Область техники
Настоящее изобретение относится к области лекарственных препаратов и, в частности, к галогензамещенному фениловому эфиру и его применению.
Уровень техники
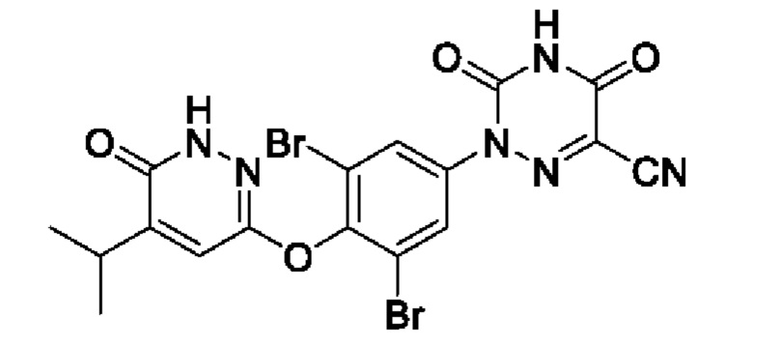
MGL-3196, хлоридзамещенное соединение фенилового эфира, представляет собой высокоселективный агонист рецептор тиреоидного гормона-бета (THR-β) со значением ЕС50 0,21 мкМ и следующей структурной формулой 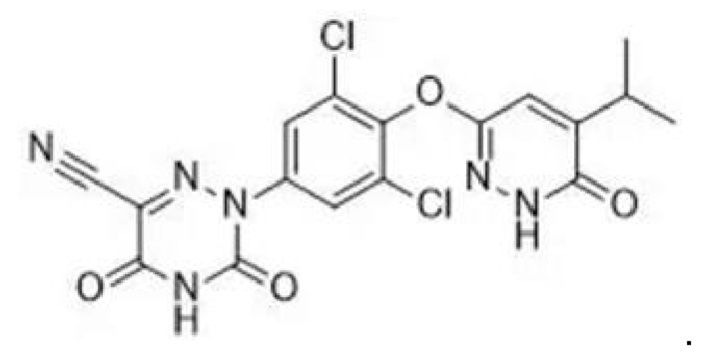 На данный момент проводятся клинические исследования в завершающей фазе, демонстрирующие терапевтический эффект при лечении дислипидемии, гиперхолестеринемии и неалкогольного стеатогепатита (НАСГ).
На данный момент проводятся клинические исследования в завершающей фазе, демонстрирующие терапевтический эффект при лечении дислипидемии, гиперхолестеринемии и неалкогольного стеатогепатита (НАСГ).
Установлено, что требуется улучшение фармакокинетических свойств и агонистической активности MGL-3196 in vivo при его использовании в качестве агониста THR-β. Поэтому одним из основных направлений исследований стала структурная модификация MGL-3196 и разработка препаратов на основе MGL-3196 с улучшенными свойствами. Однако химические модификации в различной степени приводят к изменению структуры соединения, а также его физических и химических свойств на различных уровнях, что, в свою очередь, приводит к непрогнозируемому изменению фармацевтических свойств соединения. Поэтому одной из наиболее сложных задач в данной области стала разработка способа химической модификации для получения лекарственного средства с улучшенными характеристиками.
Распространенным способом модификации является дейтерирование. Дейтерирование лекарственных средств включает замещение некоторых атомов водорода в молекуле лекарственного средства дейтерием. Поскольку дейтерий по форме и объему близок к водороду в молекулах лекарственного средства, дейтерированные лекарственные средства, как правило, сохраняют биологическую активность и селективность исходного лекарственного средства. Поскольку связь C-D является более стабильной, чем связь С-Н, разрушение связи в дейтерированных лекарственных средствах во время химической реакции менее вероятно, и, таким образом, период полувыведения может быть увеличен. Однако ввиду сложности метаболических процессов в живом организме на фармакокинетические свойства лекарственных средств in vivo влияет множество факторов, и они, соответственно, также сложны. Было показано, что изменения фармакокинетических свойств дейтерированных лекарственных средств имеют очевидные непредвиденные и непрогнозируемые последствия в сравнении с соответствующими недейтерированными лекарственными средствами. Поэтому вместо увеличения периода полувыведения дейтерирование по некоторым положениям может приводить к сокращению периода полувыведения (Scott L. Harbecon, Roger D. Tung. Deuterium in Drag Discovery and Development, P405-406) и ухудшению фармакокинетических свойств. С другой стороны, атомы водорода в некоторых положениях молекулы лекарственного средства сложно замещаются на дейтерий вследствие стерических затруднений и других причин. Поэтому дейтерирование лекарственных средств не является произвольным, а положения дейтерирования являются непрогнозируемыми.
В настоящем изобретении поставлена задача получения класса лекарственных средств с улучшенной активностью и фармакокинетическими свойствами с помощью подходящей химической модификации.
Сущность изобретения
Задачей настоящего изобретения является получение класса лекарственных средств с высокой активностью, подходящими фармакокинетическими свойствами, незначительными токсическими и побочными эффектами и хорошей метаболической стабильностью.
В настоящем изобретении предложено соединение формулы (I) или его оптический изомер, соль, пролекарство, гидрат или неводный сольват:
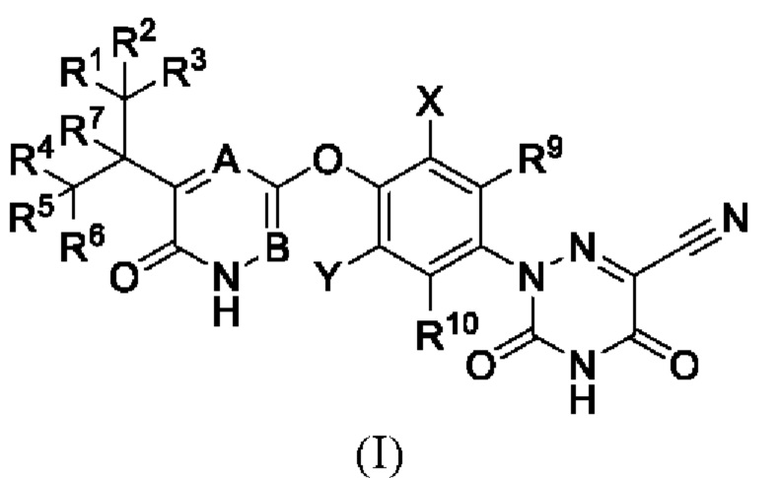
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; каждый А и В независимо выбран из группы, состоящей из N, СН и CD; каждый X и Y независимо выбран из группы, состоящей из F, Cl, Br и I;
при этом, когда В представляет собой N, а А представляет собой СН или CD, X и Y одновременно не могут быть Cl.
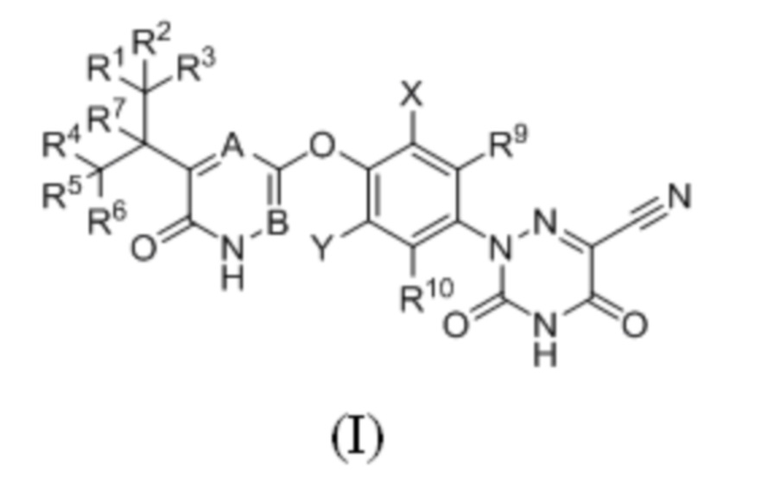
Кроме того, указанное соединение имеет структуру формулы (II):
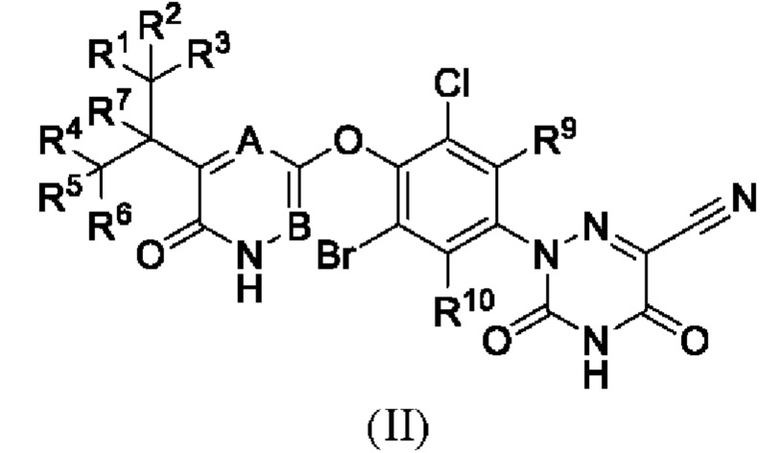
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; каждый А и В независимо выбран из группы, состоящей из N, СН и CD.
Кроме того, указанное соединение имеет структуру формулы (III):
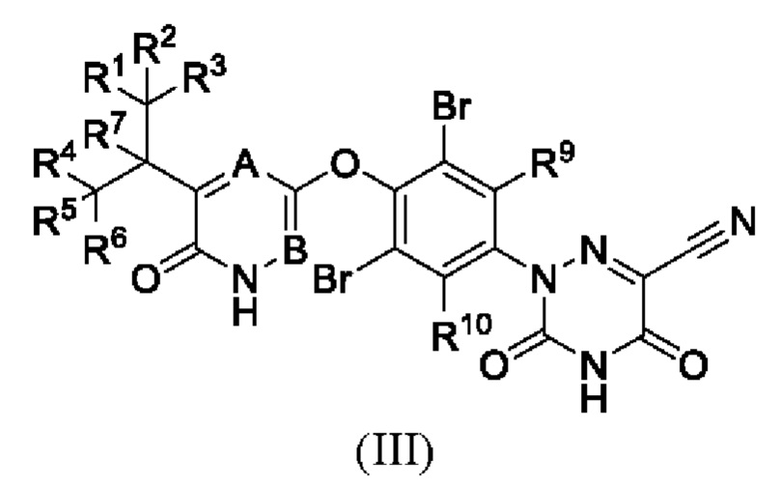
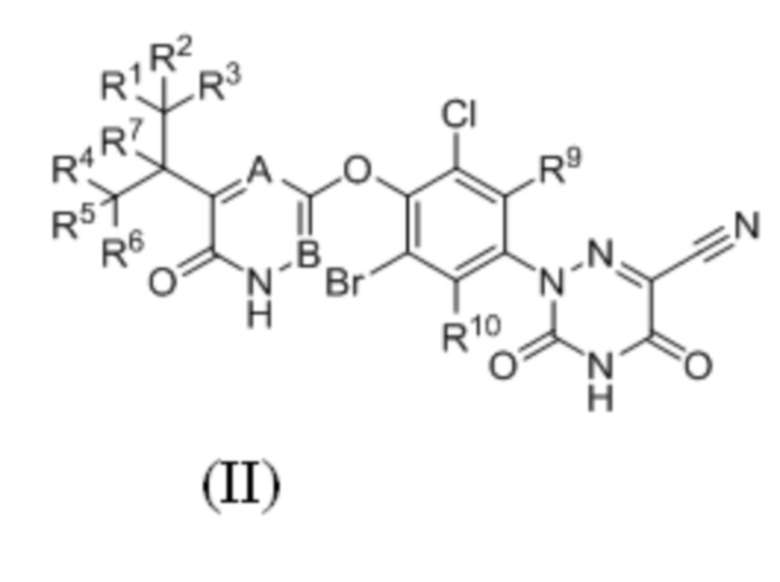
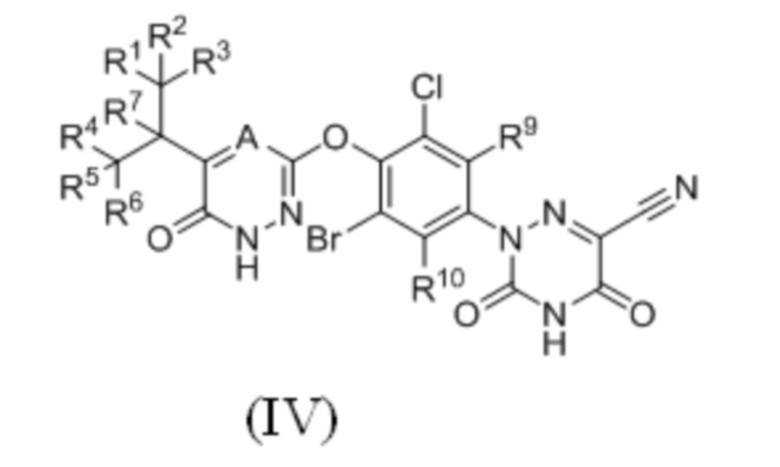
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; каждый А и В независимо выбран из группы, состоящей из N, СН и CD. Кроме того, указанное соединение имеет структуру формулы (IV):
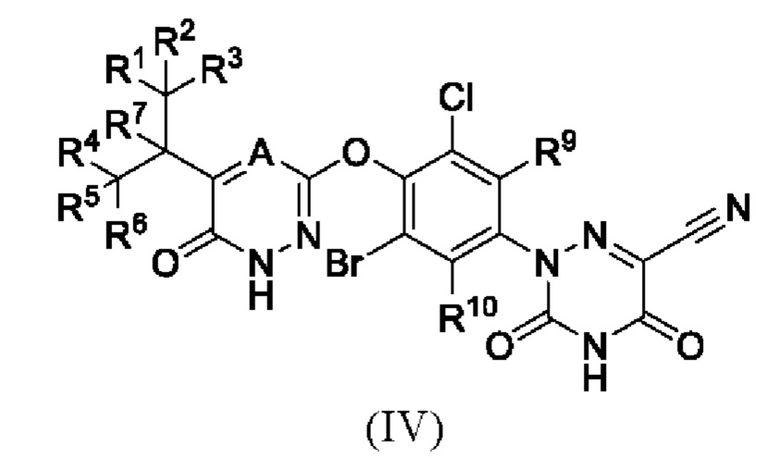
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; А выбран из группы, состоящей из N, СН и CD;
предпочтительно каждый из R7, R9 и R10 независимо выбран из Н; каждый из R1-R6 независимо выбран из Н и D; А выбран из СН и CD.
Кроме того, указанное соединение имеет структуру формулы (V):
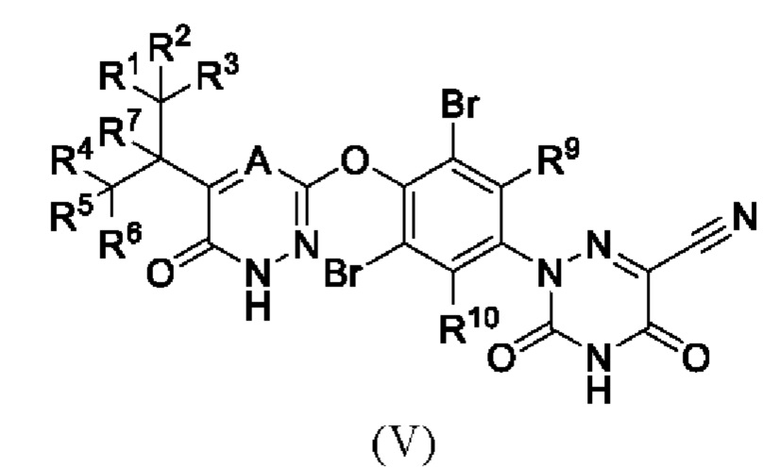
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; А выбран из группы, состоящей из N, СН и CD;
предпочтительно каждый из R7, R9 и R10 независимо выбран из Н; каждый из R1-R6 независимо выбран из Н и D; А выбран из СН и CD.
Кроме того, указанное соединение имеет структуру формулы (VI):
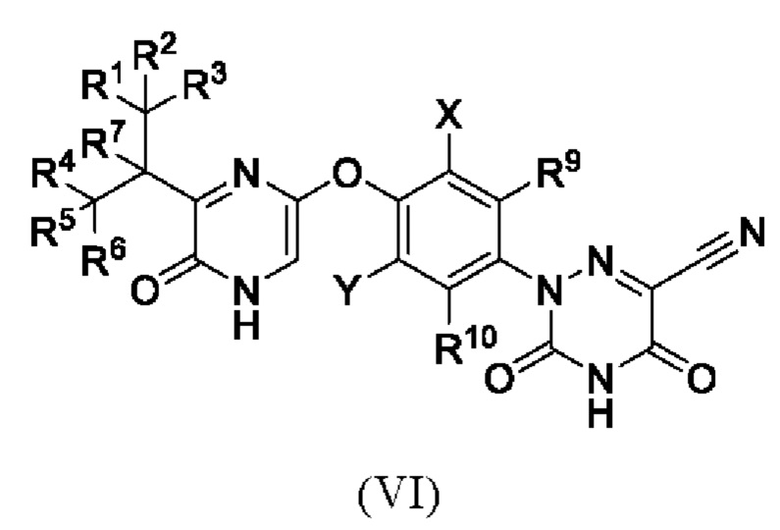
где каждый из R1-R7, R9 и R10 независимо выбран из Н и D; каждый X и Y независимо выбран из группы, состоящей из F, Cl, Br и I; предпочтительно каждый X и Y независимо выбран из группы, состоящей из Cl и Br.
Кроме того, указанное соединение выбрано из группы, состоящей из следующих соединений:
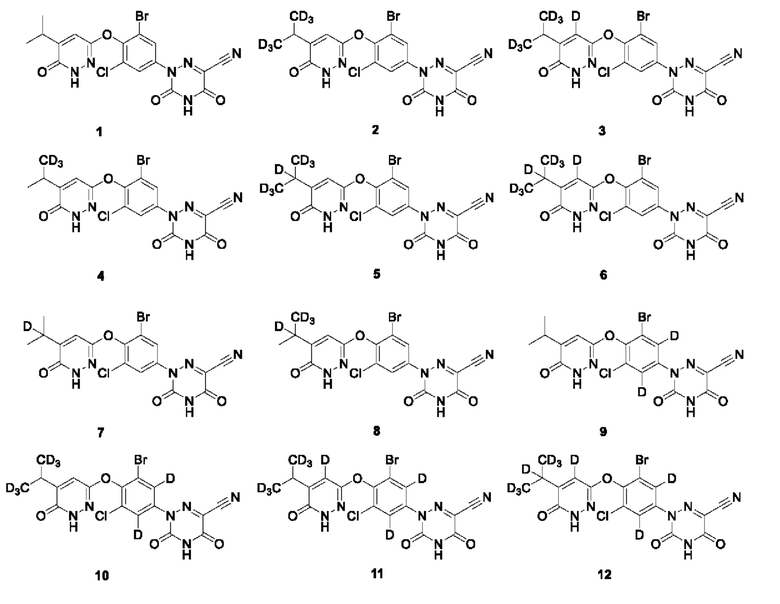
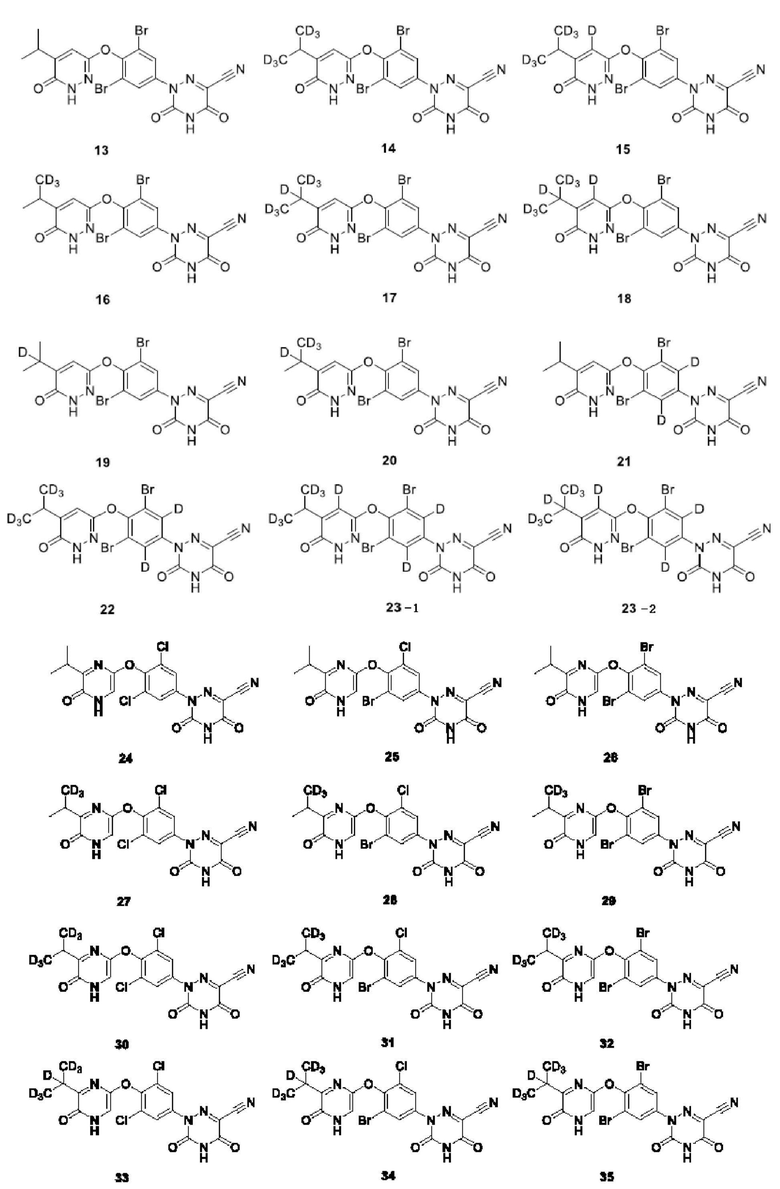
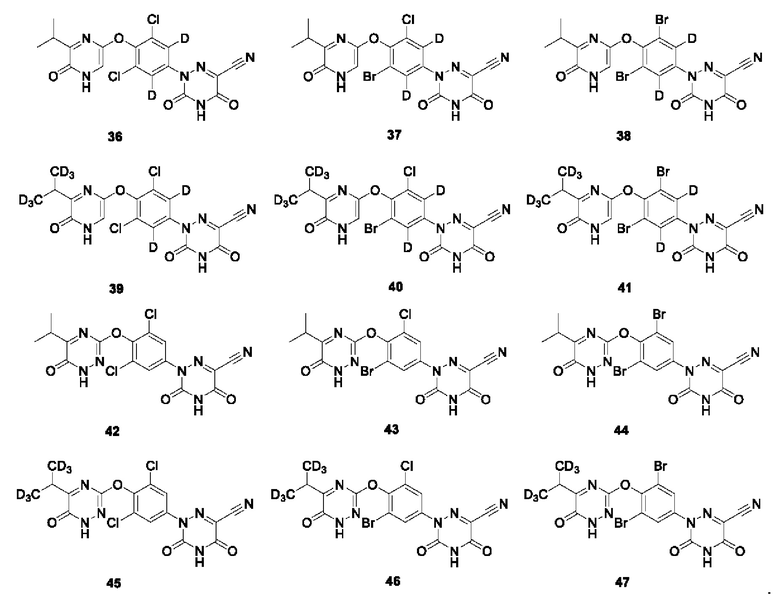
В настоящем изобретении также предложено применение указанного выше соединения или его оптического изомера, соли, пролекарства, гидрата или неводного сольвата для получения агонистов THR-β.
Кроме того, указанный агонист THR-β представляет собой лекарственное средство для снижения уровня холестерина и лечения дислипидемии и неалкогольной жировой болезни печени.
Кроме того, указанный агонист THR-β представляет собой лекарственное средство для лечения семейной гиперхолестеринемии и неалкогольного стеатогепатита. В настоящем изобретении также предложено применение указанного выше соединения или его оптического изомера, соли, пролекарства, гидрата или неводного сольвата для получения агонистов THR-α; предпочтительно указанный агонист THR-α представляет собой лекарственное средство для лечения диффузного токсического зоба. В настоящем изобретении также предложено лекарственное средство для снижения уровня холестерина и лечения дислипидемии и неалкогольной жировой болезни печени, которое представляет собой препарат, содержащий активный ингредиент, который представляет собой указанное выше соединение или его оптический изомер, соль, пролекарство, гидрат или неводный сольват, и фармацевтически приемлемые вспомогательные вещества.
В контексте настоящего изобретения термин «дейтерирование» относится к замене одного или более атомов водорода в соединении или группе дейтерием. Дейтерий может быть монозамещенным, дизамещенным, полизамещеиным или полностью замещенным. В другом предпочтительном варианте содержание изотопа дейтерия в положении замещения больше, чем природное содержание (0,015%), предпочтительно более 50%, более предпочтительно более 75%, более предпочтительно более 95%, более предпочтительно более 97%, более предпочтительно более 99% и более предпочтительно более 99,5%. В контексте данного документа термин «соединение согласно настоящему изобретению» относится к соединению формулы (I). Термин также включает различные оптические изомеры, соли, пролекарства, гидраты или неводные сольваты соединения формулы (I). Активный ингредиент, упомянутый в настоящем изобретении, относится к любому веществу или смеси, применяемой для получения лекарственных средств, обладающих фармакологической активностью или оказывающих иное прямое действие при диагностике, лечении, облегчении симптомов, лечении или профилактике заболеваний или могут оказывать влияние на функцию или структуру организма.
Фармацевтически приемлемое вспомогательное вещество обладает определенной физиологической активностью, при этом добавление вспомогательного вещества не приводит к изменению основных характеристик фармацевтической композиции при лечении заболевания, а выполняет вспомогательную роль. Эти дополнительные эффекты обусловлены только известной активностью вспомогательного вещества и соответствуют обычным вспомогательным способам лечения в области медицины. Применение вспомогательного вещества в комбинации с фармацевтической композицией согласно настоящему изобретению также находится в пределах объема испрашиваемой правовой охраны.
Термин «неводный сольват» означает сольват, отличный от гидрата.
Было установлено, что в сравнении с контрольным соединением MGL-3196, соединение формулы (I), получаемое с помощью замещения по конкретным положениям и конкретных типов замещения согласно настоящему изобретению, проявляет более высокую агонистическую активность в отношении как THR-, такβ и THR-α, и в особенности в отношении THR-β, соединение согласно настоящему изобретению обладает значительно более высокой агонистической активностью и селективностью. Кроме того, соединение согласно настоящему изобретению также демонстрирует значительно улучшенные фармакокинетические свойства и имеет хорошую перспективу применения для получения агонистов THR-β и лекарственных средств с показаниями к применению агонистов THR-β (включая дислипидемию, гиперхолестеринемию, неалкогольный стеатогепатит (НАСГ) и неалкогольную жировую болезнь печени (НАЖБП)).
Очевидно, что с учетом вышеуказанного содержания настоящего изобретения, общими техническими знаниями и стандартными средствами в данной области, без отступления от вышеуказанного основного характера изобретения, могут быть дополнительно сделаны другие различные модификации, замещения или изменения.
Вышеописанное содержание настоящего изобретения далее дополнительно проиллюстрировано с помощью конкретных примеров вариантов реализации. Однако следует отметить, что объем настоящего изобретения не ограничивается указанными примерами. Все методики, реализуемые на основании вышеуказанного содержания настоящего изобретения, входят в объем настоящего изобретения.
Примеры
Исходные материалы и приборы, указанные в настоящем изобретении, представляют собой известные изделия, доступные на рынке.
Пример 1 Синтез соединения 2
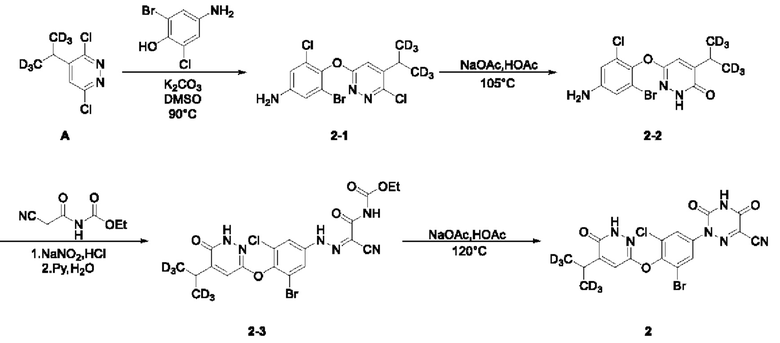
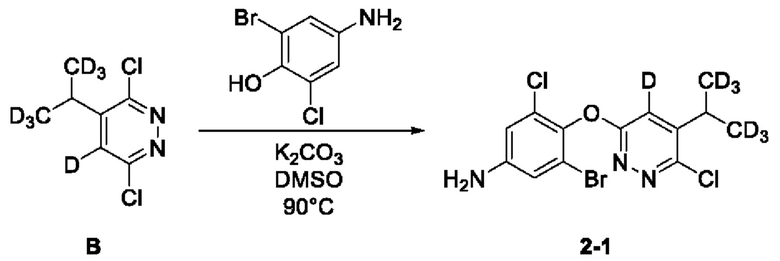
(1) Синтез соединения 3-бром-5-хлор-4-((6-хлор-5-ди(тридейтерометил)метилпиридазин-3-ил)окси)анилина (соединение 2-1)
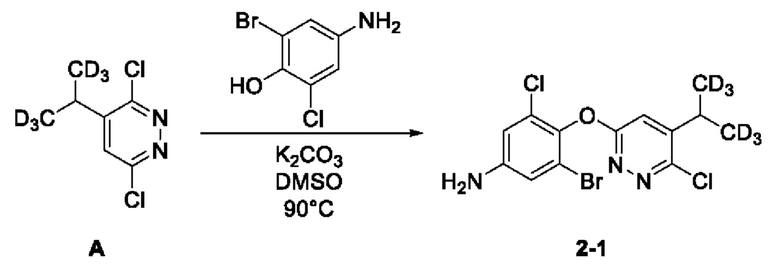
Синтез 3,6-дихлор-4-(1,1,1,3,3,3-гексадейтеропропил-2-ил) пиридазина (соединение А):
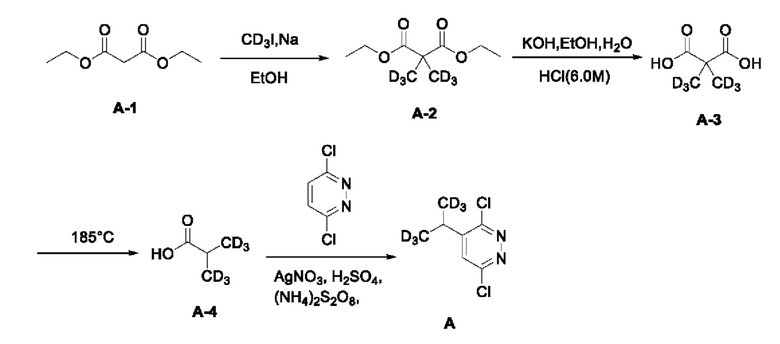
Руководствуясь способом, известным из литературы (Canadian Journal of Chemistry, 2014, 92, 305), получали 2-тридейтерометил-3,3,3-тридейтеропропионовая кислота А-4. 2-тридейтерометил-3,3,3-тридейтеропропионовую кислоту (1,4 г, 15 ммоль) взвешивали и помещали в трехгорлую круглодонную колбу объемом 100 мл, в которую добавляли 20 мл воды, а затем раствор перемешивали при комнатной температуре до тех получения прозрачного раствора. Затем в систему добавляли 3,6-дихлорпиридазин (2,2 г, 15 ммоль), а раствор дополнительно перемешивали при комнатной температуре. Затем в систему добавляли нитрат серебра (2,5 г, 15 ммоль), а систему помещали на масляную баню для нагревания и оставляли для проведения реакции при перемешивании. Когда внутренняя температура системы повышалась до 50°С, к системе по каплям добавляли концентрированную серную кислоту (3,5 мл) и после чего систему дополнительно перемешивали в течение 10 мин при указанной температуре. Затем, когда внутренняя температура системы повышалась до 60°С, к системе по каплям добавляли 6 мл водного раствора персульфата аммония (10,3 г, 45 ммоль). Когда внутренняя температура системы повышалась до 70°С, смесь оставляли для проведения реакции еще на 30 мин при перемешивании при указанной температуре. Нагревание прекращали, а систему охлаждали в естественных условиях до комнатной температуры. Затем систему переносили на ледяную водяную баню для охлаждения при перемешивании, а через 15 мин в систему добавляли раствор NaOH (6,0 М) для доведения рН системы до значения около 8. В систему добавляли этилацетат (20 мл), а раствор интенсивно перемешивали, после чего отстаивали до разделения слоев. Водную фазу экстрагировали с этилацетатом (10 мл × 3). Органическую фазу объединяли, последовательно промывали водой (10 мл × 3) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении с получением неочищенного продукта, который отделяли и очищали с помощью колоночной хроматографии, с получением 3,6-дихлор-4-(1,1,1,3,3,3-гексадейтеропропил-2-ил)пиридазина (соединение А, 1,7 г) в виде твердого вещества почти белого цвета с выходом 58%. МС (ИЭР) m/z 197,2 [М+Н]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,98 (d, J=0,8 Гц, 1H), 3,12 (s, 1H). 3-бром-5-хлор-4-ди(тридейтерометил)метилпиридазин (А, 450 мг, 2,28 ммоль) взвешивали и помещали в трехгорлую круглодонную колбу вместимостью 100 мл, в которую добавляли 10 мл диметилсульфоксида, а раствор перемешивали при комнатной температуре до получения прозрачного раствора. Систему продували аргоном, при этом операцию повторяли десять раз для обеспечения атмосферы инертного газа в системе. Затем в систему последовательно добавляли 4-амино-2-бром-6-хлорфенол (508,0 мг, 2,28 ммоль) и безводный карбонат калия (1,3 г, 9,12 ммоль). Затем систему помещали на масляную баню при 90°С, нагревали и оставляли для проведения реакции в течение ночи при перемешивании. Спустя 24 часа весь исходный материал полностью расходовался согласно данным анализа. Нагревание прекращали, а систему охлаждали в естественных условиях до комнатной температуры. В систему добавляли этилацетат (20 мл) и воду (20 мл) и интенсивно перемешивали, после чего проводили разделение слоев. Водную фазу экстрагировали этилацетатом (20 мл × 3). Органическую фазу объединяли, последовательно промывали водой (10 мл × 3) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении для удаления растворителя и получения неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением 3-бром-5-хлор-4-((6-хлор-5-ди (тридейтерометил)метилпиридазин-3-ил)окси)анилина в виде твердого вещества светло-желтовато-коричневого цвета (2-1, 463,0 мг) с выходом 52,9%. МС (ИЭР) m/z 382,0 [М+Н]+.
(2) Синтез соединения 6-(4-амино-2-бром-6-хлорфенокси)-4-ди(тридейтерометил) метилпиридазин-3(2Н)-она (соединение 2-2)
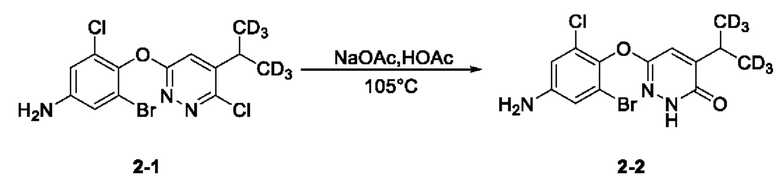
3-бром-5-хлор-4-((6-хлор-5-ди(тридейтерометил)метилпиридазин-3-ил)окси)анилин (341,0 мг, 0,89 ммоль) взвешивали и помещали в трехгорлую круглодонную колбу вместимостью 50 мл, в которую добавляли ледяную уксусную кислоту (10 мл), а затем смесь перемешивали при комнатной температуре. Затем в систему добавляли безводный ацетат натрия (256,0 мг, 3,12 ммоль). Затем систему переносили на масляную баню при 105°С, перемешивали и проводили реакцию с обратным холодильником. Через 24 часа нагревание останавливали, а систему охлаждали в естественных условиях до комнатной температуры. Растворитель удаляли на ротационном испарителе, а в систему добавляли воду (50 мл) и затем переносили на ледяную баню для охлаждения при перемешивании. Когда внутренняя температура системы снижалась до 5°С, в систему по каплям добавляли раствор гидроксида натрия (1,0 М), а рН системы доводили до около 9. После этого в систему добавляли этилацетат (30 мл), а раствор интенсивно перемешивали с последующим разделением слоев. Водную фазу экстрагировали этилацетатом (25 мл × 2). Органическую фазу объединяли, последовательно промывали один раз водой (20 мл) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении с получением твердого вещества бледно-желтого цвета. Метанол (10 мл) и раствор NaOH (1,0 М, 10 мл) последовательно добавляли в трехгорлую круглодонную колбу вместимостью 100 мл, содержащую твердое вещество, после чего систему переносили на масляную баню при 105°С для осуществления реакции с обратным холодильником. Через 16 часов нагрев останавливали, снимали систему с масляной бани и доводили до комнатной температуры. Растворитель удаляли на ротационном испарителе, а к остатку добавляли этилацетат (60 мл) и воду (40 мл). Полученный раствор интенсивно перемешивали, а затем отстаивали для разделения слоев. Водный слой подвергали экстрагировали этилацетатом (25 мл*2). Органический слой объединяли, последовательно промывали водой (20 мл*2) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Растворитель удаляли на ротационном испарителе, в результате получали неочищенный продукт, который отделяли с помощью колоночной хроматографии с получением 6-(4-амино-2-бром-6-хлорфенокси)-4-ди(тридейтерометил) метил пиридазин-3 (2Н)-она в виде твердого вещества бледно-желтого цвета (2-2, 200,0 мг) с выходом 61,5%. МС (ИЭР) m/z 364,2 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,13 (s, 1H), 7,26 (s, 1Н), 6,82 (d, J=4,0 Гц, 1H), 6,70 (d, J=4,0 Гц, 1H), 5,60 (s, 2Н), 2,99 (s, 1H).
(3) Синтез этил-(2-циано-2-(2-(3-бром-5-хлор-4-((5-ди(тридейтерометил)метил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)гидразоно)ацетил)карбамата (соединение 2-3)
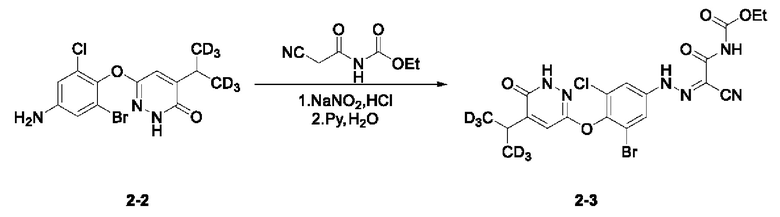
6-(4-амино-2-бром-6-хлорфенокси)-4-ди(тридейтерометил)метилпиридазин-3(2Н)-он (153 мг, 0,42 ммоль) взвешивали и помещали в трехгорлую круглодонную колбу вместимостью 50 мл, в которую добавляли воду (5,6 мл), а раствор перемешивали при комнатной температуре. Затем в систему добавляли концентрированную соляную кислоту (2,8 мл). Затем систему переносили на ледяную баню для охлаждения при перемешивании. При понижении внутренней температуры системы до 0°С в систему по каплям добавляли 0,4 мл водного раствора нитрита натрия (36,5 мг, 0,53 ммоль). Затем систему дополнительно перемешивали и осуществляли реакцию в течение 30 мин при указанной температуре. N-цианоацетилмочевину (72,0 мг, 0,46 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 25 мл, в которую добавляли воду (9,4 мл) и пиридин (2,8 мл). Полученную смесь перемешивали при комнатной температуре с получением прозрачного раствора и переносили на ледяную баню для охлаждения при перемешивании в течение дополнительных 30 мин. В систему медленно по каплям добавляли диазотирующий раствор, содержащий N-цианоацетилмочевину, а скорость добавления регулировали таким образом, чтобы внутренняя температура системы не превышала 5°С. Затем систему перемешивали и проводили реакцию на ледяной бане при указанной температуре. Через 1 ч по завершении реакции выполняли анализ с помощью тонкостенной хроматографии (ТСХ). Систему фильтровали при пониженном давлении, а осадок на фильтре несколько раз промывали небольшим количеством воды, несколько раз промывали н-гексаном и сушили с получением этил-(2-циано-2-(2-(3-бром-5-хлор-4-((5-ди(тридейтерометил)метил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)гидразоно)ацетил)карбамата в виде твердого вещества оранжево-красного цвета (2-3, 153,0 мг) с выходом 68,6%, которое в дальнейшем использовали на следующей стадии без дополнительной очистки. МС (ИЭР) m/z 531,1 [М+Н]+.
(4) Синтез соединения
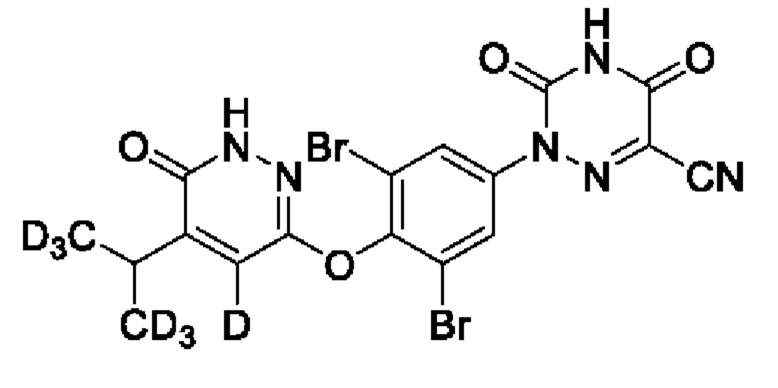
2-(3-бром-5-хлор-4-((5-дидейтерометилметил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 2)
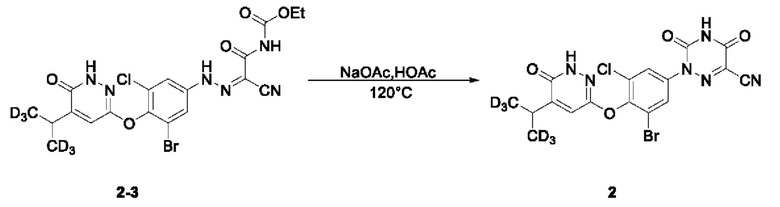
Этил(2-циано-2-(2-(3-бром-5-хлор-4-((5-ди(тридейтерометил)метил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)гидразоно)ацетил)карбамат (153 мг, 0,29 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 25 мл, в которую добавляли ледяную уксусную кислоту (5 мл), а затем смесь перемешивали при комнатной температуре. Затем в систему добавляли безводный ацетат натрия (118,0 мг, 1,44 ммоль). Затем систему переносили на масляную баню при 120°С, перемешивали и осуществляли реакцию при нагревании. По прошествии 1,5 часов весь исходный материал полностью расходовался согласно данным ТСХ. Нагревание останавливали, а систему охлаждали в естественных условиях до комнатной температуры. Затем систему переносили на ледяную баню для охлаждения при перемешивании. При понижении внутренней температуры системы до 5°С при добавлении ледяной воды полученный раствор интенсивно перемешивали в течение 20 мин. Затем раствор фильтровали при всасывании, а остаток на фильтре несколько раз промывали водой, после чего растворяли в этилацетате и сушили над безводным сульфатом натрия. Растворитель удаляли на ротационном испарителе с получением неочищенного продукта, который отделяли и очищали с помощью ТСХ с получением 2-(3-бром-5-хлор-4-((5-ди(тридейтерометил)метил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила в виде твердого вещества светло-оранжевого цвета (соединение 2, 54,0 мг) с выходом 38,6%. МС (ИЭР) m/z 485,0 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,29 (s, 1H), 12,24 (s, 1H), 7,90 (d, J=4,0 Гц, 1H), 7,82 (d, J=4,0 Гц, 1H), 7,45 (s, 1H), 3,02 (s, 1H).
Пример 2 2-(3-бром-5-хлор-4-((4-дейтеро-5-(1,1,1,3,3,3-гексадейтеропропил-2-ил)-6-оксо-1,6-дигидропиразин-3-ил)окси)фенил)-3,5-диокси-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 3)
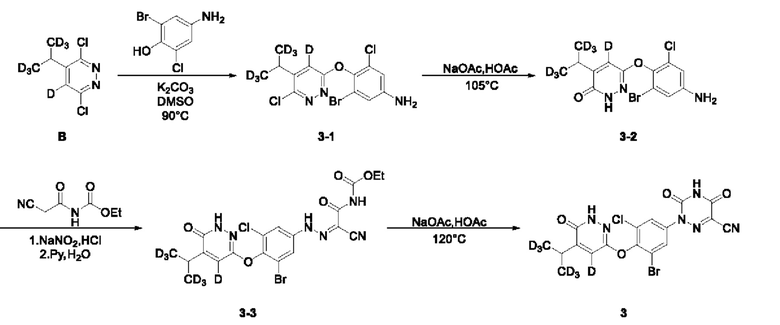
(1) Синтез соединения 3-бром-5-хлор-4-((6-хлор-5-(пропан-2-ил-1,1,1,3,3,3-гексадейтеро)пиразин-3-ил-4-дейтеро)окси)анилина
Синтез 3,6-дихлор-4-дейтеро-5-(1,1,1,3,3,3-гексадейтеропропил-2-ил)пиридазина (соединение В):
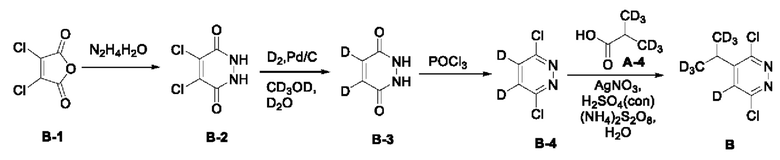
Соединение 2,3-дихлормалеиновый ангидрид В-1 (8,35 г, 50 ммоль) взвешивали и помещали в круглодонную колбу вместимостью 100 мл, в которую последовательно добавляли 40 мл воды и гидразина гидрата (2,5 г, 50 ммоль), после чего реакционный раствор нагревали с обратным холодильником и оставляли для протекания реакции в течение 4 часов с обратным холодильником. Затем реакционную смесь охлаждали до комнатной температуры и выдерживали на ледяной бане в течение 30 мин. Реакционный раствор фильтровали, а фильтрат промывали 100 мл воды и сушили с получением 4,5-дихлормалеинового гидразида (В-2, 5,0 г) с выходом 55,3%, МС (ИЭР) m/z 181,0 [М+Н]+.
Синтез 4,5-дидейтеромалеинового гидразида (В-3): Способ 1: 4,5-дихлормалеиновый гидразид (2,0 г, 11,05 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 100 мл, в которую последовательно добавляли 50 мл метанола-сЦ, 10 мл дейтерированной воды и 200 мг Pd/C. Систему 3 раза продували дейтерием, а смесь выдерживали для осуществления реакции при комнатной температуре в течение 40 ч. После этого реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении до сухого остатка. Остаток промывали 6 мл метанола и затем фильтровали. Фильтрат сушили с получением соединения 4,5-дидейтеромалеинового гидразида (1,0 г) с выходом 79%, МС (ИЭР) m/z 115,2 [М+Н]+. 13С ЯМР (101 МГц, ДМСО-d6) δ 156,76, 130,50. Способ 2: малеиновый гидразид (5,6 г, 50 ммоль) добавляли в круглодонную колбу, в которую последовательно добавляли 80 мл дейтерированной воды и 500 мг Pd/C. Систему три раза продували водородом, а затем смесь нагревали и кипятили с обратным холодильником в течение 72 ч в атмосфере водорода. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат добавляли в круглодонную колбу и повторяли описанную выше процедуру. После завершения реакции к фильтрату добавляли 100 мл метанола, а полученный раствор кипятили с обратным холодильником в течение 30 минут. Затем раствор фильтровали, и полученный фильтрат концентрировали при пониженном давлении до сухого остатка, в результате получали 2,5 г 4,5-дидейтеромалеинового гидразида с выходом 43,87%.
Синтез 4,5-дидейтеро-3,6-дихлорпиридазина (В-4)
4,5-дидейтеромалеиновый гидразид (1,0 г, 8,74 ммоль) взвешивали и помещали в круглодонную колбу вместимостью 100 мл, в которую добавляли 15 мл оксихлорида фосфора, а затем раствор кипятили с обратным холодильником при 115°С в течение 4 ч. Раствор концентрировали при пониженном давлении до сухого остатка, а затем охлаждали на ледяной бане, в которую добавляли 20 мл ледяной воды. Полученный раствор доводили до рН=9,0 с помощью аммиачной воды, а затем добавляли 30 мл дихлорметана для экстракции. Водный слой дополнительно экстрагировали один раз с помощью 20 мл дихлорметана. Органический слой объединяли, последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении до сухого остатка, в результате получали 1,2 г соединения 4,5-дидеутеро-3,6-дихлорпиридазина с выходом 90,9%. 13С ЯМР (101 МГц, ДМСО-d6) δ 156,3, 131,9 (t, J=27 Гц). МС (ИЭР) m/z 151 [М+Н]+.
Соединение 3,6-дихлор-4,5-дидетеропиридазин В-4 (604 мг, 4,0 ммоль) добавляли к 10 мл воды, к которой прибавляли соединение А-4 (372 мг, 4,0 ммоль), а затем в систему добавляли AgNO3 (680 мг, 4 ммоль) при перемешивании. При повышении температуры до 50°С в систему по каплям медленно добавляли концентрированную серную кислоту (1 мл). Затем систему нагревали до 60°С и осуществляли реакцию в течение 10 мин при указанной температуре. Персульфат аммония (2,74 г, 12 ммоль) растворяли в 6 мл воды и затем по каплям добавляли в систему. Затем систему нагревали до 70°С и осуществляли реакцию в течение еще 30 мин при указанной температуре. Анализ продуктов реакции выполняли с помощью ТСХ, а при исчезновении исходного материала нагревание останавливали. Систему переносили на ледяную баню для охлаждения. Полученный раствор доводили до рН=8,0 6 н. водным раствором NaOH и экстрагировали этилацетатом (30 мл). Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении досуха. Остаток отделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат=10:1), с получением соединения В (500 мг), с выходом 63,1%, МС (ИЭР) m/z 198,1 [М+Н]+. 3,6-дихлор-4-ди(тридейтерометил) метил-5-дейтеропиридазин В (285,0 мг, 1,44 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 25 мл, в которую добавляли 8 мл диметилсульфоксида, а раствор перемешивали при комнатной температуре до получения прозрачного раствора. Затем в систему последовательно добавляли 4-амино-2-бром-6-хлорфенол (320,0 мг, 1,44 ммоль) и безводный карбонат калия (995,0 мг, 7,20 ммоль). После этого систему продували аргоном, при этом операцию повторяли десять раз для обеспечения атмосферы инертного газа в системе. Затем систему переносили на масляную баню при 90°С, нагревали и осуществляли реакции в течение ночи при перемешивании. Через 4 часа весь исходный материал полностью расходовался согласно данным ТСХ. Нагревание прекращали, а систему охлаждали в естественных условиях до комнатной температуры. В систему добавляли этилацетат (20 мл) и воду (20 мл) и интенсивно перемешивали, после чего проводили разделение слоев. Водную фазу экстрагировали этилацетатом (30 мл × 3). Органическую фазу объединяли, последовательно промывали водой (20 мл*3) и насыщенным солевым раствором (20 мл), и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении для удаления растворителя и получения неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением 3-бром-5-хлор-4-((6-хлор-5-(пропан-2-ил-1,1,3,3,3-гексадейтеро) пиразин-3-ил-4-дейтеро)окси)анилина в виде не твердого вещества почти белого цвета (3-1, 293,0 мг) с выходом 53,0%. МС (ИЭР) m/z 383,0 [М+Н]+.
(2) Синтез 6-(4-амино-2-бром-6-хлорфенокси)-4-(пропан-2-ил-1,1,1,3,3,3-гексадейтеро)пиразин-3(2Н)-она-5-дейтерия (3-2)
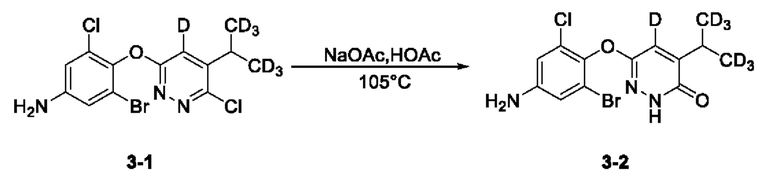
3-бром-5-хлор-4-((6-хлор-5-дидейтерометилметил-4-дейтеропиридазин-3-ил)окси)анилин (230,0 мг, 0,60 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 50 мл, в которую добавляли ледяную уксусную кислоту (10 мл), а полученную смесь перемешивали при комнатной температуре. Затем в систему добавляли безводный ацетат натрия (172,0 мг, 2,10 ммоль). Затем систему переносили на масляную баню при 105°С, перемешивали и проводили реакцию с обратным холодильником. По прошествии 22 ч нагревание прекращали, а систему охлаждали в естественных условиях до комнатной температуры. Растворитель удаляли на ротационном испарителе, а в систему добавляли воду (50 мл) и затем переносили на ледяную баню для охлаждения при перемешивании. Когда внутренняя температура системы снижалась до 5°С, в систему по каплям добавляли раствор гидроксида натрия (1,0 М), а рН системы доводили до около 9. После этого в систему добавляли этилацетат (50 мл), а раствор интенсивно перемешивали с последующим разделением слоев. Водную фазу экстрагировали этилацетатом (25 мл*2). Органическую фазу объединяли, последовательно промывали один раз водой (20 мл) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Раствор концентрировали при пониженном давлении с получением твердого вещества бледно-желтого цвета. Метанол (10 мл) и раствор NaOH (1,0 М, 10 мл) последовательно добавляли в трехгорлую круглодонную колбу вместимостью 100 мл, содержащую твердое вещество, после чего систему переносили на масляную баню при 105°С для осуществления реакции с обратным холодильником. Спустя 11 ч нагрев останавливали, удаляли систему с масляной бани и доводили до комнатной температуры. Растворитель удаляли на ротационном испарителе, а к остатку добавляли этилацетат (80 мл) и воду (50 мл). Полученный раствор интенсивно перемешивали, а затем отстаивали для разделения слоев. Водный слой подвергали экстракции этилацетатом (25 мл*2). Органический слой объединяли, последовательно промывали водой (20 мл*2) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Растворитель удаляли на ротационном испарителе, в результате получали неочищенный продукт, который отделяли с помощью колоночной хроматографии, при этом получали 6-(4-амино-2-бром-6-хлорфенокси)-4-дидейтерометилметил-5-дейтеропиридазин-3 (2Н)-он в виде твердого вещества бледно-желтого цвета (3-2, 115,0 мг) с выходом 52,5%. МС (ИЭР) m/z 365,1 [М+Н]+.
(3) Синтез соединения этил-(2-(2-(3-бром-5-хлор-4-((6-оксо-5-(пропан-2-ил-1,1,1,3,3,3-гексадейтеро)-1,6-дигидропиразин-3-ил-4-дейтеро)окси)фенил)гидразоно)-2-цианоацетил)карбамата (3-3)
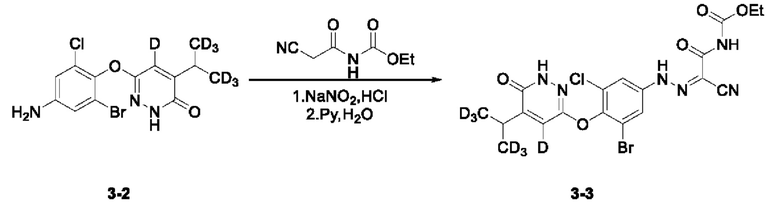
6-(4-амино-2-бром-6-хлорфенокси)-4-дидейтерометил-5-дейтеропиридазин-3(2Н)-он (100,0 мг, 0,27 ммоль) взвешивали и помещали в одногорлую круглодонную колбу вместимостью 25 мл, в которую добавляли воду (3,7 мл), а полученный раствор перемешивали при комнатной температуре. Затем в систему добавляли концентрированную соляную кислоту (1,9 мл). Затем систему переносили на ледяную баню для охлаждения при перемешивании. При снижении внутренней температуры системы до 0°С в систему по каплям добавляли 0,5 мл водного раствора нитрита натрия (24,0 мг, 0,34 ммоль). Затем систему дополнительно перемешивали и осуществляли реакцию в течение 30 мин при указанной температуре. Кроме того, взвешивали N-цианоацетилмочевину (47,0 мг, 0,30 ммоль) и помещали в одногорлую круглодонную колбу вместимостью 50 мл, в которую последовательно добавляли воду (6,3 мл) и пиридин (1,9 мл). Полученную смесь перемешивали при комнатной температуре с получением прозрачного раствора и переносили на ледяную баню для охлаждения при перемешивании в течение дополнительных 30 мин. В систему медленно по каплям добавляли диазотирующий раствор, содержащий N-цианоацетилмочевину, а скорость добавления регулировали таким образом, чтобы внутренняя температура системы не превышала 5°С. Затем систему перемешивали и проводили реакцию на ледяной бане при указанной температуре. Через 1 ч по завершении реакции выполняли анализ с помощью тонкостенной хроматографии (ТСХ). Систему фильтровали при пониженном давлении, а осадок на фильтре несколько раз промывали небольшим количеством воды, несколько раз промывали н-гексаном и сушили с получением этил (2-циано-2-(2-(3-бром-5-хлор-4-((5-дидейтерометилметил-4-дейтеро-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил) гидразоно)ацетил)карбамата (3-3,104,0 мг) с выходом 71,7%, который сразу применяли на следующей стадии без дополнительной очистки. МС (ИЭР) m/z 532,1 [М+Н]+.
(4) Синтез 2-(3-бром-5-хлор-4-((4 дейтеро-5-(1,1,1,3,3,3-гексадейтеропропил-2-ил)-6-оксо-1,6-дигидропиразин-3-ил)окси)фенил)-3,5-диокси-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 3)
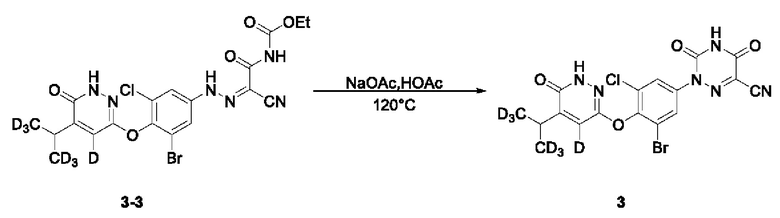
Этил (2-циано-2-(2-(3-бром-5-хлор-4-((5-дидеутерометилметил-4-дейтеро-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)гидразоно)цианоацетил)карбамат (104,0 мг, 0,20 ммоль)взвешивали и помещали в одногорлую круглодонную колбу вместимостью 25 мл, в которую добавляли ледяную уксусную кислоту (5 мл), а затем смесь перемешивали при комнатной температуре. Затем в систему добавляли безводный ацетат натрия (82,0 мг, 1,00 ммоль). Затем систему переносили на масляную баню при 120°С, перемешивали и оставляли для протекания реакцию при нагревании. Спустя 2 часа весь исходный материал полностью расходовался согласно данным ТСХ. Нагревание прекращали, и систему охлаждали в естественных условиях до комнатной температуры. Затем систему переносили на ледяную баню для охлаждения при перемешивании. При понижении внутренней температуры системы до 5°С при добавлении ледяной воды полученный раствор интенсивно перемешивали в течение 30 мин. Затем раствор фильтровали при всасывании, и остаток на фильтре несколько раз промывали небольшим количеством воды, а затем растворяли в этилацетате и сушили над безводным сульфатом натрия. Растворитель удаляли на ротационном испарителе с получением неочищенного продукта, который отделяли и очищали с помощью ТСХ с получением 2-(3-бром-5-хлор-4-((4-дейтеро-5-(1,1,1,3,3,3-гексадейтеропропил-2-ил)-6-оксо-1,6-дигидропиразин-3-ил)окси)фенил)-3,5-диокси-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила в виде твердого вещества светло-оранжевого цвета (соединение 3, 68,0 мг) с выходом 71,6%. МС (ИЭР) m/z 486,2 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 13,22 (s, 1H), 12,24 (s, 1H), 7,89 (d, J=4,0 Гц, 1H), 7,82 (d, J=4,0 Гц, 1H), 3,02 (s, 1H).
Пример 3 Синтез 2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,1,3,3,3-гексадейтеро)-1,6-дигидропиразин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрила (соединение 14)
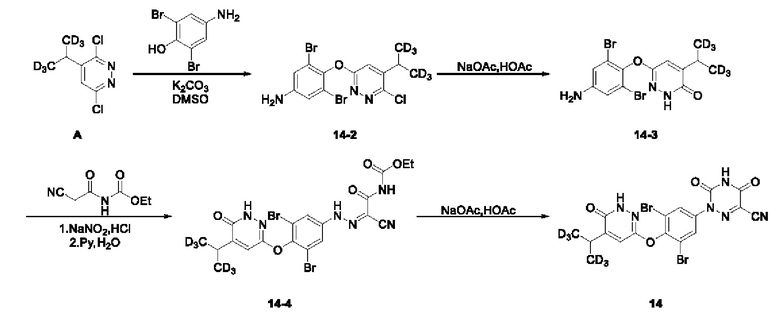
(1) Синтез 3,5-дибром-4-((6-хлор-5-(пропан-2-ил-1,1,1,3,3,3-гексадейтеро) пиридазин-3-ил)окси)анилина:
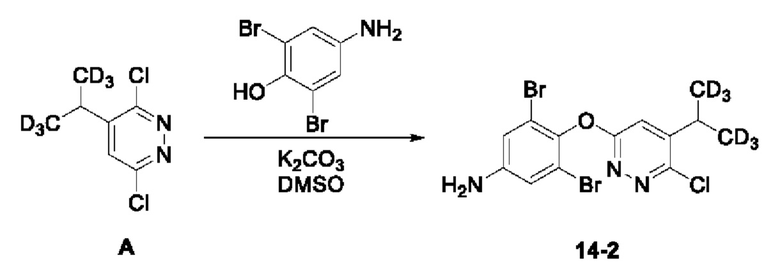
Соединение 3,6-дихлор-4-(пропан-2-ил-1,1,3,3,3-гексадейтеро)пиридазина А (330 мг, 1,67 ммоль) растворяли в 5 мл ДМСО, к которому добавляли соединение 4-амино-2,6-дибромфенол (559 мг, 2,09 ммоль), а затем в систему добавляли K2CO3 (923 мг, 6,68 ммоль)при перемешивании. Систему три раза продували Ar, а затем нагревали до 90°С и оставляли для проведения реакции в течение 3 ч. Затем систему охлаждали и добавляли воду, а полученный раствор экстрагировали этилацетатом. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали досуха при пониженном давлении. Полученный остаток отделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 5:1), с получением соединения 3,5-дибром-4-((6-хлор-5-(пропан-2-ил-1,1,3,3,3-d6) пиридазин-3-ил)окси)анилин (14-2, 210 мг) с выходом 30%, МС (ИЭР) m/z 426,0 [М+Н]+.
(2) Синтез 6-(4-амино-2,6-дибромфенокси)-4-(пропан-2-ил-1,1,1,3,3,3-d6) пиридазин-3(2Н)-она
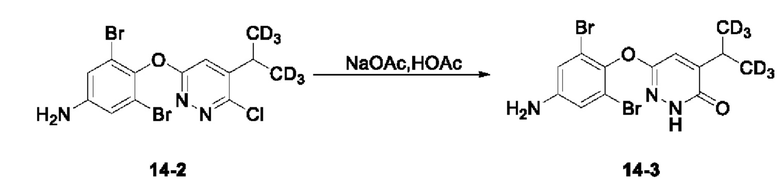
Соединение 3,5-дибром-4-((6-хлор-5-(пропан-2-ил-1,1,1,3,3,3-d6)пиридазин-3-ил)окси)анилин (185 мг, 0,43 ммоль) растворяли в 5 мл уксусной кислоты, к которой добавляли безводный ацетат натрия (142 мг, 1,7 ммоль) при перемешивании. Смесь оставляли для протекания реакции в течение ночи при 105°С и затем охлаждали до комнатной температуры. Полученный раствор концентрировали при пониженном давлении досуха, и рН остатка доводили до 9 с помощью 6 н. раствора NaOH. Раствор экстрагировали 20 мл этилацетата и водный слой дополнительно однократно экстрагировали 15 мл этилацетата. Органический слой объединяли, последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали при пониженном давлении досуха. К остатку добавляли 10 мл метанола, к которому прибавляли 10 мл 1 н. водного раствора NaOH, а смесь оставляли для осуществления реакции в течение ночи при 105°С. Реакционный раствор охлаждали, а затем большую часть метанола удаляли испарением при пониженном давлении при добавлении 20 мл этилацетата. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, а затем концентрировали досуха при пониженном давлении. Полученный остаток отделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 3:2), с получением соединения 6-(4-амино-2,6-дибромфенокси)-4-(пропан-2-ил-1,1,1,3,3,3-d6)пиридазин-3(2Н)-он (14-3, 130 мг) с выходом 73,9%, МС (ИЭР) m/z 408,0 [М+Н]+.
(3) Синтез этил-(2-циано-2-(2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,1,3,3,3-d6)-1,6-дигидропиридазин-3-ил-4-d6)окси)фенил)гидразоно)этил)карбамата
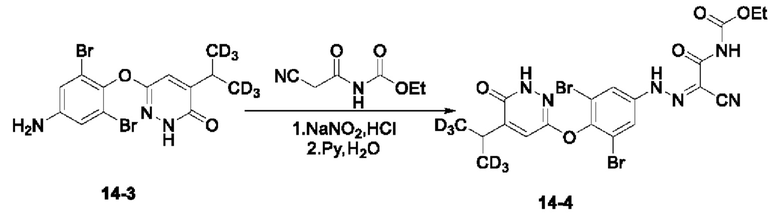
Соединение 6-(4-амино-2,6-дибромфенокси)-4-(пропан-2-ил-1,1,1,3.,3,3-d6) пиридазин-3 (2Н)-он (125 мг, 0,31 ммоль) добавляли в круглодонную колбу, в которую последовательно добавляли 4,6 мл воды и 2,2 мл концентрированной соляной кислоты, а затем систему переносили на ледяную баню и выдерживали в течение 30 мин. N-цианоацетилуретан (52 мг, 0,33 ммоль) добавляли в круглодонную колбу, в которую последовательно добавляли 8 мл воды и 2,2 мл пиридина, после чего систему помещали на ледяную баню и выдерживали в течение 30 минут. Реакционный раствор, полученный на предыдущем этапе, добавляли в реакционную систему. Затем систему выдерживали при указанной температуре и перемешивали в течение 30 мин. Полученный раствор фильтровали, а осадок на фильтре промывали 30 мл воды и сушили с получением продукта этил-(2-циано-2-(2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,3,3-d6)-1,6-дигидропиридазин-3-ил-4-d)окси)фенил)гидразоно)этил)карбамата в виде твердого вещества пурпурного цвета (14-4, 130 мг) с выходом 72,8%, МС (ИЭР) m/z 576,1 [М+Н]+.
(4) Синтез 2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,1,3,3-d6)-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-формонитрила
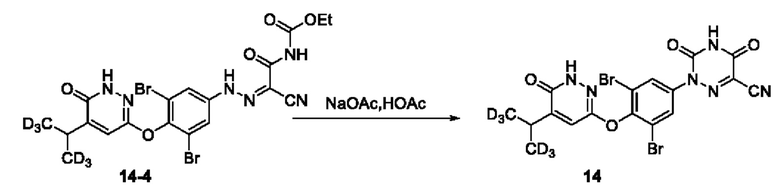
Соединение этил-(2-циано-2-(2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,1,3,3,3-d6)-1,6-дигидропиридазин-3-ил-4-d6)окси)фенил)гидразоно)этил)карбамат (125 мг, 0,22 ммоль) растворяли в 5 мл уксусной кислоты, к которой добавляли ацетат натрия (73 мг, 0,88 ммоль), а реакционный раствор кипятили с обратным холодильником при 125°С в течение 2 ч. Затем раствор охлаждали на ледяной бане и добавляли 30 мл воды, затем перемешивали в течение 20 мин. Полученный раствор фильтровали, а остаток на фильтре три раза промывали водой и сушили. Полученное твердое вещество добавляли в 3 мл этилацетата и встряхивали в течение 30 мин. Раствор фильтровали, а остаток на фильтре концентрировали досуха при пониженном давлении с получением соединения 2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,3,3,3-d6)-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диоксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-формонитрила (соединение 14, 95 мг) с выходом 81,4%, МС (ИЭР) m/z 529,0 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,44-13,12 (m, 1H), 12,23 (s, 1H), 7,92 (s, 2Н), 7,44 (s, 1H), 3,02 (s, 1H).
Пример 4 Синтез соединения 1
Соединение
2-(3-бром-5-хлор-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-дио ксо-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 1)
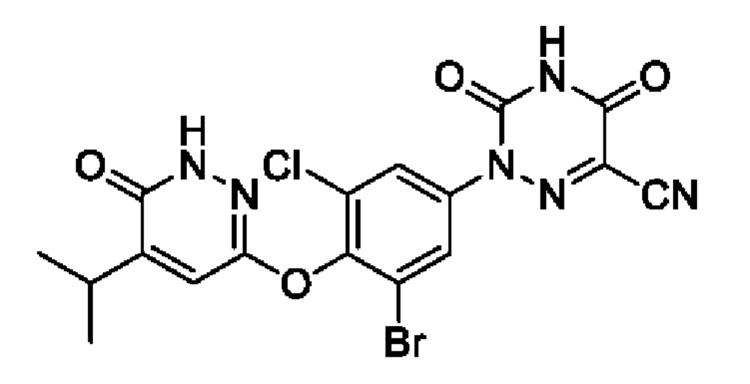
При использовании известного соединения 3,6-дихлор-4-изопропилпиперазина в качестве исходного материала (номер международной заявки РСТ, 2013045519) получали соединение 1 способом согласно примеру 1.
ЖХ/МС (ИЭР+) расч. для C17H12BrClN6O4 [М+Н]+ m/z, 477,98; наблюдаемое: 479,0, 481,0. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,29 (s, 1H), 12,22 (s, 1H), 7,88 (d, J=2,4 Гц, 1H), 7,81 (d, J=2,4 Гц, 1H), 7,44 (s, 1H), 3,05 (dt, J=13,8, 6,9 Гц, 1Н), 1,20 (d, J=6,9 Гц, 6Н).
Пример 5 Синтез соединения 13
2-(3,5-Дибром-4-((5-изопропил-6-оксо-1,6-дигидропиридазин-3-ил)окси)фенил)-3,5-диокс и-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 13)
При использовании известного соединения 3,6-дихлор-4-изопропилпиперазина в качестве исходного материала (номер международной заявки РСТ, 2013045519) получали соединение 13 способом согласно примеру 3.
ЖХ/МС (ИЭР+) рассч. для C17H12Br2N6O4 [М+Н]+m/z, 524,13; наблюдаемое значение: 523,1, 525,1.
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,16 (s, 1H), 7,91 (s, 2Н), 7,42 (s, 1H), 3,06 (dt, J=13,7, 6,9 Гц, 1H), 1,20 (d, J=6,8 Гц, 6Н).
Пример 6 Синтез соединения 15
2-(3,5-дибром-4-((6-оксо-5-(пропан-2-ил-1,1,3,3,3-d6)-1,6-дигидропиридазин-3-ил-4-d)окси)фенил)-3,5-диокси-2,3,4,5-тетрагидро-1,2,4-триазин-6-нитрил (соединение 15)
При использовании указанного выше соединения В в качестве исходного материала получали соединение 15 способом согласно примеру 3.
ЖХ/МС (ИЭР+) расч. для C17H5D7Br2N6C4 [М+Н]+m/z, 531,17; наблюдаемое значение: 530,0, 532,0.
1H ЯМР (400 МГц, DMSO-d6) δ 13,28 (s, 1H), 12,23 (s, 1H), 7,92 (s, 2Н), 3,02 (s, 1H).
Благоприятное действие настоящего изобретения продемонстрировано в следующих экспериментальных примерах.
Экспериментальный пример 1 Эксперимент на агонистическую активность соединения согласно настоящему изобретению в отношении бета-рецепторов тиреоидного гормона (THR-β)
1) Способ проведения эксперимента
Агонистическую активность соединения в отношении THR-β оценивали способом, аналогичным способу, описанному в литературе (J. Med. Chem. 2014, 57, 3912) : Готовили растворы (100Х) эталонных соединений или соединений с помощью ДМСО, а затем разбавляли в равном соотношении 1:3. Градиентный разбавленный раствор (100Х) эталонного соединения или соединения дополнительно разбавляли до концентрации 4Х при помощи IX реакционным буфером и затем добавляли на планшет. Смешанный раствор 4Х TRα-LBD или TRβ-LBD и 4Х RXRα готовили с IX реакционным буфером и добавляли на планшет для испытаний. Смешанный раствор 2Х биотипа-SRC2-2, 2Х Eu-анти-GST и 2Х стрептавидина-d2 готовили с IX реакционным буфером и добавляли на планшет для испытаний. Планшет центрифугировали при 1000 об/мин в течение 1 мин, а затем инкубировали при комнатной температуре и в темноте в течение 4 ч. Интенсивность сигнала флуоресценции при 665 нм и 615 нм оценивали на планшет-ридере En Vision 2104, а затем рассчитывали отношение 665 нм/615 нм для получения значения ЕС50 THR-β (нМ) для THR-β.
Агонистическую активность соединения согласно настоящему изобретению в отношении рецептор тиреоидного гормона α (THR-α) оценивали с помощью вышеупомянутого аналогичного способа, а дляα THR- рассчитывали значение ЕС50 THR-α (нМ), которое использовали для определения агонистической селективности соединения согласно настоящему изобретению в отношении THR-β/THR-α.
Селективность THR-β/THR-α=THR-α ЕС50 ÷ THR-β ЕС50.
2) Результаты экспериментов
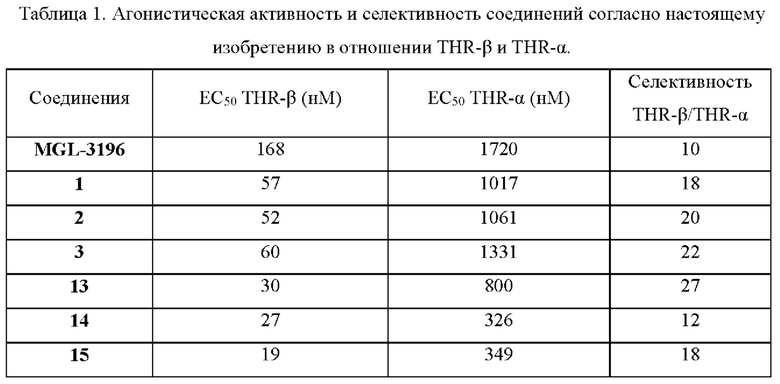
Полученные результаты приведены в таблице 1. Видно, что соединения 1, 2, 3, 13, 14 и 15 согласно настоящему изобретению обладают значительно более высокой агонистической активностью в отношении как THR-β, так и THR-α в сравнении с контрольным соединением MGL-3196, и, в частности, в отношении THR-β, при этом соединения согласно настоящему изобретению обладают значительно более высокой агонистической активностью. Кроме того, в отношении THR-β и THR-α соединения согласно настоящему изобретению также демонстрируют значительно более высокую агонистическую селективность в отношении THR-p при сравнении с контрольным соединением MGL-3196.
Экспериментальный пример 2 Фармакокинетические исследования соединений согласно настоящему изобретению на мышах
1) Экспериментальные материалы и приборы:
Полиэтиленгликоль 400 (ПЭГ400), производитель: Chengdu Kelong Chemical Reagent Factory;
Гидроксипропил-р-циклодекстрин (HP-p-CD), производитель: Shanghai Dibai Chemical Technology Co., Ltd.;
Гидроксипропилцеллюлоза низкой вязкости (HPC LF), производитель: Chengdu Yuannuo Tiancheng Technology Co., Ltd.;
Гепарин натрия, производитель: Chengdu Kelong Chemical Reagent Factory. Экспериментальные животные: мыши ICR (Chengdu Dossy Experimental Animals CO., LTD.).
2) Способ проведения эксперимента
Получение раствора исследуемого соединения
Группа в/в введения: Брали точную навеску 1,15 мг исследуемого соединения, затем добавляли DMA (0,228 мл) для растворения соединения, а затем последовательно добавляли ПЭГ 400 (1,139 мл) и 0,1 М фосфатного буфера (5,012 мл). После этого добавляли 40% HP-B-CD до конечного объема 11,39 мл. Раствор тщательно перемешивали при помощи ультразвукового вортекса. Получали прозрачный раствор с концентрацией 0,1 мг/мл.
Группа п/о применения: Точную навеску 5,06 мг исследуемого соединения, а затем для достижения конечного объема 20,04 мл добавляли 2% HPC LF (содержит 0,1% Твин 80). Раствор перемешивали с помощью ультразвукового вортекса для получения однородной суспензии с концентрацией 0,25 мг/мл.
Экспериментальные процедуры: Девять взрослых мышей ICR (3 мыши в каждой контрольной точке) не принимавших пищу в течение ночи (при этом имели свободный доступ к питьевой воде) получали исследуемое соединение при инъекции через хвостовую вену и через зонд, соответственно. В группе в/в введения через 5 мин, 15 мин, 0,5 ч, 1 ч, 2 ч, 4 ч, 8 ч, 12 ч и 24 ч после введения из подчелюстной вены собирали 0,1 мл крови, центрифугировали при 4°С в течение 5 мин для отделения плазмы и хранили при -20°С для анализа. В группе п/о применения перед применением и через 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 12 ч и 24 ч после применения из подчелюстной вены собирали 0,1 мл крови, и процедура была такой же, как и в группе в/в введения.
Затем определяли концентрацию исходного лекарственного средства в плазме методом ЖХ/МС/МС. Строили кривые зависимости концентрации в плазме от времени и рассчитывали основные фармакокинетические параметры с помощью программного обеспечения WinNonlin 6.3 (см. таблицу 2).
3) Результаты экспериментов
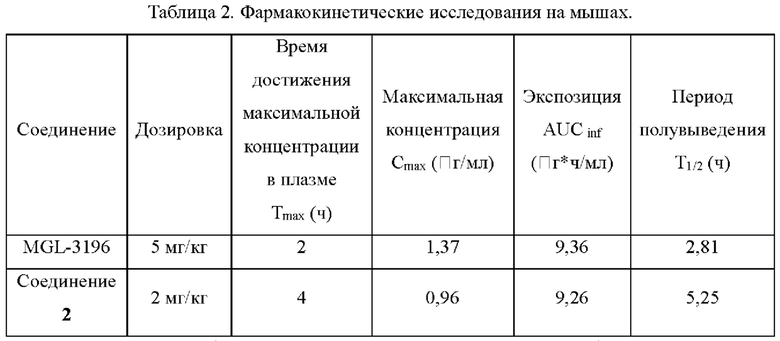
Согласно данным из таблицы 2 соединение согласно настоящему изобретению применяли в дозе 5 мг/кг у мышей, a MGL-3196 в дозе 2 мг/кг; и соединение 2 согласно настоящему изобретению в более низкой дозе показало более длительный период полувыведения, чем MGL-3196. Было показано, что соединения согласно настоящему изобретению обладают лучшими фармакокинетическими свойствами по сравнению с MGL-3196.
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его оптический изомер, соль, пролекарство, гидрат или неводный сольват. В сравнении с контрольным соединением MGL-3196, соединение формулы (I), которое получали с помощью замещения по конкретным положениям и конкретных типов замещения согласно настоящему изобретению, проявляло более высокую агонистическую активность как в отношении THR-α, так и THR-α, и особенно в отношении THR-β, соединение согласно настоящему изобретению обладало значительно более высокой агонистической активностью и селективностью. Кроме того, соединение согласно настоящему изобретению также демонстрирует значительно улучшенные фармакокинетические свойства и имеет хорошую перспективу применения для получения агонистов THR-β и лекарственных средств для показаний к применению агонистов THR-β (включая дислипидемию, гиперхолестеринемию, неалкогольный стеатогепатит и неалкогольную жировую болезнь печени).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЕЙТЕРИРОВАННОГО ДЕФАКТИНИБА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2761825C1 |
| ИНГИБИТОР FAK И КОМБИНАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ С НИМ | 2019 |
|
RU2820555C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| ПРОИЗВОДНОЕ 6-ОКСО-1,6-ДИГИДРОПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2020 |
|
RU2791534C2 |
| Соединения С,O-спиро-арил-гликозидов, их приготовление и их использование | 2016 |
|
RU2746858C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| ПРОЛЕКАРСТВЕННЫЕ СРЕДСТВА ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ | 2015 |
|
RU2693622C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемым солям, где каждый из R1-R7, R9 и R10 независимо выбран из H и D; каждый X и Y независимо выбран из группы, состоящей из Cl и Br, при этом X и Y одновременно не могут быть Cl; B представляет собой N, а A представляет собой CH или CD. Соединение обладает большей агонистической активностью в отношении THR-бета и значительно улучшенной селективностью в отношении THR-бета/THR-альфа в сравнении с соединением MGL-3196 и имеет значительно улучшенные фармакокинетические свойства. Соединение согласно настоящему изобретению имеет хорошую перспективу применения для получения агониста THR-бета и лекарственных средств для лечения заболеваний, связанных с агонистами THR-бета, например, дислипидемии, гиперхолестеринемии, неалкогольного стеатогепатита и неалкогольной жировой болезни печени. 4 н. и 8 з.п. ф-лы, 2 табл., 6 пр.
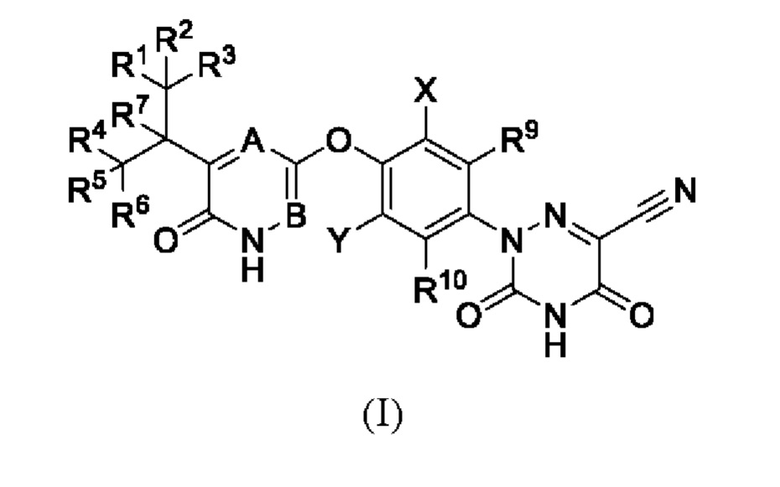
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
 ,
,
где каждый из R1-R7, R9 и R10 независимо выбран из H и D; каждый X и Y независимо выбран из группы, состоящей из Cl и Br, при этом X и Y одновременно не могут быть Cl; B представляет собой N, а A представляет собой CH или CD.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (II):
 ,
,
где каждый из R1-R7, R9 и R10 независимо выбран из H и D; B представляет собой N, а A представляет собой CH или CD.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (IV):
 ,
,
где каждый из R1-R7, R9 и R10 независимо выбран из H и D; A выбран из группы, состоящей из CH и CD;
предпочтительно каждый из R7, R9 и R10 независимо выбран из H; каждый из R1-R6 независимо выбран из H и D.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (III):
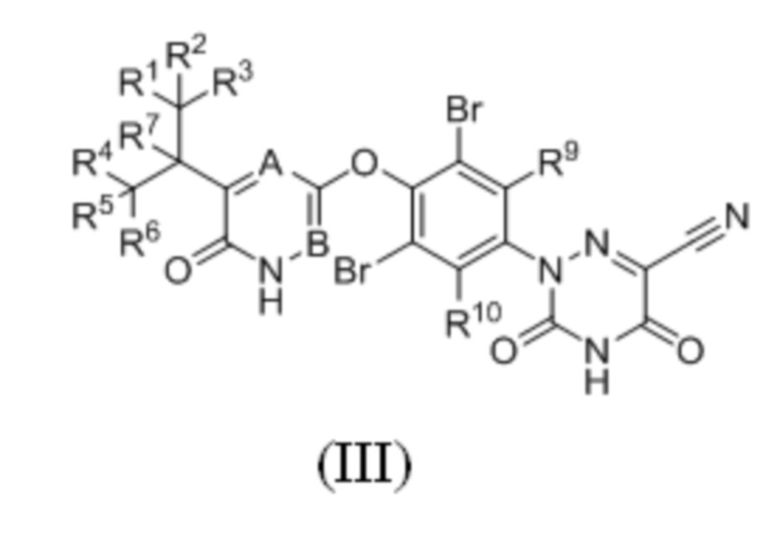 ,
,
где каждый из R1-R7, R9 и R10 независимо выбран из H и D; B представляет собой N, а A представляет собой CH или CD.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (V):
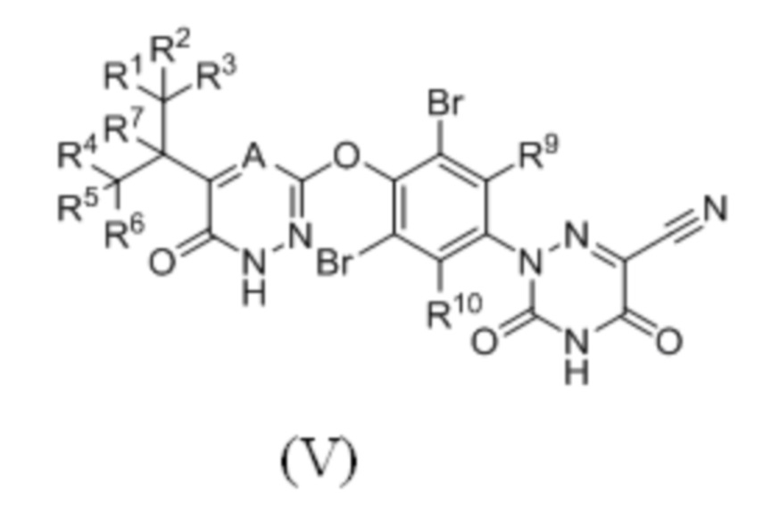 ,
,
где каждый из R1-R7, R9 и R10 независимо выбран из H и D; A выбран из группы, состоящей из CH и CD;
предпочтительно каждый из R7, R9 и R10 независимо выбран из H; каждый из R1-R6 независимо выбран из H и D.
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение выбрано из группы, состоящей из следующих соединений:
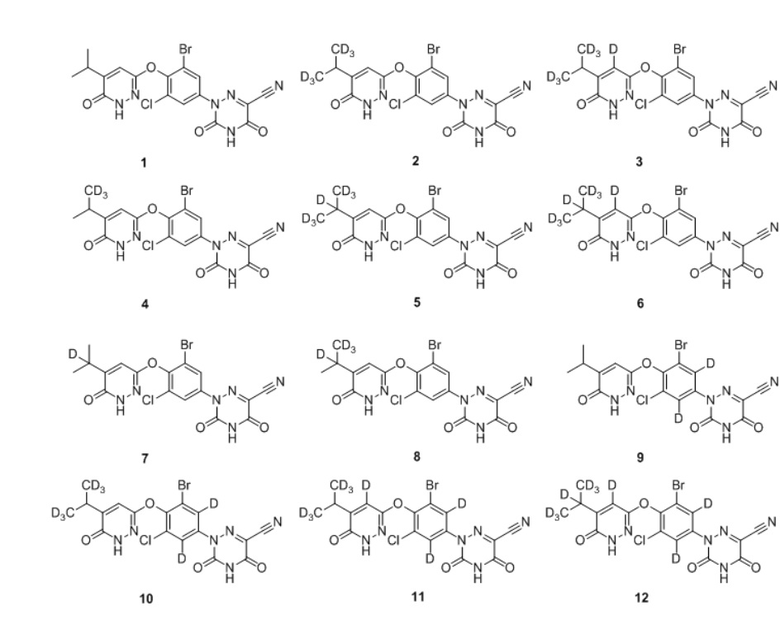
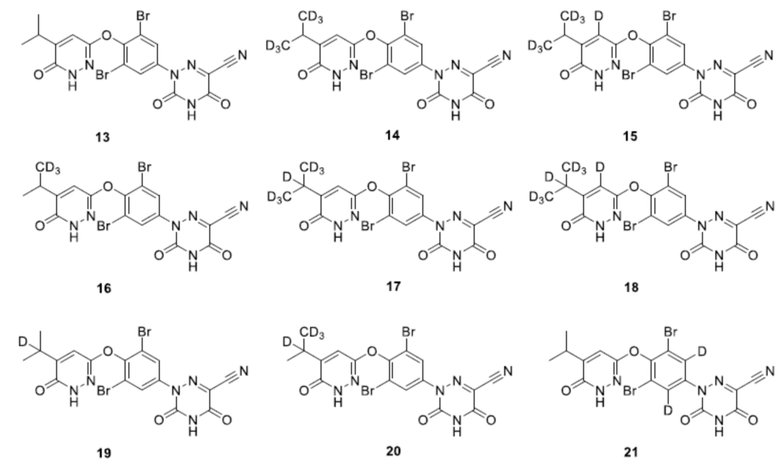
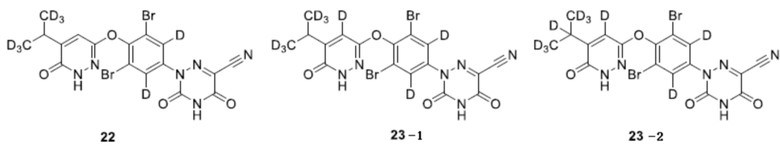 .
.
7. Применение соединения по любому из пп. 1-6 или его фармацевтически приемлемой соли для получения агонистов THR-β.
8. Применение по п. 7, отличающееся тем, что указанный агонист THR-β представляет собой лекарственное средство для снижения уровня холестерина и лечения дислипидемии и неалкогольной жировой болезни печени.
9. Применение по п. 7, отличающееся тем, что указанный агонист THR-β представляет собой лекарственное средство для лечения семейной гиперхолестеринемии и неалкогольного стеатогепатита.
10. Применение соединения по любому из пп. 1-6 или его фармацевтически приемлемой соли для получения агонистов THR-α.
11. Применение по п. 10, отличающееся тем, что указанный агонист THR-α представляет собой лекарственное средство для лечения диффузного токсического зоба.
12. Применение лекарственного средства, содержащего соединение по пп. 1-6 или его фармацевтически приемлемую соль в качестве активного ингредиента и фармацевтически приемлемые вспомогательные вещества, для снижения уровня холестерина и лечения дислипидемии и неалкогольной жировой болезни печени.
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА ТИРЕОИДНОГО ГОРМОНА | 2006 |
|
RU2379295C2 |
| WO 2019144835 A1, 01.08.2019 | |||
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| MARTHA J | |||
| KELLY ET AL, "Discovery of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (MGL-3196), a Highly Selective Thyroid Hormone Receptor β Agonist in Clinical Trials for the | |||

