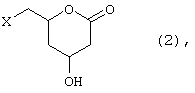
Изобретение относится к способу получения производного 2-(6-замещенной-1,3-диоксан-4-ил) уксусной кислоты формулы 1
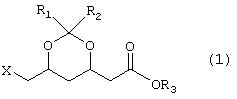
где Х обозначает уходящую группу и R1, R2 и R3, каждая независимо обозначает алкильную группу с от 1 до 3 атомами углерода, используя в качестве исходного соединение формулы 2
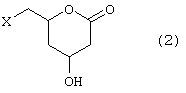
где Х является таким же, как и описано выше, используют подходящий агент ацетализации, в присутствии кислотного катализатора.
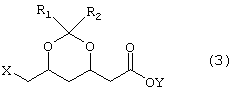
Изобретение также относится к новым соединениям формулы 1, как и солям и кислотам формулы 3, которые из них могут быть получены
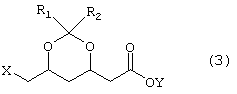
где R1 и R2 имеют описанные выше значения и где Y обозначает щелочной (щелочноземельный) металл или замещенную или незамещенную аммониевую группу или обозначает водород.
Найдено, что производное 2-(6-замещенной-1,3-диоксан-4-ил)уксусной кислоты может быть селективно и с высоким выходом получено из соответствующего соединения формулы (2), оказалось возможным получить эти продукты, которые относительно мало стабильны, в мягких условиях. Это тем более интересно, поскольку это обеспечивает простой путь через соответствующую соль, соответствующий трет.-бутиловый сложный эфир и 2-гидроксиметил-замещенное соединение в качестве промежуточных соединений, получение ингибиторов HMG-CoA редуктазы. При желании конверсия проходит (в зависимости от выбранных условий реакции) через промежуточную соль или сложный эфир с раскрытием кольца в соединении согласно формуле (2).
Дополнительным преимуществом способа по изобретению является то, что исходные соединения формулы (2) и продукты формулы 3 являются кристаллическими соединениями, что выгодно при получении продуктов с (химической или стереохимической) высокой чистотой. Это важно в особенности с точки зрения предполагаемого фармацевтического применения. Для предполагаемого применения в особенности важно производное (4R,5S)-2-(6-замещенной-1,3-диоксан-4-ил) уксусной кислоты. Оно может быть получено из соответствующей 6-замещенной-2,4,6-тридеокси-D-эритрогексозы. Следовательно, изобретение также относится к исходным соединениям формулы 1, в особенности в которых Х=Cl, и к частицам таких соединений. В особенности более чем 90 вес.% этих частиц имеет отношение длина/диаметр от 1:1,5 до 1:6, предпочтительно от 1:2 до 1:4,4 и длина частиц от 0,05 до 2 мм, в особенности от 0,1 до 1 мм.
Изобретение также относится к таким частицам. Соединение формулы II дает прозрачные кристаллические частицы с четкой точкой плавления 73-74°С. Продукты формулы 3, полученные из производного (4R,6S)-2-(6-замещенной-1,3-диоксан-4-ил)уксусной кислоты формулы 1, могут быть получены согласно изобретению с энантиомерным избытком (е.е.) более чем 95%, в особенности более 99,5% и с диастереомерным избытком (d.e.) более чем 90%, в особенности более чем 99,5%.
Примерами подходящих уходящих групп X, которые могут применяться в способе согласно изобретению, являются галогены, в особенности Cl, Br или I; тозилатные группы; мезилатные группы; ацилоксигруппы, в особенности ацетокси- и бензоилоксигруппы; арилокси-, в особенности бензилокси- или нитро-замещенная бензолсульфонильная группа. С практической точки зрения предпочтительно в качестве уходящей группы выбирают Cl.
Группы R1, R2 и R3, каждая, независимо обозначает алкильную группу с от 1 до 3 атомами углерода, предпочтительно метил или этил. На практике наиболее предпочтительно R1=R2=R3=метилу.
Примерами подходящих агентов ацетализации, которые могут применяться в способе согласно изобретению, являются диалкоксипропановые соединения с алкоксигруппами, каждая из которых предпочтительно имеет от 1 до 3 атомов углерода, например, 2,2-диметоксипропан или 2,2-диэтоксипропан; алкоксипропен, с алкоксигруппой, предпочтительно имеющей от 1 до 3 атомов углерода, например, 2-метоксипропен или 2-этоксипропен. Наиболее предпочтителен 2,2-диметоксипропан. Он может быть, при желании, образован in situ из ацетона и метанола, предпочтительно с удалением воды.
В качестве используемого кислотного катализатора могут использоваться кислотные катализаторы, известные для реакций ацетализации, предпочтительно ненуклеофильные сильные кислоты, например, сульфоновые кислоты, в особенности, п-толуолсульфоновая кислота, метансульфоновая кислота, камфарная сульфоновая кислота; неорганические кислоты с ненуклеофильным анионом, например, серная кислота, фосфорная кислота: кислотные ионообменники, например, DOWEX; или твердые кислоты, например, так называемые, гетерополикислоты.
Ацетализация может проводиться без использования отдельного растворителя, если желательно реакция также может проводиться в органическом растворителе. Примерами пригодных органических растворителей являются кетоны, в особенности ацетон, углеводороды, в особенности ароматические углеводороды, например, толуол, хлорированные углеводороды, например, метиленхлорид.
Температура, при которой проводится реакция ацетализации, предпочтительно составляет от -20°С до 60°С, в особенности от 0°С до 30°С. Реакция ацетализации предпочтительно проводится в инертной атмосфере.
Молярное отношение агента ацетализации к исходному соединению формулы (2) предпочтительно составляет от 1:1 до 20:1, в особенности от 3:1 до 5:1. При использовании органического растворителя молярное отношение предпочтительно составляет от 1:1 до 2:1.
Молярное отношение кислотного катализатора к исходному соединению формулы (2) предпочтительно составляет от 1:1 до 0,001:1, в особенности от 0,01:1 до 0,05:1.
Полученное производное 2-(6-замещенной-1,3-диоксан-4-ил) уксусной кислоты может быть впоследствии гидролизовано в присутствии основания и воды для получения соответствующей соли формулы 3
где Y обозначает щелочной метал, щелочноземельный металл или замещенную или незамещенную аммониевую группу, предпочтительно Na, Ca или тетраалкиламмониевое соединение. При желании с последующим гидролизом до уксусной кислоты формулы 3, в которой Y=H.
Гидролиз соединения формулы (3) предпочтительно проводят, по крайней мере, 1 эквивалентом основания, в особенности с от 1 до 1,5 эквивалентами основания, по отношению к соединению формулы (3). В принципе может использоваться значительный избыток, но на практике это обычно не дает никаких преимуществ.
Реакцию предпочтительно проводят при температуре от -20°С до 60°С, в особенности от 0°С до 30°С.
Гидролиз, например, может проводиться в воде, органическом растворителе, например, в спирте, в особенности метаноле или этаноле, ароматическом углеводороде, например толуоле или кетоне, в особенности ацетоне или метилизобутилкетоне (MIBK) или в смеси органического растворителя и воды, при желании, катализируемый катализатором фазового переноса или добавлением сорастворителя.
Гидролиз также может проводиться ферментативно, посредством, при желании, селективного гидролиза заданного диастереомера.
Примерами ферментов, которые могут использоваться в способе согласно изобретению, являются ферменты с липазной или экстеразной активностью, например ферменты из Pseudomonas, в особенности Pseudomonas fluorescens, Pseudomonas fragi; Burkholderia, например, Burkholderia cepacia; Chrombacterium, в особенности Chrombacterium viscosum; Bacillus, в особенности Bacillus thermocatenulatus, Bacillus licheniformis; Alcaligenes, в особенности Alcaligenes faecalis; Aspergillus, в особенности Aspergillus niger, Candida, в особенности Candida antarctica, Candida rugosa, Candida lipolytica, Candida cylindracea; Geotrlchum, в особенности Geotrlchum candidum; Humicola, в особенности Humicola lanuginosa; Penicillum в особенности Penicillum cyclopium, Penicillum roquefortii, Penicillum camembertii; Rhizomucor в особенности Rhizomucor javanicus, Rhizomucor miehei; Mucor в особенности Mucor javanicus; Rhizopus, в особенности Rhizopus oryzae, Rhizopus arhizus, Rhizopus delemar, Rhizopus niveus, Rhizopus japonicus, Rhizopus javanicus; липаза свиной поджелудочной железы, липаза проростка пшеницы, липаза поджелудочной железы коровы, эстераза печени поросенка. Предпочтительно используют фермент из Pseudomonas cepacia, Pseudomonas sp., Burkholderia cepacia, свиной поджелудочной железы, Rhizomucor miehei, Humicola lanuglnosam Candida rugorsa или Candida an tractica или субстилина. Если используют энантиоселективный фермент, при гидролизе реализуется дополнительное энантиомерное обогащение. Такие ферменты могут быть получены известными способами. Многие ферменты производят в промышленном масштабе, и они являются коммерчески доступными.
Эти получаемые соли (кислоты) являются новыми. Изобретение, таким образом, относится к этим продуктам формулы 3
где Х обозначает галоген, в особенности, Cl, Br или I, тозилатную или мезилатную группу, ацилоксигруппу с от 3 до 10 атомов углерода или нитрозамещенную бензолсульфоновую группу и Y обозначает Н, щелочной (щелочноземельный) металл или замещенную или незамещенную аммониевую группу.
Полученная соль формулы 3 может быть затем превращена в соответствующий трет.-бутиловый сложный эфир (формула 1а с R3=трет.-бутил) известным способом.
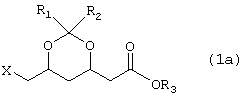
В способе согласно изобретению соединение формулы (3) может быть, например, этерифицировано с образованием соответствующего трет.-бутильного сложного эфира, при использовании следующих способов, которые в общем описаны в литературе: реакция с изобутеном и сильной кислотой, например, паратолуолсульфоновой кислотой (pTs), серной кислотой или сильным кислотным ионообменником (US-A-3325466);
реакция хлорида кислоты и трет.-бутанола, при воздействии основания, например, триэтиламина (Et3N), диметиламинопиридина (DMAP). Хлорид кислоты может быть получен с помощью, например, SOCl2, POCl3, (COCl)2 и катализирован, например, диметилформамидом (ДМФА) (J. Org. Chem. 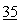 2429 (1970));
2429 (1970));
реакция хлорида кислоты с Li-трет.-бутанолатом (Org. Synth. 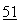 96 (1971));
96 (1971));
переэтерификация с трет.-бутилацетатом при воздействии сильной кислоты (Z. Chem. 12(7) 264 1972));
реакция соли с трет.-бутилбромидом, предпочтительно в ДМФА, диметилацетамиде (DMAA), 1-метил-2-пирролидиноне (NMP) и при использовании катализатора фазового переноса (РТС) (Tetr. Let. 34 (46) 7409 (1993));
реакция кислоты с трет.-бутанолом, 1,3-дициклогексилкарбодиимидом (DCC) и DMAP (Synth. Comm. 9, 542 (1979));
реакция кислоты с трет.-бутилтрихлорацетамидатом (Tetr. Let. 39 1557 (1998));
реакция соли с карбоксилдиимидазолом (CDI) и трет.-бутанолом;
реакция кислоты с пивалоилхлоридом и трет.-бутанолом при воздействии DMAP или N-метилморфолина (NMM) (Bull. Chem. Soc. Japan 52 (7) 1989 (1979));
реакция соли с ди-трет.-бутилдикарбонатом, DMAP и трет,-бутанолом (Synthesis 1063 (1994));
реакция кислоты с цианурхлоридом и пиридином или триэтиламином (Org Process R&D 3, 172 (1999); Heterocycles 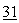 11, 2055 (1990)).
11, 2055 (1990)).
Полученный трет.-бутиловый сложный эфир 2-(6-замещенной-1,3-диоксан-4-ил)уксусной кислоты может быть затем превращен в 2-(6-гидроксиметил-1,3-диоксан-4-ил)уксусную кислоту, например, как описано в US-A-5594153 или в ЕР-А-1024139, в присутствии тетраалкиламмонийгалогенида и/или карбоновой кислоты в виде соли, через превращение в соединение формулы 1а с R3, представляющим собой трет.-бутил, и Х, представляющим собой ацилокси, например, ацетоксигруппу. Ацилоксигруппу затем можно превратить посредством сольволиза хорошо известным способом в гидроксигруппу. Сольволиз может проводиться при использовании основания (Na2СО3, К2СО3 или натрий метанолат в метаноле), при желании, одновременной отгонкой образованного метилацетата.
Трет.-бутиловый сложный эфир 2-(6-замещенной-1,3-диоксан-4-ил)уксусной кислоты представляет собой целевой промежуточный продукт при получения различных статинов, например, ZD-4522, как описано в Drugs of the future, (1999), 24(5), 511-513 М. Watanabe et al., Bioorg. & Med. Chem. (1997), 5(2), 437-444. Следовательно, изобретение обеспечивает новый, интересный путь к этим промежуточным продуктам и конечным продуктам, в особенности к статинам.
Исходные соединения формулы 2 могут быть, например, получены, как описано в WO-A-96/31615.
Изобретение будет объяснено применительно к следующим примерам, которые, однако, не ограничиваются ими.
Пример I
Получение (4R,6S)-4-гидрокси-6-хлорметил-тетрагидропиран-2-она (соединение II, охватываемое формулой 2)
К смеси 6,7 г (40 ммоль) 6-хлор-2,4,6-тридеокси-D-эритрогексозы (соединение I; полученное согласно способу, описанному в WO-A-96/31615) и 6,7 г натрийбикарбоната в 40 мл метиленхлорида и 10 мл воды при комнатной температуре в течение 45 мин добавляли 2,1 мл брома. CO2 газ выделялся, при этом рН оставался равным 5. После перемешивания в течение одного часа согласно газожидкостной хроматографии (ГЖХ) происходила полная конверсия исходного материала. Избыток брома нейтрализовали твердым Na2S2O3. После разделения фаз водную фазу экстрагировали два раза 100 мл этилацетата. Объединенные органические фазы сушили над Na2SO4 и фильтровали. После выпаривания на роторном испарителе получали 5,5 г желтого масла (82% выход соединения формулы (2), где Х=Cl, по отношению к соединению I).
1H ЯМР (200 МГц, CDCl3): δ 1,8-2,1 (м, 2Н); 2,6-2,7 (м, 2Н); 3,5-3,8 (м, 2Н (CH2Cl); 4,4 (м, 1Н); 4,9 (м, 1Н).
Пример II
Получение (4R,6S)-4-гидрокси-6-хлорметил-тетрагидропиран-2-она (Соединение II, охватываемое формулой 2)
К раствору 75 г (450 ммоль) соединения I в 390 мл воды добавляли 114 г (715 ммоль) брома при температуре 15-25°С в течение 3 часов. рН реакционной смеси поддерживали при 5-6 посредством добавления натрийкарбоната (общее количество 88 г). Избыток брома нейтрализовали бисульфатом натрия. Продукт экстрагировали из водной фазы этилацетатом (противоточной экстракцией).
Продукт кристаллизовали из этилацетата/гептана (125 г/ 62 г). После охлаждения до 0°С кристаллы отфильтровывали, промывали 50 мл гептана/этилацетата (вес.:вес.=9:1) и сушили с получением 49,2 г (67% по отношению к соединению I) соединения II в виде бесцветных иголок (т.пл. 73-74°С).
Пример III
Получение метилового сложного эфира (4R-цис)-6-(хлорметил)-2,2-диметил-1,3-диоксан-4-ид уксусной кислоты (соединение III)
5,5 г Соединения II, полученного в примере I, добавляли при комнатной температуре к 20 мл коммерчески доступного диметоксипропана и 100 мг моногидрата п-толуолсульфоновой кислоты. После перемешивания в течение одного часа при комнатной температуре ГЖХ анализ показал, что произошла полная конверсия и образовался прозрачный раствор. После добавления 500 мг NaHCO3 проводили перемешивание в течение 30 минут при комнатной температуре. После фильтрации и выпаривания на роторном испарителе получали 7,1 г соединения в виде светло-желтого масла (91% по отношению к соединению II).
1H ЯМР (200 МГц, CDCl3): δ 1,25 (дт, 1Н); 1,40 (с, 3Н); 1,47 (с, 3Н); 1,79 (дт, 1Н); 2,42 (дд, 1Н); 2,58 (дд, 1Н); 3,40 (дд, 1Н); 3,52 (дд, 1Н); 3,70 (с, 3Н); 4,1 (м, 1Н); 4,35 (м, 1Н).
Пример IV
Получение метилового сложного эфира (4R-цис)-6-(хлорметил)-2,2-диметил-1,3-диоксан-4-ил уксусной кислоты (соединение III)
К раствору 49,2 г (300 ммоль) соединения II в 100 мл толуола добавляли 47 г (450 ммоль) диметоксипропана и 850 мг моногидрата п-толуолсульфоновой кислоты (4,5 ммоль).
После перемешивания в течение одного часа при комнатной температуре ГЖХ анализ показал полную конверсию соединения II.
Толуольную фазу промывали 50 мл 0,2н раствора NaOH в воде. После выпаривания получали 67 г соединения III в виде светло-желтого масла (94% по отношению к соединению II).
Пример V
Натриевая соль (4R-цис)-(6-хлорметил)-2,2-диметил-1,3-диоксан-4-ил уксусной кислоты (соединение IV)
55 г (233 ммоль) Соединения III добавляли к 200 мл воды. 20 г 50% раствора NaOH в воде добавляли прикапыванием при комнатной температуре в течение 2 часов при рН 12. Гидролиз отслеживали при использовании ГЖХ. После 20 г рН раствора оставался постоянным. Для понижения рН до 10 использовали концентрированную соляную кислоту. Водную фазу промывали 100 мл этилацетата и упаривали при использовании роторного испарителя. Образованное масло сушили отгонкой с абсолютным этанолом и толуолом. Твердое вещество перемешивали в 200 мл ацетона, фильтровали и промывали холодным ацетоном. Выход после вакуумной сушки: 45,6 г=80% Na соли по отношению к соединению III.
1H ЯМР (200 МГц, CDCl3/CD3OD): δ 1,21 (дт, 1Н); 1,36 (с, 3Н); 1,49 (с, 3Н); 1,79 (дт, 1Н); 2,25 (дд, 1Н); 2,45 (дд, 1Н); 3,46 (м, 2Н); 4,11 (м, 1Н); 4,36 (м, 1Н).
Пример VI
Натриевая соль (4R-цис)-(6-хлорметил)-2,2-диметил-1,3-диоксан-4-ил уксусной кислоты (соединение IV)
Раствор соединения III в толуоле получали, как описано в примере IV, используя 49,2 г соединения I в качестве исходного.
Добавляли 5 г метанола и 25 мл воды. Добавляли 25 г 50% раствора NaOH в воде прикапыванием при комнатной температуре в течение 1 часа.
После перемешивания в течение 4 часов при комнатной температуре ГЖХ анализ показал, что гидролиз прошел полностью.
Избыток основания нейтрализовали до значений рН от 8,5 до 9,5 33% раствором HCl в воде. Водную фазу отделяли и сушили азеотропной перегонкой, используя 470 мл толуола, с выходом 65 г соединения IV в виде 16% вес./вес. суспензии в толуоле с KF<0,1%.
Суспензию можно было использовать для синтеза соединения V.
Пример VII
Трет-бутиловый сложный эфир (4R-цис)-(6-хлорметил)-2,2-диметил-1,3-диоксан-4-ил уксусной кислоты (соединение V)
45,5 г Натриевой соли IV (186 ммоль) добавляли к раствору 159 г ди-трет-бутилдикарбоната в 1400 мл сухого ди-трет-бутанола. После добавления 6,8 г диметиламинопиридина смесь перемешивали при 40°С в течение 16 часов. Реакционную смесь выливали в 1500 мл этилацетата и 1000 мл насыщенного аммонийхлорида. Водную фазу повторно экстрагировали 1500 мл этилацетата. Объединенные органические фазы промывали 600 мл насыщенного раствора NaCl. Органический слой сушили над Na2SO4, фильтровали и затем упаривали в вакууме с получением 51,9 г желтого масла (100% по отношению к соединению IV).
1H ЯМР (200 МГц, CDCl3): δ 1,15-1,33 (м, 1Н); 1,40 (с, 3Н); 1,45 (с, 3Н); 1,47 (с, 9Н) 1,77 (дт, 1Н); 2,33 (дд, 1Н); 2,46 (дд, 1Н); 3,40 (дд, 1Н); 3,49 (дд, 1Н); 4,08 (м, 1Н); 4,28 (м, 1Н).
Пример VIII
Трет-бутиловый сложный эфир (4R-цис)-6-[(ацетокси)метил]-2,2-диметил-1,3-диоксан-4-ил-уксусной кислоты (соединение VI)
К 33 г исходного соединения V, полученного согласно примеру VII, добавляли одной порцией 40 г тетра-н-бутиламмонийацетата, полученного путем взаимодействия ледяной уксусной кислоты, добавляемой по каплям при перемешивании к 40% водному раствору тетрабутиламмоний гидроксида в атмосфере аргона. В процессе добавления уксусной кислоты температура реакции поддерживалась ниже 35°С. При достижении рН среды 8.5 добавление прекращали и раствор концентрировали на роторном испарителе в высоком вакууме. Полученный полутвердый продукт подвергают азеотропной сушке с толуолом (4×500 мл) на роторном испарителе, затем в высоком вакууме в течение 24-48 час, получая твердый ацетат тетрабутиламмония, который и добавляли к соединению V. Процесс проводили в 200 мл ДМФА в атмосфере аргона при 100°С в течение 16 часов до завершения реакции. Реакционную смесь охлаждали до комнатной температуре и выливали в рН 7 фосфатный буфер (4 л) и экстрагировали гептаном (3×1 л). Органические слои объединяли и промывали водой (1 л), рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении до 1 л и обрабатывали нейтральным NORIT (40 г). Гетерогенный раствор кипятили на водяной бане 2 мин и горячим фильтровали через Целит на воронке Бюхнера. Остаток промывали горячим гептаном (3×250 мл). Фильтраты объединяли и концентрировали на роторном испарителе при пониженном давлении, получая 29 г соединения VI в виде твердого вещества после кристаллизации из 75 мл гептана.
1H ЯМР (200 МГц, CDCl3): δ 1,1-1,3 (дт, 1Н); 1,39 (с, 3Н); 1,45 (с, 9Н); 1,47 (с, 3Н); 1,57 (дт, 1Н); 2,08 (с, 3Н); 2,32 (дд, 1Н); 2,46 (дд, 1Н); 4,0-4,2 (м, 3Н); 4,3 (м, 1Н).
Пример IX
Трет-бутиловый сложный эфир (4R-цис)-6-[гидроксиметил]-2,2-диметил-1,3-диоксан-4-ил-уксусной кислоты (соединение VII)
Используя в качестве исходного 29 г соединения VI, полученного согласно примеру VIII, получали 25,0 г соединения VII в виде светло-желтого масла с е.е.=100%, d.e.=99,9% (согласно ГЖХ), путем добавления к 29 г соединения VI 6,9 г карбоната калия. Процесс проводят в 300 мл метанола при интенсивном перемешивании до полного гидролиза. Раствор фильтруют через воронку Бюхнера и концентрируют на роторном испарителе при комнатной температуре и пониженном давлении. Осадок растворяли в воде (250 мл) и экстрагировали эфиром (3×200 мл). Объединенные органические слои промывали водой (150 мл), рассолом (150 мл), сушили над MgSO4, фильтровали и концентрировали на роторном испарителе с получением соединения, указанного в оглавлении. Сырой продукт перегоняли, получая соединение VII со следующими характеристиками.
1H ЯМР (200 МГц, CDCl3): спектр совпадает с литературным (Synthesis 1014, 1995).
Изобретение относится к способу получения производных 2-(6-замещенной-1,3-диоксан-4-ил)уксусной кислоты формулы 1 или его соли, или кислоты:
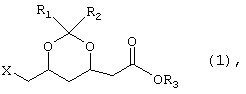
где Х означает галоген, тозилатную, мезилатную, ацилоксигруппу, арилокси- или нитро-замещенную бензолсульфонильную группу и R1, R2 и R3, каждая независимо означает C1-3 алкильную группу из соединения формулы 2:
где Х имеет вышеуказанные значения, с использованием подходящего агента ацетализации, в присутствии кислотного катализатора, и с последующим преобразованием его, при необходимости, в соответствующую соль или кислоту. Эти соединения являются промежуточными продуктами в производстве статинов-ингибиторов HMG-CoA редуктазы. Изобретения также относятся к новым исходным соединениям формулы 2 в (4R, 6S) форме и к новым соединениям формулы 1b. Технический результат - получение ценного промежуточного продукта с высоким выходом. 3 н. и 8 з.п. ф-лы.
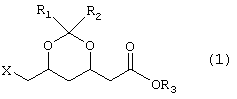
где Х обозначает галоген, тозилатную, мезилатную, ацилокси группу, арилокси- или нитрозамещенную бензолсульфонильную группу, и R1, R2 и R3, каждая, независимо обозначает алкильную группу с от 1 до 3 атомами углерода, исходя из соединения формулы 2
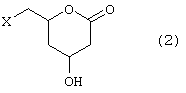
где Х является таким же, как и описано выше, с использованием подходящего агента ацетализации, в присутствии кислотного катализатора, и при необходимости, преобразованием его в соответствующую соль или кислоту.
где X, R1 и R2 являются такими же, как описано выше, и Y обозначает щелочной металл, щелочноземельный металл или замещенную или незамещенную аммониевую группу.
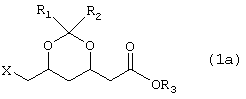
в котором Х обозначает галоген, тозилатную, мезилатную, ацилокси группу, арилокси- или нитро-замещенную бензолсульфонильную группу.
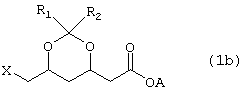
в котором X представляет галоген, тозилатную, мезилатную, ацилокси группу, арилокси- или нитро-замещенную бензолсульфонильную группу, R1, R2, каждая, независимо представляет собой алкильную группу с от 1 до 3 атомами углерода, А представляет собой алкильную группу с от 1 до 3 атомами углерода или щелочной (щелочноземельный) металл или замещенную или незамещенную аммониевую группу.
| US 5457227 А 10.10.1995 | |||
| Устройство для отделения верхнего листа от стопы и подачи его в зону обработки | 1981 |
|
SU1024139A1 |
| WO 9113876 А 19.09.1991 | |||
| Chem | |||
| Abst., v.118, № 11, 1993, 101787x | |||
| JUN-ICHISAKAKI, Tetrahedron: Asymmetry, v.2, № 5, 1991, p.343-346 F.Bennet et al, J.Chem | |||
| Soc., v.1, 1991, p.133-140 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА И ИСХОДНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2115649C1 |

