ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По данной заявке испрашивается приоритет в соответствии с китайской патентной заявкой № 201811527414.4, поданной 13 декабря 2018 г., описание которой включено в настоящий документ посредством ссылки во всей своей полноте для всех целей.
ОБЛАСТЬ ТЕХНИКИ
[0002] Настоящее изобретение относится к области фармацевтического синтеза и, в частности, к соединению, которое может служить новым агонистом рецепторов THRβ, а также к способу его получения и применению.
УРОВЕНЬ ТЕХНИКИ
[0003] Гормон щитовидной железы (TH) синтезируется в щитовидной железе в ответ на тиреотропный гормон (TSH), секретируемый гипофизом. Тироксин играет довольно важную роль в регулировании роста, развития, обмена веществ и баланса организма. Гормоны щитовидной железы в основном бывают двух типов: 3,5,3’-трийод-L-тироксин (Т3) и тироксин (T4). Человеческие тела в основном секретируют Т4. В периферических органах Т4 трансформируется дейодиназой в Т3, обладающий большей активностью. Т3 и Т4, вырабатываемые щитовидной железой, находятся под контролем отрицательной обратной связи. Тиротропин (TSH) отвечает за нормальную функцию щитовидной железы и секрецию гормонов щитовидной железы. Тиротропин синтезируется в передней доле гипофиза, и его секреция контролируется рилизинг-гормоном щитовидной железы (TRH), синтезируемым в гипоталамусе.
[0004] Гормоны щитовидной железы функционируют путем связывания с рецепторами гормонов щитовидной железы (THR). Рецептор гормона щитовидной железы принадлежит к семейству ядерных рецепторов и регулирует экспрессию генов-мишеней. Рецепторы тироидных гормонов включают два разных подтипа, то есть THRα и THRβ. THRα в основном распределяется в сердечных тканях и играет важную роль в регуляции функции сердца. Подтип THRβ в основном экспрессируется в печени и гипофизе и регулирует метаболизм холестерина и секрецию тиреотропина.
[0005] При нормальных уровнях гормоны щитовидной железы TH поддерживают вес тела, скорость метаболизма, температуру тела и настроение и отвечают за регулирование холестерина в сыворотке. Были предприняты попытки использовать гормоны щитовидной железы для регулирования уровня холестерина в сыворотке крови. Однако, учитывая возможные побочные эффекты на сердце от приема природного гормона щитовидной железы (например, тахикардия и аритмия, сердечная недостаточность и функция щитошейного ствола, метаболизм в мышцах и остеопороз), гормоны щитовидной железы не подходят для лечения высокого уровня холестерина и ожирения. Результаты исследования животных с селективным нокаутом гена THR и результаты исследований некоторых селективных лигандов THR показывают, что побочные эффекты на сердце, вызываемые этими гормонами щитовидной железы, могут быть связаны с THRα.
[0006] Путь рецептора гормона щитовидной железы регулирует метаболизм липидов, включая холестерин, триглицериды и липопротеины. Клинически показано, что снижение холестерина низкой плотности может снизить частоту сердечно-сосудистых заболеваний.
[0007] Неалкогольная жировая болезнь печени (НАЖБП) также представляет собой тип метаболического расстройства, вызванного чрезмерным накоплением триглицеридов в печени, которое может дополнительно вызывать повреждение и воспаление клеток печени, что приводит к неалкогольному стеатогепатиту (НАСГ). Пациенты с НАСГ обычно страдают диабетом 2 типа, высоким уровнем холестерина, гиперлипемией и ожирением наряду с НАСГ. Кроме того, у них более высока вероятность развития гепатоцирроза, печеночной недостаточности и, в конечном итоге, рака печени. В настоящее время количество лекарств, которые эффективно лечат НАСГ, ограничено. Учитывая функцию гормона щитовидной железы по регулированию липидного обмена, путь рецепторов щитовидной железы становится потенциальной мишенью для лечения НАСГ и НАЖБП. На животных было доказано, что аналоги гормонов щитовидной железы могут значительно снизить уровень жира в печени животных.
[0008] Селективные агонисты THRβ можно использовать для предотвращения побочных эффектов на сердце, которые являются результатом обычных агонистов рецепторов THR, и, избирательно только для активации THRβ, тем самым улучшая метаболизм липидов в клетках и достигая функции снижения холестерина и жира в крови. Однако селективные агонисты THRβ могут также ингибировать щитошейный ствол, что приводит к депрессии, усталости, остеопорозу и другим побочным эффектам. Следовательно, желательно разработать селективный агонист THRβ для активации THRβ, но также для уменьшения ингибирования щитошейного ствола, чтобы избежать побочных эффектов, сопровождающих подавление функции щитошейного ствола.
[0009] В патентах, например, WO03094845, WO2007009913, WO2010122980 и WO2011038207, описаны некоторые агонисты рецептора THR. Почти все структуры этих агонистов сконструированы и разработаны на основе природного лиганда Т3 рецептора THR. Исходя из этого, все еще желательно разработать селективные агонисты рецептора THRβ, которые не только обладают полезными терапевтическими эффектами гормонов щитовидной железы, но также позволяют избежать побочных эффектов, влияющих на сердце.
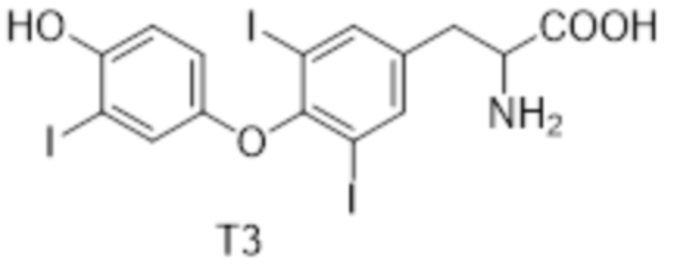
[0010] Структурная модификация осуществляется в настоящем изобретении также на основе природного лиганда Т3 рецептора THR. Авторы настоящего изобретения неожиданно обнаружили, что большинство модифицированных соединений сохраняют хорошую агонистическую активность в отношении рецептора THRβ, а некоторые соединения также обладают улучшенной селективностью в отношении THRα по сравнению с соединением сравнения 53 в ссылочных документах («Discovery of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (MGL-3196), a Highly Selective Thyroid Hormone Receptor β Agonist in Clinical Trials for the Treatment of Dyslipidemia», Martha et al., Journal of Medicinal Chemistry, 2014, 3912-3923). В то же время некоторые соединения по настоящему изобретению также проявляют весьма желательные фармакокинетические свойства. Фармакокинетические свойства некоторых предпочтительных соединений значительно лучше, чем у сравнительного соединения, что улучшает свойства готового лекарственного средства.
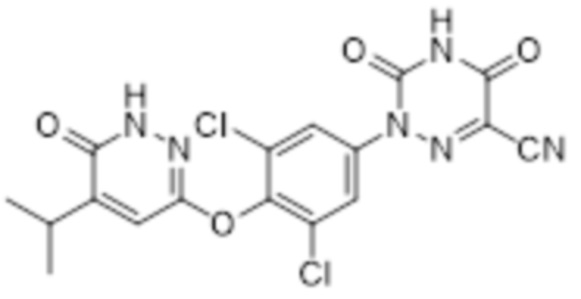 Соединение сравнения 53
Соединение сравнения 53
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0011] Для решения указанной выше технической проблемы в настоящем изобретении используются следующие технические решения:
[0012] В соответствии с одним аспектом в настоящем изобретении предложено соединение, представленное следующей формулой (I) и его фармацевтически приемлемая соль,
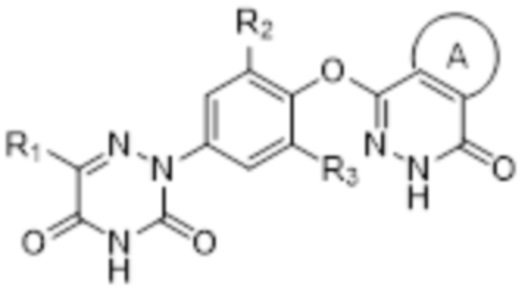
(I),
где
R1 выбран из группы, состоящей из водорода, циано, замещенного или незамещенного C1-6 алкила, или замещенного или незамещенного C3-6 циклоалкила, где заместитель выбран из группы, состоящей из атомов галогенов, гидрокси и C1-6 алкокси;
R2 и R3, каждый, независимо, выбраны из группы, состоящей из атомов галогенов или замещенного или незамещенного C1-6 алкила, где заместитель выбран из группы, состоящей из атомов галогенов, гидрокси и C1-6 алкокси;
кольцо A представляет собой замещенное или незамещенное насыщенное или ненасыщенное C5-10 алифатическое кольцо, или замещенное или незамещенное C5-10 ароматическое кольцо, где заместителем является один или более заместителей, выбранных из группы, состоящей из водорода, атомов галогена, гидрокси, -OCF3, -NH2, -NHC1-4 алкила, -N(C1-4 алкил)2, -CONH2, -CONHC1-4 алкила, -CON(C1-4 алкил)2, -NHCOC1-4 алкила, C1-6 алкила, C1-6 алкокси или C3-6 циклоалкила, и, когда имеются два заместителя, два заместителя могут образовывать кольцевую структуру вместе с присоединенным к ним атомом углерода; и
атомы галогена выбраны из группы, состоящей из F, Cl или Br.
[0013] Соединение в соответствии с настоящим изобретением имеет структуру, представленную следующей формулой (II):
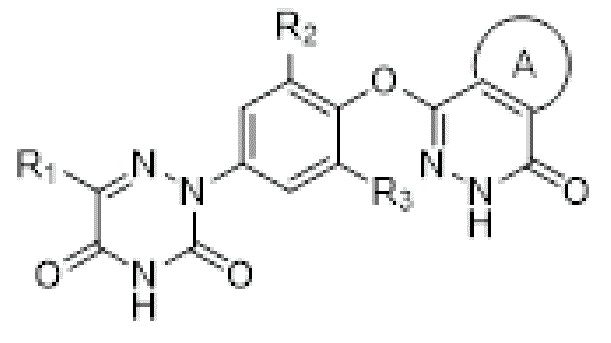
(II),
где
R1-R3 определены как описано для вышеприведенной формулы (I);
L отсутствует или выбран из группы, состоящей из -CH2- и -CH2CH2-;
R4 выбран из группы, состоящей из водорода, атомов галогена, гидрокси, -OCF3, -NH2, -NHC1-4 алкила, -N(C1-4 алкил)2, C1-6 алкила, C1-6 алкокси или C3-6 циклоалкила;
n обозначает целое число в диапазоне от 1 до 4;
m обозначает целое число в диапазоне от 1 до 4; и
когда L отсутствует, кольцо может иметь два или больше заместителей R4; и
атомы галогена выбраны из группы, состоящей из F, Cl или Br.
Предпочтительно, в структуре, представленной формулой (II), R4 выбран из группы, состоящей из водорода, атомов галогена, гидрокси, C1-3 алкила, C1-3 алкокси или C3-6 циклоалкила;
L отсутствует или выбран из группы, состоящей из -CH2- или -CH2CH2-;
n обозначает 1, 2 или 3; и
m обозначает 1 или 2.
Более предпочтительно, в структуре, представленной формулой (II), R4 выбран из группы, состоящей из водорода или C1-3 алкила;
L представляет собой -CH2- или -CH2CH2-;
n обозначает 1, 2 или 3; и
m обозначает 1 или 2.
Более предпочтительно, в структуре, представленной формулой (II), R4 выбран из группы, состоящей из водорода или C1-3 алкила;
L отсутствует;
n обозначает 1, 2 или 3; и
m обозначает 1 или 2.
[0014] Соединение в соответствии с настоящим изобретением имеет структуру, представленную следующей формулой (III):
 ,
,
где
R1-R3 определены как описано для вышеприведенной формулы (I);
R4 выбран из группы, состоящей из водорода, атомов галогена, гидрокси, -OCF3, -NH2, -NHC1-4 алкила, -N(C1-4 алкил)2, -CONH2, -CONHC1-4 алкила, -CON(C1-4 алкил)2, -NHCOC1-4 алкила, C1-6 алкила, C1-6 алкокси или C3-6 циклоалкила;
m обозначает целое число в диапазоне от 1 до 4; и
атомы галогена выбраны из группы, состоящей из F, Cl или Br.
[0015] Предпочтительно, в структуре, представленной формулой (III), R4 выбран из группы, состоящей из водорода, атомов галогена, гидрокси, -OCF3, C1-6 алкила, C1-6 алкокси или C3-6 циклоалкила; и m обозначает целое число в диапазоне от 1 до 3.
[0016] Предпочтительно, в структуре, представленной формулой (III), R4 выбран из группы, состоящей из водорода, атомов галогена или C1-3 алкила; и m обозначает 1 или 2.
[0017] Предпочтительно, в вышеуказанном соединении, имеющем структуру формулы (I), (II) или (III) в соответствии с настоящим изобретением, R1 выбран из группы, состоящей из водорода, циано, замещенного или незамещенного C1-6 алкила; более предпочтительно, R1 выбран из группы, состоящей из циано или C1-3 алкила; и, еще более предпочтительно, R1 представляет собой циано.
[0018] Предпочтительно, в вышеуказанном соединении, имеющем структуру формулы (I), (II) или (III) в соответствии с настоящим изобретением, R2 и R3, каждый, независимо, выбраны из группы, состоящей из F, Cl или Br, и более предпочтительно, оба R2 и R3 представляют собой Cl.
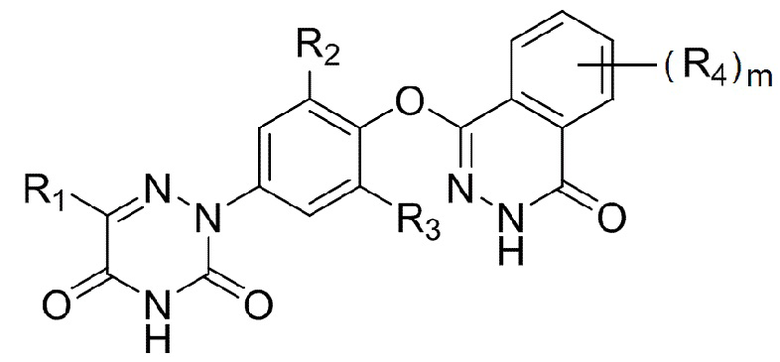
[0019] Предпочтительно, соединение и его фармацевтически приемлемая соль в соответствии с изобретением являются одним из следующих:
[0020] В соответствии с другим аспектом настоящего изобретения в настоящем изобретении предложен способ получения соединения, где способ получения включает следующие стадии:
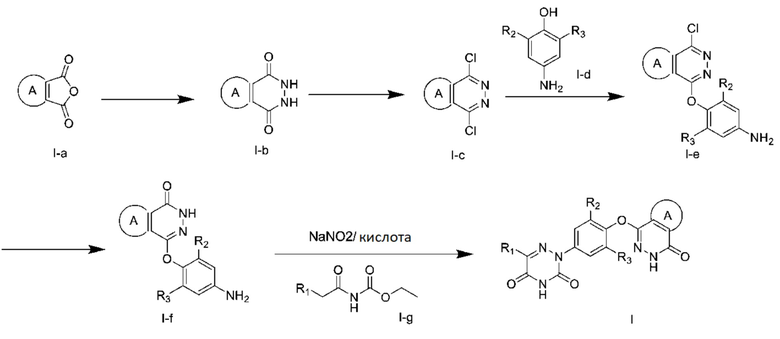
1) взаимодействие промежуточного альдегида I-a с гидрохлоридом гидразина с получением соединения общей формулы I-b;
2) нагревание полученного соединения общей формулы I-b в оксихлориде фосфора с образованием соединения общей формулы I-c;
3) подвергание полученного соединения общей формулы I-c и соединения I-d реакции конденсации при высокой температуре с получением соединения общей формулы I-e, где катализатором в этих условиях, предпочтительно, является йодид меди;
4) реакция полученного соединения общей формулы I-e в кислой или щелочной среде при высокой температуре с получением соединения общей формулы I-f; и
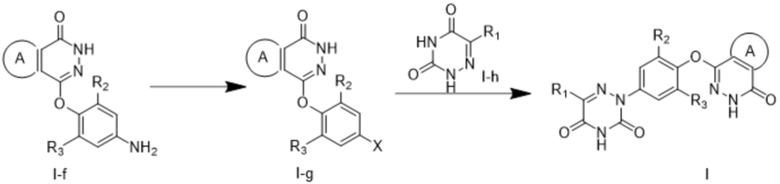
5) взаимодействие полученного соединения общей формулы I-f с нитритом натрия в водном растворе кислоты с последующим добавлением соединения Ig для дальнейшей реакции и замыкания цикла при высокой температуре с получением соединения общей формулы I, где кислота в этих условиях, предпочтительно, представляет собой соляную кислоту.
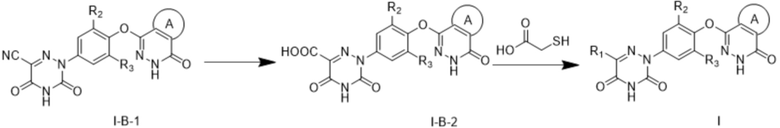
[0021] Соединение общей формулы I в соответствии с настоящим изобретением также может быть получено следующим образом:
 .
.
[0022] Соединение общей формулы I-B-1 подвергают высокотемпературному гидролизу в водном растворе кислоты с получением соединения общей формулы I-B-2, где кислота в этих условиях, предпочтительно, представляет собой соляную кислоту; и полученное соединение общей формулы I-B-2 подвергают реакции в присутствии меркаптоуксусной кислоты при высокой температуре с удалением карбокси с получением соединения общей формулы I.
[0023] Соединение общей формулы I в соответствии с настоящим изобретением также может быть получено следующим способом:
 .
.
[0024] Соединение общей формулы I-f подвергают взаимодействию с нитритом натрия в кислых условиях с образованием соединения соли диазония с последующим добавлением галогенид-аниона с получением соединения общей формулы I-g; и при катализе переходным металлом полученное соединение общей формулы I-g сочетается с промежуточным соединением I-h с получением соединения общей формулы I.
[0025] В соответствии с другим аспектом настоящего изобретения в настоящем изобретении предложено применение соединения при изготовлении терапевтического лекарственного средства от заболевания, связанного с метаболизмом.
[0026] В соответствии с другим аспектом настоящего изобретения в настоящем изобретении предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по настоящему изобретению и его фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый адъювант.
[0027] Предпочтительно, заболевание, связанное с метаболизмом, выбрано из группы, состоящей из ожирения, гиперлипидемии, гиперхолестеринемии, диабета и неалкогольного стеатогепатита (НАСГ), стеатоза печени, атеросклероза, гипотиреоза и рака щитовидной железы; и, предпочтительно, заболевание, связанное с метаболизмом, выбрано из группы, состоящей из неалкогольного стеатогепатита (НАСГ), гипотиреоза и рака щитовидной железы.
[0028] В соответствии с другим аспектом настоящего изобретения в настоящем изобретении предложен способ лечения заболевания, связанного с метаболизмом, где способ включает введение субъекту эффективного количества соединения по настоящему изобретению или фармацевтической композиции, содержащей соединение и его фармацевтически приемлемую соль в качестве активного ингредиента.
[0029] Предпочтительно, согласно способу лечения заболевания, связанного с метаболизмом, заболевание, связанное с метаболизмом, выбрано из группы, состоящей из ожирения, гиперлипидемии, гиперхолестеринемии, диабета и неалкогольного стеатогепатита (НАСГ), стеатоза печени, атеросклероза, гипотиреоза и рака щитовидной железы; и, предпочтительно, заболевание, связанное с метаболизмом, выбрано из группы, состоящей из неалкогольного стеатогепатита (НАСГ), гипотиреоза и рака щитовидной железы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0030] Настоящее изобретение подробно описывается ниже. Перед ознакомлением с описанием следует учесть, что термины, используемые в описании и прилагаемой формуле изобретения, не должны интерпретироваться как ограниченные общим значением и словарным значением, но должны интерпретироваться согласно соответствующему значению и концепциям в технических аспектах настоящего изобретения на основе принципа, согласно которому изобретатель может правильно определять термины для наилучшей интерпретации. Следовательно, предлагаемое в настоящем изобретении описание является только предпочтительным примером с целью иллюстрации и не предназначено для ограничения объема изобретения. Следовательно, следует понимать, что из него могут быть получены другие эквиваленты или улучшения, не выходящие за рамки сущности и объема настоящего изобретения.
[0031] В соответствии с настоящим изобретением, все термины, цитируемые в настоящем изобретении, имеют то же значение, что и термины, понимаемые специалистами в данной области техники, если не указано иное.
[0032] Как используется в настоящем документе, термин «соль» относится к соединению, содержащему катионы и анионы, которое может быть образовано путем протонирования участка, который может принимать протоны, и/или отщепления протонов от участка, который может поставлять протоны. Стоит отметить, что протонирование участка, который может принимать протоны, приводит к образованию катионного вещества, и его заряд уравновешивается наличием физиологических анионов, тогда как отрыв протона от участка, который может поставлять протоны, приводит к образование анионного вещества, и его заряд уравновешивается наличием физиологических катионов.
[0033] Термин «фармацевтически приемлемая соль» означает, что соль является фармацевтически приемлемой. Примеры фармацевтически приемлемой соли включают, но не ограничиваются ими: (1) кислотно-аддитивные соли, образованные с неорганической кислотой, например хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой и фосфорной кислотой; или образованные с органической кислотой, например, гликолевой кислотой, пировиноградной кислотой, молочной кислотой, малоновой кислотой, яблочной кислотой, малеиновой кислотой, фумаровой кислотой, винной кислотой, лимонной кислотой, 3-(4-гидроксибензоил)бензойной кислотой, коричной кислотой, миндальной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, 1,2-этандисульфоновой кислотой, 2-гидроксиэтансульфоновой кислотой, бензолсульфоновой кислотой, 4-хлорбензолсульфоновой кислотой, 2-нафталинсульфоновой кислотой, 4-пара-толуолсульфоновой кислотой, камфорной кислотой, додецилсерной кислотой, глюконовой кислотой, глутаминовой кислотой, салициловой кислотой и цис-муконовой кислотой; или (2) щелочно-аддитивные соли, образованные с любым основанием конъюгата вышеуказанных неорганических кислот, где основание конъюгата включает катионный компонент, выбранный из группы, состоящей из Na+, K+, Mg2+, Ca2+ и NHxR4-x+, где NHxR4-x+ (R представляет собой C1-4 алкил, и нижний индекс x обозначает целое число, выбранное из группы, состоящей из 0, 1, 2, 3 или 4) представляет собой катион в четвертичной аммониевой соли. Следует понимать, что все задействованные фармацевтически приемлемые соли включают форму присоединения растворителя (сольват) или кристаллическую форму (полиморфное вещество), как определено в настоящем изобретении для той же соли присоединения кислоты.
[0034] Термин «C1-M алкил» относится к алкилу, содержащему от 1 до M атомов углерода, например, где M обозначает целое число, имеющее следующее числовое значение: 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30. Например, термин «C1-6 алкил» относится к алкилу, содержащему от 1 до 6 атомов углерода. Примеры алкила включают, но не ограничиваются ими, низший алкил, включая метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или амил, изоамил, неопентил, гексил, гептил и октил.
[0035] Термин «ароматическая группа» относится к ароматической системе, которая может быть моноциклическим кольцом или полиароматическим кольцом, изначально конденсированным или соединенным вместе, так что по крайней мере часть конденсированных или соединенных колец образует сопряженную ароматическую систему. Арильные группы включают, но не ограничиваются ими: фенил, нафтил и тетралил. Арил может быть, необязательно, замещен, например, 1-4 арильными группами или гетероциклическими кольцами, замещенными группой, выбранной из группы, состоящей из: галогена, -CN, -OH, -NO2, амино, алкила, циклоалкила, алкенила, алкинила, алкокси, арилокси, замещенного алкокси, алкилкарбонила, алкилкарбокси, алкиламино или арилтио.
[0036] Термин «замещение» относится к тому, что упоминаемая ная группа может быть замещена одной или рядом дополнительных групп, где дополнительные группы отдельно и независимо выбраны из группы, состоящей из алкила, циклоалкила, арила, гетероарила, гетероалициклического углеводорода, гидрокси, алкокси, алкилтиола, арилтио, алкилсульфинила, арилсульфинила, алкилсульфонила, арилсульфонила, циано, галогена, карбонила, тиокарбонила, нитро, галогеналкила, фторалкила и амино, включая монозамещенные и дизамещенные аминогруппы и защищенные ими производные.
[0037] Соединение, представленное формулой (I), или его фармацевтически приемлемая соль, предложенные в настоящем изобретении, и фармацевтические композиции, содержащие это соединение, могут быть в различных формах, таких как таблетки, капсулы, порошки, сиропы, растворы, суспензии и аэрозоли, и так далее, и может быть представлено в подходящем твердом или жидком носителе или разбавителе, а также в дезинфекторе, подходящем для инъекции или инфузии.
[0038] Различные лекарственные формы фармацевтической композиции по настоящему изобретению могут быть изготовлены в соответствии со способами получения, общепринятыми в области фармацевтики. Например, стандартная доза состава препарата содержит от 0,05 до 200 мг соединения формулы (I) или его фармацевтически приемлемой соли, и, предпочтительно, стандартная доза формулы препарата содержит от 0,1 до 100 мг соединения формулы (I).
[0039] Соединение, представленное общей формулой (I), или фармацевтические композиции, предложенные в настоящем изобретении, могут быть использованы клинически у млекопитающих, включая людей и животных, посредством введения, такими путями как пероральный, назальный, трансдермальный, транспульмональный или желудочно-кишечный тракт. Пероральное введение является наиболее предпочтительным. Наиболее предпочтительная суточная доза составляет от 0,01 до 200 мг/кг веса при однократном введении или от 0,01 до 100 мг/кг веса за раздельном введении. Независимо от способа введения, оптимальная доза для человека должна определяться на основе конкретного лечения. Обычно введение начинается с небольшой дозы и ее постепенно увеличивают до тех пор, пока не будет найдена наиболее подходящая доза.
[0040] В настоящем изобретении термин «эффективное количество» может относиться к эффективному количеству для дозы и периода времени, необходимых для достижения ожидаемого эффекта. Эта эффективная доза может варьироваться по-разному в зависимости от определенных факторов, таких как вид заболевания, симптомы заболевания на момент лечения; структура конкретного органа-мишени, подлежащего введению; индивидуальный рост и вес пациента или тяжесть заболевания или симптома. Специалисты в данной области могут на собственном опыте определить эффективное количество конкретного соединения без проведения ненужных экспериментов.
[0041] Типичный состав получают путем смешивания соединения по изобретению, представленного общей формулой (I), и носителя, разбавителя или наполнителя. Подходящие носители, разбавители или наполнители хорошо известны специалистам в данной области, включая такие вещества, как углеводы, воски, водорастворимые и/или вспениваемые полимеры, гидрофильные или гидрофобные вещества, желатин, масло, растворители и воду.
[0042] Конкретный носитель, разбавитель или наполнитель, который следует использовать, выбирают в соответствии с применением и назначением соединения по настоящему изобретению. Обычно растворитель выбирают на основе растворителей, которые могут быть безопасно и эффективно дозированы млекопитающим согласно мнению специалистов в данной области. Как правило, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы или смешиваются с водой. Подходящие водные растворители включают один или несколько из следующих: вода, этанол, пропиленгликоль, полиэтиленгликоль (такой как PEG400 и PEG300) и тому подобные. Состав может также включать одно или несколько из следующих: буферы, стабилизаторы, поверхностно-активные вещества, смачивающие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксидантов, светоотражающие агенты, добавки для придания текучести, технологические добавки, красители, подсластители, ароматизаторы, вкусовые добавки или другие известные добавки, позволяющие производить или использовать лекарство в приемлемой форме.
[0043] Когда соединение формулы (I) по настоящему изобретению используется в комбинации по меньшей мере с одним другим лекарственным средством, два или более лекарственных средства можно использовать отдельно или в комбинации, предпочтительно, в виде фармацевтической композиции. Соединение формулы (I) или фармацевтическая композиция согласно настоящему изобретению могут быть введены субъекту отдельно или вместе в любых известных пероральных, внутривенных, ректальных, вагинальных, трансдермальных, других местных или системных лекарственных формах.
[0044] Указанные фармацевтические композиции могут также содержать одно или несколько из следующих: буферы, стабилизаторы, поверхностно-активные вещества, смачивающие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, светоотражающие агенты, добавок текучести, технологических добавок, красителей, подсластители, ароматизаторы, вкусовые добавки или другие известные добавки, позволяющие производить или использовать фармацевтические композиции в приемлемой форме.
[0045] Пероральный способ дозирования является предпочтительным для лекарственного средства по настоящему изобретению. Твердая лекарственная форма для перорального приема может включать капсулы, таблетки, порошки или составы в виде частиц. В твердой лекарственной форме соединение или фармацевтическая композиция согласно настоящему изобретению смешаны по меньшей мере с одним инертным наполнителем, разбавителем или носителем. Подходящие наполнители, разбавители или носители включают такие вещества, как цитрат натрия или дикальций фосфат, или крахмал, лактоза, сахароза, маннит, кремниевая кислота и тому подобное; клеи, такие как карбоксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и аравийская камедь; смачивающие агенты, такие как глицерин; разрыхлители, такие как агар, карбонат кальция, картофельный крахмал или крахмал маниоки, альгиновая кислота, специфический сложный силикат и карбонат натрия; блокаторы раствора, такие как парафин; промоторы абсорбции, такие как соединения четвертичного аммония; адсорбенты, такие как каолин и бентонит; и смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль и лаурилсульфат натрия. В случае капсул и таблеток лекарственная форма может также включать буферы. Подобные виды твердых композиций могут также использоваться в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах, в которых в качестве наполнителей используются лактоза и высокомолекулярный полиэтиленгликоль.
[0046] Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, а также сиропы и эликсиры. В дополнение к соединению или его фармацевтической композиции согласно настоящему изобретению жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в данной области, такие как вода или другие растворители; солюбилизаторы и эмульгаторы, такие как этанол, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутандиол, диметилформамид; масла (например, масло семян хлопка, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло); глицерин; тетрагидрофурфуриловый спирт; сложные эфиры жирных кислот полиэтиленгликоля и сорбитана; или смеси нескольких из этих веществ и так далее.
[0047] В дополнение к этим инертным разбавителям композиция может также включать наполнитель, такой как одно или несколько из следующих: смачивающие агенты, эмульгаторы, суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
[0048] Что касается суспензии, в дополнение к соединению, представленному общей формулой (I) или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей его, согласно настоящему изобретению, суспензия может дополнительно содержать носители, такие как суспендирующие агенты, например, этоксилированный изостеарол, полиоксиэтиленсорбит, сложный эфир сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар и трагакантовая камедь или смеси некоторых из этих веществ и так далее.
[0049] Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль, или фармацевтическая композиция, содержащая его, согласно настоящему изобретению, могут быть дозированы с использованием других лекарственных форм для местного применения, включая мази, порошки, спреи и ингаляторы. Это лекарственное средство можно смешивать в стерильных условиях с фармацевтически приемлемыми наполнителями, разбавителями или носителями и любыми необходимыми консервантами, буферами или пропеллентами. Офтальмологические составы, офтальмологические мази, порошки и растворы также входят в объем настоящего изобретения.
[0050] Кроме того, настоящее изобретения дополнительно охватывает наборы (например, фармацевтические упаковки). Предложенный набор может включать фармацевтическую композицию или соединение, описанные в настоящем изобретении, и контейнеры (например, флаконы с лекарственными средствами, ампулы, флаконы, шприцы и/или субпакеты или другие подходящие контейнеры). В некоторых вариантах осуществления предложенный набор может, необязательно. дополнительно включать второй контейнер, содержащий фармацевтический наполнитель для разбавления или суспендирования фармацевтической композиции или соединения, описанных в настоящем документе. В некоторых вариантах осуществления фармацевтическая композиция или соединение, описанные в настоящем документе, предоставленные в первом и втором контейнерах, объединены с образованием стандартной лекарственной формы.
[0051] В некоторых вариантах осуществления описанный в настоящем изобретении набор дополнительно включает инструкции по применению соединения или фармацевтической композиции, входящей в набор. Описанный в настоящем изобретении набор может также включать информацию, требуемую регулирующими органами, такими как Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA). В некоторых вариантах осуществления информация, включенная в набор, представляет собой информацию о составе. В некоторых вариантах осуществления набор и инструкции предоставляются для лечения пролиферативного заболевания у субъектов, нуждающихся в этом, и/или предотвращения пролиферативного заболевания у субъектов, которые в этом нуждаются. Описанный в настоящем изобретении набор может включать один или несколько дополнительных фармацевтических препаратов в виде отдельных композиций.
[0052] Настоящее изобретение подробно описано далее в сочетании с конкретными вариантами осуществления, но настоящее изобретение не ограничивается представленными далее вариантами осуществления, которые служат цели лучшего объяснения конкретных вариантов осуществления настоящего изобретения и не должны интерпретироваться как ограничивающие объем изобретения каким-либо образом. Условия, не указанные в вариантах осуществления изобретения, являются обычными условиями. Если не указано иное, все реагенты и устройства, используемые в следующих вариантах осуществления изобретения, являются коммерчески доступными продуктами.
[0053] Структуры соединений в следующих вариантах осуществления изобретения определяют с помощью ядерного магнитного резонанса (ЯМР) или/и масс-спектрометрии (МС). Смещение ЯМР (δ) дано в единицах 10-6 (м. д.). Для определения методом ЯМР используется ядерно-магнитный спектрометр Bruker AVANCE-400. Растворителями являются дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренним стандартом является тетраметилсилан (ТМС).
[0054] Для определения с помощью МС используют масс-спектрометр FINNIGAN LCQAd (ESI) (производитель: Thermo, модель: Finnigan LCQ advance max).
[0055] В качестве силикагелевых пластин для тонкослойной хроматографии используются силикагелевых пластины Yantai Huanghai HSGF254 или Qingdao GF254. Спецификацией силикагелевой пластины для тонкослойной хроматографии (ТСХ) является от 0,15 до 0,2 мм, и спецификацией для тонкослойной хроматографии для разделения и очистки продуктов является от 0,4 до 0,5 мм.
[0056] В колоночной хроматографии в качестве носителя обычно используют силикагель Yantai Huanghai силикагель 200-300 меш.
[0057] Когда не существует специального описания в вариантах осуществления изобретения, температура реакции составляет комнатную температуру, то есть от 20°C до 30°C.
[0058] Тонкослойная хроматография (ТСХ) используется для отслеживания протекания реакции в вариантах осуществления изобретения. Используемая система проявителя и элюентная система колоночной хроматографии, применяемая для очистки соединения, включают: A: система дихлорметана и метанола; B: система н-гексана и этилацетата; C: система петролейного эфира и этилацетата; D: система ацетона и петролейного эфира, где объемное соотношение растворителей регулируется в соответствии с различной полярностью соединений.
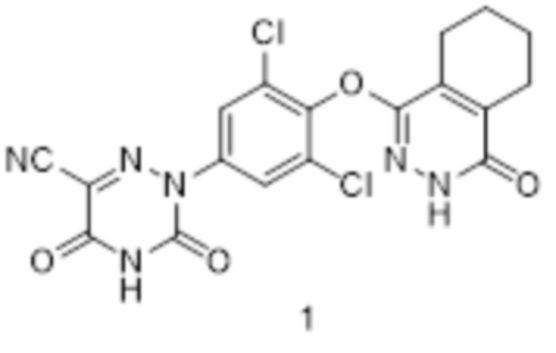
[0059] Вариант осуществления 1: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила
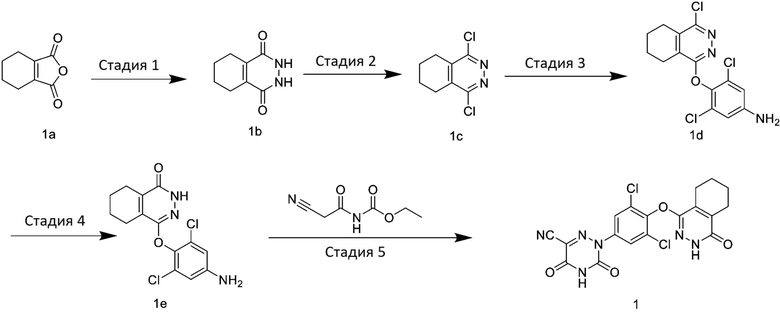
[0060] Стадия 1: Получение 2,3,5,6,7,8-гексагидрофталазин-1,4-диона (соединение 1b)
[0061] Ацетат натрия (3,69 г, 45 ммоль) и гидрохлорид гидразина (3,08 г, 45 ммоль) последовательно добавляли в раствор соединения 3,4,5,6-тетрагидрофталевого ангидрида 1a (4,56 г, 30 ммоль) в уксусной кислоте (50 мл) и воде (100 мл). После добавления смесь нагревали до температуры 100ºC и перемешивали в течение 3 часов. Реакцию останавливали и естественным образом охлаждали до комнатной температуры. Твердое вещество осаждали и отфильтровывали с получением соединения 1b (4,2 г), и продукт напрямую использовали в следующей реакции.
[0062] 1H ЯМР (400 МГц, ДМСО-d6): 11,26 (с, 2H), 2,36 (с, 4H), 1,65(с, 4H).
[0063] Стадия 2: Получение 1,4-дихлор-5,6,7,8-тетрагидрофталазина (соединение 1c)
[0064] Соединение 1b (1 г, 6,02 ммоль) растворяли в оксихлориде фосфора (8 мл). Воздух в системе 3 раза заменяли газообразным азотом. Систему нагревали до температуры 110ºC и перемешивали в течение 3 часов. Реакцию останавливали и затем охлаждали естественным путем. Реакционный раствор медленно прибавляли по каплям в ледяную воду. Смесь доводили до pH 10 с помощью 1 н водного раствора гидроксида натрия и 3 раза экстрагировали этилацетатом. Органические фазы объединяли, и органическую фазу промывали насыщенным солевым раствором и концентрировали досуха с получением соединения 1c (1,1 г). Продукт напрямую использовали в следующей реакции.
[0065] Стадия 3: Получение 3,5-дихлор-4-((4-хлор-5,6,7,8-тетрагидрофталазин-1-ил)окси)анилина (соединение 1d)
[0066] Диметилсульфоксид (8 мл) добавляли к смеси соединения 1c (1,0 г, 5,0 ммоль), 2,6-дихлор-4-аминофенола (0,93 г, 6 ммоль), карбоната калия (2,76 г, 20 ммоль). ммоль) и CuI (0,57 г, 3 ммоль). Воздух в системе 3 раза заменяли газообразным азотом. Систему нагревали до температуры 100ºC и перемешивали в течение 3 часов. После прекращения реакции ее охлаждали. Твердое вещество в реакционном растворе сначала отфильтровывали, а остаток на фильтре многократно промывали этилацетатом. К фильтрату добавляли 80 мл воды, и затем водную фазу экстрагировали этилацетатом. Органические фазы объединяли и промывали насыщенным солевым раствором. Соединение 1d (450 мг) выделяли колоночной хроматографией после фильтрации и концентрирования растворителя.
[0067] Стадия 4: Получение 4-(4-амино-2,6-дихлорфенокси)-5,6,7,8-тетрагидрофталазин-1(2H)-она (соединение 1e)
[0068] Соединение 1d (100 мг, 0,3 ммоль) растворяли в уксусной кислоте (4 мл) и добавляли ацетат натрия (200 мг, 2,5 ммоль). Смесь нагревали до температуры 120ºC и перемешивали в течение 12 часов. После прекращения реакции растворитель удаляли при пониженном давлении. Добавляли 10 мл воды, а затем добавляли 1 н водный раствор гидроксида натрия, чтобы довести pH до 8. Затем проводили экстракцию этилацетатом (10 мл×3) и органические фазы объединяли. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, соответственно, и концентрировали при пониженном давлении с получением серого твердого вещества. К полученному твердому веществу добавляли 10 мл метанола и 10 мл 1 н водного раствора гидроксида натрия, смесь нагревали до температуры 120ºC и перемешивали в течение 12 часов. После прекращения реакции метанол концентрировали при пониженном давлении. Оставшуюся водную фазу трижды экстрагировали этилацетатом, и органические фазы объединяли. После концентрирования растворителя при пониженном давлении получали соединение 1e (80 мг) путем выделения и очистки с помощью тонкослойной хроматографии.
[0069] Стадия 5: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 1)
[0070] Нитрит натрия (16 мг) растворяли в воде (0,5 мл). При охлаждении на ледяной бане этот раствор медленно по каплям добавляли к смеси соединения 1e (60 мг, 0,185 ммоль), воды (2,5 мл) и насыщенного раствора соляной кислоты (1,25 мл) и перемешивали в течение 0,5 ч на ледяной бане до тех пор, пока раствор не становился прозрачным. При этой температуре по каплям дополнительно добавляли смешанный раствор N-цианоацетилуретана (32 мг) в воде (4,2 мл) и пиридина (1,3 мл) и после добавления по каплям перемешивали в течение ночи. После прекращения реакции желтое твердое вещество отфильтровывали и промывали водой и петролейным эфиром. К полученному твердому веществу добавляли уксусную кислоту (5 мл) и ацетат натрия (160 мг, 2 ммоль). Смесь нагревали до температуры 120°C и перемешивали в течение 6 ч, затем охлаждали до комнатной температуры. Добавляли 100 мл воды для осаждения твердого вещества светло-желтого цвета. После выделения и очистки с помощью тонкослойной хроматографии (DCM:MeOH=8:1) получали соединение 1 (12,0 мг).
[0071] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,03 (с, 1H), 7,75 (с,2H), 2,68-2,63 (м, 2H), 2,45-2,39 (м, 2H), 1,80-1,67 (м, 4H).
[0072] MS m/z (ESI): 447,4 [M+1].
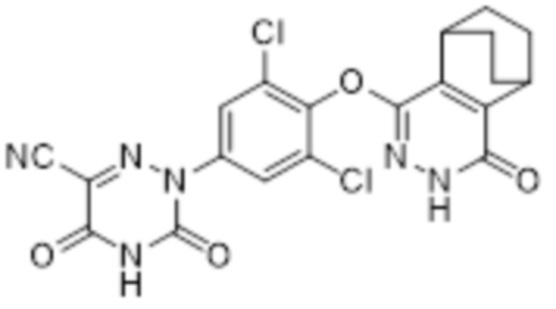
[0073] Вариант осуществления 2: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 2b).
 2
2
[0074] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид 1a на стадии 1 заменяли на ангидрид бицикло[2,2,2]окт-2-ен-2,3-дикарбоновой кислоты, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 2).
[0075] 1H ЯМР (400 МГц, ДМСО-d6): 12,13 (с, 1H), 7,77 (с,2H), 1,88-1,73 (м, 4H), 1,39-1,17 (м, 6H).
[0076] MS m/z (ESI): 473,2 [M+1].
[0077] Вариант осуществления 3: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-метанофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 3).
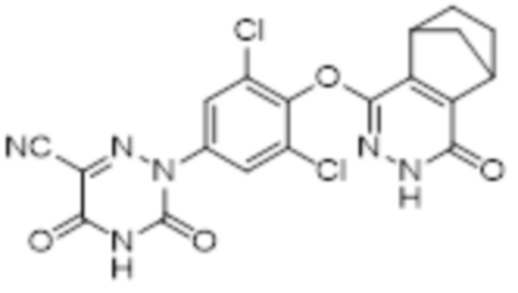 3
3
[0078] Стадия 1: Получение диметил-бицикло[2.2.1]гепта-2,5-diene-2,3-дикарбоксилата
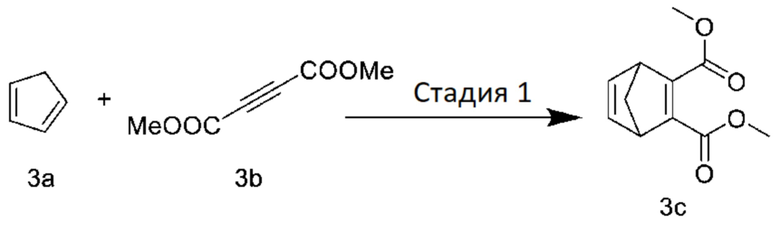
[0079] Диметилацетилендикарбоксилат (соединение 3b) (2,75 мл, 22,3 ммоль) медленно по каплям добавляли к циклопентадиену (соединение 3a) (1,475 г, 22,3 ммоль) и после добавления перемешивали в течение 2 часов при комнатной температуре. Реакцию останавливали, и получали маслянистый продукт 3c (4,0 г). Неочищенный продукт без очистки использовали непосредственно в следующей реакции.
[0080] Стадия 2: Получение диметил-бицикло[2.2.1]гепта-2-ен-2,3-дикарбоксилата:
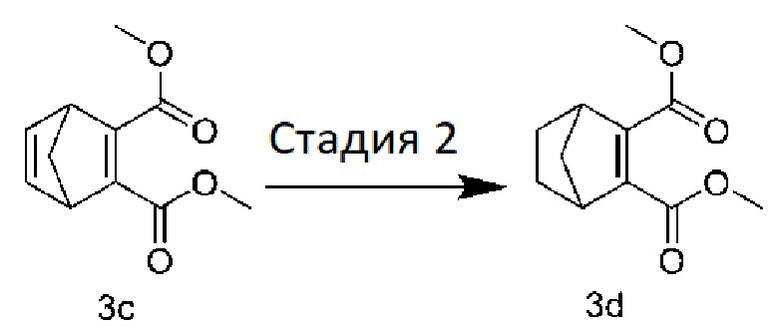
[0081] Соединение 3c (1,0 г, 4,8 ммоль), палладий на угле (0,05 г) и 15 мл ацетона помещали в реакционную колбу, и 3 раза с помощью баллона с водородом осуществляли замену газообразной среды. Систему перемешивали 1 ч при комнатной температуре. Фильтровали и растворитель концентрировали досуха при пониженном давлении с получением жидкого продукта 3d светло-зеленого цвета (0,9 г).
[0082] Стадия 3: Получение бицикло[2.2.1]гепта-2-ен-2,3-дикислоты:
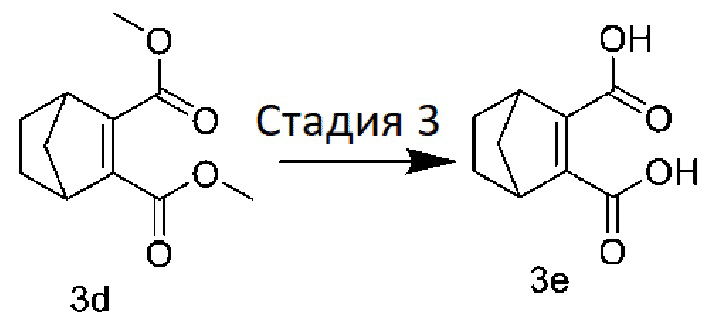
[0083] Тетрагидрофуран (20 мл) и воду (20 мл) добавляли к смеси соединения 3d (3,0 г, 14,3 ммоль) и моногидрата гидроксида лития (1,54 г, 35,8 ммоль) и перемешивали в течение 2 часов при комнатной температуре. . Раствор доводили до pH 1 с помощью 2 н разбавленной соляной кислоты, экстрагировали 3 раза этилацетатом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением твердого соединения 3e белого цвета (2,0 г).
[0084] Стадия 4: Получение 4,5,6,7-тетрагидро-4,7-метаноизофенилфуран-1,3-диона:

[0085] к соединению 3e (200 мг, 1,1 ммоль) добавляли уксусный ангидрид (10 мл). Смесь нагревали до температуры 100ºC и перемешивали в течение 2 часов. Растворитель удаляли путем концентрирования при пониженном давлении с получением сырого твердого продукта соединения 3f (200 мг).
[0086] Стадия 5: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-метанофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила
[0087] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 4,5,6,7-тетрагидроизобензофуран-1,3-дион (соединение 1a) на стадии 1 заменяли на 4,5,6,7-тетрагидро-4,7-метаноизофенилфуран 1,3-дион (соединение 3f), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-метанофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 3).
[0087] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,08 (с, 1H), 7,77 (с,2H), 2,75-2,65 (м, 2H), 1,80-1,72 (м, 4H), 1,55-1,50 (м, 2H).
[0089] MS m/z (ESI): 459,0 [M+1].
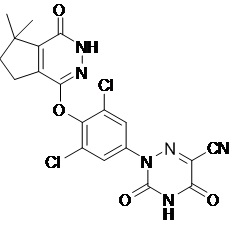
[0090] Вариант осуществления 4: Получение 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 4)
4
[0091] Стадия 1: Получение метил 3,3-диметил-2-оксоциклопентил-1-карбоксилата
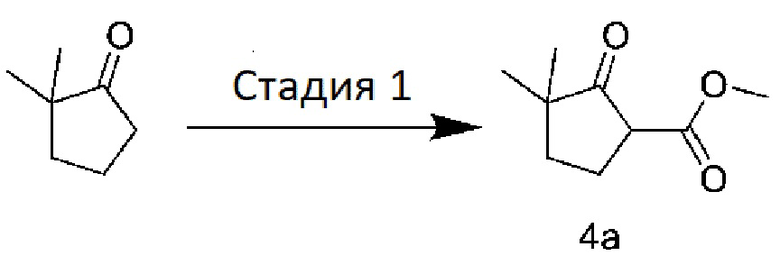
[0092] К раствору соединения 2,2-диметилциклопентан-1-она (1,12 г, 10 ммоль) и MeOH (0,5 мл) в DMC (11 мл) добавляли NaH (480 мг, 12 ммоль). Смесь нагревали до температуры 82°C, перемешивали 3 ч и охлаждали. В систему последовательно добавляли метанол (0,5 мл) и уксусную кислоту (1 мл), затем систему выливали в ледяную воду, экстрагировали дихлорметаном, сушили и подвергали удалению растворителя при пониженном давлении с получением бесцветного маслянистого вещества, соединения 4a (1,7 г), непосредственно используемого на следующей стадии.
[0093] Стадия 2: Получение метил-3,3-диметил-2-(((трифторметил)сульфо)оксо)циклопент-1-ен-1-карбоксилата.
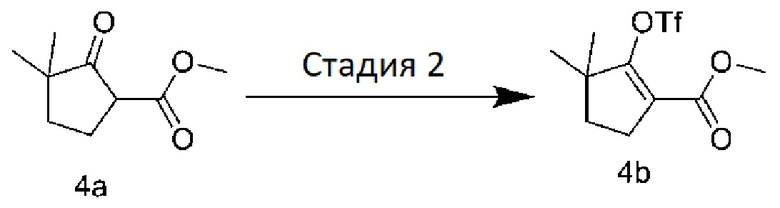
[0094] К раствору соединения 4a (1,7 г, 10 ммоль) и диизопропилэтиламина (8,2 мл, 50 ммоль) в дихлорметане (17 мл) при температуре -60°C добавляли по каплям трифторуксусный ангидрид (2 мл, 12 ммоль). После добавления смесь медленно нагревали до комнатной температуры и перемешивали в течение 16 часов. Добавляли 50 мл воды, смесь экстрагировали этилацетатом, удаляли растворитель при пониженном давлении и очищали с помощью колоночной хроматографии (РЕ:ЕА=50:1) с получением бесцветного маслянистого продукта, соединения 4b (1,8 г).
[0095] Стадия 3: Получение 2-((метоксикарбонил)-5,5-диметилциклопент-1-ен-1-карбоновой кислоты
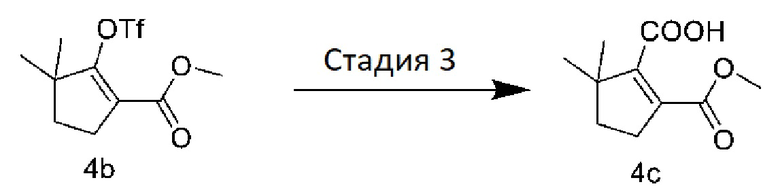
[0096] К смеси 4b (1,8 г, 5,96 ммоль), диизопропилэтиламина (1,97 мл, 11,92 ммоль), уксусного ангидрида (1,13 мл, 11,92 ммоль), формиата натрия (1,22 г, 5,96 ммоль), диацетата палладия (66,9 мг, 0,30 ммоль) и хлорида лития (758 мг, 17,88 ммоль) в атмосфере газообразного азота из баллона добавляли ДМФА (25 мл). После добавления систему перемешивали в течение 16 ч при комнатной температуре и в систему добавляли 300 мл этилацетата. Систему промывали водой и насыщенным солевым раствором по одному разу. Растворитель удаляли при пониженном давлении с получением бесцветного масла 4c (1,2 г).
[0097] Стадия 4: Получение 3,3-диметилциклопент-1-ен-1,2-дикарбоновой кислоты
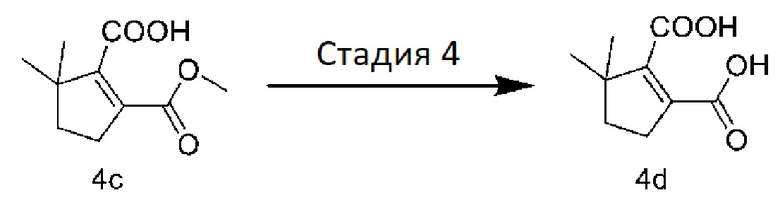
[0098] К смеси 4c (1,2 г, 6 ммоль) и моногидрата гидроксида лития (756 мг, 18 ммоль) при комнатной температуре добавляли метанол (6 мл) и воду (6 мл) и перемешивали в течение 3 часов. Метанол удаляли при пониженном давлении, затем раствор доводили до pH 1 концентрированной соляной кислотой и экстрагировали этилацетатом, и растворитель удаляли при пониженном давлении с получением твердого соединения 4d белого цвета (1,0 г).
[0099] Стадия 5: Получение 4,4-диметил-5,6-дигидро-1H-циклопентил[c]фуран-1,3(4H)-диона
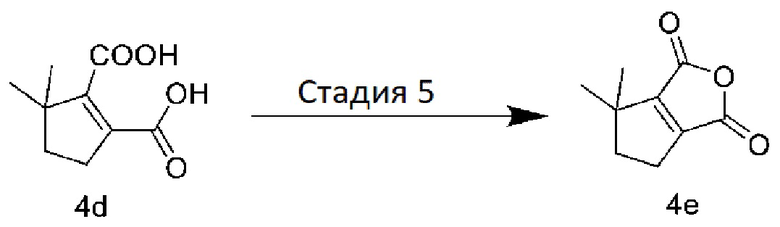
[0100] К соединению 4d (1,0 г, 5,43 ммоль) добавляли уксусный ангидрид (10 мл). Смесь нагревали до температуры 100°C, перемешивали 3 ч и охлаждали. Избыточный уксусный ангидрид удаляли при пониженном давлении с получением жидкого соединения 4e светло-коричневого цвета (875 мг), которое напрямую использовали в следующих синтезах.
[0101] Стадия 6: Получение 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила
[0102] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид (соединение 1a) на стадии 1 заменяли на 4,4-диметил-5,6-дигидро-1H-циклопентил[c]фуран-1,3(4H)-дион (соединение 4e), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 4).
[0103] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,09 (с, 1H), 7,79 (с, 2H), 2,97-2,93 (м, 2H), 2,01-1,98 (м, 2H), 1,34-1,20 (м, 6H).
[0104] MS m/z (ESI): 460,9 [M+1].
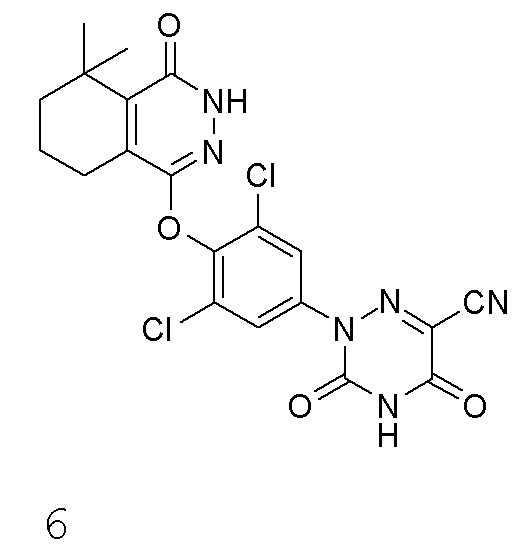
[0105] Вариант осуществления 5: Получение 2-(3,5-дихлор-4-((5-метил-4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 5)
[0106] Стадия 1: Получение метил 3-метил-2-оксоциклогексан-1-карбоксилата 5b:
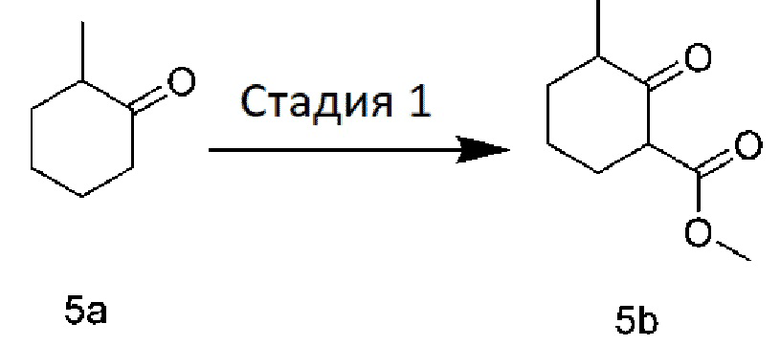
[0107] К раствору 2-метилциклогексанона (соединение 5a) (4,48 г, 40 ммоль) в дихлорметане (50 мл) добавляли NaH (содержание 60%, 1,92 г, 48 ммоль). Смесь кипятили с обратным холодильником, перемешивали в течение 3 ч и охлаждали. Смесь на глазок гасили метанолом (0,5 мл) и уксусной кислотой (1 мл). Реакционную систему выливали в ледяную воду и экстрагировали дихлорметаном. Растворитель удаляли при пониженном давлении и очисткой с помощью колоночной хроматографии получали масло (соединение 5b) (5,6 г).
[0108] Стадия 2: Получение метил 3-метил-2-(((трифторметил)сульфо)оксо)циклогексил-1-ен-1-карбоксилата
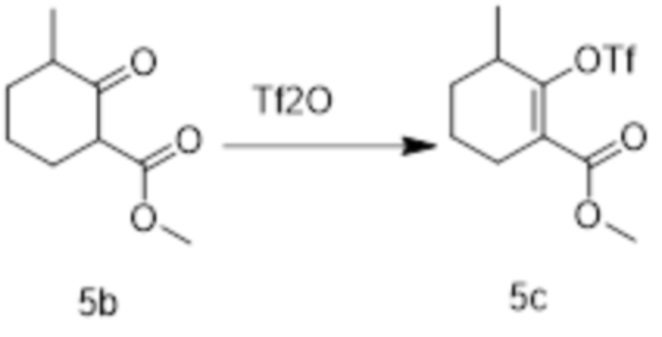
[0109] При охлаждении на ледяной бане к соединению 5b (0,85 г, 5 ммоль) в этиловом эфире (20 мл) добавляли NaH (содержание 60%, 600 мг, 25 ммоль) и перемешивали в течение 0,5 часа. Затем в систему по каплям добавляли ангидрид трифторметансульфоновой кислоты (2,8 г, 10 ммоль) и перемешивали еще в течение 1 ч при температуре 0°C. Систему гасили добавлением H2O (50 мл), доводили до pH 1 добавлением 1 н HCl и экстрагировали дихлорметаном. Растворитель удаляли при пониженном давлении, и очисткой с помощью колоночной хроматографии (PE:EA=20:1) получали бесцветное маслянистое соединение 5c (1,04 г).
[0110] Стадия 3: Получение 2-((метоксикарбонил)-6-метилциклогексил-1-ен-1-карбоновой кислоты
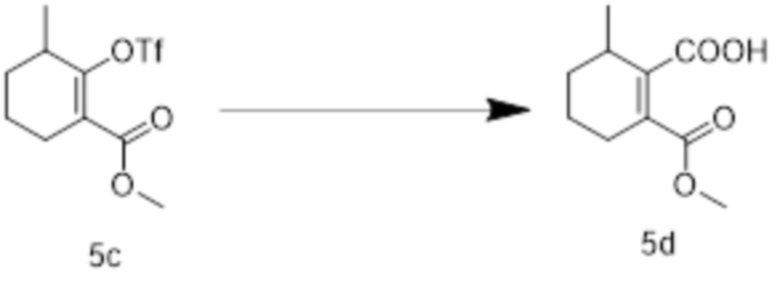
[0111] К раствору соединения 5c (0,714 г, 10,5 ммоль) и формиата натрия (0,714 г) в N, N-диметилформамиде (15 мл) в атмосфере азота последовательно добавляли диизопропилэтиламин (0,714 г, 7,00 ммоль) и уксусный ангидрид (0,903 г, 7,00 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Затем добавляли ацетат палладия (40 мг, 0,18 ммоль) и хлорид лития (445 мг, 10,50 ммоль) и систему перемешивали в течение ночи при комнатной температуре. Добавляли этилацетат (30 мл) и смесь промывали водой. Растворитель удаляли при пониженном давлении с получением маслянистого соединения 5d светло-желтого цвета (590 мг).
[0112] Стадия 4: Получение 3-метилциклогексил-1-ен-1,2-дикислоты 5e
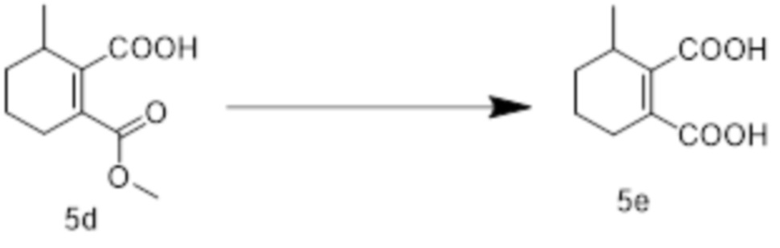
[0113] К раствору соединения 5d (0,590 г, 2,98 ммоль) в метаноле (3 мл) и воде (3 мл) добавляли моногидрат гидроксида лития (0,375 г, 8,94 ммоль) и перемешивали в течение 3 часов при комнатной температуре. Затем метанол удаляли при пониженном давлении. Систему доводили до pH 1 добавлением 1 н водного раствора соляной кислоты и трижды экстрагировали этилацетатом. Органические фазы объединяли и концентрировали при пониженном давлении для удаления растворителя с получением маслянистого соединения 5e (0,550 г).
[0114] Стадия 5: Получение 4-метил-4,5,6,7- тетрагидроизофенилфуран-1,3-диона 5f

[0115] К соединению 5e (0,550 г, 2,99 ммоль) добавляли уксусный ангидрид (6 мл). Смесь нагревали до температуры 100ºC и перемешивали в течение 2 часов. Систему охлаждали и растворитель удаляли при пониженном давлении с получением коричневого маслянистого соединения 5f (0,34 г), которое напрямую использовали в следующих синтезах.
[0116] Стадия 6: Получение 2-(3,5-дихлор-4-((5-метил-4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,3-триазин-6-нитрила (соединение 5)
[0117] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид (соединение 1a) на стадии 1 заменяли на 4-метил-4,5,6,7-тетрагидроизофенилфуран-1,3-дион (соединение 5f), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((5-метил-4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 5).
[0118] MS m/z (ESI): 460,1 [M+1]
[0119] Вариант осуществления 6: Получение 2-(3,5-дихлор-4-((5,5-диметил-4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила
[0120] Использовался путь синтеза по варианту осуществления 5, за исключением того, что исходный продукт 2-метилциклогексанон (соединение 5a) на стадии 1 заменяли на 2,2-диметилциклогексанон, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((5,5-диметил-4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 6).
[0121] MS m/z (ESI): 474,1 [M+1]
[0122] Вариант осуществления 7: Получение 2-(3,5-дихлор-4-((7-метил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентил[d]пиридазин-4-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 7)
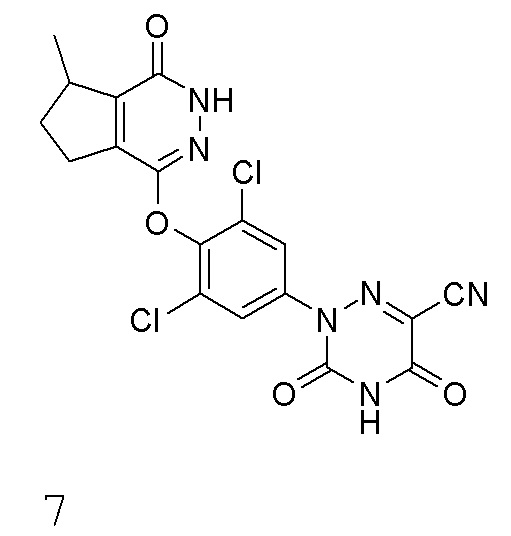
[0123] Использовался путь синтеза по варианту осуществления 5, за исключением того, что исходный продукт 2-метилциклогексанон (соединение 5a) на стадии 1 заменяли на 2-метилциклопентанон, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((7-метил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентил[d]пиридазин-4-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 7).
[0124] MS m/z (ESI): 446,0 [M+1]
[0125] Вариант осуществления 8: Получение 2-(3,5-дихлор-4-((7-этил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 8)
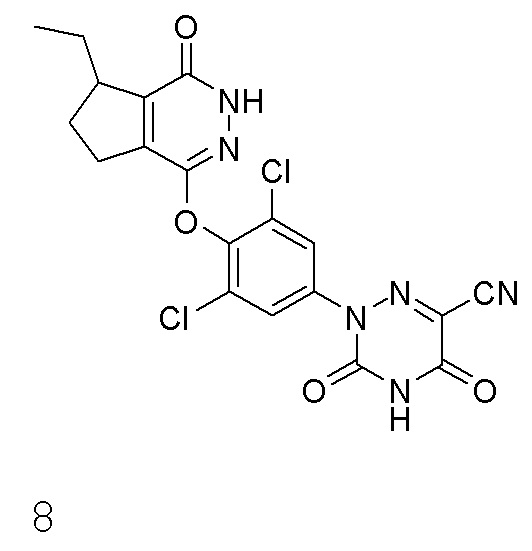
[0126] Использовался путь синтеза по варианту осуществления 5, за исключением того, что исходный продукт 2-метилциклогексанон (соединение 5a) на стадии 1 заменяли на 2-этилциклопентанон, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((7-этил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 8).
[0127] MS m/z (ESI): 460,1 [M+1]
[0128] Вариант осуществления 9: Получение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 9)
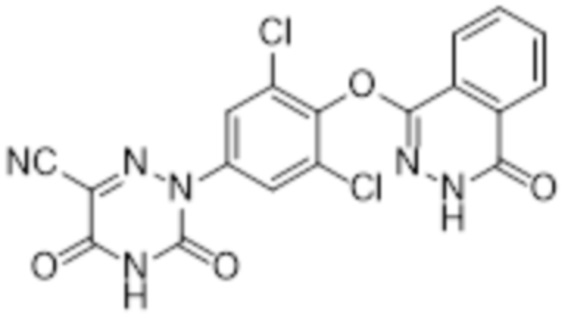
9
[0129] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 1,4-дихлор-5,6,7,8-тетрагидрофталазин (соединение 1c) на стадии 3 заменяли на 1,4-дихлорфталазин, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 9).
[0130] 1H ЯМР (400 МГц, ДМСО-d6): 12,00 (с, 1H), 8,31 (д, J=8,0 Гц, 1H), 8,26 (д, J=8,0 Гц, 1H), 8,09 (т, J=12,0 Гц 1H), 8,03 (т, J=16,0 Гц 1H), 7,81 (с, 2H).
[0131] MS m/z (ESI): 443,0 [M+1].
[0132] Вариант осуществления 10: Получение 2-(3,5-дихлор-4-((5-хлор-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 10)
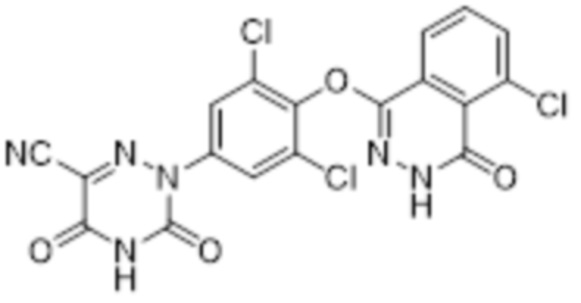
10
[0133] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид (соединение 1a) на стадии 1 заменяли на 3-хлорфталевый ангидрид, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((5-хлор-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 10)
[0134] 1H ЯМР (400 МГц, ДМСО-d6): 12,00 (с, 1H), 8,30 (д, J=8,0 Гц, 1H), 8,15 (д, J=8,0 Гц, 1H), 7,95 (м, 1H), 7,80 (с, 2H).
[0135] MS m/z (ESI): 477,0 [M+1].
[0136] Вариант осуществления 11: Получение 2-(3,5-дихлор-4-((5-метил-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 11):
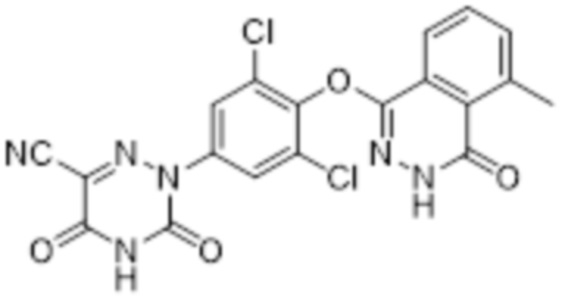
11
[0137] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид (соединение 1a) на стадии 1 заменяли на 3-метилфталевый ангидрид, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((5-метил-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 11).
[0138] MS m/z (ESI): 456,9 [M+1].
[0139] Вариант осуществления 12: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)оксо)фенил)-1,2,4-триазин-3,5(2H,4H)-диона (соединение 12):
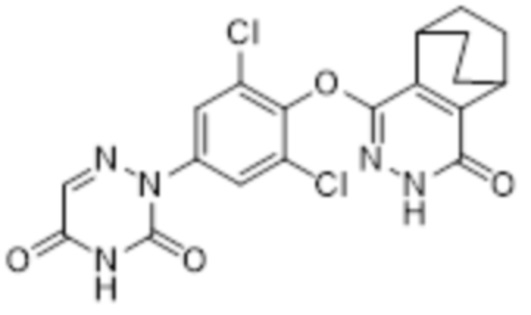
12
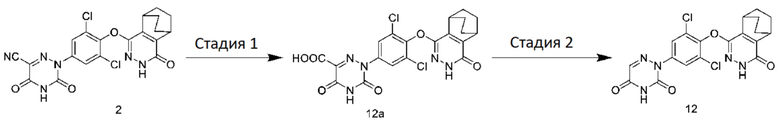
[0140] Стадия 1: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбоновой кислоты
[0141] К соединению 2 (77 мг) добавляли уксусную кислоту (4 мл) и концентрированную соляную кислоту (1 мл), соответственно, нагревали до температуры 120°C, перемешивали в течение 5 часов и охлаждали. Реакционный раствор разбавляли водой. Полученное твердое вещество фильтровали, промывали водой и промывали петролейным эфиром с получением твердого соединения 12a (35 мг), которое напрямую использовали в следующей реакции.
[0142] Стадия 2: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)оксо)фенил)-1,2,4-триазин-3,5(2H,4H)-диона
[0143] К соединению 12a (35,0 мг) добавляли меркаптоуксусную кислоту (2 мл), и смесь нагревали до температуры 170°C и перемешивали в течение 2 часов. Смесь охлаждали, доводили до pH 8 с помощью 1 н водного раствора гидроксида натрия и 3 раза экстрагировали этилацетатом. Органические фазы объединяли. Растворитель удаляли при пониженном давлении, и соединение 12 (10,0 мг) получали путем выделения и очистки с помощью тонкослойной хроматографии (DCM:MeOH=8:1).
[0144] 1H ЯМР (400 МГц, ДМСО-d6): 12,10 (с, 1H), 7,77 (с, 2H), 7,33 (с, 1H), 2,01 (м, 2H), 1,93-1,75 (м, 4H), 1,39-1,27 (м, 4H).
[0145] MS m/z (ESI): 448,0 [M+1].
[0146] Вариант осуществления 13: Получение 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентил[d]пиридазин-4-ил)оксо)фенил)-1,2,4-триазин-3,5(2H,4H)-диона (соединение 13)
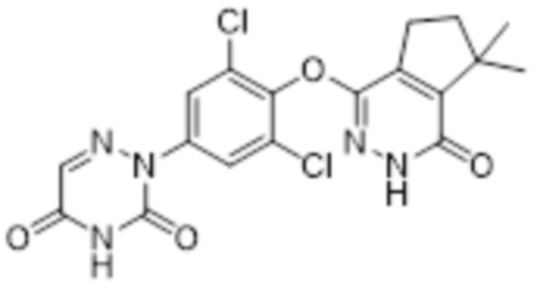
13
[0147] Использовался путь синтеза по варианту осуществления 12, за исключением того, что исходный продукт 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 2) на стадии 1 заменяли на 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентан[d]пиридазин-4-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 4), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((7,7-диметил-1-оксо-2,5,6,7-тетрагидро-1H-циклопентил[d]пиридазин-4-ил)оксо)фенил)-1,2,4-триазин-3,5(2H,4H)-дион (соединение 13).
[0148] 1H ЯМР (400 МГц, ДМСО-d6): 12,02 (с, 1H), 7,77 (с, 2H), 7,35 (с, 1H), 2,94-2,90 (м, 2H), 1,99-1,95 (м, 2H), 1,32 (d, 6H).
[0149] MS m/z (ESI): 436,0 [M+1].
[0150] Вариант осуществления 14: Получение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-1,2,4-триазин-3,5-(2H,4H)диона (соединение 14)
14
[0151] Использовался путь синтеза по варианту осуществления 12, за исключением того, что исходный продукт 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидро-5,8-этанофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 2) на стадии 1 заменяли на 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 9), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-1,2,4-триазин-3,5-(2H,4H)дион (соединение 14).
[0152] 1H ЯМР (400 МГц, ДМСО-d6): 12,00 (с, 1H), 8,30-8,23 (м, 2H), 8,09-7,99 (м, 2H), 7,18 (с, 1H).
[0153] MS m/z (ESI): 418,0 [M+1].
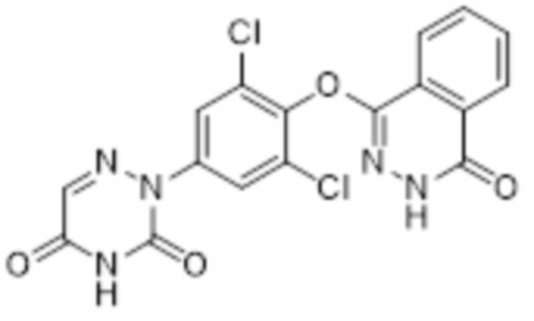
[0154] Вариант осуществления 15: Получение 2-(3,5-дихлор-4-((5-фтор-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 15)
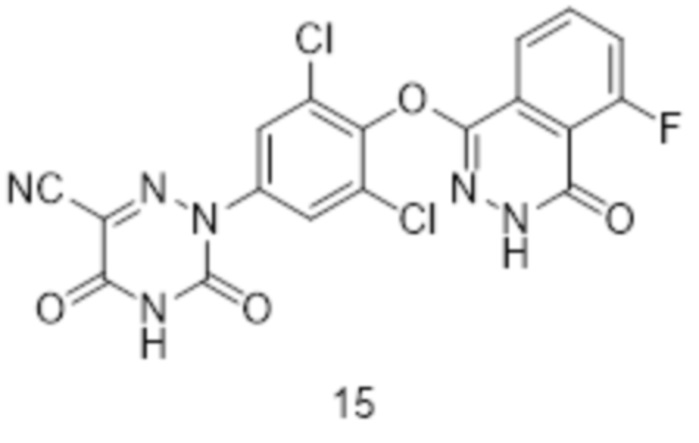
[0155] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт 3,4,5,6-тетрагидрофталевый ангидрид (соединение 1a) на стадии 1 заменяли на 3-фторфталевый ангидрид, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((5-фтор-4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 15).
[0156] MS m/z (ESI): 461,0 [M+1].
[0157] Вариант осуществления 16: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-6-метил-1,2,4-триазин-3,5(2H,4H)-диона (соединение 16)
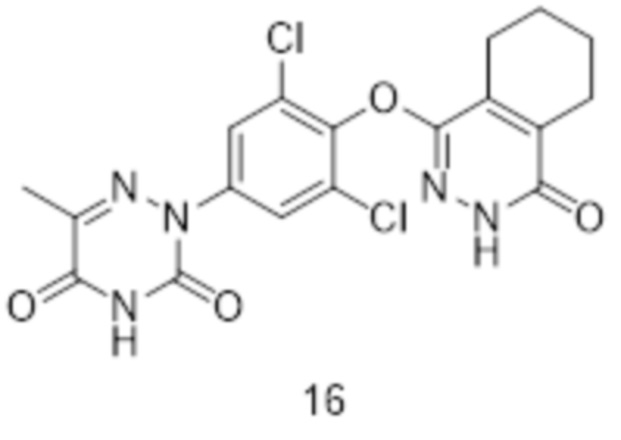
[0158] Стадия 1: Получение 4-(2,6-дихлор-4-йодфенокси)-5,6,7,8-тетрагидрофталазин-1(2H)-она (соединение 16a)
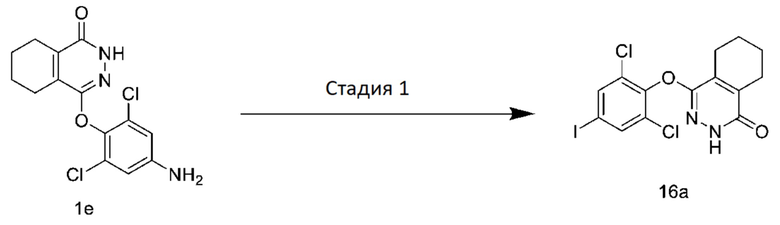
[0159] При охлаждении на ледяной бане к 1e (100 мг) последовательно добавляли воду (4 мл) и концентрированную соляную кислоту (2 мл) с последующим добавлением по каплям раствора нитрита натрия (30 мг) в воде (2 мл). мл). Реакционную смесь перемешивали в течение 1 ч на ледяной бане. По каплям добавляли йодид калия (104 мг), смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Смесь экстрагировали дихлорметаном и растворитель удаляли при пониженном давлении с получением твердого вещества 16a желтого цвета (100 мг), которое напрямую использовали в следующей реакции.
[0160] Стадия 2: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-6-метил-1,2,4-триазин-3,5(2H,4H)-диона (соединение 16)
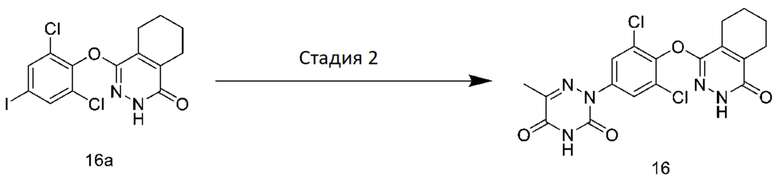
[0161] к смеси соединения 16a (20 мг), 6-азатимина (6,6 мг), йодида меди (8,9 мг) и карбоната калия (30 мг) добавляли N, N-дикарбонамид (2,0 мл), и систему нагревали до температуры 120°C и перемешивали в течение 12 часов. Систему охлаждали, фильтровали и выделяли от воды добавлением этилацетата. Растворитель удаляли из органической фазы при пониженном давлении, и соединение 16 (5,0 мг) получали путем выделения и очистки с помощью тонкослойной хроматографии (DCM:MeOH=8:1).
[0162] MS m/z (ESI): 436,1 [M+1].
[0163] Вариант осуществления 17: Получение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-6-изопропил-1,2,4-триазин-3,5(2H,4H)-диона (соединение 17)
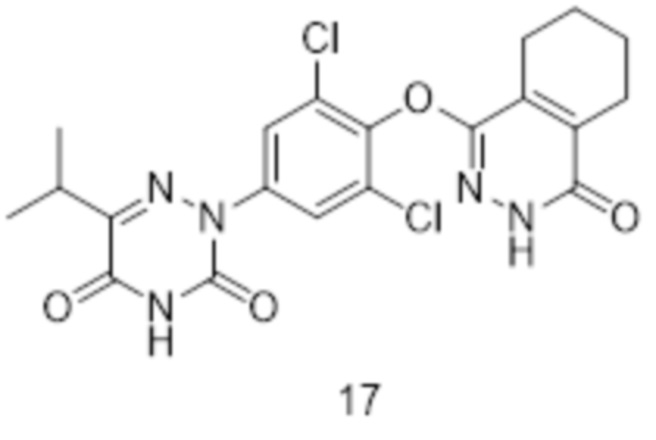
[0164] Использовался путь синтеза по варианту осуществления 16, за исключением того, что исходный продукт 6-азатимин на стадии 2 заменяли на 6-изопропил-1,2,4-триазин-3,5(2H,4H)-дион (полученный с использованием хорошо известного способа, описанного в работе «Chemistry and Biodiversity, 2012, 9(3) 536-556»), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-6-изопропил-1,2,4-триазин-3,5(2H,4H)-дион (соединение 17).
[0165] MS m/z (ESI): 464,1 [M+1].
[0166] Вариант осуществления 18: Получение 6-циклопропил-2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-1,2,4-триазин-3,5(2H,4H)-диона (соединение 18)
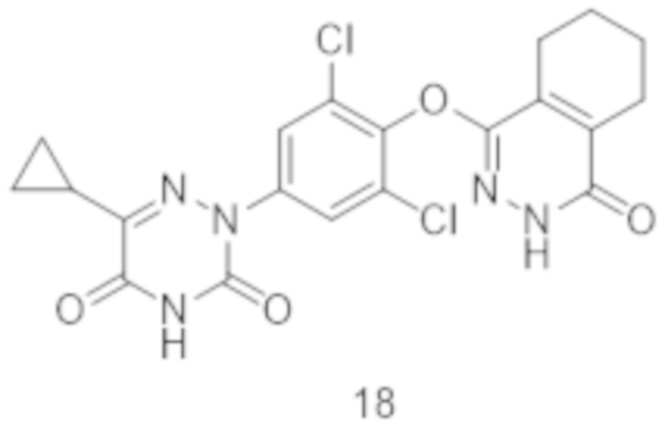
[0167] Использовался путь синтеза по варианту осуществления 16, за исключением того, что исходный продукт 6-азатимин на стадии 2 заменяли на 6-циклопропил-1,2,4-триазин-3,5(2H,4H)-дион (полученный с использованием хорошо известного способа, описанного в работе «Collection of Czechoslovak Chemical Communications, 1975, 40, 1038-1041»), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4,5,6,7,8-гексагидрофталазин-1-ил)оксо)фенил)-6-циклопропил-1,2,4-триазин-3,5(2H,4H)-дион (соединение 18).
[0168] MS m/z (ESI): 462,1 [M+1].
[0169] Вариант осуществления 19: Получение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-6-метил-1,2,4-триазин-3,5(2H,4H)-диона (соединение 19)
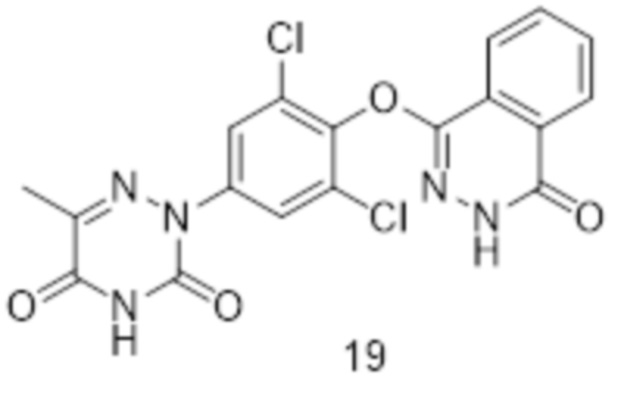
[0170] Стадия 1: Получение 3,5-дихлор-4-((4-хлорфталазин-1-ил)оксо)анилина (соединение 19a)
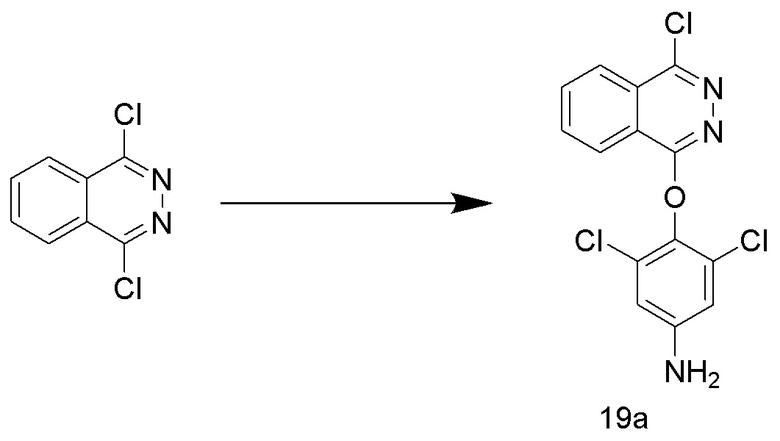
[0171] Использовался путь синтеза промежуточного соединения 1d на стадии 3 по варианту осуществления 1, за исключением того, что 1,4-дихлор-5,6,7,8-тетрагидрофталазин (соединение 1c) заменяли на 1,4-дихлорфталазин, что давало указанное в заголовке соединение 3,5-дихлор-4-((4-хлорфталазин-1-ил)оксо)анилин (соединение 19a).
[0172] Стадия 2: Получение 4-(4-амино-2,6-дихлорфенокси)фталазин-1(2H)-она (соединение 19b)
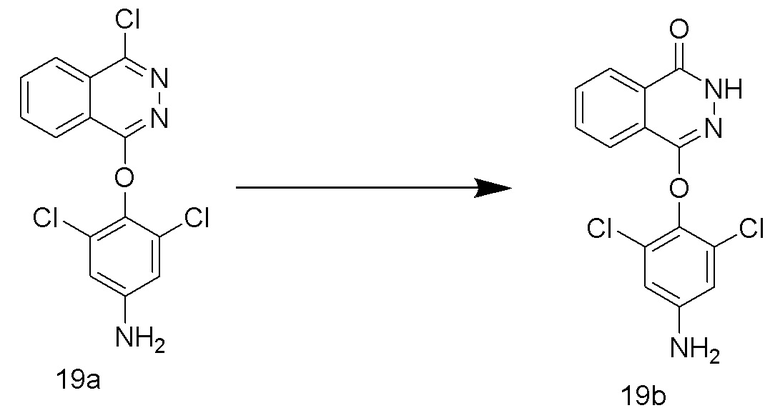
[0173] Использовался путь синтеза промежуточного соединения 1e на стадии 4 по варианту осуществления 1, за исключением того, что 3,5-дихлор-4-((4-хлор-5,6,7,8-тетрагидрофталазин-1-ил)окси)анилин (соединение 1d) заменяли на 3,5-дихлор-4-((4-хлорфталазин-1-ил)оксо)анилин (соединение 19a), что давало указанное в заголовке соединение 4-(4-амино-2,6-дихлорфенокси)фталазин-1(2H)-он (соединение 19b).
[0174] Стадия 3: Получение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-6-метил-1,2,4-триазин-3,5(2H,4H)-диона (соединение 19)
[0175] Использовался путь синтеза по варианту осуществления 16, за исключением того, что 4-(2,6-дихлор-4-йодфенокси)-5,6,7,8-тетрагидрофталазин-1(2H)-он (соединение 16a) на стадии 1 заменяли на соединение 4-(4-амино-2,6-дихлорфенокси)фталазин-1(2H)-он (соединение 19b), что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((4-оксо-3,4-дигидрофталазин-1-ил)оксо)фенил)-6-метил-1,2,4-триазин-3,5(2H,4H)-дион (соединение 19).
[0176] MS m/z (ESI): 432,0 [M+1].
[0177] Вариант осуществления 20: Получение 2-(3,5-дихлор-4-((6-метил-4-оксо-3,4-дигидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 20):
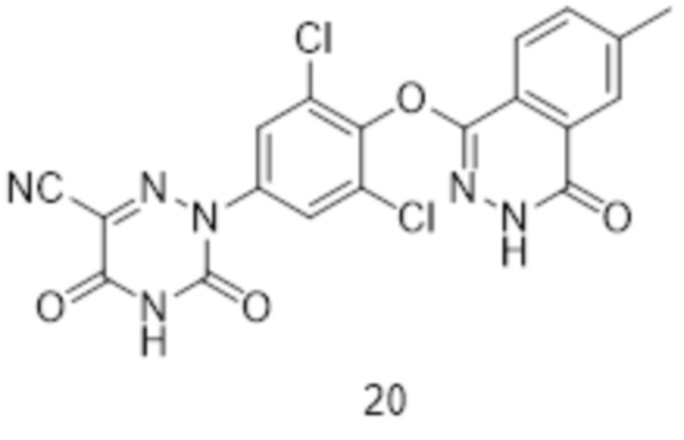
[0178] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт тетрагидрофталевый ангидрид (1a) на стадии 1 заменяли на 4-метилфталевый ангидрид, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((6-метил-4-оксо-3,4-дигидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 20).
[0179] MS m/z (ESI): 457,0 [M+1].
[0180] Вариант осуществления 21: Получение 2-(3,5-дихлор-4-((6-хлор-4-оксо-3,4-дигидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 21):
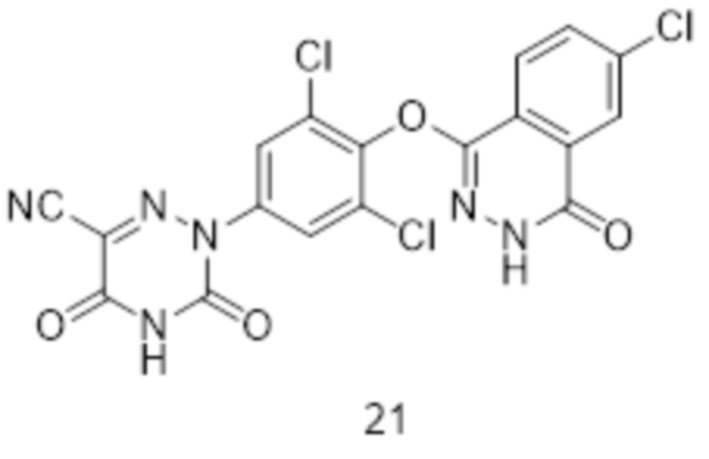
[0181] Использовался путь синтеза по варианту осуществления 1, за исключением того, что исходный продукт тетрагидрофталевый ангидрид (1a) на стадии 1 заменяли на 4-хлорфталевый ангидрид, что давало указанное в заголовке соединение 2-(3,5-дихлор-4-((6-хлор-4-оксо-3,4-дигидрофталазин-1-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 21).
[0182] MS m/z (ESI): 477,0 [M+1].
[0183] Вариант испытания 1: Исследование силы связывания соединения с TRα:
1. Основные экспериментальные материалы и аппаратура:
Микропланшет-ридер Envision 2104;
Пептид-коактиватор биотин-SRC2-2, коммерчески доступный от Sangon Biotech (Shanghai) Co., Ltd.;
TRα LBD, GST, коммерчески доступный от ThermoFisher (артикул № PV4762);
Связывающее европий антитело против глутатиона, коммерчески доступное от Cisbio (артикул № 61GSTKLB); и
Стрептавидин-D2, коммерчески доступный от Cisbio (артикул № 610SADAB)
2. Получение и обработка соединений
2.1 Приготовление исходного раствора соединения в диметилсульфоксиде
Все соединения растворяли в диметилсульфоксиде с получением 10 ммоль исходного раствора.
2.2 Хранение соединений
После растворения соединений в диметилсульфоксиде раствор можно хранить в течение 3 месяцев в эксикаторе при комнатной температуре. При длительном хранении соединения необходимо поместить в холодильник с температурой -20ºC.
3. Стадии эксперимента
3.1 Приготовление 1× реакционного буфера
3.2 Скрининг соединений:
a) 100% диметилсульфоксид использовали для разведения положительного лекарственного препарата трийодтиронина (Т3) от 10 ммоль (100×) или исследуемого соединения от 1 ммоль (100×) в равном соотношении 1:3, всего 10 концентрации.
b) разведенного до 4-кратного градиента концентрации соединения получали с 1-кратным реакционным буфером.
c) 5 мкл соединения, разведенного до 4-кратного градиента концентрации, добавляли в 384-луночный планшет для испытаний.
d) 4× TRα LBD и 4× RXRα получали с 1-кратным реакционным буфером.
e) 5 мкл 4× TRα LBD и 4× RXRα добавляли в 384-луночный планшет для испытаний.
f) 2× биотин-SRC2-2, 2× европий-связывающее антитело против глутатиона и 2× стрептавидин-d2 получали с 1-кратным реакционным буфером.
g) 10 мкл 2-кратно смешанного раствора (относится к стадии f) добавляли в 384-луночный планшет для испытаний.
h) 384-луночный планшет для испытаний центрифугировали в центрифуге при 1000 оборотов в 1 мин.
i) Инкубацию при комнатной температуре проводили в течение 1 ч в защищенном от света месте.
j) Значения сигнала флуоресценции каждой лунки 384-луночного планшета для испытаний при длине волны 665 нм и 615 нм регистрировали с помощью микропланшета-ридера Envision 2104 и рассчитывали соотношение флуоресценции 665 нм/615 нм.
4. Анализ данных
4.1 Расчет относительного соотношения (отношение665 нм/615 нм-отношениехолостое) для каждой лунки
4.2 Процент активности рассчитывался следующим образом:
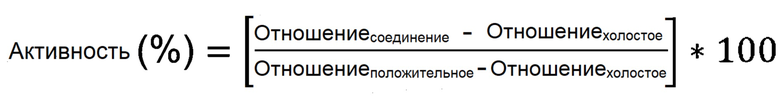
Отношениесоединение: среднее относительное отношение лунок вариантов соединений
Отношениеположительное: среднее относительное соотношение всех лунок с положительным контролем
Отношениехолостое: среднее относительное соотношение всех лунок с отрицательным контролем
4.3 Построение графика и расчет EC50:
EC50 рассчитывали с помощью Graphpad 5.0 с помощью метода нелинейной регрессии для соответствия соотношению между активностью (%) и логарифмической концентрацией соединения.
Y=низ+ верх-низ)/(1+10^ (LogEC50-X)×наклон))
X: Логарифм концентрации соединения Y: процент активности
Конкретные данные испытаний показаны в таблице 1 следующим образом.
[0184] Вариант испытания 2: Оценка агонистической активности соединения в отношение TRα
1. Основные экспериментальные материалы и аппаратура:
Микропланшет-ридер Envision 2104
Линия клеток HEK293T, коммерчески доступная от ATCC (артикул №: CRL-3216)
pGL4.35[luc2P/9XGAL4 UAS/Hygro], коммерчески доступный от Promega (артикул №: E1370)
Плазмида pBIND-TRα от Pharmaron
Плазмида pBIND-RXRα от Pharmaron
Агент трансфекции LipoLTX, коммерчески доступный от ThermoFisher (артикул №: 15338-100)
2. Приготовление соединения
2.1 Разведение соединения
Порошок соединения готовили в виде 10 ммоль исходного раствора в диметилсульфоксиде по стандартной схеме.
2.2 Хранение соединений
Все соединения, растворенные в диметилсульфоксиде, хранили в эксикаторе при комнатной температуре в течение короткого времени или хранили при температуре -20°C в течение длительного времени.
2.3 Приготовление экспериментальных соединений
2.3.1 Все испытуемые соединения разводили в диметилсульфоксиде с 3-кратным градиентом, с 10 градиентами разведения, начиная с начальной концентрации 10 мкмоль.
2.3.2 Положительный контроль трийодтиронин (Т3) разводили в диметилсульфоксиде с 3-кратным градиентом в 10 градиентах разведения, начиная с начальной концентрации 16,67 мкмоль.
2.3.3 Готовили 166,7-кратный положительный контроль (16,67 мкмоль, трийодтиронин (Т3)) и 166,7-кратный отрицательный контроль (100% диметилсульфоксид).
2.4 Планшет для соединения закрывали и встряхивали в течение 5 мин.
3 Процесс эксперимента
3.1 Приготовление клеточной суспензии и посев на чашки
a) Все клетки культивировали в соответствии со стандартной операцией ATCC, и HEK293T тестировали в период экспоненциального роста;
b) Среду отбрасывали;
c) Клетки дважды промывали фосфатным буфером;
d) Добавляли раствор трипсинизации для переваривания клеток и прекращали расщепление полной средой;
e) Клетки собирали и подсчитывали, и только когда жизнеспособность клеток превышала 90%, можно было провести эксперимент;
f) 2,5×106 клеток HEK293-LUC высевали на чашку для культивирования клеток диаметром 60 мм; и
g) Чашку для культивирования с засеянными клетками помещали в инкубатор при температуре 37ºC и 5% CO2 и культивировали в течение ночи.
3.2 Трансфекция клеток
a) Агент трансфекции LipoLTX выдерживали при комнатной температуре для установления равновесия;
b) 6 мкл агента Plus и 6 мкг ДНК были добавлены в 250 мкл среды Opti-MEMTM, не контактируя со стенкой пробирки, и равномерно перемешивали путем продувки и пипетирования с помощью пистолета для пипетирования, и
Плазмида: добавляли 2,5 мкг pBIND-TRα, 2,5 мкг pBIND-RXRα и 1 мкг плазмиды pGL4.35, соответственно;
c) 12 мкл Lipo LTX и 250 мкл среды Opti-MEMTM добавляли, не контактируя со стенкой пробирки, и равномерно перемешивали путем продувки и пипетирования с помощью пистолета для пипетирования;
d) агент, смешанный с DNA Plus (см. стадию 3.2.b), добавляли к разведенному трансфекционному агенту LipoLTX (см. стадию 3.2.c) и выдерживали в течение 15 минут при комнатной температуре;
e) агент трансфекции, смешанный с ДНК, добавляли в чашку для культивирования клеток диаметром 60 мм (см. стадию 3.1); и
f) культуральную чашку помещали в инкубатор при температуре 37°C и 5% CO2 и культивировали в течение 5 часов.
3.3 Обработка соединением
a) 150 нл разведенного соединения (см. стадию 2.3) переносили с помощью Echo550 в планшет для культивирования клеток (6007680-50, PE);
b) клетки (см. стадию 3.2) высевали в планшет для культивирования клеток 384 (6007680-50, PE) с 15000 клеток и 25 мкл среды, содержащей 5% фетальной бычьей сыворотки на лунку; и
c) клетки культивировали в течение ночи в инкубаторе при температуре 37°C и 5% CO2.
3.4 Обнаружение соединений
a) Детектор Steady-GloTM выдерживали при комнатной температуре;
b) 384 луночный планшет (см. стадию 3.3) выдерживали при комнатной температуре;
c) 25 мкл детектирующего агента Steady-GloTM на лунку добавляли в планшет для культивирования клеток (см. стадию 3.4b);
d) планшет помещали на шейкер и встряхивали в течение 5 мин в защищенном от света месте; и
e) значение хемилюминесценции определяли с помощью микропланшета-ридера Envision 2104.
4. Анализ данных
4.1 Расчет активности (%):
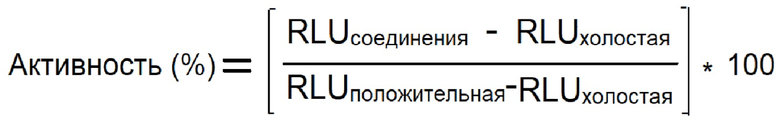
RLU: Генерируемая флуоресценция
RLUсоединения: Хорошо усредненное количество вариантов осуществления
RLUположительная: среднее значение положительного контроля
RLUхолостая: среднее значение отрицательного контроля
4.2 Расчет EC50 и построение кривой доза-эффект соединения
EC50 соединения рассчитывали с использованием Graphpad 5.0 для подбора активности (%) и логарифмической концентрации соединения.
Y=низ+ верх-низ)/(1+10^ (LogEC50-X)×наклон))
X: Логарифм концентрации соединения Y: процент ингибирования
[0185] Конкретные данные испытаний показаны в таблице 1 следующим образом.
[0186] Вариант испытания 3: Тест на силу связывания соединения с TRβ
1. Основной экспериментальный материал и аппаратура:
Микропланшет-ридер Envision 2104,
Пептид-коактиватор биотин-SRC2-2, коммерчески доступный от Sangon Biotech (Shanghai) Co., Ltd.
TRβ LBD, GST, коммерчески доступный от ThermoFisher (артикул № PV4762)
Связывающее европий антитело против глутатиона, коммерчески доступное от Cisbio (артикул № 61GSTKLB)
Стрептавидин-D2, коммерчески доступный от Cisbio (артикул № 610SADAB)
2. Получение и обработка соединений
2.1 Приготовление исходного раствора соединения в диметилсульфоксиде
Все соединения растворяли в диметилсульфоксиде с получением 10 ммоль исходного раствора.
2.2 Хранение соединений
После растворения соединений в диметилсульфоксиде раствор можно хранить в течение 3 месяцев в эксикаторе при комнатной температуре. При длительном хранении соединения необходимо поместить в холодильник с температурой -20ºC.
3. Стадии эксперимента
3.1 Приготовление 1× реакционного буфера
3.2 Скрининг соединений:
a) 100% диметилсульфоксид использовали для разведения положительного лекарственного препарата трийодтиронина (Т3) от 10 мкмоль (100×) или исследуемого соединения от 1 ммоль (100×) в равном соотношении 1:3, всего в 10 концентрациях;
b) Соединение, разведенное в 4-кратном градиенте концентрации, получали с 1-кратным реакционным буфером;
c) 5 мкл соединения, разведенного в 4-кратном градиенте концентрации, добавляли в 384-луночный планшет для испытаний;
d) 4× TRβ LBD и 4× RXRβ получали с 1x реакционным буфером;
e) 5 мкл 4× TRβ LBD и 4× RXRβ добавляли в 384-луночный планшет для испытаний;
f) 2× биотин-SRC2-2, 2× европий-связывающее антитело против глутатиона и 2× стрептавидин-d2 получали с 1-кратным реакционным буфером;
g) 10 мкл 2-кратного смешанного раствора (относится к стадии f) добавляли в 384-луночный планшет для испытаний;
h) 384-луночный планшет для испытаний центрифугировали в центрифуге при 1000 оборотов в 1 мин;
i) Инкубацию при комнатной температуре проводили в течение 1 ч в защищенном от света месте; и
j) Значения сигнала флуоресценции каждой лунки 384-луночного планшета для испытаний при длине волны 665 нм и 615 нм регистрировали с помощью микропланшета-ридера Envision 2104 и рассчитывали соотношение флуоресценции 665 нм/615 нм.
4. Анализ данных
4.1 Расчет относительного соотношения (соотношение665 нм/615 нм-соотношениехолостое) для каждой лунки
4.2 Процент активности рассчитывался следующим образом:
Отношениесоединение: среднее относительное отношение лунок вариантов соединений
Отношениеположительное: среднее относительное соотношение всех лунок с положительным контролем
Отношениехолостое: среднее относительное соотношение всех лунок с отрицательным контролем
4.3 Построение графика и расчет EC50:
EC50 рассчитывали с помощью Graphpad 5.0 с помощью метода нелинейной регрессии для соответствия соотношению между активностью (%) и логарифмической концентрацией соединения.
Y=низ+ верх-низ)/(1+10^ (LogEC50-X)×наклон))
X: Логарифм концентрации соединения Y: процент активности
[0187] Конкретные данные испытаний показаны в таблице 1 следующим образом.
Таблица 1 Связывающая активность соединения с рецептором тироксина β показана следующим образом:
[0188] Заключение: по сравнению с описанным соединением сравнения 53, некоторые из соединений согласно настоящему изобретению неожиданно показали высокую активность THRβ (<0,2 мкМ), и некоторые из соединений показали более высокую селективность к THRα, чем соединение сравнения 53.
[0189] Вариант испытания 4: Оценка агонистической активности соединения в отношении рецептора TRβ
[0190] Краткое описание эксперимента: Кодирующие последовательности TRβ-LBD и RXRα-LBD вставляли в плазмиду pBIND (Promega, E1581) соответственно. Вектор экспрессии и репортерный вектор (pGL4.35, несущий репортерный ген люциферазы, управляемый стабильным интегрированным промотором GAL4) коэкспрессировали в клетке-хозяине. Когда агонист связывается с соответствующим химерным рецептором, химерный рецептор связывается с сайтом связывания GAL4 в векторе репортерного гена и стимулирует экспрессию репортерного гена. По интенсивности сигналов хемилюминесценции определяли агонистическую активность соединения в отношении рецептора TRβ.
[0191] Экспериментальные материалы и аппараты:
Микропланшет-ридер Envision 2104
HEK293T линия клеток, коммерчески доступная от ATCC (Артикул №: CRL-3216)
pGL4.35[luc2P/9XGAL4 UAS/Hygro], коммерчески доступный от Promega (Артикул №: E1370)
Плазмида pBIND-TR β от Pharmaron
Плазмида pBIND-RXRα от Pharmaron
Агент трансфекции LipoLTX, коммерчески доступный от ThermoFisher (Артикул №: 15338-100)
Набор для обнаружения люциферазы Steady-GloTM, коммерчески доступный от Promega (Артикул №: E2520)
2.3 Приготовление экспериментальных соединений
□ Все тестируемые соединения разводили в диметилсульфоксиде с 3-кратным градиентом, с 10 градиентами разведения, начиная с начальной концентрации 10 ммоль.
□ Положительный контроль трийодтиронин (Т3) разводили в диметилсульфоксиде с 3-кратным градиентом в 10 градиентах разведения, начиная с начальной концентрации 16,67 мкмоль.
□ Готовили 166,7-кратный положительный контроль (16,67 мкмоль, трийодтиронин (Т3)) и 166,7-кратный отрицательный контроль (100% диметилсульфоксид).
□ Планшет для соединения закрывали и встряхивали в течение 5 мин.
3 Процесс эксперимента
3.1 Приготовление клеточной суспензии и посев на чашки
a) Все клетки культивировали в соответствии со стандартной операцией ATCC, и HEK293T тестировали в период экспоненциального роста;
b) Среду отбрасывали;
c) Клетки дважды промывали фосфатным буфером;
d) Добавляли раствор трипсинизации для переваривания клеток и прекращали расщепление полной средой;
e) Клетки собирали и подсчитывали, и только когда жизнеспособность клеток превышала 90%, можно было провести эксперимент;
f) 2,5×106 Клеток HEK293-LUC высевали в чашку для культивирования клеток диаметром 60 мм; и
g) Чашку для культивирования с засеянными клетками помещали в инкубатор при температуре 37°C и 5% CO2 и культивировали в течение ночи.
3.2 Трансфекция клеток
a) Агент трансфекции LipoLTX выдерживали при комнатной температуре для установления равновесия;
b) 6 мкл агента Plus и 6 мкг ДНК добавляли в 250 мкл среды Opti-MEMTM, не контактируя со стенкой пробирки, и равномерно смешивали путем продувки и пипетирования с помощью пистолета для пипетирования; плазмида: 2,5 мкг pBIND-TRβ, 2,5 мкг pBIND-RXRα и 1 мкг плазмиды pGL4.35
c) 12 мкл Lipo LTX и 250 мкл среды Opti-MEMTM добавляли, не контактируя со стенкой пробирки, и они равномерно смешивали путем продувки и пипетирования с помощью пистолета для пипетирования;
d) агент, смешанный с ДНК плюс (см. стадию 3.2.b), добавляли к разведенному агенту трансфекции LipoLTX (см. стадию 3.2.c) и выдерживали в течение 15 минут при комнатной температуре;
e) агент трансфекции, смешанный с ДНК, добавляли в чашку для культивирования клеток диаметром 60 мм (см. стадию 3.1); и
f) культуральную чашку помещали в инкубатор при температуре 37°C и 5% CO2 и культивировали в течение 5 часов.
3.3 Обработка соединением
a) 150 нл разведенного соединения (см. стадию 2.3) переносили с помощью Echo550 в планшет для культивирования клеток (6007680-50, PE);
b) клетки (см. этап 3.2) высевали в планшет для культивирования клеток 384 (6007680-50, PE) с 15000 клеток и 25 мкл среды на лунку; и
c) клетки культивировали в течение ночи в инкубаторе при температуре 37°C и 5% CO2.
3.4 Обнаружение соединений
a) Детектор Steady-GloTM помещали при комнатной температуре;
b) планшет с ячейками 384 (см. стадию 3.3) выдерживали при комнатной температуре;
c) 25 мкл детектирующего агента Steady-GloTM на лунку добавляли в планшет для культивирования клеток (см. стадию 3.4b);
d) планшет помещали на шейкер и встряхивали в течение 5 мин в защищенном от света месте; и
e) значение хемилюминесценции определяли с помощью микропланшета-ридера Envision 2104.
4. Анализ данных
4.1 Расчет активности (%):
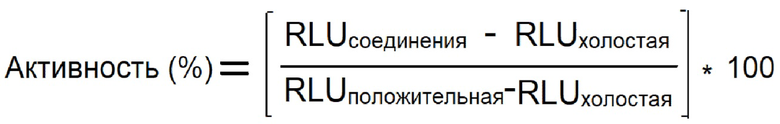
RLU: Генерируемая флуоресценция
RLUсоединения: Хорошо усредненное количество вариантов осуществления
RLUположительная: среднее значение положительного контроля
RLUхолостая: среднее значение отрицательного контроля
4.2 Расчет EC50 и построение кривой доза-эффект соединения
EC50 соединения рассчитывали с использованием Graphpad 5.0 для подбора активности (%) и логарифмической концентрации соединения.
Y=низ+ верх-низ)/(1+10^ (LogEC50-X)×наклон))
X: Логарифм концентрации соединения Y: процент ингибирования
Таблица 2: Агонистическая активность соединения настоящего изобретения в отношении бета-рецептора тироксина показана следующим образом:
[0192] Заключение: соединение по настоящему изобретению может активировать нисходящий сигнал бета-рецептора тироидного гормона.
[0193] Вариант испытания 5: Фармакокинетическая оценка:
[0194] В качестве подопытных животных использовали крыс. После внутрижелудочной инфузии соединений из варианта осуществления 4 и варианта осуществления 9 исследовали концентрации лекарственного средства в плазме крыс в разное время. Было изучено фармакокинетическое поведение соединений по настоящему изобретению у крыс и оценены характеристики фармацевтического метаболизма. В каждой группе варианта осуществления были отобраны 3 крысы-самца SD с аналогичным весом тела, и пероральная доза составляла 2 мг/кг разовой дозой. Кровь собирали через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 10 часов и 24 часа после введения дозы животным. Аналитический метод LC-MS/MS использовался для определения содержания соединений в плазме, и нижний предел количественного определения метода составлял 20 нг/мл. Программное обеспечение для анализа метакинетических данных WinNonlin 7.0 использовали для создания статистики по VT088 и данных о концентрации в плазме. Метод внекомпартментной модели (NCA) использовали для расчета фармакокинетических параметров, в частности, как показано в таблице 3 ниже.
[0195] Схема эксперимента:
[0196] Лекарства для экспериментов: соединения из варианта осуществления 4 и варианта осуществления 9.
[0197] Лабораторные животные:
[0198] Шесть здоровых крыс-самцов SD, коммерчески доступных от Shanghai Sippr-Bk Laboratory Animal Co., Ltd., с лицензией на животноводство №: SCXK (Шанхай) 2008-0016, были разделены на 2 группы по 3 в каждой группе.
[0199] Приготовление лекарственного средства: было взято определенное количество лекарственного средства и добавлено в 2% водный раствор Klucel LF+0,1% Tween 80, чтобы приготовить прозрачный раствор или однородную суспензию.
[0200] Дозировка: крысам SD давали голодать в течение ночи и вводили лекарство путем внутрижелудочной инфузии в дозе 2 мг/кг и введенном объеме 10 мл/кг каждой.
[0201] Операция: крысам вводили внутрижелудочную инфузию соединений из варианта осуществления 4 и варианта осуществления 9. По меньшей мере 0,2 мл крови собирали из хвостовой вены через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 10 ч и 24 ч до и после дозировки; затем кровь помещали в гепаринизированные пробирки для образцов, центрифугировали при температуре 4°C и 3500 об/мин в течение 10 минут для отделения плазмы. Затем пробирки с гепаринизированными образцами хранили при температуре -20°C, и крысам позволяли есть пищу через 2 часа после введения дозы.
[0202] Определение содержания исследуемых соединений в плазме крыс после внутрижелудочного вливания лекарств в различных концентрациях: образцы плазмы размораживали при комнатной температуре, отбирали по 50 мкл каждого и добавляли к 130 мкл внутреннего стандарта. рабочий раствор (1000 нг/мл, ацетонитрил, толбутамид), смесь вращали около 1 мин, а затем центрифугировали при 4°C и 13000 об/мин в течение 10 мин. Отбирали 50 мкл супернатанта и смешивали со 100 мкл 50% ацетонитрильной воды, а затем вводили для анализа ЖХ/МС/МС.
[0203] Результаты фармакокинетических параметров показаны в таблице 3.
Таблица 3: Фармацевтические данные метаболизма крыс
(мг/кг)
(ч)
рация препарата в крови
(нг/мл)
(нг∙ч/мл)
распада
(ч)
[0204] Заключение: соединения по настоящему изобретению обладают хорошей фармакокинетической абсорбцией и значительными фармакокинетическими преимуществами. По сравнению с описанным соединением сравнения 53, некоторые из соединений по настоящему изобретению неожиданно показывают более высокие значения Cmax и величины воздействия при той же дозе и препарате. Все приведенные выше результаты PK показывают, что соединения, предлагаемые в настоящем изобретении, обладают хорошими PK-свойствами и могут использоваться в качестве терапевтического лекарственного средства при заболевании, связанном с метаболизмом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГАЛОГЕНЗАМЕЩЕННОЕ ФЕНИЛАТНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЯ | 2020 |
|
RU2820475C2 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2021 |
|
RU2802282C2 |
| ПРОИЗВОДНЫЕ ТРИАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, АНТИПРОТОЗОЙНАЯ КОМПОЗИЦИЯ, ДОБАВКА В ПИЩУ ЖИВОТНЫХ, СПОСОБ ИНГИБИРОВАНИЯ ПРОТОЗОИ У ЖИВОТНЫХ | 1994 |
|
RU2146674C1 |
| ТРИАРИЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2840345C2 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА ТИРЕОИДНОГО ГОРМОНА | 2006 |
|
RU2379295C2 |
| ДИАЗЕПИНОИНДОЛЫ - ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ-IV | 1995 |
|
RU2174517C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, которые могут быть полезны в качестве агониста рецепторов THRβ. В формуле (I) R1 выбран из группы, состоящей из водорода, циано и C1-6 алкила; R2 и R3, каждый, представляют собой независимо выбранный галоген; кольцо A представляет собой замещенное или незамещенное C5-10 ароматическое кольцо, где заместитель выбран из группы, состоящей из водорода, галогена и C1-6 алкила; и галоген выбран из группы, состоящей из F, Cl и Br. Изобретение относится также к фармацевтической композиции, содержащей указанное соединение, и его применению для изготовления терапевтического лекарственного средства от заболевания, связанного с метаболизмом, связанного с бета-рецептором тироидного гормона. 3 н. и 18 з.п. ф-лы, 3 табл., 21 пр.
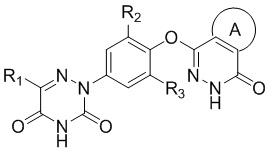 (I)
(I)
1. Соединение формулы (I) или его фармацевтически приемлемая соль
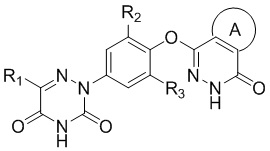 (I),
(I),
где R1 выбран из группы, состоящей из водорода, циано и C1-6 алкила;
R2 и R3, каждый, представляют собой независимо выбранный галоген;
кольцо A представляет собой замещенное или незамещенное C5-10 ароматическое кольцо, где заместитель выбран из группы, состоящей из водорода, галогена и C1-6 алкила; и
галоген выбран из группы, состоящей из F, Cl и Br.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (III)
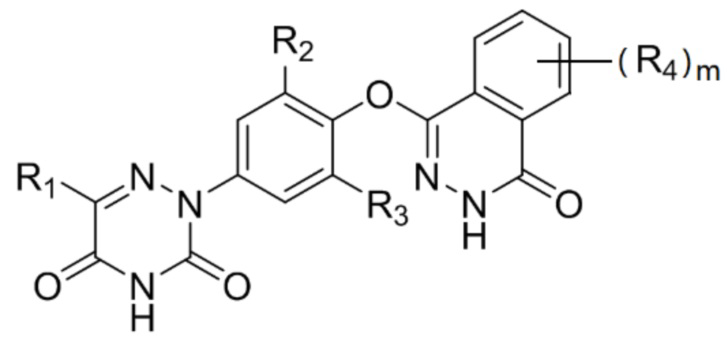 (III),
(III),
где R4 выбран из группы, состоящей из водорода, галогена и C1-6 алкила;
m обозначает целое число в диапазоне от 1 до 4; и
галоген выбран из группы, состоящей из F, Cl и Br.
3. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где R4 выбран из группы, состоящей из водорода, галогена и C1-6 алкила; и
m обозначает целое число в диапазоне от 1 до 3.
4. Соединение или его фармацевтически приемлемая соль по п. 3, где R4 выбран из группы, состоящей из водорода, галогена или C1-3 алкила; и m обозначает 1 или 2.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где R1 выбран из группы, состоящей из водорода, циано и C1-6 алкила; и галоген выбран из группы, состоящей из F, Cl и Br.
6. Соединение или его фармацевтически приемлемая соль по п. 5, где R1 выбран из группы, состоящей из циано или C1-3 алкила.
7. Соединение или его фармацевтически приемлемая соль по п. 6, где R1 представляет собой циано.
8. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-7, где R2 и R3, каждый, независимо выбраны из группы, состоящей из F, Cl или Br.
9. Соединение или его фармацевтически приемлемая соль по п. 8, где R2 и R3 оба представляют собой Cl.
10. Соединение по любому из пп. 1-9, где соединение представляет собой 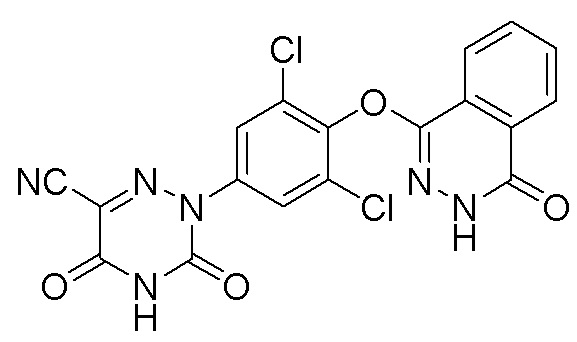 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
11. Соединение по любому из пп. 1-9, где соединение представляет собой 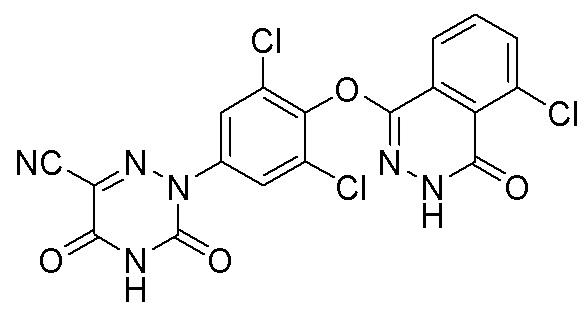 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
12. Соединение по любому из пп. 1-9, где соединение представляет собой 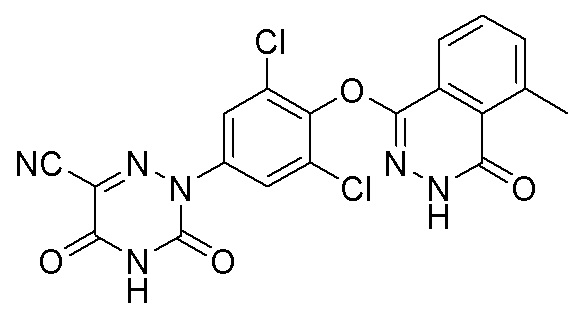 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
13. Соединение по любому из пп. 1-9, где соединение представляет собой 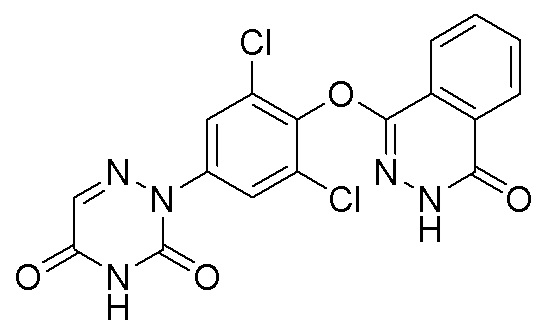 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
14. Соединение по любому из пп. 1-9, где соединение представляет собой 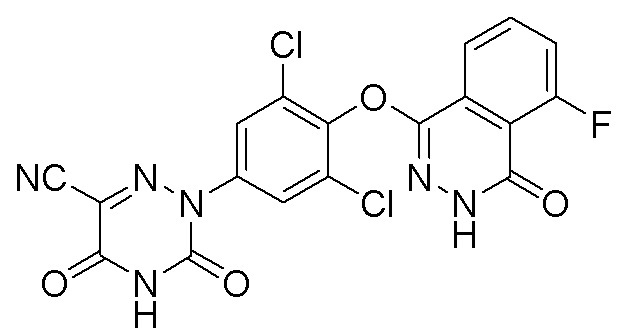 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
15. Соединение по любому из пп. 1-9, где соединение представляет собой 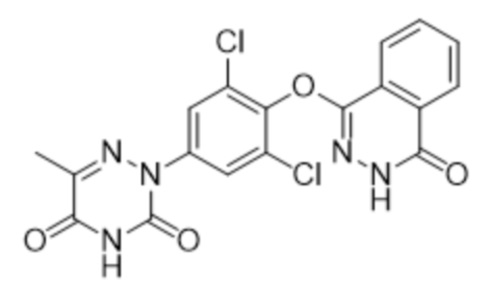 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
16. Соединение по любому из пп. 1-9, где соединение представляет собой 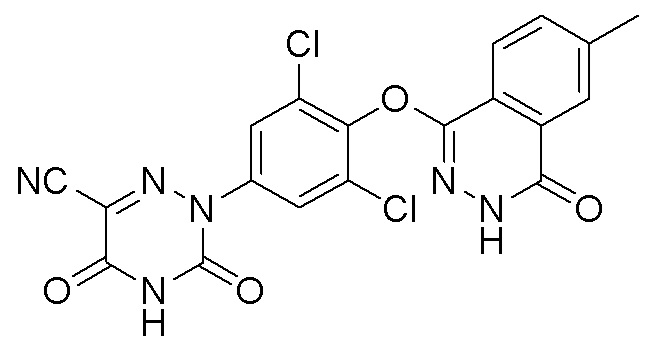 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
17. Соединение по любому из пп. 1-9, где соединение представляет собой 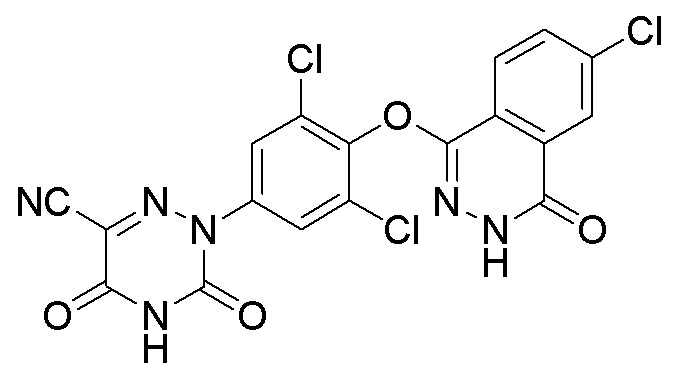 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
18. Фармацевтическая композиция, полезная в качестве агониста рецепторов THRβ, содержащая терапевтически эффективное количество соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли по любому из пп. 1-17 и фармацевтически приемлемый адъювант.
19. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-17 для изготовления терапевтического лекарственного средства от заболевания, связанного с метаболизмом, связанного с бета-рецептором тироидного гормона.
20. Применение по п. 19, где заболевание, связанное с метаболизмом, связанное с бета-рецептором тироидного гормона, выбрано из группы, состоящей из ожирения, гиперлипидемии, гиперхолестеринемии, диабета и неалкогольного стеатогепатита (НАСГ), стеатоза печени, атеросклероза, гипотиреоза и рака щитовидной железы.
21. Применение по п. 19, где заболевание, связанное с метаболизмом, связанное с бета-рецептором тироидного гормона, выбрано из группы, состоящей из неалкогольного стеатогепатита (НАСГ), гипотиреоза и рака щитовидной железы.
| US 7807674 B2, 05.10.2010 | |||
| M.J | |||
| KELLY ET AL | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| MED | |||
| CHEM., 2014, vol | |||
| Способ получения на волокне оливково-зеленой окраски путем образования никелевого лака азокрасителя | 1920 |
|
SU57A1 |

