ЗАЯВЛЕНИЕ ОБ ИССЛЕДОВАНИЯХ, ФИНАНСИРУЕМЫХ ФЕДЕРАЛЬНЫМ ПРАВИТЕЛЬСТВОМ
Правительство США владеет оплаченной лицензией на данное изобретение и правом требовать от патентообладателя предоставить лицензию другим на разумных условиях при определенных обстоятельствах, как это предусмотрено условиями гранта № 1R44AI122488-01, присуждаемого Национальным институтом изучения аллергических и инфекционных заболеваний.
ОБЛАСТЬ ТЕХНИКИ
Настоящий документ относится к соединениям, подходящим для предотвращения, лечения или ослабления вирусной инфекции.
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США № 62/574067, поданной 18 октября 2017 г., которая полностью включена в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
По данным Отдела вирусных заболеваний Армейского научно-исследовательского института им. Уолтера Рида, неаденовирусные респираторные инфекции «вызывают 25-30% случаев госпитализации по причине инфекционных заболеваний среди военнослужащих, уступая только травмам, являющимся причиной DNBI [потери от болезней и небоевых ранений] среди дислоцированных сил».
Грипп A является показательным примером. Он вносит значительный вклад в бремя военных заболеваний, а также ежегодно поражает значительную часть гражданского населения США, вызывая респираторные заболевания с серьезными осложнениями и смертностью. Каждый год 5-20% населения США заражаются сезонным гриппом, что приводит к более чем 200000 случаев госпитализаций и примерно 24000 смертей. Более того, неизбежное появление смертельного пандемического вируса гриппа А представляет серьезную угрозу, а недавние сообщения о генетических манипуляциях иллюстрируют потенциал гриппа А в качестве средства ведения биологической войны. Современные вакцины против гриппа не являются решением; их эффективность варьировалась от 10-60% за последние 12 лет. Представленная на рынке противовирусная терапия прямого действия против гриппа включает ингибиторы вирусной нейраминидазы (NA) и блокаторы М2-каналов. На этапе клинических испытаний оцениваются дополнительные противогриппозные препараты, включая перепрофилированный антипротозойный агент, который блокирует созревание вирусного гемагглютинина путем изменения его гликозилирования посредством неизвестного механизма. Недостатком современных лекарственных средств является то, что к ним быстро вырабатывается резистентность, поскольку они взаимодействуют непосредственно с вирусными белками (противовирусные препараты прямого действия, DAA). Репликация генома вирусной РНК сильно подвержена ошибкам, и такая высокая частота мутаций (генетический дрейф) в сочетании с реассортацией сегментированного вирусного генома (генетический сдвиг) приводит к быстрой эволюции устойчивых к лекарственным средствам изолятов, которые могут быть такими же заразными, как и их аналоги дикого типа. В результате существует значительная неудовлетворенная медицинская потребность в новых терапевтических стратегиях для преодоления ограничений существующих лекарственных средств. Противовирусные препараты, нацеленные на широкий спектр подтипов гриппа, и в то же время сводящие к минимуму возможность появления устойчивого к лекарственным препаратам вируса, представляют собой серьезную неудовлетворенную потребность общественного здравоохранения.
Но потребность в новых терапевтических средствах для лечения респираторных патогенов выходит далеко за рамки гриппа. Не существует лекарств для лечения коронавирусов, таких как MERS и SARS, вирусов парагриппа или аденовирусов; а инфекции РСВ лечат рибавирином, который малоэффективен (вирус-специфическое антитело также доступно, но только для профилактического применения). Кроме того, многие респираторные патогены проявляются сходными симптомами, называемыми гриппоподобным заболеванием, в связи с чем одно лекарственное средство широкого спектра действия, обладающее эффективностью в отношении различных патогенных микроорганизмов, было бы весьма полезным. Существует потребность в разработке одного фармацевтического препарата, эффективного в отношении широкого спектра вирусов, которые могут вызывать респираторные инфекции, - не только гриппа, но и коронавирусов, РСВ, вируса парагриппа, цитомегаловируса человека (HCMV) и аденовируса.
Цитомегаловирус человека (HCMV) является основной причиной врожденных дефектов и оппортунистических инфекций у людей с ослабленным иммунитетом и возможным кофактором при некоторых видах рака. Пациенты с трансплантацией органов, получающие иммуносупрессивную терапию, подвержены высокому риску вирусных инфекций; активация латентного вируса, а также первичных инфекций донора или внебольничных инфекций, может вызвать значительные осложнения, включая отторжение трансплантата, привести к увеличению тяжести заболевания и смертности. Герпесвирусы (например, HCMV, HSV-1), полиомавирусы (например, BKV и JCV), вирусы гепатита (HBV и HCV) и респираторные вирусы (например, грипп A, аденовирус) являются 4 основными вирусными классами, инфицирующими указанных пациентов. Цитомегаловирус (HCMV) является наиболее распространенным посттрансплантационным патогеном; HCMV может инфицировать большинство органов, и, несмотря на наличие противовирусных препаратов HCMV, таких как ганцикловир, нефротоксические побочные эффекты и повышение уровня лекарственной устойчивости значительно снижают выживаемость трансплантата и пациента. Кроме того, HCMV-опосредованная иммуномодуляция может реактивировать отдельные скрытые вирусы, носителями которых являются большинство взрослых. Компания FORGE Life Science, LLC ранее раскрыла тиазолсодержащие соединения, которые активны в отношении репликации HCMV в опубликованных заявках на патент WO 2016/077232 и WO 2016/077240.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
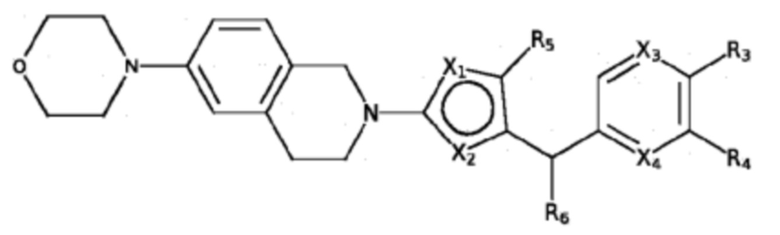
В изобретении предложены соединения, имеющие структуру формулы I:
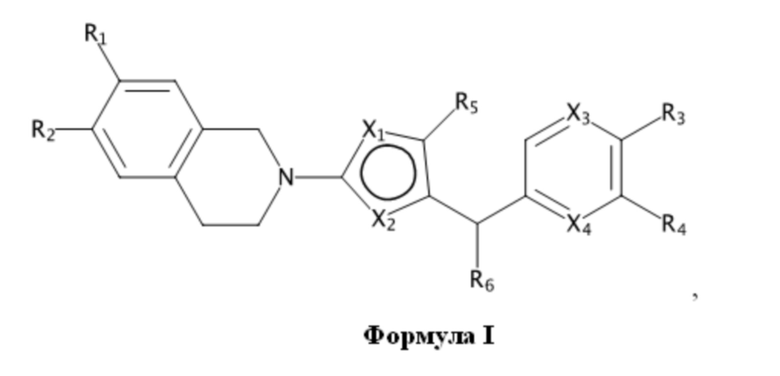
где:
один из X1 и X2 представляет собой N, и другой представляет собой S;
X3 и X4 независимо выбраны из C и N; и когда X3 представляет собой C, он необязательно замещен метилом, этилом, пропилом, изопропилом или н-пропилом;
один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-3 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8 или -OR12; и C3-6 циклоалкила, необязательно замещенного -R12, -OR12 или -NR7R8,
или R1 и R2 вместе образуют 5- или 6-членный арил или циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-3 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8 или -OR12; и C3-6 циклоалкила, необязательно замещенного -R12, -OR12 или -NR7R8;
R3 выбран из H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7, -C(O)NR7R8 и кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила;
R4 выбран из H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7, -C(O)NR7R8, кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила,
или группа R4 связана с X4 с образованием 5- или 6-членного арила или циклоалкила с 0-3 кольцевыми гетероатомами, выбранными из N, O и S, и замещенного 0-2 группами, выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила;
при условии, что:
по меньшей мере один из R3 и R4 выбран из группы, состоящей из: H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7 и -C(O)NR7R8, и
оба R3 и R4 не представляют собой H;
R5 выбран из группы, состоящей из H, метила, этила, н-пропила, изопропила, н-бутила, CF3, CH2CF3 и галогена;
R6 выбран из группы, состоящей из H, метила, этила, н-пропила, изопропила, н-бутила, CF3, CH2CF3, галогена, циклопропилметила и C1-4 алкокси;
R7 и R8 независимо, в каждом случае, выбраны из H, C1-6 алкила с прямой или разветвленной цепью, C3-6 циклоалкила, циклопропилметила и циклобутилметила; и
R12 независимо, в каждом случае, выбран из H и C1-4 алкила с прямой или разветвленной цепью;
или их фармацевтически приемлемая соль или сольват.
Соединения согласно изобретению подходят для лечения и/или предотвращения вирусных инфекций. В частности, соединения согласно изобретению являются противовирусными средствами широкого спектра действия. Например, соединения согласно изобретению блокируют репликацию двух очень разных патогенов человека с сопоставимой эффективностью, включая грипп A, быстро реплицирующийся ортомиксовирус с РНК-геномом, и HCMV, медленно реплицирующийся герпесвирус с ДНК-геномом.
В изобретении также предложены способы предотвращения, лечения и/или ослабления инфекций HCMV с помощью соединений формулы I. В изобретении также предложены способы предотвращения, лечения и/или ослабления инфекций гриппа с помощью соединений формулы I.
Если не указано иное, все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понимается специалистами в области техники, к которой относится данное изобретение. Способы и материалы описаны в настоящем документе для использования в настоящем изобретении; другие подходящие способы и материалы, известные в данной области, также могут быть использованы. Материалы, способы и примеры являются только иллюстративными и не предназначены для ограничения. Все публикации, заявки на патенты, патенты, последовательности, записи в базе данных и другие ссылки, упомянутые в настоящем документе, включены посредством ссылки во всей их полноте. В случае разночтений настоящее описание, включая определения, следует считать приоритетным.
Другие признаки и преимущества изобретения будут очевидны из следующего подробного описания и фигур, а также из формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 представлен спектр ядерного магнитного резонанса 1H ЯМР (CD3OD, 500 МГц) для соединения, полученного согласно примеру 6.
На Фиг. 2 представлен спектр ядерного магнитного резонанса 1H ЯМР (CD3OD, 500 МГц) для соединения, полученного согласно примеру 7.
На Фиг. 3 представлен спектр ядерного магнитного резонанса 1H ЯМР (CD3OD, 500 МГц) для соединения, полученного согласно примеру 8.
На Фиг. 4 представлен спектр ядерного магнитного резонанса 1H ЯМР (CDCl3, 500 МГц) для соединения, полученного согласно примеру 9.
На Фиг. 5 представлен спектр ядерного магнитного резонанса 1H ЯМР (CD3OD, 500 МГц) для соединения, полученного согласно примеру 10.
На Фиг. 6 представлен спектр ядерного магнитного резонанса 1H ЯМР (ДМСО-d6, 500 МГц) для соединения, полученного согласно примеру 16.
ПОДРОБНОЕ ОПИСАНИЕ
В настоящем документе предложены соединения, подходящие для лечения и/или предотвращения широкого спектра вирусных инфекций.
В настоящем документе предложены способы лечения или предотвращения вирусной инфекции у субъекта. Согласно некоторым вариантам реализации способы включают введение терапевтически эффективного количества одного или более соединений, предложенных в настоящем документе. Согласно некоторым вариантам реализации соединения, предложенные в настоящем документе, могут ингибировать продуцирование вируса в клетке, инфицированной вирусом. В таких вариантах реализации клетку приводят в контакт с ингибирующим продуцирование вируса количеством одного или более соединений, предложенных в настоящем документе.
В настоящем документе предложены соединения структуры формулы I:
где:
один из X1 и X2 представляет собой N, и другой представляет собой S;
X3 и X4 независимо выбраны из C и N; и когда X3 представляет собой C, он необязательно замещен метилом, этилом, пропилом, изопропилом или н-пропилом;
один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-3 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8 или -OR12; и C3-6 циклоалкила, необязательно замещенного -R12, -OR12 или -NR7R8;
или R1 и R2 вместе образуют 5- или 6-членный арил или циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-3 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8 или -OR12; и C3-6 циклоалкила, необязательно замещенного -R12, -OR12 или -NR7R8;
R3 выбран из H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7, -C(O)NR7R8, и кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила;
R4 выбран из H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7, -C(O)NR7R8, кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 0-3 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила,
или группа R4 связана с X4 с образованием 5- или 6-членного арила или циклоалкила с 0-3 кольцевыми гетероатомами, выбранными из N, O и S, и замещенного 0-2 группами, выбранными из =O, галогена; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; -C(O)-C1-6 алкила и -C(O)O-C1-6 алкила;
при условии, что:
по меньшей мере один из R3 и R4 выбран из группы, состоящей из: H, галогена, -C≡CH, -C≡N, -OH, -OCF3, -OCHF2, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -N(CH3)2, -C(O)NH2, -NHSO2R7 и -C(O)NR7R8, и оба R3 и R4 не представляют собой H;
R5 выбран из группы, состоящей из H, метила, этила, н-пропила, изопропила, н-бутила, CF3, CH2CF3 и галогена;
R6 выбран из группы, состоящей из H, метила, этила, н-пропила, изопропила, н-бутила, CF3, CH2CF3, галогена, циклопропилметила и C1-4 алкокси;
R7 и R8 независимо, в каждом случае, выбраны из H, C1-6 алкила с прямой или разветвленной цепью, C3-6 циклоалкила, циклопропилметила и циклобутилметила; и
R12 независимо, в каждом случае, выбран из H и C1-4 алкила с прямой или разветвленной цепью;
и их фармацевтически приемлемые соли или сольваты.
Соединения формулы I подходят для предотвращения, лечения и/или ослабления вирусной инфекции. В частности, указанные соединения представляют собой противовирусные средства широкого спектра действия, способные лечить широкий спектр инфекций, вызываемых вирусами, такими как грипп, коронавирусы, респираторно-синцитиальный вирус (РСВ), вирус парагриппа, цитомегаловирус человека (HCMV) и аденовирус. В частности, заявители продемонстрировали антивирусную полезность соединения формулы I широкого спектра действия, продемонстрировав способность указанных соединений блокировать репликацию двух очень разных патогенов человека с сопоставимой эффективностью - гриппа A, быстро реплицирующегося ортомиксовируса с РНК-геномом, и HCMV, медленно реплицирующегося герпесвируса с ДНК-геномом.
Согласно некоторым вариантам реализации противовирусных соединений формулы I один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с 1-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; циклопропила, циклопропилметила, циклобутила, циклопентила и циклогексила,
или R1 и R2 вместе образуют 5- или 6-членный арил, циклоалкил или циклоалкенил с 1-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8 или -OR12; циклопропила, циклопропилметила, циклобутила, циклопентила и циклогексила.
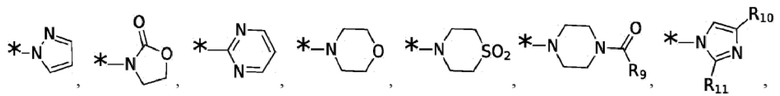
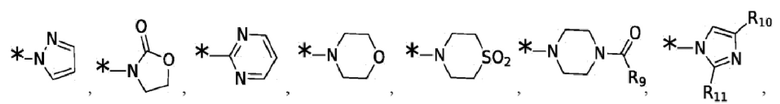
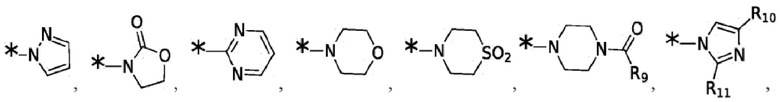
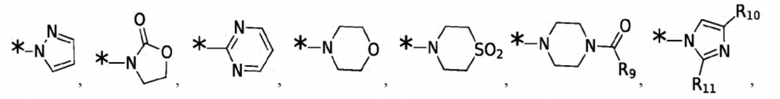
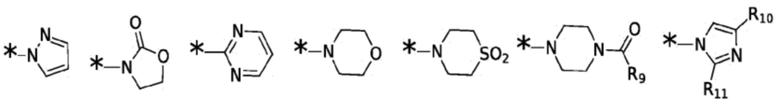
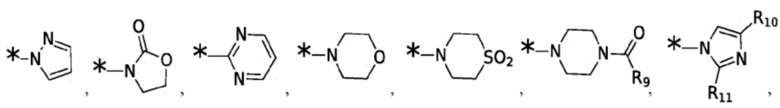
Согласно некоторым вариантам реализации противовирусных соединений формулы I R3 выбран из группы, состоящей из:
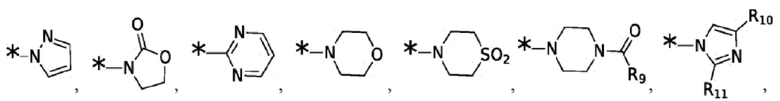
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
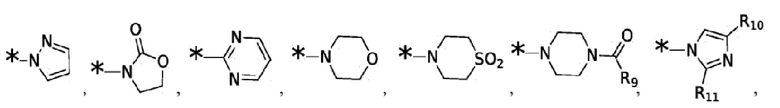
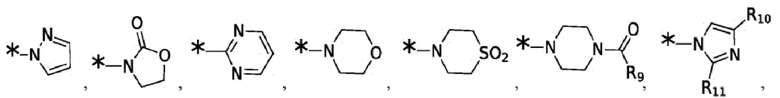
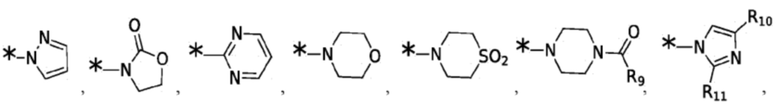
Согласно некоторым вариантам реализации противовирусных соединений формулы I R4 выбран из группы, состоящей из:
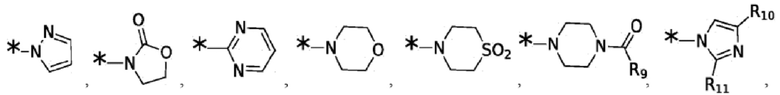
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы I
один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с по меньшей мере одним кольцевым гетероатомом N и от 0 до 2 дополнительными кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из:
=O; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или NR7R8; C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8 или -OR12; и C3-6 циклоалкила, необязательно замещенного -R12, -OR12 или -NR7R8.
Соединения данного варианта реализации включают, но не ограничиваются ими:
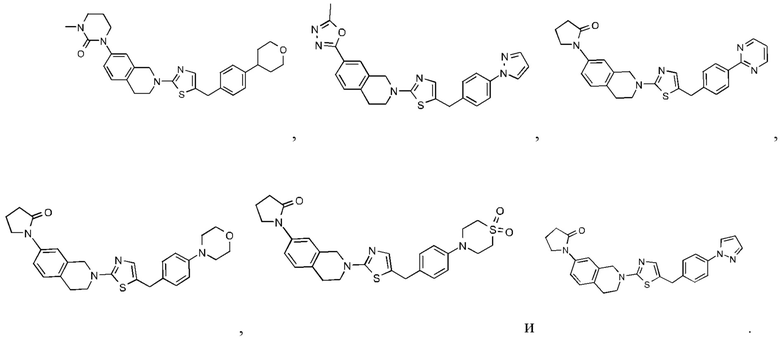
Согласно некоторым вариантам реализации соединений формулы I один из R1 и R2 представляет собой H, и другой выбран из группы, состоящей из:
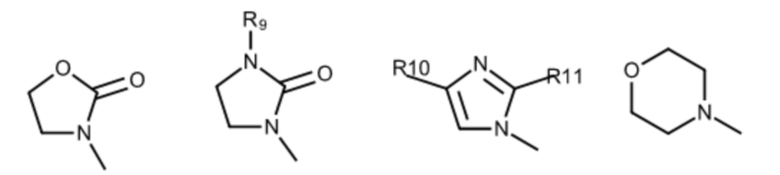
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Соединения данного варианта реализации включают, но не ограничиваются ими:
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы II:
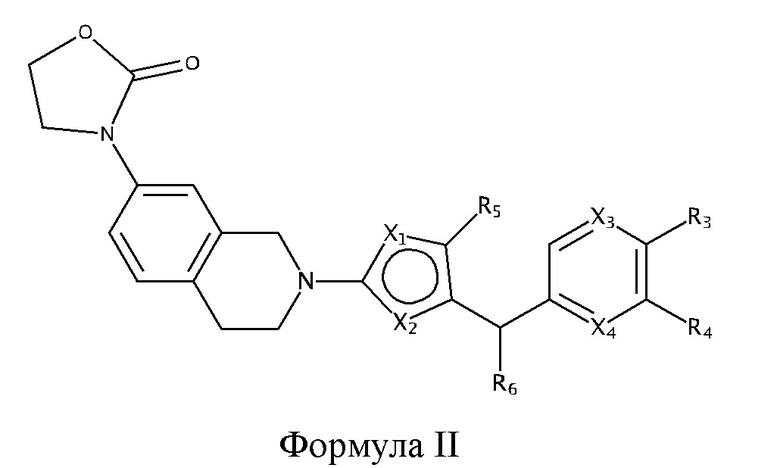
где X1, X2, X3, X4, R3, R4, R5 и R6 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы II R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы II R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы III:
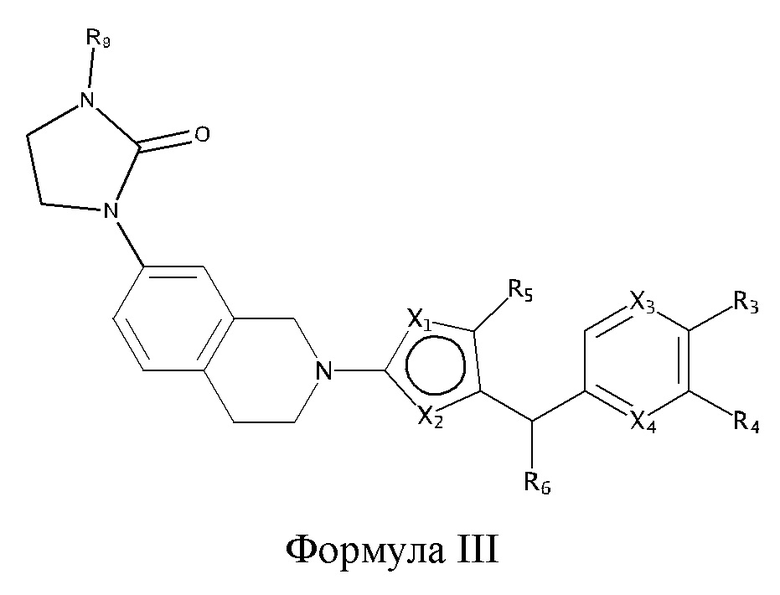
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8, и
X1, X2, X3, X4, R3, R4, R5, R6, R7, R8 и R12 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы III R3 выбран из группы, состоящей из:
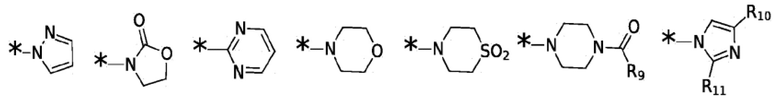
и -SO2(C1-6 алкил);
где: R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы III R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где: R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы IV:
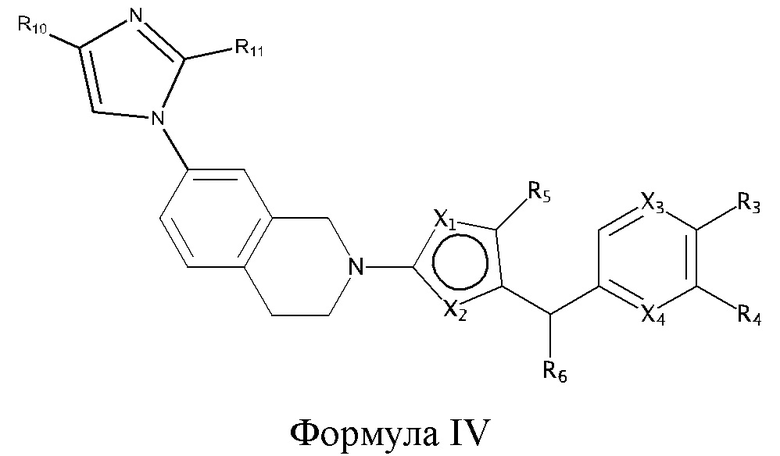
где:
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8, и
X1, X2, X3, X4, R3, R4, R5, R6, R7, R8 и R12 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы IV R3 выбран из группы, состоящей из:
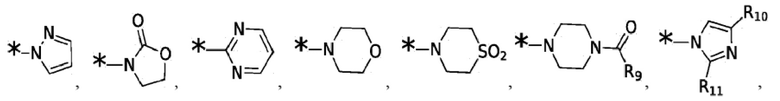
и -SO2(C1-6 алкил);
где: R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8. Согласно некоторым вариантам реализации противовирусных соединений формулы IV R4 выбран из группы, состоящей из:
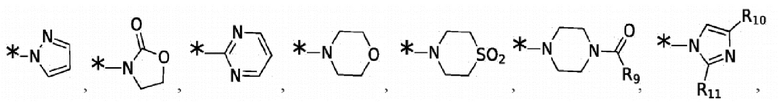
и -SO2(C1-6 алкил);
где: R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы V:
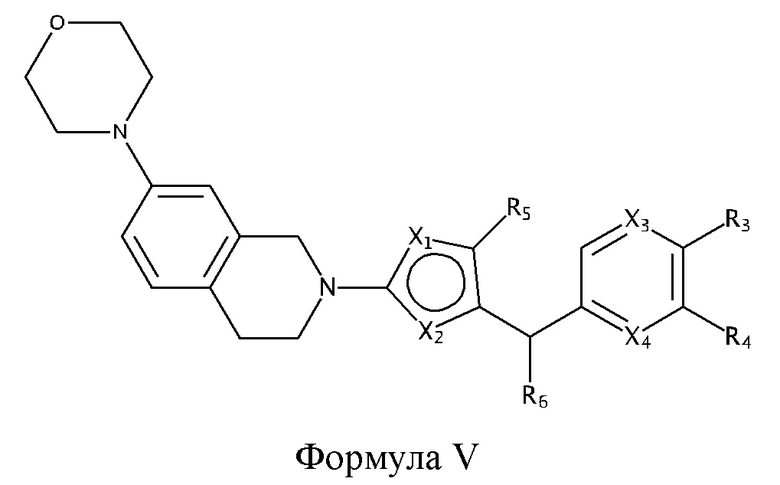
где X1, X2, X3, X4, R3, R4, R5 и R6 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы V R3 выбран из группы, состоящей из:
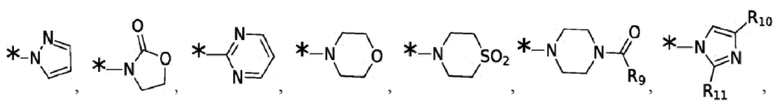
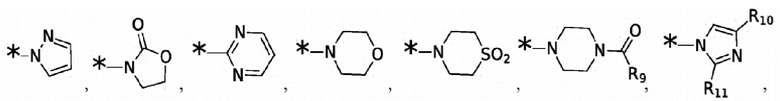
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы V R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы VI:
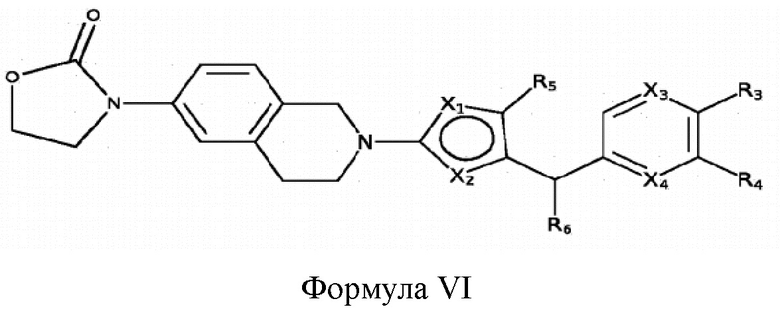
где X1, X2, X3, X4, R3, R4, R5 и R6 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы VI R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы VI R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы VII:
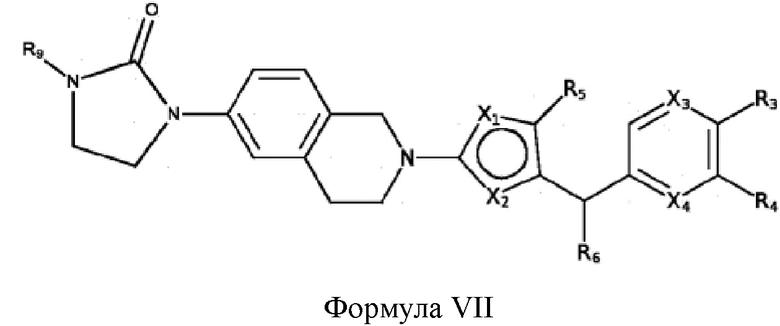
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8, и
X1, X2, X3, X4, R3, R4, R5, R6, R7, R8 и R12 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы VII R3 выбран из группы, состоящей из:
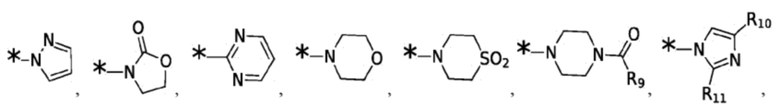
и -SO2(C1-6 алкил);
где: R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы VII R4 выбран из группы, состоящей из:
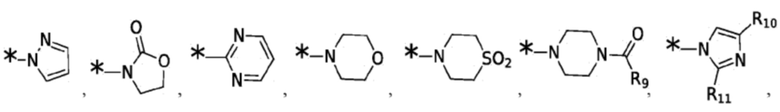
и -SO2(C1-6 алкил);
где: R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы VIII:
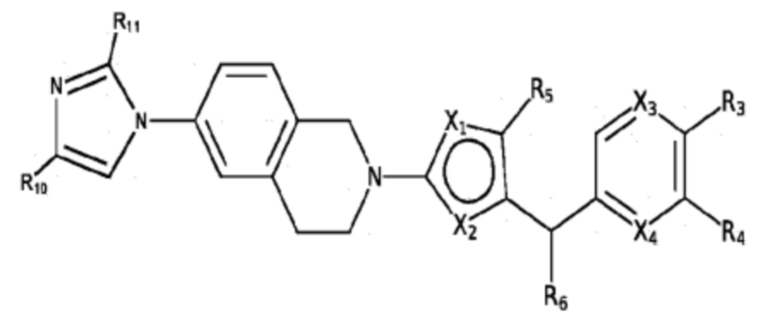
Формула VIII
где:
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8, и
X1, X2, X3, X4, R3, R4, R5, R6, R7, R8 и R12 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы VIII R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где: R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы VIII R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где: R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8.
Некоторые варианты реализации соединений формулы I представляют собой соединения формулы IX:
Формула IX
где X1, X2, X3, X4, R3, R4, R5 и R6 являются такими, как определено для формулы I. Согласно некоторым вариантам реализации противовирусных соединений формулы IX R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
Согласно некоторым вариантам реализации противовирусных соединений формулы IX R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R9 выбран из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила, циклопентила, циклогексила; C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; и C1-6 алкокси с прямой или разветвленной цепью, необязательно замещенного -NR7R8; и
R10 и R11 независимо выбраны из группы, состоящей из: H, циклопропила, циклопропилметила, циклобутила; C1-4 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12 или -NR7R8; C1-4 алкокси с прямой или разветвленной цепью, необязательно замещенного NR7R8.
В настоящем документе также предложен способ лечения или предотвращения вирусной инфекции у субъекта, включающий введение терапевтически эффективного количества соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII, формулы IX или его фармацевтически приемлемых солей или сольватов.
В настоящем документе также предложен способ ингибирования продуцирования вируса, включающий приведение инфицированной вирусом клетки в контакт с ингибирующим продуцирование вируса количеством соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII, формулы IX или его фармацевтически приемлемых солей или сольватов.
В настоящем документе также предложен способ лечения или предотвращения инфекции HCMV у субъекта путем введения терапевтически эффективного количества соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII, формулы IX или его фармацевтически приемлемых солей или сольватов.
В настоящем документе также предложен способ ингибирования продуцирования HCMV, включающий приведение инфицированной HCMV клетки в контакт с ингибирующим продуцирование вируса количеством соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII формулы IX или его фармацевтически приемлемых солей или сольватов.
В настоящем документе также предложен способ лечения или предотвращения инфекции гриппа у субъекта путем введения терапевтически эффективного количества соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII, формулы IX или его фармацевтически приемлемых солей или сольватов.
В настоящем документе также предложен способ ингибирования продуцирования гриппа, включающий приведение инфицированной гриппом клетки в контакт с ингибирующим продуцирование вируса количеством соединения формулы I, формулы II, формулы III, формулы IV, формулы V, формулы VI, формулы VII, формулы VIII, формулы IX или его фармацевтически приемлемых солей или сольватов.
В сочетании с соединениями и способами, описанными в настоящем документе, также можно вводить противовирусный агент. Агент может быть любым терапевтическим агентом, подходящим для лечения вирусной инфекции, инфекции HCMV или инфекции гриппа. Например, противовирусный агент может включать ацикловир, докозанол, рибаривин, интерфероны и тому подобное; ацетат целлюлозы, карбопол и каррагинан, плеконарил, амантидин, римантидин, фомивирсен, зидовудин, ламивудин, занамивир, осельтамивир, бривудин, абакавир, адефовир, ампренавир, арбидол, атазанавир, атрипла, цидофовир, комбивир, эдоксудин, эфавиренц, эмтрицитабин, энфувиртид, энтекавир, фамцикловир, фосампренавир, фоскарнет, фосфонет, ганцикловир, гардасил, ибацитабин, имуновир, идоксуридин, имиквимод, индинавир, инозин, ингибитор интегразы, ламивудин, лопинавир, ловирид, mk-0518, маравирок, мороксидин, нельфинавир, невирапин, нексавир, аналоги нуклеотидов и/или нуклеозидов, осельтамивир, пенцикловир, перамивир, подофиллотоксин, римантадин, ритонавир, саквинавир, ставудин, тенофовир, тенофовира дизопроксил, типранавир, трифлуридин, тризивир, тромантадин, трувада, валацикловир, валганцикловир, викривирок, видарабин, вирамидин, зальцитабин, морфолиновые олигонуклеотиды, рибозим, ингибиторы протеазы, ингибитор сборки (например, рифампицин), зидовудин, бринцидофовир, фавипиравир, нитоксанид, летермовир, марибавир, CMX157 или их комбинации или два или более противовирусных агентов.
Согласно некоторым вариантам реализации соединение, предложенное в настоящем документе, можно вводить до, после или одновременно с введением одного или более противовирусных агентов.
Предложенный в настоящем документе противовирусный агент, включая его фармацевтически приемлемую соль или сольват, может быть приобретен коммерчески или получен с использованием известных методов органического синтеза.
Способы, предложенные в настоящем документе, включают получение и применение фармацевтических композиций, которые включают соединения, предложенные в настоящем документе, и один или более фармацевтически приемлемых носителей. В настоящем документе также предложены сами композиции.
Фармацевтические композиции обычно включают фармацевтически приемлемый носитель. В настоящем документе термин «фармацевтически приемлемый носитель» включает физиологический раствор, растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и тому подобное, совместимые с фармацевтическим введением.
Фармацевтическую композицию обычно получают так, чтобы она была совместима с предполагаемым способом ее введения. Примеры способов введения включают парентеральное, например, внутривенное, внутрикожное, подкожное, пероральное (например, ингаляционное), трансдермальное (местное), трансмукозальное и ректальное введение.
Способы получения подходящих фармацевтических композиций известны в данной области техники, см., например, Remington: The Science and Practice of Pharmacy, 21-е изд., 2005; и книги из серии «Drugs and the Pharmaceutical Sciences: a Series of Textbooks and Monographs» (Dekker, Нью-Йорк). Например, растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. рН можно регулировать кислотами или основаниями, такими как соляная кислота или гидроксид натрия. Парентеральный препарат может быть заключен в ампулы, одноразовые шприцы или флаконы с многократными дозами, сделанные из стекла или пластика.
Фармацевтические композиции, подходящие для инъекций, могут включать стерильные водные растворы (если они растворимы в воде) или дисперсии и стерильные порошки для немедленного приготовления стерильных инъекционных растворов или дисперсий. Для внутривенного введения подходящие носители включают физиологический раствор, бактериостатическую воду, Cremophor EL™ (BASF, Parsippany, NJ) или физиологический раствор с фосфатным буфером (PBS). Во всех случаях композиция должна быть стерильной и должна быть текучей до такой степени, чтобы ее можно было легко вводить через шприц. Композиция должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкий полиэтиленгликоль и тому подобное) и их подходящие смеси. Надлежащую текучесть можно поддерживать, например, путем использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсии и путем использования поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и тому подобного. Во многих случаях будет предпочтительно включать в композицию изотонические агенты, например сахара, полиспирты, такие как маннит, сорбит и хлорид натрия. Длительная абсорбция инъецируемых композиций может быть достигнута включением в композицию агента, который задерживает абсорбцию, например моностеарата алюминия и желатина.
Стерильные растворы для инъекций могут быть получены путем включения соединения, предложенного в настоящем документе, в необходимом количестве в подходящем растворителе, с одним или комбинацией ингредиентов, перечисленных выше, по мере необходимости, с последующей стерилизацией фильтрованием. Как правило, дисперсии получают путем включения соединения, предложенного в настоящем документе, в стерильный носитель, который содержит основную дисперсионную среду и необходимые другие ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных растворов для инъекций предпочтительными способами получения являются вакуумная сушка и сублимационная сушка, в результате чего получают порошок соединения, предложенного в настоящем документе, плюс любой дополнительный желаемый ингредиент из его предварительно стерильно отфильтрованного раствора.
Композиции для перорального введения обычно включают инертный разбавитель или пищевой носитель. Для целей перорального терапевтического введения соединение, предложенное в настоящем документе, может быть включено в вспомогательные вещества и использоваться в форме таблеток, троше или капсул, например желатиновых капсул. Пероральные композиции также могут быть получены с использованием жидкого носителя для применения в качестве жидкости для полоскания рта. Фармацевтически совместимые связующие агенты и/или адъюванты могут быть включены в состав композиции. Таблетки, пилюли, капсулы, троше и тому подобное могут содержать любой из следующих ингредиентов или соединений аналогичной природы: связующее, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; вспомогательное вещество, такое как крахмал или лактоза, разрыхлитель, такой как альгиновая кислота, Примогель (англ. Primogel) или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Стеротес (англ. Sterotes); скользящее вещество, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор.
Для введения путем ингаляции соединения могут быть доставлены в форме аэрозольного спрея из находящегося под давлением контейнера или дозатора, который содержит подходящий пропеллент, например газ, такой как диоксид углерода, или распылитель. Такие способы включают описанные в патенте США № 6468798.
Системное введение терапевтического соединения, как описано в настоящем документе, также может осуществляться трансмукозальным или трансдермальным путем. Для трансмукозального или трансдермального введения в составе используют обеспечивающие проникновение средства, подходящие для барьера, через который нужно проникнуть. Такие обеспечивающие проникновение средства, как правило, известны в данной области и включают, например, трансмукозальное введение, детергенты, соли желчных кислот и производные фузидиевой кислоты. Трансмукозальное введение может быть достигнуто путем использования назальных спреев или суппозиториев. Для трансдермального введения соединения, предложенные в настоящем документе, могут быть получены в виде мазей, бальзамов, гелей или кремов, как это общеизвестно в данной области.
Фармацевтические композиции также могут быть получены в форме суппозиториев (например, с традиционными основами для суппозиториев, такими как масло какао и другие глицериды) или удерживающих клизм для ректальной доставки.
Кроме того, возможна интраназальная доставка, как описано, среди прочего, в Hamajima et al., Clin. Immunol. Immunopathol., 88 (2), 205-10 (1998). Также могут быть использованы липосомы (например, как описано в патенте США № 6473375) и микроинкапсулирование. Также можно использовать системы доставки биоразлагаемых нацеливаемых микрочастиц (например, как описано в патенте США № 6471996).
Согласно одному из вариантов реализации терапевтические соединения получают с носителями, которые будут защищать терапевтические соединения от быстрого выведения из организма, такие как состав с контролируемым высвобождением, включая имплантаты и микрокапсулированные системы доставки. Могут быть использованы биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Такие составы могут быть получены с использованием стандартных методик или получены коммерчески, например, от Alza Corporation и Nova Pharmaceuticals, Inc. В качестве фармацевтически приемлемых носителей также можно использовать липосомные суспензии (включая липосомы, нацеленные на выбранные клетки с моноклональными антителами к клеточным антигенам). Они могут быть получены в соответствии со способами, известными специалистам в данной области, например, как описано в патенте США № 4522811.
Фармацевтическую композицию можно вводить сразу или ее можно разделить на несколько меньших доз, вводимых через определенные промежутки времени. Понятно, что точная дозировка и длительность лечения зависят от заболевания, которое лечат, и они могут быть определены эмпирически с использованием известных протоколов тестирования или путем экстраполяции из данных испытаний in vivo или in vitro. Следует отметить, что концентрации и дозировки также могут варьироваться в зависимости от тяжести состояния, которое должно быть облегчено. Кроме того, следует понимать, что для любого конкретного пациента конкретные режимы дозирования должны корректироваться с течением времени в соответствии с индивидуальной потребностью и профессиональным суждением лица, осуществляющего или контролирующего введение композиций, и что диапазоны концентраций, изложенные в настоящем документе, представлены только для примера и не предназначены для ограничения объема или применения заявленных композиций.
Могут быть получены лекарственные формы или композиции, содержащие соединение, как описано в настоящем документе, в диапазоне от 0,005% до 100%, где остальное состоит из нетоксичного носителя. Способы получения указанных композиций известны специалистам в данной области техники. Рассматриваемые композиции могут содержать 0,001%-100% соединения, предложенного в настоящем документе, в одном варианте реализации 0,1-95%, в другом варианте реализации 75-85%.
Фармацевтические композиции могут быть включены в контейнер, упаковку или дозатор вместе с инструкциями по введению.
Как описано выше, препараты одного или более соединений, предложенных в настоящем документе, можно вводить перорально, парентерально, местно или ректально. Их, разумеется, обеспечивают в формах, подходящих для каждого способа введения. Например, их вводят в форме таблеток или капсул, путем инъекции, ингаляции, лосьона для глаз, мази, суппозитория, инфузии; местно лосьоном или мазью; и ректально через суппозитории. В некоторых вариантах реализации введение является пероральным.
Фразы «парентеральное введение» и «вводят парентерально», используемые в настоящем документе, означают способы введения, отличные от энтерального и местного введения, обычно путем инъекции, и включают, без ограничения, внутривенные, внутримышечные, внутриартериальные, интратекальные, интракапсулярные, интраорбитальные, внутрисердечные, внутрикожные, внутрибрюшинные, транстрахеальные, подкожные, субкутикулярные, внутрисуставные, субкапсулярные, субарахноидальные, интраспинальные и внутригрудинные инъекции и инфузии.
Фактические уровни дозировки активных ингредиентов в фармацевтических композициях, представленных в настоящем документе, можно варьировать, чтобы получить количество активного ингредиента, которое эффективно для достижения желаемого терапевтического ответа для конкретного пациента, композиции и способа введения без токсичности для пациента.
Концентрация соединения, предложенного в настоящем документе, в фармацевтически приемлемой смеси будет варьироваться в зависимости от нескольких факторов, включая дозировку соединения, подлежащего введению, фармакокинетические характеристики используемого соединения(й) и способа введения. Согласно некоторым вариантам реализации композиции, предложенные в настоящем документе, могут быть обеспечены в водном растворе, содержащем примерно 0,1-10% мас./об. соединения, описанного в настоящем документе, среди других веществ, для парентерального введения. Типичные диапазоны доз могут включать от примерно 0,01 до примерно 500 мг/кг массы тела в день, вводимых в 1-4 разделенных дозах. Каждая разделенная доза может содержать одинаковые или разные соединения. Дозировка будет терапевтически эффективным количеством в зависимости от нескольких факторов, включая общее состояние здоровья пациента, а также состав и способ введения выбранного соединения(й).
Хотя дозировка будет варьироваться в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести расстройства, которое нужно вылечить или предотвратить, способа введения и формы лекарственного средства, как правило, для взрослого человека рекомендуется суточная дозировка, составляющая от 0,01 до 2000 мг соединения, и ее можно вводить в единичной дозе или в разделенных дозах. Количество активного ингредиента, которое можно комбинировать с материалом носителя для получения единичной дозированной формы, обычно будет представлять собой такое количество соединения, которое оказывает терапевтический эффект.
Точное время введения и/или количество композиции, которое приведет к наиболее эффективным результатам с точки зрения эффективности лечения у определенного пациента, будет зависеть от активности, фармакокинетики и биодоступности конкретного соединения, физиологического состояния пациента (включая возраст, пол, вид и стадию заболевания, общее физическое состояние, чувствительность к определенной дозировке и тип лекарства), способа введения и т. д. Однако приведенные выше рекомендации могут быть использованы в качестве основы для точной корректировки лечения, например, определения оптимального времени и/или количества введения, которые потребуют не более чем рутинных экспериментов, заключающихся в мониторинге пациента и корректировке дозы и/или времени.
В настоящем документе также предложена комбинированная терапия, в которой один или более других терапевтических агентов вводят с соединением или фармацевтической композицией, содержащей соединение, предложенное в настоящем документе. Такая комбинированная терапия может быть достигнута путем одновременного, последовательного или раздельного введения доз отдельных компонентов лечения.
Определения
Подразумевается, что после терминов «например» и «такой как» и их грамматических эквивалентов следует фраза «и без ограничения», если явно не указано иное. Используемый в настоящем документе термин «примерно» предназначен для учета отклонений вследствие погрешности эксперимента. Все измерения, представленные в данном документе, понимаются как модифицированные термином «примерно», независимо от того, используется ли этот термин явным образом или нет, если явно не указано иное. В настоящем документе формы единственного числа включают множественное число, если контекст явно не предписывает иное.
«Субъект», используемый в настоящем документе, включает как людей, так и других животных, в частности млекопитающих. Таким образом, способы применимы как для терапии человека, так и для применения в ветеринарии. Согласно некоторым вариантам реализации пациентом является млекопитающее, например, примат. Согласно некоторым вариантам реализации пациент представляет собой человека.
«Терапевтически эффективное» количество соединения, предложенного в настоящем документе, как правило, представляет собой такое количество, которое является достаточным для предотвращения, устранения, ослабления или снижения симптомов вирусной инфекции, включая, но не ограничиваясь ими, грипп, коронавирусы, респираторно-синцитиальный вирус (RSV), вирус парагриппа, цитомегаловирус человека (HCMV) и аденовирусную инфекцию. Следует понимать, что для профилактики можно использовать концентрации, отличающиеся от используемых для лечения активного заболевания.
Количество соединения, предложенного в настоящем документе, «ингибирующее продуцирование вируса», как правило, представляет собой такое количество, которое является достаточным для достижения измеримого уменьшения количества вируса, продуцируемого клетками, контактирующими с соединением. Согласно некоторым вариантам реализации количество, «ингибирующее продуцирование вируса», представляет собой количество, которое ингибирует по меньшей мере 30% продукции вируса в необработанных клетках. Согласно некоторым вариантам реализации количество, «ингибирующее продуцирование вируса», представляет собой количество, которое ингибирует по меньшей мере 50% продукции вируса в необработанных клетках. Согласно некоторым вариантам реализации количество, «ингибирующее продуцирование вируса», представляет собой количество, которое ингибирует по меньшей мере 70% продукции вируса в необработанных клетках. Согласно некоторым вариантам реализации количество, «ингибирующее продуцирование вируса», представляет собой количество, которое ингибирует по меньшей мере 90% продукции вируса в необработанных клетках.
Термины «лечение» и «предотвращение» являются общепризнанными и включают введение одного или более соединений или фармацевтических композиций, предложенных в настоящем документе. Если их вводят до клинического проявления нежелательного состояния (например, заболевания или другого нежелательного состояния субъекта), тогда лечение является профилактическим (то есть оно защищает субъекта от развития нежелательного состояния). В данном контексте термин «предотвращать» означает замедление или предотвращение появления по меньшей мере одного симптома расстройства, как предусмотрено в настоящем документе. Например, такое предотвращение может быть рекомендовано вероятностью воздействия инфекционного агента (например, вируса) или когда у субъекта проявляются другие симптомы, которые указывают на начало развития расстройства (например, нарушение обмена веществ или сердечно-сосудистое нарушение). Альтернативно, если их вводят после проявления нежелательного состояния, лечение является терапевтическим (т.е. оно предназначено для уменьшения, ослабления или стабилизации существующего нежелательного состояния или его побочных эффектов). В данном контексте, «лечить» означает ослабление по меньшей мере одного симптома расстройства, как предусмотрено в настоящем документе.
В настоящем документе термин «соединение» включает все стереоизомеры, геометрические изомеры и таутомеры изображенных структур. Предполагается, что соединения, идентифицированные в настоящем документе по названию или структуре в виде одной конкретной таутомерной формы, включают другие таутомерные формы, если не указано иное.
Согласно некоторым вариантам реализации соединение, предложенное в настоящем документе, или его соль, по существу, является выделенным. «По существу выделенное» означает, что соединение по меньшей мере частично или по существу отделено от среды, в которой оно было образовано или обнаружено. Частичное отделение может включать, например, композицию, обогащенную соединением, предложенным в настоящем документе. Существенное отделение может включать композиции, содержащие по меньшей мере примерно 50%, по меньшей мере примерно 60%, по меньшей мере примерно 70%, по меньшей мере примерно 80%, по меньшей мере примерно 90%, по меньшей мере примерно 95%, по меньшей мере примерно 97% или по меньшей мере примерно 99% по массе соединения, предложенного в настоящем документе, или его соли. Способы выделения соединений и их солей являются общепринятыми в данной области техники.
Фраза «фармацевтически приемлемый» используется в настоящем документе для обозначения соединений, материалов, композиций и/или лекарственных форм, которые в рамках здравого медицинского суждения подходят для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерных с разумным соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к относительно нетоксичным солям присоединения неорганических и органических кислот и соединения, предложенного в настоящем документе. Указанные соли могут быть получены in situ в ходе конечного выделения и очистки соединения, предложенного в настоящем документе, или путем отдельной реакции соединения в форме его свободного основания с подходящей органической или неорганической кислотой и выделения образованной таким образом соли. Типичные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонатные соли и аминокислотные соли и тому подобное. (См., например, Berge et al. (1977) «Pharmaceutical salts», J. Pharm. Sci. 66: 1-19.)
Согласно некоторым вариантам реализации соединение, предложенное в настоящем документе, может содержать одну или несколько кислотных функциональных групп и, таким образом, способно образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли» в указанных случаях относится к относительно нетоксичным солям присоединения неорганического и органического основания и соединения, предложенного в настоящем документе. Указанные соли также могут быть получены in situ в ходе конечного выделения и очистки соединения или путем отдельной реакции очищенного соединения в форме его свободной кислоты с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Типичные соли щелочных или щелочноземельных металлов включают соли лития, натрия, калия, кальция, магния и алюминия и тому подобное. Типичные органические амины, подходящие для образования солей присоединения основания, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и тому подобное (см., например, Berge et al., выше).
Термин «сольват» означает соединение, которое дополнительно включает стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными межмолекулярными силами. Когда растворителем является вода, сольват представляет собой гидрат. Термин «фармацевтически приемлемый сольват» относится к относительно нетоксичным сольватам соединения, предложенного в настоящем документе, с использованием растворителя, который, в рамках здравого смысла медицинского суждения, подходит для использования в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерных с разумным соотношением польза/риск.
В настоящем документе термин «алкил» относится к алифатическим группам с прямой и разветвленной цепью, содержащим от 1 до 12 атомов углерода, предпочтительно 1-8 атомов углерода и более предпочтительно 1-6 атомов углерода, которые необязательно замещены одним, двумя или тремя заместители. Предпочтительные алкильные группы включают, без ограничения, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил. «C0» алкил (как в «C0-C3-алкиле») представляет собой ковалентную связь (например, «C0-гидрокарбил»). Термин «низший алкил» относится к алифатическим группам с прямой и разветвленной цепью, содержащим от 1 до 6 атомов углерода. Если не указано иное, термин «алкил» включает алкенильную, алкинильную и циклическую алкильную группы.
В настоящем документе термин «алкенил» означает ненасыщенную алифатическую группу с прямой или разветвленной цепью с одной или несколькими двойными углерод-углеродными связями, содержащую от 2 до 12 атомов углерода, предпочтительно 2-8 атомов углерода и более предпочтительно 2-6 атомов углерода, которая необязательно замещена одним, двумя или тремя заместителями. Предпочтительные алкенильные группы включают, без ограничения, этенил, пропенил, бутенил, пентенил и гексенил.
В настоящем документе термин «алкинил» означает ненасыщенную алифатическую группу с прямой или разветвленной цепью с одной или несколькими тройными углерод-углеродными связями, содержащую от 2 до 12 атомов углерода, предпочтительно 2-8 атомов углерода и более предпочтительно 2-6 атомов углерода, которая необязательно замещена одним, двумя или тремя заместителями. Предпочтительные алкинильные группы включают, без ограничения, этинил, пропинил, бутинил, пентинил и гексинил.
Термин «гетероалкил» относится к алкильной группе, как определено в настоящем документе выше, где один или несколько атомов углерода в цепи заменены гетератомом, выбранным из группы, состоящей из O, S и N.
«Арильная» группа представляет собой C6-C14 ароматический фрагмент, содержащий от одного до трех ароматических колец, которые необязательно замещены. Предпочтительно арильная группа представляет собой C6-C10 арильную группу. Предпочтительные арильные группы включают, без ограничения, фенил, нафтил, антраценил и флуоренил.
«Гетероциклильная» или «гетероциклическая» группа представляет собой кольцевую структуру, содержащую от примерно 3 до примерно 8 атомов углерода, где один или более атомов выбраны из группы, состоящей из N, O и S. Гетероциклическая группа необязательно замещена по атому углерода в одном или нескольких положениях. Гетероциклическая группа также независимо необязательно замещена по азоту алкилом, арилом, аралкилом, алкилкарбонилом, алкилсульфонилом, арилкарбонилом, арилсульфонилом, алкоксикарбонилом, аралкоксикарбонилом или по сере оксогруппой или низшим алкилом. Предпочтительные гетероциклические группы включают, без ограничения, эпокси, азиридинил, тетрагидрофуранил, пирролидинил, пиперидинил, пиперазинил, тиазолидинил, оксазолидинил, оксазолидинонил и морфолино. В некоторых предпочтительных вариантах реализации гетероциклическая группа конденсирована с арильной, гетероарильной или циклоалкильной группой. Примеры таких конденсированных гетероциклов включают, без ограничения, тетрагидрохинолин и дигидробензофуран. Специально исключены из объема этого термина соединения, имеющие соседние кольцевые атомы O и/или S.
В настоящем документе термин «гетероарил» относится к группам, содержащим от 5 до 14 атомов кольца, предпочтительно 5, 6, 9 или 10 атомов в кольце; имеющим 6, 10 или 14 π электронов, сопряженных в циклической системе; и имеющим, помимо атомов углерода, от одного до трех гетероатомов на кольцо, выбранных из группы, состоящей из N, O и S. «Гетероаралкильная» или «гетероарилалкильная» группа включает гетероарильную группу, ковалентно связанную с алкильной группой, каждая из которых независимо необязательно замещена или незамещена. Предпочтительные гетероалкильные группы включают C1-C6 алкильную группу и гетероарильную группу, содержащую 5, 6, 9 или 10 кольцевых атомов. Специально исключены из объема этого термина соединения, имеющие соседние кольцевые атомы O и/или S. Примеры предпочтительных гетероаралкильных групп включают пиридилметил, пиридилэтил, пирролилметил, пирролилэтил, имидазолилметил, имидазолилэтил, тиазолилметил и тиазолилэтил. Специально исключены из объема этого термина соединения, имеющие соседние кольцевые атомы O и/или S.
Варианты осуществления гетероциклилов и гетероарилов включают, но не ограничиваются ими, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1 2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил.
В настоящем документе, когда фрагмент (например, циклоалкил, гидрокарбил, арил, гетероарил, гетероциклил, мочевина и т.д.) описывается как «необязательно замещенный», это означает, что группа необязательно имеет от одного до четырех, предпочтительно от одного до трех, более предпочтительно один или два неводородных заместителя. Подходящие заместители включают, без ограничения, галоген, гидрокси, оксо (например, кольцевой -СН-, замещенный оксо, представляет собой -C(O)-), нитро, галогенгидрокарбил, гидрокарбил, арил, аралкил, алкокси, арилокси, амино, ациламино, алкилкарбамоил, арилкарбамоил, аминоалкил, ацил, карбокси, гидроксиалкил, алкансульфонил, аренсульфонил, алкансульфонамидо, аренсульфонамидо, аралкилсульфонамидо, алкилкарбонил, ацилокси, циано и уреидогруппы.
В настоящем документе термин «галоген» или «гало» относится к хлору, брому, фтору или йоду. В настоящем документе термин «ацил» относится к алкилкарбонильному или арилкарбонильному заместителю. Термин «ациламино» относится к амидной группе, присоединенной у атома азота (то есть R-CO-NH-). Термин «карбамоил» относится к амидной группе, присоединенной у карбонильного атома углерода (то есть NH2-CO-). Атом азота ациламино или карбамоильного заместителя необязательно дополнительно замещен. Термин «сульфонамидо» относится к сульфонамидному заместителю, присоединенному к атому серы или азота. Термин «амино» включает NH2, алкиламино, ариламино и циклические аминогруппы. В настоящем документе термин «уреидо» относится к замещенному или незамещенному фрагменту мочевины.
Фрагмент, который является замещенным, представляет собой группу, в которой один или несколько атомов водорода независимо заменены другим химическим заместителем. В качестве неограничивающего примера замещенные фенилы включают 2-фторфенил, 3,4-дихлорфенил, 3-хлор-4-фторфенил, 2-фтор-3-пропилфенил. В качестве другого неограничивающего примера замещенные н-октилы включают 2,4-диметил-5-этилоктил и 3-циклопентилооктил. В это определение включены метилены (-CH2-), замещенные кислородом с образованием карбонил-CO-).
«Незамещенный» фрагмент, как определено выше (например, незамещенный циклоалкил, незамещенный гетероарил и т. о.), означает фрагмент, как определено выше, который не имеет каких-либо необязательных заместителей, для которых в противном случае действует определение фрагмента (выше). Так, например, хотя «арил» включает фенил и фенил замещен галогеном, «незамещенный арил» не включает фенил, замещенный галогеном.
СИНТЕЗ СОЕДИНЕНИЙ СОГЛАСНО ИЗОБРЕТЕНИЮ
Соединения по настоящему изобретению (соединения формулы I) могут быть получены с использованием общей схемы реакций, представленной на схемах ниже. Используются следующие сокращения:
NMP, N-метил-2-пирролидон; КТ, комнатная температура; ДХМ, дихлорметан; ДМФА, N,N-диметилформамид; ТГФ, тетрагидрофуран; DCE, 1,2-дихлорэтан; TES или TES-H, триэтилсилан; TES, триэтоксисилан; TFA, трифторуксусная кислота; EtOAc или EA, этилацетат; М, молярный (молярность); TBAF, фторид тетрабутиламмония; трет-BuOH, трет-бутанол; MeI, йодистый метил; ДМСО, диметилсульфоксид; MeCN, ацетонитрил; XPhos, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил; МеОН, метанол; ч или час., часы; водн., водный; DME, 1,2-диметоксиэтан; нас., насыщенный; атм, атмосфера; Ac2O, уксусный ангидрид; конц., концентрированный; экв., эквиваленты; DIEA, N,N-диизопропилэтиламин; HATU, N-оксид N-[(диметиламино)-1H-1,2,3-триазоло-[4,5-b]пиридин-1-илметилен]-N-метилметанаминия гексафторфосфата; DMA, N,N-диметилацетамид; Pd2(dba)3, трис(дибензилиденацетон)дипалладий(0); S-Phos, дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин; PE, петролейный эфир; AcOK, ацетат калия; Pd(dppf)Cl2, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II); DMI, 1,3-диметил-2-имидазолидинон; преп-ТСХ, препаративная тонкослойная хроматография; трет-BuONa, трет-бутоксид натрия; трет-BuOK, трет-бутоксид калия; HMDS, гексаметилдисилазан; (Pd(OAc)2, ацетат палладия (II); EtOH, этанол; DEA, диэтиламин; AcOH, уксусная кислота; BOC2O, ди-трет-бутилдикарбонат; Et3N, триэтиламин; преп-ВЭЖХ, препаративная ВЭЖХ; TsOH, п- толуолсульфокислота; TBAB, бромид тетра-н-бутиламмония.
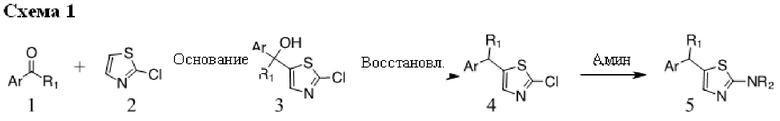
Основание, например н-BuLi или втор-BuLi, можно подвергать взаимодействию с 2-хлор-1,3-тиазолом (2) и подходящим ароматическим или гетероароматическим альдегидом или кетоном общей формулы 1 с получением соединений общей структуры 3. Соединения общей структуры 3 можно обработать подходящим восстановителем, например силаном, таким как триэтилсилан, и кислотой, такой как трифторуксусная кислота, с получением соединений общей структуры 4. Соединения общей структуры 4 можно обработать подходящим амином, например, замещенным или незамещенным 1,2,3,4-тетрагидроизохинолином, с получением соединений общей структуры 5. Будет понятно, что соединения общей структуры 5 идентичны соединениям формулы I.
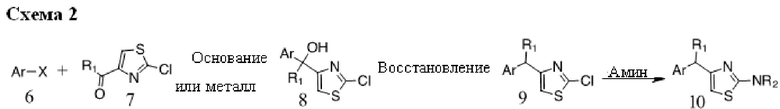
Основание, например, н-BuLi или втор-BuLi, или металл, например, Mg или Li, можно подвергать взаимодействию с подходящим галогенированным ароматическим или гетероароматическим соединением общей формулы 6, где X представляет собой Cl, Br или I, и соединениями общей структуры 7 с получением соединений общей структуры 8. Соединения общей структуры 8 можно обработать подходящим восстановителем, например силаном, таким как триэтилсилан, и кислотой, такой как трифторуксусная кислота, с получением соединений общей структуры 9. Соединения общей структуры 9 можно обработать подходящим амином, например замещенным или незамещенным 1,2,3,4-тетрагидроизохинолином, с получением соединений общей структуры 10. Будет понятно, что соединения общей структуры 10 идентичны соединениям формулы I.
Специалистам в данной области будет понятно, что могут существовать альтернативные способы синтеза для получения соединений формулы I. Следующие схемы описывают примеры таких альтернативных путей синтеза, но их не следует считать ограничивающими.
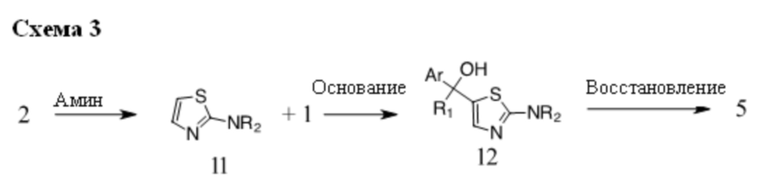
В некоторых случаях подходящий амин, например замещенный или незамещенный 1,2,3,4-тетрагидроизохинолин, можно подвергать взаимодействию с 2-хлор-1,3-тиазолом (2) с получением соединений общей структуры 11. Соединения общей структуры 11 можно подвергать взаимодействию с основанием, например, н-BuLi или втор-BuLi и соединениями общей структуры 1, с получением соединений общей структуры 12. Соединения общей структуры 12 можно обрабатывать подходящим восстановителем, например силаном, таким как триэтилсилан, и кислотой, такой как трифторуксусная кислота, с получением соединений общей структуры 5.
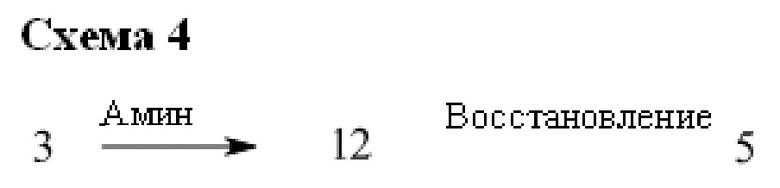
В некоторых случаях соединения общей структуры 3 можно обрабатывать подходящим амином, например замещенным или незамещенным 1,2,3,4-тетрагидроизохинолином, с получением соединений общей структуры 12. Соединения общей формулы 12 можно обрабатывать, как описано выше, с получением соединения общей формулы 5.
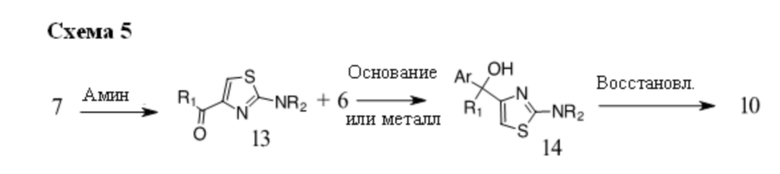
В некоторых случаях подходящий амин, например замещенный или незамещенный 1,2,3,4-тетрагидроизохинолин, можно подвергать взаимодействию с соединениями общей структуры 7 с образованием соединений общей структуры 13. Основание, например, н-BuLi или втор-BuLi или металл, например Mg или Li, можно подвергать взаимодействию с подходящими галогенированными ароматическими или гетероароматическими соединениями общей формулы 6, где X представляет собой Cl, Br или I, и соединениями общей формулы 13 с получением соединений общей формулы 14 Соединения общей формулы 12 можно обрабатывать, как описано выше, с получением соединения общей формулы 10.
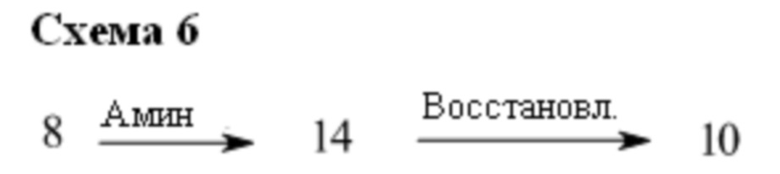
В некоторых случаях соединения общей формулы 8 можно подвергать взаимодействию с подходящим амином, например замещенным или незамещенным 1,2,3,4-тетрагидроизохинолином, с получением соединений общей формулы 14. Соединения общей формулы 14 можно обрабатывать, как описано выше, c получением соединения общей формулы 10.
Способы выполнения описанных выше реакций и процессов будут понятны для специалистов в данной области техники на основании настоящего описания или могут быть выведены по аналогии из примеров. Исходные вещества коммерчески доступны или могут быть получены способами, аналогичными описанным в приведенных ниже примерах.
Получение промежуточных соединений
Получение Промежуточного соединения 1
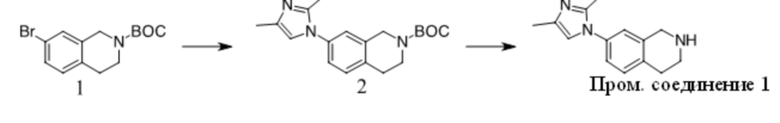
1. Смесь 1 (Key Organics, 15 г, 48,08 ммоль), 2,4-диметил-1H-имидазола (13,8 г, 144,23 ммоль), (S,S)-N,N′-диметил-1,2-диаминоциклогексана (1,37 г, 9,62 ммоль), трет-BuOK (16,15 г, 144,23 ммоль) и CuI (4,58 г, 24,04 ммоль) в NMP (150 мл) перемешивали при 160 oC в течение ночи в атмосфере N2. Смесь охлаждали до КТ, добавляли нас. водн. NaHCO3 (50 мл) и Boc2O (26,2 г, 120 ммоль) и полученную смесь перемешивали при КТ в течение ночи. Смесь концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением вещества, которое очищали с помощью преп-ВЭЖХ с получением 2 (6 г, 38% выход) в виде бледно-желтого масла. MS (ESI): вычисленная масса для C19H25N3O2 327,43, m/z найдено 327,9 [M+H]+.
2. К раствору 2 (6 г, 18,35 ммоль) в ДХМ (50 мл) добавляли TFA (50 мл). Полученную смесь перемешивали при КТ в течение ночи, концентрировали и остаток разбавляли водой, подщелачивали K2CO3 до pH = 10, экстрагировали ДХМ, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением Промежуточного соединения 1 (3,4 г, 81,5% выход) в виде бледно-желтого масла. MS (ESI): вычисленная масса для C14H17N3 227,31, m/z найдено 227,9 [M+H]+.
Получение Промежуточного соединения 2
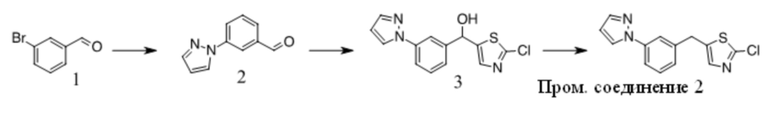
1. К раствору 1 (30 г, 163 ммоль) в сухом ДМФА (250 мл) добавляли пиразол (11,1 г, 163 ммоль), Cs2CO3 (79,2 г, 243 ммоль) и CuI (3 г, 15,8 ммоль). Полученную смесь перемешивали при 120 ºC в течение ночи. После охлаждения до КТ остаток обрабатывали водой и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 2 в виде желтого масла (12 г, 43%).
2. К раствору 2-хлортиазола (8,3 г, 70 ммоль) в сухом ТГФ (100 мл) при -78 ºC в атмосфере N2 по каплям добавляли н-BuLi (28 мл, 70 ммоль). Через 1 ч добавляли по каплям раствор 2 (12 г, 70 ммоль) в ТГФ (30 мл). Полученный раствор медленно нагревали до КТ. Реакционную смесь разбавляли раствором NH4Cl и экстрагировали EtOAc. Органические экстракты концентрировали с получением неочищенного масла, которое очищали с помощью хроматографии на силикагеле с получением 3 в виде коричневого полутвердого вещества (12,8 г, 63%).
3. К раствору 3 (12,8 г, 44 ммоль) в DCE (150 мл) добавляли TES-H (15,3 г, 132 ммоль), смесь охлаждали до 0 ºC и добавляли по каплям TFA (50 г, 0,44 моль). Полученный раствор перемешивали при 60 ºC в течение 4 ч. Остаток концентрировали и очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 2 в виде желтого масла (9,2 г, 76 %).
Альтернативное получение Промежуточного соединения 2
1. Смесь 1 (18,5 г, 100 ммоль), 1H-пиразола (6,8 г, 100 ммоль), Cs2CO3 (35,9 г, 110 ммоль), 18-краун-6 (1,9 г, 7,2 ммоль), CuI (1,9 г, 10 ммоль) в ДМФА (200 мл) перемешивали при 80 oC в течение 16 ч. Полученную смесь охлаждали, фильтровали и концентрировали, разбавляли EA, промывали водой, солевым раствором, сушили над Na2SO4, концентрировали и очищали с помощью Combi-Flash с получением 2 (9 г, 52 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H8N2O 172, m/z найдено 173 [M+H]+.
2. К раствору 2-хлортиазола (6,5 г, 55 ммоль) в ТГФ (250 мл) при -78 oC медленно добавляли н-BuLi (25 мл, 60 ммоль, 2,4 M в гексане), полученную смесь перемешивали при -78 oC в течение 1 ч. Раствор 2 (8,5 г, 50 ммоль) в ТГФ (40 мл) медленно добавляли при -78 oC, осле перемешивания в течение 1 ч реакционную смесь нагревали до КТ медленно и перемешивали в течение 20 мин. Замет ее гасили насыщенным NH4Cl. Смесь экстрагировали EA и объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и остаток очищали с помощью Combi-Flash с получением 3 (4,2 г, 29% выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C13H10ClN3OS 291, m/z найдено 292 [M+H]+.
3. К смеси 3 (4,2 г, 14,4 ммоль), TFA (16,4 г, 144 ммоль) в DCE (200 мл), добавляли TES (7,1 г, 43,3 ммоль) при 0 °C и смесь нагревали до 100 oC в течение 16 ч. Полученную смесь охлаждали до КТ, промывали водой, солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью Combi-Flash с получением продукта Промежуточного соединения 2 (2,2 г, 55 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C13H10ClN3S 275,0, m/z найдено 276 [M+H]+.
Получение Промежуточного соединения 3
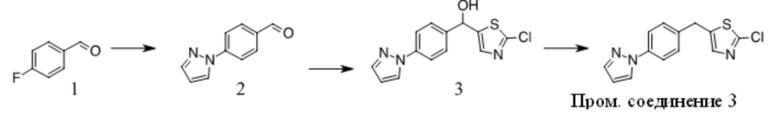
1. К раствору 1(10 г, 80,6 ммоль) в сухом ДМФА (100 мл) добавляли пиразол (5,5 г, 80,6 ммоль) и K2CO3 (12,2 г, 88,7 ммоль). Полученную смесь перемешивали при 100 ºC в течение ночи. После охлаждения до КТ смесь обрабатывали водой и экстрагировали EA. Органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали с получением неочищенного масла. Неочищенный продукт очищали путем перекристаллизации с получением 2 (4 г, 29 %). 1H ЯМР (CDCl3, 300 МГц) δ: 6,5-6,6 (s, 1 H), 7,7-7,8 (s, 1 H), 7,9-8,0 (d, 2 H), 8,0-8,1 (d, 2 H), 8,1-8,2 (s, 1 H), 10,0-10,1 (s, 1 H).
2. К раствору 2-хлортиазола (1,45 г, 12,1 ммоль) в сухом ТГФ (10 мл) при -78 ºC в атмосфере N2 по каплям добавляли н-BuLi (5 мл, 12,1 ммоль). Через 1 ч раствор 2 (1,6 г, 9,3 ммоль) добавляли по каплям при -78 ºC. Полученный раствор медленно нагревали до КТ. Реакционную смесь разбавляли раствором NH4Cl и экстрагировали EA. Органические экстракты концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 3 (1,2 г, 50 %).1H ЯМР (CDCl3, 300 МГц) δ: 6,1-6,2 (s, 1 H), 6,5-6,6 (s, 1 H), 7,2-7,3 (s, 1 H), 7,4-7,5 (d, 2 H), 7,6-7,7 (d, 2 H), 7,7-7,8 (s, 1 H), 7,9-8,0 (s, 1 H).
3. К раствору 3 (1,2 г, 4,1 ммоль) в DCE (20 мл) добавляли TES-H (1,4 г, 12,8 ммоль), смесь охлаждали до 0 ºC и добавляли по каплям TFA (4,7 г, 41 ммоль). Полученный раствор перемешивали при 60 ºC в течение 4 ч. Остаток концентрировали и очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 3 (1 г, 91 %). 1H ЯМР (CDCl3, 300 МГц) δ: 4,1-4,2 (s, 2 H), 6,4-6,5 (s, 1 H), 7,2-7,4 (m, 3 H), 7,6-7,8 (m, 3 H), 7,9-8,0 (s, 1 H).
Альтернативное получение Промежуточного соединения 3
1. К раствору 2-хлортиазола (3,85 г, 32,3 ммоль) в сухом ТГФ (80 мл) при -78 ºC в атмосфере N2 по каплям добавляли н-BuLi (14,3 мл, 35,5 ммоль). Через 1 ч раствор 2 (5,0 г, 29,1 ммоль) в ТГФ (40 мл) добавляли по каплям при -78 ºC. Полученный раствор медленно нагревали до КТ. Реакционную смесь разбавляли раствором NH4Cl и экстрагировали EtOAc. Органические экстракты концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 3 в виде светло-коричневого масла (5,9 г, 69,7 %).
2. К раствору 3 (5,9 г, 20,3 ммоль) в DCE (20 мл) добавляли TES-H (7,06 г, 60,9 ммоль), смесь охлаждали до 0 ºC и добавляли по каплям TFA (22,8 г, 0,2 моль). Полученный раствор перемешивали при 60 ºC в течение 4 ч. Остаток концентрировали и очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 3 в виде белого твердого вещества (4,84 г, 86,9 %).
Альтернативное получение Промежуточного соединения 3
1. Смесь 4-бромбензальдегида (100,0 г, 540 ммоль), 1H-пиразола (37 г, 540 ммоль), Cs2CO3 (194 г, 594 ммоль), CuI (10,3 г, 54 ммоль), 18-Краун-6 (11 г, 41 ммоль) в ДМФА (500 мл) перемешивали при 80 °C в течение 24 часов. После охлаждения до комнатной температуры, ледяную воду добавляли к смеси, которую экстрагировали EA. Органические экстракты промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением 2 (76 г, 81,7% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C10H8N2O 172,18, m/z найдено 173,0 [M+H]+.
2. К раствору 2-хлортиазола (54,2 г, 0,45 моль) в сухом (550 мл) при -78 °C по каплям добавляли н-BuLi (187,5 мл, 2,4 M, 0,45 моль). Смесь перемешивали в течение 1 ч при -78 °C и раствор 2 (65 г, 0,38 моль) в ТГФ (700 мл) добавляли по каплям при -78 °C. Полученный раствор оставляли медленно нагреваться до комнатной температуры. Реакционную смесь гасили нас. водн. NH4Cl и экстрагировали EA. Органические экстракты промывали солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением 3 (90 г, 82,6 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C13H10ClN3OS 291,76, m/z найдено 291,7 [M+H]+.
3. К раствору 3 (66 г, 0,23 моль) в TFA (330 мл) добавляли TES (148 г, 0,9 моль) при комнатной температуре. Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 1 ч. Смесь выпаривали и смесь разбавляли EA. Полученную смесь промывали нас. NaHCO3, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением Промежуточного соединения 3 (55 г, 88,7 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C13H10ClN3S 275,76, m/z найдено 275,8 [M+H]+.
Получение Промежуточного соединения 4
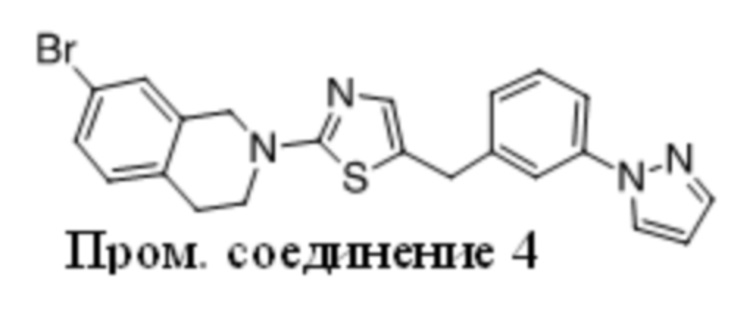
1. Смесь гидрохлорида 7-бром-1,2,3,4-тетрагидроизохинолина (Key Organics, 250 мг, 1 ммоль), Промежуточного соединения 2 (276 мг, 1 ммоль) и K2CO3 (414 мг, 3 ммоль) в ДМСО (5 мл) перемешивали при 140 °C в течение 2 часов. Затем ее охлаждали до КТ, выливали в воду, нейтрализовывали 3N HCl, экстрагировали EtOAc. Объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и полученный остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 4 (200 мг, 44% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C22H19BrN4S 451,39, m/z найдено 450,7 452,7 [M+H]+.
Получение Промежуточного соединения 5
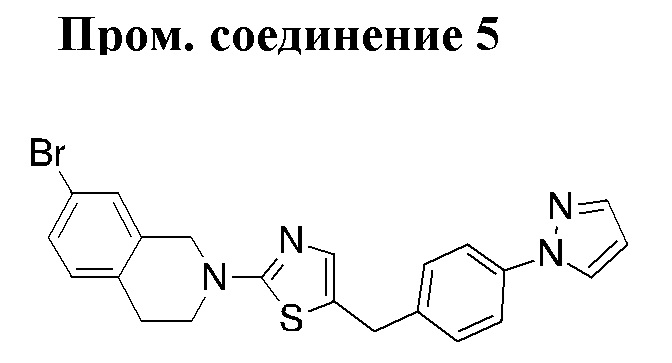
1. Смесь Промежуточного соединения 3 (1,0 г, 3,6 ммоль), Cs2CO3 (3,5 г, 10,8 ммоль) и 7-бром-1,2,3,4-тетрагидроизохинолина (Key Organics, 848 мг, 4,0 ммоль) в ДМСО (25 мл) перемешивали в атмосфере азота при 140 °C в течение 2 ч. После охлаждения до КТ, добавляли к смеси ледяную воду, которую затем экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором и сушили над Na2SO4, фильтровали, концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением Промежуточного соединения 5 (1,0 г, 61,3 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C22H19BrN4S 451,38, m/z найдено 450,7 [M+H]+.
Получение Промежуточного соединения 6
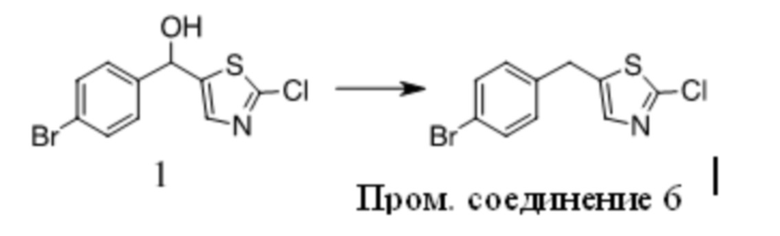
1. К раствору 2-хлортиазола (5,76 г, 48 ммоль) в сухом ТГФ (40 мл) при -78 °C в атмосфере N2 по каплям добавляли н-BuLi (2,4M, 20,0 мл, 48 ммоль). Через 1 ч добавляли по каплям раствор 4-бромбензальдегида (7,40 г, 40 ммоль) в ТГФ (40 мл). Смесь медленно нагревали до КТ и перемешивали в течение ночи. Смесь гасили нас. водн. NH4Cl и экстрагировали EtOAc. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и полученный остаток очищали с помощью хроматографии на силикагеле с получением 1 (8,00 г, 66 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H7BrClNOS 304,59, m/z найдено 305,7 [M+H]+.
2. Смесь 1 (8,00 г, 26,4 ммоль) и TES (18 мл) в TFA (50мл) перемешивали при КТ в течение 2 часов, концентрировали и остаток разбавляли нас. водн. NaHCO3. Смесь экстрагировали ДХМ и объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 6 (7,20 г, 94,7 % выход) в виде коричневого масла. MS (ESI): вычисленная масса для C10H7BrClNS 288,59, m/z найдено 289,6 [M+H]+.
Получение Промежуточного соединения 7
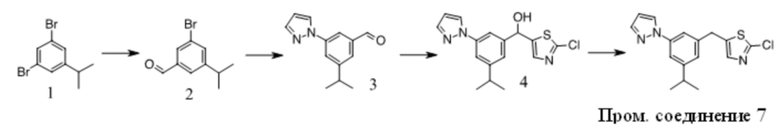
1. К раствору 1 (20 г, 71,9 ммоль) в сухом ТГФ (100 мл) при -78 °С по каплям добавляли н-BuLi (31,7 мл, 79,1 ммоль) в атмосфере N2. Через 1 ч ДМФА (1,66 г, 79,1 ммоль) добавляли по каплям при -78°С. Полученный раствор оставляли медленно нагреваться до КТ. Реакционную смесь гасили раствором NH4Cl и экстрагировали EA. Объединенные экстракты концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 2 (12 г, 73,5 %).
2. К раствору 2 (12 г, 52,9 ммоль) в сухом ДМФА (200 мл) добавляли 1H-пиразол (4,0 г, 58,1 ммоль), Cs2CO3 (19 г, 58,1 ммоль), CuI (1,2 г), 18-Краун-6 (1,2 г) и полученную смесь перемешивали при 80 ° в течение 24 ч. Смесь охлаждали до КТ, разбавляли водой и экстрагировали EA. Объединенный органический слой промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали путем перекристаллизации с получением 3 (7,3 г, 64,5 %).
3. К раствору 2-хлортиазола (4,47 г, 37,5 ммоль) в сухом ТГФ (100 мл) при -78 °С в атмосфере N2 по каплям добавляли н-BuLi (15 мл, 37,5 ммоль). Через 1 ч раствор 3 (7,3 г, 34,1 ммоль) добавляли по каплям при -78 °С. Полученную смесь оставляли медленно нагреваться до КТ. Реакционную смесь гасили раствором NH4Cl и экстрагировали EA. Органический слой концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 4 (4,6 г, 40,3 %).
4. К раствору 4 (4,6 г, 13,8 ммоль) в DCE (40 мл) при 0 °С медленно добавляли TFA (10,4 мл, 138 ммоль), TES (6,6 мл, 41,3 ммоль). Полученный раствор перемешивали при КТ в течение 3 ч. Реакционную смесь гасили H2O и экстрагировали ДХМ. Объединенные экстракты концентрировали с получением неочищенного масла и которое очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 7 (1,8 г, 41,1 %). 1H ЯМР (CDCl3, 300 МГц) δ: 1,2-1,3 (d, 6 H), 2,9-3,0 (m, 1 H), 4,1-4,2 (s, 2 H), 6,4-6,5 (s, 1 H), 7,0 (s, 1 H), 7,3-7,4 (d, 2 H), 7,4-7,5 (s, 1 H), 7,7-7,8 (s, 1 H), 7,9-8,0 (s, 1 H).
Получение Промежуточного соединения 9
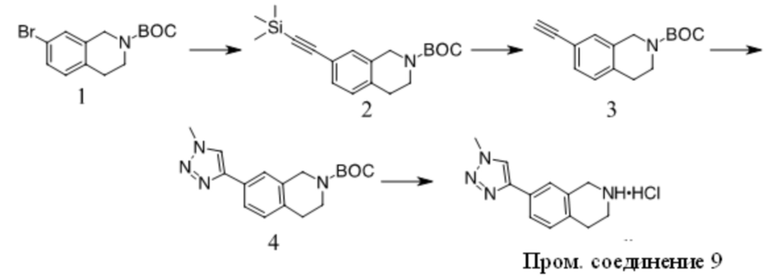
1. Смесь 1 (Key Organics, 500 мг, 1,60 ммоль), этинилтриметилсилана (236 мг, 2,40 ммоль), Pd(PPh3)2Cl2 (140 мг, 0,20 ммоль), CuI (60,8 мг, 0,32 ммоль) и Et3N (485 мг, 4,80 ммоль) в сухом ДМФА (5,00 мл) перемешивали при 60 °C в течение 1 ч. Реакционную смесь гасили водой и экстрагировали EtOAc. Объединенные органические слои промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (494 мг, 94% выход) в виде желтого масла.
2. К раствору 2 (494 мг, 1,50 ммоль) в ТГФ добавляли TBAF (1 M в ТГФ, 10 мл) и перемешивали при КТ в течение 6 ч. Смесь выливали в воду и экстрагировали EtOAc. Объединенные органические слои промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 20/1) с получением 3 (280 мг, 73%) в виде желтого твердого вещества.
3. Смесь 3 (280 мг, 1,09 ммоль), йодметана (155 мг, 1,09 ммоль), NaN3 (84,5 мг, 1,30 ммоль), CuI (207 мг, 1,09 ммоль), трет-BuOH (658 мг, 8,89 ммоль) и H2O (1,96 г, 109 ммоль) перемешивали в герметичной пробирке при 100 °C в течение ночи. Смесь охлаждали, гасили водой и экстрагировали EtOAc. Объединенные органические слои промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле с получением 4 (171 мг, 50 % выход) в виде бесцветного твердого вещества.
4. Смесь 4 (171 мг, 0,54 ммоль), HCl-диоксан (4 M, 3,00 мл, 12,0 ммоль) и ДХМ (3,00 мл) перемешивали при КТ в течение 2 ч. Смесь концентрировали с получением Промежуточного соединения 9 (100 мг, 74% выход) в виде беловатого твердого вещества.
Альтернативное получение Промежуточного соединения 9
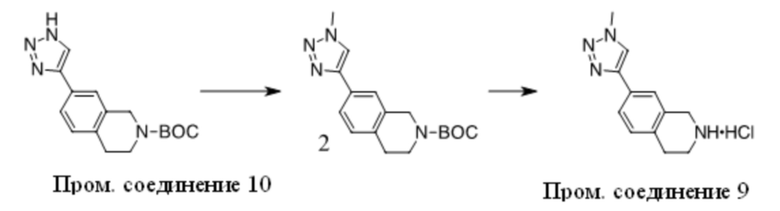
1. К раствору Промежуточного соединения 10 (2,5 г, 8,3 ммоль) в ДМФА (20 мл) при 0 ºC добавляли K2CO3 (2,3 г, 16,6 ммоль). Полученную смесь перемешивали при 0 ºC в течение 2 ч и добавляли MeI (2,6 мл) и смесь перемешивали при КТ в течение 4 ч. Смесь обрабатывали водой и экстрагировали EA. Объединенные экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали на силикагеле с получением 2 (1,4 г, 54 %).
2. К раствору 2 (1,4 г) в сухом ДХМ (20 мл) при 0 ºC добавляли HCl/Эфир (5 мл, 3 M). Полученный раствор оставляли медленно нагреваться до КТ и перемешивали в течение ночи. Реакционную смесь концентрировали с получением Промежуточного соединения 9 (1 г, 78 %)
Получение Промежуточного соединения 10
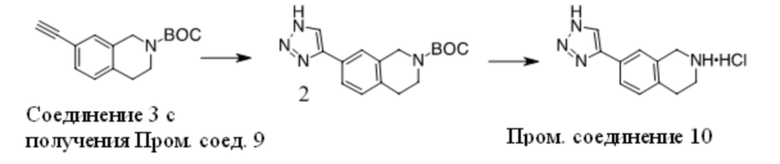
1. К раствору соединения 3 с получения Промежуточного соединения 9 (4,6 г, 17,9 ммоль) в ДМСО (40 мл) добавляли NaN3 (1,4 г, 21,5 ммоль) и NH4Cl (1,44 г, 26,8 ммоль). Полученный раствор перемешивали при 70 ° в течение ночи. Смесь обрабатывали водой и экстрагировали EA. Объединенные экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали на силикагеле с получением 2 (3,4 г, 62,2 %).
2. К раствору 2 (3,4 г) в сухом ДХМ (20 мл) при 0 ºC добавляли HCl/Эфир (10 мл, 3 M). Полученный раствор оставляли медленно нагреваться до КТ и перемешивали в течение ночи. Реакционную смесь концентрировали с получением Промежуточного соединения 10 (1,76 г, 57,1 %).
Получение Промежуточного соединения 11
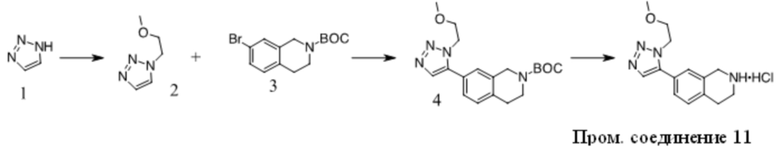
1. К суспензии 1 (1 г, 15 ммоль) и K2CO3 (3,1 г, 22,5 ммоль) в MeCN (10 мл) добавляли раствор 1-бром-2-метоксиэтана (2,5 г, 18 ммоль) в MeCN (10 мл) в течение 3 минут. Реакционную смесь перемешивали при КТ в течение 2 ч. Смесь фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии на силикагеле с получением 2 в виде желтого масла (0,66 г, 35%).
2. К раствору 3 (Key Organics, 312 мг, 1 ммоль) в ДМФА (6 мл) добавляли 2 (127 мг, 1 ммоль), Pd(OAc)2 (11 мг, 0,05 ммоль), XPhos (48 мг, 0,1 ммоль) и K2CO3 (276 мг, 2 ммоль). Смесь нагревали до 100 oC в атмосфере N2 и перемешивали в течение 20 ч. Реакционную смесь гасили водой и экстрагировали EtOAc. Объединенные экстракты концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 4 в виде желтого масла (0,108 г, 30%).
3. К раствору 4 (108 мг, 0,3 ммоль) в MeOH (10 мл) добавляли HCl/Диоксан (4 мл, 16 ммоль). Смесь перемешивали при КТ в течение 3 ч. Смесь концентрировали с получением Промежуточного соединения 11 (100 мг, 100%).
Получение Промежуточного соединения 12
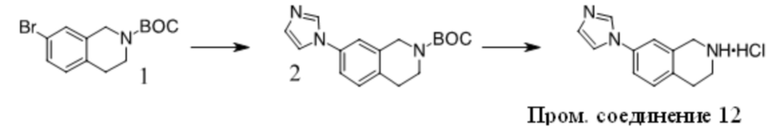
1. К раствору 1 (Key Organics, 160 мг, 0,5 ммоль) в ДМФА (3 мл) добавляли Cs2CO3 (165 мг, 0,5 ммоль), имидазол (68 мг, 1 ммоль), CuI (10 мг, 0,05 ммоль). Смесь нагревали до 100°C и перемешивали в течение 20 ч в атмосфере N2. Смесь выливали в воду и экстрагировали EtOAc. Экстракты промывали водой, солевым раствором, и сушили над Na2SO4, фильтровали и концентрировали с получением 2 (168 мг, 90%).
2. Смесь 2 (160 мг, 0,53 ммоль) и HCl/Диоксан (3 мл, 12 ммоль) перемешивали при КТ в течение 2 ч. Смесь концентрировали с получением Промежуточного соединения 12 (106 мг, 90%).
Получение Промежуточного соединения 13
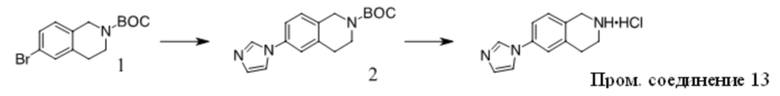
1. Промежуточное соединение 13 получали в соответствии с процедурой, описанной для Промежуточного соединения 12, за исключением того, что 2-N-BOC-6-бром-1,2,3,4-тетрагидроизохинолин 1 (Bioorg. & Med. Chem. Lett. 2018, 28, 3050) использовали вместо 2-N-BOC-7-бром-1,2,3,4-тетрагидроизохинолина.
Получение Промежуточного соединения 14
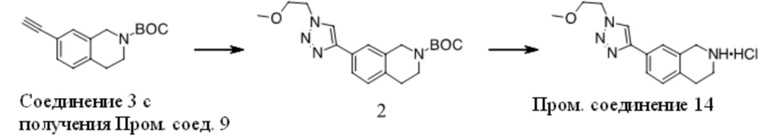
1. Смесь соединения 3 с получения Промежуточного соединения 9 (650 мг, 2,53 ммоль), 1-йод-2-метоксиэтана (471 мг, 2,53 ммоль), NaN3 (164 мг, 2,53 ммоль), CuI (482 мг, 2,53 ммоль), трет-BuOH (748 мг, 10,1 ммоль) и H2O (5,00 г, 278 ммоль) перемешивали при 100 °C в герметичной пробирке в течение ночи. Смесь охлаждали до КТ и экстрагировали EtOAc. Объединенные органические слои промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (350 мг, 39%) в виде беловатого твердого вещества.
2. Смесь 2 (350 мг, 0,98 ммоль), HCl-диоксан (4 M, 5 мл, 20,0 ммоль) и CH2Cl2 (10 мл) перемешивали при КТ в течение 2 ч. Смесь концентрировали с получением Промежуточного соединения 14 (260 мг, 90%) в виде белого твердого вещества.
Получение Промежуточного соединения 15
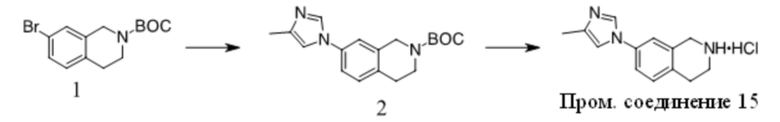
1. Смесь 1 (Key Organics, 1,00 г, 3,20 ммоль), (S,S)-N,N′-диметил-1,2-диаминоциклогексан (91,0 мг, 0,64 ммоль), 4-метил-1H-имидазол (525 мг, 6,39 ммоль), CuI (304 мг, 1,60 ммоль) и трет-BuOK (1,07 г, 9,60 ммоль) в ДМФА (5 мл) перемешивали при 120 °C в течение 4 ч. После охлаждения до КТ, смесь непосредственно очищали с помощью преп-ВЭЖХ с получением 2 (320 мг, 31% выход) в виде беловатого твердого вещества.
2. Смесь 2 (220 мг, 0,70 ммоль), HCl-диоксан (4 M, 3,00 мл, 12,0 ммоль) и CH2Cl2 (3 мл) перемешивали при КТ в течение 2 ч. Смесь концентрировали с получением Промежуточного соединения 15 (150 мг, 86% выход) в виде беловатого твердого вещества.
Получение Промежуточного соединения 16
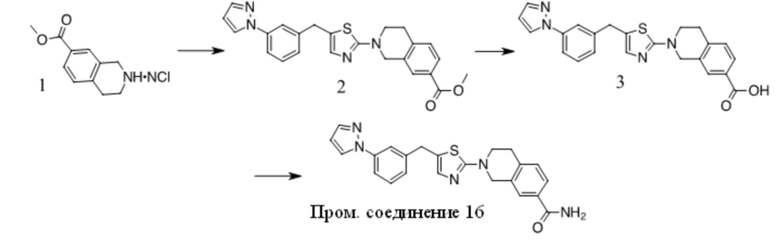
1. Смесь 1 (J. Med. Chem. 42, 1, 118-134, 750 мг), Промежуточного соединения 2 (908 мг, 3,29 ммоль), Pd2(dba)3 (290 мг, 0,33 ммоль), SPhos (132 мг, 0,33 ммоль), трет-BuOK (750 мг, 6,70 ммоль) и диоксана (10 мл) перемешивали при 100 °C в течение 4 ч. Смесь гасили водой и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и солевым раствором, сушили над Na2SO4, и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением 2 (550 мг, 37% выход за 2 стадии) в виде желтого твердого вещества.
2. Смесь 2 (550 мг, 1,28 ммоль), LiOH (61,0 мг, 2,56 ммоль), MeOH (3 мл) и H2O (3 мл) перемешивали при КТ в течение 3 ч. Смесь подкисляли 2 N HCl до pH 4 - 5, что привело к выпадению осадка. Осадок фильтровали, промывали водой и сушили с получением 3 (170 мг, 32% выход) в виде беловатого твердого вещества.
3. Смесь 3 (170 мг, 0,41 ммоль), HATU (250 мг, 0,62 ммоль), DIEA (280 мг, 2,17 ммоль), NH4Cl (134 мг, 2,5 ммоль) и ДМФА (2 мл) перемешивали при КТ в течение 4 ч. Смесь гасили водой и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и солевым раствором, сушили над Na2SO4, и концентрировали. Остаток очищали с помощью преп-ТСХ с получением Промежуточного соединения 16 (130 мг, 76% выход) в виде желтого твердого вещества.
Получение Промежуточного соединения 17
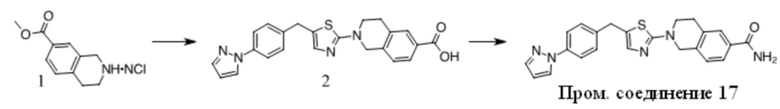
1. К раствору 1 (J. Med. Chem. 42, 1, 118-134, 227 мг, 1 ммоль) в диоксане (20 мл) добавляли Промежуточное соединение 3 (300 мг, 1,1 ммоль), Pd2(dba)3 (72 мг, 0,08 ммоль), SPhos (65 мг, 0,16 ммоль) и трет-KOBu (340 мг, 3 ммоль). Смесь нагревали до 100 oC и перемешивали в течение 20 ч в атмосфере N2. смесь охлаждали до КТ и добавляли LiOH (0,4 г, 10 ммоль) и воду (4 мл). Полученную смесь перемешивали при 60 oC в течение 2 ч, охлаждали, подкисляли 1 N HCl до pH ≈ 5. Полученную смесь экстрагировали EtOAc и органические экстракты концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 2 в виде бледно-желтого твердого вещества (0,31 г, 74 %).
2. К раствору 2 (50 мг, 0,12 ммоль) в ДМФА (2 мл) добавляли DIEA (25 мг, 0,18 ммоль), HATU (0,69 г, 0,18 ммоль) и NH4Cl (7,1 мг, 0,13 ммоль). Смесь перемешивали при КТ в течение 1 ч. Смесь обрабатывали водой и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 17 в виде белого твердого вещества (11,2 мг, 22,4 %).
Получение Промежуточного соединения 18
1. К раствору 1 (20 г, 108,1 ммоль) в сухом ДМФА (200 мл) добавляли 1H-имидазол (8,1 г, 118,9 ммоль), Cs2CO3 (38,7 г, 118,9 ммоль), CuI (2 г), 18-Краун-6 (2 г). Полученный раствор перемешивали при 80 ° в течение 24 ч. Смесь охлаждали до КТ, обрабатывали водой и экстрагировали EA. Объединенные экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали путем перекристаллизации с получением 2 (11 г, 59,2 %).
2. К раствору 2-хлортиазола (3,8 г, 32 ммоль) в сухом ТГФ (50 мл) добавляли н-BuLi (14 мл, 34,9 ммоль) по каплям при -78 ° в атмосфере N2. Через 1 ч раствор 2 (5 г, 29,1 ммоль) добавляли по каплям при -78 °. Полученный раствор оставляли медленно нагреваться до КТ. Реакционную смесь гасили раствором NH4Cl и экстрагировали EA. Объединенные экстракты концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 3 (3,5 г, 41,4 %).
3. К раствору 3 (3,5 г, 12,0 ммоль) в DCE (60 мл) при 0 ° медленно добавляли TFA (9 мл, 120 ммоль) и TES (5,8 мл, 36 ммоль). Полученный раствор перемешивали при КТ в течение 3 ч. Реакционную смесь гасили H2O и экстрагировали ДХМ. Органический слой концентрировали с получением неочищенного масла и очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 18 (1,8 г, 54,4 %).
Получение Промежуточного соединения 19
1. К раствору 1 (6,20 г, 50 ммоль) и 2,4-диметил-1H-имидазола (7,20 г, 75 ммоль) в ДМФА (100 мл) добавляли Cs2CO3 (48,70 г, 150 ммоль). Полученную смесь перемешивали при 120 oC в течение 2 часов. Ее охлаждали до КТ, фильтровали и фильтрат выливали в воду, экстрагировали CH2Cl2, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали. Остаток очищали с помощью хроматографии на силикагеле с получением 2 (1,80 г, 18% выход) в виде коричневого масла. MS (ESI): вычисленная масса для C12H12N2O 200,24, m/z найдено 200,9 [M+H]+.
2. К раствору 2-хлортиазола (1,20 г, 10 ммоль) в ТГФ (20 мл) при -78 oC по каплям добавляли н-BuLi (2,4 M, 5,5 мл) в атмосфере N2. Через 1 ч раствор 2 (1,80 г, 9 ммоль) в ТГФ (20 мл) добавляли по каплям. Реакционную смесь медленно нагревали до КТ и перемешивали в течение ночи. Смесь гасили нас. водн. NH4Cl и экстрагировали EtOAc, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали. Остаток очищали с помощью хроматографии на силикагеле с получением 3 (1,83 г, 64 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C15H14ClN3OS 319,81, m/z найдено 319,8 [M+H]+.
3. Смесь 3 (1,83 г, 5,7 ммоль), TES (9 мл) в TFA (18 мл) перемешивали при 60 oC в течение 3 ч. Затем ее концентрировали и остаток разбавляли нас. водн. NaHCO3 и экстрагировали CH2Cl2, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 19 (1,20 г, 69,0 % выход) в виде коричневого масла. MS (ESI): вычисленная масса для C15H14ClN3S 303,81, m/z найдено 303,8 [M+H]+.
Получение Промежуточного соединения 20
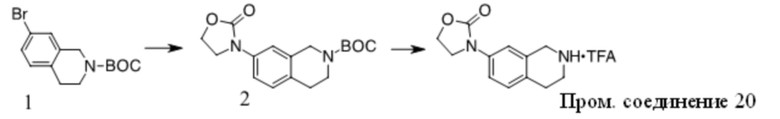
1. Смесь 1 (Key Organics, 5,68 г, 0,018 моль), оксазолидин-2-она (4,7 г, 0,054 моль), Pd2(dba)3 (8,23 г, 0,009 моль), S-phos (3,69 г, 0,009 моль) и трет-BuOK (6,0 г, 0,054 моль) в высушенном 1,4-диоксане (150 мл) перемешивали при 100 °C в течение ночи. Реакционную смесь охлаждали до КТ, фильтровали и концентрировали с получением неочищенного продукта. Остаток очищали с помощью хроматографии на силикагеле с получением 2 (5,02 г, 88 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C17H22N2O4 318,37, m/z найдено 340,8 [M+H]+.
2. К раствору 2 (5,02 г, 16 ммоль) в ДХМ (100 мл) добавляли TFA (25 мл). Реакционную смесь перемешивали при КТ в течение 6 ч. Затем реакционную смесь концентрировали с получением неочищенного продукта. Ее растворяли в ДХМ и разбавляли петролейным эфиром. Полученную суспензию фильтровали и фильтрационный осадок промывали петролейным эфиром и сушили с получением Промежуточного соединения 20 (3,50 г, 70%), MS (ESI): вычисленная масса для C14H15F3N2O3 316,28, m/z найдено 218,9 [M+H]+.
Получение Промежуточного соединения 21
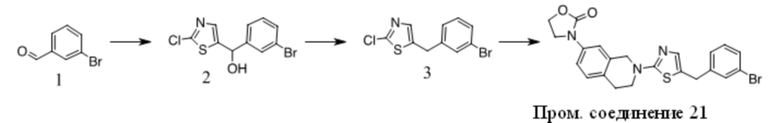
1. К раствору 2-хлортиазола (5,76 г, 48 ммоль) в сухом ТГФ (200 мл) при -78 oC по каплям добавляли н-BuLi (2,4M, 25,0 мл, 60 ммоль) в атмосфере N2. Через 0,5 ч раствор 1 (8,00 г, 43 ммоль) в ТГФ (50 мл) добавляли по каплям. Реакционную смесь медленно нагревали до КТ. Смесь гасили нас. водн. NH4Cl и экстрагировали EtOAc и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (8,50 г, 64 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H7BrClNOS 304,59, m/z найдено 305,7 [M+H]+.
2. Смесь 2 (8,50 г, 27,9 ммоль) в TES (20 мл) и TFA (60мл) перемешивали при 60 ºC в течение 2 ч. Смесь концентрировали и остаток разбавляли нас. водн. NaHCO3 и экстрагировали ДХМ и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 3 (7,00 г, 86,9 % выход) в виде коричневого масла. MS (ESI): вычисленная масса для C10H7BrClNS 288,59, m/z найдено 289,6 [M+H]+.
3. К раствору 3 (3,00 г, 10,4 ммоль) в ДМСО (30 мл) добавляли Промежуточное соединение 20 (3,45 г, 10,4 ммоль) и K2CO3 (4,31 г, 31,2 ммоль). Реакционную смесь перемешивали при 140 oC в течение 3 ч. Реакционную смесь охлаждали до КТ, выливали в ледяную воду, экстрагировали CH2Cl2, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 21 (2,10 г, 42,9 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C22H20BrN3O2S 470,39, m/z найдено 470,5 [M+H]+.
Получение Промежуточного соединения 22
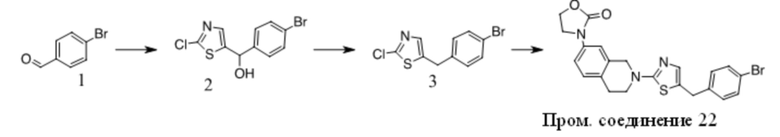
1. К раствору 2-хлортиазола (5,76 г, 48 ммоль) в сухом ТГФ (40 мл) по каплям добавляли н-BuLi (2,4M, 20,0 мл, 48 ммоль) при -78 oC в атмосфере N2. Через 1 ч добавляли по каплям раствор 1 (7,40 г, 40 ммоль) в ТГФ (40 мл). Реакционную смесь медленно нагревали до КТ и перемешивали в течение ночи. Замет ее гасили нас. водн. NH4Cl и экстрагировали EtOAc и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением 2 (8,00 г, 66 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H7BrClNOS 304,59, m/z найдено 305,7 [M+H]+.
2. Смесь (4-бромфенил)(2-хлортиазол-5-ил)метанола (8,00 г, 26,4 ммоль) и TES (18 мл) в TFA (50мл) перемешивали при КТ в течение 2 часов. Затем ее концентрировали и остаток разбавляли нас. водн. NaHCO3, экстрагировали ДХМ и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением 3 (7,20 г, 94,7 % выход) в виде коричневого масла. MS (ESI): вычисленная масса для C10H7BrClNS 288,59, m/z найдено 289,6 [M+H]+.
3. К раствору 3 (3,10 г, 11 ммоль) в ДМСО (30 мл) добавляли Промежуточное соединение 20 (3,5 г, 11 ммоль) и K2CO3 (4,56 г, 33 ммоль). Реакционную смесь перемешивали при 140 oC в течение 3 ч. Реакционную смесь охлаждали до КТ и затем выливали в ледяную воду, экстрагировали CH2Cl2, сушили над Na2SO4.Объединенные органические слои концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 22 (4,78 г, 92 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C22H20BrN3O2S 470,39, m/z найдено 470,7 [M+H]+.
Получение Промежуточного соединения 23
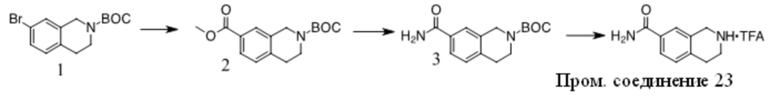
1. Смесь 1 (Key Organics, 15 г, 48 ммоль), TEA (9,7 г, 96 ммоль), и Pd(dppf)Cl2 (2,8 г, 3,84 ммоль) в MeOH (200мл) и MeCN (50мл) в бомбе продували 3x CO, затем нагревали при 100 oC в течение 24 ч под давлением 120 атм СО. Смесь охлаждали, фильтровали через целит, фильтрационный осадок промывали EA и фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (11,5г, 82,1 % выход) в виде масле. MS (ESI): вычисленная масса для C16H21NO4 291,15, m/z найдено 313,9 [M+Na]+. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,84 (d, J = 8,2 Гц, 1H), 7,82 (s, 1H), 7,22 (d, J = 7,9 Гц, 1H), 4,63 (s, 2H), 3,93 (s, 3H), 3,68 (t, J = 5,4 Гц, 2H), 2,90 (t, J = 5,5 Гц, 2H), 1,51 (s, 9H).
2. К раствору 2 (10,8 г, 37 ммоль) в MeOH (20мл) добавляли MeOH, насыщенный NH3 (250мл). Реакционную смесь перемешивали при 120 oC в течение 60 ч в бомбе, охлаждали до КТ и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 3 (8,4 г, 81,9 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C15H20N2O3 276,34, m/z найдено 298,9 [M+Na]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,89 (s, 1H), 7,69 - 7,67 (m, 2H), 7,30 (s, 1H), 7,23 (d, J = 8,0 Гц, 1H), 4,54 (s, 2H), 3,56 (t, J = 5,8 Гц, 2H), 2,81 (t, J = 5,8 Гц, 2H), 1,44 (s, 9H).
3. К раствору 3 (8,4 г, 30,3 ммоль) в ДХМ (20мл) добавляли TFA (12мл). Реакционную смесь перемешивали при КТ в течение 6 ч, концентрировали с получением неочищенного продукта. Ее растворяли в ДХМ и разбавляли PE, что привело к выпадению осадка, который собирали фильтрацией. Фильтрационный осадок промывали EA и сушили в вакууме с получением Промежуточного соединения 23 (6,3 г, 71,4%), MS (ESI): вычисленная масса для C12H13F3N2O3 290,24, m/z найдено 177,0 [M+H]+.
Получение Промежуточного соединения 24
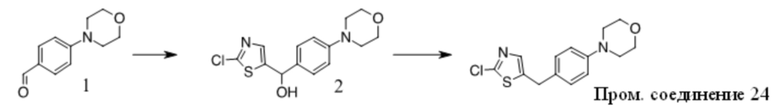
1. К раствору 2-хлортиазола (1,24 г, 10,35 ммоль) в сухом ТГФ (50 мл) при -78 oC в атмосфере N2 добавляли по каплям н-BuLi (2,4 M, 4,8 мл). Через 0,5 ч добавляли по каплям раствор 1 (Sigma-Aldrich, 1,80 г, 9,41 ммоль) в высушенном ТГФ (10 мл). Реакционную смесь медленно нагревали до КТ. Смесь гасили водн. NH4Cl и экстрагировали ДХМ и сушили над Na2SO4. Объединенные органические слои концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (2,00 г, 68,36 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C14H15ClN2O2S 310,80, m/z найдено 311,4 [M+H]+.
2. Смесь 2 (2,0 г, 6,44 ммоль), TES (10 мл) и TFA (30 мл) перемешивали при 80 oC в течение 2 ч. Смесь концентрировали и остаток промывали водн. NaHCO3 и экстрагировали ДХМ (30 мл x 3) и сушили над Na2SO4. Объединенные экстракты концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 24 (1,1 г, 57,94 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C14H15ClN2OS 294,8, m/z найдено 295,4 [M+H]+.
Получение Промежуточного соединения 25
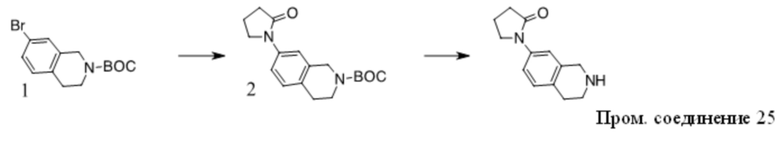
1. Смесь 1 (Key Organics, 5,0 г, 16,01 ммоль), пирролидин-2-он (4,09 г, 48,03ммоль), Pd2(dba)3 (1,17 г, 1,6 ммоль), S-Phos (1,31 г, 3,2 ммоль) и трет-BuOK (5,38 г, 48,03 ммоль) в высушенном 1,4-диоксане (200 мл) перемешивали при 100 oC в течение ночи. Смесь выливали в воду и экстрагировали ДХМ, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 2 (3,2 г, 63,17 %) в виде белого твердого вещества. Вычисленная масса для C18H24N2O3 316,40, m/z найдено 316,8 [M+H]+.
2. Смесь 2 (3,2 г, 10,11 ммоль), ДХМ (40 мл) и TFA (20 мл) перемешивали при комнатной температуре в течение 5 часов. Смесь концентрировали и остаток промывали водн. NaHCO3 и экстрагировали ДХМ, сушили над Na2SO4. Объединенные экстракты концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 25 (1,05 г, 48,02 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C13H16N2O 216,28, m/z найдено 216,8 [M+H]+.
Получение Промежуточного соединения 26
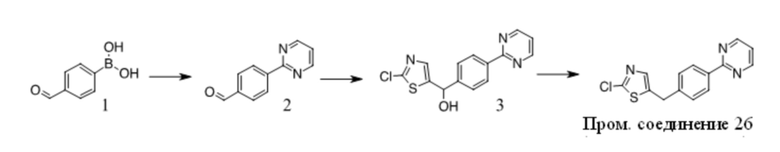
1. Смесь 1 (Sigma-Aldrich, 4 г, 26,7 ммоль), 2-бромпиримидина (3,51 г, 22,1 ммоль), NaHCO3 (6,73 г, 80,1 ммоль), Pd(PPh3)4 (766 мг, 0,663 ммоль) в DME/H2O (100 мл/50 мл) перемешивали при 90 oC в течение 18 часов в атмосфере N2. Смесь фильтровали через целит и фильтрационный осадок промывали EA и фильтрат концентрировали. Полученную смесь экстрагировали EA и объединенные экстракты промывали солевым раствором, водой и сушили Na2SO4. Смесь фильтровали и фильтрат концентрировали с получением остатка, который очищали с помощью хроматографии на силикагеле с получением 2 (3,84 г, 78,1 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C11H8N2O 184,20, m/z найдено 185,0 [M+H]+.
2. К раствору 2-хлортиазола (2,69 г, 22,7 ммоль) в сухом ТГФ (80 мл) при -78 oC в атмосфере N2 добавляли по каплям н-BuLi (2,4 M, 9,9 мл, 23,75 ммоль). Через 1 ч раствор 2 (3,8 г, 20,6 ммоль, 106 мл ТГФ) добавляли по каплям к смеси. Реакционную смесь медленно нагревали до КТ и перемешивали в течение 18 ч. Полученную смесь гасили водн. NH4Cl и экстрагировали EtOAc и объединенные экстракты сушили над Na2SO4. Органический раствор концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 3 (4,65 г, 74,2 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C14H10ClN3OS 303,76, m/z найдено 303,8 [M+H]+.
3. К раствору 3 (3 г, 9,87 ммоль) и TFA (11,3 г, 98,7 ммоль) в сухом DCE при 0 oC добавляли по каплям TES (3,42 г, 29,6 ммоль) и реакционную смесь перемешивали в течение 8 ч при 60 oC. Смесь охлаждали и концентрировали. Полученный остаток обрабатывали насыщенным NaHCO3, экстрагировали EA и объединенные экстракты промывали солевым раствором, воду и сушили над Na2SO4. Растворитель удаляли с получением неочищенного продукта, который очищали с помощью флэш-хроматографии с получением Промежуточного соединения 26 (1,2 г, 42,2 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C14H10ClN3S 287,77, m/z найдено 287,8 [M+H]+.
Получение Промежуточного соединения 27
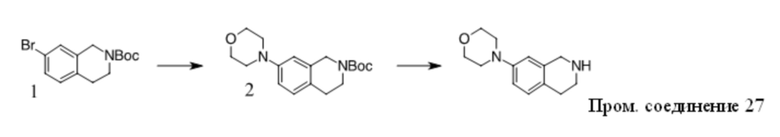
1. Смесь 1 (2,00 г, 6,4 ммоль), морфолина (1,80 г, 20,6 ммоль), Pd2(dba)3 (2,0 г, 2,2 ммоль), S-Phos (1,20 г, 3,0 ммоль) и K2CO3 (2,80 г, 20 ммоль) в высушенном 1,4-диоксане (80 мл) перемешивали при 100 oC в течение ночи. Реакционную смесь охлаждали до КТ, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 2 (1,30 г, 64%) в виде желтого масла, вычисленная масса для C18H26N2O3 318,19, m/z найдено 319,0[M+H]+.
2. Смесь 2 (1,30 г, 5 ммоль) и TES (5 мл) в TFA (10 мл) перемешивали при 50 oC в течение 2 часов. Смесь охлаждали, концентрировали и остаток обрабатывали нас. водн. NaHCO3, экстрагировали CH2Cl2 и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением Промежуточного соединения 27 (1,4 г, неочищенный продукт) в виде коричневого масла. MS (ESI): вычисленная масса для C13H18N2O 218,14, m/z найдено 219,0[M+H]+.
Получение Промежуточного соединения 28
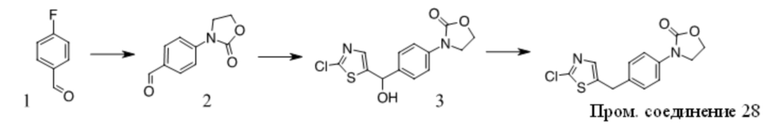
1. К раствору 1 (4,46 г, 36 ммоль) в ДМФА (100 мл) добавляли оксазолидин-2-он (2,61 г, 30 ммоль) и Cs2CO3 (11,74 г, 36 ммоль). Полученную смесь перемешивали при 120 oC в течение ночи. Смесь охлаждали до КТ, фильтровали, фильтрат выливали в воду и смесь экстрагировали EA. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, концентрировали и очищали с помощью хроматографии на силикагеле с получением 2 (4,17 г, 70 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C10H9NO3 191,19, m/z найдено 192,0 [M+H]+.
2. К раствору 2-хлортиазола (0,86 г, 7,2 ммоль) в ТГФ (25 мл) при -78 oC по каплям добавляли н-BuLi (3 мл, 7,2 ммоль). Через 1 ч добавляли по каплям раствор 2 (1,06 г, 5,5 ммоль) в ТГФ (15 мл). Реакционную смесь перемешивали 2 ч, гасили нас. водн. NH4Cl и экстрагировали EtOAc. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением 3 (неочищенный, 1,8 г) в виде желтого масла. MS (ESI): вычисленная масса для C14H12ClN3O2S 310,75, m/z найдено 311,21[M+H]+.
1. Смесь 3 (0,1 г, 0,32 ммоль) и TES (0,5 мл) в TFA (1 мл) перемешивали при КТ в течение 2 часов. Смесь концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 28 (78 мг, 82% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C13H11ClN2O2S 294,75, m/z найдено 295,19[M+H]+.
Получение Промежуточного соединения 29
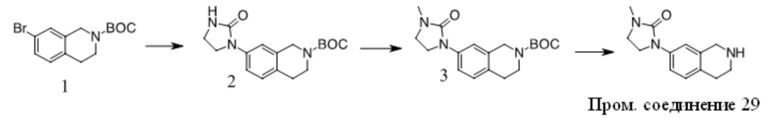
1. Смесь соединения 1 (Key Organics, 5,40 г, 17,4 ммоль), имидазолидин-2-она (4,50 г, 52,3 ммоль), трет-BuOK (5,75 г, 51,3 ммоль), CuI (2,52 г, 13,2 ммоль) и (S,S)-N,N′-диметил-1,2-диаминоциклогексана (0,83 г, 6,5 ммоль) в ДМФА (200 мл) перемешивали при 120 oC в течение ночи. Реакционную смесь охлаждали до КТ, фильтровали и концентрировали с получением 2 (5,1 г, 92%) в виде желтого масла. Вычисленная масса для C17H23N3O3 317,39, m/z найдено 317,9[M+H]+.
2. К раствору 2 (1,50 г, 4,7 ммоль) в ТГФ при 0 oC (40 мл) добавляли NaH (200 мг, 13,8 ммоль). Через 0,5 ч раствор йодметана (1,41 г, 9,9 ммоль) в ТГФ (10 мл) добавляли по каплям. Реакционную смесь медленно нагревали до КТ и перемешивали при 60 oC в течение 4 ч. Реакционную смесь гасили водой и экстрагировали EA, объединенные экстракты промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением 3 (1 г, 64%). Вычисленная масса для C18H25N3O3 331,42. m/z найдено 332,0 [M+H]+.
3. Смесь 3 (2 г, 6 ммоль) и TFA (9 мл) в ДХМ (40 мл) перемешивали при КТ в течение 2 часов. Затем ее концентрировали и остаток разбавляли нас. водн. NaHCO3, экстрагировали CH2Cl2 и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением Промежуточного соединения 29 (1,1 г) в виде коричневого масла. MS (ESI): вычисленная масса для C13H17N3O 231,3, m/z найдено 231,9 [M+H]+.
Получение Промежуточного соединения 30
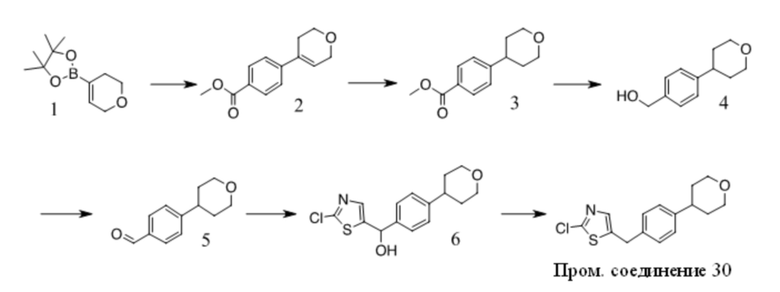
1. Сосуд, содержащий смесь метил-4-бромбензоата (1,1 г, 5,12 ммоль), 1 (Sigma-Aldrich, 1,1 г, 5,24 ммоль), Pd(dppf)Cl2 (299 мг, 0,41 ммоль) и K2CO3 (1,412 г, 10 ммоль) в 1,4-диоксане (15 мл) и H2O (1 мл), три раза продували N2 и полученную смесь нагревали до 100 oC в течение 16 ч. Ее охлаждали до КТ, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 2 (940 мг, 93% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C13H14O3 218,25, m/z найдено 219,0 [M+H]+.
2. Смесь 2 (940 мг, 4,31 ммоль) и Pd/C (250 мг) в EtOAc (40 мл) перемешивали при КТ в течение 16 ч в атмосфере H2. Смесь фильтровали и концентрировали с получением 3 (948 мг, 100% выход) в виде бесцветного масла. MS (ESI): вычисленная масса для C13H16O3 220,27, m/z найдено 221,0 [M+H]+.
3. LiAlH4 (160 мг, 4,09 ммоль) добавляли к раствору 3 (900 мг, 4,09 ммоль) в сухом ТГФ (15 мл) при 0 oC. Смесь нагревали до КТ и перемешивали в течение 2 ч, охлаждали до 0 ºC и гасили нас. водн. NH4Cl и экстрагировали EtOAc. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением 4 (765 мг, 97% выход) в виде желтого масла. MS (ESI): вычисленная масса для C12H16O2 192,26, m/z найдено 175,0 [M-OH]+.
4. К раствору 4 в ДХМ (8 мл) добавляли реактив Десса-Мартина (70 мг, 0,12 ммоль). Полученную смесь перемешивали при КТ в течение 1 ч, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 5 (15,5 мг, 78% выход) в виде желтого масла. MS (ESI): вычисленная масса для C12H14O2 190,24, m/z найдено 191,0 [M+H]+.
5. н-BuLi (1,45 мл, 3,47 ммоль, 2,4 M) добавляли по каплям к раствору 2-хлортиазола (416 мг, 3,47 ммоль) в ТГФ (3 мл) при - 78 oC. Через 30 мин добавляли по каплям раствор 5 (600 мг, 3,16 ммоль) в ТГФ (6 мл) и полученную смесь нагревали до КТ и перемешивали в течение ночи. Смесь гасили нас. водн. NH4Cl, экстрагировали EtOAc. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением 6 (860 мг, 88% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C15H16ClNO2S 309,81, m/z найдено 309,8 [M+H]+.
6. Смесь 6 (690 мг, 2,23 ммоль), TES (2 мл) и TFA (2 мл) перемешивали при КТ в течение 2 ч в атмосфере N2. Смесь концентрировали и остаток разбавляли нас. водн. NaHCO3, экстрагировали ДХМ. Объединенные экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 30 (566,6 мг, 87% выход) в виде желтого масла. MS (ESI): вычисленная масса для C15H16ClNOS 293,81, m/z найдено 294,1 [M+H]+.
Получение Промежуточного соединения 31
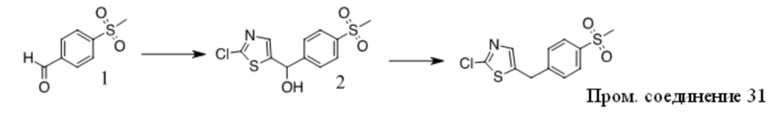
1. К раствору 2-хлортиазола (3,59 г, 29,9 ммоль) в сухом ТГФ (94 мл) при -78 oC в атмосфере N2 добавляли по каплям н-BuLi (2,4 M в гексане, 13,0 мл, 31,2 ммоль). Через 1 ч добавляли по каплям раствор 1 (Enamine, 5 г, 27,2 ммоль) в высушенном ТГФ (200 мл). Реакционную смесь нагревали до КТ и перемешивали в течение 18 ч. Смесь гасили насыщенным водн. NH4Cl и экстрагировали EtOAc. Объединенные экстракты сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (1,6 г, 19,4% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C11H10ClNO3S2 303,78, m/z найдено 303,7 [M+H]+.
2. К раствору 2 (1,6 г, 5,26 ммоль) в TFA (12 мл) добавляли по каплям TES (3 г, 26,3 ммоль) в течение 15 мин. Реакционную смесь перемешивали при 70 oC в течение 2 часов, гасили насыщенным NaHCO3 и экстрагировали EtOAc. Объединенные экстракты промывали водой, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 31 (1,3 г, 85,8 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C11H10ClNO2S2 287,78, m/z найдено 287,7 [M+H]+.
Получение Промежуточного соединения 34
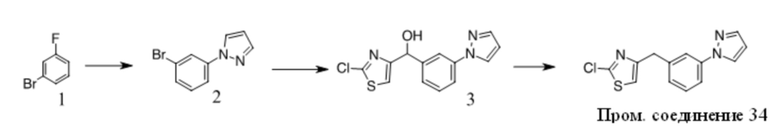
1. К раствору 1H-пиразола (5,86 г, 86,2 ммоль) в безводном ДМФА (50,0 мл) добавляли NaH (60%, 10,3 г, 258,6 ммоль) в атмосфере N2 и реакционную смесь перемешивали при 60 °C в течение 1 ч. Затем добавляли 1 (15,1 г, 86,2 ммоль) и полученную смесь перемешивали при 120 °C в течение 3 ч. Реакционную смесь гасили водой и экстрагировали EtOAc. Объединенные органические слои промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (4,0 г, 21% выход) в виде бесцветного масла.
2. К раствору 2 (2,66 г, 18,0 ммоль) в сухом ТГФ (50 мл) по каплям добавляли н-BuLi (2,5 M в гексане, 7,2 мл, 18,0 ммоль) при -78 ºC в атмосфере N2. Через 1 ч добавляли по каплям раствор 2-хлор-4-тиазолкарбоксальдегида (Sigma-Aldrich, 4,00 г, 18,02 ммоль) в ТГФ (10 мл). Полученную смесь нагревали до комнатной температуры. Реакционную смесь гасили насыщенным NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты концентрировали с получением неочищенного масла, которое очищали с помощью хроматографии на силикагеле с получением 3 (420 мг, 8% выход) в виде желтого масла.
3. К раствору 3 (400 мг, 1,44 ммоль) в TFA (10 мл) добавляли TES (3 мл) и полученную смесь перемешивали при 100 ºC в течение 2 ч. Реакционную смесь концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением Промежуточного соединения 34 (250 мг, 63% выход) в виде желтого твердого вещества.
Получение Промежуточного соединения 35
1. К смеси 1 (4 г, 22,5 ммоль) в нас. водн. NaHCO3 (50 мл) и ТГФ (50 мл) добавляли BOC2O (5,63 г, 25,8 ммоль) и полученную смесь перемешивали при КТ в течение 16 ч. Смесь концентрировали и остаток экстрагировали EtOAc, и объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением 2 (5,8 г, 93,5% выход) в виде коричневого твердого вещества. MS (ESI): вычисленная масса для C14H18N2O4 278,31, m/z найдено 301,0 [M+Na]+.
2. Смесь 2 (5,8 г, 20,9 ммоль) и Pd/C (1,2 г) в EtOAc (60 мл) перемешивали при КТ в течение 16 ч в атмосфере H2. Смесь фильтровали и концентрировали с получением 3 (5 г, 96,7% выход) в виде коричневого масла. MS (ESI): вычисленная масса для C14H20N2O2 248,33, m/z найдено 271,0 [M+Na]+.
3. Смесь 3 (2 г, 8,06 ммоль) и 3-хлорпропилизоцианат (1,16 г, 9,68 ммоль) в сухом ДХМ (20 мл) перемешивали при КТ в течение 16 ч. Затем ее концентрировали и остаток промывали смесью EA/PE (1/50), фильтровали и полученное твердое вещество сушили в вакууме с получением 4 (2,8 г, 95% выход) в виде коричневого твердого вещества. MS (ESI): вычисленная масса для C18H26ClN3O3 367,87, m/z найдено 368,0 [M+H]+.
4. К раствору 4 (2,7 г, 7,4 ммоль) в ТГФ (270 мл) при 0 oC порциями добавляли NaH (888 мг, 22,2 ммоль). Затем его нагревали до КТ и перемешивали в течение 16 ч. Объем уменьшали примерно до 40 мл и его охлаждали до 0 ºC и добавляли MeI (1,58 г, 11,1 ммоль) и полученную смесь нагревали до КТ и перемешивали в течение 16 ч. Смесь гасили водой, экстрагировали EtOAc, объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя с получением 5 (2,4 г, 96% выход) в виде желтого масла. MS (ESI): вычисленная масса для C19H27N3O3 345,44, m/z найдено 346,0 [M+H]+.
5. К раствору 5 (2,4 г, 6,96 ммоль) в ДХМ (15 мл) добавляли TFA (15 мл). После перемешивания в течение 16 ч его концентрировали и остаток промывали EtOAc, фильтровали и сушили в вакууме с получением требуемого продукта Промежуточного соединения 35 (2,2 г, 88% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C16H20F3N3O3 359,35, m/z найдено 246,0 [M-TFA+H]+.
ПРИМЕРЫ
Пример 1.
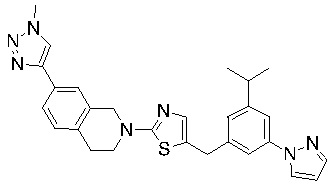
1. К раствору Промежуточного соединения 7 (346 мг, 1,09 ммоль) в ДМСО (10 мл) добавляли Промежуточное соединение 9 (329 мг, 1,31 ммоль) и K2CO3 (300 мг, 2,18 ммоль). Смесь перемешивали при 120 °C в течение ночи, охлаждали до КТ. Смесь обрабатывали водой и экстрагировали EA. Объединенные органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 90,36 мг Примера 1. 1H ЯМР (CDCl3, 300 МГц) δ: 1,3-1,4 (d, 6 H), 3,0-3,1 (m, 2 H), 3,6-3,8 (m, 2 H), 4,1 (s, 2 H), 4,2-4,3 (s, 2 H), 4,8-4,9 (s, 2 H), 6,5 (s, 1 H), 7,0-7,1 (s, 1 H), 7,1-7,2 (s, 1 H), 7,4-7,5 (s, 1 H), 7,5-7,6 (s, 1 H), 7,7.6-7,7 (d, 1 H), 7,7-7,8 (s, 1 H), 7,8-7,9 (s, 1 H) , 7,9-8,0 (s, 1 H). LC-MS: m/z=496,5 (M+1) +.
Пример 2.
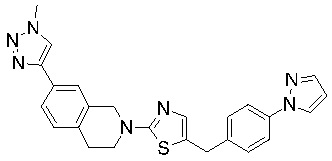
1. Смесь Промежуточного соединения 9 (100 мг, 0,40 ммоль), Промежуточного соединения 3 (121 мг, 0,44 ммоль), Pd2(dba)3 (36,6 мг, 0,04 ммоль), SPhos (16,4 мг, 0,04 ммоль) и трет-BuOK (123 мг, 1,10 ммоль) в диоксане (2,00 мл) перемешивали при 95 °C в течение 2 ч. Реакционную смесь гасили водой и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и солевым раствором, сушили над Na2SO4, и концентрировали. Остаток очищали с помощью преп-ВЭЖХ с получением Примера 2 (45,9 мг, 25% выход) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,91 (2H, t, J = 5,6 Гц), 3,67 (2H, t, J = 5,6 Гц), 4,04 (2H, s), 4,08 (3H, s), 4,60 (2H, s), 6,53 (1H, t, J = 2,0 Гц), 7,03 (1H, s), 7,25 (1H, d, J = 8,0 Гц), 7,36 (2H, d, J = 8,4 Гц), 7,63-7,73 (3H, m), 7,77 (2H, d, J = 8,4 Гц), 8,46 (2H, d, J = 2,0 Гц). Вычислен. масса: 453,2; Найденная масса: 454,2 [M + H]+.
Пример 3.
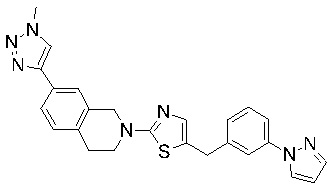
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 9 и Промежуточное соединение 2 подвергали взаимодействию с получением 11,03 мг Примера 3. (9,58 мг) 1H ЯМР (CDCl3, 300 МГц) δ: 3,0-3,1 (m, 2 H), 3,7-3,8 (m, 2 H), 4,1 (s, 2 H), 4,4.3 (s, 2 H), 4,6 (s, 2 H), 6,5 (s, 1 H), 7,0-7,1 (s, 1 H), 7,1-7,3 (m, 2 H), 7,3 (s, 1 H), 7,4 (m, 1 H), 7,5-7,7 (m, 4 H), 7,7-7,8 (s, 1 H), 7,8-7,9 (s, 1 H), 7,9-8,0 (s, 1 H). LC-MS: m/z=454,4 (M+1)+.
Пример 4.
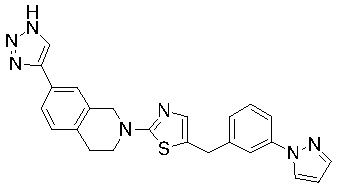
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 10 и Промежуточное соединение 2 подвергали взаимодействию с получением 11,03 мг Примера 4. 1H ЯМР (CDCl3, 300 МГц) δ: 3,0-3,1 (m, 2 H), 3,6-3,8 (m, 2 H), 4,1 (s, 2 H), 4,8 (s, 2 H), 6,5 (s, 1 H), 7,7.1-7,3 (m, 5 H), 7,4-7,5 (m, 1 H), 7,6-7,7 (m, 3 H), 7,7-7,8 (s, 1 H), 7,8-7,9 (s, 1 H), 7,9-8,0 (s, 1 H). LC-MS: m/z=440,4 (M+1)+.
Пример 5.
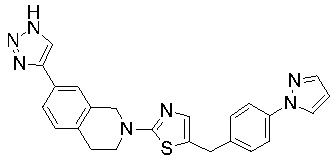
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 10 и Промежуточное соединение 3 подвергали взаимодействию с получением 30,02 мг Примера 5. 1H ЯМР (CDCl3, 300 МГц) δ: 3,0-3,1 (m, 4 H), 3,6-3,8 (m, 2 H), 4,1 (s, 2 H), 4,8-4,9 (s, 2 H), 6,4-6,5 (s, 1 H), 7,1-7,3 (m, 5 H), 7,4 (m, 1 H), 7,5-7,6 (m, 3 H), 7,7 (s, 1 H), 7,7.8 (s, 1 H), 7,9-8,0 (s, 1 H). LC-MS: m/z=440,4 (M+1)+.
Пример 6.
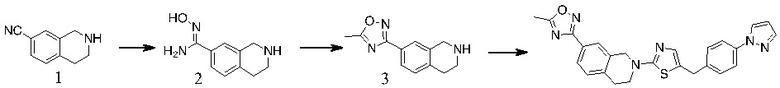
1. К раствору 1 (Sigma-Aldrich, 158 мг, 1 ммоль) в EtOH (20 мл) добавляли NH2OH (1,5 мл). Смесь нагревали с обратным холодильником в течение 20 ч. Смесь охлаждали и концентрировали с получением 2 в виде белого твердого вещества (0,19 г, 100%).
2. Смесь 2 (0,19 г, 1 ммоль) в Ac2O (10 мл) нагревали с обратным холодильником в течение 2 ч. Смесь охлаждали и концентрировали и остаток растворяли в конц. HCl (10 мл) и смесь нагревали с обратным холодильником в течение ночи. Смесь концентрировали с получением 3 в виде белого твердого вещества (0,4 г, 100%).
3. К раствору 3 (20 мг, 0,5 ммоль) в диоксане (10 мл) добавляли Промежуточное соединение 3 (250 мг, 0,9 ммоль), Pd2(dba)3 (32 мг, 0,04 ммоль), SPhos (30 мг, 0,07 ммоль) и трет-BuOK (165 мг, 0,75 ммоль). Полученную смесь нагревали до 100oC и в течение 20 ч в атмосфере N2. Смесь гасили водой и экстрагировали EtOAc. Объединенные экстракты концентрировали с получением неочищенного масла, которое очищали с помощью хроматографии на силикагеле с получением Примера 6 в виде желтого твердого вещества (40 мг, 20%). LC-MS: m/z = 455,1 (M+1)+. Спектр 1H ЯМР (CD3OD, 500 МГц) для полученного соединения представлен на Фиг. 1.
Пример 7.
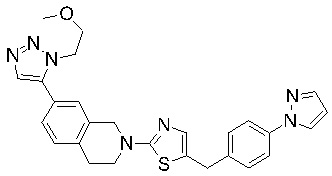
1. К раствору Промежуточного соединения 11 (82 мг, 0,3 ммоль, 1 экв.) в диоксане (20 мл) добавляли Промежуточное соединение 3 (137 мг, 0,5 ммоль, 1,6 экв.), Pd2(dba)3 (18 мг, 0,027 ммоль, 0,09 экв.), SPhos (18 мг, 0,04 ммоль, 0,13 экв.) и трет-BuOK (330 мг, 3 ммоль, 10 экв.). Смесь нагревали до 100 oC и перемешивали в течение 20 ч в атмосфере N2. Смесь гасили водой и экстрагировали EtOAc. Объединенные экстракты концентрировали с получением неочищенного масла, которое очищали с помощью хроматографии на силикагеле с получением Примера 7 в виде желтого твердого вещества (40 мг, 27 %). LC-MS: m/z = 498,2 (M+1)+. Спектр 1H ЯМР (CD3OD, 500 МГц) для полученного соединения представлен на Фиг. 2.
Пример 8.
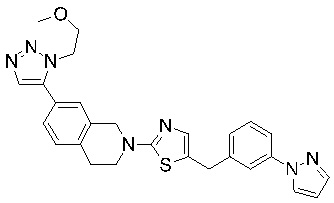
Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 2 вместо Промежуточного соединения 3 и 5 экв. трет-BuOK с получением Примера 8 в виде желтого твердого вещества (90 мг, 27 %). LC-MS: m/z = 498,2 (M+1)+. Спектр 1H ЯМР (CD3OD, 500 МГц) для полученного соединения представлен на Фиг. 3.
Пример 9.
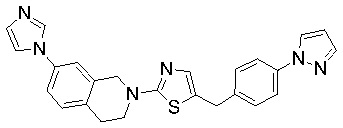
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 12 вместо Промежуточного соединения 11, 1 экв. Промежуточного соединения 3, 0,06 экв. Pd2(dba)3, 0,07 экв. SPhos и 2,4 экв. трет-BuOK с получением Примера 9 в виде белого твердого вещества (100 мг, 45,6 %). LC-MS: m/z = 439,2 (M+1)+. Спектр 1H ЯМР (CDCl3, 500 МГц) для полученного соединения представлен на Фиг. 4.
Пример 10.
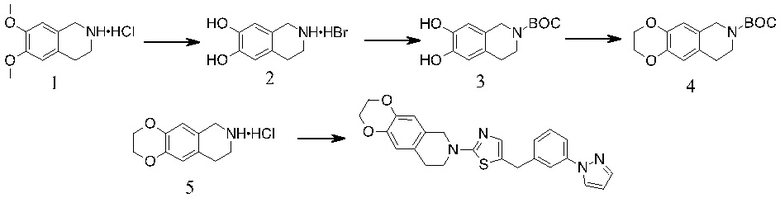
1. Смесь 1 (Sigma-Aldrich, 2,03 г, 8,86 ммоль) в 45% HBr в HOAC (15 мл) нагревали до 110 ºC и перемешивали в течение 5 ч. После охлаждения до КТ осадок фильтровали с получением 2 в виде белого твердого вещества (1,92 г, 88%).
2. BOC2O (1,88 г, 8,61 ммоль) и TEA (2,8 мл, 16,45 ммоль) в ТГФ (20 мл) добавляли по каплям к суспензии 2 (1,92 г, 7,83 ммоль) в воде (6 мл). Смесь перемешивали при КТ в течение ночи. После концентрирования остаток растворяли в EtOAc, промывали водой. Органическую смесь сушили над Na2SO4 и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии с получением 3 (1,8 г, 86,9%).
3. К раствору 3 (0,4 г, 1,5 ммоль) в диоксане (20 мл) добавляли 1,2-дибромэтан (0,31 г, 1,66 ммоль), 2 N водн. NaOH (5 мл, 10 ммоль) и TBAB (20 мг, 0,16 ммоль). Смесь нагревали до 90 °C и перемешивали в течение ночи. Реакционную смесь гасили водой и экстрагировали EtOAc. Органические экстракты сушили над Na2SO4, фильтровали и концентрировали с получением 4 (210 мг, 47,8%).
4. К раствору 4 (210 мг, 0,72 ммоль) в ДХМ (20 мл) добавляли 4 M HCl/Диоксан (3 мл, 12 ммоль). Смесь перемешивали при КТ в течение ночи, затем концентрировали с получением 5 (150 мг, 92%).
5. К раствору 5 (137 мг, 0,6 ммоль) в диоксане (10 мл) добавляли Промежуточное соединение 2 (170 мг, 0,62 ммоль), Pd2(dba)3 (55 мг, 0,06 ммоль), SPhos (50 мг, 0,12 ммоль) и трет-KOBu (135 мг, 1,2 ммоль). Смесь нагревали до 100 °C и перемешивали в течение 20 ч в атмосфере N2. Смесь гасили водой и экстрагировали EtOAc. Объединенные органические экстракты концентрировали. Полученное вещество очищали с помощью хроматографии на силикагеле с получением Примера 10 в виде белого твердого вещества (80 мг, 30%). m/z = 431,1 (M+H)+. Спектр 1H ЯМР (CD3OD, 500 МГц) для полученного соединения представлен на Фиг. 5.
Пример 11.
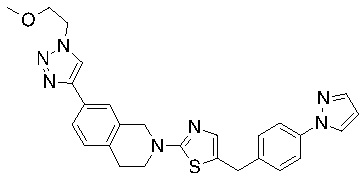
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 14 вместо Промежуточного соединения 11, 1 экв. Промежуточного соединения 3, 0,09 экв. Pd2(dba)3, 0,16 экв. SPhos и 3 экв. трет-BuOK с получением Примера 11 (75,9 мг, 35%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,92 (2H, t, J = 6,0 Гц), 3,26 (3H, s), 3,67 (2H, t, J = 6,0 Гц), 3,77 (2H, t, J = 5,2 Гц), 4,04 (2H, s), 4,56 (2H, t, J = 5,6 Гц), 4,60 (2H, s), 6,52-6,53 (1H, m), 7,03 (1H, s), 7,25 (1H, d, J = 8,0 Гц), 7,36 (2H, d, J = 8,8 Гц), 7,65-7,67 (1H, m), 7,70-7,72 (2H, m), 7,76-7,78 (2H, m), 8,45 (1H, d, J = 2,4 Гц), 8,48 (1H, s). Вычислен. масса: 497,2; Найденная масса: 498,2 [M + H]+.
Пример 12.
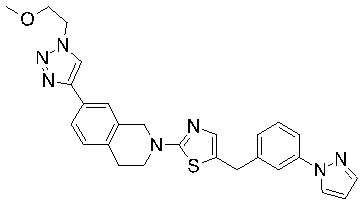
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 14 вместо Промежуточного соединения 11, 1 экв. Промежуточного соединения 2 вместо Промежуточного соединения 3, 0,09 экв. Pd2(dba)3, 0,18 экв. SPhos и 3 экв. трет-BuOK с получением Примера 12 (51,5 мг, 23%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,91 (2H, t, J = 6,0 Гц), 3,26 (3H, s), 3,67 (2H, t, J = 6,0 Гц), 3,77 (2H, t, J = 5,2 Гц), 4,08 (2H, s), 4,56 (2H, t, J = 5,2 Гц), 4,59 (2H, s), 6,54 (1H, t, J = 2,0 Гц), 7,06 (1H, s), 7,19 (1H, d, J = 7,6 Гц), 7,25 (1H, d, J = 8,0 Гц), 7,43 (1H, t, J = 8,0 Гц), 7,65-7,77 (5H, m), 8,48-8,49 (2H, m). Вычислен. масса: 497,2; Найденная масса: 498,2 [M + H]+.
Пример 13.
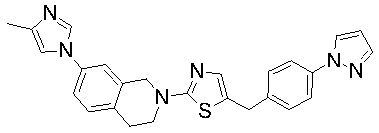
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 15 вместо Промежуточного соединения 11, 1 экв. Промежуточного соединения 3, 0,09 экв. Pd2(dba)3, 0,2 экв. SPhos и 2,8 экв. трет-BuOK с получением Примера 13 (33,4 мг, 15% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,15 (3H, s), 2,91 (2H, t, J = 5,6 Гц), 3,66 (2H, t, J = 6,0 Гц), 4,04 (2H, s), 4,60 (2H, s), 6,53 (1H, t, J = 2,0 Гц), 7,03 (1H, s), 7,29 (1H, d, J = 8,0 Гц), 7,35-7,43 (4H, m), 7,51 (1H, d, J = 2,0 Гц), 7,72 (1H, d, J = 1,6 Гц), 7,77 (2H, d, J = 8,8 Гц), 8,08 (1H, d, J = 1,2 Гц), 8,46 (1H, d, J = 2,4 Гц). Вычислен. масса:452,2; Найденная масса: 453,2 [M + H]+.
Пример 14.
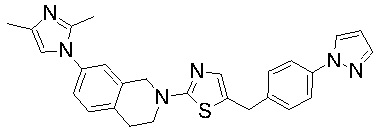
1. Смесь Промежуточного соединения 1 (100 мг, 0,38 ммоль), Промежуточного соединения 3, (115 мг, 0,42 ммоль), Pd2(dba)3 (36,6 мг, 0,04 ммоль), SPhos (32,9 мг, 0,08 ммоль) и трет-BuOK (128 мг, 1,14 ммоль) в сухом диоксане (4,00 мл) перемешивали при 90 °C в течение 4 ч. Когда реакция была завершена, ее гасили водой и экстрагировали EtOAc. Объединенные органические слои промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью преп-ВЭЖХ с получением Примера 14 (33,5 мг, 19% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,08 (3H, s), 2,22 (3H, s), 2,94 (2H, t, J = 5,6 Гц), 3,67 (2H, d, J = 5,6 Гц), 4,04 (2H, s), 4,59 (2H, s), 6,53 (1H, t, J = 2,0 Гц), 6,92 (1H, s), 7,03 (1H, s), 7,21 (1H, d, J = 8,0, 2,0 Гц), 7,30-7,32 (2H, m), 7,36 (2H, d, J = 8,4 Гц), 7,72 (1H, d, J = 2,0 Гц), 7,77 (2H, d, J = 8,8 Гц), 8,46 (1H, d, J = 2,4 Гц). Вычислен. масса: 466,2; Найденная масса: 467,2 [M + H]+.
Альтернативное получение Примера 14
1. К раствору Промежуточного соединения 1 (25 г, 110 ммоль) в ДМСО (500 мл) добавляли Промежуточное соединение 3 (40 г, 145 ммоль) и K2CO3 (45,54 г, 330 ммоль). Колбу три раза продували N2 и перемешивали при 140 oC в течение 2 часов, охлаждали до КТ, разбавляли EA, фильтровали, концентрировали и остаток очищали с помощью хроматографии на силикагеле, элюируя с получением вещества, которое было перекристаллизовано EtOAc с получением Примера 14 (21,8 г, 42,5% выход) в виде беловатого твердого вещества. MS (ESI): вычисленная масса для C27H26N6S 466,61, m/z найдено 466,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 8,46 (d, J = 2,3 Гц, 1H), 7,78 (d, J = 8,4 Гц, 2H), 7,73 (s, 1H), 7,37 (d, J = 8,4 Гц, 2H), 7,33 - 7,31 (m, 2H), 7,22 (d, J = 8,1 Гц, 1H), 7,03 (s, 1H), 6,94 (s, 1H), 6,53 (s, 1H), 4,60 (s, 2H), 4,05 (s, 2H), 3,68 (t, J = 5,9 Гц, 2H), 2,95 (t, J = 5,8 Гц, 2H), 2,23 (s, 3H), 2,09 (s, 3H).
Пример 15.
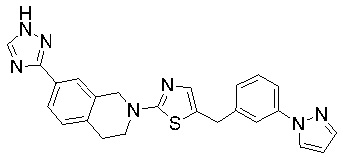
1. Смесь Промежуточного соединения 16 (130 мг, 0,31 ммоль) и ДМФА-DMA (10 мл) перемешивали при 100 °C в течение 1 ч. Смесь концентрировали и остаток растворяли в смеси EtOH (10 мл) и N2H4*H2O (2 мл), и полученную смесь перемешивали при КТ в течение 0,5 ч. Смесь гасили водой и экстрагировали EtOAc. Объединенные органические слои промывали водой и солевым раствором, сушили над Na2SO4, и концентрировали. Остаток очищали с помощью преп-ВЭЖХ с получением Примера 15 (11,0 мг, 8% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,92-2,94 (2H, m), 3,68 (2H, t, J = 6,0 Гц), 4,08 (2H, s), 4,61 (2H, s), 6,54 (1H, t, J = 2,0 Гц), 7,06 (1H, s), 7,18 (1H, d, J = 8,0 Гц), 7,25-7,30 (1H, m), 7,43 (1H, t, J = 7,6 Гц), 7,67-7,69 (1H, m), 7,73 (1H, d, J = 1,6 Гц), 7,77 (1H, s), 7,81-7,84 (2H, m), 8,49 (1H, d, J = 2,8 Гц), 8,58-8,62 (1H, m), 14,09-14,40 (1H, m). Вычислен. масса: 439,2; Найденная масса: 440,1 [M + H] +.
Пример 16.
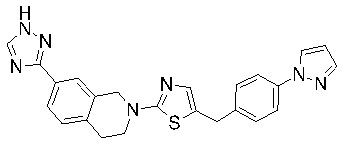
1. В соответствии с процедурой, описанной для Примера 15, Промежуточное соединение 17 (0,18 г, 0,43 ммоль) превращали в Пример 16 в виде белого твердого вещества (60 мг, 31%). LC-MS: m/z = 440,2 (M+1)+. Спектр 1H ЯМР (ДМСО-d6, 500 МГц) для полученного соединения представлен на Фиг. 6.
Пример 17.
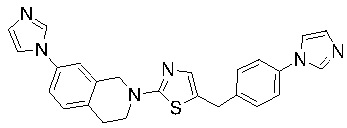
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 18 и Промежуточное соединение 12 подвергали взаимодействию с получением 19,6 мг Примера 17. 1H ЯМР (CDCl3, 300 МГц) δ: 3,0-3,1 (m, 2 H), 3,7-3,8 (m, 2 H), 4,1 (m, 2 H), 4,7-4,8 (s, 2 H), 7,0-7,1 (s, 1 H), 7,2-7,5 (m, 14 H), 7,8-7,9 (s, 2 H). LC-MS: m/z=439,4 (M+1)+.
Пример 18.
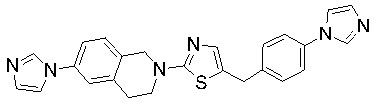
1. В соответствии с процедурой, описанной для Примера 1, 300 мг Промежуточного соединения 3 и 350 мг Промежуточного соединения 13 превращали в Пример 18 (24,62 мг). 1H ЯМР (CDCl3, 300 МГц) δ: 3,0-3,1 (m, 2 H), 3,7-3,8 (m, 2 H), 4,0-4,1 (m, 2 H), 4,6-4,7 (s, 2 H), 6,4-6,5 (s, 1 H), 6,9-7,0 (s, 1 H), 7,2 (s, 1 H), 7,3-7,4 (d, 2 H), 7,6-7,7 (d, 1 H), 7,7-7,8 (s, 1 H), 7,8-7,9 (s, 1 H)7,9-8,0 (s, 1 H). LC-MS: m/z=439,3 (M+1) +.
Пример 19.
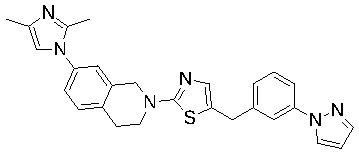
1. В соответствии с процедурой, описанной для Примера 7, с использованием Промежуточного соединения 1 вместо Промежуточного соединения 11, 1,2 экв. Промежуточного соединения 2 вместо Промежуточного соединения 3, 0,11 экв. Pd2(dba)3, 0,22 экв. SPhos и 3 экв. трет-BuOK получали Пример 19 (33,5 мг, 20% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,08 (3H, s), 2,21 (3H, s), 2,94 (2H, t, J = 5,6 Гц), 3,67 (2H, t, J = 2,0 Гц), 4,09 (2H, d, J = 4,8 Гц), 4,59 (2H, s), 6,54 (1H, t, J = 2,4 Гц), 6,91 (1H, s), 7,06-7,07 (1H, m), 7,20 (2H, t, J = 8,0 Гц), 7,29-7,35 (2H, m), 7,40-7,44 (1H, m), 7,69 (1H, d, J = 8,4 Гц), 7,73 (1H, d, J = 1,2 Гц), 7,76 (1H, s), 8,48 (1H, d, J = 2,8 Гц). Вычислен. масса: 466,2; Найденная масса: 467,3 [M + H]+.
Пример 20.
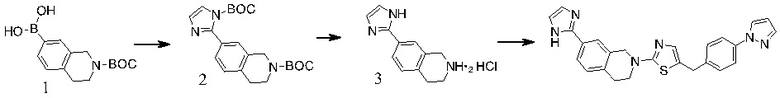
1. К смеси 1 (междун.заявка PCT (2008), WO 2008079277 A, 4 г, 14 ммоль) трет-бутил-2-бром-1H-имидазол-1-карбоксилата (FCH Group, 1,8 г, 7 ммоль) и NaOH (3 мл, 1,5 M) в толуоле (20 мл) и этаноле (2 мл) добавляли Pd(PPh3)4 (0,084 г, 0,14 ммоль) в атмосфере N2. Полученную смесь перемешивали при 120 ° в течение 24 ч. Смесь охлаждали до КТ, обрабатывали водой и экстрагировали EA. Объединенные органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла. Неочищенный продукт очищали путем перекристаллизации с получением 2 (1,1 г, 38 %).
2. Смесь 2 (1,1 г, 2,76 ммоль) в HCl/Et2O (3 M, 20 мл) перемешивали при КТ в течение ночи. Смесь фильтровали фильтрационный осадок промывали Et2O с получением 3 (0,4 г, 80 %).
3. К смеси 3 (0,3 г, 1,6 ммоль) в ДМСО (5 мл) добавляли Промежуточное соединение 3 (0,3 г, 1,1 ммоль)) и K2CO3 (0,3 г, 2,2 ммоль). Смесь перемешивали при 140 ° в течение ночи. Смесь охлаждали до КТ, обрабатывали водой и экстрагировали EA. Объединенные органические экстракты промывали водой, солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла, которое очищали с помощью хроматографии на силикагеле с получением Примера 20 (15 мг). 1H ЯМР (CDCl3, 300 МГц) δ: 2,9-3,1 (m, 2 H), 3,7-3,8 (m, 2 H), 4,0-4,1 (m, 2 H), 4,6-4,7 (s, 2 H), 6,4-6,5 (s, 1 H), 6,9-7,0 (s, 1 H), 7,1-7,2 (m, 3 H), 7,6-7,8 (m, 4 H), 7,9-8,0 (s, 1 H). LC-MS: m/z=439,4 (M+23) +
Пример 21.
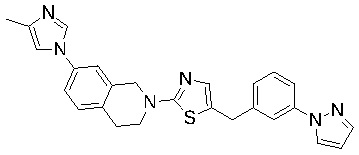
1. Смесь Промежуточного соединения 4 (190 мг, 0,42 ммоль), 4-метил-1H-имидазола (104 мг, 1,26 ммоль), (S,S)-N,N′-диметил-1,2-диаминоциклогексана (12 мг, 0,08 ммоль), трет-BuOK (141 мг, 1,26 ммоль) и CuI (40 мг, 0,21 ммоль) в NMP (5 мл) перемешивали при 140 oC в течение ночи в атмосфере N2. Смесь охлаждали до КТ, разбавляли MeOH, фильтровали, фильтрат концентрировали и очищали с помощью преп-ВЭЖХ с получением Примера 21 (20 мг, 10,5% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C26H24N6S 452,58, m/z найдено 452,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 8,49 (d, J = 2,3 Гц, 1H), 8,08 (s, 2H), 7,77 (s, 1H), 7,74 (s, 1H), 7,69 (d, J = 8,3 Гц, 1H), 7,51 (s, 1H), 7,44 - 7,41 (m, 2H), 7,29 (d, J = 8,2 Гц, 1H), 7,20 (d, J = 7,6 Гц, 1H), 7,07 (s, 1H), 6,55 (s, 1H), 4,60 (s, 2H), 4,09 (s, 2H), 3,66 (t, J = 5,9 Гц, 2H), 2,92 (t, J = 5,8 Гц, 2H), 2,16 (s, 2H).
Пример 22.
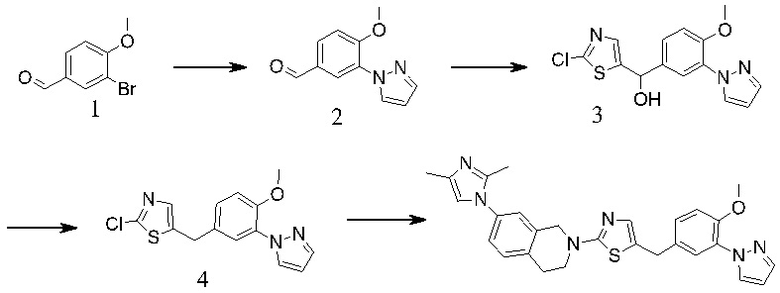
1. К раствору 1 (4,50 г, 20,93 ммоль) в ДМФА (30 мл) добавляли (R,R)-N,N′-диметил-1,2-диаминоциклогексан (0,30 г, 2,09 ммоль), 1H-пиразол (1,42 г, 20,93 ммоль), CuI (0,40 г, 2,09 ммоль) и K2CO3 (5,75 г, 41,7 ммоль). Реакционную смесь перемешивали при 150 oC в течение 18 ч. Реакционную смесь охлаждали до КТ, выливали в ледяную воду, экстрагировали CH2Cl2, сушили над Na2SO4. Объединенные органические экстракты концентрировали при пониженном давлении и очищали с помощью хроматографии на силикагеле с получением 2 (1,80 г, 42,6 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C20H18BrN3OS 202,21, m/z найдено 202,7 [M+H]+.
2. К раствору 2-хлортиазола (1,07 г, 8,9 ммоль) в сухом ТГФ (40 мл) при -78 oC по каплям добавляли н-BuLi (2,4M, 4,0 мл, 9,6 ммоль). Через 1 ч добавляли по каплям раствор 2 (1,80 г, 8,9ммоль) в ТГФ (40 мл). Реакционную смесь медленно нагревали до КТ. Смесь гасили нас. водн. NH4Cl и экстрагировали EtOAc и объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 3 (1,4 г, 48,9% выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H7BrClNOS 321,78, m/z найдено 322,4 [M+H]+.
3. Смесь 3 (1,40 г, 4,35 ммоль) в TES (5 мл) и TFA (15мл) перемешивали при 60 oC в течение 2 часов. Смесь концентрировали и остаток промывали нас. водн. NaHCO3 и экстрагировали CH2Cl2, сушили над Na2SO4.Органические экстракты концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 4 (1,2 г, 90,2 % выход) в виде желтого масла. MS (ESI): вычисленная масса для C10H7BrClNS 305,78, m/z найдено 306,2 [M+H]+.
4. К раствору 4 (0,2 г, 0,65 ммоль) в 1,4-диоксане (30 мл) добавляли Промежуточное соединение 1 (0,148 г, 0,65 ммоль) и K2CO3 (0,27 г, 1,95 ммоль). Реакционную смесь перемешивали при 120 oC в течение 5 часов. Смесь охлаждали до КТ, выливали в ледяную воду, экстрагировали CH2Cl2 и объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении. Полученный остаток очищали с помощью преп-ВЭЖХ с получением Примера 22 (18,5 мг, 5,7 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C28H28N6OS 496,63, m/z найдено 496,7 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,16 (d, J = 2,4 Гц, 1H), 7,68 (s, 1H), 7,53 (d, J = 1,6 Гц, 1H), 7,32 (d, J = 8,4 Гц, 2H), 7,28 - 7,15 (m, 3H), 7,03 (s, 1H), 6,93 (s, 1H), 6,47 (s, 1H), 4,59 (s, 2H), 4,02 (s, 2H), 3,67 (t, J = 5,8 Гц, 3H), 2,67 (t, J = 5,8 Гц, 2H), 2,95 (t, J = 5,8 Гц, 2H), 2,23 (s, 3H), 2,09 (s, 3H).
Пример 23.
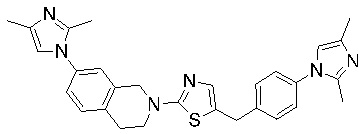
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 19 (225 мг) и Промежуточное соединение 1 (202 мг) превращали в Пример 23 в виде белого твердого вещества. MS (ESI): вычисленная масса для C29H30N6S 494,66, m/z найдено 494,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,44 (s, 4H), 7,39 (d, J = 8,8 Гц, 2H), 7,31 (d, J = 8,1 Гц, 1H), 7,21 (d, J = 8,3 Гц, 2H), 7,07 (s, 1H), 4,63 (s, 2H), 4,10 (s, 2H), 3,69 (t, J = 5,7 Гц, 2H), 2,98 (t, J = 5,6 Гц, 2H), 2,35 (d, J = 5,2 Гц, 6H), 2,19 (s, 6H).
Пример 24.
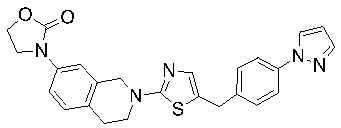
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 21, с использованием Промежуточного соединения 5 (100 мг) вместо Промежуточного соединения 4, оксазолидин-2-она (1,15 экв.) вместо 4-метил-1H-имидазола, трет-BuOK (3 экв.), CuI (0,6 экв.), (S,S)-N,N′-диметил-1,2-диаминоциклогексана (0,2 экв.) с получением Примера 24 (40 мг, 14,8 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C25H23N5O2S 457,55, m/z найдено 457,7 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,46 (d, J = 2,0 Гц, 1H), 7,78 (d, J = 8,4 Гц, 2H), 7,73 (s, 1H), 7,45 (d, J = 8,4 Гц, 1H), 7,39-7,36 (m, 3H), 7,21 (d, J = 8,4 Гц, 1H), 7,08 (s, 1H), 6,54 (s, 1H), 4,57 (s, 2H), 4,44 (t, J = 7,6 Гц, 2H), 4,08 - 4,00 (m, 4H), 3,67 (t, J = 5,6 Гц, 2H), 2,89 (t, J = 6,0 Гц, 2H).
Пример 25.
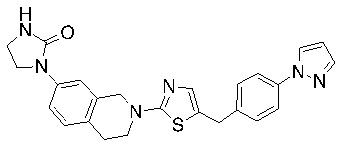
1. Указанное в заголовке соединение получали в соответствии с процедурой, описанной для Примера 21, с использованием Промежуточного соединения 5 (200 мг) вместо Промежуточного соединения 4, имидазолидин-2-она (1 экв.) вместо 4-метил-1H-имидазола, трет-BuOK (3 экв.), CuI (0,5 экв.), (S,S)-N,N′-диметил-1,2-диаминоциклогексана (0,2 экв.) с получением Примера 25 (30 мг, 14,8 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C25H24N6OS 456,56, m/z найдено 456,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,45 (d, J = 2,4 Гц, 1H), 7,76 (d, J = 8,4 Гц, 2H), 7,72 (s, 1H), 7,44 (dd, J = 8,4, 1,6 Гц, 1H), 7,38 - 7,31 (m, 3H), 7,09 (d, J = 8,4 Гц, 1H), 7,01 (s, 1H), 6,91 (s, 1H), 6,52 (s, 1H), 4,51 (s, 2H), 4,03 (s, 2H), 3,80 (t, J = 7,6 Гц, 2H), 3,63 (t, J = 6,0 Гц, 2H), 3,38 (t, J = 8,4 Гц, 2H), 2,82 (t, J = 6,0 Гц, 2H).
Пример 26.
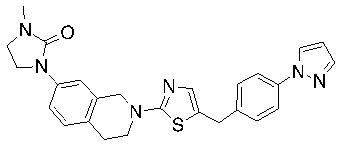
1. Смесь Промежуточного соединения 5 (100 мг, 0,22 ммоль), 1-метилимидазолидин-2-он (66 мг, 0,66 ммоль), (Pd2(dba)3) (100 мг, 0,11 ммоль), трет-BuOK (74 мг, 0,66 ммоль) и SPhos (44 мг, 0,11 ммоль) в диоксане (10 мл) перемешивали в атмосфере азота при 100 oC в течение 16 ч. Растворитель удаляли при пониженном давлении и остаток очищали с помощью преп-ВЭЖХ с получением Примера 26 (50 мг, 48,1 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C26H26N6OS 470,59, m/z найдено 470,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,45 (d, J = 2,4 Гц, 1H), 7,76 (d, J = 8,4 Гц, 2H), 7,72 (s, 1H), 7,47 (dd, J = 8,4, 2,0 Гц, 1H), 7,38 - 7,31 (m, 3H), 7,10 (d, J = 8,4 Гц, 1H), 7,01 (s, 1H), 6,52 (s, 1H), 4,51 (s, 2H), 4,03 (s, 2H), 3,74 (t, J = 7,2 Гц, 2H), 3,63 (t, J = 5,6 Гц, 2H), 3,41 (t, J = 8,4 Гц, 2H), 2,83 (t, J = 6,0 Гц, 2H), 2,75 (s, 3H).
Пример 27. 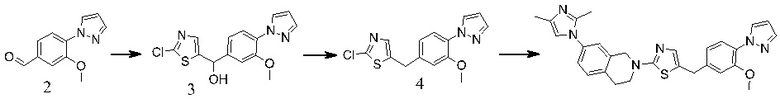
1. Смесь 4-фтор-3-метокси-бензальдегида (5,0 г, 32,4 ммоль), 1H-пиразола (3,3 г, 48,6 ммоль), K2CO3 (6,8 г, 48,6 ммоль) в ДМФА (30 мл) перемешивали в атмосфере N2 при 120 oC в течение 20 ч. После охлаждения до КТ к смеси добавляли ледяную воду, которую затем экстрагировали EA. Объединенные органические экстракты промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением 2 (3,0 г, 53,4 % выход) в виде бледно-желтого твердого вещества. MS (ESI): вычисленная масса для C11H10N2O2 202,21, m/z найдено 202,9 [M+H]+.
2. К раствору 2-хлортиазола (3,0 г, 25,1 ммоль) в сухом ТГФ (100 мл) при -78 oC по каплям добавляли н-BuLi (11,3 мл, 2,4 M, 27,2 ммоль) и перемешивали при этой температуре в течение 1 ч. Раствор 2 (4,2 г, 20,9 ммоль) добавляли по каплям при -78 oC. Полученный раствор медленно нагревали до КТ. Реакционную смесь гасили раствором NH4Cl и экстрагировали EA. Объединенные органические экстракты промывали солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением 3 (5,6 г, 83,8 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C14H12ClN3O2S 321,78, m/z найдено 321,8 [M+H]+.
3. К раствору 3 с КТ (5,6 г, 17,4 ммоль) в TFA (20 мл) добавляли TES (11,4 г, 69,6 ммоль). Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 1 ч. Смесь выпаривали и к смеси добавляли ледяную воду, которую затем экстрагировали EA. Объединенные органические экстракты промывали насыщенным раствором NaHCO3, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле 4 (4,8 г, 90,2 % выход) в виде прозрачной жидкости. MS (ESI): вычисленная масса для C14H12ClN3OS 305,78, m/z найдено 305,8 [M+H]+.
4. В соответствии с процедурой, описанной для Примера 1, 4 (200 мг) и Промежуточное соединение 1 (148 мг) превращали в Пример 27 (10 мг, 57,3 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C28H28N6OS 496,63, m/z найдено 496,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,11 (d, J = 2,4 Гц, 1H), 7,66 (s, 1H), 7,53 (d, J = 8,4 Гц, 1H), 7,36 (m, 2H), 7,28 (d, J = 8,0 Гц, 1H), 7,16 (s, 1H), 7,13 (s, 1H), 7,05 (s, 1H), 6,93 (d, J = 8,0 Гц, 1H), 6,45 (s, 1H), 4,61 (s, 2H), 4,06 (s, 2H), 3,83 (s, 3H), 3,68 (t, J = 6,0 Гц, 2H), 2,96 (t, J = 6,0 Гц, 2H), 2,31 (s, 3H), 2,15 (s, 3H).
Пример 28.
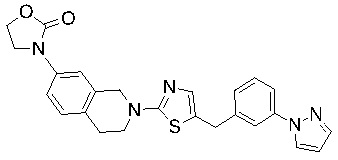
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 20 и Промежуточное соединение 2 подвергали взаимодействию с получением Примера 28 (43,2 мг, 26,2 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C25H23N5O2S 457,55, m/z найдено 457,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,48 (d, J = 2,0 Гц, 1H), 7,75 (d, J = 12,4 Гц, 2H), 7,69 (d, J = 8,0 Гц, 1H), 7,44 (dd, J = 15,8, 7,8 Гц, 2H), 7,36 (s, 1H), 7,19 (d, J = 8,0 Гц, 2H), 7,05 (s, 1H), 6,54 (s, 1H), 4,54 (s, 2H), 4,43 (t, J = 7,8 Гц, 2H), 4,08 (s, 2H), 4,03 (t, J = 8,0 Гц, 2H), 3,64 (t, J = 5,8 Гц, 2H), 2,86 (t, J = 5,6 Гц, 2H).
Пример 29.
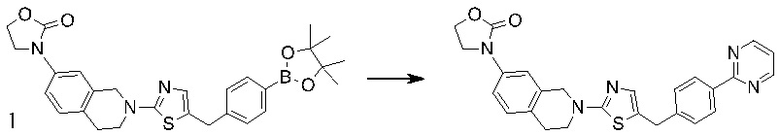
1. Смесь Промежуточного соединения 22 (329 мг, 0,48 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (203 мг, 0,8 ммоль), Pd(dppf)Cl2 (102 мг, 0,14 ммоль) и AcOK (206 мг, 2,1 ммоль) в сухом 1,4-диоксане (20 мл) перемешивали при 100 oC в течение ночи. Смесь охлаждали до КТ, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 1 (250 мг, 69,0%), вычисленная масса для C28H32BN3O4S 517,45, m/z найдено 517,6 [M+H]+.
2. К смеси 1 (100 мг, 0,19 ммоль) и 2-хлорпиримидина (23 мг, 0,20 ммоль) в 1,4-диоксане (8 мл) и H2O (2 мл) добавляли K2CO3 (80 мг, 0,57 ммоль), Pd(dppf)Cl2 (14 мг, 0,02 ммоль). Полученную смесь перемешивали при 100 oC в течение ночи. Смесь охлаждали до комнатной температуры, выливали в ледяную воду, экстрагировали EA, объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 29 (9 мг, 10%) в виде белого твердого вещества. вычисленная масса для C26H23N5O2S 469,16. m/z найдено 469,7 [M+H]+, 1H ЯМР (400 МГц, ДМСО) δ ppm 8,90 (d, J = 4,8 Гц, 2H), 8,34 (d, J = 8,1 Гц, 2H), 7,51 - 7,32 (m, 5H), 7,20 (d, J = 8,4 Гц, 1H), 7,05 (s, 1H), 4,55 (s, 2H), 4,43 (t, J = 7,9 Гц, 2H), 4,09 (s, 2H), 4,03 (t, J = 8,0 Гц, 2H), 3,65 (t, J = 5,8 Гц, 2H), 2,87 (t, J = 5,7 Гц, 2H).
Пример 30.
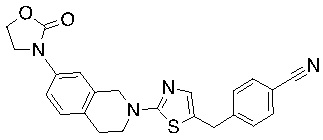
1. К раствору Промежуточного соединения 22 (180 мг, 0,38 ммоль) в DMI (6 мл) добавляли CuCN (70 мг, 0,78 ммоль) и CuI (90 мг, 0,46 ммоль). Полученную смесь нагревали до 190 oC в течение 1 ч в микроволновой печи. Реакционную смесь охлаждали до КТ, выливали в воду и экстрагировали CH2Cl2. Объединенные экстракты сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 30 (11 мг, 7%) в виде белого твердого вещества. вычисленная масса для C23H20N4O2S 416,5, m/z найдено 416,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,78 (d, J = 8,1 Гц, 2H), 7,46 (d, J = 8,1 Гц, 2H), 7,37 (s, 1H), 7,20 (d, J = 8,5 Гц, 1H), 7,04 (s, 1H), 4,55 (s, 2H), 4,46 - 4,41 (m, 2H), 4,11 (s, 2H), 4,06 - 4,01 (m, 2H), 3,65 (t, J = 5,9 Гц, 2H), 2,86 (t, J = 5,8 Гц, 2H).
Пример 31.
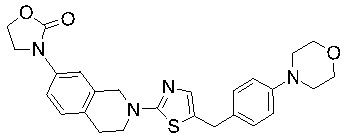
1. Смесь Промежуточного соединения 22 (240 мг, 0,5 ммоль), морфолина (131 мг, 1,5 ммоль), Pd2(dba)3 (229 мг, 0,25 ммоль), SPhos (103 мг, 0,25 ммоль) и K2CO3 (207 мг, 1,5 ммоль) в 1,4-диоксане (15 мл) перемешивали при 100 oC в течение ночи. Смесь охлаждали до КТ, выливали в воду и экстрагировали CH2Cl2. Объединенные экстракты сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 31 (10 мг,4,2%) в виде белого твердого вещества. вычисленная масса для C26H28N4O3S 476,6, m/z найдено 476,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,45 (d, J = 9,9 Гц, 1H), 7,37 (s, 1H), 7,19 (d, J = 8,4 Гц, 1H), 7,09 (d, J = 8,4 Гц, 2H), 6,95 (s, 1H), 6,87 (d, J = 8,5 Гц, 2H), 4,53 (s, 2H), 4,48 - 4,34 (m, 2H), 4,12 - 3,95 (m, 2H), 3,88 (s, 2H), 3,75 - 3,67 (m, 4H), 3,64 (t, J = 5,8 Гц, 2H), 3,08 - 3,01 (m, 4H), 2,87 (d, J = 5,9 Гц, 2H).
Пример 32.
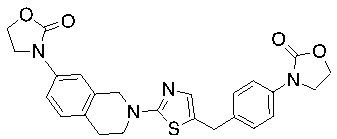
1. В соответствии с процедурой, описанной для Примера 31, с использованием трет-BuOK (170 мг) вместо K2CO3, Промежуточное соединение 22 (240 мг) и оксазолидин-2-он (170 мг) превращали в Пример 32 (10 мг, 4%) в виде белого твердого вещества. Вычисленная масса для C25H24N4O4S 476,55, m/z найдено 476,7 [M+H]+, 1H ЯМР (400 МГц, ДМСО) δ ppm 7,50 (d, J = 8,4 Гц, 2H), 7,45 (d, J = 8,2 Гц, 1H), 7,37 (s, 1H), 7,26 (d, J = 8,4 Гц, 2H), 7,20 (d, J = 8,3 Гц, 1H), 6,98 (s, 1H), 4,54 (s, 2H), 4,43 (t, J = 7,0 Гц, 4H), 4,04 (t, J = 7,1 Гц, 4H), 3,98 (s, 2H), 3,64 (t, J = 5,9 Гц, 2H), 2,86 (t, J = 5,9 Гц, 2H).
Пример 33.
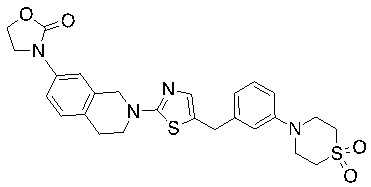
1. В соответствии с процедурой, описанной для Примера 31, с использованием Cs2CO3 (488 мг) вместо K2CO3, Промежуточное соединение 21 (220 мг) и тиоморфолина 1,1-диоксид (270 мг) превращали в Пример 33 (7,8 мг, 2,9%) в виде белого твердого вещества. Вычисленная масса для C26H28N4O4S2 524,65, m/z найдено 524,7 [M+H]+, 1H ЯМР (400 МГц, CDCl3) δ ppm 7,38 (d, J = 8,8 Гц, 2 H), 7,25 (d, J = 7,8 Гц, 1H), 7,19 (d, J= 8,4 Гц, 1H), 7,00 (s, 1H), 6,87 - 6,77 (m, 3H), 4,64 (s, 2 H), 4,51 (dd, J = 8,8, 7,0 Гц, 2 H), 4,16 (t, J = 8,0 Гц, 2H), 3,98 (s, 2 H), 3,85 (t, J = 5,2 Гц, 4 H), 3,76 (t, J = 5,8 Гц, 2H), 3,11 (t, J = 5,0 Гц, 4H), 2,96 (t, J = 5,8 Гц, 2H).
Пример 34.
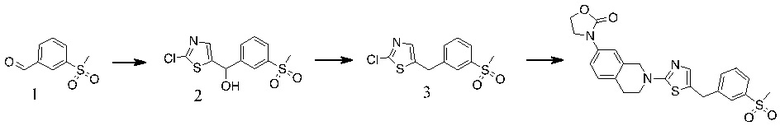
1. К раствору 2-хлортиазола (0,72 г, 6,00 ммоль) в сухом ТГФ (50 мл) при -78 oC в атмосфере N2 по каплям добавляли н-BuLi (2,4M, 2,5 мл, 6 ммоль). Через 0,5 ч добавляли по каплям раствор 1 (1,00 г, 5,43 ммоль) в сухом ТГФ (5 мл). Реакционную смесь медленно нагревали до КТ. Реакционную смесь гасили насыщенным NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (0,60 г, 36% выход) в виде желтого масла. MS (ESI): вычисленная масса для C11H10ClNO3S2 303,78, m/z найдено 304,1 [M+H]+.
2. Смесь 2 (600 мг, 1,97 ммоль) в TES 3 мл) и TFA (10мл) перемешивали при 60 oC в течение 2 ч. Смесь концентрировали и остаток разбавляли насыщенным NaHCO3, экстрагировали ДХМ и объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 3 (400 мг, 70,5 % выход) в виде коричневого масла. MS (ESI): вычисленная масса для C11H10ClNO2S2 287,78, m/z найдено 288,0 [M+H]+.
3. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 20 (229 мг) и 3 (200 мг) превращали в Пример 34 (30 мг, 9,26% выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C23H23N3O4S2 469,57, m/z найдено 469,7 [M+H]+. 1H ЯМР (301 МГц, ДМСО-d6) δ ppm 7,80 (d, J = 7,5 Гц, 2H), 7,61 (s, 2H), 7,45 (d, J = 7,2 Гц, 1H), 7,37 (s, 1H), 7,19 (d, J = 8,1 Гц, 1H), 7,05 (s, 1H), 4,54 (s, 2H), 4,42 (t, J = 7,8 Гц, 2H), 4,14 (s, 2H), 4..02 (t, J = 7,8 Гц, 2H), 3,64 (t, J = 5,4 Гц, 2H), 3,18 (s, 3H), 2,86 (t, J = 5,1 Гц, 2H).
Пример 35.
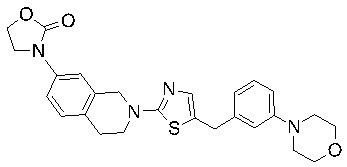
1. В соответствии с процедурой, описанной для Примера 31, с использованием Cs2CO3 (488 мг) вместо K2CO3, Промежуточное соединение 21 (235 мг) и морфолин (130 мг) превращали в Примера 35 (10,6 мг, 4,4%) в виде белого твердого вещества. Вычисленная масса для C26H28N4O3S 476,60, m/z найдено 476,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,45 (d, J = 6,0 Гц, 1H), 7,37 (s, 1H), 7,23 - 7,12 (m, 2H), 6,98 (s, 1H), 6,84 (s, 1H), 6,79 (d, J = 7,6 Гц, 1H), 6,68 (d, J = 7,6 Гц, 1H), 4,54 (s, 2H), 4,43 (t, J = 7,8 Гц, 2H), 4,03 (t, J = 7,8 Гц, 2H), 3,92 (s, 2H), 3,72 (t, J = 4,6 Гц, 4H), 3,64 (t, J = 6,0 Гц, 2H), 3,08 (t, J = 4,6 Гц, 4H), 2,86 (t, J = 5,8 Гц, 2H).
Пример 36.
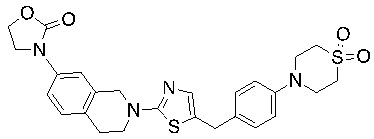
1. В соответствии с процедурой, описанной для Примера 31, с использованием Cs2CO3 (371 мг) вместо K2CO3, Промежуточное соединение 22 (180 мг) и тиоморфолина 1,1-диоксид (154 мг) превращали в Пример 36 (9 мг, 4%) в виде белого твердого вещества. Вычисленная масса для C26H28N4O4S2 524,65, m/z найдено 524,7 [M+H]+, 1H ЯМР (300 МГц, ДМСО-d6) δ ppm 7,40 (d, J = 8,7 Гц, 1H), 7,32 (s, 1H), 7,14 (d, J = 8,4 Гц, 1H), 7,07 (d, J = 8,3 Гц, 2H), 6,92 (d, J = 4,6 Гц, 3H), 4,50 (s, 2H), 4,39 (t, J = 7,8 Гц, 2H), 3,99 (t, J = 7,8 Гц, 2H), 3,85 (s, 2H), 3,69 (s, 4H), 3,60 (t, J = 5,7 Гц, 2H), 3,07 (s, 4H), 2,83 (s, 2H).
Альтернативное получение Примера 36
1. Смесь Промежуточного соединения 22 (50 г, 0,106 моль), тиоморфолина 1,1-диоксид (71,8 г, 0,531 моль), Pd(OAc)2 (12 г, 0,053 моль), SPhos (23,4 г, 0,053 моль) и Cs2CO3 (43,3 г, 0,133 моль) в высушенном 1,4-диоксане (2,8 L) перемешивали при 100 oC в течение 6 ч в атмосфере N2. Смесь разбавляли ДХМ:MeOH 10:1 (1L) и полученную суспензию фильтровали. Фильтрационный осадок промывали дважды ДХМ:MeOH=10:1 и объединенный фильтрат концентрировали. Полученный остаток растворяли ДХМ:MeOH 10:1 (200 мл), смесь нагревали с обратным холодильником в течение 30 мин и добавляли MeOH (300 мл) и смесь нагревали с обратным холодильником в течение 20 мин и суспензию фильтровали через целит. Фильтрат концентрировали и остаток обрабатывали как описано выше четыре раза. После четвертой фильтрации объединенный фильтрат концентрировали с получением неочищенного продукта в виде оранжевого твердого вещества, которое суспендировали дважды с MeCN (300 мл) с получением Примера 36 (25,9 г, 46,4%) в виде белого твердого вещества. MS (ESI): вычисленная масса для C26H28N4O4S2 524,65 m/z найдено 524,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,45 (dd, J=8,4, 2,4, 1H), 7,37 (d, J=2,1, 1H), 7,19 (d, J=8,4, 1H), 7,12 (d, J=8,7, 2H), 6,99 - 6,94 (m, 3H), 4,54 (s, 2H), 4,43 (dd, J=8,9, 7,1, 2H), 4,03 (dd, J=8,9, 7,2, 2H), 3,89 (s, 2H), 3,76 - 3,70 (m, 4H), 3,64 (t, J=6,0, 2H), 3,16 - 3,07 (m, 4H), 2,86 (t, J=5,9, 2H).
Пример 37.
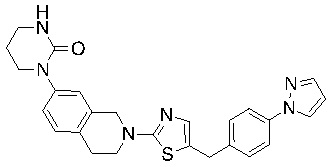
1. Смесь Промежуточного соединения 5 (225 мг, 0,50 ммоль), тетрагидропиримидин-2(1H)-она (150 мг, 1,50 ммоль), Pd2(dba)3 (92 мг, 0,1 ммоль), SPhos (82 мг, 0,2 ммоль) и трет-BuOK (168 мг, 1,5 ммоль) в 1,4-диоксане (20 мл) перемешивали при 100 oC в течение ночи. Смесь охлаждали, выливали в воду и экстрагировали ДХМ, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 37 (3,4 мг,1,4%) в виде белого твердого вещества. вычисленная масса для C26H26N6OS 470,60, m/z найдено 470,8 [M+H]+, 1H ЯМР (400 МГц, CD3OD) δ ppm 8,20 (d, J=2,8 Гц, 1H), 7,72 (d, J=1,6 Гц, 2H), 7,70 (s, 1H), 7,68 (s, 1H), 7,40 (d, J=8,8 Гц, 2H), 7,20 (d, J=9,2 Гц, 1H), 7,14 (s, 1H), 4,58 (s, 2H), 4,09 (s, 2H), 3,70 (m, 1H), 3,40 (t, J=5,8 Гц, 4H), 2,97 (t, J=5,8 Гц, 2H), 2,1 (t, J=6,0 Гц, 2H).
Соединения, перечисленные в таблице 2 ниже, были получены способом, аналогичным описанному в Примере 37.
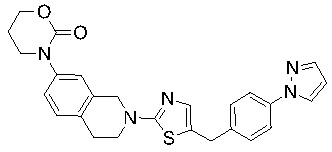
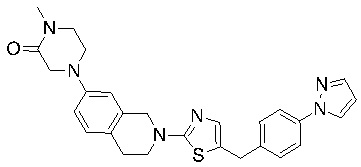
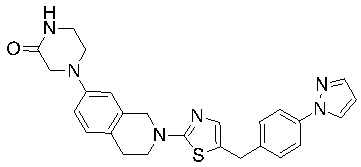
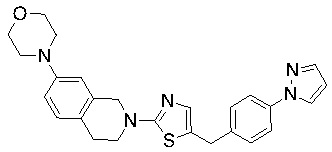
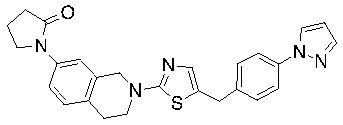
Пример 38.
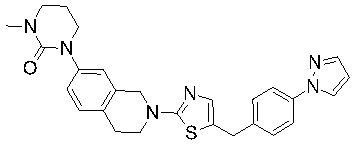
1. A раствор Примера 37 (30 мг, 0,064 ммоль) в высушенном ДМФА (3 мл) охлаждали до 0 oC, добавляли NaH (24 мг, 1 ммоль) и через 0,5 ч добавляли CH3I. Реакционную смесь нагревали до КТ и перемешивали в течение ночи. Смесь выливали в воду и экстрагировали ДХМ. Объединенные экстракты сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 38 (10,06 мг, 34,22 %) в виде белого твердого вещества. Вычисленная масса для C27H28N6OS 484,62, m/z найдено 484,8 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ = 7,92 (d, J=2,1 Гц, 1H), 7,74 (d, J=1,5 Гц, 1H), 7,66 (d, J=8,5 Гц, 2H), 7,34 (d, J=8,5 Гц, 2H), 7,13 (s, 2H), 7,08 (s, 1H), 7,01 (s, 1H), 6,54 - 6,45 (m, 1H), 4,61 (s, 2H), 4,05 (s, 2H), 3,75 (d, J=5,8 Гц, 2H), 3,71 - 3,64 (m, 2H), 3,40 (t, J=6,1 Гц, 2H), 2,95 (t, J=5,8 Гц, 2H), 2,13 (dt, J=11,9 Гц, 6,0 Гц, 2H).
Пример 39.
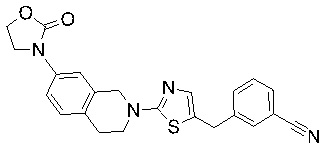
1. Смесь Промежуточного соединения 21 (170 мг, 0,36 ммоль), CuCN (39 мг, 0,72 ммоль) и CuI (168 мг, 1,44 ммоль) в DMI (2 мл) нагревали до 190 oC в микроволновой печи в течение 50 минут. Смесь охлаждали, фильтровали и твердые вещества промывали ДХМ/MeOH (10:1) и водой, водный слой экстрагировали ДХМ/MeOH (10:1). Объединенные органические экстракты промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 39 (8,5 мг, 5,7 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C23H20N4O2S 416,50, m/z найдено 416,8 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,59 - 7,53 (m, 2H), 7,50 (d, J=7,9 Гц, 1H), 7,45 (d, J=7,8 Гц, 1H), 7,42 - 7,37 (m, 2H), 7,20 (d, J=9,0 Гц, 1H), 7,00 (s, 1H), 4,64 (s, 2H), 4,51 (dd, J=8,8 Гц, 7,0 Гц, 2H), 4,06 (t, J=8,0 Гц, 4H), 3,75 (t, J=5,9 Гц, 2H), 2,97 (t, J=5,8 Гц, 2H).
Пример 40.
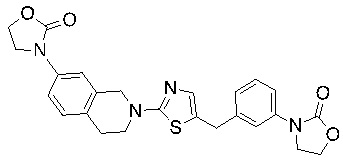
1. В соответствии с процедурой, описанной для Примера 31, с использованием Cs2CO3 (488 мг) вместо K2CO3, Промежуточное соединение 21 (235 мг) и оксазолидин-2-он (200 мг) превращали в Пример 40 (5 мг, 2%) в виде белого твердого вещества, вычисленная масса для C25H24N4O4S 476,55, m/z найдено 476,8 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,47 - 7,39 (m, 3H), 7,38 - 7,32 (m, 2H), 7,19 (d, J = 8,4 Гц, 1H), 7,05 (d, J = 7,6 Гц, 1H), 7,00 (s, 1H), 4,64 (s, 2H), 4,53 - 4,48 (m, 4H), 4,10 - 4,03 (m, 6H), 3,75 (t, J = 5,6 Гц, 2H), 2,96 (t, J = 5,8 Гц, 2H).
Пример 41.
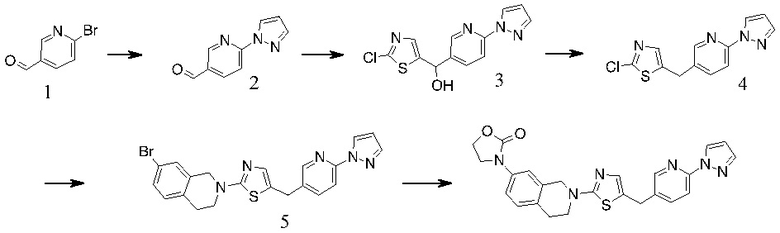
1. К раствору 1 (2,00 г, 10,75 ммоль) в ДМФА (30 мл) добавляли 1H-пиразол (1,10 г, 16,13 ммоль) и K2CO3 (4,46 г, 32,25 ммоль). Реакционную смесь перемешивали при 100 oC в течение 18 ч. Реакционную смесь охлаждали до КТ и затем выливали в ледяную воду, экстрагировали CH2Cl2 и экстракты сушили над Na2SO4. Органические экстракты концентрировали с получением остатка, который очищали с помощью хроматографии на силикагеле с получением 2 (1,00 г, 53,7 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C9H7N3O 173,18, m/z найдено 173,8 [M+H]+.
2. К раствору 2-хлортиазола (762 мг, 6,35 ммоль) в сухом ТГФ (50 мл) при -78 oC в атмосфере N2 по каплям добавляли н-BuLi (2,4 M, 2,89 мл). Через 0,5 ч добавляли по каплям раствор 2 (1,00 г, 5,77 ммоль) в высушенном ТГФ (10 мл). Реакционную смесь медленно нагревали до КТ. Смесь гасили водн. NH4Cl и экстрагировали EtOAc и экстракты сушили над Na2SO4. Органические экстракты концентрировали с получением остатка, который очищали с помощью хроматографии на силикагеле с получением 3 (800 мг, 47,36 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C12H9ClN4OS 292,74, m/z найдено 292,8 [M+H]+.
3. Смесь 3 (800 мг, 2,73 ммоль), TES (5 мл) и TFA (15 мл) перемешивали при 70 oC в течение 2 ч. Смесь концентрировали и остаток разбавляли водн. NaHCO3 и экстрагировали ДХМ и экстракты сушили над Na2SO4. Экстракты концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 4 (400 мг, 52,9 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C12H9ClN4S 276,74, m/z найдено 276,8 [M+H]+.
4. К раствору 4 (400 мг, 1,46 ммоль) в ДМСО (10 мл) добавляли 7-бром-1,2,3,4-тетрагидроизохинолин (Key Organics, 335 мг, 1,58 ммоль) и Cs2CO3 (1,54 г, 4,74 ммоль). Реакционную смесь перемешивали при 140 oC в течение 5 часов, охлаждали до КТ и выливали в ледяную воду, экстрагировали ДХМ. Объединенные экстракты сушили над Na2SO4 и экстракты концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением 5 (300 мг, 45,42 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C21H18BrN5S 452,37, m/z найдено 452,8 [M+H]+.
5. Смесь 5 (300 мг, 0,45 ммоль), оксазолидин-2-она (136 мг, 1,36 ммоль), Pd2(dba)3 (66 мг, 0,09 ммоль), SPhos (92 мг, 0,225 ммоль) и трет-BuOK (162 мг, 1,35 ммоль) в высушенном 1,4-диоксане перемешивали при 100 oC в течение ночи. Смесь охлаждали, выливали в воду и экстрагировали CH2Cl2, сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью преп-ВЭЖХ с получением Примера 41 (39,6 мг, 19,19%) в виде белого твердого вещества, вычисленная масса для C24H22N6O2S 458,54, m/z найдено 458,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,59 (dd, J=2,8, 0,6 Hz ,1H), 8,39 (d, J=1,6 Гц, 1H), 7,90- 7,84(m, 2H), 7,82 - 7,79 (m, 1H), 7,45 (dd, J=8,4 Гц, 2,4, 1H), 7,37 (d, J=2,2 Гц, 1H), 7,20 (d, J=8,4 Гц, 1H), 7,04 (s, 1H), 6,57 (dd, J=2,5 Гц, 1,7 Гц, 1H), 4,56 (s, 2H), 4,43 (dd, J=9,6 Гц, 6,4 Гц, 2H), 4,09 (s, 2H), 4,03 (dd, J=8,4 Гц, 7,0 Гц, 2H), 3,65 (t, J=6,0 Гц, 2H), 2,87 (t, J=6,0 Гц, 2H).
Пример 42.
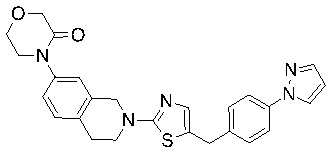
1. К раствору Промежуточного соединения 5 (225 мг, 0,50 ммоль), морфолин-3-она (171 мг, 1,50 ммоль) в высушенном 1,4-диоксане (15 мл) добавляли K2CO3 (209 мг, 1,51 ммоль), CuI (10 мг, 0,05 ммоль) и (S,S)-N,N′-диметил-1,2-диаминоциклогексан (8 мг, 0,05 ммоль). Полученный раствор перемешивали при 150 oC в течение 1,5 ч в микроволновой печи. Смесь охлаждали до КТ, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 42 (20,8 мг, 5,6%) в виде белого твердого вещества. вычисленная масса для C26H25N5O2S 471,58, m/z найдено 471,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,45 (d, J=2,4 Гц, 1H), 7,77 (d, J=8,8 Гц, 2H), 7,73 (d, J=1,6 Гц, 1H), 7,36 (d, J=8,4 Гц, 2H), 7,25 (s, 1H), 7,21 (s, 2H), 7,02 (s, 1H), 6,53 (t, J = 2,2 Гц, 1H), 4,55 (s, 2H), 4,19 (s, 2H), 4,04 (s, 2H), 3,96 (t, J = 5,0 Гц, 2H), 3,70 (t, J = 5,0 Гц, 2H), 3,66 (t, J=6,0 Гц, 2H), 2,90 (t, J=6,0 Гц, 2H).
Пример 43.
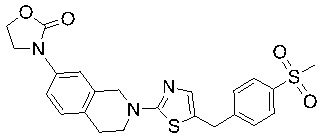
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 20 (357 мг) и Промежуточное соединение 31 (270 мг) превращали в Примера 43 (55 мг, 13% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C23H23N3O4S2 469,11, m/z найдено 69,7 [M+H]+, 1H ЯМР (400 МГц, ДМСО) δ ppm 7,87 (d, J = 8,3 Гц, 2H), 7,52 (d, J = 8,3 Гц, 2H), 7,45 (dd, J = 8,4, 2,3 Гц, 1H), 7,38 (d, J = 2,0 Гц, 1H), 7,20 (d, J = 8,4 Гц, 1H), 7,06 (s, 1H), 4,55 (s, 2H), 4,48 - 4,35 (m, 2H), 4,13 (s, 2H), 4,09 - 3,98 (m, 2H), 3,65 (t, J = 5,9 Гц, 2H), 3,19 (s, 3H), 2,87 (t, J = 5,9 Гц, 2H).
Пример 44.
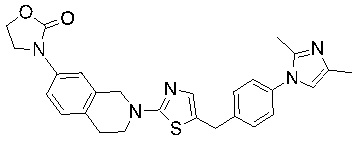
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 20 (227 мг) и Промежуточное соединение 19 (200 мг) превращали в неочищенный продукт, который очищали с помощью преп-ТСХ с получением Примера 44 (12 мг, 4% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C27H27N5O2S 485,19. m/z найдено 486,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,46 (d, J = 8,7 Гц, 1H), 7,36 (dd, J = 14,2, 7,2 Гц, 5H), 7,20 (d, J = 8,4 Гц, 1H), 7,05 (s, 1H), 6,97 (s, 1H), 4,56 (s, 2H), 4,44 (t, J = 7,9 Гц, 2H), 4,06 (d, J = 5,5 Гц, 2H), 4,03 (d, J = 7,7 Гц, 2H), 3,66 (t, J = 5,9 Гц, 2H), 2,88 (t, J = 5,6 Гц, 2H), 2,23 (s, 3H), 2,09 (s, 3H).
Пример 45.
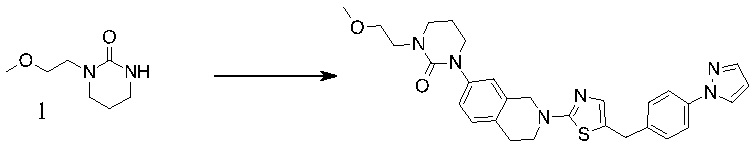
1. К смеси 2-метоксиэтан-1-амина (1 г, 13,3 ммоль) в ДМФА (100 мл) добавляли 3-хло1рпропилизоцианат (1,9 г, 16 ммоль), затем добавляли трет-BuOK (2,23 г, 20 ммоль) и полученную смесь перемешивали при КТ в течение 16 ч. Смесь концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 1 (630 мг, 30% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C7H14N2O2S 158,20, m/z найдено 159,0 [M+H]+.
2. Смесь 1 (245 мг, 1,55 ммоль), Промежуточного соединения 5 (140 мг, 0,31 ммоль), трет-BuONa (104 мг, 1,1 ммоль), Pd(OAc)2 (43 мг, 0,19 ммоль) и SPhos (78 мг, 0,19 ммоль) в 1, 4-диоксане (8 мл) продували 3x N2. Полученную смесь перемешивали при 100 oC в течение 15 часов, охлаждали до КТ, разбавляли смесью (ДХМ/MeOH, 10/1, 20 мл), фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением Примера 45 (13 мг, 8% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C29H32N6O2S 528,68, m/z найдено 528,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 8,46 (s, 1H), 7,77 (d, J = 8,3 Гц, 2H), 7,72 (s, 1H), 7,36 (d, J = 8,3 Гц, 2H), 7,00 - 7,06 (m, 3H), 7,02 (s, 1H), 6,53 (s, 1H), 4,50 (s, 2H), 4,04 (s, 2H), 3,66 - 3,58 (m, 4H), 3,45 - 3,40 (m, 6H), 3,26 (s, 3H), 2,85 (t, J = 5,8 Гц, 1H), 2,01 - 1,96 (m, 2H).
Пример 46.
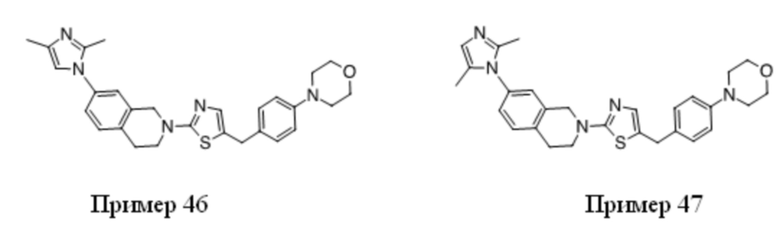
1. Смесь Промежуточного соединения 1 (150 мг, 0,66 ммоль), Промежуточного соединения 24 (233 мг, 0,79 ммоль) и K2CO3 (273 мг, 1,98 ммоль) в ДМСО (6 мл) вакуумировали и повторно наполняли N2 три раза и перемешивали при 130 oC в течение 3 ч. Смесь охлаждали до КТ, разбавляли смесь ДХМ/MeOH 20/1 (20 мл), фильтровали и фильтрат концентрировали и очищали с помощью хроматографии на силикагеле с получением неочищенного продукта. Неочищенный продукт очищали с помощью SFC (chiralpak-IB, CO2-EtOH(DEA)) с получением Примера 46 (56,38 мг, 17,6% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C28H31N5OS 485,65, m/z найдено 485,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,32 - 7,30 (m, 2H), 7,21 (d, J = 7,7 Гц, 1H), 7,10 (d, J = 8,2 Гц, 2H), 6,96 (s, 1H), 6,92 (s, 1H), 6,88 (d, J = 8,4 Гц, 2H), 4,58 (s, 2H), 3,89 (s, 2H), 3,74 - 3,71 (m, 4H), 3,66 (t, J = 5,7 Гц, 2H), 3,09 - 3,01 (m, 4H), 2,94 (t, J = 5,7 Гц, 2H), 2,23 (s, 3H), 2,08 (s, 3H) и Примера 47 (15 мг) в виде желтого/белого твердого вещества MS (ESI): вычисленная масса для C28H31N5OS 485,65, m/z найдено 485,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,35 (d, J = 8,2 Гц, 1H), 7,25 (s, 1H), 7,15 (d, J = 7,9 Гц, 1H), 7,10 (d, J = 8,4 Гц, 2H), 6,96 (s, 1H), 6,88 (d, J = 8,5 Гц, 2H), 6,62 (s, 1H), 4,60 (s, 2H), 3,89 (s, 2H), 3,74 - 3,67 (m, 6H), 3,07 - 3,05 (m, 4H), 2,98 (t, J = 5,4 Гц, 2H), 2,09 (s, 3H), 1,95 (s, 3H).
Пример 48.
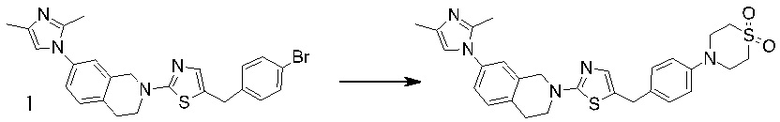
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 oC в течение 2 ч, Промежуточное соединение 1 (280 мг) и Промежуточное соединение 6 (403 мг) превращали в неочищенный продукт, которое очищали с помощью Combi-Flash с получением 1 (380 мг, 66 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C24H23BrN4S 479,4, m/z найдено 479 [M+H]+.
2. Смесь 1 (240 мг, 0,5 ммоль), 1,1-диоксида тиоморфолина (135 мг, 1 ммоль), Pd2(dba)3 (41 мг, 0,05 ммоль), SPhos (20 мг, 0,05 ммоль), трет-BuOK (122 мг, 1 ммоль) в толуоле (10 мл) перемешивали при 100 oC в течение 2 ч. Смесь охлаждали и разбавляли EA, промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью преп-ВЭЖХ с получением Примера 48 (50 мг, 22 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C28H31N5O2S2 533,7, m/z найдено 534 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,16 (s, 1H), 7,31 (d, J = 8,4 Гц, 2H), 7,22 (d, J = 8,3 Гц, 1H), 7,12 (d, J = 8,4 Гц, 2H), 6,97 (d, J = 7,4 Гц, 3H), 6,92 (s, 1H), 4,59 (s, 2H), 3,90 (s, 2H), 3,73 (s, 5H), 3,67 (t, J = 5,7 Гц, 2H), 3,11 (s, 4H), 2,94 (t, J = 5,5 Гц, 2H), 2,23 (s, 3H), 2,08 (s, 3H).
Пример 49.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 ºC в течение 5 часов, Промежуточное соединение 25 (200 мг) и Промежуточное соединение 26 (266 мг) превращали в неочищенный продукт, который очищали с помощью преп-ВЭЖХ с получением Примера 49 (82,6 мг, 19,20 %) в виде белого твердого вещества. Вычисленная масса для C27H25N5OS 467,59, m/z найдено 467,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,90 (d, J=4,8 Гц, 2H), 8,34 (d, J=8,0 Гц, 2H), 7,52 (d, J=8,4 Гц, 1H), 7,42 (m, 4H), 7,17 (d, J=8,4 Гц, 1H), 7,04 (s, 1H), 4,54 (s, 2H), 4,09 (s, 2H), 3,80 (t, J=7,0 Гц, 2H), 3,65 (t, J=5,8 Гц, 2H), 2,86 (t, J=5,4 Гц, 2H), 2,47 (d, J=7,8 Гц, 2H), 2,05 (dt, J=14,7 Гц, 7,3 Гц, 1H).
Пример 50.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 ºC в течение 5 часов и Cs2CO3 (663 мг) использовали вместо K2CO3, Промежуточное соединение 25 (235 мг) и Промежуточное соединение 24 (200 мг) превращали в неочищенный продукт, который очищали с помощью преп-ВЭЖХ с получением Примера 50 (66,2 мг, 20,51 %) в виде белого твердого вещества. Вычисленная масса для C27H30N4O2S 474,62, m/z найдено 474,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,52 (d, J=8,4 Гц, 1H), 7,45 (s, 1H), 7,17 (d, J=8,4 Гц, 1H), 7,09 (d, J=8,4 Гц, 2H), 6,95 (s, 1H), 6,88 (d, J=8,4 Гц, 2H), 4,52 (s, 2H), 3,88 (s, 2H), 3,80 (t, J=7,0 Гц, 2H), 3,73(t, J=7,0 Гц, 4H), 3,63 (t, J=5,8 Гц, 2H), 3,01(t, J=4,6 Гц, 4H), 2,86 (t, J=5,8 Гц, 2H), 2,47 (t, J=4,0 Гц, 2H), 2,09 - 1,99 (m, 2H).
Пример 51.
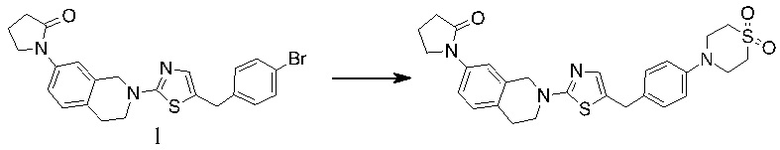
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 ºC в течение 2 ч, Промежуточное соединение 25 (380 мг) и Промежуточное соединение 6 (366 мг) превращали в неочищенный продукт, который очищали с помощью Combi-Flash с получением 1 (280 мг, 52 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C23H22BrN3OS 468,4, m/z найдено 468 [M+H]+.
2. Смесь 1 (235 мг, 0,5 ммоль), тиоморфолина 1,1-диоксида (135 мг, 1 ммоль), Pd2(dba)3 (41 мг, 0,05 ммоль), SPhos (20 мг, 0,05 ммоль), трет-BuOK (122 мг, 1 ммоль) в толуоле (10 мл) перемешивали при 100 oC в течение 2 ч. Смесь охлаждали и разбавляли EA, промывали водой, солевым раствором и сушили над Na2SO4, фильтровали, концентрировали. Полученный остаток очищали с помощью преп-ВЭЖХ с получением Примера 51 (110 мг, 42 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C27H30N4O3S2 522,7, m/z найдено 523 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,51 (d, J = 8,2 Гц, 1H), 7,45 (s, 1H), 7,16 (d, J = 8,3 Гц, 1H), 7,11 (d, J = 8,2 Гц, 2H), 6,99 - 6,93 (m, J = 4,9 Гц, 3H), 4,52 (s, 2H), 3,89 (s, 2H), 3,80 (t, J = 6,9 Гц, 2H), 3,72 (s, 4H), 3,66 - 3,59 (m, 2H), 3,10 (s, 4H), 2,85 (t, J = 5,6 Гц, 2H), 2,47 (d, J = 8,0 Гц, 2H), 2,07 - 1,99 (m, 2H).
Пример 52.
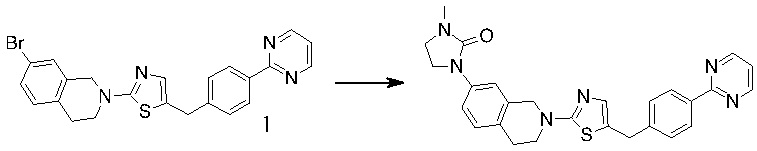
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 ºC в течение 4 ч, Промежуточное соединение 26 (300 мг) и гидрохлорид 7-бром-1,2,3,4-тетрагидроизохинолина (Key Organics, 270 мг) превращали в неочищенный продукт, который очищали с помощью флэш-хроматографии с получением 1 (380 мг 78,7 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C23H19BrN4S 463,40, m/z найдено 462,8 [M+H]+.
2. Смесь 1 (150 мг, 0,324 ммоль), 1-метилимидазолидин-2-она (162 мг, 1,62 ммоль), Pd2(dba)3 (148 мг, 0,162 ммоль), SPhos (67 мг, 0,162 ммоль) и трет-BuOK (109 мг, 0,972 моль) в высушенном 1,4-диоксане (21 мл) перемешивали при 100 oC в течение 6 ч. Смесь разбавляли ДХМ:MeOH 10:1 и фильтровали и фильтрационный осадок промывали дважды ДХМ:MeOH 10:1. Объединенный фильтрат концентрировали и остаток очищали с помощью флэш-хроматографии с получением продукта, который очищали с помощью преп-ВЭЖХ с получением Примера 52 (69 мг, 44,2%, 99,1% чистота 214 нм) в виде белого твердого вещества. MS (ESI): вычисленная масса для C27H26N6OS 482,61 m/z найдено 482,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,90 (d, J=4,6, 2H), 8,34 (d, J=7,6, 2H), 7,48 (d, J=8,2,1H), 7,42 (dd, J=14,3, 6,4, 3H), 7,33 (s, 1H), 7,11 (d, J=8,4, 1H), 7,04 (s, 1H), 4,52 (s, 2H), 4,09 (s, 2H), 3,74 (t, J=7,9, 2H), 3,64 (t, J=5,8, 2H), 3,42 (t, J=7,7, 2H), 2,83 (t, J=5,5, 2H), 2,75 (s, 3H).
Пример 53.
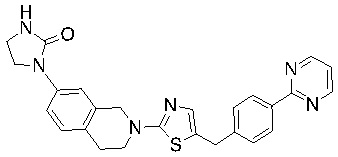
1. Смесь продукта Примера 52 Стадии 1 (150 мг, 0,324 ммоль) и имидазолидин-2-она (139 мг, 1,62 ммоль) обрабатывали, как описано в Примере 52 Стадия 2 с получением неочищенного продукта, который очищали с помощью флэш-хроматографии. Полученное вещество суспендировали со смесью PE:Гексан 1:1 с получением Примера 53 (54,3 мг, 35,8 %, 99,8% чистота 214 нм) в виде белого твердого вещества. MS (ESI): вычисленная масса для C26H24N6OS 468,58 m/z найдено 468,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,90 (d, J=4,8, 2H), 8,34 (d, J=8,0, 2H), 7,43 (dd, J=16,4, 8,5, 4H), 7,34 (s, 1H), 7,10 (d, J=8,4, 1H), 7,05 (s, 1H), 6,92 (s, 1H), 4,52 (s, 2H), 4,09 (s, 2H), 3,82 (t, J=5,8, 2H), 3,64 (t, J=5,8, 2H), 3,43 - 3,36 (m, 4H), 2,84 (t, J=5,7, 2H).
Пример 54.
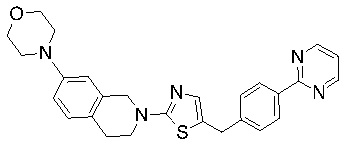
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 oC в течение 5 часов, Промежуточное соединение 26 (170 мг) и Промежуточное соединение 27 (224 мг) превращали в неочищенный продукт. Неочищенный продукт очищали с помощью преп-ВЭЖХ с получением Примера 54 (64,7 мг, 23,35 %) в виде белого твердого вещества. Вычисленная масса для C27H27N5OS 469,61, m/z найдено 469,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,90 (d, J=4,8 Гц, 2H), 8,34 (d, J=7,6 Гц, 2H), 7,51 - 7,36 (m, 3H), 7,10 - 6,98 (m, 2H), 6,86 - 6,75 (m, 2H), 4,49 (s, 2H),4,08 (s, 2H), 3,72 (t, J=4,4 Гц, 2H), 3,61 (t, J=5,6 Гц, 2H), 3,05(t, J=4,8 Гц, 4H), 2,79(t, J=5,8 Гц, 2H).
Пример 55.
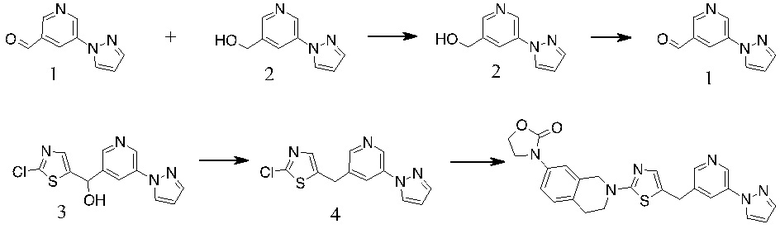
1. Смесь 5-бромникотинальдегида (2 г, 10,8 ммоль), 1H-пиразола (1,46 г, 21,5 ммоль), CuI (1,03 г, 5,4 ммоль), (S,S)-N,N′-диметил-1,2-диаминоциклогексана (307 мг, 2,16 ммоль) и Cs2CO3 (10,6 г, 32,4 ммоль) в ДМФА (40 мл) три раза продували N2 и смесь нагревали до 120 oC в течение 16 ч. Смесь охлаждали до КТ, разбавляли EtOAc, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением смеси 1 и 2 (1 г, неочищенной) в виде коричневого твердого вещества. MS (ESI): вычисленная масса для C9H7N3O 173,18, m/z найдено 174,0 [M+H]+.
2. К смеси 1 и 2 (1 г, 5,78 ммоль) в MeOH (10 мл) при 0 oC добавляли NaBH4 (330 мг, 8,67 ммоль) и смесь нагревали до КТ и перемешивали в течение 2 ч. Смесь концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 2 (470 мг, 25% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C9H9N3O 175,19, m/z найдено 176,1 [M+H]+.
3. К 2 (470 мг, 2,69 ммоль) в ДХМ (15 мл) добавляли реактив Десса-Мартина (1,48 г, 3,49 ммоль). Полученную смесь перемешивали при КТ в течение 30 мин, концентрировали и остаток очищали с помощью хроматографии на силикагеле с получением 1 (420 мг, 90% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C9H7N3O 173,18, m/z найдено 174,0 [M+H]+.
4. н-BuLi (1,32 мл, 3,16 ммоль, 2,4 M) по каплям добавляли к раствору 2-хлортиазола (378 мг, 3,16 ммоль) в ТГФ (2 мл) в атмосфере N2 при - 70 oC. После перемешивания в течение 30 мин добавляли по каплям раствор 1 (420 мг, 2,43 ммоль) в ТГФ (15 мл) и полученную смесь нагревали до КТ и перемешивали в течение ночи. Смесь гасили нас. водн. NH4Cl (20 мл), экстрагировали EtOAc и объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 3 (490 мг, 69% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C12H9ClN4OS 292,74, m/z найдено 292,8 [M+H]+.
5. К раствору 3 (490 мг, 1,68 ммоль) в ДХМ (8 мл) по каплям добавляли SOCl2 (240 мг, 2,01 ммоль). После перемешивания в течение 2 ч смесь концентрировали с получением желто-белого твердого вещества. Твердое вещество растворяли в AcOH (10 мл) и добавляли Zn порошок (546 мг, 8,4 ммоль) и полученную смесь перемешивали при КТ в течение 1 ч, нейтрализовывали нас. водн. NaHCO3 и экстрагировали EtOAc. Объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением 4 (350 мг, 75% выход) в виде белого масла. MS (ESI): вычисленная масса для C12H9ClN4S 276,74, m/z найдено 277,0 [M+H]+.
6. Смесь 4 (100 мг, 0,36 ммоль), Промежуточного соединения 20 (140 мг, 0,44 ммоль) и K2CO3 (150 мг, 1,08 ммоль) в ДМСО (10 мл) перемешивали в атмосфере азота при 120 oC в течение 2 ч, охлаждали до КТ и к смеси добавляли ледяную воду, которую затем экстрагировали EA. Объединенные органические экстракты промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью преп-ТСХ с получением Примера 55 (75 мг, 45,5 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C24H22N6O2S 458,54, m/z найдено 458,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,97 (d, J = 2,0 Гц, 1H), 8,60 (d, J = 2,0 Гц, 1H), 8,44 (s, 1H), 8,12 (s, 1H), 7,80 (s, 1H), 7,45 (d, J = 8,4 Гц, 1H), 7,36 (s, 1H), 7,18 (d, J = 8,4 Гц, 1H), 7,07 (s, 1H), 6,60 (s, 1H), 4,54 (s, 2H), 4,42 (t, J = 8,0 Гц, 2H), 4,14 (s, 2H), 4,02 (t, J = 8,0 Гц, 2H), 3,64 (t, J = 5,6 Гц, 2H), 2,86 (t, J = 5,6 Гц, 2H).
Пример 56.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 120 ºC в течение 3 ч, Промежуточное соединение 1 (100 мг) и Промежуточное соединение 28 (156 мг) превращали в неочищенный продукт, который очищали с помощью преп-ВЭЖХ с получением Примера 56 (60 мг, 0,124 ммоль, выход 28%) в виде белого твердого вещества. MS (ESI): вычисленная масса для C9H9BrO2 485,19, m/z найдено 485,9 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 7,51 ppm (d, J = 8,4 Гц, 2H), 7,36 (d, J = 8,0 Гц, 1H), 7,27 (d, J = 8,7 Гц, 3H), 7,22 (d, J = 8,1 Гц, 1H), 6,96 (s, 1H), 6,93 (s, 1H), 4,63 (s, 2H), 4,47 (t, J = 8,2 Гц, 2H), 4,09 (t, J = 8,0 Гц, 2H), 4,00 (s, 2H), 3,72 (t, J = 5,9 Гц, 2H), 3,03 (t, J = 5,8 Гц, 2H), 2,33 (s, 3H), 2,22 (s, 3H).
Пример 57.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 120 ºC в течение 2 ч, Промежуточное соединение 29 (135 мг) и Промежуточное соединение 28 (100 мг) превращали в неочищенный продукт, который очищали с помощью преп-ТСХ с получением Примера 57 (35 мг, 35,3 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C26H27N5O3S 489,59, m/z найдено 489,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,50-7,45 (m, 3H), 7,32 (s, 1H), 7,25 (d, J = 8,0 Гц, 2H), 7,10 (d, J = 8,4 Гц, 1H), 6,97 (s, 1H), 4,51-4,48 (m, 2H), 4,42 (t, J = 8,0 Гц, 2H), 4,03 (t, J = 8,4 Гц, 2H), 3,96 (s, 2H), 3,74 (t, J = 8,0 Гц, 2H), 3,62 (t, J = 5,6 Гц, 2H), 3,42 (t, J = 8,4 Гц, 2H), 2,82 (t, J = 5,6 Гц, 2H), 2,75 (s, 3H).
Пример 58.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 120 ºC в течение 3 ч, Промежуточное соединение 27 (300 мг) и Промежуточное соединение 28 (307 мг) превращали в неочищенный продукт, который очищали с помощью преп-ТСХ с получением Примера 58 (38 мг, 8% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C26H28N4O3S, 476,19. m/z найдено 476,9 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,49 (d, J = 8,3 Гц, 2H), 7,26 (d, J = 8,4 Гц, 2H), 7,03 (d, J = 8,1 Гц, 1H), 6,97 (s, 1H), 6,80 (d, J = 10,1 Гц, 2H), 4,44 (dd, J = 17,0, 9,1 Гц, 4H), 4,04 (t, J = 7,9 Гц, 2H), 3,97 (s, 2H), 3,78 - 3,64 (m, 4H), 3,60 (t, J = 5,7 Гц, 2H), 3,06 (d, J = 5,1 Гц, 4H), 2,78 (t, J = 5,7 Гц, 2H).
Пример 59.
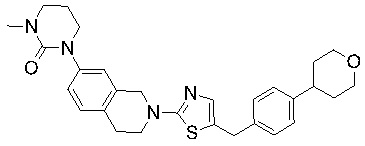
1. В соответствии с процедурой, описанной для Примера 1, Промежуточное соединение 35 (351 мг) и Промежуточное соединение 30 (250 мг) нагревали при 130 ºC в течение 2 ч. Смесь охлаждали до КТ, разбавляли смесь ДХМ/MeOH 20/1 (20 мл), фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением Примера 59 (37,5 мг, 9% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C29H34N4O2S 502,68, m/z найдено 502,9 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,20 - 7,16 (m, 4H), 7,10 - 7,04 (m, 3H), 6,98 (s, 1H), 4,49 (s, 2H), 3,95 - 3,92 (m, 4H), 3,64 - 3,59 (m, 4H), 3,44 - 3,38 (m, 2H), 3,32 - 3,31 (m, 2H), 2,84 (s, 5H), 2,76 - 2,68 (m, 1H), 2,04 - 1,98 (m, 2H), 1,67 - 1,61 (m, 4H).
Пример 60.
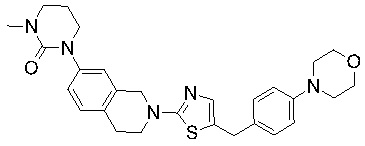
1. В соответствии с процедурой, описанной для Примера 1, с использованием Cs2CO3 (313 мг) вместо K2CO3 и нагревания смеси при 130 ºC в течение 4 ч, Промежуточное соединение 35 (100 мг) и Промежуточное соединение 24 (106 мг) превращали в неочищенный продукт, который очищали с помощью преп-ТСХ с получением Примера 60 (20 мг, 13% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса C28H33N5O2S 503,24,1H ЯМР (400 МГц, ДМСО) δ ppm 7,21 (s, 1H), 7,13 (dd, J = 9,4, 5,6 Гц, 5H), 6,92 (d, J = 8,5 Гц, 2H), 4,56 (s, 2H), 3,93 (s, 2H), 3,79 - 3,70 (m, 4H), 3,68 (t, J = 5,9 Гц, 2H), 3,64 - 3,54 (m, 2H), 3,33 (t, J = 6,0 Гц, 2H), 3,17 - 3,00 (m, 4H), 2,91 (t, J = 5,7 Гц, 2H), 2,85 (s, 3H), 2,18 - 1,93 (m, 2H).
Пример 61.
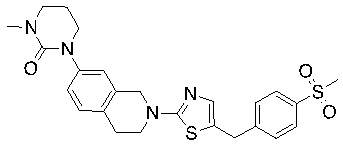
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 100 ºC в течение 3 ч, Промежуточное соединение 35 (170 мг) и Промежуточное соединение 31 (200 мг) превращали в неочищенный продукт, который очищали с помощью преп-ТСХ с получением Примера 61 (27 мг, 8% выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C25H28N4O3S2, 496,16. m/z найдено 497,0 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,87 (d, J = 8,0 Гц, 2H), 7,52 (d, J = 8,2 Гц, 2H), 7,08 (dd, J = 15,4, 7,2 Гц, 4H), 4,51 (s, 2H), 4,14 (s, 2H), 3,62 (dt, J = 15,4, 5,6 Гц, 4H), 3,32 (d, J = 9,1 Гц, 2H), 3,19 (s, 3H), 2,92 - 2,77 (m, 5H), 2,08 - 1,95 (m, 2H).
Пример 62.
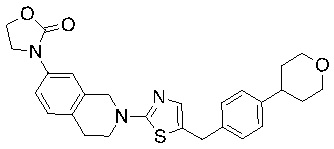
1. В соответствии с процедурой, описанной для Примера 59, за исключением того, что смесь нагревали при 130 ºC в течение 4,5 ч, Промежуточное соединение 20 (204 мг) и Промежуточное соединение 30 (150 мг) превращали в Примера 62 (56 мг, 23% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C27H29N3O3S 475,61, m/z найдено 475,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,45 (d, J = 8,1 Гц, 1H), 7,37 (s, 1H), 7,20 - 7,17 (m, 5H), 6,99 (s, 1H), 4,54 (s, 2H), 4,45 - 4,41 (m, 2H), 4,05 - 4,01 (m, 2H), 3,95 - 3,92 (m, 4H), 3,64 (t, J = 5,7 Примера 61 Гц, 2H), 3,45 - 3,39 (m, 2H), 2,86 (t, J = 5,7 Гц, 2H), 2,75 - 2,71 (m, 1H), 1,67 - 1,61 (m, 4H).
Пример 63.
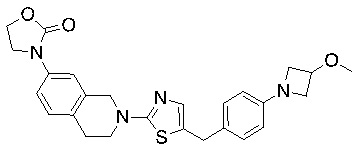
1. В соответствии с процедурой, описанной для Примера 31, с использованием Cs2CO3 (833 мг) вместо K2CO3 и продувки сосуда 3x N2, Промежуточное соединение 22 (200 мг) и гидрохлорид 3-метоксиазетидина (159 мг) нагревали при 100 ºC в течение 5 часов и смесь охлаждали до КТ, разбавляли смесью (ДХМ/MeOH, 10/1, 20 мл), фильтровали и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с получением Примера 63 (35,7 мг, 17,7% выход) в виде желто-белого твердого вещества. MS (ESI): вычисленная масса для C26H28N4O3S 476,60, m/z найдено 476,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО) δ ppm 7,45 (d, J = 8,3 Гц, 1H), 7,37 (s, 1H), 7,19 (d, J = 8,7 Гц, 1H), 7,04 (d, J = 8,1 Гц, 2H), 6,93 (s, 1H), 6,38 (d, J = 8,2 Гц, 2H), 4,53 (s, 2H), 4,43 (t, J = 7,8 Гц, 2H), 4,32 - 4,27 (m, 1H), 4,05 - 3,99 (m, 4H), 3,85 (s, 2H), 3,64 (t, J = 5,8 Гц, 2H), 3,55 - 3,52 (m, 2H), 3,23 (s, 3H), 2,86 (t, J = 5,8 Гц, 2H).
Пример 64.
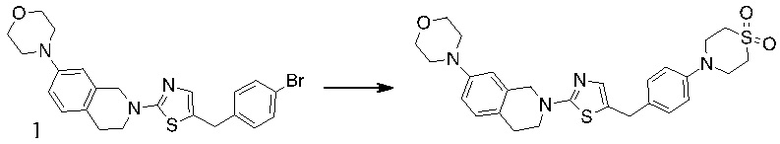
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что смесь нагревали при 130 oC в течение 3 ч, Промежуточное соединение 27 (900 мг) и Промежуточное соединение 6 (1,1 г) превращали в 1 (400 мг, 23 % выход) в виде желтого твердого вещества. MS (ESI): вычисленная масса для C23H24BrN3OS 469,08, m/z найдено 469,7 [M+H]+.
2. Смесь 1 (150 мг, 0,32 ммоль), тиоморфолина 1,1-диоксида (180 мг, 1,3 ммоль), Pd2(dba)3 (90 мг, 0,1 ммоль), SPhos (80 мг, 0,2 ммоль) и трет-BuONa (170 мг, 1,8 ммоль) в высушенном 1,4-диоксане (25 мл) перемешивали при 100 oC в течение ночи. Реакционную смесь охлаждали до КТ, фильтровали и фильтрат концентрировали с получением остатка, который очищали с помощью преп-ТСХ с получением Примера 64 (35 мг, 21%) в виде белого твердого вещества. Вычисленная масса для C27H32N4O3S2 524,19, m/z найдено 524,8 [M+H]+, 1H ЯМР (400 МГц, ДМСО) δ ppm 7,08 (d, J = 8,4 Гц, 2H), 7,00 (d, J = 8,2 Гц, 1H), 6,94 (d, J = 5,5 Гц, 3H), 6,77 (d, J = 9,1 Гц, 2H), 4,45 (s, 2H), 3,87 (s, 2H), 3,69 (m, 8H), 3,58 (t, J = 5,7 Гц, 2H), 3,04 (m, 8H), 2,76 (t, J = 5,6 Гц, 1H).
Пример 65.
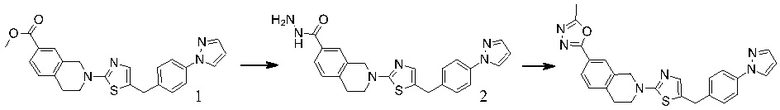
1. Смесь Промежуточного соединения 5 (200 мг, 0,44 ммоль), TEA (134 мг, 1,33 ммоль) и Pd(dppf)Cl2 (32,4 мг, 0,044 ммоль) в MeOH (2 мл) и MeCN (1 мл) в бомбе в атмосфере CO нагревали при 100oC в течение 24 ч. Смесь фильтровали через целит, фильтрационный осадок промывали EA и объединенный фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 1 (140 мг, 73,4 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C24H22N4O2S 430,53, m/z найдено 430,8 [M+H]+.
2. Смесь 1 (140 мг, 0,33 ммоль), N2H4⋅H2O (130 мг, 2,60 ммоль) в EtOH (1 мл) и ТГФ (1 мл) помещали в бомбу, которую нагревали при 80 oC в течение 5 дней. Смесь концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2 (125 мг, 89,3 % выход) в виде белого твердого вещества. MS (ESI): вычисленная масса для C23H22N6OS 430,53, m/z найдено 430,8 [M+H]+.
3. Смесь 2 (120 мг, 0,28 ммоль), триметилортоацетата (132 мг, 0,70 ммоль), NH4Cl (4,5 мг, 0,08 ммоль) в EtOH (1 мл) и ТГФ (1 мл) помещали в герметичную пробирку, которую нагревали при 80 oC в течение 18 часов. Смесь охлаждали и добавляли триметилортоацетат (132 мг, 0,70 ммоль) и NH4Cl (4,5 мг, 0,08 ммоль) и смесь нагревали при 80 oC в течение 18 часов. Смесь охлаждали и добавляли триметилортоацетат (132 мг, 0,70 ммоль) и NH4Cl (4,5 мг, 0,08 ммоль) и смесь нагревали при 80 oC в течение 18 часов. Смесь охлаждали и концентрировали. Полученный остаток очищали с помощью преп-ВЭЖХ с получением Примера 65 (30 мг, 23,7% выход, 96,2% чистота 214 нм) в виде белого твердого вещества. MS (ESI): вычисленная масса для C25H22N6OS 454,55 m/z найдено 454,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,46 (d, J=2,2, 1H), 7,85 (s, 1H), 7,80 - 7,76 (m, 3H), 7,73 (d, J=1,5, 1H), 7,40 (d, J=8,0, 1H), 7,36 (d, J=8,6, 2H), 7,04 (s, 1H), 6,53 (dd, J=2,4, 1,8, 1H), 4,65 (s, 2H), 4,05 (s, 2H), 3,70 (t, J=5,9, 2H), 2,98 (t, J=5,8, 2H), 2,58 (s, 3H).
Пример 66.
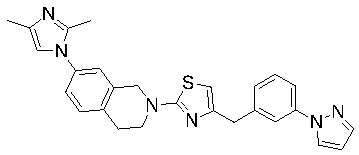
1. Смесь гидрохлоридной соли Промежуточного соединения 1 (55 мг, 0,21 ммоль), Промежуточного соединения 34 (57,7 мг, 0,21 ммоль), Pd2(dba)3 (27,5 мг, 0,03ммоль), SPhos (24,7 мг, 0,06 ммоль) и трет-BuOK (84 мг, 0,75 ммоль) в сухом диоксане (4 мл) перемешивали при 90 °C в течение 4 ч в атмосфере N2. Смесь охлаждали до КТ, выливали в воду и экстрагировали EtOAc. Объединенные органические слои промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью преп-ВЭЖХ с получением Примера 66 (1,9 мг, 2% выход) в виде белого твердого вещества. Вычислен. масса: 467,2; Найденная масса: 468,3 [M + H] +. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,08 (3H, s), 2,23 (3H, s), 2,96 (2H, t, J = 6,4 Гц),, 3,68 (2H, t, J = 6,0 Гц), 3,95 (2H, s), 4,61 (2H, s), 6,29 (1H, d, J = 1,6 Гц), 6,38 (1H, s), 6,93 (1H, s), 7,22 (1H, d, J = 7,6 Гц), 7,31-7,34 (2H, m), 7,42-7,44 (2H, m), 7,50-7,60 (5H, m).
Пример 67.
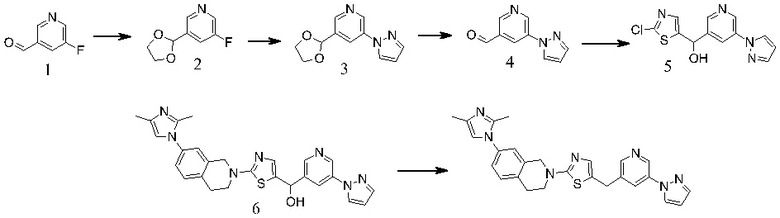
1. Смесь 1 (2,00 г, 16,0 ммоль), этандиола (1,12 г, 18,0 ммоль) и TsOH (100 мг, 0,53 ммоль) в бензоле (10 мл) нагревали с обратным холодильником в течение 18 ч и концентрировали. Остаток растворяли в Et2O и раствор промывали 10% NaHCO3. Органическую смесь сушили над Na2SO4 и фильтровали и фильтрат выпаривали с получением 2 (1,45 г, 54%) в виде бесцветного масла.
2. К раствору 1H-пиразола (1,16 г, 17,0 ммоль) в безводном ДМФА (10,0 мл) добавляли NaH (60% дисперсия в минеральном масле, 3,50 г, 87,0 ммоль) в атмосфере N2, и полученную смесь перемешивали при 60 °C в течение 2 ч. Затем добавляли по каплям раствор 2 (1,45 г, 8,57 ммоль) в ДМФА (3,0 мл) и полученную смесь перемешивали при 80 °C в течение 3 ч. Смесь охлаждали до КТ, выливали в воду и экстрагировали EtOAc. Объединенные органические экстракты промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 3 (862 мг, 58% выход) в виде бесцветного масла.
3. Смесь 3 (862 мг, 3,97 ммоль), CuSO4 (64,0 мг, 0,40 ммоль), H2O (5,00 мл) и HCO2H (20,0 мл) перемешивали при 80 °C в течение 4 ч. Смесь охлаждали, выливали в воду и подщелачивали водным K2CO3 до pH 8. Смесь экстрагировали EtOAc. Объединенные органические экстракты промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 4 (320 мг, 46% выход) в виде бесцветного масла.
4. К раствору 2-хлортиазола (221 мг, 1,85 ммоль) в сухом ТГФ (10 мл) при -78 ºC в атмосфере N2 по каплям добавляли н-BuLi (2,5 M, 0,8 мл, 2,00 ммоль). Через 1 ч добавляли по каплям раствор 4 (320 мг, 1,85 ммоль) в сухом ТГФ (3 мл). Полученный раствор медленно нагревали до КТ. Смесь разбавляли раствором NH4Cl и экстрагировали EtOAc. Органические экстракты концентрировали с получением остатка, который очищали с помощью хроматографии на силикагеле с получением 5 (173 мг, 32% выход) в виде желтого масла.
5. Смесь 5 (388 мг, 1,33 ммоль), гидрохлоридной соли Промежуточного соединения 1 (350 мг, 1,33 ммоль), Pd2(dba)3 (91,5 мг, 0,1 ммоль), SPhos (82,3 мг, 0,2 ммоль) и трет-BuOK (446 мг, 3,99 ммоль) в сухом диоксане (6 мл) перемешивали при 90 °C в течение 4 ч в атмосфере N2. Смесь охлаждали до КТ, выливали в воду и экстрагировали EtOAc. Объединенные органические экстракты промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали с помощью преп-ВЭЖХ с получением 6 (170 мг, 26% выход) в виде белого твердого вещества.
6. К раствору 6 (170 мг, 0,35 ммоль) в TFA (10 мл) добавляли TES (3 мл), и полученную смесь перемешивали при 100 ºC в течение 1 ч. Смесь концентрировали и остаток очищали с помощью преп-ВЭЖХ с получением Примера 67 (5,20 мг, 3% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 2,08 (3H, s), 2,22 (3H, s), 2,94 (2H, t, J = 5,6 Гц),, 3,67 (2H, t, J = 5,6 Гц), 4,15 (2H, s), 4,60 (2H, s), 6,60 (1H, t, J = 2,0 Гц), 6,91 (1H, s), 7,08 (1H, s), 7,22 (1H, s), 7,30 (2H, d, J = 8,0 Гц), 7,81 (1H, d, J = 1,2 Гц), 8,13 (1H, s), 8,45 (1H, d, J = 1,6 Гц), 8,60 (1H, d, J = 2,0 Гц), 8,97 (1H, d, J = 2,4 Гц).
Пример 68.
1. В соответствии с процедурой, описанной для Примера 1, за исключением того, что Cs2CO3 (1,37 мг) использовали вместо K2CO3 и смесь нагревали при 110 ºC в течение 2 ч, Промежуточное соединение 6 (400 мг) и Промежуточное соединение 35 (580 мг) превращали в 1 (600 мг, 85%) в виде коричневого масла. MS (ESI): вычисленная масса для C24H25BrN4OS 496,09. m/z найдено 496,8 [M+H]+.
2. Смесь 1 (500 мг, 1 ммоль), тиоморфолина 1,1-диоксида (405 мг, 3 ммоль), Pd(dba)2 (288 мг, 0,5 ммоль), SPhos (205 мг, 0,5 ммоль) и трет-BuONa (290 мг, 3,0 ммоль) в высушенном 1,4-диоксане (30 мл) перемешивали при 100 oC в течение ночи. Смесь охлаждали до КТ, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле. Полученное вещество очищали с помощью преп-ТСХ с получением Примера 68 (50 мг, 9%) в виде белого твердого вещества. Вычисленная масса для C28H33N5O3S2 551,2, m/z найдено 551,72 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,17 - 7,03 (m, 5H), 7,00 - 6,92 (m, 3H), 4,49 (s, 2H), 3,90 (s, 2H), 3,77 - 3,69 (m, 4H), 3,62 (m, 4H), 3,19 - 3,03 (m, 4H), 2,86 (m, 5H), 2,07 - 1,94 (m, 2H), 1,24 (s, 2H).
Оценка противовирусной активности в отношении цитомегаловируса человека (HCMV)
Чтобы оценить противовирусную активность, некоторые соединения были протестированы в отношении цитомегаловируса человека (HCMV) in vitro. Клетки MRC5 человека выращивали до конфлюэнтности (~ 1,0 × 10^4 клеток/лунку) в 96-луночном планшете в модифицированной по способу Дульбекко среде Игла (DMEM) с добавлением 10% фетальной бычьей сыворотки (FBS), 2 мМ L-глутамина, 0,1 мМ заменимых аминокислот, 10 мМ HEPES и 100 ед/мл каждого из пенициллина и стрептомицина и инфицировали вариантом HCMV, экспрессирующим pUL99, меченый mCherry (продукт позднего вирусного гена UL99), с множественностью 0,01 инфекционной единицы (IU) на клетку. Анализы были выполнены в трех повторностях. Через час среду клеток заменяли свежей средой, содержащей указанные соединения в концентрациях 25, 12,5, 6,25, 3,13, 1,56, 0,78, 0,39 мкМ или носитель, в котором растворяли соединения (ДМСО). Конечная концентрация ДМСО составляла 0,5% в каждой обработке. Выход вируса в культуре определяли через 7 дней после заражения путем количественного определения флуоресцентных (mCherry-положительных) клеток в каждой лунке с помощью флуоресцентной микроскопии. Результаты были представлены с использованием CDD Vault (CDD Vault была разработана Collaborative Drug Discovery, Inc., 1633 Bayshore Hwy, Suite 342, Burlingame, CA 94010) для расчета IC50. Результаты, полученные в отношении соединений, протестированных с помощью данного анализа, представлены в таблице. 1.
Оценка противовирусной активности в отношении гриппа
Чтобы оценить противовирусную активность, некоторые соединения были протестированы в отношении адаптированного к мышам гриппа человека (PR8) in vitro. Клетки собаки MDCK выращивали до конфлюэнтности (~1,0×104 клеток/лунку) в 96-луночном планшете в минимальной основной среде Игла (EMEM), дополненной 10% фетальной бычьей сывороткой (FBS) и 100 ед/мл каждого из пенициллина и стрептомицина. Клетки промывали в 1xPBS и инфицировали вариантом PR8, экспрессирующим mCherry, который расположен по ходу транскрипции и отделен сайтом ауторасщепления 2A от белка NS-1, с множественностью 0,01 инфекционной единицы (IU) на клетку в бессывороточной EMEM. Анализы были выполнены в трех повторностях. Через час вирусосодержащую среду в клетках заменяли свежей полной средой, содержащей указанные соединения в концентрациях 25, 12,5, 6,25, 3,13, 1,56, 0,78, 0,39 мкМ или носитель, в котором растворены соединения (ДМСО), и дополненной 2,5 мкг/мл трипсина, обработанного TPCK. Конечная концентрация ДМСО составляла 0,5% в каждой обработке. Выход вируса в культуре определяли через 3 дня после заражения путем количественного определения флуоресцентных (mCherry-положительных) клеток в каждой лунке с помощью флуоресцентной микроскопии. Результаты были представлены с использованием CDD Vault (CDD Vault была разработана Collaborative Drug Discovery, Inc., 1633 Bayshore Hwy, Suite 342, Burlingame, CA 94010) для расчета IC50. Результаты, полученные в отношении соединений, протестированных с помощью этого анализа, представлены в таблице 1.
Таблица 1
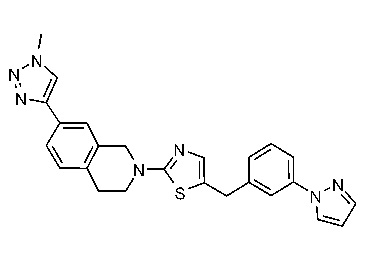
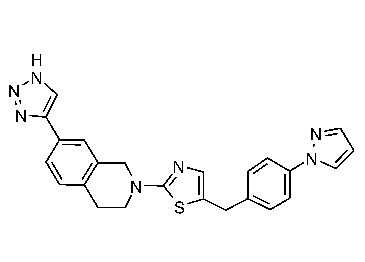
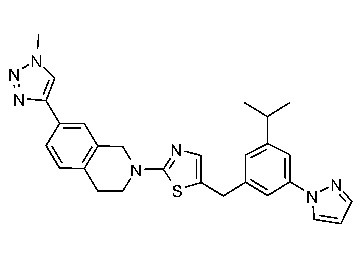
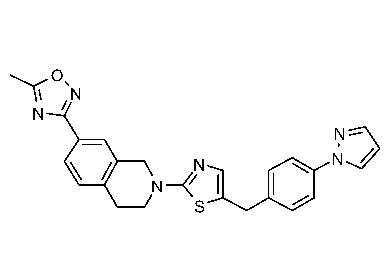
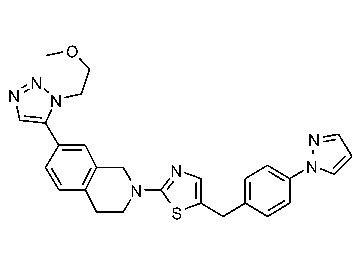
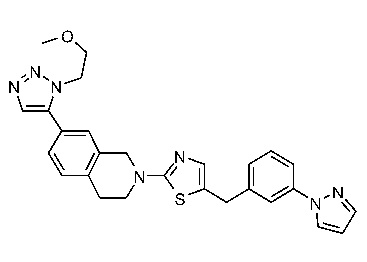
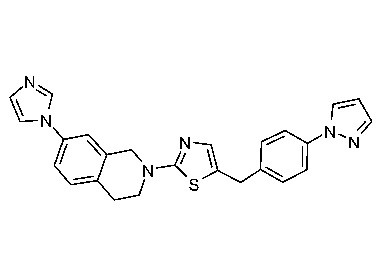
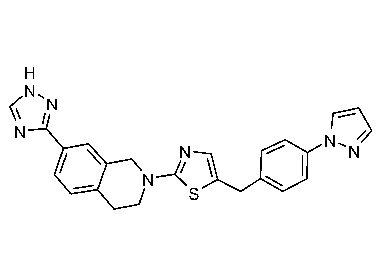
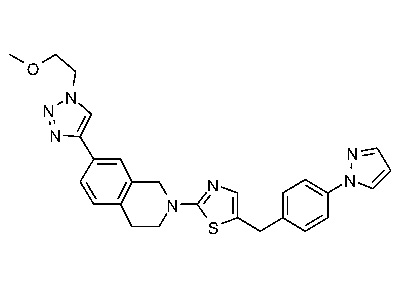
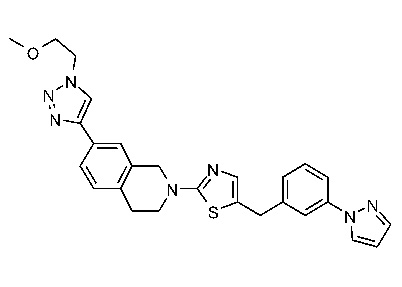
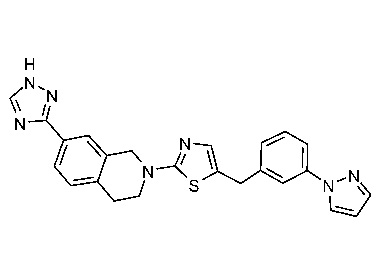
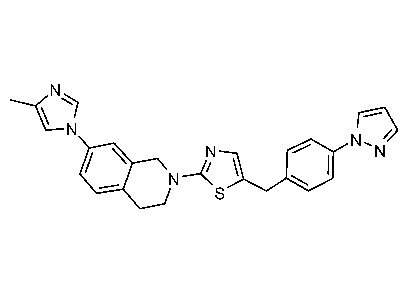
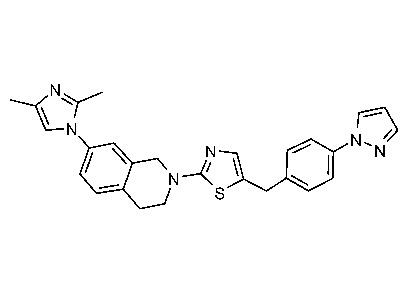
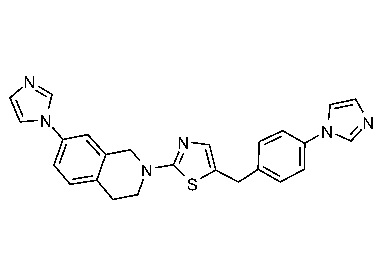
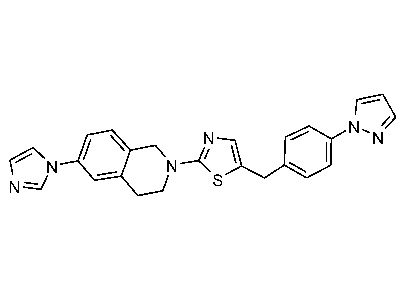
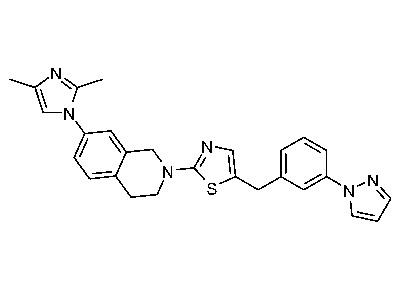
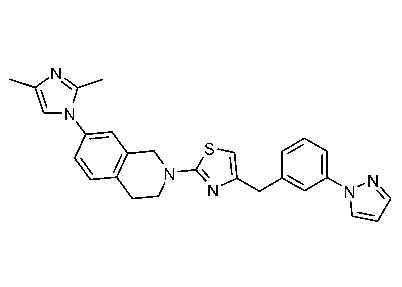
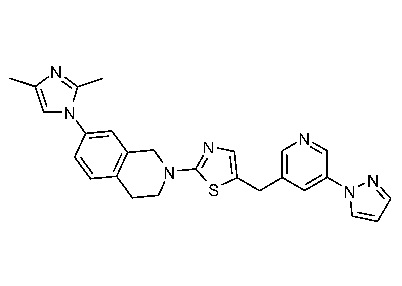
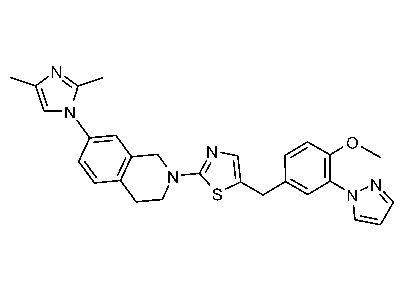
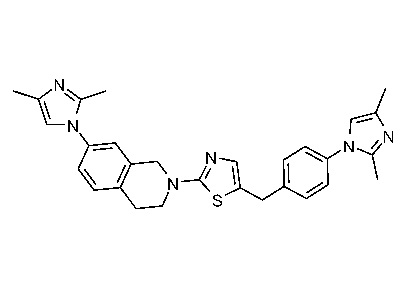
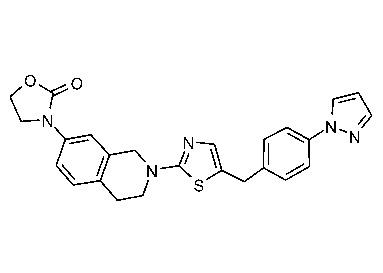
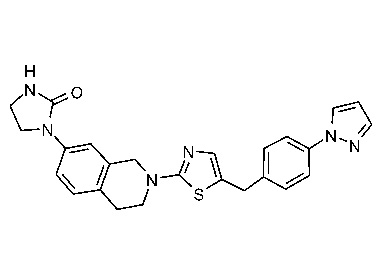
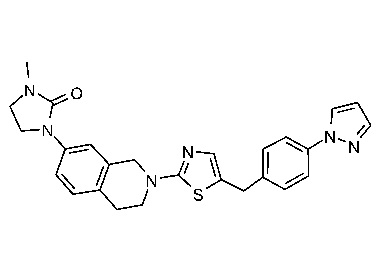
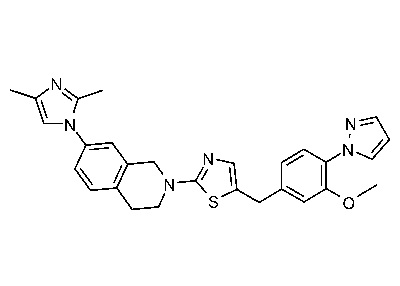
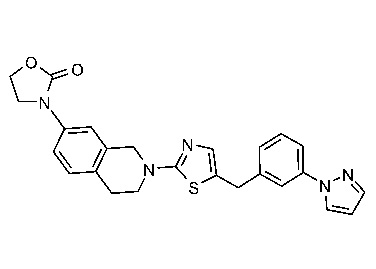
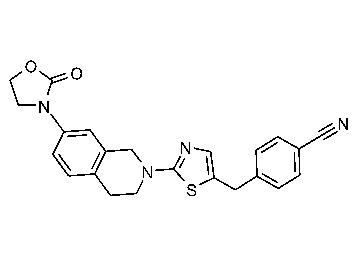
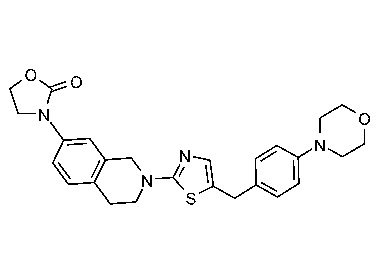
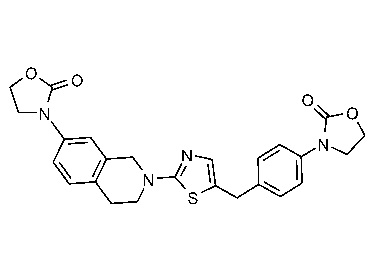
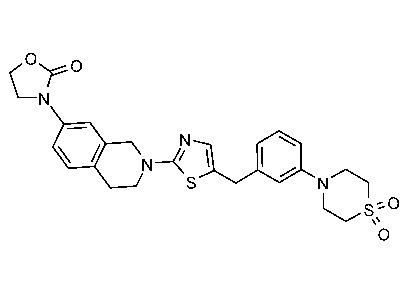
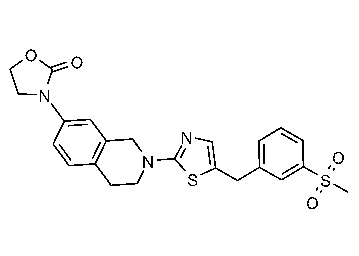
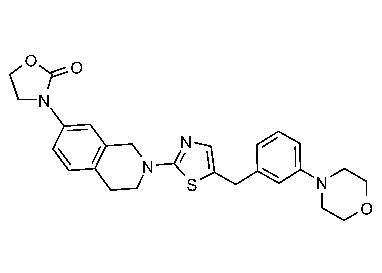
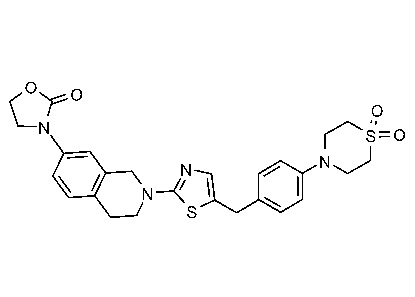
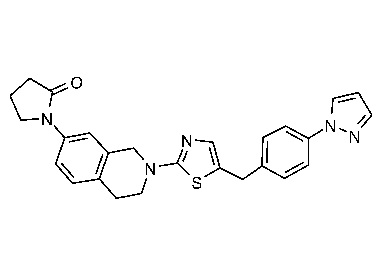
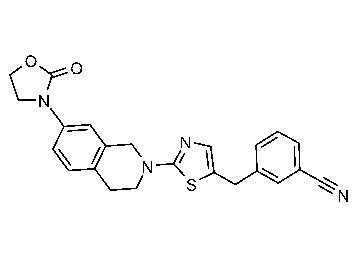
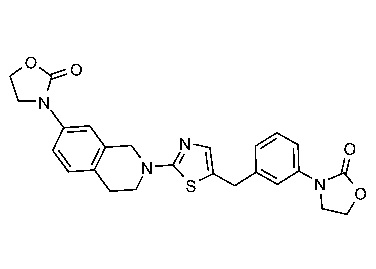
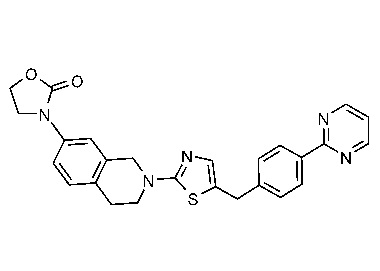
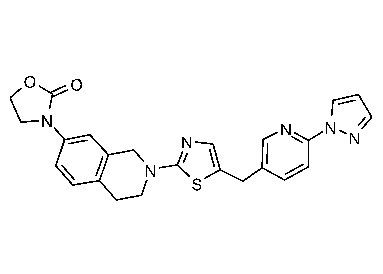
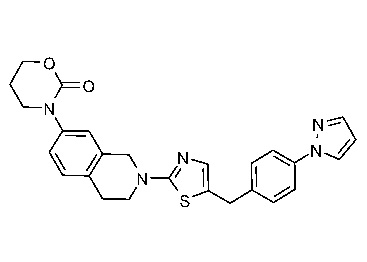
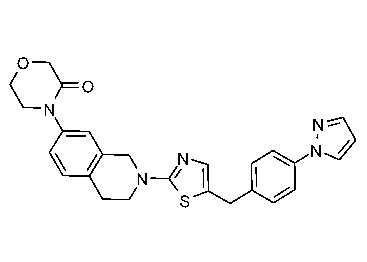
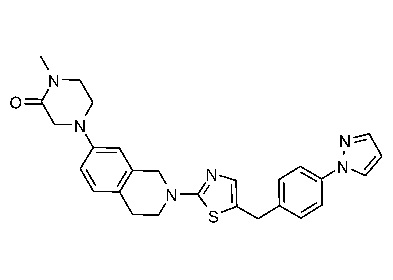
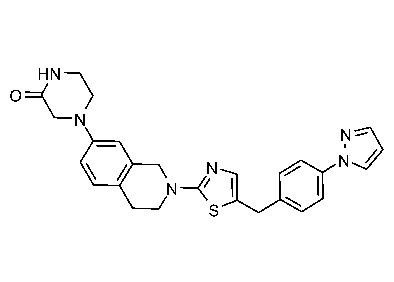
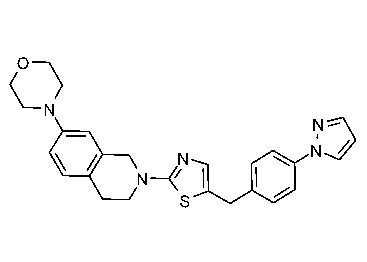
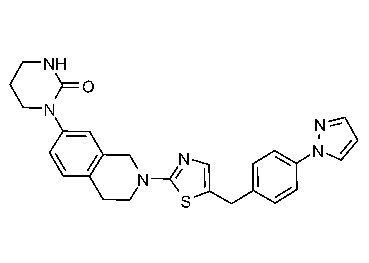
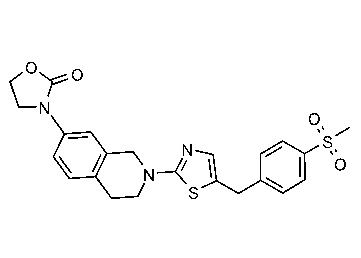
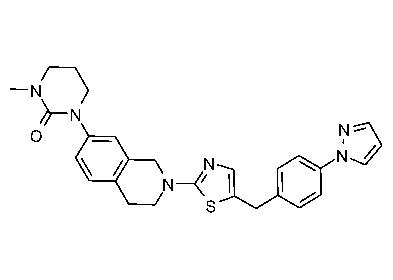
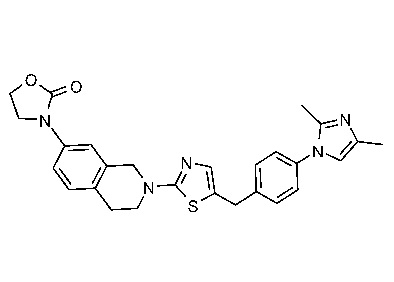
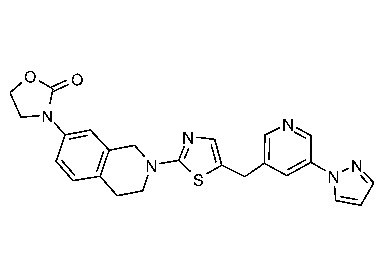
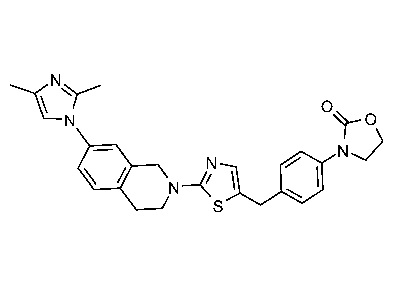
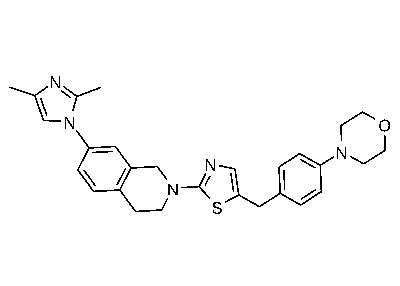
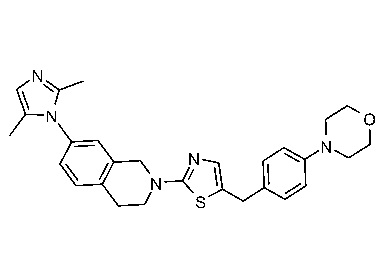
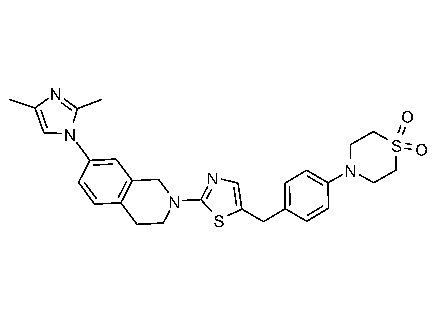
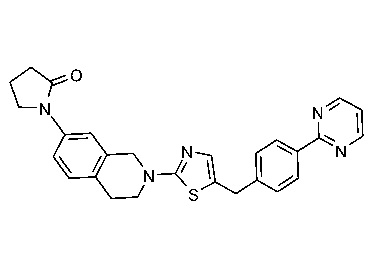
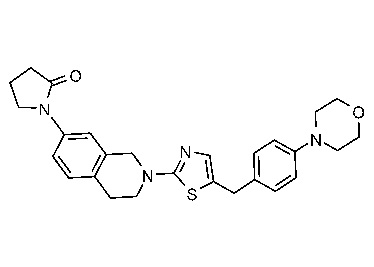
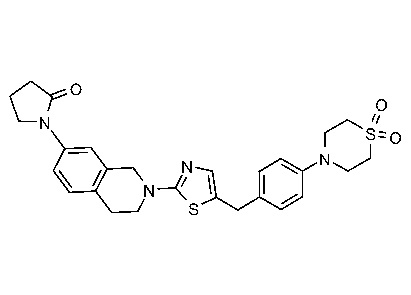
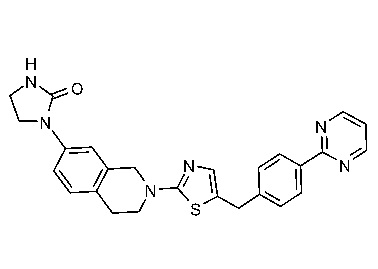
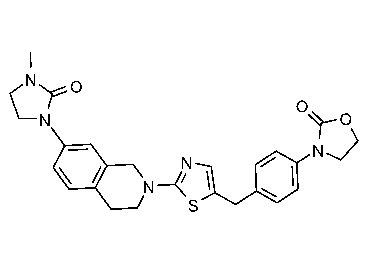

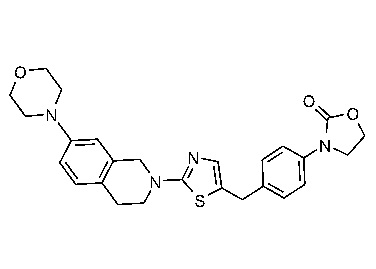
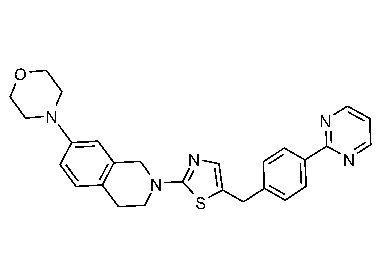
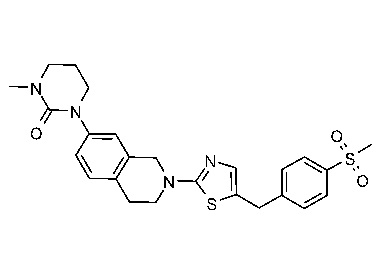
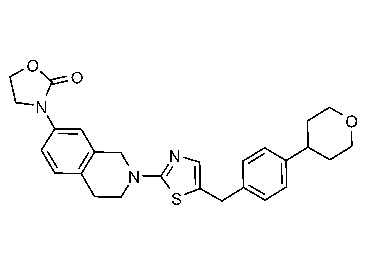
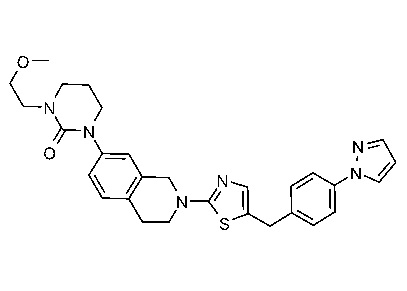
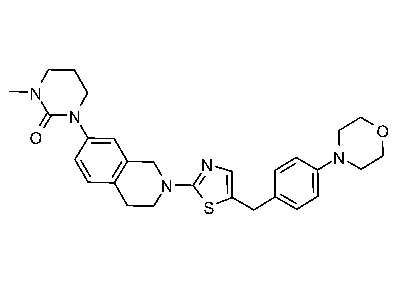
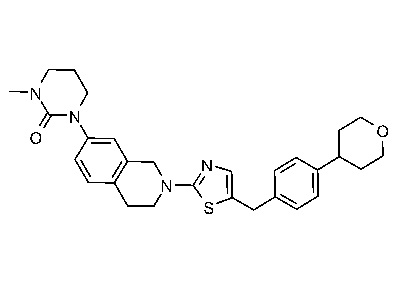
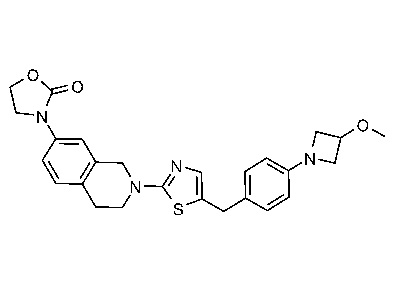
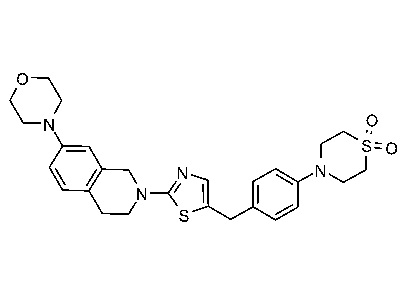

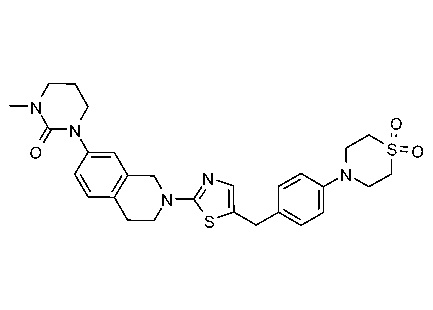
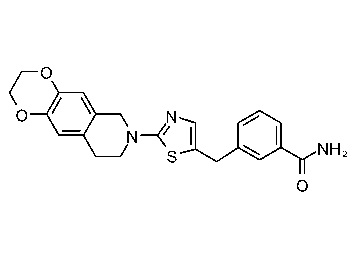
ND = не выполнено.
Некоторые соединения в таблице 1 были протестированы дополнительно, и было обнаружено, что они ингибируют репликацию RSV, штамма вируса Зика MR776 и вируса ВК в культуре клеток.
ДРУГИЕ ВАРИАНТЫ РЕАЛИЗАЦИИ
Следует понимать, что, хотя изобретение было описано в сочетании с его подробным описанием, вышеприведенное описание предназначено для иллюстрации, а не для ограничения объема изобретения, который определяется объемом прилагаемой формулы изобретения. Другие аспекты, преимущества и модификации находятся в пределах объема следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| ПРОИЗВОДНОЕ С СОЧЛЕНЕННЫМИ КОЛЬЦАМИ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2683566C1 |
| ИНГИБИТОРЫ ГЛИКОЛАТОКСИДАЗЫ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2805308C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2811969C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ MAT2A И СПОСОБЫ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2809987C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
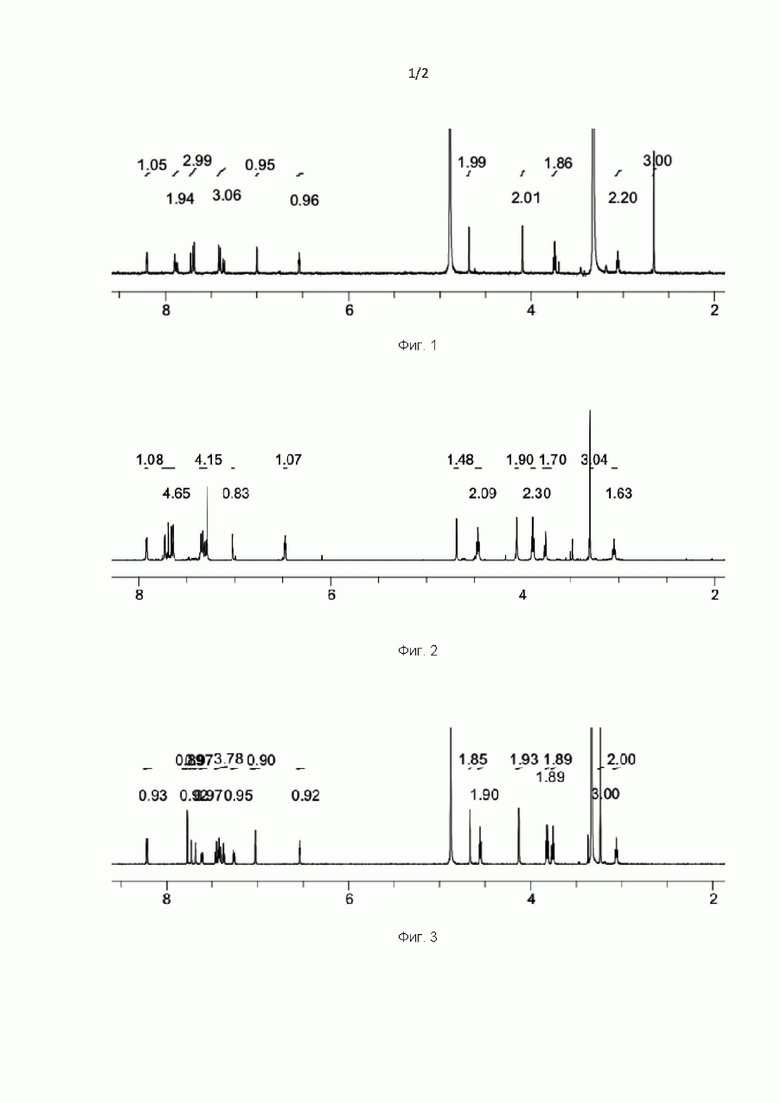
Изобретение относится к соединению формулы I и его фармацевтически приемлемой соли или сольвату, фармацевтической композиции на их основе, а также способу лечения или предотвращения вирусной инфекции и способу ингибирования продуцирования вируса с их использованием. В общей формуле I один из X1 и X2 представляет собой N, и другой представляет собой S; X3 и X4 независимо выбраны из C и N; и когда X3 представляет собой C, он необязательно замещен изопропилом; один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с 1-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из: =O, C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12; или R1 и R2 вместе образуют незамещенный или 6-членный циклоалкил с 2 кольцевыми гетероатомами, представляющими собой O; R3 выбран из H, -C≡N, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), и кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 1 или 2 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, C1-6 алкила с прямой или разветвленной цепью, и C1-6 алкокси с прямой или разветвленной цепью; R4 выбран из H, -C≡N, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -C(O)NH2, кольцевой структуры, содержащей 5-членный арил или 5- или 6-членный циклоалкил с 2 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, представляющими собой =O; R5 представляет собой H; R6 представляет собой H; и R12 независимо, в каждом случае, выбран из C1-4 алкила с прямой или разветвленной цепью. 4 н. и 18 з.п. ф-лы, 6 ил., 2 табл., 68 пр.
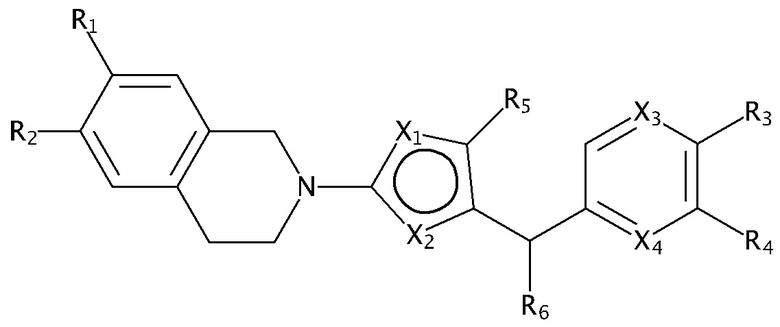
1. Соединение формулы I:
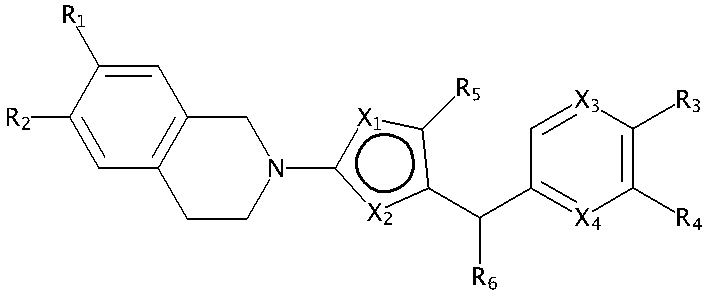 ,
,
где:
один из X1 и X2 представляет собой N, и другой представляет собой S;
X3 и X4 независимо выбраны из C и N; и когда X3 представляет собой C, он необязательно замещен изопропилом;
один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с 1-3 кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из:
=O, C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12;
или R1 и R2 вместе образуют незамещенный или 6-членный циклоалкил с 2 кольцевыми гетероатомами, представляющими собой O;
R3 выбран из H, -C≡N, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), и кольцевой структуры, содержащей 5- или 6-членный арил или 4-, 5- или 6-членный циклоалкил с 1 или 2 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, независимо выбранными из =O, C1-6 алкила с прямой или разветвленной цепью, и C1-6 алкокси с прямой или разветвленной цепью;
R4 выбран из H, -C≡N, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), -C(O)NH2, кольцевой структуры, содержащей 5-членный арил или 5- или 6-членный циклоалкил с 2 кольцевыми гетероатомами, независимо выбранными из N, O и S, и замещенный 0-2 группами, представляющими собой =O;
при условии, что:
по меньшей мере один из R3 и R4 выбран из группы, состоящей из: H, -C≡N, C1-4 алкокси с прямой или разветвленной цепью, -SO2(C1-6 алкил), и -C(O)NH2, и
оба R3 и R4 не представляют собой H;
R5 представляет собой H;
R6 представляет собой H;
и
R12 независимо, в каждом случае, выбран из C1-4 алкила с прямой или разветвленной цепью;
или его фармацевтически приемлемая соль или сольват.
2. Соединение по п. 1, отличающееся тем, что:
R3 выбран из группы, состоящей из:
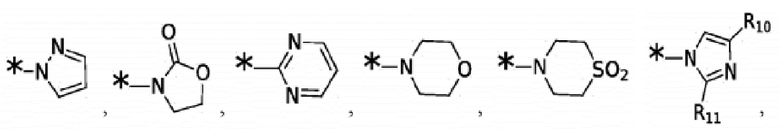
и -SO2(C1-6 алкил);
где:
R10 и R11 независимо выбраны из группы, состоящей из: H и C1-4 алкила с прямой или разветвленной цепью.
3. Соединение по п. 1, отличающееся тем, что:
R4 выбран из группы, состоящей из:
 и -SO2(C1-6 алкил).
и -SO2(C1-6 алкил).
4. Соединение по п. 1, отличающееся тем, что:
один из R1 и R2 представляет собой H, и другой представляет собой 5- или 6-членный арил или циклоалкил с по меньшей мере одним кольцевым гетероатомом N и 0-2 дополнительными кольцевыми гетероатомами, независимо выбранными из N и O, и замещенный 0-2 группами, независимо выбранными из:
=O, и C1-6 алкила с прямой или разветвленной цепью, необязательно замещенного -OR12.
5. Соединение по п. 4, отличающееся тем, что соединение формулы I представляет собой:
 .
.
6. Соединение по п. 4, отличающееся тем, что:
R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R10 и R11 независимо выбраны из группы, состоящей из: H, и C1-4 алкила с прямой или разветвленной цепью.
7. Соединение по п. 6, отличающееся тем, что соединение формулы I выбрано из группы, состоящей из:
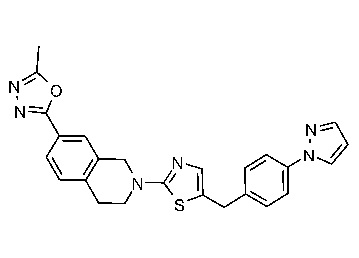
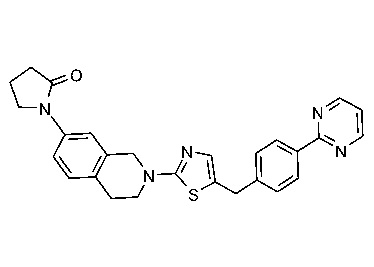 ,
, 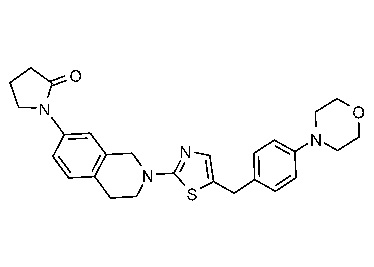 ,
, 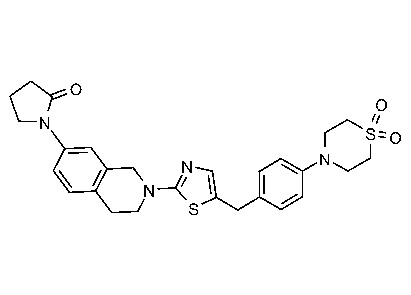 и
и
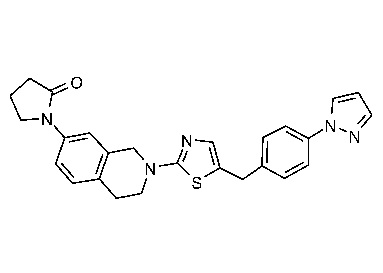 .
.
8. Соединение по п. 4, отличающееся тем, что:
R4 выбран из группы, состоящей из:
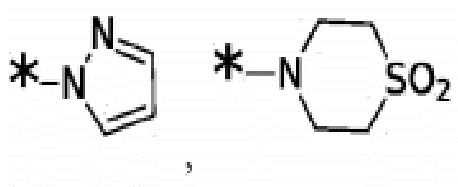 и -SO2(C1-6 алкил).
и -SO2(C1-6 алкил).
9. Соединение по п. 4, отличающееся тем, что:
один из R1 и R2 представляет собой H, и другой выбран из группы, состоящей из:
 ,
, 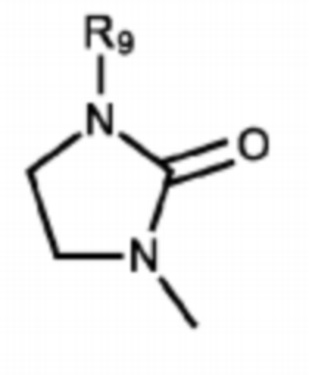 ,
, 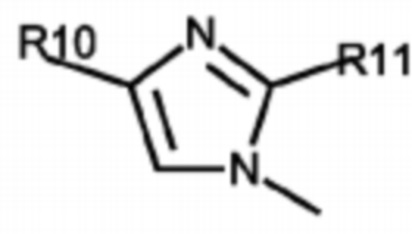 и
и  ,
,
где:
R9 выбран из группы, состоящей из: H и C1-6 алкила с прямой или разветвленной цепью;
R10 и R11 независимо выбраны из группы, состоящей из: H и C1-4 алкила с прямой или разветвленной цепью.
10. Соединение по п. 9, отличающееся тем, что соединение формулы I представляет собой:
 .
.
11. Соединение по п. 9, отличающееся тем, что:
R3 выбран из группы, состоящей из:
и -SO2(C1-6 алкил);
где:
R10 и R11 независимо выбраны из группы, состоящей из: H и C1-4 алкила с прямой или разветвленной цепью.
12. Соединение по п. 11, отличающееся тем, что соединение формулы I выбрано из группы, состоящей из:
 ,
,  ,
,
 ,
, 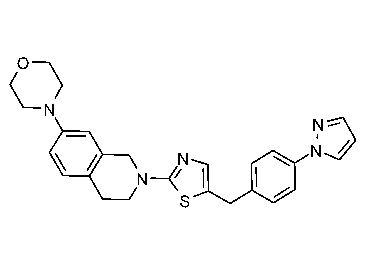 ,
,
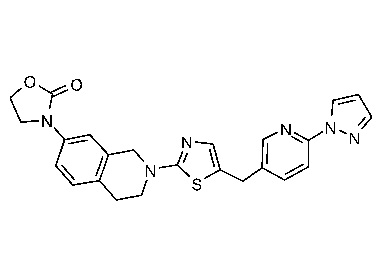 ,
, 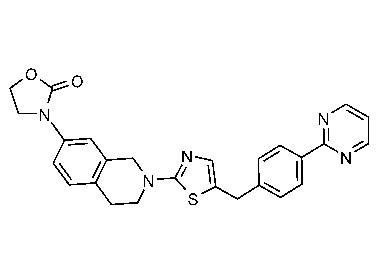 ,
,
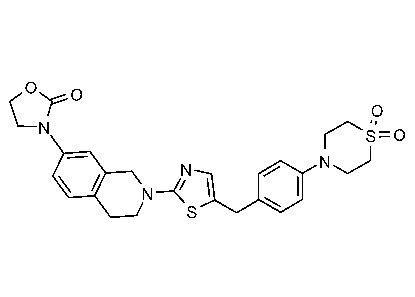 ,
, 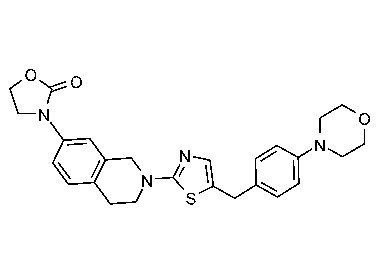 ,
,
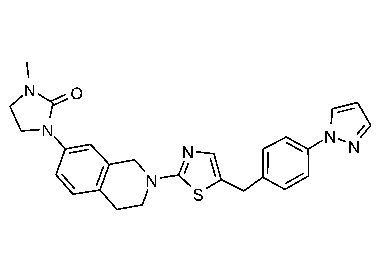 ,
, 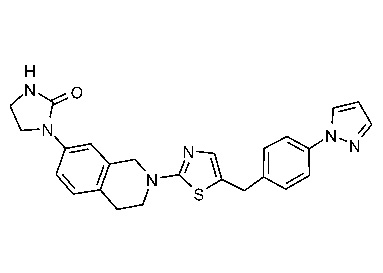 ,
,
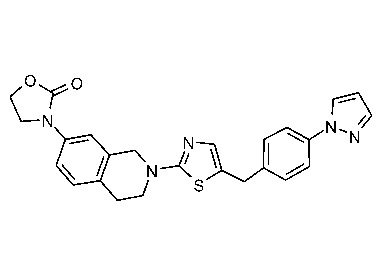 и .
и .
13. Соединение по п. 9, отличающееся тем, что:
R4 выбран из группы, состоящей из:
и -SO2(C1-6 алкил).
14. Соединение по п. 13, отличающееся тем, что соединение формулы I представляет собой:
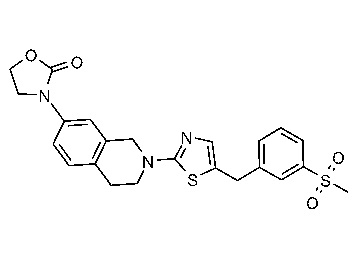
или его фармацевтически приемлемая соль или сольват.
15. Соединение по п. 1, отличающееся тем, что соединение формулы I представляет собой:
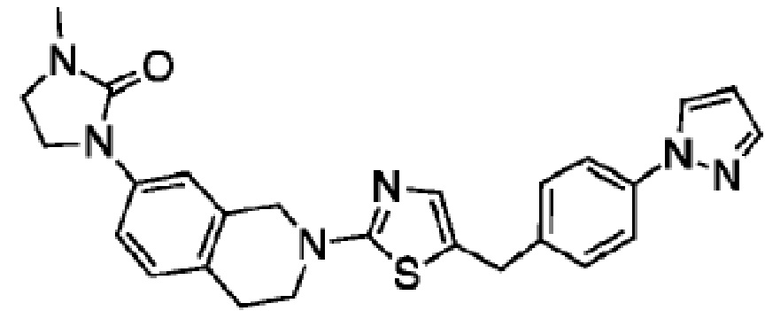
или его фармацевтически приемлемая соль или сольват.
16. Соединение по п. 1, отличающееся тем, что соединение формулы I представляет собой:
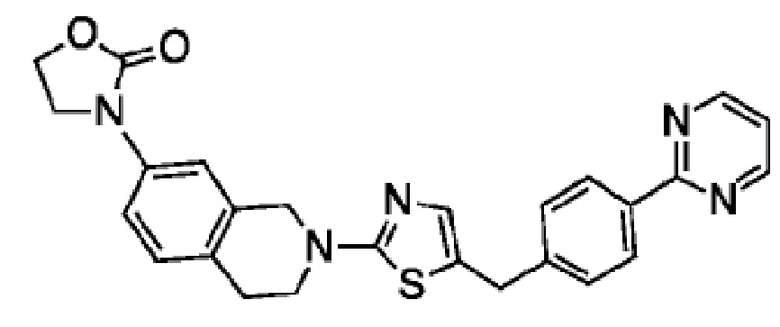
или его фармацевтически приемлемая соль или сольват.
17. Соединение по п. 1, отличающееся тем, что соединение формулы I представляет собой:
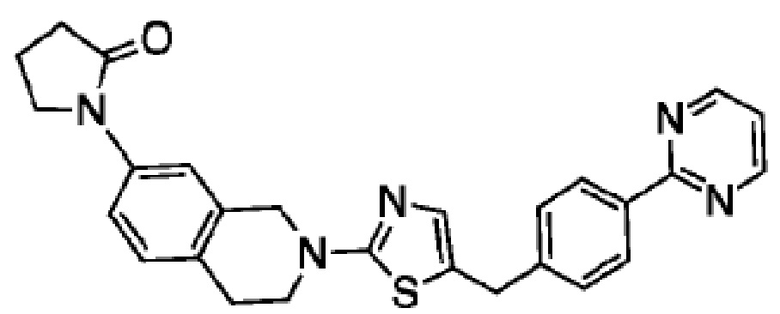
или его фармацевтически приемлемая соль или сольват.
18. Соединение по п. 1, отличающееся тем, что соединение формулы I представляет собой:
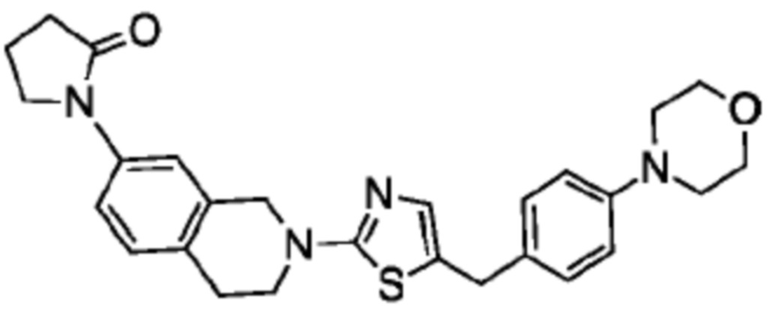
или его фармацевтически приемлемая соль или сольват.
19. Соединение по п. 1, отличающееся тем, что соединение формулы I представляет собой:
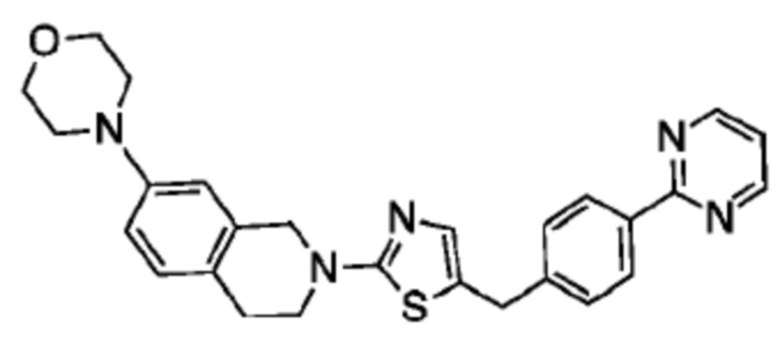
или его фармацевтически приемлемая соль или сольват.
20. Фармацевтическая композиция для лечения или предотвращения вирусной инфекции, содержащая эффективное количество соединения по любому из пп. 1 - 19 и фармацевтически приемлемый носитель.
21. Способ лечения или предотвращения вирусной инфекции у субъекта, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1 - 19.
22. Способ ингибирования продуцирования вируса, включающий приведение инфицированной вирусом клетки в контакт с ингибирующим продуцирование вируса количеством соединения по любому из пп. 1 - 19.
| WO 2016077232 A2, 19.05.2016 | |||
| WO 2016107832 A1, 07.07.2016 | |||
| US 20170226128 A1, 10.08.2017 | |||
| WO 2016177655 A1, 10.11.2016 | |||
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЛАМИДОМ ТИАЗОЛЫ, ПИРРОЛЫ И ТИОФЕНЫ | 2006 |
|
RU2425829C2 |
