ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтического синтеза и, в частности, к FGFR и его ингибитору мутаций, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Рецепторы фактора роста фибробластов (FGFR) представляют собой тирозинкиназные рецепторы, которые связываются с лигандами фактора роста фибробластов. В настоящее время обнаружены FGFR четырех типов, которые способны связываться с лигандами. Полагают, что сигнальные пути, зависимые от фактора роста фибробластов (FGF), играют важную роль во многих процессах, таких как эмбриогенез, дифференцировка тканей, заживление ран и регуляция метаболизма; также полагают, что они тесно связаны с характеристиками многих опухолей. Связывание FGF со своим рецептором вызывает димеризацию и фосфорилирование рецептора, а также стимуляцию его протеинкиназной активности, в результате чего облегчается активация целого ряда внутриклеточных путей передачи сигнала, включая Ras-MAPK, AKT-PI3K и фосфатазы C, которые играют важную роль в росте, пролиферации и выживаемости клеток.
Генетические изменения в членах семейства FGFR часто связаны с ростом, метастазированием, ангиогенезом и выживаемостью опухолей. В клинических исследованиях было показано, что многие ингибиторы FGFR вызывают клинические ответы у пациентов с аномалиями FGFR, а недавно ингибиторы FGFR также были одобрены для коммерческой реализации. Однако в клинических исследованиях было показано, что на ингибиторы FGFR быстро возникает приобретенная резистентность, что ведет к относительно небольшой выживаемости без прогрессирования. Мутации, вызывающие изменения в аминокислотах FGFR, могут приводить к резистентности к ингибиторам FGFR или снижению активности последних. Важным механизмом приобретения резистентности к ингибированию FGFR являются вторичные мутации в киназном домене FGFR, возникающие под действием ингибитора FGFR. Соответствующие точечные мутации FGFR также присутствуют в опухолях. Сообщается, что одним из главных механизмов резистентности к тирозинкиназам является мутация в «ключевых» остатках (гейткиперная мутация); при этом мутации, придающие резистентность к FGFR, были обнаружены как в клеточных системах in vitro, так и в клинических исследованиях. К числу гейткиперных мутаций относятся мутации FGFR3V555M, FGFR2V565F/V565I/V565L и другие. По данным недавно проведенных исследований, гейткиперная мутация FGFR2V565F была обнаружена у трех пациентов с холангиокарциномой, получавших терапию препаратом BGJ398; у двух из них также имелись другие мутации, которые находились в других киназных участках FGFR2. В связи с этим, для решения проблемы приобретенной резистентности, возникающей в случае клинического применения ингибиторов FGFR первого поколения, необходимо как можно быстрее разработать ингибиторы FGFR нового поколения, которые бы в течение длительного времени действовали на опухоли с мутациями генов, вовлеченных в FGFR-зависимый путь передачи сигнала. Однако такие ингибиторы FGFR второго поколения должны не только обладать такой же высокой подавляющей активностью в отношении FGFR, но и обеспечивать тот же уровень активности в отношении гейткиперных мутаций, в отношении которых активность ингибиторов FGFR первого поколения снижена.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
После проведения всесторонних и углубленных исследований авторы настоящего изобретения впервые разработали FGFR и ингибитор его мутаций, способ его получения и его применение. Ряд соединений по настоящему изобретению обладает высокой активностью в отношении мутантных форм FGFR, в особенности в отношении FGFR с гейткиперными мутациями, и в особенности в отношении FGFR3 V555M, FGFR2 V565I, FGFR2 V565F, FGFR2 V565L и гейткиперной мутации FGFR2 N550K; ожидается разработка ингибиторов FGFR нового поколения.
Первый аспект настоящего изобретения относится к соединению формулы (I), стереоизомеру или его фармацевтически приемлемой соли:
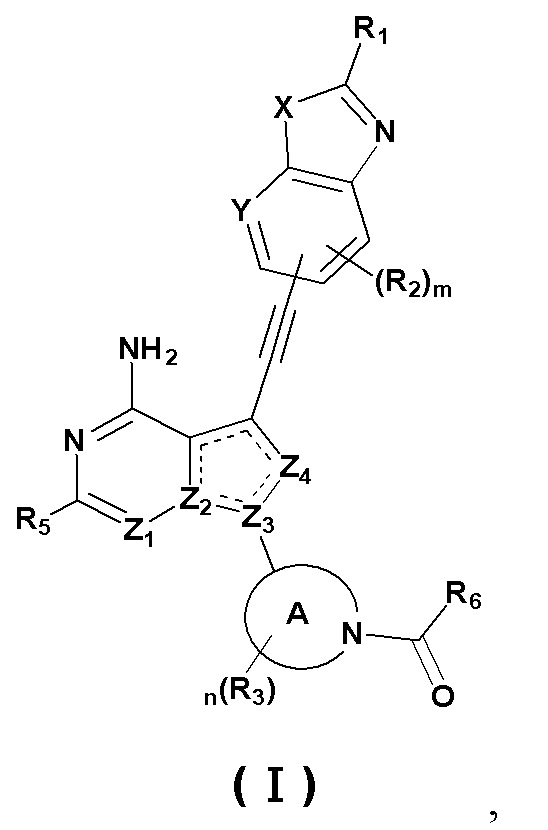
где « » представляет собой двойную связь или одинарную связь;
» представляет собой двойную связь или одинарную связь;
X представляет собой O или S; Y представляет собой CR4 или N;
Z1 и Z4 каждый независимо представляют собой N или CR7;
Z2 и Z3 каждый независимо представляют собой N или C;
кольцо А представляет собой 3-12-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
R1 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10;
R2, R3 и R4 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10;
R5 и R7 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10;
R6 выбран из группы, состоящей из C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C3-12 циклоалкила, 3-12-членного гетероциклила, -C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10;
каждый R8 независимо выбран из группы, состоящей из водорода, дейтерия, гидрокси, C1-10 алкила, C2-10 алкенила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, оксо, C1-10 алкила, C1-10 алкокси, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12 членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси и -NR11R12;
каждый R9 независимо выбран из группы, состоящей из водорода, дейтерия, C1-10 алкила, C2-10 алкенила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила и 5-10-членного гетероарила, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, оксо, циано, C1-10 алкила, C1-10 алкокси, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12-членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси и -NR11R12;
каждый R10 независимо выбран из группы, состоящей из водорода, дейтерия, гидрокси, C1-10 алкила, C1-10 алкокси, C2-10 алкенила, C2-10 алкинила, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12-членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, циано, C1-10 алкила, C1-10 алкокси, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12-членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси и -NR11R12;
R11 и R12 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, гидрокси, C1-10 алкокси, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, сульфинила, сульфонила, метилсульфонила, изопропилсульфонила, циклопропилсульфонила, п-толуолсульфонила, аминосульфонила, диметиламиносульфонила, амино, моноалкиламино, диалкиламино и C1-10 алканоила, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C1-10 алкокси, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12-членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси, амино, моноалкиламино, диалкиламино и C1-10 алканоила;
или R11 и R12, вместе с атомом азота, непосредственно присоединенным к ним, образуют 4-10-членный гетероциклил, вышеуказанная группа опционально замещена одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C1-10 алкокси, C3-12 циклоалкила, C3-12 циклоалкилокси, 3-12-членного гетероциклила, 3-12-членного гетероциклилокси, C5-10 арила, C5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси, амино, моноалкиламино, диалкиламино и C1-10 алканоила;
m равно 0, 1 или 2;
n равно 0, 1, 2, 3 или 4;
каждый r независимо принимает значения 0, 1 или 2.
В предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли R5 и R7 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, -C0-4 алкила-SF5, -C0-4 алкила-S(O)rR8, -C0-4 алкила-O-R9, -C0-4 алкила-C(O)OR9, -C0-4 алкила-C(O)R10, -C0-4 алкила-O-C(O)R10, -C0-4 алкила-NR11R12, -C0-4 алкила-C(=NR11)R10, -C0-4 алкила-N(R11)-C(=NR12)R10, -C0-4 алкила-C(O)NR11R12 и -C0-4 алкила-N(R11)-C(O)R10;
где R8, R9, R10, R11, R12 и r соответствуют таковым в соединении формулы (I).
В предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли R2, R3 и R4 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, -C0-4 алкила-SF5, -C0-4 алкила-S(O)rR8, -C0-4 алкила-O-R9, -C0-4 алкила-C(O)OR9, -C0-4 алкила-C(O)R10, -C0-4 алкила-O-C(O)R10, -C0-4 алкила-NR11R12, -C0-4 алкила-C(=NR11)R10, -C0-4 алкила-N(R11)-C(=NR12)R10, -C0-4 алкила-C(O)NR11R12 и -C0-4 алкила-N(R11)-C(O)R10;
где R8, R9, R10, R11, R12 и r соответствуют таковым в соединении формулы (I).
В предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли R6 выбран из группы, состоящей из C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, 3-6-членного гетероциклила, -C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, =O, -C0-4 алкила-SF5, -C0-4 алкила-S(O)rR8, -C0-4 алкила-O-R9, -C0-4 алкила-C(O)OR9, -C0-4 алкила-C(O)R10, -C0-4 алкила-O-C(O)R10, -C0-4 алкила-NR11R12, -C0-4 алкила-C(=NR11)R10, -C0-4 алкила-N(R11)-C(=NR12)R10, -C0-4 алкила-C(O)NR11R12 и -C0-4 алкила-N(R11)-C(O)R10;
где R8, R9, R10, R11, R12 и r соответствуют таковым в соединении формулы (I).
В предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли R1 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, -C0-4 алкила-SF5, -C0-4 алкила-S(O)rR8, -C0-4 алкила-O-R9, -C0-4 алкила-C(O)OR9, -C0-4 алкила-C(O)R10, -C0-4 алкила-O-C(O)R10, -C0-4 алкила-NR11R12, -C0-4 алкила-C(=NR11)R10, -C0-4 алкила-N(R11)-C(=NR12)R10, -C0-4 алкила-C(O)NR11R12 и -C0-4 алкила-N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, =O, -C0-4 алкила-SF5, -C0-4 алкила-S(O)rR8, -C0-4 алкила-O-R9, -C0-4 алкила-C(O)OR9, -C0-4 алкила-C(O)R10, -C0-4 алкила-O-C(O)R10, -C0-4 алкила-NR11R12, -C0-4 алкила-C(=NR11)R10, -C0-4 алкила-N(R11)-C(=NR12)R10, -C0-4 алкила-C(O)NR11R12 и -C0-4 алкила-N(R11)-C(O)R10;
где R8, R9, R10, R11, R12 и r соответствуют таковым в соединении формулы (I).
В еще одном предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли соединение формулы (I) представляет собой соединение, структура которого описывается формулой (II):
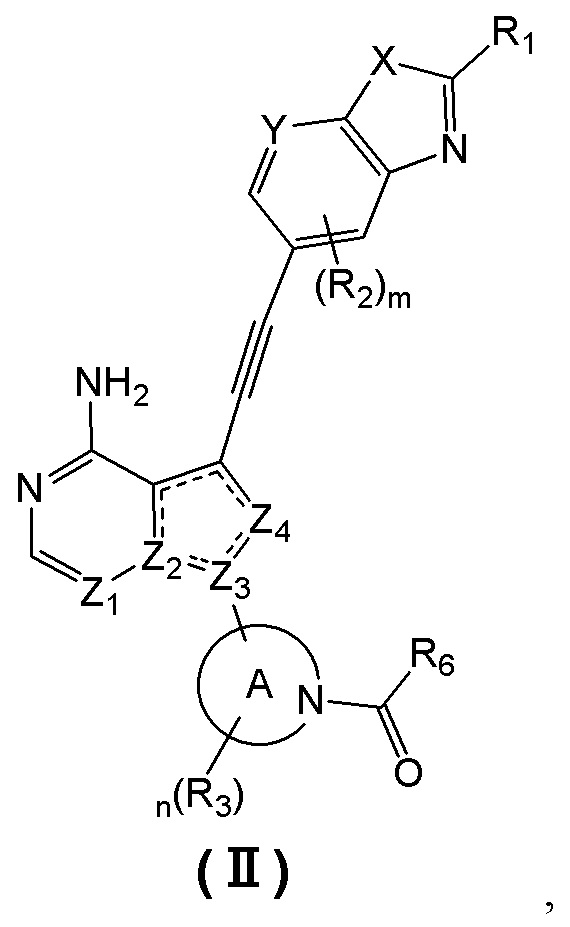
где «» представляет собой двойную связь или одинарную связь;
X представляет собой O или S; Y представляет собой CR4 или N;
Z1 и Z4 каждый независимо представляют собой N или CR7;
Z2 и Z3 каждый независимо представляют собой N или C;
кольцо А представляет собой 3-8-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
R1 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, =O, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
каждый R2 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
каждый R3 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -S(O)rR8, -CH2-O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
R4 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
R6 выбран из группы, состоящей из C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, 3-6-членного гетероциклила, -C(O)R10, -NR11R12, -C(=NR11)R10 и -N(R11)-C(=NR12)R10, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, =O, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -CH2-NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
каждый R7 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -S(O)rR8, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10, -NR11R12, -C(=NR11)R10, -N(R11)-C(=NR12)R10, -C(O)NR11R12 и -N(R11)-C(O)R10;
где R8, R9, R10, R11, R12, m, n и r соответствуют таковым в соединении формулы (I).
В еще одном предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IIIa) или формулой (IIIb):
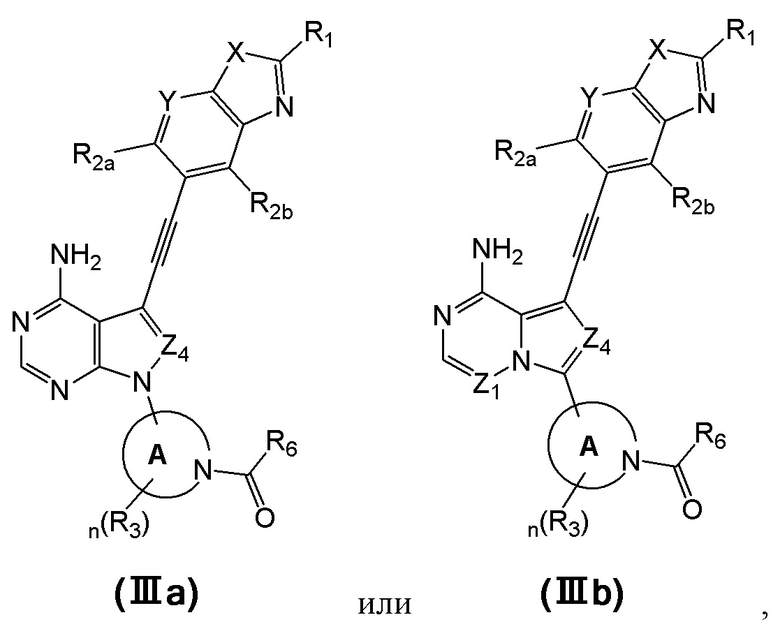
где каждый X независимо представляет собой O или S; каждый Y независимо представляет собой CH или N;
в соединении формулы (IIIb) Z1 представляет собой N или CR7;
каждый Z4 независимо представляет собой N или CR7;
каждое кольцо А независимо представляет собой 4-6-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
каждый R1 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, =O, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый из R2a и R2b независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый R3 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -CH2-O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый R6 независимо представляет собой винил, вышеуказанная группа опционально замещена одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, =O, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -CH2-NR11R12;
каждый R7 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
где R9, R10, R11, R12 и n соответствуют таковым в соединении формулы (I).
В еще одном предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли кольцо А, вместе с -(R3)n, образуют следующие структуры:
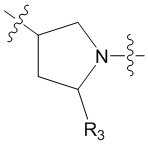 ,
, 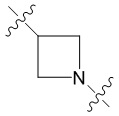 или
или 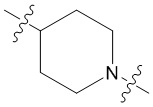 ;
;
R3 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -CH2-O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
где R9, R10, R11 и R12 соответствуют таковым в соединении формулы (I).
В еще одном предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IVa1) или формулой (IVa2):
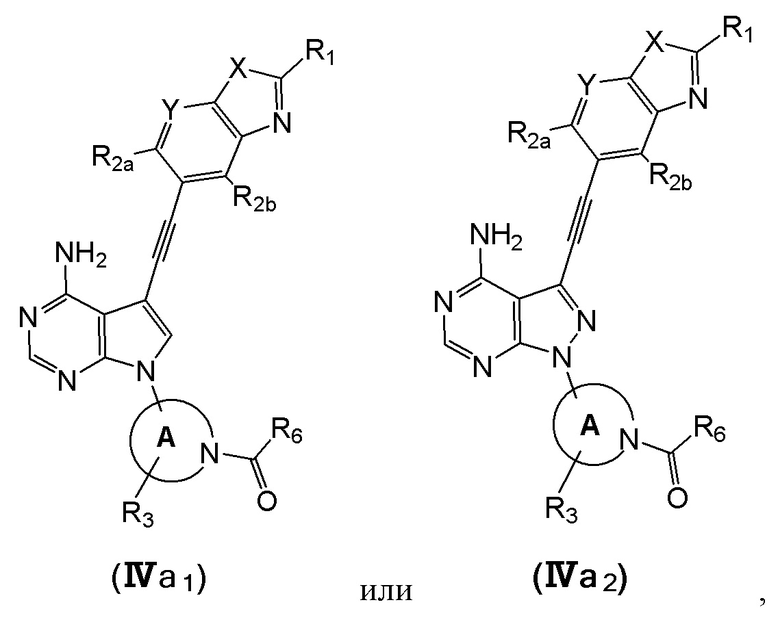
где каждый X независимо представляет собой O или S; каждый Y независимо представляет собой CH или N;
кольцо А, вместе с -R3, образуют следующие структуры:
, или ;
R3 выбран из группы, состоящей из водорода, дейтерия, галогена, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила и -CH2-O-R9;
каждый R1 независимо выбран из группы, состоящей из водорода, дейтерия, фтора, хлора, циано, C1-4 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, фтора, хлора, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, =O, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый из R2a и R2b независимо выбраны из группы, состоящей из водорода, дейтерия, фтора, хлора, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый R6 независимо представляет собой винил, вышеуказанная группа опционально замещена одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, фтора, циано, метила, этила, н-пропила и -CH2-NR11R12;
где R9, R10, R11 и R12 соответствуют таковым в соединении формулы (I).
В еще одном предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IVb1) или формулой (IVb2):
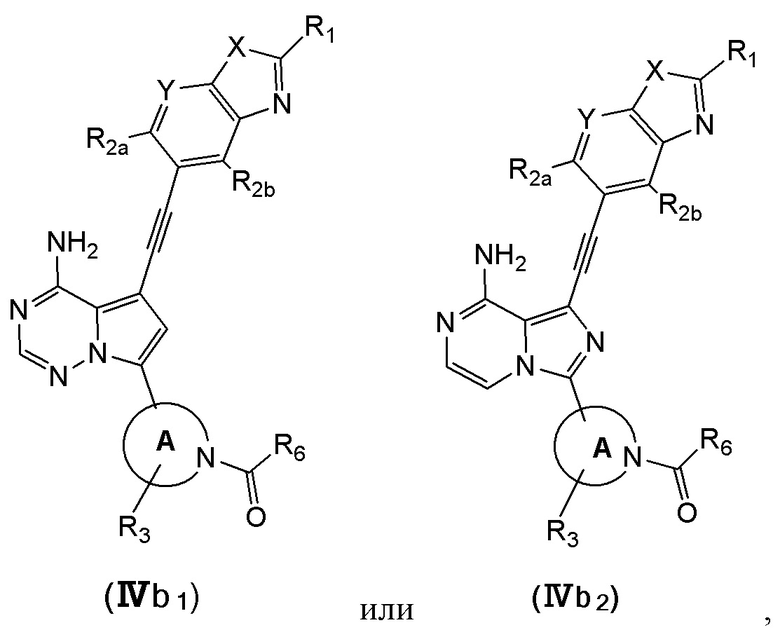
где каждый X независимо представляет собой O или S; каждый Y независимо представляет собой CH или N;
кольцо А, вместе с -R3, образуют следующие структуры:
, или ;
R3 выбран из группы, состоящей из водорода, дейтерия, галогена, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила и -CH2-O-R9;
каждый R1 независимо выбран из группы, состоящей из водорода, дейтерия, фтора, хлора, циано, C1-4 алкила, C3-6 циклоалкила, 3-6-членного гетероциклила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, фтора, хлора, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, 3-6-членного гетероциклила, =O, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый из R2a и R2b независимо выбраны из группы, состоящей из водорода, дейтерия, фтора, хлора, циано, C1-4 алкила, C1-4 галоалкила, C1-4 дейтероалкила, C3-6 циклоалкила, -SF5, -O-R9, -C(O)OR9, -C(O)R10, -O-C(O)R10 и -NR11R12;
каждый R6 независимо представляет собой винил, вышеуказанная группа опционально замещена одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, фтора, циано, метила, этила, н-пропила и -CH2-NR11R12;
где R9, R10, R11 и R12 соответствуют таковым в соединении формулы (I).
В предпочтительном варианте осуществления изобретения, в соединении формулы (I), его стереоизомере или фармацевтически приемлемой соли каждый R8 независимо выбран из группы, состоящей из водорода, дейтерия, гидрокси, C1-4 алкила, C2-4 алкенила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, оксо, C1-4 алкила, C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси и -NR11R12;
каждый R9 независимо выбран из группы, состоящей из водорода, дейтерия, C1-4 алкила, C2-4 алкенила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила и 5-8-членного гетероарила, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, оксо, циано, C1-4 алкила, C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси и -NR11R12;
каждый R10 независимо выбран из группы, состоящей из водорода, дейтерия, гидрокси, C1-4 алкила, C1-4 алкокси, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси и -NR11R12, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, циано, C1-4 алкила, C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси и -NR11R12;
R11 и R12 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, гидрокси, C1-4 алкокси, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C3-6 циклоалкила, 3-6-членного гетероциклила, C5-8 арила, 5-8-членного гетероарила, сульфинила, сульфонила, метилсульфонила, изопропилсульфонила, циклопропилсульфонила, п-толуолсульфонила, аминосульфонила, диметиламиносульфонила, амино, моноалкиламино, диалкиламино и C1-4 алканоила, вышеуказанные группы опционально замещены одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси, амино, моноалкиламино, диалкиламино и C1-4 алканоила;
или R11 и R12, вместе с атомом азота, непосредственно присоединенным к ним, образуют 4-6-членный гетероциклил, вышеуказанная группа опционально замещена одним или несколькими дополнительными заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидрокси, C1-4 алкила, C2-4 алкенила, C2-4 алкинила, C1-4 галоалкила, C1-4 дейтероалкила, C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкилокси, 3-6-членного гетероциклила, 3-6-членного гетероциклилокси, C5-8 арила, C5-8 арилокси, 5-8-членного гетероарила, 5-8-членного гетероарилокси, амино, моноалкиламино, диалкиламино и C1-4 алканоила.
В наиболее предпочтительном варианте осуществления изобретения соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль включает, помимо прочего, следующие соединения:
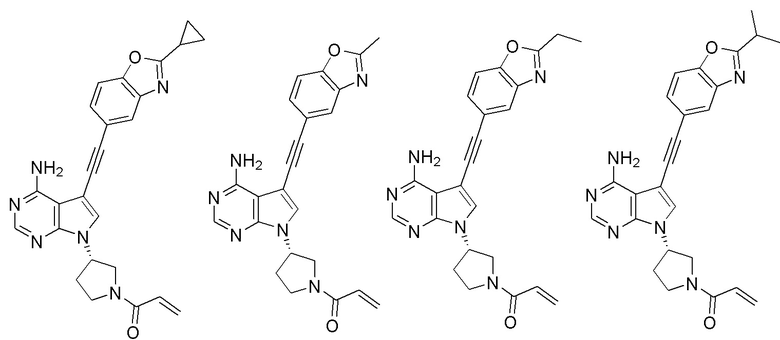
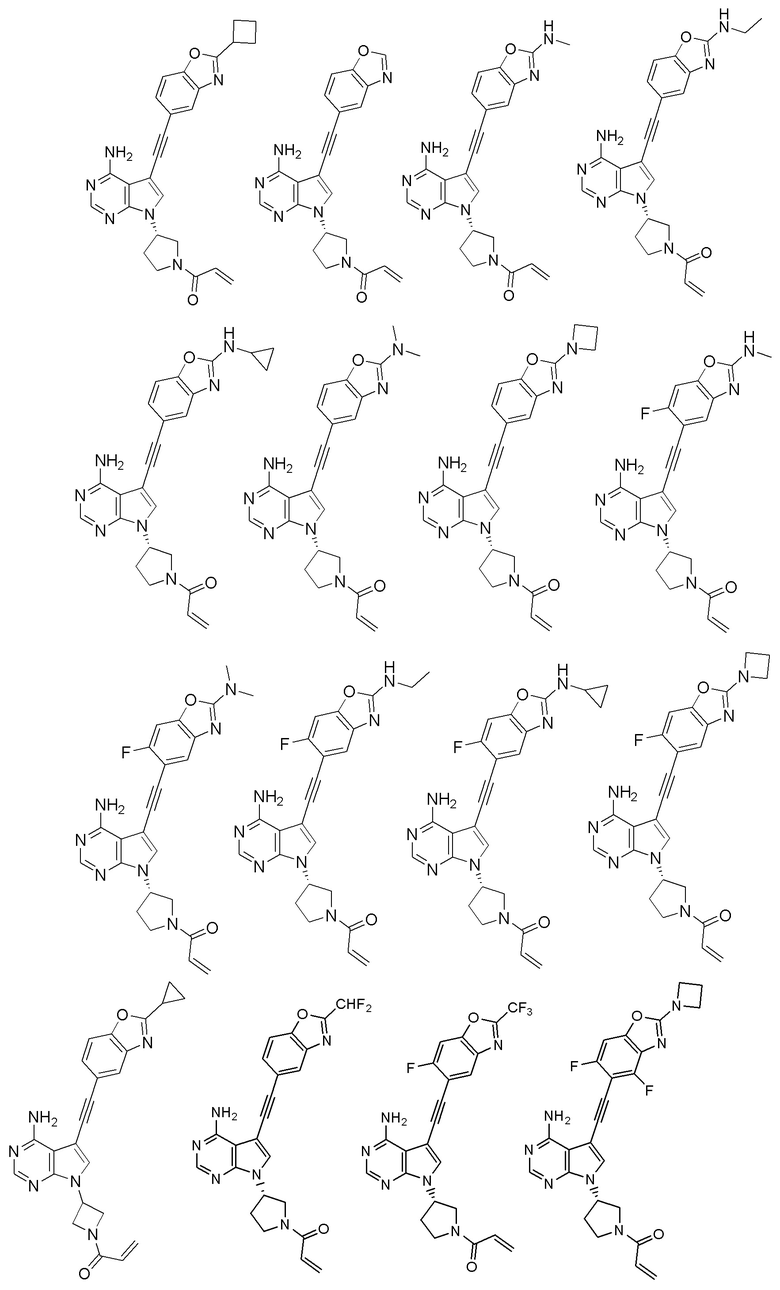
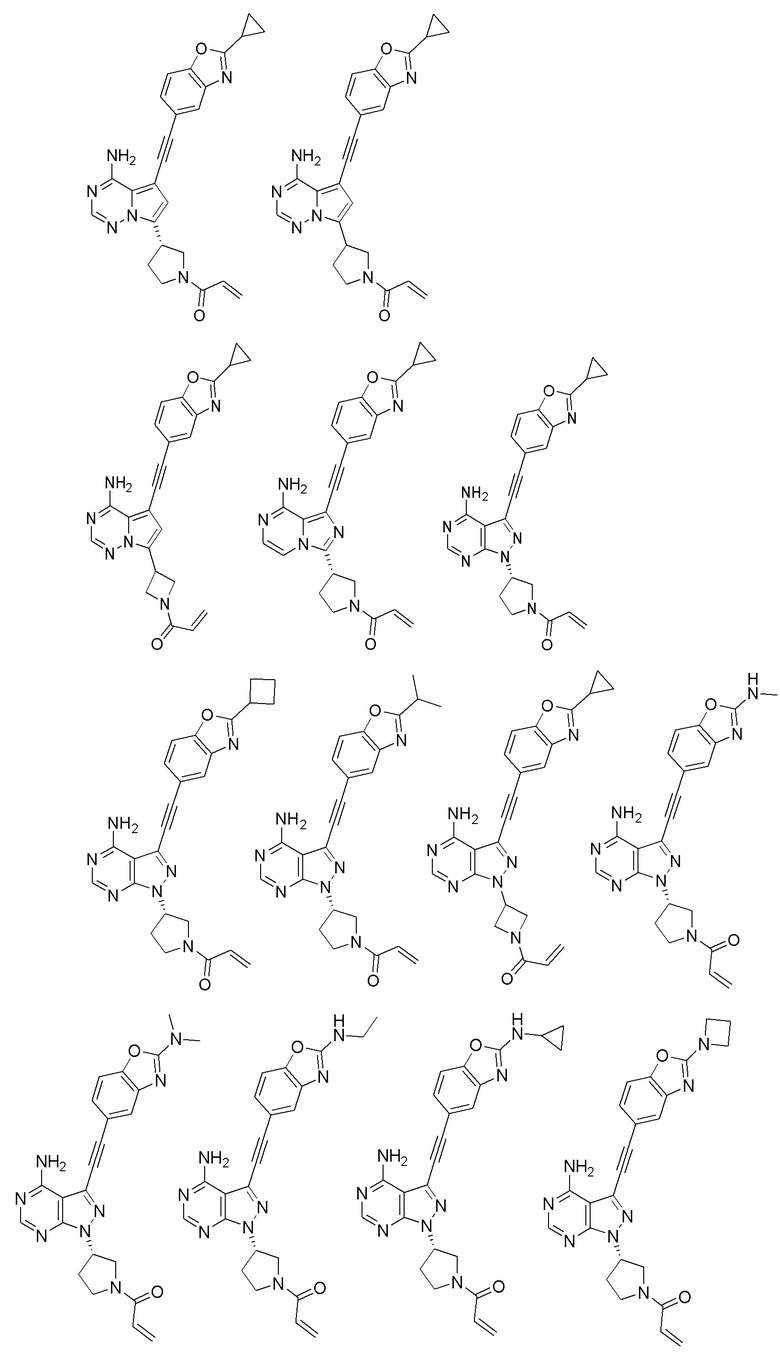
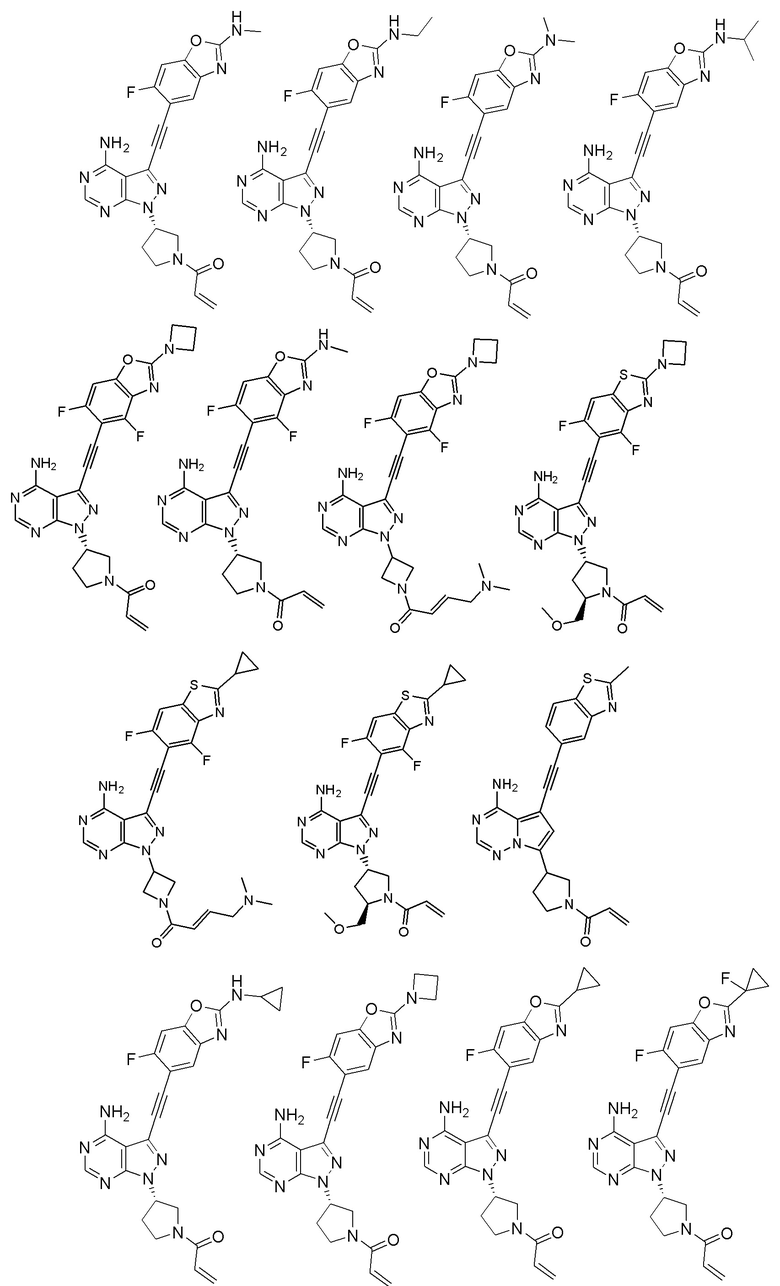
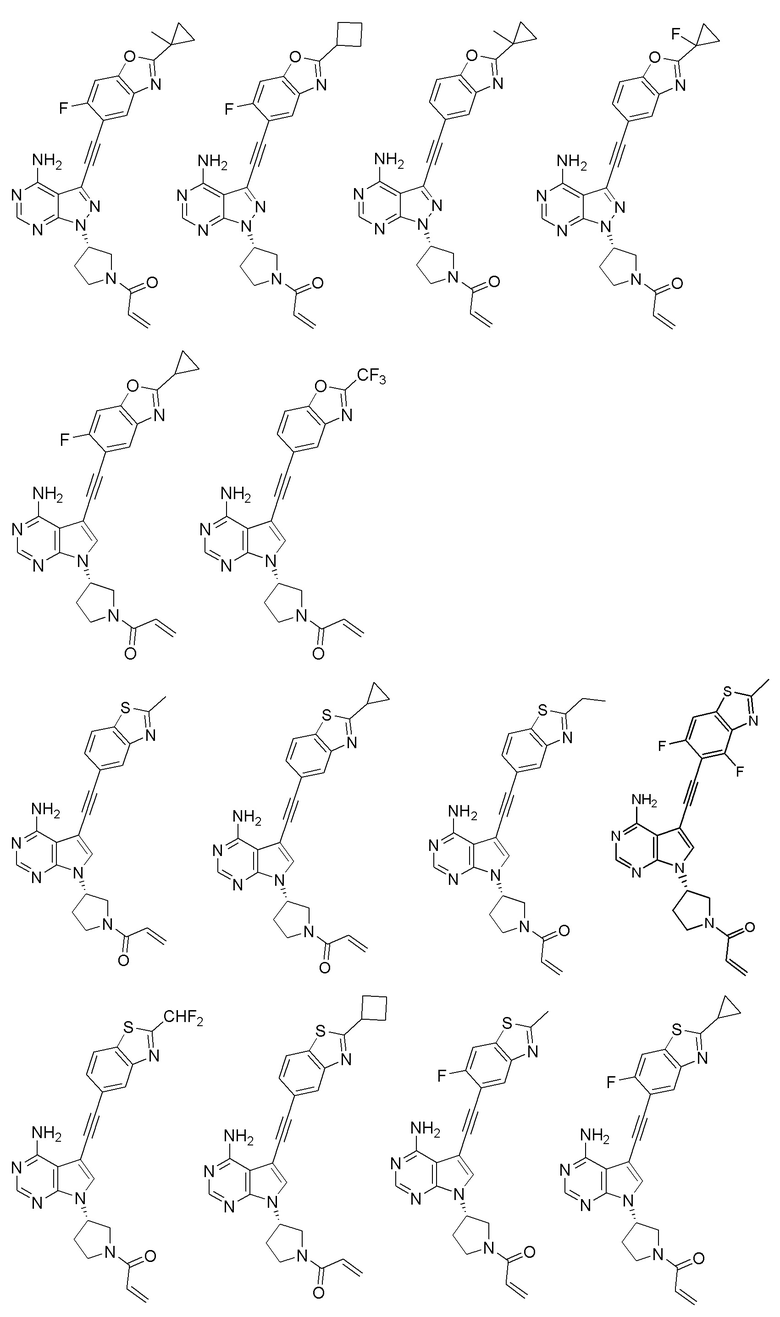
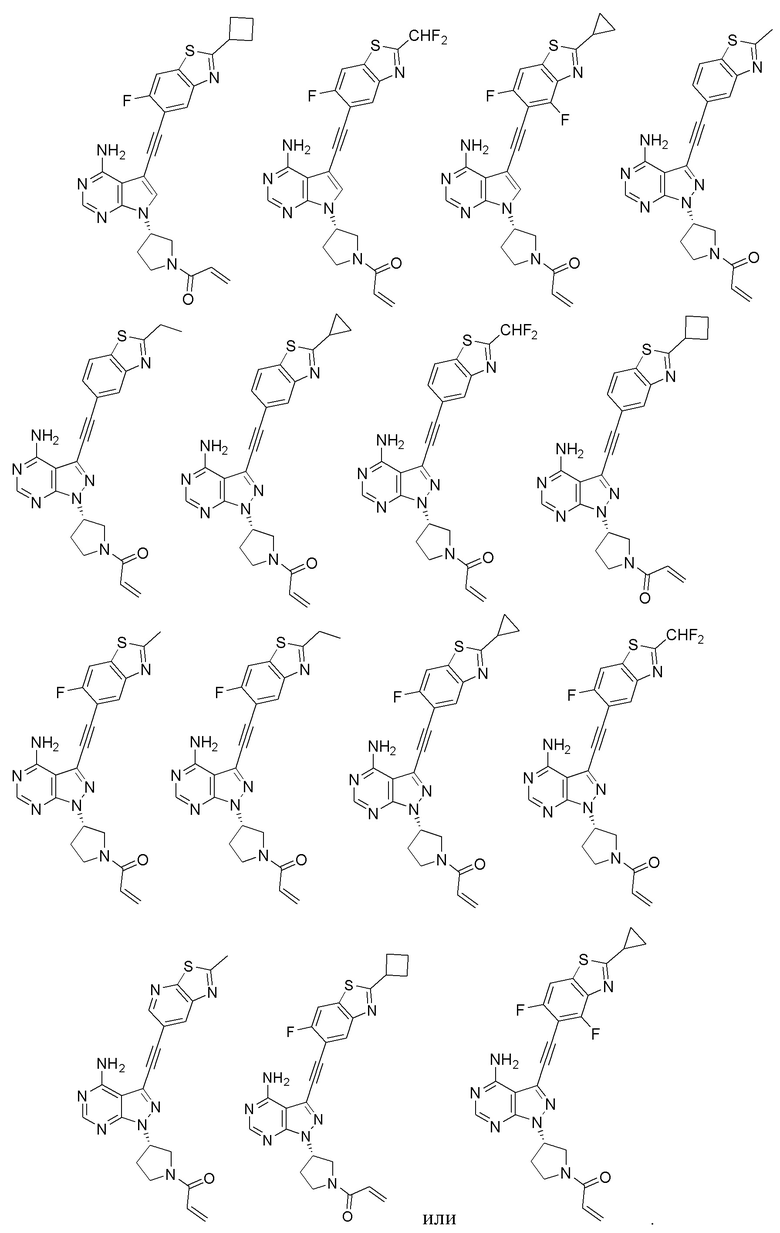
Вторым аспектом настоящего изобретения является способ получения соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли, включающий следующие стадии:
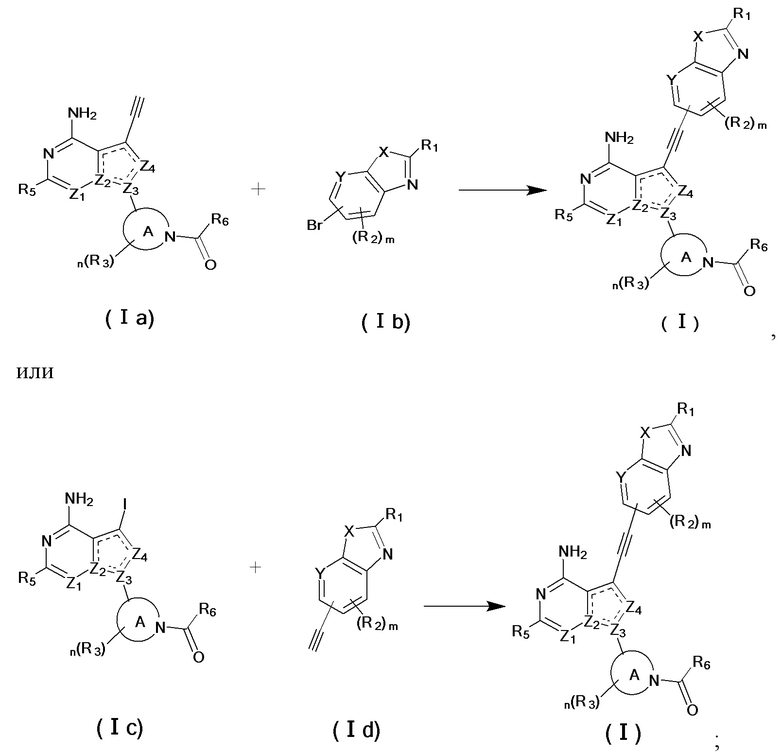 ,
,
где кольцо А, X, Y, Z1, Z2, Z3, Z4, R1, R2, R3, R5, R6, m и n соответствуют таковым в соединении формулы (I).
Третьим аспектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I), его стереоизомер или фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к применению соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли для получения лекарственного препарата, предназначенного для лечения пациента с опухолью, резистентной к ингибитору FGFR.
В предпочтительном варианте осуществления изобретения пациент с опухолью имеет мутации в FGFR V561, V565, N550, N540, V555, E566, K660 и/или V550.
В другом предпочтительном варианте осуществления изобретения пациент с опухолью имеет мутации FGFR2 V565F, V565I, V565L, V565M, N550K, N550H, E566A, E566G, K660M и/или K660Q.
В еще одном предпочтительном варианте осуществления изобретения пациент с опухолью имеет мутации FGFR3 V555M/L и/или N540K.
Настоящее изобретение также относится к использованию соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли для профилактики или лечения заболевания или состояния, опосредованного киназой FGFR.
Настоящее изобретение также относится к использованию соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли для профилактики или лечения опухоли или злокачественного новообразования, опосредованного киназой FGFR.
В предпочтительном варианте осуществления изобретения опухоль или злокачественное новообразование представляет собой рак мочевого пузыря, рак молочной железы, рак шейки матки, колоректальный рак, рак эндометрия, рак желудка, рак головы и шеи, карциному почки, рак печени, рак легких, рак яичников, рак простаты, рак пищевода, рак желчного пузыря, рак поджелудочной железы, рак щитовидной железы, рак кожи, лейкоз, множественную миелому, хроническую лимфоцитарную лимфому, T-клеточный лейкоз взрослых, B-клеточную лимфому, острый миелоцитарный лейкоз, лимфому Ходжкина или неходжкинскую лимфому, макроглобулинемию Вальденстрема, волосатоклеточную лимфому, клеточную лимфому, лимфому Беркитта, глиобластому, меланому или рабдомиосаркому.
Настоящее изобретение также относится к применению соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли для получения лекарственного препарата, предназначенного для лечения миелопролиферативного заболевания, заболевания клеток костной или хрящевой ткани и гипофосфатемии.
В настоящем изобретении миелопролиферативное заболевание представляет собой эритроцитоз, первичный тромбоцитоз или первичный миелофиброз; заболевание клеток костной или хрящевой ткани представляет собой дисплазию, ахондроплазию, карликовость, танатофорную дисплазию (ТД), синдром Аперта, синдром Крузона, синдром Джексона-Вейсса, синдром кутисовой извилины Бира-Стивенсона, синдром Пфайффера или синдром черепной мышечной атрофии; гипофосфатемия представляет собой X-сцепленный гипофосфатемический рахит, аутосомно-рецессивный гипофосфатемический рахит, аутосомно-доминантный гипофосфатемический рахит или неопластическую остеомаляцию.
Настоящее изобретение также относится к использованию соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли, или вышеуказанной фармацевтической композиции для лечения заболеваний, связанных с аберрантной экспрессией и мутацией рецептора FGFR2 или FGFR3, или аберрантной экспрессией и активностью соответствующего лиганда как селективного ингибитора FGFR2 и/или FGFR3.
Настоящее изобретение также относится к методу лечения пациента с опухолью, резистентной к ингибитору FGFR, который состоит во введении пациенту, нуждающемуся в таком лечении, соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли.
Настоящее изобретение также относится к методу лечения пациента с опухолью, несущей мутации FGFR2 V565F, V565I, V565L, V565M, N550K, N550H, E566A, E566G, K660M и/или K660Q, который состоит во введении пациенту, нуждающемуся в таком лечении, соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
После проведения всесторонних и углубленных исследований авторы настоящего изобретения впервые разработали FGFR и ингибитор его мутаций, способ его получения и его применение. Ряд соединений по настоящему изобретению обладают высокой активностью в отношении мутантных форм FGFR, в особенности в отношении FGFR с гейткиперными мутациями, и в особенности в отношении мутаций FGFR3 V555M, FGFR2 V565I, FGFR2 V565F, FGFR2 V565L и FGFR2 N550K.
Подробное описание: если не определено или не указано иное, следующие термины, используемые в описании и формуле изобретения, имеют следующие значения.
«Алкил» относится к линейным или разветвленным насыщенным алифатическим алкильным группам, предпочтительно к линейным или разветвленным алкильным группам, содержащим от 1 до 10, от 1 до 6 или от 1 до 4 атомов углерода, включая, помимо прочего, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил или их различные разветвленные изомеры, и тому подобное. «C1-10 алкил» относится к линейным алкильным или разветвленным алкильным группам, содержащим от 1 до 10 атомов углерода, «C1-4 алкил» относится к линейным алкильным или разветвленным алкильным группам, содержащим от 1 до 4 атомов углерода, «C0-8 алкил» относится к линейным алкильным или разветвленным алкильным группам, содержащим от 0 до 8 атомов углерода, а «C0-4 алкил» относится к линейным алкильным или разветвленным алкильным группам, содержащим от 0 до 4 атомов углерода.
Алкил может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Циклоалкил» или «карбоциклил» относятся к моноциклическому или полициклическому углеводородному заместителю, который является насыщенным или частично ненасыщенным. Под частично ненасыщенным циклическим углеводородом понимается циклический углеводород, который может содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одного из колец нет полностью сопряженной π-электронной системы; циклоалкил может быть моноциклическим циклоалкилом или полициклическим циклоалкилом и предпочтительно является циклоалкилом, содержащим от 3 до 12, от 3 до 8 или от 3 до 6 атомов углерода. Например, «C3-12 циклоалкил» относится к циклоалкилу, содержащему от 3 до 12 атомов углерода, а «C3-6 циклоалкил» относится к циклоалкилу, содержащему от 3 до 6 атомов углерода.
Моноциклический циклоалкил включает, помимо прочего, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное;
полициклический циклоалкил включает спироциклоалкил, конденсированный циклоалкил и мостиковый циклоалкил. «Спироциклоалкил» относится к полициклической группе, в которой атом углерода (называемый спироатомом) является общим для моноциклических колец, причем эти кольца могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одной из них нет полностью сопряженной π-электронной системы. В соответствии с числом спироатомов, общих для колец, спироциклоалкил может быть моноспироциклоалкилом, биспироциклоалкилом или полиспироциклоалкилом, включая, помимо прочего:
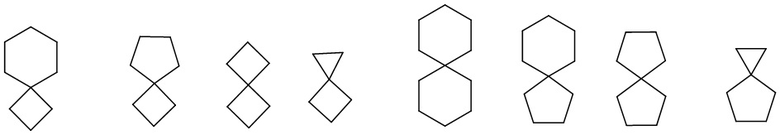 .
.
«Конденсированный циклоалкил» относится к полностью углеродной полициклической группе, в которой каждое кольцо имеет два общих соседних атома углерода с другими кольцами в системе, причем одно или несколько колец могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одной из них нет полностью сопряженной π-электронной системы. В зависимости от количества образованных колец конденсированный циклоалкил может быть бициклическим, трициклическим, тетрациклическим или полициклическим, включая, помимо прочего:
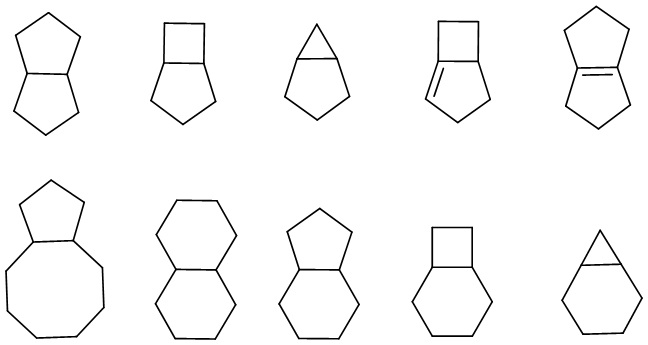 .
.
«Мостиковый циклоалкил» относится к полностью углеродной полициклической группе, в которой любые два кольца имеют два атома углерода, которые не связаны непосредственно друг с другом, причем эти кольца могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одной из них нет полностью сопряженной π-электронной системы. В зависимости от количества образованных колец мостиковый циклоалкил может быть бициклическим, трициклическим, тетрациклическим или полициклическим, включая, помимо прочего:
 .
.
Циклоалкильное кольцо может быть конденсировано с арильным, гетероарильным или гетероциклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой циклоалкил, который включает, помимо прочего, инданил, тетрагидронафтил, бензоциклогептил и тому подобное.
Циклоалкил может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Гетероциклил» или «гетероцикл» относятся к моноциклическому или полициклическому углеводородному заместителю, который является насыщенным или частично ненасыщенным. Под частично ненасыщенным циклическим углеводородом понимается циклический углеводород, который может содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одного из колец нет полностью сопряженной π-электронной системы; в гетероциклиле один или несколько (предпочтительно 1, 2, 3 или 4) атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, S(O)(=NH) и S (O)r (где r представляет собой целое число 0, 1 или 2), исключая входящие в кольцо группы -O-O-, -O-S- или -S-S-, а остальные атомы кольца представляют собой атомы углерода. Предпочтительно, гетероциклил содержит от 3 до 12, от 3 до 8 или от 3 до 6 атомов кольца; например, «3-6-членный гетероциклил» относится к циклической группе, содержащей от 3 до 6 атомов кольца, «4-6-членный гетероциклил» относится к циклической группе, содержащей от 4 до 6 атомов кольца, «4-10-членный гетероциклил» относится к циклической группе, содержащей от 4 до 10 атомов кольца, а «3-12-членный гетероциклил» относится к циклической группе, содержащей от 3 до 12 атомов кольца.
Моноциклический гетероциклил включает, помимо прочего, пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное.
Полициклический гетероциклил включает спирогетероциклил, конденсированный гетероциклил и мостиковый гетероциклил. «Спирогетероциклил» относится к полициклической гетероциклильной группе, в которой атом (называемый спироатомом) является общим для моноциклических колец, где один или несколько (предпочтительно 1, 2, 3 или 4) кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, S(O)(=NH) и S(O)r (где r представляет собой целое число 0, 1 или 2), а остальные атомы кольца являются атомами углерода. Эти кольца могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одной из них нет полностью сопряженной π-электронной системы. В соответствии с числом спироатомов, общих для колец, спирогетероциклил может быть моноспирогетероциклилом, биспирогетероциклилом или полиспирогетероциклилом, включая, помимо прочего:
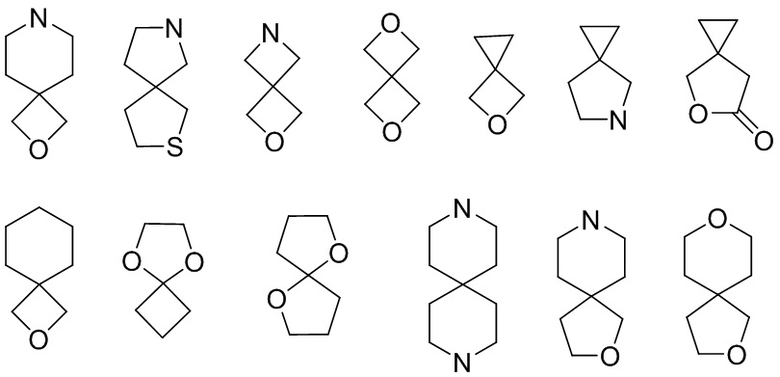 .
.
«Конденсированный гетероциклил» относится к полициклической гетероциклильной группе, в которой каждое кольцо имеет два общих соседних атома с другими кольцами в системе, причем одно или несколько (предпочтительно 1, 2, 3 или 4) колец могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одного из них нет полностью сопряженной π-электронной системы, и при этом один или несколько (предпочтительно 1, 2, 3 или 4) атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, S(O)(=NH) и S(O)r (где r представляет собой целое число 0, 1 или 2), а остальные атомы кольца представляют собой атомы углерода. В зависимости от количества образованных колец конденсированный гетероциклоакил может быть бициклическим, трициклическим, тетрациклическим или полициклическим, включая, помимо прочего:
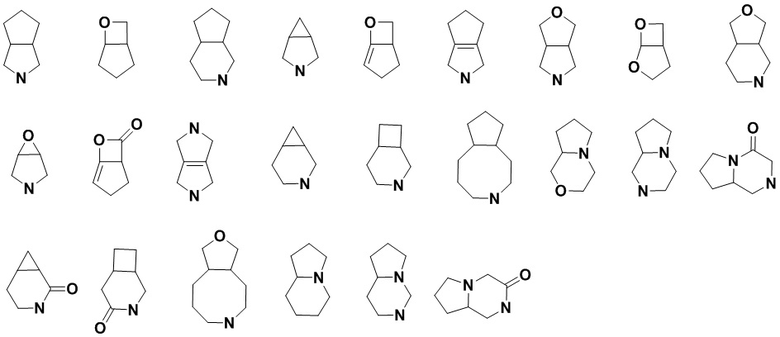 .
.
«Мостиковый гетероциклил» относится к полициклической гетероциклильной группе, в которой любые два кольца имеют два атома углерода, которые не связаны непосредственно друг с другом, причем эти кольца могут содержать одну или несколько (предпочтительно 1, 2 или 3) двойных связей, но ни у одной из них нет полностью сопряженной π-электронной системы, и при этом один или несколько (предпочтительно 1, 2, 3 или 4) атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода, S(O)(=NH) и S(O)r (где r представляет собой целое число 0, 1 или 2), а остальные атомы кольца представляют собой атомы углерода. В зависимости от количества образованных колец связанный гетероциклил может быть бициклическим, трициклическим, тетрациклическим или полициклическим, включая, помимо прочего:
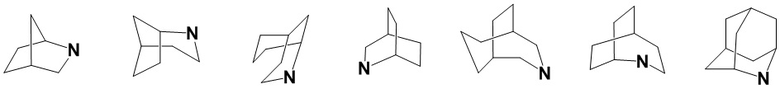 .
.
Гетероциклильное кольцо может быть конденсировано с арильным, гетероарильным или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой гетероциклил, включая, помимо прочего:
 .
.
Гетероциклил может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Арил» или «ароматическое кольцо» означает полностью углеродную моноциклическую группу или конденсированную полициклическую группу (т. е. кольца, которые имеют пару соседних атомов углерода) и полициклическую группу, имеющую сопряженную π-электронную систему (т. е. кольца с соседними парами атомов углерода), и предпочтительно представляет собой полностью углеродный арил, содержащий от 5 до 10 атомов или от 5 до 8 атомов углерода. Например, «C5-10 арил» относится к полностью углеродному арилу, содержащему от 5 до 10 атомов углерода, а «C5-8 арил» относится к полностью углеродному арилу, содержащему от 5 до 8 атомов углерода. Он включает, помимо прочего, фенил и нафтил. Арильное кольцо может быть конденсировано с гетероарильным, гетероциклильным или циклоалкильным кольцом, причем кольцо, присоединенное к исходной структуре, представляет собой арильное кольцо, включая, помимо прочего:
 .
.
«Арил» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Гетероарил» относится к гетероароматической системе, содержащей один или несколько (предпочтительно 1, 2, 3 или 4) гетероатомов, включая азот, кислород и S(O)r (где r представляет собой целое число 0, 1 или 2), и предпочтительно представляет собой гетероароматическую систему, содержащую от 5 до 10, от 5 до 8 или от 5 до 6 атомов кольца. Например, «5-8-членный гетероарил» относится к гетероароматической системе, содержащей от 5 до 8 атомов кольца, а «5-10-членный гетероарил» относится к гетероароматической системе, содержащей от 5 до 10 атомов кольца. Гетероарил включает, помимо прочего, фурил, тиофенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразоил и т. д. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклильным или циклоалкильным кольцом, причем кольцо, присоединенное к исходной структуре, представляет собой гетероарильное кольцо, включая, помимо прочего:
 .
.
«Гетероарил» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Алкенил» относится к алкилу, определенному выше, который содержит не менее двух атомов углерода и не менее одной двойной связи углерод-углерод, и предпочтительно представляет собой линейный или разветвленный алкенил, содержащий от 2 до 10 или от 2 до 4 атомов углерода. Например, «C2-10 алкенил» относится к линейному или разветвленному алкенилу, содержащему от 2 до 10 атомов углерода, а «C2-4 алкенил» относится к линейному или разветвленному алкенилу, содержащему от 2 до 4 атомов углерода. Алкенил включает, помимо прочего, винил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и тому подобное.
«Алкенил» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Алкинил» относится к алкилу, определенному выше, который содержит не менее двух атомов углерода и не менее одной тройной связи углерод-углерод, и предпочтительно представляет собой линейный или разветвленный алкинил, содержащий от 2 до 10 или от 2 до 4 атомов углерода. Например, «C2-10 алкинил» относится к линейному или разветвленному алкинилу, содержащему от 2 до 10 атомов углерода, а «C2-4 алкинил» относится к линейному или разветвленному алкинилу, содержащему от 2 до 4 атомов углерода. Алкинил включает, помимо прочего, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил и тому подобное.
«Алкинил» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Алкокси» относится к -O-алкилу, где алкил соответствует вышеприведенному определению. Например, «C1-10 алкокси» относится к алкоксигруппе, содержащей от 1 до 10 атомов углерода, а «C1-4 алкокси» относится к алкоксигруппе, содержащей от 1 до 4 атомов углерода. Алкокси включает, помимо прочего, метокси, этокси, пропокси, бутокси и тому подобное.
«Алкокси» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Циклоалкилокси» относится к -O-циклоалкилу, где циклоалкил соответствует вышеприведенному определению. Например, «C3-12 циклоалкилокси» относится к циклоалкилоксигруппе, содержащей от 3 до 12 атомов углерода, а «C3-6 циклоалкилокси» относится к циклоалкилоксигруппе, содержащей от 3 до 6 атомов углерода. Циклоалкилокси включает, помимо прочего, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т. д.
«Циклоалкилокси» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«Гетероциклилокси» относится к -O-гетероциклилу, где гетероциклил соответствует вышеприведенному определению, а гетероциклилокси включает, помимо прочего, азациклобутилокси, оксациклобутилокси, азациклопентилокси, азот, оксацилогексилокси и т. д.
«Гетероциклилокси» может быть опционально замещенным или незамещенным, и в случае если он является замещенным, заместители предпочтительно представляют собой одну или более (предпочтительно 1, 2, 3 или 4) групп, независимо выбранных из группы, состоящей из дейтерия, галогена, циано, нитро, азидо, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, C1-10 галоалкила, C1-10 дейтероалкила, C3-12 циклоалкила, 3-12-членного гетероциклила, C5-10 арила, 5-10-членного гетероарила, =O, -C0-8 алкила-SF5, -C0-8 алкила-S(O)rR8, -C0-8 алкила-O-R9, -C0-8 алкила-C(O)OR9, -C0-8 алкила-C(O)R10, -C0-8 алкила-O-C(O)R10, -C0-8 алкила-NR11R12, -C0-8 алкила-C(=NR11)R10, -C0-8 алкила-N(R11)-C(=NR12)R10, -C0-8 алкила-C(O)NR11R12 и -C0-8 алкила-N(R11)-C(O)R10.
«C1-10 алканоил» относится к одновалентной атомной группе, которая образуется после удаления гидроксигруппы из C1-10 алкильной кислоты, и также обычно обозначается как «C0-9-C(O)-», где «C0-9» относится к C0-9 алкилу. Например, «C1-C(O)-» относится к ацетилу, «C2-C(O)-» относится к пропионилу, а «C3-C(O)-» относится к бутирилу или изобутирилу.
«-C0-8 алкил-S(O)rR8» означает, что атом серы в -S(O)rR8 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8 алкил-O-R9» означает, что атом кислорода в -O-R9 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8 алкил-C(O)OR9» означает, что карбонил в -C(O)OR9 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«C0-8 алкил-C(O)R10» означает, что карбонил в -C(O)R10 присоединен к C0-8 алкилу, где C0 алкил означает, что здесь присутствует 0 атомов углерода, а C1-8 алкил соответствует вышеприведенному определению.
«-C0-8-O-C(O)R10» означает, что атом кислорода в -O-C(O)R10 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8-NR11R12» означает, что атом азота в -NR11R12 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8-C(=NR11)R10» означает, что атом углерода в -C(=NR11)R10 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8-N(R11)-C(=NR12)R10» означает, что атом азота в -N(R11)-C(=NR12)R10 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8-C(O)NR11R12» означает, что карбонил в -C(O)NR11R12 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«-C0-8-N(R11)-C(O)R10» означает, что атом азота в -N(R11)-C(O)R10 присоединен к C0-8 алкилу, где C0-8 алкил соответствует вышеприведенному определению.
«C1-10 галоалкил» относится к алкильной группе, содержащей от 1 до 10 атомов углерода, в которой алкильные атомы водорода опционально замещены атомом фтора, хлора, брома или йода, и включает, помимо прочего, дифторметил, дихлорметил, дибромметил, трифторметил, трихлорметил, трибромметил и т. д.
«C1-10 галоалкокси» относится к алкоксигруппе, содержащей от 1 до 10 атомов углерода, в которой алкильные атомы водорода опционально замещены атомом фтора, хлора, брома или йода, и включает, помимо прочего, дифторметокси, дихлорметокси, дибромметокси, трифторметокси, трихлорметокси, трибромометокси и тому подобное.
«C1-10 дейтероалкил» относится к алкильной группе, содержащей от 1 до 10 атомов углерода, в которой алкильные атомы водорода опционально замещены атомом дейтерия, и включает, помимо прочего, монодейтерометил, дидейтерометил, тридейтерометил и тому подобное.
«Галоген» относится к фтору, хлору, брому или йоду. «ПЭ» относится к петролейному эфиру. «ЭтАц» относится к этилацетату. «ДХМ» относится к дихлорметану.
«Опциональный» или «опционально» означает, что описанное далее событие или обстоятельство может происходить, но не обязательно произойдет, и что описание включает случаи, когда событие или обстоятельство происходит или не происходит, то есть случаи, когда замещение происходит или не происходит. Например, «гетероциклильная группа, опционально замещенная алкилом» означает, что алкил может присутствовать, но не обязательно присутствует, и что описание включает случаи, когда гетероциклильная группа замещена или не замещена алкилом.
Термин «замещенный» означает, что один или несколько атомов водорода в группе независимо замещены соответствующим числом заместителей. Само собой разумеется, что заместитель находится только в своем возможном химическом положении в согласии с теорией валентных химических связей, и специалисты в данной области без приложения чрезмерных усилий смогут определить (экспериментально или на основе теории), является замещение возможным или невозможным. Например, оно может быть нестабильным в случае, если аминогруппа или гидроксигруппа, имеющие свободный водород, связаны с атомом углерода, имеющим ненасыщенную связь (например, олефин).
«Стереоизомеры» относятся к изомерам, образующимся в результате различного пространственного расположения атомов в молекуле, и могут подразделяться на цис-транс изомеры и энантиомеры, а также на энантиомеры и диастереомеры. Стереоизомеры, образующиеся в результате вращения вокруг одинарных связей, называют конформационными стереоизомерами, а иногда ротамерами. Стереоизомеры, различающиеся длинами связей, углами между связями, внутримолекулярными двойными связями, кольцами и тому подобное, называют конфигурационными стереоизомерами; при этом конфигурационные стереоизомеры подразделяют на две категории. Изомеры, образующиеся в результате того, что вокруг двойной связи или одинарной связи при атоме углерода, участвующем в образовании кольца, невозможно свободное вращение, называют геометрическими изомерами, а также цис-транс изомерами; такие изомеры подразделяют на конфигурации Z и E. Например, цис-2-бутен и транс-2-бутен представляют собой пару геометрических изомеров; при этом стереоизомеры, различающиеся по оптическому вращению вследствие отсутствия антиосевой симметрии в молекуле, называют оптическими изомерами и подразделяют на конфигурации R и S. В настоящем изобретении следует понимать, что термин «стереоизомер» включает один или несколько вышеназванных энантиомеров, конфигурационных изомеров и конформационных изомеров, если специально не указано иное.
Термин «фармацевтически приемлемая соль», используемый в данном документе, относится к фармацевтически приемлемым аддитивным солям с кислотой или основанием, включая соли с неорганической или органической кислотой, которые могут быть получены методами, известными специалистам в данной области.
«Фармацевтическая композиция» относится к смеси, содержащей одно или несколько соединений, описанных в настоящем документе, или его физиологически/фармацевтически приемлемой соли, или препарату-предшественнику, а также другим химическим компонентам, например физиологически/фармацевтически приемлемым носителям и вспомогательным веществам. Цель этой фармацевтической композиции состоит в том, чтобы облегчать введение препарата в организм, улучшая всасывание действующего вещества и тем самым способствуя проявлению его биологической активности.
Далее представлено подробное объяснение настоящего изобретения со ссылкой на примеры его осуществления, которые не предназначены для ограничения настоящего изобретения; при этом настоящее изобретение не ограничивается представленными примерами.
Структура соединения по настоящему изобретению определяется с помощью ядерного магнитного резонанса (ЯМР) и/или жидкостной хроматографии в комбинации с масс-спектрометрией (ЖХ-МС). Химические сдвиги ЯМР-спектров (δ) приведены в частях на миллион (ppm). Определение по ЯМР-спектру проводят с использованием установки ядерного магнитного резонанса Bruker AVANCE-400 с гексадейтеродиметилсульфоксидом (ДМСО-d6), тетрадейтерометанолом (CD3OD) и дейтерированным хлороформом (CDCl3) в качестве растворителей, и тетраметилсиланом (ТМС) в качестве внутреннего стандарта.
Определение с помощью ЖХ-МС проводят с использованием масс-спектрометра Agilent 6120. Определение с помощью ВЭЖХ проводят с использованием жидкостного хроматографа высокого давления Agilent 1200 DAD (хроматографическая колонка Sunfire C18 150 × 4,6 мм) и жидкостного хроматографа высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150 × 4,6 мм).
В качестве пластины для тонкослойной хроматографии (ТСХ) используется пластина с силикагелем Yantai Yellow Sea HSGF254 или Qingdao GF254. В спецификации пластины с силикагелем для ТСХ установлено значение 0,15-0,20 мм, а в спецификации для разделения и очистки методом ТСХ установлено значение 0,4-0,5 мм. В качестве носителя для колоночной хроматографии обычно используют силикагель Yantai Yellow Sea размером частиц 200-300 меш.
Исходные материалы в примерах осуществления настоящего изобретения известны и имеются в продаже или могут быть синтезированы методами, известными в данной области техники, или в соответствии с ними.
Если не указано иное, то все реакции по настоящему изобретению проводят в атмосфере сухого азота или аргона с непрерывным перемешиванием на магнитной мешалке, где растворитель представляет собой сухой растворитель, а температура реакции указана в градусах Цельсия (°C).
Получение промежуточных веществ
Промежуточное вещество 1: получение 2-циклопропил-5-этинилбензо[d]оксазола
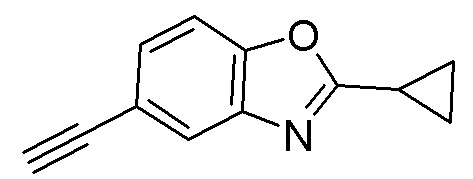
Стадия 1. Синтез N-(5-бром-2-гидроксифенил)циклопропанкарбоксамида
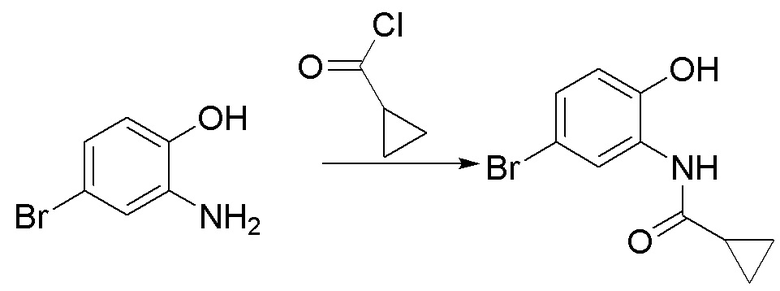
2-амино-4-бромфенол (2,0 г, 10,6 ммоль) растворяли в ДХМ (30 мл), после чего последовательно добавляли триэтиламин (1,29 г, 12,8 ммоль) и циклопропилкарбонилхлорид (1,11 г, 10,6 ммоль) при температуре 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч, разбавляли водным раствором бикарбоната натрия и экстрагировали с помощью ДХМ, органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением N-(5-бром-2-гидроксифенил)циклопропанкарбоксамида (1,34 г, выход: 49%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 256/258 [M + H]+.
Стадия 2. Синтез 5-бром-2-циклопропилбензо[d]оксазола
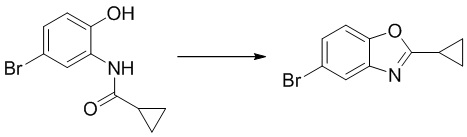
N-(5-бром-2-гидроксифенил)циклопропанкарбоксамид (1,04 г, 4,06 ммоль) растворяли в ацетонитриле (30 мл), после чего последовательно добавляли трифенилфосфин (4,26 г, 16,2 ммоль) и тетрахлорид углерода (1,25 г, 8,1 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 1 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 5-бром-2-циклопропилбензо[d]оксазола (548 мг, выход: 55%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 238/240 [M + H]+.
Стадия 3. Синтез 2-циклопропил-5-((триметилсилил)этинил)бензо[d]оксазола
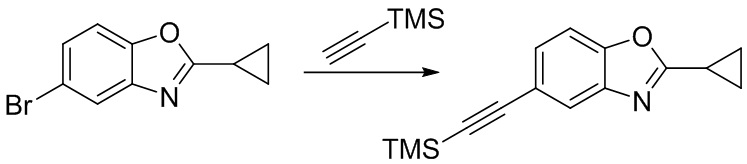
5-бром-2-циклопропилбензо[d]оксазол (200 мг, 0,84 ммоль) добавляли в ДМФ (3 мл), после чего последовательно добавляли триэтиламин (1 мл), йодид меди (16 мг, 0,08 ммоль), триметилсилилацетилен (825 мг, 8,4 ммоль) и тетракис(трифенилфосфин)палладий (49 мг, 0,04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 16 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 2-циклопропил-5-((триметилсилил)этинил)бензо[d]оксазола (204 мг, выход: 95%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 256 [M + H]+.
Стадия 4. Синтез 2-циклопропил-5-этинилбензо[d]оксазола
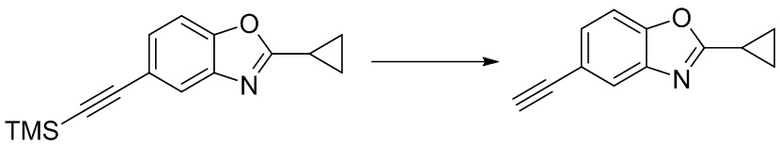
2-циклопропил-5-((триметилсилил)этинил)бензо[d]оксазол (204 мг, 0,8 ммоль) добавляли к метанолу (8 мл), а затем добавляли карбонат калия (1,1 г, 8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 2-циклопропил-5-этинилбензо[d]оксазола (112 мг, выход: 76%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 184 [M + H]+.
Промежуточные вещества 2-7 могут быть получены путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) промежуточного вещества 1:
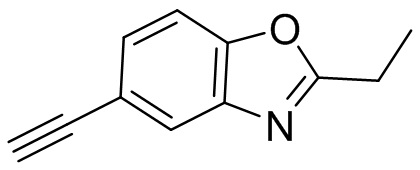
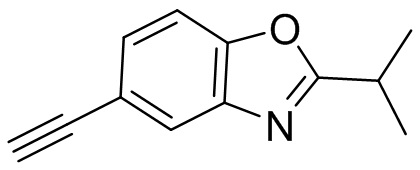
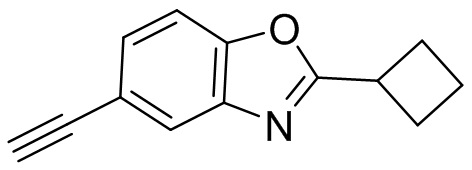
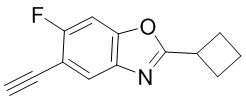
[d]оксазол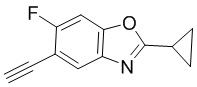
[d]оксазол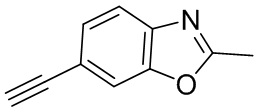
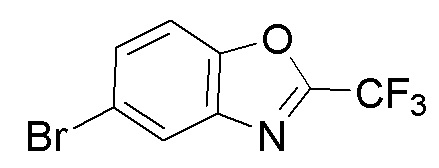
Промежуточное вещество 8: получение 5-бром-2-(трифторметил)бензо[d]оксазола
Стадия 1. Синтез N-(5-бром-2-гидроксифенил)-2,2,2-трифторацетамида
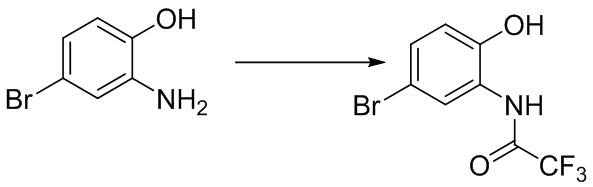
2-амино-4-бромфенол (2,0 г, 10,6 ммоль) растворяли в ДХМ (30 мл), после чего последовательно добавляли триэтиламин (1,29 г, 12,8 ммоль) и трифторуксусный ангидрид (2,23 г, 10,6 ммоль) при температуре 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли водным раствором бикарбоната натрия и экстрагировали с помощью ДХМ, и органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением N-(5-бром-2-гидроксифенил)-2,2,2-трифторацетамида (2,7 г, выход: 89%).
1H- ЯМР (400 МГц, ДМСО-d6): δ 10,6 (s, 1 ч), 10,3 (s, 1 ч), 7,55-7,54 (d, J = 4 Гц, 1 ч), 7,33-7,30 (dd, J = 4,8 Гц, 1 ч), 6,92-6,91 (d, J = 4 Гц, 1 ч).
Стадия 2. Синтез 5-бром-2-(трифторметил)бензо[d]оксазола
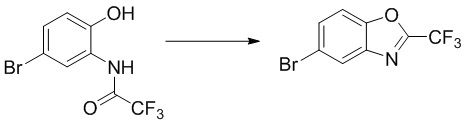
N-(5-бром-2-гидроксифенил)-2,2,2-трифторацетамид (500 мг, 1,76 ммоль) растворяли в толуоле (20 мл) и добавляли п-толуолсульфоновую кислоту (303 мг, 1,76 ммоль). Реакционную смесь перемешивали при температуре 120°C в течение 24 ч, после чего концентрировали в таком виде. Остаток разделяли с помощью колоночной хроматографии с получением 5-бром-2-(трифторметил)бензо[d]оксазола (200 мг, выход: 43%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,25-8,23 (d, J = 8 Гц, 1 ч), 7,92-7,88 (dd, J = 8,16 Гц, 1 ч), 7,79-7,77 (d, J = 8 Гц, 1 ч).
Промежуточное вещество 9: получение 5-этинил-N-метилбензо[d]оксазол-2-амина
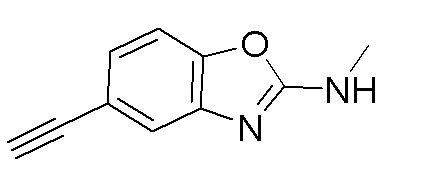
Стадия 1. Синтез 1-(5-бром-2-гидроксифенил)-3-метилтиомочевины
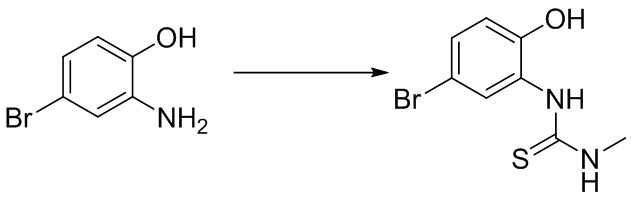
2-амино-4-бромфенол (1,0 г, 5,3 ммоль) добавляли в тетрагидрофуран (20 мл), после чего добавляли изотиоцианатометан (324 мг, 4,43 ммоль) при температуре 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 1-(5-бром-2-гидроксифенил)-3-метилтиомочевины (1,05 г, выход: 90%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 261/263 [M + H]+.
Стадия 2. Синтез 5-бром-N-метилбензо[d]оксазол-2-амина
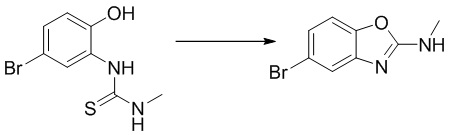
1-(5-бром-2-гидроксифенил)-3-метилтиомочевину (970 мг, 3,71 ммоль) добавляли в тетрагидрофуран (20 мл), после чего последовательно добавляли тетрабутиламмоний йодид (14 мг, 0,04 ммоль) и 30%-ный пероксид водорода (841 мг, 7,42 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли водным сульфитом натрия и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 5-бром-N-метилбензо[d]оксазол-2-амина (604 мг, выход: 72%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 227/229 [M + H]+.
Стадия 3. Синтез N-метил-5-((триметилсилил)этинил)бензо[d]оксазол-2-амина
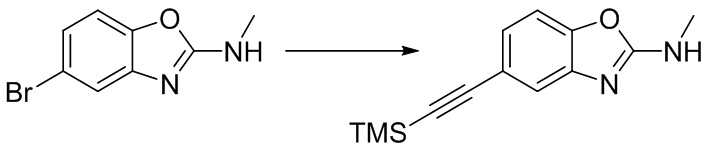
5-бром-N-метилбензо[d]оксазол-2-амин (600 мг, 2,64 ммоль) добавляли в ДМФ (5 мл), после чего последовательно добавляли триэтиламин (1,5 мл), йодид меди (50 мг, 0,26 ммоль), триметилсилилацетилен (2,6 г, 26,4 ммоль) и тетракис(трифенилфосфин)палладий (153 мг, 0,13 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 16 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением N-метил-5-((триметилсилил)этинил)бензо[d]оксазол-2-амина (230 мг, выход: 36%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 245 [M + H]+.
Стадия 4. Синтез 5-этинил-N-метилбензо[d]оксазол-2-амина
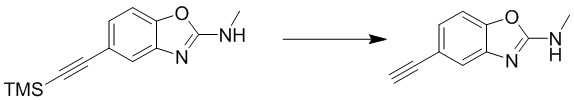
N-метил-5-((триметилсилил)этинил)бензо[d]оксазол-2-амин (100 мг, 0,4 ммоль) добавляли к метанолу (8 мл), а затем добавляли карбонат калия (566 мг, 4 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли водой и экстрагировали этилацетатом, после чего органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 5-этинил-N-метилбензо[d]оксазол-2-амина (54 мг, выход: 77%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 173 [M + H]+.
Промежуточные вещества 10-14 могут быть получены путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) промежуточного вещества 9:
вещества
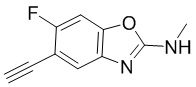
[d]оксазол-2-амин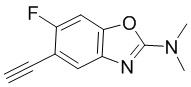
[d]оксазол-2-амин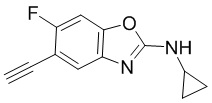
[d]оксазол-2-амин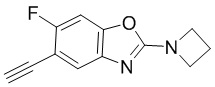
фторбензо[d]оксазол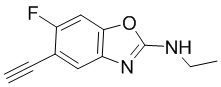
[d]оксазол-2-амин
Промежуточное вещество 15: получение 2-циклопропил-5-этинил-6-фторбензо[d]тиазола
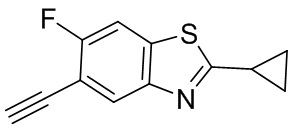
Стадия 1. Синтез N-(5-бром-2,4-дифторфенил)циклопропанкарбоксамида

5-бром-2,4-дифторанилин (1,04 г, 5,0 ммоль) растворяли в дихлорметане (20 мл), а затем добавляли циклопропилкарбонилхлорид (572 г, 5,5 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего концентрировали в таком виде. Остаток разделяли с помощью колоночной хроматографии с получением N-(5-бром-2,4-дифторфенил)циклопропанкарбоксамида (1,0 г, выход: 72%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 276/278 [M + H]+.
Стадия 2. Синтез N-(5-бром-2,4-дифторфенил)циклопропанкарботиамида

N-(5-бром-2,4-дифторфенил)циклопропанкарбоксамид (1,0 г, 3,6 ммоль) и реагент Лавессона (750 мг, 1,85 ммоль) растворяли в ацетонитриле (25 мл), после чего реакционную смесь нагревали до 85°C и перемешивали в течение ночи. По завершении реакции раствор N-(5-бром-2,4-дифторфенил)циклопропанкарботиамида использовали на следующей стадии в таком виде. Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 292/294 [M + H]+.
Стадия 3. Синтез 5-бром-2-циклопропил-6-фторбензо[d]тиазола

Трет-бутоксид натрия (1,7 г, 18 ммоль) добавляли к охлажденному раствору N-(5-бром-2,4-дифторфенил)циклопропанкарботиамида, после чего реакционную смесь нагревали до 50°C, перемешивали в течение 16 ч и концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 5-бром-2-циклопропил-6-фторбензо[d]тиазола (0,82 г, выход: 83%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 272/274 [M + H]+.
Стадия 4. Синтез 2-циклопропил-5-этинил-6-фторбензо[d]тиазола
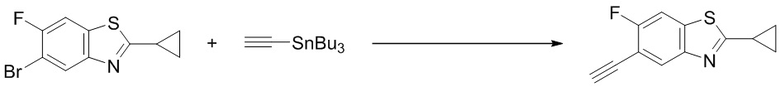
5-бром-2-циклопропил-6-фторбензо[d]тиазол (0,82 г, 3,0 ммоль) растворяли в ДМФ (15 мл), после чего добавляли трибутил(этинил)станнан (1,9 г, 6,0 ммоль) и тетракис(трифенилфосфин)палладий (173 мг, 0,15 ммоль). Реакционную смесь перемешивали в микроволновом реакторе при температуре 130°C в течение 0,5 ч, после чего концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением 2-циклопропил-5-этинил-6-фторбензо[d]тиазола (0,16 г, выход: 25%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 218 [M + H]+.
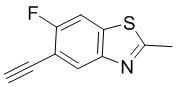
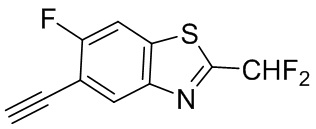
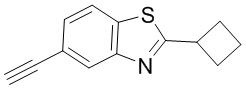
Промежуточные вещества 16-23 могут быть получены путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) промежуточного вещества 15:
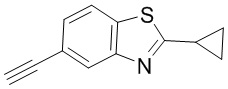
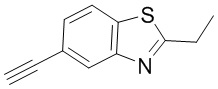
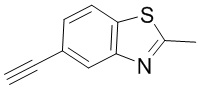
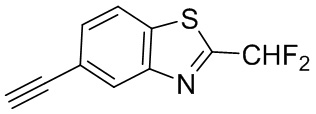
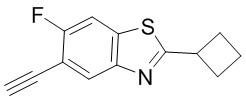
Промежуточное вещество 24: получение (S)-1-(3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
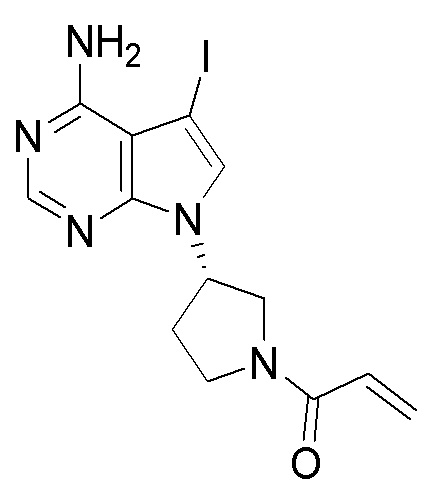
Стадия 1. Синтез трет-бутил (S)-3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-карбоксилата
5-йод-7H-пирроло[2,3-d]пиримидин-4-амин (520 мг, 2 ммоль), трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилат (561 мг, 3 ммоль) и трифенилфосфин (786 мг, 3 ммоль) растворяли в тетрагидрофуране (10 мл), а затем добавляли диизопропилазодиформиат (0,6 мл, 3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали до сухого остатка, и остаток разделяли с помощью колоночной хроматографии с получением трет-бутил (S)-3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин
-7-ил)пирролидин-1-карбоксилата (300 мг, выход: 35%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 430 [M + H]+.
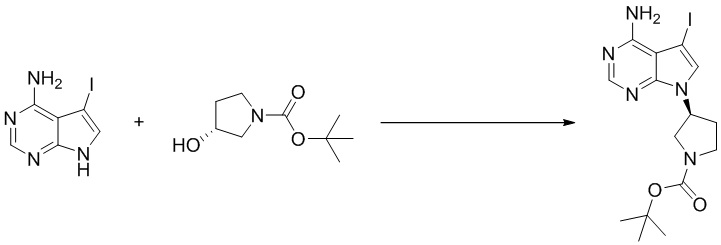
Стадия 2. Синтез (S)-5-йод-7-(пирролидин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
Трет-бутил (S)-3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-карбоксилат (300 мг, 0,69 ммоль) растворяли в дихлорметане (10 мл), а затем добавляли раствор HCl / диоксана (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После завершения реакции реакционную смесь концентрировали до сухого остатка с получением (S)-5-йод-7-(пирролидин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (230 мг, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 330 [M + H]+.
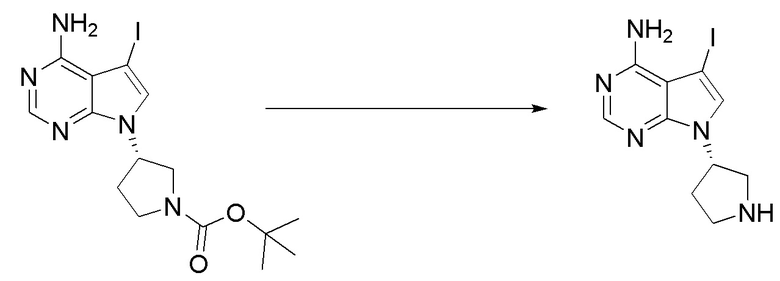
Стадия 3. Синтез (S)-1-(3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
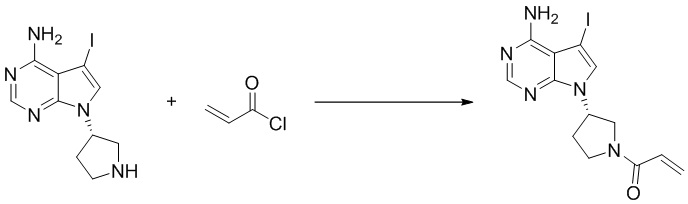
(S)-5-йод-7-(пирролидин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амин (230 мг, 0,70 ммоль) растворяли в тетрагидрофуране (4 мл) и насыщенном водном растворе бикарбоната натрия (1 мл), после чего смесь охлаждали до 0°C и добавляли акрилоилхлорид (65 мг, 0,7 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 0,5 ч. После завершения реакции реакционную смесь разделяли с помощью обращенно-фазовой хроматографии с получением (S)-1-(3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она (50 мг, выход: 18%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 384 [M + H]+.
Промежуточное вещество 25: получение (S)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-ил)проп-2-ен-1-она
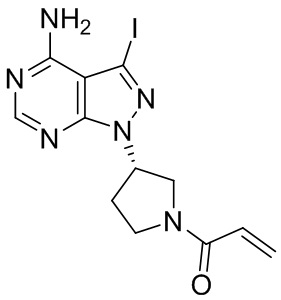
Стадия 1. Синтез трет-бутил (S)-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-карбоксилата
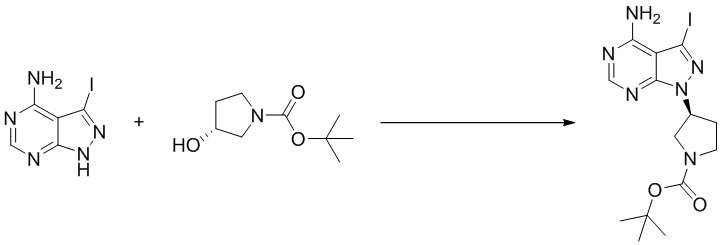
3-йод-1H-пиразоло[3,4-d]пиримидин-4-амин (1000 мг, 3,82 ммоль), трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилат (1070 мг, 5,73 ммоль) и трифенилфосфин (1500 мг, 5,73 ммоль) растворяли в тетрагидрофуране (100 мл), а затем добавляли диизопропилазодиформиат (1150 мл, 5,73 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали до сухого остатка, и остаток разделяли с помощью колоночной хроматографии с получением трет-бутил (S)-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин -1-ил)пирролидин-1-карбоксилата (1100 мг, выход: 67%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 431 [M + H]+.
Стадия 2. Синтез (S)-3-йод-1-(пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амина
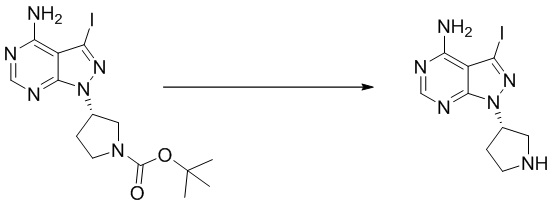
Трет-бутил (S)-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-карбоксилат (2000 мг, 4,65 ммоль) растворяли в дихлорметане (50 мл) и добавляли раствор HCl / диоксана (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции реакционную смесь концентрировали до сухого остатка с получением (S)-3-йод-1-(пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амина (1540 мг, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 331 [M + H]+.
Стадия 3. Синтез (S)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-ил)проп-2-ен-1-она
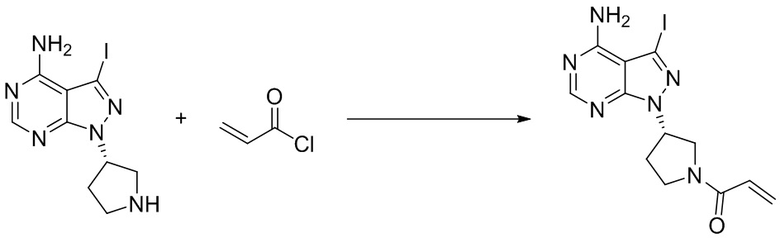
(S)-3-йод-1-(пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амин (1,54 мг, 4,65 ммоль) растворяли в тетрагидрофуране (20 мл) и насыщенном водном растворе бикарбоната натрия (5 мл), после чего смесь охлаждали до 0°C и добавляли акрилоилхлорид (473 мг, 5,11 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 1 ч. После завершения реакции реакционную смесь разделяли с помощью обращенно-фазовой хроматографии с получением (S)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-ил)проп-2-ен-1-она (500 мг, выход: 28%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 385 [M + H]+.
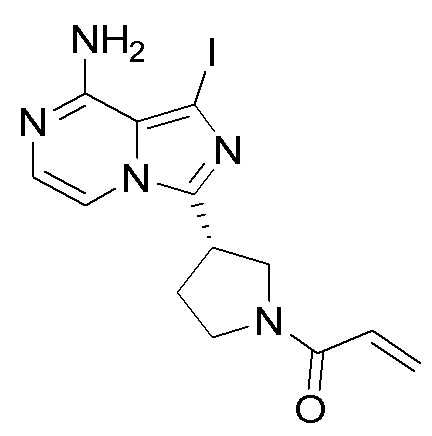
Промежуточное вещество 26: получение (S)-1-(3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-ил)проп-2-ен-1-она
Стадия 1. Синтез бензил (S)-3-(((3-хлорпиразин-2-ил)метил)карбамоил)пирролидин-1-карбоксилата
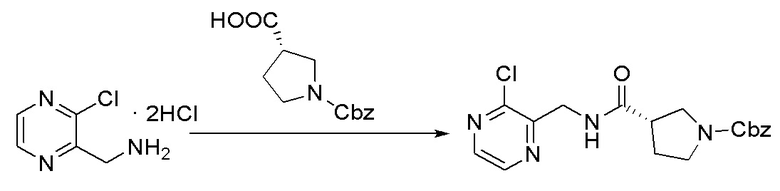
3-хлорпиразин-2-метанамин дигидрохлорид (4,45 г, 20,55 ммоль), (S)-1-((бензилокси)карбонил)пирролидин-3-карбоновая кислота (5,12 г, 20,55 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (8,2 г, 21,57 ммоль) и триэтиламин (8,3 г, 82,20 ммоль) растворяли в безводном дихлорметане (100 мл), после чего реакционную смесь перемешивали в течение ночи при комнатной температуре. Для остановки реакции добавляли насыщенный раствор бикарбоната натрия (50 мл), и полученную реакционную смесь экстрагировали три раза дихлорметаном (50 мл). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного бензил (S)-3-(((3-хлорпиразин-2-ил)метил)карбамоил)пирролидин-1-карбоксилата (8 г, выход: 95%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 375 [M + H]+.
Стадия 2. Синтез бензил (S)-3-(8-хлоримидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата
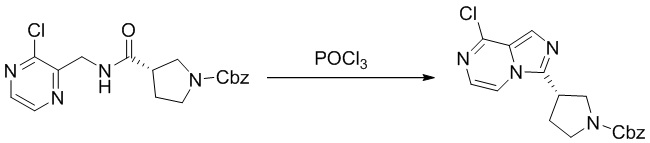
Бензил (S)-3-(((3-хлорпиразин-2-ил)метил)карбамоил)пирролидин-1-карбоксилат (8 г, 21,4 ммоль) растворяли в безводном ацетонитриле (100 мл), после чего по каплям добавляли оксихлорид фосфора (16,4 г, 107,0 ммоль) на ледяной бане. Реакционную смесь медленно нагревали до 80°C и перемешивали в течение 5 ч. Реакционную смесь охлаждали, затем по каплям добавляли насыщенный водный раствор бикарбоната натрия для остановки реакции (следя за тем, чтобы температура в реакционном сосуде не поднималась выше 5°C), и полученную реакционную смесь три раза экстрагировали этилацетатом (60 мл). Органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали над сульфатом натрия, фильтровали и концентрировали, а остаток разделяли с помощью колоночной хроматографии с получением бензил (S)-3-(8-хлоримидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата (3,8 г, выход: 50%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 357 [M + H]+.
Стадия 3. Синтез бензил (S)-3-(8-хлор-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата

Бензил (S)-3-(8-хлоримидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилат (0,65 г, 1,82 ммоль) и N-йодсукцинимид (1,43 г, 6,37 ммоль) растворяли в ДМФ (10 мл), и реакционную смесь перемешивали в течение ночи при комнатной температуре. После этого добавляли деионизованную воду, и полученную реакционную смесь два раза экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл), высушивали над сульфатом натрия, фильтровали и концентрировали, а остаток разделяли с помощью колоночной хроматографии с получением бензил (S)-3-(8-хлор-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата (0,76 г, выход: 86%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 483 [M + H]+.
Стадия 4. Синтез бензил (S)-3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата
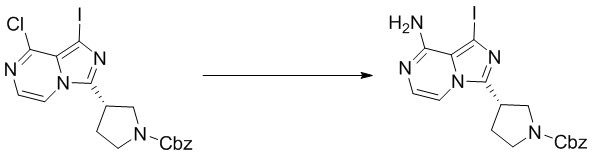
Бензил (S)-3-(8-хлор-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилат (0,76 г, 1,57 ммоль) растворяли в диоксане (10 мл), после чего в герметично закрытую пробирку добавляли концентрированный раствор аммиака (20 мл). Реакционную смесь нагревали до 80°C, перемешивали в течение ночи и затем концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением бензил (S)-3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилата (0,64 г, выход: 88%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 464 [M + H]+.
Стадия 5. Синтез (S)-1-йод-3-(пирролидин-3-ил)имидазо[1,5-а]пиразин-8-амина
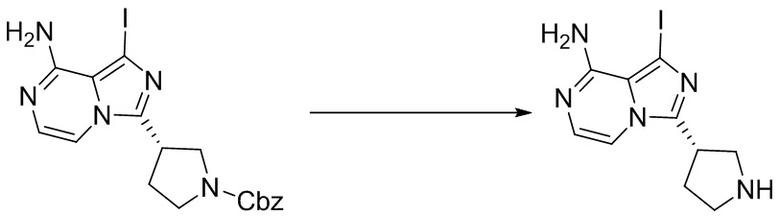
Бензил (S)-3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-карбоксилат (0,64 г, 1,38 ммоль) растворяли в дихлорметане (15 мл), затем смесь охлаждали до 0°C и по каплям добавляли триметилсилилйодид (0,7 г, 2,76 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 2 ч. Затем добавляли насыщенный раствор тиосульфата натрия для остановки реакции, и полученную реакционную смесь три раза экстрагировали дихлорметаном. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали, а остаток разделяли с помощью колоночной хроматографии с получением (S)-1-йод-3-(пирролидин-3-ил)имидазо[1,5-а]пиразин-8-амина (95 мг, выход: 21%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 330 [M + H]+.
Стадия 6. Синтез (S)-1-(3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-ил)проп-2-ен-1-она
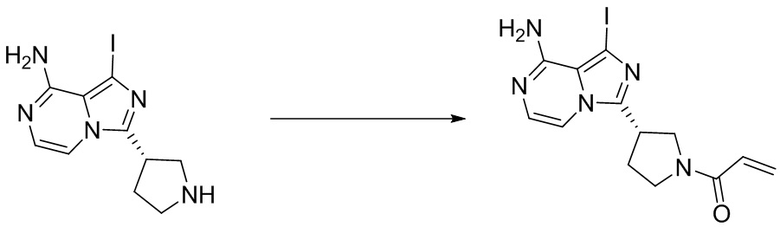
(S)-1-йод-3-(пирролидин-3-ил)имидазо[1,5-а]пиразин-8-амин (95 мг, 0,29 ммоль) растворяли в тетрагидрофуране (4 мл) и насыщенном водном растворе бикарбоната натрия (2 мл), после чего добавляли акрилоилхлорид (31 мг, 0,35 ммоль) на ледяной бане. Реакционную смесь выдерживали 30 минут на ледяной бане, после чего в таком виде разделяли с помощью обращенно-фазовой хроматографии, и полученную водную фазу лиофизировали с получением (S)-1-(3-(8-амино-1-йодимидазо[1,5-а]пиразин-3-ил)пирролидин-1-ил)проп-2-ен-1-она (72 мг, выход: 65%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 384 [M + H]+.
Промежуточное вещество 27: получение 1-(3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
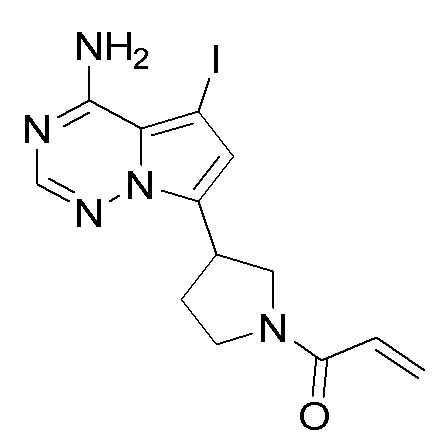
Стадия 1. Синтез 7-йодпирроло[2,1-f][1,2,4]триазин-4-амина

4-хлор-7-йодпирроло[2,1-f][1,2,4]триазин (1 г, 3,58 ммоль) растворяли в изопропаноле (10 мл), после чего в герметично закрытую пробирку добавляли концентрированный раствор аммиака (10 мл). Реакционную смесь нагревали до 70°C и перемешивали в течение 3 ч. После охлаждения реакционную смесь в таком виде концентрировали с получением неочищенного 7-йодпирроло[2,1-f][1,2,4]триазин-4-амина (1 г, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 261 [M + H]+.
Стадия 2. Синтез трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата
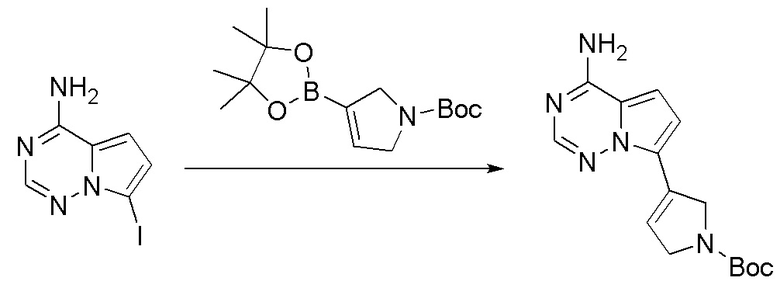
7-йодпирроло[2,1-f][1,2,4]триазин-4-амин (0,55 г, 2,11 ммоль), трет-бутил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат (0,75 г, 2,54 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид (172 мг, 0,21 ммоль) и карбонат калия (0,87 г, 6,33 ммоль) растворяли в диоксане (10 мл) и воде (2 мл), после чего реакционную смесь нагревали до 80°C, перемешивали в течение ночи и затем концентрировали. Остаток разделяли с помощью колоночной хроматографии с получением трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата (0,5 г, выход: 79%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 302 [M + H]+.
Стадия 3. Синтез трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилата
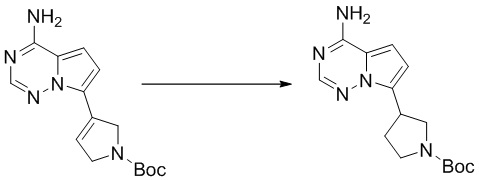
Трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат (0,5 г, 1,66 ммоль) растворяли в метаноле (10 мл), и к полученному раствору добавляли 10%-ный влажный палладий на угле (100 мг). Реакционную смесь нагревали до 50°C в атмосфере водорода, перемешивали в течение ночи и фильтровали, после чего фильтрат концентрировали. Остаток высушивали с получением трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилата (460 мг, выход: 91%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 304 [M + H]+.
Стадия 4. Синтез трет-бутил-3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилата
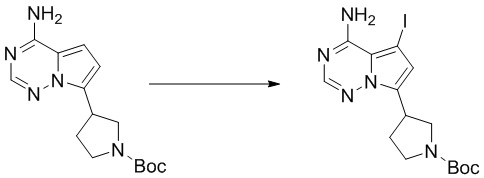
Трет-бутил-3-(4-аминопирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилат (0,46 г, 1,52 ммоль) и N-йодосукцинимид (1,02 г, 4,55 ммоль) растворяли в N,N-диметилформамиде (5 мл), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли деионизованную воду (50 мл) и высаживали большое количество твердого вещества. Смесь фильтровали, а отфильтрованный осадок дважды промывали ацетоном (10 мл) и высушивали с получением трет-бутил-3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилата (0,74 г, выход: 95%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 430 [M + H]+.
Стадия 5. Синтез 5-йод-7-(пирролидин-3-ил)пирроло[2,1-f][1,2,4]триазин-4-амина
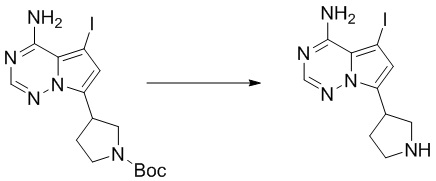
Трет-бутил-3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-карбоксилат (300 мг, 0,70 ммоль) растворяли в этилацетате (10 мл), а затем по каплям добавляли раствор 4 N HCl / диоксана (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч и концентрировали, а полученный остаток высушивали с получением 5-йод-7-(пирролидин-3-ил)пирроло[2,1-f][1,2,4]триазин-4-амина (300 мг, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 330 [M + H]+.
Стадия 6. Синтез 1-(3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
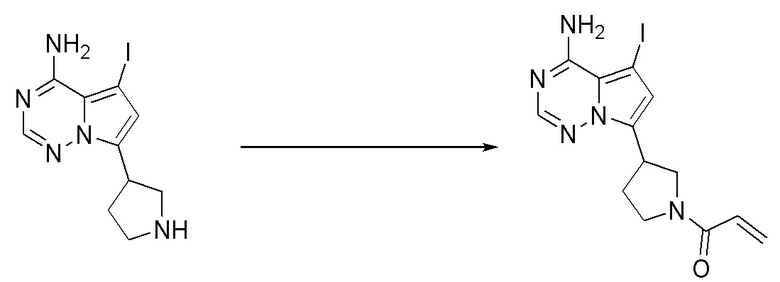
5-йод-7-(пирролидин-3-ил)пирроло[2,1-f][1,2,4]триазин-4-амин (300 мг, 0,91 ммоль) растворяли в тетрагидрофуране (10 мл) и насыщенном растворе бикарбоната натрия (5 мл), после чего смесь охлаждали до 0°C и по каплям добавляли акрилоилхлорид (90 мг, 0,99 ммоль). Реакционную смесь перемешивали в течение 30 минут, затем в таком виде разделяли с помощью обращенно-фазовой хроматографии, и полученную водную фазу лиофизировали с получением 1-(3-(4-амино-5-йодпирроло[2,1-f][1,2,4]триазин-7-ил)пирролидин-1-ил)проп-2-ен-1-она (300 мг, выход: 85%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 384 [M + H]+.
Промежуточное вещество 28: получение 2-циклопропил-5-этинил-4,6-дифторбензо[d]тиазола
Стадия 1. Синтез N-(2,4-дифторфенил)циклопропанкарбоксамида
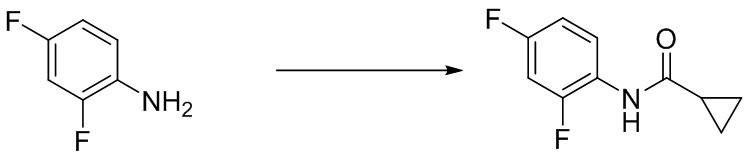
2,4-дифторанилин (10 г, 77,45 ммоль) растворяли в дихлорметане (100 мл), а затем добавляли триэтиламин (15,64 г, 154,9 ммоль). По каплям добавляли циклопропилкарбонилхлорид (8,1 г, 77,45 ммоль) на ледяной бане, после чего реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь промывали достаточным количеством насыщенного водного раствора хлорида аммония и насыщенного солевого раствора, высушивали над безводным сульфатом натрия и концентрировали; остаток разделяли с помощью колоночной хроматографии на силикагеле с получением N-(2,4-дифторфенил)циклопропанкарбоксамида (10,1 г, выход: 66%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 196 [M + H]+.
Стадия 2. Синтез N-(2,4-дифторфенил)циклопропанкарботиамида

N-(2,4-дифторфенил)циклопропанкарбоксамид (5 г, 25,36 ммоль) растворяли в ксилоле (80 мл) и добавляли реагент Лавессона (7,18 г, 17,75 ммоль). Реакционную смесь нагревали до 120°C, перемешивали в течение ночи, а затем концентрировали. Остаток разделяли в таком виде с помощью колоночной хроматографии на силикагеле с получением N-(2,4-дифторфенил)циклопропанкарботиамида (4,1 г, выход: 75%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 212 [M + H]+.
Стадия 3. Синтез 2-циклопропил-4,6-дифторбензо[d]тиазола

N-(2,4-дифторфенил)циклопропанкарботиамид (1 г, 4,69 ммоль) растворяли в растворе гидроксида натрия (1,69 г, 42,21 ммоль), после чего полученный раствор растворяли в этаноле (5 мл) и воде (10 мл) (концентрация по массе - около 30%). Полученный раствор добавляли по каплям к водному раствору феррицианида калия (6,18 г, 18,78 ммоль) в 10 мл воды, нагретому до 90°C, и реакционную смесь перемешивали в течение 2 ч, после чего нагревание сразу же прекращали. Реакционную смесь охлаждали, слегка подкисляли концентрированной хлористоводородной кислотой и экстрагировали этилацетатом. Полученный экстракт промывали насыщенным солевым раствором и концентрировали, а остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-циклопропил-4,6-дифторбензо[d]тиазола (500 мг, выход: 51%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 212 [M + H]+.
Стадия 4. Синтез 2-циклопропил-4,6-дифторбензо[d]тиазол-5-карбальдегида
2-циклопропил-4,6-дифторбензо[d]тиазол (980 мг, 4,64 ммоль) растворяли в тетрагидрофуране (15 мл), после чего смесь охлаждали до -70°C и по каплям добавляли диизопропиламид лития (2,55 мл, 5,104 ммоль, 2 M). После выдерживания смеси при этой температуре в течение 2 ч добавляли N,N-диметилформамид (1,01 г, 13,92 ммоль), затем вновь выдерживали смесь при этой температуре в течение 2 ч, после чего добавляли достаточное количество водного раствора хлорида аммония для остановки реакции. Реакционную смесь нагревали до комнатной температуры и экстрагировали этилацетатом, после чего органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-циклопропил-4,6-дифторбензо[d]тиазол-5-карбальдегида (675 мг, выход: 65%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 240 [M + H]+.
Стадия 5. Синтез 2-циклопропил-5-этинил-4,6-дифторбензо[d]тиазола

2-циклопропил-4,6-дифторбензо[d]тиазол-5-карбальдегид (122 мг, 0,51 ммоль) растворяли в метаноле (10 мл), после чего последовательно добавляли карбонат калия (211 мг, 1,53 ммоль) и диметиловый эфир (1-диазо-2-оксопропил)-фосфоновой кислоты (159 мг, 0,76 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-циклопропил-5-этинил-4,6-дифторбензо[d]тиазола (73 мг, выход: 61%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 236 [M + H]+.
Промежуточное вещество 29 может быть получено путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) промежуточного вещества 28:
вещества
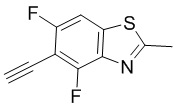
Промежуточное вещество 30: получение 2-(азетидин-1-ил)-5-этинил-4,6-дифторбензо[d]оксазола
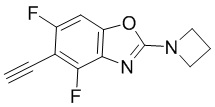
Стадия 1. Синтез 2-амино-3,5-дифторфенола

3,5-дифтор-2-нитрофенол (10 г, 57,11 ммоль) растворяли в метаноле (100 мл), а затем добавляли палладий на угле (1 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре и фильтровали, после чего фильтрат концентрировали с получением 2-амино-3,5-дифторфенола (8,5 г, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 146 [M + H]+.
Стадия 2. Синтез 4,6-дифторбензо[d]оксазол-2-тиола
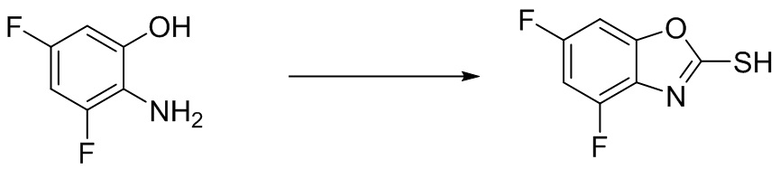
2-амино-3,5-дифторфенол (8,5 г, 58,62 ммоль) растворяли в этаноле (100 мл), а затем добавляли O-этилдисульфат калия (1,13 г, 70,34 ммоль). Реакционную смесь нагревали до температуры 95°C и выдерживали в течение ночи. Затем реакционную смесь охлаждали и фильтровали, и фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 4,6-дифторбензо[d]оксазол-2-тиола (11 г, выход: 95,31%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 188 [M + H]+.
Стадия 3. Синтез 2-хлор-4,6-дифторбензо[d]оксазола

4,6-дифторбензо[d]оксазол-2-тиол (4 г, 21,371 ммоль) растворяли в дихлорметане (50 мл), после чего добавляли оксалилхлорид (10,85 г, 85,48 ммоль) и N,N-диметилформамид (0,16 г, 2,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и концентрировали с получением 2-хлор-4,6-дифторбензо[d]оксазола (4,05 г, выход: 100%), который в таком виде использовали на следующей стадии. Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 190 [M + H]+.
Стадия 4. Синтез 2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазола

Неочищенный 2-хлор-4,6-дифторбензо[d]оксазол (4,05 г, 21,36 ммоль) растворяли в тетрагидрофуране (50 мл), после чего добавляли N,N-диизопропилэтиламин (5,51 г, 42,72 ммоль) и азетидин гидрохлорид (2,40 г, 25,63 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали. К остатку добавляли этилацетат, после чего смесь дважды промывали насыщенным водным раствором хлорида аммония, затем дважды промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, а остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазола (3,2 г, выход: 72%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 211 [M + H]+.
Стадия 5. Синтез 2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-карбальдегида
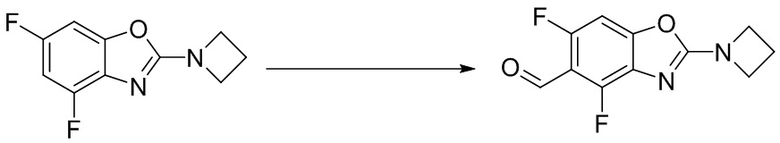
2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол (3 г, 14,28 ммоль) растворяли в тетрагидрофуране (100 мл), затем смесь охлаждали до -70°C, после чего по каплям добавляли раствор диизопропиламида лития в тетрагидрофуране (10,7 мл, 21,41 ммоль, 2 M). Реакционную смесь перемешивали при низкой температуре в течение 1 ч, а затем одной порцией добавляли N,N-диметилформамид (11,04 мл, 142,73 ммоль). После этого реакционную смесь перемешивали при низкой температуре в течение 2 ч и добавляли достаточное количество насыщенного водного раствора хлорида аммония для остановки реакции. Затем реакционную смесь доводили до комнатной температуры и дважды экстрагировали этилацетатом. Фазы этилацетата объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-карбальдегида (0,8 г, выход: 24%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 239 [M + H]+.
Стадия 6. Синтез 2-(азетидин-1-ил)-5-этинил-4,6-дифторбензо[d]оксазола

2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-карбальдегид (0,8 г, 3,40 ммоль) растворяли в метаноле (10 мл) и тетрагидрофуране (10 мл), после чего добавляли карбонат калия (1,17 г, 8,5 ммоль) и диметиловый эфир (1-диазо-2-оксопропил)-фосфоновой кислоты (1,31 г, 6,801 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали и добавляли достаточное количество насыщенного водного раствора хлорида аммония для остановки реакции. Затем реакционную смесь доводили до комнатной температуры и дважды экстрагировали этилацетатом. Фазы этилацетата объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 2-(азетидин-1-ил)-5-этинил-4,6-дифторбензо[d]оксазола (0,75 г, выход: 94%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 235 [M + H]+.
Промежуточное вещество 31: получение 5-этинил-4,6-дифтор-N-метилбензо[d]оксазол-2-амина
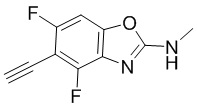
Стадия 1. Синтез 2-амино-3,5-дифторфенола

3,5-дифтор-2-нитрофенол (10 г, 57,11 ммоль) растворяли в метаноле (100 мл), а затем добавляли палладий на угле (1 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре и фильтровали, после чего фильтрат концентрировали с получением 2-амино-3,5-дифторфенола (8,5 г, выход: 100%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 146 [M + H]+.
Стадия 2. Синтез 4,6-дифторбензо[d]оксазол-2-тиола
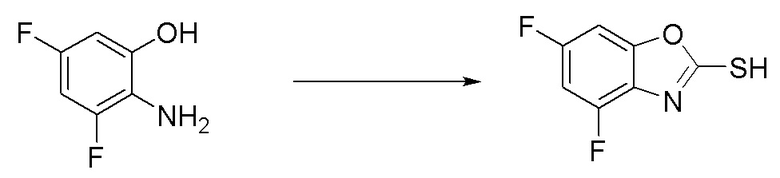
2-амино-3,5-дифторфенол (8,5 г, 58,62 ммоль) растворяли в этаноле (100 мл), а затем добавляли O-этилдисульфат калия (1,13 г, 70,34 ммоль). Реакционную смесь нагревали до температуры 95°C и выдерживали в течение ночи. Затем реакционную смесь охлаждали и фильтровали, и фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 4,6-дифторбензо[d]оксазол-2-тиола (11 г, выход: 95,31%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 188 [M + H]+.
Стадия 3. Синтез 2-хлор-4,6-дифторбензо[d]оксазола

4,6-дифторбензо[d]оксазол-2-тиол (4 г, 21,37 ммоль) растворяли в дихлорметане (50 мл), после чего добавляли оксалилхлорид (10,85 г, 85,48 ммоль) и N,N-диметилформамид (0,16 г, 2,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а после завершения реакции реакционную смесь в таком виде концентрировали с получением неочищенного 2-хлор-4,6-дифторбензо[d]оксазола (4,05 г), который в таком виде использовали на следующей стадии. Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 190 [M + H]+.
Стадия 4. Синтез 4,6-дифтор-N-метилбензо[d]оксазол-2-амина

2-хлор-4,6-дифторбензо[d]оксазол (1 г, 5,28 ммоль) растворяли в тетрагидрофуране (10 мл) и к полученному раствору добавляли раствор метиламина в этаноле (7,92 мл, 15,84 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали, а остаток в таком виде разделяли с помощью колоночной хроматографии на силикагеле с получением 4,6-дифтор-N-метилбензо[d]оксазол-2-амина (0,4 г, выход: 29%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 190 [M + H]+.
Стадия 5. Синтез 4,6-дифтор-2-(метиламино)бензо[d]оксазол-5-карбальдегида
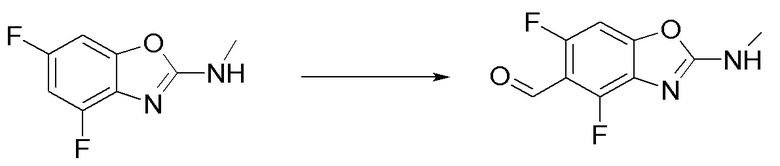
4,6-дифтор-N-метилбензо[d]оксазол-2-амин (0,25 г, 1,36 ммоль) растворяли в тетрагидрофуране (25 мл), затем смесь охлаждали до -70°C и по каплям добавляли раствор диизопропиламида лития в тетрагидрофуране (2,0 мл, 4,07 ммоль, 2 M). Реакционную смесь перемешивали при низкой температуре в течение 1 ч, а затем одной порцией добавляли N,N-диметилформамид (2,1 мл, 27,15 ммоль). После этого реакционную смесь перемешивали при низкой температуре в течение 2 ч и добавляли достаточное количество насыщенного водного раствора хлорида аммония для остановки реакции. Затем реакционную смесь доводили до комнатной температуры и дважды экстрагировали этилацетатом. Фазы этилацетата объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 4,6-дифтор-2-(метиламино)бензо[d]оксазол-5-карбальдегида (0,12 г, выход: 41%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 213 [M + H]+.
Стадия 6. Синтез 5-этинил-4,6-дифтор-N-метилбензо[d]оксазол-2-амина

4,6-дифтор-2-(метиламино)бензо[d]оксазол-5-карбальдегид (120 мг, 0,57 ммоль) растворяли в метаноле (5 мл) и тетрагидрофуране (5 мл), после чего добавляли карбонат калия (195 мг, 1,41 ммоль) и диметиловый эфир (1-диазо-2-оксопропил)-фосфоновой кислоты (217 мг, 1,13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали и добавляли достаточное количество насыщенного водного раствора хлорида аммония для остановки реакции. Затем реакционную смесь доводили до комнатной температуры и дважды экстрагировали этилацетатом. Фазы этилацетата объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 5-этинил-4,6-дифтор-N-метилбензо[d]оксазол-2-амина (110 мг, выход: 91%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 209 [M + H]+.
Промежуточное вещество 32: получение 1-((2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она
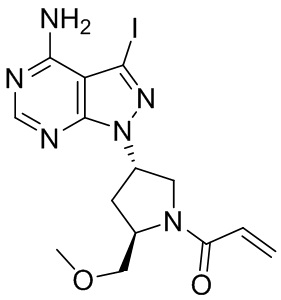
Стадия 1. Синтез 1-(трет-бутил)-2-метил-(2R,4R)-4-(бензилокси)пирролидин-1,2-дикарбоксилата
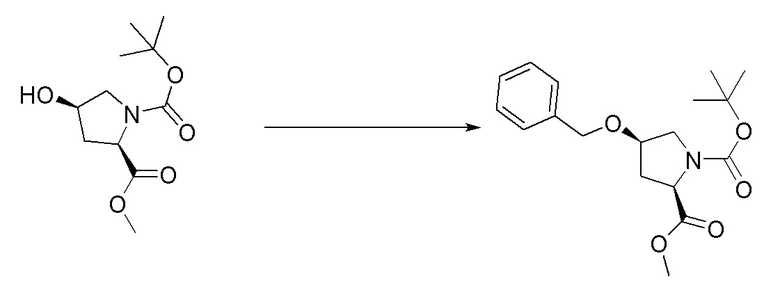
1-(трет-бутил)-2-метил(2R,4R)-4-гидроксипирролидин-1,2-дикарбоксилат (10 г, 40,77 ммоль) растворяли в N,N-диметилформамиде (120 мл), после чего смесь помещали в ледяную воду и добавляли гидрид натрия (1,63 г, 40,77 ммоль). Реакционную смесь перемешивали в течение 0,5 ч, а затем добавляли бензилбромид (6,5 мл, 54,36 ммоль). Полученную таким образом реакционную смесь перемешивали в течение ночи при комнатной температуре. По завершении реакции добавляли разбавленную хлористоводородную кислоту для остановки реакции, и полученную смесь дважды экстрагировали достаточным количеством этилацетата. Органические фазы объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 1-(трет-бутил)-2-метил-(2R,4R)-4-(бензилокси)пирролидин-1,2-дикарбоксилата (11,8 г, выход: 97%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 336 [M + H]+.
Стадия 2. Синтез трет-бутил-(2R,4R)-4-(бензилокси)-2-(гидроксиметил)пирролидин-1-карбоксилата
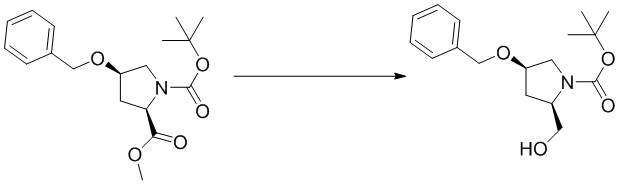
1-(трет-бутил)-2-метил-(2R,4R)-4-(бензилокси)пирролидин-1,2-дикарбоксилат (11,8 г, 26,39 ммоль) растворяли в тетрагидрофуране (150 мл), после чего добавляли боргидрид лития (0,86 г, 39,58 ммоль) на ледяной бане. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем добавляли разбавленную хлористоводородную кислоту для остановки реакции, и полученную смесь дважды экстрагировали достаточным количеством этилацетата. Органические фазы объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением трет-бутил-(2R,4R)-4-(бензилокси)-2-(гидроксиметил)пирролидин-1-карбоксилата (8 г, выход: 98%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 308 [M + H]+.
Стадия 3. Синтез трет-бутил-(2R,4R)-4-(бензилокси)-2-(метоксиметил)пирролидин-1-карбоксилата

Трет-бутил-(2R,4R)-4-(бензилокси)-2-(гидроксиметил)пирролидин-1-карбоксилат (8,00 г, 26,03 ммоль) растворяли в N,N-диметилформамиде (120 мл), после чего смесь помещали в ледяную воду и добавляли гидрид натрия (3,12 г, 78,08 ммоль). Реакционную смесь перемешивали в течение 0,5 ч, а затем добавляли йодметан (16,2 мл, 260,26 ммоль). Полученную таким образом реакционную смесь перемешивали в течение ночи при комнатной температуре. По завершении реакции добавляли разбавленную хлористоводородную кислоту для остановки реакции, и полученную смесь дважды экстрагировали достаточным количеством этилацетата. Органические фазы объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением трет-бутил-(2R,4R)-4-(бензилокси)-2-(метоксиметил)пирролидин-1-карбоксилата (8 г, выход: 95%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 322 [M + H]+.
Стадия 4. Синтез трет-бутил (2R,4R)-4-гидрокси-2-(метоксиметил)пирролидин-1-карбоксилата
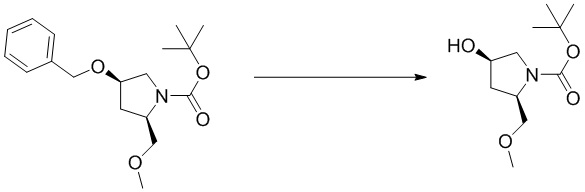
Трет-бутил-(2R,4R)-4-(бензилокси)-2-(метоксиметил)пирролидин-1-карбоксилат (1,0 г, 3,11 ммоль) растворяли в этаноле (20 мл), и к полученному раствору добавляли 10%-ный палладий на угле (0,15 г). Трижды вводили водород для замещения воздуха путем откачки, после чего реакционную смесь перемешивали в течение ночи при комнатной температуре и фильтровали. Фильтрат концентрировали с получением трет-бутил (2R,4R)-4-гидрокси-2-(метоксиметил)пирролидин-1-карбоксилата (0,7 г, выход: 99%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 232 [M + H]+.
Стадия 5. Синтез трет-бутил-(2R,4R)-2-(метоксиметил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилата
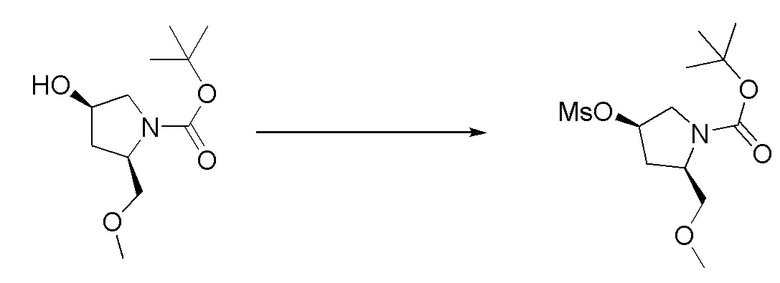
Трет-бутил (2R,4R)-4-гидрокси-2-(метоксиметил)пирролидин-1-карбоксилат (300 мг, 1,30 ммоль) растворяли в дихлорметане (10 мл) и в полученный раствор добавляли N,N-диизопропилэтиламин (1 мл, 6,49 ммоль) и метансульфонилхлорид (0,3 мл, 3,89 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего концентрировали с получением трет-бутил-(2R,4R)-2-(метоксиметил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилата (355 мг, выход: 88%); неочищенный продукт в таком виде использовали на следующей стадии без дополнительной очистки. Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 310 [M + H]+.
Стадия 6. Синтез трет-бутил-(2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-карбоксилата
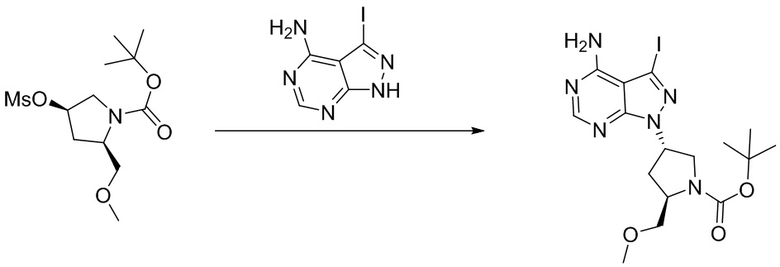
Трет-бутил-(2R,4R)-2-(метоксиметил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилат (355 мг, 1,15 ммоль) и 3-йод-1H-пиразоло[3,4-d]пиримидин-4-амин (300 мг, 1,15 ммоль) последовательно растворяли в N,N-диметилформамиде (15 мл), а затем к полученному раствору добавляли карбонат цезия (1,12 г, 3,45 ммоль). Реакционную смесь нагревали до температуры 80°C и перемешивали в течение ночи. По завершении реакции реакционную смесь охлаждали до комнатной температуры, добавляли достаточное количество насыщенного водного раствора бикарбоната натрия, после чего водную фазу трижды экстрагировали достаточным количеством этилацетата. Органические фазы объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия и фильтровали, а затем фильтрат концентрировали. Остаток разделяли с помощью колоночной хроматографии на силикагеле с получением трет-бутил-(2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-карбоксилата (456 мг, выход: 96%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 475 [M + H]+.
Стадия 7. Синтез 3-йод-1-((3S,5R)-5-(метоксиметил)пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амина
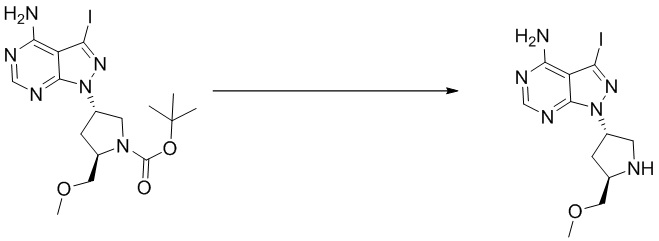
Трет-бутил-(2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-карбоксилат (456 мг, 0,96 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли раствор хлористого водорода в 1,4-диоксане (3,2 мл, 19,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. После завершения реакции реакционную смесь в таком виде концентрировали, а остаток высушивали с получением 3-йод-1-((3S,5R)-5-(метоксиметил)пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амина (300 мг, выход: 69%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 375 [M + H]+.
Стадия 8. Синтез 1-((2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она
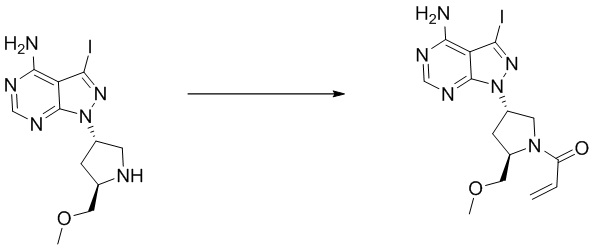
3-йод-1-((3S,5R)-5-(метоксиметил)пирролидин-3-ил)-1H-пиразоло[3,4-d]пиримидин-4-амин (300 мг, 0,67 ммоль) растворяли в тетрагидрофуране (10 мл) и воде (5 мл), после чего добавляли бикарбонат натрия (281,84 мг, 3,35 ммоль). Реакционную смесь помещали на ледяную баню и медленно по каплям добавляли раствор акрилоилхлорида (0,07 мл, 0,87 ммоль) в тетрагидрофуране (1 мл). По окончании покапельного добавления реакционную смесь перемешивали в течение 10 минут, добавляли достаточное количество насыщенного водного раствора бикарбоната натрия для остановки реакции, после чего полученную реакционную смесь трижды экстрагировали этилацетатом. Органические фазы объединяли, дважды промывали насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и остаток разделяли с помощью колоночной хроматографии на силикагеле с получением 1-((2R,4S)-4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она (180 мг, выход: 63%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 429 [M + H]+.
Промежуточное вещество 33: получение трет-бутил-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-карбоксилата
3-йод-1H-пиразоло[3,4-d]пиримидин-4-амин (2500 мг, 10,16 ммоль), трет-бутил 3-((метилсульфонил)окси)азетидин-1-карбоксилат (2550 мг, 10,16 ммоль) и карбонат цезия (4970 мг, 15,24 ммоль) растворяли в N,N-диметилформамиде (30 мл) и перемешивали реакционную смесь при 80°C в течение 16 ч. После завершения реакции добавляли воду (60 мл) и полученную реакционную смесь фильтровали с получением трет-бутил-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-карбоксилата (2000 мг, выход: 47%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 417 [M + H]+.
II. Получение соединений в качестве примеров
Пример 1. Получение (S)-1-(3-(4-амино-5-((2-циклопропилбензо[d]оксазол-5-ил)этинил)-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
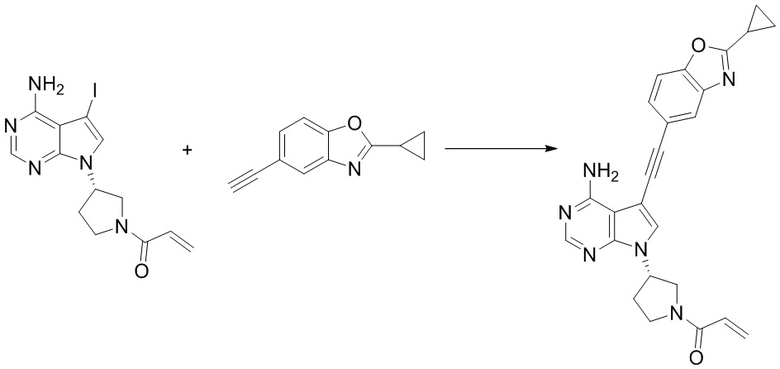
(S)-1-(3-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-он (40 мг, 0,1 ммоль), 2-циклопропил-5-этинилбензо[d]оксазол (23 мг, 0,12 ммоль), триэтиламин (0,4 мл), йодид меди (2 мг, 0,01 ммоль) и тетракис(трифенилфосфин)палладий (12 мг, 0,01 ммоль) добавляли к ДМФ (2 мл). Реакционную смесь перемешивали при температуре 80°C в течение 1 ч в атмосфере азота, после чего в таком виде разделяли с помощью обращенно-фазовой колоночной хроматографии и лиофилизировали с получением (S)-1-(3-(4-амино-5-((2-циклопропилбензо[d]оксазол-5-ил)этинил)-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она (29,5 мг, выход: 65%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 439 [M + H]+.
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,11 (s, 1H), 7,78-7,77 (d, J = 4 Гц,1H), 7,69-7,65 (d, J = 16 Гц, 1H), 7,61-7,59 (d, J = 8 Гц, 1H), 7,46-7,44 (m, 1H), 6,85-6,48 (m, 3H), 6,14-6,07 (m, 1H), 5,67-5,59 (m, 1H), 5,31-5,18 (m, 1H), 4,06-3,79 (m, 2H), 3,71-3,43 (m, 2H), 2,38-2,19 (m, 3H), 1,17-1,07 (m, 4H).
Вещества из примеров 2-75 могут быть получены путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) вещества из примера 1.
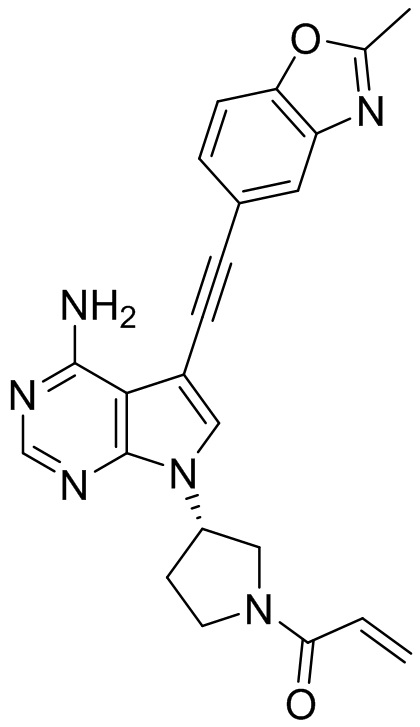
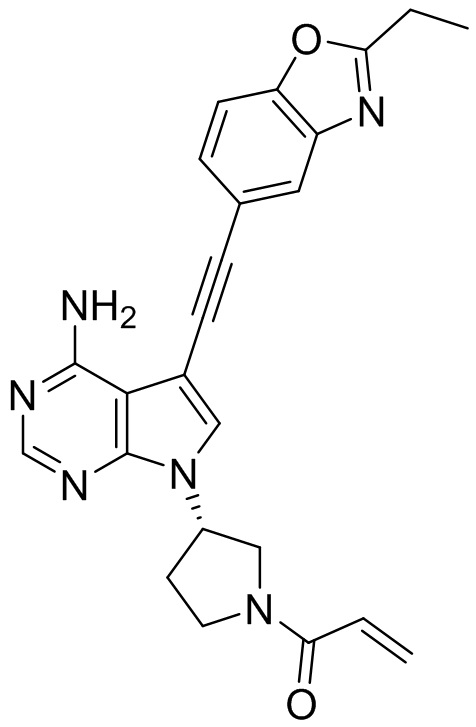
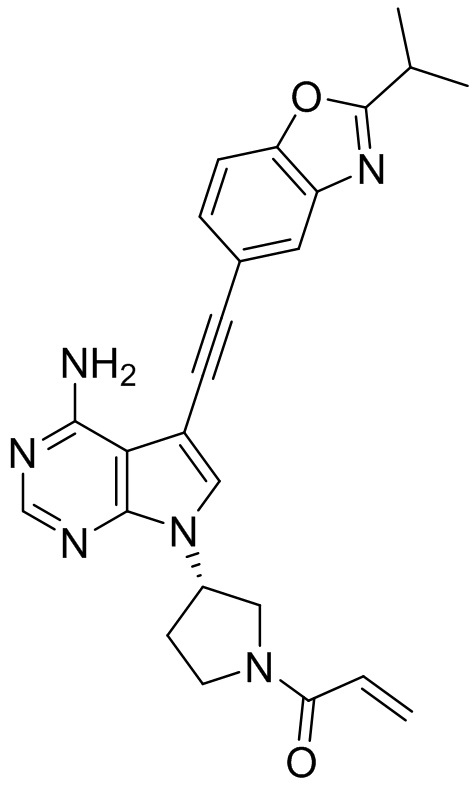
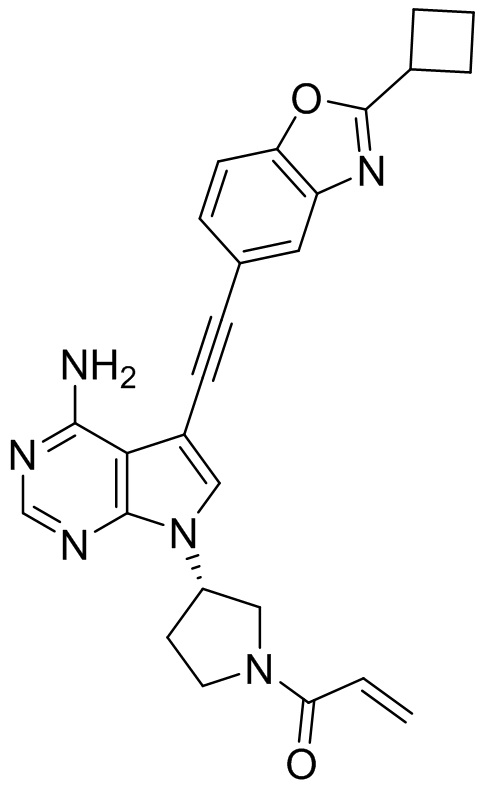
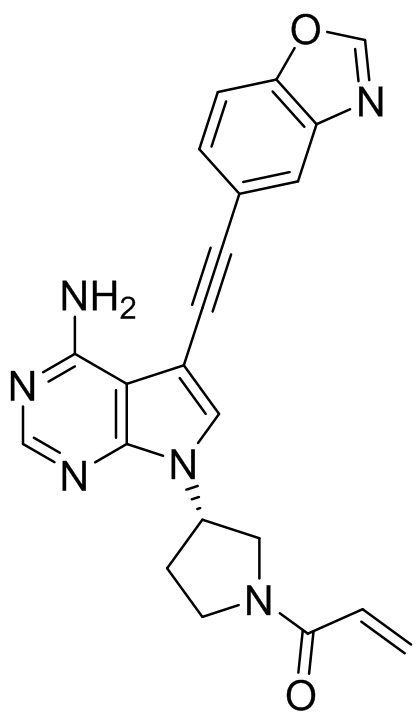
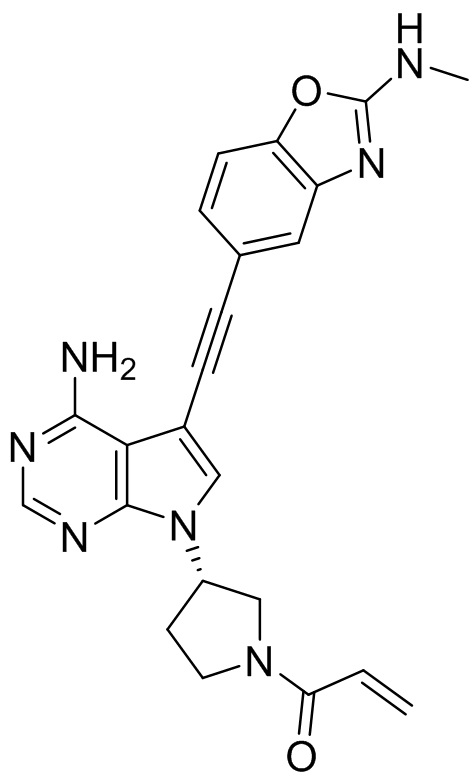
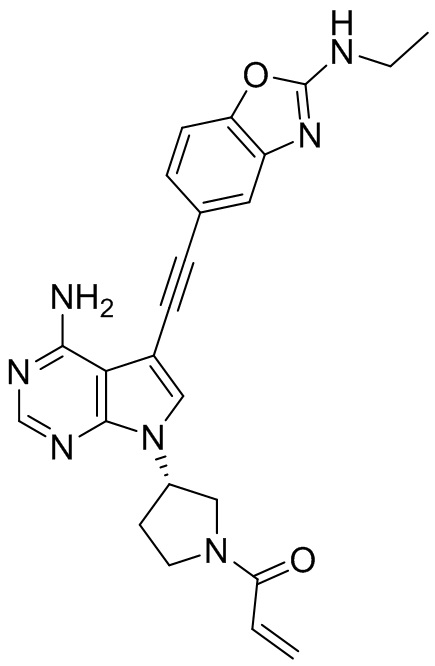
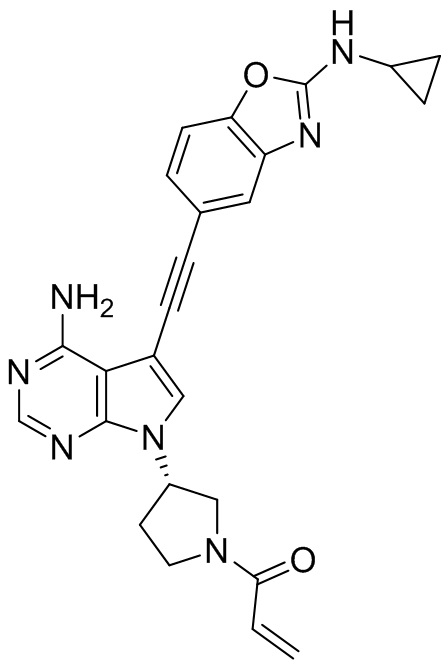
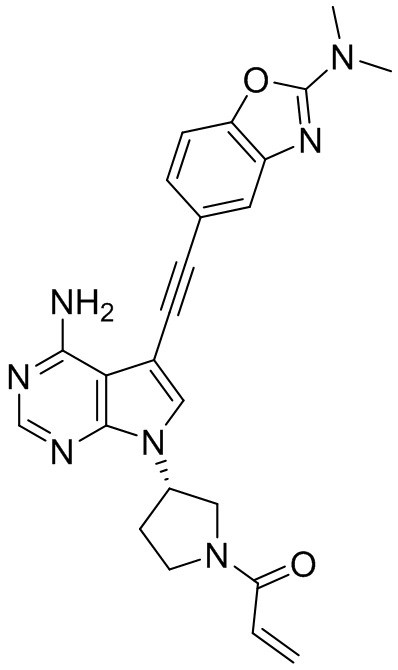
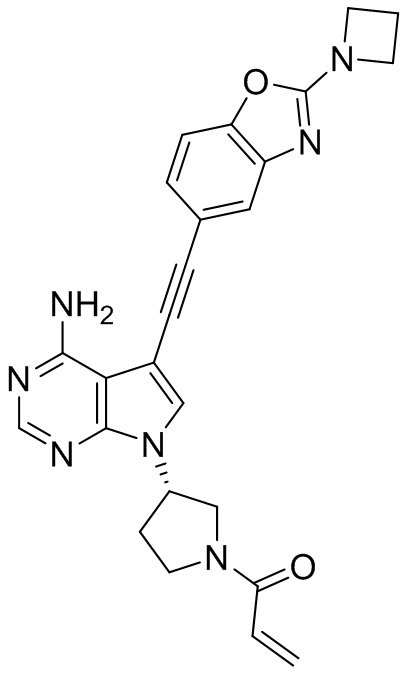
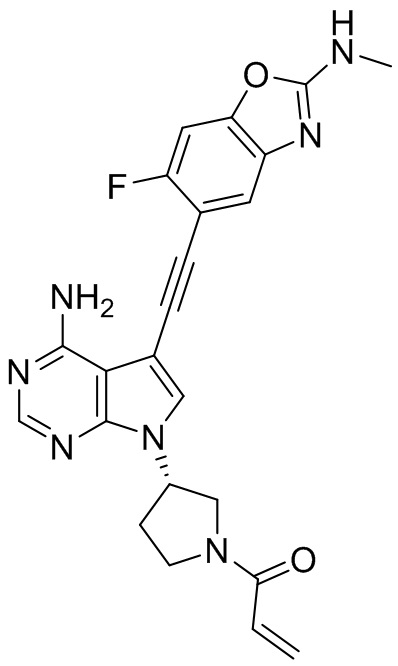
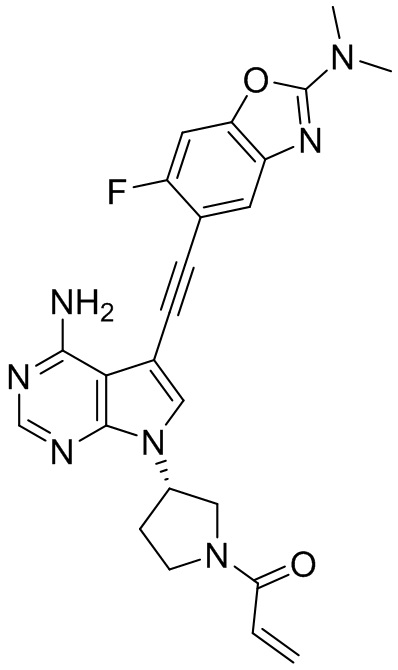
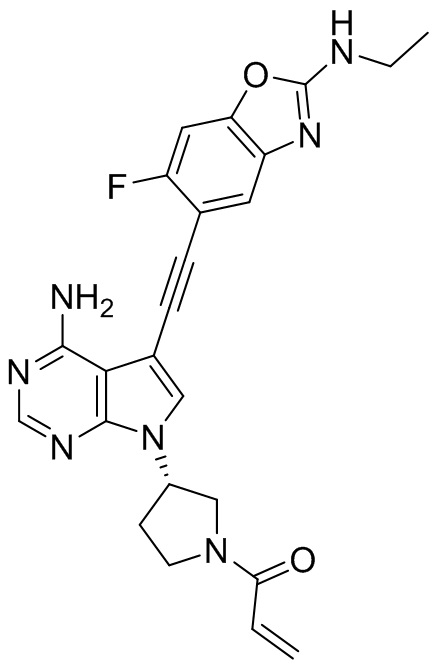
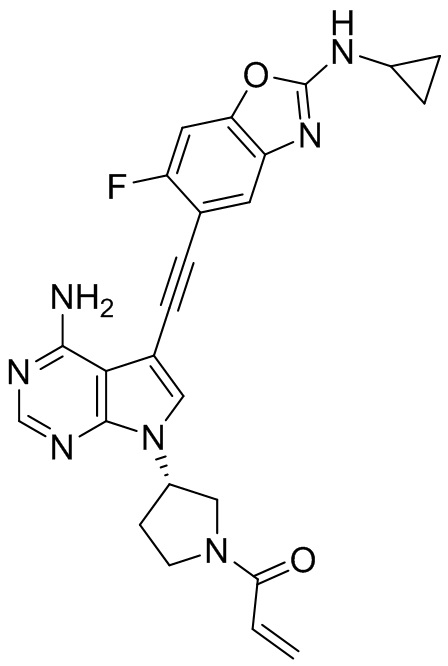
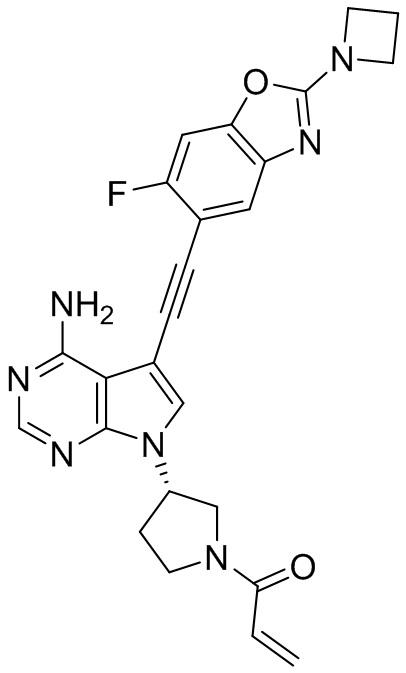
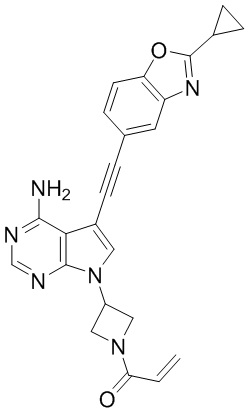
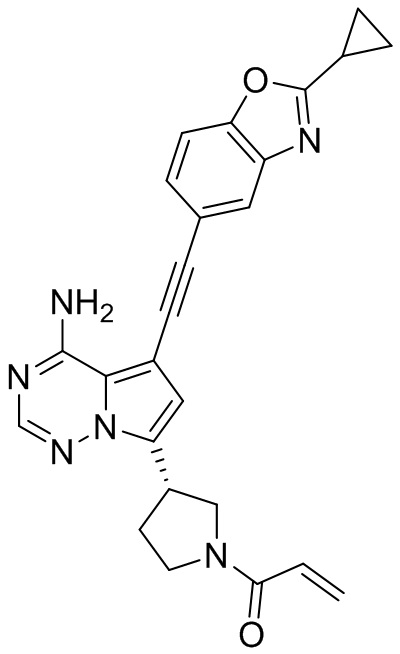
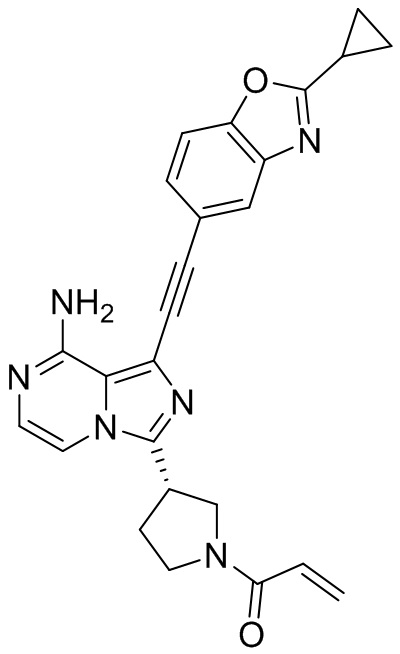
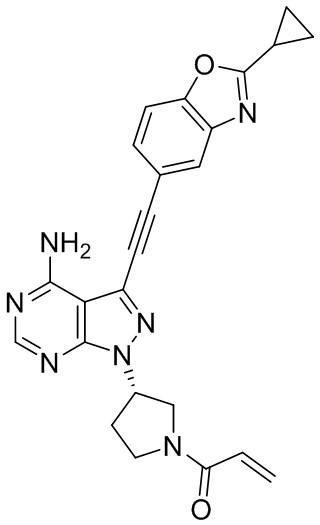
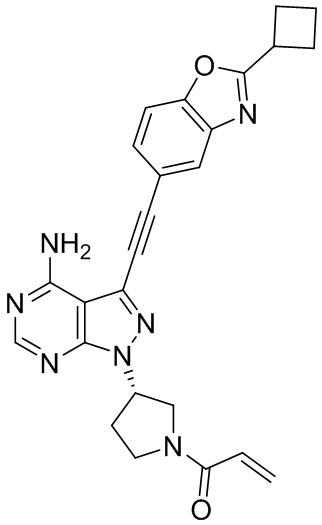
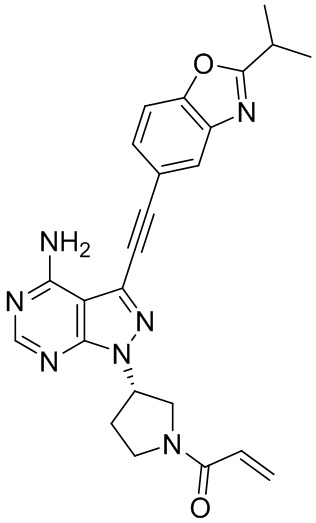
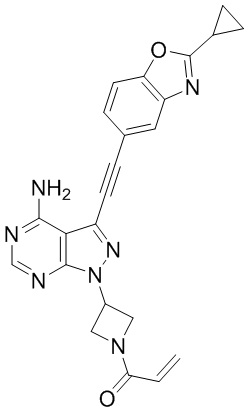
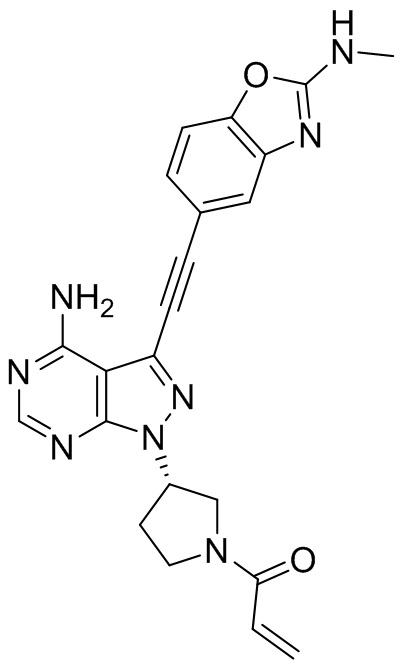
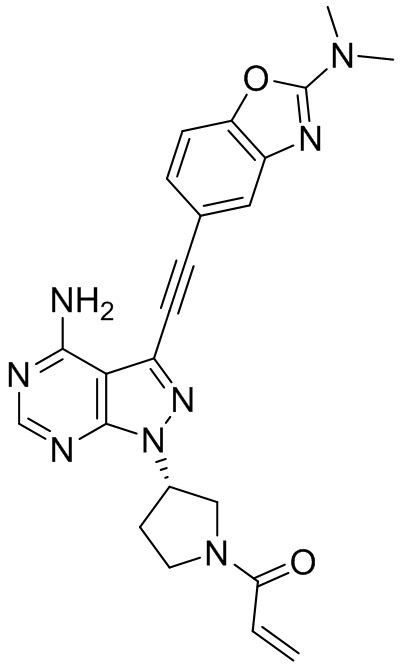
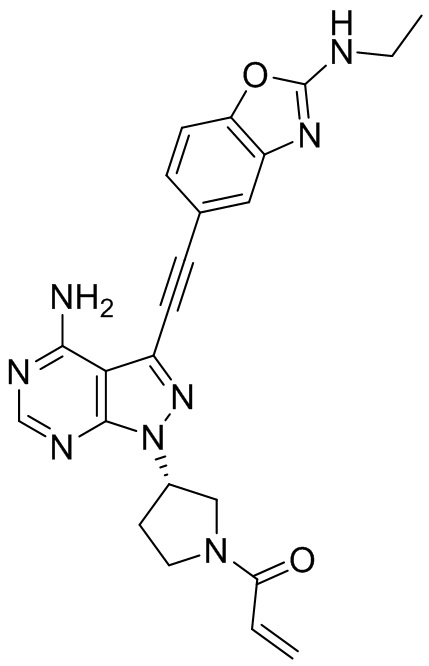
4-амино-3-((2-(этиламино)бензо[d]оксазол-5-ил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-ил)проп-2-ен-1-он
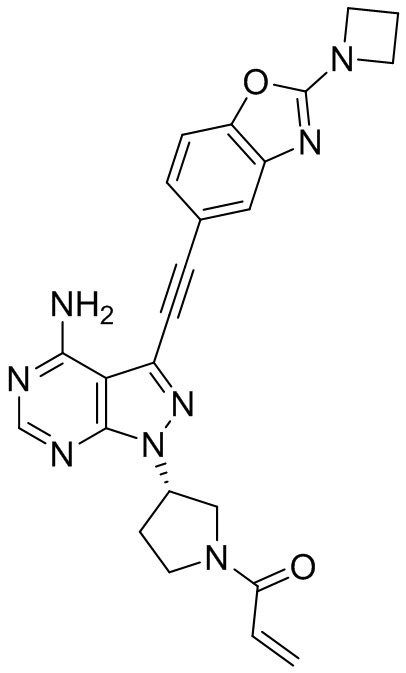
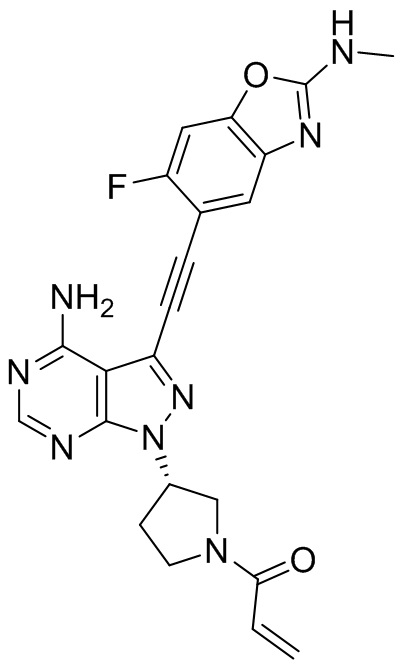
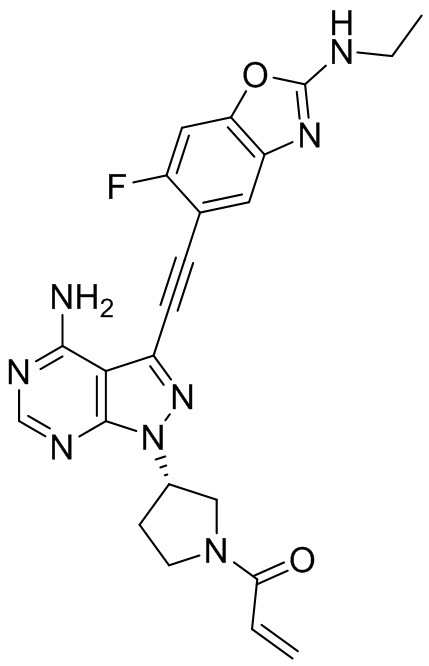
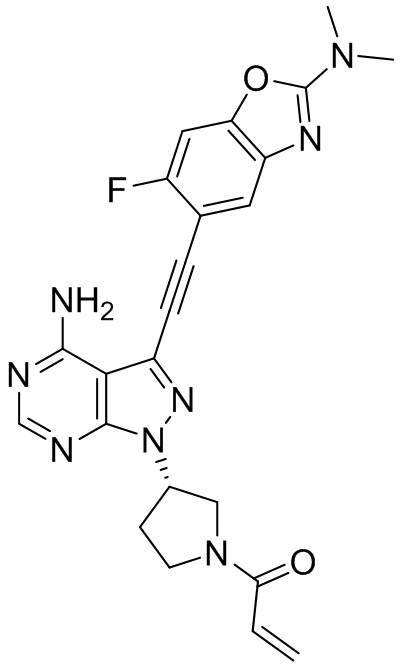
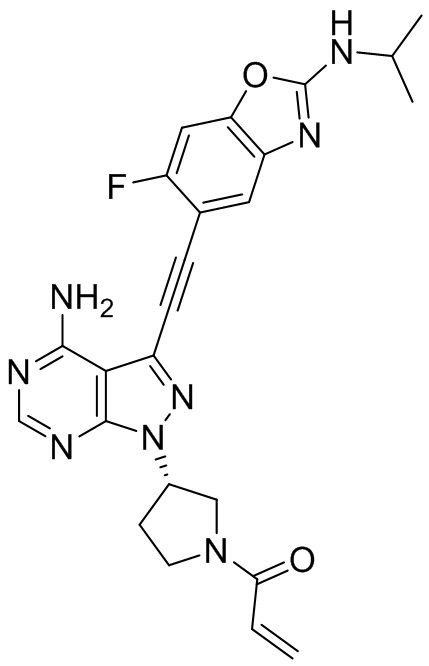
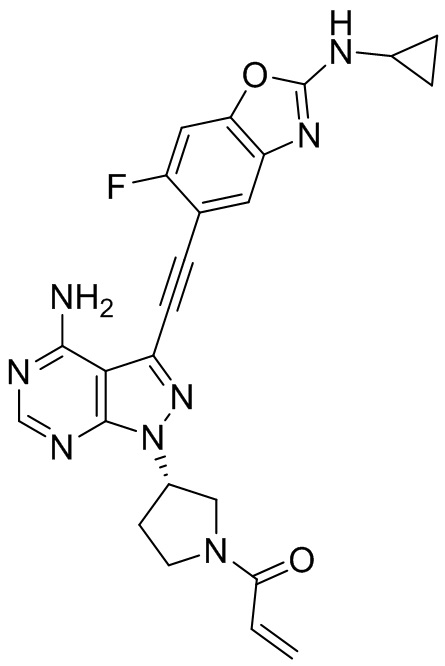
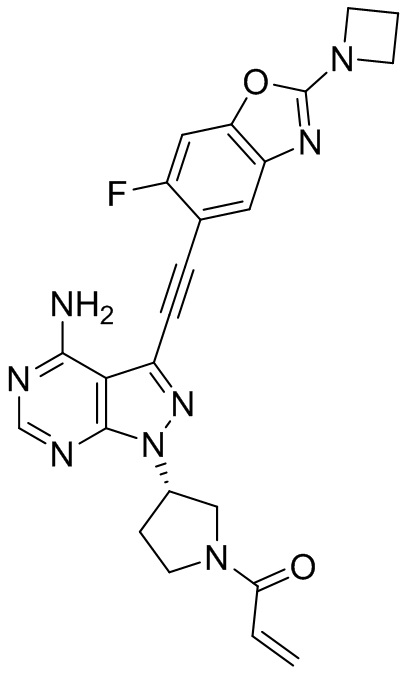
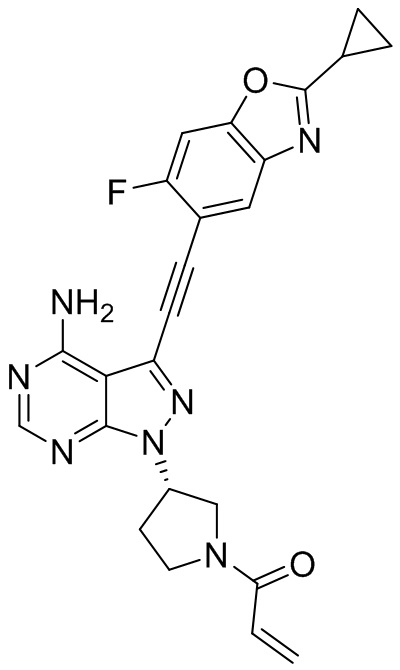
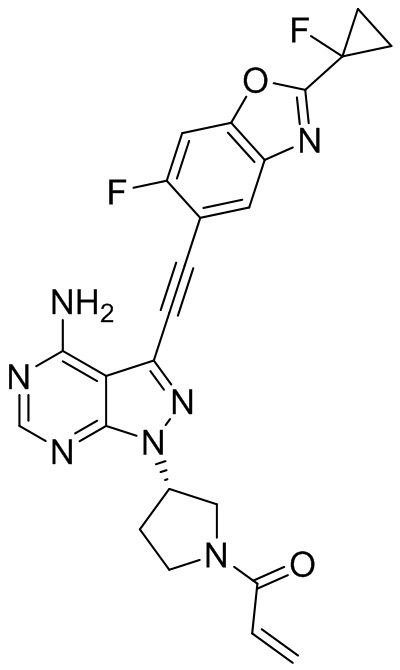
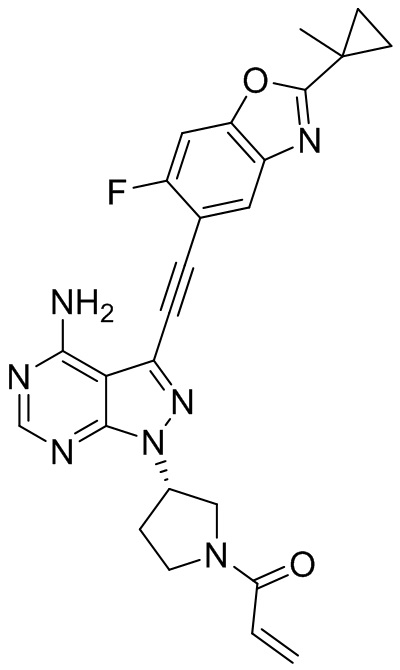
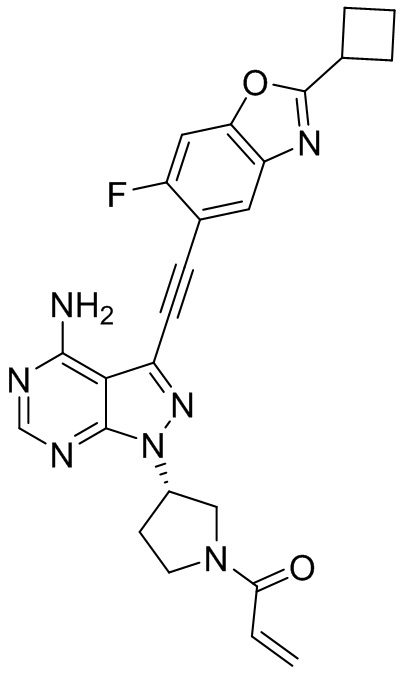
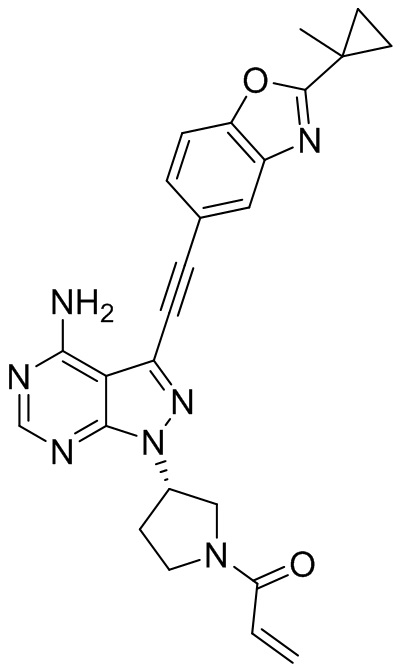
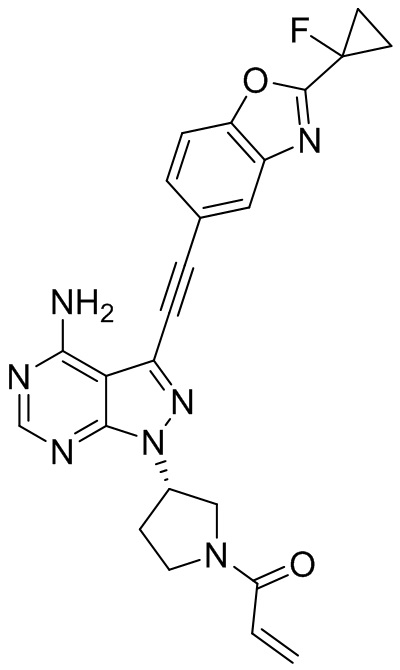
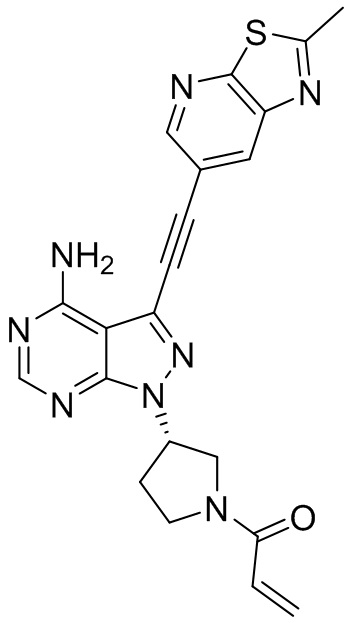

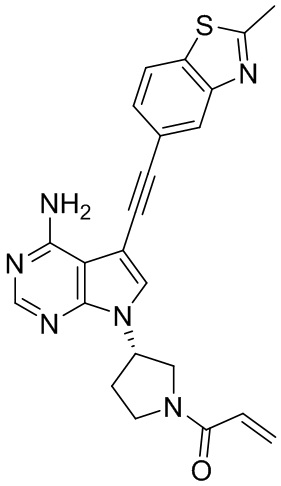
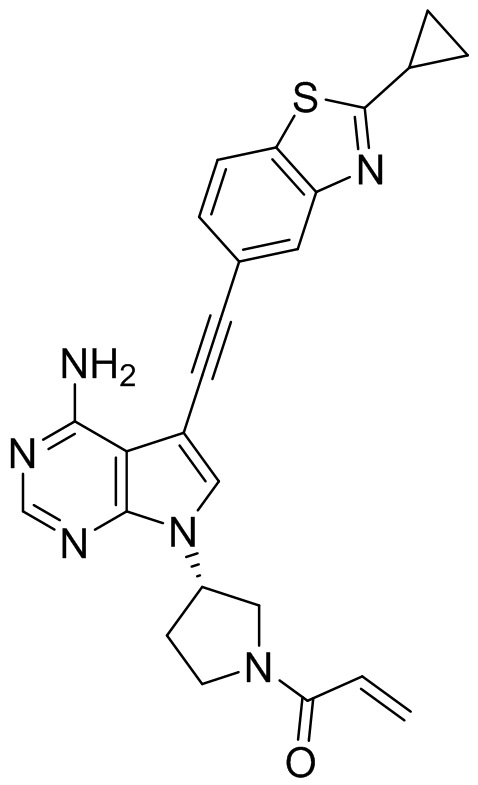
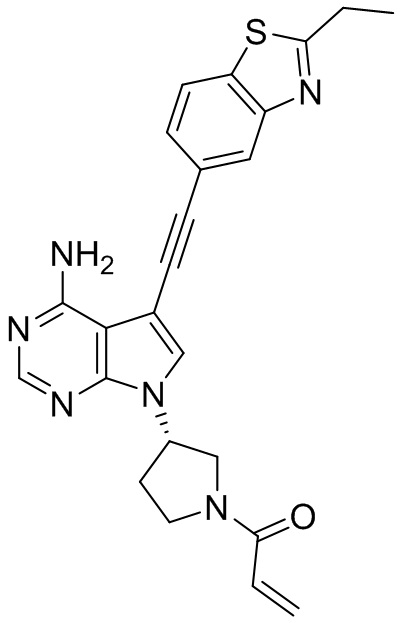
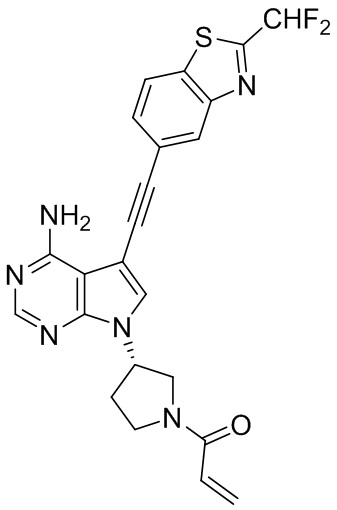
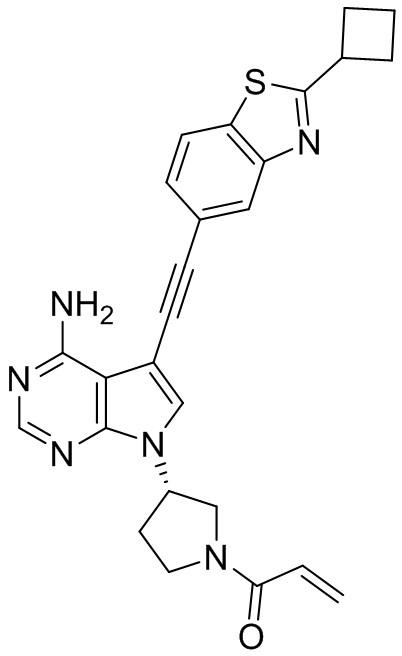
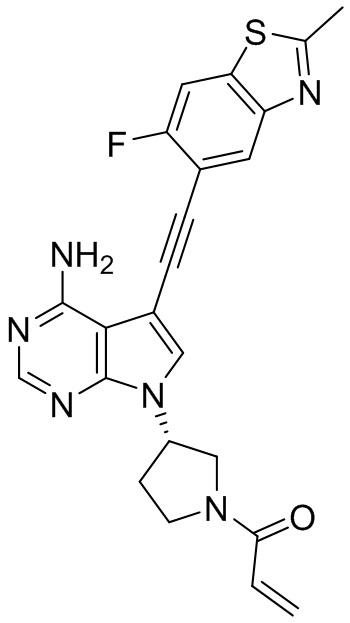
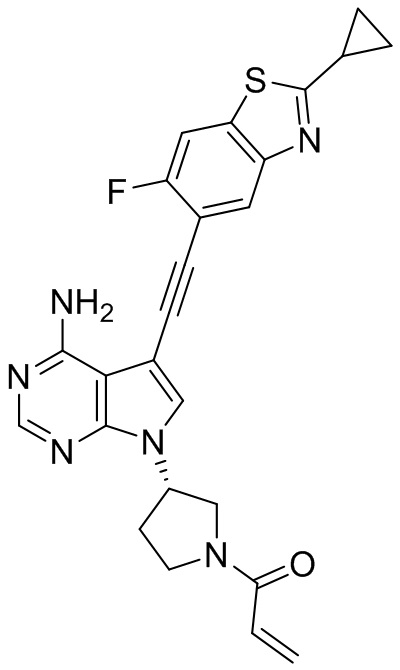
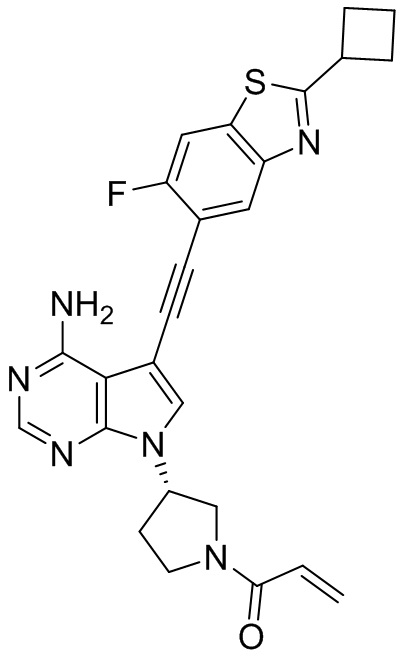
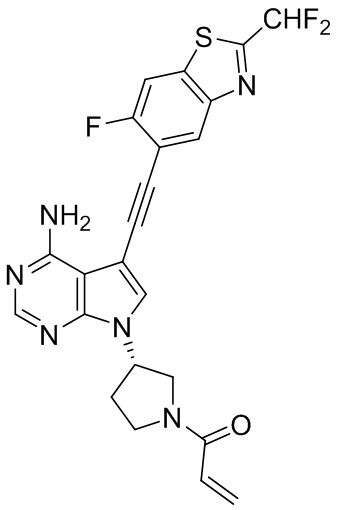
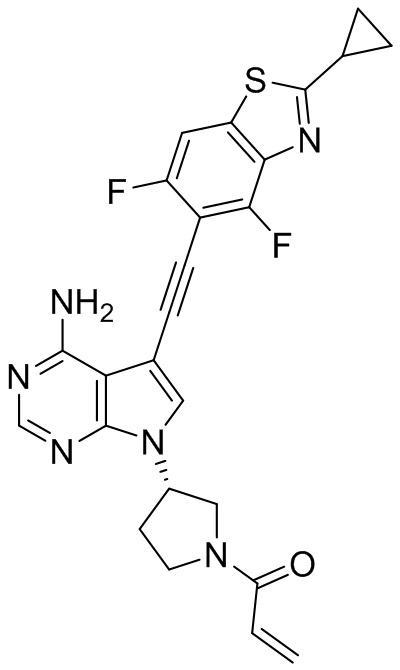
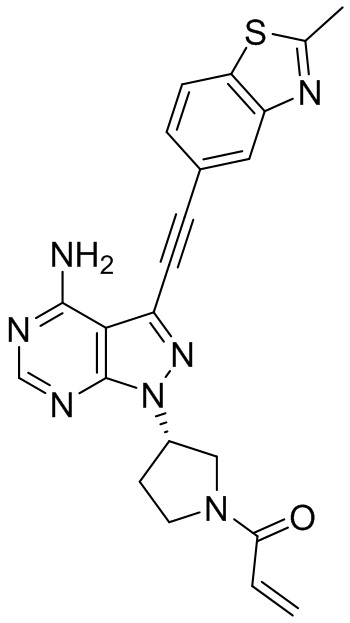
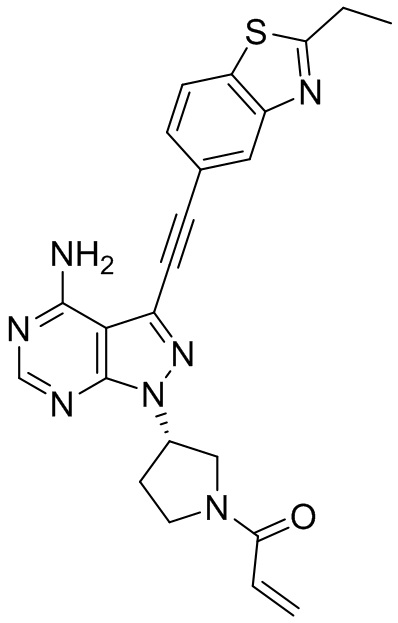
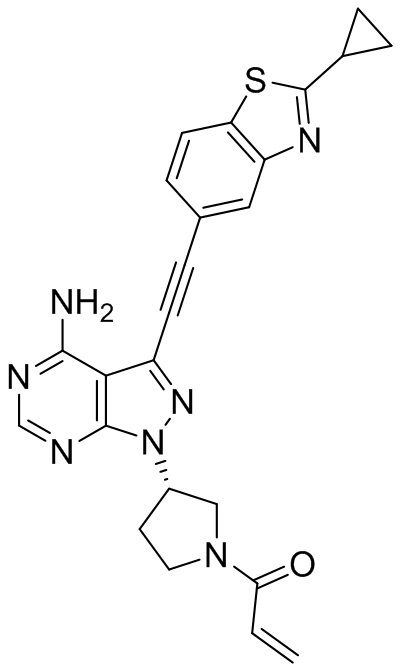
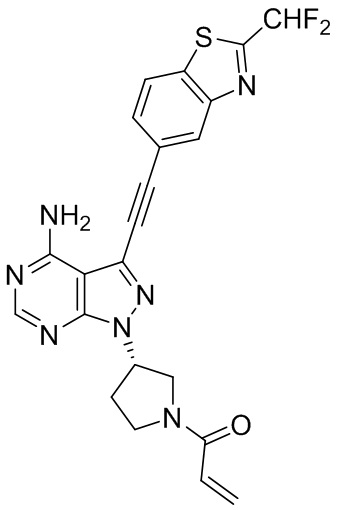
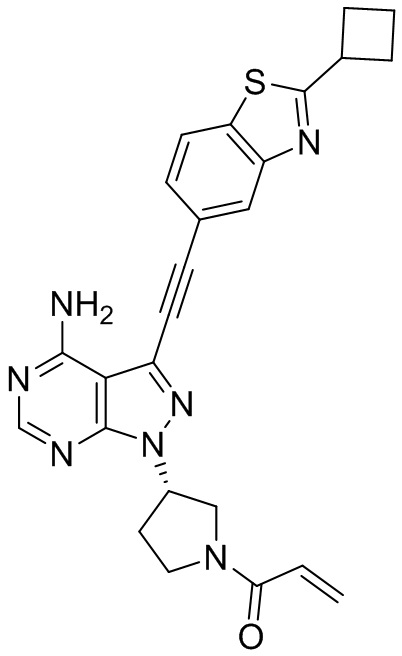
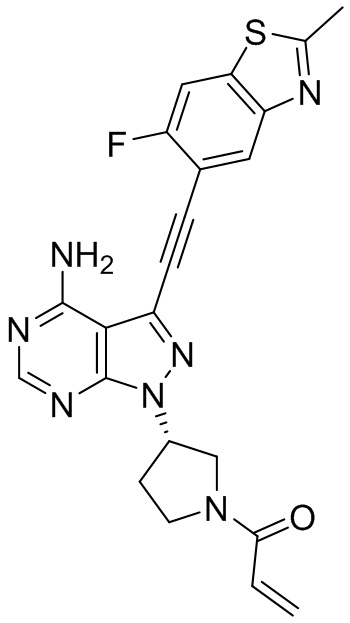
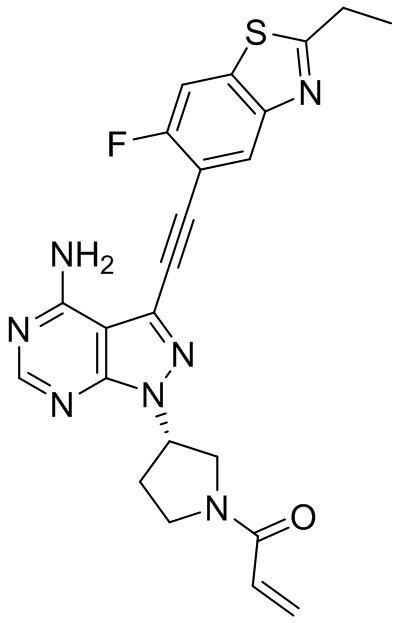
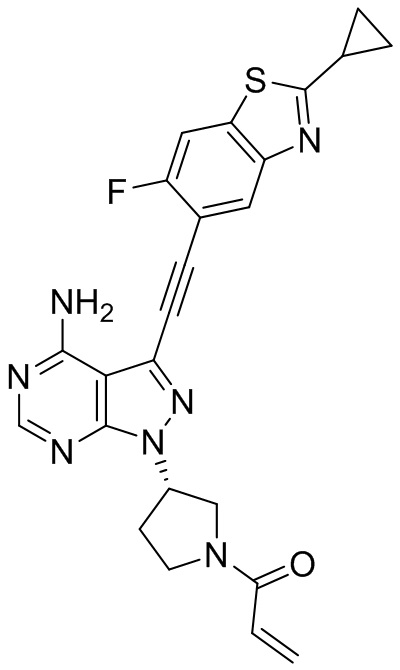
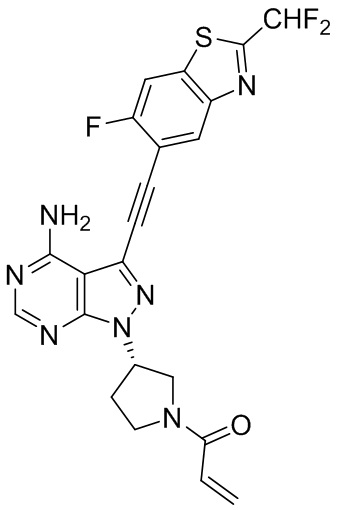
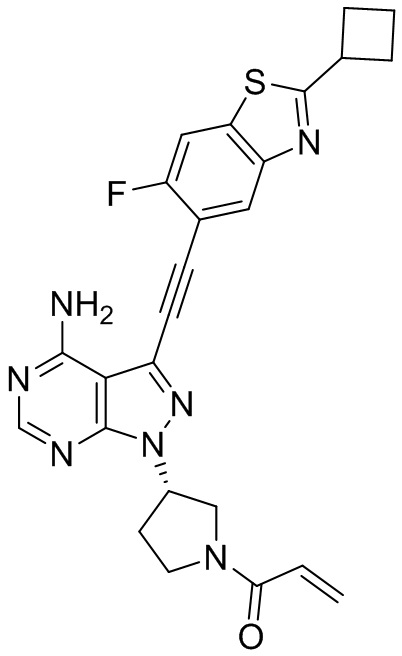
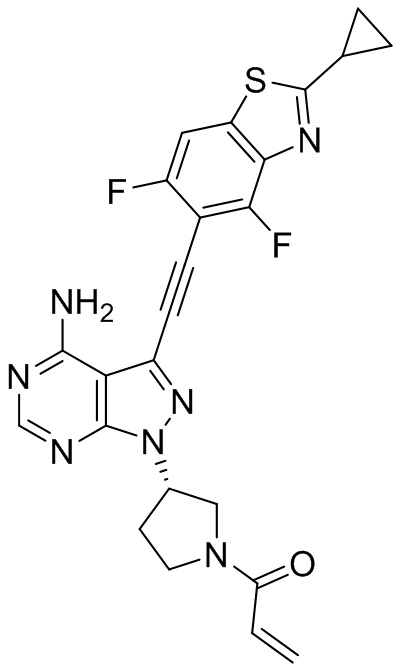
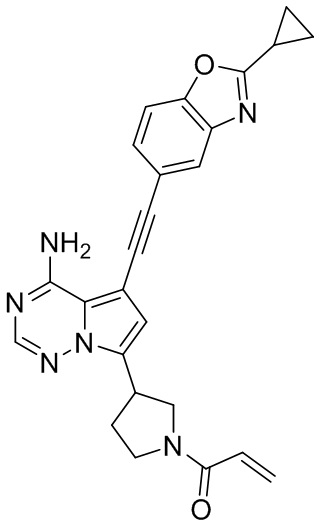
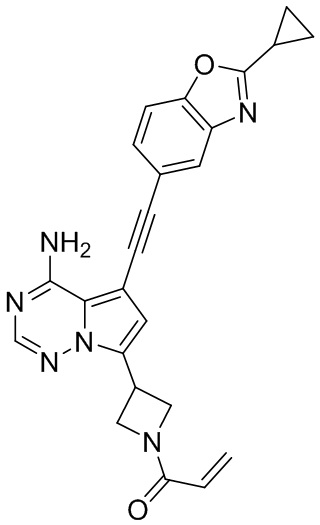
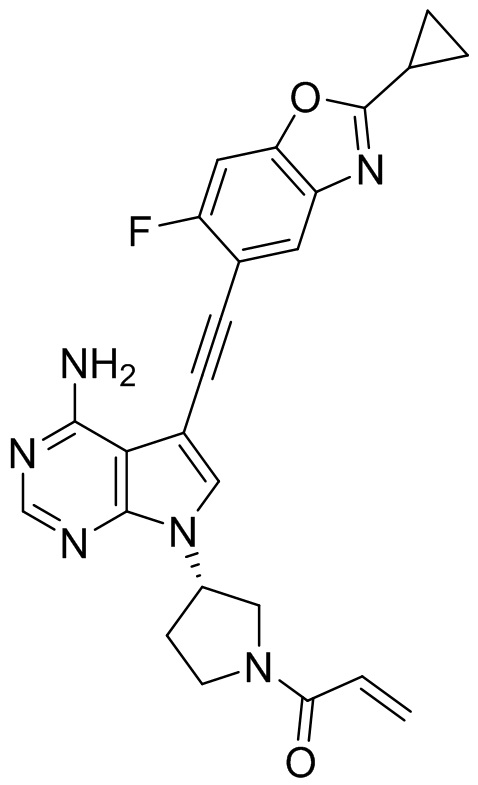
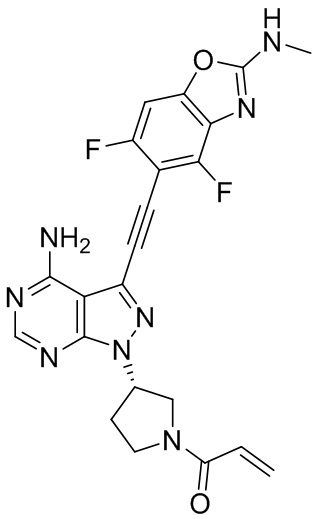
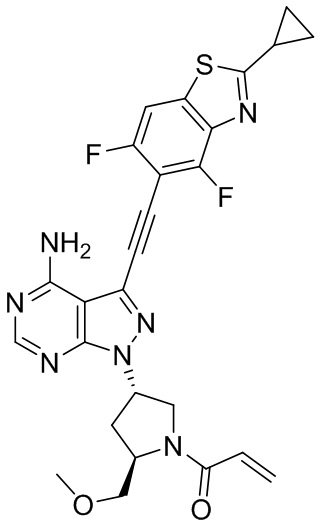
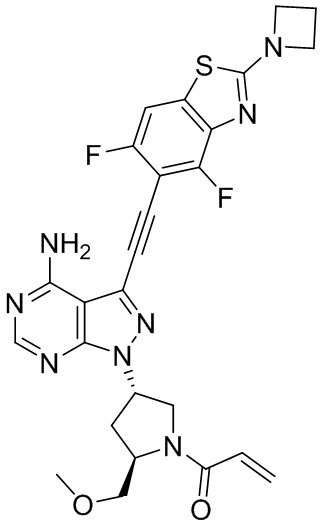
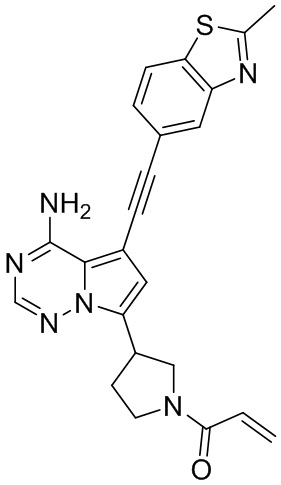
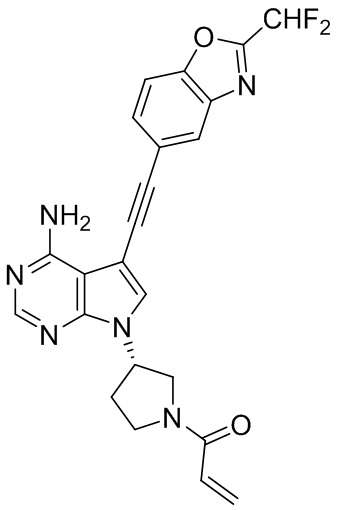
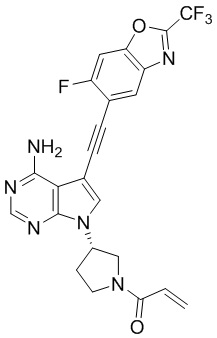
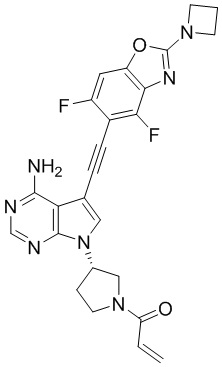
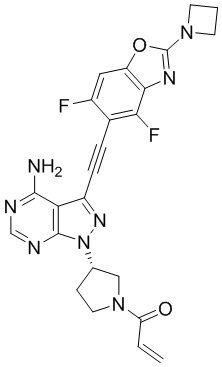
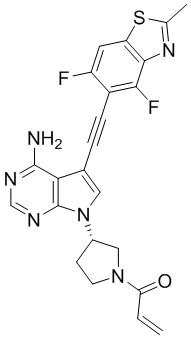
Данные, полученные методом ЯМР на ядрах 1Н, о соединениях, полученных в вышеуказанных примерах:
соединения (примера)
Пример 76. Получение (S)-1-(3-(4-амино-5-((2-(трифторметил)бензо[d]оксазол-5-ил)этинил)-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она
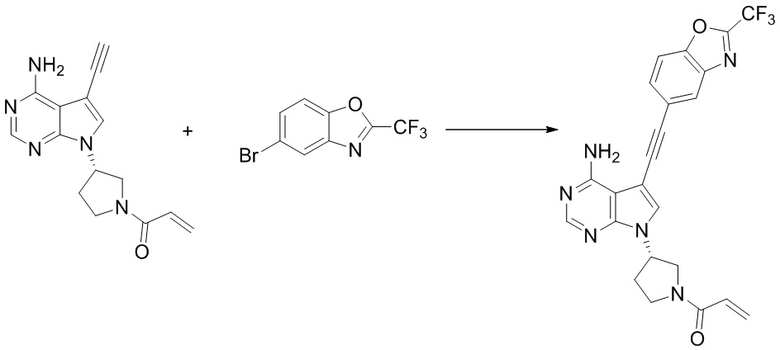
(S)-1-(3-(4-амино-5-этинил-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-он (30 мг, 0,1 ммоль), 5-бром-2-(трифторметил)бензо[d]оксазол (28 мг, 0,1 ммоль), триэтиламин (0.2 мл), йодид меди (2 мг, 0,01 ммоль) и тетракис(трифенилфосфин)палладий (12 мг, 0,01 ммоль) добавляли к ДМФ (1 мл). Реакционную смесь перемешивали при температуре 80°C в течение 12 ч в атмосфере азота, после чего в таком виде разделяли с помощью обращенно-фазовой колоночной хроматографии и лиофилизировали с получением (S)-1-(3-(4-амино-5-((2-(трифторметил)бензо[d]оксазол-5-ил)этинил)-7H-пирроло[2,3-d]пиримидин-7-ил)пирролидин-1-ил)проп-2-ен-1-она (3,5 мг, выход: 6%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 467 [M + H]+.
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,21 (s, 1H), 8,12 (s, 1H), 7,96-7,94 (d, J = 8 Гц, 1H), 7,81-7,79 (d, J = 8 Гц, 1H), 7,75-7,73 (d, 1H J = 8 Гц, 1H), 6,85-6,48 (m, 3H), 6,14-6,07 (m, 1H), 5,67-5,59 (m, 1H), 5,34-5,18 (m, 1H), 4,06-3,43 (m, 4H), 2,40-2,28 (m, 2H).
Пример 77. Получение (E)-1-(3-(4-амино-3-((2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-ил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-она
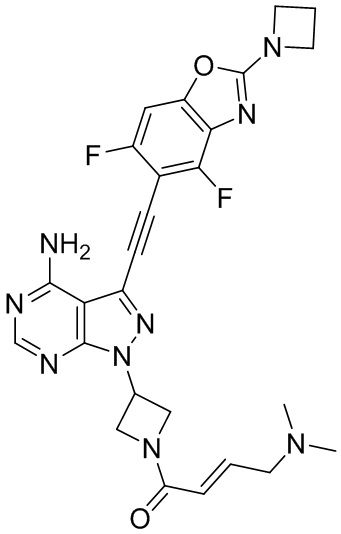
Стадия 1. Синтез 1-(азетидин-3-ил)-3-йод-1H-пиразоло[3,4-d]пиримидин-4-амина

Трет-бутил-3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-карбоксилат (2000 мг, 4,81 ммоль) растворяли в дихлорметане (20 мл) и добавляли раствор HCl / диоксана (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь концентрировали до сухого остатка с получением 1-(азетидин-3-ил)-3-йод-1H-пиразоло[3,4-d]пиримидин-4-амина (1400 мг, выход: 82,64%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 317 [M + H]+.
Стадия 2. Синтез (E)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-она
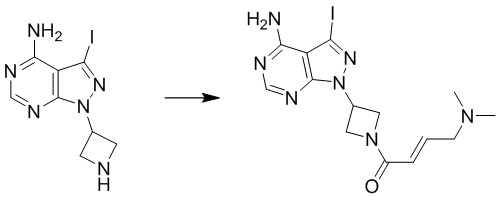
1-(азетидин-3-ил)-3-йод-1H-пиразоло[3,4-d]пиримидин-4-амин (700 мг, 1,99 ммоль) растворяли в N,N-диметилформамиде (5 мл), после чего добавляли (2E)-4-(диметиламино)бут-2-еноевую кислоту (394,59 мг, 2,38 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (1132,38 мг, 2,98 ммоль) и N,N-диизопропилэтиламин (769,79 мг, 5,96 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь разделяли с помощью обращенно-фазовой хроматографии с получением (E)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-она (400 мг, выход: 47%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 428 [M + H]+.
Стадия 3. Синтез (E)-1-(3-(4-амино-3-((2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-ил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-она
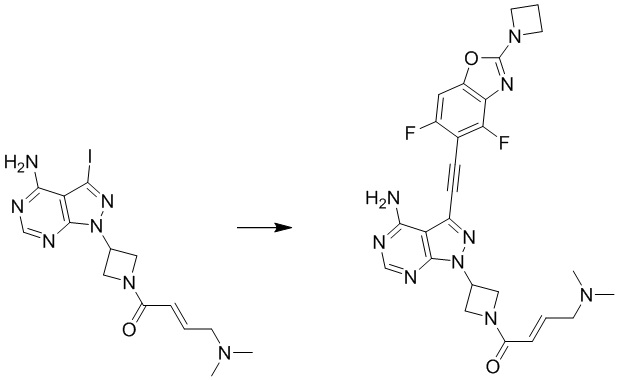
(E)-1-(3-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-он (100 мг, 0,23 ммоль), 2-(азетидин-1-ил)-5-этинил-4,6-дифтор-1,3-бензоксазол (65,77 мг, 0,28 ммоль), триэтиламин (1 мл), йодид меди (4,46 мг, 0,02 ммоль) и бис[5-(дифенилфосфиноалкил)циклопент-1,3-диен-1-ил]железо палладия дихлорид (8,56 мг, 0,01 ммоль) добавляли к ДМФ (5 мл). Реакционную смесь перемешивали при температуре 60°C в течение 2 ч в атмосфере азота, после чего в таком виде разделяли с помощью обращенно-фазовой колоночной хроматографии с получением (E)-1-(3-(4-амино-3-((2-(азетидин-1-ил)-4,6-дифторбензо[d]оксазол-5-ил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)азетидин-1-ил)-4-(диметиламино)бут-2-ен-1-она (30 мг, выход: 24%). Масс-спектрометрия c ионизацией распылением в электрическом поле (ESI-МС, m/z): 534 [M + H]+.
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,51-7,86 (m, 2H), 7,55 (d, J = 8,4 Гц, 1H), 6,77-6,28 (m, 2H), 6,11 (d, J = 15,4 Гц, 1H), 5,82-5,52 (m, 1H), 4,67 (t, J = 8,6 Гц, 1H), 4,54 (dd, J = 9,3, 5,2 Гц, 1H), 4,38 (t, J = 9,3 Гц, 1H), 4,24 (dd, J = 10,6, 5,2 Гц, 1H), 4,18 (t, J = 7,6 Гц, 4H), 2,97 (d, J = 6,2 Гц, 2H), 2,40-2,34 (m, 2H), 2,08 (s, 6H).
Вещество из примера 78 может быть получено путем отбора соответствующих исходных материалов в соответствии с методом синтеза (полного или частичного) вещества из примера 77.
соединения (примера)
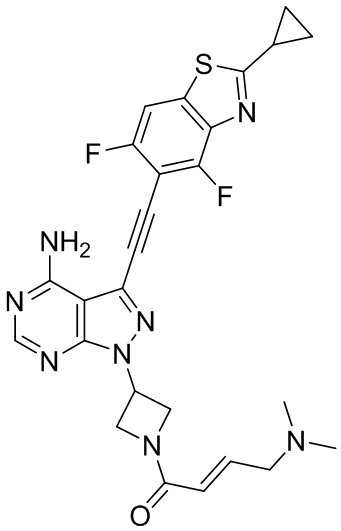
Данные, полученные методом ЯМР на ядрах 1Н, о соединениях, полученных в вышеуказанных примерах:
Оценка в ходе биологического анализа
I. Анализ пролиферации клеток в случае Tel-FGFR2 дикого типа (WT) и различных мутантных форм FGFR2 в клетках линии Baf3
Путем трансфекции клеток Baf получали клеточные линии, стабильно экспрессирующие слитый белок внутриклеточного киназного домена TEL-FGFR2 WT или слитые белки внутриклеточных доменов, содержащих мутации FGFR2 V564I, V564F, V564L, N549K и K659M. В день 1 суспензию клеток высевали в 96-луночный планшет, содержащий ростовую среду (DMEM, содержащая 1% Glutamax, 10% FBS и 1% Pen/Strep), в количестве 2000-4000 клеток / 90 мкл на лунку, после чего смесь инкубировали в течение ночи при температуре 37°C в присутствии 5% CO2. В день 2 10 мкл ростовой среды, содержащей 10-кратный основной раствор исследуемого вещества, добавляли к культуре клеток (9 концентраций; последовательное разведение с шагом 3, начиная с 1 мкМ; конечная концентрация ДМСО - 0,3%). В день 3, после инкубации при 37°C в присутствии 5% CO2 в течение 48 ч, в лунки 96-луночного планшета с клетками добавляли по 50 мкл реагента CellTiter Glo (CTG), после чего инкубировали планшет при комнатной температуре в течение 10 мин. Затем на устройстве для считывания микропланшетов с модулем обнаружения люминесценции измеряли интенсивность люминесценции в относительных световых единицах (RLU). Значения люминесценции в RLU нормировали с учетом % выживших клеток, после чего с помощью программы Prism строили кривые «концентрация-ответ» для расчета IC50 (нМ).
(FGFR2)
(FGFR2)
(FGFR2)
(FGFR2)
(FGFR2)
(FGFR2)
2. «+++++» указывает на то, что IC50 ≤ 0,5 нМ;
«++++» указывает на то, что 0,5 нМ < IC50 ≤ 5,0 нМ;
«+++» указывает на то, что 5,0 нМ < IC50 ≤ 50,0 нМ;
«++» указывает на то, что 50,0 нМ < IC50 ≤ 500,0 нМ;
«+» указывает на то, что IC50.> 500,0 нМ.
II. Анализ пролиферации клеток в случае Tel-FGFR3 дикого типа (WT) и FGFR3 V555M в клетках линии BaF3
Путем трансфекции клеток Baf получали клеточные линии, стабильно экспрессирующие слитый белок внутриклеточного киназного домена TEL-FGFR3 WT или слитые белки внутриклеточных доменов, содержащие мутацию FGFR3 V555M или FGFR3 N540K, а соединения из примеров по настоящему изобретению использовали для определения их влияния на пролиферацию клеток BaF3-Tel-FGFR3 WT, FGFR3 V555M или FGFR3 N540K. Методика анализа состояла в следующем:
1) Суспензию клеток высевали в 96-луночный планшет, содержащий ростовую среду (DMEM, содержащая 1% Glutamax, 10% FBS и 1% Pen/Strep), в количестве 2000-4000 клеток / 90 мкл на лунку, после чего смесь инкубировали в течение ночи при температуре 37°C в присутствии 5% CO2.
2) 10 мкл ростовой среды, содержащей 10-кратный основной раствор исследуемого вещества, добавляли к культуре клеток (9 концентраций; последовательное разведение с шагом 3, начиная с 1 мкМ; конечная концентрация ДМСО - 0,3%).
3) Смесь инкубировали при температуре 37°C в присутствии 5% CO2 в течение 48 ч.
4) В лунки 96-луночного планшета с клетками добавляли 50 мкл реагента CellTiter Glo (CTG) и инкубировали планшет при комнатной температуре 10 минут.
5) На устройстве для считывания микропланшетов с модулем обнаружения люминесценции измеряли интенсивность люминесценции в относительных световых единицах (RLU). Значения люминесценции в RLU нормировали с учетом % выживших клеток, после чего с помощью программы Prism строили кривые «концентрация-ответ» для расчета IC50 (нМ). Результаты анализа показаны в таблице ниже.
2. «+++++» указывает на то, что IC50 ≤ 0,5 нМ;
«++++» указывает на то, что 0,5 нМ < IC50 ≤ 5,0 нМ;
«+++» указывает на то, что 5,0 нМ < IC50 ≤ 50,0 нМ;
«++» указывает на то, что 50,0 нМ < IC50 ≤ 500,0 нМ;
«+» указывает на то, что IC50.> 500,0 нМ.
Из данных по биологической активности соединений, указанных в конкретных примерах, видно, что многие соединения по настоящему изобретению оказывают выраженное ингибирующее действие на FGFR дикого типа и мутантные формы FGFR на клеточном уровне, причем ингибирующее действие в отношении мутантных форм не является ослабленным.
Все документы, упомянутые в настоящем изобретении, включены в виде ссылок, и каждый документ цитируется индивидуально в виде ссылки. Кроме того, следует понимать, что специалисты в данной области, ознакомившись с вышеизложенной информацией по настоящему изобретению, могут внести различные модификации или изменения, и эти эквивалентные формы также входят в объем формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| ИНГИБИТОР CD73, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2787428C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2677653C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН-О-АЦИЛТРАНСФЕРАЗЫ ТИПА 1 | 2008 |
|
RU2486186C2 |
| ИНГИБИТОР FGFR, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2771526C1 |
| НОВЫЕ ДИАМИДЫ ПИРИМИДИН-4,6-ДИКАРБОНОВОЙ КИСЛОТЫ ДЛЯ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ КОЛЛАГЕНАЗ | 2003 |
|
RU2344129C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
Изобретение относится к соединению формулы (I), где значения R1, R2, R3, R5, R6, X, Y, Z1-Z4, m, n определены в формуле изобретения, его стереоизомеру или фармацевтически приемлемой соли, которое обладает высокой активностью в отношении мутантных форм FGFR. Также предложен способ получения соединения, содержащая его фармацевтическая композиция, его применение для получения лекарственного препарата, предназначенного для лечения и/или профилактики опухолей или злокачественных новообразований, опосредованных киназой FGFR, а также для лечения пациента с опухолью, резистентной к ингибитору FGFR, в частности его применение для получения лекарственного препарата, предназначенного для лечения и/или профилактики пациента с опухолью, несущей мутации V561, V565, N550, N540, V555, E566, K660 и/или V550 в FGFR-зависимом сигнальном пути. 8 н. и 15 з.п. ф-лы, 2 табл., 78 пр.
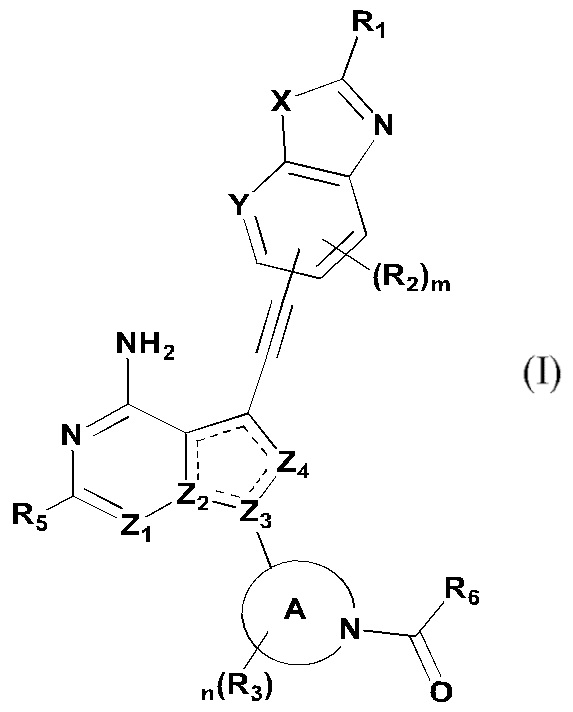
1. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль:

где  представляет собой двойную связь или одинарную связь;
представляет собой двойную связь или одинарную связь;
X представляет собой О или S;
Y представляет собой CR4 или N;
Z1 и Z4 каждый независимо представляют собой N или CR7;
Z2 и Z3 каждый независимо представляют собой N или С;
кольцо А представляет собой 4-6-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
R1 выбран из группы, состоящей из водорода, С1-10 алкила, С3-12 циклоалкила и С0-8 алкила-NR11R12, причем вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из галогена и 1-10 алкила;
R2, R3 и R4 каждый независимо выбраны из водорода, галогена, -С0-8 алкила-О-R9;
R5 и R7 представляют собой водород;
R6 выбран С2-10 алкенила, необязательно замещенного заместителем -С0-8 алкил-NR11R12;
каждый R9 независимо представляет собой С1-10 алкил;
R11 и R12 каждый независимо выбраны из группы, состоящей из водорода, С1-10 алкила и С3-12 циклоалкила;
или R11 и R12, вместе с атомом азота, непосредственно присоединенным к ним, образуют 4-10-членный гетероциклил;
m равно 0, 1 или 2;
n равно 0, 1, 2, 3 или 4;
если не определено специально, гетероатом «гетероциклила» в упомянутых выше заместителях представляет собой один атом азота.
2. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где R2, R3 и R4 каждый независимо выбраны из водорода, галогена и -С0-4 алкила-NR11R12, где R9 соответствует определению в п. 1.
3. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где R6 выбран из С2-4 алкинила, необязательно замещенного заместителем -С0-4 алкил-NR11R12, где R11 и R12 соответствуют определениям в п. 1.
4. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где R1 выбран из группы, состоящей из водорода, С1-4 алкила, С3-6 циклоалкила и -С0-4 алкила-NR11R12, причем вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из галогена и С1-4 алкила,
где R11 и R12 соответствуют определениям в п. 1.
5. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение формулы (I) представляет собой соединение, структура которого описывается формулой (II)
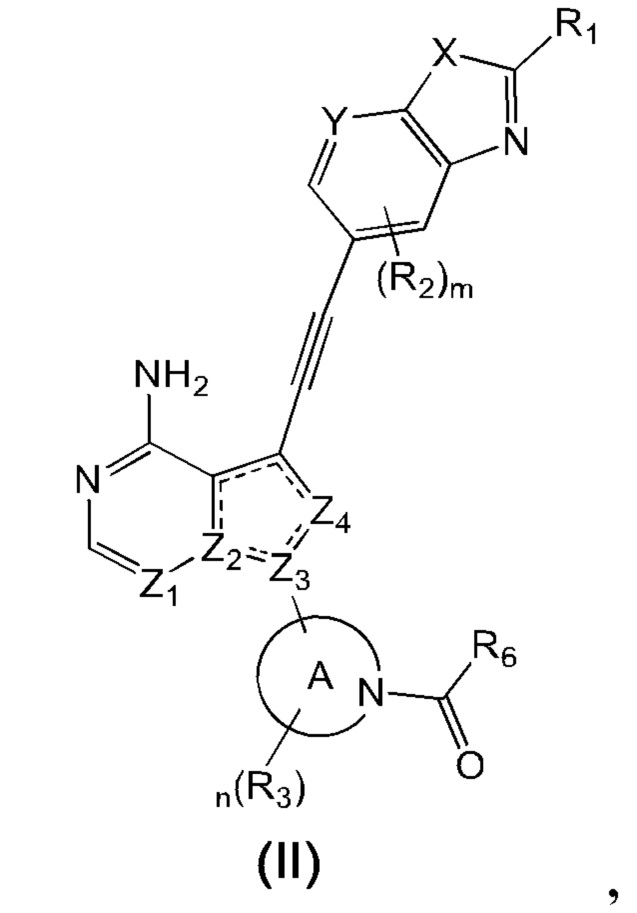
где  представляет собой двойную связь или одинарную связь;
представляет собой двойную связь или одинарную связь;
X представляет собой О или S;
Y представляет собой CR4 или N;
Z1 и Z4 каждый независимо представляют собой N или CR7;
Z2 и Z3 каждый независимо представляют собой N или С;
кольцо А представляет собой 6-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
R1 выбран из группы, состоящей из водорода, С1-4 алкила и -NR11R12, вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из галогена и С1-4 алкила;
каждый R2 независимо выбран из водорода и галогена;
каждый R3 независимо выбран из водорода и -CH2-O-R9;
R4 представляет собой водород;
R6 представляет собой С2-4 алкенил, необязательно замещенный одним или несколькими дополнительными заместителями, выбранными из -CH2-NR11R12; каждый R7 представляет собой водород,
где R9, R11, R12, тип соответствуют определениям в п. 1.
6. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IIIa) или формулой (IIIb):
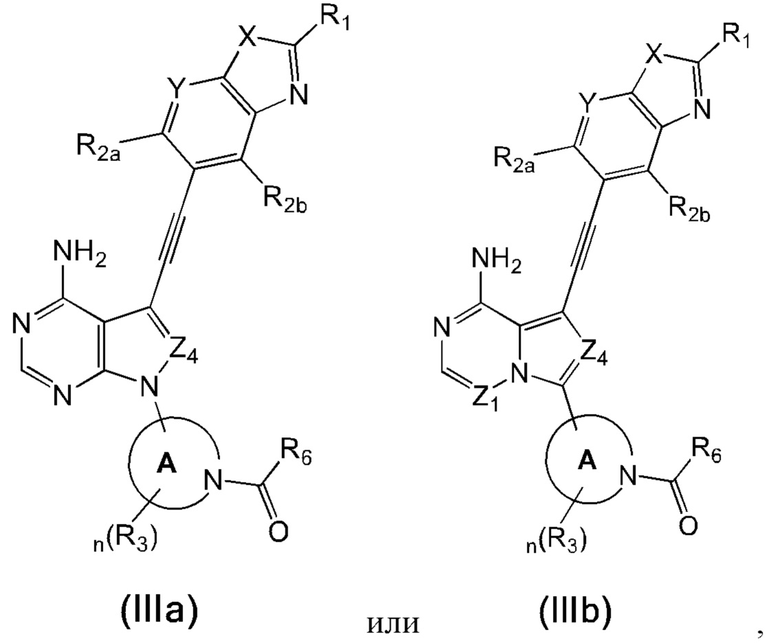
где каждый X независимо представляет собой О или S; каждый Y независимо представляет собой СН или N;
в соединении формулы (IIIb) Z1 представляет собой N или CR7;
каждый Z4 независимо представляет собой N или CR7;
каждое кольцо А независимо представляет собой 4-6-членный азотсодержащий гетероциклил, атом азота в котором связан с карбонилом;
каждый R1 независимо выбран из группы, состоящей из водорода, C1-4 алкила, С3-6 циклоалкила и -NR11R12, вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из галогена и С1-4 алкила;
каждый из R2a и R2b независимо выбран из водорода и галогена;
каждый R3 независимо выбран из водорода H-CH2-O-R9;
каждый R6 независимо представляет собой винил, необязательно замещенный одним или несколькими дополнительными заместителями, выбранными из -CH2-NR11R12; каждый R7 представляет собой водород, где R9, R11, R12 и n соответствуют определениям в п. 1.
7. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 6, где кольцо А вместе с -(R3)n образуют следующие структуры:
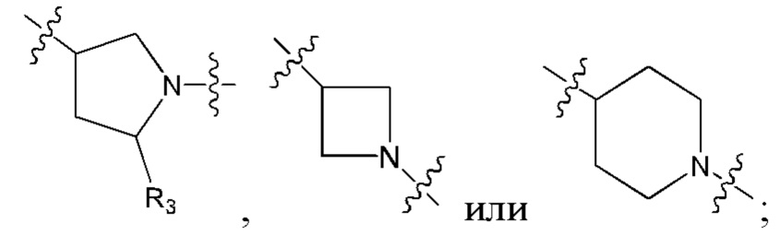
R3 выбран из группы, состоящей из водорода и -CH2-O-R9,
где R9 cooтветствует определению в п. 1.
8. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IVa1) или формулой (IVa2):
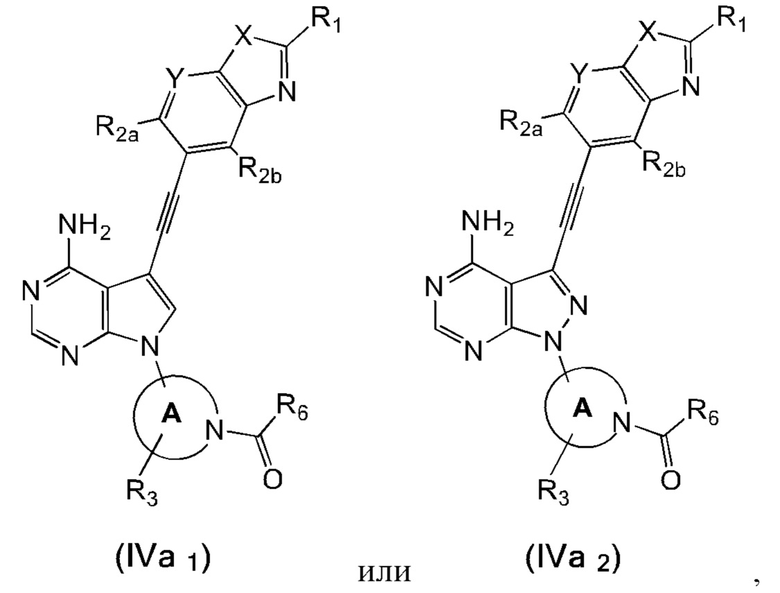
где каждый X независимо представляет собой О или S; каждый Y независимо представляет собой СН или N;
кольцо А, вместе с -R3, образуют следующие структуры:
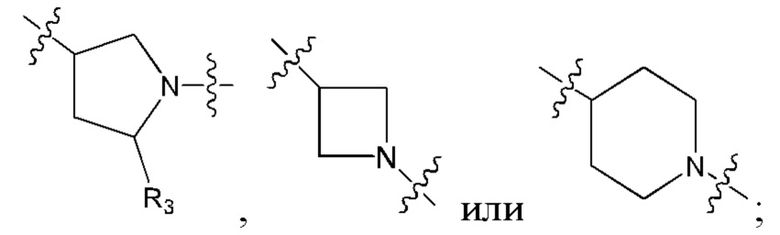
R3 выбран из группы, состоящей из водорода и -CH2-O-R9;
каждый R1 независимо выбран из группы, состоящей из водорода, С1-4 алкила, С3-6 циклоалкила и -NR11R12, вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из фтора, хлора и С1-4 алкила;
каждый из R2a и R2b независимо выбран из группы, состоящей из водорода, фтора и хлора;
каждый R6 независимо представляет собой винил, необязательно замещенный одним или несколькими дополнительными заместителями, выбранными из -CH2-NR11R12,
где R9, R11 и R12 соответствуют определениям в п. 1.
9. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение формулы (I) представляет собой соединение, структура которого описывается формулой (IVb1) или формулой (IVb2):
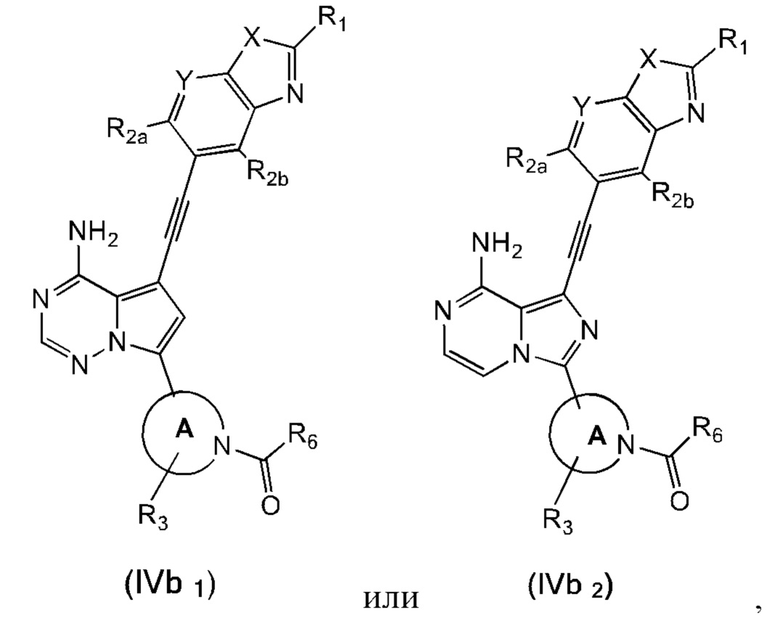
где каждый X независимо представляет собой О или S; каждый Y независимо представляет собой СН или N;
кольцо А, вместе с -R3, образуют следующие структуры:
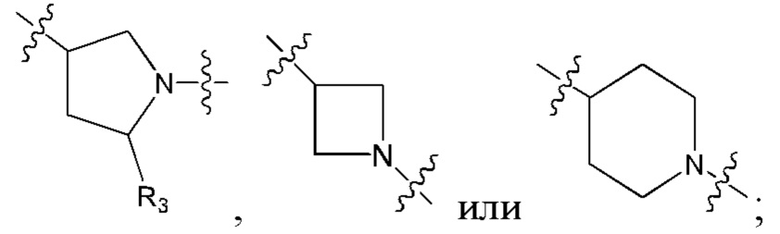
R3 выбран из водорода и -CH2-O-R9;
каждый R1 независимо выбран из группы, состоящей из водорода, С1-4 алкила, С3-6 циклоалкила и -NR11R12, вышеуказанные группы необязательно замещены одним или несколькими дополнительными заместителями, выбранными из фтора, хлора и С1-4 алкила;
каждый из R2a и R2b независимо выбран из водорода, фтора и хлора;
каждый R6 независимо представляет собой винил, необязательно замещенный одним или несколькими дополнительными заместителями, выбранными из -CH2-NR11R12,
где R9, R11 и R12 соответствуют определениям в п. 1.
10. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по п. 1, где каждый R9 независимо выбран С1-4 алкила;
R11 и R12 каждый независимо выбраны из водорода, С1-4 алкила и С3-6 циклоалкила;
или R11 и R12, вместе с атомом азота, непосредственно присоединенным к ним, образуют 4-6-членный гетероциклил.
11. Соединение формулы (I), его стереоизомер или фармацевтически приемлемая соль по любому из пп. 1-10, выбранное из следующих соединений:
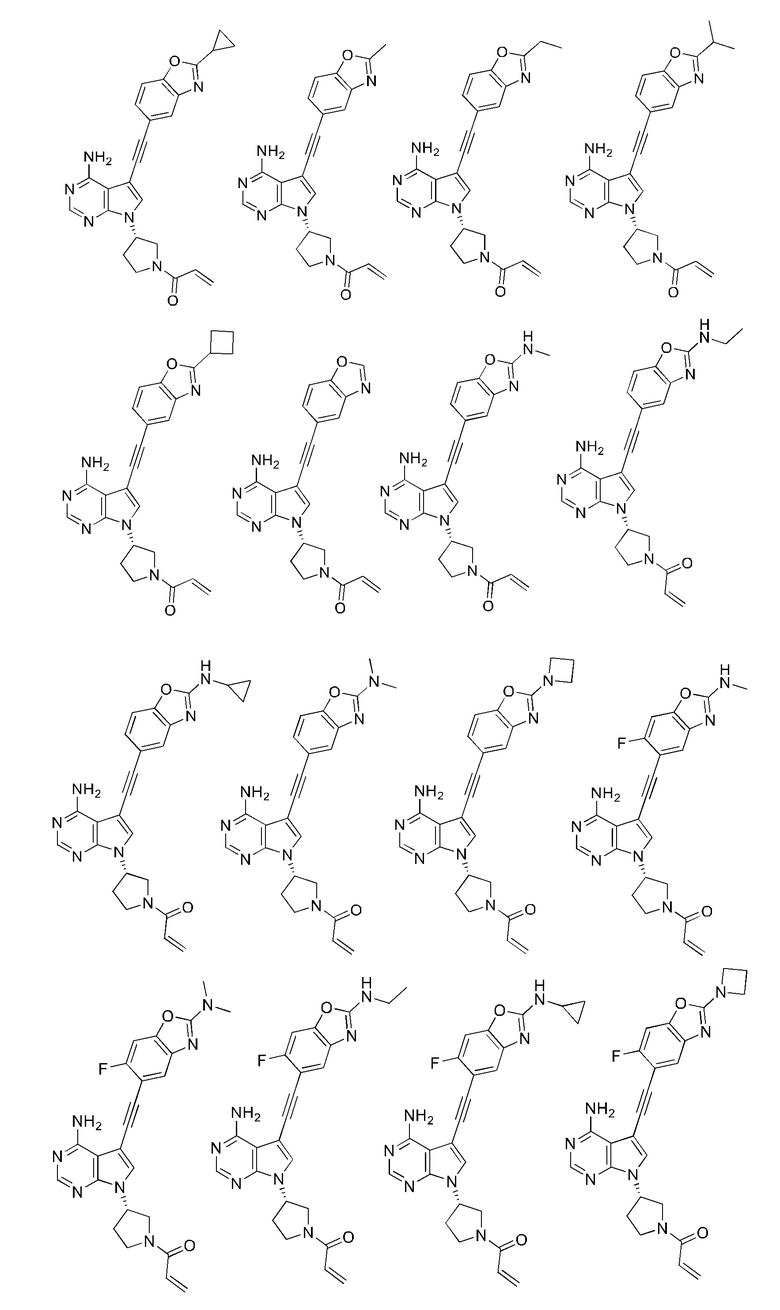
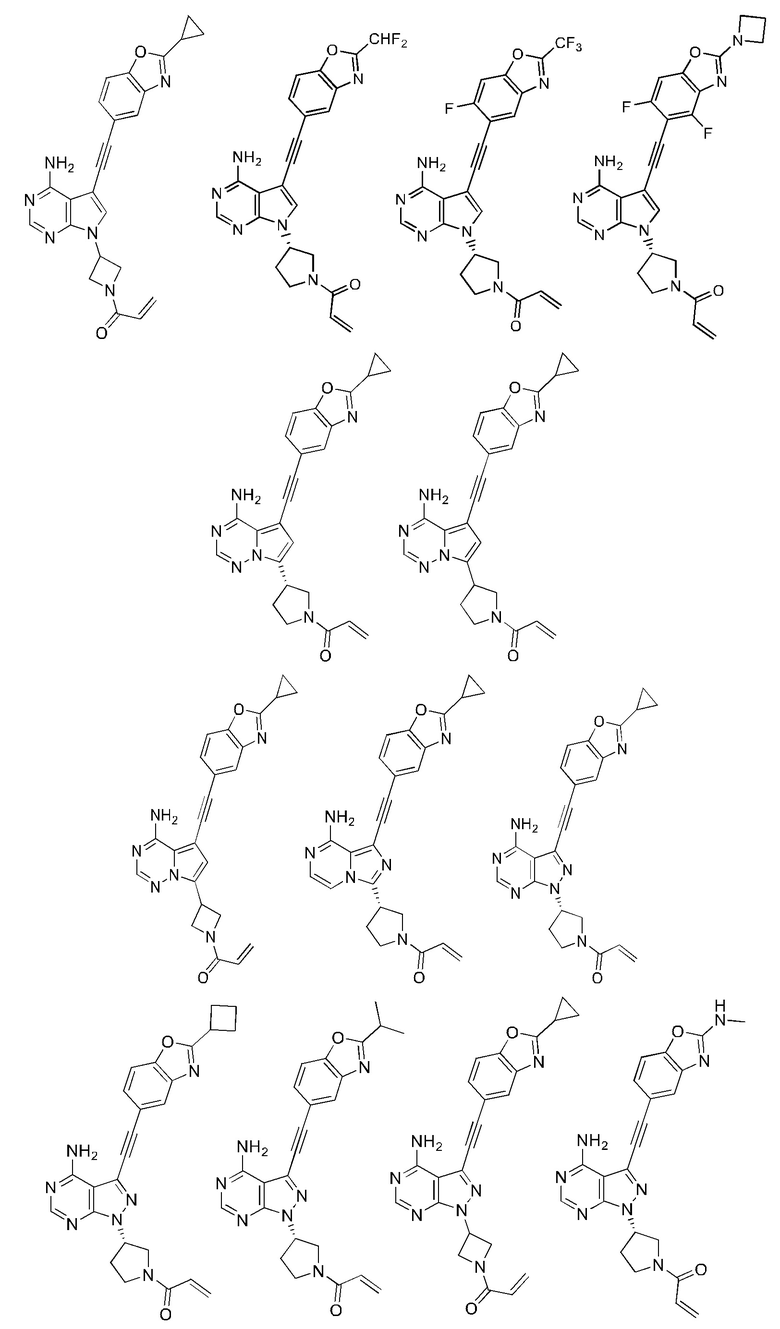
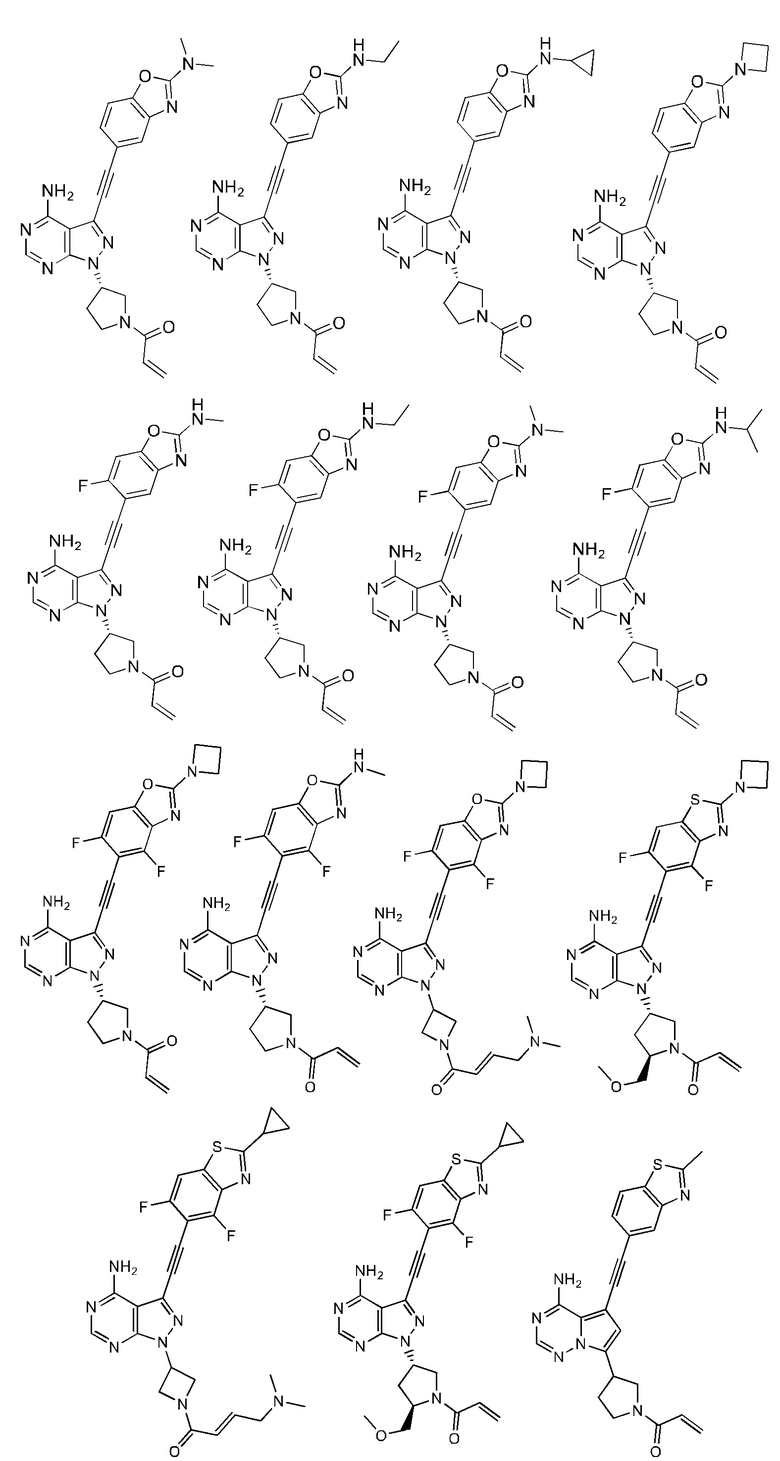
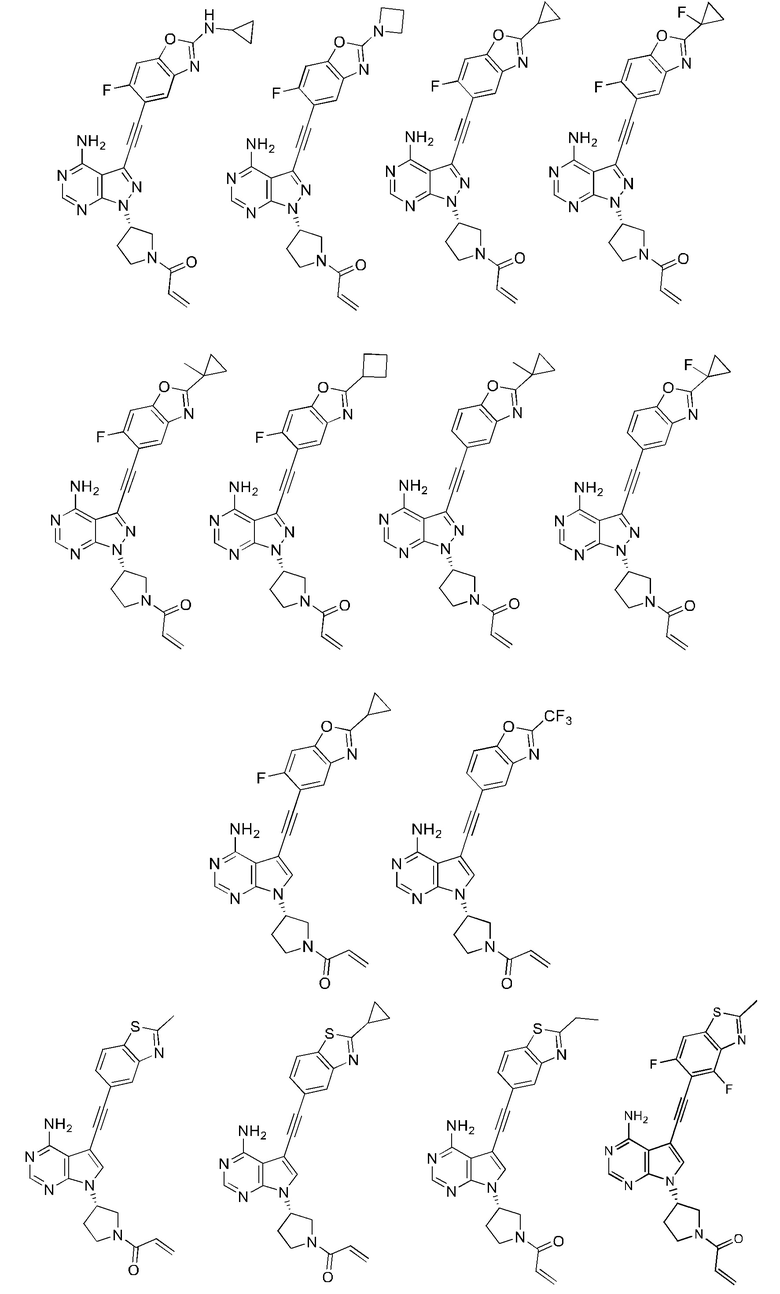
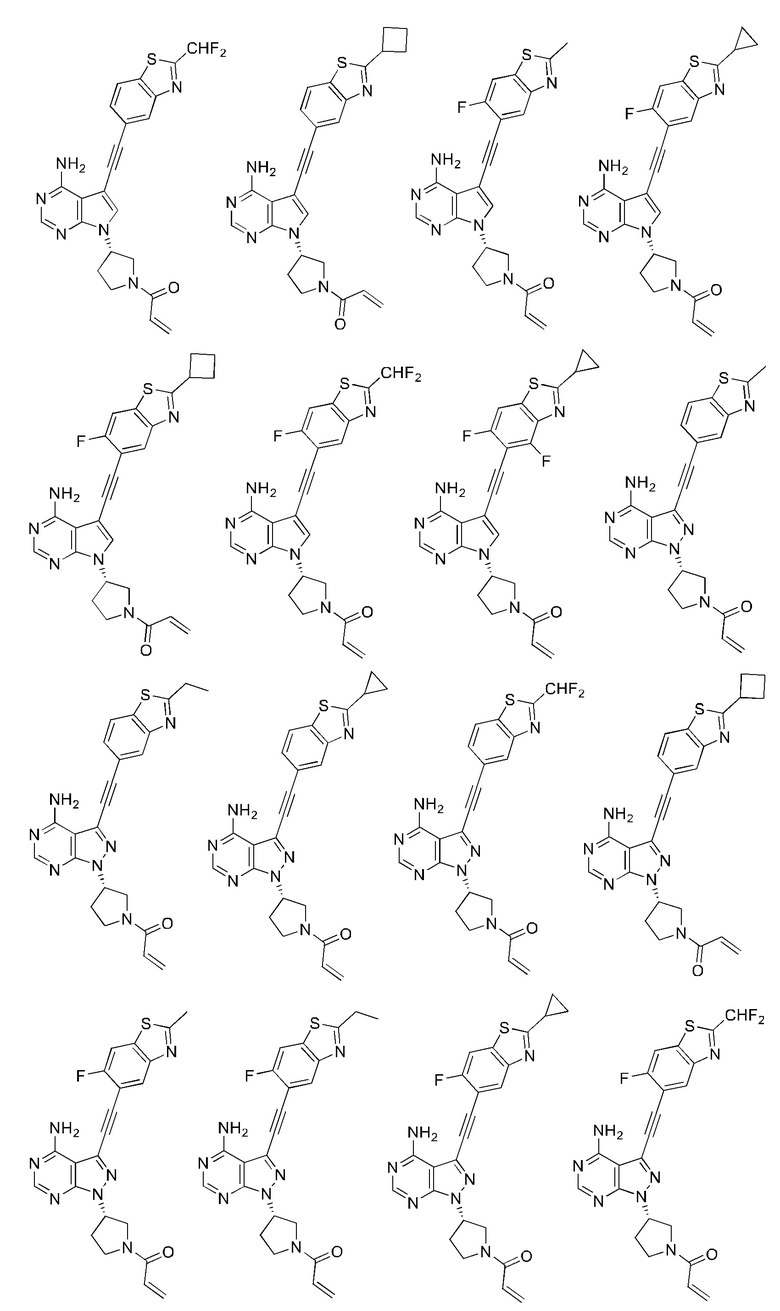
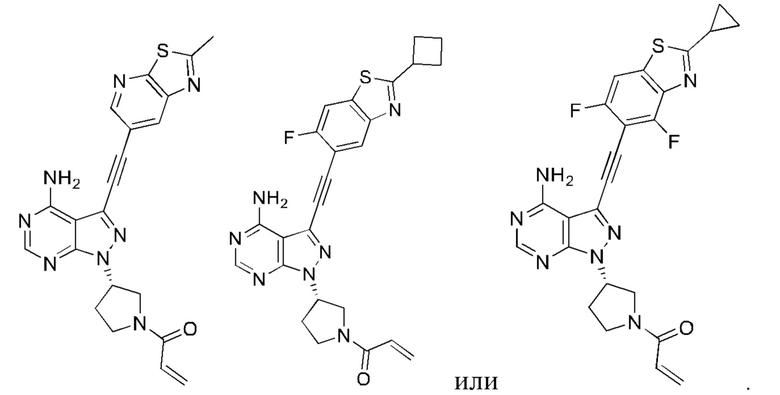
12. Способ получения соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11, включающий следующую стадию:
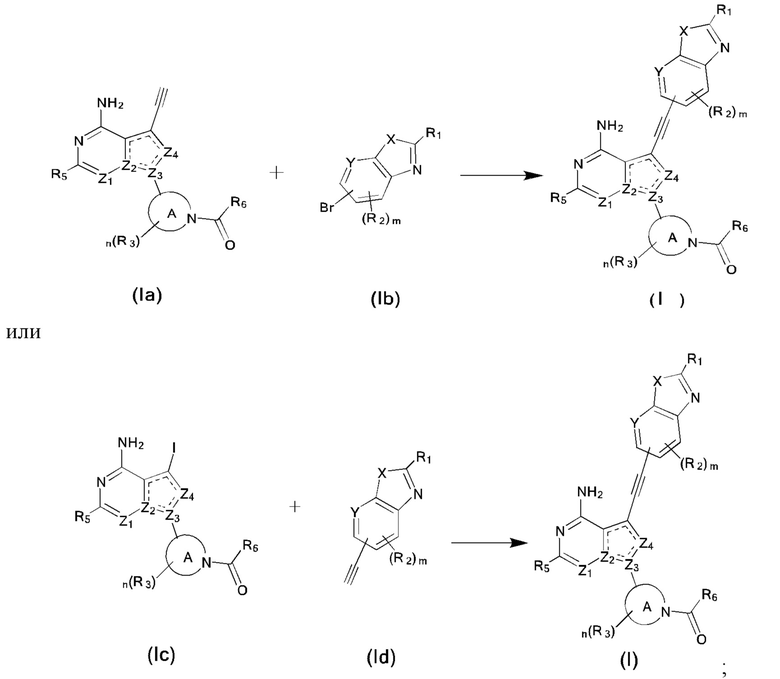
где кольцо А, X, Y, Z1, Z2, Z3, Z4, R1, R2, R3, R5, R6, m и n соответствуют определениям в п. 1.
13. Фармацевтическая композиция для профилактики или лечения заболевания или состояния, опосредованного киназой FGFR, включающая соединение формулы (I), его стереоизомер или фармацевтически приемлемую соль по любому из пп. 1-11, а также фармацевтически приемлемый носитель.
14. Применение соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11 для получения лекарственного препарата для лечения пациента с опухолью, резистентной к ингибитору FGFR.
15. Применение по п. 14, где пациент с опухолью имеет мутации в FGFR V561, V565, N550, N540, V555, Е566, K660 и/или V550.
16. Применение по п. 15, где пациент с опухолью имеет мутации FGFR2 V565F, V565I, V565L, V565M, N550K, N550H, Е566А, E566G, K660M и/или K660Q.
17. Применение по п. 15, где пациент с опухолью имеет мутации FGFR3 V555M, V555L и/или N540K.
18. Применение соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11 для профилактики или лечения заболевания или состояния, опосредованного киназой FGFR.
19. Применение соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11 для профилактики или лечения опухоли или злокачественного новообразования, опосредованного киназой FGFR.
20. Применение по п. 19, где опухоль или злокачественное новообразование представляет собой рак мочевого пузыря, рак молочной железы, рак шейки матки, колоректальный рак, рак эндометрия, рак желудка, рак головы и шеи, карциному почки, рак печени, рак легких, рак яичников, рак простаты, рак пищевода, рак желчного пузыря, рак поджелудочной железы, рак щитовидной железы, рак кожи, лейкоз, множественную миелому, хроническую лимфоцитарную лимфому, Т-клеточный лейкоз взрослых, В-клеточную лимфому, острый миелоцитарный лейкоз, лимфому Ходжкина или неходжкинскую лимфому, макроглобулинемию Вальденстрема, волосатоклеточную лимфому, клеточную лимфому, лимфому Беркитта, глиобластому, меланому или рабдомиосаркому.
21. Применение соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11 для получения лекарственного препарата для лечения заболевания, опосредованного киназой FGFR, выбранного из миелопролиферативного заболевания, заболевания клеток костной или хрящевой ткани и гипофосфатемии.
22. Применение по п. 21, где миелопролиферативное заболевание представляет собой эритроцитоз, первичный тромбоцитоз или первичный миелофиброз; заболевание клеток костной или хрящевой ткани представляет собой дисплазию, ахондроплазию, карликовость, танатофорную дисплазию (ТД), синдром Аперта, синдром Крузона, синдром Джексона-Вейсса, синдром кутисовой извилины Бира-Стивенсона, синдром Пфайффера или синдром черепной мышечной атрофии; гипофосфатемия представляет собой X-сцепленный гипофосфатемический рахит, аутосомно-рецессивный гипофосфатемический рахит, аутосомно-доминантный гипофосфатемический рахит или остеомаляцию яичников.
23. Применение соединения формулы (I), его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-11 или фармацевтической композиции по п. 13 для лечения заболеваний, связанных с аберрантной экспрессией и мутацией рецептора FGFR2 или FGFR3 или аберрантной экспрессией и активностью соответствующего лиганда как селективного ингибитора FGFR2 и/или FGFR3.
| CN 109776544 A, 21.05.2019 | |||
| CN 105461720 B, 06.08.2019 | |||
| WO 2007087395 A2, 02.08.2007 | |||
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| US 20160193210 A1, 07.07.2016 | |||
| US 20160136168 A1, 19.05.2016 | |||
| CN 107344940 A, 14.11.2017. | |||

