Область техники
Настоящее изобретение относится к аналогам 2',5'-олигоаденилата (2-5A), которые стабильны и обладают превосходной активностью (в особенности, противоопухолевой активностью).
Уровень техники
2-5A, который известен как биологическое вещество, обладающее противовирусной активностью (Pharmacol. Ther. Vol.78, №2, pp.55-113, 1998), представляет собой короткоцепочечный олигонуклеотид, состоящий из трех или более аденозиновых звеньев, в котором две 2' и 5' гидроксильные группы аденозина соединены с помощью 2',5'-фосфодисложноэфирных связей фосфата, и в котором трифосфатная группа связана с 5' концом. Когда клетки, инфицированные вирусом, подвергаются стимуляции внеклеточным интерфероном, 2-5А синтетаза индуцируется в присутствии вирусной дсРНК и 2-5А продуцируется из АТФ. 2-5А представляет собой вещество, которое превращает неактивную форму разрушающего РНК фермента, RNase L, в активную форму в клетках-хозяевах. Такая активированная RNase L ингибирует размножение вируса в клетках за счет разрушения вирусной РНК. Более того, когда клетки рака яичников Hey1B трансфицированы 2-5А, как известно, происходит специфичное расщепление последовательности 18S рРНК, что приводит к проявлению противоопухолевой активности в результате апоптоза через высвобождение цитохрома С и активации каспазы (J. Interferon Cytokine Res., 20, 1091-1100 (2000)). Таким образом, предполагают, что 2-5А действует как ингибитор размножения вируса и, более конкретно, как противовирусное лекарственное средство или противоопухолевое лекарственное средство.
Известно, что в опытах in vitro олигонуклеотид, состоящий из трех или более аденозиновых звеньев, содержащих монофосфатную группу на 5' конце и соединенных с помощью 2',5'-фосфодисложноэфирных связей, активирует RNase L (Pharmacol. Ther. Vol. 78, №2, pp.55-113, 1998; J. Biol. Chem. Vol.270, №11, pp.5963-5978). Однако сам 2-5А легко распадается до АМФ и АТФ с помощью 2'-фосфодиэстеразы и нуклеазы. Более того, 5'-фосфатная группа или 5'-трифосфатная группа закрываются, так как дефосфорилируются с помощью фосфатаз в живом организме, и теряют активность. Таким образом, в случае использования 2-5А в качестве ингибитора размножения вируса или противоопухолевого лекарства желателен аналог 2-5А, который обладает аналогичной активностью, но имеет высокую стабильность, делающую его более устойчивым к разрушению и метаболизму в живом организме.
Чтобы преодолеть указанные недостатки, были опробованы различные способы в качестве примеров модификации фосфатных групп. Примеры таких известных способов включают способ, в котором немостиковый атом кислорода, связанный с атомом фосфора фосфодисложноэфирной связи олигонуклеотида, замещен атомом серы (тиофосфатная модификация), способ, в котором указанный атом кислорода замещен метильной группой, способ, в котором указанный атом кислорода замещен атомом бора, и способ, в котором фрагмент сахара или фрагмент нуклеооснования олигонуклеотида химически модифицирован (Freier, S.M.; Altmann, K.H., Nucleic Acids Res., 25, 4429 (1997)). Известный пример такого аналога 2-5А представляет собой показанный ниже аденозиновый тетрамер, который подвергнут фосфотиоатной модификации (Carpten, J. et al. Nature Genetics, 30, 181 (2002)).
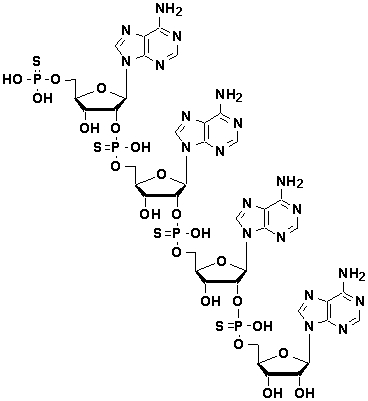
Более того, аналоги, имеющие химическую структуру, подобную химической структуре, показанной ниже, в которой модифицирован фрагмент сахара аденозина, описаны в японской патентной заявке (Kokai) № Hei 10-195098 и в японском патенте № 3420984 как аденозиновые звенья аналогов 2-5А.
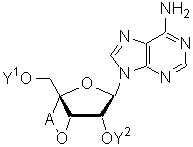
(В приведенной выше формуле, Y1 и Y2 представляют собой атом водорода или защитную группу для гидроксильной группы, и А представляет собой алкиленовую группу, содержащую от 1 до 3 атомов углерода).
Кроме того, молекулу 2-5А, связанную с помощью линкера с антисмысловой молекулой в форме олигонуклеотида, имеющего последовательность, комплементарную мРНК, вовлеченную в заболевания, использовали в качестве 2-5А антисмыслового олигонуклеотида, который ингибирует функцию мРНК (S.A. Adahet, et al., Current Medicinal Chemistry (2001), 8, 1189-1212). Высокостабильный аналог 2-5А, который устойчив к разложению и метаболизму в живом организме, пригоден в качестве фрагмента превосходного 2-5А антисмыслового олигонуклеотида и, как полагают, является полезным лекарственным средством. В частности, олигонуклеотиды, содержащие мостиковый нуклеозид, в котором атом кислорода в положении 2' и атом углерода в положении 4' фрагмента сахара связаны алкиленовой группой, как известно, могут быть полезны в качестве антисмысловых молекул (японская патентная заявка (Kokai) № Hei 10-304889, японская патентная заявка (Kokai) № 2000-297097).
Описание изобретения
Авторы настоящего изобретения проводили интенсивные исследования в течение многих лет неприродных аналогов 2-5А, которые обладают противовирусной активностью, противоопухолевой активностью или превосходной антисмысловой активностью, являются стабильными в живом организме и связаны с проявлением незначительных неблагоприятных побочных эффектов. В результате были найдены соединения, которые могут быть полезны в качестве стабильных и превосходных противовирусных лекарственных средств, противоопухолевых лекарственных средств и антисмысловых лекарственных средств, что привело к завершению настоящего изобретения.
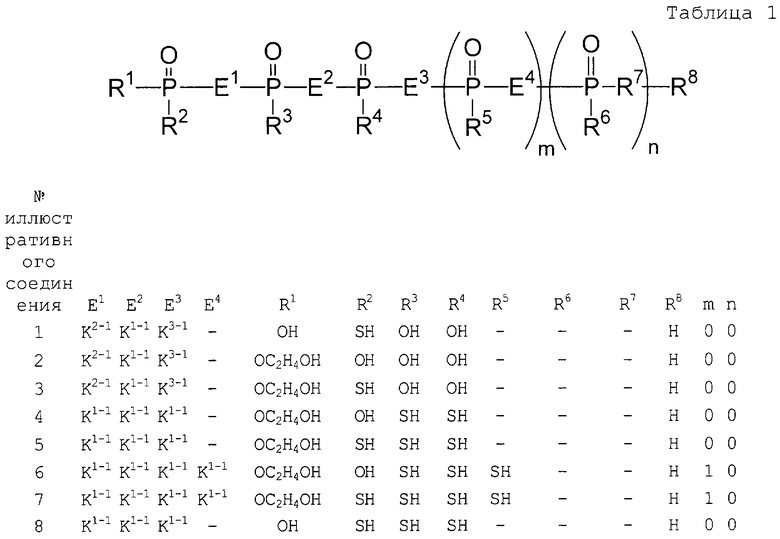
Аналог 2-5А настоящего изобретения относится к аналогу 2',5'-олигоаденилата, представленного общей формулой (1):
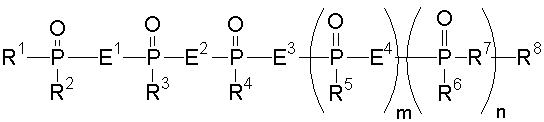
[где m равно целому числу 0 или 1; n равно целому числу от 0 до 2; R1 представляет собой алкоксигруппу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, меркаптогруппу, меркаптогруппу, защищенную с защитной группой синтеза нуклеиновой кислоты, алкилтиогруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, аминогруппу, аминогруппу, защищенную защитной группой синтеза нуклеиновой кислоты, аминогруппу, замещенную алкильной(ыми) группой(ами), содержащей(ими) от 1 до 6 атомов углерода, которая(ые) может(гут) быть замещена(ы), алкильную группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, арилоксигруппу, которая может быть замещена, или арилтиогруппу, которая может быть замещена, или группу, формулы: X1-X2-X3-S-; R2, R3, R4, R5 и R6 представляют собой гидроксильную группу, гидроксильную группу, защищенную защитной группой синтеза нуклеиновой кислоты, алкоксигруппу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, меркаптогруппу, меркаптогруппу, защищенную защитной группой синтеза нуклеиновой кислоты, алкилтиогруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, аминогруппу, аминогруппу, защищенную защитной группой синтеза нуклеиновой кислоты, аминогруппу, замещенную алкильной(ыми) группой(ами), содержащей(ими) от 1 до 6 атомов углерода, которая(ые) может(гут) быть замещена(ы), или алкильную группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена; R7 представляет собой атом кислорода, атом серы, -NH-, группу -O(CH2CH2O)q (q равно целому числу от 2 до 6), оксиалкиленоксигруппу, содержащую от 1 до 6 атомов углерода, или группу формулы: X1-X2-X3-S-; R8 представляет собой атом водорода, алкильную группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, аралкильную группу, которая может быть замещена, арильную группу, которая может быть замещена, или 5'-фосфорилированный аналог олигонуклеотида, в котором одна гидроксильная группа удалена из группы 5'-фосфорной кислоты; E1, E2, E3 и E4 являются одинаковыми или различными и представляют собой K1, K2, K3 или K4 (K1, K2, K3 и K4 представляют собой:
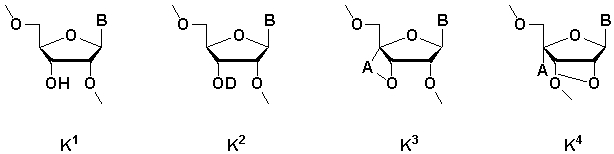
соответственно, где B представляет собой пурин-9-ильную группу или замещенную пурин-9-ильную группу, содержащую заместитель(и), выбранный(е) из приведенной ниже группы α, A представляет собой алкиленовую группу, содержащую от 1 до 4 атомов углерода, D представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, или алкенильную группу, содержащую от 2 до 6 атомов углерода, которая может быть замещена); X1 представляет собой алкильную группу, содержащую от 1 до 24 атомов углерода, которая может быть замещена, или арильную группу, которая может быть замещена, или аралкильную группу, которая может быть замещена; X2 представляет собой группу -C(=O)O-, -ОС(=O)-, -C(=O)NH-, -NHC(=O)-, -C(=O)S-, -SC(=O)-, -OC(=O)NH-, -NHC(=O)O-, -NHC(=O)NH-, -OC(=S)- или -C(=S)O-, -NHC(=S)-, -C(=S)NH-; и X3 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена] (при условии, что соединения, в которых m равно 0, n равно 1, R2, R3, R4 и R6 представляют собой гидроксильную группу, R7 представляет собой атом кислорода и R8 представляет собой 2-гидроксиэтильную группу, и соединение, в котором m равно 1, n равно 0, R1, R3, R4 и R5 представляют собой меркаптогруппу, R2 представляет собой гидроксильную группу, R8 представляет собой атом водорода и все E1, E2, E3 и E4 представляют собой K1, исключены), или его фармакологически приемлемой соли.
(Группа α)
гидроксильная группа,
гидроксильная группа, защищенная защитной группой синтеза нуклеиновой кислоты,
алкоксигруппа, содержащая от 1 до 6 атомов углерода, которая может быть замещена,
меркаптогруппа,
меркаптогруппа, защищенная защитной группой синтеза нуклеиновой кислоты,
алкилтиогруппа, содержащая от 1 до 4 атомов углерода, которая может быть замещена,
аминогруппа,
аминогруппа, защищенная защитной группой синтеза нуклеиновой кислоты,
аминогруппа, замещенная алкильной(ыми) группой(ами), содержащей(ими) от 1 до 4 атомов углерода, которая(ые) может(гут) быть замещена(ы),
алкильная группа, содержащая от 1 до 6 атомов углерода, которая может быть замещена, и
атом галогена.
Приведенный выше аналог 2',5'-олигоаденилата или его фармакологически приемлемая соль предпочтительно представляет собой:
(1) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где R1 представляет собой алкоксигруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, меркаптогруппу, меркаптогруппу, защищенную защитной группой синтеза нуклеиновой кислоты, или алкилтиогруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, или группу формулы: X1-X2-X3-S-; R2, R3, R4, R5 и R6 представляют собой гидроксильную группу, гидроксильную группу, защищенную защитной группой синтеза нуклеиновой кислоты, алкоксигруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, меркаптогруппу, меркаптогруппу, защищенную защитной группой синтеза нуклеиновой кислоты, алкилтиогруппу, содержащую от 1 до 4 атомов углерода, которая может быть замещена, или группу формулы: X1-X2-X3-S-; X1 представляет собой алкильную группу, содержащую от 10 до 24 атомов углерода, которая может быть замещена; X2 представляет собой группу -C(=O)O-, -C(=O)NH-, -C(=O)S-, -NHC(=O)O- или -C(=S)NH-; и X3 представляет собой алкиленовую группу, содержащую от 1 до 4 атомов углерода, которая может быть замещена;
(2) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где R7 представляет собой атом кислорода, группу -O(CH2CH2O)q- (q равно целому числу от 2 до 6), или оксиалкиленоксигруппу, содержащую от 1 до 6 атомов углерода; и R8 представляет собой атом водорода; алкильную группу, содержащую от 1 до 6 атомов углерода, которая может быть замещена, или 5'-фосфорилированный аналог олигонуклеотида, в котором одна гидроксильная группа удалена из группы 5'-фосфорной кислоты;
(3) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где E2 представляет собой K1;
(4) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где E1 представляет собой K2 и D представляет собой метильную группу или 2-пропенильную группу;
(5) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где E3 представляет собой K3 или K4, и A представляет собой метиленовую, этиленовую или пропиленовую группу;
(6) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где B представляет собой 6-аминопурин-9-ильную (то есть, аденинил), 6-амино-8-бромпурин-9-ильную, 6-амино-8-хлорпурин-9-ильную, 6-амино-8-фторпурин-9-ильную, 6-амино-8-метоксипурин-9-ильную, 6-амино-8-этоксипурин-9-ильную, 6-амино-8-трет-бутоксипурин-9-ильную, 6-амино-2-бромпурин-9-ильную, 6-амино-2-хлорпурин-9-ильную, 6-амино-2-фторпурин-9-ильную, 6-амино-2-метоксипурин-9-ильную, 6-амино-2-этоксипурин-9-ильную, 6-амино-2-трет-бутоксипурин-9-ильную или 2,6-диаминопурин-9-ильную группу; или
(7) аналог 2',5'-олигоаденилата или его фармакологически приемлемую соль, где B представляет собой 6-амино-9-ил (то есть аденинил) или 6-амино-8-бромпурин-9-ил.
В приведенной выше общей формуле «алкиленовая группа, содержащая от 1 до 4 атомов углерода» заместителя A может представлять собой, например, метиленовую, этиленовую, триметиленовую или тетраметиленовую группу, и предпочтительно представляет собой этиленовую или триметиленовую группу.
В приведенной выше общей формуле (1) защитная группа «гидроксильной группы, защищенной защитной группой синтеза нуклеиновой кислоты» заместителей R2, R3, R4, R5 и R6 или группы α не имеет особых ограничений, пока она может стабильно защищать гидроксильную группу при синтезе нуклеиновой кислоты, и особенно означает защитную группу, стабильную в кислых или нейтральных условиях и расщепляемую с помощью химического метода, такого как гидрогенолиз, гидролиз, электролиз или фотолиз. Такими защитными группами могут быть, например, «алифатическая ацильная группа», такая как алкилкарбонильная группа, например формил, ацетил, пропионил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, нонаноил, деканоил, 3-метилнонаноил, 8-метилнонаноил, 3-этилоктаноил, 3,7-диметилоктаноил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, 1-метилпентадеканоил, 14-метилпентадеканоил, 13,13-диметилтетрадеканоил, гептадеканоил, 15-метилгексадеканоил, октадеканоил, 1-метилгептадеканоил, нонадеканоил, эйкозаноил и генэйкозаноил; карбоксилированная алкилкарбонильная группа, например сукциноил, глутароил и адипоил; галоген(низший)алкилкарбонильная группа, например, хлорацетил, дихлорацетил, трихлорацетил и трифторацетил; (низший)алкокси(низший)алкилкарбонильная группа, например, метоксиацетил; или ненасыщенная алкилкарбонильная группа, например, (E)-2-метил-2-бутеноил;
«низшая алкильная группа», такая как, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилфенил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил и 2-этилбутил;
«низшая алкенильная группа», такая как этенил, 1-пропенил, 2-пропенил, 1-метил-2-пропенил, 1-метил-1-пропенил, 2-метил-1-пропенил, 2-метил-2-пропенил, 2-этил-2-пропенил, 1-бутенил, 2-бутенил, 1-метил-2-бутенил, 1-метил-1-бутенил, 3-метил-2-бутенил, 1-этил-2-бутенил, 3-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 1-этил-3-бутенил, 1-пентенил, 2-пентенил, 1-метил-2-пентенил, 2-метил-2-пентенил, 3-пентенил, 1-метил-3-пентенил, 2-метил-3-пентенил, 4-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил и 5-гексенил;
«ароматическая ацильная группа», такая как арилкарбонильная группа, например, бензоил, α-нафтоил и β-нафтоил; галогенарилкарбонильная группа, например, 2-бромбензоил и 4-хлорбензоил; низшая алкилированная арилкарбонильная группа, например, 2,4,6-триметилбензоил и 4-толуоил; низкая алкоксилированная арилкарбонильная группа, например 4-анизоил; карбоксилированная арилкарбонильная группа, например 2-карбоксибензоил, 3-карбоксибензоил и 4-карбоксибензоил; нитрованная арилкарбонильная группа, например, 4-нитробензоил и 2-нитробензоил; низшая алкоксикарбонилированная арилкарбонильная группа, например 2-(метоксикарбонил)бензоил; или арилированная арилкарбонильная группа, например 4-фенилбензоил;
«тетрагидропиранильная или тетрагидротиопиранильная группа», такая как тетрагидропиран-2-ил, 3-бромтетрагидропиран-2-ил, 4-метокситетрагидропиран-4-ил, тетрагидротиопиран-2-ил и 4-метокситетрагидротиопиран-4-ил;
«тетрагидрофуранильная или тетрагидротиофуранильная группа», такая как тетрагидрофуран-2-ил и тетрагидротиофуран-2-ил;
«силильная группа», такая как три(низший)алкилсилильная группа, например триметилсилил, триэтилсилил,
изопропилдиметилсилил, трет-бутилдиметилсилил,
метилдиизопропилсилил, метилди-трет-бутилсилил и
триизопропилсилил; или три(низший)алкилсилильная группа, замещенная 1 или 2 арильными группами, например дифенилметилсилил, дифенилбутилсилил, дифенилизопропилсилил и фенилдиизопропилсилил;
«низшая алкоксиметильная группа», такая как метоксиметил, 1,1-диметил-1-метоксиметил, этоксиметил, пропоксиметил, изопропоксиметил, бутоксиметил и трет-бутоксиметил;
«низшая алкоксилированная низшая алкоксиметильная группа», такая как 2-метоксиэтоксиметил;
«галоген(низший)алкоксиметил», такой как 2,2,2-трихлорэтоксиметил и бис(2-хлорэтокси)метил;
«низшая алкоксилированная этильная группа», такая как 1-этоксиэтил и 1-(изопропокси)этил;
«галогенированная этильная группа», такая как 2,2,2-трихлорэтил;
«метильная группа, замещенная 1-3 арильными группами», такая как бензил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, α-нафтилдифенилметил и 9-антрилметил;
«метильная группа, замещенная 1-3 арильными группами, арильное кольцо которых замещено низшими алкильными, низшими алкоксигруппами, галогеном или цианогруппами», такая как 4-метилбензил, 2,4,6-триметилбензил, 3,4,5-триметилбензил,
4-метоксибензил, 4-метоксифенилдифенилметил,
4,4'-диметокситрифенилметил, 2-нитробензил, 4-нитробензил,
4-хлорбензил, 4-бромбензил и 4-цианобензил;
«низшая алкоксикарбонильная группа», такая как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и изобутоксикарбонил;
«арильная группа, замещенная атомом(ами) галогена, низшей(ми) алкоксигруппой(ами) или нитрогруппой(ами)», такая как 4-хлорфенил, 2-хлорфенил, 4-метоксифенил, 4-нитрофенил и 2,4-динитрофенил;
«низшая алкоксикарбонильная группа, замещенная галогеном или три(низший)алкилсилильной(ыми) группой(ами)», такая как 2,2,2-трихлорэтоксикарбонил и 2-триметилсилилэтоксикарбонил;
«алкенилоксикарбонильная группа», такая как винилкарбонил и аллилоксикарбонил;
«аралкилоксикарбонильная группа, арильное кольцо которой может быть замещено 1 или 2 низшими алкокси или нитрогруппами», такая как бензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 2-нитробензилоксикарбонил и 4-нитробензилоксикарбонил;
«алифатическая ацилоксиметильная группа», такая как алкилкарбонилоксиметильная группа, например ацетилоксиметил,
пропионилоксиметил, бутирилоксиметил, изобутирилоксиметил,
пентаноилоксиметил, пивалоилоксиметил, валерилоксиметил,
изовалерилоксиметил, октаноилоксиметил, нонаноилоксиметил,
деканоилоксиметил, 3-метилнонаноилоксиметил,
8-метилнонаноилоксиметил, 3-этилоктаноилоксиметил,
3,7-диметилоктаноилоксиметил, ундеканоилоксиметил,
додеканоилоксиметил, тридеканоилоксиметил,
тетрадеканоилоксиметил, пентадеканоилоксиметил,
гексадеканоилоксиметил, 1-метилпентадеканоилоксиметил,
14-метилпентадеканоилоксиметил,
13,13-диметилтетрадеканоилоксиметил, гептадеканоилоксиметил,
15-метилгексадеканоилоксиметил, октадеканоилоксиметил,
1-метилгептадеканоилоксиметил, нонадеканоилоксиметил,
эйкозаноилоксиметил и генэйкозаноилоксиметил; карбоксилированная алкилкарбонилоксиметильная группа, например сукциноилоксиметил,
глутароилоксиметил и адипоилоксиметил;
галоген(низший)алкилкарбонилоксиметильная группа, например хлорацетилоксиметил, дихлорацетилоксиметил,
трихлорацетилоксиметил и трифторацетилоксиметил;
(низший)алкокси(низший)алкилкарбонилоксиметильная группа, например метоксиацетилоксиметил; или ненасыщенная алкилкарбонилоксиметильная группа, например (E)-2-метил-2-бутеноил;
«алифатическая ацилтиоэтильная группа», такая как алкилкарбонилтиоэтильная группа, например ацетилтиоэтил,
пропионилтиоэтил, бутирилтиоэтил, изобутирилтиоэтил,
пентаноилтиоэтил, пивалоилтиоэтил, валерилтиоэтил,
изовалерилтиоэтил, октаноилтиоэтил, нонаноилтиоэтил,
деканоилтиоэтил, 3-метилнонаноилтиоэтил, 8-метилнонаноилтиоэтил,
3-этилоктаноилтиоэтил, 3,7-диметилоктаноилтиоэтил,
ундеканоилтиоэтил, додеканоилтиоэтил, тридеканоилтиоэтил,
тетрадеканоилтиоэтил, пентадеканоилтиоэтил,
гексадеканоилтиоэтил, 1-метилпентадеканоилтиоэтил,
14-метилпентадеканоилтиоэтил, 13,13-диметилтетрадеканоилтиоэтил,
гептадеканоилтиоэтил, 15-метилгексадеканоилтиоэтил,
октадеканоилтиоэтил, 1-метилгептадеканоилтиоэтил,
нонадеканоилтиоэтил, эйкозаноилтиоэтил и генэйкозаноилтиоэтил;
карбоксилированная алкилкарбонилтиоэтильная группа, например сукционоилтиоэтил, глутароилтиоэтил и адипоилтиоэтил; галоген(низший)алкилкарбонилтиоэтильная группа, например хлорацетилтиоэтил, дихлорацетилтиоэтил, трихлорацетилтиоэтил и трифторацетилтиоэтил; (низший)алкокси(низший)алкилкарбонилтиоэтильная группа, например метоксиацетилтиоэтил; или ненасыщенная алкилкарбонилтиоэтильная группа, например (E)-2-метил-2-бутеноил.
Защитная группа «гидроксильной группы, защищенной защитной группой синтеза нуклеиновой кислоты» заместителей R2, R3, R4, R5 и R6 или группы α предпочтительно представляет собой «метильную группу, замещенную 1-3 арильными группами», «арильную группу, замещенную атомом(ами) галогена, низшей(ими) алкоксигруппой(ами) или нитрогруппой(ами)», «низшую алкильную группу», «низшую алкенильную группу», «алифатическую ацилоксиметильную группу» или «алифатическую ацилтиоэтильную группу», более предпочтительно бензильную группу, 2-хлорфенильную группу, 4-хлорфенильную группу, 2-пропенильную группу, пивалоилоксиметильную группу, ацилтиоэтильную группу или пивалоилтиоэтильную группу.
В приведенной выше формуле (1) «алкоксигруппа, содержащая от 1 до 6 атомов углерода, которая может быть замещена» заместителей R1, R2, R3, R4, R5, R6 или группы α может представлять собой, например, «низшую алкилоксигруппу», такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, н-пентилокси, изопентилокси, 2-метилбутокси, неопентилокси, 1-этилпропокси, н-гексилокси, изогексилокси, 4-метилпентилокси, 3-метилпентилокси, 2-метилпентилокси, 1-метилпентилокси, 3,3-диметилбутокси, 2,2-диметилбутокси, 1,1-диметилбутокси, 1,2-диметилбутокси, 1,3-диметилбутокси, 2,3-диметилбутокси и 2-этилбутоксигруппа;
«низшую алкилоксигруппу, замещенную гидроксильной(ыми) группой(ами)», такую как 1-гидроксиметилокси,
2-гидроксиэтилокси, 3-гидроксипропилокси,
4-гидроксибутилокси, 2-гидроксипропилокси,
1-метил-2-гидроксиэтилокси, 1-метил-1-гидроксиэтилокси,
1,1-диметил-2-гидроксиэтилокси, 2-гидроксибутилокси,
3-гидроксибутилокси, 1-метил-3-гидроксипропилокси и
2-метил-3-гидроксипропилоксигруппа;
«низшую алкилоксигруппу, замещенную аминогруппой(ами)», такую как 1-аминометилокси, 2-аминоэтилокси, 3-аминопропилокси,
4-аминобутилокси, 2-аминопропилокси, 1-метил-2-аминоэтилокси,
1-метил-1-аминоэтилокси, 1,1-диметил-1-аминоэтилокси,
2-аминобутилокси, 3-аминобутилокси, 1-метил-3-аминопропилокси и
2-метил-3-аминопропилоксигруппа;
«низшую алкилоксигруппу, замещенную алкоксигруппой(ами)», такую как 1-метоксиметилокси, 2-метоксиэтилокси,
3-метоксипропилокси, 4-метоксибутилокси, 2-метоксипропилокси,
1-метил-2-метоксиэтилокси, 1-метил-1-метоксиэтилокси,
1,1-диметил-2-метоксиэтилокси, 2-метоксибутилокси,
3-метоксибутилокси, 1-метил-3-метоксипропилокси,
2-метил-3-метоксипропилокси, 1-этоксиметилокси,
2-этоксиэтилокси, 3-этоксипропилокси, 4-этоксибутилокси,
2-этоксипропилокси, 1-метил-2-этоксиэтилокси,
1-метил-1-этоксиэтилокси, 1,1-диметил-2-этоксиэтилокси,
2-этоксибутилокси, 3-этоксибутилокси,
1-метил-3-этоксипропилокси и 2-метил-3-этоксипропилоксигруппа;
или
«циклоалкилоксигруппу», такую как циклопропокси, циклобутилокси, циклопентилокси, циклогексилокси, циклогептилокси, норборнилокси и адамантилоксигруппа; и предпочтительно представляет собой 2-гидроксиэтоксигруппа.
В приведенной выше общей формуле (1) «оксиалкиленоксигруппа, содержащая от 1 до 6 атомов углерода» заместителя R7 может представлять собой, например, оксиметиленокси, оксиэтиленокси, окситриметиленокси, окситетраметиленокси, оксипентаметиленокси или оксигексаметиленоксигруппу, и предпочтительно представляет собой окситетраметиленокси или оксипентаметиленоксигруппу.
В приведенной выше общей формуле (1) защитная группа «меркаптогруппы, защищенной защитной группой синтеза нуклеиновой кислоты» заместителей R1, R2, R3, R4, R5 и R6 или группы α не имеет особых ограничений, пока она способна устойчиво защищать меркаптогруппу при синтезе нуклеиновой кислоты, и конкретно означает защитную группу, устойчивую в кислых и нейтральных условиях и расщепляемую с помощью химических методов, таких как гидрогенолиз, гидролиз, электролиз или фотолиз. Такой защитной группой может быть, например, «группа, которая может образовывать дисульфид», такая как алкилтиогруппа, например, метилтио, этилтио и трет-бутилтиогруппа, или арилтиогруппа, например бензилтиогруппа, помимо групп, перечисленных в качестве защитной группы гидроксильной группы, и предпочтительно представляет собой «алифатическую ацильную группу», «ароматическую ацильную группу», «алифатическую ацилоксиметильную группу» или «алифатическую ацилтиоэтильную группу», более предпочтительно пивалоилоксиметильную группу, ацетилтиоэтильную группу или пивалоилтиоэтильную группу.
В приведенной выше общей формуле (1) «алкилтиогруппа, содержащая от 1 до 4 атомов углерода, которая может быть замещена» заместителей R1, R2, R3, R4, R5 и R6 или группы α может представлять собой, например, метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, втор-бутилтио или трет-бутилтиогруппу, и предпочтительно представляет собой метилтио или этилтиогруппу.
В приведенной выше формуле (1) защитная группа «аминогруппы, защищенной защитной группой синтеза нуклеиновой кислоты» заместителей R1 R2, R3, R4, R5 и R6 или группы α не имеет особых ограничений, пока она способна устойчиво защищать аминогруппу при синтезе нуклеиновой кислоты, и конкретно означает защитную группу, устойчивую в кислых и нейтральных условиях и расщепляемую с помощью химических методов, таких как гидрогенолиз, гидролиз, электролиз или фотолиз. Такой защитной группой может быть, например, «алифатическая ацильная группа», такая как алкилкарбонильная группа, например формил, ацетил, пропионил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, нонаноил, деканоил, 3-метилнонаноил, 8-метилнонаноил, 3-этилоктаноил, 3,7-диметилоктаноил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, 1-метилпентадеканоил, 14-метилпентадеканоил, 13,13-диметилтетрадеканоил, гептадеканоил, 15-метилгексадеканоил, октадеканоил, 1-метилгептадеканоил, нонадеканоил, эйкозаноил и генэйкозаноил; карбоксилированная алкилкарбонильная группа, например сукциноил, глутароил и адипоил; галоген(низший)алкилкарбонильная группа, например хлорацетил, дихлорацетил, трихлорацетил и трифторацетил; и (низший)алкокси(низший)алкилкарбонильная группа, например метоксиацетил; или ненасыщенная алкилкарбонильная группа, например (E)-2-метил-2-бутеноил;
«ароматическая ацильная группа», такая как арилкарбонильная группа, например бензоил, α-нафтоил и β-нафтоил; галогенарилкарбонильная группа, например 2-бромбензоил и 4-хлорбензоил; низшая алкилированная арилкарбонильная группа, например 2,4,6-триметилбензоил и 4-толуоил; низшая алкоксилированная арилкарбонильная группа, например 4-анизоил; карбоксилированная арилкарбонильная группа, например 2-карбоксибензоил, 3-карбоксибензоил и 4-карбоксибензоил; нитрованная арилкарбонильная группа, например 4-нитробензоил и 2-нитробензоил; низшая алкоксикарбонилированная арилкарбонильная группа, например 2-(метоксикарбонил)бензоил; или арилированная арилкарбонильная группа, например 4-фенилбензоил;
«низшая алкоксикарбонильная группа», такая как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и изобутоксикарбонил;
«низшая алкоксикарбонильная группа, замещенная галогеном или три(низший)алкилсилильной(ыми) группой(ами), такая как 2,2,2-трихлорэтоксикарбонил и 2-триметилсилилэтоксикарбонил;
«алкенилоксикарбонильная группа», такая как винилоксикарбонил и аллилоксикарбонил; или
«аралкилоксикарбонильная группа, арильное кольцо которой может быть замещено 1 или 2 низшими алкокси или нитрогруппами», такая как бензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 2-нитробензилоксикарбонил и 4-нитробензилоксикарбонил; и предпочтительно представляет собой «алифатическую ацильную группу» или «ароматическую ацильную группу», более предпочтительно бензоильную группу.
В приведенной выше общей формуле (1) «аминогруппа, замещенная алкильной(ыми) группой(ами), содержащей(ими) от 1 до 4 атомов углерода, которая(ые) может(гут) быть замещена(ы)» заместителей R1, R2, R3, R4, R5 и R6 или группы α может представлять собой, например, «низшую алкиламиногруппу», такую как метиламино, этиламино, пропиламино, изопропиламино, бутиламино, изобутиламино, втор-бутиламино, трет-бутиламино, диметиламино, диэтиламино, дипропиламино, диизопропиламино, дибутиламино, диизобутиламино, ди(втор-бутил)амино и ди(трет-бутил)аминогруппа;
«низшую алкиламиногруппу, замещенную гидроксильной(ыми) группой(ами), низшей(ими) алкоксигруппой(ами) или атомом(ами) галогена», такую как 1-гидроксиэтиламино, 2-гидроксиэтиламино, 1-метоксиэтиламино, 2-метоксиэтиламино, 1-бромэтиламино, 2-метоксиэтиламино, 1-хлорэтиламино и 2-хлорэтиламиногруппа; или
«низшую алкоксикарбониламиногруппу», такую как 1-метоксикарбонилэтиламино, 2-метоксикарбонилэтиламино,
1-этоксикарбонилэтиламино, 2-этоксикарбонилэтиламино,
1-пропоксикарбонилэтиламино и 1-пропоксикарбонилэтиламиногруппа; и предпочтительно представляет собой 1-гидроксиэтиламино, 2-гидроксиэтиламино, метиламино, этиламино, диметиламино, диэтиламино, диизопропиламино, 1-метоксикарбонилэтиламино или 1-этоксикарбонилэтиламиногруппу.
В приведенной выше общей формуле (1) «алкильная группа, содержащая от 1 до 6 атомов углерода, которая может быть замещена» заместителей D, R1, R2, R3, R4, R5, R6, R8 или группы α может представлять собой, например, «низшую алкильную группу», такую как метил, этил, п-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилфенил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил и 2-этилбутил;
«низшую алкильную группу, замещенную гидроксильной(ыми) группой(ами)», такую как 1-гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 4-гидроксибутил, 2-гидроксипропил, 1-метил-2-гидроксиэтил, 1-метил-1-гидроксиэтил, 1,1-диметил-2-гидроксиэтил, 2-гидроксибутил, 3-гидроксибутил, 1-метил-3-гидроксипропил и 2-метил-3-гидроксипропил;
«низшую алкильную группу, замещенную аминогруппой(ами)», такую как 1-аминометил, 2-аминоэтил, 3-аминопропил, 4-аминобутил, 2-аминопропил, 1-метил-2-аминоэтил, 1-метил-1-аминоэтил, 1,1-диметил-2-аминоэтил, 2-аминобутил, 3-аминобутил, 1-метил-3-аминопропил и 2-метил-3-аминопропил;
«низшую алкильную группу, замещенную алкоксигруппой(ами)», такую как 1-метоксиметил, 2-метоксиэтил, 3-метоксипропил, 4-метоксибутил, 2-метоксипропил, 1-метил-2-метоксиэтил, 1-метил-1-метоксиэтил, 1,1-диметил-2-метоксиэтил, 2-метоксибутил, 3-метоксибутил, 1-метил-3-метоксипропил, 2-метил-3-метоксипропил, 1-этоксиметил, 2-этоксиэтил, 3-этоксипропил, 4-этоксибутил, 2-этоксипропил, 1-метил-2-этоксиэтил, 1-метил-1-этоксиэтил, 1,1-диметил-2-этоксиэтил, 2-этоксибутил, 3-этоксибутил, 1-метил-3-этоксипропил и 2-метил-3-этоксипропил; или
«циклоалкильную группу», такую как циклопропил, циклобутил, циклопентил, циклогексил, циклопентил, норборнил и адамантил; и предпочтительно представляет собой 2-метоксиэтильную группу или 2-гидроксиэтильную группу.
В приведенной выше общей формуле (1) «алкильная группа, содержащая от 1 до 24 атомов углерода, которая может быть замещена» заместителя X1 может представлять собой, например, стеарил, 2,2-диметилстеарил, гептадецил, 2,2-диметилгептадецил, гексадецил, 2,2-диметилгексадецил, пентадецил, 2,2-диметилпентадецил, тетрадецил, 2,2-диметилтетрадецил, тридецил, 2,2-диметилтридецил, додецил, 2,2-диметилдодецил, ундецил, 2,2-диметилундецил, децил, 2,2-диметилдецил, нонил, 2,2-диметилнонил, октил, 2,2-диметилоктил, гептил, 2,2-диметилгептил, гексил, 2,2-диметилгексил, пентил, 2,2-диметилпентил, бутил, 2,2-диметилбутил, пропил, трет-бутил, этил или метил, и предпочтительно представляет собой стеарил или 2,2-диметилстеарил.
В приведенной выше общей формуле (1) «алкиленовая группа, содержащая от 1 до 6 атомов углерода, которая может быть замещена» заместителя X3 может представлять собой, например, метилен, этилен, пропилен, бутилен, 2,2-диметилэтилен, 2,2-диметилпропилен или 2,2-диметилбутилен, и предпочтительно представляет собой метилен или этилен.
В приведенной выше общей формуле (1) «арилоксигруппа, которая может быть замещена» заместителя R1 может представлять собой, например «арилоксигруппу, замещенную низшей(ими) алкильной(ыми) группой(ами), атомом(ами) галогена или нитрогруппой(ами)», такую как 2-метилфенокси, 3-метилфенокси, 4-метилфенокси, 2,6-диметилфенокси, 2-хлорфенокси, 4-хлорфенокси, 2,4-дихлорфенокси, 2,5-дихлорфенокси, 2-бромфенокси, 4-нитрофенокси и 4-хлор-2-нитрофеноксигруппа.
В приведенной выше общей формуле (1) «арильная группа, которая может быть замещена» заместителя R8 или X1 может представлять собой, например, «арильную группу, замещенную низшей(ими) алкильной(ыми) группой(ами), атомом(ами) галогена или нитрогруппой(ами)», такую как 2-метилфенил, 3-метилфенил, 4-метилфенил, 2,6-диметилфенил, 2-хлорфенил, 4-хлорфенил, 2,4-дихлорфенил, 2,5-дихлорфенил, 2-бромфенил, 4-нитрофенил и 4-хлор-2-нитрофенил.
В приведенной выше общей формуле (1) «арилтиогруппа, которая может быть замещена» заместителя R1 может представлять собой, например, «арилтиогруппу, замещенную низшей(ими) алкильной(ыми) группой(ами), атомом(ами) галогена или нитрогруппой(ами)», такую как 2-метилфенилтио,
3-метилфенилтио, 4-метилфенилтио, 2,6-диметилфенилтио,
2-хлорфенилтио, 4-хлорфенилтио, 2,4-дихлорфенилтио,
2,5-дихлорфенилтио, 2-бромфенилтио, 4-нитрофенилтио и
4-хлор-2-нитрофенилтиогруппа.
В приведенной выше общей формуле (1) «алкенильная группа, содержащая от 2 до 6 атомов углерода, которая может быть замещена» заместителя D может представлять собой, например, этенил, 1-пропенил, 2-пропенил, 1-метил-2-пропенил,
1-метил-1-пропенил, 2-метил-1-пропенил, 2-метил-2-пропенил,
2-этил-2-пропенил, 1-бутенил, 2-бутенил, 1-метил-2-бутенил,
1-метил-1-бутенил, 3-метил-2-бутенил, 1-этил-2-бутенил,
3-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил,
1-этил-3-бутенил, 1-пентенил, 2-пентенил, 1-метил-2-пентенил,
2-метил-2-пентенил, 3-пентенил, 1-метил-3-пентенил,
2-метил-3-пентенил, 4-пентенил, 1-метил-4-пентенил,
2-метил-4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил,
4-гексенил или 5-гексенил.
В приведенной выше общей формуле (1) «аралкильная группа, которая может быть замещена» заместителя R8 или X1 может представлять собой, например, «аралкильную группу», такую как бензил, α-нафтилметил, β-нафтилметил, инденилметил, фенантренилметил, антраценилметил, дифенилметил, трифенилметил, 1-фенетил, 2-фенетил, 1-нафтилэтил, 2-нафтилэтил, 1-фенилпропил, 2-фенилпропил, 3-фенилпропил, 1-нафтилпропил, 2-нафтилпропил, 3-нафтилпропил, 1-фенилбутил, 2-фенилбутил, 3-фенилбутил, 4-фенилбутил, 1-нафтилбутил, 2-нафтилбутил, 3-нафтилбутил, 4-нафтилбутил, 1-фенилпентил, 2-фенилпентил, 3-фенилпентил, 4-фенилпентил, 5-фенилпентил, 1-нафтилпентил, 2-нафтилпентил, 3-нафтилпентил, 4-нафтилпентил, 5-нафтилпентил, 1-фенилгексил, 2-фенилгексил, 3-фенилгексил, 4-фенилгексил, 5-фенилгексил, 6-фенилгексил, 1-нафтилгексил, 2-нафтилгексил, 3-нафтилгексил, 4-нафтилгексил, 5-нафтилгексил и 6-нафтилгексил; или «аралкильную группу, арильное кольцо которой замещено нитрогруппой(ами) или атомом(ами) галогена», такую как 4-хлорбензил, 2-(4-нитрофенил)этил, o-нитробензил, 4-нитробензил, 2,4-динитробензил и 4-хлор-2-нитробензил.
В приведенной выше общей формуле (1) из всех «пурин-9-ильных групп» и «замещенных пурин-9-ильных групп» заместителя B предпочтительными группами являются 6-аминопурин-9-ильная группа (то есть аденинил), 6-аминопурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-бромпурин-9-ильная группа, 6-амино-8-бромпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-хлорпурин-9-ильная группа, 6-амино-8-хлорпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-фторпурин-9-ильная группа, 6-амино-8-фторпурин-9-ильная, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-метоксипурин-9-ильная группа, 6-амино-8-метоксипурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-этоксипурин-9-ильная группа, 6-амино-8-этоксипурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-8-трет-бутоксипурин-9-ильная группа, 6-амино-8-трет-бутоксипурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 2,6-диаминопурин-9-ильная группа, 2-амино-6-хлорпурин-9-ильная группа, 2-амино-6-хлорпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 2-амино-6-фторпурин-9-ильная группа, 2-амино-6-фторпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 2-амино-6-бромпурин-9-ильная группа, 2-амино-6-бромпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 2-амино-6-гидроксипурин-9-ильная группа (то есть гуанинил), 2-амино-6-гидроксипурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-2-метоксипурин-9-ильная группа, 6-амино-2-метоксипурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-2-хлорпурин-9-ильная группа, 6-амино-2-хлорпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 6-амино-2-фторпурин-9-ильная группа, 6-амино-2-фторпурин-9-ильная группа, в которой аминогруппа защищена защитной группой синтеза нуклеиновой кислоты, 2,6-диметоксипурин-9-ильная, 2,6-дихлорпурин-9-ильная и 6-меркаптопурин-9-ильная группа, и более предпочтительными группами являются 6-бензоиламинопурин-9-ил или аденинил.
Отсутствуют особые ограничения по функциональной группе, представленной формулой "X1-X2-X3-S", при условии, что она представляет собой комбинацию, содержащую X1, X2, X3 и S, приведенных выше, и она может представлять собой, например, ацилоксиалкилтиогруппу, такую как 2-(стеароилокси)этилтио,
2-(миристоилокси)этилтио, 2-(деканоилокси)этилтио,
2-(бензоилокси)этилтио, 2-(пивалоилокси)этилтио,
2-(2,2-диметилоктадеканоилокси)этилтио,
3-(стеароилокси)пропилтио, 3-(миристоилокси)пропилтио,
3-(деканоилокси)пропилтио, 3-(бензоилокси)пропилтио,
3-(пивалоилокси)пропилтио,
3-(2,2-диметилоктадеканоилокси)пропилтио,
4-(стеароилокси)бутилтио, 4-(миристоилокси)бутилтио,
4-(деканоилокси)бутилтио, 4-(бензоилокси)бутилтио,
4-(пивалоилокси)бутилтио и
4-(2,2-диметилоктадеканоилокси)бутилтиогруппа, или
алкилкарбамоилоксиалкилтиогруппу, такую как
2-(стеарилкарбамоилокси)этилтиогруппа, или следующие соединения:
и предпочтительной является 2-стеароилоксиэтилтио или 2-(2,2-диметилоктадеканоилокси)этилтиогруппа.
В приведенной выше общей формуле (1) «атом галогена» группы α может представлять собой, например, атом фтора, атом хлора, атом брома или атом йода и предпочтительно представляет собой атом брома или атом хлора.
«Аналог 2',5'-олигоаденилата (аналог 2-5A)» означает производное неприродного типа «2',5'-олигоаденилата», в котором положение 2' и положение 5' «нуклеозидов» 3 или 4, являющихся одинаковыми или различными, связаны фосфодисложноэфирной связью или модифицированной фосфодисложноэфирной связью, и фосфорильное производное связано с 5'-концом, или фосфорильное производное необязательно связано с 2'-концом, или 5'-фосфорилированный аналог олигонуклеотида необязательно связан с 2'-концом через алкиленовый линкер. Такой аналог предпочтительно может представлять собой производное сахара, в котором модифицирован фрагмент сахара; тиоатное производное, где фосфодисложноэфирный связывающий фрагмент тионирован; фосфорильное производное, где фрагмент фосфорной кислоты на конце замещен; или пуриновое производное, где пуриновое основание замещено; и более предпочтительно представляет собой фосфорильное производное, где фрагмент фосфорной кислоты на конце замещен, производное сахара, где фрагмент сахара модифицирован, или тиоатное производное, где фосфодисложноэфирный связывающий фрагмент тионирован.
«5'-Фосфорилированный аналог олигонуклеотида, в котором одна гидроксильная группа удалена из группы 5'-фосфорной кислоты» означает производное неприродного типа «олигонуклеотида», в котором от 2 до 50 «нуклеозидов», являющихся одинаковыми или различными, связаны с помощью фосфодисложноэфирных связей, и означает производное, содержащее следующую остаточную группу:
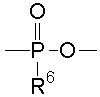
(где R6 имеет такие же значения, как определено выше) вместо гидроксильной группы у 5'-конца олигонуклеотида.
Такой аналог предпочтительно может быть производным сахара, где фрагмент сахара модифицирован; тиоатным производным, где фосфодисложноэфирный связывающий фрагмент тионирован; сложным эфиром, где фрагмент фосфорной кислоты на конце этерифицирован; или амидом, где аминогруппа на пуриновом основании амидирована; и более предпочтительно представляет собой производное сахара, в котором фрагмент сахара модифицирован, или тиоатное производное, в котором фосфодисложноэфирный связывающий фрагмент тионирован.
Выражение «его соль» означает соль соединения (1) настоящего изобретения, так как соединение может быть превращено в соль. Такая соль предпочтительно может представлять собой соль металла, такую как соль щелочного металла, например соль натрия, соль калия и соль лития; соль щелочно-земельного металла, например соль кальция и соль магния; соль алюминия, соль железа, соль цинка, соль меди, соль никеля или соль кобальта; аминовую соль, такую как неорганическая соль, например соль аммония; или органическая соль, например трет-октиламиновую соль, дибензиламиновую соль, морфолиновую соль, глюкозаминовую соль, соль сложного алкилового эфира фенилглицина, этилендиаминовую соль, N-метилглюкаминовую соль, гуанидиновую соль, диэтиламиновую соль, триэтиламиновую соль, дициклогексиламиновую соль, N,N'-дибензилэтилендиаминовую соль, хлорпрокаиновую соль, прокаиновую соль, диэтаноламиновую соль, N-бензилфенетиламиновую соль, пиперазиновую соль, тетраметиламмониевую соль и соль трис(гидроксиметил)аминометана; соль неорганической кислоты, такую как соль галогеноводорода, например гидрофторид, гидрохлорид, гидробромид и гидройодид; нитрат, перхлорат, сульфат или фосфат; или соль органической кислоты, такую как низший алкансульфонат, например метансульфонат, трифторметансульфонат и этансульфонат; арилсульфонат, например бензолсульфонат и п-толуолсульфонат; ацетат, малат, фумарат, сукцинат, цитрат, тартрат, оксалат или малеат; или соль аминокислоты, такую как соль глицина, соль лизина, соль аргинина, соль орнитина, глутамат или аспартат.
Выражение «его фармакологически приемлемая соль» означает соль аналога 2-5A настоящего изобретения, поскольку он может быть превращен в соль. Такая соль предпочтительно может представлять собой соль металла, такую как соль щелочного металла, например соль натрия, соль калия и соль лития; соль щелочно-земельного металла, например соль кальция и соль магния; соль алюминия, соль железа, соль цинка, соль меди, соль никеля или соль кобальта; аминовую соль, такую как неорганическая соль, например соль аммония; или органическая соль, например трет-октиламиновую соль, дибензиламиновую соль, морфолиновую соль, глюкозаминовую соль, соль сложного алкилового эфира фенилглицина, этилендиаминовую соль, N-метилглюкаминовую соль, гуанидиновую соль, диэтиламиновую соль, триэтиламиновую соль, дициклогексиламиновую соль, N,N'-дибензилэтилендиаминовую соль, хлорпрокаиновую соль, прокаиновую соль, диэтаноламиновую соль, N-бензилфенетиламиновую соль, пиперазиновую соль, тетраметиламмониевую соль и соль трис(гидроксиметил)аминометана; соль неорганической кислоты, такую как соль галогеноводорода, например гидрофторид, гидрохлорид, гидробромид и гидройодид; нитрат, перхлорат, сульфат или фосфат; или соль органической кислоты, такую как низший алкансульфонат, например метансульфонат, трифторметансульфонат и этансульфонат; арилсульфонат, например бензолсульфонат и п-толуолсульфонат; ацетат, малат, фумарат, сукцинат, цитрат, тартрат, оксалат или малеат; или соль аминокислоты, такую как соль глицина, соль лизина, соль аргинина, соль орнитина, глутамат или аспартат.
Конкретные соединения, относящиеся к соединениям приведенной выше формулы (1) настоящего изобретения, представлены в таблице 1. Однако соединения настоящего изобретения не ограничены только ими.
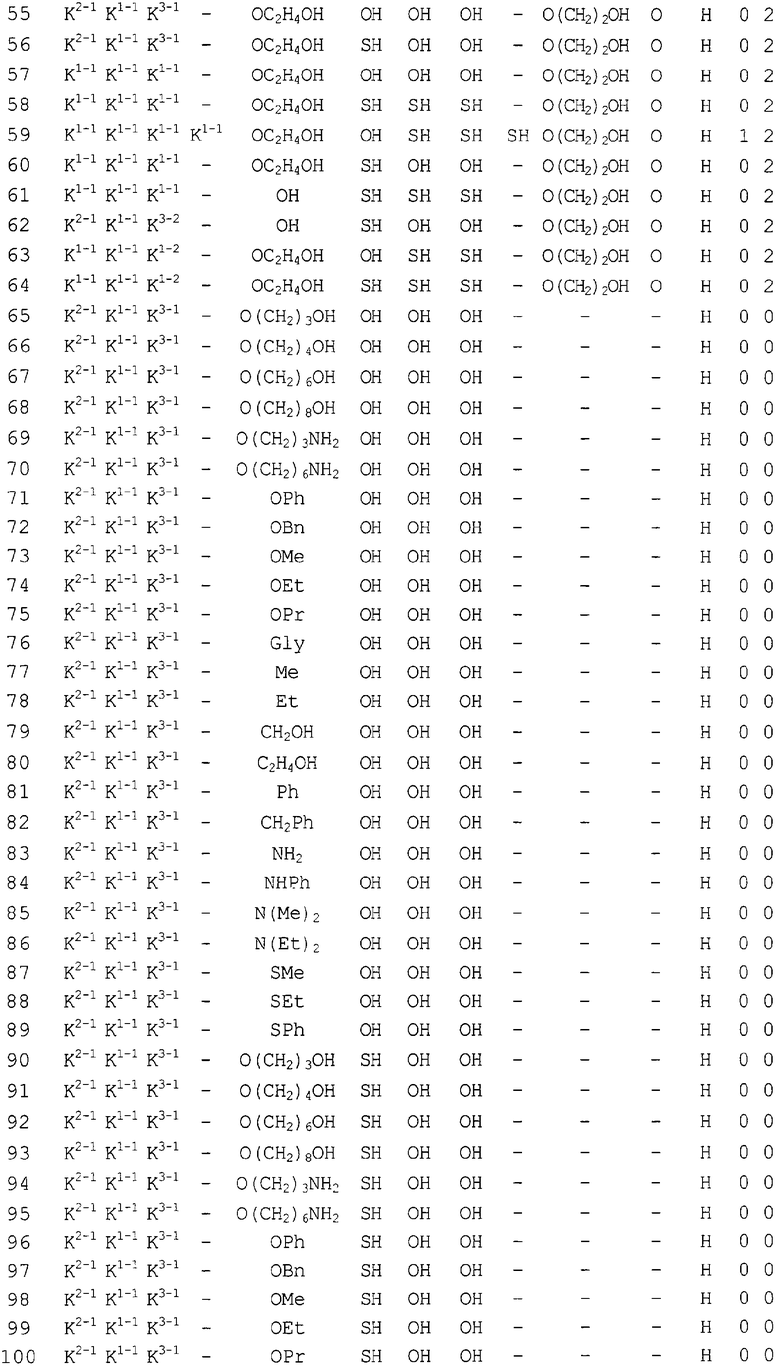
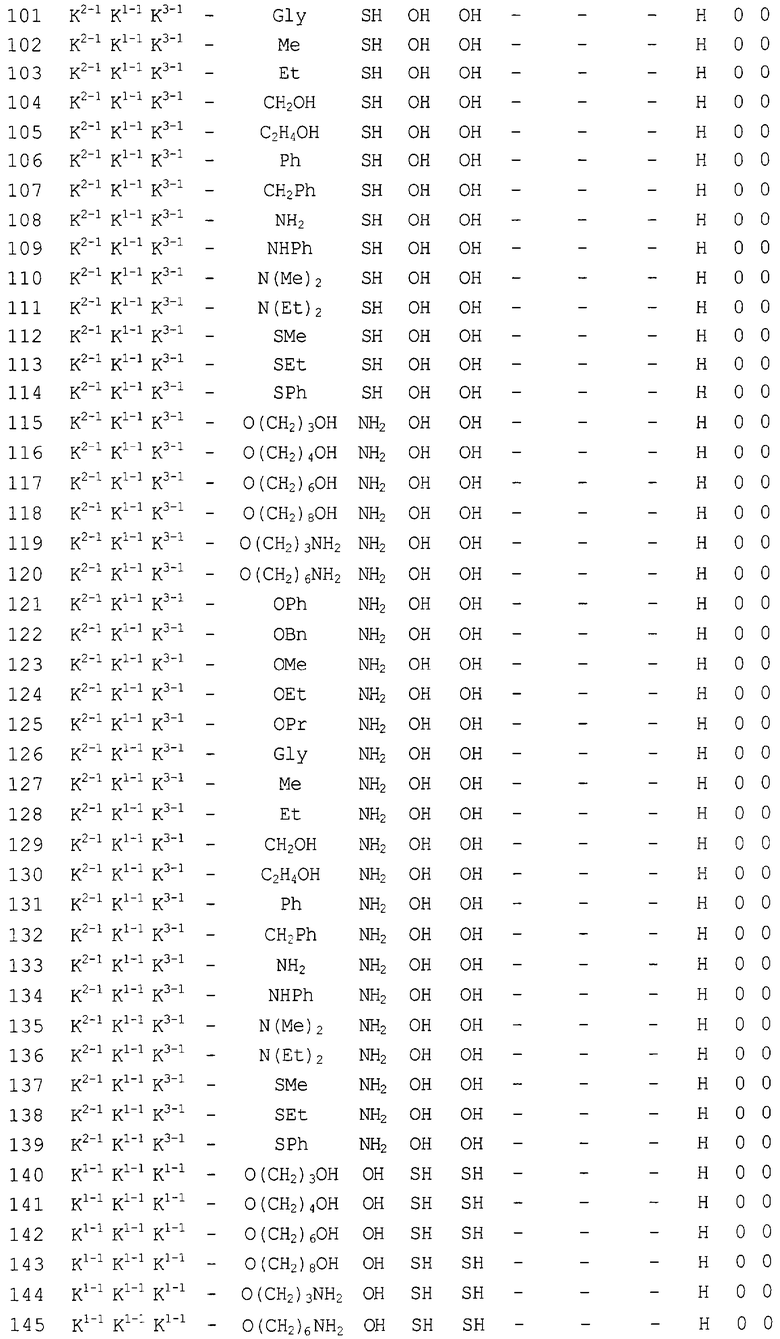
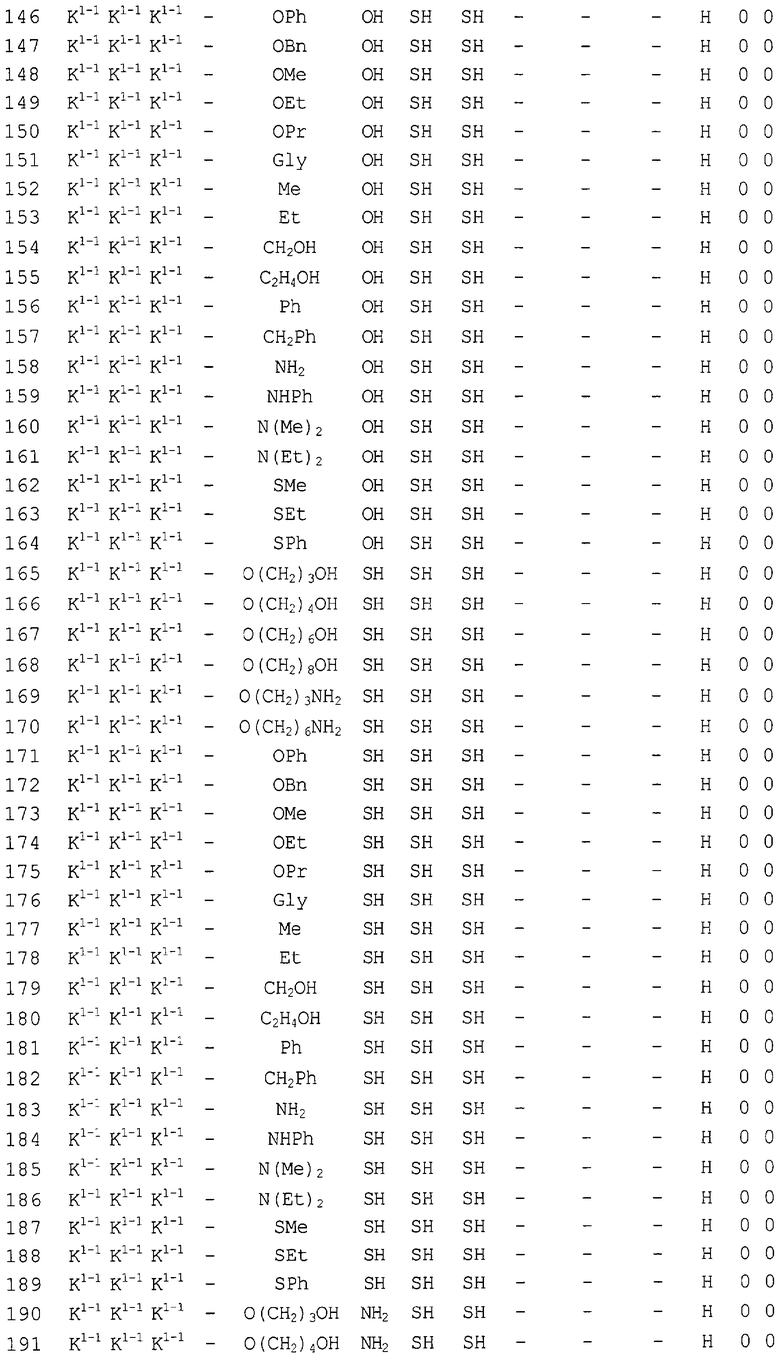
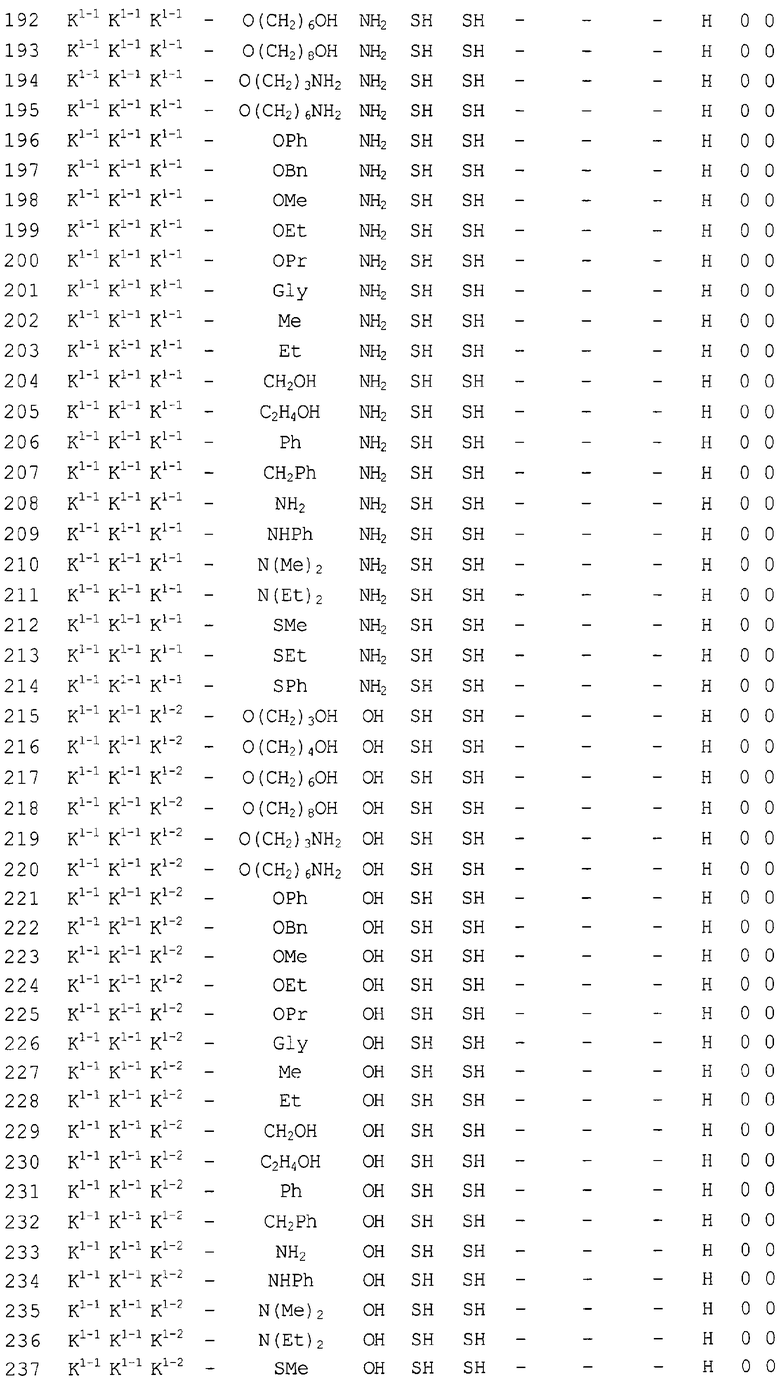
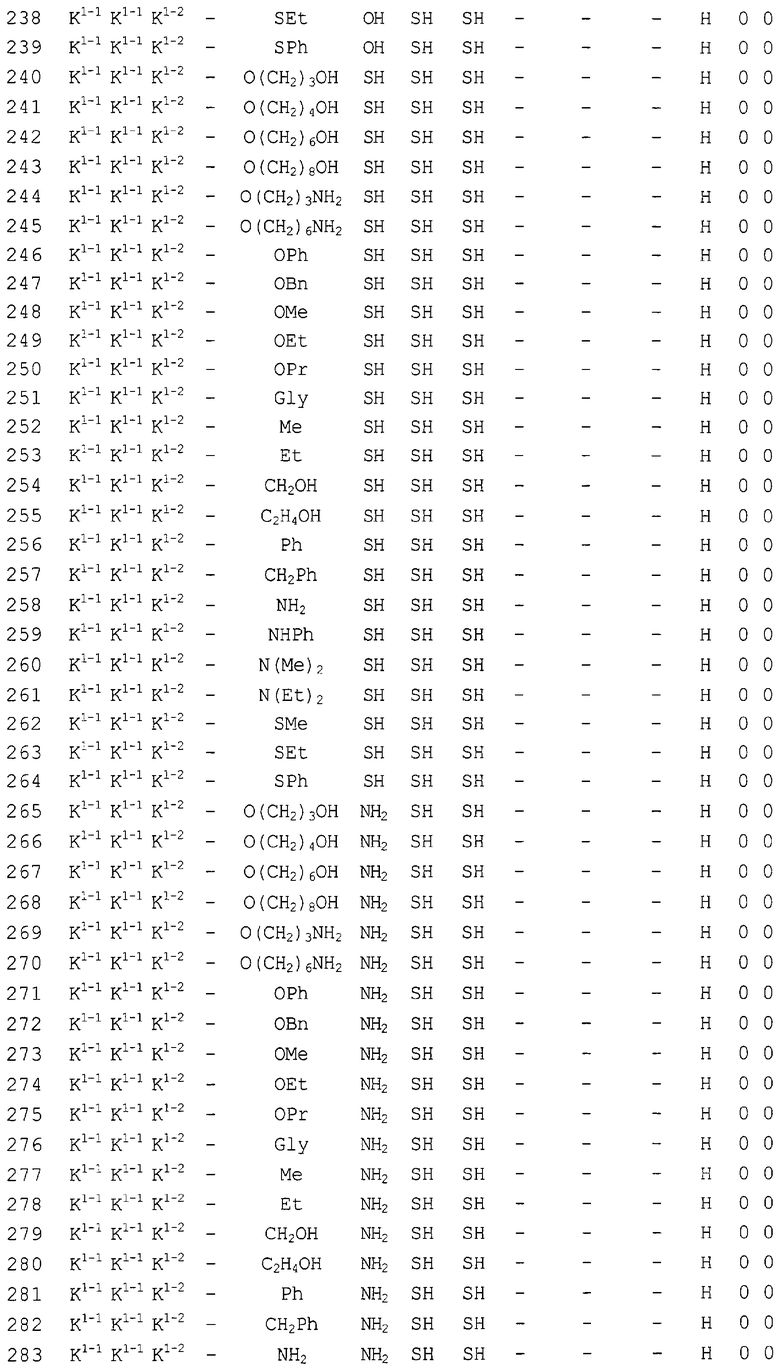
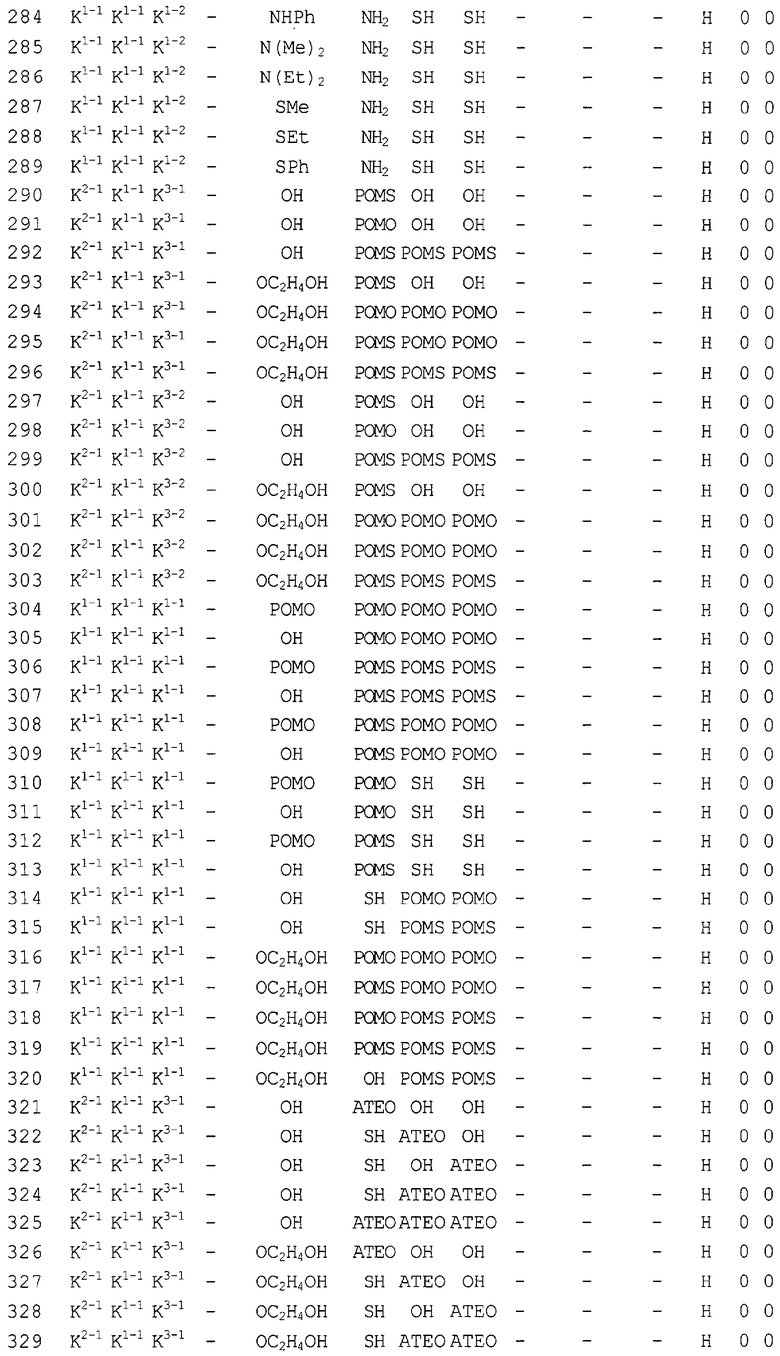
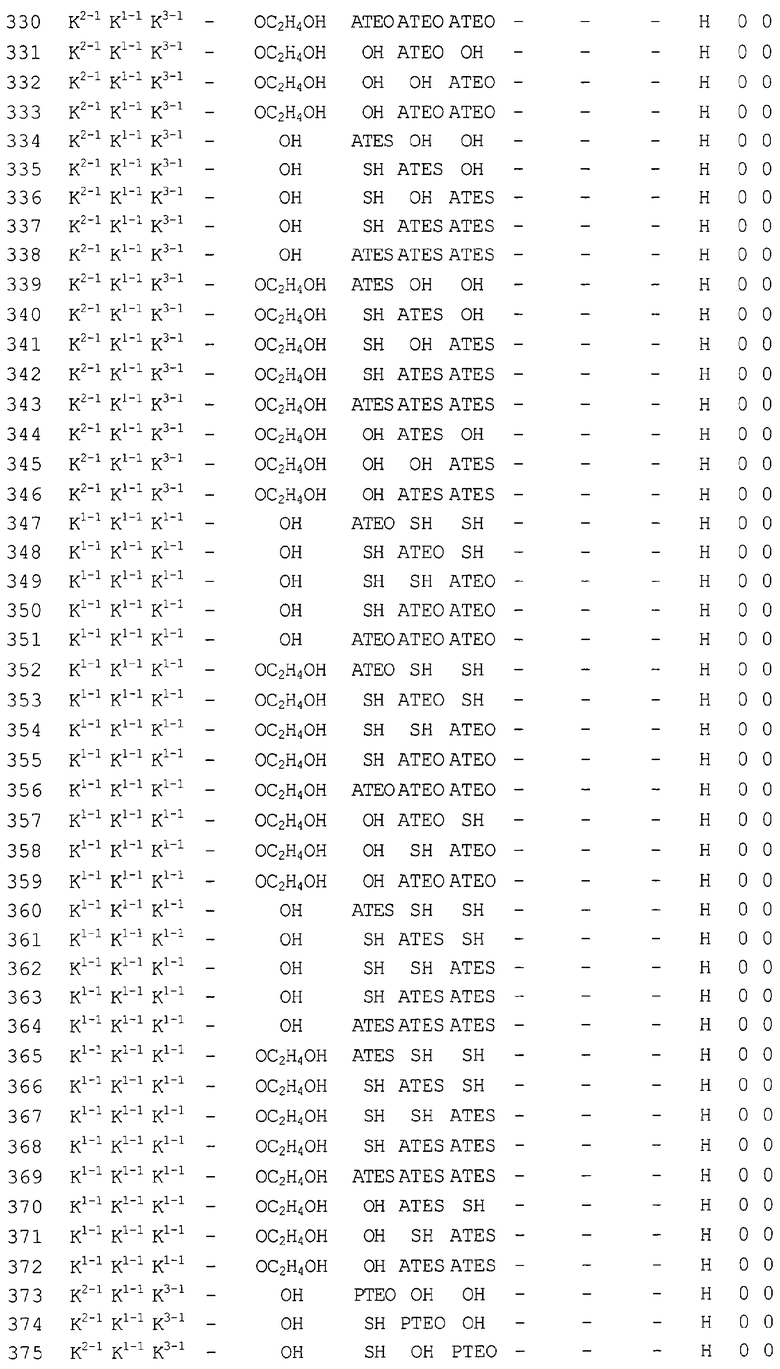
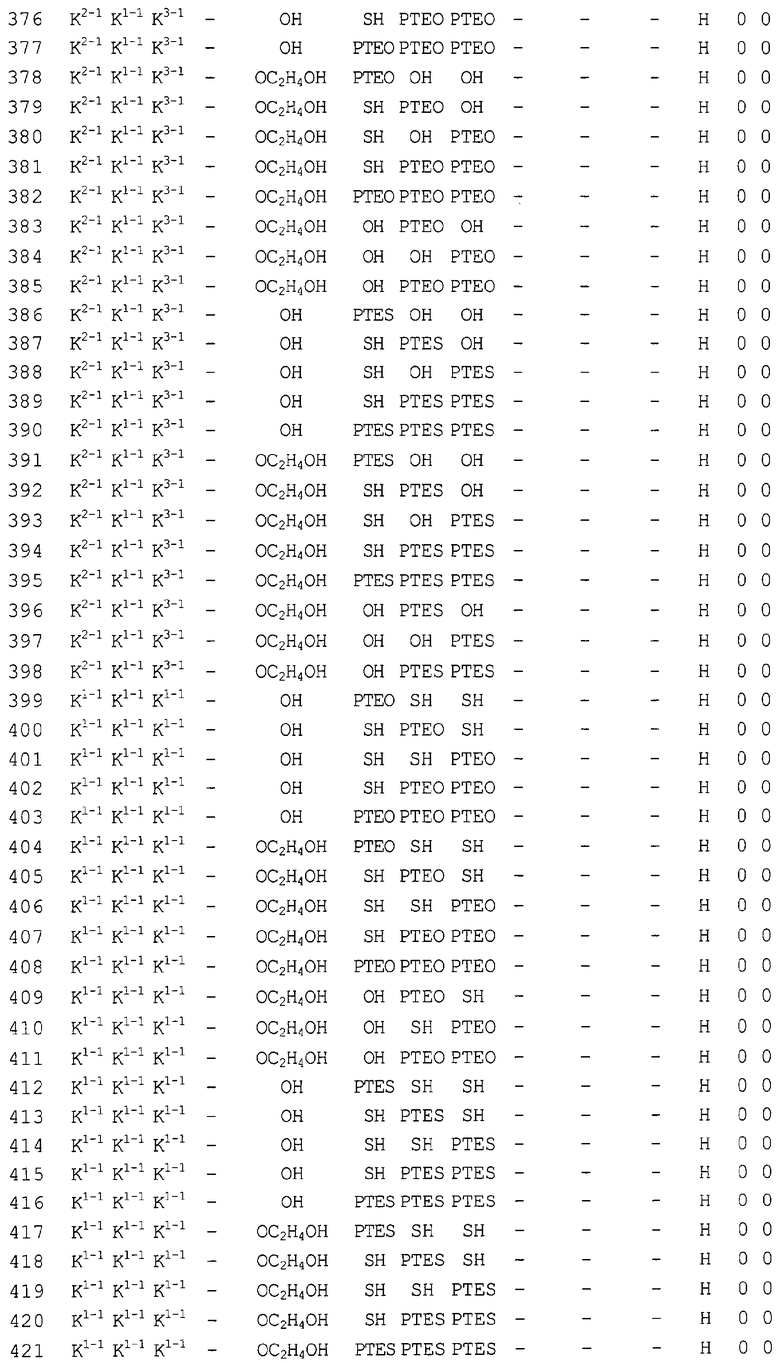








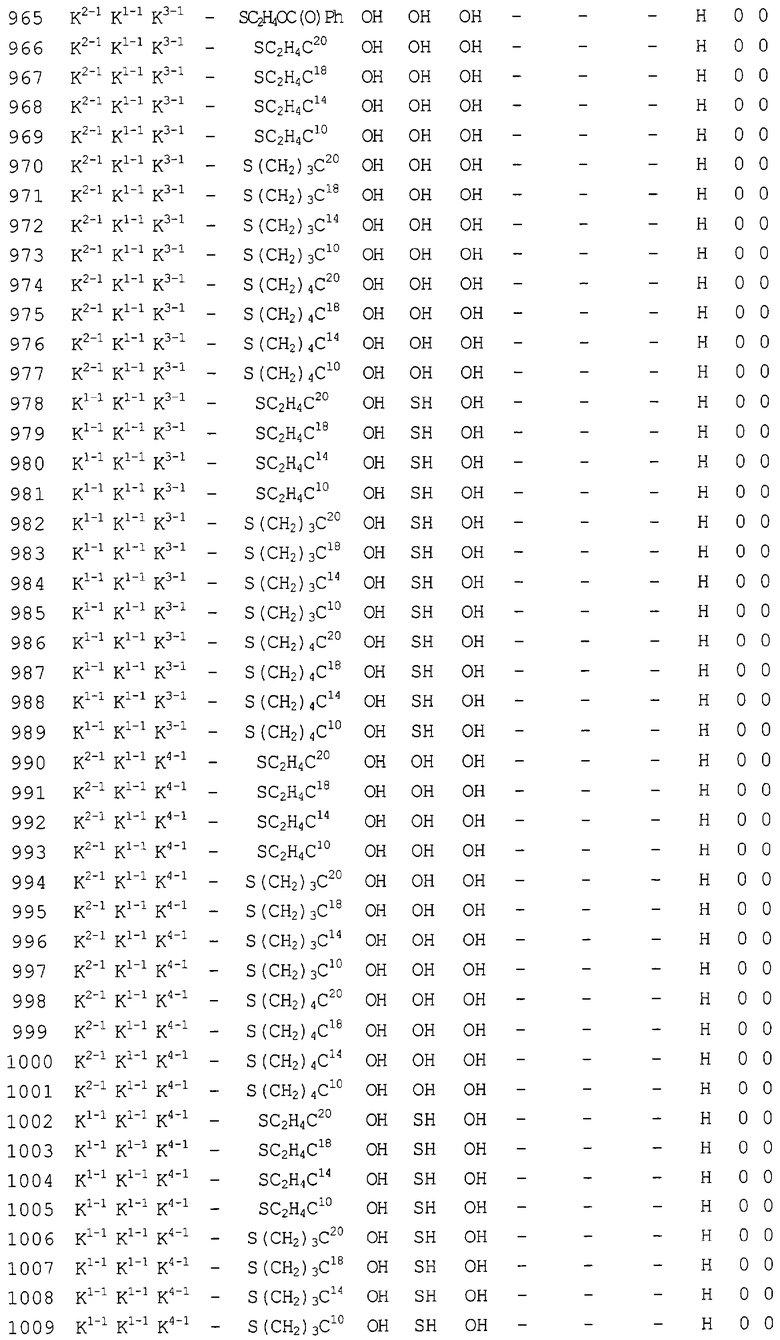
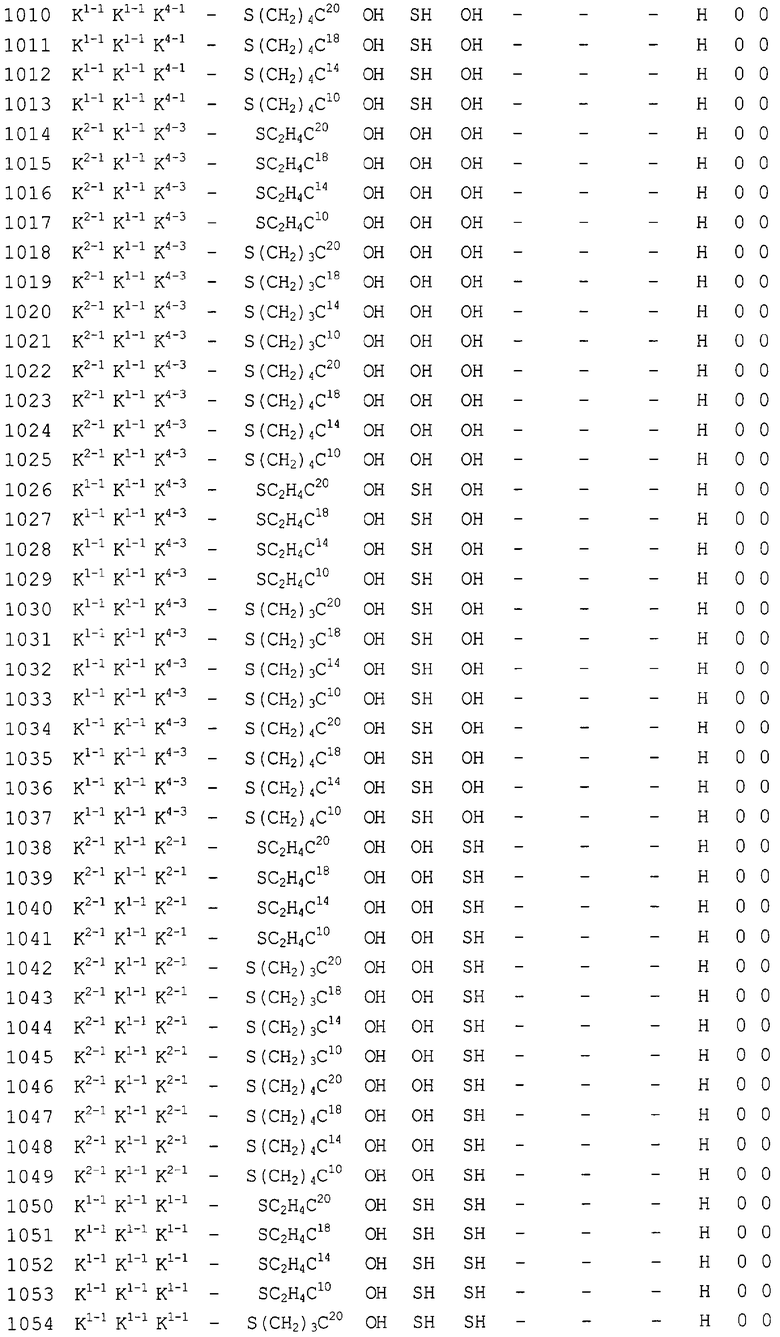
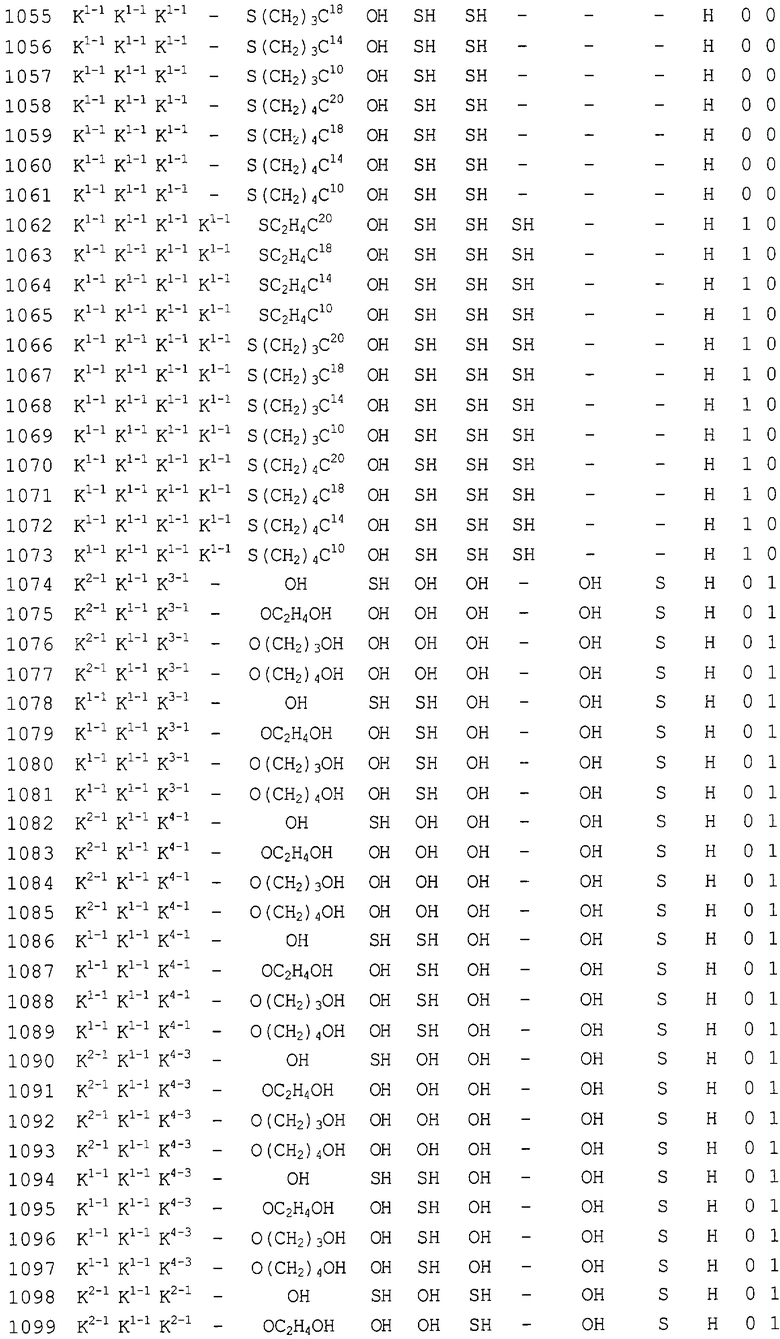
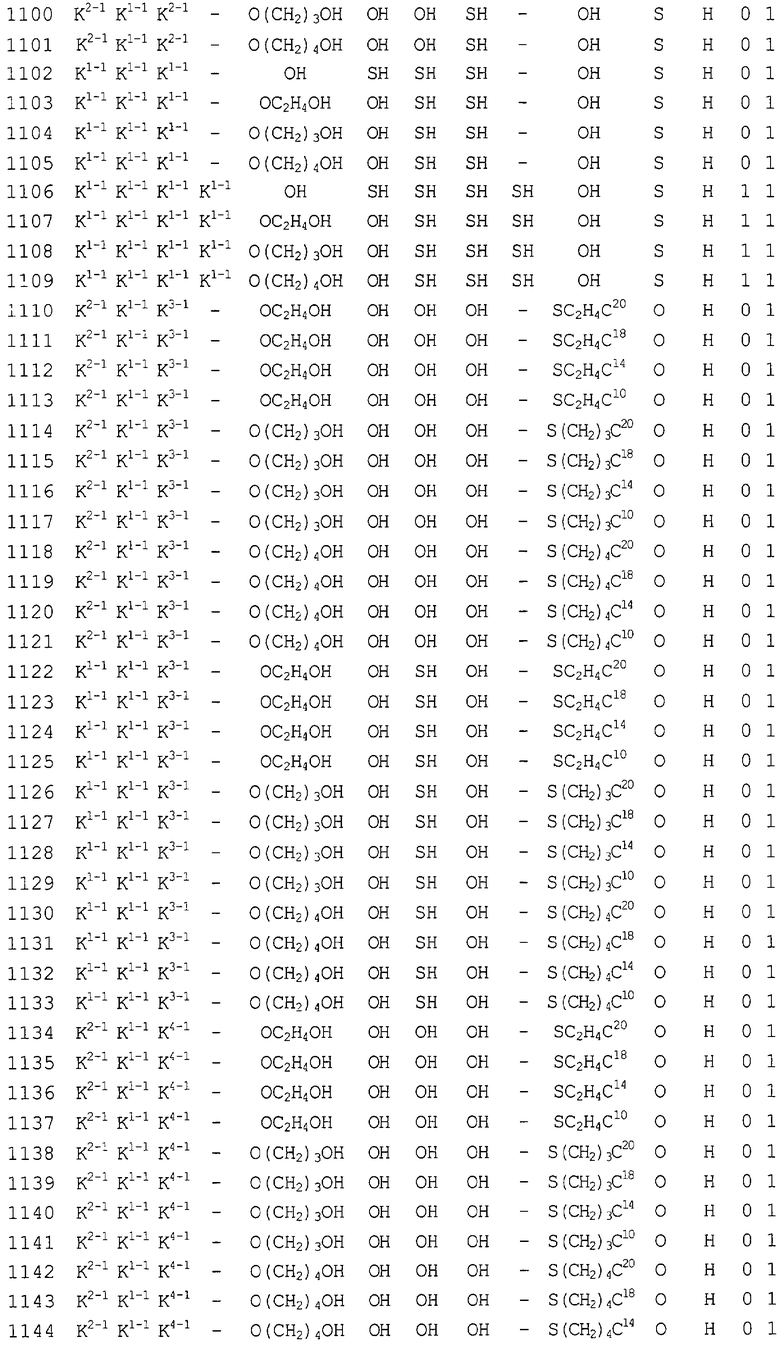
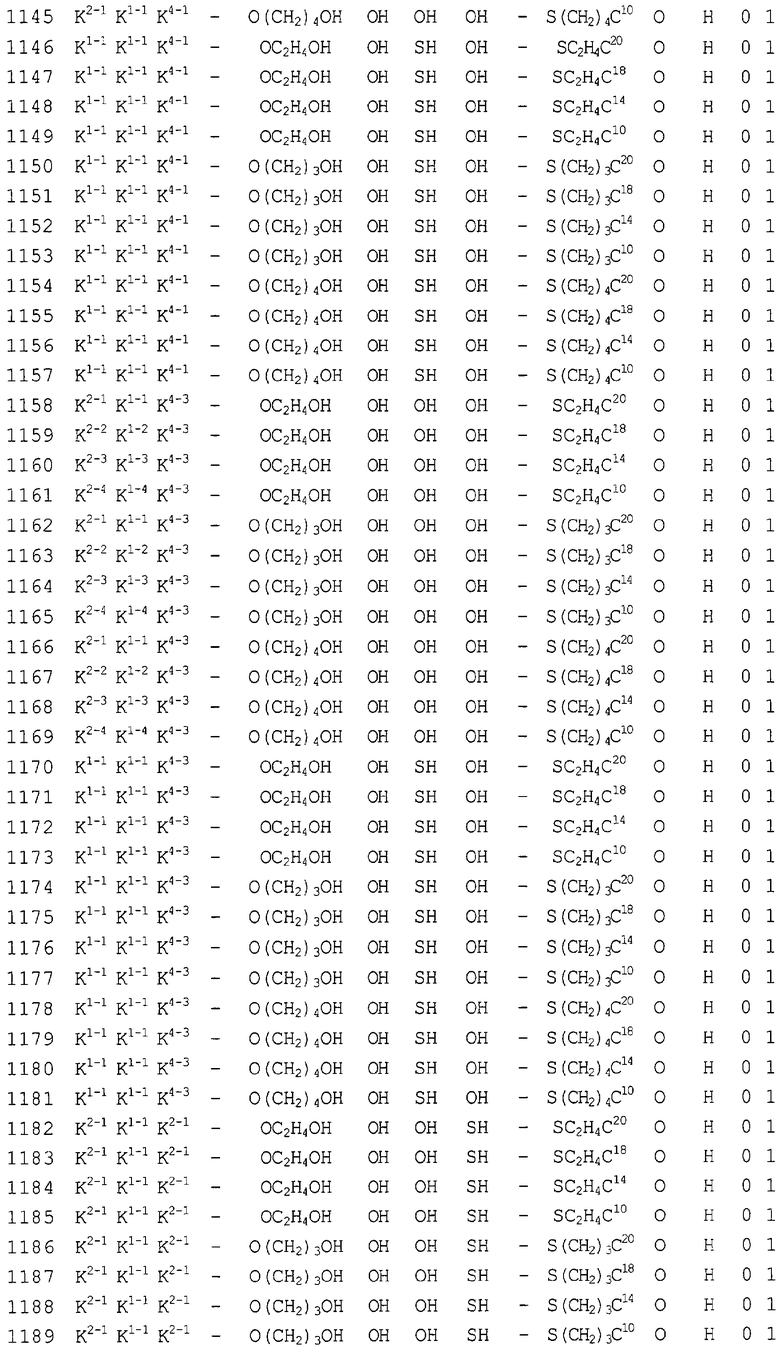
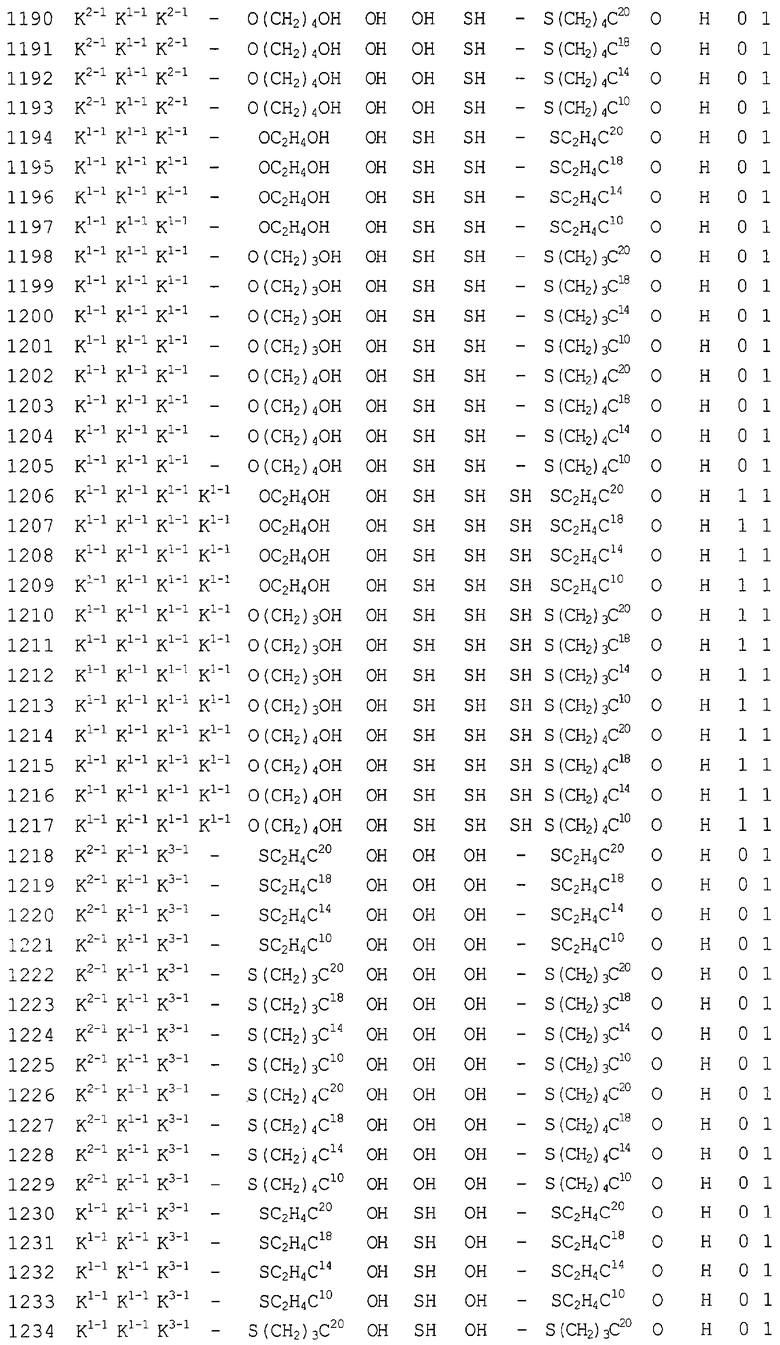
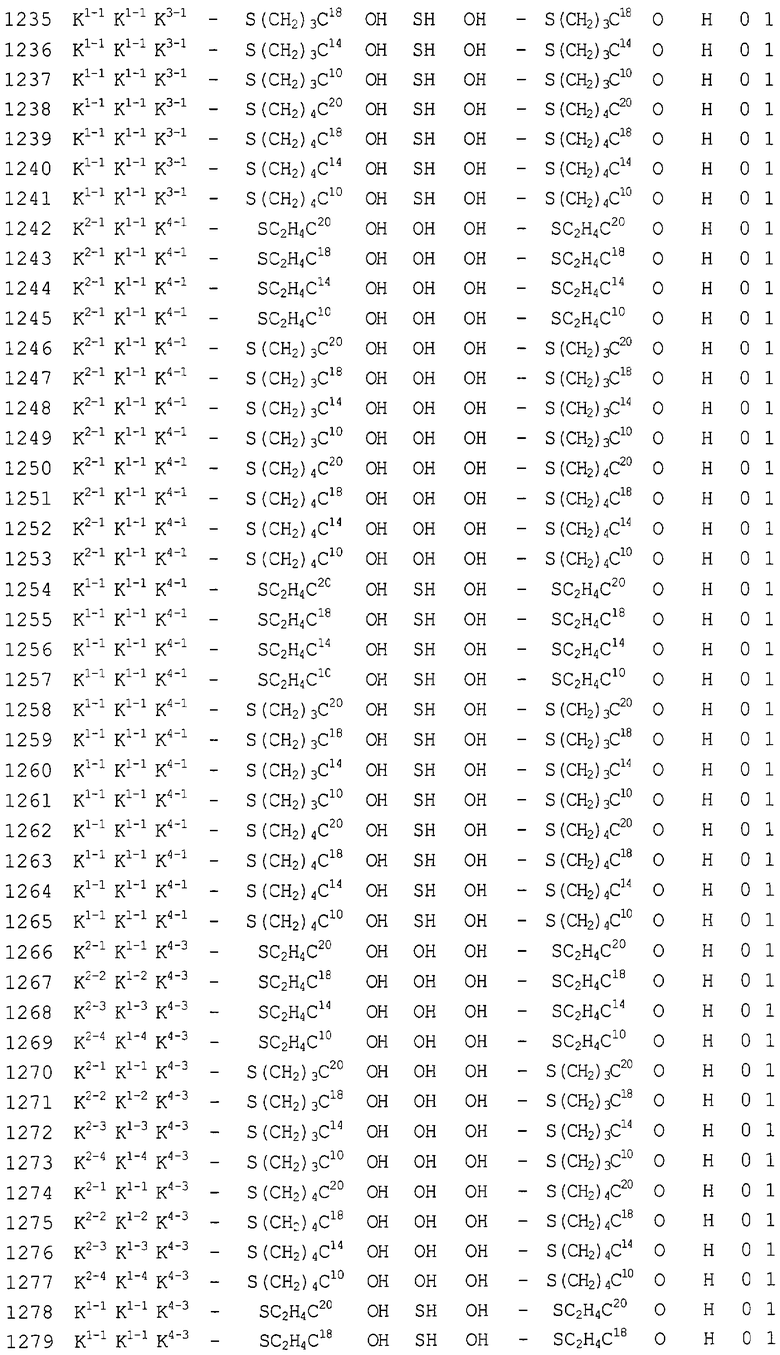
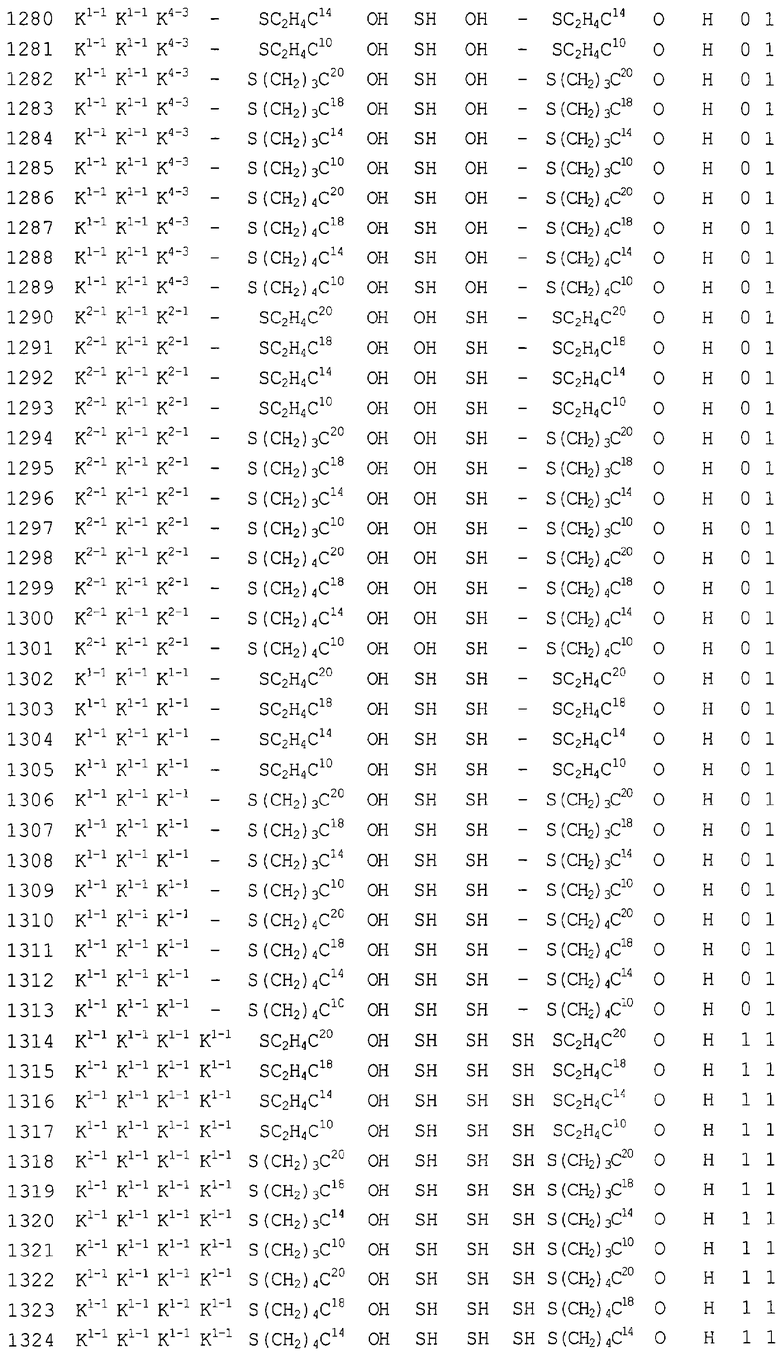
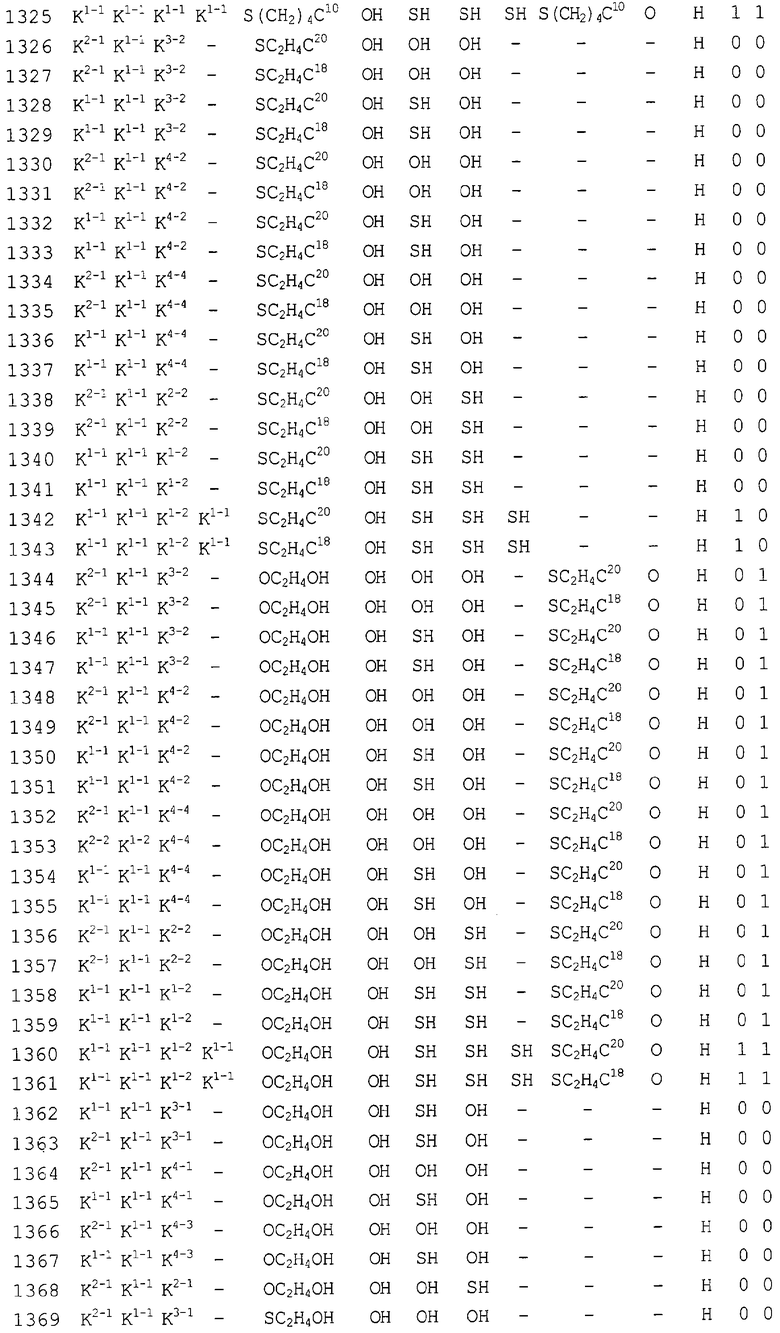
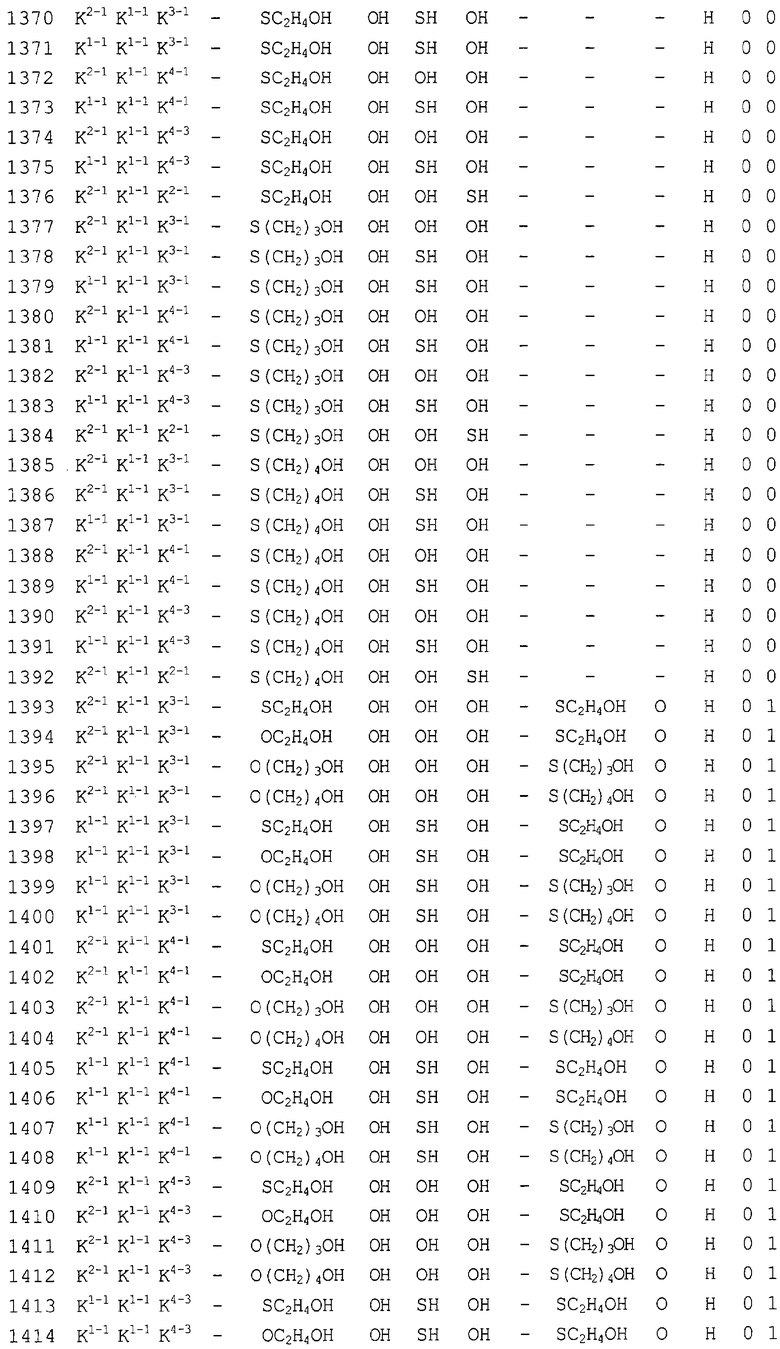
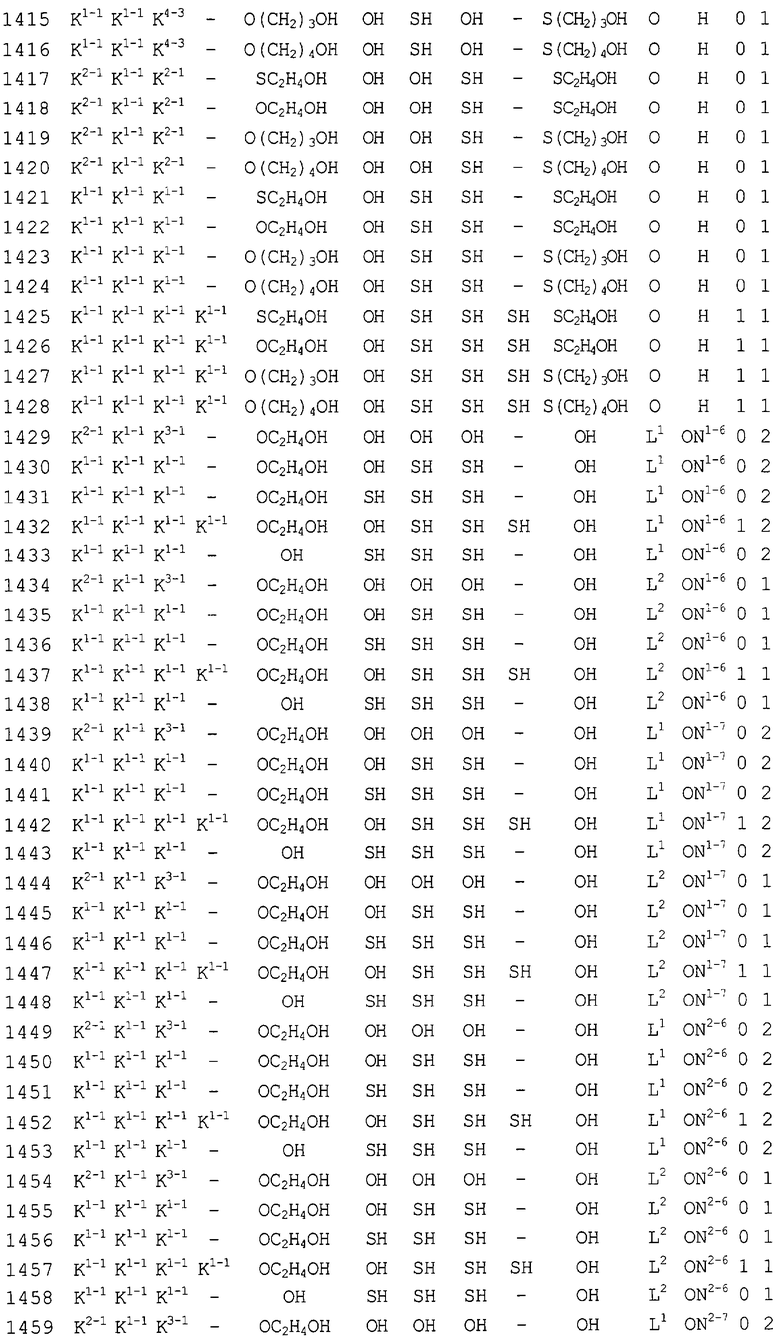
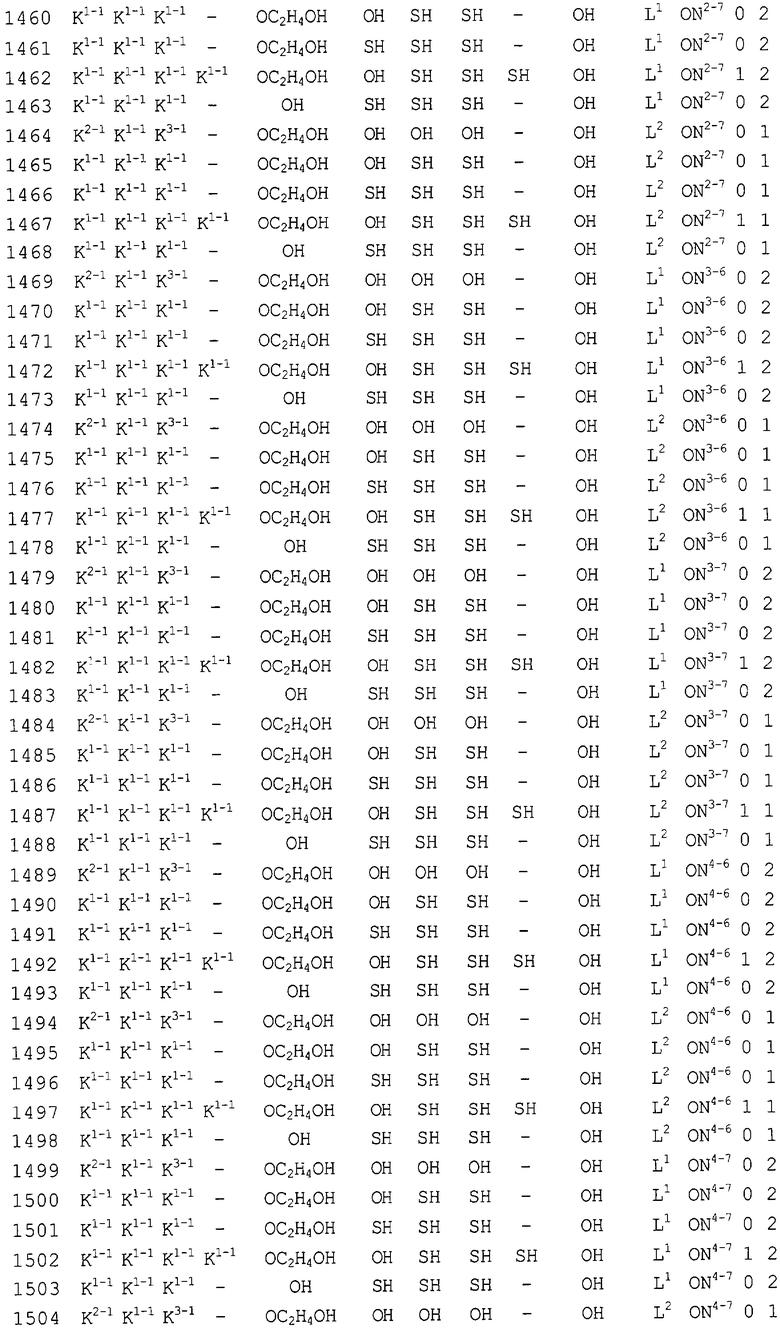
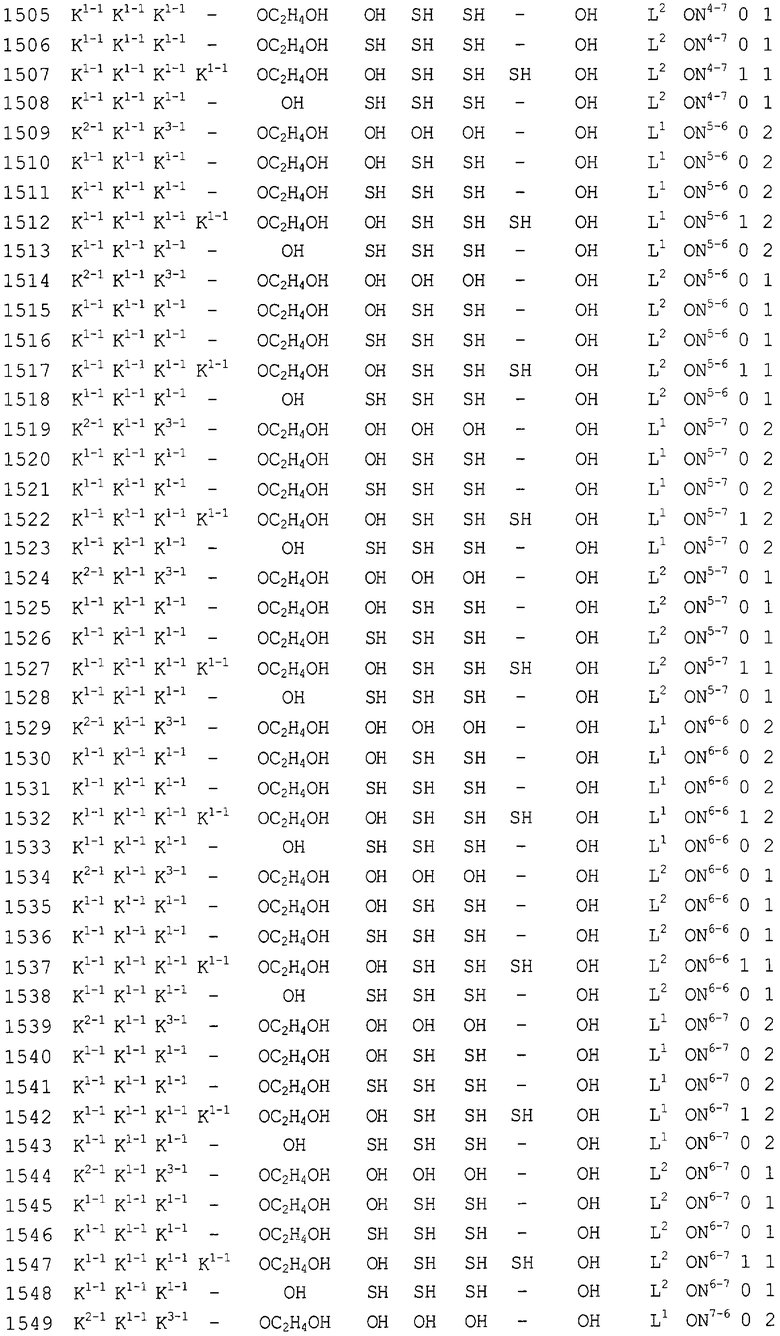
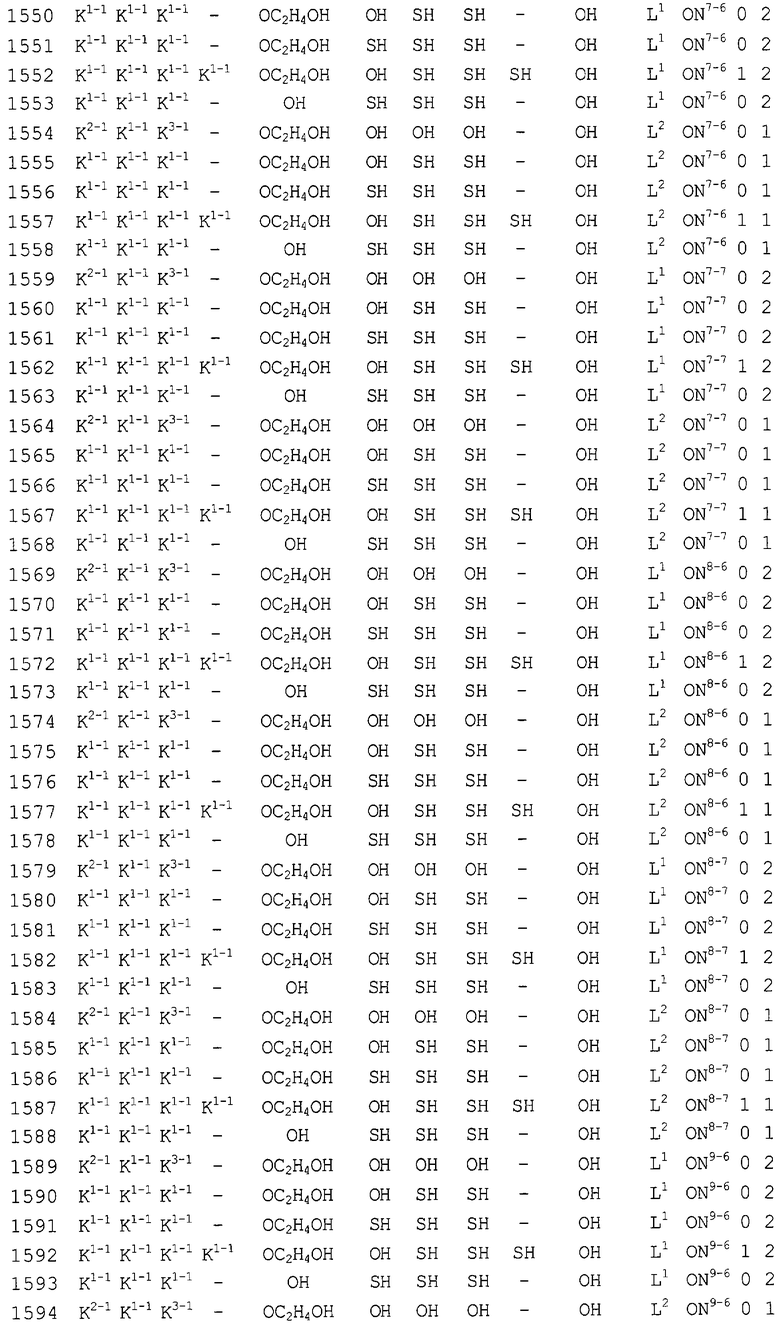

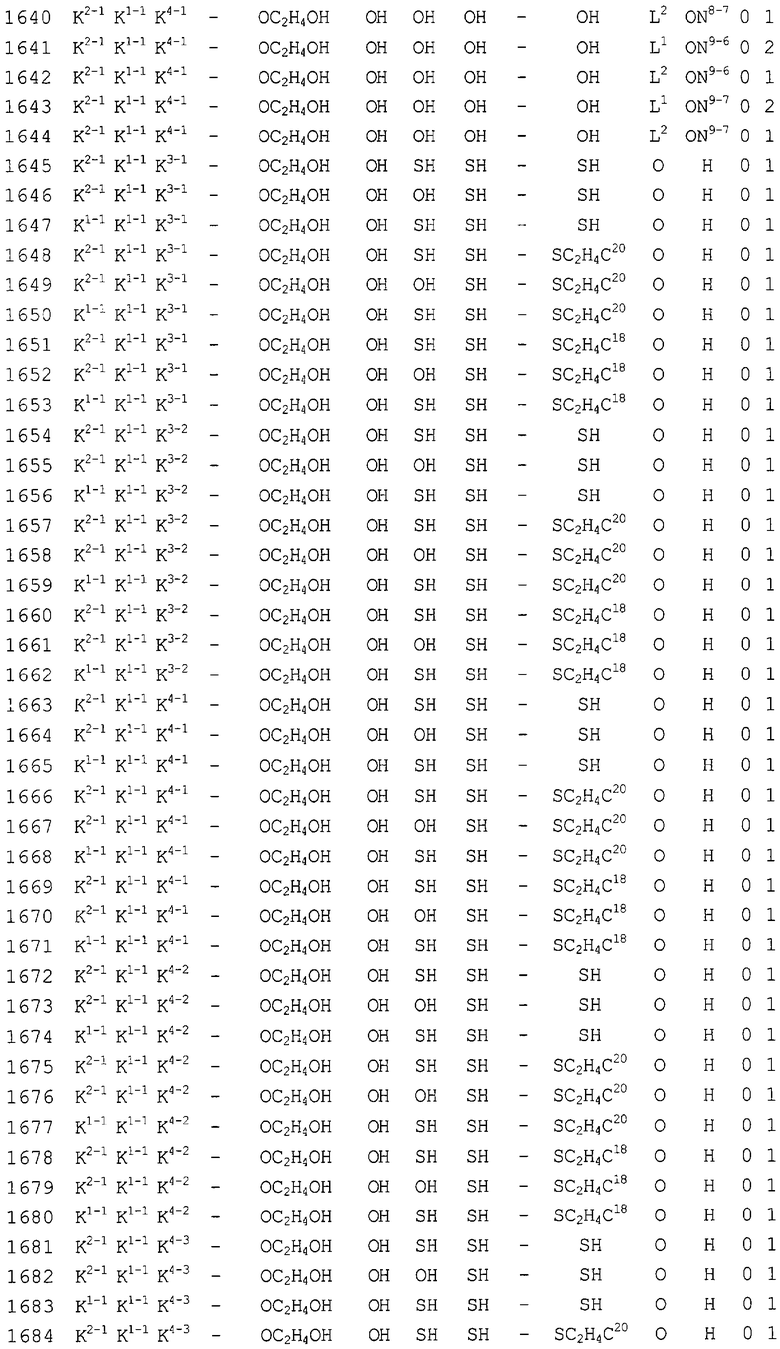
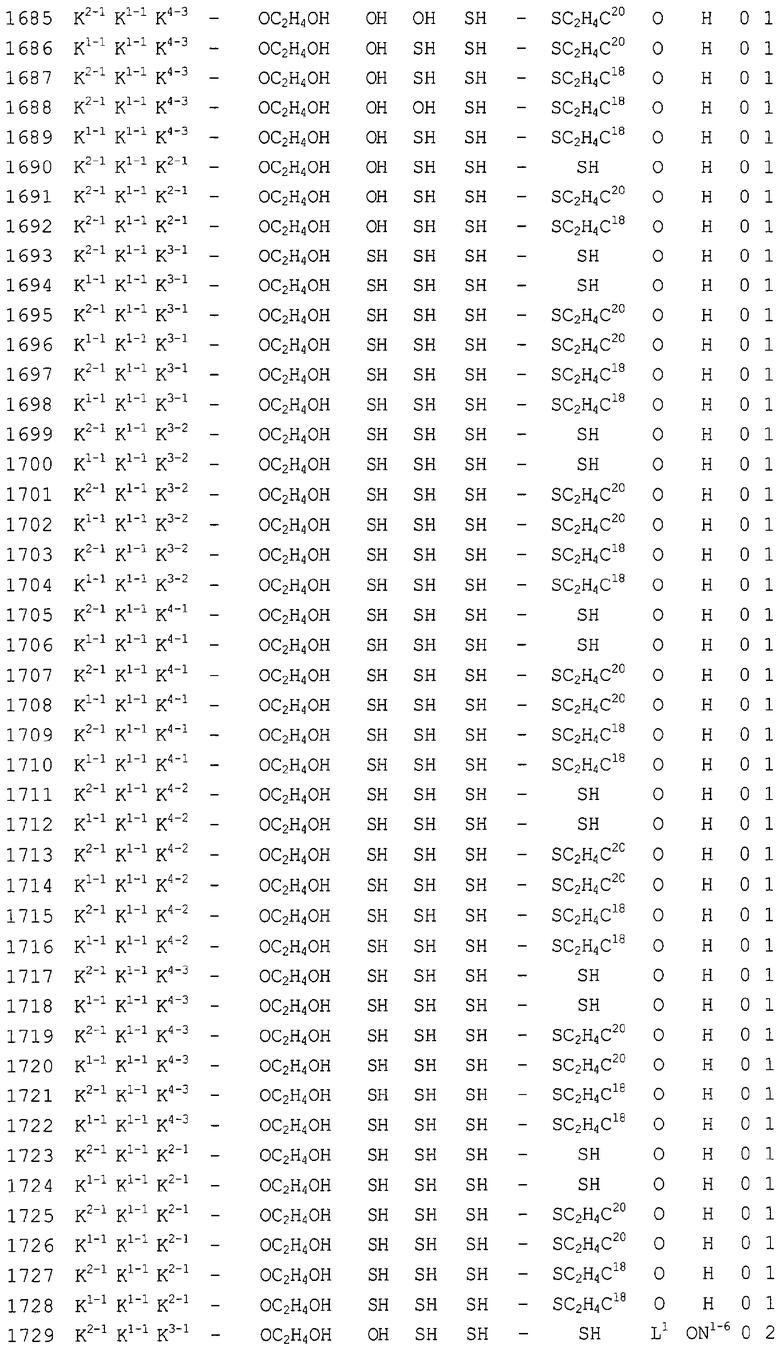




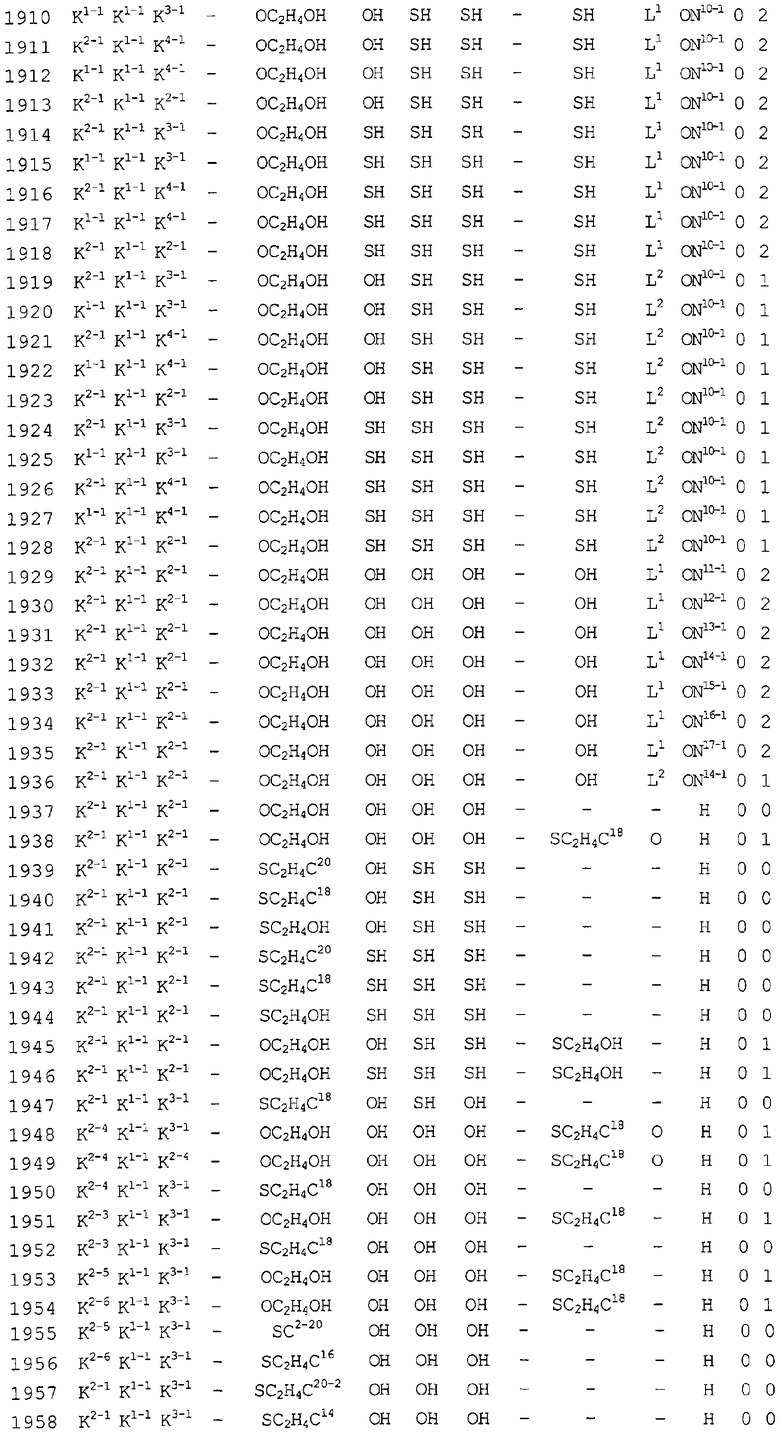
В таблице 1: Ph представляет собой фенильную группу, Bn представляет собой бензильную группу, Me представляет собой метильную группу, Et представляет собой этильную группу, Pr представляет собой н-пропильную группу и трет-Bu представляет собой трет-бутильную группу; и в таблице 1 группы, описываемые как Кх, представляют собой группы, имеющие следующую структуру.
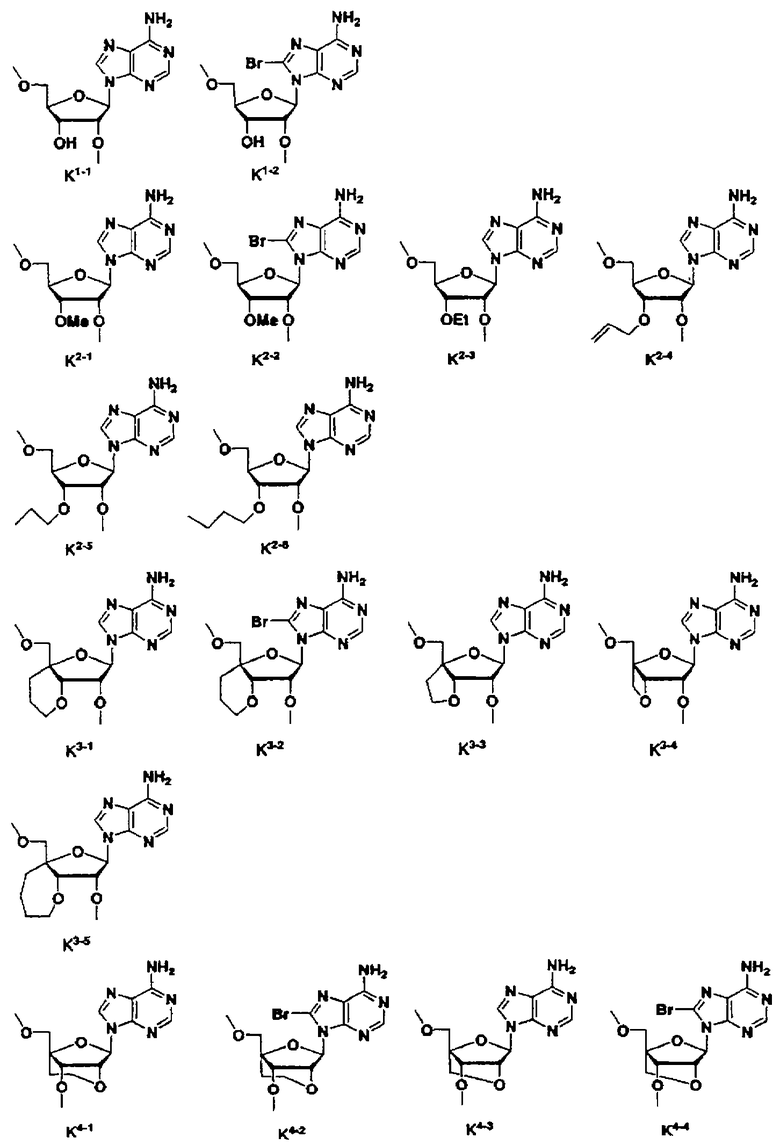
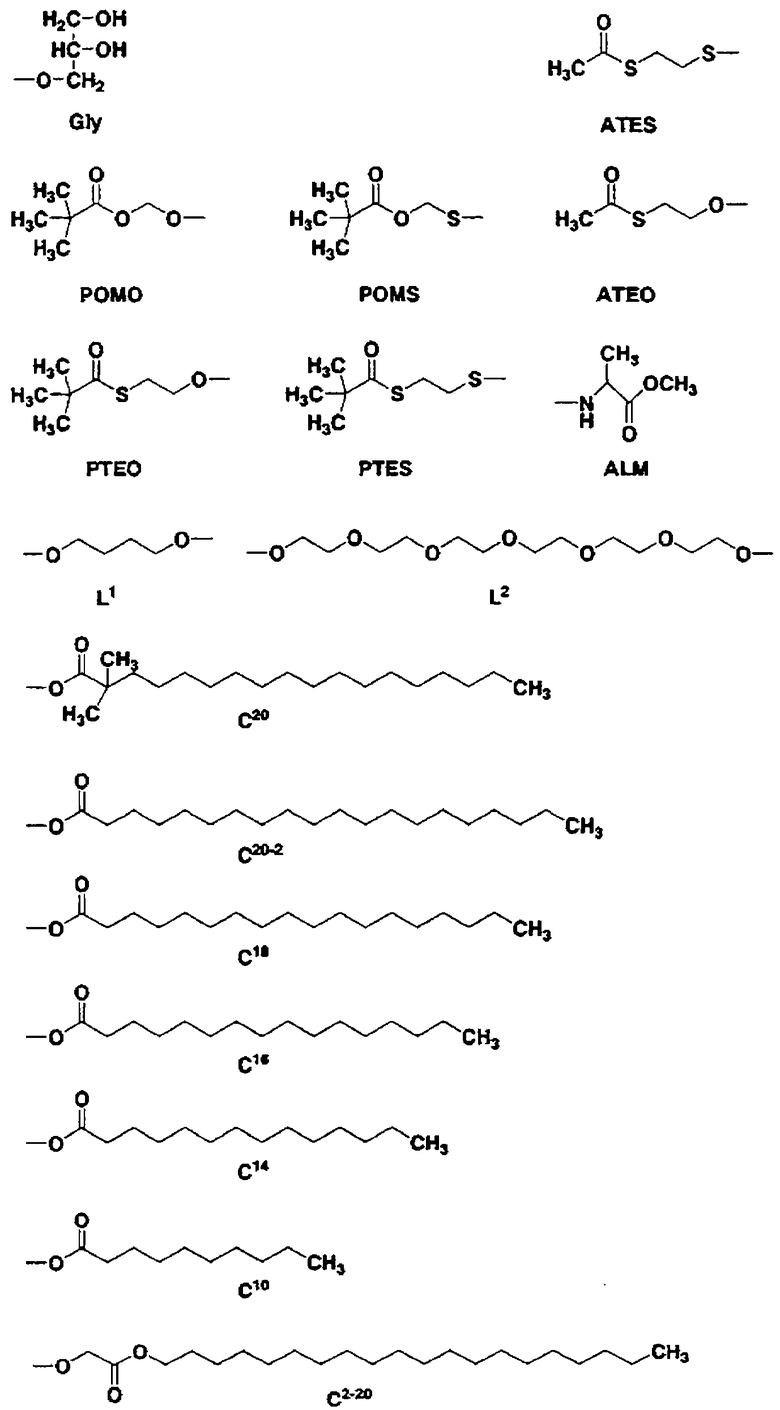
Кроме того, в таблице 1 группы, обозначенные как Gly, POMO, POMS, ATE, PTE, ALM, L1, L2, C20, C18, C14 и C10, представляют собой группы, имеющие соответственно следующие структуры.
Кроме того, в таблице 1 группы, обозначенные как ONX, представляют собой аналоги олигонуклеотида, имеющие структуры, представленные ниже и связанные с заместителем R7 на конце.


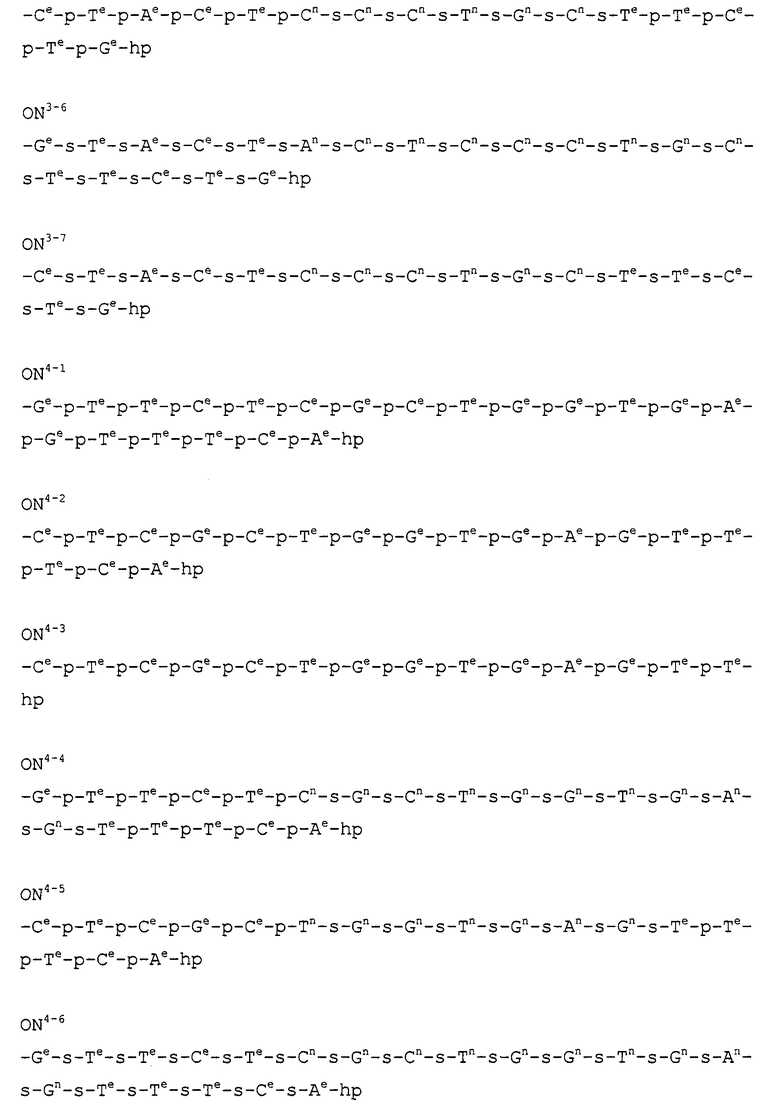
ON4-7



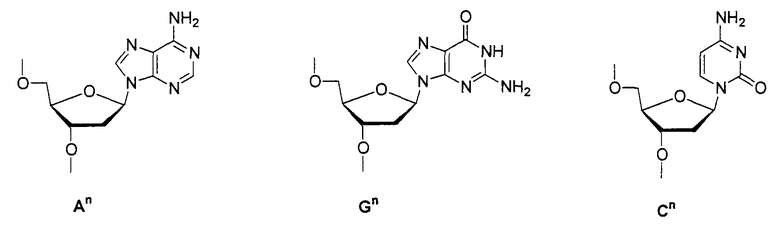
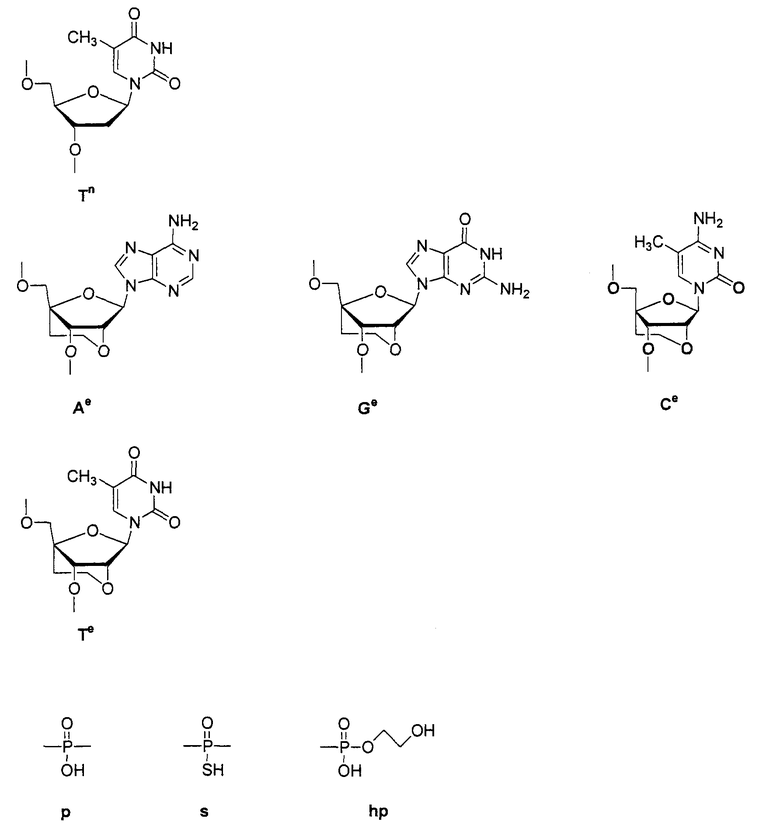
ON8-6


Кроме того, группы, обозначенные выше как An, Gn, Cn, Tn, Ae, Ge, Ce, Te, p, s и hp, представляют собой группы, имеющие следующую структуру.
В последовательностях оснований приведенных выше аналогов олигонуклеотида ON1 представляет собой последовательность в теломеразе человека (GenBank Accession № U86046, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 170-188), ON2 представляет собой последовательность в области кластера точки разрыва (BCR) мРНК человека (GenBank Accession № NM-021574.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 597-614), ON3 представляет собой последовательность в индуцируемой интерфероном зависимой от двухспиральной РНК протеинкиназы (PKR) мРНК человека (GenBank Accession № NM-002759.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 490-508), ON4 представляет собой последовательность в протеинкиназе С, альфа (PKCα) мРНК человека (GenBank Accession № NM-002737.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 2044-2063), ON5 представляет собой последовательность в молекуле межклеточной адгезии (ICAM1) мРНК человека (GenBank Accession № NM-000201.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 2100-2119), ON6 представляет собой последовательность в гене ras-трансформирующего белка человека (GenBank Accession № M38453.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 121-140), ON7 представляет собой последовательность в факторе опухолевого роста (суперсемейство TNF, член 2) (TNF) мРНК человека (GenBank Accession № NM-000594.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 279-298), ON8 представляет собой последовательность в фосфотирозилпротеинфосфатазе (PTP-1B) мРНК человека (GenBank Accession № M31724.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 951-970), ON9 представляет собой последовательность в c-raf-1 мРНК человека (GenBank Accession № NM-002880.1, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 2484-2503), и ON10 представляет собой последовательность в теломеразе мРНК человека (GenBank Accession № U86046, последовательность оснований комплементарной цепи порядковых номеров нуклеотидов 136-148).
В приведенной выше таблице 1 предпочтительными соединениями являются соединения:
1, 2, 3, 4, 5, 6, 7, 8, 13, 22, 27, 28, 31, 39, 41, 42, 50, 52, 53, 61, 63, 64, 71, 73, 77, 79, 96, 98, 102, 104, 146, 148, 152, 154, 171, 173, 177, 179, 290, 292, 293, 305, 307, 310, 311, 312, 313, 314, 316, 319, 320, 325, 330, 334, 338, 339, 343, 344, 351, 356, 364, 369, 377, 382, 386, 390, 391, 395, 396, 403, 408, 416, 421, 424, 425, 428, 438, 441, 451, 452, 453, 454, 455, 461, 462, 463, 464, 465, 471, 472, 473, 474, 475, 481, 482, 483, 484, 485, 491, 492, 493, 494, 495, 501, 502, 503, 504, 505, 511, 512, 513, 514, 515, 521, 522, 523, 524, 525, 531, 532, 533, 534, 535, 541, 542, 543, 544, 545, 551, 552, 553, 554, 555, 561, 562, 563, 564, 565, 571, 572, 573, 574, 575, 581, 582, 583, 584, 585, 591, 592, 593, 594, 595, 601, 602, 603, 604, 605, 611, 612, 613, 614, 615, 621, 622, 623, 624, 625, 631, 632, 633, 634, 635, 641, 642, 643, 644, 645, 651, 652, 653, 654, 655, 661, 662, 663, 664, 665, 671, 672, 673, 674, 675, 681, 682, 683, 684, 685, 691, 692, 693, 694, 695, 701, 702, 703, 704, 705, 711, 712, 713, 714, 715, 721, 722, 723, 724, 725, 731, 732, 733, 734, 735, 741, 742, 743, 744, 745, 751, 752, 753, 754, 755, 761, 762, 763, 764, 765, 771, 772, 773, 774, 775, 781, 782, 783, 784, 785, 791, 792, 793, 794, 795, 801, 802, 803, 804, 805, 811, 812, 813, 814, 815, 821, 822, 823, 824, 825, 831, 832, 833, 834, 835, 841, 842, 843, 844, 845, 851, 852, 853, 854, 855, 861, 862, 863, 864, 865, 871, 872, 873, 874, 875, 881, 882, 883, 884, 885, 891, 892, 893, 894, 895, 901, 902, 903, 907, 908, 909, 913, 914, 915, 919, 920, 924, 925, 926, 930, 931, 932, 936, 937, 941, 942, 943, 947, 948, 949,953, 954, 959, 960, 961, 962, 963, 966, 967, 978, 979, 990, 991, 1002, 1003, 1014, 1015, 1026, 1027, 1038, 1039, 1050, 1051, 1062, 1063, 1074, 1075, 1078, 1079, 1082, 1083, 1086, 1087, 1090, 1091, 1094, 1095, 1098, 1099, 1102, 1103, 1106, 1107, 1110, 1111, 1122, 1123, 1134, 1135, 1146, 1147, 1158, 1159, 1170, 1171, 1182, 1183, 1194, 1195, 1206, 1207, 1220, 1231, 1243, 1255, 1267, 1279, 1291, 1303, 1315, 1344, 1345, 1346, 1347, 1348, 1349, 1350, 1351, 1352, 1353, 1354, 1355, 1356, 1357, 1358, 1359, 1360, 1361, 1429, 1430, 1431, 1432, 1433, 1449, 1450, 1451, 1452, 1453, 1469, 1470, 1471, 1472, 1473, 1489, 1490, 1491, 1492, 1493, 1509, 1510, 1511, 1512, 1513, 1529, 1530, 1531, 1532, 1533, 1549, 1550, 1551, 1552, 1553, 1569, 1570, 1571, 1572, 1573, 1589, 1590, 1591, 1592, 1593, 1609, 1609, 1613, 1617, 1621, 1625, 1629, 1633, 1637, 1641, 1645, 1647, 1648, 1650, 1651, 1653, 1663, 1665, 1666, 1668, 1669, 1671, 1690, 1691, 1692, 1693, 1694, 1695, 1696, 1697, 1698, 1705, 1706, 1707, 1708, 1709, 1710, 1723, 1724, 1725, 1726, 1727, 1728, 1734, 1735, 1736, 1737, 1738, 1754, 1755, 1756, 1757, 1758, 1774, 1775, 1776, 1777, 1778 1794, 1795, 1796, 1797 1798, 1814, 1815, 1816, 1817, 1818, 1834, 1835, 1836, 1837 1838, 1854, 1855, 1856, 1857, 1858, 1874, 1875, 1876, 1877, 1878, 1894, 1895, 1896, 1897, 1898, 1914, 1915, 1916, 1917 и 1918, и более предпочтительными соединениями являются соединения 1, 2, 3, 4, 5, 8, 290, 305, 307, 338, 343, 364, 369, 390, 395, 416, 421, 451, 452, 455, 461, 462, 465, 471, 472, 475, 481, 482, 485, 491, 492, 495, 501, 502, 505, 511, 512, 515, 521, 522, 525, 531, 532, 535, 541, 542, 545, 551, 552, 555, 561, 562, 565, 571, 572, 575, 581, 582, 585, 591, 592, 595, 601, 602, 605, 611, 612, 615, 621, 622, 625, 631, 632, 635, 641, 642, 645, 651, 652, 655, 661, 662, 665, 671, 672, 675, 681, 682, 685, 691, 692, 695, 701, 702, 705, 711, 712, 715, 721, 722, 725, 731, 732, 735, 741, 742, 745, 751, 752, 755, 761, 762, 765, 771, 772, 775, 781, 782, 785, 791, 792, 795, 801, 802, 805, 811, 812, 815, 821, 822, 825, 831, 832, 835, 841, 842, 845, 851, 852, 855, 861, 862, 865, 871, 872, 875, 881, 882, 885, 891, 892, 895,953, 954, 959, 960, 961, 962, 963, 966, 967, 978, 979, 990, 991, 1002, 1003, 1014, 1015, 1026, 1027, 1038, 1039, 1050, 1051, 1062, 1063, 1075, 1079, 1083, 1087, 1091, 1095, 1099, 1103, 1107, 1110, 1111, 1122, 1123, 1134, 1135, 1146, 1147, 1158, 1159, 1170, 1171, 1182, 1183, 1194, 1195, 1206, 1207, 1429, 1430, 1449, 1450, 1469, 1470, 1489, 1490, 1509, 1510, 1529, 1530, 1549, 1550, 1569, 1570, 1589, 1590, 1648, 1650, 1651, 1653, 1666, 1668, 1669, 1671, 1691, 1692, 1695, 1696, 1697, 1698, 1707, 1708, 1709, 1710, 1725, 1726, 1727 и 1728.
Способ осуществления настоящего изобретения
Соединения (1) настоящего изобретения могут быть получены при соответствующем использовании Способа А, Способа B, Способа C, Способа D, Способа E, Способа F, Способа G и Способа H, которые приведены ниже.
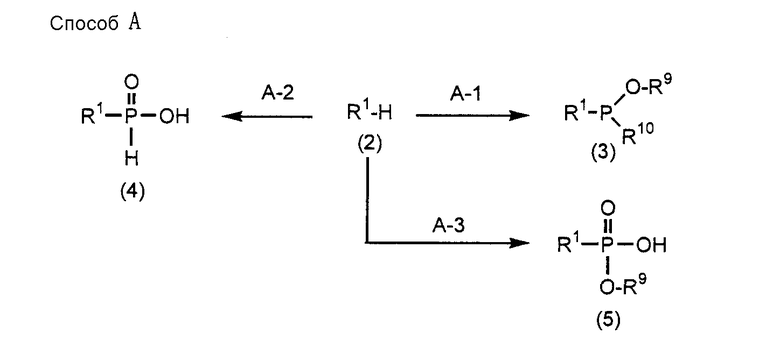
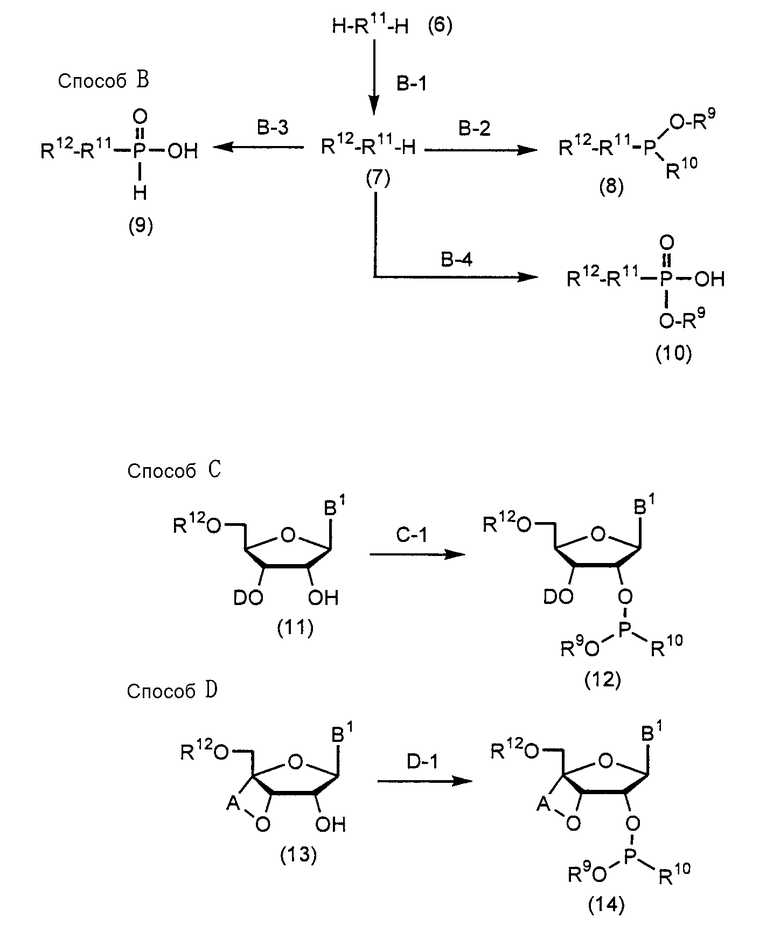
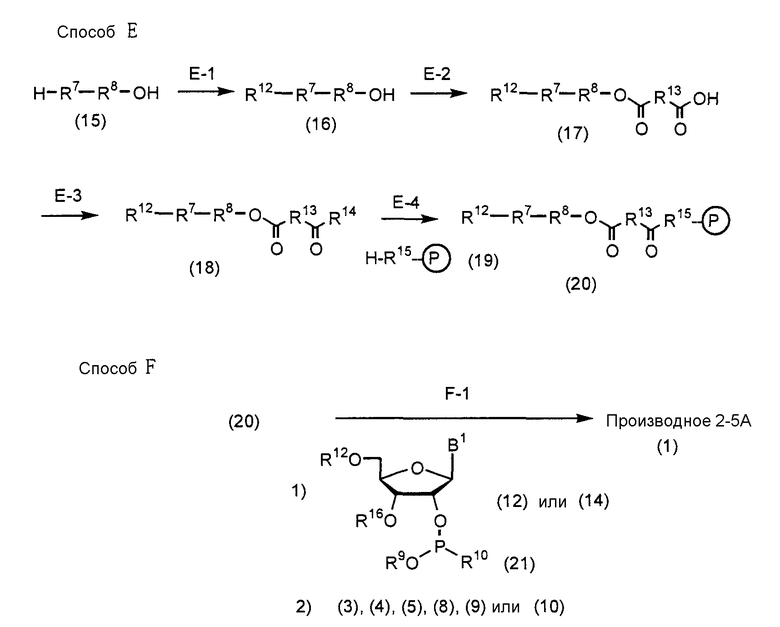
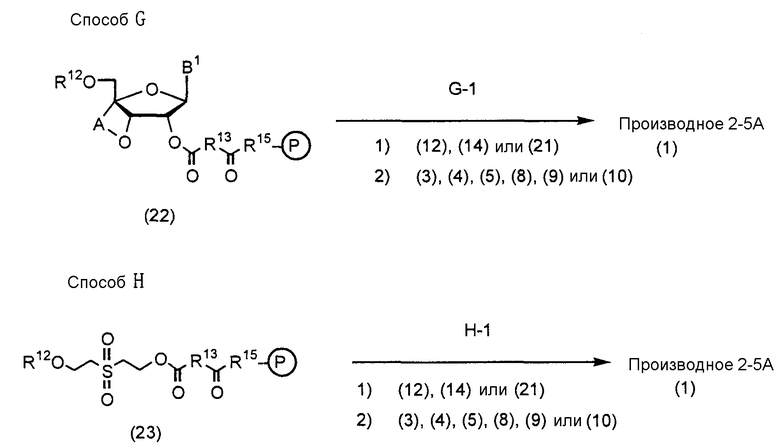
В Способе A, Способе B, Способе C, Способе D, Способе E, Способе F, Способе G и Способе H A, D, R1, R7 и R8 имеют такие же значения, как определено выше; R9 представляет собой защитную группу для защиты группы фосфорной кислоты или группы фосфористой кислоты; R10 представляет собой диалкиламиногруппу (особенно диизопропиламиногруппу или диэтиламиногруппу); R11 представляет собой группу R1, которой при синтезе аналога 2-5A требуется защитная группа; B1 представляет собой пурин-9-ильную группу или замещенную пурин-9-ильную группу, содержащую заместитель(и), выбранный(е) из группы α, но группа, замещенная аминогруппой, исключается. Заместители R12 и R16 являются одинаковыми или различными и представляют собой защитную группу; R13 представляет собой группу -(CH2)h- (h равно целому числу от 2 до 8); R14 представляет собой гидроксильную группу, фенилоксигруппу, которая может быть замещена, или этилоксигруппу, которая может быть замещена атомом галогена; R15 представляет собой атом кислорода, атом серы или NH-группу; и HR15-P (обведенная кружком) представляет собой высокомолекулярное соединение.
«Защитная группа» в определении заместителя R9 может представлять собой, например, низшую алкильную группу, такую как метил; низшую алкенильную группу, такую как 2-пропенил; циано(низшую)алкильную группу, такую как 2-цианоэтил; низшую алкоксилированную низшую алкоксиметильную группу, такую как 2-метоксиэтоксиметил; галоген(низший)алкоксиметильную группу, такую как 2,2,2-трихлорэтоксиметил и бис(2-хлорэтокси)метил; галогенированную этильную группу, такую как 2,2,2-трихлорэтил; метильную группу, замещенную арильной группой, такую как бензил; метильную группу, замещенную 1-3 арильными группами, арильное кольцо которых замещено низшим алкилом, низшей алкоксигруппой, галогеном или цианогруппой(ами), такую как 4-метилбензил, 2-нитробензил, 4-нитробензил, 4-хлорбензил, 4-бромбензил и 4-цианобензил; арильную группу, замещенную атомом(ами) галогена, низшей(ими) алкоксигруппой(ами) или нитрогруппой(ами), такую как 4-хлорфенил, 2-хлорфенил, 4-метоксифенил, 4-нитрофенил и 2,4-динитрофенил; или (низший)алкилкарбонилметильную группу, такую как пентаноилоксиметил и пивалоилоксиметил; и предпочтительно представляет собой метильную группу, 2-цианоэтильную группу, бензильную группу, 2-хлорфенильную группу, 4-хлорфенильную группу, 2-пропенильную группу или пивалоилоксиметильную группу.
«Защитная группа» в определении заместителей R12 и R16 может представлять собой, например, защитную группу «ацильного типа», включая «алифатическую ацильную группу», такую как алкилкарбонильная группа, например формил, ацетил, пропиноил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, нонаноил, деканоил, 3-метилнонаноил, 8-метилнонаноил, 3-этилоктаноил, 3,7-диметилоктаноил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, 1-метилпентадеканоил, 14-метилпентадеканоил, 13,13-диметилтетрадеканоил, гептадеканоил, 15-метилгексадеканоил, октадеканоил, 1-метилгептадеканоил, нонадеканоил, эйкозаноил и генэйкозаноил; карбоксилированная алкилкарбонильная группа, например сукциноил, глутароил и адипоил; галоген(низший)алкилкарбонильная группа, например, хлорацетил, дихлорацетил, трихлорацетил и трифторацетил; и (низший)алкокси(низший)алкилкарбонильная группа, например, метоксиацетил; или ненасыщенная алкилкарбонильная группа, например (E)-2-метил-2-бутеноил; и
«ароматическую ацильную группу», такую как арилкарбонильная группа, например бензоил, α-нафтоил и β-нафтоил; галогенарилкарбонильная группа, например 2-бромбензоил и 4-хлорбензоил; низшая алкилированная арилкарбонильная группа, например 2,4,6-триметилбензоил и 4-толуоил; низшая алкоксилированная арилкарбонильная группа, например 4-анизоил; карбоксилированная арилкарбонильная группа, например 2-карбоксибензоил, 3-карбоксибензоил и 4-карбоксибензоил; нитрованная арилкарбонильная группа, например 4-нитробензоил и 2-нитробензоил; низшая алкоксикарбонилированная арилкарбонильная группа, например 2-(метоксикарбонил)бензоил; или арилированная арилкарбонильная группа, например 4-фенилбензоил;
«низшую алкильную группу», такую как, метил, этил,
н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил,
н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил,
н-гексил, изогексил, 4-метилпентил, 3-метилпентил,
2-метилпентил, 1-метилпентил, 3,3-диметилбутил,
2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил,
1,3-диметилбутил, 2,3-диметилбутил и 2-этилбутил;
«низшую алкенильную группу», такую как этенил, 1-пропенил, 2-пропенил, 1-метил-2-пропенил, 1-метил-1-пропенил, 2-метил-1-пропенил, 2-метил-2-пропенил, 2-этил-2-пропенил, 1-бутенил, 2-бутенил, 1-метил-2-бутенил, 1-метил-1-бутенил, 3-метил-2-бутенил, 1-этил-2-бутенил, 3-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 1-этил-3-бутенил, 1-пентенил, 2-пентенил, 1-метил-2-пентенил, 2-метил-2-пентенил, 3-пентенил, 1-метил-3-пентенил, 2-метил-3-пентенил, 4-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил и 5-гексенил;
«тетрагидропиранильную или тетрагидротиопиранильную группу», такую как тетрагидропиран-2-ил, 3-бромтетрагидропиран-2-ил, 4-метокситетрагидропиран-4-ил, тетрагидротиопиран-2-ил и 4-метокситетрагидротиопиран-4-ил;
«тетрагидрофуранильную или тетрагидротиофуранильную группу», такую как тетрагидрофуран-2-ил и тетрагидротиофуран-2-ил;
«силильную группу», такую как три(низший)алкилсилильная группа, например триметилсилил, триэтилсилил,
изопропилдиметилсилил, трет-бутилдиметилсилил,
метилдиизопропилсилил, метилди-трет-бутилсилил и
триизопропилсилил; или три(низший)алкилсилильная группа, замещенная 1 или 2 арильными группами, например дифенилметилсилил, дифенилбутилсилил, дифенилизопропилсилил и фенилдиизопропилсилил;
«низшую алкоксиметильную группу», такую как метоксиметил,
1,1-диметил-1-метоксиметил, этоксиметил, пропоксиметил,
изопропоксиметил, бутоксиметил и трет-бутоксиметил;
«низшую алкоксилированную низшую алкоксиметильную группу», такую как 2-метоксиэтоксиметил;
«галоген(низший)алкоксиметил», такой как 2,2,2-трихлорэтоксиметил и бис(2-хлорэтокси)метил;
«низшую алкоксилированную этильную группу», такую как 1-этоксиэтил и 1-(изопропокси)этил;
«галогенированную этильную группу», такую как 2,2,2-трихлорэтил;
«метильную группу, замещенную 1-3 арильными группами», такую как бензил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, α-нафтилдифенилметил и 9-антрилметил;
«метильную группу, замещенную 1-3 арильными группами, арильное кольцо которых замещено низшим алкилом, низшей алкоксигруппой, галогеном или цианогруппой(ами)», такие как 4-метилбензил, 2,4,6-триметилбензил, 3,4,5-триметилбензил,
4-метоксибензил, 4-метоксифенилдифенилметил,
4,4'-диметокситрифенилметил, 2-нитробензил, 4-нитробензил,
4-хлорбензил, 4-бромбензил и 4-цианобензил;
«низшую алкоксикарбонильную группу», такую как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и изобутоксикарбонил;
«арильную группу, замещенную атомом(и) галогена, низшей(ми) алкоксигруппой(ами) или нитрогруппой(ами)», такую как 4-хлорфенил, 2-хлорфенил, 4-метоксифенил, 4-нитрофенил и 2,4-динитрофенил;
«низшую алкоксикарбонильную группу, замещенную галогеном или три(низший)алкилсилильной(ыми) группой(ами)», такими как 2,2,2-трихлорэтоксикарбонил и 2-триметилсилилэтоксикарбонил;
«алкенилоксикарбонильную группу», такую как винилоксикарбонил и арилоксикарбонил; или
«аралкилоксикарбонильную группу, арильное кольцо которой может быть замещено 1-2 низшими алкоксигруппами или нитрогруппами», такую как бензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 2-нитробензилоксикарбонил и 4-нитробензилоксикарбонил.
Далее соответствующие стадии Способа A, Способа B, Способа C, Способа D, Способа E, Способа F, Способа G и Способа H будут описаны более подробно.
(Стадия А-1)
Рассматриваемая стадия представляет собой стадию, на которой соединение (3) получают при взаимодействии соединения (2) с монозамещенным хлор(алкокси)фосфином, дизамещенным алкоксифосфином, монозамещенным хлор(бензилокси)фосфином или дизамещенным бензилоксифосфином, обычно используемыми для образования амидита, в инертном растворителе.
Растворитель, который должен быть использован, не имеет особых ограничений, пока он не влияет на реакцию, но предпочтительно растворитель может представлять собой простой эфир, такой тетрагидрофуран, диэтиловый эфир или диоксан; или галогенированный углеводород, такой как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол или дихлорбензол.
Используемый монозамещенный хлор(алкокси)фосфин может представлять собой, например, фосфин, такой как хлор(морфолино)метоксифосфин,
хлор(морфолино)цианоэтоксифосфин,
хлор(диметиламино)метоксифосфин,
хлор(диметиламино)цианоэтоксифосфин, хлор(диизопропиламино)метоксифосфин или
хлор(диизопропиламино)цианоэтоксифосфин, и предпочтительно
представляет собой хлор(морфолино)метоксифосфин,
хлор(морфолино)цианоэтоксифосфин,
хлор(диизопропиламино)метоксифосфин или
хлор(диизопропиламино)цианоэтоксифосфин.
В случае применения монозамещенного хлор(алкокси)фосфина используют восстановитель. В этом случае используемый восстановитель может представлять собой гетероциклический амин, такой как пиридин или диметиламинопиридин; или алифатический амин, такой как триметиламин, триэтиламин, диизопропиламин или диизопропилэтиламин, и предпочтительно представляет собой алифатический амин (предпочтительно диизопропилэтиламин).
Используемый дизамещенный алкоксифосфин может представлять собой, например, фосфин, такой как
бис(диизопропиламино)цианоэтоксифосфин,
бис(диэтиламино)метансульфонилэтоксифосфин,
бис(диизопропиламино)(2,2,2-трихлорэтокси)фосфин или
бис(диизопропиламино)(4-хлорфенилметокси)фосфин,
и предпочтительно представляет собой
бис(диизопропиламино)цианоэтоксифосфин.
В случае применения дизамещенного алкоксифосфина используют кислоту или органическую соль. В этом случае кислота, которую необходимо использовать, представляет собой тетразол, уксусную кислоту или п-толуолсульфоновую кислоту, и органическая соль, которую необходимо использовать, представляет собой диизопропиламиновую соль тетразола, диизопропиламиновую соль уксусной кислоты или диизопропиламиновую соль п-толуолсульфоновой кислоты, предпочтительно тетразол или диизопропиламиновую соль тетразола.
Используемый монозамещенный хлор(бензилокси)фосфин может представлять собой, например, фосфин, такой как
хлор(морфолино)бензилоксифосфин,
хлор(диметиламино)метоксифосфин,
хлор(диметиламино)бензилоксифосфин или
хлор(диизопропиламино)бензилоксифосфин, и предпочтительно представляет собой хлор(диизопропиламино)бензилоксифосфин.
В случае применения монозамещенного хлор(бензилокси)фосфина используют восстановитель. В этом случае используемый восстановитель может представлять собой гетероциклический амин, такой как пиридин или диметиламинопиридин; или алифатический амин, такой как триметиламин, триэтиламин, диизопропиламин или диизопропилэтиламин, и предпочтительно представляет собой алифатический амин (предпочтительно диизопропилэтиламин).
Используемый дизамещенный бензилоксифосфин может представлять собой, например, фосфин, такой как бис(диизопропиламино)бензилоксифосфин или бис(диэтиламино)бензилоксифосфин, и предпочтительно представляет собой бис (диизопропиламино)бензилоксифосфин.
В случае применения дизамещенного бензилоксифосфина используют кислоту или органическую соль. В этом случае кислота, которую необходимо использовать, представляет собой тетразол, уксусную кислоту или п-толуолсульфоновую кислоту, и органическая соль, которую необходимо использовать, представляет собой диизопропиламиновую соль тетразола, диизопропиламиновую соль уксусной кислоты или диизопропиламиновую соль п-толуолсульфоновой кислоты, предпочтительно тетразол или диизопропиламиновую соль тетразола.
Температура реакции не имеет особых ограничений, но обычно составляет от 0 до 80°С, предпочтительно соответствует комнатной температуре.
Хотя время реакции меняется в зависимости от исходных веществ, используемых реагентов и температуры, обычно оно составляет от 5 минут до 30 часов; и в случае, когда реакцию проводят при комнатной температуре, время реакции предпочтительно составляет от 30 минут до 10 часов.
По окончании реакции требуемое соединение (3) настоящей реакции получают, например, после соответствующей нейтрализации реакционной смеси и удаления любого нерастворимого вещества, если оно присутствует, фильтрованием, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой над безводным сульфатом магния или др., и отгонкой растворителя. Полученное таким образом требуемое соединение может быть дополнительно очищено обычными методами, такими как перекристаллизация, переосаждение или хроматография, если это необходимо.
(Стадия A-2)
Рассматриваемая стадия представляет собой стадию, на которой соединение (4) получают путем введения соединения (2) в реакцию с трис-(1,2,4-триазолил)фосфитом в инертном растворителе (предпочтительно в галогенированном углеводороде, таком как метиленхлорид) и добавления в реакционную смесь воды, чтобы вызвать H-фосфонирование.
Температура реакции не имеет особых ограничений, но обычно составляет от -20 до 100°С, предпочтительно от 10 до 40°С.
Хотя время реакции меняется в зависимости от исходных веществ, используемых реагентов и температуры, обычно оно составляет от 5 минут до 30 часов; и в случае, когда реакцию проводят при комнатной температуре, время реакции предпочтительно составляет 30 минут.
По окончании реакции требуемое соединение (4) настоящей реакции получают, например, после соответствующей нейтрализации реакционной смеси и удаления любого нерастворимого вещества, если оно присутствует, фильтрованием, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой над безводным сульфатом магния или др., и отгонкой растворителя. Полученное таким образом требуемое соединение может быть дополнительно очищено обычными методами, такими как перекристаллизация, переосаждение или хроматография, если это необходимо.
(Стадия A-3)
Рассматриваемая стадия представляет собой стадию, на которой соединение (5) получают путем введения соединения (2) в реакцию с бис(1,2,4-триазолил)арилфосфатом,
бис(1,2,4-триазолил)бензилфосфатом,
бис(1,2,4-триазолил)-2-цианоэтилфосфатом,
бис(1,2,4-триазолил)(2,2,2-трихлорэтил)фосфатом или
бис(1,2,4-триазолил)(2-пропенил)фосфатом в инертном растворителе (предпочтительно в галогенированном углеводороде, таком как метиленхлорид), и добавления в реакционную смесь воды для получения сложного фосфорного диэфира.
Используемый бис(1,2,4-триазолил)арилфосфат может представлять собой, например, бис(1,2,4-триазолил)фенилфосфат, бис(1,2,4-триазолил)(2-хлорфенил)фосфат, бис(1,2,4-триазолил)(4-хлорфенил)фосфат, бис(1,2,4-триазолил)-(2-нитрофенил)фосфат или бис(1,2,4-триазолил)(4-нитрофенил)фосфат, и предпочтительно представляет собой бис(1,2,4-триазолил)(2-хлорфенил)фосфат или бис(1,2,4-триазолил)(4-хлорфенил)фосфат.
Температура реакции не имеет особых ограничений, но обычно составляет от -20 до 100°С, предпочтительно от 10 до 40°С.
Хотя время реакции меняется в зависимости от исходных веществ, используемых реагентов и температуры, обычно оно составляет от 5 минут до 30 часов; и в случае, когда реакцию проводят при комнатной температуре, время реакции предпочтительно составляет 30 минут.
По окончании реакции требуемое соединение (5) настоящей реакции получают, например, после соответствующей нейтрализации реакционной смеси и удаления любого нерастворимого вещества, если оно присутствует, фильтрованием, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой над безводным сульфатом магния или др., и отгонкой растворителя. Полученное таким образом требуемое соединение может быть дополнительно очищено обычными методами, такими как перекристаллизация, переосаждение или хроматография, если это необходимо.
(Стадия B-1)
Рассматриваемая стадия представляет собой стадию, на которой соединение (7) получают путем введения соединения (6) в реакцию с защитным реагентом в присутствии основного катализатора в инертном растворителе.
Используемый растворитель предпочтительно может представлять собой ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол или дихлорбензол; сложный эфир, такой как формиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диметиловый эфир диэтиленгликоля; кетон, такой как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединение, такое как нитроэтан или нитробензол; нитрил, такой как ацетонитрил или изобутиронитрил; амид, такой как формамид, диметилформамид (ДМФА), диметилацетамид или гексаметилфосфортриамид; сульфоксид, такой как диметилсульфоксид или сульфолан; алифатический третичный амин, такой как триметиламин, триэтиламин или N-метилморфолин; или ароматический амин, такой как пиридин или пиколин; и более предпочтительно представляет собой галогенированный углеводород (предпочтительно метиленхлорид) или ароматический амин (предпочтительно пиридин).
Используемый защитный реагент не имеет особых ограничений, пока он пригоден для последующего синтеза нуклеиновой кислоты и может быть снят в кислых или нейтральных условиях, и предпочтительно может представлять собой триарилметилгалогенид, такой как тритилхлорид, монометокситритилхлорид или диметокситритилхлорид; или простой эфир триарилметанола, такой как диметокситритил-О-трифлат.
В случае применения триарилметилгалогенида в качестве защитного реагента обычно используют основание. В этом случае используемое основание может представлять собой гетероциклический амин, такой как пиридин, диметиламинопиридин или пирролидинопиридин; или алифатический третичный амин, такой как триметиламин или триэтиламин; и предпочтительно представляет собой пиридин, диметиламинопиридин или пирролидинопиридин.
В случае применения жидкого основания в качестве растворителя, поскольку само основание действует как восстановитель, также нет необходимости добавлять основание.
Температура реакции меняется в зависимости от исходных веществ, используемых реагентов и растворителя, и обычно составляет от 0 до 150°С, предпочтительно от 20 до 100°С. Хотя время реакции меняется в зависимости от исходных веществ, используемого растворителя и температуры, обычно оно составляет от 1 до 100 часов, предпочтительно от 2 до 24 часов.
По окончании реакции требуемое соединение (7) рассматриваемой реакции получают, например, концентрированием реакционной смеси, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой над безводным сульфатом магния или др., и отгонкой растворителя.
Полученное соединение может быть дополнительно очищено обычными методами, такими как, например, перекристаллизация или колоночная хроматография на силикагеле, если это необходимо.
(Стадия B-2)
Рассматриваемая стадия представляет собой стадию, на которой соединение (8) получают путем введения соединения (7), полученного на стадии B-1, в реакцию с монозамещенным хлор(алкокси)фосфином, дизамещенным алкоксифосфином, монозамещенным хлор(бензилокси)фосфином или дизамещенным бензилоксифосфином, которые обычно используют для образования амидита, в инертном растворителе.
Рассматриваемую стадию проводят аналогично стадии (A-1).
(Стадия B-3)
Рассматриваемая стадия представляет собой стадию, на которой соединение (9) получают путем введения соединения (7), полученного на стадии B-1, в реакцию с трис-(1,2,4-триазолил)фосфитом в инертном растворителе (предпочтительно в галогенированном углеводороде, таком как метиленхлорид), с последующим добавлением воды для проведения H-фосфонирования.
Рассматриваемую стадию проводят аналогично стадии (A-2).
(Стадия B-4)
Рассматриваемая стадия представляет собой стадию, на которой соединение (8) получают введением соединения (7), полученного на стадии B-1, в реакцию с
бис(1,2,4-триазолил)арилфосфатом,
бис(1,2,4-триазолил)бензилфосфатом,
бис(1,2,4-триазолил)-2-цианоэтилфосфатом,
бис(1,2,4-триазолил)(2,2,2-трихлорэтил)фосфатом или
бис(1,2,4-триазолил)(2-пропенил)фосфатом в инертном растворителе (предпочтительно в галогенированном углеводороде, таком как метиленхлорид), с последующим добавлением воды для получения сложного фосфорного диэфира.
Рассматриваемую стадию проводят аналогично стадии A-3.
(Стадия C-1)
Рассматриваемая стадия представляет собой стадию, на которой соединение (12) получают путем введения соединения (11) в реакцию с замещенным хлор(алкокси)фосфином, дизамещенным алкоксифосфином, монозамещенным хлор(бензилокси)фосфином или дизамещенным бензилоксифосфином, которые обычно используют для образования амидита, в инертном растворителе.
Соединение (11) представляет собой соединение, в котором нуклеозид вводят в реакцию с алкилгалогенидом, таким как метилйодид, или алкенилгалогенидом, таким как аллилбромид в присутствии гидрида натрия в соответствии со способом, описанным в заявке PCT/US94/10131, с получением 3'-замещенного соединения, и затем 5'-гидроксильную группу и аминогруппу основного фрагмента защищают с помощью защитных групп. Например, 3'-O-аллиладенозин (№ по каталогу: RP-3101) может быть закуплен у компании ChemGene Industries, и 5'-O-диметокситритил-3'-O-аллил-N-бензоиладенозин может быть получен из него путем введения защитных групп с использованием известных опубликованных способов.
Рассматриваемую стадию проводят аналогично стадии A-1.
Из соединений (12), например, 5'-O-диметокситритил-3'-O-метил-N-бензоиладенозин-2'-O-(2-цианоэтил-N,N-диизопропилфосфорамидит) (№ по каталогу: ANP-2901) может быть закуплен у компании ChemGene Industries.
(Стадия D-1)
Рассматриваемая стадия представляет собой стадию, на которой соединение (14) получают путем введения соединения (13) в реакцию с монозамещенным хлор(алкокси)фосфином, дизамещенным алкоксифосфином, монозамещенным хлор(бензилокси)фосфином или дизамещенным бензилоксифосфином, которые обычно используют для образования амидита, в инертном растворителе.
Соединение (13) представляет собой то же самое соединение, что и соединение (20), описанное в Способе F японской патентной заявки (Kokai) № 2002-249497, или соединение, описанное в японской патентной заявке (Kokai) № Hei 10-195098, где Y1 представляет собой защитную группу и Y2 представляет собой атом водорода.
Рассматриваемую стадию проводят аналогично стадии (A-1).
(Стадия E-1)
Рассматриваемая стадия представляет собой стадию, на которой соединение (16) получают путем введения соединения (15) в реакцию с защищающим реагентом в присутствии основного катализатора в инертном растворителе.
Рассматриваемую стадию проводят аналогично стадии (B-1).
(Стадия E-2)
Рассматриваемая стадия представляет собой стадию, на которой соединение (17) получают путем введения соединения (16), полученного на стадии E-1, в реакцию с ангидридом дикарбоновой кислоты в инертном растворителе.
Используемый растворитель не имеет особых ограничений, пока он не ингибирует реакцию и растворяет исходное вещество до определенной степени, и может представлять собой, например, ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как метиленхлорид или хлороформ; простой эфир, такой как тетрагидрофуран, диоксан или диметоксиэтан; амид, такой как диметилформамид, диметилацетамид или гексаметилфосфортриамид; сульфоксид, такой как диметилсульфоксид; кетон, такой как ацетон или метилэтилкетон; гетероциклический амин, такой как пиридин; или нитрил, такой как ацетонитрил; и предпочтительно представляет собой галогенированный углеводород, такой как метиленхлорид.
Восстановитель, который необходимо использовать, может представлять собой пиридин, такой как пиридин, диметиламинопиридин или пирролидинопиридин, и предпочтительно представляет собой диметиламинопиридин.
Ангидрид дикарбоновой кислоты, который должен быть использован, не имеет особых ограничений, пока он представляет собой ангидрид α,ω-алкилдикарбоновой кислоты, содержащей от 3 до 16 атомов углерода, и предпочтительно может представлять собой янтарный ангидрид.
Хотя температура реакции и время реакции меняются в зависимости от используемого ангидрида кислоты и восстановителя, в случае использования янтарного ангидрида и использования в качестве восстановителя диметиламинопиридина, реакцию проводят при комнатной температуре в течение 30 минут.
По окончании реакции требуемое соединение выделяют из реакционной смеси обычными методами. Например, после соответствующей нейтрализации реакционной смеси и удаления любого нерастворимого вещества, если оно присутствует, целевое соединение получают фильтрованием, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой экстракта над безводным сульфатом магния или др., и отгонкой растворителя. Полученное таким образом требуемое соединение может быть дополнительно очищено обычными методами, такими как перекристаллизация, переосаждение или хроматография, если это необходимо.
(Стадия E-3)
Рассматриваемая стадия представляет собой стадию, на которой активный сложный эфир (18) образуется при взаимодействии карбоксильной группы соединения (17), содержащего свободную гидроксильную группу, с образующим эфир реагентом в инертном растворителе, и последующем взаимодействии с фенолом, который может быть замещен.
Растворитель, который необходимо использовать, не имеет особых ограничений, пока он не ингибирует реакцию, и может представлять собой, например, ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол или дихлорбензол; сложный эфир, такой как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; кетон, такой как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединение, такое как нитроэтан или нитробензол; нитрил, такой как ацетонитрил или изобутиронитрил; амид, такой как формамид, диметилформамид (ДМФА), диметилацетамид или гексаметилфосфортриамид; или сульфоксид, такой как диметилсульфоксид или сульфолан; и предпочтительно представляет собой галогенированный углеводород (предпочтительно метиленхлорид) или амид (предпочтительно диметилформамид).
Фенол, который необходимо использовать, не имеет особых ограничений, пока он может быть использован в качестве активного сложного эфира, и указанный фенол может представлять собой 4-нитрофенол, 2,4-динитрофенол, 2,4,5-трихлорфенол, 2,3,4,5,6-пентахлорфенол или 2,3,5,6-тетрафторфенол, и предпочтительно представляет собой пентахлорфенол.
Образующий эфир реагент, который необходимо использовать, может представлять собой, например, N-гидроксисоединение, такое как N-гидроксисукцинимид, 1-гидроксибензотриазол или N-гидрокси-5-норбонен-2,3-дикарбоксиимид; соединение диимидазола, такое как 1,1'-оксалилдиимидазол или N,N'-карбонилдиимидазол; дисульфидное соединение, такое как 2,2'-дипиридилдисульфид; соединение янтарной кислоты, такое как N,N'-дисукцинимидилкарбонат; соединение фосфинхлорида, такое как N,N'-бис(2-оксо-3-оксазолидинил)фосфинхлорид; оксалатное соединение, такое как N,N'-дисукцинимидилоксалат (DSO), N,N-дифталимидилоксалат (DPO),
N,N'-бис(норборненилсукцинимидил)оксалат (BNO),
1,1'-бис(бензотриазолил)оксалат (BBTO),
1,1'-бис(6-хлорбензотриазолил)оксалат (BCTO) или
1,1'-бис(6-трифторметилбензотриазолил)оксалат (BTBO); или карбодиимид, такой как дициклогексилкарбодиимид (DCC); и предпочтительно представляет собой диимидазольное соединение или карбодиимид (в особенности DCC).
Хотя температура реакции и время реакции меняются в зависимости от образующего эфир реагента и типа используемого растворителя, реакцию проводят при температуре от 0 до 100°С в течение от 5 до 50 часов, и, в частности, в том случае, когда используют пентахлорфенол и DCC в ДМФА, реакцию проводят при комнатной температуре в течение 18 часов.
По окончании реакции требуемое соединение выделяют из реакционной смеси обычными методами. Например, после соответствующей нейтрализации реакционной смеси и удаления любого нерастворимого вещества, если оно присутствует, целевое соединение получают фильтрованием, добавлением воды и несмешивающегося органического растворителя, такого как этилацетат, с последующей промывкой водой, отделением органического слоя, содержащего требуемое соединение, сушкой экстракта над безводным сульфатом магния или др., и отгонкой растворителя. Полученное таким образом требуемое соединение может быть дополнительно очищено обычными методами, такими как перекристаллизация, переосаждение или хроматография, если это необходимо.
(Стадия Е-4)
Рассматриваемая стадия представляет собой стадию, на которой высокомолекулярное производное (20), которое может быть использовано в качестве носителя для синтеза олигонуклеотида, получают путем введения соединения (18), содержащего активированную карбоксильную группу, полученного на стадии Е-3, в реакцию с высокомолекулярным веществом (19), таким как стекло с контролируемым размером пор (CPG), связанное с аминогруппой, гидроксильной группой, сульфгидрильной группой или др. через алкиленовую группу, в инертном растворителе.
Высокомолекулярное вещество (19), используемое на рассматриваемой стадии, не имеет особых ограничений, пока оно используется в качестве носителя, но необходимо проверять размер частиц носителя, размер площади поверхности трехмерной решетчатой структуры, соотношение положений гидрофильных групп, химический состав, предел прочности при сжатии и т.д.
Используемый носитель может представлять собой полисахаридное производное, такое как целлюлоза, декстран или агароза; синтетический полимер, такой как полиакриламидный гель, полистирольная смола или полиэтиленгликоль; или неорганическое вещество, такое как силикагель, пористое стекло или оксид металла. Более конкретно, носитель может представлять собой коммерчески доступный носитель, такой как аминопропил-CPG, длинноцепочечный аминоалкил-CPG (производимые CPG Inc.), Cosmoseal NH2, Cosmoseal Diol (производимые Nacalai Tesque), носитель на основе СРС-диоксида кремния, покрытого силаном (CPC-Silica Carrier Silane Coated), аминопропил-CPG-550 Å, аминопропил-CPG-1400 Å, монометиловый эфир полиэтиленгликоля 5000 (производимые Furuka Inc.), смола на основе п-алкоксибензилового спирта, аминометиловая смола, гидроксиметиловая смола (производимые Kokusan Kagaku Inc.) и монометиловый эфир полиэтиленгликоля 14000 (производимый Union Carbide Inc.), но не ограничиваясь только ими.
Кроме того, функциональная группа, связанная с носителем, предпочтительно представляет собой аминогруппу, сульфгидрильную группу или гидроксильную группу.
Растворитель, используемый на рассматриваемой стадии, не имеет особых ограничений, пока он не ингибирует реакцию и растворяет исходное вещество до определенной степени, и растворитель предпочтительно может представлять собой ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол или дихлорбензол; сложный эфир, такой как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; кетон, такой как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединение, такое как нитроэтан или нитробензол; нитрил, такой как ацетонитрил или изобутиронитрил; амид, такой как формамид, диметилформамид (ДМФА), диметилацетамид или гексаметилфосфортриамид; или сульфоксид, такой как диметилсульфоксид или сульфолан; и предпочтительно представляет собой галогенированный углеводород (предпочтительно метиленхлорид) или амид (предпочтительно диметилформамид).
Температура реакции обычно составляет от -20 до 150°С, предпочтительно от 0 до 50°С. Время реакции меняется в зависимости от используемых исходных веществ, растворителя и температуры реакции, но обычно время реакции составляет от 1 до 200 часов, предпочтительно от 24 до 100 часов. По окончании реакции требуемое соединение выделяют из реакционной смеси обычными методами. Например, требуемое соединение получают путем выделения высокомолекулярного носителя из реакционной смеси фильтрованием, промывкой органическим растворителем, таким как метиленхлорид, и сушкой при пониженном давлении.
(Стадия F-1)
Рассматриваемая стадия представляет собой стадию, на которой аналог 2-5A (1) получают на автоматическом синтезаторе ДНК обычными способами с использованием CPG (20), полученного на стадии E-4, используя соединения (3), (8), (12) и (14), полученные на стадии A-1, B-2, C-1 или D-1, и коммерчески доступного фосфорамидитного реагента (21).
Аналог 2-5A, имеющий целевую нуклеотидную последовательность, может быть синтезирован в соответствии со способом, описанным в литературе (Nucleic Acids Research, 12, 4539 (1984)), и руководством, прилагаемым к синтезатору, фосфорамидитным способом с использованием синтезатора ДНК, например, модели 392 компании Perkin Elmer Inc.
Соединение (21), например, 5'-O-диметокситритил-3'-O-(трет-бутилдиметилсилил)-N-бензоиладенозин-2'-O-(2-цианоэтил-N,N-диизопропилфосфорамидит) может быть закуплен у компании ChemGene Inc. (№ по каталогу: ANP-5681).
На рассматриваемой стадии амидитный реагент для соединений (3), (8), (12), (14) и (21) активируют с использованием кислого катализатора с образованием трисложноэфирной связи фосфорной кислоты, и он может быть окислен до сложного триэфира фосфорной кислоты с использованием соответствующего окисляющего агента, или его переводят в сложный триэфир тиофосфорной кислоты с использованием подходящего тионирующего агента.
Кислое вещество, используемое в качестве катализатора в реакции конденсации рассматриваемой стадии, может представлять собой кислое вещество, такое как тетразол, и предпочтительно представляет собой тетразол или этилтиотетразол. Окисляющий агент, используемый в реакции окисления рассматриваемой стадии, не имеет особых ограничений, если он обычно используется в реакциях окисления, и предпочтительно представляет собой неорганический окисляющий агент на основе металла, такой как оксид марганца, то есть перманганат калия или диоксид марганца; оксид рутения, то есть тетраоксид рутения; соединение селена, то есть диоксид селена; соединение железа, то есть хлорид железа; соединение осмия, то есть тетраоксид осмия; соединение серебра, то есть оксид серебра; соединение ртути, то есть ацетат ртути; оксид свинца, то есть оксид свинца или тетраоксид свинца; соединение хромовой кислоты, то есть хромат калия, комплекс (хромовая кислота)-(серная кислота) или комплекс (хромовая кислота)-(пиридин); или соединение церия, то есть церид нитрата аммония (CAN); неорганический окисляющий агент, такой как молекула галогена, то есть молекула хлора, молекула брома или молекула йода; перйодная кислота, то есть перйодат натрия; озон; перекись водорода; соединение азотистой кислоты, то есть азотистая кислота; соединение хлористой кислоты, например, хлорит калия или хлорит натрия; или соединение надсерной кислоты, то есть персульфат калия или персульфат натрия; или органический окисляющий агент, такой как реагент, используемый при окислении в ДМСО (комплекс диметилсульфоксида с дициклогексилкарбодиимидом, оксалилхлорид, уксусный ангидрид или пентаоксид фосфора, или комплекс пиридин-(триоксид серы); пероксид, такой как трет-бутилгидропероксид; стабильный катион, такой как трифенилметильный катион; имид янтарной кислоты, такой как имид N-бромянтарной кислоты; соединение хлорноватистой кислоты, такое как трет-бутилгидрохлорит; соединение азодикарбоновой кислоты, такое как сложный эфир азодикарбоновой кислоты; дисульфид, такой как диметилдисульфид, дифенилдисульфид или дипиридилдисульфид, и трифенилфосфин; сложный эфир азотистой кислоты, такой как метилнитрит; тетрагалогенид углерода, например тетрабромид углерода; или соединение хинона, например 2,3-дихлор-5,6-дициано-п-бензохинон (DDQ); предпочтительно йод.
Используемый растворитель не имеет особых ограничений, пока он не ингибирует реакцию и растворяет исходное вещество до определенной степени, и предпочтительно представляет собой ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как метиленхлорид или хлороформ; простой эфир, такой как тетрагидрофуран, диоксан или диметоксиэтан; амид, такой как диметилформамид, диметилацетамид или гексаметилфосфортриамид; сульфоксид, такой как диметилсульфоксид; спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол или изоамиловый спирт; разбавленная кислота, такая как водная серная кислота; разбавленное основание, такое как водный гидроксид натрия; вода; кетон, такой как ацетон или метилэтилкетон; гетероциклический амин, такой как пиридин; или нитрил, такой как ацетонитрил; предпочтительно гетероциклический амин (предпочтительно пиридин), нитрил (предпочтительно ацетонитрил), простой эфир (предпочтительно тетрагидрофуран) или галогенированный углеводород (предпочтительно метиленхлорид).
Кроме того, в том случае, когда соединение тионировано, если это желательно, производное тиоата может быть получено способом, описанным в литературе (Tetrahedron Letters, 32, 3005 (1991), J. Am Chem. Soc., 112, 1253 (1990)), с использованием реагента, такого как сера, тетраэтилтиурамдисульфид (TETD, Applied Biosystems Inc., или реагент Beaucage (Millipore Inc.)), для получения тиоата при взаимодействии с фосфитом.
Температура реакции обычно составляет от 0 до 150°С, предпочтительно от 10 до 60°С. Время реакции меняется в зависимости от исходного вещества, используемого растворителя и температуры реакции, но обычно составляет от 1 минуты до 20 часов, предпочтительно от 1 минуты до 1 часа.
В том случае, когда соединение Н-фосфоновой кислоты (4) или (9), полученное на стадии A-2 или B-3, конденсируют с образованием трисложноэфирной связи фосфорной кислоты на рассматриваемой стадии, то после его конденсации, например, в присутствии конденсирующего агента, такого как пивалоилхлорид, и восстановителя с образованием дисложноэфирной связи Н-фосфоновой кислоты, связь Н-фосфоновая кислота может быть превращена в дисложноэфирную связь фосфорной кислоты с использованием окислителя.
Растворитель, используемый на рассматриваемой стадии, не имеет особых ограничений, пока он не ингибирует реакцию, но предпочтительно используют безводный ацетонитрил. В качестве реагента, используемого как конденсирующий агент, используют хлорангидрид карбоновой кислоты или фосфорной кислоты, и предпочтительно используют пивалоилхлорид.
Окисляющий агент для окисления ODN, содержащего связь H-фосфорная кислота, до ODN фосфодисложноэфирного типа не имеет особых ограничений, если он обычно используется для реакций окисления, и может представлять собой неорганический окисляющий агент на основе металла, такой как оксид марганца, например перманганат калия или диоксид марганца; оксид рутения, например тетраоксид рутения; соединение селена, например диоксид селена; соединение железа, например хлорид железа; соединение осмия, например тетраоксид осмия; соединение серебра, например оксид серебра; соединение ртути, например ацетат ртути; оксид свинца, например оксид свинца или тетраоксид свинца; соединение хромовой кислоты, например хромат калия, комплекс (хромовая кислота)-(серная кислота) или комплекс (хромовая кислота)-(пиридин); или соединение церия, например церид нитрата аммония (CAN); неорганический окисляющий агент, такой как молекула галогена, например молекула хлора, молекула брома или молекула йода; перйодная кислота, например перйодат натрия; озон; перекись водорода; соединение азотистой кислоты, например азотистая кислота; соединение хлористой кислоты, например хлорит калия или хлорит натрия; или соединение надсерной кислоты, например персульфат калия или персульфат натрия; или органический окисляющий агент, такой как реагент, используемый при окислении в ДМСО (комплекс диметилсульфоксида с дициклогексилкарбодиимидом, оксалилхлорид, уксусный ангидрид или пентаоксид фосфора, или комплекс пиридин-триоксид серы); пероксид, такой как трет-бутилгидропероксид; стабильный катион, такой как трифенилметильный катион; имид янтарной кислоты, такой как имид N-бромянтарной кислоты; соединение хлорноватистой кислоты, такое как трет-бутилгипохлорит; соединение азодикарбоновой кислоты, такое как метилазодикарбоксилат; дисульфид, такой как диметилдисульфид, дифенилдисульфид или дипиридилдисульфид, и трифенилфосфин; сложный эфир азотистой кислоты, такой как метилнитрит; тетрагалогенид углерода, например тетрабромид углерода; или соединение хинона, например 2,3-дихлор-5,6-дициано-п-бензохинон (DDQ); предпочтительно использование молекулы йода.
Восстановитель, который должен быть использован, может представлять собой гетероциклический амин, такой как пиридин или диметиламинопиридин; или алифатический амин, такой как триметиламин, триэтиламин или диизопропилэтиламин; и предпочтительно представляет собой алифатический амин (в особенности диизопропилэтиламин). Температура реакции не имеет особых ограничений, но обычно составляет от -50 до 50°С, предпочтительно соответствует комнатной температуре.
Время реакции меняется в зависимости от исходных веществ, используемого реагента и температуры, но обычно оно составляет от 5 минут до 30 часов; и предпочтительно в случае, когда реакцию проводят при комнатной температуре, время реакции составляет 30 минут.
Растворитель для реакции образования метоксиметиламинофосфатной группы не имеет особых ограничений, пока он не ингибирует реакцию, но тетрахлорид углерода, который обычно используется в качестве реагента, используют в количестве, принятом для растворителя.
Температура реакции не имеет особых ограничений в интервале от -50 до 100°С, но в случае, когда реакцию проводят при комнатной температуре, время реакции составляет от 1 до 10 часов.
Кроме того, в случае, когда фосфодисложноэфирное соединение (5) или (10), полученное на стадии A-3 или B-4, конденсируют на рассматриваемой стадии с образованием фосфатной трисложноэфирной связи, используемый растворитель на данной стадии не имеет особых ограничений, пока он не ингибирует реакцию, но предпочтительно используют ароматический амин, такой как пиридин.
Конденсирующий агент, используемый при конденсации, может представлять собой дициклокарбодиимид (DCC), мезитиленсульфохлорид (Ms-Cl), триизопропилбензолсульфохлорид, триазолид мезитиленсульфоновой кислоты (MST), 3-нитротриазолид мезитиленсульфоновой кислоты (MSNT), тетразолид триизопропилбензолсульфоновой кислоты (TPS-Te), нитроимидазолид триизопропилбензолсульфоновой кислоты (TPS-NI) или пиридилтетразолид триизопропилбензолсульфоновой кислоты, и предпочтительно представляет собой MSNT, TPS-Te и TPS-NI.
Температура реакции особенно не ограничена в интервале от -10 до 100°С, но обычно реакцию проводят при комнатной температуре.
Время реакции меняется в зависимости от используемого растворителя и температуры реакции, но в случае, когда используют пиридин в качестве реакционного растворителя, и реакцию проводят при комнатной температуре, время реакции составляет 30 минут.
Отщепление от CPG в случае, когда аналог 2-5 связан с CPG, и удаление защитных групп, отличных от замещающего фрагмента на 5'-конце, описанных ниже, может быть осуществлено общеизвестным способом (J. Am. Chem. Soc., 103, 3185, (1981)).
Полученный неочищенный аналог 2-5A может быть идентифицирован очисткой с использованием хроматографической колонки с обращенной фазой и анализом чистоты очищенного продукта с помощью ВЭЖХ.
Длина цепи полученного таким образом олигонуклеотидного аналога обычно составляет от 2 до 50, предпочтительно от 10 до 30 нуклеозидных звеньев.
(Стадия G-1)
Рассматриваемая стадия служит для получения аналога 2-5A (1) на автоматическом синтезаторе ДНК с помощью обычных способов с использованием CPG (22), с использованием соединений (3), (4), (5), (8), (9), (10), (12) или (14), полученных на стадии A-1, A-2, A-3, B-2, B-3, B-4, C-1 или D-1, и (21).
CPG (22) является тем же самым соединением, что и соединение (24), описанное в способе G японской патентной заявки (Kokai) № 2002-249497, и рассматриваемую стадию проводят аналогично стадии F-1.
(Стадия H-1)
Рассматриваемая стадия представляет собой стадию, на которой аналог 2-5A (1) получают на автоматическом синтезаторе ДНК обычными способами с использованием CPG (23), с использованием соединений (3), (4), (5), (8), (9), (10), (12) или (14), полученных на стадии A-1, A-2, A-3, B-2, B-3, B-4, C-1 или D-1, и (21).
CPG (23) является тем же самым соединением, что и соединение (4), описанное в японской патентной заявке (Kokai) № Hei 7-53587, и рассматриваемую стадию проводят аналогично стадии F-1.
Кроме того, в аналоге 2-5A (1), в случае, когда любой из заместителей R1, R2, R3, R4, R5, R6 и R7 представляет собой меркаптогруппу, после синтеза и очистки аналога 2-5A (1) Способом F, G или H, в меркаптогруппу может быть введен заместитель реакцией с соединением, имеющим галогенидную группу, в присутствии основания в инертном растворителе.
Галоген может представлять собой, например, атом фтора, атом хлора, атом брома или атом йода, и предпочтительно представляет собой атом хлора, атом брома или атом йода.
Используемое соединение, содержащее галогенидную группу, не имеет особых ограничений, пока это соединение представляет собой соединение, содержащее галогенидную группу, которая может быть введена в реакцию с группой тиофосфорной кислоты, и может представлять собой, например, «алкилгалогенид, который может быть замещен», такой как этилгалогенид, пропилгалогенид, бутилгалогенид, 2-галогенэтанол, 3-галогенпропанол или 4-галогенбутанол;
«ацилоксиалкилгалогенид», такой как
2-(стеароилокси)этилгалогенид, 2-(миристоилокси)этилгалогенид,
2-(деканоилокси)этилгалогенид, 2-(бензоилокси)этилгалогенид,
2-(пивалоилокси)этилгалогенид,
2-(2,2-диметилоктадеканоилокси)этилгалогенид,
3-(стеароилокси)пропилгалогенид,
3-(миристоилокси)пропилгалогенид,
3-(деканоилокси)пропилгалогенид, 3-(бензоилокси)пропилгалогенид,
3-(пивалоилокси)пропилгалогенид,
3-(2,2-диметилоктадеканоилокси)пропилгалогенид,
4-(стеароилокси)бутилгалогенид, 4-(миристоилокси)бутилгалогенид,
4-(деканоилокси)бутилгалогенид, 4-(бензоилокси)бутилгалогенид,
4-(пивалоилокси)бутилгалогенид или
4-(2,2-диметилоктадеканоилокси)бутилгалогенид;
«алкилкарбамоилоксиалкилгалогенид», такой как 2-стеарилкарбамоилоксиэтилгалогенид; или одно из следующих соединений:
Из приведенных выше соединений 2-стеароилоксиэтилгалогенид и 2-(2,2-диметилоктадеканоилокси)этилгалогенид являются предпочтительными.
Из соединений, содержащих указанные галогенидные группы, соединения, содержащие сложноэфирную группу (-OC(=O)- или -C(=O)O-), карбаматную группу (-NHC(=O)O- или -OC(=O)NH-), амидную группу (-NHC(=O)- или -C(=O)NH-), сложную тиоэфирную группу (-SC(=O)- или -C(=O)S-), группу мочевины (-NHC(=O)NH-), сложноэфирную группу тиокарбоновой кислоты (-OC(=S)- или -C(=S)O-) или амидную группу тиокарбоновой кислоты (-NHC(=S)- или -C(=S)NH-), могут быть получены в присутствии основания или конденсирующего агента конденсацией соединения галогенангидрида кислоты или соединения карбоновой кислоты с соединением, содержащим спиртовую группу; конденсацией соединения галогенида сложного эфира муравьиной кислоты с соединением, содержащим аминогруппу; конденсацией соединения галогенангидрида кислоты или карбоновой кислоты c соединением, содержащим аминогруппу; конденсацией галогенангидрида кислоты или карбоновой кислоты с соединением, содержащим тиольную группу; конденсацией соединения, содержащего два вида аминогруппы с фосгеном; конденсацией соединения тиокарбоновой кислоты с соединением, содержащим спиртовую группу; или конденсацией соединения тиокарбоновой кислоты с соединением, содержащим аминогруппу.
Используемое основание может представлять собой гетероциклический амин, такой как пиридин или диметиламинопиридин; или алифатический амин, такой как триметиламин, триэтиламин или диизопропиламин; и предпочтительно представляет собой гетероциклический амин (предпочтительно пиридин).
Не существует особенного ограничения для используемого растворителя при условии, что он не ингибирует реакцию и растворяет исходное вещество до определенной степени, и он может представлять собой воду; амид, такой как диметилформамид, диметилацетамид или гексаметилфосфортриамид; сульфоксид, такой как диметилсульфоксид; гетероциклический амин, такой как пиридин; нитрил, такой как ацетонитрил; или смесь указанных растворителей; и предпочтительно представляет собой диметилформамид.
Температура реакции не имеет особых ограничений в интервале от -50 до 100°С, но обычно реакцию проводят при комнатной температуре. Время реакции меняется в зависимости от вещества, используемого реагента и температуры реакции, но обычно составляет от 10 до 100 часов.
Скорость реакции также может быть соответствующим образом повышена при добавлении йодида, такого как йодид тетрабутиламмония.
Вместо используемого CPG (23), который используется в способе H, может быть синтезирован 2-5А антисмысловой олигонуклеотид конденсацией фосфорамидита, служащего в качестве линкера, такого как DMT-бутанол-CED-фосфорамидит (ChemGene) или Spacer-фосфорамидит 18 (GlenResearch), с CPG, к которому присоединен олигонуклеотид, имеющий желаемую антисмысловую последовательность, который защищен защитной группой; затем проводят процедуру рассматриваемой стадии. Например, в случае «CPG, к которому присоединен олигонуклеотид, защищенный защитной группой», может быть синтезирован модифицированный олигонуклеотид, в котором атом кислорода в положении 2' фрагмента сахара присоединен мостиковой связью к атому углерода в положении 4' с помощью алкиленовой группы способом, описанным в японской патентной заявке (Kokai) № Hei 10-304889 или в японской патентной заявке (Kokai) № 2000-297097. Кроме того, модифицированный олигонуклеотид, содержащий 2'-O-метоксиэтоксигруппу, может быть синтезирован, если обратиться к следующим литературным источникам (Teplove, M. et al., Nat. Struct. Biol. (1999), 6, 535; Zhang H. et al., Nature Biotech. (2000), 18, 862), и модифицированный олигонуклеотид, содержащий 3'-аминогруппу, может быть синтезирован, если обратиться к следующим литературным источникам (Gryaznov, S.M. et al., Proc. Natl. Acad. Sci. USA 1995, 92, 5798; Tereshko, V. et al., J. Am. Chem. Soc. 1998, 120, 269).
Противоопухолевая активность (цитоцидная активность) соединений настоящего изобретения может быть изучена путем добавления соединений настоящего изобретения к раковым клеткам в среде и выращивания клеток с последующим подсчетом числа жизнеспособных клеток с использованием метода оценки MTT (Tim Mosmann, J. Immunological Methods, 1983: 65, 55-63), метода оценки MTS (Rotter, B.A., Thompson, B.K., Clarkin, S., Owen, T.C. Nat. Toxins 1993; 1(5): 303-7), метода оценки XTT (Meshulam, T., Levitz, S.M., Christin, L., Diamond, R.D. J. Infect. Dis. 1995; 172(4): 1153-6) или окрашивания трипановым синим.
Противовирусная активность соединений настоящего изобретения может быть изучена при использовании системы культуры инфицированных клеток, таких как клетки HeLa, клетки MDCK, клетки MRC-5 и др., путем добавления соединений настоящего изобретения к вирусным клеткам, таким как вирус коровьей оспы, вирус гриппа или цитомегаловирус, в среде или до или после инфицирования, выдерживания в течение определенного времени и последующего измерения степени ингибирования размножения вируса с использованием метода подсчета антителообразующих клеток, с помощью которого определяют инфекционный титр вируса (Kobayashi, N., Nagata, K. Virus Experimental Protocols, Medical View Publishing), или методом ELISA, с помощью которого определяют уровень вирусного антигена (Okuno, Y., Tanaka, K., Baba, K., Maeda, A., Kunita, N., Ueda, J. Clin. Microbiol., June 1, 1990; 28(6): 1308-13).
Введение аналогов 2-5A общей формулы (1) настоящего изобретения может включать, например, пероральное введение с помощью таблеток, капсул, гранул, порошков или сиропов, или парентеральное введение с помощью инъекций или суппозиториев. Такие препараты получают известными способами с использованием вспомогательных веществ, таких как эксципиенты (которые включают, например, органические эксципиенты, такие как производные сахаров, например лактозу, сахарозу, глюкозу, маннит и сорбит; производные крахмала, например кукурузный крахмал, картофельный крахмал, α-крахмал и декстрин; производные целлюлозы, например кристаллическую целлюлозу; аравийскую камедь; декстран; и пуллулан; и неорганические эксципиенты, такие как силикатные производные, например легкий кремниевый ангидрид, синтетический алюмосиликат, силикат кальция и алюмо(метасиликат)магния; фосфаты, например гидрофосфат кальция; карбонаты, например карбонат кальция; и сульфаты, например, сульфат кальция), лубриканты (которые могут включать, например, стеариновую кислоту и ее соли с металлами, такие как стеариновая кислота, стеарат кальция и стеарат магния; тальк; коллоидный диоксид кремния; воски, такие как пчелиный воск и спермацет; борную кислоту; адипиновую кислоту; сульфаты, такие как сульфат натрия; гликоль; фумаровую кислоту; бензоат натрия; DL-лейцин; натриевые соли алифатических кислот; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; кремниевые кислоты, такие как кремниевый ангидрид; и приведенные выше производные крахмала), связующие вещества (которые могут включать, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон, Macrogol и соединения, аналогичные приведенным выше эксципиентам), дезинтегрирующие агенты (которые могут включать, например, производные целлюлозы, такие как низкозамещенная гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, кальцийкарбоксиметилцеллюлоза и внутренне поперечно сшитая натрийкарбоксиметилцеллюлоза; и химически модифицированные крахмалы/целлюлозы, такие как карбоксиметилкрахмал, натрий-карбоксиметилкархмал и поперечно сшитый поливинилпирролидон), стабилизаторы (которые могут представлять собой параоксибензоаты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; бензалконийхлорид; фенолы, такие как фенол и крезол; тимеросал; дегидроуксусная кислота; и сорбиновая кислота), корригирующие вкус и запах агенты (которые могут включать, например, подслащивающие вещества, подкислители, отдушки или т.д., которые обычно находят применение), разбавители и т.д.
Хотя используемое количество соединения меняется в зависимости симптомов, возраста, способа введения и т.д., желательно введение от одного раза до нескольких раз в день, и в случае перорального введения предпочтительно вводить 0,01 мг/кг массы тела (предпочтительно 0,1 мг/кг массы тела) за один раз в качестве нижней границы и 1000 мг/кг массы тела (предпочтительно 100 мг/кг массы тела) в качестве верхней границы, и в случае внутривенного введения предпочтительно вводить 0,001 мг/кг массы тела (предпочтительно 0,01 мг/кг массы тела) за один раз в качестве нижней границы и 100 мг/кг массы тела (предпочтительно 10 мг/кг массы тела) в качестве верхней границы соответственно симптомам.
Кроме того, препараты могут быть использованы в комбинации с другими противоопухолевыми агентами, например химикатами типа нитрозомочевины, такими как 5FU, AraC, ACNU или BCNU, цисплатин, дауномицин, адриамицин, митомицин C, винкристин и таксол.
Далее настоящее изобретение объяснено более подробно с помощью примеров, справочных примеров и примеров испытаний.
Наилучший способ осуществления изобретения
(Пример 1)
Синтез соединения примера 1 (Пример соединения № 4)
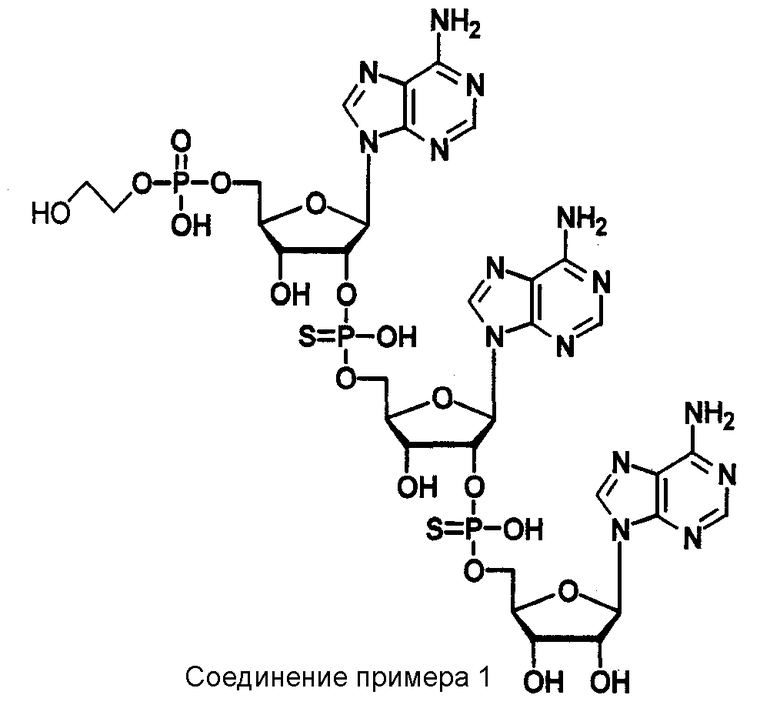
В качестве синтезатора ДНК используют синтезатор ДНК/РНК ABI Модель 392 (Applied Biosystems). Растворители, реагенты и концентрации фосфорамидита в каждом цикле синтеза являются такими же, что и в случае общего синтеза природных олигонуклеотидов, и продукты Applied Biosystems используют в случае реагентов и растворителей, не являющихся фосфорамидитом и сульфурирующим агентом. Аналог 5'-O-DMTr-рибоаденозина, Bz-Аденозин-РНК-500 (Glen Research) (2,0 мкмоль), связанный с подложкой CPG, используют в качестве исходного вещества. Синтез проводят с использованием синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4) с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) используют в качестве фосфорамидита в циклах 1 и 2, тогда как в цикле 3 используют соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592. Для окисления или в качестве сульфурирующего агента в циклах 1 и 2 используют ксантангидрид (Tokyo Kasei Kogyo), тогда как в цикле 3 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Сульфурирование (циклы 1 и 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
Окисление (цикл 3): йод/вода/пиридин/тетрагидрофуран; 15 с.
После синтеза защищенного аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-13% CH3CN (линейный градиент, 30 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 20,9, 22,7, 25,4 и 28,0 мин, что соответствует четырем диастереомерам. Соединение настоящего изобретения элюируются приблизительно через 10,55 мин при анализе с помощью ионообменной ВЭЖХ (колонка (Tosoh DEAE-2SW (4,6×150 мм)); раствор A (20% ацетонитрил), раствор B (20% ацетонитрила и 67 мМ фосфатного буфера, 2 М NaCl); раствор B 5→60% (15 мин, линейный градиент); 60°С; 1 мл/мин). (Выход: 457 нмоль при измерении с помощью УФ с использованием рассчитанного значения ε=39400 (260 нм) аденозинового триммера)), λмакс (H2O)=258,3 нм, ESI-Масс-спектр (отрицательный): 1080,1 [M-H]-.
(Пример 2)
Синтез соединения примера 2 (Пример соединения № 1)
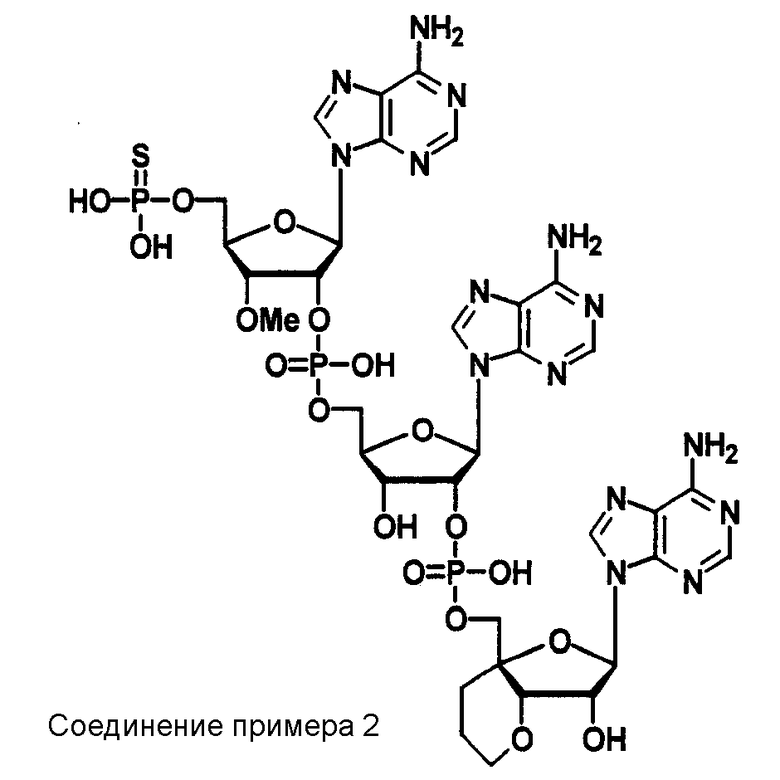
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) № 2002-249497 (2,0 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 2 используют 5'-DMT-3'-(O-метил)аденозин(bz)-2'-фосфорамидит (ChemGene), в цикле 3 используют химический фосфорилирующий реагент II (Glen Research). При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют йод, тогда как в цикле 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру, в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 30 мин); 40°С; 10 мл/мин; 254 нм), собирают фракцию, которая элюируется через 16,7 мин. Соединение настоящего изобретения элюируется приблизительно через 9,46 мин при анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-25% CH3CN (линейный градиент, 14 мин); 60°С; 10 мл/мин). (Выход: 445 нмоль при измерении с помощью УФ при 260 нм), λмкс (H2O)=258,2 нм, ESI-Масс-спектр (отрицательный): 1074,15 [M-H]-.
(Пример 3)
Синтез соединения примера 3 (Пример соединения № 5)
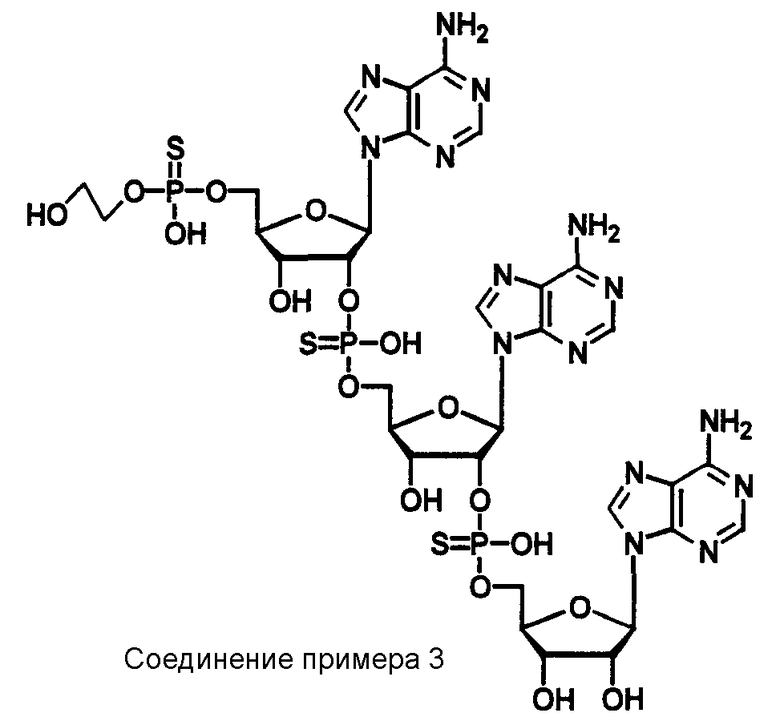
Синтез проводят с использованием Bz-Аденозин-РНК 500 (Glen Research Co.) (2,0 мкмоль) в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в циклах 1 и 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 3 используют соединение примера 8а, описанное в японской патентной заявке (Kokai) № Hei 11-246592. В циклах 1, 2 и 3 в качестве сульфурирующего агента используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 1, 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру с все еще незатронутой группой 5'-DMTr, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 60% CH3CN (изократно); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 9,5 и 11,8 мин как диастереомеры. После отгонки растворителя при пониженном давлении добавляют 80%-ную водную уксусную кислоту, смесь оставляют стоять в течение 30 мин, и при этом удаляется группа DMTr. После отгонки растворителя добавляют смесь концентрированный водный аммиак-этанол (4:1) и смесь оставляют стоять в течение 30 мин. После отгонки растворителя доводят pH до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.), с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 30 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 16,5-19,1 мин. Соединение настоящего изобретение элюируется приблизительно через 8,8-9,8 мин при анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-25% CH3CN (линейный градиент, 14 мин); 60°С; 10 мл/мин). (Выход: 565 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,2 нм, ESI-Масс-спектр (отрицательный): 1096,1 [M-H]-.
(Пример 4)
Синтез соединения примера 4 (Пример соединения № 8)
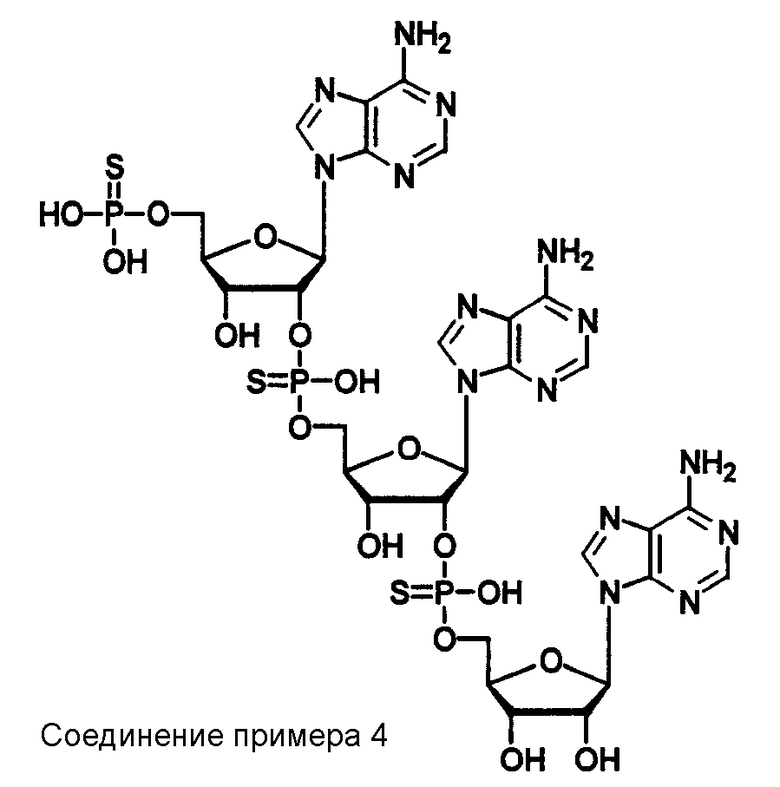
Синтез проводят с использованием Bz-Аденозин-РНК 500 (Glen Research Co.) (2,0 мкмоль) в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в циклах 1 и 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 3 используют химический фосфорилирующий реагент II (Glen Research Co.). В циклах 1, 2 и 3 в качестве сульфурирующего агента используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 1, 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 6-25% CH3CN (линейный градиент, 30 мин); 40°С; 10 мл/мин; 254 нм), собирают четыре фракции, которые элюируются через 13,3, 13,7, 13,9 и 14,4 мин, что соответствует четырем диастереомерам. Соединение настоящего изобретение элюируется приблизительно через 7,2-8,0 мин при анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-20% CH3CN (линейный градиент, 10 мин); 60°C; 10 мл/мин). (Выход: 252 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,0 нм, ESI-Масс-спектр (отрицательный): 1052,1 [M-H]-.
(Пример 5)
Синтез соединения примера 5 (Пример соединения № 290)
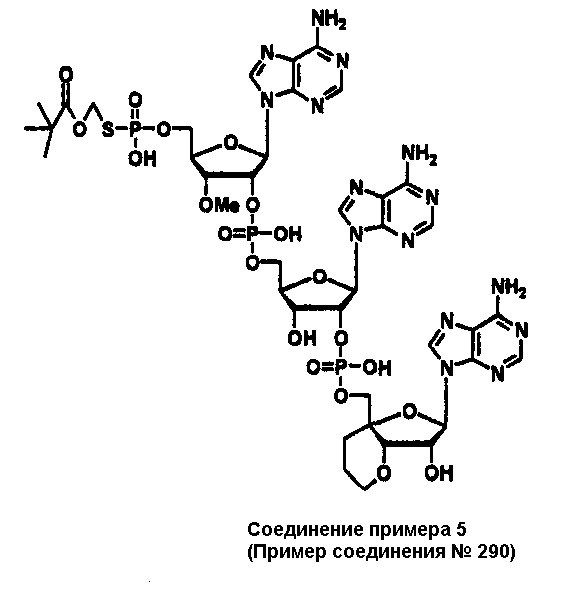
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мкл пивалоилоксиметилхлорида (Tokyo Kasei Kogyo), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-42% CH3CN (линейный градиент, 10 мин); 60°С; 10 мл/мин; 254 нм), собирают фракцию через 5,6 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-42% CH3CN (линейный градиент, 14 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,52 мин. (Выход: 4,8 нмоль), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1188,2 [M-H]-.
(Пример 6)
Синтез соединения примера 6 (Пример соединения № 334)
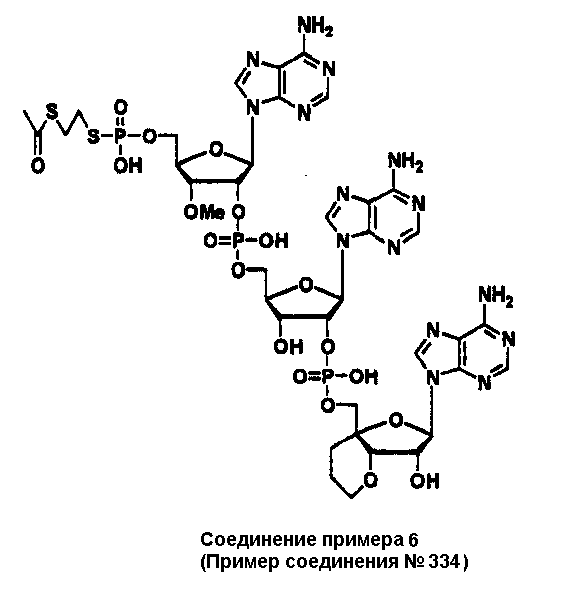
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мкл S-(2-бромэтилового) эфира тиоуксусной кислоты (Bauer, L. et al. J. Org. Chem. 1965, 30, 949-951), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 10 мл/мин; 254 нм), собирают фракцию через 4,5 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,25 мин. (Выход: 14 нмоль), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1176,2 [M-H]-.
(Пример 7)
Синтез соединения примера 7 (Пример соединения № 953)
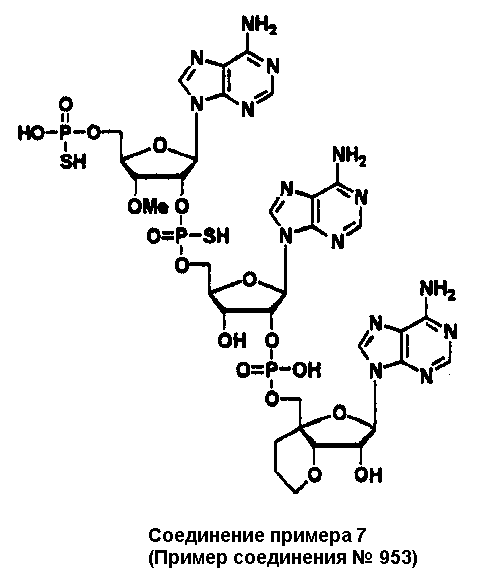
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) № 2002-249497 (2,0 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 2 используют 5'-DMT-3'-(O-метил)аденозин-(N-bz)-2'-фосфорамидит (ChemGene) и в цикле 3 используют химический фосфорилирующий реагент II (Glen Research). При окислении или в качестве сульфурирующего агента в цикле 1 используют йод и в циклах 2 и 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 1): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 12,1 и 13,0 мин, что соответствует двум диастереомерам. Соединение настоящего изобретение элюируется приблизительно через 8,66 и 8,98 мин при анализе помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин). (Выход: 768 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI Масс-спектр (отрицательный): 1090,2 [M-H]-.
(Пример 8)
Синтез соединения примера 8 (Пример соединения № 954)
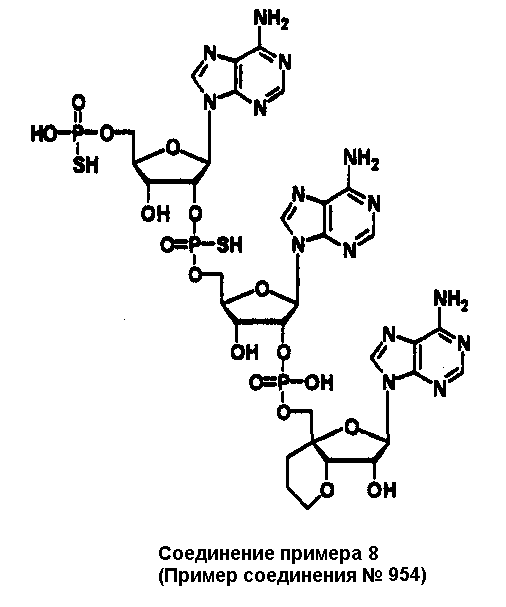
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) № 2002-249497 (2,0 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в циклах 1 и 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 3 используют химический фосфорилирующий реагент II (Glen Research). При окислении или в качестве сульфурирующего агента в цикле 1 используют йод и в циклах 2 и 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 1): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 11,5 и 12,4 мин, что соответствует двум диастереомерам. Соединение настоящего изобретение элюируется приблизительно через 8,28 и 8,60 мин при анализе помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин). (Выход: 718 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1076,1 [M-H]-.
(Пример 9)
Синтез соединения примера 9 (Пример соединения № 955)
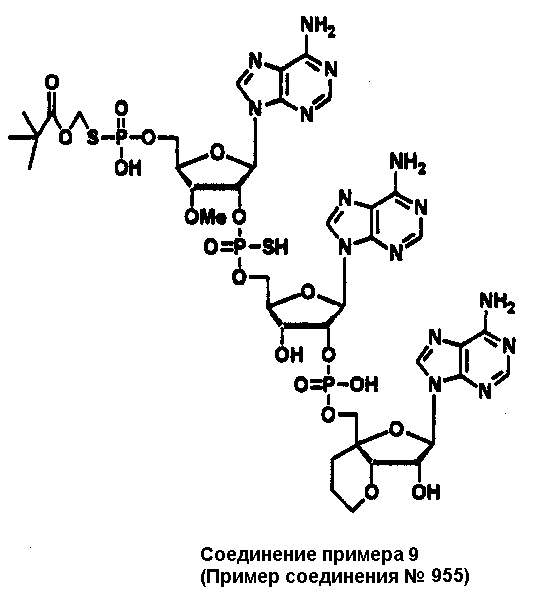
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 7 и добавляют 1 мкл пивалоилоксиметилхлорида (Tokyo Kasei Kogyo), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 10 мл/мин; 254 нм), собирают фракции через 5,5 и 5,6 мин, что соответствует двум диастереомерам. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,42 и 7,56 мин. (Выход: 1,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1204,2 [M-H]-.
(Пример 10)
Синтез соединения примера 10 (Пример соединения № 956)
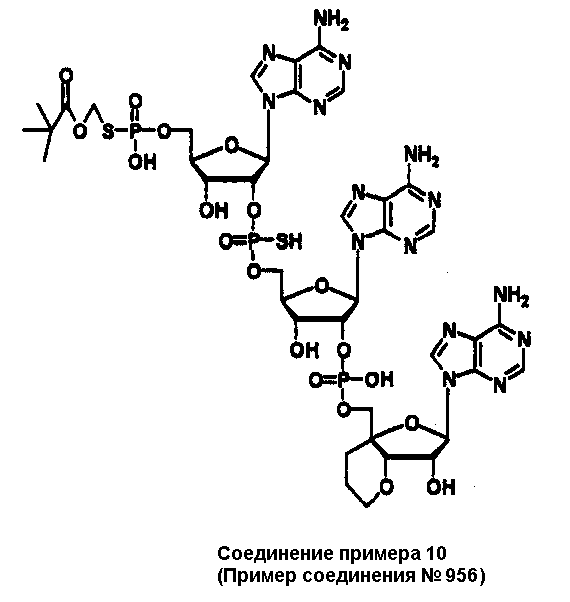
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 8 и добавляют 1 мкл пивалоилоксиметилхлорида (Tokyo Kasei Kogyo), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракции через 5,7 и 5,9 мин, что соответствует двум диастереомерам. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,68 и 7,85 мин. (Выход: 11 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1191,20 [M-H]-.
(Пример 11)
Синтез соединения Примера 11 (Пример соединения № 957)
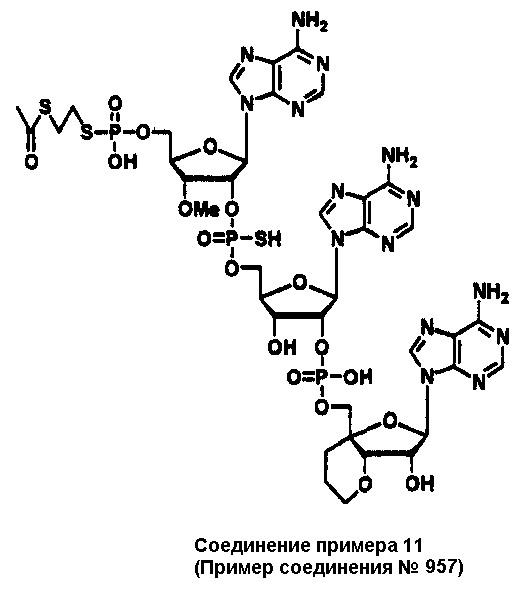
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 7 и добавляют 1 мкл S-(2-бромэтилового) эфира тиоуксусной кислоты (Bauer, L. et al. J. Org. Chem. 1965, 30, 949-951), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракции через 4,7 и 4,8 мин, что соответствует двум диастереомерам. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°C; 1 мл/мин) соединение настоящего изобретения элюируется через 6,57 и 6,75 мин. (Выход: 20 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1192,1 [M-H]-.
(Пример 12)
Синтез соединения примера 12 (Пример соединения № 958)
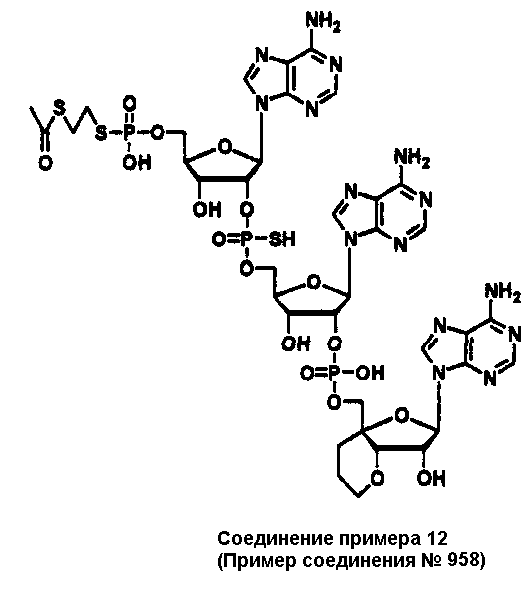
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 8 и добавляют 1 мкл S-(2-бромэтилового) эфира тиоуксусной кислоты (Bauer, L. et al. J. Org. Chem. 1965, 30, 949-951), приблизительно 1 мг йодида тетрабутиламмония (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракции через 4,9 и 5,1 мин, что соответствует двум диастереомерам. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-43% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 6,83 и 7,04 мин. (Выход: 6,7 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1178,1 [M-H]-.
(Пример 13)
Синтез соединения примера 13 (Пример соединения № 964)
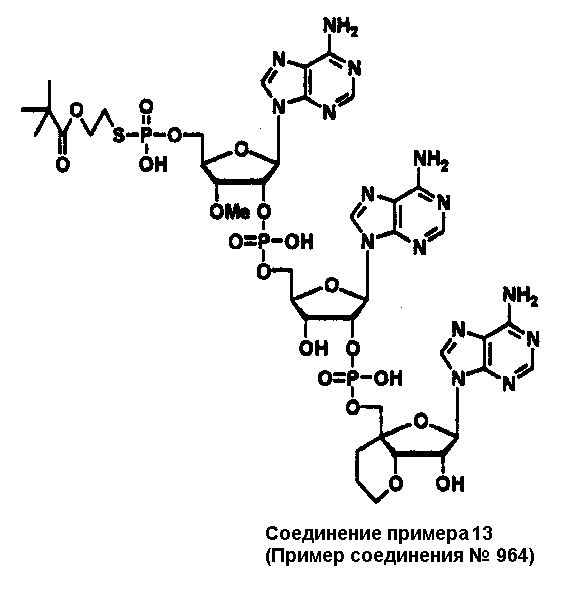
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мкл 2-(пивалоилокси)этилбромида (способ получения, описанный в публикации EP0395313) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 4,3 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,48 мин. (Выход: 19,1 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,6 нм, FAB-Масс-спектр (отрицательный): 1202 [M-H]-.
(Пример 14)
Синтез соединения примера 14 (Пример соединения № 965)
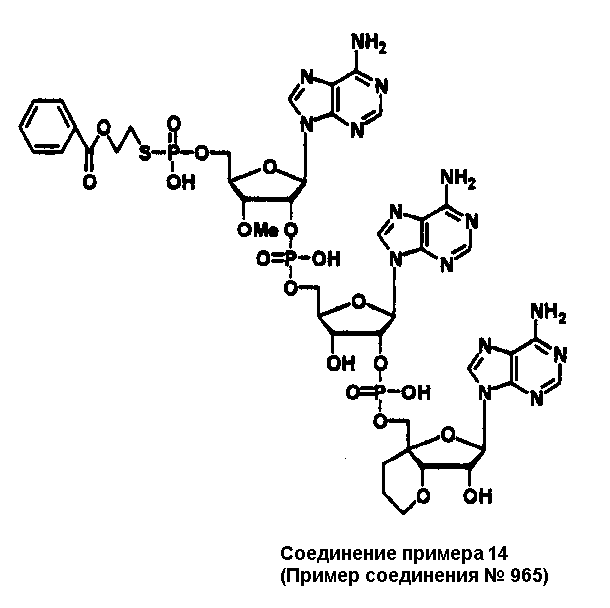
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мкл 2-(бензоилокси)этилбромида (Tokyo Kasei Kogyo) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 7,0 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,44 мин. (Выход: 19,7 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,7 нм, FAB-Масс-спектр (отрицательный): 1222 [M-H]-.
(Пример 15)
Синтез соединения примера 15 (Пример соединения № 967)
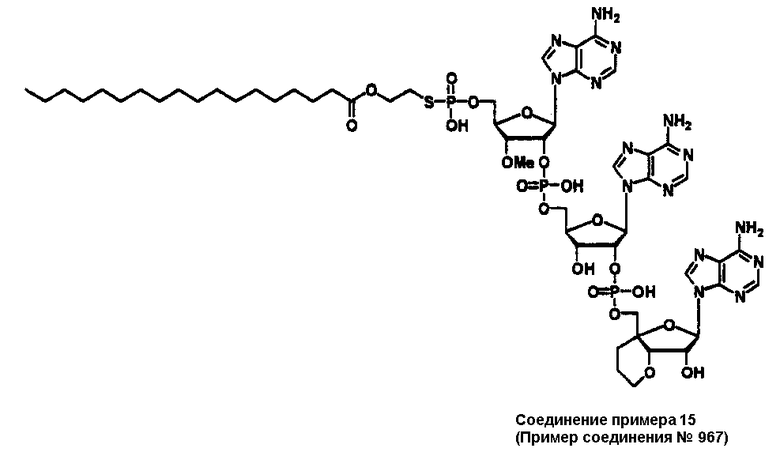
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 8,2 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 14,62 мин. (Выход: 14,9 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=260,1 нм, FAB-Масс-спектр (отрицательный): 1384 [M-H]-.
(Пример 16)
Синтез соединения примера 16 (Пример соединения № 968)
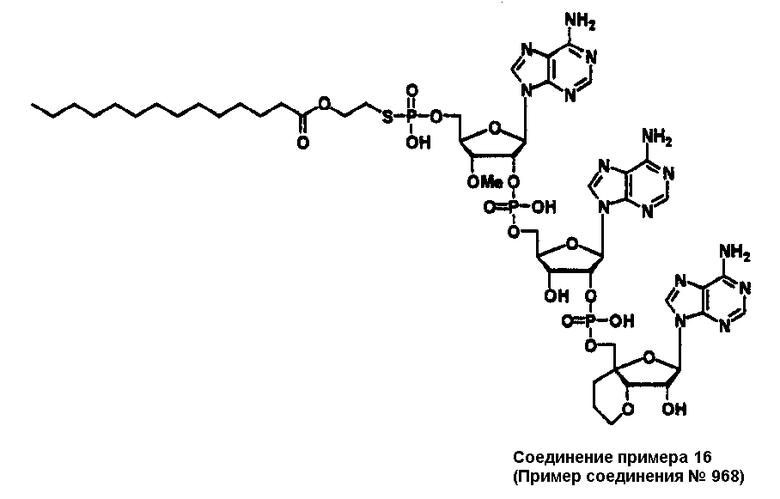
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мг 2-(миристоилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, после чего протекает взаимодействие смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 6,3 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 12,57 мин. (Выход: 13,1 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259,7 нм, FAB-Масс-спектр (отрицательный): 1328 [M-H]-.
(Пример 17)
Синтез соединения примера 17 (Пример соединения № 969)
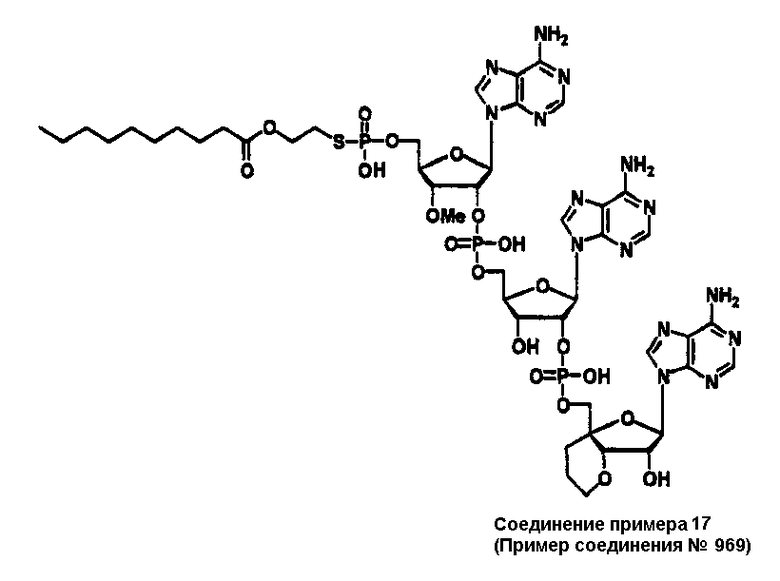
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 2 и добавляют 1 мг 2-(деканоилокси)этилбромида (Devinsky, Ferdinand et al., Collect. Czech. Chem. Commun. 49, 12, 1984, 2819-2827) и 1 мкл триэтиламина, после чего реакция смеси протекает при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 50 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 4,3 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-80% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 10,36 мин. (Выход: 19,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,2 нм, FAB-Масс-спектр (отрицательный): 1272 [M-H]-.
(Пример 18)
Синтез соединения примера 18 (Пример соединения № 1074)
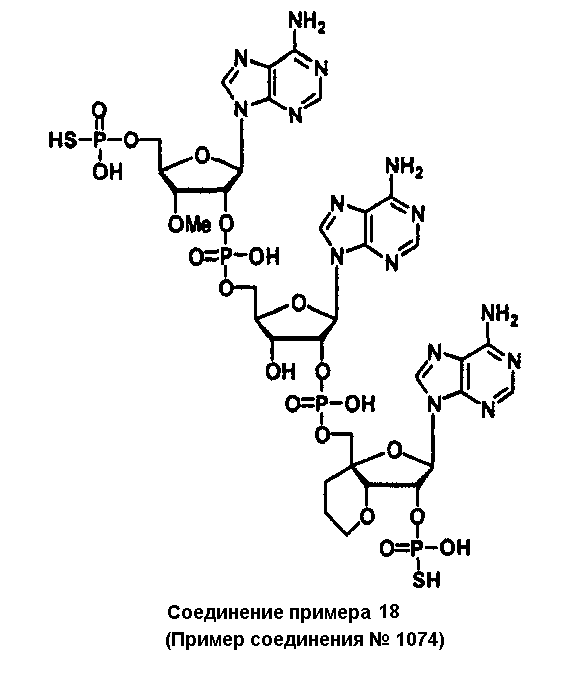
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (2,0 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 16, описанное в японской патентной заявке (Kokai) № 2002-249497, в качестве фосфорамидита в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene) и соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. Для окисления или в качестве сульфурирующего агента в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo) и в циклах 2, 3 и 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 2, 3 и 4): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (цикл 1): ксантангидрид (0,02М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-17% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 254 нм), собирают фракцию, которая элюируется через 14,9 мин. Соединение настоящего изобретения элюируется приблизительно через 6,77 мин при анализе ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин). (Выход: 1440 нмоль при измерении помощью УФ при 260 нм), λмакс (H2O)=258,5 нм, ESI-Масс-спектр (отрицательный): 1198,1 [M-H]-.
(Пример 19)
Синтез соединения примера 19 (Пример соединения № 1075)
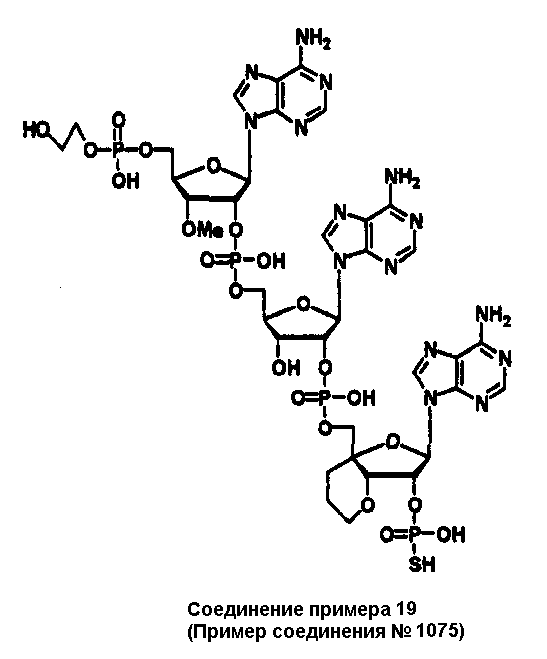
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (2,0 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 16, описанное в японской патентной заявке (Kokai) № 2002-249497, в качестве фосфорамидита в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene) и химический фосфорилирующий реагент II (Glen Research) используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 1 и 4 используют ксантангидрид (Tokyo Kasei Kogyo) и в циклах 2 и 3 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 2 и 3):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1 и 4): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-17% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 254 нм), собирают фракцию, которая элюируется через 15,5 мин. Соединение настоящего изобретения элюируется приблизительно через 8,63 мин при анализе ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин). (Выход: 1482 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,2 нм, ESI-Масс-спектр (отрицательный): 1170,1 [M-H]-.
(Пример 20)
Синтез соединения примера 20 (Пример соединения № 1937)
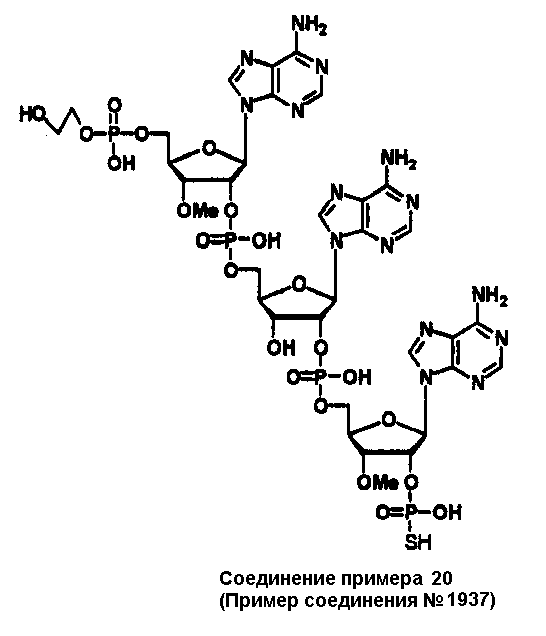
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (1,0 мкмоль). В качестве фосфорамидита в циклах 1 и 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene), в качестве фосфорамидита в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 4 используют соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592. В качестве окислителя используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление: йод/вода/пиридин/тетрагидрофуран; 15 с.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, при котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 2,5-10% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракцию, которая элюируется через 4,8 мин. Соединение настоящего изобретения элюируется приблизительно через 3,16 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-10% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин). (Выход: 95 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=256,2 нм, ESI-Масс-спектр (отрицательный): 1171,9 [M-H]-.
(Пример 21)
Синтез соединения примера 21 (Пример соединения № 1099)
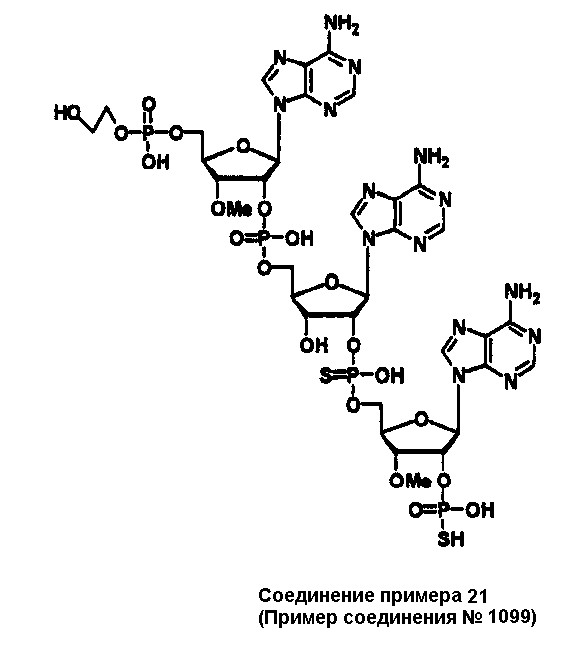
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (1,0 мкмоль). В качестве фосфорамидита в циклах 1 и 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene), в качестве фосфорамидита в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 4 используют соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592. Для окисления или в качестве сульфурирующего агента ксантангидрид (Tokyo Kasei Kogyo) используют в циклах 1 и 2 и йод используют в циклах 3 и 4.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 3 и 4):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1 и 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-10% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракции, которые элюируются через 6,0 и 6,4 мин. Соединение настоящего изобретения элюируется приблизительно через 4,89 и 5,43 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-10% CH3CN (линейный градиент, 10 мин); 60°C; 2 мл/мин). (Выход: 54 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,0 нм, ESI-Масс-спектр (отрицательный): 1189 [M-H]-.
(Пример 22)
Синтез соединения примера 22 (Пример соединения № 1110)
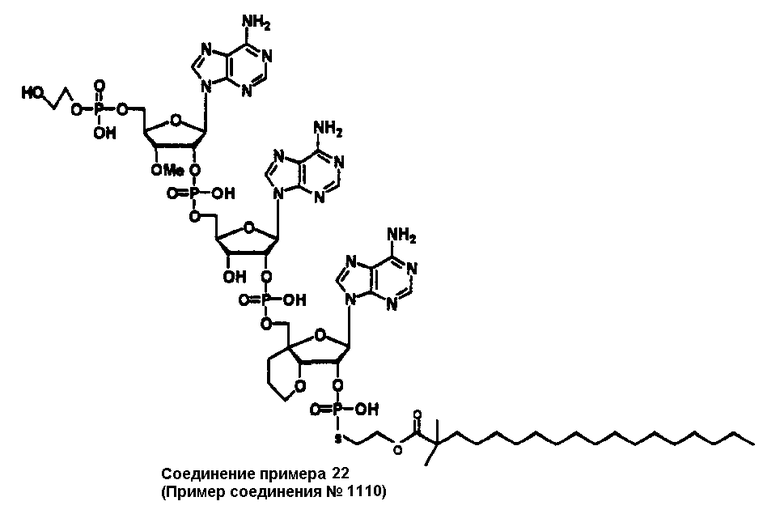
В безводном дихлорметане (10 мл) растворяют 500 мг (1,6 ммоль) 2,2-диметилоктадекановой кислоты (Roth, Bruce D. et al., J. Med. Chem. 1992, 35(9), 1609-17) и добавляют 350 мг (1,8 ммоль) дициклогексилкарбодиимида (DCC) и 140 мкл (2 ммоль) 2-бромэтанола, затем смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь очищают на колонке с силикагелем (элюирование смесью растворителей гексан-этилацетат, (7:1)), получая 230 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, который используют на следующей стадии синтеза.
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 19 и добавляют 3 мг 2-(2,2-диметилоктадеканоилокси)этилбромида и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 33-80% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 6,7 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 9,65 мин. (Выход: 24,5 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259,3 нм, ESI-Масс-спектр (отрицательный): 1537,3 [M-H]-.
(Пример 23)
Синтез соединения примера 23 (Пример соединения № 1111)
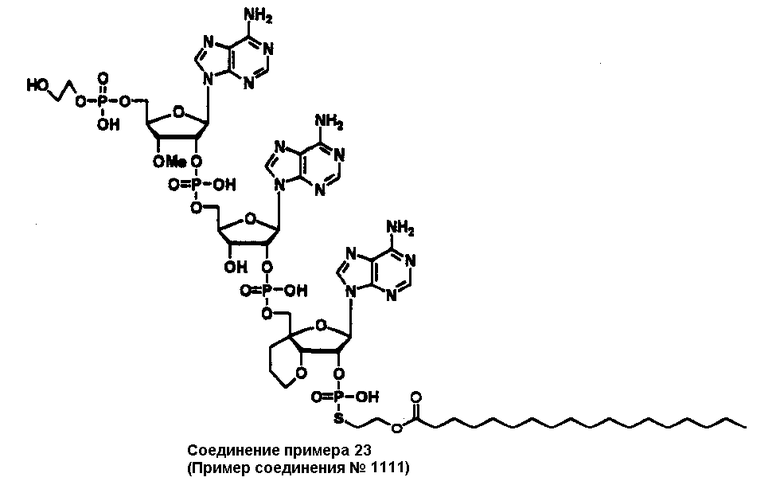
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 19 и добавляют 3 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 52-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 1,9 мин. При анализе ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 15-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 10,06 мин. (Выход: 39,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,2 нм, ESI-Масс-спектр (отрицательный): 1508,4 [M-H]-.
(Пример 24)
Синтез соединения примера 24 (Пример соединения № 1112)
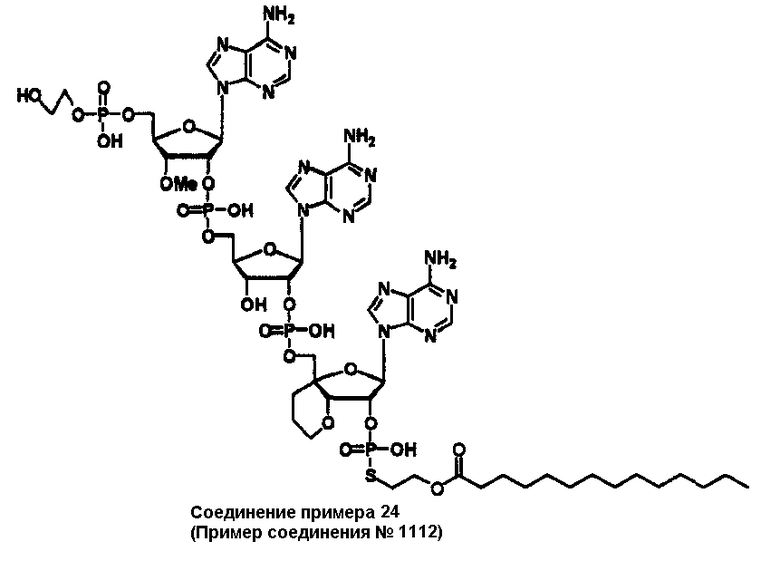
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 19 и добавляют 3 мг 2-(миристоилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 33-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 4,1 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 15-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 8,75 мин. (Выход: 54,3 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,0 нм, ESI-Масс-спектр (отрицательный): 1452,4 [M-H]-.
(Пример 25)
Синтез соединения примера 25 (Пример соединения № 1113)
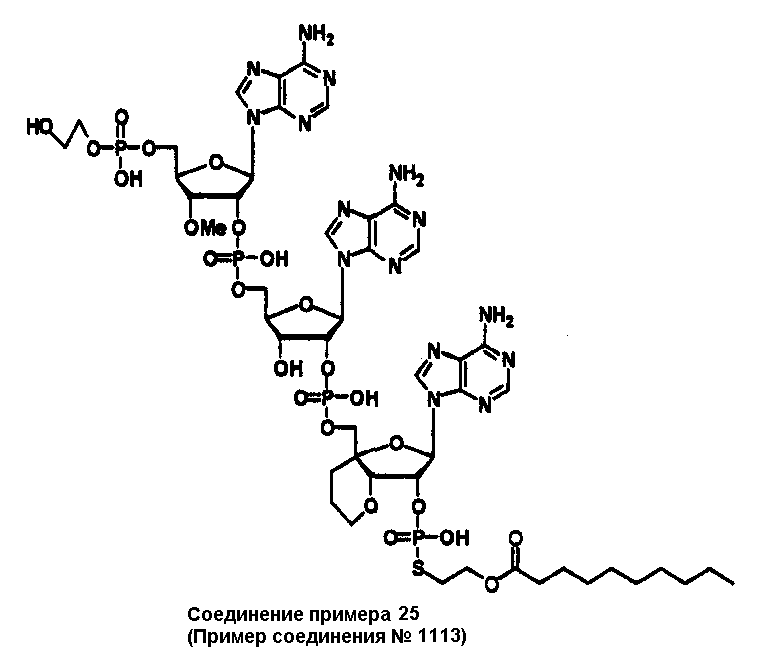
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 19 и добавляют 3 мг 2-(деканоилокси)этилбромида (Devinsky, Ferdinand et al., Collect. Czech. Chem. Commun. 49, 12, 1984, 2819-2827) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 15-62% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 6,2 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 15-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,29 мин. (Выход: 50,9 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,4 нм, ESI-Масс-спектр (отрицательный): 1396,3 [M-H]-.
(Пример 26)
Синтез соединения примера 26 (Пример соединения № 1938)
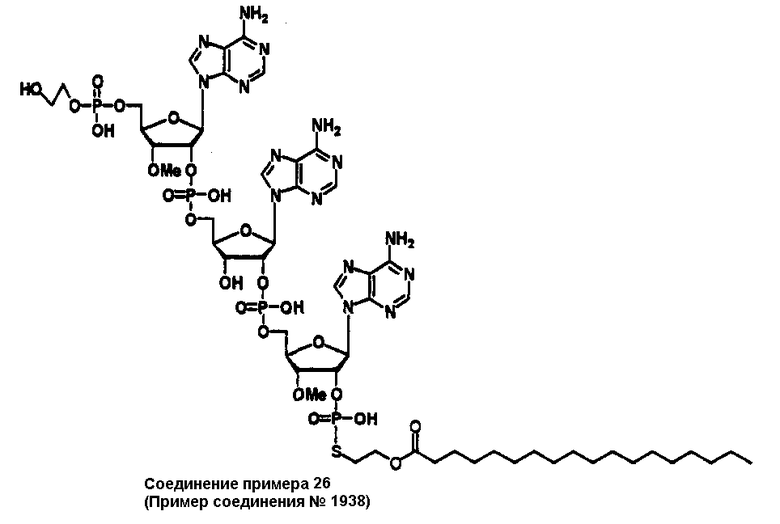
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 20 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 100 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 28-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 5,3 мин. При анализе ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,51 мин. (Выход: 14,9 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1483,2 [M-H]-.
(Пример 27)
Синтез соединения примера 27 (Пример соединения № 1183)
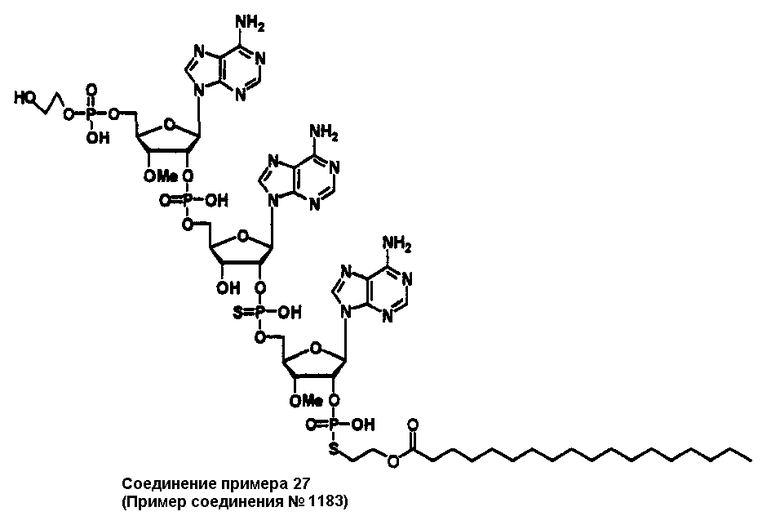
В 30 мкл безводного ДМФА растворяют 30 нмоль соединения примера 21 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 100 мкл воды и водный слой промывают три раза 100 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 28-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 5,2 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,57 мин. (Выход: 16,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1498,5 [M-H]-.
(Пример 28)
Синтез соединения примера 28 (Пример соединения № 1219)
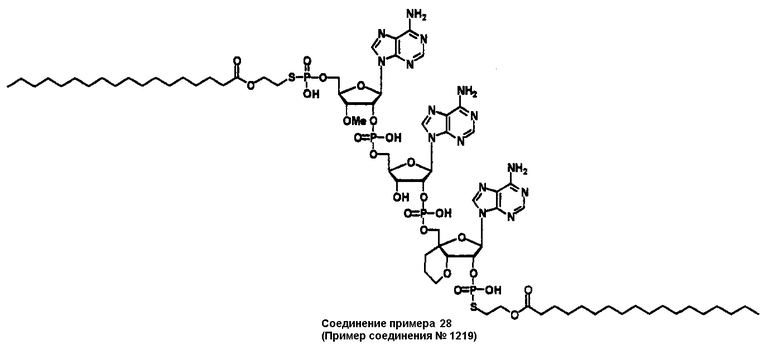
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 18 и добавляют 3 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 72-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 3,3 мин. (Выход: 8,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=260,5 нм, ESI-Масс-спектр (отрицательный): 1791,4 [M-H]-.
(Пример 29)
Синтез соединения примера 29 (Пример соединения № 1220)
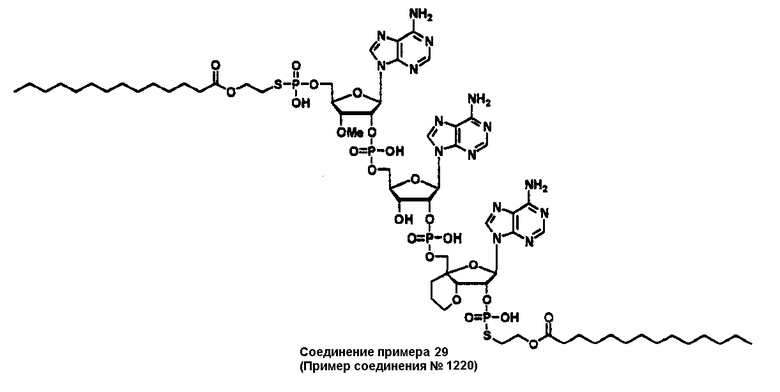
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 18 и добавляют 3 мг 2-(миристоилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 52-100% CH3CN (линейный градиент, 10 мин); 60°C; 2 мл/мин; 254 нм), собирают фракцию через 3,7 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 15-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 10,96 мин. (Выход: 41,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259,1 нм, ESI-Масс-спектр (отрицательный): 1679,5 [M-H]-.
(Пример 30)
Синтез соединения примера 30 (Пример соединения № 1221)
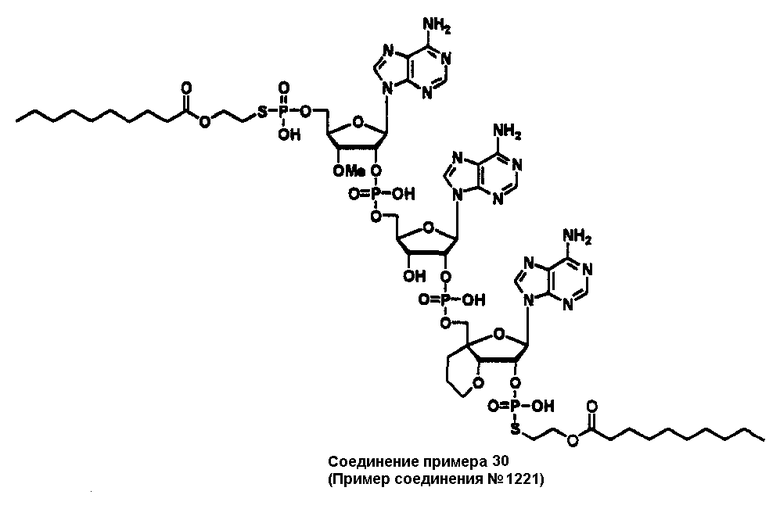
В 100 мкл безводного ДМФА растворяют 100 нмоль соединения примера 18 и добавляют 3 мг 2-(деканоилокси)этилбромида (Devinsky, Ferdinand et al., Collect. Czech. Chem. Commun. 49, 12, 1984, 2819-2827) и 3 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 34-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 4,5 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 15-100% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 8,92 мин. (Выход: 46,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259,3 нм, ESI-Масс-спектр (отрицательный): 1566,3 [M-H]-.
(Пример 31)
Синтез соединения примера 31 (Пример соединения № 1362)
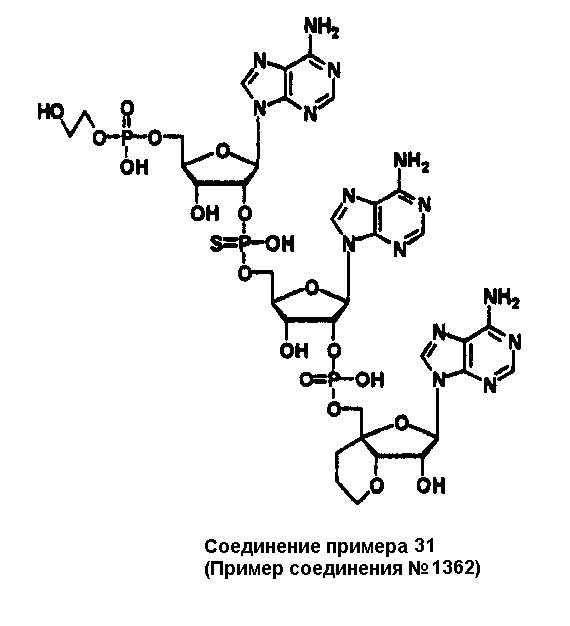
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) № 2002-249497, (2,0 мкмоль) с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в циклах 1 и 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 3 используют соединение примера 8а, описанное в японской патентной заявке (Kokai) № Hei 11-246592. Для окисления или в качестве сульфурирующего агента в циклах 1 и 3 используют йод и в цикле 2 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 1 и 3):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (цикл 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 30 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 10,9 и 12,0 мин, что соответствует двум диастереомерам. Соединение настоящего изобретения элюируется приблизительно через 8,09 и 8,50 мин при анализе помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин). (Выход: 749 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1104,2 [M-H]-.
(Пример 32)
Синтез соединения примера 32 (Пример соединения № 1363)
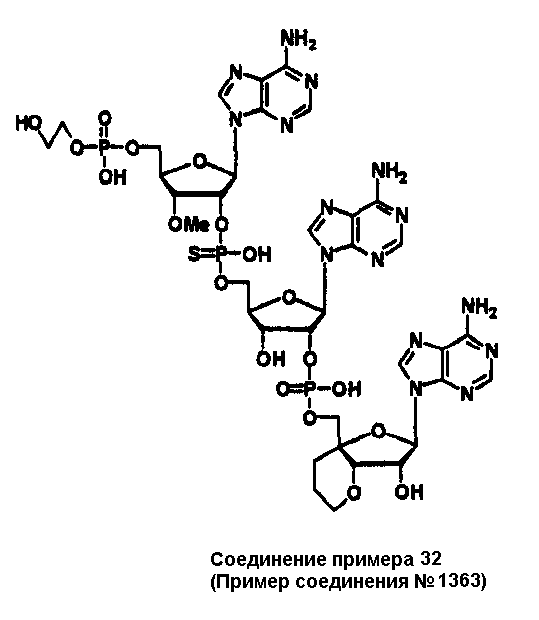
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной публикации (Kokai) № 2002-249497, (2,0 мкмоль) с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 2 используют 5'-DMT-3'-(O-метил)аденозин-(N-bz)-2'-фосфорамидит (ChemGene) и в цикле 3 используют соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592. При окислении или в качестве сульфурирующего агента йод используют в циклах 1 и 3 и ксантангидрид (Tokyo Kasei Kogyo) используют в цикле 2.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 1 и 3):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (цикл 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 10 мин); 40°С; 10 мл/мин; 254 нм), собирают фракции, которые элюируются через 11,5 и 12,7 мин, что соответствует двум стереоизомерам. Соединение настоящего изобретения элюируется приблизительно при 8,48 и 8,97 мин при анализе ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°C; 1 мл/мин). (Выход: 555 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1118,2 [M-H]-.
(Пример 33)
Синтез соединения примера 33 (Пример соединения № 1369)
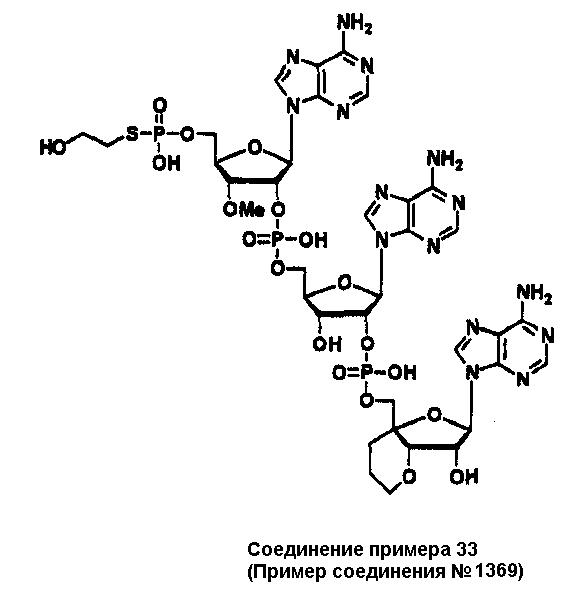
В 50 мкл безводного ДМФА растворяют 100 нмоль соединения примера 2 и добавляют 2 мкл 2-бромэтанола (Tokyo Kasei Kogyo) и 2 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 200 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-25% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 6,4 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 12,54 мин. (Выход: 20,3 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,1 нм, ESI-Масс-спектр (отрицательный): 1118,2 [M-H]-.
(Пример 34)
Синтез соединения примера 34 (Пример соединения № 1394)
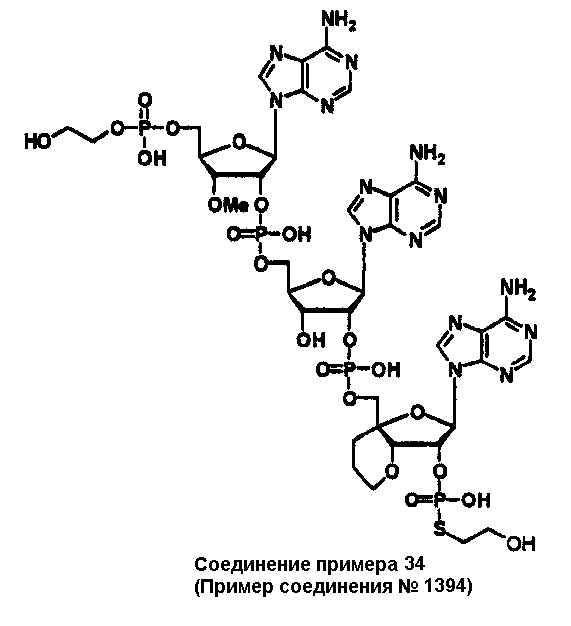
В 50 мкл безводного ДМФА растворяют 100 нмоль соединения примера 19 и добавляют 2 мкл 2-бромэтанола (Tokyo Kasei Kogyo) и 2 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 200 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-25% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин; 254 нм), собирают фракцию через 6,0 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 0-15% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 11,60 мин. (Выход: 48,2 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,0 нм, ESI-Масс-спектр (отрицательный): 1242,2 [M-H]-.
(Пример 35)
Синтез соединения примера 35 (Пример соединения № 1645)
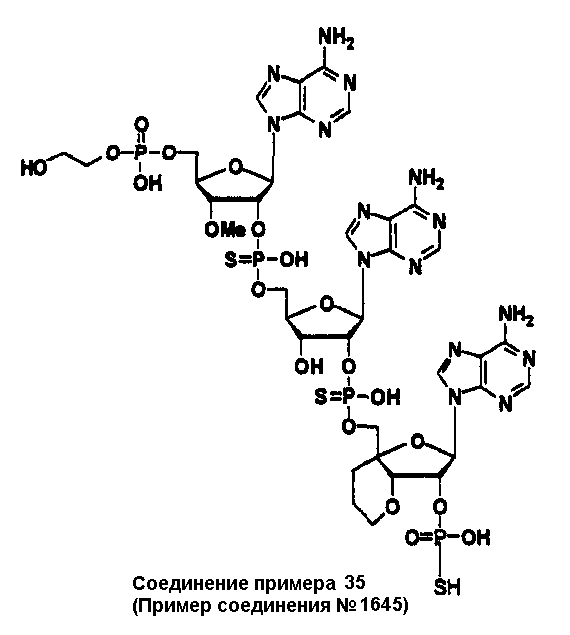
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,5 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 16, описанное в японской патентной заявке (Kokai) № 2002-249497, в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene) и соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. При окислении или в качестве сульфурирующего агента в циклах 1, 2 и 3 используют ксантангидрид (Tokyo Kasei Kogyo) и в цикле 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 4): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1, 2 и 3): ксантангидрид (0,02М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и оставшийся остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин) собирают фракции, которые элюируются через 4,8-5,2 мин, что соответствует двум диастереомерам.
После упаривания растворителя при пониженном давлении к оставшемуся остатку добавляют 1 мл водной хлористо-водородной кислоты (0,01 н.), чтобы точно довести значение рН до 2,0, с последующей реакцией при 30°С в течение 5 ч для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом, получают желаемое соединение. При анализе соединения настоящего изобретения ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин) соединение настоящего изобретения элюируется через 4,72 и 5,06 мин. Выход: 70 нмоль при измерении с помощью УФ при 260 нм), λmax (H2O)=258 нм, ESI-Масс-спектр (отрицательный); 1230,1 [M-H]-.
(Пример 36)
Синтез соединения примера 36 (Пример соединения № 1646)
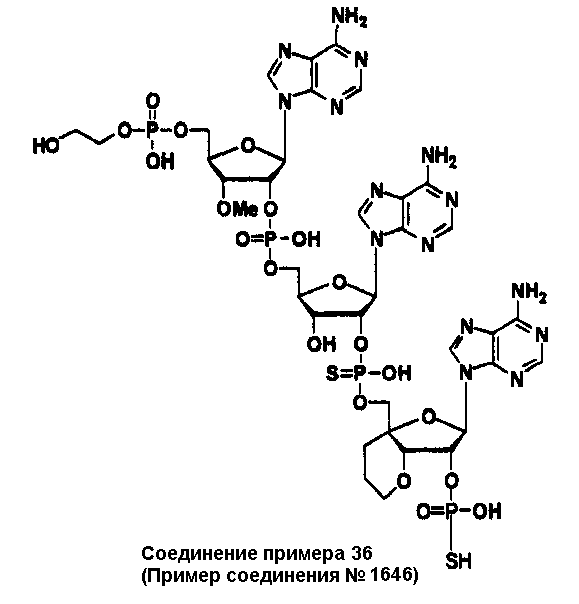
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,5 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 16, описанное в японской патентной заявке (Kokai) № 2002-249497, в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene) и соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют ксантангидрид (Tokyo Kasei Kogyo) и в циклах 3 и 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (циклы 3 и 4):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1 и 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и оставшийся остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракции, которые элюируются через 4,7-5,0 мин, что соответствует двум диастереомерам.
После упаривания растворителя при пониженном давлении к оставшемуся остатку добавляют 1 мл водной хлористо-водородной кислоты (0,01 н.), чтобы точно довести значение рН до 2,0, с последующей реакцией при 30°С в течение 5 ч для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом, получают желаемое соединение. При анализе соединения настоящего изобретения ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 5-20% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин) соединение настоящего изобретения элюируется через 4,12 и 4,44 мин. (Выход: 95 нмоль при измерении с помощью УФ при 260 нм), λmax (H2O)=258 нм, ESI-Масс-спектр (отрицательный); 1214,2 [M-H]-.
(Пример 37)
Синтез примера 37 (Пример соединения № 1648)
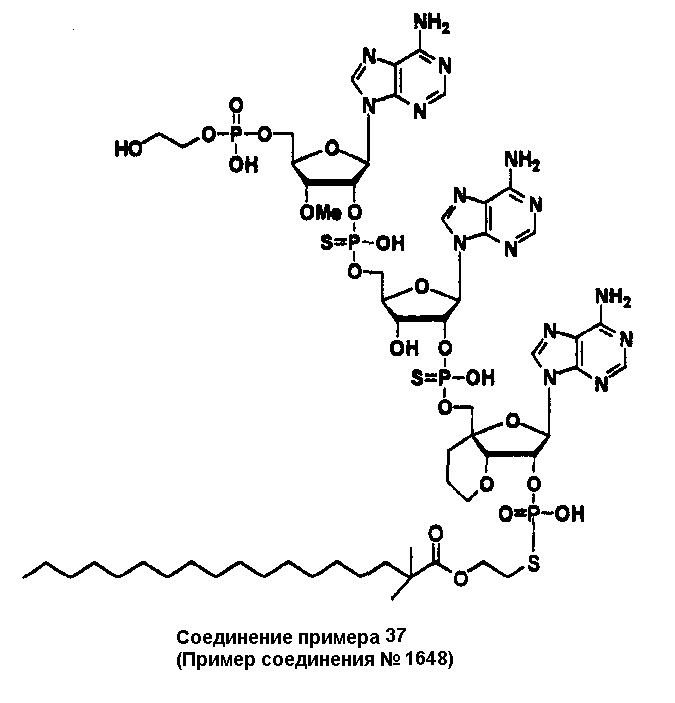
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 35 и добавляют 1 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, описанного в примере 22, и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,4 мин. (Выход: 12,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1568,3 [M-H]-.
(Пример 38)
Синтез соединения примера 38 (Пример соединения № 1649)
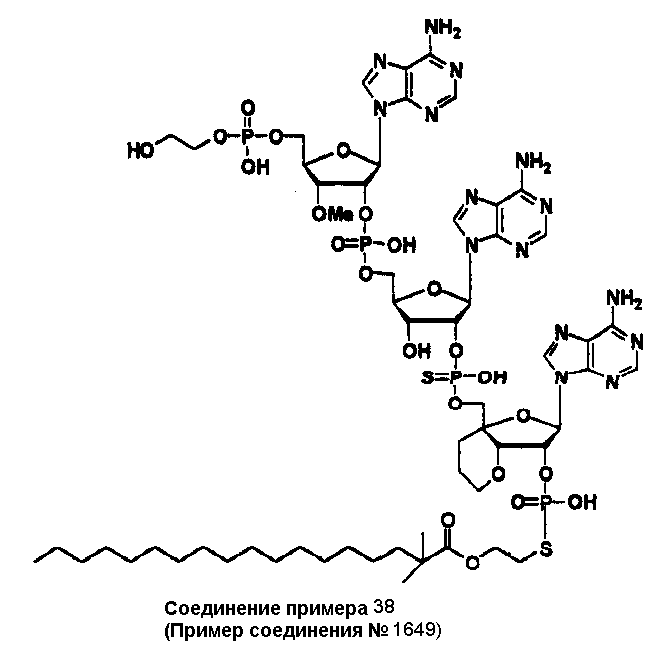
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 36 и добавляют 1 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, описанного в примере 22, и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,4 мин. Выход: 21,8 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1552,3 [M-H]-.
(Пример 39)
Синтез соединения примера 39 (Пример соединения № 1651)
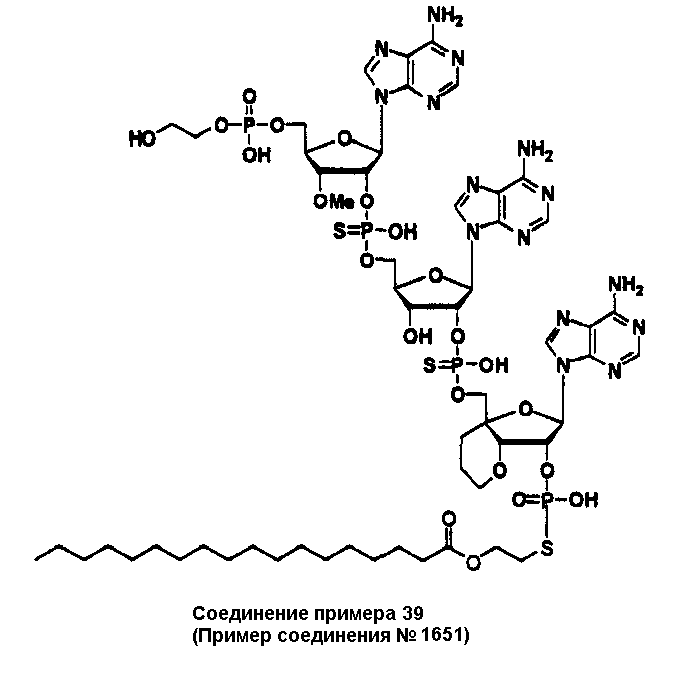
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 35 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°C; 2 мл/мин), собирают фракцию через 6,7 мин. (Выход: 11,4 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1540,3 [M-H]-.
(Пример 40)
Синтез соединения примера 40 (Пример соединения № 1652)
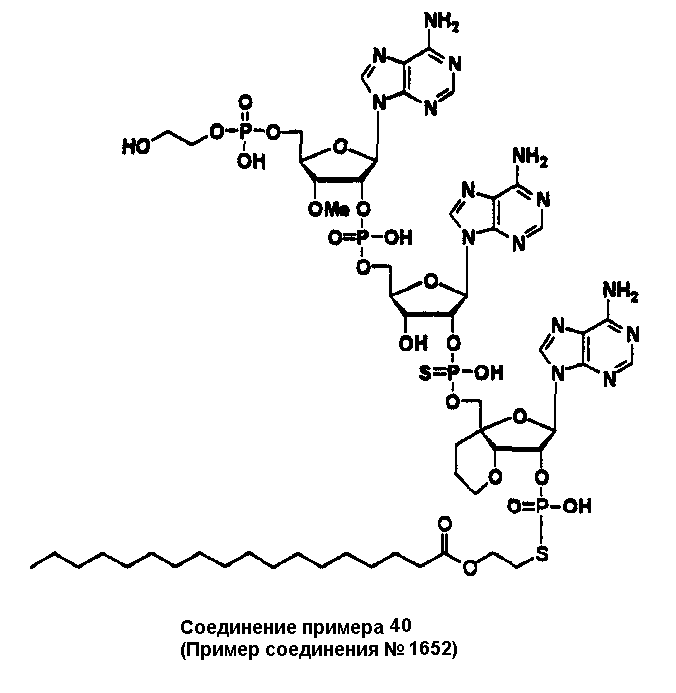
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 36 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 7,0 мин. (Выход: 23,1 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1524,3 [M-H]-.
(Пример 41)
Синтез соединения примера 41 (Пример соединения № 1663)
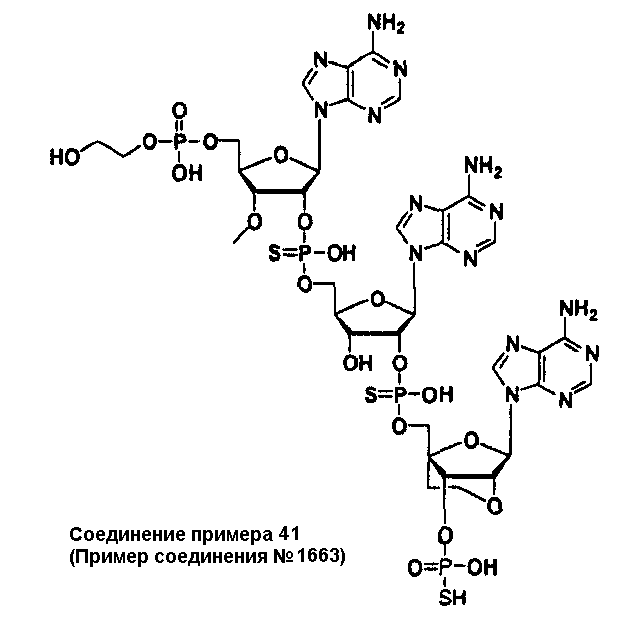
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,5 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 14, описанное в японском патенте № 3420984, в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин-(N-bz)-2'-фосфорамидит (ChemGene) и соединение примера 8a, описанное в патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. При окислении или в качестве сульфурирующего агента в циклах 1, 2 и 3 используют ксантангидрид (Tokyo Kasei Kogyo) и в цикле 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 4): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1, 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и точно доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение 5 ч при 30°С для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом с получением желаемого соединения. Продукт выходит через 3,75, 4,12, 4,53 и 4,76 мин при анализе с помощью ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-20% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин). Выход: 111 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1216,1 [M-H]-.
(Пример 42)
Синтез соединения примера 42 (Пример соединения № 1664)
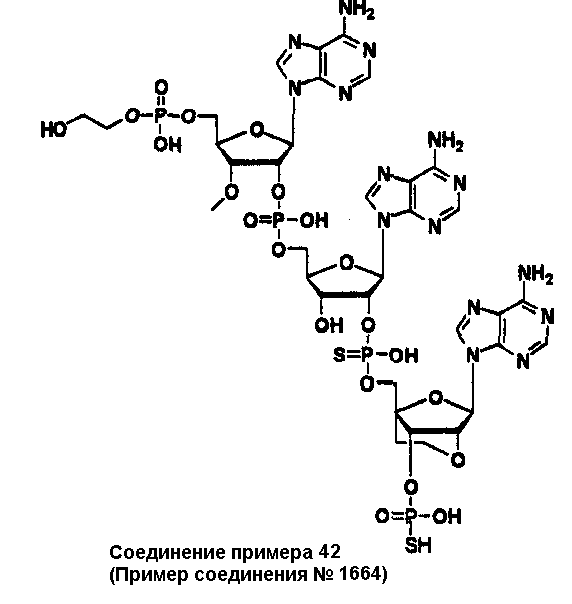
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,5 мкмоль). В качестве фосфорамидита в цикле 1 используют соединение примера 14, описанное в японском патенте № 3420984, в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), в цикле 3 используют 5'-DMT-3'-(O-метил)аденозин-(N-bz)-2'-фосфорамидит (ChemGene) и соединение примера 8a, описанное в патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 1 и 2 используют ксантангидрид (Tokyo Kasei Kogyo) и в циклах 3 и 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 3 и 4):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1 и 2): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и оставшийся остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°C; 2 мл/мин), собирают фракции, которые элюируются через 4,6-4,9 мин, что соответствует двум диастереомерам.
После упаривания растворителя при пониженном давлении к оставшемуся остатку добавляют 1 мл водной хлористо-водородной кислоты (0,01 н.), чтобы точно довести значение рН до 2,0, с последующей реакцией при 30°С в течение 5 ч для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом, получают желаемое соединение. При анализе соединения настоящего изобретения ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-20% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин) соединение настоящего изобретения элюируется через 2,40 и 3,01 мин. (Выход: 127 нмоль при измерении с помощью УФ при 260 нм), λmax (H2O)=258 нм, ESI-Масс-спектр (отрицательный); 1200,15 [M-H]-
(Пример 43)
Синтез соединения примера 43 (Пример соединения № 1666)
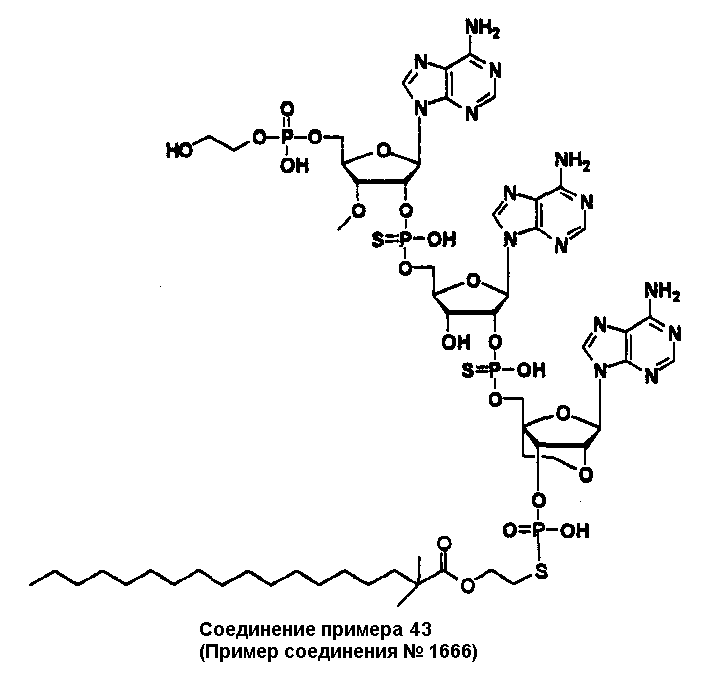
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 41 и добавляют 1 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, описанного в примере 22, и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,9 мин. (Выход: 9,5 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1554,3 [M-H]-.
(Пример 44)
Синтез соединения примера 44 (Пример соединения № 1667)
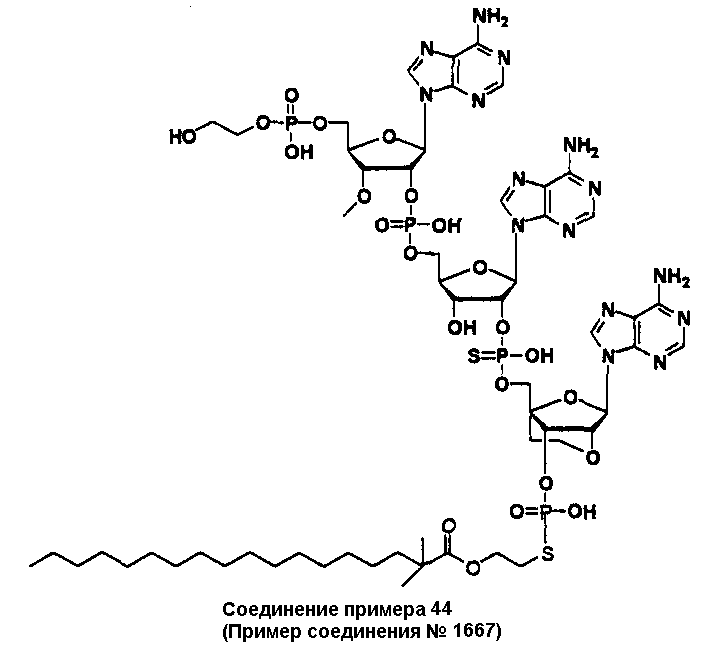
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 42 и добавляют 1 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, описанного в примере 22, и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,9 мин. (Выход: 4,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1538,3 [M-H]-.
(Пример 45)
Синтез соединения примера 45 (Пример соединения № 1669)
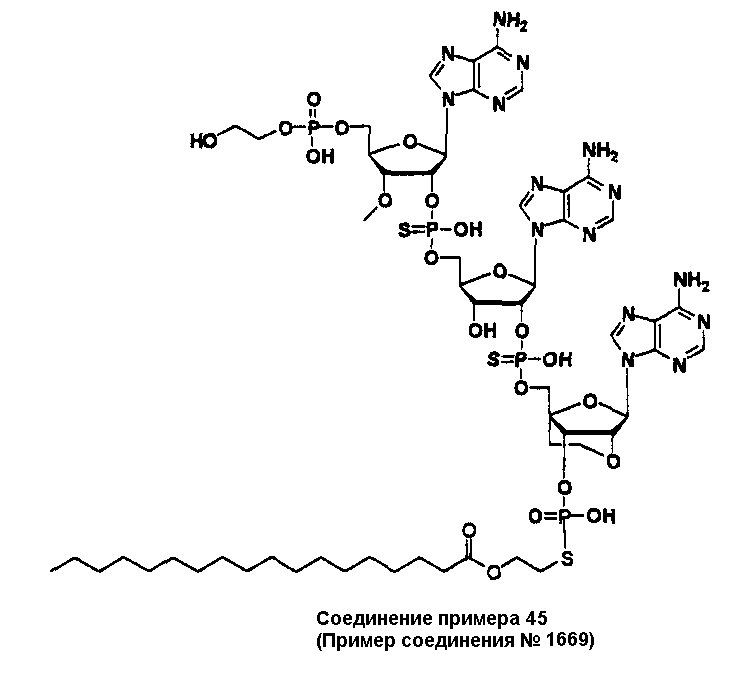
В 100 мкл безводного ДМФА растворяют 80 нмоль соединения примера 41 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл пиридина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 500 мкл воды и водный слой промывают три раза 500 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,6 мин. (Выход: 40,4 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1526,3 [M-H]-.
(Пример 46)
Синтез соединения примера 46 (Пример соединения № 1670)
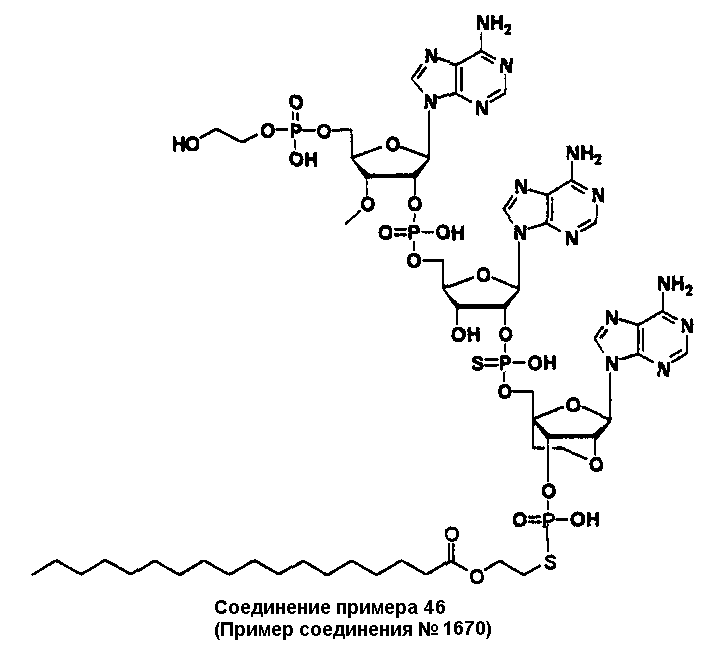
В 100 мкл безводного ДМФА растворяют 80 нмоль соединения примера 42 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл пиридина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 500 мкл воды и водный слой промывают три раза 500 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,6 мин. Выход: 41,9 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=259 нм, ESI-Масс-спектр (отрицательный): 1510,29 [M-H]-.
(Пример 47)
Синтез соединения примера 47 (Пример соединения № 1690)
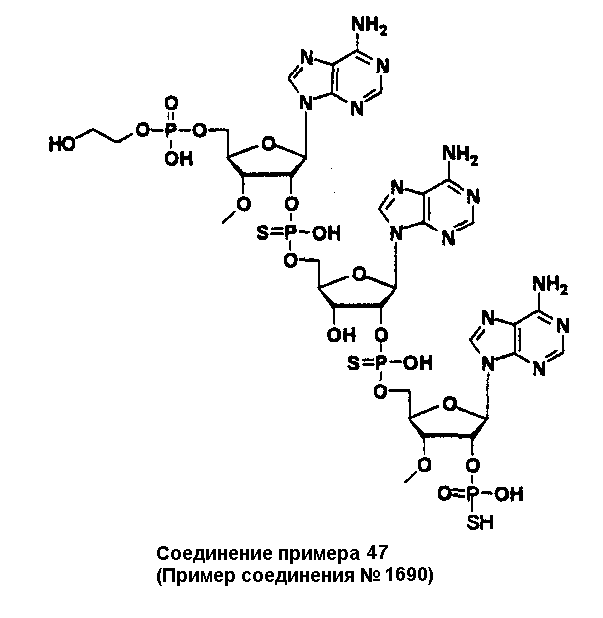
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,5 мкмоль). В качестве фосфорамидита в циклах 1 и 3 используют 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene), в цикле 2 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и в цикле 4 используют соединение примера 8a, описанное в японской патентной заявке (Kokai) № Hei 11-246592. При окислении или в качестве сульфурирующего агента ксантангидрид (Tokyo Kasei Kogyo) используют в циклах 1, 2 и 3 и йод используют в циклах 3 и 4.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 4):
йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1, 2 и 3): ксантангидрид (0,02 М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и оставшийся остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракции, которые элюируются через 4,7-5,1 мин, что соответствует двум диастеремерам.
После упаривания растворителя при пониженном давлении к оставшемуся остатку добавляют 1 мл водной хлористо-водородной кислоты (0,01 н.), чтобы точно довести значение рН до 2,0, с последующей реакцией при 30°С в течение 5 ч для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом, получают желаемое соединение. При анализе соединения настоящего изобретения ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-20% CH3CN (линейный градиент, 10 мин); 60°C; 2 мл/мин) соединение настоящего изобретения элюируется через 3,67, 4,01, 4,15 и 4,55 мин. (Выход: 140 нмоль при измерении с помощью УФ при 260 нм), λmax (H2O)=258 нм, ESI-Масс-спектр (отрицательный); 1204,1 [M-H]-
(Пример 48)
Синтез соединения примера 48 (Пример соединения № 1691)
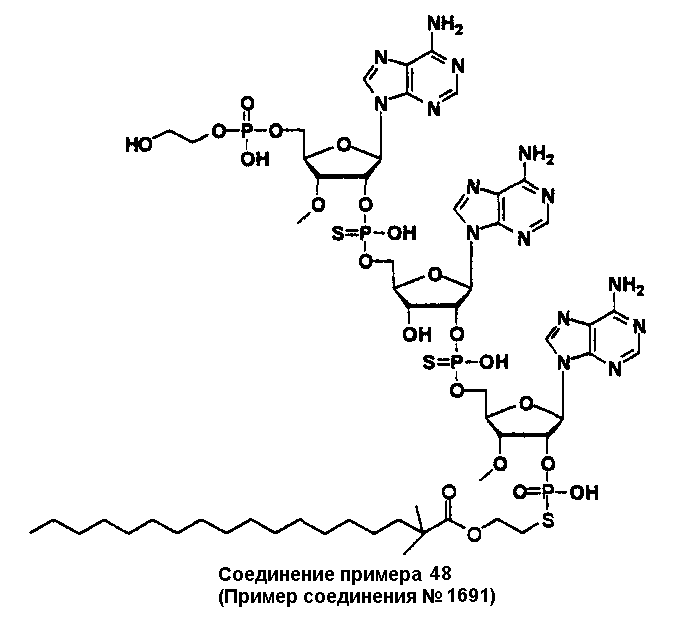
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 47 и добавляют 1 мг 2-(2,2-диметилоктадеканоилокси)этилбромида, описанного в примере 22, и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают с помощью ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 7,0 мин. Выход: 2,5 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1542,3 [M-H]-.
(Пример 49)
Синтез соединения примера 49 (Пример соединения № 1692)
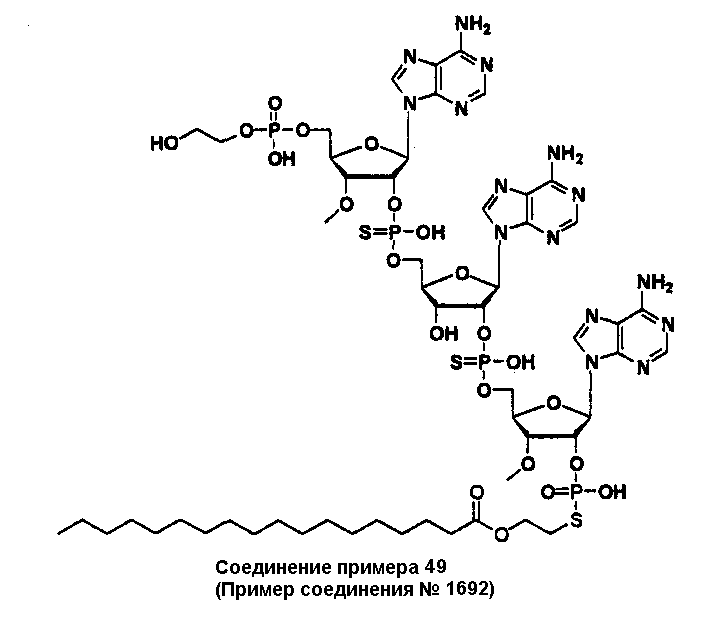
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 47 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл триэтиламина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 300 мкл воды и водный слой промывают три раза 200 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,8 мин. (Выход: 29,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258 нм, ESI-Масс-спектр (отрицательный): 1514,3 [M-H]-.
(Пример 50)
Синтез соединения примера 50 (Пример соединения № 1929)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Ge-p-Ae-p-Ge-p-Ae-p-Ce-p-Cn-p-Cn-p-Tn-p-Gn-p-An-p-An-p-Cn-p-An-p-Gn-p-Tn-p-Te-p-Ge-p-Ae-p-Te-p-Ce-hp
Аналог 2-5A, содержащий целевую последовательность, синтезируют путем сочетания различных фосфорамидитов в определенном порядке, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием синтезатора ДНК, программы синтеза для обычного синтеза 1 мкмоль РНК и 1 мкмоль соединения, описанного в примере 12b патентной заявки (Kokai) № Hei 7-87982, в качестве твердофазной подложки.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление: йод/вода/пиридин/тетрагидрофуран; 15 с.
В качестве фосфорамидитов, используемых для синтеза фрагмента антисмыслового олигонуклеотида, используют фосфорамидит аденина (dAbz), фосфорамидит гуанина (dGibu), фосфорамидит цитозина (dCbz) и фосфорамидит тимина (T) (Applied Biosystems) в случае последовательности, эквивалентной природным типам нуклеотидов, тогда как соединения Примеров 14, 27, 22 и 9, описанные в японском патенте № 3420984, используют для синтеза последовательности, эквивалентной неприродным типам нуклеотидов (Ae, Ge, Ce, Te). DMT-Бутанол-CED-фосфорамидит (ChemGene) используют в случае фосфорамидита, эквивалентного L1, и 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene), 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene), 5'-DMT-3'-(O-метил)аденозин(N-bz)-2'-фосфорамидит (ChemGene) и фосфорамидит примера 8a, описанный в патентной заявке (Kokai) № Hei 11-246592, сочетают в определенном порядке.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и добавляют к остатку 1 мл тригидрофторида триэтиламина, после чего перемешивают при комнатной температуре. Через 24 часа добавляют 200 мкл H2O, затем добавляют 10 мл 1-бутанола, выдерживают в течение 1 ч при -20°С и центрифугируют с получением осадка, похожего на пеллету. После осторожной промывки EtOH, пеллету растворяют в 150 мкл H2O и затем подвергают электрофорезу на 15% денатурированном акриламидном геле (1 раз, TBE-раствор (7 M мочевина, 0,89 М Tris, борная кислота, раствор ЭДТУК (pH 8,3, Takara Shuzo), 600 В, 60 мин). Полосу в геле, которая поглощает УФ, отрезают и элюируют из геля 1 мл элюирующего буфера (0,5 М ацетата аммония, 10 мМ ацетата магния, 1 мМ ЭДТУК (pH 8,0), 0,1% SDS). Остальной гель фильтруют, добавляют 4 мл EtOH к фильтрату, которому затем дают стоять в течение 1 ч при -20°С, после чего центрифугируют с получением осадка, похожего на пеллету. Полученный продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 M водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 4,02 мин. (Выход: 16,0 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259 нм.
(Пример 51)
Синтез соединения пример 51 (Соединение примера № 1930)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Te-p-Ce-p-Te-p-Te-p-Ge-p-Gn-p-Tn-p-Tn-p-Gn-p-Tn-p-An-p-An-p-Gn-p-An-p-Gn-p-Ae-p-Ge-p-Ae-p-Ge-p-Ae-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 3,61 мин. (Выход: 15,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=257 нм.
(Пример 52)
Синтез соединения примера 52 (Пример соединения № 1931)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Te-p-Te-p-Ce-p-Ae-p-Ge-p-Gn-p-Cn-p-Cn-p-Tn-p-Cn-p-Cn-p-An-p-Tn-p-An-p-Tn-p-Ge-p-Ge-p-Ae-p-Ae-p-Te-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 3,66 мин. (Выход: 7,8 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=259 нм.
(Пример 53)
Синтез соединения примера 53 (Пример соединения № 1932)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Ge-p-Te-p-Te-p-Ce-p-Te-p-Cn-p-Gn-p-Cn-p-Tn-p-Gn-p-Gn-p-Tn-p-Gn-p-An-p-Gn-p-Te-p-Te-p-Te-p-Ce-p-Ae-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 8-12% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракцию через 8,0-10,0 мин. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-29% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,54 мин. (Выход: 37,6 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,9 нм.
(Пример 54)
Синтез соединения примера 54 (Пример соединения № 1933)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Ge-p-Ae-p-Te-p-Ge-p-Ge-p-An-p-An-p-An-p-Tn-p-Cn-p-Tn-p-Cn-p-Tn-p-Gn-p-Cn-p-Ce-p-Ge-p-Ce-p-Ae-p-Te-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 3,74 мин. Выход: 131 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=262 нм.
(Пример 55)
Синтез соединения примера 55 (Пример соединения № 1934)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Ae-p-Te-p-Ge-p-Ge-p-Ce-p-An-p-Cn-p-Cn-p-Tn-p-Cn-p-Tn-p-Tn-p-Gn-p-Tn-p-Gn-p-Ge-p-Ae-p-Ce-p-Ce-p-Ae-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 3,83 мин. (Выход: 5,5 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=261 нм.
(Пример 56)
Синтез соединения примера 56 (Пример соединения № 1935)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L1-P(=O)(OH)-L1-p-Ce-p-Ae-p-Ge-p-Ce-p-Ce-p-An-p-Tn-p-Gn-p-Gn-p-Tn-p-Cn-p-Cn-p-Cn-p-Cn-p-Cn-p-Ce-p-Ce-p-Ce-p-Ae-p-Ae-hp
Указанное в заголовке соединение получают аналогично способу примера 50. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 9-25% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 3,25 мин. (Выход: 55 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=266 нм.
(Пример 57)
Синтез соединения примера 57 (Пример соединения № 1936)
HOC2H4O-P(=O)(OH)-K2-1-P(=O)(OH)-K1-1-P(=O)(OH)-K2-1-P(=O)(OH)-L2-p-Ge-p-Te-p-Te-p-Ce-p-Te-p-Cn-p-Gn-p-Cn-p-Tn-p-Gn-p-Gn-p-Tn-p-Gn-p-An-p-Gn-p-Te-p-Te-p-Te-p-Ce-p-Ae-hp
Указанное в заголовке соединение получают аналогично способу примера 50. В данном случае Spacer-фосфорамидит 18 (Glen Research Inc.) используют в качестве фосфорамидита, соответствующего L2. Указанное соединение очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 8-12% CH3CN (линейный градиент, 10 мин); 60°С; 2 мл/мин), собирают фракцию через 8,0-10,0 мин. При анализе ВЭЖХ с обращенной фазой (колонка (Tosoh superODS (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 5-29% CH3CN (линейный градиент, 10 мин); 60°С; 1 мл/мин) соединение настоящего изобретения элюируется через 7,52 мин. (Выход: 59,5 нмоль при измерении с помощью УФ при 260 нм), λмакс (H2O)=258,6 нм.
(Пример 58)
Синтез соединения примера 58 (Пример соединения № 1103)
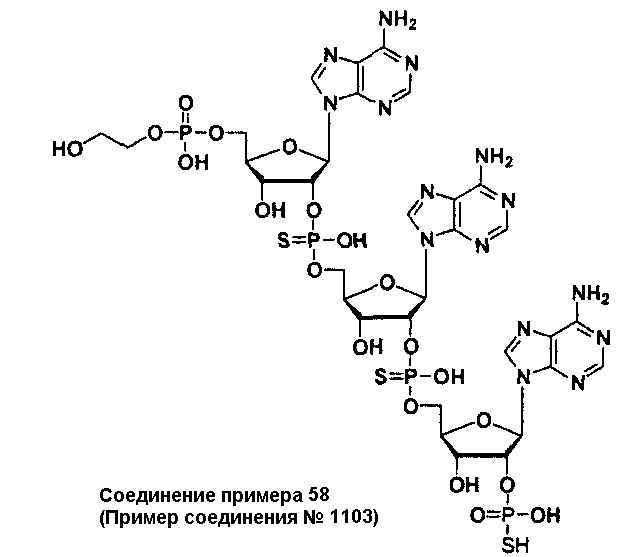
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат-CPG (Glen Research) (0,2 мкмоль). В качестве фосфорамидита в циклах 1, 2 и 3 используют 3'-tBDСилилрибоаденозин(N-bz)фосфорамидит (ChemGene) и соединение примера 8a, описанное в патентной заявке (Kokai) № Hei 11-246592, используют в цикле 4. При окислении или в качестве сульфурирующего агента в циклах 1, 2 и 3 используют ксантангидрид (Tokyo Kasei Kogyo) и в цикле 4 используют йод.
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 85 с.
2) Сочетание: фосфорамидит (приблизительно 25 экв.)/ацетонитрил, тетразол/ацетонитрил; 10-20 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 15 с.
4) Окисление (цикл 4): йод/вода/пиридин/тетрагидрофуран; 15 с.
Сульфурирование (циклы 1, 2 и 3): ксантангидрид (0,02М)/ацетонитрилпиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5A, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr сохранена, вместе с отщеплением олигомера от подложки цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1). Затем растворитель отгоняют при пониженном давлении и остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракции, которые элюируются через 6,0-6,7 мин, что соответствует четырем диастереомерам.
После упаривания растворителя при пониженном давлении к оставшемуся остатку добавляют 1 мл водной хлористо-водородной кислоты (0,01 н.), чтобы точно довести значение рН до 2,0, с последующей реакцией при 30°С в течение 5 ч для удаления группы DMTr и силильной группы. После нейтрализации водным аммиаком снятые силанол и DMTrOH удаляют экстракцией этилацетатом. При анализе оставшегося водного раствора помощью ВЭЖХ с обращенной фазой (колонка (Merck chromolith (4,6×50 мм)); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 0-20% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракции, которые элюируются через 4,6 и 5,4 мин, получают требуемое соединение. (Выход: 38 нмоль при измерении с помощью УФ при 260 нм), λmax (H2O)=259 нм.
(Пример 59)
Синтез соединения примера 59 (пример соединения №1195)
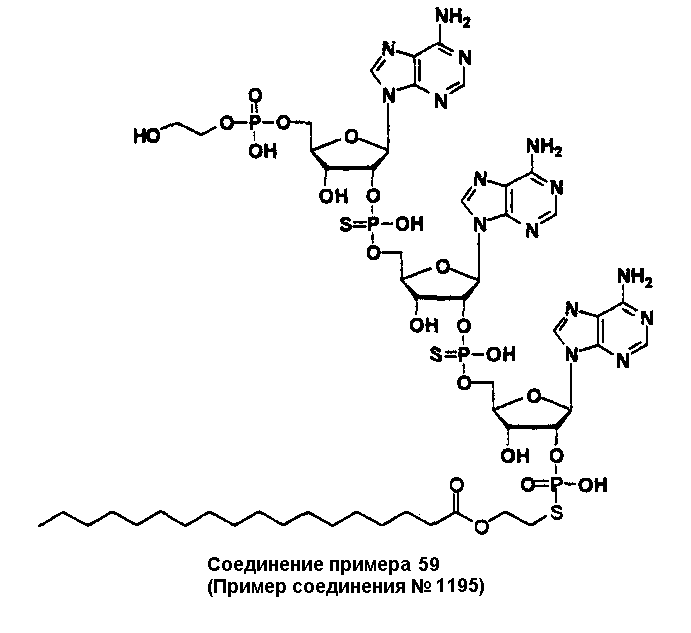
В 40 мкл безводного ДМФА растворяют 40 нмоль соединения примера 58 и добавляют 1 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) и 1 мкл пиридина, с последующей реакцией смеси при комнатной температуре в течение ночи. По окончании реакции добавляют 500 мкл воды и водный слой промывают три раза 500 мкл AcOEt. После упаривания водного слоя остаток очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (Merck chromolith (4,6×50 мм); 0,1 М водный триэтиламинацетат (TEAA), pH 7; 24-100% CH3CN (линейный градиент, 8 мин); 60°С; 2 мл/мин), собирают фракцию через 6,8 мин. Выход: 8,9 нмоль при измерении с помощью УФ при 260 нм, λмакс (H2O)=259 нм.
(Пример 60)
Синтез соединения примера 60 (Пример соединения №1947)
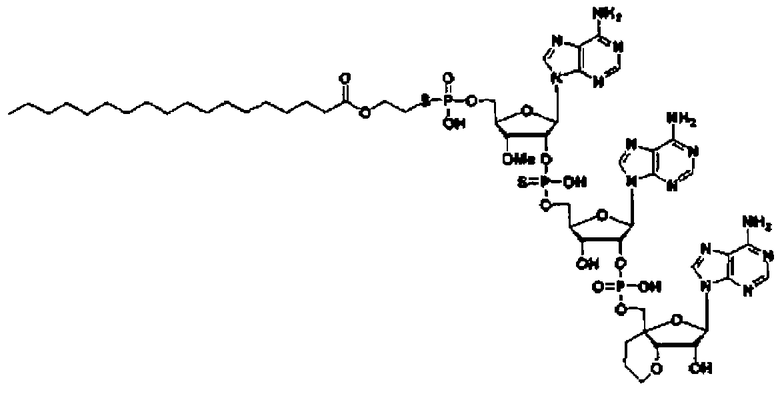
11 мг соединения примера 7 и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,6 мл Н2O, и добавляют 135 мг 2-(стеароилокси) этилбромида (Ackerman et al., J, Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение 18,5 ч. Затем органический растворитель упаривают при пониженном давлении, и остаток очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil Prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 50-80% (линейный градиент, 20 мин); 40°С; 10 мл/мин), и фракции, которые элюируются через 16,6 мин, собирают. Эту фракцию концентрируют в вакууме, и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (2,5 мл каждая) и элюируют H2O. Фракцию сушат вымораживаем, и продукт получают в виде натриевой соли. Выход: 109 OD, ESI-MS (отрицательный); 1400,45 [M-H]-. При анализе с помощью ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; B% 0-30% (линейный градиент, 5 минут) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), соединение элюируется через 11,53 мин.
(Пример 61)
Синтез соединения примера 61 (Пример соединения №1948)
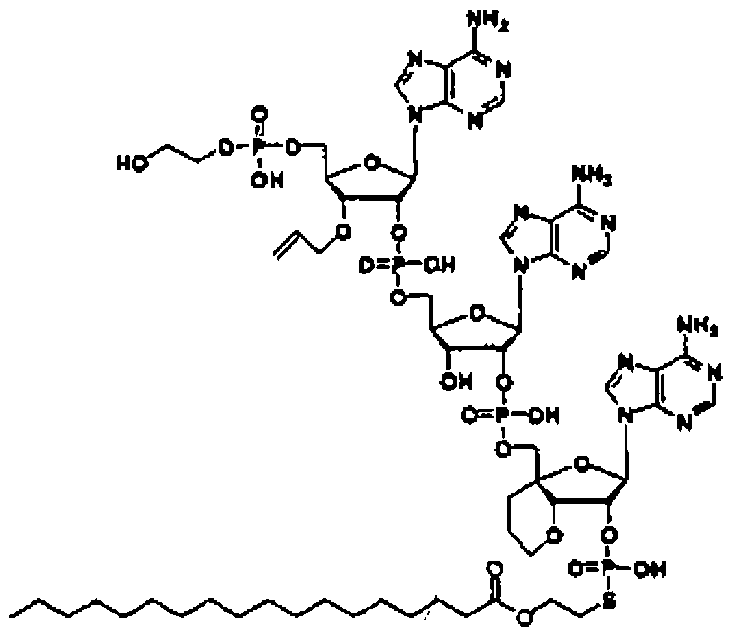
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 1 мкмоль РНК. 3'-Фосфат CPG (Glen Research) (40 мкмоль) используют в качестве твердофазного носителя. Соединение примера 16, описанное в японской патентной заявке (Kokai) №2002-249497, используют в качестве фосфорамидита в цикле 1, 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene, ANP-5681) используют в качестве фосфорамидита в цикле 2, 5'-DMT-3'-пропаргил(N-bz)-2'-фосфорамидит (ChemGene, ANP-9751) используют в цикле 3, и соединение примера 8а, описанное в японской патентной заявке (Kokai), используют в цикле 4. При окислении или в качестве сульфурирующего агента в циклах 2, 3 и 4 используют йод, тогда как в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2):
йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру, в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией в течение ночи при комнатной температуре для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-52,5% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 260 нм), собирают фракцию, которая элюируется через 13,2 мин. Соединение настоящего изобретения элюируется приблизительно через 3,92 мин при анализе с помощью ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор A: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (H2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2О, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 60% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-9% (линейный градиент, 18 мин); 40°С; 10 мл/мин), и собирают фракции, содержащие продукт. Фракцию концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (8 мл каждая) и элюируют Н2О. Фракцию сушат вымораживанием и продукт получают в виде натриевой соли. Выход: 8,9 мг, ESI-Масс-спектр (отрицательный): 1535 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,65 мин.
(Пример 62)
Синтез соединения примера 62 (Пример соединения №1949)
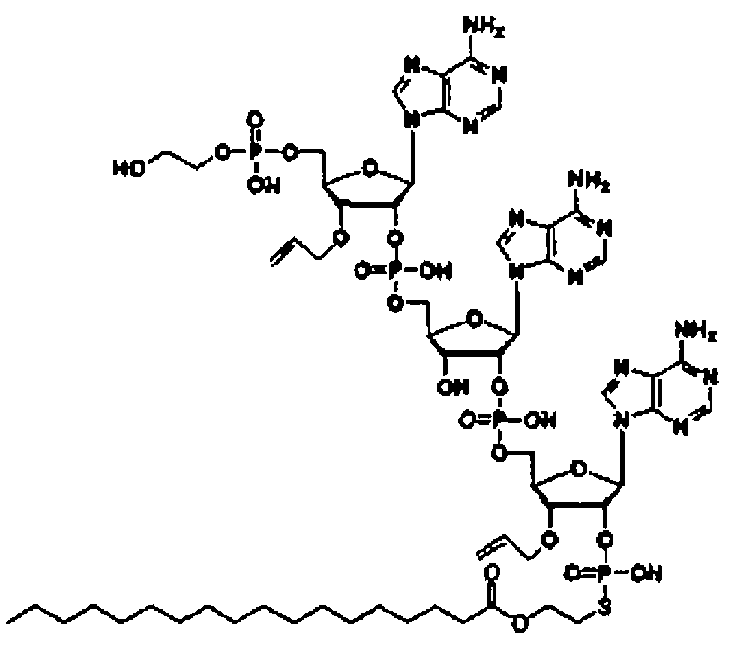
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат CPG (Glen Research) (40 мкмоль). 5'-DMT-3'-пропаргиладенозин(N-bz)-2'-фосфорамидит (ChemGene, ANP-9751) используют в цикле 1 и 3, 8'-tBDсилилрибоаденозин(N-bz)-фосфорамидит (ChemGene, ANP-5681) используют в качестве фосфорамидита в цикле 2, и соединение примера 8а, описанное в японской патентной заявке (Kokai) № Hei 11-246592 используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 2, 3 и 4 используют йод, и в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан;
195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М) /ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-52,5% CH3CN (линейный градиент, 20 мин); 40°C; 10 мл/мин; 260 нм), собирают фракцию, которая элюируется через 13,4 мин. Соединение настоящего изобретения элюируется приблизительно через 3,89 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл H2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-4% (линейный градиент, 18 мин); 40°С; 10 мл/мин), и собирают фракции, содержащие продукт. Фракцию концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2О. Фракцию сушат вымораживанием и продукт получают в виде натриевой соли. Выход: 11 мг, ESI-Масс-спектр (отрицательный): 1535 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор A: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,77 мин.
(Пример 63)
Синтез соединения примера 63 (Пример соединения №1950)
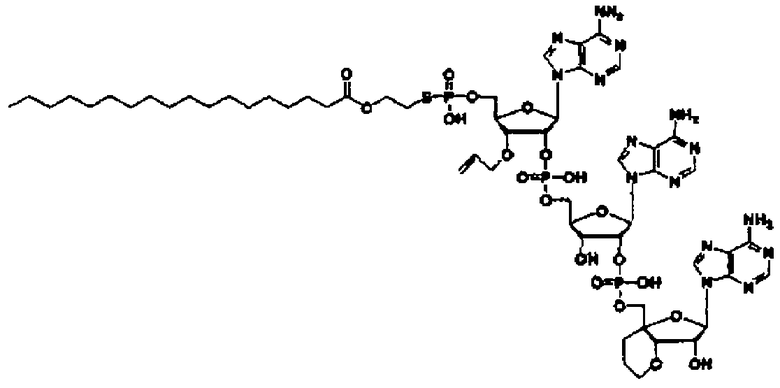
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) №2002-249497 (40 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene, ANP-5681), в цикле 2 используют 5'-DMT-3'пропаргиладенозин(N-bz)-2'-фосфорамидит (ChemGene, ANP-9751), в цикле 3 используют 5'-фосфорилирующий реагент (Phosphalink reagent, Applied Biosystems). При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют йод, тогда как в цикле 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан;
195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2):
йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-52,5% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 260 нм), и собирают фракцию, которая элюируется через 17,8 мин. Соединение настоящего изобретения элюируется приблизительно через 3,98 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (H2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, при пониженном давлении, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 0-9% (линейный градиент, 18 мин); 40°С; 10 мл/мин), и собирают фракции, содержащие продукт. Фракцию концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2O. Фракцию сушат сублимацией, продукт получают в виде натриевой соли. Выход: 6,6 мг, ESI-Macc-спектр (отрицательный): 1411 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,97 мин.
(Пример 64)
Синтез соединения Примера 64 (пример соединения №1951)
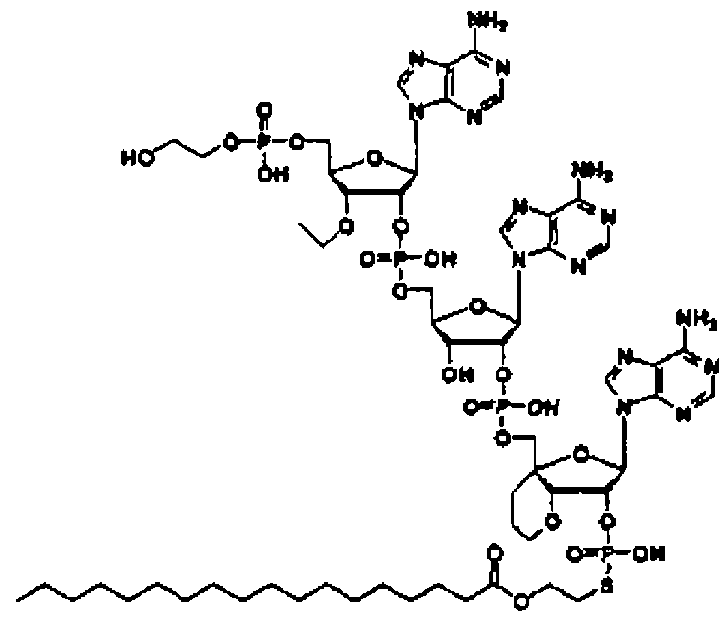
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат CPG (Glen Research) (40 мкмоль). Соединение Примера 16, описанное в японской патентной заявке (Kokai) №2002-249497, используют в качестве фосфорамидита в цикле 1, 3'-tBDсилилрибоаденозин(N-bz)-фосфорамидит (ChemGene, ANP-5681) используют в качестве фосфорамидита в цикле 2, соединение ссылочного примера 4 используют в цикле 3, и соединение примера 8а, описанное в японской патентной заявке (Kokai) №Hei 11-246592, используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 2, 3 и 4 используют йод, и в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2):
йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к полученному остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-47,75% CH3CN (линейный градиент, 15 мин); 40°С; 10 мл/мин; 260 нм), собирают фракцию, которая элюируется через 11,9 мин. Соединение настоящего изобретения элюируется приблизительно через 3,71 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
256 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; В% 0-10% (линейный градиент, 10 мин); 40°С; 10 мл/мин), и собирают фракции, которые элюируются приблизительно в диапазоне 13,9 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2O. Фракцию сушат сублимацией и продукт получают в виде натриевой соли. Выход: 8,9 мг, ESI-Macc-спектр (отрицательный): 1523 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,72 мин.
(Пример 65)
Синтез соединения примера 65 (Пример соединения №1952)
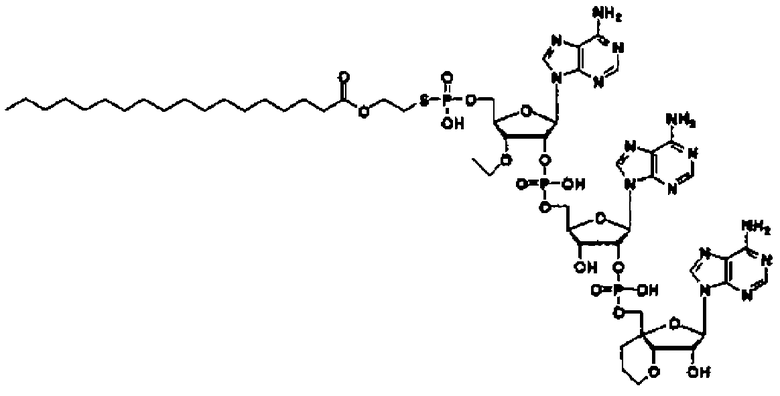
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) №2002-249497 (40 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene, ANP-5681), в цикле 2 используют соединение ссылочного примера 4, и в цикле 3 используют 5'-фосфорилирующий реагент (Phosphalink reagent, Applied Biosystems). При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют йод, тогда как в цикле 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание; фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-47,75% CH3CN (линейный градиент, 20 мин); 40°С; 10 мл/мин; 260 нм), и собирают фракцию, которая элюируется приблизительно через 12,4 мин. Соединение настоящего изобретения элюируется приблизительно через 3,89 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор A: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
266 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 2-12% (линейный градиент, 15 мин); 40°С; 10 мл/мин), и собирают фракции, которые элюируются приблизительно через 16,3 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2О. Фракцию сушат сублимацией продукт получают в виде натриевой соли. Выход: 8,2 мг, ESI-Macc-спектр (отрицательный): 1399 [М-Н]-
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор A: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,99 мин.
(Пример 66)
Синтез соединения примера 66 (Пример соединения №1953)
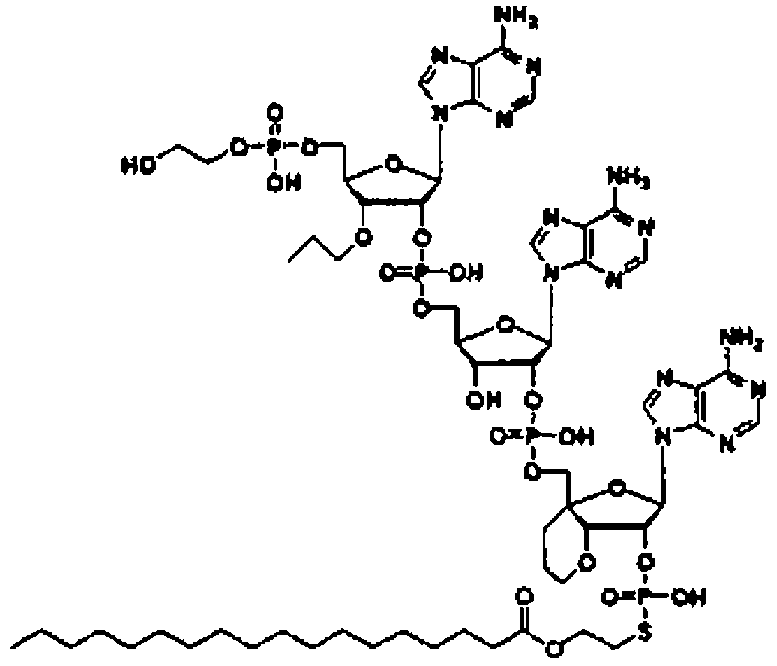
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат CPG (Glen Research) (40 мкмоль). Соединение Примера 16, описанное в японской патентной заявке (Kokai) №2002-249497, используют в качестве фосфорамидита в цикле 1, 3'-1 ВОсилилрибоаденозин(N-bz)-фосфорамидит (ChemGene, ANP-5681) используют в качестве фосфорамидита в цикле 2, соединение ссылочного примера 8 используют в цикле 3, и соединение примера 8а, описанное в японской патентной заявке (Kokai) №Hei 11-246592, используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 2, 3 и 4 используют йод, и в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2):
йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-47,75% CH3CN (линейный градиент, 15 мин); 40°С; 10 мл/мин; 260 нм), собирают фракцию, которая элюируется приблизительно через 12,8 мин. Соединение настоящего изобретения элюируется через 4,11 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5%СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-10% (линейный градиент, 15 мин); 40°С; 10 мл/мин), и собирают фракции, которые элюируются приблизительно через 13,4 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2О. Фракцию сушат сублимацией и продукт получают в виде натриевой соли. Выход: 8,9 мг, ESI-Macc-спектр (отрицательный): 1537 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,85 мин.
(Пример 67)
Синтез соединения примера 67 (Пример соединения №1954)
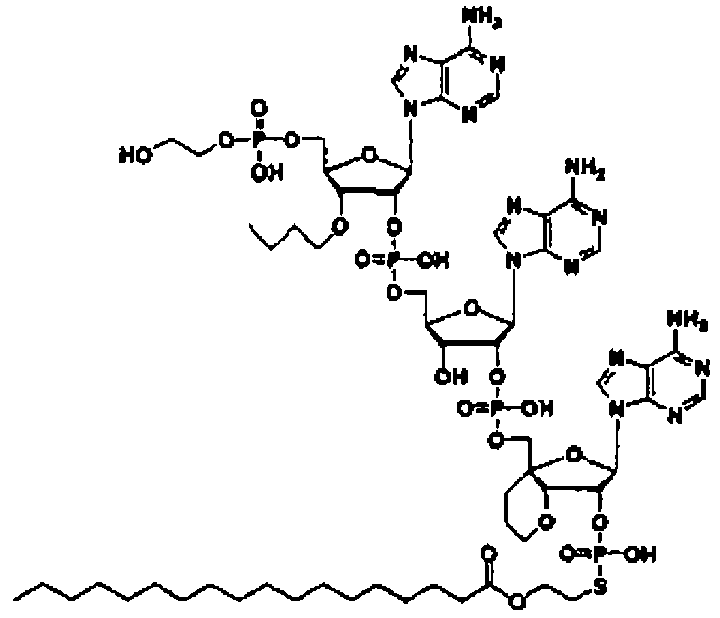
Синтез проводят с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве твердофазного носителя используют 3'-фосфат CPG (Glen Research)(40 мкмоль). Соединение Примера 16, описанное в японской патентной заявке (Kokai) №2002-249497, используют в качестве фосфорамидита в цикле 1, 3'-tBDсилилрибоаденозин(N-bz)-фосфорамидит (ChemGene, ANP-5681) используют в качестве фосфорамидита в цикле 2, соединение ссылочного примера 12 используют в цикле 3, и соединение примера 8а, описанное в японской патентной заявке (Kokai) №Hei 11-246592, используют в цикле 4. Для окисления или в качестве сульфурирующего агента в циклах 2, 3 и 4 используют йод, и в цикле 1 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан;
195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (TEAA), рН 7; 5-47,75% CH3CN (линейный градиент, 15 мин); 40°С; 10 мл/мин; 260 нм), собирают фракции, которые элюируются приблизительно через 13,9 мин. Соединение настоящего изобретения элюируется через 4,40 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5%СН3CN, 0,1 М TEAA (рН 7,0), раствор В: CH3CN; B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-10% (линейный градиент, 15 мин); 40°С; 10 мл/мин), и собирают фракции, которые элюируются приблизительно через 14,0 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2O. Фракцию сушат сублимацией и продукт получают в виде натриевой соли. Выход: 7,7 мг, ESI-Macc-спектр (отрицательный): 1551 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,87 мин.
(Пример 68)
Синтез соединения примера 68 (Пример соединения №1955)
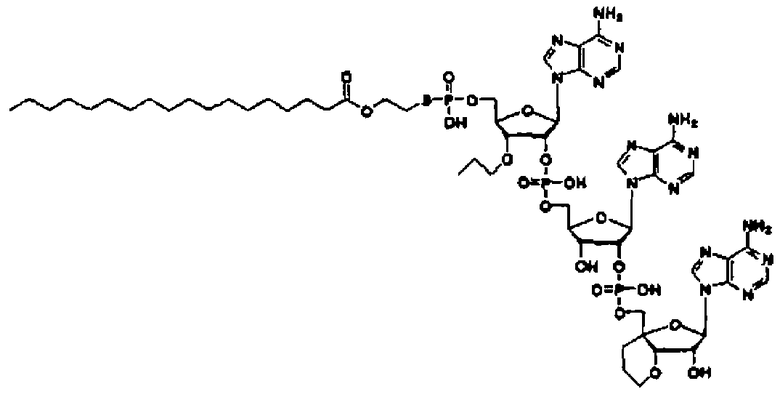
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) №2002-249497 (40 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene, ANP-5681), в цикле 2 используют соединение ссылочного примера 8, и в цикле 3 используют 5'-фосфорилирующий реагент (Phosphalink reagent, Applied Biosystems). При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют йод, тогда как в цикле 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование: трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к оставшемуся остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-47,75% СН3CN (линейный градиент, 15 мин); 40°С; 10 мл/мин; 260 нм), и собирают фракцию, которая элюируется приблизительно через 13,5 мин. Соединение настоящего изобретения элюируется через 4,00 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2О, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 5-15% (линейный градиент, 15 мин); 40°С; 10 мл/мин), и собирают фракции, которые элюируются приблизительно через 17,1 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2O. Фракцию сушат сублимацией, и продукт получают в виде натриевой соли. Выход: 2,5 мг, ESI-Macc-спектр (отрицательный): 1413 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 11,19 мин.
(Пример 69)
Синтез соединения примера 69 (Пример соединения №1956)
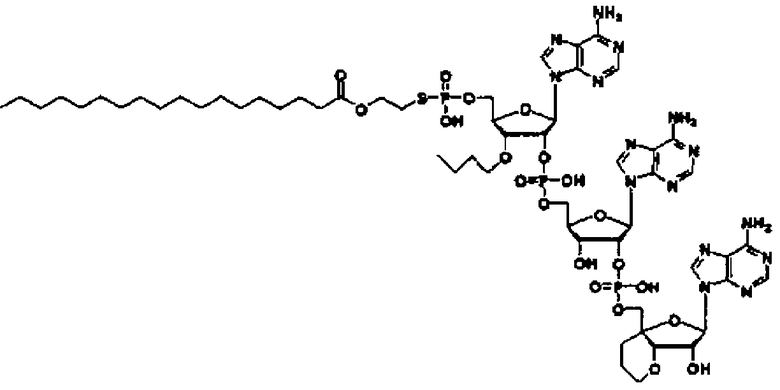
Синтез проводят с использованием в качестве аналога 5'-O-DMTr-рибоаденозина, связанного с подложкой CPG, соединения примера 17, описанного в японской патентной заявке (Kokai) №2002-249497 (40 мкмоль), с помощью синтезатора ДНК, основываясь на одном цикле конденсации, включающем приведенные ниже стадии 1)-4), с использованием программы синтеза для обычного синтеза 10 мкмоль РНК. В качестве фосфорамидита в цикле 1 используют 3'-tBDсилилрибоаденозин(N-bz)фосфорамидит (ChemGene, ANP-5681), в цикле 2 используют соединение ссылочного примера 12, и в цикле 3 используют 5'-фосфорилирующий реагент (Phosphalink reagent, Applied Biosystems). При окислении или в качестве сульфурирующего агента в циклах 1 и 2 используют йод, тогда как в цикле 3 используют ксантангидрид (Tokyo Kasei Kogyo).
Цикл конденсации:
1) Детритилирование; трихлоруксусная кислота/дихлорметан; 195 с.
2) Сочетание: фосфорамидит (приблизительно 10 экв.)/ацетонитрил, тетразол/ацетонитрил; 15 мин.
3) Кэппирование: 1-метилимидазол/тетрагидрофуран, уксусный ангидрид/пиридин/тетрагидрофуран; 65 с.
4) Окисление (циклы 1 и 2): йод/вода/пиридин/тетрагидрофуран; 70 с.
Сульфурирование (цикл 3): ксантангидрид (0,02 М)/ацетонитрил'пиридин (смешанный растворитель, 9:1); 15 мин.
После синтеза аналога 2-5А, имеющего целевую структуру в состоянии, в котором группа 5'-DMTr удалена, вместе с отщеплением олигомера от подложки, цианоэтильную группу, служащую в качестве защитной группы на атоме фосфора, и бензоильную группу на адениновом основании удаляют обработкой смесью концентрированного водного аммиака и этанола (3:1) при 55°С в течение 18 ч. Затем растворитель отгоняют при пониженном давлении и доводят рН до 2,0 путем добавления водной хлористо-водородной кислоты (2 н.) к полученному остатку, с последующей реакцией при комнатной температуре в течение ночи для удаления силильной группы. После нейтрализации водным аммиаком и отгонки растворителя продукт очищают ВЭЖХ с обращенной фазой (Shimadzu Seisakusho LC-VP; колонка (GL Science Inertsil Prep-ODS (20×250 мм)); 0,1 М водный триэтиламинацетат (ТЕАА), рН 7; 5-47,75% CH3CN (линейный градиент, 15 мин); 40°С; 10 мл/мин; 260 нм), и собирают фракции, которые элюируются приблизительно через 13,8 мин. Соединение настоящего изобретения элюируется через 4,10 мин при анализе ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN; В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), λмакс (Н2O)=258 нм.
250 OD продукта и 48,1 мкл триэтиламина растворяют в смеси 5 мл ацетонитрила и 0,5 мл Н2O, и добавляют 135 мг 2-(стеароилокси)этилбромида (Ackerman et al., J. Am. Chem. Soc., 78, 1956, 6025) в 5,5 мл ацетонитрила, с последующим взаимодействием смеси при 42°С в течение ночи. Органический растворитель затем отгоняют при пониженном давлении, и остаток промывают гексаном. Водный слой смешивают с МеОН и затем смесь наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil prep ODS (20×250 мм)); раствор А: 5% CH3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN; B% 10-30% (линейный градиент, 20 мин); 40°С; 10 мл/мин; 260 нм), и собирают фракции, которые элюируются приблизительно через 30,5 мин. Фракции концентрируют в вакууме и остаток вводят в две последовательные ионообменные колонки Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая) и элюируют Н2O. Фракцию сушат сублимацией, и продукт получают в виде натриевой соли. Выход: 5,9 мг, ESI-Macc-спектр (отрицательный): 1427 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: B% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 11,30 мин.
(Пример 70)
Синтез соединения примера 70 (Пример соединения №1957)
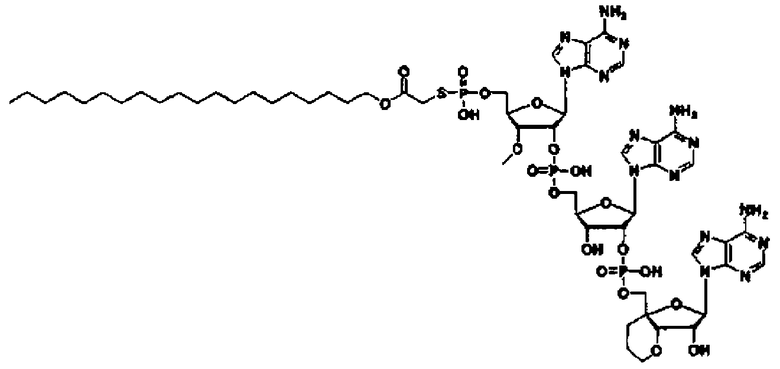
5,1 мг соединения ссылочного примера 13, растворенного в 0,8 мл ацетонитрила, растворяют в смеси 2 мг соединения Примера 2, 8 мкл пиридина, 0,72 мл ацетонитрила и 80 мкл H2O, с последующей реакцией смеси при 44°С в течение 3 часов. Затем органический растворитель отгоняют при пониженном давлении, и остаток промывают гексаном. Водный слой смешивают с МеОН и затем наносят на Sephadex LH-20 (GE healthcare) хроматограмму для гель-фильтрации и очищают. После концентрирования фракции, содержащей продукт, в вакууме, продукт дополнительно очищают с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 40-80% (линейный градиент, 10 мин); 60°С; 2 мл/мин), и фракции, которые элюируются приблизительно через 7,6 мин, собирают. Выход: 3,83 OD, ESI-Macc-спектр (отрицательный): 1413 [М-Н]-.
При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 40-80% (линейный градиент, 10 мин); 60°С; 2 мл/мин), продукт элюируется через 8,02 мин.
(Пример 71)
Синтез соединения примера 71 (Пример соединения №1958)
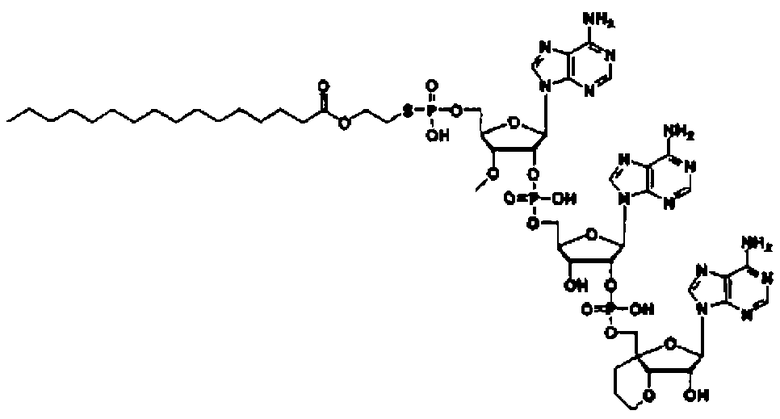
10 мг соединения Примера 2 и 81 мг 2-(стеароилокси)этилбромида растворяют в смеси 4 мл ацетонитрила и 160 мкл воды, и добавляют 13 мкл триэтиламина, с последующей реакцией смеси при 42°С в течение 4,5 часов. Органический растворитель отгоняют при пониженном давлении, и остаток очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil Prep ODS (30×250 мм)); 0,1 М ТЕАА, рН 7,0; 60% CH3CN (изократический); 40°С; 20 мл/мин; 260 нм, собирают фракции, содержащие продукт, и концентрируют в вакууме. Остаток вводят в две последовательные ионообменные колонки с Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая), и элюируют Н2O. Фракцию сушат сублимацией, и продукт получают в виде натриевой соли. Выход: 2,7 мг, ESI-Macc-спектр: 1356,50 (М-Н). При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: СН3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 10,30 мин.
(Пример 72)
Синтез соединения примера 72 (Пример соединения №966)
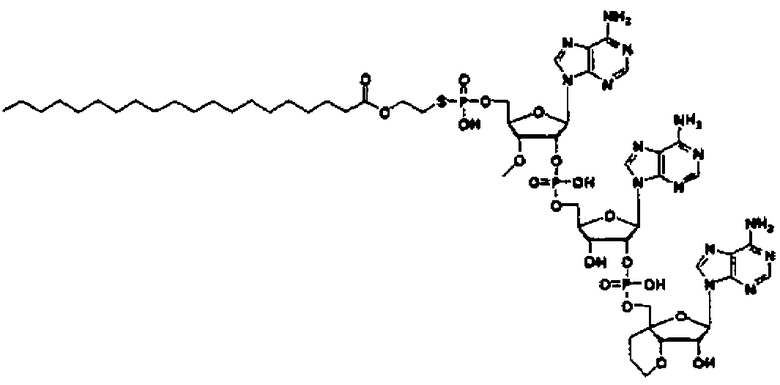
10 мг соединения примера 2 и 20 мг 2-(стеароилокси)этилбромида растворяют в смеси 4 мл ацетонитрила и 160 мкл H2O, и добавляют 20 мкл триэтиламина, с последующей реакцией смеси при 42°С в течение 4,5 часов. Затем органический растворитель отгоняют при пониженном давлении, и остаток очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil Prep-ODS (30×250 мм)); 0,1 М ТЕАА, рН 7,0; 60% CH3CN (изократический); 40°C; 20 мл/мин; 260 нм, и собирают фракции, содержащие продукт, и концентрируют в вакууме. Остаток вводят в две последовательные ионообменные колонки с Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая), и элюируют Н2O. Фракцию сушат сублимацией, и продукт получают в виде натриевой соли. Выход: 3,1 мг, ESI-Macc-спектр: 1412 (М-Н). При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); раствор А: 5% СН3CN, 0,1 М ТЕАА (рН 7,0), раствор В: CH3CN: В% 0-30% (линейный градиент, 5 мин) и 30-80% (линейный градиент, 5 мин); 60°С; 2 мл/мин), продукт элюируется через 11,50 мин.
(Пример 73)
Синтез соединения примера 73 (Пример соединения №968):
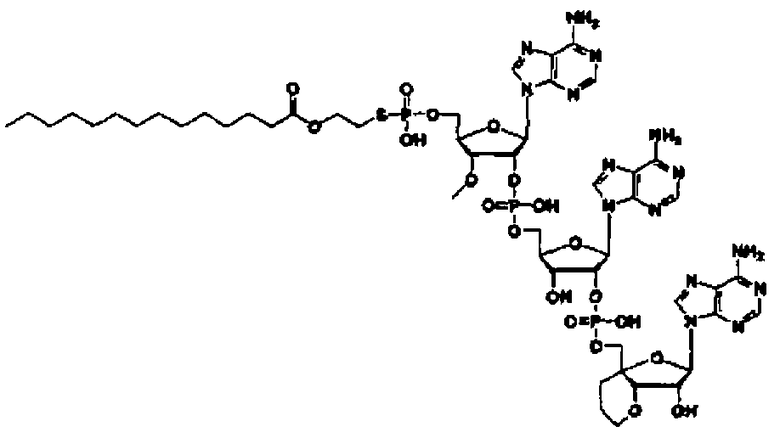
10 мг соединения примера 2 и 20 мг 2-(стеароилокси)этилбромида растворяют в смеси 4 мл ацетонитрила и 160 мкл H2O и добавляют 20 мкл триэтиламина, с последующей реакцией смеси при 42°С в течение 4,5 часов. Затем органический растворитель отгоняют при пониженном давлении, и остаток очищают с помощью ВЭЖХ с обращенной фазой (колонка (GL Science Inertsil Prep-ODS (30×250 мм)); 0,1 М ТЕАА, рН 7,0; 60-80% CH3CN (линейный градиент, 20 мин); 40°С; 20 мл/мин; 260 нм, собирают фракции, содержащие продукт, и концентрируют в вакууме. Остаток вводят в две последовательные ионообменные колонки с Dowex 50WX8-200 в пиридиниевой и натриевой форме (3 мл каждая), и элюируют Н2O. Фракцию сушат сублимацией, и продукт получают в виде натриевой соли. Выход: 1,8 мг, ESI-Macc-спектр: 1328 (М-Н). При анализе продукта с помощью ВЭЖХ с обращенной фазой (колонка (Merck Chromolith (4,6×150 мм)); 0,1 М ТЕАА, рН 7,0, 43-62% CH3CN (линейный градиент, 10 мин) 60°С; 2 мл/мин), продукт элюируется через 5,94 мин.
(Ссылочный пример 1)
Синтез соединения ссылочного примера 1.
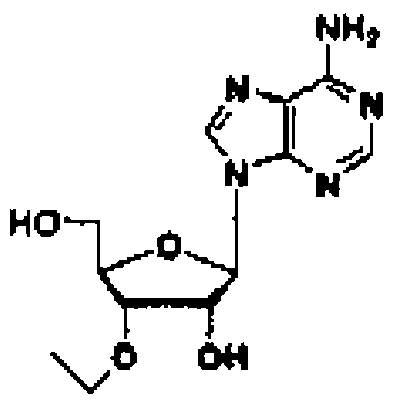
К раствору аденозина (15,0 г, 56,1 ммоль) в диметилсульфоксиде (150 мл) добавляют КОН (8,18 г, 146 ммоль) и этилбромид (10,6 мл, 141 ммоль), и смесь перемешивают при комнатной температуре в течение 3 часов. К реакционной смеси добавляют ацетон (200 мл), затем смесь фильтруют для удаления нерастворимых веществ. Фильтрат упаривают в вакууме, и остаток очищают хроматографией на силикагеле (СН3Cl2:МеОН=9:1). Продукт дополнительно очищают с помощью хроматографии на DOWEX1-2 (EtOH:H2O=l:1) для отделения другого изомера с получением продукта (1,56 г, 9,4%). FAB-MS (mNBA): 296 [М+Н]+.
(Ссылочный пример 2)
Синтез соединения ссылочного примера 2.
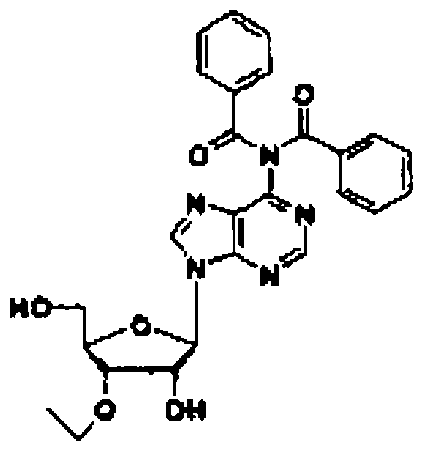
К раствору соединения ссылочного примера 1 (700 мг, 2,37 ммоль) в безводном пиридине (5 мл), добавляют триметилсилилхлорид (1,0 мл, 7,88 ммоль). После перемешивания при 0°C в течение 3 часов добавляют бензоилхлорид (1,0 мл, 8,61 ммоль). После перемешивания при комнатной температуре в течение 1,5, часов при 0°С добавляют Н2О (1 мл). После перемешивания в течение 15 мин добавляют концентрированный аммиак (2,0 мл) и перемешивают в течение 30 мин. Реакционную смесь концентрируют в вакууме, и остаток очищают хроматографией на силикагеле (СН2Cl2:МеОН=100:2) с получением продукта (720 мг, 80%). 1H-ЯМР (400 МГц, CDCl3) δ 1,31(3Н, т, 7,0 Гц), 3,61(1Н, дд, 7,0 и 9,0 Гц); 3,85(2Н, м), 3,78(1Н, дд, 7,0 и 9,0 Гц), 3,98(1Н, д; 1,5 Гц), 4,01(1Н, д; 1,5 Гц), 4,21(1Н, дд, 0,8 и 5,9 Гц), 4,35(1Н, д, 1,6 Гц), 4,95(1Н, ушир.), 7,2-7, 9 (10Н, м), 8,10(1Н, с), 8,60(1Н, с). FAB-MS (mNBA): 504 [М+Н]+.
(Ссылочный пример 3)
Синтез соединения ссылочного примера 3.
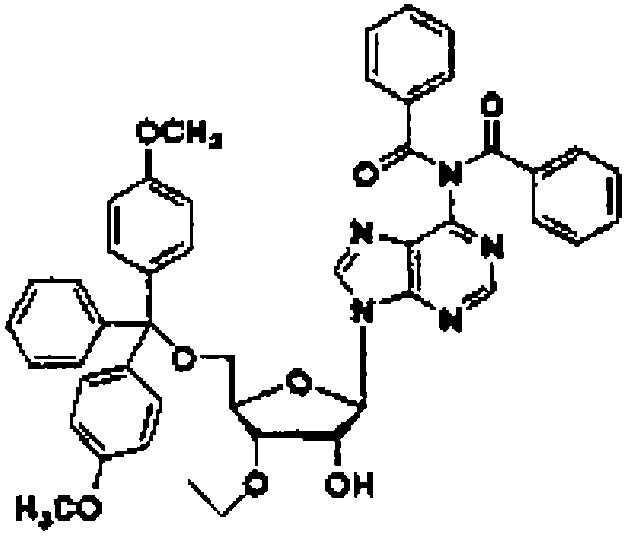
К раствору соединения Ссылочного примера 2 (700 мг, 1,39 ммоль) в сухом СН2Cl2 (8 мл) добавляют пиридин (1,0 мл) и диметокситритилхлорид (612 мг, 1,81 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 80 мин. После гашения МеОН (1 мл) в СН2Cl2 (10 мл), реакционную смесь промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и затем упаривают в ваккуме. Остаток очищают хроматографией на силикагеле (гексан:AcOEt=1:1) с получением продукта (983 мг, 84%). FAB-MS (mNBA): 806 [М+Н]+.
(Ссылочный пример 4)
Синтез соединения ссылочного примера 4.
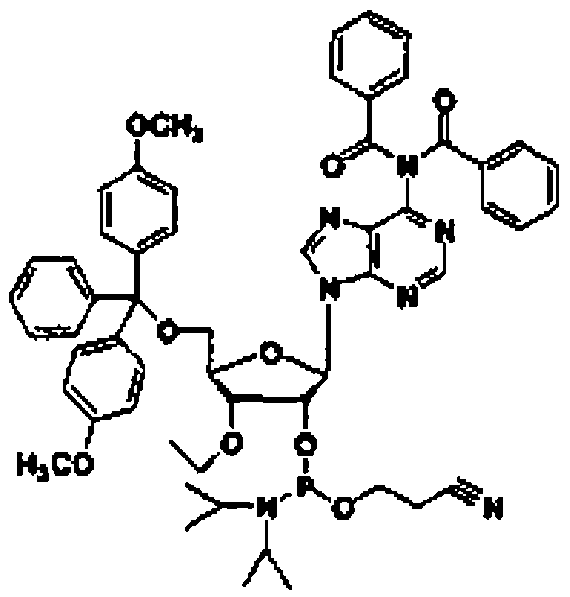
К раствору соединения ссылочного примера 3 (400 мг, 0,497 ммоль) в сухом СН2Cl2 (4 мл) добавляют диизопропиламин (180 мкл, 1,03 ммоль) и N,N-диизопроил-2-цианоэтилхлорфосфорамидит (130 мкл, 0,538 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После разбавления AcOEt (10 мл) реакционную смесь промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и затем упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:AcOEt=2:3) с получением продукта (367 мг, 73%). 1H-ЯМР (400 МГц, CDCl3) δ 1,1-1,2(15Н, м), 2,4(2Н, м), 3,73(3Н, с), 3,74(3Н, с), 3,3-4,0(8Н, м), 4,1(1Н, м), 4,2(1Н, м), 5,0(1Н, м), 6,25(1Н, д, 4,3 Гц), 6,79(4Н, д, 8,6 Гц), 7,1-7,9(19Н, ь), 8,37(1Н, с), 8,56(1Н, с). FAB-MS (mNBA): 1006 [М+Н]+.
(Ссылочный пример 5)
Синтез соединения ссылочного примера 5.
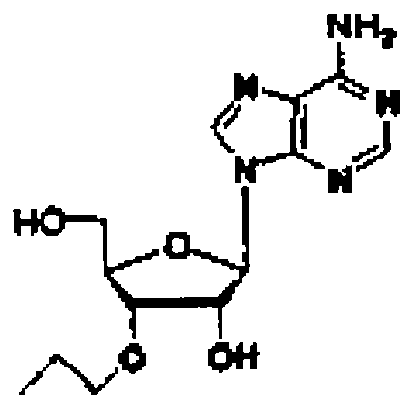
К раствору аденозина (15,0 г, 56,1 ммоль) в диметилсульфоксиде (100 мл) добавляют КОН (8,18 г, 146 ммоль) и 1-йодпропан (10,2 мл, 105 ммоль), и смесь перемешивают при комнатной температуре в течение 12 часов. К реакционной смеси добавляют ацетон (200 мл), и смесь фильтруют для удаления нерастворимых веществ. Фильтрат упаривают в вакууме, и остаток очищают хроматографией на силикагеле (СН2Cl3:МеОН=9:1). Продукт дополнительно очищают хроматографией на DOWEX1-2 (EtOH:H2O=1:1) для отделения другого изомера с получением продукта (752 мг, 4,3%). FAB-MS (mNBA): 310 [М+Н]+.
(Ссылочный пример 6)
Синтез соединения ссылочного примера 6.
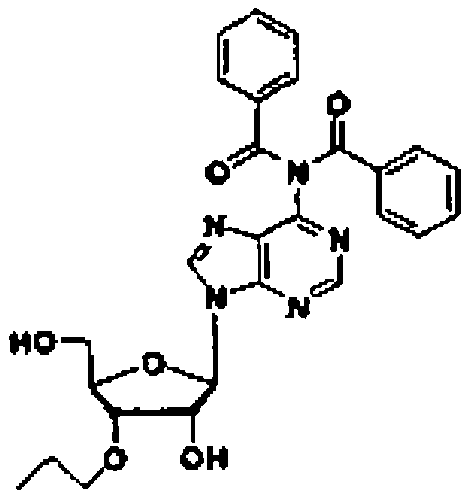
К раствору соединения ссылочного примера 5 (700 мг, 2,26 ммоль) в безводном пиридине (7 мл) добавляют триметилсилилхлорид (1,5 мл, 11,8 ммоль). После перемешивания при 0°С в течение 45 мин добавляют бензоилхлорид (1,3 мл, 11,2 ммоль). После перемешивания при комнатной температуре в течение 1,5 часов, добавляют при 0°С Н2O (1,5 мл). После перемешивания в течение 15 мин добавляют концентрированный аммиак (1,5 мл) и перемешивают в течение 30 мин. Реакционную смесь концентрируют в вакууме, и остаток очищают хроматографией на силикагеле (CH2Cl2:MeOH=100:1) с получением продукта (1,05 г, 90%). 1H-ЯМР (400 МГц, CDCl3) δ 0,99(3Н, т, 7,4 Гц), 1,70(2Н, дт, 7,4 и 11 Гц), 3,12(1Н, д, 9,4 Гц), 3,51(1Н, дт, 6,5 и 9,0 Гц), 3,66(1Н, дт, 6,5 и 9,0 Гц), 3,72(1Н, д, 11 Гц), 3,99(1Н, д, 13 Гц), 4,19(1Н, дд, 1,2 и 5.6 Гц), 4,35(1Н, д, 1,2 Гц), 4,96(1Н, ушир.), 5,53(1Н, ушир.), 5,79(1Н, д, 7,0 Гц), 7,2-7,9(10Н, м), 8,11(1Н, с), 8,59(1Н, с). FAB-MS (mNBA): 518 [М+Н]+.
(Ссылочный пример 7)
Синтез соединения ссылочного примера 7.

К раствору соединения ссылочного примера 6 (1,04 г, 2,0 ммоль) в сухом СН2Cl2 (8 мл) добавляют пиридин (2,0 мл) и диметокситритилхлорид (885 мг, 2,61 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После гашения МеОН (2 мл) в СН2Cl2 (10 мл) реакционную смесь промывают насыщенным раствором NaCO3 и рассолом, сушат над MgSO4, затем упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:AcOEt=1:1) с получением продукта (1, 3 г, 79%). 1H-ЯМР 400 МГц, CDCl3) δ 0,93(1Н, т, 7,6 Гц), 1,62(2Н, дт, 7,6 и 12 Гц), 3,25(1Н, д, 7,0 Гц), 3,29(1Н, дд, 3,9 и 10 Гц), 3,50(2Н, м), 3,75(6Н, с), 4,19(1Н, дд, 4.0 и 5,4 Гц), 4,25(1Н, дд, 4,0 и 7,8 Гц), 4,84(1Н, дд, 5,4 и 12 Гц), 6,02(1Н, д, 5,4 Гц), 6,77(4Н, д, 9,0 Гц), 7,1-7,9(19Н, м), 8,25(1Н, с), 8,56(1Н, с). FAB-MS (mNBA): 820 [М+Н]+.
(Ссылочный пример 8)
Синтез соединения ссылочного примера 8.
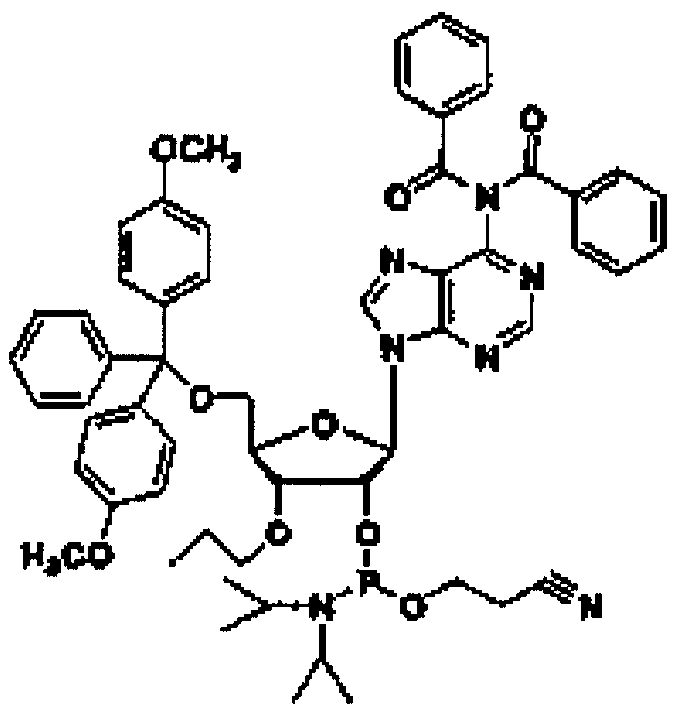
К раствору соединения ссылочного примера 7 (200 мг, 0,244 ммоль) в сухом СН2Cl2 (2 мл) добавляют N,N-диизопропил-2-цианоэтилхлорфосфорамидит (65 мкл, 0,291 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После разбавления AcOEt (10 мл) реакционную смесь промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и затем упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:AcOEt=2:3) с получением продукта (305 мг, количественный). 1H-ЯМР (400 МГц, CDCl3) δ 1,0-1,2(15Н, м), 1,6(2Н, м), 2,4(2Н, м), 3,72(3Н, с), 3,73(3Н, с), 3,3-3,9(8Н, м), 4,1(1Н, м), 4,2(1Н, м), 4,9(1Н, м), 6,23(1Н, д, 4,3 Гц), 6,77(4Н, д, 9,0 Гц), 7,1-7,9(19Н, м), 8,34(1Н, с), 8,56(1Н, с). FAB-MS (mNBA): 1020 [М+Н]+.
(Ссылочный пример 9)
Синтез соединения ссылочного примера 9.
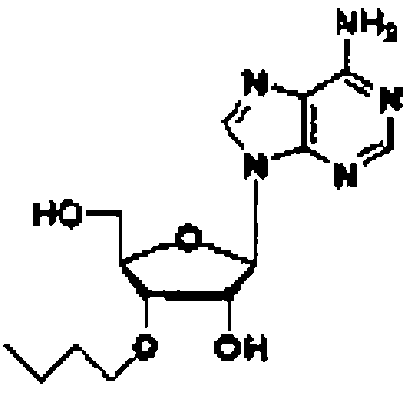
К раствору аденозина (16,0 г, 56,1 ммоль) в диметилсульфоксиде (100 мл) добавляют КОН (8,18 г, 146 ммоль) и 1-бромбутан (10,5 мл, 97,8 ммоль), и смесь перемешивают при комнатной температуре в течение 4 часов. К реакционной смеси добавляют ацетон (200 мл), и смесь фильтруют для удаления нерастворимых веществ. Фильтрат упаривают в вакууме, и остаток очищают хроматографией на силикагеле (CH2Cl2:MeOH=9:1). Продукт дополнительно очищают хроматографией на DOWEX1-2 (EtOH:H2O=1:1) для отделения другого изомера с получением продукта (204 мг, 1,6%). FAB-MS (mNBA): 324 [М+Н]+.
(Ссылочный пример 10)
Синтез соединения ссылочного примера 10
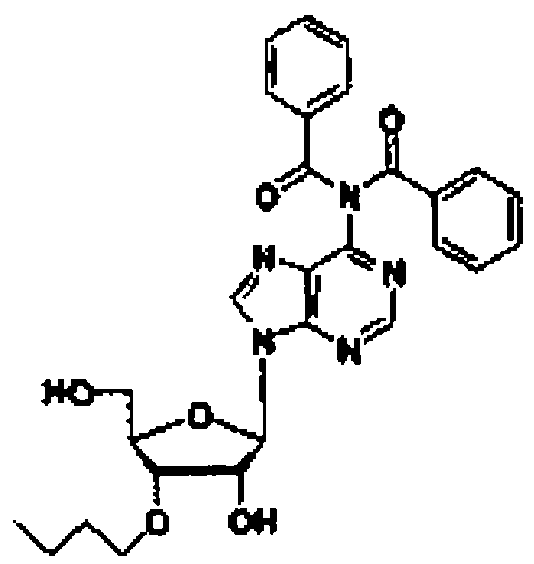
К раствору соединения ссылочного примера 9 (290 мг, 0,897 ммоль) в безводном пиридине (3 мл) добавляют гриметилсилихлорид (0,5 мл, 3,94 ммоль). После перемешивания при 0°С в течение 45 мин. Добавляют бензоилхлорид (0,5 мл, 4,31 ммоль). После перемешивания при комнатной температуре в течение 1 часа добавляют Н2O (0,5 мл) при 0°С. После перемешивания в течение 15 мин добавляют концентрированный аммиак (0,5 мл) и перемешивают в течение 30 мин. Реакционную смесь концентрируют в вакууме, и остаток очищают хроматографией на силикагеле (CH2Cl2:MeOH=100:1) с получением продукта (345 мг, 72%). FAB-MS (mNBA): 532 [М+Н]+.
(Ссылочный пример 11)
Синтез соединения ссылочного примера 11
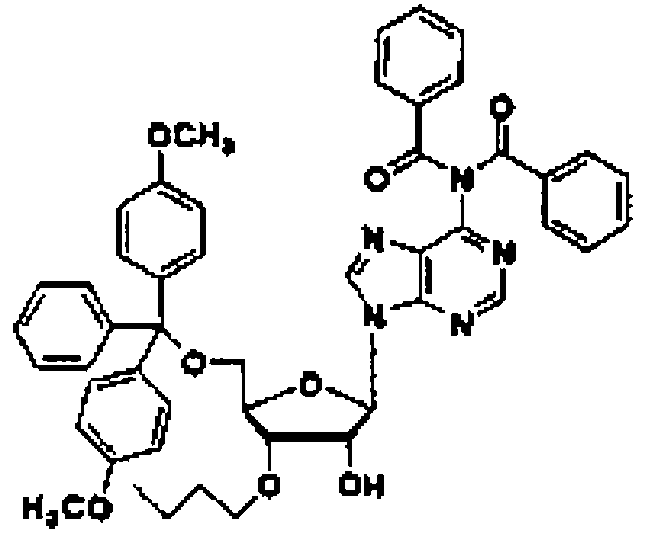
К раствору соединения ссылочного примера 10 (340 мг, 0,64 ммоль) в сухом СН2Cl2 (3 мл) добавляют пиридин (0,5 мл) и диметокситритилхлорид (282 мг, 0,832 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1,5 ч. После гашения МеОН (2 мл) в СН2Cl2 (10 мл) реакционную смесь промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и затем упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:АсОЕ1=1:1) с получением продукта (422 мг, 79%). 1H-ЯМР (400 МГц, CDCl3) δ 0,92(3Н, т, 7,5 Гц), 1,38(2Н, м), 1,56(2Н, м), 3,24(1Н, д, 7,0 Гц), 3,29(1Н, дд, 4,0 и 11 Гц), 3,53(2Н, м), 3,75(6Н, с), 4,18(1Н, дд, 3,9 и 5,5 Гц), 4,24(1Н, м, 3,9 и 8,2 Гц), 4,84(1Н, дд, 5,5 и 12 Гц), 6,01(1Н, д, 5,1 Гц), 6,77(4Н, д, 8,6 Гц), 7,1-7,9(19Н, м), 8,24(1Н, с), 8,56(1Н,с). FAB-MS (mNBA): 834 [М+Н]+.
(Ссылочный пример 12)
Синтез соединения ссылочного примера 12
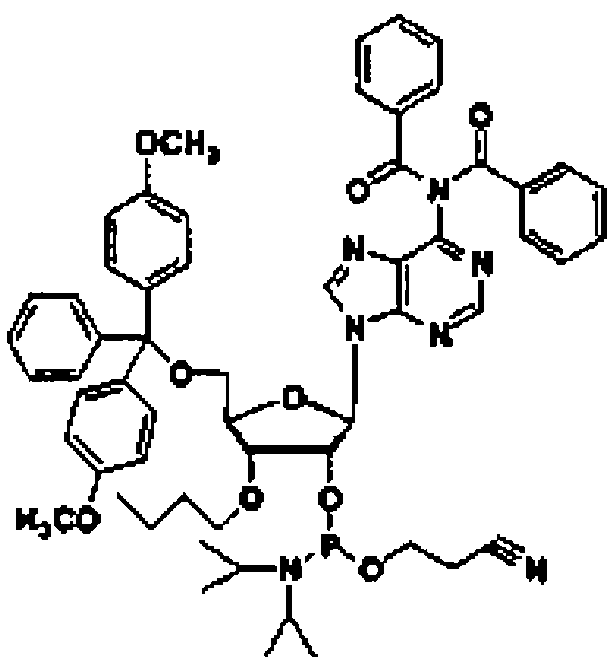
К раствору соединения ссылочного примера 11 (200 мг, 0,244 ммоль) в сухом CH2Cl2 (2 мл) добавляют N,N-диизопропил-2-цианоэтилхлорфосфорамидит (64 мкл, 0,287 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После разбавления AcOEt (10 мл) реакционную смесь промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и затем упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:AcOEt=2:3) с получением продукта (206 мг, 83%). 1H-ЯМР 400 МГц, CDCl3) δ 0,9-1,2(15Н, м), 1,3 (2Н, м), 1,5(2Н, м), 2,4(2Н, м), 3,73(6Н, с), 3,3-3,9(8Н, м), 4,1(1Н, м), 4,2(1Н, м), 5,0(1Н, м), 6,23(1Н, д, 4,3 Гц), 6,76(4Н, д, 8,6 Гц), 7,1-7,9(19Н, м), 8,33(1Н, с), 8,56(1Н, с). FAB-MS (mNBA): 1034 [М+Н]+.
(Ссылочный пример 13)
Синтез соединения ссылочного примера 13
К раствору 1-эйкозанола (1,19 г, 4,00 ммоль) в сухом СН2Cl2 (5 мл) и сухом ТГФ (5 мл) добавляют триэтиламин (840 мкг, 6,00 ммоль). К смеси при 0°С добавляют 2-бромацетилбромид (440 мкг, 5,00 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. После гашения Н2O при 0°С реакционную смесь разбавляют СН2Cl2 (10 мл), затем промывают насыщенным раствором NaHCO3 и рассолом, сушат над MgSO4 и упаривают в вакууме. Остаток очищают хроматографией на силикагеле (гексан:СН2Cl2=1:1) с получением продукта (1,39 г, 83%). 1H-ЯМР (400 МГц, CDCl3) δ 0,88(3Н, т, 7,0 Гц), 1,25(34Н, с), 1,66(2Н, м), 3,82(2Н, с), 4,16(2Н, т, 6,7 Гц). FAB-MS (mNBA): 419 [M+H]+.
(Пример испытания 1)
Измерение цитотоксической активности аналогов 2-5A (анализ MTT)
Клетки линии А549 клеток рака легких человека высевают с плотностью 800 клеток/200 мкл в 96-луночном планшете с использованием для среды RPMI1640 (Gibco BRL) (содержащей 10% телячьей фетальной сыворотки (Hyclone)), затем выдерживают в течение ночи в 5% CO2 при 37°С. Каждый аналог 2-5A добавляют в каждую лунку так, чтобы конечная концентрация составляла 10 мкМ, затем выдерживают 72 ч (3 дня). После выдерживания в течение 72 ч (3 дня) в каждую лунку добавляют MTT (бромид 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия) в 50 мкл аликвотах при концентрации MTT/RPMI1640 5 мг/мл, с последующим дополнительным выдерживанием в течение 4 ч. Через 4 ч среду удаляют и в каждую лунку добавляют 150 мкл диметилсульфоксида. После встряхивания в течение 5 мин измеряют УФ-поглощение при 540 нм для определения отношения числа жизнеспособных клеток группы с дозой соединения к числу жизнеспособных клеток группы с необработанными клетками через 72 ч после добавления испытуемого соединения.
Цитотоксическая активность во время добавления рассматриваемых соединений (10 мкМ) в отношении клеток А549 представлена на графике. На этом графике природный тип 2-5A означает 3 mer, 2',5'-олигоаденилат с 5'-монофосфатной группой, имеющий представленную ниже структуру (Imai, J. and Torrence, P.F., J. Org. Chem., 1985, 50(9), 1418-1426).
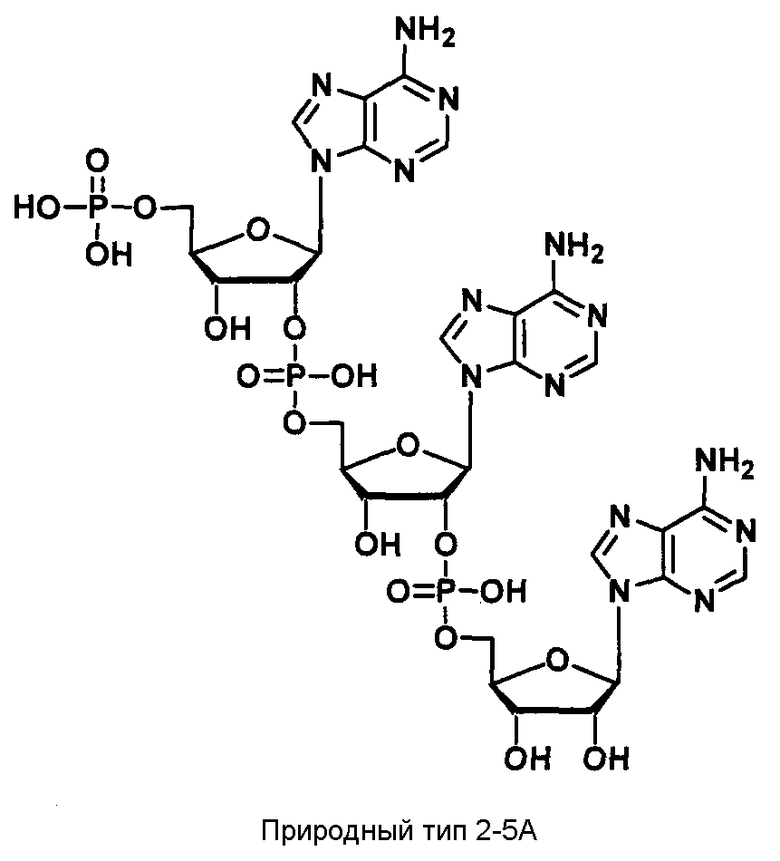
Цитотоксическая активность на клетках A549 в период добавления изучаемых соединений (10 мкМ)
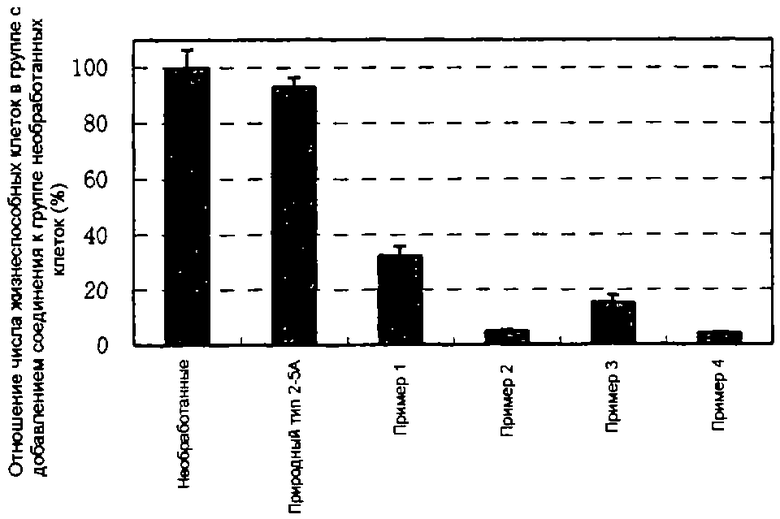
Как видно из графика, в период добавления к среде в концентрации 10 мкМ, в отличие от природного типа 2-5A, не проявляющего никакого цитотоксического действия против клеток линии А549 клеток рака легких человека, испытуемые соединения показывают превосходную цитотоксическую активность.
(Пример испытания 2)
Измерение цитотоксической активности аналогов 2-5A (анализ MTT)
Клетки линии А549 клеток рака легких человека высевают с плотностью 800 клеток/200 мкл в 96-луночном планшете с использованием для среды RPMI1640 (Gibco BRL) (содержащей 10% телячьей фетальной сыворотки (Hyclone)), затем выдерживают в течение ночи в 5% CO2 при 37°С. Каждый аналог 2-5A добавляют в каждую лунку так, чтобы конечная концентрация составляла 0,001-10 мкМ, затем выдерживают 72 ч (3 дня) (n=3 или 4). После выдерживания в течение 72 ч (3 дня) в каждую лунку добавляют MTT (бромид 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия) в 50 мкл аликвотах при концентрации MTT/RPMI1640 5 мг/мл, с последующим дополнительным выдерживанием в течение 4 ч. Через 4 ч среду удаляют и в каждую лунку добавляют 150 мкл диметилсульфоксида. После встряхивания в течение 5 мин измеряют УФ-поглощение при 540 нм для определения относительного показателя числа жизнеспособных клеток группы с дозой соединения к числу жизнеспособных клеток группы с необработанными клетками, затем рассчитывают концентрацию IC50, при которой ингибируется рост клеток на 50%.
В таблице показаны ингибирующие на 50% рост клеток концентрации испытуемых соединений в отношении клеток A549.
Как видно из данных приведенной выше таблицы, в отличие от природного типа 2-5A, не проявляющего никакого цитотоксического действия против клеток линии А549 клеток рака легких человека даже при концентрации 10 мкМ, испытуемые соединения показывают превосходную цитотоксическую активность.
(Пример испытания 3)
Измерение цитотоксической активности аналогов 2-5А (анализ МТТ)
Клеточные линии НСТ-15 карциномы толстой кишки высевают с плотностью 2000 клеток/200 мкл в 96-луночном планшете с использованием для среды RPMI1640 (Gibco BRL) (содержащей 10% телячьей фетальной сыворотки (Hyclone)), затем выдерживают в течение ночи в 5% CO2 при 37°С. Каждый аналог 2-5А добавляют в каждую лунку так, чтобы конечная концентрация составляла 0,001-10 мкМ, затем выдерживают 72 ч (3 дня) (n=3 или 4). После выдерживания в течение 72 ч (3 дня) в каждую лунку добавляют МТТ (бромид 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия) в 50 мкл аликвотах при концентрации MTT/RPMI1640 1 мг/мл, с последующим дополнительным выдерживанием в течение 4 ч. Через 4 ч среду удаляют и в каждую лунку добавляют 150 мкл диметилсульфоксида. После встряхивания в течение 5 мин измеряют УФ-поглощение при 540 нм для определения отношения числа жизнеспособных клеток группы с дозой соединения к числу жизнеспособных клеток группы с необработанными клетками, с последующим вычислением концентрации IC50, при которой ингибируется рост клеток на 50%.
В таблице показаны ингибирующие на 50% рост клеток концентрации испытуемых соединений (мкМ) в отношении клеток НТС-15.
Ингибирующие на 50% рост клеток концентрации испытуемых соединений относительно клеток НТС-15.
(Пример испытания 4)
Измерение активности аналогов 2-5А в отношении вируса коровьей оспы.
Клетки HeLa, линия клеток цервикального рака человека, высеянные в 24-луночном планшете, выращивают до конфлюэнтности в минимальной основной среде (MEM), содержащей 10% телячьей фетальной сыворотки (FBS), при 37°С в термостате с 5% CO2. После удаления среды добавляют 250 мкл среды, содержащей 2,5% FBS, a через 24 часа среду заменяют 200 мкл свежей среды, содержащей 2,5% FBS.
Для исследования противовирусной активности добавляют 70 бляшкообразующих единиц (БОЕ) штамма WR вируса коровьей оспы в 50 мкл MEM, содержащей 2,5% FBS, и клетки инкубируют еще в течение 2 часов. Затем добавляют 250 мкл различных концентраций соединения примера 23 в той же самой среде, и продолжают инкубацию в течение 24 часов. После удаления среды клетки промывают один раз забуференным фосфатом физиологическим раствором (FBS) и фиксируют 500 мкл этанола в течение 10 мин. После удаления этанола клетки высушивают. Добавляют 100 мкл кроличьего иммуноглобулина против вируса коровьей оспы (Virostat), и затем инкубируют в течение 1 часа при комнатной температуре. Клетки промывают три раза PBS, и добавляют 100 мкл козьих антител против кроличьего иммуноглобулина Ig (Dako Oytomation A/S, Denmark) и клетки инкубируют в течение 1 часа при комнатной температуре. Вновь, после промывки клеток три раза, добавляют 100 мкл растворимых комплексов кроличьих антител с пероксидазой хрена и пероксидазу хрена (Rabbit PAP, Dako Cytomation A/S, Denmark) с последующей инкубацией в течение часа при комнатной температуре. Клетки промывают три раза PBS, и добавляют 100 мкл системы DAKO®LIQUID DAB SUBSTRATE-CHROMOGEN SYSTEM (Dako Cytomation A/S, Denmark), и клетки промывают водопроводной водой. Количество окрашенных очагов, содержащих инфицированные вирусом коровьей оспы клетки, подсчитывают визуально. Количество очагов в лунках, обработанных соединением, (в %) вычисляют, принимая количество очагов в неинфицированных лунках за 100%, и затем определяют концентрация IC50 соединения, которая снижает на 50% количество очагов.
Для исследования цитотоксичности соединения примера 23 в отношении клеток HeLa вместо добавления вируса коровьей оспы добавляют 50 мкл MEM, содержащей 2,5% FBS, и клетки инкубируют в течение 2 часов. Затем добавляют 250 мкл различных концентраций соединения примера 23 в той же самой среде, и инкубацию продолжают еще 24 часа. После удаления среды клетки промывают один раз PBS. Добавляют 800 мкл свежей среды, содержащей 2,5% FBS и 200 мкл XTT/PMS, и клетки инкубируют в течение 1 часа в термостате с 5% CO2. Раствор XTT/PMS получают следующим образом. Натриевую соль 2,3-бис[2-метокси-4-нитро-5-сульфофенил]-2Н-тетразолий-5-карбоксанилида (ХТТ, Sigma-Aldrich Co.) растворяют в MEM до получения концентрации 1 мг/мл. Добавляют 5 мкл 5 мМ феназинметосульфата (PMS, Sigma-Aldrich Co.), растворенного в PBS, на 1 л раствора ХТТ. Раствор XTT/PMS получают путем смешения растворов ХТТ и PMS непосредственно перед использованием. Измеряют оптическую плотность при 450 нм для определения относительного показателя числа жизнеспособных клеток группы с дозой соединения к числу жизнеспособных клеток группы с необработанными клетками, затем рассчитывают концентрацию IC50, при которой ингибируется рост клеток на 50%.
В таблице показаны концентрации IC50 (мкМ) для противовирусной активности и для цитотоксичности.
[Промышленная применимость]
Соединения настоящего изобретения обладают стабильностью и превосходной активностью (особенно противоопухолевой активностью) и могут быть полезны в качестве фармацевтического лекарственного средства (особенно в качестве противоопухолевых агентов).
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИРАЛЬНЫЕ РЕАГЕНТЫ ДЛЯ ПОЛУЧЕНИЯ ГОМОГЕННЫХ ОЛИГОМЕРОВ | 2016 |
|
RU2791532C2 |
| ПЕПТИД ЗВЕЗДЧАТКИ STELLARIA MEDIA L., ОБЛАДАЮЩИЙ АНТИФУНГАЛЬНОЙ АКТИВНОСТЬЮ | 2015 |
|
RU2603058C1 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЕПТИДОВ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ К РЕЦЕПТОРАМ ИНТЕГРИНОВ | 2002 |
|
RU2303042C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НЕОПТЕРИНА В КРОВИ | 2006 |
|
RU2313092C1 |
| АГЕНТЫ ДЛЯ ВИЗУАЛИЗАЦИИ | 2004 |
|
RU2355702C2 |
| КАТИОННЫЕ ОЛИГОНУКЛЕОТИДЫ, АВТОМАТИЗИРОВАННЫЕ СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2451022C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pЕТ22b(+)/SLURP-1, КОДИРУЮЩАЯ БЕЛОК SLURP-1, И ШТАММ БАКТЕРИЙ Escherichia coli BL21(DE3)/pET22b(+)/SLURP-1-ПРОДУЦЕНТ БЕЛКА SLURP-1 ЧЕЛОВЕКА | 2010 |
|
RU2453602C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНКИНАЗ | 2000 |
|
RU2260592C9 |
| СЕЛЕКТИВНЫЕ ЦИКЛОПЕПТИДЫ | 2001 |
|
RU2239642C1 |
| ВАРИАНТ АПРОТИНИНА | 1997 |
|
RU2197983C2 |
Изобретение относится к новым аналогам 2',5'-олигоаденилата, представленными общей формулой (1), или их фармакологически приемлемым солям, обладающим противовирусной и противоопухолевой активностью, к фармацевтической композиции на их основе и к их применению в качестве лекарственного средства и при получении лекарственного средства.
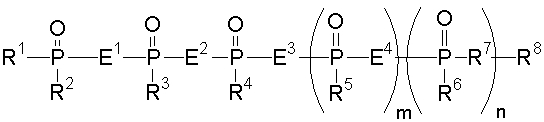
где Е1 представляет собой К2, Е2 представляет собой К1, Е3 представляет собой К2 или К3, и Е4 представляет собой К1, К2, К3 или К4, при этом К1, К2, К3 и К4 представляют собой:
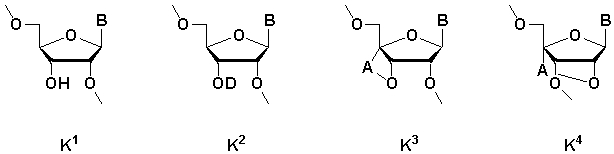
Значения заместителей R1-R8, m, n, А, В, D указаны в формуле изобретения. 6 н. и 4 з.п. ф-лы, 4 табл.
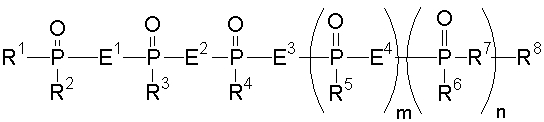
где m равно 0;
n равно О или 1;
R1 представляет собой алкоксигруппу, содержащую от 1 до 6 атомов углерода, замещенную гидроксильной группой, меркаптогруппу, алкилтиогруппу, содержащую от 1 до 4 атомов углерода, замещенную гидроксильной группой, или группу формулы Х1-Х2-Х3-S-; R2, R3, R4, R5 и R6 представляют собой гидроксильную группу, меркаптогруппу, алкилтиогруппу, содержащую от 1 до 4 атомов углерода, замещенную гидроксильной группой, или группу формулы Х1-Х2-Х3-S-;
R7 представляет собой атом кислорода, атом серы, группу -O(CH2CH2O)q (q равно целому числу от 2 до 6) или оксиалкиленоксигруппу, содержащую от 1 до 6 атомов углерода;
R представляет собой атом водорода или 5'-фосфорилированный аналог олигонуклеотида, в котором одна гидроксильная группа удалена из группы 5'-фосфорной кислоты;
Е1 представляет собой К2, Е2 представляет собой К', Е представляет собой К2 или К3, и Е4 представляет собой К1, К2, К3 или К4 (К1, К2, К3 и К4 представляют собой
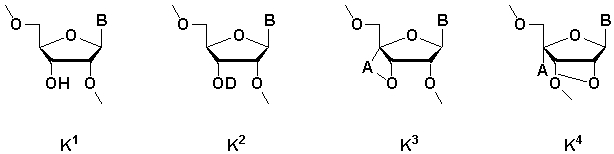
соответственно, где В представляет собой аденил, А представляет собой алкиленовую группу, содержащую от 1 до 4 атомов углерода, D представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода или алкенильную группу, содержащую от 2 до 6 атомов углерода;
X1 представляет собой алкильную группу, содержащую от 1 до 24 атомов углерода, или фенил;
X2 представляет собой группу -С(=O)O-, -ОС(=O)- или -C(=O)S-;
Х3 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода или его фармакологически приемлемая соль.
X1 представляет собой алкильную группу, содержащую от 10 до 24 атомов углерода;
Х3 представляет собой алкиленовую группу, содержащую от 1 до 4 атомов углерода.
| S.NAIK et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Nucleic Acids Research, 1998, т | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| Прибор для задней заточки радиальных режущих кромок на конических концах инструментов | 1917 |
|
SU1522A1 |
| S.S.JONES et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Journal of the American Chemical Society, 1979, т | |||
| Приспособление для записи звуковых явлений на светочувствительной поверхности | 1919 |
|
SU101A1 |

