ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Эта не являющаяся предварительной заявка на патент испрашивает приоритет предварительной заявки на патент США №62/687930, поданной 21 июня 2018 г., и предварительной заявки на патент США №62/719896, поданной 20 августа 2018 г., каждая из которых включена в данное описание посредством ссылки во всей своей полноте и для всех назначений.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Эта заявка включает посредством ссылки Перечень последовательностей, представленный вместе с этой заявкой в виде текстового файла с названием P34807-WO_SL.txt, созданного 12 июня 2019 г. и имеющего размер 16,4 килобайт.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Согласно данному изобретению предложены твердые формы 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола и способы их применения в лечении рака. Кроме того, в данной заявке описаны способы получения конденсированных трициклических соединений, содержащих замещенную фенильную или пиридинильную группировку.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Конденсированные трициклические соединения, содержащие замещенную фенильную или пиридинильную группировку, включенные в объем настоящего изобретения, полезны в качестве агентов, направленных на рецепторы эстрогенов ("ER").
ER представляет собой лиганд-активируемый регулирующий транскрипцию белок, который опосредует индуцирование ряда биологических эффектов путем своего взаимодействия с эндогенными эстрогенами. Эндогенные эстрогены включают 17β(бета)-эстрадиол и эстроны. Обнаружено, что ER имеет две изоформы, ER-α (альфа) и ER-β (бета). Эстрогены и рецепторы эстрогенов вовлечены в ряд заболеваний или состояний, таких как рак молочной железы, рак легкого, рак яичников, рак толстой кишки, рак предстательной железы, рак эндометрия, рак матки, а также другие заболевания или состояния. ERα-направленные агенты обладают особой активностью в условиях метастазирующего течения заболевания и приобретенной устойчивости. ERα-направленные агенты описаны в публикации заявки США под номером 2016/0175289.
Полезные способы получения конденсированных трициклических соединений, содержащих замещенную фенильную или пиридинильную группировку, описаны в публикации заявки США под номером 2016/0175289. Однако существует потребность в усовершенствованных способах получения ERα-направленных агентов.
Идентификация и выбор твердой формы фармацевтического соединения является весьма сложной задачей. Различия в твердых формах таких соединений влияют как на физические, так и химические свойства и могут воздействовать на процесс обработки, стабильность, биодоступность, состав и способ хранения фармацевтических соединений. Не существует никакой надежной прогнозируемости свойств твердой формы и ее полезности в качестве кристаллического или аморфного твердого вещества. Кристаллические твердые вещества могут считаться полезными, например, для обеспечения физической или химической стабильности, в то время как аморфные твердые вещества могут считаться полезными, например, в плане улучшения растворимости и повышения биодоступности.
Смеси кристаллических веществ возникают как следствие полиморфизма. Априори невозможно предсказать, существуют ли кристаллические формы соединения, не говоря уже о том, могут ли быть получены или выделены кристаллические формы. Jones et al., 2006, Pharmaceutical Cocrystals: An Emerging Approach to Physical Property Enhancement" MRS Bulletin, 31: 875-879 (в настоящее время, как правило, невозможно путем вычислений предсказать количество наблюдаемых полиморфов даже у простейших молекул). Такое количество возможных твердых форм обуславливает наличие разных химических и физических свойств у фармацевтического соединения и может оказать сильное влияние на разработку, стабильность продукта и продвижение его на рынок.
Рецептор эстрогенов ("ER") представляет собой лиганд-активируемый регулирующий транскрипцию белок, который опосредует индуцирование ряда биологических эффектов путем своего взаимодействия с эндогенными эстрогенами. Эндогенные эстрогены включают 17β(бета)-эстрадиол и эстроны. Обнаружено, что ER имеет две изоформы, ER-α (альфа) и ER-β (бета). Эстрогены и рецепторы эстрогенов вовлечены в ряд заболеваний или состояний, таких как рак молочной железы, рак легкого, рак яичников, рак толстой кишки, рак предстательной железы, рак эндометрия, рак матки, а также другие заболевания или состояния. Существует потребность в новых ERα-направленных агентах, которые обладают активностью в условиях метастазирующего течения заболевания и приобретенной устойчивости. Соответственно, сохраняется необходимость в противораковых видах терапии с участием конкретных твердых форм.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение предусматривает решения вышеупомянутых проблем и других проблем данной области техники.
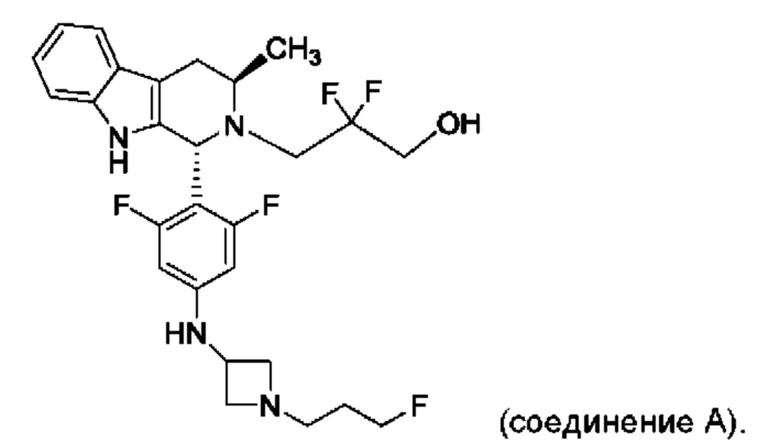
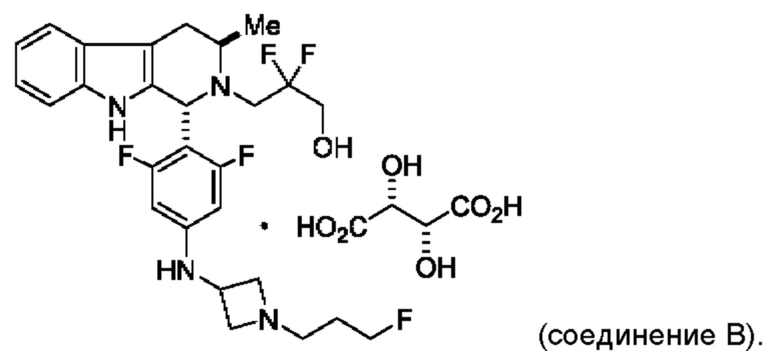
Согласно одному из аспектов данного изобретения предложено соединение, соединение В, имеющее название 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола тартрат, которое описано в данной заявке.
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая картину дифракции рентгеновских лучей на порошке, содержащую пики при 19,32; 20,26; 21,63; 23,28 или 24,81±0,1°2θ (±0,1°2θ).
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая картину дифракции рентгеновских лучей на порошке, содержащую пики при 11,49; 12,54; 19,16; 19,42 или 24,67±0,1°2θ (±0,1°2θ).
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая по существу такую картину дифракции рентгеновских лучей на порошке, как показано на ФИГ. 10 или ФИГ. 14.
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая картину дифракции рентгеновских лучей на порошке, содержащую пики при 11,31; 15,70; 16,54; 19,10 или 22,76±0,1°2θ.
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая картину дифракции рентгеновских лучей на порошке, содержащую пики при 12,52; 15,90; 19,66; 20,65 или 24,99±0,1°2θ.
Согласно другому аспекту данного изобретения предложена кристаллическая форма соединения В, имеющая картину дифракции рентгеновских лучей на порошке, содержащую пики при 11,46; 12,51; 19,29; 19,42 или 20,23±0,1°2θ.
Согласно другому аспекту данного изобретения предложено аморфное твердое вещество, содержащее соединение А.
Кроме того, согласно данному изобретению предложены фармацевтические композиции, содержащие соединение В или его кристаллическую соль. Такие соединения и фармацевтические композиции могут быть использованы в способах лечения рака, указанных в данном описании.
Согласно другому аспекту данного изобретения предложен способ получения соединения формулы (IV) или его соли, указанных в данном описании. Способ включает (1) приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (I), описанного в данной заявке, органического растворителя и тионилхлорида с образованием соединения формулы (IIa), описанного в данной заявке, и (2) приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (IIa), катализатора, окислителя и растворителя с образованием соединения формулы (II), описанного в данной заявке. Способ дополнительно включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (II) и соединения формулы (III), описанного в данной заявке, в органическом растворителе с образованием соединения формулы (IV) или его соли, описанного(ой) в данной заявке.
Согласно другому аспекту данного изобретения предложен способ получения соединения формулы (VIII) или его фармацевтически приемлемой соли, описанного(ой) в данной заявке. Способ включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (IV), описанного в данной заявке, соединения формулы (V), описанного в данной заявке, или соединения формулы (X), описанного в данной заявке, и органического растворителя с образованием соединения формулы (VI), описанного в данной заявке. Способ дополнительно включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (VI), органического растворителя и соединения формулы (VII), описанного в данной заявке, или его соли с образованием соединения формулы (VIII) или его соли.
Согласно другому аспекту данного изобретения предложен способ получения соединения формулы (VIII) или его фармацевтически приемлемой соли, описанного(ой) в данной заявке. Способ включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (IX), описанного в данной заявке, или соединения формулы (X), описанного в данной заявке, соединения формулы (IV), описанного в данной заявке, и органического растворителя с образованием соединения формулы (VIII) или его соли, описанного(ой) в данной заявке.
Согласно еще одному аспекту данного изобретения предложен способ получения соединения формулы (IX) или его соли, описанного(ой) в данной заявке. Способ включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (X), описанного в данной заявке, соединения формулы (VII) или его соли, описанного(ой) в данной заявке, органического растворителя и катализатора с образованием соединения формулы (XI) или его соли, описанного(ой) в данной заявке.
Согласно еще одному аспекту данного изобретения предложен способ получения соединения формулы (III) или его соли, описанного(ой) в данной заявке. Способ включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (XII), описанного в данной заявке, соединения В и органического растворителя с образованием соединения формулы (XIII), описанного в данной заявке.
Согласно еще одному аспекту данного изобретения предложено соединение формулы (XVI), описанное в данной заявке.
Более того, согласно данному изобретению предложен способ получения соединения, имеющего формулу (XX), причем данный способ включает приведение в контакт соединения формулы (XXI), описанного в данной заявке, с белком трансаминазой с образованием соединения формулы (3). Соединение формулы (3) приводят в контакт с соединением формулы (II), описанным в данной заявке, с образованием соединения формула (XX).
Воплощения настоящего изобретения можно понять более полно, обратившись к подробному описанию и примерам, которые предназначены для иллюстрации неограничивающих воплощений.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На ФИГ. 1 изображена картина дифракции рентгеновских лучей на порошке (XRPD) для Формы А соединения В.
На ФИГ. 2 представлены результаты термогравиметрического анализа (TGA) и дифференциальной сканирующей калориметрии (DSC) для Формы А соединения В.
На ФИГ. 3 показано полученное с применением PLM (микроскопия в поляризованном свете) изображение для Формы А соединения В.
На ФИГ. 4 изображена картина XRPD для Формы В соединения В.
На ФИГ. 5 представлены результаты TGA и DSC для Формы В соединения В.
На ФИГ. 6 показан спектр 13С ттЯМР (твердотельный ядерный магнитный резонанс) для Формы В соединения В.
На ФИГ. 7 показан спектр 19F ттЯМР для Формы В соединения В.
На ФИГ. 8 показан график сорбции/десорбции воды для Формы В соединения В.
На ФИГ. 9а показано изображение, полученное с применением сканирующей электронной микроскопии (SEM); на ФИГ. 9b показано полученное с применением PLM изображение для Формы В соединения В; на Фиг. 9с показано распределение частиц по размерам (PSD) для Формы В соединения В.
На ФИГ. 10 изображена картина XRPD для Формы С соединения В по сравнению с Формой А и Формой В соединения В. Было обнаружено, что Форма С представляет собой смесь Формы А и Формы В.
На ФИГ. 11 представлены результаты TGA и DSC для Формы С соединения В.
На ФИГ. 12 изображена картина XRPD для Формы D соединения В.
На ФИГ. 13 представлены результаты TGA и DSC для Формы D соединения В.
На ФИГ. 14 изображена картина XRPD для Формы Е соединения В.
На ФИГ. 15 представлены результаты TGA и DSC для Формы Е соединения В.
На ФИГ. 16 изображена картина XRPD для Формы F соединения В.
На ФИГ. 17 показан график динамической сорбции паров (DVS) для Формы F соединения В.
На ФИГ. 18 представлены результаты DSC для Формы F соединения В.
На ФИГ. 19 изображена картина XRPD для Формы G соединения В.
На ФИГ. 20 представлены результаты TGA и DSC для Формы G соединения В.
На ФИГ. 21 показано наложение картин XRPD для Формы А, Формы В, Формы С, Формы D, Формы F и Формы G соединения В.
На ФИГ. 22 изображена картина XRPD, полученная в анализе свойств сжимаемости для Формы В соединения В.
На ФИГ. 23 показан спектр 19F ттЯМР, полученный в анализе свойств сжимаемости для Формы В соединения В.
На ФИГ. 24 представлены результаты DSC, полученные в анализе свойств сжимаемости для Формы В соединения В.
На ФИГ. 25 показаны пути фазового превращения для Форм А, В, D и F соединения В.
На ФИГ. 26 показаны пути фазового превращения при получении Формы В соединения В из Формы F.
На ФИГ. 27а изображена картина XRPD для Формы 1 соединения С; на ФИГ. 27b изображена картина XRPD для Формы 2 соединения С.
На ФИГ. 28 представлены результаты TGA и DSC для Формы 1 соединения С.
На ФИГ. 29 показано полученное с применением PLM изображение для Формы 1 соединения С.
На ФИГ. 30 изображена картина XRPD для Формы М соединения D.
На ФИГ. 31 представлены результаты TGA и DSC для Формы М соединения D.
На ФИГ. 32 изображена картина XRPD для аморфной формы соединения А.
На ФИГ. 33 представлены результаты TGA и DSC для аморфной формы соединения А.
На ФИГ. 34 показана жизнеспособность клеток в клеточной линии ER+ рака молочной железы в случае применения соединения В по сравнению с GDC-0810 и GDC-0927.
На ФИГ. 35 показано влияние на объем опухоли соединения В в дозе 0,1 мг/кг и 1 мг/кг по сравнению с GDC-0927 в дозе 100 мг/кг.
На ФИГ. 36а представлены результаты сканирования методом компьютерной томографии (СТ), а на ФИГ. 36b представлены результаты сканирования методом FES-PET (позитронно-эмиссионная томография с использованием 18F-фторэстрадиола) пациента с раком молочной железы, получавшего лечение соединением В.
На ФИГ. 37а представлены результаты СТ-сканирования, а на ФИГ. 37b представлены результаты FES-PET-сканирования второго пациента с раком молочной железы, получавшего лечение соединением В.
ПОДРОБНОЕ ОПИСАНИЕ
Если не указано иное, то все технические и научные термины, использованные в данном описании, имеют то же самое значение, которое обычно понимается специалистом средней квалификации в области техники, к которой данное изобретение относится. См., например, Singleton et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY, 2nd ed., J. Wiley & Sons (New York, NY 1994); Sambrook et al., MOLECULAR CLONNING, A LABORATORY MANUAL, Cold Springs Harbor Press (Cold Springs Harbor, NY, 1989). Любые способы, устройства и материалы, аналогичные или эквивалентные таковым, описанным в данной заявке, можно использовать при практическом применении данного изобретения.
Следующие далее определения приведены для облегчения понимания некоторых терминов, часто используемых в данном описании, и не предназначены для ограничения объема настоящего изобретения. Все ссылки, упомянутые в данном описании, включены посредством ссылки во всей своей полноте.
Как использовано в данном описании и если не указано иное, термины "примерно" и "приблизительно," при ссылке на дозы, количества или массовые проценты ингредиентов в композиции или лекарственной форме, означают дозу, количество или процент по массе, которые, как признано специалистом средней квалификации в данной области техники, обеспечивают фармакологический эффект, эквивалентный эффекту, получаемому в результате применения указанных дозы, количества или процента по массе. Эквивалентные доза, количество или процент по массе могут находиться в пределах 30%, 20%, 15%, 10%, 5%, 1% или меньше от указанных дозы, количества или процента по массе.
Как использовано в данном описании и если не указано иное, термины "примерно" и "приблизительно", при ссылке на численное значение или диапазон значений, используемые для характеристики конкретной твердой формы, описанной в данной заявке (например, при ссылке на значения пиков XRPD), указывают на то, что это значение или этот диапазон значений могут отклоняться от заданного значения в степени, которая считается разумной для специалиста средней квалификации в данной области техники, при этом все еще описывая твердую форму. В одном из воплощений значение положения пиков XRPD может варьировать на величину до ±0,1°2θ (или ±0,05 градуса 2θ), все еще описывая конкретный пик XRPD.
Как использовано в данном описании и если не указано иное, кристалл, который является "чистым", означает, что он по существу не содержит других кристаллических или аморфных твердых веществ или других химических соединений и содержит менее чем примерно 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,4%, 0,3%, 0,2%, 0,1%, 0,05% или 0,01% одной или нескольких других твердых форм из расчета по массе. Обнаружение других твердых форм может быть выполнено, например, с применением дифракционного анализа, термического анализа, органического элементного анализа и/или спектроскопического анализа. Обнаружение других химических соединений может быть выполнено, например, с применением масс-спектрометрического анализа, спектроскопического анализа, термического анализа, органического элементного анализа и/или хроматографического анализа.
Если не указано иное, термины "сольват" и "сольватированный", использованные в данном описании, относятся к твердой форме вещества, содержащей растворитель. Термины "гидрат" и "гидратированный" относятся к сольвату, где растворителем является вода. Термины "сольват" и "сольватированный", использованные в данном описании, также могут относиться к сольвату соли, сокристалла или молекулярного комплекса. Термины "гидрат" и "гидратированный", использованные в данном описании, также могут относиться к гидрату соли, сокристалла или молекулярного комплекса.
Термин "фармацевтически приемлемый" относится к разбавителю, эксципиенту или носителю в композиции, совместимому с другим(ими) ингредиентом(ами) данной композиции и не оказывающему вредного воздействия на своего реципиента.
Соединение А обозначает соединение, имеющее структуру:
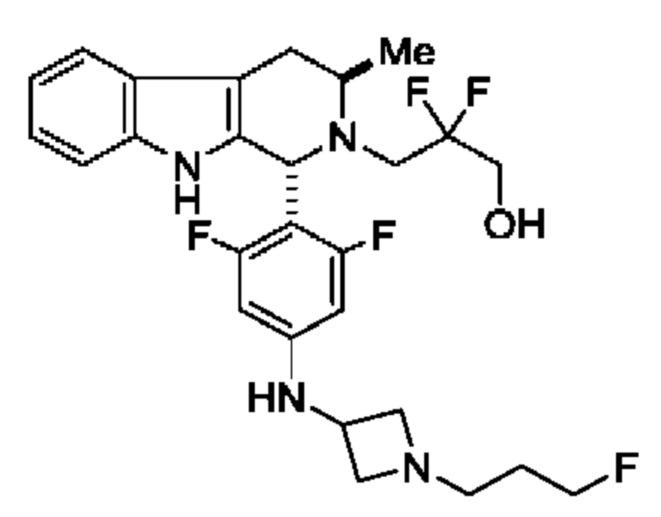
и имеющее название 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ол, включая его фармацевтически приемлемую соль. Соединение А может представлять собой соль винной кислоты, описанную в данной заявке (например, соединение В). Соединение А может представлять собой соль фумаровой кислоты, описанную в данной заявке (например, соединение С). Соединение А может представлять собой малонатную соль, описанную в данной заявке (например, соединение D).
Термин "твердая форма" относится к физической форме, которая предпочтительно не находится в жидком или газообразном состоянии. Твердая форма может представлять собой кристаллическую форму или их смесь. В некоторых воплощениях твердая форма может представлять собой жидкий кристалл. В некоторых воплощениях твердой формой соединения А является Форма А, Форма В, Форма С, Форма D, Форма Е, Форма F, Форма G, Форма 1 или Форма 2, аморфное твердое вещество или их смесь. В одном из воплощений твердой формой соединения А является тартратная соль. В другом воплощении твердой формой соединения А является фумаратная соль или их смесь. Твердой формой может быть кристаллическая форма, определенная в данном описании.
Термин "в форме кристалла" или "кристаллическая форма" относится к твердой форме, которая является кристаллической. В некоторых воплощениях кристаллическая форма соединения, описанного в данной заявке, может по существу не содержать аморфных твердых веществ и/или других кристаллических форм. В некоторых воплощениях кристаллическая форма соединения, описанного в данной заявке, может содержать одно или несколько аморфных твердых веществ и/или одну или несколько других кристаллических форм в количестве меньше примерно 1%, меньше примерно 2%, меньше примерно 3%, меньше примерно 4%, меньше примерно 5%, меньше примерно 6%, меньше примерно 7%, меньше примерно 8%, меньше примерно 9%, меньше примерно 10%, меньше примерно 15%, меньше примерно 20%, меньше примерно 25%, меньше примерно 30%, меньше примерно 35%, меньше примерно 40%, меньше примерно 45% или меньше примерно 50% по массе. В некоторых воплощениях кристаллическая форма, описанная в данной заявке, является чистой. В некоторых воплощениях чистота кристаллической формы соединения, описанного в данной заявке, может составлять примерно 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91% или 90%.
Термин "аморфный" или "аморфное твердое вещество" относится к твердой форме, которая по существу не является кристаллической, как определено посредством дифракции рентгеновских лучей. В частности, термин "аморфное твердое вещество" описывает неупорядоченную твердую форму, то есть твердую форму, у которой отсутствует дальний порядок кристаллической решетки. В некоторых воплощениях аморфное твердое соединение, описанное в данной заявке, может по существу не содержать других аморфных твердых веществ и/или кристаллических форм. В некоторых воплощениях аморфное твердое вещество может быть чистым. В некоторых воплощениях чистота аморфного твердого соединения, описанного в данной заявке, может составлять примерно 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91% или 90%.
Термин "лечение", использованный в данном описании, означает облегчение, полностью или частично, расстройства, заболевания или состояния либо одного или более симптомов, ассоциированных с расстройством, заболеванием или состоянием, либо замедление или приостановку дальнейшего прогрессирования или ухудшения этих симптомов, либо облегчение или устранение причин(ы) самого расстройства, заболевания или состояния. В одном из воплощений расстройство представляет собой рак.
Термин "эффективное количество" или "терапевтически эффективное количество" относится к количеству соединения, описанного в данной заявке, с применением которого можно лечить или предотвращать расстройство, заболевание или состояние или его симптомы, изложенные в данном описании.
Определение в данном описании термина "пациент" или "субъект" включает животных, таких как млекопитающие, в том числе, но не ограничиваясь этим, приматы (например, люди), крупный рогатый скот, овцы, козы, лошади, собаки, кошки, кролики, крысы, мыши, обезьяны, куры, индюки, перепелки или морские свинки и тому подобные, в одном из воплощений это млекопитающее, в другом воплощении это человек. В одном из воплощений, субъектом является человек, имеющий рак или риск развития рака.
Использованные в данном описании термины "группировка" и "заместитель" относятся к атому или группе связанных химическими связями атомов, который(ая) присоединен(а) к другому атому или другой молекуле посредством одной или нескольких химических связей с образованием тем самым части молекулы.
Использованный в данном описании термин "алкил" относится к алифатической насыщенной углеводородной группировке с прямой или разветвленной цепью, имеющей 1-20 атомов углерода. В конкретных воплощениях алкил имеет 1-10 атомов углерода. В конкретных воплощениях алкил имеет 1-6 атомов углерода. Алкильные группы возможно могут быть независимо замещены одним или несколькими заместителями, описанными в данной заявке.
Использованный в данном описании термин "замещенный" относится к замене по меньшей мере одного атома водорода в соединении или группировке на другой заместитель или другую группировку. Примеры таких заместителей включают, без ограничения, атом галогена, -ОН, -CN, оксо, алкокси, алкил, алкилен, арил, гетероарил, галогеналкил, галогеналкокси, циклоалкил и гетероцикл. Например, термин "галогеналкил" относится к тому обстоятельству, что один или более атомов водорода в алкиле (который определен ниже) заменен(ы) одним или более атомами галогена (например, трифторметил, дифторметил, фторметил, хлорметил и т.д.). В одном из воплощений термин "замещенный", использованный в данном описании, может относиться к замене по меньшей мере одного атома водорода в соединении или группировке, описанном(ой) в данной заявке, на атом галогена или алкил.
Использованный в данном описании термин "алкилен" относится к линейному или разветвленному насыщенному двухвалентному углеводородному радикалу из одного-двенадцати атомов углерода и, согласно другому аспекту, из одного-шести атомов углерода, при этом алкиленовый радикал возможно может быть независимо замещен одним или более заместителями, описанными в данной заявке. Примеры включают, но не ограничиваются этим, метилен, этилен, пропилен, 2-метилпропилен, пентилен и тому подобное.
Использованный в данном описании термин "алкокси" относится к группе формулы -O-R', где R' представляет собой алкильную группу. Алкоксигруппы возможно могут быть независимо замещены одним или более заместителями, описанными в данной заявке. Примеры алкоксигруппировок включают метокси, этокси, изопропокси и трет-бутокси.
Использованный в данном описании термин "арил" относится к циклической ароматической углеводородной группировке, имеющей моно-, би- или трициклическое ароматическое кольцо из 5-16 атомов углерода в кольце. Бициклические арильные кольцевые системы включают конденсированные бициклические системы, имеющие два конденсированных пятичленных арильных кольца (обозначенные как 5-5), имеющие пятичленное арильное кольцо и конденсированное шестичленное арильное кольцо (обозначенные как 5-6 и как 6-5), и имеющие два конденсированных шестичленных арильных кольца (обозначенные как 6-6). Арильная группа возможно может быть замещена так, как определено в данном описании. Примеры арильных группировок включают, но не ограничиваются этим, фенил, нафтил, фенантрил, флуоренил, инденил, пенталенил, азуленил и тому подобное. Термин "арил" также включает в себя частично гидрированные производные циклической ароматической углеводородной группировки при условии, что по меньшей мере одно кольцо в этой циклической ароматической углеводородной группировке является ароматическим, при этом каждое из них является возможно замещенным.
Использованный в данном описании термин "гетероарил" относится к ароматической гетероциклической моно-, би- или трициклической кольцевой системе из 5-16 атомов в кольце, содержащем 1, 2, 3 или 4 гетероатома, выбранных из N, О и S, причем остальные атомы в кольце представляют собой атомы углерода. В некоторых воплощениях моноциклические гетероарильные кольца могут быть 5-6-членными. Бициклические гетероарильные кольцевые системы включают конденсированные бициклические системы, имеющие два конденсированных пятичленных гетероарильных кольца (обозначенные как 5-5), имеющие пятичленное гетероарильное кольцо и конденсированное шестичленное гетероарильное кольцо (обозначенные как 5-6 и как 6-5), и имеющие два конденсированных шестичленных гетероарильных кольца (обозначенные как 6-6). Гетероарильная группа возможно может быть замещена так, как определено в данном описании. Примеры гетероарильных группировок включают пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридазинил, пиримидинил, триазинил, изоксазолил, бензофуранил, изотиазолил, бензотиенил, бензотиофенил, индолил, азаиндолил, изоиндолил, изобензофуранил, бензимидазолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, бензооксадиазолил, бензотиадиазолил, бензотриазолил, пуринил, хинолинил, изохинолинил, хиназолинил, хиноксалинил, пирролопиридинил, фуропиридинил, тиенопиридинил, пирролопиридазинил, пирролопиримидинил, пирролопиразинил, тиенопиридазинил, тиенопиримидинил, тиенопиразинил, фуропиридазинил, фуропиримидинил и фуропиразинил.
Использованные в данном описании термины "галоген", "атом галогена" и "галогенид", которые могут быть использованы взаимозаменяемо, относятся к атому фтора, хлора, брома или йода как заместителю.
Использованный в данном описании термин "галогеналкил" относится к алкильной группе, причем один или более атомов водорода в алкильной группе заменены одинаковыми или разными атомами галогена, в частности, атомами фтора и/или хлора. Примеры галогеналкила включают монофтор-, дифтор- или трифтор-метил, -этил или -пропил, например 3,3,3-трифторпропил, 2-фторэтил, 2,2,2-трифторэтил, фторметил, дифторметил или трифторметил.
Использованный в данном описании термин "гидроксиалкил" относится к алкильной группе, при этом один или более чем один атом водорода данной алкильной группы заменен на гидроксильную группировку. Примеры включают спирты и диолы.
Использованный в данном описании термин "гетероалкил" относится к алкилу с прямой или разветвленной цепью, который определен в данном описании, имеющему от 2 до 14 атомов углерода, от 2 до 10 атомов углерода или от 2 до 6 атомов углерода в цепи, один или более чем один из которых заменен на гетероатом, выбранный из S, О, Р и N. Неограничивающие примеры гетероалкилов включают простые алкиловые эфиры, вторичные и третичные алкиламины, амиды и ал кил сульфиды.
Использованный в данном описании термин "циклоалкил" означает насыщенную или частично ненасыщенную карбоциклическую группировку, имеющую моно-, би- (в том числе мостиковые бициклические) или трициклические кольца и от 3 до 10 атомов углерода в кольце. Циклоалкильная группировка возможно может быть замещена одним или несколькими заместителями. В конкретных воплощениях циклоалкил содержит от 3 до 8 атомов углерода (т.е. представляет собой (С3-С8)циклоалкил). В других конкретных воплощениях циклоалкил содержит от 3 до 6 атомов углерода (т.е. представляет собой (С3-С6)циклоалкил). Примеры циклоалкильных группировок включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и их частично ненасыщенные (циклоалкенильные) производные (например, циклопентенил, циклогексенил и циклогептенил), бицикло[3.1.0]гексанил, бицикло[3.1.0]гексенил, бицикло[3.1.1]гептанил и бицикло[3.1.1]гептенил. Циклоалкильная группировка может быть присоединена по типу "спироциклоалкила", например, "спироциклопропила": 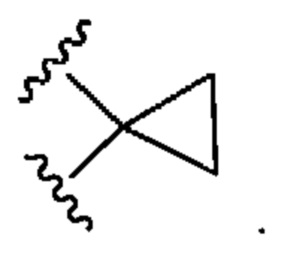
Использованные в данном описании термины "гетероцикл" или "гетероциклил" относятся к 4-, 5-, 6- и 7-членной моноциклической, 7-, 8-, 9- и 10-членной бициклической (в том числе мостиковой бициклической) или 10-, 11-, 12-, 13-, 14- и 15-членной бициклической гетероциклической группировке, которая является насыщенной или частично ненасыщенной и имеет один или несколько (например, 1, 2, 3 или 4) гетероатомов, выбранных из атомов кислорода, азота и серы, в кольце, при этом остальные атомы в кольце представляют собой атомы углерода. В некоторых воплощениях гетероцикл представляет собой гетероциклоалкил. В конкретных воплощениях гетероцикл или гетероциклил относится к 4-, 5-, 6- или 7-членному гетероциклу. Применительно к атому в гетероциклическом кольце, атом азота или серы также может быть в окисленной форме, а атом азота может быть замещен одним или более чем одним (С1-С6)алкилом или одной или более чем одной группой. К гетероциклу может быть присоединена его боковая группа по любому гетероатому или атому углерода, в результате присоединения в которому образуется стабильная структура. Любой из атомов в гетероциклическом кольце возможно может быть замещен одним или более заместителями, описанными в данной заявке. Примеры таких насыщенных или частично ненасыщенных гетероциклов включают, без ограничения, тетрагидрофуранил, тетрагидротиенил, пирролидинил, пирролидонил, пиперидинил, пирролинил, тетрагидрохинолинил, тетрагидроизохинолинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил. Термин "гетероцикл" также включает в себя группы, в которых гетероцикл сконденсирован с одним или более чем одним арильным, гетероарильным или циклоалкильным кольцом, таким как индолинил, 3Н-индолил, хроманил, азабицикло[2.2.1]гептанил, азабицикло[3.1.0]гексанил, азабицикпо[3.1.1]гептанил, октагидроиндолил или тетрагидрохинолинил.
Если не указано иное, термин "атом водорода" или "гидро" относится к группировке атома водорода (-Н), а не к Н2.
Использованный в данном описании термин "органический растворитель" относится к любому неводному полярному апротонному растворителю, полярному протонному растворителю и неполярному растворителю.
Использованный в данном описании термин "полярный органический растворитель" относится как к полярным апротонным растворителям, так и к полярным протонным растворителям, за исключением воды.
Использованный в данном описании термин "полярный апротонный растворитель" относится к любому полярному растворителю, не обладающему способностью отдавать протоны. Примеры включают, без какого-либо ограничения, 2-метилтетрагидрофуран, тетрагидрофуран, этилацетат, пропилацетат (например, изопропилацетат), ацетон, диметилсульфоксид, N,N-диметилформамид, ацетонитрил, N,N-диметилацетамид, N-метилпирролидон, гексаметилфосфорамид и пропиленкарбонат.
Использованный в данном описании термин "полярный протонный растворитель" относится к любому полярному растворителю, обладающему способностью отдавать протоны. Примеры включают, без ограничения, метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол, муравьиную кислоту, нитрометан и уксусную кислоту. Органический полярный протонный растворитель исключает любое эффективное количество воды.
Использованный в данном описании термин "неполярный растворитель" относится к растворителям, в структуре которых имеются связи между атомами со схожими значениями электроотрицательности, такими как атом углерода и водорода, благодаря чему электрический заряд на молекуле распределяется равномерно. Неполярные растворители характеризуются как имеющие низкую диэлектрическую проницаемость. Примеры включают, без ограничения, пентан, гексан, гептан, циклопентан, метил-трет-бутиловый эфир (МТВЕ), диэтиловый эфир, толуол, бензол, 1,4-диоксан, четыреххлористый углерод, хлороформ и дихлорметан (DCM). В некоторых воплощениях неполярный растворитель имеет диэлектрическую проницаемость меньше 2, примеры которого включают, без ограничения, пентан, гексан и гептан. По сравнению с другими неполярными растворителями DCM демонстрирует некоторую степень полярности на уровне связей (т.е. между атомами углерода и хлора), но только небольшую степень полярности на молекулярном уровне вследствие "отмены" полярности, обусловленной симметрией.
Использованный в данном описании термин "антирастворитель" относится к растворителю, в котором упоминаемое соединение является слабо растворимым и которое индуцирует выпадение в осадок или кристаллизацию указанного соединения из раствора.
Использованный в данном описании термин "кислотный катализатор" относится к такому кислотному катализатору, как кислота Бренстеда, кислота Льюиса или катализатор Бренстеда-Лоури, но этим не ограничиваясь. Неограничивающие примеры кислотных катализаторов включают уксусную кислоту, ледяную уксусную кислоту, трифторуксусную кислоту, бензойную кислоту, пивалевую кислоту, дифенилфосфорную кислоту, трифлатную кислоту, муравьиную кислоту, винную кислоту, фумаровую кислоту, малоновую кислоту, салициловую кислоту, п-толуолсульфоновую кислоту, серную кислоту, соляную кислоту, фосфорную кислоту, метансульфоновую кислоту, камфорсульфоновую кислоту, нафталинсульфоновую кислоту, монтмориллонитовую составляющую глины К-10 и смолу на основе Амберлист и их комбинации.
Использованный в данном описании термин "амин-защитная группа" относится к любой известной защитной группе, которая блокирует или защищает функциональную группу аминов. Амин-защитные группы в объеме данного изобретения включают, без ограничения, 1-хлорэтилкарбамат (ACD); 4-метоксибензолсульфонамид; ацетамид (Ас); бензиламин (Bn); бензилоксикарбамат (CBz); формамид; метилкарбамат; трифторацетамид; трет-бутоксикарбамат (Boc); п-метоксибензилкарбонил (MeOZ); 9-флуоренилметоксикарбонил (FMOC); бензоил (Bz); п-метоксибензил (РМВ); 3,4-диметоксибензил (DMPM); п-метоксифенил (РМР); тозил (Ts) и трихлорэтилхлорформиат (Troc). Для описания амин-защитных групп и их применения см. P. G. М. Wuts and Т. W. Greene, Greene's Protective Groups in Organic Synthesis, 4th edition, Wiley-lnterscience, New York, 2006.
Использованный в данном описании термин "альдегид-защитная группа" относится к любому известному заместителю, присоединяемому к альдегидной группе, который блокирует или защищает карбонильную группу альдегидной функциональной группы. Подходящие защитные группы для альдегидной функциональной группы включают, но не ограничиваются этим, (а) циклические ацетали и кетали, (b) циклические моно- или ди-тиоацетали или -кетали или другие производные, такие как имины, гидразоны, цианогидрин, оксимы или семикарбазоны, например, диалкил- или диарилацетали либо 1,3-дитиан, (с) циклические имины, такие как замещенные метиленовые производные или N,N'-диметилимидазолидин. Некоторые неограничивающие примеры альдегид-защитных групп включают 1,3-дитиан, 1,3-дитиолан, диэтилацеталь, диметилацеталь, ацеталь этиленгликоля, ацеталь неопентилгликоля, триметилсилилцианогидрин и триалкил-ортоформиаты, такие как триэтил-ортоформиат. Для описания альдегид-защитных групп и их применения см. Wuts и Greene.
Используемый здесь термин «уходящая группа» относится к атому или группе атомов, которые замещаются в химической реакции как стабильные частицы.
Использованный в данном описании термин "уходящая группа" относится к атому или группе атомов, который(ая) замещаются в химической реакции как стабильные структуры. Подходящие уходящие группы хорошо известны в данной области техники, например, см. March's Advanced Organic Chemistry, 5-ое изд., под ред.: Smith М.В. and March J., John Wiley & Sons, New York: 2001 и Т.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991, полные содержания которых тем самым включены посредством ссылки. Такие уходящие группы включают, но не ограничиваются этим, галоген, алкокси, сульфонилокси, возможно замещенный алкилсульфонил, возможно замещенный алкенилсульфонил, возможно замещенный арилсульфонил и диазониевые группировки. Примеры таких уходящих групп включают хлор, иод, бром, фтор, метансульфонил (мезил), тозил, трифлат, нитро-фенилсульфонил (нозил) и бром-фенилсульфонил (брозил).
Термин "катализаторы на основе переходного металла" в объеме данного изобретения включают, без ограничения, катализаторы на основе палладия, платины, золота, рутения, родия и иридия. Неограничивающие примеры подходящих катализаторов включают: хлорид (2-бифенил)ди-трет-бутилфосфин-золота(I) ("JohnPhos"), хлорид 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил-золота(I) ("XPhos AuCI"), бис(трифторметансульфонил)имид 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил-золота(I) ("XPhos AuNTf2"), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]палладий(II) ("палладацикл на основе XPhos"), аддукт хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2-аминоэтилфенил)]палладия(II) и метил-трет-бутилового эфира ("палладацикл на основе SPhos"), трет-BuXPhos-палладия(II) фенетиламин-хлорид ("tBuXPhos Pd первого поколения (G1)"), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) ("XPhos Pd G2"), хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(И) ("SPhos Pd G2"), хлор(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) ("RuPhos Pd G2"), хлор[(2-дициклогексилфосфино-2',6'-бис(N,N-диметиламино)-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладий(II) ("CPhos-Pd-G2"), [(2-дициклогексилфосфино-2',6'-бис(N,N-диметиламино)-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат ("CPhos-Pd-G3"), [(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат ("tBuXPhos-Pd-G3"), (2-дицикпогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат ("RuPhos-Pd-G3"), (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат ("XPhos-Pd-G3"), [(2-ди-циклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]паллад ия(II) метансульфонат ("BrettPhos-Pd-G3"), [(2-{бис[3,5-бис(трифторметил)фенил]фосфин-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат ("JackiePhos-Pd-G3"), трет-бутил-BrettPhos-Pd-G3, [трет-бутил-BrettPhos-Pd (аллил)]OTf) и их комбинации.
Как использовано в данном описании, термин "неорганические кислоты" относится к таким кислотам, как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, азотная кислота, борная кислота, фосфорная кислота и их комбинации, но этим не ограничивается.
Как использовано в данном описании, термин "органические кислоты" относится к таким кислотам, как: уксусная кислота; трифторуксусная кислота; фенилуксусная кислота; пропионовая кислота; стеариновая кислота; молочная кислота; аскорбиновая кислота; малеиновая кислота; гидроксималеиновая кислота; изетионовая кислота; янтарная кислота; валериановая кислота; фумаровая кислота; малоновая кислота; пировиноградная кислота; щавелевая кислота; гликолевая кислота; салициловая кислота; олеиновая кислота; пальмитиновая кислота; лауриновая кислота; пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота; альфа-гидрокси кислота, такая как миндальная кислота, лимонная кислота или винная кислота; цистеинсупьфиновая кислота; аминокислота, такая как аспарагиновая кислота, глутаровая кислота или глутаминовая кислота; ароматическая кислота, такая как бензойная кислота, 2-ацетоксибензойная кислота, нафтойная кислота или коричная кислота; сульфоновая кислота, такая как лаурилсульфоновая кислота, п-толуолсульфоновая кислота, метансульфоновая кислота, бензолсульфоновая кислота или этансульфоновая кислота; цистеин-сульфоновая кислота; и их комбинации, но этим не ограничивается.
Как использовано в данном описании, термин "неорганические основания" относится к таким основаниям, как гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид аммония, гидроксид магния, карбонат натрия, карбонат калия и их комбинации, но этим не ограничивается.
Использованный в данном описании термин "органическое основание" относится к органическому соединению, содержащему один или более атомов азота и действующему в качестве основания. Примеры органических оснований включают, но не ограничиваются этим, основания, представляющие собой третичные амины. Примеры органических оснований включают, но не ограничиваются этим, 1,8-диазабицикло[5.4.0]ундец-7-ен ("DBU"), N-метилморфолин (NMM), диизопропилэтиламин (DIPEA), триэтиламин (TEA), трет-бутилат (например, трет-бутилат натрия, калия, кальция или магния).
Соединения по настоящему изобретению могут присутствовать в солевой форме, которая охватывает фармацевтически приемлемые соли и фармацевтически неприемлемые соли. Использованный в данном описании термин "фармацевтически приемлемые соли" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, и не являются нежелательными ни в биологическом, ни в других отношениях. Помимо фармацевтически приемлемых солей соединения по настоящему изобретению могут быть в форме фармацевтически неприемлемых солей, которые могут быть полезны в качестве промежуточного соединения для выделения или очистки указанных соединений.
Типичные полученные присоединением кислоты соли соединений по настоящему изобретению включают, но не ограничиваются этим, соли: сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, биартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат (мезилат), этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). При образовании фармацевтически приемлемой соли может происходить включение остатка другой молекулы, такого как ацетат-ион, сукцинат-ион или другой противоион. Противоион может представлять собой любую органическую или неорганическую группировку, которая стабилизирует заряд на исходном соединении. Кроме того, фармацевтически приемлемая соль в своей структуре может иметь более одного заряженного атома. В случаях, когда частью фармацевтически приемлемой соли являются несколько заряженных атомов, могут присутствовать несколько противоионов. Поэтому, фармацевтически приемлемая соль может иметь один или более чем один заряженный атом и/или один или более чем один противоион.
Типичные полученные присоединением основания соли соединений по настоящему изобретению включают, но не ограничиваются этим, неорганические соли, образованные из катионов натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца и алюминия. Органические соли, образованные из катионов, включают соли первичных, вторичных и третичных аминов; замещенных аминов, в том числе природных замещенных аминов; циклических аминов; основных ионообменных смол; изопропиламина; триметиламина; диэтиламина; триметиламина; трипропиламина; этаноламина; 2-диэтиламиноэтанола; триметамина; дициклогексиламина; лизина; аргинина; гистидина; кофеина; прокаина; гидрабамина; холина; бетаина; этилендиамина; глюкозамина; метилглюкамина; теобромина; пуринов; пиперазина; пиперидина; N-этилпиперидина и полиаминных смол.
Соединения по настоящему изобретению также могут быть сольватированы, а именно, гидратированы. Сольватация может осуществляться в ходе процесса получения или может происходить, например, вследствие гигроскопических свойств исходно безводных соединений. В данном контексте "сольват" относится к ассоциации или комплексу одной или нескольких молекул растворителя и соединения по изобретению. Неограничивающие примеры растворителей, которые образуют сольваты, включают, но не ограничиваются этим, воду, изопропанол, этанол, метанол, диметилсульфоксид (DMSO), этилацетат (EtOAc), уксусную кислоту (АсОН) и этаноламин.
Соединения, имеющие одинаковую молекулярную формулу, но отличающиеся природой или последовательностью связывания своих атомов либо организацией своих атомов в пространстве, обозначаются термином "изомеры". Изомеры, которые отличаются организацией своих атомов в пространстве, обозначаются термином "стереоизомеры". Диастереомеры представляют собой стереоизомеры с противоположной конфигурацией по отношению к одному или нескольким хиральных центрам, которые не являются энантиомерами. Стереоизомеры, несущие один или более асимметрических центров, которые не совпадают с зеркальными отображениями друг друга, называются "энантиомерами". Когда соединение имеет асимметрический центр, например, если атом углерода связан с четырьмя разными группами, возможна пара энантиомеров. Энантиомер может быть охарактеризован согласно абсолютной конфигурации своего(их) асимметрического(ых) центра или центров и описан согласно правилам последовательного старшинства Кана, Ингольда и Прелога для присвоения конфигурации R и S или согласно способу вращения молекулой плоскости поляризованного света с обозначением как правовращающий или левовращающий (т.е. как (+)- или (-)-изомеры, соответственно). Хиральное соединение может существовать либо в виде отдельного энантиомера, либо в виде их смеси. Смесь, содержащую энантиомеры в равной пропорции называют "рацемической смесью". В некоторых воплощениях соединение обогащено по меньшей мере примерно на 90% (по массе) одним диастереомером или энантиомером. В других воплощениях соединение обогащено по меньшей мере примерно на 95%, 98% или 99% (по массе) одним диастереомером или энантиомером.
Некоторые соединения по настоящему изобретению обладают асимметрическими атомами углерода (оптическими центрами) или двойными связями; подразумевается, что все рацематы, диастереомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) включены в объем настоящего изобретения.
Соединения по изобретению могут содержать асимметрические или хиральные центры и ввиду этого существовать в разных стереоизомерных формах. Подразумевается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. В некоторых случаях стереохимическая конфигурация не была определена или была присвоена ориентировочно. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов) используют префиксы D и L или R и S. Для обозначения знака направления вращения плоскополяризованного света соединением применяют префиксы d и l или (+) и (-), при этом (-) или l означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер также может быть определен как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь с соотношением энантиомеров 50:50 называется рацемической смесью или рацематом, которая может образовываться, если не соблюдалось никакой стереоизбирательности или стереоспецифичности в химической реакции или химическом процессе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных разновидностей, не обладающей оптической активностью. Энантиомеры можно выделить по отдельности из рацемической смеси методом хирального разделения, таким как сверхкритическая жидкостная хроматография (SFC). Присвоение конфигурации при хиральных центрах в разделенных энантиомерах может быть ориентировочным, тогда как стереохимическая конфигурация является однозначно установленной, например, по данным рентгеноструктурного анализа кристаллов.
Как использовано в данном описании, "по существу" относится по меньшей мере к 90%, по меньшей мере к 95%, по меньшей мере к 98% или по меньшей мере к 99%.
В представленном в данной заявке описании, если есть несоответствие между изображенной структурой и названием, приданным этой структуре, то изображенная структура имеет приоритет. Кроме того, если стереохимическая конфигурация структуры или части структуры не указана, например, жирными клиновидными или пунктирными линиями, то следует интерпретировать, что данная структура или часть структуры охватывает все ее стереоизомеры. Однако, в некоторых случаях, когда имеется более одного хирального центра, данные структуры и названия могут быть представлены в виде одиночных энантиомеров для облегчения описания относительной стереохимической конфигурации.
Если не указано иное, термины "соединение данной формулы" или "соединение формулы" либо "соединения данной формулы" или "соединения формулы" относятся к любому соединению, выбранному из класса соединений, определенных данной формулой (включая любую фармацевтически приемлемую соль любого такого соединения, если не указано иное).
Согласно одному из аспектов данного изобретения предложен способ получения соединения формулы (IV) или его соли:
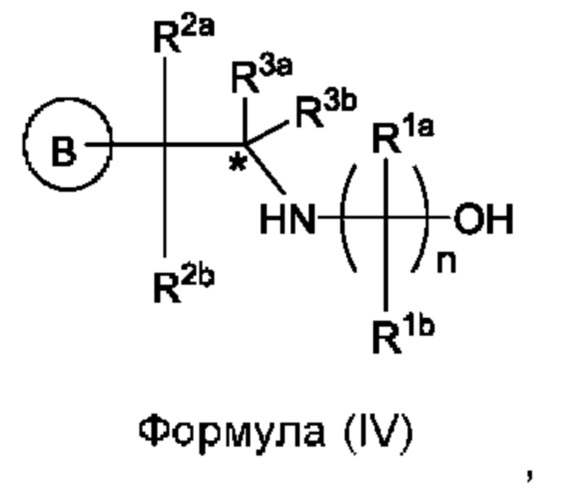
при этом способ включает стадии:
(а) приведения во взаимодействие содержащихся в реакционной смеси соединения формулы (I), органического растворителя и тионилхлорида с образованием соединения формулы (IIa) в соответствии с приведенной ниже стадией 1 и приведения во взаимодействие содержащихся в реакционной смеси соединения формулы (IIa), катализатора, окислителя и растворителя с образованием соединения формулы (II) в соответствии с приведенной ниже стадией 2,
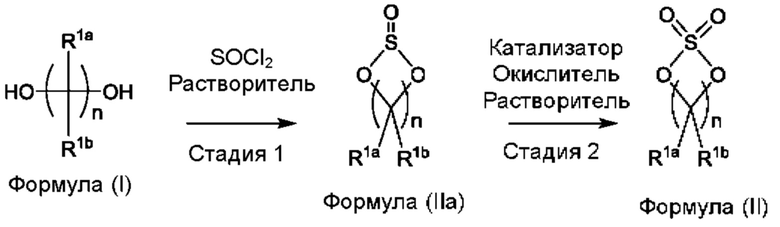
где каждый из R1a и R1b независимо представляет собой атом водорода, галогена, С1-3алкил, С1-3галогеналкил, С1-3алкокси, -CN, С3-6циклоалкил или С3-6 спироциклоалкил, и
n равно целому числу 2 или 3; и
(b) приведения во взаимодействие содержащихся в реакционной смеси соединения формулы (II) и соединения формулы (III) в органическом растворителе с образованием соединения формулы (IV) или его соли в соответствии с приведенной ниже стадией 3,
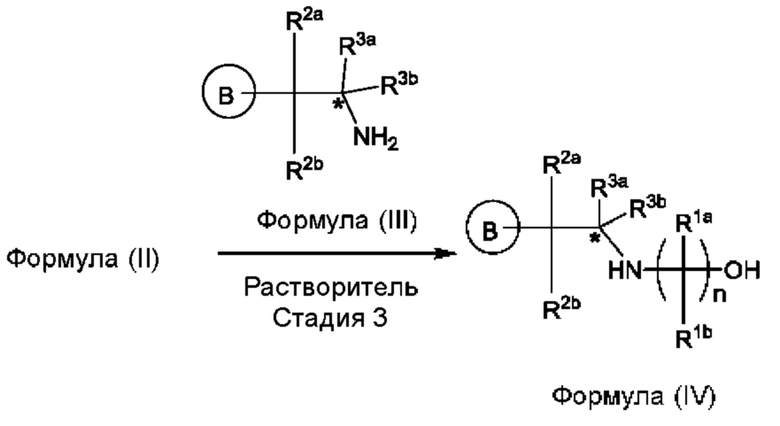
где В представляет собой замещенный или незамещенный индолил, бензофуранил, бензотиофенил, индазолил, азаиндолил, бензимидазолил, пирролопиридинил, фуропиридинил, тиенопиридинил, пирролопиридазинил, пирролопиримидинил, пирролопиразинил, тиенопиридазинил, тиенопиримидинил, тиенопиразинил, фуропиридазинил, фуропиримидинил или фуропиразинил,
каждый из R2a и R2b независимо представляет собой атом водорода, галогена, -ОН, С1-3алкил, С1-3 галогеналкил, С1-3алкокси, С1-3 гидроксиалкил, -CN, С3-6циклоалкил или С3-6 спироциклоалкил,
R3a и R3b независимо представляют собой атом водорода, С1-3алкил, C1-3галогеналкил, С1-3гидроксиалкил, -CN, С3-6циклоалкил, С3-6гетероциклоалкил, фенил, С3-6гетероарил или С3-6спироциклоалкил, и
звездочка обозначает хиральный центр, когда R3a и R3b являются разными.
В одном из воплощений В представляет собой замещенный индолил, бензофуранил, бензотиофенил, индазолил, азаиндолил, бензимидазолил, пирролопиридинил, фуропиридинил, тиенопиридинил, пирролопиридазинил, пирролопиримидинил, пирролопиразинил, тиенопиридазинил, тиенопиримидинил, тиенопиразинил, фуропиридазинил, фуропиримидинил или фуропиразинил.
В другом воплощении В представляет собой незамещенный индолил, бензофуранил, бензотиофенил, индазолил, азаиндолил, бензимидазолил, пирролопиридинил, фуропиридинил, тиенопиридинил, пирролопиридазинил, пирролопиримидинил, пирролопиразинил, тиенопиридазинил, тиенопиримидинил, тиенопиразинил, фуропиридазинил, фуропиримидинил или фуропиразинил.
В одном из воплощений В представляет собой замещенный или незамещенный индолил, бензофуранил или бензотиофенил. В другом воплощении В представляет собой индолил, бензофуранил или бензотиофенил, замещенный одним или несколькими атомами галогена или С1-3алкилами, описанными в данной заявке. В еще одном воплощении В представляет собой замещенный или незамещенный пирролопиридазинил, пирролопиримидинил или пирролопиразинил. В другом воплощении В представляет собой пирролопиридазинил, пирролопиримидинил или пирролопиразинил, замещенный одним или несколькими атомами галогена или С1-3алкилами, описанными в данной заявке. В еще одном воплощении В представляет собой замещенный или незамещенный индолил. В одном из предпочтительных воплощений В представляет собой незамещенный индолил. В одном из воплощений В представляет собой замещенный индолил (например, замещенный одним или несколькими атомами галогена или С1-3алкилами, описанными в данной заявке). В другом предпочтительном воплощении В представляет собой замещенный индолил, содержащий замещение по меньшей мере одной группировкой, выбранной из группы, состоящей из метила, Cl и F. В еще одном воплощении В представляет собой бензофуранил или замещенный бензофуранил, содержащий замещение по меньшей мере одной группировкой, выбранной из группы, содержащей метил, Cl и Fl.
В может быть соответственно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из атома фтора, хлора, С1-3алкила, С1-3галогеналкила, -CN, -ОН, С1-3алкокси и С1-3гидроксиалкила. В одном из воплощений В представляет собой индолил, замещенный галогеном (например, F или Cl).
В одном из воплощений R1a и R1b каждый независимо представляет собой атом водорода, галогена, С1-3алкил, С1-3 галогеналкил, С1-3алкокси, -CN или С3-6циклоалкил. В другом воплощении R1a и R1b каждый независимо представляет собой атом водорода, -F, -Cl, -ОН, -CN, -СН3, -CF3, -CHF2, -CH2F или спироциклопропил. В одном из воплощений R1a и R1b независимо представляют собой атом F или водорода. В предпочтительном воплощении R1a и R1b каждый независимо представляет собой атом водорода, -F или -СН3. В другом предпочтительном воплощении R1a и R1b каждый независимо представляет собой атом водорода, -F или циклопропил. В одном из воплощений n равно 3.
В одном из воплощений:
 относится к формуле
относится к формуле 
R2a и R2b каждый независимо представляет собой атом водорода, галогена, -ОН, С1-3алкил, С1-3галогеналкил, С1-3алкокси, С1-3гидроксиалкил, -CN или С3-6циклоалкил. В некоторых воплощениях R2a и R2b каждый представляет собой атом водорода. В одном из воплощений R1a и R1b каждый независимо представляет собой -F или СН3, a R2a и R2b каждый независимо представляет собой атом водорода. В одном из воплощений В представляет собой индолил, бензофуранил или бензотиофенил, R1a и R1b каждый независимо представляет собой -F или СН3, a R2a и R2b каждый независимо представляет собой атом водорода.
R3a и R3b каждый независимо представляет собой атом водорода, С1-3алкил, С1-3галогеналкил, С1-3гидроксиалкил, -CN, С3-6циклоалкил, С3-6гетероциклоалкил, фенил или С3-6гетероарил. В одном из воплощений R3a и R3b каждый независимо представляет собой атом водорода или -СН3.
Звездочка в формуле (IV) обозначает хиральный центр, когда R3a и R3b являются разными. Таким образом, в некоторых воплощениях R3a и R3b являются разными и представляют собой атом водорода или -СН3.
В одном из воплощений соединение формулы (I) представляет собой:

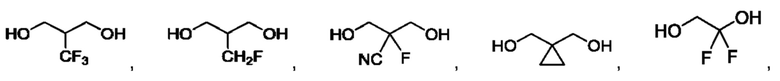
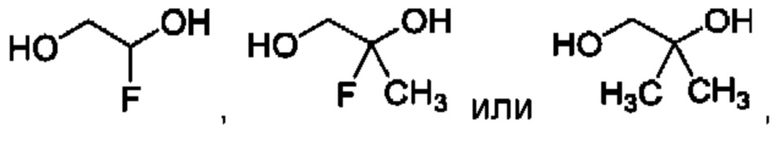
в том числе его стереоизомеры.
В конкретном воплощении соединение формулы (I) представляет собой:
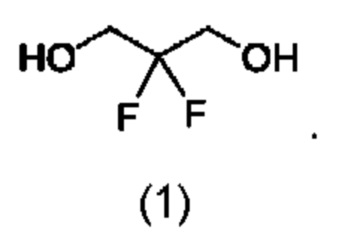
В другом воплощении соединение формулы (I) представляет собой:
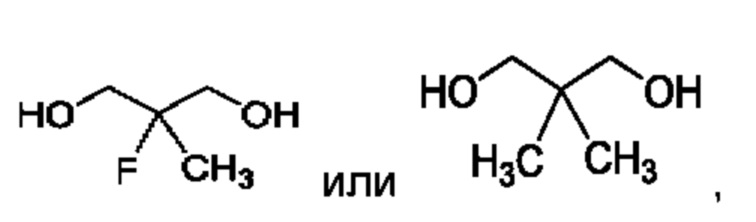
в том числе его стереоизомеры.
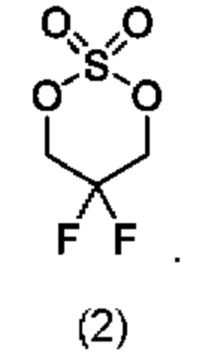
В одном из воплощений соединение формулы (II) представляет собой:
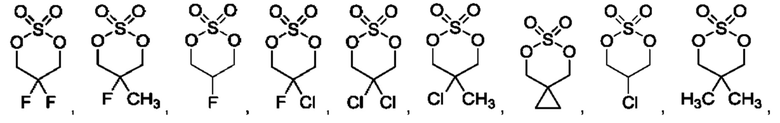
в том числе его стереоизомеры.
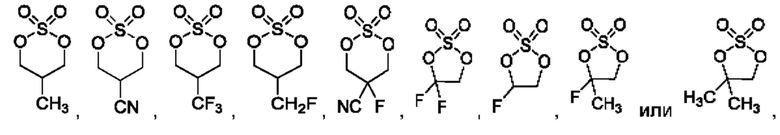
В конкретном воплощении соединение формулы (II) представляет собой:
В некоторых конкретных воплощениях соединение формулы (II) представляет собой:
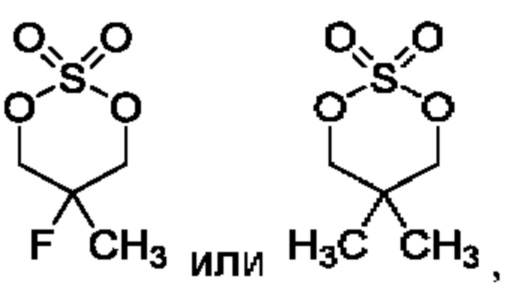
в том числе его стереоизомеры.
В одном из воплощений соединение формулы (III) представляет собой:
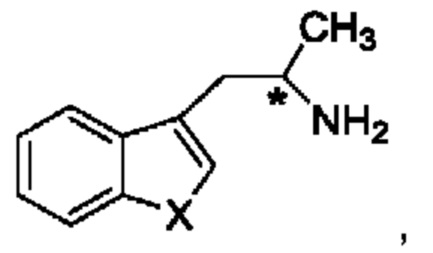
в том числе его стереоизомеры, где X представляет собой -NH-, -N-(незамещенный С1-С3алкил), -О- или -S-.
В конкретном воплощении соединение формулы (III) представляет собой:
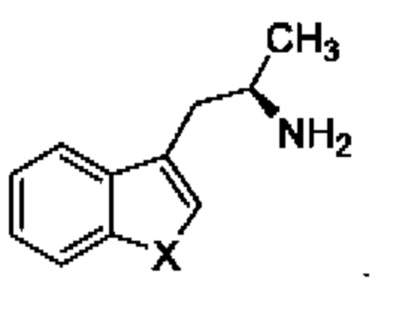
В другом конкретном воплощении соединение формулы (III) представляет собой:
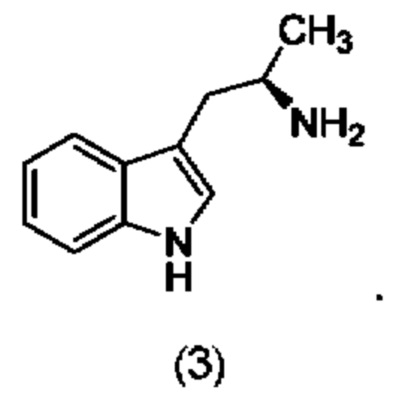
В одном из воплощений соединение формулы (IV) представляет собой:
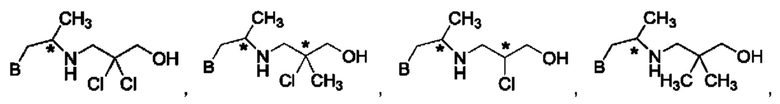
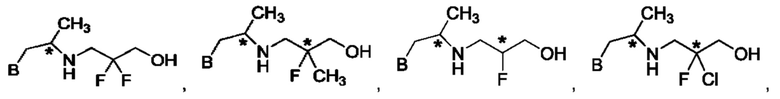
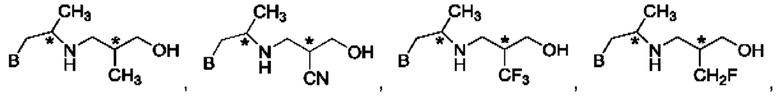
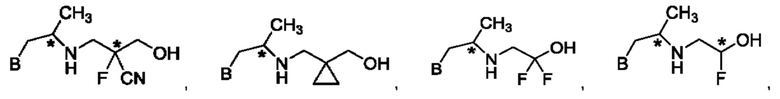
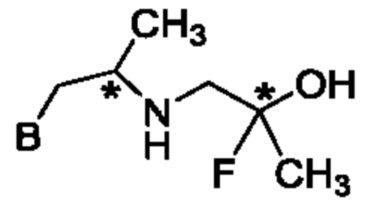 или
или 
либо их соль, в том числе их стереоизомеры; и при этом звездочка обозначает хиральный центр.
В конкретном воплощении соединение формулы (IV) представляет собой:
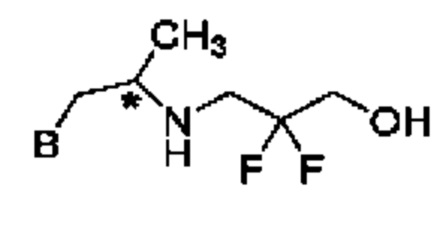 или
или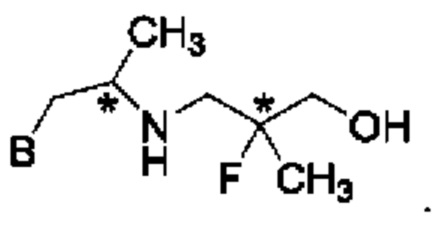
В другом конкретном воплощении соединение формулы (IV) представляет собой:
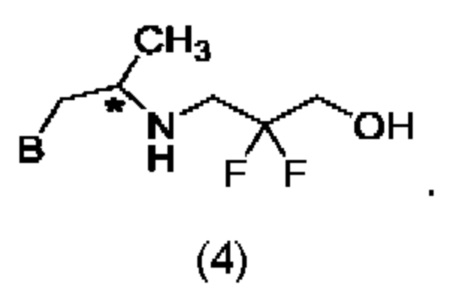
В конкретном воплощении соединение формулы (I) представляет собой:
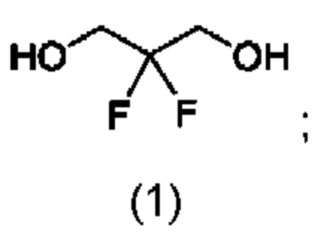
соединение формулы (II) представляет собой
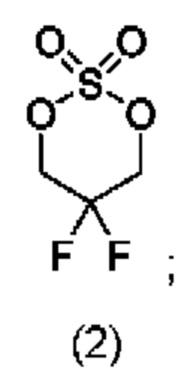
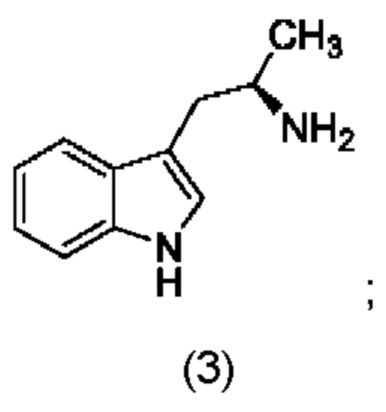
соединение формулы (III) представляет собой
 и
и
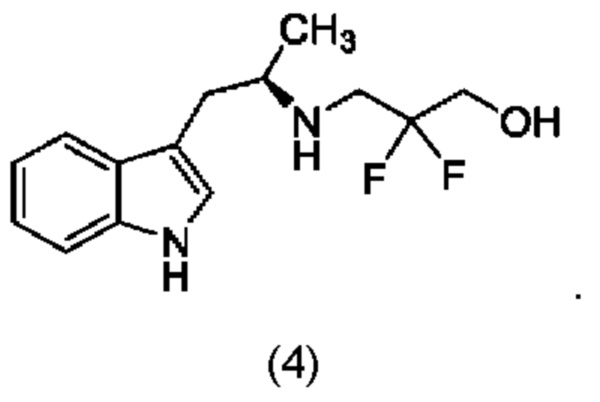
соединение формулы (IV) представляет собой
На стадии 1 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (I), органический растворитель и тионилхлорид с образованием соединения формулы (IIa). В одном из воплощений соединение формулы (I) представляет собой соединение 1. В одном из воплощений органический растворитель представляет собой неполярный растворитель или полярный растворитель. В одном из воплощений растворитель является неполярным. Неограничивающие примеры подходящих неполярных растворителей включают пентан, циклопентан, гексан, циклогексан, бензол, толуол, 1,4-диоксан, хлороформ, диэтиловый эфир, дихлорметан ("DCM") и их комбинации. В одном из воплощений растворителем является DCM. В одном из воплощений концентрация соединения формулы (I) в растворителе может соответственно составлять примерно 25 г/л, примерно 50 г/л, примерно 100 г/л, примерно 150 г/л, примерно 200 г/л, примерно 250 г/л и вплоть до концентрации, приближающейся к насыщению при данной температуре реакции, и на основании этих концентраций составлены диапазоны, такие как от примерно 100 г/л до примерно 250 г/л. Соотношение эквивалентов тионилхлорида и соединения формулы (I) соответственно составляет примерно 1:1, примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,5:1 или примерно 2:1, и на основании этих соотношений составлены диапазоны, такие как от примерно 1,1:1 до примерно 1,5:1. В одном из воплощений температура реакции будет ниже температуры дефлегмации реакционной смеси. В одном из воплощений реакцию осуществляют при температуре дефлегмации. Например, если растворитель представляет собой DCM, то температура реакции соответственно может составлять от примерно 25°С до примерно 40°С. Продолжительность реакции строго не ограничивается, и реакцию обычно продолжают до тех пор, пока превращение соединения формулы (I) в соединение формулы (IIa) по существу не будет завершено, что определяется посредством хроматографии (например, тонкослойной хроматографии (TLC), газовой хроматографии (GC) или высокоэффективной жидкостной хроматографии (HPLC)).
После завершения реакции можно провести гашение реакционной смеси. В некоторых таких воплощениях гашение реакционной смеси можно провести, используя холодную воду. В таких воплощениях фазы могут быть разделены на водную фазу и органическую фазу, содержащую соединение формулы (IIa). Для извлечения дополнительного количества соединения формулы (IIa) можно один или несколько раз провести экстракцию водной фазы органическим растворителем.
На стадии 2 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (IIa), катализатор, окислитель и растворитель с образованием соединения формулы (II). В одном из воплощений органическая фаза или объединенные органические фазы со стадии 1, содержащая(ие) соединение формулы (IIa), используют в качестве источника соединения формулы (IIa) для стадии 2. В одном из воплощений катализатором является металлический катализатор окислительно-восстановительных реакций. Неограничивающие примеры подходящих катализаторов включают NiCl2, RuCl3, CoCl2, FeCl3, FeCl2 и MnCl2. Неограничивающие примеры подходящих окислителей включают NalO4, NaOCl и оксон. Подходящие органические растворители включают неполярные и полярные растворители, которые рассмотрены в других местах в данной заявке. В общем случае окислитель присутствует в эквивалентном избытке по отношению к соединению формулы (IIa), например, соотношение окислителя и соединения формулы (IIa) может составлять 1,1:1; 1,5:1; 2:1; 2,5:1; 3:1; 3,5:1; 4:1; 4,5:1 или 5:1. Реакционная смесь на стадии 2 может дополнительно содержать воду. В таких воплощениях соотношение объемов воды и органического растворителя, используемого в реакционной смеси на стадии 1, может составлять примерно 9:1, примерно 5:1, примерно 3:1, примерно 2:1, примерно 1:1, примерно 1:2, примерно 1:3, примерно 1:5 или примерно 1:9, и на их основе составлены диапазоны, такие как от примерно 2:1 до примерно 1:2. Температура реакции на стадии 2 соответственно может составлять примерно 25°С, примерно 15°С, примерно 5°С, примерно 0°С, примерно -5°С или примерно -10°С, и на их основе составлены диапазоны, такие как от примерно -10°С до примерно 10°С. В одном из воплощений органическую(ие) фазу(ы), содержащую(ие) соединение формулы (IIa), катализатор и воду, объединяют и охлаждают до температуры реакции. Затем в течение некоторого периода времени добавляют окислитель, поддерживая температуру, примерно равную температуре реакции.
После завершения реакции реакционная смесь со стадии 2 может быть разделена на водную фазу и органическую фазу, содержащую соединение формулы (II) в растворе. В некоторых возможных воплощениях перед выполнением фазы разделения можно провести фильтрование реакционной смеси, как например, через вспомогательное фильтрующее средство (например, целит). Для извлечения дополнительного количества соединения формулы (II) можно один или несколько раз провести экстракцию водной фазы органическим растворителем.
В другом воплощении органическая(ие) фаза(ы) со стадии 2 может/могут быть обработана(ы) методами, известными специалистам в данной области техники. Например, органические фазы можно промыть основанием, например, водным раствором Na2SO3. Возможно, что органические фазы далее могут быть подвергнуты сушке, как например, с использованием раствора рассола и/или путем добавления твердого сушильного агента, такого как CaCl2, MgSO4 или Na2SO4. Твердые высушивающие средства могут быть соответственно удалены фильтрованием. В одном из воплощений для последующей реакции может быть использован раствор соединения формулы (II). В одном из воплощений соединение формулы (II) можно выделить из раствора методами, известными в данной области техники, например, посредством дистилляции, концентрирования, осаждения (как например, путем добавления антирастворителя или подведения рН) и/или кристаллизации. В некоторых таких воплощениях органическая(ие) фаза(ы) может/могут быть сконцентрирована(ы) посредством дистилляции или отгонки для уменьшения объема, например, по меньшей мере на 25%, 50%, 100% или больше. Затем можно осуществить осаждение/кристаллизацию соединения формулы (II) из раствора посредством добавления антирастворителя, после чего можно выполнить дальнейшее концентрирование. В одном из воплощений антирастворитель представляет собой неионный С4-8растворитель, такой как пентан, гексан или гептан. Соединение формулы (II) в виде твердых веществ можно собирать методами, известными в данной области техники, такими как фильтрация или центрифугирование. Твердые вещества можно подвергнуть сушке, например, в условиях частичного вакуума, с получением твердого соединения формулы (II). Выход соединения формулы (II) исходя из соединения формулы (I) для стадий 1 и 2 составляет по меньшей мере 60%, по меньшей мере 70% или по меньшей мере 75%. В одном из воплощений соединение формулы (II) представляет собой соединение 2.
На стадии 3 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (II), соединение формулы (III) и органический растворитель с образованием соединения формулы (IV). В одном из воплощений органический растворитель представляет собой полярный апротонный растворитель. Неограничивающие примеры подходящих растворителей включают тетрагидрофуран, этилацетат, ацетон, диметилформамид, ацетонитрил ("ACN"), диметилсульфоксид, нитрометан и пропиленкарбонат. В одном из воплощений растворитель представляет собой ACN. Мольное соотношение между соединением формулы (II) и соединением формулы (III) соответственно составляет примерно 1:1, примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,4:1, примерно 1,5:1 или выше, и на основании этих соотношений составлены диапазоны, такие как от 1:1 до 1,3:1. Концентрация соединения формулы (II) в растворителе соответственно составляет примерно 10 г/л, примерно 25 г/л, примерно 50 г/л, примерно 75 г/л, примерно 100 г/л, примерно 125 г/л, примерно 150 г/л и вплоть до концентрации, приближающейся к насыщению при данной температуре реакции, и на основании этих концентраций составлены диапазоны, такие как от примерно 50 г/л до примерно 150 г/л. Кислотный катализатор может представлять собой кислотный катализатор, описанный в данной заявке в других местах. В некоторых воплощениях кислотный катализатор представляет собой серную кислоту, п-толуолсульфоновую кислоту (п-TsOH) или метансульфоновую кислоту или их комбинации. В одном из воплощений кислотный катализатор представляет собой п-толуолсульфоновую кислоту. Соотношение эквивалентов кислотного катализатора и соединения формулы (II) соответственно составляет примерно 0,75:1, примерно 0,9:1, примерно 1:1, примерно 1,05:1, примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,4:1, примерно 1,5:1 или выше, и на их основе составлены диапазоны, такие как от примерно 1:1 до примерно 1,2:1. В одном из воплощений соединение формулы (III) представляет собой соединение 3.
В некоторых воплощениях стадии 3 соединение формулы (II), соединение формулы (III), органический растворитель и основание объединяют с образованием смеси. Основанием соответственно может быть умеренно сильное основание, неограничивающие примеры которого включают трет-бутилат калия, триметиламин, бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, гидроксид натрия, гидроксид аммония и их комбинации. Смесь можно нагревать при перемешивании до температуры реакции, обычно до температуры в диапазоне от 2°С до примерно 30°С ниже температуры дефлегмации и вплоть до температуры дефлегмации, и выдерживать в течение некоторого времени, достаточного для существенного завершения образования продукта реакции, содержащего соединение формулы (III). В случае использования ACN в качестве растворителя температура реакции соответственно составляет примерно 65°С, примерно 70°С, примерно 75°С или примерно 80°С. Затем смесь продуктов реакции можно охладить, например, до температуры ниже 50°С, 40°С или 30°С, и возможно подвергнуть фильтрованию для удаления твердых примесей. Твердые вещества возможно могут быть подвергнуты промывке растворителем для извлечения дополнительного количества продукта реакции. Затем добавляют кислоту (например, п-TsOH) и воду. Соотношение объемов органического растворителя и воды может составлять 25:1, 15:1, 10:1, 5:1, 2:1 или 1:1, и на их основе составлены диапазоны, такие как от примерно 15:1 до примерно 5:1. Смесь можно нагревать при перемешивании до температуры реакции, обычно до температуры в диапазоне от 2°С до примерно 20°С ниже температуры дефлегмации и вплоть до температуры дефлегмации, и выдерживать в течение некоторого времени, достаточного для существенного завершения образования соединения формулы (IV), что определяется посредством хроматографии (например, TLC, GC или HPLC). После завершения реакции можно провести гашение реакционной смеси, как например, холодной водой (например, с температурой меньше 10°С или меньше 5°С). Значение рН погашенной реакционной смеси затем можно подвести с использованием основания до значения выше 7, как например, до рН приблизительно 8, до рН приблизительно 9, до рН приблизительно 10 или до рН приблизительно 11. В одном из воплощений основание представляет собой водный раствор основания, такого как бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, гидроксид натрия или гидроксид аммония.
После завершения реакции реакционная смесь со стадии 3 может быть разделена на водную фазу и органическую фазу, содержащую соединение формулы (IV) в растворе. В некоторых возможных воплощениях перед выполнением фазы разделения можно провести фильтрование реакционной смеси, как например, через вспомогательное фильтрующее средство (например, целит). Для извлечения дополнительного количества соединения формулы (IV) можно один или несколько раз провести экстракцию водной фазы органическим растворителем. В одном из воплощений растворитель является апротонным растворителем. В конкретном воплощении растворителем для экстракции соответственно является изопропилацетат ("i-PrOAc").
В некоторых воплощениях органическую(ие) фазу(ы) со стадии 3 можно обработать, например, путем промывки этих органических фаз водой. Возможно, что органические фазы могут быть подвергнуты сушке, как например, с использованием раствора рассола и/или путем добавления твердого сушильного агента, такого как CaCl2, MgSO4 или Na2SO4. Твердые высушивающие средства могут быть соответственно удалены фильтрованием и собранное высушивающее средство возможно может быть промыто растворителем для извлечения из него соединения формулы (IV). В таких воплощениях органическая(ие) фаза(ы) может/могут быть сконцентрирована(ы) посредством дистилляции в условиях частичного вакуума или отгонки с образованием оставшегося количества соединения формула (IV). Затем оставшееся количество соединения формулы (IV) можно растворить в органическом растворителе при температуре ниже температуры дефлегмации. Далее к раствору соединения формула (IV) можно добавить антирастворитель, такой как неполярный органический растворитель, описанный в других местах в данной заявке, при охлаждении, например, до температуры меньше примерно 10°С, для осаждения/кристаллизации соединения формулы (IV) из раствора. Соединение формулы (IV) в виде твердых веществ можно собирать методами, известными в данной области техники, такими как фильтрация или центрифугирование и возможно промывать антирастворителем. Твердые вещества можно подвергнуть сушке, например, в условиях частичного вакуума, с получением твердого соединения формулы (IV). Выход соединения формулы (IV) исходя из соединения формулы (II) для стадии 3 составляет по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96% или по меньшей мере 97%. Чистота соединения формулы (IV) составляет по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99%.
Один из аспектов изобретения относится к способу получения соединения формулы (VIII) или его соли:
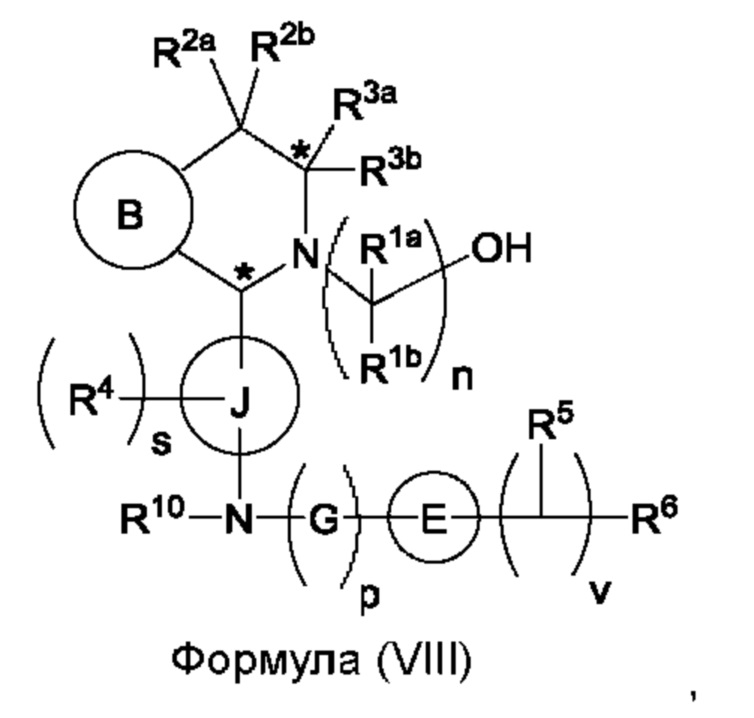
при этом данный способ включает стадии:
(а) приведения во взаимодействие содержащихся в реакционной смеси соединения формулы (IV), соединения формулы (V) или соединения формулы (X) и органического растворителя с образованием соединения формулы (VI) в соответствии с приведенной ниже стадией 1,
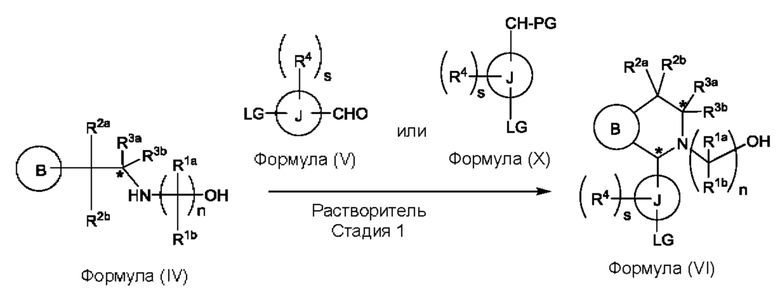
где В представляет собой замещенный или незамещенный индолил, бензофуранил, бензотиофенил, азаиндолил, индазолил, бензимидазолил, пирролопиридинил, фуропиридинил, тиенопиридинил, пирролопиридазинил, пирролопиримидинил, пирролопиразинил, тиенопиридазинил, тиенопиримидинил, тиенопиразинил, фуропиридазинил, фуропиримидинил или фуропиразинил;
каждый из R1a и R1b независимо представляет собой атом водорода, фтора, хлора, -ОН, С1-3алкил, С1-3галогеналкил, С1-3алкокси, С1-3гидроксиалкил и -CN, С3-6циклоалкил или С3-6спироциклоалкил,
n равно целому числу 2 или 3,
каждый из R2a и R2b независимо представляет собой атом водорода, галогена, -ОН, С1-3алкил, С1-3галогеналкил, С1-3алкокси, С1-3гидроксиалкил, -CN, С3-6циклоалкил или С3-6спироциклоалкил,
R3a и R3b независимо представляют собой атом водорода, С1-3алкил, С1-3галогеналкил, С1-3алкокси, -CN, С3-6циклоалкил, С3-6гетероциклоалкил, фенил, С3-6 гетероарил или С3-6спироциклоалкил,
J представляет собой фенил или пиридинил;
каждый R4 независимо представляет собой атом водорода, галогена или С1-3алкил,
s равно целому числу от 0 до 2,
LG представляет собой уходящую группу,
LG и СНО локализованы в пара-положении относительно друг друга в J в соединении формулы (V),
PG представляет собой альдегид-защитную группу,
LG и CH-PG локализованы в пара-положении относительно друг друга в J в соединении формула (X), и
каждая звездочка независимо обозначает хиральный центр, при этом атом углерода, несущий R3a и R3b, представляет собой хиральный центр, когда R3a и R3b являются разными; и
(b) приведения во взаимодействие содержащихся в реакционной смеси соединения формулы (VI), органического растворителя и соединения формулы (VII) или его соли с образованием соединения формулы (VIII) или его соли в соответствии с приведенной ниже стадией 2,
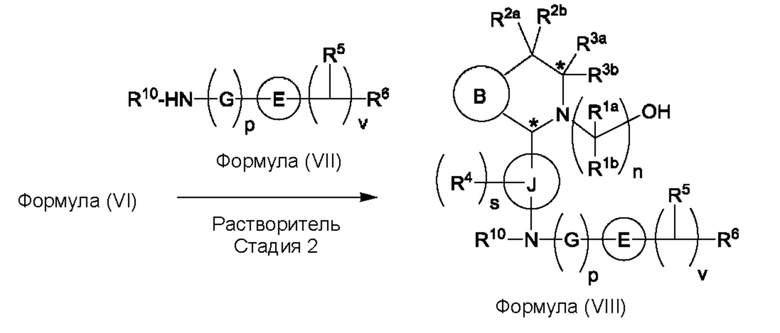
где G представляет собой С1-3алкил,
р равно 0 или 1,
Е представляет собой замещенный или незамещенный азетидинил или пирролидинил,
каждый R5 независимо представляет собой атом водорода, галогена, -ОН, -CN, С1-5алкокси или С1-5гидроксиалкил,
v равно целому числу от 1 до 5,
R6 представляет собой атом галогена или -CN; и
R10 представляет собой атом водорода или С1-3алкил.
В, R1a, R1b, n, R2a, R2b, R3a, R3b и звездочка (*) являются такими, как определено в данном описании.
В одном из предпочтительных воплощений J представляет собой фенил.
В другом воплощении J представляет собой пиридинил.
В одном из воплощений каждый R4 независимо представляет собой атом водорода или галогена. В предпочтительном воплощении каждый R4 представляет собой атом фтора. В одном из воплощений s равно 1 или 2. В одном из воплощений s равно 2. В одном из предпочтительных воплощений каждый R4 представляет собой атом фтора и s равно 2.
В одном из воплощений G представляет собой метилен или этилен.
В одном из воплощений р равно 0.
В одном из воплощений каждый R5 независимо представляет собой атом водорода, галогена, -ОН или -CN. В одном из предпочтительных воплощений каждый R5 представляет собой атом водорода. В одном из воплощений v равно 2. В другом воплощении v равно 3. В другом воплощении v равно 5. В предпочтительном воплощении каждый R5 представляет собой атом водорода, и v равно 3.
В одном из предпочтительных воплощений R6 представляет собой атом галогена. В одном из воплощений R6 представляет собой атом F. В другом воплощении R6 представляет собой -CN.
В одном из воплощений R10 представляет собой атом водорода или метил. В предпочтительном воплощении R10 представляет собой атом водорода.
В одном из воплощений Е представляет собой азетидинил. В другом воплощении Е представляет собой пирролидинил.
В одном из воплощений Е имеет следующую структуру:

В одном из воплощений Е представляет собой азетидинил следующей структуры:

В одном из воплощений Е имеет следующую структуру:
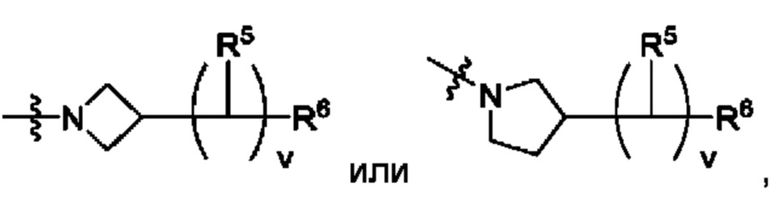
где R5 представляет собой Н, v равно 2 или 3, и R6 представляет собой атом галогена.
В одном из воплощений Е представляет собой азетидинил следующей структуры:
В одном из воплощений соединение формулы (VIII) представляет собой соль, полученную присоединением кислоты. Такая соль с кислотой может представлять собой фармацевтически приемлемую соль. В некоторых таких воплощениях соединение формулы (VIII) представляет собой соль фармацевтически приемлемой кислоты. В некоторых конкретных воплощениях соединение формулы (VIII) представляет собой соль фармацевтически приемлемой органической кислоты. В предпочтительном воплощении соединение формулы (VIII) представляет собой фармацевтически приемлемую соль винной кислоты. В одном из воплощений соединение формулы (VIII) представляет собой соединение А, описанное в данной заявке. В другом воплощении соединение формулы (VIII) представляет собой соединение А (тартратную соль соединения А), описанного в данной заявке. В другом воплощении соединение формулы (VIII) представляет собой фармацевтически приемлемую соль фумаровой кислоты. В еще одном воплощении соединение формулы (VIII) представляет собой соединение В (фумаратную соль соединения А), описанного в данной заявке.
В одном из воплощений соединение формулы (VIII) представляет собой любую из следующих структур или их фармацевтически приемлемой соли:
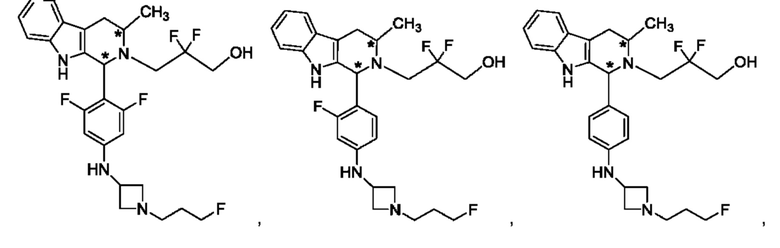
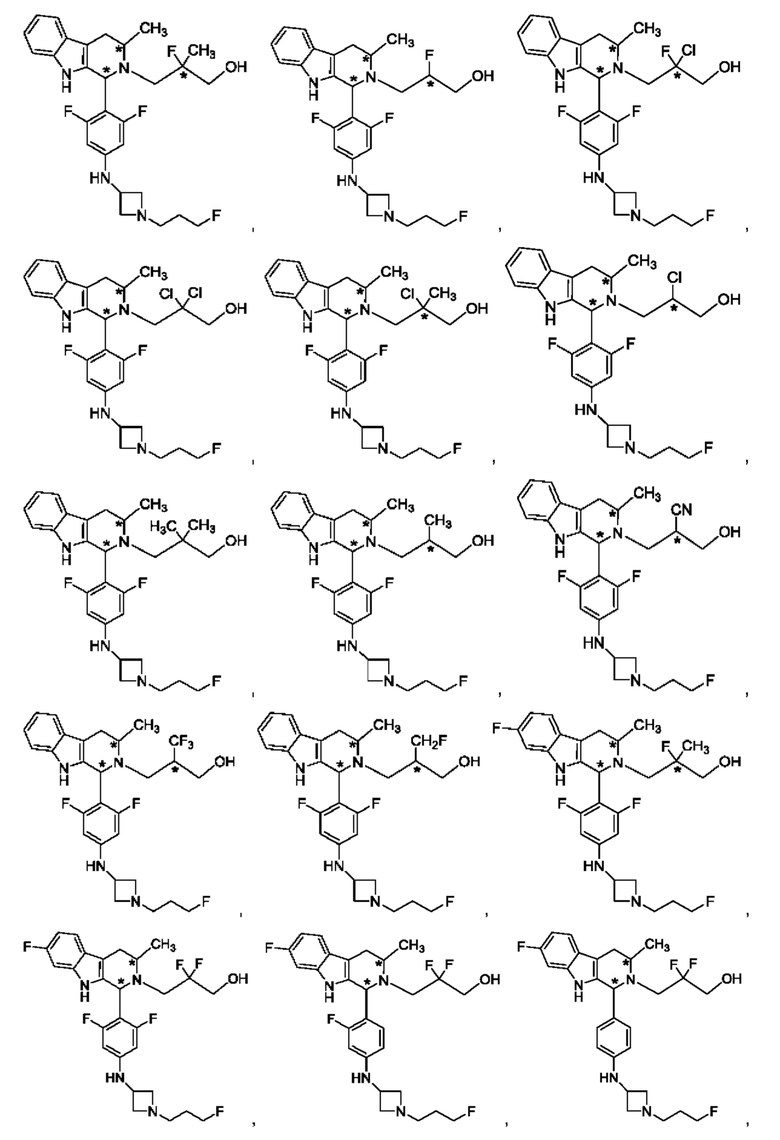
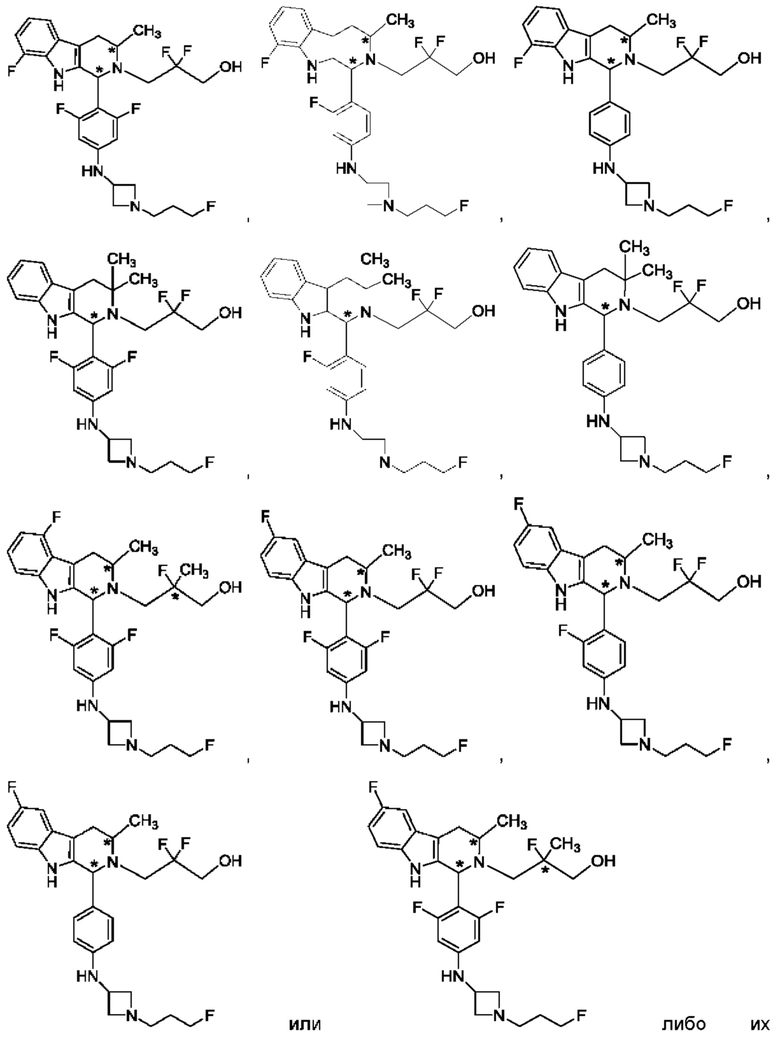
фармацевтически приемлемой соли, и в том числе их стереоизомеров.
В одном из воплощений соединение формулы (VIII) представляет собой соединение следующей структуры или его фармацевтически приемлемую соль:
В одном из воплощений соединение формулы (VIII) представляет собой соединение следующей структуры:
В одном из воплощений соединение формулы (VIII) представляет собой соединение следующей структуры:
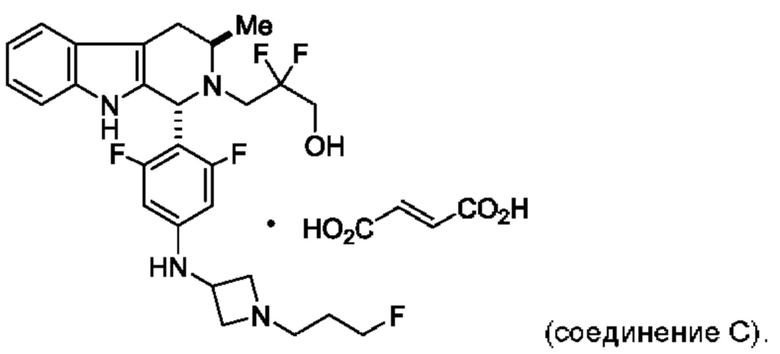
Стадия 1 способа синтеза соединений формулы (VIII) включает приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (IV), соединения формулы (V) или соединения формулы (X) и органического растворителя с образованием соединения формулы (VI), указанного в данном описании. В одном из воплощений LG представляет собой атом брома. В предпочтительном воплощении, при взаимодействии с участием соединений формулы (IV) и формулы (V), LG и СНО локализованы в J в пара-положении относительно друг друга. В предпочтительном воплощении, при взаимодействии соединений формулы (IV) и формулы (X), LG и CH-PG локализованы в пара-положении относительно друг друга в J.
Соединение формулы (IV) является таким, как описано в данной заявке. В одном из воплощений соединение формулы (V) представляет собой любое из следующих соединений или их соли:
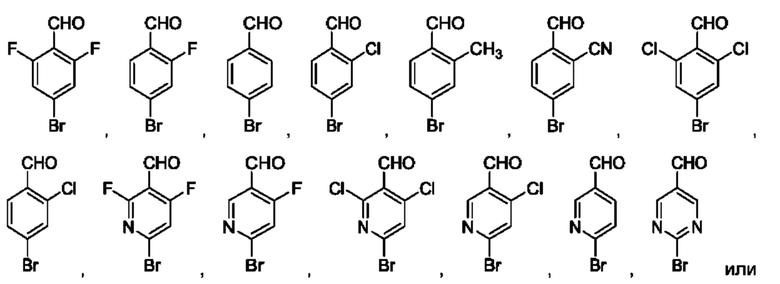
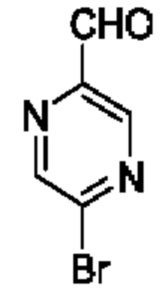
либо их соль.
В некоторых воплощениях соединение формулы (X) соответствует любой приведенной выше структуре формулы (V) или ее соли, но при этом альдегидная группировка (-СНО) представляет собой защищенную группировку структуры -СН-PG, где PG представляет собой альдегид-защитную группу, определенную в других местах данного описания.
В некоторых воплощениях соединение формулы (VI) представляет собой любую из следующих структур:
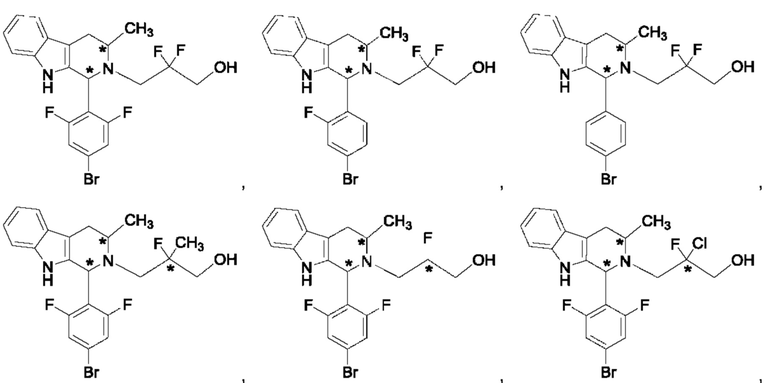
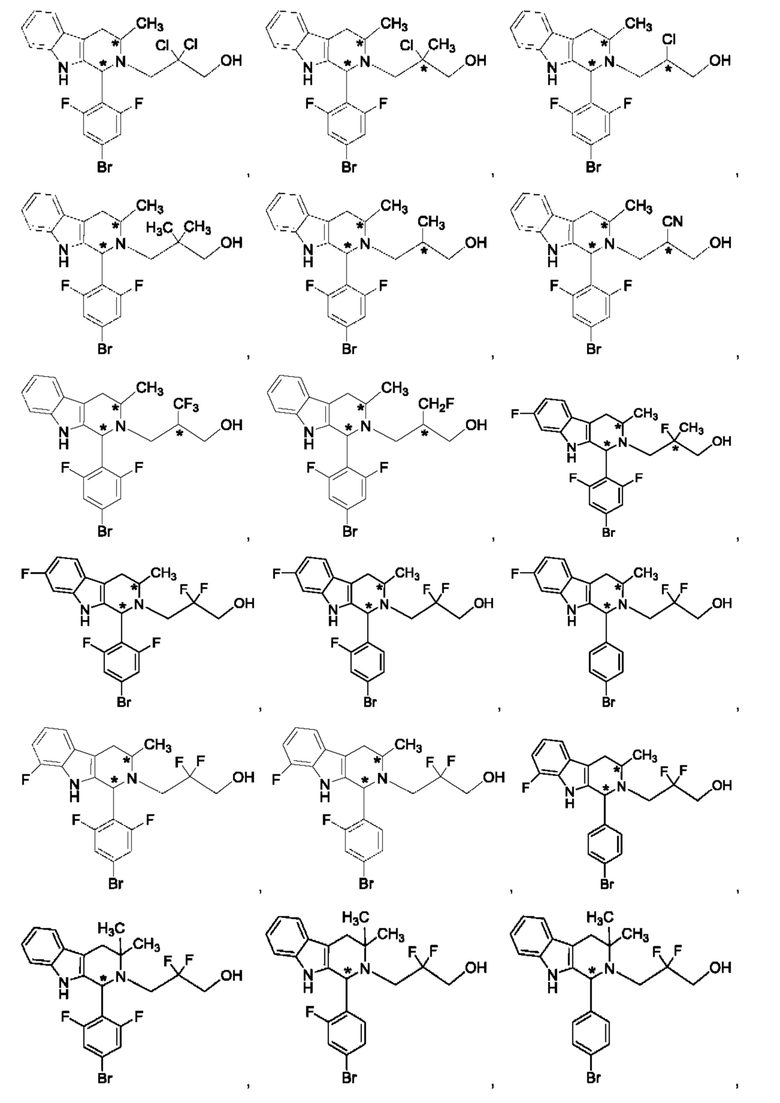
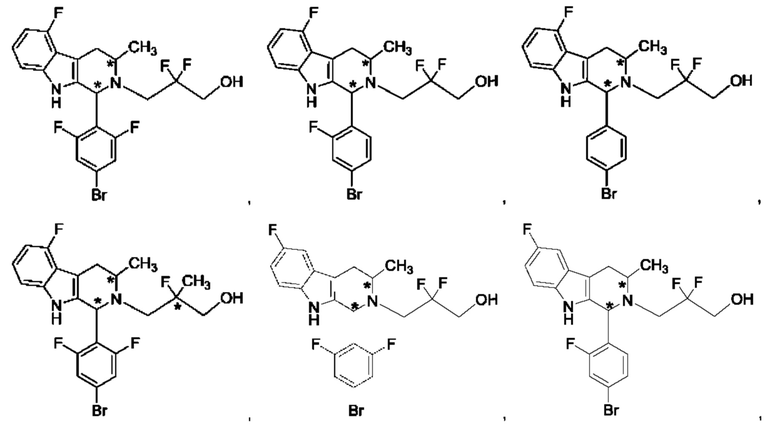
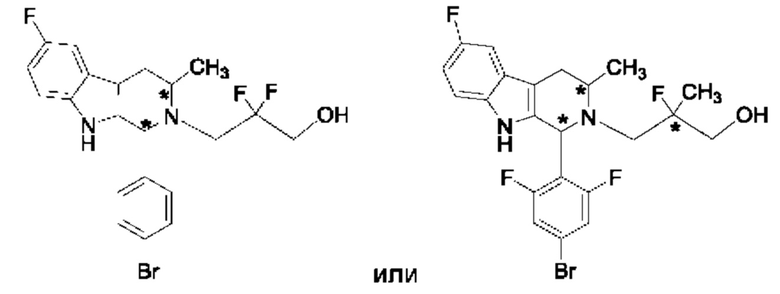
либо их соль, в том числе их стерео изо меры.
На стадии 2 синтеза соединений формулы (VIII), указанных в данном описании, соединение формулы (VII) может представлять собой любую из следующих структур:
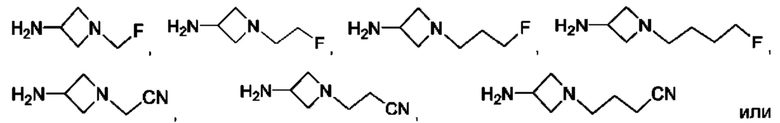
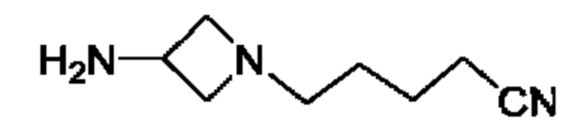
либо их соль.
В одном из воплощений соль представляет собой этан-дисульфонат (например, соль этан-1,2-дисульфонат).
В одном из воплощений соединение формулы (VII) имеет структуру
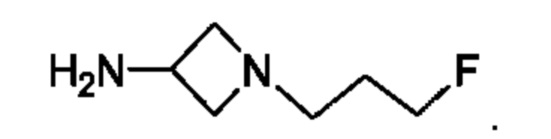
Соединение (7) может быть получено в соответствии с примерами, приведенными в данном описании, такими как, например, пример 4 или пример 4а.
В одном из воплощений соединение 7 получают в соответствии с приведенной ниже схемой:
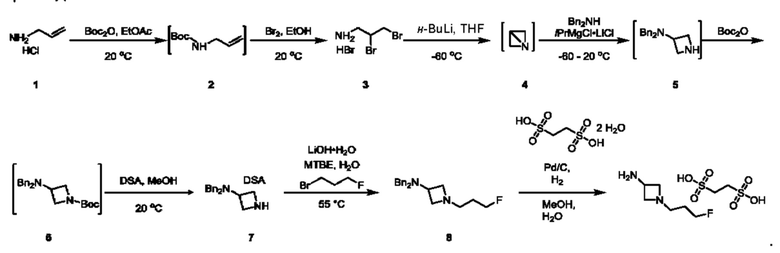
На стадии 1 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (IV), соединение формулы (V) или соединение формулы (X) и органический растворитель с образованием соединения формулы (VI). В одном из воплощений органический растворитель представляет собой полярный протонный растворитель, неполярный растворитель, полярный апротонный растворитель или их комбинацию, как описано в других местах в данной заявке. В некоторых неограничивающих воплощениях растворителем является толуол. В одном из воплощений растворителем является ацетонитрил, метилэтилкетон или метилтетрагидрофуран. В одном из воплощений реакционная смесь со стадии 1 дополнительно содержит кислотный катализатор, описанный в данной заявке в других местах. В некоторых таких неограничивающих воплощениях кислотным катализатором является уксусная кислота. Мольное соотношение между соединением (IV) и соединением (V) или соединением (X) соответствует примерно стехиометрическим количествам, составляющим от примерно 0,95:1 до примерно 1,05:1, или, в некоторых воплощениях, соединение (V) или соединение (X) присутствует в незначительном молярном избытке. Кислотный катализатор обычно присутствует в стехиометрическом избытке, например, при соотношении эквивалентов катализатора и соединения формулы (IV), составляющем примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,4:1, примерно 1,5:1, примерно 1,6:1, примерно 1,7:1, примерно 1,8:1, примерно 2:1 или выше, и на их основе составлены диапазоны, такие как от примерно 1,2:1 до примерно 1,8:1.
В некоторых воплощениях стадии 1 реакционную смесь можно нагревать при перемешивании до температуры реакции в диапазоне от 2°С до примерно 30°С ниже температуры дефлегмации и вплоть до температуры дефлегмации и выдерживать в течение некоторого времени, достаточного для существенного завершения реакция с целью получения соединения формулы (VI), что определяется посредством хроматографии (например, TLC, GC или HPLC). В случае использования толуола в качестве растворителя температура реакции соответственно составляет примерно 65°С, примерно 70°С, примерно 75°С или примерно 80°С. Затем смесь продуктов реакции можно охладить и возможно разбавить дополнительным количеством растворителя. Далее смесь продуктов реакции можно погасить, используя основание, такое как водный раствор основания, описанного в других местах в данной заявке. Затем органическая фаза смеси продуктов реакции со стадия 1, содержащая соединение формулы (VI), может быть выделена и, в некоторых воплощениях, обработана методами, известными в данной области техники, такими как промывка водой и/или раствором рассола, как описано в других местах в данной заявке, с последующим выделением продукта в виде твердого вещества. В одном из воплощений смесь продуктов реакции можно обработать активированным углем, после чего осуществить фильтрование и возможно промывку осадка на фильтре из активированного угля растворителем. Как описано в других местах в данной заявке: (1) органическая(ие) фаза(ы), содержащая(ие) соединение формулы (VI), может/могут быть сконцентрирована(ы) посредством дистилляции или отгонки для уменьшения объема, например, по меньшей мере на 25%, 50%, 100% или больше; и (2) затем можно осуществить осаждение/кристаллизацию соединения формулы (IV) из раствора посредством добавления антирастворителя, после чего можно выполнить дальнейшее концентрирование.
В некоторых других воплощениях стадии 1 реакционную смесь на стадии 1 нагревают при температуре дефлегмации в течение промежутка времени, достаточного для практически полного завершения реакции, что определяется посредством хроматографии (например, TLC, GC или HPLC). Затем реакционную смесь можно охладить и подвести значение рН, используя основание, такое как водный раствор основания, до рН, при котором соединение формулы (VI) осаждается из раствора.
В любом из различных воплощений стадии 1 соединение формулы (VI) в виде твердых веществ можно собирать методами, известными в данной области техники, такими как фильтрация или центрифугирование. Твердые вещества можно подвергнуть сушке, например, в условиях частичного вакуума, с получением твердого соединения формулы (VI). Выход соединения формулы (VI) исходя из соединения формулы (V) для стадий 1 и 2 составляет по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90% или по меньшей мере 95%.
На стадии 2 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (VI), соединение формулы (VII) и органический растворитель с образованием соединения формулы (VIII). В некоторых воплощениях органический растворитель представляет собой полярный апротонный растворитель, описанный в других местах в данной заявке. В некоторых неограничивающих воплощениях растворителем является ACN. Реакционная смесь на стадии 2 может дополнительно содержать основание, такое как органическое основание. Неограничивающие примеры органических оснований включают DBU, NMM, DIPEA и TEA. В некоторых таких воплощениях основанием является DBU. Реакционная смесь на стадии 2 может дополнительно содержать катализатор, такой как катализатор на основе переходного металла. В некоторых воплощениях катализатор представляет собой Pd-катализатор. Мольное соотношение между соединением (VII) и соединением (VI) составляет примерно 0,95:1, примерно 1:1, примерно 1,05:1, примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,4:1 или примерно 1,5:1, и на их основе составлены диапазоны, такие как от примерно 1:1 до примерно 1,4:1. Обычно основание присутствует в стехиометрическом избытке, например, в соотношении эквивалентов основания и соединения формулы (VI), составляющем примерно 1,1:1, примерно 2:1, примерно 3:1, примерно 4:1, примерно 5:1, примерно 6:1, примерно 7:1 или примерно 8:1, и на их основе составлены диапазоны, такие как от примерно 3:1 до примерно 7:1. Реакционную смесь можно нагревать при перемешивании до температуры реакции, обычно до температуры в диапазоне от 2°С до примерно 30°С ниже температуры дефлегмации и вплоть до температуры дефлегмации, и выдерживать в течение некоторого времени, достаточного для существенного завершения реакция, что определяется посредством хроматографии (например, TLC, GC или HPLC). В случае использования ACN в качестве растворителя температура реакции соответственно составляет примерно 65°С, примерно 70°С, примерно 75°С или примерно 80°С.
По завершении смесь продуктов реакции можно соответствующим образом охладить и возможно разбавить органическим растворителем. В одном из воплощений смесь продуктов реакции разбавляют неполярным растворителем, описанным в других местах в данной заявке. Одним из неограничивающих примеров подходящего неполярного растворителя является МТВЕ. Смесь продуктов реакции со стадии 2 может быть обработана методами, известными специалистам в данной области техники, включая промывку водой и промывку рассолом. В некоторых таких неограничивающих воплощениях такая обработка может включать промывку водным раствором хлорида аммония, рассолом и водой. В некоторых воплощениях смесь продуктов реакции со стадии 2 может быть приведена в контакт с поглотителем ионов металлов, известным в данной области техники, таким как, например и без ограничения, SiliaMetS Thiol. Затем можно провести фильтрование смеси продуктов реакции для удаления твердых веществ, после чего выделить из нее соединение формулы (VIII).
В некоторых воплощениях смесь продуктов реакции со стадии 2 может быть сконцентрирована, например, посредством вакуумной дистилляции или отгонки, и разбавлена органическим растворителем, таким как спирт (например, этанол), как например, на стадии смены растворителя. Затем к разбавленному раствору соединения формулы (VIII) можно добавить кислоту, после чего охладить для осуществления кристаллизации соединения формулы (VIII) в виде соли с кислотой. В некоторых конкретных воплощениях кислота представляет собой винную кислоту, и соединение формулы (VIII) представляет собой соль винной кислоты. В некоторых таких воплощениях кислота представляет собой (2R-3R)-винную кислоту (L-(+)-винную кислоту). Согласно другому аспекту кислота представляет собой (2S-3S)-винную кислоту (D-(-)-винную кислоту). В некоторых таких воплощениях растворитель содержит органический растворитель. Данное кристаллическое вещество может быть собрано путем центрифугирования или фильтрования, возможно промыто растворителем и возможно подвергнуто сушке.
В некоторых других воплощениях соединение формулы (VIII) можно выделить из смеси продуктов реакции со стадии 2, используя методы, описанные в других местах в данной заявке, включая: (1) дистилляцию, концентрирование, осаждение (как например, путем добавления антирастворителя или подведения рН) и/или кристаллизацию; (2) сбор твердых веществ путем центрифугирования или фильтрования; (3) возможную промывку собранных твердых веществ; и (4) сушку.
Выход соединения формулы (VIII) на стадии 2 либо в виде свободного основания, либо соли, полученной присоединением кислоты, составляет по меньшей мере 80%, по меньшей мере 85% или по меньшей мере 90%.
Другой аспект изобретения относится к способу получения соединения формулы (VIII) или его соли, причем соединение формулы (VIII) является таким, как описано в других местах в данной заявке. Способ получения соединения формулы (VIII) по этому воплощению включает стадию 1 реакции, изображенную ниже:
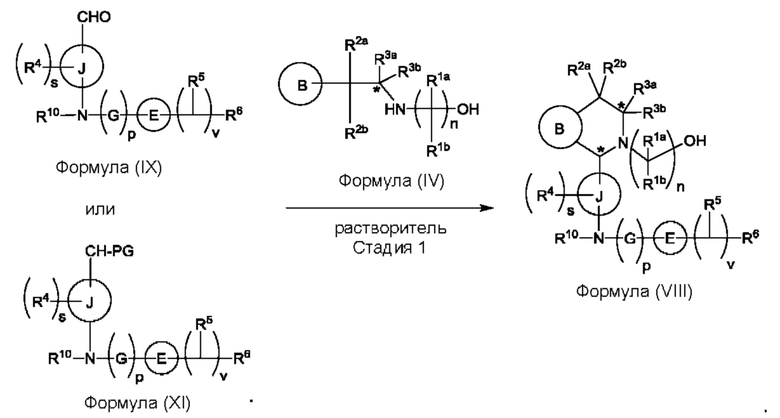
Каждая переменная из В, R1a, R1b, n, R2a, R2b, R3a, R3b, R4, s, J, R5, v, R6, R10, G, p, E, PG и звездочки является такой, как описано в других местах в данной заявке. Группировка СНО и атом азота, соединяющий J и G, локализованы в пара-положении относительно друг друга в J. Группировка CH-PG и атом азота, соединяющий J и G, локализованы в пара-положении относительно друг друга в J.
В одном из воплощений соединение формулы (IX) представляет собой:
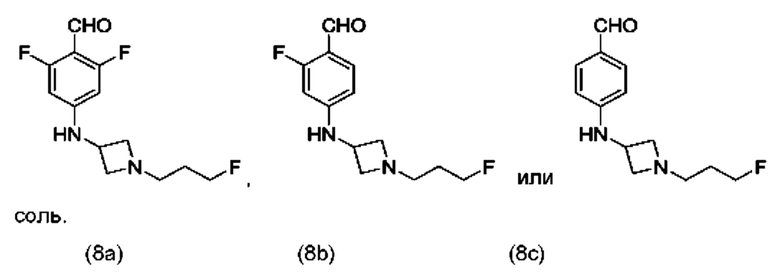 либо их
либо их
В одном из предпочтительных воплощений соединение формулы (IX) представляет собой соединение (8а). В некоторых воплощениях соединение формулы (XI) соответствует любой из приведенных выше структур формулы (IX) или их соли, но при этом альдегид (-СНО) представляет собой защищенную группировку структуры -CH-PG, где PG представляет собой альдегид-защитную группу, определенную в данном описании в других местах.
На стадии 1 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (IX) или формулы (XI), соединение формулы (IV) и органический растворитель с образованием соединения формулы (VIII) или его соли. В одном из воплощений органический растворитель представляет собой полярный растворитель или представляет собой полярный протонный растворитель. Реакционная смесь на стадии 1 дополнительно содержит кислотный катализатор, описанный в данной заявке в других местах. В конкретном воплощении кислотный катализатор представляет собой винную кислоту или фумаровую кислоту. В одном из воплощений кислотным катализатором является винная кислота. В другом воплощении кислотным катализатором является фумаровая кислота. Неограничивающие примеры подходящих растворителей включают н-бутанол, изопропиловый спирт, н-пропанол, изопропанол, этанол, метанол и их комбинации. В некоторых конкретных воплощениях растворителем является этанол. В одном из воплощений концентрация соединения формулы (IX) в растворителе может соответственно составлять примерно 25 г/л, примерно 50 г/л, примерно 100 г/л, примерно 150 г/л, примерно 200 г/л, примерно 250 г/л и вплоть до концентрации, приближающейся к насыщению при данной температуре реакции, и на основании этих концентраций составлены диапазоны, такие как от примерно 100 г/л до примерно 250 г/л. Мольное соотношение между соединением формулы (IX) или формулы (XI) и соединением формулы (IV) соответственно составляет примерно 0,25:1, примерно 0,3:1, примерно 0,4:1, примерно 0,5:1, примерно 0,6:1, примерно 0,7:1, примерно 0,8:1, примерно 0,9:1, примерно 0,95:1, примерно 1:1, примерно 1,05:1, примерно 1,1:1 или примерно 1,2:1, и на основании этих соотношений составлены диапазоны, такие как от примерно 0,95:1 до примерно 1,05:1. В одном из воплощений соединение формулы (IX) или формулы (XI) и соединение формулы (IV) присутствуют приблизительно в стехиометрических количествах. В одном из воплощений температура реакции будет ниже температуры дефлегмации реакционной смеси. В некоторых других воплощениях реакцию осуществляют при температуре дефлегмации. Например, если растворителем является этанол, то температура реакции соответственно может составлять от примерно 50°С до примерно 75°С. Продолжительность реакции строго не ограничивается, и реакцию обычно продолжают до тех пор, пока превращение соединения формулы (IX) или формулы (XI) в соединение формулы (VIII) по существу не будет завершено, что определяется посредством хроматографии (например, TLC, GC или HPLC).
В одном из воплощений соединение формулы (VIII) может быть образовано в виде соли кислоты. Подходящие кислоты включают органические кислоты и неорганические кислоты, описанные в других местах в данной заявке. В одном из воплощений кислота представляет собой органическую кислоту. В предпочтительном воплощении кислотой является винная кислота. В другом воплощении кислотой является фумаровая кислота. В одном из воплощений соединение формулы (VIII) в виде свободного основания может быть растворено в подходящем растворителе, таком как полярный протонный растворитель (например, спирте, таком как метанол или этанол) при повышенной температуре, после чего добавлена кислота. Обычно кислота находится в стехиометрическом избытке по сравнению с соединением формулы (VIII). В некоторых таких воплощениях значение температуры и/или концентрации раствора соединения формулы (VIII) регулируют с целью поддержания концентрации ниже насыщения и тем самым предотвращения осаждения и/или кристаллизации соединения формулы (VIII). После добавления кислоты в данный раствор возможно может быть добавлена затравка кристаллической соли соединения формулы (VIII) в виде соли указанной кислоты. В любом из различных воплощений раствор охлаждают при перемешивании с образованием соли кристаллического соединения формулы (VIII). Затем соль может быть собрана методами, известными в данной области техники, такими как фильтрование или центрифугирование. В одном из воплощений соль собирают фильтрованием. Собранная соль соединения формулы (VIII) возможно может быть промыта, например, использованным для растворения растворителем, и затем высушена, например, в условиях частичного вакуума.
В конкретном воплощении реакционная смесь на стадии 1, указанная выше для синтеза соединения формулы (VIII), содержит винную кислоту в качестве кислотного катализатора, кристаллическую соль соединения формулы (VIII) с винной кислотой и спиртовой растворитель (например, этанол); смесь продуктов реакции со стадии 1 разбавляют спиртовым растворителем; и полученную суспензию охлаждают при перемешивании с образованием кристаллической соли соединения формулы (VIII) с винной кислотой. В другом воплощении реакционная смесь на стадии 1 содержит фумаровую кислоту в качестве кислотного катализатора, кристаллическую соль соединения формулы (VIII) с фумаровой кислотой и растворитель. Выход соединения формулы (VIII) на стадии 1 в виде соли винной кислоты составляет по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90% или по меньшей мере 95%. В таких воплощениях реакционная схема для стадии 1 является такой, как приведено ниже:
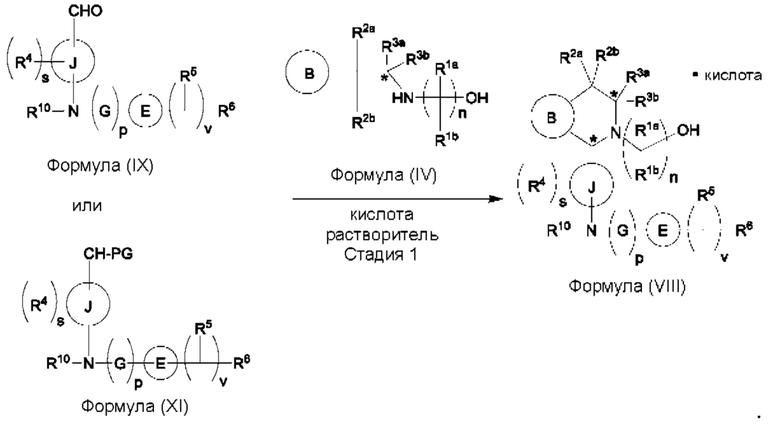
Еще один аспект изобретения относится к способу получения соединения формулы (IX) или его соли. Способ получения соединения формулы (IX) включает две стадии реакции, изображенные ниже:
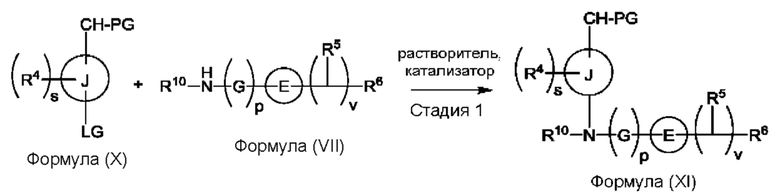
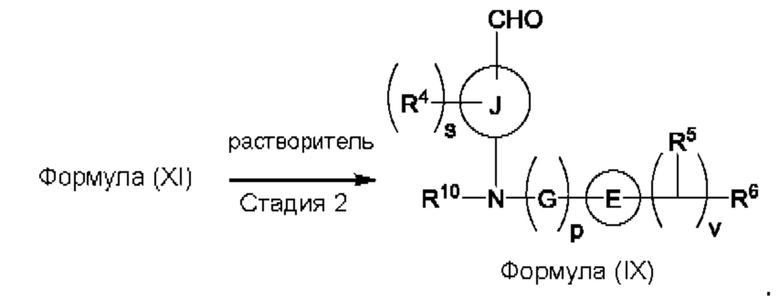
R4, s, LG, PG, R5, v, R6, R10, G, p, J и E являются такими, как описано в других местах в данной заявке.
В данном описании определены альдегид-защитные группы, и их неограничивающие примеры включают 1,3-дитиан, 1,3-дитиолан, диэтилацеталь, диметилацеталь, ацеталь этиленгликоля, ацеталь неопентилгликоля, триметилсилилцианогидрин и триэтилортоформиат.
На стадии 1 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (X), соединение формулы (VII) или его соль, органический растворитель и катализатор с образованием соединения формулы (XI). В одном из воплощений растворитель представляет собой неполярный растворитель, описанный в других местах в данной заявке. Неограничивающие примеры подходящих растворителей включают пентан, гексан, гептан, циклопентан, МТВЕ, диэтиловый эфир, толуол, 2-метилтетрагидрофуран (2-MeTHF), бензол, 1,4-диоксан, четыреххлористый углерод, хлороформ, дихлорметан и их комбинации. В некоторых примерах растворителем является толуол. В одном из воплощений катализатор представляет собой катализатор на основе переходного металла, описанный в данной заявке. В одном из воплощений неограничивающие примеры катализаторов на основе переходного металла включают катализаторы на основе палладия, платины, золота, рутения, родия и иридия. Неограничивающие примеры подходящих катализаторов включают JohnPhos, XPhos AuCI, XPhos AuNTf2, палладацикл на основе XPhos, палладацикл на основе SPhos, tBuXPhos Pd G1, Xphos Pd G2, SPhos Pd G2, RuPhos Pd G2, CPhos-Pd-G2, CPhos-Pd-G3, tBuXPhos-Pd-G3, RuPhos-Pd-G3, XPhos-Pd-G3, BrettPhos-Pd-G3, JackiePhos-Pd-G3, трет-бутил-BrettPhos-Pd-G3, [трет-бутил-BrettPhos-Pd (аллил)]OTf) и их комбинации. В некоторых воплощениях катализатором является BrettPhos-Pd-G3. В некоторых воплощениях реакционная смесь на стадии 1 дополнительно содержит органическое основание, описанное в других местах в данной заявке. В некоторых таких воплощениях основание представляет собой трет-бутилат, такой как трет-бутилат натрия или калия. Мольное соотношение между соединением формулы (VII) и соединением формулы (X) соответственно составляет примерно 1:1, примерно 1,1:1, примерно 1,2:1, примерно 1,3:1, примерно 1,5:1 или примерно 2:1, и на основании этих соотношений составлены диапазоны, такие как от примерно 1,1:1 до примерно 1,5:1.
В некоторых воплощениях стадии 1 реакционную смесь можно нагревать при перемешивании до температуры реакции в диапазоне от 2°С до примерно 30°С ниже температуры дефлегмации и вплоть до температуры дефлегмации и выдерживать в течение некоторого времени, достаточного для существенного завершения реакции получения соединения формула (XI), что определяется посредством хроматографии (например, TLC, GC или HPLC). В случае использования толуола в качестве растворителя температура реакции соответственно составляет примерно 50°С, примерно 55°С, примерно 60°С, примерно 65°С, примерно 70°С, примерно 75°С или примерно 80°С. Продолжительность реакции строго не ограничивается, и реакцию обычно продолжают до тех пор, пока превращение соединений формул (VII) и (X) в соединение формулы (XI) по существу не будет завершено, что определяется посредством хроматографии (например, TLC, GC или HPLC). После завершения реакции смесь продуктов реакции можно соответствующим образом погасить. В некоторых воплощениях реакцию на стадии 1 можно гасить водой. Если для гашения реакция используют воду, то органическую фазу, содержащую соединение формулы (XI) в растворе, можно выделить и возможно промыть по меньшей мере один раз водой. В некоторых воплощениях смесь продуктов реакции со стадии 1 может быть приведена в контакт с поглотителем ионов металлов, известным в данной области техники, таким как, например и без ограничения, SiliaMetS Thiol. Затем можно провести фильтрование смеси продуктов реакции для удаления твердых веществ.
На стадии 2 с соединения формулы (XI) удаляют защитную группу с образованием соединения формулы (IX) путем объединения раствора соединения (XI) в органическом растворителе (например, толуоле) с кислотой и водой. Как правило, кислота присутствует в эквивалентном избытке, например, в соотношении эквивалентов кислоты и соединения (XI), составляющем 1,01:1; 1,05:1; 1,1:1; 1,15:1; 1,2:1 или выше. В одном из воплощений температура, при которой осуществляют удаление защиты, не является очень критичной и может соответственно представлять собой комнатную температуру. После удаления защиты выполняют разделение органической фазы и водной фазы (содержащей соединение формулы (IX)). Органическая фаза возможно может быть промыта водой. Водную(ые) фазу(ы) можно обработать основанием, таким как неорганическое основание (например, NaOH или KOH), объединить с затравочными кристаллами соединения формулы (IX). Это основание может быть соответственно добавлено в эквивалентном избытке. Образуется суспензия кристаллического соединения формулы (IX), возможно при охлаждении. Соединение формулы (IX) в виде твердых веществ может быть собрано методами, известными в данной области техники, такими как фильтрация или центрифугирование. Твердые вещества могут быть подвергнуты сушке, например, в условиях частичного вакуума, с получением твердого соединения формулы (IX). Выход соединения формулы (IX) исходя из соединения формулы (XI) для стадий 1 и 2 составляет по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80% или по меньшей мере 85%.
Один из аспектов изобретения относится к способу получения соединения формулы (III) или его соли. Данный способ получения соединения формулы (III) включает две стадии реакции, изображенные ниже:
(1) приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (XII), соединения В и органического растворителя с образованием соединения формулы (XIII) в соответствии с приведенной ниже стадией 1,
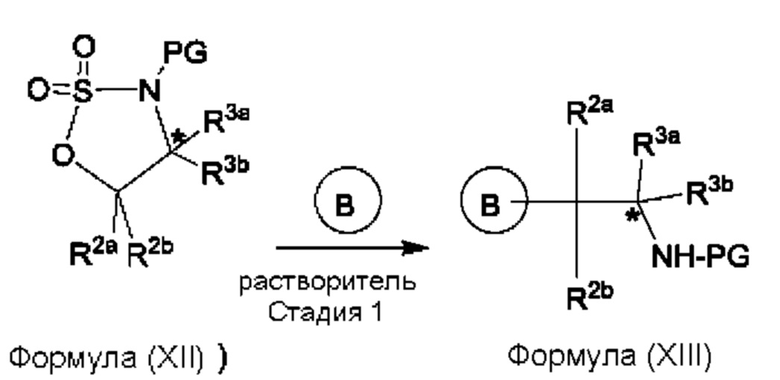
где PG представляет собой амин-защитную группу; и
(2) удаление защиты с соединения формулы (XIII) с образованием соединения формулы (III) в соответствии с приведенной ниже стадией 2,
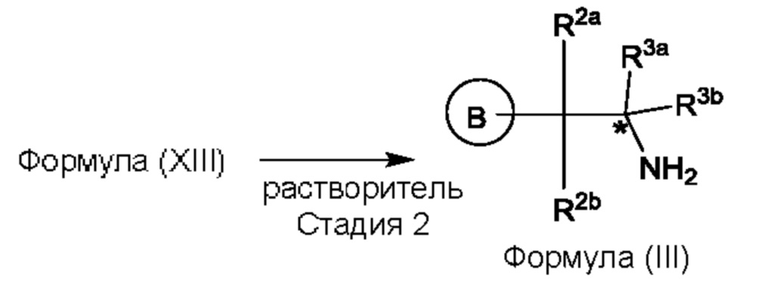
где R2a, R2b, R3a, R3b, В и звездочка являются такими, как описано в других местах в данной заявке.
Неограничивающие примеры защитных групп амина включают ACD, Ас, Bn, CBz, трифторацетамид, Вое, MeOZ, FMOC, Bz, РМВ, DMPM, РМР, Ts и Troc. В одном из воплощений PG представляет собой Boc.
В одном из воплощений соединение формулы (III) представляет собой соединение структуры
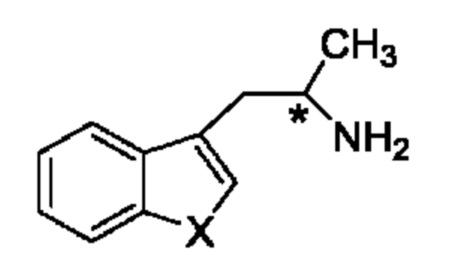
или его соль, описанные в данной заявке.
В некоторых других воплощениях соединение формулы (III) представляет собой соединение структуры
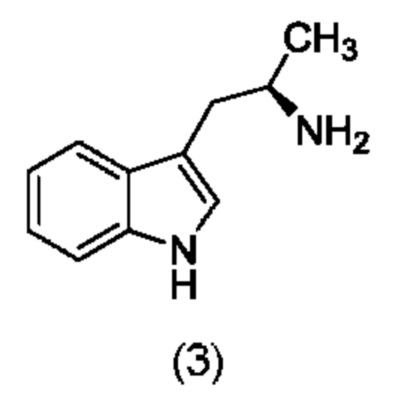
или его соль, описанные в других местах в данной заявке.
На стадии 1 приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (XII), соединение В и органический растворитель с образованием соединения формулы (XIII). В одном из воплощений органический растворитель представляет собой неполярный растворитель или полярный растворитель. В одном из воплощений растворитель является неполярным. Неограничивающие примеры подходящих растворителей включают пентан, циклопентан, гексан, циклогексан, бензол, толуол, 1,4-диоксан, хлороформ, диэтиловый эфир, DCM и их комбинации. В одном из воплощений растворителем является DCM. В некоторых воплощениях концентрация соединения В в растворителе может соответственно составлять примерно 10 г/л, примерно 25 г/л, примерно 50 г/л, примерно 75 г/л, примерно 100 г/л, примерно 125 г/л, примерно 150 г/л, примерно 175 г/л или примерно 200 г/л и вплоть до концентрации, приближающейся к насыщению при температуре реакции, и на основании этих концентраций составлены диапазоны, такие как от примерно 25 г/л до примерно 125 г/л. Соотношение эквивалентов соединения В и соединения формулы (XII) составляет примерно 0,75:1, примерно 0,9:1, примерно 1:1, примерно 1,25:1, примерно 1,5:1, примерно 1,75:1 или примерно 2:1, а их диапазоны составляют, например, от приблизительно 1,25:1 до приблизительно 1,75:1. Реакционная смесь на стадии 1 может дополнительно содержать подходящий алкилирующий агент. Неограничивающие примеры алкилирующих реагентов включают алкиллитий (например, метиллитий) и такие соединения, как магнийорганические галогениды, такие как метилмагния хлорид (например, в THF). Соотношение эквивалентов соединения В и алкилирующего реагента соответственно составляет примерно 0,75:1, примерно 0,8:1, примерно 0,85:1, примерно 0,9:1, примерно 0,95:1, примерно 1:1, примерно 1,05:1, примерно 1,1:1, примерно 1,15:2, примерно 1,2:1, примерно 1,3:1, примерно 1,4:1, примерно 1,5:1 или выше, и на их основе составлены диапазоны, такие как от примерно 1,1 до примерно 1,3:1. В одном из воплощений реакционная смесь дополнительно содержит катализатор алкилирования. В некоторых таких воплощениях такой катализатор представляет собой катализатор на основе переходного металла. В некоторых таких воплощениях переходным металлом является медь. В конкретном воплощении катализатор соответственно представляет собой галогенид переходного металла, такой как галогенид меди(I) (например, CuCl). В одном из воплощений температура реакции составляет примерно 25°С, примерно 20°С, примерно 15°С, примерно 10°С, примерно 5°С, примерно 0°С, примерно -5°С, примерно -10°С, примерно -15°С, примерно -20°С, примерно -25°С, примерно -30°С, примерно -35°С, примерно -40°С, примерно -45°С или примерно -50°С, и на их основе составлены диапазоны, такие как от примерно -20°С до примерно 0°С. В одном из воплощений продолжительность реакции строго не ограничивается, и реакцию обычно продолжают до тех пор, пока превращение соединения В и соединения формулы (XII) в соединение формулы (XIII) по существу не будет завершено, что определяется посредством хроматографии (например, TLC, GC или HPLC).
После завершения реакции можно выполнить гашение данной реакции, в частности, например, путем добавления водного раствора кислоты. В одном из воплощений кислота может представлять собой органическую кислоту, описанную в других местах в данной заявке, одним из неограничивающих примеров которой является лимонная кислота. Органическую фазу, содержащую соединение формулы (XIII), затем можно подвергнуть обработке с целью высушивания соединения. В некоторых таких воплощениях подвергнутая гашению смесь продуктов реакции может быть разделена на водную фазу и органическую фазу, содержащую соединение формулы (XIII). Водную фазу можно один или несколько раз промыть органическим растворителем, таким как растворитель, использованный для образования реакционной смеси (например, выполнить две промывки, каждый раз одним объемом растворителя, сравнимым с объемом реакционной смеси). Органические фазы можно объединить и промыть один или несколько раз рассолом (например, выполнить две промывки рассолом, каждый раз одним объемом рассола, сравнимым с объемом реакционной смеси). Затем подвергнутые промывке органические фазы могут быть объединены при перемешивании с активированным углем и с твердым сушильным агентом (например, Na2SO4). И активированный уголь, и твердый сушильный агент могут быть удалены фильтрованием или центрифугированием. Затем все собранные твердые вещества возможно могут быть промыты дополнительным количеством растворителя для извлечения из него соединения формулы (XIII).
В одном из воплощений в качестве исходного вещества для стадии 2 можно использовать раствор соединения формулы (XIII). В одном из воплощений может быть получено твердое соединение формулы (XIII). В таких воплощениях собранный раствор соединения формулы (XIII) в органическом растворителе может быть сконцентрирован в условиях частичного вакуума с образованием неочищенного соединения формулы (XIII). Альтернативно, можно осуществить осаждение/кристаллизацию твердого соединения формулы (XIII) из раствора путем добавления антирастворителя, такого как неполярный растворитель (например, 2-х объемов гептана, сравнимым с объемом растворителя в реакционной смеси). Твердые вещества можно собрать фильтрованием или центрифугированием, а собранные твердые вещества можно промыть антирастворителем. Для окончательного получения соединения формулы (XIII) твердые вещества можно подвергнуть сушке в условиях частичного вакуума при температуре меньше 40°С. Выход соединения формулы (XIII) исходя из соединения формулы (XII) обычно составляет по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60% или по меньшей мере 65%. Чистота по данным HPLC (в процентах площади) составляет по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99%.
В некоторых воплощениях стадии 1 соединение В, металлический катализатор и алкилирующий агент объединяют в первом объеме растворителя при температуре реакции, указанной выше. Объем растворителя составляет соответственно от примерно 30% до примерно 80% от общего объема растворителя, используемого для стадии 1. После этого в течение некоторого периода времени добавляют раствор соединения формулы (XII) в оставшейся части растворителя при температуре реакции с получением реакционной смеси. Затем температуру реакционной смеси поддерживают в течение промежутка времени, достаточного для практически полного завершения образования соединения формула (XIII), после чего осуществляют гашение и обработку с целью получения раствора соединения формулы (XIII) или высушенного соединения формулы (XIII).
На стадии 2 с соединения формулы (XIII) удаляют защитную группу с образованием соединения формулы (III). В любом из различных воплощений удаление защиты с соединения формулы (XIII) соответственно осуществляют в растворе посредством добавления кислоты. В одном из воплощений твердое соединение формулы (XIII) растворяют в полярном протонном растворителе, описанном в других местах в данной заявке (таком как метанол, этанол или изопропанол). Концентрация соединения формулы (XIII) в растворителе составляет примерно 25 г/л, примерно 50 г/л, примерно 75 г/л, примерно 100 г/л, примерно 125 г/л, примерно 150 г/л, примерно 175 г/л или примерно 200 г/л, и на их основе составлены диапазоны, такие как от примерно 50 г/л до примерно 150 г/л. Температура, при которой осуществляют добавление кислоты, не является очень критичной.
В некоторых воплощениях стадии 2 кислота представляет собой неорганическую кислоту, описанную в других местах в данной заявке. Неограничивающим примером подходящей неорганической кислоты является HCI. В одном из воплощений соотношение эквивалентов кислоты и соединения формулы (XIII) составляет примерно 1,5:1, примерно 2,5:1, примерно 5:1, примерно 7,5:1, примерно 10:1, примерно 12,5:1 или примерно 15:1, и на их основе составлены диапазоны, такие как от примерно 5:1 до примерно 15:1. После добавления кислоты раствор выдерживают при данной температуре с перемешиванием до тех пор, пока по существу не будет завершено удаление защиты с соединения формулы (XIII) с образованием соединения формулы (III). В одном из воплощений может быть выполнена смена растворителей от полярного протонного растворителя к неполярному растворителю (описанному в других местах в данной заявке) или полярному апротонному растворителю (описанному в других местах в данной заявке). В некоторых таких воплощениях раствор соединения формулы (III) может быть сконцентрирован в условиях частичного вакуума и экстрагирован неполярным или полярным апротонным растворителем. В некоторых таких воплощениях растворителем для экстракции является DCM. После экстракции значение рН водной фазы может быть подведено до соответствия сильно основному раствору (т.е. больше рН 11) путем использования подходящего основания, такого как водный раствор гидроксида натрия. Далее, после подщелачивания, можно провести по меньшей мере один раз дополнительное экстрагирование водной фазы с использованием растворителя для экстракции. Затем органическую(ие) фазу(ы) можно подвергнуть сушке, как например, с использованием рассола и/или над твердым высушивающим средством, после чего провести фильтрование для удаления любых твердых веществ. Для извлечения дополнительного количества соединения формулы (III) собранные твердые вещества возможно могут быть промыты. Твердое соединение можно выделить из раствора в органическом растворителе методами, известными в данной области техники. Например, раствор можно досуха сконцентрировать в условиях частичного вакуума. Альтернативно, раствор можно сконцентрировать, после чего добавить антирастворитель с образованием твердого соединения формулы (III), которое может быть собрано фильтрованием или центрифугированием, промыто и подвергнуто сушке. Выход соединения формулы (III) исходя из соединения формулы (XIII) обычно составляет по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 97%. Чистота по данным HPLC (в процентах площади) составляет по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 96%.
В некоторых других воплощениях стадии 2 кислота представляет собой органическую кислоту, описанную в других местах в данной заявке. Неограничивающие примеры подходящих органических кислот включают сульфоновые кислоты и камфорсульфоновую кислоту (CSA) (например, L-(-)-CSA). В случае CSA соотношение эквивалентов CSA и соединения формулы (XIII) составляет примерно 1,5:1, примерно 2:1, примерно 2,5:1, примерно 5:1, примерно 7,5:1 или примерно 10:1, и на их основе составлены диапазоны, такие как от примерно 2:1 до примерно 4:1. После добавления кислоты раствор может быть нагрет и выдержан при повышенной температуре (например, от примерно 35°С до примерно 60°С) с перемешиванием для завершения реакции удаления защиты и образования полученной присоединением кислоты соли соединения формулы (XIII). Затем раствор/суспензию соли, полученной присоединением кислоты, можно охладить, как например, до температуры меньше примерно 5°С, с образованием суспензии полученной присоединением кислоты соли соединения формулы (XIII). Соль может быть собрана фильтрованием или центрифугированием и возможно промыта растворителем. Затем данную соль можно подвергнуть сушке в условиях частичного вакуума с получением твердой соли соединения формулы (XIII), такой как его соль с L-(-)-CSA. Выход соли соединения формулы (III) исходя из соединения формулы (XIII) обычно составляет по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 92% или по меньшей мере 95%. Чистота по данным HPLC (в процентах площади) составляет по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96% или по меньшей мере 97%. Соль соединения формулы (XIII) можно растворить в воде и рН подвести до значения выше 13, например, приблизительно 14, с использованием сильного основания, в результате чего образуется суспензия, содержащая твердое соединение формулы (III) в виде свободного основания. Твердое вещество может быть собрано фильтрованием или центрифугированием и возможно промыто охлажденной водой. Затем твердые вещества могут быть подвергнуты сушке в условиях частичного вакуума с получением высушенного соединения формулы (III) в виде свободного основания. Выход соединения формулы (III) исходя из соединения формулы (XIII) обычно составляет по меньшей мере 80%, по меньшей мере 85% или по меньшей мере 90%. Чистота по данным HPLC (в процентах площади) составляет по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96% или по меньшей мере 97%.
В одном из воплощений соединение формулы (VIII), полученное согласно данному изобретению, далее перекристаллизовывают. В одном из воплощений перекристаллизация включает перекристаллизацию соединения формулы (VIII) в 2-стадийном способе. Данный способ включает нагревание суспензии, содержащей соединение формулы (VIII) в смеси метанол/этанол, дистилляцию с метанолом и охлаждение смеси. В одном из воплощений смесь метанол/этанол представляет собой смесь 95:5, 90:10, 85:15 или 80:20 метанол/этанол. В другом воплощении смесь этанол/метанол представляет собой смесь 90:10 метанол/этанол. Смесь можно нагревать при температуре выше примерно 50°С, например, примерно 55°С, 60°С или 65°С. Охлаждение может быть осуществлено примерно до комнатной температуры. В одном из воплощений охлаждение осуществляют примерно до 20°С, 25°С или 30°С. Можно провести фильтрование и сушку данного раствора.
В другом воплощении перекристаллизация включает перекристаллизацию соединения формулы (VIII) из МТВЕ. К суспензии, содержащей соединение формулы (VIII) в МТВЕ, добавляют основание, такое как NaOH или KOH. Смесь перемешивают, например, при 15°С, 20°С, 25°С или 30°С, возможно фильтруют и подвергают дистилляции с этанолом.
В некоторых воплощениях способа получения соединения формулы (III) или его соли данный способ дополнительно включает получение соединения формулы (XII) или его соли.
Способ получения соединения формулы (XII) включает стадии реакции 3а и 3b, показанные ниже:
(1) приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (XIV), тионилхлорида и органического растворителя с образованием соединения формулы (XV) в соответствии с приведенной ниже стадией 3а
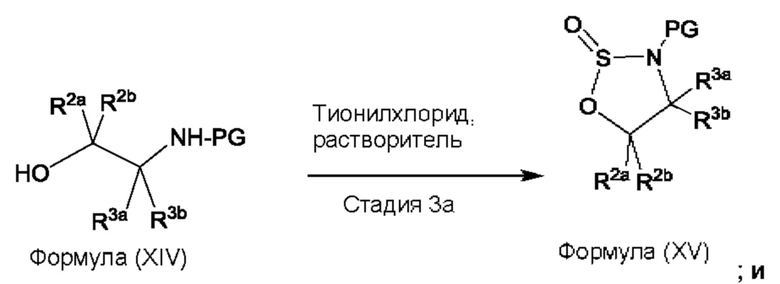
(2) приведение во взаимодействие содержащихся в реакционной смеси соединения формулы (XV), катализатора, окисляющего агента и органического растворителя с образованием соединения формулы (XII) в соответствии с приведенной ниже стадией 3b
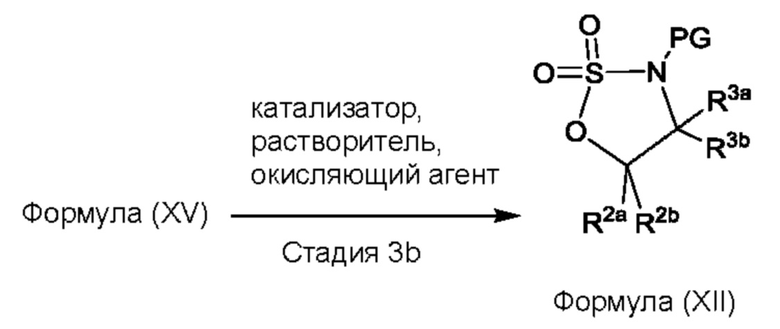
R2a, R2b, R3a, R3b и защитная группа атома азота PG являются такими, как описано в данной заявке.
На стадии 3а приводят во взаимодействие содержащиеся в реакционной смеси соединение формулы (XIV), тионилхлорид и органический растворитель с образованием соединения формулы (XV). В одном из воплощений органический растворитель представляет собой неполярный растворитель или полярный растворитель, описанный в других местах в данной заявке. В одном из воплощений растворитель является неполярным растворителем, описанным в других местах в данной заявке. Неограничивающие примеры подходящих растворителей включают пентан, циклопентан, гексан, циклогексан, бензол, толуол, 1,4-диоксан, хлороформ, диэтиловый эфир, DCM и их комбинации. В некоторых конкретных воплощениях растворителем является DCM. В одном из воплощений концентрация соединения формулы (XIV) в растворителе может соответственно составлять примерно 10 г/л, примерно 25 г/л, примерно 50 г/л, примерно 75 г/л, примерно 100 г/л, примерно 125 г/л, примерно 150 г/л, примерно 175 г/л или примерно 200 г/л и вплоть до концентрации, приближающейся к насыщению при температуре реакции, и на основании этих концентраций составлены диапазоны, такие как от примерно 25 г/л до примерно 125 г/л. Соотношение эквивалентов тионилхлорида и соединения формулы (XIV) составляет примерно 1:1, примерно 1,25:1, примерно 1,5:1, примерно 1,75:1, примерно 2:1, примерно 2,25:1 или примерно 2,5:1, а их диапазоны составляют, например, от приблизительно 1,25:1 до приблизительно 1,75:1. В одном из воплощений температура реакции составляет примерно 10°С, примерно 5°С, примерно 0°С, примерно -5°С или примерно -10°С, и на их основе составлены диапазоны, такие как от примерно -5°С до примерно 5°С. В одном из воплощений продолжительность реакции строго не ограничивается, и реакцию обычно продолжают до тех пор, пока превращение соединения формулы (XIV) по существу не будет завершено, что определяется посредством хроматографии (например, TLC, GC или HPLC). Реакционная смесь на стадии 3а может дополнительно содержать основание, неограничивающим примером которого является имидазол. В таких воплощениях соотношение эквивалентов тионилирующего реагента и тионилхлорида может составлять примерно 1:1, примерно 2:1, примерно 3:1 или примерно 4:1. Реакционная смесь на стадии За может дополнительно содержать основание, такое как органическое основание, описанное в других местах в данной заявке. В некоторых таких воплощениях основанием может быть TEA. В таких воплощениях соотношение эквивалентов основания и соединения формулы (XIV) составляет примерно 1,25:1, примерно 1,5:1, примерно 1,75:1, примерно 2:1, примерно 2,25:1, примерно 2,5:1, примерно 3:1, примерно 3.5:1 или примерно 4:1, и на их основе составлены диапазоны, такие как от примерно 1,5:1 до примерно 2,5:1.
В некоторых особых воплощениях стадии 3а готовят раствор основания (например, имидазола) в растворителе, куда добавляют при перемешивании тионилхлорид, поддерживая температуру реакции. Затем при перемешивании с поддержанием температуры реакции можно добавить соединение формулы (XIV) в растворителе, после чего добавить основание при перемешивании, поддерживая температуру реакции. Затем реакционную смесь выдерживают при температуре реакции с перемешиванием до тех пор, пока по существу не будет завершено превращение соединения формулы (XIV) с образованием смеси продуктов реакции, содержащей соединение формулы (XV).
В любом из различных воплощений стадии 3а смесь продуктов реакции может быть обработана методами, известными специалистам в данной области техники. Например, можно провести гашение реакционной смеси на стадии 3а охлажденной водой (например, от 0,25 до 2 объемов воды из расчета на один объем органического растворителя в смеси продуктов реакции на стадии 3а). Органическую фазу, содержащую соединение формулы (XV) в растворе, можно выделить, а выделенную водную фазу можно экстрагировать органическим растворителем для извлечения дополнительного количества соединения формулы (XV). Можно объединить органические фазы и промыть водным раствором кислоты, водным раствором основания и рассолом. Неограничивающим примером водного раствора кислоты является раствор слабой кислоты, такой как лимонная кислота. Неограничивающим примером раствора основания является раствор слабого основания, такого как бикарбонат натрия.
На стадии 3b реакционную смесь, содержащую соединение формулы (XV) в растворе в органическом растворителе, объединяют с катализатором и окисляющим агентом, и проводят взаимодействие с образованием смеси продуктов реакции, содержащей соединение формулы (XII). В одном из воплощений катализатор представляет собой металлический катализатор окислительно-восстановительных реакций, описанный в данной заявке в других местах. Неограничивающие примеры подходящих катализаторов включают NiCl2, RuCl3, CoCl2, FeCl3, FeCl2 и MnCl2. Неограничивающие примеры подходящих окислителей включают NalO4, NaOCl и оксон. В некоторых воплощениях реакционная смесь дополнительно содержит воду. Соотношение объемов воды и органического растворителя соответственно составляет примерно 0,25:1, примерно 0,5:1, примерно 0,75:1, примерно 1:1, примерно 1,25:1, примерно 1,50:1, примерно 1,75:1, примерно 2:1 или примерно 2,5:1, и на их основе составлены диапазоны, такие как от примерно 0,5:1 до примерно 1,5:1. Температура реакции соответственно составляет от примерно 5°С до примерно 50°С. Реакционную смесь выдерживают при температуре реакции с перемешиванием до тех пор, пока по существу не будет завершено превращение соединения формулы (XV) с образованием смеси продуктов реакции, содержащей соединение формулы (XII), что определяется посредством хроматографии (например, TLC, GC или HPLC).
Смесь продуктов реакции со стадии 3b можно обработать методами, известными специалистам в данной области техники. Например, органическая фаза, содержащая соединение формулы (XII) в растворе, может быть выделена, а в отношении водной фазы возможно может быть выполнено фильтрование и затем экстрагирование органическим растворителем для извлечения соединения формулы (XII). Органическая(ие) фаза(ы) возможно может/могут быть промыта(ы) восстанавливающим агентом (например, тиосульфатом натрия) и рассолом. Органическую(ие) фазу(ы) можно подвергнуть сушке с использованием твердого высушивающего средства (например, сульфата натрия), после чего отфильтровать для удаления твердых веществ и возможно промыть их органическим растворителем. Твердое соединение формулы (XII) можно выделить из раствора в органическом растворителе методами, известными в данной области техники. Например, раствор можно досуха сконцентрировать в условиях частичного вакуума. Альтернативно, раствор можно сконцентрировать, после чего добавить антирастворитель с образованием твердого соединения формулы (XII), которое может быть собрано фильтрованием или центрифугированием, промыто и подвергнуто сушке. Выход соединения формулы (XII) исходя из соединения (XIV) обычно составляет по меньшей мере 85%, по меньшей мере 90% или по меньшей мере 92%. Чистота по данным HPLC (в процентах площади) составляет по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99%.
Хиральные первичные амины могут быть образованы с использованием стереоспецифичной трансаминазы либо посредством асимметрического трансаминирования прохирального кетона, либо посредством кинетического разделения рацемического амина. Трансаминирование представляет собой равновесную реакцию с участием пары амин и кетон (донор и акцептор), и в применяемых условиях реакции происходит смещение равновесия в сторону образования целевого хирального амина. Согласно одному из таких аспектов донором аминогруппы является аланин или 2-пропиламин. В одном из воплощений акцептором аминогруппы является пируват или ацетон.
Таким образом, далее согласно данному изобретению предложен способ получения соединения формулы (XX) или его соли, при этом превращение хирального триптамина включает использование превращения с участием фермента:
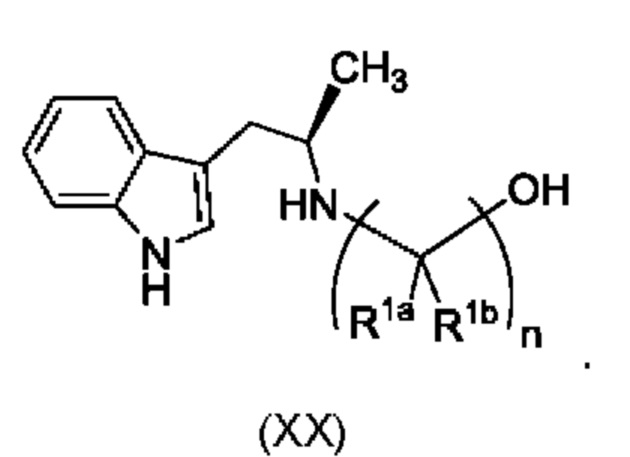
Способ получения соединения формулы (XX) включает приведение в контакт соединения формулы (XXI)
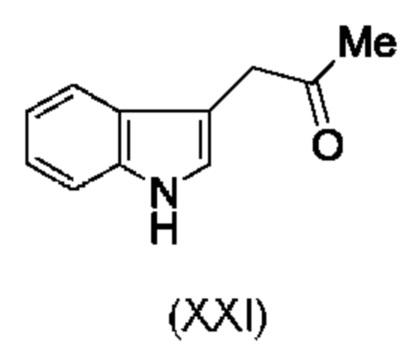
с белком трансаминазой с образованием соединения формулы (3):
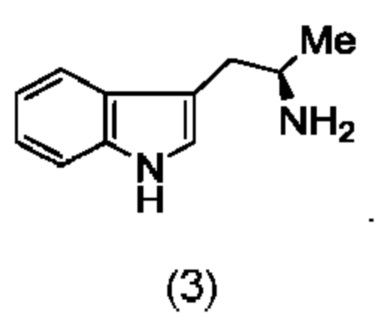
Данный способ дополнительно предусматривает присутствие одного или более доноров аминогруппы. В одном из воплощений донором аминогруппы является аланин или изопропиламин.
Приведение в контакт, описанное выше, может быть осуществлено в смешанных водно-органических системах растворителей. Например, реакция с участием трансаминазы может быть осуществлена в водном буфере с органическим сорастворителем, таком как циклогексан, метил циклогексан, изооктан, DMSO, ацетонитрил или ацетон. Такие сорастворители могут быть представлены в количестве 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45 или 50% (об./об.).
Смешанная органическая/водная система растворителей (микро-водная реакционная система) в определенных случаях может представлять собой один или более чем один органический растворитель, содержащий небольшое количество водного буфера. В одном из воплощений приведение в контакт осуществляют в простом трет-бутил-метиловом эфире (ТВМЕ), простом дибутиловом эфире, простом циклопентил-метиловом эфире (СРМЕ), толуоле, этилацетате, бутилацетате, изопропилацетате, бутилбутирате, этилбутирате или изобутилацетате, содержащем меньше 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2 или 1% (об./об.) водного буфера. В случае проведения реакции в органическом растворителе трансаминаза может быть иммобилизована на твердой подложке.
В другом воплощении степень превращения соединения (XXI) в соединение (3) составляет значение, превышающее по меньшей мере 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98% или 99%. Согласно другому аспекту степень превращения превышает 90%. В другом воплощении степень превращения превышает 95%.
В результате восстановительного аминирования, выполненного с использованием трансаминазы, может быть получено соединение формулы (3) с энантиомерным избытком (ЕЕ) по меньшей мере 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100%. В определенных случаях под действием трансаминазы соединение формулы (XXI) превращается в соединение формулы (3) с ЕЕ более 99%.
В одном из воплощений асимметрическое трансаминирование соединения формулы (XXI) осуществляют, используя приведенную ниже схему:
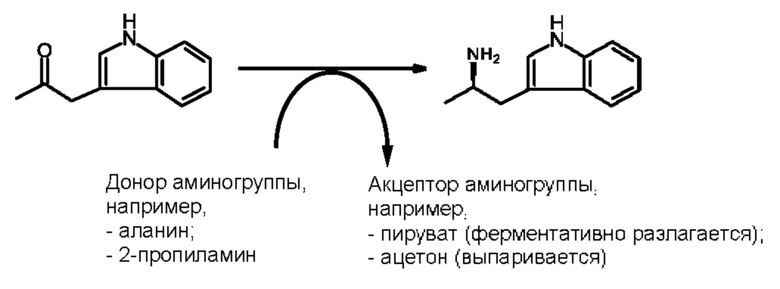
Соединение формулы (3) приводят в контакт с соединением формулы (II), описанным в данной заявке,
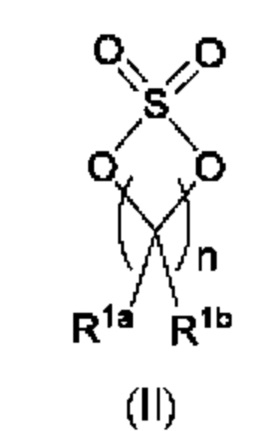
с образованием соединения формула (XX).
R1a, R1b и n являются такими, как описано в других местах в данной заявке. В одном из воплощений R1a и R1b независимо представляют собой атом фтора.
В одном из воплощений соединение формулы (XX) имеет структуру:
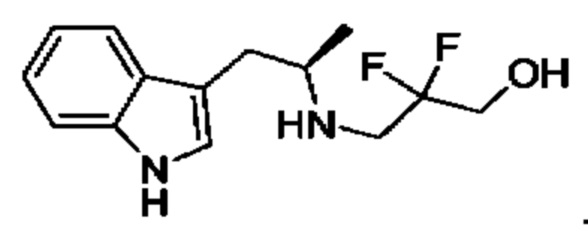
Белок трансаминаза может быть выбран из Таблицы 6. Согласно одному из таких аспектов трансаминаза является (R)-селективной. В одном из воплощений трансаминаза представляет собой ТА-Р2-А01. В одном из воплощений белок трансаминаза выбран из (З)-энантиоселективной трансаминазы с SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3 и SEQ ID NO: 4.
Кроме того, согласно данному изобретению предложены способы ферментативного синтеза соединения формулы (3) посредством кинетического разделения рацемического амина в присутствии акцептора аминогруппы и трансаминазы. Данная реакция может быть проведена в условиях, описанных в данной заявке, например, в смешанных водных/органических системах растворителей, описанных в данной заявке. Согласно одному из таких аспектов реакцию проводят в водном буфере, содержащем ацетонитрил. В результате осуществления такого кинетического разделения может быть получено соединение формулы (3) с ЕЕ по меньшей мере 99%, для чего необходима степень превращения выше 50%. Кинетическое разделение включает применение (S)-селективной трансаминазы, описанной в данной заявке для проведения кинетического разделения. В одном из воплощений трансаминаза выбрана из Таблицы 6.
Одно из воплощений изобретения относится к соединению формулы (XVI):
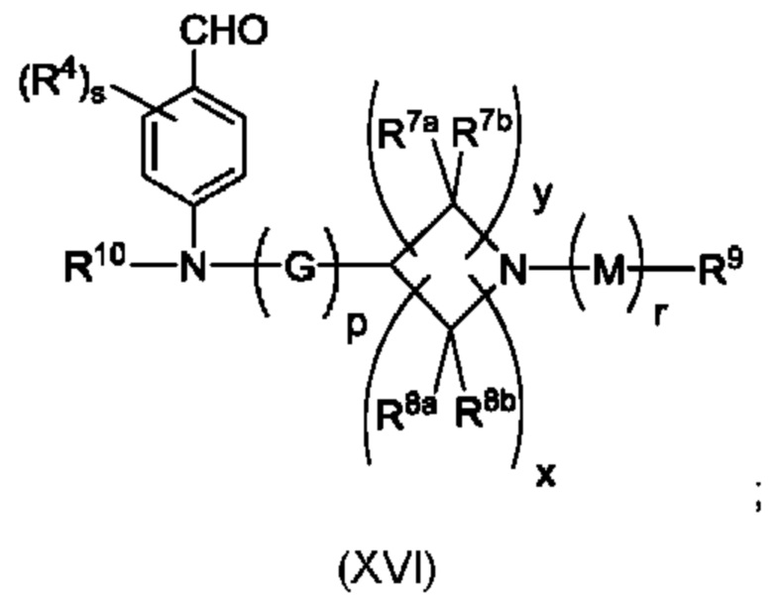
R4, s, R10, G и р являются такими, как определено в данном описании;
каждый из R7a, R7b, R8a и R8b независимо представляет собой атом водорода, галогена, С1-3алкил, С1-3галогеналкил, С1-3гидроксиалкил или -CN. В одном из воплощений каждый из R7a, R7b, R8a и R8b независимо представляет собой атом водорода, фтора, -СН3 или -CN. В одном из воплощений каждый из R7a, R7b, R8a и R8b представляет собой атом водорода;
у равно целому числу 1 или 2, х равно целому числу 1 или 2, а сумма х и у равна 2 или 3;
М представляет собой С1-5алкил;
r равно 0 или 1; и
R9 представляет собой атом галогена или -CN.
В одном из воплощений М представляет собой -СН2СН2СН2-, р равно 1, и R9 представляет собой атом фтора.
В одном из воплощений М представляет собой -СН2СН2СН2-, р равно 1, и R9 представляет собой -CN.
В одном из воплощений соединение формулы (XVI) представляет собой:
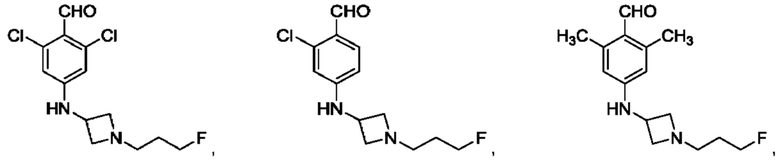
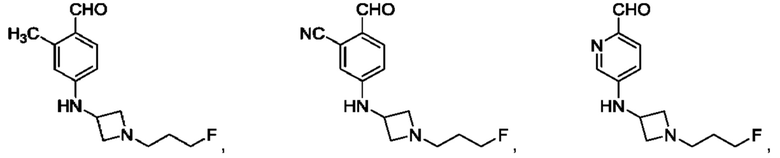
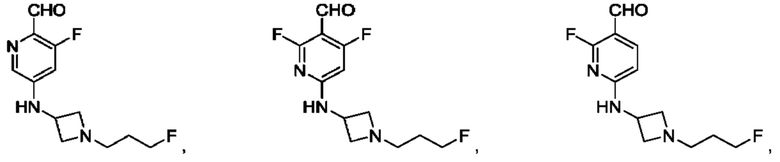
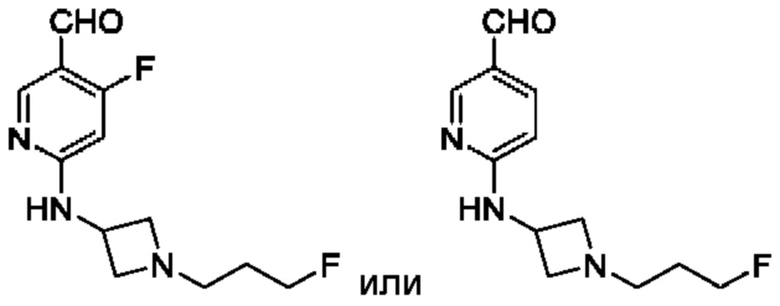
либо их соль.
В конкретном воплощении соединение формулы (XVI) представляет собой:
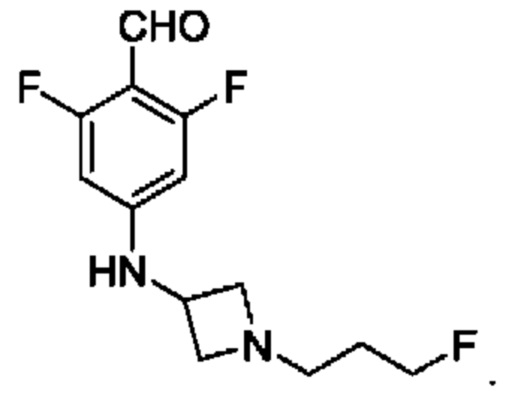
Согласно другому аспекту данного изобретения предложено соединение, имеющее структуру:
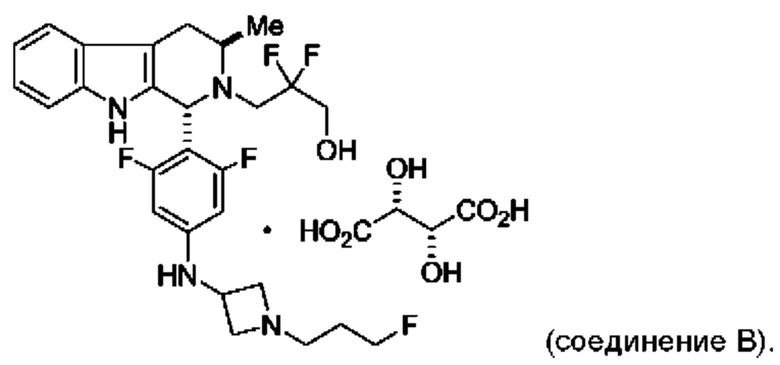
Согласно другому аспекту данного изобретения предложен способ получения соединения, имеющего формулу:
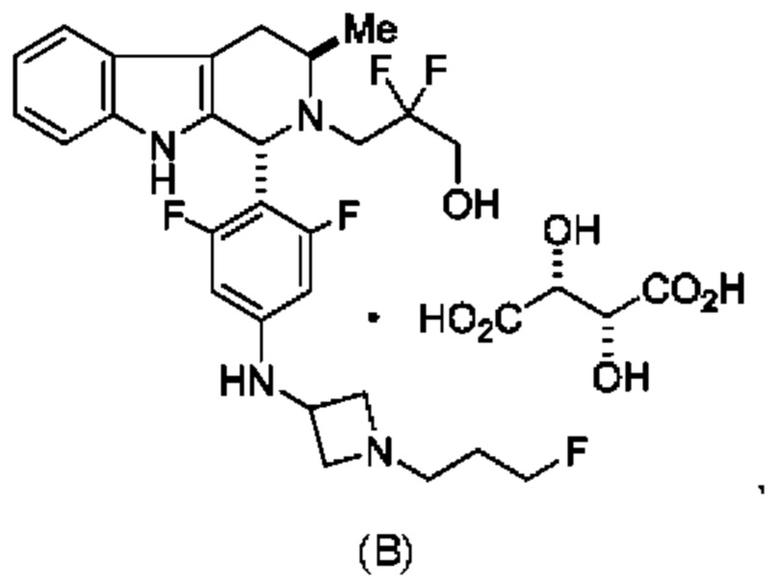
при этом данный способ осуществляют так, как представлено на приведенной ниже схеме А, и в соответствии с указанными воплощениями и воплощениями, представленными в данном описании.
Схема А:
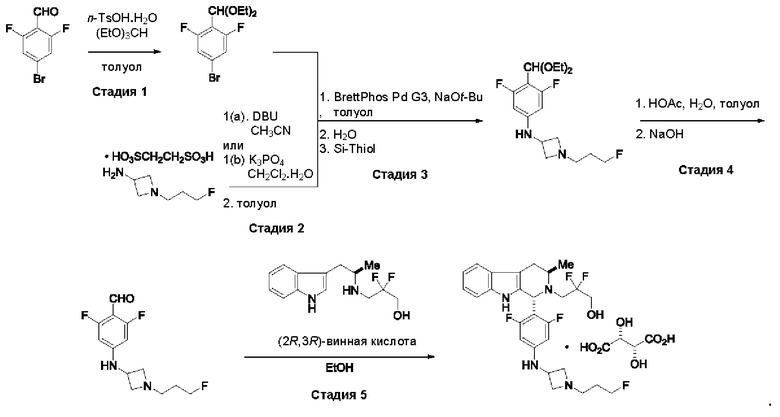
BrettPhos Pd G3 означает [(2-ди-циклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат,
В еще одном воплощении изобретения предложен способ получения соединения, имеющего формулу:
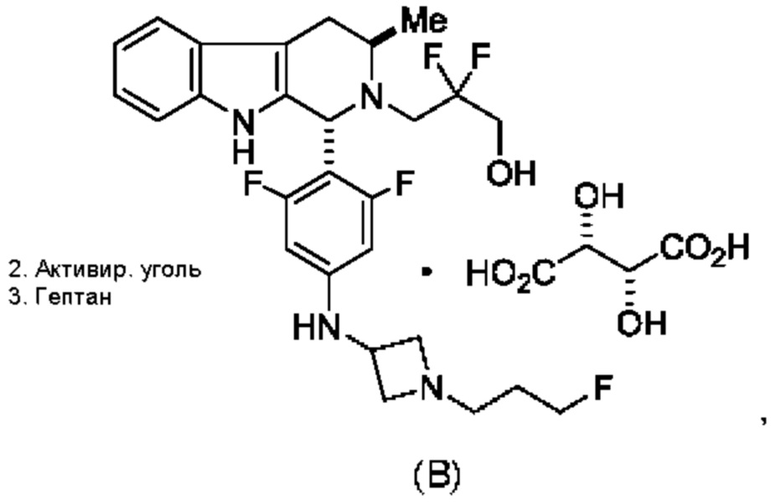
при этом данный способ осуществляют так, как представлено на приведенной ниже схеме В, и в соответствии с указанными воплощениями и воплощениями, представленными в данном описании.
Схема В:
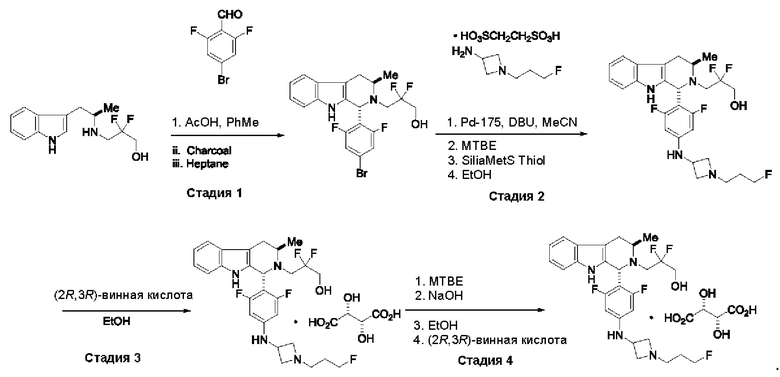
Приведенные выше способы синтеза соединения В (например, на схеме А и В) могут дополнительно включать перекристаллизацию согласно схеме С или D, показанной ниже.
Схема С:
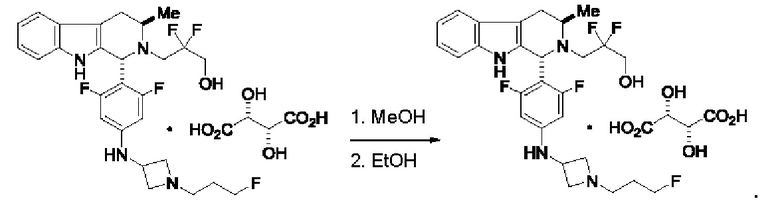
Схема D:
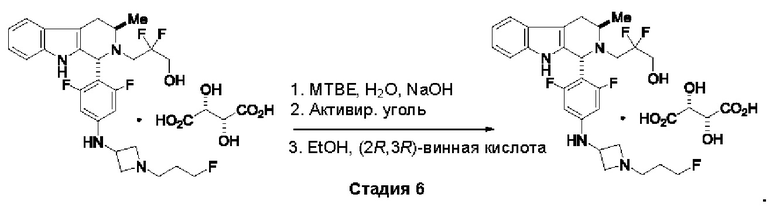
В одном из воплощений соединения (3) и (4), описанные в данной заявке и представленные на схеме А и схеме В и, возможно, схеме С или D, показанным выше, синтезируют согласно приведенной ниже схеме Е и в соответствии с указанными воплощениями и воплощениями, представленными в данном описании.
Схема Е:
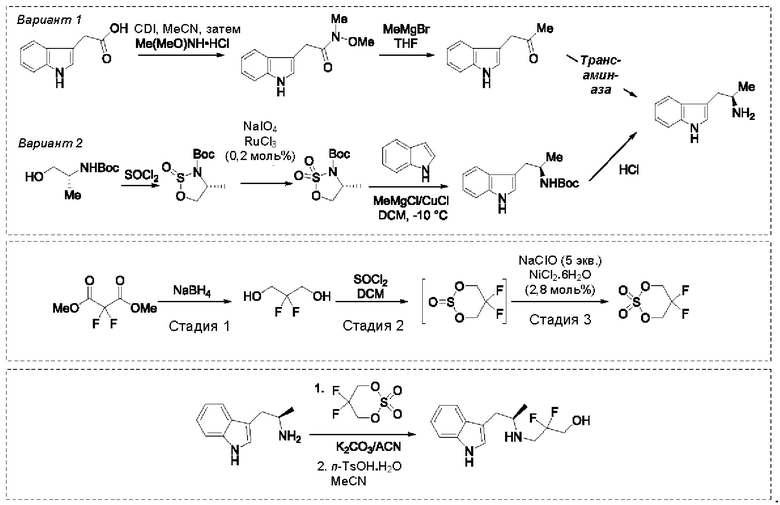
Кроме того, согласно данному изобретению предложен способ получения соединения формулы (XXIII) или его соли:
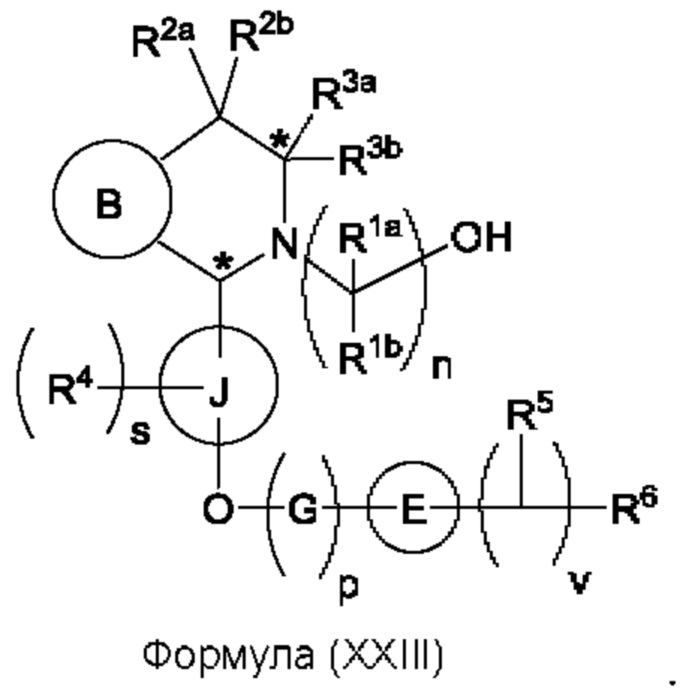
В, R1a, R1b, n, R2a, R2b, R3a, R3b, J, R4, s, r; R5, v, R6, E и звездочки являются такими, как определено в данном описании.
В одном из воплощений соединение формулы (XXIII) представляет собой соль с кислотой. В таких воплощениях соединение формулы (VIII) представляет собой соль кислоты. В предпочтительном воплощении соединение формулы (XXIII) представляет собой соль винной кислоты. В некоторых воплощениях соединение формулы (XXIII) представляет собой соль фумаровой кислоты.
В одном из воплощений соединение формулы (XXIII) представляет собой любую из следующих структур или их соль с винной кислотой:
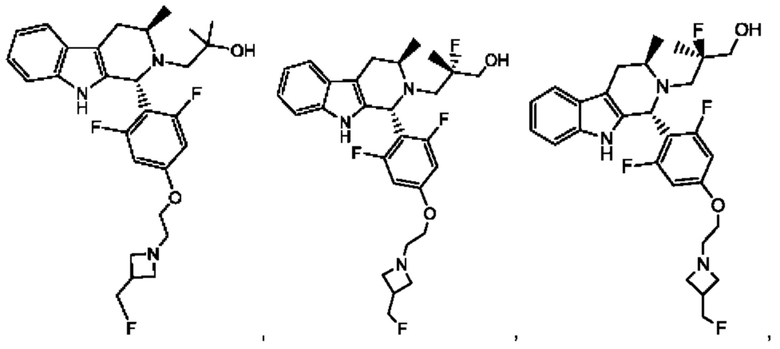
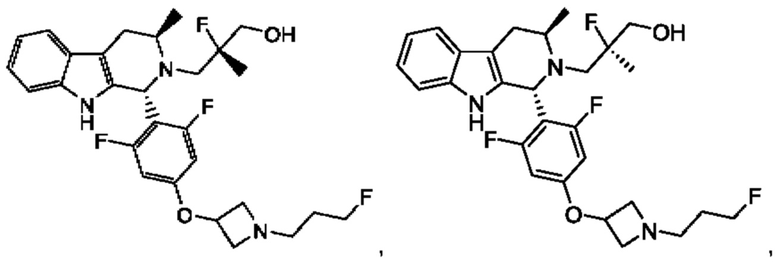
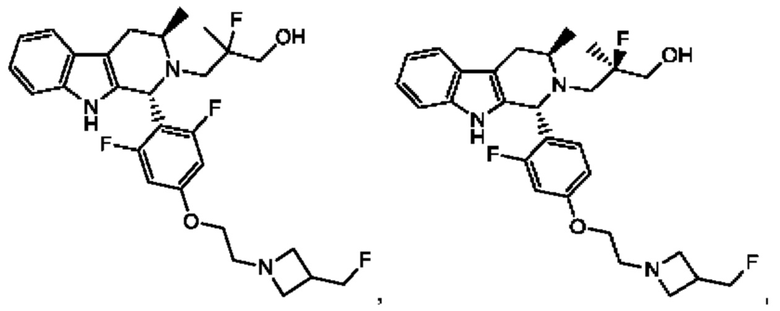
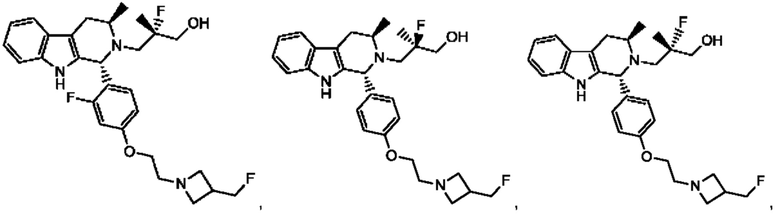
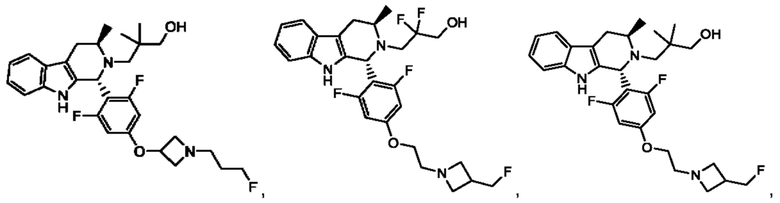
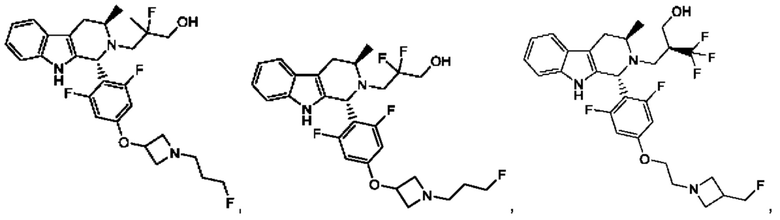
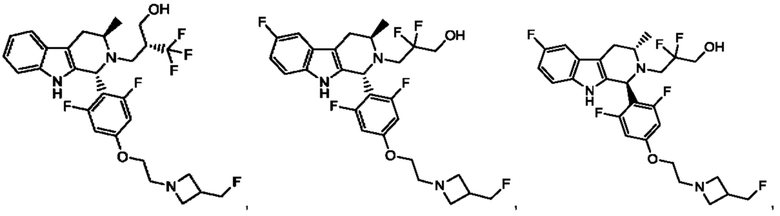
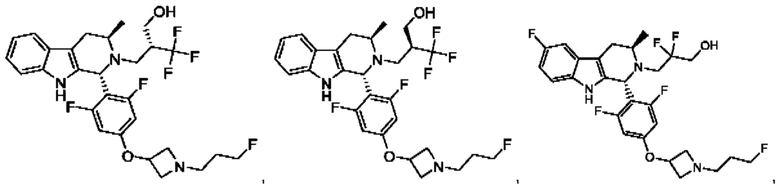
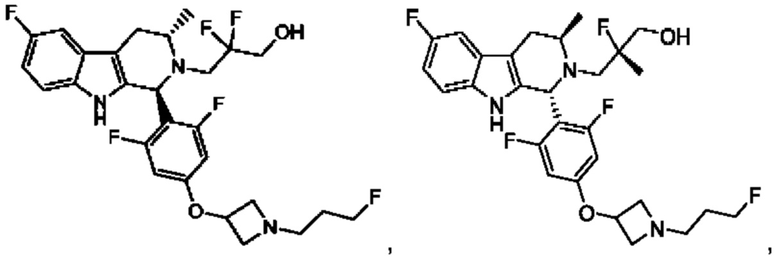
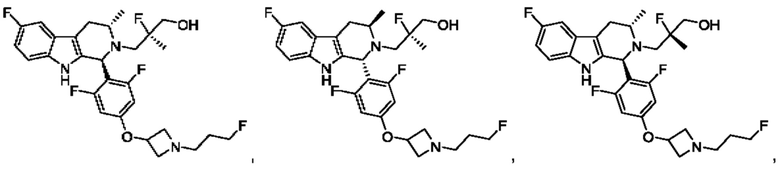
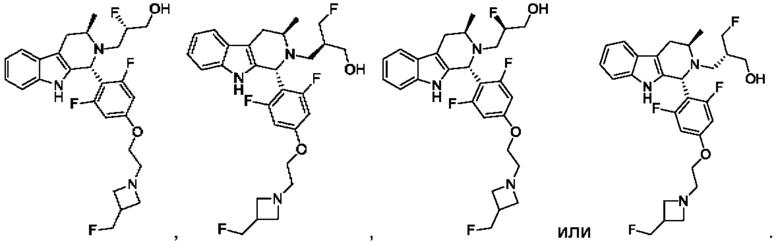
В другом воплощении приведенные выше соединения представляют собой соль фумаровой кислоты.
В одном из воплощений соединение формулы (XXIII) представляет собой соединение следующей структуры или его фармацевтически приемлемую соль:
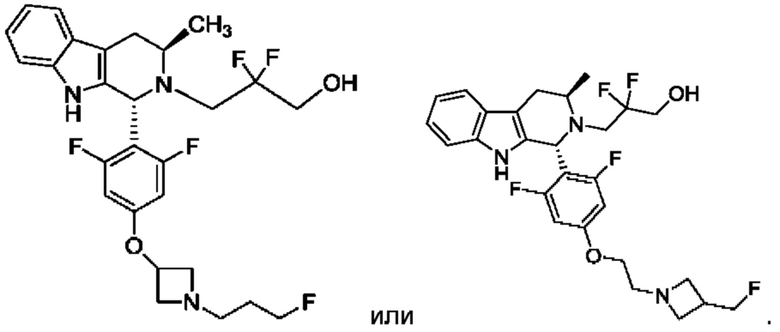
Способ получения соединения формулы (XXIII) включает две стадии реакции, изображенные ниже:
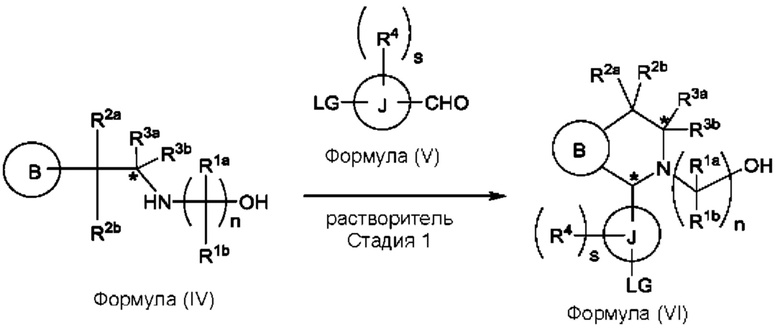
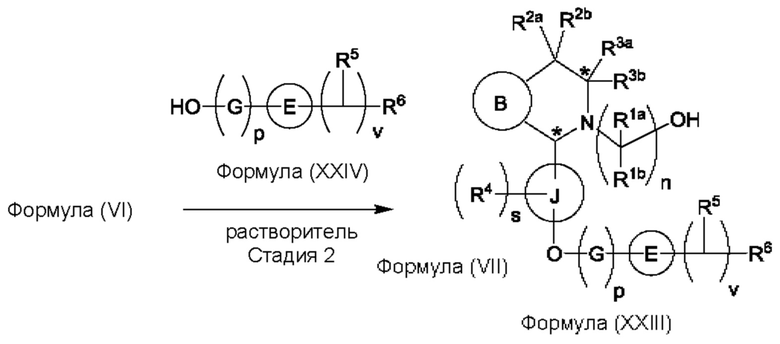
LG является таким, как определено в данном описании.
Переменные формул (IV), (V) и (VI) являются такими, как описано в данной заявке.
В одном из воплощений соединение формулы (XXIV) представляет собой любую из следующих структур или их соль:


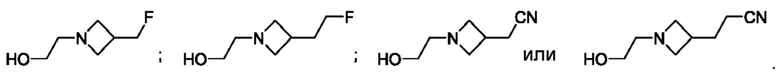
Одно из воплощений изобретения относится к способу получения соединения формулы (XXIII) или его соли, при этом соединение формулы (XXIII) является таким, как описано в данной заявке. Способ получения соединения формулы (XXIII) согласно этому воплощению включает реакционную стадию 1, изображенную ниже:
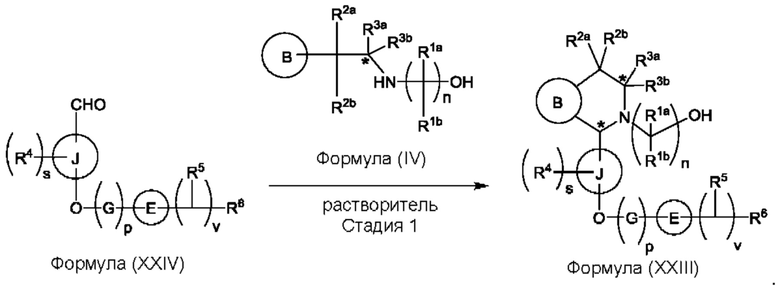
Каждая переменная из В, R1a, R1b, n, R2a, R2b, R3a, R3b, R4, s, J, R5, v, R6, G, p, E и звездочки является такой, как описано в других местах в данной заявке. Группировка СНО и атом азота, соединяющий J и G, локализованы в J в пара-положении относительно друг друга.
В некоторых воплощениях соединение формулы (XXIV) представляет собой:
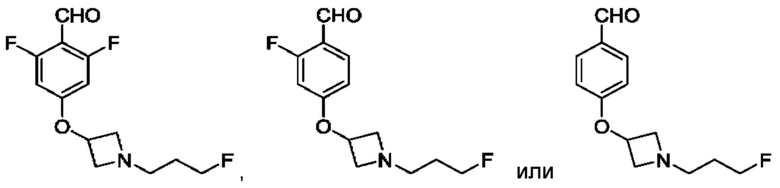 либо их соль.
либо их соль.
Одно из воплощений изобретения относится к способу получения соединения формулы (XXVII) или его соли.
Способ получения соединения формулы (XXVII) включает две стадии реакции, изображенные ниже:
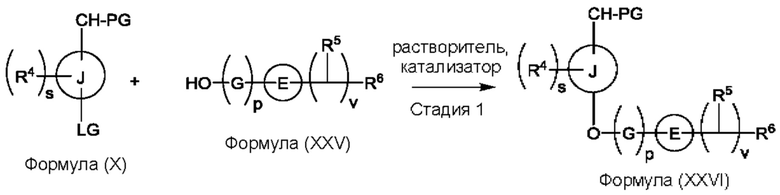
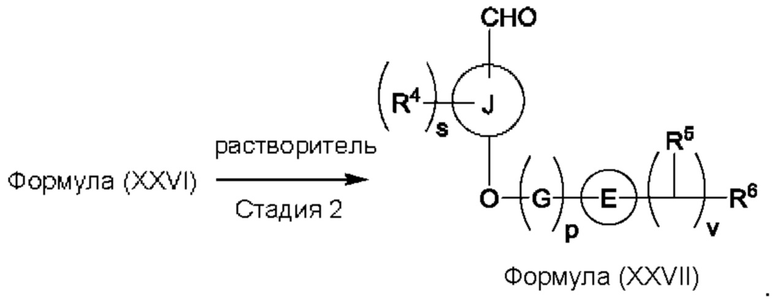
R4, s, LG, R5, v, R6, G, p, E и PG являются такими, как описано в данной заявке.
Кроме того, согласно данному изобретению предложены твердые формы, композиции, содержащие такие твердые формы, и способы применения таких твердых форм соединения А:
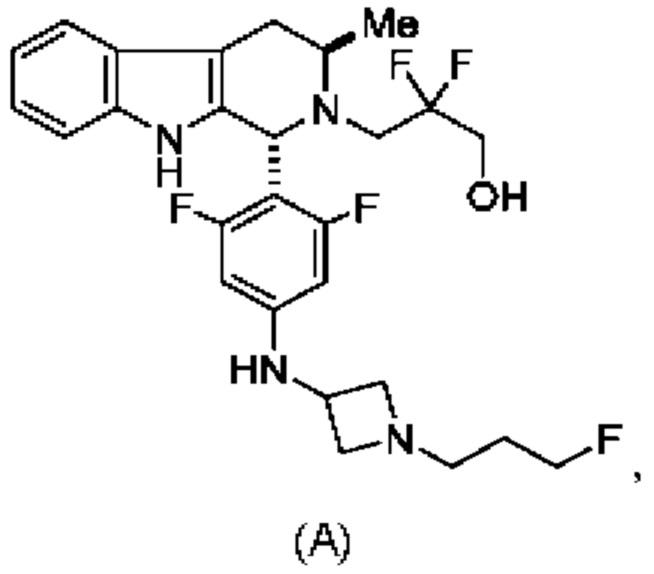
и имеющего название 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ол, включая его фармацевтически приемлемую соль.
В одном из воплощений данного изобретения предложены твердые формы соединения А. Соединение А может представлять собой свободное основание, описанное в данной заявке, существующее в аморфной твердой форме. В другом воплощении соединение А представляет собой кристаллическое вещество, описанное в данной заявке. В еще одном воплощении соединение А представляет собой кристаллическую тартратную соль, имеющую структуру:
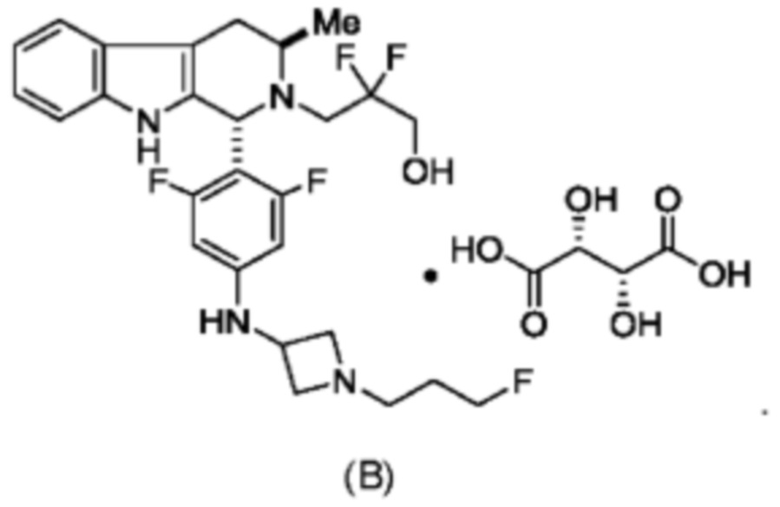
Согласно другому аспекту данного изобретения предложены кристаллические твердые формы соединения А в виде фумаратной соли, имеющей структуру:
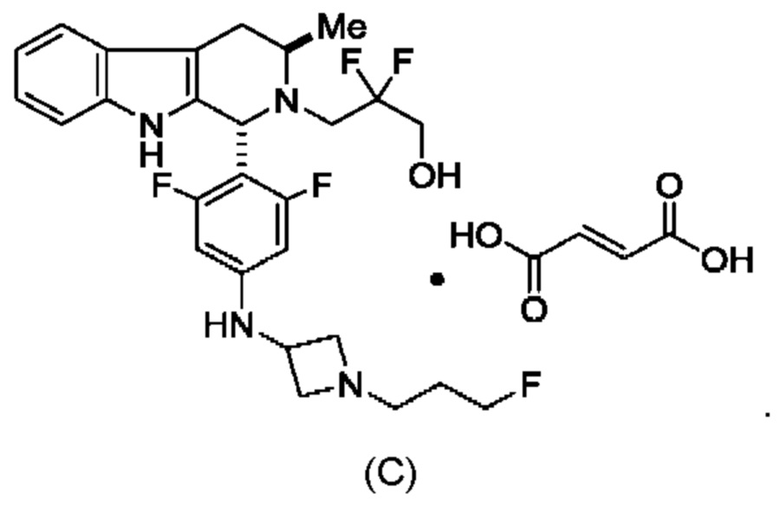
Согласно еще одному аспекту данного изобретения предложены твердые формы соединения А в виде малонатных солей, имеющих структуру:
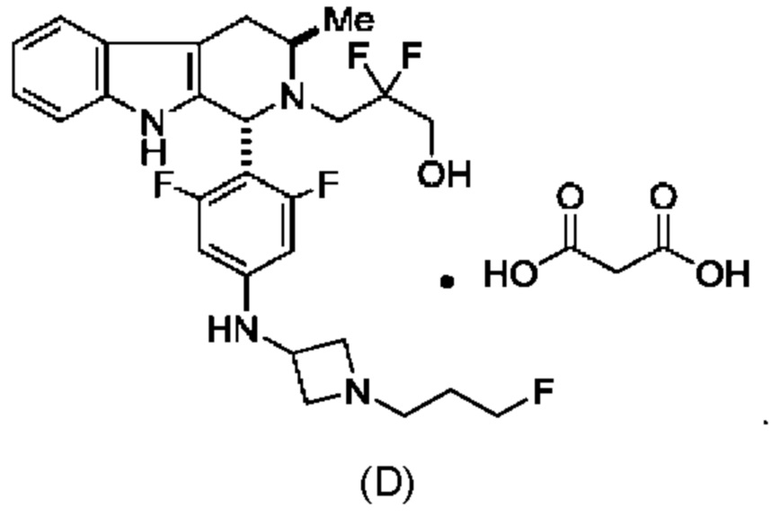
Твердые формы, описанные в данной заявке, могут быть кристаллическими. В другом воплощении твердая форма представляет собой однокомпонентную твердую форму. Твердые формы, описанные в данной заявке, могут представлять собой сольваты, гидраты, безводные формы или соли, указанные в данном описании. В одном из воплощений твердые формы, описанные в данной заявке, содержат тартратные соли. В другом воплощении твердые формы, описанные в данной заявке, содержат безводные формы соединения В. В другом воплощении твердые формы, описанные в данной заявке, содержат фумаратные соли. В еще одном воплощении твердые формы, описанные в данной заявке, содержат фосфатные соли или другие соли.
Без связи с какой-либо конкретной теорией твердые формы можно охарактеризовать по физическим свойствам, таким как, например, стабильность, растворимость и скорость растворения, плотность, сжимаемость, прочность, строение, расщепляемость, адгезивность, растворимость, поглощение влаги, электрические свойства, поведение при термическом воздействии, реакционная способность в твердом состоянии, физическая стабильность и химическая стабильность, затрагивающим конкретные процессы (например, выход, фильтрацию, промывку, сушку, измельчение, смешивание, таблетирование, текучесть, растворение, технологию приготовления композиции и лиофилизацию), которые делают определенные твердые формы подходящими для приготовления твердой лекарственной формы. Такие свойства можно определить, используя конкретные методы аналитической химии, включая методы анализа твердого состояния (например, дифракцию рентгеновских лучей, микроскопию, спектроскопию и термический анализ), описанные в данной заявке.
Твердые формы, описанные в данной заявке, в том числе солевые формы, кристаллические формы и аморфные твердые вещества, могут быть охарактеризованы рядом методов, включая, например, дифракцию рентгеновских лучей на монокристалле, дифракцию рентгеновских лучей на порошке (XRPD), микроскопию (например, сканирующую электронную микроскопию (SEM)), термический анализ (например, дифференциальную сканирующую калориметрию (DSC), динамическую сорбцию паров (DVS), термогравиметрический анализ (TGA) и микроскопию для высокотемпературных исследований), спектроскопию (например, инфракрасную спектроскопию, спектроскопию комбинационного рассеяния и твердотельного ядерного магнитного резонанса), сверхвысокоэффективную жидкостную хроматографию (UHPLC), ядерный магнитный резонанс на протонах (спектр).
Методы, применяемые для определения характеристик кристаллических форм и аморфных твердых веществ, включают, например, термогравиметрический анализ (TGA), дифференциальную сканирующую калориметрию (DSC), рентгеновскую порошковую дифрактометрию (XRPD), рентгеновскую дифрактометрию на монокристалле, колебательную спектроскопию, например, инфракрасную (ИК) спектроскопию и спектроскопию комбинационного рассеяния, спектроскопию ядерного магнитного резонанса (ЯМР) в твердом состоянии и растворе (в том числе 1Н-ЯМР и F-ЯМР), сканирующую электронную микроскопию (SEM), электронную кристаллографию и количественный анализ, анализ размера частиц (PSA), анализ площади поверхности, исследования растворимости и исследования растворения.
Чистота твердых форм, предложенных в данном изобретении, может быть определена стандартными аналитическими методами, такими как тонкослойная хроматография (TLC), электрофорез в гелях, газовая хроматография, сверхвысокоэффективная жидкостная хроматография (UHPLC) и масс-спектрометрия (MS).
Соединение В, Форма А
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма А. Форма А представляет собой кристаллическую твердую форму соединения В. В одном из воплощений Форма А представляет собой сольват соединения В с ацетоном. В одном из воплощений Форма А представляет собой кристаллическую тартратную соль сольвата соединения А с ацетоном.
В другом воплощении Форму А соединения В получают путем суспендирования в ацетоне с последующим упариванием. Форма А может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма А, представляет собой тартратную соль соединения А (т.е. соединение В) и является кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD твердой формы, предложенной в данной заявке, например, Формы А, является по существу такой, как показано на ФИГ. 1. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма А, имеет один или несколько характерных пиков XRPD при приблизительно 4,64; 8,26; 9,28; 11,18; 11,49; 11,96; 12,54; 13,77; 14,22; 14,61; 15,09; 15,56; 16,01; 17,35; 18,55; 18,84; 19,32; 19,82; 20,26; 21,34; 21,63; 21,92; 22,52; 22,97; 23,28; 23,54; 23,94; 24,81 или 25,96 ±0,1°2θ, изображенных, например, на ФИГ. 1, и приведенных в Таблице 16 данного описания. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма А, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 пиков XPRD при приблизительно 4,64; 8,26; 9,28; 11,18; 11,49; 11,96; 12,54; 13,77; 14,22; 14,61; 15,09; 15,56; 16,01; 17,35; 18,55; 18,84; 19,32; 19,82; 20,26; 21,34; 21,63; 21,92; 22,52; 22,97; 23,28; 23,54; 23,94; 24,81 или 25,96 ±0,1°2θ, изображенных, например, на ФИГ. 1, и приведенных в Таблице 16 данного описания. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма А, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать, шестнадцать, семнадцать, восемнадцать, девятнадцать, двадцать или все из характерных пиков XRPD, указанных в Таблице 16.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма А, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 12,54; 14,61; 16,01; 19,32; 20,26; 21,63; 23,28; 23,54; 23,94 или 24,81 ±0,1°2θ, изображенных, например, на ФИГ. 1, и приведенных в Таблице 16 данного описания. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма А, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 19,32; 20,26; 21,63; 23,28 или 24,81 ±0,1°2θ, изображенных, например, на ФИГ. 1, и приведенных в Таблице 16 данного описания. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма А, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 19,32; 20,26; 21,63; 23,28 или 24,81 ±0,05°2θ, изображенных, например, на ФИГ. 1, и приведенных в Таблице 16 данного описания.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма А, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 2. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 7,2% от общей массы образца до приблизительно 125°С.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма А, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 2, содержащую событие десольватации при примерно 124°С и событие плавления, имеющее начальную температуру примерно 164°С и максимальную температуру в пике примерно 171°С.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма А, имеющая изображение, полученное с применением микроскопии в поляризованном свете, изображенное на ФИГ. 3.
В еще одном воплощении Форма А является чистой. В некоторых воплощениях чистая Форма А по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях чистота Формы А составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма В
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма В. Форма В представляет собой кристаллическую твердую форму соединения В. В одном из воплощений Форма В представляет собой безводную форму соединения В. В другом воплощении Форма В представляет собой безводную форму тартратной соли соединения А.
В одном из воплощений Форму В соединения В получают путем суспендирования соединения B в этилацетате при КТ в течение примерно 24 часов. В другом воплощении Форму В соединения В получают путем суспендирования соединения B в смеси ацетон : вода (например, 90:10, 95:5, 96:4, 97:3, 99:1 об./об.) при примерно 50°С в течение примерно 6 часов. В еще одном воплощении Форму В соединения В получают путем суспендирования соединения В в этаноле.
В определенных случаях Форму В соединения В получают путем суспендирования соединения В в системе растворителей, содержащей ≥95% ацетона (например, смеси ≥95:5 ацетон : вода). Затем Форму В соединения В выделяют из суспензии, например, путем центрифугирования или фильтрации. В другом воплощении Форму В соединения В получают либо из Формы А, либо из Формы F, как описано в данной заявке. В одном из воплощений Форму А соединения В ресуспендируют в этаноле (например, 100%-ном этаноле) в течение 10, 12, 16 или 24 часов (например, в течение ночи), получая Форму В. В одном из воплощений Форма F преобразуется с образованием Формы D, как описано в данной заявке, в присутствии воды. Затем эту смесь можно подвергнуть суспендированию в неразбавленном этаноле (например, при приблизительно 50°С) либо в смеси 95:5 или 97:3 ацетон : вода (об./об.) с образованием Формы В. В смеси возможно может быть добавлена затравка кристалла Формы В.
Форма В может быть получена в соответствии со способами и примерами, описанными в данной заявке. Таким образом, согласно данному изобретению предложен способ получения Формы В, причем способ включает суспендирование соединения В в системе растворителей, содержащей ацетон или водные смеси ацетона в воде (например, 50%, 90%, 95%, 96%, 97%, 98% и 99% (об./об.) ацетона). В одном из воплощений система растворителей для кристаллизации Формы В содержит ≥95% ацетона. В одном из воплощений система растворителей для Формы В содержит смесь 96:4 ацетон : вода. Смеси можно подвергнуть суспендированию при КТ в течение примерно 120 ч. Можно выполнить фильтрование и анализ данных смесей так, как описано в данной заявке (например, по XRPD). В одном из воплощений Форму В получают в соответствии со способами и/или примерами, указанными в данном описании.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма В, представляет собой тартратную соль соединения А и является кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD твердой формы, предложенной в данной заявке, например, Формы В, является по существу такой, как показано на ФИГ. 4. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма В, имеет один или несколько характерных пиков XRPD при приблизительно 7,68; 11,49; 12,54; 14,24; 15,30; 15.55; 16,01; 16,63; 17,37; 18,24; 19,16; 19,42; 19,89; 20,24; 21,81; 22,52; 22,99; 23,25; 23,57; 24,67; 25,07; 25,91 ±0,1°2θ, изображенных, например, на ФИГ. 4, и приведенных в данном описании в Таблице 17. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма В, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XPRD при приблизительно 7;68; 11,49; 12,54; 14,24; 15,30; 15,55; 16,01; 16,63; 17,37; 18,24; 19,16; 19,42; 19,89; 20,24; 21,81; 22,52; 22,99; 23,25; 23,57; 24,67; 25,07; 25,91 ±0,1°2θ, изображенных, например, на ФИГ. 4, и приведенных в данном описании в Таблице 17. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма В, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать или все из характерных пиков XRPD, приведенных в Таблице 17.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма В, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 11,49; 12,54; 15,30; 15,55; 19,16; 19,42; 20,24; 23,25; 24,67 или 25,91 ±0,1°2θ, изображенных, например, на ФИГ. 4. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма В, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,49; 12,54; 19,16; 19,42 или 24,67 ±0,1°2θ, изображенных, например, на ФИГ. 4. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма В, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,49; 12,54; 19,16; 19,42 или 24,67 ±0,05°2θ, изображенных, например, на ФИГ. 4.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма В, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 5. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 3,5% от общей массы образца.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма В, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 5, содержащую эндотермическое превращение с начальной температурой примерно 171°С и максимальной температурой в пике примерно 179°С.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма В, имеющая спектр 13С- и 19F-ЯМР по существу такой, как изображено на ФИГ. 6 и ФИГ. 7, соответственно.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма В, имеющая профиль сорбции-десорбции воды, изображенный на ФИГ. 8. Твердая форма, например, Форма В соединения В, абсорбирует примерно 1,2% (масс./масс.) влаги вплоть до достижения относительной влажности (RH) 98% при примерно 25°С.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма В, имеющая изображение, полученное с применением сканирующей электронной микроскопии (SEM), и полученное с применением PLM изображение, изображенные на ФИГ. 9а и ФИГ. 9b, соответственно. Образец содержит плотные сферические агрегаты.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма В, имеющая распределение частиц по размерам (PSD), изображенное на ФИГ. 9с.
В еще одном воплощении твердая форма, например, Форма В, остается по существу неизмененной после сжатия, как описано в данной заявке. На ФИГ. 22, ФИГ. 23 и ФИГ. 24, соответственно, показаны результаты XRPD, 19F-ттЯМР и DSC для Формы В соединения В и выполнено сравнение соединения до и после сжатия, как описано в данной заявке.
В еще одном воплощении Форма В является по существу чистой. В некоторых воплощениях чистая Форма В по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях по существу чистая Форма В по существу не содержит Формы А, Формы D или Формы F. В некоторых воплощениях чистота Формы В составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма С
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма С. Форма С представляет собой кристаллическую твердую форму соединения В. В одном из воплощений Форма С представляет собой сольват соединения В с THF.
В одном из воплощений Форму С соединения В получают путем суспендирования соединения B в THF. Затем можно выполнить фильтрование данной смеси. Форма С может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма С, представляет собой тартратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений картина XRPD является по существу такой, как показано на ФИГ. 10.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма С, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 11. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 6,8% от общей массы образца.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма С, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 11, содержащую эндотермическое превращение с начальной температурой примерно 118°С и максимальной температурой в пике примерно 125°С.
В еще одном воплощении Форма С является чистой. В некоторых воплощениях чистота Формы С составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма D
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма D. Форма D представляет собой кристаллическую твердую форму соединения В. В одном из воплощений Форма D представляет собой гидрат соединения В. В другом воплощении Форма D представляет собой моногидрат соединения В.
В одном из воплощений Форму D соединения В получают путем суспендирования соединения B в 100%-ном этаноле в течение примерно 48 ч. Затем можно выполнить фильтрование данной смеси.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма D, представляет собой тартратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В другом воплощении Форма D представляет собой гидрат соединения В. В одном из воплощений XRPD твердой формы, предложенной в данной заявке, например, Формы D, является по существу такой, как показано на ФИГ. 12. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма D, имеет один или несколько характерных пиков XRPD при приблизительно 7,32; 10,99; 11,31; 12,18; 13,23; 13,48; 14,11; 14,66; 15,14; 15,70; 16,03; 16,21; 16,54; 17,24; 17,63; 18,11; 18,34; 19,10; 20,20; 20,58; 21,16; 21,47; 21,89; 22,76; 23,33 или 23,56 ±0,1°2θ, изображенных, например, на ФИГ. 12, и приведенных в Таблице 19. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма D, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XRPD при приблизительно 7,32; 10,99; 11,31; 12,18; 13,23; 13,48; 14,11; 14,66; 15,14; 15,70; 16,03; 16,21; 16,54; 17,24; 17,63; 18,11; 18,34; 19,10; 20,20; 20,58; 21,16; 21,47; 21,89; 22,76; 23,33 или 23,56 ±0,1°2θ, изображенных, например, на ФИГ. 12, и приведенных в Таблице 19. В еще одном воплощении твердая форма, описанная в данной заявке, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать или все из характерных пиков XRPD, указанных в Таблице 19.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма D, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 11,31; 15,70; 16,54; 19,10; 20,58; 21,16; 21,47; 21,89; 22,76 или 23,33 ±0,1°2θ, изображенных, например, на ФИГ. 12. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма D, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,31; 15,70; 16,54; 19,10 или 22,76 ±0,1°2θ, изображенных, например, на ФИГ. 12. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма D, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,31; 15,70; 16,54; 19,10 или 22,76 ±0,05°2θ, изображенных, например, на ФИГ. 12.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма D, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 13. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 1,4% от общей массы образца до приблизительно 150°С.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма D, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 13, демонстрирующую эндотермическое превращение, имеющее начальную температуру примерно 55°С и максимальную температуру в пике примерно 82°С, затем второе эндотермическое превращение с начальной температурой примерно 165°С и максимальной температурой в пике примерно 172°С.
В еще одном воплощении Форма D является чистой. В некоторых воплощениях Форма D по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях Форма D по существу не содержит Формы А, Формы В или Формы F. В некоторых воплощениях чистота Формы D составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма Е
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма Е. Форма Е представляет собой твердую форму соединения В. В одном из воплощений Форма Е представляет собой сольват соединения В с DMSO.
В одном из воплощений Форму Е соединения В получают путем суспендирования соединения B в DMSO и добавления изопропилацетата (IPAc), в течение примерно 24 часов. Затем можно провести фильтрование данной смеси. Форма Е может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма Е, представляет собой тартратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD является по существу такой, как показано на ФИГ. 14.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма Е, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 15. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 8,3% от общей массы образца.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма Е, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 15, содержащую первую эндотерму с начальной температурой примерно 126°С и максимальной температурой в пике примерно 134°С и второе эндотермическое превращение с начальной температурой примерно 143°С и максимальной температурой в пике примерно 147°С.
В еще одном воплощении Форма Е является чистой. В некоторых воплощениях Форма Е по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях чистота Форма Е составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма F
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма F. Форма F представляет собой кристаллическую твердую форму соединения В. В одном из воплощений Форма F представляет собой безводную форму соединения В. В другом воплощении Форма F представляет собой безводную форму тартратной соли соединения А.
В одном из воплощений Форму F соединения В получают путем суспендирования соединения B в 100%-ном этаноле при КТ или при 50°С в течение примерно 8, 10, 12, 15, 20 или 25 часов (например, в течение ночи). В другом воплощении Форма F может быть получена путем суспендирования соединения B в смеси этанол/вода (например, 65:35 об./об.). Суспендирование соединения В с целью получения Формы F возможно может включать внесение затравки Формы В, описанной в данной заявке. Для получения Формы F можно провести фильтрование данной суспензии. Форма F также может быть получена путем суспендирования при КТ в 100%-ной деионизированной (DI) воде, смеси 1:1 ацетон/вода или 100%-ном ацетоне. Суспензии Формы F в чистых растворителях могут быть выдержаны при КТ. В одном из воплощений Форма F может быть получена из смеси 1:1 ацетон : вода с перемешиванием при 5°С. В еще одном воплощении Форма F может быть получена путем суспендирования соединения B в смесях 95:5 ацетон : вода и 97:3 ацетон : вода при 50°С в течение примерно 2 часов и охлаждения до КТ. Форма F может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма F, представляет собой тартратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD твердой формы, предложенной в данной заявке, например, Формы F, является по существу такой, как показано на ФИГ. 16. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма F, имеет один или несколько характерных пиков XRPD при приблизительно 3,92; 10,54; 11,72; 12,52; 14,22; 15,40; 15,54; 15,90; 16,48; 16,84; 17,29; 18,26; 18,47; 19,39; 19,66; 20,00; 20,50; 20,65; 21,16; 21,28; 21,95; 22,97; 23,49; 23,70; 23,94; 24,31; 24,67 или 24,99 ±0,1°2θ, изображенных, например, на ФИГ. 16, и приведенных в данном описании в Таблице 20. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма F, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XPRD при приблизительно 3,92; 10,54; 11,72; 12,52; 14,22; 15,40; 15,54; 15,90; 16,48; 16,84; 17,29; 18,26; 18,47; 19,39; 19,66; 20,00; 20,50; 20,65; 21,16; 21,28; 21,95; 22,97; 23,49; 23,70; 23,94; 24,31; 24,67 или 24,99 ±0,1°2θ, изображенных, например, на ФИГ. 16, и приведенных в данном описании в Таблице 20. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма F, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать, двадцать, двадцать или все из характерных пиков XRPD, указанных в Таблице 20.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма F, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 12,52; 15,90; 19,66; 20,65 или 24,99 ±0,1°2θ, изображенных, например, на ФИГ. 16. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма F, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 12,52; 15,90; 19,66; 20,65 или 24,99 ±0,1°2θ, изображенных, например, на ФИГ. 16. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма F, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 12,52; 15,90; 19,66; 20,65 или 24,99 ±0,05°2θ, изображенных, например, на ФИГ. 16.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма F, имеющая изотермы кривой DVS, по существу соответствующие репрезентативным изотермам кривой DVS, изображенным на ФИГ. 17.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма F, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 18, содержащую эндотермическое превращение с начальной температурой примерно 162°С и максимальной температурой в пике примерно 167°С.
В еще одном воплощении, Форма F является чистой. В некоторых воплощениях чистая Форма F по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях чистота Формы F составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение В, Форма G
В некоторых воплощениях данного изобретения предложена твердая форма соединения В, обозначенная как Форма G. Форма G представляет собой твердую форму соединения В. В одном из воплощений Форма G представляет собой метанольный сольват соединения В. В другом воплощении Форма F представляет собой сольват с метанолом тартратной соли соединения А.
В одном из воплощений Форму G соединения В получают путем суспендирования соединения B в метаноле и медленного выпаривания метанола. Можно выполнить фильтрование данной смеси. Форма G может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма G, представляет собой тартратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD твердой формы, предложенной в данной заявке, например, Формы G, является по существу такой, как показано на ФИГ. 19. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма G, имеет один или несколько характерных пиков XRPD при приблизительно 7,65; 11,46; 12,51; 15,27; 15,51; 16,00; 17,34; 18,21; 19,11; 19,29; 19,42; 19,84; 20,23; 21,31; 21,57; 21,79; 22,49; 22,97; 23,22; 24,65; 25,04 или 25,88 ±0,1°2θ, изображенных, например, на ФИГ. 19, и приведенных в данном описании в Таблице 21. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма G, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XPRD при приблизительно 7,65; 11,46; 12,51; 15,27; 15,51; 16,00; 17,34; 18,21; 19,11; 19,29; 19,42; 19,84; 20,23; 21,31; 21,57; 21,79; 22,49; 22,97; 23,22; 24,65; 25,04 или 25,88 ±0,1°2θ, изображенных, например, на ФИГ. 19, и приведенных в данном описании в Таблице 21. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма G, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать, двадцать или все из характерных пиков XRPD, указанных в Таблице 21.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма G, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 11,46; 12,51; 15,27; 16,00; 19,29; 19,42; 20,23; 22,49; 22,97 или 24,65 ±0,1°2θ, изображенных, например, на ФИГ. 19. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма G, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,46; 12,51; 19,29; 19,42 или 20,23 ±0,1°2θ, изображенных, например, на ФИГ. 19. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма G, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 11,46; 12,51; 19,29; 19,42 или 20,23 ±0,05°2θ, изображенных, например, на ФИГ. 19.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма G, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 20. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 2% от общей массы образца.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма G, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 20, содержащую эндотермическое превращение с начальной температурой примерно 173°С и максимальной температурой в пике примерно 178°С.
В еще одном воплощении Форма G является чистой. В некоторых воплощениях чистая Форма G по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях чистая Форма G по существу не содержит Формы В, Формы D или Формы Е. В некоторых воплощениях чистота Формы G составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
Соединение С, Форма 1
В некоторых воплощениях данного изобретения предложена твердая форма соединения С, обозначенная как Форма 1. Форма 1 представляет собой кристаллическую твердую форму соединения С.
В одном из воплощений Форму 1 соединения С получают путем суспендирования соединения С в смеси изоамиловый спирт/вода в соотношении примерно 3:1 в течение примерно 1,5 часа при примерно 55°С. Жидкую часть смеси затем выпаривают в токе азота и при пониженном давлении. В другом воплощении Форму 1 соединения С получают путем суспендирования соединения С в смеси этанол/гептан в соотношении примерно 3:8 при КТ. В еще одном воплощении Форму 1 соединения С получают путем суспендирования соединения С в смеси этанол/гептан в соотношении примерно 1:1 при КТ. Форма 1 может быть получена в соответствии со способами и примерами, описанными в данной заявке.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма 1, представляет собой фумаратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD является по существу такой, как показано на ФИГ. 27а. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один или несколько характерных пиков XRPD при приблизительно 7,58; 10,59; 11,44; 11,84; 12,5; 14,44; 15,45; 15,78; 16,09; 17,55; 18,92; 19,69; 19,86; 20,23; 21,35; 22,04; 23,16; 23,89; 24,23; 24,67; 25,23 или 25,93 ±0,1°2θ, изображенных, например, на ФИГ. 27а, и приведенных в Таблице 36 данного описания. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XPRD при приблизительно 7,58; 10,59; 11,44; 11,84; 12,5; 14,44; 15,45; 15,78; 16,09; 17,55; 18,92; 19,69; 19,86; 20,23; 21,35; 22,04; 23,16; 23,89; 24,23; 24,67; 25,23 или 25,93 ±0,1°2θ, изображенных, например, на ФИГ. 27а, и приведенных в Таблице 36 данного описания. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма 1, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать, двадцать или все из характерных пиков XRPD, указанных в Таблице 36.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 10,59; 15,45; 15,78; 16,09; 18,92; 19,69; 19,86; 21,35; 23,16 или 24,23 ±0,1°2θ, изображенных, например, на ФИГ. 27а. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 16,09; 18,92; 19,69; 19,86 или 23,16 ±0,1°2θ, изображенных, например, на ФИГ. 27а. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 16,09; 18,92; 19,69; 19,86 или 23,16 ±0,05°2θ, изображенных, например, на ФИГ. 27а.
В одном из воплощений, описанных в данной заявке, предложена твердая форма, например, Форма 1, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA, изображенной на ФИГ. 28. В некоторых воплощениях кристаллическая форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 2% от общей массы образца.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма 1, имеющая по существу такую термограмму DSC, как изображено на ФИГ. 28, содержащую эндотермическое превращение с начальной температурой примерно 167°С и максимальной температурой в пике примерно 172°С.
В еще одном воплощении Форма 1 является чистой. В некоторых воплощениях чистая Форма 1 по существу не содержит других твердых форм, например, аморфного твердого вещества. В некоторых воплощениях чистота Формы 1 составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5% или не меньше, чем примерно 99,9%.
В другом воплощении, описанном в данной заявке, предложена твердая форма, например, Форма 1, имеющая полученное с применением PLM изображение, изображенное на ФИГ. 29.
Соединение С, Форма 2
В некоторых воплощениях данного изобретения предложена твердая форма соединения С, обозначенная как Форма 2. Форма 2 представляет собой кристаллическую твердую форму соединения С.
В одном из воплощений твердая форма, предложенная в данной заявке, например, Форма 2, представляет собой фумаратную соль соединения А и является по существу кристаллической, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений XRPD является по существу такой, как показано на ФИГ. 27b. В другом воплощении твердая форма, предложенная в данной заявке, например, Форма 2, имеет один или несколько характерных пиков XRPD при приблизительно 11,52; 11,87; 15,55; 16,04; 16,51; 17,32; 18,36; 19,00; 19,43; 19,87; 20,24; 21,35; 22,03; 23,23; 23,91; 25,43 или 26,03 ±0,1°2θ, изображенных, например, на ФИГ. 27b, и приведенных в Таблице 37 данного описания. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 2, имеет по меньшей мере 3, по меньшей мере 5, по меньшей мере 7 или по меньшей мере 10 характерных пиков XPRD при приблизительно 11,52; 11,87; 15,55; 16,04; 16,51; 17,32; 18,36; 19,00; 19,43; 19,87; 20,24; 21,35; 22,03; 23,23; 23,91; 25,43 или 26,03 ±0,1°2θ, изображенных, например, на ФИГ. 27b, и приведенных в Таблице 37 данного описания. В еще одном воплощении твердая форма, описанная в данной заявке, например, Форма 1, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, пятнадцать или все из характерных пиков XRPD, указанных в данном описании в Таблице 37.
В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре, пять, шесть, семь, восемь, девять или десять характерных пиков XRPD при приблизительно 11,87; 15,55; 16,04; 16,51; 17,32; 19,43; 19,87; 20,24; 23,23 или 23,91 ±0,1°2θ, изображенных, например, на ФИГ. 27b. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 15,55; 19,43; 19,87; 20,24 или 23,23 ±0,1°2θ, изображенных, например, на ФИГ. 27b. В еще одном воплощении твердая форма, предложенная в данной заявке, например, Форма 1, имеет один, два, три, четыре или пять характерных пиков XRPD при приблизительно 15,55; 19,43; 19,87; 20,24 или 23,23 ±0,05°2θ, изображенных, например, на ФИГ. 27b.
Аморфная форма соединения А в виде свободного основания
В некоторых воплощениях данного изобретения предложена аморфная твердая форма соединения А в виде свободного основания.
В одном из воплощений предложена аморфная твердая форма соединения А в виде свободного основания, как показано измерениями по картине дифракции рентгеновских лучей на порошке (XRPD). В одном из воплощений картина XRPD является по существу такой, как показано на ФИГ. 32.
В одном из воплощений, описанных в данной заявке, предложена аморфная твердая форма соединения А в виде свободного основания, имеющая термограмму TGA, по существу соответствующую репрезентативной термограмме TGA и DSC, изображенной на ФИГ. 33. В некоторых воплощениях аморфная форма демонстрирует термограмму TGA, соответствующую общей потере массы приблизительно 9,3% от общей массы образца.
В еще одном воплощении аморфная твердая форма соединения А является чистой. В некоторых воплощениях чистая аморфная твердая форма соединения А по существу не содержит других твердых форм, например, кристаллических веществ, описанных в данной заявке. В некоторых воплощениях чистота составляет не меньше, чем примерно 95%, не меньше, чем примерно 96%, не меньше, чем примерно 97%, не меньше, чем примерно 98%, не меньше, чем примерно 98,5%, не меньше, чем примерно 99%, не меньше, чем примерно 99,5%, или не меньше, чем примерно 99,9%.
Фармацевтические композиции
Соединения, описанные в данной заявке, можно вводить, например, перорально, внутримышечно, подкожно, внутривенно, интрадермально, чрескожно, интраартериально, внутрибрюшинно, в область поражения, в интракраниальное пространство, внутрь сустава, в предстательную железу, интраплеврально, интратрахеально, интратекально, интраназально, интравагинально, интраректально, местно, внутрь опухолей, перитонеально, субконъюнктивально, внутрипузырно мукозально, в перикардиальную полость, интраумбиликально, интраокулярно, внутриглазнично, интравитреально (например, путем интравитреальной инъекции), посредством глазных капель, местно, трансдермально, парентерально, посредством ингаляции, посредством инъекции, посредством имплантации, посредством инфузии, посредством непрерывной инфузии, путем непосредственного локализованного перфузионного промывания целевых клеток, с помощью катетера, посредством лаважа, в композициях в виде кремов или в липидных композициях. Соединения, описанные в данной заявке, могут быть приготовлены в составе фармацевтических композиций, предложенных в данном изобретении, подходящих для перорального введения. В другом воплощении соединение, описанное в данной заявке, можно вводить внутримышечно.
В одном из воплощений описанные в данной заявке соединения вводят в составе фармацевтических композиций, которые могут быть введены субъекту перорально или парентерально. Фармацевтические композиции на основе соединений, описанных в данной заявке, могут быть приготовлены в виде пероральных лекарственных форм, таких как, например, капсулы, микрокапсулы, таблетки (покрытые и не покрытые оболочкой таблетки), гранулы, порошки, пилюли или суппозитории. На основе соединений, описанных в данной заявке, могут быть приготовлены композиции для местного или парентерального применения, при этом соединение растворяют или иным образом суспендируют в растворе, подходящем для инъекций, суспензий, сиропов, кремов, мазей, гелей, спреев, растворов и эмульсий.
Фармацевтические композиции, описанные в данной заявке, включают один или более чем один фармацевтически приемлемый эксципиент, такой как, но не ограничиваясь этим: сахароза, крахмал, маннит, сорбит, лактоза, глюкоза, целлюлоза, тальк, фосфат кальция или карбонат кальция, целлюлоза, метил целлюлоза, гидроксиметилцеллюлоза, гидроксипропилцеллюлоза, карбоксиметил целлюлоза, гидроксипропил крахмал, полипропилпирролидон, поливинилпирролидон, желатин, аравийская камедь, полиэтиленгликоль (ПЭГ), крахмал, бикарбонат натрия, цитрат кальция, стеарат магния, лаурилсульфат натрия, бензоат натрия, бисульфит натрия, метилпарабен, пропилпарабен, лимонная кислота, цитрат натрия или уксусная кислота, поливинилпирролидон, стеарат алюминия, вода и масло какао. Использование таких эксципиентов в качестве, например, разбавителей, связующих веществ, смазывающих веществ и разрыхлителей, хорошо известно в данной области техники.
Фармацевтические композиции, описанные в данной заявке, включают в себя эффективное количество соединения, описанного в данной заявке (например, соединения А, соединения В, соединения С, соединения D или их твердой формы). Доза соединения, описанного в данной заявке, может быть мерой конкретного количества соединения (например, количества стандартной дозы) или может быть измерена как функция, например, массы тела пациента. В одном из воплощений описанное в данной заявке соединение вводят в количестве, эквивалентном примерно 0,1; 0,5; 0,75; 1; 2; 3; 4; 5; 10; 15; 20; 30; 50; 75; 100; 200 или 250 мг/кг. В другом воплощении описанное в данной заявке соединение вводят в количестве от примерно 0,1 мг/кг до примерно 1 мг/кг; от примерно 0,5 мг/кг до примерно 2 мг/кг; от примерно 1 мг/кг до примерно 5 мг/кг; от примерно 3 мг/кг до примерно 10 мг/кг; от примерно 8 мг/кг до примерно 15 мг/кг; или от примерно 15 мг/кг до примерно 30 мг/кг. В еще одном воплощении описанное в данной заявке соединение вводят в количестве меньше примерно 100 мг/кг, меньше примерно 50 мг/кг, меньше примерно 30 мг/кг, меньше примерно 10 мг/кг или меньше примерно 1 мг/кг.
В одном из воплощений описанное в данной заявке соединение вводят в количестве примерно 1, 5, 10, 20, 25, 30, 50, 60, 75, 90, 100, 120, 150 или 250 мг. В другом воплощении описанное в данной заявке соединение вводят в количестве в количестве примерно 10 мг. В еще одном воплощении описанное в данной заявке соединение вводят в количестве примерно 30 мг. В еще одном воплощении описанное в данной заявке соединение вводят в количестве примерно 90 мг. В одном из воплощений фармацевтическую композицию, содержащую описанное в данной заявке соединение, вводят в установленном выше количестве один раз в сутки (QD). Соединение может представлять собой соединение В или его твердую форму (например, Форму А, Форму В, Форму С, Форму D, Форму Е, Форму F или Форму G). В другом воплощении соединение представляет собой твердую форму соединения В (например, Форму В, Форму D или Форму F). В одном из воплощений соединение представляет собой соединение С либо Форму 1 или Форму 2 соединения С. В другом воплощении соединение представляет собой соединение D или твердую форму, описанные в данной заявке.
В другом воплощении описанное в данной заявке соединение вводят в количестве от примерно 1 мг до примерно 10 мг; от примерно 10 мг до примерно 30 мг; от примерно 10 мг до примерно 90 мг; от примерно 30 мг до примерно 90 мг; или от примерно 90 мг до примерно 250 мг. В одном из воплощений вводимое соединение представляет собой соединение B в количестве примерно 1,10, 30, 50, 90, 100 или 150 мг. Дозы соединения, описанного в данной заявке, могут быть представлены в виде однократной дозы (например, единственной таблетки или капсулы с заданным дозировочным количеством или могут быть представлены в виде многократных доз, принимаемых в течение некоторого периода времени (например, 2-х или более таблеток или капсул, равных данному дозировочному количеству). Соединение может представлять собой соединение В или его твердую форму (например, Форму А, Форму В, Форму С, Форму D, Форму Е, Форму F или Форму G). В другом воплощении соединение представляет собой твердую форму соединения В (например, Форму В, Форму D или Форму F). В одном из воплощений соединение представляет собой соединение С либо Форму 1 или Форму 2 соединения С. В другом воплощении соединение представляет собой соединение D или твердую форму, описанные в данной заявке.
Фармацевтические композиции, описанные в данной заявке, можно вводить один раз в сутки (QD); два раза в сутки (BID), три раза в сутки (TID), один раз каждые двое суток (Q2D), каждые трое суток (Q3D) или один раз в неделю. Кроме того, дозы фармацевтических композиций, предложенных в данном изобретении, содержащих соединение, описанное в данной заявке, можно вводить до приема пищи (ас (от лат. ante cibum)), после приема пищи (рс (от лат. post cibum)) или с пищей. В одном из воплощений описанное в данной заявке соединение вводят QD в течение периода лечения (периода времени, в течение которого пациенту, описанному в данной заявке, вводят лекарственное средство) с последующим периодом покоя (периодом времени, в течение которого пациенту, описанному в данной заявке, лекарственное средство не вводят). В периоды покоя можно проводить введение противораковых средств, отличающихся от соединения, описанного в данной заявке. В одном из воплощений на основе описанного в данной заявке соединения готовят композицию для перорального введения, предложенную в данном изобретении, и вводят QD в течение 20-28 суток с последующим 3-10-дневным периодом покоя. В другом воплощении соединение вводят QD без какого-либо периода покоя.
Предпочтительно, на основе описанного в данной заявке соединения готовят композицию для перорального введения. Пероральное введение может способствовать соблюдению больным режима и схемы приема соединения (например, в случае приготовления в виде фармацевтической композиции), тем самым повышая приверженность режиму терапии и ее эффективность. Пероральные фармацевтические композиции, содержащие соединение, описанное в данной заявке, включают, но не ограничиваются этим, таблетки (например, имеющие покрытие, не имеющие покрытия и жевательные) и капсулы (например, твердые желатиновые капсулы, мягкие желатиновые капсулы, капсулы с энтеросолюбильным покрытием и капсулы с длительным высвобождением). Таблетки могут быть приготовлены путем прямого прессования, путем влажного гранулирования или путем сухого гранулирования. Пероральные фармацевтические композиции, содержащие соединение, описанное в данной заявке, могут быть приготовлены, что очевидно в данной области техники, для достижения задержанного или пролонгированного высвобождения. В одном из воплощений на основе соединения В или твердой формы, описанных в данной заявке (например, Формы В, Формы D или Формы F) готовят композиции в виде таблетки или капсулы для перорального введения в количестве, указанном в данном описании.
Кроме того, согласно данному изобретению предложены соединения, имеющие формулы:
Соединения М1, М2, М3 и М4 можно считать метаболитами и/или продуктами деградации соединения В, в том числе твердых форм, описанных в данной заявке. В определенных случаях такие соединения можно обнаружить в композициях, описанных в данной заявке, причем хранение таких композиций осуществляли в течение заданного периода времени при относительной влажности (RH) примерно 50%, 55%, 60%, 65%, 70%, 75%, 80% или выше. Такие соединения также можно обнаружить при повышенных температурах, составляющих примерно 30°С, 35°С, 40°С, 45°С или примерно 50°С. В одном из воплощений соединение М1, М2, М3 или М4 обнаруживают в композиции, описанной в данной заявке, причем данная композиция содержит менее чем примерно 30 мг соединения В или его твердой формы. В определенных случаях такие соединения обнаруживают в композиции, причем такая композиция представлена в виде таблетки без покрытия, описанной в данной заявке.
В некоторых воплощениях композиции, описанные в данной заявке (например, Форма А, Форма В, Форма С, Форма D, Форма Е, Форма F или Форма G), содержат менее 0,01%; 0,02%; 0,03%; 0,04; 0,05; 0,1; 0,15; 0,75; 0,2; 0,225; 0,25; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9 или 1% (масс/масс.) одного или более чем одного из М1, М2, М3 или М4. В одном из воплощений композиция, описанная в данной заявке, содержит менее чем примерно 0,5% (масс/масс.) одного или более чем одного из М1, М2, М3 или М4.
Способы лечения рака
Соединения и твердые формы, описанные в данной заявке, можно вводить в эффективном количестве (например, в количестве, описанном в данной заявке) для лечения рака. Следует понимать, что способы, изложенные в данном описании, также включают лечение фармацевтической композицией, описанной в данной заявке, содержащей соединение (например, соединение В или его твердую форму), описанное в данной заявке, и один или более чем один фармацевтически приемлемый эксципиент.
В одном из воплощений данного изобретения предложен способ лечения рака путем введения эффективного количества соединения В, описанного в данной заявке, пациенту, имеющему рак. В одном из воплощений соединение В представляет собой твердую форму, описанную в данной заявке (например, Форму А, Форму В, Форму С, Форму D, Форму Е, Форму F или Форму G).
Согласно другому аспекту предложенную в данном изобретении Форму А соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму В соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму С соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму D соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму Е соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму F соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак, как указано в данном описании.
Согласно другому аспекту предложенное в данном изобретении Форму G соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно другому аспекту предложенную в данном изобретении аморфную некристаллическую форму соединения А или соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно другому аспекту данного изобретения предложен способ лечения рака легкого, рака яичников, рака эндометрия, рака предстательной железы, рака матки или рака молочной железы путем введения эффективного количества соединения В или твердой формы, описанной в данной заявке, пациенту, имеющему указанный рак. В одном из воплощений рак представляет собой рак яичников или рак эндометрия. В одном из воплощений рак представляет собой рак молочной железы. В одном из воплощений таких способов соединение В представляет собой твердую форму, описанную в данной заявке (например, Форму А, Форму В, Форму С, Форму D, Форму Е, Форму F или Форму G).
Согласно следующему аспекту Форму А соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму В соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму С соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму D соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму Е соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму F соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту Форму G соединения В можно вводить так, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Согласно следующему аспекту аморфную некристаллическую форму соединения А или соединения В можно вводить, как описано в данной заявке, для лечения пациента, имеющего рак легкого, рак яичников, рак эндометрия, рак предстательной железы, рак матки или рак молочной железы, как указано в данном описании.
Кроме того, согласно данному изобретению предложены способы лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы соединения В, как описано в данной заявке. В одном из воплощений предложен способ лечения рака молочной железы у такого пациента путем введения эффективного количества твердой формы соединения В, как описано в данной заявке. В одном из воплощений способ включает лечение рака молочной железы у пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы В соединения В, как описано в данной заявке. В одном из воплощений способ включает лечение рака молочной железы у пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества чистого соединения В (например, по существу не содержащего другой твердой формы, описанной в данной заявке, например, по существу не содержащего Формы D и/или Формы F). Согласно другому аспекту способ включает лечение рака молочной железы у пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы D соединения В, как описано в данной заявке. Согласно другому аспекту способ включает лечение рака молочной железы у пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы F соединения В, как описано в данной заявке. Согласно еще одному аспекту способ включает лечение рака молочной железы пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы А соединения В, как описано в данной заявке. Согласно еще одному аспекту способ включает лечение рака молочной железы у пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы С соединения В, как описано в данной заявке. Согласно еще одному аспекту способ включает лечение рака молочной железы пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы Е соединения В, как описано в данной заявке. Согласно еще одному аспекту способ включает лечение рака молочной железы пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества Формы G соединения В, как описано в данной заявке. Согласно еще одному аспекту способ включает лечение рака молочной железы пациента, имеющего рак молочной железы, путем введения пациенту эффективного количества аморфной некристаллической формы соединения В, как описано в данной заявке.
Соединения, описанные в данной заявке (например, соединение В или его твердую форму, описанные в данной заявке) можно использовать при изготовлении лекарственного средства для применения в лечении рака молочной железы, как описано в данной заявке.
Способы лечения рака молочной железы, предложенные в данном изобретении, включают лечение, при котором рак молочной железы может представлять собой позитивный по рецепторам гормонов рак молочной железы (например, ER+ рак молочной железы), HER2-позитивный рак молочной железы, HER2-негативный рак молочной железы или тройной негативный рак молочной железы (TNBC).
В одном из воплощений рак молочной железы представляет собой HER2-негативный рак молочной железы. HER2-негативный рак молочной железы может быть определен в данном описании, например, баллом в иммуногистохимическом (IHC) исследовании HER2, равным 0 или 1+, либо IHC баллом 2+ с сопутствующими отрицательными результатами тестирования гибридизации in situ с флуоресцентным, хромогенным окрашиванием или с окрашиванием серебром, указывающими на отсутствие амплификации HER2-гена, либо соотношением HER2/CEP17<2,0, либо по результатам местных клинических протоколов. В одном из воплощений рак молочной железы представляет собой ER+/HER2- рак молочной железы. Развитие рака молочной железы может быть определено стадией 0, I, II, III или IV, как известно в данной области техники.
В другом воплощении рак молочной железы представляет собой локально распространенный или метастазирующий рак молочной железы (mBC).
В одном из воплощений соединение В или его твердую форму (например, Форму В, Форму D или Форму F) можно вводить в качестве компонента адъювантной терапии. В другом воплощении соединение В или его твердую форму (например, Форму В, Форму D или Форму F) можно вводить в качестве компонента неоадъювантной терапии.
До лечения соединением или твердой формой, как описано в данной заявке, пациенты с раком молочной железы, описанные в данной заявке, могут находиться в пременопаузе. До лечения соединением или твердой формой, как описано в данной заявке, пациенты с раком молочной железы, описанные в данной заявке, могут находиться в постменопаузе.
Способы, предложенные в данном изобретении, включают введение эффективного количества соединения В или твердой формы, описанных в данной заявке, пациенту в количестве, указанном в данном описании. Эффективное количество может составлять, например, количество приблизительно равное 10 мг, 30 мг, 50 мг, 90 мг, 100 мг, 125 мг или 250 мг. В одном из воплощений способов, предложенных в данном изобретении, соединение В или твердую форму, описанные в данной заявке, вводят перорально. В одном из воплощений соединение В или его твердую форму вводят в виде таблетки (например, таблетки, покрытой или не покрытой оболочкой). В другом воплощении соединение В или его твердую форму вводят в виде капсулы. Таким образом, согласно данному изобретению предложены композиции, подходящие для введения пациенту с раком молочной железы, при этом такие композиции содержат соединение В или твердую форму, описанные в данной заявке, в количестве примерно 10 мг, 30 мг, 50 мг, 90 мг, 100 мг, 125 мг или 250 мг в виде таблетки или капсулы, как указано в данном описании. При введении в соответствии со способами, предложенными в данном изобретении, соединение В или его твердая форма могут быть чистыми, как описано в данной заявке.
Пациенты, к которым применимы описанные выше способы, могли ранее получать лечение с использованием одного или более противораковых средств или лучевой терапии. Например, в одном из воплощений пациент мог ранее получать лечение (например, терапию 1-ой, 2-ой, 3-ей или последующих линий) доксорубицином, ПЭГилированным липосомальным доксорубицином, эпирубицином, паклитакселом, стабилизированным альбумином паклитакселом, доцетакселом, 5-фторурацилом, циклофосфамидом, цисплатином, карбоплатином, винорелбином, капецитабином, гемцитабином, иксабепилоном, эрибулином, олапарибом, метотрексатом, анастрозолом, экземестаном, торемифеном, летрозолом, тамоксифеном, 4-гидрокситамоксифеном, ралоксифеном, дролоксифеном, триоксифеном, кеоксифеном, флутамидом, нилутамидом, бикалутамидом, лапатинибом, винбластином, гозерелином, лейпролидом, пэгфилграстимом, филграстимом или венетоклаксом.
В другом воплощении пациент мог ранее получать лечение (например, терапию 1-ой, 2-ой, 3-ей или последующих линий) ингибитором протеинкиназы В (AKT), ингибитором циклинзависимой киназы 4/6 (CDK4/6), ингибитором поли(АДФ-рибоза)-полимеразы (PARP) или ингибитором ароматазы. В одном из воплощений ингибитором AKT является ипатасертиб (GDC-0068). В одном из воплощений ингибитором CDK4/6 является абемацикпиб, рибоциклиб или палбоцикпиб. В определенных случаях пациент мог ранее получать лечение: (1) абемацикпибом, рибоциклибом или палбоциклибом; (2) ипатасертибом; (3) эверолимусом или фулвестрантом; (4) трастузумаба эмтанзином, трастузумабом, пертузумабом или атезолизумабом; либо (5) алемтузумабом, бевацизумабом, цетуксимабом, панитумумабом, ритуксимабом, тозитумомабом или их комбинацией. Пациенты, описанные в данной заявке, могли перенести хирургическую операцию до лечения соединением В или его твердой формой.
В другом воплощении описываемый в данной заявке пациент может плохо поддаваться лечению одним или более чем одним видом противораковой терапии.
Например, описываемый в данной заявке пациент может плохо поддаваться лечению ингибиторами ароматазы. В другом примере описываемый в данной заявке пациент может плохо поддаваться лечению селективным супрессором рецепторов эстрогенов (SERD), таким как, например, фулвестрант. В еще одном примере пациент может плохо поддаваться лечению одним или более чем одним препаратом для эндокринной терапии, таким как кломифен, торемифен, ралоксифен, анордрин, базедоксифен, бропарестрол, циклофенил, лазофоксифен, ормелоксифен, аколбифен, элацестрант, бриланестрант, кломифеноксид, дролоксифен, этакстил или оспемифен. В другом воплощении пациент может плохо поддаваться лечению абемациклибом, анастрозолом, экземестаном, фулвестрантом, гозерелином, летрозолом, лейпрорелином, мегестролом, палбоциклибом, тамоксифеном или торемифеном. В другом примере пациент может плохо поддаваться лечению трастузумаба эмтанзином, трастузумабом, пертузумабом, атезолизумабом, пембролизумабом, дурвалумабом, авелумабом или ниволумабом.
Соединения, описанные в данной заявке, также могут быть использованы в способах, включающих ингибирование ER-альфа у пациента. Такие способы включают введение количества соединения, описанного в данной заявке, (например, соединения А или соединения В, включая их твердые формы, описанные в данной заявке) пациенту.
Комбинированные терапии
Соединения и твердые формы, описанные в данной заявке, можно вводить в комбинации с одним или более чем одним противораковым средством. Введение "в комбинации", как указано в данном описании, включает последовательное введение (в любом порядке) соединения, описанного в данной заявке, и одного или более чем одного противоракового терапевтического средства, а также одновременное введение. Соответственно, согласно данному изобретению предложены способы лечения рака молочной железы у пациента, имеющего рак молочной железы, причем такие способы включают введение соединения В или твердой формы, описанных в данной заявке, в комбинации с одним или более чем одним дополнительным противораковым терапевтическим средством. В одном из воплощений противораковая терапия включает доксорубицин, ПЭГилированный липосомальный доксорубицин, эпирубицин, пакпитаксел, стабилизированный альбумином пакпитаксел, доцетаксел, 5-фторурацил, циклофосфамид, цисплатин, карбоплатин, винорелбин, капецитабин, гемцитабин, иксабепилон, эрибулин, олапариб, метотрексат, анастрозол, экземестан, торемифен, летрозол, тамоксифен, 4-гидрокситамоксифен, ралоксифен, дролоксифен, триоксифен, кеоксифен, флутамид, нилутамид, бикалутамид, лапатиниб, винбластин, гозерелин, лейпролид, пэгфилграстим, филграстим или венетокпакс.
В одном из воплощений данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с доксорубицином, ПЭГилированным липосомальным доксорубицином, эпирубицином, пакпитакселом, стабилизированным альбумином пакпитакселом, доцетакселом, 5-фторурацилом, циклофосфамидом, цисплатином, карбоплатином, винорелбином, капецитабином, гемцитабином, иксабепилоном, эрибулином, олапарибом, метотрексатом, анастрозолом, экземестаном, торемифеном, летрозолом, тамоксифеном, 4-гидрокситамоксифеном, ралоксифеном, дролоксифеном, триоксифеном, кеоксифеном, флутамидом, нилутамидом, бикалутамидом, лапатинибом, винбластином, гозерелином, лейпролидом, пэгфилграстимом, филграстимом или венетоклаксом.
Согласно другому аспекту данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с пакпитакселом, стабилизированным альбумином пакпитакселом, метотрексатом, анастрозолом, эксеместаном, торемифеном, летрозолом, тамоксифеном, 4-гидрокситамоксифеном, ралоксифеном, дролоксифеном, триоксифеном, кеоксифеном или венетоклаксом. Согласно еще одному аспекту данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с фулвестрантом, паклитакселом, стабилизированным альбумином пакпитакселом, кломифеном, торемифеном, ралоксифеном, анордрином, базедоксифеном, бропарестролом, циклофенилом, лазофоксифеном, ормелоксифеном, аколбифеном, элацестрантом, брилан бриланестрантом, кломифеноксидом, дролоксифеном, этакстилом или оспемифеном.
Согласно еще одному аспекту данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с ингибитором CDK4/6, ингибитором PARP или ингибитором ароматазы.
Согласно следующему аспекту данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, как описано в данной заявке, причем способ включает введение эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с ингибитором CDK4/6, при этом ингибитор CDK4/6 представляет собой абемациклиб, рибоциклиб или палбоциклиб. В предпочтительном воплощении способ включает введение соединения В или твердой формы, описанных в данной заявке, в комбинации с палбоциклибом. В еще одном воплощении способ включает введение соединения В или твердой формы, описанных в данной заявке, в комбинации с абемацикпибом или рибоциклибом. Согласно другому аспекту данного изобретения предложен набор, содержащий (1) соединение В или его твердую форму в стандартной лекарственной форме; (2) ингибитор CDK4/6 (например, палбоцикпиб) во второй стандартной лекарственной форме; и контейнер, содержащий каждую из лекарственных форм.
Доза абемациклиба может составлять от 50 мг до 500 мг в сутки или от 150 мг до 450 мг в сутки, и такое введение может быть осуществлено по схеме ежесуточно в 28-суточных циклах или по схеме меньше, чем 28 суток, из расчета на 28-суточные циклы, как например, 21 сутки из расчета на 28-суточный цикл, или 14 суток из расчета на 28-суточный цикл, или 7 суток из расчета на 28-суточные циклы. В одном из воплощений абемациклиб вводят один раз в сутки или предпочтительно по схеме BID, при этом введение осуществляют пероральным путем. В случае введения по схеме BID дозы могут разделены интервалом продолжительностью 4 часа, 8 часов или 12 часов. В некоторых воплощениях абемациклиб вводят BID перорально в дозе 150 мг, при этом каждую дозу вводят с интервалом примерно 12 ч. В некоторых воплощениях дозу абемацикпиба вводят в соответствии с инструкцией по применению.
Доза рибоцикпиба может составлять от 200 мг до 1000 мг в сутки или от 250 мг до 750 мг в сутки, и такое введение может быть осуществлено по схеме ежесуточно в 28-суточных циклах или по схеме меньше, чем 28 суток, из расчета на 28-суточные циклы, как например, 21 сутки из расчета на 28-суточный цикл, или 14 суток из расчета на 28-суточный цикл, или 7 суток из расчета на 28-суточные циклы. В одном из воплощений рибоциклиб вводят один раз в сутки, при этом введение осуществляют пероральным путем. В некоторых воплощениях дозу рибоциклиба вводят в соответствии с инструкцией по применению.
Доза палбоциклиба может составлять от 25 мг до 250 мг в сутки, или от 50 мг до 125 мг в сутки, или от 75 мг до 125 мг в сутки, или от 75 мг в сутки до 100 мг в сутки либо 125 мг в сутки. Такое введение может быть осуществлено по схеме ежесуточно в 28-суточных циклах или по схеме меньше, чем 28 суток, из расчета на 28-суточные циклы, как например, 21 сутки из расчета на 28-суточный цикл, или 14 суток из расчета на 28-суточный цикл, или 7 суток из расчета на 28-суточные циклы. В одном из воплощений палбоцикпиб вводят один раз в сутки, при этом введение осуществляют пероральным путем. В некоторых воплощениях дозу палбоциклиба вводят в соответствии с инструкцией по применению.
Согласно другому аспекту, предложенные в данном изобретении способы, изложенные в данном описании, включают введение эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с ингибитором ароматазы (AI), при этом AI представляет собой летрозол, анастрозол, экземестан или тестолактон.
Согласно еще одному аспекту данного изобретения предложен способ лечения рака молочной железы у пациента, имеющего рак молочной железы, путем введения эффективного количества соединения В или твердой формы, описанных в данной заявке, в комбинации с противораковым иммунотерапевтическим средством (например, антителом). В одном из воплощений соединение В или твердую форму, описанные в данной заявке, вводят в комбинации с трастузумаба эмтанзином, трастузумабом, пертузумабом, атезолизумабом, пембролизумабом, дурвалумабом, авелумабом или ниволумабом либо их комбинацией. В одном из воплощений соединение В или твердую форму, описанные в данной заявке, вводят в комбинации с противораковым иммунотерапевтическим средством, содержащим ингибитор белка 1 программируемой клеточной смерти (PD-1) или лиганда PD-1 (PD-L1), при этом противораковое иммунотерапевтическое средство представляет собой атезолизумаб, пембролизумаб или ниволумаб.
Введение соединения, описанного в данной заявке (например, соединения В или твердой формы, описанных в данной заявке) приводит к возникновению у пациентов неблагоприятных эффектов (АЕ), характеризующихся как имеющие степень 2 или ниже. В одном из воплощений пациент, которому вводили соединение В или твердую форму, описанные в данной заявке, имеет АЕ со степенью 2 или ниже.
Примеры
Следующие далее примеры представлены для иллюстрации, а не для ограничения.
В данной заявке описан синтез соединений. Все реагенты и растворители приобретали у коммерческих поставщиков и использовали без какой-либо дополнительной очистки. Использовали безводный растворитель (дихлорметан). Дополнительную очистку имеющихся в продаже растворителей не проводили.
Все реакции проводили во флаконах с завинчивающимися крышками, оснащенных тефлоновыми прокладками, в атмосфере азота.
Колоночную флэш-хроматографию проводили, используя прибор Teledyne Isco CombiFlash(R) Rf, оснащенный предварительно упакованными диоксидом кремния картриджами RediSepRf Gold.
Если не указано иное, опубликованные значения выхода приведены для выделенного вещества и скорректированы на предмет присутствия оставшихся растворителей.
Соединения характеризовали по результатам одного или более анализов: 1H ЯМР, 13С ЯМР, точек плавления и HRMS (масс-спектрометрия высокого разрешения) и HPLC (например, для подтверждения чистоты).
Спектры 1H- и 13С-ядерного магнитного резонанса регистрировали на приборе Bruker 400 МГц при температуре окружающей среды. Все 1H ЯМР-спектры измеряли в числе частей на миллион (млн-1) относительно сигнала остаточного хлороформа (7.26 млн-1) или диметилсульфоксида (2.50 млн-1) в дейтерированном растворителе, если не указано иное. Данные для 1H ЯМР представлены следующим: химический сдвиг, мультиплетность (br = уширенный сигнал, перекрыв. = перекрывание, s = синглет, d = дублет, t = триплет, q = квартет, р = пентет, m = мультиплет), константы спин-спинового взаимодействия и интегрирование. Все 13С ЯМР спектры приведены в млн-1 относительно дейтерохлороформа (77.06 млн-1) или дейтерированного диметилсульфоксида (39.53 млн-1) и получены с полным расщеплением 1H-сигнала, если не указано иное. HPLC-анализы проводили на системе для HPLC Agilent 1260 Infinity с ультрафиолетовым (УФ) детектором при 220 нм, используя колонку Асе Super C18. Точки плавления получали, используя устройство для определения точек плавления В-540 от Buchi, и не корректировали. Данные масс-спектрометрии высокого разрешения (HRMS) получали на масс-спектрометре Thermo Scientific Orbitrap Fusion.
Примеры 1-3. Алкилирование индолов
Алкилирование индолов осуществляли согласно последовательности, состоящей из: введения защитной группы Boc (пример 1), образования сульфамида (пример 2) и алкилирования индола (пример 3).
Пример 1. Введение защитной группы Boc
Пример 1. Общая реакционная схема для введения защитной группы Boc Введение защитной группы Boc осуществляли согласно приведенной далее общей реакционной схеме, где ra и rb соответствуют различным функциональным группам в реакциях введения защитных групп в приведенном далее примере 1, и где звездочки представляют хиральный центр:
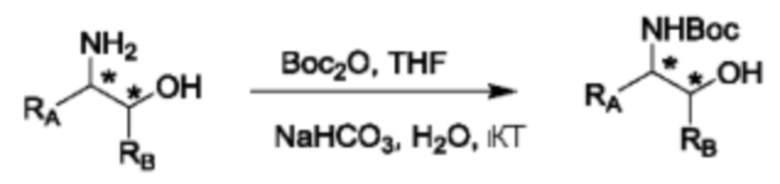
Пример 1А. Получение трет-бутил-(S)-(2-гидрокси-1-(4-метоксифенил)этил)карбамата
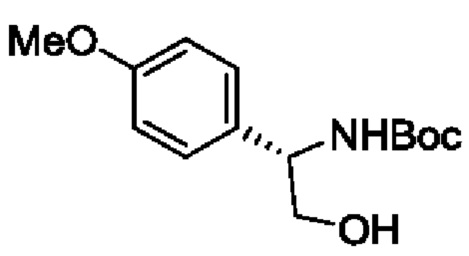
Методику, приведенную выше на общей реакционной схеме 1, выполняли так, как описано ниже. К суспензии (S)-2-амино-2-(4-метоксифенил)этан-1-ола гидрохлорида (1,04 г; 5,10 ммоль; 100 моль%) в THF (4,4 мл) добавляли Boc2O (1,21 мл; 5,61 ммоль; 110 моль%), NaHCO3 (451 мг; 5,10 ммоль; 100 моль%) и воду (4,4 мл) при KT. Раствор перемешивали при KT в течение 18 ч и экстрагировали iPrOAc (20 мл × 2). Органический слой промывали насыщенным рассолом (20 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, получая продукт без дополнительной очистки. Приведенное значение для выхода соединения из примера 1А скорректировано с учетом остаточного растворителя по данным 1H ЯМР. В результате реакции получали трет-бутил-(S)-(2-гидрокси-1-(4-метоксифенил)этил)карбамат (1,36 г; выход 100%) в виде белого твердого вещества. Т.пл.: 139,0-139,9°С; FTIR (инфракрасная (спектроскопия) с преобразованием Фурье) (для чистого вещества, см-1) 3370, 2984, 2837, 1681, 1613, 1512, 1461; 1H ЯМР (400 МГц, CDCl3): δ 7.24-7.20 (m, 2H), 6.92-6.86 (m, 2H), 5.10 (d, J=7,2 Гц, 1H), 4.72 (br, 1H), 3.82 (t, J=5,6 Гц, 2H), 3.80 (s, 3H), 2.35 (br, 1H), 1.43 (s, 9Н); 13С ЯМР (100 МГц, CDCl3): δ 159.1, 156.2, 131.6, 127.7, 114.2, 80.0, 66.8, 56.4, 55.3, 28.4.
Пример 1В. Получение трет-бутил-(R)-(1-циклопропил-2-гидроксиэтил)карбамата
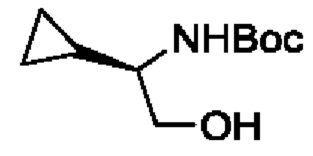
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 1А, выполняли с использованием (R)-2-амино-2-цикпопропилэтан-1-ола (1,16 г; 11,5 ммоль; 100 моль%), получая трет-бутил-(R)-(1-циклопропил-2-гидроксиэтил)карбамат (2,31 г; выход 100%) в виде белого твердого вещества. Т.пл.: 70,0-70,8°С; FTIR (для чистого вещества, см-1) 3358, 2974, 2937, 1682, 1521, 1366; 1H ЯМР (400 МГц, CDCl3): δ 4.80 (br, 1H), 3.80 (ddd, J=10,8; 6,8; 3,2 Гц, 1Н), 3.67 (ddd, J=10,8; 6,0; 4,8 Гц, 1H), 2.94 (dtd, J=9,6; 6,4; 3,2 Гц, 1H), 2.81 (br, 1H), 1.45 (s, 9Н), 0.85 (dtt, J=9,6; 8,0; 4,8 Гц, 1H), 0.60-0.47 (m, 2H), 0.44-0.25 (m, 2H); 13С ЯМР (100 МГц, CDCl3): δ 156.6, 79.7, 66.3, 57.9, 28.4, 13.0, 3.3, 2.9.
Пример 1C. Получение трет-бутил-((1R,2S)-2-гидрокси-2,3-дигидро-1Н-инден-1-ил)карбамата
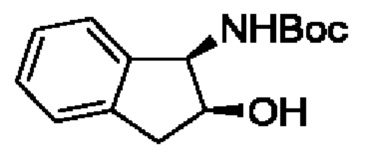
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 1А, выполняли с использованием (1R,2S)-1-амино-2,3-дигидро-1Н-инден-2-ола (5,15 г; 34,5 ммоль; 100 моль%), получая трет-бутил-((1R,2S)-2-гидрокси-2,3-дигидро-1Н-инден-1-ил)карбамат (8,61 г; выход 100%) в виде белого твердого вещества. Т.пл.: 67,3-68,4°С; FTIR (для чистого вещества, см-1) 3428, 3350, 2983, 2933, 1688, 1509; 1H ЯМР (400 МГц, CDCl3): δ 7.31-7.26 (m, 1H), 7.26-7.18 (m, 3H), 5.17 (br, 1H), 5.05 (br, 1H), 4.57 (ddd, J=7,2; 4,8; 2,0 Гц, 1Н), 3.12 (dd, J=16,8; 5,2 Гц, 1H), 2.91 (dd, J=16,8; 2,4 Гц, 1H), 2.31 (d, J=4,8 Гц, 1H), 1.50 (s, 9Н); 13С ЯМР (100 МГц, CDCl3): δ 156.3, 140.9, 139.9, 128.2, 127.1, 125.3, 124.5, 79.9, 73.6, 58.9, 39.4, 28.4.
Пример 1D. Получение трет-бутил-(3)-(1-(3-фторфенил)-2-гидроксиэтил)карбамата
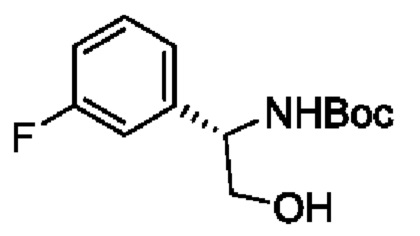
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 1А, выполняли с использованием (S)-2-амино-2-(3-фторфенил)этан-1-ола (1,36 г; 8,73 ммоль; 100 моль%), получая трет-бутил-(S)-(1-(3-фторфенил)-2-гидроксиэтил)карбамат (2,23 г; выход 100%) в виде белого твердого вещества. Т.пл.: 106,5-107,9°С; FTIR (для чистого вещества, см-1) 3251, 3059, 2977, 2901, 1671, 1587, 1543; 1H ЯМР (400 МГц, CDCl3): δ 7.33 (ddd, J=7,6; 7,6; 6,0 Гц, 1Н), 7.12-7.07 (m, 1Н), 7.05-6.95 (m, 2H), 5.24 (br, 1Н), 4.77 (br, 1Н), 3.93-3.77 (m, 2H), 2.02 (br, 1Н), 1.44 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 163.0 (d, 1JCF=246 Гц), 156.0, 142.5,130.2, 3JCF=9 Гц), 122.2, 4JCF=3 Гц), 114.5 (d, 2JCF=21 Гц), 113.6 (d, 2JCF=21 Гц), 80.2, 66.2, 56.3, 28.3; 19F ЯМР (CDCl3, 376 МГц): δ -112.4.
Пример 1Е. Получение (S)-(1-(3-фторфенил)-2-гидроксиэтил)карбамата
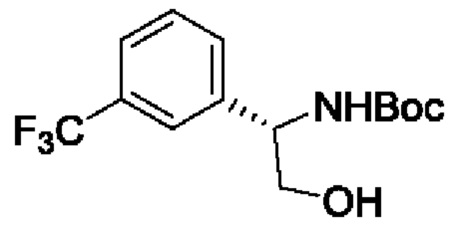
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 1А, выполняли с использованием (S)-2-амино-2-(3-(трифторметил)фенил)этан-1-ола гидрохлорида (1,00 г; 4,12 ммоль; 100 моль%), получая (S)-(1-(3-фторфенил)-2-гидроксиэтил)карбамат (1,26 г; выход 100%) в виде белого твердого вещества. Т.пл.: 50,3-52,5°С; FTIR (для чистого вещества, см-1) 3368, 3254, 2979, 2939, 1691, 1510, 1453, 1333; 1H ЯМР (400 МГц, CDCl3): δ 7.59-7.54 (m, 2H), 7.53-7.45 (m, 2H), 5.31 (d, J=6,4 Гц, 1Н), 4.83 (br, 1Н), 3.92 (ddd, J=11,2; 6,8; 4,0 Гц, 1H), 3.88-3.79 (m, 1H), 1.94 (br, 1H), 1.43 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 156.1, 141.1, 130.9 (q, 2JCF=32 Гц), 130.1, 129.0, 124.3 (q, 3JCF=4 Гц), 124.1 (q, 1JCF=270 Гц), 123.4 (q, 3JCF=4 Гц), 80.3, 65.8, 56.3, 28.2; 19F ЯМР (CDCl3, 376 МГц): δ -62.6.
Пример 2. Образование сульфамидатов
Пример 2. Общая реакционная схема образования сульфамидатов
Образование сульфамидатов осуществляли согласно приведенной далее общей реакционной схеме, где RA и rb соответствуют различным функциональным группам в реакциях приведенного далее примера 2, и где звездочки представляют хиральный центр:
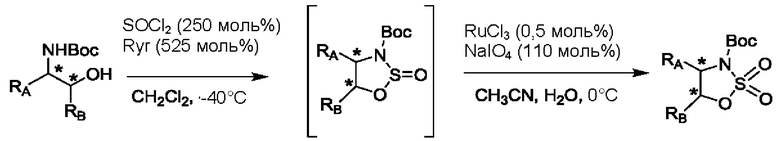
Пример 2А. Получение трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
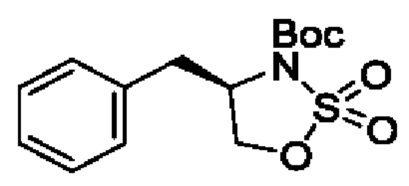
Методику, приведенную выше на общей реакционной схеме 2, выполняли так, как описано ниже. К охлажденному (-40°С) раствору SOCl2 (10,9 мл; 149 ммоль; 250 моль%) в CH2Cl2 (60,0 мл) добавляли раствор трет-бутил-(R)-(1-гидрокси-3-фенилпропан-2-ил)карбамата (15,0 г; 59,7 ммоль; 100 моль%) в CH2Cl2 (60,0 мл) в течение 60 мин при -40°С. Затем к реакционной смеси добавляли пиридин (25,3 мл; 313 ммоль; 525 моль%) в течение 30 мин при -40°С. Реакционную смесь перемешивали при -40°С в течение 2 ч, растворитель заменяли на смесь CH2Cl2/iPrOAc (1:1), фильтровали. Фильтрат промывали насыщенным раствором рассола (20 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Остаток растворяли в CH3CN (60,0 мл) при 0°С. В реакционную смесь добавляли NaIO4 (14,0 г; 65,7 ммоль; 110 моль%), RuCl3 (61,9 мг; 0,298 ммоль; 0,5 моль%) и воду (60,0 мл) при 0°С и перемешивали в течение 15 мин. Затем реакционную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 2 ч, экстрагировали iPrOAc (20 мл), промывали насыщенным раствором NaHCO3 (15 мл), насыщенным раствором рассола (15 мл), сушили (Na2SO4), фильтровали, очищали хроматографией на SiO2 Конкретный градиент, использованный для каждого образца, включен в характеристики. Все приведенные значения для выхода скорректированы с учетом остаточного растворителя по данным 1H ЯМР. В результате реакции, описанной в примере 2А, получали трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (18,7 г; выход 56%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 134,4-135,0°С; FTIR (для чистого вещества, см-1) 3261, 2979, 2903, 1712, 1673, 1540; 1H ЯМР (400 МГц, CDCl3): δ 7.38-7.20 (m, 5H), 4.49-4.40 (m, 2H), 4.35-4.28 (m, 1 H), 3.37 (dd, J=14,0; 4,0 Гц, 1H), 2.98-2.87 (m, 1H), 1.56 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 148.5, 135.2, 129.5, 129.1, 127.5, 85.6, 68.8, 58.6, 37.9, 28.0.
Пример 2 В. Получение трет-бутил-(3)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
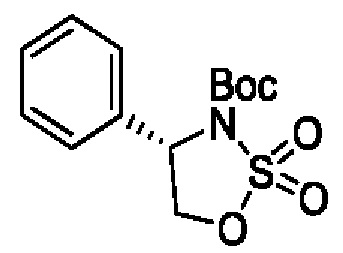
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(2-гидрокси-1-фенилэтил)карбамата (10,0 г; 42,1 ммоль; 100 моль%), получая соединение трет-бутил-(3)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (5,23 г; выход 42%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 144,3-145,0°С; FTIR (для чистого вещества, см-1) 2976, 1722, 1458, 1377; 1H ЯМР (400 МГц, CDCl3): δ 7.44-7.35 (m, 5H), 5.28 (dd, J=6,4; 4,0 Гц, 1Н), 4.87 (dd, J=9,2; 6,4 Гц, 1Н), 4.39 (dd, J=9,2; 4,4 Гц, 1Н), 1.42 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 148.3, 137.0, 129.2, 129.1, 126.2, 85.5, 71.8, 60.8, 27.8.
Пример 2С. Получение трет-бутил-(S)-4-(4-метоксифенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
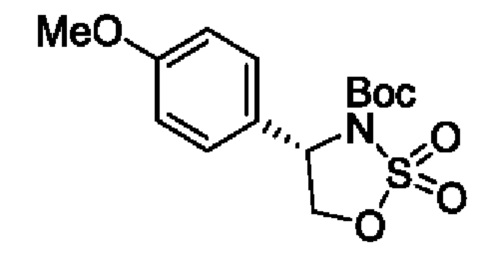
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(2-гидрокси-1-(4-метоксифенил)этил)карбамата (1,45 г; 5,42 ммоль; 100 моль%), получая соединение трет-бутил-(3)-4-(4-метоксифенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (1,05 г; выход 59%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 151,6-153,0°С; FTIR (для чистого вещества, см-1) 2979, 2933, 2838, 1721, 1636, 1510, 1457; 1H ЯМР (400 МГц, CDCl3): δ 7.37-7.32 (m, 2H), 6.95-6.90 (m, 2H), 5.24 (dd, J=6,8; 4,4 Гц, 1Н), 4.84 (dd, J=9,2; 6,8 Гц, 1H), 4.39 (dd, J=9,2; 4,4 Гц, 1H), 3.82 (s, 3H), 1.44 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 160.2, 148.3, 128.9, 127.7, 114.6, 85.5, 72.0, 60.5, 55.4, 27.9.
Пример 2D. Получение трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
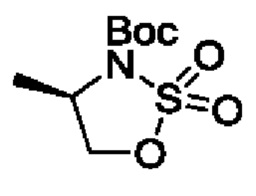
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(R)-(1-гидроксипропан-2-ил)карбамата (5,00 г; 28,5 ммоль; 100 моль%), получая соединение трет-бутил-(R)-4-метил-1,2,3-о ксатиазолидин-3-карбоксилат-2,2-диоксид (3,85 г; выход 57%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. FTIR (для чистого вещества, см-1) 3245, 2982, 1719, 1402, 1329; 1H ЯМР (400 МГц, CDCl3): δ 4.66 (dd, J=9,2; 6,0 Гц, 1H), 4.41 (qdd, J=6,4; 6,0; 2,8 Гц, 1Н), 4.19 (dd, J=9,2; 2,8 Гц, 1Н), 1.54 (s, 9H), 1.50 (d, J=6,4 Гц, 3Н); 13С ЯМР (100 МГц, CDCl3): δ 148.5, 85.4, 71.4, 53.8, 28.0, 18.3.
Пример 2Е. Получение трет-бутил-(R)-4-изопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
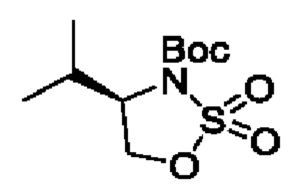
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(R)-(1-гидрокси-3-метилбутан-2-ил)карбамата (5,00 г; 24,6 ммоль; 100 моль%), получая соединение трет-бутил-(R)-4-изопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (3,93 г; выход 60%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 104,8-105,8°С; 1H ЯМР (400 МГц, CDCl3): δ 4.55 (dd, J=9,6; 6,4 Гц, 1Н), 4.38 (dd, J=9,6; 2,0 Гц, 1Н), 4.17 (ddd, J=6,4; 5,2; 1,6 Гц, 1Н), 2.24 (qqd, J=6,8; 6,8; 5,2 Гц, 1H), 1.53 (s, 9H), 1.00 (d, J=6,8 Гц, 3Н), 0.95 (d, J=6,8 Гц, 3Н); 13С ЯМР (100 МГц, CDCl3): δ 149.1, 85.3, 67.0, 62.0, 30.0, 27.9, 18.0, 16.4.
Пример 2F. Получение трет-бутил-(R)-4-циклопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
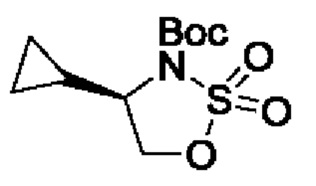
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(R)-(1-циклопропил-2-гидроксиэтил)карбамата (2,31 г; 11,5 ммоль; 100 моль%), получая трет-бутил-(R)-4-цикпопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (1,36 г; выход 45%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 52,7-55,7°С; FTIR (для чистого вещества, см-1) 2977, 1734, 1460, 1363; 1H ЯМР (400 МГц, CDCl3): δ 4.64 (dd, J=9,2; 6,0 Гц, 1H), 4.40 (dd, J=8,8; 2,0 Гц, 1H), 3.77 (ddd, J=9,2; 6,0; 2,0 Гц, 1Н), 1.54 (s, 9H), 1.35-1.23 (m, 1H), 0.74-0.65 (m, 2H), 0.63-0.54 (m, 1H), 0.29-0.20 (m, 1H); 13С ЯМР (100 МГц, CDCl3): δ 148.9, 85.3, 71.1, 61.6, 27.9, 14.3, 4.4, 1.7.
Пример 2G. Получение трет-бутил-(3aR,8aS)-8,8а-дигидроиндено[1,2-d][1,2,3]оксатиазол-3(3аН)-карбоксилат-2,2-диоксида
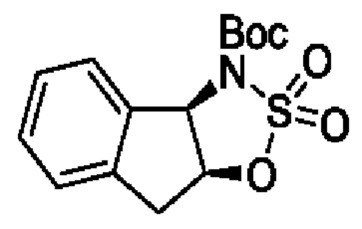
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-((1R,2S)-2-гидрокси-2,3-дигидро-1Н-инден-1-ил)карбамата (9,12 г; 36,6 ммоль; 100 моль%), получая трет-бутил-(3aR,8aS)-8,8а-дигидроиндено[1,2-d][1,2,3]оксатиазол-3(3аН)-карбоксилат-2,2-диоксид (7,50 г; выход 66%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 134,2-135,0°С; FTIR (для чистого вещества, см-1) 2988, 2937, 1732, 1462, 1375; 1H ЯМР (400 МГц, CDCl3): δ 7.62-7.57 (m, 1H), 7.39-7.24 (m, 3H), 5.71 (d, J=5,6 Гц, 1H), 5.50 (dt, J=6,0; 3,2 Гц, 1H), 3.38 (d, J=3,2 Гц, 1H), 1.62 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 149.6, 138.4, 137.9, 129.9, 128.4, 126.2, 125.2, 85.7, 82.2, 65.0, 36.5, 28.0.
Пример 2Н. Получение трет-бутил-(S)-5-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
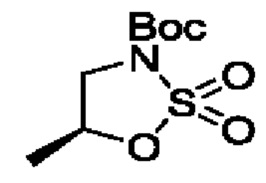
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(2-гидроксипропил)карбамата (6,25 г; 35,7 ммоль; 100 моль%), получая соединение трет-бутил-(S)-5-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (6,10 г; выход 72%) в виде белого твердого вещества. Градиент для колонки: 0-5% СН3ОН в CH2Cl2. Т.пл.: 116,9-118,2°С; FTIR (для чистого вещества, см-1) 3370, 2956, 2938, 2837, 1681, 1512, 1461, 1366; 1H ЯМР (400 МГц, CDCl3): δ 5.00-4.90 (m, 1H), 4.06 (dd, J=9,6; 5,6 Гц, 1H), 3.63 (dd, J=9,6; 9,2 Гц, 1H), 1.56 (d, J=6,4 Гц, 3H), 1.53 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 148.6, 85.3, 76.2, 51.7, 27.9, 18.0.
Пример 21. Получение трет-бутил-(S)-4-(3-фторфенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида

Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(1-(3-фторфенил)-2-гидроксиэтил)карбамата (1,45 г; 5,68 ммоль; 100 моль%), получая трет-бутил-(S)-4-(3-фторфенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (0,702 г; выход 39%) в виде белого твердого вещества. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 112,9-114,3°С; FTIR (для чистого вещества, см-1) 2976, 1722, 1636, 1594, 1458; 1H ЯМР (400 МГц, CDCl3): δ 7.44-7.36 (m, 1H), 7.24-7.18 (m, 1H), 7.17-7.11 (m, 1H), 7.11-7.05 (m, 1H), 5.28 (dd, J=6,8; 3,6 Гц, 1Н), 4.88 (dd, J=9,2; 6,8 Гц, 1H), 4.39 (dd, J=9,2; 3,6 Гц, 1H), 1.47 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 163.2 (d, 1JCF=246 Гц), 148.2, 139.6 (d, 3JCF=7 Гц), 131.1 (d, 3JCF=8 Гц), 121.8 (d, 4JCF=3 Гц), 116.2 (d, 2JCF=21 Гц), 113.4 (d, 2JCF=22 Гц), 86.0, 71.6, 60.2 (d, 4JCF=3 Гц), 27.9; 19F ЯМР (CDCl3, 376 МГц): δ -111.0.
Пример 2J. Получение трет-бутил-(3)-4-(3-(трифторметил)фенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида
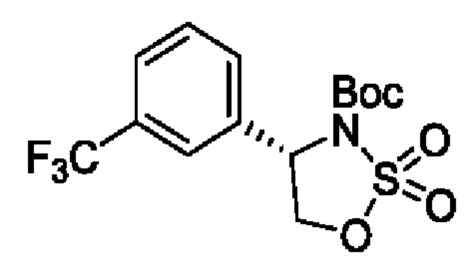
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(1-(3-фторфенил)-2-гидроксиэтил)карбамата (1,00 г; 3,28 ммоль; 100 моль%), получая трет-бутил-(S)-4-(3-(трифторметил)фенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксид (0,528 г; выход 44%) в виде белого твердого вещества. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 92,0-92,6°С; FTIR (для чистого вещества, см-1) 2989, 1720, 1463, 1373, 1325; 1H ЯМР (400 МГц, CDCl3): δ 7.70-7.61 (m, 3H), 7.61-7.55 (m, 1H), 5.35 (dd, J=6,8; 4,0 Гц, 1H), 4.92 (dd, J=9,2; 6,8 Гц, 1H), 4.41 (dd, J=9,2; 3,6 Гц, 1H), 1.46 (s, 9H); 13С ЯМР (100 МГц, CDCl3): δ 148.2, 138.2, 131.8 (q, 2JCF=32 Гц), 130.1, 129.4, 126.1 (q, 3JCF=4 Гц), 123.7 (q, 1JCF=270 Гц), 123.4 (q, 3JCF=4 Гц), 86.2, 71.4, 60.2, 27.8; 19F ЯМР (CDCl3, 376 МГц): δ -62.8.
Пример 2K. Получение трет-бутил-(3)-4-фенил-1,2,3-оксатиазинан-3-карбоксилат-2,2-диоксида
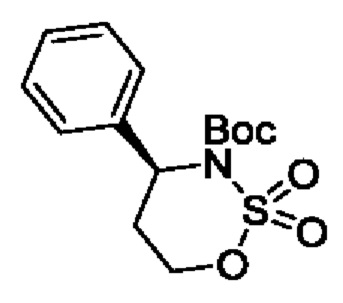
Методику, приведенную на общей реакционной схеме из вышеупомянутого примера 2А, выполняли с использованием трет-бутил-(S)-(3-гидрокси-1-фенилпропил)карбамата (2,00 г; 7,96 ммоль; 100 моль%), получая трет-бутил-(S)-4-фенил-1,2,3-оксатиазинан-3-карбоксилат-2,2-диоксид (1,26 г; выход 51%) в виде белого твердого вещества. Градиент для колонки: 0-50% iPrOAc. Т.пл.: 128,6-129,9°С; FTIR (для чистого вещества, см-1) 2986, 1727, 1449, 1367; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 94:6): δ 7.48-7.35 (m, 4H), 7.35-7.23 (m, 1H), 5.65 (dd, J=4,4; 4,4 Гц; 0,94Н), 5.53 (dd, J=11,2; 4,4 Гц; 0,06Н), 4.70 (ddd, J=10,4; 7,2; 2,8 Гц; 0,94Н), 4.51 (ddd, J=8,8; 4,4; 4,4 Гц; 0,06Н), 4.40 (ddd, J=10,4; 10,4; 6,8 Гц, 1H), 2.80-2.68 (m, 1H), 2.66-2.56 (m, 1H), 1.41 (s; 8,46H), 1.12 (s; 0,54H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1, 143.0, 128.4 (128.6), 127.0 (127.3), 126.3 (125.4), 78.1 (84.3), 73.6 (70.9), 50.5 (60.0), 36.0, 28.2 (27.4).
Пример 3. Алкилирование индолов
Хиральные триптамины часто встречаются в фармакологии ввиду их значительной биологической активности в центральной нервной системе. Помимо этого, хиральная группировка триптаминов служит в качестве предшественника синтеза многих важных с медицинской точки зрения индолсодержащих алкалоидов и содержится во многих биологически активных природных продуктах и фармацевтических средствах.
В частности, стереоконтролируемый синтез тетрагидро-β-карболинсодержащих соединений, таких как описанные в данной заявке, часто основывается на диастереоселективной реакции Пикте-Шпенглера, которая в свою очередь требует применения энантиомерно чистых триптаминов в качестве исходного вещества. Последние обычно получают нестереоселективным образом согласно многостадийной последовательности с участием опасных нитроалкановых реагентов. Таким образом, разработка простого подхода для региоселективного алкилирования, заключающегося в применении легко доступных незащищенных индолов в качестве нуклеофилов и хиральных электрофилов на основе аминов, таких как хиральные азиридины и циклические сульфамидаты, предоставляет удобный способ получения этих полезных хиральных каркасов.
Азиридины как электрофилы были применены для осуществления С3-селективного алкилирования в присутствии кислот Льюиса; однако этот способ применим только для синтеза β-замещенных триптаминов, поскольку замещение происходит предпочтительно при более замещенном атоме углерода. Раскрытый в данной заявке индолсодержащий купрат низшего порядка в комбинации с хиральными циклическими сульфамидатами с успехом обеспечил возможность практического получения как α-, так и β-замещенных хиральных триптаминов, обладающих высокой региоселективностью.
Было обнаружено, что в результате взаимодействия с хиральными азиридинами как электрофилами всегда проходило образование смеси α- и β-замещенных триптаминов, в то время как взаимодействие с циклическими сульфамидатами как электрофилами однозначно проходило с замещением по связи С-O.
В противоположность описанным в литературе случаям, предполагающим предпочтительное алкилирование по Соположению, когда в качестве основания использовали реагенты Гриньяра, первоначальные попытки приводили к более низким, чем ожидалось, выходам и плохой сайт-селективности. Без связи с какой-либо конкретной теорией отметим, что были опробованы более мягкие индолсодержащие нуклеофилы, поскольку они по всей вероятности предпочтительнее могли бы взаимодействовать как углерод-центрированные нуклеофилы. Изучали различные добавки, включая соли Си и Zn. Интересно, что система в виде смеси галогенидов, как например, MeMgCl в комбинации с CuBr или Cul либо MeMgBr в комбинации с CuCl, была гораздо менее эффективной, чем система на основе только хлорида. Другие соли меди, такие как CuCl2, CuCN, CuTC (тиофен-2-карбоксилат меди(I)) или Cu(SCN), также уступали CuCl.
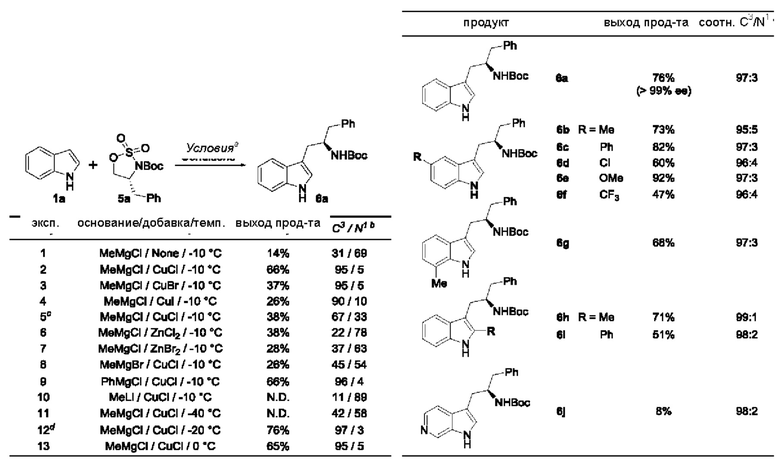
Применение CuCl в каталитических количествах было недопустимым и приводило к значительному снижению выхода и уменьшению селективности. Примечательно, что неудачное изменение региоселективности наблюдали, когда вместо CuCl использовали галогениды цинка (см. выше). То же самое наблюдали, когда в качестве основания вместо MeMgCl использовали MeLi, в то время как применение другого реагента Гриньяра было полностью допустимо Проводили оценку влияния температуры реакции, и было установлено, что реакция замещения оптимально протекала при приблизительно -20°С. При температуре ниже -30°С наблюдали значительное снижение региоселективности, предположительно вследствие не завершения реакции образования купрата.
Cu-опосредуемое алкилирование индолов допускает ряд замещений как на индолсодержащем нуклеофиле, так и на циклическом сульфамидате, обеспечивая получение С3-алкилированных индолсодержащих продуктов с выходом от умеренного до хорошего и с превосходной региоселективностью. Например, индолы с электрондонорными или электронакцепторными заместителями полностью вступали в реакцию (приведенные выше типичные соединения 6b-6g), и также достаточно эффективно "работали" стерически напряженные субстраты (приведенные выше типичные соединения 6h и 6i).
С другой стороны, по всей видимости возможно, что азаиндолы являются плохими субстратами. В результате использования 6-азаиндола в стандартных реакционных условиях получали продукт алкилирования с выходом только 8%, хотя и со сравнимой региоселективностью (см., например, приведенное выше типичное соединение 6j). При использовании других азаиндолов, таких как индазол и 7-азаиндол, не удавалось получить каких-либо желаемых алкилированных продуктов.
В данной реакции были успешно протестированы различные сульфамидаты. Как арил-, так и алкил-замещенные сульфамидаты, полученные из соответствующих аминоспиртов согласно двухстадийной последовательности реакций, полностью превращались в соответствующие α-замещенные хиральные триптамины. Аналогичным образом, реакция алкилирования также успешно протекала при участии 6-членного циклического сульфамидата с образованием гомолога триптамина с хорошим выходом и региоселективностью. Этот способ алкилирования также может быть применен для получения β-замещенных и α,β-дизамещенных триптаминов (см. приведенные выше типичные соединения 6s и 6t). В этих случаях индолсодержащий нуклеофил добавляли, при полной стереоспецифичности, к соответствующему циклическому сульфамидату с инверсией стереохимической конфигурации при атоме кислорода, связанном с атомом углерода. Целесообразность этого способа алкилирования продемонстрирована в описании данной заявки.
Пример 3. Общая реакционная схема алкилирования индолов
Алкилирование индолов осуществляли в соответствии со следующей реакционной схемой, где RA и rb соответствуют различным функциональным группам в реакциях, представленных далее в примере 3, и где звездочки представляют хиральный центр:
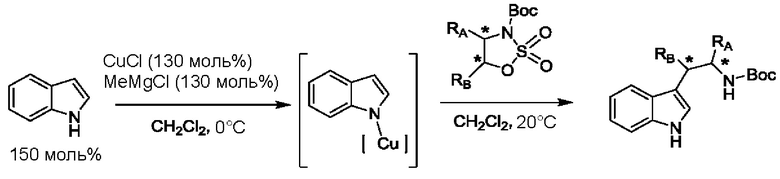
Пример 3А. Получение трет-бутил-(R)-(1-(1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамата
Методику, приведенную выше на общей реакционной схеме примера 3, выполняли так, как приведено ниже. К охлажденной (0°С) смеси индола (280 мг; 2,39 ммоль; 150 моль%) и CuCl (193 мг; 1,95 ммоль; 130 моль%) в CH2Cl2 (3,0 мл) добавляли MeMgCl (3,0 М раствор в THF; 0,65 мл; 1,95 ммоль; 130 моль%) в течение 10 мин при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч и охлаждали до -20°С. К реакционной смеси добавляли раствор трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида (500 мг; 1,60 ммоль; 100 моль%) в CH2Cl2 (2,0 мл) в течение 30 мин при -20°С. Затем реакционную смесь перемешивали при -20°С в течение 18 ч, гасили 10%-ным водным раствором лимонной кислоты (5,0 мл) при 0°С, фильтровали, экстрагировали CH2Cl2 (10,0 мл × 2), промывали насыщенным рассолом (20,0 мл × 2), сушили (Na2SO4), фильтровали, очищали хроматографией на SiO2. Конкретный градиент, использованный для каждого образца, включен в характеристики. Все приведенные значения для выхода скорректированы с учетом остаточного растворителя по данным 1H ЯМР. В результате реакции, описанной в примере 3А, получали трет-бутил-(R)-(1-(1H-индол-3-ил)-3-фенилпропан-2-ил)карбамат (424 мг; выход 76%) в виде белого твердого вещества. Соотношение С3/N1 составляло 97:3. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 152,1-153,2°С; FTIR (для чистого вещества, см-1) 3418, 3402, 3376, 2974, 2911, 1684, 1522; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.78 (br, 1H), 7.46 (d, J=8,0 Гц, 1Н), 7.32 (d, J=8,0 Гц, 1H), 7.28-7.21 (m, 2H), 7.19-7.10 (m, 4H), 7.05 (ddd, J=8,4; 7,2; 1,2 Гц, 1H), 6.95 (ddd, J=8,0; 6,8; 1,2 Гц, 1H), 6.76 (d, J=8,4 Гц, 0,85Н), 6.34 (d, J=9,2 Гц, 0,15Н), 3.97-3.83 (m, 1H), 2.90-2.65 (m, 4H), 1.29 (s, 7,65H), 1.12 (s, 1,35H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1, 139.6, 136.1, 129.0, 128.0, 127.5, 125.8, 123.2, 120.8, 118.3, 118.1, 111.4, 111.3, 77.3, 52.6, 39.9, 30.4, 28.2 (27.8).
Пример 3В. Получение трет-бутил-(S)-(2-(1Н-индол-3-ил)-1-фенилэтил)карбамата
Общую реакцию в соответствии с примером 3А проводили между индолом (294 мг; 2,51 ммоль; 150 моль%) и трет-бутил-(S)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (500 мг; 1,67 ммоль; 100 моль%), получая трет-бутил-(S)-(2-(1Н-индол-3-ил)-1-фенилэтил)карбамат (398 мг; выход 71%) в виде белого твердого вещества. Соотношение С3/N1 составляло 97:3. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 134,6-135,2°С; FTIR (для чистого вещества, см-1) 3416, 3401, 3371, 2980, 2909, 1683, 1524; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.74 (br, 1H), 7.54 (d, J=7,6 Гц, 1Н), 7.41 (d, J=8,4 Гц, 1Н), 7.38-7.25 (m, 5H), 7.20 (dd, J=6,8; 7,2 Гц, 1Н), 7.06 (dd, J=7,6; 7,2 Гц, 1Н), 7.02 (s, 1Н), 6.98 (dd, J=7,6; 7,2 Гц, 1Н), 4.89-4.74 (m, 1Н), 3.08 (dd, J=14,8; 8,8 Гц, 1Н), 2.99 (dd, J=14,4; 6,0 Гц, 1Н), 1.31 (s, 7,65H), 1.08 (s, 1,35Н); 13С ЯМР (100 МГц, DMSO-d6): δ 155.0, 144.5, 136.0, 128.0, 127.3, 126.5, 126.4, 123.2, 120.8, 118.3, 118.2, 111.3, 111.3, 77.6, 55.0, 32.8, 28.2.
Пример 3С. Получение трет-бутил-(S)-(2-(1Н-индол-3-ил)-1-(4-метоксифенил)этил)карбамата
Общую реакцию в соответствии с примером 3А проводили между индолом (264 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(S)-4-(4-метоксифенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (495 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(S)-(2-(1H-индол-3-ил)-1-(4-метоксифенил)этил)карбамат (378 мг; выход 69%) в виде белого твердого вещества. Соотношение С3/N1 составляло 98:2. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 173,3-176,5°С; FTIR (для чистого вещества, см-1) 3402, 3326, 2979, 2925, 2904, 1690, 1611, 1506; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.72 (br, 1Н), 7.54 (d, J=8,0 Гц, 1Н), 7.39-7.28 (m, 2H), 7.24 (d, J=8,8 Гц, 2Н), 7.05 (ddd, J=8,4; 7,2; 1,2 Гц, 1Н), 7.02-6.94 (m, 2H), 6.84 (d, J=8,4 Гц, 2H), 4.88-4.57 (m, 1Н), 3.72 (s, 3H), 3.06 (dd, J=14,8; 8,4 Гц, 1Н), 2.96 (dd, J=14,8; 6,4 Гц, 1Н), 1.31 (s, 7.65H), 1.11 (s, 1,35H); 13С ЯМР (100 МГц, DMSO-d6): δ 157.9, 154.9, 136.4, 136.0, 127.5, 127.3, 123.2, 120.7, 118.3, 118.1, 113.4, 111.4, 111.2, 77.5, 55.0, 54.4, 32.8, 28.2.
Пример 3D. Получение трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамата
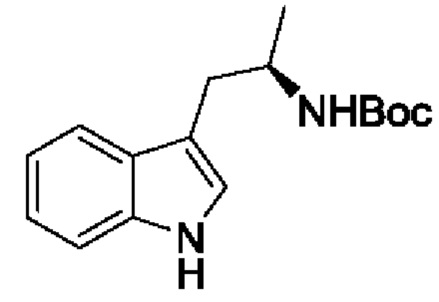
Общую реакцию в соответствии с примером 3А проводили между индолом (370 мг; 3,16 ммоль; 150 моль%) и трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (500 мг; 2,11 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамат (406 мг; выход 70%) в виде белого твердого вещества. Соотношение С3/N1 составляло 99:1. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 82,2-84,3°С; FTIR (для чистого вещества, см-1) 3416, 3401, 3366, 2974, 2963, 1684, 1524; 1H ЯМР (400 МГц, DMSO-d6): δ 10.77 (br, 1 H), 7.56 (d, J=7,6 Гц, 1H), 7.33 (ddd, J=8,0; 1,2; 0,8 Гц, 1H), 7.10 (d, J=2,4 Гц, 1 H), 7.05 (ddd, J=8,0; 6,8; 1,2 Гц, 1 H), 6.97 (ddd, J=8,0; 6,8; 1,2 Гц, 1 H), 6.71 (d, J=8,4 Гц, 1 H), 3.82-3.66 (m, 1 H), 2.87 (dd, J=14,0; 6,0 Гц, 1 H), 2.65 (dd, J=14,0; 7,6 Гц, 1Н), 1.38 (s, 9H), 1.01 (d, J=6,8 Гц, 3Н); 13С ЯМР (100 МГц, DMSO-d6): δ 155.0, 136.1, 127.5, 123.1, 120.7, 118.4, 118.1, 111.6, 111.2, 77.3, 46.8, 32.2, 28.3, 20.1.
Пример 3Е. Получение трет-бутил-(R)-(1-(1Н-индол-3-ил)-3-метилбутан-2-ил)карбамата
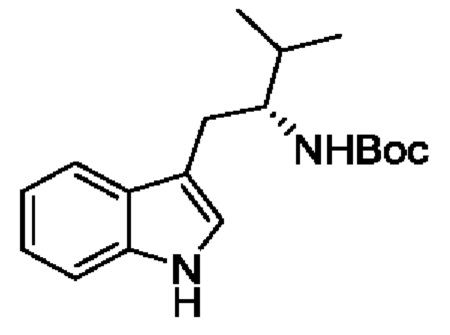
Общую реакцию в соответствии с примером 3А проводили между индолом (331 мг; 2,83 ммоль; 150 моль%) и трет-бутил-(R)-4-изопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (500 мг; 1,88 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(1Н-индол-3-ил)-3-метилбутан-2-ил)карбамат (335 мг; выход 59%) в виде белого твердого вещества. Соотношение С3/N1 составляло 94:6. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 145,1-146,9°С; FTIR (для чистого вещества, см-1) 3417, 3402, 3362, 2978, 1686, 1526; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.72 (br, 1Н), 7.51 (d, J=8,0 Гц, 1Н), 7.31 (ddd, J=8,0; 1,2; 0,8 Гц, 1Н), 7.07 (d, J=2,0 Гц, 1Н), 7.04 (ddd, J=8,0; 6,8; 1,2 Гц, 1Н), 6.96 (ddd, J=8,0; 6,8; 1,2 Гц, 1Н), 6.59 (d, J=9,2 Гц, 0,85Н), 6.15 (d, J=10,0 Гц, 0,15H), 3.64-3.53 (m, 1H), 2.80 (dd, J=14,8; 5,2 Гц, 1H), 2.68 (dd, J=14,8:8,8 Гц, 1Н), 1.77-1.65 (m, 1H), 1.32 (s, 7,65H), 1.12 (s, 1,35H), 0.95-0.82 (m, 6H); 13С ЯМР (100 МГц, DMSO-d6): δ 155.6, 136.1, 127.5, 122.7, 120.7, 118.3, 118.0, 112.0, 111.2, 77.1, 55.6, 31.4, 28.3, 27.1, 19.4, 17.7.
Пример 3F. Получение трет-бутил-(S)-(1-циклопропил-2-(1Н-индол-3-ил)этил)карбамата
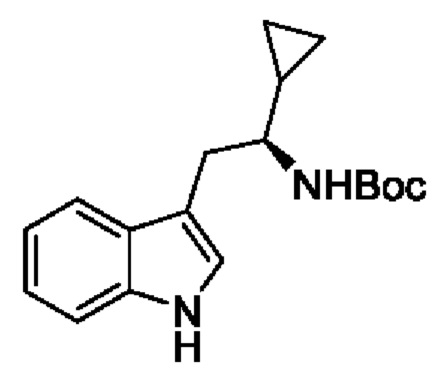
Общую реакцию в соответствии с примером 3А проводили между индолом (264 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-цикпопропил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (395 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(3)-(1-цикпопропил-2-(1Н-индол-3-ил)этил)карбамат (292 мг; выход 65%) в виде белого твердого вещества. Соотношение С3/N1 составляло 99:1. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 128,8-130,5°С; FTIR (для чистого вещества, см-1) 3414, 3400, 3362, 2981, 2937, 1683, 1525; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 9:1): δ 10.73 (br, 1H), 7.52 (d, J=7,6 Гц, 1H), 7.31 (ddd, J=8,0; 1,2; 0,8 Гц, 1H), 7.07 (d, J=2,4 Гц, 1H), 7.04 (ddd, J=8,0; 6,8; 1,2 Гц, 1Н), 6.95 (ddd, J=8,0; 6,8; 1,2 Гц, 1H), 6.66 (d, J=8,4 Гц, 0,9Н), 6.26 (s, 0,1H), 3.30-3.18 (m, 1H), 2.91 (dd, J=14,4; 5,6 Гц, 1H), 2.85 (dd, J=14,4; 8,0 Гц, 1H), 1.33 (s, 8,1H), 1.17 (s, 0,9H), 0.96-0.83 (m, 1H), 0.42-0.32 (m, 1H), 0.32-0.24 (m, 2H), 0.15-0.01 (m, 1H); 13С ЯМР (100 МГц, DMSO-d6): δ 155.3, 136.0, 127.7, 122.9, 120.6, 118.4, 118.0, 111.6, 111.2, 77.2, 54.2, 30.4, 28.2, 16.0, 3.0, 1.9.
Пример 3G. Получение трет-бутил-((1S,2S)-2-(1Н-индол-3-ил)-2,3-дигидро-1Н-инден-1-ил)карбамата
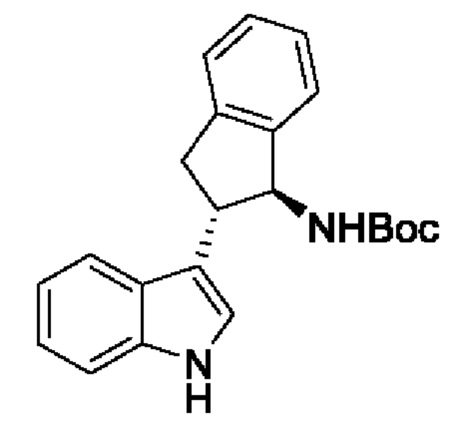
Общую реакцию в соответствии с примером 3А проводили между индолом (282 мг; 2,41 ммоль; 150 моль%) и трет-бутил-(3aR,8aS)-8,8а-дигидроиндено[1,2-d][1,2,3]оксатиазол-3(3аН)-карбоксилат-2,2-диоксидом (500 мг; 1,61 ммоль; 100 моль%), получая трет-бутил-((1S,2S)-2-(1H-индол-3-ил)-2,3-дигидро-1Н-инден-1-ил)карбамат (304 мг; выход 54%) в виде белого твердого вещества. Соотношение С3/N1 составляло 95:5. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 155,7-157,1°С; FTIR (для чистого вещества, см-1) 3387, 3351, 2980, 2938, 1691, 1500; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 87:13): 10.85 (br, 1H), 7.61 (d, J=8,0 Гц, 1H), 7.42-7.32 (m, 2H), 7.30-7.20 (m, 4H), 7.17 (dd, J=8,0; 4,0 Гц, 1H), 7.07 (ddd, J=8,0:6,8; 1,2 Гц, 1H), 6.97 (ddd, J=8,0; 6,8; 1,2 Гц, 1H), 5.18 (dd, J=9,2; 9,2 Гц, 1H), 3.69 (dd, J=18,4; 9,6 Гц, 0,87Н), 3.64-3.53 (m, 0,13H), 3.37 (dd, J=15,2; 8,0 Гц, 0,87Н), 3.30-3.23 (m, 0,13H), 3.13-3.04 (m, 0,13H), 2.99 (dd, J=15,2; 10,4 Гц, 0,87Н), 1.38 (s, 7,83H), 1.12 (s, 1,17H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.9, 144.5, 141.3, 136.6, 127.2, 127.1, 126.4, 124.4, 123.3, 121.7, 120.9, 119.1, 118.2, 115.5, 111.4, 77.7, 60.8, 44.0, 37.2, 28.2 (27.7).
Пример 3Н. Получение трет-бутил-(S)-(2-(1Н-индол-3-ил)пропил)карбамата
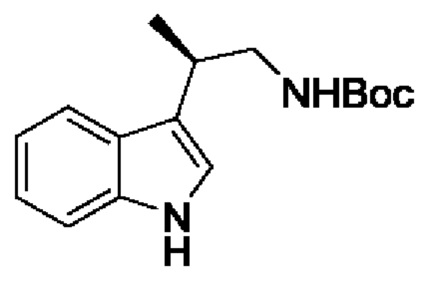
Общую реакцию в соответствии с примером 3А проводили между индолом (264 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(S)-5-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (356 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(S)-(2-(1Н-индол-3-ил)пропил)карбамат (319 мг; выход 78%) в виде бесцветной жидкости. Соотношение С3/N1 составляло 93:7. Градиент для колонки: 0-50% iPrOAc в гептане. FTIR (для чистого вещества, см-1) 3412, 3327, 2971, 2930, 1685, 1508, 1456; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 9:1): δ 10.78 (br, 1H), 7.58 (d, J=7,6 Гц, 1H), 7.33 (ddd, J=8,0; 1,2; 0,8 Гц, 1H), 7.10 (d, J=2,0 Гц, 1Н), 7.05 (ddd, J=8,0; 6,8; 1,2 Гц, 1H), 6.96 (ddd, J=8,0; 6,8; 1,2 Гц, 1H), 6.86 (dd, J=6,0; 6,0 Гц, 0,9Н), 6.51 (s, 0,1H), 3.29 (ddd, J=13,2; 5,6; 5,6 Гц, 1H), 3.18-3.05 (m, 1H), 2.97 (ddd, J=13,2; 8,8; 6,0 Гц, 1H), 1.41 (s, 0,9H), 1.38 (s, 8,1H), 1.25 (d, J=6,8 Гц, 3Н); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.7, 136.4, 126.6, 121.0, 120.8, 118.6, 118.1, 117.9, 111.4, 77.4, 46.8, 30.9, 28.3 (28.2), 18.4 (18.2).
Пример 31. Получение трет-бутил-(R)-(1-(3-фторфенил)-2-(1Н-индол-3-ил)этил)карбамата
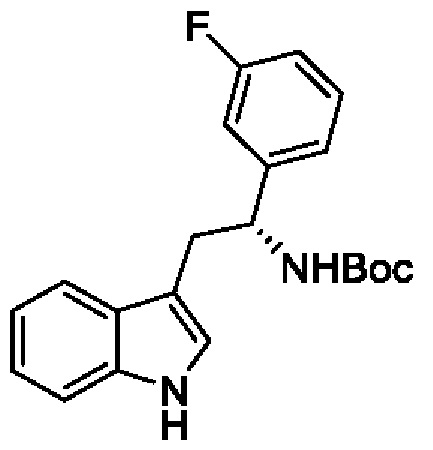
Общую реакцию в соответствии с примером 3А проводили между индолом (263 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(S)-4-(3-фторфенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (475 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(3-фторфенил)-2-(1Н-индол-3-ил)этил)карбамат (382 мг; выход 72%) в виде белого твердого вещества. Соотношение С3/N1 составляло 99:1. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 148,6-151,2°С; FTIR (для чистого вещества, см-1) 3414, 3398, 3363, 3055, 2981, 1682, 1591, 1527; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 9:1): δ 10.75 (br, 1H), 7.53 (d, J=7,6 Гц, 1H), 7.44 (d, J=8,4 Гц, 1H), 7.37-7.26 (m, 2H), 7.20-7.11 (m, 2H), 7.08-6.93 (m, 4H), 4.85-4.65 (m, 1H), 3.06 (dd, J=14,4; 8,4 Гц, 1H), 2.98 (dd, J=14,4; 6,4 Гц, 1H), 1.30 (s, 8,1H), 1.08 (s, 0,9Н); 13С ЯМР (100 МГц, DMSO-d6): δ 162.2 (d, 1JCF=243 Гц), 155.0, 147.5 (d, 3JCF=7 Гц), 136.0, 129.9 (d, 3JCF=8 Гц), 127.2, 123.3, 122.6, 120.8, 118.3, 118.2, 113.3 (d, 2JCF=21 Гц), 113.0 (d, 2JCF=21 Гц), 111.3, 111.0, 77.8, 54.7, 32.5, 28.2; 19F ЯМР (DMSO-d6, 376 МГц): δ -113.6.
Пример 3J. Получение трет-бутил-(R)-(2-(1Н-индол-3-ил)-1-(3-(трифторметил)фенил)этил)карбамата

Общую реакцию в соответствии с примером 3А проводили между индолом (176 мг; 1,50 ммоль; 150 моль%) и трет-бутил-(S)-4-(3-(трифторметил)фенил)-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (368 мг; 1,00 ммоль; 100 моль%), получая трет-бутил-(R)-(2-(1Н-индол-3-ил)-1-(3-(трифторметил)фенил)этил)карбамат (320 мг; выход 79%) в виде белого твердого вещества. Соотношение C3/N1 составляло 98:2. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 99,3-101,4°С; FTIR (для чистого вещества, см-1) 3414, 3399, 3361, 2982, 2936, 1683, 1523; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 88:12): δ 10.76 (br, 1H), 7.80-7.45 (m, 6H), 7.31 (d, J=8,0 Гц, 1Н), 7.08-7.00 (m, 2H), 6.96 (ddd, J=8,0; 7,2; 1,2 Гц, 1H), 5.11-4.69 (m, 1H), 3.09 (dd, J=14,4; 8,4 Гц, 1Н), 3.00 (dd, J=14,4; 6,4 Гц, 1H), 1.30 (s, 7,9H), 1.07 (s, 1,1H); 13С ЯМР (100 МГц, DMSO-d6): δ 155.1, 145.8, 136.1, 130.8, 129.0, 128.9 (q, 2JCF=32 Гц), 127.3, 124.4 (q, 1JCF=270 Гц), 123.3 (q, 3JCF=4 Гц), 123.0, 122.8 (q, 3JCF=4 Гц), 120.8, 118.3, 118.2, 111.3, 110.8, 77.9, 55.0, 32.5, 28.2; 19F ЯМР (DMSO-d6, 376 МГц): δ -61.0.
Пример 3K. Получение трет-бутил-(S)-(3-(1Н-индол-3-ил)-1-фенилпропил)карбамата
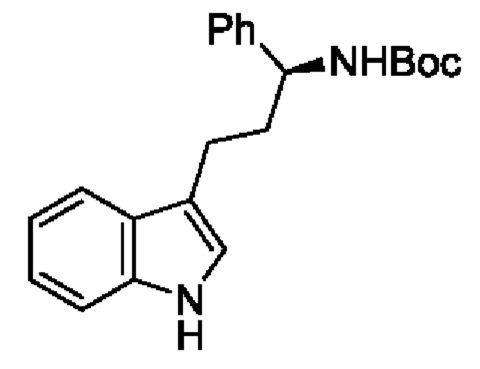
Общую реакцию в соответствии с примером 3А проводили между индолом (264 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(S)-4-фенил-1,2,3-оксатиазинан-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(S)-(3-(1Н-индол-3-ил)-1-фенилпропил)карбамат (401 мг; выход 73%) в виде белого твердого вещества. Соотношение С3/N1 составляло 98:2. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 121,7-123,2°С; FTIR (для чистого вещества, см-1) 3390, 2979, 2929, 2859, 1681, 1507, 1457, 1364; 1H ЯМР (400 МГц, CDCl3) (смесь ротамеров, 80:20): δ 8.04 (br, 0,2Н), 7.96 (br, 0,8Н), 7.66 (d, J=8,0 Гц, 0,2Н), 7.52 (d, J=8,0 Гц, 0,8Н), 7.40-7.22 (m, 6H), 7.22-7.15 (m, 1H), 7.15-7.06 (m, 1H), 7.00 (br, 1H), 4.88 (br, 0,8H), 4.75 (br, 1H), 4.53 (br, 0,2H), 3.52-3.35 (m, 0,4H), 3.32-3.20 (m, 0,4H), 2.87-2.67 (m, 1,6H), 2.16 (d, J=8,8 Гц, 1,6Н), 1.42 (s, 9H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.4 (156.2), 143.0, 136.4 (136.6), 128.6, 127.3, 127.2, 126.4, 121.8 (122.0), 121.5(120.8), 119.0(119.2), 118.7, 115.3, 111.2 (111.3), 79.5, 54.7 (46.7), 37.2 (31.6), 28.4 (29.7), 21.9 (18.7).
Пример 3L. Получение трет-бутил-(R)-(1-(5-метил-1H-индол-3-ил)-3-фенилпропан-2-ил)карбамата
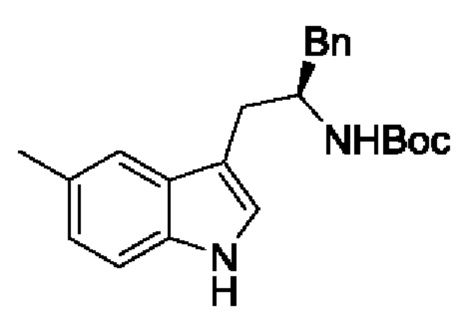
Общую реакцию в соответствии с примером 3А проводили между 5-метил-1H-индолом (295 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(5-метил-1H-индол-3-ил)-3-фенилпропан-2-ил)карбамат (401 мг; выход 73%) в виде белого твердого вещества. Соотношение С3/N1 составляло 92:8. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 123,0-123,8°С; FTIR (для чистого вещества, см-1) 3413, 3368, 2974, 2927, 1685, 1524; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.63 (br, 1H), 7.30-7.13 (m, 7H), 7.07 (d, J=2,0 Гц, 1H), 6.87 (dd, J=8,4; 1,6 Гц, 1H), 6.74 (d, J=8,8 Гц, 0,85Н), 6.34 (d, J=7,6 Гц, 0,15Н), 3.96-3.80 (m, 1H), 2.86-2.64 (m, 4H), 2.36 (s, 3H), 1.30 (s, 7,65H), 1.16 (s, 1,35H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1, 139.6, 134.5, 129.1, 128.0, 127.7, 126.4, 125.8, 123.2, 122.3, 117.9, 111.0, 110.9, 77.2, 52.7, 40.0, 30.2, 28.2 (27.8), 21.3.
Пример 3М. Получение трет-бутил-(R)-(1-(7-метил-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамата
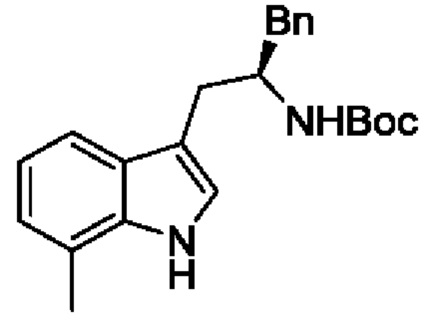
Общую реакцию в соответствии с примером 3А проводили между 7-метил-1H-индолом (295 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(7-метил-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамат (371 мг; выход 68%) в виде белого твердого вещества. Соотношение С3/N1 составляло 97:3. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 124,4-125,9°С; FTIR (для чистого вещества, см-1) 3417, 3407, 3370, 2974, 2926, 1683, 1524; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.75 (br d, J=2,0 Гц, 1Н), 7.34-7.21 (m, 3H), 7.21-7.09 (m, 4H), 6.90-6.83 (m, 2H), 6.74 (d, J=8,8 Гц, 0,85Н), 6.34 (d, J=7,6 Гц, 0,15Н), 3.98-3.84 (m, 1H), 2.91-2.65 (m, 4H), 2.44 (s, 3H), 1.31 (s, 7,65Н), 1.14 (s, 1,35Н); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.2, 139.6, 135.7, 129.0, 128.0, 127.2, 125.8, 122.9, 121.3, 120.3, 118.4, 115.9, 111.9, 77.3, 52.6, 39.9, v30.5, v28.2 (27.8), 16.7.
Пример 3N. Получение трет-бутил-(R)-(1-(5-хлор-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамата
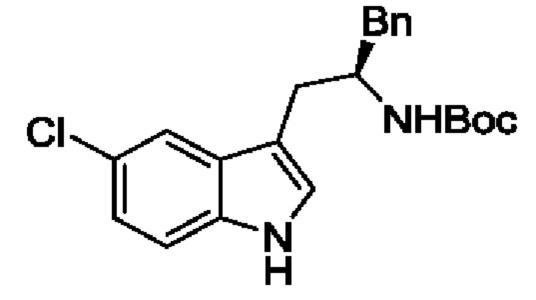
Общую реакцию в соответствии с примером 3А проводили между 5-хлор-1Н-индолом (341 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(5-хлор-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамат (345 мг; выход 60%) в виде белого твердого вещества. Соотношение С3/N1 составляло 96:4. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 70,5-73,9°С; FTIR (для чистого вещества, см-1) 3417, 3368, 2980, 2928, 1684, 1518; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.99 (br, 1H), 7.49 (d, J=2,0 Гц, 1Н), 7.34 (d, J=8,8 Гц, 1H), 7.30-7.22 (m, 2H), 7.22-7.13 (m, 4H), 7.04 (dd, J=8,4; 2,0 Гц, 1H), 6.77 (d, J=8,8 Гц, 0,85Н), 6.34 (d, J=8,8 Гц, 0,15Н), 3.90-3.76 (m, 1H), 2.83-2.62 (m, 4H), 1.27 (s, 7,65H), 1.10 (s, 1,35Н); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1, 139.5, 134.6, 129.0, 128.8, 128.0, 125.8, 125.2, 122.9, 120.6, 117.7, 112.8, 111.5, 77.2, 52.9, 40.0, 30.1, 28.2 (27.7).
Пример 3O. Получение трет-бутил-(R)-(1-фенил-3-(5-(трифторметил)-1Н-индол-3-ил)пропан-2-ил)карбамата
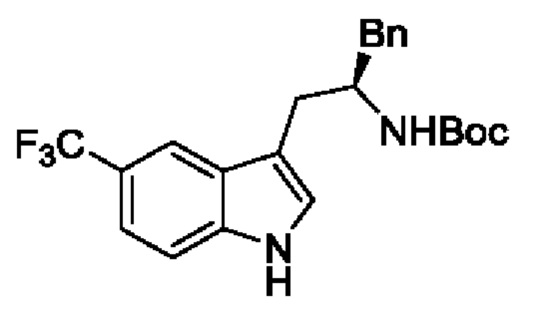
Общую реакцию в соответствии с примером 3А проводили между 5-(трифторметил)-1H-индолом (417 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-фенил-3-(5-(трифторметил)-1Н-индол-3-ил)пропан-2-ил)карбамат (297 мг; выход 47%) в виде белого твердого вещества. Соотношение С3/N1 составляло 96:4. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 130,9-131,8°С; FTIR (для чистого вещества, см-1) 3417, 3368, 2980, 2929, 1684, 1519; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 11.26 (br, 1Н), 7.84 (s, 1H), 7.50 (d, J=8,4 Гц, 1Н), 7.37-7.22 (m, 4H), 7.22-7.10 (m, 3H), 6.80 (d, J=8,8 Гц, 0,85Н), 6.36 (d, J=9,2 Гц, 0,15Н), 3.99-3.77 (m, 1Н), 2.86 (d, J=6,8 Гц, 2H), 2.76 (d, J=6,8 Гц, 2H), 1.22 (s, 7,65H), 1.04 (s, 1,35H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1 (154.6), 139.5, 137.6 (137.7), 129.1, 128.0, 126.9, 125.8, 125.7 (q, 1JCF=269 Гц), 125.6, 119.1 (q, 2JCF=32 Гц), 117.1 (q, 3JCF=4 Гц), 116.1 (q, 3JCF=4 Гц), 112.9, 111.9, 77.2, 53.0 (53.7), 40.3 (41.0), 29.9 (30.9), 28.1 (27.6); 19F ЯМР (DMSO-d6, 376 МГц): δ -58.1.
Пример 3Р. Получение трет-бутил-(R)-(1-(5-метокси-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамата
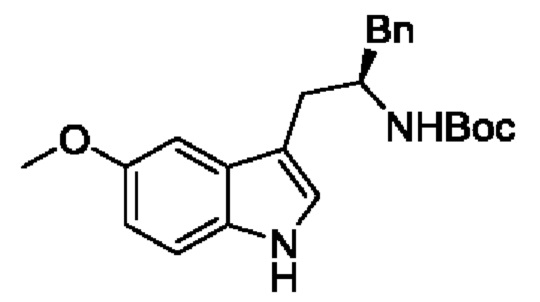
Общую реакцию в соответствии с примером 3А проводили между 5-метокси-1H-индолом (331 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(5-метокси-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамат (523 мг; выход 92%) в виде белого твердого вещества. Соотношение С3/N1 составляло 97:3. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 123,2-123,9°С; FTIR (для чистого вещества, см-1) 3368, 2975, 2933, 1692, 1680, 1516; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.60 (s, 1H), 7.32-7.24 (m, 2H), 7.24-7.13 (m, 4H), 7.08 (d, J=2,4 Гц, 1H), 6.91 (d, J=2,4 Гц, 0.85Н), 6.83 (br, 0,15H), 6.77 (d, J=8,8 Гц, 0,85Н), 6.70 (dd, J=8,8; 2,4 Гц, 1H), 6.34 (d, J=9,2 Гц, 0,15Н), 3.94-3.80 (m, 1H), 3.72 (s, 3H), 2.85-2.65 (m, 4H), 1.29 (s, 7,65H), 1.13 (s, 1,35Н); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.2, 152.9, 139.6, 131.3, 129.1, 128.0, 127.8, 125.8, 123.8, 111.9, 111.3, 110.8, 100.3, 77.3, 55.3, 52.8, 40.2, 30.2, 28.2 (27.8).
Пример 3Q. Получение трет-бутил-(R)-(1-фенил-3-(5-фенил-1Н-индол-3-ил)пропан-2-ил)карбамата
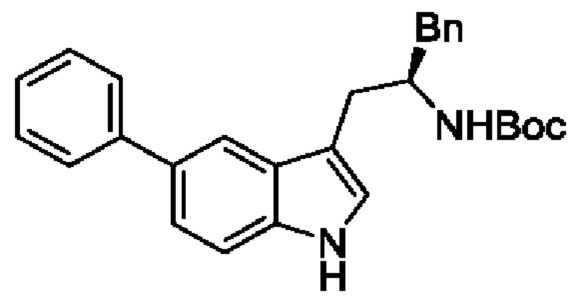
Общую реакцию в соответствии с примером 3А проводили между 5-фенил-1Н-индолом (435 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-фенил-3-(5-фенил-1Н-индол-3-ил)пропан-2-ил)карбамат (523 мг; выход 82%) в виде белого твердого вещества. Соотношение С3/N1 составляло 97:3. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 68,9-70,8°С; FTIR (для чистого вещества, см-1) 3427, 3411, 3392, 3310, 2977, 2929, 1715, 1696, 1506; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 85:15): δ 10.85 (br, 1H), 7.70 (s, 1H), 7.67-7.60 (m, 2H), 7.48-7.33 (m, 4H), 7.32-7.24 (m, 3H), 7.23-7.15 (m, 4H), 6.80 (d, J=8,8 Гц, 0,85Н), 6.39 (d, J=9,2 Гц, 0,15H), 4.03-3.84 (m, 1H), 2.87 (d, J=6,8 Гц, 2H), 2.77 (d, J=6,8 Гц, 2H), 1.24 (s, 7,65H), 1.09 (s, 1,35H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.1, 142.0, 139.6, 135.8, 130.7, 129.1, 128.6, 128.1, 128.0, 126.6, 126.0, 125.8, 124.0, 120.1, 116.6, 112.2, 111.6, 77.2, 53.1, 40.4, 30.1, 28.2 (27.8).
Пример 3R. Получение трет-бутил-(R)-(1-(2-метил-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамата
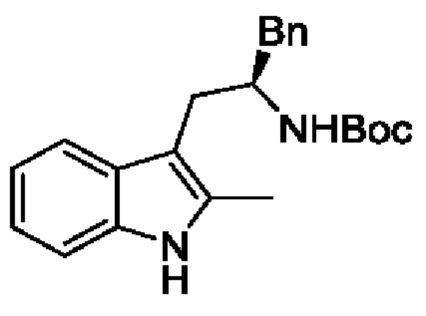
Общую реакцию в соответствии с примером 3А проводили между 2-метил-1H-индолом (295 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-(2-метил-1Н-индол-3-ил)-3-фенилпропан-2-ил)карбамат (386 мг; выход 71%) в виде белого твердого вещества. Соотношение С3/N1 составляло 99:1. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 117,6-119,0°С; FTIR (для чистого вещества, см-1) 3440, 3385, 2984, 2930, 1684, 1524; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 83:17): δ 10.67 (br, 1H), 7.39 (d, J=7,6 Гц, 0,83Н), 7.33 (d, J=8,0 Гц, 0,17Н), 7.28-7.18 (m, 3H), 7.18-7.07 (m, 3H), 6.96 (ddd, J=8,0; 7,2; 1,2 Гц, 1H), 6.90 (ddd, J=8,0; 8,0; 1,2 Гц, 1H), 6.73 (d, J=8,8 Гц, 0,83Н), 6.30 (d, J=8,8 Гц, 0,17Н), 3.94-3.74 (m, 1H), 2.85 (dd, J=14,0; 6,4 Гц, 1H), 2.75-2.62 (m, 3H), 2.32 (s, 3H), 1.27 (s, 7,47H), 1.04 (s, 1,53H); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): 155.1, 139.8, 135.2, 132.4, 128.9, 128.7, 128.0, 125.7, 119.8, 118.0, 117.5, 110.2, 107.4, 77.2, 53.4, 39.4, 29.9, 28.2 (27.7), 11.4.
Пример 3S. Получение трет-бутил-(R)-(1-фенил-3-(2-фенил-1Н-индол-3-ил)пропан-2-ил)карбамата
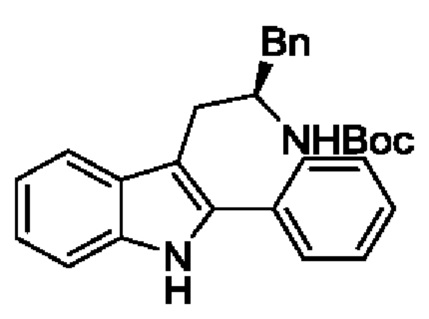
Общую реакцию в соответствии с примером 3А проводили между 2-фенил-1H-индолом (435 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1 -фенил-3-(2-фенил-1Н-индол-3-ил)пропан-2-ил)карбамат (324 мг; выход 51%) в виде белого твердого вещества. Соотношение С3/N1 составляло 98:2. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 178,5-178,8°С; FTIR (для чистого вещества, см-1) 3380, 3354, 3376, 2981, 2932, 1683, 1508; 1H ЯМР (400 МГц, DMSO-d6) (смесь ротамеров, 82:18): δ 11.14 (s, 1H), 7.69 (d, J=7,6 Гц, 1H), 7.60 (d, J=7,2 Гц, 2Н), 7.44 (dd, J=7,2; 7,2 Гц, 2Н), 7.35 (dd, J=7,2; 7,2 Гц, 2Н), 7.21 (dd, J=7,2; 7,2 Гц, 2Н), 7.17-6.97 (m, 5H), 6.79 (d, J=9,2 Гц, 0,82Н), 6.38 (d, J=9,6 Гц, 0,18H), 4.15-3.90 (m, 1H), 3.06 (dd, J=14,0; 6,8 Гц, 1H), 2.96 (dd, J=14,4; 7,2 Гц, 1H), 2.64 (d, J=6,8 Гц, 2Н), 1.21 (s, 7,40H), 0.97 (s, 1,60Н); 13С ЯМР (100 МГц, DMSO-d6) (ротамеры): δ 155.0, 139.5, 136.0, 134.7, 133.0, 129.2, 128.9, 128.6, 128.0, 127.8, 127.1, 125.8, 121.3, 119.2, 118.5, 111.0, 109.2, 77.2, 53.4, 40.2, 28.1 (27.6).
Пример 3Т. Получение трет-бутил-(R)-(1-фенил-3-(1H-пирроло[2,3-с]пиридин-3-ил)пропан-2-ил)карбамата
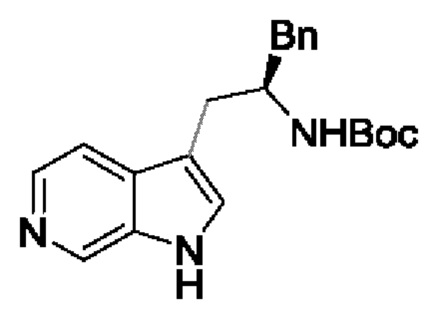
Общую реакцию в соответствии с примером 3А проводили между 1H-пирроло[2,3-с]пиридином (266 мг; 2,25 ммоль; 150 моль%) и трет-бутил-(R)-4-бензил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксидом (470 мг; 1,50 ммоль; 100 моль%), получая трет-бутил-(R)-(1-фенил-3-(1Н-пирроло[2,3-с]пиридин-3-ил)пропан-2-ил)карбамат (40 мг; выход 8%) в виде белого твердого вещества. Соотношение С3/N1 составляло 98:2. Градиент для колонки: 0-50% iPrOAc в гептане. Т.пл.: 196,2-197,0°С; FTIR (для чистого вещества, см-1) 3360, 2978, 2931, 1685, 1625, 1524; 1H ЯМР (400 МГц, CDCl3) (смесь ротамеров, 90:10): δ 8.56 (br, 1H), 8.28 (s, 1 H), 7.57 (d, J=6,4 Гц, 1H), 7.38-7.31 (m, 2Н), 7.30-7.25 (m, 2H), 7.25-7.20 (m, 2Н), 6.70 (br, 1H), 5.44 (br, 0,10H), 5.24 (d, J=5,6 Гц, 0,90Н), 4.52 (dd, J=13,2; 8,4 Гц, 1Н), 4.36 (dd, J=13,6; 4,8 Гц, 1Н), 4.26-4.14 (m, 1H), 3.00 (dd, J=14,0; 7,6 Гц, 1H), 2.91 (dd, J=14,0; 6,8 Гц, 1Н), 1.27 (s, 8,10H), 1.11 (s, 0,90H).
Пример 4
Получение азетидиниламинов
Получение соединений формулы (VII) проводили в соответствии с приведенной ниже общей схемой:
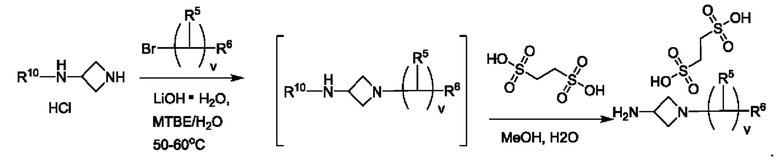
R5, v, R6 и R10 являются такими, как описано в данной заявке.
Пример 4А
Получение 1-(3-фторпропил)азетидин-3-амин-этан-1,2-дисульфоната
1-(3-Фторпропил)азетидин-3-амин-этан-1,2-дисульфонат получали в соответствии с приведенной далее реакционной схемой:
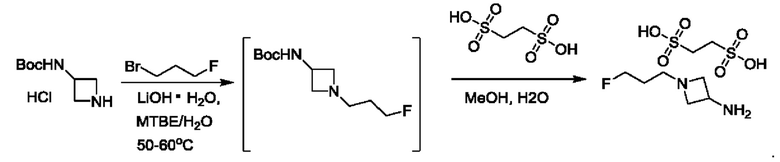
трет-Бутил-азетидин-3-илкарбамата гидрохлорид (109,7 кг; 1,0 экв.) растворяли в МТВЕ (793,4 кг) и добавляли 1-бром-3-фторпропан (82,3 кг). Добавляли МТВЕ (14 кг) и воду (530 кг), затем LiOH⋅H2O (66,0 кг) при 15-25°С, после чего перемешивали при 50-60°С. После завершения реакции органическую фазу отделяли и затем объединяли с водным раствором 1,2-этандисульфоновой кислоты дигидрата (247,8 кг) при 0-5°С.Полученную смесь перемешивали в течение 30 мин при 15-20°С. Водную фазу отделяли и к органической фазе добавляли 1,2-этандисульфоновой кислоты дигидрат (56,85 кг). Полученную смесь перемешивали в течение 5 ч при 35-40°С до полного удаления защитных групп. Затем добавляли МеОН (884,1 кг) при 35-40°С и смесь перемешивали в течение 2 ч при 35-40°С. После охлаждения до комнатной температуры реакционную смесь перемешивали в течение 4 ч. Твердое вещество собирали фильтрованием и промывали водн. раствором МеОН. Промытое твердое вещество сушили при 35-42°С при пониженном давлении, получая 106,8 кг 1-(3-фторпропил)азетидин-3-амин-этан-1,2-дисульфоната (выход 63%). 1Н ЯМР (400 МГц, DMSO-d6) δ 9.92 (s, 0,7Н), 9.63 (s, 0,3H), 8.35 (s, 3Н), 4.52 (dt, J=47,1; 5,6 Гц, 2Н), 4.43-4.08 (m, 5Н), 3.36 (s, 3Н), 2.74 (s, 4Н), 2.06-1.77 (m, 2Н).
Пример 4 В
Получение 1-(3-фторпропил)азетидин-3-амин-этан-1,2-дисульфоната в химических реакциях со снятием напряжения
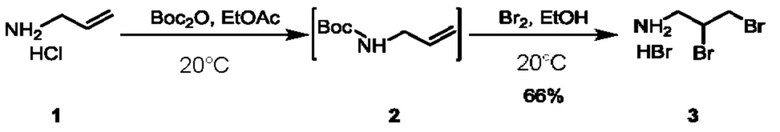
Получение 2,3-дибромпропан-1-амина гидробромида (3)
К соединению 1 (20,0 г; 1,0 экв.; 50 масс. % в воде) добавляли раствор K2CO3 (29,55 г; 1,0 экв.) в воде (100 мл) и раствор Boc2O (93,32 г; 2,0 экв.) в EtOAc (100 мл) при 0°С. Реакционную смесь перемешивали при 20°С в течение 15 ч. Органический слой отделяли и растворитель заменяли на EtOH (100 мл), получая раствор соединения 2 в EtOH. Затем его добавляли в охлажденный (0°С) раствор Br2 (71,74 г; 2,1 экв.) в EtOH (60 мл) при 0°С. Реакционную смесь перемешивали при 20°С в течение 16 ч, фильтровали, промывали МТВЕ (60 мл), сушили под вакуумом при 40°С в течение 15 ч, получая соединение 3 в виде бесцветного твердого вещества (43,01 г; 66%): 1Н ЯМР (400 МГц, CD3OD) δ 4.55-4.47 (m, 1Н), 4.01 (dd, J=10,9; 4,6 Гц, 1Н), 3.86 (dd, J=10,9; 8,7 Гц, 1Н), 3.71 (dd, J=13,9; 3,2 Гц, 1Н), 3.39-3.33 (m, 1Н).
Получение N,N-дибензилазетидин-3-амин-этан-1,1-дисульфоната
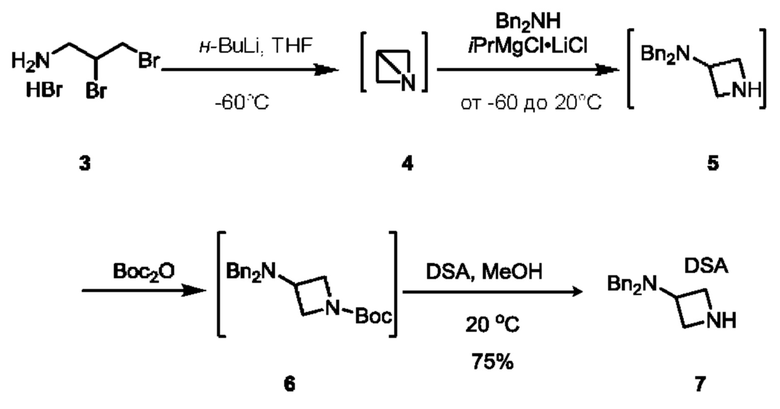
К раствору Bn2NH (33,12 г; 1,0 экв.) в THF (330 мл) добавляли iPrMgCl⋅LiCl (1,3 М раствор в THF; 130 мл; 1,0 экв.) при 20°С и перемешивали при 20°С в течение 5 ч, получая раствор Bn2NMgCl⋅LiCl в THF. В отдельный реактор загружали соединение 3 (50,0 г; 1,0 экв.) в THF (500 мл), охлаждали до -60°С. К суспензии добавляли H-BuLi (201 мл; 2,5 М раствор в н-гексане; 3,0 экв.) при -60°С и перемешивали при -60°С в течение 2 ч, получая раствор соединения 4 в THF. Раствор Bn2NMgCl⋅LiCl добавляли в раствор соединения 4 при -60°С, нагревали до 20°С и перемешивали при 20°С в течение 12 ч, получая раствор соединения 5 в THF. Реакционную смесь охлаждали до 0°С, добавляли раствор Boc2O (73,28 г; 2,0 экв.) в THF (200 мл) при 0°С, перемешивали при 20°С в течение 2 ч, охлаждали до 0°С, гасили раствором АсОН (20,16 г; 2,0 экв.) в H2O (383 мл). Органический слой промывали 5%-ным раствором Na2S04 (150 мл). Объединенный водный слой повторно экстрагировали МТВЕ (100 мл × 2), получая раствор соединения 6. В этот раствор добавляли раствор этандисульфоновой кислоты (40,0 г; 1,5 экв.) в МеОН (225 мл) при 20°С и перемешивали при 40°С в течение 20 ч. Суспензию фильтровали, промывали THF (100 мл), сушили под вакуумом (40°С) в течение 20 ч, получая соединение 7 (69,21 г; выход 75%) в виде белого твердого вещества: 1Н ЯМР (400 МГц, D2O) δ 7.57-7.44 (m, 10Н), 4.66 (t, J=8,2 Гц, 1Н), 4.36 (s, 4Н), 4.10-3.95 (m, 4Н), 3.22 (s, 4Н). MS ([М+Н]+) для C17H21N2: рассчитано 253,17, обнаружено 253,00.
Получение N,N-дибензил-1-(3-фторпропил)азетидин-3-амина (8)
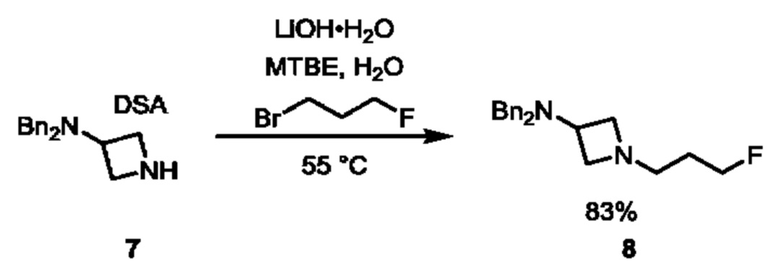
К раствору соединения 7 (40,0 г; 1,0 экв.) в МТВЕ (200 мл) и H2O (200 мл) добавляли 1-бром-3-фторпропан (17,20 г; 1,5 экв.) и LiOH⋅H2O (15,18 г; 4,0 экв.). Реакционную смесь нагревали до 55°С и перемешивали при 55°С в течение 20 ч, охлаждали до 20°С. Органический слой отделяли и промывали 5%-ным раствором Na2SO4 (120 мл × 2). Путем концентрирования раствора в МТВЕ под вакуумом можно получить соединение 8 в виде белого твердого вещества (22,3 г; выход 83%): 1Н ЯМР (400 МГц, DMSO-d6) δ 7.35-7.22 (m, 10Н), 4.45 (t, J=6,0 Гц, 1 Н), 4.33 (t, J=6,0 Гц, 1Н), 3.45-3.39 (m, 4Н), 3.28-3.17 (m, 3Н), 2.67-2.56 (m, 2Н), 2.35 (t, J=7,0 Гц, 2Н), 1.64-1.48 (m, 2Н). MS ([М+Н]+) для C20H26FN2: рассчитано 313,21; обнаружено 313,10.
Получение 1-(3-фторпропил)азетидин-3-амина этан-1,2-дисульфоната
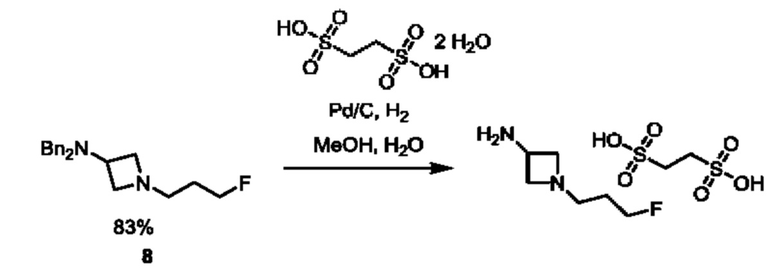
К раствору соединения 8 (2,00 г; 1,0 экв.) в МеОН (20,0 мл) добавляли раствор этандисульфоновой кислоты дигидрата (1,23 г; 1,0 экв.) в H2O (10,0 мл) при 0°С. Реакционную смесь нагревали до 30°С, пропускали через картридж с активированным углем. Затем в реакционную смесь загружали Pd/C (0,40 г; 0,20 экв.), перемешивали при 45°С под давлением Н2 40 ф/кв.дюйм (275,8 кПа), фильтровали, концентрировали до объема 5 мл; вносили МеОН (20,0 мл), перемешивали при 20°С в течение 3 ч, фильтровали, промывали МеОН (2 мл), получая указанное в заголовке соединение (1,36 г; выход 93%) в виде белого твердого вещества: 1Н ЯМР (400 МГц, D2O) δ 4.70-4.57 (m, 3Н), 4.57-4.31 (m, 4Н), 3.51 (t, J=7,2 Гц, 2Н), 3.24 (s, 4Н), 2.11-1.97 (m, 2Н); 19F ЯМР (376 МГц, D2O) δ - 219.59.
Пример 5
Получение (R)-1-(1Н-индол-3-ил)пропан-2-амина
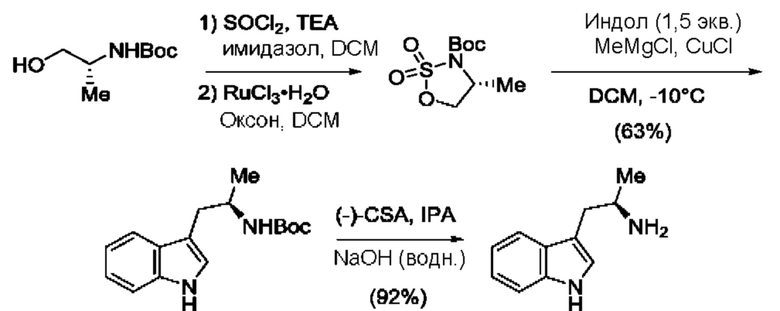
К раствору имидазола (102,8 г; 1,51 моль; 1,5 экв.) в DCM (1,33 л) по каплям добавляли SOCl2 (179,3 г; 1,51 моль; 1,5 экв.) при температуре от -5 до 0°С в атмосфере N2 в течение 30 мин. Реакционную смесь перемешивали в течение 0,5 ч при температуре от -5 до 0°С. По каплям добавляли трет-бутил-(R)-(1-гидроксипропан-2-ил)карбамат (Вос-аланинол) (177,7 г; 1,01 моль; 1,0 экв.) в DCM (1,33 л; 7,5 об.) при температуре от -5 до 0°С в течение 1 ч. Реакционную смесь перемешивали в течение 0,5 ч при температуре от -2 до 0°С, затем по каплям добавляли триэтиламин (204 г; 2,02 моль; 2 экв.) при температуре от -5 до 0°С. Полученную смесь перемешивали в течение 0,5 ч или до полного расходования N-Boc-аланинола, что подтверждали данными GC-анализа. К реакционной смеси добавляли воду (1,3 л) при 0-20°С. Фазы разделяли и водную фазу экстрагировали DCM (1,3 л). Органические фазы объединяли и последовательно промывали 10 масс. %-ным раствором лимонной кислоты (1,3 л), водным раствором NaHCO3 (1,3 л) и рассолом (1 л). Органическую фазу охлаждали до 0-10°С, после чего добавляли воду (3,1 л) и RuCl3⋅H2O (2,66 г) и затем добавляли оксон (927,1 г; 1,51 моль; 1,5 экв.). Реакционную смесь постепенно нагревали до 22°С и выдерживали в течение 3,5 ч или до полного расходования сульфамидитного промежуточного соединения, что подтверждали данными GC-анализа. Фазы разделяли и водную фазу фильтровали через набивку целита (50 г), затем промывали DCM (1,3 л). Фильтрат экстрагировали DCM (1,3 л), органические фазы объединяли и затем промывали насыщенным раствором Na2S2O3 (1,3 л) и рассолом (1 л × 2). Органическую фазу сушили с использованием Na2SO4 (50 г) и затем фильтровали, промывая DCM (200 мл). Объединенные фильтрат и промывки концентрировали под вакуумом при 30°С в течение 1 ч и затем дополнительно сушили под высоким вакуумом, получая 220 г трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида с чистотой более 99 масс. % и с фактическим выходом (скорректированным) 91,8% за 2 стадии. 1Н ЯМР (400 МГц, CDCl3): δ 4.66 (dd, J=9,2; 6,0 Гц, 1Н), 4.41 (qdd, J=6,4; 6,0; 2,8 Гц, 1Н), 4.19 (dd, J=9,2; 2,8 Гц, 1Н), 1.54 (s, 9Н), 1.50 (d, J=6,4 Гц, 3Н).
Другие реакции окисления обычно проводили в соответствии с методикой, приведенной непосредственно выше, варьируя окислитель, катализатор и растворитель. Результаты приведены ниже в Таблице 1, где "эксп." относится к номеру эксперимента, "продукт" относится к трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксиду, a "SM" относится к являющемуся исходным веществом Вос-аланинолу, "LC А%" относится к чистоте, выраженной в процентах площади по данным жидкостной хроматографии, и "ND" означает "не определено". Температура, при которой проводили реакции в экспериментах 1 и 3-9, составляла 0-25°С, а температура, при которой проводили реакции в эксперименте 2, составляла 0-40°С. Продолжительность реакций в экспериментах 1-4 и 7 составляла 4 часа, продолжительность реакций в экспериментах 5 и 6 составляла 8 часов, а продолжительность реакций в экспериментах 8 и 9 составляла 18 часов.
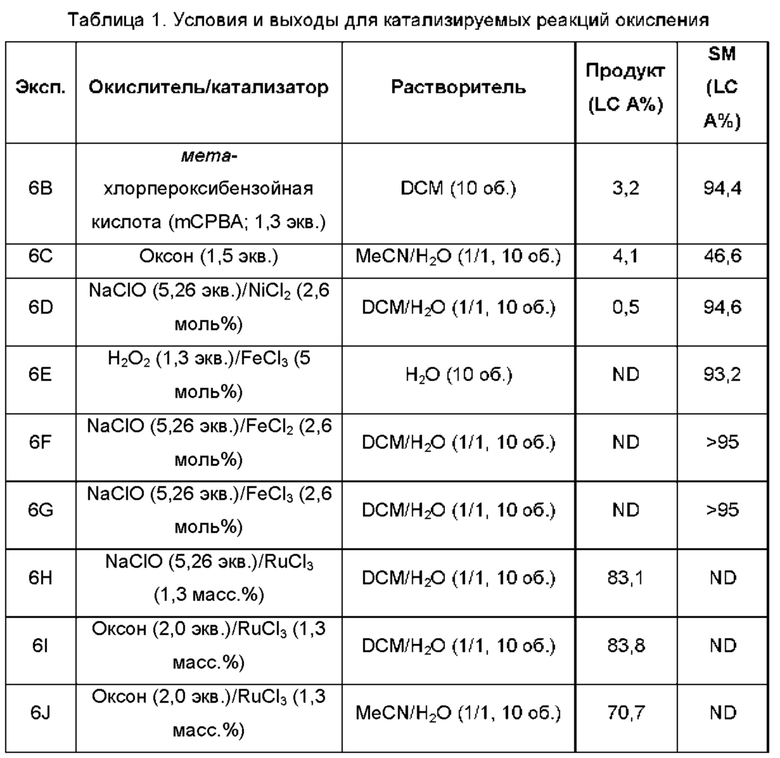
К смеси индола (7,4 г; 63,2 ммоль; 1,5 экв.) и CuCl (5,4 г; 54,7 ммоль; 1,3 экв.) в DCM (60 мл) добавляли MeMgCl (3,0 М раствор в THF; 18,7 мл; 56 ммоль; 1,33 экв.) при -15°С в атмосфере N2 в течение 10 мин. Полученную светло-желтую смесь перемешивали при -20°С в течение 10 мин и затем по каплям добавляли раствор трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида (10,0 г; 42,1 ммоль; 1,0 экв.) в DCM (40 мл) при -10°С в атмосфере N2 в течение 30 мин. Смесь перемешивали при -10°С в течение 2 часов или до завершения реакции, что подтверждали данными TLC (смесь 5/1 РЕ/ЕА (петролейный эфир/этил ацетат), по исчезновению трет-бутил-(R)-4-метил-1,2,3-оксатиазолидин-3-карбоксилат-2,2-диоксида) или по данными GC. Реакцию гасили путем добавления 10%-ного раствора лимонной кислоты (100 мл), поддерживая внутреннюю температуру меньше 5°С. Фазы разделяли и водную фазу экстрагировали DCM (100 мл × 2). Органические фазы объединяли и промывали рассолом (100 мл × 2), затем к органической фазе добавляли активированный уголь (5 г) и Na2SO4 (10 г). Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем фильтровали. Осадок на фильтре промывали DCM (50 мл × 2). Фильтрат и промывку объединяли и концентрировали под вакуумом при 30°С, получая неочищенный трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамат (16,9 г; 40,9 LC А%). К неочищенному трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамату добавляли гептан (200 мл) и смесь перемешивали при комнатной температуре в течение 1 часа, за это время в осадок постепенно выпадало беловатое твердое вещество. Это твердое вещество собирали фильтрованием и осадок на фильтре промывали гептаном (17 мл × 3). Твердое вещество сушили под высоким вакуумом при 25°С, получая 7,0 г трет-бутил-(К)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамата (25,5 ммоль; 97 А%, 99 масс. %). 1Н ЯМР (400 МГц, DMSO-d6) δ 10.76 (s, 1Н), 7.55 (d, J=7,9 Гц, 1Н), 7.32 (dt, J=8,1; 1,0 Гц, 1Н), 7.09 (d, J=2,3 Гц, 1Н), 7.05 (ddd, J=8,2; 7,0; 1,2 Гц, 1Н), 6.96 (ddd, J=8,0; 6,9; 1,1 Гц, 1Н), 6.71 (d, J=8,0 Гц, 1Н), 3.76-3.69 (m, 1Н), 2.86 (dd, J=13,9; 5,8 Гц, 1Н), 2.64 (dd, J=14,1; 7,7 Гц, 1Н), 1.37 (s, 9Н), 1.00 (d, J=6,6 Гц, 3Н).
Другие реакции алкилирования индолов обычно проводили в соответствии с методикой, приведенной непосредственно выше, варьируя разновидности катализатора на основе меди и стехиометрическое соотношение для реагента Гриньяра. Результаты приведены ниже в Таблице 2, где "эксп." относится к номеру эксперимента, "CuX" относится к разновидностям катализатора на основе меди, "экв." относится к эквивалентам, "продукт" относится к трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамату, "N1" относится к побочному продукту, в котором алкилирован индоламин, "ВА" относится к побочному продукту бисалкилу, "AY" относится к выходу по данным LC-анализа, "А %" относится к чистоте, выраженной в процентах площади по данным жидкостной хроматографии, и "ND" означает "не определено".
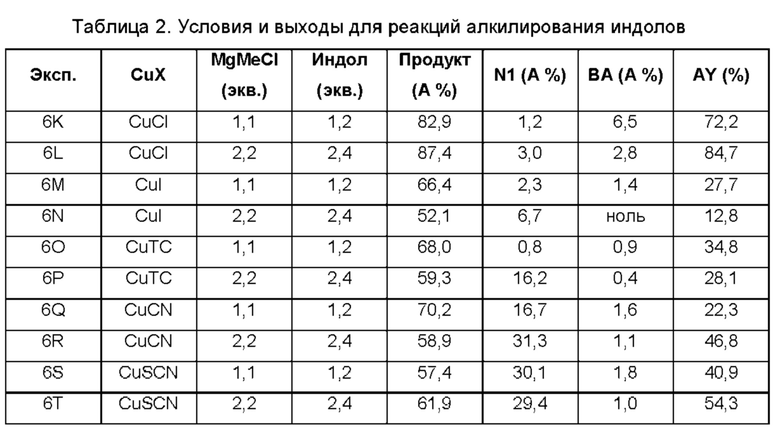
Другие реакции алкилирования индолов обычно проводили в соответствии с методикой, приведенной непосредственно выше, варьируя температуру реакции. Результаты приведены ниже в Таблице 3, где "эксп." относится к номеру эксперимента, "G темп." относится к температуре в°С, при которой проводили добавление реагента Гриньяра, "S темп." относится к температуре в°С, при которой проводили добавление сульфамидата, "продукт" относится к трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамату, "N1" относится к побочному продукту, в котором алкилирован индоламин, "ВА" относится к побочному продукту бисалкилу, "АУ относится к выходу по данным LC-анализа, "А %" относится к чистоте, выраженной в процентах площади по данным жидкостной хроматографии.

К раствору трет-бутил-(R)-(1-(1Н-индол-3-ил)пропан-2-ил)карбамата (0,5 г; 1,8 ммоль; 92 А%) в МеОН (5 мл; 10 об.) по каплям при 0°С добавляли HCl в МеОН (10 М раствор; 1,8 мл; 18 ммоль) в атмосфере N2 в течение 10 мин. Полученный раствор перемешивали при 0°С в течение 2 ч, затем при 25-30°С в течение 1 ч. Раствор концентрировали под вакуумом при 30°С, разбавляли водой (10 мл) и экстрагировали DCM (10 мл × 2). Значение рН водной фазы подводили до примерно 13, используя 1 М водный раствор NaOH при температуре 0-10°С, и затем экстрагировали DCM (20 мл × 4). Органическую фазу сушили над Na2SO4 (10 г). Осушитель отфильтровывали и осадок на фильтре промывали DCM (10 мл). Объединенные фильтрат и промывку концентрировали под вакуумом при 30°С, получая 0,31 г неочищенного (R)-1-(1Н-индол-3-ил)пропан-2-амина с 95,8 LC А% и энантиомерной чистотой 99,2% и с фактическим выходом 97%. 1Н ЯМР: 400 МГц (CDCl3): δ 1.20 (d, J=6 Гц, 3Н), 1.56 (s, br, 2Н), 2.69 (dd, J=14; 8 Гц, 1Н), 2.91 (dd, J=14; 4 Гц, 1Н), 3.30-3.33 (m, 1Н), 7.05 (s, 1Н), 7.14 (t, J=7 Гц, 1Н), 7.22 (t, J=7 Гц, 1Н), 7.38 (d, J=8 Гц, 1 Н), 7.64 (d, J=8 Гц, 1Н), 8.27 (s, 1Н).
Пример 6
Получение (R)-3-((1-(1Н-индол-3-ил)пропан-2-ил)амино)-2,2-дифторпропан-1-ола
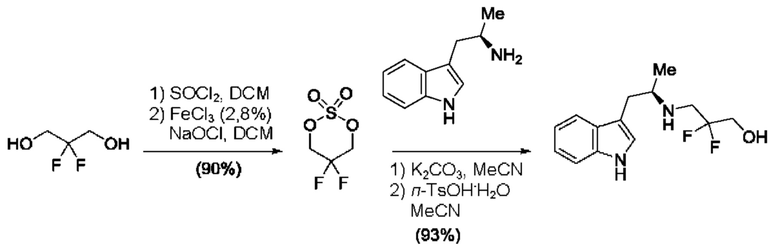
К раствору 2,2-дифторпропан-1,3-диола (51 кг; 90 масс. %; 409,5 моль; 1 экв.) в DCM (332 кг; 5 об.) добавляли тионилхлорид (58,4 кг; 491,4 моль; 1,2 экв.) при 20-25°С в течение 2 ч. Реакционную смесь нагревали до 30-35°С, перемешивали в течение 2 ч и для гашения реакции добавляли ледяную воду (255 кг). Фазы разделяли и водную фазу экстрагировали DCM. Органические фазы объединяли и промывали водой. К неочищенной органической фазе добавляли воду (255 кг) и FeCl3 (1,6 кг) и двухфазную смесь охлаждали до -3°С. Затем по каплям добавляли гипохлорит натрия (NaClO (8,1 масс. %); 680 кг; 1556 моль; 3,8 экв.) в течение 5 ч при температуре от -5 до 1°С и реакционную смесь перемешивали в течение 1 ч при 0°С. Реакционную смесь фильтровали через целит. Фазы разделяли и водную фазу экстрагировали DCM. Органические фазы объединяли и промывали водн. раствором Na2SO3 и рассолом, затем сушили над Na2SO4. Осушитель отфильтровывали и фильтрат концентрировали до объема прибл. 100 л. К полученной суспензии добавляли гептан (80 кг) и смесь далее концентрировали до объема прибл. 100 л. Твердые вещества собирали фильтрованием и сушили под вакуумом, получая 52,01 кг 5,5-дифтор-1,3,2-диоксатиан-2,2-диоксида в виде беловатых кристаллов (выход 72% за две стадии из 2,2-дифторпропан-1,3-диола). 1Н ЯМР (400 МГц, DMSO-d6) δ 5.14 (t, J=10,8 Гц, 1Н).
Для получения 5,5-дифтор-1,3,2-диоксатиан-2,2-диоксида оценивали различные другие катализаторы обычно в соответствии с методикой, приведенной непосредственно выше. Результаты представлены ниже в Таблице 4, где "эксп." означает номер эксперимента, "диол" означает 2,2-дифторпропан-1,3-диол, "продукт" означает 90 масс.%-ный 5,5-дифтор-1,3,2-диоксатиан-2,2-диоксид, а "выход" означает выход в анализе. Каждую реакцию гасили 5 объемами воды.
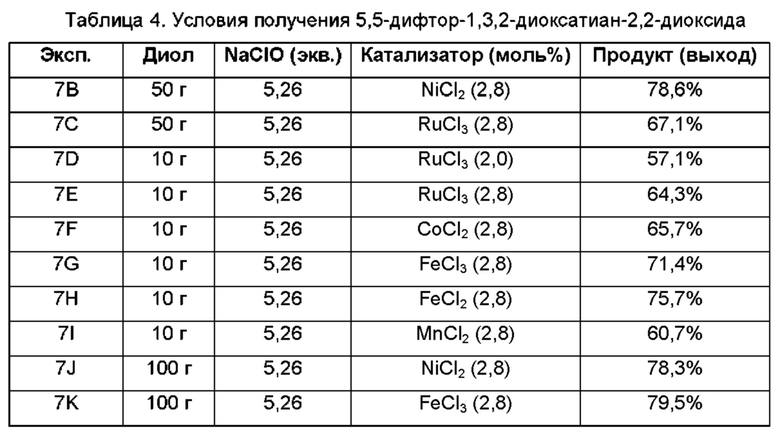
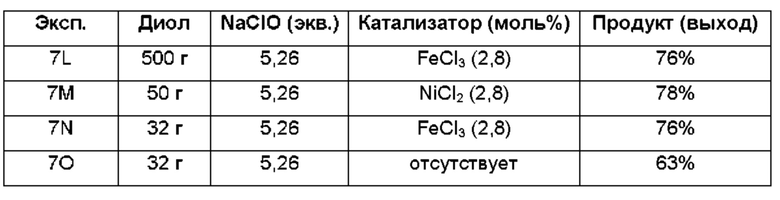
В колбу емкостью 2 л помещали (R)-1-(1Н-индол-3-ил)пропан-2-амин (99 масс. %; 100 г; 568,2 ммоль; 1 экв.), 5,5-дифтор-1,3,2-диоксатиан-2,2-диоксид (97,9 масс. %; 108 г; 608 ммоль; 1,07 экв.), K2CO3 (55 г; 398 ммоль; 0,7 экв.) и ацетонитрил (1 л; 10 об.). Полученную смесь нагревали до 80°С и перемешивали в течение 4 ч. Реакционную смесь охлаждали до 35°С и фильтровали. Осадок на фильтре промывали ацетонитрилом (100 мл × 2) и к объединенному фильтрату добавляли п-TsOH моногидрат (119 г; 625,6 ммоль; 1,1 экв.) и воду (100 мл; 1 об.). Полученную двухфазную смесь нагревали до 80°С и перемешивали в течение 3 ч. Затем смесь выливали в 1 л ледяной воды и рН смеси подводили до 9 насыщенным раствором Na2CO3 (350 мл) при температуре ниже 5°С. Фазы разделяли и водную фазу экстрагировали i-PrOAc (500 мл × 2). Органические фазы объединяли и промывали водой (500 мл) и рассол (500 мл × 2), затем сушили над Na2SO4 (30 г). Осушитель отфильтровывали и осадок на фильтре промывали i-PrOAc (100 мл × 2). Фильтрат и промывку объединяли и концентрировали под вакуумом при 40°С. Остаток растворяли в i-PrOAc (200 мл) и нагревали до 60°С. По каплям добавляли гептан (800 мл) при 60°С и затем полученную смесь перемешивали в течение 10 мин. Смесь медленно охлаждали до 30-35°С в течение 2 ч и затем дополнительно охлаждали до 0°С. После перемешивания в течение 10 мин, твердые вещества собирали фильтрованием и осадок на фильтре промывали гептаном (100 мл × 2). Полученное в результате этого твердое вещество сушили под вакуумом при 45°С, получая (R)-3-((1-(1Н-индол-3-ил)пропан-2-ил)амино)-2,2-дифторпропан-1-ол (155 г; 99,2 LC А%; 96 масс. %; фактический выход 97,6%). 1Н ЯМР (400 МГц, DMSO-d6) δ 10.79 (s, 1Н), 7.51 (dd, J=7,9; 1,2 Гц, 1Н), 7.33 (dt, J=8,2; 1,0 Гц, 1Н), 7.13 (d, J=2,3 Гц, 1Н), 7.05 (ddd, J=8,2; 6,9; 1,3 Гц, 1Н), 6.96 (ddd, J=8,0; 6.9; 1,2 Гц, 1Н), 5.35 (t, J=6,3 Гц, 1Н), 3.61 (td, J=13,8; 5,2 Гц, 2Н), 3.03-2.91 (m, 3Н), 2.83 (dd, J=14,0; 5,7 Гц, 1Н), 2.59 (dd, J=14,0; 7,2 Гц, 1Н), 1.69 (s, 1Н),0.96 (d, J=6,2 Гц, 3Н).
Пример 7
Получение 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола (2R,3R)-2,3-дигидроксисукцината
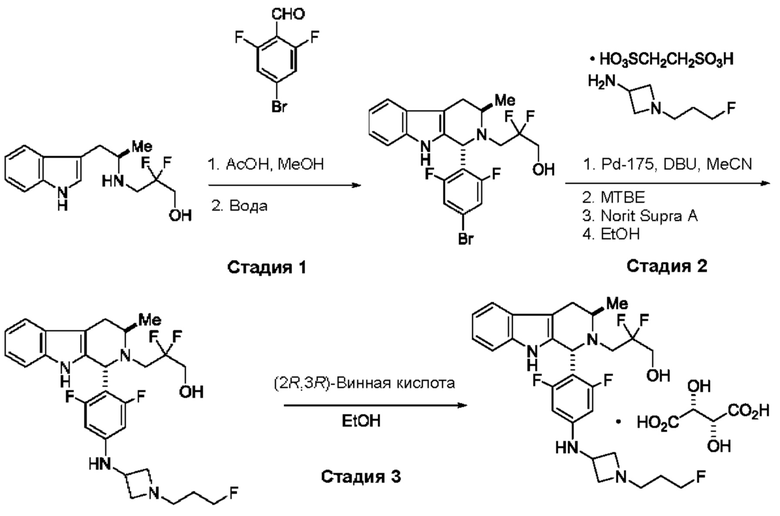
На стадии 1 в колбе с устройством для перемешивания объединяли 4-бром-2,6-дифторбензальдегид (131,79 г; 596,35 ммоль), (R)-3-((1-(1 Н-индол-3-ил)пропан-2-ил)амино)-2,2-дифторпропан-1-ол (160 г; 596,35 ммоль), уксусную кислоту (51,26 мл; 894,52 ммоль) и толуол. Реакционную смесь нагревали до 75°С и выдерживали в течение ночи, охлаждали и затем разбавляли толуолом. Полученный раствор затем гасили водным раствором карбоната калия, промывали рассолом и водой и обрабатывали активированным углем. После фильтрования раствор концентрировали и осуществляли кристаллизацию из смеси толуол/гептан, получая 3-((1R,3R)-1 -(4-бром-2,6-дифторфенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ол с выходом 81% в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ 10.59 (s, 1Н), 7.45-7.36 (m, 3Н), 7.20 (d, J=8,1 Гц, 1 Н), 7.05-6.92 (m, 2Н), 5.29 (t, J=6,1 Гц, 1 Н), 5.21 (s, 1Н), 3.71-3.55 (m, 1Н), 3.53-3.38 (m, 2Н), 3.17 (q, J=15,2 Гц, 1Н), 2.89 (ddd, J=15,3; 4,8; 1,5 Гц, 1Н), 2.71-2.55 (m, 2Н), 1.08 (d, J=6,5 Гц, 3Н). MS ([М+Н]+) рассчитано для C21H19BrF4N2O 470,06; обнаружено 470,80.
Альтернативно, 4-бром-2,6-дифторбензальдегид (67,2 г; 304 ммоль), (R)-3-((1-(1 Н-индол-3-ил)пропан-2-ил)амино)-2,2-дифторпропан-1-ол (80,0 г; 298 ммоль), уксусную кислоту (25,6 мл; 447 ммоль) и метанол объединяли в колбе с перемешиванием. Смесь кипятили с обратным холодильником в течение 24 часов, затем охлаждали. Твердые вещества выпадали в осадок из полученного раствора в результате добавления водного раствора карбоната калия. Полученную суспензию фильтровали и промывали, получая 3-((1R,3R)-1-(4-бром-2,6-дифторфенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ол в виде бледно-желтого твердого вещества с выходом 97%.
На стадиях 2 и 3, 3-((1R,3R)-1-(4-бром-2,6-дифторфенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ол (100 г; 212,2 ммоль), 1-(3-фторпропил)азетидин-3-амина этан-1,2-дисульфонат (82,09 г; 254,6 ммоль), ацетонитрил, DBU (1061 ммоль; 161,5 г; 159,4 мл) объединяли в колбе с перемешиванием, затем добавляли Pd-175 (5,304 ммоль; 4,144 г). Реакционную смесь нагревали до 75°С в течение 2 часов, охлаждали, концентрировали и затем разбавляли метил-трет-бутиловым эфиром. Этот полученный раствор обрабатывали водным раствором хлорида аммония, рассолом и водой, затем очищали, используя SiliaMetS Thiol. После фильтрования раствор концентрировали, разбавляли этанолом и осуществляли кристаллизацию с использованием винной кислоты, получая после фильтрования и промывки 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола (2R,3R)-2,3-дигидроксисукцинат в виде желтого твердого вещества с выходом 90%. 1Н ЯМР (400 МГц, DMSO-d6) δ 10.52 (s, 1Н), 7.38 (dd, J=7,5; 1,3 Гц, 1Н), 7.21-7.16 (m, 1Н), 7.02-6.90 (m, 2Н), 6.82 (d, J=6,9 Гц, 1Н), 6.18-6.08 (m, 2Н), 5.07 (s, 1Н), 4.53 (t, J=5,8 Гц, 1Н), 4.42 (t, J=5,9 Гц, 1Н), 4.18 (s, 2Н), 4.14-4.05 (m, 1Н), 3.93 (ddt, J=9,1; 7,0; 3,5 Гц, 2Н), 3.74-3.60 (m, 1Н), 3.51-3.32 (m, 2Н), 3.21-3.02 (m, 3Н), 2.88-2.73 (m, 3Н), 2.70-2.51 (m, 2Н), 1.83-1.66 (m, 2Н), 1.09-1.05 (m, 3Н). MS ([М+Н]+) рассчитано для C27H31F5N4O 522,24; обнаружено 523,00.
Пример 8
Получение 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола (2R,3R)-2,3-дигидроксисукцината
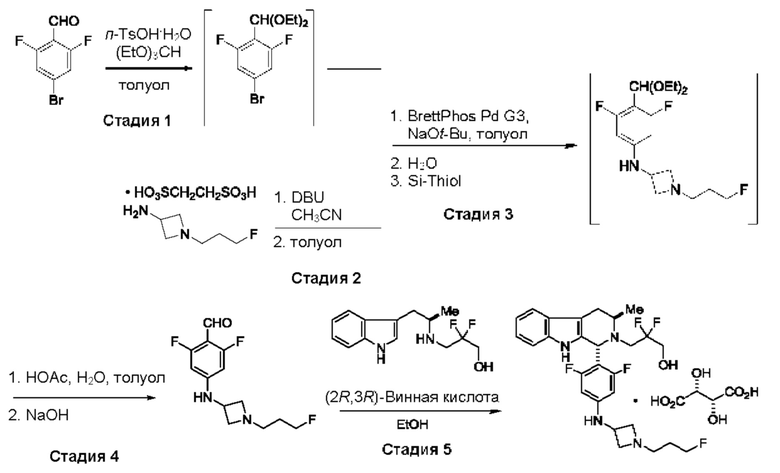
На стадиях 1-4 в реактор загружали 4-бром-2,6-дифторбензальдегид (75,0 г; 0,339 моль; 100 моль%), п-толуолсульфоновой кислоты моногидрат (162 мг; 0,000849 моль; 0,250 моль%) и толуол (225 мл). В течение 15 мин при КТ добавляли триэтилортоформиат (55,4 г; 62,1 мл; 0,373 моль; 110 моль%), смесь перемешивали при КТ в течение 1 ч и подвергали дистилляции, получая раствор 5-бром-2-(диэтоксиметил)-1,3-дифторбензола в толуоле. В другой реактор загружали 1-(3-фторпропил)азетидин-3-амина 2-(триоксиданилтио)этан-1-сульфонат (131,3 г; 0,407 моль; 120 моль%) и ацетонитрил (656 мл). В течение 15 мин при КТ добавляли DBU (124,0 г; 122,8 мл; 0,814 моль; 240 моль%) и смесь перемешивали при КТ в течение 2 ч, подвергали дистилляции с толуолом и фильтровали, получая раствор 1-(3-фторпропил)азетидин-3-амина в толуоле, который загружали вместе с NaOfBu (39,14 г; 0,407 моль; 120 моль %) в раствор 5-бром-2-(диэтоксиметил)-1,3-дифторбензола в толуоле. Смесь барботировали, загружали BrettPhos Pd G3 (3,076 г; 0,00309 моль; 1,00 моль %), барботировали, нагревали при 60°С в течение 18 ч, охлаждали, гасили водой и дважды промывали водой. Загружали SiliaMets Thiol (20,0 г) и смесь нагревали при 50°С в течение 2 ч, охлаждали и фильтровали, получая раствор N-(4-(диэтоксиметил)-3,5-дифторфенил)-1-(3-фторпропил)азетидин-3-амина в толуоле. Добавляли уксусную кислоту (21,4 мл; 0,373 моль; 110 моль %) и воду (300 мл) и смесь выдерживали при КТ в течение 2 ч. Водную фазу отделяли, обрабатывали водным раствором NaOH (50 масс. %; 21,5 мл; 0,407 моль; 120 моль%) и вносили затравку 2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)бензальдегида (923 мг; 0.00339 моль; 1,00 моль %). Полученные твердые вещества фильтровали, промывали водой и сушили, получая 2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)бензальдегид (77,9 г; выход 84,3%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10.05 (dd, J=1,2 Гц, 1Н), 6.05-5.97 (m, 2Н), 5.03 (d, J=6,4 Гц, 1Н), 4.54 (t, J=6,0 Гц, 1Н), 4.42 (t, J=6,0 Гц, 1Н), 4.14-4.03 (m, 1Н), 3.70 (td, J=6,8; 1,6 Гц, 2Н), 2.99-2.93 (m, 2Н), 2.59 (t, J=7,2 Гц, 2Н), 1.83-1.66 (m, 2Н). MS: рассчит. для C13H15F3N2O [М+Н]+=273,1; обнаружено=273,0.
На стадии 5 в реактор загружали (R)-3-((1-(1Н-индол-3-ил)пропан-2-ил)амино)-2,2-дифторпропан-1-ол (6,60 кг; 24,60 моль; 100 моль %), 2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)бензальдегид (6,70 кг; 24,60 моль; 100 моль%), L-винную кислоту (5,54 кг; 36,90 моль; 150 моль %) и этанол (39,6 л). Реакционную смесь нагревали при 70°С в течение 2 ч, вносили затравку 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола (2R,3R)-2,3-дигидроксисукцината (0,0827 кг; 0,123 моль; 0,5 моль %), перемешивали при 70°С в течение 2 суток, гасили этанолом (26,4 л), охлаждали и фильтровали для сбора твердого вещества. Твердое вещество промывали этанолом и сушили под вакуумом, получая 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола (2R,3R)-2,3-дигидроксисукцинат (12,7 кг; выход 78%) в виде беловатого твердого вещества.
Различные кислоты, отличные от винной кислоты, а также винную кислоту в качестве примера сравнения оценивали в реакции замыкания цикла на стадии 5, в общем случае в соответствии с методикой, непосредственно описанной выше. Результаты приведены ниже в Таблице 5, где "эксп." относится к номеру эксперимента, а "превращение" относится к степени превращения в конденсированную трициклическую кольцевую структуру. Применение соли винной кислоты привело к существенному увеличению выхода конечного выхода. Кроме того, применение соли винной кислоты свело к минимуму образование олигомеров в качестве побочных продукт во время синтеза, что привело к получению чистого продукта и меньшей эпимеризации конечного соединения.
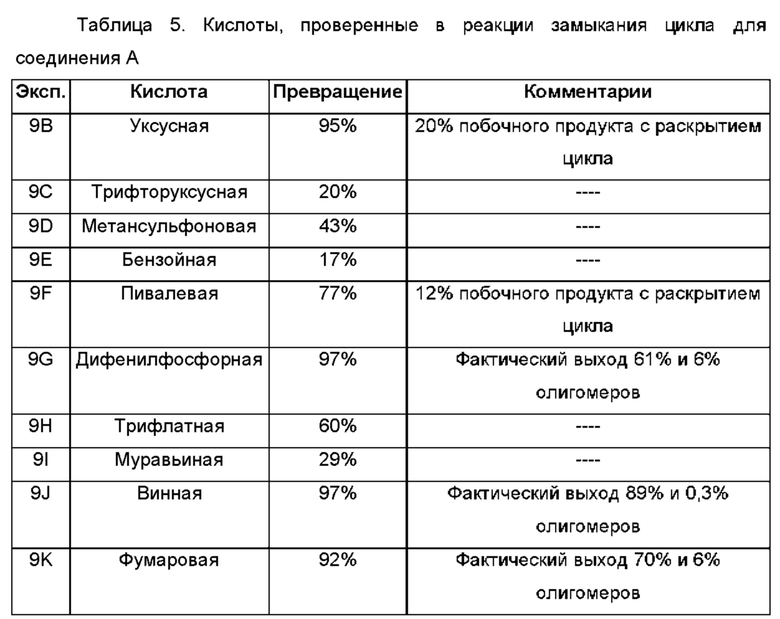
Пример 9
Перекристаллизация соединения В
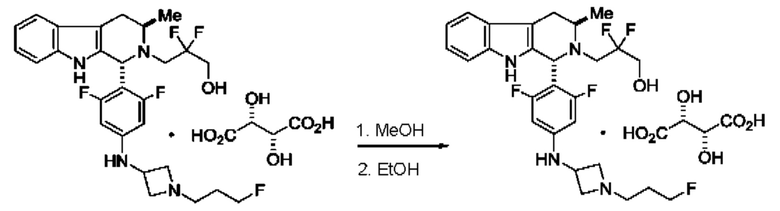
В реактор емкостью 250 мл загружали неочищенное соединение В (4,00 г; 5,95 ммоль) в смеси МеОН (90,0 мл) и EtOH (10,0 мл). Суспензию нагревали до 60°С, и она становилась гомогенной. В раствор добавляли затравку соединения В (200 мг; 0,297 ммоль; 5 моль %). Суспензию подвергали дистилляции с EtOH, охлаждали до 25°С в течение 1 ч, выдерживали при 25°С в течение 1 ч, фильтровали, сушили, получая соединение В (3,83 г; выход 91%) в виде беловатого твердого вещества.
Пример 10
Перекристаллизация соединения В
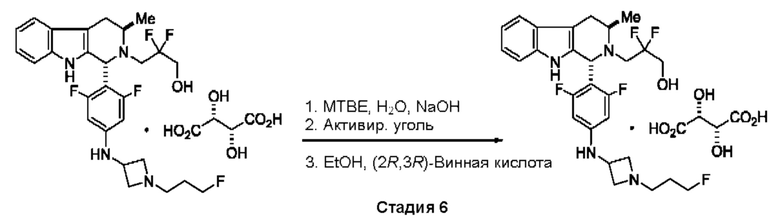
Пример 10А. В реактор емкостью 30 л загружали неочищенное соединение В (1,26 кг; 1,88 моль; 100 моль%) и МТВЕ (6,30 л; 5,00 мл/г). В эту суспензию добавляли раствор NaOH (154 г; 3,85 моль; 205 моль%) в воде (6,30 л; 5,00 мл/г). Реакционную смесь перемешивали при 20°С в течение 30 мин. Верхний органический слой дважды промывали водой (2,53 л; 2,00 мл/г), пропускали через картридж с активированным углем, подвергали дистилляции с EtOH, получая раствор в EtOH соединения В в форме свободного основания.
Пример 10В. В реактор емкостью 20 л загружали /.-винную кислоту (296 г; 1,97 моль; 105 моль%), EtOH (2,52 л; 2,00 мл/г) и нагревали до 70°С. В реактор емкостью 20 л переносили 20%-ный раствор соединения В в форме свободного основания. В этот раствор добавляли затравку соединения В (25,2 г; 0,0375 моль; 2 моль%). Оставшийся раствор соединения В в форме свободного основания переносили в реактор емкостью 20 л в течение 1 ч, выдерживали при 70°С в течение 30 мин, охлаждали до 20°С в течение 5 ч, выдерживали при 20°С в течение минимум 2 ч, фильтровали, три раза промывали EtOH (2,52 л; 2,00 мл/г), сушили, получая соединение В (1,12 кг; 89%) в виде беловатого твердого вещества.
Пример 11
Предложенные в данном изобретении соединения характеризовали методами масс-спектрометрии и/или ЯМР, известными в данной области техники. Для соединений из этого примера, если не указано иное, подтверждена стереохимическая конфигурация.
Соединение (М1)
Соединение (М2)
Соединение (М3)
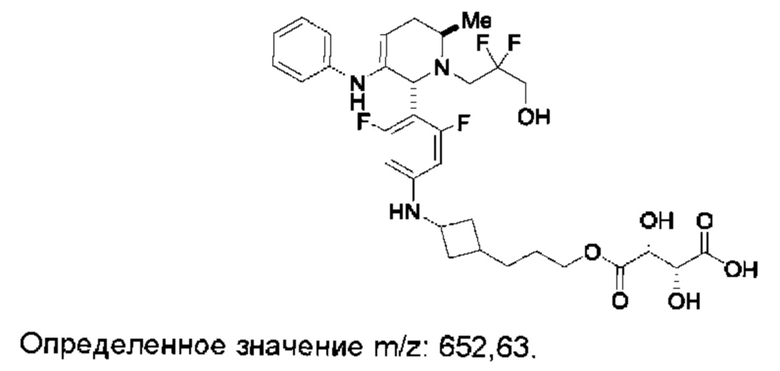
Соединение (М4)
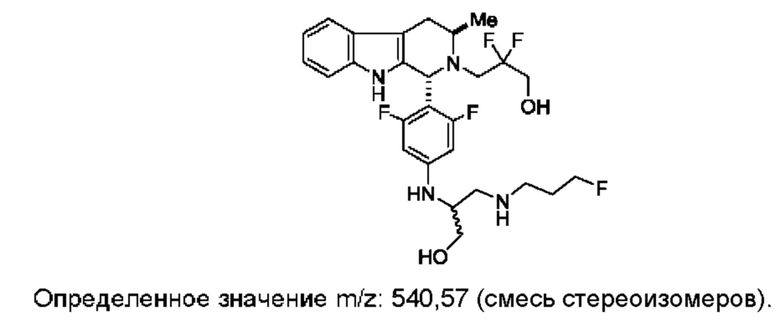
Пример 12
Превращения с участием ферментов
Скрининг трансаминаз в режиме кинетического разделения рацемического альфа-метилтриптамина выявил ферменты, обладающие значительной (R)-энантиоселективностью при использовании подхода асимметрического восстановительного аминирования и значительной (8)-энантиоселективностью при использовании подхода в режиме кинетического разделения. Типичные трансаминазы приведены в Таблице 6.

Асимметрическое восстановление с применением трансаминазы ТА-Р2-А07 проводили в буфере; использовали изопропиламин и органические сорастворители, такие как DMSO или ацетонитрил, в масштабе 5 г. Соединение (XXI) через 1 сутки превращалось в соединение (3), описанное в данной заявке, со степенью превращения выше 95%, с энантиомерным избытком (ЕЕ) выше 99%.
Восстановительное аминирование также проводили в органических растворителях, содержащих небольшие количества буфера и изопропиламина, причем трансаминаза ТА-Р2-А07 была иммобилизована на твердой подложке. Соединение (XXI) через 4 недели превращалось в соединение (3) со степенью превращения выше 95% и ЕЕ выше 99%. Применение органического растворителя сделало возможным увеличение загрузок субстратов до 5% (масс./масс.) включительно.
Кинетическое разделение проводили в присутствии трансаминазы АТА12 в (1) цельных клетках; (2) не содержащем клеток лизате или (3) неочищенном лиофилизате в буфере, содержащем пируват и органический сорастворитель, такой как ацетонитрил. Достигали энантиомерного избытка выше 99%. Достигали превращения в соединение (3) со степенью, равной или превышающей 50%. Истощение по нежелательному энантиомеру осуществляли посредством окислительного дезаминирования в сторону образования кетона.
Идентификация последовательностей для отобранных ТА
3FCR Y59F/Y87F/T231G: аминокислотная последовательность (SEQ ID NO: 1)

3FCR Y59W/Y87F/T231A: аминокислотная последовательность (SEQ ID NO: 2)

3FCR Y59F/S86A/Y87F/Y152F/T231A/I234M/L382M: аминокислотная последовательность (SEQ ID NO: 3)

3HMU1264V: аминокислотная последовательность (SEQ ID NO: 4)

Остатки, выделенные жирным шрифтом и подчеркиванием соответствуют мутациям по сравнению с природной последовательностью.
Пример 13
Характеристика соединения А в форме свободного основания, соли и полиморфа
Сокращения
MIBK Метилизобутилкетон
ACN Ацетонитрил
EtOAc Этил ацетат
EtOH Этанол
МТВЕ Метил-трет-бутиловый простой эфир
IPAc Изопропилацетат
MeTHF Метилтетрагидрофуран
СРМЕ Циклопентил-метиловый простой эфир
МеОН Метанол
THF Тетрагидрофуран
IPA Изопропиловый спирт
DMSO Диметилсульфоксид
DMC Диметилкарбонат
MEK Метилэтилкетон
DCM Дихлорметан
Скрининг полиморфов соединения в форме свободного основания. Для идентификации возможных полиморфных форм соединения А в виде свободного основания проводили автоматизированный высокопроизводительный скрининг (HTS) в 96-луночных планшетах с использованием системы Symyx СМ2 (Freeslate Inc., СА). Соединение А вносили в каждую лунку (по 8 мг/лунка), используя автоматический дозатор порошка, после чего добавляли по 800 мкл растворителя (чистого или смеси растворителей) и суспензию перемешивали в течение 2 часов при 50°С. Сначала соединение А вносили в лунки планшета для суспензий, используя либо этилацетат (в случае тартрата), либо MIBK (в случае фумарата) для поддержания его твердой формы. Из этого эталонного планшета отфильтровывали супернатант и распределяли на три отдельных планшета для проведения упаривания, осаждения посредством добавления антирастворителя и контролируемого охлаждения в течение 8-10 ч от 50°С до 20°С. Во всех случаях оставшиеся количества растворителей либо выпаривали, либо сливали и твердое вещество исследовали с использованием микроскопии в поляризованном свете и XRPD.
Пример 14
Скрининг солей. Согласно приблизительным данным относительно растворимости формы свободного основания и списку желательных кислот для скрининга использовали пять систем растворителей. Сначала приблизительно 20 мг аморфного соединения в форме свободного основания диспергировали в 0,5 мл выбранного растворителя в стеклянном флаконе и затем добавляли соответствующую кислоту при соотношении молярных зарядов 1:1. В случае HCl предпринимали попытки получения избыточного молярного соотношения 2:1 (кислота/свободное основание) ввиду наличия у соединения двух основных функциональных группы. Смеси перемешивали при КТ в течение ночи. Полученные твердые вещества анализировали посредством XRPD. Прозрачные растворы, полученные после смешивания, перемешивали при 5°С в течение 2 суток, затем в каждый из прозрачных растворов в ACN/H2O (19:1; об./об.) добавляли по 0,5 мл H2O, тогда как в другой системе растворителей в каждый из прозрачных растворов добавляли по 0,5 мл н-гептана, после чего перемешивали при 5°С в течение приблизительно 3 суток и конечные прозрачные растворы переносили для медленного упаривания при КТ, чтобы идентифицировать как можно больше вариантов кристаллов.
В первом раунде скрининга были выявлены варианты кристаллических форм, приведенные в Таблице 7. В случае тартрата и фумарата проводили дополнительное масштабирование в количестве от 50 мг до 1,5 г с целью дальнейшего определения характеристик. Тестирование в разных системах растворителей и в разных условиях позволило а) детально охарактеризовать разные полиморфные формы, полученные в виде вариантов при проведении данного скрининга, и b) определить условия ингибирования и контроля образования цис-эпимеров. Содержание эпимеров варьировало от менее 1% и до 22% в разных партиях соединения в форме свободного основания.
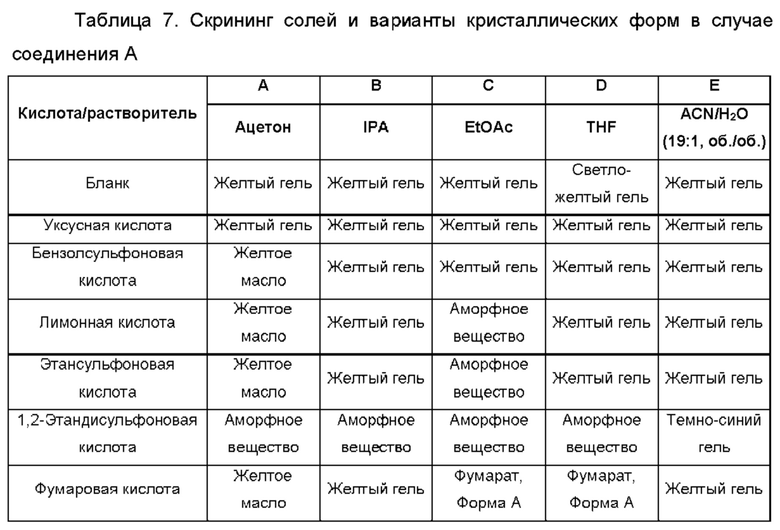
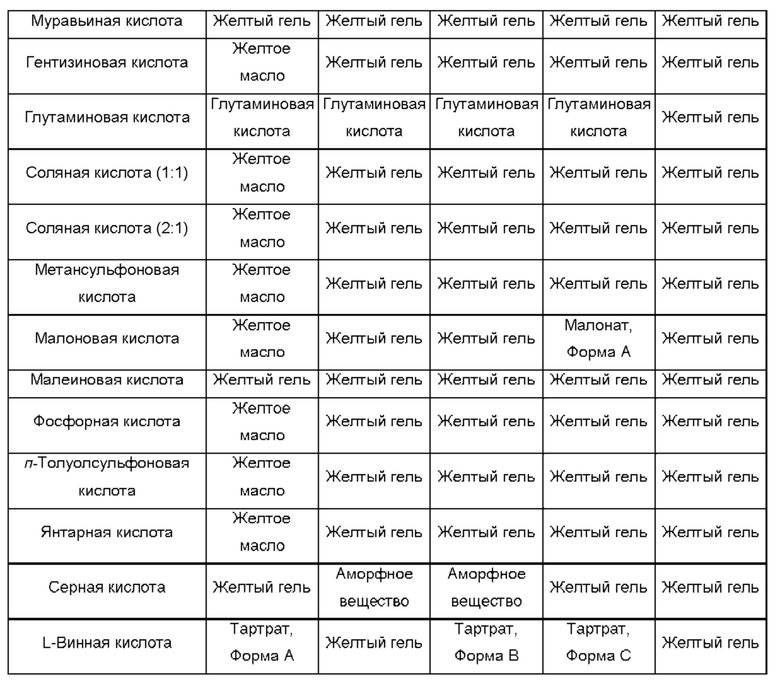
Методы
Рентгеновская порошковая дифрактометрия (XRPD) в условиях окружающей среды. Картины XRPD получали, используя порошковый рентгеновский дифрактометр PANalytical Empyrean (PANalytical Inc., Lelyweg, Netherlands). Образец порошка упаковывали в кремниевый держатель с нулевым фоном, и измерения проводили в режиме отражения (в конфигурации Брэгга-Брентано). Прибор был оборудован источником Cu-Ka-излучения с напряжением и током трубки 45 кВ и 40 мА, соответственно. Данные собирали при температуре окружающей среды в диапазоне от 3,0 до 40,0°2θ, используя размер шага 0,0263°, с частотой вращения 8 с. На пути падающего луча располагали щель Соллера (0,02°), антирассеивающую щель с фиксированной установкой 1°, маску падающего луча с фиксированной установкой 10 мм и программируемую щель расходимости, работающую в автоматическом режиме. Для линейных детекторов использовали вырезающую диафрагму. На пути дифрагированного луча располагали щель Соллера (0,02°), программируемую щель расходимости, работающую в автоматическом режиме, и никелевый фильтр N-β. Детектор PIXcel 1D использовали в режиме линейного сканирующего детектора (1D). Данные анализировали, используя имеющееся в продаже программное обеспечение (JADE®, версию 9, Materials Data Inc., Livermore, CA).
Анализ сорбции воды. Примерно 5-6 мг образца порошка помещали в поддон для образцов автоматического анализатора сорбции воды (Q5000SA, ТА Instruments, Newcastle, DE) при 25°С и скорости потока азота 200 мл/мин. Сначала образец "сушили" при RH 0% в течение в общей сложности 400 минут (при 60°С, затем при 25°С), после чего его выдерживали в условиях постепенного повышения RH в диапазоне 0-90% с шагом 10%, с временем выдерживания 200 минут для каждого значения RH. После этого выполняли постепенное снижение RH с шагом 10% обратно до значения RH 0%, используя тот же протокол.
Дифференциальная сканирующая калориметрия (DSC). Анализу подвергали приблизительно 3-8 мг образца порошка, используя прибор для DSC Q2000™ (ТА Instruments, New Castle, DE), оборудованный рефрижераторным охлаждающим устройством. Образцы упаковывали в негерметичные поддоны (алюминиевые поддоны Tzero™) и обычно нагревали в диапазоне 0-200°С при скорости 10°С/мин с продувкой сухим азотом. Калибровку прибора проводили, используя сапфир (базовая линия) и индий (температура и постоянная ячейки). Данные анализировали, используя имеющееся в продаже программное обеспечение (Universal Analysis 2000, версию 4.7А, ТА Instruments).
Термогравиметрия (TGA). В термогравиметрическом анализаторе (Discovery TGA, ТА Instruments) нагревали по 3-5 мг образцов соединения А в открытом алюминиевом поддоне при температуре от КТ до 350°С со скоростью нагревания 10°С/мин с продувкой сухим азотом. Калибровку температуры проводили, используя Alumel® и никель. Для калибровки по массе использовали стандартные разновесы по 100 мг и 1 г.
Микроскопия в поляризованном свете (PLM). Образцы диспергировали в силиконовом масле и рассматривали с использованием кросс-поляризационных фильтров микроскопа Leica DM 4000 В с улучшенным видеоизображением, оборудованного камерой высокого разрешения на приборах с зарядовой связью (CCD) и предметодержателем, снабженным приводом (Clemex Technologies Inc., Longueuil, Quebec, Canada), при 200-кратном увеличении. Микрофотоснимки получали, используя программное обеспечение Clemex Vision РЕ (Clemex Technologies Inc., Longueuil, Quebec, Canada).
Сканирующая электронная микроскопия (SEM). Образец порошка напыляли на держатель образцов для SEM и затем исследовали, используя настольный сканирующий электронный микроскоп (SEM) Phenom (Nanoscience Instruments, Inc., AZ). Микроснимки получали при разном увеличении.
Распределение частиц по размерам (PSD). Анализ размера частиц проводили с использованием прибора Mastersizer 2000 от Malvern, оборудованного приставкой для влажного диспергирования Hydros 2000SM (Malvern Instruments Ltd., Malvern, UK). Приблизительно 40 мг активного фармацевтического ингредиента (API) отвешивали во флакон и добавляли 1 мл 0,1%-ного Спана 85 в гептане. Содержимое флакона обрабатывали ультразвуком в течение 10 секунд, примерно 0,5 мл добавляли в пробоотборник при скорости перемешивания 1500 об./мин и PSD определяли при затемнении 10-20%. Ввиду наличия в образце больших кластеров, которые быстро осаждались из суспензии, диспергирующее вещество заменяли на 0,2%-ный Спан 85 в гептане для стабилизации суспензии. Для разрушения этих кластеров применяли внешнюю обработку ультразвуком 2, проводя два следующих друг за другом воздействия продолжительностью 5 минут, и полученные с применением PLM изображения собирали до и после ультразвуковой обработки. Чтобы обеспечить гомогенность, перед сбором данных аликвоту перемешивали в пробоотборнике в течение 2 минут. Прибор дважды промывали изопропиловым спиртом (IPA) и один раз гептаном, после чего заполняли 0,1%-ным Спаном 85 в гептане для каждого образца. После проведения анализа последнего образца прибор один раз промывали IPA. Приведены данные для повторов с 5-минутной ультразвуковой обработкой.
Анализ удельной поверхности по методу Брунауэра-Эммета-Теллера (BET). Измерение площади поверхности проводили, используя прибор ASAP 2460 от Micromeritics с приставкой Smart VacPrep от Micromeritics (Micromeritics Instrument Corp., GA). Образец массой от 500 мг до 1 г отвешивали в пустую пробирку ASAP 2460 и помещали в систему Smart VacPrep, дегазировали в течение 24 часов в условиях окружающей среды и затем подвергали адсорбции газа криптона при 25°С, и выдерживали под давлением 100 мм Hg (13,3 кПа). Измерение по 11 точкам выполняли при относительном давлении в диапазоне 0,050-0,300 и анализ данных проводили, используя предоставленное поставщиком программное обеспечение MicroActive.
Спектроскопия твердотельного ядерного магнитного резонанса (ттЯМР). Все эксперименты с 13С ттЯМР (при скорости вращения 8 кГц) проводили, используя прибор от Bruker (500 МГц) (Bruker BioSpin GmbH, Karlsruhe, Germany). Данные для 13C получали, используя последовательность импульсов с кросс-поляризацией при полном подавлении боковых полос (CP/TOSS). Для усреднения сигнала собирали 1-2 тысячи сканограмм. Использовали время контакта 2 мс и время ожидания восстановления 5 секунд. Для развязки применяли последовательность Spinal 64 с длительностью импульса 5,3 микросекунды. Использовали длительность 1Н 90-градусного импульса 2,9 микросекунды. Все эксперименты с 19F ттЯМР (при скорости вращения 14 кГц) проводили, используя прибор от Bruker (500 МГц). Данные для 19F получали, используя последовательности импульсов с CP и прямой поляризацией. Для усреднения сигнала собирали 64-256 тысяч сканограмм. Использовали время контакта 750 микросекунд и время ожидания восстановления 7 секунд. Использовали длительность 1Н 90-градусного импульса 3,54 микросекунды.
Пример 15
Предварительное определение характеристик соединения А в форме свободного основания. Было обнаружено, что соединение А в форме свободного основания является аморфным веществом. Перед скринингом характеристики исходного вещества, соединения А в форме свободного основания, определяли, используя XRPD, TGA и модулированную DSC (mDSC). Как показывают результаты определения характеристик, представленные на ФИГ. 32 и ФИГ. 33, исходное соединение А было аморфным, демонстрируя уменьшение массы на 9,3% вплоть до 220°С в TGA и отсутствие какого-либо существенного сигнала стеклования при проведении mDSC. Соединение А в форме свободного основания в виде белого твердого вещества получали путем добавления антирастворителя в МеОН/H2O (3:20, об./об.) со встряхиванием и высушивания на воздухе в течение приблизительно 7 суток.
Скрининг полиморфов. Оценку растворимости соединения А в форме свободного основания выполняли в 16 растворителях при КТ (Таблица 8). Эксперименты по скринингу полиморфов проводили, используя разные методы кристаллизации из растворов или методы перевода в твердую фазу. Использованные методы и идентифицированные кристаллические формы суммированы в Таблице 9.
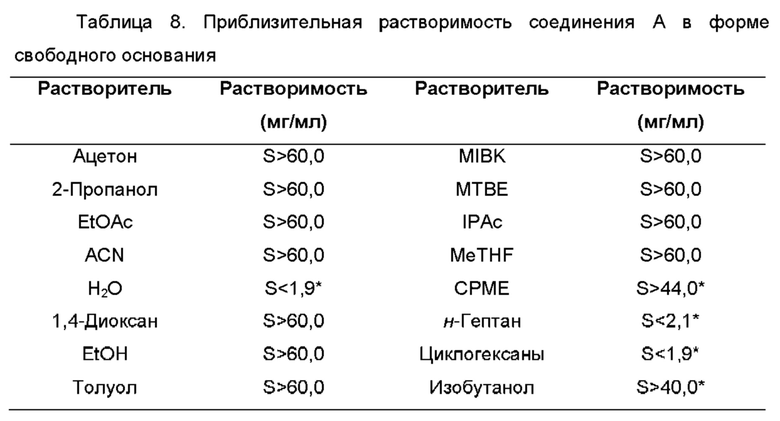
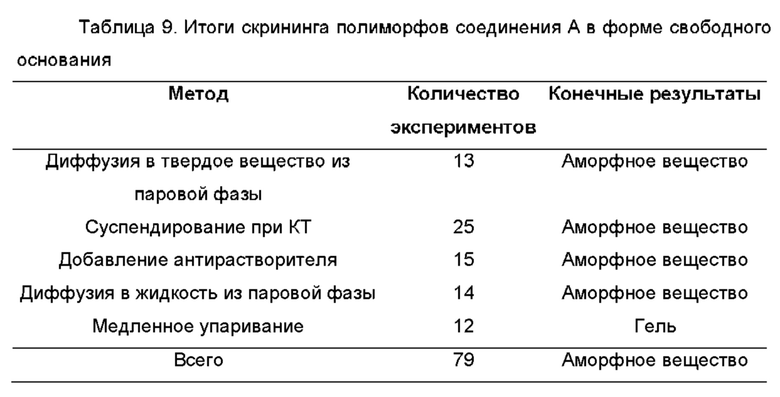
Диффузия в твердое вещество из паровой фазы. Эксперименты по диффузии в твердое вещество из паровой фазы проводили, используя 13 растворителей разных видов. В каждом эксперименте отвешивали примерно по 15 мг исходного вещества во флакон емкостью 3 мл, который помещали во флакон емкостью 20 мл, содержащий 2 мл соответствующего летучего растворителя. Флакон емкостью 20 мл герметично закрывали крышкой и выдерживали при КТ в течение 11 суток, чтобы пары растворителя могли взаимодействовать с твердым образцом. Извлеченные твердые вещества тестировали посредством XRPD. Как суммировано в Таблице 10, получали только масло или аморфное вещество.

Превращение суспензии при КТ. Эксперименты по превращению суспензии проводили при КТ в разных системах растворителей. В стеклянном флаконе емкостью 1,5 мл суспендировали примерно 40 мг исходного вещества в 0,3 мл растворителя. После перемешивания все прозрачные растворы переносили на 5°С, после чего проводили медленное упаривание при КТ в течение 3 суток. Итоговые результаты указывали на получение только аморфного вещества, геля или масла.


Добавление антирастворителя. В общей сложности проводили 15 экспериментов с добавлением антирастворителя. В стеклянный флакон емкостью 20 мл отвешивали примерно 30 мг исходного вещества и растворяли в 0,15 мл соответствующего растворителя при КТ. Антирастворитель добавляли постепенно до появления осадка или до тех пор, пока общее количество антирастворителя не достигало 12 мл, при этом образец перемешивали с помощью магнитной мешалки. Для проведения XRPD-анализа осадок извлекали. Если образования твердого вещества не наблюдали, то прозрачные растворы перемешивали с помощью магнитной мешалки при 5°С в течение ночи и затем упаривали при КТ. Результаты, приведенные в Таблице 12, показали, что образовывались только аморфное вещество или гель.

Диффузия в жидкость из паровой фазы. Эксперименты по диффузии в жидкость из паровой фазы проводили для 14 условий (Таблица 13). В каждый стеклянный флакон емкостью 3 мл отвешивали примерно по 30 мг исходного вещества. Добавляли соответствующий растворитель, получая раствор. Флакон помещали в стеклянный флакон емкостью 20 мл, содержащий 3 мл соответствующего антирастворителя, который герметично закрывали и выдерживали при КТ, давая парам возможность в течение примерно 11 суток взаимодействовать с раствором. Для проведения XRPD-анализа осадок извлекали. Прозрачные растворы переносили для упаривания при КТ.

Медленное упаривание. Эксперименты по медленному упариванию проводили для 12 условий. В каждом эксперименте в стеклянный флакон емкостью 3 мл отвешивали примерно по 20 мг исходного вещества, затем добавляли соответствующий растворитель или смесь растворителей, получая прозрачный раствор. После этого флакон закрывали парафильмом с 3-4 крошечными отверстиями и хранили при 4°С, давая раствору возможность медленно упариваться. Как приведено в Таблице 14, получали только гель.

В Таблице 15 обобщены формы, которые получали в результате проведения вручную скрининга, как описано в данной заявке.
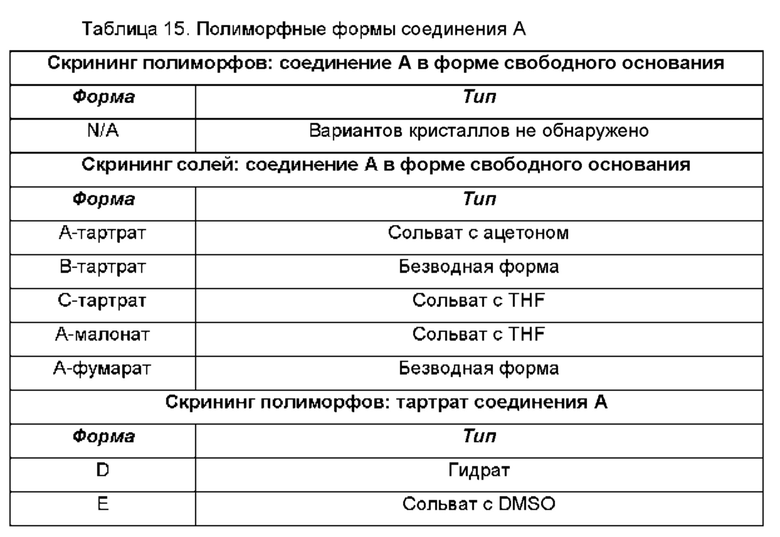
Пример 16
Определение характеристик Формы А соединения В. Форму А соединения В получали в масштабе 200 мг посредством происходящей в процессе реакции кристаллизации (reactive crystallization) из раствора в ацетоне, что подтверждается данными XRPD (ФИГ. 1). Как показано на ФИГ. 2а и ФИГ. 2b, при проведении TGA наблюдали уменьшение массы примерно на 7,2% вплоть до 125°С, а результатом DSC стало обнаружение одной эндотермы при 124,3°С (температура начала разложения). С использованием 1Н ЯМР было показано, что молярное соотношение составляло 0,98 (кислота/свободное основание), и был обнаружен ацетон в количестве 5,6% (молярное отношение к свободному основанию составляло 0,69). Для дальнейшего изучения Формы А проводили эксперименты с нагреванием. Не наблюдали никакого изменения формы после нагревания Формы А до 90°С, но после нагревания до 140°С происходило превращение образца в аморфное вещество. После нагревания образца Формы А до 90°С по-прежнему наблюдали значительное количество ацетона (4,5%). Образец Формы А состоял в основном из тонкодисперсных частиц и некоторого количества агрегатов (ФИГ. 3). На основании полученных данных Форма А соединения В представляет собой сольват с ацетоном, и потеря растворителя происходит одновременно с плавлением.
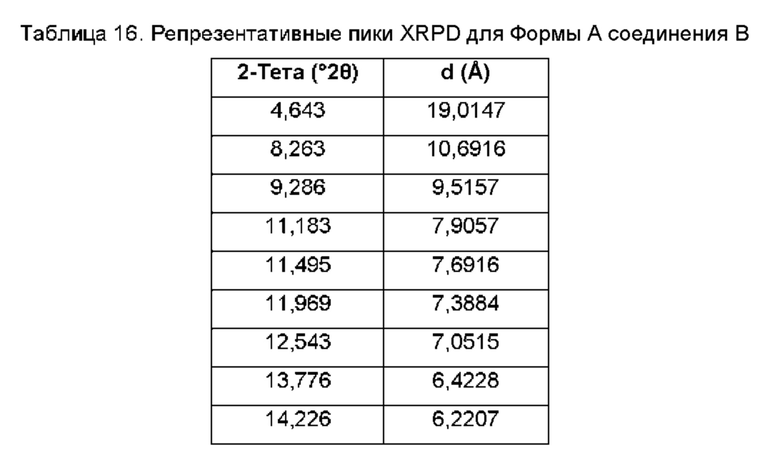
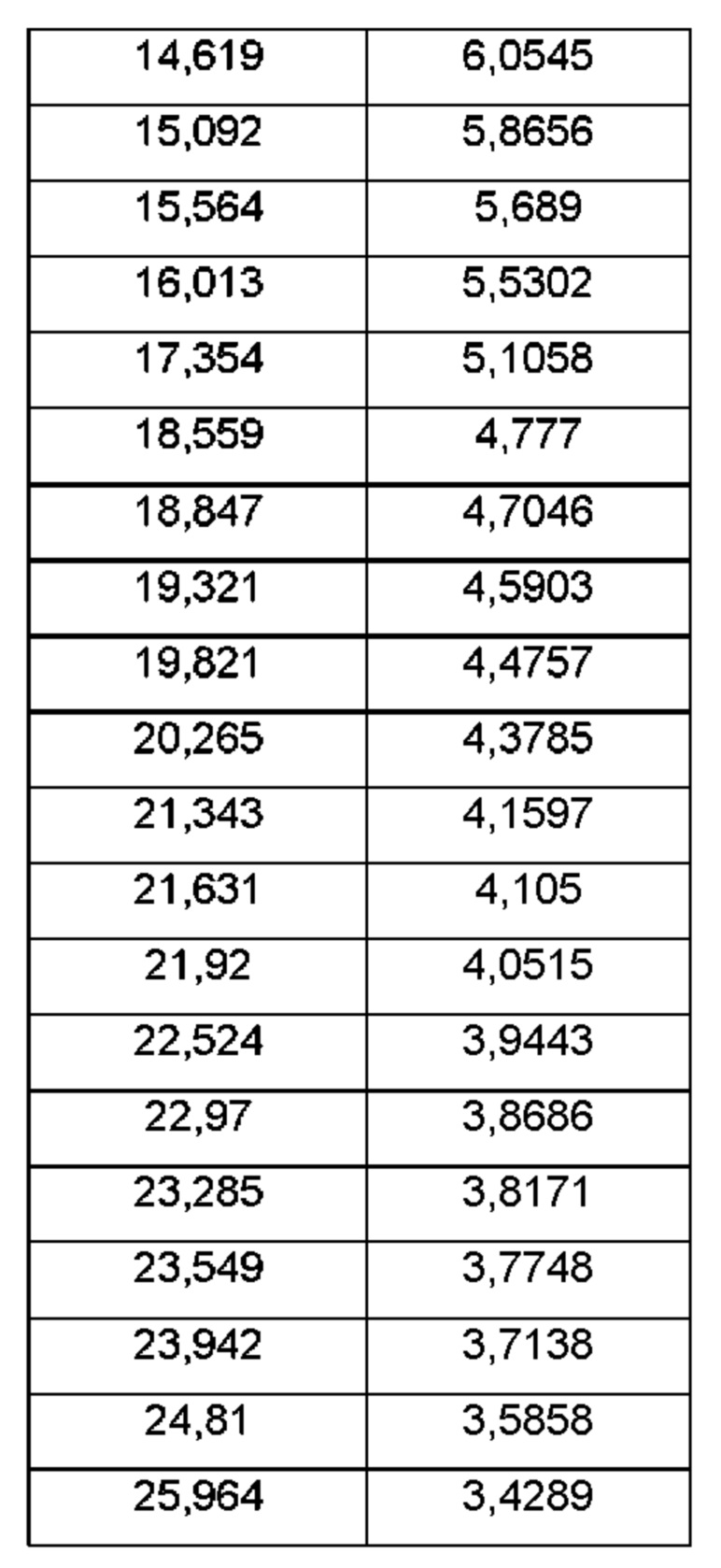
Получение Формы А соединения В. В стеклянный флакон емкостью 5 мл отвешивали примерно 57,5 мг винной кислоты и добавляли 2,0 мл ацетона, получая прозрачный раствор. К прозрачному раствору кислоты в этом флаконе емкостью 5 мл добавляли 2,0 мл концентрированного раствора соединения в форме свободного основания в ацетоне (примерно 100 мг/мл) в молярном соотношении 1:1 и перемешивали при КТ. В качестве затравки добавляли примерно 1 мг Формы А соединения В, и раствор становился мутным. Образец перемешивали в течение ночи, после чего проводили измерения с использованием XRPD, как описано в данной заявке. Полученная картина соответствовала Форме А соединения В. Затем суспензию перемешивали при КТ в течение еще 24 часов и осадок на фильтре сушили при 50°С в течение 1,5 ч. Получено 170,4 мг с выходом приблизительно 65,3%.
Пример 17
Определение характеристик Формы В соединения В. Форму В соединения В получали в масштабе 200 мг посредством происходящей в процессе реакции кристаллизации из раствора в EtOAc, что подтверждается данными XRPD (ФИГ. 4). Как показано на ФИГ. 5а и ФИГ. 5b, при проведении TGA наблюдали ограниченное уменьшение массы примерно на 1,3% вплоть до 140°С, а результатом DSC стало обнаружение одной эндотермы при 156,7°С (температура начала разложения). С использованием 1Н ЯМР стехиометрическое соотношение было определено как 1,08 (кислота/свободное основание), и был обнаружен EtOAc в количестве 1,4% (молярное отношение к свободному основанию составляло 0,11). На основании полученных характеристик Форма В соединения В представляет собой безводную форму.
Характеристики твердого состояния указывают на то, что Форма В является по существу кристаллической (XRPD), а результаты TGA указывают на наличие небольшого количества распределенного по поверхности растворителя, составляющего приблизительно 2% по данным начального уменьшения массы в диапазоне от КТ до 100°С, с началом плавления приблизительно при 163°С. Начало плавления после неглубокой эндотермы (испарение остаточного растворителя) происходит при 163°С. На основании производной кривой уменьшения массы было обнаружено очень раннее начало уменьшения массы (2,4% масс./масс.) вплоть до 100°С, что может быть связано с распределенным по поверхности растворителем. Общее уменьшение массы, включая уменьшение при плавлении, составляет 3,5% (масс./масс). Было обнаружено, что Форма В является слегка гигроскопичной и характеризуется поглощением влаги, составляющим 1,2% масс/масс, при RH 90% и 25°С, как показано на профиле сорбции-десорбции воды на ФИГ. 8. Спектры 13С (ФИГ. 6) и 19F ттЯМР (ФИГ. 7) дополнительно указывали на образование Формы В. Полученные с использованием SEM (ФИГ. 9а, при 500-кратном увеличении) и PLM (ФИГ. 9b, при 200-кратных увеличениях) изображения соединения В показывают, что Форма В содержит плотные сферические агрегаты.
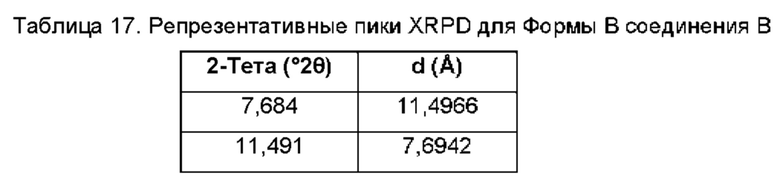
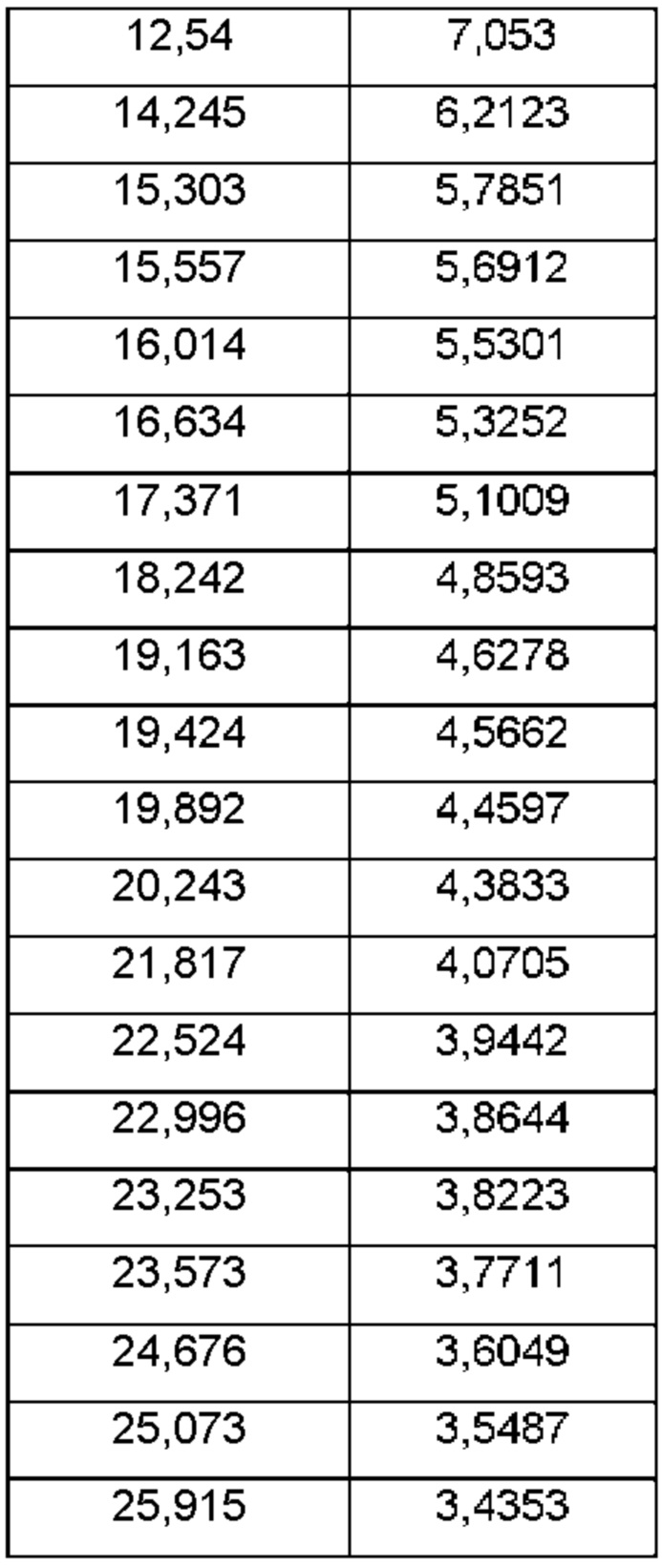
Предварительный анализ устойчивости к внешним воздействиям Формы В соединения В. Картины XRPD для солей соединения В (фумарата и тартрата) после выдерживания при 40°C/RH 75% в течение одного месяца в открытом состоянии, показали, что данная твердая форма в этих условиях не претерпела изменений.
Несмотря на то что никакой корреляции между содержанием эпимеров, изменяющимся в диапазоне 0,56-0,72%, и свойствами разных солевых форм в твердом состоянии не наблюдали, данные ттЯМР, XRPD и DSC указывают на возможность получения смеси форм, существующих для фумаратных солей, и что регулирование содержания форм любой из солей требовало значительных дальнейших усилий с учетом вариабельности в структурных данных и точках плавления при изменении условий кристаллизации. В тартратной соли отсутствовали примеси, обнаруженные в образце фумаратной соли в количестве 0,18%, после выдерживания в условиях 40°C/RH 75% в течение одного месяца.
Анализ сжатия Формы В соединения В. На ФИГ. 22, ФИГ. 23 и ФИГ. 24 демонстрируется влияние сжатия на тартратную Форму В соединения В. Проводили анализ брикетов массой 250 мг для чистой Формы В соединения В. Данные как ттЯМР, так и XRPD (ФИГ. 23 и ФИГ. 22, соответственно) показывают, что эта форма остается неизменной при сжатии. При сжатии наблюдали уменьшение времени релаксации 19F T1, что связано с возникновением разупорядоченности, хотя очень незначительной, судя по спектрам 19F ттЯМР для подвергнутой сжатию Формы В соединения В, показанным на ФИГ. 23. Значения времен релаксации 19F T1 включены в Таблицу 18 для исходной и подвергнутой сжатию Формы В соединения В. Как видно на термограмме DSC (ФИГ. 24), сжатие не влияло на точку плавления Формы В соединения В. Сравнительные XRPD-данные, полученные до и после выдерживания в условиях "ускоренного старения" при 30°C/RH 65% и 40°C/RH 75% в течение вплоть до 6 месяцев (в открытом состоянии), показали, что Форма В соединения В остается неизменной после выдерживания в этих условиях.
Испытание стабильности методом "ускоренного старения

XRPD образца после воздействия условий "ускоренного старения" в течение одного месяца. На XRPD Формы В соединения В, выдержанной при 40°C/RH 75% в течение одного месяца, показано, что Форма В соединения В остается неизмененной в условиях внешнего воздействия, включая повышенную температуру и влажность.
Получение Формы В соединения В. В стеклянный флакон емкостью 5 мл отвешивали примерно 58,0 мг винной кислоты и добавляли 2,0 мл EtOAc. Кислота оставалась нерастворенной. В этот флакон емкостью 5 мл добавляли примерно 2,0 мл концентрированного раствора соединения в форме свободного основания в EtOAc (примерно 100 мг/мл) в молярном соотношении 1:1 и раствор перемешивали при КТ. В качестве затравки добавляли примерно 1 мг Формы В, и раствор оставался прозрачным. Раствор перемешивали в течение ночи и отбирали образец для проведения XRPD. Полученная картина соответствовала Форме В соединения В. Суспензию перемешивали при 50°С в течение еще 2 суток. Суспензию центрифугировали и осадок на фильтре сушили при 50°С в течение 2 ч. Получено 144,5 мг с выходом примерно 56,1%.
Пример 18
Получение Формы С соединения В. Форму С получали в масштабе 200 мг посредством происходящей в процессе реакции кристаллизации из раствора в THF, что подтверждается данными XRPD (ФИГ. 10). Как показано на ФИГ. 11а и ФИГ. 11b, при проведении TGA наблюдали уменьшение массы примерно на 6,8% вплоть до 130°С, а результатом DSC стало обнаружение одной эндотермы при 118,1°С (температура начала разложения). Стехиометрическое соотношение было определено как 1,02 (кислота/свободное основание), и был обнаружен THF в количестве 9,7%.
Получение Формы С соединения В. В стеклянный флакон емкостью 3 мл помещали примерно 56,9 мг винной кислоты и добавляли 1,0 мл THF, получая прозрачный раствор. К прозрачному раствору кислоты добавляли 2,0 мл концентрированного раствора соединения в форме свободного основания в THF (примерно 100 мг/мл) в молярном соотношении 1:1 и перемешивали при КТ. В качестве затравки добавляли примерно 1 мг Формы С, и раствор становился слегка мутным. Суспензию перемешивали в течение ночи и отбирали образец для проведения XRPD. Полученная картина соответствовала образованию Формы С. Суспензию перемешивали при КТ в течение еще 24 часов, осадок на фильтре сушили при 50°С в течение 1,5 часа. Суспензию центрифугировали для сбора твердых веществ. Получено 161,4 мг с выходом примерно 62,7%.
Пример 19
Получение Формы D соединения В. Образцы Формы D получали путем быстрого упаривания в MeOH/DCM и суспендирования в H2O при КТ, соответственно. Картина XRPD для Формы D показана на ФИГ. 12. Результаты TGA и DSC для образца Формы D представлены на ФИГ. 13а и ФИГ. 13b, соответственно, и демонстрируют уменьшение массы на 3,5% до 150°С и эндотерму при 73,0°С перед плавлением/разложением при 163,9°С (температура начала разложения). Изменение формы с образованием Формы F наблюдали после нагревания Формы D до 150°С, охлаждения до 30°С в атмосфере азота и затем выдерживания в условиях окружающей среды. С использованием 1Н ЯМР не было обнаружено никакого значительного количества растворителя МеОН или DCM, примененного в способе получения, и дополнительно было определено, что стехиометрическое соотношение L-винной кислоты к свободному основанию составляет 1,0. Форма D представляет собой гидрат.
Чтобы провести оценку физической стабильности Формы D при разных значениях влажности, собирали данные DVS для образца Формы D при 25°С после уравновешивания образца при влажности, соответствующей условиям окружающей среды (RH 80%). В процессе десорбции при проведении тестирования с использованием DVS наблюдали плато при переходе от RH 20% (поглощение влаги 2,25%) до RH 80% (поглощение влаги 2,72%), что позволяет предположить, что дегидратация водной Формы D происходила, когда соответствующее значение влажности составляло меньше 20%. Кроме того, поскольку теоретически рассчитанное содержание воды в моногидрате равно 2,6%, то Форма D, следовательно, может быть моногидратом.
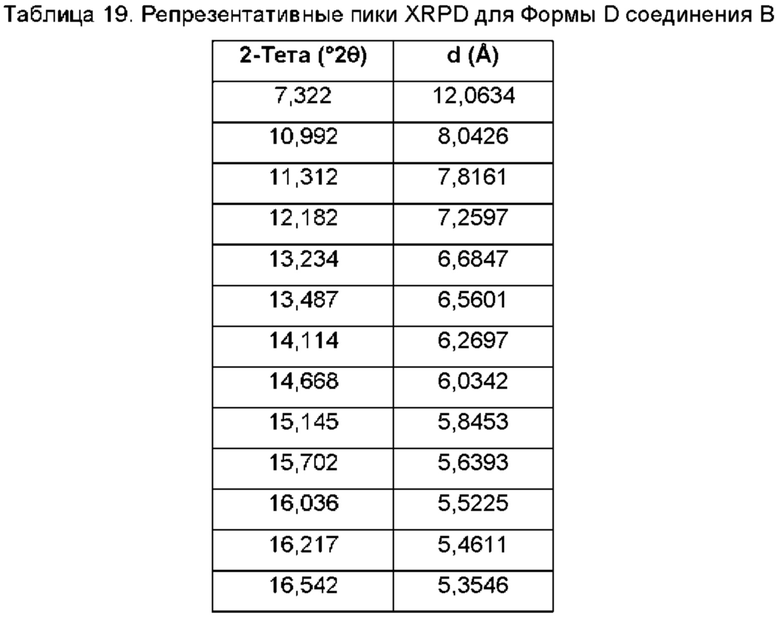
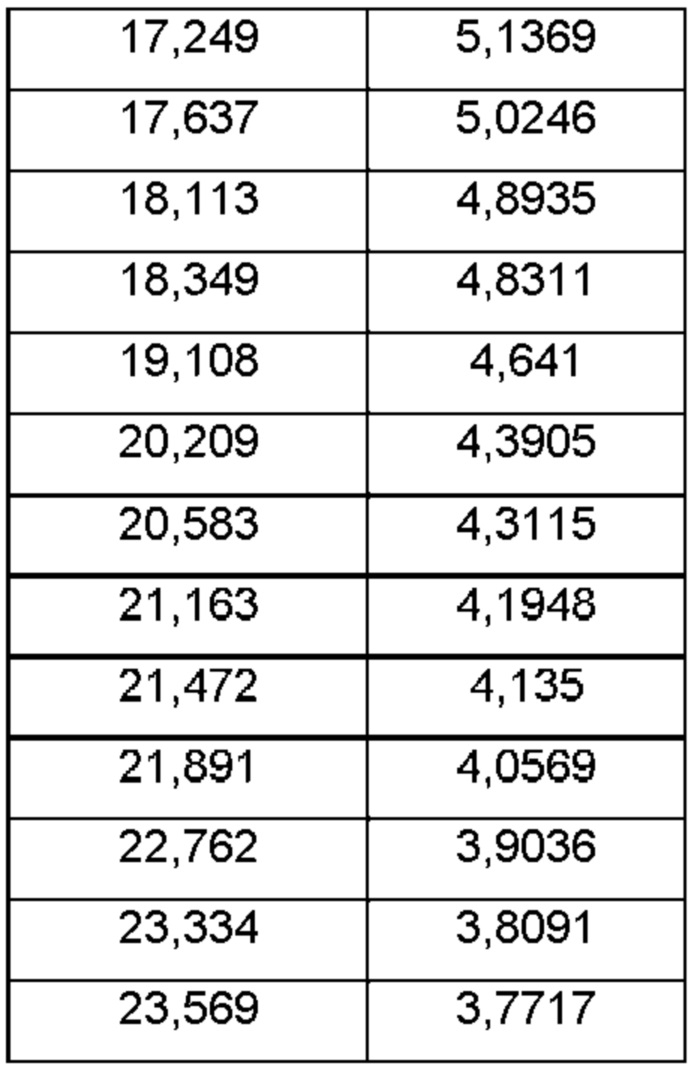
Пример 20
Получение Формы Е соединения В. Форму Е получали в результате DMSO-опосредуемой кристаллизации, добавляя IPAc в раствор в DMSO, и картина ее XRPD показана на ФИГ. 14. Кривые TGA и DSC (ФИГ. 15а и ФИГ. 15b, соответственно) показали значительное, на 8,3%, уменьшение массы до 140°С и эндотерму при 126,3°С перед плавлением/разложением при 142,6°С (температура начала разложения). Спектр 1Н ЯМР показал наличие 0,7 эквивалента DMSO (приблизительно 9,4 масс. %), и было отмечено отсутствие какого-либо значительного количества IPAc. Было определено, что стехиометрическое соотношение L-винной кислоты к свободному основанию составляет 1,0. Форма Е представляет собой сольват в DMSO.
Пример 21
Получение Формы F соединения В. Форму F получали посредством нагревания образца Формы D до 150°С, охлаждения до 30°С в атмосфере азота и выдерживания в условиях окружающей среды. Картина XRPD для Формы F приведена на ФИГ. 16. Результаты DSC-анализа (ФИГ. 18) показали, что Форма F была кристаллической с эндотермой при 164,2°С (температура начала разложения). С использованием 1Н ЯМР было определено, что стехиометрическое соотношение L-винной кислоты к свободному основанию составляет 1,0, и не было обнаружено никакого значительного сигнала от растворителя.
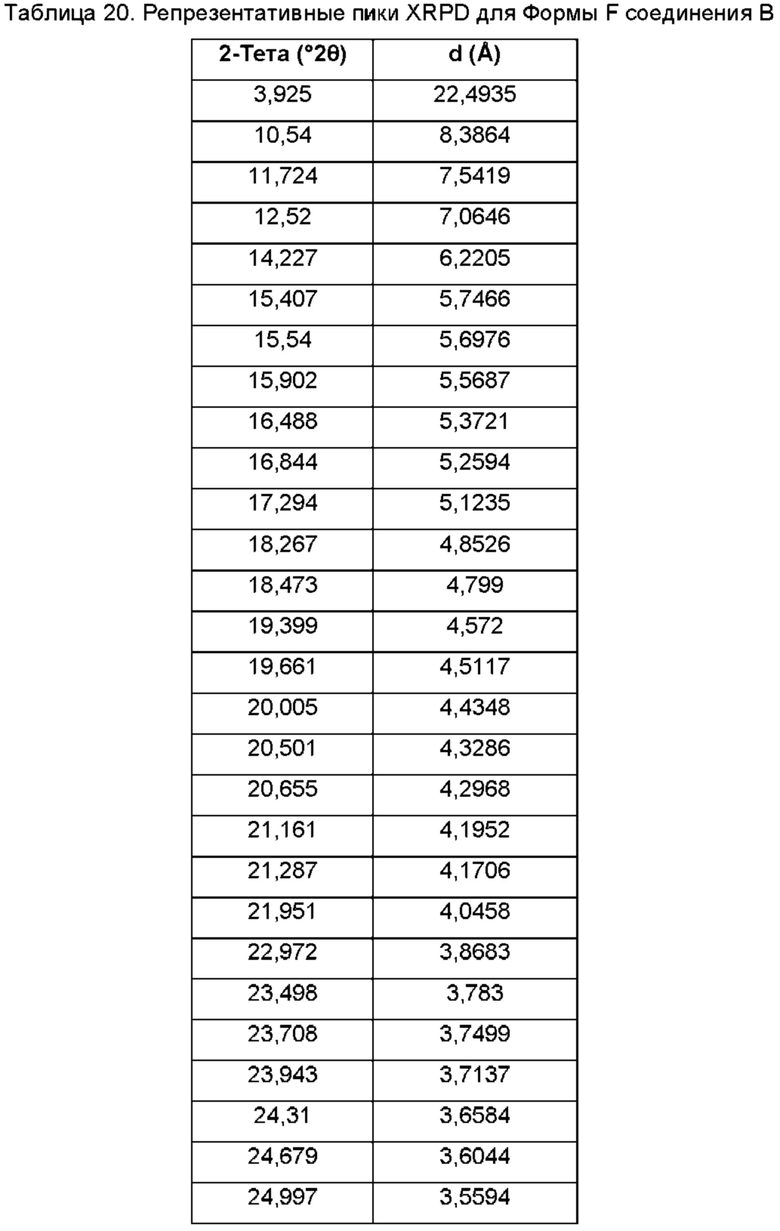
Чтобы дополнительно охарактеризовать Форму F, проводили исследование Формы D с использованием XRPD с переменной температурой (VT-XRPD), при этом температуру повышали до 150°С и снижали обратно до 30°С в атмосфере азота. В таких условиях образование Формы F наблюдали после дегидратации образца Формы D при повышенных температурах, что указывает на то, что Форма F представляла собой безводную форму.
Чтобы провести оценку физической стабильности Формы F при разных значениях влажности, собирали данные о динамической сорбции паров (DVS) для Формы F при 25°С после уравновешивания образца при влажности, соответствующей условиям окружающей среды (RH 80%). Наблюдали поглощение влаги, составляющее приблизительно 1,9%, вплоть до RH 80%, что позволяет предположить, что Форма F является слегка гигроскопичной.
Получение Формы G соединения В. Форму G получали путем медленного упаривания в МеОН при КТ. Картина XRPD для Формы G приведена на ФИГ. 19. Результаты TGA и DSC представлены на ФИГ. 20а и ФИГ. 20b, соответственно, и на них продемонстрировано уменьшение массы на 3,3% до 150°С и пик плавления/разложения при 170,4°С (температура начала разложения). Спектр 1Н ЯМР показал наличие МеОН в количестве 0,49 эквивалента (что соответствует приблизительно 3,0 масс. %). Было определено, что стехиометрическое соотношение L-винной кислоты к свободному основанию составляет 1,0. Форма G представляет собой сольват с МеОН.
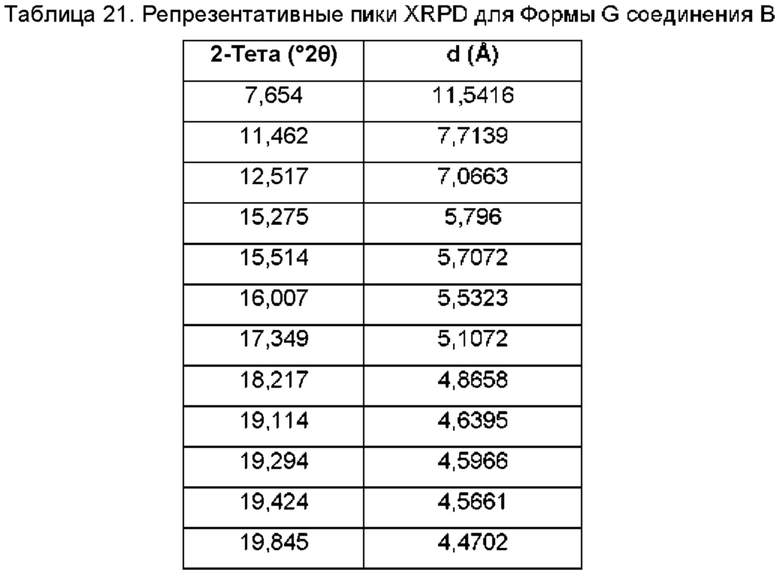
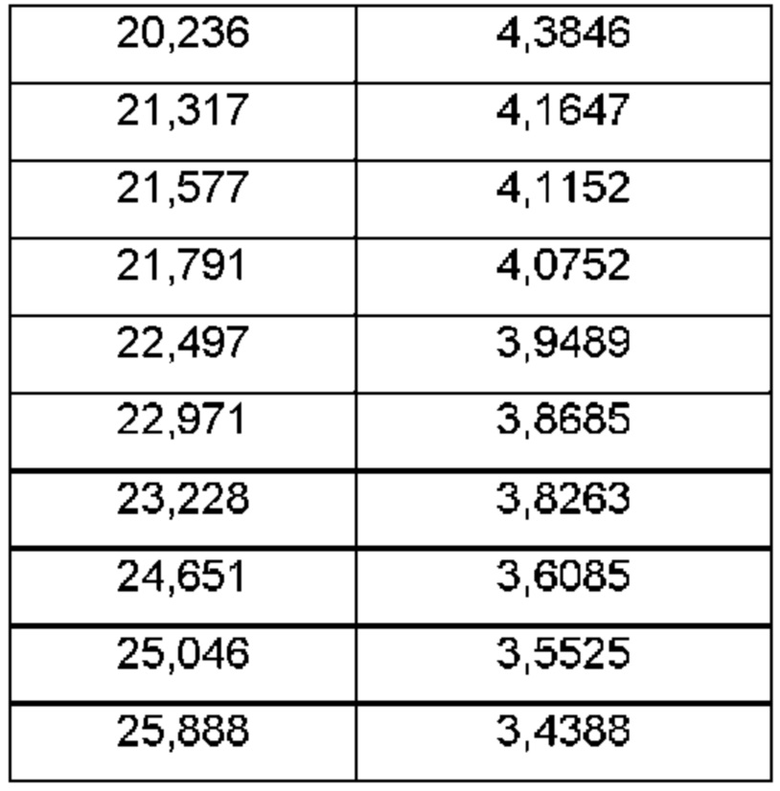
Пример 22
Скрининг полиморфов L-тартрата. Оценку растворимости Формы В соединения B выполняли в 20 растворителях при КТ (Таблица 22). В каждый стеклянный флакон емкостью 3 мл добавляли приблизительно по 2 мг твердого вещества. Затем в эти флаконы постепенно добавляли растворители, приведенные в Таблицах 6-7, до тех пор, пока твердые вещества не растворялись или общий объем не достигал 1 мл. Результаты, приведенные в Таблице 22, использовали как руководство для выбора растворителя при скрининге полиморфов. Эксперименты по скринингу полиморфов проводили, используя разные методы кристаллизации из раствора или методы перевода в твердую фазу. Использованные методы и идентифицированные кристаллические формы суммированы в Таблице 23.
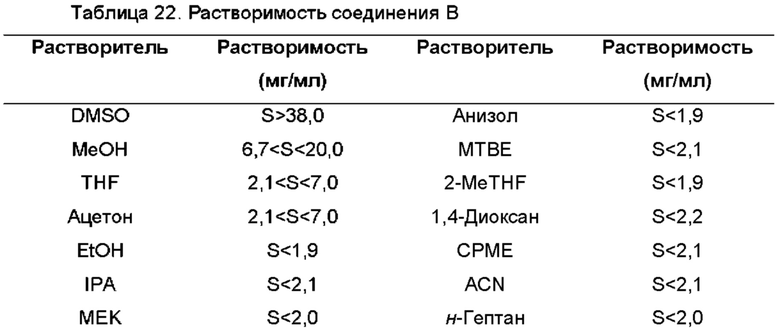
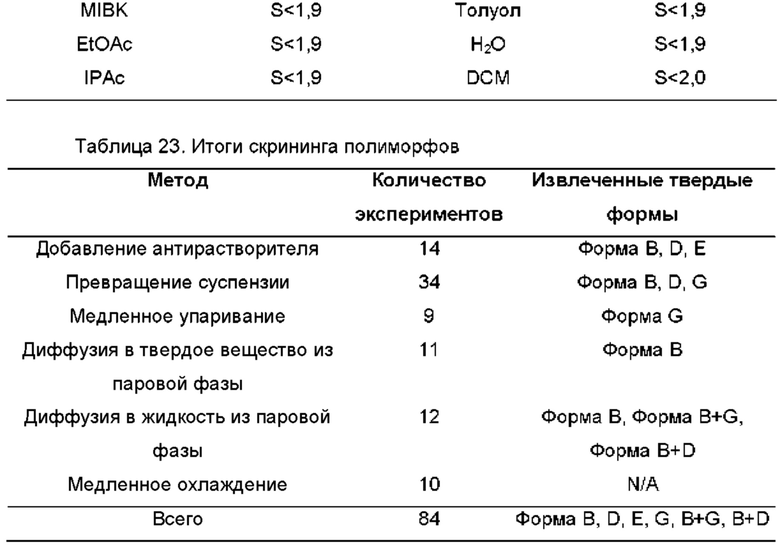
Пример 23
Добавление антирастворителя. В общей сложности проводили 14 экспериментов с добавлением антирастворителя. В каждом эксперименте в стеклянный флакон емкостью 20 мл отвешивали примерно 15 мг Формы В соединения В, после чего добавляли 0,3-1,0 мл соответствующего растворителя. Затем эту смесь перемешивали на магнитной мешалке со скоростью 500 об/мин при КТ, получая прозрачный раствор. После этого в раствор добавляли соответствующий антирастворитель, вызывая выпадение осадка, или до тех пор, пока общее количество антирастворителя не достигало 10,0 мл. Прозрачные растворы переводили в суспензию при 5°С. Затем, если никакого выпадения осадка не происходило, раствор переносили для выполнения быстрого упаривания при КТ. Для проведения XRPD-анализа твердые вещества извлекали. Результаты, приведенные в Таблице 24, показали получение Форм В, D и Е соединения В.

Пример 24
Превращение суспензии при КТ. Эксперименты по превращению суспензии проводили при КТ в разных системах растворителей. В каждом эксперименте в стеклянном флаконе емкостью 1,5 мл суспендировали примерно по 20 мг Формы В соединения B в 0,3 мл соответствующего растворителя. После перемешивания суспензии на магнитной мешалке в течение 17 суток при КТ извлекали оставшиеся твердые вещества для проведения XRPD-анализа. Результаты, приведенные в Таблице 25, указывали на получение Форм В, D и G соединения В.


Пример 25
Превращение суспензии при повышенных температурах. Эксперименты по превращению суспензии проводили при 50°С и 70°С в разных системах растворителей. В каждом эксперименте в стеклянном флаконе емкостью 1,5 мл суспендировали примерно 20 мг Формы В соединения B в 0,3 мл соответствующего растворителя. После перемешивания суспензии на магнитной мешалке в течение 17 суток при 50°С и 70°С извлекали оставшиеся твердые вещества для проведения XRPD-анализа. Результаты, приведенные в Таблице 26, указывали на получение Формы В соединения В.

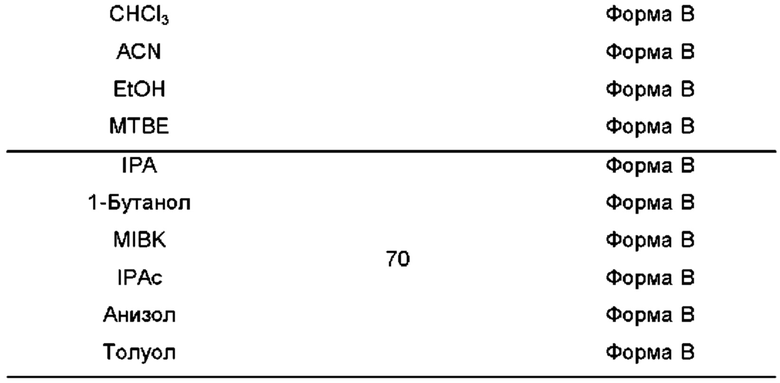
Пример 26
Медленное упаривание. Эксперименты по медленному упариванию проводили для 9 условий. В каждом эксперименте в стеклянный флакон емкостью 3 мл отвешивали примерно по 15 мг Формы В соединения В, затем добавляли соответствующий растворитель или смесь растворителей, получая прозрачный раствор. После этого флакон закрывали парафильмом с 3-4 крошечными отверстиями и хранили при КТ, давая раствору возможность медленно упариваться. Извлеченные твердые вещества тестировали посредством XRPD. Как приведено в Таблице 27, получали Форму G соединения В.

Пример 27
Диффузия в твердое вещество из паровой фазы. Эксперименты по диффузии в твердое вещество из паровой фазы проводили, используя 11 растворителей. В каждом эксперименте отвешивали примерно 15 мг Формы В соединения B во флакон емкостью 3 мл, который помещали во флакон емкостью 20 мл, содержащий 4 мл соответствующего растворителя. Флакон емкостью 20 мл герметично закрывали крышкой и выдерживали при КТ в течение 14 суток, чтобы пары растворителя могли взаимодействовать с твердым образцом. Извлеченные твердые вещества тестировали посредством XRPD. Результаты, приведенные в Таблице 28, указывали на получение Формы В соединения В.
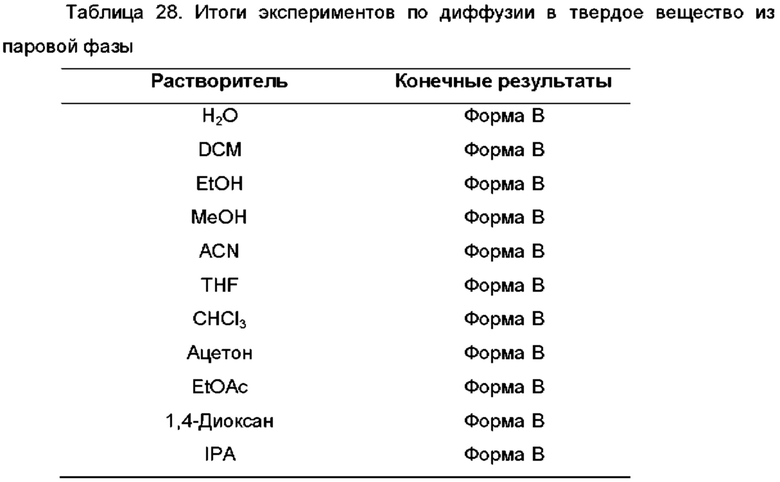
Пример 28
Диффузия в жидкость из паровой фазы. Проводили двенадцать экспериментов по диффузии в жидкость из паровой фазы. В каждом эксперименте во флаконе емкостью 3 мл растворяли примерно по 15 мг Формы В соединения B в 0,5-1,0 мл соответствующего растворителя, получая прозрачный раствор. После этого данный раствор помещали во флакон емкостью 20 мл, содержащий 4 мл соответствующего антирастворителя. Флакон емкостью 20 мл герметично закрывали крышкой и выдерживали при КТ, что давало парам растворителя возможность взаимодействовать с раствором в течение достаточного промежутка времени. Твердые вещества извлекали для XRPD-анализа. Результаты, приведенные в Таблице 29, указывали на получение Формы В соединения В и смеси Форм B+G/B+D.
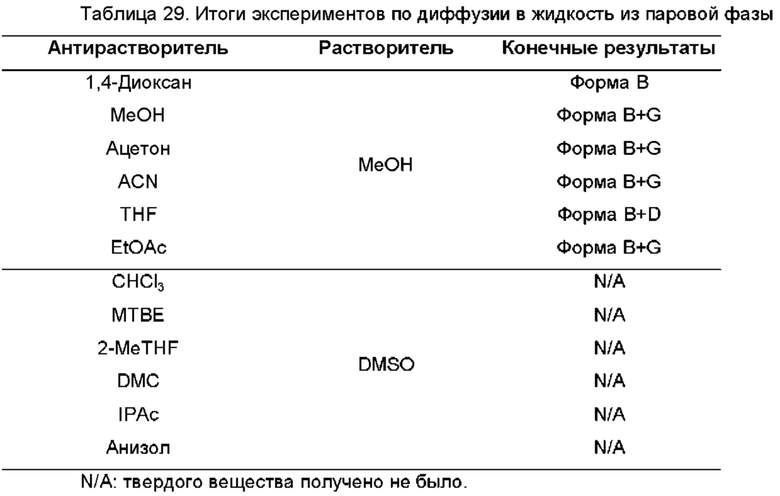
Пример 29
Медленное охлаждение. Эксперименты по медленному охлаждению проводили в 10 системах растворителей. В каждом эксперименте в стеклянном флаконе емкостью 3 мл при КТ суспендировали примерно по 20 мг Формы В соединения B в 1,0 мл соответствующего растворителя. Суспензию переводили во взвесь при 50°С, используя магнитную мешалку со скоростью 500 об./мин. Образец уравновешивали при 50°С в течение 2 ч и фильтровали, используя нейлоновую мембрану с размером пор 0,45 мкм. После этого фильтрат медленно охлаждали от 50°С до 5°С со скоростью 0,1°С/мин. Обобщенные результаты указывали на то, что твердого вещества обнаружено не было.
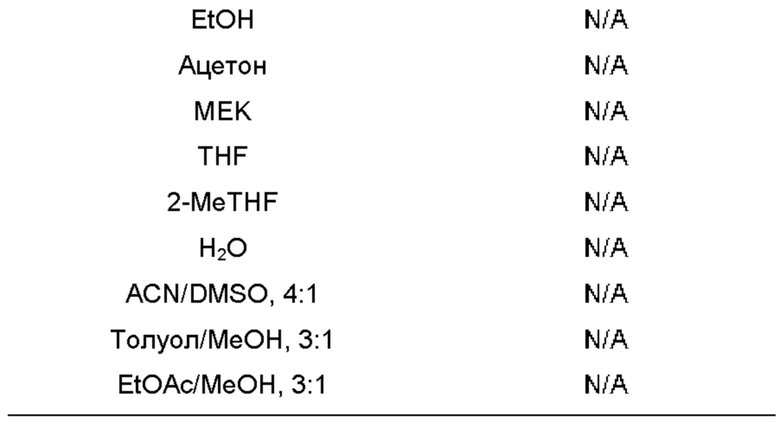
Пример 30
Фазовое превращение. Кристаллизация соединения В из растворителей, таких как этилацетат, этанол, ацетон или их смесь с водой, может приводить к образованию еще одной из следующих форм: безводной Формы В, безводной Формы F, Формы D в виде моногидрата и Формы А в виде сольвата с ацетоном. Картины XRPD для этих четырех твердых форм показаны на ФИГ. 21. Поэтому было важно знать физическую стабильность этих форм, чтобы определить перспективу полного фазового превращения и регулировать процесс кристаллизации для получения желаемой Формы В. Ввиду непосредственной близости значений точек плавления для Форм F и В (начало при 160-164°С для Формы F по сравнению с 166-175°С для Формы В) превращение форм протекало медленно в экспериментах по конкурентному преобразованию суспензии (competitive slurry bridging) при КТ, и в результате получали смесь обеих форм. В итоге были проведены исследования растворимости в условиях равновесия для обеих форм в этаноле через 17 ч при 25, 35 и 50°C с целью определения соотношения их термодинамической стабильности.
Было обнаружено, что Форма F обладает более высокой растворимостью при этих трех температурах, что является подтверждением большей термодинамической стабильности Формы B в диапазоне 25-50°С. График Вант-Гоффа зависимости растворимости Форм В и F от температуры показал, что эти две безводные формы были связаны энантиотропно, с температурой перехода приблизительно 19°С. В случае сравнения стабильности гидрата и безводной формы было обнаружено, что Форма В остается стабильной при активности воды (aw) aw<0,2 (КТ), выше которой была стабильной Форма D в виде гидрата. Однако, в экспериментах по преобразованию суспензии было обнаружено, что Форма В является кинетически стабильной вплоть до aw, равной 0,4 (КТ), в течение вплоть до 36 суток. Это является подтверждением того, что хотя критическое значение aw для превращения формы в гидрат является низким, существует значительный кинетический барьер для превращения Формы B в Форму D.
Образцы после суспендирования и кристаллизации. Форма В + Форма А
Готовили смеси в отношении 1:1 партий соединения В, содержащих (1) смесь Формы А/сольвата с ацетоном и Формы В; и (2) Форму В, и добавляли в 100%-ный ацетон и смеси ацетона и воды (90% ацетона, 95, 96, 97, 98 и 99% ацетона, об./об.). Образцы суспендировали при КТ в течение 120 ч, фильтровали и далее анализировали с использованием XRPD.
Форма F
Форму F суспендировали в 100%-ном этаноле при КТ и 50°С в течение ночи. В одну из суспензий при КТ вносили затравку Формы В, тогда как в другую затравку не вносили. В суспензию, перемешиваемую при 50°С, вносили затравку Формы В. Образцы фильтровали и анализировали посредством XRPD. Образцы Формы F суспендировали по отдельности при КТ в 100%-ной DI воде, смеси 1:1 ацетон/вода и 100%-ном ацетоне. Суспензии Формы F в чистых растворителях выдерживали при КТ, тогда как раствор Формы F готовили в смеси 1:1 ацетон : вода, которую таким образом перемешивали при 5°C с целью кристаллизации/осаждения при охлаждении. Через 24 ч все образцы фильтровали и анализировали посредством XRPD. Форму F также суспендировали в смесях 95:5 ацетон : вода и 97:3 ацетон : вода при 50°С в течение 2 ч, после чего суспензии охлаждали до КТ, фильтровали и анализировали посредством XRPD. Окончательно, Форму F перекристаллизовывали из смеси 95:5 ацетон : вода, в которую была внесена затравка Формы В при 50°С.
Форма D
Форму D суспендировали при КТ в течение 48 ч в 100%-ном этаноле. После этого образец фильтровали и анализировали посредством XRPD.
Результаты
Разработка калориметрического метода оценки чистоты. При переходе с этанола на смесь ацетон/вода в качестве растворителя для кристаллизации произошло заметное улучшение чистоты, при этом содержание олигомеров снизилось в среднем на 5% (масс./масс.) с получением приемлемого выхода. Изменение содержания олигомеров, а также выход зависели от состава растворителей в суспензии. Увеличение содержания воды улучшало чистоту, но уменьшало выход. При заданном составе растворителей для точек начала плавления и содержания олигомеров в % постоянно получали противоположные результаты, что указывало на вероятное влияние чистоты на точку плавления, при этом при более высоком %-ном содержании олигомеров точка плавления снижается, и наоборот. С учетом этого наблюдения несколько партий суспензий, полученных из смеси ацетон/вода, анализировали с использованием DSC и тестировали на предмет содержания олигомеров с использованием SEC, и строили кривую корреляции. Для всех этих образцов собирали данные XRPD, чтобы убедиться их соответствию Форме В соединения В. С использованием аппроксимации данных экспонентой делали оценки чистоты для нескольких партий, в том числе для образца в масштабе 20 г и с контролем в ходе технологического процесса (IPC). Для линейного приближения между экспериментально измеренным и прогнозируемым содержанием олигомеров получали значение R2, равное 0,96. Учитывая, что это были твердые/порошкообразные образцы с присущими им проблемами однородности, это высокое значение R2 обеспечивало уверенность в надежности корреляции между точкой начала плавления и чистотой.
Форму В соединения В соответственно получали из 100%-ного этанола (в условиях суспендирования или кристаллизации). Эксперименты проводили и в других условиях, включая условия суспендирования или кристаллизации из смеси этанол/вода и условия суспендирования в смеси ацетон/вода (≥95% ацетона). В зависимости от параметров условий суспендирования или кристаллизации получали несколько других твердых форм соединения В, а именно, Форму F (этанол/вода), Форму А/растворитель ацетон (≥95% ацетона) и смеси Форм А и В или Форм А и F. В условиях, когда получали Форму F, предполагалось образование промежуточного гидрата (Формы D), поскольку величины aw в смесях этанол/вода, используемых для этих образцов, значительно превышали 0,2 (КГ), при этом гидрат является термодинамически стабильным. В Таблице 31 показаны условия кристаллизации вместе с полученными формами. Поскольку в нескольких случаях получались смеси форм, то была реализована стратегия регулирования форм с целью получения Формы B в конечной твердой фазе.
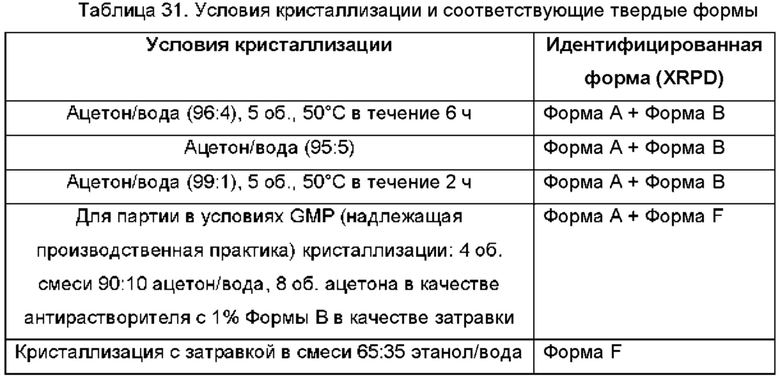
Поскольку система ацетон/вода оказалась наиболее эффективной для удаления олигомеров из соединения В, то проводили эксперимент по конкурентному преобразованию суспензии, используя смеси 1:1 двух отдельных партий соединения В, одна из которых представляла собой по большей части Форму А с некоторым количеством Формы В, а второй была партия, которую суспендировали в смесях ацетон/вода разного состава при КТ в течение 120 ч. Сольват с ацетоном получали в виде стабильной формы в случае содержания ацетона ≥95%, в то время как в случае содержания ацетона 90% получали смесь Формы В и гидрата Формы D. Поскольку желаемый уровень ацетона находился в диапазоне от 96 до 95% (с точки зрения уровней олигомеров), то существовала возможность получения любой из Форм А, В или D в результате заключительной кристаллизации.
Стратегия регулирования получения форм: превращение Форм А и F с образованием Формы В. На ФИГ. 2b показаны термограммы DSC для Формы А в виде сольвата с ацетоном. Первая эндотерма при 124°С указывает на потерю растворителя и испарение, в то время как вторая эндотерма при 164°С указывает на плавление соответствующей безводной формы. Данные XRPD подтверждали, что эта безводная форма представляет собой Форму В, когда Форму А нагревали до 152°С. Форму А ресуспендировали в 100%-ном этаноле в течение ночи для превращения в Форму В. Таким образом, был предложен двухстадийный способ суспендирования для облегчения регулирования образования форм соединения В с целью получения Формы B в том случае, если на первой стадии суспендирования в смеси ацетон/вода образуется либо Форма А, либо смесь Форм А и В, в зависимости от условий суспендирования. Это может гарантировать, что независимо от изменения формы на первой стадии суспендирования, конечной формой соединения В будет Форма В, которая будет выделена после второй стадии суспендирования.
В отличие от случая использования сольвата с ацетоном, Форма F, полученная в результате кристаллизации из смеси 65:35 этанол : вода с затравкой Формы В, демонстрировала только одну эндотерму плавления при 162°С. Ввиду почти идентичных значений растворимости Форм F и B в этаноле при 25°С (0,23 мг/мл для Формы В по сравнению с 0,26 мг/мл для Формы F), термодинамически обусловленная движущая сила для превращения метастабильной Формы F в стабильную Форму В является незначительной, и поэтому процесс превращения Формы F в Форму В будет медленным, даже в присутствии Формы B в качестве затравки. Независимо от внесения затравки и температуры процесс превращения Формы F в Форму В протекает медленно, и через 12 часов получают смесь обеих форм.
Чтобы способствовать превращению Формы F в Форму B в более короткие сроки, был выбран путь с образованием сольвата, при этом Форма F должна быть превращена в Форму В посредством образования сольвата с ацетоном или гидрата, которые впоследствии будут подвергнуты десольватированию с образованием Формы В. Форму F суспендировали в течение ночи в чистой воде, ацетоне и смеси 1:1 ацетон : вода без какого-либо внесения затравки. Форма F остается неизмененной в 100%-ном ацетоне, но превращается в гидрат (Форму D) в присутствии воды. Когда этот гидрат суспендируют в чистом этаноле, получается смесь Формы D и Формы F, что указывает на склонность гидрата кдесольватации с превращением в метастабильную безводную форму. Для Формы F, суспендированной в смесях 95:5 ацетон : вода и 97:3 ацетон : вода при 50°С, продемонстрировано появление Формы В, но это превращение является неполным. После перекристаллизации Формы F из смеси 95:5 ацетон : вода с использованием затравки Формы В при 50°С наблюдали полное превращение Формы F в Форму В. На ФИГ. 25 и ФИГ. 26 представлены схематичные изображения превращения между Формами А, В, D и F.
Термодинамическое соотношение между безводными Формой В и Формой F в условиях конкуренции при суспендировании (slurry competition) между безводными Формами В и F. Чтобы определить термодинамическую стабильность, связанную с Формами В и F, проводили эксперименты по конкурентному преобразованию суспензии в системе растворителей ацетон/EtOH/EtOAc при КТ (25±3°С) и 50°С, как указано в Таблице 32. Смесь Форм В и F в равном массовом соотношении суспендировали в насыщенных растворах Формы B в смеси ацетон/EtOH/EtOAc и затем перемешивали на магнитной мешалке при заданной температуре. После суспендирования в течение примерно 4-11 суток оставшиеся твердые вещества извлекали для определения характеристик посредством XRPD. Обнаруживали смесь Форм В и F, что указывает на медленный переход между Формами В и F.
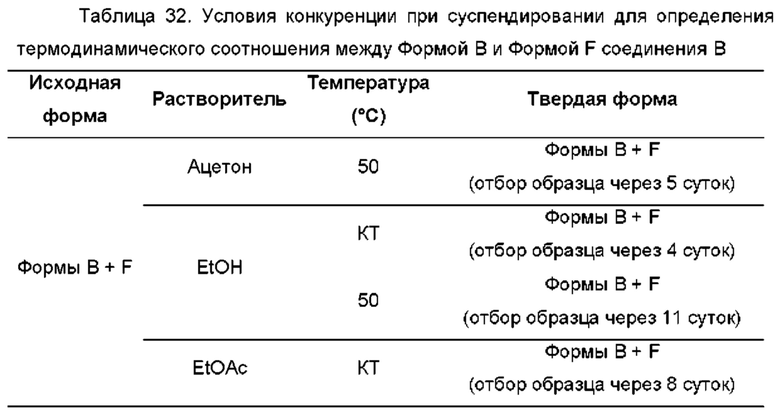
Соотношение термодинамических стабильностей между безводными Формами В и F определяли в условиях конкуренции при суспендировании и посредством измерения растворимости в условиях равновесия. В результате во всех экспериментах по конкуренции при суспендировании обнаруживали смесь Форм В и F, что указывает на медленный переход между Формами В и F. Без связи с какой-либо конкретной теорией можно сказать, что низкая растворимость Формы В и Формы F может быть вызвана их низкой растворимостью в исследуемых растворителях (ацетон/EtOH/EtOAc). Ввиду этого, для определения соотношения их термодинамических стабильностей измеряли растворимость в условиях равновесия (через 17 часов) в EtOH при 25°С, 35°С и 50°С, соответственно. По сравнению с Формой В Форма F демонстрировала более высокую растворимость в EtOH при всех трех тестированных температурах, что указывает на то, что в диапазоне от 25°С до 50°С Форма В термодинамически более стабильна, чем Форма F.
Соотношение между безводной формой В и водной формой D в зависимости от aw определяли в условиях конкуренции при суспендировании при различной активности воды при КТ. Форму D обнаруживали при aw от 0,6 до 0,8 через одну неделю и в воде через 36 суток. Форму В наблюдали обнаруживали в EtOH (aw<0,2) через одну неделю. Смесь Форм D и В наблюдали обнаруживали в системах с aw от 0,2 до 0,4 после перемешивания при КТ в течение 36 суток. Для дальнейшего подтверждения соотношения термодинамических стабильностей между Формой В и Формой D в системах с aw 0,2 и 0,4 при КТ определяли их растворимость в условиях равновесия (через 24 часа) в соответствующих условиях. По сравнению с образцами, где изначально присутствовала Форма В, более низкую растворимость обнаруживали в образцах, где изначально присутствовала Форма D (наличие кристаллических форм конечных, присутствующих в ограниченном количестве твердых веществ не проверяли), в обеих системах с aw равной 0,2 и 0,4.
Измерение растворимости в условиях равновесия для безводных форм В и F. Для дальнейшего определения соотношения термодинамических стабильностей между Формами В и F проводили эксперименты по измерению растворимости в условиях равновесия в EtOH при 25°С, 35°С и 50°С, соответственно. Подробные методики обобщены следующим образом: Формы В и F в виде твердых веществ суспендировали в 0,4 мл EtOH при заданных температурах и перемешивали на магнитной мешалке в течение 17 ч (750 об/мин). После центрифугирования определяли в фильтрате концентрацию и, используя HPLC, чистоту этих форм в виде свободного основания. Кристаллическую форму оставшихся твердых веществ проверяли посредством XRPD.
По сравнению с Формой В Форма F показала более высокую растворимость при 25°С, 35°С и 50°С в EtOH (Таблица 33). Согласно результатам XRPD, после тестирования растворимости никакого изменения формы не наблюдали, что указывает на то, что Форма В по всей вероятности является термодинамически более стабильной, чем Форма F в диапазоне температур от 25°С до 50°С.
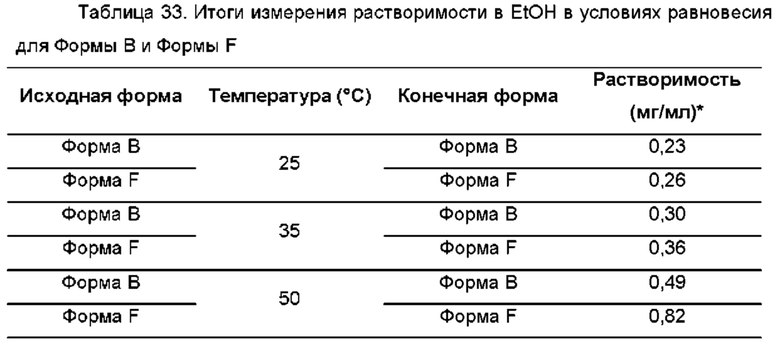
Определение критического значения активности воды для соотношения Форм В и D. Чтобы определить критическое значение активности воды для соотношения безводной Формы В и водной Формы D, проводили эксперименты по конкуренции при суспендировании в условиях с различной активностью воды при КТ, как указано в Таблице 34. Смесь Форм В и D в равном массовом соотношении суспендировали в насыщенных растворах Формы B в смеси EtOH-вода (с различными значениями aw) и затем перемешивали на магнитной мешалке при КТ. После суспендирования в течение примерно 7-36 суток оставшиеся твердые вещества извлекали для определения характеристик посредством XRPD.

Форму D обнаруживали при aw 0,6 и 0,8 после суспендирования в течение одной недели и в воде через 36 суток. Форму В обнаруживали в EtOH после суспендирования в течение одной недели. Смесь Форм D и В обнаруживали в системах с aw 0,2 и 0,4 после перемешивания при КТ в течение 36 суток.
Измерение растворимости Форм В и D. Для дальнейшего подтверждения соотношения термодинамических стабильностей между Формой В и Формой D в условиях с aw 0,2 и 0,4 при КТ определяли растворимость в условиях равновесия, приведенных в Таблице 35.

Безводную Форму В и водную Форму D в виде твердых веществ суспендировали в 0,5 мл целевой системы растворителей (aw=0,2/0,4), соответственно, и перемешивали на магнитной мешалке в течение 24 ч (750 об./мин). Суспензию фильтровали и в фильтрате определяли концентрацию этих форм в виде свободного основания. Более низкую растворимость наблюдали в случае образцов, где изначально присутствовала Форма D. Водная Форма D оказывается термодинамически более стабильной, чем безводная форма В, когда aw≥0,2 при КТ (25±3°С).
Пример 31
Форма М в виде малоната
После скрининга получали один вариант в виде малоната с низкой степенью кристалличности, Форму М в виде малоната. Картина XRPD для нее показана на ФИГ. 30. При проведении TGA наблюдали уменьшение массы на 4,1% вплоть до 110°С, а результатом DSC стало обнаружение нескольких эндотерм (ФИГ. 31). С использованием 1Н ЯМР стехиометрическое соотношение было определено как 0,50 (кислота/свободное основание), и был обнаружен THF в количестве 6,2% (молярное отношение к свободному основанию составляло 0,58). Поскольку наблюдается несколько эндотерм и значительное количество THF, то Форма М в виде малоната представляет собой сольват с THF, но никакого дальнейшего определения характеристик не проводили ввиду низкой степени ее кристалличности.
Пример 32
Форма 1 в виде фумарата
После скрининга получали один кристаллический вариант в виде фумарата, Форму 1 в виде фумарата. При проведении TGA наблюдали уменьшение массы на 0,9% вплоть до 150°С, а результатом DSC (ФИГ. 28) стало обнаружение одного эндотермического пика плавления при 164,3°С (температура начала разложения). С использованием 1Н ЯМР стехиометрическое соотношение было определено как 0,88 (кислота/свободное основание), и был обнаружен EtOAc в количестве 1,5% (молярное отношение к свободному основанию составляло 0,11).
Получение Формы 1 в виде фумарата. В стеклянный флакон емкостью 5 мл отвешивали примерно 44,7 мг фумаровой кислоты и добавляли 2,0 мл EtOAc. Кислота не растворялась. В этот флакон емкостью 5 мл добавляли примерно 2,0 мл концентрированного раствора соединения в форме свободного основания в EtOAc (примерно 100 мг/мл) в молярном соотношении 1:1 и перемешивали при КТ. В качестве затравки добавляли примерно 3 мг Формы 1 в виде фумарата, и раствор становился мутным. Суспензию перемешивали в течение ночи и отбирали образец для XRPD-анализа (ФИГ. 27) с получением картины, соответствующей Форме 1 в виде фумарата. Суспензию перемешивали при 50°С в течение еще 3 суток для повышения степени кристалличности, затем центрифугировали. Осадок на фильтре сушили при 50°С в течение 4 ч. Получено 186,6 мг с выходом приблизительно 76,2%.
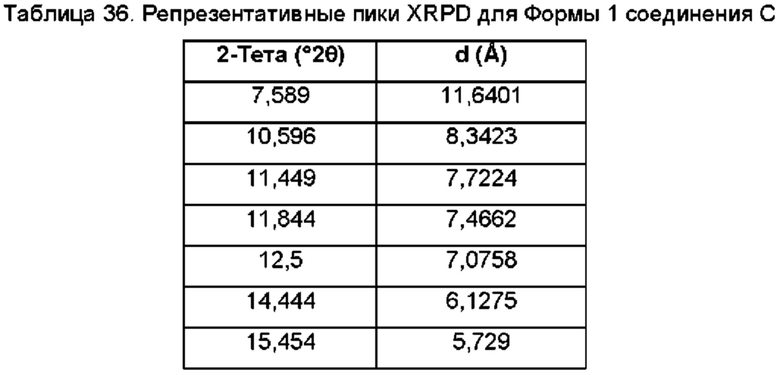
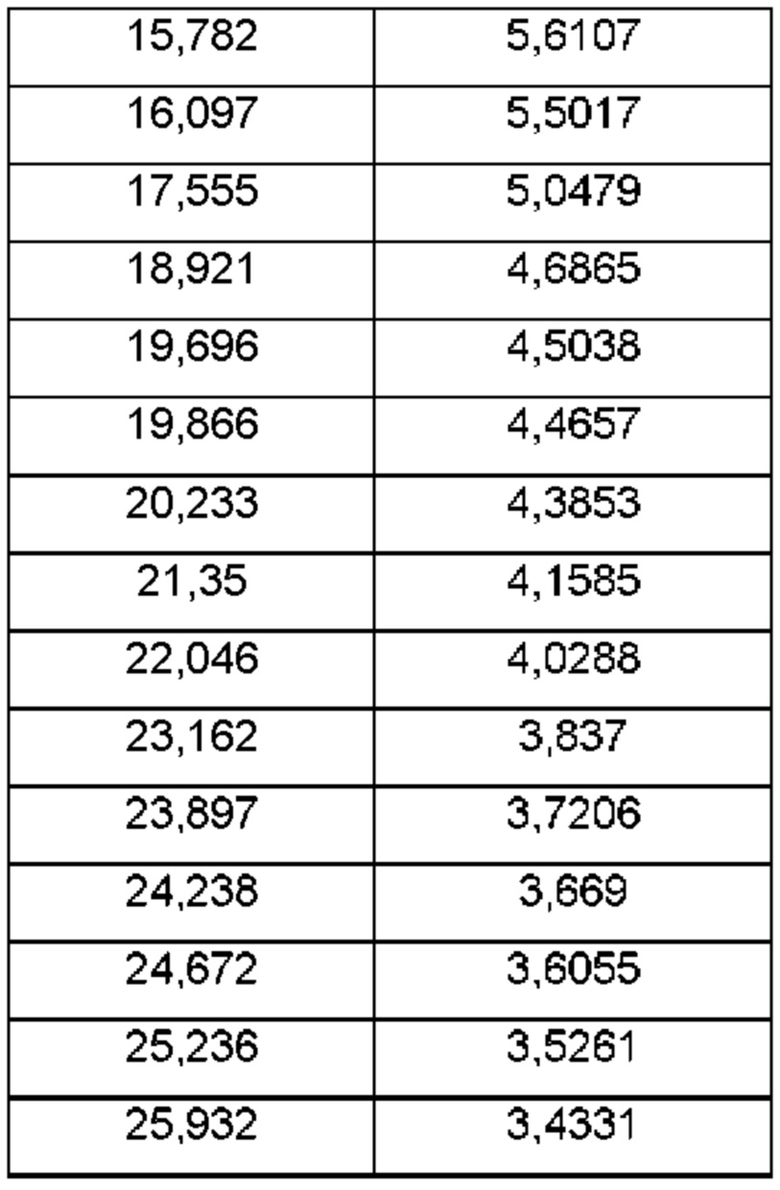
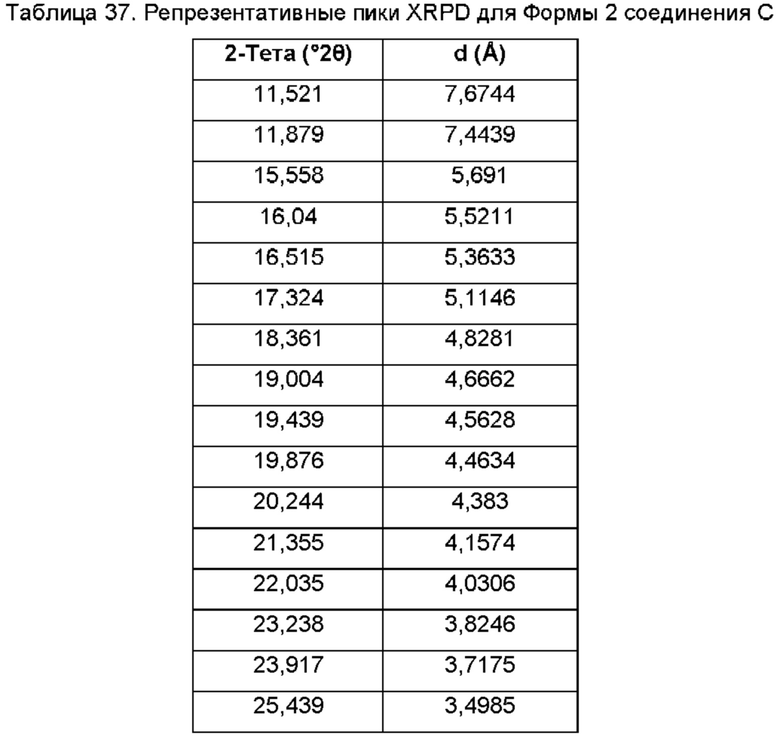
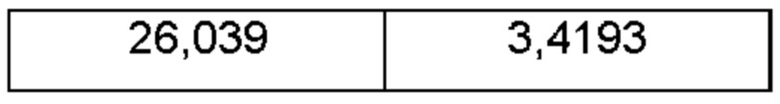
Резюме. Соединение А в форме свободного основания идентифицировали как аморфную твердую форму с уменьшением массы <0,5% до разложения и с точкой плавления, составляющей примерно 87°С. Ее гигроскопичность при RH 95% составляла <1%, и она характеризовалась содержанием эпимеров <1%. Растворимость ее при 37°С составляла примерно 6,6 мг/мл. Картина XRPD для этой аморфной формы приведена на ФИГ. 32. Форма 1 соединения С представляла собой кристаллическое вещество с уменьшением массы <0,5% до разложения и с точкой плавления, составляющей примерно 165-174°С. В зависимости от растворителя получали несколько форм соединения С. Их гигроскопичность при RH 95% составляла <1,5%, и они характеризовались содержанием эпимеров <1%. Растворимость их при 37°С составляла примерно 6,6 мг/мл. Форма В соединения В представляла собой кристаллическое вещество с уменьшением массы <0,5% до разложения и с точкой плавления, составляющей примерно 168°С. В безводной форме В была представлена единственная форма. Ее гигроскопичность при RH 95% составляла <1%, и она характеризовалась содержанием эпимеров <2,5%. Растворимость ее при 37°С составляла примерно 5,9 мг/мл. Было обнаружено, что безводная форма соединения В, Форма В, является стабильной чистой кристаллической формой, обладающей свойствами, более благоприятными, чем свойства форм D, Е и F, описанных в данной заявке.
Пример 33
Оценка безопасности, фармакокинетики и активности соединения В. Рак молочной железы представляет собой наиболее часто встречающийся рак, диагностируемый у женщин, с общей заболеваемостью, по оценкам составляющей 1,67 миллиона новых случаев, зарегистрированных в 2012 году (Ferlay et al., 2013). На рак молочной железы приходится приблизительно 15% (приблизительно 522000 случаев) от всех случаев смертей от рака.
Приблизительно в 80% всех случаев рака молочной железы экспрессируется фактор - рецептор эстрогенов (ER), и в подавляющем большинстве из них рост и прогрессирование опухолей зависят от ER. Модулирование активности и/или синтеза эстрогенов является основой терапевтических подходов при лечении женщин с ER-положительным раком молочной железы. Однако, несмотря на эффективность доступных средств эндокринной терапии, таких как антагонисты ER (например, тамоксифен), ингибиторы ароматазы (например, анастозол, летрозол и экземестан) и полные антагонисты/супрессоры ER (например, фулвестрант), у многих пациентов в конечном итоге возникает рецидив или развивается устойчивость к этим агентам, и поэтому требуется проведение дальнейшего лечения для оптимального контроля заболевания.
Несмотря на возникновение невосприимчивости к ингибиторам ароматазы или тамоксифену, рост и выживаемость резистентных опухолевых клеток остаются зависимыми от передачи сигнала с участием ER; ввиду этого пациенты с ER-положительным раком молочной железы по-прежнему могут отвечать на эндокринную терапию второй или третьей линии после проведения предшествующей терапии (Di Leo et al., 2010; Baselga et al., 2012). Появляется все больше доказательств того, что в случае устойчивости к эндокринной терапии ER может участвовать в передаче сигнала лиганд-независимым образом посредством использования входных сигналов для других сигнальных путей (Miller et al., 2010; Van Tine et al., 2011). Без связи с какой-либо конкретной теорией отметим, что агент с двойным механизмом действия (антагонизмом в отношении ER плюс деградацией ER) имеет возможность направленно воздействовать как на лиганд-зависимую, так и на лиганд-независимую ER-опосредованную передачу сигнала и, следовательно, улучшать результаты лечения на поздних стадиях ER-положительного рака молочной железы. Кроме того, в недавних исследованиях идентифицированы мутации в гене ESR1 (т.е. гене, который кодирует ERα), затрагивающие лиганд-связывающий домен (LBD) в ER (Segal и Dowsett, 2014). В доклинических моделях показано, что мутантный ER может управлять транскрипцией и пролиферацией в отсутствие эстрогена, что является подтверждением возможности мутантных по LBD форм ER участвовать в опосредовании клинической устойчивости к некоторым видам эндокринной терапии (Li et al., 2013; Robinson et al., 2013; Toy et al., 2013). Антагонисты ER, эффективные в отношении этих лиганд-независимых, конститутивно активных мутантных форм ER-рецепторов, могут обладать значительным терапевтическим преимуществом.
Таким образом, существует потребность в новых ER-направленных видах терапии, характеризующихся повышенной противоопухолевой активностью, для дальнейшего замедления прогрессирования заболевания и/или преодоления устойчивости к доступным в настоящее время средствам эндокринной терапии и в конечном итоге для увеличения продолжительности жизни женщин с ER-положительным раком молочной железы.
Соединение В представляет собой сильнодействующий, обладающий биодоступностью при пероральном введении, низкомолекулярный терапевтический агент, разрабатываемый для лечения пациентов с ER-положительным раком молочной железы. Предложенное в данном изобретении соединение В, в том числе его твердые формы (например, Форма В), представляют собой стабильные соединения с благоприятными для продолжения фармацевтических разработок свойствами. Без связи с какой-либо конкретной теорией отметим, что соединение В, по-видимому, проявляет антагонистическое действие в отношении эффектов, оказываемым эстрогенами, путем конкурентного связывания с LBD в ER как дикого типа, так и в его мутантных формах, с эффективностью, соответствующей наномолярным концентрациям. Без связи с какой-либо конкретной теорией отметим, что после связывания соединение В индуцирует переход LBD ER в неактивную конформацию, что следует из результатов измерений по замещению коактиваторных пептидов. Без связи с какой-либо конкретной теорией отметим, что помимо наличия у соединения В прямых антагонистических свойств, механизм его действия заключается в снижении уровней белка ERα за счет опосредуемой протеасомами деградации. Гипотетически предполагается, что деградация ER делает возможным полное подавление ER-опосредуемой передачи сигнала, что не достигается при применении ER-направленных терапевтических средств первого поколения, таких как тамоксифен, которые проявляют частично агонистическое действие. Соединение В эффективно ингибирует пролиферацию многих клеточных линий ER-положительного рака молочной железы in vitro, включая клетки, сконструированные для экспрессии клинически релевантных мутантных форм ER.
В случае in vivo соединение В демонстрировало дозозависимую противоопухолевую активность в моделях ксенотрансплантата ER-положительного рака молочной железы, в том числе в модели ксенотрансплантата, полученного от пациента, несущего мутацию в ESR1 (ER.Y537S). Было обнаружено, что эффективный диапазон доз составлял 0,1-10 мг/кг/сутки, и все дозы переносились хорошо. Фулвестрант, при введении в соответствии с клинически релевантной схемой введения, был менее эффективен, чем соединение В, при оценке в моделях ксенотрансплантата. Таким образом, в доклинических исследованиях для соединения В продемонстрирована надежная активность на моделях ER-положительного рака молочной железы в случае заболевания с вовлечением гена ESR1 дикого типа и мутантных форм ESR1.
Пример 34
Анализ эффективности соединения В in vitro и in vivo. Соединение В демонстрирует более полную деградацию ER и супрессию ER-опосредуемого пути по сравнению как с GDC-0927, так и с GDC-0810. Кроме того, соединение В имеет улучшенные свойства с точки зрения метаболизма и фармакокинетики лекарственных средств (DMPK) по сравнению как с GDC-0927, так и с GDC-0810, что обуславливает такую же эффективность in vivo, как и у GDC-0927, но при введении в 100 раз более низких дозах (например, в дозе 1 мг или 10 мг). (См. ФИГ. 34 и ФИГ. 35).
Фармакокинетика и метаболизм. После однократного внутривенного (в.в.) введения крысам, собакам и обезьянам было обнаружено, что соединение В характеризуется клиренсом в степени от низкой до умеренной, большим объемом распределения и конечным периодом полувыведения 7-24 часа. В случае крыс и собак биодоступность при пероральном введении была умеренной (41%-55%), а в случае обезьян низкой (17%). Данные in vitro показали, что связывание соединения В с белками плазмы крови является сильным среди всех видов, изменяясь от 98% до 99% связывания.
Эксперименты по идентификации метаболитов in vitro показали, что глюкуронизация, опосредуемая изоформой 1А4 УДФ(уридиндифосфат)-глюкуронозилтрансферазы (UGT1A4), является основным путем метаболизма соединения В in vitro. Вклад изоформ семейства цитохрома Р450 (CYP450) был незначительным и включал вклад как CYP3A4, так и CYP2C9. Исследования ингибирования CYP in vitro в микросомах печени человека и исследования индукции в гепатоцитах человека подтвердили наличие возможности межлекарственных взаимодействий в степени от низкой до умеренной. Соединение В напрямую ингибировало CYP3A4, при этом значения концентраций, вызывающих 50%-ное ингибирование (IC50), составляли 6,5 мкМ (1'-гидроксилирование мидазолама) и 26 мкМ (6β-гидроксилирование тестостерона); значения IC50 в случае ингибирования CYP2B6 и CYP2C8 составляли 13 мкМ и 21 мкМ, соответственно. Соединение В демонстрировало слабое метаболизм-зависимое ингибирование CYP2C9.
Токсикология. Для характеристики профиля безопасности соединения B в доклинических испытаниях проводили четырехнедельные, соответствующие стандартам надлежащей лабораторной практики (GLP), исследования токсичности при повторном пероральном введении самкам крыс и обезьян совместно с комплексными оценками неврологической (у крыс, обезьян), дыхательной (у обезьян) и сердечно-сосудистой (у обезьян) функций.
Исследовании переносимости соединения В проводили на крысах при типичных уровнях доз (10, 30 и 100 мг/кг) с обнаружением нежелательных эффектов, в большинстве случаев связанных с почками и печенью, при дозе 100 мг/кг. В исследовании на обезьянах (20, 60 и 200 мг/кг) максимальной переносимой дозой (MTD) считалась доза 60 мг/кг, поскольку высокая доза 200 мг/кг оказалась недопустимой. Неблагоприятные эффекты наблюдали в основном при высоком уровне доз, составляющем 200 мг/кг, и непереносимость связывали с повреждением и истощением почек и печени.
Как в случае крыс, так и обезьян наблюдали дозозависимый ассоциированный с приемом лекарственных средств фосфолипидоз (PLD) во многих органах с содержанием, которое превышало ожидаемые при введении соответствующей фазе I начальной дозы для человека (по меньшей мере в 44 раза и 6 раз на основании площади под кривой (AUC) зависимости концентрации от времени, соответственно), при этом неблагоприятные в отношении органов эффекты ограничивались действием на почки и печень. При введении крысам дозы 10 мг/кг (18-кратный фактор превышения содержания) PLD не отмечали, но число случаев его возникновения и их тяжесть возрастали для доз от 30 до 100 мг/кг. У обезьян дозовосприимчивый PLD имел место при всех дозах, но был ограничен минимальными изменениями в легком при дозе 20 мг/кг (6-кратный фактор превышения содержания). Такие значения кратности превышения содержания подтверждают, что риск PLD-ассоциированной токсичности для людей при приеме начальной дозы, соответствующей фазе I, является низким.
Возможность перенесения связанных с PLD результатов доклинических исследований на пациентов не является надежной, но может быть обоснована (Reasor et al., 2006). Для таких лекарственных средств, как тамоксифен и палбоциклиб, не продемонстрировано каких-либо связанных с применением в клинике опасений, несмотря на их относящиеся к PLD данные, полученные в доклинических исследованиях. И хотя при введении соединения В отмечали связь с PLD во многих тканях как в случае крыс, так и обезьян, в этих исследованиях с применением оптической микроскопии доказательств вовлечения в этот процесс таких важных органов, как сердце или глаза, и нейронов, обнаружено не было (Chatman et al., 2009).
После 28-суточного перорального введения крысам и обезьянам увеличение содержания соединения B в системном кровотоке было пропорционально дозе. С учетом характера и обратимости клинических признаков, клинической патологии и данных гистопатологии высокотоксичная доза для 10% животных (STD10) в случае крыс была определена как 100 мг/кг, с соответствующими значениями максимальной концентрации в плазме крови (Cmax) и AUC для интервала от 0 до 24 часов (AUC0-24), составляющими 6560 нг/мл и 143000 нг⋅ч/мл, соответственно. В случае обезьян самая высокая неопасная токсичная доза была определена как 60 мг/кг/сутки, с соответствующими значениями Cmax и AUC0-24, составляющими 841 нг/мл и 16200 нг⋅ч/мл, соответственно, на основании клинических признаков и показателей смертности, которые отмечаются в случае дозы 200 мг/кг/сутки.
Таким образом, результаты завершенных в настоящее время доклинических фармакологических исследований токсичности и безопасности характеризуют соединение В как надежное в плане профиля токсичности и обосновывают его введение раковым пациентам в фазе I испытания.
Введение соединения B в качестве монотерапии отдельно взятым агентом. В доклинических исследованиях для соединения В продемонстрирована надежная активность на моделях ER-положительного рака молочной железы в случае заболевания с вовлечением гена ESR1 дикого типа и мутантных форм ESR1. Безопасность, фармакокинетическую (ФК), фармакодинамическую (ФД) активность и предварительную противоопухолевую активность соединения B в качестве отдельно взятого агента анализировали в фазе Ia/Ib много центрового открытого исследования на пациентах с локально распространенным или метастазирующим ER-положительным раком молочной железы. Пациентов включали в испытание на этапе повышения дозы с последующим участием на этапе расширения. В ходе повышения дозы отдельно взятого агента проводили оценку групп при возрастающих уровнях доз с целью определения MTD или максимальной вводимой дозы (MAD).
После установления значений MTD или MAD для отдельно взятого агента в испытание может быть включена "группа повышения дозы", получающая лечение соединением В (в дозах, равных или ниже MTD либо MAD) или соединением B в комбинации с палбоциклибом. Кроме того, пациенты будут включены в испытание на этапе расширения и будут получать лечение, в дозах, равных или ниже MTD либо MAD для отдельно взятого агента, отдельно взятым соединением В или в комбинации с палбоциклибом и/или агонистом рилизинг-фактора лютеинизирующего гормона (LHRH). Расширения дозы для отдельно взятого агента можно оценивать по двум разным уровням доз для соединения B вместе с агонистом LHRH и без него.
Мониторинг состояния пациентов на предмет нежелательных явлений проводили во время окна оценки дозолимитирующей токсичности (DLT), определенного как промежуток времени от -7-х до 28-х суток цикла 1 (в группах с отдельно взятым агентом). Для оценки DLT степень токсичности классифицировали в соответствии со Стандартными терминологическими критериями нежелательных явлений Национального института рака, версией 4.0 (NCI СТСАЕ v4.0).
Пациенты были включены в испытание на этапе повышения дозы соединения В как отдельно взятого агента, которая состоит из периода скрининга, вводного периода ФК исследования, периода лечения и периода последующего наблюдения безопасности. К непрерывному ежесуточному введению приступали во время периода лечения, начинающегося с 1-х суток цикла 1. Начальная доза соединения В как отдельно взятого агента составляла 10 мг, и ее вводили в рот пациентам в первой группе непрерывно в течение 28-суточных циклов. Для каждой последующей группы будет проведено повышение дозы вплоть до 200% от уровня предыдущей дозы, пока не будут обнаружены проблемы, связанные с безопасностью (т.е. либо будет отмечена DLT, либо будет обнаружен какой-либо пациент с клинически значимой токсичностью степени ≥ 2, либо профиль общих нежелательных явлений не будет соответствовать 200%-ным приращениям). После обнаружения проблем, связанных с безопасностью, повышение дозы не будет превышать 100%-ных приращений.
Соединение В имеет период полувыведения примерно 40 часов. Как отмечено выше, содержание соединения В повышалось пропорционально увеличению дозы от 10 до 30 мг с нормальной изменчивостью.
Лечение шести пациентов начинали с введения начальной дозы, составляющей 10 мг QD, в течение 28-суточных циклов, как описано в данной заявке. Как отмечено в приведенной ниже Таблице 38, все пациенты, получавшие лечение с FES-PET-сканированием, имели качественно практически полные (NC) или полные ответы (CR). У получавших лечение пациентов не обнаружено случаев DLT, серьезных нежелательных явлений (SAE), нежелательных явлений особого значения (AESI) или клинически значимых зафиксированных в лаборатории нарушений. Все соответствующие нежелательные явления (АЕ) представляли собой явления 1-ой степени или 2-ой степени.
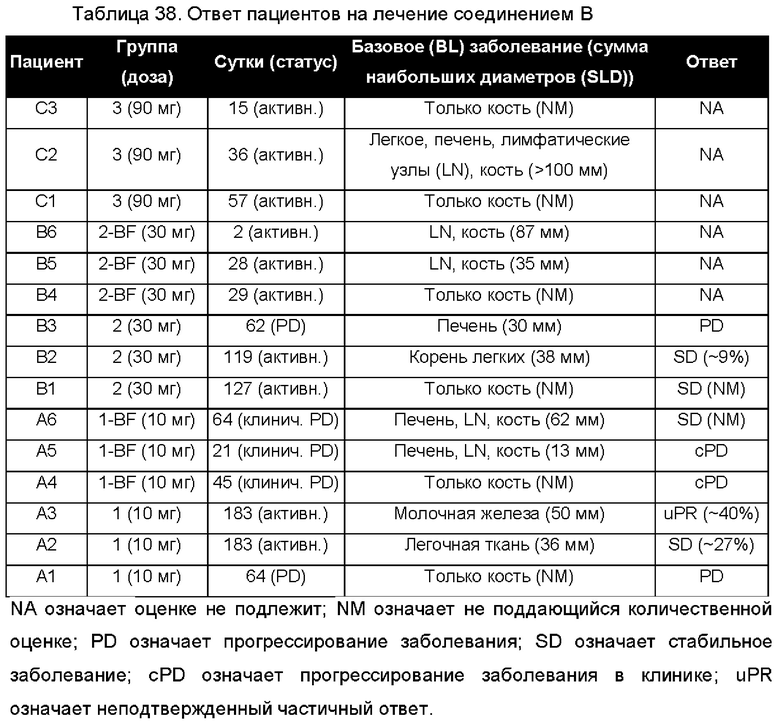
Одному подвергаемому лечению пациенте был поставлен диагноз ER+PR+рака молочной железы (PR означает рецептор прогестерона). Ранее, перед включением в лечение и лечением, этот пациент перенес операцию и лечение противораковыми средствами, в том числе терапию с применением селективных модуляторов рецепторов эстрогенов (SERM) и терапию с применением AI. Пациент получал лечение соединением B в дозе 10 мг и показал ответ на лечение по окончании 3 циклов, указанный на ФИГ. 36а и ФИГ. 36b.
Другому пациенту был поставлен диагноз ранней стадии положительного по рецептору гормона (HR+) рака молочной железы, и до этого он подвергался хирургическому вмешательству и лечению противораковыми агентами, включая цитотоксические средства, ингибиторы CDK4/6 и AI. Этот пациент получал лечение соединением B в дозе 10 мг и показал ответ на лечение по окончании 3 циклов, указанный на ФИГ. 37а и ФИГ. 37b, после включения в испытание согласно данному изобретению в марте 2018 года с назначением дозы 10 мг.
Получившие лечение пациенты в настоящее время демонстрируют хорошую переносимость соединения В, введенного в дозах 10 мг и 30 мг, и имеют АЕ только 1-ой и 2-ой степени. Как правило, содержание соединения B в плазме крови возрастало пропорционально увеличению доз от 10 до 30 мг после однократного введения. Содержание в стационарном состоянии, по-видимому, возрастает в большей степени по сравнению с пропорциональным увеличением дозы от 10 мг до 30 мг. Предполагаемый период полувыведения, составляющий примерно 40 часов, позволяет осуществлять дозирование один раз в сутки.
В этом представленном в письменном виде описании используются примеры для раскрытия изобретения, в том числе его лучший вариант, а также для того, чтобы дать возможность любому специалисту в данной области техники применить изобретение на практике, включая создание и использование любых устройств или систем и выполнение любых включенных методов. Патентоспособный объем изобретения определяется формулой изобретения и может включать другие примеры, которые встречаются специалистам в данной области. Подразумевается, что такие другие примеры включены в объем формулы изобретения, если они имеют структурные элементы, которые не отличаются от буквальной формулировки формулы изобретения, или если они включают эквивалентные структурные элементы с несущественными отличиями от буквальных формулировок формулы изобретения.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> GENENTECH, INC.
<120> ТВЕРДЫЕ ФОРМЫ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-
ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-B]ИНДОЛ-2-ИЛ)-
2,2-ДИФТОРПРОПАН-1-ОЛА И СПОСОБЫ ПОЛУЧЕНИЯ КОНДЕНСИРОВАННЫХ
ТРИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ЗАМЕЩЕННУЮ ФЕНИЛЬНУЮ ИЛИ
ПИРИДИНИЛЬНУЮ ГРУППИРОВКУ, В ТОМ ЧИСЛЕ СПОСОБЫ ИХ ПРИМЕНЕНИЯ
<130> P34807-WO
<140>
<141>
<150> 62/719,896
<151> 2018-08-20
<150> 62/687,930
<151> 2018-06-21
<160> 4
<170> PatentIn version 3.5
<210> 1
<211> 457
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="описание искусственной последовательности:
синтетический полипептид"
<400> 1
Met Leu Lys Asn Asp Gln Leu Asp Gln Trp Asp Arg Asp Asn Phe Phe
1 5 10 15
His Pro Ser Thr His Leu Ala Gln His Ala Arg Gly Glu Ser Ala Asn
20 25 30
Arg Val Ile Lys Thr Ala Ser Gly Val Phe Ile Glu Asp Arg Asp Gly
35 40 45
Thr Lys Leu Leu Asp Ala Phe Ala Gly Leu Phe Cys Val Asn Val Gly
50 55 60
Tyr Gly Arg Gln Glu Ile Ala Glu Ala Ile Ala Asp Gln Ala Arg Glu
65 70 75 80
Leu Ala Tyr Tyr His Ser Phe Val Gly His Gly Thr Glu Ala Ser Ile
85 90 95
Thr Leu Ala Lys Met Ile Leu Asp Arg Ala Pro Lys Asn Met Ser Lys
100 105 110
Val Tyr Phe Gly Leu Gly Gly Ser Asp Ala Asn Glu Thr Asn Val Lys
115 120 125
Leu Ile Trp Tyr Tyr Asn Asn Ile Leu Gly Arg Pro Glu Lys Lys Lys
130 135 140
Ile Ile Ser Arg Trp Arg Gly Tyr His Gly Ser Gly Leu Val Thr Gly
145 150 155 160
Ser Leu Thr Gly Leu Glu Leu Phe His Lys Lys Phe Asp Leu Pro Val
165 170 175
Glu Gln Val Ile His Thr Glu Ala Pro Tyr Tyr Phe Arg Arg Glu Asp
180 185 190
Leu Asn Gln Thr Glu Glu Gln Phe Val Ala His Cys Val Ala Glu Leu
195 200 205
Glu Ala Leu Ile Glu Arg Glu Gly Ala Asp Thr Ile Ala Ala Phe Ile
210 215 220
Gly Glu Pro Ile Leu Gly Gly Gly Gly Ile Val Pro Pro Pro Ala Gly
225 230 235 240
Tyr Trp Glu Ala Ile Gln Thr Val Leu Asn Lys His Asp Ile Leu Leu
245 250 255
Val Ala Asp Glu Val Val Thr Gly Phe Gly Arg Leu Gly Thr Met Phe
260 265 270
Gly Ser Asp His Tyr Gly Leu Glu Pro Asp Ile Ile Thr Ile Ala Lys
275 280 285
Gly Leu Thr Ser Ala Tyr Ala Pro Leu Ser Gly Ser Ile Val Ser Asp
290 295 300
Lys Val Trp Lys Val Leu Glu Gln Gly Thr Asp Glu Asn Gly Pro Ile
305 310 315 320
Gly His Gly Trp Thr Tyr Ser Ala His Pro Ile Gly Ala Ala Ala Gly
325 330 335
Val Ala Asn Leu Lys Leu Leu Asp Glu Leu Asn Leu Val Ser Asn Ala
340 345 350
Gly Glu Val Gly Ala Tyr Leu Asn Ala Thr Met Ala Glu Ala Leu Ser
355 360 365
Gln His Ala Asn Val Gly Asp Val Arg Gly Glu Gly Leu Leu Cys Ala
370 375 380
Val Glu Phe Val Lys Asp Arg Asp Ser Arg Thr Phe Phe Asp Ala Ala
385 390 395 400
Asp Lys Ile Gly Pro Gln Ile Ser Ala Lys Leu Leu Glu Gln Asp Lys
405 410 415
Ile Ile Ala Arg Ala Met Pro Gln Gly Asp Ile Leu Gly Phe Ala Pro
420 425 430
Pro Phe Cys Leu Thr Arg Ala Glu Ala Asp Gln Val Val Glu Gly Thr
435 440 445
Leu Arg Ala Val Lys Ala Val Leu Gly
450 455
<210> 2
<211> 457
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="описание искусственной последовательности:
синтетический полипептид"
<400> 2
Met Leu Lys Asn Asp Gln Leu Asp Gln Trp Asp Arg Asp Asn Phe Phe
1 5 10 15
His Pro Ser Thr His Leu Ala Gln His Ala Arg Gly Glu Ser Ala Asn
20 25 30
Arg Val Ile Lys Thr Ala Ser Gly Val Phe Ile Glu Asp Arg Asp Gly
35 40 45
Thr Lys Leu Leu Asp Ala Phe Ala Gly Leu Trp Cys Val Asn Val Gly
50 55 60
Tyr Gly Arg Gln Glu Ile Ala Glu Ala Ile Ala Asp Gln Ala Arg Glu
65 70 75 80
Leu Ala Tyr Tyr His Ser Phe Val Gly His Gly Thr Glu Ala Ser Ile
85 90 95
Thr Leu Ala Lys Met Ile Leu Asp Arg Ala Pro Lys Asn Met Ser Lys
100 105 110
Val Tyr Phe Gly Leu Gly Gly Ser Asp Ala Asn Glu Thr Asn Val Lys
115 120 125
Leu Ile Trp Tyr Tyr Asn Asn Ile Leu Gly Arg Pro Glu Lys Lys Lys
130 135 140
Ile Ile Ser Arg Trp Arg Gly Tyr His Gly Ser Gly Leu Val Thr Gly
145 150 155 160
Ser Leu Thr Gly Leu Glu Leu Phe His Lys Lys Phe Asp Leu Pro Val
165 170 175
Glu Gln Val Ile His Thr Glu Ala Pro Tyr Tyr Phe Arg Arg Glu Asp
180 185 190
Leu Asn Gln Thr Glu Glu Gln Phe Val Ala His Cys Val Ala Glu Leu
195 200 205
Glu Ala Leu Ile Glu Arg Glu Gly Ala Asp Thr Ile Ala Ala Phe Ile
210 215 220
Gly Glu Pro Ile Leu Gly Ala Gly Gly Ile Val Pro Pro Pro Ala Gly
225 230 235 240
Tyr Trp Glu Ala Ile Gln Thr Val Leu Asn Lys His Asp Ile Leu Leu
245 250 255
Val Ala Asp Glu Val Val Thr Gly Phe Gly Arg Leu Gly Thr Met Phe
260 265 270
Gly Ser Asp His Tyr Gly Leu Glu Pro Asp Ile Ile Thr Ile Ala Lys
275 280 285
Gly Leu Thr Ser Ala Tyr Ala Pro Leu Ser Gly Ser Ile Val Ser Asp
290 295 300
Lys Val Trp Lys Val Leu Glu Gln Gly Thr Asp Glu Asn Gly Pro Ile
305 310 315 320
Gly His Gly Trp Thr Tyr Ser Ala His Pro Ile Gly Ala Ala Ala Gly
325 330 335
Val Ala Asn Leu Lys Leu Leu Asp Glu Leu Asn Leu Val Ser Asn Ala
340 345 350
Gly Glu Val Gly Ala Tyr Leu Asn Ala Thr Met Ala Glu Ala Leu Ser
355 360 365
Gln His Ala Asn Val Gly Asp Val Arg Gly Glu Gly Leu Leu Cys Ala
370 375 380
Val Glu Phe Val Lys Asp Arg Asp Ser Arg Thr Phe Phe Asp Ala Ala
385 390 395 400
Asp Lys Ile Gly Pro Gln Ile Ser Ala Lys Leu Leu Glu Gln Asp Lys
405 410 415
Ile Ile Ala Arg Ala Met Pro Gln Gly Asp Ile Leu Gly Phe Ala Pro
420 425 430
Pro Phe Cys Leu Thr Arg Ala Glu Ala Asp Gln Val Val Glu Gly Thr
435 440 445
Leu Arg Ala Val Lys Ala Val Leu Gly
450 455
<210> 3
<211> 457
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="описание искусственной последовательности:
синтетический полипептид"
<400> 3
Met Leu Lys Asn Asp Gln Leu Asp Gln Trp Asp Arg Asp Asn Phe Phe
1 5 10 15
His Pro Ser Thr His Leu Ala Gln His Ala Arg Gly Glu Ser Ala Asn
20 25 30
Arg Val Ile Lys Thr Ala Ser Gly Val Phe Ile Glu Asp Arg Asp Gly
35 40 45
Thr Lys Leu Leu Asp Ala Phe Ala Gly Leu Phe Cys Val Asn Val Gly
50 55 60
Tyr Gly Arg Gln Glu Ile Ala Glu Ala Ile Ala Asp Gln Ala Arg Glu
65 70 75 80
Leu Ala Tyr Tyr His Ala Phe Val Gly His Gly Thr Glu Ala Ser Ile
85 90 95
Thr Leu Ala Lys Met Ile Leu Asp Arg Ala Pro Lys Asn Met Ser Lys
100 105 110
Val Tyr Phe Gly Leu Gly Gly Ser Asp Ala Asn Glu Thr Asn Val Lys
115 120 125
Leu Ile Trp Tyr Tyr Asn Asn Ile Leu Gly Arg Pro Glu Lys Lys Lys
130 135 140
Ile Ile Ser Arg Trp Arg Gly Phe His Gly Ser Gly Leu Val Thr Gly
145 150 155 160
Ser Leu Thr Gly Leu Glu Leu Phe His Lys Lys Phe Asp Leu Pro Val
165 170 175
Glu Gln Val Ile His Thr Glu Ala Pro Tyr Tyr Phe Arg Arg Glu Asp
180 185 190
Leu Asn Gln Thr Glu Glu Gln Phe Val Ala His Cys Val Ala Glu Leu
195 200 205
Glu Ala Leu Ile Glu Arg Glu Gly Ala Asp Thr Ile Ala Ala Phe Ile
210 215 220
Gly Glu Pro Ile Leu Gly Ala Gly Gly Met Val Pro Pro Pro Ala Gly
225 230 235 240
Tyr Trp Glu Ala Ile Gln Thr Val Leu Asn Lys His Asp Ile Leu Leu
245 250 255
Val Ala Asp Glu Val Val Thr Gly Phe Gly Arg Leu Gly Thr Met Phe
260 265 270
Gly Ser Asp His Tyr Gly Leu Glu Pro Asp Ile Ile Thr Ile Ala Lys
275 280 285
Gly Leu Thr Ser Ala Tyr Ala Pro Leu Ser Gly Ser Ile Val Ser Asp
290 295 300
Lys Val Trp Lys Val Leu Glu Gln Gly Thr Asp Glu Asn Gly Pro Ile
305 310 315 320
Gly His Gly Trp Thr Tyr Ser Ala His Pro Ile Gly Ala Ala Ala Gly
325 330 335
Val Ala Asn Leu Lys Leu Leu Asp Glu Leu Asn Leu Val Ser Asn Ala
340 345 350
Gly Glu Val Gly Ala Tyr Leu Asn Ala Thr Met Ala Glu Ala Leu Ser
355 360 365
Gln His Ala Asn Val Gly Asp Val Arg Gly Glu Gly Leu Met Cys Ala
370 375 380
Val Glu Phe Val Lys Asp Arg Asp Ser Arg Thr Phe Phe Asp Ala Ala
385 390 395 400
Asp Lys Ile Gly Pro Gln Ile Ser Ala Lys Leu Leu Glu Gln Asp Lys
405 410 415
Ile Ile Ala Arg Ala Met Pro Gln Gly Asp Ile Leu Gly Phe Ala Pro
420 425 430
Pro Phe Cys Leu Thr Arg Ala Glu Ala Asp Gln Val Val Glu Gly Thr
435 440 445
Leu Arg Ala Val Lys Ala Val Leu Gly
450 455
<210> 4
<211> 464
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> источник
<223> /примечание="описание искусственной последовательности:
синтетический полипептид"
<400> 4
Met Ala Thr Ile Thr Asn His Met Pro Thr Ala Glu Leu Gln Ala Leu
1 5 10 15
Asp Ala Ala His His Leu His Pro Phe Ser Ala Asn Asn Ala Leu Gly
20 25 30
Glu Glu Gly Thr Arg Val Ile Thr Arg Ala Arg Gly Val Trp Leu Asn
35 40 45
Asp Ser Ala Leu Ala Gln Lys Leu Ala Glu Leu Ala Gly Leu Trp Cys
50 55 60
Val Asn Ile Gly Tyr Gly Arg Asp Glu Leu Ala Glu Val Ala Ala Arg
65 70 75 80
Gln Met Arg Glu Leu Pro Tyr Tyr Asn Thr Phe Phe Lys Thr Thr His
85 90 95
Val Pro Ala Ile Ala Leu Ala Gln Lys Leu Ala Glu Leu Ala Pro Gly
100 105 110
Asp Leu Asn His Val Phe Phe Ala Gly Gly Gly Ser Glu Ala Asn Asp
115 120 125
Thr Asn Ile Arg Met Val Arg Thr Tyr Trp Gln Asn Lys Gly Gln Pro
130 135 140
Glu Lys Thr Val Ile Ile Ser Arg Lys Asn Ala Tyr His Gly Ser Thr
145 150 155 160
Val Ala Ser Ser Ala Leu Gly Gly Met Ala Gly Met His Ala Gln Ser
165 170 175
Gly Leu Ile Pro Asp Val His His Ile Asn Gln Pro Asn Trp Trp Ala
180 185 190
Glu Gly Gly Asp Met Asp Pro Glu Glu Phe Gly Leu Ala Arg Ala Arg
195 200 205
Glu Leu Glu Glu Ala Ile Leu Glu Leu Gly Glu Asn Arg Val Ala Ala
210 215 220
Phe Ile Ala Glu Pro Val Gln Gly Ala Gly Gly Val Ile Val Ala Pro
225 230 235 240
Asp Ser Tyr Trp Pro Glu Ile Gln Arg Ile Cys Asp Lys Tyr Asp Ile
245 250 255
Leu Leu Ile Ala Asp Glu Val Val Cys Gly Phe Gly Arg Thr Gly Asn
260 265 270
Trp Phe Gly Thr Gln Thr Met Gly Ile Arg Pro His Ile Met Thr Ile
275 280 285
Ala Lys Gly Leu Ser Ser Gly Tyr Ala Pro Ile Gly Gly Ser Ile Val
290 295 300
Cys Asp Glu Val Ala His Val Ile Gly Lys Asp Glu Phe Asn His Gly
305 310 315 320
Tyr Thr Tyr Ser Gly His Pro Val Ala Ala Ala Val Ala Leu Glu Asn
325 330 335
Leu Arg Ile Leu Glu Glu Glu Asn Ile Leu Asp His Val Arg Asn Val
340 345 350
Ala Ala Pro Tyr Leu Lys Glu Lys Trp Glu Ala Leu Thr Asp His Pro
355 360 365
Leu Val Gly Glu Ala Lys Ile Val Gly Met Met Ala Ser Ile Ala Leu
370 375 380
Thr Pro Asn Lys Ala Ser Arg Ala Lys Phe Ala Ser Glu Pro Gly Thr
385 390 395 400
Ile Gly Tyr Ile Cys Arg Glu Arg Cys Phe Ala Asn Asn Leu Ile Met
405 410 415
Arg His Val Gly Asp Arg Met Ile Ile Ser Pro Pro Leu Val Ile Thr
420 425 430
Pro Ala Glu Ile Asp Glu Met Phe Val Arg Ile Arg Lys Ser Leu Asp
435 440 445
Glu Ala Gln Ala Glu Ile Glu Lys Gln Gly Leu Met Lys Ser Ala Ala
450 455 460
<---
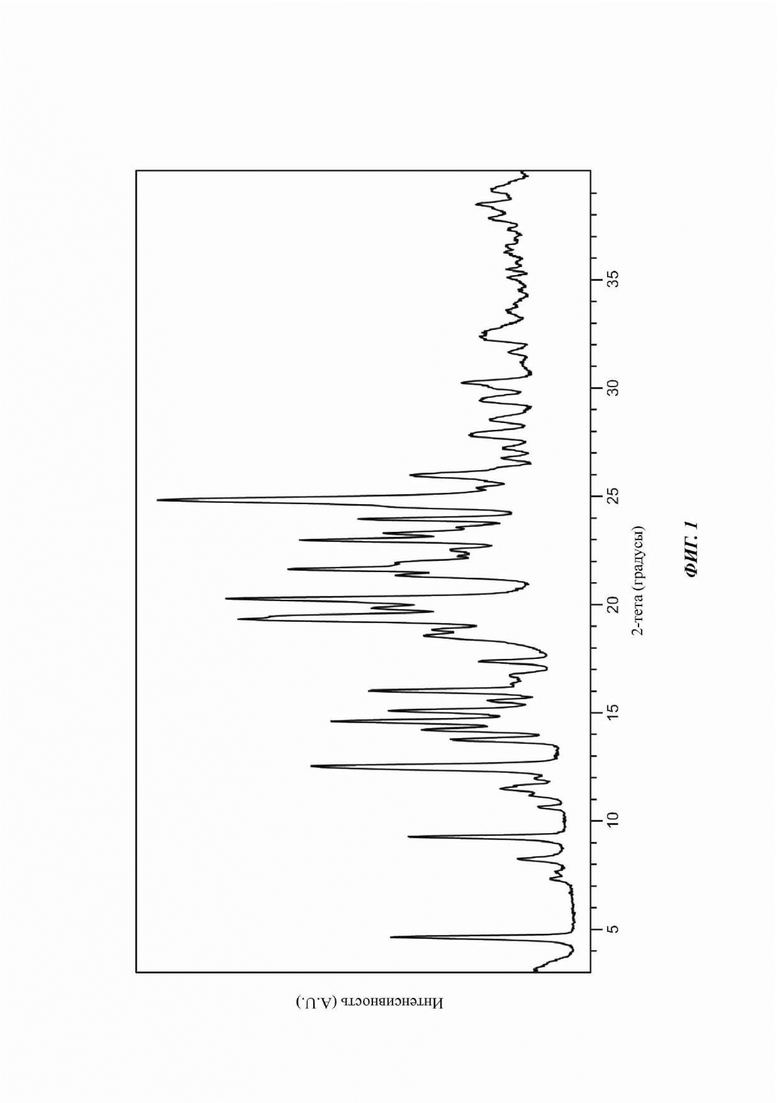

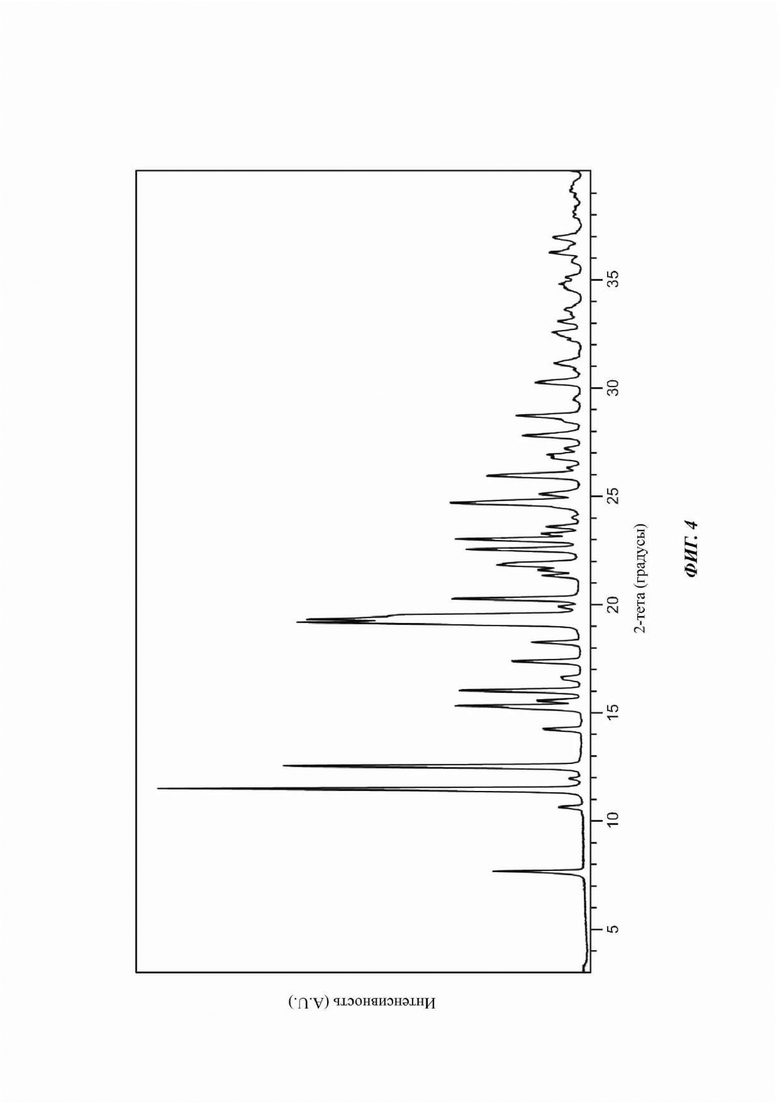
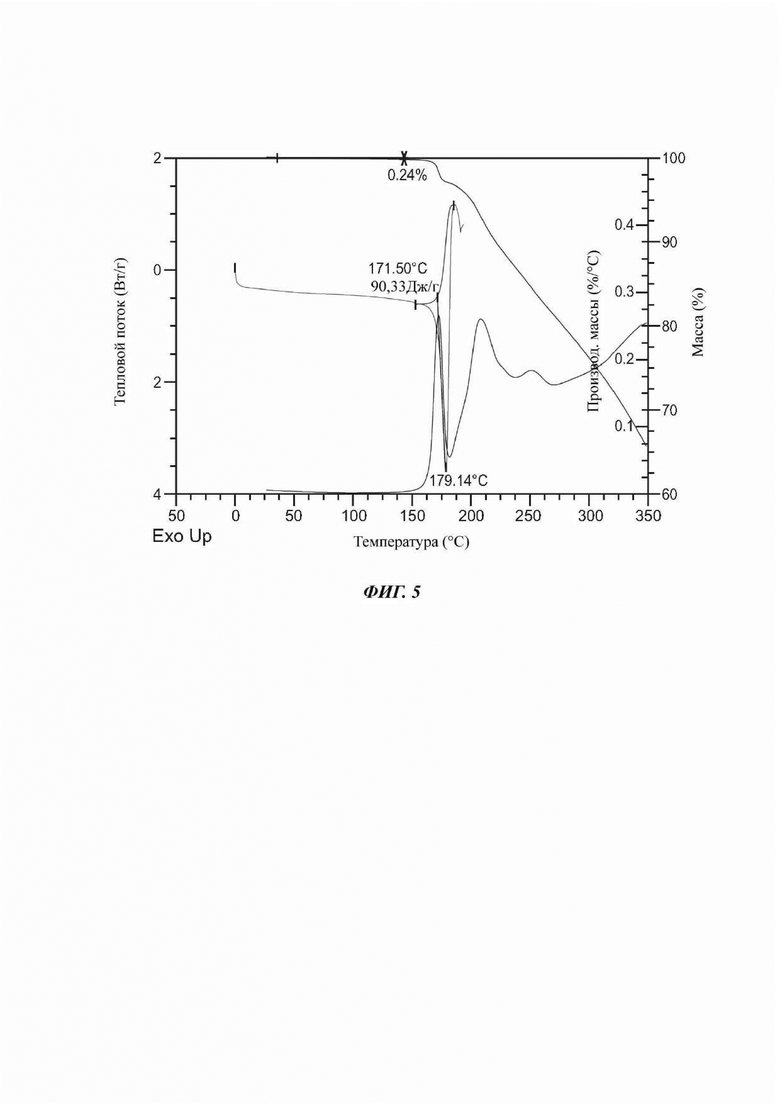
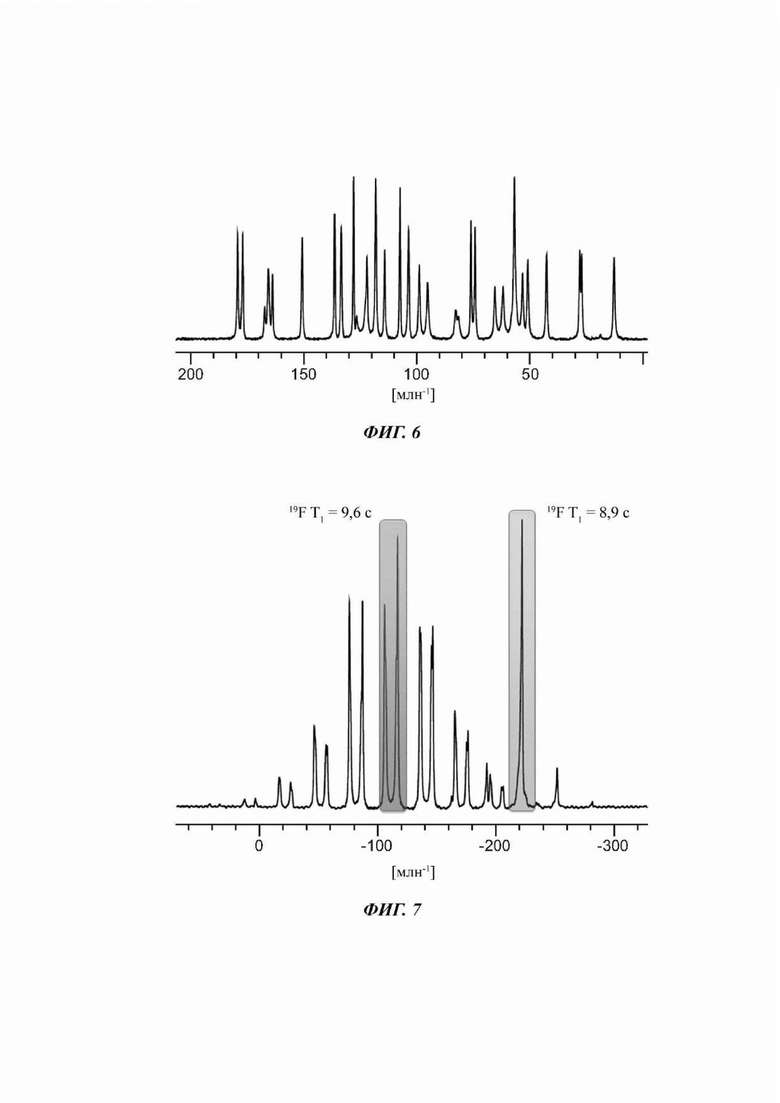
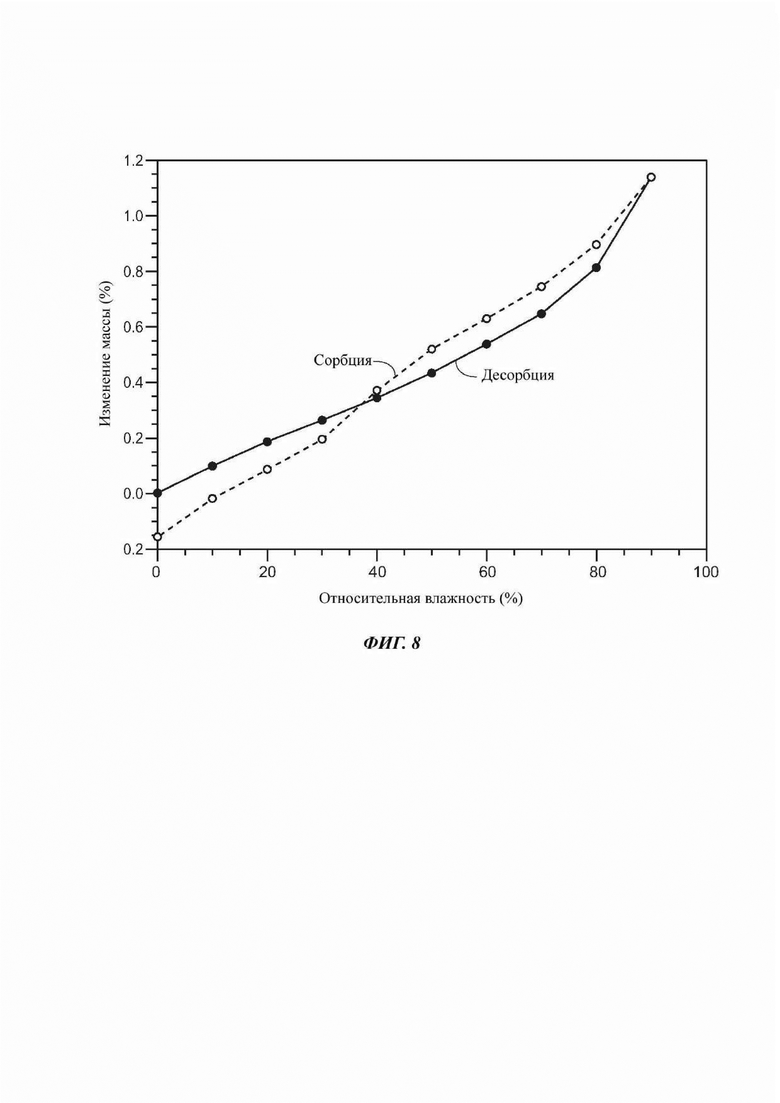
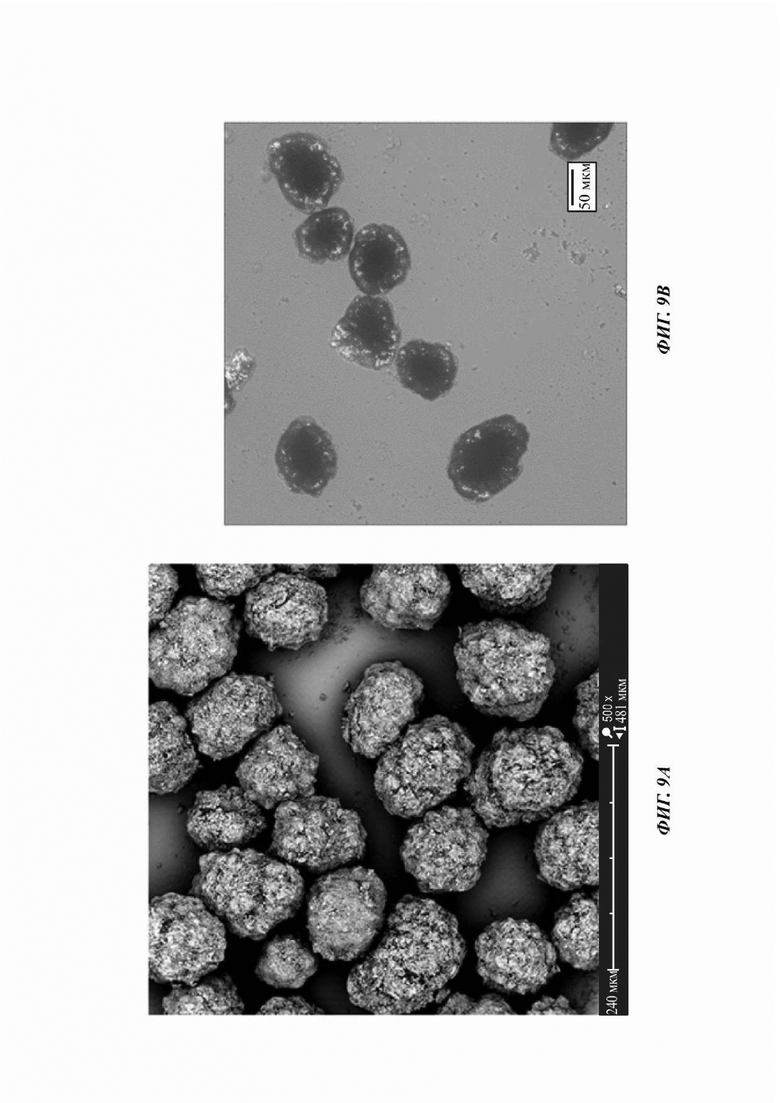
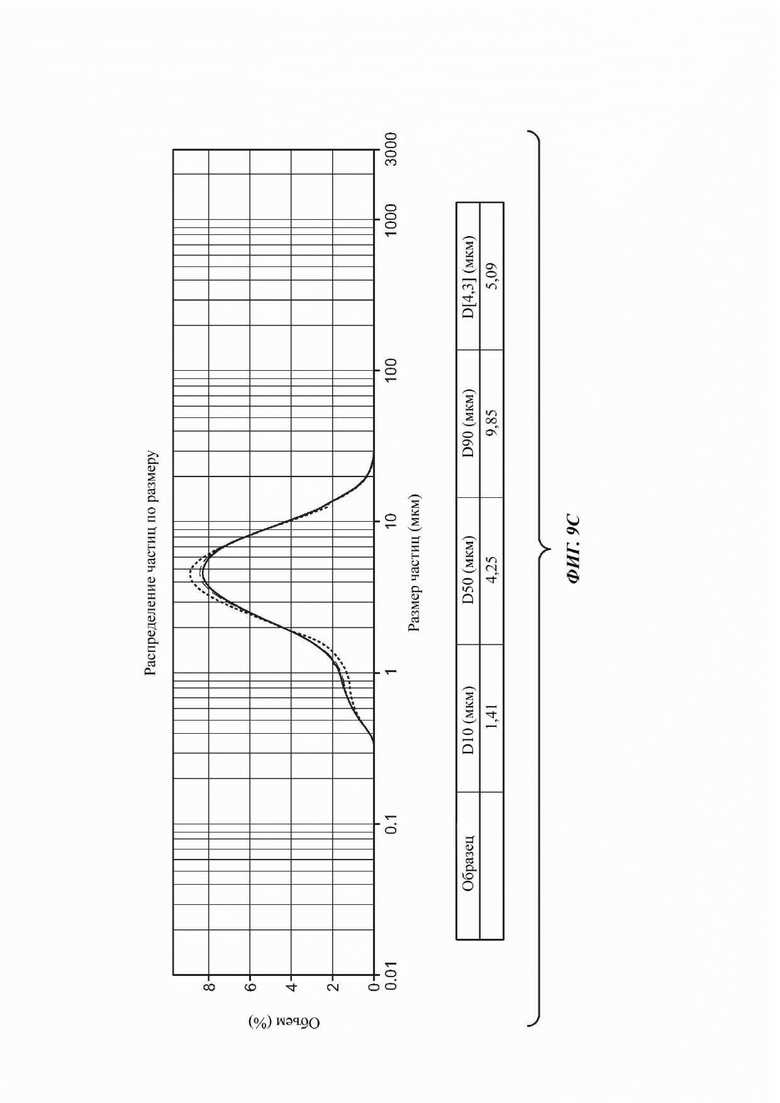
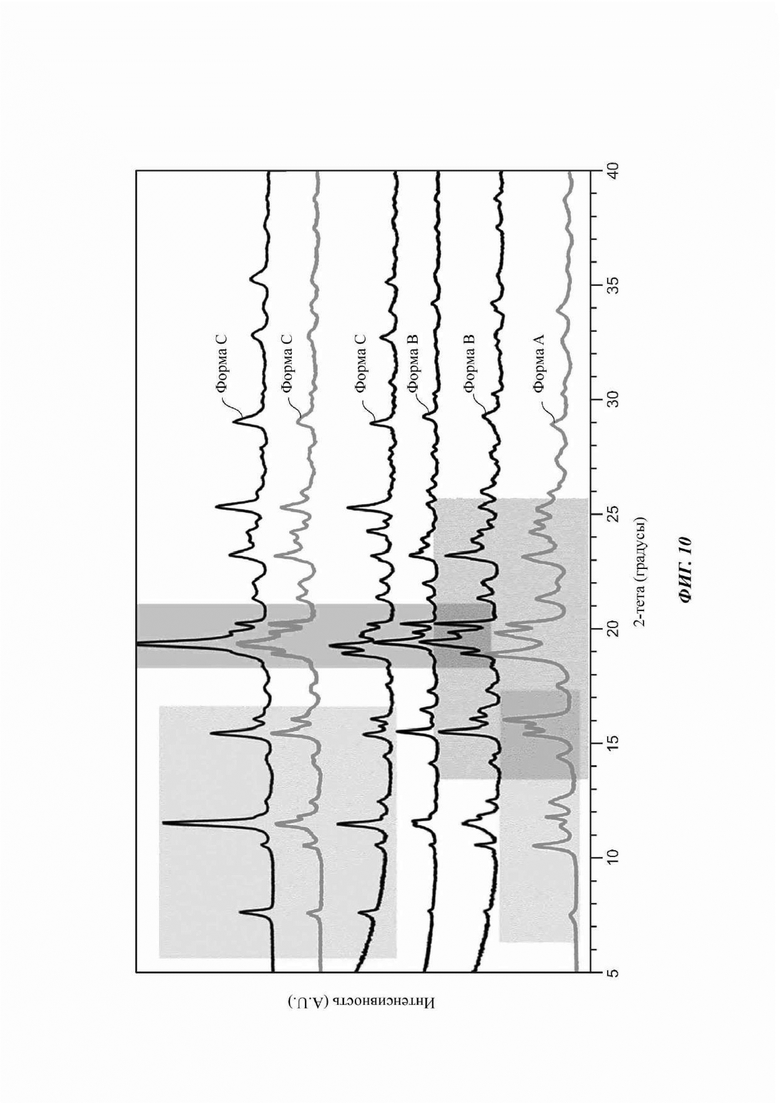
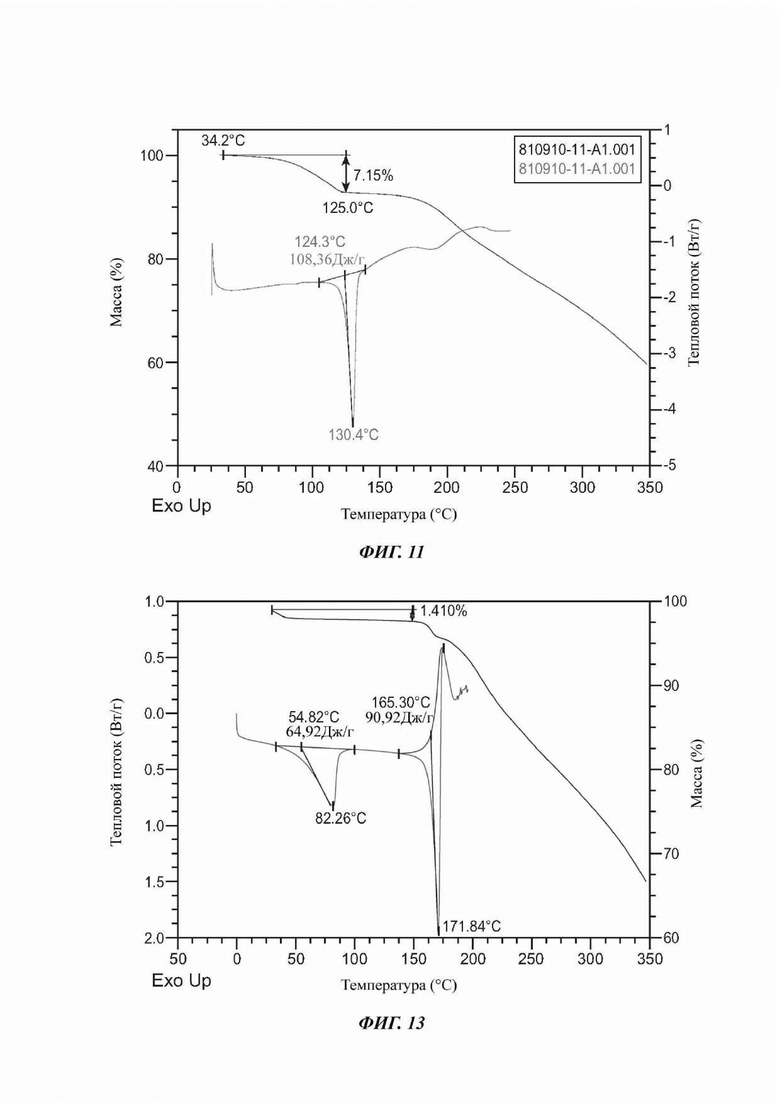
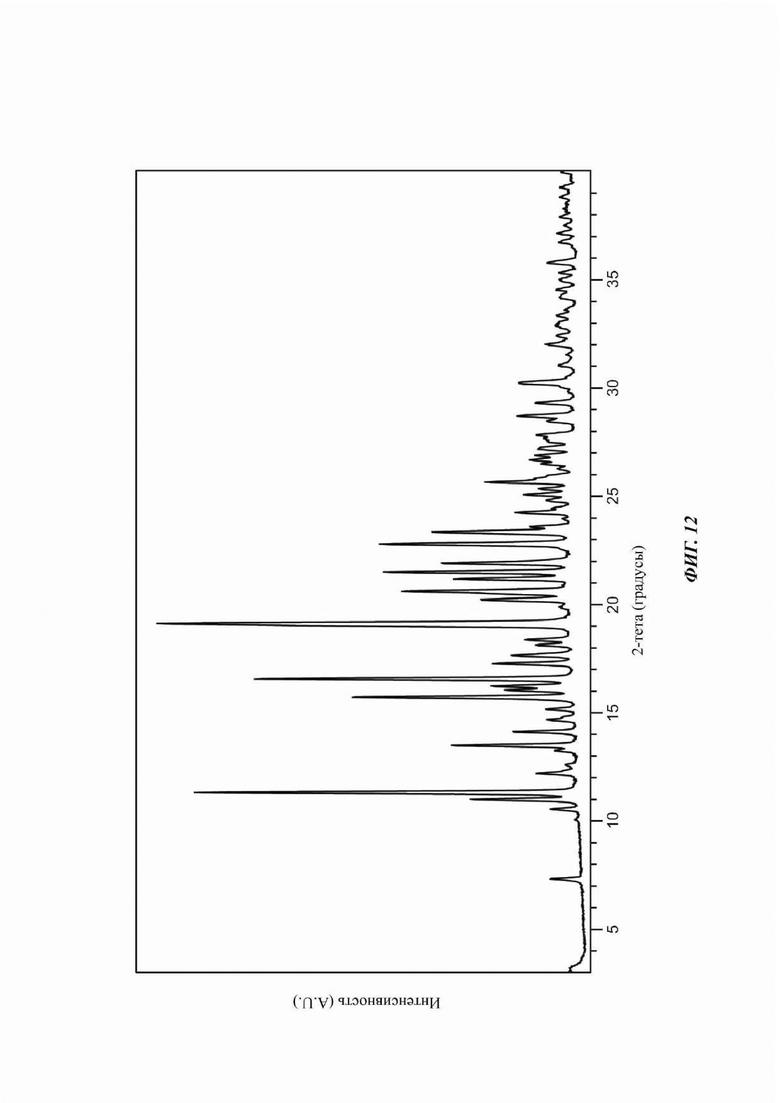
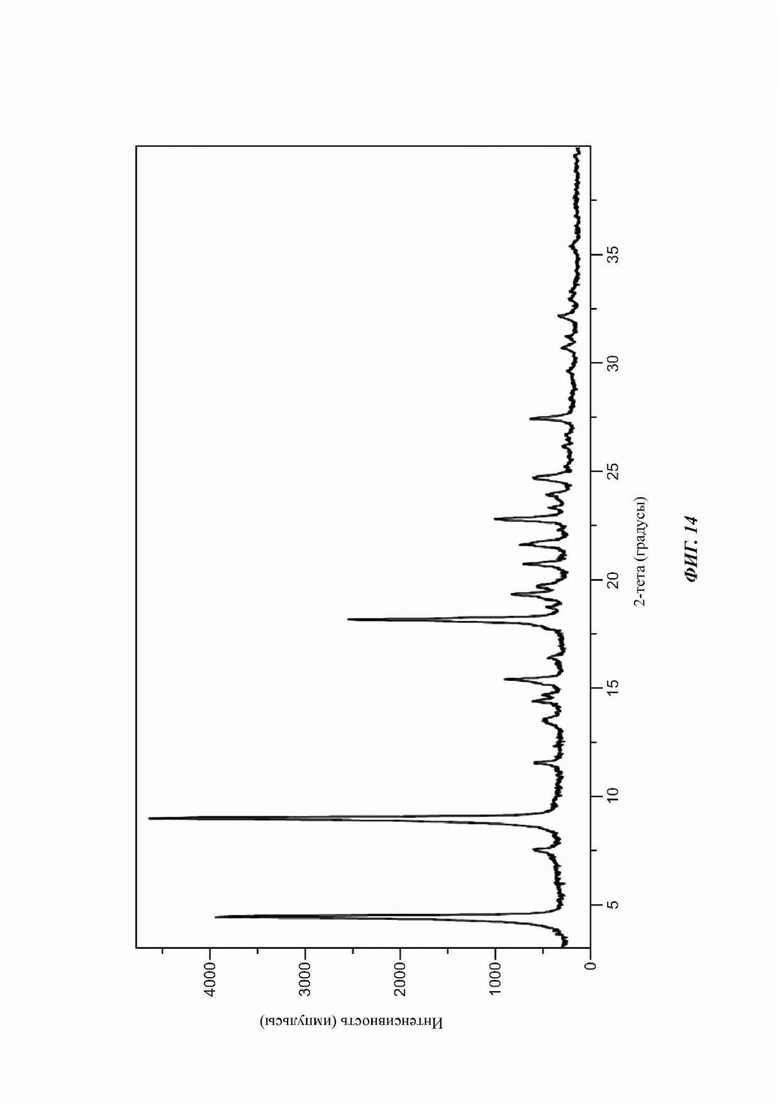
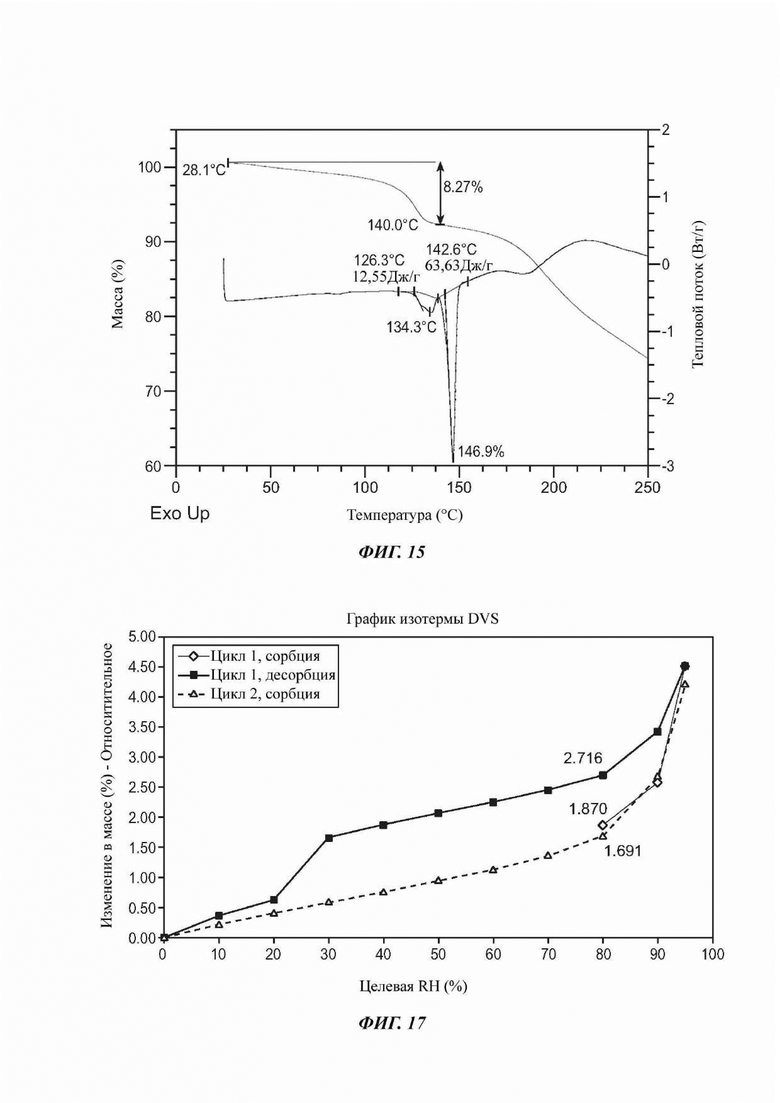
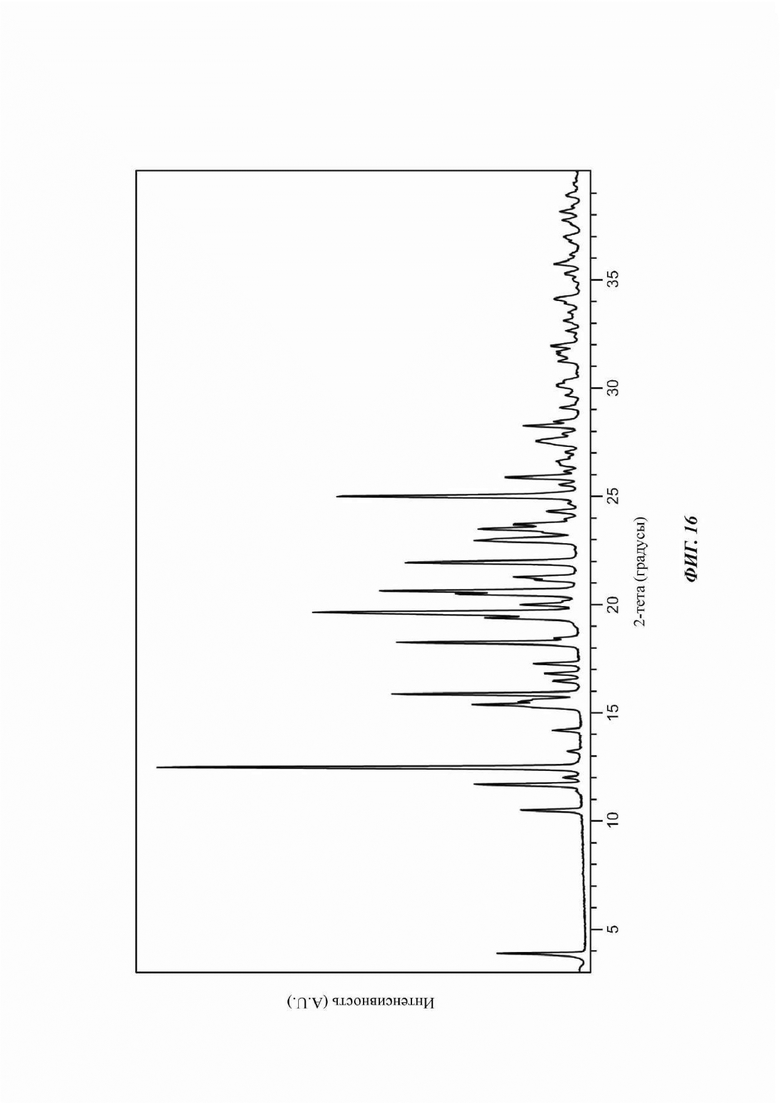
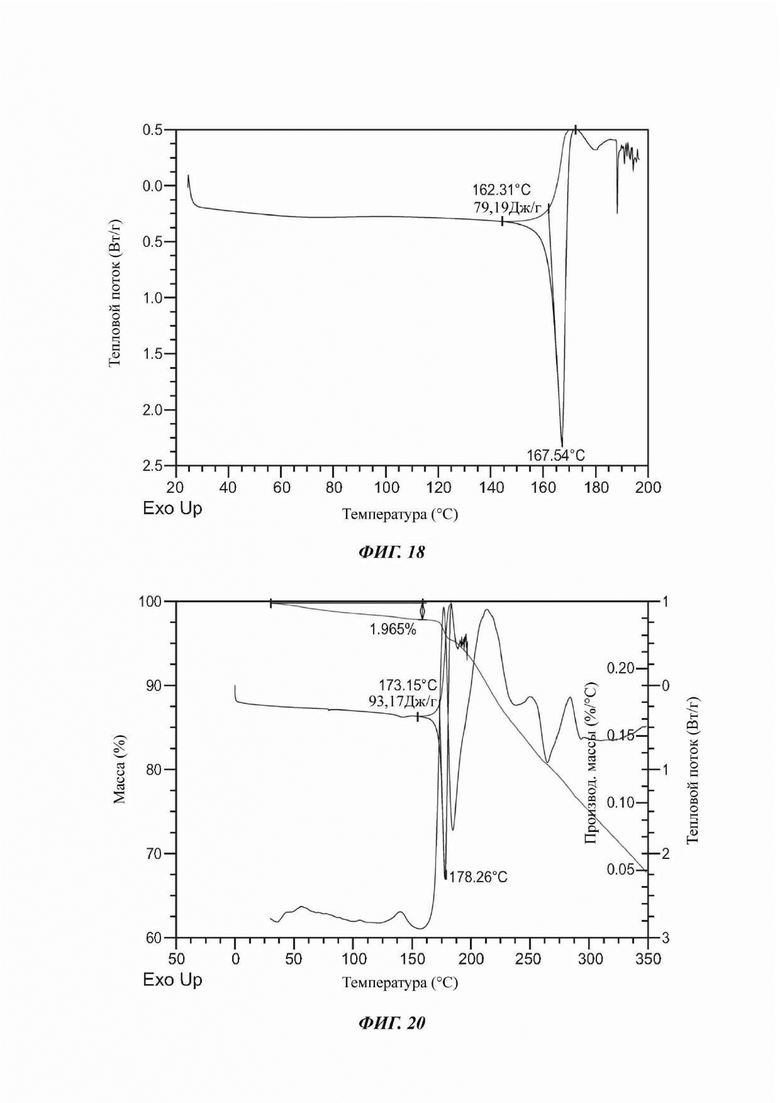
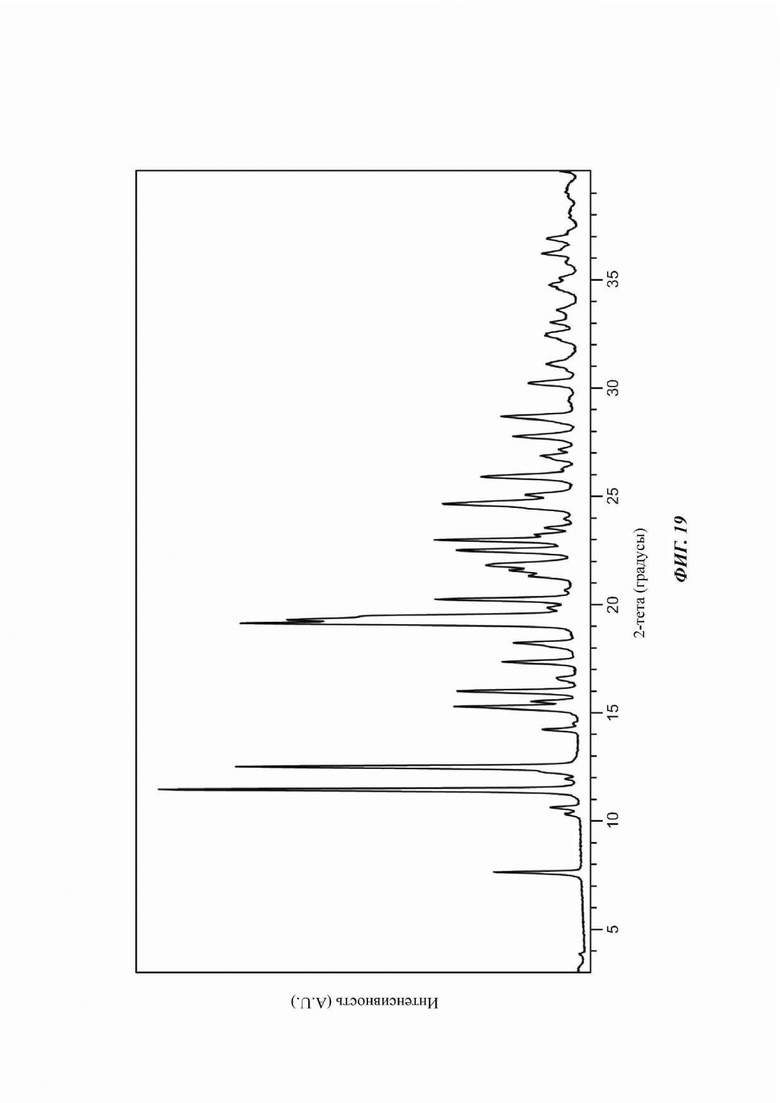
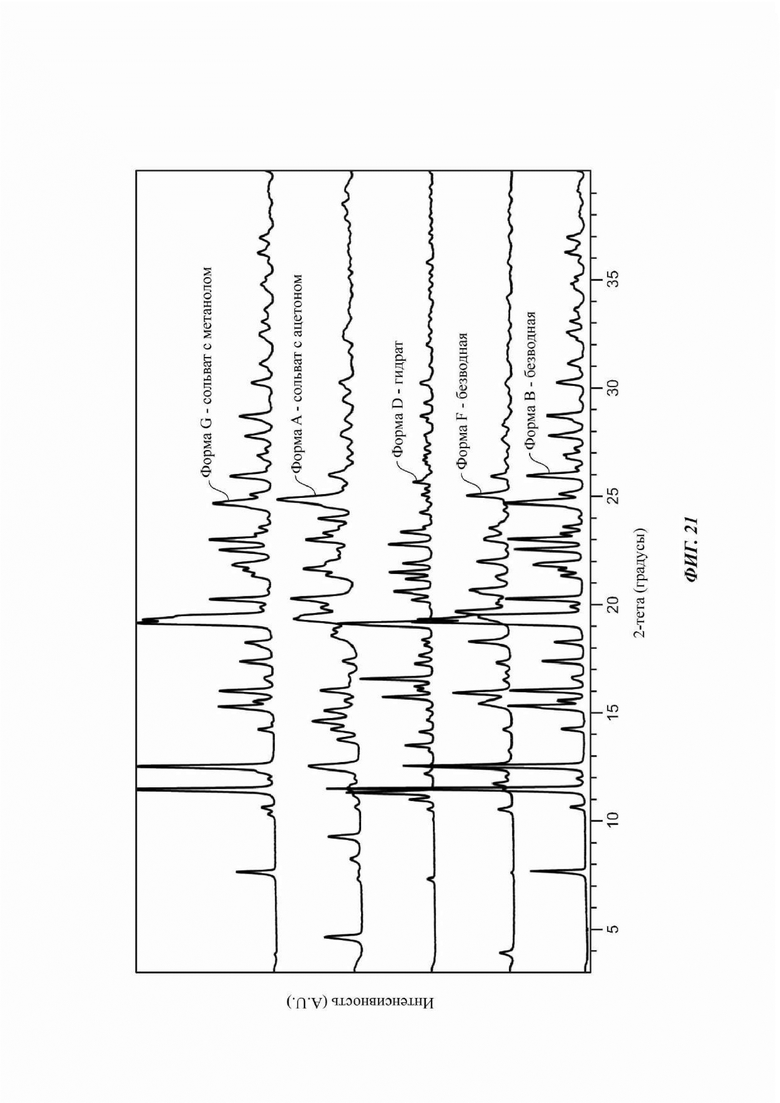
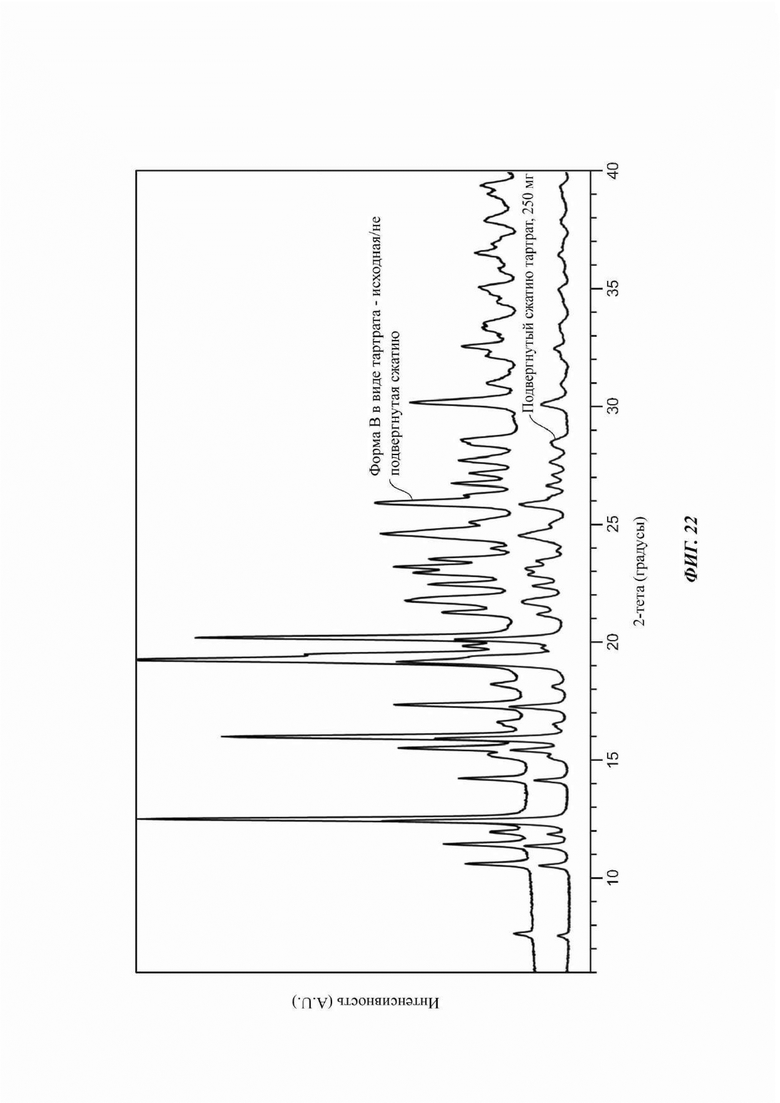
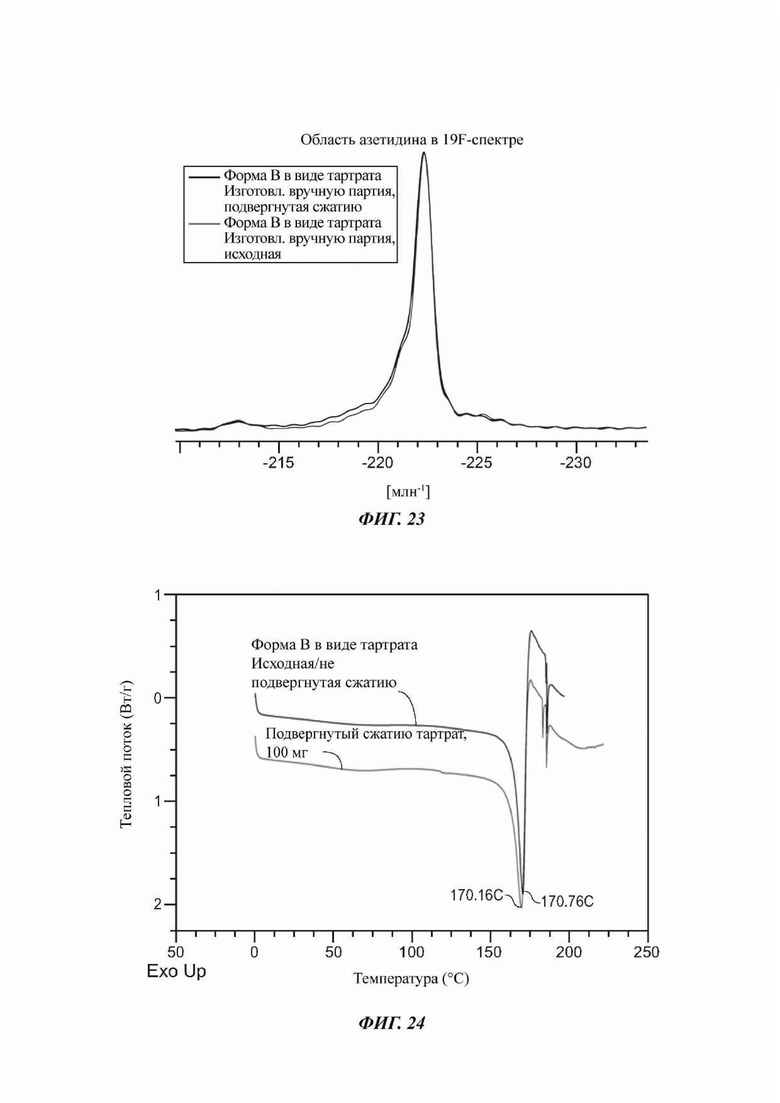
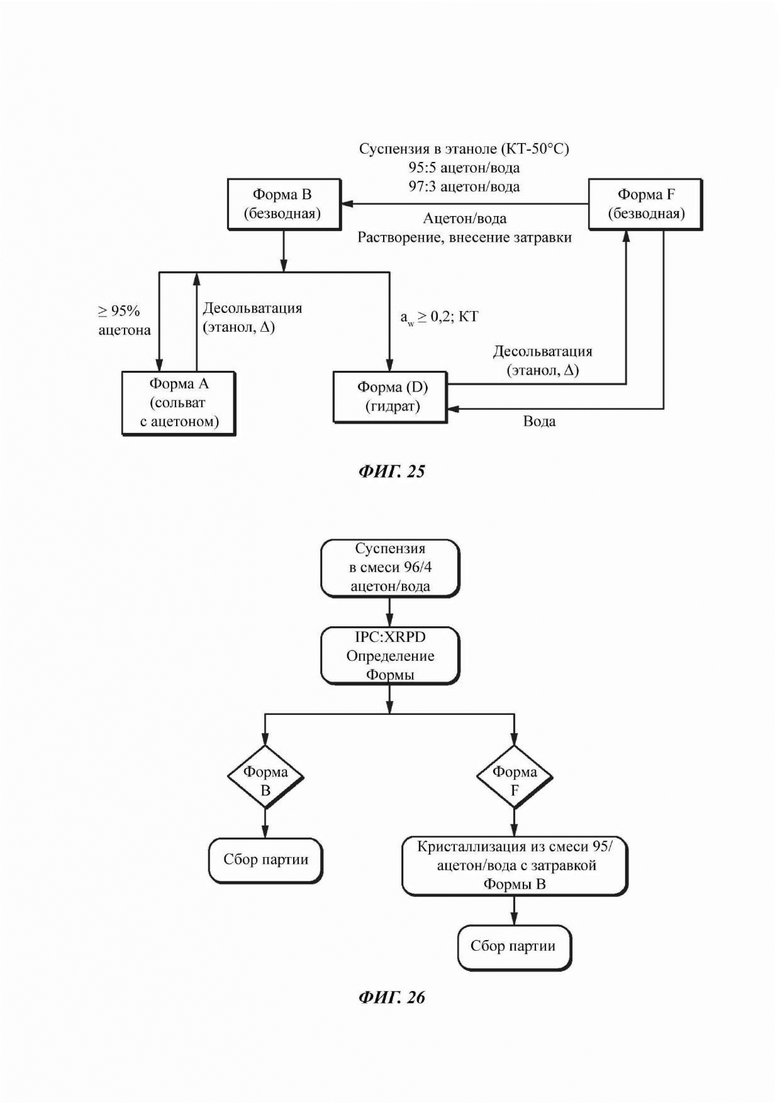
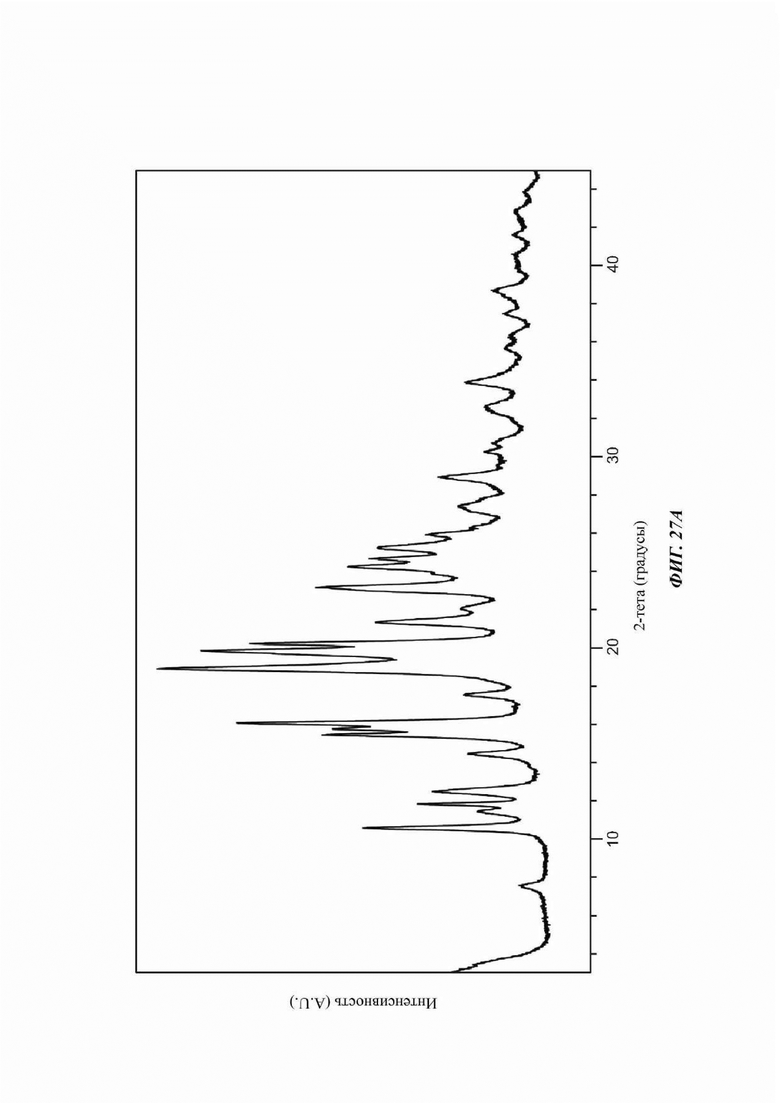
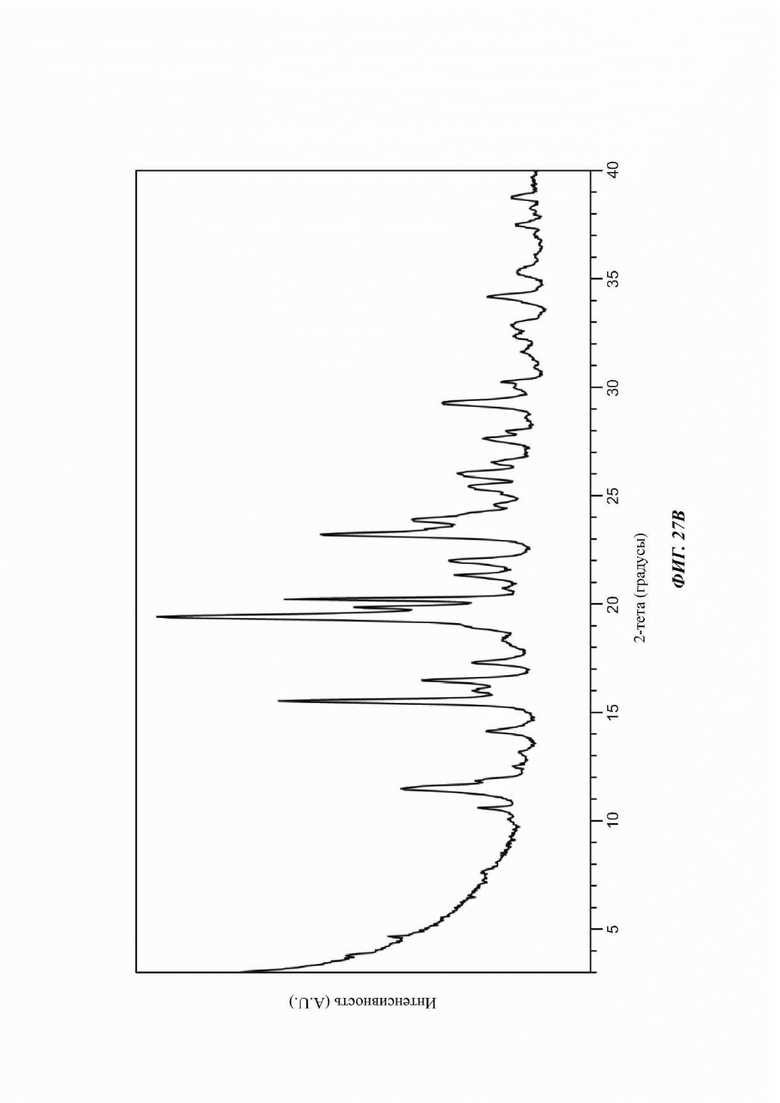
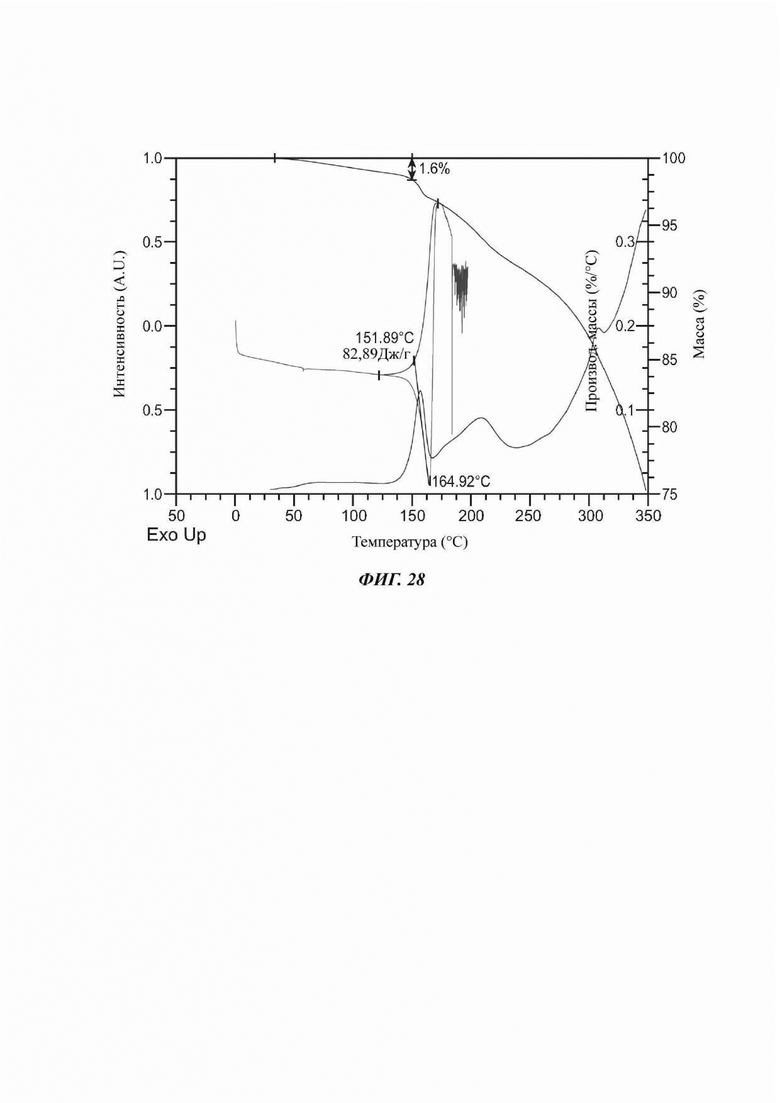

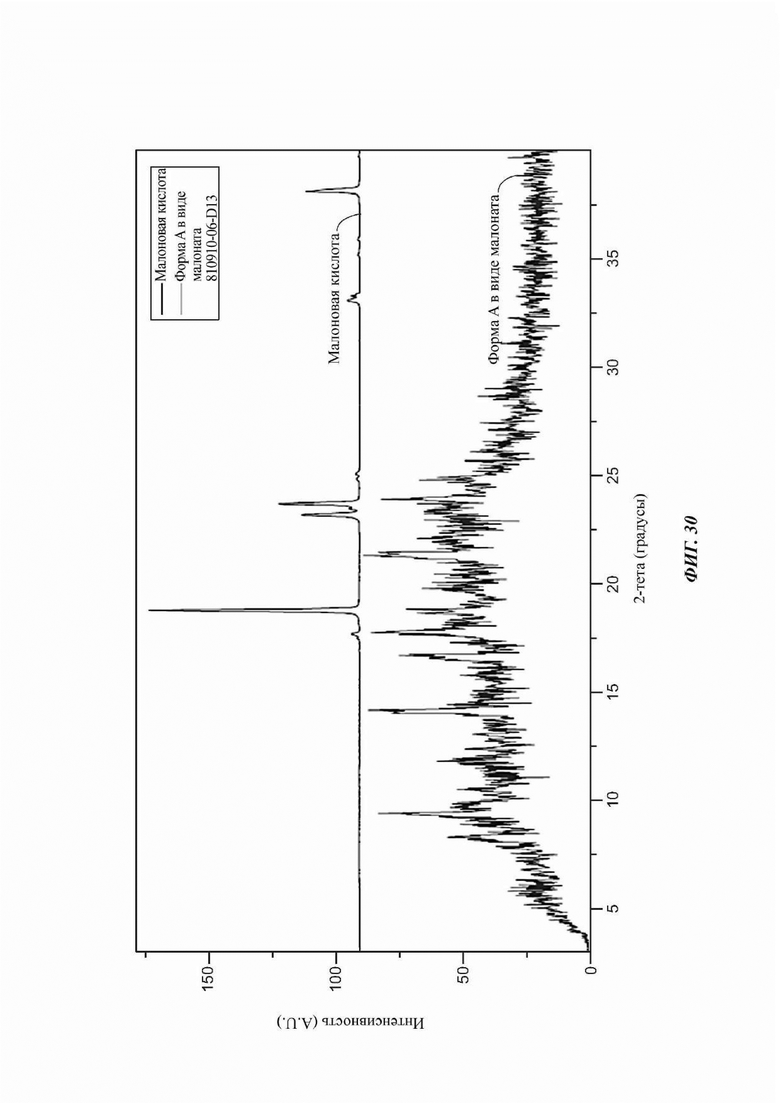
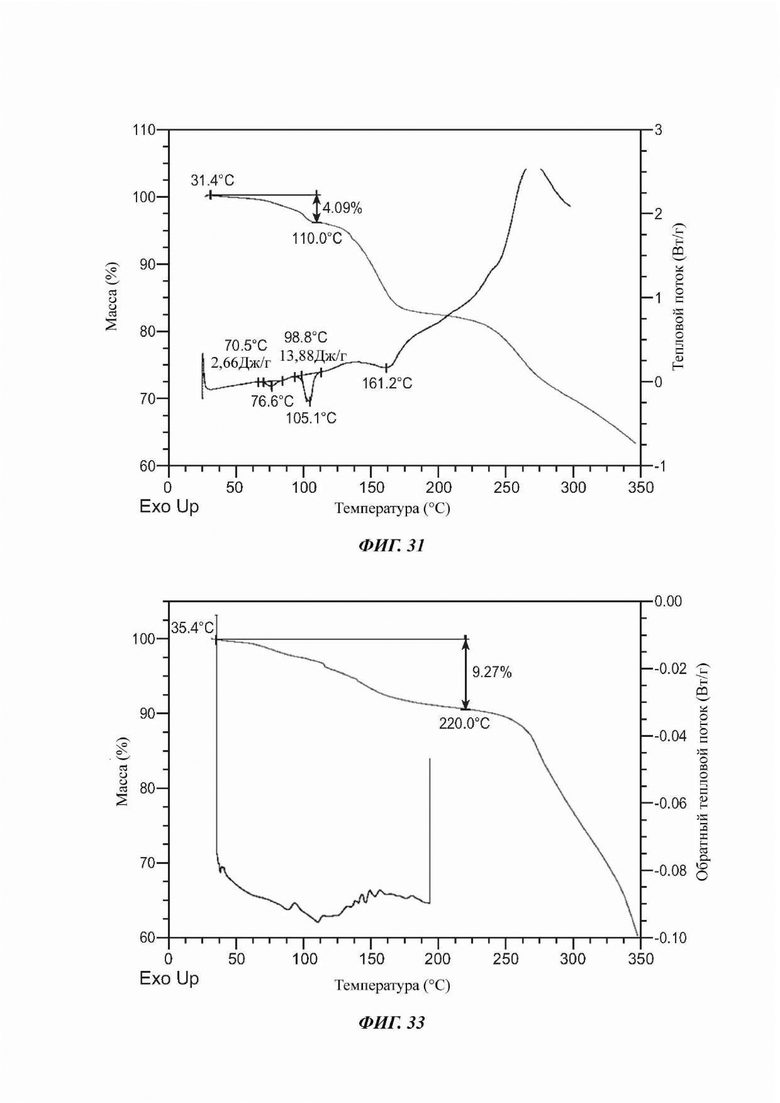
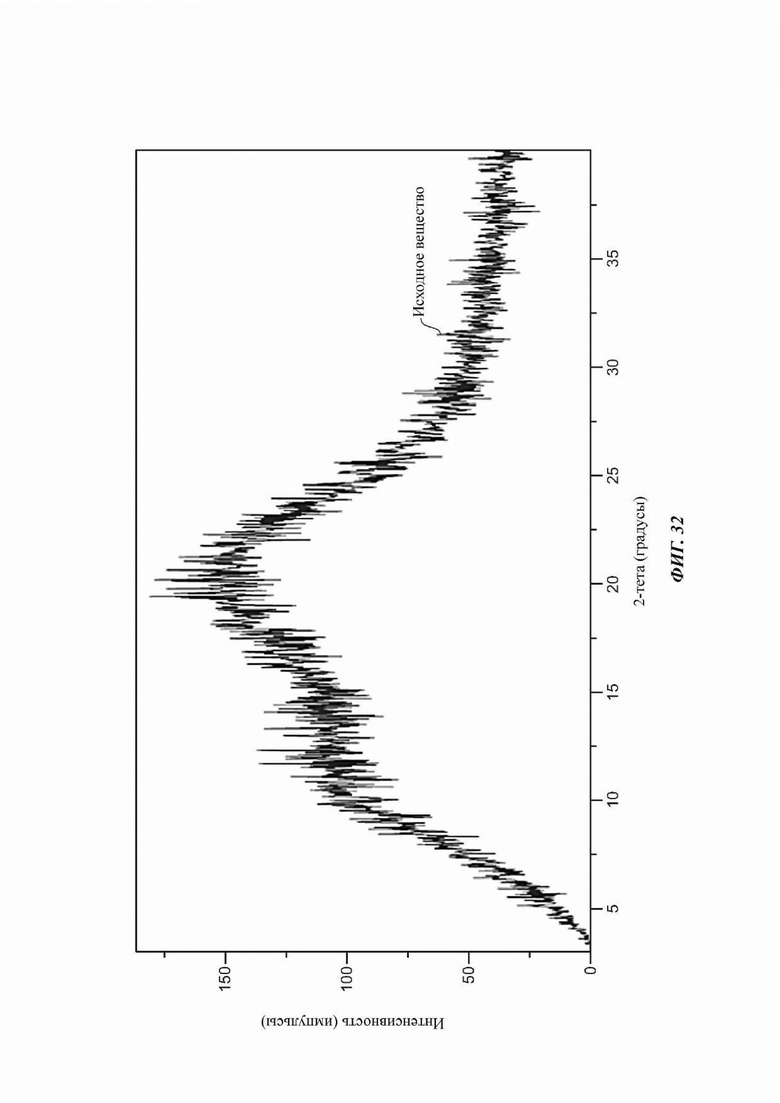
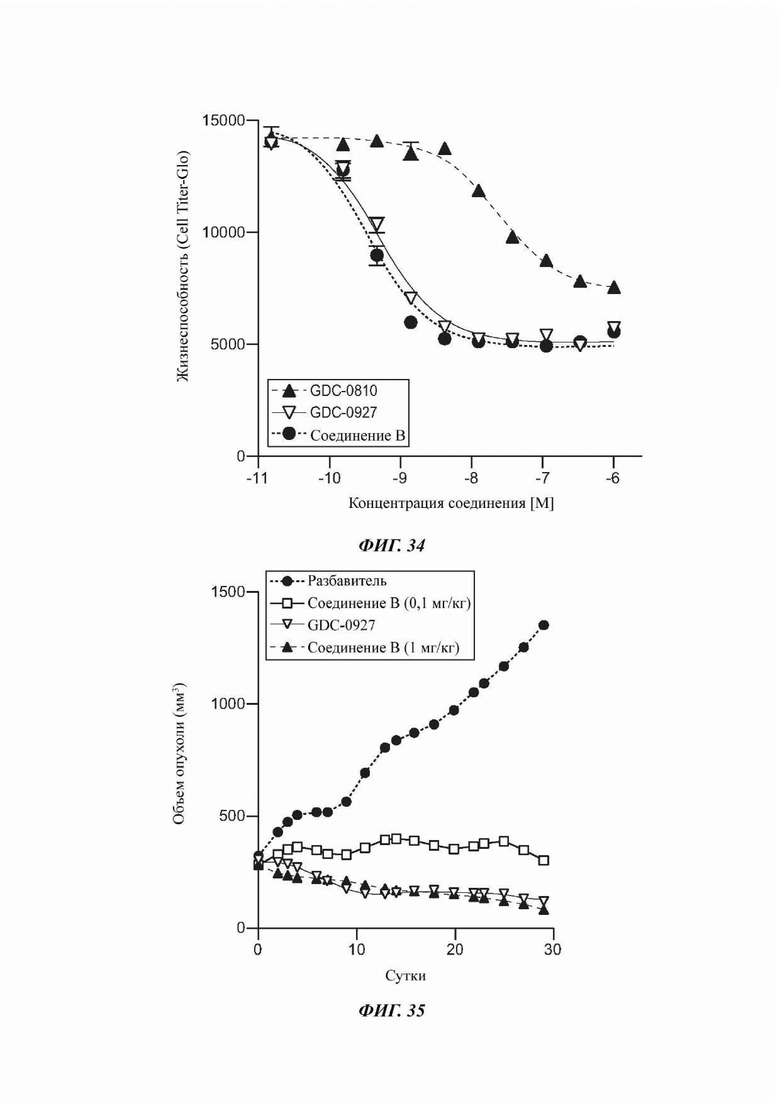
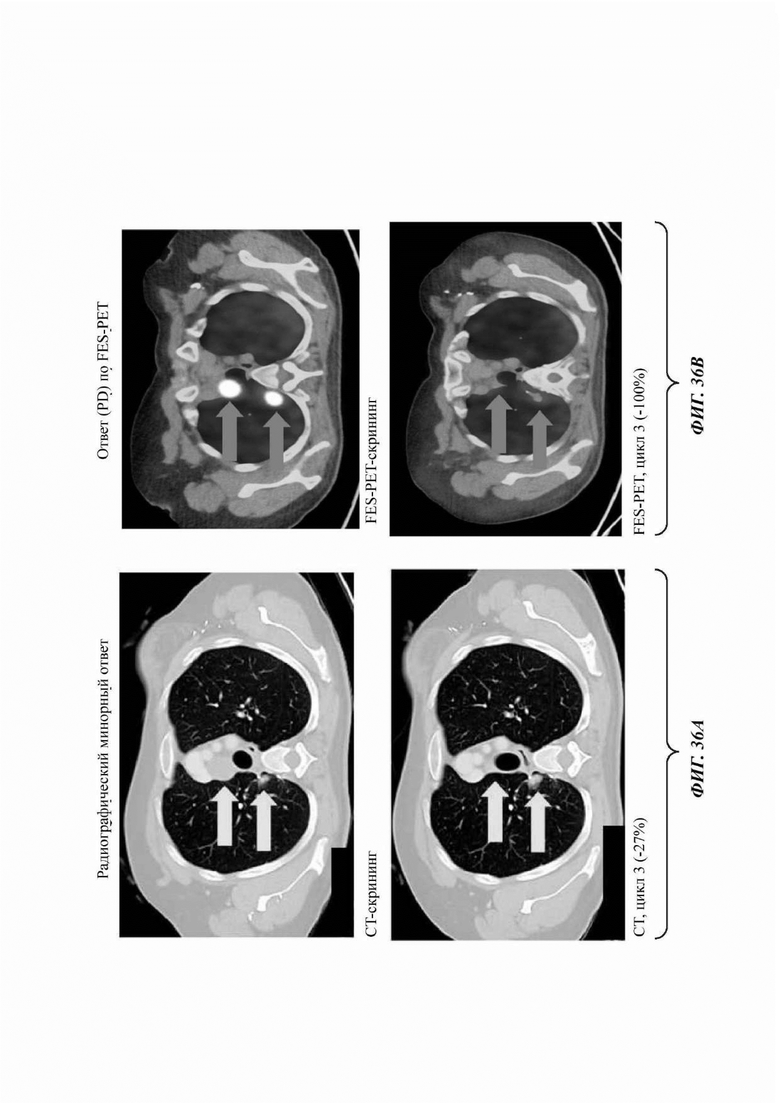
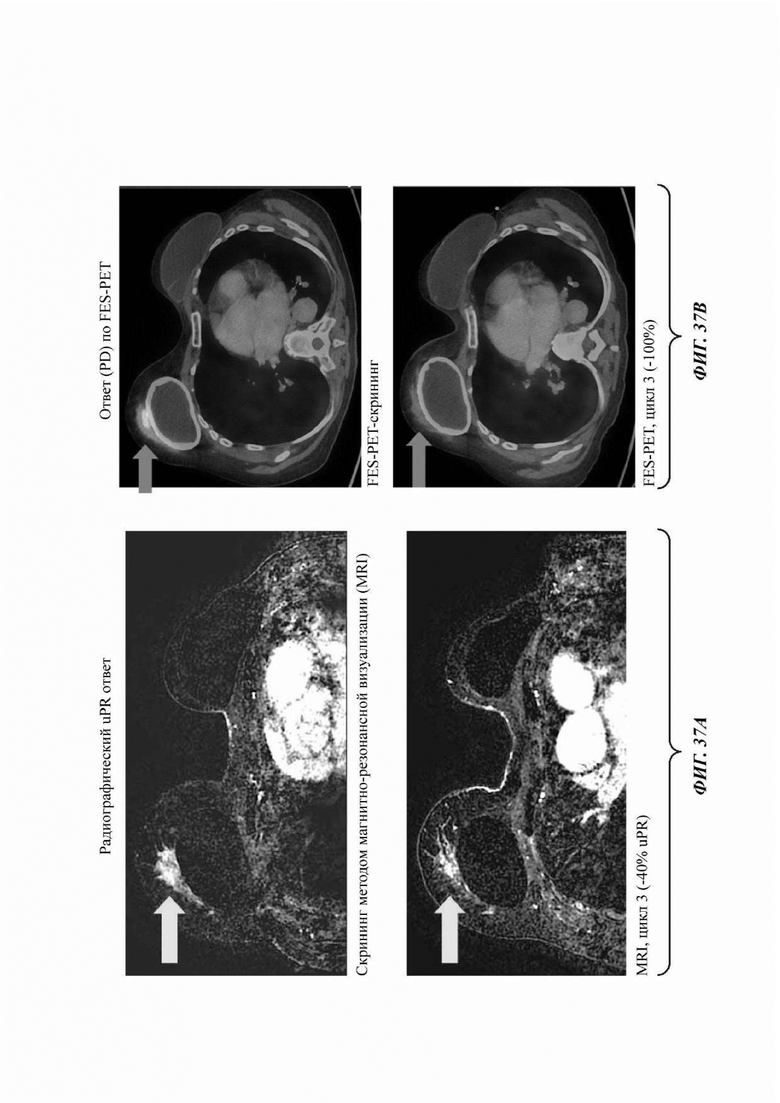
Изобретение относится к способу получения соединения В,
 и включающему стадии:
и включающему стадии:
 Технический результат: разработаны способы получения тартрата 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола, который может быть использован в лечении рака, 2 н. и 3 з.п. ф-лы, 38 табл., 34 пр., 39 ил.
Технический результат: разработаны способы получения тартрата 3-((1R,3R)-1-(2,6-дифтор-4-((1-(3-фторпропил)азетидин-3-ил)амино)фенил)-3-метил-1,3,4,9-тетрагидро-2Н-пиридо[3,4-b]индол-2-ил)-2,2-дифторпропан-1-ола, который может быть использован в лечении рака, 2 н. и 3 з.п. ф-лы, 38 табл., 34 пр., 39 ил.
1. Способ получения соединения В,
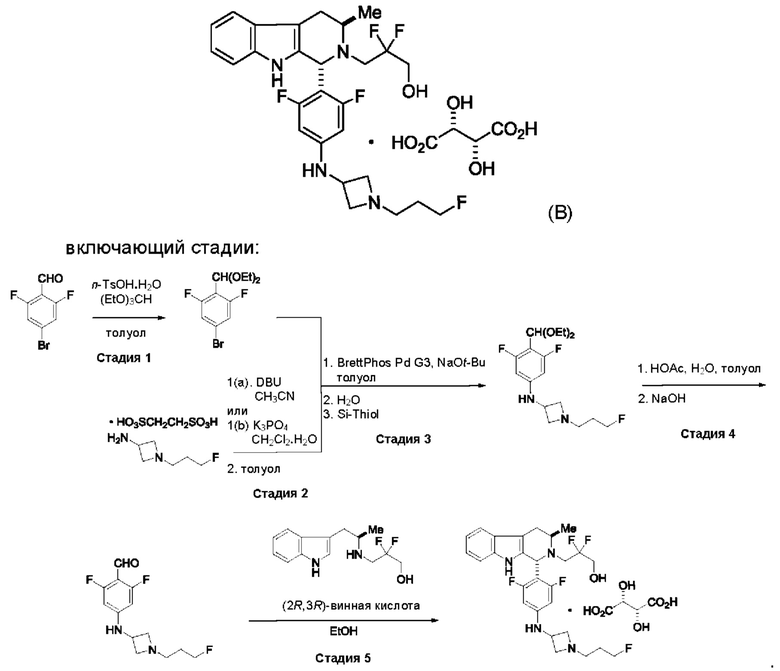
2. Способ получения соединения В,
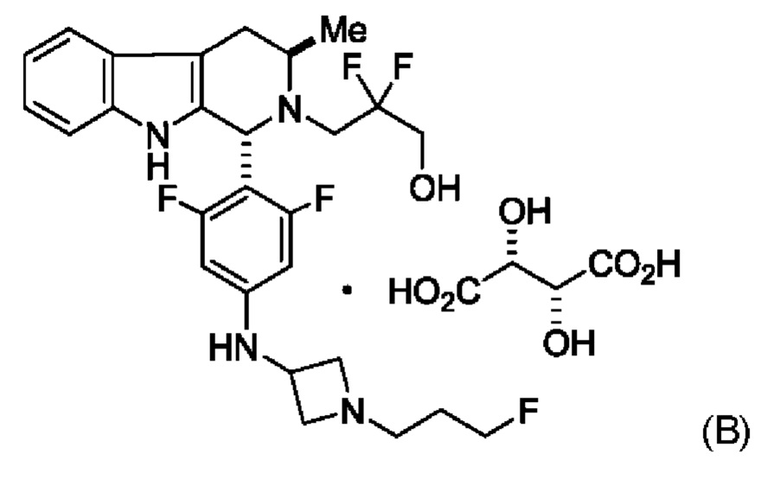
включающий стадии:
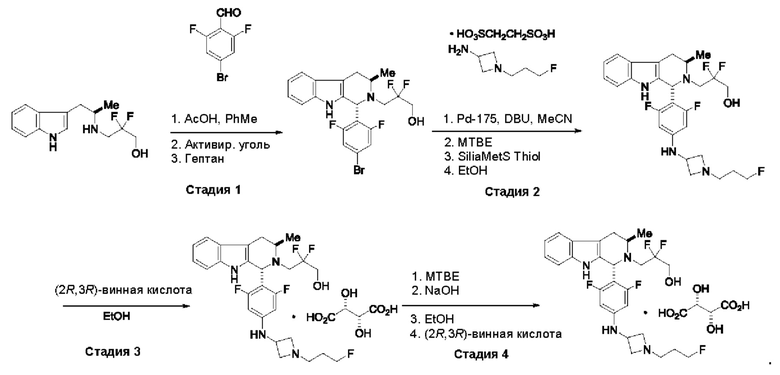
3. Способ по п. 1 или 2, дополнительно включающий перекристаллизацию соединения В в метаноле и этаноле:
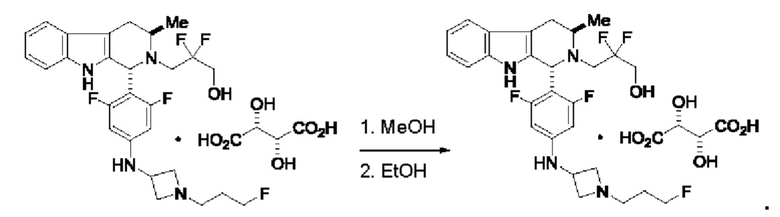
4. Способ по п. 1 или 2, дополнительно включающий перекристаллизацию соединения В в МТВЕ (метил-трет-бутиловый эфир), воде, NaOH и в этаноле:
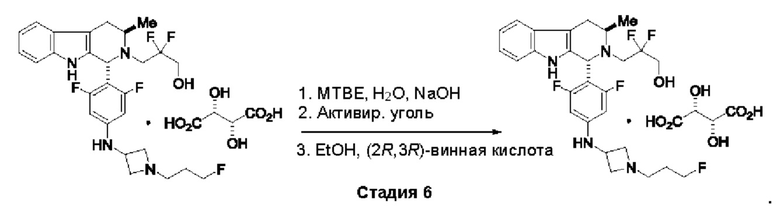
5. Способ по любому из пп. 1-4, где индолил-содержащие промежуточные соединения синтезируют в соответствии со стадиями:
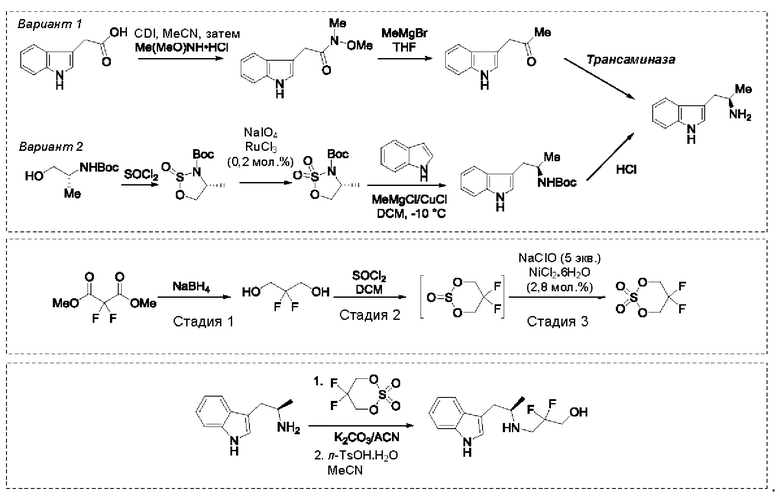
| WO 2016097072 A1, 23.06.2016 | |||
| Abu T.M.Serajuddin: "Salt formation to improve drug solubility", Advanced Drug Reviews, 2007, vol.59, p.603-616 (DOI:10.1016/J.ADDR.2007.05.010), табл.2, реферат | |||
| М.R.CAIRA, Crystalline polymorphism of organic compounds, TOPICS IN CURRENT CHEMISTRY, Springer Verlag Berlin Heidelberg, 1998, V.198, p.163-208 с.165, |
