РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашиваются приоритет и преимущества Предварительной заявки США No. 61/702137, поданной 17 сентября 2012 г., и Предварительной заявки США No. 61/790432, поданной 15 марта 2013 г., полное содержание которых приведено в настоящем документе в качестве ссылки.
ВКЛЮЧЕНИЕ В КАЧЕСТВЕ ССЫЛКИ СПИСКА ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Полное содержание текстового файла, названного «41245-522001WO_ST25.txt», который был создан 16 сентября 2013 г. и имеет размер 4 KB, таким образом, приведено в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ
Гормоны щитовидной железы являются критическими для нормального роста и развития и для поддержания метаболического гомеостаза (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Уровни циркулирующих гормонов щитовидной железы жестко регулируются по механизмам обратной связи в гипоталамо/гипофизарно/тиреоидной (HPT) оси. Дисфункция щитовидной железы, приводящая к гипотиреозу или гипертиреозу, явно демонстрирует, что гормоны щитовидной железы оказывают сильные эффекты на сердечную функцию, массу тела, метаболизм, скорость метаболизма, температуру тела, холестерин, кости, мышцы и поведение.
Биологическая активность гормонов щитовидной железы опосредована рецепторами гормонов щитовидной железы (TR или THR) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TR принадлежат к суперсемейству, известному как ядерные рецепторы. TR формируют гетеродимеры с ретиноидным рецептором, действующие как индуцируемые лигандом факторы транскрипции. TR имеют связывающий лиганд домен, ДНК-связывающий домен и аминоконцевой домен, и регулируют экспрессию гена посредством взаимодействия с отвечающими элементами ДНК и с различными ядерными коактиваторами и корепрессорами. Рецепторы гормонов щитовидной железы происходят от двух отдельных генов, α и β. Эти продукты различных генов образуют множество форм соответствующих им рецепторов посредством дифференциального процессинга РНК. Главными изоформами тиреоидных рецепторов являются α1, α2, β1 и β2. Рецепторы гормонов щитовидной железы α1, β1 и β2 связывают гормоны щитовидной железы. Показано, что подтипы рецепторов гормонов щитовидной железы могут отличаться по их вкладу в конкретные биологические ответы. Недавние исследования позволяют предполагать, что TRβ1 играет важную роль в регуляции TRH (высвобождающего тиреотропин гормона) и в регуляции воздействий гормонов щитовидной железы на печень. TRβ2 играет важную роль в регуляции TSH (тиреостимулирующего гормона) (Abel et. al., J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TRβ1 играет важную роль в регуляции частоты сердечных сокращений (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al., Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
Предприняты усилия для синтеза аналогов гормонов щитовидной железы, обладающих увеличенной избирательностью для рецептора гормонов щитовидной железы бета и/или тканеспецифическим действием. С такими миметиками гормонов щитовидной железы можно получать желательные снижения массы тела, уровней липидов, холестерина и липопротеинов, с уменьшенным влиянием на сердечно-сосудистую функцию или нормальную функцию гипоталамо/гипофизарно/тиреоидной оси (см., например, Joharapurkar et al., J. Med. Chem., 2012, 55 (12), pp 5649-5675). Разработка аналогов гормонов щитовидной железы, позволяющих избегать нежелательных эффектов гипертиреоза и гипотиреоза при сохранении в то же время предоставляющих преимущества эффектов гормонов щитовидной железы, может открывать новые подходы к лечению пациентов с метаболическим заболеванием, таким как ожирение, гиперлипидемия, гиперхолестеринемия, диабет и другие нарушения и заболевания, такие как стеатоз печени и NASH, атеросклероз, сердечно-сосудистые заболевания, гипотиреоз, рак щитовидной железы, заболевания щитовидной железы, устойчивость к гормонам щитовидной железы и родственные нарушения и заболевания.
Настоящее изобретение, частично, относится к способам синтеза аналогов гормонов щитовидной железы, таких как пиридазиноновые соединения, и их пролекарств. Идеальный способ синтеза аналогов гормонов щитовидной железы и их пролекарств может, например, предоставлять соединения-продукты с высокой чистотой и высоким выходом. Настоящее изобретение направлено на предоставление одного или нескольких из этих желательных признаков.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ОПИСАНИЯ
В настоящем описании описан способ синтеза, который можно использовать для получения 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7»), соединения, которое является пригодным в качестве промежуточного соединения для получения пиридазиноновых соединений в качестве аналогов гормонов щитовидной железы, следующим образом:
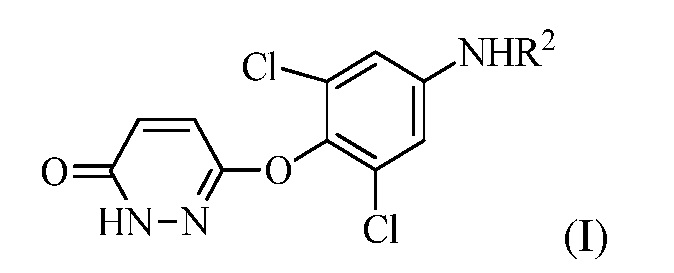
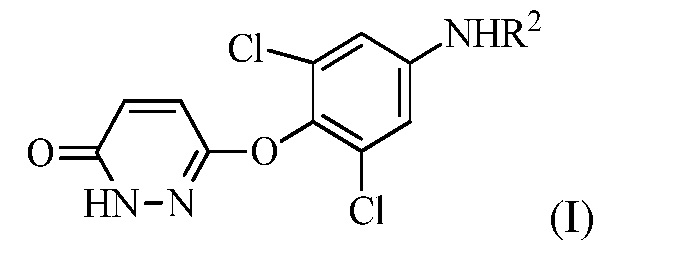
(a) контакт R1MgX или R1Li с соединением формулы (I):
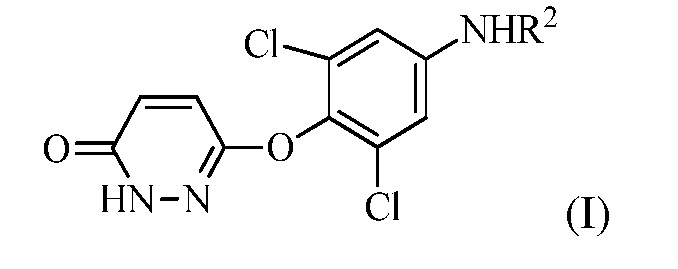
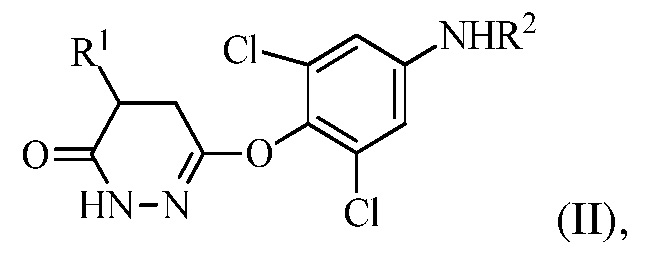
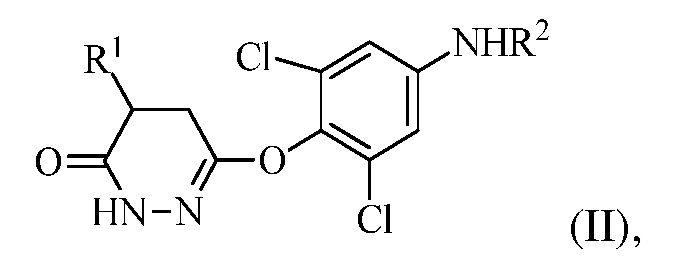
для получения соединения формулы (II):
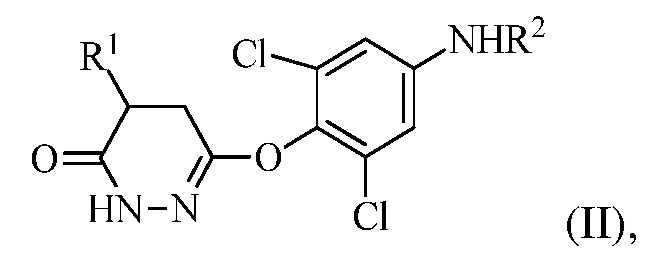
в которой R1 представляет собой изопропил или изопропенил, X представляет собой гало, и R2 представляет собой H или защитную группу для амина; и
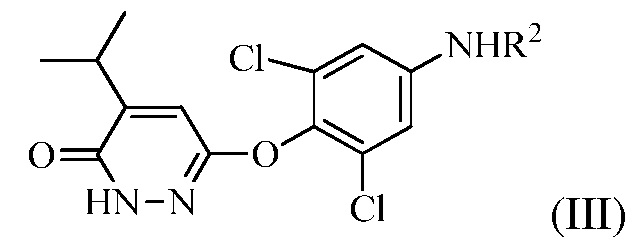
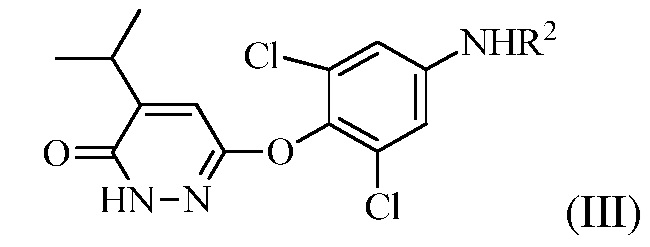
(b) перевод соединения формулы (II) в соединение формулы (III):
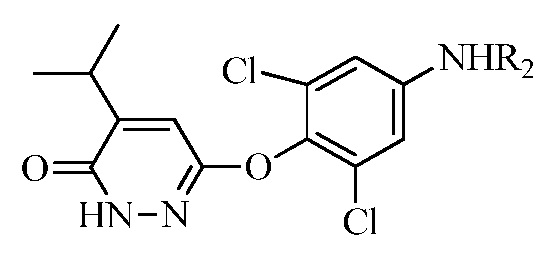 (III)
(III)
в присутствии основания, когда R1 представляет собой изопропенил, или в присутствии окисляющего средства, когда R1 представляет собой изопропил.
На стадии (a) растворитель может представлять собой апротонный органический растворитель, такой как THF, диэтиловый эфир, толуол или диоксан, температура реакции может составлять 0-60°C, 20-50°C, 30-45°C, или 35-45°C, время реакции может составлять от 10 мин до 10 часов, 1-8 часов или 3-5 часов, и количество реагента Гриньяра (R1MgX) может составлять 3-10 эквивалентов или 3-6 эквивалентов соединения формулы (I).
На стадии (b) основание используют для изомеризации соединения формулы (II). Оно может представлять собой органическое основание или неорганическое основание. Примеры оснований включают, но без ограничения, триэтиламин, пиридин, KOH, NaOH и карбонаты. Изомеризации можно также достигать в других условиях, например, обработкой кислотой или нагреванием в апротонном растворителе.
Также, на стадии (b), окисляющее средство не является конкретно ограниченным. Например, можно использовать бром в уксусной кислоте или проприоновой кислоте.
Защитные группы для амина включают, в качестве неограничивающих примеров, замещенный алкил, ацил (например, бензоил или ацетил) и силил. Защитные группы для гидрокси- и амина обсуждают в T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed., John Wiley and Sons (1991).
В одном варианте осуществления стадию (a) проводят посредством контакта R1MgX с соединением формулы (I), в котором R1 представляет собой изопропенил, и X представляет собой Br. Растворитель, используемый в этой реакции, может представлять собой THF с соотношением объема к массе THF к соединению формулы (I) в диапазоне между 7 и 30 (или между 7 и 15). Эту стадию можно проводить в присутствии кислоты Льюиса (например, галогенида лития).
В одном варианте осуществления стадию (a) проводят посредством контакта R1MgX с соединением формулы (I), в котором R1 представляет собой изопропил, и X представляет собой Cl. Растворитель, используемый в этой реакции, может представлять собой THF с соотношением объема к массе THF к соединению формулы (I) в диапазоне между 7 и 30 (или между 7 и 15). Эту стадию можно проводить в присутствии кислоты Льюиса (например, галогенида лития).
В одном варианте осуществления основание на стадии (b) представляет собой гидроксид металла (например, гидроксид калия).
В одном варианте осуществления окисляющее средство на стадии (b) представляет собой бром, и стадию (b) проводят в присутствии кислоты.
В одном варианте осуществления группа R2 в формуле (I) и в формуле (II) представляет собой ацетил или бензоил. В следующем варианте осуществления R2 представляет собой бензоил.
В одном варианте осуществлении способ дополнительно включает получение соединения формулы (I) посредством контакта 3,6-дихлорпиридазина с 2,6-дихлор-4-аминофенолом для образования 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина, гидролиза 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина и защиты аминогруппы 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина либо до, либо после гидролиза для получения соединения формулы (I). Контакт 3,6-дихлорпиридазина с 2,6-дихлор-4-аминофенолом проводят в полярном апротонном растворителе (например, диметилацетамиде (DMAC)) в присутствии основания (например, CS2CO3) при температуре реакции между 60 и 120°C (например, приблизительно 65°C). Кроме того, можно включать стадию очистки. То есть, перед стадией (a), соединение формулы (I) очищают в кислом растворе при температуре между 80 и 100°C.
В одном варианте осуществления способ дополнительно включает стадию (c) удаления, когда присутствует, защитной группы для амина R2 соединения формулы (III) для получения 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она.
В одном варианте осуществления, соединение, например, промеж. 7, полученное способом, описанным в настоящем документе, обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5%, или более 99,8%.
В одном варианте осуществления соединение, т.е., 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-он, полученное способом, описанным в настоящем документе, имеет менее 1,5% 6-(4-амино-2,6-дихлорфенокси)-5-изопропилпиридазин-3(2H)-она, например, менее 1,0% 6-(4-амино-2,6-дихлорфенокси)-5-изопропилпиридазин-3(2H)-она, или менее 0,5% 6-(4-амино-2,6-дихлорфенокси)-5-изопропилпиридазин-3(2H)-она.
В другом варианте осуществления, соединение, полученное описанным выше способом, является свободным от 6-(4-амино-2,6-дихлорфенокси)-5-изопропилпиридазин-3(2H)-она.
Способ синтеза по этому изобретению может дополнительно включать следующую стадию для синтеза пиридазиноновых соединений в качестве аналогов гормонов щитовидной железы и их пролекарств:
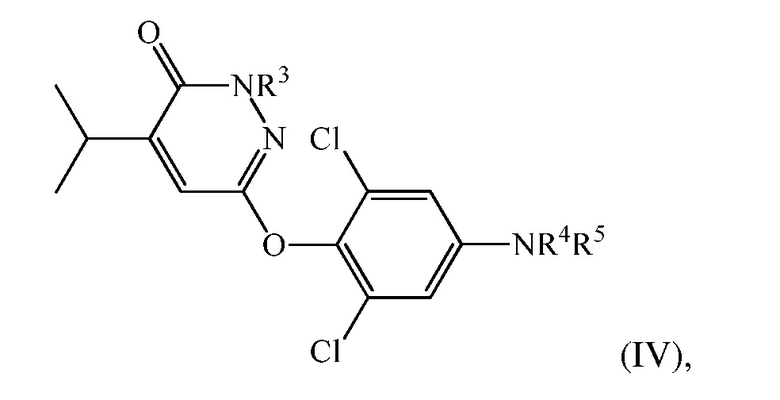
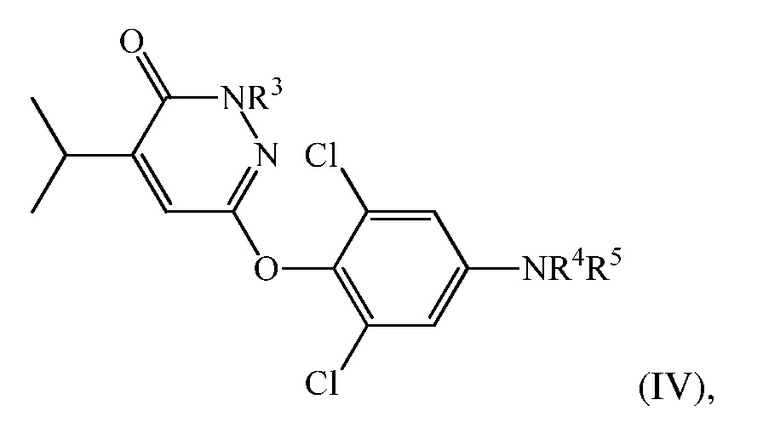
(d) перевод 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она в соединение формулы (IV):
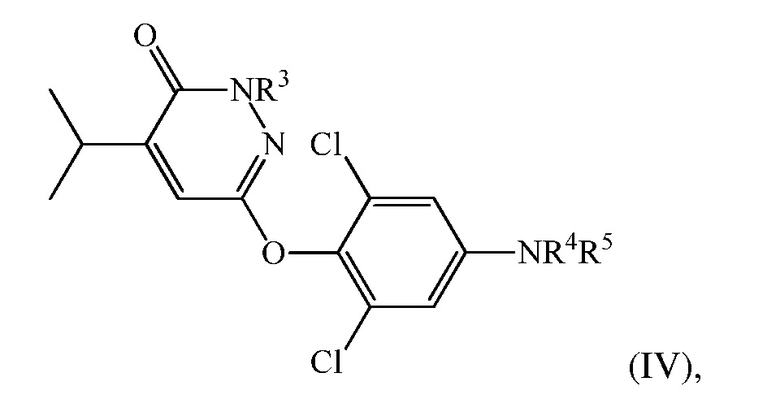
где
R3 представляет собой H или CH2Ra, в котором Ra представляет собой гидроксил, О-связанную аминокислоту, -OP(O)(OH)2 или -OC(O)-Rb, где Rb представляет собой низший алкил, алкокси, алкилкислоту, циклоалкил, арил, гетероарил или -(CH2)n-гетероарил, и n представляет собой 0 или 1;
R4 представляет собой H, и R5 представляет собой CH2COOH, C(O)CO2H, или их сложный эфир или амид, или R4 и R5 вместе представляют собой -N=C(Rc)-C(O)-NH-C(O)-; в котором Rc представляет собой H или циано.
В одном варианте осуществления соединение формулы (IV) представляет собой 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрил («соединение A») и вышеуказанную стадию проводят посредством контакта 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она с этил(2-цианоацетил)карбаматом и нитритом металла с последующей обработкой с помощью ацетата калия в DMAC.
В одном варианте осуществления способ дополнительно включает получение морфологической формы 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила («Соединения A») (формы I), характеризующейся картиной рентгеновской порошковой дифракции, включающей пики при 2θ приблизительно 10,5, 18,7, 22,9, 23,6 и 24,7 градусов.
В одном варианте осуществления, соединение формулы (IV) представляет собой формулу (V)
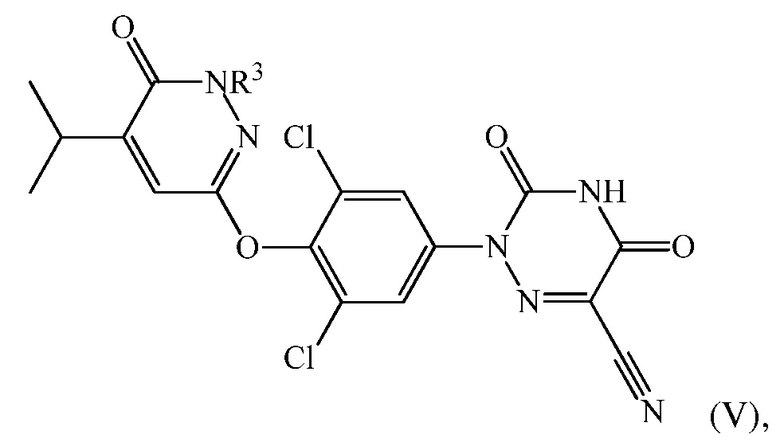
где R3 представляет собой CH2Ra, и стадию (d) проводят посредством контакта 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она с этил(2-цианоацетил)карбаматом с последующей обработкой с помощью ацетата калия в DMAC для получения 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила («Соединения A») и перевода соединения A в соединение формулы (V) подходящим образом, например, с использованием одного из способов, описанных в Патенте США 8076334.
В одном варианте осуществления соединение формулы (IV), например, 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрил («соединение A»), полученное способом, описанным в настоящем документе, обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, полученных способом, описанным в настоящем документе, отличных от соединения формулы (IV), таких как побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5% или менее 0,2%.
В одном варианте осуществления соединение формулы (IV), полученное способом, описанным в настоящем документе, представляет собой соединение A в форме I, и обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5%, или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, полученных способом, описанным в настоящем документе, отличных от соединения A, таких как побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5%, или менее 0,2%.
В одном варианте осуществления соединение формулы (IV) полученное способом, описанным в настоящем документе, представляет собой соединение A в форме I, и форма I обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, полученных способом, описанным в настоящем документе, отличных от формы I, таких как другие морфологические формы соединения A, побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5%, или менее 0,2%.
В одном варианте осуществления композиция, содержащая соединение формулы (IV), такое как соединение A, полученное способом, описанным в настоящем документе, имеет менее 1,5% (например, менее 1,0%, например, менее 0,5%) соответствующего региоизомера β-изопропилпиридазин-3(2H)-она (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, региоизомера соединения A β-изопропилпиридазин-3(2H)-она).
В одном варианте осуществления композиция, содержащая соединение формулы (IV), такое как соединение A, полученное способом, описанным в настоящем документе, является свободным от соответствующего региоизомера β-изопропилпиридазин-3(2H)-она (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6- дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, региоизомера соединения A β-изопропилпиридазин-3(2H)-она).
В одном варианте осуществления композиция, содержащая соединение формулы (IV), такое как соединение A, полученное способом, описанным в настоящем документе, имеет менее 1,5% (например, менее 0,1%) тяжелого металла, например, серебра.
В одном варианте осуществления композиция, содержащая соединение формулы (IV), такое как соединение A, полученное способом, описанным в настоящем документе, является свободным от тяжелого металла, например, серебра, золота или платины.
Способы синтеза, описанные в настоящем документе, предоставляют преимущества по сравнению с предшествующими способами, такими как способы, описанные в Патенте США 7452882. Например, общий выход 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-l,2,4-триазин-6-карбонитрила («соединения A») является сильно увеличенным (например, >40% по сравнению с ~9% при получении способом, описанным в Патенте США 7452882).
Также, региоселективность синтеза является гораздо лучшей. Кроме того, в новых способах предлагают более простую переработку, например, более простые фильтрации. Наконец, тяжелых металлов не используют в способах, описанных в настоящем документе для соединения A. В отличие от этого, серебро использовали в способе, описанном в Патенте США 7452882, что делало необходимым обработку для извлечения с помощью смолы.
В другом аспекте изобретение относится к композиции, содержащей более 85% соединения формулы (IV), менее 1,5% соответствующего региоизомера β-изопропилпиридазин-3(2H)-она (т.е., 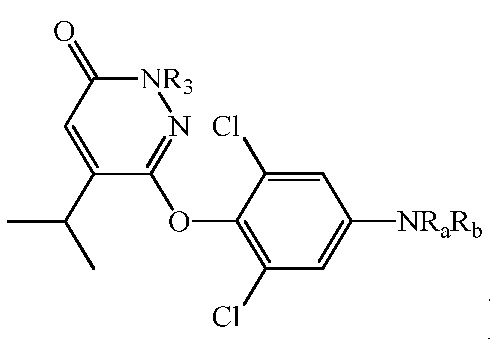 ), и/или менее 1,5% тяжелого металла.
), и/или менее 1,5% тяжелого металла.
В одном варианте осуществления соединение формулы (IV), например, соединение A, обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, содержащей соединение формулы (IV), отличных от соединения формулы (IV), таких как побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5% или менее 0,2%.
В одном варианте осуществления соединение формулы (IV) представляет собой соединение A в форме I, и обладает чистотой более 85%, например, более 86%, более 90%, более 20 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5%, или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, содержащей соединение A, отличных от соединение A, таких как побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5% или менее 0,2%.
В одном варианте осуществления соединение формулы (IV) представляет собой соединение A в форме I, и форма I обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%. Например, содержание примесей (т.е., любых компонентов композиции, содержащей форму I, отличных от формы I, таких как другие морфологические формы соединения A, побочные продукты, исходный материал, остатки растворителя, тяжелые металлы и т.д.) составляет менее 15%, менее 14%, менее 10%, менее 8%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1,5%, менее 1%, менее 0,8%, менее 0,5% или менее 0,2%.
В одном варианте осуществления соединение формулы (IV), такое как соединение A, имеет менее 1,5% (например, менее 1,0%, например, менее 0,5%) соответствующего региоизомера β-изопропилпиридазин-3(2H)-она (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, региоизомера соединения A β-изопропилпиридазин-3(2H)-она).
В одном варианте осуществления соединение формулы (IV), такое как соединение A, является свободным от соответствующего региоизомера β-изопропилпиридазин-3(2H)-она (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, региоизомера соединения A β-изопропилпиридазин-3(2H)-она).
В одном варианте осуществления соединение формулы (IV), такое как соединение A, имеет менее 1,5% (например, менее 1,0%, например, менее 0,5%) тяжелого металла, например, серебра, золота или платины.
В одном варианте осуществления соединение формулы (IV), такое как соединение A, полученное способом, описанным в настоящем документе, является свободным от тяжелого металла, например, серебра.
Кроме того, изобретение относится к морфологической форме 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила («соединения A») (формы I), характеризующейся картиной рентгеновской порошковой дифракции («XRPD»), включающей пики при 2θ приблизительно 10,5, 18,7, 22,9, 23,6 и 24,7 градусов.
В одном варианте осуществления форма I характеризуется картиной рентгеновской порошковой дифракции, дополнительно включающей пики при 2θ приблизительно 8,2, 11,2, 15,7 16,4, 17,7, 30,0 и 32,2 градусов.
В одном варианте осуществления форма I характеризуется картиной рентгеновской порошковой дифракции, включающей пики при 2θ приблизительно 8,2, 10,5, 18,7, 22,9, 23,6 и 24,7 градусов.
В одном варианте осуществления форма I характеризуется картиной рентгеновской порошковой дифракции, включающей пики при 2θ приблизительно 8,2, 10,5, 11,2, 15,7 16,4, 17,7, 18,7, 22,9, 23,6 и 24,7 градусов.
В одном варианте осуществления форма I характеризуется картиной рентгеновской порошковой дифракции, включающей пики при 2θ приблизительно 8,2, 10,5, 11,2, 15,7 16,4, 17,7, 18,7, 22,9, 23,6, 24,7, 30,0 и 32,2 градусов.
В одном варианте осуществления форма I характеризуется картиной рентгеновской порошковой дифракции, по существу сходной с картиной, приведенной на ФИГ. 1.
В другом аспекте в настоящем описании описан способ получения формы I. Способ включает смешивание образца, содержащего соединение A (например, либо неочищенного, либо очищенного препарата соединения A) с органическим растворителем, таким как спирт (например, этанол), кетон (например, метилизобутилкетон, т.е., MIBK), или водным раствором, включающим спирт или кетон. Например, полученную смесь (например, взвесь или суспензию), содержащую исходное соединение A и растворитель, нагревают до первой температуры, и затем охлаждают до второй температуры, которая ниже первой температуры. Предпочтительно, органический растворитель представляет собой этанол. Исходное соединение A, вступающее в перевод формы, может представлять собой сольват, такой как гидрат (например, моногидрат или дигидрат), или сольват органического растворителя (например, диметилацетамида, этанола или MIBK). Альтернативно, исходное соединение A может представлять собой ансольват (например, ангидрат).
В одном варианте осуществления способ осуществляют посредством нагревания соединение A с органическим растворителем до повышенной температуры (например, приблизительно 60-110°C или приблизительно 80°C) для получения взвеси или суспензии, с последующим охлаждением (например, до температуры приблизительно 0-60°C, приблизительно 40-60°C, приблизительно 45-55°C, или при приблизительно комнатной температуре) для получения формы I соединения A. Например, органический растворитель представляет собой этанол, и взвесь, содержащую соединение A, можно охлаждать до температуры более приблизительно 40°C для получения формы I. Например, органический растворитель представляет собой MIBK, и взвесь, содержащую соединение A, можно охлаждать до комнатной температуры для получения формы I.
В другом варианте осуществления суспензию в этаноле соединения A нагревают до повышенной температуры (например, приблизительно 80°C) и затем охлаждают до температуры не ниже приблизительно 40°C (например, приблизительно 45-55°C), фильтруют (например, при приблизительно 45-55°C), промывают нагретым (например, до 45-55°C) этанолом и высушивают, например, при 45-55°C для получения формы I соединения A, которая является в основном свободной от любого сольвата соединения A, такого как этанолсольват. Например, форма I соединения A, как получено, обладает содержанием этанолсольвата <5% (например, <2%, <1%, <0,5% или <0,1%).
В одном варианте осуществления способ дополнительно включает, после охлаждения смеси, фильтрацию смеси. Стадию фильтрации можно проводить при температуре между приблизительно 0°C и приблизительно 60°C (например, приблизительно 40-60°C, приблизительно 45-55°C, или при приблизительно комнатной температуре) для получения отфильтрованного осадка.
В одном варианте осуществления способ дополнительно включает, после фильтрации смеси, промывку отфильтрованного осадка. Стадию промывки можно проводить при температуре между приблизительно 0°C и приблизительно 60°C (например, приблизительно 40-60°C, приблизительно 45-55°C или при приблизительно комнатной температуре) с помощью органического растворителя (например, спирта, такого как этанол) для получения промытого отфильтрованного осадка.
В одном варианте осуществления способ дополнительно включает, после промывки отфильтрованного осадка, сушку промытого отфильтрованного осадка. Стадию сушки можно проводить при температуре между приблизительно 0°C и приблизительно 60°C (например, приблизительно 40-60°C, приблизительно 45-55°C, или при приблизительно комнатной температуре) для получения формы I соединения A.
В одном варианте осуществления форма I обладает чистотой более 91%, например, более 92,5%, более 95%, более 96%, более 97%, или более 97,5%.
В одном варианте осуществления форма I обладает чистотой более 98%, например, более 98,5%, более 99%, более 99,2%, более 99,5%, или более 99,8%.
В другом аспекте описание относится к соединениям, таким как
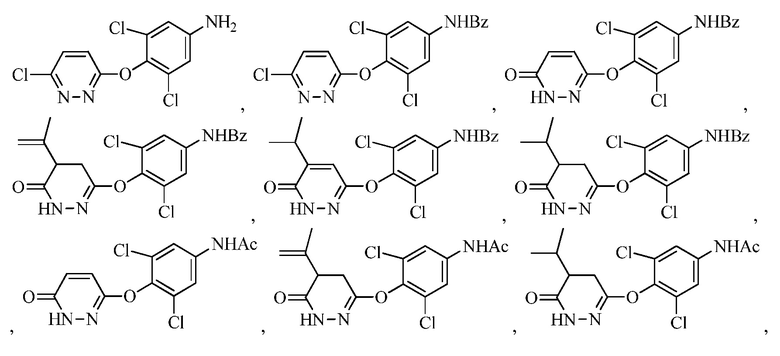
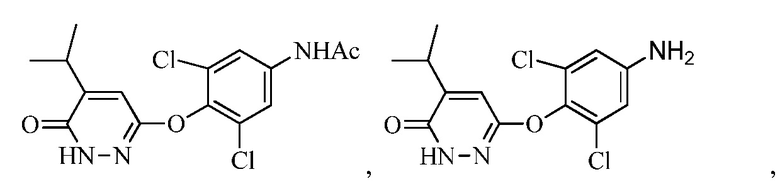 и к их солям, например, пригодным для синтеза 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7»).
и к их солям, например, пригодным для синтеза 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7»).
Описание относится также к способу лечения устойчивости к гормонам щитовидной железы (RTH) у нуждающегося в этом субъекта. Способ включает введение субъекту, имеющему по меньшей мере одну мутацию TRβ, терапевтически эффективного количества соединения формулы (IV):
где
R3 представляет собой H или CH2Ra, в которой Ra представляет собой гидроксил, О-связанную аминокислоту, -OP(O)(OH)2
или
-OC(O)-Rb, где Rb представляет собой низший алкил, алкокси, алкилкислоту, циклоалкил, арил, гетероарил или
-(CH2)n-гетероарил, и n представляет собой 0 или 1;
R4 представляет собой H, и R5 представляет собой CH2COOH, C(O)CO2H, или их сложный эфир или амид, или R4 и R5 вместе представляют собой -N=C(Rc)-C(O)-NH-C(O)-; в котором Rc представляет собой H или циано.
Устойчивость к гормонам щитовидной железы (RTH) представляет собой синдром, характеризующийся гипочувствительностью различных тканей к гормонам щитовидной железы, и вызван в первую очередь аутосомными доминантными мутациями до THRβ. См. Shi et al., Biochemistry 2005, 44, 4612-4626.
В одном варианте осуществления соединение, используемое в вышеуказанном способе, представляет собой 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрил («соединение A»), например, соединение A в форме I.
В одном варианте осуществления субъект, подлежащий лечению вышеописанным способом, страдает ожирением, гиперлипидемией, гиперхолестеринемией, диабетом, неалкогольным стеатогепатитом, жировой инфильтрацией печени, заболеванием костей, изменением тиреоидной оси, атеросклерозом, сердечно-сосудистым нарушением, тахикардией, гиперкинетическим поведением, гипотиреозом, зобом, расстройством дефицита внимания с гиперактивностью, нарушением обучаемости, задержкой в умственном развитии, потерей слуха, запаздывающим старением костей, неврологическим или психиатрическим заболеванием, или раком щитовидной железы.
В одном варианте осуществления мутация THRβ выбрана из группы, состоящей из замены треонином (T) остатка аланина (A) дикого типа в положении аминокислоты 234 из SEQ ID NO: 1 (A234T); замены глутамином (Q) остатка аргинина (R) дикого типа в положении аминокислоты 243 из SEQ ID NO: 1 (R243Q); замены гистидином (H) остатка аргинина (R) дикого типа в положении аминокислоты 316 из SEQ ID NO: 1 (R316H); и замены треонином (T) остатка аланина (A) дикого типа в положении аминокислоты 317 из SEQ ID NO: 1 (A317T). В другом варианте осуществления соединение, используемое в способе, восстанавливает активность мутантного THRβ.
В одном варианте осуществления чистоты соединения формулы (IV), такого как соединение A, достигают посредством ресуспендирования неочищенного соединения из пригодного растворителя, описанного в настоящем документе. В другом варианте осуществления соединение не является сольватом (например, гидратом).
В одном варианте осуществления соединение формулы (IV), например, соединение A, обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%.
В одном варианте осуществления соединение формулы (IV) представляет собой соединение A в форме I, и обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%.
В одном варианте осуществления соединение формулы (IV) представляет собой соединение A в форме I, и форма I обладает чистотой более 85%, например, более 86%, более 90%, более 92,5%, более 95%, более 96%, более 97%, более 97,5%, более 98%, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%.
В одном варианте осуществления соединение формулы (IV), такое как соединение A, имеет менее 1,5% (например, менее 1,0%, например, менее 0,5%) соответствующего β-изопропилпиридазин-3(2H)-онового региоизомера (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, β-изопропилпиридазин-3(2H)-онового региоизомера соединения A).
В одном варианте осуществления соединение формулы (IV), такое как соединение A, является свободным от соответствующего β-изопропилпиридазин-3(2H)-онового региоизомера (например, 2-(3,5-дихлор-4-((4-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила, β-изопропилпиридазин-3(2H)-онового региоизомера соединения A).
В одном варианте осуществления соединение формулы (IV), такое как соединение A, имеет менее 1,5% (например, менее 1,0%, например, менее 0,5%) тяжелого металла, например, серебра, золота или платины.
В одном варианте осуществления субъект представляет собой млекопитающее. В другом варианте осуществления субъект представляет собой человека.
Описание, кроме того, относится к способу определения способности субъекта отвечать на соединение формулы (IV) или его фармацевтически приемлемую соль, где способ включает:
(a) получение образца от субъекта; и
(b) детекцию мутации в рецепторе гормонов щитовидной железы («TR»), где присутствие мутации указывает на то, что субъект является отвечающим на соединения или их фармацевтически приемлемые соли.
В одном варианте осуществления соединение формулы (IV) представляет собой 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрил («соединение A»).
В одном варианте осуществления TR представляет собой TRβ.
В одном варианте осуществления субъект, подвергаемый лечению способом по этому изобретению, страдает ожирением, гиперлипидемией, гиперхолестеринемией, диабетом, неалкогольным стеатогепатитом, жировой инфильтрацией печени, заболеванием костей, изменением тиреоидной оси, атеросклерозом, сердечно-сосудистым нарушением, тахикардией, гиперкинетическим поведением, гипотиреозом, зобом, расстройством дефицита внимания с гиперактивностью, нарушением обучаемости, задержкой в умственном развитии, потерей слуха, запаздывающим старением костей, неврологическим или психиатрическим заболеванием или раком щитовидной железы.
В одном варианте осуществления способ для определения способности отвечать на соединение формулы (IV) можно использовать вместе со способом для лечения устойчивости к гормонам щитовидной железы. То есть, перед лечением, субъекта тестируют для определения способности отвечать на соединение.
Другие признаки и преимущества настоящего изобретения очевидны из подробного описания, примеров и формулы изобретения.
Краткое описание рисунков
Фигура 1 представляет собой рентгеновскую порошковую дифрактограмму (XRPD) 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила («Соединения A»), формы I.
Фигура 2 представляет собой диаграмму дифференциальной сканирующей калориметрии (DSC) соединения A, формы I.
Фигуры 3А и 3B представляют собой изображения моделирования в MacPymol для показа T3 и соединения A в THRβ, соответственно.
Фигура 4 представляет собой изображение моделирования в MacPymol для показа наложенных T3 и соединения A в THRβ.
Фигура 5A представляет собой изображение моделирования в MacPymol для показа полярных взаимодействий между T3 и THRβ дикого типа, где T3 взаимодействует с Arg320 очень специфически.
Фигура 5B представляет собой изображение моделирования в MacPymol для показа полярных взаимодействий между соединением A и THRβ дикого типа, где соединение A взаимодействует с Arg320 и Arg316.
Фигура 6 представляет собой изображение моделирования в MacPymol для показа того, что мутации приводят к множеству изменений в полярной области связывающего лиганд домена («LBD»).
Фигура 7A представляет собой изображение моделирования в MacPymol для показа взаимодействий между T3 и мутантами THRβ: Ala234Thr, Arg243Gln, Arg316His, Ala317Thr.
Фигура 7B представляет собой изображение моделирования в MacPymol для показа взаимодействий между соединением A и мутантами THRβ: Ala234Thr, Arg243Gln, Arg316Flis, Ala317Thr; указывающее на то, что, по сравнению с T3, отрицательно заряженный гетероцикл в соединении A вмещает мутации лучше.
Фигуры 8A и 8B представляют собой изображения моделирования в MacPymol T3 и соединения A в мутанте Arg316His, соответственно. Взаимодействие T3-Arg320, вероятно, является более слабым из-за поворота Arg320 от лиганда в мутанте, в то время как соединение A сохраняет обеспечивающее преимущество взаимодействие с Arg320 и хорошо расположено для того, чтобы группа CN формировала пи-катионное взаимодействие с мутантным His316.
Фигуры 9A и 9B представляют собой изображения моделирования в MacPymol соединения A в WT THRβ и мутантном Arg316His, соответственно.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как используют в описании и прилагаемой формуле изобретения, формы единственного числа «a», «an» и «the» включают объекты ссылки во множественном числе, если контекст явно не требует иного. Таким образом, например, ссылка на «реагент» включает не только один реагент, но также комбинацию или смесь из двух или более различных реагентов, ссылка на «заместитель» включает один заместитель, так же как два или более заместителей, и т.п.
Как применяют в настоящем документе, фразы «например», «для примера», «такой как» или «включая» предназначены для введения примеров, которые дополнительно проясняют более общий объект. Эти примеры представлены только в качестве вспомогательного средства для понимания описания, и не предназначены для ограничения каким-либо образом. Более того, как применяют в настоящем документе, термины «можно», «возможно», «необязательно» или «можно, необязательно» означают, что описанное затем условие может возникать или может не возникать, так что описание включает случаи, когда условие возникает, и случаи, когда оно не возникает. Например, фраза «необязательно присутствует» означает, что объект может присутствовать или может не присутствовать, и, таким образом, описание включает случаи, когда объект присутствует, и случаи, когда объект не присутствует.
В описании и формуле настоящего изобретения, следующую терминологию используют в соответствии с определениями, указанными ниже.
Как применяют в настоящем документе, сокращение «TR» или «THR» относится к рецептору гормонов щитовидной железы. Нуклеиновые кислоты и полипептиды TR из различных видов (например, человека, крысы, курицы и т.д.) описаны ранее. См., например, R. L. Wagner et al. (2001), Molecular Endocrinology 15(3): 398-410; J. Sap et al. (1986), Nature 324:635-640; C. Weinberger et al. (1986), Nature 324:641-646; и C.C.Tompson et al. (1986), Science 237:1610-1614; полное содержание каждого из которых приведено в настоящем документе в качестве ссылки. Аминокислотная последовательность TRβ человека представлена, например, No. доступа в Genbank P10828.2, содержание которого приведено в настоящем документе в качестве ссылки.
Остатки в положениях 234, 243, 316 и 317 TRβ человека подчеркнуты в SEQ ID NO: 1. Часть нуклеотидной последовательности TRβ человека, кодирующая вышеуказанную аминокислотную последовательность, представляет собой SEQ ID NO: 2. Нуклеотидная последовательность TRβ человека представлена, например, посредством No. доступа в Genbank NM_000461.4, содержание которого приведено в настоящем документе в качестве ссылки.
Как применяют в настоящем документе, фраза «имеющий формулу» или «имеющий структуру» не предназначена, чтобы являться ограничивающей, и ее используют таким же образом, как обычно используют термин «содержащий». Термин «независимо выбранный из» применяют в настоящем документе для обозначения того, что перечисленные элементы, например, группы R или т.п., могут являться одинаковыми или различными.
Термин «алкил», как применяют в настоящем документе, относится к разветвленной или неразветвленной насыщенной углеводородной группе, как правило, хотя не обязательно, содержащей от 1 до приблизительно 24 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, т-бутил, октил, децил и т.п., так же как к циклоалкильным группам, таким как циклопентил, циклогексил и т.п. Как правило, хотя не обязательно, алкильные группы в настоящем документе могут содержать от 1 до приблизительно 18 атомов углерода, и такие группы могут содержать от 1 до приблизительно 12 атомов углерода. Термин «низший алкил» подразумевает алкильную группу от 1 до 6 атомов углерода, например, 1, 2, 3, 4, 5 или 6 атомов углерода. «Замещенный алкил» относится к алкилу, замещенному одной или несколькими группами заместителей, и термины «содержащий гетероатом алкил» и «гетероалкил» относятся к алкильному заместителю, в котором по меньшей мере один атом углерода заменен на гетероатом, как более подробно описано ниже.
Термин «алкенил», как применяют в настоящем документе, относится к линейной, разветвленной или циклической углеводородной группе из от 2 до приблизительно 24 атомов углерода, содержащей по меньшей мере одну двойную связь, такой как этенил, н-пропенил, изопропенил, н-бутенил, изобутенил, октенил, деценил, тетрадеценил, гексадеценил, эйкозенил, тетракозенил и т.п. Как правило, хотя снова не обязательно, алкенильные группы в настоящем документе содержат от 2 до приблизительно 18 атомов углерода, и например, могут содержать от 2 до 12 атомов углерода. Термин «низший алкенил» подразумевает алкенильную группу из 2-6 атомов углерода.
Термин «замещенный алкенил» относится к алкенилу, замещенному одной или несколькими группами заместителя, и термины «содержащий гетероатом алкенил» и «гетероалкенил» относятся к алкенилу, в котором по меньшей мере один атом углерода заменен на гетероатом, например, N, P, O или S.
Термин «алкинил», как применяют в настоящем документе, относится к линейной или разветвленной углеводородной группе из 2-24 атомов углерода, содержащей по меньшей мере одну тройную связь, такой как этинил, н-пропинил и т.п. Как правило, хотя снова не обязательно, алкинильные группы в настоящем документе могут содержать от 2 до приблизительно 18 атомов углерода, и такие группы могут далее содержать 2-12 атомов углерода. Термин «низший алкинил» подразумевает алкинильную группу из 2-6 атомов углерода. Термин «замещенный алкинил» относится к алкинилу, замещенному одной или несколькими группами заместителей, и термины «содержащий гетероатом алкинил» и «гетероалкинил» относятся к алкинилу, в которой по меньшей мере один атом углерода заменен на гетероатом.
Термин «алкокси», как применяют в настоящем документе, подразумевает алкильную группу, связанную через одну концевую простую эфирную связь; то есть, группу «алкокси» можно представить как -O-алкил, где алкил является таким, как определено выше. Группа «низший алкокси» подразумевает группу алкокси, содержащую 1-6 атомов углерода, и включает в себя, например, метокси, этокси, н-пропокси, изопропокси, т-бутилокси и т.д. Заместители, определенные как «C1-C6 алкокси» или «низший алкокси» в настоящем документе, могут, например, содержать 1-3 атома углерода, и в качестве дополнительного примера, такие заместители могут содержать 1 или 2 атома углерода (т.е., метокси и этокси).
Термин «алкилкислота» относится к кислому заместителю, который находится на алкильной группе, такому как -(CH2)OCOOH, в котором о представляет собой целое число между 1 и 6. Алкильная группа может являться либо линейной, либо разветвленной.
Термин «арил», как применяют в настоящем документе, и если не указано иначе, относится к ароматическому заместителю, как правило, хотя не обязательно, содержащему 5-30 атомов углерода и содержащему одно ароматическое кольцо или несколько ароматических колец, конденсированных вместе, связанных напрямую или связанных опосредованно (так что различные ароматические кольца связаны с общей группой, такой как группа метилена или этилена). Арильные группы могут, например, содержать 5-20 атомов углерода, и в качестве дополнительного примера, арильные группы могут содержать 5-12 атомов углерода. Например, арильные группы могут содержать одно ароматическое кольцо или два конденсированных или связанных ароматических кольца, например, фенил, нафтил, бифенил, дифениловый эфир, дифениламин, бензофенон и т.п. «Замещенный арил» относится к арильной группе, замещенной одной или несколькими группами заместителя, и термины «содержащий гетероатом арил» и «гетероарил» относятся к арильному заместителю, в котором по меньшей мере один атом углерода заменен на гетероатом, как более подробно описано ниже. Если не указано иначе, термин «арил» включает в себя кольца, которые являются незамещенными, замещенными и/или имеют содержащие гетероатом ароматические заместители.
Термин «аралкил» относится к алкильной группе с арильным заместителем, и термин «алкарил» относится к арильной группе с алкильным заместителем, где «алкил» и «арил» являются такими, как определено выше. Как правило, аралкильные и алкарильные группы в настоящем документе содержат 6-30 атомов углерода. Аралкильные и алкарильные группы могут, например, содержать 6-20 атомов углерода, и в качестве дополнительного примера, такие группы могут содержать 6-12 атомов углерода.
Термин «амино» применяют в настоящем документе для обозначения группы -NZ1Z2, где Z1 и Z2 представляют собой водород или неводородные заместители, где неводородные заместители включают в себя, например, алкил, арил, алкенил, аралкил и их замещенные и/или содержащие гетероатом варианты.
Термины «гало» и «галоген» используют в общепринятом смысле для обозначения заместителя хлора, брома, фтора или иода.
Термин «содержащий гетероатом», как в «содержащей гетероатом алкильной группе» (называемой также «гетероалкильной» группой) или «содержащей гетероатом арильной группой» (называемой также «гетероарильной» группой) относится к молекуле, связи или заместителю, в которых один или несколько атомов углерода заменены на атом, отличный от углерода, например, азот, кислород, серу, фосфор или кремний, как правило, азот, кислород или серу. Сходным образом, термин «гетероалкил» относится к алкильному заместителю, который является содержащим гетероатом, термин «гетероцикл» или «гетероциклический» относится к циклической группе, которая является содержащей гетероатом, термины «гетероарил» и «гетероароматический», соответственно, относятся к «арильным» и «ароматическим» заместителям, которые являются содержащими гетероатом, и т.п. Примеры гетероалкильных групп включают алкоксиарил, замещенный алкилсульфанилом алкил, N-алкилированный аминоалкил, и т.д. Примеры гетероарильных заместителей включают пирролил, пирролидинил, пиридинил, хинолинил, индолил, фурил, пиримидинил, имидазолил, 1,2,4-триазолил, тетразолил и т.д., и примерами содержащих гетероатом алициклических групп являются пирролидинo, морфолино, пиперазинo, пиперидинo, тетрагидрофуранил и т.д.
«Гидрокарбил» относится к одновалентным радикалам гидрокарбила, содержащим от 1 до приблизительно 30 атомов углерода, включая от 1 до приблизительно 24 атомов углерода, дополнительно включая от 1 до приблизительно 18 атомов углерода, и дополнительно включая приблизительно 1-12 атомов углерода, включая линейные, разветвленные, циклические, насыщенные и ненасыщенные соединения, такие как алкильные группы, алкенильные группы, арильные группы и т.п. «Замещенный гидрокарбил» относится к гидрокарбилу, замещенному одной или несколькими группами заместителя, и термин «содержащий гетероатом гидрокарбил» относится к гидрокарбилу, в котором по меньшей мере один атом углерода заменен на гетероатом.
Термин «О-связанная аминокислота» обозначает любую аминокислоту, природную или синтетическую, связанную с молекулой через кислород карбоксильной группы аминокислоты, предпочтительно, через карбоксильную группу карбокси-конца аминокислоты.
Как применяют в настоящем документе, термин «защитная группа» означает, что конкретная функциональная группа, например, O, S, или N, временно блокирована, так что реакцию можно проводить избирательно в другом реакционноспособном участке многофункционального соединения. В предпочтительных вариантах осуществления защитная группа вступает в реакцию избирательно с хорошим выходом для получения защищенного субстрата, являющегося устойчивым к защищенным реакциям; защитная группа должна избирательно удаляться с хорошим выходом посредством легко доступных, предпочтительно, нетоксичных реагентов, не атакующих другие функциональные группы; защитная группа образует легко отделяемое производное (более предпочтительно, без образования новых стереогенных центров); и защитная группа обладает минимумом дополнительной функциональности во избежание дополнительных участков реакции. Как подробно описано в настоящем документе, можно использовать защитные группы для кислорода, серы, азота и углерода. Например, в конкретных вариантах осуществления, можно использовать конкретные иллюстративные защитные группы для кислорода. Эти защитные группы для кислорода включают, но без ограничения, метиловые эфиры, замещенные метиловые эфиры (например, MOM (метоксиметиловый эфир), MTM (метилтиометиловый эфир), BOM (бензилоксиметиловый эфир), и PMBM (п-метоксибензилоксиметиловый эфир)), замещенные этиловые эфиры, замещенные бензиловые эфиры, силиловые эфиры (например, TMS (триметилсилиловый эфир), TES (триэтилсилиловыйэфир), TIPS (триизопропилсилиловый эфир), TBDMS (т-бутилдиметилсилиловый эфир), трибензилсилиловый эфир и TBDPS (т-бутилдифенилсилиловый эфир), сложные эфиры (например, формат, ацетат, бензоат (Bz), трифторацетат и дихлорацетат), карбонаты, циклические ацетали и кетали. В других конкретных иллюстративных вариантах осуществления используют защитные группы для азота. Защитные группы для азота, так же как способы защиты и снятия защиты, известны в данной области. Защитные группы для азота включают, но без ограничения, карбаматы (включая метил-, этил- и замещенные этилкарбаматы (например, Troc), амиды, циклические производные имидов, N-алкил- и N-ариламины, производные иминов и производные енамина. В других вариантах осуществления можно использовать конкретные иллюстративные защитные группы для серы. Защитные группы для серы включают в себя, но без ограничения, защитные группы для кислорода, описанные выше, так же как алифатическую карбоновую кислоту (например, акриловую кислоту), малеинимид, винилсульфонил и, необязательно, замещенную малеиновую кислоту. Другие конкретные иллюстративные защитные группы подробно описаны в настоящем документе, однако, следует понимать, что настоящее изобретение не предназначено для ограничения этими защитными группами; наоборот, множество дополнительных эквивалентных защитных групп можно легко идентифицировать с использованием вышеуказанных критериев и использовать по настоящему изобретению. Кроме того, множество защитных групп описано в «Protective Groups in Organic Synthesis» Third Ed. Greene, T.W. and Wuts, P.G., Eds., John Wiley & Sons, New York: 1999, полное содержание которого таким образом приведено в настоящем документе в качестве ссылки.
Под «замещенным», как в «замещенный гидрокарбил», «замещенный алкил», «замещенный арил» и т.п., как упомянуто в некоторых из вышеуказанных определений, понимают, что в гидрокарбиле, алкиле, ариле или другой группе, по меньшей мере один атом водорода, связанный с атомом углерода (или другим), заменен на один или несколько неводородных заместителей. Примеры таких заместителей включают, без ограничения, функциональные группы и гидрокарбиловые группы C1-C24 алкил (включая C1-C18 алкил, дополнительно включая C1-C12 алкил и дополнительно включая C1-C6 алкил), C2-C24 алкенил (включая C2-C18 алкенил, дополнительно включая C2-C12 алкенил и дополнительно включая С2-С6 алкенил), C2-C24 алкинил (включая C2-C18 алкинил, дополнительно включая C2-C12 алкинил и дополнительно включая С2-С6 алкинил), C5-C30 арил (включая C5-C20 арил и дополнительно включая C5-C12 арил), и С6-С30 аралкил (включая С6-С20 аралкил и дополнительно включая C6-C12 аралкил).
Под «функциональной группой», как упомянуто в некоторых из вышеуказанных определений, понимают неводородную группу, содержащую одну или несколько неуглеводородных функциональных групп. Примеры функциональных групп включают, без ограничения: гало, гидроксил, сульфгидрил, C1-C24 алкокси, C2-C24 алкенилокси, C2-C24 алкинилокси, C5-C20 арилокси, ацил (включая C2-C24 алкилкарбонил (-CO-алкил) и С6-С20 арилкарбонил (-CO-арил)), ацилокси (-O-ацил), C2-C24 алкоксикарбонил (-(CO)-O-алкил), С6-С20 арилоксикарбонил (-(CO)-O-арил), галокарбонил (-CO)-X, где X представляет собой гало), C2-C24 алкилкарбонато (-O-(CO)-O-алкил), С6-С20 арилкарбонато (-O-(CO)-O-арил), карбокси (-COOH), карбоксилато (-COO-), карбамоил (-(CO)-NH2), монозамещенный C1-C24 алкилкарбамоил (-(CO)-NH(C1-C24 алкил)), двузамещенный алкилкарбамоил (-(CO)-N(C1-C24 алкил)2), монозамещенный арилкарбамоил (-(CO)-NH-арил), тиокарбамоил (-(CS)-NH2), карбамидо (-NH-(CO)-NH2), циано (-C≡N), изоциано (-N+≡C-), цианато (-O-C≡N), изоцианато (-O-N+≡C-), изотиоцианато (-S-C≡N), азидо (-N=N+=N»), формил (-(CO)-H), тиоформил (-(CS)-H), амино (-NH2), моно- и ди-(C1-C24 алкил)-замещенный амино, моно- и ди-(C5-C20 арил)-замещенный амино, C2-C24 алкиламидо (-NH-(CO)-алкил), C5-C20 ариламидо (-NH-(CO)-арил), имино (-CR=NH, где R=водород, C1-C24 алкил, C5-C20 арил, С6-С20 алкарил, С6-С20 аралкил и т.д.), алкилимино (-CR=N(алкил), где R=водород, алкил, арил, алкарил и т.д.), арилимино (-CR=N(арил), где R=водород, алкил, арил, алкарил и т.д.), нитро (-NO2), нитрозо (-NO), сульфо (-SO2-OH), сульфонато (-SO2-O-), C1-C24 алкилсульфанил (-S-алкил; называемый также «алкилтио»), арилсульфанил (-S-арил; называемый также «арилтио»), C1-C24 алкилсульфинил (-(SO)-алкил), C5-C20 арилсульфинил (-(SO)-арил), C1-C24 алкилсульфонил (-SO2-алкил), C5-C20 арилсульфонил (-SO2-арил), фосфоно (-Р(O)(ОН)2), фосфонато (-Р(O)(O-)2), фосфинато (-P(O)(O-)), фосфо (-PO2) и фосфино (-PH2), моно- и ди-(C1-C24 алкил)-замещенный фосфино, моно- и ди-(C5-C20 арил)-замещенный фосфино; и гидрокарбиловые группы C1-C24 алкил (включая C1-C18 алкил, дополнительно включая C1-C12 алкил и дополнительно включая C1-С6 алкил), C2-C24 алкенил (включая C2-C18 алкенил, дополнительно включая C2-C12 алкенил и дополнительно включая С2-С6 алкенил), C2-C24 алкинил (включая C2-C18 алкинил, дополнительно включая C2-C12 алкинил и дополнительно включая С2-С6 алкинил), C5-C30 арил (включая C5-C20 арил и дополнительно включая C5-C12 арил), и С6-С30 аралкил (включая С6-С20 аралкил и дополнительно включая C6-C12 аралкил). Кроме того, вышеупомянутые функциональные группы можно, если конкретная группа позволяет, далее замещать одной или несколькими функциональными группами или одной или несколькими гидрокарбиловыми группами, такими как группы, конкретно перечисленные выше. Аналогично, вышеупомянутые гидрокарбиловые группы можно далее замещать одной или несколькими функциональными группами или дополнительными гидрокарбиловыми группами, такими как конкретно перечисленные группы.
Термин «экономизация способа» относится к сжатию многоступенчатого способа до меньшего количества стадий или элементарных операций. Элементарная операция включает преобразования, но также включает стадии обработки и выделения. Центрифугирование, фильтрация, дистилляция, декантация, преципитация/кристаллизация и упаковка являются примерами элементарных операций. Существует большое количество примеров экономизации и других улучшений способов в литературе (см., например, J. Org. Chem., 2007, 72, 9757-9760).
Следует понимать, что некоторые из вышеупомянутых определений могут перекрываться, так что некоторые химические группы могут попадать под более чем одно определение.
Когда термин «замещенный» появляется перед списком возможных замещенных групп, подразумевают, что термин применяют к каждому члену этой группы. Например, фразу «замещенный алкил и арил» следует интерпретировать как «замещенный алкил и замещенный арил».
Описание относится к способам синтеза соединения, например, соединения, пригодного в качестве промежуточного соединения для синтеза пиридазиноновых соединений в качестве аналогов гормонов щитовидной железы. Пиридазиноновые соединения в качестве аналогов гормонов щитовидной железы, так же как их пролекарства, описаны, например, в Патентах США 7452882, 7807674 и 8076334.
В частности, изобретение относится к способу получения 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7») или его соли, где способ включает:
(a) контакт R1MgX или R1Li с соединением формулы (I):
для получения соединения формулы (II):
в котором R1 представляет собой изопропил или изопропенил, X представляет собой гало, и R2 представляет собой H или защитную группу для амина; и
(b) перевод соединения формулы (II) в соединение формулы (III):
в присутствии основания, когда R1 представляет собой изопропенил или в присутствии окисляющего средства, когда R1 представляет собой изопропил.
В настоящем описании описан также способ синтеза пиридазиноновых соединений в качестве аналогов гормонов щитовидной железы, так же как их пролекарств. Такие соединения включают соединения, описанные в Патентах США 7452882, 7807674 и 8076334. В частности, в описании описан способ получения соединения формулы (IV) или его фармацевтически приемлемой соли:
где
R3 представляет собой H или CH2Ra, в котором Ra представляет собой гидроксил, О-связанную аминокислоту, -OP(O)(OH)2
или
-OC(O)-Rb, где Rb представляет собой низший алкил, алкокси, алкилкислоту, циклоалкил, арил, гетероарил, или
-(CH2)n-гетероарил, и n представляет собой 0 или 1;
R4 представляет собой H, и R5 представляет собой CH2COOH, C(O)CO2H, или их сложный эфир или амид, или R4 и R5 вместе представляют собой -N=C(Rc)-C(O)-NH-C(O)-; в котором Rc представляет собой H или циано. Способ включает:
(a) контакт R1MgX или R1Li с соединением формулы (I):
для получения соединения формулы (II):
в котором R1 представляет собой изопропил или изопропенил, X представляет собой гало, и R2 представляет собой H или защитную группу для амина; и
(b) перевод соединения формулы (II) в соединение формулы (III):
в присутствии основания, когда R1 представляет собой изопропенил, или в присутствии брома и кислоты, когда R1 представляет собой изопропил,
(с) удаление защитной группы для амина R2 соединения формулы (III), когда присутствует, для получения 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она;
и, необязательно,
(d) перевод 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она в соединение формулы (IV) в подходящих условиях.
Настоящее изобретение также относится к подробным способам синтеза различных описанных соединений по настоящему изобретению в соответствии со следующими схемами, и как показано в примерах.
На протяжении описания, когда композиции описаны как имеющие, включающие или содержащие конкретные компоненты, предусматривают, что композиции также состоят в основном из, или состоят из перечисленных компонентов. Подобным образом, когда методы или способы описаны как имеющие, включающие или содержащие конкретные стадии способа, способы также состоят в основном из, или состоят из перечисленных стадий способа. Кроме того, следует понимать, что порядок стадий или порядок проведения конкретных действий является несущественным, при условии, что изобретение остается осуществимым. Более того, две или более стадии или действия можно проводить одновременно.
Способы синтеза по изобретению могут являться устойчивыми к широкому множеству функциональных групп, таким образом, можно использовать различные замещенные исходные материалы. Способы, как правило, предоставляют желательное конечное соединение в конце или ближе к концу общего процесса, хотя может быть желательным в конкретных случаях далее переводить соединение в его фармацевтически приемлемую соль, сложный эфир или пролекарство.
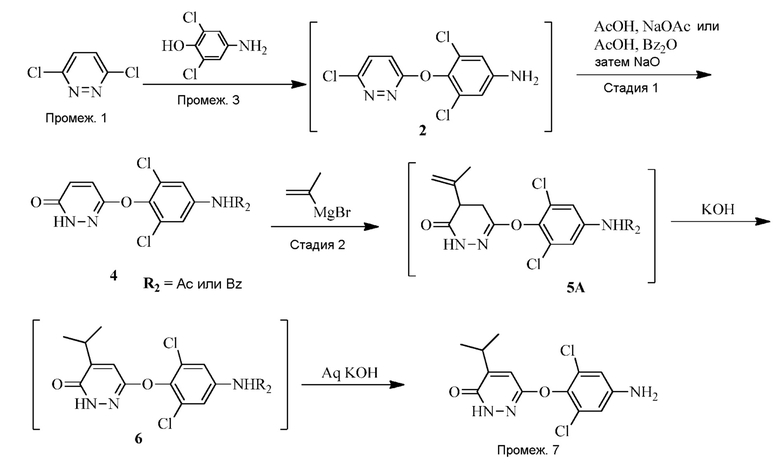
В вариантах осуществления 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-он («Промеж. 7») получают в соответствии со схемой 1 или 2 ниже.
Схема 1: Синтез 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она (Промеж. 7) с изопропиловым реагентом Гриньяра (iPrMgX).
Схема 1
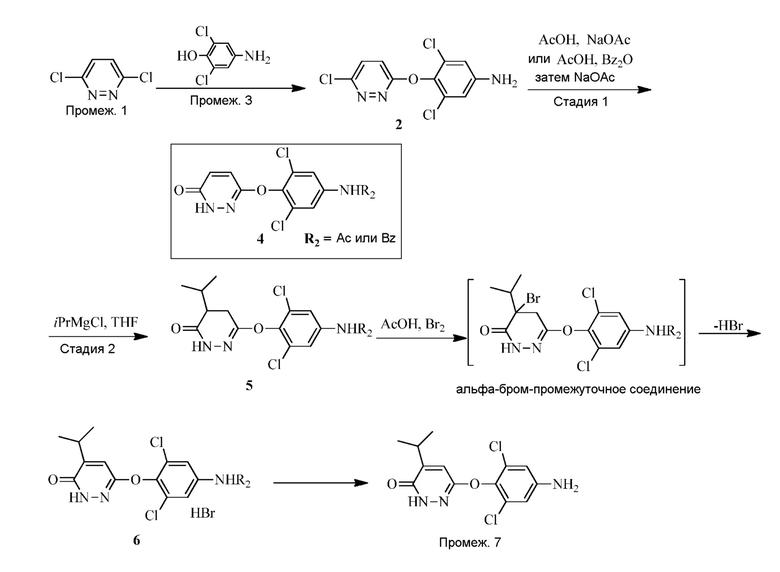
Схема 2: Синтез 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она (Промеж. 7) с изопропиловым реагентом Гриньяра.
Схема 2
Стадия 1: Синтез 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина (соединения 2) и N-(3,5-дихлор-4-((6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)бензамида или N-(3,5-дихлор-4-((6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)ацетамида (соединения 4)
Соединение 2 получают посредством контакта 3,6-дихлорпиридазина с 2,6-дихлор-4-аминофенолом в присутствии небольшого количества пригодного основания, такого как карбонат металла (например, карбонат цезия или калия) или алкоксид металла (например, т-бутоксид калия) в пригодном органическом растворителе (например, DMSO или DMAC) при подходящей температуре реакции (например, 60-120°C) до завершения реакции, как правило, приблизительно 3-30 часов, например, приблизительно 3-15 часов.
Соединение 4 получают посредством защиты соединения 2 с помощью пригодного реагента для защиты амина (такого как бензойный ангидрид или бензойный хлорид) с последующей обработкой защищенного промежуточного соединения с помощью ацетата натрия в присутствии пригодного органического растворителя (такого как уксусная кислота) при подходящей температуре реакции (например, 100-120°C) до завершения реакции, как правило, приблизительно 2-20 часов, например, приблизительно 5-15 часов. Неочищенный продукт очищают с помощью пригодного растворителя (например, смеси воды и уксусной кислоты) при пригодной температуре (например, 88-100°C). Защищенное ацетатом соединение 4 можно получать подверганием соединения 2 воздействию гидролизующих условий.
Стадия 2: Синтез N-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)бензамида или N-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)ацетамида (соединения 6) и 6-(4-амино-2,6-дихлорфенокси)-4- изопропилпиридазин-3(2H)-она (Промеж. 7)
Соединение 6 получают посредством контакта соединения 4 с изопропиловым реагентом Гриньяра в пригодном органическом растворителе (таком как тетрагидрофуран или диоксан) с последующей стадией окисления. Стадию окисления можно проводить в присутствии окисляющего реагента, такого как бром, в пригодном органическом растворителе, таком как уксусная кислота, при подходящей температуре реакции (например, 60-90°C) до завершения реакции, как правило, приблизительно 2-10 часов, например, приблизительно 2-5 часов.
Следует понимать, что реакция снятия защиты является необходимой для завершения трансформации соединения 6 до промеж. 7. В частности, N-защитную группу (т.е., ацетил или бензоил) необходимо удалять для получения свободного амино, присутствующего на промеж. 7. Таким образом, в одном варианте осуществления, промеж. 7 получают посредством снятия защиты соединения 6 (где R2 представляет собой Bz) с помощью основания, такого как гидроксид металла (например, KOH или NaOH) или карбонат металла (например, карбонат натрия). В другом варианте осуществления, промеж. 7 получают посредством снятия защиты соединения 6 (где R2 представляет собой Ac) с помощью кислоты, такой как трифторуксусная кислота.
Альтернативно, соединение 7 получают посредством контакта соединения 4 с изопропениловым реагентом Гриньяра в пригодном органическом растворителе (таком как тетрагидрофуран или 2-метил-THF) с последующей изомеризацией (например, из 5A до 6) и снятием защиты посредством обработки основанием, таким как гидроксид металла (например, KOH). Стадию изомеризация/снятия защиты проводят при подходящей температуре реакции (например, 60-90°C) до завершения реакции, как правило, приблизительно 10-60 часов, например, приблизительно 16 часов при 90°C.
Реакцию Гриньяра можно проводить в присутствии кислоты Льюиса, такой как LiCl или LiBr, при подходящей температуре реакции (например, от комнатной температуры до 40°C) до завершения реакции, как правило, приблизительно 2-10 часов, например, приблизительно 2-5 часов.
В вариантах осуществления синтез соединения 5 или 5A приводит к улучшенному выходу промеж. 7 по сравнению с другими способами, известными в данной области. Например, синтез 5 или 5A приводит к выходу более 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85% или более 90%.
В вариантах осуществления реакция Гриньяра улучшает региоселективность, приводящую к значительно меньшему количеству β-изопропилового региоизомера соединения 6, т.е.,
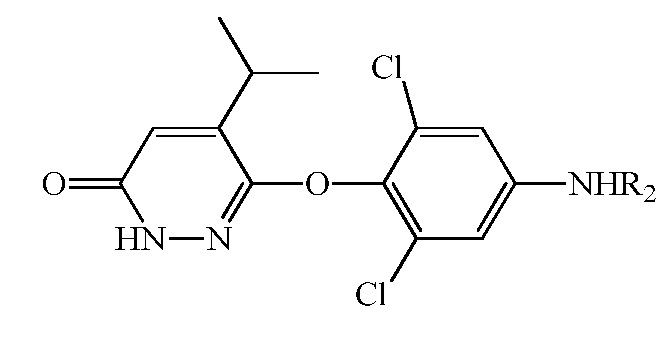 и таким образом, к более чистому промеж. 7.
и таким образом, к более чистому промеж. 7.
В одном варианте осуществления, перевод 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7») в соединение A осуществляют в соответствии со схемой 3 ниже.
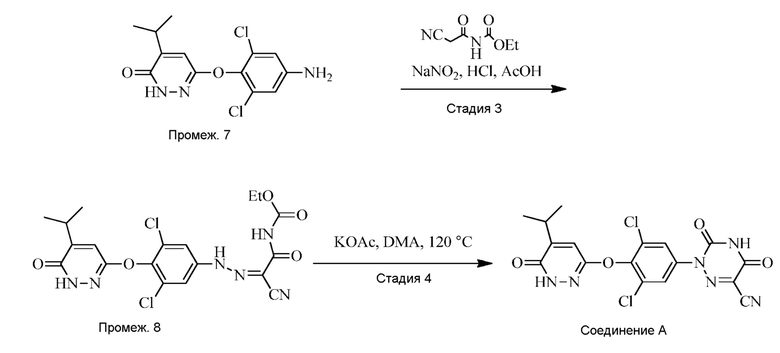
Стадия 3: Синтез 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она (Промеж. 8)
Промеж. 8 получают посредством контакта 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она с этил(2-цианоацетил)карбаматом и нитритом металла, таким как нитрит натрия, в присутствии кислоты (такой как HCl) в пригодном растворителе (например, смеси уксусной кислоты и воды) при подходящей температуре реакции (например, ниже 10°C) до завершения реакции.
Стадия 4: Синтез 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-l,2,4-триазин-6-карбонитрила (соединения A)
Соединение A получают посредством контакта промеж. 8 и основания, такого как ацетат натрия или ацетат калия, в пригодном растворителе (например, DMAC) при подходящей температуре реакции (например, при приблизительно 120°C) до завершения реакции.
В вариантах осуществления перевод 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она («Промеж. 7») в соединение формулы (IV), отличное от MGL-3916 (такое как его пролекарства) проводят в условиях, описанных, например, в Патентах США 7452882, 7807674 и 8076334, полное содержание которых, таким образом, приведено в качестве ссылки.
Способы синтеза, описанные в настоящем документе, приводят к превосходной региоселективности, с введением по Гриньяру изопропениловой или изопропиловой группы в отличие от образования биарилового эфира в способе синтеза, ранее описанном, например, в Патенте США 7452882, дающем плохую региоселективность. Кроме того, посредством экономизации от образования биарилового эфира до бензамидной защиты, способы, описанные в настоящем документе, исключают выделение продукта биарилового эфира, которое являлось практически почти невозможным из-за периодов времени фильтрации более 1 недели на партию при синтезе этого продукта в килограммовых количествах.
Настоящее изобретение относится к соединениям с высокой чистотой и/или в конкретной морфологической форме (например, форме I), к композициям, описанным в настоящем документе, и к способам лечения или предотвращения ожирения, гиперлипидемии, гиперхолестеринемии, диабета, неалкогольного стеатогепатита, жировой инфильтрации печени, заболевания костей, изменения тиреоидной оси, атеросклероза, сердечно-сосудистого нарушения, тахикардии, гиперкинетического поведения, гипотиреоза, зоба, расстройства дефицита внимания с гиперактивностью, нарушения обучаемости, задержки в умственном развитии, потери слуха, запаздывающего старения костей, неврологического или психиатрического заболевания, или рака щитовидной железы.
Следует понимать, что способы, описанные в настоящем документе, являются пригодными как для крупномасштабных, так и для мелкомасштабных получений желаемых соединений. В предпочтительных вариантах осуществления способов, описанных в настоящем документе, аналоги гормонов щитовидной железы можно получать в крупном масштабе, например, в масштабе промышленного производства, а не в экспериментальном/лабораторном масштабе. Например, способ периодического типа в соответствии со способами из описания, позволяет получение партий по меньшей мере 1 г, или по меньшей мере 5 г, или по меньшей мере 10 г, или по меньшей мере 100 г, или по меньшей мере 1 кг, или по меньшей мере 100 кг аналогов гормонов щитовидной железы. Более того, способы позволяют получение аналога гормона щитовидной железы, обладающего чистотой по меньшей мере 98%, или по меньшей мере 98,5%, как измерено посредством ВЭЖХ.
Фармацевтические композиции
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение формулы IV в комбинации по меньшей мере с одним фармацевтически приемлемым наполнителем или носителем.
«Фармацевтическая композиция» представляет собой состав, содержащий соединение по настоящему изобретению в форме, пригодной для введения субъекту. В одном варианте осуществления фармацевтическая композиция находится в нерасфасованной партии или в единичной дозированной форме. Единичная дозированная форма представляет собой любую из множества форм, включая, например, капсулу, пакет для IV вливания, таблетку, одиночный насос в аэрозольном ингаляторе или флакон. Количество активного ингредиента (например, состав описанного соединения или его соли, гидрата, сольвата или изомера) в единичной дозе композиции представляет собой эффективное количество и меняется в соответствии с конкретным предусмотренным видом лечения. Специалисту в данной области понятно, что иногда является необходимым осуществлять общепринятые изменения дозы в зависимости от возраста и состояния пациента. Доза может также зависеть от способа введения. Предусмотрено множество способов, включая пероральный, пульмональный, ректальный, парентеральный, чрескожный, подкожный, внутривенный, внутримышечный, внутрибрюшинный, ингаляционный, буккальный, подъязычный, интраплевральный, интратекальный, интраназальный и т.п. Лекарственные формы для местного или чрескожного введения соединения по этому изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и средства для ингаляции. В одном варианте осуществления активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые являются необходимыми.
Как применяют в настоящем документе, фраза «фармацевтически приемлемый» относится к тем соединениям, материалам, композициям, носителям и/или лекарственным формам, которые являются, в рамках тщательной медицинской оценки, пригодными для использования в контакте с тканями человека и животных без избыточной токсичности, раздражения, аллергического ответа или других проблем или осложнений, соразмерно с целесообразным соотношением польза/риск.
«Фармацевтически приемлемый наполнитель или носитель» обозначает наполнитель или носитель, являющийся пригодным для получения фармацевтической композиции, которая является в основном безопасной, нетоксичной и не является ни биологически, ни иным образом нежелательной, и включает в себя наполнитель, приемлемый для ветеринарного использования, так же как для фармацевтического использования для человека. «Фармацевтически приемлемый наполнитель», как используют в описании и формуле изобретения, включает в себя как один, так и более одного такого наполнителя.
Фармацевтическую композицию по изобретению составляют так, чтобы она была совместимой с предназначенным для нее способом введения. Примеры способов введения включают парентеральное, например, внутривенное, внутрикожное, подкожное, пероральное (например, ингаляцию), чрескожное (местное), и трансмукозальное введение. Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного введения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и средства для корректировки тоничности, такие как хлорид натрия или декстроза. pH можно корректировать с помощью кислот или оснований, таких как соляная кислота или гидроксид натрия. Препарат для парентерального введения можно заключать в ампулы, одноразовые шприцы или флаконы для множественных доз, изготовленные из стекла или пластика.
Термин «терапевтически эффективное количество», как применяют в настоящем документе, относится к количеству лекарственного средства для лечения, облегчения или предотвращения идентифицированного заболевания или состояния, или для проявления поддающегося детекции терапевтического или ингибирующего эффекта. Эффект можно детектировать посредством любого способа анализа, известного в данной области. Точное эффективное количество для субъекта может зависеть от массы тела, роста и состояния здоровья субъекта; характера и степени состояния; и лекарственного средства или комбинации лекарственных средств, выбранных для введения. Терапевтически эффективные количества для данной ситуации можно определять посредством общепринятых экспериментов, находящихся в пределах компетенции специалистов в данной области, и суждений клинициста. В предпочтительном аспекте, заболевание или состояние, подлежащее лечению, представляет собой нарушение метаболизма.
В практическом осуществлении способа по настоящему изобретению, эффективное количество любого одного соединения по этому изобретению или комбинации любых соединений по этому изобретению или их фармацевтически приемлемых солей или эфиров, вводят посредством любого из обычных и приемлемых способов, известных в данной области, либо отдельно, либо в комбинации. Соединения или композиции можно, таким образом, вводить перорально (например, в щечный карман), подъязычно, парентерально (например, внутримышечно, внутривенно или подкожно), ректально (например, посредством суппозиториев или промывок), чрескожно (например, электропорацией кожи) или посредством ингаляции (например, посредством аэрозоля), и в форме твердых, жидких или газообразных доз, включая таблетки и суспензии. Введение можно проводить в однократной единичной дозированной форме при непрерывной терапии или при терапии в однократной дозе по потребности. Терапевтическая композиция может также находиться в форме масляной эмульсии или дисперсии в сочетании с липофильной солью, например, памовой кислоты, или в форме биоразлагаемой композиции с замедленным высвобождением для подкожного или внутримышечного введения.
Пригодные фармацевтические носители для получения композиций в настоящем документе, могут представлять собой твердые вещества, жидкости или газы; таким образом, композиции могут принимать форму таблеток, пилюль, капсул, суппозиториев, порошков, покрытых кишечнорастворимой оболочкой или других защищенных составов (например, связанных на ионообменных смолах или упакованных в липидно-белковых везикулах), составов с замедленным высвобождением, растворов, суспензий, эликсиров, аэрозолей и т.п. Носитель можно выбирать из различных масел, включая масла из нефти, животного, растительного или синтетического происхождения, например, арахисового масла, соевого масла, минерального масла, кунжутного масла и т.п. Вода, солевой раствор, водная декстроза и гликоли являются предпочтительными жидкими носителями, в частности (при изотоничности с кровью) для пригодных для инъекций растворов. Например, составы для внутривенного введения содержат стерильные водные растворы активного ингредиента(ингредиентов), полученные растворением твердого активного ингредиента(ингредиентов) в воде для получения водного раствора, и приданием раствору стерильности. Пригодные фармацевтические наполнители включают крахмал, целлюлозу, тальк, глюкозу, лактозу, тальк, желатин, солод, рис, муку, мел, диоксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое снятое молоко, глицерин, пропиленгликоль, воду, этанол и т.п. Композиции можно подвергать воздействию общепринятых фармацевтических добавок, таких как консерванты, стабилизаторы, увлажняющие средства или эмульгаторы, соли для коррекции осмотического давления, буферы и т.п. Пригодные фармацевтические носители и их составы описаны в Remington's Pharmaceutical Sciences by E. W. Martin. Такие композиции, в любом случае, содержат эффективное количество активного соединения вместе с пригодным носителем для получения надлежащей лекарственной формы для надлежащего введения реципиенту.
Фармацевтические препараты могут также содержать консервирующие средства, солюбилизирующие средства, стабилизаторы, увлажняющие средства, эмульгаторы, подсластители, окрашивающие средства, придающие вкус средства, соли для изменения осмотического давления, буферы, покрывающие средства или антиоксиданты. Они могут также содержать другие терапевтически важные вещества, включая дополнительные активные ингредиенты, отличные от ингредиентов формулы I.
Соединения по настоящему изобретению являются пригодными в качестве лекарственных средств для лечения устойчивости к гормонам щитовидной железы (RTH) у субъекта, имеющего по меньшей мере одну мутацию TRβ. Субъект может страдать заболеванием, таким как ожирение, гиперлипидемия, гиперхолестеринемия, диабет, неалкогольный стеатогепатит, жировая инфильтрация печени, заболевание костей, изменение тиреоидной оси, атеросклероз, сердечно-сосудистое нарушение, тахикардия, гиперкинетическое поведение, гипотиреоз, зоб, расстройство дефицита внимания с гиперактивностью, нарушение обучаемости, задержка в умственном развитии, потеря слуха, запаздывающее старение костей, неврологическое или психиатрическое заболевание или рак щитовидной железы.
Терапевтически эффективное количество или доза соединения по этому изобретению могут меняться в широких пределах, и их можно определять способом, известным в данной области. Например, лекарственное средство можно дозировать в соответствии с массой тела. Такую дозу можно корректировать по индивидуальным требованиям в каждом конкретном случае, включая конкретное вводимое соединение(соединения), способ введения, подвергающееся лечению состояние, так же как подвергающегося лечению пациента. В другом варианте осуществления лекарственное средство можно вводить в фиксированных дозах, например, в дозе, не скорректированной в соответствии с массой тела. Как правило, в случае перорального или парентерального введения взрослым людям, ежесуточная доза от приблизительно 0,5 мг до приблизительно 1000 мг должна являться подходящей, хотя верхний предел можно превышать по показаниям. Доза составляет предпочтительно от приблизительно 5 мг до приблизительно 400 мг в сутки. Предпочтительная доза может составлять от приблизительно 20 мг до приблизительно 100 мг а сутки. Ежесуточную дозу можно вводить в форме однократной дозы или в дробных дозах, или в случае парентерального введения, ее можно вводить в форме непрерывной инфузии.
Эффективное количество лекарственного средства представляет собой количество, обеспечивающее поддающееся объективной оценке улучшение, как отмечено клиницистом или другим квалифицированным наблюдателем. Как применяют в настоящем документе, термин «способ эффективной дозы» относится к количеству активного соединения для получения желательного биологического эффекта у субъекта или в клетке.
Фармацевтические композиции можно заключать в контейнер, упаковку или диспенсер вместе с инструкциями для введения.
Соединения по настоящему изобретению являются способными к дальнейшему формированию солей. Все эти формы предусмотрены в объеме заявленного изобретения.
Как применяют в настоящем документе, «фармацевтически приемлемые соли» относятся к производным соединений по настоящему изобретению, где исходное соединение модифицируют получением их солей с кислотами или основаниями. Примеры фармацевтически приемлемых солей включают в себя, но без ограничения, соли неорганических или органических кислот из основных остатков, таких как амины, щелочи, или органические соли из кислых остатков, таких как карбоновые кислоты и т.п. Фармацевтически приемлемые соли включают в себя общепринятые нетоксичные соли или соли четверичного аммония исходного соединения, образованного, например, из нетоксичных неорганических или органических кислот. Например, такие общепринятые нетоксичные соли включают в себя, но без ограничения, соли, полученные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной, 2-гидроксиэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновойй, бензойной, бикарбоновой, карбоновой, лимонной, эдетовой, этандисульфоновой, 1,2-этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексилрезорциновой, гидрабаминовой, бромистоводородной, соляной, иодоводородной, гидроксималеиновой, гидроксинафтойной, изэтионовой, молочной, лактобионовой, лаурилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, напсиловой, азотной, щавелевой, памовой, пантотеновой, фенилуксусной, фосфорной, полигалктуроновой, пропионовой, салициловой, стеариновой, субуксусной, янтарной, сульфаминовой, сульфаниловой, серной, дубильной, виннокаменной, толуолсульфоновой и часто встречающихся аминокислот, например, глицина, аланина, фенилаланина, аргинина и т.д.
Другие примеры фармацевтически приемлемых солей включают гексановую кислоту, циклопентанпропионовую кислоту, пировиноградную кислоту, малоновую кислоту, 3-(4-гидроксибензоил)бензойную кислоту, коричную кислоту, 4-хлорбензолсульфоновую кислоту, 2-нафталинсульфоновую кислоту, 4-толуолсульфоновую кислоту, камфорсульфоновую кислоту, 4-метилбицикло-[2,2,2]-окт-2-ен-1-карбоновую кислоту, 3-фенилпропионовую кислоту, триметилуксусную кислоту, третичную бутилуксусную кислоту, муконовую кислоту и т.п. Настоящее изобретение относится также к солям, образованным, когда кислая часть, присутствующая в исходном соединении, либо заменена на ион металла, например, ион щелочного металла, ион щелочноземельного металла, или ион алюминия; либо скоординирована с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин, диэтиламин, диэтиламиноэтанол, этилендиамин, имидазол, лизин, аргинин, морфолин, 2-гидроксиэтилморфолин, дибензилэтилендиамин, триметиламин, пиперидин, пирролидин, бензиламин, тетраметилгидроксид аммония и т.п.
Следует понимать, что все ссылки на фармацевтически приемлемые соли включают в себя формы с присоединением растворителя (сольваты) или кристаллические формы (полиморфы), как определено в настоящем документе, той же самой соли.
Соединения по настоящему изобретению можно также получать в форме эфиров, например, фармацевтически приемлемых эфиров. Например, функциональную группу карбоновой кислоты в соединении можно переводить в ее соответствующий сложный эфир, например, метиловый, этиловый или другой сложный эфир. Также, группу спирта в соединении можно переводить в его соответствующий эфир, например, ацетатный, пропионатный или другой сложный эфир.
Соединения по настоящему изобретению можно также получать в форме пролекарств, например, фармацевтически приемлемых пролекарств. Термины «пролекарственное средство» и «пролекарство» используют взаимозаменяемо в настоящем документе, и они обозначают любое соединение, высвобождающее активное исходное лекарственное средство in vivo. Поскольку известно, что пролекарства улучшают многочисленные желательные свойства лекарственных средств (например, растворимость, биодоступность, промышленное производство и т.д.), соединения по настоящему изобретению можно доставлять в форме пролекарства. Таким образом, настоящее изобретение предназначено, чтобы включать пролекарства заявленных в настоящее время соединений, способы их доставки и содержащие их композиции. «Пролекарства» предназначены, чтобы включать любые ковалентно связанные носители, высвобождающие активное исходное лекарственное средство по настоящему изобретению in vivo, когда такое пролекарство вводят субъекту. Пролекарства по настоящему изобретению получают посредством модификации функциональных групп, присутствующих в соединении, таким образом, что модификации отщепляются, либо при общепринятой манипуляции, либо in vivo, до исходного соединения. Пролекарства включают соединения по настоящему изобретению, где гидрокси, амино, сульфгидрильная, карбокси или карбонильная группа связана с любой группой, которую можно отщеплять in vivo для образования свободной гидроксильной, свободной амино, свободной сульфгидрильной, свободной карбокси или свободной карбонильной группы, соответственно.
Примеры пролекарств включают, но без ограничения, сложные эфиры (например, производные ацетата, диалкиламиноацетатов, форматов, фосфатов, сульфатов и бензоата) и карбаматы (например, N,N-диметиламинокарбонил) функциональных гидроксигрупп, сложные эфиры (например, этиловые сложные эфиры, морфолиноэтаноловые сложные эфиры) карбоксильных функциональных групп, N-ацильные производные (например, N-ацетил) N-основания Манниха, основания Шиффа и енаминоны функциональных аминогрупп, оксимы, ацетали, кетали и еноловые сложные эфиры кетоновых и альдегидных функциональных групп в соединениях по изобретению, и т.п., См. Bundegaard, H., Design of Prodrugs, p1-92, Elesevier, New York-Oxford (1985).
Соединения или их фармацевтически приемлемые соли, сложные эфиры или пролекарства, вводят перорально, назально, чрескожно, пульмонально, ингаляционно, буккально, подъязычно, внутрибрюшинно, подкожно, внутримышечно, внутривенно, ректально, интраплеврально, интратекально и парентерально. В одном варианте осуществления соединение вводят перорально.
Специалисту в данной области понятны преимущества конкретных способов введения.
Режим дозирования с использованием соединений выбирают в соответствии со множеством факторов, включая тип, вид, возраст, массу, пол и медицинское состояние пациента; тяжесть состояния, подлежащего лечению; способ введения; функцию почек и печени пациента; и конкретное применяемое соединение или его соль. Специалист в области терапии или ветеринарии может легко определять и предписывать эффективное количество лекарственного средства, необходимое для лечения состояния, противодействия состоянию или ареста прогрессирования состояния.
Способы получения составов и введения для описанных соединений по изобретению можно обнаружить в Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, PA (1995). В одном варианте осуществления, соединения, описанные в настоящем документе, и их фармацевтически приемлемые соли, используют в фармацевтических препаратах в комбинации с фармацевтически приемлемым носителем или разбавителем. Пригодные фармацевтически приемлемые носители включают инертные твердые наполнители или разбавители и стерильные водные или органические растворы. Соединения могут присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения желательной величины дозы в диапазоне, описанном в настоящем документе.
Изобретение относится к способу лечения или облегчения симптома устойчивости к гормону щитовидной железы у субъекта посредством введения субъекту, экспрессирующему мутантный TRβ, содержащий мутацию в связывающем лиганд домене, терапевтически эффективного количества соединения формулы (IV), такого как соединение A, например, его форма I.
Описание относится также к способу определения способности субъекта, обладающего устойчивостью к гормонам щитовидной железы (RTH), отвечать на соединение формулы (IV), описанное в настоящем документе, посредством получения образца от субъекта; и детекции по меньшей мере одной мутации TRβ (например, мутации в гене или мутации в связывающем лиганд домене полипептида TRβ, например, полипептида, как определено в SEQ ID NO: 1); и присутствие указанной мутации указывает на то, что субъект является способным отвечать на соединение формулы (IV), такое как соединение A, например, его форма I. Способ может дополнительно включать лечение субъекта, имеющего мутацию, посредством введения терапевтически эффективного количества соединения формулы (IV), такого как соединение A, например, его формы I.
В одном варианте осуществления субъект, который способен или может являться способным отвечать на соединение формулы (IV), такое как соединение A, страдает ожирением, гиперлипидемией, гиперхолестеринемией, диабетом, неалкогольным стеатогепатитом, жировой инфильтрацией печени, заболеванием костей, изменением тиреоидной оси, атеросклерозом, сердечно-сосудистым нарушением, тахикардией, гиперкинетическим поведением, гипотиреозом, зобом, расстройством дефицита внимания с гиперактивностью, нарушением обучаемости, задержкой в умственном развитии, потерей слуха, запаздывающим старением костей, неврологическим или психиатрическим заболеванием или раком щитовидной железы.
Кроме того, описание относится также к способу, включающему определение присутствия мутации в гене TRβ в образце от субъекта; и выбор, на основании присутствия мутации в гене TRβ, терапии, включающей введение терапевтически эффективного количества соединения формулы (IV), такого как соединение A, например, его форма I.
Описание относится также к способу, включающему амплификацию нуклеиновой кислоты в образце от субъекта с помощью праймера, комплементарного последовательности нуклеиновой кислоты мутантного TRβ, содержащей мутацию в гене TRβ в последовательности нуклеиновой кислоты, как определено в SEQ ID NO: 2; определение присутствия амплифицированной нуклеиновой кислоты, и выбор, на основании присутствия амплифицированной нуклеиновой кислоты, терапии, включающей введение терапевтически эффективного количества соединения формулы (IV), или лечение субъекта посредством введения терапевтически эффективного количества соединения формулы (IV) на основании присутствия амплифицированной нуклеиновой кислоты.
Мутантный TRβ, описанный в настоящем документе, представляет собой мутантный полипептид TRβ или последовательность нуклеиновой кислоты, кодирующую мутантный полипептид TRβ.
В одном варианте осуществления мутантный TRβ содержит одну или несколько мутаций в положениях аминокислот 234, 243, 316 и 317 из SEQ ID NO: 1. Более предпочтительно, мутация выбрана из группы, состоящий из замены треонином (T) остатка аланина (A) дикого типа в положении аминокислоты 234 из SEQ ID NO: 1 (A234T); замены глутамином (Q) остатка аргинина (R) дикого типа в положении аминокислоты 243 из SEQ ID NO: 1 (R243Q); замены гистидином (H) остатка аргинина (R) дикого типа в положении аминокислоты 316 из SEQ ID NO: 1 (R316H); и замены треонином (T) остатка аланина (A) дикого типа в положении аминокислоты 317 из SEQ ID NO: 1 (A317T).
В одном варианте осуществления мутантный TRβ содержит последовательность нуклеиновой кислоты, кодирующую мутантный полипептид TRβ, имеющий одну или несколько мутаций в положениях аминокислот 234, 243, 316 и 317 из SEQ ID NO: 1. Последовательность нуклеиновой кислоты, кодирующей мутантный полипептид или фрагмент пептида TRβ, являющуюся характерной для мутантного полипептида TRβ, можно детектировать с использованием любого пригодного способа. Например, последовательность нуклеиновой кислоты, кодирующую мутантный полипептид TRβ, можно детектировать с использованием повторного секвенирования полного генома или повторного секвенирования области-мишени (последнее известно также как направленное повторное секвенирование) с использованием подходящим образом выбранных источников ДНК и праймеров для полимеразной цепной реакции (ПЦР) в соответствии со способами, хорошо известными в данной области. См., например, Bentley (2006) Curr Opin Genet Dev. 16:545-52, и Li et al. (2009) Genome Res 19:1124-32. Способ, как правило, и в общем, включает стадии очистки геномной ДНК, амплификации ПЦР для амплификации интересующей области, циклического секвенирования, очистки реакционной смеси для секвенирования, капиллярного электрофореза и анализа данных. Праймеры для ПЦР высокого качества для перекрывания интересующей области разработаны с использованием инструментов для дизайна праймеров in silico. Циклическое секвенирование представляет собой простой способ, в котором последовательные циклы денатурации, отжига и удлинения в термоциклере приводят к линейной амплификации продуктов удлинения. Продукты, как правило, останавливают с помощью флуоресцентной метки, которая идентифицирует концевое нуклеотидное основание как G, A, T или C. Не включившиеся терминирующие красители и соли, которые могут конкурировать за ввод в капиллярный электрофорез, удаляют промывкой. В ходе капиллярного электрофореза, продукты реакции циклического секвенирования мигрируют через капилляры, заполненные полимером. Отрицательно заряженные фрагменты ДНК разделяют по размерам, поскольку они движутся через капилляры к положительному электроду. После электрофореза, программное обеспечение для сбора данных создает файл для образца с необработанными данными. С использованием нижестоящих программных приложений, проводят дальнейший анализ данных для перевода собранных данных цветных изображений в соответствующие нуклеотидные основания. Альтернативно или дополнительно, способ может включать использование основанного на микромассивах связывания и/или секвенирования области-мишени геномной ДНК. Наборы, реагенты и способы для подбора подходящих праймеров для ПЦР и проведения повторного секвенирования являются коммерчески доступными, например, из Applied Biosystems, Agilent, и NimbleGen (Roche 30 Diagnostics GmbH). Для использования по настоящему изобретению, праймеры для ПЦР можно подбирать так, чтобы амплифицировать, например, по меньшей мере относящуюся к делу часть последовательности нуклеиновой кислоты, кодирующую мутантный полипептид TRβ, имеющий одну или несколько мутаций в положениях аминокислот 234, 243, 316 и 317 из SEQ ID NO: 1.
Альтернативно или дополнительно, последовательность нуклеиновой кислоты, кодирующую мутантный полипептид TRβ, можно детектировать с использованием Саузерн-блоттинга в соответствии со способами, хорошо известными в данной области.
В конкретных вариантах осуществления способы по изобретению включают стадию проведения анализа для детекции мутанта TRβ в образце от субъекта. Как применяют в настоящем документе, «образец от субъекта» относится к любому пригодному образцу, содержащему клетки или компоненты клеток, полученные или происходящие от субъекта. В одном варианте осуществления образец представляет собой образец крови. В одном варианте осуществления образец представляет собой образец биопсии, полученный, например, из щитовидной железы.
Описание относится также к комплексу лиганд-мутантный TRβ, содержащему: мутантный полипептид TRβ и соединение формулы (IV). Например, мутантный полипептид TRβ, формирующий комплекс, содержит одну или несколько мутаций в положениях аминокислот 234, 243, 316 и 317 из SEQ ID NO: 1. Например, соединение, формирующее комплекс, представляет собой соединение A.
Кроме того, описание относится к комплексу праймер-нуклеиновая кислота, содержащему: последовательность нуклеиновой кислоты мутантного TRβ, и праймер для ПЦР, комплементарный последовательности нуклеиновой кислоты мутантного TRβ, где мутантная последовательность нуклеиновой кислоты содержит мутацию в гене EZH2 в последовательности нуклеиновой кислоты, как определено в SEQ ID NO: 2.
Полное содержание всех патентов, патентных заявок и публикаций, упомянутых в настоящем документе, таким образом, приведено в качестве ссылки. Однако, когда содержание патента, патентной заявки или публикации, содержащей специальные определения, приведено в качестве ссылки, эти специальные определения следует понимать как применимые к включенным патенту, патентной заявке или публикации, в которых они обнаружены, а не к остальному тексту этой заявки, в частности, к формуле изобретения этой заявки.
Следует понимать, что в то время как изобретение описано в отношении предпочтительных конкретных вариантов его осуществления, предшествующее описание, так же как последующие примеры, предназначены для иллюстрации, а не ограничения объема изобретения. Специалистам в данной области понятно, что различные изменения можно осуществлять и эквиваленты можно заменять без отклонения от объема изобретения, и далее, что другие аспекты, преимущества и модификации очевидны специалистам в области, к которой относится изобретение.
Все проценты и соотношения, применяемые в настоящем документе, если не указано иначе, представлены по массе. Другие признаки и преимущества настоящего изобретения очевидны из различных примеров. Представленные примеры иллюстрируют различные компоненты и способы, пригодные в практическом осуществлении настоящего изобретения. Примеры не ограничивают заявленное изобретение. На основании настоящего описания специалист в данной области может идентифицировать и использовать другие компоненты и способы, пригодные для практического осуществления настоящего изобретения.
Примеры
Если не указано иначе, аналитические устройства и параметры, использованные для соединений, описанных в примерах, являются следующими:
Данные XRPD собирали на рентгеновском порошковом дифрактометре (CubiX-Pro XRD) с излучением Cu Kα (45 кВ, 40 мА) 2-тета (2θ) от 3 до 45 градусов при скорости сканирования 0,12 градусов/мин и размере шага 0,020 градусов.
Образец помещали в Si ультра-микродержатели для образцов с возвратом к нулю. Анализ проводили с использованием ширины излучения 10 мм, и следующие параметры выставляли в аппаратном оборудовании/программном обеспечении:
Рентгеновская трубка: Cu KV, 45 кВ, 40 мА
Детектор: X’Celerator
Первичная щель ASS: фиксированная 1°
Щель расходимости (Prog): автоматическая - 5 мм длина излучения
Щели Соллера: 0,02 радиан
Щель рассеяния (PASS): автоматическая - 5 мм наблюдаемая длина
Диапазон сканирования: 3,0-45,0°
Режим сканирования: непрерывный
Размер шага: 0,02°
Время на шаг: 10 с
Активная длина: 2,54°
После анализа данные переводили от регулируемых до фиксированных щелей с использованием программного обеспечения X’Pert HighScore Plus со следующими параметрами:
Фиксированный размер щели расходимости: 1,00°, 1,59 мм
Точка пересечения: 44,3° омега
В примерах, описанных ниже, если не указано иначе, соединение 4 представляет собой защищенное бензоилом соединение.
Пример 1: Получение N-(3,5-дихлор-4-((6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)бензамида (соединения 4, где R2 представляет собой бензоил)
В 1 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, обратным холодильником и входом/выходом N2, загружали 3,6-дихлорпиридазин (100 г, 0,672 моль, 1 масса), 4-амино-2,6-дихлорфенол (122 г, 0,686 моль, 1,02 эквивалента) и DMAC (500 мл, 5 объемов). В полученный раствор загружали карбонат цезия (251 г, 0,771 моль, 1,15 эквивалента), и суспензию нагревали до 110°C. После 3 час при этой температуре, температуру партии понижали до 70°C и перемешивали при этой температуре в течение 16 час. Анализ 1H ЯМР (DMSO) показал, что почти весь дихлорпиридазин израсходован, и реакция, по-видимому, завершена. Партию охлаждали до комнатной температуры и переносили в 3 л, круглодонную колбу с помощью EtOAc (2 л, 20 объемов). Добавляли силикагель (100 г, 1 масса), и суспензию встряхивали в течение 30 мин и фильтровали. Реактор и осадок промывали с помощью EtOAc (500 мл, 5 объемов), пока элюированный фильтрат не стал бесцветным. Полученный фильтрат обрабатывали 10% водным NaCl (2 л, 20 объемов), двухфазную смесь встряхивали в течение 30 мин, и нижний водный слой выбрасывали. Верхний органический слой концентрировали до сухого состояния под пониженным давлением. EtOAc (100 мл, 1 объемов) добавляли к осадку и концентрировали до сухого состояния под пониженным давлением с получением неочищенного соединения 2 (251 г, выход 128%) в форме масла. Анализ ВЭЖХ показал чистоту 93,4%. Анализ 1H ЯМР (DMSO) был согласованным с заданной структурой и показал присутствие ≈25% DMAC и 2% EtOAc.
Другие условия для синтеза соединения 2 описаны в таблицах 1-3 ниже.
Обобщение параметров реакции для соединения 2
(г)
DMSO 5 объемов
KOtBu 1,1 эквивалента Промеж. 3 1 эквивалент
85°C
DMSO 5 объемов
KOtBu 1,1 эквивалента Промеж. 3 1 эквивалент
85°C
DMAC 5 объемов
KOtBu 1,1 эквивалента
Промеж. 3 1 эквивалент
85°C
DMAC 5 объемов
KOtBu 1,1 эквивалент
Промеж. 3 1 эквивалент
85°C
(г)
KOtBu 1,1 эквивалента
Промеж. 3 1 эквивалент
85°C
Обобщение параметров реакции для соединения 2
(час)
1 эквивалент Промеж. 1
1,02 эквивалента Промеж. 3
85°C
DMSO
1 эквивалент Промеж. 1
1,02 эквивалента Промеж. 3
120°C
1 эквивалент Промеж. 1
1,02 эквивалент Промеж. 3
120°C
1 эквивалент Промеж. 1
1,02 эквивалента Промеж. 3
120°C
42%
DMAC
1,15 эквивалента Cs2CO3
5 объемов DMAC
от 110°C до 70°C
2,25
3,25
19
Обобщение параметров реакции для соединения 2 (все реакции происходят в DMAC)
(измельченный)
Неочищенное соединение 2 выше пропитывали уксусной кислотой (1,48 л, 7,5 объемов) и добавляли бензойный ангидрид (168 г, 0,741 моль, 1,1 эквивалента). Полученную смесь нагревали до 100°C и после 35 мин при этой температуре, количество соединения 2 составляло 0,8%. Добавляли ацетат натрия (110 г, 2 эквивалента), и температуру увеличивали до 110°C. После 14,5 час при этой температуре, анализ ВЭЖХ реакционной смеси показал отсутствие оставшегося промежуточного соединения, и реакция, по-видимому, была завершена. Партию охлаждали до 75°C и воду (1,5 л, 7,7 объемов) добавляли в течение периода 1 час при поддержании в то же время температуры партии между 72-75°C. Партию охлаждали до 21°C и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок последовательно промывали водой (1 л, 5 объемов). После сушки собранного твердого вещества в вакуумной печи при 50°C в течение 16 час, выход неочищенного соединения 4 составлял 195 г (77%). Анализ ВЭЖХ (способ B, 220 нм) показал чистоту 91,6%.
ВЭЖХ способ B:
Колонка: Waters Sunfire C18, 3,5 мкМ, 4,6 × 150 мм
Скорость потока: 1,0 мл/мин.
Подвижная фаза A: 0,05% TFA в воде
Подвижная фаза B: 0,05% TFA в H2O
Разбавитель: 50:50 MeCN/H2O
Анализ 1H ЯМР (DMSO) был согласованным с заданной структурой и показал содержание уксусной кислоты 1%. Бензоилхлорид также использовали для защиты вместо бензойного ангидрида. Когда использовали бензоилхлорид, использовали основания, такие как карбонат цезия или карбонат калия, и реакцию проводили при комнатной температуре.
Другие условия для синтеза соединения 4 описаны в таблицах 4 и 5 ниже.
Защита/гидролиз соединения 2 (выходы указаны от промеж. 1) (бензоильная защитная группа)
(час)
(% AUC)
1,15 эквивалента Cs2CO3 3 объема DMSO
2. Bz20
3. Уксусная кислота, 115°C
2. 19 час
3. 20,5 час
2. 70,7
3. 68,8
2 эквивалента NaOAc 110°C
2 эквивалента NaOAc
100-110°C
Обобщение параметров реакции для соединения 4 (ацетатная защитная группа)
115°C
115°C
115°C
2-стадия
Очистка соединения 4: в 5 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, обратным холодильником и входом/выходом N2, загружали неочищенное соединение 4 (100 г, 1 масса) и уксусную кислота (2 л, 20 объемов). Взвесь встряхивали и нагревали до 95°C, и происходило растворение. Воду (2 л, 20 объемов) добавляли в течение периода 2,75 час при поддержании температуры партии ≈95°C, и происходила преципитация. Полученную взвесь нагревали при 95°C в течение других 30 мин до удаления нагрева. После того, как партия достигала температуры окружающей среды, ее перемешивали при этой температуре в течение ночи для удобства и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок последовательно промывали водой (1 л, 10 объемов). Собранное белое твердое вещество сушили в вакуумной печи при 40°C до постоянной массы 91 г (91%). Анализ ВЭЖХ высушенного твердого вещества показал чистоту 98,0%. Анализ 1H ЯМР (DMSO) был согласованным с заданной структурой и показал содержание уксусной кислоты 0,3%. В таблице 6 ниже перечислены другие условия для очистки соединения 4.
Очистка соединения 4 (R2=Bz)
(час)
выхода
по ВЭЖХ
(% AUC)
20 объемов AcOH
20 объемов воды
88-100°C
20 объемов AcOH
20 объемов воды
95°C
20 объемов AcOH
20 объемов воды
95°C
12 объемов AcOH
10 объемов воды 100-110°C
Пример 2: Получение 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она (промеж. 7)
В 4 л, четырехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, входом/выходом N2 и обратным холодильником, загружали 4 (95 г, 0,253 моль, 1 масса), THF (665 мл, 7 объемов), и LiCl (32,3 г, 0,759 моль, 3 эквивалента). Полученную суспензию нагревали до 35°C, и раствор бромида изопропенилмагния (0,5M в THF, 1,72 л, 0,859 моль, 3,4 эквивалента) добавляли в течение периода 80 мин при поддержании температуры партии между 35-45°C. После нагревания полученной взвеси при 40°C в течение 3 час, анализ ВЭЖХ показал превращение 87%). Добавляли дополнительный раствор бромида изопропенилмагния (0,5M в THF, 51 мл, 0,026 моль, 0,1 эквивалента), и взвесь встряхивали при 40-43°C в течение следующих 90 мин. Анализ ВЭЖХ показал превращение 92,9%, и реакция, по-видимому, была завершена. Нагревание удаляли, реакционную смесь охлаждали до 14°C, и 3н водную HC1 (380 мл, 4 объема) добавляли медленно в течение 15 мин при поддержании температуры партии ниже 26°C, после этого времени все твердые вещества растворялись. Нижний водный слой удаляли и проводили экстракцию с помощью THF (350 мл, 3,7 объема). После удаления нижнего водного слоя, объединенные органические слои концентрировали под пониженным давлением приблизительно до 5 объемов по отношению к 4. В полученный раствор загружали 10% (масс./масс.) водного KOH (532 мл, 5,6 объемов), и смесь нагревали до 85°C при отгонке в то же время THF с использованием устройства для молекулярной дистилляции. Партию поддерживали при 85°C в течение 11 час, и нагревание удаляли. Партию охлаждали до температуры окружающей среды в течение ночи для удобства. Анализ ВЭЖХ (способ A ниже) полученной взвеси показал превращение 99% в промеж. 7, и реакция, по-видимому, была завершена.
ВЭЖХ способ A
Колонка: Waters Sunfire C18, 3,5 мкМ, 4,6×150 мм
Скорость потока: 1,0 мл/мин.
Подвижная фаза A: 0,05% TFA в воде
Подвижная фаза B: 0,05% TFA в H2O
Разбавитель: 50:50 MeCN/H2O
Температуру партии доводили до 48°C, и 3н водную HCl (152 мл, 1,6 объемов) добавляли в течение 35 мин для доведения pH до 7,5-8,0 при поддержании в то же время температуры партии 46-48°C. Нагревание удаляли, и взвесь охлаждали до 30°C. Анализ 1H ЯМР (DMSO) показал молярное соотношение промеж. 7/THF 1,0:0,22 (Приложение 14). Партию фильтровали при 30°C через шагреневую фильтровальную бумагу, и реактор и осадок последовательно промывали водой (475 мл, 5 объемов). Бежевое твердое промеж. 7 сушили в вакуумной печи при 40°C до постоянной массы 81,6 г (выход 102%). Анализ Карла Фишера показал содержание воды 0,8%. Анализ 1H ЯМР (DMSO) был согласованным с заданной структурой и показал содержание THF 0,4%. Анализ ВЭЖХ показал чистоту 92,6%. В таблицах 7-10 ниже представлены обобщения параметров реакции для получения промеж. 7.
Обобщение циклов изопропенилирования с реагентом Гриньяра
(г)
7 объемов THF
3,4 эквивалента реагента Гриньяра
3 эквивалента LiCl
40°C
7 объемов THF
3,4 эквивалента реагента Гриньяра
3 эквивалента LiCl
40°C
7 объемов THF
3,5 эквивалента реагента Гриньяра
3 эквивалент LiCl
40°C
3 эквивалента LiCl
8 объемов THF
3,4 эквивалента реагента Гриньяра 1,5М в MeTHF
40°C
3 эквивалента LiCl
15 объемов THF
2 эквивалента t-BuMgCl 2M в THF
1,7 эквивалента реагента Гриньяра 1,5M в MeTHF
40°C
3 эквивалента LiCl
15 объемов THF
3,6 эквивалента реагента Гриньяра
0,5M в THF
40°C
3 эквивалента LiCl
13 объемов THF
3,7 эквивалента реагента Гриньяра 1,5M в MeTHF
40°C
3 эквивалента LiBr
13 объемов THF
3,7 эквивалента реагента Гриньяра 1,5M в MeTHF
40°C
5 эквивалентов LiCl
13 объемов THF
3,7 эквивалента реагента Гриньяра 0,5M в THF
40°C
Обобщение циклов изопропенилирования с реагентом Гриньяра
20 объемов THF
3,3 эквивалента iPrMgCl
30°C
20 объемов THF
6 эквивалентов iPrMgCl
40°C
20 объемов THF
4 эквивалента iPrMgCl
20°C
20 объемов диоксана
4,1 эквивалента iPrMgCl
40°C
8 эквивалентов iPrMgCl
30 объемов THF
25-42°C
2 эквивалента LiCl
4 эквивалента iPrMgCl
27 объемов THF
3 эквивалента LiCl
5 эквивалентов iPrMgCl
25 объемов THF
3 эквивалента LiCl
4 эквивалента iPrMgCl
10 объемов THF
3 эквивалент LiCl
4,1 эквивалента iPrMgCl
7 объемов THF
3 эквивалента LiCl
3,5 эквивалента iPrMgCl
7 объемов THF
3 эквивалента LiCl
3,2 эквивалента iPrMgCl
7 объемов THF
3 эквивалента LiCl
3,4 эквивалента iPrMgCl
10 объемов THF
3 эквивалента LiCl
3,4 эквивалента iPrMgCl
10 объемов THF
Обобщение окисления бромом пиридазинонового соединения 5
(г)
Br2 2 эквивалента
AcOH 10 объемов
90°C
Br2 1,5 эквивалента
AcOH 7 объемов
90°C
Br2 1,5 эквивалента
AcOH 7 объемов
90°C
Br2 5 эквивалентов
90°C
2-стадия
2-стадия
AcOH 10 объемов
Br2 1,5 эквивалента
90°C
AcOH 10 объемов
Br2 1,5 эквивалента
60°C
2-стадия
2-стадия
Br2 1,5 эквивалента
60°C
2-стадия
Обобщение снятия защиты соединения 6 для получения промеж. 7
(%AUC)
Вода (10 объемов)
(бензоил)
Вода (10 объемов)
(6 эквивалентов)
MeOH (10 объемов)
(10 эквивалентов)
MeOH (2,5 объема)
5 объемов)
5 объемов)
Пример 3: Получение (Z)-этил(2-циано-2-(2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)гидразоно)ацетил)карбамата (Промеж. 8)
В 2 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, входом/выходом N2, загружали промеж. 7 (75,0 г, 0,239 моль, 1 масса), уксусную кислоту (600 мл, 8 объемов), воду (150 мл, 2 объема) и концентрированную HC1 (71,3 мл, 0,95 объема). Полученную тонкую взвесь охлаждали до 6°C, и раствор NaNO2 (16,8 г, 0,243 моль, 1,02 эквивалента) в воде (37,5 мл, 0,5 объемов) добавляли в течение периода 10 мин при поддержании в то же время температуры партии ниже 10°C. После дополнительных 10 мин встряхивания между 5-10°C, анализ ВЭЖХ показал полное превращение промеж. 7 в диазониевое промежуточное соединение. Раствор NaOAc (54,5 г, 0,664 моль, 2,78 эквивалента) в воде (225 мл, 3 объема) добавляли в течение периода 6 мин при поддержании в то же время температуры партии ниже 10°C. Немедленно добавляли N-цианоацетилуретан (37,9 г, 0,243 моль, 1,02 эквивалента), охлаждение удаляли, и партию естественным образом нагревали до 8°C в течение 35 мин. Анализ ВЭЖХ показал полное израсходование диазониевого промежуточного соединения, и реакция, по-видимому, была завершена. Партию естественным образом нагревали до 21°C и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок последовательно промывали водой (375 мл, 5 объемов) дважды. Собранное оранжевое твердое вещество сушили в вакуумной печи при 35°C в течение 64 час с получением неочищенного промеж. 8 (104,8 г, 91%).
В 1 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой и входом/выходом N2, загружали неочищенное промеж. 8 (104,4 г, 1 масса) и уксусную кислоту (522 мл, 5 объемов). Полученную взвесь нагревали до 50°C и держали при этой температуре в течение 1,5 час. Партию естественным образом охлаждали до 25°C в течение 2 час и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок последовательно промывали водой (522 мл, 5 объемов), и осадок кондиционировали в вакууме в течение 1,75 час. Светло-оранжевое твердое вещество сушили до постоянной массы в вакуумной печи при 40°C с получением 89,9 г (78% от промеж. 7) желательного продукта. 1H ЯМР (DMSO) был согласованным с заданной структурой.
Пример 4: Получение 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила (соединения A)
В 2 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, входом/выходом N2 и обратным холодильником, загружали промеж. 8 (89,3 г, 0,185 моль, 1 масса), DMAC (446 мл, 5 объемов), и КОAc (20,0 г, 0,204 моль, 1,1 эквивалента). Смесь нагревали до 120°C и держали при этой температуре в течение 2 час. Анализ ВЭЖХ показал полное превращение в соединение A. Температуру партии доводили до 18°C в течение 1 час, и добавляли уксусную кислоту (22,3 мл, 0,25 объемов). Температуру партии доводили до 8°C, и воду (714 мл, 8 объемов) добавляли в течение 1 час; образовывалась оранжевая взвесь. Партию фильтровали через шагреневую фильтровальную бумагу и позволяли кондиционирование осадка в течение ночи под N2 без вакуума для удобства. Предварительно смешанный раствор 1:1 ацетона/воды (445 мл, 5 объемов) загружали в колбу и добавляли к осадку в качестве промывки с подключенным вакуумом. После 2 час кондиционирования осадка в вакууме, его переносили в чистую 1 л, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой и входом/выходом N2. Загружали этанол (357 мл, 4 объема) и ацетон (357 мл, 4 объема), и полученную взвесь нагревали до 60°C; происходило растворение. Воду (890 мл, 10 объемов) добавляли в течение периода 90 мин при поддержании в то же время температуры партии между 55-60°C. Полученной взвеси позволяли охлаждаться до 25°C и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок промывали последовательно раствором 1:1 EtOH/воды (446 мл, 5 объемов). Осадок кондиционировали в течение ночи под N2 без вакуума для удобства. Трещины в осадке сглаживали и подключали вакуум. Осадок промывали водой (179 мл, 2 объема) и высушивали в вакуумной печи при 45°C до постоянной массы 70,5 г (87%, неочищенное соединение A). Анализ ВЭЖХ показал чистоту 94,8%.
В 500 мл, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, входом/выходом N2 и обратным холодильником, загружали неочищенное соединение A (70,0 г) и MIBK (350 мл, 5 объемов). Оранжевую взвесь нагревали до 50°C и держали при этой температуре в течение 2 час. Партию естественным образом охлаждали до 23°C и фильтровали через шагреневую фильтровальную бумагу. Реактор и осадок последовательно промывали MIBK (35 мл, 0,5 объемов) дважды. Собранные твердые вещества высушивали в вакуумной печи при 45°C до постоянной массы 58,5 г (84%). Это твердое вещество загружали в 500 мл, трехгорлую, круглодонную колбу, оборудованную верхней мешалкой, термопарой, входом/выходом N2 и обратным холодильником. Добавляли этанол (290 мл, 5 объемов), и взвесь нагревали с обратным холодильником. После 3,5 часов кипячения с обратным холодильником, XRPD показала, что твердое вещество соответствовало форме I, и нагревание удаляли. После достижения 25°C партию фильтровали через фильтровальную бумагу, и реактор и осадок последовательно промывали EtOH (174 мл, 3 объема). Коричневое твердое соединение A сушили в вакуумной печи при 40°C до постоянной массы 50,4 г (87%, 64% от промеж. 8). Анализ ВЭЖХ показал чистоту 99,1%. 1H ЯМР (DMSO) был согласованным с заданной структурой.
Пример 5: Масштабирование получения 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила (соединения A)
Партию соединения A в более крупном масштабе синтезировали в соответствии со схемой ниже. Условия на схеме ниже являлись сходными с условиями, описанными в примерах 1-4 выше.
Синтез 4: В 50 л стеклянный сосуд с рубашкой (продутый N2) загружали 3,6-дихлорпиридазин (2,00 кг), 4-амино-2,6-дихлорфенол (2,44 кг) и N,N-диметилацетамид (10,0 л). Партию 3 раза продували под вакуумом (26 в Hg (88050 Па))/азотом (1 PSIG (6895 Па)). Добавляли карбонат цезия (5,03 кг), и температуру партии доводили от 22,3°C до 65,0°C в течение 3,5 часов. Партию держали при 65,0°C в течение 20 часов. В этой точке анализ 1H ЯМР показал 3,34% 3,6-дихлорпиридазина относительно 2. Температуру партии доводили до 21,5°C и к партии добавляли этилацетат (4,00 л). Партию встряхивали в течение 10 минут и затем фильтровали через 18” нутч-фильтр, оборудованный полипропиленовой фильтровальной тканью. Фильтрация занимала 15 минут. Этилацетат (5,34 л) загружали в сосуд и переносили на фильтр в качестве промывки. Затем партию вручную ресуспендировали в фильтре перед повторным включением вакуума. Этот процесс повторяли еще 2 раза, и отфильтрованный осадок кондиционировали в течение 10 минут. Фильтрат загружали в 100-л сосуд, содержащий (16,0 л) ранее подготовленного 15% хлорида натрия в H2O. Партию встряхивали в течение 5 минут и затем позволяли разделение в течение 35 минут. Граница фаз была невидимой, так что удаляли расчетные 23 л нижней водной фазы. 16,0 л 15% хлорида натрия в H2O добавляли к партии. Партию встряхивали в течение 6 минут и затем позволяли разделение в течение 7 минут. Граница фаз была видимой на ~19 л, и нижнюю водную фазу удаляли. 17,0 л 15% хлорида натрия в H2O добавляли к партии. Партию встряхивали в течение 7 минут и затем позволяли разделение в течение 11 минут. Нижнюю водную фазу удаляли. Сосуд подключали для вакуумной дистилляции, и партию концентрировали от 17,0 л до 8,0 л в течение 2 часов 20 минут при поддержании температуры партии около 21°C. Бензойный ангидрид (3,19 кг) и уксусную кислоту (18,0 л) загружали в сосуд. Сосуд подключали для вакуумной дистилляции, и партию концентрировали от 28,0 л до 12,0 л в течение 2 суток (в течение ночи сохраняли при 20°C) при сохранении температуры партии между 20 и 55°C. В этой точке анализ 1H ЯМР показал молярное соотношение уксусной кислоты к этилацетату 1,0:0,015. Уксусную кислоту (4,0 л) загружали в партию, и партию подвергали дистилляции до 12 л. Анализ 1H ЯМР показал молярное соотношение уксусной кислоты к этилацетату 1,0:0,0036. Уксусную кислоту (20,0 л) загружали в партию, и температуру партии доводили до 70,0°C. Из партии отбирали образцы для анализа ВЭЖХ, и соединение 2 составляло 0,16%. Ацетат натрия (2,20 кг) добавляли к партии, и температуру партии доводили от 72,4°C до 110,0°C. Через 18,5 часов анализ ВЭЖХ не показал детектированного промеж. В. Температуру партии доводили от 111,3 до 74,7°C и DI воду (30,0 л) добавляли к партии в течение 2 часов. Температуру партии доводили до 20,5°C и затем фильтровали с использованием 24” нутч-фильтра из хастеллоя, оборудованного полипропиленовой фильтровальной тканью. Ранее подготовленный раствор 1:1 уксусной кислоты в DI H2O (10,0 л) загружали в сосуд и встряхивали в течение 5 минут. Смыв переносили на фильтр, и затем партию вручную ресуспендировали в фильтре перед повторным включением вакуума. DI H2O (10,0 л) загружали в сосуд и затем переносили на фильтр. Партию вручную ресуспендировали в фильтре перед повторным включением вакуума. DI H2O (10,0 л) загружали непосредственно на фильтр, и затем партию вручную ресуспендировали в фильтре перед повторным включением вакуума. Отфильтрованному осадку позволяли кондиционироваться в течение 18 часов с получением 14,4 кг соединения 4. Анализ ВЭЖХ показал чистоту 93,7%. Этот влажный отфильтрованный осадок переносили для очистки. В 100 л стеклянный сосуд с рубашкой (продутый N2) загружали неочищенное соединение 4 (влажный отфильтрованный осадок 14,42 кг), уксусную кислоту (48,8 л) и запускали мешалку. Загружали DI H2O (1,74 л). Температуру партии (взвеси) доводили от 18,1 до 100,1°C в течение 4,25 часов. Партию держали при 100,1-106,1°C в течение 1 часа и затем доводили до 73,1°C. DI H2O (28,0 л) добавляли к партии в течение 1 час, поддерживая температуру партии между 73,1 и 70,3°C. Температуру партии далее доводили от 70,3°C до 25,0°C в течение ночи. Партию фильтровали с использованием 24” нутч-фильтра из хастеллоя, оборудованного полипропиленовой фильтровальной тканью. Фильтрация занимала 13 минут. Раствор DI H2O (9,00 л) и уксусной кислоты (11,0 л) подготавливали и добавляли в 100 л сосуд. Смесь встряхивали в течение 5 минут и затем переносили на отфильтрованный осадок. DI H2O (20,0 л) загружали в сосуд, встряхивали в течение 6 минут и затем переносили на отфильтрованный осадок. DI H2O (20,0 л) загружали в сосуд, встряхивали в течение 9 минут и затем переносили на отфильтрованный осадок. Партии позволяли кондиционироваться в течение 3 суток и затем переносили в лотки для сушки в вакуумной печи. Через 3 суток при 50°C и 28”/Hg (94820 Па), для партии получили 74% выход (3,7 кг) соединения 4 в форме грязно-белого твердого вещества. Спектр 1H ЯМР был согласованным с заданной структурой, анализ ВЭЖХ показал чистоту 98,87%, и анализ KF показал 0,14% H2O.
Синтез промеж. 7: В 100-л стеклянный сосуд с рубашкой (продутый N2) загружали тетрагидрофуран (44,4 л). Мешалку включали (125 об./мин) и загружали соединение 4 (3,67 кг), затем хлорид лития (1,26 кг). Наблюдали, что температура партии составляла 26,7°С, и раствор был янтарным. Добавляли 1,64 молярный раствор бромида изопропенилмагния в 2-метил-THF (21,29 кг) в течение 2 часов, поддерживая партию между 24,3 и 33,6°C. Партию встряхивали при 24,5°C в течение 17 часов, в этой точке анализ ВЭЖХ показал 9% соединения соединения. Во 2-й 100-л стеклянный сосуд с рубашкой (продутый N2) загружали 3н хлороводород (18,3 л). Партию переносили в сосуд, содержащий 3н HCl, на 25 минут, поддерживая температуру партии между 20 и 46°C. Наблюдали двухфазный раствор. Партию после остановки реакции переносили обратно в 1-й 100-л сосуд для остановки реакции в оставшемся небольшом количестве осадка. THF (2,00 л) использовали в качестве промывки. Наблюдали температуру партии 40,9°С, и ее встряхивали при 318 об./мин в течение 45 минут. Температуру партии доводили до 21,8°С, и позволяли разделение слоев. Разделение занимало 10 минут. Нижнюю водную фазу удаляли (~26,0 л). Раствор хлорида натрия (1,56 кг) в DI воде (14,0 л) получали и добавляли к партии. Это встряхивали при 318 об./мин в течение 10 минут, и мешалку останавливали. Разделение занимало 3 минут. Нижнюю водную фазу удаляли (~16,0 л). Партию подвергали вакуумной дистилляции от 58,0 л до 18,4 л с использованием ~24”/Hg (81270 Па) и температуры рубашки 50-55°C. Раствор гидроксида калия (2,30 кг) в DI воде (20,7 л) подготавливали в 72-л круглодонной колбе. Сосуд подключали для атмосферной дистилляции с использованием 2 дистилляционных головок, и партию переносили в 72-л флакон. THF (0,75 л) использовали в качестве промывки. Объем партии составлял ~41,0 л, температуру доводили до 64,1°C, и дистилляцию начинали с помощью обдувки N2. Нагревание продолжали для доведения температуры партии до 85,4°C во время дистилляции, в этой точке 72-л сосуд подключали для кипячения с обратным холодильником (объем партии составлял приблизительно 28,0 л в конце дистилляции). Партию держали при 85°C в течение 13 часов, в этой точке анализ ВЭЖХ показал 0,3% соединение 6A. Нагревание останавливали, и партию переносили в 100-л стеклянный сосуд с рубашкой. Наблюдали твердые вещества. Температуру партии доводили от 70,6°C до 56,7°C. Ранее приготовленный раствор гидрокарбоната натрия (2,82 кг) в DI воде (35,0 л) добавляли в течение 80 минут, сохраняя температуру партии между 56,7 и 46,7°C. pH партии в конце добавления составлял 9,8. Партию держали при 46,7-49,0°C в течение 40 минут и затем охлаждали до 25,0°C. Партию фильтровали с использованием 18” нутч-фильтра из нержавеющей стали. DI воду (18,4 л) загружали в сосуд и переносили на фильтр. Отфильтрованный осадок вручную ресуспендировали на фильтре и затем жидкости удаляли. Этот процесс повторяли еще раз, и отфильтрованный осадок имел толщину 3” (7,62 см). Отфильтрованный осадок кондиционировали на фильтре в течение 3 суток, переносили в лотки для сушки и сушили в вакуумной печи при 45°C с получением 2,93 кг промеж. 7 (выход 95%) с чистотой по ВЭЖХ 87,6%.
часов, поддерживая партию между 24,3 и 33,6°C. Партию встряхивали при 24,5°C в течение 17 часов, в этой точке анализ ВЭЖХ показал 9% соединения соединения. Во 2-й 100-л стеклянный сосуд с рубашкой (продутый N2) загружали 3н хлороводород (18,3 л). Партию переносили в сосуд, содержащий 3н HCl, на 25 минут, поддерживая температуру партии между 20 и 46°C. Наблюдали двухфазный раствор. Партию после остановки реакции переносили обратно в 1-й 100-л сосуд для остановки реакции в оставшемся небольшом количестве осадка. THF (2,00 л) использовали в качестве промывки. Наблюдали температуру партии 40,9°С, и ее встряхивали при 318 об./мин в течение 45 минут. Температуру партии доводили до 21,8°С, и позволяли разделение слоев. Разделение занимало 10 минут. Нижнюю водную фазу удаляли (~26,0 л). Раствор хлорида натрия (1,56 кг) в DI воде (14,0 л) получали и добавляли к партии. Это встряхивали при 318 об./мин в течение 10 минут, и мешалку останавливали. Разделение занимало 3 минут. Нижнюю водную фазу удаляли (~16,0 л). Партию подвергали вакуумной дистилляции от 58,0 л до 18,4 л с использованием ~24”/Hg (81270 Па) и температуры рубашки 50-55°C. Раствор гидроксида калия (2,30 кг) в DI воде (20,7 л) подготавливали в 72-л круглодонной колбе. Сосуд подключали для атмосферной дистилляции с использованием 2 дистилляционных головок, и партию переносили в 72-л флакон. THF (0,75 л) использовали в качестве промывки. Объем партии составлял ~41,0 л, температуру доводили до 64,1°C, и дистилляцию начинали с помощью обдувки N2. Нагревание продолжали для доведения температуры партии до 85,4°C во время дистилляции, в этой точке 72-л сосуд подключали для кипячения с обратным холодильником (объем партии составлял приблизительно 28,0 л в конце дистилляции). Партию держали при 85°C в течение 13 часов, в этой точке анализ ВЭЖХ показал 0,3% соединение 6A. Нагревание останавливали, и партию переносили в 100-л стеклянный сосуд с рубашкой. Наблюдали твердые вещества. Температуру партии доводили от 70,6°C до 56,7°C. Ранее приготовленный раствор гидрокарбоната натрия (2,82 кг) в DI воде (35,0 л) добавляли в течение 80 минут, сохраняя температуру партии между 56,7 и 46,7°C. pH партии в конце добавления составлял 9,8. Партию держали при 46,7-49,0°C в течение 40 минут и затем охлаждали до 25,0°C. Партию фильтровали с использованием 18” нутч-фильтра из нержавеющей стали. DI воду (18,4 л) загружали в сосуд и переносили на фильтр. Отфильтрованный осадок вручную ресуспендировали на фильтре и затем жидкости удаляли. Этот процесс повторяли еще раз, и отфильтрованный осадок имел толщину 3” (7,62 см). Отфильтрованный осадок кондиционировали на фильтре в течение 3 суток, переносили в лотки для сушки и сушили в вакуумной печи при 45°C с получением 2,93 кг промеж. 7 (выход 95%) с чистотой по ВЭЖХ 87,6%.
Синтез промеж. 8: В 100 л стеклянный сосуд с рубашкой (продутый N2 и присоединенный к щелочному скрубберу) загружали кислую кислоту (13,0 л). Промеж. 7 (2,85 кг) загружали в сосуд и запускали мешалку. N-цианоацетилуретан (1,56 кг) и DI воду (5,70 л) загружали в сосуд. Температуру партии доводили от 17,0°C до 5,5°C, и наблюдали тонкую взвесь. В этой точке добавляли 37% хлороводород (2,70 л) в течение 10 минут, сохраняя температуру партии между 4,8°C и 8,8°C. Ранее приготовленный раствор нитрита натрия (638 г) в DI воде (1,42 л) добавляли в течение 26 минут, сохраняя температуру партии между 5,8°C и 8,7°C. Коричневый газ наблюдали в пространстве крышки сосуда во время добавления. Анализ ВЭЖХ не показал детектированного промеж. 7. В этой точке ранее приготовленный раствор ацетата натрия (2,07 кг) в DI воде (8,50 л) добавляли в течение 47 минут, сохраняя температуру партии между 5,5°C и 9,5°C. После добавления наблюдали тонкий слой оранжевого осадка на стенке сосуда немного выше уровня партии. Температуру партии доводили от 9,4°C до 24,5°C и держали при 25°C (±5°C) в течение 12 часов. Партию фильтровали с использованием 24” нутч-фильтра из хастеллоя, оборудованного полипропиленовой фильтровальной тканью плотного переплетения. Фильтрация занимала 30 минут. Сосуд промывали 14,3 л 1:1 кислой кислотой/DI водой. Оранжевый осадок на реакторе отмылся при промывке. Смыв переносили на фильтр, где партию ресуспендировали вручную. Снова подключали вакуум, чтобы убрать смыв. 2-ю промывку 1:1 кислой кислотой/DI водой проводили, как выше, и партию кондиционировали на фильтре в течение 26 часов. Анализ ВЭЖХ влажного отфильтрованного осадка показал чистоту 90,4%. Партию сушили до постоянной массы 3,97 кг (выход 91%) в вакуумной печи при 45°C и 28”/Hg (94820 Па).
Получение DMAC сольвата соединения A
В 100 л стеклянный сосуд с рубашкой, продутый N2, загружали промеж. 8 (3,90 кг) и ацетат калия (875 г). N,N-диметилацетамид (DMAC, 18,3 л) загружали в сосуд и запускали мешалку. Температуру партии доводили до 115°C в течение 2 час. Через 2 час при 115°C из партии отбирали образцы, и анализ ВЭЖХ показал 0,27% оставшегося промеж. 8. Температуру партии доводили до 25,0°C в течение ночи. Уксусную кислоту (975 мл) добавляли к партии, и партию дополнительно встряхивали в течение 3 час. Партию переносили в бутыль, и сосуд промывали дочиста с помощью 800 мл DMAC. Партию переносили обратно в 100 л сосуд с использованием вакуума через 10 мкм встроенный фильтр и использовали промывку DMAC (1,15 л). Фильтрация являлась быстрой в начале, но медленной в конце, засоряя фильтр. Температуру партии доводили до 11,1°C и DI воду (35,1 л) добавляли в течение 2 час 20 мин, сохраняя температуру партии между 5-15°C. Партию держали в течение 1 час и фильтровали с использованием 18” нутч-фильтра, оборудованного полипропиленовой фильтровальной тканью плотного переплетения. Фильтрация занимала 15 час. Промывку 1:1 этанолом/DI водой (19,5 л) загружали в сосуд, охлаждали до 10°C, и переносили на отфильтрованный осадок. Осадку позволяли кондиционироваться под N2 и вакуумом в течение 8 час и переносили в лотки для сушки. Партию сушили в вакуумной печи при 45°C и 28”/Hg (94820 Па) с получением 89% выхода (3,77 кг) DMAC сольвата соединения A в форме оранжевого/коричневого твердого вещества. Спектр 1H ЯМР был согласованным с заданной структурой, и анализ Карла Фишера показал 0,49% H2O. XRPD показала ожидаемую форму, т.е., DMAC сольват соединения A. Термогравиметрический анализ (TGA) показал потерю 16% массы. Анализ ВЭЖХ показал чистоту 93,67%.
Получение неочищенного соединения A
В 100 л стеклянный сосуд с рубашкой, продутый N2, загружали DMAC сольват соединения A (3,75 кг) и этанол (15,0 л). Запускали мешалку и добавляли ацетон (15,0 л). Температуру партии доводили от 10,6°C до 60,0°C в течение 1 час. В этой точке партия находилась в растворе. DI воду добавляли к партии в течение 1,5 час, сохраняя температуру партии при 60±5°C. Партию держали при 60±5°C в течение 1 час и охлаждали до 23,5°C. Присоединяли 18” нутч-фильтр, оборудованный полипропиленовой тканью плотного переплетения (0,67 CFM), и партию фильтровали. Фильтрация занимала 15 час. Промывку 1:1 этанолом/DI водой (19,5 л) загружали в сосуд и переносили на отфильтрованный осадок. Отфильтрованному осадку позволяли кондиционироваться под N2 и вакуумом в течение 8 час и переносили в лотки для сушки. Партию сушили в вакуумной печи при 45°C и 28”/Hg (94820 Па) в течение пяти суток с получением 94% выхода (2,90 кг) соединения A в форме порошкообразного коричневого твердого вещества. Спектр 1H ЯМР был согласованным с заданной структурой, и анализ Карла Фишера показал 6,6% H2O. XRPD показала ожидаемую форму дигидрата. TGA показал потерю 6,7% массы. Анализ ВЭЖХ показал чистоту 96,4% (AUC).
Очистка неочищенного соединения A
В 50 л стеклянный сосуд с рубашкой, продутый N2, загружали неочищенное соединение A (2,90 кг) и метилизобутилкетон (14,5 л). Запускали мешалку, и температуру партии доводили от 20,2°C до 50,4°C в течение 1,5 час. Партию держали при 50°C (±5°C) в течение 1 час и охлаждали до 20-25°C. Партию держали при 20-25°C в течение 2,5 час. Присоединяли 18” нутч-фильтр, оборудованный полипропиленовой тканью плотного переплетения (0,67 CFM), и партию фильтровали. Фильтрация занимала 20 мин. Метилизобутилкетон (MIBK, 1,45 л) загружали в сосуд и переносили на отфильтрованный осадок. Отфильтрованный осадок ресуспендировали вручную, и жидкости прокачивали с помощью вакуума. Метилизобутилкетон (2,90 л) загружали на отфильтрованный осадок, и отфильтрованный осадок ресуспендировали вручную. Жидкости прокачивали с помощью вакуума, и отфильтрованный осадок кондиционировали с помощью вакуума и азота в течение 15 час. Отфильтрованный осадок высушивали до коричневого твердого диска 18” × 1 ” (45,72×3,81 см). Его разламывали вручную и пропускали через кофейные мельницы с получением 76% выхода (2,72 кг) MGL-3196 MIBK сольвата в форме коричневого порошкообразного твердого вещества. Не было необходимости в сушке в печи. Спектр 1H ЯМР был согласованным с заданной структурой, и анализ Карла Фишера показал <0,1% H2O. XRPD показала ожидаемую форму MIBK сольвата. TGA показал потерю 17,3% массы. Анализ ВЭЖХ показал чистоту 98,5%.
” (45,72×3,81 см). Его разламывали вручную и пропускали через кофейные мельницы с получением 76% выхода (2,72 кг) MGL-3196 MIBK сольвата в форме коричневого порошкообразного твердого вещества. Не было необходимости в сушке в печи. Спектр 1H ЯМР был согласованным с заданной структурой, и анализ Карла Фишера показал <0,1% H2O. XRPD показала ожидаемую форму MIBK сольвата. TGA показал потерю 17,3% массы. Анализ ВЭЖХ показал чистоту 98,5%.
Пример 6: Перевод соединения A в форму I
Очищенное соединение A (4802 г) в форме 1:1 MIBK сольвата, полученное из промеж. 8, как описано в примере 5 выше, добавляли в 100 л реактор с рубашкой вместе с 24 литрами этанола. Полученную взвесь нагревали до 80±5°C (с обратным холодильником) в течение 1 час 25 мин; смесь перемешивали при этой температуре в течение 4 час 25 мин. Анализ фильтрованных твердых веществ через 2 час 55 мин показал, что превращение форм было завершено, со спектрами XRPD, согласующимися с формой I. Смесь охлаждали до 20±5°C в течение 45 мин и перемешивали при этой температуре в течение 15 мин. Взвесь фильтровали, и отфильтрованный осадок промывали дважды предварительно фильтрованным этанолом (2×4,8 л). Влажный отфильтрованный осадок (4,28 кг) сушили в вакууме при 40±5°C в течение 118 час с получением 3390 г соединения A, формы I.
Исследование рентгеновской порошковой дифракции проводили для различных партий соединения A, морфологической формы I, полученной способом, описанным выше. XRPD после тонкого измельчения подтвердила форму 1.
Данные для формы I представлены в таблице 11 ниже, и дифрактограммы формы I представлены в качестве фиг. 1.
(Å)
(счет)
Обнаружено, что форма I имеет начало плавления приблизительно 321°C, с последующим распадом после плавления по DSC (фигура 2).
Пример 7: Получение формы I соединения A: перевод сольвата соединения A в форму I
В 50 л стеклянный сосуд с рубашкой, продутый N2, загружали MIBK сольват соединения A (2,72 кг) из примера 5 выше и этанол (13,6 л). Запускали мешалку, и температуру партии доводили от 16,8°C до 79,4°C в течение 1,3 час. Партию держали при 79,5°C в течение 2 час и отбирали образцы для анализа XRPD. XRPD показала форму I, и партию охлаждали до 24,9°C в течение 1 час и 10 мин. Присоединяли 18” нутч-фильтр, оборудованный полипропиленовой тканью плотного переплетения (0,67 CFM), и партию фильтровали. Фильтрация занимала 4 мин. Этанол (2,8 л) загружали в сосуд и переносили на отфильтрованный осадок. Отфильтрованный осадок ресуспендировали вручную и жидкости прокачивали с помощью вакуума. Этанол (2,80 л) загружали на отфильтрованный осадок и отфильтрованный осадок ресуспендировали вручную. Жидкости прокачивали с помощью вакуума, и отфильтрованный осадок кондиционировали с помощью вакуума и азота в течение 1 час. Отфильтрованный осадок переносили в тарелки сушилки и высушивали при 45°C и 28”/Hg (94820 Па) в течение одних суток с получением 89% выхода (1,96 кг) соединения A в форме светло-желтого твердого вещества. Анализ ВЭЖХ показал чистоту 99,6%. Анализ XRPD был согласован с формой I. При тонком измельчении 300 г этого материла в 2” струйной мельнице получили 284 г (95% выход) тонко измельченного соединения A. Анализ XRPD подтвердил, что тонко измельченное соединение A сохраняет форму I.
DMAC сольват соединения A можно переводить, через дигидрат и MIBK сольват, в форму I, как описано в примере 7. Альтернативно, DMAC сольват переводили напрямую в форму I с 75% выходом (выход рассчитывали по промежуточному соединению 8) посредством его нагревания с 8 объемами этанола до 80°C в течение 2 часов с последующим охлаждением до комнатной температуры и фильтрованием. В другой реакции образец соединения A, представлявший собой смесь DMAC сольвата и дигидрата, переводили в форму I с выходом 69% посредством его нагревания с 8 объемами MIBK до 80°C с последующим охлаждением до комнатной температуры.
Моделирование взаимодействия между соединением A и рецептором гормонов щитовидной железы
Кристаллические структуры получали из базы данных белков RCSB (номера ID: 1N46, 1NQ0, 1NQ1, 1NQ2 и 1NUO). Совместные кристаллические структуры белков выравнивали с использованием MacPymol для Mac OS X (Copyright 2006 DeLano Scientific LLC.; в настоящее время продукт Schrodinger Inc.). MacPymol использовали также для всех анализов взаимодействий лиганд-белок и для получения фигур 3-9. На этих фигурах показано, что, в общем, соединение A лучше способно вмещать структурные варианты мутантов THRβ. Например, в мутантном Arg316His, Arg316 мутирован до His и Arg320 немного отодвинут от лиганда. В результате, специфическое взаимодействие между Arg320 и T3 является менее оптимальным в мутанте Arg316His.
В отличие от этого, большой, способный к отрицательной поляризации гетероцикл в соединении A формирует предоставляющие преимущество взаимодействия, не нарушаемые мутацией Arg316His. Иными словами, соединение A, имеющее больший, более способный к поляризации гетероцикл, сохраняет предоставляющие преимущество взаимодействия с Arg320 и мутированным His316. См., например, фигуры 8 и 9. Результаты являются сходными для других мутаций.
В таблице ниже перечислены биохимические свойства конкретных мутантов TRβ. Другие мутанты и их свойства можно обнаружить, например, в M. Adams et al., J Clin Invest. 1994; 94(2): 506-515, B.R. Huber et al., Mol Endocrinol, 2003, 17(4):643-652; и B.R. Huber et al., Mol Endocrinol, 2003, 17(1): 107-116, полное содержание каждого из которых, таким образом, приведено в качестве ссылки.
ЭКВИВАЛЕНТЫ
Изобретение можно осуществлять в других конкретных формах без отклонения от его содержания или существенных характеристик. Вышеуказанные варианты осуществления, таким образом, следует рассматривать как во всех отношениях иллюстративные, а не ограничивающие изобретение, описанное в настоящем документе. Объем изобретения, таким образом, указан в прилагаемой формуле изобретения, а не в предшествующем описании, и все изменения, возникающие в пределах смысла и объема эквивалентности формулы изобретения, предназначены для включения в него
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Hoffmann-La Roche, Inc.
F. Hoffmann-La Roche Ltd.
Madrigal Pharmaceuticals, Inc.
<120> СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ
<130> 41245-522001WO
<150> US 61/702,137
<151> 2012-09-17
<150> US 61/790,432
<151> 2013-03-15
<160> 2
<170> PatentIn версии 3.5
<210> 1
<211> 259
<212> PRT
<213> Homo sapiens
<400> 1
Glu Leu Gln Lys Ser Ile Gly His Lys Pro Glu Pro Thr Asp Glu Glu
1 5 10 15
Trp Glu Leu Ile Lys Thr Val Thr Glu Ala His Val Ala Thr Asn Ala
20 25 30
Gln Gly Ser His Trp Lys Gln Lys Arg Lys Phe Leu Pro Glu Asp Ile
35 40 45
Gly Gln Ala Pro Ile Val Asn Ala Pro Glu Gly Gly Lys Val Asp Leu
50 55 60
Glu Ala Phe Ser His Phe Thr Lys Ile Ile Thr Pro Ala Ile Thr Arg
65 70 75 80
Val Val Asp Phe Ala Lys Lys Leu Pro Met Phe Cys Glu Leu Pro Cys
85 90 95
Glu Asp Gln Ile Ile Leu Leu Lys Gly Cys Cys Met Glu Ile Met Ser
100 105 110
Leu Arg Ala Ala Val Arg Tyr Asp Pro Glu Ser Glu Thr Leu Thr Leu
115 120 125
Asn Gly Glu Met Ala Val Thr Arg Gly Gln Leu Lys Asn Gly Gly Leu
130 135 140
Gly Val Val Ser Asp Ala Ile Phe Asp Leu Gly Met Ser Leu Ser Ser
145 150 155 160
Phe Asn Leu Asp Asp Thr Glu Val Ala Leu Leu Gln Ala Val Leu Leu
165 170 175
Met Ser Ser Asp Arg Pro Gly Leu Ala Cys Val Glu Arg Ile Glu Lys
180 185 190
Tyr Gln Asp Ser Phe Leu Leu Ala Phe Glu His Tyr Ile Asn Tyr Arg
195 200 205
Lys His His Val Thr His Phe Trp Pro Lys Leu Leu Met Lys Val Thr
210 215 220
Asp Leu Arg Met Ile Gly Ala Cys His Ala Ser Arg Phe Leu His Met
225 230 235 240
Lys Val Glu Cys Pro Thr Glu Leu Phe Pro Pro Leu Phe Leu Glu Val
245 250 255
Phe Glu Asp
<210> 2
<211> 780
<212> ДНК
<213> Homo sapiens
<400> 2
gagctgcaga agtccatcgg gcacaagcca gagcccacag acgaggaatg ggagctcatc 60
aaaactgtca ccgaagccca tgtggcgacc aacgcccaag gcagccactg gaagcaaaaa 120
cggaaattcc tgccagaaga cattggacaa gcaccaatag tcaatgcccc agaaggtgga 180
aaggttgact tggaagcctt cagccatttt acaaaaatca tcacaccagc aattaccaga 240
gtggtggatt ttgccaaaaa gttgcctatg ttttgtgagc tgccatgtga agaccagatc 300
atcctcctca aaggctgctg catggagatc atgtcccttc gcgctgctgt gcgctatgac 360
ccagaaagtg agactttaac cttgaatggg gaaatggcag tgacacgggg ccagctgaaa 420
aatgggggtc ttggggtggt gtcagacgcc atctttgacc tgggcatgtc tctgtcttct 480
ttcaacctgg atgacactga agtagccctc cttcaggccg tcctgctgat gtcttcagat 540
cgcccggggc ttgcctgtgt tgagagaata gaaaagtacc aagatagttt cctgctggcc 600
tttgaacact atatcaatta ccgaaaacac cacgtgacac acttttggcc aaaactcctg 660
atgaaggtga cagatctgcg gatgatagga gcctgccatg ccagccgctt cctgcacatg 720
aaggtggaat gccccacaga actcttcccc cctttgttct tggaagtgtt cgaggattag 780
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| АГОНИСТ РЕЦЕПТОРОВ THRβ И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2019 |
|
RU2809040C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА ТИРЕОИДНОГО ГОРМОНА | 2006 |
|
RU2379295C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ ПРОИЗВОДНЫХ ТРЕТ-БУТИЛ 4-((1R,2S,5R)-6-(БЕНЗИЛОКСИ)-7-ОКСО-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИДО)ПИПЕРИДИН-1-КАРБОКСИЛАТА | 2014 |
|
RU2689339C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ФЕНИЛАТНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЯ | 2020 |
|
RU2820475C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(ЦИКЛОПРОПИЛМЕТОКСИ)-N-(3,5-ДИХЛОР-1-ОКСИДО-4-ПИРИДИЛ)-5-МЕТОКСИПИРИДИН-2-КАРБОКСАМИДА | 2013 |
|
RU2635094C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ МЕК И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2414455C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| ПРОИЗВОДНЫЕ 4-ХИНОЛИНИЛА, ОБЛАДАЮЩИЕ АНТИГЕЛИКОБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1993 |
|
RU2130933C1 |
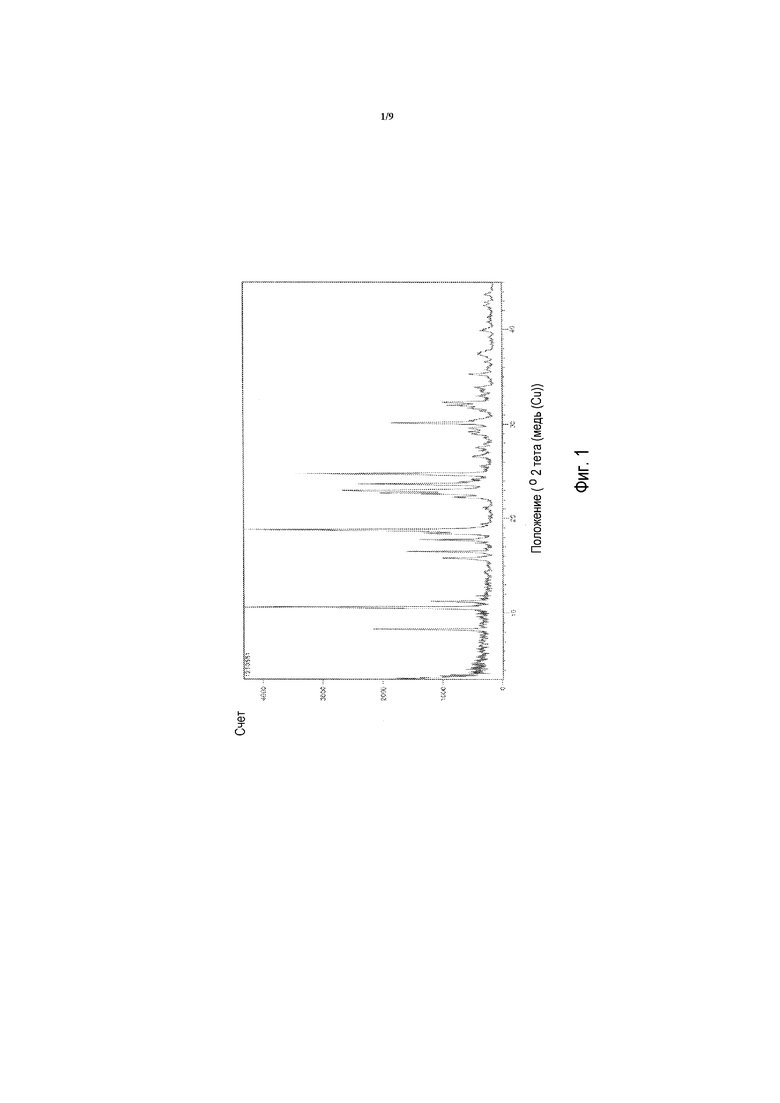

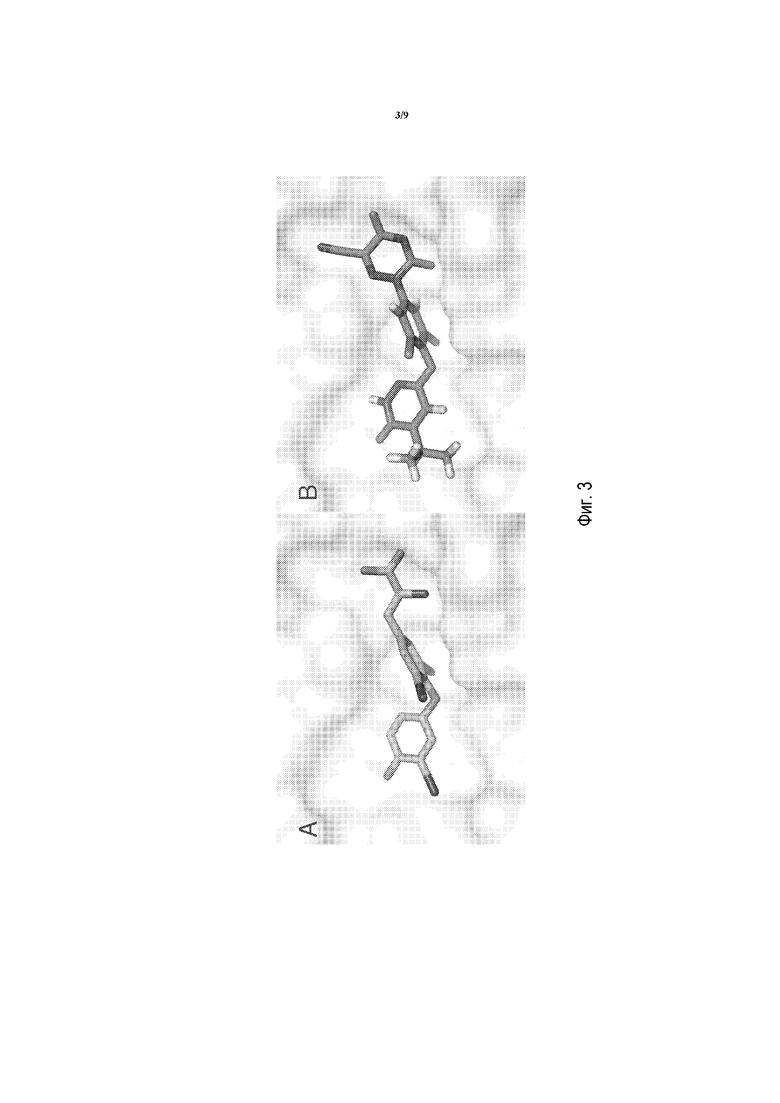
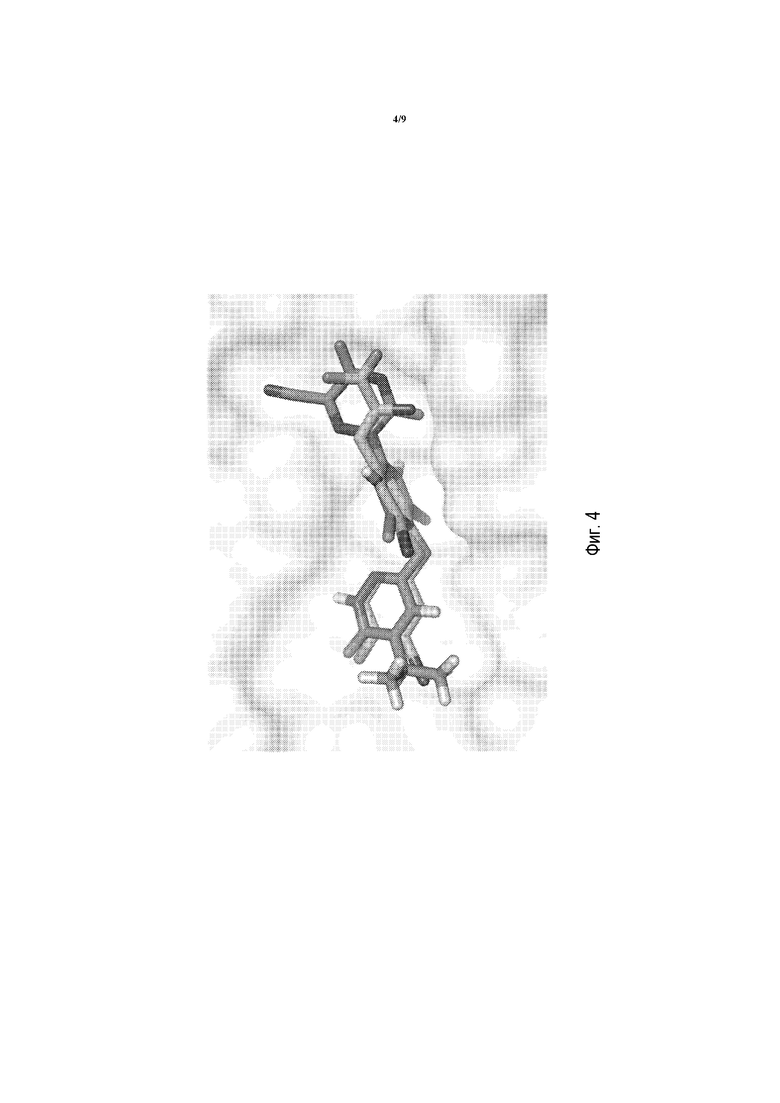
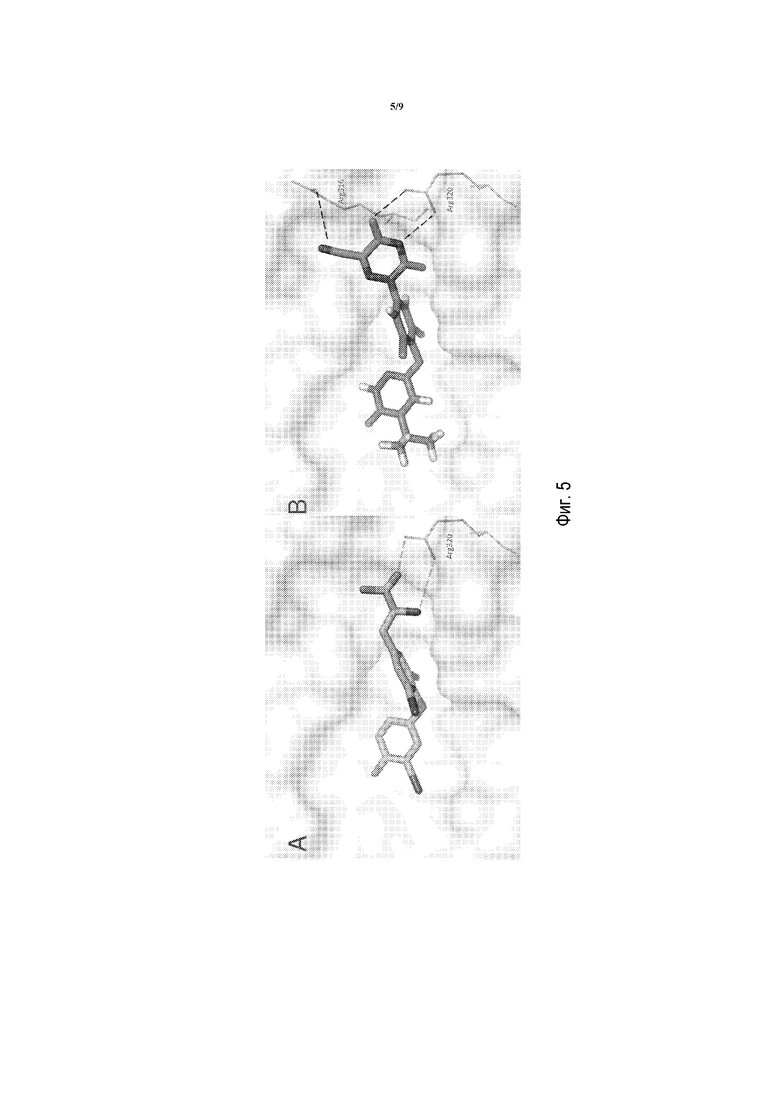
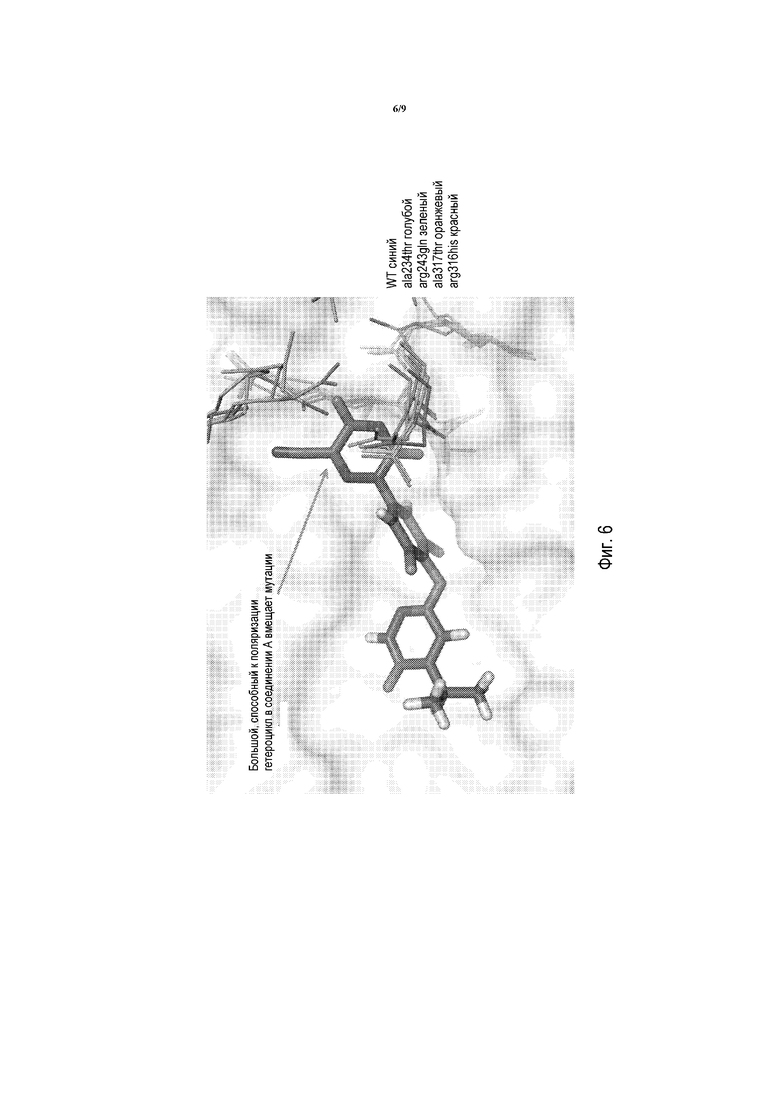
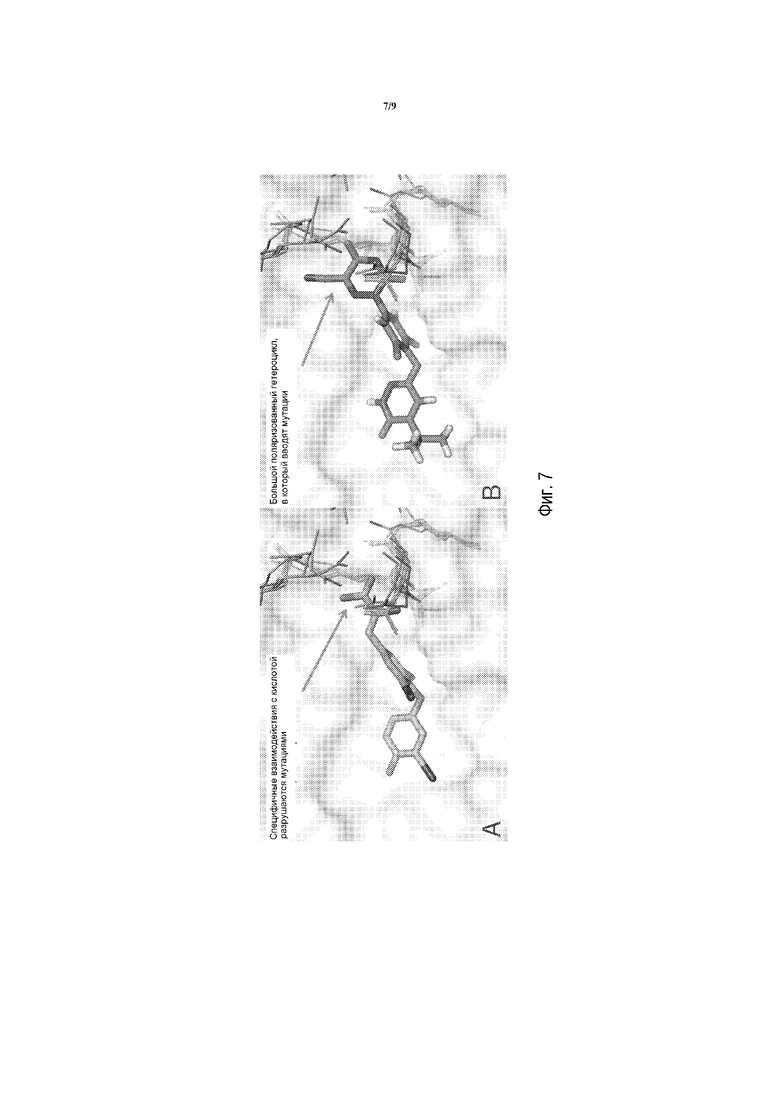
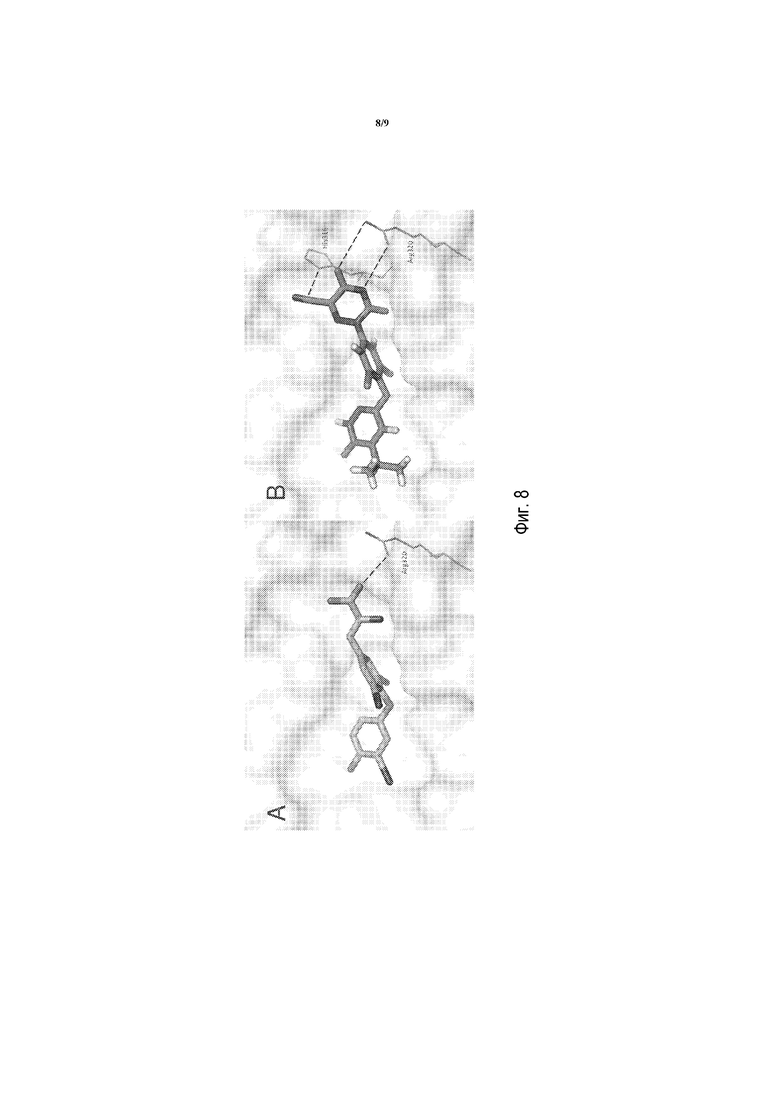
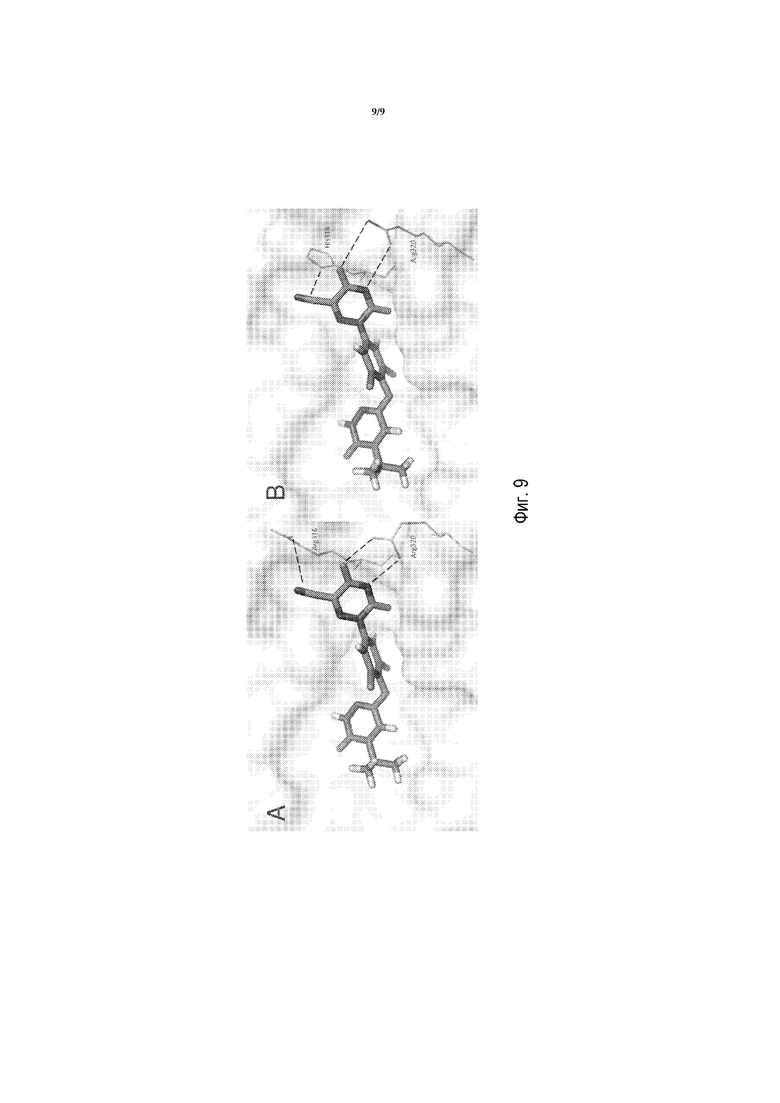
Изобретение относится к способу синтеза для получения соединения пиридазинона. Способ включает: (a) приведение в контакт R1MgX с соединением формулы (I)
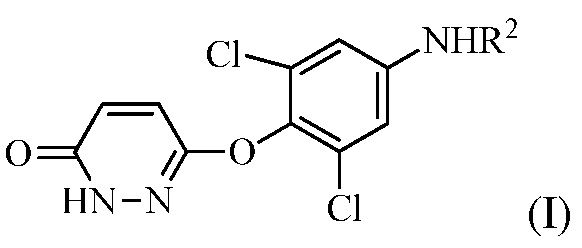
с получением соединения формулы (II)
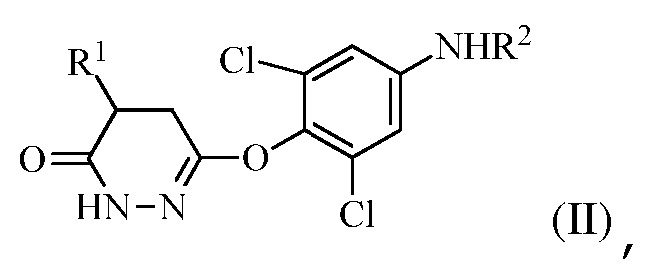
в которой R1 представляет собой изопропил или изопропенил, X представляет собой галоген и R2 представляет собой H или защитную группу для амина; и (b) перевод соединения формулы (II) в соединение формулы (III)
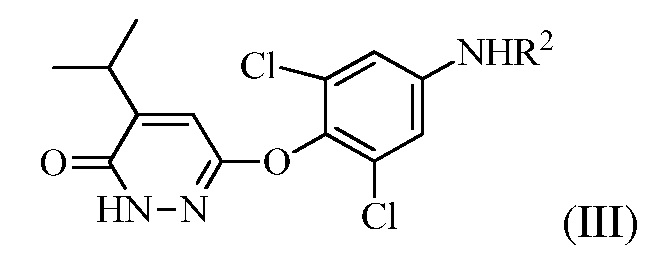
в присутствии основания, когда R1 представляет собой изопропенил, или в присутствии окисляющего средства, когда R1 представляет собой изопропил. Также предложены соединения, пригодные для осуществления указанного способа. Предложенный способ позволяет крупномасштабное получение пиридазиноновых соединений, обладающих высокой чистотой, с высоким выходом. 2 н. и 22 з.п. ф-лы, 9 ил., 12 табл., 7 пр.
1. Способ синтеза для получения соединения пиридазинона, включающий:
(a) приведение в контакт R1MgX с соединением формулы (I)
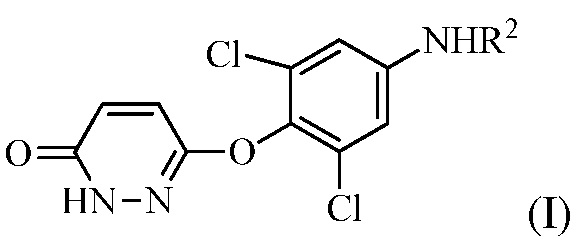
с получением соединения формулы (II)
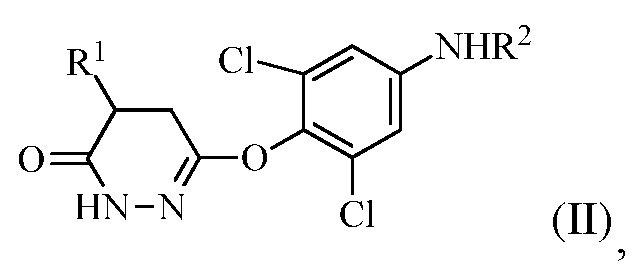
в которой R1 представляет собой изопропил или изопропенил, X представляет собой галоген и R2 представляет собой H или защитную группу для амина; и
(b) перевод соединения формулы (II) в соединение формулы (III)
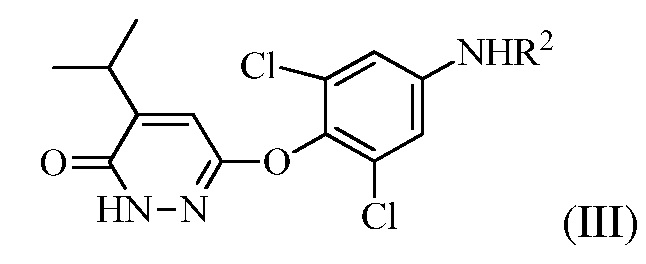
в присутствии основания, когда R1 представляет собой изопропенил, или в присутствии окисляющего средства, когда R1 представляет собой изопропил.
2. Способ по п.1, дополнительно включающий: (c) удаление защитной группы для амина R2 соединения формулы (III), когда присутствует, с получением 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она.
3. Способ по п.1, где стадию (a) осуществляют посредством приведения в контакт R1MgX с соединением формулы (I), в котором R1 представляет собой изопропенил и X представляет собой Br.
4. Способ по п.1, где основание на стадии (b) представляет собой гидроксид металла.
5. Способ по п.1, где R2 представляет собой ацетил или бензоил.
6. Способ по п.1, где соединение формулы (I) представляет собой
 или
или 
7. Способ по п.1, в котором соединение формулы (II) представляет собой
 ,
, 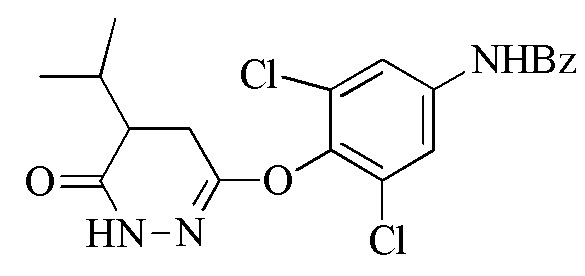 ,
, 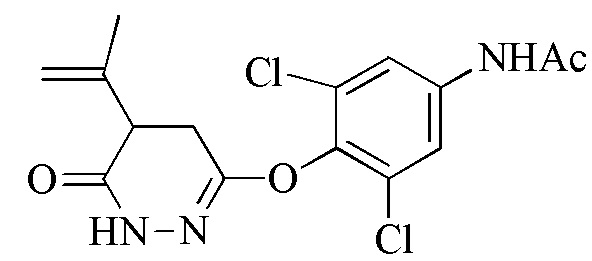 или
или 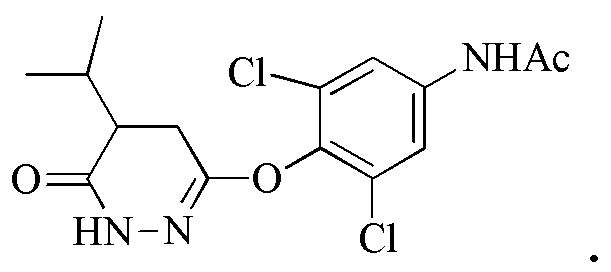
8. Способ по п.1, где соединение формулы (III) представляет собой
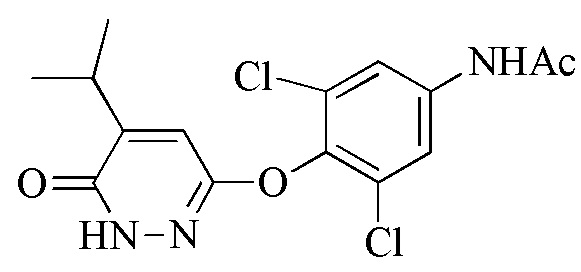 или
или 
9. Способ по п.1, дополнительно включающий получение соединения формулы (I) путем приведения в контакт 3,6-дихлорпиридазина с 2,6-дихлор-4-аминофенолом в полярном апротонном растворителе в присутствии основания при температуре реакции от 60 до 120°C с образованием 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина, гидролиза 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина и защиты аминогруппы 3,5-дихлор-4-((6-хлорпиридазин-3-ил)окси)анилина либо до, либо после гидролиза для образования соединения формулы (I).
10. Способ по п.9, в котором полярный апротонный растворитель представляет собой диметилацетамид (DMAC), основание представляет собой Cs2CO3, а температура реакции составляет 65°C.
11. Способ по п.2, дополнительно включающий: (d) превращение 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она в соединение формулы (IV)
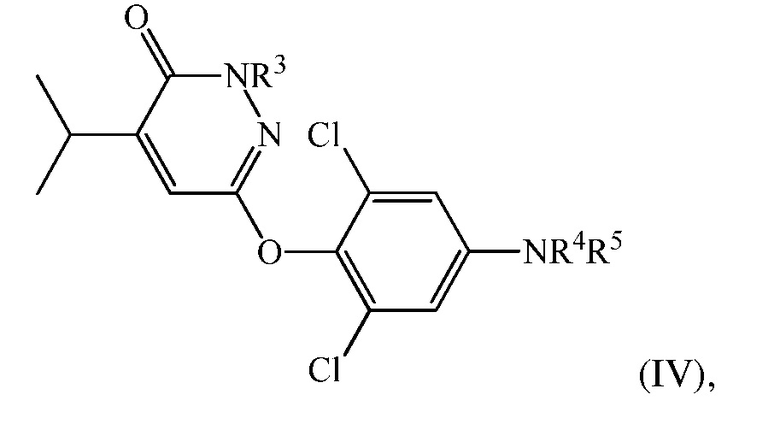
где R3 представляет собой H или CH2Ra, в котором Ra представляет собой гидроксил, О-связанную аминокислоту, -OP(O)(OH)2 или -OC(O)-Rb, где Rb представляет собой низший алкил, алкокси, алкилкислоту, циклоалкил, арил, гетероарил или -(CH2)n-гетероарил, и n представляет собой 0 или 1;
R4 представляет собой H; и
R5 представляет собой CH2COOH, C(O)CO2H или их сложный эфир или амид; или R4 и R5 вместе представляют собой -N=C(Rc)-C(O)-NH-C(O)-, в котором Rc представляет собой H или циано.
12. Способ по п.3, где стадию (a) проводят в THF с соотношением объема к массе THF к соединению формулы (I) в диапазоне между 7 мл/г и 15 мл/г.
13. Способ по п.3, где стадию (a) проводят в присутствии кислоты Льюиса.
14. Способ по п.13, где кислота Льюиса представляет собой галогенид лития.
15. Способ по п.1, где стадию (a) проводят посредством контакта R1MgX c соединением формулы (I), в котором R1 представляет собой изопропил и X представляет собой Cl.
16. Способ по п.15, где стадию (a) проводят в THF с соотношением объема к массе THF к соединению формулы (I) в диапазоне между 7 мл/г и 30 мл/г.
17. Способ по п.4, где гидроксид металла представляет собой гидроксид калия.
18. Способ по п.1, где окисляющее средство на стадии (b) представляет собой бром и стадию (b) проводят в присутствии кислоты.
19. Способ по п.5, где R2 представляет собой бензоил.
20. Способ по п.9, дополнительно включающий очистку соединения формулы (I) до стадии (a) в кислом растворе при температуре между 80 и 100°C.
21. Способ по п.9, где приведение в контакт 3,6-дихлорпиридазина с 2,6-дихлор-4-аминофенолом проводят в полярном апротонном растворителе в присутствии основания при температуре реакции между 60 и 120°C.
22. Способ по п.11, где соединение формулы (IV) представляет собой 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрил («Соединение A») и стадию (d) проводят посредством контакта 6-(4-амино-2,6-дихлорфенокси)-4-изопропилпиридазин-3(2H)-она с этил(2-цианоацетил)карбаматом и нитритом металла с последующей обработкой с помощью ацетата калия в DMAC.
23. Способ по п.11, дополнительно включающий получение кристаллической формы 2-(3,5-дихлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-карбонитрила («Соединения A»), характеризующейся картиной рентгеновской порошковой дифракции, включающей пики при 2θ 10,5, 18,7, 22,9, 23,6 и 24,7 градусов.
24. Соединение, пригодное для применения в способе синтеза по п. 1, где соединение представляет собой:
 ,
,  ,
, 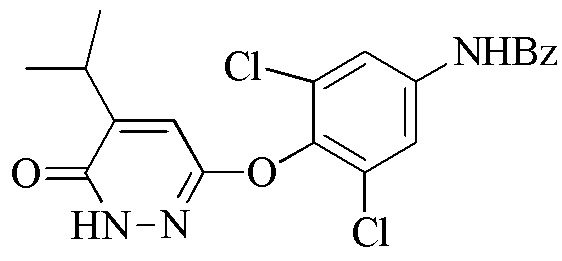 ,
, 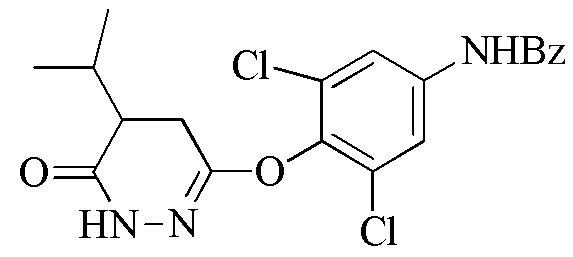 ,
, 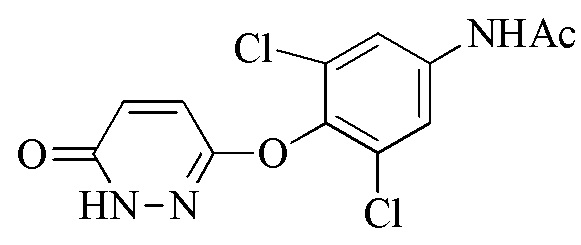 ,
, 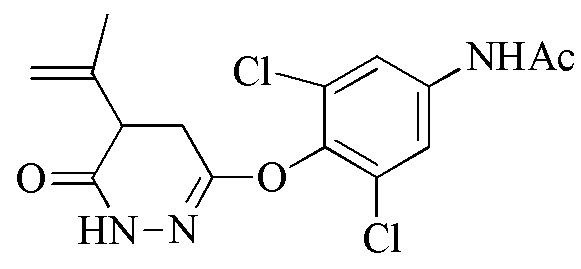 ,
,  ,
,  , или его соль.
, или его соль.
| WO 2007009913 A1, 25.01.2007 | |||
| WO 2009037172 A1, 26.03.2009 | |||
| БЕНЗИЛПИРИДАЗИНОНЫ КАК ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ | 2004 |
|
RU2344128C2 |
