Настоящее изобретение относится к способу получения порошкообразных пористых кристаллических силикатов металлов.
Силикаты представляют собой соединения, образованные тетраэдрами SiO4, которые могут соединяться друг с другом различными способами. Силикатные структуры этого класса могут включать другие элементы, например, металлы, и тогда их называют силикатами металлов. Важным примером указанных силикатов металлов являются цеолиты.
Цеолиты представляют собой кристаллические силикаты, например, алюмосиликаты, в которых трехмерное соединение тетраэдров силикатов (SiO4-) и других структурных единиц (например, тетраэдров AlO4-) приводит к образованию регулярных структур, характеризующихся полостями и порами. Существуют различные типы цеолитов, которые называются в соответствии с типом их структуры. Общую информацию, относящуюся к цеолитам, прежде всего к типам кристаллической структуры известных цеолитов, можно найти в энциклопедии Ullmann's Encyclopedia of Industrial Chemistry (энциклопедия Ульмана промышленной химии), в главе "Zeolites" (цеолиты), опубликованной онлайн 15.04.2012, DOI: 10.1002/14356007.a28_475.pub2.
Благодаря уникальной структуре своих пор, цеолиты обладают интересными каталитическими свойствами, и их можно использовать, например, в качестве окислительных катализаторов.
Синтетические цеолиты в большинстве случаев получают гидротермальным синтезом в присутствии матрицы, формирующей пористую структуру. Например, в патенте CN 101348263 А описан способ получения цеолитов, характеризующихся соотношением Si/Al от 50 до 5000 и размером частиц от 30 до 200 мкм, который включает следующие технологические стадии: (1) получение реакционной смеси, включающей источники кремния и источники алюминия, а также гидроксид металла, (2) реакция гидролиза, (3) последующее высушивание смеси при распылении, при этом получают алюмосиликатные микросферы, (4) гидротермальная реакция микросфер, полученных, как описано выше, в присутствии воды и органического амина при температуре от 160 до 200°С и кристаллизация полученного цеолита, и (5) промывка, (6) высушивание и (7) прокаливание продукта при температуре от 350 до 800°С.
В патенте США 4410501 А описан способ получения силикалита титана. Силикалит титана получают при (1) формировании синтетического геля из гидролизующегося соединения кремния, например, тетраэтилортосиликата, и гидролизующегося соединения титана в присутствии гидроксида тетра-н-пропиламмония при 175°С, (2) последующем гидротермальном синтезе, гидролизе и кристаллизации указанной реакционной смеси. После завершения кристаллизации кристаллы (3) отделяют фильтрованием, (4) промывают, (5) высушивают и наконец (6) прокаливают при 550°С в течение 6 ч.
В патенте ЕР 814058 А1 описано получение различных цеолитов из соответствующих пирогенных смешанных оксидов кремния и металла. Смешанные оксиды кремния и металла получают (1) гидротермальным синтезом при температуре от 100 до 220°С в присутствии матрицы, выбранной из аминов, соединений аммония и гидроксидов щелочных металлов/щелочноземельных металлов, с последующим (2) фильтрованием, (3) промывкой водой и (4) прокаливанием, например, при температуре 550°С в течение 4 ч. В конкретном варианте методом высушивания при распылении получают предварительно сформованный гранулированный материал смешанного оксида, содержащий матрицу, который затем подвергают гидротермальному синтезу, отделяют фильтрованием, промывают и прокаливают.
В патенте CN 1482062 описан способ получения силикалита-1 титана, согласно которому проводят гидротермальную реакцию твердого силикагеля с неорганическим источником титана. Способ включает следующие стадии: (1) импрегнирование твердого силикагеля солью Ti(SO4)2, (2) прокаливание, (3) гидротермальный синтез с использованием силикагеля и смеси Ti(SO4)2 + ТРАОН + вода, (4) фильтрование, (5) промывка, (6) высушивание, (7) прокаливание.
Все описанные выше способы включают время затратные многостадийные методики. Более конкретно, фильтрование и промывка образующегося цеолита необходимы для удаления примесей, попадающих из сопутствующих реагентов или непрореагировавших реагентов и т.п. Это приводит к образованию значительных количеств загрязненной воды, которая в большинстве случаев содержит вредные для присутствующих в воде организмов вещества, которые сложно утилизировать, такие как соли тетраалкиламмония. В связи с этим, крайне необходимо разработать процесс, который, во-первых, включает меньшее число технологических стадий по сравнению со стандартным процессом, и, во-вторых, решает проблему утилизации производственных отходов.
Указанные проблемы решены способом получения порошкообразного пористого кристаллического силиката металла, включающим следующие стадии:
а) гидротермальный синтез в водной смеси, включающей (А) по меньшей мере один источник кремния, (Б) по меньшей мере один источник металла и (В) по меньшей мере одну минерализирующую добавку, при этом получают водную суспензию, содержащую пористый кристаллический силикат металла в качестве продукта реакции, и
б) прокаливание продукта реакции, отличающееся тем, что
прокаливание проводят с использованием пламенного спрей-пиролиза при адиабатической температуре сжигания в диапазоне 450-2200°С, при этом суспензию, содержащую ≤70 мас. % твердых веществ, которую получают на стадии а), распыляют в пламени, создаваемом при сжигании топлива в присутствии кислорода, при этом формируется порошкообразный пористый кристаллический силикат металла.
Неожиданно было установлено, что структура силиката металла, полученного на стадии а) способа по настоящему изобретению, сохраняется в ходе термической обработки при относительно высоких температурах на стадии б), указанное прежде всего справедливо для пористой структуры цеолитов. Известно, что упорядоченная пористая структура, например, цеолитов разрушается при повышенных температурах. Например, согласно данным статьи Advanced Materials Research, тт. 287-290, сс. 317-321 (2011), силикалит-1 титана претерпевает необратимые структурные изменения даже при 650°С, что может негативно влиять на каталитические свойства таких материалов.
Порошкообразный пористый кристаллический силикат металла, получаемый способом по настоящему изобретению, предпочтительно характеризуется цеолитной структурой.
Цеолиты представляют собой кристаллические силикаты, например, алюмосиликаты, в которых трехмерное присоединение тетраэдров силикатов (SiO4-) и других структурных единиц (например, тетраэдров AlO4-) через атомы кислорода приводит к образованию регулярных структур, содержащих полости и поры. Существуют различные типы цеолитов, которые называют в соответствии с их типом структуры. Общую информацию, относящуюся к цеолитам, прежде всего к типам кристаллической структуры известных цеолитов, можно найти в энциклопедии Ullmann's Encyclopedia of Industrial Chemistry (энциклопедия Ульмана промышленной химии), в главе "Zeolites" (цеолиты), опубликованной онлайн 15.04.2012, DOI: 10.1002/14356007.a28_475.pub2.
Порошкообразный пористый кристаллический силикат металла, получаемый способом по настоящему изобретению, предпочтительно характеризуется цеолитной структурой с кристаллической структурой типа LTA, MFI, FAU, MOR, MEL или MWW. Более предпочтительно, силикаты металлов характеризуются кристаллической структурой типа MFI и MEL. Соответствующий тип кристаллической структуры можно определить методом рентгеноструктурного анализа (XRD) с использованием дифракции рентгеновских лучей. Типы структуры микро- и мезопористых материалов установлены Международной ассоциацией по цеолитам (International Zeolite Association, IZA, www.iza-online.org).
Порошкообразный пористый кристаллический силикат металла, получаемый способом по настоящему изобретению, предпочтительно содержит микро- и мезопоры. Согласно определению ГОРАС (Международный союз теоретической и прикладной химии) микропоры характеризуются диаметром менее 2 нм, а диаметр мезопор составляет от 2 нм до 50 нм.
Общий состав порошкообразных пористых кристаллических силикатов металлов обычно описывается следующей формулой:
(SiO2)1-x(AmOn)x,
А означает металл с валентностью р, выбранный из группы, состоящей из Ti, Al, Zr, Fe, Sn, Ge, In и В, индексы тип означают число атомов, где m×р=2n, x означает число от 0,0001 до 0,25, предпочтительно от 0,001 до 0,2 и прежде всего от 0,005 до 0,1. В случае нескольких различных металлов А, х соответственно относится к общей сумме всех оксидов металлов. А предпочтительно означает элементы титан (Ti), алюминий (Al), цирконий (Zr), железо (Fe), олово (Sn) и бор (В), Ge (германий), In (индий), предпочтительным является титан (Ti).
Порошкообразный пористый кристаллический силикат металла, получаемый способом по настоящему изобретению, предпочтительно может представлять собой силикат титана, боросиликат, силикат циркония, алюмосиликат или силикат железа. Прежде всего предпочтительным является силикат титана, прежде всего силикалит-1 титана (структура MFI) или силикалит-2 титана (структура MEL).
Источником кремния, используемым в способе по настоящему изобретению, в принципе может быть любое соединение, которое содержит диоксид кремния или смешанный оксид, содержащий диоксид кремния, или из которого может образовываться диоксид кремния в результате окисления или термического и/или гидролитического разложения. Однако предпочтительными являются соединения, которые содержат аморфный диоксид кремния или аморфный смешанный оксид, содержащий диоксид кремния, или из которых может образовываться аморфный диоксид кремния при окислении или термическом и/или гидролитическом разложении. Такой источник кремния можно, например, выбрать из группы, состоящей из пирогенного диоксида кремния, осажденного диоксида кремния, диоксида кремния, полученного золь-гель процессом, и из смесей указанных соединений.
Пирогенный диоксид кремния, также называемый высокодисперсный диоксид кремния, получают с использованием пламенного гидролиза или пламенного окисления. Указанное включает окисление или гидролиз гидролизующихся или окисляющихся исходных материалов, как правило, в водороднокислородном пламени. Исходные материалы, которые можно использовать для пирогенных способов, включают органические и неорганические вещества. Прежде всего пригоден тетрахлорид кремния. Получаемый таким образом гидрофильный диоксид кремния является аморфным. Высокодисперсный диоксид кремния обычно находится в агрегированной форме. Термин "агрегированная" следует понимать таким образом, что исходно формирующиеся так называемые первичные частицы образуют между собой прочные внутренние связи в дальнейшем ходе реакции с образованием трехмерной сетки. Первичные частицы в значительной степени не содержат пор, и на их поверхности присутствуют свободные гидроксильные группы.
Диоксид кремния, получаемый осаждением (осажденный диоксид кремния), образуется, например, в ходе реакции растворов жидкого стекла (силикатов натрия) с неорганическими кислотами.
Золь-гель процесс представляет собой процесс получения неметаллических неорганических или гибридных полимерных материалов из коллоидных дисперсий, называемых золями.
Исходными материалами для получения золей в большинстве случаев являются алкоксиды металлов или кремния. Гидролиз таких исходных материалов и конденсация образующихся реакционноспособных соединений являются важными основными реакциями золь-гель процесса. Пригодными источниками кремния для золь-гель процессов являются прежде всего тетраалкилортосиликаты, где алкил предпочтительно выбирают из группы, состоящей из метила, этила, пропила и бутила. Наиболее предпочтительным тетраалкилортосиликатом является тетраэтилортосиликат.
Источником металла, используемым в способе по настоящему соединению, может быть любое соединение, которое содержит оксид металла или металлсодержащий смешанный оксид, или из которого может образовываться соответствующий оксид металла или смешанный оксид в результате окисления или термического и/или гидролитического разложения. Источниками металлов, используемыми в контексте настоящего изобретения, являются источники следующих элементов: титан (Ti), алюминий (Al), цирконий (Zr), железо (Fe), олово (Sn), германий (Ge), индий (In) и бор (В), прежде всего предпочтителен титан.
Специалист в данной области техники по своему усмотрению выбирает пригодные источники кремния и металла. В принципе, специалист в данной области техники выбирает из следующих комбинаций: а) источник кремния и источник металла находятся в жидкой форме, б) источник кремния находится в твердой форме, а источник металла находится в жидкой форме, в) источник кремния и источник металла оба входят в состав одного вещества.
В данном контексте термин "в жидкой форме" означает, что источник кремния и/или источник металла представлен в форме жидкости или раствора.
Источники кремния в твердой форме можно, например, выбрать из группы, состоящей из пирогенного диоксида кремния, осажденного диоксида кремния, диоксида кремния, полученного золь-гель процессом, а также из их смесей. Предпочтительным является диоксид кремния с высокой степенью очистки, полученный осаждением, или пирогенный диоксид кремния.
Диоксид кремния высокой степени очистки, полученный осаждением, представляет собой диоксид кремния, полученный осаждением, который содержит
а) алюминий не более 1 част./млн,
б) бор не более 0,1 част./млн,
в) кальций не более 0,3 част./млн,
г) железо не более 0,6 част./млн,
д) никель не более 0,5 част./млн,
ж) фосфор не более 0,1 част./млн,
з) титан не более 1 част./млн,
и) цинк не более 0,3 част./млн,
где общая сумма указанных выше элементов, а также натрия и калия составляет менее 5 част./млн. Такой диоксид кремния с высокой степенью очистки можно получить способом, описанным в заявке WO 2010/037702. Содержание заявки WO 2010/037702 в полном объеме включено в настоящее описание в качестве ссылки.
Источник кремния и источник металла оба могут присутствовать в одном веществе различным образом. Примерами таких веществ являются смешанные оксиды металла и кремния, допированный оксидом металла диоксид кремния, металл-импрегнированный диоксид кремния, силикат металла, допированный металлом тетраалкилортосиликат, а также их смеси. Вещества такого типа предпочтительно являются аморфными. Предпочтительно таким веществом является аморфный диоксид кремния, допированный оксидом металла, аморфный диоксид кремния, импрегнированный металлом, или аморфный смешанный оксид металла и кремния.
"Смешанный оксид металла и кремния " содержит наряду с SiO2 один или более оксидов металлов, предпочтительно выбранных из группы, состоящей из GeO2, In2O3, Al2O3, TiO2, В2О3, SnO2, ZrO2 или Fe2O3. Смешанные оксиды металла и кремния можно получить любым пригодным способом, например, пламенным пиролизом, совместным осаждением, золь-гель процессом. Смешанные оксиды металла и кремния описаны, например, в патентах ЕР 0814058 или DE 102007049742.
"Допированный оксидом металла диоксид кремния" можно получить любым известным способом, например, пламенным пиролизом или способом импрегнирования с последующим прокаливанием.
"Металл-импрегнированный диоксид кремния" можно получить любым известным способом импрегнирования, например, способами "пропитки по влагоемкости".
В предпочтительном варианте способа по настоящему изобретению на стадии а) компонент (А) и компонент (Б) оба присутствуют в составе одного вещества, и указанное вещество выбирают из группы, состоящей из аморфного смешанного оксида кремния и металла, аморфного диоксида кремния, допированного оксидом металла, аморфного диоксида кремния, импрегнированного металлом, силиката металла, металл-допированного тетраалкилортосиликата, а также из их смесей. Более предпочтительно компонентом (А) в данном контексте является аморфный диоксид кремния, допированный оксидом металла, аморфный диоксид кремния, импрегнированный металлом, или аморфный смешанный оксид кремния и металла.
В другом предпочтительном варианте способа по настоящему изобретению на стадии а) компонент (А) присутствует в твердой форме, а компонент (Б) в жидкой форме. Более предпочтительно компонент (А) в данном контексте выбирают из группы, состоящей из пирогенного диоксида кремния, осажденного диоксида кремния, диоксида кремния, полученного золь-гель процессом, а также из их смесей. Наиболее предпочтительно компонентом (А) в данном контексте является диоксид кремния с высокой степенью очистки, полученный осаждением, или пирогенный диоксид кремния.
Гидротермальный синтез порошкообразных пористых кристаллических силикатов металлов сам по себе известен. То же самое относится к гидротермальному синтезу цеолитов, прежде всего цеолитов металлов. Следовательно, в принципе стадия а) способа по настоящему изобретению не ограничивается конкретными параметрами. Напротив, пригодны также все реагенты и параметры, которые известны специалистам в данной области техники для гидротермального синтеза пористых кристаллических силикатов металлов.
На стадии а) способа по настоящему изобретению при проведении гидротермального синтеза в водной смеси, включающей (А) по меньшей мере один источник кремния, (Б) по меньшей мере один источник металла и (В) по меньшей мере одну минерализирующую добавку, в качестве продукта реакции получают водную суспензию, содержащую пористый кристаллический силикат металла. Гидротермальный синтез, также называемый гидротермальным ростом кристаллов, представляет собой кристаллизацию из водных смесей при температурах выше 100°С до приблизительно выше 300°С и повышенном давлении вплоть до приблизительно 100 бар. Указанное означает, что рост из водных смесей является пригодным для веществ, которые в обычных условиях умеренно растворимы. Предпочтительно стадию а) способа по настоящему изобретению проводят при температуре от 100 до 250°С, более предпочтительно при температуре от 100 до 200°С, при автогенном давлении, создаваемом в реакторе высокого давления, например, в автоклаве. Давление, устанавливаемое в ходе гидротермального синтеза на стадии а) способа по настоящему изобретению, может находиться в диапазоне от 1,05 до 50 бар. Предпочтительно давление находится в диапазоне от 1,5 до 30 бар, более предпочтительно в диапазоне от 2 до 20 бар.
В указанных выше условиях осуществления способа по настоящему изобретению продолжительность проведения стадии а) способа по настоящему изобретению обычно составляет менее 12 ч. Предпочтительно, продолжительность стадии а) находится в диапазоне от 0,1 до 6 ч, более предпочтительно в диапазоне от 0,5 до 4 ч.
Минерализующие добавки, используемые в большинстве случаев, представляют собой HF или органические или неорганические основания, такие как гидроксиды четвертичного аммония, например, гидроксид тетрапропиламмония, или неорганические гидроксиды, такие как гидроксид натрия, гидроксид калия, гидроксид аммония или гидроксид кальция. Указанные добавки служат для повышения растворимости источников кремния и источников металлов, а также для достижения значения рН, оптимального для формирования кристаллов. Кроме того, можно также использовать соединения, которые в качестве минерализующих добавок могут образовывать комплексы с источниками металлов и источниками кремния. Гидротермальный синтез, как правило, проводят в щелочной среде при рН выше 7. Предпочтительно рН находится в диапазоне от 8 до 14, более предпочтительно в диапазоне от 9 до 13.
Для оптимального проведения стадии а) способа по настоящему изобретению водная смесь может дополнительно включать пригодные затравочные кристаллы. Затравочные кристаллы указанного типа известны специалисту в данной области техники.
В способе по настоящему изобретению водная смесь на стадии а) может дополнительно содержать матрицу, выбранную из группы, состоящей из аминов, соединений четвертичного аммония, спиртов и смесей указанных соединений. Матрица представляет собой соединение, которое при включении в кристаллическую решетку продукта в ходе гидротермального синтеза определяет кристаллическую структуру образующегося силиката металла. Примеры пригодных аминов включают: три(н-пропил)амин, ди(н-пропил)амин, н-пропиламин, ди(н-бутил)амин, этилендиамин, пропаноламин, этаноламин, хинуклидин и их замещенные производные, аммоний, соли аммония, морфолин, 1,5-диаминопентан, 1,6-диаминогексан, дипропилентриамин, дигексаметилентриамин, триэтилентетрамин, диэтилентриамин, галогениды 1-алкил-4-азобицикло[2.2.2]октан-4-оксида. Примеры пригодных спиртов включают: глицерин, гександиол, пентандиол.
Прежде всего предпочтительным является использование соединений тетраалкиламмония, таких как гидроксиды тетраалкиламмония, прежде всего гидроксида тетра-н-пропиламмония (ТРАОН), например, для получения силикалита-1 титана (структура MFI), гидроксида тетра-н-бутиламмония, например, для получения силикалита-2 титана (структура MEL), а также гидроксида тетраэтиламмония. Соединения четвертичного аммония предпочтительно используют в форме водных растворов.
Молярное соотношение количества матрицы, используемой на стадии а) способа по настоящему изобретению, и количества используемого кремния в принципе не ограничено. Предпочтительно, соотношение составляет ≥0,12 молей матрицы и менее <0,20 молей кремния.
Термин "пламенный спрей-пиролиз" известен специалисту в данной области техники и относится к процессу окислительной термической конверсии жидкого исходного материала, распределенного в виде высокодисперсных частиц в потоке газа при распылении, или суспензии в пламени, образующемся при сжигании топлива в присутствии кислорода. Пламенный спрей-пиролиз является общепринятым способом получения оксидов металлов, который описан, например, в заявке WO 2017/001366 А1 и в патенте США 2002/0041963 А1. Например, в заявке WO 2017/001366 А1 описан процесс данного типа для получения порошкообразных оксидов металлов с использованием пламенного спрей-пиролиза, в ходе которого силоксан-содержащий аэрозоль подают напрямую в пламя в реакторе, где он превращается в диоксид кремния.
Примеры топлива, используемого на стадии б) способа по настоящему изобретению, включают водород, метан, этан, пропан, бутан, природный газ и их смеси. Указанное топливо предпочтительно подают в газообразном состоянии в реактор, пригодный для проведения стадии б).
Кислород можно подавать в форме любого кислородсодержащего газа. Предпочтительным является использование воздуха.
Адиабатическая температура горения является стандартным параметром в специальной области техники для характеризации процесса горения по меньшей мере одного топлива и окислителя, состояние которых известно до начала горения. Соответственно, адиабатическую температуру горения, например, в соответствующем реакторе, можно рассчитывать способом, известным специалисту в данной области техники, исходя из известных параметров процесса, таких как температуры предварительного нагрева, величины удельного массового расхода и т.п.
Адиабатическая температура горения при проведении стадии б) способа по настоящему изобретению находится в диапазоне от 450 до 2200°С. Прежде всего предпочтительно адиабатическая температура горения находится в диапазоне от 450 до 1600°С, более предпочтительно в диапазоне от 500 до 1400°С, еще более предпочтительно в диапазоне от 500 до 1200°С, еще более предпочтительно в диапазоне от 550 до 1000°С, наиболее предпочтительно в диапазоне от 600 до 900°С.
В зависимости от участка проведения измерений, фактическая температура, создаваемая в пламени, изменяется в относительно широком диапазоне. Например, на стадии б) способа по настоящему изобретению температура, измеренная на 1,5 м ниже участка воспламенения может составлять по меньшей мере 300°С, предпочтительно температура на 1,5 м ниже участка воспламенения находится в диапазоне от 400 до 800°С.
Среднее время нахождения суспензии, полученной на стадии а), в реакторе при проведении стадии б) может составлять от 1 мс до 100 с. Предпочтительно среднее время нахождения находится в диапазоне от 0,1 с до 10 с, более предпочтительно в диапазоне от 0,5 с до 5 с. Расчет упомянутого выше среднего времени нахождения в реакторе (<t>, [с]) проводят с использованием общего объема газа, подаваемого в реактор в единицу времени (Vt, [м3/с (STP)]), и объема реактора (VR, [м3]).
<t>=VR/vt
Упомянутые выше температуры и среднее время нахождения на стадии б) способа по настоящему изобретению предпочтительно выбирают таким образом, чтобы окислительное разложение органической матрицы происходило на указанной стадии, при этом исключая повреждение пористой структуры получаемого продукта под действием избыточно высоких температур. Таким образом, если устанавливается относительно высокая температура пламени, которая в случае длительного применения может вызывать необратимые изменения пористой структуры силиката металла, целесообразно выбирать относительно короткое время нахождения в пламени.
Содержание твердых веществ wFT (мас. %) в водной суспензии, полученной на стадии а), можно рассчитать из общей массы указанной суспензии (MS) и массы воды в указанной суспензии (MH2O):
WFT=(MS-MH2O)/MS*100%
Содержание твердых веществ в суспензии, полученной на стадии а), составляет ≤70 мас. %. Предпочтительно содержание твердых веществ находится в диапазоне от 10 мас. % до 70 мас. %, более предпочтительно в диапазоне от 10 мас. % до 60 мас. %, наиболее предпочтительно в диапазоне от 20 мас. % до 50 мас. %. Содержание твердых веществ более 70 мас. % может вызвать технические сложности при распылении и проведении пламенного спрей-пиролиза на стадии б) способа по настоящему изобретению, в то время как содержание твердых веществ менее 10 мас. % может оказывать негативное действие на экономическую эффективность процесса из-за избыточного больших количеств воды, подлежащей испарению. Специалисту в данной области техники известны способы регулирования содержания твердых веществ, например, реагенты можно использовать в пригодной концентрации или суспензию можно соответствующим образом разбавить.
Средний диаметр (d50) частиц силиката металла в водной дисперсии, которую получают на стадии а) способа по настоящему изобретению, предпочтительно составляет менее 500 нм и более предпочтительно менее 400 нм. Средний диаметр частиц силиката металла можно определить, например, с использованием динамического лазерного светорассеяния (DLS).
Потери при прокаливании (в мас. %) определены немецким промышленным стандартом DIN 18128:2002-12 как доля органических соединений в образце. При прокаливании из образца удаляется органический компонент, например, присутствующий углерод окисляется и удаляется в виде диоксида углерода. Согласно DIN 18128:2002-12, потери при прокаливании порошкообразного пористого кристаллического силиката металла, получаемого способом по настоящему изобретению, предпочтительно составляют менее 5 мас. %, более предпочтительно менее 3 мас. %, наиболее предпочтительно менее 2 мас. %.
Если используют матрицу, по меньшей мере частичное термическое и/или окислительное разложение матрицы предпочтительно происходит на стадии б) способа по настоящему изобретению. Более предпочтительно, согласно настоящему изобретению, разлагается более 70 мас. % используемой матрицы, наиболее предпочтительно более 90 мас. %.
Удельная поверхность порошкообразных пористых кристаллических силикатов металлов, получаемых способом по настоящему изобретению, может составлять ≥20 м2/г, предпочтительно от 30 до 800 м2/г, более предпочтительно от 50 до 700 м2/г, наиболее предпочтительно от 70 до 600 м2/г. Удельную поверхность, также называемую просто площадью поверхности BET, определяют согласно DIN 9277:2014 адсорбцией азота согласно методу Брунауера-Эммета-Теллера (Brunauer-Emmett-Teller, BET).
Совокупный объем азота, десорбируемого из пор, рассчитывают методом BJH, описанным в статье Barret, Joyner и Halenda, Journal of the American Chemical Society, 73, cc. 373-380 (1951).
Способ по настоящему изобретению обеспечивает получение кристаллических силикатов металлов в порошкообразной форме. Соответственно, для применения в качестве катализаторов окисления при необходимости их можно переводить в пригодную для применения форму, например, микрогранул, сфер, таблеток, литых цилиндров, полых цилиндров или сотовых пузырей известными способами формования порошкообразных катализаторов, например, прессованием, грануляцией, высушиванием при распылении или экструзией.
В другом варианте осуществления способа по настоящему изобретению после стадии б) следует стадия в), включающая следующие стадии:
(1) добавление воды, при этом получают водную суспензию пористого кристаллического силиката металла,
(2) смешивание суспензии, полученной на стадии (1), с гранулирующими вспомогательными веществами,
(3) прессование, грануляция, высушивание при распылении или экструзия, при этом получают пористый кристаллический силикат металла в форме микрогранул, сфер, таблеток, литых цилиндров, полых цилиндров или сотовых пузырей.
Размер частиц таких формованных изделий предпочтительно находится в диапазоне от 0,1 мм до 10 мм.
Для смешивания и формования можно использовать все известные смешивающие и формующие устройства и способы, а также использовать все стандартные гранулирующие средства. Известные формующие устройства такого типа описаны, например, в энциклопедии Ульмана промышленной химии (Ullmann's  der Technischen Chemie, Ullmann's Encyclopedia of Industrial Chemistry, 4-е изд., т.2, с. 295 и далее, (1972)). Предпочтительным является использование одно- и двухшнековых экструдеров или экструзионных прессов. В данном случае возможно получение множества известных геометрических форм, например, литых цилиндров, полых цилиндров, звезд и т.п. Однако также можно получать сотовые пузыри.
der Technischen Chemie, Ullmann's Encyclopedia of Industrial Chemistry, 4-е изд., т.2, с. 295 и далее, (1972)). Предпочтительным является использование одно- и двухшнековых экструдеров или экструзионных прессов. В данном случае возможно получение множества известных геометрических форм, например, литых цилиндров, полых цилиндров, звезд и т.п. Однако также можно получать сотовые пузыри.
Водную суспензию, получаемую на стадии а) способа по настоящему изобретению, распыляют в ходе проведения стадии б), то есть распыляют в виде высокодисперсных частиц в окружающем газе, и таким образом формируют аэрозоль, трехфазную смесь твердое вещество/жидкость/газ, состоящую из газа и распределенных в нем жидких капель, которые в свою очередь содержат твердые частицы. Газ, используемый для распыления водной суспензии, может содержать кислород и/или по меньшей мере одно из вышеупомянутых топлив и/или по меньшей мере один инертный газ, например, азот. Для распыления предпочтительно использовать N2, Н2 или воздух, прежде всего предпочтителен воздух.
Аэрозоль, полученный на стадии а) при распылении водной суспензии, предпочтительно включает капли жидкости, характеризующиеся среднечисловым диаметром не более 2 мм, более предпочтительно не более 1 мм, наиболее предпочтительно не более 0,5 мм. Такой среднечисловой размер капель жидкости в аэрозоле может рассчитать, например, специалист в данной области техники на основании результатов определения размеров, регистрируемых используемыми приборами, соответствующих скоростей потока, свойств жидкости и газа и других параметров. В другом варианте, среднечисловой размер капель жидкости в аэрозоле, полученном на стадии а), можно измерить напрямую с использованием метода лазерной дифракции. Полученное измеренное распределение размера капель используют для определения среднего диаметра d50, который в качестве среднечислового размера частиц соответствует размеру капли, не превышает 50% всех частиц.
Распыление водной суспензии, которое осуществляется на стадии б) способа по настоящему изобретению, можно проводить с использованием различных установок и приборов, которые известны специалисту в данной области техники для указанной цели. Например, в данном контексте возможно применение центробежных распылителей, ротационных распылителей, ультразвуковых распылителей, однофазных, двухфазных или многофазных форсунок, а также различных инжекторных систем или аналогичных систем. Предпочтительно, согласно способу по настоящему изобретению, водную суспензию на стадии б) распыляют в пламени по меньшей мере через одну форсунку.
Кислород, используемый на стадии б) способа по настоящему изобретению, можно подавать в реактор пламенного спрей-пиролиза в нескольких участках. Например, используемую суспензию можно распылять в первый газовый поток, включающий воздух, в то время как основную часть воздуха (первичный воздух) подают в пламя в качестве второго газового потока параллельно направлению потока подаваемой распыляемой суспензии, а третий газовый поток (вторичный воздух) можно подавать тангенциально (под прямыми углами к направлению потока подаваемой распыляемой суспензии), например, чтобы исключить отложение материала. Аналогичным образом целесообразно подавать топливо в реактор в нескольких участках, например, в основной поток (поток первичного топлива) наряду с потоком первичного воздуха и вторичным потоком (поток вторичного топлива, внешнее топливо), например, для стабилизации создаваемого пламени.
Прежде всего целесообразно при проведении стадии б) способа по настоящему изобретению, количество кислорода превышает общее количество все горючих компонентов реакционной смеси. В данном контексте реакционная смесь означает суспензию, превращенную на стадии б), наряду с газообразными компонентами, используемыми на стадии б). Горючие компоненты указанной реакционной смеси включают, например, используемые топлива и матрицы. Индекс λ (лямбда) представляет собой соотношение, полученное при делении количества кислорода, присутствующего в реакционной смеси, на количество кислорода, необходимого для полного сжигания всех горючих компонентов реакционной смеси, при этом каждое количество выражают в моль/ч. Предпочтительно λ устанавливают на уровне от 1 до 10, более предпочтительно λ выбирают на уровне от 2 до 6.
Кислород и топливо, используемые при проведении стадии б) способа по настоящему изобретению, можно подавать в предварительно нагретой форме. В данном контексте диапазон пригодных температур составляет от 50 до 400°С. Суспензию, получаемую на стадии а) способа по настоящему изобретению, можно также подавать в пламя, предварительно нагретую до температуры от 50 до 300°С. Более предпочтительно указанную суспензию можно использовать на стадии б) напрямую после получения на стадии а) без охлаждения.
Соотношение общего объема газа, используемого на стадии б), выраженное в стандартных кубических метрах, и количества используемой водной суспензии, выраженного в кг, предпочтительно составляет от 0,1 до 100 м3/кг (нормальные условия), более предпочтительно от 0,5 до 50 м3/кг (нормальные условия), наиболее предпочтительно от 1 до 10 м3/кг (нормальные условия).
Использование способа по настоящему изобретению более предпочтительно позволяет получать титансодержащие цеолиты типа силикалита-1 титана и силикалита-2 титана, которые можно использовать в качестве катализаторов в окислительных реакциях с пероксидом водорода. Более конкретно, такие титансодержащие цеолиты можно использовать в качестве катализаторов при эпоксидировании олефинов с использованием водного раствора пероксида водорода.
Примеры
Пример 1
Получение неочищенной суспензии гидротермальным синтезом
Синтез цеолита силикалита-1 титана (TS-1, структура типа MFI) проводили в реакторе высокого давления (объем 3 м3) соответствующим способом, описанным в примере 1 патента ЕР 0814058 В1. В качестве источника кремния использовали аморфный диоксид кремния с высокой степенью очистки (фирма-производитель Evonik Industries), а в качестве источника титана использовали водный раствор гидроксида тетрапропиламмония титана (раствор Ti-TPA), где содержание TiO2 составляет 19,0 мас. %. Раствор Ti-TPA получали следующим образом.
Деионизированную воду (90,1 кг), 40% водный раствор гидроксида тетрапропиламмония (167,3 кг, фирма-производитель Sachem) и тетраэтилортотитанат (141,6 кг, фирма-производитель Connect Chemicals GmbH) перемешивали в течение 1 ч при 40°С в закрытом сосуде. Экзотермическое протекание реакции приводило к повышению температуры приблизительно на 25°С. Затем этанол отгоняли при 80°С при скорости 30 л/ч. Требуемое содержание ТЮ2 в полученном растворе Ti-TPA составляло 19,0 мас. %. После охлаждения раствор Ti-TPA использовали в синтезе TS-1.
В реактор высокого давления сначала загружали диоксид кремния с высокой степенью очистки (500 кг, фирма Evonik Industries), 40% водный раствор гидроксида тетрапропиламмония (382 кг, фирма Sachem), раствор Ti-TPA (193 кг), затравочные кристаллы силикалита-1 (10 кг) и деионизированную воду (1800 кг). Смесь перемешивали в замкнутом реакторе высокого давления при скорости перемешивания 50 об./мин в течение 3 ч при 170°С. Время нагрева до 170°С составляло 180 мин, после охлаждения в течение 150 мин синтез останавливали. Перемешивание при скорости перемешивания 50 об./мин продолжали с начала синтеза до его завершения.
Затравочные кристаллы силикалита-1 получали гидротермальным синтезом в реакторе высокого давления с использованием диоксида кремния с высокой степенью очистки (500 кг, фирма Evonik Industries), 40% водного раствора гидроксида тетрапропиламмония (400 кг, фирма Sachem) и деионизированной воды (1800 кг). Смесь перемешивали в замкнутом реакторе высокого давления при скорости перемешивания 50 об./мин в течение 3 ч при 160°С. Время нагрева до 160°С составляло 180 мин, после охлаждения в течение 150 мин синтез останавливали. Перемешивание при скорости перемешивания 50 об./мин продолжали с начала синтеза до его завершения.
Пример сравнения 1
Стандартная обработка после гидротермального синтеза
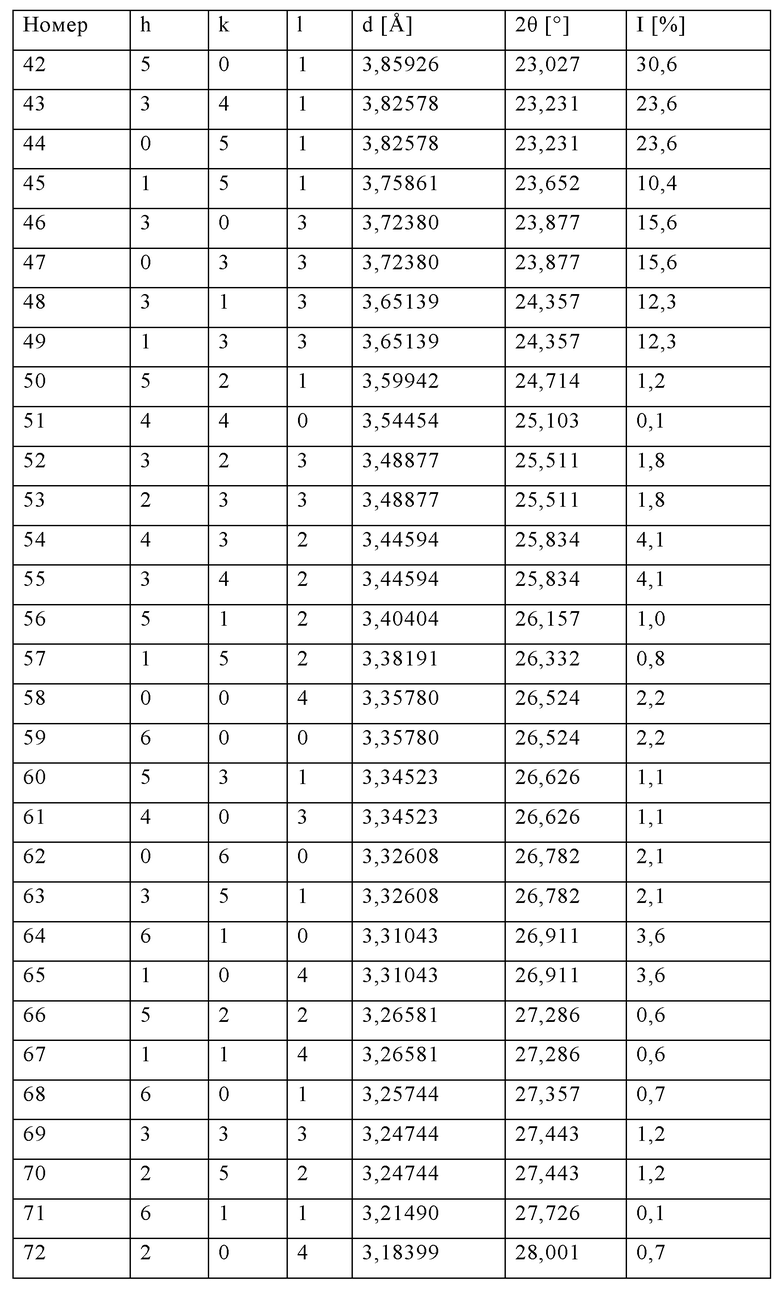
Уксусную кислоту (60 мас. %) добавляли в неочищенную суспензию, описанную в примере 1, до рН=7, полученный осадок отделяли фильтрованием на фильтр-прессе и промывали дистиллированной водой. Полученное твердое вещество сушили с использованием метода высушивания при распылении при температуре на входном отверстии 420°С и скорости распыления 1700 мин-1 (температура на выходном отверстии составляла 110°С). Затем частично высушенный порошок прокаливали при температуре максимум 650°С в трубчатой печи в течение 2 ч. Полученный таким образом продукт характеризовался площадью поверхности BET 470 м2/г, при этом потери при прокаливании (измеренные при 550°С) составляли 0,65%. По результатам рентгеноструктурного анализа (XRD, фиг.1) продукт характеризовался кристаллической структурой TS-1 (код идентификации в Международном центре дифракционных данных ICDD: 01-089-8099). По данным пороструктурного анализа с использованием азота методом расчета распределения пор по размерам BJH, объем пор составлял 0,23 мл/г.
Пример 2
Обжиг с распылением после гидротермального синтеза (600°С)
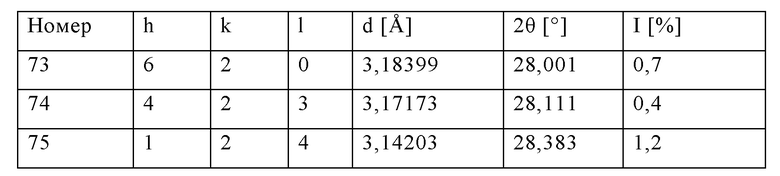
Неочищенную суспензию (16 кг/ч), описанную в примере 1, распыляли на опытной установке в потоке азота (скорость потока 18 м3/ч) для распыления через двухфазное сопло с внутренним диаметром 2 мм и щелью 1 мм. Пламя водород/воздух поддерживали в потоке водорода (10 м3/ч) и потоке первичного воздуха (45 м3/ч). Для исключения отложений материалов в перпендикулярном направлении подавали вторичный воздух (скорость потока 25 м3/ч). Температуру, измеряемую на 1,5 м ниже участка воспламенения, поддерживали при 600°С за счет незначительного изменения потока водорода. Адиабатическая температура сгорания в реакторе составляла приблизительно 680°С. Время нахождения в реакторе составляло приблизительно 1,1 с. Отходящие газы, содержащие прокаленный цеолит, направляли через охлаждаемую водой зону охлаждения (температура охлаждающего агента: 25°С) с диаметром 100 мм и длиной 6 м и затем собирали в патронных фильтрах при максимум 250°С. При последующей очистке патронных фильтров можно собирать готовый прокаленный продукт (4,35 кг/ч). Площадь поверхности BET полученного таким образом продукта составляла 499 м2/г, а потери при прокаливании (измеренные при 550°С) составляли 1,35 мас. %. Результаты XRD (фиг.2) свидетельствовали о том, что продукт характеризовался кристаллической структурой TS-1 (код идентификации ICDD: 01-089-8099). По данным пороструктурного анализа с использованием азота методом BJH, объем пор составлял 0,3 мл/г.
Пример сравнения 2 (отрицательный пример)
Обжиг с распылением после гидротермального синтеза (400°С)
Неочищенную суспензию (15 кг/ч), описанную в примере 1, распыляли на опытной установке в потоке азота (скорость потока 18 м3/ч) для распыления через двухфазное сопло с внутренним диаметром 2 мм и щелью 1 мм. Пламя водород/воздух поддерживали в потоке водорода (8 м3/ч) и потоке первичного воздуха (45 м3/ч). Температуру, измеряемую на 1,5 м ниже участка воспламенения, поддерживали при 400°С за счет незначительного изменения потока водорода. Адиабатическая температура горения в реакторе составляла приблизительно 544°С. Время нахождения в реакторе составляло приблизительно 1,35 с. Отходящие газы, содержащие прокаленный цеолит, направляли через охлаждаемую водой зону охлаждения (температура охлаждающего агента: 25°С) с диаметром 100 мм и длиной 6 м и затем собирали в патронных фильтрах при максимум 250°С. При последующей очистке патронных фильтров можно собирать готовый прокаленный продукт (4,4 кг/ч). Площадь поверхности BET полученного таким образом продукта составляла 240 м2/г, а потери при прокаливании (измеренные при 550°С) составляли 9,0 мас. %. Из-за значительных потерь при прокаливании полученный продукт был непригоден для дальнейшей переработки с целью получения конечного продукта и использовании его в контрольной реакции НРРО.
Пример 3
Обжиг с распылением после гидротермального синтеза (700°С)
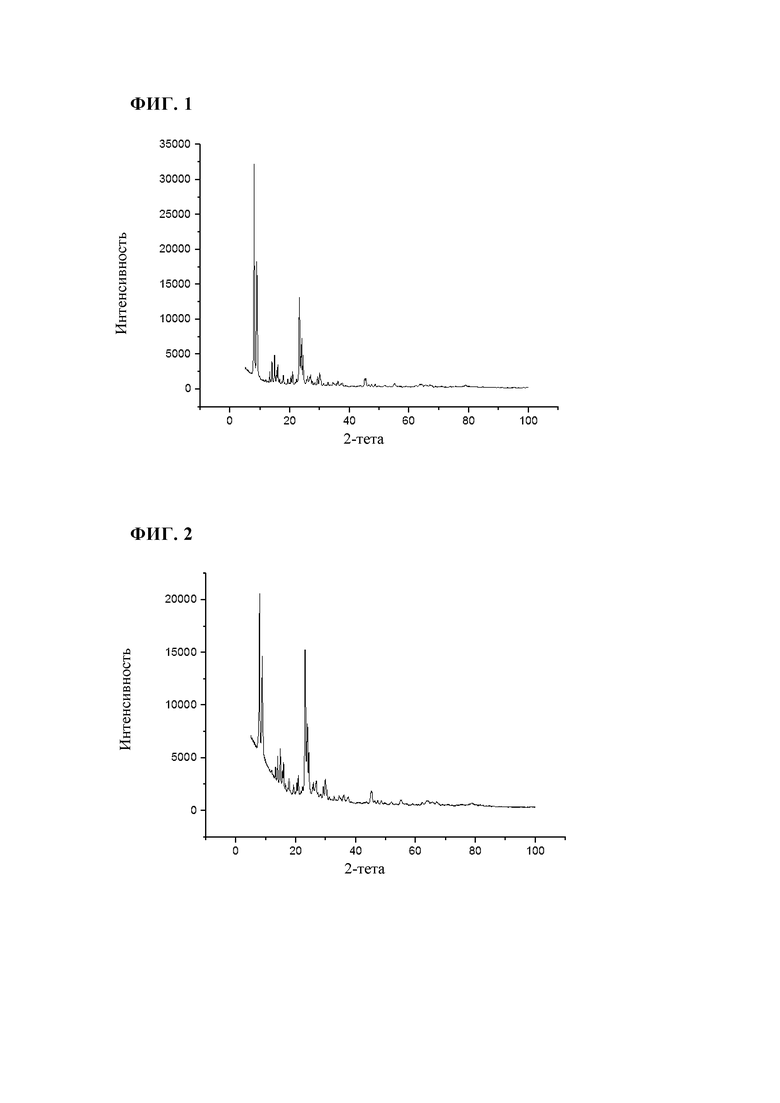
Неочищенную суспензию (30 кг/ч), описанную в примере 1, распыляли на опытной установке в потоке азота (скорость потока 18 м3/ч) для распыления через двухфазное сопло с внутренним диаметром 2 мм и щелью 1 мм. Пламя водород/воздух поддерживали в потоке водорода (9,1 м3/ч) и потоке первичного воздуха (27 м3/ч). Температуру, измеряемую на 1,5 м ниже участка воспламенения, поддерживали при 700°С за счет незначительного изменения потока водорода. Адиабатическая температура горения в реакторе составляла приблизительно 750°С. Время нахождения в реакторе составляло приблизительно 1,1 с. Отходящие газы, содержащие прокаленный цеолит, направляли через охлаждаемую водой зону охлаждения (поток Н2О со скоростью 10 л/ч, поток воздуха со скоростью 4 м3/ч) с диаметром 100 мм и длиной 6 м и затем собирали в патронных фильтрах при максимум 250°С. При последующей очистке патронных фильтров можно собирать готовый прокаленный продукт (8,7 кг/ч). Площадь поверхности BET полученного таким образом продукта составляла 506 м2/г, а потери при прокаливании (измеренные при 550°С) составляли 1,1 мас. %. Результаты XRD (фиг.3) свидетельствовали о том, что продукт характеризовался кристаллической структурой TS-1 (код идентификации ICDD: 01-089-8099). По данным пороструктурного анализа с использованием азота методом BJH, объем пор составлял 0,3 мл/г.
Пример 4
Обжиг с распылением после гидротермального синтеза (800°С)
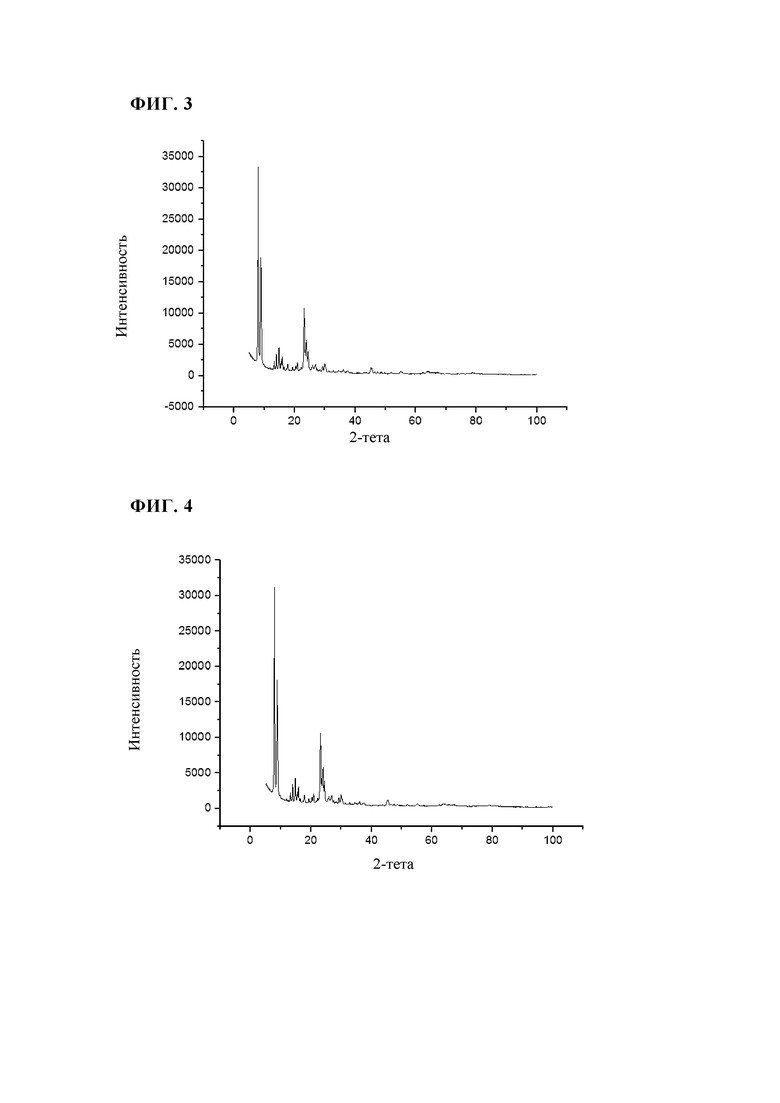
Неочищенную суспензию (14 кг/ч), описанную в примере 1, распыляли на опытной установке в потоке азота (скорость потока 18 м3/ч) для распыления через двухфазное сопло с внутренним диаметром 2 мм и щелью 1 мм. Пламя водород/воздух поддерживали в потоке водорода (12,2 м3/ч) и потоке первичного воздуха (40 м3/ч). Температуру, измеряемую на 1,5 м ниже участка воспламенения, поддерживали при 800°С за счет незначительного изменения потока водорода. Адиабатическая температура горения в реакторе составляла приблизительно 830°С. Время нахождения в реакторе составляло приблизительно 0,85 с. Отходящие газы, содержащие прокаленный цеолит, направляли через охлаждаемую водой зону охлаждения (температура охлаждающего агента: 25°С) с диаметром 100 мм и длиной 6 м и затем собирали в патронных фильтрах при максимум 250°С. При последующей очистке патронных фильтров можно собирать готовый прокаленный продукт (4,2 кг/ч). Площадь поверхности BET полученного таким образом продукта составляла 477 м2/г, а потери при прокаливании (измеренные при 550°С) составляли 0,87 мас. %. Результаты XRD (фиг.4) свидетельствовали о том, что продукт характеризовался кристаллической структурой TS-1 (код идентификации ICDD: 01-089-8099). По данным пороструктурного анализа с использованием азота методом BJH, объем пор составлял 0,3 мл/г.
Пример сравнения 3
Формование порошкообразного цеолита, описанного в примере сравнения 1
Порошок (1200 г), полученный в примере сравнения 1, перемешивали с метилгидроксиэтилцеллюлозой (75 г, Tylose МН1000), продуктом Licowax С (75 г), с коллоидным раствором кремниевой кислоты (1000 г, Koestrosol 0830 AS) и деионизированной водой (350 г) на высокоскоростной мешалке фирмы Eirich. Полученную массу экструдировали с использованием экструдера (НВ-Feinmechanik LTW 63) через перфорированную тарелку с диаметром 3,2 мм. Затем экструдаты сушили в сушильном шкафу при 80°С в течение 1 ч и прокаливали в муфельной печи при 570°С в течение 12 ч.
Пример 5
Формование порошкообразного цеолита, описанного в примере 2
Порошок (1200 г), полученный в примере 2, смешивали с метилгидроксиэтилцеллюлозой (75 г, Tylose МН1000), продуктом Licowax С (75 г), с коллоидным раствором кремниевой кислоты (1000 г, Koestrosol 0830 AS) и деионизированной водой (350 г) на высокоскоростной мешалке фирмы Eirich. Полученную массу экструдировали с использованием экструдера (НВ-Feinmechanik LTW 63) через перфорированную тарелку с диаметром 3,2 мм. Затем экструдаты сушили в сушильном шкафу при 80°С в течение 1 ч и прокаливали в муфельной печи при 570°С в течение 12 ч.
Пример 6
Каталитическое испытание с использованием катализатора, описанного в примере сравнения 3
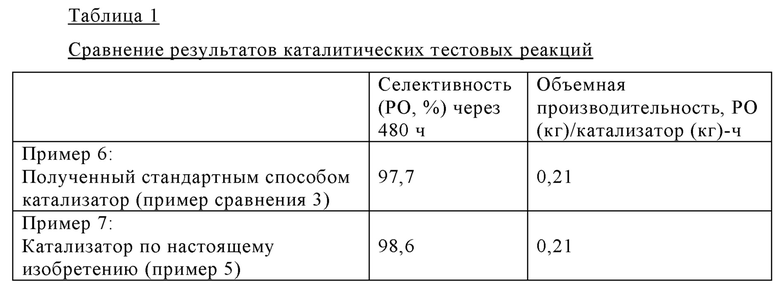
Эпоксидирование пропена с использованием пероксида водорода (60%) проводили в двух реакторах с псевдоожиженным слоем, каждый из которых содержал катализатор (9 г), описанный в примере сравнения 3, в форме экструдатов. Реакторы устанавливали последовательно (реактор 1 → реактор 2) и эксплуатировали в восходящем потоке. Первый питающий поток с общей скоростью потока 20 г/ч, содержащий метанол, пероксид водорода и воду, и второй питающий поток с общей скоростью потока 20 г/ч, содержащий пропен, оба подавали в первый реактор. Давление реактора поддерживали при 25 бар с использованием расположенного ниже по потоку клапана для поддержания давления во втором реакторе. Объем реакционной смеси, поступающей из второго реактора с псевдоожиженным слоем, увеличивали при нормальном давлении. В полученной газовой фазе определяли содержание пропена, пропиленоксида и кислорода, а в полученной жидкой фазе определяли содержание пропиленоксида и пероксида водорода. Исходная селективность в отношении пропиленоксида после завершения 23-часового цикла реакции составляла 91,1%. Через 480 ч селективность в отношении пропиленоксида составляла 97,7%.
Пример 7
Каталитическое испытание с использованием катализатора, описанного в примере 5
Каталитическое эпоксидирование пропена проводили, как описано в примере 6, но с использованием катализатора, полученного в примере 5.
Исходная селективность в отношении пропиленоксида после завершения 25-часового цикла реакции составляла 93,5%. Через 480 ч селективность в отношении пропиленоксида составляла 98,6%.
Как показано в примерах 2-4 в сравнении с примером сравнения 1, способ по настоящему изобретению включает значительно меньшее количество технологических стадий по сравнению со стандартным процессом. Кроме того, в указанном способе решены проблемы утилизации сточных вод, которые, как правило, образуются при фильтровании и очистке продукта после гидротермального синтеза. Неожиданно было установлено, что получаемые силикалиты титана, после пламенного спрей-пиролиза характеризуются пористостью, сравнимой с пористостью полученного стандартным способом силикалита титана.
Как следует из примеров 6 и 7, а также из данных, представленных в табл.1, полученный стандартным способом катализатор (пример сравнения 3) и катализатор, который получен из силиката металла согласно настоящему изобретению (пример 5), после эксплуатации в течение 480 ч в ходе эпоксидирования пропилена в пропиленоксид (РО), оба являются высоко активными и селективными. Катализатор, который получен из силиката металла согласно настоящему изобретению, в действительности характеризуется повышением селективности в отношении пропиленоксида на 0,9% по сравнению с полученным стандартным способом катализатором при сравнимой объемной производительности. Таким образом, с использованием катализатора, который получен из силикалита металла согласно настоящему изобретению, определенно можно значительно повысить выход продукта, пропиленоксида, при расчете на единицу времени и объем реактора.
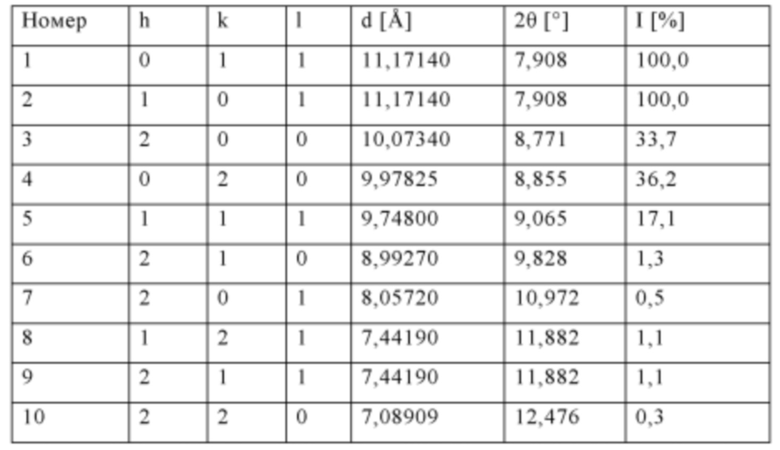
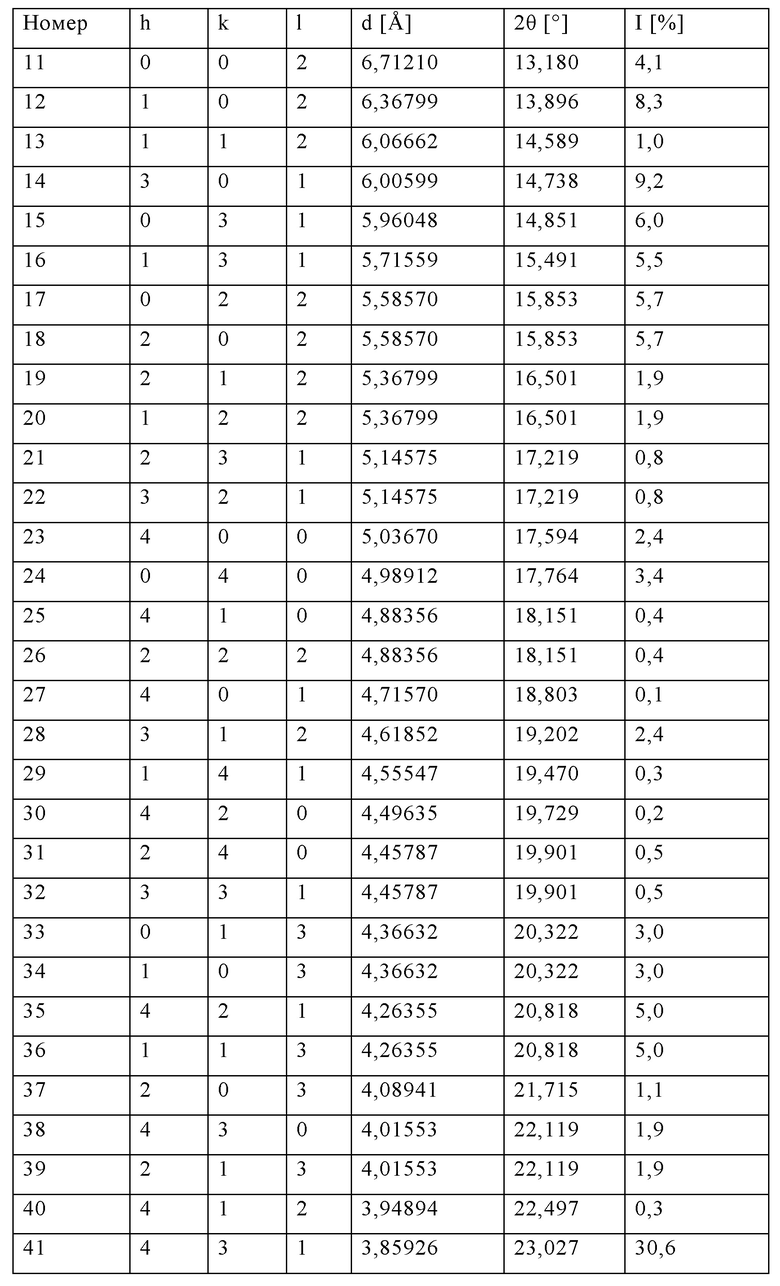
Данные кристаллографического анализа силикалита-1 титана (источник: база данных Международного дифракционного центра ICDD)
Код идентификации: 01-089-8099
Название соединения: силикалит титана
Код ICSD (Inorganic Crystal Structure Database, база данных неорганических кристаллических веществ): 88413
Ссылка: Lamberti С., Bordiga S., Zecchina A., Carati A., Fitch A.N., Artioli G., Petrini G., Salvalaggio M., Marra G.L., J. Catal., 183, c. 222 (1999).
Перечень рефлексов
Изобретение относится к способу получения порошкообразных пористых кристаллических силикатов металлов. Способ получения порошкообразного пористого кристаллического цеолита силикалита-1 титана включает стадии: а) гидротермальный синтез в водной смеси, включающей (А) аморфный диоксид кремния, (Б) водный раствор гидроксида тетрапропиламмония титана и (В) водный раствор гидроксида тетрапропиламмония, при этом получают водную суспензию, содержащую пористый кристаллический силикат металла в качестве продукта реакции, и б) прокаливание продукта реакции, при этом прокаливание проводят с использованием пламенного спрей-пиролиза при адиабатической температуре горения в диапазоне 450-2200°С. Далее суспензию, содержащую ≤70 мас. % твердых веществ, полученную на стадии а), распыляют в пламени, создаваемом при горении топлива в присутствии кислорода, при этом получают порошкообразный пористый кристаллический силикалит-1 титана. Технический результат заключается в разработке способа получения порошкообразных пористых кристаллических силикатов металлов, который включает меньшее число технологических стадий по сравнению со стандартным процессом и решает проблему утилизации производственных отходов. 8 з.п. ф-лы, 4 ил., 2 табл., 10 пр.
1. Способ получения порошкообразного пористого кристаллического цеолита силикалита-1 титана,
включающий следующие стадии:
а) гидротермальный синтез в водной смеси, включающей (А) аморфный диоксид кремния, (Б) водный раствор гидроксида тетрапропиламмония титана и (В) водный раствор гидроксида тетрапропиламмония, при этом получают водную суспензию, содержащую пористый кристаллический силикат металла в качестве продукта реакции, и
б) прокаливание продукта реакции, отличающееся тем, что
прокаливание проводят с использованием пламенного спрей-пиролиза при адиабатической температуре горения в диапазоне 450-2200°С, при этом суспензию, содержащую ≤70 мас. % твердых веществ, которую получают на стадии а), распыляют в пламени, создаваемом при горении топлива в присутствии кислорода, при этом получают порошкообразный пористый кристаллический силикалит-1 титана.
2. Способ по п. 1, отличающийся тем, что пористый кристаллический силикат металла характеризуется цеолитной структурой с кристаллической структурой типа LTA, MFI, FAU, MOR, MEL или MWW.
3. Способ по п. 1 или 2, отличающийся тем, что топливо выбирают из группы, состоящей из водорода, метана, этана, пропана, бутана, природного газа и их смесей.
4. Способ по любому из пп. 1-3, отличающийся тем, что среднее время нахождения суспензии, полученной на стадии а), в ходе конверсии указанной суспензии на стадии б) находится в диапазоне от 0,1 с до 10 с.
5. Способ по любому из пп. 1-4, отличающийся тем, что согласно DIN 18128:2002-12 потери при прокаливании пористого кристаллического силиката металла составляют менее 5 мас. %.
6. Способ по любому из пп. 1-5, отличающийся тем, что водная смесь, полученная на стадии а), дополнительно включает пригодные затравочные кристаллы.
7. Способ по любому из пп. 1-6, отличающийся тем, что водная смесь, полученная на стадии а), дополнительно содержит матрицу, выбранную из группы, состоящей из аминов, соединений четвертичного аммония, спиртов и смесей указанных соединений.
8. Способ по любому из пп. 1-7, отличающийся тем, что стадию а) проводят при температуре от 100 до 250°С при автогенном давлении, создаваемом в реакторе высокого давления.
9. Способ по любому из пп. 1-8, отличающийся тем, что после стадии б) проводят формование на стадии в), включающее стадии:
(1) добавление воды, при этом получают водную суспензию порошкообразного пористого кристаллического силиката металла,
(2) смешивание суспензии, полученной на стадии (1), с гранулирующими вспомогательными веществами,
(3) прессование, грануляция, высушивание при распылении, грануляция распылением или экструзия, при этом получают пористый кристаллический силикат металла в форме микрогранул, сфер, таблеток, литых цилиндров, полых цилиндров или сотовых пузырей.
| EP 0814058 А1, 29.12.1997 | |||
| ВЫСОКОКРЕМНЕЗЕМНЫЙ ЦЕОЛИТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2313487C1 |
| СПОСОБ ПОЛУЧЕНИЯ СИНТЕТИЧЕСКОГО ЦЕОЛИТА | 1987 |
|
RU2005691C1 |
| КРИСТАЛЛИЧЕСКИЙ СИНТЕТИЧЕСКИЙ МАТЕРИАЛ НА ОСНОВЕ ОКСИДА КРЕМНИЯ И ОКСИДА ТИТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1980 |
|
RU2076775C1 |
| ПОРОШОК СМЕШАННОГО ОКСИДА КРЕМНИЯ И ТИТАНА, ЕГО ДИСПЕРСИЯ И ТИТАНСОДЕРЖАЩИЙ ЦЕОЛИТ НА ЕГО ОСНОВЕ | 2007 |
|
RU2415081C2 |
| US 2017253547 A1, 07.09.2017 | |||
| US 5919430 A, 06.07.1999. | |||
