Изобретение относится к области генной инженерии и биотехнологии, в частности к вариантам способа получения препарата большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli с высоким выходом фермента в растворимой форме.
Полимераза - ключевой фермент, отвечающий за репликацию ДНК в живых организмах. Известны ДНК-зависимые ДНК-полимеразы из различных источников: бактерий, дрожжей, человека. В зависимости от источника полимеразы отличаются по своим свойствам: наличию или отсутствию корректирующей активности, по уровню термостабильности, по оптимальной температуре синтеза цепи, процессивности и другим.
Наиболее известные и широко применяемые полимеразы были выделены из термофильных и гипер-термофильных бактерий и архей. Оптимум работы таких ферментов лежит в диапазоне 70-75°С, при этом они характеризуются достаточно длительным временем полужизни при температурах 90-100°С. Наиболее известная Taq-полимераза из термофильных бактерий Thermus aquaticus сохраняет 50% своей активности даже через 1,5 часа инкубации при температуре 95°С [Eom, S. Н. Structure of Taq polymerase with DNA at the polymerase active site / S. H. Eom, J. Wang, and T. A. Steitz // Nature.- 2003. - vol. 382, no. 6588, pp. 278-81, https://doi.org/10.1038/2278a0, Karantzeni, I. Comparative thermal denaturation of Thermus aquaticus and Escherichia coli type 1 DNA polymerases / Karantzeni, I. et al. // Biochemical Journal, - 2003. - vol. 374, no. 3, pp. 785-92, https://doi.org/10.1042/bj20030323]. Другим интересным ферментом является Pfu-полимераза из гипертермофильных архей Pyrococcus furiosus, она не только обладает длительным периодом полужизни при температуре 95°С, но и обладает корректирующей активностью [Cline, J. PCR Fidelity of Pfu DNA Polymerase and Other Thermostable DNA Polymerases. / Cline, J., Braman, J. C. and Hogrefe, H. H. // Nucleic Acids Research, vol. 24, no. 18, Sept. 1996, pp. 3546-51, https://doi.org/10.1093/nar/24.18.3546]. Корректирующую активность полимераз обеспечивает наличие 3'-5' экзонуклеазной активности у фермента. Кроме Pfu-полимеразы корректирующую активность проявляют такие полимеразы, как, например, Vent-полимераза, Deep Vent-полимераза и UlTima-полимераза [Cline, J. PCR Fidelity of Pfu DNA Polymerase and Other Thermostable DNA Polymerases. / Cline, J., Braman, J. C. and Hogrefe, H. H. // Nucleic Acids Research, vol. 24, no. 18, Sept. 1996, pp. 3546-51, https://doi.org/10.1093/nar/24.18.3546].
Кроме 3'-5' экзонуклеазной активности полимеразы могут обладать 5'-3' экзонуклеазной активностью, которая необходима для ник-трансляции или репарации ДНК. Одним из таких ферментов является ДНК-полимераза I из бактерий Geobacillus stearothermophilus (ранее классифицировалась как Bacillus stearothermophilus). Этот фермент в отличие от Taq-полимеразы относится к умеренным термофилам и обладает температурным оптимумом 60-65°С и характеризуется большей процессивностью по сравнению с Taq-полимеразой. Bst-полимераза является большим фрагментом ДНК-полимеразы I из бактерий G.stearothermophilus, его длина составляет 592 а.к., и этот фермент не имеет 5'-3' экзонуклеазной активности и характеризуется более высокой полимеразной активностью. При этом Bst-полимераза обладает вытесняющей активностью. Благодаря такому свойству Bst-полимераза (большой фрагмент ДНК-полимеразы I из бактерий G.stearothermophilus) нашла свое применение в том числе в методе петлевой изотермической амплификации LAMP.
Кроме классической ПЦР известны различные модификации метода на основе изотермической амплификации, такие как амплификация по типу «катящегося кольца» (RCA), геликаза-зависимая амплификация (HDA), петлевая амплификация (LAMP), амплификация с замещением цепей (SDA), метод амплификации молекул рибосомальной РНК (NASBA) и другие.
В методе петлевой изотермической амплификации LAMP используется система из 4-6 праймеров, за счет чего повышается специфичность реакции [Notomi, Tsugunori, et al. Loop-Mediated Isothermal Amplification (LAMP): Principle, Features, and Future Prospects. / Notomi, Tsugunori, et al. // Journal of Microbiology. - 2000. - vol. 53, no. 1, pp. 1-5. https://doi.org/10.1007/s12275-015-4656-9.]. Использование Bst-полимеразы, обладающей оптимумом каталитической активности при 65°С и вытесняющей активностью, позволяет проводить реакцию амплификации в изотермическом режиме, и обеспечивает сокращение времени реакции с 1,5 часов до 20-30 мин. Также в этом случае не требуется шаг предварительной денатурации молекулы ДНК. Сам метод основан на дизайне специфической системы праймеров таким образом, чтобы амплификация приводила к образованию множества конкатемеров инвертированных повторов целевого фрагмента. Образующиеся ампликоны формируют разнообразные пространственные структуры, содержащие множество петель или «шпилек». Длина получаемых конкатемеров может достигать 20 т.п.н. и больше. При этом накопление целевого продукта происходит лавинообразно, из-за чего количественная оценка в такой системе представляет большие сложности. К недостаткам такой системы можно отнести достаточно сложный дизайн праймеров, очень ограниченную возможность детекции нескольких мишеней в одной пробирке и высокую опасность контаминации. При этом к достоинствам метода стоит отнести высокую эффективность, специфичность, простоту, скорость постановки реакции и возможность использования различных методов детекции, в том числе, не требующих специального оборудования (колориметрический или турбодиметрический) [Becherer, L. Loop-Mediated Isothermal Amplification (LAMP) Review and Classification of Methods for Sequence-Specific Detection. / Becherer, L. et al. // Analytical Methods. - 2020. - vol. 12, no. 6, pp. 717-46. https://doi.org/10.1039/C9AY02246E.]. Современные диагностические системы на основе LAMP с флуоресцентной детекцией обладают специфичностью и чувствительностью сопоставимыми с классическим ПЦР, при этом являются гораздо более быстрым и простым методом.
В 1972 году J. Stenesh и B.A. Roe, а в 1981 O.K. Kaboev и соавторы описали способ выделения и свойства фермента ДНК-полимеразы I из бактерий Bacillus stearothermophilus (позже классифицированы как Geobacillus stearothermophilus) [Stenesh, J., and В. A. Roe. DNA Polymerase from Mesophilic and Thermophilic Bacteria. / Stenesh, J. and Roe, B. A. // Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis. - 1972. - Vol. 272, no. 2, 1972, pp. 156-66, https://doi.org/10.1016/0005-2787(72)90240-7. Kaboev, О.K., et al. Purification and Properties of Deoxyribonucleic Acid Polymerase from Bacillus Stearothermophilus. / Kaboev, О.K., et al. // Journal of Bacteriology, vol. 145, no. 1, 1981, pp. 21-26, https://doi.org/10.1128/jb.145.1.21-26.1981]. Выделенный в работах фермент обладал и 3'-5', и 5'-3' экзонуклеазной активностью. Выделение фермента из организма-хозяина не является оптимальным подходом: каждый организм обладает индивидуальными особенностями и требует подбора специфических сред и условий культивирования.
Другие авторы в 1987 году предложили способ получения Bst-полимеразы (большого фрагмента ДНК-полимеразы I из бактерий G.stearothermophilus) за счет отщепления N-концевого домена, несущего 5'-3' экзонуклеазную активность, с помощью расщепления полипептидной цепи под действием субтилизина [Ye, S. Y., and G. F. Hong. Heat-Stable DNA Polymerase I Large Fragment Resolves Hairpin Structure in DNA Sequencing. / Ye, S.Y. and Hong, G.F. // Scientia Sinica. Series B, Chemical, Biological, Agricultural, Medical & Earth Sciences. - 1987. - Vol. 30, no. 5, May 1987, pp. 503-06.]. Такой способ получения фермента является довольно трудоемким за счет дополнительных стадий обработки субтилизином и очистки.
Из уровня техники известен способ получения Bst-полимеразы в виде рекомбинантного фермента в клетках E.coli. Такой подход требует клонирования гена, кодирующего соответствующую последовательность Bst-полимеразы, с последующей экспрессией в клетках Е.coli [Shamsuddin S. PCR-Based Gene Synthesis, Cloning, Expression, Purification and Characterization of Bst DNA Polymerase in E. Coli Cells. / Shamsuddin S, Suppan M. // Current Synthetic and Systems Biology. 2015. - Vol. 03, no. 03, https://doi.org/10.4172/2332-0737.1000126.]. Преимуществом такого способа получения фермента является использование хорошо изученных и известных систем экспрессии и методов клонирования. Авторы использовали вектор рЕТ28а, с помощью которого получали рекомбинантный белок, содержащий С-концевой полигистидиновый (6xHis) тэг. Однако в данной работе исследователи получали полноразмерную версию Bst-полимеразы, которая не подходит для применения в методе петлевой изотермической амплификации LAMP.
В другой работе авторы экспрессировали в клетках E.coli большой фрагмент Bst-полимеразы [Ma, Yi, et al. Enhancement of Polymerase Activity of the Large Fragment in DNA Polymerase I from Geobacillus Stearothermophilus by Site-Directed Mutagenesis at the Active Site. / Ma, Yi, et al. // BioMed Research International. 2016. - Vol. 2016, 2016, pp. 1-8, https://doi.org/10.1155/2016/2906484.]. Последовательность гена была клонирована из геномной ДНК Geobacillus stearothermophilus с использованием специфических праймеров, содержащих фланкирующие сайты рестрикции для последующего клонирования в экспрессионный вектор. Авторы использовали вектор рЕТ21а, с помощью которого получали рекомбинантный белок, содержащий С-концевой полигистидиновый (6xHis) тэг. Экспрессию проводили в клетках E.coli BL21 (DE3). В результате авторами был получен активный фермент, обладающий полимеразной активностью. Однако в работе не была показана возможность использования данной версии в методе LAMP, а также не была оценена эффективность полученного фермента в реакции LAMP.
Из уровня техники известен способ получения большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli, применимого в реакции изотермической амплификации LAMP [Li, P. et al. Cloning and Purification of Large Fragment of DNA Polymerase I from Geobacillus Stearothermophilus and It's Application in Isothermal DNA Amplification. / Li, P. et al. // Biotechnology. Theory and Practice. - 2017. - https://doi.org/10.11134/btp.1.2017.6.]. В работе авторы клонировали ген фермента, используя геномную ДНК Geobacillus stearothermophilus в качестве источника гена. Ген клонировали в экспрессионный вектор рЕТ28с(+) таким образом, что при экспрессии белок получался с полигистидиновым тэгом на N-конце. К недостаткам работы следует отнести то, что для получения достаточного количества геномной ДНК исследователям вынуждены были провести культивирование Geobacillus stearothermophilus, что требует применения специфических сред и условий культивирования. Кроме того, авторы показали работоспособность фермента в условиях LAMP, при этом детекцию продукта проводили с помощью интеркалирующего флуоресцентного красителя, однако образование продукта детектировали не по кривым накопления сигнала в режиме реального времени с помощью амплификатора с детекцией в режиме реального времени, а по конечной точке визуально (в условиях облучения пробы УФ-светом). Такой подход не позволяет точно и достоверно оценить уровень активности фермента, например, в сравнении со стандартизованным препаратом и препаратом с известной активностью. Также к недостаткам работы можно отнести не слишком высокий выход фермента, всего 1,5 мг с литра культуральной среды.
В другой работе [Agustriana, Eva, et al. Optimized Expression of Large Fragment DNA Polymerase I from Geobacillus Stearothermophilus in Escherichia Coli Expression System. / Agustriana, Eva, et al. // Preparative Biochemistry & Biotechnology. - 2022. - pp. 1-10, https://doi.org/10.1080/10826068.2022.2095573.] исследователи получали большой фрагмент Bst-полимеразы в виде рекомбинантного белка из клеток E.coli, несущего мутацию Gly510Leu, повышающую эффективность полимеризации. В данной работе ген получали методом синтеза, при этом конструкция гена была разработана в рамках патента CN 106399299 «Способ повышения активности крупнофрагментной ДНК-полимеразы Geobacillus Stearothermophilus (BST) путем точечной мутации и применения» (дата подачи 29.09.2016, дата публикации 15.02.20017). В работе проводили оптимизацию протокола экспрессии белка: варьировали концентрации индуктора ИПТГ, время культивирования после индукции, значение оптической плотности культуры клеток перед индукцией, скорость перемешивания и аэрацию при культивировании культуры клеток в ферментере. Авторам удалось получить выход фермента на уровне 11,7 мг/л. Следует отметить, что в данной работе не показана возможность использования полученного фермента в реакции изотермической амплификации и не проведена оценка активности фермента в LAMP.
Также из уровня техники известно несколько способов получения как полноразмерного фермента, так и большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli [патент US 5814506, дата подачи 08.02.1995, дата публикации 29.09.1998; патент CN 106399299 дата подачи 29.09.2016, дата публикации 15.02.20017]. Однако в данных решения не показана возможность использования полученного фермента в реакции изотермической амплификации и не проведена оценка активности фермента в LAMP.
Система экспрессии на основе клеток E.coli является наиболее популярной и удобной за счет своей простоты, невысокой стоимости, достаточно быстрого роста клеток (в сравнении с эукариотическими системами) и простоты масштабирования.
В связи с этим актуальной является задача разработки эффективного метода получения большого фрагмента (БФ) Bst-полимеразы в растворимой форме в виде рекомбинантного белка в клетках E.coli, с высоким выходом и оценка возможности применения полученного фермента в реакциях петлевой изотермической амплификации LAMP.
Технический результат заявляемого изобретения направлен на разработку вариантов эффективного способа получения большого фрагмента (БФ) Bst-полимеразы в растворимой форме, с высоким выходом. При этом ферментный препарат должен проявлять высокий уровень ферментативной активности и должен обладать вытесняющей активностью, что позволяет эффективно применять его в методе петлевой изотермической амплификации LAMP.
Технический результат достигается за счет разработки уникальных нуклеотидных последовательностей, кодирующих БФ-Bst-полимеразу, подбора вектора для экспрессии и варьирования условий культивирования.
Нуклеотидную последовательность (SEQ ID NO: 51), кодирующую аминокислотную последовательность БФ-Bst-полимеразы (SEQ ID NO: 1), получают методом сборки из длинных перекрывающихся праймеров, так называемым «методом лесенки» [Xiong, A. S. A Simple, Rapid, High-Fidelity and Cost-Effective PCR-Based Two-Step DNA Synthesis Method for Long Gene Sequences./ Xiong, A. S. et al. // Nucleic Acids Research. 2004. - Vol. 32, no. 12, pp. e98-e98, https://doi.org/10.1093/nar/gnh094.]. Первоначально проводят обратную трансляцию аминокислотной последовательности БФ-Bst-полимеразы (SEQ ID NO: 1) с использованием соответствующего программного обеспечения, например, программы VectorNTI, а затем оптимизируют нуклеотидный состав с учетом частоты встречаемости кодонов для бактериальной системы экспрессии на основе клеток E.coli. На концах нуклеотидной последовательности (SEQ ID NO: 51) введены сайты рестрикции NdeI - на 5'-конце, и KpnI и XhoI - на 3'-конце, для последующего переклонирования в экспрессионный вектор рЕТ16.
Разработанная нуклеотидная последовательность Bst-pol-NHis (SEQ ID NO: 51) содержит 4 кодирующих триплета с частотой встречаемости меньше 10. Замена от одного до четырех триплетов приводит к увеличению выхода целевого белка в растворимой форме. Замены в нуклеотидную последовательность Bst-pol-NHis SEQ ID NO: 51 вводили с помощью мутагенеза с использованием набора QuikChange II Site-Directed Mutagenesis Kit (Agilent, США) или любого аналогичного коммерчески-доступного набора в соответствии с инструкцией производителя. Для этого использовали праймеры для мутагенеза, несущие целевые замены, SEQ ID NO: 52-59 (таблица 3). В результате была получена нуклеотидная последовательность Bst-pol-NHis_m4 (SEQ ID NO: 60).
Панель олигонуклеотидов для синтеза нуклеотидной последовательности SEQ ID NO: 51 in-vitro разрабатывается с помощью программы [Owczarzy, R., et al. IDT SciTools: A Suite for Analysis and Design of Nucleic Acid Oligomers. / Owczarzy, R., et al. // Nucleic Acids Research. -2008. - Vol. 36, no. Web Server, pp. W163-69, https://doi.org/10.1093/nar/gkn198.], реализованной в виде веб-сервиса (https://eu.idtdna.com/calc/analyzer). Последовательности праймеров приведены в таблице 1. 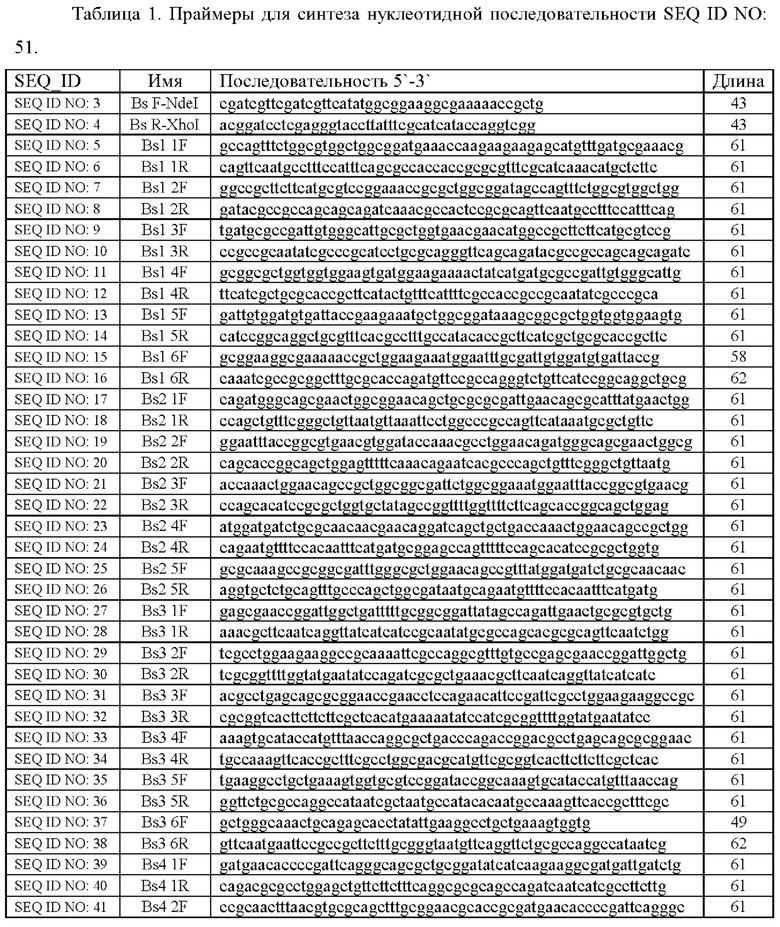

Итоговые нуклеотидные последовательности: Bst-pol-NHis (SEQ ID NO: 51) и Bst-pol-NHis_m4 (SEQ ID NO: 60), дают возможность получения целевого фермента с полигистидиновым тэгом на N-конце (SEQ ID NO: 2), что позволяет использовать высокоэффективный способ очистки белков методом аффинной хроматографии с использованием Ni-содержащих носителей, например, Ni-NTA агарозы.
Экспрессия проводится в клетках E.coli BL21 DE3 на питательной среде Лурии-Бертани (LB) в присутствии ампициллина, в качестве индуктора экспрессии используется изопропил-β-D-тиогалактозид (ИПТГ). Синтез белка в клетках проводится в течение 4 часов после добавления индуктора при температуре от 23°С до 37°С. Выделение целевого белка производится из фракции растворимых белков, очистка проводится методом хроматографии в две стадии.
Для получения стартовой культуры клетки-продуценты наращивают при 37°С в течение 12-16 часов, затем клетки переносят в питательную среду LB, содержащую 100 мкг/мл ампициллина, 1% глюкозы и 2,5% глицерина. Также клетки-продуценты после разведения культивируют при 37°С до достижения оптической плотности OD600≈1,2-1,5, затем культуру клеток охлаждают до выбранной температуры от 23°С до 37°С, после чего в питательную среду добавляют изопропил-β-D-тиогалактозид (ИПТГ) и инкубируют культуру еще 4 часа при выбранной температуре (23°С или 37°С). Две различные температуры: 23°С и 37°С используют для оптимизации условий культивирования. При культивировании при 23°С перед индукцией среда предварительно охлаждалась до 23°С.
Клетки ресуспендируют в буфере, содержащем 50 мМ Трис/HCl, 1 мг/мл лизоцим, рН 8.0 и инкубируют на льду 30 мин, затем добавляют равный объем лизирующего буфера 50 мМ Трис/HCl рН 8.0 и проводят дезинтеграцию клеток под действием ультразвука. Затем проводят центрифугирование при 6000 g в течение 30 минут.
Первый этап хроматографической очистки проводят методом металл-хелатной хроматографии. Элюцию проводят в серии буферных растворов, содержащих 50 мМ Трис/HCl, 100 мМ KCl, рН 8.0 при различных концентрациях имидазола: 60 мМ, 120 мМ, 300 мМ и 800 мМ. Второй этап хроматографии проводят методом ион-обменной хроматографии: для этого фракцию с наибольшим содержанием целевого белка по данным белкового электрофореза разбавляют в 10 раз буфером, содержащим 50 мМ Трис/HCl, рН 8.0, 5% глицерин. Элюцию проводят в серии буферных растворов, содержащих содержащим 50 мМ Трис/HCl, рН 8.0, 5% глицерин при различных концентрациях KCl: 100 мМ, 300 мМ, 500 мМ и 1 М.
В качестве сорбента на первом этапе хроматографической очистки предпочтительно использовать коммерчески-доступный сорбент Chelating Sepharose FF или его аналог. В качестве сорбента на втором этапе хроматографической очистки предпочтительно использовать коммерчески-доступный сорбент Capto Q или его аналог.
Препараты фермента БФ-Bst-полимеразы, полученные согласно данному изобретению возможно использовать в реакциях петлевой изотермической амплификации LAMP для выявления нуклеиновых кислот различных патогенов.
Заявляемое изобретение является результатом работы в рамках разработки ферментов, применяющихся в различных методах молекулярной диагностики, проделанной в ФБУН ЦНИИ Эпидемиологии Роспотребнадзора (Москва, Россия).
Краткое описание чертежей
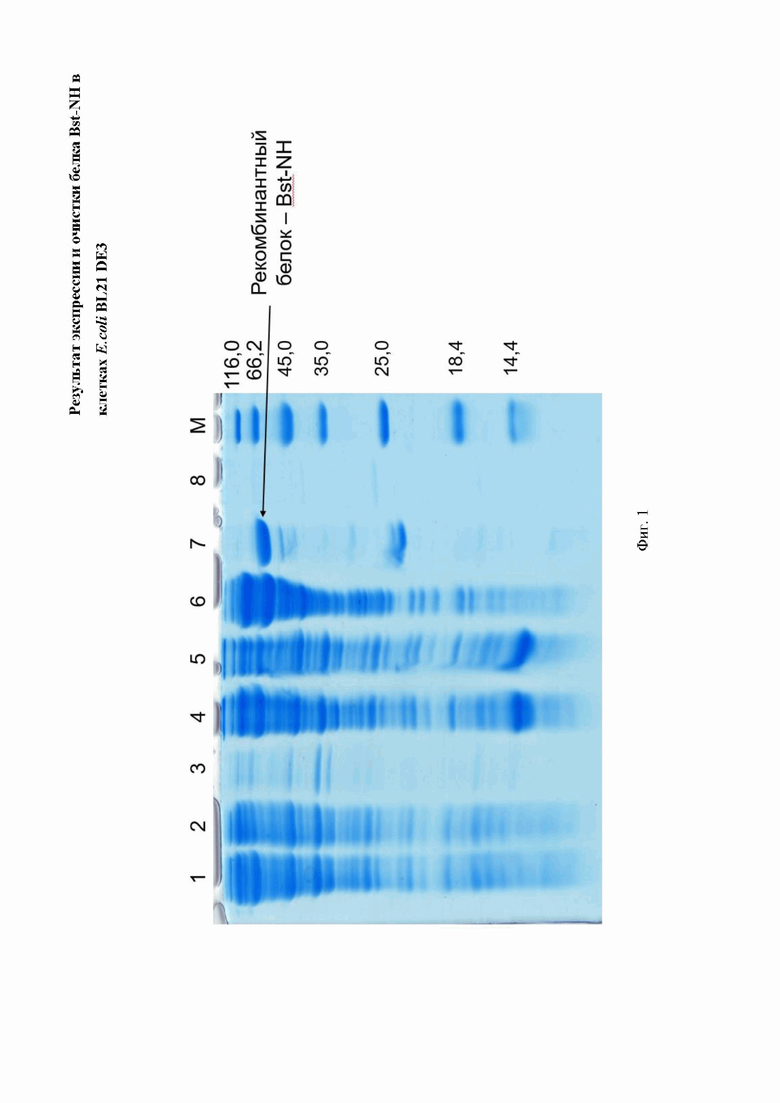
Фиг. 1. Электрофореграмма различных фракций, полученных при экспрессии фермента Bst-NH в клетках Е. coli BL21(DE3) и его очистке методом металл-хелатной хроматографии, где цифрами 1-8 обозначены:
1 - тотальная фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК рЕТ16_Bst_NHis, несущей ген, содержащий последовательность SEQ ID NO: NO: 2, индуцированной добавлением ИПТГ, культивированной при температуре 37°С в течение 4 часов;
2 - тотальная фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК pET16_Bst_NHis, несущей ген, содержащий последовательность SEQ ID NO: NO: 2, не индуцированной добавлением ИПТГ, культивированной при температуре 37°С в течение 4 часов;
3 - нерастворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК pET16_Bst_NHis, несущей ген, содержащий последовательность SEQ ID NO: NO: 2, индуцированной добавлением ИПТГ, культивированной при температуре 37°С в течение 4 часов;
4 - растворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК pET16_Bst_NHis, несущей ген, содержащий последовательность SEQ ID NO: NO: 2, индуцированной добавлением ИПТГ, культивированной при температуре 37°С в течение 4 часов;
5 - фракция, полученная при нанесении белка из растворимой цитоплазмы штамма-продуцента рЕТ16_Bst_NHis/BL21(DE3), на колонку для металл-хелатной хроматографии (проскок);
6 - препарат Bst-NH, полученный при очистке методом металл-хелатной хроматографии, элюция буферным раствором, содержащим 60 мМ имидазола;
7 - препарат Bst-NH, полученный при очистке методом металл-хелатной хроматографии, элюция буферным раствором, содержащим 300 мМ имидазола;
8 - препарат Bst-NH, полученный при очистке методом металл-хелатной хроматографии, элюция буферным раствором, содержащим 1 М имидазола;
М - маркер молекулярных масс.
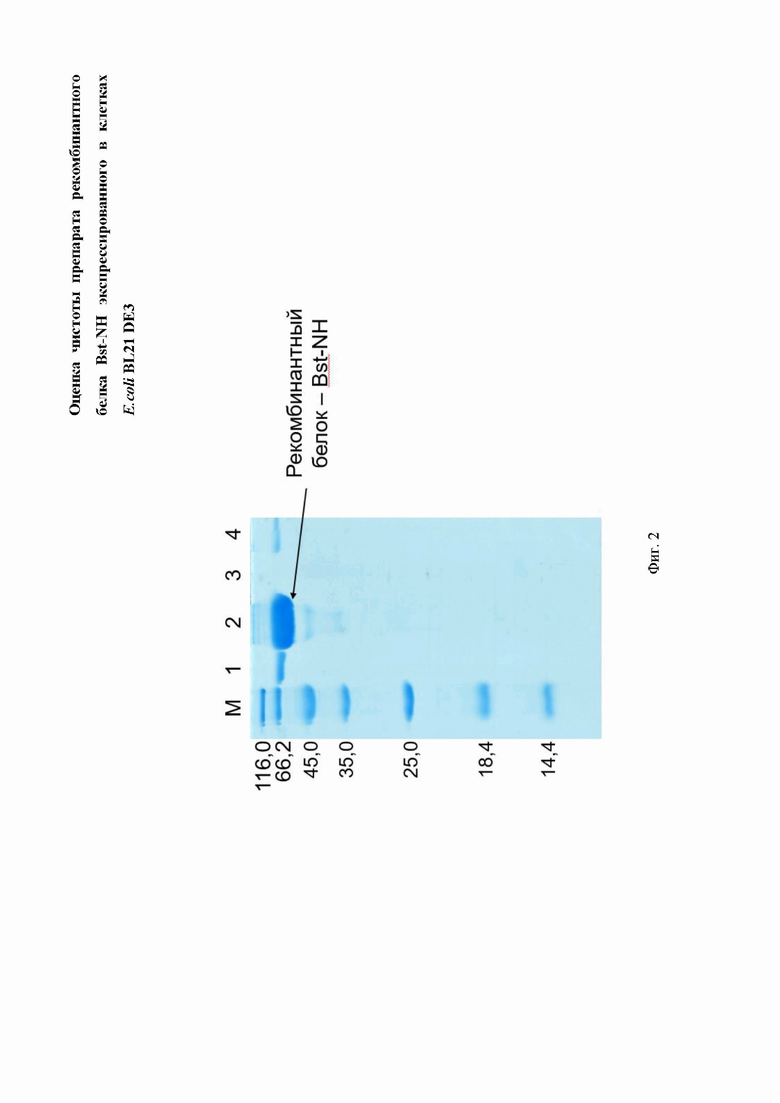
Фиг. 2. Электрофореграмма препаратов белка, содержащих рекомбинантный фермента Bst-NH, полученного в результате ион-обменной хроматографии; где цифрами 1-4 обозначены:
1 - препарат Bst 2.0 фирмы NEB стандарт сравнения;
2 - препарат Bst-NH, полученный в результате ион-обменной хроматографии, элюция буферным раствором, содержащим 0,3 М хлорида натрия;
3 - препарат Bst-NH, полученный в результате ион-обменной хроматографии, элюция буферным раствором, содержащим 1 М хлорида натрия;
4 - препарат Bst-NH, полученный в результате ион-обменной хроматографии, элюция буферным раствором, содержащим 0,5 М хлорида натрия;
М - маркер молекулярных масс.
Фиг. 3. Электрофореграмма различных фракций, полученных при экспрессии фермента Bst-NHm4 в клетках Е. coli BL21(DE3) и его очистке методом металл-хелатной хроматографии, где цифрами 1-14 обозначены:
1 - растворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК pET16_Bst_NHism4, несущей ген, содержащий последовательность SEQ ID NO: NO: 51, индуцированной добавлением ИПТГ, культивированной при температуре 23°С в течение 4 часов;
2 - нерастворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК pET16_Bst_NHism4, несущей ген, содержащий последовательность SEQ ID NO: NO: 51, индуцированной добавлением ИПТГ, культивированной при температуре 23°С в течение 4 часов;
3-14 - различные фракции препарата Bst-NHm4, полученные при очистке методом металл-хелатной хроматографии, элюция буферным раствором в градиенте концентраций имидазола;
М - маркер молекулярных масс.
Фиг. 4. Электрофореграмма препаратов белка, содержащих рекомбинантный фермент Bst-NHm4, полученного в результате экспрессии в клетках Е.coli BL21(DE3) и двух стадий очистки; где цифрами 1-5 обозначены:
1 - тотальная фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК рЕТ16_Bst_NHis_m4, несущей ген, содержащий последовательность SEQ ID NO: NO: 51, индуцированной добавлением ИПТГ, культивированной при температуре 23°С в течение 4 часов;
2 - растворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК рЕТ16_Bst_NHis_m4, несущей ген, содержащий последовательность SEQ ID NO: NO: 51, индуцированной добавлением ИПТГ, культивированной при температуре 23°С в течение 4 часов;
3 - нерастворимая фракция Е.coli BL21(DE3), трансформированной плазмидной ДНК рЕТ16_Bst_NHis_m4, несущей ген, содержащий последовательность SEQ ID NO: NO: 51, индуцированной добавлением ИПТГ, культивированной при температуре 23°С в течение 4 часов;
4 - тотальная фракция Bst-NHm4 после металл-хелатной аффинной хроматографии;
5 - тотальная фракция Bst-NHm4 после гель-фильтрационной хроматографии;
М - маркер молекулярных масс.
Фиг. 5. Профиль флуоресценции в реальном времени для реакции петлевой изотермической амплификации (LAMP), на котором посредством графика визуализировано накопление целевых ампликонов в модельной системе с использованием рекомбинантного фермента Bst-NH и коммерческого фермента сравнения Bst 2.0 (NEB):
- по вертикали указаны значения нормализированного флуоресцентного сигнала, создаваемого красителем;
- по горизонтали указан номер цикла;
- на кривых 1-4 показан использованный фермент, а также количество копий нуклеиновой кислоты на реакцию, а именно:
1 - Bst 2.0 (NEB) 1000 копий/реакция;
2 - Bst 2.0 (NEB) 100 копий/реакция;
3 - Bst-NH 1000 копий/реакция;
4 - Bst-NH 100 копий/реакция;
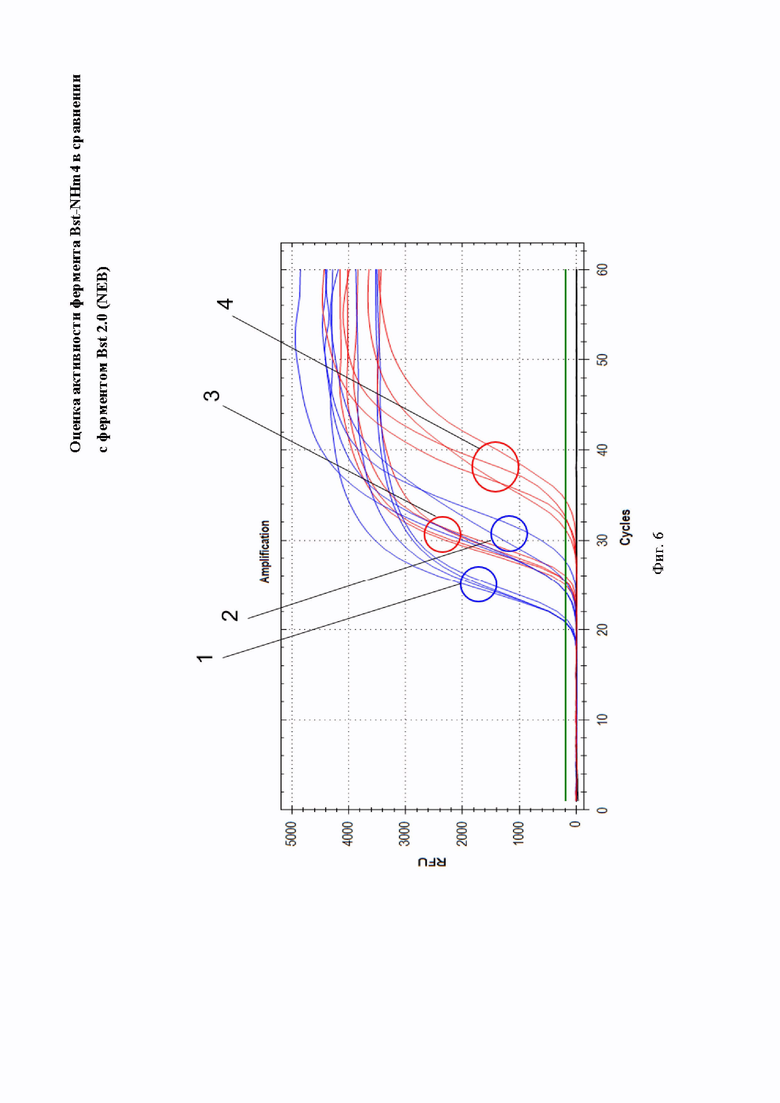
Фиг. 6. Профиль флуоресценции в реальном времени для реакции петлевой изотермической амплификации (LAMP), на котором посредством графика визуализировано накопление целевых ампликонов в модельной системе с использованием рекомбинантного фермента Bst-NH и коммерческого фермента сравнения Bst 2.0 (NEB):
- по вертикали указаны значения нормализированного флуоресцентного сигнала, создаваемого красителем;
- по горизонтали указан номер цикла;
- на группах кривых 1-4 показан использованный фермент, а также количество копий нуклеиновой кислоты на реакцию, а именно:
1 - Bst-NHm4 1000 копий/реакция;
2 - Bst-NHm4 100 копий/реакция;
3 - Bst 2.0 (NEB) 1000 копий/реакция;
4 - Bst 2.0 (NEB) 100 копий/реакция.
Реализация заявляемого изобретения поясняется следующими примерами:
Пример 1. Дизайн нуклеотидных последовательностей БФ-Bst-полимеразы из Geobacillus stearothermophilus и панели олигонуклеотидов для их сборки
Нуклеотидную последовательность SEQ ID NO: 51, кодирующую аминокислотную последовательность БФ-Bst-полимеразы (SEQ ID NO: 1), получили методом сборки из длинных перекрывающихся праймеров, так называемым «методом лесенки» [Xiong, A. S. A Simple, Rapid, High-Fidelity and Cost-Effective PCR-Based Two-Step DNA Synthesis Method for Long Gene Sequences./ Xiong, A. S. et al. // Nucleic Acids Research. - 2004. - Vol. 32, no. 12, pp. e98-e98, https://doi.org/10.1093/nar/gnh094.]. На первом этапе провели обратную трансляцию аминокислотной последовательности БФ-Bst-полимеразы (SEQ ID NO: 1) с использованием программы VectorNTI, а затем провели оптимизацию нуклеотидного состава с учетом частоты встречаемости кодонов для бактериальной системы экспрессии на основе клеток E.coli. На концах нуклеотидной последовательности SEQ ID NO: 51, ввели сайты рестрикции Ndel - на 5'-конце, и KpnI и XhoI - на 3'-конце, для последующего переклонирования в экспрессионный вектор рЕТ16.
Панель олигонуклеотидов для синтеза нуклеотидной последовательности in-vitro разрабатывали с помощью программы [Owczarzy, R., et al. IDT SciTools: A Suite for Analysis and Design of Nucleic Acid Oligomers. / Owczarzy, R., et al. // Nucleic Acids Research. -2008. - Vol. 36, no. Web Server, pp. W163-69, https://doi.org/10.1093/nar/gkn198.], реализованной в виде веб-сервиса (https://eu.idtdna.com/calc/analyzer). Последовательности праймеров приведены в таблице 1.
Разработанная нуклеотидная последовательность Bst-pol-NHis (SEQ ID NO: 51) содержит 4 кодирующих триплета с частотой встречаемости меньше 10. Замена от одного до четырех триплетов приводит к увеличению выхода целевого белка в растворимой форме. Замены указаны в таблице 2, нумерация нуклеотидных остатков приводится относительно SEQ ID NO: 51.

Замены в нуклеотидную последовательность Bst-pol-NHis (SEQ ID NO: 51) вводили с помощью мутагенеза с использованием набора QuikChange II Site-Directed Mutagenesis Kit (Agilent, США). Для этого использовали праймеры для мутагенеза, несущие целевые замены, SEQ ID NO: 52-59 (таблица 3). Реакцию мутагенеза проводили согласно инструкции производителя. Наличие целевых мутаций подтверждали с помощью секвенирования методом Сэнгера. В результате была получена нуклеотидная последовательность Bst-pol-NHis-m4 (SEQ ID NO: 60).

Синтезированные нуклеотидные последовательности Bst-pol-NHis (SEQ ID NO: 51) и Bst-pol-NHis-m4 (SEQ ID NO: 60) дают возможность получения целевого фермента с полигистидиновым тэгом на N-конце (SEQ ID NO: 2).
Пример 2. Получение нуклеотидных последовательностей БФ-Bst-полимеразы из Geobacillus stearothermophilus методом ПЦР и их клонирование
Нуклеотидная последовательность Bst-pol-NHis (SEQ ID NO: 51) была получена методом сборки из длинных перекрывающихся праймеров (таблица 1). Синтез нуклеотидной последовательности проводили методом ПЦР с использованием высокоточной полимеразы КАРА HiFi полимеразы фирмы KapaBiosystems. На первом этапе проводили сборку четырех фрагментов нуклеотидной последовательности, а затем фрагменты попарно сшивали между собой. Для сборки фрагмента 1 (Bs1) использовали праймеры SEQ ID NO: 5-16, для фрагмента 2 (Bs2) праймеры SEQ ID NO: 17-26, для фрагмента 3 (Bs3) - праймеры SEQ ID NO: 27-38, для фрагмента 4 (Bs4) - праймеры SEQ ID NO: 39- 50.
Для сборки каждого фрагмента проводятся 5-6 последовательных раундов ПЦР. Общий объем реакционной смеси 25 мкл. Для первого раунда сборки нуклеотидной последовательности компоненты ПЦР смешиваются следующим образом:
(a) 9,5 мкл реактива ddH2O (ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Россия):
(b) 5 мкл реактива 10х KAPApol buffer (KapaBiosystems);
(c) 10 мкл смеси, содержащей:
- олигонуклеотидные праймеры X_1F и X_1R по 1,6 мкМ;
- dNTPs - 0,44 мМ.
(d) 0,5 мкл реактива KAPApol (KapaBiosystems).
Программа амплификации состояла из 3 циклов: денатурация при 95°С - 20 сек., отжиг - 30 сек. при 60°С, элонгация при 72°С - 10 мин.
Все последующие раунды сборки проводились идентичным способом. Компоненты ПЦР смешиваются следующим образом:
(a) 9,0 мкл реактива ddH2O (ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Россия):
(b) 5 мкл реактива 10х KAPApol buffer (KapaBiosystems);
(c) 10 мкл смеси, содержащей:
- олигонуклеотидные праймеры X_nF и X_nR - по 0,6 мкМ; (где n - число от 2 до 6)
- dNTPs - 0,44 мМ.
(d) 0,5 мкл ампликона из ПЦР-смеси предыдущего раунда амплификации;
(e) 0,5 мкл реактива KAPApol (KapaBiosystems).
Программа амплификации состояла из 6 циклов: денатурация при 95°С - 20 сек., отжиг - 30 сек. при 60°С, элонгация при 72°С - 1 мин; затем еще 10 циклов: денатурация при 95°С - 20 сек., элонгация при 72°С - 1 мин.
Полученный в результате проведения всех раундов сборки ампликон очищали от побочных продуктов амплификации выделением из геля. На первом этапе проводят разделение фрагментов ДНК с помощью электрофореза в агарозном геле. Затем полоску геля, содержащую нужный продукт реакции вырезают. Выделение ДНК из геля проводят
набором фирмы «QIAGEN» (Германия) согласно прилагаемой методике фирмы-производителя.
На втором этапе сборки полученные ампликоны сшивали попарно между собой с помощью ПЦР. Компоненты ПЦР смешиваются следующим образом:
(a) 9,5 мкл реактива ddH2O (ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Россия):
(b) 5 мкл реактива 10х KAPApol buffer (KapaBiosystems);
(c) 10 мкл смеси, содержащей:
- концевые олигонуклеотидные праймеры - по 0,6 мкМ;
- dNTPs - 0,44 мМ.
(d) 5 мкл очищенного ампликона;
(e) 0,5 мкл реактива KAPApol (KapaBiosystems).
Программа амплификации состояла из 25 циклов: денатурация при 95°С - 30 сек., отжиг - 30 сек. при 60°С, элонгация при 72°С - 1 мин.
Для сшивки фрагментов Bs1 и Bs2 использовали праймеры Bs1 6F (SEQ ID NO: 15) и Bs2 5R (SEQ ID NO: 26) с получением фрагмента (Bs12). Для сшивки фрагментов Bs3 и Bs4 использовали праймеры Bs3 6F (SEQ ID NO: 38) и Bs4 6R (SEQ ID NO: 50) с получением фрагмента Bs34. Для сшивки фрагментов Bs12 и Bs34 использовали праймеры Bs F-NdeI (SEQ ID NO: 3) и Bs R-XhoI (SEQ ID NO: 4).
Полученный в результате проведения всех раундов сборки ампликон очищали от побочных продуктов амплификации выделением из геля. На первом этапе проводили разделение фрагментов ДНК с помощью электрофореза в агарозном геле. Затем полоску геля, содержащую нужный продукт реакции вырезали. Выделение ДНК из геля провели набором фирмы «QIAGEN» (Германия) согласно прилагаемой методике фирмы-производителя. Очищенный ампликон инкубировали 30 мин при 72°С в присутствии Taq-полимеразы и 0,44 мМ смеси дезоксинуклеотидов dNTPs для того, чтобы получить выступающие А-концы для последующего клонирования продукта вектор pGEM-T (Promega). Целевой продукт лигируют в вектор pGEM-T (Promega). Наличие целевой последовательности и ее корректность подтверждают с помощью секвенирования методом Сэнгера.
Замены в нуклеотидную последовательность Bst-pol-NHis SEQ ID NO: 51 вводили с помощью мутагенеза с использованием набора QuikChange II Site-Directed Mutagenesis Kit (Agilent, США). Для этого использовали праймеры для мутагенеза, несущие целевые замены (таблица 3). Реакцию мутагенеза проводили согласно инструкции производителя. Наличие целевых мутаций подтверждали с помощью секвенирования методом Сэнгера. В результате была получена нуклеотидная последовательность Bst-pol-NHis-m4 SEQ ID NO: 60.
Подготовленный описанным способом материал, содержащий уникальные нуклеотидные последовательности, используют для получения экспрессионных конструкций.
В результате получены плазмидные ДНК, содержащий одну из последовательностей большого фрагмента Bst-полимеразы:
1. pBst_NHis_Tv (содержит нуклеотидную последовательность SEQ ID NO: 51);
2. pBst_NHis_m4_Tv (содержит нуклеотидную последовательность SEQ ID NO: 60).
Пример 3. Получение экспрессионных конструкций, кодирующих БФ-Bst-полимеразу.
Для клонирования фрагментов, несущих целевые гены, проводили рестрикцию плазмид, содержащих целевые последовательности, и вектора для экспрессии рЕТ16. Рестрикцию плазмидной ДНК проводили в течение 1-2 ч в соответствующем буфере при 37°С в суммарном объеме 20 мкл. Для гидролиза 1-2 мкл плазмидной ДНК (300 нг) брали 2-4 ед. рестриктазы. Полноту протекания реакции контролировали, разделяя фрагменты в 1% агарозном геле. Затем проводили попарное лигирование целевых вставок векторной ДНК и трансформацию плазмид в клетки E.coli DH10B. Единичные клоны, содержащие целевую вставку определяли методом ПЦР с универсальными праймерами T7-forward и T7-reverse (Гентерра, Россия). Выделяли плазмидную ДНК и подтверждали корректность встраивания и наличие целевой последовательности с помощью секвенирования методом Сэнгера.
Таким образом, были получены экспрессионные конструкции, содержащие последовательности различных вариантов гена Bst-полимеразы.
1. pET16_Bst_NHis (содержит нуклеотидную последовательность SEQ ID NO: 51);
2. рЕТ16_Bst_NHis_m4 (содержит нуклеотидную последовательность SEQ ID NO: 60).
Пример 4. Экспрессия БФ-Bst-полимеразы из Geobacillus stearothermophilus в клетках Е.coli
Экспрессионную конструкцию рЕТ16_Bst_NHis, содержащую последовательность SEQ ID NO: 51, или рЕТ16_Bst_NHis_m4, содержащую нуклеотидную последовательность SEQ ID NO: 60, вводят в клетки Е.coli BL21 DE3 методом химической трансформации. Аналитическая экспрессия проводилась на питательной среде Лурии-Бертани (LB), содержащей триптон, дрожжевой экстракт и хлорид натрия, в присутствии ампициллина и 1% глюкозы, в качестве индуктора экспрессии используется изопропил-β-D-тиогалактозид (ИПТГ) с конечной концентрацией 0,5 мМ. Синтез белка в клетках проводился в течение 4 часов после добавления индуктора. Выделение целевого белка производился из фракции растворимых белков, очистка проводится методом хроматографии в две стадии.
Для получения стартовой культуры клетки-продуценты наращивали при 37°С в течение 12-16 часов, затем клетки перенесли в питательную среду LB, содержащую 100 мкг/мл ампициллина, 1% глюкозы и 2,5% глицерина. Также клетки-продуценты после разведения культивировали при 37°С до достижения оптической плотности OD600≈1,2-1,5, после чего в питательную среду добавляли ИПТГ и инкубировали культуру еще 4 часа при температуре 37°С.
Аналогичным образом проводили культивирование при температуре 23°С, снижение температуры проводили перед добавлением индуктора и поддерживали при дальнейшем культивировании в течении 4 часов.
Клетки ресуспендировали в буфере, содержащем 50 мМ Трис HCl, 1 мг/мл лизоцим, рН 8.0 и инкубировали на льду 30 мин, затем добавляли равный объем лизирующего буфера 50 мМ Трис HCl рН 8.0 и проводили дезинтеграцию клеток под действием ультразвука. Затем проводят центрифугирование при 6000 g в течение 30 минут.
Первый этап хроматографической очистки проводили методом металл-хелатной хроматографии. Элюцию проводили в серии буферных растворов, содержащих 50 мМ Трис HCl, 100 мМ KCl, рН 8.0 при различных концентрациях имидазола: 60 мМ, 120 мМ, 300 мМ и 800 мМ.
Второй этап хроматографии проводили методом ион-обменной хроматографии: для этого фракцию с наибольшим содержанием целевого белка по данным белкового электрофореза разбавляли в 10 раз буфером, содержащим 50 мМ Трис HCl, рН 8.0, 5% глицерин. Элюцию проводили в серии буферных растворов, содержащих 50 мМ Трис/HCl, рН 8.0, 5% глицерин при различных концентрациях NaCl: 100 мМ, 300 мМ, 500 мМ и 1 М.
В качестве сорбента на первом этапе хроматографической очистки использовали сорбент Chelating Sepharose FF. В качестве сорбента на втором этапе хроматографической очистки использовали сорбент Capto Q.
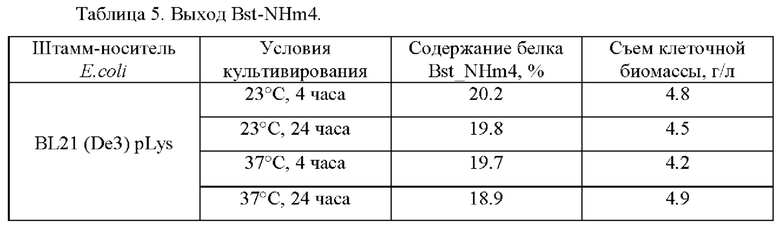
При экспрессии рЕТ16_Bst_NHis (содержит нуклеотидную последовательность SEQ ID NO: 51) в клетках E.coli BL21 DE3 было показано наличие белка (SEQ ID NO: 2), соответствующего расчетной массе (69 кДа) (Фиг. 1). В результате использования указанного протокола очистки фермента были получены препараты белка с чистотой более 95% (Фиг. 2). Концентрация фермента в полученном растворе составляла 1,65-1,8 мг/мл для разных препаратов. При оптимизации условий культивирования использовались две различные температуры: 23°С и 37°С.При культивировании при 23°С перед индукцией среда предварительно охлаждалась до 23°С.Понижение температуры культивирования приводило к небольшому увеличению доли целевого белка в клетке (таблица 4, где показано содержание фермента Bst-NH в штаммах-продуцентах относительно общего белка клетки. Данные получены при использовании программы Image Lab (версия 5.2.1), Bio-Rad, США).) и повышению выхода фермента.

При экспрессии рЕТ16_Bst_NHis_m4 (содержит нуклеотидную последовательность SEQ ID NO: 60) в клетках E.coli BL21 DE3 было показано наличие белка, соответствующего расчетной массе (69 кДа) (Фиг. 3). В результате использования указанного протокола очистки фермента были получены препараты белка с чистотой более 95% (Фиг. 4). Концентрация фермента в полученном растворе составляла 2,8-3,1 мг/мл для разных препаратов. При оптимизации условий культивирования использовались две различные температуры: 23°С и 37°С.При культивировании при 23°С перед индукцией среда предварительно охлаждалась до 23°С.Понижение температуры культивирования приводило к небольшому увеличению доли целевого белка в клетке и повышению выхода фермента. Следует отметить, что при экспрессии рЕТ16_Bst_NHis_m4 в клетках E.coli BL21 DE3 за счет удаления четырех кодирующих кодонов с частотой меньше 10 удалось увеличить долю рекомбинантного белка до 20% (23°С, 4 часа культивирования) (таблица 5, где показано содержание фермента Bst-NHm4 в штаммах-продуцентах относительно общего белка клетки. Данные получены при использовании программы Image Lab (версия 5.2.1), Bio-Rad, США)).
В результате проведения экспрессии и хроматографической очистки получены препараты большого фрагмента Bst-полимеразы следующего состава:
1. рекомбинантный большой фрагмент Bst-полимеразы из Geobacillus stearothermophilus (Bst_NH, аминокислотная последовательность SEQ ID NO: 2, нуклеотидная последовательность SEQ ID NO: 51), белок с полигистидиновым тэгом на N-конце в буферном растворе, содержащем 50 мМ Трис-HCl, 100 мМ NaCl, 1 мМ ЭДТА, 20% глицерин, рН 8.0.
2. рекомбинантный большой фрагмент Bst-полимеразы из Geobacillus stearothermophilus (Bst_NHm4, аминокислотная последовательность SEQ ID NO: 2, нуклеотидная последовательность SEQ ID NO: 60), белок с полигистидиновым тэгом на N-конце с четырьмя мутантными кодирующими триплетами, в буферном растворе, содержащем 50 мМ Трис-HCl, 100 мМ NaCl, 1 мМ ЭДТА, 20% глицерин, рН 8.0.
Пример 5. Сравнение эффективности полученных ферментных препаратов в условиях LAMP
Сравнение каталитических характеристик полученных ферментных препаратов проводили в условиях реакции изотермической амплификации LAMP. Реакцию проводили в реакционной смеси следующего состава: 20 мМ Трис-HCl (рН 8,8), 10 мМ KCl, 8 мМ MgSO4, 10 мМ (NH4)2SO4, 0,1% Твин 20, по 1,4 мМ каждого дНТФ, по 0,2 мкМ внешних праймеров F3 и В3, по 1,6 мкМ внутренних праймеров FIP и BIP, по 0,4 мкМ петлевых праймеров LF и LB, 1 мкМ SYTO 9 (Thermo Fisher Scientific, США). Амплификацию проводили при 65°С в амплификаторе CFX96 (Bio-Rad Laboratories, США), детекцию продуктов амплификации проводили с помощью флуоресцентной детекции в режиме реального времени, в состав реакционной смеси входил интеркалирующий краситель SYTO 9, съемка флуоресцентного сигнала проходила каждые 30 с (продолжительность одного «цикла» амплификации).
Свойства полученных препаратов сравнивали с коммерчески-доступным ферментом сравнения - Bst 2.0 DNA Polymerase (120000 U/ml) кат.M0537M лот 10067327 (New England BioLabs, США) (далее Bst 2.0). Производитель рекомендует использовать рабочую концентрацию фермента Bst 2.0 - 320 Ед/мл. В качестве единицы активности принимается такое количество фермента, которое катализирует включение 25 нмоль дНТФ в состав продукта реакции за 30 мин при температуре 65°С. Выравнивание ферментов по активности проводили с использованием набора реагентов EvaEZ™ Fluorometric Polymerase Activity Assay Kit (Biotium, США) в соответствии с инструкцией фирмы-производителя.
В качестве системы праймеров использовали смесь IT-mix SARS-CoV-2 из набора реагентов для качественного определения РНК SARS-CoV-2 в биологическом материале методом обратной транскрипции и изотермической амплификации (АмплиСенс®SARS-CoV-2-IT, ФБУН ЦНИИ Эпидемиологии, Россия). В качестве модельной матрицы в реакцию брали контрольные плазмиды, несущие детектируемые участки нуклеотидной последовательности возбудителя. В эксперименте использовали две концентрации матрицы: 100 копий в реакцию и 1000 копий в реакцию.
Результаты проведенных экспериментов показывают, что препарат фермента Bst_NH (аминокислотная последовательность SEQ ID NO: 2, нуклеотидная последовательность SEQ ID NO: 51) обладает каталитической активностью, может использоваться в реакциях изотермической амплификации LAMP, однако, уступает по своей эффективности ферменту сравнения Bst 2.0 (Фиг. 5). При том, что уровень разгорания сигнала сопоставим, показатель Ct для Bst_NH выше, особенно в случае образца с низкой концентрацией.
Для препарата Bst_NHm4 (аминокислотная последовательность SEQ ID NO: 2, нуклеотидная последовательность SEQ ID NO: 60) также было показано наличие каталитической активности, на Фиг. 6 продемонстрировано, что полученный препарат не только может применяться в реакциях изотермической амплификации LAMP, но и сопоставим по уровню своей эффективности с ферментом сравнения Bst 2.0 и позволяют уверенно детектировать в реакционной смеси модельный образец с концентрацией до 100 копий в реакции.
Препараты фермента БФ-Bst-полимеразы, полученные согласно данному изобретению возможно использовать в реакциях петлевой изотермической амплификации LAMP для выявления нуклеиновых кислот различных патогенов.
Таким образом, заявляемое изобретение позволяет получать большой фрагмент Bst-полимеразы в клетках E.coli со стабильно высоким выходом фермента, характеризующийся достаточно высокой полимеразной активностью, обладающий вытесняющей активностью, что позволяет применять его в реакциях петлевой изотермической амплификации на основе метода LAMP в различных областях техники, связанных с выявлением нуклеиновых кислот патогенных для человека организмов.



Изобретение относится к области биотехнологии. Описаны способы получения большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli с высоким выходом фермента в растворимой форме. Способы включают этапы синтеза нуклеотидной последовательности, получения экспрессионной конструкции, экспрессии большого фрагмента Bst-полимеразы и получения препарата большого фрагмента Bst-полимеразы. Технический результат заключается в разработке вариантов эффективного способа получения большого фрагмента Bst-полимеразы в растворимой форме с высоким выходом. 2 н. и 3 з.п. ф-лы, 6 ил., 5 табл., 5 пр.
1. Способ получения большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli с высоким выходом фермента в растворимой форме, включающий:
(i) синтез нуклеотидной последовательности SEQ ID NO: 51 методом ПЦР, очищение полученного ампликона, и лигирование полученного целевого продукта в вектор pGEM-T;
(ii) получение экспрессионной конструкции, содержащей нуклеотидную последовательность SEQ ID NO: 51 и кодирующей большой фрагмент Bst-полимеразы, где последовательно проводят рестрикцию плазмидной ДНК и вектора для экспрессии pET16, попарное лигирование целевых вставок и векторной ДНК и трансформацию плазмид в клетки E.coli DH10B, выделение плазмидной ДНК;
(iii) экспрессию большого фрагмента Bst-полимеразы из Geobacillus stearothermophilus в клетках E.coli на питательной среде Лурии-Бертани в присутствии ампициллина, где экспрессионную конструкцию, содержащую нуклеотидную последовательность SEQ ID NO: 51 вводят в клетки E.coli BL21 DE3 методом химической трансформации, в качестве индуктора экспрессии используется изопропил-β-D-тиогалактозид, синтез белка в клетках проводят при температуре 23°С и 37°С в течение 4 часов после добавления индуктора, выделение целевого белка производят из фракции растворимых белков, а очистку проводят методом хроматографии в две стадии.
(iiii) получение препарата большого фрагмента Bst-полимеразы Bst_NH следующего состава: рекомбинантный большой фрагмент Bst-полимеразы из Geobacillus stearothermophilus - белок с аминокислотной последовательностью SEQ ID NO: 2, кодирующийся нуклеотидной последовательностью SEQ ID NO: 51, и полигистидиновым тэгом на N-конце в буферном растворе, содержащем 50 мМ Трис-HCl, 100 мМ NaCl, 1 мМ ЭДТА, 20% глицерин, pH 8.0.
2. Способ получения большого фрагмента Bst-полимеразы в виде рекомбинантного белка в клетках E.coli с высоким выходом фермента в растворимой форме, включающий:
(i) получение нуклеотидной последовательности SEQ ID NO: 60 путем ввода замен в нуклеотидную последовательность SEQ ID NO: 51 с использованием праймеров для мутагенеза SEQ ID NO: 52 – 59;
(ii) получение экспрессионной конструкции, содержащей нуклеотидную последовательность SEQ ID NO: 60 и кодирующей большой фрагмент Bst-полимеразы, где последовательно проводят рестрикцию плазмидной ДНК и вектора для экспрессии pET16, попарное лигирование целевых вставок векторной ДНК и трансформацию плазмид в клетки E.coli DH10B, выделение плазмидной ДНК;
(iii) экспрессию большого фрагмента Bst-полимеразы из Geobacillus stearothermophilus в клетках E.coli на питательной среде Лурии-Бертани в присутствии ампициллина, где экспрессионную конструкцию, содержащую нуклеотидную последовательность SEQ ID NO: 60 вводят в клетки E.coli BL21 DE3 методом химической трансформации, в качестве индуктора экспрессии используется изопропил-β-D-тиогалактозид, синтез белка в клетках проводят при температуре 23°С и 37°С в течение 4 часов после добавления индуктора, выделение целевого белка производят из фракции растворимых белков, а очистку проводят методом хроматографии в две стадии.
(iiii) получение препарата большого фрагмента Bst-полимеразы Bst_NHm4 следующего состава: рекомбинантный большой фрагмент Bst-полимеразы из Geobacillus stearothermophilus - белок с аминокислотной последовательностью SEQ ID NO: 2, кодирующийся нуклеотидной последовательностью SEQ ID NO: 60, и полигистидиновым тэгом на N-конце с четырьмя мутантными кодирующими триплетами, в буферном растворе, содержащем 50 мМ Трис-HCl, 100 мМ NaCl, 1 мМ ЭДТА, 20% глицерин, pH 8.0.
3. Способ получения большого фрагмента Bst-полимеразы по п. 1, где нуклеотидную последовательность SEQ ID NO: 51 получают путем обратной трансляции аминокислотной последовательности большого фрагмента Bst-полимеразы с аминокислотной последовательностью SEQ ID NO: 1, а на концах нуклеотидной последовательности SEQ ID NO: 51 вводят сайты рестрикции NdeI – на 5`-конце, и KpnI и XhoI – на 3`-конце.
4. Способ получения большого фрагмента Bst-полимеразы по п. 1, где для синтеза нуклеотидной последовательности SEQ ID NO: 51 используют панель олигонуклеотидных праймеров SEQ ID NO: 3 – 50.
5. Способ получения большого фрагмента Bst-полимеразы по п. 1, при котором ПЦР проводят в два этапа, при этом на первом этапе проводят сборку четырех фрагментов нуклеотидной последовательности SEQ ID NO: 51, где для сборки фрагмента 1 (Bs1) используют праймеры SEQ ID NO: 5-16, для фрагмента 2 (Bs2) – праймеры SEQ ID NO: 17-26, для фрагмента 3 (Bs3) - праймеры SEQ ID NO: 27-38, для фрагмента 4 (Bs4) - праймеры SEQ ID NO: 39- 50, а на втором этапе ПЦР полученные фрагменты сшивают, при этом для сшивки фрагментов Bs1 и Bs2 используют праймеры SEQ ID NO: 15 (Bs1 6F) и SEQ ID NO: 26 (Bs2 5R) с получением фрагмента Bs12, для сшивки фрагментов Bs3 и Bs4 используют праймеры SEQ ID NO: 3 (Bs3 6F 8) и SEQ ID NO: 50 (Bs4 6R) с получением фрагмента Bs34, для сшивки полученных фрагментов Bs12 и Bs34 используют праймеры SEQ ID NO: 3 (Bs F-NdeI) и SEQ ID NO: 4 (Bs R-XhoI).
| CN 106399299 B 29.01.2019 | |||
| DE 69627080 D1 08.05.2003 | |||
| ТЕРМОСТАБИЛЬНАЯ ДНК-ПОЛИМЕРАЗА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, ФРАГМЕНТ ВЫДЕЛЕННОЙ ДНК, СПОСОБ АМПЛИФИКАЦИИ ДНК, СПОСОБ ВВЕДЕНИЯ МЕТКИ В ДНК И СПОСОБ ОБРАТНОЙ ТРАНСКРИПЦИИ | 1997 |
|
RU2197524C2 |
| Вращательный станок для разведочного бурения | 1932 |
|
SU33101A1 |

