Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения арил-2-тетразол-2-илкетона формулы 1а с улучшенной селективностью.
[Формула 1a]
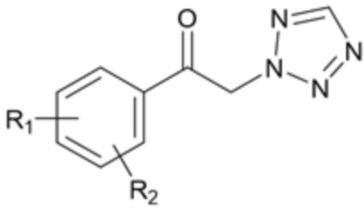
где R1 и R2 являются такими, как определено в данном документе.
Уровень техники
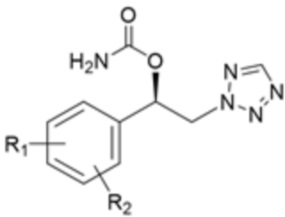
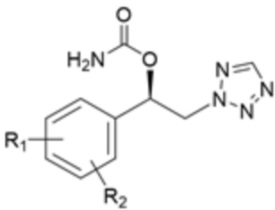
(R)-1-арил-2-тетразолилэтиловый сложный эфир карбаминовой кислоты (далее также называемый «карбаматное соединение») применяется при лечении заболеваний ЦНС, в частности, тревожности, депрессии, судорог, эпилепсии, мигрени, маниакальной депрессии, злоупотребления наркотиками, курения, синдрома дефицита внимания и гиперактивности (СДВГ), ожирения, нарушений сна, нейропатической боли, инсульта, когнитивных нарушений, нейродегенерации, мышечного спазма вследствие инсульта и тому подобного, в связи с его противосудорожным действием.
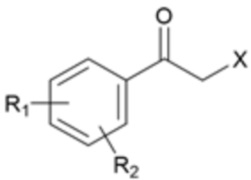
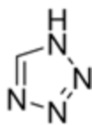
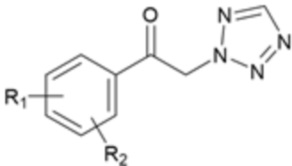
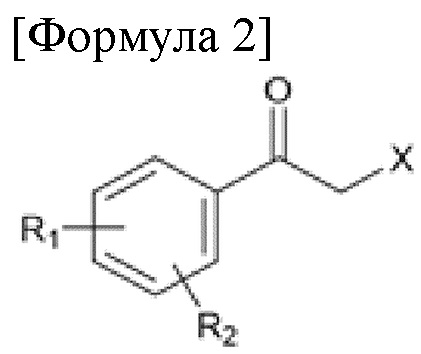
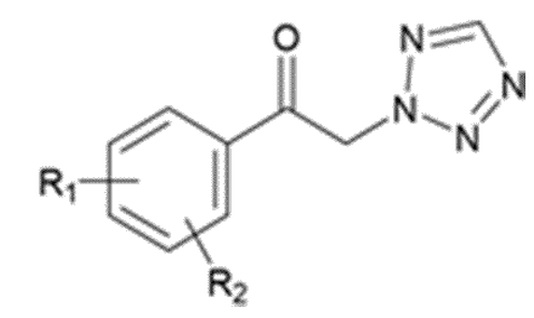
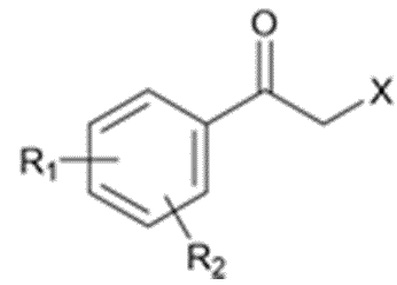
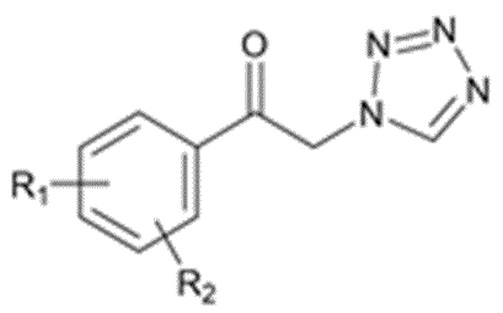
Карбаматное соединение получают из соединения формулы 1а, которое получают в результате реакции замещения соединения формулы 2 и соединения формулы 3, как промежуточный продукт. Обычно основание добавляют к соединению формулы 2, и затем туда добавляют раствор тетразола для осуществления реакции замещения. Однако в данном случае соединение формулы 1b, которое является позиционным изомером, в дополнение к желаемому соединению формулы 1a получают совместно в виде смеси с помощью реакции замещения (WO 2011/046380).
[Формула 2]
[Формула 3]
[Формула 1a]
[Формула 1b]
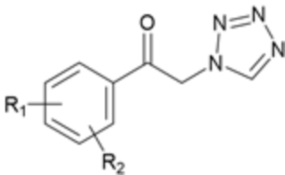
(В данных формулах каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода; и X является уходящей группой)
Кроме того, когда соединение формулы 2 и соединение формулы 3 подвергают реакции замещения, селективность реакции азота №1 соединения формулы 3 лучше, чем селективность реакции азота №2, и таким образом, соединение формулы 1b получают с превосходящей селективностью относительно соединения формулы 1а (Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), (7), 1157-63; 1986). Таким образом, в соответствии с общепринятым способом, соединение формулы 1a, используемое для получения карбаматного соединения, образуется в меньшей степени, чем соединение формулы 1b, и соответственно, выход является низким при получении карбаматного соединения с использованием соединения формулы 2 и соединения формулы 3 в качестве исходных материалов.
В связи с этим, существует альтернативный способ селективного получения только соединения той же формы, что и замещенное азотом № 2, путем синтеза тетразольной кольцевой формы (Chem. Pharm. Bull. 30(9) 3450-3452; 1982). Однако могут возникнуть проблемы, связанные с тем, что его трудно использовать в коммерческих целях, поскольку используется материал на основе диазометана, который является взрывоопасным во время реакции, и в качестве исходного материала используется 2 эквивалента или более диизопропиламида лития.
Таким образом, существует необходимость в разработке способа, в котором арил-2-тетразол-2-илкетон формулы 1а может быть получен из соединения формулы 2 и соединения формулы 3 с лучшей селективностью, чем арил-2-тетразол 1-илкетон формулы 1b, и в результате, который может быть внедрен в промышленное использование, при получении арил-2-тетразол-2-илкетона формулы 1а и карбаматного соединения с высоким выходом.
Сущность изобретения
Техническая задача
Задачей настоящего изобретения является предложить коммерчески доступный способ, способный повысить выход карбаматного соединения за счет более селективного синтеза арил-2-тетразол-2-илкетона, который используется в качестве промежуточного продукта карбаматного соединения, при крупномасштабном производстве.
Решение задачи
Один аспект настоящего изобретения предусматривает способ получения соединения формулы 1a, который включает стадию реакции соединения формулы 2 с солью соединения формулы 3:
[Формула 1a]
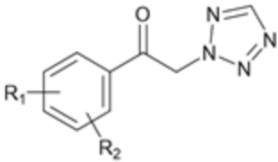
[Формула 2]
[Формула 3]
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода; и X является уходящей группой.
Другой аспект настоящего изобретения предусматривает способ повышения селективности получения соединения формулы 1a путем использования соли соединения формулы 3 при синтезе соединения формулы 1a и соединения формулы 1b из соединения формулы 2 и соединения формулы 3:
[Формула 1a]
[Формула 1b]
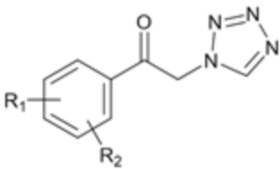
[Формула 2]
[Формула 3]
где R1, R2 и X являются такими, как определено выше.
Другой аспект настоящего изобретения предусматривает способ получения соединения формулы 4, включающий: (1) реакцию соединения формулы 2 с солью соединения формулы 3; (2) выделение соединения формулы 1а из смеси, полученной в результате реакции на стадии (1); и (3) восстановление соединения формулы 1а, выделенного на стадии (2), и карбамирование восстановленного соединения формулы 1а:
[Формула 1a]
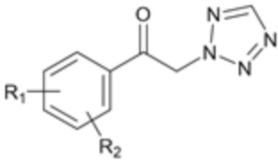
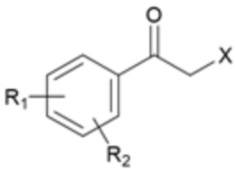
[Формула 2]
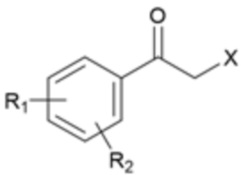
[Формула 3]
[Формула 4]
где R1, R2 и X являются такими, как определено выше.
Другой аспект настоящего изобретения предусматривает способ выделения соединения формулы 1a из смеси, содержащей соединение формулы 1a и соединение 1b, путем термообработки смеси, содержащей соединение химической формулы 1a и соединение формулы 1b, с получением соединения формулы 5 и удалением данного соединения:
[Формула 1a]
[Формула 1b]
[Формула 5]
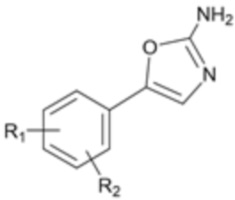
где R1 и R2 являются такими, как определено выше.
Другой аспект настоящего изобретения предусматривает способ получения соединения формулы 1а, включающий: реакцию соединения формулы 2 с солью соединения формулы 3; и очистку продукта реакции соли соединения формулы 3 и соединения формулы 2, где стадия очистки включает процесс кристаллизации или процесс термообработки:
[Формула 1a]
[Формула 2]
[Формула 3]
где R1, R2 и X являются такими, как определено выше.
Полезные эффекты изобретения
Согласно настоящему изобретению выход карбаматных соединений может быть значительно улучшен в результате более селективного синтеза арил-2-тетразол-2-илкетона при крупномасштабном производстве, который используется в качестве промежуточного продукта карбаматного соединения, с помощью простого процесса.
Краткое описание чертежей
На фиг.1 представлены результаты ВЭЖХ соотношения для смеси соединения формулы 1a и соединения формулы 1b в реакционной смеси, полученной в примере 1.
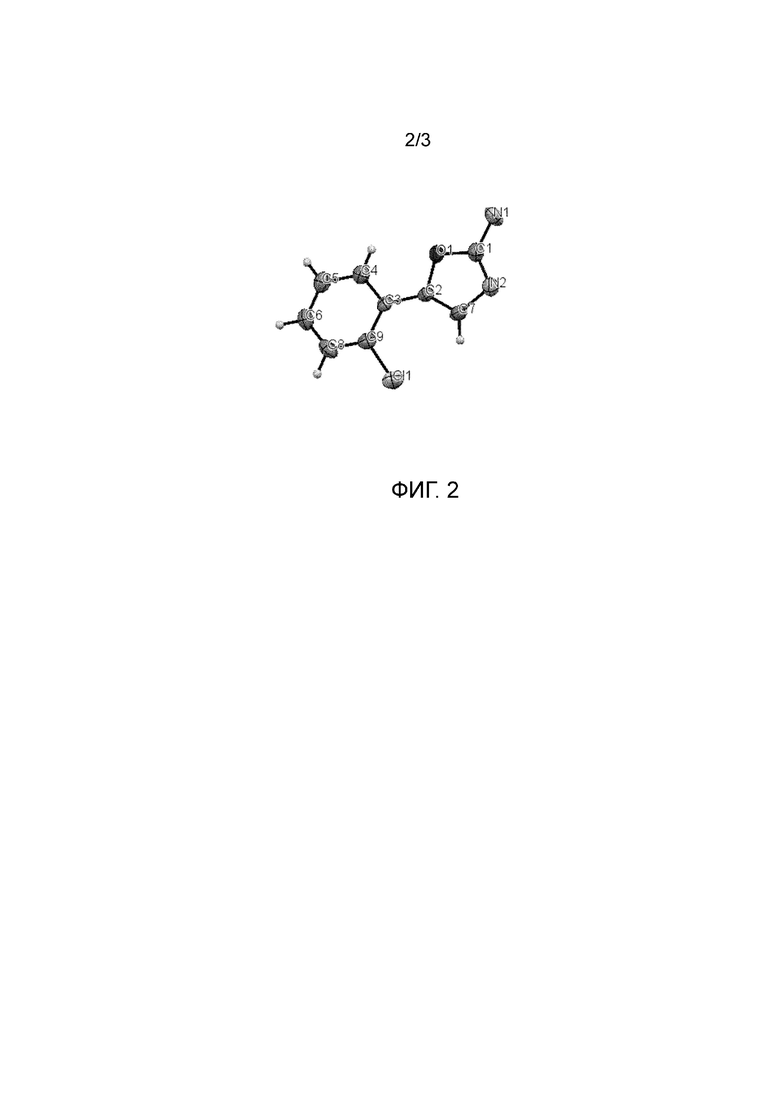
На фиг.2 представлено ORTEP-изображение (Oak Ridge Thermal Ellipsoid Plot) структуры 5-(2-хлорфенил)оксазол-2-амина, полученного в примере 11.
На фиг.3 представлены результаты ВЭЖХ соотношения для соединения формулы 4, преобразованного из соединения формулы 1b и соединения формулы 1a в реакционной смеси, полученной в примере 11.
Варианты осуществления изобретения
Далее настоящее изобретение описывается подробно.
Способ получения соединения формулы 1а в соответствии с одним аспектом настоящего изобретения включает стадию реакции соединения формулы 2 с солью соединения формулы 3:
[Формула 1a]
[Формула 2]
[Формула 3]
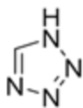
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода, и в частности, выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-4 атомов углерода, алкила, включающего 1-4 атомов углерода, и алкокси, включающего 1-4 атомов углерода; и
X является уходящей группой, и в частности, выбран из галогенидов, таких как хлорид, бромид и тому подобное, и сульфонатов, такие как мезилат, тозилат, 4-нитрофенилсульфонат и тому подобное.
Соль соединения формулы 3 получают путем реакции соединения формулы 3 с основанием, и соль может представлять собой неорганическую соль или органическую соль.
В одном варианте осуществления неорганическая соль соединения формулы 3 может представлять собой соль металла, в частности соль щелочного металла, и даже более конкретно соль лития, соль натрия, соль калия или соль цезия.
В одном варианте осуществления неорганическая соль соединения формулы 3 может быть получена путем реакции соединения формулы 3 с неорганическим основанием. Неорганическое основание может представлять собой гидроксид металла (например, гидроксид лития, гидроксид натрия, гидроксид калия и т.д.) или карбонат металла (например, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия и т.д.), но этим не ограничивается.
В одном варианте осуществления органическая соль соединения формулы 3 может быть получена путем реакции соединения формулы 3 с органическим основанием. Органическое основание может представлять собой аминное соединение (например, триэтиламин, диизопропилэтиламин и т.д.), но этим не ограничивается.
Реакцию между соединением формулы 3 и основанием можно проводить при комнатной температуре, и реакционным растворителем может быть вода, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, этилацетат, изопропилацетат, н-бутилацетат, дихлорметан, хлороформ, 1,4-диоксан, C1-C4 низший спирт (например, метанол, этанол, пропанол, бутанол), по отдельности или в сочетании.
В соответствии с одним вариантом осуществления, соединение формулы 3 может вступать в реакцию с основанием в реакционном растворителе, и затем соль соединения формулы 3 может быть выделена из продукта реакции соединения формулы 3 и основания. В соответствии с другим вариантом осуществления выделенную соль соединения формулы 3 можно ввести в реакцию с соединением формулы 2.
В соответствии с другим вариантом осуществления после реакции соединения формулы 3 с основанием, соединение формулы 2 может быть добавлено к продукту реакции для реакции с солью соединения формулы 3.
Реакцию между солью соединения формулы 3 и соединением формулы 2 можно проводить при комнатной температуре, и реакционным растворителем может быть вода, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, этилацетат, изопропилацетат, н-бутилацетат, дихлорметан, хлороформ, 1,4-диоксан, C1-C4 низший спирт (например, метанол, этанол, пропанол, бутанол), по отдельности или в сочетании.
Продукт реакции соли соединения формулы 3 и соединения формулы 2, полученный, как описано выше, представляет собой смесь, включающую соединение формулы 1a и соединение формулы 1b:
[Формула 1a]
[Формула 1b]
где R1 и R2 являются такими, как определено выше.
Таким образом, для отделения соединения формулы 1а и соединения формулы 1b друг от друга в продукте реакции, способ получения соединения формулы 1a может также включать очистку продукта реакции соли соединения формулы 3 и соединения формулы 2.
В одном варианте осуществления стадия очистки может включать процесс кристаллизации, и в частности, процесс кристаллизации может включать первый процесс кристаллизации и второй процесс кристаллизации.
В одном варианте осуществления процесс кристаллизации может быть процессом, в котором первый растворитель для кристаллизации (например, вода, C1-C4 низший спирт, простой диэтиловый эфир, простой трет-бутилметиловый эфир, простой изопропиловый эфир, пентан, гексан, циклогексан, гептан и их смесь) добавляют к продукту реакции соли соединения формулы 3 и соединения формулы 2, соединение формулы 1b кристаллизуют, и затем отфильтровывают и отделяют («первый процесс кристаллизации»), второй растворитель для кристаллизации (например, ацетон, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, этилацетат, изопропилацетат, н-бутилацетат, дихлорметан, хлороформ, 1,4-диоксан, C1-C4 низший спирт или их смесь) добавляют к оставшемуся фильтрату, и соединение формулы 1a кристаллизуют, отфильтровывают и отделяют («второй процесс кристаллизации»).
В одном варианте осуществления процесс промывки и концентрирования может быть дополнительно проведен перед добавлением первого растворителя для кристаллизации, если это необходимо.
В одном варианте осуществления стадия очистки может включать процесс термообработки. Благодаря процессу термообработки соединение формулы 1b может быть преобразовано в соединение формулы 5.
В одном варианте осуществления процесс термообработки может проводиться при давлении от примерно 1 атмосферы до 50 атмосфер (0,10-5,07 МПа). Давление измеряют для внутреннего давления реагента, и давление может зависеть от изменения температуры в реагенте.
В одном варианте осуществления процесс термообработки может проводиться при температуре реакции от 100°C до 250°C, предпочтительно от 150°C до 220°C.
В одном варианте осуществления процесс термообработки может проводиться в течение от 10 мин до 40 ч, предпочтительно от 20 мин до 24 ч. Однако время реакции может быть соответствующим образом скорректировано в соответствии с температурой реакции.
В одном варианте осуществления процесс термообработки может представлять собой стадию нагревания продукта реакции соли соединения формулы 3 и соединения формулы 2 для селективного преобразования только соединения формулы 1b в соединение формулы 5 (при этом, поскольку соединение формулы 1a является термически стабильным по сравнению с соединением формулы 1b, степень разложения или реакции соединения формулы 1a за счет термообработки является чрезвычайно низкой по сравнению с соединением формулы 1b). Нагревание может осуществляться в присутствии растворителя (например, ацетона, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, этилацетата, изопропилацетата, н-бутилацетата, дихлорметана, хлороформа, 1,4-диоксана, C1-C4 низшего спирта или их смеси).
После процесса термообработки может быть дополнительно проведен процесс промывки водным раствором кислоты. Благодаря процессу промывки соединение формулы 5, которое преобразуется из соединения формулы 1b, удаляют.
В одном варианте осуществления водный раствор кислоты может быть раствором сильной кислоты, такой как соляная кислота или серная кислота, или раствором слабой кислоты, такой как уксусная кислота или лимонная кислота, но этим не ограничивается.
[Формула 5]
где R1 и R2 являются такими, как определено выше.
В соответствии с настоящим изобретением, когда соединение формулы 3 получают в виде соли и затем подвергают реакции с соединением формулы 2, соединение формулы 1а может быть получено с повышенной селективностью по сравнению с общепринятым способом, в котором соединение формулы 2, соединение формулы 3 и основание реагируют одновременно. В частности, отношение соединения формулы 1a к соединению формулы 1b составляло 4:6 в общепринятом способе, но отношение может составлять примерно 6:4 в соответствии со способом настоящего изобретения.
Поскольку селективность соединения формулы 1a повышается в соответствии со способом настоящего изобретения по сравнению с общепринятым способом, как описано выше, выходы соединения формулы 1а и получаемого из него карбаматного соединения также могут значительно повыситься. В частности, их выходы повышаются по меньшей мере примерно на 70%, и в частности, примерно от 70% до примерно 100%.
Таким образом, другой аспект настоящего изобретения относится к способу повышения селективности соединения формулы 1a путем использования соли соединения формулы 3 при синтезе соединения формулы 1a и соединения формулы 1b из соединения формулы 2 и соединения формулы 3.
Кроме того, еще один аспект настоящего изобретения относится к получению соединения формулы 1а, включающему: реакцию соединения формулы 2 с солью соединения формулы 3; и очистку продукта реакции соли соединения формулы 3 и соединения формулы 2, при этом стадия очистки включает процесс кристаллизации или процесс термообработки:
[Формула 1a]
[Формула 2]
[Формула 3]
где R1, R2 и X являются такими, как определено выше.
Кроме того, еще один аспект настоящего изобретения относится к способу получения соединения формулы 4, включающему: (1) реакцию соединения формулы 2 с солью соединения формулы 3; (2) выделение соединения формулы 1а из смеси, полученной в результате реакции на стадии (1); и (3) восстановление соединения формулы 1а, выделенного на стадии (2), и карбамирование восстановленного соединения формулы 1а:
[Формула 4]
где R1 и R2 являются такими, как определено выше.
Реакция соединения формулы 2 и соединения формулы 3 такая же, как описано выше.
Выделение соединения формулы 1а может включать стадию очистки, описанную выше.
На стадии восстановления и карбамирования процесс восстановления может осуществляться с использованием фермента оксидоредуктазы, который суспендирован в реакционной смеси или иммобилизован общепринятым способом. Фермент может применяться в полностью очищенном состоянии, в частично очищенном состоянии или в клетках микроорганизмов, где он экспрессирован. Сами клетки могут быть в нативном состоянии, в состоянии с нарушенной проницаемостью мембраны или в лизированном состоянии. Специалистам в данной области будет понятно, что, когда способ настоящего изобретения осуществляется с использованием фермента в клеточном состоянии, это позволяет значительно снизить затраты, что является предпочтительным. Наиболее предпочтительно, фермент экспрессирован в E. coli и используется в виде суспензии нативных клеток.
Ферментативное восстановление соединения формулы 1а может быть осуществлено в реакционной смеси, содержащей соединение формулы 1а, оксидоредуктазу, NADH или NADPH в качестве кофактора, косубстрат и подходящий буфер. Оксидоредуктазу можно использовать для восстановления соединения формулы 1а с высокой конверсией и энантиомерной селективностью с использованием полипептидов, обладающих оксидоредуктазной активностью. Энантиомерный избыток спирта R-конфигурации, образующегося при энантиомерном селективном ферментативном восстановлении, составляет, по меньшей мере, примерно 89%, предпочтительно, по меньшей мере, примерно 95% и наиболее предпочтительно, по меньшей мере, примерно 99%.
На стадии восстановления и карбамирования способ введения карбамоильной группы может, например, включать введение карбамоильной группы путем использования смеси неорганического цианата-органической кислоты, изоцианата-воды или карбонильного соединения-аммиака.
При карбамировании с помощью неорганического цианата-органической кислоты, спиртовое соединение (R)-конфигурации, преобразованное из соединения формулы 1a способом восстановления, может быть растворено в органическом растворителе, например, в простом диэтиловом эфире, тетрагидрофуране, 1,4-диоксане, ацетонитриле, дихлорметане, хлороформе или их смеси, и затем туда могут быть добавлены неорганический цианат, такой как цианат натрия, и органическая кислота, такая как метансульфоновая кислота или уксусная кислота, которые составляют 1-4 эквивалентов, и реакцию можно проводить при температуре реакции от примерно -10°С до примерно 70°С.
В способе использования изоцианата-воды, от 1 до 4 эквивалентов изоцианата, например, хлорсульфонилизоцианата, трихлорацетилизоцианата, триметилсилилизоцианата или тому подобного, может быть добавлено к раствору спиртового соединения, имеющего (R)-конфигурацию, полученного восстановлением соединения формулы 1a в органическом растворителе, например, в простом диэтиловом эфире, тетрагидрофуране, 1,4-диоксане, ацетонитриле, дихлорметане, хлороформе или их смеси, и реагируют при температуре реакции от примерно -50°C до 40°C, и затем туда могут быть последовательно добавлены 1-20 эквивалентов воды без какой-либо очистки для осуществления гидролиза.
В способе использования карбонильного соединения-аммиака, от 1 до 4 эквивалентов карбонильного соединения, например, 1,1’-карбонилдиимидазола, карбамоилхлорида, N, N’-дисукцинимидилкарбоната, фосгена, трифосгена, хлороформиата или тому подобного, добавляли к раствору спиртового соединения в (R)-конфигурации, полученного восстановлением соединения формулы 1a в органическом растворителе, например, в простом диэтиловом эфире, тетрагидрофуране, 1,4-диоксане, ацетонитриле, дихлорметане, хлороформе или их смеси, и затем последовательно добавляли от 1 до 10 эквивалентов аммиака без очистки при температуре реакции примерно от -10°C до 70°C.
В дополнение к этому, еще один аспект настоящего изобретения относится к способу выделения соединения формулы 1a из смеси, содержащей соединение формулы 1a и соединение 1b, путем термообработки смеси, содержащей соединение химической формулы 1a и соединение формулы 1b, с получением соединения формулы 5 и удалением данного соединения.
Процесс термообработки является таким же, как описано выше.
В одном варианте осуществления удаление соединения формулы 5 может быть осуществлено путем промывки водным раствором кислоты. Водный раствор кислоты может быть раствором сильной кислоты, такой как соляная кислота или серная кислота, или раствором слабой кислоты, такой как уксусная кислота или лимонная кислота, но этим не ограничивается.
Далее настоящее изобретение будет подробно описано со ссылкой на следующие примеры. Однако они приведены только для лучшего понимания настоящего изобретения, и объем настоящего изобретения ими не ограничивается.
Примеры
Пример 1
Тетразол (0,165 г) растворяли в метаноле (9 мл) и добавляли туда карбонат калия (0,538 г) при комнатной температуре. Продукт реакции перемешивали при комнатной температуре в течение примерно 15 мин. После подтверждения того, что углекислый газ больше не образуется, добавляли н-бутилацетат (9 мл), удаляли метанол перегонкой при пониженном давлении и добавляли н-бутилацетат. После добавления 2-бром-2'-хлорацетофенона (0,50 г) к реакционному раствору, продукт реакции перемешивали при 50°C в течение 12 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ подтверждали, что селективность соединения формулы 1b, соответствующего формуле 1b, составляла 40%, и селективность соединения формулы 1a, соответствующего формуле 1a, составляла 60%. Условия ВЭЖХ были следующими и использовались в примерах ниже:
Колонка была представлена Phenomenex Luna C18, 5 мкм, 4,6×250 мм, и температура колонки составляла 35°C. Подвижная фаза представляла собой смесь ацетонитрил:вода в соотношении 6:4 и содержала 0,1% трифторуксусной кислоты, и ее пропускали в течение 10 мин при 2,0 мл/мин в изократических условиях. Длину волны фиксировали на уровне 245 нм, и время достижения пика составляло 2,36 мин для соединения формулы 1а, 1,94 мин для соединения формулы 1b и 4,01 мин для 2-бром-2'-хлорацетофенона.
Результаты ВЭЖХ представлены на фиг.1.
Пример 2
Тетразол (0,165 г) растворяли в н-бутилацетате (9 мл) и карбонат калия (0,538 г) добавляли при комнатной температуре. Продукт реакции перемешивали при комнатной температуре в течение примерно 24 ч. После подтверждения того, что углекислый газ больше не образуется, 2-бром-2'-хлорацетофенон (0,50 г) добавляли к реакционному раствору и продукт реакции перемешивали при 50°C в течение 12 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 60%.
Пример 3
Тетразол (1,40 г) и карбонат калия (1,38 г) добавляли в воду (10 мл) и перемешивали при 100°С в течение примерно 1 ч при нагревании с обратным холодильником. Температуру понижали до комнатной, воду отгоняли и продукт реакции разбавляли в 20 мл этанола. Смесь перемешивали при 80°С в течение 2 ч, и затем температуру доводили до комнатной температуры. Примерно 10 мл этанола удаляли перегонкой при пониженном давлении и перемешивали в течение 2 ч, затем фильтровали и сушили в атмосфере азота с получением калиевой соли тетразола (1,70 г). Полученную калиевую соль тетразола (0,153 г) добавляли к н-бутилацетату (1,8 мл), добавляли туда 2-бром-2′-хлорацетофенон (0,30 г), и реакционную смесь перемешивали при 50°С в течение 12 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 62%.
Пример 4
Калиевую соль тетразола (0,509 г), полученную в примере 3, добавляли к 2-метилтетрагидрофурану (6 мл), к ним добавляли 2-бром-2'-хлорацетофенон (1,00 г), и реакционную смесь перемешивали при 50°C в течение 22 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 57%.
Пример 5
Тетразол (1,40 г) и карбонат цезия (3,26 г) добавляли в воду (10 мл) и перемешивали при 100°С в течение примерно 1 ч при нагревании с обратным холодильником. Температуру понижали до комнатной, воду отгоняли и реакционную смесь высушивали под вакуумом с получением цезиевой соли тетразола (1,685 г). Полученную цезиевую соль тетразола (0,285 г) добавляли к н-бутилацетату (1,8 мл), добавляли туда 2-бром-2′-хлорацетофенон (0,30 г), и реакционную смесь перемешивали при 50°С в течение 12 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 56%.
Пример 6
Тетразол (1,40 г) и карбонат натрия (0,68 г) добавляли в воду (10 мл) и перемешивали при 100°С в течение примерно 1 ч при нагревании с обратным холодильником. Температуру понижали до комнатной, воду отгоняли и реакционную смесь высушивали под вакуумом с получением натриевой соли тетразола (1,53 г). Полученную натриевую соль тетразола (0,156 г) добавляли к н-бутилацетату (1,8 мл), добавляли туда 2-бром-2′-хлорацетофенон (0,30 г), и реакционную смесь перемешивали при 50°С в течение 12 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 63%.
Сравнительный пример 1
2-Бром-2′-хлорацетофенон (86,0 г), карбонат калия (30,5 г) и 35% раствор тетразола в ДМФА (81,0 г) добавляли в этилацетат (245 мл) и перемешивали при 55°С в течение 2 ч. После того, как температура была доведена до комнатной температуры, с помощью ВЭЖХ было подтверждено, что селективность соединения формулы 1a составляла 42%.
Пример 7
2-Бром-2'-хлорацетофенон (13,0 г) подвергали взаимодействию с калиевой солью тетразола (6,62 г) и изопропилацетатом (117 мл), и затем промывали разбавленной соляной кислотой и насыщенным солевым раствором для удаления вторично образованного бромида калия. Отделенный слой изопропилацетата полностью концентрировали, замещали простым трет-бутилметиловым эфиром, перемешивали при нагревании с обратным холодильником в течение примерно 1 ч, и затем медленно охлаждали до 15°C. Когда соединение формулы 1b было в достаточной степени осаждено, продукт реакции отфильтровывали для получения соединения формулы 1b (4,1 г, включая соединение формулы 1a) в виде твердого вещества. Для сведения, фильтрат анализировали с помощью ВЭЖХ, и было подтверждено, что фильтрат состоял из 6,0 г соединения формулы 1a и 0,74 г соединения формулы 1b.
Соединения формулы 1b: 1H ЯМР (CDCl3) 8,86 (с, 1H), 7,77 (д, 1H), 7,40-7,62 (м, 3H), 5,97 (с, 2H)
Пример 8
Раствор смеси 6,0 г соединения формулы 1a и 0,74 г соединения формулы 1b, полученный в качестве фильтрата в примере 7, концентрировали при пониженном давлении для максимально возможного удаления растворителя, и при замещении изопропиловым спиртом, соединение формулы 1a растворяли в изопропиловом спирте (45 мл), перемешивали в течение примерно 1 ч при 60°C и медленно охлаждали до 10°C. Соединение формулы 1а отфильтровывали после достаточного осаждения, дважды промывали охлажденным изопропиловым спиртом (13 мл) и один раз н-гептаном (26 мл), чтобы получить соединение формулы 1а (5,55 г) в виде твердого вещества с чистотой по ВЭЖХ 94,2%.
Соединения формулы 1a: 1H ЯМР (CDCl3) 8,62 (с, 1H), 7,72 (д, 1H), 7, 35-7, 55 (м, 3H), 6,17 (с, 2H)
Пример 9
После реакции 2-бром-2'-хлорацетофенона (17,1 г) с калиевой солью тетразола (8,71 г) и изопропилацетатом (130 мл) добавляли гептан (165 мл) для получения вторично образованного бромида калия и соединения формулы 1b в виде твердого вещества одновременно. После перемешивания при 60°C в течение 1 ч реакционную смесь медленно охлаждали до примерно 8,5°C. Когда бромид калия и соединение формулы 1b были в достаточной степени осаждены, их отфильтровывали для получения бромида калия и соединения формулы 1b (всего 14 г) в виде твердых веществ. Для сведения, фильтрат анализировали с помощью ВЭЖХ, и было подтверждено, что фильтрат состоял из 9,5 г соединения формулы 1a и 1,4 г соединения формулы 1b.
Пример 10
Раствор смеси 9,5 г соединения формулы 1a и 1,4 г соединения формулы 1b, полученный в качестве фильтрата в примере 9, концентрировали при пониженном давлении для максимально возможного удаления растворителя, и замещали изопропиловым спиртом. Соединение формулы 1a перемешивали в течение примерно 1 ч при 60°C для растворения в изопропиловом спирте (96 мл), и затем медленно охлаждали до 10°C. Когда соединение формулы 1a осаждалось в достаточной степени, твердое вещество отфильтровывали и дважды промывали холодным изопропиловым спиртом (17 мл) и один раз гептаном (34 мл). Соединение формулы 1а (7,6 г) получали в виде твердого вещества.
Пример 11
Смесь, в которой 2'-хлорфенил-2-тетразол-2-илкетон (0,3 г) и 2'-хлорфенил-2-тетразол-1-илкетон (0,2 г) были растворены в изопропилацетате (3 мл), нагревали при 150°C в течение 24 ч, температуру доводили до комнатной, и степень пиролиза подтверждали с помощью ВЭЖХ. С помощью ВЭЖХ было подтверждено, что 99,9% 2′-хлорфенил-2-тетразол-1-илкетона разлагается и превращается в 5-(2-хлорфенил)оксазол-2-амин, и 88,2% 2'-хлорфенил-2-тетразол-2-илкетона остается без разложения. Полученный 5-(2-хлорфенил)оксазол-2-амин промывали 1 н. HCl для удаления данного соединения.
Колонка была представлена Phenomenex Luna C18, 5 мкм, 4,6×250 мм, и температура колонки составляла 35°C. Подвижная фаза представляла собой смесь ацетонитрил:вода в соотношении 6:4 и содержала 0,1% трифторуксусной кислоты, и ее пропускали в течение 10 мин при 2,0 мл/мин в изократических условиях. Длину волны фиксировали на уровне 245 нм, и время достижения пика составляло 2,36 мин для соединения формулы 1а, 1,94 мин для соединения формулы 1b и 1,23 мин для 5-(2-хлорфенил)оксазол-2-амина формулы 5.
5-(2-Хлорфенил)оксазол-2-амин: ЖХМС [M+H]=195,0 г/моль
Структура 5-(2-хлорфенил)оксазол-2-амина была подтверждена ORTEP-изображением и представлена на фиг.2.
Результаты ВЭЖХ представлены на фиг.3.
Пример 12
Смесь, в которой 2′-хлорфенил-2-тетразол-2-илкетон (0,3 г) и 2′-хлорфенил-2-тетразол-1-илкетон (0,2 г) были растворены в изопропанoле, (3 мл), пропускали в трубчатый реактор непрерывного действия при 210°С в течение 20 мин, и затем температуру доводили до комнатной, и степень пиролиза подтверждали с помощью ВЭЖХ. Было подтверждено, что 99,9% 2′-хлорфенил-2-тетразол-1-илкетона разлагается и превращается в 5-(2-хлорфенил)оксазол-2-амин, и 90% 2'-хлорфенил-2-тетразол-2-илкетона остается без разложения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (R)-1-АРИЛ-2-ТЕТРАЗОЛИЛЭТИЛОВОГО ЭФИРА КАРБАМИНОВОЙ КИСЛОТЫ | 2010 |
|
RU2539983C2 |
| СПОСОБ ПОЛУЧЕНИЯ (R)-1-АРИЛ-2-ТЕТРАЗОЛИЛЭТИЛОВОГО ЭФИРА КАРБАМИНОВОЙ КИСЛОТЫ | 2009 |
|
RU2508290C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-(6-ЗАМЕЩЕННОЙ-1,3-ДИОКСАН-4-ИЛ)УКСУСНОЙ КИСЛОТЫ | 2001 |
|
RU2266903C2 |
| КОМБИНАЦИИ АЛЬФА-2-ДЕЛЬТА-ЛИГАНДА С СЕЛЕКТИВНЫМ ИНГИБИТОРОМ ЦИКЛООКСИГЕНАЗЫ-2 | 2003 |
|
RU2286151C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ВЫДЕЛЕНИЯ 2-АЦИЛАМИНО-3-ДИФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2520215C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2764667C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2602083C2 |
| 2-АМИНОБЕНЗОТИАЗОЛЫ В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ РЕЦЕПТОРОВ CB | 2004 |
|
RU2344132C2 |
| НОВЫЕ АНАЛОГИ 2`, 5`-ОЛИГОАДЕНИЛАТА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2311422C2 |
| КАРБОКСИЛ- ИЛИ ГИДРОКСИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА | 2008 |
|
RU2493153C2 |


Настоящее изобретение относится к химико-фармацевтической промышленности, конкретно к способу синтеза арил-2-тетразол-2-илкетона формулы 1а, применяемому в качестве промежуточного продукта для крупномасштабного производства (R)-1-арил-2-тетразолилэтилового сложного эфира карбаминовой кислоты, используемого при лечении заболеваний ЦНС. Способ получения соединения формулы 1а включает реакцию соединения формулы 2 с солью соединения формулы 3, где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода, и X является уходящей группой. Техническим результатом изобретения является предоставление способа с повышенным выходом карбаматного соединения за счет более селективного синтеза арил-2-тетразол-2-илкетона. 3 н. и 21 з.п. ф-лы, 3 ил., 12 пр.


1. Способ получения соединения формулы 1а
[Формула 1a]
 ,
,
включающий реакцию соединения формулы 2 с солью соединения формулы 3:
[Формула 2]
 ,
,
[Формула 3]
 ,
,
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода; и
X является уходящей группой.
2. Способ получения соединения формулы 1а
[Формула 1a]
,
включающий реакцию соединения формулы 2 с солью соединения формулы 3; и
очистку продукта реакции соли соединения формулы 3 и соединения формулы 2, где стадия очистки включает процесс кристаллизации или процесс термообработки;
[Формула 2]
,
[Формула 3]
,
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода; и
X является уходящей группой.
3. Способ по п.1 или 2, в котором соль соединения формулы 3 получают путем реакции соединения формулы 3 с основанием.
4. Способ по п.3, в котором соль представляет собой одно или более, выбранное из группы, состоящей из соли лития, соли натрия, соли калия или соли цезия.
5. Способ по п.3, в котором основание представляет собой неорганическое основание или органическое основание.
6. Способ по п.5, в котором неорганическое основание представляет собой гидроксид металла или карбонат металла, и органическое основание представляет собой аминное соединение.
7. Способ по п.6, в котором гидроксид металла выбран из гидроксида лития, гидроксида натрия и гидроксида калия; карбонат металла выбран из карбоната лития, карбоната натрия, карбоната калия и карбоната цезия; и аминное соединение выбрано из триэтиламина и диизопропилэтиламина.
8. Способ по п.3, в котором соединение формулы 3 вводят в реакцию с основанием в реакционном растворителе, и затем соль соединения формулы 3 выделяют из продукта реакции соединения формулы 3 и основания.
9. Способ по п.8, в котором выделенную соль соединения формулы 3 вводят в реакцию с соединением формулы 2.
10. Способ по п.3, в котором после реакции соединения формулы 3 с основанием соединение формулы 2 добавляют к продукту реакции для реакции с солью соединения формулы 3.
11. Способ по п.2, в котором процесс кристаллизации включает первый процесс кристаллизации и второй процесс кристаллизации.
12. Способ по п.11, в котором растворитель первого процесса кристаллизации выбран из группы, состоящей из воды, C1-C4 низшего спирта, простого диэтилового эфира, простого трет-бутилметилового эфира, простого изопропилового эфира, пентана, гексана, циклогексана, гептана и их смеси.
13. Способ по п.11, в котором растворитель второго процесса кристаллизации выбран из группы, состоящей из ацетона, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, этилацетата, изопропилацетата, н-бутилацетата, дихлорметана, хлороформа, 1,4-диоксана, C1-C4 низшего спирта или их смеси.
14. Способ по п.1 или 2, в котором уходящая группа выбрана из группы, состоящей из хлорида, бромида, мезилата, тозилата и 4-нитрофенилсульфоната.
15. Способ выделения соединения формулы 1a из смеси, содержащей соединение формулы 1a и соединение формулы 1b, включающий процесс термообработки смеси, содержащей соединение формулы 1a и соединение формулы 1b:
[Формула 1a]
,
[Формула 1b]
 ,
,
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, галогена, перфторалкила, включающего 1-8 атомов углерода, алкила, включающего 1-8 атомов углерода, тиоалкокси, включающего 1-8 атомов углерода, и алкокси, включающего 1-8 атомов углерода.
16. Способ по п.2 или 15, в котором процесс термообработки представляет собой стадию преобразования соединения формулы 1b в соединение формулы 5
[Формула 5]
 ,
,
где R1 и R2 являются такими, как определено в п.15.
17. Способ по п.2 или 15, в котором процесс термообработки проводят в присутствии растворителя.
18. Способ по п.17, в котором растворитель представляет собой ацетон, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, этилацетат, изопропилацетат, н-бутилацетат, дихлорметан, хлороформ, 1,4-диоксан, C1-C4 низший спирт или их смесь.
19. Способ по п.2 или 15, в котором процесс термообработки проводят при давлении от 1 до 50 атм (0,10-5,07 МПа).
20. Способ по п.2 или 15, в котором процесс термообработки проводят при температуре реакции от 100 до 250°C.
21. Способ по п.2 или 15, в котором процесс термообработки проводят от 10 мин до 40 ч.
22. Способ по п.2 или 15, который дополнительно включает стадию промывки продукта процесса термообработки водным раствором кислоты.
23. Способ по п.22, в котором водный раствор кислоты представляет собой водный раствор соляной кислоты, серной кислоты, уксусной кислоты или лимонной кислоты.
24. Способ по п.22, в котором соединение формулы 5 удаляют на стадии промывки водным раствором кислоты,
[Формула 5]
,
где R1 и R2 являются такими, как определено в п.15.
| A.S | |||
| KRYLOV et al., Russian Journal of Organic Chemistry, 2014, pp.892-894 | |||
| СПОСОБ ПОЛУЧЕНИЯ (R)-1-АРИЛ-2-ТЕТРАЗОЛИЛЭТИЛОВОГО ЭФИРА КАРБАМИНОВОЙ КИСЛОТЫ | 2009 |
|
RU2508290C2 |
| S.A.F | |||
| ROSTOM et al., Bioorganic & Medicinal Chemistry, 2009, pp | |||
| Безударный, винтовой прибор для разгонки зазоров железнодорожных рельсов | 1925 |
|
SU2410A1 |
| JP 58140083 A, 19.08.1983. | |||

