Предпосылки изобретения
Техническая область изобретения
Данное изобретение относится к способу получения ε-капролактама из циклогексаноноксима, причем ε-капролактам полезен в качестве исходного вещества для получения нейлона-6. Точнее, данное изобретение относится к способу получения ε-капролактама из циклогексаноноксима, где способ включает описанный далее первый или второй аспект: в первом аспекте способ включает контактирование циклогексаноноксима со специфическим твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима и получения таким образом ε-капролактама или во втором аспекте способ включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, где реакция перегруппировки осуществляется в присутствии специфического производного многоатомного спирта.
Преимущество способа данного изобретения состоит не только в том, что ε-капролактам может быть получен с высокой селективностью и с высоким выходом, но и в том, что катализатор, используемый в способе данного изобретения, может иметь подходящую морфологию для коммерческого применения, и обладает также прекрасной механической прочностью, поэтому данный катализатор может использоваться в различных процессах каталитических реакций, таких как каталитические процессы, использующие реактор с неподвижным слоем, реактор с псевдоожиженным слоем и реактор с движущимся слоем, и катализатор может использоваться в реакционном процессе, который осуществляется в течение длительного времени с часто повторяющимся реакционным циклом и регенерацией катализатора. Поэтому способ данного изобретения является стабильным для получения ε-капролактама в течение длительного времени.
Предшествующий уровень
ε-Капролактам известен как очень важное химическое вещество для получения различных химических продуктов, таких как нейлон-6. Б настоящее время коммерческое получение ε-капролактама проводится главным образом с использованием способа, который включает контактирование циклогексаноноксима с олеумным катализатором (то есть дымящей серной кислотой) в жидкой фазе для осуществления реакции перегруппировки циклогексаноноксима и получения таким образом ε-капролактама. Однако проблемы данного способа обусловлены тем, что в качестве побочного продукта образуется большое количество сульфата аммония, и реакционная аппаратура подвержена коррозии под действием олеума. Эти проблемы все еще не решены.
С другой стороны, что касается способа получения ε-капролактама, который включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе, имелось большое количество предложений по его усовершенствованию.
В публикации Japanese Patent Application Laid-Open Specification № Sho 57-139062 (соответствующей Патенту США №4359421) описывается способ получения ε-капролактама с применением цеолита в качестве твердого кислотного катализатора, где отношение диоксид кремния/оксид алюминия в цеолите равно 12 или более и индекс проницаемости находится в интервале от 1 до 12, то есть цеолит представляет собой любой из цеолита ZSM-5 и других специфических ZSM цеолитов.
В публикации Unexamined Japanese Patent Application Laid-Open Specification № Sho 62-123167 (соответствующей Патенту США №4709024) описывается способ получения ε-капролактама с применением кристаллического цеолитного катализатора, который имеет индекс проницаемости в интервале от 1 до 12 и в котором атомное отношение Si/Al равно 500 или более и количество внешней кислоты (количество кислотных сайтов, наблюдаемых на кристаллической поверхности) составляет 5 микроэквивалентов/г или менее.
Однако все эти способы имеют обычный недостаток, обусловленный только применением цеолита (который является активной породой) в качестве катализатора.
На практике при осуществлении процессов на основе реакций в газовой фазе в промышленном масштабе очень редко в качестве катализатора используется один цеолит. В большинстве случаев цеолит используется в виде формованного изделия с подходящей структурой, которое получают формованием смеси цеолита с обычным связующим веществом, таким как диоксид кремния, диоксид кремния-оксид алюминия или оксид алюминия. Это обусловлено тем, что цеолит является поликристаллическим и поэтому очень плохо поддается формованию в желаемую структуру.
Однако при проведении реакции перегруппировки циклогексаноноксима для получения ε-капролактама с применением описанного выше цеолита в форме формованного изделия, полученного формованием смеси цеолита с обычным связующим веществом, таким как диоксид кремния, диоксид кремния-оксид алюминия или оксид алюминия, возникает проблема в связи с тем, что поскольку обычные связующие вещества не являются инертными к реакционной системе указанной реакции перегруппировки, они ускоряют побочные реакции (например, побочные реакции с получением пека и смолы), сокращая таким образом срок службы катализатора и значительно снижая активность катализатора и селективность реакции получения ε-капролактама.
Для решения этой проблемы в публикации Unexamined Japanese Patent Application Laid-Open Specification №2000-202296 (соответствующей EP 1002577 А1) предлагается способ получения катализатора, который в качестве основных компонентов включает MFI цеолит и кремнистый лиганд и который подходит для применения в реакции перегруппировки для превращения оксима в соответствующий амид. В частности, способ, предложенный в данном патентном документе, включает связывание частиц цеолита размером менее одного микрона с лигандом, полученным кислотным гидролизом (то есть гидролизом, осуществляемым в кислотном растворе) алкоксида кремния. Однако недостатком данного способа является то, что получение катализатора должно осуществляться в строго контролируемых условиях, таких как значение рН и степень дисперсности цеолита, что усложняет процесс получения.
В публикации Japanese Patent №3023581 (соответствующей Патенту США №5407881) описывается способ, в котором в качестве катализатора используется формованное изделие из цеолита, прочность которого улучшена обработкой, при которой формованное изделие из цеолита pentasil типа, полученное без использования любого неорганического связующего вещества, подвергается контактированию со щелочным раствором, имеющим значение рН в интервале от 9 до 13, при температуре от 30 до 100°С. Однако, когда такой катализатор, не содержащий неорганического связующего вещества, как описано выше, используется в течение длительного времени, во время реакции он становится порошкообразным, вероятно, вследствие недостаточной механической прочности. Еще одним недостатком данного способа является то, что когда катализатор, не содержащий неорганического связующего вещества, подвергается дополнительной обработке для улучшения его механической прочности, способ получения становится громоздким.
Помимо описанных выше способов были заявлены способы получения ε-капролактама в газовой фазе, в которых реакция перегруппировки осуществляется при одновременном присутствии цеолита в качестве катализатора и специфического соединения.
Например, в публикации ЕР 0380364 А2 описывается способ, в котором реакция в газовой фазе для получения ε-капролактама из циклогексаноноксима проводится при одновременном присутствии твердого кислотного катализатора и соединения, представленного формулой R1-O-R2 (где R1 представляет собой низшую алкильную группу, которая может быть замещена атомом фтора, R2 представляет собой атом водорода, низшую алкильную группу или фенильную группу). То есть в данном способе реакция получения ε-капролактама проводится при совместном присутствии твердого кислотного катализатора и простого эфира или низшего спирта.
Кроме того, в публикации Unexamined Japanese Patent Application Laid-Open Specification № Hei 10-87611 описывается способ получения ε-капролактама, который включает контактирование циклогексаноноксима с катализатором, выбранным из группы, включающей ZSM-5 цеолит с атомным отношением Si/Al менее 100, морденитный (mordenite) цеолит и цеолит Y типа, в газовой фазе в присутствии алифатического спирта, содержащего семь или более атомов углерода.
Однако, что касается упомянутых выше патентных документов, в которых описывается применение специфического соединения в сочетании с цеолитом, во всех этих патентных документах описывается применение в качестве катализатора одного цеолита, а не катализатора в форме практически полезного формованного изделия, полученного формованием цеолита с использованием походящего связующего вещества.
Кроме того, в практике получение ε-капролактама реакцией перегруппировки циклогексаноноксима в большинстве случае несущий газ используется для улучшения контакта между исходным веществом и катализатором. В способе данного изобретения необходимо отделять несущий газ, используемый в реакции, от описанного выше специфического соединения (такого как эфирное производное и/или низший спирт), используемого в сочетании с цеолитом.
Однако при использовании эфирного соединения и/или низшего спирта возникает проблема, которая состоит в том, что данные соединения обычно имеют низкую температуру кипения, так что трудно отделить несущий газ от этих соединений.
С другой стороны, при использовании высшего спирта возникает другая проблема, которая заключается в том, что более высокий спирт обычно имеет слишком высокую температуру кипения, которая близка к температуре кипения циклогексаноноксима, поэтому трудно отделить циклогексаноноксим от более высокого спирта.
Краткое описание изобретения
В данной ситуации заявители данного изобретения провели глубокие исследования для разработки улучшенного способа получения ε-капролактама контактированием циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, данный способ не имеет указанных выше недостатков предшествующего уровня и может использоваться для стабильного и эффективного получения ε-капролактама с высоким выходом в течение длительного времени.
В результате авторами данного изобретения было установлено, что указанная задача может быть достигнута способом, включающим контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, где твердый кислотный катализатор получен кальцинированием высушенного предшественника катализатора, причем предшественник катализатора содержит цеолит, кристаллический глинистый минерал и, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид, где неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы (термин “Периодическая таблица”, используемый в данном изобретении, описан в номенклатуре ИЮПАК (Международный союз теоретической и прикладной химии) (1989)), и где неорганический оксид отличен от оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале. Исходя из этого открытия было составлено данное изобретение.
Заявителями данного изобретения было также установлено, что с помощью специфического способа получения ε-капролактама, в котором указанная выше реакция перегруппировки осуществляется при совместном присутствии твердого кислотного катализатора и специфического производного многоатомного спирта, может предотвращаться дезактивация катализатора и может быть повышена селективность катализатора в направлении получения ε-капролактама. Указанный специфический способ включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, где реакция перегруппировки осуществляется в присутствии производного многоатомного спирта, представленного формулой R1-O-R2-OH (где R1 представляет собой C1-C5 алкильную группу или фенильную группу и R2 представляет собой C2-C5 алкиленовую группу). На основе этого нового открытия было составлено данное изобретение.
Описанные выше и другие объекты, отличительные признаки и преимущества данного изобретения станут понятными из приведенного далее подробного описания и прилагаемых рисунков и формулы изобретения.
Краткое описание фигур
Фигура 1 представляет график, показывающий сравнение конверсий циклогексаноноксима, полученных в примере 16 и в сравнительном примере 5;
Фигура 2 представляет график, показывающий сравнение селективностей получения ε-капролактама примера 16 и сравнительного примера 5;
Фигура 3 представляет график, показывающий сравнение конверсий циклогексаноноксима, полученных в примере 17 и в сравнительном примере 6;
Фигура 4 представляет график, показывающий сравнение селективностей получения ε-капролактама примера 17 и сравнительного примера 6;
Фигура 5 представляет диаграмму, показывающую классификацию кристаллических глинистых минералов, где кристаллические глинистые минералы, которые могут содержаться в предшественнике катализатора (перед кальцинированием) для катализатора, используемого в первом аспекте данного изобретения, представлены в точечной рамке.
Подробное описание изобретения
В первом аспекте данного изобретения представлен способ получения ε-капролактама, который включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, где твердый кислотный катализатор получен кальцинированием высушенного предшественника катализатора, причем предшественник катализатора включает цеолит, кристаллический глинистый минерал и, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид, где неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы, и где неорганический оксид отличен от оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале.
Во втором аспекте данного изобретения представлен способ получения ε-капролактама, который включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, где реакция перегруппировки осуществляется в присутствии производного многоатомного спирта, представленного формулой R1-O-R2-OH (где R1 представляет собой C1-C5 алкильную группу или фенильную группу и R2 представляет собой С2-С5 алкиленовую группу).
Для простоты понимания данного изобретения существенные отличительные признаки и различные предпочтительные воплощения данного изобретения перечислены ниже.
1. Способ получения ε-капролактама, который включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, причем твердый кислотный катализатор получен кальцинированием высушенного предшественника катализатора, который включает цеолит, кристаллический глинистый минерал и, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид, где неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы, и где неорганический оксид отличен от оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале.
2. Способ по п.1, представленному выше, где цеолит представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 10 или более, металлосиликат с атомным отношением Si/металл, равным 10 или более, и силикалит.
3. Способ по п.1 или 2, представленным выше, где цеолит представляет собой MFI цеолит.
4. Способ по любому из пп.1-3, представленных выше, где цеолит представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей MFI силикалит и ZSM-5 цеолит.
5. Способ по любому из пп.1-4, представленных выше, где кристаллический глинистый минерал представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей каолиновый минерал, тальк, монтмориллонит и пирофиллит.
6. Способ по любому из пп.1-5, представленных выше, где неорганический оксид представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей диоксид кремния, диоксид кремния-оксид алюминия и оксид алюминия.
7. Способ по любому из пп.1-6, представленных выше, где количество кристаллического глинистого минерала в высушенном предшественнике катализатора составляет от 5 до 50 мас.% из расчета на общую массу цеолита, кристаллического глинистого минерала и, по меньшей мере, одного соединения, выбранного из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид.
8. Способ по любому из пп.1-7, представленных выше, где реакция перегруппировки циклогексаноноксима проводится в условиях, где температура реакции находится в интервале от 200 до 500°С, давление реакции находится в интервале от 0,01 до 1 МПа, часовая объемно-массовая скорость указанного циклогексаноноксима находится в интервале от 0,01 до 100 час-1.
9. Способ по любому из пп.1-8, представленных выше, где реакция перегруппировки осуществляется посредством процесса в псевдоожиженном слое.
10. Способ по любому из пп.1-9, представленных выше, где часть катализатора, используемого в реакции перегруппировки, постоянно или периодически извлекается из реактора реакции перегруппировки, после чего извлеченный катализатор регенерируется в атмосфере кислородсодержащего газа или инертного газа, и регенерированный катализатор возвращается на повторную переработку в реактор.
11. Способ получения ε-капролактама, который включает контактирование циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима, причем реакция перегруппировки осуществляется в присутствии производного многоатомного спирта, представленного формулой R1-O-R2-OH, где R1 представляет собой C1-C5 алкильную группу или фенильную группу, R2 представляет собой С2-С5 алкиленовую группу.
12. Способ по п.11, представленному выше, где твердый кислотный катализатор представляет собой цеолит или цеолитсодержащий катализатор.
13. Способ по п.12, представленному выше, где цеолит представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 10 или более, металлосиликат с атомным отношением Si/металл, равным 10 или более, и силикалит.
14. Способ по п.12 или 13, представленным выше, где цеолит представляет собой MFI цеолит.
15. Способ по любому из пп.11-14, представленных выше, где производное многоатомного спирта представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей этиленгликольмонометиловый эфир, этиленгликольмоноэтиловый эфир, пропиленгликольмонометиловый эфир и пропиленгликольмоноэтиловый эфир.
16. Способ по любому из пп.11-14, представленных выше, где производное многоатомного спирта представляет собой этиленгликольмонометиловый эфир.
17. Способ по любому из пп.11-16, представленных выше, где реакция перегруппировки циклогексаноноксима проводится в условиях, где температура реакции находится в интервале от 200 до 500°С, давление реакции находится в интервале от 0,01 до 1 МПа, часовая объемно-массовая скорость указанного циклогексаноноксима находится в интервале от 0,01 до 100 час-1.
18. Способ по любому из пп.11-17, представленных выше, где реакция перегруппировки осуществляется посредством процесса в псевдоожиженном слое.
19. Способ по любому из пп.11-18, представленных выше, где часть катализатора, используемого в реакции перегруппировки, непрерывно или периодически извлекается из реактора реакции перегруппировки, после чего извлеченный катализатор регенерируется в атмосфере кислородсодержащего газа или инертного газа, и регенерированный катализатор возвращается на повторную переработку в реактор.
Далее данное изобретение описывается подробно. В способе согласно первому аспекту данного изобретения необходимо, чтобы использовался твердый кислотный катализатор, где твердый кислотный катализатор получен кальцинированием высушенного предшественника катализатора, включающего в качестве первого компонента цеолит, в качестве второго компонента кристаллический глинистый минерал и в качестве третьего компонента, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид, где неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы, и где неорганический оксид отличен от оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале.
Для улучшения способности к формованию и механической прочности твердого кислотного катализатора помимо указанных выше компонентов высушенный предшественник твердого кислотного катализатора может содержать дополнительный компонент, такой как графит, в количестве от 1 до 5 мас.% из расчета на общую массу первого, второго и третьего компонентов высушенного предшественника катализатора перед кальцинированием.
Цеолит в качестве первого компонента высушенного предшественника катализатора представляет собой пористый кристаллический силикат, который, в свою очередь, представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат, металлосиликат и силикалит, которые описаны ниже. Кроме того, к цеолиту данного изобретения относится пористый кристаллический фосфат (такой как SAPO-5 или SAPO-11), который имеет по существу такую же структуру, что и цеолит.
Алюмосиликат данного изобретения представляет собой соединение, состоящее, главным образом, из структурных единиц (SiO4)4- и структурных единиц (AlО4)5- (далее в описании обе структурные единицы часто обозначаются как TO4, что означает тетроксид атома Т с тетраэдрической структурой, в которой атомы кислорода расположены в вершинах тетраэдра), где каждая частица ТО4 содержит четыре атома кислорода при вершинах тетраэдра и окружена четырьмя соседними структурными единицами ТО4, поэтому эти структурные единицы являются трехмерно-связанными с образованием кристалла. Кристалл является пористым, и диаметры отверстий пор кристалла находятся в интервале от примерно 0,4 до примерно 0,8 нм. Такой кристалл действует как молекулярное сито.
Металлосиликат данного изобретения имеет по существу такую же пористую кристаллическую структуру, что и алюмосиликат, с тем отличием, что кристаллическая структура металлосиликата дополнительно включает структурные единицы ТО4, каждая из которых содержит в качестве Т-атома атом металла, отличный от атома Si и Аl. Примеры таких атомов металлов включают титан (Ti), бор (В), железо (Fe), цинк (Zn), галлий (Ga), хром (Сr), кобальт (Со), цирконий (Zr), ванадий (V), медь (Сu), ниобий (Nb) и бериллий (Be). Среди указанных элементов бор (В) обычно не рассматривается как металл; однако в данном изобретении бор может вводиться в кристаллическую решетку металлосиликата в качестве Т-атома.
Силикалит данного изобретения обладает по существу такой же пористой кристаллической структурой, что и алюмосиликат, но с тем отличием, что кристаллическая структура силикалита не содержит структурных единиц ТO4 с атомом Аl или любого другого металла, указанного выше, в качестве Т-атома, но включает только структурные единицы ТO4, содержащие в качестве Т-атома атом Si.
Пористый кристаллический фосфат, определенный в данном изобретения, состоит, главным образом, из структурных единиц (АlO4)5-и структурных единиц (РO4)3-, которые представляют собой структурные единицы TO4 пористого кристаллического фосфата. В пористом кристаллическом фосфате структурные единицы (AlO4)5- и (РO4)3- связаны друг с другом с образованием трехмерной кристаллической структуры. Обычно пористые кристаллические фосфаты обозначают общей формулой АlРO4-n (где n представляет собой положительное целое число и показывает идентификационный номер, который обычно используется для идентификации типа пористого кристаллического фосфата в соответствии с кристаллической структурой). Среди пористых кристаллических фосфатов фосфаты, содержащие структурные единицы (AlO4)5- и (РO4)3-, а также фрагменты ТO4, включающие в качестве Т-атома атомы Si, называют SAPO-n (где n представляет собой целое положительное число и показывает идентификационный номер, который обычно используется для идентификации типа пористого кристаллического фосфата в соответствии с кристаллической структурой), а фосфаты, которые дополнительно содержат фрагменты ТО4, включающие атом любого металла из Ga, Mg, Mn, Fe, Co, Zn и т.п. в качестве Т-атома, называют МеАРО. Пористый кристаллический фосфат имеет структуру, подобную структурам цеолитов, как описано выше.
Среди данных соединений предпочтительными являются алюмосиликат, металлосиликат и силикалит.
Более предпочтительно цеолит представляет собой, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 10 или более, металлосиликат с атомным отношением Si/Me, равным 10 или более, и силикалит. Еще более предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 100 или более, металлосиликат с атомным отношением Si/металл, равным 100 или более, и силикалит. Еще более предпочтительно использовать алюмосиликат с атомным отношением Si/Al, равным 250 или более, металлосиликат с атомным отношением Si/Me, равным 250 или более, и силикалит. Еще более предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 500 или более, металлосиликат с атомным отношением Si/металл, равным 500 или более, и силикалит. Наиболее предпочтительно использовать алюмосиликат с атомным отношением, равным 1000 или более, металлосиликат с массовым отношением Si/Me, равным 1000 или более, и силикалит.
Ниже представлены конкретные примеры указанных выше цеолитов.
Примеры алюмосиликатов включают цеолит А-типа, цеолит Х-типа, цеолит Y-типа и цеолит L-типа, оффретит (offretite zeolite), морденит, ферриерит (ferrierite) и ZSM-5 (см. Examined Japanese Patent Application Publication № Sho 46-10064 (соответствует Патенту Канады №902334)), цеолит ZSM-11 (см. Examined Japanese Patent Application Publication № Sho 53-23280 (соответствует Патенту США №3709979)), цеолит ZSM-12 (см. Examined Japanese Patent Application Publication № Sho 52-16079 (соответствует Патенту США №3832449)), цеолит ZSM-23 (“ZEOLITES” 5, pp.352-354, А.С. Rohrman Jr. et al. (1985)), цеолит β-типа (см. Патент США №3308069) и цеолит МСМ-22 (см. Патент США №4954325).
Примеры металлосиликатов включают титаносиликат (см. Патент США №4410501), боросиликат (см. ЕР 7081 В1 (соответствует Патенту США №4456582)).
Примеры силикалитов включают силикалит, описанный в Патенте США №4061724.
Примеры пористых кристаллических фосфатов со структурой, подобной структуре цеолита, включают SAPO-5 (см. Unexamined Japanese Patent Application Laid-Open Specification № Sho 59-35018 (соответствует Патенту США №4440871)), SAPO-11 (см. Unexamined Japanese Patent Application Laid-Open Specification № Sho 59-35018 (соответствует Патенту США №4440871)).
Каждый из указанных выше патентных документов, описывающих различные цеолиты, содержит описание рентгенограммы цеолита и способа получения цеолита.
Что касается цеолита А-типа, цеолита Х-типа, цеолита Y-типа, цеолита L-типа, оффретита, морденита, ферриерита, цеолита ZSM-5 и цеолита ZSM-11, их состав и структуры описаны в публикации “An Introduction to Zeolite Molecular Sieves”, A. Dyer, pp.12-37, U.K., 1988.
Среди перечисленных выше цеолитов предпочтительно использовать силикалит, цеолит ZSM-5, титаносиликат, боросиликат, цеолит β-типа и ферриерит, более предпочтительно использовать цеолит со структурой MFI-типа, такой как силикалит MFI-типа, цеолит ZSM-5 или титаносиликат MFI-типа.
Термин “MFI” представляет собой кодовое название в соответствии с номенклатурой IUPAC, используемое для определения структуры цеолита. Подробное описание структуры, соответствующей кодовому названию, приведено в публикации “Atlas of zeolite framework types”, 5th edition, p.13, Ch. Baerlocher, W.M. Meier and D.H. Cison (2001). Рентгенограммы цеолитов MFI-типа представлены в статье R.V. Ballmoos and J.B. Higgins, “Collection of simulated XRD powder patterns for zeolites” (“ZEOLITES”, vol.10, №5, June 1990, pp.442S-445S).
В качестве наиболее предпочтительных цеолитов могут быть указаны алюмосиликаты с атомным отношением Si/Al, равным 1000 или более, металлосиликат MFI-типа с атомным отношением Si/металл, равным 1000 или более, и силикалит MFI-типа.
Данные цеолиты могут обрабатываться для введения ионов водорода, ионов различных металлов, различных соединений металлов и т.п. обычными способами, такими как метод ионного обмена, метод импрегнирования и метод адсорбции. Среди этих методов предпочтительным является метод ионного обмена.
Что касается типа и количества металла, который может вводиться в цеолит, особых ограничений нет, и тип и количество металла могут выбираться подходящим образом при отсутствии неблагоприятного воздействия на реакцию, проводимую в соответствии со способом данного изобретения. В результате экспериментов, проведенных заявителями данного изобретения, было установлено, что, когда в цеолит вводится атом металла 11 группы Периодической таблицы (то есть Сu, Аg и Аu) указанным выше способом, таким как ионный обмен, срок службы полученного катализатора может продлеваться. Среди этих металлов наиболее предпочтителен Аg.
В способе в соответствии с первым аспектом данного изобретения примеры кристаллических глинистых минералов, используемых в качестве второго компонента, включают кристаллические глинистые минералы, представленные в точечной рамке на Фигуре 5.
Далее описаны глинистые минералы, используемые в данном изобретении.
Глинистые минералы состоят из основных структурных единиц двух видов: тетраэдрической структурной единицы SiO4, в которой четыре иона О2- координированно связаны с ионом Si4+, и октаэдрической структурной единицы Аl(ОН)6, в структуре которой шесть ионов ОН- или О2-координированно связаны с ионом Аl3+ (при условии, что имеется также октаэдрическая структурная единица, в структуре которой ион Аl3+ структурной единицы Аl(ОН)6 замещен на ион Mg2+ или Fe2+). Листовая структура, включающая множество тетраэдрических структурных единиц SiO4, двухмерно связанных друг с другом, как известно, называется “тетраэдрический лист”, а листовая структура, включающая множество октаэдрических структурных единиц Аl(ОН)6, двухмерно связанных друг с другом, как известно, называется “октаэдрическим листом”.
В глинистом минерале слои такого тетраэдрического листа и октаэдрического листа наложены друг на друга.
В соответствии со слоистой структурой, включающей такие листы, выделяют три типа глинистых минералов. В частности, выделяют “глинистый минерал 1:1”, “глинистый минерал 2:1” и “глинистый минерал смешанного слоя”.
Глинистый минерал 1:1 представляет собой глинистый минерал со структурой (1:1 структура), включающей множество элементарных слоев, каждый из которых состоит из одного тетраэдрического листа, связанного с одним октаэдрическим листом. Глинистый минерал 2:1 представляет собой глинистый минерал со структурой (2:1 структура), включающей множество элементарных слоев, каждый из которых состоит из одного октаэдрического листа, соединенного с двумя тетраэдрическими листами. Глинистый минерал смешанного слоя представляет собой глинистый минерал, включающий сочетание, по меньшей мере, двух различных глинистых минералов описанных выше структур.
Конкретные примеры глинистых минералов 1:1 включают каолиновый минерал, имеющий теоретический химический состав Al2Si2O5(ОН)4, и серпентиновый минерал, имеющий теоретический химический состав Mg3Si2O5(OH)4. Примеры каолиновых минералов включают каолинит, дикит, накрит и галлуазит, содержащие включенные молекулы воды. Примеры серпентиновых минералов включают хризотил, лизардит (lizardite) и антигорит (antigorite).
Конкретные примеры глинистых минералов 2:1 включают пирофиллит, имеющий теоретический химический состав Al2Si4O10(ОН)2, тальк, имеющий теоретический химический состав Мg3Si4O10(ОН)2, смектит (smectite), представленный монтмориллонитом, вермикулит (vermiculite), минерал слюдистой глины и хлорит (chlorite). Дополнительные примеры глинистых минералов 2:1 включают сепиолит (sepiolite) и палигорскит (palygorskite), каждый из которых имеет структуру, в которой листы тетраэдрического SiO4 инвертированы с образованием пор.
Конкретные примеры глинистых минералов смешанного слоя включают минерал смешанного слоя, включающий сочетание слюды и смектита, и минерал смешанного слоя, включающий сочетание каолина и монтмориллонита.
Обычно некоторые природные глинистые минералы помимо кристаллического глинистого минерала содержат аморфный компонент, такой как аморфный диоксид кремния.
Оксиды (такие как SiO2 и Аl2O3), образующие кристаллический глинистый минерал, используемый в качестве второго компонента твердого кислотного катализатора, который применяется в способе в соответствии с первым аспектом данного изобретения, могут образовывать кристаллическую структуру, и, следовательно, кристаллический глинистый минерал имеет структуру, в которой такие оксиды равномерно распределены с образованием кристалла. С другой стороны, неорганический оксид (включая SiO2, Аl2O3 и т.п.), используемый в качестве третьего компонента (который описан подробно ниже), не имеет структуры с упорядоченным распределением таких оксидов (SiO2, Аl2O3 и т.п.). Благодаря такому отличию, оксиды, содержащиеся во втором компоненте, и неорганический оксид, используемый в качестве третьего компонента, можно отличить друг от друга. То есть неорганический оксид, используемый в качестве третьего компонента, представляет собой неорганический оксид, отличный от кристаллических оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале.
Глинистый минерал, используемый в данном изобретении, предпочтительно содержит кристаллический глинистый минерал в количестве 50% или более, более предпочтительно 90 мас.% или более. Когда используется неорганический оксид, отличный от оксида, присутствующего в кристаллическом глинистом минерале, этот “другой” оксид рассматривается как часть неорганического оксида, используемого в качестве третьего компонента, и, соответственно, принимается во внимание при расчете массовой доли второго компонента твердого кислотного катализатора.
Описанные выше кристаллические глинистые минералы используются отдельно или в сочетании. Кристаллические глинистые минералы могут быть любыми из минералов природной глины и минералов синтетической глины.
Кроме того, перед применением для получения твердого кислотного катализатора кристаллический глинистый минерал может подвергаться предварительной обработке, такой как кальцинирование, ионный обмен или кислотная обработка. Среди таких кристаллических глинистых минералов предпочтительными являются каолиновый минерал, пирофиллит, тальк и монтмориллонит. Более предпочтительным является каолиновый минерал. И еще более предпочтителен каолинит.
Кристаллический глинистый минерал добавляется, прежде всего, для улучшения прочности и термостойкости каталитического формованного изделия. Кроме того, применение кристаллического глинистого минерала в качестве второго компонента твердого кислотного катализатора реакции перегруппировки циклогексаноноксима для получения ε-капролактама в способе данного изобретения (далее реакция перегруппировки для простоты называется “целевая реакция”) исключительно эффективно улучшает селективность процесса получения ε-капролактама и увеличивает срок службы катализатора, особенно улучшает селективность, по сравнению с применением катализатора, не содержащего кристаллического глинистого минерала (то есть катализатора, содержащего только цеолит и неорганический оксид, включающий оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы).
Для целевой реакции наличие алюминия является особенно неблагоприятным, поскольку алюминий способствует образованию побочных продуктов. Тем не менее в данном изобретении благоприятный эффект может быть получен при применении, например, каолинового минерала, содержащего примерно 40 мас.% оксида алюминия. В самом деле, такой благоприятный эффект, полученный при применении каолинового минерала, является абсолютно неожиданным.
С другой стороны, каталитическое формованное изделие, включающее только цеолит и кристаллический глинистый минерал, не проявляет достаточной прочности.
Третий компонент высушенного предшественника катализатора, который должен подвергаться кальцинированию для получения твердого кислотного катализатора, используемого в способе согласно первому аспекту данного изобретения, представляет собой, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид (далее в описании данное соединение часто называется как “неорганический оксид и/или оксидобразующее соединение”). Третий компонент высушенного предшественника катализатора добавляется в качестве связующего вещества с целью улучшения прочности каталитического формованного изделия. Неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы. Элементами 4 группы Периодической таблицы являются титан (Ti), цирконий (Zr) и гафний (Hf); элементами 13 группы Периодической таблицы являются бор (В), алюминий (Аl), галлий (Ga), индий (In) и таллий (Тl); элементами 14 группы Периодической таблицы являются углерод (С), кремний (Si), германий (Ge), олово (Sn) и свинец (Рb). Предпочтительными являются оксиды Si, Al и Ti. Точнее, предпочтительными являются диоксид кремния, диоксид кремния-оксид алюминия, оксид алюминия, диоксид кремния-оксид титана, оксид титана, диоксид кремния-оксид циркония и оксид циркония; более предпочтительными являются диоксид кремния, диоксид кремния-оксид алюминия и оксид алюминия; еще более предпочтительным является диоксид кремния. Данные оксиды могут использоваться отдельно или в сочетании. Как описано выше, неорганический оксид, используемый в качестве третьего компонента, является неорганическим оксидом, отличным от оксидов, содержащихся в упорядоченной кристаллической форме в цеолите и кристаллическом глинистом минерале.
Конкретные примеры соединений, которые при кальцинировании образуют неорганические оксиды, включают гидроксиды, такие как Аl(ОН)3, Ti(OH)4 и Zr(OH)4, и соли, такие как Аl(NО3)3 и т.п.
Что касается различия между способом получения катализатора, используемого в способе согласно первому аспекту данного изобретения, и способом, описанным в Unexamined Japanese Parent Application Laid-Open Specification №2000-202296 (где кремнистый лиганд, который получен кислотным гидролизом алкоксида кремния, используется в качестве связующего вещества для цеолита), различие заключается в том, что способ, описанный в Unexamined Japanese Patent Application Laid-Open Specification №2000-202296, заключается в осуществлении кислотного гидролиза алкоксида кремния, в то время как при получении катализатора, используемого в способе данного изобретения, не возникает проблемы даже когда неорганический оксид и соединение, которое образует неорганический оксид при кальцинировании, являются основными. Следовательно, способ получения катализатора, используемый в данном изобретении, является преимущественным в том, что велика свобода выбора исходных веществ и что не требуется строгое контролирование значения рН, что облегчает получение катализатора.
Что касается соотношения между цеолитом в качестве первого компонента, кристаллическим глинистым минералом в качестве второго компонента и неорганическим оксидом и/или оксидобразующим соединением в качестве третьего компонента, предпочтительно использовать цеолит в качестве первого компонента в количестве от 20 до 80 мас.%, кристаллический глинистый минерал в качестве второго компонента в количестве от 5 до 50 мас.% и неорганический оксид и/или оксидсодержащее соединение в качестве третьего компонента в количестве от 15 до 30 мас.% в каждом случае из расчета на общую массу первого, второго и третьего компонентов высушенного предшественника катализатора. Более предпочтительно использовать цеолит в качестве первого компонента в количестве от 30 до 70 мас.%, кристаллический глинистый минерал в качестве второго компонента в количестве от 15 до 35 мас.% и неорганический оксид и/или оксидобразующее соединение в качестве третьего компонента в количестве от 15 до 35 мас.%. Когда количество неорганического оксида и/или оксидобразующего соединения в качестве третьего компонента составляет менее 15 мас.%, полученное формованное каталитическое изделие имеет тенденцию проявлять низкую механическую прочность. Когда количество кристаллического глинистого минерала в качестве второго компонента составляет менее 5 мас.%, затруднено получение формованного каталитического изделия для получения желаемого эффекта улучшения селективности.
Далее описан способ получения катализатора, используемого в способе согласно первому аспекту данного изобретения. Что касается способа получения катализатора, то здесь нет особого ограничения. Однако обычно предпочтительно использовать способ, который включает две стадии: (1) стадию получения исходной смеси для получения катализатора, включающей цеолит в качестве первого компонента, кристаллический глинистый минерал в качестве второго компонента и неорганический оксид и/или оксидобразующее соединение в качестве третьего компонента; и (2) стадию, на которой исходная смесь для получения катализатора подвергается формованию, сушке и кальцинированию.
Далее описаны стадии (1) и (2).
(1) Стадия получения исходной смеси для получения катализатора
Исходная смесь для получения катализатора включает цеолит в качестве первого компонента, кристаллический глинистый минерал в качестве второго компонента и неорганический оксид и/или оксидобразующее соединение в качестве третьего компонента.
В качестве цеолита может использоваться цеолит, полученный гидротермическим синтезом, осуществляемым с использованием стандартного способа, описанного, например, в ЕР 0380364 А1. Может также использоваться коммерчески доступный цеолит.
Когда цеолит, полученный гидротермическим способом, содержит органический направляющий агент (то есть органическое соединение, которое используется для образования остова цеолита, такое как гидроксид тетрапропиламмония), предпочтительно, чтобы органический шаблон цеолита разлагался с помощью кальцинирования перед применением, где кальцинирование для разложения органического шаблона проведено тепловой обработкой цеолита в печи, такой как электропечь или трубчатая печь, при температуре в интервале от 400 до 700°С в течение 1-24 часов в атмосфере, содержащей кислород или азот.
Если необходимо, цеолит может использоваться в виде цеолита, подвергшегося ионному обмену, где ионный обмен проведен обработкой цеолита водным раствором неорганической соли, такой как NH4NO3, NH4Cl или АgNО3. Предпочтительно, чтобы ионный обмен проводился способом, в котором к водному раствору неорганической соли добавляется цеолит в количестве от 1 до 30 мас.% из расчета на массу водного раствора неорганической соли, и полученная смесь выдерживается при температуре в интервале от 15 до 90°С в течение 1-10 часов с последующей фильтрацией полученной реакционной смеси, и цикл ионного обмена и фильтрации затем повторяется 1-3 раза. Когда ион аммония превращается в ион водорода, предпочтительно, чтобы конверсия иона аммония в ион водорода выполнялась посредством тепловой обработки цеолита в печи, такой как описаны выше, при температуре в интервале от 400 до 600°С в течение 1-10 часов в атмосфере воздуха.
Что касается кристаллического глинистого минерала, может использоваться непосредственно коммерчески доступный кристаллический глинистый минерал. Альтернативно, перед применением коммерчески доступный кристаллический глинистый минерал может подвергаться кальцинированию при температуре в интервале от 500 до 1200°С.
Что касается исходного вещества для получения неорганического оксида, предпочтительно использовать золь, гель или т.п., содержащий составные элементы неорганического оксида.
Примеры таких исходных веществ включают водный раствор силиката, золь диоксида кремния, силикагель (который получен, например, добавлением к водному раствору силиката натрия кислоты, такой как серная кислота, соляная кислота или азотная кислота), алюмосиликатный золь, золь доксида алюминия, золь оксида титана и золь оксида циркония. Предпочтительными являются водный раствор силиката натрия, золь диоксида кремния, алюмосиликатный золь и золь оксида алюминия и более предпочтительными являются водный раствор силиката натрия и золь диоксида кремния.
Золь диоксида кремния, золь оксида алюминия или т.п., используемые в качестве исходного вещества для получения неорганического оксида, в качестве загрязняющей примеси обычно содержит следовое количество металла, такого как натрий. Поэтому неорганический оксид, который содержится в высушенном предшественнике катализатора, полученном сушкой исходной смеси, содержащей такое исходное вещество, обычно содержит следовое количество металла. Что касается типа и количества такого металла, конкретного ограничения нет. Однако предпочтительно, чтобы количество металла в качестве примеси было небольшим, насколько это возможно, для предупреждения неблагоприятного воздействия на целевую реакцию.
Следовое количество металла может быть удалено из высушенного предшественника катализатора кислотной обработкой высушенного предшественника катализатора перед кальцинированием. Что касается применяемой кислоты, то здесь ограничения нет; однако предпочтительными являются азотная кислота, серная кислота и соляная кислота.
Предпочтительно, чтобы исходная смесь для получения катализатора предоставлялась в форме гомогенной дисперсии, содержащей компоненты высушенного предшественника катализатора.
В качестве предпочтительного примера способа получения исходной смеси для получения катализатора может использоваться следующий процесс. Предопределенное количество воды добавляется к исходному веществу, по меньшей мере, одного соединения, выбранного из группы, включающей неорганический оксид и соединение, которое при кальцинировании образует неорганический оксид, и полученная смесь перемешивается предпочтительно с использованием перемешивающего устройства, такого как гомогенизатор или т.п., при скорости вращения от 2000 до 10000 об/мин в течение от 15 минут до 1 часа при комнатной температуре. Затем к смеси добавляется цеолит и кристаллический глинистый минерал предпочтительно с последующим перемешиванием полученной смеси по существу при условиях перемешивания, указанных выше.
При получении исходной смеси для получения катализатора добавление воды не является обязательным. Однако для улучшения диспергируемости каждого компонента предпочтительно добавлять воду в количестве, которое составляет от 0,5 до 20, более преимущественно от 1 до 10 масс твердых веществ, содержащихся в исходной смеси для получения катализатора, то есть общей массы цеолита, кристаллического глинистого минерала и неорганического оксида (где при использовании золя диоксида кремния или т.п., или соединения, которое при кальцинировании образует неорганический оксид, его количество выражается через количество неорганического оксида, полученного из него). Кроме того, получение исходной смеси для катализатора, содержащей воду, может осуществляться с помощью способа, в котором цеолит и кристаллический глинистый минерал диспергируются в воде с последующим перемешиванием полученной водной смеси с помощью гомогенизатора или т.п. по существу при условиях перемешивания, описанных выше, для получения таким образом суспензии (водной смеси цеолит/кристаллический глинистый минерал), и полученная суспензия добавляется к остатку исходных веществ.
С другой стороны, когда используется водный раствор силиката натрия, предпочтительно, чтобы предопределенное количество водного раствора силиката натрия добавлялось к 10-15 мас.% водному раствору серной кислоты с получением геля SiO2 и к полученной смеси добавлялась указанная водная смесь цеолит/кристаллический глинистый минерал.
В любом случае важно гомогенно диспергировать цеолит, кристаллический глинистый минерал и неорганический оксид и/или соединение, которое при кальцинировании образует неорганический оксид, в исходной смеси для получения катализатора.
(2) Стадия, на которой исходная смесь для получения катализатора подвергается формованию, сушке и кальцинированию
Исходная смесь, полученная на предыдущей стадии, подвергается формованию, сушке и кальцинированию для получения таким образом катализатора, используемого в способе данного изобретения. Каждая операция проводится способом, который выбран подходящим образом в соответствии с видом реакции перегруппировки в способе данного изобретения. Типичные примеры формования, сушки и кальцинирования описаны ниже.
<Реакция в псевдоожиженном слое>
В данном случае способ получения катализатора включает следующие стадии: распылительную сушку полученной исходной смеси (с получением мелкозернистого предшественника катализатора с частицами сферической формы, подходящего для применения в реакторе с псевдоожиженным слоем) и кальцинирование полученного высушенного предшественника катализатора.
На стадии распылительной сушки распыление исходной смеси может осуществляться центрифугированием, методом распыления двухфазного потока с использованием сопла (two-phase flow nozzle method) или методом распыления под высоким давлением (high pressure nozzle method). В качестве источника тепла для сушки предпочтительно использовать воздух, нагретый паром, электронагревателем и т.п. Предпочтительно, чтобы температура в распылительной сушилке на входе в секцию сушки составляла от 150 до 500°С. Нагретый таким образом воздух контактирует с исходной смесью, движущейся противотоком или параллельным потоком, вызывая испарение влаги в исходной смеси с получением высушенного каталитического порошка, сформованного в частицы сферической формы диаметром от примерно 20 до примерно 150 мкм. Полученный высушенный мелкодисперсный предшественник катализатора подвергается кальцинированию в воздухе с использованием печи, такой как электропечь или трубчатая печь, при температуре в интервале от 500 до 1000°С в течение от 1 часа до 48 часов, предпочтительно при температуре в интервале от 600 до 800°С в течение от 1 до 10 часов.
<Реакция в неподвижном слое>
В этом случае способ получения катализатора включает следующие стадии: сушку или полусушку полученной выше исходной смеси; формование полученного высушенного или полувысушенного предшественника катализатора в цилиндрическую, трубчатую или гранулированную форму или т.п. с применением метода формования, такого как метод экструзионного формования, метод таблетирования или метод компрессионного формования; и кальцинирование полученного сформованного изделия из высушенного предшественника катализатора.
В случае экструзионного формования исходная смесь для получения катализатора подвергается тепловой обработке для обезвоживания смеси до уровня, при котором экструзионное формование может выполняться подходящим образом (то есть для обезвоживания смеси до содержания влаги от 10 до 40 мас.%), и затем полученная наполовину высушенная исходная смесь подвергается экструзионному формованию с использованием аппарата экструзионного формования для получения твердого изделия, сформованного в частицы цилиндрической формы диаметром примерно 1-5 мм и длиной примерно 2-10 мм. Полученное формованное изделие подвергается сушке с использованием сушилки при температуре в интервале от 80 до 200°С в течение от 1 до 48 часов для получения высушенного предшественника катализатора, и высушенный предшественник катализатора затем кальцинируется по существу в условиях, описанных выше.
В случае использования метода таблетирования или метода компрессионного формования исходная смесь для получения катализатора распыляется на стальную пластину, предварительно нагретую до температуры в интервале от 100 до 200°С, для испарения влаги, содержащейся в смеси, и получения исходной высушенной смеси в виде смешанного порошка (сухого порошка) с последующим формованием полученного сухого порошка таблетированием (то есть способом, в котором сухой порошок прессуется в формованное изделие между “пестиком” и “ступой” аппарата таблетирования) или компрессией с использованием аппарата таблетирования или аппарата компрессионного формования для получения формованного изделия из высушенного предшественника катализатора. Для улучшения способности к формованию сухого порошка (предшественника катализатора) к сухому порошку может добавляться графит или т.п., где количество графита составляет от 1 до 5 мас.%, предпочтительно от 2 до 3 мас.% из расчета на массу сухого порошка. В методе таблетирования предпочтительно формование сухого порошка (предшественника катализатора) в цилиндрическую или трубчатую форму с диаметром каждой от 2 до 5 мм и длиной от 3 до 10 мм. В случае метода компрессионного формования формованное изделие из высушенного предшественника катализатора может обрабатываться способом, в котором формованное изделие может распыляться с получением предшественника катализатора в виде макрочастиц, и полученный предшественник катализатора в виде макрочастиц классифицируется как способ получения частицами желаемого размера (например, в интервале от 0,3 до 3 мм). Полученный таким образом высушенный предшественник катализатора подвергается кальцинированию по существу в условиях, описанных выше.
Способ, соответствующий первому аспекту данного изобретения, использует полученный таким образом катализатор, то есть катализатор, который получен кальцинированием высушенного предшественника катализатора, включающего цеолит в качестве первого компонента, кристаллический глинистый минерал в качестве второго компонента и неорганический оксид и/или оксидобразующее соединение в качестве третьего компонента. Способ согласно первому аспекту данного изобретения является преимущественным не только потому, что формованное каталитическое изделие проявляет удовлетворительную механическую прочность, но и потому, что достигается высокая селективность получения ε-капролактама.
Далее описывается способ согласно второму аспекту данного изобретения.
В способе согласно второму аспекту данного изобретения предпочтительные примеры твердых кислотных катализаторов включают твердые аморфные кислотные катализаторы, такие как алюмосиликатный катализатор, магниевосиликатный катализатор и алюмоборный катализатор, твердые некристаллические кислотные катализаторы с регулярной мезопористой структурой, такие как МСМ-41 (см. US Р. №5098684 и публикацию J. Am. Chem. Soc., 114, pp.10834-10843, 1992), и твердые кристаллические кислотные катализаторы, такие как цеолиты.
Когда в качестве твердого кислотного катализатора используется цеолит, может использоваться цеолит, описанный в первом аспекте данного изобретения, а именно: алюмосиликат, металлосиликат и силикалит. Среди них более предпочтительно использовать силикалит, цеолит ZSM-5, титаносиликат, боросиликат, цеолит β-типа и ферриерит. Еще более предпочтительными являются цеолиты, имеющие структуру MFI-типа, такие как силикалит MFI-типа, цеолит ZSM-5 и титаносиликат MFI-типа.
С другой стороны, что касается атомного отношения Si/металл, более предпочтительно применять, по меньшей мере, одно соединение, выбранное из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 10 или более, металлосиликат с атомным отношением Si/металл, равным 10 или более, и силикалит.
Еще более предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 100 или более, металлосиликат с атомным отношением Si/металл, равным 100 или более, и силикалит. Особенно предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 250 или более, металлосиликат с атомным отношением Si/металл, равным 250 или более, и силикалит. Еще более предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 500 или более, металлосиликат с атомным отношением Si/металл, равным 500 или более, и силикалит. Наиболее предпочтительно использовать, по меньшей мере, один представитель, выбранный из группы, включающей алюмосиликат с атомным отношением Si/Al, равным 1000 или более, металлосиликат с атомным отношением Si/металл, равным 1000 или более, и силикалит.
Кроме того, при использовании цеолита катализатор может представлять собой любой катализатор, состоящий только из цеолита, и цеолитный катализатор, содержащий традиционное связующее вещество.
В способе согласно второму аспекту данного изобретения, когда твердый кислотный катализатор включает, например, цеолит и связующее вещество, исключительно преимущественно применять те же самые второй и третий компоненты, что и в способе согласно первому аспекту данного изобретения, то есть кристаллический глинистый минерал и, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид (в том числе оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы) и соединение, которое при кальцинировании образует неорганический оксид.
То есть твердый кислотный катализатор, используемый в способе согласно первому аспекту данного изобретения, является предпочтительным в качестве твердого кислотного катализатора, используемого в способе согласно второму аспекту данного изобретения. Другими словами, предпочтительно, чтобы состав твердого кислотного катализатора, используемого в способе согласно второму аспекту данного изобретения, был таким же, что и состав твердого кислотного катализатора, используемого в способе согласно первому аспекту данного изобретения.
В способе второго аспекта данного изобретения производное многоатомного спирта в реакции перегруппировки представляет собой соединение формулы R1-O-R2-OH, где R1 представляет собой C1-C5 алкильную группу или фенильную группу, R2 представляет собой C2-C5 алкиленовую группу.
Конкретные примеры таких производных многоатомных спиртов включают этиленгликольмонометилэфир, этиленгликольмоноэтилэфир, этиленгликольизопропилэфир, этиленгликольмонобутилэфир, этиленгликольизобутилэфир, этиленгликольизоамилэфир, этиленгликольмонофенилэфир, пропиленгликольмонометилэфир, пропиленгликольмоноэтилэфир, пропиленгликольизопропилэфир, пропиленгликольмонобутилэфир, пропиленгликольизобутилэфир, 1-метокси-2-пропанол, 1-метокси-2-бутанол, 3-метокси-1-бутанол, 3-метокси-3-метилбутанол и т.п.
Более предпочтительно использовать, по меньшей мере, одно соединение, выбранное из группы, включаощей этиленгликольмонометилэфир, этиленгликольмоноэтилэфир, пропиленгликольмонометилэфир и пропиленгликольмоноэтилэфир. Еще более предпочтительным является этиленгликольмонометилэфир.
Данные соединения могут использоваться отдельно или в смеси. Кроме того, часть атомов водорода в R1 может замещаться атомами фтора. Эти производные многоатомных спиртов являются растворителями, обычно используемыми в промышленности, и являются легкодоступными.
Благодаря присутствию производного многоатомного спирта в системе реакции перегруппировки могут быть достигнуты преимущественные эффекты снижения подавления каталитической активности, и селективность реакции получения ε-капролактама значительно улучшается.
Что касается способа доставки производного многоатомного спирта в реакционную систему, то ограничений здесь нет. Производное многоатомного спирта может подаваться в реакционную систему отдельно от циклогексаноноксима. Альтернативно, подача производного многоатомного спирта может осуществляться способом, при котором циклогексаноноксим растворяется в производном многоатомного спирта, и полученный раствор подается в реакционную систему. Поскольку производное многоатомного спирта является хорошим растворителем для циклогексаноноксима, предпочтительно, чтобы подача производного многоатомного спирта осуществлялась указанным альтернативным способом (то есть способом, при котором циклогексаноноксим растворяется в производном многоатомного спирта и полученный раствор подается в реакционную систему).
Что касается количества производного многоатомного спирта, присутствующего в реакционной системе, то ограничений здесь нет.
Однако предпочтительно, чтобы производное многоатомного спирта присутствовало в количестве, которое составляет от 0,1 до 30 значений массы циклогексаноноксима, более преимущественно от 0,5 до 20 значений массы циклогексаноноксима, еще более преимущественно от 1 до 10 значений массы циклогексаноноксима.
Если необходимо, в реакционной системе могут находиться и другие органические соединения. Примеры таких “других” органических соединений включают ароматические соединения, такие как бензол и толуол, и алифатические спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, н-амиловый спирт, н-гексанол, н-гептанол и н-октанол. Дополнительные примеры таких органических соединений включают нитрилы, амиды, простые эфиры и кетоны. Когда используются такие дополнительные органические соединения, количество таких соединений доводится до уровня, который не превышает количество производного многоатомного спирта.
Кроме того, вода, основное вещество (такое как метиламин) и ε-капролактам (который является целевым продуктом) также могут подаваться в реакционную систему. Добавлением этих веществ в реакционную систему может подавляться отравление катализатора. Когда такие соединение подаются в реакционную систему, предпочтительно, чтобы количество воды составляло от 0,05 до 1,0 моль на моль циклогексаноноксима и чтобы количество амина (такого как метиламин) и ε-капролактама составляло от 0,05 до 0,5 моль на моль циклогексаноноксима.
В способе согласно первому аспекту данного изобретения в реакционную систему, содержащую катализатор, может подаваться один циклогексаноноксим. Однако предпочтительно, чтобы реакция в способе согласно первому аспекту данного изобретения проводилась в присутствии производного многоатомного спирта, используемого в способе согласно второму аспекту данного изобретения. Это обусловлено тем, что при таком осуществлении способа согласно первому аспекту данного изобретения побочные реакции, протекающие с получением в качестве продуктов смолы и пека, могут подавляться, и срок службы катализатора может значительно увеличиваться, позволяя получать ε-капролактам стабильно с высоким выходом в течение длительного времени.
Кроме того, если это желательно, “другие органические соединения”, как описано выше в связи с методом согласно второму аспекту данного изобретения, могут присутствовать в реакционной системе в способе согласно первому аспекту данного изобретения.
Далее описываются способ получения ε-капролактама данного изобретения контактированием циклогексаноноксима с твердым кислотным катализатором в газовой фазе и способ регенерации катализатора, используемого в реакции. Это описание применимо к способам согласно первому и второму аспектам данного изобретения.
Реакция осуществляется следующим образом. Твердый кислотный катализатор вводится в реактор, такой как реактор с неподвижным слоем катализатора, реактор с движущимся слоем или реактор с псевдоожиженным слоем. Циклогексаноноксим (который предварительно выпарен в испарителе или который выпаривается после введения в реактор) контактирует с твердым кислотным катализатором в реакторе в газовой фазе в подходящих условиях реакции.
Наиболее предпочтительно осуществлять реакцию с использованием твердого кислотного катализатора, как описано в способе согласно первому аспекту данного изобретения, в присутствии производного многоатомного спирта, как описано в способе согласно второму аспекту данного изобретения.
Далее описываются температура реакции и давление реакции. Термин “температура реакции” означает температуру, при которой осуществляется реакция перегруппировки циклогексаноноксима данного изобретения с получения ε-капролактама. Термин “давление” реакции означает давление, при котором проводится данная реакция.
Температура реакции предпочтительно составляет от 200 до 500°С, более предпочтительно от 300 до 450°С, еще более предпочтительно от 330 до 380°С. Когда температура реакции ниже 200°С, скорость реакции имеет тенденцию быть неудовлетворительной. Когда температура реакции выше 500°С, вероятно разложение циклогексаноноксима.
Давление реакции предпочтительно составляет от 0,01 до 1 МПа, более предпочтительно от 0,03 до 0,5 МПа, еще более предпочтительно от 0,06 до 0,3 МПа.
Часовая объемно-массовая скорость подачи циклогексаноноксима (WHSV) составляет от 0,01 до 100 час-1, более предпочтительно от 0,1 до 10 час-1.
Часовую объемно-массовую скорость подачи циклогексаноноксима (WHSV) представляет формула WHSV=F/C (час-1), где F - количество подаваемого циклогексаноноксима (кг/час), С - масса катализатора (кг).
Реакция может осуществляться без применения несущего газа; однако предпочтительно применять, например, газообразный азот, азообразный аргон, газообразный диоксид углерода и газообразный водород. Что касается количества несущего газа, то конкретных ограничений здесь нет. Однако предпочтительно, чтобы несущий газ присутствовал в количестве, при котором концентрация циклогексаноноксима в газообразном потоке исходного сырья, включающем несущий газ, составляла примерно от 1 до 20 об.%.
При дезактивации катализатора в процессе реакции перегруппировки он подвергается активации (регенерации) для регенерации каталитической активности.
Активация осуществляется способом, при котором сам воздух или газ, полученный разбавлением воздуха инертным газом (таким как газообразный азот, газообразный диоксид углерода или газообразный аргон; предпочтительно газообразный азот) до желаемой концентрации кислорода (примерно, от 1 до 10 об.%), или инертный газ пропускают через аппарат для прокаливания катализатора, поддерживаемый при определенной температуре и в течение времени, достаточного для того, чтобы смола, пек или углеродистые соединения, которые скопились на поверхности катализатора, сгорели, разложились и улетучились. В данном случае предпочтительно, чтобы температура составляла от 400 до 700°С. Время выдерживания при температуре регенерации обычно составляет от 0,5 до 48 часов.
Реакция, используемая в данном изобретении (целевая реакция), может проводиться с помощью различных реакторов для каталитических процессов, таких как реактор с неподвижным слоем катализатора, реактор с движущимся слоем катализатора и реактор с псевдоожиженным слоем катализатора. Углеродистые соединения, скопившиеся на поверхности катализатора в процессе целевой реакции, значительно дезактивируют катализатор. Следовательно, предпочтительно проводить реакцию способом, при котором может периодически осуществляться указанная операция регенерации для активации изношенного катализатора. Например, в процессе с реактором с неподвижным слоем катализатора предпочтительно, чтобы реакция осуществлялась с использованием двух или более реакторов так, чтобы целевая реакция и регенерация катализатора попеременно осуществлялись в каждом реакторе и чтобы реакция перегруппировки и регенерация катализатора проводились одновременно в отдельных реакторах.
С другой стороны, в процессе с использованием реактора с псевдоожиженным слоем катализатора предпочтительно, чтобы часть катализатора, используемого в реакции перегруппировки, непрерывно или периодически извлекалась из реактора для регенерации, после чего извлеченный катализатор подвергался регенерации с использованием аппарата регенерации, в котором катализатор нагревается и выдерживается при подходящей температуре, как описано выше, в атмосфере кислородсодержащего газа или инертного газа, чтобы вызвать сгорание, разложение или испарение углеродистых веществ для удаления углеродистых веществ и регенерации катализатора. Предпочтительно также, чтобы регенерированный катализатор непрерывно или периодически возвращался на повторную переработку в реактор. В частности, поскольку реакция перегруппировки, которая приводит к превращению циклогексаноноксима в ε-капролактам, является экзотермической реакцией, наиболее предпочтителен процесс с использованием реактора с псевдоожиженным слоем катализатора, в котором относительно легко осуществляется контроль температуры реакции.
Выделение и очистка полученного ε-капролактама из газообразной реакционной смеси могут осуществляться охлаждением/конденсацией газообразной реакционной смеси и выделением полученного конденсата с последующей экстракцией, отгонкой или кристаллизацией.
Что касается производного многоатомного спирта, который используется в данной реакции, производное многоатомного спирта может выделяться из газообразной реакционной смеси и снова использоваться.
Наилучший способ воплощения данного изобретения
Данное изобретение далее будет описано более подробно со ссылкой на приведенные ниже ссылочные примеры, примеры и сравнительные примеры, которые не следует рассматривать как ограничение области данного изобретения.
В приведенных ниже ссылочных примерах, примерах и сравнительных примерах определение различных характеристик проводится следующими методами.
(1) Получение рентгенограммы мелкозернистого цеолита
Мелкозернистый цеолит анализируется с помощью аппаратуры, описанной ниже, и на основе полученной рентгенограммы определяется кристаллическая структура цеолита.
Определение кристаллической структуры цеолита проводится в соответствии с информацией, представленной в публикации R.V. Ballmoos and J.B. Higgins, "Collection of simulated XRD powder patterns for zeolites", ("ZEOLITES" vol.10, №5, June 1990).
Аппарат: рентгеновская установка (RAD-IIIA, изготовлена и предоставлена Rigaku Corporation, Japan).
Условия измерения: СuКα-радиация.
Напряжение рентгеновской трубки 40 кВ.
Ток рентгеновской трубки 30 мА.
Угол измерения 2θ: 5-45°.
(2) Определение атомного отношения Si/Al и атомного отношения Si/Ti цеолита.
К 50 г 5 н. водного раствора NaOH добавляют 0,2 г цеолита. Полученную смесь переносят в тефлоновый микробаллон, и микробаллон герметично закрывают. Микробаллон нагревают на масляной бане и выдерживают при температуре 150°С в течение 12-70 часов для полного растворения цеолита в растворе NaOH. Полученный раствор, содержащий растворенный в нем цеолит, разбавляют ионнообменной водой. (Степень разведения, подходящую для описанного далее измерения с использованием индуктивно соединенного плазменного эмиссионного спектрометра (далее называемого ICP-спектрометром), изменяется в зависимости от состава цеолита и т.п. Таким образом, полученный раствор, содержащий растворенный в нем цеолит, разбавляют примерно в 5-100 раз, как это подходит для описанного далее измерения с помощью ICP). Концентрации кремния, алюминия и титана определяют с помощью ICP, используя описанный ниже ICP-спектрометр в указанных ниже условиях, и атомное отношение Si/Al и атомное отношение Si/Ti цеолита вычисляют из концентраций кремния, алюминия и титана.
ICP-спектрометр и условия ICP-спектрометрии являются следующими:
- ICP-спектрометр: JOBIN YVON (JY138 ULTRACE) (изготовлен и поставлен Rigaku Corporatiom, Япония).
- Условия ICP
Длина волны для измерения концентрации кремния: 251,60 нм;
Длина волны для измерения концентрации алюминия: 396,152 нм;
Длина волны для измерения концентрации титана: 334,94 нм;
Мощность плазмы: 1,0 кВт;
Скорость потока распыляющего газа: 0,28 л/мин;
Скорость потока газа в капсуле: 0,3-0,8 л/мин;
Скорость потока охлаждающего газа: 13 л/мин.
(3) Анализ продукта реакции газовой хроматографией
Реакционную смесь, содержащую полученный ε-капролактам, анализируют газовой хроматографией (ГХ) с использованием описанного ниже аппарата в описанных ниже условиях, и исходя из состава реакционной смеси оцениваются результаты реакции (то есть вычисляются степень конверсии циклогексаноноксима и селективность получения ε-капролактама).
Аппарат ГХ и условия проведения ГХ являются следующими:
Аппарат: Газовый хроматограф модели GC-17A, изготовлен и поставлен Shimadzu Corporation, Япония.
Колонка: Капиллярная колонка HR-20M (внутренний диаметр: 0,25 нм, длина: 50 м, толщина мембраны: 0,25 мкм).
Предобработка образца: Перед проведением анализа образца методом газовой хроматографии (реакционная смесь) 3 г растворителя, используемого в реакции получения ε-капролактама, и 0,15 г этилбензола в качестве внутреннего стандарта добавляют к 1 г образца (растворитель и образец аккуратно взвешивают).
Количество образца, вводимого в колонку: 1 мкл.
Профиль повышения температуры: Температуру выдерживают на уровне 100°С в течение 5 минут, повышают до 240° со скоростью 10°С/мин и затем поддерживают на уровне 240°С в течение 42 минут.
Отношение деления потока: 100/1.
Скорость потока несущего газа (азота): 200 мл/мин.
Пламенно-ионизационный детектор (flame ionization detector - FID): Давление подаваемого воздуха 50 кПа (примерно 500 мл/мин), давление подаваемого водорода 60 кПа (примерно 50 мл/мин).
(4) Определение показателя истирания формованного катализатора
Показатель истирания обычно используется для оценки механической прочности мелкозернистых катализаторов, таких как катализаторы, используемые в реакциях в псевдоожиженном слое катализатора. Показатель истирания определяют, подвергая мелкозернистый катализатор испытанию на истирание в специфических условиях, и данный показатель определяется как степень увеличения количества частиц с размером 10 мкм или менее в полученном катализаторе. Меньший показатель истирания соответствует более высокому сопротивлению истирания.
Далее описан конкретный пример определения показателя истирания.
25 г мелкозернистого катализатора обрабатывают для получения содержания воды 10 мас.%, и полученный катализатор загружают в цилиндрический аппарат для измерения показателя истирания (включающей нижнюю цилиндрическую часть длиной 27,5 дюймов с внутренним диаметром 1,5 дюйма, верхнюю цилиндрическую часть длинной 22 дюйма с внутренним диаметром 5 дюймов и емкость для сбора тонкоизмельченных частиц, которая размещена на верхней цилиндрической части). Затем в аппарат через 3 отверстия (каждое диаметром 1/64 дюйма), расположенных на дне аппарата, со скоростью 425 л/час в течение 5 часов подается увлажненный воздух для циркуляции катализатора в аппарате с целью его истирания. Перед циркуляцией катализатора в аппарате определения показания истирания и после нее катализатор исследуется с помощью оптического аппарата измерения гранулометрического состава, и определяется разность содержания тонкоизмельченных частиц (размером 10 мкм или менее) в катализаторе до циркуляции катализатора и после нее.
Показатель истирания (I.D.) вычисляется с помощью формулы
I.D.=(A-B)/(C-B)×100,
где А: масса (г) частиц тонкого помола (каждая размером 19 мкм или менее) в катализаторе после 5 часов циркуляции,
В: масса (г) частиц тонкого помола (каждая размером 19 мкм или менее) в катализаторе до 5 часов циркуляции и
С: масса (г) катализатора, загруженного в аппарат для определения показателя истирания.
В приведенных далее ссылочных примерах 1-19 получают катализаторы, используемые в примерах и сравнительных примерах.
Составы и характеристики каждого из кристаллических глинистых минералов, используемых в ссылочных примерах, указаны ниже.
(1) Каолинит
Водный каолин "ASP072" (торговое название; произведен и поставлен Engelhard Corporation, США). Содержание каолинита: примерно 100 мас.%.
Состав, мас.%:
Аl2O3 38,5
SiO2 45,4
TiO2 1,6
Прочие металлсодержащие компоненты 0,9
Кристаллизационная вода 13,6
Средний диаметр частиц: 0,3 мкм.
(2) Каолинит
Кальцинированный каолинит "SATINTONE SP33" (торговое название, произведен и поставлен Engelgard Corporation, США).
Содержание каолинита: примерно 100 мас.%.
Состав, мас.%:
Аl2O3 44,3
SiO2 52,2
ТiO2 1,8
Прочие металлсодержащие компоненты 1,2
Кристаллизационная вода 0,5
Средний диаметр частиц: 1,4 мкм.
(3) Пирофиллит
"5М" (торговое название; произведен и поставлен Tsuchiya Kaolin Co., Ltd., Japan).
Содержание пирофиллита: примерно 60 мас.% (остальное составляет, главным образом, аморфный диоксид кремния).
Состав, мас.%:
Аl2O3 17,0
SiO2 78,0
Прочие металлсодержащие компоненты 0,4
Кристаллизационная вода 4,6
Средний диаметр частиц: 0,3 мкм.
(4) Каолинит
Водный каолин "ASP600" (торговое название; произведен и поставлен Engelgard Corporation, США). Содержание каолинита: примерно 100 мас.%.
Состав, мас.%:
Аl2O3 38,5
SiO2 45,4
TiO2 1,6
Прочие металлсодержащие компоненты 0,9
Кристаллизационная вода 13,6
Средний диаметр частиц: 0,6 мкм.
(5) Тальк
"MICRO АСЕ К-1" (торговое название, произведен и поставлен Nippon Talc Co., Ltd., Япония). Содержание талька: примерно 100 мас.%.
Состав, мас.%:
МgО 30,7
SiO2 60,1
Прочие металлсодержащие компоненты 1,6
Кристаллизационная вода 5,3
Ссылочный пример 1: получение катализатора (А)
К 130 г тетраэтилортосиликата добавляют 278 г этанола и затем 291 г 10 мас.% водного раствора гидроксида тетрапропиламмония. Полученный раствор перемешивают с использованием гомогенизатора со скоростью вращения 5000 об/мин в течение 30 минут, затем переносят в автоклав объемом 1 л и осуществляют гидротермический синтез при 105-110°С в течение 150 часов при перемешивании со скоростью вращения 500 об/мин с получением суспензии. Полученную суспензию фильтруют через фильтр и полученный остаток промывают до нейтральной реакции, затем промытый осадок сушат при 120°С в течение 12 часов, получая таким образом кристаллическое вещество белого цвета. Полученное кристаллическое вещество кальцинируют в воздухе в электропечи при температуре в интервале от 500 до 550°С в течение 6 часов, и полученный кальцинированный продукт (цеолит) подвергают рентгенографическому анализу, как описано выше.
Рентгенографический анализ показывает, что кальцинированный продукт проявляет характеристические пики при 10,99, 9,87, 3,83, 3,79, 3,73 и 3,69 в единицах 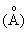 межплоскостных расстояний.
межплоскостных расстояний.
С другой стороны, на странице 444S статьи R.V. Ballmoos, J.B. Higgins, “Collection of simulated XRD powder patterns for zeolites” (“ZEOLITES”, vol.10, №5, June 1990) показано, что на рентгенограмме MFI цеолита имеют место характеристические пики при 11,1±0,2, 10,0±0,2, 3,85±0,07, 3,80±0,05, 3,73±0,05, 3,71±0,05 в значениях (d), то есть межплоскостных расстояниях (d).
Поскольку указанные результаты анализа полученного цеолита хорошо согласуются с известными значениями (d) MFI цеолита, полученный цеолит идентифицируется как MFI цеолит.
Атомное отношение Si/Al полученного цеолита определяют в соответствии с описанным выше методом. Установлено, что содержание Аl в цеолите равно 10 м.д. или менее, и атомное отношение равно 45000 или более (таким образом, определяют, что цеолит по существу не содержит Аl). Поэтому цеолит идентифицируется как силикалит.
Цеолит добавляют к 7,5 мас.% водному раствору нитрата аммония, получая таким образом суспензию с содержанием твердых веществ 10 мас.%, полученную суспензию подвергают ионному обмену при 80°С в течение 2 часов. После этого суспензию фильтруют. Затем цикл ионного обмена и фильтрования повторяют дважды, осадок промывают водой, сушат при 120°С в течение 12 часов и кальцинируют при 500°С в течение 4 часов, получая таким образом силикалит Н-типа. (Термин “Н-типа”, используемый в данном описании, означает, что цеолит подвергался ионному обмену в водном растворе нитрата аммония и затем кальцинированию с получением таким образом цеолита, несущего ионы водорода. Далее в описании термин “силикалит Н-типа” означает цеолит, полученный синтетическим способом, как описано в данном ссылочном примере 1).
К 33,3 г золя диоксида кремния "SNOWTEX® STN30" (торговое название; произведен и поставлен Nissan Chemical Industries Ltd., Япония; содержание SiO2 в золе диоксида кремния составляет 30 мас.%, содержание натрия равно 700 м.д. (масс.) и значение рН равно 10, то есть сильно основный) добавляют 137 г очищенной воды, 20 г силикалита Н-типа и 10 г каолинита "ASP072" (торговое название), и полученную смесь тщательно перемешивают с получением суспензии. Полученную суспензию распыляют с использованием небольшого аэрозольного устройства на горячую пластину, предварительно нагретую до 200°С, получая таким образом высушенный предшественник мелкозернистого катализатора. Полученный таким образом мелкозернистый порошок кальцинируют при 800°С в течение 6 часов, получая таким образом катализатор (А). Высушенный мелкозернистый предшественник катализатора имеет следующий состав: 50 мас.% силикалита Н-типа, 25 мас.% SiO2 и 25 мас.% каолинита.
Для удобства состав высушенного предшественника катализатора перед кальцинированием также называют “состав катализатора (А)”. Это применимо ко всем следующим ссылочным примерам 2-19.
Ссылочный пример 2: получение катализатора (В) (катализатор, не содержащий кристаллических глинистых минералов)
Катализатор (В) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 66,7 г золя диоксида кремния "SNOWTEX® STN30" (торговое название; произведен и поставлен Nissan Chemical Industries Ltd., Япония) добавляют 113 г очищенной воды и 20 г силикалита Н-типа. Полученный таким образом катализатор (В) имеет следующий состав: 50 мас.% силикалита Н-типа и 50 мас.% SiO2.
Ссылочный пример 3: получение катализатора (С)
Катализатор (С) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что используют золь диоксида кремния (называемый далее “золь диоксида кремния высокой чистоты) с очень низким содержанием загрязняющих металлов (золь диоксида кремния высокой чистоты получают в соответствии с методом, описанным в рабочих примерах публикации Unexamined Japanese Patent Application Laid-Open Specification № Hei 4-231319), в котором содержание SiO2 равно 30 мас.%, содержание натрия равно 120 м.д. (масс.) или менее и значение рН равно 10 (то есть золь диоксида кремния является сильно основным). Полученный таким образом катализатор (С) имеет следующий состав: 50 мас.% силикалита Н-типа, 25 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 4: получение катализатора (D) (катализатор, не содержащий кристаллических глинистых минералов)
Катализатор (D) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 66,7 г золя диоксида кремния высокой чистоты добавляют 113 г очищенной воды и 20 г силикалита Н-типа. Полученный таким образом катализатор (D) имеет следующий состав: 50 мас.% силикалита Н-типа и 50 мас.% SiO2.
Ссылочный пример 5: получение катализатора (Е)
Катализатор (Е) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 25,0 г золя диоксида кремния высокой чистоты добавляют 23,8 г золя оксида алюминия "Alumina sol 200" (торговое название, произведен и поставлен Nissan Chemical Industries Ltd., Япония; золь оксида алюминия содержит 10,5 мас.% Al2O2 и имеет значение рН от 4 до 6, то есть является слабо кислотным), 121 г очищенной золы, 20 г силикалита Н-типа и 10 г каолинита "ASP072" (торговое название), и температура кальцинирования равна 700°С. Полученный таким образом катализатор (Е) имеет следующий состав: 50 мас.% силикалита Н-типа, 19 мас.% SiO2, 6 мас.% Al2O3 и 25 мас.% каолинита.
Ссылочный пример 6: получение катализатора (F) (катализатор, не содержащий кристаллических глинистых минералов)
Катализатор (F) получают по существу в соответствии с методикой ссылочного примера 5, с тем отличием, что к 50,0 г золя диоксида кремния высокой чистоты добавляют 47,6 г золя оксида алюминия "Alumina sol 200" (торговое название, произведен и поставлен Nissan Chemical Industries Ltd., Япония), 82 г очищенной воды, 20 г силикалита Н-типа. Полученный таким образом катализатор (F) имеет следующий состав: 50 мас.% силикалита Н-типа, 38 мас.% SiO2, 12 мас.% Аl2O3.
Ссылочный пример 7: получение катализатора (G)
Катализатор (G) получают по существу в соответствии с методикой ссылочного примера 5, с тем отличием, что к 95,2 г золя оксида алюминия "Alumina sol 200" (торговое название, произведен и поставлен Nissan Chemical Industries Ltd., Япония) добавляют 275 г очищенной воды, 20 г силикалита Н-типа и 10 г каолинита "ASP072" (торговое название). Полученный таким образом катализатор (G) имеет следующий состав: 50 мас.% силикалита Н-типа, 25 мас.% Al2O3 и 25 мас.% каолинита.
Ссылочный пример 8: получение катализатора (Н) (катализатор, не содержащий кристаллических глинистых минералов)
Катализатор (Н) получают по существу в соответствии с методикой ссылочного примера 5, с тем отличием, что к 190 г золя оксида алюминия "Alumina sol 200" (торговое название, произведен и поставлен Nissan Chemical Industries Ltd., Япония) добавляют 190 г очищенной воды и 20 г силикалита Н-типа. Полученный таким образом катализатор (Н) имеет следующий состав: 50 мас.% силикалита Н-типа, 50 мас.% Al2O3.
Ссылочный пример 9: получение катализатора (I)
В ссылочном примере 9 получают катализатор (I) данного изобретения, и в данном катализаторе содержание кристаллического глинистого минерала отлично от содержания в катализаторах ссылочных примеров 1, 3, 5 и 7. В частности, катализатор (I) получают следующим образом.
Катализатор (I) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 53,3 г золя диоксида кремния высокой чистоты добавляют 123 г очищенной воды, 20 г силикалита Н-типа и 4 г каолинита "ASP072" (торговое название). Полученный таким образом катализатор (I) имеет следующий состав: 50 мас.% силикалита Н-типа, 40 мас.% SiO2 и 10 мас.% каолинита.
Ссылочный пример 10: Получение катализатора (J)
Катализатор (J) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 33,3 г золя диоксида кремния высокой чистоты добавляют 137 г очищенной воды, 20 г силикалита Н-типа и 10 г талька "MICRO АСЕ К-1" (торговое название, произведен и поставлен Nippon Talc Co., Ltd., Япония). Полученный таким образом катализатор (J) имеет следующий состав: 50 мас.% силикалита Н-типа, 25 мас.% SiO2 и 25 мас.% талька.
Ссылочный пример 11: получение катализатора (К)
Катализатор (К) получают по существу в соответствии с методикой ссылочного примера 1, с тем отличием, что к 33,3 г золя диоксида кремния высокой чистоты добавляют 137 г очищенной воды, 20 г силикалита Н-типа и 10 г монтмориллонита "К-10" (торговое название; произведен и поставлен Sigma-Aldrich Corporation, США). Полученный таким образом катализатор (К) имеет следующий состав: 50 мас.% силикалита Н-типа, 25 мас.% SiO2 и 25 мас.% монтмориллонита.
Ссылочный пример 12: получение катализатора (L)
10 г каолинита "ASP072" (торговое название) и 18 г силикалита Н-типа добавляют к водному раствору (сильно основному раствору), содержащему 47,4 г силиката натрия (сорт: JIS Special №3) (произведен и поставлен Fuji Chemical Co., Ltd., Япония; содержание SiO2 25,2 мас.%, содержание Аl2O3 0,01 мас.%, мольное отношение SiO2/Na2O 3,3), 71 г очищенной воды и 6,0 г серной кислоты. Полученный раствор тщательно перемешивают с использованием гомогенизатора со скоростью вращение 5000 об/мин, получая таким образом суспензию. Полученную суспензию распыляют, используя небольшое аэрозольное устройство, на пластину, предварительно нагретую до 200°С с получением таким образом высушенного мелкозернистого предшественника катализатора. Полученный таким образом высушенный мелкозернистый предшественник катализатора добавляют к 1 М водному раствору азотной кислоты с получением суспензии с содержанием твердых вещества 10 мас.%, полученную суспензию выдерживают при 25°С в течение 1 часа, затем промывают водой, сушат и кальцинируют при 600°С в течение 5 часов, получая катализатор (L). Полученный катализатор (L) имеет следующий состав: 45 мас.% силикалита Н-типа, 30 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 13: получение катализатора (М)
Катализатор (М) получают по существу в соответствии с методикой ссылочного примера 12, с тем отличием, что используют 10 г каолинита "SATINTONE SP33" (торговое название). Полученный таким образом катализатор (М) имеет следующий состав: 45 мас.% силикалита Н-типа, 30 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 14: получение катализатора (N)
Катализатор (N) получают по существу в соответствии с методикой ссылочного примера 12, с тем отличием, что используют 10 г пирофиллита "5М" (торговое название). Полученный таким образом катализатор (N) имеет следующий состав: 45 мас.% силикалита Н-типа, 40 мас.% SiO2 и 15 мас.% пирофиллита.
Ссылочный пример 15: получение катализатора (О)
Катализатор (О) получают по существу в соответствии с методикой ссылочного примера 12, с тем отличием, что используют 10 г каолинита "ASP600" (торговое название). Полученный таким образом катализатор (О) имеет следующий состав: 45 мас.% силикалита Н-типа, 30 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 16: получение катализатора (Р)
К 130 г тетраэтилортосиликата добавляют 78 г этанола и затем водный раствор, содержащий 65 г очищенной воды и 0,0886 г изопропоксида титана, и 291 г 10 мас.% водного раствора гидроксида тетрапропиламмония. Полученный водный раствор используют в гидротермическом синтезе и затем обрабатывают по существу в соответствии с методикой ссылочного примера 1, получая таким образом белое кристаллическое вещество. Полученное кристаллическое вещество кальцинируют по существу в условиях, описанных в ссылочном примере 1, и полученный кальцинированный продукт (цеолит) подвергают рентгенографическому анализу, как описано выше.
Рентгенографический анализ показывает, что кальцинированный продукт проявляет характеристические пики при 10,99, 9,87, 3,83, 3,79, 3,73 и 3,69 в единицах  межплоскостных расстояний (d).
межплоскостных расстояний (d).
Поскольку приведенные выше результаты анализа полученного цеолита хорошо согласуются с известными значениями (d) MFI цеолита, которые описаны в указанной выше статье, полученный цеолит идентифицируется как цеолит MFI.
Атомное отношение Si/Ti полученного цеолита определяют в соответствии с описанным выше методом. Устанавливают, что цеолит имеет атомное отношение Si/Ti, равное 1900. Следовательно, цеолит идентифицируют как титаносиликат. Далее цеолит подвергают ионному обмену по существу в условиях, описанных в ссылочном примере 1, получая таким образом титаносиликат Н-типа.
Затем катализатор (Р) получают по существу в соответствии со ссылочным примером 3, с тем отличием, что вместо силикалита Н-типа полученный выше титаносиликат Н-типа смешивают с каолинитом "АSР072" (торговое название) и золем диоксида кремния высокой чистоты. Полученный таким образом катализатор (Р) имеет следующий состав: 50 мас.% титаносиликата Н-типа, 25 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 17: получение катализатора (Q)
К 130 г тетраэтилортосиликата добавляют 278 г этанола, водный раствор, полученный растворением 0,197 г 14-18-гидрата сульфата алюминия в 13 г очищенной воды, и 291 г 10 мас.% водного раствора гидроксида тетрапропиламмония. Полученный водный раствор подвергают гидротермическому синтезу и последующей обработке по существу способом, описанным в ссылочном примере 1, с получением белого кристаллического вещества. Полученное кристаллическое вещество кальцинируют по существу в соответствии с методикой ссылочного примера 1, и полученный кальцинированный продукт подвергают рентгенографическому анализу, как описано выше.
Рентгенографический анализ показывает, что кальцинированный продукт проявляет характеристические пики при 10,99, 9,87, 3,83, 3,79, 3,73 и 3,69 в единицах 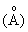 межплоскостных расстояний (d).
межплоскостных расстояний (d).
Поскольку приведенные выше результаты анализа полученного цеолита хорошо согласуются с известными (d) значениями MFI цеолита, которые описаны в указанной выше статье, полученный цеолит идентифицируется как цеолит MFI.
Атомное отношение Si/Al полученного цеолита определяют в соответствии с описанным выше методом. Устанавливают, что цеолит имеет атомное отношение Si/Ti, равное 1000. Таким образом, цеолит идентифицируют как цеолит ZSM-5. Далее цеолит подвергают ионному обмену по существу в соответствии с методикой ссылочного примера 1, получая таким образом цеолит ZSM-5 Н-типа (с атомным отношение Si/Al, равным 1000).
Затем получают катализатор (Q) по существу в соответствии со ссылочным примером 3, с тем отличием, что вместо силикалита Н-типа используют полученный выше цеолит ZSM-5 (с атомным отношением Si/Al, равным 1000), который смешивают с каолинитом "ASP072" (торговое название) и золем диоксида кремния высокой чистоты, и температура кальцинирования составляет 600°С. Полученный таким образом катализатор (Q) имеет следующий состав: 50 мас.% цеолита ZSM-5 Н-типа (с атомным отношением Si/Al, равным 1000), 25 мас.% SiO2 и 25 мас.% каолинита.
Ссылочный пример 18: получение катализатора (R)
Цеолит β-типа "СР814В-50" (торговое название; произведен и поставлен Zeolyst International, США; атомное отношение Si/Al цеолита равно 25) подвергают рентгенографическому анализу. Анализ показывает, что цеолит проявляет характеристические пики при 11,58, 6,56, 4,16, 3,94 и 3,01, выраженные в 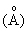 межплоскостных расстояний (d).
межплоскостных расстояний (d).
С другой стороны, в Патенте США №3308069 показано, что в рентгенограмме порошкового цеолита β-типа присутствуют характеристические пики при 11,4±0,2, 6,7±0,2, 4,25±0,1, 3,97±0,1 и 3,0±0,1 в единицах (d), т.е. межплоскостных расстояний (d).
Поскольку указанные выше известные значения (d) цеолита β-типа хорошо согласуются с результатами анализа цеолита, используемого в данном примере, используемый цеолит идентифицируют как цеолит β-типа.
Цеолит добавляют к 1 М водному раствору хлорида аммония, получая таким образом суспензию с содержанием твердых веществ 10 мас.%, и полученную суспензию подвергают ионному обмену при 70°С в течение 3 часов с последующим фильтрованием и промывкой водой, сушкой при 120°С в течение 12 часов и кальцинированием при 500°С в течение 4 часов, в результате получают цеолит Н-β-типа.
Затем катализатор (R) получают по существу в соответствии с методикой ссылочного примера 17, с тем отличием, что вместо цеолита ZSM-5 Н-типа (с атомным отношением Si/Al, равным 1000) полученный выше цеолит Н-β смешивают с каолинитом "ASP072" (торговое название) и золем диоксида кремния высокой чистоты. Полученный таким образом катализатор (R) имеет следующий состав: 50 мас.% цеолита Н-β-типа, 25 мас.% SiО2 и 25 мас.% каолинита.
Ссылочный пример 19: получение катализатора (S)
Ферриерит (цеолит) "СР914" (торговое название; произведен и поставлен Zeolyst International, США; атомное отношение Si/Al 28) подвергают рентгенографическому анализу. Анализ показывает, что цеолит проявляет характеристические пики при 9,38, 5,62, 3,97, 3,53 и 3,45 в единицах 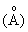 межплоскостных расстояний (d).
межплоскостных расстояний (d).
С другой стороны, в публикации R.V. Ballmoos and J.B. Higgins, "Collection of simulated XRD powder patterns for zeolites" ("ZEOLITES, vol.10, №5, June 1990) показано, что в порошковой рентгенограмме ферриерита наблюдаются характеристические пики при 9,58±0,2, 5,82±0,2, 4,00±0,1, 3,53±0,05 и 3,49±0,05 в единицах величин (d), то есть межплоскостных расстояний (d).
Поскольку указанные известные значения (d) ферриерита хорошо согласуются с результатами анализа цеолита, используемого в данном примере, цеолит идентифицируют как ферриерит.
Ферриерит подвергают ионному обмену по существу в соответствии с методикой ссылочного примера 18, получая таким образом ферриерит Н-типа.
Затем катализатор (S) получают по существу в соответствии с методикой ссылочного примера 17, с тем отличием, что вместо цеолита ZSM-5 Н-типа (с атомным отношением Si/Al, равным 1000) используют полученный выше ферриерит Н-типа, который смешивают с каолинитом "ASP072" (торговое название) и золем диоксида кремния высокой чистоты, получая таким образом катализатор (S). Полученный таким образом катализатор (S) имеет следующий состав: 50 мас.% ферриерита Н-типа, 25 мас.% SiO2 и 25 мас.% каолинита.
Пример 1
Катализатор (А) формуют прессованием и распыляют в частицы размером от 0,5 до 1,5 мм. 1,5 г частиц полученного катализатора помещают в реактор из кварцевого стекла (длинной 40 см и внутренним диаметром 12 мм) для применения в качестве реактора с неподвижным слоем катализатора, затем содержимое реактора нагревают до 400°С и продувают потоком азота в течение часа со скоростью при этой температуре в течение часа 200 Ncc/мин (где Ncc означает объем (куб.см. - сс), измеренный при нормальных температуре и давлении, то есть при 0°С и 1 атм). Затем в реакторе создают условия, при которых сохраняются атмосферное давление и температура 350°С, и пропускают поток азота со скоростью 70 Ncc/мин, и в реактор подают исходный раствор (который представляет собой 35,7 мас.% раствор циклогексаноноксима в метаноле) со скоростью 8,4 г/час. Часовая объемно-массовая скорость подачи циклогексаноноксима в реакторе равна 2,0 час-1 (4,0 час-1, как определено из расчета на массу цеолитового компонента в качестве наиболее активной породы). Полученную газообразную реакционную смесь пропускают через конденсатор с температурой примерно 3°С с получением жидкой реакционной смеси, и полученную жидкую реакционную смесь собирают в ловушке, охлажденной льдом или льдом сухого этанола. Полученную реакционную смесь анализируют газовой хроматографией, как описано выше. Результаты представлены в таблице 1.
Конверсию циклогексаноноксима и селективность получения ε-капролактама вычисляют следующим образом:
Конверсия (%) циклогексаноноксима: [(О-R)/О]×100,
Селективность (%) получения ε-капролактама: [L/(О-R)]×100,
где О - количество молей циклогексаноноксима, поступающий в реактор (моль/час);
R - количество молей непрореагировавшего циклогексаноноксима (моль/час);
L - количество молей полученного ε-капролактама (моль/час).
Сравнительный пример 1
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (В). Результаты представлены в таблице 1.
Пример 2
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (С). Результаты представлены в таблице 1. Сравнение результатов, полученных в примере 2, с результатами, полученными в примере 1, показывает, что при получении катализатора с использованием золя диоксида кремния высокой чистоты (как в примере 2) селективность получения целевого продукта улучшается.
Сравнительный пример 2
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (D). Результаты представлены в таблице 1.
Пример 3
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (Е). Результаты представлены в таблице 1.
Катализатор (Е), содержащий оксид алюминия в качестве неорганического оксида, проявляет меньшую селективность, чем катализатор (С), не содержащий оксида алюминия, но катализатор (Е) проявляет улучшенную механическую прочность формованного каталитического изделия по сравнению с катализатором (С). Для измерения механической прочности каждый катализатор отдельно подвергают формованию прямым прессованием при одном и том же давлении в цилиндрическую форму с диаметром 3 мм и длиной 4 мм. Твердость каждого катализатора, формованного в цилиндрическую форму, определяют с использованием аппарата определения твердости Кийя. То есть катализатор (Е) проявляет свойство, которое является преимущественным для коммерческого применения.
Сравнительный пример 3
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (F). Результаты представлены в таблице 1.
Пример 4
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (G). Результаты представлены в таблице 1.
По сравнению с катализатором (Е) катализатор (G) проявляет меньшую селективность, но катализатор (G), содержащий большее количество оксида алюминия, проявляет большую механическую прочность в формованном каталитической изделии по сравнению с прочностью катализатора (Е). (Твердость каждого катализатора изменяют по существу так же, как и в примере 3). То есть катализатор (G) проявляет свойство, преимущественное для коммерческого применения.
Сравнительный пример 4
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (Н). Результаты представлены в таблице 1.
Сравнительный пример 5
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (I). Результаты представлены в таблице 1.
Пример 6
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (J). Результаты представлены в таблице 2.
Пример 7
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (К). Результаты представлены в таблице 2.
Пример 8
Реакцию проводят по существу в условиях, описанных в примере 1, используя 1,50 г катализатора (L), с тем отличием, что 35,7 мас.% раствор циклогексаноноксима в этиленгликольмонометилэфире (исходный раствор) подают в реактор со скоростью 8,4 г/час и скорость подачи газообразного азота изменяют до 106 Ncc/мин. Часовая объемно-массовая скорость в реакторе составляет 2,0 час-1 (4,5 час-1 из расчета на цеолитовый компонент как наиболее активную породу). Результаты представлены в таблице 3.
Пример 9
Реакцию проводят по существу в условиях, описанных в примере 8, используя катализатор (М). Результаты представлены в таблице 3.
Пример 10
Реакцию проводят по существу в условиях, описанных в примере 8, используя катализатор (N). Результаты представлены в таблице 3.
Пример 11
Реакцию проводят по существу в условиях, описанных в примере 8, используя катализатор (О). Результаты представлены в таблице 3.
Пример 12
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (Р). Результаты представлены в таблице 4.
Пример 13
Реакцию проводят по существу в условиях, описанных в примере 1, используя катализатор (Q). Результаты представлены в таблице 4.
Пример 14
Реакцию проводят по существу в условиях, описанных в примере 1, используя 1,2 г катализатора (R), с тем отличием, что 9,0 мас.% раствор циклогексаноноксима в 1-гексаноле (исходный раствор) подают в реактор со скоростью потока 4,4 г/час и скорость потока газообразного азота изменяют до 20 Ncc/мин. Часовая объемно-массовая скорость подачи составляет 0,33 час-1 (0,66 час-1 из расчета на массу цеолитового компонента как наиболее активной породы). Результаты представлены в таблице 4.
Пример 15
Реакцию проводят по существу в условиях, описанных в примере 1, используя 1,5 г катализатора (S), с тем отличием, что 5,0 мас.% раствор циклогексаноноксима в 1-гексаноле (исходный раствор) подают в реактор со скоростью потока 5,0 г/час и скорость потока газообразного азота изменяют до 50 Ncc/мин. Часовая объемно-массовая скорость подачи в реакторе составляет 0,17 час-1 (0,33 час-1 из расчета на массу цеолитового компонента как наиболее активной породы). Результаты представлены в таблице 4.
Пример 16
Силикалит Н-типа, полученный в ссылочном примере 1, формуют прямым прессованием и затем распыляют. Из полученных частиц выбирают частицы размером от 0,5 до 1,5 мм для получения катализатора, включающего только цеолит. Реакцию проводят по существу в условиях, описанных в примере 1, используя полученный катализатор, но с тем отличием, что 35,7 мас.% раствор циклогексаноноксима в этиленгликольмонометилэфире (исходный раствор) подают в реактор со скоростью потока 4,2 г/час и скорость потока газообразного азота изменяют до 53 Ncc/мин. Часовая объемно-массовая скорость подачи составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Циклогексаноноксим в газах, подаваемых в реактор, составляет 7,0 об.%. Результаты представлены в таблице 5 и на Фигурах 1 и 2.
Сравнительный пример 5
Реакцию проводят по существу в условиях, описанных в примере 1, используя 0,375 г того же катализатора, что и в примере 16, но с тем отличием, что 35,7 мас.% раствор циклогексаноноксима в 1-пропаноле (исходный раствор) подают в реактор со скоростью потока 4,2 г/час и скорость потока газообразного азота изменяют до 49 Ncc/мин. Часовая объемно-массовая скорость подачи составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Концентрация циклогексаноноксима в газах, подаваемых в реактор, составляет 7,0 об.%. Результаты представлены в таблице 5 и на Фигурах 1 и 2.
Данный сравнительный пример проводят в таких же условиях, что и пример 16, для сравнения способа данного изобретения (где используют производное многоатомного спирта) со способом, использующим одноатомный спирт, содержащим такое число атомов углерода, что и производное многоатомного спирта, используемое в примере 16.
Пример 17
Реакцию проводят по существу в условиях, описанных в примере 1, используя 0,375 г катализатора примера 16, но с тем отличием, что 45,0 мас.% раствор циклогексаноноксима в этиленгликольмонометилэфире (исходный раствор) подают в реактор со скоростью потока 3,3 г/час и скорость потока газообразного азота изменяют до 57 Ncc/мин. Часовая объемно-массовая скорость в реакторе составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Концентрация циклогексаноноксима в газах, подаваемых в реактор, составляет 6,9 об.%. Результаты представлены в таблице 5 и на Фигурах 3 и 4.
Сравнительный пример 6
Реакцию проводят по существу в условиях, описанных в примере 1, используя 0,375 г катализатора примера 16, но с тем отличием, что 45,0 мас.% раствор циклогексаноноксима в смешанном растворителе, состоящем из спирта (27 мас.% метанола) и простого эфира (73 мас.% метил-трет-бутилового эфира) (мольное соотношение спирт/эфир 1:1) подают в реактор со скоростью потока 3,3 г/час и скорость потока газообразного азота изменяют до 54 Ncc/мин. Часовая объемно-массовая скорость в реакторе составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Концентрация циклогексаноноксима в газах, подаваемых в реактор, составляет 7,2 об.%. Результаты представлены в таблице 5 и на Фигурах 3 и 4.
Данный сравнительный пример проводят в тех же условиях, что и в примере 17, для сравнения способа данного изобретения (который использует производное многоатомного спирта) со способом, использующим (1:1 из расчета на мольное соотношение) смешанный растворитель, включающий одноатомный спирт и простой эфир.
Пример 18
Реакцию проводят по существу в условиях, описанных в примере 1, используя 0,375 г катализатора примера 16, но с тем отличием, что 20,0 мас.% раствор циклогексаноноксима в этиленгликольмоноэтилэфире (исходный раствор) подают в реактор со скоростью потока 7,5 г/час и скорость потока газообразного азота изменяют до 42 Ncc/мин. Часовая объемно-массовая скорость в реакторе составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Концентрация циклогексаноноксима в газах, подаваемых в реактор, составляет 6,9 об.%. Результаты представлены в таблице 5.
Сравнительный пример 7
Реакцию проводят по существу в условиях, описанных в примере 1, используя 0,375 г катализатора, используемого в примере 16, но с тем отличием, что 20,0 мас.% раствор циклогексаноноксима в 1-гексаноле (исходный раствор) подают в реактор со скоростью потока 7,5 г/час и скорость потока газообразного азота изменяют до 44 Ncc/мин. Часовая объемно-массовая скорость в реакторе составляет 4,0 час-1 из расчета на массу цеолитового компонента как наиболее активной породы. Концентрация циклогексаноноксима в газах, подаваемых в реактор, составляет 7,0 об.%. Результаты представлены в таблице 5.
Пример 19
Данный пример представляет пример процесса получения катализатора для применения в реакции в псевдоожиженном слое и пример реакции в псевдоожиженном слое.
К 2,370 г силиката натрия (сорт: JIS Special - №3) (произведен и поставлен Fuji Chemical Co., Ltd, Япония; содержание SiО2 25,2 мас.%, содержание Аl2O3 0,01 мас.%, мольное отношение SiO2/Na2O 3,3; сильно основный) добавляют 3,570 г очищенной воды и 300 г серной кислоты и полученную смесь тщательно перемешивают, охлаждая льдом, и к полученной смеси добавляют 500 г каолинита "ASP072" (торговое название) и 900 г силикалита Н-типа. Полученную смесь перемешивают в течение часа с использованием гомогенизатора со скоростью 5000 об/мин, получая таким образом суспензию. Полученную суспензию сушат с использованием распылительной сушилки ("Mobile Minor™; изготовлена и поставлена Niro Japan Co., Ltd., Япония), получая таким образом высушенный предшественник мелкозернистого катализатора. Сушку распылением проводят с использованием двухфазного сопла в условиях, когда температура на входе и на выходе из секции сушки аппарата равна 250°С и 100°С соответственно и скорость подачи суспензии равна 2 кг/час. Часть полученного таким образом высушенного предшественника мелкозернистого катализатора сушат при 110°С и затем добавляют к 1 М водному раствору азотной кислоты для получения суспензии с содержанием твердых веществ 10 мас.%. Полученную суспензию выдерживают при 25°С в течение 1 часа, затем промывают водой, сушат при 110°С в течение 12 часов и кальцинируют при 600°С в течение 5 часов, получая таким образом мелкозернистый катализатор (Т). Полученный катализатор имеет следующий состав: 45 мас.% силикалита Н-типа, 30 мас.% SiO2 и 25 мас.% каолинита. Мелкозернистый катализатор (Т) состоит, главным образом, из сферических частиц диаметром в интервале от 50 до 100 мкм, что видно в оптический микроскоп.
Показатель истирания (I.D.) катализатора (Т) равен 0,2 мас.%, как определено описанным выше способом. То есть катализатор, используемый в данном изобретении, проявляет предельно высокую стойкость к истиранию и обладает практичной морфологией и механической прочностью, подходящей для применения в реакции с псевдоожиженным слоем катализатора.
40,0 г полученного катализатора (Т) помещают в реактор из кварцевого стекла (длиной 60 см и внутренним диаметром 25 мм) для применения в качестве реактора с псевдоожиженным слоем катализатора, и затем катализатор переводят в псевдоожиженное состояние при 350°С в течение 1 часа с помощью потока газообразного азота, подаваемого со скоростью 393 Ncc/мин. Затем в реактор подают 35,7 мас.% раствора циклогексаноноксима в этиленгликольмонометилэфире (исходный раствор) со скоростью потока 30,8 г/час, что позволяет реактору работать под атмосферным давлением. Часовая объемно-массовая скорость подачи в реакторе равна 0,27 час-1 (0,61 час-1 из расчета на цеолитовый компонент как наиболее активную породу). Результаты реакции представлены в таблице 6.
Результаты, полученные в примере 19, показывают, что при использовании способа данного изобретения (в котором используется твердый кислотный катализатор специфического состава и/или реакция перегруппировки проводится в присутствии производного многоатомного спирта) в реакции, реализованной посредством процесса в псевдоожиженном слое, который является предпочтительным видом способа данного изобретения, катализатор проявляет прекрасные технологические характеристики, как и в случае, где способ данного изобретения используется в процессе в неподвижном слое катализатора (примеры 1-18).
Промышленная применимость
При получении ε-капролактама контактированием циклогексаноноксима с твердым кислотным катализатором в газовой фазе для осуществления реакции перегруппировки циклогексаноноксима при применении твердого кислотного катализатора, имеющего специфический состав, и/или при проведении реакции перегруппировки в присутствии специфического производного многоатомного спирта целевой ε-капролактам может быть получен из циклогексаноноксима с высокой селективностью и высоким выходом.
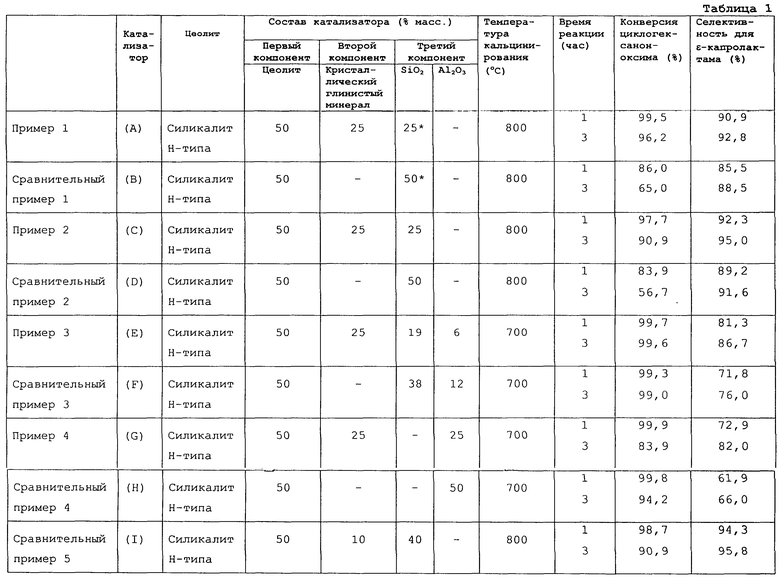

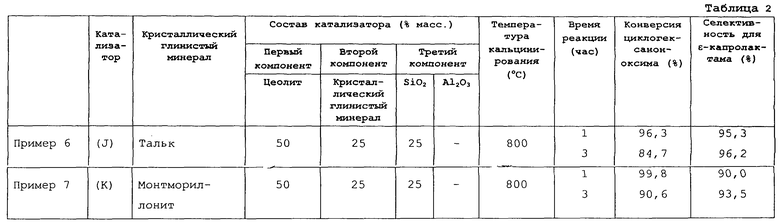

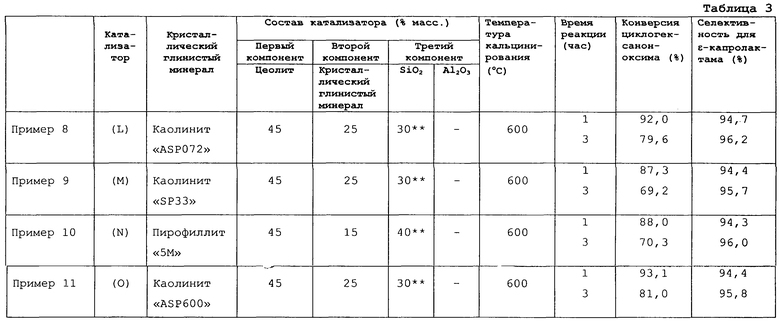

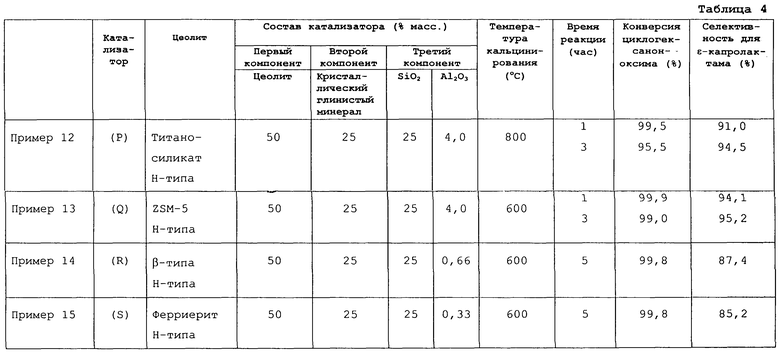

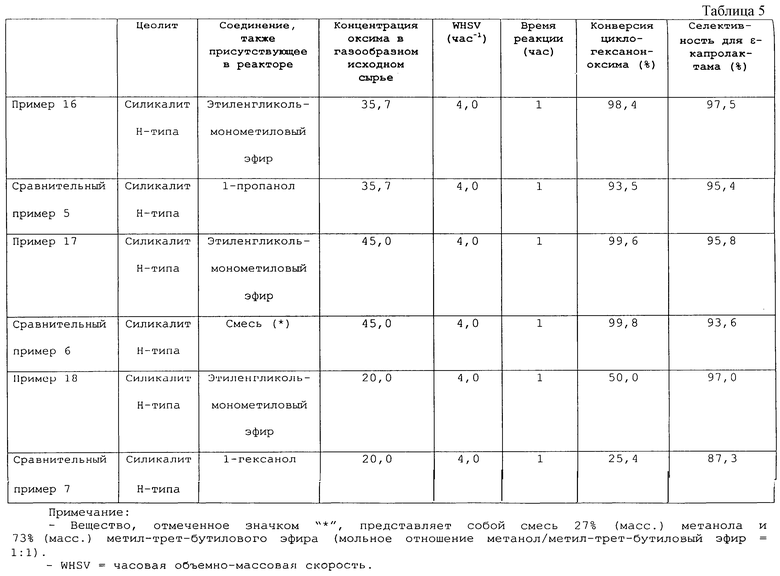
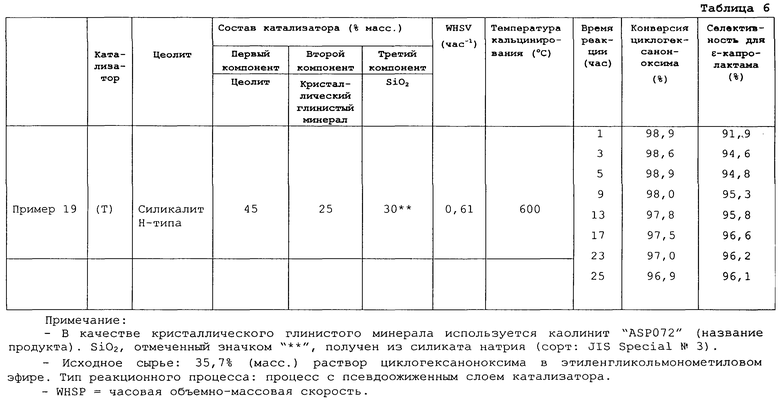
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНА С ОДНОВРЕМЕННЫМ ПОЛУЧЕНИЕМ ПРЕДШЕСТВЕННИКА НЕЙЛОНА | 2005 |
|
RU2359964C2 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ОКСИМОВ, СПОСОБ ГИДРОКСИЛИРОВАНИЯ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ И КАТАЛИТИЧЕСКИЙ СПОСОБ ОКИСЛЕНИЯ УГЛЕВОДОРОДОВ | 1993 |
|
RU2107545C1 |
| СФЕРИЧЕСКИЕ КАТАЛИЗАТОРЫ ДЛЯ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ В ЛЕГКИЕ ОЛЕФИНЫ | 2003 |
|
RU2307863C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ ГАЛОИДАЛКИЛОВ В ЭТИЛЕН И ПРОПИЛЕН | 2020 |
|
RU2815789C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕЛАМИНА | 2008 |
|
RU2535350C2 |
| ГАЗОФАЗНАЯ И ЖИДКОФАЗНАЯ КАТАЛИТИЧЕСКАЯ ПЕРЕГРУППИРОВКА БЕКМАНА ОКСИМОВ С ПОЛУЧЕНИЕМ ЛАКТАМОВ | 2012 |
|
RU2609779C2 |
| Способ очистки амидов, получаемых из оксимов при перегруппировке Бекмана | 1991 |
|
SU1834885A3 |
| КАТАЛИЗАТОР ДЛЯ КАТАЛИТИЧЕСКОГО КРЕКИНГА УГЛЕВОДОРОДА, КОТОРЫЙ ПРИМЕНЯЮТ ПРИ ПОЛУЧЕНИИ ЛЕГКОГО ОЛЕФИНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2494809C2 |
| СПОСОБ ОЧИСТКИ ЭПСИЛОН-КАПРОЛАКТАМА | 2007 |
|
RU2427570C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРОВ НА ЦЕОЛИТНОЙ ОСНОВЕ ДЛЯ ПРЕОБРАЗОВАНИЯ ОКСИГЕНАТОВ В НИЗШИЕ ОЛЕФИНЫ | 2011 |
|
RU2548916C2 |
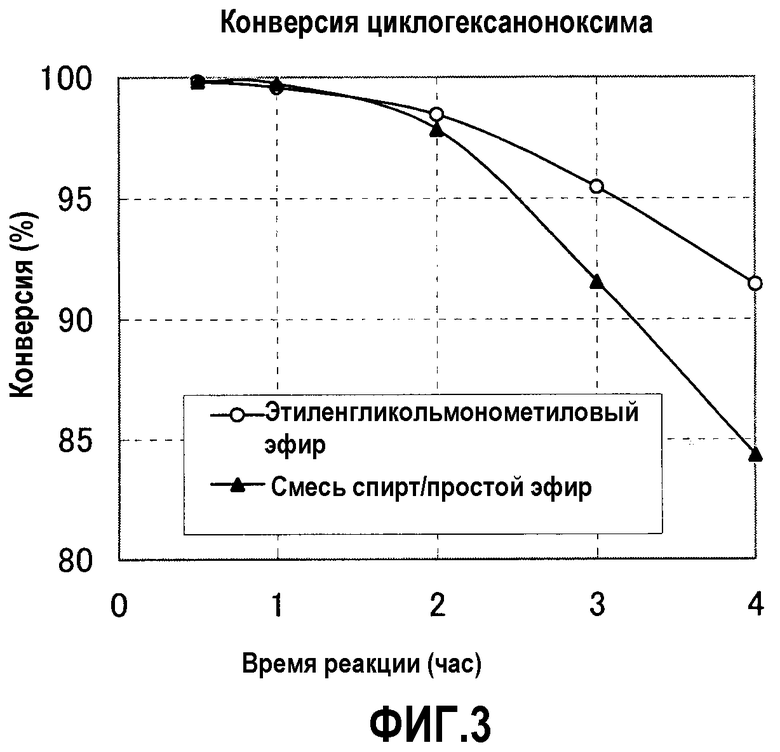
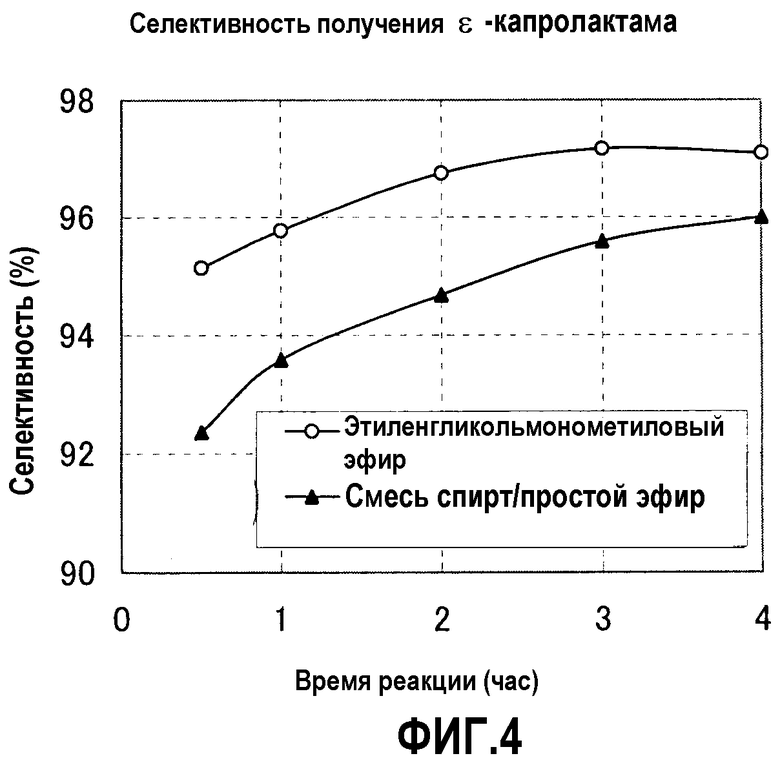
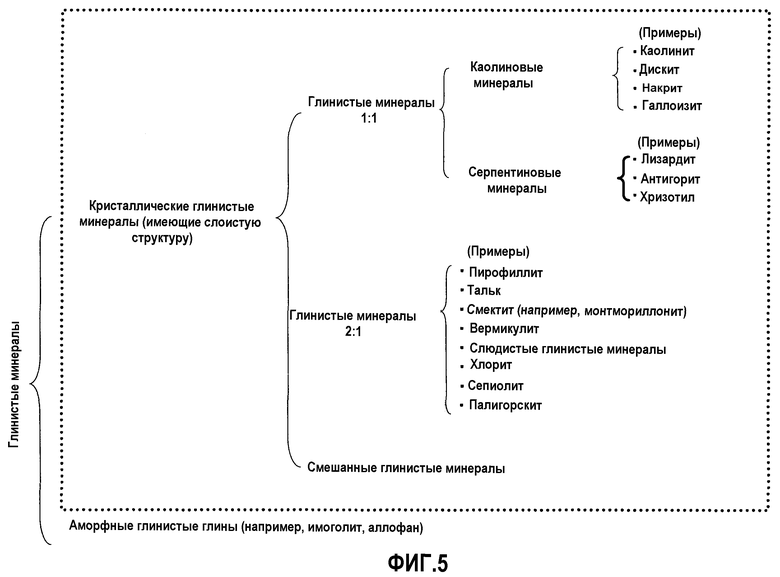
Изобретение относится к способу получения ε-капролактама перегруппировкой циклогексаноноксима с твердым кислотным катализатором в газовой фазе. Указанный кислотный катализатор получают кальцинированием высушенного предшественника катализатора. В первом аспекте способа предшественник катализатора включает цеолит, кристаллический глинистый минерал и, по меньшей мере, одно соединение, выбранное из группы, включающей неорганический оксид и соединение, которое образует неорганический оксид при кальцинировании. Неорганический оксид включает оксид, по меньшей мере, одного элемента, выбранного из группы, включающей элементы 4, 13 и 14 групп Периодической таблицы, причем неорганический оксид отличен от оксидов, содержащихся в кристаллической форме в цеолите и кристаллическом глинистом минерале. Во втором аспекте реакция перегруппировки проводится в присутствии производного многоатомного спирта формулы R1-O-R2-OH, где R1 представляет собой С1-С5 алкильную группу или фенильную группу и R2 представляет собой С2-С5 алкиленовую группу. Предпочтительно перегруппировка циклогексаноноксима проводится в псевдоожиженном слое при температуре реакции от 200 до 500° С, давлении от 0,01 до 1 МПа, при часовой объемно-массовой скорости циклогексаноноксима в интервале от 0,01 до 100 час-1. Часть катализатора, используемого в реакции перегруппировки, может непрерывно или периодически извлекаться из реактора, после чего извлеченный катализатор регенерируется в атмосфере кислородсодержащего газа или инертного газа и возвращается в указанный реактор. Технический результат - повышение выхода ε-κапролактама при высокой селективности реакции и увеличении срока службы катализатора, обладающего высокой механической прочностью. 2 с. и 17 з.п.ф-лы, 6 табл., 5 ил.
R1-O-R2-OH,
где R1 представляет собой C1-C5 алкильную группу или фенильную группу;
R2 представляет собой C2-C5 алкиленовую группу.
Приоритет по пунктам:
| US 5741904 A, 21.04.1998.JP 7-324070 A, 12.12.1995.RU 2053227 C1, 27.01.1996.RU 2035453 C1, 20.05.1995.JP 6-107627 A, 19.04.1994.EP 057136 A, 18.11.1993. |

