Настоящее изобретение относится к соединениям, обладающим ингибирующей активностью в отношении Янус-киназы и, в частности, ингибирующей активностью в отношении TYK2-киназы, содержащим их фармацевтическим композициям и их применению в лечении различных заболеваний, таких как аутоиммунные заболевания.
Уровень техники
Протеинкиназы представляют собой большое семейство структурно родственных ферментов, которые отвечают за контроль самых разнообразных процессов трансдукции сигнала в клетке (Hardie and Hanks (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA). Киназы могут быть классифицированы по семействам в зависимости от субстратов, которые они фосфорилируют (например, тирозиновые протеинкиназы, серин/треониновые протеинкиназы, липидкиназы и т.д.). Были идентифицированы мотивы последовательностей, которые в целом соответствуют каждому семейству этих киназ (например, Hanks and Hunter, FASEB J., (1995) 9. 576-596; Knighton et al., Science, (1991) 253, 407-414; Hiles et al., Cell, (1992) 70, 419-429; Kunz et al., Cell, (1993) 73, 585-596; Garcia-Bustos et al., EMBO J., (1994) 13, 2352-2361).
Протеинкиназы могут быть охарактеризованы по механизмам регуляции. Эти механизмы включают, например, аутофосфорилирование, трансфосфорилирование другими киназами, белок-белковые взаимодействия, белок-липидные взаимодействия и белок-полинуклеотидные взаимодействия. Отдельно взятую протеинкиназу может регулировать более чем один механизм.
Киназы регулируют множество различных клеточных процессов, включающих, не ограничиваясь перечисленным, пролиферацию, дифференцировку, апоптоз, подвижность, транскрипцию, трансляцию и другие процессы сигнализации, путем добавления фосфатных групп к целевым белкам. Эти события фосфорилирования действуют в качестве молекулярных переключателей «включено/выключено», которые могут модулировать или регулировать биологическую функцию целевого белка. Фосфорилирование целевых белков происходит в ответ на разнообразные внеклеточные сигналы (гормоны, нейротрансмиттеры, факторы роста и дифференцировки и т.д.), события клеточного цикла, стрессы, обусловленные окружающей средой или питанием, и т.д. Соответствующая протеинкиназа функционирует в сигнальных путях, активируя или инактивируя (прямо или непрямо), например, метаболический фермент, регуляторный белок, рецептор, белок цитоскелета, ионный канал или насос, или фактор транскрипции. Неконтролируемая передача сигналов вследствие нарушенного контроля фосфорилирования белков задействована в целом ряде заболеваний, включая, например, воспаление, рак, аллергию/астму, заболевания и состояния иммунной системы, заболевания и состояния центральной нервной системы и ангиогенез.
Семейство Янус-киназ (JAK) представляет собой семейство внутриклеточных нерецепторных тирозинкиназ, размер которых варьирует в пределах от 120 до 140 кДа, передающих опосредованные цитокинами сигналы через путь JAK-STAT. Семейство JAK играет роль в цитокин-зависимой регуляции пролиферации и функции клеток, участвующих в иммунном ответе. В настоящее время известны четыре представителя семейства JAK у млекопитающих: JAK1, JAK2, JAK3 и TYK2. JAK1, JAK2 и TYK2 экспрессируются повсеместно, в то время как JAK3 экспрессируется в миелоидных и лимфоидных линиях. Представители семейства JAK представляют собой нерецепторные протеинкиназы, которые ассоциированы со многими гемопоэтиновыми цитокинами, рецепторным тирозинкиназами и рецепторами, сопряженными с G-белками (GPCR).
Каждый белок JAK-киназы обладает киназным доменом и каталитически неактивным псевдокиназным доменом. JAK-белки связываются с цитокиновыми рецепторами через свои аминоконцевые домены FERM (белок полосы 4.1, эзрин, радиксин, моезин). После связывания цитокинов с их рецепторами JAK-белки активируются и фосфорилируют указанные рецепторы, создавая таким образом сайты докинга для сигнальных молекул, в частности, для представителей семейства переносчиков сигнала и активаторов транскрипции (STAT) (Yamaoka et al., 2004. The Janus kinases (Jaks). Genome Biology 5(12): 253).
JAK1, JAK2 и TYK2 экспрессируются у млекопитающих повсеместно. TYK2 активирует зависимую от переносчика сигнала и активатора транскрипции (STAT) экспрессию генов и функциональные ответы рецепторов интерлейкина-12, интерлейкина-23 и интерферонов типов I и III (Papp et al., The New England Journal of Medicine, 12 сентября 2018 года, DOI: 10.1056/NEJMoa1806382, с цитируемыми источниками). Эти цитокиновые пути принимают участие в патологических процессах, ассоциированных с иммуноопосредованными нарушениями, включая псориаз, и, как сообщается (Papp и соавт., выше), отличаются от ответов, управляемых Янус-киназой (JAK) 1 (JAK1), JAK1 и JAK3 в комбинации, JAK2 или другими сигнальными киназами.
Интерлейкин-23 (ИЛ-23), состоящий из двух субъединиц, p19 и p40, считается необходимым для выживания и распространения Th17-клеток, которые продуцируют провоспалительные цитокины, такие как ИЛ-17A, ИЛ-17F, ИЛ-6 и фактор некроза опухолей α (ФНО-α) (см. заявку WO2014/07466 и цитируемые там источники). Сообщается, что эти цитокины имеют решающее значение как посредники в патобиологии ряда аутоиммунных заболеваний, включая ревматоидный артрит, рассеянный склероз, воспалительное заболевание кишечника и волчанку.
ИЛ-23 действует через гетеродимерный рецептор, который состоит из ИЛ-12Rβ1 и ИЛ-23R.
ИЛ-12, наряду с такой же субъединицей p40, как у ИЛ-23, содержит субъединицу p35 и действует через гетеродимерный рецептор, который состоит из ИЛ-12R1β и ИЛ-12Rβ2. ИЛ-12 необходим для развития Th1-клеток и секреции интерферона-y (ИФН-y) - цитокина, который играет критически важную роль в иммунитете, стимулируя экспрессию главного комплекса гистосовместимости (MHC), переключение класса B-клеток на подклассы иммуноглобулина G (IgG) и активацию макрофагов (Gracie, J. A. et al., «Interleukin-12 induces interferon-gamma-dependent switching of IgG alloantibody subclass», Eur. J. Immunol, 26: 1217-1221 (1996); Schroder, K. et al., «Interferon-gamma: an overview of signals, mechanisms and functions», J. Leukoc. Biol, 75(2): 163-189 (2004)).
TYK2 связывается с субъединицей ИЛ-12Rβ1 в составе рецепторов к ИЛ-12 и ИЛ-23.
О важности цитокинов, содержащих p40, для аутоиммунитета свидетельствует открытие того, что мыши с дефицитом p40, p19 или ИЛ-23R защищены от заболевания в моделях рассеянного склероза, ревматоидного артрита, воспалительного заболевания кишечника, волчанки и псориаза, среди прочих (Kyttaris, V.C. et al., «Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice», J. Immunol, 184:4605-4609 (2010); Hong, K. et al., «IL-12, independently of IFN-gamma, plays a crucial role in the pathogenesis of a murine psoriasis like skin disorder», J. Immunol, 162:7480-7491 (1999); Hue, S. et al., «Interleukin-23 drives innate and T cell-mediated intestinal inflammation», J. Exp. Med., 203:2473-2483 (2006); Cua, D.J. et al., «Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain», Nature, 421 :744-748 (2003); Murphy, C.A. et al., «Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation», J. Exp. Med, 198: 1951-1957 (2003)).
Роль TYK2 в биологическом ответе на цитокины была охарактеризована с применением мутантной человеческой линии клеток, устойчивой к воздействию интерферонов типа I (ИФН), и демонстрацией того, что отвечаемость на ИФНα можно восстановить путем генетической комплементации TYK2 (Velazquez et al., 1992. Cell 70, 313-322). Дальнейшие исследования in vitro выявили участие TYK2 в сигнальных путях множества других цитокинов, вовлеченных как во врожденный, так и в приобретенный иммунитет. При этом в исследовании на TYK2-/- мышах обнаружены менее выраженные иммунологические дефекты, чем было спрогнозировано (Karaghiosoff et al., 2000. Immunity 13, 549-560; Shimoda et al., 2000. Immunity 13, 561-671). Неожиданным образом, мыши с дефицитом TYK2 демонстрируют только сниженную отвечаемость на ИФНα/β и нормальный сигнал в ответ на интерлейкин 6 (ИЛ-6) и интерлейкин 10 (ИЛ-10), оба из которых активируют TYK2 in vitro. И напротив, как было продемонстрировано, TYK2 необходима для передачи сигналов ИЛ-12, и отсутствие TYK2 приводит к дефектной активации STAT4 и неспособности T-клеток этих мышей дифференцироваться в Th1-клетки, которые продуцируют ИФН-y. В соответствии с вовлеченностью TYK2 в опосредование биологических эффектов ИФН типа I и ИЛ-12, TYK2-/- мыши были более восприимчивы к вирусным и бактериальным инфекциям.
Первый пациент с аутосомно-рецессивным дефицитом TYK2 был описан в публикации Minegishi et al., 2006. Immunity 25, 745-755.
Гомозиготная делеция четырех пар оснований (GCTT с нуклеотида 550 в гене TYK2) и, как следствие, мутация со сдвигом рамки считывания в кодирующей ДНК пациента вводила преждевременный стоп-кодон и приводила к усечению белка TYK2 по аминокислоте 90. Фенотип этой нулевой мутации в клетках человека был гораздо тяжелее, чем было спрогнозировано в исследованиях на клетках мыши, где отсутствовал TYK2. У пациента проявлялись клинические признаки, напоминающие первичный иммунодефицит при синдроме гипериммуноглобулинемии Е (HIES), который включает рецидивирующие абсцессы кожи, атопический дерматит, значительное повышение уровня IgE в сыворотке крови и восприимчивость к многочисленным оппортунистическим инфекциям.
В отличие от данных, сообщаемых о TYK2-/- мышах, было обнаружено нарушение сигнализации широкого ряда цитокинов, что указывает на неизбыточность роли TYK2 человека в функционировании ИФН типа I, ИЛ-6, ИЛ-10, ИЛ-12 и ИЛ- 23. Также наблюдался дисбаланс в дифференцировке хелперных T-клеток, при этом T-клетки пациента демонстрировали чрезвычайную склонность к развитию Th2-клеток, продуцирующих ИЛ-4, и нарушенную дифференцировку Th1-клеток. Безусловно, указанные дефекты сигнализации цитокинов могли являться причиной многих из описанных клинических проявлений, например, атопического дерматита и повышенных уровней IgE (повышенная активность Th2), увеличения частоты встречаемости вирусных заболеваний (дефицит ИФН), инфицирования внутриклеточными бактериями (дефицит ИЛ-12/Th1) и внеклеточными бактериями (дефицит ИЛ-6 и ИЛ-23/Th17).
Последующих семерых пациентов с дефицитом TYK2 из пяти семей и четырех различных этнических групп идентифицировали Kreins с соавторами (Kreins et al., стр. 1-22, The Journal of Experimental Medicine, опубликован 24 августа 2015 года). Эти пациенты являлись гомозиготами по одной из пяти нулевых мутаций. Сравнив данные, полученных Minegishi et al., с данными, полученными от семи последующих пациентов с дефицитом TYK2, Kreins et al. сделали вывод о том, что основным клиническим фенотипом дефицита TYK2 являются микобактериальные и/или вирусные инфекции, вызванные нарушением ответной реакции на ИЛ-12 и ИФН-α/β, а нарушения ответа на ИЛ-6 и HIES не являются неотъемлемыми характеристиками дефицита TYK2 у людей.
Растущий объем данных полногеномного поиска ассоциаций позволяет предположить, что однонуклеотидные полиморфизмы (SNP) в гене TYK2 оказывают значимое влияние на предрасположенность к аутоиммунным заболеваниям.
Менее эффективные варианты TYK2 ассоциированы с защитой от системной красной волчанки, СКВ (SLE) (TYK2 rs2304256 и rsl2720270, Sigurdsson et al., 2005. Am. J. Hum. Genet. 76, 528-537; Graham et al., 2007. Rheumatology 46, 927-930; Hellquist et al., 2009. J. Rheumatol. 36, 1631-1638; Jarvinen et al., 2010. Exp. Dermatol. 19, 123-131) и рассеянного склероза (MS) (rs34536443, Ban et al., 2009. Eur. J. Hum. Genet. 17, 1309-1313; Mero et al., 2009. Eur. J. Hum. Genet. 18, 502-504), в то время как предсказанные мутации с приобретением функции повышают предрасположенность к воспалительному заболеванию кишечника (ВЗК) (rs280519 и rs2304256, Sato et al., 2009. J. Clin. Immunol. 29, 815-825).
У человека, как сообщается (см. WO2014074661 с цитируемыми источниками), индивидуумы, экспрессирующие неактивный вариант TYK2, защищены от рассеянного склероза и, возможно, других аутоиммунных нарушений, а полногеномные исследования ассоциаций выявили, что другие варианты TYK2 ассоциированы с такими аутоиммунными нарушениями, как болезнь Крона, псориаз, системная красная волчанка и ревматоидный артрит, что дополнительно демонстрирует важность TYK2 для аутоиммунитета.
В подтверждение вовлеченности TYK2 в иммунопатологические болезненные процессы, было показано, что мыши B10.D1, несущие миссенс-мутацию в псевдокиназном домене TYK2, что приводит к отсутствию кодируемого белка TYK2, устойчивы как к аутоиммунному артриту (коллаген-индуцированный артрит, CIA), так и к экспериментальному аутоиммунному энцефаломиелиту, EAE (Shaw et al., 2003, PNAS 100, 11594-11599; Spach et al., 2009. J. Immunol. 182, 7776-7783). Помимо этого, недавнее исследование показало, что TYK2-/- мыши абсолютно устойчивы к EAE, индуцированному миелиновым олигодендроцитарным гликопротеином (MOG) (Oyamada et al., 2009. J. Immunol. 183, 7539-7546). Устойчивость у этих мышей сопровождалась отсутствием CD4 T-клеток, инфильтрирующих спинной мозг, неспособностью к сигнализации через ИЛ-12R и ИЛ-23R и, следовательно, неспособностью к повышающей регуляции энцефалитогенных уровней ИФН-y и ИЛ-17.
Было определено, что сверхэкспрессия TYK2-киназы задействована в развитии некоторых болезненных состояний. Например, повышенные уровни TYK2 были обнаружены у пациентов, страдающих прогрессирующим легочным саркоидозом (Schischmanoff et al., Sarcoidosis Vasc. Diffuse., 2006, 23(2), 101-7).
Таким образом, имеющиеся данные убедительно демонстрируют, что TYK2 играет важнейшие роли как во врожденном, так и в приобретенном иммунитете. Недостаток экспрессии TYK2 проявляется в ослабленной сигнализации многочисленных провоспалительных цитокинов и выраженном дисбалансе в дифференцировке хелперных T-клеток. Помимо этого, данные поиска генетических ассоциаций подтверждают, что TYK2 является геном перекрестной предрасположенности к аутоиммунным заболеваниям. В совокупности эти доводы позволяют предложить TYK2 в качестве мишени для лечения воспалительных и аутоиммунных заболеваний.
В литературных источниках сообщалось о нескольких ингибиторах семейства JAK, которые могут быть полезны в медицинской сфере (Ghoreschi et al., 2009. Immunol Rev, 228:273-287). Было выдвинуто предположение, что селективный ингибитор TYK2, который с большей эффективностью ингибирует TYK2, чем JAK2, может обладать благоприятными терапевтическими свойствами, поскольку ингибирование JAK2 может вызывать анемию (Ghoreschi et al., 2009. Nature Immunol. 4, 356-360).
Papp с соавторами (Papp et al. The New England Journal of Medicine, 12 сентября 2018 года, DOI: 10.1056/NEJMoa1806382) представили результаты, полученные в клинических исследованиях фазы II BMS-986165, селективного ингибитора TYK2 для перорального приема, при лечении псориаза и пришли к выводу, что полученные результаты указывают на терапевтическую полезность.
В WO2014/074661 (Bristol-Myers Squibb) раскрыт класс пиридазиновых и триазиновых амидов в качестве ингибиторов TYK2, которые подходят для модуляции ИЛ-12, ИЛ-23 и/или ИФНα. Предполагается, что указанные соединения будут полезны в лечении различных воспалительных и аутоиммунных заболеваний.
В WO2016/027195 (Pfizer) раскрыт ряд соединений аминопиримидинила, которые обладают ингибирующей активностью в отношении JAK-киназы, в том числе активностью в отношении TYK2-киназы.
В WO2012/000970 (Cellzome) раскрыт ряд триазолопиридинов в качестве ингибиторов TYK2-киназы. Заявка WO2011/113802 (компания Roche) раскрывает ряд имидазопиридинов в качестве ингибиторов TYK2-киназы. Свойства JAK-киназ и их релевантность в качестве терапевтических мишеней также раскрыты в WO2008/156726, WO2009/155156, WO2010/005841 и WO2010/011375, все на имя компании Merck.
В WO2010/055304 и EP2634185 (обе на имя компании Sareum) раскрыто семейство замещенных оксазолкарбоксамидов для применения в профилактике или лечении аутоиммунных заболеваний и, в частности, рассеянного склероза. Соединения, раскрытые в WO2010/055304, описаны как ингибиторы fms-подобной тирозинкиназы 3 (FLT3). Ингибирующий эффект оксазолкарбоксамидов в отношении киназ также раскрыт в международной патентной заявке WO2008/139161 (Sareum).
В WO2015/032423 (компания Sareum) раскрыто применение подгруппы соединений оксазолкарбоксамида в качестве ингибиторов TYK2-киназы. Указанные соединения, согласно описанию, полезны в лечении воспалительных и иммунологических нарушений, таких как аутоиммунные заболевания.
В WO2018/073438 (компания Sareum) раскрыто применение подгруппы соединений оксазолкарбоксамида, которые обладают ингибирующей активностью в отношении TYK2-киназы, для применения в лечении T-клеточных лимфобластных лейкозов и рака (такого как рак системы кроветворения), при котором выживание раковых клеток зависит от Янус-киназы TYK2.
Определенные соединения, раскрытые в WO2015/032423 и WO2018/073438, включают амид 2-(2-хлор-6-фтор-фенил)-5-[4-(морфолин-4-карбонил)- фениламино]-оксазол-4-карбоновой кислоты (соединение A) и амид 2-(2,6-дихлор-фенил)-5-[4-(морфолин-4-карбонил)-фениламино]-оксазол-4-карбоновой кислоты (соединение B).
Изобретение
Настоящее изобретение относится к небольшой группе оксазолкарбоксамидов, которые обладают улучшенной активностью против TYK2-киназы и избирательностью в отношении нее, а также улучшенными фармакокинетическими свойствами по сравнению с соединениями, раскрытыми в WO2015/032423 и WO2018/073438, и, в частности, по сравнению с вышеупомянутыми соединением A и соединением B.
Таким образом, согласно первому варианту осуществления (вариант осуществления 1.1) настоящее изобретение обеспечивает соединение, которое имеет формулу (1):

либо представляет собой ее соль или таутомер; где R1 представляет собой водород или фтор.
Конкретные соединения по настоящему изобретению представлены во вариантах осуществления 1.2-1.9 ниже.
1.2 Соединение согласно варианту осуществления 1.1, в котором R1 представляет собой водород; указанное соединение имеет формулу (2):
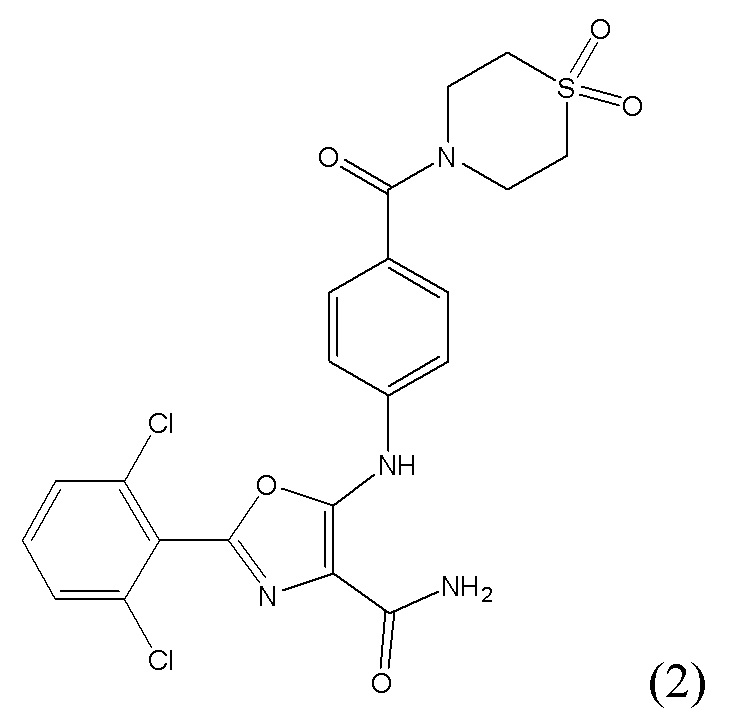
либо представляет собой ее соль или таутомер.
1.3 Соединение согласно варианту осуществления 1.1, в котором R1 представляет собой фтор; указанное соединение имеет формулу (3):
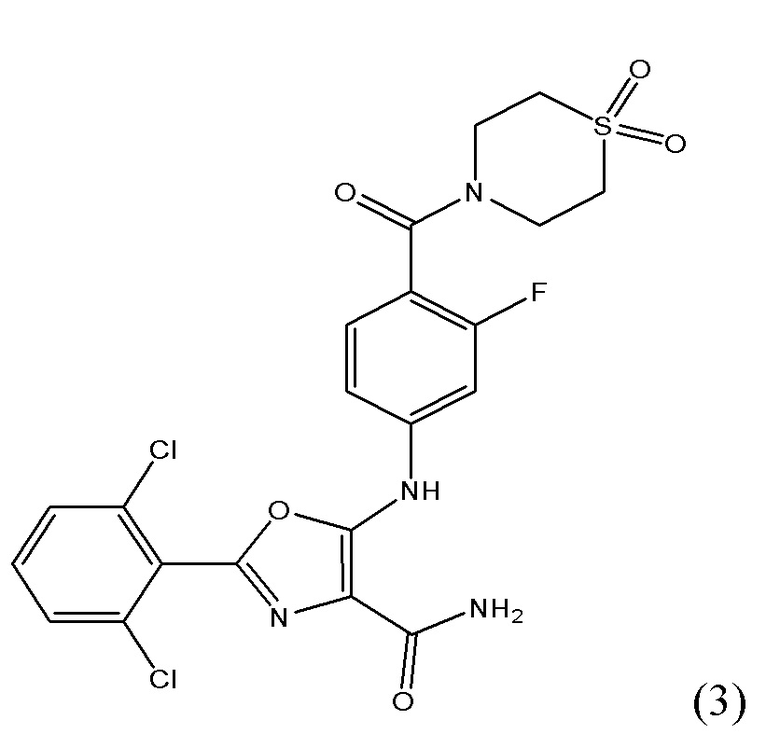
либо представляет собой ее соль или таутомер.
Соединения формул (1), (2) и (3) содержат субъединицы оксазола и анилина, обе из которых являются только слабоосновными. Следовательно, указанные соединения обычно представлены скорее в несолевой форме, а не в виде солей. Таким образом, согласно другому варианту осуществления (вариант осуществления 1.4), настоящее изобретение обеспечивает соединение согласно любому из вариантов осуществления 1.1-1.3, причем данное соединение представлено в несолевой форме.
При определенных обстоятельствах могут быть получены кислые соли с сильными кислотами, такими как соляная, серная и фосфорная, но такие соли предположительно будут, как правило, нестабильными. В тех случаях, когда могут быть получены соли, их можно синтезировать из исходного соединения традиционными химическими способами, такими как способы, описанные в публикации «Pharmaceutical Salts: Properties, Selection», and Use, P. Heinrich Stahl (ред.), Camille G. Wermuth (ред.), ISBN: 3-90639-026-8, тв. переплет, 388 стр., август 2002 г. В общем случае такие соли можно получить путем проведения реакции данного соединения в форме свободного основания с кислотой в воде или в органическом растворителе, или в смеси этих двух растворителей; обычно используют неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
В случаях, когда могут быть получены соли, они могут представлять собой фармацевтически приемлемые соли; примеры фармацевтически приемлемых солей рассмотрены в публикации Berge et al., 1977, «Pharmaceutically Acceptable Salts,» J. Pharm. Sci., Vol. 66, сс. 1-19. Однако в качестве промежуточных форм могут быть также получены соли, которые не являются фармацевтически приемлемыми и которые впоследствии можно преобразовать в фармацевтически приемлемые соли. Такие фармацевтически неприемлемые солевые формы, которые могут быть полезны, например, при очищении или разделении соединений согласно настоящему изобретению, также включены в настоящее изобретение.
Изотопы
Соединения для применения согласно настоящему изобретению, определенные в любом из вариантов осуществления 1.1-1.4, могут содержать одну или более изотопных замен, причем упоминание конкретного элемента подразумевает все изотопы данного элемента. Например, упоминание водорода подразумевает 1H, 2H (D) и 3H (T). Аналогичным образом, упоминание углерода и кислорода подразумевает 12C, 13C и 14C, а также 16O и 18O, соответственно.
Аналогичным образом, упоминание конкретной функциональной группы также подразумевает варианты изотопов, если из контекста не следует иное.
Изотопы могут быть радиоактивными или нерадиоактивными. В одном варианте осуществления настоящего изобретения (вариант осуществления 1.5) соединение согласно любому из вариантов осуществления 1.1-1.4 не содержит радиоактивных изотопов. Такие соединения предпочтительны для терапевтического применения. При этом в другом варианте осуществления (вариант осуществления 1.6) соединение по любому из вариантов осуществления 1.1-1.4 может содержать один или более радиоизотопов. Соединения, которые содержат такие радиоизотопы, могут быть полезны в диагностическом контексте.
Сольваты
Соединения для применения по определению в любом из вариантов осуществления 1.1-1.4 могут образовывать сольваты.
Предпочтительными сольватами являются сольваты, образованные путем внедрения в твердотельную структуру (например, в кристаллическую структуру) соединений по настоящему изобретению молекул нетоксичного фармацевтически приемлемого растворителя (называемого ниже сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем перекристаллизации соединений по настоящему изобретению с применением растворителя или смеси растворителей, которые содержат сольватирующий растворитель. В каждом конкретном случае можно определить, образовался сольват или нет, подвергнув кристаллы указанного соединения анализу с применением хорошо известных и стандартных методик, таких как термогравиметрический анализ, ТГА (TGE), дифференциальная сканирующая калориметрия, ДСК (DSC) и рентгеновская кристаллография.
Cольваты могут быть стехиометрическими или нестехиометрическими сольватами.
Особенно предпочтительными сольватами являются гидраты, причем примеры гидратов включают гемигидраты, моногидраты и дигидраты.
Таким образом, в других вариантах осуществления, 1.7 и 1.8, настоящее изобретение обеспечивает:
1.7 Соединение согласно любому из вариантов осуществления 1.1-1.6, причем данное соединение представлено в форме сольвата.
1.8 Соединение согласно варианту осуществления 1.7, причем сольват представляет собой гидрат.
Для более подробного рассмотрения сольватов, а также способов, применяемых для их получения и определения их характеристик, см. публикацию Bryn et al., Solid-State Chemistry of Drugs, второе издание, опубликованное компанией SSCI, Уэст-Лафайетт, штат Индиана, США, 1999, ISBN 0-967-06710-3.
В альтернативном варианте соединение по настоящему изобретению может быть безводным, а не существовать в виде гидрата. Следовательно, в другом варианте осуществления (вариант осуществления 1.9) соединение по определению в любом из вариантов осуществления 1.1-1.6, представлено в безводной форме.
Биологическая активность
Соединения формул (1), (2) и (3) по определению в вариантах осуществления 1.1-1.9 являются сильнодействующими селективными ингибиторами TYK2-киназы. Ингибирующую активность указанных соединений в отношении TYK2-киназы можно определить с применением анализов, описанных в примерах ниже.
Экспериментальные данные, полученные для соединений (2) и (3), демонстрируют, что соединения по настоящему изобретению обладают существенными преимуществами по сравнению с наиболее сходным по структуре соединением (соединением B) из WO2015/032423, Таким образом, оба соединения (2) и (3) более активны, чем наиболее близкое из известных соединений (соединение B) в исследовании ингибирования TYK2-киназы, а также оба они обладают большей селективностью, чем соединение B, в отношении TYK2-киназы по сравнению с киназами JAK1, JAK2 и JAK3, Более того, соединения (2) и (3) обладают меньшей предрасположенностью к воздействию на калиевые каналы сердца человека (hERG) по сравнению с известным из уровня техники сравнительным соединением B. Помимо этого, в анализе стабильности в гепатоцитах соединения (2) и (3) продемонстрировали сниженную скорость клиренса и, следовательно, более длительный период полужизни, чем у сравнительного соединения B.
В совокупности эти данные указывают на то, что соединения (2) и (3) не только представляют собой более сильнодействующие и более селективные ингибиторы TYK2-киназы, чем сравнительное соединение B, но и, более того, обладают лучшими фармакокинетическими свойствами, чем соединение B.
Ингибирующая активность этих соединений в отношении TYK2-киназы может быть использована в разнообразных способах лечения заболеваний, в которых TYK2 принимает участие в развитии или прогрессировании заболевания. Разнообразные варианты применения указанных соединений обычно включают приведение соединений в контакт с TYK2-киназой. Ингибирование TYK2-киназы может иметь место или in vitro, или in vivo.
Соответственно, в других вариантах осуществления настоящее изобретение обеспечивает:
2.1 Способ ингибирования TYK2-киназы, который включает приведение в контакт с TYK2-киназой эффективно ингибирующего TYK2-киназу количества соединения по определению в любом из вариантов осуществления 1.1-1.9.
2.2 Способ согласно варианту осуществления 2.1, в котором ингибирование TYK2-киназы происходит in vitro.
2.3 Способ согласно варианту осуществления 2.1, в котором ингибирование TYK2-киназы происходит in vivo.
2.4 Соединение по определению в любом из вариантов осуществления 1.1-1.9, для применения в качестве ингибитора TYK2-киназы.
2.5 Соединение по определению в любом из вариантов осуществления 1.1-1.9, для применения в медицине.
Ингибирование TYK2-киназы в предпочтительном варианте происходит in vivo в качестве части терапевтического лечения заболевания или состояния, в которое вовлечена TYK2-киназа.
Соединения по настоящему изобретению представляют собой селективные ингибиторы TYK2 и значимо более активны в отношении TYK2-киназы, чем в отношении киназ JAK2 и JAK3. Указанные соединения обладают относительно слабой активностью в отношении широкого спектра других киназ и, в частности, тех киназ, которые представляют собой общепризнанные мишени противоопухолевой химиотерапии. Таким образом, указанные соединения обладают, например, относительно малой активностью в отношении Chk1-киназы, Аврора-киназ, протеинкиназы B (Akt) и циклинзависимых киназ (CDK-киназ), которые задействованы в прохождении клеточного цикла. Отсутствие активности в отношении киназ, которые обычно рассматриваются как противоопухолевые мишени, благоприятно для соединений, которые могут применяться, например, при длительном лечении воспалительных и аутоиммунных заболеваний.
Выдвинуто предположение о том, что соединения по настоящему изобретению, исходя из их ингибирующей активности в отношении TYK2, могут быть полезны в лечении по меньшей мере некоторых из заболеваний и нарушений, рассматриваемых ниже, в том числе воспалительных заболеваний или состояний, иммунологических заболеваний или состояний, аутоиммунных заболеваний, аллергических заболеваний или нарушений, отторжения трансплантатов (отторжения аллогенных трансплантатов), болезни «трансплантат против хозяина»; в лечении сепсиса и септического шока.
В контексте настоящего изобретения аутоиммунное заболевание представляет собой заболевание, которое по меньшей мере частично вызвано иммунной реакцией организма в отношении его собственных компонентов, например, белков, липидов или ДНК. Примерами органоспецифических аутоиммунных нарушений являются инсулинозависимый диабет (I типа), который поражает поджелудочную железу, тиреоидит Хашимото и болезнь Грейвса, которые поражают щитовидную железу, пернициозная анемия, которая поражает желудок, болезнь Кушинга и болезнь Аддисона, которые поражают надпочечники, хронический активный гепатит, который поражает печень; синдром поликистозных яичников (СПКЯ), целиакия, псориаз, воспалительное заболевание кишечника (ВЗК), волчаночный нефрит (воспаление почки) и анкилозирующий спондилит. Примерами органонеспецифических аутоиммунных нарушений являются ревматоидный артрит, рассеянный склероз, системная красная волчанка и тяжелая миастения. Диабет I типа возникает в результате селективной клеточной цитотоксической реакции аутореактивных T-клеток в отношении бета-клеток островков Лангерганса, продуцирующих инсулин. Другие воспалительные или иммунные заболевания и нарушений, страдающие которыми пациенты могут получить пользу от лечения соединениями по настоящему изобретению, включают: кожное воспаление, возникшее в результате радиоактивного облучения; астму; аллергическое воспаление; хроническое воспаление; воспалительное офтальмологическое заболевание; синдром сухого глаза, ССГ (DES, также известный как сухой кератоконъюнктивит или синдром слезной дисфункции); увеит (например, хроническая прогрессирующая или рецидивирующая формы неинфекционного увеита); очаговая алопеция; первичный билиарный цирроз; и системный склероз.
Ревматоидный артрит (РА) представляет собой хроническое прогрессирующее инвалидизирующее воспалительное заболевание, которое поражает приблизительно 1% населения земного шара. РА представляет собой симметричный полиартикулярный артрит, который поражает в первую очередь мелкие суставы кистей и стоп. Наряду с воспалением синовиальной оболочки, выстилки суставов, агрессивный фронт ткани, называемой паннусом, внедряется в локальные структуры сустава и разрушает их (Firestein 2003, Nature 423:356-361).
Воспалительное заболевание кишечника (ВЗК) характеризуется хроническим рецидивирующим воспалением кишечника. ВЗК подразделяют на фенотипы болезни Крона и язвенного колита. Болезнь Крона чаще всего затрагивает терминальный отдел подвздошной кишки и ободочную кишку и является трансмуральной и периодической. Напротив, при язвенном колите воспаление является непрерывным и ограничивается слоем слизистой оболочки прямой и ободочной кишки. Приблизительно в 10% случаев, когда затронуты только прямая и ободочная кишка, невозможно окончательно классифицировать заболевание как болезнь Крона или язвенный колит, и такие случаи обозначают как «неуточненный колит». Оба заболевания включают внекишечные воспаления кожи, глаз или суставов. Повреждения, индуцированные нейтрофилами, могут быть предотвращены путем применения ингибиторов миграции нейтрофилов (Asakura et al., 2007, World J. Gastroenterol. 13(15):2145-9).
Псориаз представляет собой хронический воспалительный дерматоз, который поражает приблизительно 2% населения. Он характеризуется красными сквамозными областями на коже, которые обычно появляются на волосистой части головы, локтях и коленях, и может быть ассоциирован с тяжелым артритом. Очаговые поражения вызваны аномальной пролиферацией кератиноцитов и инфильтрацией воспалительных клеток в дерму и эпидермис (Schon et al., 2005, New Engl. J. Med. 352: 1899-1912).
Системная красная волчанка (СКВ) представляет собой хроническое воспалительное заболевание, обусловленное опосредованной T-клетками активацией B-клеток, которая приводит к гломерулонефриту и почечной недостаточности. СКВ у человека характеризуется на ранних стадиях распространением долгоживущих аутореактивных CD4+ клеток памяти (D'Cruz et al., 2007, Lancet 369(9561):587-596).
Отторжение трансплантата (отторжение аллогенного трансплантата) включает, не ограничиваясь перечисленным, острое и хроническое отторжение аллотрансплантата, например, после трансплантации почки, сердца, печени, легкого, костного мозга, кожи и роговицы. Известно, что T-клетки играют основополагающую роль в специфическом иммунном ответе, вызывающем отторжение аллотрансплантата. В случае сверхострого, острого и хронического отторжения трансплантата органа может проводиться лечение. Сверхострое отторжение возникает в течение нескольких минут после трансплантации. Острое отторжение преимущественно возникает в течение шести-двенадцати месяцев после трансплантации. Сверхострое и острое отторжение обычно обратимы при лечении иммунодепрессивными средствами. Хроническое отторжение, характеризующееся постепенной потерей функции органа, является постоянной угрозой для реципиентов трансплантатов, поскольку может возникать в любое время после трансплантации.
Болезнь «трансплантат против хозяина», БТПХ (GVHD) представляет собой основное осложнение при аллогенной трансплантации костного мозга (BMT). БТПХ обусловлена T-клетками донора, которые распознают и реагируют на отличия реципиента в системе комплекса гистосовместимости, что приводит к значимым морбидности и смертности.
Легочный саркоидоз представляет собой относительно редкое воспалительное нарушение неизвестной этиологии, однако, как было показано, ассоциированный с повышенными уровнями TYK2, и обычно развивается у взрослых в возрасте 20-50 лет. Легочный саркоидоз характеризуется небольшими узелками, или гранулемами в легких, которые обычно излечиваются и исчезают сами по себе. Тем не менее, в случае тех гранулем, которые не излечиваются, ткань может оставаться воспаленной и превращаться в рубцовую, или фиброзную. Легочный саркоидоз может перерастать в легочный фиброз, который деформирует структуру легких и может создавать помехи для дыхания.
Таким образом, в других вариантах осуществления настоящее изобретение обеспечивает:
2.6 Способ лечения заболевания или состояния у субъекта, который нуждается в этом, причем указанное заболевание или состояние выбрано из: аутоиммунного заболевания, воспалительного заболевания или состояния, иммунологического заболевания или состояния, аллергического заболевания или нарушения, отторжения трансплантата или болезни «трансплантат против хозяина», или заболевания или состояния, выбранного из сепсиса и септического шока, причем указанное заболевание или состояние восприимчиво к ингибированию TYK2, причем указанный способ включает введение субъекту эффективно ингибирующего TYK2 количества соединения по определению в любом из вариантов осуществления 1.1-1.9.
2.7 Соединение по определению в любом из вариантов осуществления 1.1-1.9 для применения в лечении заболевания или состояния, причем указанное заболевание или состояние выбрано из: аутоиммунного заболевания, воспалительного заболевания или состояния, иммунологического заболевания или состояния, аллергического заболевания или нарушения, отторжения трансплантата и болезни «трансплантат против хозяина»; или для применения в лечении сепсиса или септического шока, причем указанное заболевание или состояние восприимчиво к ингибированию TYK2.
2.8 Применение соединения по определению в любом из вариантов осуществления 1.1-1.9, для получения медикамента для лечения заболевания или состояния, выбранного из: аутоиммунного заболевания, воспалительного заболевания или состояния, иммунологического заболевания или состояния, аллергического заболевания или нарушения, отторжения трансплантата и болезни «трансплантат против хозяина»; или для применения в лечении сепсиса или септического шока, причем указанное заболевание или состояние восприимчиво к ингибированию TYK2.
2.9 Способ лечения аутоиммунного заболевания у субъекта, который нуждается в этом, причем указанный способ включает введение субъекту эффективно ингибирующего TYK2 количества соединения по определению в любом из вариантов осуществления 1.1-1.9, таким образом, чтобы ингибировать TYK2-киназу у субъекта и тем самым блокировать воспалительный процесс, ассоциированный с аутоиммунным заболеванием, или уменьшить его степень.
2.10 Соединение по определению в любом из вариантов осуществления 1.1-1.9 для применения в способе лечения аутоиммунного заболевания у субъекта, который нуждается в этом, причем указанный способ включает введение субъекту эффективно ингибирующего TYK2 количества указанного соединения таким образом, чтобы ингибировать TYK2-киназу у субъекта и тем самым блокировать воспалительный процесс, ассоциированный с аутоиммунным заболеванием, или уменьшить его степень.
2.11 Применение соединения по определению в любом из вариантов осуществления 1.1-1.9 для получения медикамента для лечения аутоиммунного заболевания у субъекта, который нуждается в этом, путем введения субъекту эффективно ингибирующего TYK2 количества указанного соединения таким образом, чтобы ингибировать TYK2-киназу у субъекта и тем самым блокировать воспалительный процесс, ассоциированный с аутоиммунным заболеванием, или уменьшить его степень.
2.12 Способ лечения заболевания или состояния у субъекта, который нуждается в этом, причем указанное заболевание или состояние представляет собой такое заболевание или состояние, которое характеризуется или вызвано (по меньшей мере частично) сверхэкспрессией (повышенной экспрессией) TYK2-киназы или ассоциировано с ней, причем указанный способ включает введение субъекту эффективно ингибирующего TYK2 количества соединения по любому из вариантов осуществления 1.1-1.9.
2.13 Соединение по определению в любом из вариантов осуществления 1.1-1.9 для применения в лечении заболевания или состояния у субъекта, который нуждается в этом, причем указанное заболевание или состояние представляет собой такое заболевание или состояние, которое характеризуется или вызвано (по меньшей мере частично) сверхэкспрессией (повышенной экспрессией) TYK2-киназы или ассоциировано с ней.
2.14 Способ, соединение для применения или применение согласно любому из вариантов осуществления 2.6-2.13, причем указанное заболевание или состояние является аутоиммунным заболеванием.
2.15 Способ, соединение для применения или применение согласно любому из вариантов осуществления 2.6-2.13, причем указанное заболевание или состояние является аутоиммунным заболеванием, отличным от рассеянного склероза.
2.16 Способ, соединение для применения или применение согласно любому из вариантов осуществления 2.6-2.13, причем указанное заболевание или состояние является псориазом.
2.17 Способ, соединение для применения или применение согласно любому из вариантов осуществления 2.6-2.13, причем указанное заболевание или состояние является псориатическим артритом.
2.18 Способ согласно варианту осуществления 2.6, причем указанное заболевание или состояние является рассеянным склерозом.
Активность соединений по настоящему изобретению в качестве ингибиторов TYK2 может быть измерена при помощи анализа, представленного в примерах ниже, и уровень активности, демонстрируемый заданным соединением, может быть определен через значение IC50. Соединения по настоящему изобретению имеют значения IC50 в отношении TYK2-киназы менее 5 наномоль/л. Таким образом, для соединения, в котором R1 представляет собой водород (соединение (2)), IC50 в отношении TYK2 составляет 1,9 наномоль/л, в то время как для соединения, в котором R1 представляет собой фтор (соединение (3)), IC50 в отношении TYK2 составляет 4,7 наномоль/л.
Преимущество соединений по настоящему изобретению состоит в том, что они демонстрируют селективность в отношении TYK2-киназы по сравнению с другими киназами семейства JAK.
Например, в биохимических анализах соединение, в котором R1 представляет собой водород (соединение (2)), обладает приблизительно 25-кратно большей селективностью в отношении TYK2 по сравнению с JAK2 и 110-кратно большей в отношении TYK2 по сравнению с JAK3.
Соединение, в котором R1 представляет собой фтор (соединение (3)), обладает приблизительно 32-кратно большей селективностью в отношении TYK2 по сравнению с JAK2 и 164-кратно большей селективностью в отношении TYK2 по сравнению с JAK3.
Пригодность соединений для применения в лечении псориаза может быть определена путем тестирования действия соединений на индуцированное имиквимодом псориазоподобное воспаление кожи у мышей: см., например, публикации Mori et al., Kobe J. Med. Sci., Vol. 62, № 4, сс. E79-E88, 2016; van der Fits et al., The Journal of Immunology, 2009; 182: 5836-5845; и Lin et al., PLOS ONE | DOI:10.1371/journal.pone.0137890 10 сентября 2015 года. Таким образом, имиквимод можно применять местно у мышей (например, наносить на ухо мыши), чтобы индуцировать псориазоподобное воспаление и шелушение, и сравнить уровни воспаления и шелушения у мышей (либо на участках тела мышей), которые получали также лечение соединением по настоящему изобретению или контролем, не содержащим имиквимод.
Способы получения соединений формулы (1)
Соединения по настоящему изобретению могут быть получены способами, описанными в последующих параграфах и в примерах ниже.
Соединения формулы (1) могут быть получены в результате последовательности реакций, показанных на схеме 1.
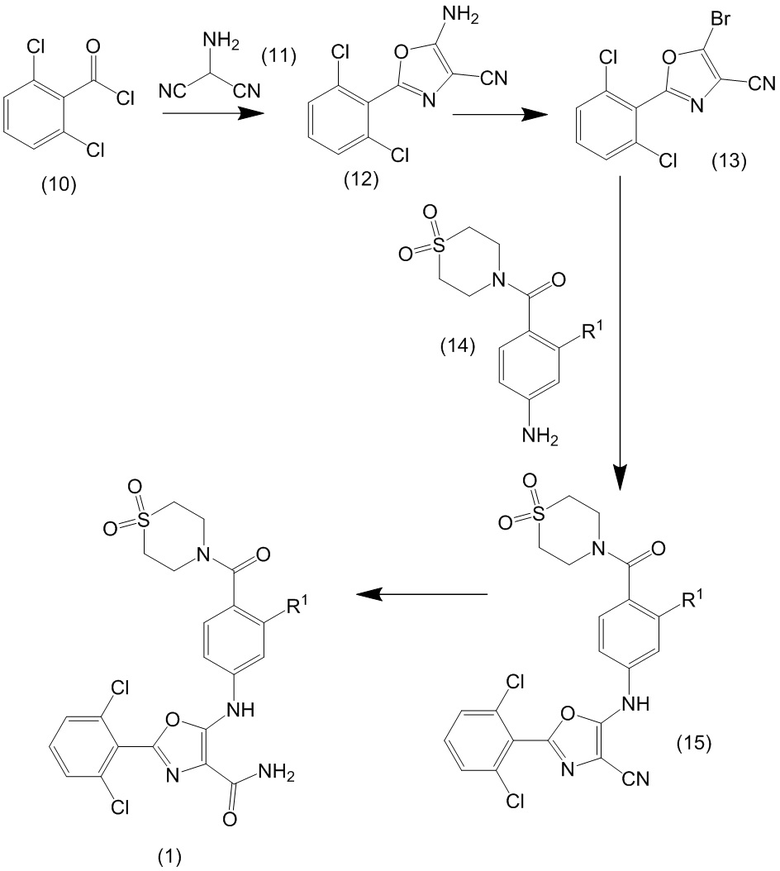
Схема 1
На первом этапе последовательности реакций проводят реакцию хлорида 2,6-дихлорбензоила (10) с аминомалононитрилом (11) (например, его п-толуолсульфонатной солью) в полярном апротонном растворителе, таком как N-метилпирролидон, NMP (NMP), с получением нитрила аминооксазола (12). Реакцию обычно проводят при повышенной температуре, например, в диапазоне 90-115 °C.
Нитрил аминооксазола (12) преобразуют в соответствующее бромзамещенное соединение (13) путем безметалловой реакции Зандмейера с применением трет-бутилового нитрита в качестве диазотирующего фактора в присутствии соединения, служащего донором галогена, такого как бром-(триметил)силан в дибромметане. Реакцию обычно осуществляют в защитной атмосфере (например, азоте) при температуре около 0 °C.
Бромзамещенное соединение (13) реагирует с замещенным анилином (14) в катализируемой палладием реакции аминирования по Бухвальду-Хартвигу с получением промежуточного цианосоединения (15). В реакции применяют палладиевый(0) катализатор, такой как бис(дибензилиденацетон)палладий(0) (Pd(dba)2), в полярном апротонном растворителе, таком как диоксан, в присутствии подходящего фосфинового лиганда, такого как 1,1′-бис(дифенилфосфино)ферроцен (DPPF, dppf) или (5-дифенил-фосфанил-9,9-диметил-ксантен-4-ил)-дифенил-фосфан, и основания, такого как карбонат калия или карбонат цезия. Реакцию обычно проводят при повышенной температуре (например, от 95-125 °C), например, в закрытой пробирке, используя микроволновой нагрев.
Промежуточное цианосоединение (15) подвергают гидролизу в мягких кислотных условиях (например, с применением серной кислоты при температуре около 0 °C) с получением соединения формулы (1).
Способы получения соединений формулы (1) и ключевых синтетических промежуточных соединений, а также новые синтетические промежуточные соединения сами по себе составляют еще один аспект изобретения. Таким образом, в дополнительных вариантах осуществления (варианты осуществления 3.1-3.5) согласно настоящему изобретению предложены:
3.1 Способ получения соединения формулы (1) по определению в настоящем документе, причем указанный способ включает гидролиз соединения формулы (15):

где R1 соответствует определению в настоящем документе, в кислотных условиях (например, с применением серной кислоты).
3.2 Способ получения соединения формулы (15) по определению в настоящем документе, причем указанный способ включает реакцию соединения формулы (13) с соединением формулы (14):

в присутствии палладиевого (0) катализатора (такого как Pd(dba)2), фосфинового лиганда (такого как DPPF) и основания (такого как карбонат калия).
3.3 Новое синтетическое промежуточное соединение формулы (15), приведенной в настоящем документе.
3.4 Новое синтетическое промежуточное соединение согласно варианту осуществления 3.3, где R1 представляет собой водород.
3.5 Новое синтетическое промежуточное соединение согласно варианту осуществления 3.3, где R1 представляет собой фтор.
Фармацевтические составы
Хотя активное соединение может вводиться отдельно, предпочтительно предоставлять его в виде фармацевтической композиции (например, состава), которая содержит по меньшей мере одно активное соединение по настоящему изобретению совместно с одним или большим количеством фармацевтически приемлемых вспомогательных веществ, таких как носители, адъюванты, разбавители, наполнители, буферы, стабилизаторы, консерванты, смазывающие вещества или другие материалы, хорошо известные специалистам в данной области техники, и, необязательно, другие терапевтические или профилактические средства.
Термин «фармацевтически приемлемый» в настоящем документе относится к соединениям, материалам, композициям и/или дозированным лекарственным формам, которые, исходя из здравого медицинского суждения, подходят для применения в контакте с тканями субъекта (например, человека) без избыточной токсичности, раздражения, аллергического ответа или других проблем или осложнений, соответствующим разумному соотношению выгоды и риска. Каждое вспомогательное вещество также должно быть «приемлемым» в смысле совместимости с другими ингредиентами состава.
Фармацевтические композиции могут быть представлены в любой форме, подходящей для перорального, парентерального, местного, интраназального, офтальмологического, ушного, ректального, интравагинального или трансдермального введения. Если композиции предназначены для парентерального введения, они могут быть входить в составы для внутривенного, внутримышечного, интраперитонеального, подкожного введения или для прямой доставки в целевой орган или ткань путем инъекции, инфузии или другого способа доставки.
Фармацевтические дозированные лекарственные формы, подходящие для перорального введения, включают таблетки, капсулы, каплеты, пилюли, пастилки, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, облатки или пластыри и буккальные пластыри.
Составы с фармацевтическими композициями, которые содержат соединения формул (1), (2) и (3) или их фармацевтически приемлемые соли, могут быть приготовлены в соответствии с известными технологиями, см., например, руководство: Remington’s Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания, США.
Таким образом, таблетированные композиции могут содержать однократную дозу активного соединения совместно с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например: лактоза, сахароза, сорбит или маннит; и/или полученный не из сахара разбавитель, такой как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производное, такое как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза и крахмалы, такие как кукурузный крахмал. Таблетки могут также содержать такие стандартные ингредиенты, как связующие и гранулирующие средства, такие как поливинилпирролидон, разрыхлители (например, способные к набуханию сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие вещества (например, стеараты), консерванты (например, парабены), антиоксиданты (например, бутилгидрокситолуол (БГТ, BHT)), буферизующие агенты (например, фосфатный или цитратный буферы) и шипучие агенты, такие как смеси цитрат/бикарбонат. Такие вспомогательные вещества хорошо известны и не нуждаются в подробном рассмотрении в настоящем документе.
Составы в форме капсул могут быть твердыми желатиновыми или мягкими желатиновыми, и могут содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть сформированы из животного желатина или из его синтетических или растительных эквивалентов.
Твердые лекарственные формы (например, таблетки, капсулы и т.д.) могут быть покрыты или не покрыты оболочкой, но обычно имеют оболочку, например, защитную пленочную оболочку (например, воск или лак) или оболочку, контролирующую высвобождение. Оболочка (например, полимер типа Eudragit ™) может быть разработана для высвобождения активного компонента в желаемом месте в желудочно-кишечном тракте. Соответственно, оболочка может быть выбрана так, чтобы она разлагалась в условиях с определенным значением pH в желудочно-кишечном тракте, тем самым селективно высвобождая указанное соединение в желудке, или в подвздошной или двенадцатиперстной кишке.
Вместо оболочки или в дополнение к ней лекарственное средство может быть заключено в твердую матрицу, содержащую агент, контролирующий высвобождение, например, агент для задержки высвобождения, которое может быть адаптировано для селективного высвобождения соединения в условиях варьирующей кислотности или щелочности в желудочно-кишечном тракте. В альтернативном варианте материал матрицы или замедляющая высвобождение оболочка могут принимать форму эродируемого полимера (например, полимера малеинового ангидрида), который практически непрерывно эродирует по мере того, как лекарственная форма проходит через желудочно-кишечный тракт. В другом альтернативном варианте активное соединение может быть включено в состав с системой доставки, которая обеспечивает осмотический контроль высвобождения соединения. Составы с осмотическим высвобождением и другие составы с отсроченным высвобождением или замедленным высвобождением могут быть получены в соответствии со способами, хорошо известным специалистам в данной области техники.
Композиции для локального применения включают мази, кремы, спреи, пластыри, гели, жидкие капли и вкладыши (например, внутриглазные вкладыши). Составы с такими композициями могут быть получены в соответствии с известными способами.
Композиции для парентерального введения обычно представлены в виде стерильных водных или масляных растворов или тонких суспензий, или могут быть предоставлены в виде тонкодисперсного стерильного порошка для восстановления стерильной водой для инъекций непосредственно перед применением.
Составы с композициями для парентерального введения могут быть приготовлены для введения в виде отдельных единиц дозы или могут быть приготовлены для введения путем инфузии.
Примеры составов для ректального или интравагинального введения включают пессарии и суппозитории, которые могут быть, например, сформированы из пластичного или воскового материала определенной формы, содержащего активное соединение.
Композиции для ингаляционного введения могут принимать форму ингалируемых порошковых композиций, либо жидких или порошковых спреев и могут вводиться в стандартной форме при помощи порошковых ингаляторов или дозирующих устройств для аэрозолей. Такие устройства хорошо известны. Порошкообразные композиции для ингаляционного введения обычно содержат активное соединение совместно с инертным твердым порошковым разбавителем, таким как лактоза.
Соединения по изобретениям обычно представлены в стандартной дозированной лекарственной форме и, таким образом, обычно содержат достаточное количество соединения, чтобы обеспечить желаемый уровень биологической активности. Например, состав, предназначенный для перорального введения, может содержать от 0,1 миллиграмма до 2 граммов активного ингредиента, чаще от 10 миллиграммов до 1 грамма, например, 50-500 миллиграммов.
Активное соединение вводят пациенту, который нуждается в этом (например, пациенту - человеку или пациенту - животному), в количестве, достаточном для достижения желаемого терапевтического эффекта.
Способы лечения
Выдвинуто предположение, что соединения формул (1), (2) и (3) по определению в описании любого из вариантов осуществления 1.1-1.9 будут полезны для применения в профилактике или лечении воспалительных заболеваний или состояний, иммунологических заболеваний или состояний, аллергических заболеваний или нарушений, отторжения трансплантатов и болезни «трансплантат против хозяина». Примеры таких болезненных статусов и состояний изложены выше.
Указанные соединения обычно вводят в количествах, терапевтически или профилактически полезных и в целом нетоксичных. Однако в определенных ситуациях (например, при жизнеугрожающих заболеваниях) полезные эффекты от введения соединения формулы (1), (2) или (3) могут перевесить ущерб от каких-либо токсических эффектов или побочных эффектов, и в этом случае может быть сочтено целесообразным введение соединения в количествах, ассоциированных с некоторой степенью токсичности.
Указанные соединения можно вводить в течение продолжительного периода, чтобы поддерживать полезные терапевтические эффекты, или можно вводить их только в течение короткого периода. В альтернативном варианте их можно вводить в пульсирующем или непрерывном режиме.
Соединение формулы (1), (2) или (3), как правило, вводят субъекту, который нуждается в таком введении, например, пациенту-человеку.
Типичная суточная доза соединения может составлять до 1000 мг в день, например, варьировать в диапазоне 0,01-10 миллиграммов на килограмм массы тела, чаще 0,025-5 миллиграммов на килограмм массы тела, например, до 3 миллиграммов на килограмм массы тела, и, более типично, 0,15-5 миллиграммов на килограмм массы тела, хотя при необходимости могут вводиться более высокие или более низкие дозы.
В качестве примера, начальная стартовая доза, составляющая 12,5 мг, может вводиться 2-3 раза в день. Дозировка может повышаться на 12,5 мг в день каждые 3-5 дней, пока не будет достигнута максимальная переносимая и эффективная доза для индивидуума, что определяет лечащий врач. В конечном счете, количество вводимого соединения соотносят с природой заболевания или физиологического состояния, которое подлежит лечению, а также с полезными терапевтическими эффектами и наличием или отсутствием побочных эффектов, вызванных данным режимом дозирования, и определяют на усмотрение врача.
Соединения формул (1), (2) и (3) могут вводиться в качестве единственного терапевтического агента или могут вводиться в составе комбинированной терапии с одним или большим количеством других соединений, таких как стероиды, интерфероны, апремиласт (при псориазе) или метотрексат (при ревматоидном артрите).
Способы диагностики
Перед введением соединения согласно настоящему изобретению можно провести скрининг пациента, чтобы определить, будет ли восприимчиво заболевание или состояние, которым пациент страдает или может страдать, к лечению соединением, обладающим активностью в отношении TYK2.
Соответственно, согласно дополнительным вариантам осуществления (4.1-4.3) настоящее изобретение обеспечивает:
4.1 Соединение по любому из вариантов осуществления 1.1-1.9 для применения в лечении или профилактике болезненного статуса или состояния у пациента, который был подвергнут скринингу и определен как страдающий заболеванием или состоянием или находящийся в группе риска развития заболевания или состояния, которое может быть восприимчиво к лечению соединением, обладающим активностью в отношении TYK2-киназы.
4.2 Применение соединения по любому из вариантов осуществления 1.1-1.9 для получения медикамента для лечения или профилактики болезненного статуса или состояния у пациента, который был подвергнут скринингу и определен как страдающий заболеванием или состоянием или находящийся в группе риска развития заболевания или состояния, которое может быть восприимчиво к лечению соединением, обладающим активностью в отношении TYK2-киназы.
4.3 Способ диагностики и лечения болезненного статуса или состояния, опосредованного TYK2-киназой, причем указанный способ включает (i) скрининг пациента для определения того, может ли быть ли заболевание или состояние, которым пациент страдает или может страдать, восприимчиво к лечению соединением, обладающим активностью в отношении указанной киназы; и (ii) если указано, что заболевание или состояние у пациента, соответственно, является восприимчивым, последующее введение пациенту эффективно ингибирующего TYK2 количества соединения по любому из вариантов осуществления 1.1-1.9.
У субъекта (например, пациента) можно провести диагностический тест для детекции маркера, указывающего на наличие заболевания или состояния, в которое вовлечена TYK2, или маркера, указывающего на предрасположенность к указанному заболеванию или состоянию. Например, может быть проведен скрининг субъектов на генетические маркеры, указывающие на предрасположенность к развитию аутоиммунного или воспалительного заболевания.
Генетический маркер может содержать конкретный аллель или однонуклеотидный полиморфизм гена TYK2, указывающий на предрасположенность к аутоиммунному заболеванию, такому как рассеянный склероз (см., например, публикацию Ban et al., European Journal of Human Genetics (2009), 17, 1309-1313), или воспалительному заболеванию кишечника, такому как болезнь Крона (см. публикацию Sato et al., J. Clin. Immunol. (2009), 29:815-825). Генетический маркер может, например, представлять собой однонуклеотидный полиморфизм в гене TYK2, или может представлять собой гаплотип, включающий однонуклеотидный полиморфизм в гене TYK2 и полиморфизм в другом гене.
Диагностические тесты обычно проводят на биологическом образце, выбранном из образцов крови, биопсийных образцов, биоптатов стула, мокроты, анализа хромосом, плевральной жидкости, перитонеальной жидкости или мочи.
Способы идентификации генетических маркеров, таких как однонуклеотидные полиморфизмы, хорошо известны. Примеры подходящих способов идентификации таких маркеров описаны в публикациях Ban et al. и Sato et al., см. выше.
ПРИМЕРЫ
Далее изобретение будет проиллюстрировано, но не ограничено, путем обращения к конкретным вариантам осуществления, описанным в приведенных ниже примерах.
Сокращения
В примерах ниже использованы следующие сокращения:
Аналитические условия
ЯМР-спектры регистрировали на спектрометре Bruker при 400 МГц.
ВЭЖХ-разделение осуществляли с использованием колонок Phenomenex LUNA-C18(2), 2 × 50 мм, размер частиц 5 мкм.
ПРИМЕР 1
2-(2,6-дихлорфенил)-5-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]-оксазол-4- карбоксамид
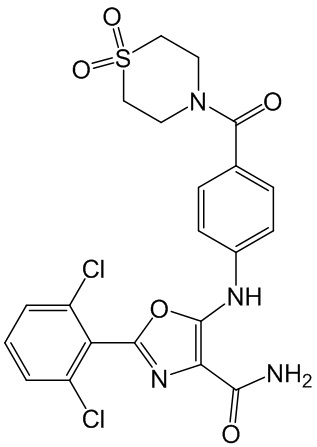
1A. Получение 5-амино-2-(2,6-дихлорфенил)-оксазол-4-карбонитрила

Хлорид 2,6-дихлорбензоила (10 г, 47,74 ммоль) медленно добавляли к раствору п-толуолсульфоната аминомалононитрила (13,3 г, 52,51 ммоль) в NMP (50 мл). Реакционную смесь нагревали до 110 °C в течение 14 часов, а затем останавливали реакцию водой (100 мл) и полученное твердое вещество собирали путем фильтрации. Неочищенный продукт растворяли в этилацетате (100 мл) и промывали водой (40 мл × 2), и органический слой высушивали над Na2SO4. Растворитель удаляли с получением титульного соединения (19 г, неочищенное) в форме твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3): δ: 7,37 - 7,35 (m, 2H), 7,29-7,26 (m, 1H), 6,19 (s, 2H).
1B. Получение 5-бром-2-(2,6-дихлорфенил)-оксазол-4-карбонитрила

К раствору 5-амино-4-циано-2-(2,6-дихлорфенил)-оксазола (9,0 г, 35,42 ммоль) в CH2Br2 (50 мл) добавляли бром(триметил)силан (13,56 г, 88,55 ммоль). Затем очень медленно добавляли трет-BuONO (36,53 г, 354,20 ммоль) при 0°C в защитной атмосфере N2 и смесь перемешивали при 0 °C в течение 2,5 часов. Затем реакционную смесь концентрировали при пониженном давлении для удаления CH2Br2, добавляли воду (H2O 100 мл) и полученную смесь экстрагировали DCM (100 мл x 3). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 50/1 до 10:1). Титульное соединение (8 г, выход 71,03%) получали в форме твердого вещества белого цвета.
1C. Получение 4-(4-нитробензоил)-1,1-диоксо-1,4-тиазинана

К смеси 4-нитробензойной кислоты (5 г, 29,92 ммоль) и гидрохлорида 1,1-диоксида 1,4-тиазинана (5,1 г, 29,92 ммоль) в DMF (50 мл) добавляли HOBt (6,1 г, 44,88 ммоль), EDCI (8,6 г, 44,88 ммоль), Et3N (6,1 г, 59,84 ммоль) одной порцией при 15°C в атмосфере N2. Смесь перемешивали при 15°C в течение 14 часов. Реакционную смесь разбавляли насыщенным раствором Na2CO3 (300 мл) и экстрагировали EtOAc (150 мл x 3). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (6,5 г, неочищенное) в форме твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3): δ : 8,27 (d, J=8,8 Гц, 2H), 7,55 (d, J=8,8 Гц, 2H), 4,33-3,75 (m, 4H), 3,22-2,75 (m, 4H).
1D. Получение 4-(4-аминобензоил)-1,1-диоксо-1,4-тиазинана
К раствору 4-(4-нитробензоил)-1,1-диоксо-1,4-тиазинана (5,5 г, 19,35 ммоль) в MeOH (100 мл) добавляли Pd/C (1,0 г, 19,35 ммоль) в атмосфере N2. Суспензию дегазировали под вакуумом и продували H2 несколько раз, а затем перемешивали в атмосфере H2 (15 фунт./кв.дюйм) при 15°C в течение 14 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением титульного соединения (4,5 г, выход 91,45%) в форме твердого вещества белого цвета.
1H ЯМР (400 МГц, (CDCl3): δ : 7,36-7,26 (m, 2H), 6,80-6,61 (m, 2H), 4,26-4,08 (m, 4H), 4,06-3,88 (m, 2H), 3,21-2,95 (m, 4H)
1E. Получение 2-(2,6-дихлорфенил)-5-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбонитрила

1,4-Диоксан (13 мл) добавляли к смеси 5-бром-4-циано-2-(2,6-дихлорфенил)-оксазола (500 мг, 1,57 ммоль), 4-(4-аминобезноил)-1,1-диоксо-1,4-тиазинана (399,25 мг, 1,57 ммоль) и Pd(dba)2 (90,28 мг, 157 мкмоль), DPPF (130,56 мг, 235,5 мкмоль), K2CO3 (976,45 мг, 7,07 ммоль) в реакционной пробирке, которую закрывали и подвергали микроволновому нагреву при 120°C в течение 4 часов. Полученную реакционную смесь фильтровали и концентрировали в вакууме, и добавляли воду (30 мл) перед экстрагированием DCM (50 мл x 3). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 10/1 до 2/3). Титульное соединение (110 мг, выход 14,26%) получали в форме твердого вещества коричневого цвета.
1F. Получение (2,6-дихлорфенил)-5-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбоксамида
Смесь 2-(2,6-дихлорфенил)-5-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбонитрила (100 мг, 203,52 мкмоль) в H2SO4 (1 мл) при 0 °C перемешивали при 15 °C в течение 2 часов в атмосфере N2. Анализ ЖХ-МС по прошествии указанного времени указывал на то, что реакция подошла к завершению, следовательно, реакцию останавливали льдом при 0 °C, а затем реакционную смесь фильтровали. Фильтрат экстрагировали EtOAc (30 мл: 10 мл x 3) и объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством препаративной ВЭЖХ (условия TFA). Титульное соединение, (2,6-дихлорфенил)-5-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)-анилино]оксазол-4-карбоксамид (25 мг, выход 24%, чистота 99,61%), получали в форме твердого вещества желтого цвета.
1H ЯМР (400 МГц, (CDCl3): δ : 9,05 (s, 1H), 7,50-7,48 (m, 2H), 7,46-7,44 (m, 3H), 7,41 - 7,38 (m, 2H), 6,50 (s, 1H), 5,38 (s, 1H), 4,12 (s, 4H), 3,07 (s, 4H).
МС (ESI): масса, вычисл. для C21 H18 Cl 2N4O5S 508.0408.04 , найденное m/z , 509,0 [M+H] +.
ПРИМЕР 2
2-(2,6-дихлорфенил)-5-[2-фтор-4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]-оксазол-4-карбоксамид
2A. Получение 4-(2-фтор-4-нитробензоил)-1,1-диоксо-1,4-тиазинана

К смеси 2-фтор-4-нитробензойной кислоты (5 г, 27 ммоль) и 1,1-диоксида 1,4-тиазинана (5,1 г, 29,7 ммоль, HCl) в DMF (50 мл) добавляли HOBt (5,47 г, 40,5 ммоль), EDCI (7,77 г, 40,5 ммоль) и Et3N (5,47 г, 54 ммоль) одной порцией при 15 °C в атмосфере N2. Полученную смесь перемешивали при 15°C в течение 14 часов, после чего ТХС (петролейный эфир / этилацетат = 1:1, Rf = 0,1) показывала, что исходное вещество в виде карбоновой кислоты полностью израсходовано и сформировалось одно новое пятно, что указывает на полную конверсию в желаемый продукт. Затем реакционную смесь разбавляли насыщенным раствором Na2CO3 (300 мл) и экстрагировали EtOAc (150 мл x 3). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (7 г, неочищенное) в форме твердого вещества желтого цвета.
1H ЯМР: 400 МГц CDCl3: δ 8,17 (d, J = 8,0 Гц, 1H), 8,06 (d, J = 8,4 Гц, 1H), 7,67-7,63 (m, 1H), 4,32 (s, 2H), 3,82 (s, 2H), 3,21 (s, 2H), 3,11 (s, 2H).
2B. Получение 4-(2-фтор-4-аминобензоил)-1,1-диоксо-1,4-тиазинана

К раствору 4-(2-фтор-4-нитробензоил)-1,1-диоксо-1,4-тиазинана (7 г, 23,2 ммоль) в MeOH (100 мл) добавляли Pd/C (3 г, чистота 10%). Суспензию дегазировали под вакуумом и продували H2 несколько раз. Затем смесь перемешивали в атмосфере H2 (15 фунт./кв.дюйм) при 15 °C в течение 12 часов, и по истечении этого времени ТХС (петролейный эфир / этилацетат = 1/1, Rf = 0,3) показывала, что исходное вещество в виде нитро-фенила полностью израсходовано и сформировалось одно пятно, соответствующее новому продукту. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением титульного соединения (5 г, неочищенный продукт) в форме твердого вещества желтого цвета.
1H ЯМР: 400 МГц CDCl3: δ 7,16-7,12 (m, 1H), 6,43-6,41 (m, 1H), 6,31-6,27 (m, 1H), 4,35 (s, 2H), 4,11-3,86 (m, 4H), 3,10 (s, 4H)
2C. Получение 2-(2,6-дихлорфенил)-5-[2-фтор-4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбонитрила

К раствору 5-бром-2-(2,6-дихлорфенил)-оксазол-4-карбонитрила (2 г, 6,29 ммоль) (пример 1A) в 1,4-диоксане (40 мл) добавляли 4-(2-фтор-4-аминобензоил)-1,1-диоксо-1,4-тиазинан (1,88 г, 6,92 ммоль), Cs2CO3 (4,10 г, 12,6 ммоль), и (5-дифенил-фосфанил-9,9-диметил-ксантен-4-ил)-дифенил-фосфан (364 мг, 629 мкмоль). Суспензию дегазировали под вакуумом и продували N2 несколько раз. Затем добавляли (1E,4E)-1,5-дифенилпента-1,4-диен-3-он : палладий (288 мг, 315 мкмоль) и продували N2 несколько раз. Затем реакционную смесь нагревали до 100 °C и перемешивали в течение 12 часов, и по истечении этого времени ТСХ (петролейный эфир / этилацетат = 1/1, Rf = 0,9) показывала, что бром-циано-оксазол полностью израсходован и сформировалось одно пятно, соответствующее новому продукту, что указывает на полную конверсию в желаемый продукт. Реакционную смесь фильтровали и осадок на фильтре промывали EtOAc (300 мл). Затем фильтрат концентрировали с получением неочищенного продукта, который очищали посредством колоночной хроматографии (SiO2, петролейный эфир / этилацетат = от 20/1 до 0:1) с получением титульного соединения (1,1 г, 2,16 ммоль, выход 34,3%) в форме твердого вещества желтого цвета.
2D. Получение (2,6-дихлорфенил)-5-[2-фтор-4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбоксамида
Смесь 2-(2,6-дихлорфенил)-5-[2-фтор-4-(1,1-диоксо-1,4-тиазинан-4-карбонил)анилино]оксазол-4-карбонитрила (0,2 г, 393 мкмоль) в H2SO4 (2 мл) дегазировали и продували N2 3 раза, а затем смесь перемешивали при 20 °C в течение 1 часа в атмосфере N2 , и по истечении этого времени анализ ВЭЖХ и ЖХ-МС свидетельствовал о том, что исходное вещество было полностью израсходовано. Остаток выливали в ледяную H2O 50 мл и экстрагировали EtOAc 60 мл (20 мл x 3). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством преп-ВЭЖХ (колонка: Phenomenex Luna C18, 250 x 50мм, 10 мкм: подвижная фаза: [вода (0,1% TFA-ACN]; B%: 20%-50%, 20 минут) с получением неочищенного продукта. Неочищенный продукт обрабатывали NaHCO3 (водн.), экстрагировали DCM (20 мл), высушивали и концентрировали, затем растворяли в смеси MeCN/вод, для проведения лиофилизации с получением титульного соединения (60,3 мг, 113 мкмоль, выход 28,8%, чистота 98,9%) в форме твердого вещества желтого цвета.
1H ЯМР: 400 МГц CDCl3: δ 9,11 (s, 1H), 7,51-7,42 (m, 4H), 7,18-7,14 (m, 1H), 7,14-7,12 (m, 1H), 6,54 (s, 1H), 5,45 (s, 1H), 4,27 (s, 2H), 3,87 (s, 2H), 3,17-3,06 (m, 4H).
ПРИМЕР 3
Биологическая активность
(i) Анализы ингибирования киназ TYK2 и JAK
Соединения по настоящему изобретению были исследованы на способность ингибировать TYK2-киназу и другие JAK-киназы. Активность соединений сопоставляли с активностями соединения A (амид 2-(2-хлор-6-фтор-фенил)-5-[4-(морфолин-4-карбонил)-фениламино]-оксазол-4-карбоновой кислоты) и соединения B (амид 2-(2,6-дихлор-фенил)-5-[4-(морфолин-4-карбонил)-фениламино]-оксазол-4-карбоновой кислоты):
 Соединение A
Соединение A  Соединение B
Соединение B
которые представляют собой соединения из примеров 25 и 29, соответственно, в каждой из WO 2015/032423 и WO2018/073438.
Субстраты и киназы, которые использовали в анализах, идентифицированы ниже в таблице 2.
Анализы с киназами проводили в Reaction Biology Corp., Малверн, штат Пенсильвания, США, с применением общей процедуры, которая описана ниже. Концентрация АТФ в анализах составляла 100 мкМ, а максимальные концентрации соединений составляли 10 мкМ.
Отметим, что данные для киназ TYK2 и JAK в таблице 7 на странице 61 WO 2015/032423 формировались с применением анализа, в котором концентрация АТФ составляла 10 мкМ, в то время как в анализе, описанном в протоколе ниже, использовали концентрацию АТФ 100 мкМ, как указано выше.
Анализ:
1) Указанный субстрат готовят в свежеприготовленном базовом реакционном буфере (20 мМ Hepes, pH 7,5, 10 мМ MgCl2, 1 мМ ЭГТК, 0,02% Brij35, 0,02 мг/мл БСА, 0,1 мМ Na3VO4, 2 мМ ДТТ, 1% ДМСО).
2) Вносят кофакторы (1,5 мМ CaCl2, 16 мкг/мл кальмодулин, 2 мМ MnCl2) в описанный выше раствор субстрата
3) В раствор субстрата вносят заданную киназу и аккуратно перемешивают
4) В киназную реакционную смесь вносят тестируемое соединение в ДМСО в варьирующих концентрациях
5) 33P-АТФ (конечная удельная активность 0,01 мкКи/мкл) вносят в реакционную смесь, чтобы инициировать реакцию
6) Киназную реакционную смесь инкубируют в течение 120 минут при комнатной температуре
7) Реакционные смеси наносят на ионообменную фильтровальную бумагу P81 (Whatman # 3698-915)
8) Несвязанный фосфат удаляют, тщательно промывая фильтры в 0,75% фосфорной кислоте.
9) Сигнал 33P определяли при помощи Typhoon Phosphorimager (компания GE Healthcare). После вычитания фона, определенного в контрольных реакциях, которые содержали неактивный фермент, определяли значения IC50 с применением функции нелинейной регрессии в Prism (программное обеспечение Graphpad).
Субстраты:
AXLtide = [KKSRGDYMTMQIG]
JAK3tide = [Ac-GEEEEYFELVKKKK-NH2]
pEY = поли Glu-Tyr [Glu:Tyr (4:1), молекулярная масса = 5,000 - 20,000]
Результаты приведены ниже в таблице 3.
Хотя было продемонстрировано, что все протестированные соединения обладают хорошей ингибирующей активностью в отношении TYK2, данные иллюстрируют, что оба соединения по настоящему изобретению (соединения (2) и (3)) являются более сильнодействующими и более селективными в отношении TYK2 (в частности, к TYK2 по сравнению с JAK2 и JAK3), чем соединения, известные из уровня техники A и B.
(ii) Анализ ингибирования цитохрома P450
Склонность соединений (2) и (3) к потенциальному межлекарственному взаимодействию тестировали путем анализа их способности ингибировать различные изоформы цитохрома P450. Соединение, известное из предшествующего уровня техники B (см. пример 3 выше), также тестировали в качестве сравнительного примера.
Анализируемые соединения, полученные и последовательно разведенные в ДМСО, инкубировали в шести концентрациях (конечная концентрация ДМСО 1%) с объединенными микросомами печени человека в присутствии маркерного субстрата для каждой изоформы и определяли их воздействие на метаболизм маркерных субстратов. Инкубацию (в 96-луночных планшетах) осуществляли при 37 °C в 0,1 М трис-буфере, pH 7,4, причем реакции инициировали путем добавления кофактора НАДФH (конечная концентрация 1 мМ).
В заданные моменты времени реакции прекращали ацетонитрилом, содержащим аналитический внутренний стандарт, образцы центрифугировали и анализировали на метаболиты маркерного субстрата посредством масс-спектрометрии (ЖХ-МС/МС). Аналитические сигналы нормировали на внутренний стандарт и сравнивали с соответствующими контрольными растворителями, чтобы определить количество метаболита, образованного из маркерных субстратов, относительно этих «неингибированных» контрольных образцов.
Результаты, которые сообщались в виде процента ингибирования и значений IC50 (концентрации, приводящей к уменьшению образования маркерного метаболита на 50%), вычисляли с применением нелинейного сигмоидального уравнения «доза-ответ» (BioBook):
% ингибирования = наименьшее значение + (наибольшее значение - наименьшее значение)/(1+10^((Log IC50 - X)*HillSlope))
где X = логарифмированная концентрация.
Исследованные изоформы CYP450 и соответствующие им маркерные субстраты приведены в таблице 4.
Результаты исследования приведены в таблице 5.
Хотя все протестированные соединения показывают хорошие профили ингибирования CYP, данные иллюстрируют, что соединения по настоящему изобретению (соединения (2) и (3)) обладают лучшими профилями ингибирования CYP (т.е. ингибируют проанализированные изоформы CYP в меньшей степени), чем сравнительное соединение B, в частности, в отношении CYP2C8 и CYP2D6.
(iii) Анализ ингибирования канала hERG
Потенциальную способность соединений к ингибированию калиевого канала hERG определяли с применением стабильно трансфицированной линии клеток hERG-HEK на автоматизированной платформе для электрофизиологических тестов Sophion Qube. Исследование выполняли при комнатной температуре, а регистрацию следового тока hERG от отдельных клеток осуществляли при помощи QChips с одним отверстием.
Мощность (IC50) тестируемых соединений в отношении ингибирования канала hERG определяли по кривой «доза-ответ», построенной на основе 8 концентрациях тестируемого соединения с количеством репликатов до 4 для каждой концентрации.
Соединения в указанной концентрации добавляли в тестовую лунку дважды, чтобы обеспечить полный обмен внешнего буфера с исследуемым соединением. В общей сложности соединение наносили на лунку в течение >7 минут.
Результаты приведены ниже в таблице 6.
Все три протестированных соединения показали относительно низкую активность в отношении hERG, но результаты демонстрируют, что соединения по настоящему изобретению (соединение (2) и соединение (3)) обладают даже меньшей предрасположенностью к воздействию на hERG, чем сравнительное соединение (B), известное из уровня техники.
(iv) Анализ стабильности в гепатоцитах
Соединения (2) и (3) по настоящему изобретению и соединение B, известные из уровня техники, тестировали в анализе стабильности в гепатоцитах с использованием объединенных гепатоцитов мыши (самец линии CD-1), крысы (самец линии Спрег-Доули), собаки (самец бигля) и человека (любого пола). Тестируемые и контрольные соединения инкубировали с гепатоцитами при 37 °C. Аликвоты отбирали в 6 временных точках на протяжении периода 1 час. Образцы центрифугировали и анализировали супернатант на исходное соединение с помощью масс-спектрометрии (ЖХ-МС/МС).
Количество оставшегося соединения (выраженное в %) определяли исходя из отклика МС для каждого образца по отношению к отклику для образцов с T=0 и использовали для определения периода полужизни и собственного клиренса соединения.
Результаты приведены ниже в таблице 7.
мкл/мин
/106 клеток
мин.
мкл/мин
/106 клеток
мин.
мкл/мин
/106 клеток
мин.
мкл/мин
/106 клеток
мин.
соединение B
В то время как сравнительное соединение B демонстрирует хороший период полужизни у человека (более 2 часов), данные в таблице 7 указывают на то, что соединения (2) и (3) по настоящему изобретению обладают значимо сниженной скоростью клиренса во всех четырех анализах на стабильность в гепатоцитах по сравнению со сравнительным соединением (B) из предшествующего уровня техники и, следовательно, период полужизни (T1/2) соединений по настоящему изобретению даже более продолжителен во всех четырех анализах, чем период полужизни сравнительного соединения (B).
(v) Ингибирование pSTAT3
Соединения (2) и (3) по настоящему изобретению и сравнительные соединения A и B, известные из уровня техники, тестировали на ингибирование pSTAT3 в ответ на стимуляцию ИЛ-22 в лишенных сыворотки клетках HT29.
Клетки HT29 лишали сыворотки на ночь перед получением разведений четырех тестируемых соединений для получения 9-точечной полулогарифмической кривой разведений доз с максимальной концентрацией 10 мкМ, плюс контроль с растворителем. Клетки HT29 инкубировали с анализируемыми соединениями на протяжении 20 минут при 37°C. На протяжении следующих 15 минут клетки HT29 инкубировали с 10 нг/мл ИЛ-22 человека, после чего клетки фиксировали 4% ПФА на протяжении 10 минут и 90% метанолом на протяжении 30 минут, после чего метили антителом фосфо-STAT3Y705 (CST #9145). Клетки промывали 3 раза 0,5% раствором БСА/ФСБ, после чего инкубировали с вторичными антителами против АТ кролика Alexa-488.
Среднюю интенсивность флуоресценции фосфо-STAT3 в одиночных клетках анализировали с применением проточной цитометрии с применением инструмента Intellicyt iQue и программного обеспечения FlowJo. IC50 определяли с применением четырех-параметрического анализа после удаления фонового сигнала и нормированием по контролю - ДМСО.
Результаты приведены ниже в таблице 8.
Хотя было продемонстрировано, что как сравнительное соединение B, так и соединение (2) обладают значениями IC50 для ингибирования pSTAT3 менее 100 нМ, значение IC50 для соединения (2) было значительно ниже, чем для сравнительного соединения B.
(vi) Анализ с первичными CD4CD45RO+ клетками человека
Ингибирование продуцирования ИЛ-17F и фосфорилирования STAT3 соединениями (2) и (3) и сравнительным соединением B измеряли в клетках Th17, полученных из CD4CD45RO+ клеток периферической крови человека.
Свежие клетки CD4CD45RO+ периферической крови человека покупали у Generon, Великобритания; 3 отдельных флакона от 3 разных добровольцев для экспериментальных репликатов. Клетки культивировали в среде для T-клеток (Thermo Fisher), которая содержала 10 нг/мл рекомбинантного человеческого ИЛ-1B (R&D Systems), ИЛ-23 (R&D Systems), трансформирующий фактор роста B1 (ТФР-B1, TGF-B1) (R&D Systems) и 50 нг/мл ИЛ-6 (R&D Systems) совместно с магнитными гранулами Dynabeads анти-CD3/CD28. Указанные клетки культивировали на протяжении 11 дней, чтобы индуцировать размножение Th17-клеток. Прежде чем высевать клетки для анализа, их выращивали на протяжении ночи в среде для T-клеток с добавлением сыворотки человека (1%). Среду удаляли и заменяли на RPMI без добавок за 4 часа до исследования.
Чтобы измерить уровни ИЛ-17F, 200 000 клеток высевали в 96-луночный планшет и преинкубировали с соединениями на протяжении 30 минут, после чего стимулировали рекомбинантным ИЛ-23 в концентрации 6,25 нг/мл и рекомбинантным ИЛ-1B человека в концентрации 0,1 нг/мл на протяжении 48 часов. Супернатанты удаляли и измеряли уровни ИЛ-17F с применением коммерчески доступного набора для ИФА ELISA (компания Thermo Fisher; BMS2037-2).
Чтобы измерить уровни pSTAT3, 200 000 клеток высевали в 96-луночный планшет и преинкубировали с соединениями на протяжении 30 минут, после чего стимулировали рекомбинантным ИЛ-23 в концентрации 12,5 нг/мл на протяжении 15 минут и затем лизировали с применением буфера для лизиса клеток. Уровни pSTAT3 в лизатах измеряли с применением коммерчески доступного набора ELISA (Thermo Fisher; 85-86102-11).
ИФА ELISA проводили в соответствии с инструкциями производителей, а поглощение считывали с применением считывающего устройства для микропланшетов (Thermo Fisher; Varioskan). Данные нормировали по ответным реакциям в необработанных образцах с применением формулы:
% в контроле = ((конц-я в стимулированном образце - конц-я в нестимулированном образце) X 100)/( конц-я в стимулированном контроле - конц-я в нестимулированном контроле)
Для вычисления уровней IC50 применяли Graphpad Prism 8.1.0 с использованием нелинейной 4-параметрической логистической модели регрессии (4PL).
Результаты приведены ниже в таблицах 9A и 9B:
(нМ)
(нМ)
В то время как все проанализированные соединения продемонстрировали ингибирование продуцирования ИЛ17-F и фосфорилирования STAT3, оба анализа демонстрируют, что соединение (2) более активно, чем сравнительное соединение B и соединение (3).
Сравнительные данные - Выводы
Данные, полученные в описанных выше анализах (i)-(vi), указывают на то, что соединения по настоящему изобретению обладают выраженными преимуществами по сравнению с наиболее сходным по структуре соединением (соединением B) из WO2015/032423.
Таким образом, оба соединения (2) и (3) более активны, чем соединение B в анализах ингибирования TYK2-киназы и оба обладают большей селективностью в отношении TYK2 по сравнению с киназами JAK1, JAK2 и JAK3, чем соединение B.
Соединения (2) и (3) обладают несколько более благоприятными свойствами по сравнению со сравнительным соединением (B), известным из уровня техники, в анализах с цитохромом P450, в первую очередь в анализах с CYP2C8 и CYP2C9.
Соединения (2) и (3) обладают меньшей предрасположенностью к воздействию на hERG по сравнению со сравнительным соединением (B), известным из уровня техники.
В анализах на стабильность в гепатоцитах соединения (2) и (3) продемонстрировали значительно сниженную скорость клиренса и, следовательно, более продолжительный период полужизни, чем сравнительное соединение B.
Кроме того, соединение (2) является более мощным ингибитором фосфорилирования STAT3 в клетках HT29, стимулированных ИЛ22, и в Th17-клетках по сравнению со сравнительным соединением B.
И наконец, соединение (2) демонстрирует большее ингибирование продуцирования ИЛ-17F в Th17-клетках по сравнению со сравнительным соединением B.
В совокупности эти данные указывают на то, что соединения (2) и (3) являются высокомощными селективными ингибиторами TYK2-киназы и обладают превосходными фармакокинетическими свойствами.
ПРИМЕР 4
Фармацевтические составы
(i) Лекарственная форма в виде таблетки
Таблетированную композицию, содержащую соединение формулы (2) или формулы (3) или их фармацевтически приемлемую соль, получают путем смешивания 50 мг соединения со 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве смазывающего вещества, и прессуют ее для формирования таблетки известным способом.
(ii) Лекарственная форма в виде капсулы
Лекарственную форму в виде капсулы получают путем смешивания 100 мг соединения формулы (2) или формулы (3) или их фармацевтически приемлемой соли со 100 мг лактозы и наполнения полученной смесью стандартных непрозрачных твердых желатиновых капсул.
(iii) Лекарственная форма для подкожного введения
Композицию для подкожного введения получают путем смешивания соединения формулы (2) или формулы (3) или их фармацевтически приемлемой соли с кукурузным маслом фармацевтической степени чистоты с получением концентрации 5 мг/мл. Композицию стерилизуют и заполняют ею подходящую емкость.
Эквиваленты
Приведенные выше примеры представлены с целью иллюстрации изобретения и не должны истолковываться как каким-либо образом ограничивающие объем изобретения. Совершенно очевидно, что многочисленные модификации и изменения могут быть введены в конкретные варианты осуществления изобретения, описанные выше и проиллюстрированные в примерах, без отступления от принципов, лежащих в основе настоящего изобретения. Подразумевается, что все такие модификации и изменения предусмотрены настоящим документом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-ФЕНИЛ-ОКСАЗОЛ-4-КАРБОКСАМИДА, МОДУЛИРУЮЩИЕ АКТИВНОСТЬ JAK И TYK2 КИНАЗ | 2013 |
|
RU2652795C2 |
| АМИНОПИРАЗОЛЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2017 |
|
RU2757218C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АМИНОИМИДАЗОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS-КИНАЗ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2772463C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ АКТИВНОСТИ TYK2 | 2021 |
|
RU2833524C1 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| ТРИАЗОЛПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ JAK, И СПОСОБЫ | 2009 |
|
RU2560153C2 |
| ПИРРОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2563644C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АНАЛОГИ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ JAK | 2012 |
|
RU2564419C1 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
Группа изобретений относится к органической химии и включает соединение формулы (1), где R1 представляет собой водород или фтор, его фармацевтически приемлемые соли, применение, способ лечения, фармацевтическую композицию, способ получения соединения формулы (1) путем гидролиза соединения формулы (15) в кислотных условиях. Технический результат – соединение формулы (1), обладающее ингибирующей активностью в отношении киназы TYK2. 9 н. и 14 з.п. ф-лы, 9 табл., 4 пр.
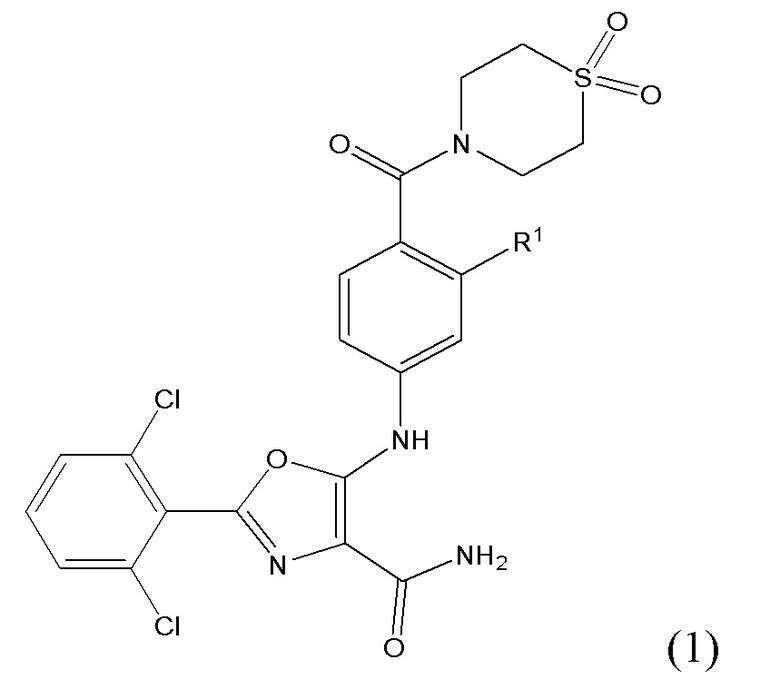

1. Соединение, которое имеет формулу (1):
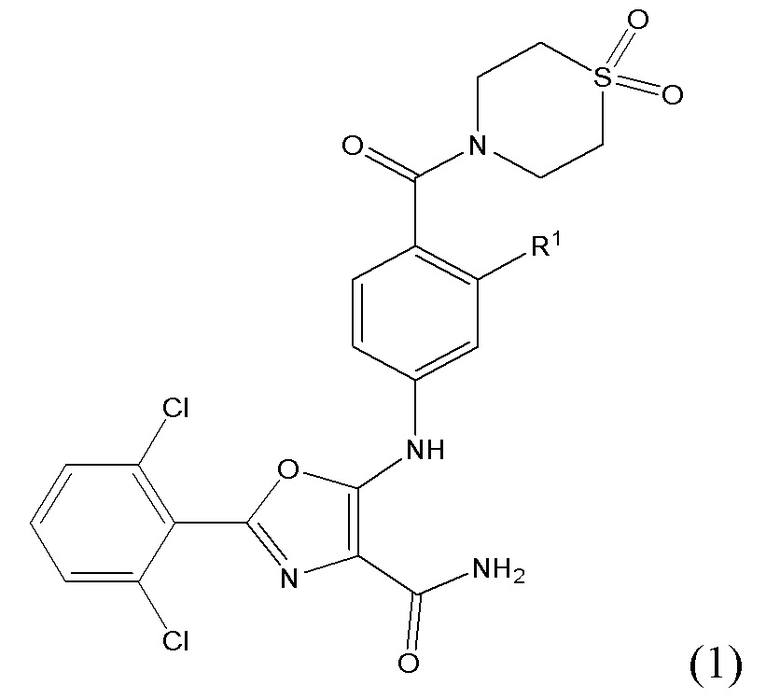
или его фармацевтически приемлемая соль,
где R1 представляет собой водород или фтор.
2. Соединение по п. 1, в котором R1 представляет собой водород, причем указанное соединение имеет формулу (2):
или его фармацевтически приемлемая соль.
3. Соединение по п. 1, в котором R1 представляет собой фтор, причем указанное соединение имеет формулу (3):

или его фармацевтически приемлемая соль.
4. Соединение по п. 1, которое представляет собой несолевую форму.
5. Соединение по п. 2, которое представляет собой несолевую форму.
6. Соединение по п. 3, которое представляет собой несолевую форму.
7. Применение соединения по любому из пп. 1-6 в качестве ингибитора TYK2.
8. Способ лечения заболевания или состояния, которое будет восприимчиво к лечению, соединением, обладающим активностью против киназы TYK2, включающий введение соединения по любому из пп. 1-6.
9. Способ по п. 8, где заболевание или состояние представляет собой воспалительное нарушение.
10. Способ по п. 8, где заболевание или состояние представляет собой иммунное нарушение.
11. Способ по п. 8, где заболевание или состояние представляет собой аутоиммунное заболевание.
12. Применение эффективного количества соединения по любому из пп. 1-6 для получения медикамента для лечения заболевания или состояния, которое будет восприимчиво к лечению соединением, обладающим активностью против киназы TYK2.
13. Применение по п. 12, где заболевание или состояние представляет собой воспалительное нарушение.
14. Применение по п. 12, где заболевание или состояние представляет собой иммунное нарушение.
15. Применение по п. 12, где заболевание или состояние представляет собой аутоиммунное состояние.
16. Способ лечения или предотвращения болезненного статуса или состояния у пациента, который прошел скрининг и для которого было определено, что он страдает или находится в группе риска заболевания или состояния, которое будет восприимчиво к лечению соединением, обладающим активностью против киназы TYK2, включающий введение эффективного количества соединения по любому из пп. 1-6.
17. Применение эффективного количества соединения по любому из пп. 1-6 в получении медикамента для лечения или профилактики болезненного статуса или состояния у пациента, который прошел скрининг и для которого было определено, что он страдает или находится в группе риска заболевания или состояния, которое будет восприимчиво к лечению соединением, обладающим активностью против киназы TYK2.
18. Фармацевтическая композиция для заболевания или состояния, которое будет восприимчиво к лечению, соединением, обладающим активностью против киназы TYK2, содержащая эффективное количество соединения по любому из пп. 1-6 и фармацевтически приемлемое вспомогательное вещество.
19. Фармацевтическая композиция по п. 18, содержащая дополнительное терапевтическое средство.
20. Способ получения соединения по любому из пп. 1-6, включающий гидролиз соединения формулы (15):
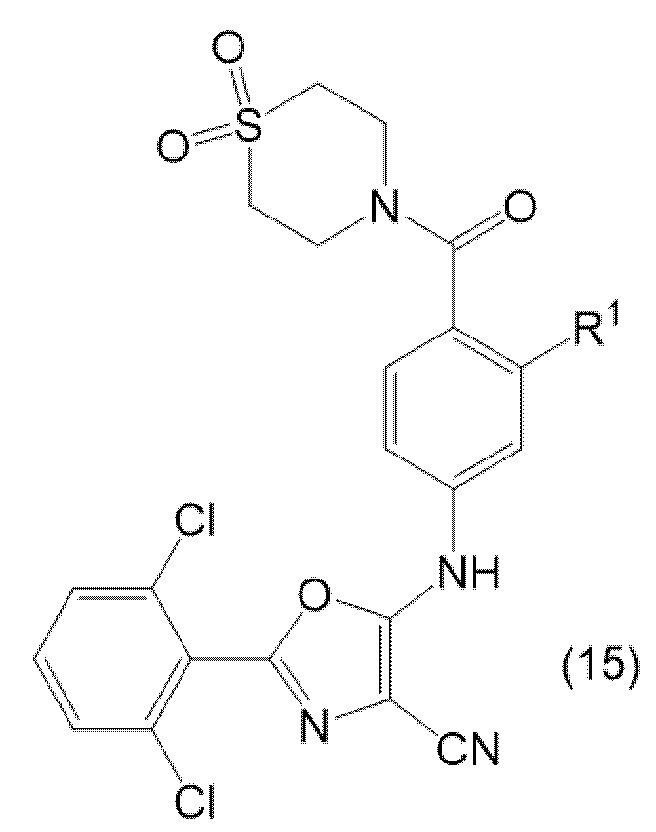
где R1 представляет собой водород или фтор,
в кислотных условиях.
21. Способ по п. 20, в котором R1 представляет собой водород.
22. Соединение формулы (15):
где R1 представляет собой водород или фтор.
23. Соединение по п. 22, в котором R1 представляет собой водород.
| WO 2015032423 A1, 12.03.2015 | |||
| КАСКАДНЫЙ УСИЛИТЕЛЬ СВЧ | 2016 |
|
RU2634185C1 |
| WO 2008139161 A1, 20.11.2008 | |||
| МОДУЛЯТОРЫ КИНАЗ AURORA И FLT3 | 2013 |
|
RU2643809C2 |

