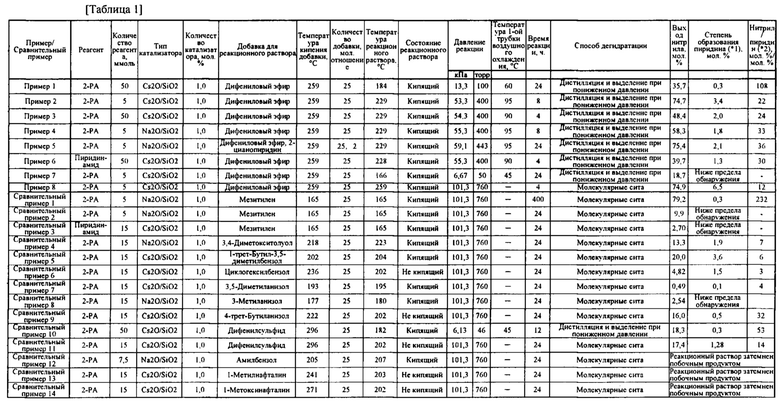

(* 1) Степень образования пиразина (мол. %) в примере 6 и в сравнительном примере 3
(*2) Степень образования нитрила/пиразина (мол. %/мол. %) в примере 6 и в сравнительном примере 3
[0067]
Как описано выше, реакция дегидратации в примерах 1-8 с применением дифенилового эфира в качестве добавки в реакционному раствору обеспечивала образование ароматического нитрильного соединения в качестве целевого соединения с высоким выходом, с одновременным подавлением образования пиридина и аналогичных соединений в качестве побочных продуктов. В частности, в примерах 1-6, в которых температуру реакционного раствора регулировали до 170-230°С, подтверждено достижение высокого выхода нитрильного соединения и уменьшение образование побочного продукта.
Напротив, в сравнительных примерах, в которых не использовали дифениловый эфир и применяли условия реакции, отличные от условий в данных примерах, получали низкий выход ароматического нитрильного соединения (см. фиг. 3, где представлены результаты одного из примеров и сравнительного примера, демонстрирующего относительно высокий выход). В некоторых сравнительных примерах образование пиридина было подавлено, но даже в таких сравнительных примерах выход нитрильного соединения был низким. В сравнительном примере 1 выход нитрильного соединения был высоким, но потребовалось слишком продолжительное время реакции, и поэтому данный сравнительный пример 1 был хуже примеров по настоящему изобретению.
[0068]
Для оценки катализаторов проводили контрольные испытания, в которых изменяли только тип катализатора, пригодного в реакции дегидратации. В контрольных испытаниях тип добавки для реакционного раствора был отличным от ее типа в примере 1 или т.п. Испытание проводили в условиях реакции, соответствующих температуре кипения добавки для реакционного раствора. Результаты представлены в Таблице 2.
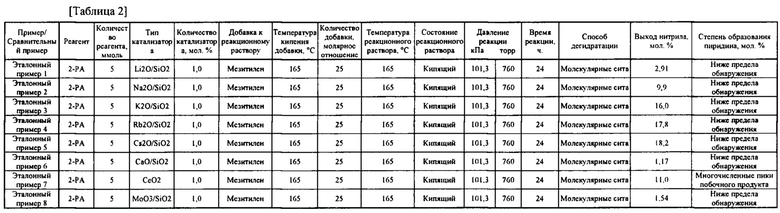
[0069]
Как показано в таблице 2, при использовании, в частности, Cs2O, Rb2O, K2O или Na2O в качестве катализатора для реакции дегидратации согласно настоящему изобретению, было подтверждено селективное получение ароматического нитрильного соединения с высоким выходом.
[0070]
(Пример 9)
3-Горлую круглодонную колбу, используемую в качестве реактора, оснащали магнитной мешалкой, добавляли катализатор Cs2O/SiO2 (10 г (Cs: 5 ммоль)), 2-пиколинамид (61 г (0,5 моль); производства компании Tokyo Chemical Industry Co., Ltd.) и дифениловый эфир (2125 г (12,5 моль); производства компании Tokyo Chemical Industry Co., Ltd.). Реакционное дистилляционное устройство собирали по существу так, как в примере 1.
Реакцию проводили в таких же условиях, как в примере 2, с получением реакционного раствора, содержащего 38,5 г 2-цианопиридина.
Реакционное дистилляционное устройство использовали для перегонки реакционного раствора при давлении 1,3 кПа с получением 33,5 г 2-цианопиридина. В результате анализа, проведенного с помощью ПИД-ГХ, установлена его чистота, составившая 99,9%.
[0071]
Было подтверждено, что в том случае, если, как описано выше, использован дифениловый эфир, имеющий температуру кипения выше температуры кипения получаемого ароматического нитрильного соединения и ниже температуры кипения ароматического амидного соединения, используемого в качестве реагента, и температура реакционного раствора отрегулирована с помощью регулятора давления, то скорость реакции существенно увеличивается, сокращая время реакции, целевое соединение образуется селективно и с высоким выходом, и ароматическое нитрильное соединение легко выделяется.
[0072]
(Пример 20)
Далее описаны примеры способов получения карбонатного сложного эфира с применением цианопиридина (реакции получения карбонатного сложного эфира). Использовали 2-цианопиридин, полученный в примере 9. Сначала прокаливали СеО2 (HSA20; производства компании Solvay) при 600°С в течение 3 часов в атмосфере воздуха с получением порошкообразного твердого катализатора. Автоклав (реактор) объемом 190 мл оснащали магнитной мешалкой, добавляли твердый катализатор (0,17 г (1 ммоль)), бутанол (7,4 г (100 ммоль); производства компании Wako Pure Chemical Industries, Ltd.), бочковое технологическое масло B-28AN (5 г) в качестве растворителя и 2-цианопиридин (5,2 г (50 ммоль)). Воздух в автоклаве три раза продували CO2, а затем в автоклав закачивали СО2 до давления 5 МПа. Температуру автоклава повышали до 132°С с помощью ленточного нагревателя, используя нагревательную мешалку для перемешивания. Время, когда температура достигала целевой температуры, считали временем начала реакции. В процессе реакции давление достигало 8 МПа. Температуру реакционного раствора повышали до 132°С, как описано выше, и продолжали реакцию в течение 24 часов. Затем автоклав охлаждали водой. После охлаждения автоклава до комнатной температуры стравливали давление в автоклаве. Раствор в автоклаве разбавляли в два раза ацетоном и добавляли к нему 1-гексанол в качестве вещества внутреннего стандарта. Полученное вещество анализировали с помощью ПИД-ГХ. Таким образом, получали дибутилкарбонат.
[0073]
(Примеры 21-53)
В примерах 21-53 получали карбонатный сложный эфир из спирта и СО2, используя 2-цианопиридин в условиях, в которой меньше мере один из следующих параметров: наличие/отсутствие растворителя, тип растворителя, количество растворителя, время реакции, тип и концентрация спирта (реагента), и тип и количества катализатора, - отличался от данного параметра в примере 20. В частности, условия, отличные от условий в примере 20, представляли собой тип и количество растворителя в примерах 21-24 и 47, время реакции в примерах 25-28, значение отношения спирт/2-цианопиридин в качестве реагентов в примерах 29-32 и 48, количество катализатора в примерах 33-36, тип катализатора в примерах 37-40, температура реакционного раствора в примерах 41-46, давление реакции в примерах 49 и 50, и тип и количество спирта в качестве реагента в примерах 41-53.
[0074]
В таблице 3 представлены результаты примеров получения карбонатного сложного эфира.
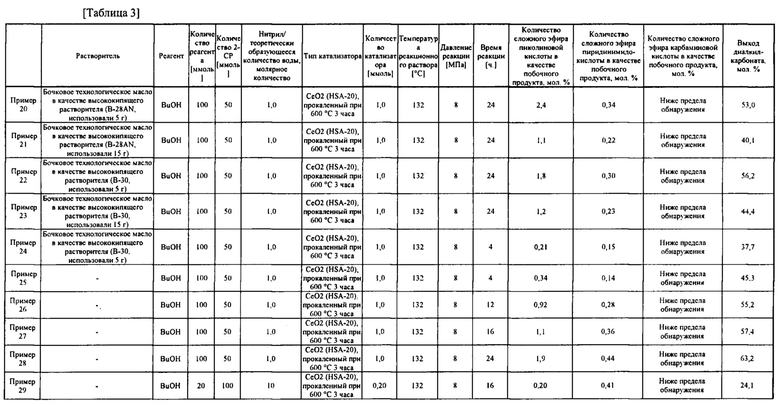
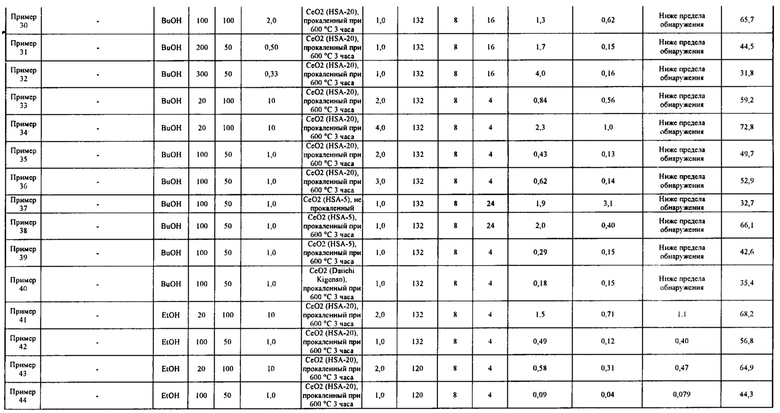
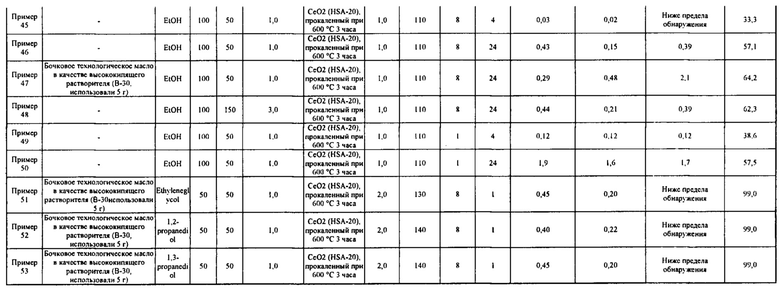
[0075]
Как описано выше, подтверждено, что в примерах 20-53 карбонатный сложный эфир получен с высоким выходом за короткое время реакции, составляющее 24 часа или менее, при этом реакция гидратации между побочным продуктом, водой, и ароматическим цианосоединением протекала одновременно с реакцией образования карбонатного сложного эфира.
[0076]
(Пример 54)
Далее описан пример выделения катализатора из реакционного раствора с карбонатным сложным эфиром. Карбонатный сложный эфир получали с помощью производственного устройства, изображенного на фиг. 1. Сначала прокаливали СеО2 (концентрация примесей: 0,02% или менее; производства компании Daiichi Kigenso Kagaku Kogyo Co., Ltd.) при 600°C в течение 3 часов в атмосфере воздуха с получением порошкообразного твердого катализатора. В автоклав (реактор) объемом 1,9 л, оснащенный мешалкой, добавляли твердый катализатор (1,72 г (10 ммоль)), бутанол (74,1 г (1 моль); производства компании Wako Pure Chemical Industries, Ltd.), бочковое технологическое масло B-28AN (50 г) в качестве растворителя и 2-цианопиридин (52,1 г (0,5 моль)). Воздух в автоклаве три раза продували СО2, а затем в автоклав закачивали СО2 до давления 5 МПа. Температуру автоклава повышали до 132°С с помощью керамического нагревателя, перемешивая вещества в автоклаве. Время, когда температура достигала целевой температуры, считали временем начала реакции. В процессе реакции давление достигало 8 МПа.
Температуру реакционного раствора повышали до 132°С, как описано выше, и продолжали реакцию в течение 24 часов. Затем давление в автоклаве понижали до атмосферного давления. В среднюю часть дистилляционной колонны, имеющей сниженное давление 2,7 кПа, вводили реакционный раствор и осуществляли простую перегонку. Из верхней части дистилляционной колонны выделяли смесь BuOH, дибутилкарбонат, 2-цианопиридин и 2-пиколинамид. Из нижней части дистилляционной колонны выделяли катализатор и бочковое технологическое масло.
В автоклав (реактор) объемом 1,9 л, оснащенный мешалкой, добавляли катализатор и растворитель, выделенный так, как описано выше, бутанол (74,1 г (1 моль); производства компании Wako Pure Chemical Industries, Ltd.) и 2-цианопиридин (52,1 г (0,5 моль)). Воздух в автоклаве три раза продували СО2, а затем в автоклав закачивали СО2 до давления 5 МПа. Температуру автоклава повышали до 132°С с помощью керамического нагревателя, перемешивая вещества в автоклаве. Время, когда температура достигала целевой температуры, считали временем начала реакции. В процессе реакции давление достигало 8 МПа. После продолжения реакции в течение 24 часов автоклав охлаждали водой. После охлаждения автоклава до комнатной температуры давление в автоклаве понижали и брали образец части реакционного раствора. Образец реакционного раствора разбавляли в два раза ацетоном и добавляли к нему 1-гексанол в качестве вещества внутреннего стандарта. Полученное вещество анализировали с помощью ПИД-ГХ. Выход дибутилкарбоната составлял 54 мол. %.
Реакционный раствор перегоняли в порядке, представленном на фиг. 1, с получением 40 г дибутилкарбоната. Анализ ПИД-ГХ показал, что чистота составляет 99,9%.
Подтверждено получение карбонатного сложного эфира с высоким выходом по реакции получения карбонатного сложного эфира, проводимой с применением использованного и регенерированного катализатора.
[0077]
Как описано выше, было подтверждено, что даже в реакции получения карбонатного сложного эфира, в случае использования растворителя, имеющего температуру кипения выше температуры кипения ароматического карбоксамида, компоненты можно отделить друг от друга лишь с помощью перегонки, без необходимости в стадии жидкостно-твердофазного выделения катализатора. Таким образом, реализован эффективный способ.
[0078]
Предпочтительные варианты реализации настоящего изобретения подробно описаны выше со ссылкой на прилагаемые графические материалы. Настоящее изобретение не ограничено ни одним из приведенных вариантов реализации. Специалисты в той области техники, к которой относится настоящее изобретение, могут предположить любой измененный или модифицированный пример в пределах технологического объема, определяемого формулой изобретения, и такой измененный или модифицированный пример в установленном порядке входит в технологический объем настоящего изобретения.
ПЕРЕЧЕНЬ ССЫЛОЧНЫХ ПОЗИЦИЙ
[0079]
1 Реактор для получения карбонатного сложного эфира
2 Колонна выделения катализатора
3 Колонна выделения агента дегидратации
4. Колонна выделения амида
5 Колонна выделения карбонатного сложного эфира
6 Реактор регенерации нитрила
7 Колонна выделения воды
8 Декомпрессионный насос
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО НИТРИЛЬНОГО СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО ЭФИРА | 2019 |
|
RU2804510C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРА УГОЛЬНОЙ КИСЛОТЫ | 2019 |
|
RU2783523C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНЫХ СЛОЖНЫХ ЭФИРОВ И КАТАЛИТИЧЕСКАЯ КОНСТРУКЦИЯ ДЛЯ ПОЛУЧЕНИЯ КАРБОНАТНЫХ СЛОЖНЫХ ЭФИРОВ | 2019 |
|
RU2796601C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ С ИСПОЛЬЗОВАНИЕМ ДИАРИЛКАРБОНАТА | 2008 |
|
RU2523201C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛМЕТАНА ИЛИ ЕГО ПРОИЗВОДНЫХ | 1998 |
|
RU2182896C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2494088C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ | 2006 |
|
RU2409552C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРА МУРАВЬИНОЙ КИСЛОТЫ ИЛИ МЕТАНОЛА И КАТАЛИЗАТОР ЭТОГО СПОСОБА | 2001 |
|
RU2231521C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2489418C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛЬДЕГИДОВ C-C | 1988 |
|
RU2005714C1 |
Изобретение относится к способу получения ароматического нитрильного соединения. Способ включает реакцию дегидратации для дегидратации ароматического амидного соединения. В указанной реакции дегидратации используют дифениловый эфир. Реакцию дегидратации проводят при кипении дифенилового эфира. Дифениловый эфир имеет температуру кипения выше температуры кипения ароматического нитрильного соединения и температуры кипения воды и ниже температуры кипения ароматического амидного соединения. Предлагаемый способ позволяет уменьшить количество стадий, повысить скорость и сократить время реакции. Изобретение относится также к способу получения карбонатного сложного эфира с использованием указанного выше способа, что позволяет обеспечить регенерацию ароматического амидного соединения. 2 н. и 10 з.п. ф-лы, 3 ил., 3 табл., 54 пр.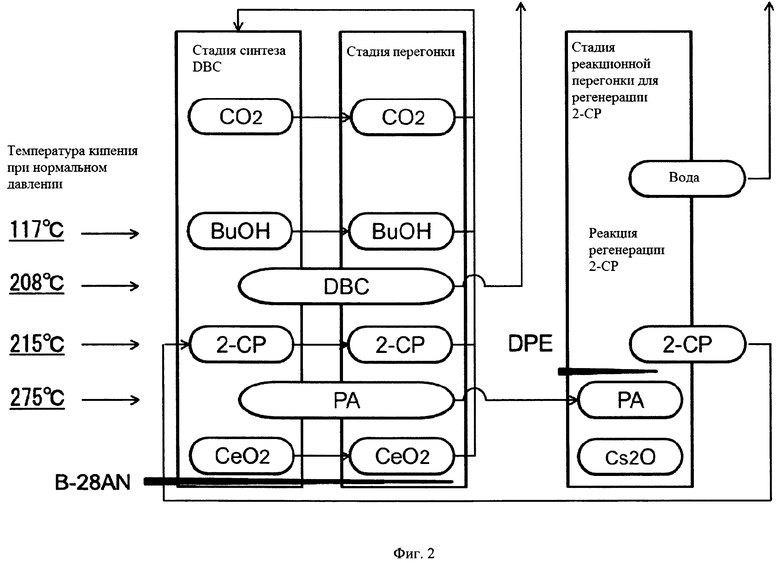
1. Способ получения ароматического нитрильного соединения, включающий:
реакцию дегидратации для дегидратации ароматического амидного соединения,
где в указанной реакции дегидратации используют дифениловый эфир, где реакцию дегидратации проводят при кипении дифенилового эфира и где дифениловый эфир имеет температуру кипения выше температуры кипения ароматического нитрильного соединения и температуры кипения воды и ниже температуры кипения ароматического амидного соединения.
2. Способ получения ароматического нитрильного соединения по п. 1, отличающийся тем, что реакцию дегидратации осуществляют при пониженном давлении.
3. Способ получения ароматического нитрильного соединения по любому из пп. 1 и 2, отличающийся тем, что реакционный раствор реакции дегидратации имеет температуру, составляющую 170 °С или выше, но ниже 230 °С.
4. Способ получения ароматического нитрильного соединения по любому из пп. 1-3, отличающийся тем, что ароматическое амидное соединение включает пиридинкарбоксамид или пиразинамид и ароматическое нитрильное соединение включает цианопиридин или цианопиразин.
5. Способ получения ароматического нитрильного соединения по любому из пп. 1-4, отличающийся тем, что в реакции дегидратации используют катализатор, содержащий цезий.
6. Способ получения карбонатного сложного эфира, включающий:
первую стадию реакции, включающую реакцию получения карбонатного сложного эфира посредством взаимодействия спирта, содержащего от 1 до 6 атомов углерода, и диоксида углерода в присутствии ароматического нитрильного соединения с образованием карбонатного сложного эфира и воды, и реакцию гидратации для гидратации полученной водой ароматического нитрильного соединения с образованием ароматического амидного соединения; и
вторую стадию реакции, осуществляемую после отделения ароматического амидного соединения от реакционной системы после первой стадии реакции, включающую регенерацию ароматического амидного соединения в ароматическое нитрильное соединение посредством реакции дегидратации для дегидратации ароматического амидного соединения в присутствии дифенилового эфира;
при этом по меньшей мере часть ароматического нитрильного соединения, регенерированного на второй стадии реакции, используют на первой стадии реакции,
где реакцию дегидратации проводят при кипении дифенилового эфира и где дифениловый эфир имеет температуру кипения выше температуры кипения ароматического нитрильного соединения и температуры кипения воды и ниже температуры кипения ароматического амидного соединения.
7. Способ получения карбонатного сложного эфира по п. 6, отличающийся тем, что реакцию дегидратации для дегидратации ароматического амидного соединения проводят при температуре реакционного раствора 170 °С или выше и ниже 230 °С.
8. Способ получения карбонатного сложного эфира по любому из пп. 6 или 7, отличающийся тем, что ароматическое амидное соединение включает пиридинкарбоксамид или пиразинамид и ароматическое нитрильное соединение включает цианопиридин или цианопиразин.
9. Способ получения карбонатного сложного эфира по любому из пп. 6-8, отличающийся тем, что в реакции дегидратации используют катализатор, содержащий цезий.
10. Способ получения карбонатного сложного эфира по любому из пп. 6-9, отличающийся тем, что в реакции получения карбонатного сложного эфира используют катализатор, содержащий CeO2.
11. Способ получения карбонатного сложного эфира по любому из пп. 6-10, отличающийся тем, что на первой стадии реакции используют растворитель, имеющий температуру кипения выше температуры кипения получаемого ароматического амидного соединения.
12. Способ получения карбонатного сложного эфира по п. 11, отличающийся тем, что указанный растворитель содержит по меньшей мере один из диалкилбензола, алкилнафталина и дифенилбензола.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| S | |||
| ENTHALER ET AL., Copper-Catalyzed Dehydration of Primary Amides to Nitriles, CATALYSIS LETTERS, 2011, vol | |||
| Топливник с глухим подом | 1918 |
|
SU141A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ ДУТЬЯ ПАРАМИ ИЛИ ГАЗАМИ В ДУГОВОМ ГЕНЕРАТОРЕ НЕЗАТУХАЮЩИХ КОЛЕБАНИЙ | 1921 |
|
SU1079A1 |
| S | |||
| ENTHALER, Straightforward Uranium-Catalyzed Dehydration of Primary Amides to Nitriles, CHEMISTRY: А EUROPEAN JOURNAL, 2011, vol | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |
| Борона с подвижными зубьями | 1925 |
|
SU9316A1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2494088C2 |

