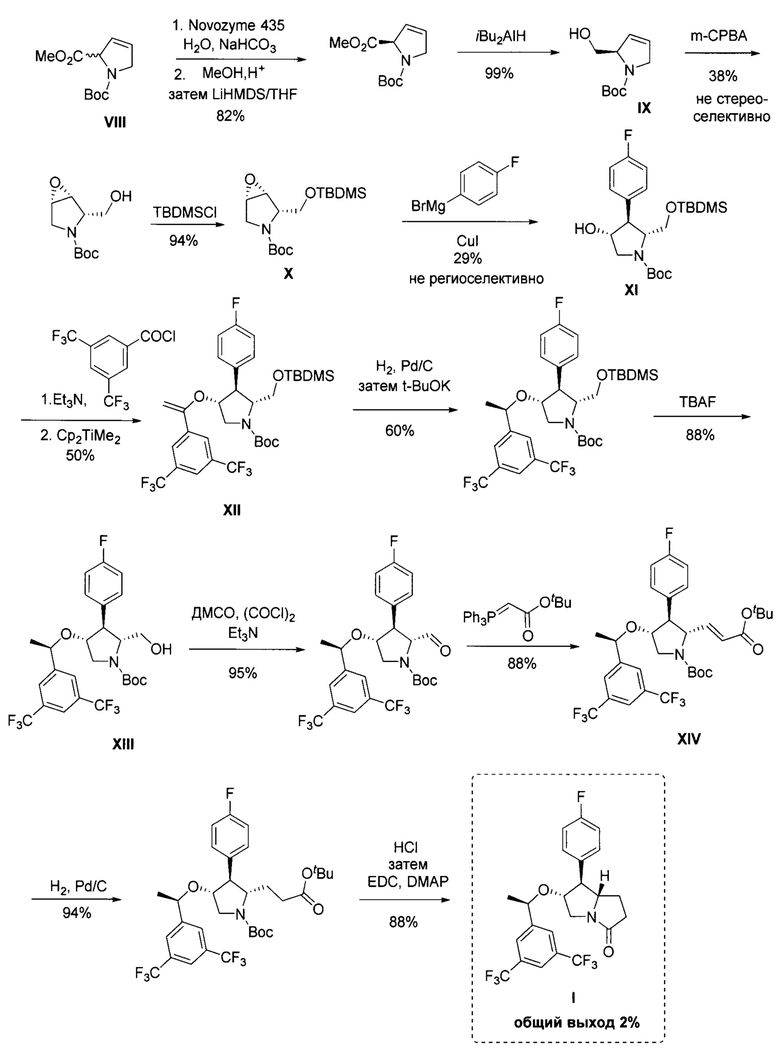
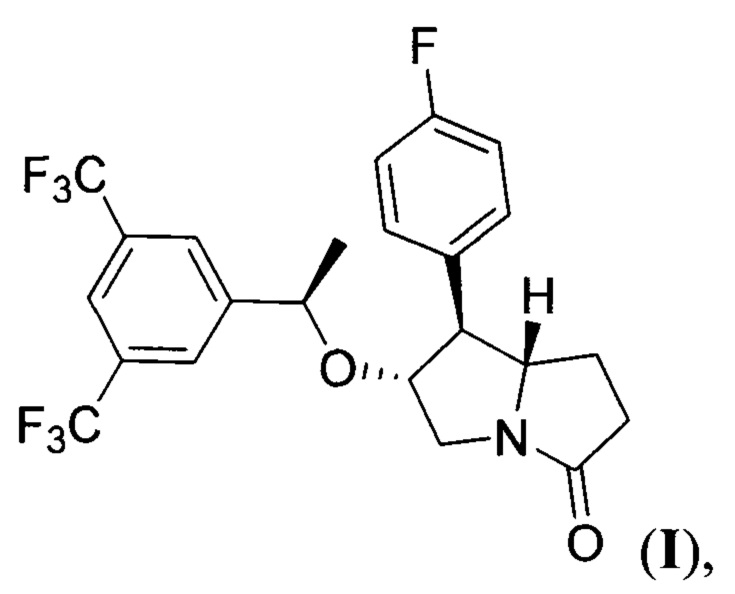
Настоящее изобретение относится к области органической химии и фармакологии, а именно к способу получения (6R,7S,7aS)-6-((7R)-1-(3,5-бис (трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она (соединение I), имеющего формулу:
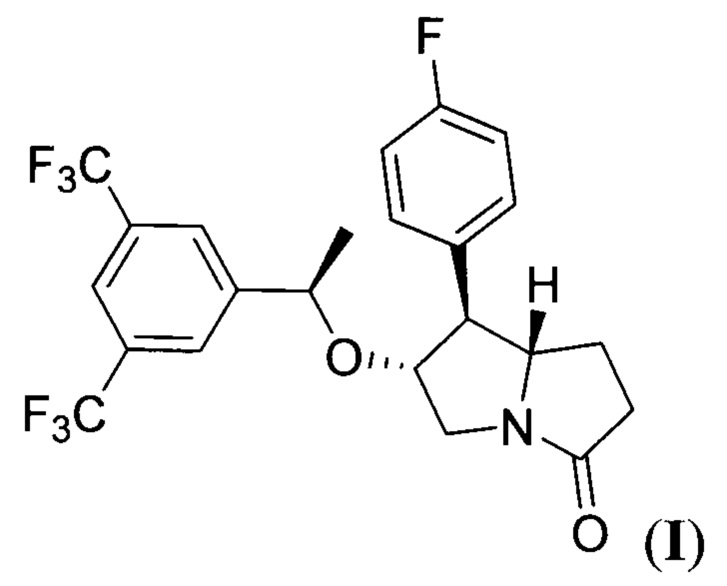
Соединение I является известным высокоактивным антагонистом человеческого NK1 рецептора и рассматривается в качестве прототипа лекарственного препарата для лечения тошноты и рвоты, связанной с применением противоопухолевых химиотерапевтических средств. Антагонисты человеческих нейрокининовых рецепторов NK1 широко применяются в медицине в качестве препаратов для лечения и профилактики рефрактерной и отсроченной тошноты и рвоты при химиотерапии (М.А. Ibrahim, С.V. Preuss, Antiemetic Neurokinin-1 Receptor Blockers, StatPearls Publishing, Treasure Island (FL), 2021; B. Rapoport, T. Smit, Expert Opin. Drug Saf. 2017, 16, 697-710). Примерами клинически применяемых препаратов этого типа являются апрепитант, его водорастворимая форма фосапрепитант, ролапитант и др.
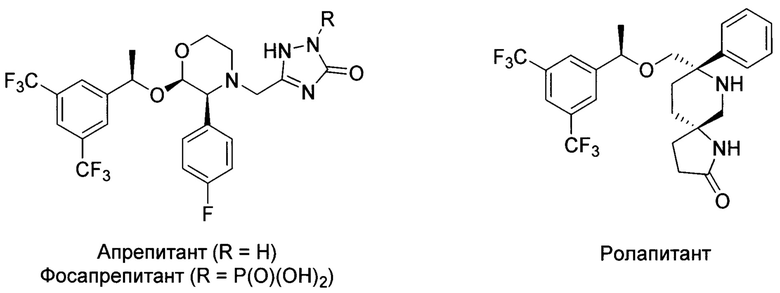
Однако используемые препараты имеют побочные эффекты (головная боль, усталость, запоры, нейтропения и ослабление естественного иммунитета), а также имеют большой риск лекарственных взаимодействий (Н.А. Шумилова, С.И. Павлова, Acta Medica Eurasica, 2020, 2, 91-99). В связи с этим, активно проводятся исследования по разработке более активных и селективных фармсубстанций, имеющих более высокое сродство к человеческому NKi рецептору, лучше проникающих в ткани головного мозга и неактивных по отношению к другим биологическим мишеням (R. Recio, P. Lerena, Е. Pozo, J.  V. Valdivia, М. Pernia Leal, В. Mouillac,
V. Valdivia, М. Pernia Leal, В. Mouillac,  N. Khiar,
N. Khiar, 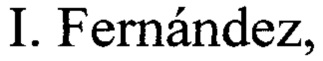 J. Med. Chem. 2021, 64, 10350-10370; G. J. Morriello, S. G. Mills, T. Johnson, M. Reibarkh, G. Chicchi, J. DeMartino, M. Kurtz, P. Davies, K. L. C. Tsao, S. Zheng, X. Tong, E. Carlson, K. Townson, F. D. Tattersall, A. Wheeldon, S. Boyce, N. Collinson, N. Rupniak, S. Moore, R. J. DeVita, Bioorg. Med. Chem. Lett. 2010, 20, 2007-2012). Одним из перспективных в этом отношении соединений является (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-он (соединение I), разработанное и запатентованное фармацевтической компанией Мерк (WO2007/087224 А2). В биологических исследованиях in vitro и in vivo соединение I показало очень высокую аффинность к человеческому NK1 рецептору в сочетании с улучшенной способностью проникать в ткани головного мозга по сравнению с апрепитантом (G.J. Morriello, R.J. DeVita, S.G. Mills, J.R. Young, P. Lin, G. Doss, G.G. Chicchi, J. DeMartino, M.M. Kurtz, K.-L. C. Tsao, E. Carlson, K. Townson, A. Wheeldon, S. Boyce, N. Collinson, N. Rupniak, S. Moore, Bioorg. Med. Chem. 2008,16, 2156-2170).
J. Med. Chem. 2021, 64, 10350-10370; G. J. Morriello, S. G. Mills, T. Johnson, M. Reibarkh, G. Chicchi, J. DeMartino, M. Kurtz, P. Davies, K. L. C. Tsao, S. Zheng, X. Tong, E. Carlson, K. Townson, F. D. Tattersall, A. Wheeldon, S. Boyce, N. Collinson, N. Rupniak, S. Moore, R. J. DeVita, Bioorg. Med. Chem. Lett. 2010, 20, 2007-2012). Одним из перспективных в этом отношении соединений является (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-он (соединение I), разработанное и запатентованное фармацевтической компанией Мерк (WO2007/087224 А2). В биологических исследованиях in vitro и in vivo соединение I показало очень высокую аффинность к человеческому NK1 рецептору в сочетании с улучшенной способностью проникать в ткани головного мозга по сравнению с апрепитантом (G.J. Morriello, R.J. DeVita, S.G. Mills, J.R. Young, P. Lin, G. Doss, G.G. Chicchi, J. DeMartino, M.M. Kurtz, K.-L. C. Tsao, E. Carlson, K. Townson, A. Wheeldon, S. Boyce, N. Collinson, N. Rupniak, S. Moore, Bioorg. Med. Chem. 2008,16, 2156-2170).
В патенте (WO2007/087224 A2) описан многостадийный способ получения соединения I из коммерчески доступного рацемического 1-(трет-бутил) 2-метил 2,5-дигидро-1Н-пиррол-1,2-дикарбоксилата VIII. Соединение VIII в результате энзиматического расщепления и восстановления превращают в энантиомерно чистый спирт IX, который далее эпоксидируют и силилируют с образованием эпоксида X. Далее реакцией эпоксида X с пара-фторфенилмагнийбромидом получают соответствующий арил-замещенный пирролидин XI, который превращают в пролинол XIII с помощью последовательного ацилирования бис(трифторметил)бензоилхлоридом, реакцией с диметилтитаноценом, каталитического гидрирования алкенового фрагмента в полупродукте XII и снятия силильной защитной группы. Пролинол XIII далее трансформируют в функционализированный пирролидин XIV путем окисления первичной гидроксильной группы методом Сверна и олефинированием по Виттигу. Заключительным этапом синтеза является каталитическое гидрирование кратной связи, гидролиз сложноэфирной группы и замыкание пирролидонового фрагмента. Аналогичный метод синтеза описан в статье тех же авторов (G.J. Morriello, R.J. DeVita, S.G. Mills, J.R. Young, P. Lin, G. Doss, G.G. Chicchi, J. DeMartino, M.M. Kurtz, K.-L.C. Tsao, E. Carlson, K. Townson, A. Wheeldon, S. Boyce, N. Collinson, N. Rupniak, S. Moore, Bioorg. Med. Chem. 2008, 16, 2156-2170). Процесс протекает по следующей схеме:
Описанный способ получения соединения I имеет недостатки, связанные с большим числом стадий (14 стадий), низкой регио- и стереоселективностью ключевых реакций, необходимостью использования дополнительных стадий введения и удаления защитных групп, образованием большого числа побочных продуктов, и, как следствие, низким суммарным выходом продукта на коммерчески доступные соединения - всего 2%. Эти обстоятельства делают невозможным практическое применение этого способа для получения продукта I в количествах, необходимых для расширенных клинических испытаний.
Технической задачей предлагаемого изобретения является упрощение технологии получения соединения I за счет сокращения количества стадий, а также повышение его выхода.
Поставленная техническая задача достигается предложенным способом получения (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она формулы:
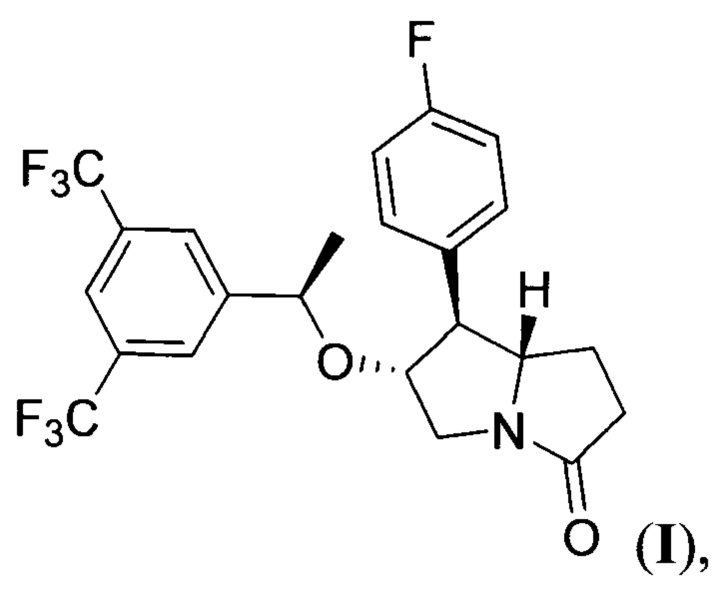
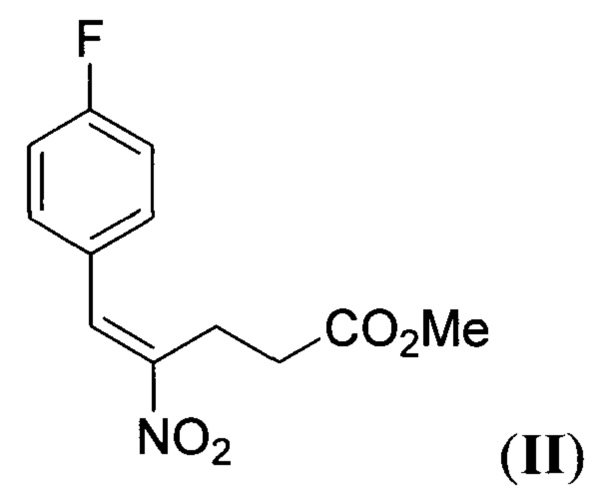
заключающимся в том, что метиловый эфир 4-нитромасляной кислоты подвергают взаимодействию с 4-фторбензальдегидом при кипячении в органическом растворителе в присутствии в качестве катализатора первичного амина, полученный при этом метил (E)-5-(4-фторфенил)-4-нитропент-4-еноат (нитроалкен II) формулы:
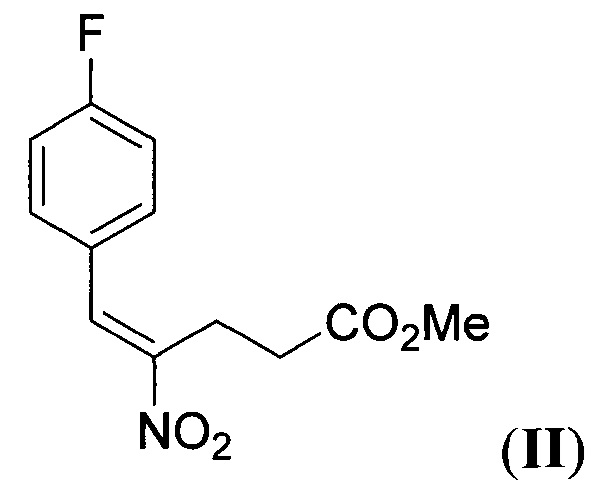
обрабатывают (Z)-2-(((1S,2R)-2-фенилциклогексил)окси)винил карбоксилатом (виниловый эфир III) общей формулы:
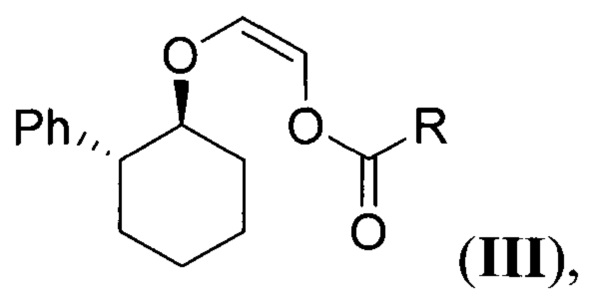 где R - низший алкил,
где R - низший алкил,
в присутствии тетрахлорида олова при пониженной температуре в среде апротонного растворителя с последующим гидролизом полученного 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(ацилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат IV) общей формулы
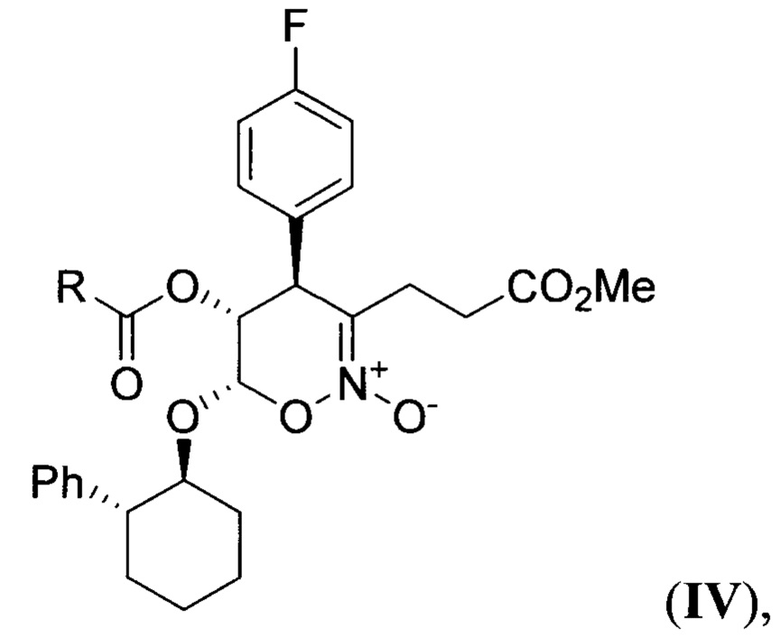 где R - низший алкил,
где R - низший алкил,
в основных условиях с образованием 4-(4-фторфенил)-5-гидрокси-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат V) формулы:
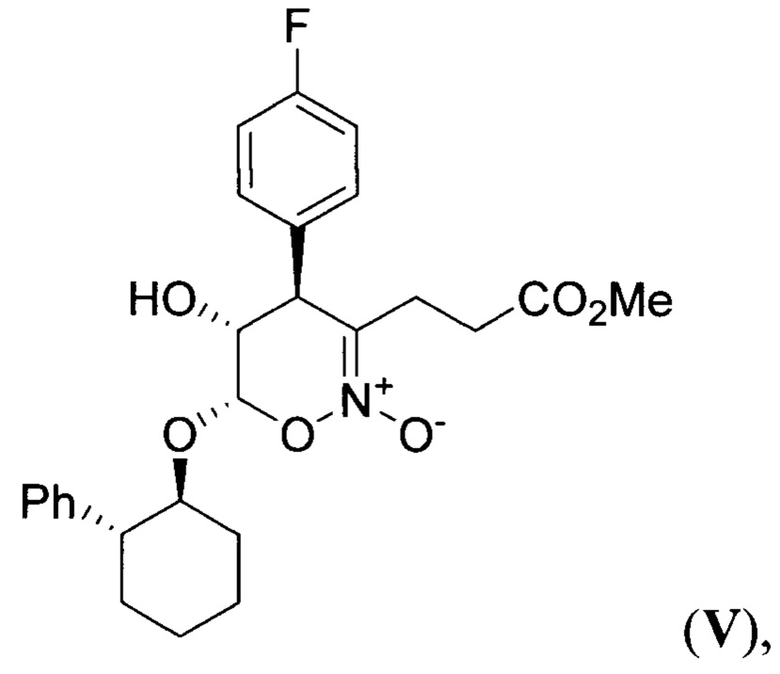
который подвергают гидрированию при повышенной температуре и повышенном давлении водорода в присутствии платинового катализатора в среде протонного растворителя и образующийся при этом 7-(4-фторфенил)-6-гидрогексагидро-3Н-пирролизин-3-он (пирролизидинон VI) формулы:
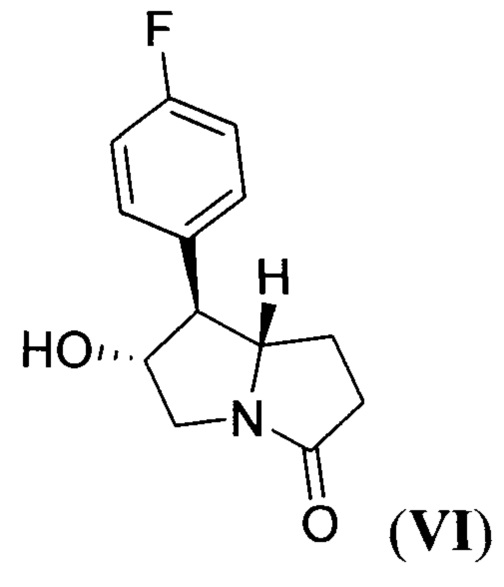
алкилируют 1-(3,5-бис(трифторметил)фенил)этил 2,2,2-трихлорацетимидатом (имидат VII) формулы:
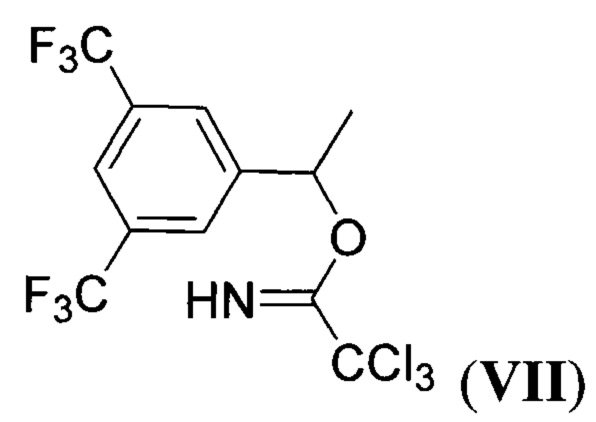
при пониженной температуре в апротонном растворителе в присутствии сильной протонной кислоты с последующим выделением целевого продукта. Предложенный новый способ получения соединения I заключается в последовательности следующих химических стадий: 1) конденсация метилового эфира 4-нитромасляной кислоты и 4-фторбензальдегида с образованием нитроалкена II, 2) циклоприсоединение нитроалкена II с виниловым эфиром III с образованием циклического нитроната IV; 3) гидролиз сложноэфирной группы в продукте IV с образованием циклического нитроната V; 4) восстановительная рециклизация циклического нитроната V с образованием пирролизидинона VI; 5) алкилирование пирролизидинона VI с помощью имидата VII. Процесс протекает по следующей схеме:
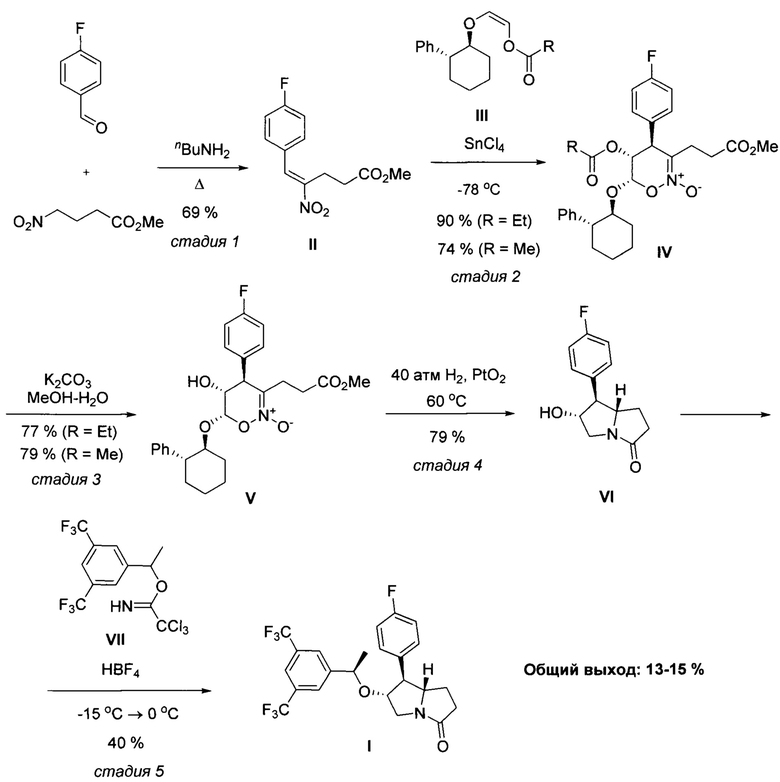
В качестве первичного амина на стадии 1 используют, например, н-бутиламин, в качестве апротонного растворителя на стадии 2 используют, например, хлористый метилен; в качестве основных условий на стадии 3 используют, например, раствор поташа в смеси вода - низкомолекулярный спирт (например, метанол, этанол); в качестве платинового катализатора на стадии 4 используют преимущественно диоксид платины или другие гомо/гетерогенные соединения платины, в качестве протонного растворителя на стадии 4 используют, например, уксусную кислоту или низкомолекулярный спирт; в качестве сильной протонной кислоты на стадии 5 используют, например, тетрафтороборную кислоту, а в качестве апротонного растворителя на стадии 5 используют, например, 1,2-дихлорэтан.
Первую стадию процесса - получение нитроалкена II - необходимо проводить при кипячении в подходящем органическом растворителе, например, толуоле, спирте и др. в присутствии катализатора - первичного амина, например, н-бутиламина. В отсутствии катализатора выход продукта II заметно снижается из-за низкой конверсии исходных соединений.
Вторую стадию процесса - циклоприсоединение нитроалкена II с виниловым эфиром III - необходимо проводить при пониженной температуре, например, в диапазоне от -30 до -90°С, преимущественно -78°С, в присутствии промотора - тетрахлорида олова. Повышение температуры приводит к падению выхода продукта IV из-за осмоления. В отсутствие тетрахлорида олова процесс не протекает.
Четвертую стадию процесса - трансформацию циклического нитроната V в пирролизидинон VI - проводят в автоклаве при повышенной температуре, например, при 40-70°С, и давлении водорода от 20 до 60 атм, преимущественно при 60°С и 40 атм. Снижение температуры процесса до комнатной и давления водорода до атмосферного приводит к понижению выхода продукта из-за неполной конверсии циклического нитроната V. Пятую стадию процесса - алкилирование пирролизидинона VI имидатом VII - необходимо проводить при пониженной температуре, например, в диапазоне от -15 до 0°С. При более низкой температуре скорость процесса снижается, при более высокой - выход продукта снижается из-за осмоления.
Разработанный способ обладает выраженными достоинствами по сравнению с описанным в литературе способом, поскольку позволяет получать целевой антагонист I с общим выходом до 15% из коммерчески доступного сырья в существенно меньшее число стадий. Сокращение количества стадий синтеза соединения I и существенное повышение его выхода было достигнуто за счет применения принципиально иной схемы синтеза, ранее не применявшейся для получения соединений этого типа.
Реакции, используемые в предлагаемом способе получения соединения I, в литературе не описаны. Найденный способ превращения циклического нитроната V в пирролизидинон VI в условиях каталитического гидрирования на платиновом катализаторе является ключевым, новым, не описанным в литературе. Таким образом, заявляемое изобретение соответствует критерию «новизна» и «изобретательский уровень».
Использующиеся в качестве стартовых соединений метиловый эфир 4-нитромасляной кислоты и 4-фторбензальдегид являются коммерчески доступными соединениями. Другие исходные вещества для предлагаемого способа получения антагониста I - виниловый эфир III и имидат VII - могут быть синтезированы из коммерчески доступных реагентов по описанным в литературе методикам в граммовых и килограммовых количествах как показано на схемах ниже.
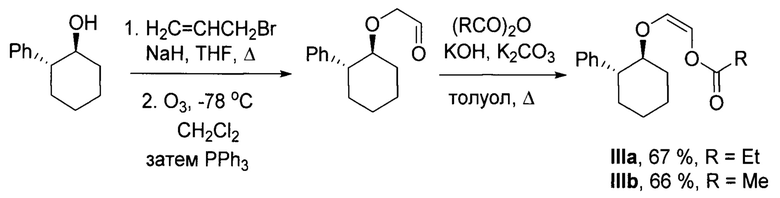
Виниловые эфиры типа III получают из коммерчески доступного (+)-(1S,2R)-2-фенил-1-циклогексанола в три стадии путем аллилирования с последующим озонолизом и ацилированием образующегося альдегида (V.S. Dorokhov, Y.V. Nelyubina, S.L. Ioffe, A.Y. Sukhorukov, J. Org. Chem. 2020, 85, 11060-11071). Процесс протекает по следующей общей схеме:
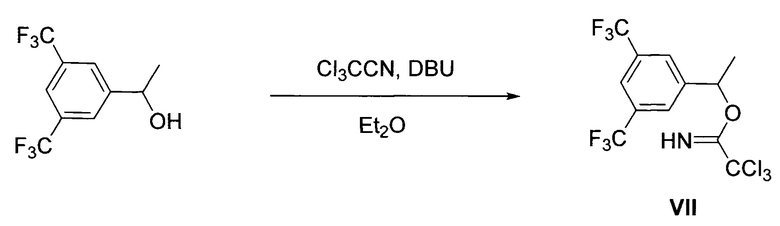
Имидат VII получают взаимодействием коммерчески доступного 1-(3,5-бис(трифторметил)фенил)этан-1-ола с трихлорацетонитрилом в присутствии диазабициклоундецена (ДБУ) (J. Jiang, J.L. Bunda, G.A. Doss, G.G.Chicchi, M.M. Kurtz, K.-L. C. Tsao, X. Tong, S. Zheng, A. Upthagrove, K. Samuel, R. Tschirret-Guth, S. Kumar, A. Wheeldon, E.J. Carlson, R. Hargreaves, D. Burns, T. Hamill, C. Ryan, S.M. Krause, W. Eng, R.J. DeVita, S.G. Mills, J. Med. Chem. 2009, 52, 3039-3046). Процесс протекает по следующей общей схеме:
Техническим результатом предлагаемого изобретения является создание простого и эффективного способа получения соединения I - известного антагониста человеческих нейрокининовых рецепторов - с существенно меньшим числом стадий (5 стадий) и высоким выходом (13 - 15%). Изобретение иллюстрируется примерами, не ограничивающими его объем.
Пример 1
Стадия 1. Получение метил (E)-5-(4-фторфенил)-4-нитропент-4-еноата (нитроалкена II).
4-Фторбензальдетид (1.10 г, 8.84 ммоль) и метил-4-нитробутират (1.00 г, 6.80 ммоль) растворяли в толуоле (4 мл). Добавляли н-бутиламин (0.134 мл, 1.40 ммоль) и кипятили раствор в течение 6 часов. Затем добавляли дополнительную порцию н-бутиламина (0.068 мл, 0.70 ммоль) и 4-фторбензальдегида (0.42 г, 3.40 ммоль) и кипятили в течение 3 часов. Упаривали растворитель при пониженном давлении, продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (20:1 ->10:1) в качестве элюента с получением метил (E)-5-(4-фторфенил)-4-нитропент-4-еноата II в виде желтого масла с выходом 1.19 г (69%). 1Н ЯМР (300 МГц, CDCl3): δ 8.06 (с, 1Н), 7.45 (дд, J=8.6, 5.3 Гц, 2Н), 7.15 (т, J=8.6 Гц, 2Н), 3.67 (с, 3Н), 3.30-3.04 (м, 2Н), 2.77-2.48 (м, 2Н). 13С{1H} ЯМР (75 МГц, CDCl3): δ 172.3, 163.8 (д, J=252.8 Гц), 149.8, 134.1, 132.0 (д, J=8.7 Гц), 128.0 (д, J=3.5 Гц), 116.5 (д, J=21.9 Гц), 52.0, 31.8, 22.9. Масс-спектр высокого разрешения: m/z теор. [C12H12FNO4Na]+ 276.0634, эксп. 276.0643 [М+Na]+.
Стадия 2. Получение 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(пропионилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклического нитроната IVa).
Полученный на предыдущей стадии нитроалкен II (0.71 г, 2.81 ммоль) и энантиомерно чистый (Z)-2-(((1S,2R)-2-фенилциклогексил)окси)винил-пропиолат (виниловый эфир IIIa) (1.0 г, 3.65 ммоль) растворяли в сухом дихлорметане (12.5 мл) в атмосфере аргона. Реакционную смесь охлаждали до -78°С. Добавляли по каплям раствор тетрахлорида олова (987 мкл, 8.43 ммоль) и перемешивали в течение 75 минут. Далее в реакционную смесь добавляли этилацетат (150 мл) и насыщенный раствор карбоната натрия в воде (150 мл). Экстрагировали этилацетатом (2 раза по 100 мл), экстракт промывали насыщенным раствором хлорида натрия в воде (100 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (5:1 - 3:1) в качестве элюента, с получением 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(пропионилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклического нитроната IVa) в виде белого твердого вещества с выходом 1.33 г (90%). Тпл=60-62°С (кристаллизуется из смеси диэтиловый эфир/гексан). 1Н ЯМР (300 МГц, CDCl3): δ 7.33 (т, J=7.5 Гц, 2Н), 7.28-7.14 (м, 3Н), 7.12-7.07 (м, 2Н), 7.02-6.96 (м, 2Н), 5.66 (д, J=3.1 Гц, 1Н), 4.93 (дд, J=10.1, 3.1 Гц, 1Н), 4.18 (ддд, J=14.2, 10.4, 4.1 Гц, 1Н), 3.59-3.54 (м, 4Н), 2.73-2.61 (м, 1Н), 2.24-2.15 (м, 3Н), 2.14-1.96 (м, 2Н), 1.93-1.82, 1.78-1.65, 1.52-1.24 (м, 3Н, 2Н, 4Н), 1.01 (т, J=7.6 Гц, 3Н). 13С{1Н} ЯМР (75 МГц, CDCl3): δ 173.4, 172.7, 162.6 (д, J=247.8 Гц), 143.7, 132.3 (д, J=3.3 Гц), 130.4 (д, J=8.2 Гц), 128.6, 127.4, 126.4, 121.4, 116.2 (д, J=21.7 Гц), 95.0, 78.1,70.3,51.6, 50.3, 43.7, 35.1, 30.8, 27.7, 27.3, 26.0, 24.6, 9.0. 19F ЯМР (282.48 МГц, CDCl3): δ - 113.25 (м, CF). Масс-спектр высокого разрешения: m/z теор. [C29H35FNO7]+ 528.2387, эксп. 528.2392 [М+Н]+. Угол оптического вращения = +232.8° (с=1, этиацетат, 29°С). Энантиомерный избыток >99% (ВЭЖХ). Стадия 3. Получение 4-(4-фторфенил)-5-гидрокси-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклического нитроната V).
Циклический нитронат IVa (358 мг, 0.679 ммоль), полученный по предыдущей методике, растворяли в 8 мл метанола и добавляли 8 мл 0.1 М водного раствора карбоната калия. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Затем упаривали растворитель при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (3:1 → 1:1) в качестве элюента, с получением циклического нитроната V в виде белого твердого вещества с выходом 247 мг (77%). Тпл=100-102°С. 1Н ЯМР (300 МГц, CDCl3): δ 7.34 (т, J=7.4 Гц, 2Н), 7.28-7.15 (м, 3Н), 7.11-6.99 (м, 4Н), 5.49 (д, J=3.4 Гц, 1Н), 4.26 (м, 1Н), 3.86 (ушир, 1Н), 3.59 (с, 3Н), 3.22 (д, J=9.2 Гц, 1Н), 2.68 (ддд, J=11.2, 10.7, 3.4 Гц, 1Н), 2.38 (м, 1Н), 2.17-2.04 (м, 2Н), 1.96-1.91 (м, 4Н), 1.81-1.65 и 1.51-1.29 (м, 2Н и 5H). 13C{1H} ЯМР (75 МГц, CDCl3): δ 172.6, 162.6 (д, J=247.5 Гц), 143.7, 133.2, 130.4 (д, J=8.1 Гц), 128.7, 127.4, 126.5, 121.7, 116.2 (д, J=21.8 Гц), 96.8, 77.8, 69.4, 51.7, 50.6, 47.8, 35.0, 30.7, 27.9, 26.3, 26.0, 24.6.19F ЯМР (282.46 МГц): δ - 113.65 (м, CF). Масс-спектр высокого разрешения: m/z теор. [C26H31FNO6]+ 472.2136, эксп. 472.2130 [М+Н+]. Угол оптического вращения = +235.9° (с=1, хлороформ, 29°С). Энантиомерный избыток >99.5% (ВЭЖХ).
Стадия 4. Получение 7-(4-фторфенил)-6-гидрогексагидро-3Н-пирролизин-3-она(пирролизидинон VI)
Циклический нитронат V (209 мг, 0.444 ммоль) растворяли в уксусной кислоте (2 мл), добавляли оксид платины (IV) (20 мг). Гидрировали в автоклаве при 60°С и давлении водорода 40 бар в течение 3 часов. Катализатор отделяли фильтрованием, промывали этанолом (5 раз по 4 мл), и объединенный раствор упаривали при пониженном давлении. Затем остаток растворяли в толуоле (2 мл) и кипятили в течение 10 минут. Растворитель упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:1), и затем этилацетата и метанола (10:1) в качестве элюента, с получением пирролизидинона VI в виде твердого светло-красного вещества с выходом 82 мг (79%). Тпл=117-119°С. 1Н ЯМР (300 МГц, CDCl3): δ 7.25 (дд, J=8.5, 5.4 Гц, 2Н), 7.07 (т, J=8.5 Гц, 2Н), 4.57 (ддд, J=7.5, 7.3, 6.4 Гц, 1Н), 3.97 (ддд, J=9.8, 7.3, 7.2 Гц, 1Н), 3.58 (дд, J=12.3, 7.5 Гц, 1Н), 3.50 (дд, J=12.3, 6.4 Гц, 1Н), 3.07 (ушир, 1Н), 2.73 (м, 1Н), 2.65-2.59 (м, 1Н), 2.47-2.37 (м, 1Н), 2.29-2.18 (м, 1Н), 1.95-1.82 (м, 1Н), 13С{1H} ЯМР (75 МГц, CDCl3): δ 175.2, 162.4 (д, J=246 Гц), 133.3, 129.2 (д, J=7.9 Гц), 116.0 (д, J=21.3 Гц), 79.8, 65.4, 59.2, 49.1, 33.6, 26.2. 19F ЯМР (282 МГц, CDCl3): δ - 114.96 (м, CF). Масс-спектр высокого разрешения: m/z теор. [C13H15FNO2]+ 236.1089, эксп. 236.1081 [М+Н+]. Угол оптического вращения = -4.8° (с=1, хлороформ, 22°С). Энантиомерный избыток >99.5% (ВЭЖХ).
Стадия 5. Получение (6R,7S,7aS)-6-((R)-1-(3,5-бис-(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она (соединения I).
Пирролизидинон VI (36 мг, 0.153 ммоль) и имидат VII (185 мг, 0.460 ммоль) растворяли в сухом дихлорэтане (1.5 мл). Добавляли молекулярные сита AW-300 (15 мг), предварительно высушенные при пониженном давлении. Реакционную смесь перемешивали 20 минут при комнатной температуре и затем охлаждали до -15°С. Затем по каплям добавляли HBF4⋅Et2O (0.25 мл, 1.84 ммоль), после этого реакционную смесь отогревали до 0°С и перемешивали в течение 24 часов. Затем добавляли дополнительную порцию имидата VII (62 мг, 0.154 ммоль) и HBF4⋅Ft2O (0.125 мл, 0.92 ммоль), и реакционную смесь перемешивали в течение 72 часов при 0°С. Реакцию гасили добавлением насыщенного водного раствора гидрокарбоната натрия (1.5 мл) и метанола (1.5 мл), перемешивая в течение 10 минут при комнатной температуре. Затем отделяли органический слой, водный слой экстрагировали дихлорметаном (2 раза по 3 мл). Экстракт сушили сульфатом натрия и упаривали при пониженном давлении. Продукт I выделяли методом колоночной хроматографии (элюент - смесь гексана и этилацетата). В результате получали 29 мг (40%) соединения I в виде прозрачного маслообразного вещества. 1Н ЯМР (300 МГц, CDCl3): δ 7.72 (с, 1Н), 7.45 (с, 2Н), 7.10-7.03 (м, 2Н), 7.02-6.94 (м, 2Н), 4.47 (кв, J=6.4 Гц, 1Н), 4.17 (ддд, J=8.3, 7.3, 5.8 Гц, 1Н), 3.89 (ддд, J=10.0, 6.9, 3.9 Гц, 1Н), 3.71 (дд, J=11.8, 5.8 Гц, 1Н), 3.60 (дд, J=11.8, 7.3 Гц, 1Н), 2.83 (дд, J=10.0, 8.3 Гц, 1Н), 2.68 (ддд, J=16.9, 9.8, 2.7 Гц, 1Н), 2.50 (ддд, J=16.9, 9.8, 2.7 Гц, 1Н), 2.29-2.14 (м, 1H), 1.97-1.80 (м, 1Н), 1.41 (д, J=6.5 Гц, 3Н). 13С{1H} ЯМР (75 МГц, CDCl3): δ 175.2, 162.4 (д, J=246.6 Гц), 145.9, 132.6 (д, J=3.3 Гц), 131.9 (кв, J=33.4 Гц), 128.8 (д, J=8.0 Гц), 126.1, 123.2 (кв, J=272.5 Гц), 121.8, 116.0 (д, J=21.5 Гц), 85.1, 76.6, 65.1, 57.9, 47.4, 33.2, 25.4, 24.7. 19F ЯМР (282 МГц, CDCl3): δ -62.9 (с, CF3), -114.9 (м, CF). Масс-спектр высокого разрешения: m/z теор. [C23H21F7NO2]+ 476.1459, эксп. 476.1455 [М+Н+]. Угол оптического вращения +53.4° (с=1, этиацетат, 24°С). Энантиомерный избыток >96% (ВЭЖХ). Общий выход соединения I на 5 стадий составляет 15%.
Пример 2
Аналогично примеру 1 получали соединение I, но на стадии 2, нитроалкен II (0.7 г, 2.78 ммоль), полученный на стадии 1, растворяли в сухом дихлорметане (10.0 мл) в атмосфере аргона. Реакционную смесь охлаждали до -78°С, добавляли по каплям раствор тетрахлорида олова (0.99 мкл, 8.43 ммоль) и перемешивали в течение 10 минут. Затем при -78°С в реакционную смесь добавляли по каплям раствор энантиомерно чистого (Z)-2-(((1S,2R)-2-фенилциклогексил)окси)винилацетата (винилового эфира IIIb) (0.94 г, 3.61 ммоль) в сухом дихлорметане (5 мл) и перемешивали в течение 1.5 часов. Реакцию гасили добавлением 1М раствора гидроксида натрия в метаноле (30 мл), отогревая до комнатной температуры. Полученную реакционную смесь экстрагировали дихлорметаном (2 раза по 100 мл), экстракт промывали насыщенным раствором хлорида натрия в воде (100 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (5:1→3:1) в качестве элюента с получением 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(ацетилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклического нитроната IVb) в виде белого твердого вещества с выходом 1.06 г (74%). Тпл=60-62°С. 1Н ЯМР (300 МГц, CDCl3): δ 7.38 - 7.21 (м, 5Н), 7.11 (дд, J=8.4, 5.2 Гц, 2Н), 7.02 (дд, J=8.4 Гц, 2Н), 6.68 (д, J=3.2 Гц, 1Н), 4.95 (дд, J=10.1, 3.2 Гц, 1Н), 4.22 (м, 1Н), 3.61 (с, 3Н), 3.58(д, J=10.1 Гц, 1H), 2.69 (ддд, J=11.5, 10.5, 3.5 Гц, 1Н), 2.24-2.20 (м, 1Н), 1.98-2.16 (м, 2Н), 1.95 (с, 3Н), 1.93-1.28 (м, 9Н). 13C{1H} ЯМР (75 МГц, CDCl3): δ 173.6, 169.8, 162.1 (д, J=249 Гц), 143.6, 132.3, 130.3 (д, J=9.2 Гц), 128.5, 127.3, 126.3, 121.3, 116.3 (д, J=21.4 Гц), 94.8, 78.1, 70.5, 51.5, 50.2, 43.7, 35.0, 30.7, 27.7, 25.6, 25.9, 24.5, 20.5. 19F ЯМР (282.5 МГц): -113.26 (Ar-F). Масс-спектр высокого разрешения: m/z теор. [C28H33FNO7]+ 514.2233, эксп. 514.2236 [М+Н]+. Угол оптического вращения = +234.2° (с=0.5, хлороформ, 22°С). А стадию 3 получения циклического нитроната V проводили с использованием полученного циклического нитроната IVb.
Циклический нитронат IVb (80 мг, 0.156 ммоль) растворяли в 4 мл метанола и добавляли 4 мл 0.1 М водного раствора карбоната калия. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Затем упаривали метанол при пониженном давлении, после чего полученную реакционную смесь экстрагировали дихлорметаном (3 раза по 30 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (3:1→1:1) в качестве элюента, с получением 4-(4-фторфенил)-5-гидрокси-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклического нитроната V) в виде белого твердого вещества с выходом 58 мг (79%). Спектральные характеристики продукта совпадают с указанными в примере 1. Общий выход соединения I с учетом 5-ти стадий его получения составляет 13%.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения замещенных 3-арилпирролов | 2024 |
|
RU2831117C1 |
| Производные изохинолинона, способ их получения и фармацевтическая композиция для профилактики или лечения заболеваний, связанных с поли(АДФ-рибоза)полимеразой-1, содержащая их в качестве активного ингредиента | 2020 |
|
RU2815480C1 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| Способ получения α-диазокарбонильных соединений в водной среде | 2018 |
|
RU2686489C1 |
| СИНТЕЗ И ПРОТИВОРАКОВАЯ АКТИВНОСТЬ ПРОИЗВОДНЫХ АРИЛ И ГЕТЕРОАРИЛХИНОЛИНОВ | 2011 |
|
RU2584688C2 |
| ЗАМЕЩЕННЫЕ БИАРИЛЬНЫЕ СОЕДИНЕНИЯ ИЛИ ЗАМЕЩЕННЫЕ ПИРИДИНЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1997 |
|
RU2195443C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
Изобретение относится к способу получения (6R,7S,7aS)-6-((R)-1-(3,5-бис (трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она (соединение I). Способ осуществляют согласно последовательности стадий: 1) конденсация метилового эфира 4-нитромасляной кислоты и 4-фторбензальдегида при кипячении в органическом растворителе в присутствии в качестве катализатора первичного амина с образованием метил (E)-5-(4-фторфенил)-4-нитропент-4-еноата (нитроалкен II), 2) циклоприсоединение нитроалкена II с (7)-2-(((1S,2R)-2-фенилциклогексил)окси)винилкарбоксилатом (виниловый эфир III) в присутствии тетрахлорида олова при пониженной температуре в среде апротонного растворителя с образованием циклического 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(ацилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат IV); 3) гидролиз сложноэфирной группы в полученном циклическом нитронате IV в основных условиях с образованием циклического 4-(4-фторфенил)-5-гидрокси-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат V); 4) восстановительная рециклизация полученного циклического нитроната V в процессе гидрирования при повышенных температуре и давлении водорода в присутствии платинового катализатора в среде протонного растворителя с образованием 7-(4-фторфенил)-6-гидрогексагидро-3Н-пирролизин-3-он (пирролизидинон VI); 5) алкилирование полученного пирролизидинона VI с помощью 1-(3,5-бис(трифторметил)фенил)этил 2,2,2-трихлорацетимидата (имидат VII) при пониженной температуре в апротонном растворителе в присутствии сильной протонной кислоты с последующим выделением целевого продукта. Технический результат – усовершенствование способа получения соединения I, являющегося антагонистом человеческого NK1 рецептора, за счет уменьшения числа стадий и увеличения общего выхода продукта. 5 з.п. ф-лы, 2 пр.
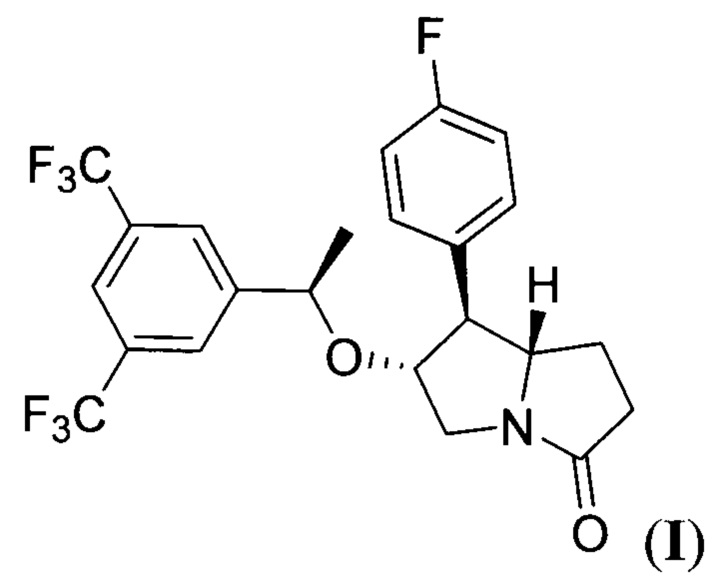
1. Способ получения (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она формулы:
отличающийся тем, что метиловый эфир 4-нитромасляной кислоты подвергают взаимодействию с 4-фторбензальдегидом при кипячении в органическом растворителе в присутствии в качестве катализатора первичного амина, полученный при этом метил (E)-5-(4-фторфенил)-4-нитропент-4-еноат (нитроалкен II) формулы:
обрабатывают (Z)-2-(((1S,2R)-2-фенилциклогексил)окси)винилкарбоксилатом (виниловый эфир III) общей формулы:
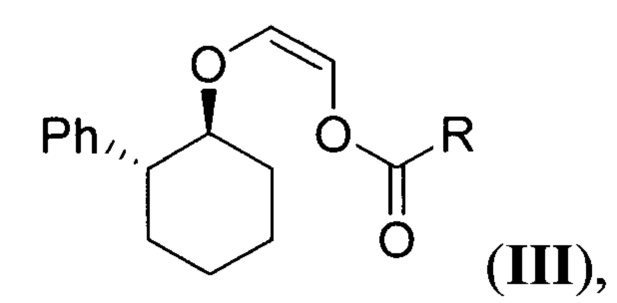 где R - низший алкил,
где R - низший алкил,
в присутствии тетрахлорида олова при пониженной температуре в среде апротонного растворителя с последующим гидролизом полученного 4-(4-фторфенил)-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5-(ацилокси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат IV) общей формулы:
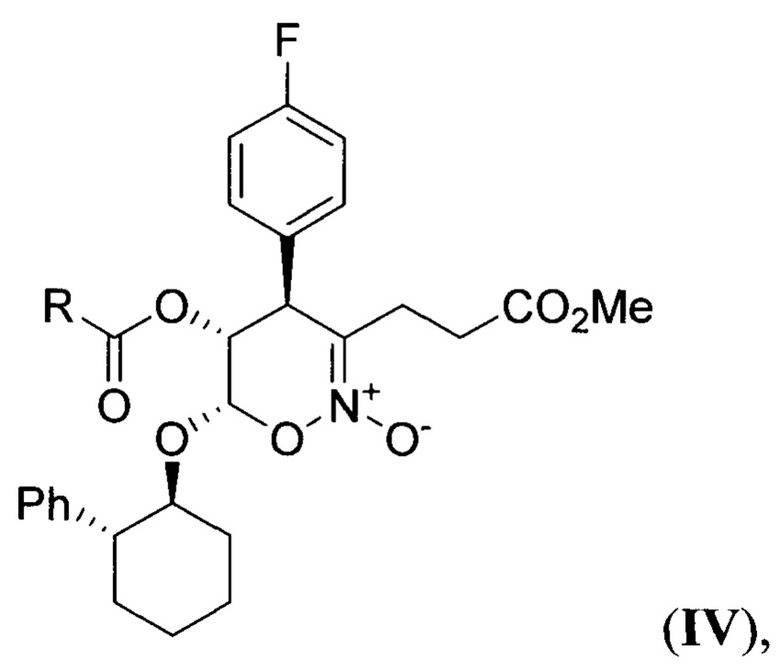 где R - низший алкил,
где R - низший алкил,
в основных условиях с образованием 4-(4-фторфенил)-5-гидрокси-3-(3-метокси-3-оксопропил)-6-((2-фенилциклогексил)окси)-5,6-дигидро-4Н-1,2-оксазин-2-оксида (циклический нитронат V) формулы:
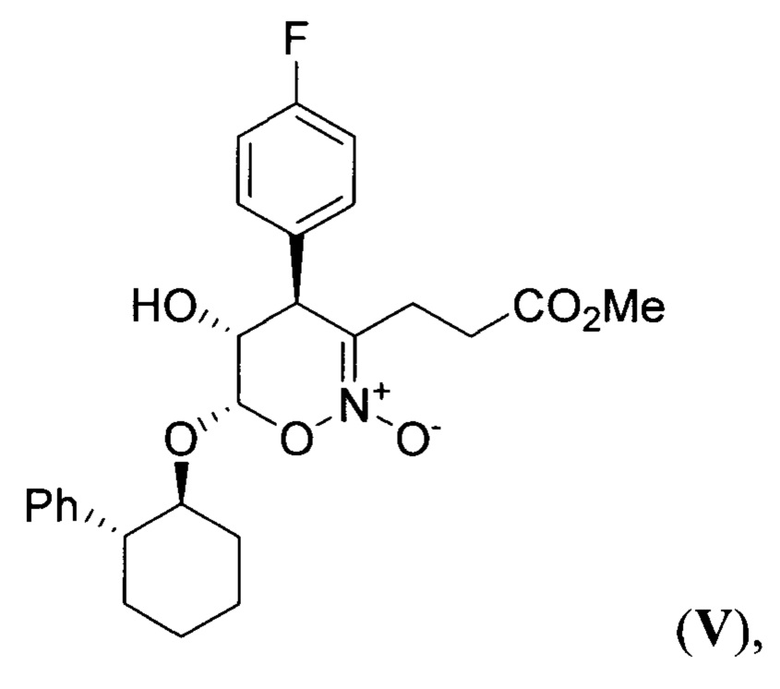
который подвергают гидрированию при повышенной температуре и повышенном давлении водорода в присутствии платинового катализатора в среде протонного растворителя и образующийся при этом 7-(4-фторфенил)-6-гидрогексагидро-3Н-пирролизин-3-он (пирролизидинон VI) формулы:
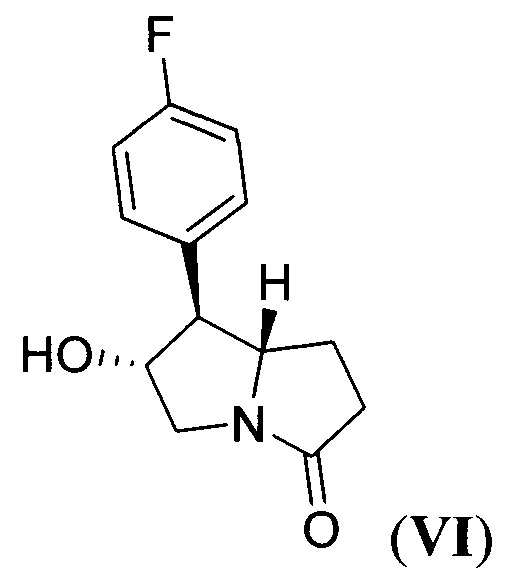
алкилируют 1-(3,5-бис(трифторметил)фенил)этил 2,2,2-трихлорацетимидатом (имидат VII) формулы:
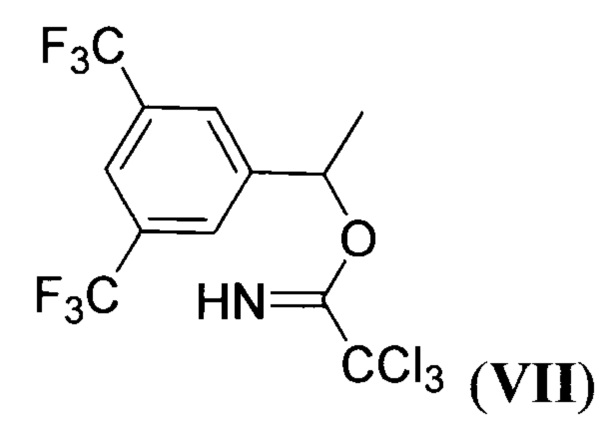
при пониженной температуре в апротонном растворителе в присутствии сильной протонной кислоты с последующим выделением целевого продукта.
2. Способ по п. 1, отличающийся тем, что на стадии получения нитроалкена II в качестве органического растворителя используют, например, толуол, а в качестве первичного амина, например, н-бутиламин.
3. Способ по п. 1, отличающийся тем, что на стадии получения циклического нитроната IV качестве апротонного растворителя используют, например, дихлорметан, и процесс проводят, преимущественно, при температуре -78°С.
4. Способ по п. 1, отличающийся тем, что на стадии получения циклического нитроната V в качестве основных условий для проведения гидролиза используют, например, раствор поташа в смеси вода - низкомолекулярный спирт, например, метанол, этанол.
5. Способ по п. 1, отличающийся тем, что на стадии получения пирролизидинона VI в процессе гидрирования в качестве платинового катализатора используют, преимущественно, диоксид платины, в качестве протонного растворителя используют, например, уксусную кислоту, и процесс проводят в автоклаве, преимущественно, при температуре 60°С и давлении водорода 40 атм.
6. Способ по п. 1, отличающийся тем, что на стадии получения целевого соединения I в качестве сильной протонной кислоты используют, например, комплекс тетрафтороборной кислоты с диэтиловым эфиром, а в качестве апротонного растворителя используют, например, 1,2-дихлорэтан, и процесс алкилирования проводят при пониженной температуре, преимущественно, в диапазоне от -15 до 0°С.
| WO 2007087224 A2, 02.08.2007 | |||
| Gregori J | |||
| Morriello et al., Fused bicyclic pyrrolizinones as new scaffolds for human NK1 antagonists | |||
| Bioorganic & Medicinal Chemistry, 2008, 16, pp.2156-2170 | |||
| Petr A | |||
| Zhmurov et al., A Novel Entry to 3,4,5-Trisubstituted 2-Pyrrolidones from Isoxazoline-N-oxides | |||
| Synlett, 2018, 29, pp.A-D | |||
| Alexey Yu | |||
| Sukhorukov et |

