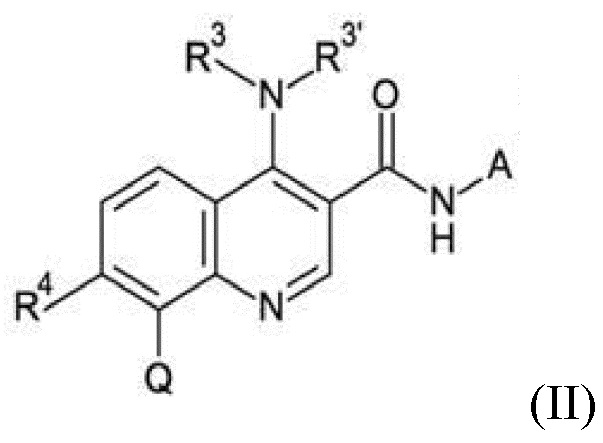
Настоящее изобретение относится к новому способу получения хинолиновых соединений общей формулы (II):
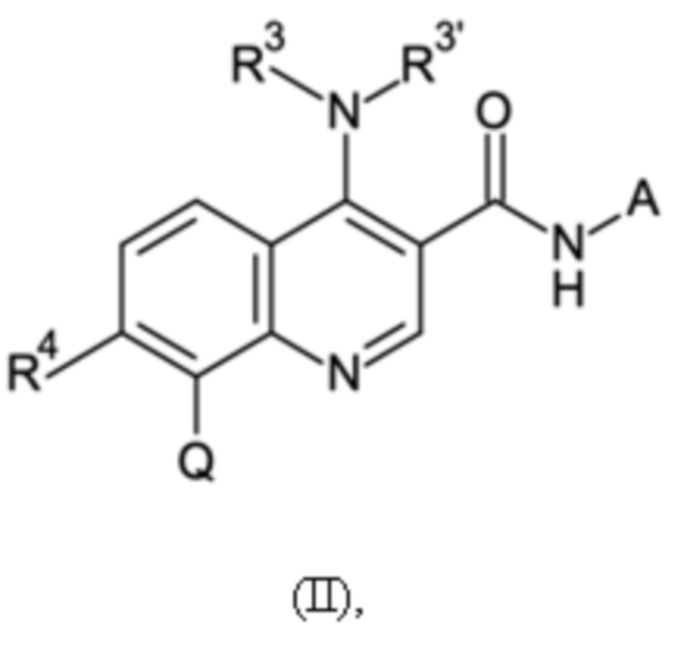
в котором
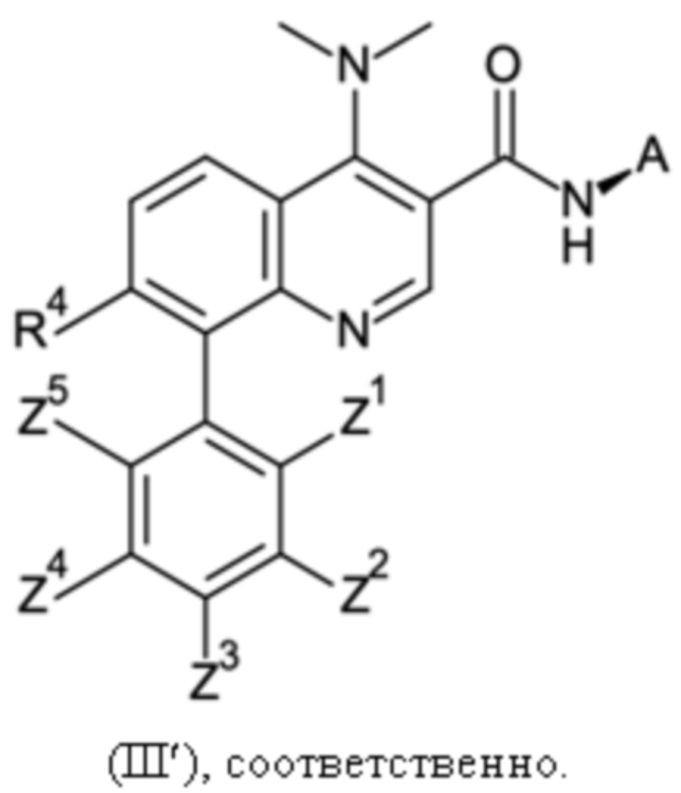
R3 и R3' могут иметь значение водорода, C1-C3-алкила, или вместе с атомом азота, с которым они связаны, образуют морфолинильное кольцо, R4 может иметь значение водорода или галогена, Q может иметь значения фенила, который может быть замещен от 1 до 5 заместителями Z1 - Z5, которые выбраны из водорода, галогена, C1-C4-алкила и C1-C4-галогеналкила, имеющего от 1 до 5 атомов галогена, и A представляет собой группу, выбранную из
Настоящее изобретение также относится к промежуточным соединениям нового способа согласно настоящему изобретению.
Уровень техники
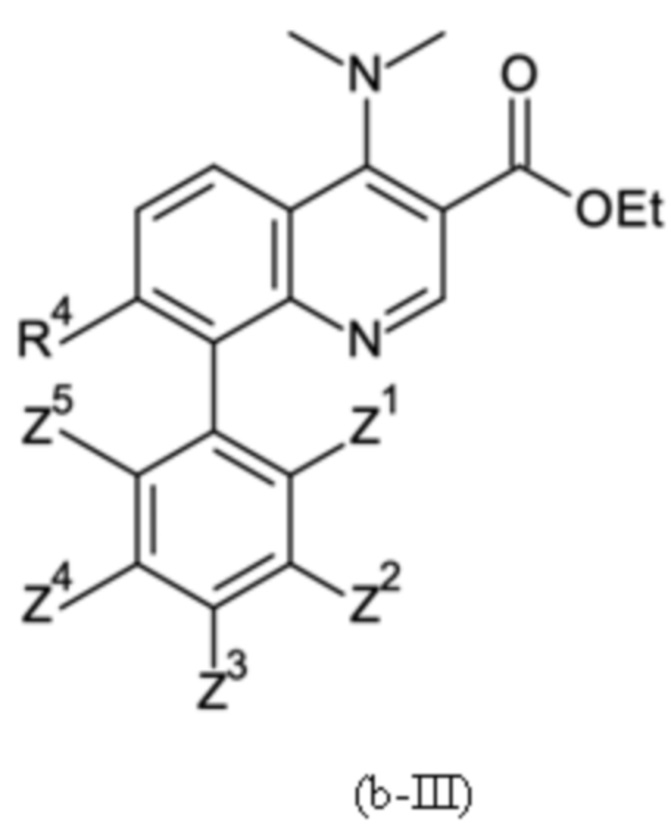
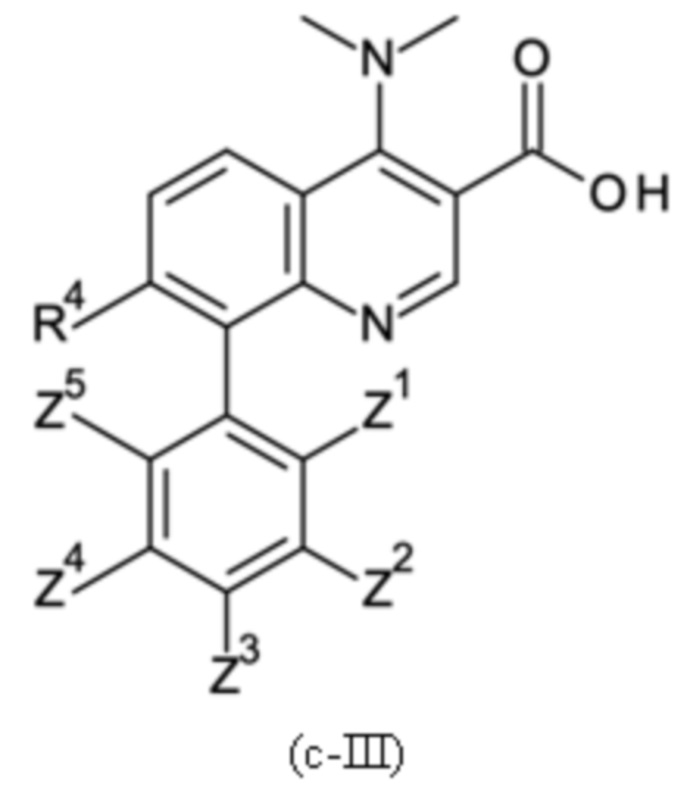
Настоящее изобретение относится к новому и улучшенному способу получения хинолиновых соединений общей формулы (II) выше, таких как, в частности, хинолиновые соединения согласно формуле (III):
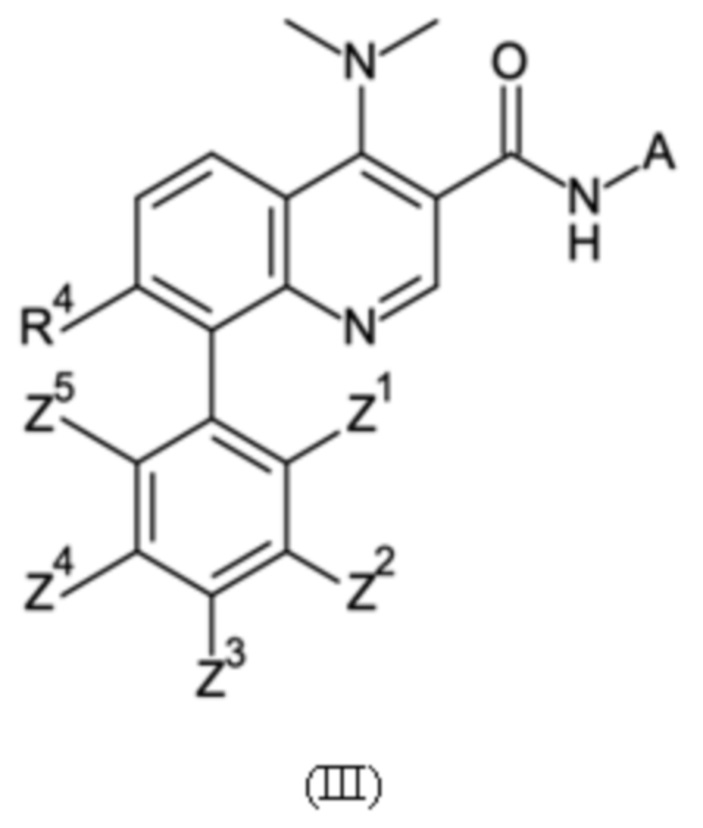
в которой A, R4 и Z1 - Z5 имеют значения, как определено в описании настоящего изобретения, а также промежуточным соединениям указанного нового способа.
Соединения общей формулы (II) и (III), которые могут быть получены способом согласно настоящему изобретению, демонстрируют ценный фармакологический спектр действия и, как было обнаружено, эффективно взаимодействуют с Slo-1 и поэтому могут использоваться для контроля, лечения и/или профилактики гельминтных инфекций, в частности желудочно-кишечных и внекишечных гельминтных инфекций.
Определенные хинолинкарбоксамиды описаны в JP2008-214323А в качестве агентов, подходящих для лечения и/или профилактики кожных заболеваний, таких как обыкновенные угри, дерматит или тому подобное.
В WO2017103851 описаны хинолин-3-карбоксамиды в качестве ингибиторов H-PGDS, полезные для лечения атеросклероза, псориаза, синусита и мышечной дистрофии Дюшенна.
Кроме того, в неопубликованной международной заявке PCT/EP2017/078319 раскрыты производные хинолина, охватываемые общей формулой (II) и (III) согласно настоящему изобретению, и способ получения таких соединений, включающий 6 стадий способа. С помощью описанного в ней 6-ти стадийного способа может быть достигнут общий выход менее 37%.
Таким образом, задача настоящего изобретения состояла в обеспечении улучшенного способа получения соединений формулы (II) и (III), как описано в настоящем документе. Усовершенствованный способ должен, в частности, обеспечивать повышенный пространственно-временной выход и, таким образом, быть особенно подходящим для обеспечения получения более высоких выходов за более короткое время и применять оптимизированные условия способа, которые, в частности, являются легко масштабируемыми, рентабельными, экологически безопасными и обеспечивают последовательное получение соединений с высокой степенью чистоты.
Эта задача была решена путем предоставления нового способа согласно настоящему изобретению, включающего 4 стадии способа со стадией дегидроксиаминирования, стадией сочетания Сузуки, стадией омыления и дополнительной стадией амидного сочетания. Новый способ может дополнительно включать предварительную стадию циклоконденсации с использованием реагента Итона для получения исходных соединений (I) согласно настоящему изобретению.
В уровне техники описано дегидроксиаминирование, например, в Bioorg. Med. Chem. Lett. 2003, p 1487-1490 или в WO 2013/118071 A1 и соответствующей заявке US 2013/210844.
Циклоконденсация с применением термического способа и реагента Итона была описана в J. Am. Chem. Soc. 1946, p 1204-1208, Org. Proc. Res. Dev. 2006, p 493-499, Org. Proc. Res. Dev. 2014, p 1482-1491 или в EP1258252 и US2003/0144507.
В частности, согласно неопубликованной международной заявке PCT/EP2017/078319 благодаря новому способу согласно настоящему изобретению возможно повысить выход стадии сочетания (стадия B-d) от 66% до 90% путем замены связывающего агента HATU ([бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния-3-оксид гексафторфосфата), как используется в неопубликованной международной заявке PCT/EP2017/078319. Улучшенная стадия дегидроксиаминирования (стадия B-a) согласно настоящему изобретению обеспечивает значительное сокращение времени реакции с 27 часов до 6 часов с по существу тем же выходом, обеспечивая новое сокращенное дегидроксиаминирование, в котором до сих пор проводимая дополнительная стадия дехлораминирования может быть сокращена до реакции в одном сосуде, таким образом предотвращая выделение нестабильного промежуточного соединения хлора и тем самым увеличивая пространственно-временной выход. Кроме того, возможно дополнительно увеличить пространственно-временной выход, выполняя предшествующую стадию циклоконденсации (стадия A) при улучшенной химической активации вместо термической активации, тем самым достигая увеличения выхода исходного соединения (I) от 68 % до 90%.
Как указано выше, по сравнению с 6-ти стадийным способом, описанным в неопубликованной международной заявке PCT/EP2017/078319, оказалось возможным повысить общий выход способа получения соединений согласно формуле (II) или (III) от около 37% до более чем 59% и в то же время сократить количество стадий синтеза и общее время способа, что приводит к значительному улучшению пространственно-временного выхода.
Описание настоящего изобретения
Согласно первому аспекту настоящее изобретение относится к способу получения соединения согласно формуле (II) выше из соединения согласно формуле (I):
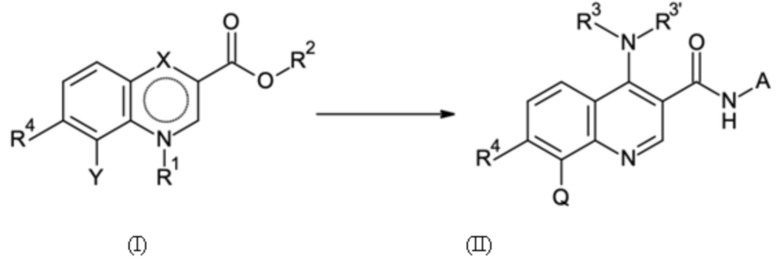
где
Y имеет значение галогена или Q;
X имеет значение C=O, C-OH или C-NR3R3';
 означает ароматическую кольцевую систему или, в случае, когда X представляет собой C=O, действующие двойные связи в кольцевой системе;
означает ароматическую кольцевую систему или, в случае, когда X представляет собой C=O, действующие двойные связи в кольцевой системе;
R1 отсутствует в случае, когда X представляет собой C-OH или C-NR3R3', или представляет собой атом водорода в случае, когда X представляет собой C=O;
R2 имеет значение водорода или C1-C3-алкила;
R3 и R3' независимо имеют значение водорода или C1-C3-алкила, или
R3 и R3' вместе с атомом азота, с которым они связаны, образуют морфолинильное кольцо
R4 имеет значение водорода или галогена;
Q имеет значение фенила, замещенного от 1 до 5 заместителями Z1 - Z5, где
Z1 - Z5 могут быть независимо выбраны из водорода, галогена, C1-C4-алкила и C1-C4-галогеналкила, имеющего от 1 до 5 атомов галогена; и
A представляет собой группу, выбранную из
где в способе группы Q, NR3R3' и NH-A в формуле (II) получают посредством реакции групп R2, X и Y в формуле (I) посредством стадий B-a, B-b, B-c и B-d, которые можно проводить в любом порядке, при условии, что стадию B-d не проводят перед стадией B-c:
Стадия B-a:
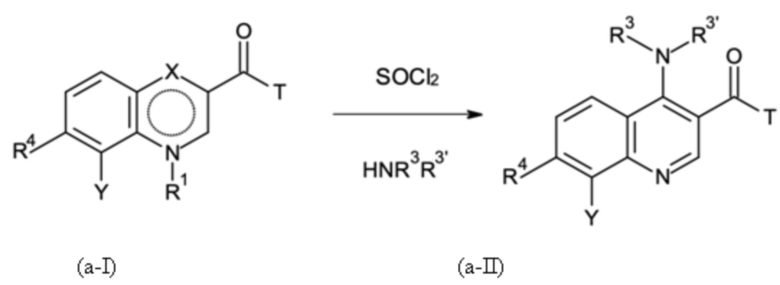
где T означает группу -O-R2 или -NH-A, причем R2 представляет собой водород или C1-C3-алкил; и
где X представляет собой C=O, и R1 представляет собой водород, или X представляет собой C-OH, и R1 отсутствует, в соответствии с формулами (a-I-a) и (a-I-b):
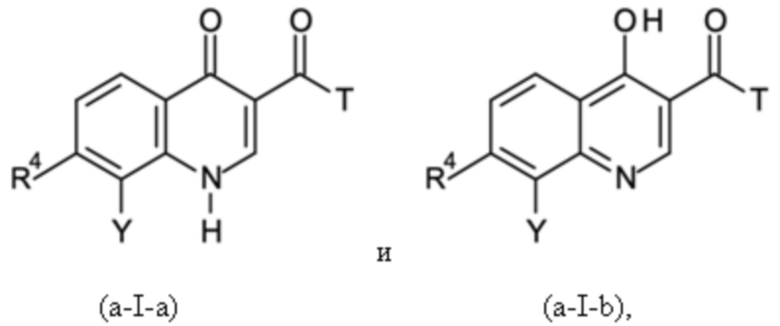
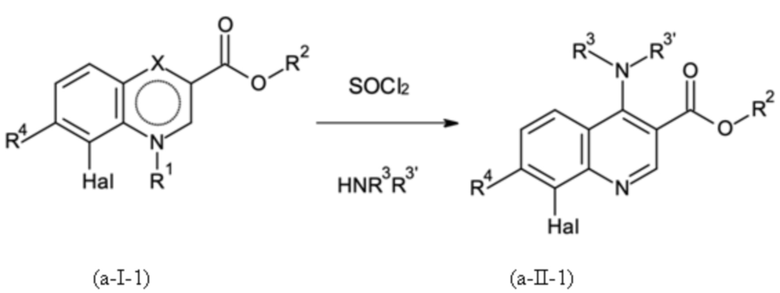
и где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2);
Стадия B-b:

где Y представляет собой галоген, T означает группу -O-R2 или -NH-A, причем R2 представляет собой водород или C1-C3-алкил, и G означает соединение бора, подходящее для осуществления реакции Сузуки, которое может быть определено следующим образом:
G представляет собой соединение бора общей формулы
(Q)nB(OH)3-n
где
n = 0, 1, 2 или 3
или
G представляет собой соединение бора общей формулы
(Q)4B- M+
где
M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
QBF3-M+
где
M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
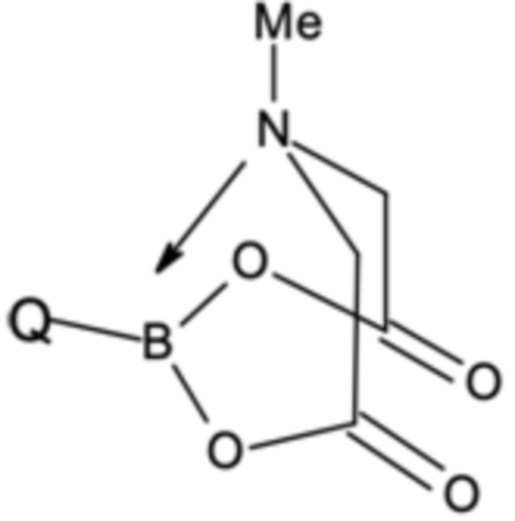
Стадия B-c:
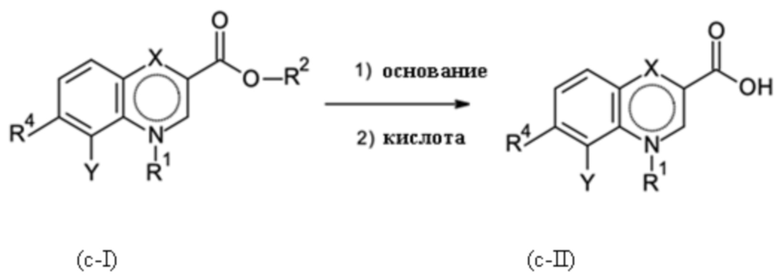
где R2 представляет собой C1-C3-алкил; где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочноземельного металла, а также любому карбонату щелочного металла и карбонату щелочноземельного металла. Кислота соответствует любой минеральной кислоте.
Стадия B-d:
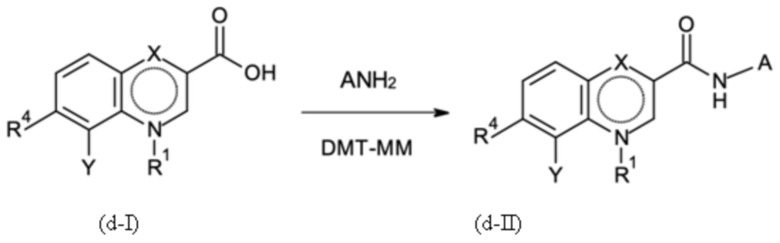
где указанную стадию способа B-d проводят с применением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиния хлорида (DMT-MM) в качестве связывающего агента;
и где на стадиях реакции B-a - B-d оставшиеся заместители имеют значение, соответствующее соответствующей стадии способа. Это означает, что в зависимости от соответствующего порядка стадий способа B-a, B-b, B-c и B-d, в частности заместители Y, X, T или R2 соответствующих исходных соединений, как определено выше, имеют значения, полученные в результате соответствующей предшествующей стадии способа.
Как указано выше, в способе согласно настоящему изобретению, на самом деле, стадии реакции B-a, B-b, B-c и B-d могут быть проведены в любом порядке, однако, при условии, что стадию B-d не проводят перед стадией B-c.
Согласно второму аспекту настоящее изобретение относится к способу, как определено выше, который может включать предшествующую стадию A для получения соединения согласно формуле (I):
Стадия A:
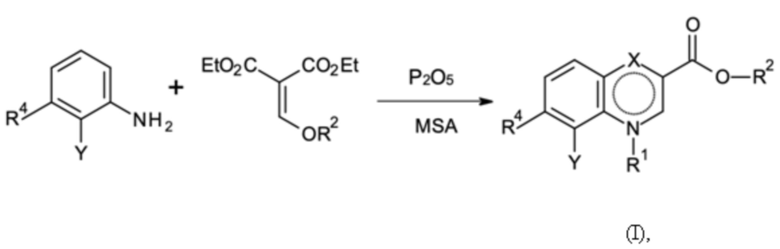
где Y, R4, R1 и R2 имеют значение, как определено выше, и
где указанную стадию способа A проводят с применением P2O5 в абсолютном количестве > 1 эквивалентов P2O5 и в количестве от 15 до 25 мас. % относительно метансульфоновой кислоты (MSA).
Определение стадий способа
Стадия способа A:
На предшествующей стадии способа A согласно второму аспекту настоящего изобретения анилин конденсируется с соединением диэтил-2-(R2-оксиметилен)пропандиоат в присутствии P2O5 и метансульфоновой кислоты (MSA). Неожиданно обнаружено, что хемоселективность указанной реакции зависит от абсолютного количества P2O5 и от количества относительно MSA.
Абсолютное количество ≥ 1 эквивалентов, предпочтительно > 1 эквивалентов, более предпочтительно от 1,5 до 3,5 эквивалентов, предпочтительно от 2,0 до 3,0 эквивалентов, более предпочтительно около 2,5 эквивалентов P2O5 применяют на стадии способа A согласно настоящему изобретению для достижения повышенных выходов, в частности выходов до 90 %.
Кроме того, на стадии способа A количество P2O5 относительно количества MSA составляет от 7,0 до 23,0 мас. %, предпочтительно от 15,0 до 23,0 мас. %, более предпочтительно около 23,0 мас. %.
Неожиданно оказалось, что предшественник циклизации нестабилен и разлагается при использовании слишком низких концентраций P2O5 или в чистой метансульфоновой кислоте.
Стадия способа A включает получение предшественника циклизации посредством конденсации анилина с соединением 2-(R2-оксиметилен)пропандиоат, которая может быть проведена при температурах от 70 до 140°C, предпочтительно от 80 до 130°C, более предпочтительно от 90 до 120°C. Реакцию предпочтительно проводят при от 50 мбар до 1 атм. Предпочтительно, на указанной стадии остаточный этанол удаляют с получением отличной хемоселективности на последующей стадии циклизации.
Полученный предшественник циклизации может быть использован в последующей реакции в виде расплава или разбавлен в инертном растворителе. Если расплав применяют, требуется температура реакции не менее 80°С. Предпочтительно, промежуточный конденсат применяют в разбавленном виде. Разбавление можно проводить в любом подходящем растворителе, включая, например, толуол, хлорбензол, ксилен, анизол, мезитилен, 1,2-дихлорбензол и т.д. или их смеси. Предпочтительными растворителями являются толуол, хлорбензол и ксилен, причем толуол является наиболее предпочтительным.
Предпочтительно толуол выбирают в качестве инертного растворителя. Разбавление промежуточного конденсата, например, в толуоле (до 27 мас. %), обеспечивает преимущество технически лучшего выполнения добавления реагента Итона и предотвращения его кристаллизации.
Концентрированный реагент Итона готовят отдельно, предпочтительно одновременно с получением предшественника циклизации.
Затем предшественник циклизации и реагент Итона объединяются для активации реакции циклоконденсации. При этом предшественник циклизации может быть добавлен к реагенту Итона или наоборот. Предпочтительно добавлять предшественник циклизации к приготовленному реагенту Итона.
Последующая реакция циклизации может быть проведена при температуре от 30 до 110°C, предпочтительно от 60 до 100°C, более предпочтительно от 70 до 90°C.
Общее количество пентоксида фосфора можно разделить на две или более порций, вплоть до восьми порций, и добавлять порционно.
Соединение (I) может быть выделено частичной нейтрализацией кислого маточного раствора, например, с содовым щелоком, тогда как нейтрализация должна контролироваться, чтобы избежать омыления продукта до соответствующей карбоновой кислоты.
Полученный осадок на фильтре может быть промыт и высушен.
Стадия способа B-a:
На стадии способа B-a способа согласно настоящему изобретению сокращенное дегидроксиаминирование проводят путем реакции в одном сосуде без необходимости выделения нестабильного промежуточного соединения хлора в результате дегидроксихлорирования. Это может быть неожиданно достигнуто при использовании стехиометрических количеств тионилхлорид (SOCl2).
При этом для первой активирующей стадии дегидроксихлорирования тионилхлорид дозируют к исходному соединению. Предпочтительно, тионилхлорид дозируют к исходному соединению при температуре от 80 до 110°С, предпочтительно при температуре от 85 до 105°С, более предпочтительно от 90 до 100°С. В этих условиях реакции реакция оказалась особенно контролируемой и эффективной.
Предпочтительно тионилхлорид добавляют в количествах от 1,15 до 2,30 эквивалентов, предпочтительно от 1,15 до 1,50 эквивалентов, более предпочтительно от 1,15 до 1,30 эквивалентов.
Реакцию предпочтительно проводят с каталитическими количествами N,N-диметилформамида (DMF) или N,N-диэтилформамида (DEF), N,N-ди-н-бутилформамида (DBF), N,N-диизопропилформамида (DIF), предпочтительно с DEF, DBF или DIF, более предпочтительно с DIF.
Предпочтительными количествами катализатора являются от 0,8 до 5,0 мол. %, более предпочтительно от 0,8 до 3,0 мол. %, даже более предпочтительно от 0,8 до 1,5 мол. %.
Остаточный тионилхлорид и хлорид водорода отгоняют, а затем желаемый амин добавляют к реакционной смеси, содержащей промежуточное соединение хлора. Амин может быть использован в газообразной форме или в водном растворе, причем последний является предпочтительным. Особенно предпочтительно использовать диметиламин. При этом дехлораминирование промежуточного соединения хлора может быть осуществлено путем добавления только амина. Предпочтительно, соединение амина, такое как предпочтительно диметиламин, добавляют в количестве ≥ 1,35 эквивалентов, предпочтительно от 1,35 до 2,70 эквивалентов, более предпочтительно от 1,35 до 1,50 эквивалентов.
Добавление соединения амина, такого как, предпочтительно, диметиламин, предпочтительно проводят при температурах от 20 до 60°C, предпочтительно от 30 до 50°C, более предпочтительно при около 40°C.
Реакция может быть проведена при от 1,0 до 6,0 бар, предпочтительно от 1,0 до 3,0 бар, более предпочтительно при атмосферном давлении.
Нейтрализация образовавшейся соляной кислоты может быть проведена для уменьшения минимального количества необходимого амина. Затем значение pH следует регулировать так, чтобы оно составляло около от 8 до 10, предпочтительно около от 9 до 10. В частности, значение pH не должно превышать 10, чтобы избежать регенерации соединения (I) или омыления сложного эфира.
Для нейтрализации и корректировки значения pH можно использовать обычные щелочные соединения, такие как растворы гидроксида натрия, калия или лития, или водные растворы щелочных карбонатов и карбонатов щелочноземельных металлов. В принципе, любое основание, способное регулировать и поддерживать значение pH от 8 до 10, предпочтительно от 9 до 10, является подходящим, при условии, что основание является таким же или менее нуклеофильным, чем соединение амина, используемое для аминирования, такое как, например, диметиламин или морфолин. Соответственно, также можно использовать триалкиламины для регулирования значения pH. Предпочтительно водный раствор гидроксида натрия применяют. В частности, было неожиданно обнаружено, что новая стадия способа B-a не требует использования сухих аминных соединений, например, сухого диметиламина, а может проводиться в щелочном водном растворе, как описано в настоящем документе.
Полученный продукт может быть выделен в чистом виде путем экстракции в водную соляную кислоту и кристаллизации при нейтрализации, например, с содовым щелоком.
Стадию способа B-a можно проводить в подходящем растворителе, таком как толуол, хлорбензол, ксилен, мезитилен, 1,2-дихлорбензол и т.д. Предпочтительными растворителями являются толуол, хлорбензол и ксилен, причем толуол является особенно предпочтительным.
Неожиданно было обнаружено, что посредством этой конкретной стадии способа B-a согласно настоящему изобретению можно исключить одну дополнительную стадию способа по сравнению со способом, известным из неопубликованной международной заявки PCT/EP2017/078319. В частности, неожиданно было обнаружено, что при этом стала возможной реакция гидролитического нестабильного промежуточного соединения в водных условиях с превосходным выходом.
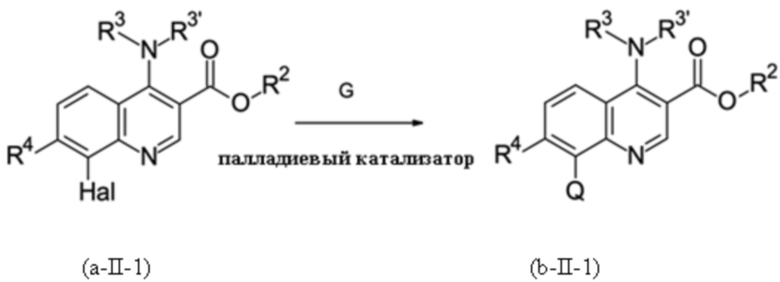
Стадия способа B-b:
На стадии способа B-b способа согласно настоящему изобретению проводят сочетание Сузуки с применением подходящего соединения бора G и палладиевого катализатора.
Подходящими катализаторами обычно являются все палладиевые катализаторы, которые обеспечивают реакцию сочетания Сузуки. Примерами являются предварительные катализаторы на основе палладия (0) и палладия (II), такие как PdCl2, Pd(OAc)2 (Ac = ацетат), Pd(NO3)2, Pd(acac)2 (acac = ацетилацетонат), Pd(dba)2 (dba = дибензилиденацетон) или палладий на носителе, таком как активированный уголь, без или в комбинации с фосфиновым лигандом L, и предварительно образованные катализаторы на основе палладия (0) и палладия (II), такие как PdCl2(L)2 или Pd(L)4.
Предпочтительными являются Pd(OAc)2, Pd(acac)2 и PdCl2(L)2, без или в комбинации с фосфиновым лигандом. Наиболее предпочтительным является Pd(acac)2 в комбинации с фосфиновым лигандом.
Подходящими лигандами L обычно являются все фосфиновые лиганды, которые, как известно, дают активные палладиевые катализаторы для реакции сочетания Сузуки. Предпочтительными являются монодентатфосфиновые лиганды P(Ar)n(алкил)3-n с n = 0, 1, 2 или 3, такие как PPh3, P(o-толил)3, P(o-анизил)3, P(п-анизил)3, P(нBu)3, P(трет-Bu)3, P(адамантил)2Ph, PPh2(трет-Bu) или PPh(трет-Bu)2. Предпочтительными являются P(трет-Bu)3, PPh2(трет-Bu) и PPh(трет-Bu)2. Наиболее предпочтительным является P(трет-Bu)3.
Можно использовать тетрафторборатную соль для лучшей работы с чувствительным к воздуху свободным фосфином. Однако также можно использовать раствор свободного фосфина, например, в толуоле, если вся работа выполняется в инертной атмосфере, например, в атмосфере аргона.
Молярное соотношение лиганда L и палладия может варьироваться в широких пределах. Предпочтительно использовать соотношение L/Pd от 0,5 до 10; более предпочтительным является соотношение от 1 до 6.
На стадии способа B-b может быть использован широкий спектр растворителей, таких как толуол, ксилены, хлорбензол, дихлорбензолы, гептан, циклогексан, метил-циклогексан, 1,4-диоксан, тетрагидрофуран, 2-метил-тетрагидрофуран, 2,5-диметил-тетрагидрофуран, метил-трет-бутиловый простой эфир, циклопентил-метиловый простой эфир, метанол, этанол, пропанол, бутанол, ацетонитрил, бутиронитрил, N,N-диметилформамид, вода или смеси этих растворителей. Предпочтительными являются толуол, ксилен, хлорбензол, метанол, этанол, метил-трет-бутиловый простой эфир, вода и смеси этих растворителей. Наиболее предпочтительной является смесь метил-трет-бутилового простого эфира и воды.
На стадии способа B-b может быть использован широкий спектр неорганических и органических оснований. Предпочтительными являются неорганические основания, такие как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид кальция, карбонат лития, карбонат натрия, карбонат калия, карбонат кальция, фторид натрия и фторид калия. Более предпочтительными являются гидроксид натрия, гидроксид калия, карбонат натрия и карбонат калия.
На стадии способа B-b согласно настоящему изобретению соединение бора G представляет собой соединение бора общей формулы
(Q)nB(OH)3-n
с
n =,1,2 или 3
или
G представляет собой соединение бора общей формулы
(Q)4B- M+
с
M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
QBF3-M+
с
M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
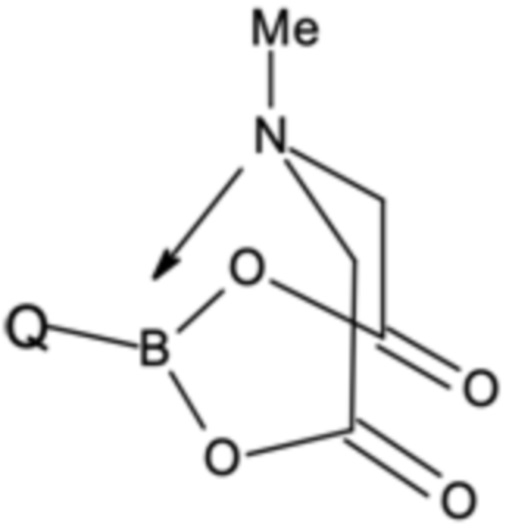
При этом соединения бора могут быть выбраны из подходящих бороновых кислот, бориновых кислот, боратов, боринатов или MIDA-защищенных сложных эфиров бороната (MIDA = N-метилиминодиуксусная кислота). Предпочтительно, соединение бора G представляет собой соединение бора, которое выбрано из (3,5-дихлорфенил)бороновой кислоты, бис(3,5-дихлорфенил)бориновой кислоты, три(3,5-дихлорфенил)бора; тетра(3,5-дихлорфенил)бората натрия, (3,5-дихлорфенил)трифторбората калия или 2-(3,5-дихлорфенил)-6-метил-1,3,6,2-диоксазаборокан-4,8-диона (MIDA боронат). Наиболее предпочтительными являются бис(3,5-дихлорфенил)бориновая кислота и (3,5-дихлорфенил)бороновая кислота.
На стадии способа B-b для достижения высоких выходов может использоваться удивительно низкая загрузка катализатора. Предпочтительными являются загрузки катализатора от 0,1 до 1 мол. % относительно исходного вещества. Более предпочтительными являются загрузки катализатора от 0,2 до 0,8 мол. %. Наиболее предпочтительными являются загрузки катализатора от 0,3 до 0,6 мол. %.
Стадия способа B-b согласно настоящему изобретению должна предпочтительно контролироваться так, чтобы количество остаточного палладия уменьшалось до ≤ 200 частей на миллион, предпочтительно < 200 частей на миллион, более предпочтительно количество остаточного палладия уменьшают до ≤ 100 частей на миллион, наиболее предпочтительно < 100 частей на миллион. Неожиданно было обнаружено, что посредством стадии способа B-b согласно настоящему изобретению можно достигнуть таких низких остаточных количеств палладия. При необходимости, дальнейшее снижение может быть достигнуто путем применения последующей стадии перекристаллизации.
Также возможно дальнейшее уменьшение остаточного палладия путем применения последующей экстракции водным раствором ацетилцистеина. При этом может быть достигнуто эффективное удаление палладия для предотвращения частичного гидродехлорирования на последующей стадии омыления (например, стадия B-c). Соответственно, эта дополнительная обработка ацетилцистеином особенно пригодна в случае проведения стадии способа B-b перед стадией способа B-c согласно настоящему изобретению.
Можно дополнительно уменьшить остаточное содержание палладия, применяя обработку ацетилцистеином и/или стадию перекристаллизации.
Кроме того, оказалось, что неожиданно с помощью стадии способа B-b согласно настоящему изобретению можно также добиться снижения содержания PCB80 от > 4000 частей на миллион до <500 частей на миллион.
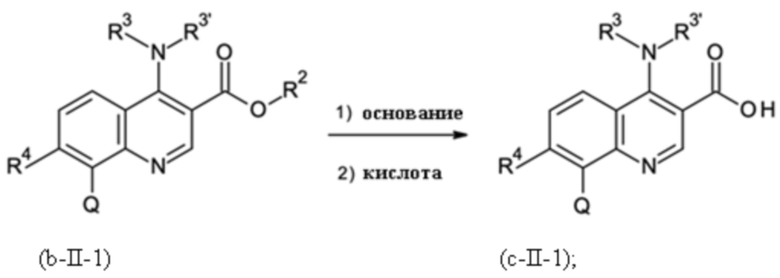
Стадия способа B-c:
На стадии способа B-c способа согласно настоящему изобретению проводят омыление группы [-(C=O)-O-C1-C3-алкил].
При этом омыление можно проводить, применяя общие условия омыления. В частности, можно использовать обычные гидроксидные соединения, такие как гидроксиды щелочных металлов и гидроксиды щелочноземельных металлов, такие как, в частности, гидроксид натрия, калия или лития. Предпочтительно применяют гидроксид натрия.
Помимо этих оснований можно использовать карбонаты щелочных металлов и карбонаты щелочноземельных металлов, такие как, в частности, карбонат натрия, калия, лития, магния или кальция.
Реакцию предпочтительно проводят в смеси воды/спирта при температуре 20-70°С. В качестве спиртов могут быть использованы алифатические спирты. Предпочтительно, этанол и метанол применяют.
На стадии способа B-c особенно предпочтительно использовать сложноэфирное соединение в качестве исходного соединения с остаточным содержанием палладия ≤ 200 частей на миллион, предпочтительно ≤ 100 частей на миллион. При использовании сложноэфирных соединений в качестве исходного соединения с более высокими уровнями палладия на стадии омыления B-c могут наблюдаться значительные количества гидродехлорированного сложного эфира и гидродехлорированной омыленной карбоновой кислоты.
Предпочтительно омыление проводят при интенсивном перемешивании.
Кроме того, предпочтительно, на стадии B-c переносить и выделять омыленный продукт в виде соли, главным образом в виде соли HCl. Было обнаружено, что по сравнению с формой свободной карбоновой кислоты омыленного продукта стадии B-c соль проявляет повышенную стабильность при хранении и применении на последующих стадиях реакции и проявляет меньшую склонность к разложению.
Стадия способа B-d:
На стадии способа B-d способа согласно настоящему изобретению проводят дополнительное связывание группы A, как определено выше.
Предпочтительно, на стадии B-d исходное соединение применяют в форме соли, предпочтительно в форме HCl соли, как описано в контексте стадии способа B-c выше.
На стадии способа B-d согласно настоящему изобретению 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиния хлорид (DMT-MM) применяют в качестве связывающего агента. Неожиданно было обнаружено, что применение DMT-MM для связывания вместо, например, TBTU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония тетрафторбората) или HATU (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфата), как описано в неопубликованной международной заявке PCT/EP2017/078319, является предпочтительным. В частности, применение DMT-MM вместо HATU неожиданно повышает выход от 66 % до 90 %. Кроме того, DMT-MM, как применяется в способе согласно настоящему изобретению, является гораздо более экономически эффективным и технически осуществимым.
Реакция сочетания может быть проведена с использованием выделенного связывающего реагента DMT-MM. Однако неожиданно оказалось, что реакцию сочетания также можно проводить, получая DMT-MM из 2-хлор-4,6-диметокси-1,3,5-триазина (CDMT) и N-метилморфолина (NMM) in situ.
Концентрация выделенного связывающего реагента CMT-MM должна быть выше из-за неблагоприятного энергетического потенциала -1110 Дж/г при начальной температуре 120°C, в то время как получение DMT-MM in situ позволяет уменьшить количество соответственно полученного ДМТ-ММ. Таким образом, на стадии способа B-d согласно настоящему изобретению особенно предпочтительно получать связывающий агент DMT-MM in situ из NMM и CDMT.
Для предотвращения кристаллизации продукта после добавления соединения A-NH2 можно проводить дробление кристаллов.
Стадию способа B-d можно проводить с применением подходящего растворителя, такого как толуол, хлорбензол, анизол, тетрагидрофуран и т.д. Предпочтительными растворителями являются толуол и хлорбензол, причем толуол является наиболее предпочтительным.
Предпочтительно, количество аминного структурного блока составляет от 1,05 до 1,4 эквивалентов, предпочтительно от 1,1 до 1,3 эквивалентов, более предпочтительно от 1,1 до 1,2 эквивалентов.
Предпочтительно, количество NMM составляет от 5,0 до 15,0 эквивалентов, предпочтительно от 5,0 до 10,0 эквивалентов, более предпочтительно от 5,0 до 7,5 эквивалентов.
Предпочтительно, количество CDMT составляет от 1,25 до 2,00 эквивалентов, предпочтительно от 1,25 до 1,80 эквивалентов, более предпочтительно от 1,25 до 1,35 эквивалентов.
Предпочтительно реакцию проводят при температурах от 20 до 60°C, предпочтительно от 25 до 50°C, более предпочтительно от 30 до 40°C.
Выделение продукта со стадии B-d предпочтительно проводят при возврате флегмы в трет-бутилметиловом простом эфире, что ускоряет отгонку растворителей реакции.
Другие аспекты способа согласно настоящему изобретению:
Продукт, полученный в результате способа согласно настоящему изобретению, может быть подвергнут дополнительным стадиям обработки, включая, например, стадии промывки, очистки, перекристаллизации и сушки, которые в общем известны специалисту в данной области.
Соединения и промежуточные соединения, полученные согласно способу согласно настоящему изобретению, могут потребовать очистки. Очистка органических соединений хорошо известна специалисту в данной области, и может быть несколько способов очистки одного и того же соединения. В некоторых случаях очистка не требуется. В некоторых случаях соединения могут быть очищены путем кристаллизации. В некоторых случаях примеси могут быть исключены с использованием подходящего растворителя. В некоторых случаях соединения могут быть очищены с помощью хроматографии, в частности колоночной флэш-хроматографии, с использованием, например, предварительно заполненных силикагелем картриджей, например, Biotage SNAP картриджи KP-Sil® или KP-NH®, в комбинации со системой Biotage autopurifier (SP4® или Isolera Four®) и элюентами, такими как градиенты гексан/этилацетат или дихлорметан/метанол. В некоторых случаях соединения могут быть очищены препаративной ВЭЖХ с использованием, например, автоочистителя Waters, оснащенного детектором на диодной матрице и/или масс-спектрометром с ионизацией распылением в реальном времени, в сочетании с подходящей предварительно набитой обращеннофазовой колонкой и элюентами, такими как градиент воды и ацетонитрила, который может содержать добавки, такие как трифторуксусная кислота, муравьиная кислота или водный аммиак.
В некоторых случаях способы очистки, как описано выше, могут обеспечить те соединения согласно настоящему изобретению, которые обладают достаточно щелочной или кислотной функциональностью в форме соли, как например, в случае соединения согласно настоящему изобретению, которое является достаточно основным, трифторацетат или формиатную соль, например, или, в случае соединения согласно настоящему изобретению, которое является достаточно кислотным, например, аммониевая соль. Соль этого типа может быть либо превращена в ее свободное основание, либо в свободную кислотную форму, соответственно, различными способами, известными специалисту в данной области, или ее можно использовать в качестве солей в последующих биологических анализах. Следует понимать, что конкретная форма (например, соль, свободное основание и т.д.) соединения согласно настоящему изобретению, как выделено и как описано в настоящей заявке, не необязательно находятся в единственной форме, в которой указанное соединение может быть применено в биологическом анализе для количественной оценки конкретной биологической активности.
Продукты (соединения и промежуточные продукты), полученные в результате способа согласно настоящему изобретению, могут быть превращены в любую соль, помимо фармацевтически приемлемых солей, любым способом, известным специалисту в данной области. Аналогично, любая соль соединения или промежуточного соединения согласно настоящему изобретению может быть превращена в свободное соединение любым способом, известным специалисту в данной области.
Также возможно получать изотопные варианты соединений согласно настоящему изобретению способами, описанными в настоящем документе, путем замещения реагента на изотопный вариант указанного реагента, предпочтительно на дейтерий-содержащий реагент. В зависимости от желаемых сайтов дейтерирования, в некоторых случаях дейтерий из D2O может быть включен либо непосредственно в соединения, либо в реагенты, которые полезны для синтеза таких соединений. Дейтерий в виде газа также является полезным реагентом для включения дейтерия в молекулы. Каталитическое дейтерирование олефиновых связей и ацетильных связей представляет собой быстрый путь для включения дейтерия. Катализаторы на основе металлов (то есть Pd, Pt, и Rh) в присутствии дейтерия в виде газа могут быть использованы для непосредственного обмена дейтерия на водород в функциональных группах, содержащих углеводороды. Различные дейтерированные реагенты и синтетические строительные блоки коммерчески доступны от таких компаний, таких как, например, C/D/N Isotopes, Quebec, Canada; Cambridge Isotope Laboratories Inc., Andover, MA, USA; и CombiPhos Catalysts, Inc., Princeton, NJ, USA.
Посредством способа согласно настоящему можно получить желаемый продукт с высоким выходом и высокой чистотой.
Кроме того, можно получить желаемый продукт в энантиомерном избытке > 80 %, предпочтительно > 85 %, предпочтительно > 90 %, более предпочтительно > 95 или даже до 99%.
Кроме того, можно получить желаемый продукт, имеющим очень низкие содержания PCB80 ≤ 1,0 частей на миллион, предпочтительно ≤ 0,5 частей на миллион, более предпочтительно ≤ 0,1 частей на миллион и очень низкое остаточное содержание палладия ≤ 20 частей на миллион, предпочтительно ≤ 10 частей на миллион.
Определения
В контексте настоящего изобретения термин «замещенный» означает, что один или более атомов водорода при обозначенном атоме или группе замещены заместителем, выбранным из указанной группы, при условии, что нормальная валентность обозначенного атома в существующих условиях не будет превышена. Допускаются комбинации заместителей и/или переменных.
Термин «необязательно замещенный» означает, что количество заместителей может быть равным или отличным от нуля. Если не указано иное, возможно, что необязательно замещенные группы замещены таким большим количеством необязательных заместителей, которые могут быть присоединены путем замещения атома водорода атомом, не являющимся водородом, при любой доступном атоме углерода или атоме азота. Как правило, число необязательных заместителей, если они присутствуют, может быть 1, 2, 3, 4 или 5, в частности 1, 2 или 3.
Как применено в описании настоящего изобретения, термин «один или более», например, в определении заместителей соединений согласно настоящему изобретению, означает «1, 2, 3, 4 или 5, особенно 1, 2, 3 или 4, более конкретно 1, 2 или 3, еще более конкретно 1 или 2».
Как применяется в настоящей заявке, положение, через которое соответствующий заместитель соединен с остальной частью молекулы, может в показанной структуре быть изображено знаком «решетка» (#) или пунктирной линией в указанном заместителе.
Термин «содержащий» при использовании в описании настоящего изобретения включает «состоящий из».
Если в описании настоящего изобретения любой признак сопровождается «как указано в описании настоящего изобретения» или «как описано в описании настоящего изобретения», это означает, что он может упоминаться или описываться в любом месте в описании настоящего изобретения.
Термины, упомянутые в описании настоящего изобретения, имеют следующие значения:
Термин “атом галогена” означает атом фтора, хлора, брома или иода, в частности атом фтора, хлора или брома.
Термин «C1C4-алкил» означает линейную или разветвленную насыщенную одновалентную углеводородную группу, имеющую 1, 2, 3 или 4 атома углерода, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил, или их изомер. Термин «C1C3-алкил» означает линейную или разветвленную насыщенную одновалентную углеводородную группу, имеющую 1, 2 или 3 атома углерода, например, метильная, этильная, н-пропильная или изопропильная группа.
Термин «C1C4-галогеноалкил» означает линейную или разветвленную насыщенную одновалентную углеводородную группу, в которой термин «C1C4-алкил» имеет значение, как определено выше, и в которой один или более атомов водорода замещены идентично или различно, атомом галогена. В частности, указанный атом галогена представляет собой атом фтора. Более конкретно, все указанные атомы галогена представляют собой атомы фтора («C1C4-фторалкил»). Указанная C1C4-галогеналкильная группа представляет собой, например, фторметил, дифторметил, трифторметил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3,3,3-трифторпропил или 1,3- дифторпропан-2-ил.
В общем, и если не указано иное, гетероарильные или гетероариленовые группы включают все их возможные изомерные формы, например, таутомеры и позиционные изомеры относительно точки связи с остальной частью молекулы.
Термин “C1-C4”, как применяется в настоящем документе, например, в контексте определения “C1C4алкил” или “C1C4галогеналкил”, означает алкильную группу, имеющую конечное чисто атомов углерода от 1 до 4, т.е. 1, 2, 3 или 4 атома углерода.
Когда задан диапазон значений, указанный диапазон охватывает каждое значение и поддиапазон в указанном диапазоне.
Например:
"C1-C4" охватывает C1, C2, C3, C4, C1-C4, C1-C3, C1-C2, C2-C4, C2-C3, и C3-C4;
"C1-C3" охватывает C1, C2, C3, C1-C3, C1-C2 и C2-C3;
Как применяется в описании настоящего изобретения, термин “уходящая группа” означает атом или группу атомов, которая замещается в ходе химической реакции в виде стабильной группы, взятой вместе со связывающими электронами. В частности, такая уходящая группа выбрана из группы, содержащей: галогенид, в частности фторид, хлорид, бромид или иодид, (метилсульфонил)окси, [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (фенилсульфонил)окси, [(4-метилфенил)сульфонил]окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
Заместитель оксо в контексте настоящего изобретения означает атом кислорода, который связан с атомом углерода через двойную связь, как например, образуя группу [-(C=O)-].
Если в описании настоящего изобретения используется множественная форма слова соединения, соли, полиморфы, гидраты, сольваты и тому подобное, это означает также одно соединение, соль, полиморф, изомер, гидрат, сольват или тому подобное.
Под «стабильным соединением» или «стабильной структурой» понимается соединение, которое является достаточно устойчивым, чтобы выдержать выделение до полезной степени чистоты из реакционной смеси и состава в эффективный терапевтический агент.
Соединения согласно настоящему изобретению необязательно содержат один или более асимметричных центров, в зависимости от расположения и природы различных желаемых заместителей. Возможно, что в конфигурации присутствует один или более асимметричных атома углерода (R) или (S), что может привести к рацемическим смесям в случае одного асимметричного центра, и диастереомерным смесям в случае множества асимметричных центров. В некоторых случаях возможно, что асимметрия также присутствует из-за ограниченного вращения вокруг данной связи, например, центральной связи, примыкающей к двум замещенным ароматическим кольцам указанных соединений.
Предпочтительными соединениями являются соединения, которые обеспечивают более желательную биологическую активность. Отделенные, чистые или частично очищенные изомеры и стереоизомеры, или рацемические или диастереомерные смеси соединений согласно настоящему изобретению также включены в объем настоящего изобретения. Очистка и разделение таких материалов могут быть выполнены стандартными методами, известными в данной области техники.
Предпочтительными изомерами являются те, которые обеспечивают более желательную биологическую активность. Эти отделенные, чистые или частично очищенные изомеры или рацемические смеси соединений согласно настоящему изобретению также включены в объем настоящего изобретения. Очистка и разделение таких материалов могут быть выполнены стандартными методами, известными в данной области техники.
Оптические изомеры могут быть получены путем разделения рацемических смесей в соответствии с обычными способами, например, путем образования диастереоизомерных солей с использованием оптически активной кислоты или основания или образования ковалентных диастереомеров. Примерами подходящих кислот являются винная, диацетилвинная, дитолуоилвинная и камфоросульфоновая кислота. Смеси диастереоизомеров могут быть разделены на их отдельные диастереомеры на основе их физических и/или химических различий с помощью способов, известных в данной области, например, путем хроматографии или фракционной кристаллизации. Оптически активные основания или кислоты затем высвобождаются из отделенных диастереомерных солей. Различные способы разделения оптических изомеров включают применение хиральной хроматографии (например, ВЭЖХ колонки с применением хиральной фазы), с или без обычной дериватизации, необязательно выбранной для максимизации разделения энантиомеров. Стабильные ВЭЖХ колонки с применением хиральной фазы являются коммерчески доступными, такие как произведенные компанией Daicel, например, Chirасel OD и Chirасel OJ, например, среди многих других, которые все доступны для выбора рутинным путем. Ферментативные разделения, с или без дериватизации, также применяются. Оптически активные соединения согласно настоящему изобретению могут подобным образом быть получены посредством хиральных синтезов, применяя оптически активные исходные вещества.
Чтобы различать различные типы изомеров друг от друга, делается ссылка на Правила IUPAC часть E (Pure Appl Chem 45, 11-30, 1976).
Настоящее изобретение включает все возможные стереоизомеры соединений и промежуточных соединений согласно настоящему изобретению, в виде отдельных стереоизомеров, или в виде любой смеси указанных стереоизомеров, например, (R) - или (S) - изомеров в любом соотношении. Выделение одного стереоизомера, например, одного энантиомера или одного диастереомера соединения согласно настоящему изобретению, достигается с помощью любого подходящего способа из уровня техники, такого как хроматография, особенно хиральная хроматография, например.
Кроме того, соединения и промежуточные соединения согласно настоящему изобретению могут существовать в виде таутомеров.
Настоящее изобретение включает все возможные таутомеры соединений и промежуточных соединений согласно настоящему изобретению в виде отдельных таутомеров, или в виде любой смеси указанных таутомеров, при любом соотношении.
Кроме того, соединения согласно настоящему изобретению могут существовать как N-оксиды, которые определяются тем, что по меньшей мере один атом азота соединений согласно настоящему изобретению окислен. Настоящее изобретение включает все такие возможные N-оксиды.
Настоящее изобретение также охватывает полезные формы соединений или промежуточных соединений согласно настоящему изобретению, такие как метаболиты, гидраты, сольваты, пролекарства, соли, в частности фармацевтически приемлемые соли и/или сопреципитаты.
Соединения или промежуточные соединения согласно настоящему изобретению могут существовать в виде гидрата или в виде сольвата, когда соединения или промежуточные соединения согласно настоящему изобретению содержат полярные растворители, в частности воду, метанол или этанол, например, в качестве структурного элемента кристаллической решетки соединений. Количество полярных растворителей, в частности воды, может находиться при стехиометрическом или нестехиометрическом соотношении. В случае стехиометрических сольватов, например, гидрата, геми-, (полу-), моно-, сескви-, ди-, три-, тетра-, пента- и т.д. сольваты или гидраты, соответственно, возможны. Настоящее изобретение включает все такие гидраты или сольваты.
Кроме того, соединения и промежуточные соединения согласно настоящему изобретению могут существовать в свободной форме, например, в виде свободного основания или в виде свободной кислоты, или в виде цвиттериона, или могут существовать в форме соли. Указанной солью может быть любая соль, либо органическая, либо неорганическая аддитивная соль, в частности любая фармацевтически приемлемая органическая или неорганическая аддитивная соль, которая стандартным образом применяется в фармацевтике, или которая применяется, например, для выделения или очистки соединений согласно настоящему изобретению.
Термин “фармацевтически приемлемая соль" относится к неорганической или органической соли кислотного добавления соединения согласно настоящему изобретению. Например, смотрите S. M. Berge, et al. “Pharmaceutical Salts,” J. Pharm. Sci. 1977, 66, 1-19.
Подходящая фармацевтически приемлемая соль соединений согласно настоящему изобретению может представлять собой, например, соль кислотного добавления соединения согласно настоящему изобретению, несущего атом азота в цепи или в кольце, например, которое является достаточно основным, как например соль кислотного добавления с неорганической кислотой или “минеральной кислотой”, такой как соляная, бромистоводородная, иодистоводородная, серная, сульфаминовая, бисерная, фосфорная или азотная кислота, например, или с органической кислотой, такой как муравьиная, уксусная, ацетоуксусная, пировиноградная, трифторуксусная, пропионовая, масляная, гексановая, гептановая, ундекановая, лауриновая, бензойная, салициловая, 2- (4-гидроксибензоил)-бензойная, камфорная, коричная, циклопентанпропионовая, диглюконовая, 3-гидрокси-2-нафтойная, никотиновая, памовая, пектиновая, 3-фенапропионовая, пивалиновая, 2-гидроксиэтансульфоновая, итаконовая, трифторметансульфоновая, додецилсульфоновая, этансульфоновая, бензолсульфоновая, пара-толуолсульфоновая, метансульфоновая, 2-нафталинсульфоновая, нафталиндисульфоновая, камфорсульфоновая, лимонная, винная, стеариновая, молочная, щавелевая, малоновая, янтарная, яблочная, адипиновая, альгиновая, малеиновая, фумаровая, D-глюконовая, миндальная, аскорбиновая, глюкогептановая, глицерофосфорная, аспарагиновая, сульфосалициловая или тиоциановая кислота, например.
Кроме того, другой подходящей фармацевтически приемлемой солью соединения согласно настоящему изобретению, которое является достаточно кислотным, является соль щелочного металла, например соль натрия или калия, соль щелочноземельного металла, например кальция, магния или стронция, или соль алюминия или цинка, или аммониевая соль, производная от аммиака или от органического первичного, вторичного или третичного амина, имеющего от 1 до 20 атома углерода, как например этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, диэтиламиноэтанол, трис(гидроксиметил)аминометан, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, 1,2-этилендиамин, N-метилпиперидин, N-метил-глюкамин, N,N-диметил-глюкамин, N-этил-глюкамин, 1,6-гександиамин, глюкозамин, саркозин, серинол, 2-амино-1,3-пропандиол, 3-амино-1,2-пропандиол, 4-амино-1,2,3-бутантриол, или соль с четвертичным ионом аммония, имеющую от 1 до 20 атомов углерода, как например тетраметиламмоний, тетраэтиламмоний, тетра(н-пропил)аммоний, тетра(н-бутил)аммоний, N-бензил-N,N,N-триметиламмоний, холин или бензалконий.
Специалистам в данной области техники также понятно, что кислотные аддитивные соли заявленных соединений могут быть получены реакцией соединений с подходящей неорганической или органической кислотой с помощью любого из ряда известных способов. Альтернативно, соли щелочных и щелочноземельных металлов кислотных соединений согласно настоящему изобретению получают путем взаимодействия соединений согласно настоящему изобретению с соответствующим основанием с помощью множества известных способов.
Настоящее изобретение включает все возможные соли соединений или промежуточных соединений согласно настоящему изобретению в виде отдельных солей, или в виде любой смеси указанных солей, в любом соотношении.
В описании настоящего изобретения, в частности в Экспериментальной части, для синтеза промежуточных соединений и примеров согласно настоящему изобретению, когда соединение упоминается в форме соли с соответствующим основанием или кислотой, точный стехиометрический состав указанной солевой формы, в виде, полученным посредством соответствующего способа получения и/или очистки, в большинстве случаев неизвестен.
Если не указано иное, суффиксы в химических названиях или структурных формулах, относящихся к солям, такие как «гидрохлорид», «трифторацетат», «натриевая соль» или «х HCl», «x CF3COOH», «x Na+», например, означают форму соли, где стехиометрия формы соли не уточняется.
Это относится аналогичным образом к случаям, когда промежуточные соединения или примерные соединения или их соли были получены посредством описанных способов получения и/или очистки, в виде сольватов, таких как гидраты, с (если определено) неизвестным стехиометрическим составом.
Кроме того, настоящее изобретение включает все возможные кристаллические формы, или полиморфы соединений согласно настоящему, изобретению либо в виде отдельного полиморфа, либо в виде смеси более одного полиморфа, при любом соотношении.
Другие варианты осуществления настоящего изобретения
Согласно третьему аспекту настоящее изобретение относится к способу получения соединения согласно формуле (II), как описано выше, где стадии способа проводят в следующем порядке с заместителями, имеющими конкретные значения, как определено в настоящем документе:
Стадия B-a:
где R2 представляет собой C1-C3-алкил, и R3, R3' и R4 имеют значение, как определено выше; и
где X представляет собой C=O, и R1 представляет собой водород, или X представляет собой C-OH, и R1 отсутствует, в соответствии с формулами (a-I-c) и (a-I-d):
и где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2); с последующей
Стадией B-b:
где G означает соединение бора, подходящее для осуществления реакции Сузуки с G, имеющим значения, как определено выше; с последующей
Стадией B-c:
где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочноземельного металла, а также любому карбонату щелочного металла и карбонату щелочноземельного металла; кислота соответствует любой минеральной кислоте; с последующей
Стадией B-d:
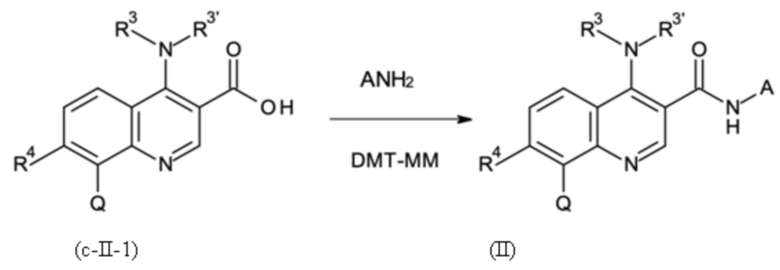
где A имеет значение, как определено выше; и
где указанную стадию способа B-d проводят с применением DMT-MM в качестве связывающего агента.
Согласно четвертому аспекту настоящее изобретение относится к способу, как определено в настоящем документе, где стадии способа проводятся в следующем порядке и представлены следующими формулами:
Стадия B-a:
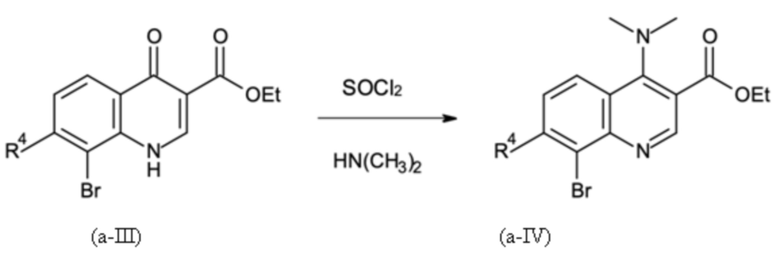
где R4 имеет значение, как определено выше, и где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2); с последующей
Стадией B-b:
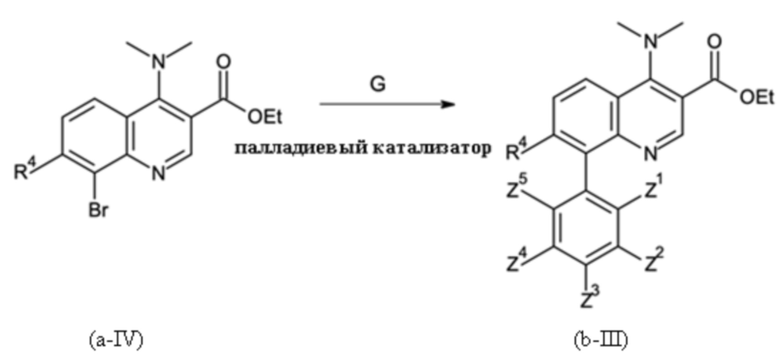
где G означает соединение бора, подходящее для осуществления реакции Сузуки с G, имеющим значения, как определено выше причем
Q представляет собой фенил, который может быть замещен от 1 до 5 заместителями Z1 - Z5, где
Z1 - Z5 независимо друг от друга выбраны из водорода, галогена, C1-C4-алкила и C1-C4-галогеналкила, имеющего от 1 до 5 атомов галогена; с последующей
Стадией B-c:
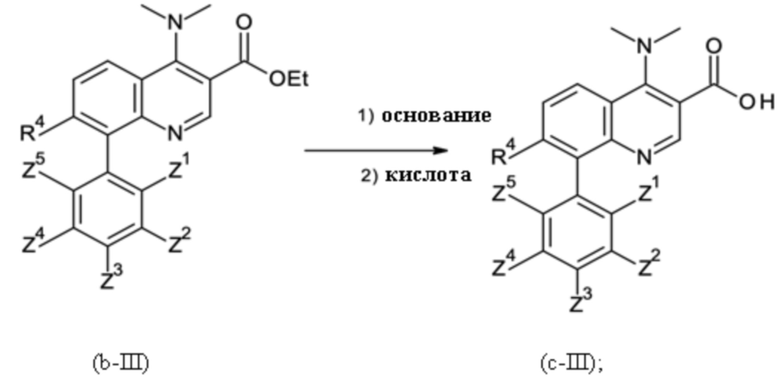
где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочноземельного металла, а также любому карбонату щелочного металла и карбонату щелочноземельного металла. Кислота соответствует любой минеральной кислоте, с последующей
Стадией B-d:
где A выбрана из группы, как определено выше, и
где DMT-MM применяют в качестве связывающего агента.
Согласно пятому аспекту настоящее изобретение относится к способу, как определено в настоящем документе, где
R4 выбран из водорода и фтора;
Z1, Z3 и Z5 представляют собой водород,
Z2 и Z4 представляют собой хлор, и
A представляет собой группу
Согласно шестому аспекту настоящее изобретение относится к способу, как определено в настоящем документе, где группа A выбрана из группы, состоящей из
Посредством применения соединений с группой, как определено в шестом аспекте выше, возможно получать соединения (и промежуточные соединения) согласно настоящему изобретению в соответствующей энантиомерной форме, такие как, в частности соединение (II), (d-II) или (III) в энантиомерной форме
Согласно седьмому аспекту настоящее изобретение относится к способу, описанному в настоящем документе, где одна или более стадий способа B-a, B-b, B-c и B-d далее определяются одним или более из следующих условий способа:
Другие предпочтительные условия способа на стадии B-a:
Предпочтительно, на стадии B-a применяют стехиометрические количества тионилхлорида.
Предпочтительно, на стадии B-a устанавливают значение рН от 8 до 10.
Другие предпочтительные условия способа на стадии B-b:
Предпочтительно, на стадии B-b палладиевый катализатор Pd(acac)2 применяют в количестве ≥ 0,3 мол. %.
Предпочтительно, на стадии B-b количество остаточного палладия уменьшают до ≤ 200 частей на миллион.
Более предпочтительно на стадии B-b количество остаточного палладия уменьшают до ≤ 100 частей на миллион.
Предпочтительно, на стадии B-b проводят последующую стадию экстракции ацетилцистеина и/или перекристаллизации.
Другие предпочтительные условия способа на стадии B-c:
Предпочтительно, на стадии B-c содержание палладия сложноэфирного соединения, применяемого в качестве исходного соединения, составляет ≤ 200 частей на миллион.
Более предпочтительно на стадии B-c содержание палладия сложноэфирного соединения, применяемого в качестве исходного соединения, составляет ≤ 100 частей на миллион.
Предпочтительно, на стадии B-c омыление сложноэфирного соединения проводят посредством применения NaOH.
Предпочтительно, на стадии B-c омыленный продукт переносят и выделяют в виде соли.
Более предпочтительно омыленный продукт переносят и выделяют в виде HCl соли.
Другие предпочтительные условия способа на стадии B-d:
Предпочтительно, на стадии B-d исходное соединение применяют в форме HCl соли.
Предпочтительно, на стадии B-d связывающий агент DMT-MM получают in situ из NMM и CDMT.
Согласно другому аспекту настоящее изобретение относится к способу, описанному в настоящем документе, где стадия способа A дополнительно определяется одним или более из следующих условий способа.
Другие предпочтительные условия способа на стадии A:
Предпочтительно, на стадии A предпочтительное промежуточное соединение в качестве предшественника циклизации применяют на последующей стадии разбавленным в инертном растворителе.
Более предпочтительно, указанное промежуточное соединение в качестве предшественника циклизации применяют разбавленным в толуоле.
Предпочтительно, на стадии A общее количество пентоксида фосфора добавляют по частям в виде двух или более порций.
Более предпочтительно, общее количество пентоксида фосфора добавляют по частям в виде до восьми порций.
Согласно другому аспекту настоящего изобретения стадию способа A проводят согласно следующим формулам:
Согласно восьмому аспекту настоящее изобретение относится к способу, описанному в настоящем документе, получения соединений формул
и/или
Согласно другим аспектам согласно настоящему изобретению можно проводить способ, как описано в настоящем документе, со следующим порядком стадий способа B-a, B-b, B-c и B-d, каждая, как определено в настоящем документе:
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-b, с последующей стадией способа B-a, с последующей стадией способа B-c, с последующей стадией способа B-d.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-b, с последующей стадией способа B-c, с последующей стадией способа B-d, с последующей стадией способа B-a.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-b, с последующей стадией способа B-c, с последующей стадией способа B-a, с последующей стадией способа B-d.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-d, с последующей стадией способа B-a, с последующей стадией способа B-b.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-d, с последующей стадией способа B-b, с последующей стадией способа B-a.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-a, с последующей стадией способа B-b, с последующей стадией способа B-d.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-a, с последующей стадией способа B-d, с последующей стадией способа B-b.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-b, с последующей стадией способа B-a, с последующей стадией способа B-d.
Способ согласно настоящему изобретению, где стадии способа проводят в порядке, начиная со стадии способа B-c, с последующей стадией способа B-b, с последующей стадией способа B-d, с последующей стадией способа B-a.
В соответствии с другим аспектом согласно настоящему изобретению, как указано выше, стадию способа А, как определено выше, можно проводить перед любым из этих альтернативных порядков способа выше. Дополнительные стадии способа, включающие, например, дальнейшую промывку, очистку, перекристаллизационную сушку и т.д., как указано выше в разделе «Другие аспекты способа согласно изобретению уровню», конечно могут быть выполнены аналогично тому, как описано в настоящем документе.
Согласно девятому аспекту, настоящее изобретение относится к способу получения промежуточных соединений согласно формулам
как определено выше, и/или промежуточных соединений согласно формулам
посредством проведения стадии способа B-a, как определено в настоящем документе, с последующим выделением и при необходимости очисткой полученных соединений. Здесь выделение в частности означает извлечение в твердой форме.
Согласно другому аспекту настоящее изобретение относится к способу получения промежуточного соединения согласно формуле
посредством проведения стадии способа A, как определено в настоящем документе, с последующим выделением и при необходимости очисткой полученных соединений. Здесь выделение в частности означает извлечение в твердой форме.
Согласно десятому аспекту, настоящее изобретение относится к способу получения промежуточных соединений согласно формулам
как определено выше, и/или промежуточных соединений согласно формулам
где Z1 - Z5 имеют значение, как определено выше,
посредством проведения стадии способа B-b, как определено в настоящем документе, с последующим выделением и при необходимости очисткой полученных соединений. Здесь выделение в частности означает извлечение в твердой форме.
В соответствии с одиннадцатым аспектом, настоящее изобретение относится к способу получения промежуточных соединений согласно формулам
как определено выше, и/или промежуточных соединений согласно формулам
где Z1 - Z5 имеют значения, как определено в любом из предшествующих пунктов,
посредством проведения стадии способа B-c, как определено в настоящем документе, с последующим выделением и при необходимости очисткой полученных соединений. Здесь выделение в частности означает извлечение в твердой форме, предпочтительно в виде соли минеральной кислоты, предпочтительно в виде соли HCl.
В соответствии с двенадцатым аспектом настоящее изобретение относится к способу получения промежуточных соединений согласно формуле
как определено выше, посредством проведения стадии способа B-d, как определено в настоящем документе, с последующим выделением и при необходимости очисткой полученных соединений. Здесь выделение в частности означает извлечение в твердой форме. Специалисту в данной области техники ясно, что в указанном восьмом аспекте промежуточное соединение может быть получено только, если указанную стадию B-d не проводят в качестве конечной стадии способа, что приводит к соединениям формулы (II) или (III) согласно настоящему изобретению.
Согласно конкретному варианту осуществления в соответствии с девятым-двенадцатым аспектом промежуточные соединения получают посредством применения соединений с группой, как определено в шестом аспекте выше, таким образом, обеспечивая соответствующие промежуточные соединения, как определено выше, в соответствующей энантиомерной форме.
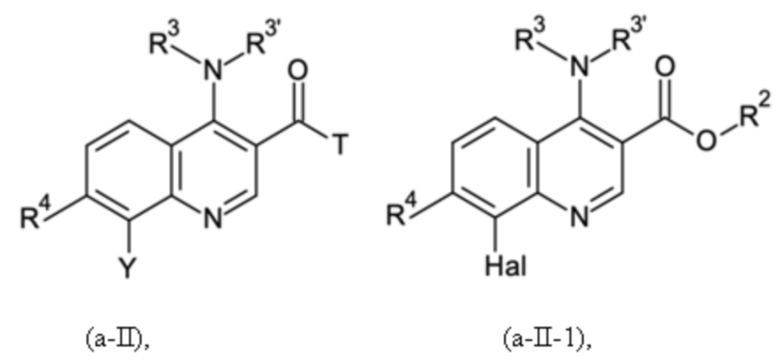
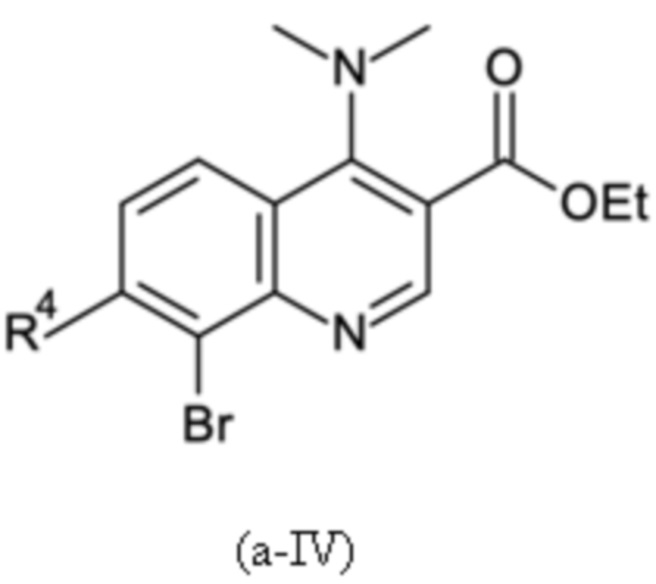
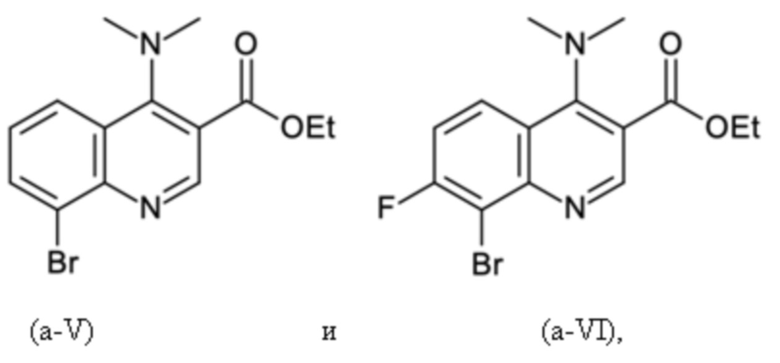
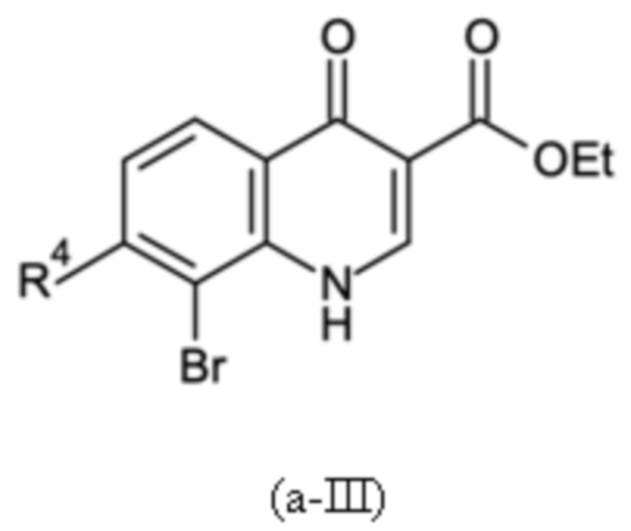
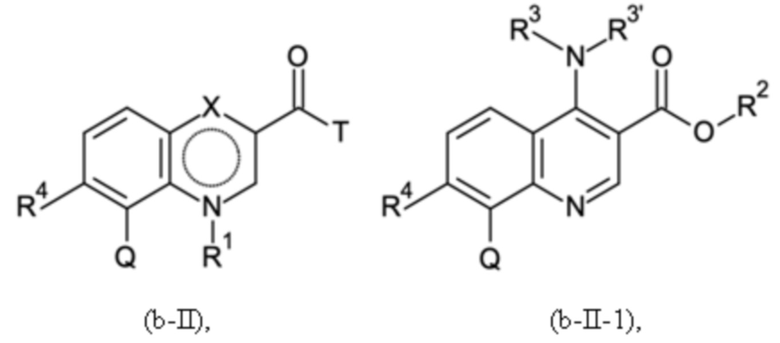
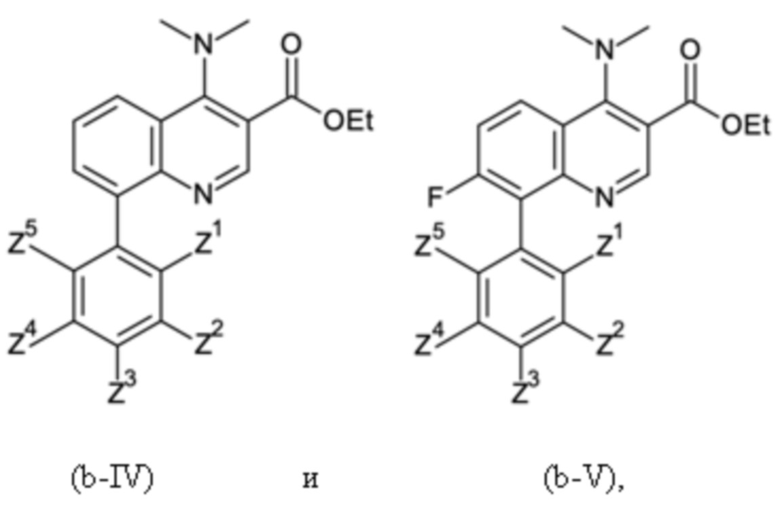
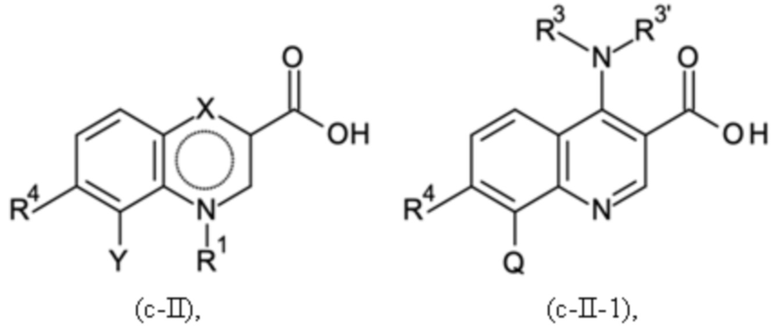
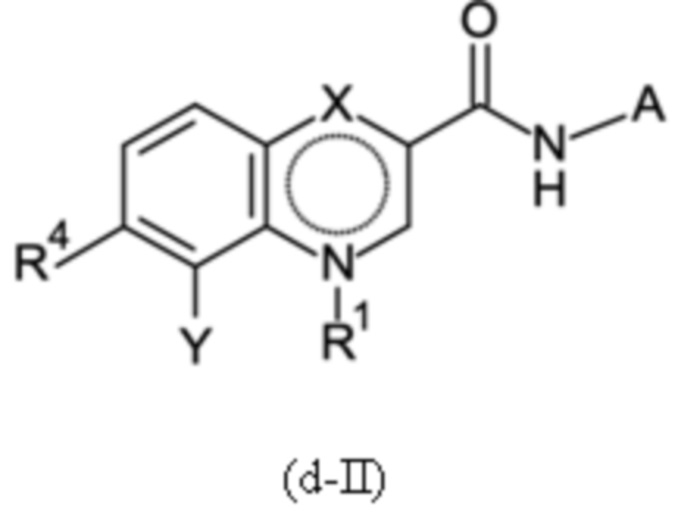
Согласно тринадцатому аспекту, настоящее изобретение относится к промежуточным соединениям согласно любой из формул (a-I-1), (a-II), (a-II-1), (a-III), (a-IV), (a-V), (a-VI), (b-II), (b-II-1), (b-III), (b-IV), (b-V), (c-II), (c-II-1), (c-III), (c-IV), (c-V) и (d-II), как определено выше, или их соответствующим энантиомерным формам (получаемым посредством применения соединения с группой, как определено в шестом аспекте выше).
Согласно четырнадцатому аспекту, настоящее изобретение относится к применению промежуточных соединений, как определено выше, или получаемым способом, как определено выше, для получения соединений формулы (II), (III), (IV) и/или (V) или их соответствующих энантиомерных форм (получаемых посредством применения соединения с группой, как определено в шестом аспекте выше), таких как, в частности, соединения формулы (IV') и/или (V'), как определено выше.
Согласно пятнадцатому аспекту, настоящее изобретение относится к применению промежуточных соединений согласно формулам (a-I-1), (a-II), (a-II-1), (a-III), (a-IV), (a-V) и (a-VI), как определено выше, или получаемым способом, как определено выше, для получения промежуточных соединений согласно формулам (b-II), (b-II-1), (b-III), (b-IV), (b-V), (c-II), (c-II-1), (c-III), (c-IV), (c-V) и (d-II), каждое, как определено выше, а также их соответствующих энантиомерных форм (получаемых посредством применения соединения с группой, как определено в шестом аспекте выше).
Согласно шестнадцатому аспекту, настоящее изобретение относится к применению промежуточных соединений согласно формулам (b-II), (b-II-1), (b-III), (b-IV) и (b-V), как определено выше, или получаемым способом, как определено выше, для получения промежуточных соединений согласно формулам (a-I-1), (a-II), (a-II-1), (a-III), (a-IV), (a-V), (a-VI) (c-II), (c-II-1), (c-III), (c-IV), (c-V) и (d-II), каждое, как определено выше, а также их соответствующих энантиомерных форм (получаемых посредством применения соединения с группой, как определено в шестом аспекте выше).
Согласно семнадцатому аспекту, настоящее изобретение относится к применению промежуточных соединений согласно формулам (c-II), (c-II-1), (c-III), (c-IV) и (c-V), как определено выше, или получаемым способом, как определено выше, для получения промежуточных соединений согласно формулам (a-I-1), (a-II), (a-II-1), (a-III), (a-IV), (a-V), (a-VI), (b-II), (b-II-1), (b-III), (b-IV), (b-V) и (d-II), каждое, как определено выше, а также их соответствующих энантиомерных форм (получаемых посредством применения соединения с группой, как определено в шестом аспекте выше).
Согласно восемнадцатому аспекту, настоящее изобретение относится к применению промежуточных соединений согласно формуле (d-II), как определено выше, или получаемым способом, как определено выше, для получения промежуточных соединений согласно формулам (a-I-1), (a-II), (a-II-1), (a-III), (a-IV), (a-V), (a-VI), (b-II), (b-II-1), (b-III), (b-IV), (b-V), (c-II), (c-II-1), (c-III), (c-IV) и (c-V), каждое, как определено выше, а также их соответствующих энантиомерных форм (получаемых посредством применения соединения с группой, как определено в шестом аспекте выше).
Экспериментальная часть
Различные аспекты изобретения, описанные в настоящей заявке, иллюстрируются следующими примерами, которые никоим образом не предназначены для ограничения настоящего изобретения.
Экспериментальная часть - общая часть
Все реагенты, синтез которых не описан в экспериментальной части, либо являются коммерчески доступными, либо являются известными соединениями, либо могут быть получены из известных соединений известными способами специалистом в данной области техники.
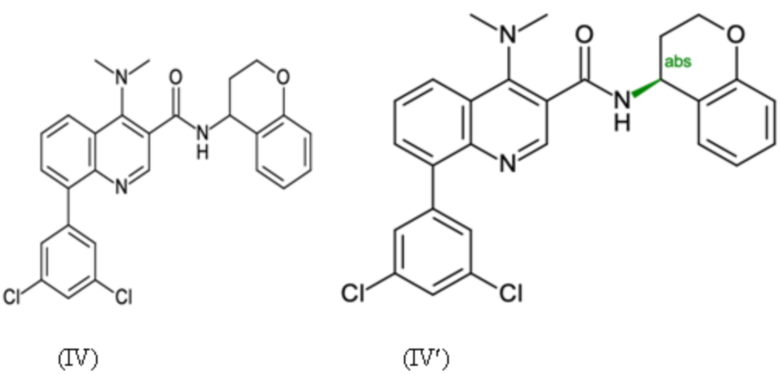
Пример 1 - Синтез N-[(4S)-хроман-4-ил]-8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксамида
Целевое соединение примера 1 представляет собой соединение, имеющее формулу (IV'), которое получают с помощью пяти стадий, начиная с 2-броманилина и стадии способа A, с последующими стадиями способа B-a, B-b, B-c и B-d в этом конкретном порядке:
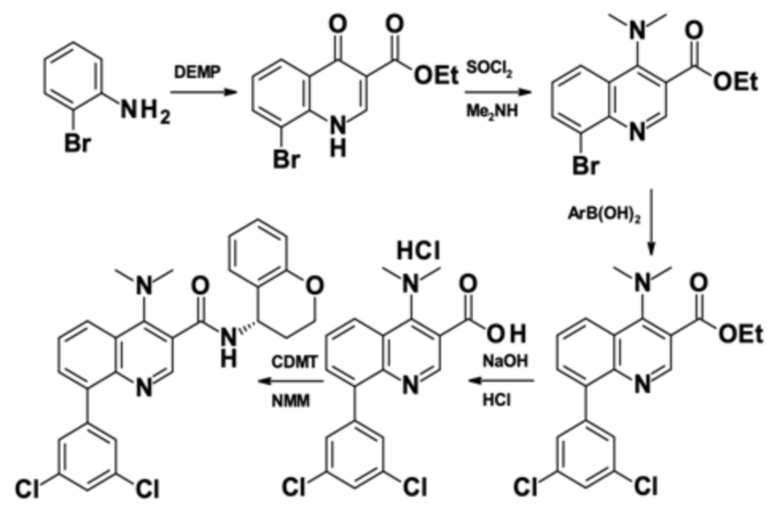
Стадия способа A:
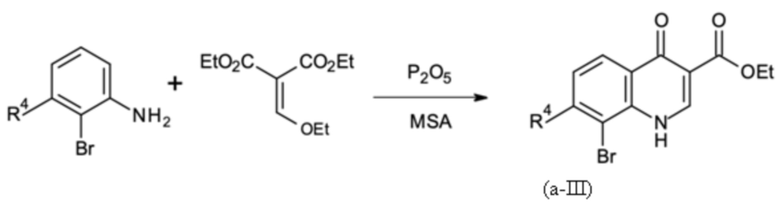
На первой стадии 2-броманилин конденсируется с диэтил 2-(этоксиметилен)пропандиоатом (DEMP) с получением этил 8-бром-4-оксо-1H-хинолин-3-карбоксилата с выходом 90% (96 мас. % чистота).
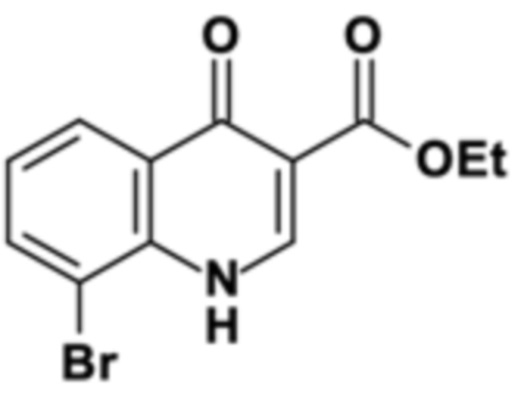
В четырехгорлую круглодонную колбу А (4000 мл), снабженную нагреваемой капельной воронкой, обратным холодильником, механической мешалкой и термометром, помещали 3,383 кг метансульфоновой кислоты. Кислоту нагревали до внутренней температуры 125°С. К кислоте добавляли 0,508 кг пентоксида фосфора. После полного растворения (1 час) еще одну порцию 0,508 кг пентоксида фосфора дозировали в раствор в течение 2 часов. Во время процесса растворения в другую двухгорлую круглодонную колбу B (1000 мл), снабженную дистилляционной головкой и термометром, загружали 0,660 кг диэтил 2-(этоксиметилен)пропандиоата и 0,500 кг 2-броманилина. Смесь нагревали до внутренней температуры 120°С при перемешивании и этанол отгоняли в течение 4 часов до тех пор, пока измерение с помощью ВЭЖХ не показало полное превращение в предшественник циклизации, и 125 мл этанола собрали. Затем давление снижали до 70 мбар для удаления остаточных 25 мл этанола из смеси. После этого содержимое сосуда В заполняли в капельную воронку сосуда А, которая была предварительно нагрета до температуры рубашки 100°С. Затем предшественник дозировали в колбу В в течение 1 часа при внутренней температуре 80°С. Полученный темный раствор дополнительно перемешивали в течение еще одного часа, пока измерение методом ВЭЖХ не показало полное превращение. Затем темный раствор добавляли на 7,500 кг льда, и полученную суспензию перемешивали до получения суспензии желтого цвета. К суспензии при перемешивании добавляли 3,524 кг содового щелока (33 мас. %), так чтобы внутренняя температура не превышала 30°С. После этого твердое вещество отфильтровывали и промывали с 3,000 кг деионизированной воды до получения промывочного раствора с нейтральным значением pH. Твердое вещество затем промывали с 1,950 кг ацетонитрила до тех пор, пока промывочный раствор не стал светлее до желтоватого цвета. Твердое вещество светло-желтого цвета затем сушили в вакууме.
Как определено посредством Q-ЯМР анализа оставшиеся 0,792 кг твердого вещества содержали 96 мас. % этил 8-бром-4-оксо-1H-хинолин-3-карбоксилата, что соответствует 0,760 кг чистого продукта и выходу 90%.
1H-ЯМР (DMSO-d6, 400 МГц) δ (частей на миллион) = 11,62 (bs, 1H), 8,45 (d, J = 8,0 Гц, 1H), 8,18 (d, J = 8,0 Гц, 1H), 8,05 (d, J = 8,0 Гц, 1H), 7,36 (dd, J = 8,0, 8,0 Гц, 1H), 4,24 (q, J = 7,0 Гц, 2H), 1,29 (t, J = 7,0 Гц, 3H).
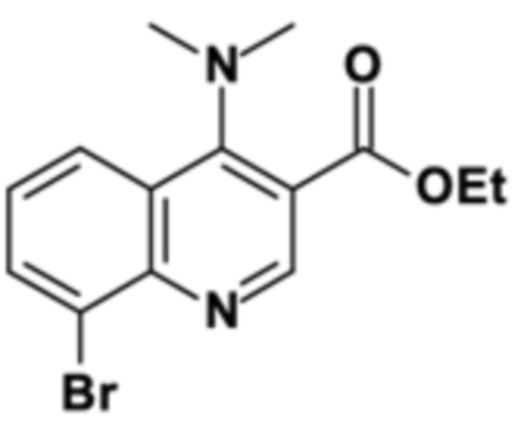
Стадия способа B-a:
Этил 8-бром-4-оксо-1H-хинолин-3-карбоксилат подвергали дегидроксихлорированию с тионилхлоридом и последующему амидированию с диметиламином на второй стадии с получением этил 8-бром-4-(диметиламино)хинолин-3-карбоксилата с выходом 93% (96 мас. % чистота) сокращенным образом.
В четырехгорлую круглодонную колбу (4000 мл), оборудованную капельной воронкой, обратным холодильником с уравнителем давления, механической мешалкой и термометром, помещали
500,0 г этил-8-бром-4-оксо-1H-хинолин-3-карбоксилата (чистота: 96,7 мас. %,) 1,65 г N,N-диизопропилформамида и 1500,0 г толуола. Суспензию нагревали до 100°C внутренней температуры при аккуратном перемешивании. После достижения температуры 223,4 г тионилхлорид дозировали в смесь в течение 1,5 ч. После завершения дозирования, смесь перемешивали в течение еще 1,5 ч до тех пор, пока измерение ВЭЖХ не показало полное превращение хлор-замещенного промежуточного соединения. Затем в общем 250 мл остаточного тионилхлорида, хлорида водорода и немного толуол отгоняли с получением хорошо смешиваемого темного раствора, который охлаждали до внутренней температуры 40°С. Обратный холодильник заменяли на рН-электрод. Затем 248,4 г диметиламина (40 мас. % в воде) дозировали в раствор в течение 30 минут. Значение рН устанавливали с в общем 365,0 г содового щелока (15 мас. %) для поддержания в диапазоне 9-10. Смесь перемешивали в течение еще 2,0 ч при 40°С, пока измерение ВЭЖХ не показало полное превращение. Затем смесь охлаждали до 25°С и затем добавляли к смеси 600 мл толуола и 1000 мл деионизированной воды. После разделения фаз органическую фазу дважды промывали, каждый раз 600 мл полуконцентрированного солевого раствора (13 мас. %). Объединенные водные фазы отбрасывали. Органическую фазу затем экстрагировали один раз смесью 100 мл 20 мас. % водной соляной кислоты и 400 мл деионизированной воды и второй раз смесью 100 мл 20 мас. % водной соляной кислоты и 200 мл деионизированной воды. Органическую фазу затем отбрасывали. Наконец объединенные водные фазы нейтрализовали добавлением в общем 640 г 15 мас. % содового щелка до достижения рН = 10 с полным осаждением продукта. Твердое вещество отфильтровывали и промывали в целом 2500 мл деионизированной воды до тех пор, пока промывочный раствор не стал свободным от галогенида. Твердое вещество сушили в вакууме до получения светло-желтого цвета.
Как определено посредством Q-ЯМР анализа оставшиеся 0,506 кг твердого вещества содержали 96,6 мас. % этил 8-бром-4-(диметиламино)хинолин-3-карбоксилата, что соответствует 0,489 кг чистого продукта и выходу 93%.
1H-ЯМР (DMSO-d6, 600 МГц) δ (частей на миллион) = 8,83 (s, 1H), 8,22 (d, J = 8,0 Гц, 1H), 8,15 (d, J = 8,0 Гц, 1H), 7,50 (dd, J = 8,0, 8,0 Гц, 1H), 4,40 (q, J = 7,0 Гц, 2H), 3,06 (s, 6H), 1,37 (t, J = 7,0 Гц, 3H).
Стадия способа B-b:
Этил 8-бром-4-(диметиламино)хинолин-3-карбоксилат затем сочетали с 3,5-дихлорфенилбороновой кислотой посредством реакции сочетания Сузуки, получая этил 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксилат:
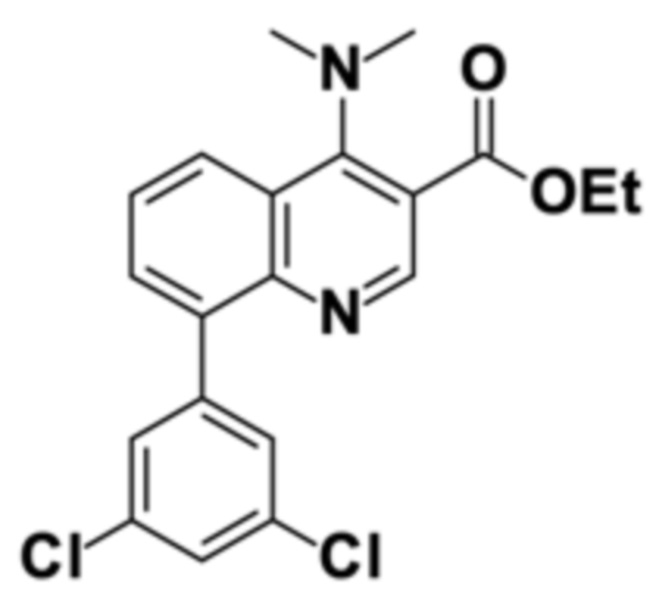
В четырехгорлую круглодонную колбу (4000 мл), оборудованную обратным холодильником с уравнителем давления, механической мешалкой и термометром, помещали в указанном порядке 167,9 г этил 8-бром-4-(диметиламино)хинолин-3-карбоксилата (чистота: 96,9), 2,375 кг MTBE (3,209 L), 0,687 кг H2O, 139,2 г K2CO3 и 100 г 3,5-дихлорфенилбороновой кислоты. Реакционную смесь продували аргоном при перемешивании в течение 30 минут. После этого добавляли 0,920 г Pd(acac)2 и 0,876 г HP(t-Bu)3BF4. Реакционную смесь нагревали до внутренней температуры 54°С (с обратным холодильником), перемешивая при слабой продувке аргоном. Через 6 часов измерение ВЭЖХ показало полное превращение. Смесь охлаждали до 20°С. После разделения фаз и выделения фазы пульпы в водный слой органическую фазу сушили над 15 г MgSO4, и осушитель отфильтровывали. Из раствора отгоняли 1500 л MTBE. К раствору затем добавляли 425 мл EtOH. Остаточный MTBE отгоняли при 70°С и давлении окружающей среды с получением раствора продукта в EtOH. Раствор постепенно охлаждали до 22°С, что приводило к кристаллизации продукта. Твердое вещество фильтровали и промывали с помощью 100 мл ледяного EtOH. После этого желтое твердое вещество сушили в вакууме. Анализ содержания продукта с помощью методики внутреннего стандарта ВЭЖХ объединенного маточного раствора и промывочного раствора показал 24,79 г этил 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксилата в фильтрате, что соответствует выходу 13%.
Как определено посредством Q-ЯМР анализа оставшиеся 0,165 кг твердого вещества содержали 99 мас. % этил 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксилата, что соответствует 0,163 кг чистого продукта и выходу 83%.
1H-NMR (DMSO-d6, 600 МГц): δ = 8,77 (s, 1H), 8,28 (dd, J = 8,5; 1,4 Гц, 1H), 7,8 (dd, J = 7; 1,4 Гц, 1H), 7,7 (dd, J = 8,5; 7 Гц; 1H), 7,6 (m, 3H), 4,4 (q, J = 7 Гц, 2H), 3,08 (s, 6H), 1,36 (t, J = 7 Гц, 3H) частей на миллион.
Стадия способа B-c:
Это промежуточное соединение подвергали омылению с применением 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоновой кислоты гидрохлорида:
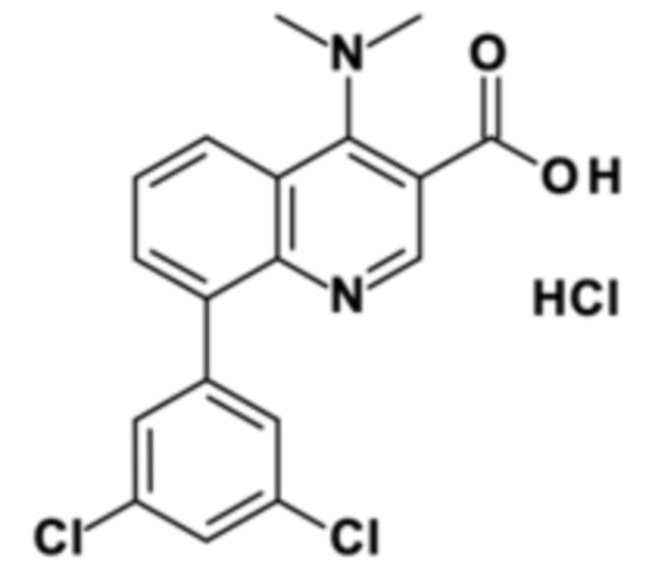
В четырехгорлую круглодонную колбу (500 мл), оборудованную механической мешалкой, капельной воронкой и термометром, помещали 0,048 кг этил 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксилата (чистота: 99 мас. %), 0,175 л EtOH (138,3 г). Густую желтоватую суспензию нагревали до внутренней температуры 50°С и перемешивали механической мешалкой. В течение 1 ч через капельную воронку добавляли 0,096 л водного NaOH (10% в воде). Смесь перемешивали при 50°C в течение 8 ч до тех пор, пока анализ ВЭЖХ не показал полное превращение.
После завершения реакции 0,125 кг летучих веществ удаляли дистилляцией при внешнем нагревании при 42°С и давлении 80 мбар. К оставшемуся остатку 0,158 г белой суспензии при перемешивании при 25°С добавляли 0,080 л деионизированной. вода. Суспензию охлаждали до 5°С при перемешивании и рН доводили до рН 1 путем добавления 0,092 кг HCl (20% в воде). Твердое вещество отфильтровывают при 5°С и промывали с помощью 0,050 л ледяной деионизированной воды. Затем твердое вещество промывали с помощью 0,050 л ацетона. Затем осадок на фильтре сушили в вакууме при 40°С, и продукт получали в виде белого твердого вещества.
Как определено посредством Q-ЯМР анализа оставшиеся 0,048 кг твердого вещества содержали 95 мас. % 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоновой кислоты гидрохлорида, что соответствует 0,046 кг чистого 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоновой кислоты гидрохлорида и выходу 95%.
1H-ЯМР (DMSO-d6, 400 МГц) δ (частей на миллион) = 8,57 (s, 1H), 8,42 (dd, J = 8,5, 1,19 Гц, 1H), 7,87 (dd, J = 7,23, 1,19 Гц, 1H), 7,78 (t, J = 1,83 Гц, 1H), 7,7 (m, 1H), 7,63 (d, J = 1,91 Гц, 2H), 3,44 (s, 6H).
Стадия способа B-d:
Наконец, 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоновой кислоты гидрохлорид амидировали с помощью (S)-хроманамин⋅HCl при применении CDMT и NMM с 90% выходом (>99 мас. % чистота).

В четырехгорлую круглодонную колбу (500 мл), оборудованную механической мешалкой, капельной воронкой и термометром, добавляли 20,0 г 8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоновой кислоты гидрохлорида (95 мас. %), 11,0 г CDMT и 160 мл толуола. При дозировании 25,0 г NMM в течение 25 мин суспензию нагревали до внутренней температуры 35°С и поддерживали впоследствии при внешнем нагревании. Суспензия стала желтой и стала более вязкой в конце добавления. Однако вязкость впоследствии уменьшалась при перемешивании. После 30-минутного перемешивания одной порцией добавляли 10,4 г (S)-хроман-4-амина гидрохлорида. После этого суспензию перемешивали в течение еще 8 часов, пока анализ ВЭЖХ не показал превращение 99%. После завершения реакции реакционную смесь охлаждали до комнатной температуры (20-22°С) и переносили в одногорлую круглодонную колбу (1000 мл). Затем добавляли 320 мл метилциклогексана, суспензию охлаждали до 0°C и перемешивали при этой температуре в течение 30 минут. Затем твердое вещество фильтровали через стеклянную фритту (пор. 3). Осадок на фильтре промывали 100 мл ледяного ацетонитрила, 100 мл водного раствора гидроксида натрия (5 мас. %) и 300 мл деионизированной воды. Твердое вещество сушили в вакууме (40°С, 70 мбар). После этого полное количество 24,2 г сухого твердого вещества суспендировали в 120 мл MTBE. Несколько кристаллов желаемого полиморфа добавляли в качестве затравки. Затем суспензию перемешивали при внешнем нагревании при 56°С в течение 4 часов. Наконец, суспензию снова охлаждали до 22°С, твердое вещество отфильтровывали и промывали 20 мл MTBE. Затем продукт сушили в вакууме (40°С, 50-10 мбар) в течение 2 часов. Продукт был получен в виде белого твердого вещества.
Как определено посредством Q-ЯМР и Q-HPLC анализа оставшиеся 22,8 г твердого вещества содержали 99,1 мас. % (S)-N-(хроман-4-ил)-8-(3,5-дихлорфенил)-4-(диметиламино)хинолин-3-карбоксамид, что соответствует 21,6 г чистого продукта и выходу 90%.
1H-ЯМР (DMSO-d6, 400 МГц) δ (частей на миллион) = 9,10 (d, J = 8,0 Гц), 8,63 (s, 1H), 8,24 (d, J = 8,0 Гц, 1H), 7,80 (d, J = 8,0 Гц, 1H), 7,67-7,63 (m, 4H), 7,37 (d, J = 8,0 Гц, 1H), 7,17 (dd, J = 8,0, 8,0 Гц, 1H), 6,94 (dd, J = 8,0, 8,0 Гц, 1H), 6,80 (d, J = 8,0 Гц, 1H), 5,79-5,72 (m, 1H), 4,21-4,32 (m, 2H), 3,07 (s, 6H), 2,25-2,15 (m, 1H), 2,09-2,00 (m, 1H).
Таким образом, целевое соединение (IV') может быть получено с общим выходом 59% за пять стадий, начиная с 2-броманилина.
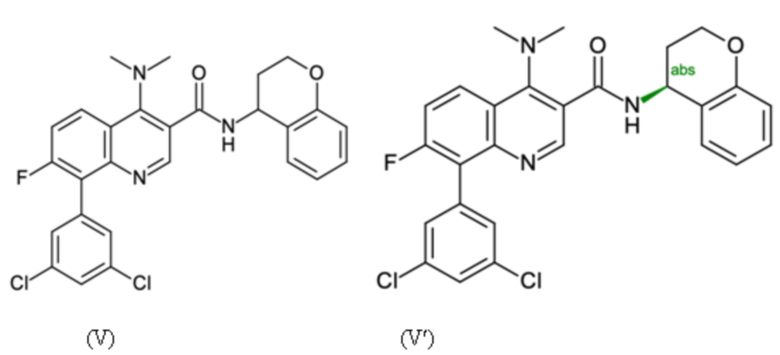
Пример 2 - Синтез N-[(4S)-хроман-4-ил]-8-(3,5-дихлорфенил)-4-(диметиламино)-7-фтор-хинолин-3-карбоксамида
Целевое соединение примера 2 представляет собой соединение, имеющее формулу (V'), которое получают таким же образом, как описано в примере 1, начиная с 2-бром-3-фтор-анилина на стадии способа А.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АЗАХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2018 |
|
RU2773290C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2019 |
|
RU2794894C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2764667C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830113C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2019 |
|
RU2794895C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2018 |
|
RU2781426C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2602083C2 |
| СПОСОБ ПОЛУЧЕНИЯ NO-ДОНОРНЫХ СОЕДИНЕНИЙ, ТАКИХ КАК NO-ДОНОРНЫЙ ДИКЛОФЕНАК | 2003 |
|
RU2322434C2 |
Группа изобретений относится к органической химии и включает способ получения соединений формулы (II), эффективно взаимодействующих с Slo-1 и применяемых для лечения и/или профилактики гельминтных инфекций. Значения заместителей в формуле (II) являются такими, как определено в формуле изобретения. Также группа изобретений относится к способу получения промежуточных соединений и их применению в способе получения соединения формулы (II). Способ получения соединений формулы (II) осуществляют посредством реализации стадий В-а, В-b, B-c, B-d, приведенных в формуле изобретения, при условии, что стадию B-d не проводят перед стадией B-c. Группа изобретений позволяет достичь высокого выхода и чистоты соединений общей формулы (II). 10 н. и 36 з.п. ф-лы, 2 пр.
1. Способ получения соединения согласно формуле (II) из соединения согласно формуле (I):
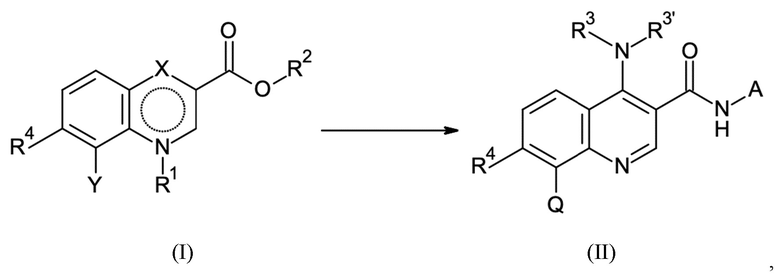
в котором Y имеет значение галогена или Q;
X имеет значение C=O, C-OH или C-NR3R3';
 означает ароматическую кольцевую систему или в случае, когда X представляет собой C=O, действующие двойные связи в кольцевой системе;
означает ароматическую кольцевую систему или в случае, когда X представляет собой C=O, действующие двойные связи в кольцевой системе;
R1 отсутствует в случае, когда X представляет собой C-OH или C-NR3R3', или представляет собой атом водорода в случае, когда X представляет собой C=O;
R2 имеет значение водорода или C1-C3-алкила;
R3 и R3' независимо имеют значение водорода или C1-C3-алкила или
R3 и R3' вместе с атомом азота, с которым они связаны, образуют морфолинильное кольцо;
R4 имеет значение водорода или галогена;
Q имеет значение фенила, замещенного от 1 до 5 заместителями Z1 - Z5, где
Z1 - Z5 могут быть независимо выбраны из водорода, галогена, C1-C4-алкила и C1-C4-галогеналкила, имеющего от 1 до 5 атомов галогена; и
A представляет собой группу, выбранную из
где в способе группы Q, NR3R3’ и NH-A в формуле (II) получают посредством реакции групп R2, X и Y в формуле (I) посредством стадий B-a, B-b, B-c и B-d, которые можно проводить в любом порядке, при условии, что стадию B-d не проводят перед стадией B-c:
Стадия B-a:
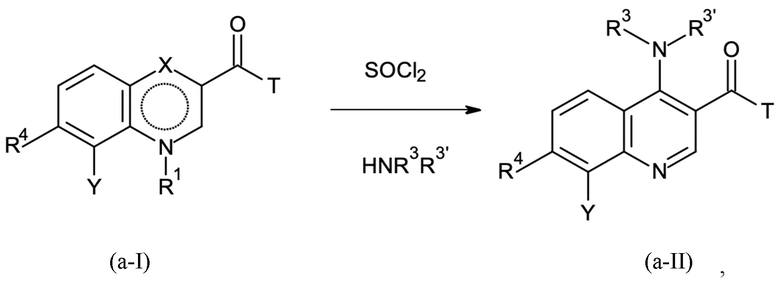
где T означает группу -O-R2 или -NH-A, причем R2 представляет собой водород или C1-C3-алкил; и
где X представляет собой C=O и R1 представляет собой водород или X представляет собой C-OH и R1 отсутствует, в соответствии с формулами (a-I-a) и (a-I-b):
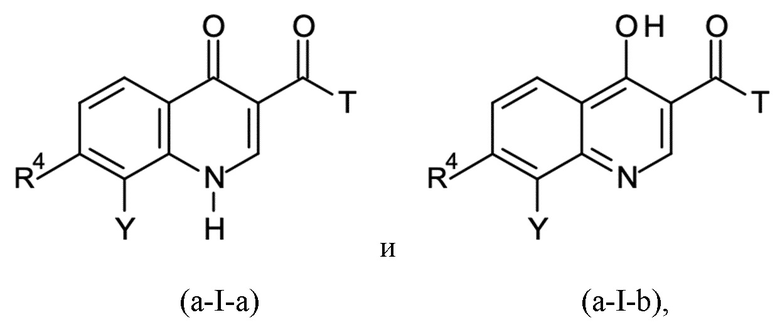
и где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2);
Стадия B-b:
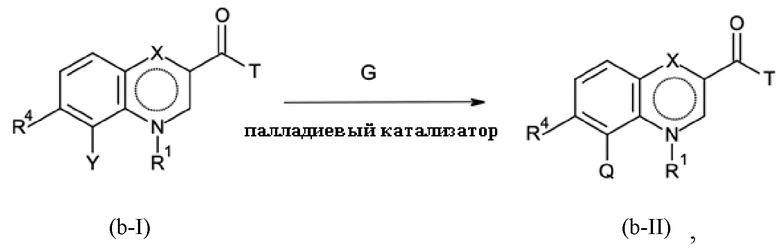
где Y представляет собой галоген, T означает группу -O-R2, -OH или -NH-A и G означает соединение бора, подходящее для осуществления реакции Сузуки с G, имеющим значения как определено далее:
G представляет собой соединение бора общей формулы
(Q)nB(OH)3-n
с n = 1, 2 или 3
или
G представляет собой соединение бора общей формулы
(Q)4B- M+
с M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
QBF3-M+
с M = литий, натрий или калий,
или
G представляет собой соединение бора общей формулы
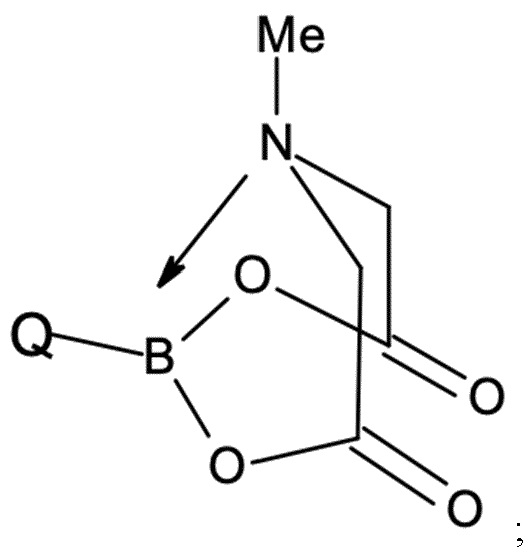
Стадия B-c:
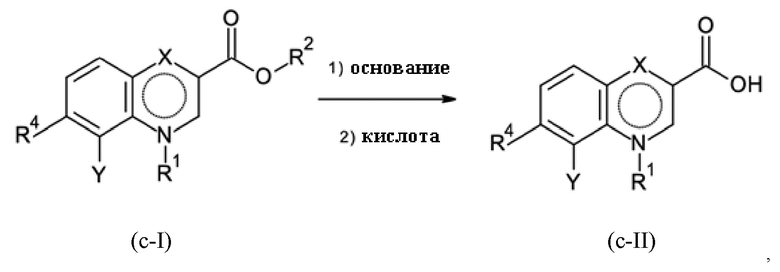
где R2 представляет собой C1-C3-алкил; где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочноземельного металла, а также любому карбонату щелочного металла и карбонату щелочно-земельного металла, кислота соответствует любой минеральной кислоте;
Стадия B-d:
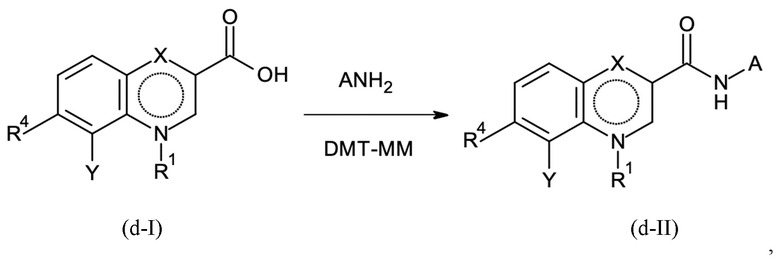
где указанную стадию способа B-d проводят с применением DMT-MM в качестве связывающего агента;
и где на стадиях реакции B-a - B-d оставшиеся заместители имеют значение, соответствующее соответствующей стадии способа.
2. Способ по п. 1, который дополнительно включает предшествующую стадию A для получения соединения согласно формуле (I):
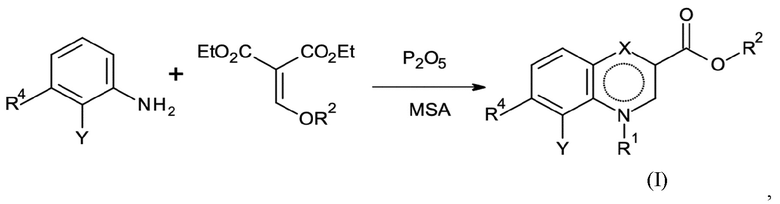
где P2O5 присутствует в реакционной смеси в абсолютном количестве > 1 экв. P2O5 и в количестве от 7,0 до 23,0 мас.% относительно метансульфоновой кислоты (MSA).
3. Способ по п. 1, где стадии способа проводят в следующем порядке:
Стадия B-a:
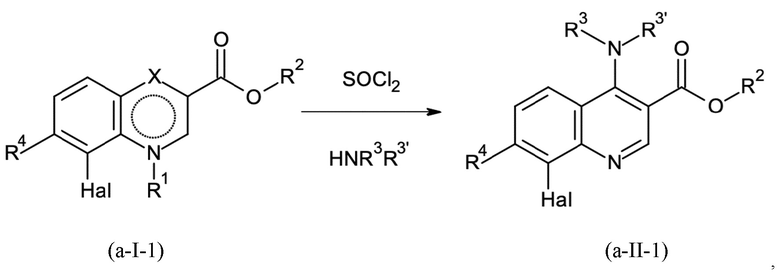
где R2 представляет собой C1-C3-алкил и R3, R3’ и R4 имеют значение, как определено в п. 1 или 2, и
где X представляет собой C=O и R1 представляет собой водород или X представляет собой C-OH и R1 отсутствует, в соответствии с формулами (a-I-c) и (a-I-d):
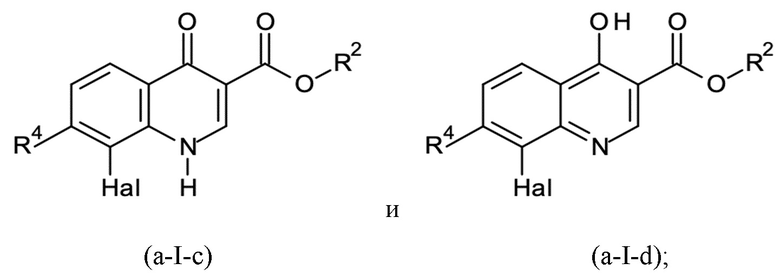
и где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2); с последующей
Стадией B-b:
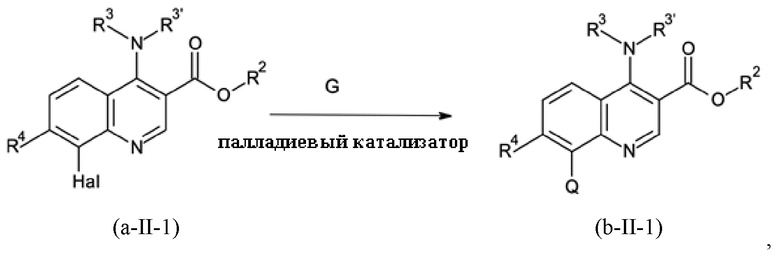
где G означает соединение бора, подходящее для осуществления реакции Сузуки, со значениями, как определено в п. 1 или 2; с последующей
Стадией B-c:
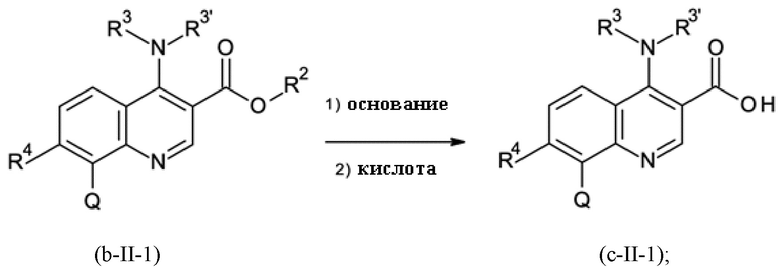
где R2 представляет собой C1-C3-алкил; где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочно-земельного металла, а также любому карбонату щелочного металла и карбонату щелочно-земельного металла, кислота соответствует любой минеральной кислоте;
с последующей
Стадией B-d:
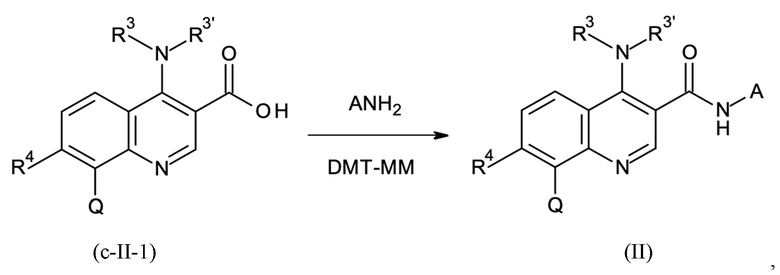
где A имеет значения, как определено в п. 1 или 2; и
где указанную стадию способа B-d проводят с применением DMT-MM в качестве связывающего агента.
4. Способ по п. 3, который представлен следующими формулами:
Стадия B-a:
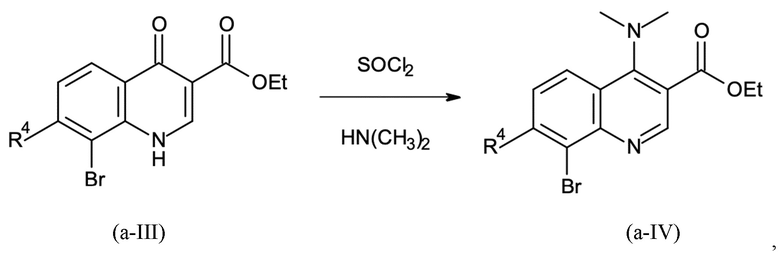
где указанную стадию способа B-a проводят с применением тионилхлорида (SOCl2); с последующей
Стадией B-b:
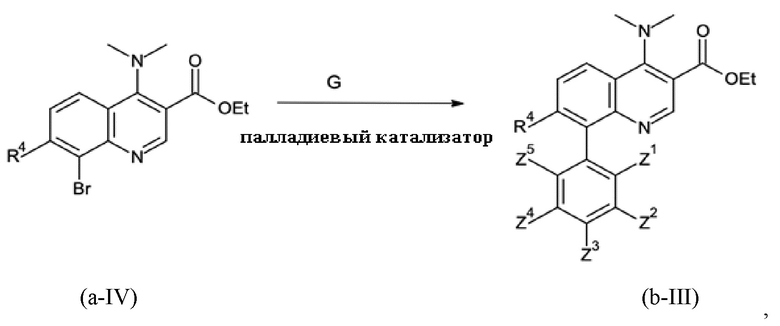
где G означает соединение бора, подходящее для осуществления реакции Сузуки со значениями, как определено в п. 1 или 2, и причем
Q представляет собой фенил, который может быть замещен от 1 до 5 заместителями Z1 - Z5, где
Z1 - Z5 независимо выбраны из водорода, галогена, C1-C4-алкила и C1-C4-галогеналкила, имеющего от 1 до 5 атомов галогена; с последующей
Стадией B-c:
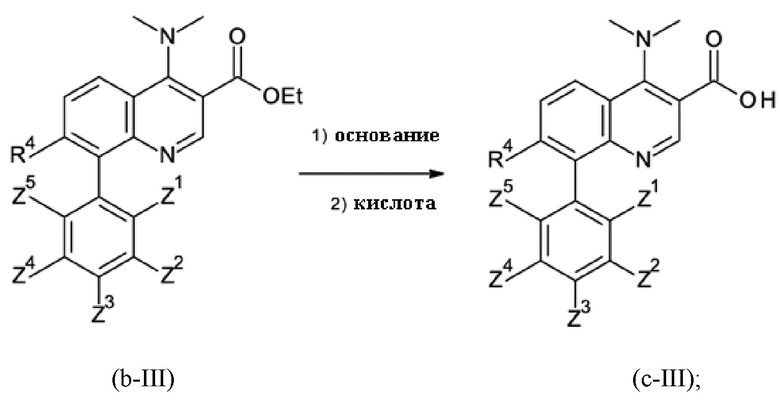
где основание соответствует любому гидроксиду щелочного металла и гидроксиду щелочно-земельного металла, а также любому карбонату щелочного металла и карбонату щелочно-земельного металла, кислота соответствует любой минеральной кислоте; с последующей
Стадией B-d:

где A выбрана из группы, как определено в п. 1, и
где указанную стадию способа B-d проводят с применением DMT-MM в качестве связывающего агента.
5. Способ по п.1, где
R4 выбирают из водорода и фтора,
Z1, Z3 и Z5 представляют собой водород,
Z2 и Z4 представляют собой хлор и
A представляет собой группу
6. Способ по п.1, где группа A выбрана из группы, состоящей из
получая таким образом соединение (II), (d-II) или (III) в энантиомерной форме
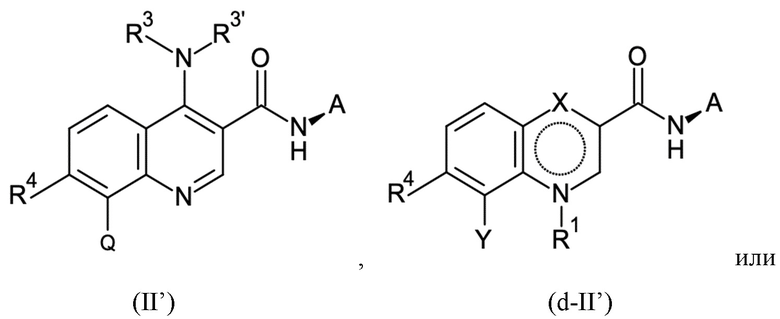
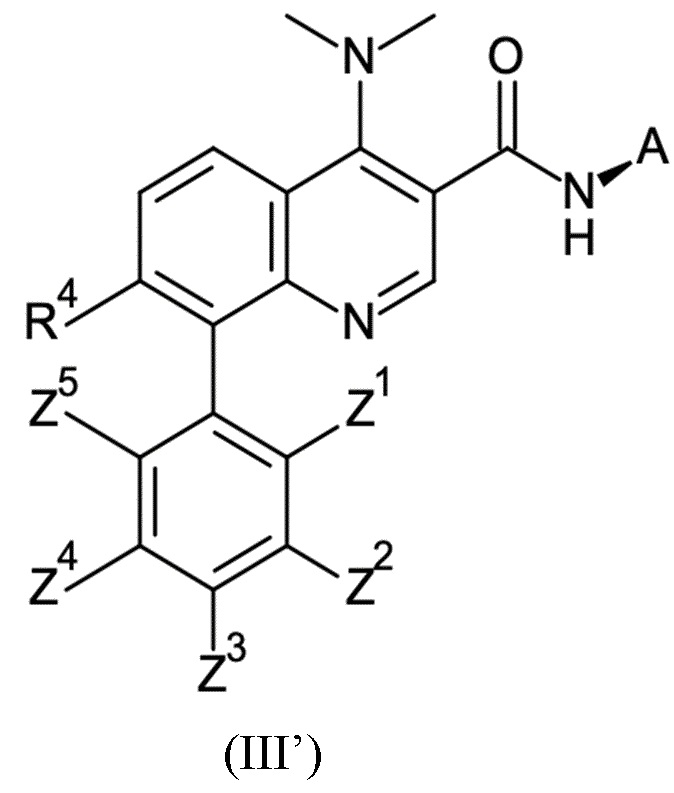
соответственно.
7. Способ по любому из пп. 1 - 6, где одна или более стадий способа B-a, B-b, B-c и B-d дополнительно определяется одним или более из следующих условий способа:
- на стадии B-a применяют стехиометрические количества тионилхлорида;
- на стадии B-a количество тионилхлорида составляет от 1,15 до 2,30 эквивалентов;
- на стадии B-a устанавливают значение рН от 8 до 10;
- на стадии B-a применяют растворитель, который выбран из группы, состоящей из толуола, хлорбензола или ксилена;
- на стадии B-a применяют катализатор, выбранный из группы, состоящей из DMF, DEF, DBF или DIF;
- на стадии B-a катализатор применяют в количествах от 0,8 до 5,0 мол.%;
- на стадии B-a соединение амина представляет собой диметиламин;
- на стадии B-a соединение амина добавляют в количестве ≥ 1,35 эквивалентов;
- на стадии B-b палладиевый катализатор применяют в количестве ≥ 0,3 мол.%;
- на стадии B-b количество остаточного палладия уменьшают до ≤ 200 частей на миллион;
- на стадии B-b палладиевый катализатор выбирают из группы, состоящей из, Pd(OAc)2, Pd(acac)2 и PdCl2(L)2, без или в комбинации с фосфиновым лигандом L;
- на стадии B-b, если присутствует, фосфиновый лиганд L представляет собой монодентатфосфиновый лиганд P(Ar)n(алкил)3-n с n = 0, 1, 2 или 3;
- на стадии B-c содержание палладия в сложноэфирном соединении составляет ≤ 200 частей на миллион;
- на стадии B-c омыление сложноэфирного соединения проводят посредством применения NaOH;
- на стадии B-c получают HCl соль полученного продукта;
- на стадии B-d исходное соединение применяют в форме HCl соли;
- на стадии B-d применяют растворитель, который выбран из группы, состоящей из толуола или хлорбензола;
- на стадии B-d связывающий агент DMT-MM применяют в виде выделенного соединения DMT-MM или получают in situ из NMM и CDMT;
- на стадии B-d исходное соединение применяют в количестве от 1,05 до 1,4 эквивалентов;
- на стадии B-d применяют количество от 5,0 до 15,0 эквивалентов NMM;
- на стадии B-d применяют количество от 1,25 до 2,00 эквивалентов CDMT;
- на стадии A предпочтительное промежуточное соединение в качестве предшественника циклизации применяют на последующей стадии разбавленным в инертном растворителе;
- на стадии A применяют абсолютное количество от 1,5 до 3,5 эквивалентов P2O5;
- на стадии A количество P2O5 относительно количества MSA составляет от 7,0 до 23,0 мас.%;
- на стадии A общее количество пентоксида фосфора добавляют по частям в виде двух или более порций.
8. Способ по п. 7, где на стадии B-a количество тионилхлорида составляет от 1,15 до 1,50 эквивалентов.
9. Способ по п. 7, где на стадии B-a количество тионилхлорида составляет от 1,15 до 1,30 эквивалентов.
10. Способ по п. 7, где на стадии B-a применяют толуол.
11. Способ по п. 7, где на стадии B-a применяют катализатор, выбранный из группы, состоящей из DEF, DBF или DIF.
12. Способ по п. 7, где на стадии B-a применяют катализатор DIF.
13. Способ по п. 7, где на стадии B-a катализатор применяют в количествах от 0,8 до 3,0 мол.%.
14. Способ по п. 7, где на стадии B-a катализатор применяют в количествах от 0,8 до 1,5 мол.%.
15. Способ по п. 7, где на стадии B-a соединение амина добавляют в количестве от 1,35 до 2,70 эквивалентов.
16. Способ по п. 7, где на стадии B-a соединение амина добавляют в количестве от 1,35 до 1,50 эквивалентов.
17. Способ по п. 7, где на стадии B-b палладиевый катализатор применяют в количестве от 0,3 до 0,6 мол.%.
18. Способ по п. 7, где на стадии B-b количество остаточного палладия уменьшают до ≤ 100 частей на миллион.
19. Способ по п. 7, где на стадии B-b палладиевый катализатор представляет собой Pd(acac)2 в комбинации с фосфиновым лигандом L.
20. Способ по п. 7, где на стадии B-b, если присутствует, фосфиновый лиганд L представляет собой PPh3, P(о-толил)3, P(о-анизил)3, P(п-анизил)3, P(нBu)3, P(трет-Bu)3, P(адамантил)2Ph, PPh2(трет-Bu) или PPh(трет-Bu)2.
21. Способ по п. 7, где на стадии B-b, если присутствует, фосфиновый лиганд L представляет собой P(трет-Bu)3, PPh2(трет-Bu) или PPh(трет-Bu)2.
22. Способ по п. 7, где на стадии B-b, если присутствует, фосфиновый лиганд L представляет собой P(трет-Bu)3.
23. Способ по п. 7, где на стадии B-c содержание палладия в сложноэфирном соединении составляет ≤ 100 частей на миллион.
24. Способ по п. 7, где на стадии B-d применяют толуол.
25. Способ по п. 7, где на стадии B-d исходное соединение применяют в количестве от 1,1 до 1,3 эквивалентов.
26. Способ по п. 7, где на стадии B-d исходное соединение применяют в количестве от 1,1 до 1,2 эквивалентов.
27. Способ по п. 7, где на стадии B-d применяют количество от 5,0 до 10,0 эквивалентов NMM.
28. Способ по п. 7, где на стадии B-d применяют количество от 5,0 до 7,5 эквивалентов NMM.
29. Способ по п. 7, где на стадии B-d применяют количество от 1,25 до 1,80 эквивалентов CDMT.
30. Способ по п. 7, где на стадии B-d применяют количество от 1,25 до 1,35 эквивалентов CDMT.
31. Способ по п. 7, где на стадии A предпочтительное промежуточное соединение в качестве предшественника циклизации применяют на последующей стадии разбавленным в толуоле, хлорбензоле или ксилене.
32. Способ по п. 7, где на стадии A предпочтительное промежуточное соединение в качестве предшественника циклизации применяют на последующей стадии разбавленным в толуоле.
33. Способ по п. 7, где на стадии A применяют абсолютное количество от 2,0 до 3,0 эквивалентов P2O5.
34. Способ по п. 7, где на стадии A применяют абсолютное количество около 2,5 эквивалентов P2O5.
35. Способ по п. 7, где на стадии A количество P2O5 относительно количества MSA составляет от 15,0 до 23,0 мас.%.
36. Способ по п. 7, где на стадии A количество P2O5 относительно количества MSA составляет около 23,0 мас.%.
37. Способ, как определено в любом из пп. 1 - 36, для получения соединения согласно формуле (IV), (IV’), (V) и/или (V’):
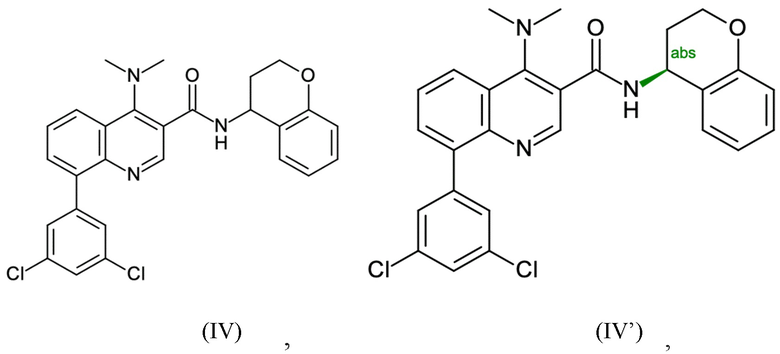
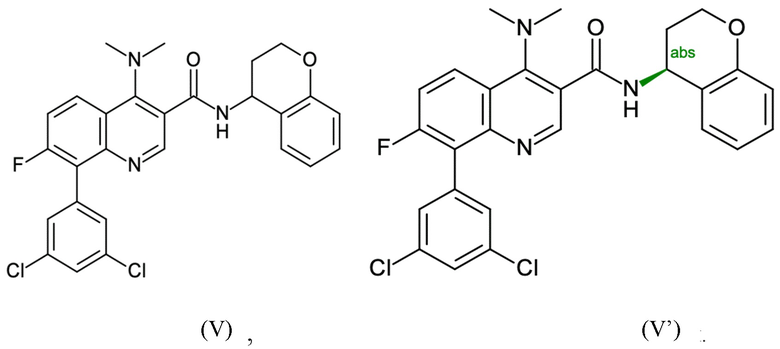
38. Способ получения промежуточных соединений согласно формуле (a-II-1), как определено в любом из пп. 1 и 3 - 6,
посредством проведения стадии способа B-a, как определено в любом из пп. 1 - 36, с последующим выделением и при необходимости очисткой полученных соединений.
39. Способ получения промежуточных соединений согласно формуле (a-IV), как определено в любом из пп. 1 и 3 - 6, и промежуточных соединений
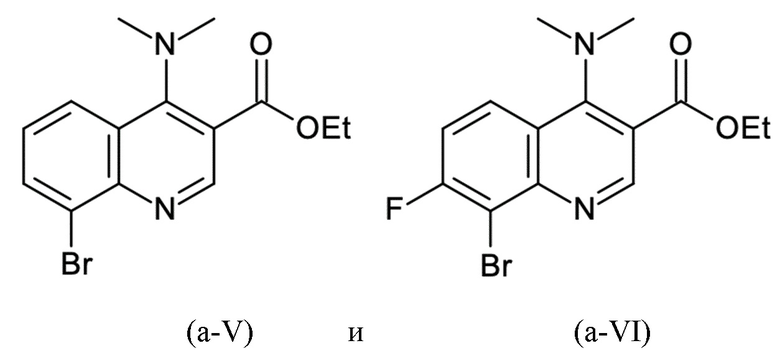
посредством проведения стадии способа B-a, как определено в любом из пп. 1 - 36, с последующим выделением и при необходимости очисткой полученных соединений.
40. Способ получения промежуточных соединений согласно формуле (b-III), как определено в любом из пп. 1 и 3 – 6,
посредством проведения стадии способа B-b, как определено в любом из пп. 1 - 36, с последующим выделением и при необходимости очисткой полученных соединений.
41. Способ получения промежуточных соединений
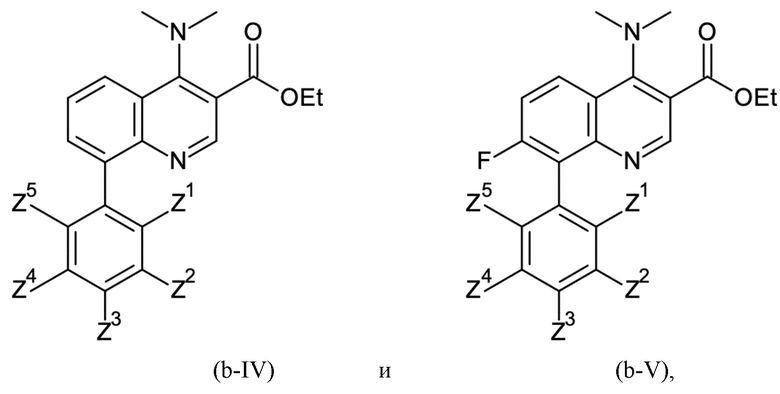
где Z1 - Z5 имеют значения, как определено в любом из предшествующих пунктов,
посредством проведения стадии способа B-b, как определено в любом из пп. 1 - 36, с последующим выделением и при необходимости очисткой полученных соединений.
42. Способ получения промежуточных соединений согласно формуле (c-II-1), как определено в любом из пп. 1 и 3 – 6,
посредством проведения стадии способа B-c, как определено в любом из пп. 1 - 36,
с последующим выделением и при необходимости очисткой полученных соединений.
43. Способ получения промежуточных соединений согласно формуле (c-III), как определено в любом из пп. 1 и 3 – 6, и промежуточных соединений
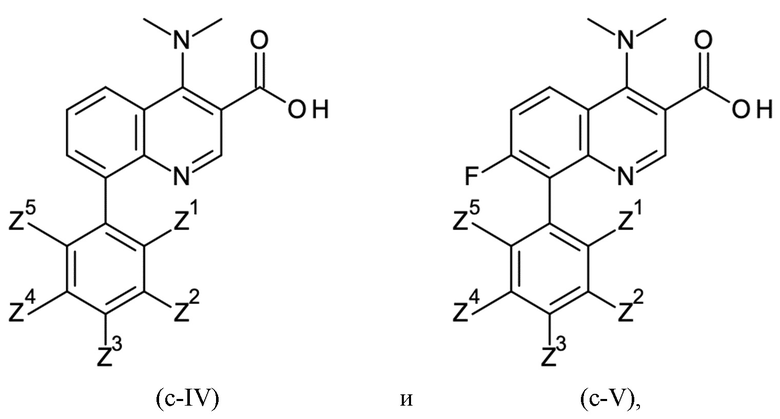
где Z1 - Z5 имеют значения, как определено в любом из предшествующих пунктов,
посредством проведения стадии способа B-c, как определено в любом из пп. 1 - 36,
с последующим выделением и при необходимости очисткой полученных соединений.
44. Способ получения промежуточных соединений, как определено формулой (d-II) в п. 1, посредством проведения стадии способа B-d, как определено в любом из пп. 1 - 36, при условии, что стадию B-d не проводят в качестве конечной стадии способа, с последующим выделением и при необходимости очисткой полученных соединений.
45. Применение промежуточных соединений согласно любой из формул (a-I-1), (a-II), (b-II), (c-II) и (d-II), как определено в любом из предшествующих пунктов, для получения соединений формулы (II), (III), (IV) или (V) или их соответствующей энантиомерной формы.
46. Применение промежуточных соединений согласно формуле (a-IV), как определено в любом из предшествующих пунктов, или получаемых способом по п. 39 для получения промежуточных соединений согласно формулам (b-III) и (c-III), как определено в любом из предшествующих пунктов;
или
промежуточных соединений согласно формуле (b-III), как определено в любом из предшествующих пунктов, или получаемых способом по п. 40 для получения промежуточных соединений согласно формулам (c-III), как определено в любом из предшествующих пунктов.
| WO 2015078800 A1, 04.06.2015 | |||
| RU 2008144806 A, 20.05.2010 | |||
| ПРОИЗВОДНЫЕ 5-ГИДРАЗИНОХИНОЛОНА, КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ИНФЕКЦИОННОГО ЗАБОЛЕВАНИЯ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ | 1993 |
|
RU2126000C1 |
| SANJAY K | |||
| SRIVASTAVA et al., Quinolones: Novel Probes in Antifilarial Chemotheraphy | |||
| JOURNAL OF MEDICINAL CHEMISTRY, 2000, vol.43, no.11, p.2275-2279 | |||
| CYRUS J | |||
| OHNMACHT et al., Antimalarials | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| alpha-Dibutylaminomethyl-and | |||
