Область техники, к которой относится настоящее изобретения
Настоящее изобретение относится к гетероциклическим соединениям, полезным для модуляции TYK2, чтобы вызвать ингибирование передачи сигнала. Соединения обеспечивают улучшенные фармакокинетические свойства у животных.
Предпосылки создания настоящего изобретения
Тирозинкиназа 2 (TYK2) является нерецепторной тирозиновой протеинкиназой, принадлежащей к семейству Янус-киназы (JAK), и было показано, что она имеет решающее значение в регуляции каскада передачи сигнала после рецепторов для IL-12, IL-23 и интерферонов I типа.
Тандемные киназные домены являются отличительной чертой JAK. JH1 представляет собой канонический белковый тирозинкиназный домен, тогда как JH2 классифицируют как псевдокиназный домен. Структура семейства JAK показана на фиг. 1.
Последние биохимические и структурные данные позволяют предположить, что псевдокиназный домен TYK2 имеет низкий уровень каталитической активности и негативно регулирует активность киназного домена.
Когда цитокиновые рецепторы связывают цитокины, запускается фосфорилирование TYK2 и других членов его семейства JAK1 и/или JAK2, которые связаны с внутриклеточными областями, что приводит к активации передачи сигнала и факторов активации транскрипции (STAT) путем димеризации. Затем димеризованные STAT мигрируют внутрь ядра и регулируют экспрессию и транскрипцию родственных генов для полной передачи сигналов от клеточной мембраны к ядру. Таким образом, JAK передают опосредованные цитокинами сигналы через путь JAK-STAT и играют важную роль во многих клеточных функциях, цитокин-зависимой регуляции клеточной пролиферации, дифференцировке, апоптозе, иммунном ответе и т.д. Мыши с дефицитом TYK2 устойчивы к экспериментальным моделям колита, псориаза и рассеянного склероза, что демонстрирует важность TYK2-опосредованной передачи сигналов при аутоиммунитете и связанных с ним нарушениях.
У людей, индивидуумы, экспрессирующие неактивный вариант TYK2, защищены от рассеянного склероза и, возможно, других аутоиммунных нарушений. В полногеномных исследованиях выявили, что другие варианты TYK2 связаны с аутоиммунными нарушениями, такими как болезнь Крона, псориаз, системная красная волчанка и ревматоидный артрит, что дополнительно демонстрирует важность TYK2 в аутоиммунитете.
Мыши, нокаутные по TYK2, имеют нормальное количество эритроцитов и способны выживать. Отсутствие экспрессии TYK2 проявляется в ослаблении передачи сигналов различных провоспалительных цитокинов и тяжелом дисбалансе клеточной дифференцировки Т-хелперов. Данные генетических исследований подтверждают, что TYK2 является общим геном восприимчивости для аутоиммунных заболеваний. Пути, регулируемые TYK2, были подтверждены антителотерапией для лечения заболеваний. Например, устекинумаб, воздействующий на IL-12/IL-23, для лечения псориаза, и анифролумаб, воздействующий на рецепторы интерферона I типа, для лечения системной красной волчанки (СКВ), продемонстрировали значительную эффективность в клинических испытаниях.
TYK2 связан с некоторыми видами рака за счет корреляции между аномальной выживаемостью клеток с острым лимфоцитарным лейкозом (Т-ОЛЛ) и активацией TYK2. Что касается онкогена Т-ОЛЛ, 88% клеточных линий Т-ОЛЛ и 63% клеток Т-ОЛЛ, полученных от пациентов, зависели от TYK2 в экспериментах с нокаутом гена (Sanda et. al., Cancer Disc. 2013, 3, 564-77). Селективный ингибитор TYK2 NDI-031301 индуцировал апоптоз для ингибирования роста человеческих клеточных линий Т-ОЛЛ и показал хорошую безопасность и эффективность в мышиной модели с опухолевыми клетками KOPT-K1 с Т-ОЛЛ (Akahane et. al., British J. Haematol. 2017, 177,271-82), что демонстрирует перспективность селективных ингибиторов TYK2 для лечения Т-ОЛЛ. Таким образом, TYK2 является одной из горячих мишеней для лечения воспалительных заболеваний, аутоиммунных заболеваний и рака (Alicea-Velazquez et. al, Curr. Drug Targets 2011, 12, 546-55).
TYK2 и другие члены семейства JAK структурно имеют киназный домен JH1 (JAK Homology 1) и смежный псевдокиназный домен JH2 (JAK Homology 2). JH2 может связывать АТФ, но не выполняет каталитическую функцию, а вместо этого негативно регулирует киназную активность JH1 (Staerk et al., J. Biol. Chem. 2015, 280, 41893-99). Из-за высокого сходства последовательности киназного домена JH1 среди семейства JAK (JAK1, JAK2, JAK3 и TYK2) сложно разработать селективный ингибитор JH1 TYK2 без ингибирования JH1 JAK1, JAK2 или JAK3. Большинство ингибиторов JAK, которые связываются с киназным доменом JAK, в том числе тофацитиниб, руксолитиниб, барицитиниб, упадацитиниб и т.д., не очень избирательны среди членов семейства JAK и проявляют дозозависимые клинические эффекты, такие как анемия. Разработка высокоселективных ингибиторов TYK2 остается привлекательной для фармацевтических компаний. Основываясь на структурных различиях между АТФ-связывающими «карманами» в JH1 и JH2 TYK2, компания Bristol-Myers Squibb разработала высокоселективное JH2-связывающее средство BMS-986165, которое ингибирует только физиологические функции, опосредованные TYK2, без связывания с киназными доменами (JH1) JAK. BMS-986165 в настоящее время находится в фазе III клинических испытаний для аутоиммунных заболеваний (Wrobleski et. al., J. Med. Chem. 2019, 62, 8973-95). Структура BMS-986165 показана ниже (WO 2014/074661):
Остается потребность в разработке новых соединений, которые избирательно связываются с псевдокиназным доменом (JH2) TYK2 с минимальным связыванием с киназными доменами семейств JAK, в частности, JAK.2.
Краткое описание чертежей
На фиг. 1 показана общая вторичная структура семейства JAK (JAK.1, JAK2, JAK3 и TYK2).
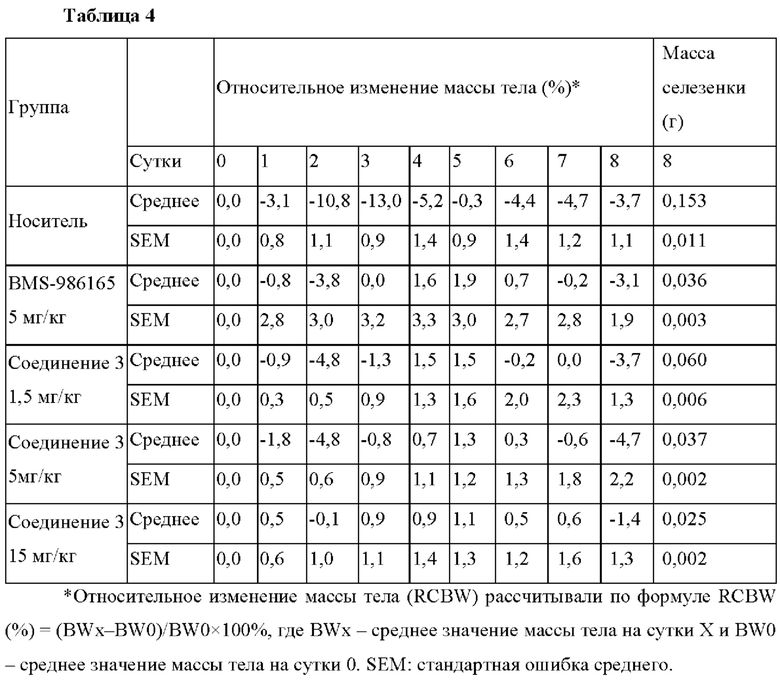
На фиг. 2 показаны эффективности при колите при воспалительном заболевании кишечника, индуцированном антителом к CD40, in vivo на модели животных. Относительные процентные изменения массы тела животных, получавших носитель, эталонное соединение и соединение 3 в трех различных дозах, нанесены на график в зависимости от числа суток после лечения.
Подробное описание настоящего изобретения
Авторы изобретения открыли селективные ингибиторы TYK.2, нацеленные не на каталитически активный центр TYK.2, а на псевдокиназный домен TYK.2 (JH2). Настоящее изобретение относится к соединениям 1-8 и их фармацевтически приемлемым солям или пролекарственным средствам. Соединения 1-8 являются селективными связывающими средствами для JH2 TYK.2. Связываясь с псевдокиназным доменом (JH2), соединения 1-8 интибируют каталитическую активность киназы TYK2, ингибируют фосфорилирование белка и проявляют значительное ингибирующее действие на физиологическую функцию TYK2. Соединения 1-8 либо слабо связываются, либо не связываются с киназным доменом (JH1) TYK2. Соединения 1-8 селективно ингибируют киназную активность TYK2 путем связывания с JH2 и обладают низкой ингибирующей активностью по отношению к киназной активности других членов семейства JAK. Селективность соединений 1-8 в отношении ингибирования TYK2 по сравнению с другими членами семейства JAK (JAK1, JAK2 и JAK3) сводит к минимуму такие эффекты, как анемия. Показано, что соединения 1-8 обладают превосходными фармакокинетическими свойствами in vivo у животных.
«Фармацевтически приемлемые соли», как применяют в настоящем документе, представляют собой соли, сохраняющие желаемую биологическую активность исходного соединения и не оказывающие нежелательного токсикологического действия. Фармацевтически приемлемые соли включают различные кристаллические полиморфы, а также аморфные формы различных солей. Фармацевтически приемлемые соли настоящих основных гетероциклических соединений могут быть получены с неорганическими кислотами или органическими кислотами.
«Пролекарственное средство», как применяют в настоящем документе, относится к соединению, которое при введении индивидууму подвергается химическому превращению посредством метаболических или химических процессов с образованием соединения 1-8 и/или его соли. Любое соединение, которое будет преобразовано in vivo в биоактивное средство соединения 1-8, является пролекарственным средством в пределах объема изобретения. Различные формы пролекарственных средств хорошо известны в данной области.
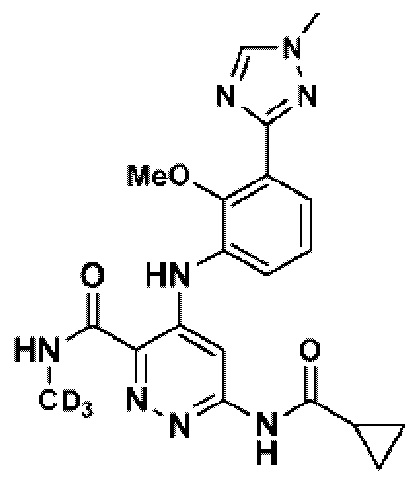
Соединение 1 имеет три-дейтерированный метил на триазольном кольце и три-дейтерированный метиламид.
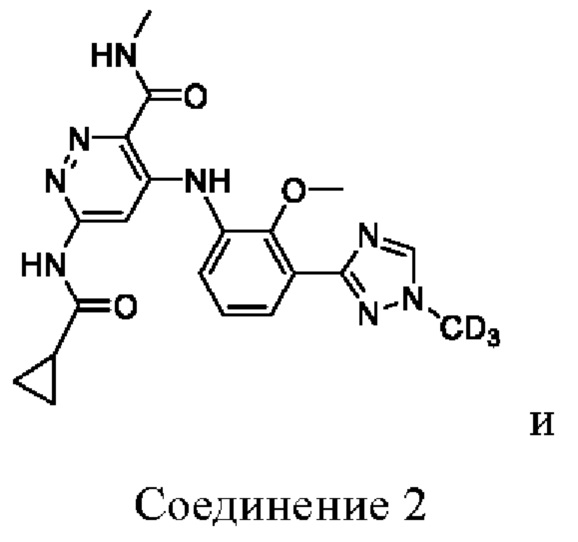
Соединение 2 имеет три-дейтерированный метил на триазольном кольце.
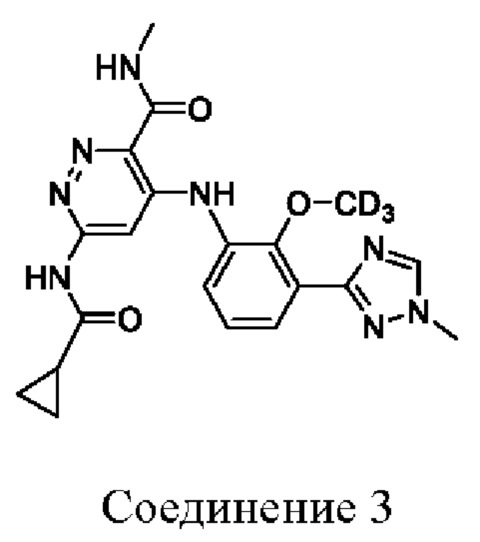
Соединение 3 имеет тридейтерированную метоксигруппу на бензольном кольце.
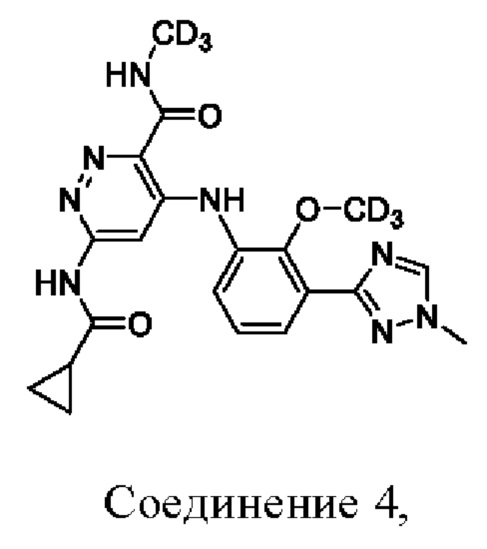
Соединение 4 имеет три-дейтерированную метоксигруппу на бензольном кольце и три-дейтерированный метиламид.
Соединение 5 имеет (S)-6-(2,2-дифторциклопропан-1-карбоксамидо) и три-дейтерированный метиламид.
Соединение 6 имеет (S)-6-(2,2-дифторциклопропан-1-карбоксамидо) без какой-либо замены дейтерием.
Соединение 7 имеет (R)-6-(2,2-дифторцикло пропан-1-карбоксамидо) и три-дейтерированный метиламид.
Соединение 8 имеет (R)-6-(2,2-дифторциклопропан-1-карбоксамидо) без какой-либо замены дейтерием.
Соединения 1-5 и 7 имеют несколько замен дейтерием на метиле для улучшения фармакокинетических (PK) свойств. Соединения 5-8 имеют дифтор на циклопропане. Соединения по настоящему изобретению обладают низкой активностью связывания с киназными доменами JAK и обладают высокой ингибирующей активностью в отношении клеточных функций TYK2, например, за счет ингибирования секреции γ-интерферона и IL-23. Соединения по настоящему изобретению обеспечивают хорошую биодоступность при пероральном введении. Соединения по настоящему изобретению безопасны для применения и эффективны при лечении воспалительного заболевания кишечника (ВЗК), что было продемонстрировано на модели колита, вызванного антителом к CD40 (ВЗК) у мышей, которые не показали значительной потери массы тела после лечения соединением 2, 3 и 5.
Фармацевтические композиции
Настоящее изобретение относится к фармацевтическим композициям, содержащим один или более фармацевтически приемлемых носителей и активное соединение соединений 1-8 или их фармацевтически приемлемую соль. Активное соединение или его фармацевтически приемлемая соль в фармацевтических композициях в основном находится в количестве около 0,01-20%, или 0,05-20%, или 0,1-20%, или 0,2-15%, или 0,5-10% или 1-5% (масс/масс.) для местного состава; примерно 0,1-5% для инъекционного состава, 0,1-5% для пластыря, около 1-90% для таблеточного состава и 1-100% для капсульного состава.
В одном из вариантов осуществления активное соединение вводят в любой приемлемый носитель, включая кремы, гели, лосьоны или другие типы суспензий, которые могут стабилизировать активное соединение и доставлять его в пораженный участок для местного применения. В другом варианте осуществления фармацевтическая композиция может иметь лекарственную форму, такую как таблетки, капсулы, гранулы, мелкозернистые гранулы, порошки, текстуры, суппозитории, растворы для инъекций, пластыри или т.п. Вышеупомянутую фармацевтическую композицию можно получать общепринятыми способами.
Фармацевтически приемлемые носители, представляющие собой неактивные ингредиенты, могут быть выбраны специалистами в данной области с использованием общепринятых критериев. Фармацевтически приемлемые носители в качестве неограничивающих примеров включают растворы на неводной основе, суспензии, эмульсии, микроэмульсии, мицеллярные растворы, гели и мази. Фармацевтически приемлемые носители могут также содержать ингредиенты, которые в качестве неограничивающих примеров включают физиологический раствор и водные растворы электролитов; ионные и неионные осмотические средства, такие как хлорид натрия, хлорид калия, глицерин и декстроза; регуляторы рН и буферы, такие как соли гидроксида, фосфата, цитрата, ацетата, бората; и троламин; антиоксиданты, такие как соли, кислоты и/или основания бисульфита, сульфита, метабисульфита, тиосульфита, аскорбиновая кислота, ацетилцистеин, цистеин, глутатион, бутилированный гидроксианизол, бутилированный гидрокситолуол, токоферол, и аскорбилпальмитат; поверхностно-активные вещества, такие как лецитин, фосфолипиды, включая в качестве неограничивающих примеров фосфатидилхолин, фосфатидилэтаноламин и фосфатидилинозитол; полоксамеры и полоксамины, полисорбаты, такие как полисорбат 80, полисорбат 60 и полисорбат 20, простые полиэфиры, такие как полиэтиленгликоли и полипропиленгликоли; поливинилы, такие как поливиниловый спирт и повидон; производные целлюлозы, такие как метилцеллюлоза, гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза и гидроксипропилцеллюлоза и их соли; производные нефти, такие как минеральное масло и белый вазелин; жиры, такие как ланолин, арахисовое масло, пальмовое масло, соевое масло; моно-, ди- и триглицериды; полимеры акриловой кислоты, такие как карбоксиполиметиленгель и гидрофобно модифицированный сшитый акрилатный сополимер; полисахариды, такие как декстраны, и гликозаминогликаны, такие как гиалуронат натрия. Такие фармацевтически приемлемые носители могут быть защищены от бактериального загрязнения с помощью хорошо известных консервантов, которые в качестве неограничивающих примеров включают хлорид бензалкония, этилендиаминтетрауксусную кислоту и ее соли, хлорид бензетония, хлоргексидин, хлорбутанол, метилпарабен, тимеросал и фенилэтиловый спирт, или можно формулировать их как состав без консервантов для одноразового или многократного применения.
Например, таблеточный или капсульный состав активного соединения, может содержать другие вспомогательные вещества, не обладающие биологической активностью и не вступающие в реакцию с активным соединением. Вспомогательные вещества для таблетки или капсулы могут включать наполнители, связывающие средства, смазочные средства и способствующие скольжению средства, дезинтегрирующие средства, увлажнители и модификаторы скорости высвобождения. Связывающие средства способствуют слипанию частиц состава и имеют важное значение для состава таблеток. Примеры вспомогательных веществ для таблетки или капсулы включают, без ограничений указанными, карбоксиметилцеллюлозу, целлюлозу, этилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу, камедь карайи, крахмал, камедь трагаканта, желатин, стеарат магния, диоксид титана, поли(акриловую кислоту) и поливинилпирролидон. Например, состав для таблетки может содержать неактивные ингредиенты, такие как коллоидный диоксид кремния, кросповидон, гипромеллоза, стеарат магния, микрокристаллическая целлюлоза, полиэтиленгликоль, крахмалгликолят натрия и/или диоксид титана. Состав для капсулы может содержать неактивные ингредиенты, такие как желатин, стеарат магния и/или диоксид титана.
Например, состав пластыря с активным соединением может содержать некоторые неактивные ингредиенты, такие как 1,3-бутиленгликоль, аминоацетат дигидроксиалюминия, диэдетат натрия, D-сорбит, желатин, каолин, метилпарабен, полисорбат 80, повидон, пропиленгликоль, пропилпарабен, натриевая карбоксиметилцеллюлоза, натрия полиакрилат, винная кислота, диоксид титана и очищенная вода. Состав пластыря может также содержать усилитель проницаемости кожи, такой как сложные эфиры лактата (например, лауриллактат) или моноэтиловый эфир диэтиленгликоля.
Составы для местного применения, содержащие активное соединение, могут иметь форму геля, крема, лосьона, жидкости, эмульсии, мази, спрея, растворителя и суспензии. Неактивные ингредиенты в составах для местного применения, например, включают, без ограничений указанными, моноэтиловый эфир диэтиленгликоля (смягчающее средство/усилитель проницаемости), ДМСО (усилитель растворимости), силиконовый эластомер (модификатор реологии/текстуры), каприловый/каприновый триглицерид (смягчающее средство), октисалат (смягчающее средство/УФ-фильтр), силиконовый флюид (смягчающее средство/разбавитель), сквален (смягчающее средство), подсолнечное масло (смягчающее средство) и диоксид кремния (загуститель).
Способ применения
Автор изобретения продемонстрировал, что настоящие соединения специфически связываются с псевдокиназным доменом TYK2 (JH2) и значительно ингибируют физиологическую функцию TYK2 в клетках NK92. Соединения также проявляют отличные фармакокинетические свойства у крыс.
Настоящее изобретение относится к способу профилактики или лечения TYK2-опосредованных заболеваний, включая в качестве неограничивающих примеров аутоиммунные заболевания, воспалительные заболевания (включая воспаление тонкой кишки и воспаление толстой кишки), различные виды рака, заболевания кожи, сахарный диабет, глазные заболевания, нейродегенеративные заболевания, аллергические реакции, астму, другие обструктивные заболевания легких и отторжение трансплантата, и т.д. Способ особенно подходит для лечения воспалительного заболевания кишечника, псориаза и системной красной волчанки (СКВ). Способ предусматривает введение нуждающемуся в этом пациенту эффективного количества соединения по настоящему изобретению или его пролекарственного средства, его фармацевтически приемлемой соли. «Эффективное количество», как применяют в настоящем документе, представляет собой количество, эффективное для лечения заболевания путем улучшения патологического состояния или уменьшения симптомов заболевших.
Фармацевтическую композицию по настоящему изобретению можно применять путем местного введения и системного введения. Местное введение включает в себя местное применение. Системное введение включает пероральный (включая буккальный или сублингвальный), парентеральный (такой как внутривенный, внутримышечный, подкожный или ректальный) и другие системные пути введения. При системном введении активное соединение сначала достигает плазмы, а затем распределяется в тканях-мишенях. Местное введение и пероральное введение являются предпочтительными путями введения по настоящему изобретению.
Дозировка композиции может варьироваться в зависимости от степени повреждения и индивидуальной реакции каждого пациента. При системном введении концентрации доставляемого активного соединения в плазме могут варьировать, но в основном составляют 1×10-10-1×10-4 моль/литр, и предпочтительно 1×10-8-1×10-5 моль/литр.
В одном из вариантов осуществления композицию наносят местно на пораженный участок и втирают в него. Композицию применяют местно, по меньшей мере, 1 или 2 раза в сутки, или 3-4 раза в сутки, в зависимости от состояния здоровья и от того, является ли патология хронической или острой. В основном, состав для местного применения содержит примерно 0,01-20%, или 0,05-20%, или 0,1-20%, или 0,2-15%, 0,5-10 или 1-5% (масс./масс.) активного соединения. Активное соединение проходит через кожу и доставляется к участку дискомфорта.
В одном из вариантов осуществления фармацевтическую композицию вводят индивидууму перорально. Дозировка для перорального введения составляет в основном, по меньшей мере, 0,1 мг/кг/сутки и менее чем 1000 мг/кг/сутки. Например, дозировка для перорального введения составляет от 0,5 мг до 1 г, от 1 мг до 700 мг или от 5 мг до 300 мг соединения в сутки.
Специалистам в данной области понятно, что широкий спектр механизмов доставки также подходит для настоящего изобретения.
Настоящее изобретение предназначено для лечения таких млекопитающих индивидуумов, как люди, лошади и собаки. Настоящее изобретение особенно подходит для лечения людей.
Следующие примеры дополнительно иллюстрируют настоящее изобретение. Эти примеры предназначены только для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие.
ПРИМЕРЫ
Примеры 1-8 иллюстрируют синтез настоящих соединений. Продукт на каждой стадии реакции получают с помощью способов разделения, известных в данной области, включая в качестве неограничивающих примеров экстракцию, фильтрацию, дистилляцию, кристаллизацию и хроматографическое разделение. Исходные вещества и химические реагенты, необходимые для синтеза, могут быть синтезированы традиционным способом по литературным данным (доступен поиск в SciFinder) или приобретены.
Структуру соединения определяют с помощью ядерно-магнитного резонанса (ЯМР) или масс-спектрометрии (МС). ЯМР измеряли на ЯМР-спектрометре Bruker ASCEND-400. Растворителями были дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) или дейтерированный метанол (CD3OD). Внутренним стандартом был тетраметилсилан (ТМС). Химический сдвиг приведен в единицах в формате 10-6 (м.д.).
МС измеряли с использованием масс-спектрометра Agilent SQD (ИЭР) (производитель: Agilent, модель: 6120).
ВЭЖХ измеряли с использованием высокоэффективной жидкостной хроматографии Agilent 1260 DAD (колонка Poroshell120 ЕС-С18, 50×3,0 мм, 2,7 мкм) или высокоэффективной жидкостной хроматографии Waters Arc (Sunfire С18, 150×4,6 мм, колонка 5 мкм).
Тонкослойную хроматографию (ТСХ) проводили с использованием пластины из силикагеля Qingdao Ocean GF254. Технические характеристики ТСХ для мониторинга реакции и разделения/очистки продуктов представляют собой 0,15-0,2 мм и 0,4-0,5 мм толщиной, соответственно.
Колоночную хроматографию проводили в основном с использованием силикагеля Qingdao Ocean 200-(300 меш) в качестве носителя.
Известные исходные вещества, используемые в настоящем масштабе, могут быть синтезированы в соответствии с известными в данной области способами или могут быть приобретены у ABCR GmbH & Со, Acros Organics, Sigma-Aldrich Chemical Company, Accela ChemBio Inc., Beijing Ouhe chemicals и других компаний.
В приведенных ниже примерах, если не указано иное, все реакции проводили в атмосфере аргона или азота.
Реакцию гидрирования, как правило, запускали в реакторе, который вакуумировали, заправляли водородом и повторяли цикл три раза.
Микроволновую реакцию проводили с использованием микроволнового реактора СЕМ Discover-SP.
В приведенных ниже примерах, если не указано иное, температура реакции представляла собой комнатную температуру от 20°С до 30°С.
За ходом реакции следили с помощью прибора Agilent LCMS (1260/6120). Можно также проводить мониторинг с помощью ТСХ. Система растворителей для ТСХ представляла собой А: систему дихлорметана и метанола; В: систему петролейного эфира и этилацетата; С: систему, показанную в примерах. Объемное соотношение растворителей корректировали в соответствии с полярностью соединения.
Система элюентов для колоночной хроматографии и ТСХ, используемая в процессе очистки соединений, включала А: систему дихлорметана и метанола; В: систему петролейного эфира и этилацетата; С: систему, показанную в примерах. Объемное соотношение растворителей корректировали в соответствии с полярностью соединения, и для корректировки можно было добавить небольшое количество триэтиламина и кислотного или основного реагента.
Очистка соединения также может быть проведена с использованием автоматизированной препаративной системы, ориентированной на масс-спектрометрию Waters (преп-ВЭЖХ с масс-детектором SQD2). В зависимости от полярности соединения применяли соответствующий профиль элюирования ацетонитрил/вода (содержащий 0,1% трифторуксусной кислоты или муравьиной кислоты) или ацетонитрил/вода (содержащий 0,05% гидроксида аммония) для промывания колонки для высокоэффективной жидкостной хроматографии (XBridge-C18, 19×150 мм, 5 мкм) при скорости потока 20 мл/мин.
Пример 1. 6-(циклопропанкарбоксамидо)-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид (1)
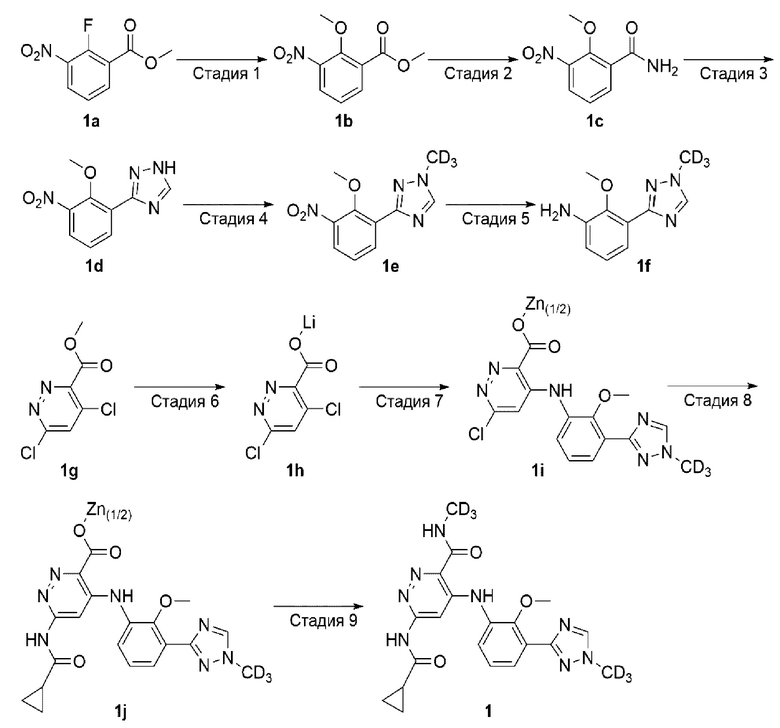
Стадия 1
Метил 2-метокси-3-нитробензоат (1b)
К раствору метил 2-фтор-3-нитробензоата 1а (10 г, 50 ммоль) в метаноле (50 мл) при комнатной температуре добавляли метоксид натрия (12,6 г, 70 ммоль). После перемешивания при комнатной температуре в течение 4 часов раствор разбавляли водой (200 мл), а затем экстрагировали этилацетатом (3×60 мл). Органические фазы объединяли, промывали насыщенным раствором соли (2×100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении для получения целевого соединения 1b (10 г, твердое вещество) с выходом 98%.
МС масса/заряд (ИЭР): 212 [М+1]
Стадия 2
2-метокси-3-нитробензамид (1с)
К раствору метил 2-метокси-3-нитробензоата 1b (10 г, 47 ммоль) в метаноле (40 мл) при комнатной температуре добавляли гидроксид аммония (20 мл). После перемешивания при комнатной температуре в течение 48 часов растворитель удаляли при пониженном давлении для получения целевого соединения 1с (неочищенное, 10 г, твердое вещество). Неочищенный продукт применяли на следующей стадии без дополнительной очистки. МС масса/заряд (ИЭР): 197 [М+1]
Стадия 3
3-(2-метокси-3-нитрофенил)-1H-1,2,4-триазол (1d)
Раствор 2-метокси-3-нитробензамида 1с (10 г, 51 ммоль) в N,N-диметилформамид диметилацетале (50 мл) нагревали до 95°С и перемешивали в течение 2 часов. После охлаждения до комнатной температуры растворитель удаляли при пониженном давлении и остаток растворяли в этаноле (30 мл) с получением раствора А. Гидрат гидразина (25 мл) медленно добавляли к смеси уксусной кислоты (35 мл) и этанола (150 мл) при 0°С с последующим добавлением раствора А. После постепенного нагревания до комнатной температуры и перемешивания в течение 12 часов растворитель удаляли при пониженном давлении. Остаток диспергировали в воде (400 мл) и фильтровали. Полученное твердое вещество отмывали водой и сушили с получением целевого соединения 1d (6 г, твердое вещество) с выходом 55%.
МС масса/заряд (ИЭР): 221 [М+1]
Стадия 4
3-(2-метокси-3-нитрофенил)-1 -(метил-d3)-1Н-1,2,4-триазол (1е)
К смеси 3-(2-метокси-3-нитрофенил)-1-(метил-d3)-1H-1,2,4-триазола 1d (1,2 г, 5,3 ммоль), карбоната калия (2,2 г, 16 ммоль) и N,N-диметилформамида (10 мл) добавляли дейтерированный йодометан (1 г, 6,9 ммоль). После перемешивания при комнатной температуре в течение 12 часов полученный раствор очищали с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 1е (530 мг, твердое вещество) с выходом 42%.
МС масса/заряд (ИЭР): 238 [М+1]
Стадия 5
2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)анилин (1f)
К раствору 3-(2-метокси-3-нитрофенил)-1-(метил-d3)-1H-1,2,4-триазола 1е (530 мг, 1,61 ммоль) в метаноле (10 мл) добавляли 10% палладий на углероде (50 мг). Реакционную смесь перемешивали в атмосфере водорода в течение 12 часов, а затем фильтровали. Фильтрат концентрировали досуха при пониженном давлении с получением целевого продукта 1f (430 мг, твердое вещество). Продукт применяли в следующей реакции без дополнительной очистки.
МС масса/заряд (ИЭР): 208 [М+1]
Стадия 6
4,6-дихлорпиридазин-3-карбоксилат лития (1h)
К смеси метил 4,6-дихлорпиридазин-3-карбоксилата 1g (5 г, 24,15 ммоль), диизопропилэтиламина (9,4 г, 72,5 ммоль), ацетонитрила (13,5 мл) и воды (3,5 мл) добавляли бромид лития (6,3 г, 72,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов и фильтровали. Полученное твердое вещество отмывали ацетонитрилом (8 мл) и сушили в вакууме с получением целевого соединения 1h (4,53 г, твердое вещество) с выходом 90%.
МС масса/заряд (ИЭР): 193 [М+1]
Стадия 7
6-хлор-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат цинка (1i)
К смеси 4,6-дихлорпиридазин-3-карбоксилата лития 1h (380 мг, 1,9 ммоль), 2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)анилина 1f (471 мг, 2,27 ммоль), изопропанола (0,5 мл) и воды (5 мл) добавляли ацетат цинка (350 мг, 1,9 ммоль) при комнатной температуре. Смесь нагревали до 65°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой (30 мл), перемешивали в течение 30 минут и фильтровали. Твердое вещество отмывали водой (2×30 мл) и тетрагидрофураном (2×30 мл) и сушили в вакууме с получением целевого соединения 1i (490 мг, твердое вещество) с выходом 71%.
МС масса/заряд (ИЭР): 364 [М+1]
Стадия 8
Метил 6-(циклопропанкарбоксамидо)-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат (1j)
К смеси 6-хлор-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилата цинка 1i (490 мг, 1,15 ммоль), циклопропанкарбоксамида (300 мг, 3,45 ммоль), (2R)-1-[(1R)-1-[бис(1,1-диметилэтил)фосфино]этил]-2-(дициклогексилфосфино)ферроцена (63 мг, 0,115 ммоль), ацетата палладия (25 мг, 0,0575 ммоль), толуола (9 мл) и ацетонитрила (5 мл), добавляли последовательно карбонат калия (320 мг, 7,8 ммоль) и 1,8-диазабициклоундек-7-ен (180 мг, 1,5 ммоль) последовательно. Полученную смесь перемешивали при 80°С в течение 72 часов в атмосфере азота. После охлаждения до комнатной температуры растворитель удаляли при пониженном давлении и остаток очищали с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 1j (560 мг, твердое вещество) с выходом 99%.
МС масса/заряд (ИЭР): 413 [М+1]
Стадия 9
6-(циклопропанкарбоксамидо)-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид (1)
Смесь метил 6-(циклопропанкарбоксамидо)-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилата 1j (280 мг, 0,68 ммоль), дейтерированного метиламина гидрохлорида (60 мг, 0,81 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорида (181 мг, 0,95 ммоль), 1-гидроксибензотриазола(53 мг, 0,34 ммоль), ацетонитрила (3 мл), N-метилпирролидона и N-метилимидазола (41 мг, 0,5 ммоль) нагревали до 65°С и перемешивали в течение 1 часа. После охлаждения до комнатной температуры растворитель удаляли при пониженном давлении и остаток очищали с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 1 (44 мг, твердое вещество) с выходом 15%.
МС масса/заряд (ИЭР): 429 [М+1]
1H ЯМР (400 МГц, DMSO-d6) δ 11,32 (s, 1Н), 10,97 (s, 1Н), 9,13 (s, 1Н), 8,56 (s, 1Н), 8,15 (s, 1Н), 7,65 (dd, J=7,8, 1,5 Гц, 1H), 7,54-7,46 (m, 1H), 7,32-7,22 (m, 1H), 3,72 (s, 3H), 2,12-2,03 (m, 1H), 0,88-0,73 (m, 4H).
Пример 2. 6-(циклопропанкарбоксамидо)-4-((2-метокси-3-(1-(метил-d3)-1H-1,2,4-триазол-3-ил)фенил)амино)-N-метилпиридазин-3-карбоксамид (2)
Соединение 2 синтезировали в соответствии со способами из примера 1, за исключением того, что гидрохлорид метиламина (CH3NH2⋅HCl) применяли в стадии 9 вместо дейтерированного гидрохлорида метиламина (CD3NH2⋅HCl).
МС масса/заряд (ИЭР): 426 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,32 (s, 1Н), 10,97 (s, 1Н), 9,22-9,11 (m, 1Н), 8,56 (s, 1Н), 8,15 (s, 1Н), 7,66 (dd, J=7,8, 1,5 Гц, 1Н), 7,51 (dd, J=8,0, 1,5 Гц, 1Н), 7,31-7,22 (m, 1Н), 3,72 (s, 3Н), 2,86 (d, J=4,8 Гц, 3Н), 2,15-2,01 (m, 1Н), 0,87-0,75 (m, 4Н).
Пример 3. 6-(циклопропанкарбоксамидо)-4-((2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-метилпиридазин-3-карбоксамид (3)
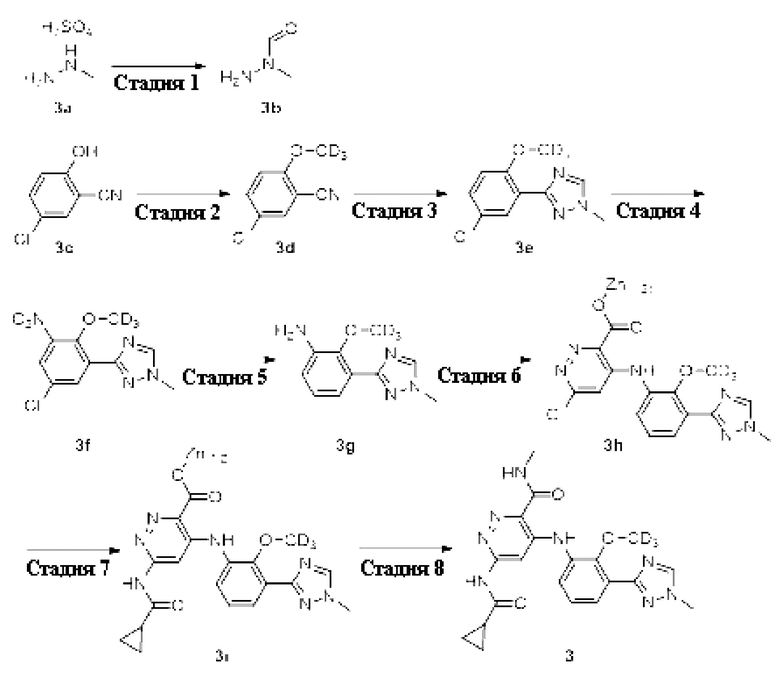
Стадия 1
N-метилформогидразид
К раствору метилгидразинсульфата 3а (40 г, 277 ммоль) в метаноле (250 ил) при комнатной температуре добавляли метоксид натрия (100 г, 554 ммоль). Полученную смесь перемешивали в течение 24 часов и фильтровали. Затем к фильтрату добавляли метилформиат (17 г, 277 ммоль) и перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли при пониженном давлении с получением целевого соединения 5b (22 г, неочищенное). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
МС масса/заряд (ИЭР): 75 [М+1]
Стадия 2
5-хлор-2-(метокси-d3)бензонитрил (3d)
К смеси 5-хлор-2-гидроксибензонитрила 3с (4 г, 26 ммоль), карбоната калия (7,3 г, 53 ммоль) и N,N-диметилформамида (30 мл) добавляли дейтерированный метилйодид при комнатной температуре (10 г, 78 ммоль). Полученную смесь нагревали по 70°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли водой (200 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические фазы промывали насыщенным раствором соли (2×100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении с получением целевого соединения 3d (4,3 г, твердое вещество) с выходом 97%.
МС масса/заряд (ИЭР): 171 [М+1]
Стадия 3
3-(5-хлор-2-(метокси-d3)фенил)-1 -метил-1H-1,2,4-триазол сульфат (3е)
К раствору трет-бутоксида калия (11,3 г, 101 ммоль) в тетрагидрофуране (30 мл) при 0°С последовательно добавляли 5-хлор-2-(метокси-d3)бензонитрил 3d (4,3 г, 25,3 ммоль) и раствор N-метилформилгидразида (4,1 г, 58 ммоль) в тетрагидрофуране (20 мл). После перемешивания при комнатной температуре в течение 12 часов добавляли воду (50 мл), нагревали до 40°С и перемешивали в течение 40 минут. После охлаждения до комнатной температуры, органическую фазу разделяли, промывали насыщенным раствором соли (40 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Осадок растворяли в этилацетате (40 мл). В полученный раствор медленно добавляли концентрированную серную кислоту (5 г) при комнатной температуре и перемешивали в течение 12 часов. Затем смесь фильтровали и сушили с получением целевого соединения 3е (5,6 г, твердое вещество) с выходом 83%.
МС масса/заряд (ИЭР): 227 [М+1]
Стадия 4
3-(5-хлор-2-(метокси-d3)-3-нитрофенил)-1-метил-1Н-1,2,4-триазол (3f)
К раствору 3-(5-хлор-2-(метокси-d3)фенил)-1-метил-1H-1,2,4-триазолсульфата 3е (5,6 г, 24,7 ммоль) в серной кислоте (25 г) добавляли азотную кислоту (2 г) при 0°С. Полученный раствор постепенно нагревали до комнатной температуры, перемешивали в течение 12 часов, а затем снова охлаждали до 0°С. Воду (67 мл) и метанол (47 мл) добавляли в раствор при 0°С, затем нагревали до комнатной температуры и перемешивали в течение часа. Раствор нагревали до 40°С и к нему добавляли гидроксид аммония (42 мл). Раствор охлаждали до 20°С, перемешивали в течение 2 часов, а затем фильтровали. Твердое вещество промывали водой (2×30 мл) и сушили в вакууме с получением целевого соединения 3f (3,37 г, твердое вещество) с выходом 50%.
МС масса/заряд (ИЭР): 272 [М+1]
Стадия 5
2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)анилин (3g)
К раствору 3-(5-хлор-2-(метокси-d3)-3-нитрофенил)-1-метил-1H-1,2,4-триазола 3f (3,37 г, 12,25 ммоль) в метаноле (10 мл) добавляли 10% палладий на углероде (400 мг) и бикарбонат натрия (1,6 г, 25 ммоль). Полученную смесь перемешивали в атмосфере водорода в течение 12 часов, а затем фильтровали. Фильтрат концентрировали досуха при пониженном давлении и полностью растворяли в дихлорметане (25 мл). Полученную смесь фильтровали, а фильтрат концентрировали досуха при пониженном давлении с получением целевого соединения 3g (2,35 г, твердое вещество) с выходом 92%.
МС масса/заряд (ИЭР): 208 [М+1]
Стадия 6
6-хлор-4-((2-(метокси-d3)-3-(1-метил-1Н-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат цинка (3h)
К смеси 4,6-дихлорпиридазин-3-карбоксилата лития 1h (3 г, 15,1 ммоль), 2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)анилина 3g (2,35 г, 11,3 ммоль), изопропанола (2,5 мл) и воды (18 мл) при комнатной температуре добавляли ацетат цинка (2,5 г, 13,6 ммоль). Смесь нагревали до 65°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры смесь разбавляли водой (20 мл), перемешивали в течение 30 минут и фильтровали. Твердое вещество промывали водой (2×30 мл) и тетрагидрофураном (2×30 мл) и сушили в вакууме с получением целевого соединения 3h (4,3 г, твердое вещество) с выходом 100%.
МС масса/заряд (ИЭР): 364 [М+1]
Стадия 7
Метил 6-(циклопропанкарбоксамидо)-4-((2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат (3i)
Смесь 6-хлор-4-((2-(метокси-d3)-3-(1-метил-1Н-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилата цинка 3h (4,3 г, 11 ммоль), циклопропанкарбоксамида (2,4 г, 27,56 ммоль), (2R)-1-[(1R)-1-[бис(1,1-диметилэтил)фосфино]этил]-2-(дициклогексилфосфино)ферроцена (600 мг, 1,1 ммоль), ацетата палладия (125 мг, 0,55 ммоль), толуола (34 мл), ацетонитрила (17 мл), карбоната калия (3,1 г, 22 ммоль) и 1,8-диазабициклоундек-7-ена (1,7 г, 11 ммоль) нагревали до 80°С в атмосфере азота и перемешивали в течение 12 часов. После охлаждения до комнатной температуры последовательно добавляли водную уксусную кислоту (50%, 17 мл) и ледяную уксусную кислоту (40 мл). После перемешивания при комнатной температуре в течение часа полученную гомогенную смесь промывали петролейным эфиром (2×20 мл). Добавляли воду (50 мл) и выдерживали смесь при комнатной температуре в течение 4 часов, а затем фильтровали. Твердое вещество последовательно промывали водным раствором ацетонитрила (50%, 20 мл) и ацетонитрилом (20 мл), а затем сушили в вакууме при 65°С в течение 30 минут с получением целевого продукта 3i (3 г, твердое вещество) с выходом 66%.
МС масса/заряд (ИЭР): 413 [М+1]
Стадия 8
6-(циклопропанкарбоксамидо)-4-((2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-метилпиридазин-3-карбоксамид
Смесь метил 6-(циклопропанкарбоксамидо)-4-((2-(метокси-d3)-3-(1-метил-1Н-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилата 3i (1,5 г, 3,38 ммоль), гидрохлорида метиламина (280 мг, 4,0 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорида (900 мг, 4,73 ммоль), 1-гидроксибензотриазола (230 мг, 1,7 ммоль), ацетонитрила (3 мл), N-метилпирролидона (3 мл) и N-метилимидазола (200 мг, 2,4 ммоль) нагревали до 65°С и перемешивали в течение 12 часов. После завершения реакции ее гасили водой (1,5 мл) и ацетонитрилом (4,5 мл). Полученную смесь выдерживали при 65°С в течение часа и при 0°С в течение 3 часов, а затем фильтровали. Твердое вещество последовательно промывали водным раствором ацетонитрила (33%, 4,5 мл) и ацетонитрилом (4,5 мл), сушили в вакууме при 65°С в течение 8 часов с получением целевого продукта 3 (811 мг, твердое вещество) с выходом 56%.
МС масса/заряд (ИЭР): 426 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,32 (s, 1Н), 10,97 (s, 1Н), 9,24-9,08 (m, 1Н), 8,57 (s, 1Н), 8,15 (s, 1Н), 7,66 (dd, J=7,8, 1,5 Гц, 1Н), 7,52 (dd, J=7,9, 1,5 Гц, 1Н), 7,33-7,20 (m, 1Н), 3,96 (s, 3Н), 2,87 (d, J=4,8 Гц, 3Н), 2,14-2,01 (m, 1Н), 0,91-0,73 (m, 4Н).
Соединение 3 можно превратить в соль HCl следующим способом:
В реакционную колбу добавляли соединение 3 (5,00 г, 11,752 ммоль) и ДМСО (27 мл). Полученную смесь нагревали до 50~55°С при перемешивании до полного растворения твердого вещества до гомогенного раствора. Затем к смеси добавляли концентрированную соляную кислоту (36%~38%, 1,18 г), а затем воду (3 мл) и кристаллическую затравку (25 мг). Полученную смесь перемешивали при 50~55°С в течение получаса, охлаждали до 35~40°С, добавляли изопропанол (60 мл) капельно в течение 0,5~1,0 часа, и перемешивали при 35~40°С в течение получаса. Смесь медленно охлаждали до 20-25°С в течение часа, перемешивали в течение ночи, а затем фильтровали. Отфильтрованный осадок отмывали изопропанолом (2×15 мл) и сушили при пониженном давлении при 65°С в течение ночи с получением моносоли HCl соединения 3 (4,5 г, твердое вещество) с выходом 83%.
МС масса/заряд (ИЭР): 426 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 13,72 (brs, 1Н), 12,13 (s, 1Н), 11,40 (s, 1Н), 9,22 (q, J=4,5 Гц, 1H), 8,87 (s, 1H), 8,00 (s, 1H), 7,78 (dd, J=7,9, 1,5 Гц, 1H), 7,61 (dd, J=8,0, 1,4 Гц, 1H), 7,35 (t, J=7,9 Гц, 1H), 4,01 (s, 3H), 2,89 (d, J=4,8 Гц, 3Н), 2,14-2,00 (m, 1H), 1,00-0,84 (m, 4H).
Пример 4. 6-(циклопропанкарбоксамидо)-4-((2-(метокси-d3)-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид (4)
Соединение 4 синтезировали в соответствии со способами из примера 3, за исключением того, что применяли дейтерированный гидрохлорид метиламина (CD3NH2⋅HCl) на этапе 8 вместо гидрохлорида метиламина (CH3NH2⋅HCl).
МС масса/заряд (ИЭР): 429 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,32 (s, 1Н), 10,98 (s, 1Н), 9,14 (s, 1Н), 8,57 (s, 1Н), 8,15 (s, 1H), 7,66 (dd, J=7,8,1,6 Гц, 1H), 7,52 (dd, J=7,9, 1,5 Гц, 1H), 7,32-7,21 (m, 1H),3,96 (s, 3H), 2,14-2,03 (m, 1H), 0,89-0,75 (m, 4H).
Пример 5. (S)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид (5)
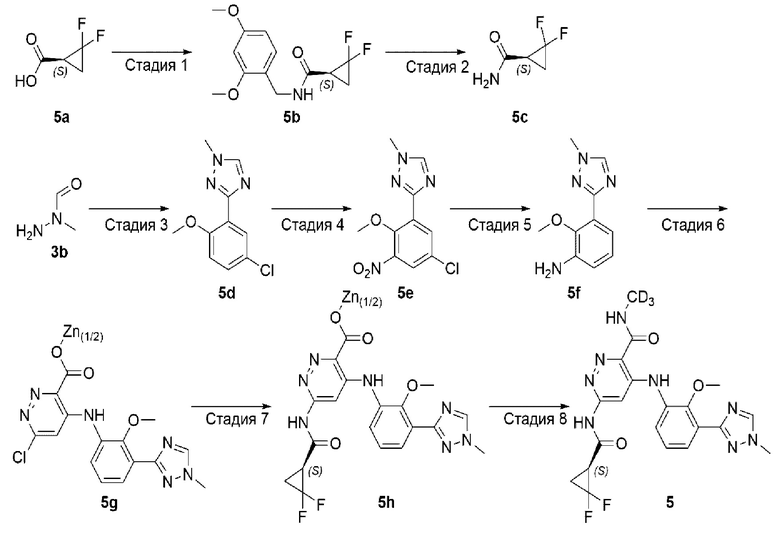
Стадия 1
(S)-N-(2,4-диметоксибензил)-2,2-дифторциклопропан-1-карбоксамид (5b)
К смеси (5)-2,2-дифторциклопропан-1-карбоновой кислоты 5а (1,5 г, 12,3 ммоль), HATU (5,7 г, 15 ммоль), диизопропиламина (4,8 г, 37 ммоль) и N,N-диметилформамида (15 мл) добавляли 2,4-диметоксибензиламин (4,0 г, 24,4 ммоль). После перемешивания при комнатной температуре в течение 3 часов растворитель удаляли при пониженном давлении и очищали с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 5b (4,4 г, твердое вещество).
МС масса/заряд (ИЭР): 272 [М+1]
Стадия 2
(S)-2,2-дифторциклопропан-1-карбоксамид (5с)
Раствор (S)-N-(2,4-диметоксибензил)-2,2-дифторциклопропан-1-карбоксамида 5b в трифторуксусной кислоте (10 мл) нагревали до 70°С и перемешивали в течение часа. После охлаждения до комнатной температуры смесь концентрировали досуха и очищали колоночной хроматографией с силикагелем (дихлорметан/метанол от 100/0 до 9/1) с получением целевого соединения 5d (1,4 г, твердое вещество) с выходом 93% в двух стадиях.
МС масса/заряд (ИЭР): 122 [М+1]
Стадия 3
3-(5-хлор-2-метоксифенил)-1-метил-1H-1,2,4-триазол (5d)
К раствору трет-бутоксида калия (34 г, 290 ммоль) в тетрагидрофуране (200 мл) при 0°С последовательно добавляли 5-хлор-2-метокси-бензонитрил (20 г, 120 ммоль) и метилформилгидразид 3b (22 г, неочищенный). После перемешивания при комнатной температуре в течение 72 часов добавляли воду (500 мл) и смесь экстрагировали этилацетатом (3×300 мл). Органические фазы объединяли, промывали насыщенным раствором соли (2×300 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении с получением целевого соединения 5d (17,1 г, твердое вещество) с выходом 88%.
МС масса/заряд (ИЭР): 224 [М+1]
Стадия 4
3-(5-хлор-2-метокси-3-нитрофенил)-1-метил-1Н-1,2,4-триазол (5е)
К раствору 3-(5-хлор-2-метоксифенил)-1-метил-1Н-1,2,4-триазола 5d (16,13 г, 72 ммоль) в концентрированной серной кислоте (72 г) добавляли концентрированную азотную кислоту (8,5 г, 87 ммоль) при 0°С. После перемешивания в течение 2 часов полученный раствор добавляли к смеси воды (250 г) и метанола (150 г) при 0°С. Затем смесь доводили до рН>7 с помощью гидроксида аммония и фильтровали. Твердое вещество промывали водой (2×100 мл) и очищали хроматографией с колонкой с силикагелем (петролейный эфир/этилацетат от 100/0 до 3/7) с получением целевого продукта 5е (17,1 г, твердое вещество) с выходом 88%.
МС масса/заряд (ИЭР): 269 [М+1]
Стадия 5
2-метокси-3-(1-метил- 2,4-триазол-3-ил)анилин (5f)
К раствору 3-(5-хлор-2-метоксифенил)-1-метил-1Н-1,2,4-триазола 5е (17 г, 63 ммоль) в метаноле добавляли 10% палладий на углероде (3 г) и бикарбонат, натрия (10,5 г, 126 ммоль). Смесь перемешивали в атмосфере водорода в течение 5 часов, а затем фильтровали. Фильтрат концентрировали досуха при пониженном давлении и очищали остаток с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 5f (8,8 г, твердое вещество) с выходом 68%.
МС масса/заряд (ИЭР): 205 [М+1]
Стадия 6
6-хлор-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат цинка (5g)
К смеси 4,6-дихлорпиридазин-3-карбоксилата лития 1h (4,53 г, 22,87 ммоль), 2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)анилина 5f (5,6 г, 27,44 ммоль), изопропанола (4,5 мл) и воды (34 мл) добавляли ацетат цинка (4,2 г, 22,87 ммоль). Полученную смесь нагревали до 65°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры смесь разбавляли водой (30 мл), выдерживали в течение 30 минут, а затем фильтровали. Твердое вещество промывали водой (2×30 мл) и тетрагидрофураном (2×30 мл) и сушили в вакууме с получением целевого соединения 5g (7,6 г, твердое вещество) с выходом 93%.
МС масса/заряд (ИЭР): 361 [М+1]
Стадия 7
(S)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилат цинка (5h)
Смесь 6-хлор-4-((2-метокси-3-(1-метил-1Н-1,2,4-триазол-3-ил)фенил)амино)пиридазин-3-карбоксилата цинка 5g (1,6 г, 3,93 ммоль), (5)-2,2-дифторциклопропан-1-карбоксамида 5с (1,2 г, 9,8 ммоль), (2R)-1-[(1R)-1-[бис(1,1-диметилэтил)фосфино]этил]-2-(дициклогексилфосфино)ферроцена (220 мг, 0,393 ммоль), ацетата палладия (44 мг, 0,196 ммоль), толуола (18 мл), ацетонитрила (11 мл), карбоната калия (1,1 г, 7,8 ммоль) и 1,8-диазабициклоундек-7-ена (600 мг, 3,93 ммоль) перемешивали при 80°С в течение 72 часов в атмосфере азота. После охлаждения до комнатной температуры смесь разбавляли уксусной кислотой (27 мл) и водой (9 мл), а полученный раствор отмывали петролейным эфиром (2×30 мл). Затем добавляли воду (50 мл) и оставляли на 3 часа. Смесь фильтровали и твердое вещество сушили в вакууме с получением целевого соединения 5h (1,1 г, твердое вещество) с выходом 62%.
МС масса/заряд (ИЭР): 446 [М+1]
Стадия 8
(S)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид
К смеси (5)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил))фенил)амино)пиридазин-3-карбоксилата цинка 5h (1,1 г, 2,46 ммоль), дейтерированного гидрохлорида метиламина (210 мг, 2,95 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (660 мг, 3,44 ммоль), 1-гидроксибензотриазола (190 мг, 1,23 ммоль), ацетонитрила (6 мл) и N-метилпирролидона (6 мл) добавляли N-метилимидазол (141 мг, 1,72 ммоль). Реакционную смесь нагревали до 65°С и перемешивали в течение часа. После охлаждения до комнатной температуры смесь концентрировали досуха при пониженном давлении и очищали осадок с помощью обратно-фазовой препаративной ВЭЖХ с получением целевого соединения 5 (420 мг, твердое вещество) с выходом 37%.
МС масса/заряд (ИЭР): 462 [М+1]
1H ЯМР (400 МГц, DMSO-d6) δ 11,52 (s, 1Н), 11,01 (s, 1Н), 9,18 (s, 1Н), 8,58 (s, 1H), 8,09 (s, 1H), 7,67 (dd, J=7,8, 1,6 Гц, 1H), 7,53 (dd, J=7,9, 1,5 Гц, 1H), 7,33-7,23 (m, 1H), 3,95 (s, 3H), 3,73 (s, 3H), 3,13 - 2,97 (m, 1H), 2,10-1,95 (m, 2H).
Пример 6. (S)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-1,2,4-триазол-3-ил)фенил)амино)-N-метилпиридазин-3-карбоксамид (6)
Соединение 6 синтезировали в соответствии со способами из примера 5, за исключением того, что дейтерированный гидрохлорид метиламина (CD3NH2⋅HCl) на стадии 8 заменяли гидрохлоридом метиламина (CH3NH2⋅HCl).
МС масса/заряд (ИЭР): 459 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,52 (s, 1Н), 11,01 (s, 1Н), 9,20 (d, J=4,8 Гц, 1Н), 8,56 (s, 1Н), 8,09 (s, 1Н), 7,67 (dd, J=7,8, 1,6 Гц, 1Н), 7,52 (dd, J=8,0, 1,5 Гц, 1Н), 7,33-7,23 (m, 1Н), 3,95 (s, 3Н), 3,73 (s, 3Н), 3,11-2,98 (m, 1Н), 2,86 (d, J=4,8 Гц, 3Н), 2,10-1,94 (m, 2Н).
Пример 7. (R)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1Н-1,2,4-триазол-3-ил)фенил)амино)-N-(метил-d3)пиридазин-3-карбоксамид (7)
Соединение 7 синтезировали в соответствии со способами из примера 5, за исключением того, что (S)-2,2-дифторциклопропан-1-карбоновая кислота (5а) на стадии 1 была заменена на (R)-2,2-дифторциклопропан-1-карбоновую кислоту.
МС масса/заряд (ИЭР): 462 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,52 (s, 1Н), 11,01 (s, 1Н), 9,18 (s, 1Н), 8,56 (s, 1Н), 8,09 (s, 1Н), 7,67 (dd, J=7,8, 1,6 Гц, 1Н), 7,53 (dd, J=7,9, 1,5 Гц, 1Н), 7,28 (m, 1Н), 3,95 (s, 2Н), 3,73 (s, 3Н), 3,11-2,99 (m, 1Н), 2,10-1,95 (m, 2Н).
Пример 8. (R)-6-(2,2-дифторциклопропан-1-карбоксамидо)-4-((2-метокси-3-(1-метил-1H-l,2,4-триазол-3-ил)фенил)амино)-N-метилпиридазин-3-карбоксамид (8)
Соединение 8 синтезировали в соответствии со способами из примера 5, за исключением того, что (i) (S)-2,2-дифторциклопропан-1-карбоновая кислота (5а) на стадии 1 была заменена на (R)-2,2-дифторциклопропан-1-карбоновую кислоту, кислоты, и (ii) дейтерированный гидрохлорид метиламина (CD3NH2⋅HCl) на стадии 8 был заменен гидрохлоридом метиламина (CH3NH2⋅HCl).
МС масса/заряд (ИЭР): 459 [М+1]
1Н ЯМР (400 МГц, DMSO-d6) δ 11,52 (s, 1Н), 11,01 (s, 1Н), 9,27-9,16 (m, 1Н), 8,56 (s, 1Н), 8,09 (s, 1Н), 7,67 (dd, J=7,8, 1,6 Гц, 1Н), 7,52 (dd, J=8,0, 1,5 Гц, 1Н), 7,33-7,24 (m, 1Н), 3,95 (s, 2Н), 3,73 (s, 2Н), 3,11-2,99 (m, 1Н), 2,86 (d, J=4,8 Гц, 3Н), 2,08-1,95 (m, 2Н).
Пример 9. Анализ ферментативной активности киназного домена JAK2
Влияние соединений по настоящему изобретению на ферментативную активность киназного домена рекомбинантной JAK2 (JH1) оценивали путем определения уровня фосфорилирования субстрата в киназной реакции с использованием набора для детекции HTRF-киназного анализа (Cisbio, кат.номер 62TK0PEC) (таблица 1).
Экспериментальный способ в общих чертах описан ниже:
Реакционный буфер, содержащий следующие компоненты: буфер для фермента (1х), 5 мМ MgCl2, 1 мМ ДТТ и 0,01% Brij35 из набора; белок киназного домена рекомбинантной человеческой JAK2 (Carna Biosciences, кат. номер 08-045), разведенный реакционным буфером до концентрации 0,15 нг/мкл; реакционный раствор субстрата, содержащий 2,5 мкМ АТФ и биотинилированный субстрат тирозинкиназы, разбавленный до 0,25 мкМ реакционным буфером; раствор для детекции, содержащий 0,1 нг/мкл меченного Eu3+ антитела (Cisbio, кат. номер 61T66KLB) и 12,5 нМ меченного стрептавидином XL665 (Cisbio, кат. номер 610SAXLB) в реакционном буфере.
Тестируемое соединение растворяли до 1 мМ в ДМСО с последующим серийным 4-кратным разведением ДМСО до минимальной концентрации 61 нМ. Каждую концентрацию дополнительно разбавляли реакционным буфером в 40 раз.
В 384-луночный планшет для анализа (Corning, кат. номер 3674) добавляли 4 мкл раствора соединения и 2 мкл раствора киназы JAK2. Смесь инкубировали при комнатной температуре в течение 15 минут, а затем добавляли 4 мкл реакционного раствора субстрата. После дальнейшего инкубирования при комнатной температуре в течение 30 минут, добавляли в реакционную смесь равный объем (10 мкл) раствора для детекции и оставляли при комнатной температуре в течение 30 минут. Затем применяли спектрофотометр Envision для планшетов (Perkin Elmer) для измерения хода реакции при 620 нм и 665 нм. Соотношение поглощений при 665 нм и 620 нм положительно коррелирует со степенью фосфорилирования субстрата, таким образом, выявляя активность киназы JAK2. В этом эксперименте группа без белка киназы JAK2 представляла собой группу со 100% ингибированием, а группа с белком киназы JAK2, но без тестируемого соединения, представляла собой группу с 0% ингибирования. Процент ингибирования активности киназы JAK2 тестируемым соединением рассчитывают по следующей формуле:
Процент ингибирования=100-100×(соотношениесоединение-соотношение100% ингибирование)/(соотношение0% ингибирования-отношение100% ингибирование)
Значение IC50 тестируемого соединения рассчитывают из 8 точек концентрации с использованием программного продукта XLfit (ID Business Solutions Ltd., UK) по следующей формуле:
Y=Низ + (Верх - Низ)/(1+10(logIC50-X)×коэффициент наклона)
где Y представляет собой процент ингибирования, X представляет собой логарифм концентрации тестируемого соединения, Низ представляет собой значение нижнего плато S-образной кривой, Верх представляет собой значение верхнего плато S-образной кривой, а коэффициент наклона представляет собой коэффициент наклона кривой.
Пример 10. Анализ ферментативной активности киназного домена TYK2
Влияние соединений по настоящему изобретению на ферментативную активность киназного домена рекомбинантной TYK2 (JH1) оценивали путем определения уровня фосфорилирования субстрата в киназной реакции с использованием набора для детекции HTRF-киназного анализа (Cisbio, кат. номер 62TK0PEC) (таблица 1).
Экспериментальный способ в общих чертах описан ниже:
Реакционный буфер, содержащий следующие компоненты: буфер для фермента (1×), 5 мМ MgCl2, 1 мМ ДТТ, 10 нМ SEB (Cisbio, кат. номер 61SEBALB), 0,625 мМ EGTA и 0,01% Brij35 из набора; белок киназного домена (JH1) рекомбинантной человеческой TYK2 (Carna Biosciences, кат. номер 08-147), разведенный реакционным буфером до концентрации 0,25 нг/мкл; реакционный раствор субстрата, содержащий 11,25 мкМ АТФ и биотинилированный субстрат тирозинкиназы, разбавленный до 0,5 мкМ реакционным буфером; раствор для детекции, содержащий 0,1 нг/мкл меченного Eu3+ антитела (Cisbio, кат. номер 61T66KLB) и 25 нМ меченного стрептавидином XL665 (Cisbio, кат. номер 610SAXLB) в реакционном буфере.
Тестируемое соединение растворяли до 1 мМ в ДМСО с последующим серийным 4-кратным разведением ДМСО до минимальной концентрации 61 нМ. Каждую концентрацию дополнительно разбавляли реакционным буфером в 40 раз.
В 384-луночный планшет для анализа (Corning, кат. номер 3674) добавляли 4 мкл раствора соединения и 2 мкл раствора киназы TYK2. Смесь инкубировали при комнатной температуре в течение 15 минут, а затем добавляли 4 мкл реакционного раствора субстрата. После дальнейшего инкубирования при комнатной температуре в течение 40 минут, добавляли в реакционную смесь равный объем (10 мкл) раствора для детекции и оставляли при комнатной температуре в течение 30 минут. Затем применяли спектрофотометр Envision для планшетов (Perkin Elmer) для измерения хода реакции при 620 нм и 665 нм. Соотношение поглощений при 665 нм и 620 нм положительно коррелирует со степенью фосфорилирования субстрата, таким образом, выявляя активность киназы TYK2. В этом эксперименте группа без белка киназы TYK2 представляла собой группу со 100% ингибированием, а группа с белком киназы TYK2, но без тестируемого соединения, представляла собой группу с 0% ингибирования. Процент ингибирования активности киназы TYK2 тестируемым соединением рассчитывают по следующей формуле:
Процент ингибирования = 100-100×(соотношениесоединение-соотношение100% ингибирование)/(соотношение0%ингибирования-отношение100% ингибирование)
Значение IC50 тестируемого соединения рассчитывают из 8 точек концентрации с использованием программного продукта XLfit (ID Business Solutions Ltd., UK) no следующей формуле:
Y=Низ + (Верх - Низ)/(1+10(logIC50-X)×коэффициент наклона)
где Y представляет собой процент ингибирования, X представляет собой логарифм концентрации тестируемого соединения, Низ представляет собой значение нижнего плато S-образной кривой, Верх представляет собой значение верхнего плато S-образной кривой, а коэффициент наклона представляет собой коэффициент наклона кривой.
Пример 11. Анализ связывания псевдокиназного домена TYK2 Связывание соединений по настоящему изобретению с псевдокиназным доменом TYK2 (JH2) определяли с помощью биохимического анализа флуоресценции переноса с временным разрешением (TR-FRET) путем конкуренции с коммерческим меченым флуоресцеином зондом (маркер киназы 178, конъюгированный с Alexa-Fluor 647) (таблица 1).
Экспериментальный способ в общих чертах описан ниже:
Связывающий буфер содержит 20 мМ Hepes рН 7,5, 150 мМ NaCl, 10 мМ MgCl2, 0,015% Brij35, 2 мМ DTT, 0,625 мМ EGTA и 100 мМ KF. Домен JH2 TYK2 (аминокислоты 556-871 в полноразмерном белке) экспрессировали и очищали при помощи платформы для очистки и идентификации белков в университете Цинхуа. Тестируемое соединение растворяли до 0,1 мМ в ДМСО с последующим серийным 4-кратным разведением ДМСО до минимальной концентрации 61 нМ. Каждую концентрацию дополнительно разбавляли реакционным буфером в 40 раз.
В 384-луночный планшет для анализа (Corning, кат. номер 4512) добавляли 5 мкл раствора соединения и 5 мкл раствора домена JH2 TYK2 (160 нМ). Смесь инкубировали при комнатной температуре в течение 30 минут, а затем добавляли 10 мкл смеси меченого флуоресцеином зонда (ThermoFisher, кат. номер PV5593) (20 нМ) и меченного GST-Europium (Eu) антитела (Cisbio, кат. номер 61GSTKLA) (40 нг/мл). После дальнейшей инкубации при комнатной температуре в течение 30 минут измеряли сигнал HTRF (соотношение интенсивности флуоресценции при длине волны испускания 615 нм и 665 нм для акцептора флуоресцеина и донора европия, соответственно) на спектрофотометре Envision для чтения планшетов (Perkin Elmer). Процент ингибирования рассчитывали путем сравнения с положительным контролем без тестируемого соединения и отрицательным контролем без белка по следующей формуле:
% ингибирования = 100-100×(сигналсоединение-сигналотрицательный контроль)/(сигналположительный контроль-сигналотрицательный контроль)
Значение IC50 тестируемого соединения рассчитывают из 8 точек концентрации с использованием программного продукта XLfit (ID Business Solutions Ltd., UK) no следующей формуле:
Y=Низ + (Верх - Низ)/(1+10(1ogIC50-X)×коэффициент наклона)
где Y представляет собой процент ингибирования, X представляет собой логарифм концентрации тестируемого соединения, Низ представляет собой значение нижнего плато S-образной кривой, Верх представляет собой значение верхнего плато S-образной кривой, а коэффициент наклона представляет собой коэффициент наклона кривой.
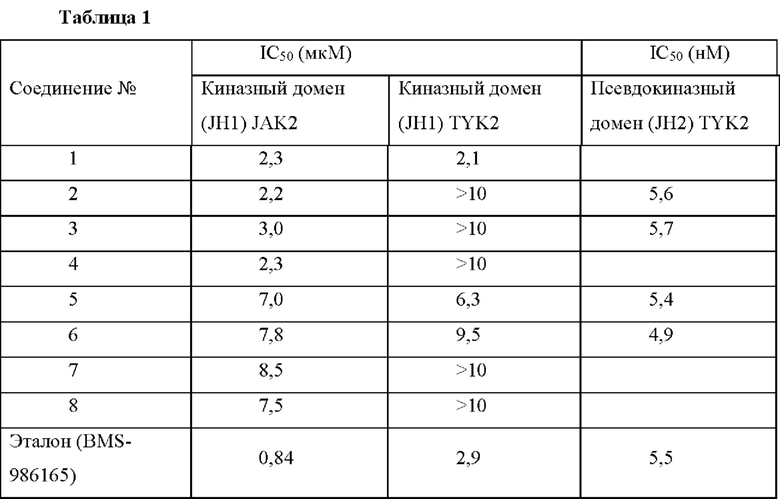
Соединения по настоящему изобретению обладают слабой или низкой ингибирующей активностью в отношении киназных доменов JAK2 или TYK2. Таблица 1 показывает, что соединения 2, 3, 4, 7 и 8 имели IC50 >10 мкМ для прямого ингибирования киназной активности, тогда как эталонное соединение имело более низкую IC50 2,9 мкМ. Тестируемые соединения и эталонные соединения показали сильное связывание с JH2 TYK2 (IC50 в диапазоне нМ).
Пример 12. Ингибирование IL-12-индуцированной секреции IFN-γ в клетках NK92
Влияние соединений по настоящему изобретению на секрецию IFN-γ, индуцированную TYK2, в клетках NK92 оценивали с помощью твердофазного иммуноферментного анализа (ИФА) (таблица 2).
Рецептор IL-12 в основном экспрессируется в активированных Т-клетках, NK-клетках (NK92 - NK-клеточная линия), дендритных клетках и В-клетках. Связываясь с IL-12, он активирует пути передачи сигнала JAK2/TYK2 внутри NK-клеток и Т-лимфоцитов, индуцируя тем самым секрецию IFN-γ.
Экспериментальный способ в общих чертах описан ниже:
Тестируемое соединение растворяют до 2,5 мкМ в ДМСО с последующим серийным 4-кратным разведением ДМСО до минимальной концентрации 0,31 мкМ. Каждую концентрацию дополнительно разводят в 50 раз средой MEM а, не содержащей FBS (Gibco, кат. номер 12561-056).
Клетки NK92 (Nan jing Cobioer, кат.номер СВР60980) культивируют на полной среде МЕМα, содержащей 12,5% FBS (Ausbian, кат.номер VS500T), 12,5% лошадиную сыворотку (Gibco, кат. номер 16050-122), 0,02 мМ фолиевую кислоту (Sigma, кат. номер F8758), 0,2 мМ инозитол (Sigma, кат. номер 17850), 0,55 мМ β-меркаптоэтанол (Gibco, кат. номер 21985-023), 200 Ед/мл IL-2 (R&D Systems, кат.номер 202- 1L), и 100 Ед/мл пенициллина (ThermoFisher, кат. номер 15140122). При покрытии 80-90% поверхности культурального контейнера клетки диспергируют и высевают на 96-луночный планшет (ThermoFisher, кат. номер 167425) по 100000 клеток на лунку (80 мкл полной среды МЕМα без IL-2). Затем 96-луночный планшет инкубируют в течение ночи в инкубаторе при 37°С/5% СО2.
После инкубации в течение ночи в каждую лунку добавляют 10 мкл тестируемого соединения и 10 мкл 50 нг/мл IL-12 (R&D Systems, кат. номер 219-1L), осторожно перемешивают и инкубируют 96-луночный планшет в инкубаторе 37°С/5% СО2 еще 24 часа. Планшеты центрифугируют при 800 об./мин. в течение 10 минут при комнатной температуре и 50 мкл супернатанта из каждой лунки переносят в другой 96-луночный планшет (Sigma, кат. номер CLS3695), покрытый антителом к IFN-γ. Количество секреции IFN-γ определяют согласно инструкции из набора Human IFN-gamma DuoSet ELISA (R&D Systems, кат. номер DY285B). В эксперименте группа с IL-12, в которой тестируемое соединение заменено на среду МЕМα, является нестимулированной контрольной группой (100% ингибирование), а группа с IL-12 и 0,2% ДМСО является стимулированной группой (0% ингибирование). Процент ингибирования IL-12-индуцированной секреции IFN-γ в клетках NK-92 с помощью тестируемого соединения рассчитывают по следующей формуле:
Процент ингибирования = 100-100×(сигналсоединение-сигналнестимулированный контроль)/(сигналстимулированный контроль-сигналнестимулированный контроль)
Значение IC50 тестируемого соединения рассчитывают из 8 точек концентрации с использованием программного продукта XLfit (ID Business Solutions Ltd., UK) no следующей формуле:
Y=Низ + (Верх - Низ)/(1+10(1ogIC50-X)×коэффициент наклона)
где Y представляет собой процент ингибирования, X представляет собой логарифм концентрации тестируемого соединения, Низ представляет собой значение нижнего плато S-образной кривой, Верх представляет собой значение верхнего плато S-образной кривой, а коэффициент наклона представляет собой коэффициент наклона кривой.
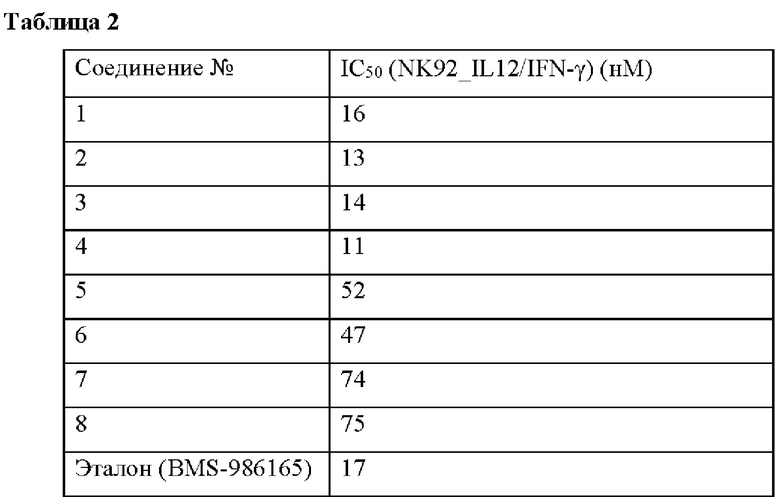
Соединения по настоящему изобретению оказывают значительное ингибирующее действие на секрецию IFN-γ, индуцированную TYK2 в клетках NK92.
Пример 13. Определение фармакокинетнки у крыс in vivo
Оценивали фармакокинетику соединения 3 по настоящему изобретению и эталонного соединения BMS-986165. Соединение 3 имеет OCD3 на бензольном кольце, тогда как эталонное соединение имеет CD3 на амидной группе. Метальная группа, как правило, лабильна in vivo, подвергается гидролизу амидазой в случае метиламида и окислительному деметилированию с помощью CYP в случае метокси и метилтриазола. Замена метила на тридейтерированный метил улучшает биодоступность и экспозицию соединения in vivo и обеспечивает лучшую эффективность соединения при той же дозе.
Соединение 3 и эталонное соединение в растворе 0,5 мг/мл, содержащем 5% N,N-диметилацетамида + 20% солутола + 75% физиологического раствора, перорально вводили трем самцам крыс Sprague Dawley в дозе 5 мг/кг. Образцы крови собирали через 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения. Концентрации соединения в плазме количественно определяли путем ЖХ-МС/МС с использованием масс-спектрометра API-4500. Предел количественного определения (LOQ) анализа составил 1 нг/мл. Фармакокинетические (PK) параметры, рассчитанные некомпартментным способом с использованием WinNonlin, представлены в таблице 3. Результаты показывают, что соединение 3 по настоящему изобретению имеет параметры распределения in vivo лучше, чем эталонное соединение.

Пример 14. Оценка эффективности in vivo на модели колита, индуцированного антителом к CD40, у животных
Самок мышей CB17-Scid (возраст 8-10 недель, 18-20 г) из лаборатории Beijing Vital River случайным образом разделили на 5 групп (n=8 в группе). На сутки 0 индуцировали у мышей колит путем однократной интраперитонеальной инъекции 100 мкг моноклонального антитела к CD40 FGK4.5 (BioXCell, кат.номер ЕВ0016-2) в PBS. Начиная с суток с 0 по сутки 7, мышам в группах с лечением перорально вводили 0, 1,5, 5, 15 мг/кг соединения 3 или 5 мг/кг BMS-986165 в носителе ДМСО/солутол/ПЭГ-400. (10:5:30) два раза в день, при этом мышам в группе с носителем перорально вводили указанный выше носитель. Ежедневно мышей взвешивали и отслеживали признаки колита, включая потерю массы тела и сопутствующие жидкий стул и диарею. На сутки 8 всех животных подвергали эвтаназии. Ткани селезенки собирали и взвешивали. Результаты показывают, что соединение 3 в дозах 1,5 мг/кг, 5 мг/кг и 15 мг/кг и эталонное соединение в дозе 5 мг/кг значительно защищали мышей от колита, предотвращая потерю массы тела (фиг. 2, таблица 4) и увеличение селезенки (таблица 4) по сравнению с мышами в группе с носителем.
Следует понимать, что вышеизложенное описывает предпочтительные варианты осуществления настоящего изобретения и что в нем могут быть сделаны модификации без отклонения от объема настоящего изобретения, изложенного в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| БИСУЛЬФАТ ИНГИБИТОРА ЯНУС-КИНАЗЫ (JAK) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2665680C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| АЛКИНИЛ-ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2017 |
|
RU2729069C1 |

Изобретение относится к соединениям, выбранным из:
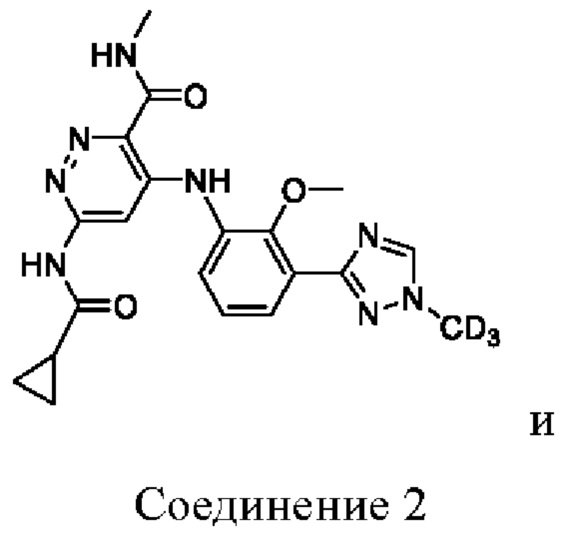
 или их фармацевтически приемлемым солям, которые являются селективными связывающими средствами для JH2 TYK2 и проявляют значительное ингибирующее действие на физиологическую функцию TYK2. Технический результат: получены новые соединения, селективно связывающие средства для JH2 TYK2 и проявляющие значительное ингибирующее действие на физиологическую функцию TYK2, а также обладающие превосходными фармакокинетическими свойствами in vivo. 3 з.п. ф-лы, 2 ил., 4 табл., 14 пр.
или их фармацевтически приемлемым солям, которые являются селективными связывающими средствами для JH2 TYK2 и проявляют значительное ингибирующее действие на физиологическую функцию TYK2. Технический результат: получены новые соединения, селективно связывающие средства для JH2 TYK2 и проявляющие значительное ингибирующее действие на физиологическую функцию TYK2, а также обладающие превосходными фармакокинетическими свойствами in vivo. 3 з.п. ф-лы, 2 ил., 4 табл., 14 пр.
1. Соединение, выбранное из:
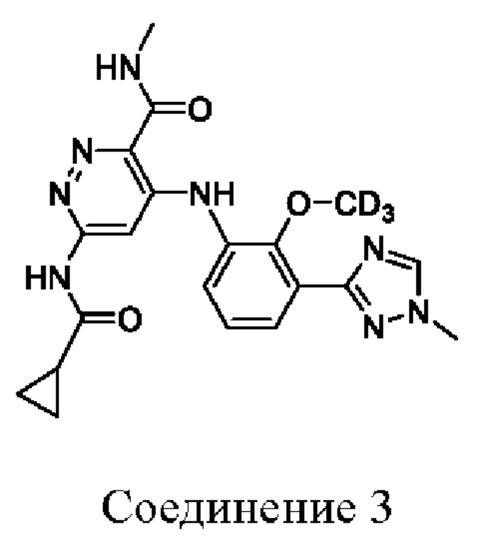

или его фармацевтически приемлемая соль.
2. Соединение по п. 1, которое представляет собой Соединение 3 или его фармацевтически приемлемую соль.
3. Соединение по п. 1, которое представляет собой Соединение 2 или его фармацевтически приемлемую соль.
4. Соединение по п. 1, которое представляет собой Соединение 4 или его фармацевтически приемлемую соль.
| RU 2021118958 A, 28.02.2023 | |||
| WO 2014074661 A1, 15.05.2014 | |||
| Wrobleski, Stephen T | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Journal of Medicinal Chemistry, 62, 18, 2019, стр | |||
| РЕЗЦОВЫЙ ПАТРОН ДЛЯ ИСПОЛЬЗОВАНИЯ В КОРООБДИРНЫХ СТАНКАХ | 1927 |
|
SU8973A1 |
| WO 2015069310 | |||

