Область техники
Изобретение относится к области органической химии, фармакологии и медицины и касается нового способа синтеза 5-(1-(3,5-бис(трифторметил)фенокси)этил) циклогексан-1,3-диона и его S-энантиомера, которое может быть использовано для изготовления лекарственных средств для лечения болезни двигательных нейронов.
Уровень техники
Болезнь двигательных нейронов (БДН) - группа дегенеративных заболеваний центральной нервной системы, при которых происходит поражение как верхних (моторная кора головного мозга), так и нижних (передние рога спинного мозга и ядра черепных нервов) двигательных нейронов. Гибель клеток нервной системы приводит к параличам и последующей атрофии мышц. Формы заболеваний (боковой амиотрофический склероз, первичный латеральный склероз, прогрессирующая мышечная атрофия, прогрессирующий бульбарный паралич, псевдобульбарный паралич, спинальная мышечная атрофия) отличаются скоростью прогрессирования и разными видами повреждения двигательных нейронов.
Известно, что к разрушению двигательных нейронов при БДН может приводить неправильная свертываемость белка mSOD1 и накопление патологического белка TDP-43 в нервных тканях. Такое скопление белков мешает нейронам работать; транспортировать в них питательные вещества, повреждает митохондрии, нарушает синтез белка. Вследствие этого процесса, нейроны разрушаются и не передают информацию к мышцам, что приводит к их параличу и атрофии.
Исследование биологической активности 5-(1-(3,5-бис(трифторметил)фенокси) этил)циклогексан-1,3-диона, разработанного исследователями из США (WO2011059821A2, [1]), показало его способность снижать токсичность mSOD1, восстанавливать митохондрии и эндоплазматический ретикулум клеток нейронов, где происходит синтез и транспорт белков, что в конечном итоге улучшает функционирование двигательных нейронов. Проведенные исследования показали, что 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-дион способен остановить дегенерацию нейронов и по эффективности превосходит существующие лекарственные средства, одобренные для лечения БДН (Genç B. et al. Improving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology // Clin Transl Med . 2021 Feb;11(2):e336. doi: 10.1002/ctm2.336, [2]).
Согласно [1] (схемы 4 и 5), при получении соединения 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона на первом этапе из 3,5-бис(трифторметил)фенола под воздействием алкилирующего агента получают соответствующий этиловый эфир, который затем восстанавливают в присутствии подходящего восстанавливающего агента (например, гидрида металла, такого как LiAIH4), до соответствующего спирта. Его дальнейшее ацилирование и омыление с последующим окислением обеспечивает получение конечного продукта.
В статье Zhang Y. et al. Chiral cyclohexane 1,3-diones as inhibitors of mutant SOD1-dependent protein aggregation for the treatment of ALS // ACS medicinal chemistry letters. - 2012. Т.3. №. 7 pp 584-587 ([3]) авторами изобретения [1] был предложен альтернативный способ синтеза хиральных циклогексан-1,3-дионов, в том числе 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона, являющийся ближайшим аналогом настоящего изобретения. Исходными компонентами для синтеза являлись этиллактат и 3,5-дитрифторметил фенол, из которых по реакции Мицунобу был получен эфир. Далее последовательно были получены альдегид, енон и конечный дион через сопряжённое присоединение по Михаэлю. Данное решение имеет некоторые недостатки, а именно: невысокий выход конечного продукта (53%) и высокую длительность определенных стадий синтеза.
Раскрытие изобретения
Задачей изобретения является разработка метода получения 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона и его S-энантиомера, обеспечивающего ускорение производства и повышение выхода целевого продукта.
Поставленная задача решается путем разработки способа получения 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона, имеющего структурную формулу
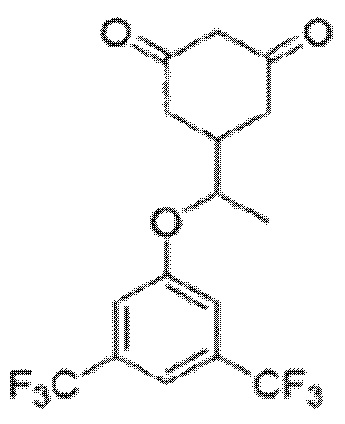
его рамемической смеси или энантиомерно чистой (S)-формы, включающего следующие стадии:
(а) взаимодействие 2-(3,5-бис(трифторметил)фенокси)пропаналя в виде рацемической смеси или энантиомерно чистой (S)-формы с (карбэтоксиметилен)трифенилфосфораном с получением этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы;
(б) взаимодействие полученного этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата с трет-бутилацетоацетатом и трет-бутилатом калия с получением целевого соединения в виде, соответственно, рацемата или его энантиомерно чистой (S)-формы,
согласно схеме:
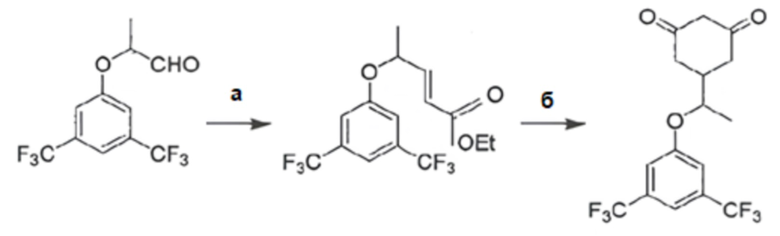 .
.
В некоторых вариантах изобретения для получения энантиомерно чистой (S)-формы 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона проводят его выделение из полученного рацемата 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона.
В частных вариантах изобретения выделение энантиомерно чистой (S)-формы 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона осуществляют методом хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ), методом превращения рацемата в смесь диастереомеров или методом кинетического расщепления. В предпочтительных вариантах выделение энантиомерно чистой (S)-формы осуществляют методом хиральной ВЭЖХ.
В предпочтительных вариантах изобретения реакции на стадиях (а) и (б) проводят в полярном апротонном растворителе.
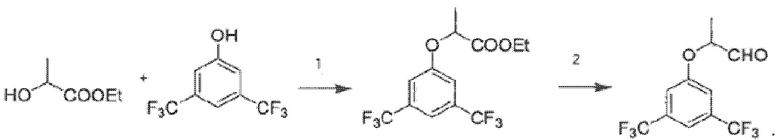
В некоторых вариантах изобретения 2-(3,5-бис(трифторметил)фенокси)пропаналь в виде рацемической смеси или энантиомерно чистой (S)-формы, используемый на стадии (а), получают путем
взаимодействия на первом (1) этапе 3,5-бис(трифторметил)фенола с этиллактатом в виде рацемической смеси или энантиомерно чистой (S)-формы с получением этил-2-(3,5-бис(трифторметил)фенокси)пропионата по реакции Мицунобу в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы;
проведения на втором (2) этапе реакции восстановления полученного этил-2-(3,5-бис(трифторметил)фенокси)пропионата с получением 2-(3,5-бис(трифторметил)фенокси)пропаналя в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы,
согласно схеме:
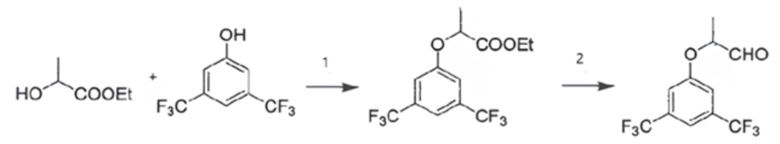 .
.
В предпочтительных вариантах изобретения на этапе (1) взаимодействие 3,5-бис(трифторметил)фенола с этиллактатом проводят в присутствии трифенилфосфина в полярном апротонном растворителе.
В предпочтительных вариантах изобретения на этапе (2) реакцию восстановления проводят в полярном апротонном растворителе, а в качестве восстановителя используют гидрид диизобутилалюминия.
В частных вариантах изобретения полярный апротонный растворитель выбирается независимо и представляет собой тетрагидрофуран, 1,4-диоксан или диметилсульфоксид.
В частных вариантах изобретения очистку промежуточного и целевого продукта на каждом из этапов (а), (б) и (1) осуществляют с помощью флэш-хроматографии.
В результате осуществления изобретения достигаются следующие технические результаты:
• разработан новый способ получения 5-(1-(3,5-бис(трифторметил) фенокси)этил)циклогексан-1,3-диона и его S-энантиомера - соединения, потенциально высоко эффективного для лечения болезни двигательных нейронов, в том числе бокового амиотрофического склероза, - путем изменения пути синтеза сырье-эфир-альдегид-кетон-продукт на сырьё-эфир-альдегид-эфир-продукт;
• разработанный способ обеспечивает повышение выхода целевого продукта до более 80%;
• разработанный способ обеспечивает существенное ускорение синтеза целевого соединения.
Краткое описание рисунков
Фиг.1 ЯМР-спектр синтезированного соединения ((S)-(5-(1-(3,5-бис(трифторметил) фенокси)этил)циклогексан-1,3-диона).
Термины и определения
Если иное не оговаривается, все технические и научные термины, используемые в данной заявке, имеют то же самое значение, которое понятно для специалистов в данной области. Ссылки на методики, используемые при описании данного изобретения, относятся к хорошо известным методам, включая изменения этих методов и замену их эквивалентными методами, известными специалистам.
В документах данного изобретения термины «включает», «включающий» и т.п., а также «содержит», «содержащий» и т.п. интерпретируются как означающие «включает, помимо всего прочего» (или «содержит, помимо всего прочего»). Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «и/или» означает один, несколько или все перечисленные элементы.
Термин «необязательный» или «необязательно» или «опциональный» или «опционально», используемый в данном документе, означает, что описываемое впоследствии событие или обстоятельство может, но не обязательно, произойти, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, в которых оно не происходит.
Термин «энантиомерно чистый» означает соединения, имеющие энантиомерный избыток не менее 95% (т.е. минимум 95% одного энантиомера и максимум 5% другого энантиомера) до энантиомерного избытка 100% (т.е. 100% одного энантиомера и нуль другого), в частности, соединения, имеющие энантиомерный избыток от 96 до 100% и особенно энантиомерный избыток от 98 до 100%. Энантиомерно чистые формы соединений могут быть легко получены из энантиомерно чистых изомерных форм исходных материалов при условии, что последующие реакции проходят стереоспецифически; или могут быть получены путем разделения рацемата.
Термин «энантиомерно чистая (S)-форма» означает избыток (S)-формы энантиомера (S-энантиомера).
Термины «рацемическая смесь» и «рацемат», «(±)» - обозначают эквимолярную смесь двух энантиомерных молекул, не обладающую оптической активностью.
Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе.
Подробное описание изобретения
Соединение (±)-5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-дион, и в большей степени его S-энантиомер, т.е. (S)-5-(1-(3,5-бис(трифторметил)фенокси) этил)циклогексан-1,3-дион
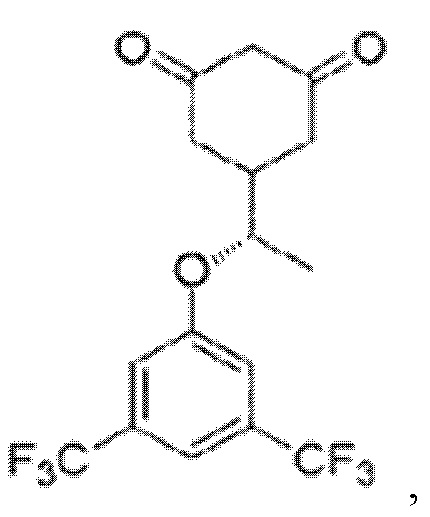
направлено на лечение болезни двигательных нейронов, таких как боковой амиотрофический склероз, первичный латеральный склероз, прогрессирующая мышечная атрофия, прогрессирующий бульбарный паралич, псевдобульбарный паралич, спинальная мышечная атрофия. Способность преодолевать гематоэнцефалический барьер, низкая токсичность и высокая эффективность в остановке деградации двигательных нейронов являются весомыми предпосылками для перспективного применения данного соединения в медицине и использования для изготовления лекарственных средств для лечения болезни двигательных нейронов.
Настоящее изобретение направлено на разработку нового метода получения (±)-5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона и его S-энантиомера.
Предлагаемый способ включает следующие стадии:
(а) взаимодействие 2-(3,5-бис(трифторметил)фенокси)пропаналя с (карбэтоксиметилен)трифенилфосфораном с получением этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата;
(б) взаимодействие полученного этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата с трет-бутилацетоацетатом и трет-бутилатом калия с получением целевого соединения.
Синтез энантиомерно чистой (S)-формы осуществляют при использовании в качестве исходного компонента на этапе (а) 2-(3,5-бис(трифторметил)фенокси)пропаналя в энантиомерно чистой (S)-форме, все дальнейшие реакции при этом проходят стереоспецифически.
Исходное соединение 2-(3,5-бис(трифторметил)фенокси)пропаналь может быть получено любыми известными методами. В некоторых предпочтительных вариантах изобретения 2-(3,5-бис(трифторметил)фенокси)пропаналь может быть получен согласно методу, описанному в документе [3], следующим образом:
(1) взаимодействие 3,5-бис(трифторметил)фенола с этиллактатом с получением этил-2-(3,5-бис(трифторметил)фенокси)пропионата по реакции Мицунобу;
(2) реакция восстановления полученного этил-2-(3,5-бис(трифторметил) фенокси)пропионата с получением 2-(3,5-бис(трифторметил)фенокси)пропаналя.
Синтез энантиомерно чистой (S)-формы осуществляют при использовании в качестве исходного компонента на этапе (1) этиллактата в энантиомерно чистой (S)-форме, все дальнейшие реакции при этом проходят стереоспецифически.
Таким образом, в предпочтительных вариантах воплощения предлагаемый метод включает следующие этапы:
1. взаимодействие 3,5-бис(трифторметил)фенола с этиллактатом с получением этил-2-(3,5-бис(трифторметил)фенокси)пропионата по реакции Мицунобу;
2. реакция восстановления полученного этил-2-(3,5-бис(трифторметил) фенокси)пропионата с получением 2-(3,5-бис(трифторметил)фенокси)пропаналя;
3. взаимодействие 2-(3,5-бис(трифторметил)фенокси)пропаналя с (карбэтоксиметилен)трифенилфосфораном с получением этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата;
4. взаимодействие (±)-этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата с трет-бутилацетоацетатом и трет-бутилатом калия в тетрагидрофуране в качестве растворителя с получением целевого соединения.
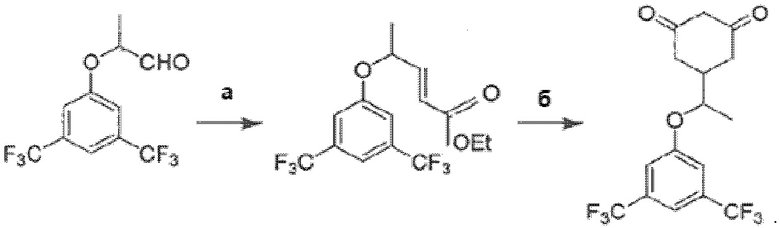
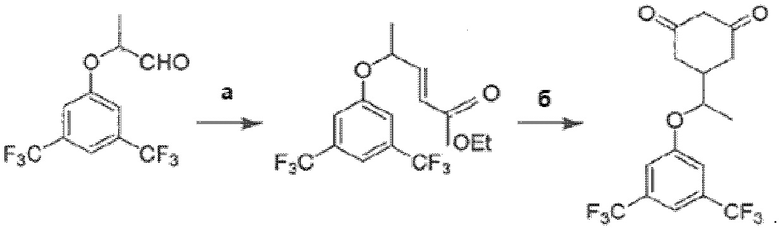
Как это представлено на схеме ниже:
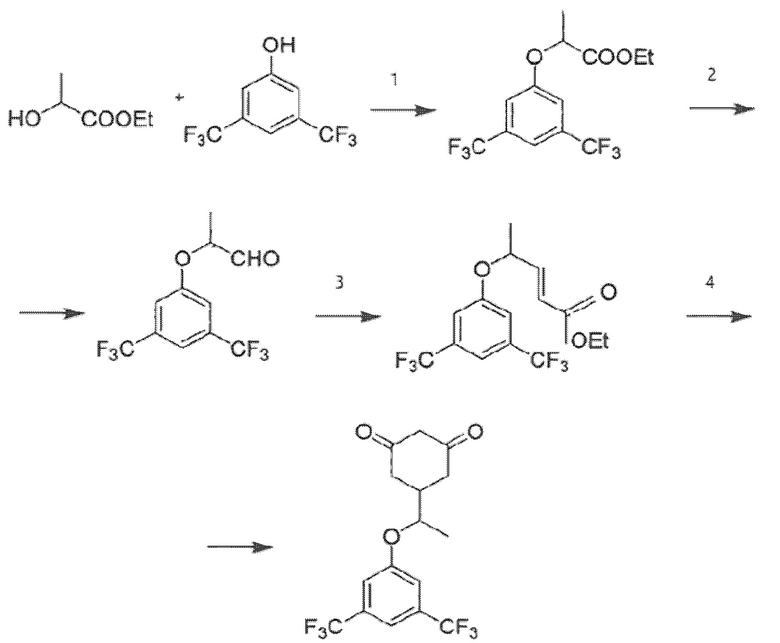
Синтез энантиомерно чистой (S)-формы осуществляют при использовании в качестве исходного компонента на первом этапе этиллактата в энантиомерно чистой (S)-форме, все дальнейшие реакции при этом проходят стереоспецифически.
При использовании в качестве исходных компонентов соединений в виде рацемической смеси синтез проходит не стереоспецифически. В результате такого синтеза получают рацемат 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона. Несмотря на то, что рацемат обладает необходимыми качествами для его применения в лечении БДН, как показали проведенные исследования (см, например, WO2023056307A1), S-энантиомер является более перспективным для применения в медицине. S-энантиомер может быть выделен из полученного рацемата любыми способами, хорошо известными специалистам в данной области (например, такими как хиральная ВЭЖХ, превращение рацемата в смесь диастереомеров, кинетическое расщепление и др.).
Таким образом, предлагаемое изобретение позволяет упростить и улучшить существующую методику синтеза рацемата 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона и получение его энатиомерно чистой (S)-формы.
Главные отличия метода по изобретению от известной методики, описанной в [3], заключаются в следующем:
• Согласно методике, описанной в [3], путь синтеза представляет собой сырье-эфир-альдегид-кетон-продукт. В способе по изобретению, третья стадия (альдегид-кетон) была изменена на стадию альдегид-эфир. В результате был получен эфир ((±)-этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноат) вместо кетона. Достигается это заменой 1-трифенилфосфоранилиден-2-пропанона на (карбэтоксиметилен)трифенилфосфоран, что обусловлено необходимостью получения эфира для проведения последующей стадии.
• Четвертая стадия (кетон-продукт) была изменена в способе по изобретению на стадию эфир-продукт. Для этого на четвертой стадии вместо диэтилмалоната, этилата натрия и этанола использовались трет-бутилацетоацетат, тетрагидрофуран и трет-бутилат калия. Это позволило сократить длительность стадии с 12 часов до 1 часа. При этом выход готового продукта составил 83% вместо 53% согласно известной методике.
Предлагаемый способ получения 5-(1-(3,5-бис(трифторметил)фенокси) этил)циклогексан-1,3-диона более подробно описан ниже в разделе примеры. Следует понимать, что перечисленные методы и приемы способа не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. Также следует понимать, что приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Пример 1. Получение энантиомерно чистой (S)-формы 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона.
Стадия 1.
Получение эфира - (S)-этил-2-(3,5-бис(трифторметил)фенокси)пропионата, М=330 г/моль (Реакция Мицунобу).
В стеклянный реактор с рубашкой загружают 118 г (0,45 моль) трифенилфосфина в 750 мл полярного апротонного растворителя (тетрагидрофурана (ТГФ)), последовательно при перемешивании приливают 92 г (0,4 моль) исходного 3,5-бис(трифторметил)фенола и 48 г (0,4 моль) исходного (S)-этиллактата.
Затем смесь охлаждают до 0°C (с помощью криостата) и добавляют по каплям 70 мл диэтилазодикарбоксилата (DEAD) со скоростью дозирования - 2 капли/сек.
Реакционную среду 24 часа при температуре +20°C в инертной среде (аргон).
Затем из реакционной системы отгоняют растворитель с помощью роторного испарителя (60 rpm, полный вакуум, температура бани +50°C) до получения сухого твёрдого осадка.
Далее добавляют 200 мл диэтилового эфира (+4°C) и интенсивно перемешивают, выпавший осадок фильтруют с помощью воронки Шотта, закреплённой в колбе Бунзена объёмом 1000 мл. Оставшийся раствор упаривают в роторном испарителе на 50% и убирают в холодильник на -20°С на 1 час. Выпавший осадок вновь фильтруют при тех же условиях. Процедуру повторяют (3-4 раза) до тех пор, пока не получат однородный раствор без осадка.
Полученный гомогенный раствор упаривают с помощью роторного испарителя до полной отгонки растворителя.
Далее проводят флэш-хроматографию. На воронку Шотта, закреплённую в колбе Бунзена, влажным способом наносят силикагель для хроматографии (0,060-0,200 мм, 60 А) в гексане. Полученный эфир растворяют в гексане в соотношении 1:2 и наносят сверху на фильтр через слой стеклянной ваты. Ждут, пока растворитель пройдёт через фильтр в колбу Бунзена.
Далее проводят флэш-хроматографию элюентом гексан-этилацетат в соотношении 10:1. Отбирают фракции объёмом по 100 мл каждая. Каждую фракцию анализируют с помощью тонкослойной хроматографии (далее - ТСХ) на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат - 5:1. Собирают фракции до прекращения наблюдения пятна продукта на тонкослойной хроматографии.
Полученные фракции объединяют и совместно упаривают с помощью роторного испарителя. Выход: 126 г (95%)
Стадия 2.
Получение альдегида - (S)-2-(3,5-бис(трифторметил)фенокси)пропаналя, М=286 г/моль.
В стеклянный реактор с рубашкой загружают раствор 126 г полученного на первой стадии эфира в 450 мл полярного апротонного растворителя (ТГФ). Раствор охлаждают до минус -78°C и добавляют при постоянном перемешивании по каплям в течение 2 часов раствор 51,5 мл гидрида диизобутилалюминия (DIBAL) в 120 мл полярного апротонного растворителя (ТГФ). Раствор перемешивают дополнительно 60 минут при строгом поддержании температуры -78°C.
Подготовка пробы для проведения тонкослойной хроматографии (ТСХ) на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат - 5:1. Для этого отбирают 0,75 мл реакционной смеси в эппендорф, добавляют 2 капли диизопропилэтиламина (далее - DIPEA), 0,75 мл дистиллированной воды и встряхивают. Верхнюю (водную) фазу отбирают пипеткой Пастера для ТСХ на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат - 5:1. По результату ТСХ судят об исчезновении характерного пятна спирта, в противном случае реакционную смесь перемешивают далее и повторяют проведение ТСХ.
Далее дают реакционной смеси нагреться до 0°C, затем дважды промывают порциями в делительной воронке, охлаждённым до +5-8°C 1М раствором HCl (400 мл), собирают органическую фазу. Собранную органическую фазу промывают насыщенным раствором NaCl до получения pH водной фазы не более 6.
Органическую фазу сушат в эксикаторе над безводным сульфатом магния в течение 12 часов. Далее растворитель отгоняют в роторном испарителе с получением требуемого альдегида. Затем жидкость извлекают и дают охладиться до комнатной температуры, при этом происходит кристаллизация в сухой продукт. Выход: 122 г (97%).
Стадия 3.
Получение эфира - (S)-этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата. M=356 г/моль.
В стеклянный реактор загружают 122 г полученного на 3 стадии альдегида в 150 мл полярного апротонного растворителя (сухого ТГФ), добавляют 165 г (карбэтоксиметилен)трифенилфосфорана (C22H21O2P). Смесь перемешивают при температуре +20°C в течение 12 часов. Контроль за протеканием реакции осуществляют методом ТСХ (сравнивают реакционную массу с исходным альдегидом) на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат - 5:1.
Затем растворитель полностью упаривают с помощью роторного испарителя и добавляют 500 мл холодного диэтилового эфира для выпадения осадка. Осадок отфильтровывают с помощью фильтра Шотта и колбы Бунзена.
Далее проводят флэш-хроматографию. На мелкопористый фильтр Шотта, закреплённый в колбе Бунзена, влажным способом наносят силикагель для хроматографии (0,060-0,200 мм, 60 А) в гексане таким образом, чтобы высота слоя силикагеля составила 8 см. Сверху фильтра на слой стеклянной ваты наносят 8 г продукта, растворенного в гексане в соотношении 1:2.
Проводят хроматографию полученного раствора с помощью элюента гексан: этилацетат =10:1 объёмом 1000 мл при этом выходят побочные продукты - верхнее пятно (контроль по ТСХ на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат - 5:1), затем применяют элюент гексан:этилацетат 7:1 объёмом 3000 мл при этом выделяют продукт (контроль по ТСХ на пластинах Sorbifil ПТСХ-АФ-А в системе гексан:этилацетат = 5:1). Полученную фракцию упаривают с помощью роторного испарителя. Выход: 110 г (90%).
Стадия 4.
Получение дикетона - (S)-5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона. M=368 г/моль.
В стеклянный реактор с рубашкой загружают 67,2 г (0,6 моль, 2 экв.) трет-бутилацетоацетата в 450 мл полярного апротонного растворителя (ТГФ). Суспензию охлаждают до 0°С и добавляют медленно 65 г (0,3 моль, 1 экв.) трет-бутилата калия. Суспензию интенсивно перемешивают 30 минут при 0°С. Затем добавляют 110 г (0,3 моль) исходного соединения (полученный на 3 стадии эфир) и перемешивают 20 минут при комнатной температуре (контроль по ТСХ на пластинах Sorbifil ПТСХ-АФ-А, элюент - хлористый метилен). После, в реакционную смесь при охлаждении (0°C) добавляют 200 мл дистиллированной воды и 18% раствор соляной кислоты до рН=4.
Вышеописанная стадия 4 способа по изобретению занимает 1 час, в то время как в аналоге время реакции на стадии 4 (кетон-продукт) составило 12 часов, при этом выход продукта составил 53%.
Смесь экстрагируют этилацетатом (3×200 мл), затем сушат в эксикаторе над безводным сульфатом магния в течение 12 часов.
Растворитель отгоняют в роторном испарителе.
Полученный полупродукт разбавляют 2Н раствором гидроксида натрия (5 экв.) и нагревают в колбе на 1000 мл до +40°С. Смесь перемешивают на магнитной мешалке 4 часа при температуре +40 °С, затем добавляют по каплям концентрированный раствор 1Н соляной кислоты до рН=1 и нагревают до 90°С. Смесь перемешивают 2 часа при 90°С, затем охлаждают до температуры +20°С, разбавляют 1000 мл этилацетата и порционно экстрагируют в делительной воронке. Собирают из воронки органический слой и сушат в эксикаторе над безводным сульфатом магния в течение 12 часов.
Растворитель отгоняют в роторном испарителе.
При флэш-хроматографии наносят раствор продукта в хлористом метилене (1:10). Продукт элюируют смесью растворителей хлористый метилен : метанол в соотношении 200:1 объем 1000 мл, далее 100:1 - 1000 мл и 50:1 - 1000 мл. Из последней фракции выделяют вещество, обогащённое целевым продуктом чистотой 99 %. Чистота продукта была подтверждена методами ВЭЖХ-ВПМС/МС (HPLC-TOFMS) (m/z = 369,09275 [M+H]+; m/z = 391,07476 [M+Na]+) и ЯМР (см. Фиг.1). Исследованная субстанция была представлена S-изомером с чистотой 107,4%. Количество примеси составило менее 1%, при этом примесь не являлась R-изомером. Массово-зарядное соотношение для примеси было оценено как 378,0649. Выход: 91 г (83%).
Несмотря на то что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.

Настоящее изобретение относится к области фармакологии и медицины, конкретно к методу синтеза 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона в виде рацемата или его S-энантиомера, который может быть использован для изготовления лекарственных средств для лечения болезни двигательных нейронов. Способ получения включает: (а) взаимодействие 2-(3,5-бис(трифторметил)фенокси)пропаналя в виде рацемической смеси или энантиомерно чистой (S)-формы с (карбэтоксиметилен)трифенилфосфораном с получением этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы и (б) взаимодействие полученного этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата с трет-бутилацетоацетатом и трет-бутилатом калия с получением целевого соединения в виде, соответственно, рацемата или его энантиомерно чистой (S)-формы, согласно схеме:
Техническим результатом изобретения является обеспечение способа получения с ускоренным синтезом и повышенным выходом целевого продукта. 8 з.п. ф-лы, 1 ил., 1 пр.
1. Способ получения 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона формулы
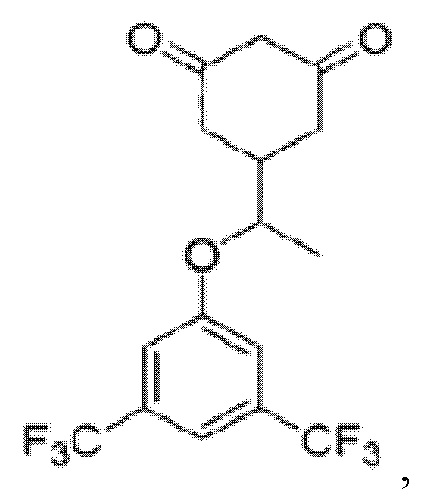
в виде его рацемата или энантиомерно чистой (S)-формы, включающий следующие стадии:
(а) взаимодействие 2-(3,5-бис(трифторметил)фенокси)пропаналя в виде рацемической смеси или энантиомерно чистой (S)-формы с (карбэтоксиметилен)трифенилфосфораном с получением этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы;
(б) взаимодействие полученного этил-5-(3,5-бис(трифторметил)фенокси)гекс-3-еноата с трет-бутилацетоацетатом и трет-бутилатом калия с получением целевого соединения в виде, соответственно, рацемата или его энантиомерно чистой (S)-формы,
согласно схеме:
2. Способ по п.1, в котором для получения энантиомерно чистой (S)-формы 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона проводят его выделение из полученного рацемата 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона.
3. Способ по п.2, в котором выделение энантиомерно чистой (S)-формы 5-(1-(3,5-бис(трифторметил)фенокси)этил)циклогексан-1,3-диона осуществляют методом хиральной ВЭЖХ, методом превращения рацемата в смесь диастереомеров или методом кинетического расщепления.
4. Способ по п.1, в котором на реакции на стадиях (а) и (б) проводят в полярном апротонном растворителе.
5. Способ по п.1, в котором 2-(3,5-бис(трифторметил)фенокси)пропаналь в виде рацемической смеси или энантиомерно чистой (S)-формы получают путем
взаимодействия на первом (1) этапе 3,5-бис(трифторметил)фенола с этиллактатом в виде рацемической смеси или энантиомерно чистой (S)-формы с получением этил-2-(3,5-бис(трифторметил)фенокси)пропионата по реакции Мицунобу в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы;
проведения на втором (2) этапе реакции восстановления полученного этил-2-(3,5-бис(трифторметил)фенокси)пропионата с получением 2-(3,5-бис(трифторметил)фенокси)пропаналя в виде, соответственно, рацемической смеси или энантиомерно чистой (S)-формы,
согласно схеме:
6. Способ по п.5, в котором на этапе (1) взаимодействие 3,5-бис(трифторметил)фенола с этиллактатом проводят в присутствии трифенилфосфина в полярном апротонном растворителе.
7. Способ по п.5, в котором на этапе (2) реакцию восстановления проводят в полярном апротонном растворителе, а в качестве восстановителя используют гидрид диизобутилалюминия.
8. Способ по любому из пп.4, 6 или 7, в котором полярный апротонный растворитель выбирается независимо и представляет собой тетрагидрофуран, 1,4-диоксан или диметилсульфоксид.
9. Способ по любому из пп.1-7, в котором очистку промежуточного и целевого продукта на каждом из этапов (а), (б) и (1) осуществляют с помощью флэш-хроматографии.
| Y | |||
| Zhang et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 2011059821 A2, 19.05.2011 | |||
| W | |||
| Zhang et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

