УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения порошкообразных пористых кристаллических силикатов металлов.
Термин "силикат" означает соединения, образованные тетраэдрическим SiO4, тетраэдры которого могут быть соединены друг с другом различным образом. Силикатные структуры этого типа, содержащие металлы, называются силикатами металлов. Важными примерами силикатов металлов являются цеолиты.
Цеолиты представляют собой кристаллические силикаты, например, алюмосиликаты, в которых трехмерное соединение силикатных тетраэдров (SiO4-) и других структурных фрагментов (например, тетраэдров AlO4-) обеспечивает получение регулярных структур, содержащих каналы и поры. Существуют разные типы цеолитов, которые называют в соответствии с типом их структуры. Общие сведения, относящиеся к цеолитам, в особенности, к типам кристаллических структур известных цеолитов, приведены в публикации Ullmann's Encyclopedia of Industrial Chemistry, "Zeolites", опубл. в интернете 15 апреля 2012 г., DOI: 10.1002/14356007.a28_475.pub2.
Вследствие их особой пористой структуры цеолиты обладают интересными характеристиками и их можно использовать, например, в качестве катализаторов окисления.
Синтетические цеолиты можно получить с помощью гидротермального синтеза в присутствии образующей пористую структуру матрицы. Так, например, в CN 101348263 А раскрыт способ получения цеолитов, обладающих отношением Si/Al, составляющим от 50 до 5000, и размером частиц, равным от 30 до 200 мкм, который включает следующие стадии способа: (1) получение реакционной смеси, содержащей источники кремния и источники алюминия, и гидроксид металла; (2) реакция гидролиза; (3) последующая распылительная сушка смеси с получением микросфер алюмосиликата; (4) гидротермальная реакция предварительно полученных микросфер в присутствии воды и органического амина при температуре, равной от 160 до 200°С, и кристаллизация полученного цеолита; и (5) промывка, (6) сушка и (7) и прокаливание цеолита при температуре, равной от 350 до 800°С.
В US 4410501 А раскрыт способ получения силикалита титана. Силикалит титана получают путем (1) получения геля для синтеза из гидролизующегося содержащего кремний соединения, например, тетраэтилортосиликата, гидролизующегося содержащего титан соединения в присутствии гидроксида тетра-н-пропиламмония при 175°С, (2) последующих гидротермального синтеза, гидролиза и кристаллизации реакционной смеси. После завершения кристаллизации кристаллы (3) удаляют фильтрованием, (4) промывают, (5) сушат и в заключение (6) прокаливают при 550°С в течение 6 ч.
В ЕР 814058 А1 раскрыто получение различных цеолитов из соответствующих пирогенных смешанных оксидов металлов и кремния. Смешанные оксиды металлов и кремния получают путем (1) гидротермального синтеза при температуре, равной от 100 до 220°С, в присутствии матрицы, выбранной из числа следующих: амины, соединения аммония и гидроксиды щелочных металлов/щелочноземельных металлов, затем проводят (2) фильтрование, (3) промывку водой и (4) прокаливание, например, при температуре, равной 550°С, в течение 4 ч. В предпочтительном варианте осуществления путем распылительной сушки получают предварительно сформованный содержащий матрицу гранулированный смешанный оксидный материал, который затем обрабатывают путем гидротермального синтеза, фильтруют, промывают и прокаливают.
В CN 1482062 раскрыт способ получения силикалита титана-1, в котором твердый силикагель вводят в гидротермальную реакцию с неорганическим источником титана. Способ включает следующие стадии: (1) пропитка твердого силикагеля с помощью Ti(SO4)2, (2) прокаливание, (3) гидротермальный синтез с использованием силикагеля и Ti(SO4)2 + ТПА-ОН (гидроксид тетрапропиламмония) + вода, (4) (осаждение и) фильтрование, (5) промывка, (6) сушка, (7) прокаливание.
Использующиеся в процедурах предшествующего уровня техники стадии способа, проводимые после гидротермального синтеза, являются дорогостоящими и требуют больших затрат времени. В частности, промывка неочищенного пористого кристаллического силиката металла для удаления органического материала, осадившегося во время гидротермального синтеза, является трудоемкой и приводит к образованию существенных количеств сточных вод, часто содержащих вещества, которые являются вредными для находящихся в воде организмов и которые затруднительно удалить, такие как соли тетраалкиламмония (образующиеся во время осаждения). Кроме того, сушка и прокаливание промытого пористого кристаллического силиката металла, проводимые в конце процедуры, являются дорогостоящими и требуют существенных количеств времени и энергии.
Согласно настоящему изобретению было обнаружено, что осаждение, фильтрование, промывку, сушку и прокаливание неочищенного пористого кристаллического силиката металла, полученного после гидротермального синтеза, можно не проводить, если вместо этого материал обрабатывают с помощью подходящей процедуры пламенного пиролиза. Это не было раскрыто или предложено в предшествующем уровне техники и это является особенно удивительным, принимая во внимание тот известный факт, что упорядоченная пористая структура кристаллических силикатов металлов разрушается при повышенных температурах. Так, например, силикалит титана-1 претерпевает необратимые изменения в структуре при температурах выше 650°С (см., например, Advanced Materials Research Vol.287-290, 2011, p.317-321), температура внутри пламени в устройстве для пламенного пиролиза, а также температура во время сгорания органического остатка, осадившегося на частицы неочищенного пористого кристаллического силиката металла во время пламенного пиролиза, является существенно более высокой.
Согласно настоящему изобретению было обнаружено, что пламенный пиролиз можно провести с использованием материалов, неочищенных пористых кристаллических силикатов металлов, полученных после гидротермального синтеза, таким образом, что сохраняется их упорядоченная пористая кристаллическая структура силиката металла и без оказания неблагоприятного воздействия на их каталитические характеристики.
Описание изобретения
В частности, согласно настоящему изобретению было обнаружено, что порошкообразный пористый кристаллический силикат металла можно получить способом, включающим следующие стадии:
(a) гидротермальный синтез с использованием водной смеси, содержащей:
(A) источник кремния,
(B) источник металла, и
(C) вспомогательный компонент,
с получением водной суспензии продукта реакции 1, содержащей неочищенный пористый кристаллический силикат металла; и
(b) пламенный распылительный пиролиз продукта реакции 1, где водную суспензию, полученную на стадии (а), распыляют в пламя, образующееся при сгорании топлива, в присутствии кислорода с получением порошкообразного пористого кристаллического силиката металла;
где водная суспензия, содержащая продукт реакции 1, полученная на стадии (а), обладает содержанием твердых веществ, составляющим ≤70 мас.%; и где максимальная эффективная температура, Тэфф, до которой во время пламенного пиролиза нагревается не менее 90 мас.% пористого кристаллического силиката металла, находится в диапазоне Тмин<Тэфф.<Тмакс., и
где Тмин равна 750°С, и
где Тмакс равна 1250°С, и
где источником металла (В) является источник титана (Ti), железа (Fe) или алюминия (Al), и
где вспомогательный компонент (С) выбран из группы, состоящей из следующих: органические основания, четвертичные гидроксиды аммония и их смеси.
Гидротермальный синтез
Гидротермальный синтез, также называющийся гидротермальным выращиванием кристаллов, представляет собой процедуру, предназначенную для кристаллизации из водных смесей при температурах, находящихся в диапазоне от примерно 100 до примерно 300°С, и при повышенном давлении, равном вплоть до примерно 100 бар, которую можно использовать для реагентов и продуктов, умеренно растворимых в водном растворе при температуре ниже 100°С. Гидротермальный синтез порошкообразных пористых кристаллических силикатов металлов и, в частности, цеолитов хорошо известен в данной области техники. Проведение стадии (а) способа, предлагаемого в настоящем изобретении, т.е. гидротермального синтеза с использованием водной смеси, содержащей (А) источник кремния, (В) источник металла и (С) вспомогательный компонент, приводит к получению водной суспензии продукта реакции 1, содержащей неочищенный пористый кристаллический силикат металла. Предпочтительно, если стадию (а) способа, предлагаемого в настоящем изобретении, проводят при температуре, равной от 100 до 250°С, более предпочтительно от 100 до 200°С, при автогенном давлении, созданном в выдерживающем давление реакторе, например, в автоклаве. Давление, установившееся во время гидротермального синтеза, проводимого на стадии (а) способа, предлагаемого в настоящем изобретении, может находиться в диапазоне от 1,05 до 50 бар. Предпочтительно, если давление находится в диапазоне от 1,5 до 30 бар; более предпочтительно, если давление находится в диапазоне от 2 до 20 бар. Указанные выше условия проведения реакции обычно позволяют специалисту в данной области техники провести стадию (а) способа, предлагаемого в настоящем изобретении, в течение менее 12 ч, предпочтительно, в течение от 0,1 до 6 ч, более предпочтительно в течение от 0,5 до 4 ч.
Гидротермальный синтез обычно проводят в щелочной среде при значении рН, превышающем 7. В соответствии с настоящим изобретением гидротермальный синтез проводят при значении рН, предпочтительно находящемся в диапазоне от 8 до 14; более предпочтительно, находящемся в диапазоне от 9 до 13.
Для проведения гидротермального синтеза пористых кристаллических силикатов металлов обычно необходимо использование вспомогательных компонентов, облегчающих растворение источников кремния и металлов и регулирование значения рН до подходящего для образования кристаллов. Кроме того, вспомогательный компонент представляет собой матрицу, которая вследствие включения в кристаллическую решетку продукта во время гидротермального синтеза определяет кристаллическую структуру образовавшегося силиката металла. Для способа, предлагаемого в настоящем изобретении подходящими являются только такие вспомогательные компоненты, которые подвергаются термическому и/или окислительному разрушению во время пламенного распылительного пиролиза, проводимого на стадии (b). В способе, предлагаемом в настоящем изобретении, предпочтительно, если степень разрушения вспомогательного компонента составляет более 70 мас.%, наиболее предпочтительно, если степень разрушения составляет более 90 мас.%. Соответствующие вспомогательные компоненты хорошо известны специалистам в данной области техники.
Типичными примерами вспомогательных компонентов, подходящих для способа, предлагаемого в настоящем изобретении, которые можно использовать для облегчения растворения источников кремния и металлов и регулирования значения рН, являются неорганические или органические основания, такие как четвертичные гидроксиды аммония, диамины, диолы и их смеси. Типичные примеры вспомогательных компонентов, подходящих для способа, предлагаемого в настоящем изобретении, которые можно использовать для содействия образованию кристаллической структуры силиката металла (матрицы), являются четвертичные гидроксиды аммония, диамины, диолы и их смеси, точнее, гидроксид тетраэтиламмония, гидроксид тетрапропиламмония, гидроксид тетрабутиламмония, гидроксид тетрапентиламмония, 1,6-диаминогексан, 1,2-пентандиол и их смеси.
В предпочтительных вариантах осуществления в способах, предлагаемых в настоящем изобретении, используют один или большее количество следующих вспомогательных компонентов, предназначенных для содействия образованию кристаллической структуры силиката металла (матрицы): гидроксид тетраэтиламмония, гидроксид тетрапропиламмония, гидроксид тетрабутиламмония, гидроксид тетрапентиламмония, 1,6-диаминогексан, 1,2-пентандиол и их смеси.
В особенно предпочтительных способах, предлагаемых в настоящем изобретении, в качестве вспомогательного компонента используют гидроксид тетрапропиламмония. Четвертичные аммониевые соединения предпочтительно использовать в виде водных растворов.
В предпочтительном варианте осуществления в способах, предлагаемых в настоящем изобретении, для содействия образованию силикалита титана-1 (структура типа MFI) используют гидроксид тетра-н-пропиламмония (ТПА-ОН).
В другом предпочтительном варианте осуществления в способах, предлагаемых в настоящем изобретении, для содействия образованию силикалита титана-2 (структура типа MEL) используют гидроксид тетра-н-бутиламмония.
Для специалистов в данной области техники должно быть очевидно, что в соответствии с настоящим изобретением вспомогательные компоненты, предназначенные для гидротермального синтеза, необходимо выбирать таким образом, что обеспечивается (i) облегчение растворения источников кремния и металлов, (ii) регулирование значения рН, а также (iii) содействие образованию кристаллической структуры силиката металла. Это можно осуществить с помощью одного вспомогательного компонента, способного выполнять все три функции ((i), (ii) и (iii)), или, альтернативно, с помощью более, чем одного вспомогательного компонента, каждый из которых выполняет некоторые из группы функций ((i), (ii) и (iii)).
На отношение количества молей полного количества вспомогательных компонентов, использующихся для содействия образованию кристаллической структуры силиката металла (матрица), к количеству молей кремния, использующихся на стадии (а) способа, предлагаемого в настоящем изобретении, по существу не налагаются ограничения. Предпочтительно, если выбрано молярное отношение, находящееся в следующем диапазоне: 0,12 ≤ количество молей матрицы/количество молей кремния < 0,20.
Для обеспечения оптимального режима проведения стадии (а) способа, предлагаемого в настоящем изобретении, водная смесь может дополнительно содержать подходящие затравочные кристаллы. Подходящие затравочные кристаллы и методики их получения известны специалистам в данной области техники. В предпочтительном варианте осуществления к реакционной смеси, использующейся на стадии (а) способа, предлагаемого в настоящем изобретении, добавляют затравочные кристаллы силикалита-1 или затравочные кристаллы силикалита титана-1 для содействия образованию кристаллов силикалита титана-1 (структура типа MFI). В другом предпочтительном варианте осуществления к реакционной смеси, использующейся на стадии (а) способа, предлагаемого в настоящем изобретении, добавляют затравочные кристаллы силикалита-2 или затравочные кристаллы силикалита титана-2 для содействия образованию кристаллов силикалита титана-2 (структура типа MEL).
Источником кремния, использующимся в способе, предлагаемом в настоящем изобретении, по существу может являться любое соединение, которое включает диоксид кремния или содержащий кремний смешанный оксид, или способно его образовывать в результате окисления или термического и/или гидролитического разрушения. Однако предпочтение отдается соединениям, включающим аморфный диоксид кремния или аморфный содержащий кремний смешанный оксид, или способным образовывать такие соединения в результате окисления или термического и/или гидролитического разрушения. Соответствующий источник кремния может быть выбран, например, из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси. В предпочтительных способах, предлагаемых в настоящем изобретении, используют компонент (А), выбранный из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси.
Пирогенный диоксид кремния, также называющийся тонкодисперсным диоксидом кремния, получают путем гидролиза в пламени или окисления в пламени. Это включает окисление или гидролиз гидролизующихся или окисляющихся исходных веществ, обычно в водородном/кислородном пламени. Исходные вещества, которые можно использовать для проведения пирогенных реакций, включают органические и неорганические вещества. Особенно подходящим является тетрахлорид кремния. Полученный таким образом гидрофильный диоксид кремния является аморфным. Тонкодисперсный диоксид кремния обычно получают в агрегированной форме. "Агрегированный" означает, что первичные частицы, т.е. частицы, образовавшиеся на начальной стадии реакции, на последующих стадиях реакции образуют друг с другом сильные связи, это в конечном счете приводит к образованию трехмерной сетки. Первичные частицы в основном не содержат пор и содержат на поверхности свободные гидроксигруппы. Содержание воды в таком источнике кремния - тонкодисперсном диоксиде кремния обычно составляет менее 5,0 мас.%.
С другой стороны, осажденный диоксид кремния, также называющийся силикагелем, представляет собой диоксид кремния, полученный по методикам осаждения, например, в результате реакции растворимого стекла (силикаты натрия) с неорганическими кислотами. Содержание воды в таком силикагеле обычно находится в диапазоне от 0,5 до 80 мас.% в зависимости от условий проведения сушки. Сушку можно провести по разным методикам (например, с использованием или без использования нагретого воздуха) в течение периода времени, составляющего от секунд (быстрая сушка) до часов (медленная сушка). Высушенный гель называется ксерогелем (содержание воды ≤40 мас.%), невысушенный гель называется гидрогелем (содержание воды >40 мас.%).
Золь-гелевая технология представляет собой технологию получения неметаллических неорганических или гибридных-полимерных материалов из коллоидных дисперсий, называющихся золями. Исходными веществами для золь-гелевого синтеза часто являются алкоксиды металлов или кремния. Гидролиз таких исходных веществ и реакция конденсации образовавшихся реакционноспособных веществ являются важными базовыми реакциями в золь-гелевой технологии. Источниками кремния, особенно подходящими для использования в золь-гелевой технологии, являются тетраалкилортосиликаты, в которых алкил предпочтительно выбран из группы, состоящей из следующих: метил, этил, пропил и бутил. Наиболее предпочтительным тетраалкилортосиликатом является тетраэтилортосиликат.
Источником металла, использующимся в способе, предлагаемом в настоящем изобретении, может являться любое соединение, включающее оксид металла или содержащий металл смешанный оксид, или способное образовывать соответствующий оксид металла или смешанный оксид в результате окисления или термического и/или гидролитического разрушения. В контексте настоящего изобретения использующимися источниками металла являются источники титана (Ti), алюминия (Al) и/или железа (Fe), особенно предпочтительным является источник титана.
Специалист в данной области техники может свободно выбрать подходящие источники кремния и металла. В принципе, специалист в данной области техники может выбрать из числа следующих комбинаций: (а) источник кремния и источник металла оба находятся в жидкой форме, (b) источник кремния находится в твердой форме и источник металла находится в жидкой форме, (с) источник кремния и источник металла объединены с получением одного компонента. Термин "в жидкой форме" означает, что источник кремния и/или источник металла находятся в форме жидкости или раствора.
Источники кремния, находящиеся в твердой форме, могут быть выбраны, например, из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси. Предпочтение отдается обладающему высокой чистотой диоксиду кремния, полученному путем осаждения, или пирогенному диоксиду кремния.
Обладающий высокой чистотой диоксид кремния, полученный путем осаждения, представляет собой диоксид кремния, полученный путем осаждения и обладающий следующими содержаниями:
- алюминий: менее 1 част./млн,
- бор: менее 0,1 част./млн,
- кальций: менее 0,3 част./млн,
- железо: менее 0,6 част./млн,
- никель: менее 0,5 част./млн,
- фосфор: менее 0,1 част./млн,
- титан: менее 1 част./млн,
- цинк: менее 0,3 част./млн,
где суммарное количество указанных выше элементов, а также натрия и калия составляет менее 5 част./млн. Такой обладающий высокой чистотой диоксид кремния можно получить, например, способом, раскрытым в WO 2010/037702.
Источник кремния и источник металла можно объединить с получением одного компонента по разным методикам. В случае использования в качестве источника кремния силикагеля объединение с источником металла (например, пропитку) можно провести с использованием ксеро- или гидрогеля. Примерами таких объединенных компонентов являются смешанные оксиды металлов и кремния, легированный оксидом металла диоксид кремния, пропитанный металлом диоксид кремния, силикат металла, легированный металлом тетраалкилортосиликат и их смеси. Предпочтительно, если объединенные компоненты такого типа являются аморфными. Предпочтительно, если такими объединенными компонентами являются аморфные диоксиды кремния, легированные оксидом металла, аморфные диоксиды кремния, пропитанные металлом, или аморфные смешанные оксиды металлов и кремния.
"Аморфный смешанный оксид металла и кремния" в дополнение к SiO2 содержит один или большее количество оксидов металлов, предпочтительно выбранных из группы, состоящей из следующих: Al2O3, TiO2 и Fe2O3. Смешанные оксиды металлов и кремния можно получить по любой подходящей методике, например, с помощью пламенного пиролиза, совместного осаждения, золь-гелевой технологии. Смешанные оксиды металлов и кремния раскрыты, например, в ЕР 0814058 и DE 102007049742.
"Легированный оксидом металла диоксид кремния" можно получить по нескольким методикам, хорошо известным специалистам в данной области техники, например, по методикам пламенного пиролиза или пропитки с последующим прокаливанием.
"Пропитанный металлом диоксид кремния" можно получить по нескольким методикам пропитки, хорошо известным специалистам в данной области техники, например, по методикам "пропитки по влагоемкости".
В предпочтительном варианте осуществления способа, предлагаемого в настоящем изобретении, на стадии (а) компонент (А) и компонент (В) объединяют с получением одного компонента и этот компонент выбран из группы, состоящей из следующих: аморфный смешанный оксид металла и кремния, аморфный диоксид кремния, легированный оксидом металла, аморфный диоксид кремния, пропитанный металлом, силикат металла, легированный металлом тетраалкилортосиликат и их смеси. Более предпочтительно, если компонентом (А) является аморфный диоксид кремния, легированный оксидом металла, аморфный диоксид кремния, пропитанный металлом, или аморфный смешанный оксид металла и кремния.
В другом предпочтительном варианте осуществления способа, предлагаемого в настоящем изобретении, на стадии (а) компонент (А) находится в твердой форме и компонент (В) находится в жидкой форме. В это случае более предпочтительно, если компонент (А) выбран из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси. В это случае наиболее предпочтительно, если компонентом (А) является обладающий высокой чистотой диоксид кремния, полученный путем осаждения пирогенного диоксида кремния.
Водная суспензия продукта реакции 1, содержащая неочищенный пористый кристаллический силикат металла, обладает содержанием твердых веществ, составляющим ≤70 мас.%. Содержание твердых веществ, СТВ (мас.%), можно рассчитать на основании полной массы этой суспензии (Мс) и массы воды, содержащейся в этой суспензии (Мн2о):
СТВ=(Мс-МН2о)/Мс×100%
Предпочтительно, если содержание твердых веществ находится в диапазоне от 10 до 70 мас.%; более предпочтительно в диапазоне от 10 до 60 мас.%; наиболее предпочтительно в диапазоне от 20 до 50 мас.%. Содержание твердых веществ, превышающее 70 мас.%, приводит к техническим затруднениям во время проведения пламенного распылительного пиролиза на стадии (b) способа, предлагаемого в настоящем изобретении, тогда как содержание твердых веществ, составляющее менее 10 мас.%, оказывает неблагоприятное воздействие на рентабельность способа вследствие чрезмерно большого количества воды, которую необходимо выпаривать. Специалисту в данной области техники известны методики регулирования содержания твердых веществ; например, можно использовать реагенты при подходящих концентрациях или суспензию можно разбавить.
Пламенный распылительный пиролиз
Термин "пламенный распылительный пиролиз" хорошо известен специалистам в данной области техники и он означает реакцию термоокислительного превращения жидкого исходного вещества, мелкораспыленного в потоке газа, проводимую путем распыления суспензии в пламя, образующееся при сгорании топлива, в присутствии кислорода. Пламенный распылительный пиролиз является известным способом получения оксидов металлов, описанным, например, в WO 2017/001366 А1 и US 2002/0041963 А1. Так, например, в WO 2017/001366 А1 раскрыт способ такого типа, предназначенный для получения порошкообразных оксидов металлов с помощью пламенного распылительного пиролиза, в котором содержащий силоксан аэрозоль вводят непосредственно в пламя в реакторе, в котором он превращается в диоксид кремния.
Для проведения способа пламенного распылительного пиролиза, предлагаемого в настоящем изобретении, необходимо использование горючего топлива. Примеры такого топлива включают водород, метан, этан, пропан, бутан, влажный, сухой или синтетический природный газ (ПГ) и их смеси. Предпочтительно, если топливо подают в реактор в газообразном состоянии. Однако если в качестве топлива используют метан, этан, пропан, бутан, влажный, сухой или синтетический природный газ (ПГ), то количество водной суспензии, распыляемой в пламя, необходимо уменьшить по сравнению со случаем использования водорода в качестве топлива. Соответственно, в способах пламенного распылительного пиролиза, предлагаемых в настоящем изобретении, в качестве топлива предпочтительно использовать водород, чтобы обеспечить равномерную температуру пламени и подходящий профиль вязкости.
Кислород можно подавать в реактор в виде любого газа, содержащего кислород. В контексте настоящего изобретения предпочтение отдается использованию воздуха.
Средняя продолжительность пребывания материала суспензии, полученной на стадии (а), в реакторе во время проведения стадии (b), может составлять от 1 мс до 100 с. Предпочтительно, если средняя продолжительность пребывания находится в диапазоне от 0,1 до 10 с; более предпочтительно в диапазоне от 0,5 до 5 с. Расчет указанной выше средней продолжительности пребывания в реакторе (<t>, [с]) проводят с использованием значений для полного объема газа, подаваемого в реактор в единицу времени (Vt, [м3/с (СТД (стандартные температура и давление))]) и объема реактора (VR, [м3]). <t> = VR/Vt. На стадии (b) способа, предлагаемого в настоящем изобретении, среднюю продолжительность пребывания выбирают таким образом, чтобы на этой стадии произошло окислительное разрушение органического остатка, но не произошло повреждение пористой структуры полученного продукта.
Водную суспензию, полученную на стадии (а) способа, предлагаемого в настоящем изобретении, распыляют во время проведения стадии (b), т.е. мелко распыляют в окружающем газе, и, таким образом, получают аэрозоль, трехфазную смесь твердое вещество/жидкость/газ, содержащую газ с мелкораспыленными в нем капельками жидкости, которые, в свою очередь, содержат твердые частицы. Газ, использующийся для распыления водной суспензии, может включать кислород и/или по меньшей мере одно из указанных выше топлив, и/или по меньшей мере один инертный газ, например, азот. Предпочтение отдается использованию N2, Н2 или воздуха, особое предпочтение отдается использованию воздуха.
Аэрозоль, образовавшийся на стадии (b) при распылении водной суспензии, предпочтительно содержит капельки жидкости, обладающие среднечисловым диаметром, равным не более 2 мм, более предпочтительно не более 1 мм, наиболее предпочтительно не более 0,5 мм. Среднечисловой диаметр капелек жидкости, содержащихся в аэрозоле, зависит от размеров использующейся установки, соответствующих скоростей потоков, характеристик жидкости и газа и других параметров, и специалисты в данной области техники могут его рассчитать с помощью численного моделирования с использованием стандартного программного обеспечения для моделирования (например, Ansys Fluent). Альтернативно, среднечисловой диаметр капелек, содержащихся в аэрозоле, образовавшемся на стадии (b), можно определить непосредственно с помощью дифракции лазерного излучения. Полученное распределение капелек по размерам используют для определения медианного размера d50, который отражает такой размер капельки, который не превышает 50% всех капелек, и является среднечисловым диаметром капелек.
Распыление водной суспензии, которое проводят на стадии (b) способа, предлагаемого в настоящем изобретении, можно осуществить с помощью разных установок и приборов, хорошо известных специалистам в данной области техники. Так, например, можно использовать дисковые распылители, центробежные распылители, ультразвуковые распылители, однофазные, двухфазные или многофазные сопла и разные системы для впрыскивания или сходные системы. Предпочтительно, если на стадии (b) способа, предлагаемого в настоящем изобретении, водную суспензию распыляют в пламя по меньшей мере через одно сопло.
Кислород, необходимый для проведения стадии (b) способа, предлагаемого в настоящем изобретении, можно подавать в реактор для пламенного распылительного пиролиза в нескольких положениях. Так, например, суспензию можно распылять в первый поток газа, содержащий воздух, тогда как наиболее значительную часть воздуха (первичный воздух) подают в пламя в виде второго потока газа параллельно направлению потока суспензии, и третий поток газа (вторичный воздух) можно подавать под углом (например, перпендикулярно направлению потока суспензии), например, для предотвращения образования отложений материала. Аналогичным образом, благоприятной может являться подача топлива в реактор в нескольких положениях, например, главный поток (поток первичного топлива) можно подавать вместе с потоком первичного воздуха и второй поток (поток вторичного топлива, наружного топлива) можно подавать, например, для стабилизации пламени.
Особенно предпочтительно, если при проведении стадии (b) способа, предлагаемого в настоящем изобретении, количество кислорода является избыточным по отношению к полному количеству всех горючих компонентов реакционной смеси. Реакционная смесь означает суспензию, подвергающуюся превращению на стадии (b), вместе с газообразными компонентами, использующимися на стадии (b). Горючие компоненты этой реакционной смеси включают, например, использующиеся топливо и матрицы. Показатель λ (лямбда) описывает отношение количества кислорода, содержащегося в реакционной смеси, к количеству кислорода, необходимому для завершения сгорания всех горючих компонентов, содержащихся в реакционной смеси, все количества указаны в моль/ч. Предпочтительно, если значение показателя λ установлено так, что оно находится в диапазоне от 1 до 10; более предпочтительно, от 2 до 6.
Кислород и топливо, использующиеся во время проведения стадии (b) способа, предлагаемого в настоящем изобретении, можно подавать в предварительно нагретом виде. Подходящий диапазон температур составляет от 50 до 400°С. Суспензию, полученную на стадии (а) способа, предлагаемого в настоящем изобретении, также можно подавать в пламя предварительно нагретой до температуры, равной от 50 до 300°С. Более предпочтительно, если для проведения пламенного распылительного пиролиза в соответствии со стадией (b) суспензию, полученную на стадии (а) способа, предлагаемого в настоящем изобретении, можно использовать непосредственно после получения, т.е. без охлаждения.
Отношение полного объема газа, использующегося на стадии (b), выраженное в стандартных кубических метрах, к количеству использующейся водной суспензии, выраженному в кг, предпочтительно составляет от 0,1 до 100 м3 (СТД)/кг, более предпочтительно от 0,5 до 50 м3 (СТД)/кг, наиболее предпочтительно от 1 до 10 м3 (СТД)/кг.
Порошкообразный пористый кристаллический силикат металла, получаемый способом, предлагаемым в настоящем изобретении, предпочтительно обладает цеолитной структурой. Цеолиты представляют собой кристаллические силикаты, например, алюмосиликаты, в которых трехмерное соединение силикатных тетраэдров (SiO4-) и других структурных фрагментов (например, тетраэдров AlO4-) через атомы кислорода обеспечивает получение регулярных структур, содержащих каналы и поры. Существуют разные типы цеолитов, которые называют в соответствии с типом их структуры. Общие сведения, относящиеся к цеолитам, в особенности, к типам кристаллических структур известных цеолитов, приведены в публикации Ullmann's Encyclopedia of Industrial Chemistry, chapter "Zeolites", опубл. в интернете 15.04.2012, DOI: 10.1002/14356007.a28 475.pub2.
Предпочтительно, если порошкообразный пористый кристаллический силикат металла, получаемый способом, предлагаемым в настоящем изобретении, обладает цеолитной структурой и кристаллической структурой типа LTA, MFI, FAU, MOR, MEL или MWW. Наиболее предпочтительно, если порошкообразный пористый кристаллический силикат металла, получаемый способом, предлагаемым в настоящем изобретении, обладает цеолитной структурой типа MFI или MEL. Тип кристаллической структуры можно определить с помощью структурного анализа с использованием рентгенографии (РГГ). Типы структур микро- и мезопористых цеолитных материалов утверждены Международной цеолитной ассоциацией (IZA, www.iza-online.org).
Предпочтительно, если порошкообразный пористый кристаллический силикат металла, получаемый способом, предлагаемым в настоящем изобретении, содержит микро- и мезопоры. В соответствии с определением IUPAC (Международный союз теоретической и прикладной химии) микропоры обладают диаметром, равным менее, 2 нм, и мезопоры обладают диаметром, равным от 2 до 50 нм.
Обычный состав порошкообразных пористых кристаллических силикатов металлов в большинстве случаев является следующим:
(SiO2)1-x(AmOn)x,
А обозначает элемент, обладающий валентностью р, выбранный из группы, состоящей из следующих Ti, Al и Fe; m и n обозначают количества атомов, где m×р равно 2n; х обозначает число, равное от 0,0001 до 0,25, предпочтительно от 0,001 до 0,2 и особенно предпочтительно от 0,005 до 0,1. В случае нескольких разных металлов А, х означает полное суммарное количеству всех оксидов металлов соответственно. Предпочтительно, если А выбран из числа следующих титан (Ti), алюминий (Al), железо (Fe), особое предпочтение отдается титану (Ti).
Предпочтительно, если порошкообразным пористым кристаллическим силикатом металла, получаемым способом, предлагаемым в настоящем изобретении, является силикат титана, алюмосиликат или силикат железа. Особое предпочтение отдается силикату титана, в особенности, силикалиту титана-1 (структура типа MFI) и силикалиту титана-2 (структура типа MEL).
Медианный диаметр частиц (d50) силиката металла, содержащегося в водной дисперсии, которая получена на стадии (а) способа, предлагаемого в настоящем изобретении, предпочтительно равен менее 500 нм и более предпочтительно менее 400 нм. Медианный диаметр частиц силиката металла можно определить, например, с помощью динамического лазерного светорассеяния (ДСР).
Порошкообразные пористые кристаллические силикаты металлов, полученные способом, предлагаемым в настоящем изобретении, могут обладать удельной площадью поверхности, составляющей ≥20 м2/г, предпочтительно от 30 до 800 м2/г, более предпочтительно от 50 до 700 м2/г, наиболее предпочтительно от 70 до 600 м2/г. Удельную площадь поверхности, также называющуюся просто БЭТ-поверхностью, определяют в соответствии со стандартом DIN 9277:2014 по адсорбции азота в соответствии с методикой Брунауэра-Эммета-Теллера. Суммарный объем пор, измеренный по десорбции азота, и объем микропор рассчитывают по методике БДХ (Баррета-Джойнера-Халенды) (BARRETT, JOYNER and HALENDA, Journal of the American Chemical Society, 73:373-380, 1951).
Потери при прокаливании (выраженные в мас.%) определены в стандарте DIN 18128:2002-12, как показатель доли органических веществ в образце. В ходе процедуры озоления происходит удаление органического компонента, содержащегося в образце; например, содержащийся углерод окисляется и выделяется в виде диоксида углерода. Потери при прокаливании порошкообразного пористого кристаллического силиката металла, полученного способом, предлагаемым в настоящем изобретении, определенные в соответствии со стандартом DIN 18128:2002-12, предпочтительно составляют менее 5 мас.%, более предпочтительно менее 3 мас.%, наиболее предпочтительно менее 2 мас.%.
В предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором пористый кристаллический силикат металла обладает цеолитной структурой типа MFI.
В другом предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором вспомогательный компонент (С) выбран из группы, состоящей из следующих: четвертичные гидроксиды аммония, диамины, диолы и их смеси, и в котором источником металла (В) является источник титана (Ti).
В другом предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором вспомогательный компонент (С) выбран из группы, состоящей из следующих: гидроксид тетраэтиламмония, гидроксид тетрапропиламмония, гидроксид тетрабутиламмония, гидроксид тетрапентиламмония, 1,6-диаминогексан, 1,2-пентандиол и их смеси, и в котором источником металла (В) является источник титана (Ti).
В другом предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором вспомогательным компонентом (С) является гидроксид тетрапропиламмония, и в котором источником металла (В) является источник титана (Ti), и в котором пористый кристаллический силикат титана обладает цеолитной структурой типа MFI.
В другом предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором
- компонент (А) выбран из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси,
и в котором
- источником металла (В) является источник титана (Ti),
и в котором
- вспомогательный компонент (С) выбран из группы, состоящей из следующих: органические основания, четвертичные гидроксиды аммония и их смеси, и в котором
- пористый кристаллический силикат металла обладает цеолитной структурой типа MFI или MEL, и в котором
- топливом, использующимся для пламенного распылительного пиролиза, является водород.
В другом предпочтительном варианте осуществления настоящее изобретение относится к способу, в котором
- компонент (А) и компонент (В) объединяют с получением одного компонента и этот компонент выбран из группы, состоящей из следующих: аморфный смешанный оксид металла и кремния, аморфный диоксид кремния, легированный оксидом металла, аморфный диоксид кремния, пропитанный металлом, силикат металла, легированный металлом тетраалкилортосиликат и их смеси,
и в котором
- источником металла (В) является источник титана (Ti),
и в котором
- вспомогательный компонент (С) выбран из группы, состоящей из следующих: органические основания, четвертичные гидроксиды аммония и их смеси,
и в котором
- пористый кристаллический силикат металла обладает цеолитной структурой типа MFI или MEL,
и в котором
- топливом, использующимся для пламенного распылительного пиролиза, является водород.
Максимальная эффективная температура, Тэфф.
Максимальная эффективная температура, Тэфф, является максимальной температурой, до которой во время пламенного пиролиза нагревается пористый кристаллический силикат металла в каждой из капелек, полученных на стадии (b). Максимальная эффективная температура, Тэфф, зависит от ряда переменных, таких как размеры использующейся установки, скорости потоков, характеристики жидкости и газа и т.п. Ее рассчитывают с помощью стандартного молекулярно-динамического моделирования (например, с помощью программного обеспечения Ansys Fluent), как это описано ниже.
В соответствии с настоящим изобретением максимальную эффективную температуру, Тэфф, до которой во время пламенного пиролиза нагревается не менее 90 мас.% пористого кристаллического силиката металла, регулируют таким образом, что окислительное разрушение органического вещества, содержащегося в продукте реакции 1, протекает в основном полностью (т.е. удалено >70%, предпочтительно >90% органического вещества, содержащегося в продукте реакции 1), но не происходит повреждение пористой структуры продукта.
Для обеспечения этого условия максимальную эффективную температуру, Тэфф, необходимо регулировать следующим образом: Тмин.<Тэфф.<Тмакс, где Тмин.=750°С и Тмакс=1250°С,
в предпочтительных вариантах осуществления Тмин. и Тмакс. выбраны таким образом, что Тмин=800°С и Тмакс=1200°С,
в других предпочтительных вариантах осуществления Тмин. и Тмакс. выбраны таким образом, что Тмин=850°С и Тмакс=1100°С.
Расчет Тэфф с помощью стандартного молекулярно-динамического моделирования (например, с помощью программного обеспечения Ansys Fluent).
Описанное ниже моделирование можно провести с помощью стандартного программного обеспечения для моделирования (например, Ansys Fluent), таким образом, можно облегчить расчет эффективной температуры, до которой нагревается пористый кристаллический силикат металла, содержащийся в каждой из множества капелек, проходящих через зону пламенного пиролиза. Максимальная температура, полученная для каждой капельки, является максимальной эффективной температурой, Тэфф., до которой нагревается пористый кристаллический силикат металла, содержащийся в этой капельке. В соответствии с настоящим изобретением максимальную эффективную температуру, Тэфф., до которой во время пламенного пиролиза нагревается не менее 90 мас.% пористого кристаллического силиката металла, необходимо регулировать следующим образом: Тмин.<Тэфф.<Тмакс. При проведении моделирования необходимо учитывать наличие в капельках воды, силиката и остаточных органических компонентов.
Непрерывная фаза (газовая):
При проведении моделирования газовую фазу рассматривают, как идеальные газы. Теплопроводность, вязкость и теплоемкость, ср, смесей газов рассчитывают с использованием закона смешения при известном массовом составе. Используют характеристики чистых компонентов (например, Н2, Н2О (пар), СО2, О2, N2), приведенные в базах данных для материалов (например, в базе данных Ansys Fluent). Коэффициент диффузии массы для каждого компонента, содержащегося в газовой фазе, рассчитывают на основании кинетической теории газов, все необходимые параметры приведены в общедоступных базах данных для материалов (например, в базе данных Ansys Fluent). В качестве исходных переменных используют массовую скорость потока топлива и массовую скорость потока воздуха. Для учета турбулентности используют стандартную модель k-s. Для моделирования излучения в газовой фазе используют модель дискретных ординат с угловой дискретностью, например, разделения тета: 4; разделения фи: 4; тета-пиксели: 1; фи-пиксели: 1; стенка: непрозрачная, внутренняя излучающая способность: 1; коэффициент теплообмена с окружающей средой: например, 5 Вт/м2/K; температура окружающей среды: например, 300 K. Модель сгорания: конечная скорость (одна стадия)/турбулентное рассеивание. Так, например, при использовании Н2 в качестве компонента топлива: Н2 + 0,5 О2 → Н2О; кинетика протекания реакции: константа скорости Аррениуса: предэкспоненциальный множитель = 9,87×108, энергия активации = 3,1×107 Дж/кмоль, показатель степени для Н2 и О2 составляет 1, для Н2О он составляет 0. Состав при смешивании: А=4, В=0,5. Дисперсная фаза (частица):
Частицы/характеристики: Плотность частиц, ρр, и ср частиц рассчитывают с использованием закона смешения для всех компонентов. Энергии активации, если они не приведены в базе данных, можно получить путем аппроксимации данных, полученных в экспериментах, проводимых с помощью дифференциальной сканирующей калориметрии (ДСК). Теплоту реакции Нреакц. можно рассчитать с использованием значении энтальпии в стандартном состоянии и ср.
Движение частицы:
Траектории частицы рассчитывают с использованием подхода Эйлера-Лагранжа, так называемой модели дискретной фазы (МДФ) (например, с помощью программного обеспечения Ansys Fluent), которую используют в качестве решающей программы для моделирования. Жидкую фазу рассматривают, как непрерывную, и решают уравнения Навье-Стокса, тогда как дисперсную фазу моделируют путем отслеживания большого количества частиц на рассчитанном поле потока. Между дисперсной фазой и жидкой фазой происходит обмен импульсом, массой и энергией. Поскольку дисперсная фаза в этом случае представляет собой обладающую меньшим объемом фракцию, взаимодействием между частицами можно пренебречь.
Траекторию находящейся в дискретной фазе частицы предварительно рассчитывают путем интегрирования уравнения для равновесия сил, действующих на частицу, которое записано в системе координат Лагранжа. В этом уравнении для равновесия сил уравнивают инерцию частицы и силы, действующие на частицу, и его можно записать следующим образом:

где 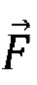 обозначает дополнительное ускорение (сила/единица массы частицы),
обозначает дополнительное ускорение (сила/единица массы частицы), 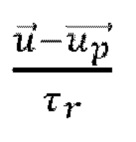 обозначает силу сопротивления в пересчете на единицу массы частицы и:
обозначает силу сопротивления в пересчете на единицу массы частицы и:

τr обозначает время релаксации частицы, 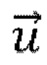 обозначает скорость жидкой фазы,
обозначает скорость жидкой фазы, 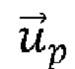 обозначает скорость частицы, μ обозначает вязкость жидкости и dp обозначает диаметр частицы, при этом число Рейнольдса рассчитывают следующим образом:
обозначает скорость частицы, μ обозначает вязкость жидкости и dp обозначает диаметр частицы, при этом число Рейнольдса рассчитывают следующим образом:

Нагревание или охлаждение частиц с использованием инертного газа: Нагревание или охлаждение с использованием инертного газа проводят в том случае, когда температура капельки ниже температуры испарения, Тисп., или растворитель и органический остаток, содержащиеся в капельке, израсходованы, т.е. капельки становятся сухими частицами. В этом случае температуру частицы рассчитывают с использованием следующего уравнения:
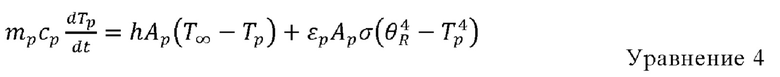
mp = масса частицы (кг),
ср = теплоемкость частицы (Дж/(кг⋅К)),
Ар = площадь поверхности частицы (м3),
T∞ = локальная температура в непрерывной фазе (К),
h = коэффициент конвективной теплопередачи (Вт/(м2⋅К)),
εр = излучательная способность частиц (безразмерная величина),
σ = постоянная Стефана-Больцмана (5,67×10-8 Вт/(м2⋅К4)),
θR = температура излучения,  .
.
При моделировании излучение частицы не учитывают вследствие небольшого объема фракции:

Коэффициент теплопередачи h рассчитывают с использованием формулы Ранца и Маршалла:

dp = диаметр частицы,
k∞ = удельная теплопроводность непрерывной фазы,
Pr = число Прандтля для непрерывной фазы, cpμ/k∞.
Перенос тепла и массы во время испарения:
После того, как в капельке обеспечена температура испарения, Тисп., начинается испарение капельки и оно продолжается до обеспечения в капельке температуры кипения или до израсходования содержащегося в капельке растворителя.
Температура капельки меняется в соответствии с тепловым равновесием и устанавливается взаимосвязь между изменением физической теплоты в капельке и переносом конвективной и скрытой теплоты между капелькой и непрерывной фазой:
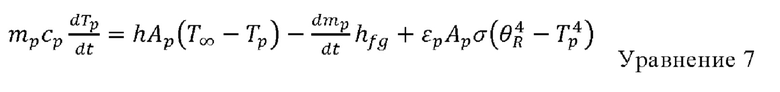
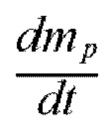 = скорость испарения (кг/с),
= скорость испарения (кг/с),
hƒg = скрытая теплота воды (Дж/кг).
Предполагают, что скорость испарения регулируется градиентом диффузии, причем поток пара капельки в газовую фазу связан с разностью концентраций пара на поверхности капельки и в объеме газа:

Ni = мольный поток пара (кмоль/(м2⋅К)),
kc = коэффициент переноса массы (м/с),
Ci, s = концентрация пара на поверхности (кмоль/м3),
Ci, ∞ = концентрация пара в объема газа (кмоль/м3).
Концентрацию пара на поверхности капельки рассчитывают, предполагая, что парциальное давление пара на границе раздела равно давлению насыщенного пара, Рнас., при температуре капельки, Тр.

R обозначает универсальную газовую постоянную.
Концентрацию пара в объеме газа определяют путем решения следующего уравнения переноса для частиц i:

Xi = мольная доля частиц i в объеме,
р = локальное абсолютное давление (Па),
T∞ = локальная объемная температура в газе (К).
Коэффициент переноса массы, kc, рассчитывают с использованием формулы для числа Шервуда:

Di, m = коэффициент диффузии пара в объеме (м2/с),
Sc = число Шмидта, 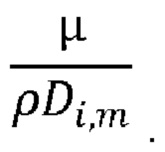
Скорость испарения 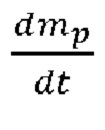 рассчитывают следующим образом:
рассчитывают следующим образом:

Mw, i = молекулярная масса пара (кг/кмоль),
Ар = площадь поверхности капельки (м3).
Перенос тепла и массы во время кипения:
После того, как в капельке обеспечена температура кипения, Ткип., используют уравнение для скорости кипения, принимая во внимание тот факт, что температура капельки во время кипения остается постоянной, и при рассмотрении фазового превращения из жидкой фазы в газовую фазу:


k∞ = теплопроводность газа, Вт/(м⋅К),
ср = теплоемкость газа, Дж/(кг⋅К),
hƒg = скрытая теплота, Дж/кг,
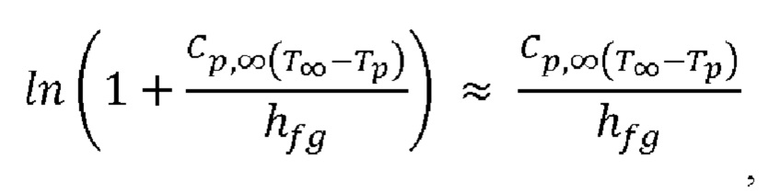
если значение 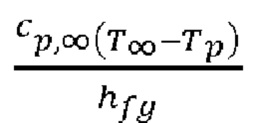 является малым.
является малым.
Перенос тепла и массы во время реакции/горения:
После выпаривания всего количества воды инициируется реакция горения и она протекает до израсходования всех органических остатков или до вылета частиц из использующейся для расчета области через выходное отверстие.
При реакции на поверхности расходуются окислительные частицы, находящиеся в газовой фазе; т.е. это обеспечивает появление (отрицательного) члена при решении уравнения переноса для этих частиц. Аналогичным образом, реакция на поверхности является источником появления частиц в газовой фазе: продукт гетерогенной реакции на поверхности появляется в газовой фазе в виде определенных химических частиц. При реакции на поверхности также расходуется или вырабатывается энергия в количестве, обусловленном определенной теплотой реакции.
Тепловое равновесие в частице во время реакции на поверхности описывается следующим образом:
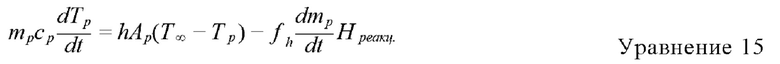
Нреакц. = тепло, выделяющееся при реакции на поверхности (Дж/кг),
h является таким, как определено для случая нагревания с использованием инертного газа,
ƒh = коэффициент.
Только часть (l-ƒh) энергии, полученной при реакции на поверхности, является источником тепла в уравнении сохранения энергии в газовой фазе, часть этого тепла, ƒh, непосредственно поглощает частица. В случае сжигания угля рекомендуют устанавливать ƒh, равным 0,3, если продуктом сгорания угля является СО2, такое же значение можно использовать в моделировании, предлагаемом в настоящем изобретении.
Формование продукта
Способ, предлагаемый в настоящем изобретении, обеспечивает получение пористых кристаллических силикатов металлов в форме порошка. Для использования в качестве катализаторов этому порошку можно придать подходящую форму, например, получить микрогранулы, сферы, таблетки, сплошные цилиндры, полые цилиндры или сотовидные структуры, по известным методикам формования порошкообразных катализаторов, например, путем прессования, гранулирования, распылительной сушки, распылительного гранулирования или экструзии.
Таким образом, другим объектом настоящего изобретения является способ, предлагаемый в настоящем изобретении, в котором после стадии (b) проводят стадию формования (с), включающую следующие подстадии:
(1) добавление воды для получения водной суспензии порошкообразного пористого кристаллического силиката металла,
(2) смешивание суспензии, полученной на подстадии (1), с гранулирующими средствами,
(3) прессование, гранулирование, распылительная сушка, распылительное гранулирование и/или экструзия продукта, полученного на подстадии (2), для получения пористого кристаллического силиката металла в форме микрогранул, сфер, таблеток, сплошных цилиндров, полых цилиндров или сотовидных структур.
Предпочтительно, если размер таких сформованных изделий находится в диапазоне от 0,1 до 10 см.
Для проведения смешивания и формования можно использовать все известные устройства и методики для смешивания и формования и можно использовать все стандартные гранулирующие средства. Известные устройства для формования такого типа описаны, например, в публикации Ullmann's  der Technischen Chemie [Ullmann's Encyclopedia of Industrial Chemistry], 4th edition, volume 2, page 295 ff., 1972. Предпочтение отдается использованию одно- и двухшнековых экструдеров или экструзионного пресса. Можно изготовить многочисленные известные геометрические тела, например, сплошные цилиндры, полые цилиндры, звездообразные тела и т.п. Однако также можно изготовить сотовидные структуры.
der Technischen Chemie [Ullmann's Encyclopedia of Industrial Chemistry], 4th edition, volume 2, page 295 ff., 1972. Предпочтение отдается использованию одно- и двухшнековых экструдеров или экструзионного пресса. Можно изготовить многочисленные известные геометрические тела, например, сплошные цилиндры, полые цилиндры, звездообразные тела и т.п. Однако также можно изготовить сотовидные структуры.
В предпочтительном варианте осуществления способ, предлагаемый в настоящем изобретении, применяют для получения содержащих титан цеолитов типа силикалита титана-1 и силикалита титана-2, которые можно использовать например, в качестве катализаторов в реакциях окисления с использованием пероксида водорода. Точнее, такие содержащие титан цеолиты можно использовать в качестве катализаторов для эпоксидирования олефинов с использованием водного раствора пероксида водорода.
ПРИМЕРЫ
Пример 1: Получение неочищенной суспензии с помощью гидротермального синтеза
Синтез цеолита - силикалита титана-1 (СТ-1; структура типа MFI) проводили в реакторе высокого давления объемом 3 м3 по соответствующей методике, описанной в примере 1, приведенном в ЕР 0814058 В1. Использующимся источником кремния являлся аморфный обладающий высокой чистотой диоксид кремния (изготовитель: Evonik Resource Efficiency GmbH) и использующимся источником титана являлся водный раствор титан-гидроксид тетрапропиламмония (раствор Ti-ТПА), обладающий содержанием ТЮ2, составляющим 19,0 мас.%. Раствор Ti-ТПА получали следующим образом:
90,1 кг Деионизированной воды, 167,3 кг 40% водного раствора гидроксида тетрапропиламмония (изготовитель: Sachem) и 141,6 кг тетраэтилортотитаната (изготовитель: Connect Chemicals GmbH) перемешивали в закрытом сосуде при 40°С в течение 1 ч. Экзотермичность реакции приводила к повышению температуры примерно на 25°С. Затем путем отгонки проводили удаление этанола, образовавшегося при 80°С, при скорости отгонки, равной 30 л/ч. Целевой объем полученного раствора Ti-ТПА являлся таким, что содержание TiO2 составляло 19,0 мас.%. После охлаждения раствор Ti-ТПА использовали для синтеза СТ-1.
В реактор высокого давления сначала помещали: 500 кг обладающего высокой чистотой диоксида кремния (Evonik Industries), 382 кг 40% водного раствора гидроксида тетрапропиламмония (изготовитель: Sachem), 193 кг раствора Ti-ТПА, 10 кг затравочных кристаллов силикалита-1 и 1800 кг деионизированной воды. Смесь перемешивали в закрытом реакторе высокого давления при скорости перемешивающего устройства, равной 50 об/мин, при 170°С в течение 3 ч. Продолжительность нагревания до 170°С составляла 180 мин; после охлаждения в течение 150 мин синтез прекращали. Перемешивание при скорости, равной 50 об/мин, продолжали от начала до завершения синтеза.
Затравочные кристаллы силикалита-1 получали путем проводимого в реакторе высокого давления гидротермального синтеза с использованием 500 кг обладающего высокой чистотой диоксида кремния (Evonik Resource Efficiency GmbH), 400 кг 40% водного раствора гидроксида тетрапропиламмония (изготовитель: Sachem) и 1800 кг деионизированной воды. Смесь перемешивали в закрытом реакторе высокого давления при скорости, равной 50 об/мин, при 160°С в течение 3 ч. Продолжительность нагревания до 160°С составляла 180 мин; после охлаждения в течение 150 мин синтез прекращали. Перемешивание при скорости, равной 50 об/мин, продолжали от начала до завершения синтеза.
Пример 2: Обычная обработка, проводимая после гидротермального синтеза
К неочищенной суспензии, описанной в примере 1, добавляли уксусную кислоту (60 мас.%) до обеспечения рН=7 и полученный осадок отфильтровывали с помощью фильтр-пресса и промывали дистиллированной водой. Полученные твердые вещества сушили путем распылительной сушки при температуре на входе, равной 420°С, и при скорости распылителя, равной 1700 мин-1 (температура на выходе равна 110°С). Затем частично высушенный порошок прокаливали во вращающейся трубчатой печи при температуре, не превышающей 650°С, в течение 2 ч. Полученный таким образом продукт обладал БЭТ-поверхностью, равной 470 м2/г, и потерями при прокаливании (определяли при 550°С), составляющими 0,65%. Анализ с помощью РГГ (фиг. 1) показал, что полученный продукт обладал кристаллической структурой, соответствующей структуре силикалита титана-1 (СТ-1) (идентификационный номер ICDD (Международный центр дифракционных данных): 01-089-8099). С помощью анализа объема пор, проводимого с использованием азота по методике БДХ, получали объем пор, равный 0,23 мл/г.
Пример 3 (отрицательный пример): Распылительное прокаливание, проводимое после гидротермального синтеза (Тэфф.=650°С)
Неочищенную суспензию (15 кг/ч), полученную в примере 1, распыляли с помощью экспериментальной установки с использованием 18 м3/ч азота для распыления через двухфазное сопло, обладающее внутренним диаметром, равным 2 мм, и щелью размером 1 мм. Пламя водород/воздух содержало 8 м3/ч водорода и 45 м3/ч первичного воздуха. Количество азота составляло 18 м3/ч и количество вторичного воздуха составляло 25 м3 1 ч. Температуру, определенную в положении, находящемся на 1,5 м ниже положения горения, устанавливали равной 400°С путем незначительного изменения потока водорода.
Адиабатическая температура сгорания в реакторе равнялась примерно 544°С. Средняя продолжительность пребывания частицы в реакторе составляла 1,35 с. Отходящие газы, содержащие прокаленный, направляли в зону охлаждения (температура охлаждающей среды: 25°С), обладающую диаметром, равным 100 мм, и длиной, равной 6 м, и затем собирали с помощью фильтрационных свечей при максимальной температуре, равной 250°С. Путем последующей очистки фильтрационных свечей можно было собрать готовый прокаленный продукт (4,4 кг/ч). Полученный таким образом продукт обладал потерями при прокаливании (определяли при 550°С), составляющими 8,6%, это явно показывало, что он являлся неподходящим для последующей обработки (формования), поскольку слишком большое количество органического остатка оставалось на поверхности продукта (потери при прокаливании явно превышали предельное значение, равное 5%). Анализ с помощью РГГ (фиг. 2) показал, что продукт обладал кристаллической структурой, соответствующей структуре СТ-1 (идентификационный номер ICDD: 01-089-8099).
Подробное описание моделирования:
Исходные параметры для газовой фазы:
Распыляющий воздух: 18 нм3/ч,
Первичный воздух: 45 нм3/ч,
Вторичный воздух: 25 нм3/ч,
Н2: 8 нм3/ч,
Модель турбулентности: стандартная модель k-ε.
Модель излучения в газовой фазе: модель дискретных ординат с угловой дискретностью: разделения тета: 4; разделения фи: 4; тета-пиксели: 1; фи-пиксели: 1;
Граничные условия для стенок: непрозрачные, внутренняя излучающая способность: 1;
Коэффициент теплообмена с окружающей средой (только для наружной стенки): 5 Вт/м2/K;
Температура окружающей среды: 300 K.
Модель сгорания: конечная скорость/турбулентное рассеивание.
Н2 + 0,5 O2 → H2O
Кинетика протекания реакции:
Константа скорости Аррениуса: предэкспоненциальный множитель = 9,87×108, энергия активации = 3,1×107 Дж/кмоль, показатель степени для Н2 и O2 составляет 1, для H2O он составляет 0.
Состав при смешивании: А=4, В=0,5.
Характеристики/модель характеристик:
Газ считали идеальным газом. Теплопроводность, вязкость и теплоемкость, ср, смеси газов рассчитывали с использованием закона смешения при известном массовом составе. Использовали характеристики чистых компонентов, Н2, H2O (пар), CO2, O2, N2, приведенные в базах данных для материалов Ansys Fluent. Коэффициент диффузии массы для каждого компонента, содержащегося в газовой фазе, рассчитывали на основании кинетической теории газов, все необходимые параметры приведены в базе данных Ansys Fluent.
Исходные параметры для фазы частицы:
Частицы/характеристики: Плотность частиц, рр, и ср частиц рассчитывали с использованием закона смешения для всех компонентов.
Частицы 1: H2O, исходная массовая доля: 60,3% ρ=998 кг/м2, ср=4182 Дж/(кг⋅К), скрытая теплота воды = 2263037 Дж/кг, температура испарения, Тисп=284 K, температура кипения: Ткип=373 K, давление насыщенного пара Рнас.(Тр): кусочно-линейное приближение с использованием 32 точек в диапазоне Т=274-647 K.
Частицы 2: ТПА-ОН, исходная массовая доля: 9,7%,
ρ=1000 кг/м2, ср=3600 Дж/(кг⋅К),
Энтальпия в стандартном состоянии, Н0=-2,12×108 Дж/кмоль (для расчета теплоты реакции, Нреакц).
Частицы 3: Силикат, исходная массовая доля: 30%,
ρ=2660 кг/м2, ср=1052 Дж/(кг⋅К), считали инертным,
Массовый поток суспензии 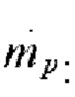 исходный: 15 кг/ч,
исходный: 15 кг/ч,
Диаметр (dp): исходный диаметр частицы: 22 мкм,
Группы частиц: 100 (массовый поток каждой группы, 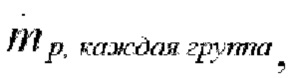 равен
равен 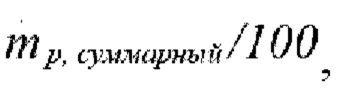 существенная разница при использовании 100 и 1000 групп отсутствовала, поэтому использовали 100 групп),
существенная разница при использовании 100 и 1000 групп отсутствовала, поэтому использовали 100 групп),
Количество экспериментов: 10 (случайное отслеживание с использованием дискретной модели случайного блуждания вследствие влияния турбулентности, отслеживание 10 частиц в каждой группе, моделировали траектории всего 1000 частиц с массовым потоком 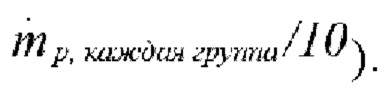 Константа шкалы времени: 0,15 (использовали для случайного отслеживания).
Константа шкалы времени: 0,15 (использовали для случайного отслеживания).
Кинетика протекания реакции:
ТПА-ОН + 18,75 O2 14,5 H2O + CO2 + N2
Константа скорости Аррениуса: предэкспоненциальный множитель = 0,2,
Энергия активации: 8×107 Дж/кмоль
Показатель степени для O2: 1.
Движение частицы рассчитывали с использованием уравнений 1-3.
Температуры частиц, которые нагревали с использованием инертного газа, которые испарялись и горели, рассчитывали с использованием уравнений 4-15.
На фиг. 5 представлена зависимость температуры частицы от продолжительности пребывания для 3 из 1000 типичных рассчитанных траекторий частиц (100 групп×10 экспериментов в каждой группе), полученных в примере 3.
Пример 4: Распылительное прокаливание, проводимое после гидротермального синтеза (Тэфф.=1000°С)
Неочищенную суспензию (25 кг/ч), полученную в примере 1, распыляли с помощью экспериментальной установки с использованием 18 м3/ч воздуха для распыления через двухфазное сопло, обладающее внутренним диаметром, равным 2 мм, и щелью размером 1 мм. Пламя водород/воздух содержало 8,5 м3/ч водорода и 27 м3/ч первичного воздуха. Количество азота составляло 18 м3/ч и количество вторичного воздуха составляло 25 м3/ч. Температуру, определенную в положении, находящемся на 1,5 м ниже положения горения, устанавливали равной 700°С путем незначительного изменения потока водорода. Адиабатическая температура сгорания в реакторе равнялась примерно 750°С.
Средняя продолжительность пребывания частицы в реакторе составляла примерно 1,1 с. Отходящие газы, содержащие прокаленный, направляли в зону охлаждения (температура охлаждающей среды: 25°С), обладающую диаметром, равным 100 мм, и длиной, равной 6 м, и затем собирали с помощью фильтрационных свечей при максимальной температуре, равной 250°С. Путем последующей очистки фильтрационных свечей можно было собрать готовый прокаленный продукт (7,3 кг/ч). Полученный таким образом продукт обладал БЭТ-поверхностью, составляющей 489 м2/г, и потерями при прокаливании (определяли при 550°С), составляющими 0,3%. Анализ с помощью РГГ (фиг. 3) показал, что продукт обладал кристаллической структурой, соответствующей структуре СТ-1 (идентификационный номер ICDD: 01-089-8099).
Моделирование эффективной температуры частицы проводили так же, как в примере 3.
На фиг. 6 представлена зависимость температуры частицы от продолжительности пребывания для 3 из 1000 типичных рассчитанных траекторий частиц (100 групп×10 экспериментов в каждой группе), полученных в примере 4.
Пример 5: (отрицательный пример): Распылительное прокаливание, проводимое после гидротермального синтеза (Тэфф.=1300°С)
Неочищенную суспензию (15 кг/ч), описанную в примере 1, распыляли с помощью экспериментальной установки с использованием 18 м3/ч воздуха для распыления через двухфазное сопло, обладающее внутренним диаметром, равным 2 мм, и щелью размером 1 мм. Пламя водород/воздух содержало 17,4 м3/ч водорода и 40 м3/ч первичного воздуха. Количество азота составляло 18 м3/ч и количество вторичного воздуха составляло 25 м3/ч. Температуру, определенную в положении, находящемся на 1,5 м ниже положения горения, устанавливали равной 950°С путем незначительного изменения потока водорода.
Адиабатическая температура сгорания в реакторе равнялась примерно 980°С.
Средняя продолжительность пребывания частицы в реакторе составляла примерно 0,9 с. Отходящие газы, содержащие прокаленный, направляли в зону охлаждения (температура охлаждающей среды: 25°С), обладающую диаметром, равным 100 мм, и длиной, равной 6 м, и затем собирали с помощью фильтрационных свечей при максимальной температуре, равной 250°С. Путем последующей очистки фильтрационных свечей можно было собрать готовый прокаленный продукт (4,4 кг/ч). Полученный таким образом продукт обладал БЭТ-поверхностью, составляющей 429 м2/г, и потерями при прокаливании (определяли при 550°С), составляющими 0,6%. Анализ с помощью РГГ (фиг. 4) показал некоторые незначительные признаки повреждения структуры СТ-1 (идентификационный номер ICDD: 01-089-8099). Значение БЭТ-поверхности и результаты, полученные с помощью РГГ, показали, что структура повреждена и в результате этого площадь поверхности уменьшена примерно на 15% (по сравнению с образцом примера 4) и поэтому полученный продукт являлся неподходящим для последующей обработки, т.е. формования, и использования в контрольных реакциях ПВ-ПО (проводимое с использованием пероксида водорода превращение в пропиленоксид).
Моделирование эффективной температуры частицы проводили так же, как в примере 3.
На фиг. 7 представлена зависимость температуры частицы от продолжительности пребывания для 3 из 1000 типичных рассчитанных траекторий частиц (100 групп×10 экспериментов в каждой группе), полученных в примере 5.
Пример 6: Формование порошкообразного цеолита, полученного в примере 2 (обычная обработка)
В смесителе Eirich порошок, полученный в примере 2 (1200 г), смешивали с 75 г метилгидроксиэтилцеллюлозы (Tylose МН1000), 75 г Licowax С, 1000 г раствора золя диоксида кремния (Koestrosol 0830 AS) 350 г деионизированной воды. Полученную массу экструдировали с помощью экструдера (НВ-Feinmechanik LTW 63) через пластину с отверстиями диаметром 3,2 мм. Затем экструдаты сушили в сушильном шкафу при 80°С в течение 1 ч и прокаливали в муфельной печи при 570°С в течение 12 ч.
Пример 7: Формование порошкообразного цеолита, полученного в примере 4 (обработка с помощью пламенного распылительного пиролиза)
В смесителе Eirich порошок, полученный в примере 4 (1200 г) смешивали с 75 г метилгидроксиэтилцеллюлозы (Tylose МН1000), 75 г Licowax С, 1000 г раствора золя диоксида кремния (Koestrosol 0830 AS) и 350 г деионизированной воды. Полученную массу экструдировали с помощью экструдера (НВ-Feinmechanik LTW 63) через пластину с отверстиями диаметром 3,2 мм. Затем экструдаты сушили в сушильном шкафу при 80°С в течение 1 ч и прокаливали в муфельной печи при 570°С в течение 12 ч.
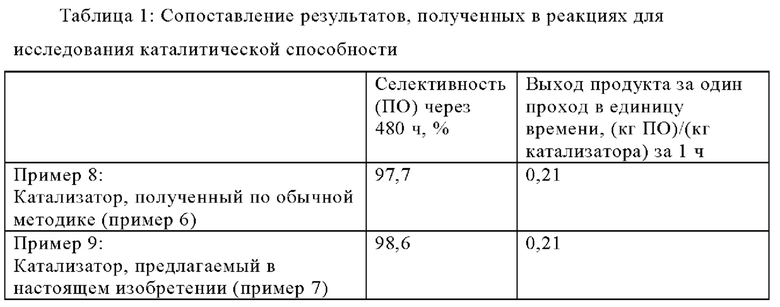
Пример 8: Исследование каталитической способности катализатора, полученного в сравнительном примере 6 (обычная обработка)
Эпоксидирование пропена проводили с использованием двух реакторов с неподвижным слоем, каждый содержал 9 г катализатора, полученного в примере 6, находящегося в форме экструдатов. Реакторы были расположены последовательно (реактор 1 → реактор 2) и работали в режиме восходящего потока. Первый поток сырья, содержащий метанол, пероксид водорода (60 мас.%) и воду, при полной скорости потока, равной 20 г/ч, и второй поток сырья, содержащий пропилен при скорости потока, равной 20 г/ч, подавали в первый реактор. Давление при проведении реакции поддерживали равным 25 бар с помощью обратного клапана давления, расположенного после второго реактора. Давление в реакционной смеси, выходящей из второго реактора с неподвижным слоем, сбрасывали до давления окружающей среды. Полученную газовую фазу анализировали для определения количеств пропилена, пропиленоксида и кислорода и полученную жидкую фазу анализировали для определения количеств пропиленоксида и пероксида водорода. Начальная селективность образования пропиленоксида после проведения реакции в течение 23 ч составляла 91,1%. Через 480 ч селективность образования пропиленоксида составляла 97,7%.
Пример 9: Исследование каталитической способности катализатора, полученного в примере 7 (обработка с помощью пламенного распылительного пиролиза)
Эпоксидирование пропена проводили таким же образом, как в примере 8, но использовали катализатор, полученный в примере 7.
Начальная селективность образования пропиленоксида после проведения реакции в течение 25 ч составляла 93,5%. Через 480 ч селективность образования пропиленоксида составляла 98,6%.
С помощью примеров 3-5 и при их сопоставлении с примером 2, показано, что способ, предлагаемый в настоящем изобретении включает намного меньшее количество стадий способа, чем обычный способ. Кроме того, в способе, раскрытом в настоящем изобретении, устранены затруднения, связанные с удалением сточных вод, обычно образующихся во время фильтрования и очистки продукта после проведения гидротермального синтеза. Неожиданно оказалось, что после проведения пламенного распылительного пиролиза полученные силикалиты титана обладают пористостью, сравнимой с пористостью силикалита титана, полученного по обычной методике.
Как видно из примеров 8 и 9 (результаты приведены в таблице 1), катализатор, полученный по обычной методике (пример 6), а также катализатор, полученный в соответствии с настоящим изобретением (пример 7), оба обладают высокой активностью и обеспечивают высокую селективность при проведении реакции эпоксидирования пропилена с получением пропиленоксида (ПО) после проведения реакции в течение 480 ч. Однако катализатор, полученный в соответствии с настоящим изобретением, обеспечивает селективность образования пропиленоксида, которая даже на 0,9% выше, чем обеспечиваемая обычным катализатором, и в то же самое время обеспечивает сравнимые выходы продукта за один проход в единицу времени. Таким образом, использование катализаторов - силикалитов титана-1, полученных в соответствии с настоящим изобретением, обеспечивает возможность существенного увеличения выхода продукта - пропиленоксида в единицу времени и в пересчете на объем реактора.
Пример 10
Описанные ниже измененные по сравнению с описанной в примере 1 методики синтеза проводили в лабораторном автоклаве объемом 1 л и затем проводили обработку в соответствии с методикой, описанной в примере 4, при Тэфф.=1000°С для подтверждения того, что проводимый при подходящих условиях распылительный пиролиз можно использовать для разных продуктов синтеза без разрушения кристаллической структуры.
Общее описание:
Цеолит получали по следующей методике: В типичном эксперименте источник металла, источник кремния, вспомогательный компонент, воду и необязательно затравочные кристаллы-золь, помещали в изготовленный из нержавеющей стали автоклав (Btichi, V (объем) = 1,1 см3, D (диаметр) = 8,4 см, Н (высота) = 20,3 см, электрическое нагревание) и осторожно перемешивали.
Альтернативно, источник кремния объединяли с источником металла (пропитывали) до проведения синтеза путем обработки ксеро- или гидрогеля диоксида кремния жидким содержащим титан раствором, таким как раствор титанилсульфата, оксалата титана, лактата титана (или другими содержащими титан растворами) и получали пропитанный металлом диоксид кремния, также называющийся ксерогелем диоксида кремния-диоксида титана или гидрогелем диоксида кремния-диоксида титана. В приведенных в настоящем изобретении примерах для пропитки гидрогеля диоксида кремния использовали титанилсульфат (необязательно с проведением последующей стадии сушки для уменьшения содержания воды). После объединения гидрогель необязательно можно высушить с получением ксерогеля для изменения содержания воды в материале. Ксеро- или гидрогель диоксида кремния-диоксида титана добавляли в автоклав вместе с другими компонентами.
Для синтеза силикалита титана-1 (структура типа MFI) необязательно можно использовать затравочные кристаллы силикалита-1 или силикалита титана-1 (или их смесь). Для синтеза силикалита титана-2 (структура типа MEL) необязательно можно использовать затравочные кристаллы силикалита-2 или силикалита титана-2 (или их смесь).
Автоклав герметизировали, затем смесь подвергали гидротермальной обработке (скорость нагревания: 1 К/мин) и перемешивали при 250-450 об/мин.
Затем автоклав охлаждали до комнатной температуры при скорости охлаждения, равной примерно 1 К/мин, и получали содержащую цеолит водную суспензию.
Неочищенную суспензию, полученную после гидротермального синтеза, обрабатывали и анализировали в соответствии с методикой, описанной в примере 4, при Тэфф.=1000°С.
a) Для получения силикалита титана-1 (структура типа MFI) синтез проводили путем добавления в автоклав 120 г тонкодисперсного порошкообразного диоксида кремния, 50 г оксалата титана, 20 г затравочных кристаллов силикалита-1 в водном растворе, 100 г гидроксида тетрапропиламмония (40% водный раствор) и 200 г воды. В соответствии с приведенным выше общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. Содержание воды в источнике кремния составляло <5 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
b) Для получения силикалита титана-1 (структура типа MFI) синтез проводили путем добавления в автоклав 300 г тетраэтилортосиликата, >99%, 8 г тетраэтилортотитаната (35%TiO2), 130 г гидроксида тетрапропиламмония (40% водный раствор) и 250 г воды. В соответствии с приведенным выше общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
c) Для получения силикалита титана-1 (структура типа MFI) синтез проводили путем добавления в автоклав 250 г гидрогеля диоксида кремния-диоксида титана (2,5 мас.%TiO2, содержание воды: 60-80 мас.%), 115 г гидроксида тетрапропиламмония (40% водный раствор), 20 г затравочных кристаллов силикалита титана-1 и 250 г воды. Как описано выше, до проведения синтеза источник кремния (гидрогель диоксида кремния) необходимо объединить с титанилсульфатом (50% в водном растворе) с получением гидрогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим 60-80%. В соответствии с общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
d) Синтез проводили так же, как в примере с), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим 50-70 мас.%, и 300 г воды. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
e) Синтез проводили так же, как в примере с), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим 50-60 мас.%, и 300 г воды. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
f) Синтез проводили так же, как в примере с), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим 30-50 мас.%, и 300 г воды. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
g) Синтез проводили так же, как в примере с), но с использованием ксерогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим 10-30 мас.%, и 350 г воды. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
h) Синтез проводили так же, как в примере с), но с использованием ксерогеля диоксида кремния-диоксида титана, обладающего содержанием воды, составляющим <10 мас.%, и 450 г воды. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
i) Синтез проводили так же, как в примере d), но автоклав нагревали до 180°С и перемешивание продолжали в течение 60 мин. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
j) Синтез проводили так же, как в примере е), но автоклав нагревали до 180°С и перемешивание продолжали в течение 60 мин. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
k) Синтез проводили так же, как в примере е), но с использованием 100 г гидроксида тетрапропиламмония (35% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
l) Синтез проводили так же, как в примере е), но с использованием 150 г гидроксида тетрапропиламмония (20% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
m) Синтез проводили так же, как в примере е), но с использованием 200 г 1,6-диаминогексана и без использования гидроксида тетрапропиламмония (40% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
n) Синтез проводили так же, как в примере m), но с использованием 100 г 1,6-диаминогексана и 50 г гидроксида тетрапропиламмония (40% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
о) Синтез проводили так же, как в примере m), но с использованием 200 г 1,2-пентадиола вместо 1,6-диаминогексана. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
р) Синтез проводили так же, как в примере n), но с использованием 100 г 1,2-пентадиола вместо 1,6-диаминогексана. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
q) Синтез проводили так же, как в примере n), но с использованием 50 г 1,2-пентадиола, 50 г 1,6-диаминогексана и 50 г гидроксида тетрапропиламмония (40% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
r) Синтез проводили так же, как в примере а), но с использованием 200 г 1,6-диаминогексана и без использования гидроксида тетрапропиламмония (40% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
s) Синтез проводили так же, как в примере а), но с использованием 200 г 1,2-пентадиола и без использования гидроксида тетрапропиламмония (40% водный раствор). На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
t) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 3,7 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
u) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 3,5 мас.% TiO2. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
v) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 3,1 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
w) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 2,8 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
х) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 2,3 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
у) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 1,8 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
z) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 1,5 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
аа) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 1,0 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
bb) Синтез проводили так же, как в примере е), но с использованием гидрогеля диоксида кремния-диоксида титана, обладающего содержанием TiO2, составляющим 0,5 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-1.
сс) Для получения силикалита титана-2 (структура типа MEL) синтез проводили путем добавления в автоклав 250 г гидрогеля диоксида кремния-диоксида титана (2,5 мас.% TiO2), 115 г гидроксида тетрапропиламмония (40% водный раствор), 20 г затравочных кристаллов силикалита-2 и 300 г воды. В соответствии с приведенным выше общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. Содержание воды в предшественнике диоксида кремния-диоксида титана составляло 50-60 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита титана-2 (как указано в базе данных ICDD).
dd) Для получения силикалита железа-1 (структура типа MFI) синтез проводили путем добавления в автоклав 250 г ксерогеля диоксида кремния, 115 г гидроксида тетрапропиламмония (40% водный раствор), 20 г затравочных кристаллов силикалита-1, 30 г цитрата железа-аммония и 350 г воды. В соответствии с приведенным выше общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. Содержание воды в предшественнике диоксида кремния составляло 20-30 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков силикалита железа-1 (как указано в базе данных ICDD).
ее) Для получения силикалита алюминия-железа-1, также называющегося железо-ZSM-S, (структура типа MFI) синтез проводили путем добавления в автоклав 250 г ксерогеля диоксида кремния, 115 г гидроксида тетрапропиламмония (40% водный раствор), 20 г затравочных кристаллов силикалита-1, 30 г цитрата железа-аммония, 50 г нитрата алюминия и 350 г воды. В соответствии с приведенным выше общим описанием автоклав нагревали до 160°С и смесь перемешивали в течение 180 мин, затем охлаждали. Содержание воды в предшественнике диоксида кремния составляло 20-30 мас.%. На рентгенограмме видны пики, находящиеся в положениях, соответствующих положениям пиков железо-ZSM-S (как указано в базе данных ICDD).
Кристаллографические данные для силикалита титана-1 (источник: база данных ICDD):
Идентификационный номер: 01-089-8099,
Название соединения: диоксид кремния-титана,
Код ICSD (база данных "Структуры неорганических кристаллов"): 88413,
Публикация: Lamberti, С., Bordiga, S., Zecchina, A., Carati, A., Fitch, A.N., Artioli, G., Petrini, G., Salvalaggio, M., Marra, G.L., J. Catal, 183, 222, (1999).
Перечень отражений:

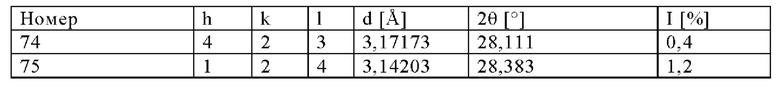
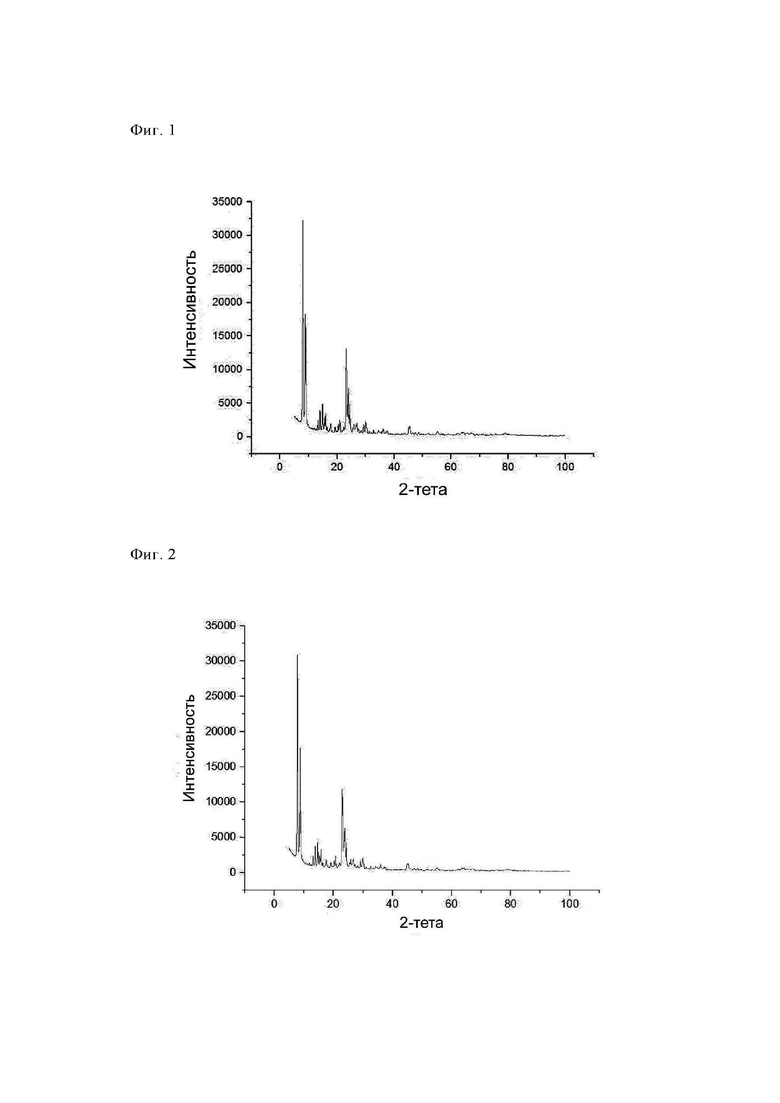
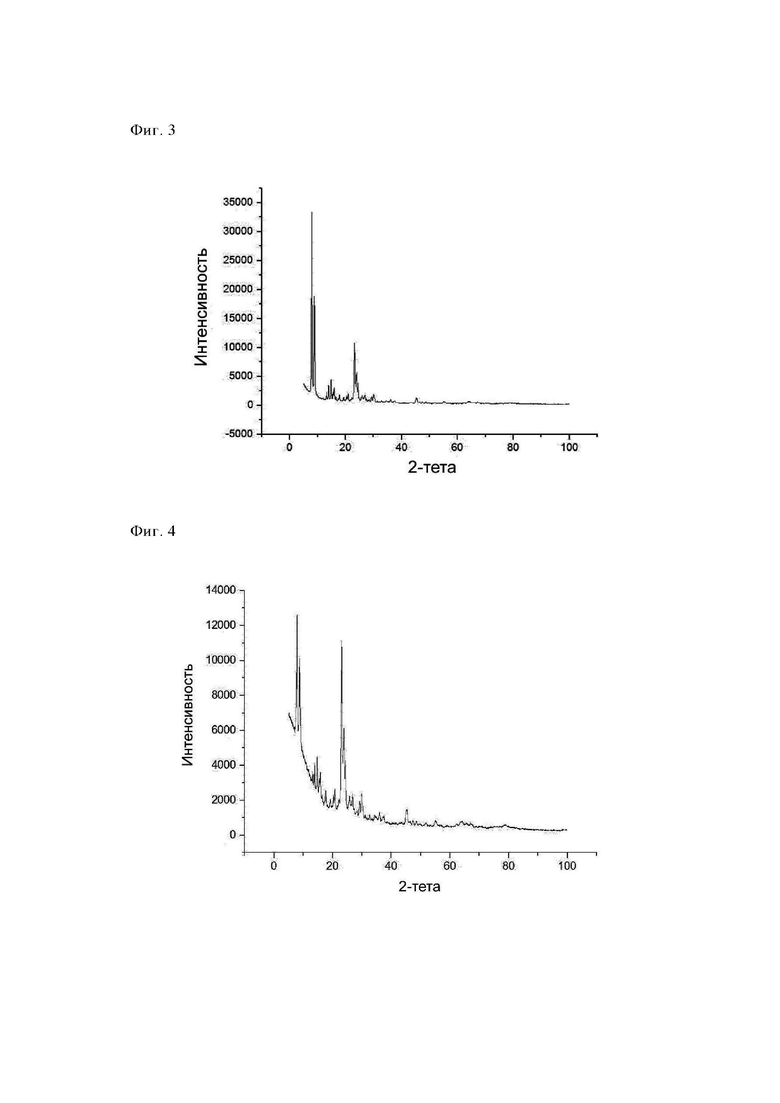
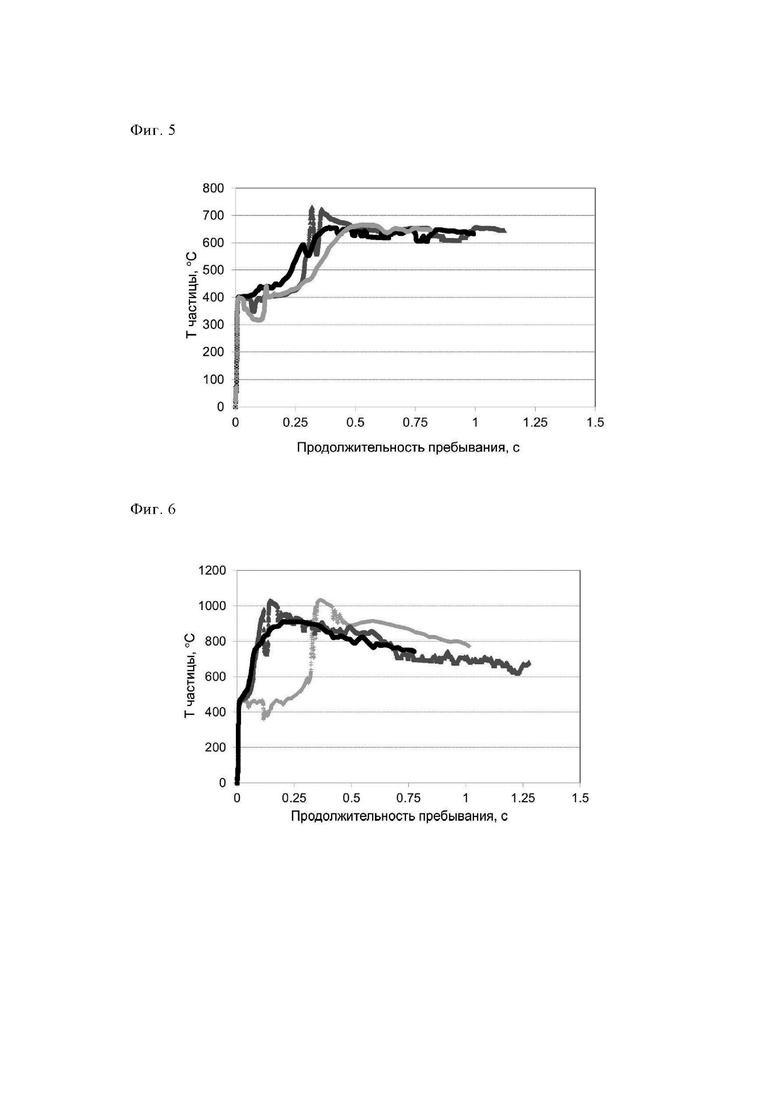
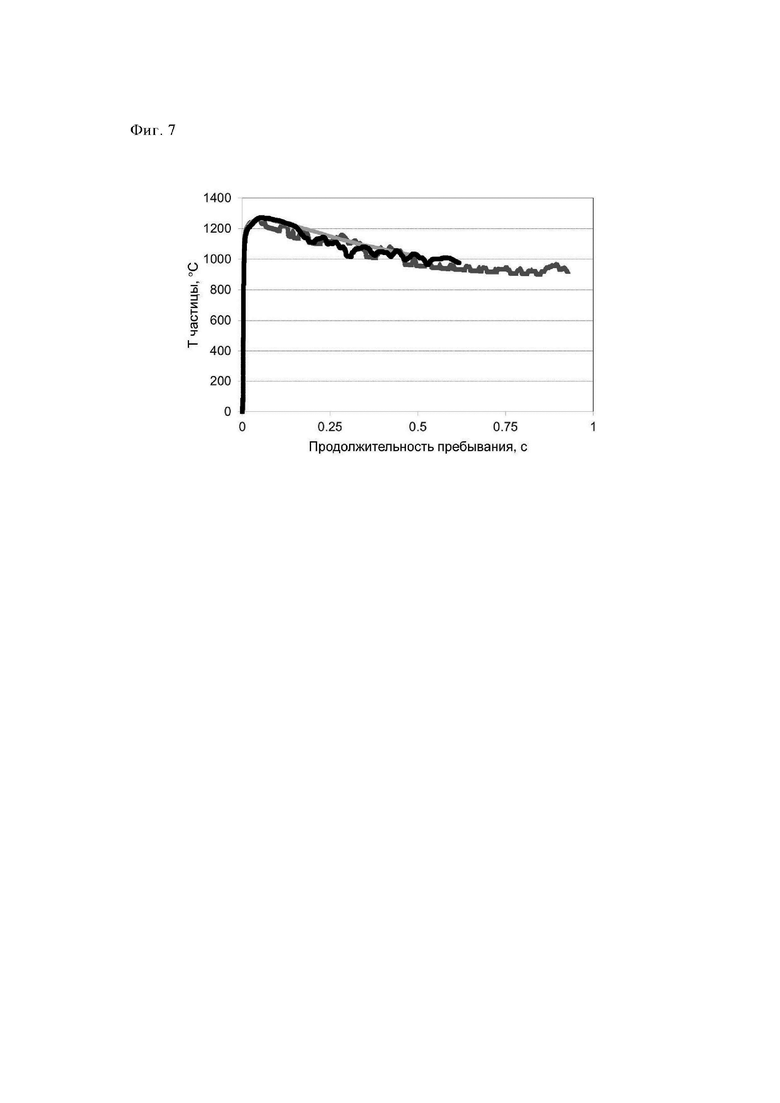
Изобретение может быть использовано при получении синтетических цеолитов. Способ получения порошкообразного пористого кристаллического силиката металла включает гидротермальный синтез с использованием водной смеси, содержащей источник кремния, источник металла, и вспомогательный компонент, с получением водной суспензии продукта реакции, содержащей неочищенный пористый кристаллический силикат металла. Источник кремния выбирают из группы, состоящей из пирогенного диоксида кремния, осажденного диоксида кремния, диоксида кремния, полученного по золь-гелевой технологии, и их смеси. Источником металла является источник титана, железа или алюминия. Водная суспензия содержит твердые вещества в количестве ≤70 мас.%. Затем осуществляют пламенный распылительный пиролиз продукта реакции, при котором полученную водную суспензию распыляют в пламя, образующееся при сгорании топлива, в присутствии кислорода с получением порошкообразного пористого кристаллического силиката металла. В качестве топлива используют водород. Максимальная эффективная температура Тэфф, до которой во время пламенного пиролиза нагревается не менее 90 мас.% пористого кристаллического силиката металла, находится в диапазоне Тмин.<Тэфф<Тмакс, где Тмин равна 750°С, Тмакс равна 1250°С. Изобретение позволяет сократить время и энергию, затрачиваемые при получении порошкообразного пористого силиката металла, при сохранении упорядоченной пористой кристаллической структуры силиката металла и его каталитических характеристик. 15 з.п. ф-лы, 7 ил., 1 табл., 10 пр.
1. Способ получения порошкообразного пористого кристаллического силиката металла, включающий следующие стадии:
(a) гидротермальный синтез с использованием водной смеси, содержащей:
(A) источник кремния, который выбирают из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси,
(B) источник металла, и
(C) вспомогательный компонент,
с получением водной суспензии продукта реакции 1, содержащей неочищенный пористый кристаллический силикат металла, причем гидротермальный синтез проводят при температуре, равной от 100 до 250°С, при автогенном давлении, созданном в выдерживающем давление реакторе, и при значении рН, превышающем 7, в течение менее 12 ч; и
(b) пламенный распылительный пиролиз продукта реакции 1, где водную суспензию, полученную на стадии (а), распыляют в пламя, образующееся при сгорании топлива, в присутствии кислорода с получением порошкообразного пористого кристаллического силиката металла; при этом топливом, использующимся для пламенного распылительного пиролиза, является водород; причем водная суспензия, содержащая продукт реакции 1, полученная на стадии (а), обладает содержанием твердых веществ, составляющим ≤70 мас.%; и причем максимальная эффективная температура Тэфф, до которой во время пламенного пиролиза нагревается не менее 90 мас.% пористого кристаллического силиката металла, находится в диапазоне Тмин<Тэфф<Тмакс, и
где Тмин равна 750°С, и
где Тмакс равна 1250°С, и
при этом источником металла (В) является источник титана (Ti), железа (Fe) или алюминия (Al), и
при этом вспомогательный компонент (С) выбирают из группы, состоящей из гидроксида тетрапропиламмония, 1,6-диаминогексана и 1,2-пентандиола для получения силикалита титана-1, и гидроксида тетрапропиламмония для получения силикалита титана-2, силикалита железа-1 и силикалита алюминия-железа-1.
2. Способ по п. 1, в котором на стадии (а) компонент (А) и компонент (В) объединяют с получением одного компонента и этот компонент выбран из группы, состоящей из следующих: аморфный смешанный оксид металла и кремния, аморфный диоксид кремния, легированный оксидом металла, аморфный диоксид кремния, пропитанный металлом, силикат металла, легированный металлом тетраалкилортосиликат и их смеси.
3. Способ по любому из пп. 1, 2, в котором источником металла (В) является источник титана (Ti).
4. Способ по п. 1, в котором
компонент (А) выбирают из группы, состоящей из следующих: пирогенный диоксид кремния, осажденный диоксид кремния, диоксид кремния, полученный по золь-гелевой технологии, и их смеси, и при этом источником металла (В) является источник титана (Ti), и при этом вспомогательный компонент (С) выбирают из группы, состоящей из гидроксида тетрапропиламмония, 1,6-диаминогексана и 1,2-пентандиола для получения силикалита титана-1, и гидроксида тетрапропиламмония для получения силикалита титана-2, и при этом
пористый кристаллический силикат металла обладает структурой цеолита типа MFI или MEL, и при этом
топливом, использующимся для пламенного распылительного пиролиза, является водород.
5. Способ по п. 1, в котором
компонент (А) и компонент (В) объединяют с получением одного компонента, и этот компонент выбран из группы, состоящей из следующих: аморфный смешанный оксид металла и кремния, аморфный диоксид кремния, легированный оксидом металла, аморфный диоксид кремния, пропитанный металлом, силикат металла, легированный металлом тетраалкилортосиликат и их смеси, и при этом источником металла (В) является источник титана (Ti), и при этом вспомогательный компонент (С) выбирают из группы, состоящей из гидроксида тетрапропиламмония, 1,6-диаминогексана и 1,2-пентандиола для получения силикалита титана-1, и гидроксида тетрапропиламмония для получения силикалита титана-2, и при этом
пористый кристаллический силикат металла обладает цеолитной структурой типа MFI или MEL, и при этом
топливом, использующимся для пламенного распылительного пиролиза, является водород.
6. Способ по любому из пп. 1-5, в котором вспомогательным компонентом является гидроксид тетрапропиламмония.
7. Способ по любому из пп. 1-6, в котором Тмин равна 800°С, и в котором Тмакс равна 1200°С.
8. Способ по любому из пп. 1-6, в котором Тмин. равна 850°С, и в котором Тмакс равна 1100°С.
9. Способ по любому из пп. 1-8, в котором использующаяся на стадии (а) водная смесь дополнительно содержит затравочные кристаллы.
10. Способ по любому из пп. 1-3 и 6-9, в котором пористый кристаллический силикат металла обладает цеолитной структурой типа MFI или MEL.
11. Способ по любому из пп. 1-3 и 6-10, в котором пористый кристаллический силикат металла обладает цеолитной структурой типа MFI.
12. Способ по любому из пп. 1-3 и 6-11, в котором вспомогательный компонент (С) выбирают из группы, состоящей из гидроксида тетрапропиламмония, 1,6-диаминогексана и 1,2-пентандиола для получения силикалита титана-1, и гидроксида тетрапропиламмония для получения силикалита титана-2, и при этом источником металла (В) является источник титана (Ti).
13. Способ по любому из пп. 1-3 и 6-12, в котором вспомогательным компонентом (С) является гидроксид тетрапропиламмония, и при этом источником металла (В) является источник титана (Ti), и при этом пористый кристаллический силикат титана обладает цеолитной структурой типа MFI.
14. Способ по любому из пп. 6-13, в котором топливом, использующимся для пламенного распылительного пиролиза, является водород.
15. Способ по любому из пп. 1-14, в котором полученный таким образом пористый кристаллический силикат металла обладает потерями при прокаливании, составляющими менее 5 мас.%.
16. Способ по любому из пп. 1-15, в котором после стадии (b) проводят стадию формования (с), включающую следующие подстадии:
(1) добавление воды для получения водной суспензии порошкообразного пористого кристаллического силиката металла,
(2) смешивание суспензии, полученной на подстадии (1), с гранулирующими средствами,
(3) прессование, гранулирование, распылительная сушка, распылительное гранулирование и/или экструзия продукта, полученного на подстадии (2), для получения пористого кристаллического силиката металла в форме микрогранул, сфер, таблеток, сплошных цилиндров, полых цилиндров или сотовидных структур.
| US 2017253547 A1, 07.09.2017 | |||
| Способ получения высококремнеземного цеолита | 1989 |
|
SU1721013A1 |
| НЕОРГАНИЧЕСКИЕ ОКСИДЫ С МЕЗОПОРИСТОСТЬЮ ИЛИ СО СМЕШАННОЙ МЕЗО- И МИКРОПОРИСТОСТЬЮ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2248934C2 |
| US 2014301942 A1, 09.10.2014 | |||
| US 6106803 A, 22.08.2000 | |||
| US 5919430 A, 09.07.1999. | |||

