Настоящая заявка испрашивает приоритеты и преимущества на основании следующих предыдущих заявок, поданных в Национальное управление интеллектуальной собственности Китая: патентной заявки на изобретение с заявкой №202010292186.8, озаглавленной «FUSED TRICYCLIC COMPOUND AS EGFR INHIBITOR)), поданной 14 апреля 2020 года, патентной заявки на изобретение с заявкой №202010852717.4, озаглавленной «FUSED TRICYCLIC COMPOUND AS EGFR INHIBITOR)), поданной 22 августа 2020 года, патентной заявки на изобретение с заявкой №202110175424.1, озаглавленной «FUSED TRICYCLIC COMPOUND AS EGFR INHIBITOR)), поданной 9 февраля 2021 года, и патентной заявки на изобретение с заявкой №202110312259.X, озаглавленной «FUSED TRICYCLIC COMPOUND AS EGFR INHIBITOR)), поданной 24 марта 2021 года; которые в полном объеме включены в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтической химии и, в частности, относится к новому соединению в качестве селективного ингибитора EGFR, фармацевтической композиции, содержащей предложенное соединение, подходящему промежуточному соединению для получения указанного соединения и способу лечения клеточного пролиферативного заболевания, например рака, с помощью соединения согласно настоящему изобретению.
УРОВЕНЬ ТЕХНИКИ
Рак легкого представляет собой раковое заболевание с самой высокой заболеваемостью и смертностью, которое серьезно угрожает здоровью и жизни человека. Рак легкого в основном подразделяют на мелкоклеточный рак легкого (МРЛ) и не мелкоклеточный рак легкого (NSCLC), из которых около 80% представляет собой NSCLC.
EGFR, т.е. рецептор эпидермального фактора роста, широко распространен на поверхности клеток млекопитающего, таких как эпителиальные клетки, фибробласты, глиальные клетки и другие клетки. Сигнальный путь EGFR играет важную роль в физиологических процессах, таких как рост, пролиферация и дифференциация клеток. Мутация EGFR также является одним из наиболее распространенных типов мутации у пациентов с NSCLC, особенно в азиатских популяциях, которая может возникать у от 40% до 50% субъектов. Поэтому EGFR всегда был одной из самых популярных мишеней в области разработки и исследований лекарственных препаратов.
В настоящее время на рынке присутствуют ингибиторы EGFR первого, второго и третьего поколений. Ингибиторы EGFR первого поколения представляют собой обратимые препараты направленного действия, такие как гефитиниб, эрлотиниб и икотиниб. Вторым поколением ингибиторов EGFR являются необратимые препараты направленного действия, такие как афатиниб и дакомитиниб. Хотя лекарственные препараты направленного действия первого и второго поколения обладают значительным лечебным эффектом, большинство пациентов все еще страдают от лекарственной устойчивости после применения лекарственных препаратов в течение 1-2 лет. Среди пациентов с устойчивостью к ингибитору EGFR 50% лекарственной устойчивости связано с мутацией Т790М. Препарат третьего поколения омисертиниб, нацеленный на EGFR, может преодолевать опухолевую резистентность, вызванную мутацией Т790М, и обеспечивает лучшую выживаемость большему количеству пациентов с раком легкого. Однако такой целенаправленно воздействующий препарат третьего поколения также неизбежно вызывает лекарственную устойчивость, при этом такая лекарственная устойчивость в основном вызвана мутацией C797S. Мутация C797S, которая проявляется посредством мутации цистеина в серии, нарушает связывание белка EGFR с лекарственными препаратами направленного действия третьего поколения, что тем самым не позволяет предотвратить фосфорилирование белка EGFR и активацию нижестоящих сигнальных путей. В настоящее время не существует разработанного лечения для двух цис-тройиых мутаций, Del19/T790M/C797S и L858R/T790M/C797S, в основном появляющихся после возникновения устойчивости к омисертинибу, клиническая потребность является крайне актуальной, и настоящее изобретение основано на решении такой проблемы.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является обеспечение класса трициклических соединений, таких как селективные ингибиторы EGFR, фармацевтической композиции, содержащей такие соединения, подходящего промежуточного продукта для получения указанных соединений и применение таких соединений для получения лекарственного средства для лечения рака.
В настоящем изобретении предложено соединение, представленное формулой (I'''), или его стереоизомер, таутомер или фармацевтически приемлемая соль, пролекарство, гидрат, сольват и изотопно-меченное производное
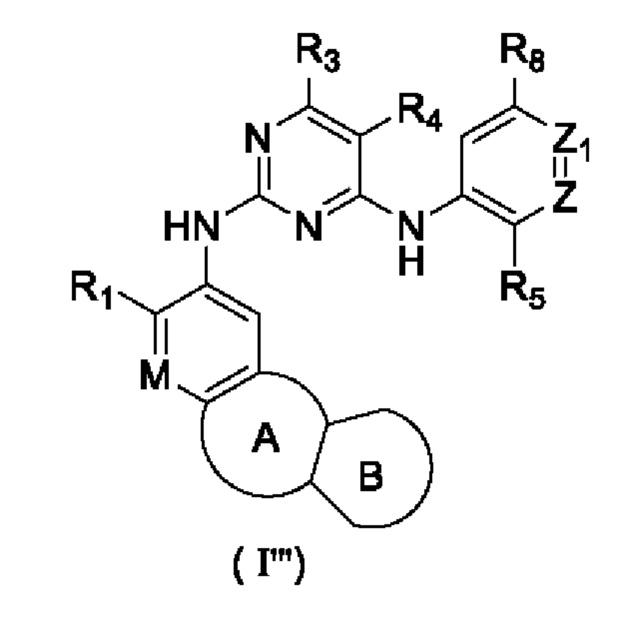
где
R1 выбирают из Н, галогена, -CN, С1-6 алкила, С1-6 алкокси, С1-6 галогеналкила, С1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, С2-6 алкинилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и С2-6 алкиниламино;
М выбирают из N и CRa; Ra представляет собой Н, галоген, C1-6 алкил, С3-6 циклоалкил, C1-6 гетероалкил или С1-6 галогеналкил;
Z выбирают из N и CR6;
Z1 выбирают из N и CR7;
или Ra циклизируется с R1 с образованием замещенного или незамещенного 5-8-членного гетероциклила или 5-8-членного карбоциклила;
кольцо А выбирают из замещенного или незамещенного 5-8-членного гетероциклила или 5-8-членного карбоциклила;
кольцо В отсутствует или выбрано из арила или 5-6-членного гетероарила и 4-8-членного гетероциклоалкила или С4-8 циклоалкила, которые необязательно содержат в качестве заместителя один или более R2;
каждый R2 независимо выбирают из Н, галогена, -CN, -C(=O)Rb, -C(=O)NRbRc, -S(=O)2Rb, -S(=O)(=NRc)Rb, -NH2, -OH, -SH, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С5-6 арила, С5-6 арилалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и -(CH2)rNRcRd, r необязательно выбирают из 0, 1, 2 и 3; при этом С3-6 циклоалкил, 3-6-членный гетероциклоалкил, С5-6 арил, С5-6 арилалкил, С3-6 циклоалкилокси, 3-6-членный гетероциклоалкилокси, С2-6 алкенилокси, С1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членный гетероциклоалкиламино или С2-6 алкениламино в R2 необязательно содержит в качестве заместителя один или более C1-3 алкил или C1-3 алкокси;
каждый из R3 и R4 независимо выбирают из Н, галогена, -CN, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-8 циклоалкила, 3-8-членного гетероциклоалкила, C1-6 алкиламино, С1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R3 циклизируется с R4 с образованием арила, С4-7 циклоалкила, 5-7-членного гетероциклоалкила или 5-6-членного гетероарила;
R5 выбирают из замещенного или незамещенного -NH2, -C(=O)NRbRc, -S(=O)2Rb, -P(=O)RbRc, -P(=O)RbNRcRd, -P(=O)RbORc, -P(=O)ORbORc, -P(=S)RbRc, -P(=S)RbNRcRd, -P(=S)RbORc, -P(=S)ORbORc, -S(=O)2NRbRc, RbS(=O)2NRc-, -N=S(=O)RbRc или RbN=S(=O)(Rc)-, -NRbC(O)Rc и RcS(=NRb)(=O)NRd-;
каждый из Rb, Rc и Rd независимо выбирают из H, -CN, С1-3 алкила, C1-3 галогеналкила, С3-6 циклоалкила, С4-6 гетероциклоалкила, С5-10 арила и 5-10-членного гетероарила;
или Rb и Rc циклизируются с атомами, к которым они оба присоединены, с образованием 5-6-членного гетероциклоалкила, незамещенного или необязательно содержащего в качестве заместителя один или более С1-3 алкил или С1-3 алкокси;
каждый из R6, R7 и R8 независимо выбирают из Н, галогена, -CN, -ОН, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С2-6 алкенила, С2-6 алкинила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)(Rb)-, -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2;
или R6 циклизируется с R7 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила;
или R7 циклизируется с R8 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила.
В некоторых вариантах реализации настоящего изобретения R1 выбирают из Н, галогена, -CN, C1-6 алкила, C1-6 алкокси, С1-6 галогеналкила и C1-6 галогеналкокси, предпочтительно R1 выбирают из Н, галогена, -CN, C1-6 алкила и C1-6 алкокси.
В некоторых вариантах реализации настоящего изобретения М выбирают из N и CRa; Ra представляет собой Н, галоген, С1-3 алкил, С3-6 циклоалкил или С1-3 галогеналкил, М предпочтительно выбирают из N и СН.
В некоторых вариантах реализации настоящего изобретения кольцо А выбирают из замещенного или незамещенного 5-8-членного карбоциклила и 5-8-членного гетероциклила, содержащего 1 или 2 гетероатома, выбранных из О, S и N; в некоторых вариантах реализации кольцо А может содержать двойную связь; в некоторых вариантах реализации 1 или 2 кольцевых атома в кольце А могут быть необязательно заменены на -С(=O), -N(=O), -S(=O) и -S(=O)2, кольцо А также может необязательно содержать в качестве заместителя одну или более групп Rx, при этом Rx выбирают из Н, -ОН, -CN, -NH2, галогена, C1-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, С3-8 циклоалкила, 3-8-членного гетероциклоалкила, С3-6 циклоалкил-C1-6 алкила-, 3-8-членного гетероциклоалкил-О-6 алкила-, арил-С1-6 алкила-, С5-13 спироциклила и 5-13-членного спирогетероциклила; при этом С3-6 циклоалкил, 3-8-членный гетероциклоалкил, С3-6 циклоалкил-С1-6 алкил-, 3-8-членный гетероциклоалкил-С1-6 алкил-, арил-С1-6 алкил-, С5-13 спироциклил и 5-13-членный спироциклил-членный спирогетероциклил необязательно содержат в качестве заместителя один или более Ry; Ry выбирают из Н, галогена, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси, С3-6 циклоалкила, 4-8-членного гетероциклоалкила, С3-6 циклоалкил-С1-6 алкила-, 4-8-членного гетероциклоалкил-С1-6 алкила-, 5-10-членного арила и 5-10-членного гетероарила.
В некоторых вариантах реализации настоящего изобретения кольцо В представляет собой арил или 5-6-членный гетероарил, необязательно содержащий в качестве заместителя один или более R2; арил и 5-6-членный гетероарил могут представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил, пиридазинил или триазинил.
В некоторых вариантах реализации настоящего изобретения каждый R2 независимо выбирают из Н, галогена, -CN, -C(=O)Rb, -S(=O)2Rb, -S(=O)(=NRc)Rb, -NH2, -OH, -SH, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С5-6 арила, С5-6 арилалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и -(CH2)rNRcRd, при этом r необязательно выбирают из 0, 1, 2 и 3; при этом 3-6-членный гетероциклоалкил или С5-6 арилалкил в R2 необязательно содержит в качестве заместителя один или более С1-3 алкил или С1-3 алкокси.
В некоторых вариантах реализации настоящего изобретения каждый из R3 и R4 независимо выбирают из Н, галогена, -CN, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, С1-6 галогеналкокси и С3-6 циклоалкила; или R3 циклизируется с R4 с образованием фенила, С4-7 циклоалкила и 5-7-членного гетероциклоалкила или 5-6-членного гетероарила, содержащего 1 или 2 гетероатома, выбранных из О, S и N; 5-6-членный гетероарил предпочтительно может представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил, пиридазинил или триазинил.
В некоторых вариантах реализации настоящего изобретения R5 выбирают из замещенного или незамещенного -NH2, -C(=O)NRbRc, -S(=O)2Rb, -P(=O)RbRc, -P(=O)RbNRcRd, -P(=O)RbORc, -P(=O)ORbORc, -P(=S)RbRc, -P(=S)RbNRcRd, -P(=S)RbORc, -P(=S)ORbORc, -S(=O)2NRbRc, RbS(=O)2NRc-, -N=S(=O)RbRc, RbN=S(=O)(Rc)-, -NRbC(O)Rc; или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)(Rb)-, -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2.
В некоторых вариантах реализации настоящего изобретения каждый из Rb, Rc и Rd независимо выбирают из Н, С1-3 алкила, С1-3 галогеналкила и С3-6 циклоалкила; или Rb и Rc циклизируются с атомами, к которым они оба присоединены, с образованием 5-6-членного гетероциклоалкила, незамещенного или необязательно содержащего в качестве заместителя один или более С1-3 алкил или С1-3 алкокси.
В некоторых вариантах реализации настоящего изобретения каждый из R6 и R7 независимо выбирают из Н, галогена, -CN, -ОН, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С2-6 алкенила, С2-6 алкинила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино; или R6 циклизируется с R7 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила; 5-6-членный гетероарил предпочтительно может представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил, пиридазинил или триазинил.
В некоторых вариантах реализации настоящего изобретения R8 представляет собой Н.
В настоящем изобретении предложено соединение, представленное формулой (I''), или его стереоизомер, таутомер или фармацевтически приемлемая соль, пролекарство, гидрат, сольват и изотопно-меченное производное
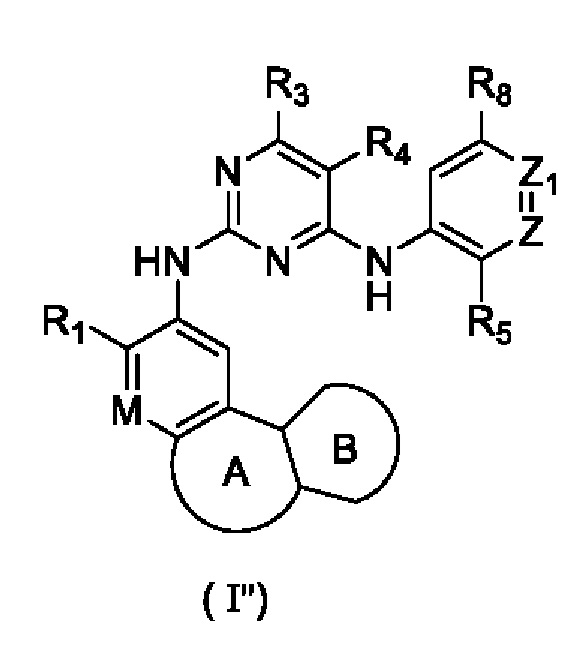
где
R1 выбирают из Н, галогена, -CN, С1-6 алкила, С1-6 алкокси, С1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, С2-6 алкинилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и С2-6 алкиниламино;
М выбирают из N и CRa; Ra представляет собой Н, галоген, C1-3 алкил, С3-6 циклоалкил или С1-3 галогеналкил;
Z выбирают из N и CR6;
Zi выбирают из N и CR7;
или Ra циклизируется с R1 с образованием замещенного или незамещенного 5-8-членного гетероциклила;
кольцо А выбирают из замещенного или незамещенного 5-8-членного гетероциклила или 5-8-членного карбоциклила;
кольцо В представляет собой арил или 5-6-членный гетероарил, необязательно содержащий в качестве заместителя один или более R2; арил и 5-6-членный гетероарил могут представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил, пиридазинил или триазинил;
каждый R2 независимо выбирают из Н, галогена, -CN, -C(=O)Rb, -S(=O)2Rb, -S(=O)(=NRc)Rb, -NH2, -ОН, -SH, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С5-6 арила, С5-6 арилалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, C1-6 алкиламино, С1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и -(CH2)rNRcRd, при этом г необязательно выбирают из 0, 1, 2 и 3;
каждый из R3 и R4 независимо выбирают из Н, галогена, -CN, C1-6 алкила, С1-6 алкокси, C1-6 галогеналкила, С1-6 галогеналкокси, С3-8 циклоалкила, 3-8-членного гетероциклоалкила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R3 циклизируется с R4 с образованием арила, С4-7 циклоалкила, 5-7-членного гетероциклоалкила или 5-6-членного гетероарила;
R5 выбирают из замещенного или незамещенного -NH2, -C(=O)NRbRc, -S(=O)2Rb, -P(=O)RbRc, -P(=O)RbNRcRd, -P(=O)RbORc, -P(=O)ORbORc, -P(=S)RbRc, -P(=S)RbNRcRd, -P(=S)RbORc, -P(=S)ORbORc, -S(=O)2NRbRc, RbS(=O)2NRc-, -N=S(=O)RbRc или RbN=S(=O)(Rc)- и -NRbC(O)Rc;
каждый из Rb, Rc и Rd независимо выбирают из H, -CN, С1-3 алкила, С1-3 галогеналкила, С3-6 циклоалкила, С4-6 гетероциклоалкила, С5-10 арила и 5-10-членного гетероарила;
или Rb и Rc циклизируются с атомами, к которым они оба присоединены, с образованием 5-6-членного гетероциклоалкила;
каждый из R6, R7 и R8 независимо выбирают из Н, галогена, -CN, -ОН, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С2-6 алкенила, С2-6 алкинила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)(Rb)-, -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2;
или R6 циклизируется с R7 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила;
или R7 циклизируется с R6 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила.
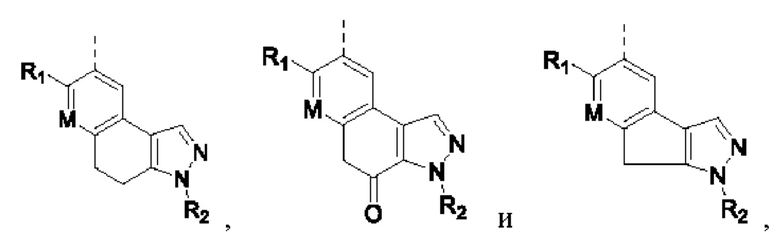
В настоящем изобретении предложено соединение, представленное формулой (I'), или его стереоизомер, таутомер или фармацевтически приемлемая соль, пролекарство, гидрат, сольват и изотопно-меченное производное
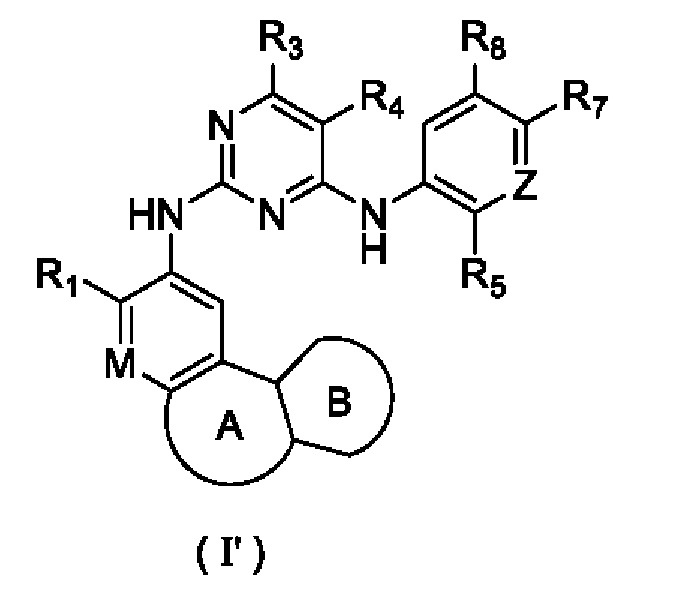
где
R1 выбирают из Н, галогена, -CN, С1-6 алкила, С1-6 алкокси, C1-6 галогеналкила, С1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, С2-6 алкинилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и С2-6 алкиниламино;
М выбирают из N и CRa; Ra представляет собой Н, галоген, С1-3 алкил, С3-6 циклоалкил или С1-3 галогеналкил;
Z выбирают из N и CR6;
или Ra циклизируется с R1 с образованием замещенного или незамещенного 5-8-членного гетероциклила;
кольцо А выбирают из замещенного или незамещенного 5-8-членного гетероциклила или 5-8-членного карбоциклила;
кольцо В представляет собой арил или 5-6-членный гетероарил, необязательно содержащий в качестве заместителя один или более R2; арил и 5-6-членный гетероарил могут представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил или пиридазинил;
каждый R2 независимо выбирают из Н, галогена, -CN, -C(=O)Rb, -S(=O)2Rb, -S(=O)(=NRc)Rb, -NH2, -ОН, -SH, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С5-6 арила, С5-6 арилалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
каждый из R3 и R4 независимо выбирают из Н, галогена, -CN, C1-6 алкила, С1-6 алкокси, C1-6 галогеналкила, С1-6 галогеналкокси, С3-6 циклоалкила, 3-8-членного гетероциклоалкила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R3 циклизируется с R4 с образованием арила, С4-7 циклоалкила, 5-7-членного гетероциклоалкила или 5-6-членного гетероарила;
R5 выбирают из замещенного или незамещенного -NH2, -C(=O)NRbRc, -S(=O)2Rb, -P(=O)RbRc, -P(=O)RbNRcRd, -P(=O)RbORc, -P(=O)ORbORc, -P(=S)RbRc, -P(=S)RbNRcRd, -P(=S)RbORc, -P(=S)ORbORc, -S(=O)2NRbRc, RbS(=O)2NRc-, -N=S(=O)RbRc и RbN=S(=O)(Rc)-;
каждый из Rb, Rc и Rd независимо выбирают из H, -CN, С1-3 алкила, С1-3 галогеналкила, С3-6 циклоалкила, С4-6 гетероциклоалкила, С5-10 арила и 5-10-членного гетероарила;
каждый из R6, R7 и R8 независимо выбирают из Н, галогена, -CN, -ОН, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, С1-6 галогеналкокси, С2-6 алкенила, С2-6 алкинила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и Сг-6 алкениламино;
или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)Rb-;
или R7 циклизируется с R7 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила;
или R7 циклизируется с R8 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила.
В настоящем изобретении предложено соединение, представленное формулой (I), или его стереоизомер, таутомер или фармацевтически приемлемая соль, пролекарство, гидрат, сольват и изотопно-меченное производное
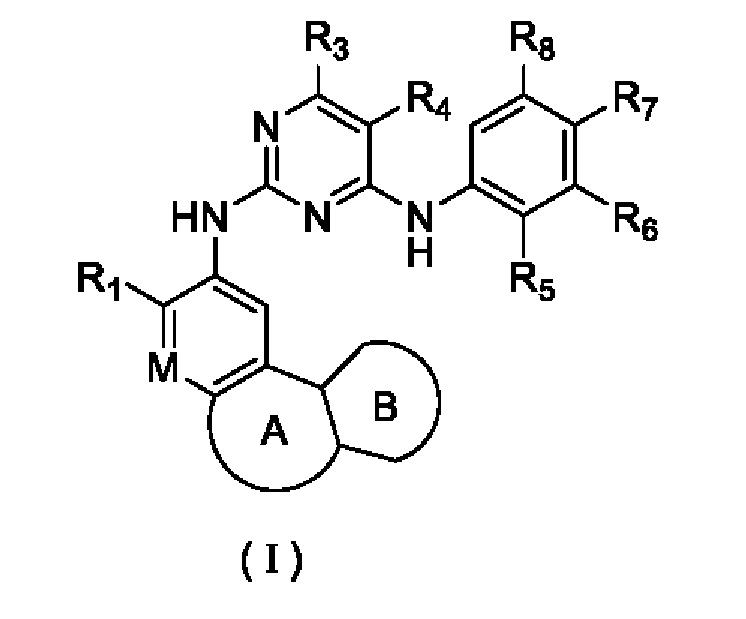
где
R1 выбирают из Н, галогена, -CN, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
М выбирают из N и CRa;
Ra представляет собой Н, галоген, C1-3 алкил, С3-6 циклоалкил или d-з галогеналкил;
или Ra циклизируется с R1 с образованием замещенного или незамещенного 5-8-членного гетероциклила;
кольцо А выбирают из замещенного или незамещенного 4-8-членного гетероциклила и 5-8-членного карбоциклила;
кольцо В представляет собой арил или 5-6-членный гетероарил, необязательно содержащий в качестве заместителя один или более R2; арил и 5-6-членный гетероарил могут представлять собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил или пиридазинил;
каждый R2 независимо выбирают из Н, галогена, -CN, -C(=O)Rb, -S(=O)2Rb, -S(=O)(=NRc)Rb, -NH2, -ОН, -SH, C1-6 алкила, С1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси и С2-6 алкенилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
каждый из R3 и R4 независимо выбирают из Н, галогена, -CN, C1-6 алкила, С1-6 алкокси, С1-6 галогеналкила, С1-6 галогеналкокси, С3-6 циклоалкила, 3-8-членного гетероциклоалкила, C1-6 алкиламино, С1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R3 циклизируется с R4 с образованием арила, С4-7 циклоалкила, 5-7-членного гетероциклоалкила или 5-6-членного гетероарила;
R5 выбирают из замещенного или незамещенного -NH2, -C(=O)NRbRc, -S(=O)2Rb, -P(=O)RbRc, -P(=O)RbNRcRd, -S(=O)2NRbRc, RbS(=O)2NRc-, -N=S(=O)RbRc, и RbN=S(=O)(Rc)-;
каждый из Rb, Rc и Rd независимо выбирают из H, -CN, С1-3 алкила, С1-3 галогеналкила, С3-6 циклоалкила, С4-6 гетероциклоалкила, С5-10 арила и 5-10-членного гетероарила;
каждый из R6, R7 и R8 независимо выбирают из Н, галогена, -CN, -ОН, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С2-6 алкенила, С2-6 алкинила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино и С2-6 алкениламино;
или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)Rb-;
или R6 циклизируется с R7 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила;
или R7 циклизируется с R8 с образованием С4-6 циклоалкила, 4-6-членного гетероциклоалкила, арила и 5-6-членного гетероарила.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении,
R1 выбирают из Н, галогена, -CN, С1-3 алкила, С1-3 алкокси, С1-3 галогеналкила и С1-3 галогеналкокси;
М выбирают из N и СН;
кольцо А выбирают из замещенного или незамещенного 5-8-членного гетероциклила или 5-8-членного карбоциклила;
кольцо В представляет собой 5-6-членный гетероарил, необязательно содержащий в качестве заместителя один или более R2; 5-6-членный гетероарил представляет собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил или пиридазинил;
каждый R2 независимо выбирают из Н, С1-4 алкила, С1-4 галогеналкила, -(CH2)rNRcRd, 3-6-членного гетероциклоалкила и С5-6 арилалкила, при этом г необязательно выбирают из О, 1, 2 и 3, и 3-6-членный гетероциклоалкил и С5-6 арилалкил необязательно содержат в качестве заместителя один или более С1-3 алкил или С1-3 алкокси;
каждый из R3 и R4 независимо выбирают из Н, галогена, С1-4 алкила, С1-4 галогенированного алкила и С3-6 циклоалкила;
или R3 циклизируется с R4 с образованием 5-6-членного гетероарила; 5-6-членный гетероарил представляет собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил или пиридазинил;
R5 выбирают из -C(=O)NRbRc, -P(=O)RbRc, -P(=S)RbRc, -S(=O)2NRbRc, RbS(=O)2NRc- и -NRbC(O)Rc;
каждый из Rb, Rc и Rd независимо выбирают из H, С1-3 алкила, С1-3 алкокси и С3-6 циклоалкила;
или Rb и Rc циклизируются с атомами, к которым они оба присоединены, с образованием 5-6-членного гетероциклоалкила, незамещенного или необязательно содержащего в качестве заместителя один или более С1-3 алкил или С1-3 алкокси;
или R5 циклизируется с R6 с образованием 4-7-членного кольца, содержащего -P(=O)(Rb)-, -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2;
каждый из R6, R7 и R8 независимо выбирают из Н, галогена и С1-3 алкила;
или R8 выбирают из Н, R6 циклизируется с R7 с образованием 5-6-членного гетероарила; 5-6-членный гетероарил представляет собой пирролил, фуранил, тиенил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, имидазолил, триазолил, фенил, пиримидинил, пиридил, пиразинил или пиридазинил.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R1 выбирают из Н, галогена, -CN, С1-3 алкила, С1-3 алкокси, С1-3 галогеналкила и С1-3 галогеналкокси; R1 предпочтительно выбирают из Н, метила, этила, н-пропила, изопропила, н-бутила, метокси, этокси, н-пропокси, изопропокси, н-бутокси, трифторметила, трифторметокси, трихлорметила, трихлорметокси и 2,2,2-трифторэтокси; и более предпочтительно R1 выбирают из метокси.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, М выбирают из N и СН, предпочтительно М выбирают из СН.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R2 выбирают из Н, метила, этила, изопропила, дифторметила, трифторметила, -CH2CH2N(СН3)СН3, 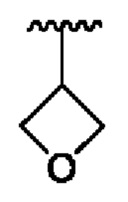 и
и 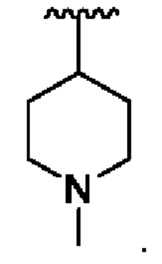
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R2 выбирают из Н, метила, этила, изопропила, дифторметила и трифторметила; предпочтительно R2 выбирают из метила.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, каждый из R3 и R4 независимо выбирают из Н, F, Cl, Br, CN, метила, этила, изопропила, метокси, этокси, изопропокси, дифторметила, трифторметила, 2,2,2-трифторэтила и циклопропила; предпочтительно каждый из R3 и R4 независимо выбирают из Н, F, О, Br, метила, дифторметила, трифторметила и циклопропила; предпочтительно R3 выбирают из Н, и R4 выбирают из Cl, Br, метила и трифторметила.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R3 выбирают из Н, и R4 выбирают из Н, F, О, Br, метила, этила, дифторметила, трифторметила и циклопропила.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R3 выбирают из Н, и R4 выбирают из О, Br и метила.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R3 выбирают из Н, и R4 выбирают из Br.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R3 циклизируется с R4 с образованием тиофенового кольца и пиррольного кольца, при этом тиофеновое кольцо и пиррольное кольцо могут необязательно содержать в качестве заместителя С1-4 алкил.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R5 выбирают из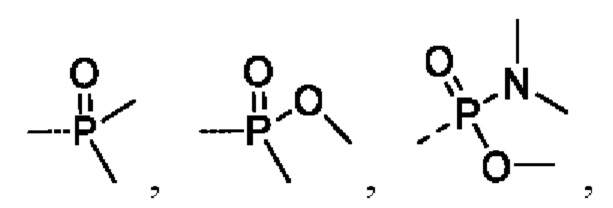
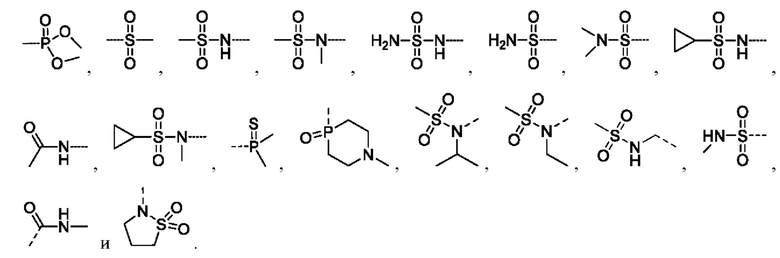
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R5 выбирают из 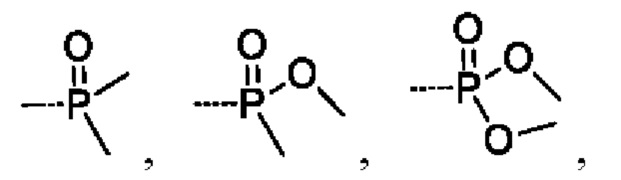
в соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R5 выбирают из 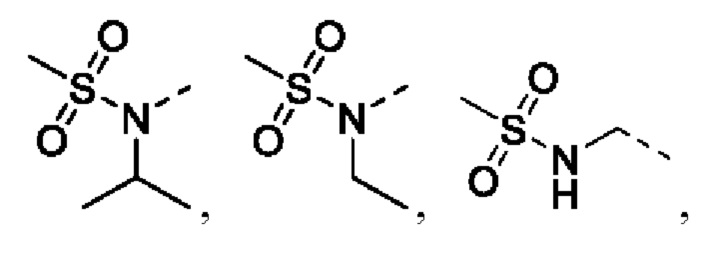
предпочтительно R5 выбирают из 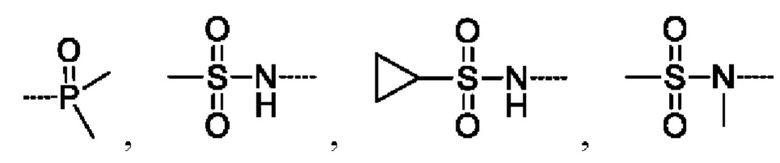 и
и 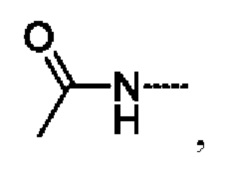
предпочтительно R5 выбирают из 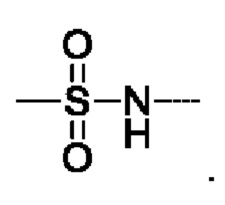
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R5 циклизируется с R6 с образованием 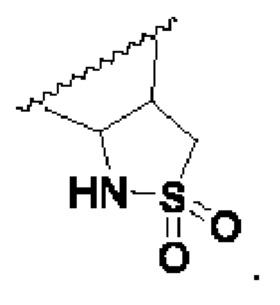
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, каждый из R6, R7 и R8 независимо выбирают из Н, метила и галогена; предпочтительно каждый из них независимо выбирают из Н и метила.
В соединениях формулы (I'"), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R6, R7 и R8 выбирают из Н.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, R6 и R8 выбирают из Н, и R7 выбирают из F.
В соединениях формулы (I'"), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, R6 независимо циклизируется с R7 или R7 независимо циклизируется с R8 с образованием циклобутана, циклопентана, тетрагидропиррольного кольца, тетрагидрофуранового кольца, тетрагидропиранового кольца, тиофенового кольца, имидазольного кольца, пиразольного кольца, пиррольного кольца, оксазольного кольца, тиазольного кольца, изоксазольного кольца, пиперазинового кольца, изотиазольного кольца, бензольного кольца, пиридинового кольца, пиперидинового кольца, пиримидинового кольца, пиридазинового кольца и пиразинового кольца; предпочтительно R6 независимо циклизируется с R7 или R7 независимо циклизируется с R8 с образованием циклобутана, пиридинового кольца и пиразинового кольца; и более предпочтительно R6 циклизируется с R7 с образованием пиразинового кольца.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'),
представленных в настоящем изобретении, структурное звено 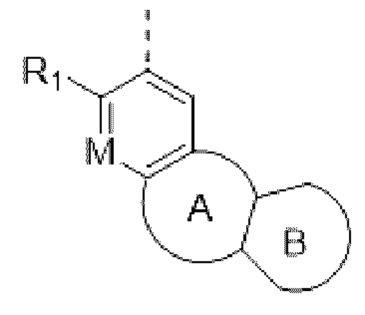 или
или 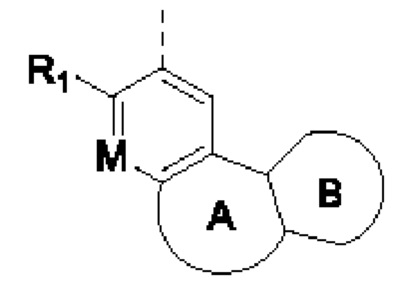 выбирают из
выбирают из 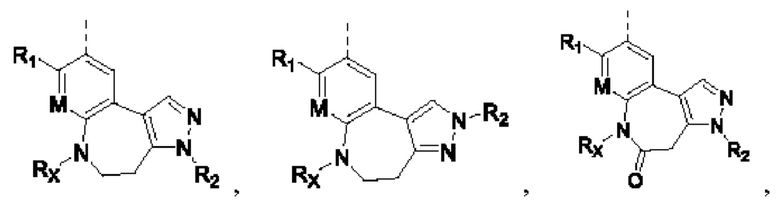
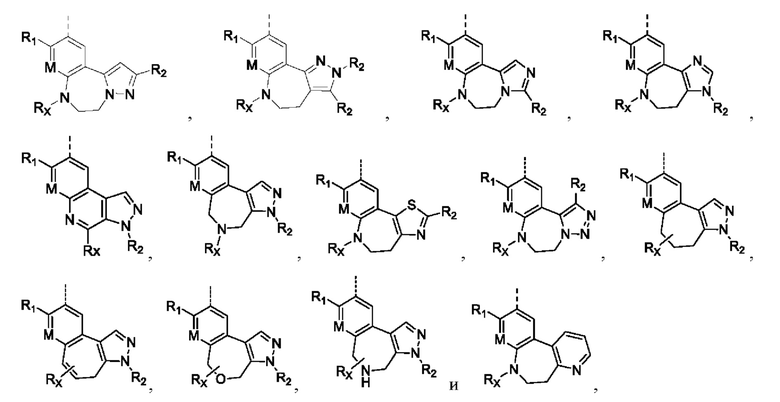
при этом R1, М и R2 определены выше; Rx выбирают из Н, -ОН, -CN, -NH2, галогена, C1-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, С3-8 циклоалкила, 3-8-членного гетероциклоалкила, С3-6 циклоалкил-C1-6 алкила-, 3-8-членного гетероциклоалкил-C1-6 алкила-, арил-C1-6 алкила-, С5-13 спироциклила и 5-13-членного спирогетероциклила; при этом С3-8 циклоалкил, 3-8-членный гетероциклоалкил, С3-8 циклоалкил-С1-6 алкил-, 3-8-членный гетероциклоалкил-C1-6 алкил-, арил-C1-6 алкил-, С5-13 спироциклил и 5-13-членный спироциклил-членный спирогетероциклил необязательно содержат в качестве заместителя один или более Ry; Ry выбирают из Н, галогена, С1-6 алкила, С1-6 галогеналкила, С1-6 алкокси, С3-8 циклоалкила, 4-8-членного гетероциклоалкила, С3-8 циклоалкил-С1-6 алкила-, 4-8-членного гетероциклоалкил-C1-6 алкила-, 5-10-членного арила и 5-10-членного гетероарила.
Когда Rx присоединен непосредственно к атому N, Rx не представляет собой -ОН, -NH2 и галоген.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из Н, -ОН, -CN, -NH2, галогена, С1-6 алкилкарбонила, C1-6 алкила, С1-6 галогеналкила, С3-8 циклоалкила, 3-8-членного гетероциклоалкила, С3-8 циклоалкил-С1-6 алкила-, 3-8-членного гетероциклоалкил-C1-6 алкила-; при этом С3-8 циклоалкил, 3-8-членный гетероциклоалкил, С3-8 циклоалкил-C1-6 алкил-, 3-8-членный гетероциклоалкил-C1-6 алкил- и арил-C1-6 алкил- необязательно содержат в качестве заместителя один или более Ry; Ry выбирают из Н, галогена, С1-6 алкила, C1-6 галогеналкила, C1-6 алкокси, С3-8 циклоалкила, 4-8-членного гетероциклоалкила, С3-8 циклоалкил-C1-6 алкила-, 4-8-членного гетероциклоалкил-C1-6 алкила-, 5-10-членного арила и 5-10-членного гетероарила.
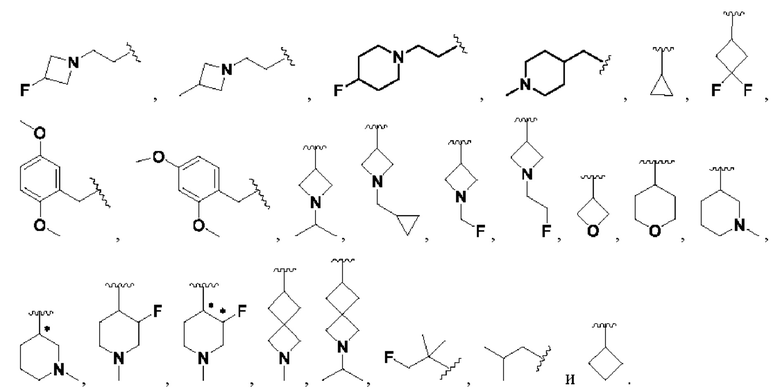
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из Н, -ОН, -CN, -NH2, галогена, С3-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкил-С1-4 алкила-, 3-6-членного гетероциклоалкил-См алкила-, арил-C1-6 алкила- и 7-11-членного спирогетероциклила, при этом С3-6 циклоалкил, 3-6-членный гетероциклоалкил, С3-6 циклоалкил-С1-4 алкил-, 3-6-членный гетероциклоалкил-С1-4 алкил-, арил-C1-6 алкил- и спирогетероциклил необязательно содержат в качестве заместителя один или более Ry; Ry выбирают из Н, галогена, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси и С3-6 циклоалкил-C1-6 алкила-; предпочтительно Ry выбирают из Н, F, метила, этила, изопропила, метокси,  и FCH2CH2-.
и FCH2CH2-.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из Н, -ОН, -CN, -NH2, F, метила, этила, изопропила, трифторэтила, метилкарбонила, 
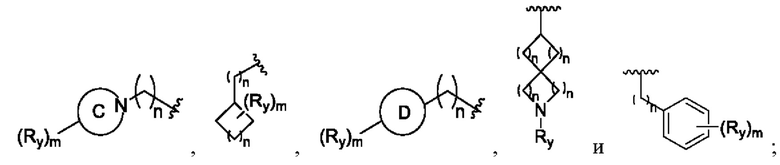 кольцо С представляет собой 4-8-членный гетероциклоалкил, кольцо D представляет собой 4-8-членный гетероциклоалкил, содержащий кислород; тип независимо представляют собой 0, 1, 2 или 3; Ry определен выше.
кольцо С представляет собой 4-8-членный гетероциклоалкил, кольцо D представляет собой 4-8-членный гетероциклоалкил, содержащий кислород; тип независимо представляют собой 0, 1, 2 или 3; Ry определен выше.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из Н, -ОН, -CN, -NH2, метила, этила, изопропила, метилкарбонила,  кольцо С представляет собой 4-8-членный гетероциклоалкил; тип независимо представляют собой 0, 1, 2 или 3.
кольцо С представляет собой 4-8-членный гетероциклоалкил; тип независимо представляют собой 0, 1, 2 или 3.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из Н, -ОН, -CN, -NH2, F, метила, этила, изопропила, трифторэтила, метилкарбонила, 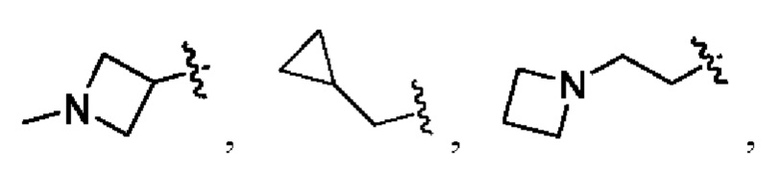
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, Rx выбирают из Н, метила, этила, изопропила, 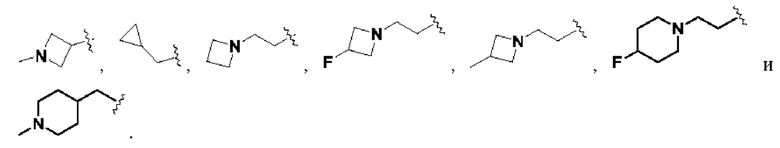
В соединениях формулы (I'"), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, R2 выбирают из Н, метила, этила, изопропила, 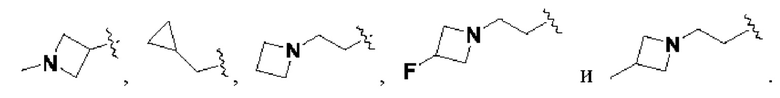
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, Rx выбирают из Н, метила, этила и изопропила.
В соединениях формулы (I'"), формулы (I''), формулы (I') или формулы (I), представленных в настоящем изобретении, Rx выбирают из метила и изопропила.
В соединениях формулы (I'"), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, структурное звено 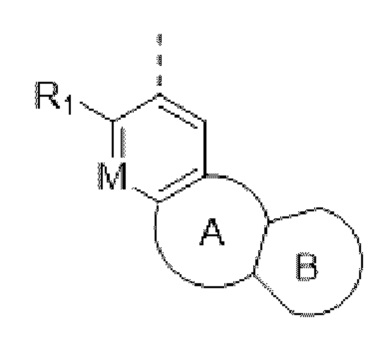 или
или 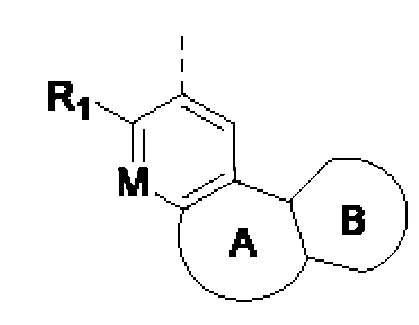 выбирают из
выбирают из 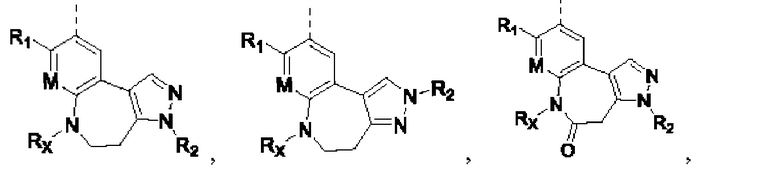
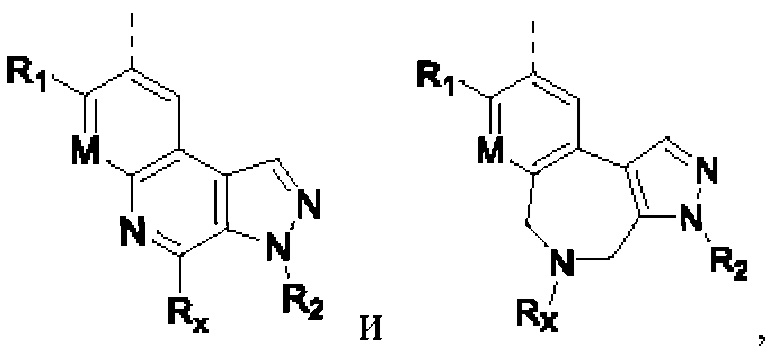 R1, М, Rx и R2 определены выше.
R1, М, Rx и R2 определены выше.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, структурное звено 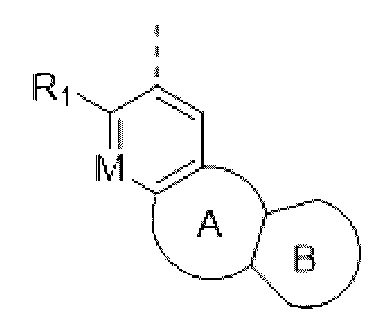 или
или 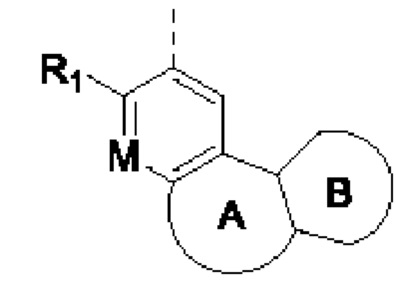 выбирают из
выбирают из 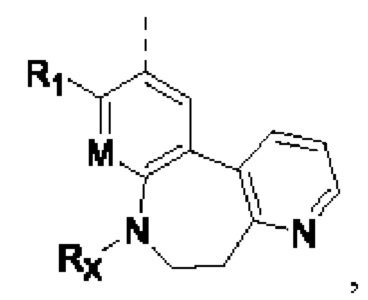 и R1, М и Rx определены выше.
и R1, М и Rx определены выше.
В соединениях формулы (I'''), формулы (I''), формулы (I') или формулы (I'), представленных в настоящем изобретении, структурное звено  или
или  выбирают из
выбирают из 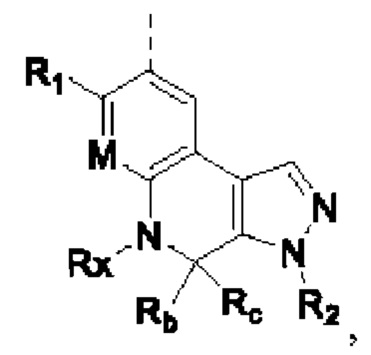 при этом R1, М Rx, Rb, Rc и R2 определены выше,
при этом R1, М Rx, Rb, Rc и R2 определены выше,
предпочтительно структурное звено  выбирают из
выбирают из 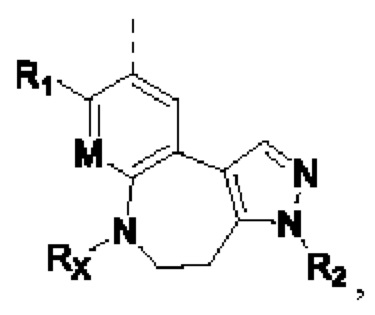 при этом М, R1, R2 и Rx определены выше.
при этом М, R1, R2 и Rx определены выше.
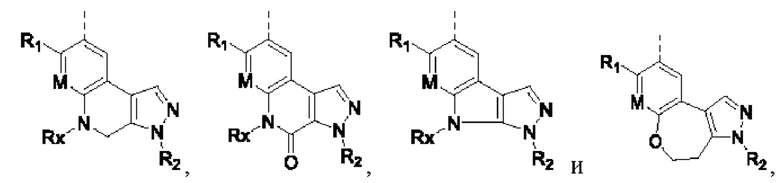
Соединения или их стерео изомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, предпочтительно выбирают из
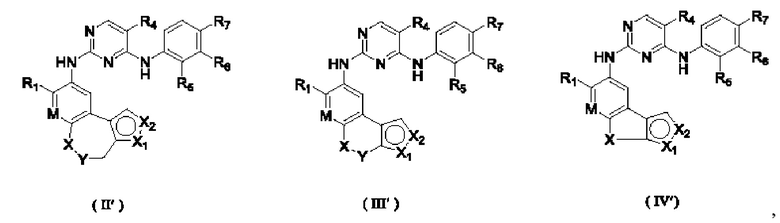 где каждый из X и Y независимо выбирают из -С(=O)-, -С=С-, -NR.X-, -О-, -CR9R10-, -S(=O)- и -S(=O)2-;
где каждый из X и Y независимо выбирают из -С(=O)-, -С=С-, -NR.X-, -О-, -CR9R10-, -S(=O)- и -S(=O)2-;
каждый из X1 и Х2 независимо выбирают из N и NR2;
каждый из R9 и R10 независимо выбирают из Н, галогена, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила и 3-8-членного гетероциклоалкила;
R1, R2, R4, R5, R6, R7, Rx и M определены выше.
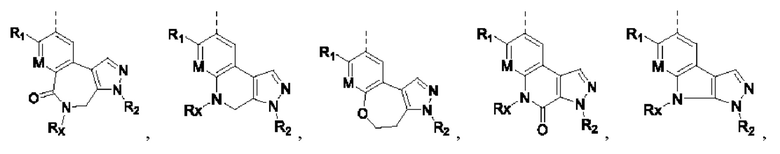
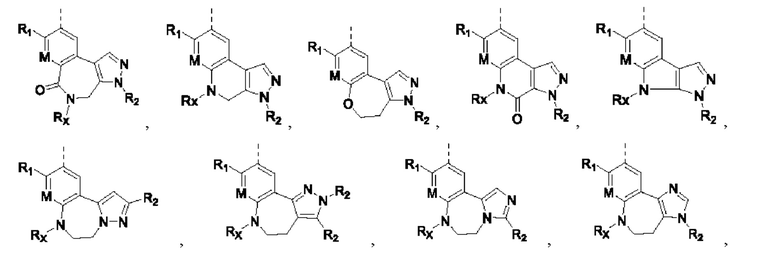
Соединения или их стерео изомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, предпочтительно выбирают из
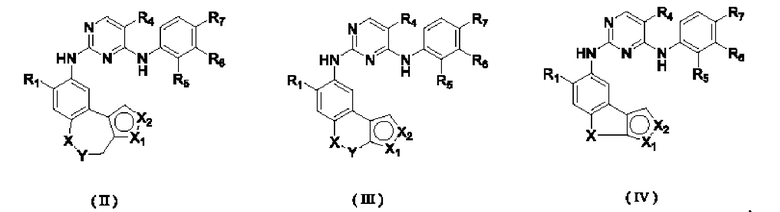
где
каждый из X и Y независимо выбирают из -С(=O)-, -NRX-, -О-, -CR9R10-, -S(=O)- и -S(=O)2-;
каждый из X1 и Х2 независимо выбирают из N и NR2;
каждый из R9 и R10 независимо выбирают из Н, галогена, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, С3-8 циклоалкила и 3-8-членного гетероциклоалкила;
R1, R2, R4, R5, R6, R7 и Rx определены выше.
Соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, предпочтительно выбирают из
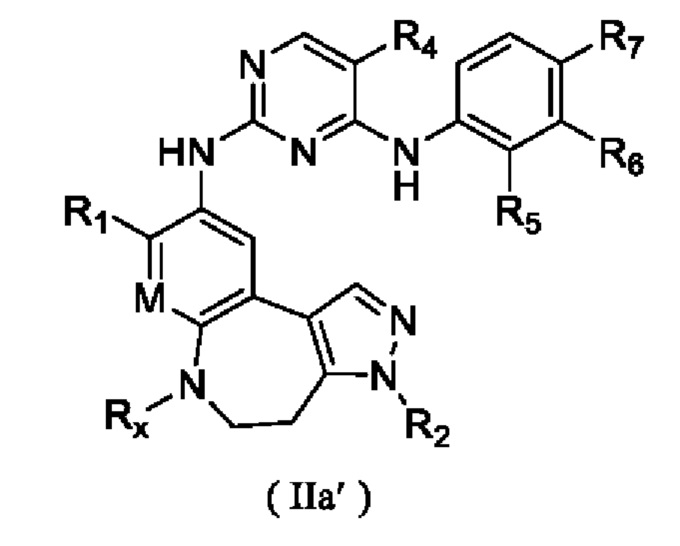
где R1, R2, R4, R5, R6, R7, Rx и М определены выше.
Соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, предпочтительно выбирают из
где R1, R2, R4, R5, R6, R7 и Rx определены выше.
Еще более предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, выбирают из
где R1, R2, R4, R5, Rx и М определены выше.
Еще более предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, выбирают из
где R1, R2, R4, R5 и Rx определены выше.
Еще более предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, выбирают из
где R4, R5, Rx и М определены выше.
Еще более предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, выбирают из
где R4, R5 и Rx определены выше.
Еще более предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные, описанные выше, выбирают из
где R5 и Rx определены выше.
В некоторых вариантах реализации настоящего изобретения соединение формулы (I''') или его стереоизомер, таутомер, фармацевтически приемлемую соль, пролекарство, гидрат, сольват и изотопно-меченное производное выбирают из соединения формулы (I'''-А)
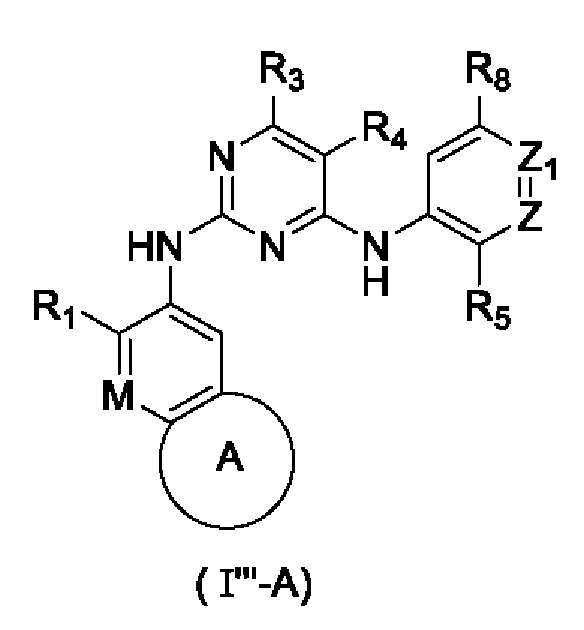
где R1, М, R3, R4, R5, R8, кольцо A, Z и Z1 определены выше.
В некоторых вариантах реализации настоящего изобретения соединение формулы (I''') или его стереоизомер, таутомер, фармацевтически приемлемую соль, пролекарство, гидрат, сольват и изотопно-меченное производное выбирают из соединения формулы (I'''-В),
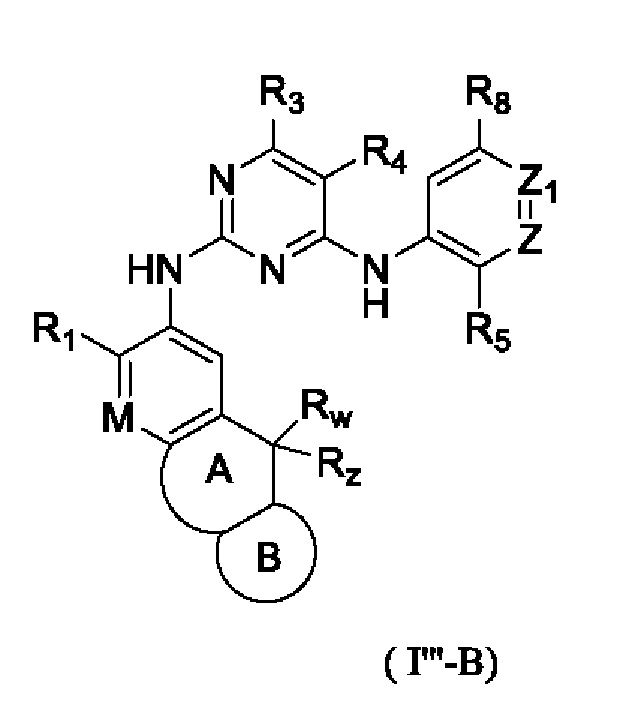
где R1, М, R3, R4, R5, R6, кольцо А, кольцо В, Z и Z1 определены выше;
каждый из Rw и Rz независимо выбирают из Н, -ОН, -CN, -NH2, галогена, C1-6 алкила, C1-6 гетероалкила, C1-6 галогеналкила, C1-6 галогеналкокси, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, С3-6 циклоалкилокси, 3-6-членного гетероциклоалкилокси, С2-6 алкенилокси, С2-6 алкинилокси, C1-6 алкиламино, C1-6 галогеналкиламино, С3-6 циклоалкиламино, 3-6-членного гетероциклоалкиламино, С2-6 алкениламино и С2-6 алкиниламино.
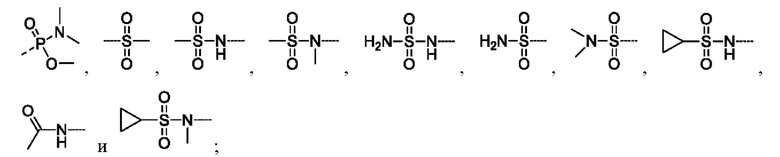
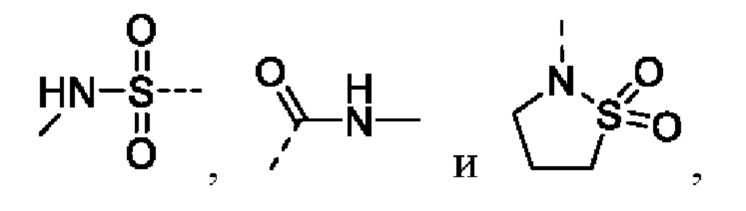
Наиболее предпочтительно, соединения или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, пролекарства, гидраты, сольваты и изотопно-меченные производные включают, но не ограничиваются ими:
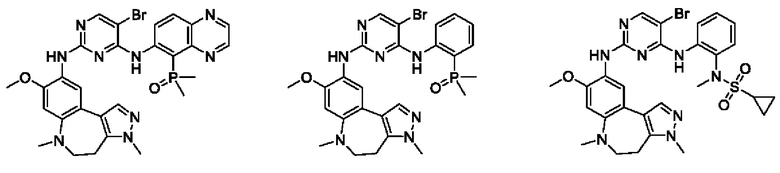
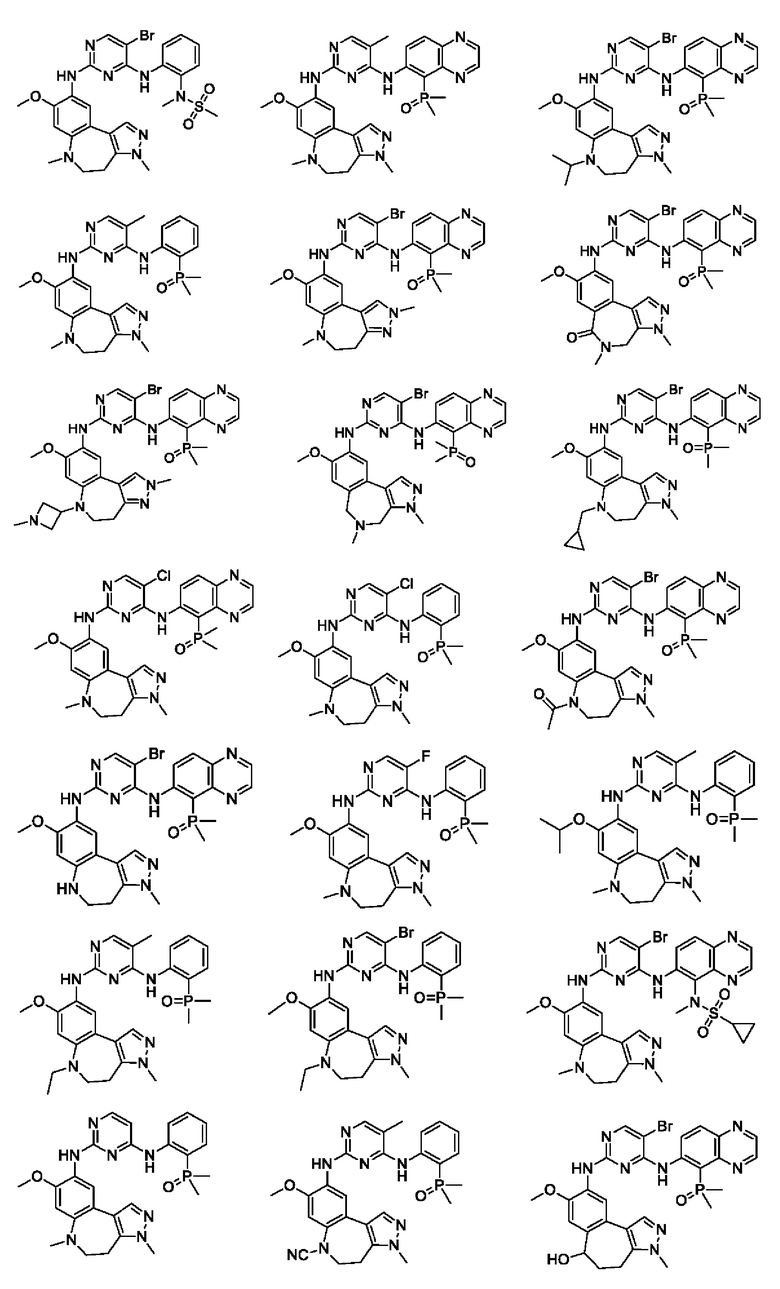
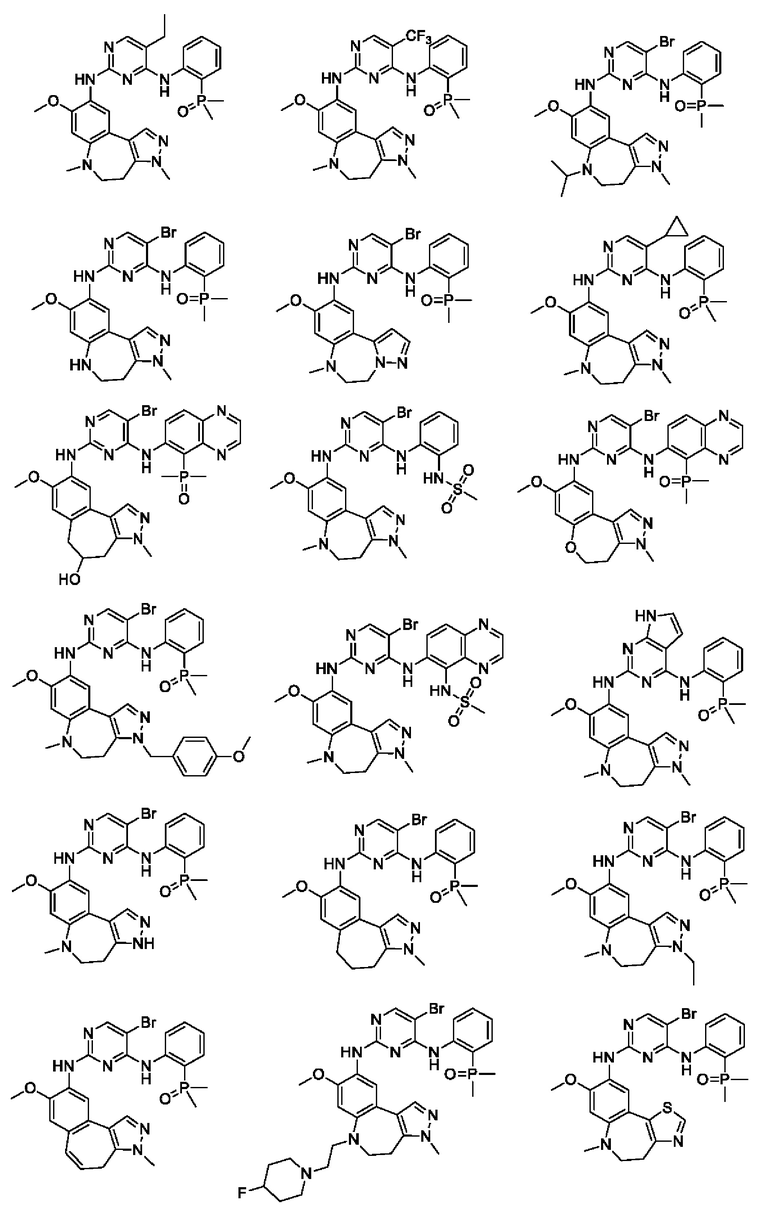
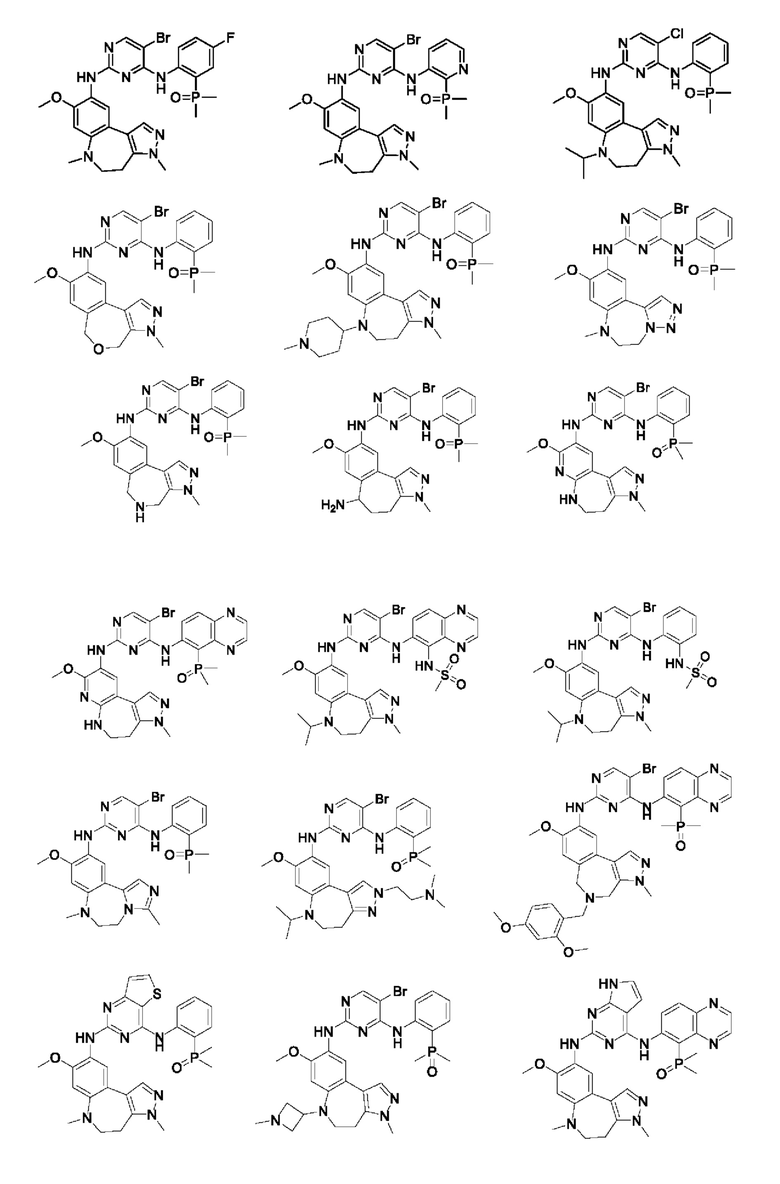
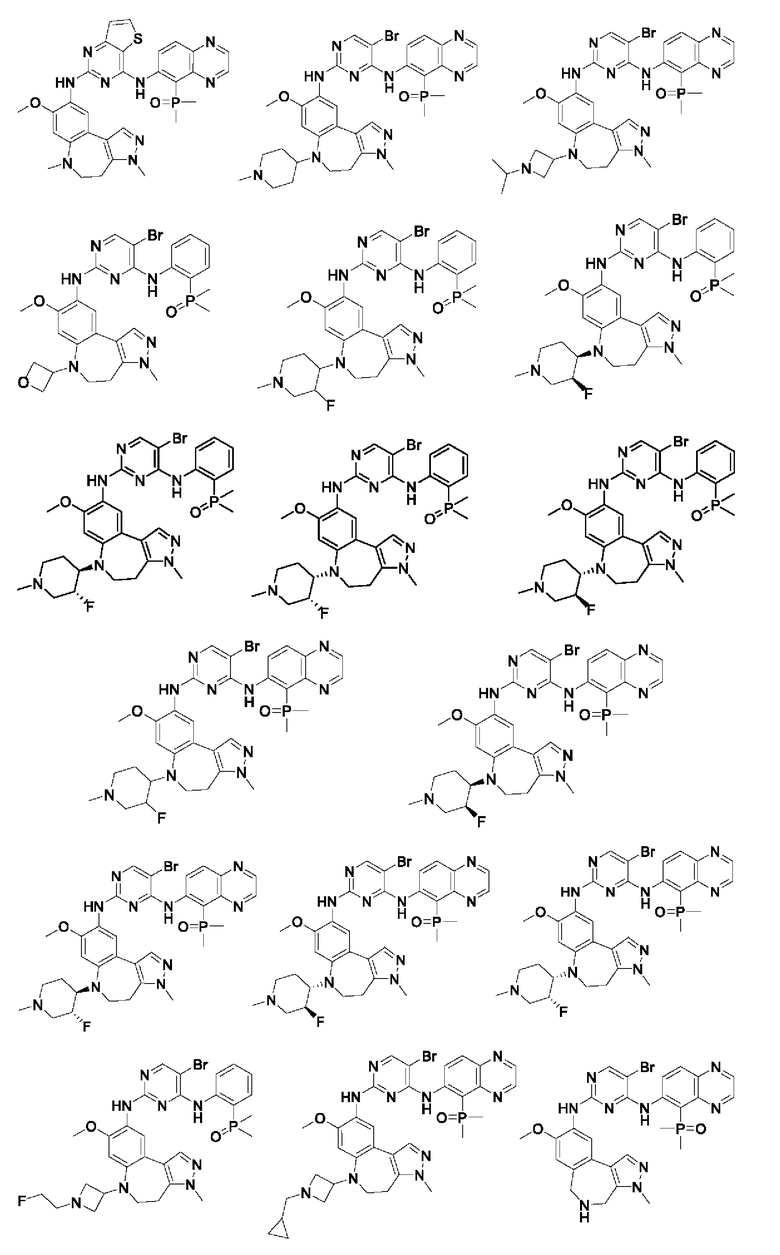
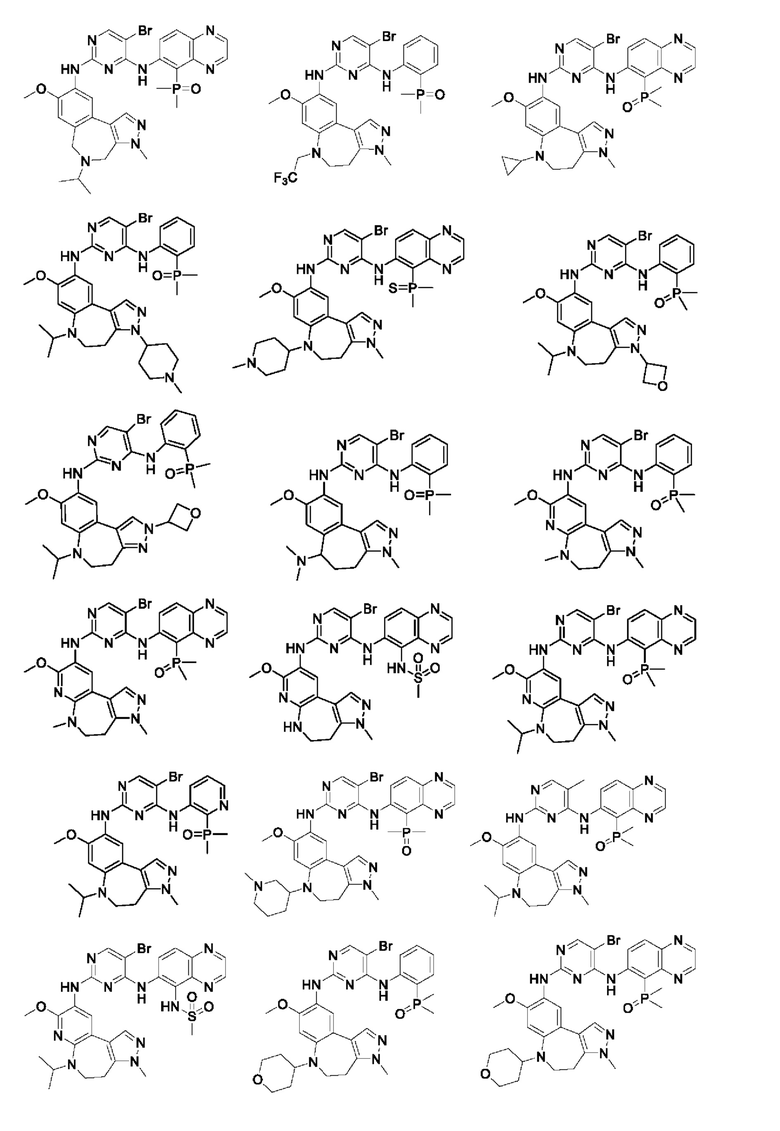
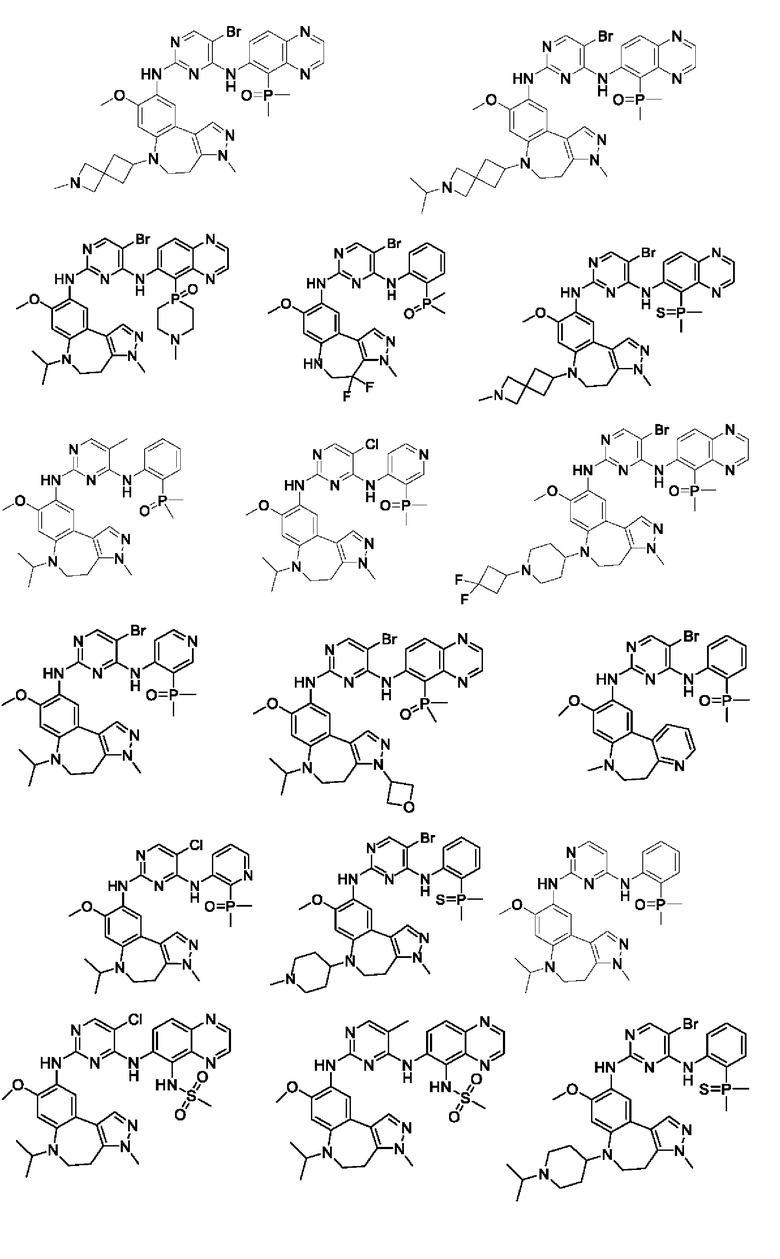
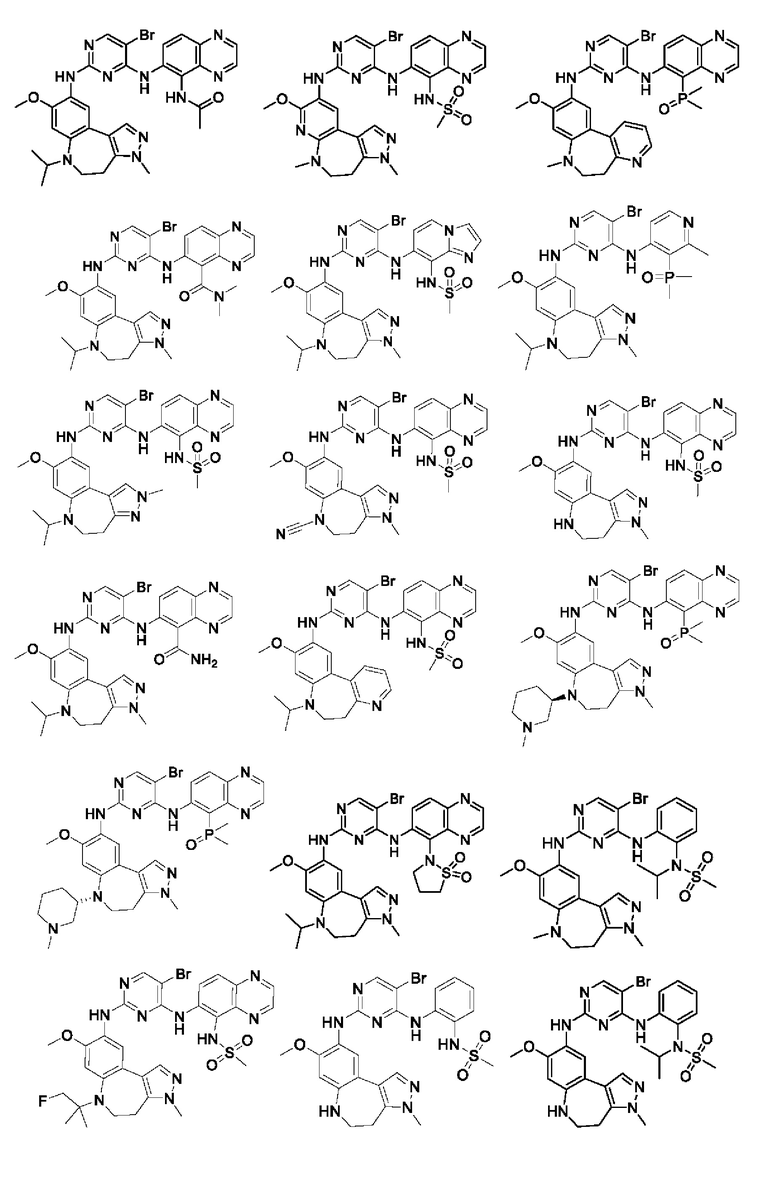
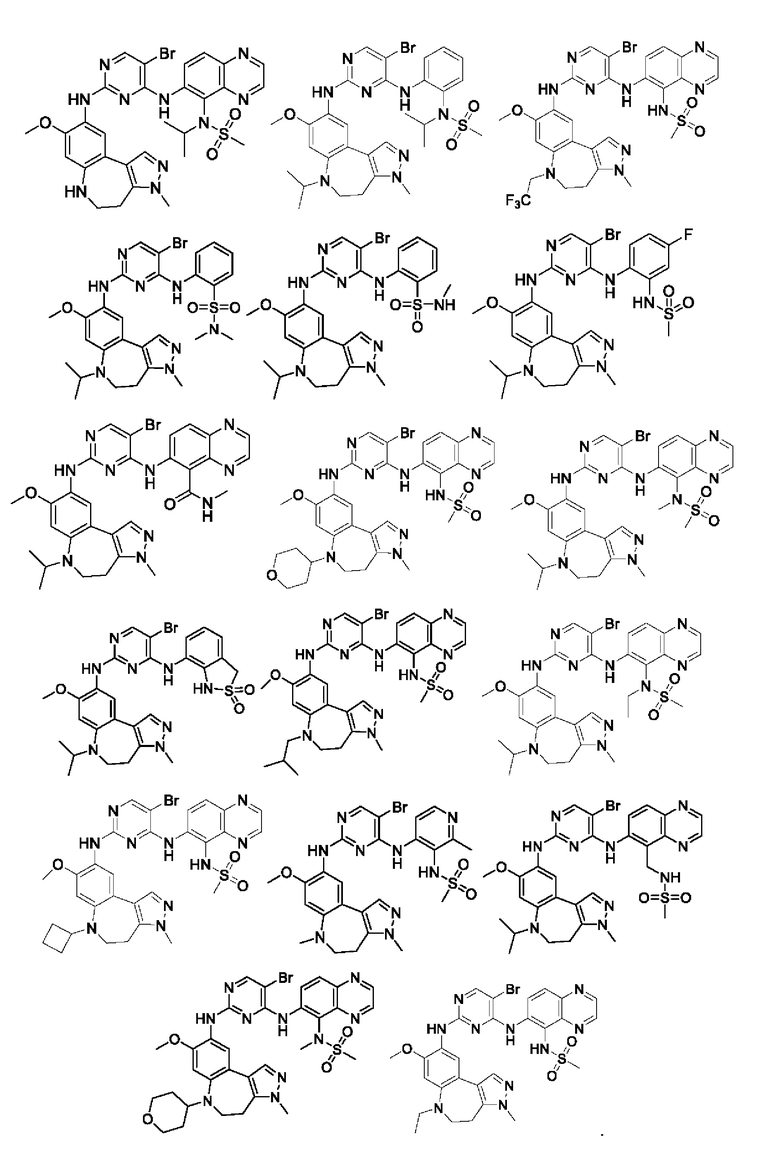
В настоящем изобретении также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли, описанных выше, и фармацевтически приемлемый носитель, разбавитель и вспомогательное вещество. Фармацевтические композиции, предложенные в настоящем изобретении, можно получить для обеспечения конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение. В частности, способы введения включают пероральное введение в формах, например, таблеток, капсул (в том числе составов с замедленным высвобождением или с установленным временем высвобождения), пилюль, порошков, гранул, эликсиров, настоек, суспензий (в том числе наносуспензий, микросуспензий, высушенных распылением дисперсий), сиропов и эмульсий; сублингвальное введение; введение в формах для приема внутрь; парентеральное введение, например, путем подкожной, внутривенной, внутримышечной или интрастернальной инъекции или с применением инфузионных методов (например, в виде стерильного водного раствора для инъекций или неводных растворов или суспензий для инъекций); назальное введение, в том числе введение в слизистую оболочку носа, например, с помощью спрея для ингаляций; местное введение в форме, например, кремов или мазей; или ректальное введение в форме, например, суппозиториев. Указанные формы могут быть введены сами по себе, но, как правило, их вводят с фармацевтическим носителем, выбранным в соответствии с выбранным путем введения и стандартной фармацевтической практикой.
«Фармацевтически приемлемые носители» относятся к наполнителям, общепринятым в данной области техники для доставки биологически активных веществ животным, в частности млекопитающим, и включают, например, адъюванты, вспомогательные вещества, такие как разбавители, консерванты, наполнители, модификаторы вязкости, разрыхлители, смачивающие средства, эмульгаторы, суспендирующие средства, подсластители, вкусоароматические вещества, ароматизаторы, антибактериальные средства, противогрибковые средства, смазывающие вещества и диспергирующие средства, в зависимости от способа введения и природы лекарственной формы. Фармацевтически приемлемые носители получают в пределах компетенции обычного специалиста в данной области техники на основе ряда факторов, включающих, но не ограниченных ими: тип и характер полученных активных веществ, субъект, которому должна быть введена композиция, содержащая указанные вещества, предполагаемый путь введения композиции и целевое терапевтическое показание. Фармацевтически приемлемые носители включают как водные, так и неводные среды, и различные твердые и полутвердые лекарственные формы. Такие носители включают множество различных ингредиентов и добавок в дополнение к активным веществам, при этом указанные дополнительные ингредиенты, включенные в состав по различным причинам (например, для стабилизации активных веществ и адгезивов), хорошо известны обычному специалисту в данной области техники.
В качестве рекомендации общего порядка, при применении для обеспечения заданного эффекта суточная доза для перорального введения каждого активного ингредиента составляет примерно от 0,001 до 5000 мг в сутки, или примерно от 1 до 500 мг, или примерно от 1 до 250 мг, или примерно от 1 до 150 мг, или примерно от 0,5 до 100 мг, или примерно от 1 до 50 мг; наиболее предпочтительная внутривенная доза во время инфузии с постоянной скоростью составляет примерно от 0,01 до 10 мг/кг/мин. Соединения согласно настоящему изобретению можно вводить в одной суточной дозе, или общую суточную дозу можно вводить в дробных дозах 2, 3 или 4 раза в сутки.
Режим дозирования соединений согласно настоящему изобретению, конечно, будет меняться в зависимости от таких известных факторов, как фармакодинамические характеристики конкретного вещества и способ и путь его введения, вид, возраст, пол, состояние здоровья, медицинское состояние и масса тела субъекта, характер и степень симптомов, вид дополнительной терапии, частота применения терапии, способ введения, функция почек и печени пациента и требуемый эффект. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинации будет зависеть от вида, массы тела, возраста и индивидуального состояния субъекта, состояния или заболевания, подлежащего лечению, или его тяжести. Врач, клиницист или ветеринар обычной квалификации может легко определить эффективное количество каждого активного ингредиента, которое необходимо для предотвращения, лечения или ингибирования прогрессирования состояния или заболевания.
В настоящем описании также предложено применение соединения или его фармацевтически приемлемой соли, описанных выше, или фармацевтической композиции для получения лекарственного средства для лечения опухоли.
EGFR (рецептор эпидермального фактора роста) широко распространен на поверхности клеток млекопитающих, таких как эпителиальные клетки, фибробласты и глиальные клетки. Сигнальный путь EGFR играет важную роль в физиологических процессах, таких как рост, пролиферация и дифференциация клеток. Мутация EGFR также является одним из наиболее распространенных типов мутаций у пациентов с NSCLC и может встречаться, в частности, у от 40% до 50% людей в азиатской популяции, соответственно, в некоторых вариантах реализации соединения согласно настоящему изобретению можно использовать для лечения рака с высокой экспрессией EGFR. Указанные раковые заболевания включают лимфому неходжкинскую лимфому, рак яичника, рак шейки матки, рак предстательной железы, колоректальный рак, рак молочной железы, рак поджелудочной железы, глиому, глиобластому меланому, лейкоз, рак желудка, рак эндометрия, рак легкого, печеночноклеточный рак, рак желудка, желудочно-кишечную стромальную опухоль (GIST), острый миелогенный лейкоз (AML), холангиокарциному, рак почки, рак щитовидной железы, анапластическую крупноклеточную лимфому, мезотелиому множественную миелому и меланому.
В настоящем изобретении также предложен способ лечения рака, включающий введение пациенту терапевтически эффективного количества соединения формулы (I'''), формулы (I''), формулы (I') или формулы (I) или его фармацевтически приемлемой соли, описанных выше, или фармацевтической композиции, описанной выше. Указанные раковые заболевания включают лимфому, неходжкинскую лимфому, рак яичника, рак шейки матки, рак предстательной железы, колоректальный рак, рак молочной железы, рак поджелудочной железы, глиому, глиобластому, меланому, лейкоз, рак желудка, рак эндометрия, рак легкого, печеночноклеточный рак, рак желудка, желудочно-кишечную стромальную опухоль (GIST), острый миелогенный лейкоз (AML), холангиокарциному, рак почки, рак щитовидной железы, анапластическую крупноклеточную лимфому, мезотелиому, множественную миелому и меланому.
Соединение формулы (I'''), формулы (I''), формулы (I') или формулы (I) или его фармацевтически приемлемую соль или фармацевтическую композицию, предложенные в настоящем изобретении, применяют для лечения раковых заболеваний, в том числе лимфомы, неходжкинской лимфомы, рака яичника, рака шейки матки, рака предстательной железы, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, лейкоза, рака желудка, рака эндометрия, рака легкого, печеночноклеточного рака, рака желудка, желудочно-кишечной стромальной опухоли (GIST), острого миелогенного лейкоза (AML), холангиокарциномы, рака почки, рака щитовидной железы, анапластической крупноклеточной лимфомы, мезотелиомы, множественной миеломы и меланомы.
В некоторых вариантах реализации настоящего изобретения рак представляет собой рак легкого.
В настоящем изобретении также предложено промежуточное соединение, представленное формулой (V), при этом указанное промежуточное соединение или его фармацевтически приемлемую соль выбирают из:
где R11 представляет собой -NH2 или -NO2;
структурное звено  определено выше;
определено выше;
еще более предпочтительно, промежуточное соединение формулы (V) или его фармацевтически приемлемую соль выбирают из
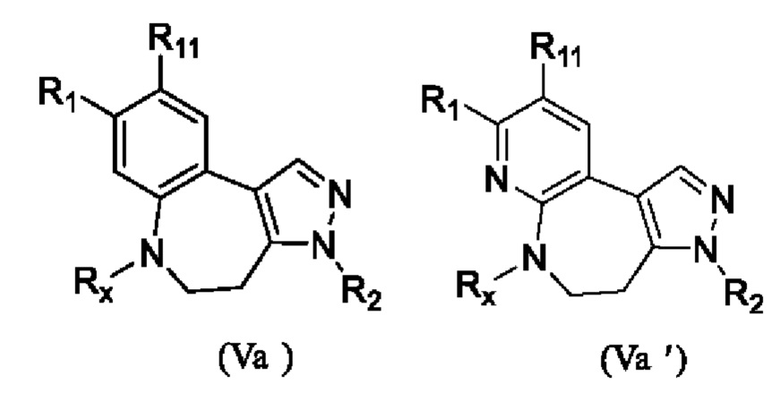
где R1, R2, R11 и Rx определены выше.
Еще более предпочтительно, промежуточное соединение, представленное формулой (V), или его фармацевтически приемлемую соль выбирают из
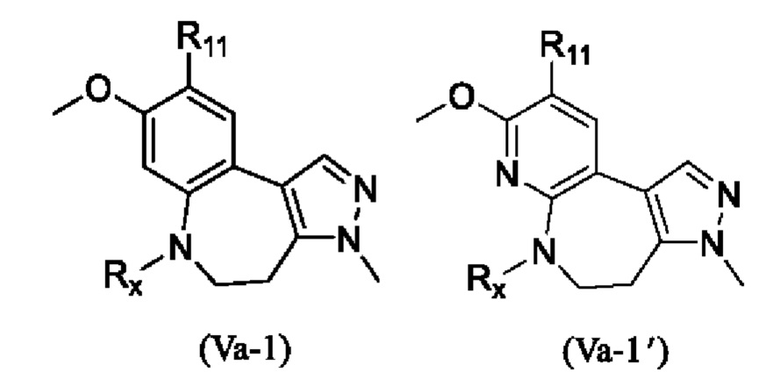
где R11 и Rx определены выше.
Технические эффекты
Соединение согласно настоящему изобретению демонстрирует хорошее ингибирующее действие на киназу EGFR (L858R/T790M/C797S) и слабое ингибирующее действие на киназу EGFR дикого типа, что указывает на то, что соединение согласно настоящему изобретению обладает относительно хорошей киназной активностью и селективностью.
Соединение согласно настоящему изобретению оказывает хорошее ингибирующее действие на клеточную пролиферацию линии клеток с тройной мутацией EGFR Ba/F3 Del19/T790M/C797S и линии клеток с тройной мутацией EGFR Ba/F3 L858R/T790M/C797S и относительно слабое ингибирующее действие на линию клеток дикого типа А431 EGFR, что указывает на то, что соединения согласно настоящему изобретению обладают хорошей клеточной жизнеспособностью и селективностью.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет кривую роста опухоли (мм3) для каждой группы животных в эксперименте по исследованию эффективности in vivo.
Фиг. 2 представляет собой кривую массы тела (г) для каждой группы животных в эксперименте по исследованию эффективности in vivo.
ОПИСАНИЕ И ОПРЕДЕЛЕНИЕ
Если не указано иное, подразумевают, что следующие термины и выражения, применяемые в настоящей заявке, имеют следующие значения. Конкретный термин или выражение, если специально не указано иное, не следует рассматривать как неопределенный или неясный, но следует понимать в соответствии с его общепринятым значением.
Термин «фармацевтически приемлемый» относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения подходят для приведения в контакт с тканями людей и животных без проявления чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений и соответствуют разумному соотношению польза/риск.
Термин «фармацевтически приемлемые соли» относится к производным соединений согласно настоящему изобретению, которые получают с применением относительно нетоксичных кислот или оснований. Указанные соли можно получить во время синтеза, разделения, очистки соединений или путем взаимодействия свободной формы очищенных соединений с подходящей кислотой или основанием. Когда соединения содержат относительно кислые функциональные группы, указанные соединения могут взаимодействовать с щелочными металлами, гидроксидами щелочноземельных металлов или органическими аминами с получением солей присоединения оснований, в том числе катионов на основе щелочных металлов и щелочноземельных металлов, таких как катионы натрия, лития, калия, кальция и магния, а также нетоксичных катионов аммония, четвертичного аммония и амина. Когда соединения содержат относительно основные функциональные группы, указанные соединения взаимодействуют с органическими кислотами или неорганическими кислотами с получением солей присоединения кислоты.
Соединения, предложенные в настоящем документе, также включают пролекарственные формы, которые означают соединения, которые быстро превращаются in vivo в исходное соединение согласно приведенным формулам и которые превращаются в соединения согласно настоящему изобретению посредством химических или биохимических процессов в среде in vivo или in vitro, например, посредством гидролиза в крови.
Соединения согласно настоящему изобретению могут существовать в несольватированных формах и сольватированных формах, в том числе в гидратированных формах. В общем случае, сольватированные формы эквивалентны несольватированным формам и должны быть включены в объем настоящего изобретения.
Соединения согласно настоящему изобретению имеют геометрические изомеры и стереоизомеры, такие как цис-транс-шомеры, энантиомеры, диастереоизомеры и их рацемические и другие смеси, которые все включены в объем настоящего изобретения.
Термин «энантиомер» относится к стереоизомерам, которые являются зеркальными отражениями друг друга.
Термин «диастереоизомер» относится к стереоизомерам, молекулы которых имеют два или более хиральных центра и не являются зеркальными отражениями друг друга.
Термин «цис-транс-жомер» относится к конфигурации, возникающей в результате неспособности одинарной связи кольцевого атома углерода или двойной связи в молекуле свободно вращаться.
Если не указано иное, абсолютную конфигурацию стереогенного центра изображают в виде клиновидной сплошной связи  и клиновидной пунктирной связи
и клиновидной пунктирной связи  а относительную конфигурацию стереогенного центра изображают в виде прямой сплошной связи
а относительную конфигурацию стереогенного центра изображают в виде прямой сплошной связи  и прямой пунктирной связи
и прямой пунктирной связи 
 означает, что атом углерода представляет собой хиральный атом углерода, при этом указанная структура изображает оптически чистое соединение, в котором пространственная конфигурация атома углерода представляет собой (R) конфигурацию или (S) конфигурацию и их смесь.
означает, что атом углерода представляет собой хиральный атом углерода, при этом указанная структура изображает оптически чистое соединение, в котором пространственная конфигурация атома углерода представляет собой (R) конфигурацию или (S) конфигурацию и их смесь.
Стереоизомеры соединений согласно настоящему изобретению можно получить с помощью хирального синтеза или хиральных реагентов или других общепринятых способов. Например, один энантиомер соединения согласно настоящему изобретению можно получить методами асимметричного катализа, или методами хиральной вспомогательной дериватизации, или методами хирального разделения, т.е. соединение с одной пространственной конфигурацией получают из смеси или путем применения хиральных исходных материалов. В настоящем изобретении разделение оптически чистых соединений обычно осуществляют с помощью препаративной хроматографии и с применением хиральных хроматографических колонок, обеспечивающих разделение хиральных соединений.
Термин «оптически чистый» или «энантиомерно обогащенный» означает, что изомер или энантиомер присутствует в количестве, составляющем более или равном 60%, или более или равном 70%, или более или равном 80%, или более или равном 90%, или более или равном 95%, или более или равном 96%, или более или равном 97%, или более или равном 98%, или более или равном 99%, или более или равном 99,5%, или более или равном 99,6%, или более или равном 99,7%, или более или равном 99,8%, или более или равном 99,9%.
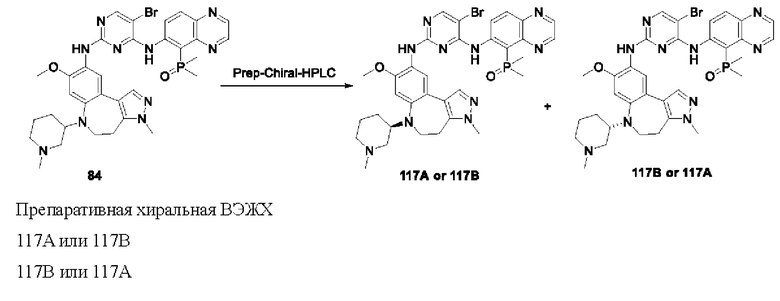
Абсолютную пространственную конфигурацию соединения можно подтвердить посредством способа, общепринятого в данной области техники. Например, абсолютную конфигурацию соединения также можно подтвердить с помощью рентгеновской дифракции монокристаллов, хиральной структуры исходных материалов и механизма реакции асимметричного синтеза. Соединения, помеченные в настоящем документе как «абсолютная конфигурация не определена», обычно отделяют от рацемата с получением отдельных изомеров с помощью хиральной препаративной ВЭЖХ или СФХ (сверхкритическая флюидная хроматография) с последующим определением характеристик и исследованием.
Например, на приведенной ниже формуле изображено отделение соединений 117А и 117 В от соединения 84 в виде энантиомеров друг друга; однако абсолютные пространственные конфигурации соединений 117Аи 117 В не определены.
Настоящее изобретение также включает меченые изотопами соединения, в том числе изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 11C, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F и 36Cl. Соединения согласно настоящему изобретению, содержащие перечисленные выше изотопы и/или другие изотопы других атомов, находятся в пределах объема настоящего изобретения.
Термин «фармацевтически приемлемые носители» относится к наполнителям, общепринятым в данной области техники для доставки биологически активных веществ животным, в частности млекопитающим, и включает, например, адъюванты, вспомогательные вещества, такие как разбавители, консерванты, наполнители, модификаторы вязкости, разрыхлители, смачивающие средства, эмульгаторы, суспендирующие средства, подсластители, вкусоароматические вещества, ароматизаторы, антибактериальные средства, противогрибковые средства, смазывающие вещества и диспергирующие средства, в зависимости от способа введения и природы лекарственной формы. Фармацевтически приемлемые носители получают в пределах компетенции обычного специалиста в данной области техники на основе ряда факторов, включающих, но не ограниченных ими: тип и характер полученных активных веществ, субъект, которому должна быть введена композиция, содержащая указанные вещества, предполагаемый путь введения композиции и целевое терапевтическое показание. Фармацевтически приемлемые носители включают как водные, так и неводные среды, и различные твердые и полутвердые лекарственные формы. Такие носители содержат множество различных ингредиентов и добавок в дополнение к активным веществам, при этом указанные дополнительные ингредиенты, включенные в состав по различным причинам (например, для стабилизации активных веществ и адгезивов), хорошо известны обычному специалисту в данной области техники.
Термин «вспомогательное вещество» относится в общем случае к носителю, разбавителю и/или среде, необходимой для получения эффективной фармацевтической композиции.
Термин «эффективное профилактическое или терапевтическое количество» относится к количеству соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, достаточному для лечения расстройства при разумном соотношении польза/риск, применимом к любому медицинскому вмешательству и/или профилактике. Однако следует понимать, что общее суточное количество соединения формулы I или его фармацевтически приемлемой соли и композиции согласно настоящему изобретению будет определено лечащим врачом в рамках здравого медицинского суждения. Для любого данного пациента конкретный терапевтически эффективный уровень дозы будет зависеть от множества факторов, в том числе расстройства, подлежащего лечению, и тяжести такого расстройства, активности конкретного применяемого соединения, конкретной применяемой композиции, возраста, массы тела, общего состояния здоровья, пола и диеты пациента, времени введения, пути введения и скорости экскреции конкретного применяемого соединения, продолжительности лечения, лекарственных препаратов, используемых в комбинации или одновременно с конкретным применяемым соединением, и аналогичных факторов, хорошо известных в области медицины. Например, в данной области техники известно, что дозу соединения начинают вводить на уровне ниже уровня, необходимого для достижения требуемого терапевтического эффекта, а затем постепенно увеличивают дозу до достижения требуемого терапевтического эффекта. В общем случае, дозировка соединения формулы I или его фармацевтически приемлемой соли согласно настоящему изобретению может составлять для млекопитающих, в частности, людей, от примерно 0,001 до примерно 1000 мг/кг массы тела в сутки, например, от примерно 0,01 до примерно 100 мг/кг массы тела в сутки и от примерно 0,01 до примерно 10 мг/кг массы тела в сутки.
Термин «необязательно замещенный» означает, что атом может содержать или не содержать заместитель. Если не указано иное, тип и количество заместителей может быть произвольным при условии, что это является химически достижимым. Например, термин «необязательно содержит в качестве заместителя один или более R.2» означает, что атом может содержать или не содержать в качестве заместителя один или более R.2.
Когда любой переменный параметр (например, R2) встречается в составе или структуре соединения более одного раза, указанный переменный параметр независимо определяют в каждом случае. Например, если группа содержит в качестве заместителя от 0 до 2 R2, указанная группа может необязательно содержать в качестве заместителя до двух R2, при этом определение R2 в каждом случае является независимым.
Когда линкерная группа изображена с применением цифры 0, например, -O(СН2)nCH3, n=O указывает на то, что указанная линкерная группа представляет собой одинарную связь, т.е. -ОСН3.
Когда заместитель может образовывать поперечные связи с двумя атомами в кольце, указанный заместитель может быть связан с любым атомом в кольце. Например, структурное звено 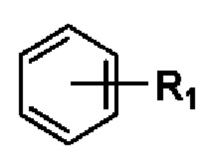 означает, что фенильное кольцо может содержать заместитель R1 в любом положении.
означает, что фенильное кольцо может содержать заместитель R1 в любом положении.
Когда при перечислении заместителей не указано, каким атомом они присоединены к соединениям, включенным в общую химическую структурную формулу, но конкретно не упомянутым, такие заместители могут быть связаны через любой их атом. Например, пиразол в качестве заместителя означает, что любой из атомов углерода в пиразольном кольце присоединен к замещенной группе;  или
или  присутствующий в такой структуре, указывает, что атом представляет собой связывающий атом, например,
присутствующий в такой структуре, указывает, что атом представляет собой связывающий атом, например, 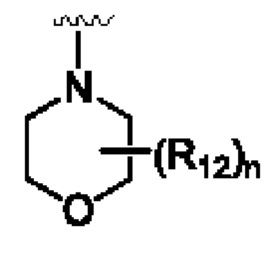 или
или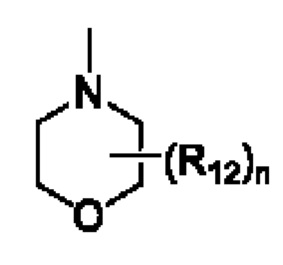 оба указывают, что атомы N в морфолиновом кольце представляют собой связывающие атомы.
оба указывают, что атомы N в морфолиновом кольце представляют собой связывающие атомы.
Если не указано иное, «кольцо» относится к насыщенному, частично насыщенному или ненасыщенному моноциклическому и полициклическому кольцу, в том числе спироциклическому, бициклическому или мостиковому кольцу. Типичные «кольца» включают замещенный или незамещенный циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил, арил или гетероарил. Термин «гетеро» обозначает замещенный или незамещенный гетероатом, а также окисленную форму гетероатома, при этом указанный гетероатом в общем случае выбран из N, О и S, при этом окисленная форма гетероатома в общем случае включает NO, SO и S(O)2, при этом атомы азота могут содержать заместитель, т.е. NR. (R представляет собой Н или другой заместитель, определенный в настоящем документе); количество атомов в кольце в общем случае определяют как количество членов кольца, например, «3-6-членный гетероциклоалкил» относится к кольцу из 3-6 атомов, расположенных вокруг кольца, при этом каждое кольцо необязательно содержит от 1 до 3 гетероатомов, т.е., N, О, S, NO, SO, S(O)2 или NR, при этом каждое кольцо необязательно содержит в качестве заместителя R, при этом R представляет собой группу, определенную в настоящем документе.
Если не указано иное, термин «карбоциклическое кольцо» или «карбоциклил» означает стабильную циклическую структуру, состоящую из атомов углерода, которая может быть моноциклической, бициклической или трициклической и которая может быть насыщенной, частично ненасыщенной или ненасыщенной (ароматической). Любое из перечисленных выше карбоциклических колец может быть сконденсировано с одним или более ароматическими кольцами и ароматическими гетероциклическими кольцами с образованием полициклических колец, таких как бициклические и трициклические кольца.
Например, в структурном звене 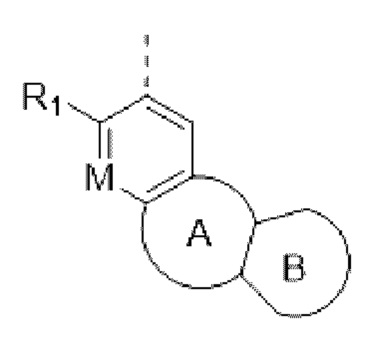 или
или 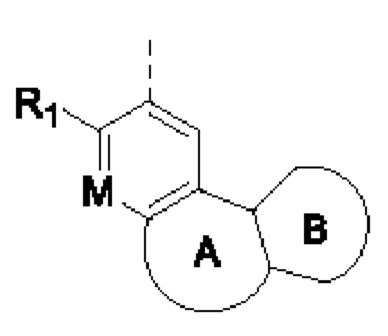 кольцо А может представлять собой 5-8-членный карбоциклил, примеры которого включают, но не ограничиваются ими,
кольцо А может представлять собой 5-8-членный карбоциклил, примеры которого включают, но не ограничиваются ими, 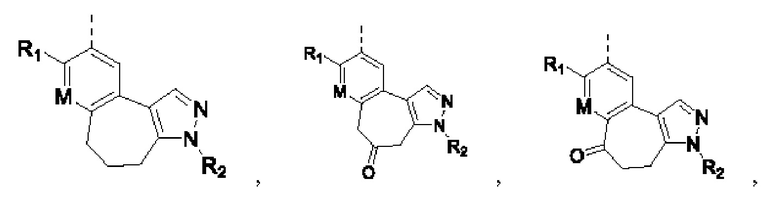
 при этом R1, R2, Rx и М представляют собой группы, определенные в настоящем документе.
при этом R1, R2, Rx и М представляют собой группы, определенные в настоящем документе.
Если не указано иное, термин «гетероциклическое кольцо» или «гетероциклил» означает стабильное моноциклическое, бициклическое или трициклическое кольцо, содержащее гетероатом или группу гетероатомов, при этом указанные кольца могут быть насыщенными, частично ненасыщенными или ненасыщенными (ароматическими) и могут содержать атомы углерода и 1, 2, 3 гетероатома, независимо выбранные из N, О, S, NO, SO, S(O)2 и NR, при этом любое из перечисленных выше гетероциклических колец может быть сконденсировано с одним или более ароматическими кольцами и ароматическими гетероциклическими кольцами с образованием полициклических колец, таких как бициклические и трициклические кольца. Например, в структурном звене 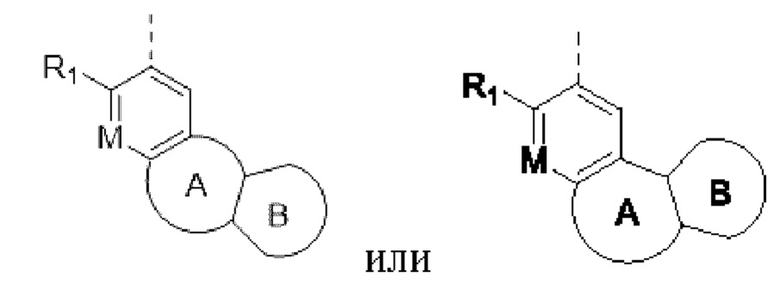 кольцо А может представлять собой 5-8-членный гетероциклил, примеры которого включают, но не ограничиваются ими,
кольцо А может представлять собой 5-8-членный гетероциклил, примеры которого включают, но не ограничиваются ими, 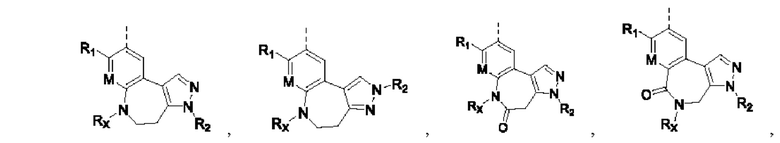
 при этом R1, R2, RK и М представляют собой группы, определенные в настоящем документе.
при этом R1, R2, RK и М представляют собой группы, определенные в настоящем документе.
Если не указано иное, термин «арил» относится к ненасыщенному и обычно ароматическому гидрокарбилу, который может представлять собой одно кольцо или множество колец, сконденсированных вместе, предпочтительно С5-10 ар илу, более предпочтительно С5-8 арилу и наиболее предпочтительно моноциклическому С5-6 арилу, примеры арила включают, но не ограничиваются ими, фенил и нафтил.
Если не указано иное, термин «гетероарил» означает стабильный моноциклический или полициклический ароматический углеводород, содержащий по меньшей мере один гетероатом (N, О, S, NO, SO, S(O)2 или NR), предпочтительно 5- или 6-членный моноциклический гетероарил. Примеры гетероарила включают, но не ограничиваются ими, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил и пиримидинил.
Если не указано иное, термин «алкил» относится к линейному или разветвленному насыщенному гидрокарбилу, предпочтительно C1-6 алкилу и более предпочтительно С1-3 алкилу; примеры алкила включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, бутил, изобутил, пентил, изопентил, неопентил, н-гексил и т.п.
Если не указано иное, термин «гетероалкил» относится к алкильной группе, в которой один или более атомов углерода замещены гетероатомом, выбранным из В, О, N и S, при этом атомы азота и серы необязательно являются окисленными, гетероатом азота необязательно является кватернизованным, в том числе, но не ограничиваясь ими, к «алкокси», «алкиламино», «алкилтио» и т.п.; примеры «гетероалкила» включают, но не ограничиваются ими, -ОСН3, -ОСН2СН3, -ОСН2СН2СН3, -ОСН(СН3)2, -N(CH3)2, -СН2-СН2-О-СНз, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -S(O)2-CH3, -CH2-CH2-S(O)2-CH3 и т.п.
Если не указано иное, «алкенил» относится к алкильной группе, содержащей одну или более углерод-углеродных двойных связей, предпочтительно С2-8 алкенилу, при этом примеры алкенила включают, но не ограничиваются ими, этенил, пропенил, бутенил, пентенил, гексенил и т.п.
Если не указано иное, «алкинил» относится к алкильной группе, содержащей одну или более углерод-углеродных тройных связей, предпочтительно С2-8 алкинилу, при этом примеры алкинила включают, но не ограничиваются ими, этинил, пропинил, бутинил, пентинил и т.п.
Если не указано иное, термин «галоген» относится к атому фтора, хлора, брома или
иода.
Если не указано иное, термин «галогеналкил» относится к алкильной группе, содержащей один или более атомов водорода, замещенных атомом галогена, предпочтительно C1-6 галогеналкилу и более предпочтительно С1-3 галогеналкилу; при этом примеры галогеналкила включают, но не ограничиваются ими, монофторметил, дифторметил, трифторметил, трихлорметил, трибромметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил и т.п.
Если не указано иное, термин «алкокси» относится к алкильной группе, присоединенной через кислородный мостик, т.е. к группе, полученной в результате замещения атома водорода в гидроксильной группе алкильной группой, предпочтительно к C1-6 алкокси и более предпочтительно С1-3 алкокси. Примеры алкокси включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентокси, неопентокси и н-гексокси.
Если не указано иное, термин «циклоалкилокси» относится к циклоалкильной группе, присоединенной через кислородный мостик, т.е. к группе, полученной в результате замещения атома водорода в гидроксильной группе циклоалкильной группой. Циклоалкилокси предпочтительно представляет собой 3-7-членный, 4-7-членный или 5-7-членный циклоалкилокси. Примеры циклоалкилокси включают, но не ограничиваются ими, циклопропилокси, циклобутилокси, циклопентилокси и циклогексилокси.
Если не указано иное, термин «галогеналкокси» относится к алкокси группе, в которой один или более атомов водорода замещены атомом галогена. Примеры галогеналкокси включают, но не ограничиваются ими, трифторметокси, трихлорметокси, 2,2,2-трифторэтокси и 2,2,2-трихлорэтокси.
Если не указано иное, «циклоалкил» относится к насыщенному моноциклическому или полициклическому гидрокарбилу. Циклоалкил предпочтительно представляет собой 3-8-членный моноциклический алкил и более предпочтительно 3-6-членный моноциклический алкил, при этом примеры таких моноциклических алкилов включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Если не указано иное, «гетероциклоалкил» относится к моногетероциклоалкилу и полигетероциклоалкилу содержащему в кольце определенное количество гетероатомов, при этом указанные гетероатомы обычно выбраны из N, О, S, NO, SO, S(O)2 и NR. Гетероциклоалкил предпочтительно представляет собой 3-8-членный моноциклический гетероциклоалкил и более предпочтительно 3-6-членный моноциклический гетероциклоалкил, при этом примеры таких моноциклических гетероциклоалкилов включают, но не ограничиваются ими, оксиранил, тетрагидропирролил, пиперидил, пиперазинил, морфолинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, 1,3-диоксолан, 1,4-диоксан и т.п.
Если не указано иное, «спироциклил» относится к бициклическому или полициклическому гидрокарбилу, два моноциклических кольца которого имеют общий атом углерода. Спироциклил предпочтительно представляет собой 5-13-членную спирокольцевую группу, 6-12-членный спироциклил или 7-11-членный спироциклил, при этом 6-12-членный спироциклил означает углеводородную группу, состоящую из от 6 до 12 атомов в спироциклической скелетной структуре, при этом примеры спироциклила включают, но не ограничиваются ими, спиро[2,2]пентил, спиро[2,3]гексил, спиро[2,4]гептил, спиро[2,5]октил, спиро[2,6]нонил, спиро[3,3]гептил, спиро[3,4]октил, спиро[3,5]нонил, спиро[3,6]децил, спиро[4,4]нонил, спиро[4,5]децил, спиро[4,6]ундецил, спиро[5,5]ундецил, спиро[5,6]додецил, спиро[6,6]тридецил и спиро[6,7]тетрадецил.
Если не указано иное, «спирогетероциклил» относится к спироциклической группе, в которой один или более атомов углерода в спироциклической скелетной структуре замещены гетероатомом, выбранным из N, О и S. Спирогетероциклил предпочтительно представляет собой 5-13-членный спирогетероциклил, 6-12-членный спирогетероциклил или 7-11-членный спирогетероциклил. Примеры спирогетероциклила включают, но не ограничиваются ими, 2-окса-7-азаспиро[5.3]нонан-7-ил, 2-окса-7-азаспиро[4.4]нонан-7-ил, 2-окса-6-азаспиро[3.3]гептан-6-ил, 2-окса-8-азаспиро[4.5]декан-8-ил, 1,4,9-триазаспиро[5,5]ундекан-9-ил, 3-окса-9-азаспиро[5,5]ундекан-9-ил, 2,6-диазаспиро[3,3]гептан-2-ил, 2,7-диазаспиро[5,3]нонан-7-ил, 2,7-диоксаспиро[5,3]нонил, 3,9-диазаспиро[5,5]ундекан-3-ил, 1-окса-4,9-диазаспиро[5,5]ундекан-9-ил, 1-окса-4,8-диазаспиро[5,4]декан-8-ил, 3-азаспиро[5.5]ундекан-3-ил, 7-азаспиро[3.5]декан-7-ил, 1-окса-4,9-диазаспиро[5.5]ундекан-4-ил, 6-окса-2,9-диазаспиро[4.5] декан-9-ил, 9-окса-2,6-диазаспиро [4.5] декан-6-ил, 3-азаспиро [5.5]ундекан-3-ил и 4-окса-1,9-диазаспиро[5.5]ундекан-9-ил.
Термин «4-7-членное кольцо, содержащее P(=O)Rb» относится к группе, имеющей структуру 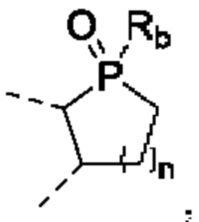 где n представляет собой 0, 1, 2 или 3; Rb представляет собой группу, определенную в настоящем документе, при этом 4-7-членное кольцо, содержащее -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2, представляет собой структуру
где n представляет собой 0, 1, 2 или 3; Rb представляет собой группу, определенную в настоящем документе, при этом 4-7-членное кольцо, содержащее -P(=S)(Rb)-, -N(Rb)S(=O)2-, -S(=O)2N(Rb)- или -S(=O)2, представляет собой структуру  где n представляет собой 0, 1, 2 или 3; Rb представляет собой группу, определенную в настоящем документе.
где n представляет собой 0, 1, 2 или 3; Rb представляет собой группу, определенную в настоящем документе.
Особо отмечается, что все комбинации заместителя и/или его варианта допустимы только в том случае, если указанные комбинации могут приводить к образованию стабильного соединения.
В примерах настоящего изобретения указанное в заголовке соединение получает название после превращения посредством структуры соединения с помощью Chemdraw. Если название соединения не соответствует структуре соединения, название соединения можно определить путем интеграции соответствующей информации и пути реакции; при этом данная структурная формула соединения будет иметь преимущественную силу, если название соединения не может быть подтверждено другими способами.
В настоящем изобретении способы получения некоторых соединений относятся к способам получения аналогичных соединений, описанных выше. Специалистам в данной области техники будет понятно, что при применении или ссылке на способ получения, указанный в настоящем документе, соотношение при подаче реагентов, реакционного растворителя, температуры реакции и т.п. можно корректировать соответствующим образом в зависимости от реагентов.
Соединения, описанные в настоящем документе, можно получить различными способами синтеза, хорошо известными специалистам в данной области техники, включая конкретные варианты реализации, перечисленные ниже, варианты реализации, полученные путем их комбинаций с другими способами химического синтеза, и их эквиваленты, известные специалистам в данной области техники. Предпочтительные варианты реализации включают, но не ограничиваются ими, примеры настоящего изобретения.
Ниже приведены сокращения и их соответствующие химические названия, применяемые в примерах настоящего изобретения
ПОДРОБНОЕ ОПИСАНИЕ
Структуру соединений согласно настоящему изобретению определяли с помощью ядерного магнитного резонанса (ЯМР) и/или жидкостной хроматографии-масс-спектрометрии (ЖХ-МС). Химический сдвиг ЯМР (δ) приведен в частях на миллион (ppm). Определение методом ЯМР проводили с помощью ЯМР-спектрометров (Bruker AVANCE III HD 400 и Bruker AVANCE III HD 300) с применением дейтерированного диметилсульфоксида (ДМСО-d6), дейтерированного метанола (CD3OD) и дейтерированного хлороформа (CDCl3) в качестве растворителей и тетраметилсилана (TMS) в качестве внутреннего стандарта.
Определение методом ЖХ-МС осуществляли с применением масс-спектрометра SHIMADZU LCMS-2020 (в качестве источника ионов использовали электрораспылительную ионизацию). Определение методом ВЭЖХ проводили с применением жидкостной хроматографии высокого давления SHIMADZU LC-20 АР XR и SPD-M20A.
Для тонкослойной хроматографии (ТСХ) использовали пластину с силикагелем Xinnuo Chemical GF254 (Yantai) согласно спецификациям от 0Д5 мм до 0,20 мм, а в качестве носителя в колоночной хроматографии использовали в общем случае силикагель Yuchen Chemical размером от 200 до 300 меш.
Исходные материалы, применяемые в примерах настоящего изобретения, являются известными и коммерчески доступными или могут быть синтезированы с помощью или в соответствии со способами, известными в данной области техники.
Пример 1:
Получение промежуточного соединения 1А
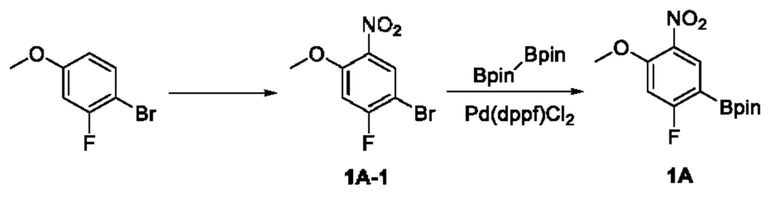
Соединение 1А-1:
3-Фтор-4-броманизол (20 г, 98 ммоль) растворяли в концентрированной серной кислоте (80 мл) при 0°С. Затем к реакционной жидкости отдельными порциями добавляли нитрат калия (9,86 г, 98 ммоль) и последовательно перемешивали реакционную систему при указанной температуре в течение 30 минут. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость медленно выливали в ледяную воду (500 г) для гашения реакции. Смесь экстрагировали этилацетатом (200 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (200 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этил ацетат = 5/1) с получением соединения 1А-1 (11,2 г).
1Н ЯМР (300 МГц, CDCl3) δ 8,19 (d, J=7,2 Гц, 1Н), 6,91 (d, J=9,9 Гц, 1H), 3,99 (s, 3Н).
Промежуточное соединение 1А:
Соединение 1А-1 (3 г, 12 ммоль) растворяли в 1,4-диоксане (20 мл) при комнатной температуре в атмосфере азота. Затем в реакционную жидкость последовательно добавляли бис(пинаколато)дибор (3,3 г, 13 ммоль), ацетат калия (2,4 г, 24 ммоль) и дихлор [1,1'-бис(дифенилфосфино)ферроцен] палладий (II) (0,88 г, 1,2 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 5/1) с получением соединения 1А (1,5 г).
1H ЯМР (300 МГц, CDCl3) δ 8,36 (d, J=6,0 Гц, 1H), 6,76 (d, J=10,5 Гц, 1H), 4,00 (s, 3H), 1,37 (s, 12H).
Получение промежуточного соединения IB
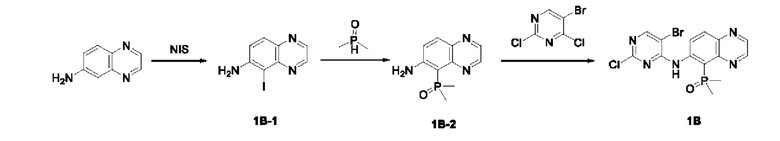
Соединение 1B-1:
6-Аминохиноксалин (59 г, 406,4 ммоль) растворяли в N,N-диметилформамиде (600 мл) и отдельными порциями добавляли N-йодсукцинимид (100,6 г, 447,1 ммоль) при комнатной температуре. Реакционную систему перемешивали при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (3000 мл). Смесь экстрагировали этилацетатом (1000 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (500 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/2) с получением соединения 1В-1 (50 г).
МС (ИЭР) масса/заряд: 272,0 [М+Н]+.
Соединение 1В-2:
Соединение 1В-1 (40 г, 147,6 ммоль) растворяли в N,N-диметилформамиде (400 мл) при комнатной температуре в атмосфере азота. Затем к описанной выше реакционной жидкости последовательно добавляли диметилфосфиноксид (17,3 г, 221,4 ммоль), ацетат палладия (3,3 г, 14,7 ммоль), 4,5-бис-дифенилфосфино-9,9-диметилксантен (12,8 г, 22,1 ммоль) и N,N-диизопропилэтиламин (38,1 г, 295,1 ммоль). Реакционную жидкость нагревали до 120°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и фильтровали, после чего фильтрационный осадок промывали этанолом (100 мл × 3); фильтрат концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 1В-2 (30 г).
МС (ИЭР) масса/заряд: 222,0 [М+Н]+.
Промежуточное соединение 1В:
Соединение 1В-2 (5 г, 22,6 ммоль) растворяли в N,N-диметилформамиде (100 мл) при комнатной температуре в атмосфере азота. Затем к описанной выше реакционной жидкости отдельными порциями добавляли при 0°С гидрид натрия (60%, 1,99 г, 49,8 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут; затем к реакционной жидкости при 0°С добавляли 2,4-дихлор-5-бромпиримидин (6,18 г, 27,1 ммоль), нагревали указанную реакционную жидкость до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (600 мл). Смесь экстрагировали этилацетатом (200 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (500 мл), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 1В (3 г).
Получение промежуточного соединения 1С
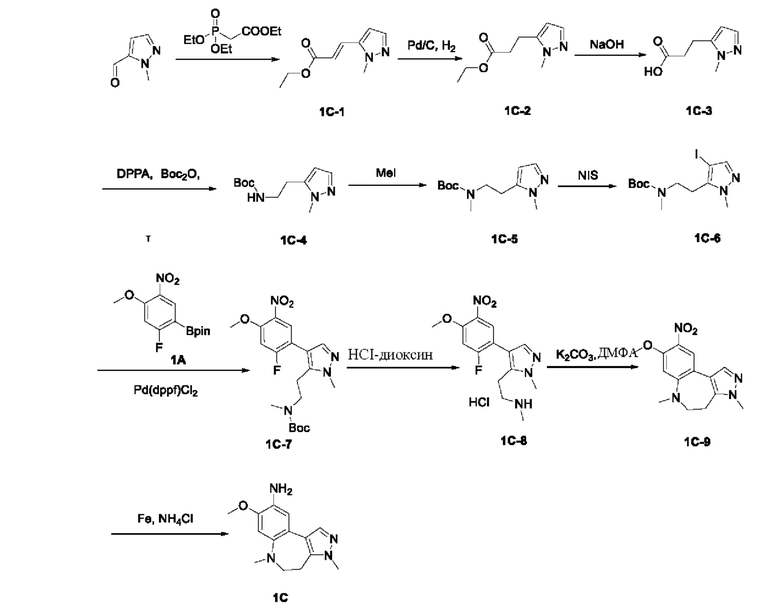
Соединение 1С-1:
Триэтилфосфорилацетат (61,1 г, 273 ммоль) растворяли в тетрагидрофуране (120 мл). Затем к реакционной жидкости в атмосфере азота при 0°С добавляли гидрид натрия (60%, 10,9 г, 273 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут. К реакционной жидкости добавляли раствор 1-метил-Ш-пиразол-5-карбальдегида (20 г, 182 ммоль) в тетрагидрофуране (100 мл). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1,5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (100 мл). Смесь экстрагировали этилацетатом (300 мл × 3), после чего органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этил ацетат = 1/1) с получением соединения 1С-1 (33 г).
МС (ИЭР) масса/заряд: 181,2 [М+Н]+.
Соединение 1С-2:
Соединение 1С-1 (33 г, 183,1 ммоль) растворяли в этаноле (500 мл). К реакционной жидкости добавляли влажный палладий на углероде (10%, 6,6 г). После того, как реакционную систему продували 3 раза с помощью водорода, реакционную жидкость перемешивали при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали этилацетатом (30 мл × 3) и концентрировали полученный фильтрат при пониженном давлении с получением соединения 1С-2 (33 г).
МС (ИЭР) масса/заряд: 183,1 [М+Н]+.
Соединение 1С-3:
Соединение 1С-2 (32 г, 175,6 ммоль) растворяли в этаноле (320 мл). Затем к реакционной жидкости добавляли гидроксид натрия (1 Н, 352 мл). Реакционную систему нагревали до 70°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления этанола. Водную фазу доводили до рН=3 с помощью разбавленной соляной кислоты (1Н) и этилацетата (100 мл × 3), после чего органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 1С-3 (25 г).
МС (ИЭР) масса/заряд: 155,1 [М+Н]+.
Соединение 1С-4:
Соединение 1С-3 (5 г, 32,4 ммоль) и дифенилфосфорилазид (сокращенно DPPA, 9,8 г, 35,7 ммоль) растворяли в трет-бутаноле (80 мл) в атмосфере азота. Затем к реакционной жидкости добавляли триэтиламин (13,1 г, 129,7 ммоль) и ди-трет-бутилдикарбонат (21,2 г, 97,3 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления насыщенного бикарбоната натрия (100 мл). Смесь экстрагировали этилацетатом (200 мл × 3), после чего органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 1С-4 (5,8 г).
МС (ИЭР) масса/заряд: 226,1 [М+Н]+.
Соединение 1С-5:
Соединение 1С-4 (500 мг, 2,22 ммоль) растворяли в N,N-диметилформамиде (4 мл) при 0°С в атмосфере азота. Затем к описанной выше реакционной жидкости добавляли гидрид натрия (60%, 69 мг, 1,7 ммоль). Реакционную жидкость последовательно перемешивали при 0°С в течение 30 минут. Затем к реакционной жидкости добавляли йодметан (329 мг, 2,3 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (20 мл). Смесь экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 15/1) с получением соединения 1С-5 (450 мг).
МС (ИЭР, масса/заряд): 240,1 [М+Н]+.
Соединение 1С-6:
Соединение 1С-5 (400 мг, 1,7 ммоль) растворяли в ацетонитриле (2 мл). Затем к реакционной жидкости добавляли при 0°С N-йодсукцинимид (451 мг, 2,0 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 1С-6 (600 мг).
МС (ИЭР, масса/заряд): 366,0 [М+Н]+.
Соединение 1С-7:
Соединение 1С-6 (664 мг, 1,8 ммоль) и соединение 1А (450 мг, 1,5 ммоль) растворяли в атмосфере азота в смешанном растворителе, состоящем из диоксана (6 мл) и воды (1,2 мл). Затем к реакционной жидкости добавляли дихлор[1,1'-бис(дифенилфосфино)ферроцен] палладий (II) (124 мг, 0,15 ммоль) и карбонат натрия (321 мг, 3,0 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (20 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/2) с получением соединения 1С-7 (440 мг).
МС (ИЭР) масса/заряд: 409,2 [М+Н]+.
Соединение 1С-8:
Соединение 1С-7 (430 мг, 1,1 ммоль) растворяли в растворе соляной кислоты в диоксане (4 М, 4 мл) и последовательно перемешивали полученную смесь при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении с получением соединения 1С-8 (400 мг).
МС (ИЭР) масса/заряд: 309,2 [М+Н]+.
Соединение 1С-9:
Соединение 1С-8 (100 мг, 0,3 ммоль) и карбонат калия (120 мг, 0,9 ммоль) растворяли в N,N-диметилформамиде (2 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и непосредственно очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 40% до 80% в течение 30 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 1С-9 (50 мг).
МС (ИЭР) масса/заряд: 289,2 [М+Н]+.
Промежуточное соединение 1С:
Соединение 1С-9 (50 мг, 0,17 ммоль) растворяли в смешанном растворителе, состоящем из этанола (1 мл) и воды (0,2 мл). Затем к описанной выше реакционной жидкости добавляли железный порошок (48 мг, 0,9 ммоль) и хлорид аммония (14 мг, 0,26 ммоль), после чего реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и фильтровали, фильтрационный осадок промывали этанолом (10 мл × 5) и концентрировали фильтрат при пониженном давлении с получением промежуточного соединения 1С (50 мг).
МС (ИЭР) масса/заряд: 259,2 [М+Н]+.
Получение соединения 1
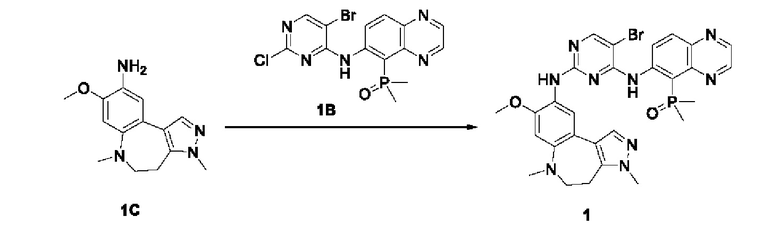
Соединение 1С (40 мг, 0,16 ммоль) и соединение 1В (64 мг, 0,16 ммоль) растворяли в N-метилпирролидиноне (2 мл). Затем к описанной выше реакционной жидкости добавляли метансульфоновую кислоту (45 мг, 0,47 ммоль). Реакционную жидкость нагревали до 100°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 10% до 50% в течение 20 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 1 (20 мг).
МС (ИЭР, масса/заряд): 634,2, 636,2 [М+Н]+.
1H ЯМР (300 МГц, CD3OD) δ 8,82 (d, J=2,1 Гц, 1H), 8,76-8,67 (m, 2Н), 8,25 (s, 1Н), 8,09 (s, 1H), 7,52 (d, J=9,6 Гц, 1H), 6,89 (s, 1H), 6,66 (s, 1H), 3,92 (s, 3Н), 3,69 (s, 3Н), 3,30 (br s, 2Н), 3,06 (br s, 5Н), 2,17 (d, J=14,4 Гц, 6Н).
Пример 2:
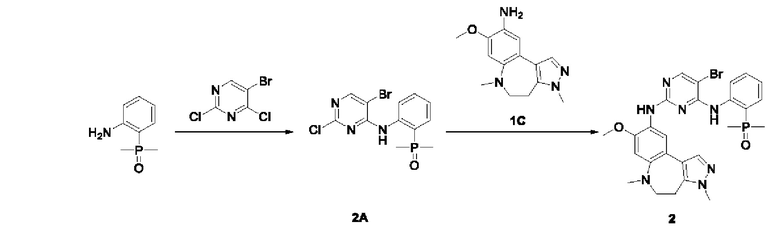
Соединение 2А:
5-Бром-2,4-дихлорпиримидин (2 г, 8,8 ммоль) растворяли в N,N-диметилформамиде (30 мл) и добавляли последовательно безводный карбонат калия (3,64 г, 26,3 ммоль) и 2-(диметилфосфинокси)анилин (1,48 г, 8,8 ммоль). Реакционную жидкость нагревали до 60°С и последовательно перемешивали в течение 12 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (150 мл). Смесь экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 2А (2,96 г).
МС (ИЭР) масса/заряд: 360,0, 362,0 [М+Н]+.
Соединение 2
Соединение 2 (13 мг) получали путем замены исходного материала на соединение 2А (100 мг, 0,28 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 582,3, 584,3 [М+Н]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,96 (s, 1H), 8,38 (s, 1H), 8,26 (s, 1H), 8,16 (s, 1Н), 7,71 (s, 1H), 7,57 (s, 1H), 7,50-7,45 (m, 1H), 7,08-6,99 (m, 2H), 6,58 (s, 1H), 3,77 (s, 3Н), 3,75 (s, 3Н), 3,19 (t, J=5,2 Гц, 2H), 3,03-3,00 (m, 5H), 1,75 (d, J=13,6 Гц, 6H).
Пример 3:
Получение промежуточного соединения ЗА
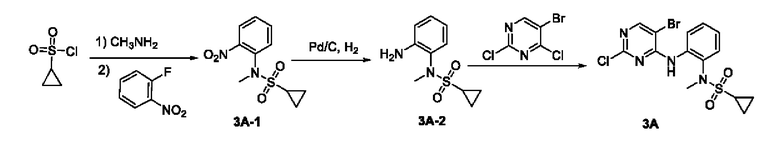
Соединение 3А-1:
Раствор метиламина в тетрагидрофуране (2 М, 106,7 мл, 213,4 ммоль) и триэтиламине (10,8 г, 106,7 ммоль) растворяли в дихлорметане (80 мл). Затем к реакционной жидкости добавляли по каплям циклопропилсульфонилхлорид (10 г, 71,1 ммоль) при 0°С. Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 24 часов. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в ацетонитриле (100 мл). Затем к реакционной жидкости добавляли карбонат цезия (31,1 г, 95,4 ммоль) и о-нитрофторбензол (15,5 г, 95,4 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 3А-1 (14 г).
МС (ИЭР) масса/заряд: 257,0 [М+Н]+.
Соединение 3А-2:
Соединение 3А-1 (14 г, 55 ммоль) растворяли в смешанном растворителе, состоящем из этанола (50 мл) и этилацетата (50 мл). Затем к реакционной жидкости добавляли влажный палладий на углероде (10%, 600 мг). После того, как реакционную систему продували с применением водорода (1 атм) 3 раза, реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали этилацетатом (30 мл × 3) и концентрировали полученный фильтрат при пониженном давлении с получением соединения 3А-2 (12 г).
МС (ИЭР) масса/заряд: 227,1 [М+Н]+.
Промежуточное соединение 3А:
Соединение 3А (9 г) получали путем замены исходного материала на соединение 3А-2 (12 г, 53 ммоль) согласно способу получения соединения 2А в примере 2. МС (ИЭР, масса/заряд): 416,9, 418,9 [М+Н]+.
Получение соединения 3
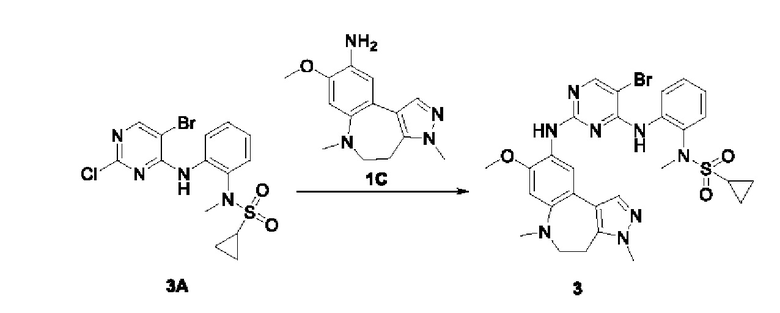
Соединение 3 (96 мг) получали путем замены исходного материала на соединение 3А (80 мг, 0,3 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 638,9, 640,9 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 8,30-8,13 (m, 4Н), 7,72 (s, 1H), 7,60-7,55 (m, 2Н), 7,06-7,01 (m, 2Н), 6,60 (s, 1H), 3,77 (s, 3Н), 3,75 (s, 3Н), 3,22-3,19 (m, 5Н), 3,04-3,01 (m, 5Н), 2,89-2,81 (m, 1Н), 1,07-1,05 (m, 2Н), 0,88 (s, 2Н).
Пример 4:
Получение промежуточного соединения 4А
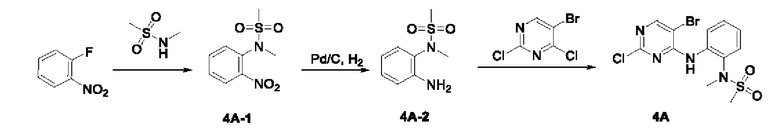
Соединение 4А-1:
Метилсульфонилметиламин (34,8 г, 319 ммоль) и о-фторнитробензол (30 г, 212,6 ммоль) растворяли в ацетонитриле (400 мл). Затем к описанной выше реакционной жидкости добавляли карбонат цезия (138,6 г, 425 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (300 мл). Смесь экстрагировали этилацетатом (300 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (150 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением соединения 4А-1 (30 г).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,97-7,95 (m, 1H), 7,87-7,75 (m, 2Н), 7,67-7,58 (m, 1H), 3,29 (s, 3Н), 3,06 (s, 3Н).
Соединение 4А-2:
Соединение 4А-2 (12 г) получали путем замены исходного материала на соединение 4А-1 (15 г, 65 ммоль) согласно способу получения соединения 3А-2 из соединения 3А-1 в примере 3.
МС (ИЭР, масса/заряд): 201,0 [М+Н]+.
Промежуточное соединение 4А:
Промежуточное соединение 4А (3 г) получали путем замены исходного материала на соединение 4А-2 (5 г, 25 ммоль) согласно способу получения соединения 2А в примере 2. МС (ИЭР, масса/заряд): 390,9, 392,9 [М+Н]+.
Получение соединения 4
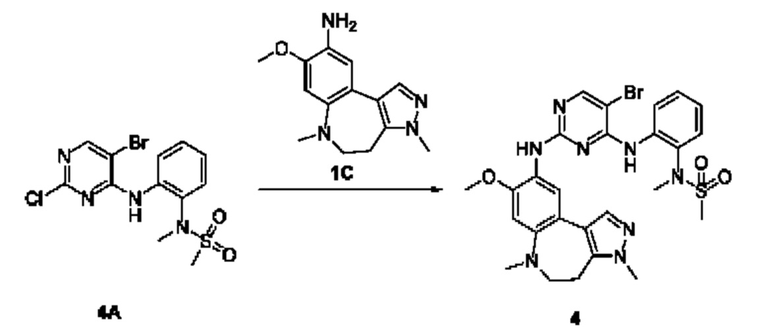
Соединение 4 (25 мг) получали путем замены исходного материала на соединение 4А (152 мг, 0,4 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 613,1, 615,1 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,29-8,25 (m, 3Н), 8,19 (s, 1Н), 7,72 (s, 1H), 7,55-7,53 (m, 2Н), 7,07-6,93 (m, 2Н), 6,60 (s, 1H), 3,77 (s, 3Н), 3,76 (s, 3Н), 3,22-3,19 (m, 5Н), 3,11 (s, 3Н), 3,04-3,01 (m, 5Н).
Пример 5:
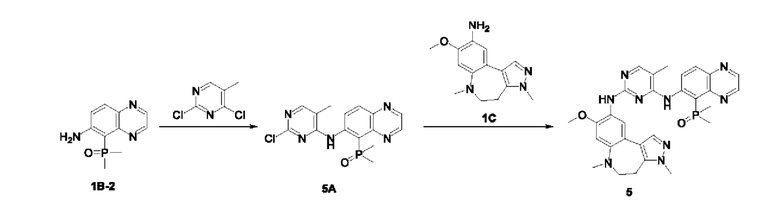
Промежуточное соединение 5А:
Соединение 5А (4,2 г) получали путем замены исходного материала на 2,4-дихлор-5-метилпиримидин (5,53 г, 34 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 348,3, 350,3 [М+Н]+.
Соединение 5
Соединение 5 (53 мг) получали путем замены исходного материала на соединение 5А (80 мг, 0,23 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 570,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,56 (s, 1H), 9,15 (dd, J=9,6, 4,4 Гц, 1H), 8,67-8,66 (m, 2H), 8,31-8,23 (m, 2H), 7,87 (s, 1H), 7,59 (d, J=9,2 Гц, 1H), 7,23 (s, 1H), 6,57 (s, 1H), 3,90 (s, 3H), 3,75 (s, 3H), 3,33 (t, J=5,6 Гц, 2H), 3,08-3,05 (m, 5H), 2,28 (s, 3H), 2,13 (s, 3H), 2,09 (s, 3H).
Пример 6:
Получение промежуточного соединения 6А
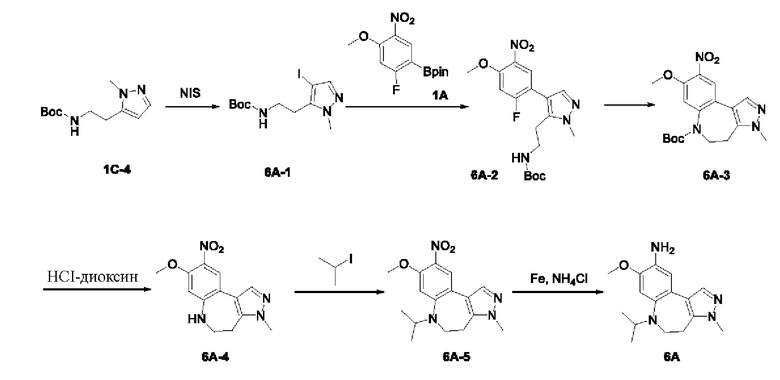
Соединение 6А-1:
Соединение 6А-1 (3,57 г) получали путем замены исходного материала на соединение 1С-4 (3,5 г, 15,5 ммоль) согласно способу получения соединения 1С-6 из соединения 1С-5 в примере 1.
МС (ИЭР, масса/заряд): 352,0 [М+Н]+.
Соединение 6А-2:
Соединение 6А-2 (2,8 г) получали путем замены исходного материала на соединение 6А-1 (3,4 г, 9,7 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 395,3 [М+Н]+.
Соединение 6А-3:
Соединение 6А-3 (1,2 г) получали путем замены исходного материала на соединение 6А-2 (2,7 г, 6,8 ммоль) согласно способу получения соединения 1С-9 из соединения 1С-8 в примере 1.
МС (ИЭР, масса/заряд): 375,2 [М+Н]+.
Соединение 6А-4:
Соединение 6А-4 (0,84 г) получали путем замены исходного материала на соединение 6А-3 (1,2 г, 3,3 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 275,0 [М+Н]+.
Соединение 6А-5:
Соединение 6А-4 (300 мг, 1,1 ммоль) и карбонат цезия (1,07 г, 3,3 ммоль) растворяли в N,N-диметилформамиде (6 мл). Затем к описанной выше реакционной жидкости добавляли 2-йодпропан (1,86 г, 10,9 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (30 мл). Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 6А-5 (75 мг).
МС (ИЭР, масса/заряд): 317,2 [М+Н]+.
Промежуточное соединение 6А:
Промежуточное соединение 6А (48 мг) получали путем замены исходного материала на соединение 6А-5 (75 мг, 0,24 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 287,2 [М+Н]+.
Получение соединения 6
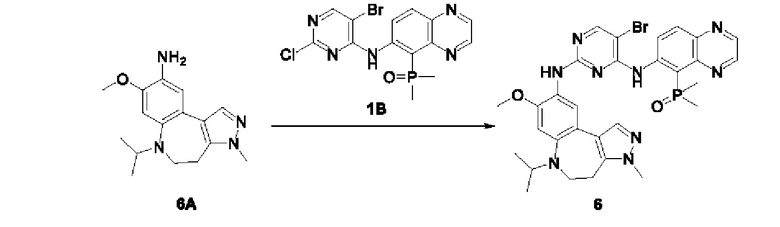
Соединение 6 (19 мг) получали путем замены исходного материала на соединение 6А (48 мг, 0,17 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 662,3, 664,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,47 (s, 1H), 8,88 (dd, J=9,2, 4,0 Гц, 1H), 8,69 (d, J=2,0 Гц, 1Н), 8,67 (d, J=2,0 Гц, 1H), 8,37 (s, 1H), 8,27 (s, 1H), 7,63 (s, 1H), 7,35 (s, 1H), 7,00 (s, 1H), 6,56 (s, 1H), 3,88 (s, 4H), 3,70 (s, 3H), 3,35-3,27 (m, 2H), 3,00-2,91 (m, 2H), 2,15 (s, 3H), 2,12 (s, 3H), 1,31 (d, J=6,4 Гц, 6H).
Пример 7:
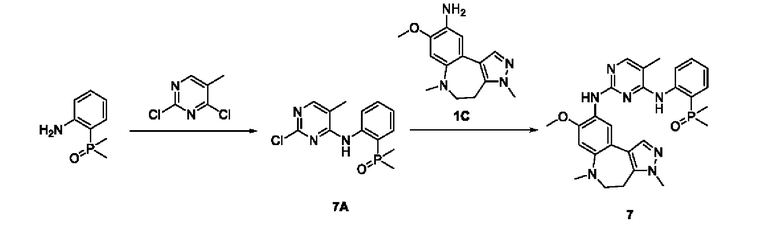
Промежуточное соединение 7А:
Соединение 7А (300 мг) получали путем замены исходного материала на 2,4-дихлор-5-метилпиримидин (578 мг, 3,55 ммоль) и 2-(диметилфосфорил)анилин (300 мг, 2,96 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 296,1, 298,1 [М+Н]+.
Соединение 7
Формиат соединения 7 (111 мг) получали путем замены исходного материала на соединение 7А (80 мг, 0,27 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 518,2 [М+Н]+.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (dd, J=8,4, 4,4 Гц, 1H), 8,32 (s, 1H), 7,91 (s, 1Н), 7,73 (s, 1H), 7,55-7,49 (m, 1H), 7,38 (s, 1H), 7,20-7,16 (m, 1H), 7,10-7,06 (m, 1H), 6,66 (s, 1H), 3,87 (s, 3Н), 3,80 (s, 3Н), 3,27 (t, J=5,6 Гц, 2H), 3,06-3,03 (m, 5H), 2,17 (s, 3Н), 1,89 (s, 3Н), 1,85 (s, 3Н).
Пример 8:
Получение промежуточного соединения 8А
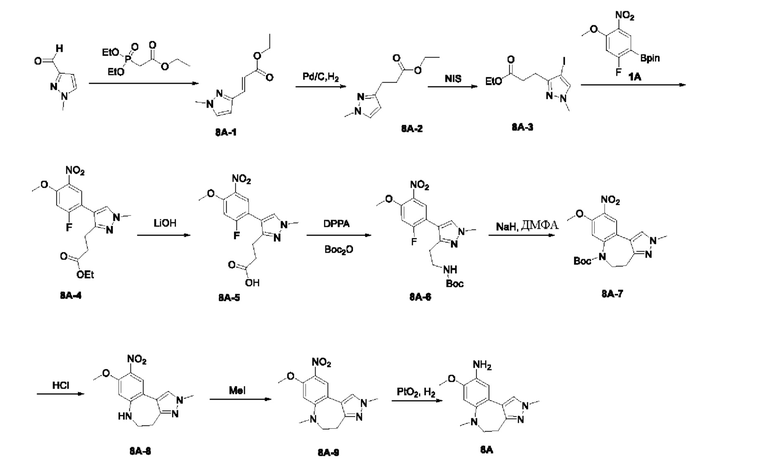
Соединение 8А-1:
Соединение 8А-1 (16 г) получали путем замены исходного материала на 1-метил-Ш-пиразол-3-карбальдегид (10 г, 90,8 ммоль) согласно способу получения соединения 1С-1 в примере 1.
МС (ИЭР, масса/заряд): 181,2 [М+Н]+.
Соединение 8А-2:
Соединение 8А-2 (16 г) получали путем замены исходного материала на соединение 8А-1 (16,8 г, 93,2 ммоль) согласно способу получения соединения 1С-2 из соединения 1С-1 в примере 1.
МС (ИЭР, масса/заряд): 183,1 [М+Н]+.
Соединение 8А-3:
Соединение 8А-2 (3 г, 16,5 ммоль) растворяли в гексафторизопропаноле (20 мл). Затем к описанной выше реакционной жидкости добавляли N-йодсукцинимид (7,4 г, 32,9 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в воде (100 мл), экстрагировали смесь дихлорметаном (3 × 100 мл), после чего органические фазы объединяли, промывали насыщенным солевым раствором (150 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 40/1) с получением соединения 8А-3 (4,5 г).
МС (ИЭР, масса/заряд): 309,1 [М+Н]+.
Соединение 8А-4:
Соединение 8А-4 (470 мг) получали путем замены исходного материала на соединение 8А-3 (750 мг, 2,4 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 352,2 [М+Н]+.
Соединение 8А-5:
Соединение 8А-4 (380 мг, 1,1 ммоль) растворяли в смешанном растворителе, состоящем из тетрагидрофурана (10 мл) и воды (2 мл). Затем к описанной выше реакционной жидкости добавляли гидроксид лития (130 мг, 5,4 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, к реакционной жидкости добавляли воду (20 мл) для разбавления и доводили до рН=2 с помощью 1 N соляной кислоты. Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3); затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении с получением соединения 8А-5 (330 мг).
МС (ИЭР, масса/заряд): 324,1 [М+Н]+.
Соединение 8А-6:
Соединение 8А-6 (260 мг) получали путем замены исходного материала на соединение 8А-5 (350 мг, 1,1 ммоль) согласно способу получения соединения 1С-4 из соединения 1С-3 в примере 1.
МС (ИЭР, масса/заряд): 395,1 [М+Н]+.
Соединение 8А-7:
Соединение 8А-6 (260 мг, 0,66 ммоль) растворяли в N,N-диметилформамиде (10 мл). Затем к описанной выше реакционной жидкости добавляли гидрид натрия (60%, 40 мг, 1 ммоль) при 0°С. Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (30 мл). Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 8А-7 (230 мг).
МС (ИЭР, масса/заряд): 375,2 [М+Н]+.
Соединение 8А-8:
Соединение 8А-8 (160 мг) получали путем замены исходного материала на соединение 8А-7 (240 мг, 0,64 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 275,1 [М+Н]+.
Соединение 8А-9:
Соединение 8А-8 (240 мг, 0,88 ммоль) растворяли в N,N-диметилформамиде (10 мл). Затем к реакционной жидкости добавляли при 0°С гидрид натрия (60%, 350 мг, 8,75 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут, после чего добавляли йодметан (1,2 г, 8,5 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (30 мл). Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 8А-9 (140 мг).
МС (ИЭР, масса/заряд): 289,1 [М+Н]+.
Промежуточное соединение 8А:
Соединение 8А-9 (70 мг, 0,24 ммоль) растворяли в этаноле (5 мл). Затем к реакционной жидкости добавляли диоксид платины (20 мг). После того, как реакционную систему продували 3 раза с помощью водорода, реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали этилацетатом (10 мл × 3) и концентрировали полученный фильтрат при пониженном давлении с получением соединения 8А (60 мг). МС (ИЭР) масса/заряд: 259,1 [М+Н]+.
Получение соединения 8
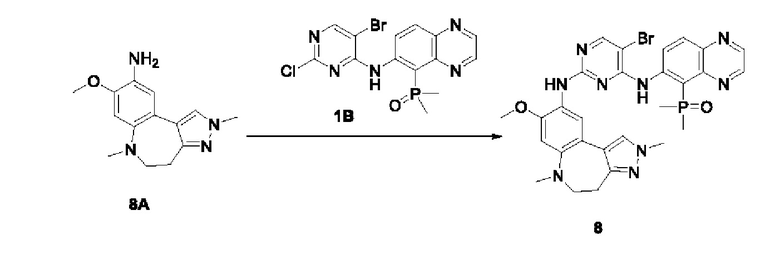
Соединение 8 (37 мг) получали путем замены исходного материала на соединение 8А (70 мг, 0,17 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 634,2, 636,2 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 12,67 (s, 1H), 8,83-8,79 (m, 3Н), 8,36 (s, 1H), 8,25 (s, 1Н), 7,77 (s, 1H), 7,65 (s, 1Н), 7,56 (s, 1H), 6,59 (s, 1H), 3,79 (s, 3Н), 3,57 (s, 3Н), 3,22-3,18 (m, 2Н), 3,01 (s, 3Н), 2,98-2,94 (m, 2Н), 2,04 (s, 3Н), 1,99 (s, 3Н).
Пример 9:
Получение промежуточного соединения 9А
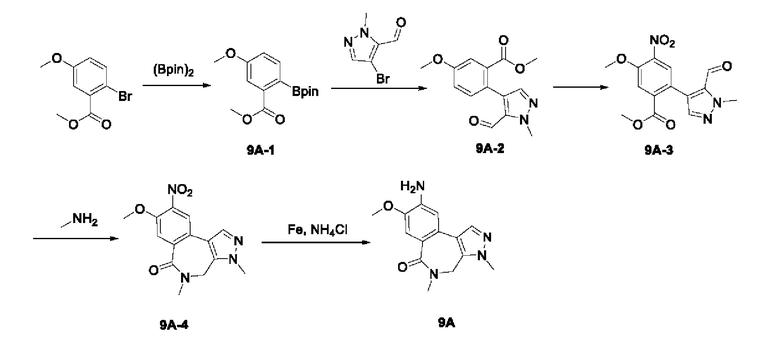
Соединение 9А-1:
Соединение 9А-1 (10 г) получали путем замены исходного материала на метил-2-бром-5-метоксибензоат (10 г, 40,8 ммоль) согласно способу получения соединения 1А из соединения 1А-1 в примере 1.
МС (ИЭР, масса/заряд): 293,1 [М+Н]+.
Соединение 9А-2:
Соединение 9А-2 (4,7 г) получали путем замены исходного материала на соединение 9А-1 (10 г, 34,2 ммоль) и 4-бром-1-метил-пиразол-5-карбальдегид (6,47 г, 34,2 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 275,1 [М+Н]+.
Соединение 9А-3:
Соединение 9А-2 (2,4 г, 8,75 ммоль) растворяли в концентрированной серной кислоте (25 мл) при 0°С. Затем к реакционной жидкости отдельными порциями добавляли нитрат калия (973 мг, 9,63 ммоль), после чего реакционную жидкость последовательно перемешивали при указанной температуре в течение 10 минут. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость медленно выливали в ледяную воду (100 г) для гашения реакции. Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/2) с получением соединения 9А-3 (650 мг).
МС (ИЭР, масса/заряд): 320,1 [М+Н]+.
Соединение 9А-4:
Соединение 9А-3 (1,4 г, 4,4 ммоль) и раствор метиламина в тетрагидрофуране (2 М, 8,77 мл, 17,54 ммоль) растворяли в метаноле (10 мл). Затем к описанной выше реакционной жидкости добавляли ледяную уксусную кислоту (276 мг, 4,6 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. К описанной выше реакционной жидкости добавляли борогидрид натрия (332 мг, 8,77 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3); после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 9А-4 (540 мг).
МС (ИЭР, масса/заряд): 303,1 [М+Н]+.
Промежуточное соединение 9А:
Соединение 9А (80 мг) получали путем замены исходного материала на соединение 9А-4 (100 мг, 0,33 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 273,1 [М+Н]+.
Получение соединения 9
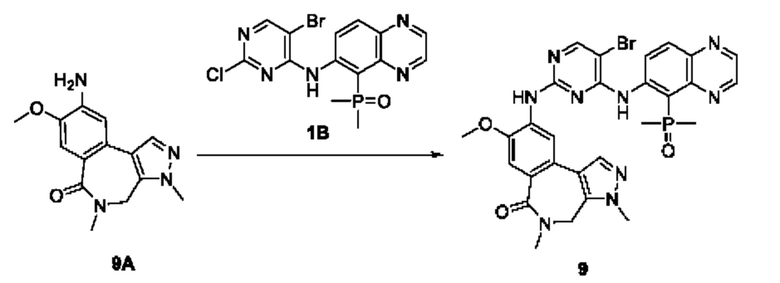
Соединение 9
Соединение 9 (25 мг) получали путем замены исходного материала на соединение 9А (60 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 648,1, 650,1 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 12,65 (s, 1Н), 8,88 (dd, J=9,2, 3,6 Гц, 1H), 8,74 (s, 2H), 8,43 (s, 1H), 8,34 (s, 1H), 7,85 (s, 1H), 7,57 (s, 2H), 6,98 (s, 1H), 4,34 (s, 2H), 3,99 (s, 3H), 3,93 (s, 3H), 3,26 (s, 3H), 2,17 (s, 3H), 2,13 (s, 3H).
Пример 10:
Получение промежуточного соединения 10
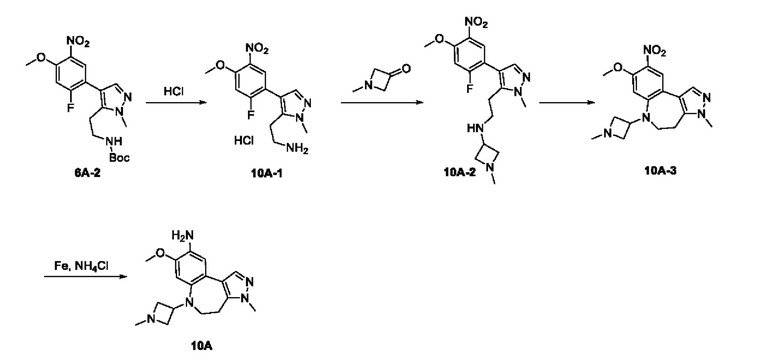
Соединение 10А-1:
Соединение 10А-1 (260 мг) получали путем замены исходного материала на соединение 6А-2 (300 мг, 0,76 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 295,2 [М+Н]+.
Соединение 10А-2:
Соединение 10А-1 (370 мг, 1,12 ммоль) и 1-метил-3-азетидинон (95 мг, 1,12 ммоль) растворяли в дихлорметане (7 мл). Затем к реакционной жидкости добавляли триэтиламин (453 мг, 4,48 ммоль) и тетраизопропилтитанат (636 мг, 2,24 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. К описанной выше реакционной жидкости добавляли борогидрид натрия (85 мг, 2,24 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 5% до 40% в течение 20 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 10А-2 (70 мг).
МС (ИЭР) масса/заряд: 364,2 [М+Н]+.
Соединение 10А-3:
Соединение 10А-3 (50 мг) получали путем замены исходного материала на соединение 10А-2 (70 мг, 0,19 ммоль) согласно способу получения соединения 1С-9 из соединения 1С-8 в примере 1.
МС (ИЭР, масса/заряд): 344,1 [М+Н]+.
Промежуточное соединение 10А:
Соединение 10А (50 мг) получали путем замены исходного материала на соединение 10А-3 (50 мг, 0,15 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 314,2 [М+Н]+.
Получение соединения 10
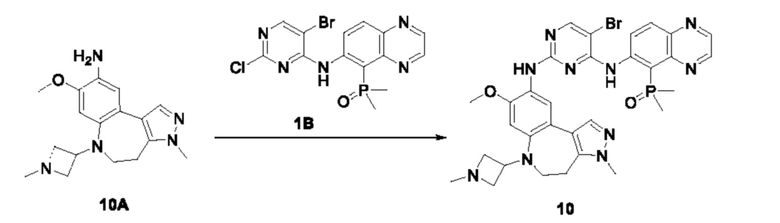
Соединение 10 (8 мг) получали путем замены исходного материала на соединение 10А (50 мг, 0,12 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 689,3, 691,3 [М+Н]+.
1Н ЯМР (400 МГц, CD3OD) δ 8,82 (d, J=2,0 Гц, 1H), 8,73 (d, J=2,0 Гц, 1H), 8,66-8,63 (m, 1H), 8,52 (br s, 1H), 8,25 (s, 1H), 8,18 (s, 1H), 7,47 (d, J=9,6 Гц, 1H), 6,84 (s, 1H), 6,24 (s, 1H), 4,57-4,54 (m, 1H), 4,32-4,28 (m, 2H), 3,89 (s, 3H), 3,85-3,82 (m, 2H), 3,68 (s, 3H), 3,31-3,30 (m, 2H), 3,13-3,10 (m, 2H), 2,80 (s, 3H), 2,17 (s, 3H), 2,13 (s, 3H).
Пример 11:
Получение промежуточного соединения 11А
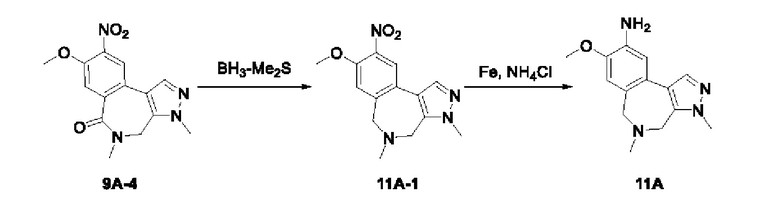
Соединение 11А-1:
Соединение 9А-4 (100 мг, 0,33 ммоль) растворяли в тетрагидрофуране (2 мл) в атмосфере азота. Затем к описанной выше реакционной жидкости добавляли раствор борана-диметилсульфида (251 мг, 3,31 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления метанола (5 мл). Смесь нагревали до 60°С, последовательно перемешивали в течение 1 часа и охлаждали до 0°С; белое твердое вещество осаждали, фильтровали и высушивали с получением соединения 11А-1 (60 мг).
МС (ИЭР, масса/заряд): 289,1 [М+Н]+.
Промежуточное соединение 11А:
Соединение 11А (100 мг) получали путем замены исходного материала на соединение 11А-1 (125 мг, 0,43 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 259,1 [М+Н]+.
Получение соединения 11
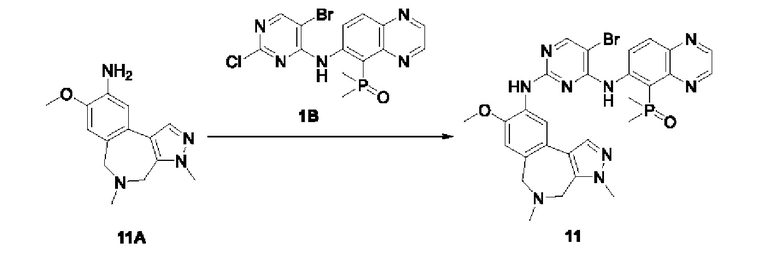
Соединение 11 (20 мг) получали путем замены исходного материала на соединение 11А (90 мг, 0,35 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 634,2, 636,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,55 (s, 1Н), 8,88 (dd, J=9,6, 4,2 Гц, 1H), 8,74-8,72 (m, 2Н), 8,52 (s, 1H), 8,34 (s, 1H), 7,64 (d, J=9,3 Гц, 1Н), 7,56 (s, 1H), 7,00 (s, 1H), 6,74 (s, 1H), 4,16 (s, 2Н), 3,94 (s, 3Н), 3,93 (s, 2Н), 3,75 (s, 3Н), 2,55 (s, 3Н), 2,19 (s, 3Н), 2,14 (s, 3Н).
Пример 12:
Получение промежуточного соединения 12А
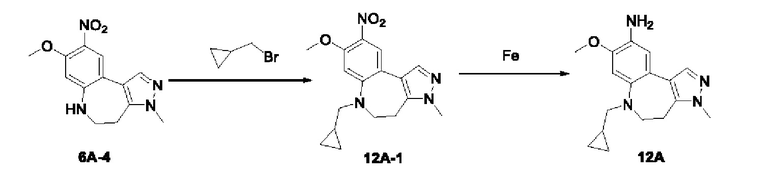
Соединение 12А-1:
Соединение 12А-1 (160 мг) получали путем замены исходного материала на соединение 6А-4 (150 мг, 0,55 ммоль) и (бромметил)циклопропан (736 мг, 5,45 ммоль) согласно способу получения соединения 8А-9 из соединения 8А-8 в примере 8.
МС (ИЭР, масса/заряд): 329,2 [М+Н]+.
Промежуточное соединение 12А:
Соединение 12А (70 мг) получали путем замены исходного материала на соединение 12А-1 (80 мг, 0,24 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 299,1 [М+Н]+.
Получение соединения 12
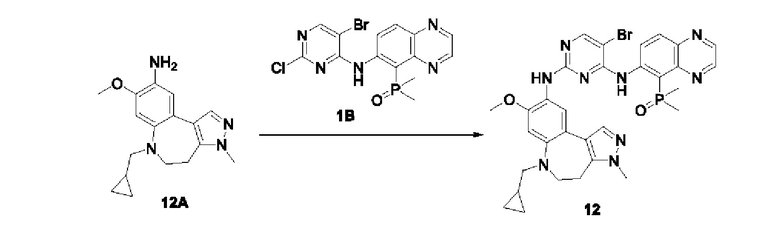
Соединение 12 (30 мг) получали путем замены исходного материала на соединение 12А (70 мг, 0,24 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 674,3, 676,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,51 (s, 1Н), 8,89 (dd, J=9,6, 3,9 Гц, 1H), 8,71-8,68 (m, 2Н), 8,37 (s, 1H), 8,27 (s, 1H), 7,66-7,63 (m, 1H), 7,36 (s, 1H), 7,04 (s, 1H), 6,56 (s, 1H), 3,90 (s, 3Н), 3,74 (s, 3Н), 3,48-3,45 (m, 2Н), 3,17 (d, J=6,3 Гц, 2H), 3,07-3,04 (m, 2Н), 2,17 (s, 3Н), 2,12 (s, 3Н), 1,13-1,07 (m, 1H), 0,67-0,61 (m, 2Н), 0,30-0,27 (m, 2Н).
Пример 13:
Получение промежуточного соединения 13А
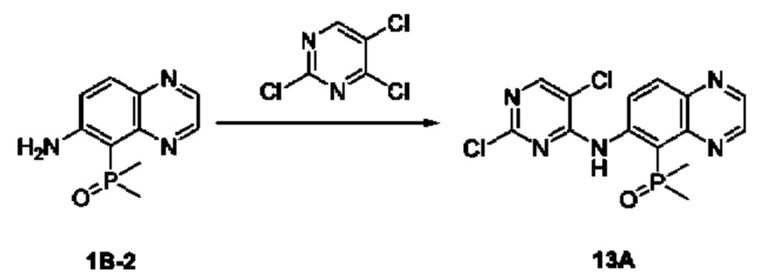
Соединение 13А (4,5 г) получали путем замены исходного материала на 2,4,5-трихлорпиримидин (5,37 г, 29,3 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 367,9, 369,9 [М+Н]+.
Получение соединения 13
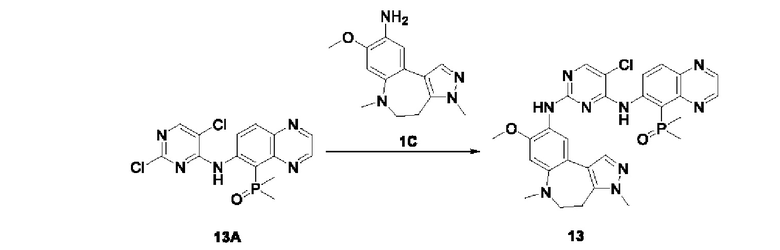
Соединение 13 (73 мг) получали путем замены исходного материала на соединение 13А (100 мг, 0,27 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 590,3, 592,3 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 12,84 (s, 1H), 8,96 (s, 1H), 8,82 (dd, J=11,4, 1,8 Гц, 2Н), 8,42 (s, 1H), 8,19 (s, 1H), 7,72 (s, 1H), 7,59 (s, 1H), 7,52 (s, 1H), 6,65 (s, 1H), 3,80 (s, 3Н), 3,73 (s, 3Н), 3,24-3,20 (m, 2Н), 3,04 (br s, 5Н), 2,04 (s, 3Н), 1,99 (s, 3Н).
Пример 14:
Получение соединения 14
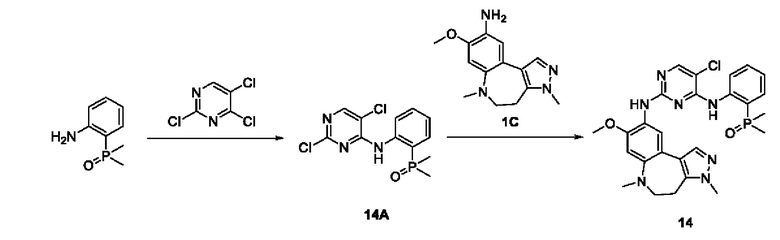
Соединение 14А:
Соединение 14А (14,4 г) получали путем замены исходного материала на 2,4,5-трихлорпиримидин (13 г, 71 ммоль) и 2-(диметилфосфорил)анилин (10 г, 59 ммоль) согласно способу получения соединения 2А в примере 2.
МС (ИЭР, масса/заряд): 316,0, 318,0 [М+Н]+.
Соединение 14
Соединение 14 (80 мг) получали путем замены исходного материала на соединение 14А (80 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 538,3, 540,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,88 (s, 1H), 8,62 (dd, J=8,4, 4,5 Гц, 1H), 8,43 (s, 1H), 8,13 (s, 1H), 7,38 (s, 1H), 7,32-7,17 (m, 3H), 7,02-6,97 (m, 1H), 6,57 (s, 1H), 3,92 (s, 3H), 3,84 (s, 3H), 3,32-3,28 (m, 2H), 3,06 (br s, 5H), 1,88 (s, 3H), 1,84 (s, 3H).
Пример 15:
Получение промежуточного соединения 15А
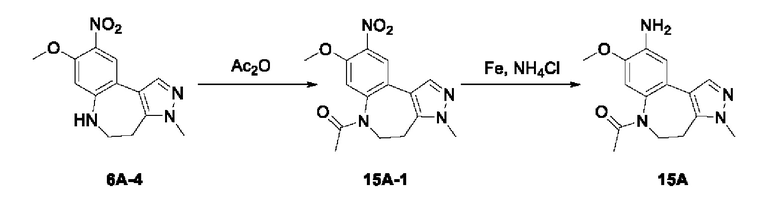
Соединение 15А-1:
Соединение 6А-4 (100 мг, 0,37 ммоль) растворяли в уксусном ангидриде (1,5 мл). Реакционную жидкость нагревали до 140°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 15А-1 (90 мг).
МС (ИЭР, масса/заряд): 317,2 [М+Н]+.
Промежуточное соединение 15А:
Соединение 15А (66 мг) получали путем замены исходного материала на соединение 15А-1 (80 мг, 0,25 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 287,1 [М+Н]+.
Получение соединения 15
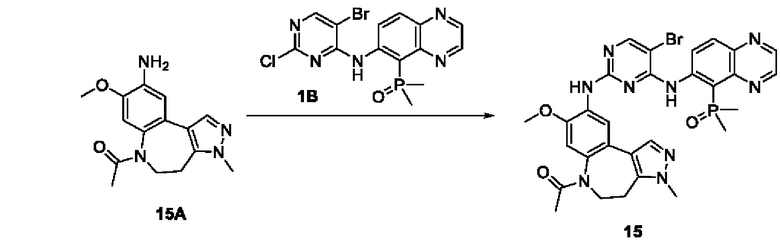
Соединение 15 (17 мг) получали путем замены исходного материала на соединение 15А (50 мг, 0,17 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 662,2, 664,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,62 (s, 1Н), 8,86 (dd, J=9,6, 4,2 Гц, 1H), 8,75-8,73 (m, 2H), 8,61 (s, 1H), 8,36 (s, 1H), 7,70-7,63 (m, 2H), 6,99 (s, 1H), 6,73 (s, 1H), 4,85 (dd, J=13,2, 5,7 Гц, 1H), 3,94 (s, 3H), 3,71 (s, 3H), 3,53-3,41 (m, 1H), 3,13-3,03 (m, 1H), 2,94-2,89 (m, 1H), 2,21-2,14 (m, 6H), 1,99 (s, 3H).
Пример 16:
Получение промежуточного соединения 16А
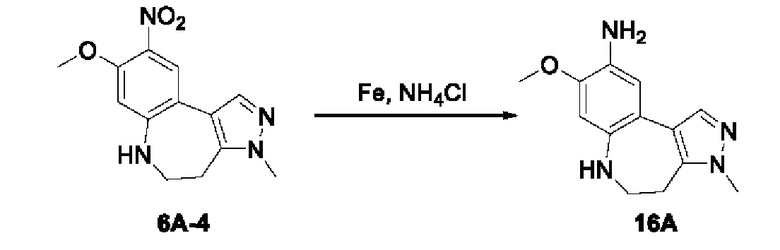
Соединение 16А (70 мг) получали путем замены исходного материала на соединение 6А-4 (100 мг, 0,25 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 245,2 [М+Н]+.
Получение соединения 16
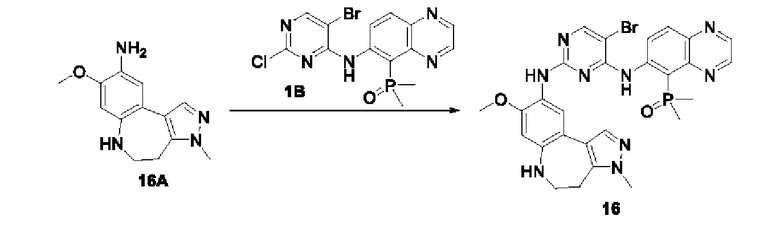
Соединение 16 (13 мг) получали путем замены исходного материала на соединение 16А (60 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 620,2, 622,2 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 12,65 (s, 1H), 8,84-8,79 (m, 3Н), 8,34 (s, 1H), 8,24 (s, 1H), 7,63 (s, 2Н), 7,53 (s, 1H), 6,58 (s, 1H), 5,93 (s, 1H), 3,71 (s, 3Н), 3,70 (s, 3Н), 3,28-3,23 (m, 2Н), 2,98-2,95 (m, 2Н), 2,03 (s, 3Н), 1,99 (s, 3Н).
Пример 17:
Получение соединения 17
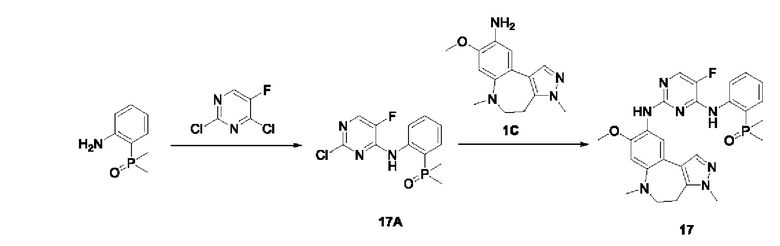
Соединение 17 А:
Соединение 17А (1,9 г) получали путем замены исходного материала на 2,4-дихлор-5-фторпиримидин (2 г, 11,98 ммоль) и 2-(диметилфосфорил)анилин (2,03 г, 11,98 ммоль) согласно способу получения соединения 2А в примере 2.
МС (ИЭР, масса/заряд): 300,0, 302,0 [М+Н]+.
Соединение 17
Соединение 17 (21 мг) получали путем замены исходного материала на соединение 17А (60 мг, 0,2 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 522,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 11,41 (s, 1Н), 8,82-8,76 (m, 1H), 8,44 (s, 1H), 8,00 (s, 1H), 7,56 (s, 1H), 7,34-7,20 (s, 3H), 7,00 (s, 1H), 6,59 (s, 1H), 3,92 (s, 3H), 3,84 (s, 3H), 3,32-3,27 (m, 2H), 3,07 (br s, 5H), 1,86 (m, 6H).
19F ЯМР (282 МГц, CDCl3) δ -164,46.
Пример 18:
Получение промежуточного соединения 18А
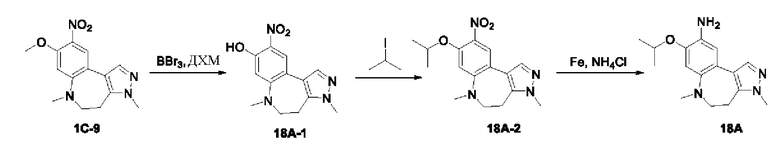
Соединение 18А-1
Соединение 1С-9 (100 мг, 0,35 ммоль) растворяли в дихлорметане (2 мл) в атмосфере азота при 0°С. Затем к описанной выше реакционной жидкости добавляли трибромид бора (174 мг, 0,69 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (10 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 8А-1 (50 мг).
МС (ИЭР, масса/заряд): 275,1 [М+Н]+.
Соединение 18А-2:
Соединение 18А-1 (75 мг, 0,27 ммоль) и карбонат калия (113 мг, 0,82 ммоль) растворяли в N,N-диметилформамиде (20 мл). К реакционной жидкости добавляли при 0°С 2-йодопропан (139 мг, 0,82 ммоль). Реакционную систему нагревали до 60°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (20 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 18А-2 (70 мг).
МС (ИЭР, масса/заряд): 317,1 [М+Н]+.
Промежуточное соединение 18А:
Соединение 18А (55 мг) получали путем замены исходного материала на соединение 18А-2 (70 мг, 0,22 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 287,1 [М+Н]+.
Получение соединения 18
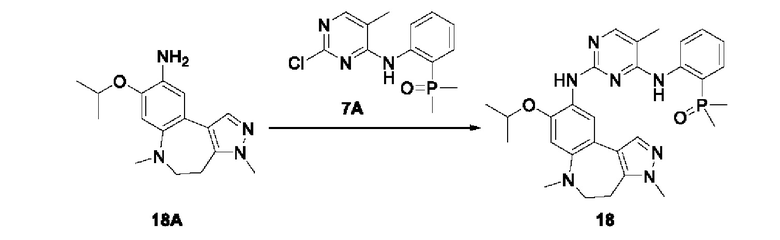
Соединение 18 (20 мг) получали путем замены исходного материала на соединение 18А (50 мг, 0,18 ммоль) и соединение 7А (52 мг, 0,18 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 546,0 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,49 (s, 1Н), 8,74 (dd, J=8,7, 4,5 Гц, 1H), 8,48 (s, 1H), 7,92 (s, 1H), 7,52 (s, 1H), 7,37 (s, 1H), 7,26-7,19 (m, 1H), 7,16-7,10 (m, 1H), 6,96-6,90 (m, 1H), 6,59 (s, 1H), 4,62-4,54 (m, 1H), 3,83 (s, 3H), 3,29 (t, J=5,4 Гц, 2H), 3,04 (t, J=5,7 Гц, 5H), 2,22 (s, 3H), 1,87 (s, 3H), 1,83 (s, 3H), 1,41 (s, 3H), 1,39 (s, 3H).
Пример 19:
Получение промежуточного соединения 19А
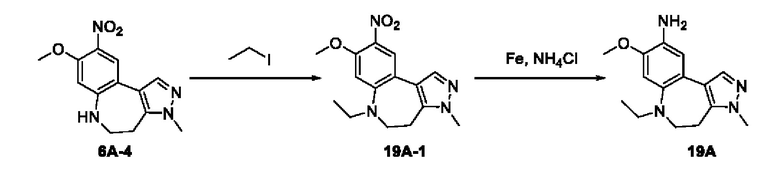
Соединение 19А-1:
Соединение 19А-1 (150 мг) получали путем замены исходного материала на соединение 6А-4 (200 мг, 0,73 ммоль) и йодэтан (341 мг, 2,19 ммоль) согласно способу получения соединения 8А-9 из соединения 8А-8 в примере 8.
МС (ИЭР, масса/заряд): 303,2 [М+Н]+.
Промежуточное соединение 19А:
Соединение 19А (100 мг) получали путем замены исходного материала на соединение 19А-1 (150 мг, 0,5 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 273,1 [М+Н]+.
Получение соединения 19
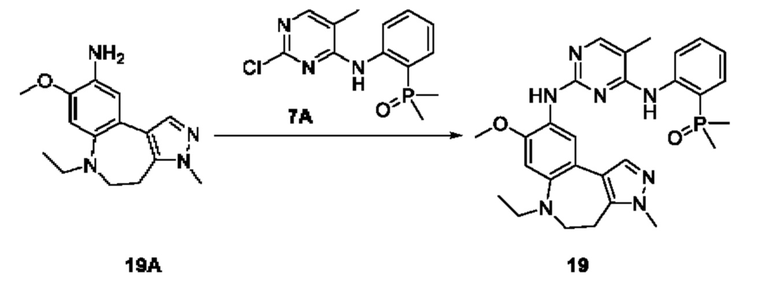
Соединение 19 (33 мг) получали путем замены исходного материала на соединение 19А (60 мг, 0,22 ммоль) и соединение 7А (65 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 532,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,49 (s, 1H), 8,74 (dd, J=8,7, 4,5 Гц, 1H), 8,45 (s, 1H), 7,92 (s, 1H), 7,50 (s, 1H), 7,44 (s, 1H), 7,25-7,14 (m, 1H), 6,97-6,92 (m, 2H), 6,57 (s, 1H), 3,89 (s, 3H), 3,83 (s, 3H), 3,39-3,29 (m, 4H), 3,01-2,98 (m, 2H), 2,22 (s, 3H), 1,87 (s, 3H), 1,82 (s, 3H),
1.31 (t, J=6,9 Гц, 3Н).
Пример 20:
Получение соединения 20
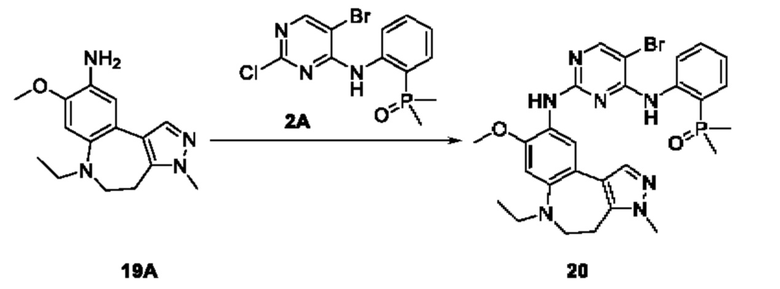
Соединение 20 (33 мг) получали путем замены исходного материала на соединение 19А (60 мг, 0,22 ммоль) и соединение 2А (79 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 596,2, 598,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,61 (s, 1H), 8,50 (dd, J=8,6, 4,5 Гц, 1H), 8,41 (s, 1H), 8,23 (s, 1H), 7,33-7,20 (m, 4H), 7,03-6,97 (m, 1H), 6,57 (s, 1H), 3,89 (s, 3H), 3,83 (s, 3H), 3,39-3.32 (m, 4H), 3,01-2,97 (m, 2H), 1,88 (s, 3H), 1,84 (s, 3H), 1,31 (t, J=7,2 Гц, 3Н).
Пример 21:
Получение промежуточного соединения 21А
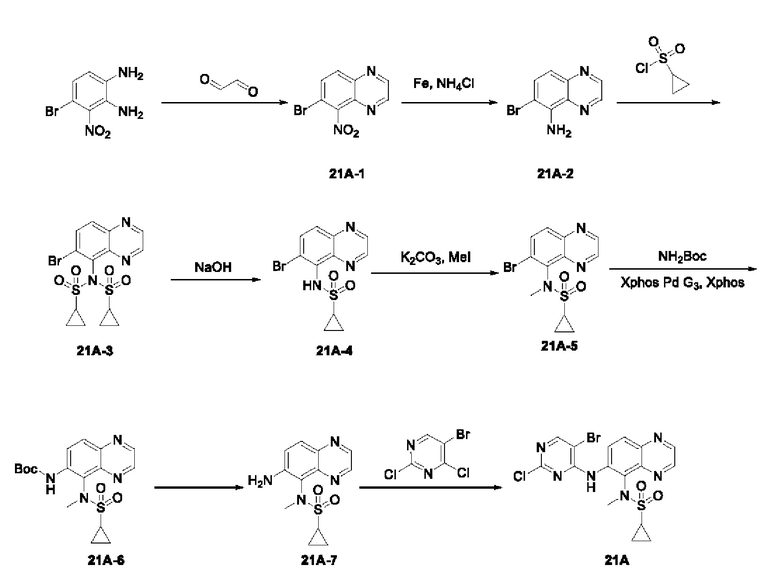
Соединение 21А-1:
4-бром-3-нитро-1,2-фенилендиамин (10 г, 43 ммоль) и водный раствор глиоксаля (40%, 15,01 г, 103 ммоль) растворяли в воде (400 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость охлаждали до комнатной температуры. Осажденное твердое вещество отфильтровывали, промывали водой (20 мл × 3) и высушивали с получением неочищенного соединения 21А-1 (13,4 г).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,16 (d, J=1,8 Гц, 1H), 9,11 (d, J=2,1 Гц, 1H), 8,30 (d, J=5,4 Гц, 2Н).
Соединение 21А-2:
Соединение 21А-2 (5,3 г) получали путем замены исходного материала на соединение 21А-1 (11 г, 43,3 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 223,8, 225,8 [М+Н]+.
Соединение 21А-3:
Соединение 21А-2 (3,7 г, 17 ммоль) растворяли в N,N-диметилформамиде (50 мл) в атмосфере азота. Затем к реакционной жидкости добавляли при 0°С гидрид натрия (60%, 2,6 г, 66 ммоль) и последовательно перемешивали указанную реакционную жидкость в течение 30 минут, после чего добавляли по каплям циклопропилсульфонилхлорид (7 г, 50 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (50 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 21А-3 (1,5 г).
МС (ИЭР, масса/заряд): 431,8, 433,8 [М+Н]+.
Соединение 21А-4:
Соединение 21А-3 (2,2 г, 5,1 ммоль) растворяли в метаноле (25 мл) с последующим добавлением к реакционной жидкости гидроксида натрия (8,1 г, 102 ммоль). Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении, добавляли воду (50 мл) для разбавления и доводили до рН=6 с помощью концентрированной соляной кислоты. Смесь экстрагировали хлороформом/изопропанолом (3/1, 80 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 21А-4 (0,94 г).
МС (ИЭР, масса/заряд): 327,9, 329,9 [М+Н]+.
Соединение 21А-5:
Соединение 21А-4 (930 мг, 2,8 ммоль) и безводный карбонат калия (789 мг, 5,7 ммоль) растворяли в N,N-диметилформамиде (10 мл). Затем к описанной выше реакционной жидкости добавляли йодметан (603 мг, 4,3 ммоль) при 0°С.Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (80 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 21А-5 (930 мг).
МС (ИЭР, масса/заряд): 341,9, 343,9 [М+Н]+.
Соединение 21А-6:
Соединение 21А-5 (950 мг, 2,8 ммоль), трет-бутилкарбамат (488 мг, 4,2 ммоль), карбонат цезия (1,8 г, 5,6 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (235 мг, 0,28 ммоль) и 2-дициклогексилфосфоний-2,4,6-триизопропилбифенил (132 мг, 0,28 ммоль) растворяли в 1,4-диоксане (10 мл) в атмосфере азота. Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 21А-6 (810 мг).
МС (ИЭР, масса/заряд): 379,0 [М+Н]+.
Соединение 21А-7:
Соединение 21А-6 (810 мг, 2,1 ммоль) растворяли в растворе хлористого водорода в этилацетате (4 М, 10 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении, добавляли воду (10 мл) для разбавления и доводили до рН=8 с помощью насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении с получением неочищенного соединения 21А-7 (660 мг).
МС (ИЭР, масса/заряд): 279,2 [М+Н]+.
Промежуточное соединение 21А:
Соединение 21А (700 мг) получали путем замены исходного материала на соединение 21А-7 (620 мг, 2,2 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 468,9, 470,9 [М+Н]+.
Получение соединения 21
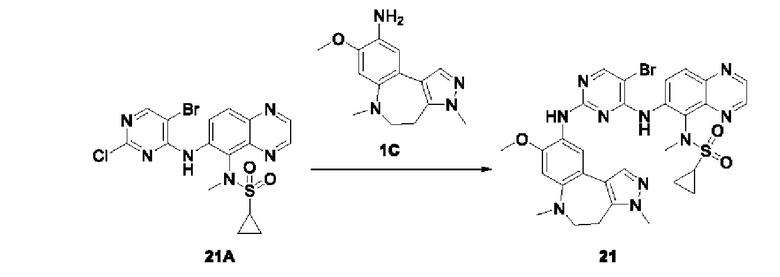
Соединение 21 (29 мг) получали путем замены исходного материала на соединение 21А (ПО мг, 0,23 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 691,3, 693,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,35 (s, 1H), 8,85 (d, J=1,6 Гц, 1H), 8,79 (d, J=1,6 Гц, 1H), 8,43 (d, J=9,2 Гц, 1H), 8,23-7,93 (m, 2H), 7,58 (d, J=9,2 Гц, 1H), 7,02 (s, 1H), 3,93 (s, 3H), 3,74 (s, 3H), 3,61-3,46 (m, 4H), 3,19-2,98 (m, 4H), 2,93-2,89 (m, 2H), 2,79-2,71 (m, 1H), 1,37-1,29 (m, 1H), 1,17-1,08 (m, 1H), 0,83-0,75 (m, 1H), 0,68-0,58 (m, 1H).
Пример 22:
Получение соединения 22
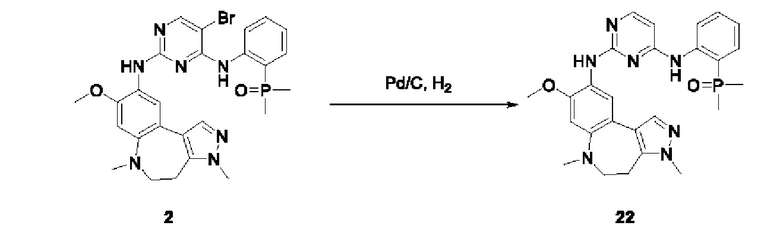
Соединение 2 (70 мг, 0,12 ммоль) растворяли в метаноле (6 мл). К реакционной жидкости добавляли палладий на углероде (10%, 25 мг). После 3-разовой продувки с применением водорода реакционную систему последовательно перемешивали при комнатной температуре в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, палладий на углероде отфильтровывали, фильтрат концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 22 (27 мг).
МС (ИЭР, масса/заряд): 504,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,91 (s, 1H), 8,65-8,44 (m, 2Н), 8,30 (s, 1H), 7,96 (d, J=6,3 Гц, 1H), 7,54 (s, 1H), 7,25-7,17 (m, 1H), 7,13-7,08 (m, 1H), 6,98-6,92 (m, 1H), 6,58 (s, 1H), 6,16 (d, J=6,0 Гц, 1H), 3,91 (s, 3Н), 3,84 (s, 3Н), 3,33-3,29 (m, 2Н), 3,07 (s, 3Н), 3,06-3,04 (m, 2Н), 1,86 (s, 3Н), 1,82 (s, 3Н).
Пример 23:
Получение промежуточного соединения 23А
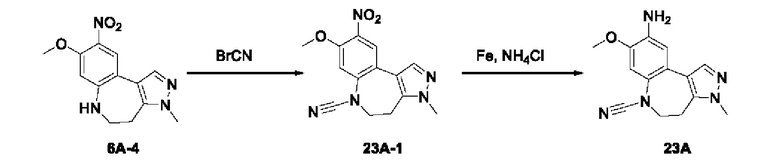
Соединение 23 А-1:
Соединение 6А-4 (100 мг, 0,36 ммоль) растворяли в N,N-диметилформамиде (3 мл) в атмосфере азота. Затем к реакционной жидкости добавляли при 0°С гидрид натрия (60%, 22 мг, 0,5 ммоль) и последовательно перемешивали указанную реакционную жидкость в течение 30 минут, после чего добавляли цианогенбромид (386 мг, 3,6 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; расход: 35 мл/мин; градиент: повышение содержания ацетонитрила от 10% до 80% в течение 20 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 23А-1 (65 мг).
МС (ИЭР, масса/заряд): 300,2 [М+Н]+.
Промежуточное соединение 23А:
Соединение 23А (50 мг) получали путем замены исходного материала на соединение 23А-1 (55 мг, 0,18 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 270,2 [М+Н]+.
Получение соединения 23:
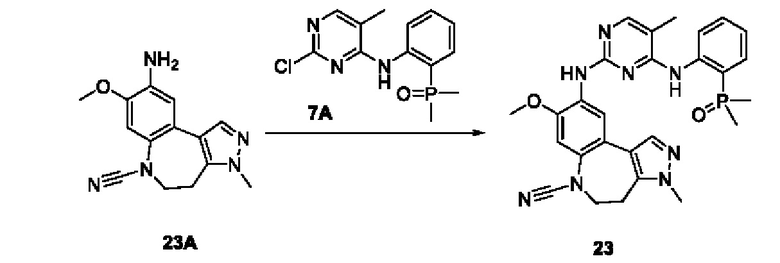
Соединение 23А (50 мг, 0,18 ммоль), соединение 7А (38 мг, 0,13 ммоль), карбонат цезия (121 мг, 0,37 ммоль), трис(дибензилиденацетон)дипалладий (0) (17 мг, 0,019 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (11 мг, 0,019 ммоль) растворяли в 1,4-диоксане (1 мл) в атмосфере азота. Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 23 (17 мг). МС (ИЭР, масса/заряд): 529,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,51 (s, 1H), 8,78 (s, 1H), 8,69-8,65 (m, 1H), 7,99 (s, 1H), 7,42 (s, 2H), 7,33-7,26 (m, 2H), 7,05-7,00 (m, 1H), 6,91 (s, 1H), 3,95 (s, 3Н), 3,86 (s, 3Н), 3,85-3,81 (m, 2H), 3,23 (t, J=5,7 Гц, 2H), 2,24 (s, 3Н), 1,89 (s, 3Н), 1,84 (s, 3Н).
Пример 24:
Получение промежуточного соединения 24А
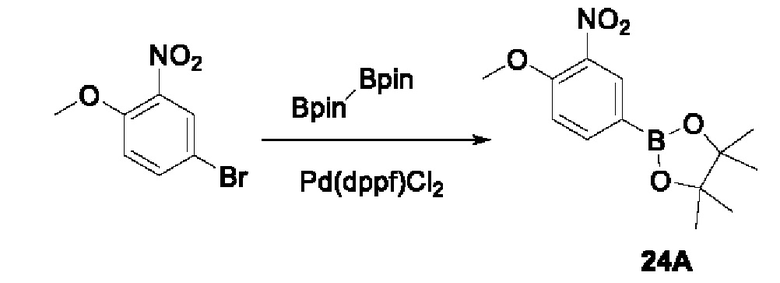
Соединение 24А (19,7 г) получали путем замены исходного материала на 4-бром-2-нитроанизол (20 г, 86,16 ммоль) согласно способу получения соединения 1А из промежуточного соединения 1А-1 в примере 1.
1Н ЯМР (300 МГц, CDCl3) δ 8,27 (d, J=1,5 Гц, 1Н), 7,96 (dd, J=8,4, 1,8 Гц, 1Н), 7,08 (d, J=8,4 Гц, 1H), 4,00 (s, 3Н), 1,36 (s, 12Н).
Получение промежуточного соединения 24 В
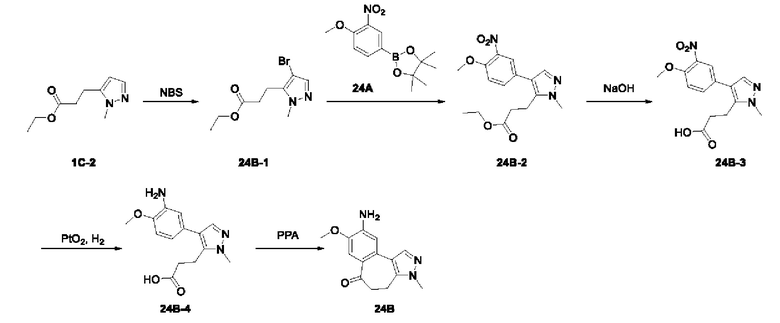
Соединение 24В-1:
Соединение 1С-2 (6 г, 32,9 ммоль) растворяли в дихлорметане (100 мл). Затем к описанной выше реакционной жидкости добавляли N-бромсукцинимид (8,8 г, 49,4 ммоль) при 0°С. Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в воду (100 мл) для гашения реакции. Смесь экстрагировали дихлорметаном (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3)5 затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 24В-1 (8,5 г).
МС (ИЭР) масса/заряд: 261,1, 263,1 [М+Н]+.
Соединение 24В-2:
Соединение 24В-2 (2 г) получали путем замены исходного материала на соединение 24В-1 (2 г, 7,6 ммоль) и соединение 24А (2,57 г, 9,2 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 334,1 [М+Н]+.
Соединение 24В-3:
Соединение 24В-2 (2 г, 6,0 ммоль) растворяли в этаноле (20 мл). Затем к реакционной жидкости добавляли водный раствор гидроксида натрия (2 N, 9 мл). Реакционную систему последовательно перемешивали при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и доводили до рН=2 с помощью 1 N водного раствора соляной кислоты. Осажденное твердое вещество отфильтровывали и высушивали с получением соединения 24В-3 (1,5 г).
МС (ИЭР, масса/заряд): 306,2 [М+Н]+.
Соединение 24В-4:
Соединение 24В-3 (1,1 г, 3,6 ммоль) растворяли в смешанном растворителе, состоящем из этанола (25 мл) и тетрагидрофурана (25 мл). Затем к реакционной жидкости добавляли диоксид платины (221 мг, 0,97 ммоль). После того, как реакционную систему продували 3 раза с помощью водорода, реакционную жидкость перемешивали при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали дихлорметаном (30 мл × 3) и концентрировали полученный фильтрат при пониженном давлении с получением соединения 24В-4 (0,95 г).
МС (ИЭР, масса/заряд): 276,2 [М+Н]+.
Промежуточное соединение 24 В:
Соединение 24В-4 (1 г, 3,6 ммоль) растворяли в полифосфорной кислоте (15 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 48 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в ледяную воду (80 г). Смесь экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 24 В (0,65 г).
МС (ИЭР) масса/заряд: 258,2 [М+Н]+.
Получение соединения 24
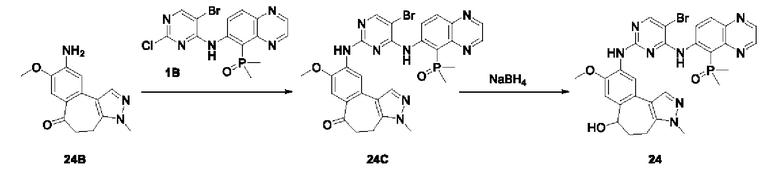
Соединение 24С:
Соединение 24С (200 мг) получали путем замены исходного материала на соединение 24 В (250 мг, 0,97 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 633,3, 635,2 [М+Н]+.
Соединение 24
Соединение 24С (100 мг, 0,16 ммоль) растворяли в метаноле (2 мл). К реакционной жидкости добавляли при 0°С борогидрид натрия (12 мг, 0,3 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, к реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл). Смесь экстрагировали этилацетатом (20 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 24 (60 мг).
МС (ИЭР) масса/заряд: 634,9, 636,9 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,68 (s, 1H), 8,87 (dd, J=9,6, 4,2 Гц, 1H), 8,74-8,72 (m, 2Н), 8,43 (s, 1H), 8,30 (s, 1H), 7,60 (s, 2Н), 7,05 (s, 1H), 7,01 (s, 1Н), 4,90 (d, J=8,1 Гц, 1H), 3,95 (s, 3H), 3,73 (s, 3H), 3,09-2,98 (m, 2H), 2,47-2,28 (m, 2H), 2,18 (s, 3H), 2,14 (s, 3H).
Пример 25:
Получение соединения 25
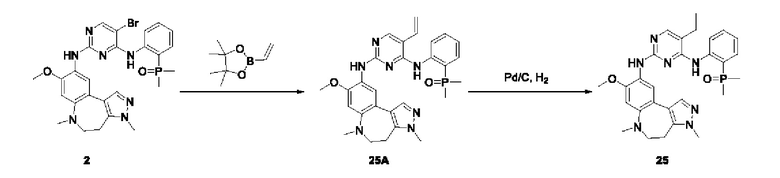
Соединение 25А:
Соединение 2 (120 мг, 0,21 ммоль), сложный пинаколовый эфир винилбороновой кислоты (63 мг, 0,41 ммоль), ацетат палладия (5 мг, 0,02 ммоль), 2-бициклогексилфосфино-2',6'-димезоксибифенил (13 мг, 0,03 ммоль) и фосфат калия (131 мг, 0,62 ммоль) растворяли в атмосфере азота в смешанном растворителе, состоящем из 1,4-диоксана (3 мл) и воды (0,6 мл). Реакционную систему нагревали до 85°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 25А (70 мг).
МС (ИЭР, масса/заряд): 530,3 [М+Н]+.
Соединение 25
Соединение 25А (70 мг, 0,13 ммоль) растворяли в этаноле (5 мл). К реакционной жидкости добавляли влажный палладий на углероде (10%, 50 мг). После того, как реакционную систему продували 3 раза с помощью водорода, реакционную жидкость перемешивали при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали этанолом (5 мл × 3) и концентрировали полученный фильтрат при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 25 (53 мг).
МС (ИЭР, масса/заряд): 532,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,82 (s, 1H), 8,94 (s, 1H), 8,64 (dd, J=8,1, 4,5 Гц, 1H), 8,17 (s, 1H), 7,80 (s, 1H), 7,42 (s, 1H), 7,25-7,18 (m, 1H), 7,08-6,96 (m, 1H), 6,96-6,90 (m, 1H), 6,58 (s, 1H), 3,89 (s, 3H), 3,83 (s, 3H), 3,32-3,29 (m, 2H), 3,07-3,04 (m, 5H), 2,63 (q, J=7,5 Гц, 2Н), 1,86 (s, 3Н), 1,82 (s, 3Н), 1,30 (t, J=7,5 Гц, 3Н).
Пример 26.
Получение соединения 26
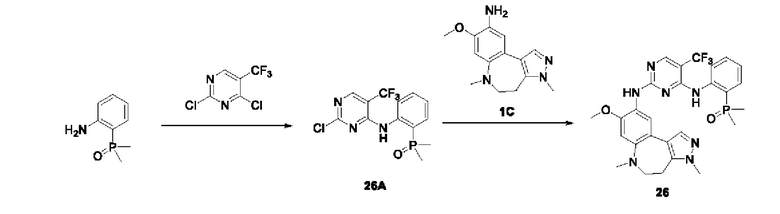
Соединение 26А:
2,4-Дихлор-5-трифторметилпиримидин (2 г, 9,2 ммоль) растворяли в N,N-диметилформамиде (20 мл) и последовательно добавляли безводный карбонат калия (1,5 г, 11,1 ммоль) и 2-(диметилфосфинокси)анилин (1,6 г, 9,2 ммоль). Реакционную жидкость нагревали до 80°С и последовательно перемешивали в течение 12 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (150 мл). Смесь экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 26А (0,35 г).
МС (ИЭР) масса/заряд: 350,1, 352,1 [М+Н]+.
Соединение 26
Соединение 26 (41 мг) получали путем замены исходного материала на соединение 26А (108 мг, 0,31 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 572,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,35 (s, 1H), 8,40-8,32 (m, 3Н), 7,49 (s, 1Н), 7,37-7,29 (m, 1Н), 7,17-6,98 (m, 2Н), 6,56 (s, 1Н), 3,97 (s, 3Н), 3,83 (s, 3Н), 3,31-3,27 (m, 2Н), 3,05-3,02 (m, 5Н), 1,88 (s, 3Н), 1,83 (s, 3Н).
19F ЯМР (282 МГц, CDCl3) δ-60,82.
Пример 27:
Получение соединения 27
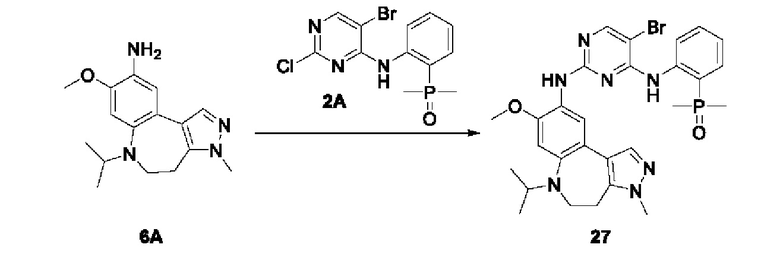
Соединение 6А (80 мг, 0,28 ммоль) и соединение 2А (101 мг, 0,28 ммоль) растворяли в N-метилпирролидиноне (2 мл). Затем к описанной выше реакционной жидкости добавляли метансульфоновую кислоту (81 мг, 0,84 ммоль). Реакционную систему нагревали до 95°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 80 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 50 мл/мин; градиент: повышение содержания ацетонитрила от 15% до 55% в течение 25 минут; длина волны детектирования: 254 нм. Продукт собирали и концентрировали при пониженном давлении с получением соединения 27 (60 мг).
МС (ИЭР, масса/заряд): 610,2, 612,2 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 10,93 (s, 1H), 8,39 (s, 1H), 8,22 (s, 1H), 8,16 (s, 1Н), 7,72 (s, 1H), 7,56 (s, 1H), 7,52-7,44 (m. 1H), 7,07-6,99 (m, 2Н), 6,57 (s, 1H), 3,98-3,89 (m, 1H), 3,75 (br s, 6Н), 3,22-3,18 (m, 2Н), 2,96-2,92 (m, 2Н), 1,78 (s, 3Н), 1,73 (s, 3Н), 1,29 (s, 3Н), 1,27 (s, 3Н).
Пример 28:
Получение соединения 28
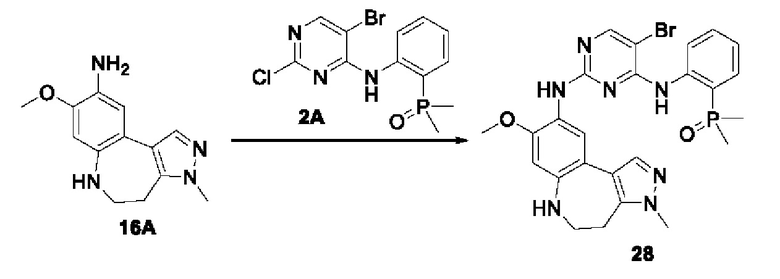
Соединение 28 (15 мг) получали путем замены исходного материала на соединение 2А (162 мг, 0,45 ммоль) и соединение 16А (110 мг, 0,45 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 568,2, 570,2 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 10,95 (s, 1H), 8,40 (s, 1H), 8,18 (s, 1H), 8,14 (s, 1Н), 7,63 (s, 1H), 7,61 (s, 1H), 7,51-7,44 (m, 1H), 7,07-6,99 (m, 2H), 6,53 (s, 1H), 5,87 (s, 1H), 3,73 (s, 3H), 3,68 (s, 3H), 3,26-3,24 (m, 2H), 2,98-2,94 (m, 2H), 1,77 (s, 3H), 1,73 (s, 3H).
Пример 29.
Получение промежуточного соединения 29
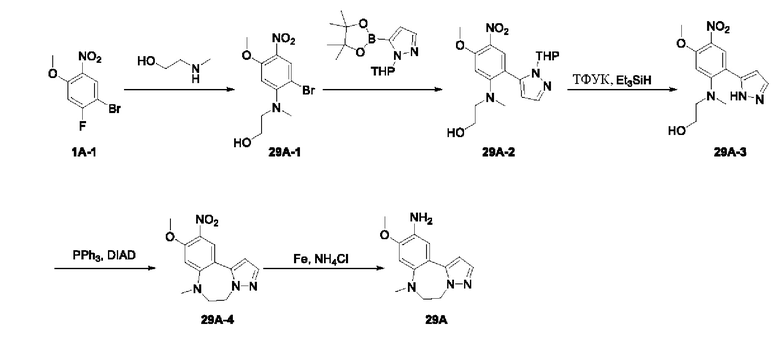
Соединение 29А-1:
Соединение 1А-1 (1 г, 4 ммоль), карбонат калия (1,11 г, 8 ммоль) и N-метилэтаноламин (0,45 г, 6 ммоль) растворяли в N,N-диметилформамиде (20 мл). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (80 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 29А-1 (1 г).
МС (ИЭР) масса/заряд: 305,0, 307,0 [М+Н]+.
Соединение 29А-2:
Соединение 29А-2 (0,15 г) получали путем замены исходного материала на соединение 29А-1 (1 г, 3,3 ммоль) и сложный пинаколовый эфир 1-(2-тетрагидропиранил)-1H-пиразол-5-бороновой кислоты (1,37 г, 4,92 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 377,3 [М+Н]+.
Соединение 29А-3:
Соединение 29А-2 (300 мг, 0,8 ммоль) растворяли в дихлорметане (3 мл). Затем к реакционной жидкости добавляли трифторуксусную кислоту (1 мл) и триэтилсилан (185 мг, 1,6 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 29А-3 (90 мг).
МС (ПЭР) масса/заряд: 292,9 [М+Н]+.
Соединение 29А-4:
Трифенилфосфин (129 мг, 0,5 ммоль) растворяли в тетрагидрофуране (2 мл) в атмосфере азота. К реакционной жидкости добавляли при 0°С диэтилазодикарбоксилат (97 мг, 0,56 ммоль) и последовательно перемешивали указанную реакционную жидкость при 0°С в течение 1 часа (с получением мутной жидкости молочного цвета). Соединение 29А-3 (90 мг, 0,31 ммоль) добавляли к описанной выше реакционной жидкости и последовательно перемешивали реакционную жидкость при комнатной температуре в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 29А-4 (35 мг).
МС (ИЭР) масса/заряд: 275,1 [М+Н]+.
Промежуточное соединение 29А:
Соединение 29А (20 мг) получали путем замены исходного материала на соединение 29А-4 (35 мг, 0,13 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 245,2 [М+Н]+.
Получение соединения 29
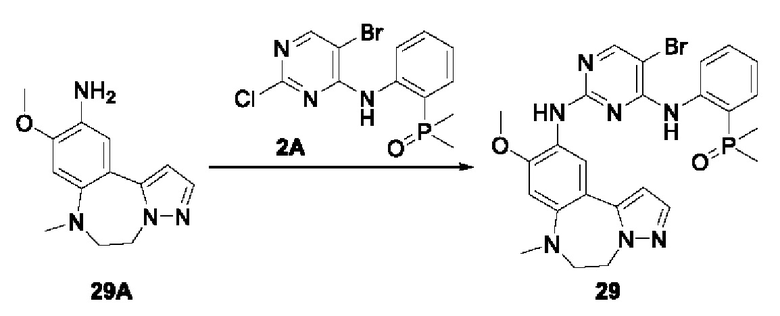
Соединение 29 (7,6 мг) получали путем замены исходного материала на соединение 2А (51,66 мг, 0,14 ммоль) и соединение 29А (35 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 568,2, 570,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,58 (s, 1H), 8,39 (s, 1H), 8,34 (s, 1H), 8,23 (s, 1H), 7,39 (s, 1H), 7,30 (s, 1H), 7,23 (s, 1H), 7,01 (s, 2H), 6,56 (s, 1H), 6,04 (s, 1H), 4,46-4,42 (m, 2H), 3,95 (s, 3H), 3,63-3,59 (m, 2H), 2,98 (s, 3H), 1,87 (s, 3H), 1,83 (s, 3H).
Пример 30:
Получение соединения 30
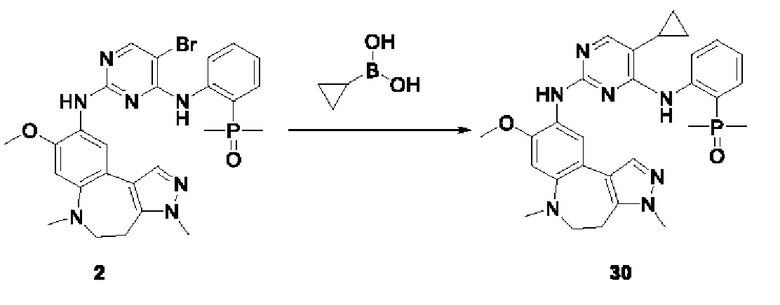
Соединение 2 (300 мг, 0,52 ммоль), циклопропилбороновую кислоту (133 мг, 1,55 ммоль), фосфат калия (328 мг, 1,55 ммоль), ацетат палладия (12 мг, 0,05 ммоль) и трициклогексилфосфин (14 мг, 0,05 ммоль) растворяли в атмосфере азота в смешанном растворителе, состоящем из толуола (2 мл) и воды (0,4 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 24 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, к реакционной жидкости добавляли воду (10 мл). Смесь экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 30 (91 мг).
МС (ИЭР) масса/заряд: 544,3 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,43 (s, 1H), 8,72-8,67 (m, 1H), 8,52 (s, 1H), 7,95 (s, 1H), 7,38 (s, 1H), 7,33-7,30 (m, 1H), 7,27-7,15 (m, 2H), 7,00-6,93 (m, 1H), 6,56 (s, 1H), 3,91 (s, 3H), 3,83 (s, 3H), 3,31-3,27 (m, 2H), 3,06-3,02 (m, 5H), 1,87 (s, 3H), 1,83 (s, 3H), 1,77-1,70 (m, 1H), 1,10-0,99 (m, 2H), 0,67-0,55 (m, 2H).
Пример 31:
Получение промежуточного соединения 31А
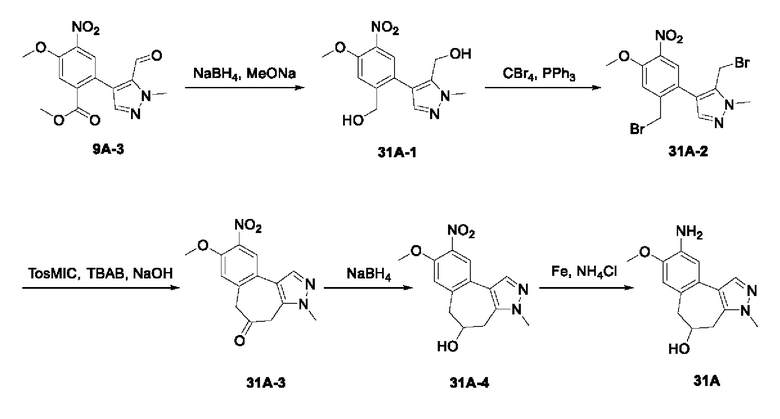
Соединение 31А-1:
Соединение 9А-3 (600 мг, 1,88 ммоль) и метоксид натрия (5 мг, 0,09 ммоль) растворяли в метаноле (12 мл). Затем к реакционной жидкости добавляли борогидрид натрия (284 мг, 7,52 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали, фильтрат концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 31А-1 (550 мг).
МС (ИЭР) масса/заряд: 294,1 [М+Н]+.
Соединение 31А-2:
Соединение 31А-1 (850 мг, 2,9 ммоль) и тетрабромметан (3,84 г, 11,59 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота. Затем к реакционной жидкости добавляли трифенилфосфин (3,04 г, 11,59 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 31А-2 (130 мг).
МС (ИЭР) масса/заряд: 418,0, 420,0, 422,0 [М+Н]+.
Соединение 31А-3:
Соединение 31А-2 (880 мг, 2,1 ммоль), бромид тетрабутиламмония (162 мг, 0,5 ммоль) и тозилметилизоцианид (451 мг, 2,3 ммоль) растворяли в дихлорметане (24 мл). Затем к реакционной жидкости добавляли при 0°С раствор гидроксида натрия (428 мг, 10,71 ммоль) в воде (6 мл). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, две фазы разделяли и экстрагировали водную фазу дихлорметаном (20 мл). Органические фазы объединяли и добавляли водный раствор соляной кислоты (7 мл, 37%), после чего последовательно перемешивали реакционную жидкость при комнатной температуре в течение 3 часов. Две фазы разделяли, и органическую фазу последовательно промывали водой (40 мл), насыщенным бикарбонатом натрия (40 мл) и солевым раствором (40 мл), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 31А-3 (360 мг).
МС (ИЭР) масса/заряд: 288,1 [М+Н]+.
Соединение 31А-4:
Соединение 31А-3 (100 мг, 0,35 ммоль) растворяли в метаноле (4 мл). Затем к реакционной жидкости добавляли при 0°С борогидрид натрия (20 мг, 0,5 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 31А-4 (85 мг).
МС (ИЭР) масса/заряд: 290,1 [М+Н]+.
Промежуточное соединение 31А:
Соединение 31А (65 мг) получали путем замены исходного материала на соединение 31А-4 (85 мг, 0,29 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 260,1 [М+Н]+.
Получение соединения 31
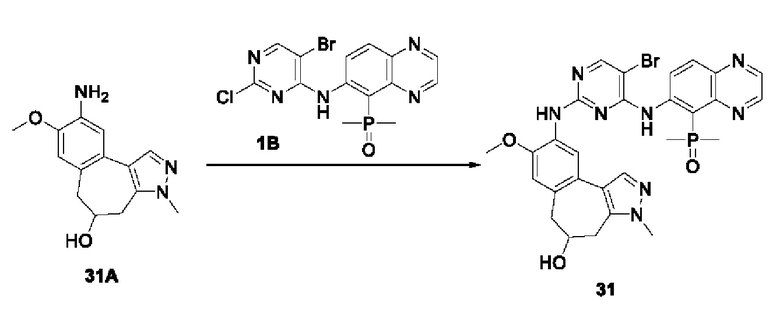
Соединение 31 (30 мг) получали путем замены исходного материала на соединение 31А (65 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 635,1, 637,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,97 (s, 1H), 8,87-8,70 (m, 3Н), 8,43 (s, 1H), 8,21 (s, 1Н), 8,14 (s, 1H), 7,59 (d, J=9,6 Гц, 1H), 7,10 (s, 1H), 6,80 (s, 1H), 4,49-4,47 (m, 1H), 3,92 (s, 3H), 3,77 (s, 3H), 3,24-2,95 (m, 5H), 2,17 (s, 3H), 2,13 (s, 3H).
Пример 32.
Получение промежуточного соединения 32А
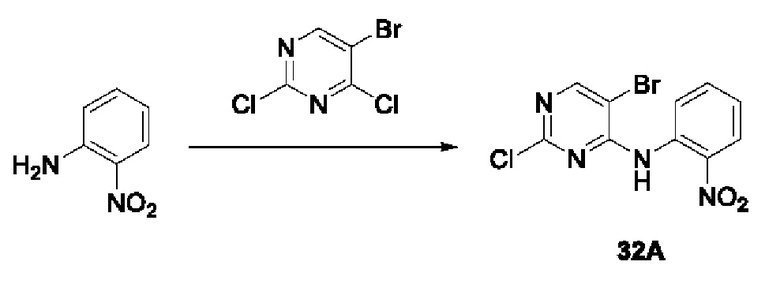
Соединение 32А (3,2 г) получали путем замены исходного материала на 2-нитроанилин (5,0 г, 36,2 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 328,8, 330,8 [М+Н]+.
Получение соединения 32
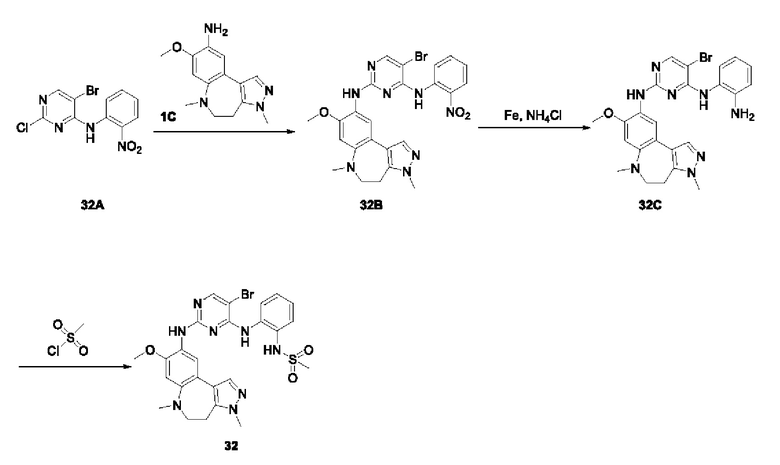
Соединение 32В:
Соединение 32В (270 мг) получали путем замены исходного материала на соединение 32А (200 мг, 0,61 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 551,0, 553,0 [М+Н]+.
Соединение 32С:
Соединение 32С (80 мг) получали путем замены исходного материала на соединение 32В (130 мг, 0,24 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 521,2, 523,2 [М+Н]+.
Соединение 32
Соединение 32С (40 мг, 0,08 ммоль) растворяли в пиридине (1,5 мл) при 0°С. Затем к реакционной жидкости добавляли метансульфонилхлорид (27 мг, 0,24 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 80 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 50 мл/мин; градиент: повышение содержания ацетонитрила от 5% до 40% в течение 30 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 32 (21 мг).
МС (ИЭР, масса/заряд): 599,0, 601,0 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 8,17 (s, 1Н), 8,13 (s, 1H), 7,68-7,59 (m, 1H), 7,49 (s, 1H), 7,39 (d, J=7,5 Гц, 1H), 7,17-7,12 (m, 3H), 6,52 (s, 1H), 3,89 (s, 3H), 3,85 (s, 3H), 3,27 (s, 2H), 3,04 (br s, 5H), 2,91 (s, 3H).
Пример 33.
Получение промежуточного соединения 33А
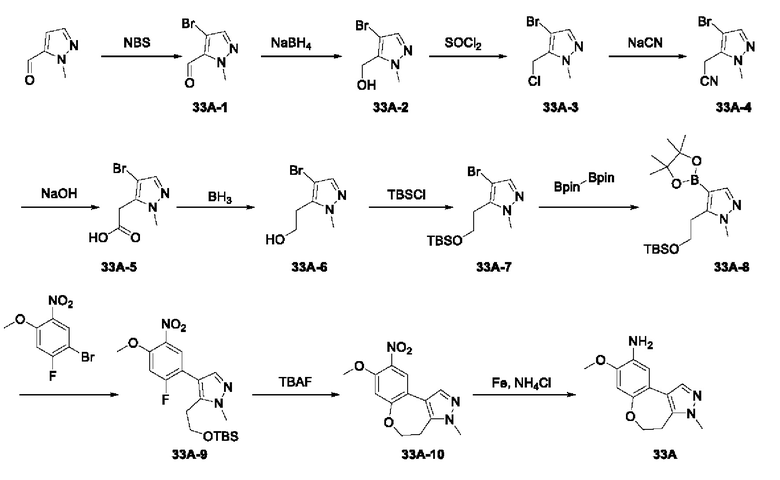
Соединение 33 А-1:
Соединение 33А-1 (80 г) получали путем замены исходного материала на 1-метил-1Н-пиразол-5-карбальдегид (59,7 г, 542 ммоль) согласно способу получения соединения 24В-1 из промежуточного соединения 1С-2 в примере 24.
1H ЯМР (300 МГц, CDCl3) δ 9,91 (s, 1H), 7,54 (s, 1Н), 4,18 (s, 3Н).
Соединение 33А-2:
Соединение 33А-1 (60 г, 317,4 ммоль) растворяли в этаноле (500 мл) при 0°С. Затем к реакционной жидкости отдельными порциями добавляли борогидрид натрия (6 г, 158,7 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. В смесь добавляли воду (300 мл) для разбавления и доводили до рН=6 с помощью концентрированной соляной кислоты. Смесь экстрагировали этилацетатом (300 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (200 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 33А-2 (60 г).
МС (ИЭР) масса/заряд: 191,2, 193,2 [М+Н]+.
Соединение 33А-3:
Соединение 33А-2 (60 г, 314 ммоль) растворяли в дихлорметане (500 мл) при 0°С. Затем к реакционной жидкости добавляли по каплям тионилхлорид (18,68 г, 157 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. В смесь добавляли дихлорметан (300 мл) для разбавления и медленно добавляли по каплям к насыщенному водному раствору бикарбоната натрия (300 мл). Смесь экстрагировали дихлорметаном (300 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (300 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 33А-3 (62 г).
МС (ИЭР) масса/заряд: 209,0, 211,0 [М+Н]+.
Соединение 33А-4:
Соединение 33А-3 (62 г, 296 ммоль) и цианид натрия (17,41 г, 355 ммоль) растворяли в диметилсульфоксиде (300 мл). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в насыщенный бикарбонат натрия (500 мл). Смесь экстрагировали этилацетатом (300 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (300 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 33А-4 (15 г).
МС (ИЭР) масса/заряд: 200,0, 202,0 [М+Н]+.
Соединение 33А-5:
Соединение 33А-4 (5 г, 25 ммоль) и гидроксид натрия (10 г, 250 ммоль) растворяли в воде (100 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и доводили до рН=1 с помощью концентрированной соляной кислоты. Смесь экстрагировали этилацетатом (200 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (150 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 33А-5 (5 г).
МС (ИЭР) масса/заряд: 218,7, 220,7 [М+Н]+.
Соединение 33А-6:
Соединение 33А-5 (1 г, 4,6 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота. Затем к реакционной жидкости добавляли раствор борана в тетрагидрофуране (1 М, 10 мл). Реакционную систему нагревали до 40°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и медленно добавляли к метанолу (40 мл), нагревали до 60°С и последовательно перемешивали в течение 30 минут. Реакционную жидкость охлаждали до комнатной температуры и затем концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 33А-6 (0,8 г).
МС (ИЭР) масса/заряд: 205,0, 207,0 [М+Н]+.
Соединение 33А-7:
Соединение 33А-6 (300 мг, 1,46 ммоль,) и имидазол (120 мг, 1,76 ммоль) растворяли в N,N-диметилформамиде (3 мл). Затем к реакционной жидкости добавляли при 0°С трет-бутилдиметилсилилхлорид (265 мг, 1,76 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (20 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (10 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/метил-трет-бутиловый эфир = 2/1) с получением соединения 33А-7 (140 мг).
МС (ИЭР) масса/заряд: 319,1, 321,1 [М+Н]+.
Соединение 33А-8:
Соединение 33А-7 (330 мг, 1,03 ммоль) растворяли в тетрагидрофуране (5 мл) в атмосфере азота. Затем к реакционной жидкости добавляли при -78°С н-бутиллитий (2,5 М, 0,6 мл, 1,5 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 1 часа. К реакционной жидкости добавляли пинаколовый эфир изопропоксибороновой кислоты (230 мг, 1,2 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (10 мл). Смесь экстрагировали этилацетатом (20 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (10 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/метил-трет-бутиловый эфир = 1/1) с получением соединения 33А-8 (300 мг).
МС (ИЭР) масса/заряд: 367,2 [М+Н]+.
Соединение 33А-9:
Соединение 33А-9 (100 мг) получали путем замены исходного материала на соединение 33А-8 (100 мг, 0,273 ммоль) и соединение 1А-1 (55 мг, 0,22 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 410,2 [М+Н]+.
Соединение 33А-10:
Соединение 33А-9 (100 мг, 0,24 ммоль) растворяли в тетрагидрофуране (2 мл). Затем к реакционной жидкости добавляли раствор фторида тетрабутиламмония в тетрагидрофуране (1 М, 1 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 33А-10 (40 мг).
МС (ИЭР) масса/заряд: 276,1 [М+Н]+.
Промежуточное соединение 33А:
Соединение 33А (30 мг) получали путем замены исходного материала на соединение 33А-10 (40 мг, 0,15 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 246,1 [М+Н]+.
Получение соединения 33
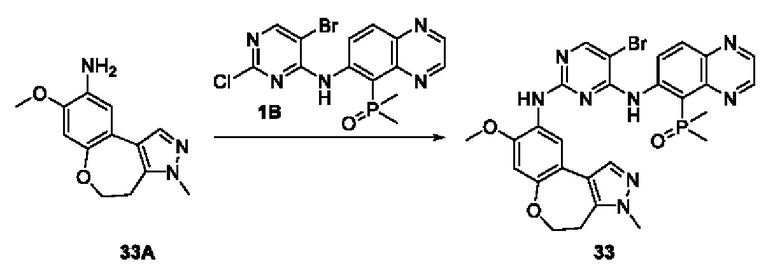
Соединение 33 (20 мг) получали путем замены исходного материала на соединение 33A (25 мг, 0,1 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 621,1, 623,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,66 (s, 1H), 8,91-8,86 (m, 1H), 8,73-8,70 (m, 2H), 8,34 (s, 1H), 8,29 (s, 1H), 7,72 (d, J=9,3 Гц, 1H), 7,58 (s, 1H), 6,99 (s, 1H), 6,63 (s, 1H), 4,40-4,36 (m, 2H), 3,88 (s, 3H), 3,73 (s, 3H), 3,19-3,15 (m, 2H), 2,18 (s, 3H), 2,13 (s, 3H).
Пример 34.
Получение промежуточного соединения 34А
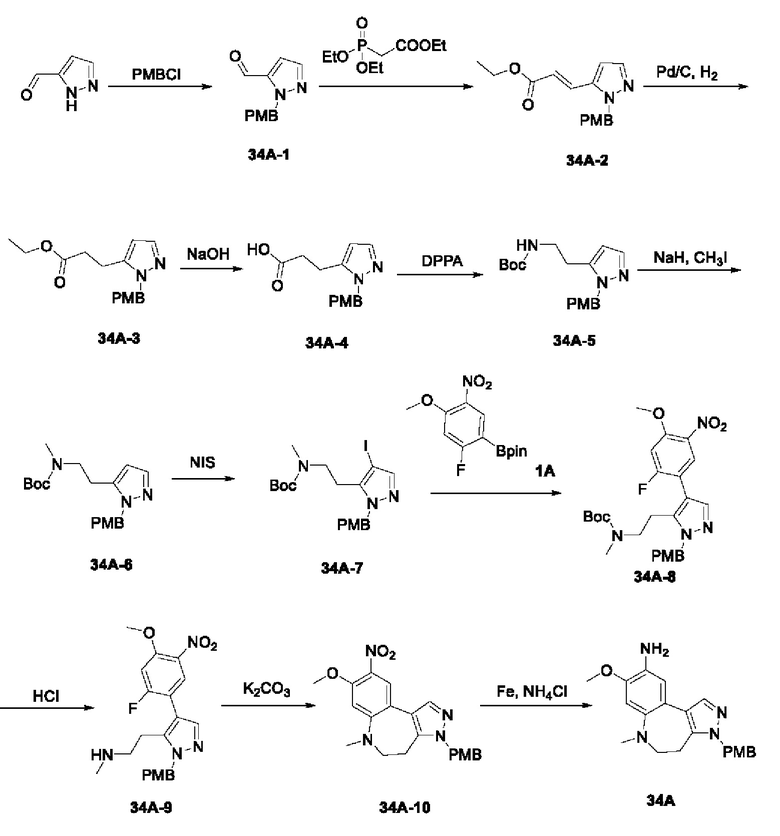
Соединение 34А-1:
1H-пиразол-5-карбальдегид (3,7 г, 38 ммоль) и фосфат калия (16 г, 77 ммоль) растворяли в N,N-диметилформамиде (50 мл). Затем к реакционной жидкости добавляли п-метоксибензилхлорид (РМВ-Cl, 6 г, 38 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в воду (150 мл). Смесь экстрагировали этилацетатом (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 34А-1 (4 г).
МС (ИЭР) масса/заряд: 217,2 [М+Н]+.
Соединение 34А-2:
Соединение 34А-2 (2,6 г) получали путем замены исходного материала на соединение 34А-1 (2 г, 9 ммоль) согласно способу получения соединения 1С-1 в примере 1. МС (ИЭР, масса/заряд): 287,1 [М+Н]+.
Соединение 34А-3:
Соединение 34А-3 (2,4 г) получали путем замены исходного материала на соединение 34А-2 (2,6 г, 9 ммоль) согласно способу получения соединения 1С-2 из соединения 1С-1 в примере 1.
МС (ИЭР, масса/заряд): 289,1 [М+Н]+.
Соединение 34А-4:
Соединение 34А-4 (2 г) получали путем замены исходного материала на соединение 34А-3 (2,6 г, 9 ммоль) согласно способу получения соединения 1С-3 из соединения 1С-2 в примере 1.
МС (ИЭР, масса/заряд): 259,1 [М-Н]-.
Соединение 34А-5:
Соединение 34А-5 (1,8 г) получали путем замены исходного материала на соединение 34А-4 (2 г, 7,7 ммоль) согласно способу получения соединения 1С-4 из соединения 1С-3 в примере 1.
МС (ИЭР, масса/заряд): 332,2 [М+Н]+.
Соединение 34А-6:
Соединение 34А-6 (1,6 г) получали путем замены исходного материала на соединение 34А-5 (1,8 г, 5,4 ммоль) согласно способу получения соединения 1С-5 из соединения 1С-4 в примере 1.
МС (ИЭР, масса/заряд): 346,3 [М+Н]+.
Соединение 34А-7:
Соединение 34А-7 (2,1 г) получали путем замены исходного материала на соединение 34А-6 (2 г, 5,8 ммоль) согласно способу получения соединения 1С-6 из соединения 1С-5 в примере 1.
МС (ИЭР, масса/заряд): 472,0 [М+Н]+.
Соединение 34А-8:
Соединение 34А-8 (1,9 г) получали путем замены исходного материала на соединение 34А-7 (1,3 г, 4,2 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 515,3 [М+Н]+.
Соединение 34А-9:
Соединение 34А-9 (1 г) получали путем замены исходного материала на соединение 34А-8 (1,9 г, 3,7 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 415,2 [М+Н]+.
Соединение 34А-10:
Соединение 34А-10 (0,8 г) получали путем замены исходного материала на соединение 34А-9 (1 г, 2,4 ммоль) согласно способу получения соединения 1С-9 из соединения 1С-8 в примере 1.
МС (ИЭР, масса/заряд): 395,3 [М+Н]+.
Соединение 34А:
Соединение 34А (200 мг) получали путем замены исходного материала на соединение 34А-10 (300 мг, 0,76 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 365,2 [М+Н]+.
Получение соединения 34
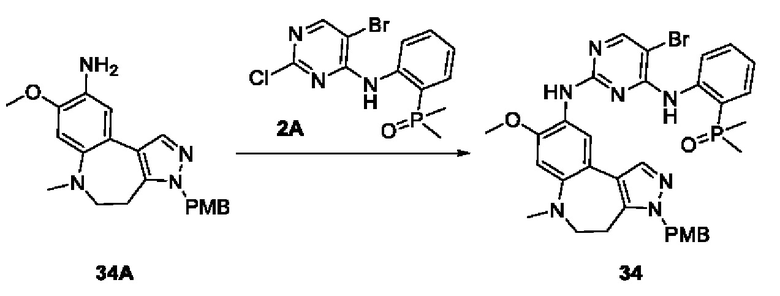
Соединение 34 (200 мг) получали путем замены исходного материала на соединение 34А (202 мг, 0,56 ммоль) и соединение 2А (200 мг, 0,56 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 688,3, 690,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,84 (s, 1H), 8,69-8,65 (m, 1H), 8,31 (s, 1H), 8,27 (s, 1H), 7,32-7,27 (m, 2H), 7,20-7,17 (m, 3Н), 7,05-7,00 (m, 1H), 6,91-6,88 (m, 2H), 6,61 (s, 1H), 5,16 (s, 2H), 3,97 (s, 3H), 3,87 (s, 3H), 3,39-3,37 (m, 2H), 3,26-3,23 (m, 2H), 3,11 (s, 3H), 1,93 (s, 3H), 1,89 (s, 3H).
Пример 35:
Получение промежуточного соединения 35А
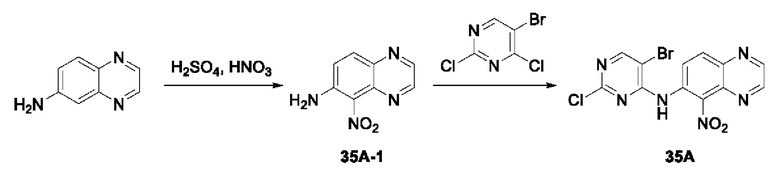
Соединение 35А-1:
6-Аминохиноксалин (10 г, 68,89 ммоль) растворяли в концентрированной серной кислоте (20 мл). К реакционной жидкости при 0°С добавляли отдельными порциями нитрат калия (9,054 г, 89,55 ммоль) и последовательно перемешивали реакционную систему при указанной температуре в течение 30 минут. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в ледяную воду (100 г). Реакционную жидкость доводили до рН=8 с помощью 1 М водного раствора гидроксида натрия. Смесь экстрагировали этилацетатом (200 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3); затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 35А-1 (2 г).
МС (ИЭР) масса/заряд: 191,2 [М+Н]+.
Промежуточное соединение 35А:
Соединение 35А (40 мг) получали путем замены исходного материала на соединение 35А-1 (50 мг, 0,26 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 381,0, 383,0 [М+Н]+.
Получение соединения 35
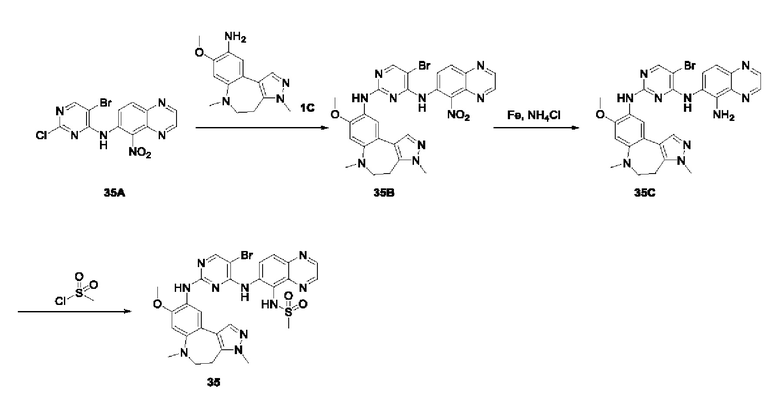
Соединение 35В:
Соединение 35В (130 мг) получали путем замены исходного материала на соединение 35А (200 мг, 0,52 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 603,0, 605,0 [М+Н]+.
Соединение 35С:
Соединение 35С (76 мг) получали путем замены исходного материала на соединение 35В (130 мг, 0,22 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 573,1, 575,1 [М+Н]+.
Соединение 35
Соединение 35 (33 мг) получали путем замены исходного материала на соединение 35С (65 мг, 0,11 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 650,9, 652,9 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,13 (s, 1H), 8,91-8,69 (m, 2H), 8,59 (d, J=9,3 Гц, 1H), 8,27 (s, 2H), 7,59 (s, 2H), 7,39 (s, 1H), 6,75 (s, 1H), 6,53 (s, 1H), 3,91 (s, 3H), 3,69 (s, 3H), 3,31 (s, 2H), 3,03 (br s, 5H), 2,96 (s, 3H).
Пример 36:
Получение промежуточного соединения 36А
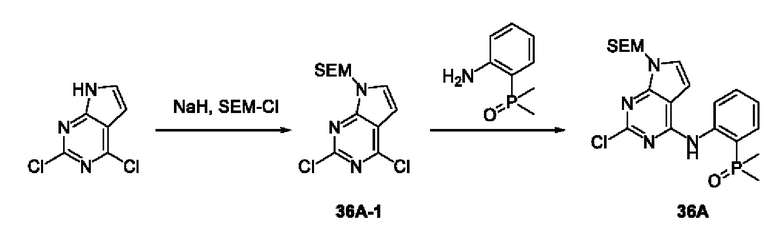
Соединение 36А-1:
2,4-Дихлор-7H-пирроло[2,3-d]пиримидин (2,5 г, 13,3 ммоль) растворяли в N,N-диметилформамиде (25 мл) в атмосфере азота. Затем к реакционной жидкости при 0°С добавляли отдельными порциями гидрид натрия (60%, 0,69 г, 17,3 ммоль) и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут. К реакционной жидкости добавляли 2-(триметилсилил)этоксиметилхлорид (2,88 г, 17,3 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 5/1) с получением соединения 36А-1 (3,9 г).
МС (ИЭР) масса/заряд: 318,1, 320,1 [М+Н]+.
Промежуточное соединение 36А:
Соединение 36А (1,1 г) получали путем замены исходного материала на соединение 36А-1 (1,1 мг, 3,55 ммоль) и 2-(диметилфосфорил)анилин (0,5 г, 2,96 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 451,2, 452,2 [М+Н]+.
Получение соединения 36
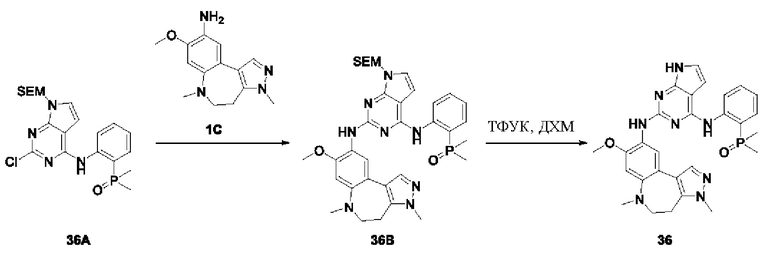
Соединение 36В:
Соединение 36А (200 мг, 0,44 ммоль), соединение 1С (126 мг, 0,49 ммоль), трис(дибензилиденацетон)дипалладий (41 мг, 0,044 ммоль), 2-дициклогексилфосфоний-2',4',6'-триизопропилбифенил (32 мг, 0,07 ммоль) и карбонат калия (123 мг, 0,89 ммоль) растворяли в трет-бутаноле (8 мл) в атмосфере азота. Реакционную систему нагревали до 110°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 36В (260 мг).
МС (ИЭР) масса/заряд: 673,4 [М+Н]+.
Соединение 36
Соединение 36В (260 мг, 0,39 ммоль) растворяли в дихлорметане (5 мл). Затем к описанной выше реакционной жидкости добавляли трифторуксусную кислоту (1,5 мл), после чего последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1,5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли метанол (5 мл) и карбонат калия (300 мг) и последовательно перемешивали полученную реакционную жидкость при комнатной температуре в течение 30 минут. Реакционную жидкость фильтровали и концентрировали фильтрат при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 36 (45 мг).
МС (ИЭР) масса/заряд: 543,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 11,26 (s, 1H), 9,69 (s, 1H), 8,89 (dd, J=8,4, 4,4 Гц, 1H), 8,50 (s, 1H), 7,62 (s, 1H), 7,41 (s, 1H), 7,30-7,14 (m, 2Н), 7,00-6,85 (m, 1Н), 6,79 (d, J=3,6 Гц, 1H), 6,56 (d, J=4,0 Гц, 2H), 3,90 (s, 3H), 3,82 (s, 3H), 3,29 (t, J=5,6 Гц, 2H), 3,04 (d, J=5,6 Гц, 2H), 3,00 (s, 3H), 1,86 (s, 3H), 1,82 (s, 3H).
Пример 37:
Получение соединения 37
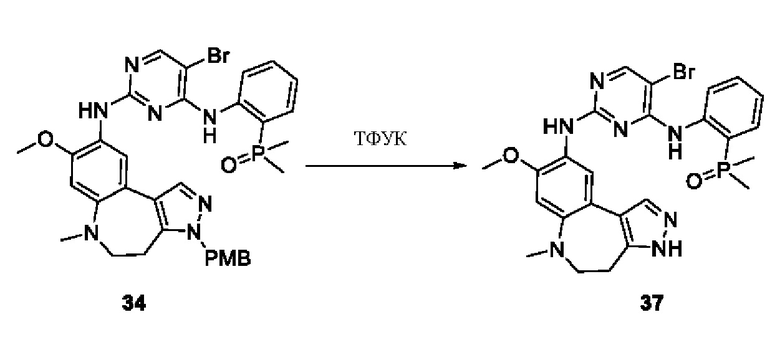
Соединение 34 (40 мг, 0,06 ммоль) растворяли в трифторуксусной кислоте (1 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 6 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток получали при высоком давлении (условия получения: колонка с обращенной фазой: колонка XBridge Shield RP18 OBD, 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,1% муравьиная кислота), и подвижная фаза В: ацетонитрил; расход: 25 мл/мин; градиент: повышение содержания подвижной фазы В от 10% до 90% в течение 28 минут; длина волны детектирования: 254 нм) с получением соединения 37 (10 мг).
МС (ИЭР) масса/заряд: 568,0, 570,0 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,71 (s, 1H), 8,45-8,43 (m, 2Н), 8,22 (s, 1H), 7,55 (s, 1Н), 7,35-7,30 (m, 3Н), 7,18-7,11 (m, 1H), 7,02-6,98 (m, 1H), 6,61 (s, 1H), 3,92 (s, 3Н), 3,31-3,06 (m, 7H), 1,89 (s, 3Н), 1,85 (s, 3Н).
Пример 38:
Получение промежуточного соединения 38А
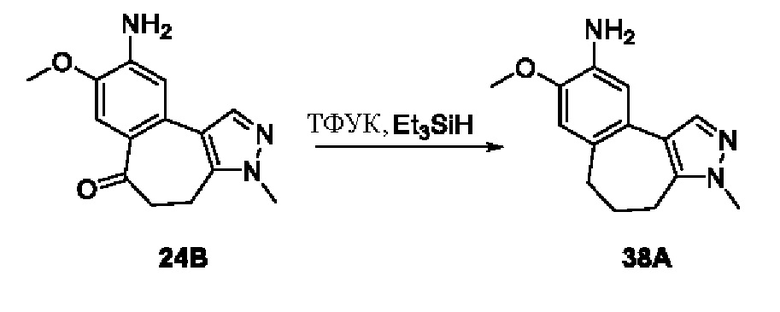
Соединение 24В (100 мг, 0,39 ммоль) и триэтилсилан (226 мг, 1,9 ммоль) растворяли в трифторуксусной кислоте (4 мл). Реакционную систему нагревали до 60°С и последовательно перемешивали в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 5% до 95% в течение 20 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 38А (90 мг).
МС (ИЭР, масса/заряд): 244,2 [М+Н]+.
Получение соединения 38
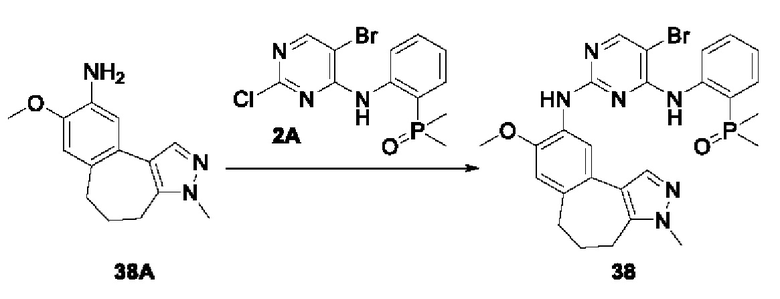
Соединение 38 (10 мг) получали путем замены исходного материала на соединение 38А (90 мг, 0,37 ммоль) и соединение 2А (133 мг, 0,37 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 567,2, 569,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10,80 (s, 1H), 8,45-8,42 (m, 1H), 8,31 (s, 1H), 8,18 (s, 1H), 7,52 (s, 1H) 7,32-7,28 (m, 1H), 7,18-7,12 (m, 2H), 7,03-6,99 (m, 1H), 6,66 (s, 1H), 3,88 (s, 3H), 3,78 (s, 3H), 2,93-2,89 (m, 2H), 2,81-2,79 (m, 2H), 2,14-2,07 (m, 2H), 1,86 (s, 3H), 1,83 (s, 3H).
Пример 39:
Получение соединения 39
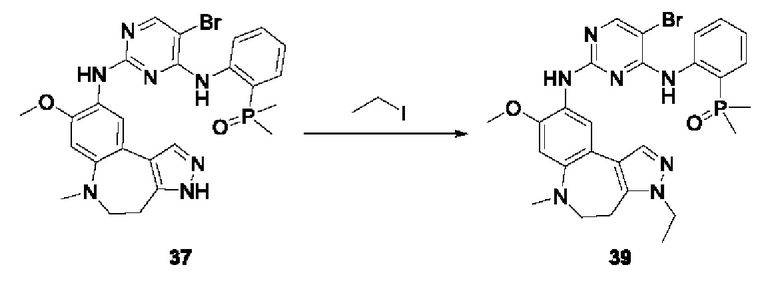
Соединение 37 (100 мг, 0,18 ммоль) и карбонат калия (49 мг, 0,35 ммоль) растворяли в N,N-диметилформамиде (2 мл). К реакционной жидкости добавляли йодтан (41 мг, 0,26 ммоль). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и добавляли воду (10 мл). Смесь экстрагировали этилацетатом (10 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (10 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 39 (10 мг).
МС (ИЭР) масса/заряд: 595,9, 597,9 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,78 (s, 1Н), 8,63 (dd, J=8,4, 4,5 Гц, 1H), 8,30 (s, 1H), 8,22 (s, 1H), 7,26-7,18 (m, 2H), 7,10 (s, 1H), 6,98-6,93 (m, 1H), 6,54 (s, 1H), 3,99 (q, J=7,2 Гц, 2H), 3,89 (s, 3H), 3,30 (br s, 2H), 3,15 (br s, 2H), 3,03 (s, 3H), 1,87 (s, 3H), 1,82 (s, 3H), 1,37 (t, J=7,2 Гц, 3H).
Пример 40:
Получение промежуточного соединения 40А
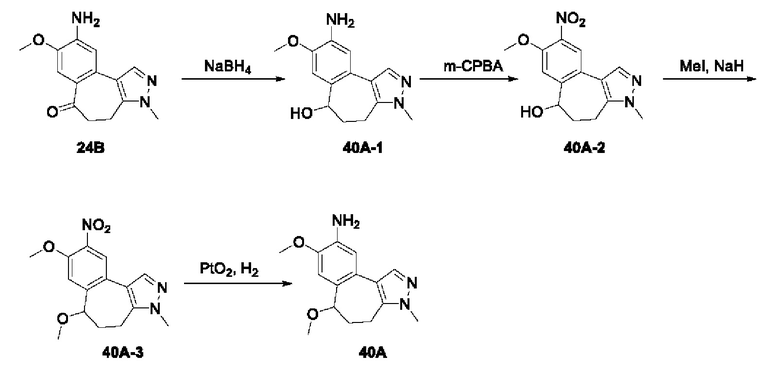
Соединение 40А-1:
Соединение 40А-1 (400 мг) получали путем замены исходного материала на соединение 24В (700 мг, 2,72 ммоль) согласно способу получения соединения 24 из соединения 24С в примере 24.
МС (ИЭР, масса/заряд): 260,1 [М+Н]+.
Соединение 40А-2:
Соединение 40А-1 (400 мг, 1,54 ммоль) растворяли в 1,2-дихлорэтане (8 мл). Затем к реакционной жидкости добавляли м-хлорпероксибензойную кислоту (799 мг, 4,63 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и добавляли воду (20 мл). Смесь экстрагировали дихлорметаном (20 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 40А-2 (160 мг).
МС (ИЭР, масса/заряд): 290,1 [М+Н]+.
Соединение 40А-3:
Соединение 40А-2 (75 мг, 0,26 ммоль) растворяли в N,N-диметилформамиде (2 мл) в атмосфере азота. Затем к реакционной жидкости отдельными порциями добавляли гидрид натрия (60%, 31 мг, 0,78 ммоль) при 0°С и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут. Затем к реакционной жидкости добавляли йодметан (110 мг, 0,78 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (20 мл). Смесь экстрагировали этилацетатом (20 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 40А-3 (75 мг).
МС (ИЭР) масса/заряд: 304,1 [М+Н]+.
Промежуточное соединение 40А:
Соединение 40А (65 мг) получали путем замены исходного материала на соединение 40А-3 (75 мг, 0,25 ммоль) согласно способу получения соединения 8А из соединения 8А-9 в примере 8.
МС (ИЭР, масса/заряд): 274,1 [М+Н]+.
Получение соединения 40
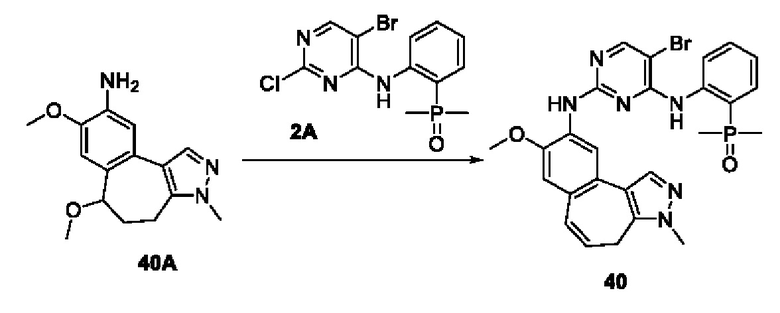
Соединение 40 (10 мг) получали путем замены исходного материала на соединение 40А (65 мг, 0,24 ммоль) и соединение 2А (86 мг, 0,24 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 564,9, 566,9 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 10,70 (s, 1H), 8,53-8,34 (m, 2Н), 8,23 (s, 1H), 7,76 (s, 1Н), 7,34-7,28 (m, 1H), 7,16-7,12 (m, 2H), 7,03-6,99 (m, 1H), 6,74 (s, 1H), 6,51 (d, J=11,2 Гц, 1H), 5,82-5,76 (m, 1H), 3,92 (s, 3H), 3,86 (s, 3H), 3,33 (d, J=6,0 Гц, 2H), 1,87 (s, 3H), 1,84 (s, 3H).
Пример 41:
Получение промежуточного соединения 41А
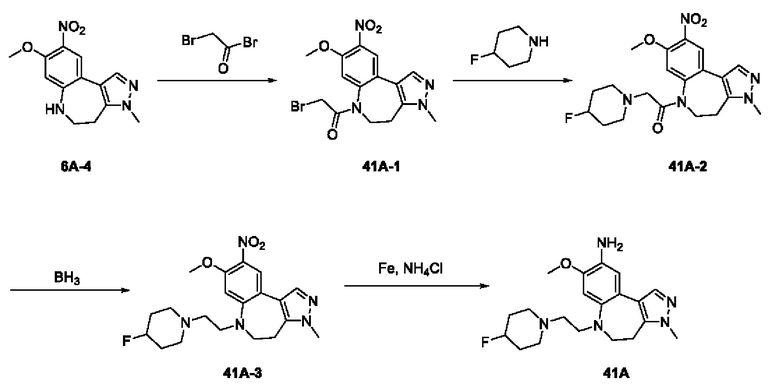
Соединение 41А-1:
Соединение 6А-4 (500 мг, 1,8 ммоль) и триэтиламин (553 мг, 5,5 ммоль) растворяли в дихлорметане (10 мл). Затем к реакционной жидкости добавляли при 0°С бромацетилбромид (736 мг, 3,65 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (50 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 41А-1 (720 мг).
МС (ИЭР) масса/заряд: 395,0, 397,0 [М+Н]+.
Соединение 41А-2:
Соединение 41А-1 (670 мг, 1,7 ммоль) и диизопропилэтиламин (657 мг, 5,09 ммоль) растворяли в N,N-диметилформамиде (15 мл). Затем к реакционной жидкости добавляли гидрохлорид 4-фторпиперидина (355 мг, 2,54 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (50 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 41А-2 (690 мг).
МС (ИЭР) масса/заряд: 417,9 [М+Н]+.
Соединение 41А-3:
Соединение 41А-3 (298 мг) получали путем замены исходного материала на соединение 41А-2 (450 мг, 1,08 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 404,1 [М+Н]+.
Промежуточное соединение 41А:
Соединение 41А (90 мг) получали путем замены исходного материала на соединение 41А-3 (100 мг, 0,25 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 374,1 [М+Н]+.
Получение соединения 41
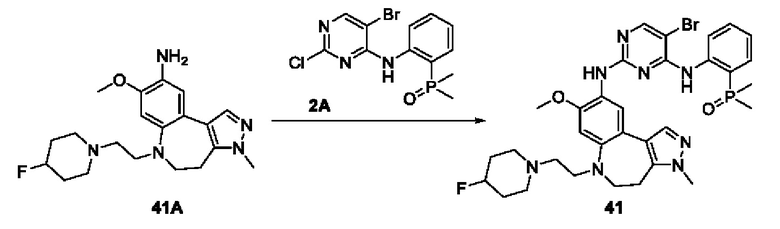
Соединение 41 (64 мг) получали путем замены исходного материала на соединение 41А (90 мг, 0,24 ммоль) и соединение 2А (87 мг, 0,24 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 697,1, 699,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,57 (s, 1H), 8,53-8,39 (m, 2Н), 8,24 (s, 1H), 7,39-7,29 (m, 2H), 7,28-7,16 (m, 2H), 7,05-6,94 (m, 1H), 6,59 (s, 1H), 4,88-4,72 (m, 1H), 3,91 (s, 3H), 3,84 (s, 3H), 3,75-3,70 (m, 2H), 3,39-3,35 (m, 2H), 3,10-2,65 (m, 8H), 2,23-1,91 (m, 4H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 42.
Получение промежуточного соединения 42А
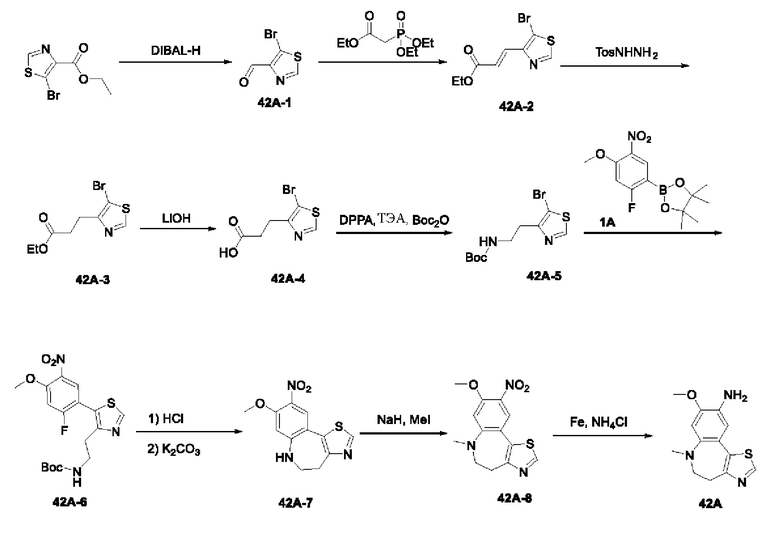
Соединение 42А-1:
Этил-5-бромтиазол-4-формиат (10 г, 42,36 ммоль) растворяли в дихлорметане (80 мл) в атмосфере азота. Затем к реакционной жидкости добавляли раствор гидрида диизобутилалюминия в толуоле (1,5 М, 56,5 мл, 84,74 ммоль) при -78°С и последовательно перемешивали реакционную жидкость при указанной температуре в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, к реакционной жидкости добавляли насыщенный водный раствор виннокислого натрий-калия (200 мл) и дихлорметан (100 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. Смесь экстрагировали дихлорметаном (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 42А-1 (6 г).
МС (ИЭР, масса/заряд): 191,9, 193,9 [М+Н]+.
Соединение 42А-2:
Соединение 42А-2 (7,4 г) получали путем замены исходного материала на соединение 42А-1 (6 г, 31,25 ммоль) согласно способу получения соединения 1С-1 в примере 1.
МС (ИЭР, масса/заряд): 261,7, 263,7 [М+Н]+.
Соединение 42А-3:
Соединение 42А-2 (7,4 г, 28,23 ммоль), ацетат натрия (4,63 г, 56,46 ммоль) и п-толуолсульфонил гидр азид (21,03 г, 112,9 ммоль) растворяли в этаноле (80 мл). Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 5/1) с получением соединения 42А-3 (6,4 г).
МС (ИЭР) масса/заряд: 263,9, 265,9 [М+Н]+.
Соединение 42А-4:
Соединение 42А-3 (3,3 г, 12,49 ммоль) растворяли в смешанном растворителе, состоящем из тетрагидрофурана (30 мл) и метанола (10 мл). К реакционной жидкости добавляли гидроксид лития (1,2 г, 49,97 ммоль) и воду (10 мл). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. К реакционной жидкости добавляли воду (20 мл) для разбавления и доводили до рН=4 с помощью 1 М соляной кислоты. Смесь экстрагировали этилацетатом (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 42А-4 (2,5 г).
МС (ИЭР) масса/заряд: 234,0, 236,0 [М-Н]-.
Соединение 42А-5:
Соединение 42А-5 (2,5 г) получали путем замены исходного материала на соединение 42А-4 (3 г, 12,7 ммоль) согласно способу получения соединения 1С-4 из соединения 1С-3 в примере 1.
МС (ИЭР, масса/заряд): 306,8, 308,8 [М+Н]+.
Соединение 42А-6:
Соединение 42А-6 (2,8 г) получали путем замены исходного материала на соединение 42А-5 (2,5 г, 8,14 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 398,1 [М+Н]+.
Соединение 42А-7:
Соединение 42А-6 (1 г, 2,52 ммоль) растворяли в дихлорметане (10 мл). Затем к реакционной жидкости добавляли раствор хлористого водорода в этилацетате (4 М, 5 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в N,N-диметилформамиде (10 мл) и добавляли к реакционной жидкости карбонат калия (1,74 г, 12,6 ммоль). Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 42А-7 (0,4 г).
МС (ИЭР) масса/заряд: 277,9 [М+Н]+.
Соединение 42А-8:
Соединение 42А-8 (117 мг) получали путем замены исходного материала на соединение 42А-7 (150 мг, 0,54 ммоль) согласно способу получения соединения 8А-9 из соединения 8А-8 в примере 8.
МС (ИЭР, масса/заряд): 292,2 [М+Н]+.
Промежуточное соединение 42А:
Соединение 42А (92 мг) получали путем замены исходного материала на соединение 42А-8 (117 мг, 0,4 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 262,3 [М+Н]+.
Получение соединения 42
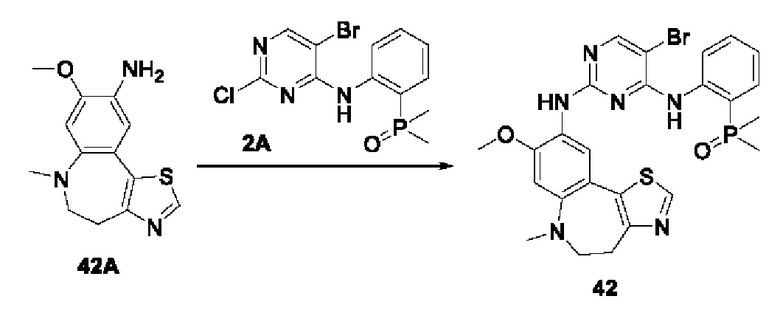
Соединение 42 (35 мг) получали путем замены исходного материала на соединение 42А (70 мг, 0,27 ммоль) и соединение 2А (96 мг, 0,27 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1. МС (ИЭР, масса/заряд): 585,1, 587,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,75 (s, 1H), 8,47 (s, 1H), 8,45-8,40 (m, 1H), 8,37 (s, 1Н), 8,22 (s, 1H), 7,55 (s, 1H), 7,26-7,21 (m, 1H), 7,15-7,09 (m, 1H), 6,97-6,92 (m, 1H), 6,54 (s, 1H), 3,93 (s, 3H), 3,36 (br s, 4H), 3,08 (s, 3H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 43:
Получение промежуточного соединения 43А
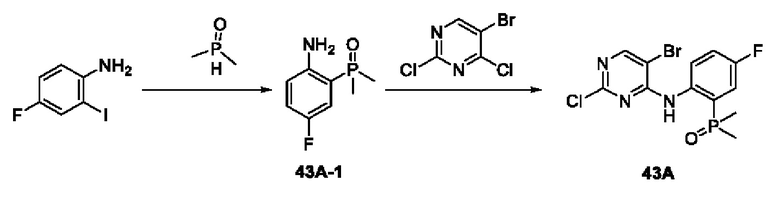
Соединение 43А-1
2-Йод-4-фторанилин (3 г, 12,66 ммоль), диметилфосфиноксид (1,18 г, 15,2 ммоль), ацетат палладия (142 мг, 0,63 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (549 мг, 0,95 ммоль) и диизопропилэтиламин (3,27 г, 25,32 ммоль) растворяли в N,N-диметилформамиде (30 мл) в атмосфере азота. Реакционную систему нагревали до 120°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 43А-1 (0,65 г).
МС (ИЭР) масса/заряд: 187,9 [М+Н]+.
Промежуточное соединение 43А:
Соединение 43А (200 мг) получали путем замены исходного материала на соединение 43А-1 (450 мг, 2,4 ммоль) согласно способу получения соединения 2А в примере 2.
МС (ИЭР, масса/заряд): 377,6, 379,6 [М+Н]+.
Получение соединения 43
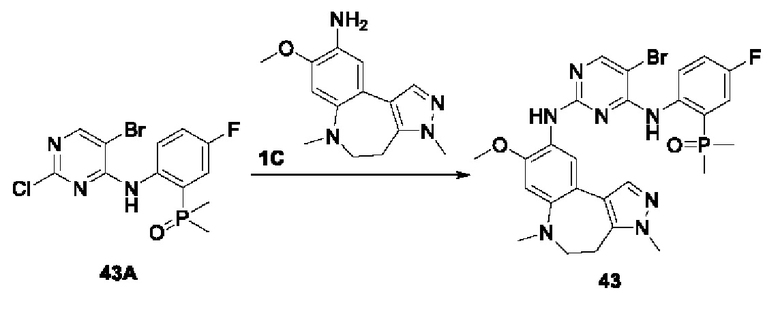
Соединение 43 (10 мг) получали путем замены исходного материала на соединение 43А (33 мг, 0,087 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 600,2, 602,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,39 (s, 1H), 8,36-8,30 (m, 2Н), 8,20 (s, 1H), 7,46 (s, 1Н), 7,18 (s, 1H), 7,01-6,93 (m, 1H), 6,77-6,71 (m, 1H), 6,57 (s, 1H), 3,92 (s, 3H), 3,84 (s, 3H), 3,32 (br s, 2H), 3,06 (br s, 5H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 44:
Получение промежуточного соединения 44А
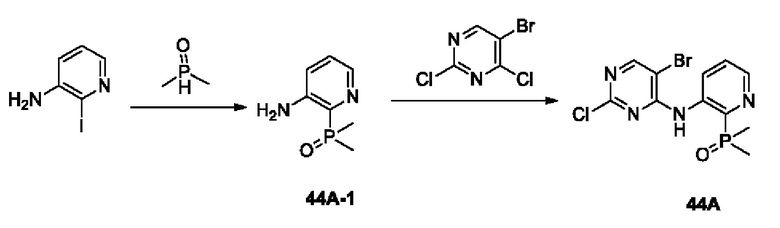
Соединение 44А-1:
Соединение 44А-1 (300 мг) получали путем замены исходного материала на 2-йод-3-аминопиридин (500 мг, 2,27 ммоль) согласно способу получения соединения 43А в примере 43.
МС (ИЭР, масса/заряд): 171,1 [М+Н]+.
Промежуточное соединение 44А:
Соединение 44А (730 мг) получали путем замены исходного материала на соединение 44А-1 (800 мг, 4,7 ммоль) согласно способу получения соединения 2А в примере 2.
МС (ИЭР, масса/заряд): 360,9, 362,9 [М+Н]+.
Получение соединения 44
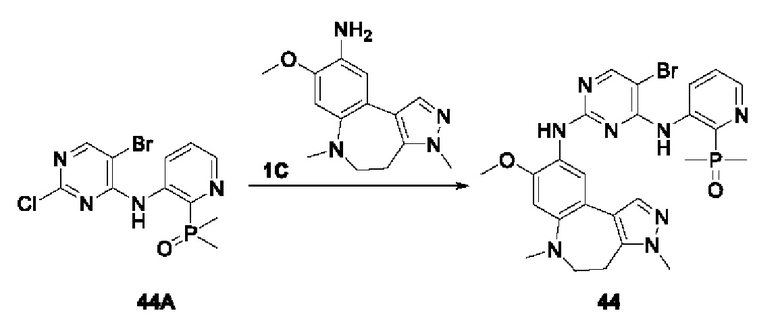
Соединение 44 (33 мг) получали путем замены исходного материала на соединение 44А (80 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 583,1, 585,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 11,04 (s, 1H), 9,01-8,96 (m, 1H), 8,29 (s, 1H), 8,24 (s, 1H), 8,19-8,17 (m, 1H), 7,48 (s, 1H), 7,19 (s, 1H), 6,96 (s, 1H), 6,58 (s, 1H), 3,91 (s, 3H), 3,85 (s, 3H), 3,32 (br s, 2H), 3,07 (br s, 5H), 1,91 (s, 3H), 1,86 (s, 3H).
Пример 45:
Получение соединения 45
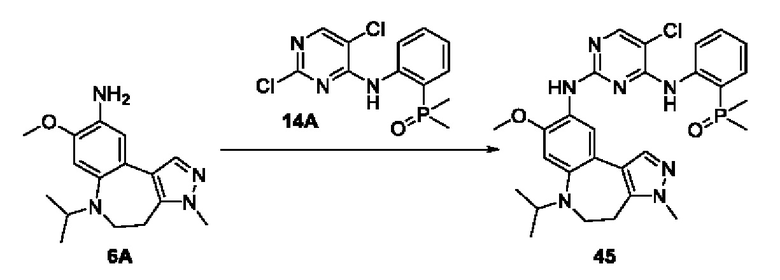
Соединение 45 (49 мг) получали путем замены исходного материала на соединение 14А (60 мг, 0,19 ммоль) и соединение 6А (49 мг, 0,19 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 566,1, 568,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,79 (s, 1H), 8,64-8,59 (m, 1H), 8,42 (s, 1H), 8,13 (s, 1H), 7,38 (s, 1H), 7,31-7,23 (m, 1H), 7,19 (s, 2H), 7,01-6,95 (m, 1H), 6,58 (s, 1H), 3,88 (br s, 4H), 3,82 (s, 3H), 3,30 (br s, 2H), 2,95 (br s, 2H), 1,87 (s, 3H), 1,83 (s, 3H), 1,33 (s, 3H), 1,27 (s, 3H).
Пример 46:
Получение промежуточного соединения 46А
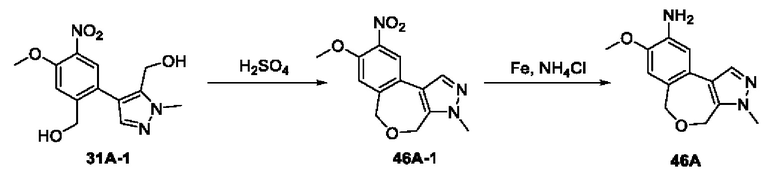
Соединение 46А-1:
Соединение 31А-1 (180 мг, 0,61 ммоль) растворяли в 50% водном растворе серной кислоты (5 мл). Реакционную систему нагревали до 85°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и доводили до рН=8 с применением 1 М гидроксида натрия. Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 46А-1 (35 мг).
МС (ИЭР) масса/заряд: 276,0 [М+Н]+.
Промежуточное соединение 46А:
Соединение 46А (35 мг) получали путем замены исходного материала на соединение 46А-1 (45 мг, 0,16 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 246,1 [М+Н]+.
Получение соединения 46
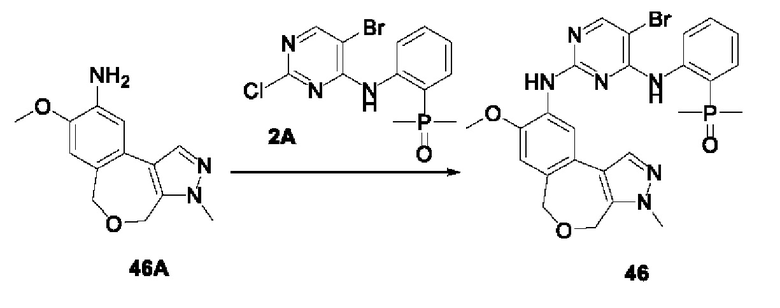
Соединение 46 (19 мг) получали путем замены исходного материала на соединение 46А (35 мг, 0,14 ммоль) и соединение 2А (51 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 569,2, 571,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 10,55 (s, 1H), 8,56 (s, 1H), 8,42 (dd, J=8,4, 4,4 Гц, 1H), 8,26 (s, 1H), 7,57 (s, 1H), 7,37-7,30 (m, 1H), 7,25-7,21 (m, 1H), 7,10 (s, 1H), 7,08-6,99 (m, 1H), 6,71 (s, 1H), 5,04 (s, 2H), 4,64 (s, 2H), 3,90 (s, 3H), 3,74 (s, 3H), 1,88 (s, 3H), 1,85 (s, 3H).
Пример 47:
Получение промежуточного соединения 47А
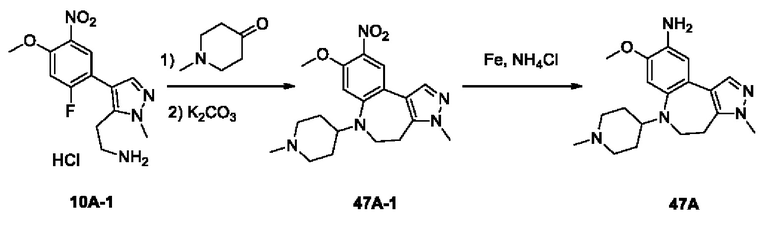
Соединение 47А-1:
Соединение 10А-1 (200 мг, 0,61 ммоль), N-метил-4-пиперидон (342 мг, 3,03 ммоль) и ацетат натрия (99 мг, 1,21 ммоль) растворяли в метаноле (3 мл) и реакционную жидкость перемешивали при комнатной температуре в течение 30 минут. К реакционной жидкости добавляли уксусную кислоту (73 мг, 1,21 ммоль) и цианоборогидрид натрия (76 мг, 1,21 ммоль). Реакционную систему последовательно перемешивали при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в N,N-диметилформамиде (3 мл) и добавляли к реакционной жидкости карбонат калия (200 мг). Реакционную жидкость нагревали до 110°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и выливали в воду (30 мл). Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 47А-1 (160 мг).
МС (ИЭР) масса/заряд: 372,2 [М+Н]+.
Промежуточное соединение 47А:
Соединение 47А (130 мг) получали путем замены исходного материала на соединение 47А-1 (160 мг, 0,43 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 342,2 [М+Н]+.
Получение соединения 47
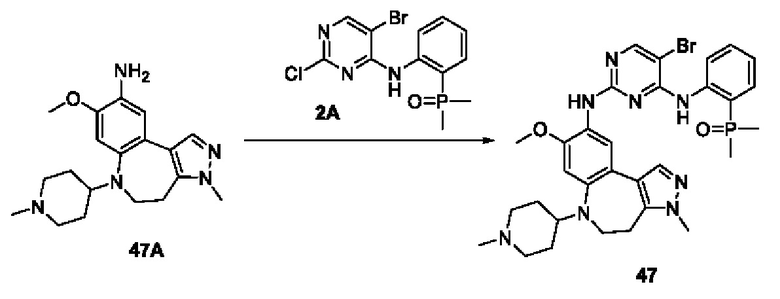
Соединение 47 (44 мг) получали путем замены исходного материала на соединение 47А (130 мг, 0,38 ммоль) и соединение 2А (137 мг, 0,38 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 665,0, 667,0 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,57 (s, 1H), 8,49-8,45 (m, 2Н), 8,24 (s, 1H), 7,35-7,17 (m, 4H), 7,03-6,98 (m, 1H), 6,52 (s, 1H), 3,86 (s, 3H), 3,80 (s, 3H), 3,52-3,45 (m, 1H), 3,43-3,36 (m, 4H), 2,94 (t, J=5,7 Гц, 2H), 2,59 (s, 3H), 2,57-2,50 (m, 2H), 2,31-2,15 (m, 2H), 2,02-1,92 (m, 2H), 1,86 (s, 3H), 1,82 (s, 3H).
Пример 48:
Получение промежуточного соединения 48А
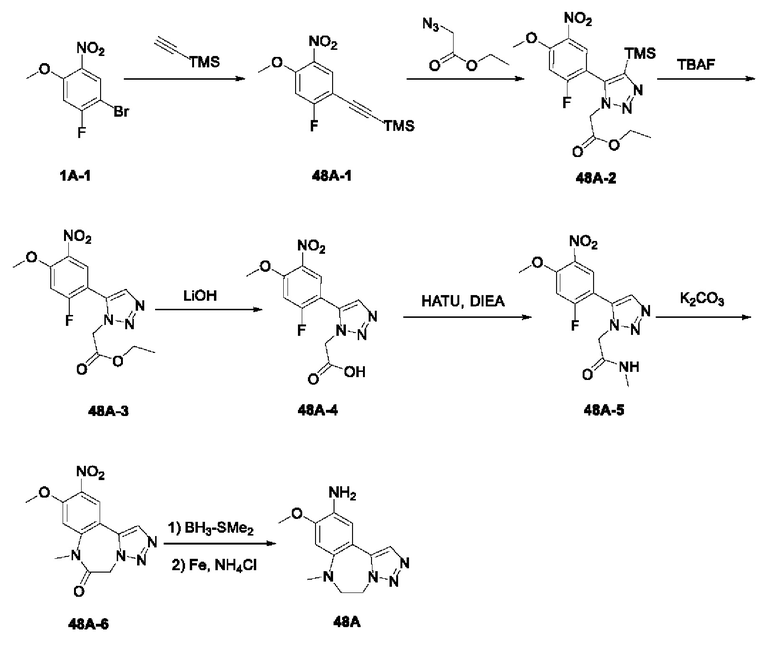
Соединение 48А-1:
Соединение 1А-1 (2 г, 8,0 ммоль), триметилсилилацетилен (2,36 г, 24 ммоль), тетракис(трифенилфосфин)палладий (1,85 г, 1,6 ммоль) и йодид меди(I) (0,15 г, 0,8 ммоль) растворяли в триэтиламине (20 мл) в атмосфере азота. Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 15/1) с получением соединения 48А-1 (1,73 г).
1H ЯМР (400 МГц, CDCl3) δ 8,07 (d, J=7,2 Гц, 1H), 6,79 (d, J=10,4 Гц, 1H), 3,97 (s, 3H), 0,26 (s, 9H).
Соединение 48A-2:
Соединение 48A-1 (2,6 г, 9,7 ммоль) и этилазидоацетат (1,26 г, 9,7 ммоль) растворяли в толуоле (30 мл). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 2 дней. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением соединения 48А-2 (2,8 г). МС (ИЭР, масса/заряд): 397,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 7,95 (d, J=7,5 Гц, 1H), 6,96 (d, J=10,5 Гц, 1H), 5,03 (s, 2H), 4,20 (q, J=7,2 Гц, 2H), 4,07 (s, 3H), 1,25 (t, J=7,2 Гц, 3H), 0,23 (s, 9H).
Соединение 48A-3:
Соединение 48A-2 (2,8 г, 7,1 ммоль) растворяли в тетрагидрофуране (20 мл) при 0°С. Затем к реакционной жидкости добавляли фторид тетрабутиламмония (1,85 г, 7,1 ммоль). Реакционную жидкость нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 48А-3 (1 г).
МС (ИЭР, масса/заряд): 325,1 [М+Н]+.
Соединение 48А-4:
Соединение 48А-4 (200 мг) получали путем замены исходного материала на соединение 48А-3 (300 мг, 0,9 ммоль) согласно способу получения соединения 42А-4 из соединения 42А-3 в примере 42.
МС (ИЭР, масса/заряд): 297,1 [М+Н]+.
Соединение 48А-5:
Соединение 48А-4 (300 мг, 1,0 ммоль) и гексафторфосфат 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония (501 мг, 1,3 ммоль) растворяли в N,N-диметилформамиде (5 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 30 минут. Затем к реакционной жидкости добавляли гидрохлорид метиламина (137 мг, 2,0 ммоль) и диизопропилэтиламин (524 мг, 4,1 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; расход: 35 мл/мин; градиент: повышение содержания ацетонитрила от 20% до 70% в течение 20 минут; длина волны детектирования 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 48А-5 (100 мг).
МС (ИЭР, масса/заряд): 310,1 [М+Н]+.
Соединение 48А-6:
Соединение 48А-5 (100 мг, 0,3 ммоль) и карбонат калия (89 мг, 0,6 ммоль) растворяли в N,N-диметилформамиде (1 мл). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры. Реакционную жидкость очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; расход: 35 мл/мин; градиент: повышение содержания ацетонитрила от 20% до 60% в течение 20 минут; длина волны детектирования 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 48А-6 (45 мг).
МС (ИЭР, масса/заряд): 290,1 [М+Н]+.
Промежуточное соединение 48:
Соединение 48А-6 (45 мг, 0,17 ммоль) растворяли в тетрагидрофуране (2 мл). Затем к реакционной жидкости добавляли диметилсульфид борана (59 мг, 0,78 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем медленного добавления метанола (1 мл) и концентрировали при пониженном давлении. Остаток растворяли в этаноле (2 мл) и воде (0,4 мл). Затем добавляли железный порошок (43 мг, 0,78 ммоль) и хлорид аммония (13 мг, 0,23 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и фильтровали, после чего концентрировали фильтрат при пониженном давлении с получением неочищенного соединения 48А (40 мг).
МС (ИЭР, масса/заряд): 246,2 [М+Н]+.
Получение соединения 48
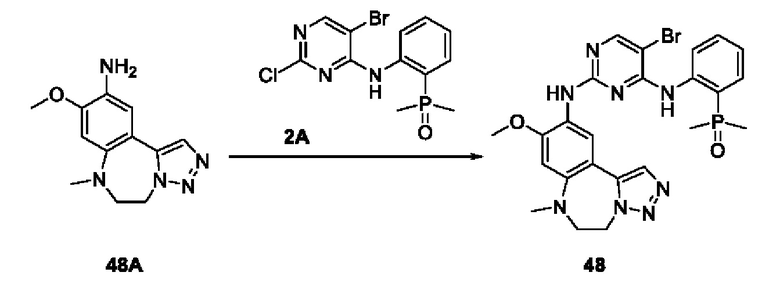
Соединение 48 (25 мг) получали путем замены исходного материала на соединение 48А (40 мг, 0,16 ммоль) и соединение 2А (41 мг, 0,1 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 568,9, 570,9 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,67 (s, 1H), 8,49 (s, 1H), 8,32 (s, 1H), 8,23 (s, 1H), 7,40-7,33 (m, 1H), 7,55 (m, 1H), 7,15-7,03 (m, 3H), 6,53 (s, 1H), 4,77-4,67 (m, 2H), 3,97 (s, 3H), 3,60-3,57 (m, 2H), 3,09 (s, 3H), 1,91 (s, 3H), 1,87 (s, 3H).
Пример 49:
Получение промежуточного соединения 49А
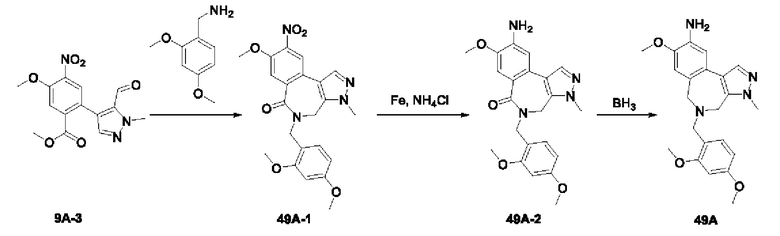
Соединение 49А-1:
Соединение 49А-1 (410 мг) получали путем замены исходного материала на 2,4-диметоксибензиламин (786 мг, 4,7 ммоль) согласно способу получения соединения 9А-4 из соединения 9А-3 в примере 9.
МС (ИЭР, масса/заряд): 439,1 [М+Н]+.
Соединение 49А-2:
Соединение 49А-2 (308 мг) получали путем замены исходного материала на соединение 49А-1 (380 мг, 0,87 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 409,2 [М+Н]+.
Промежуточное соединение 49А:
Соединение 49А (212 мг) получали путем замены исходного материала на соединение 49А-1 (250 мг, 0,61 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 395,2 [М+Н]+.
Получение соединения 49
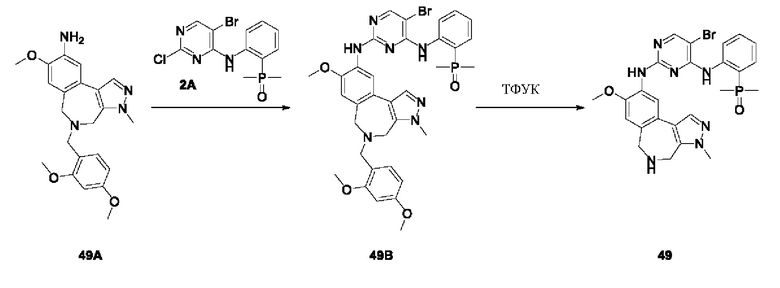
Соединение 49В:
Соединение 49В (117 мг) получали путем замены исходного материала на соединение 49А (100 мг, 0,25 ммоль) и соединение 2А (91 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 718,1, 720,1 [М+Н]+.
Соединение 49
Соединение 49 (26 мг) получали путем замены исходного материала на соединение 49В (100 мг, 0,14 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 568,1,1, 570,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,60 (s, 1H), 8,56 (s, 1H), 8,47 (dd, J=8,7, 4,5 Гц, 1H), 8,27 (s, 1H), 7,50 (s, 1H), 7,38-7,31 (m, 1H), 7,25 (d, J=8,1 Гц, 1H), 7,19 (s, 1H), 7,09-7,04 (m, 1H), 6,70 (s, 1H), 4,27 (s, 2H), 3,97 (s, 2H), 3,92 (s, 3H), 3,79 (s, 3H), 1,90 (s, 3H), 1,85 (s, 3H).
Пример 50:
Получение промежуточного продукта 50А
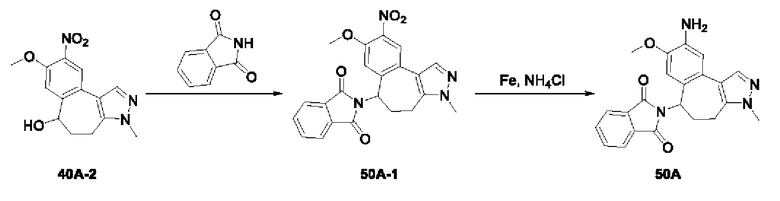
Соединение 50А-1:
Соединение 40А-2 (150 мг, 0,52 ммоль) и трифенилфосфин (204 мг, 0,78 ммоль) растворяли в тетрагидрофуране (5 мл) в атмосфере азота. Затем к реакционной жидкости добавляли при 0°С диэтилазодикарбоксилат (135 мг, 0,78 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 50А-1 (70 мг).
МС (ИЭР, масса/заряд): 419,0 [М+1]+.
Промежуточное соединение 50А:
Соединение 50А (56 мг) получали путем замены исходного материала на соединение 50А-1 (65 мг, 0,16 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 389,2 [М+Н]+.
Получение соединения 50
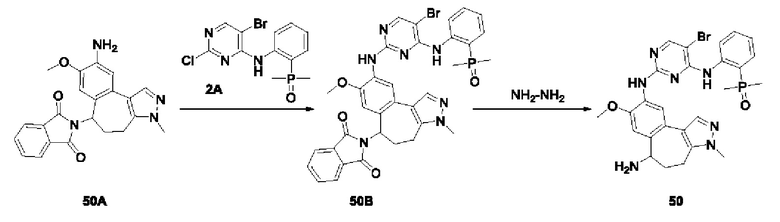
Соединение 50В:
Соединение 50В (38 мг) получали путем замены исходного материала на соединение 50А (56 мг, 0,14 ммоль) и соединение 2А (52 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 712,1, 714,1 [М+Н]+.
Соединение 50:
Соединение 50В (38 мг, 0,05 ммоль) растворяли в метаноле (2 мл). Затем к реакционной жидкости добавляли гидрат гидразина (27 мг, 0,54 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 15% до 55% в течение 25 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 50 (8 мг).
МС (ИЭР, масса/заряд): 582,1, 584,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,53 (s, 1H), 8,55 (s, 1H), 8,40 (s, 1H), 8,23 (s, 1H), 8,22 (s, 1H), 7,50 (s, 1H), 7,19 (s, 1H), 7,21 (s, 2H), 7,03 (s, 1H), 6,85 (s, 1H), 4,35 (s, 1H), 3,89 (s, 3Н), 3,75 (s, 3Н), 2,91 (br s, 2H), 2,22 (br s, 2H), 2,24 (br s, 2H), 1,88 (s, 3H).
Пример 51:
Получение промежуточного соединения 51А
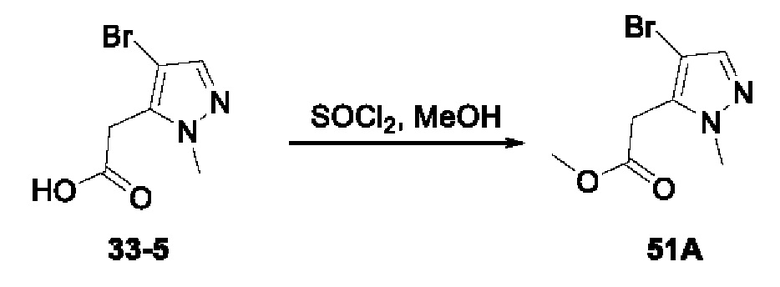
Соединение 33-5 (4,9 г, 22,37 ммоль) растворяли в метаноле (50 мл). Затем к реакционной жидкости добавляли по каплям тионилхлорид (3,99 г, 33,56 ммоль) при 0°С. Реакционную жидкость нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и добавляли воду (50 мл). Реакционную жидкость доводили до рН=7 с помощью гидроксида натрия (1 N) при 0°С. Смесь экстрагировали дихлорметаном (50 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 51А (4,4 г).
МС (ИЭР) масса/заряд: 233,1, 235,1 [М+Н]+.
Получение промежуточного соединения 51В
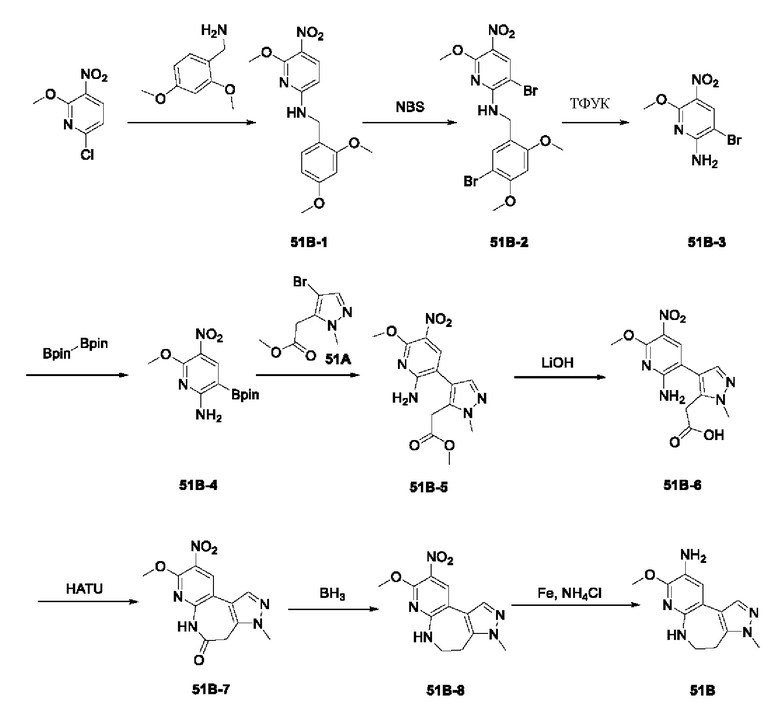
Соединение 51В-1:
Соединения 2-метокси-3-нитро-6-хлорпиридин (5 г, 26,5 ммоль) и 2,4-диметоксибензиламин (4,43 г, 26,5 ммоль) растворяли в ацетонитриле (50 мл) и N,N-диметилформамиде (25 мл). Затем к реакционной жидкости добавляли триэтиламин (2,68 г, 26,5 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (200 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 51В-1 (6 г).
МС (ИЭР) масса/заряд: 320,1 [М+Н]+.
Соединение 51В-2:
Соединение 51В-1 (6 г, 18,79 ммоль) растворяли в N,N-диметилформамиде (50 мл). Затем к реакционной жидкости добавляли N-бромсукцинимид (6,69 г, 37,58 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (300 мл). Смесь экстрагировали этилацетатом (300 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (200 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 51В-2 (7 г).
МС (ИЭР) масса/заряд: 475,9, 477,9, 479,9 [М+Н]+.
Соединение 51В-3:
Соединение 51В-3 (3 г) получали путем замены исходного материала на соединение 51В-2 (6,5 г, 13,63 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 248,1, 250,1 [М+Н]+.
Соединение 51В-4:
Соединение 51В-4 (1,5 г) получали путем замены исходного материала на соединение 51В-3 (3 г, 12,1 ммоль) согласно способу получения соединения 1А из соединения 1А-1 в примере 1.
МС (ИЭР, масса/заряд): 296,1 [М+Н]+.
Соединение 51В-5:
Соединение 51В-4 (1 г, 3,39 ммоль), соединение 51А (658 мг, 2,82 ммоль), метансульфонато(три-трет-бутилфосфино)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (162 мг, 0,28 ммоль), три-трет-бутилфосфин тетрафторборат (82 мг, 0,28 ммоль) и фосфат калия (1199 мг, 5,65 ммоль) растворяли в смешанном растворителе, состоящемиз диоксана(20 мл) и воды (0,2 мл), в атмосфере азота. Реакционную систему нагревали до 60°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/3) с получением соединения 51В-5 (760 мг).
МС (ИЭР) масса/заряд: 322,2 [М+Н]+.
Соединение 51В-6:
Соединение 51В-5 (260 мг, 0,81 ммоль) растворяли в смешанном растворителе, состоящем из тетрагидрофурана (4 мл), метанола (2 мл) и воды (2 мл). К реакционной жидкости добавляли гидроксид лития (39 мг, 1,62 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении и доводили до рН=3 с помощью 1 N соляной кислоты. Смесь экстрагировали этилацетатом (20 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного соединения 51В-6 (170 мг).
МС (ИЭР) масса/заряд: 308,1 [М+Н]+.
Соединение 51В-7:
Соединение 51В-6 (400 мг, 1,3 ммоль) растворяли в N,N-диметилформамиде (5 мл). К реакционной жидкости добавляли гексафторфосфат 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония (594 мг, 1,56 ммоль) и N,N-диизопропилэтиламин (505 мг, 3,91 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (20 мл). Смесь экстрагировали этилацетатом (20 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного соединения 51В-7 (300 мг).
МС (ИЭР) масса/заряд: 290,1 [М+Н]+.
Соединение 51В-8:
Соединение 51В-8 (140 мг) получали путем замены исходного материала на соединение 51В-7 (170 мг, 0,59 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 276,1 [М+Н]+.
Соединение 51В:
Соединение 51В (53 мг) получали путем замены исходного материала на соединение 51В-8 (140 мг, 0,51 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 246,1 [М+Н]+.
Получение соединения 51
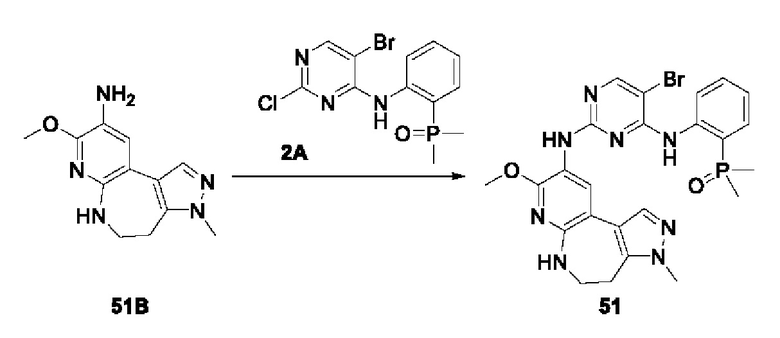
Соединение 51 (14 мг) получали путем замены исходного материала на соединение 51В (57 мг, 0,23 ммоль) и соединение 2А (84 мг, 0,23 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 569,1, 571,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,59 (s, 1H), 8,60 (s, 1H), 8,44 (dd, J=8,4, 4,5 Гц, 1H), 8,23 (s, 1H), 7,39-7,19 (m, 4H), 7,06-7,01 (m, 1H), 3,94 (s, 3H), 3,82 (s, 3H), 3,48 (t, J=5,1 Гц, 2H), 3,02 (t, J=5,1 Гц, 2H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 52:
Получение соединения 52
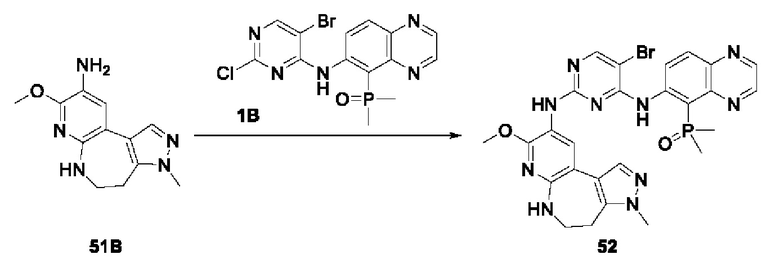
Соединение 52 (25 мг) получали путем замены исходного материала на соединение 51В (80 мг, 0,33 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 621,1, 623,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,52 (s, 1H), 8,88 (dd, J=9,6, 4,2 Гц, 1H), 8,72-8,69 (m, 2H), 8,53 (s, 1H), 8,30 (s, 1H), 7,76 (s, 1H), 7,07 (s, 2H), 3,95 (s, 3H), 3,74 (s, 3H), 3,51 (t, J=5,1 Гц, 2H), 3,03 (t, J=5,1 Гц, 2H), 2,18 (s, 3H), 2,13 (s, 3H).
Пример 53:
Получение соединения 53
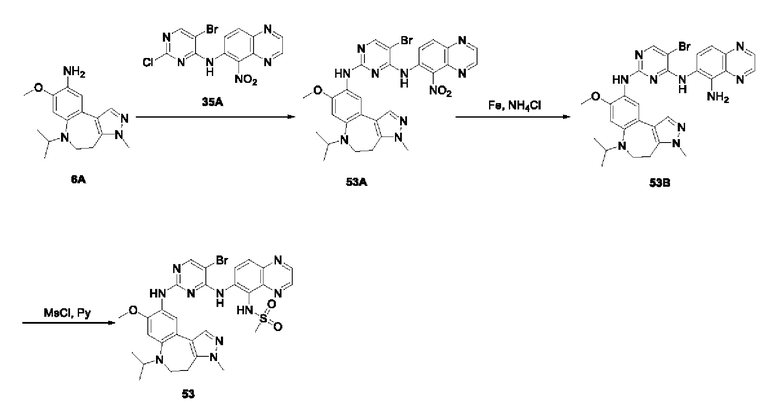
Соединение 53А:
Соединение 6А (2,7 г, 9,43 ммоль) и соединение 35А (3,6 г, 9,43 ммоль) растворяли в N-метилпирролидиноне (30 мл). Затем к описанной выше реакционной жидкости добавляли метансульфоновую кислоту (2,72 г, 28,28 ммоль). Реакционную систему нагревали до 95°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и очищали с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 330 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 70 мл/мин; градиент: повышение содержания ацетонитрила от 10% до 50% в течение 20 минут; длина волны детектирования: 254 нм. Продукт собирали и концентрировали при пониженном давлении с получением соединения 53А (3,4 г).
МС (ИЭР, масса/заряд): 631,2, 633,2 [М+Н]+.
Соединение 53В:
Соединение 53А (3,4 г, 5,38 ммоль) растворяли в смешанном растворителе, состоящем из этанола (40 мл) и воды (8 мл). Затем к описанной выше реакционной жидкости добавляли железный порошок (1,50 г, 26,92 ммоль) и хлорид аммония (0,86 г, 16,15 ммоль), после чего реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 53В (2,8 г).
МС (ИЭР, масса/заряд): 601,2, 603,2 [М+Н]+.
Соединение 53:
Соединение 53В (2,8 г, 4,66 ммоль) растворяли в пиридине (20 мл) при 0°С. Затем к реакционной жидкости добавляли метансульфонилхлорид (1,07 г, 9,31 ммоль). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры, гасили путем добавления воды (3 мл) и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) и очищали полученный неочищенный продукт с применением колонки с обращенной фазой С18. Условия очистки: хроматографическая колонка: 330 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 70 мл/мин; градиент: повышение содержания ацетонитрила от 5% до 60% в течение 30 минут; длина волны детектирования: 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 53 (2,2 г).
МС (ИЭР, масса/заряд): 679,2, 681,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,11 (s, 1H), 8,84-8,79 (m, 2Н), 8,60 (d, J=9,3 Гц, 1H), 8,33 (s, 1H), 8,28 (s, 1H), 7,58 (s, 2H), 6,74 (s, 1H), 6,54 (s, 1H), 3,89 (s, 4H), 3,67 (s, 3H), 3,30 (br s, 2H), 2,95 (br s, 5H), 1,31 (d, J=6,3 Гц, 6H).
1Н ЯМР (300 МГц, ДМСО-d6) δ 9,89 (brs, 1H), 8,94 (d, J=1,8 Гц, 1H), 8,85 (d, J=2,l Гц, 1H), 8,81 (s, 1H), 8,68 (brs, 1H), 8,35 (s, 1H), 8,27 (s, 1H), 7,74 (s, 1H), 7,49 (s, 1H), 7,40 (s, 1H), 6,58 (s, 1H), 3,99-3,91 (m, 1H), 3,76 (s, 3H), 3,71 (s, 3H), 3,22 (t, J=5,4 Гц, 2H), 3,01 (s, 3H), 2,94 (t, J=5,4 Гц, 2H), 1,29 (d, J=6,3 Гц, 6H).
Пример 54:
Получение соединения 54
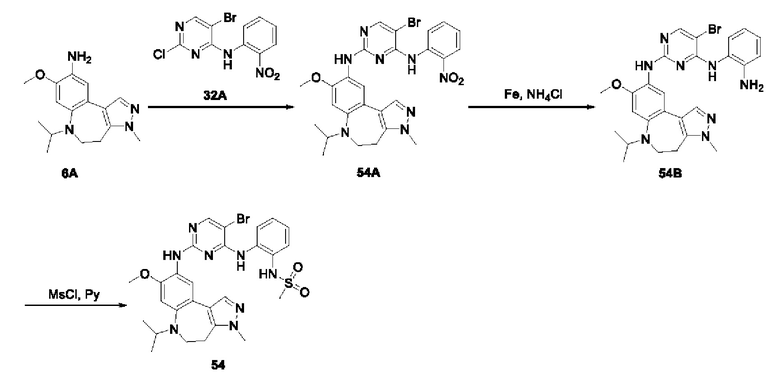
Соединение 54А:
Соединение 54А (110 мг) получали путем замены исходного материала на соединение 6А (130 мг, 0,46 ммоль) и соединение 32А (150 мг, 0,46 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 579,3, 581,3 [М+Н]+.
Соединение 54В:
Соединение 54В (89 мг) получали путем замены исходного материала на соединение 54А (100 мг, 0,17 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 549,3, 551,3 [М+Н]+.
Соединение 54:
Соединение 54 (47 мг) получали путем замены исходного материала на соединение 54В (80 мг, 0,15 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 627,2, 629,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 8,19 (s, 1Н), 8,16 (s, 1H), 7,64 (dd, J=7,5, 1,5 Гц, 1H), 7,45 (s, 1H), 7,38 (d, J=7,5 Гц, 1H), 7,32-7,30 (m, 1H), 7,21-7,12 (m, 4H), 6,53 (s, 1H), 3,92-3,67 (m, 7H), 3,29-3,26 (m, 2H), 2,94-2,90 (m, 5H), 1,31 (d, J=6,6 Гц, 6H).
Пример 55:
Получение промежуточного соединения 55А
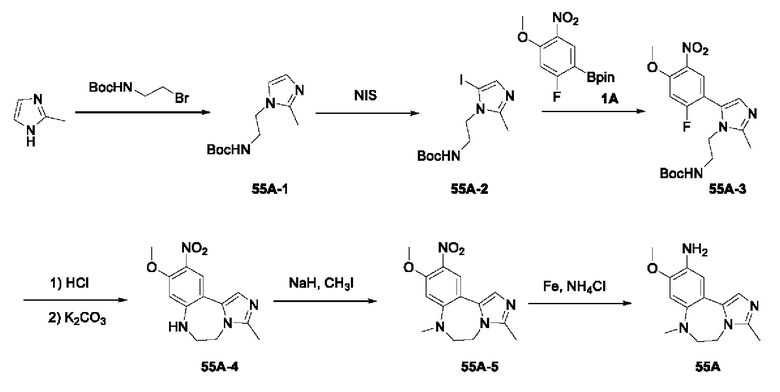
Соединение 55А-1:
2-Метилимидазол (3 г, 36,5 ммоль) и безводный карбонат калия (10,1 г, 73,1 ммоль) растворяли в ацетонитриле (25 мл). Затем к реакционной жидкости добавляли N-трет-бутоксикарбонил-бромэтиламин (8,2 г, 36,5 ммоль). Реакционную систему нагревали до 70°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 120 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; скорость потока: 60 мл/мин; градиент: повышение содержания ацетонитрила от 30% до 80% в течение 30 минут; длина волны детектирования: 220 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 55А-1 (1,7 г).
МС (ИЭР) масса/заряд: 226,3 [М+Н]+.
Соединение 55А-2:
Соединение 55А-1 (2,5 г, 11,1 ммоль) растворяли в ацетонитриле (50 мл). Затем к реакционной жидкости добавляли N-йодсукцинимид (3,8 г, 16,7 ммоль). Реакционную жидкость нагревали до 120°С с помощью СВЧ-реактора и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 120 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 10 мМ бикарбонат аммония) и ацетонитрил; скорость потока: 60 мл/мин; градиент: повышение содержания ацетонитрила от 30% до 80% в течение 30 минут; длина волны детектирования: 220 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 55А-2 (1,1 г).
МС (ИЭР) масса/заряд: 352,1 [М+Н]+.
Соединение 55А-3:
Соединение 55А-2 (900 мг, 2,56 ммоль), соединение 1А (990 мг, 3,33 ммоль) и безводный карбонат калия (708 мг, 5,13 ммоль) растворяли в 1,4-диоксане (8 мл) и воде (2 мл). Затем к реакционной жидкости добавляли дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий (167 мг, 0,26 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 55А-3 (300 мг).
МС (ИЭР) масса/заряд: 395,2 [М+Н]+.
Соединение 55А-4:
Соединение 55А-4 (130 мг) получали путем замены исходного материала на соединение 55А-3 (270 мг, 0,68 ммоль) согласно способу получения соединения 42А-7 из соединения 42А-6 в примере 42.
МС (ИЭР, масса/заряд): 274,8 [М+Н]+.
Соединение 55А-5:
Соединение 55А-5 (90 мг) получали путем замены исходного материала на соединение 55А-4 (130 мг, 0,47 ммоль) согласно способу получения соединения 8А-9 из соединения 8А-8 в примере 8.
МС (ИЭР, масса/заряд): 289,1 [М+Н]+.
Промежуточное соединение 55А:
Соединение 55А (70 мг) получали путем замены исходного материала на соединение 55А-5 (90 мг, 0,31 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 259,2 [М+Н]+.
Получение соединения 55
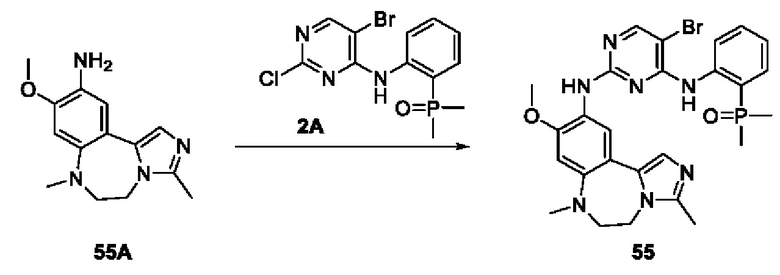
Соединение 55 (37 мг) получали путем замены исходного материала на соединение 55А (72 мг, 0,28 ммоль) и соединение 2А (101 мг, 0,28 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 582,0, 584,0 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 10,91 (s, 1H), 8,34-8,30 (m, 1H), 8,22 (s, 1H), 8,17 (s, 1H), 7,57 (s, 1H), 7,53-7,45 (m, 1H), 7,03-6,97 (m, 2Н), 6,77 (s, 1H), 6,66 (s, 1H), 4,03 (t, J=5,4 Гц, 2Н), 3,82 (s, 3Н), 3,41 (t, J=5,4 Гц, 2Н), 2,90 (s, 3Н), 2,37 (s, 3Н), 1,75 (d, J=13,5 Гц, 6Н).
Пример 56:
Получение промежуточного соединения 56А
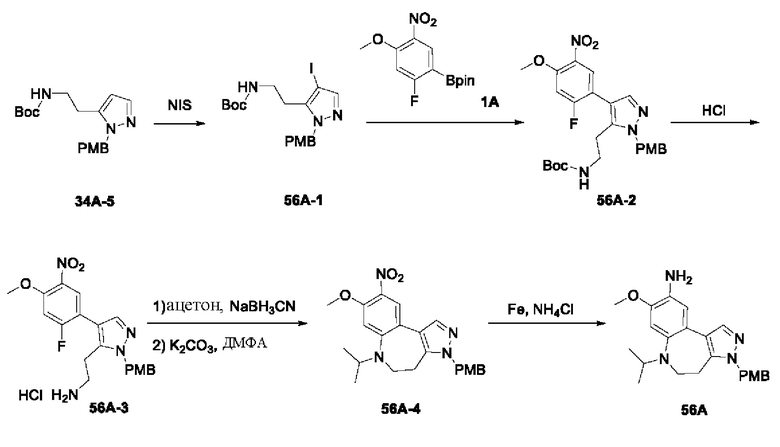
Соединение 56А-1:
Соединение 56А-1 (12,5 г) получали путем замены исходного материала на соединение 34А-5 (10 г, 30,2 ммоль) согласно способу получения соединения 1С-6 из соединения 1С-5 в примере 1.
МС (ИЭР, масса/заряд): 458,1 [М+Н]+.
Соединение 56А-2:
Соединение 56А-2 (10 г) получали путем замены исходного материала на соединение 56А-1 (12,1 г, 26,5 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 501,2 [М+Н]+.
Соединение 56А-3:
Соединение 56А-3 (2,4 г) получали путем замены исходного материала на соединение 56А-2 (3 г, 6,0 ммоль) согласно способу получения соединения 1С-9 из соединения 1С-8 в примере 1.
МС (ИЭР, масса/заряд): 401,0 [М+Н]+.
Соединение 56А-4:
Соединение 56А-3 (2,4 г, 6,0 ммоль) и ацетон (0,7 г, 12,0 ммоль) растворяли в метаноле (20 мл). Затем к реакционной жидкости добавляли безводный ацетат натрия (1,5 г, 18,0 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 30 минут. Затем к реакционной жидкости добавляли ледяную уксусную кислоту (0,72 г, 12,0 ммоль) и цианоборогидрид натрия (0,75 г, 12,0 ммоль). Реакционную систему последовательно перемешивали при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в N,N-диметилформамиде (30 мл); затем к реакционной жидкости добавляли безводный карбонат калия (4,2 г, 30,5 ммоль). Реакционную систему нагревали до 110°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (150 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 56А-4 (2,12 г).
МС (ИЭР) масса/заряд: 423,2 [М+Н]+.
Промежуточное соединение 56А:
Соединение 56А (1,67 г) получали путем замены исходного материала на соединение 56А-4 (2,12 г, 5,02 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 393,3 [М+Н]+.
Получение соединения 56
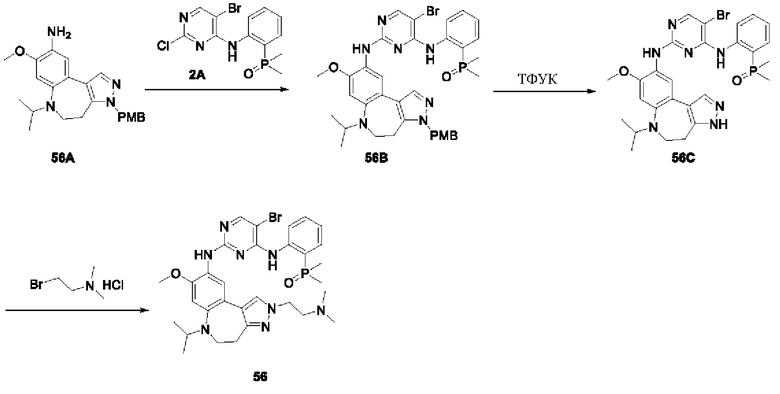
Соединение 56В:
Соединение 56В (2,88 г) получали путем замены исходного материала на соединение 55А (2,4 г, 6,12 ммоль) и соединение 2А (1,98 г, 5,5 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 716,3, 718,3 [М+Н]+.
Соединение 56С:
Соединение 56С (1,2 г) получали путем замены исходного материала на соединение 56В (2,88 г, 4,02 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 596,2, 598,2 [М+Н]+.
Соединение 56:
Соединение 56С (100 мг, 0,17 ммоль) и безводный карбонат калия (232 мг, 1,68 ммоль) растворяли в N,N-диметилформамиде (2 мл). Затем к реакционной жидкости добавляли гидробромид 2-бром-N,N-диметилэтиламина (195 мг, 0,84 ммоль). Реакционную систему нагревали до 120°С и последовательно перемешивали в течение 2 дней. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и получали под высоким давлением, при этом условия получения были следующими: препаративная колонка: колонка XSelect CSH Prep С18 OBD, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,1% муравьиная кислота), и подвижная фаза В: ацетонитрил; расход: 25 мл/мин; градиент: повышение содержания подвижной фазы В от 25% до 70% в течение 25 минут; длина волны детектирования: 254/220 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 56 (3 мг).
МС (ИЭР, масса/заряд): 667,2, 669,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,58 (s, 1H), 8,51 (s, 1H), 8,42 (s, 1H), 8,23 (s, 1Н), 7,45 (s, 1H), 7,29-7,19 (m, 3H), 6,98-6,95 (m, 1H), 6,57 (s, 1H), 4,28-4,26 (m, 2H), 3,93-3,89 (m, 4H), 3,33-3,30 (m, 2H), 3,02-2,96 (m, 4H), 2,41 (s, 6H), 1,86 (d, J=13,2 Гц, 6H), 1,33 (s, 3H), 1,31 (s, 3H).
Пример 57:
Получение соединения 57
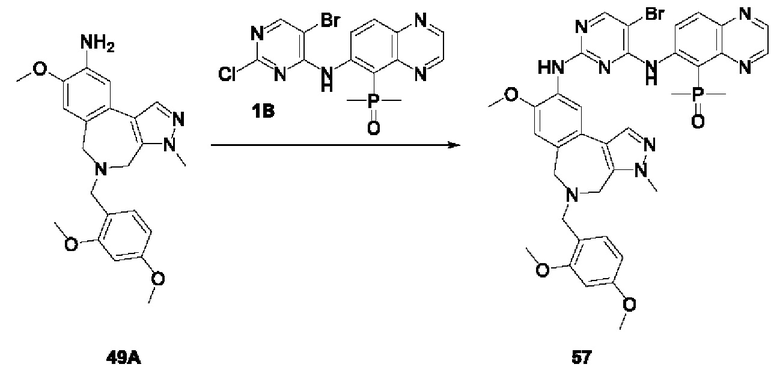
Соединение 57:
Соединение 57 (133 мг) получали путем замены исходного материала на соединение 49А (100 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 770,3, 772,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,56 (s, 1H), 8,95 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,73-8,70 (m, 2Н), 8,53 (s, 1H), 8,35 (s, 1H), 7,74 (d, J=9,6 Гц, 1Н), 7,49 (s, 1Н), 7,25-7,18 (m, 1Н), 7,03 (s, 1H), 6,58 (s, 1H), 6,51-6,45 (m, 2Н), 4,17 (s, 2Н), 4,06 (s, 2Н), 3,90 (s, 3Н), 3,84 (s, 3Н), 3,78 (s, 3Н), 3,71 (s, 2Н), 3,64 (s, 3Н), 2,19 (s, 3Н), 2,15 (s, 3Н).
Пример 58:
Получение соединения 58
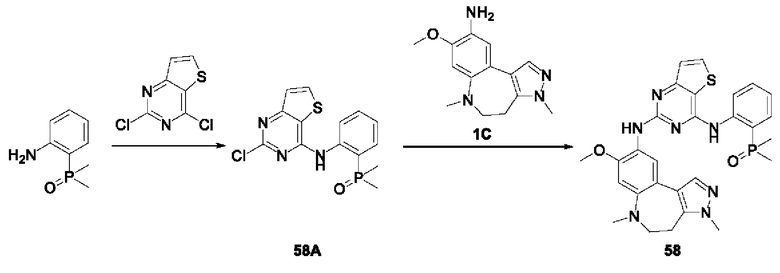
Соединение 58А:
Соединения 2-(диметилфосфорил)анилин (1,37 г, 8,1 ммоль) и 2,4-дихлортиофен[3,2-d]пиримидин (1,99 г, 9,72 ммоль) растворяли в N,N-диметилформамиде (20 мл) и добавляли к реакционному раствору диизопропилэтиламин (2,09 г, 16,2 ммоль). Реакционную систему нагревали до 110°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (80 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением соединения 58А (1,82 г).
МС (ИЭР) масса/заряд: 338,1, 340,1 [М+Н]+.
Соединение 58:
Соединение 58А (100 мг, 0,3 ммоль), соединение 1С (76 мг, 0,3 ммоль) и безводный карбонат калия (82 мг, 0,59 ммоль) растворяли в трет-бутаноле (3 мл) в атмосфере азота. Затем к реакционной жидкости добавляли трис(дибензилиденацетон)дипалладий (27 мг, 0,03 ммоль) и 2-дициклогексилфосфоний-2',4',6'-триизопропилбифенил (21 мг, 0,04 ммоль). Реакционную систему нагревали до 110°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 58 (103 мг).
МС (ИЭР) масса/заряд: 560,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 11,38 (s, 1H), 8,96-8,87 (m, 1H), 8,69 (s, 1Н), 7,71 (m, J=5,4 Гц, 1H), 7,66 (s, 1H), 7,61 (s, 1H), 7,28 (s, 1H), 7,26 (s, 1H), 7,25-7,19 (m, 1H), 7,03-6,97 (m, 1H), 6,59 (s, 1H), 3,93 (s, 3H), 3,85 (s, 3H), 3,32 (t, J=5,4 Гц, 2H), 3,10-3,03 (m, 5H), 1,90 (s, 3H), 1,86 (s, 3H).
Пример 59:
Получение соединения 59
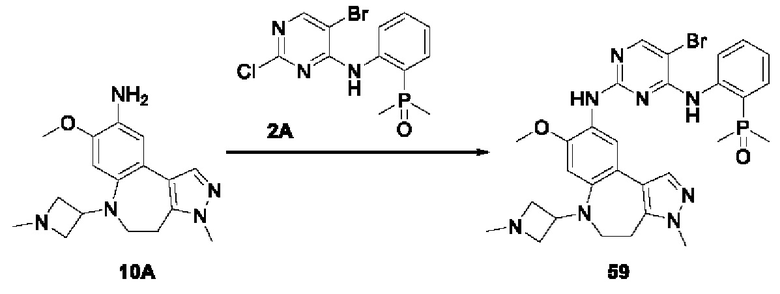
Соединение 59 (12 мг) получали путем замены исходного материала на соединение 10А (71 мг, 0,23 ммоль) и соединение 2А (65 мг, 0,18 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 637,2, 639,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,59 (s, 1H), 8,57 (s, 1H), 8,50-8,41 (m, 2Н), 8,26 (s, 1H), 7,39 (s, 1H), 7,37-7,31 (m, 1H), 7,27-7,20 (m, 1H), 7,08-6,99 (m, 1H), 6,23 (s, 1H), 4,54 (t, J=7,2 Гц, 1H), 4,33 (t, J=8,1 Гц, 2H), 3,89 (s, 3H), 3,84 (s, 3H), 3,45 (t, J=8,1 Гц, 2H), 3,28 (t, J=6,0 Гц, 2H), 3,02 (t, J=6,0 Гц, 2H), 2,65 (s, 3H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 60:
Получение промежуточного соединения 60А
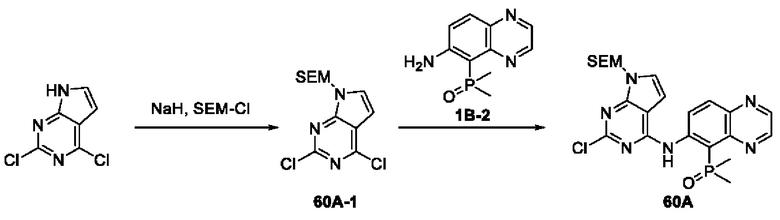
Соединение 60А-1:
Гидрид натрия (60%, 0,69 г, 17,25 ммоль) растворяли в N,N-диметилформамиде (25 мл) в атмосфере азота при 0°С. Затем к реакционной жидкости отдельными порциями добавляли 2,4-дихлор-7H-пирроло[2,3-d]пиримидин (2,5 г, 13,3 ммоль), после чего реакционную жидкость последовательно перемешивали при указанной температуре в течение 10 минут. После добавления к реакционной жидкости 2-(триметилсилил)этоксиметилхлорида (2,88 г, 17,29 ммоль) и последовательного перемешивания при указанной температуре в течение 10 минут реакционную жидкость нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (80 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 5/1) с получением соединения 60А-1 (3,9 г).
МС (ИЭР) масса/заряд: 318,1, 320,1 [М+Н]+.
Промежуточное соединение 60А:
Соединение 60А (3 г) получали путем замены исходного материала на соединение 60А-1 (5,76 г, 18,08 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 503,3, 505,3 [М+Н]+.
Получение соединения 60
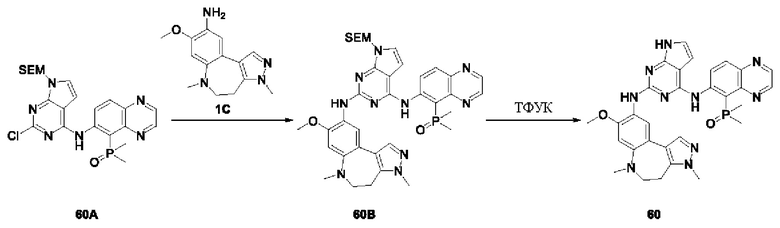
Соединение 60В:
Соединение 60В (70 мг) получали путем замены исходного материала на соединение 60А (80 мг, 0,16 ммоль) согласно способу получения соединения 58 из соединения 58А и соединения 1С в примере 58.
МС (ИЭР, масса/заряд): 725,4 [М+Н]+.
Соединение 60:
Соединение 60 (21 мг) получали путем замены исходного материала на соединение 60В (148 мг, 0,2 ммоль) согласно способу получения соединения 36 из соединения 36В в примере 36.
МС (ИЭР, масса/заряд): 595,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 13,04 (s, 1H), 9,77 (dd, J=9,6, 4,2 Гц, 1Н), 8,68 (m, 4Н), 7,87 (d, J=9,6 Гц, 1H), 7,64 (s, 1Н), 7,16 (s, 1H), 6,87 (dd, J=3,6, 2,1 Гц, 1H), 6,74 (dd, J=3,6, 2,1 Гц, 1H), 6,61 (s, 1H), 3,96 (s, 3Н), 3,83 (s, 3Н), 3,34 (t, J=5,4 Гц, 2Н), 3,11-3,06 (m, 5Н), 2,18 (s, 3Н), 2,13 (s, 3Н).
Пример 61:
Получение соединения 61
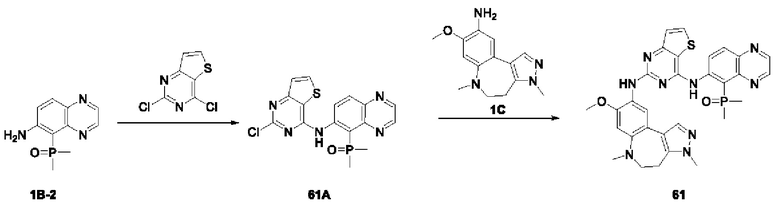
Соединение 61А:
Соединение 61А (1 г) получали путем замены исходного материала на 2,4-дихлортиофен[3,2-d]пиримидин (1,85 г, 9,04 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 390,0, 392,0 [М+Н]+.
Соединение 61:
Соединение 61 (46 мг) получали путем замены исходного материала на соединение 61А (100 мг, 0,26 ммоль) согласно способу получения соединения 58 из соединения 58А и соединения 1С в примере 58.
МС (ИЭР, масса/заряд): 612,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 13,22 (s, 1H), 9,54 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,72-8,70 (m, 2Н), 8,65 (s, 1H), 7,83-7,76 (m, 2Н), 7,57-7,51 (m, 2Н), 7,30 (s, 1H), 6,62 (s, 1H), 3,95 (s, 3Н), 3,82 (s, 3Н), 3,36 (t, J=5,4 Гц, 2Н), 3,13-3,07 (m, 5Н), 2,19 (s, 3Н), 2,14 (s, 3Н).
Пример 62:
Получение соединения 62
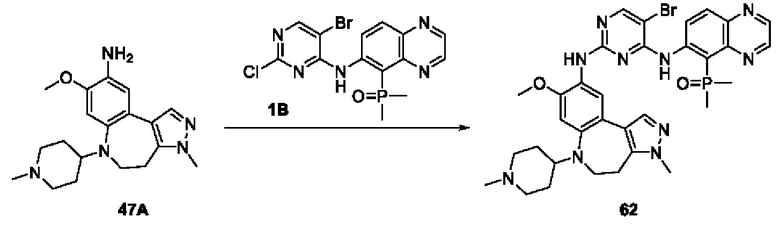
Соединение 62:
Соединение 62 (42 мг) получали путем замены исходного материала на соединение 47А (100 мг, 0,29 ммоль) и соединение 1В (100 мг, 0,24 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 717,3, 719,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,63 (s, 1H), 8,86-8,78 (m, 3Н), 8,26 (s, 1H), 8,20 (s, 1H), 7,74 (s, 1H), 7,55 (s, 1H), 7,45 (s, 1H), 6,58 (s, 1H), 3,76 (s, 3Н), 3,70 (s, 3Н), 3,28-3,26 (m, 2H), 2,96-2,86 (m, 4H), 2,23 (s, 3Н), 2,18-2,06 (m, 3Н), 2,04 (s, 3Н), 1,99 (s, 3Н), 1,90-1,80 (m, 4H).
Пример 63:
Получение промежуточного соединения 63А
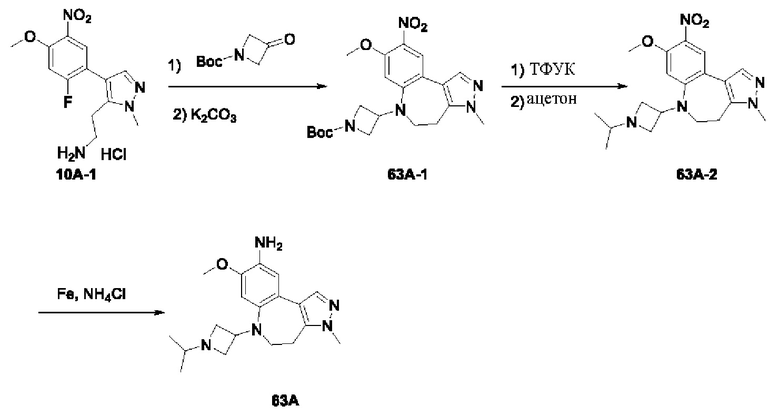
Соединение 63А-1:
Соединение 63А-1 (0,9 г) получали путем замены исходного материала на трет-бутил-3-оксоазетидин-1-карбоксилат (2,2 г, 12,9 ммоль) согласно способу получения соединения 47А-1 из соединения 10А-1 в примере 47.
МС (ИЭР, масса/заряд): 430,2 [М+Н]+.
Соединение 63А-2:
Соединение 63А-1 (300 мг, 0,7 ммоль) растворяли в дихлорметане (5 мл); затем к реакционной жидкости добавляли трифторуксусную кислоту (1 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в метаноле (5 мл). Затем к реакционной жидкости добавляли ацетон (203 мг, 3,49 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 30 минут. К описанной выше реакционной жидкости добавляли цианоборогидрид натрия (88 мг, 1,4 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 63А-2 (150 мг).
МС (ИЭР) масса/заряд: 372,2 [М+Н]+.
Промежуточное соединение 63:
Соединение 63А (87 г) получали путем замены исходного материала на соединение 63А-2 (150 мг, 0,4 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 342,2 [М+Н]+.
Получение соединения 63
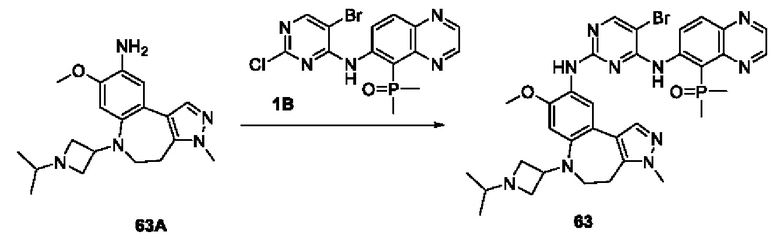
Соединение 63 (64 мг) получали путем замены исходного материала на соединение 63А (87 мг, 0,26 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 717,2, 719,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,50 (s, 1H), 8,87 (dd, J=9,2 Гц, 4,4 Гц, 1H), 8,71 (d, J=1,6 Гц, 1H), 8,68 (d, J=1,6 Гц, 1H), 8,53 (s, 1H), 8,3 (s, 1H), 7,63 (s, 1H), 7,32 (s, 1H), 6,99 (s, 1Н), 6,27 (s, 1H), 4,53-4,44 (m, 1Н), 4,16 (t, J=8,0 Гц, 2Н), 3,88 (s, 3Н), 3,72 (s, 3Н), 3,30-3,20 (m, 4Н), 3,00 (t, J=6,0 Гц, 2Н), 2,79-2,71 (m, 1H), 2,16 (s, 3Н), 2,12 (s, 3Н), 1,11 (s, 3Н), 1,09 (s, 3Н).
Пример 64:
Получение соединения 64
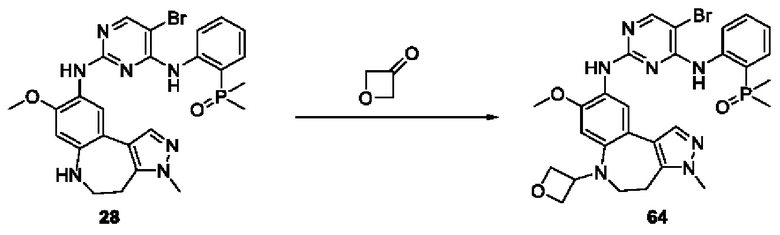
Соединение 28 (100 мг, 0,18 ммоль) и 3-оксетанон (63 мг, 0,88 ммоль) растворяли в метаноле (3 мл) и перемешивали полученную реакционную жидкость при комнатной температуре в течение 30 минут. К реакционной жидкости добавляли уксусную кислоту (21 мг, 0,35 ммоль) и цианоборогидрид натрия (22 мг, 0,35 ммоль). Реакционную систему последовательно перемешивали при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 64А (64 мг).
МС (ИЭР, масса/заряд): 624,2, 626,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,67 (s, 1H), 8,50-8,40 (m, 2Н), 8,23 (s, 1H), 7,39 (s, 1H), 7,36-7,30 (m, 1H), 7,27-7,15 (m, 2H), 7,06-6,98 (m, 1H), 6,01 (s, 1H), 4,96 (t, J=6,3 Гц, 2H), 4,86-4,76 (m, 1H), 4,71 (t, J=6,0 Гц, 2H), 3,86 (s, 3H), 3,84 (s, 3H), 3,37 (t, J=6,0 Гц, 2H), 3,07 (t, J=6,0 Гц, 2H), 1,89 (s, 3H), 1,85 (s, 3H).
Пример 65:
Получение промежуточного соединения 65А
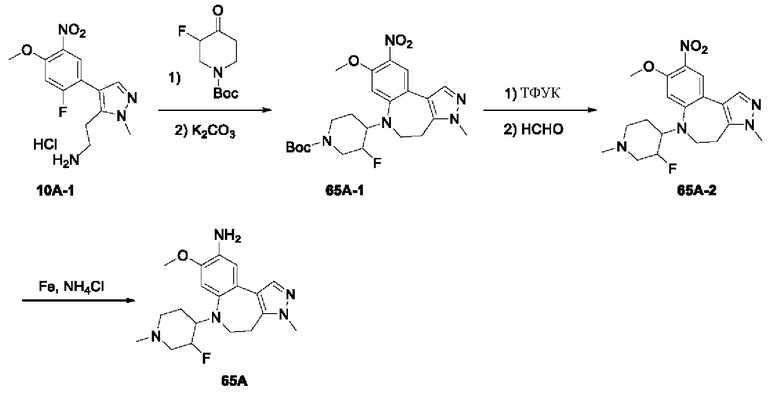
Соединение 65А-1:
Соединение 65А-1 (50 мг) получали путем замены исходного материала на трет-бутил-3-фтор-4-оксопиперидин-1-карбоксилат (828 мг, 3,81 ммоль) согласно способу получения соединения 47А-1 из соединения 10А-1 в примере 47.
МС (ИЭР, масса/заряд): 476,2 [М+Н]+.
Соединение 65А-2:
Соединение 65А-2 (200 мг) получали путем замены исходного материала на водный раствор формальдегида (40%, 200 мг, 2,67 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 390,2 [М+Н]+.
Промежуточное соединение 65А:
Соединение 65А (64 мг) получали путем замены исходного материала на соединение 65А-2 (110 мг, 0,28 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 360,2 [М+Н]+.
Получение соединения 65
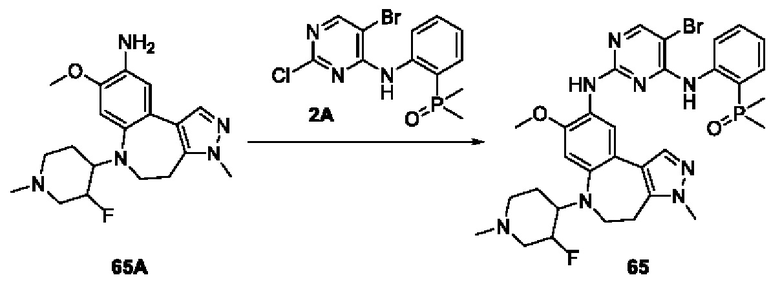
Соединение 65 (11 мг) получали путем замены исходного материала на соединение 65А (30 мг, 0,08 ммоль) и соединение 2А (30 мг, 0,08 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 683,2, 685,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,53 (s, 1H), 8,48-8,42 (m, 1H), 8,24 (s, 1H), 8,21 (s, 1H), 7,45 (s, 1H), 7,36-7,29 (m, 1H), 7,26-7,14 (m, 2H), 7,04-7,97 (m, 1H), 6,59 (s, 1H), 4,87 (d, J=48,0 Гц, 1H), 3,88 (s, 3H), 3,83 (s, 3H), 3,73-3,71 (m, 1H), 3,55-3,05 (m, 5H), 2,96-2,92 (m, 1H), 2,53-2,29 (m, 6H), 1,90 (s, 3H), 1,85 (s, 3H), 1,83-1,76 (m, 1H).
19F ЯМР (282 МГц, CDCl3) δ -196,73.
Пример 66:
Получение соединений 66A и 66В
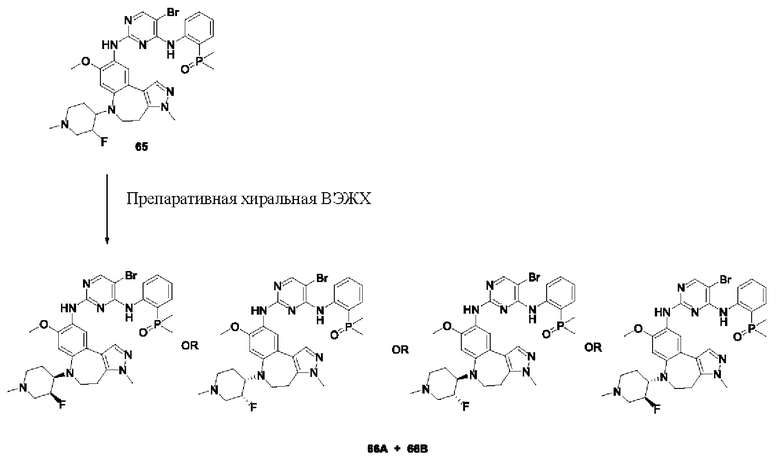
Соединение 65 (168 мг, 0,25 ммоль) получали и отделяли с помощью хиральной колонки, при этом условия получения были следующими: хиральная колонка: Lux 5 мкм Cellulose-4, 2,12×25 см, 5 мкм; подвижная фаза А: н-гексан (10 мМ раствор аммиака в метаноле), и подвижная фаза В: этанол; скорость потока: 20 мл/мин; элюирование: повышение содержания подвижной фазы В до 50% в течение 51 минут; длина волны детектирования: 205/260 нм; (время удерживания)1: 22 минут; (время удерживания)2: 34,5 мин. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 66А (15 мг, энантиомерный избыток 100%) со временем удерживания RT1 22 мин и соединение 66В (12 мг, энантиомерный избыток 99,7%) со временем удерживания RT2 34,5 мин.
Соединение 66А и соединение 66В были энантиомерами друг друга, абсолютная конфигурация не определялась.
66А: МС (ИЭР, масса/заряд): 683,3, 685,3 [М+Н]+.
66В: МС (ИЭР, масса/заряд): 683,2, 685,2 [М+Н]+.
66А: 1H ЯМР (300 МГц, CDCl3) δ 10,83 (s, 1H), 8,56-8,47 (m, 2Н), 8,30 (s, 1H), 7,75 (s, 1H), 7,43-7,36 (m, 1H), 7,27 (t, J=8,1 Гц, 1H), 7,14-7,06 (m, 1H), 6,69 (s, 1H), 5,01 (d, J=48,0 Гц, 1H), 3,96 (s, 3H), 3,89 (s, 3H), 3,84-3,31 (m, 6H), 3,23-3,93 (m, 3H), 2,88-2,64 (m, 5H), 1,96 (s, 3H), 1,91 (s, 3H).
19F ЯМР (282 МГц, CDCl3) δ -196,86.
66В: 1H ЯМР (300 МГц, CDCl3) δ 10,72 (s, 1H), 8,58-8,51 (m, 2H), 8,31 (s, 1H), 7,45 (s, 1H), 7,42-7,36 (m, 1H), 7,28 (t, J=8,1 Гц, 1H), 7,12-7,05 (m, 1H), 6,68 (s, 1H), 5,01 (d, J=48,0 Гц, 1H), 3,95 (s, 3H), 3,89 (s, 3H), 3,84-3,31 (m, 6H), 3,23-3,93 (m, 3H), 2,88-2,64 (m, 5H), 1,96 (s, 3H), 1,91 (s, 3H).
19F ЯМР (282 МГц, CDCl3) δ -196,86.
Пример 67:
Получение соединения 67
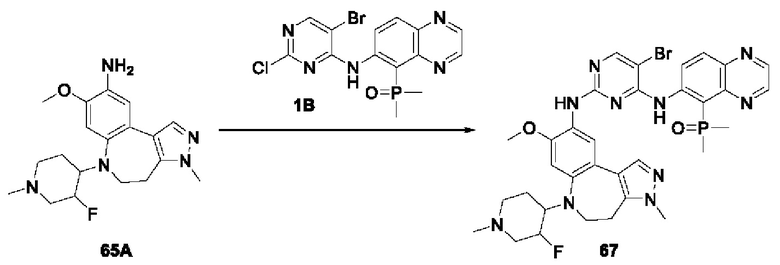
Соединение 67 (19 мг) получали путем замены исходного материала на соединение 65А (30 мг, 0,08 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 735,2, 737,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,48 (s, 1H), 8,88 (dd, J=8,4 Гц, 1,8 Гц, 1H), 8,73-8,69 (m, 2Н), 8,30 (s, 1H), 8,18 (s, 1H), 7,66-7,61 (m, 1H), 7,44 (s, 1Н), 7,01 (s, 1H), 6,66 (s, 1Н), 4,89 (d, J=48,3 Гц, 1H), 3,89 (s, 3H), 3,77-3,75 (m, 1H), 3,72 (s, 3H), 3,55-3,04 (m, 5H), 3,00-2,96 (m, 1H), 2,54-2,28 (m, 6H), 2,18 (d, J=4,8 Гц, 3Н), 2,14 (d, J=4,8 Гц, 3Н), 1,81 (d, J=12,9 Гц, 1H).
19F ЯМР (282 МГц, CDCl3) δ -196,72.
Пример 68:
Получение соединения 68
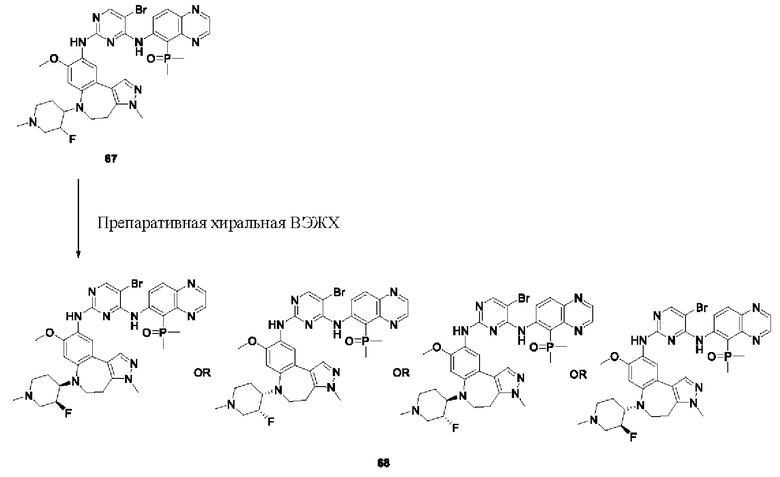
Соединение 67 (50 мг, 0,07 ммоль) получали с помощью хиральной колонки, при этом условия получения были следующими: хиральная колонка: CHIRALPAK IC, 2×25 см, 5 мкм; подвижная фаза А: н-гексан: метил-трет-бутиловый эфир=1:1 (0,5% 2 М раствор аммиака в метаноле), и подвижная фаза В: этанол; скорость потока: 20 мл/мин; элюирование: повышение содержания подвижной фазы В до 30% в течение 25 минут; длина волны детектирования: 260/210 нм; время удерживания: 23,47 мин. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 68 (11 мг, то есть, одного из четырех возможных отделенных энантиомеров; энантиомерный избыток 100%) с временем удерживания, составляющим 23,47 мин.
МС (ИЭР, масса/заряд): 735,2, 737,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,51 (s, 1H), 8,89 (dd, J=9,3 Гц, 5,1 Гц, 1H), 8,73-8,69 (m, 2Н), 8,43 (s, 1H), 8,31 (s, 1H), 7,69-7,61 (m, 1H), 7,35 (s, 1H), 7,03 (s, 1H), 6,60 (s, 1H), 4,89 (d, J=49,5 Гц, 1H), 3,89 (s, 3Н), 3,85-3,75 (m, 1H), 3,73 (s, 3Н), 3,55-3,35 (m, 1H), 3,34-3,22 (m, 2Н), 3,20-3,05 (m, 2Н), 2,99-2,90 (m, 1H), 2,55-2,26 (m, 6Н), 2,18 (d, J=4,8 Гц, 3Н), 2,14 (d, J=4,8 Гц, 3Н), 1,86-1,85 (m, 1H).
Пример 69:
Получение промежуточного соединения 69А
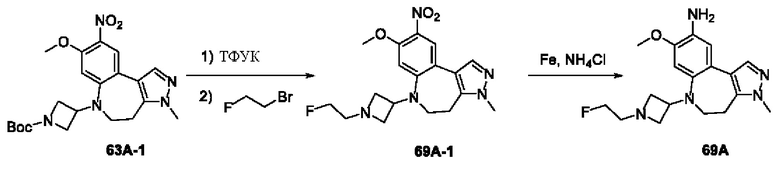
Соединение 69А-1:
Соединение 63А-1 (250 мг, 0,58 ммоль) растворяли в дихлорметане (3 мл). Затем к описанной выше реакционной жидкости добавляли трифторуксусную кислоту (0,5 мл), после чего последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток растворяли в N,N-диметилформамиде (3 мл) с последующим добавлением к реакционной жидкости безводного карбоната калия (482 мг, 3,5 ммоль) и 1-бром-2-фторэтана (370 мг, 2,9 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (20 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 69А-1 (100 мг).
МС (ИЭР, масса/заряд): 376,2 [М+Н]+.
Промежуточное соединение 69А:
Соединение 69А (60 мг) получали путем замены исходного материала на соединение 69А-1 (100 мг, 0,27 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 346,2 [М+Н]+.
Получение соединения 69
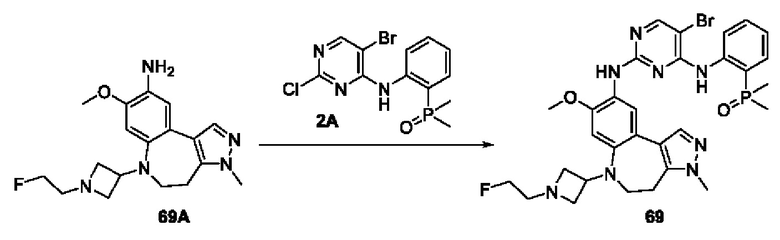
Соединение 69 (20 мг) получали путем замены исходного материала на соединение 69А (60 мг, 0,17 ммоль) и соединение 2А (52 мг, 0,15 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 669,2, 671,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 10,54 (s, 1H), 8,51-8,39 (m, 2Н), 8,24 (s, 1H), 7,46 (s, 1Н), 8,38-7,17 (m, 3Н), 7,06-6,97 (m, 1H), 6,25 (s, 1H), 4,70-4,62 (m, 1H), 4,55-4,44 (m, 2H), 4,19-4,07 (m, 2H), 3,88 (s, 3H), 3,84 (s, 3H), 3,43-3,41 (m, 2H), 3,31-3,21 (m, 2H), 3,11-2,92 (m, 4H), 1,90 (s, 3H), 1,86 (s, 3H).
Пример 70:
Получение промежуточного соединения 70А
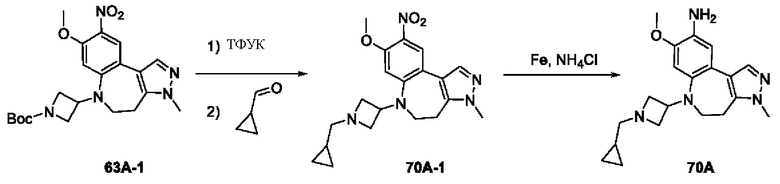
Соединение 70А-1:
Соединение 70А-1 (430 мг) получали путем замены исходного материала на циклопропанкарбоксальдегид (230 мг, 3,3 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 384,2 [М+Н]+.
Промежуточное соединение 70А:
Соединение 70А (120 мг) получали путем замены исходного материала на соединение 70А-1 (150 мг, 0,39 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 354,2 [М+Н]+.
Получение соединения 70

Соединение 69 (55 мг) получали путем замены исходного материала на соединение 70А (120 мг, 0,34 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 729,2, 731,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,52 (s, 1H), 8,88 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,73-8,69 (m, 2Н), 8,52 (s, 1H), 8,32 (s, 1H), 7,65 (d, J=9,6 Гц, 1Н), 7,37 (s, 1Н), 7,00 (s, 1 Н), 6,27 (s, 1Н), 4,65-4,53 (m, 1H), 4,37-4,29 (m, 2Н), 3,91 (s, 3Н), 3,74 (s, 3Н), 3,41 (t, J=8,1 Гц, 2Н), 3,29 (t, J=5,7 Гц, 2Н), 3,02 (t, J=5,7 Гц, 2Н), 2,70 (d, J=7,2 Гц, 2Н), 2,18 (s, 3Н), 2,14 (s, 3Н), 0,982-0,840 (m, 1H), 0,625-0,542 (m, 2Н), 0,267-0,194 (m, 2Н).
Пример 71:
Получение соединения 71
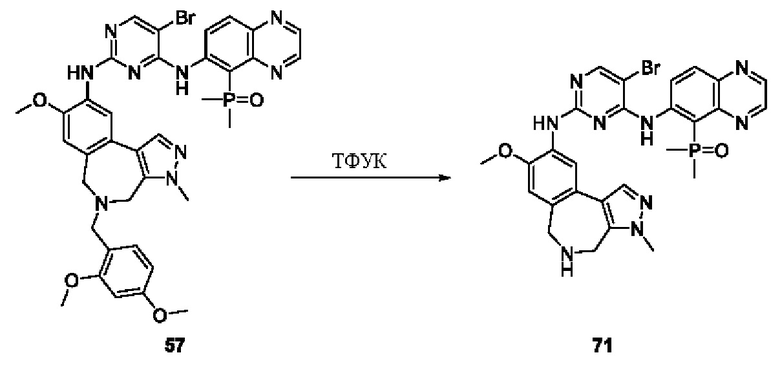
Соединение 71:
Соединение 71 (12 мг) получали путем замены исходного материала на соединение 57 (133 мг, 0,17 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 620,1,1, 622,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,53 (s, 1H), 8,88-8,83 (m, 1H), 8,74-8,70 (m, 2H), 8,52 (s, 1H), 8,33 (s, 1H), 7,68-7,65 (m, 1H), 7,54 (s, 1H), 6,99 (s, 1H), 6,77 (s, 1H), 4,36 (s, 2H), 4,04 (s, 2H), 3,93 (s, 3H), 3,75 (s, 3H), 2,18 (s, 3H), 2,13 (s, 3H).
Пример 72:
Получение соединения 72
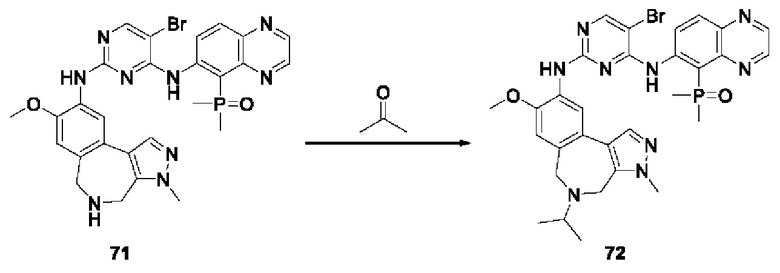
Соединение 71 (146 мг, 0,24 ммоль), ледяную уксусную кислоту (71 мг, 1,18 ммоль) и ацетон (137 мг, 2,35 ммоль) растворяли в дихлорметане (5 мл) и последовательно перемешивали полученную реакционную жидкость при комнатной температуре в течение 30 минут. Затем к описанной выше реакционной жидкости добавляли цианоборогидрид натрия (249 мг, 1,18 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (10 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 72 (98 мг).
МС (ИЭР, масса/заряд): 662,2, 664,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,56 (s, 1H), 8,95 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,73-8,70 (m, 2Н), 8,49 (s, 1H), 8,34 (s, 1H), 7,68 (d, J=9,9 Гц, 1H), 7,51 (s, 1H), 6,99 (s, 1H), 6,73 (s, 1Н), 4,23 (s, 2Н), 3,95-3,92 (m, 5Н), 3,76 (s, 3Н), 3,23-3,12 (m, 1Н), 2,19 (s, 3Н), 2,14 (s, 3Н), 1,28 (s, 3Н), 1,26 (s, 3Н).
Пример 73:
Получение промежуточного соединения 73А
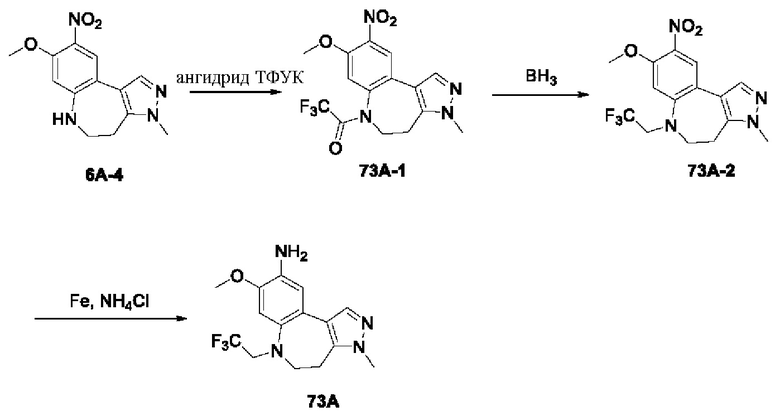
Соединение 73А-1:
Соединение 6А-4 (200 мг, 0,73 ммоль) растворяли в дихлорметане (5 мл); затем к реакционной жидкости добавляли трифторуксусный ангидрид (766 мг, 3,65 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 73А-1 (250 мг).
МС (ИЭР, масса/заряд): 371,2 [М+Н]+.
Соединение 73А-2:
Соединение 73А-2 (93 мг) получали путем замены исходного материала на соединение 73А-1 (168 мг, 0,45 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 357,1 [М+Н]+.
Промежуточное соединение 73А:
Соединение 73А (70 мг) получали путем замены исходного материала на соединение 73А-2 (93 мг, 0,26 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 327,2 [М+Н]+.
Получение соединения 73
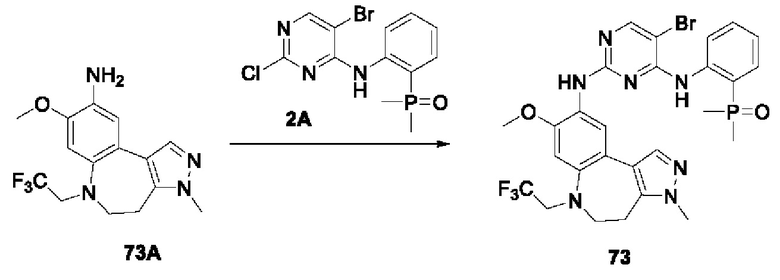
Соединение 73 (43 мг) получали путем замены исходного материала на соединение 73А (70 мг, 0,21 ммоль) и соединение 2А (77 мг, 0,21 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 650,2, 652,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,61 (s, 1H), 8,52-8,44 (m, 2Н), 8,25 (s, 1H), 7,38-7,19 (m, 3Н), 7,06-6,98 (m, 1H), 6,67 (s, 1H), 3,90 (s, 3H), 3,84 (s, 3H), 3,83-3,75 (m, 2H), 3,49 (t, J=5,7 Гц, 2H), 3,05 (t, J=5,7 Гц, 2H), 1,89 (s, 3H), 1,85 (s, 3H).
19F ЯМР (282 МГц, CDCl3) δ -70,35.
Пример 74:
Получение промежуточного соединения 74А
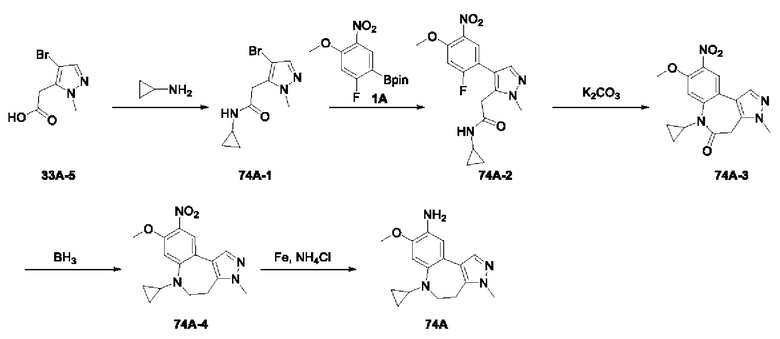
Соединение 74А-1:
Соединение 33А-5 (500 мг, 2,28 ммоль) растворяли в N,N-диметилформамиде (5 мл); затем в реакционную жидкость добавляли гексафторфосфат 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония (1042 мг, 2,74 ммоль) и последовательно перемешивали при комнатной температуре в течение 30 минут. К реакционной жидкости добавляли циклопропиламин (196 мг, 3,42 ммоль) и диизопропилэтиламин (738 мг, 5,71 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (30 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 15/1) с получением соединения 74А-1 (500 мг).
МС (ИЭР, масса/заряд): 258,0, 260,0 [М+Н]+.
Соединение 74А-2:
Соединение 74А-1 (700 мг, 2,71 ммоль), соединение 1А (967 мг, 3,25 ммоль) и безводный фосфат калия (1,1 г, 5,42 ммоль) растворяли в диоксане (10 мл) и воде (1 мл) в атмосфере азота с последующим добавлением к полученной реакционной жидкости метансульфонато(три-трет-бутилфосфино)(2'-амино-1,1'-бифенил-2-ил)палладия (II) (155 мг, 0,27 ммоль) и тетрафторбората три-трет-бутилфосфина (79 мг, 0,27 ммоль). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 15/1) с получением соединения 74А-2 (700 мг).
МС (ИЭР, масса/заряд): 349,2 [М+Н]+.
Соединение 74А-3:
Соединение 74А-2 (680 мг, 1,95 ммоль) растворяли в N,N-диметилформамиде (10 мл); затем к реакционной жидкости добавляли безводный карбонат калия (1079 мг, 7,81 ммоль). Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (30 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (30 мл × 3); затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 15/1) с получением соединения 74А-3 (230 мг).
МС (ИЭР, масса/заряд): 329,2 [М+Н]+.
Соединение 74А-4:
Соединение 74А-4 (110 мг) получали путем замены исходного материала на соединение 74А-3 (240 мг, 0,73 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 315,2 [М+Н]+.
Соединение 74А:
Соединение 74А (80 мг) получали путем замены исходного материала на соединение 74А-4 (110 мг, 0,35 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 285,3 [М+Н]+.
Получение соединения 74
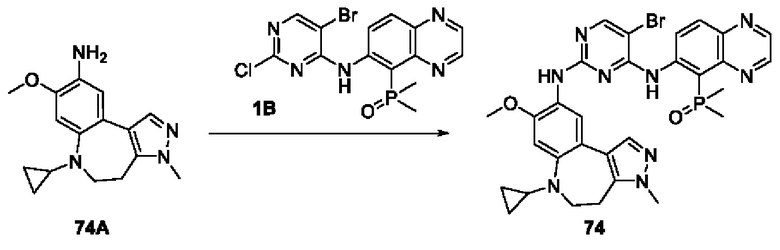
Соединение 74 (15 мг) получали путем замены исходного материала на соединение 74А (70 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 660,2, 662,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,58 (s, 1H), 8,95 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,72-8,68 (m, 2Н), 8,33 (s, 1H), 8,27 (s, 1H), 7,70-7,58 (m, 1H), 7,45 (s, 1H), 7,00 (s, 1H), 6,90 (s, 1H), 3,93 (s, 3Н), 3,71 (s, 3Н), 3,53 (t, J=5,7 Гц, 2Н), 3,00 (t, J=5,7 Гц, 2Н), 2,80-2,72 (m, 1H), 2,17 (s, 3Н), 2,13 (s, 3Н), 0,84-0,77 (m, 2Н), 0,61-0,55 (m, 2Н).
Пример 75:
Получение соединения 75
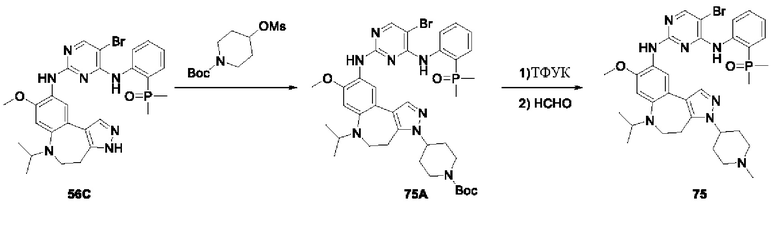
Соединение 75А:
Соединение 75А (250 мг) получали путем замены исходного материала на трет-бутил-4-метансульфонилоксипиперидин-1-карбоксилат (281 мг, 1,01 ммоль) согласно способу получения соединения 56 из соединения 56С в примере 56.
МС (ИЭР, масса/заряд): 779,4, 781,4 [М+Н]+.
Соединение 75:
Соединение 75 (7 мг) получали путем замены исходного материала на водный раствор формальдегида (37%, 177 мг, 2,18 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 693,4, 695,4 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,59 (s, 1H), 8,54 (s, 1H), 8,28 (s, 1Н), 7,46 (s, 1H), 7,38-7,28 (m, 2H), 7,28-7,15 (m, 2H), 7,04-6,95 (m, 1H), 6,63 (s, 1H), 4,13 (s, 1H), 4,04-3,91 (m, 4H), 3,37 (t, J=5,7 Гц, 2H), 3,32-3,15 (m, 3H), 3,03 (t, J=5,7 Гц, 2H), 2,60-2,31 (m, 6H), 2,22-2,02 (m, 2H), 1,95 (s, 3H), 1,90 (s, 3H), 1,40 (s, 3H), 1,38 (s, 3H).
Пример 76:
Получение промежуточного соединения 76А
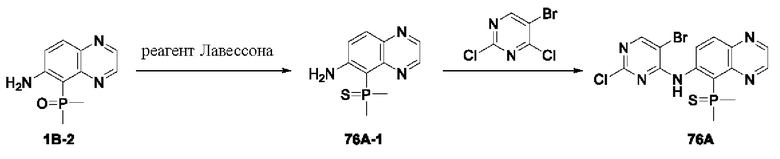
Соединение 76А-1:
Соединение 1В-2 (5 г, 22,81 ммоль) растворяли в толуоле (125 мл) в атмосфере азота; затем к реакционной жидкости добавляли реагент Лавессона (18,45 г, 45,62 ммоль). Реакционную систему нагревали до 95°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением соединения 76А-1 (3 г).
МС (ИЭР, масса/заряд): 238,2 [М+Н]+.
Соединение 76А:
Соединение 76А (2 г) получали путем замены исходного материала на соединение 76А-1 (3 г, 12,64 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 428,0, 430,0 [М+Н]+.
Получение соединения 76
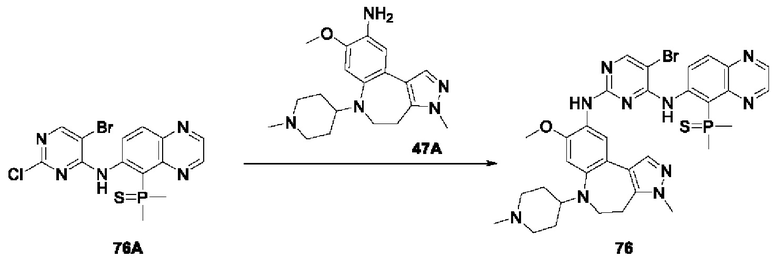
Соединение 76 (2 мг) получали путем замены исходного материала на соединение 76А (109 мг, 0,25 ммоль) и соединение 47А(87 мг, 0,25 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 733,2, 735,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 11,72 (s, 1H), 8,78 (d, J=1,8 Гц, 1H), 8,71 (d, J=1,8 Гц, 1H), 8,42 (s, 1H), 8,35 (s, 1H), 8,23 (dd, J=9,6 Гц, 4,2 Гц, 1H), 7,65 (d, J=9,3 Гц, 1H), 7,48 (s, 1H), 6,59-6,49 (m, 1H), 6,46 (s, 1H), 3,88 (s, 3H), 3,61 (s, 3H), 3,55-3,44 (m, 3H), 3,39 (t, J=5,7 Гц, 2H), 2,89 (t, J=5,7 Гц, 2H), 2,73-2,60 (m, 5H), 2,51 (s, 3H), 2,46 (s, 3H), 2,37-2,21 (m, 2H), 2,03-1,93 (m, 2H).
Пример 77:
Получение соединений 77А и 77В
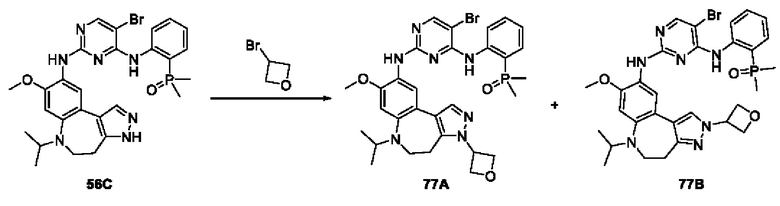
Соединения 77А (6 мг) и 77В (4 мг) получали путем замены исходного материала на 3-бромоксетан (103 мг, 0,75 ммоль) согласно способу получения соединения 56 из соединения 56С в примере 56.
77А: МС (ИЭР, масса/заряд): 652,2, 654,2 [М+Н]+.
77В: МС (ИЭР, масса/заряд): 652,2, 654,2 [М+Н]+.
77А: 1Н ЯМР (400 МГц, CDCl3) δ 10,56 (s, 1H), 8,50-8,42 (m, 2Н), 8,21 (s, 1Н), 7,47 (s, 1H), 7,31-7,25 (m, 1H), 7,21 (s, 1H), 7,15 (s, 1H), 6,93 (t, J=7,6 Гц, 1H), 6,54 (s, 1H), 5,43-5,34 (m, 1H), 5,23 (t, J=6,4 Гц, 2H), 5,00 (t, J=6,4 Гц, 2H), 3,93-3,84 (m, 4H), 3,26 (t, J=5,6 Гц, 2H), 2,86 (t, J=5,6 Гц, 2H), 1,87 (s, 3H), 1,84 (s, 3H), 1,30 (s, 3H), 1,28 (s, 3H).
77В: 1H ЯМР (400 МГц, CDCl3) δ 10,77 (s, 1H), 8,66 (dd, J=8,4 Гц, 4,4 Гц, 1H), 8,34 (s, 1H), 8,22 (s, 1H), 7,43 (s, 1H), 7,26-7,18 (m, 2H), 7,06 (s, 1H), 6,90 (t, J=8,0 Гц, 1H), 6,56 (s, 1H), 5,27-5,17 (m, 1H), 4,96-4,86 (m, 4H), 3,94-3,86 (m, 4H), 3,31 (t, J=5,2 Гц, 2H), 3,10 (t, J=5,2 Гц, 2H), 1,87 (s, 3H), 1,84 (s, 3H), 1,30 (s, 3H), 1,29 (s, 3H).
Пример 78:
Получение соединения 78
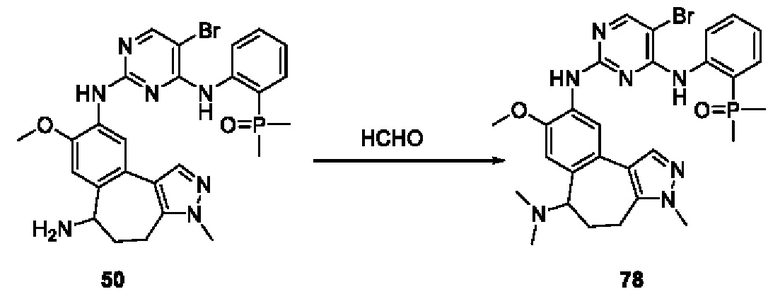
Соединение 78 (17 мг) получали путем замены исходного материала на соединение 50 (40 мг, 0,07 ммоль) и параформальдегид (31 мг, 0,35 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 610,2, 612,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 10,57 (s, 1H), 8,39 (dd, J=8,8 Гц, 4,4 Гц, 1H), 8,26 (s, 1H), 8,23 (s, 1H), 7,55 (s, 1H), 7,36-7,29 (m, 1H), 7,22 (t, J=7,6 Гц, 1H), 7,14 (s, 1H), 7,04 (t, J=7,6 Гц, 1H), 6,78 (s, 1H), 3,97-3,90 (m, 4Н), 3,79 (s, 3Н), 3,18-3,05 (m, 1H), 3,04-3,94 (m, 1Н), 2,69-2,58 (m, 1H), 2,52 (s, 6H), 2,42-2,31 (m, 1H), 1,88 (s, 3H), 1,85 (s, 3H).
Пример 79:
Получение соединения 79
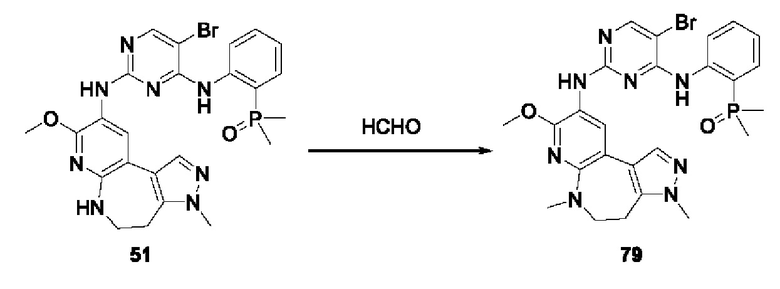
Соединение 79 (35 мг) получали путем замены исходного материала на соединение
51 (50 мг, 0,09 ммоль) и водный раствор формальдегида (40%, 13 мг, 0,18 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 583,2, 585,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,69 (s, 1H), 8,58 (s, 1H), 8,45 (dd, J=8,7 Гц, 4,5 Гц, 1H), 8,21 (s, 1Н), 7,35-7,29 (m, 1H), 7,27-7,14 (m, 3H), 7,06-6,98 (m, 1H), 3,99 (s, 3H), 3,83 (s, 3H), 3,38 (t, J=5,4 Гц, 2H), 3,16 (s, 3H), 3,03 (t, J=5,4 Гц, 2H), 1,88 (s, 3H), 1,84 (s, 3H).
Пример 80:
Получение соединения 80
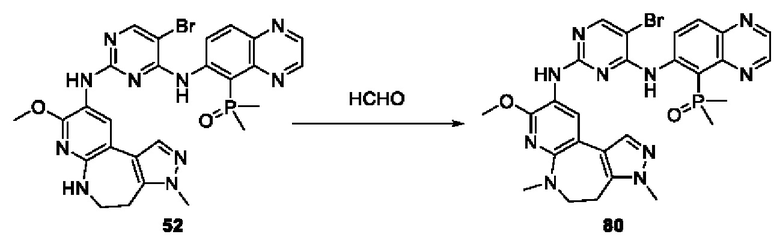
Соединение 80 (33 мг) получали путем замены исходного материала на соединение 52 (100 мг, 0,16 ммоль) и водный раствор формальдегида (40%, 24 мг, 0,32 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 635,2, 637,2 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-Л) δ 8,84-8,77 (m, 2Н), 8,72 (s, 1Н), 8,53 (s, 1H), 8,25 (s, 1H), 8,00 (s, 1H), 7,57 (s, 1H), 3,85 (s, 3Н), 3,71 (s, 3Н), 3,33 (t, J=4,2 Гц, 2Н), 3,15 (s, 3Н), 3,03 (t, J=4,2 Гц, 2Н), 2,03 (s, 3Н), 1,98 (s, 3Н).
Пример 81:
Получение промежуточного соединения 81А
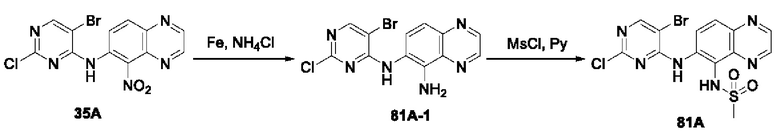
Соединение 81А-1:
Соединение 81А-1 (2 г) получали путем замены исходного материала на соединение 35А (2,4 г, 6,3 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 351,1, 353,1 [М+Н]+.
Соединение 81А:
Соединение 81А (300 мг) получали путем замены исходного материала на соединение 81А-1 (590 мг, 1,68 ммоль) согласно способу получения соединения 32 из соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 429,0, 431,0 [М+Н]+.
Получение соединения 81
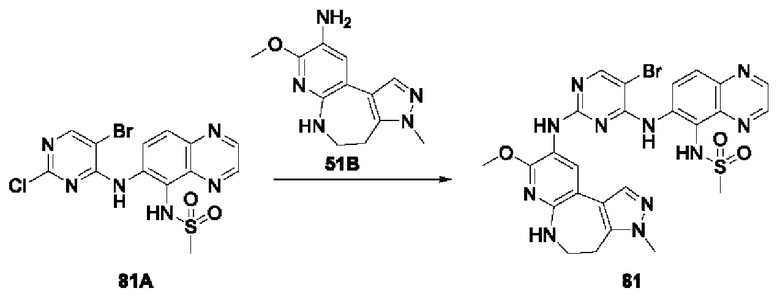
Соединение 81А (88 мг, 0,2 ммоль), соединение 51В (50 мг, 0,2 ммоль) и хлорид аммония (109 мг, 2 ммоль) растворяли в этаноле (2 мл). Реакционную систему нагревали до 85°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 81 (40 мг).
МС (ИЭР, масса/заряд): 638,1, 640,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,14 (s, 1H), 8,86-8,79 (m, 2H), 8,56 (d, J=9,6 Гц, 1H), 8,43 (s, 1H), 8,28 (s, 1H), 7,68 (s, 1H), 7,58 (s, 1H), 7,06 (s, 1H), 3,94 (s, 3H), 3,70 (s, 3H), 3,48 (t, J=4,8 Гц, 2H), 2,99 (t, J=4,8 Гц, 2H), 2,95 (s, 3H).
Пример 82:
Получение соединения 82
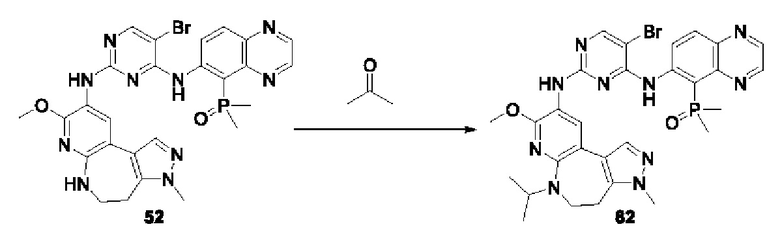
Соединение 82 (20 мг) получали путем замены исходного материала на соединение 52 (85 мг, 0,14 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 663,2, 665,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,63 (s, 1H), 8,84-8,77 (m, 2Н), 8,74 (s, 1H), 8,51 (s, 1H), 8,25 (s, 1H), 8,02 (s, 1H), 7,58 (s, 1H), 7,53 (s, 1H), 4,59-4,43 (m, 1H), 3,83 (s, 3Н), 3,72 (s, 3Н), 3,27 (t, J=5,2 Гц, 2Н), 2,95 (t, J=5,2 Гц, 2Н), 2,02 (s, 3Н), 1,99 (s, 3Н), 1,31 (s, 3Н), 1,29 (s, 3Н).
Пример 83:
Получение соединения 83
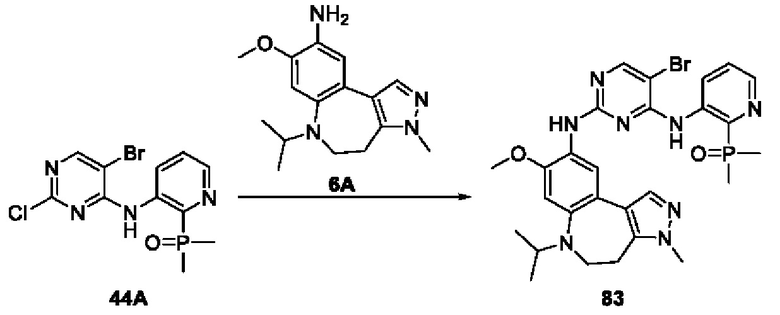
Соединение 83 (33 мг) получали путем замены исходного материала на соединение 44А (126 мг, 0,35 ммоль) и соединение 6А (100 мг, 0,35 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 611,2, 613,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 11,03 (s, 1Н), 9,04-8,96 (m, 1H), 8,30 (s, 1H), 8,24 (s, 1H), 8,18 (d, J=4,5 Гц, 1H), 7,50 (s, 1H), 7,12 (s, 1H), 6,99-6,91 (m, 1H), 6,58 (s, 1H), 3,98-3,90 (m, 1H), 3,88 (s, 3H), 3,84 (s, 3H), 3,32 (t, J=4,8 Гц, 2H), 2,97 (t, J=4,8 Гц, 2H), 1,90 (s, 3H), 1,86 (s, 3H), 1,35 (s, 3H), 1,32 (s, 3H).
Пример 84:
Получение промежуточного соединения 84А
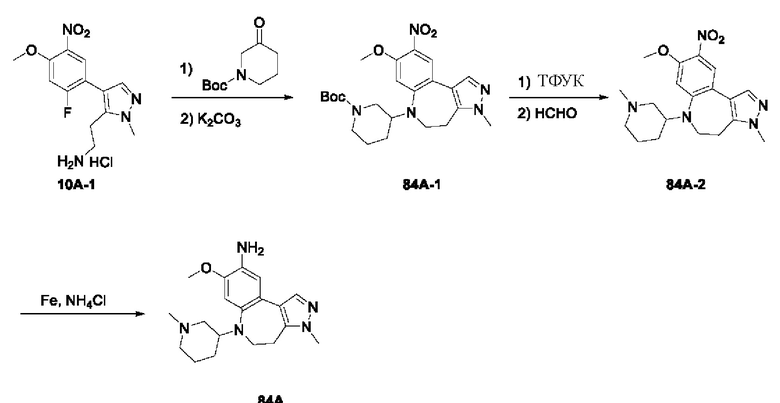
Соединение 84А-1:
Соединение 84А-1 (500 мг) получали путем замены исходного материала на трет-бутил-3-оксопиперидин-1-карбоксилат (1693 мг, 8,5 ммоль) согласно способу получения соединения 47А-1 из соединения 10А-1 в примере 47.
МС (ИЭР, масса/заряд): 458,1 [М+Н]+.
Соединение 84А-2:
Соединение 84А-2 (80 мг) получали путем замены исходного материала на водный раствор формальдегида (40%, 82 мг, 1,1 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 372,2 [М+Н]+.
Промежуточное соединение 84А:
Соединение 84А (45 мг) получали путем замены исходного материала на соединение 84А-2 (80 мг, 0,22 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 342,2 [М+Н]+.
Получение соединения 84
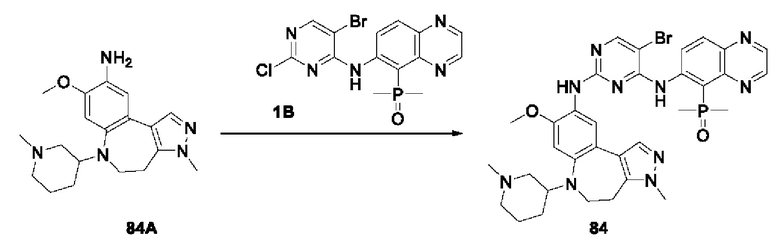
Соединение 84 (8 мг) получали путем замены исходного материала на соединение 84а (70 мг, 0,21 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 717,2, 719,2 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 12,62 (s, 1H), 8,86-8,71 (m, 3Н), 8,37 (s, 1H), 8,26 (s, 1Н), 7,76 (s, 1H), 7,55 (s, 1Н), 7,44 (s, 1H), 6,60 (s, 1H), 3,77 (s, 3Н), 3,70 (s, 3Н), 3,65-3,59 (m, 2Н), 3,09-3,00 (m, 2Н), 2,97-2,88 (m, 2Н), 2,84-2,77 (m, 1H), 2,28 (s, 3Н), 2,20 (t, J=10,5 Гц, 1H), 2,04 (s, 3Н), 1,99 (s, 3Н), 1,96-1,87 (m, 2Н), 1,74-1,53 (m, 3Н).
Пример 85:
Получение соединения 85
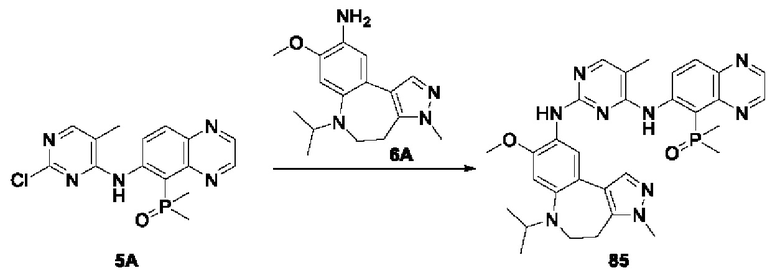
Соединение 85 (46 мг) получали путем замены исходного материала на соединение 6А (100 мг, 0,35 ммоль) и соединение 5А (109 мг, 0,31 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 598,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,36 (s, 1H), 9,20 (dd, J=9,6 Гц, 4,4 Гц, 1H), 8,66-8,64 (m, 2Н), 8,38 (s, 1H), 7,95 (s, 1H), 7,76-7,56 (m, 2Н), 7,21 (s, 1H), 6,58 (s, 1H), 3,96-3,89 (m, 1H), 3,38 (s, 3Н), 3,73 (s, 3Н), 3,32 (t, J=5,6 Гц, 2Н), 2,97 (t, J=5,6 Гц, 2Н), 2,28 (s, 3Н), 2,13 (s, 3Н), 2,10 (s, 3Н), 1,33 (s, 3Н), 1,31 (s, 3Н).
Пример 86:
Получение промежуточного соединения 86А
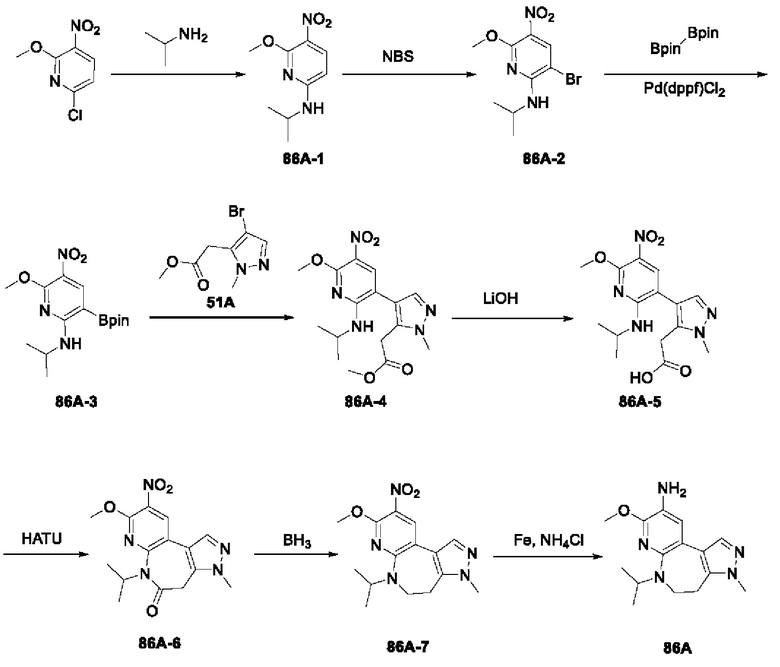
Соединение 86А-1: 91
Соединение 86А-1 (3 г) получали путем замены исходного материала на изопропиламин (1,57 г, 26,5 ммоль) согласно способу получения соединения 51В-1 из 2-метокси-3-нитро-6-хлорпиридина в примере 51.
МС (ИЭР, масса/заряд): 212,1 [М+Н]+.
Соединение 86А-2:
Соединение 86А-2 (3,81 г) получали путем замены исходного материала на соединение 86А-1 (3 г, 14,2 ммоль) согласно способу получения соединения 24В-1 из соединения 1С-2 в примере 24.
МС (ИЭР, масса/заряд): 290,0, 292,0 [М+Н]+.
Соединение 86А-3:
Соединение 86А-3 (3,54 г) получали путем замены исходного материала на соединение 86А-2 (5,3 г, 18,3 ммоль) согласно способу получения соединения 1А из соединения 1А-1 в примере 1.
МС (ИЭР, масса/заряд): 338,3 [М+Н]+.
Соединение 86А-4:
Соединение 86А-4 (1,7 г) получали путем замены исходного материала на соединение 86А-3 (3,54 г, 10,5 ммоль) согласно способу получения соединения 51В-5 из соединения 51В-4 в примере 51.
МС (ИЭР, масса/заряд): 364,1 [М+Н]+.
Соединение 86А-5:
Соединение 86А-5 (1,6 г) получали путем замены исходного материала на соединение 86А-4 (1,7 г, 4,68 ммоль) согласно способу получения соединения 51В-6 из соединения 51В-5 в примере 51.
МС (ИЭР, масса/заряд): 350,3 [М+Н]+.
Соединение 86А-6:
Соединение 86А-6 (1,4 г) получали путем замены исходного материала на соединение 86А-5 (1,72 г, 4,92 ммоль) согласно способу получения соединения 51В-7 из соединения 51В-6 в примере 51.
МС (ИЭР, масса/заряд): 332,3 [М+Н]+.
Соединение 86А-7:
Соединение 86А-7 (1,3 г) получали путем замены исходного материала на соединение 86А-6 (1,4 г, 4,23 ммоль) согласно способу получения соединения 11А-1 из соединения 9А-4 в примере 11.
МС (ИЭР, масса/заряд): 318,1 [М+Н]+.
Соединение 86А:
Соединение 86А (0,9 г) получали путем замены исходного материала на соединение 86А-7 (1,35 г, 4,25 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 288,2 [М+Н]+.
Получение соединения 86
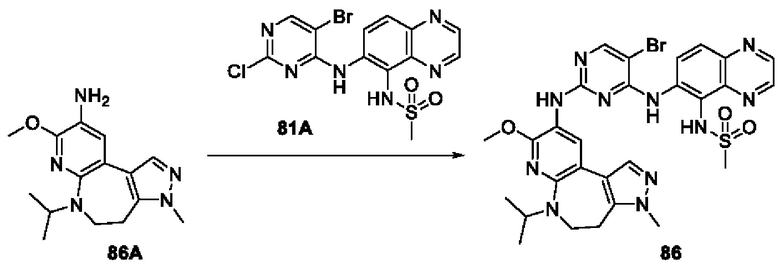
Соединение 86 (56 мг) получали путем замены исходного материала на соединение 86А (75 мг, 0,26 ммоль) и соединение 81А (112 мг, 0,26 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 680,2, 682,2 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d) δ 10,00 (s, 1H), 9,02 (d, J=5,1 Гц, 1H), 8,93 (d, J=5,1 Гц, 1H), 8,86 (s, 1H), 8,70 (s, 1H), 8,58 (s, 1H), 8,34 (s, 1H), 8,08 (s, 1H), 7,66-7,40 (m, 2Н), 4,67-4,52 (m, 1H), 3,88 (s, 3Н), 3,80 (s, 3Н), 3,35 (t, J=5,4 Гц, 2H), 3,08 (s, 3Н), 3,03 (t, J=5,4 Гц, 2H), 1,38 (s, 3Н), 1,36 (s, 3Н).
Пример 87:
Получение промежуточного соединения 87А
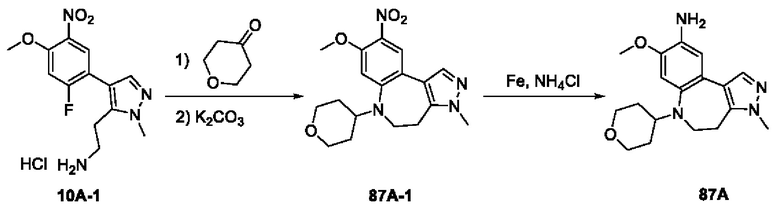
Соединение 87А-1:
Соединение 87А-1 (163 мг) получали путем замены исходного материала на 4-оксациклогексанон (204 мг, 2,04 ммоль) согласно способу получения соединения 47А-1 из соединения 10А-1 в примере 47.
МС (ИЭР, масса/заряд): 359,2 [М+Н]+.
Промежуточное соединение 87А:
Соединение 87А (130 мг) получали путем замены исходного материала на соединение 87А-1 (163 мг, 0,46 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 329,3 [М+Н]+.
Получение соединения 87
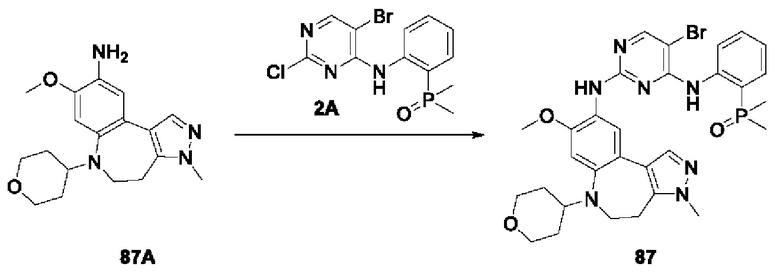
Соединение 87 (26 мг) получали путем замены исходного материала на соединение 87А (50 мг, 0,15 ммоль) и соединение 2А (55 мг, 0,15 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 652,2, 654,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,67 (s, 1H), 8,48 (dd, J=8,1 Гц, 4,2 Гц, 1H), 8,42 (s, 1H), 8,22 (s, 1H), 7,43 (s, 1H), 7,35-7,25 (m, 2H), 7,23-7,15 (m, 1H), 7,04-6,96 (m, 1H), 6,57 (s, 1H), 4,13-4,04 (m, 2H), 3,88 (s, 3H), 3,83 (s, 3H), 3,66-3,36 (m, 5H), 3,00-2,93 (m, 2H), 1,97-1,86 (m, 7H), 1,84 (s, 3H).
Пример 88:
Получение соединения 88
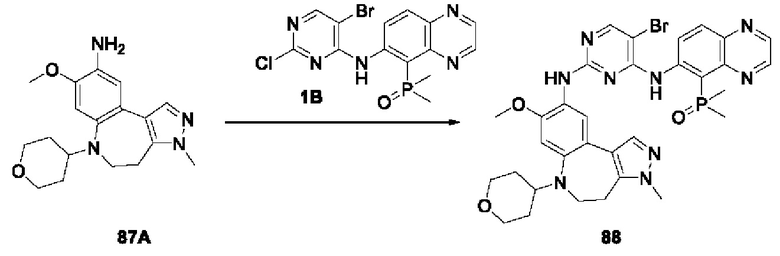
Соединение 88 (45 мг) получали путем замены исходного материала на соединение 87А (50 мг, 0,15 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 704,2, 706,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,54 (s, 1H), 8,89 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,73-8,68 (m, 2Н), 8,40 (s, 1H), 8,30 (s, 1H), 7,70-7,60 (m, 1H), 7,41 (s, 1H), 7,03 (s, 1H), 6,58 (s, 1H), 4,14-4,03 (m, 2Н), 3,89 (s, 3Н), 3,72 (m, 3Н), 3,67-3,56 (m, 1H), 3,55-3,40 (m, 4Н), 3,03-2,93 (m, 2Н), 2,18 (s, 3Н), 2,13 (s, 3Н), 2,00-1,83 (m, 4Н).
Пример 89:
Получение соединения 89
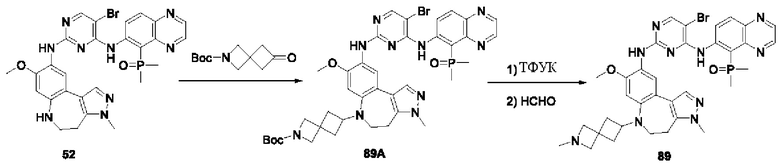
Соединение 89А:
Соединение 89А (88 мг) получали путем замены исходного материала на соединение 52 (150 мг, 0,24 ммоль) и трет-бутил-6-оксо-2-азаспиро[3,3]гептан-2-карбоксилат (102 мг, 0,48 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 815,2, 817,2 [М+Н]+.
Соединение 89:
Соединение 89 (16 мг) получали путем замены исходного материала на соединение 89А (50 мг, 0,06 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 729,2, 731,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,50 (s, 1H), 8,87 (dd, J=9,6 Гц, 4,0 Гц, 1H), 8,72-8,65 (m, 2H), 8,48 (s, 1H), 8,28 (s, 1H), 7,59 (s, 1H), 7,25 (s, 1H), 6,98 (s, 1H), 6,20 (s, 1H), 4,04-3,94 (m, 3H), 3,86 (s, 3H), 3,84-3,79 (m, 2H), 3,72 (s, 3H), 3,20 (t, J=6,0 Гц, 2H), 2,99 (t, J=6,0 Гц, 2H), 2,72-2,64 (m, 5H), 2,30-2,23 (m, 2H), 2,15 (s, 3H), 2,11 (s, 3H).
Пример 90:
Получение соединения 90
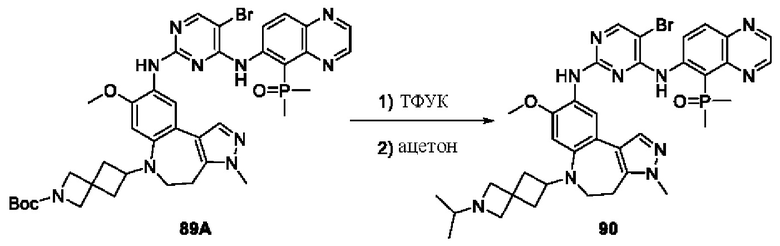
Соединение 90 (4 мг) получали путем замены исходного материала на соединение 89А (50 мг, 0,06 ммоль) и ацетон (11 мг, 0,14 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 757,2, 759,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 12,49 (s, 1H), 8,88 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,75-8,64 (m, 2Н), 8,53 (s, 1Н), 8,38 (s, 1H), 8,30 (s, 1H), 7,67-7,53 (m, 1Н), 6,98 (s, 1H), 6,22 (s, 1H), 4,09-3,92 (m, 3Н), 3,87 (s, 3Н), 3,83-3,77 (m, 2Н), 3,73 (s, 3Н), 3,26-3,14 (m, 2Н), 3,07-3,92 (m, 3Н), 2,75-2,67 (m, 2Н), 2,30-2,55 (m, 2Н), 2,18 (s, 3Н), 2,13 (s, 3Н), 1,26 (s, 3Н), 1,24 (s, 3Н).
Пример 91: получение промежуточного соединения 91А
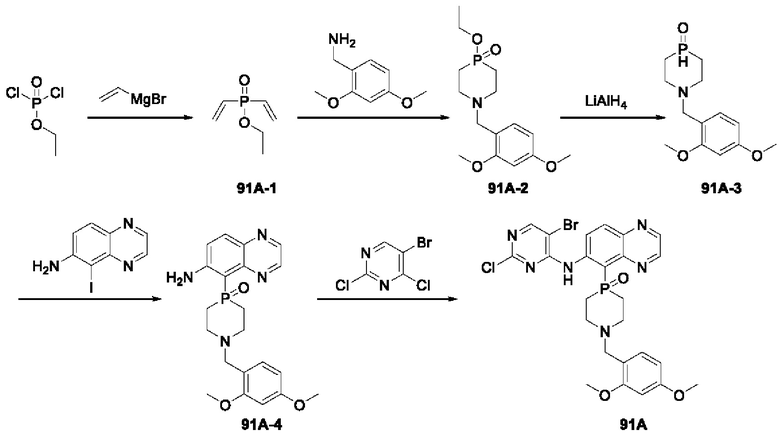
Соединение 90А-1:
Этилдихлорфосфат (5 г, 30,69 ммоль) растворяли в дихлорметане в атмосфере азота и охлаждали реакционную жидкость до -78°С, добавляли раствор бромида винилмагния в тетрагидрофуране (1 М, 68 мл, 68 ммоль) и последовательно перемешивали при указанной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления 1 N соляной кислоты (100 мл). Смесь экстрагировали этилацетатом (150 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (150 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 90А-1 (1,1 г).
МС (ИЭР) масса/заряд: 147,3 [М+Н]+.
Соединение 91А-2:
Соединение 91А-1 (1,4 г, 9,58 ммоль) растворяли в этаноле (10 мл) с последующим добавлением к реакционной жидкости 2,4-диметоксибензиламина (1,92 г, 11,45 ммоль). Реакционную систему нагревали до 90°С и последовательно перемешивали в течение 4 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 91А-2 (2 г).
МС (ИЭР) масса/заряд: 314,1 [М+Н]+.
Соединение 91А-3:
Соединение 91А-2 (1,1 г, 3,51 ммоль) растворяли в безводном тетрагидрофуране (15 мл) в атмосфере азота; затем к реакционной жидкости при 0°С добавляли раствор алюмогидрида лития в тетрагидрофуране (1 М, 2,81 мл, 2,81 ммоль) и последовательно перемешивали при указанной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (0,1 мл), 15% водного раствора гидроксида натрия (0,1 мл) и воды (0,3 мл). Реакционную жидкость последовательно перемешивали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 91А-3 (0,27 г).
МС (ИЭР) масса/заряд: 270,1 [М+Н]+.
Соединение 91А-4:
Соединение 91А-4 (200 мг) получали путем замены исходного материала на соединение 91А-3 (220 мг, 0,82 ммоль) согласно способу получения соединения 1В-2 из соединения 1В-1 в примере 1.
МС (ИЭР, масса/заряд): 413,1 [М+Н]+.
Промежуточное соединение 91А:
Соединение 91А (78 мг) получали путем замены исходного материала на соединение 91А-4 (150 мг, 0,36 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 603,1, 605,1 [М+Н]+.
Получение соединения 91
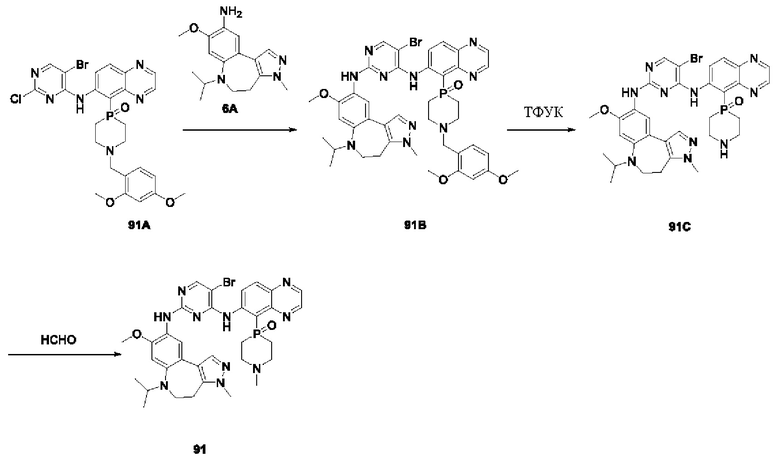
Соединение 91В:
Соединение 91В (50 мг) получали путем замены исходного материала на соединение 91А (78 мг, 0,13 ммоль) и соединение 6А (37 мг, 0,13 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 853,4, 855,4 [М+Н]+.
Соединение 91С:
Соединение 91С (35 мг) получали путем замены исходного материала на соединение 91В (50 мг, 0,06 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 703,3, 705,3 [М+Н]+.
Соединение 91:
Соединение 91 (10 мг) получали путем замены исходного материала на соединение 91С (35 мг, 0,05 ммоль) и водный раствор формальдегида (40%, 19 мг, 0,25 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 717,2, 719,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 11,72 (s, 1H), 9,12 (d, J=1,8 Гц, 1H), 8,83-8,78 (m, 1H), 8,77 (d, J=1,8 Гц, 1H), 8,34 (s, 1H), 8,31 (s, 1H), 7,76-7,64 (m, 1H), 7,33 (s, 1H), 6,91 (s, 1H), 6,60 (s, 1H), 3,93-3,80 (m, 9H), 3,72-3,65 (m, 4H), 3,38-3,28 (m, 2H), 3,01-2,92 (m, 5H), 2,47-2,30 (m, 2H), 1,34 (s, 3H), 1,32 (s, 3H).
Пример 92:
Получение промежуточного продукта 92
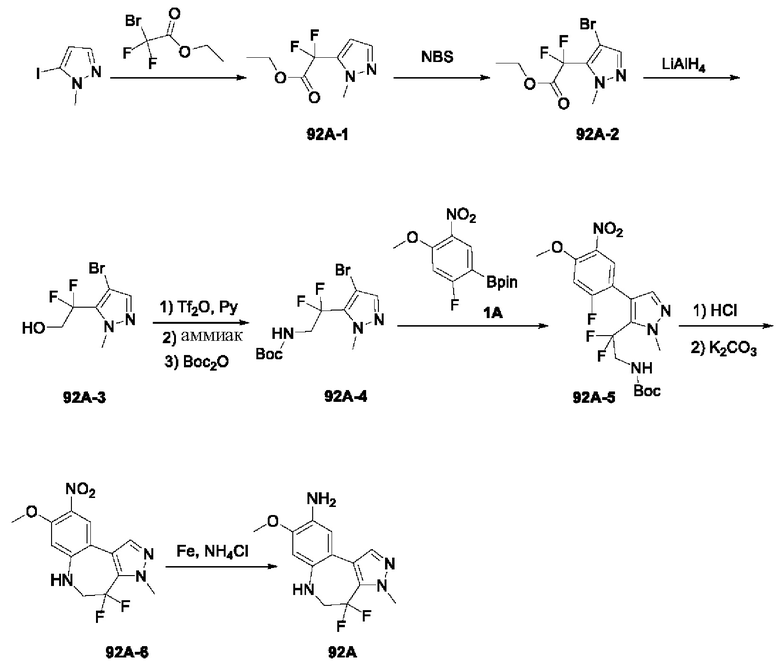
Соединение 92А-1:
5-Йод-1-метил-1Н-пиразол (9,5 г, 45,67 ммоль) и этил(бромдифтор)ацетат (13,91 г, 68,51 ммоль) растворяли в диметилсульфоксиде (100 мл); затем к реакционной жидкости добавляли медный порошок (5,80 г, 91,35 ммоль). Реакционную систему нагревали до 80°С и перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (500 мл). Смесь экстрагировали этилацетатом (200 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (200 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/метил-трет-бутиловый эфир = 1/1) с получением соединения 92А-1 (6,22 г).
МС (ИЭР) масса/заряд: 205,3 [М+Н]+.
Соединение 92А-2:
Соединение 92А-1 (2,95 г, 14,45 ммоль) растворяли в ацетонитриле (50 мл); затем к реакционной жидкости добавляли N-бромсукцинимид (5,66 г, 31,79 ммоль). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/метил-трет-бутиловый эфир = 1/1) с получением соединения 92А-2 (3,26 г).
МС (ИЭР) масса/заряд: 283,0, 285,0 [М+Н]+.
Соединение 92А-3:
Соединение 92А-3 (1,6 г) получали путем замены исходного материала на соединение 92А-2 (3,76 г, 13,28 ммоль) согласно способу получения соединения 91А-3 из соединения 91А-2 в примере 91.
МС (ИЭР, масса/заряд): 241,0, 243,0 [М+Н]+.
Соединение 92А-4:
Соединение 92А-3 (1,8 г, 7,47 ммоль) и пиридин (1,3 г, 16,430 ммоль) растворяли в ацетонитриле (15 мл); затем к реакционной жидкости добавляли по каплям ангидрид трифторметансульфоновой кислоты (4,21 г, 14,97 ммоль) при 0°С. После нагревания реакционной жидкости до комнатной температуры и последовательного перемешивания в течение 1 часа к реакционной жидкости добавляли водный раствор аммиака (12 мл), после чего полученную реакционную систему нагревали до 50°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане (30 мл); затем к реакционной жидкости добавляли триэтиламин (1,52 г, 15 ммоль) и ди-трет-бутилдикарбонат (8,18 г, 37,49 ммоль) и последовательно перемешивали при комнатной температуре в течение 2 часов. Реакционную жидкость гасили путем добавления воды (80 мл). Смесь экстрагировали этилацетатом (100 мл × 2), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/метил-трет-бутиловый эфир = 1/1) с получением соединения 92А-4 (1,6 г).
МС (ИЭР) масса/заряд: 340,0, 342,0 [М+Н]+.
Соединение 92А-5:
Соединение 92А-5 (700 мг) получали путем замены исходного материала на соединение 92А-4 (650 мг, 1,91 ммоль) и соединение 1А (681 мг, 2,29 ммоль) согласно способу получения соединения 51В-5 из соединения 51В-4 в примере 51.
МС (ИЭР, масса/заряд): 431,2 [М+Н]+.
Соединение 92А-6:
Соединение 92А-6 (387 мг) получали путем замены исходного материала на соединение 92А-5 (700 мг, 1,63 ммоль) согласно способу получения соединения 42А-7 из соединения 42А-6 в примере 42.
МС (ИЭР, масса/заряд): 311,0 [М+Н]+.
Промежуточное соединение 92А:
Соединение 92А (100 мг) получали путем замены исходного материала на соединение 92А-6 (120 мг, 0,39 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 281,1 [М+Н]+.
Получение соединения 92
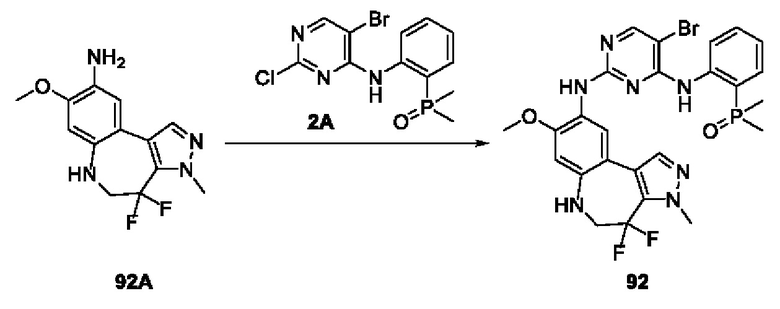
Соединение 92 (38 мг) получали путем замены исходного материала на соединение 92А (100 мг, 0,36 ммоль) и соединение 2А (129 мг, 0,36 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 604,1, 606,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,60 (s, 1H), 8,53 (s, 1H), 8,43 (dd, J=9,6 Гц, 4,2 Гц, 1H), 8,25 (s, 1Н), 7,36-7,30 (m, 2H), 7,26-7,15 (m, 2H), 7,04-6,96 (m, 1H), 6,43 (s, 1H), 4,10 (s, 3H), 3,89 (s, 3H), 3,59 (t, J=12,0 Гц, 2H), 1,89 (s, 3H), 1,85 (s, 3H).
19F ЯМР (282 МГц, CDCl3) δ -92,73.
Пример 93:
Получение соединения 93
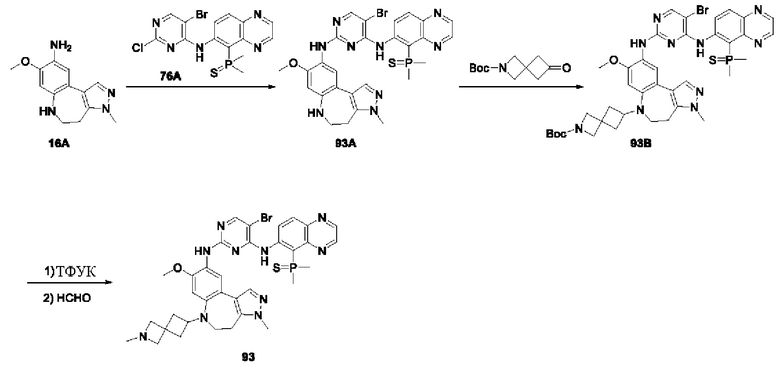
Соединение 93А:
Соединение 93А (50 мг) получали путем замены исходного материала на соединение 16А (48 мг, 0,19 ммоль) и соединение 76А (100 мг, 0,23 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 636,2, 638,2 [М+Н]+.
Соединение 93В:
Соединение 93В (62 мг) получали путем замены исходного материала на соединение 93А (50 мг, 0,08 ммоль) и трет-бутил-6-оксо-2-азаспиро[3,3]гептан-2-карбоксилат (33 мг, 0,16 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 831,2, 833,2 [М+Н]+.
Соединение 93:
Соединение 93 (7 мг) получали путем замены исходного материала на соединение 93В (62 мг, 0,08 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 745,2, 747,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 11,74 (s, 1H), 8,77 (d, J=1,6 Гц, 1H), 8,69 (d, J=1,6 Гц, 1H), 8,32-8,29 (m, 2H), 8,21 (dd, J=9,6 Гц, 4,8 Гц, 1H), 7,65-7,58 (m, 1H), 7,39 (s, 1H), 6,52 (s, 1H), 6,14 (s, 1H), 4,49-4,22 (m, 2H), 4,01-3,95 (m, 1H), 3,85 (s, 3H), 3,63 (s, 3H), 3,27-3,05 (m, 2H), 2,99-2,89 (m, 2H), 2,79 (s, 3H), 2,49 (s, 3H), 2,45 (s, 3H), 2,17-2,08 (m, 1H), 2,01-1,95 (m, 1H), 1,29-1,19 (m, 3H), 0,91-0,80 (m, 1H).
Пример 94:
Получение соединения 94
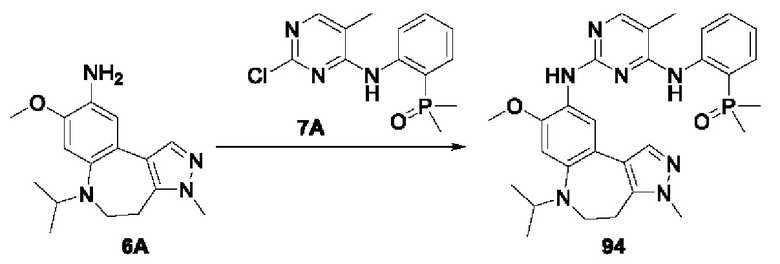
Соединение 94 (20 мг) получали путем замены исходного материала на соединение 6А (70 мг, 0,24 ммоль) и соединение 7А (72 мг, 0,24 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 546,3 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 10,94 (s, 1H), 8,66 (dd, J=9,6 Гц, 4,8 Гц, 1H), 8,23 (s, 1H), 7,76 (s, 1H), 7,42 (s, 1H), 7,27-7,18 (m, 1H), 7,11-7,02 (m, 1H), 7,00-6,93 (m, 1H), 6,59 (s, 1H), 3,99-3,90 (m, 1H), 3,88 (s, 3H), 3,83 (s, 3H), 3,32 (t, J=5,4 Гц, 2H), 2,96 (t, J=5,4 Гц, 2H), 2,23 (s, 3H), 1,87 (s, 3H), 1,82 (s, 3H), 1,35 (s, 3H), 1,33 (s, 3H).
Пример 95:
Получение соединения 95
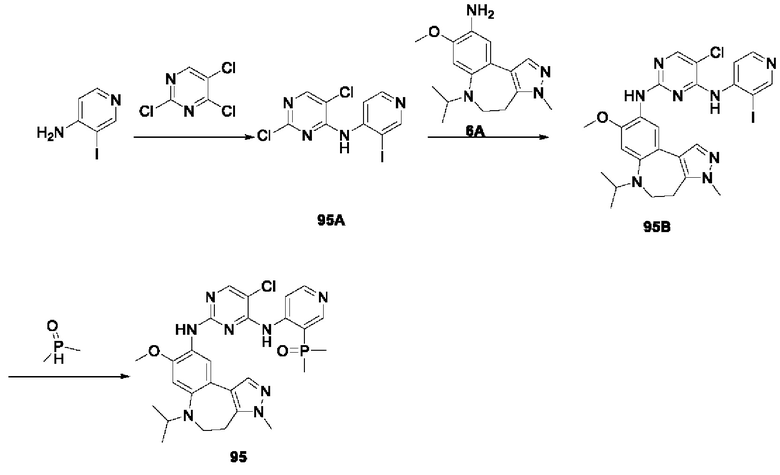
Соединение 95А:
Соединение 95А (290 мг) получали путем замены исходного материала на 4-амино-3-йодпиридин (500 мг, 2,27 ммоль) и 2,4,5-трихлорпиримидин (414 мг, 2,27 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 366,9, 368,9 [М+Н]+.
Соединение 95В:
Соединение 95В (65 мг) получали путем замены исходного материала на соединение 6А (100 мг, 0,35 ммоль) и соединение 95А (128 мг, 0,35 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 617,1, 619,1 [М+Н]+.
Соединение 95:
Соединение 95 (20 мг) получали путем замены исходного материала на соединение 95В (60 мг, 0,1 ммоль) согласно способу получения соединения 1В-2 из соединения 1В-1 в примере 1.
МС (ИЭР, масса/заряд): 567,2, 569,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 11,43 (s, 1H), 8,77 (t, J=5,4 Гц, 1Н), 8,35 (d, J=9,0 Гц, 1H), 8,28 (s, 1H), 8,20 (s, 1H), 8,06 (s, 1H), 7,55 (s, 1H), 7,21 (s, 1H), 6,60 (s, 1H), 4,00-3,91 (m, 1H), 8,89 (s, 3H), 8,84 (s, 3H), 3,33 (t, J=5,4 Гц, 2H), 2,98 (t, J=5,4 Гц, 2H), 1,83 (s, 3H), 1,89 (s, 3H), 1,35 (s, 3H), 1,33 (s, 3H).
Пример 96:
Получение промежуточного соединения 96А
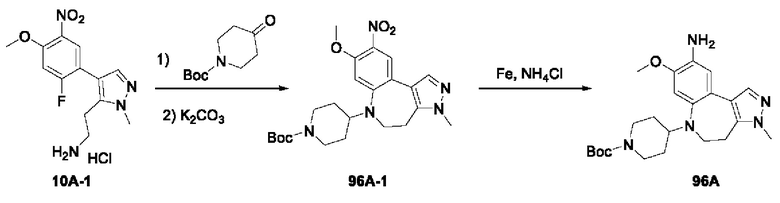
Соединение 96А-1:
Соединение 96А-1 (0,9 г) получали путем замены исходного материала на N-трет-бутоксикарбонил-4-пиперидон (1,35 г, 6,8 ммоль) согласно способу получения соединения 47А-1 из соединения 10А-1 в примере 47.
МС (ИЭР, масса/заряд): 458,3 [М+Н]+.
Соединение 96А:
Соединение 96А (750 мг) получали путем замены исходного материала на соединение 96А-1 (900 мг, 1,97 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 427,1 [М+Н]+.
Получение соединения 96
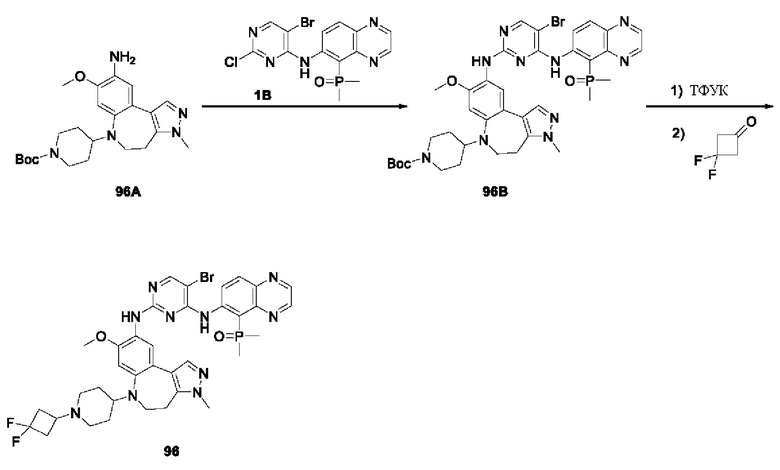
Соединение 96В:
Соединение 96В (65 мг) получали путем замены исходного материала на соединение 96А (100 мг, 0,23 ммоль) и соединение 1В (106 мг, 0,26 ммоль) согласно способу получения соединения 81 из соединения 81А и соединения 51В в примере 81.
МС (ИЭР, масса/заряд): 803,4, 805,4 [М+Н]+.
Соединение 96:
Соединение 96 (3 мг) получали путем замены исходного материала на соединение 96В (65 мг, 0,08 ммоль) и 3,3-дифторциклобутанон (46 мг, 0,43 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 793,2, 795,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,48 (s, 1H), 8,89 (dd, J=9,6 Гц, 4,8 Гц, 1H), 8,73-8,68 (m, 2Н), 8,40 (s, 1H), 8,30 (s, 1H), 7,70-7,60 (m, 1H), 7,37 (s, 1H), 7,00 (s, 1H), 6,54 (s, 1H), 3,88 (s, 3Н), 7,72 (s, 3Н), 3,49-3,39 (m, 3Н), 3,12-3,01 (m, 2Н), 2,97 (t, J=5,4 Гц, 2Н), 2,81-2,66 (m, 3Н), 2,62-2,44 (m, 2Н), 2,18 (s, 3Н), 2,13 (s, 3Н), 2,04-1,92 (m, 3Н), 1,34-1,24 (m, 2Н), 0,95-0,83 (m, 1Н).
Соединение 97:
Получение соединения 97
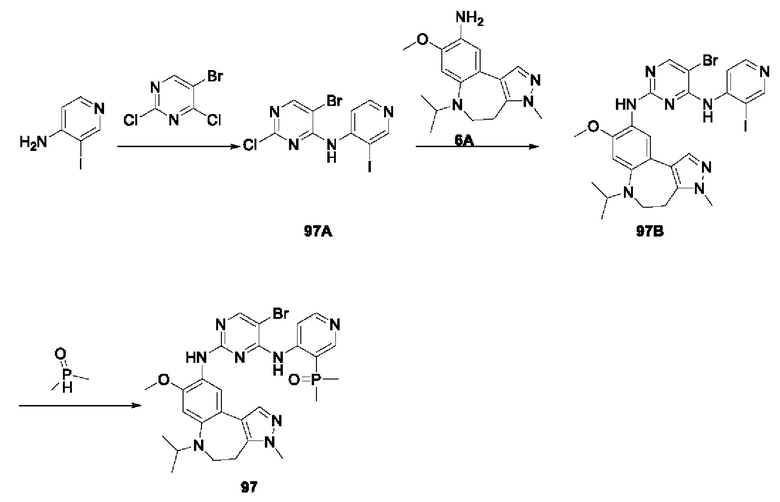
Соединение 97А:
Соединение 97А (130 мг) получали путем замены исходного материала на 4-амино-3-йодпиридин (500 мг, 2,27 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 410,8, 412,8 [М+Н]+.
Соединение 97В:
Соединение 97B (71 мг) получали путем замены исходного материала на соединение 6А (91 мг, 0,32 ммоль) и соединение 97А (130 мг, 0,32 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 661,0, 663,0 [М+Н]+.
Соединение 97:
Соединение 97 (23 мг) получали путем замены исходного материала на соединение 97B (70 мг, 0,11 ммоль) согласно способу получения соединения 1В-2 из соединения 1В-1 в примере 1.
МС (ИЭР, масса/заряд): 611,2, 613,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 11,23 (s, 1Н), 8,73-8,66 (m, 1H), 8,35 (d, J=8,4 Гц, 1H), 8,32-8,25 (m, 2H), 8,07 (s, 1H), 7,51 (s, 1H), 7,17 (s, 1H), 6,60 (s, 1H), 4,00-3,91 (m, 1H), 3,89 (s, 3H), 3,83 (s, 3H), 3,33 (t, J=5,4 Гц, 2H), 2,98 (t, J=5,4 Гц, 2H), 1,93 (s, 3H), 1,89 (s, 3H), 1,35 (s, 3H), 1,32 (s, 3H).
Пример 98:
Получение соединения 98
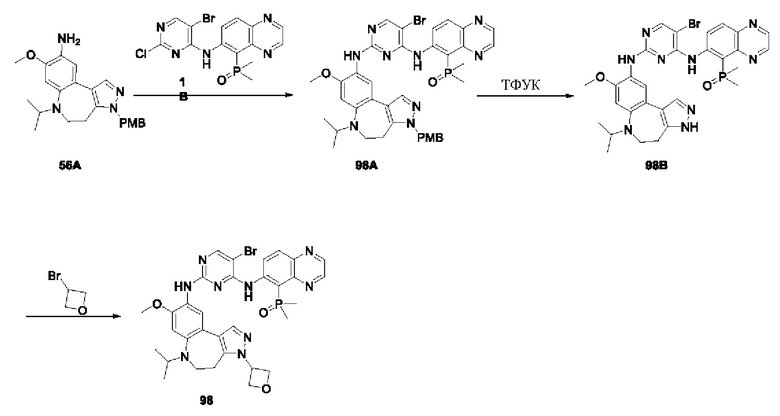
Соединение 98А:
Соединение 98А (336 мг) получали путем замены исходного материала на соединение 56А (300 мг, 0,76 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 768,2, 770,2 [М+Н]+.
Соединение 98В:
Соединение 98В (265 мг) получали путем замены исходного материала на соединение 98А (326 мг, 0,42 ммоль) согласно способу получения соединения 37 из соединения 34 в примере 37.
МС (ИЭР, масса/заряд): 648,0, 650,0 [М+Н]+.
Соединение 98:
Соединение 98 (50 мг) получали путем замены исходного материала на соединение 98В (260 мг, 0,4 ммоль) и 3-бромоксетан (165 мг, 1,2 ммоль) согласно способу получения соединения 56 из соединения 56С в примере 56.
МС (ИЭР, масса/заряд): 704,2, 706,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,57 (s, 1H), 9,02 (dd, J=9,3 Гц, 5,2 Гц, 1H), 8,71 (d,J=2,1 Гц, 1H), 8,67 (d, J=2,1 Гц, 1H), 8,46 (s, 1H), 8,29 (s, 1H), 7,69-7,59 (m, 1H), 7,36 (s, 1H), 7,18 (s, 1H), 6,58 (s, 1H), 5,41-5,29 (m, 1H), 5,14 (t, J=6,6 Гц, 2Н), 4,98 (t, J=6,6 Гц, 2Н), 3,98-3,86 (m, 4Н), 3,31 (t, J=5,4 Гц, 2Н), 2,88 (t, J=5,4 Гц, 2Н), 2,19 (s, 3Н), 2,14 (s, 3Н), 1,33 (s, 3Н), 1,31 (s, 3Н).
Соединение 99:
Получение промежуточного соединения 99А
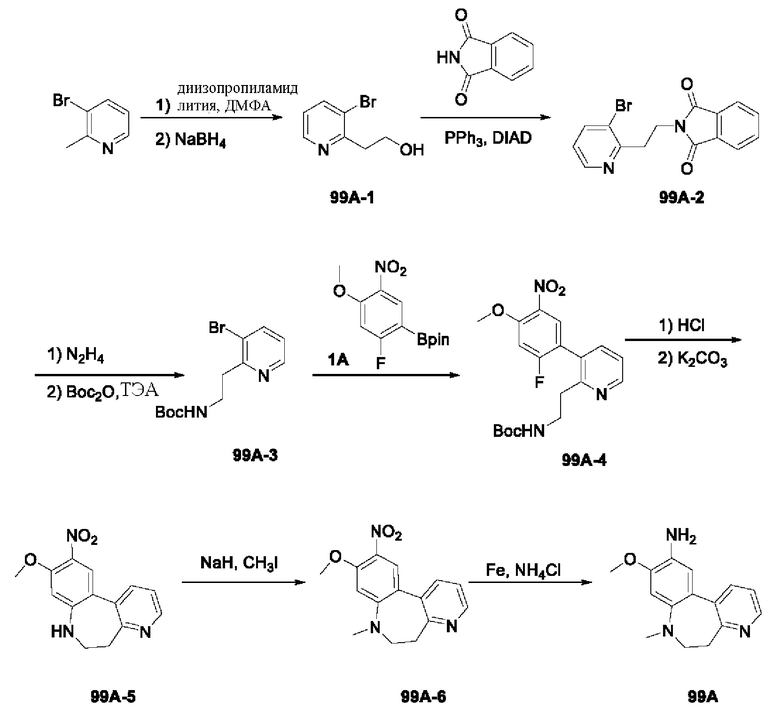
Соединение 99А-1:
2-Метил-3-бромпиридин (5 г, 28,9 ммоль) растворяли в тетрагидрофуране (80 мл) в атмосфере азота; затем в реакционную жидкость при -78°С медленно по каплям добавляли раствор диизопропиламида лития в тетрагидрофуране (2 М, 14,5 ммоль, 29 ммоль) и последовательно перемешивали при указанной температуре в течение 1 часа. В реакционную жидкость добавляли по каплям N,N-диметилформамид (2,11 г, 28,9 ммоль) и последовательно перемешивали при указанной температуре в течение 30 минут. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления метанола (30 мл). Затем к реакционной жидкости добавляли ледяную уксусную кислоту (1,74 г, 28,9 ммоль) и борогидрид натрия (1,31 г, 34,68 ммоль), нагревали до комнатной температуры и последовательно перемешивали в течение 10 минут, после чего гасили путем добавления насыщенного водного раствора бикарбоната натрия (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 99А-1 (4,5 г).
МС (ИЭР, масса/заряд): 201,8, 203,8 [М+Н]+.
Соединение 99А-2:
Соединение 99А-2 (2,5 г) получали путем замены исходного материала на соединение 99А-1 (2 г, 9,89 ммоль) согласно способу получения соединения 51А-1 из соединения 40А-2 в примере 51.
МС (ИЭР, масса/заряд): 330,9, 332,9 [М+Н]+.
Соединение 99А-3:
Соединение 99А-2 (2,5 г, 7,55 ммоль) растворяли в этаноле (30 мл) с последующим добавлением к реакционной жидкости гидрата гидразина (1,51 г, 30,19 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и осажденное твердое вещество отфильтровывали и высушивали. Фильтрационный осадок растворяли в дихлорметане (30 мл) и добавляли к реакционной жидкости триэтиламин (1,81 г, 17,88 ммоль) и ди-трет-бутилкарбонат (2,93 г, 13,43 ммоль), после чего последовательно перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость гасили путем добавления воды (80 мл). Смесь экстрагировали этилацетатом (80 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (80 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 99А-3 (2,5 г).
МС (ИЭР, масса/заряд): 300,9, 302,9 [М+Н]+.
Соединение 99А-4:
Соединение 99А-4 (470 мг) получали путем замены исходного материала на соединение 99А-3 (800 мг, 2,65 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 392,1 [М+Н]+.
Соединение 99А-5:
Соединение 99А-5 (400 мг) получали путем замены исходного материала на соединение 99А-4 (700 мг, 1,78 ммоль) согласно способу получения соединения 42А-7 из соединения 42А-6 в примере 42.
МС (ИЭР, масса/заряд): 272,2 [М+Н]+.
Соединение 99А-6:
Соединение 99А-6 (230 мг) получали путем замены исходного материала на соединение 99А-5 (400 мг, 1,66 ммоль) согласно способу получения соединения 8А-9 из соединения 8А-8 в примере 8.
МС (ИЭР, масса/заряд): 286,3 [М+Н]+.
Промежуточное соединение 99А:
Соединение 99А (160 мг) получали путем замены исходного материала на соединение 99А-6 (230 мг, 0,81 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 256,3 [М+Н]+.
Получение соединения 99
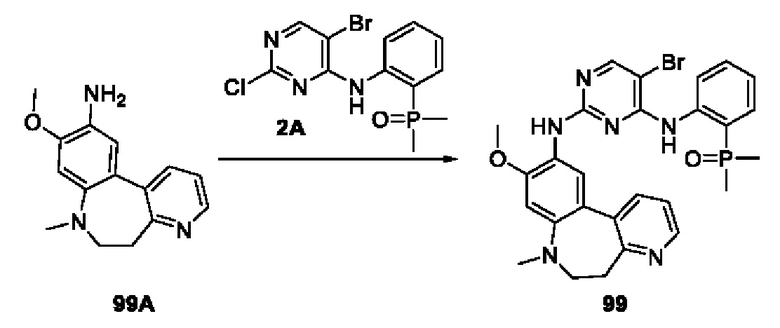
Соединение 99 (20 мг) получали путем замены исходного материала на соединение 99А (35 мг, 0,14 ммоль) и соединение 2А (50 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 579,1, 581,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10,55 (s, 1H), 8,46-8,34 (m, 1H), 8,33-8,26 (m, 1H), 8,22 (s, 1H), 8,20 (s, 1H), 7,49-7,35 (m, 2H), 7,24-7,15 (m, 1H), 7,11 (dd, J=6,4, 6,8 Гц, 1H), 6,87-6,76 (m, 2H), 6,57 (s, 1H), 3,96 (s, 3H), 3,59 (t, J=6,4 Гц, 2H), 3,03 (t, J=6,4 Гц, 2H), 2,79 (s, 3H), 1,83 (s, 3H), 1,80 (s, 3H).
Пример 100:
Получение соединения 100
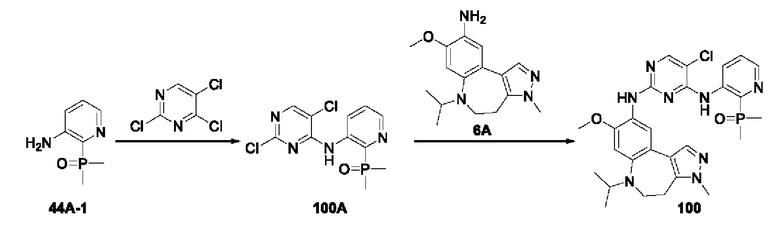
Соединение 100А:
Соединение 100А (75 мг) получали путем замены исходного материала на соединение 44А-1 (1 г, 5,88 ммоль) и 2,4,5-трихлорпиримидин (2,16 г, 11,75 ммоль) согласно способу получения соединения 2А в примере 2.
МС (ИЭР, масса/заряд): 317,2, 319,2 [М+Н]+.
Соединение 100:
Соединение 100 (20 мг) получали путем замены исходного материала на соединение 100А (70 мг, 0,22 ммоль) и соединение 6А (63 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 567,2, 569,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 11,20 (s, 1Н), 9,09-9,04 (m, 1H), 8,28 (s, 1H), 8,16 (d,J=4,4 Гц, 1H), 8,12 (s, 1H), 7,52 (s, 1H), 7,15 (s, 1H), 6,93 (s, 1H), 6,57 (s, 1H), 3,96-3,88 (m, 1H), 3,87 (s, 3H), 3,83 (s, 3H), 3,30 (t, J=5,6 Гц, 2H), 2,96 (t, J=5,6 Гц, 2H), 1,88 (s, 3H), 1,85 (s, 3H), 1,32 (s, 3H), 1,31 (s, 3H).
Пример 101:
Получение промежуточного соединения 101А
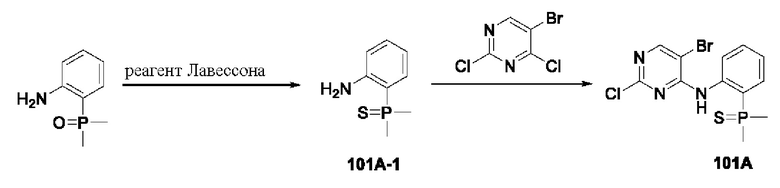
Соединение 101А-1:
Соединение 101А-1 (770 мг) получали путем замены исходного материала на 2-(диметилфосфорил)анилин (5 г, 29,6 ммоль) согласно способу получения соединения 76А-1 из соединения 1В-2 в примере 76.
МС (ИЭР, масса/заряд): 186,2 [М+Н]+.
Промежуточное соединение 101А:
Соединение 101А (330 г) получали путем замены исходного материала на соединение 101А-1 (770 мг, 4,16 ммоль) согласно способу получения соединения 2А из 2-(диметилфосфино)анилина в примере 2.
МС (ИЭР, масса/заряд): 376,0, 378,0 [М+Н]+.
Получение соединения 101
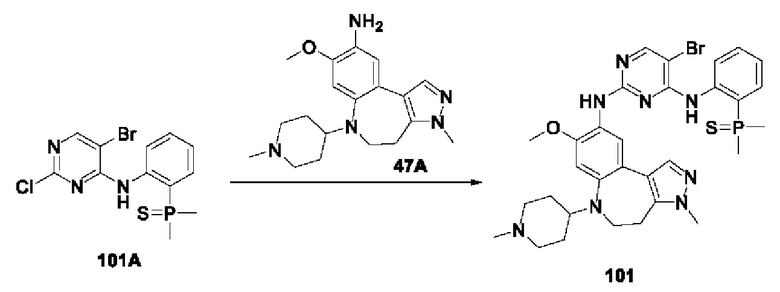
Соединение 101 (88 мг) получали путем замены исходного материала на соединение 101А (80 мг, 0,22 ммоль) и соединение 47А (76 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 681,2, 683,2 [М+Н]+.
1H ЯМР (300 МГц, CD3OD) δ 8,25 (s, 1H), 7,80-7,60 (m, 3Н), 7,44-7,13 (m, 3Н), 6,65 (s, 1H), 3,89 (s, 3Н), 3,87-3,78 (m, 4Н), 3,63 (d, J=12,6 Гц, 2Н), 3,38 (t, J=5,1 Гц, 2Н), 3,28-3,16 (m, 2Н), 3,03 (t, J=5,1 Гц, 2Н), 2,92 (s, 3Н), 2,24-2,13 (m, 4Н), 2,11 (s, 3Н), 2,06 (s, 3Н).
Пример 102:
Получение соединения 102
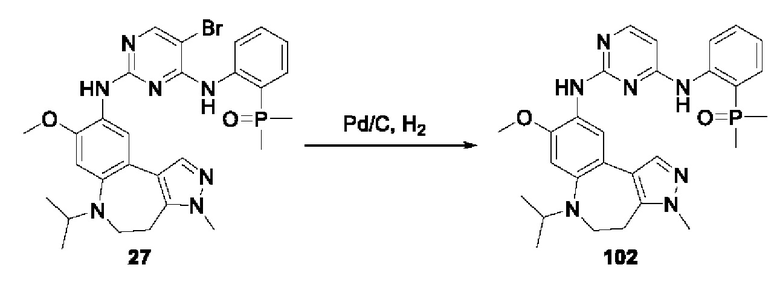
Соединение 102 (30 мг) получали путем замены исходного материала на соединение 27 (120 мг, 0,2 ммоль) согласно способу получения соединения 22 из соединения 2 в примере 22.
МС (ИЭР, масса/заряд): 532,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 11,02 (s, 1H), 9,09 (s, 1H), 8,54 (dd, J=8,4 Гц, 4,4 Гц, 1H), 8,15 (s, 1Н), 7,88 (d, J=6,0 Гц, 1Н), 7,51 (s, 1H), 7,22-7,14 (m, 1Н), 7,06-6,98 (m, 1H), 6,96-6,90 (m, 1Н), 6,58 (s, 1Н), 6,12 (d, J=6,4 Гц, 1H), 3,98-3,88 (m, 1H), 3,85 (s, 3Н), 3,81 (s, 3Н), 3,30 (t, J=5,6 Гц, 2Н), 2,95 (t, J=5,6 Гц, 2Н), 1,83 (s, 3Н), 1,80 (s, 3Н), 1,33 (s, 3Н), 1,31 (s, 3Н).
Пример 103:
Получение промежуточного соединения 103А
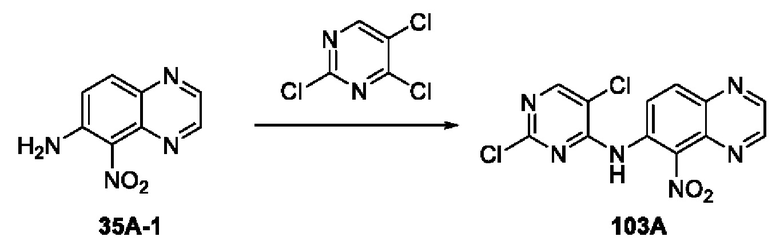
Соединение 103А (720 мг) получали путем замены исходного материала на соединение 35А-1 (700 мг, 3,68 ммоль) и 2,4,5-трихлорпиримидин (1350 мг, 7,36 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере. МС (ИЭР, масса/заряд): 337,0, 339,0 [М+Н]+.
Получение соединения 103
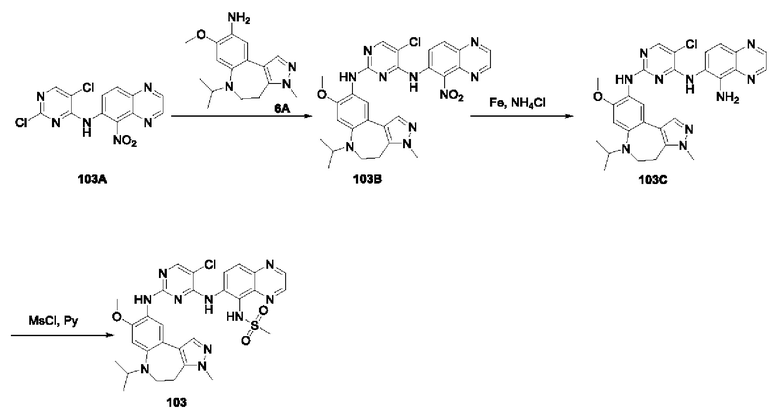
Соединение 103В:
Соединение 103В (100 мг) получали путем замены исходного материала на соединение 103А (150 мг, 0,46 ммоль) и соединение 6А (127 мг, 0,46 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 587,0, 589,0 [М+Н]+.
Соединение 103С:
Соединение 103С (80 мг) получали путем замены исходного материала на соединение 103В (100 мг, 0,17 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 557,3, 559,3 [М+Н]+.
Соединение 103:
Соединение 103 (20 мг) получали путем замены исходного материала на соединение 103С (50 мг, 0,09 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 635,3, 637,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,16 (s, 1H), 8,81 (d, J=2,0 Гц, 1H), 8,78 (d, J=2,0 Гц, 1H), 8,63 (d, J=9,2 Гц, 1H), 8,32 (s, 1H), 8,17 (s, 1H), 7,63-7,51 (m, 2H), 7,30 (s, 1H), 6,77 (s, 1H), 6,53 (s, 1H), 3,93-3,80 (m, 4H), 3,66 (s, 3H), 3,33-3,24 (m, 2H), 2,96-2,86 (m, 5H), 1,30 (s, 3H), 1,28 (s, 3H).
Пример 104:
Получение промежуточного соединения 104А
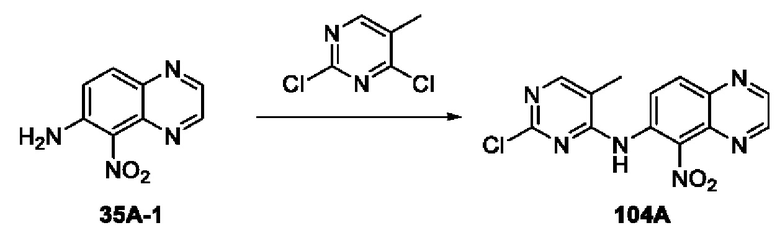
Соединение 104А (510 мг) получали путем замены исходного материала на соединение 35А-1 (500 мг, 2,63 ммоль) и 2,4-дихлор-5-метилпиримидин (857 мг, 5,26 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 317,2, 319,2 [М+Н]+.
Получение соединения 104
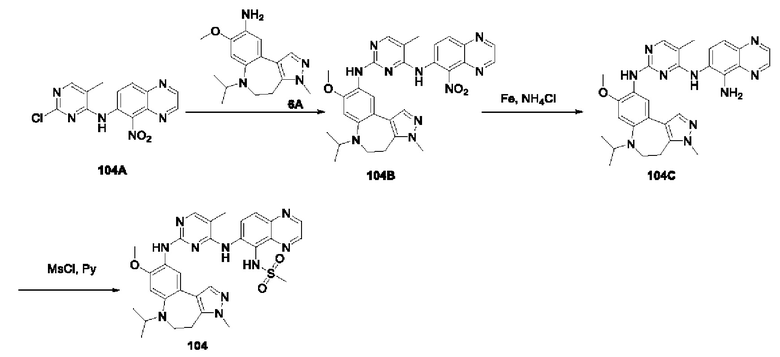
Соединение 104В:
Соединение 104В (80 мг) получали путем замены исходного материала на соединение 104А (100 мг, 0,32 ммоль) и соединение 6А (90 мг, 0,32 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 567,3 [М+Н]+.
Соединение 104С:
Соединение 104С (65 мг) получали путем замены исходного материала на соединение 104В (75 мг, 0,13 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 536,8 [М+Н]+.
Соединение 104:
Соединение 104 (20 мг) получали путем замены исходного материала на соединение 104С (50 мг, 0,09 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 615,4 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 8,81-8,77 (m, 2H), 8,75 (d, J=1,6 Гц, 1H), 8,69 (d, J=9,6 Гц, 1H), 8,29 (s, 1H), 7,95 (s, 1H), 7,86 (s, 1H), 7,51 (d, J=9,2 Гц, 1H), 6,84 (s, 1H), 6,53 (s, 1H), 3,92-3,84 (m, 4H), 3,66 (s, 3H), 3,29 (t, J=5,6 Гц, 2H), 2,95-2,87 (m, 5H), 2,25 (s, 3H), 1,30 (s, 3H), 1,28 (s, 3H).
Пример 105:
Получение соединения 105
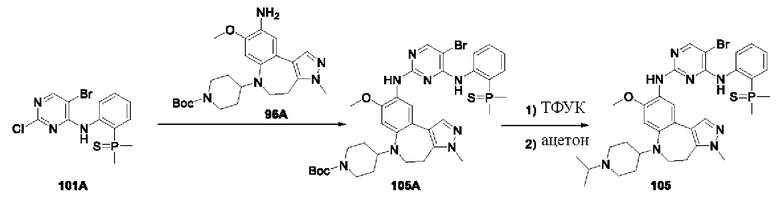
Соединение 105А:
Соединение 105А (146 мг) получали путем замены исходного материала на соединение 96А (227 мг, 0,53 ммоль) и соединение 101А (200 мг, 0,53 ммоль) согласно способу получения соединения 81 из соединения 81А и соединения 51В в примере 81.
МС (ИЭР, масса/заряд): 767,2, 769,2 [М+Н]+.
Соединение 105:
Соединение 105 (8 мг) получали путем замены исходного материала на соединение 105А (70 мг, 0,09 ммоль) согласно способу получения соединения 63А-2 из соединения 63А-1 в примере 63.
МС (ИЭР, масса/заряд): 709,3, 711,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,32 (s, 1H), 8,38 (s, 1H), 8,25 (s, 1H), 7,95 (dd, J=8,4 Гц, 3,2 Гц, 1H), 7,53-7,46 (m, 1H), 7,32 (s, 1H), 7,26-7,21 (m, 1H), 7,13-7,05 (m, 1H), 6,90 (s, 1H), 6,48 (s, 1H), 3,85 (s, 3Н), 3,78 (s, 3Н), 3,51-3,23 (m, 6Н), 2,94 (t, J=5,6 Гц, 2H), 2,61 (t, J=12,8 Гц, 2H), 2,40-2,32 (m, 2Н), 2,10 (s, 3Н), 2,06 (s, 3Н), 2,03-1,95 (m, 2Н), 1,28 (s, 3Н), 1,26 (s, 3Н).
Пример 106:
Получение соединения 106
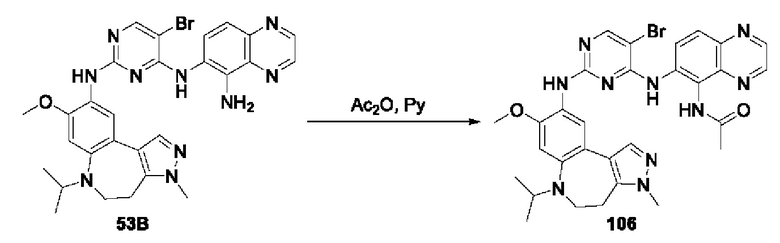
Соединение 106 (63 мг) получали путем замены исходного материала на соединение 53В (100 мг, 0,17 ммоль) и уксусный ангидрид (34 мг, 0,33 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 643,2, 645,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,36 (s, 1H), 9,19 (s, 1H), 8,80 (d, J=2,1 Гц, 1H), 8,76 (d, J=2,1 Гц, 1H), 8,31 (s, 1H), 8,22 (s, 1H), 8,19 (d, J=6,3 Гц, 1H), 7,50 (d, J=9,6 Гц, 1H), 7,38 (s, 1H), 6,62-6,31 (m, 2H), 3,94-3,78 (m, 4H), 3,62 (s, 3H), 3,26 (t, J=5,4 Гц, 2H), 2,87 (t, J=5,4 Гц, 2H), 2,48 (s, 3H), 1,30 (s, 3H), 1,27 (s, 3H).
Пример 107:
Получение соединения 107
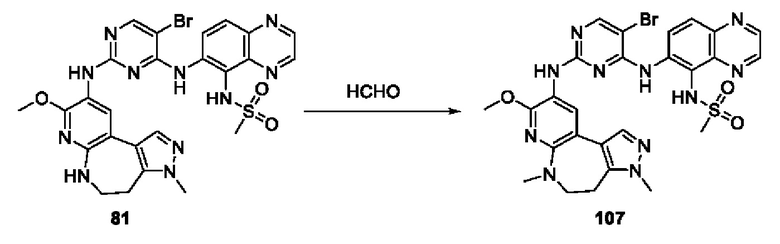
Соединение 107 (10 мг) получали путем замены исходного материала на соединение 81 (40 мг, 0,06 ммоль) и водный раствор формальдегида (40%, 10 мг, 0,13 ммоль) согласно способу получения соединения 72 из соединения 71 в примере 72.
МС (ИЭР, масса/заряд): 652,1, 654,1 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,16 (s, 1H), 8,82 (m, 2Н), 8,52 (d, J=9,4 Гц, 1H), 8,38 (s, 1H), 8,26 (s, 1H), 7,61 (s, 2H), 7,24 (s, 1H), 6,74 (s, 1H), 3,99 (s, 3H), 3,71 (s, 3H), 3,44-3,34 (m, 2H), 3,16 (s, 3H), 3,00 (t, J=5,1 Гц, 2H), 2,95 (s, 3H).
Пример 108:
Получение соединения 108
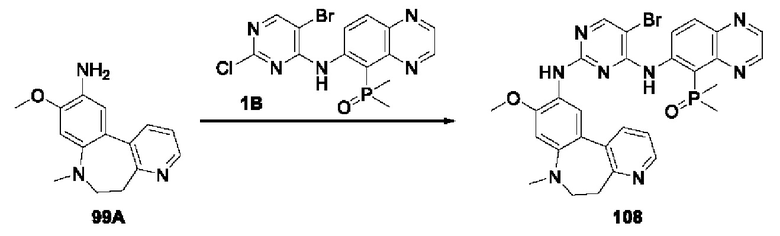
Соединение 108 (40 мг) получали путем замены исходного материала на соединение 99А (80 мг, 0,31 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 631,1, 633,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 12,48 (s, 1H), 8,83 (dd, J=8,4, 5,2 Гц, 1H), 8,76 (d, J=2,1 Гц, 1H), 8,71 (d, J=1,8 Гц, 1H), 8,31-8,27 (m, 3Н), 7,70 (d, J=9,3 Гц, 1H), 7,43 (s, 1H), 7,32 (s, 1H), 6,75 (s, 1H), 6,67 (s, 1H), 3,99 (s, 3Н), 3,60 (t, J=6,6 Гц, 2H), 3,02 (t, J=6,6 Гц, 2H), 2,81 (s, 3Н), 2,16 (s, 3Н), 2,11 (s, 3Н).
Пример 109:
Получение промежуточного соединения 109А
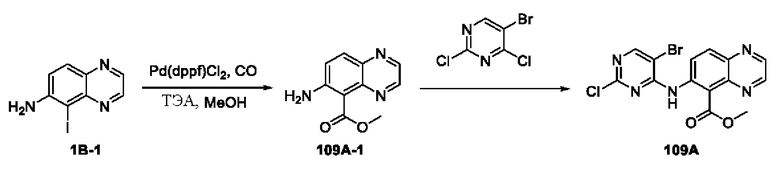
Соединение 109А-1:
Соединение 1В-1 (6,9 г, 25,46 ммоль) и триэтиламин (12,88 г, 127,28 ммоль) растворяли в метаноле (100 мл) с последующим добавлением к реакционной жидкости дихлор[1,1'-бис(дифенилфосфино)ферроцен] палладия (II) (1,86 г, 2,55 ммоль). В реакционную жидкость вводили монооксид углерода (50 атм) и нагревали до 70°С, после чего последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и удаляли газ при пониженном давлении, затем концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/10) с получением соединения 109А-1 (1,5 г).
МС (ИЭР) масса/заряд: 204,3 [М+Н]+.
Промежуточное соединение 109А:
Соединение 109А (0,5 г) получали путем замены исходного материала на соединение 109А-1 (1,5 г, 7,38 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 394,0, 396,0 [М+Н]+.
Получение соединения 109
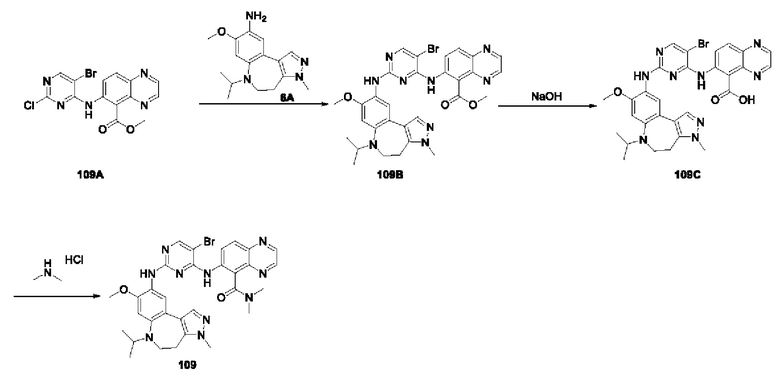
Соединение 109В:
Соединение 109В (400 мг) получали путем замены исходного материала на соединение 109А (350 мг, 0,89 ммоль) и соединение 6А (254 мг, 0,89 ммоль) согласно способу получения соединения 81 из соединения 81А и соединения 51В в примере 81.
МС (ИЭР, масса/заряд): 644,2, 646,2 [М+Н]+.
Соединение 109С:
Соединение 109С (150 мг) получали путем замены исходного материала на соединение 109В (400 мг, 0,62 ммоль) согласно способу получения соединения 1С-3 из соединения 1С-2 в примере 1.
МС (ИЭР, масса/заряд): 630,1, 632,1 [М+Н]+.
Соединение 109:
Соединение 109 (7 мг) получали путем замены исходного материала на соединение 109С (50 мг, 0,08 ммоль) согласно способу получения соединения 48А-5 из соединения 48А-4 в примере 48.
МС (ИЭР, масса/заряд): 657,2, 659,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 8,84 (d, J=1,8 Гц, 1Н), 8,74 (d, J=1,8 Гц, 1H), 8,65 (d, J=9,3 Гц, 1H), 8,50 (s, 1H), 8,42 (s, 1H), 8,25 (s, 1H), 7,65 (s, 1H), 7,35 (s, 1H), 6,90 (s, 1H), 6,58 (s, 1H), 3,99-3,91 (m, 4H), 3,68 (s, 3H), 3,53-3,48 (m, 1H), 3,35 (s, 3H), 3,18-3,11 (m, 1H), 3,04-2,99 (m, 1H), 2,93 (s, 3H), 2,89-2,83 (m, 1H), 1,36-1,29 (m, 6H).
Пример 193:
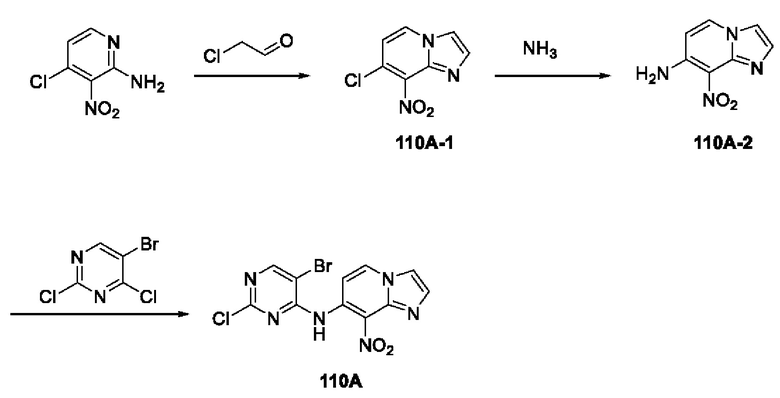
Соединение 110А-1:
2-Амино-3-нитро-4-хлорпиридин (3 г, 17,29 ммоль) и водный раствор хлорацетальдегида (40%, 5,09 г, 25,928 ммоль) растворяли в 1,4-диоксане (10 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, осажденное твердое вещество отфильтровывали и промывали фильтрационный осадок диэтиловым эфиром (5 мл × 3). Фильтрационный осадок высушивали с получением соединения 110А-1 (2,8 г).
МС (ИЭР, масса/заряд): 198,3, 200,3 [М+Н]+.
Соединение 110А-2:
Соединение 110А-1 (800 мг, 4,05 ммоль) растворяли в метанольном аммиаке (7 М, 20 мл) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 110А-2 (100 мг).
МС (ИЭР, масса/заряд): 179,2 [М+Н]+.
Промежуточное соединение 110А:
Соединение 110А (480 мг) получали путем замены исходного материала на соединение 110А-2 (500 мг, 2,81 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 368,9, 370,9 [М+Н]+.
Получение соединения 110
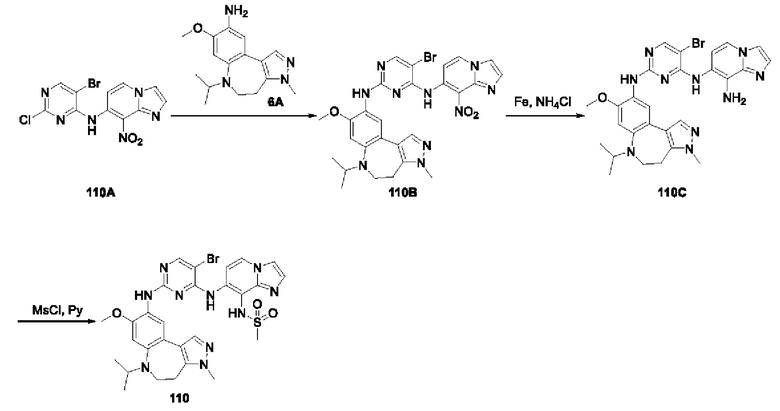
Соединение 110В:
Соединение 110В (160 мг) получали путем замены исходного материала на соединение 110А (290 мг, 0,79 ммоль) и соединение 6А (150 мг, 0,52 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 619,1, 621,1 [М+Н]+.
Соединение 110С:
Соединение 110С (130 мг) получали путем замены исходного материала на соединение 110В (150 мг, 0,24 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 589,3, 591,3 [М+Н]+.
Соединение 110:
Соединение 110 (16 мг) получали путем замены исходного материала на соединение 110С (150 мг, 0,25 ммоль) согласно способу получения соединения 32 из промежуточного соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 667,2, 669,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 8,90 (s, 1H), 8,31 (s, 1H), 8,23 (s, 1H), 8,03 (d, J=5,4 Гц, 1H), 7,52 (s, 2H), 7,30 (s, 2H), 7,23 (s, 1H), 6,55 (s, 1H), 3,86 (s, 4H), 3,78 (s, 3H), 3,32-3,23 (m, 2H), 3,17 (s, 3H), 2,97-2,90 (m, 2H), 1,31 (s, 3H), 1,30 (s, 3H).
Пример 195:
Получение промежуточного соединения 111A
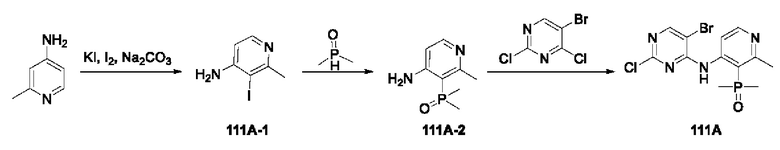
Соединение 111А-1:
2-Метил-4-аминопиридин (6 г, 55,48 ммоль) и карбонат натрия (4,12 г, 38,84 ммоль) растворяли в воде (30 мл) с последующим добавлением к реакционной жидкости иодида калия (11,97 г, 72,13 ммоль) и иода (11,27 г, 44,39 ммоль) отдельными порциями. Реакционную систему нагревали до 100°С и последовательно перемешивали в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры. Смесь экстрагировали этилацетатом (50 мл × 3), после чего органические фазы объединяли, сначала промывали с помощью насыщенного водного раствора тиосульфата натрия (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 111А-1 (0,83 г).
МС (ИЭР, масса/заряд): 235,0 [М+Н]+.
Соединение 111А-2:
Соединение 111А-2 (238 мг) получали путем замены исходного материала на соединение 111А-1 (400 мг, 1,71 ммоль) согласно способу получения соединения 1В-2 из соединения 1В-1 в примере 1.
МС (ИЭР, масса/заряд): 185,3 [М+Н]+.
Промежуточное соединение 111А:
Соединение 111А (27 мг) получали путем замены исходного материала на соединение 111А-2 (238 мг, 1,29 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 375,0, 377,0 [М+Н]+.
Получение соединения 111
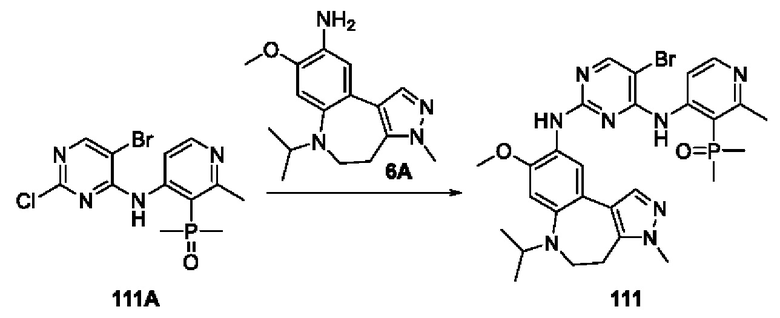
Соединение 111 (6 мг) получали путем замены исходного материала на соединение 111А (22 мг, 0,06 ммоль) и соединение 6А (17 мг, 0,06 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 625,2, 627,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 12,33 (s, 1Н), 8,51 (dd, J=6,0, 3,2 Гц, 1H), 8,27 (s, 2H), 7,94 (s, 1H), 7,44 (s, 1H), 7,11 (s, 1H), 6,56 (s, 1H), 3,93-3,86 (m, 1H), 3,86 (s, 3H), 3,81 (s, 3H), 3,29 (t, J=5,2 Гц, 2H), 2,95 (t, J=5,6 Гц, 2H), 2,59 (s, 3H), 2,01 (d, J=13,2 Гц, 6H), 1,31 (d, J=6,4 Гц, 6H).
Пример 112:
Получение промежуточного соединения 112А
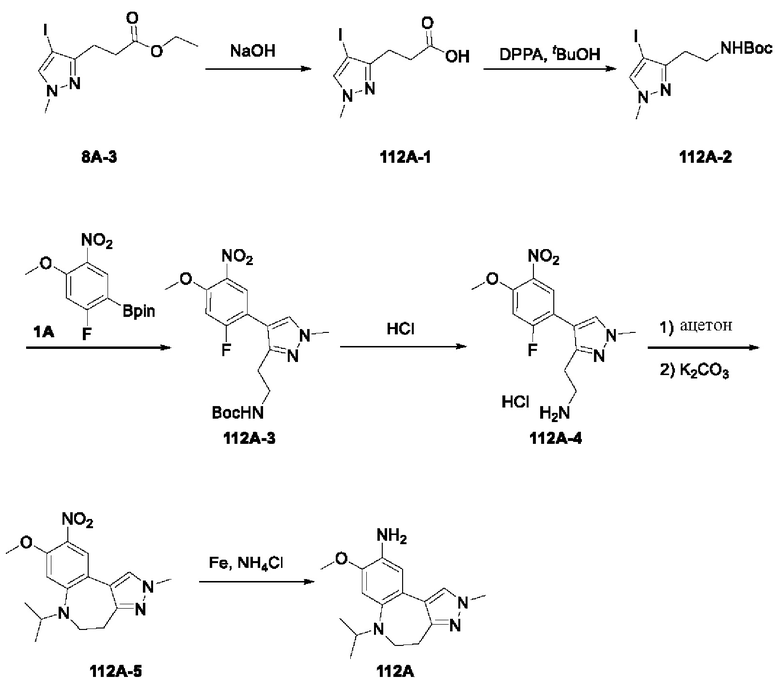
Соединение 112А-1:
Соединение 112А-1 (1,19 г) получали путем замены исходного материала на соединение 8А-3 (2,05 г, 6,65 ммоль) согласно способу получения соединения 1С-3 из соединения 1С-2 в примере 1.
МС (ИЭР, масса/заряд): 280,9 [М+Н]+.
Соединение 112А-2:
Соединение 112А-2 (952 мг) получали путем замены исходного материала на соединение 112А-1 (1,2 г, 4,29 ммоль) согласно способу получения соединения 1С-4 из соединения 1С-3 в примере 1.
МС (ИЭР, масса/заряд): 352,1 [М+Н]+.
Соединение 112А-3:
Соединение 112А-3 (885 мг) получали путем замены исходного материала на соединение 112А-2 (930 мг, 2,65 ммоль) согласно способу получения соединения 1С-7 из соединения 1С-6 в примере 1.
МС (ИЭР, масса/заряд): 395,2 [М+Н]+.
Соединение 112А-4:
Соединение 112А-4 (645 мг) получали путем замены исходного материала на соединение 112А-3 (885 мг, 2,24 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 295,1 [М+Н]+.
Соединение 112А-5:
Соединение 112А-5 (260 мг) получали путем замены исходного материала на соединение 112А-4 (330 мг, 1,12 ммоль) согласно способу получения соединения 56А-4 из соединения 56А-3 в примере 56.
МС (ИЭР, масса/заряд): 317,3 [М+Н]+.
Промежуточное соединение 112А:
Соединение 112А (160 мг) получали путем замены исходного материала на соединение 112А-5 (260 мг, 0,82 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 287,3 [М+Н]+.
Получение соединения 112
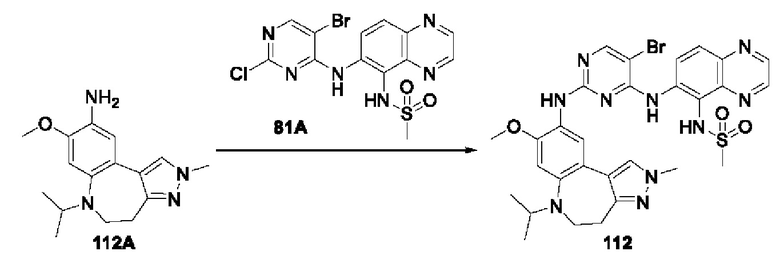
Соединение 112 (36 мг) получали путем замены исходного материала на соединение 112А (57 мг, 0,2 ммоль) и соединение 81А (86 мг, 0,2 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 679,2, 681,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 9,08 (s, 1H), 8,83 (d, J=2,0 Гц, 1H), 8,79 (d, J=2,0 Гц, 1H), 8,73 (d, J=9,6 Гц, 1H), 8,26 (s, 1H), 8,12 (s, 1H), 7,72 (s, 1H), 7,52 (s, 1H), 7,23 (s, 1H), 7,04 (s, 1H), 6,53 (s, 1H), 3,86 (s, 4H), 3,49 (s, 3Н), 3,33-3,27 (m, 2H), 3,10-3,02 (s, 2H), 2,93 (s, 3Н), 1,30 (d, J=6,8 Гц, 6H).
Пример 113:
Получение соединения 113
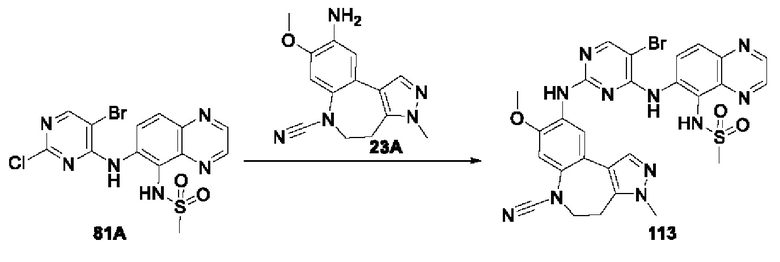
Соединение 113 (13 мг) получали путем замены исходного материала на соединение 81А (80 мг, 0,19 ммоль) и соединение 23А (50 мг, 0,19 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 662,1, 664,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,21 (s, 1H), 8,89-8,83 (m, 2Н), 8,49-8,46 (m, 2Н), 8,32 (s, 1H), 7,66-7,62 (m, 3Н), 6,88 (s, 1H), 6,61 (s, 1H), 3,93 (s, 3H), 3,82 (t, J=5,4 Гц, 2H), 3,68 (s, 3H), 3,18 (t, J=5,7 Гц, 2H), 2,98 (s, 3H).
Пример 114:
Получение соединения 114
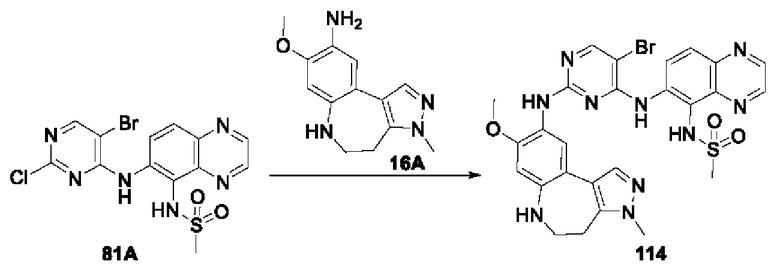
Соединение 114 (3 мг) получали путем замены исходного материала на соединение 81А (123 мг, 0,29 ммоль) и соединение 16А (70 мг, 0,29 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 637,2, 639,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,10 (s, 1Н), 8,82-8,76 (m, 2H), 8,55 (d, J=9,2 Гц, 1H), 8,23-8,17 (m, 2H), 7,71 (s, 1H), 7,57 (s, 1H), 7,37 (s, 1H), 6,67 (s, 1H), 6,25 (s, 1H), 3,82 (s, 3H), 3,64 (s, 3H), 3,46-3,37 (m, 2H), 2,98-2,94 (m, 5H).
Пример 115:
Получение соединения 115
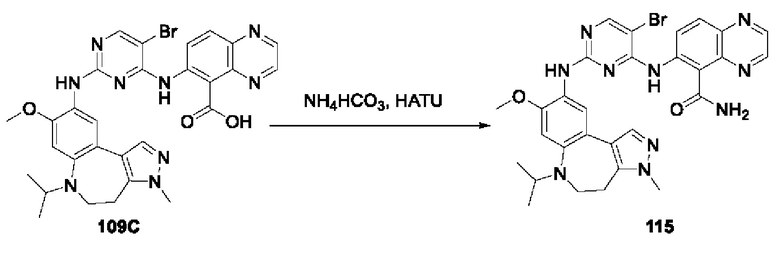
Соединение 115 (5 мг) получали путем замены исходного материала на соединение 109С (110 мг, 0,17 ммоль) и бикарбонат аммония (28 мг, 0,35 ммоль) согласно способу получения соединения 48А-5 из соединения 48А-4 в примере 48.
МС (ИЭР, масса/заряд): 629,1, 631,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 13,45 (s, 1H), 10,97 (s, 1H), 9,24 (d, J=9,6 Гц, 1H), 8,78-8,76 (m, 2H), 8,41 (s, 1H), 8,33 (s, 1H), 7,69-7,65 (m, 1H), 7,36 (s, 1H), 7,12 (s, 1H), 6,60 (s, 1H), 6,17 (s, 1H), 3,91 (s, 4H), 3,74 (s, 3H), 3,36-3,30 (m, 2H), 3,02-2,94 (m, 2H), 1,35 (s, 3H), 1,33 (s, 3H).
Пример 116:
Получение промежуточного соединения 116А
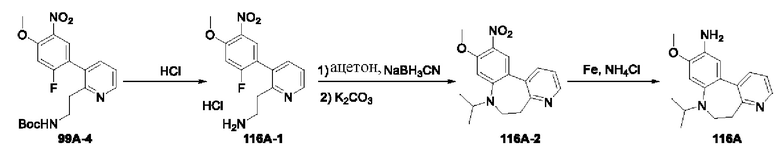
Соединение 116А-1:
Соединение 116А-1 (750 мг) получали путем замены исходного материала на соединение 99А-4 (1 г, 2,56 ммоль) согласно способу получения соединения 1С-8 из соединения 1С-7 в примере 1.
МС (ИЭР, масса/заряд): 291,1 [М+Н]+.
Соединение 116А-2:
Соединение 116А-2 (370 мг) получали путем замены исходного материала на соединение 116А-1 (500 мг, 1,53 ммоль) согласно способу получения соединения 56А-4 из соединения 56А-3 в примере 56.
МС (ИЭР, масса/заряд): 314,3 [М+Н]+.
Промежуточное соединение 116А:
Соединение 116А (55 мг) получали путем замены исходного материала на соединение 116А-2 (70 мг, 0,22 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 284,1 [М+Н]+.
Получение соединения 116
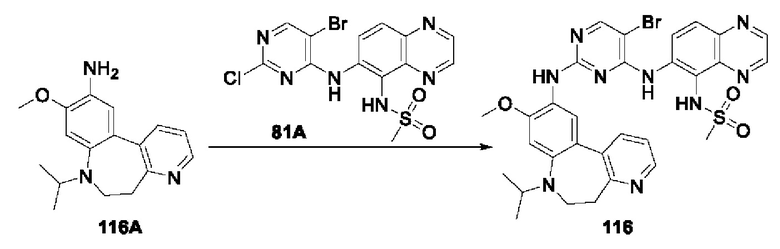
Соединение 116 (10 мг) получали путем замены исходного материала на соединение 116А (50 мг, 0,18 ммоль) и соединение 81А (76 мг, 0,18 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 676,3, 678,3 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,13 (s, 1H), 8,89 (d, J=2,1 Гц, 1H), 8,81 (d, J=2,1 Гц, 1H), 8,58 (d, J=9,3 Гц, 1H), 8,29 (s, 1H), 8,25 (d, J=4,8 Гц, 1H), 8,09 (s, 1H), 7,57 (d, J=10,5 Гц, 1H), 7,48 (s, 1H), 7,43 (s, 1H), 7,24 (d, J=8,1 Гц, 1H), 6,76 (s, 2H), 3,94 (s, 3H), 3,66-3,53 (m, 3H), 3,04 (t,J=6,6 Гц, 2H), 2,93 (s, 3H), 1,15 (s, 3H), 1,13 (s, 3H).
Пример 117:
Получение соединений 117A и 117B
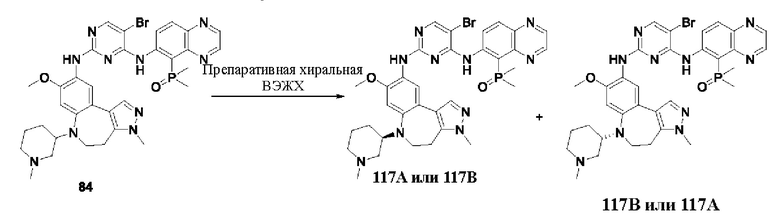
Соединение 84 (60 мг, 0,25 ммоль) получали с помощью хиральной колонки, при этом условия получения были следующими: CHIRAL ART Cellulose-SB, 2×25 см, 5 мкм; подвижная фаза А: н-гексан (10 мМ раствор аммиака в метаноле), и подвижная фаза В: этанол; скорость потока: 20 мл/мин; элюирование: повышение содержания подвижной фазы В до 20% в течение 16 минут; длина волны детектирования: 262 нм; (время удерживания)1: 10,7 минут; (время удерживания)2: 13,2 мин. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 117А (18 мг, энантиомерный избыток 100%) со временем удерживания 10,7 мин и соединения 117B (18 мг, энантиомерный избыток 98,4%) со временем удерживания 13,2 мин.
117А: МС (ИЭР, масса/заряд): 717,2, 719,2 [М+Н]+.
117B: МС (ИЭР, масса/заряд): 717,2, 719,2 [М+Н]+.
117А: 1H ЯМР (300 МГц, CDCl3) δ 12,49 (s, 1H), 8,88 (dd, J=9,0 Гц, 4,8 Гц, 1H), 8,72 (d, J=1,8 Гц, 1H), 8,69 (d, J=1,8 Гц, 1H), 8,44 (s, 1H), 8,31 (s, 1H), 7,66 (d, J=9,9 Гц, 1H), 7,38 (s, 1H), 6,99 (s, 1Н), 6,67 (s, 1Н), 4,04-3,88 (m, 4Н), 3,73 (s, 3Н), 3,50-3,38 (m, 3Н), 3,31-3,23 (m, 1H), 3,02 (t, J=5,4 Гц, 2Н), 2,59 (s, 3Н), 2,37-2,27 (m, 2Н), 2,18 (s, 3Н), 2,14 (s, 3Н), 2,09-2,00 (m, 2Н), 1,95-1,85 (m, 1H), 1,61-1,44 (m, 1Н).
117B: 1Н ЯМР (300 МГц, CDCl3) δ 12,52 (s, 1H), 8,89 (dd, J=9,0 Гц, 4,8 Гц, 1H), 8,73 (d, J=1,8 Гц, 1H), 8,70 (d, J=1,8 Гц, 1H), 8,42 (s, 1Н), 8,31 (s, 1H), 7,64 (d, J=9,9 Гц, 1H), 7,36 (s, 1H), 6,98 (s, 1Н), 6,69 (s, 1Н), 4,00-3,86 (m, 4Н), 3,73 (s, 3Н), 3,51-3,39 (m, 3Н), 3,30-3,21 (m, 1H), 3,04 (t, J=5,4 Гц, 2Н), 2,56 (s, 3Н), 2,37-2,25 (m, 2Н), 2,16 (s, 3Н), 2,14 (s, 3Н), 2,09-2,02 (m, 2Н), 1,92-1,83 (m, 1H), 1,60-1,43 (m, 1Н).
Пример 118:
Получение промежуточного соединения 118А
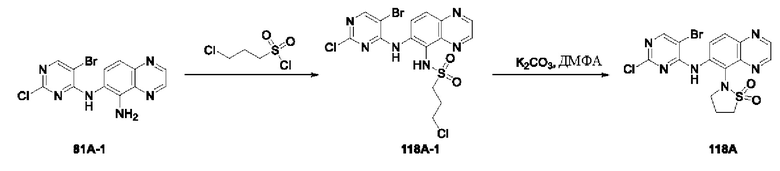
Соединение 118А-1:
Соединение 81А-1 (600 мг, 1,71 ммоль) растворяли в пиридине (25 мл). Затем к реакционной жидкости добавляли при 0°С 3-хлорпропилсульфонилхлорид (453 мг, 2,56 ммоль). Реакционную систему нагревали до 40°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и дистиллировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 118А-1 (456 мг).
МС (ИЭР, масса/заряд): 490,9, 492,9, 494,9 [М+Н]+.
Соединение 118А:
Соединение 118А-1 (200 мг, 0,41 ммоль) и безводный карбонат калия (112 мг, 0,81 ммоль) растворяли в N,N-диметилформамиде (3 мл). Реакционную систему нагревали до 50°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 118А (150 мг). МС (ИЭР) масса/заряд: 454,9, 456,9 [М+Н]+.
Получение соединения 118
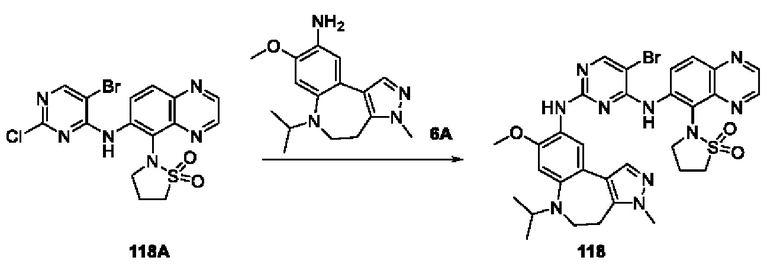
Соединение 118 (25 мг) получали путем замены исходного материала на соединение 118А (42 мг, 0,09 ммоль) и соединение 6А (32 мг, 0,11 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 705,2, 707,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 8,94 (s, 1Н), 8,90-8,79 (m, 2H), 8,73 (d, J=1,8 Гц, 1H), 8,28 (s, 2H), 7,63 (s, 1H), 7,41-7,34 (m, 1H), 7,11 (s, 1H), 6,59 (s, 1H), 4,39-4,27 (m, 1H), 3,90 (s, 6H), 3,74 (s, 3H), 3,55-3,46 (m, 2H), 3,22-3,93 (m, 3H), 2,81-2,64 (m, 2H), 1,33 (s, 6H).
Пример 119:
Получение промежуточного соединения 119А
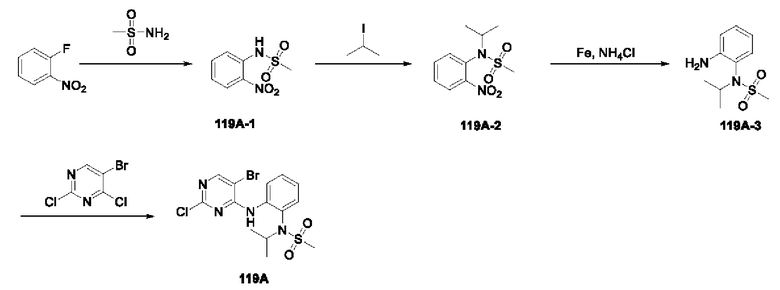
Соединение 119А-1:
о-Нитрофторбензол (4 г, 28,35 ммоль) и метилсульфонамид (4,04 г, 42,52 ммоль) растворяли в N,N-диметилформамиде (20 мл) при комнатной температуре в атмосфере азота. Затем к реакционной жидкости отдельными порциями добавляли трет-бутоксид калия (6,36 г, 56,7 ммоль), после чего реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 119А-1 (3 г).
МС (ИЭР) масса/заряд: 215,1 [М-Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,79 (s, 1H), 8,29 (dd, J=8,4, 1,5 Гц, 1H), 7,91 (dd,J=8,4, 1,5 Гц, 1H), 7,74-7,68 (m, 1H), 7,29-7,23 (m, 1H), 3,18 (s, 3Н).
Соединение 119А-2:
Соединение 119А-1 (3 г, 13,88 ммоль) и 2-йодпропан (7,08 г, 41,63 ммоль) растворяли в N,N-диметилформамиде (20 мл). Затем к реакционной жидкости добавляли карбонат цезия (13,56 г, 41,63 ммоль), после чего реакционную систему нагревали до 80°С и последовательно перемешивали в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении; полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 119А-2 (2,5 г).
1H ЯМР (300 МГц, CDCl3) δ 7,90 (dd, J=7,8, 1,8 Гц, 1Н), 7,65 (dd, J=7,8, 1,8 Гц, 1H), 7,58 (dd, J=7,8, 1,8 Гц, 1H), 7,51 (dd, J=7,8, 1,5 Гц, 1H), 4,36-4,27 (m, 1H), 3,15 (s, 3Н), 1,32 (d, J=6,6 Гц, 3Н), 1,09 (d, J=6,6 Гц, 3Н).
Соединение 119А-3:
Соединение 119А-3 (1,85 г) получали путем замены исходного материала на соединение 119А-2 (2,5 г, 8,96 ммоль) согласно способу получения соединения 1С из промежуточного соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 229,2 [М+Н]+.
Промежуточное соединение 119А:
Соединение 119А (200 мг) получали путем замены исходного материала на соединение 119А-3 (1,5 г, 6,57 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
MC (ИЭР, масса/заряд): 418,9, 420,9 [М+Н]+.
Получение соединения 119
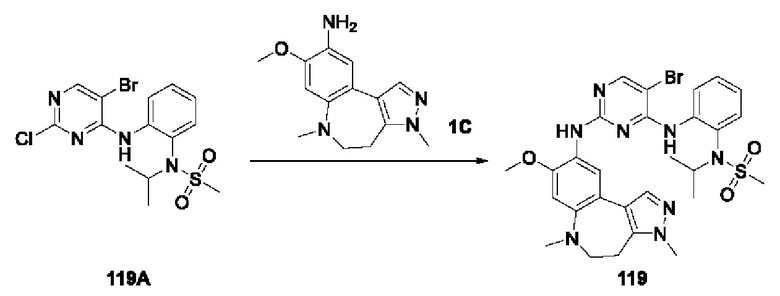
Соединение 119 (5 мг) получали путем замены исходного материала на соединение 119А (40 мг, 0,095 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 641,2, 643,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (s, 1H), 8,34-8,31 (m, 2Н), 8,15 (s, 1H), 7,68-7,44 (m, 2H), 7,27 (s, 1H), 7,10-7,02 (m, 2H), 6,56 (s, 1H), 4,69-4,62 (m, 1H), 3,89 (s, 3H), 3,83 (s, 3H), 3,33-3,25 (m, 2H), 3,08-2,98 (m, 8H), 1,32 (d, J=6,8 Гц, 3Н), 1,06 (d, J=6,8 Гц, 3Н).
Пример 120:
Получение промежуточного соединения 120А
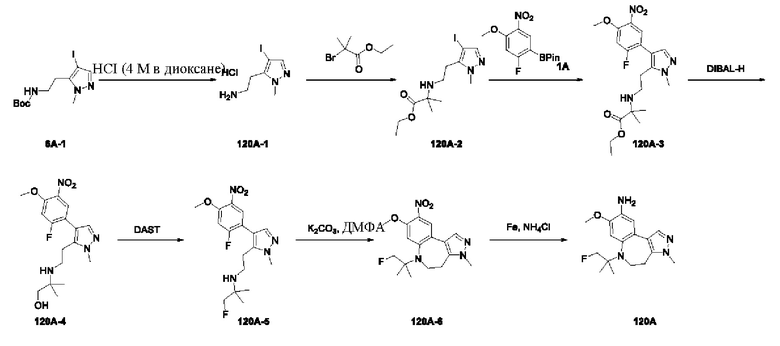
Соединение 120А-1:
Соединение 6А-1 (5 г, 14,24 ммоль) растворяли в дихлорметане (40 мл) и добавляли к реакционной жидкости раствор хлористого водорода в диоксане (4 М, 40 мл). Реакционную систему последовательно перемешивали при комнатной температуре в течение 1,5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении с получением соединения 120А-1 (3,6 г).
МС (ИЭР, масса/заряд): 252,2 [М+Н]+.
Соединение 120А-2:
Соединение 120А-1 (500 мг, 1,74 ммоль) растворяли в ацетонитриле (5 мл). Затем к реакционной жидкости добавляли безводный карбонат калия (719 мг, 5,2 ммоль), иодид натрия (131 мг, 0,87 ммоль) и этил-2-бромизобутаноат (679 мг, 3,48 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/этилацетат = 1/1) с получением соединения 120А-2 (381 мг).
МС (ИЭР, масса/заряд): 366,1 [М+Н]+.
Соединение 120А-3:
Соединение 120А-2 (500 мг, 1,68 ммоль) и соединение 1А (827,47 мг, 2,36 ммоль) и карбонат калия (465 мг, 3,37 ммоль) растворяли в смешанном растворителе, состоящем из 1,4-диоксана (5 мл) и воды (1 мл), в атмосфере азота. Затем к реакционной жидкости добавляли бутилди-1-адамантилфосфин (60 мг, 0,17 ммоль) и хлор[(ди(1-адамантил)-N-бутилфосфин)-2-(2-аминобифенил)]палладий (II) (113 мг, 0,17 ммоль). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 3 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/этилацетат = 1/1) с получением соединения 120А-3 (460 мг).
МС (ИЭР, масса/заряд): 409,2 [М+Н]+.
Соединение 120А-4:
Соединение 120А-3 (2 г, 4,9 ммоль) растворяли в дихлорметане (30 мл) в атмосфере азота. К реакционной жидкости добавляли раствор гидрида диизобутилалюминия в толуоле (1,5 М, 8,17 мл, 12,25 ммоль) при -78°С и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 часов. Реакционную систему медленно нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора хлорида аммония (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл х 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 120А-4 (0,75 г).
МС (ИЭР, масса/заряд): 367,1 [М+Н]+.
Соединение 120А-5:
Соединение 120А-4 (700 мг, 1,91 ммоль) растворяли в дихлорметане (7 мл) в атмосфере азота. К реакционной жидкости медленно по каплям добавляли трифторид диэтиламиносеры (3080 мг, 19,11 ммоль) при -78°С и последовательно перемешивали реакционную жидкость при указанной температуре в течение 30 минут. Реакционную систему медленно нагревали до комнатной температуры и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления насыщенного водного раствора бикарбоната натрия (20 мл). Смесь экстрагировали этилацетатом (20 мл × 3), органические фазы объединяли, сначала промывали насыщенным солевым раствором (20 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 120А-5 (164 мг).
МС (ИЭР, масса/заряд): 369,4 [М+Н]+.
Соединение 120А-6:
Соединение 120А-6 (104 мг) получали путем замены исходного материала на соединение 120А-5 (150 мг, 0,41 ммоль) согласно способу получения соединения 1С-9 из соединения 1С-8 в примере 1.
МС (ИЭР, масса/заряд): 349,1 [М+Н]+.
Промежуточное соединение 120А:
Соединение 120А (86 мг) получали путем замены исходного материала на соединение 120А-6 (104 мг, 0,3 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 319,1 [М+Н]+.
Получение соединения 120
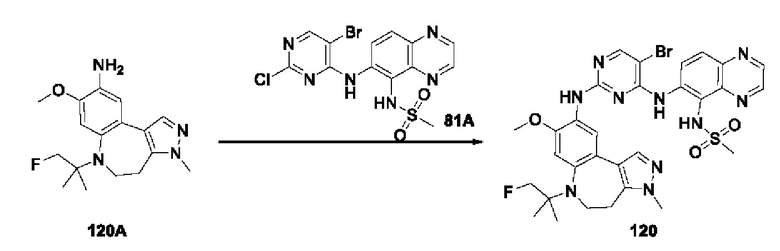
Соединение 120А (60 мг, 0,19 ммоль) и соединение 81А (73 мг, 0,17 ммоль) растворяли в этаноле (1,5 мл). Реакционную жидкость нагревали до 150°С с помощью СВЧ-реактора и последовательно перемешивали в течение 40 минут. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1) с получением соединения 120 (26 мг).
МС (ИЭР) масса/заряд: 711,2, 713,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,14 (s, 1H), 8,84-8,81 (m, 2H), 8,60 (d, J=9,3 Гц, 1H), 8,28 (s, 2H), 7,60 (s, 1H), 7,55 (s, J=9,3 Гц, 1H), 7,40 (s, 1H), 6,82 (s, 1H), 6,59 (s, 1H), 3,88 (s, 3H), 3,69 (s, 3H), 3,53 (s, 1H), 3,46-3,42 (m, 3H), 3,10-3,02 (m, 2H), 2,95 (s, 3H), 1,35 (s, 3H), 1,28 (s, 3H).
19F ЯМР (282 МГц, CDCl3) δ -140,58.
Пример 121:
Получение промежуточного соединения 121А
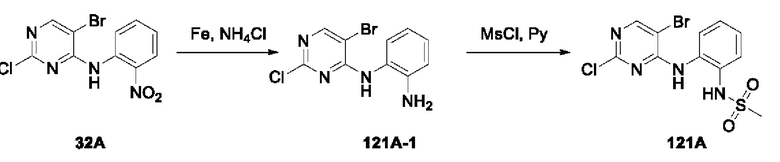
Соединение 121А-1:
Соединение 121А-1 (380 мг) получали путем замены исходного материала на соединение 32А (500 мг, 1,52 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 298,9, 300,9 [М+Н]+.
Соединение 121А:
Соединение 121А (150 мг) получали путем замены исходного материала на соединение 121А-1 (200 мг, 0,67 ммоль) согласно способу получения соединения 32 из соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 376,9, 378,9 [М+Н]+.
Получение соединения 121
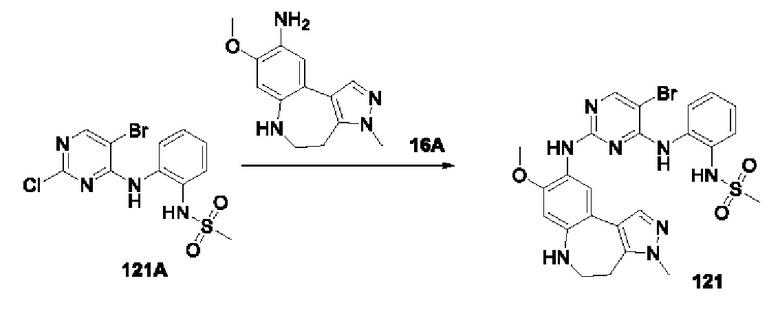
Соединение 121 (22 мг) получали путем замены исходного материала на соединение 121А (70 мг, 0,19 ммоль) и соединение 16А (54 мг, 0,22 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 585,1, 587,1 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 10,02 (br s, 1H), 8,34 (s, 1H), 8,16-8,12 (m, 3Н), 7,65 (s, 1H), 7,56 (s, 1H), 7,28 (dd, J=7,8, 1,8 Гц, 1H), 7,03-6,92 (m, 2Н), 6,51 (s, 1Н), 5,85 (s, 1H), 3,73 (s, 3Н), 3,67 (s, 3Н), 3,27-3,21 (m, 2Н), 2,98 (s, 3Н), 2,96-2,94 (m, 2Н).
Пример 122:
Получение соединения 122
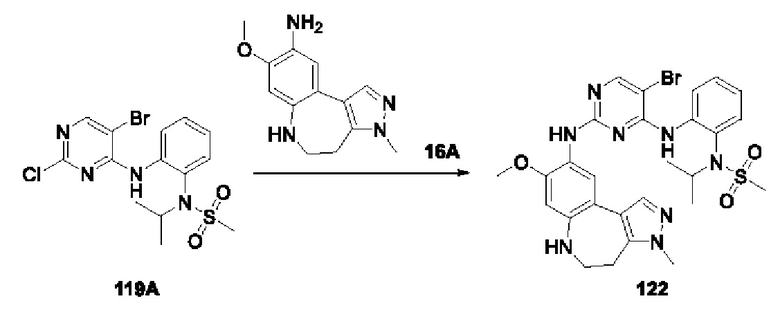
Соединение 122 (15 мг) получали путем замены исходного материала на соединение 119А (60 мг, 0,14 ммоль) и соединение 16А (35 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 627,2, 629,2 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 8,34 (s, 1H), 8,25 (s, 1Н), 8,18 (s, 1H), 8,07 (s, 1Н), 7,67 (s, 1H), 7,52 (s, 1Н), 7,41-7,38 (m, 1Н), 7,09-7,02 (m, 2Н), 6,54 (s, 1H), 5,88 (dd, J=4,2 Гц, 1H), 4,49-4,40 (m, 1H), 3,73 (s, 3Н), 3,68 (s, 3Н), 3,29-3,24 (m, 2Н), 3,14 (s, 3Н), 2,99-2,95 (m, 2Н), 1,18 (d, J=6,9 Гц, 3Н), 0,98 (d, J=6,9 Гц, 3Н).
Пример 209:
Получение соединения 209
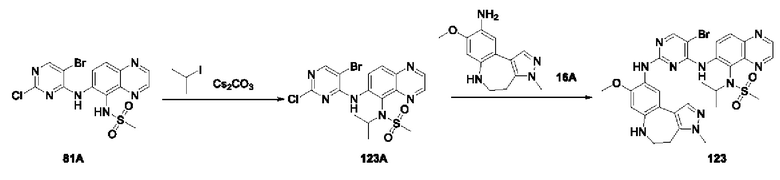
Соединение 123А:
Соединение 81А (200 мг, 0,47 ммоль) и 2-йодпропан (237 мг, 1,39 ммоль) растворяли в диметилсульфоксиде (3 мл). Затем к реакционной жидкости добавляли карбонат цезия (455 мг, 1,4 ммоль), после чего реакционную систему нагревали до 80°С и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и непосредственно очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 10% до 60% в течение 30 минут; длина волны детектирования 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 123А (70 мг).
МС (ИЭР) масса/заряд: 470,9, 472,9 [М+Н]+.
Соединение 123:
Соединение 123 (20 мг) получали путем замены исходного материала на соединение 123А (70 мг, 0,15 ммоль) и соединение 16А (36 мг, 0,15 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 679,2, 681,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 8,82-8,74 (s, 3Н), 8,64 (d, J=6,9 Гц, 1H), 8,22 (s, 2Н), 7,83-7,63 (m, 2Н), 6,86 (s, 1H), 6,31 (s, 1Н), 4,54-4,47 (m, 1H), 3,84 (s, 3Н), 3,68 (s, 3Н), 3,57-3,49 (m, 1H), 3,42-3,35 (m, 1H), 3,23 (s, 3Н), 3,06-2,97 (m, 2H), 1,35 (d, J=4,8 Гц, 3Н), 1,26 (d, J=4,8 Гц, 3Н).
Пример 124:
Получение соединения 124
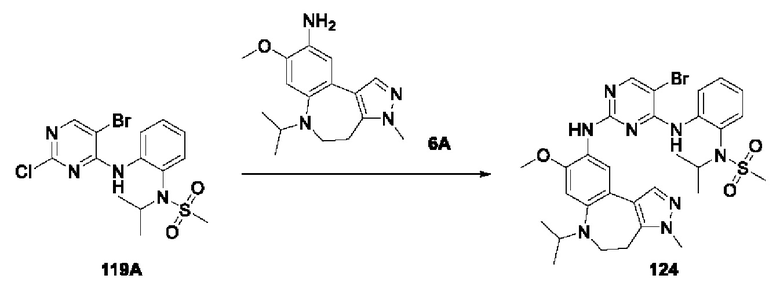
Соединение 124 (2 мг) получали путем замены исходного материала на соединение 119А (60 мг, 0,14 ммоль) и соединение 6А (41 мг, 0,14 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 669,3, 671,3 [М+Н]+.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,44-8,38 (m, 1H), 8,35-8,30 (m, 1H), 8,28 (s, 1H), 8,19 (s, 1H), 7,75 (s, 1H), 7,46 (s, 1H), 7,41-7,378 (m, 1H), 7,07-7,01 (m, 2Н), 6,58 (s, 1H), 4,49-4,43 (m, 1H), 3,98-3,92 (m, 1H), 3,75 (s, 6Н), 3,24-3,19 (m, 2Н), 3,13 (s, 3Н), 2,96-2,93 (m, 2Н), 1,29 (d, J=6,6 Гц, 6Н), 1,19 (d, J=6,6 Гц, 3Н), 0,98 (d, J=6,6 Гц, 3Н).
Пример 125:
Получение соединения 125
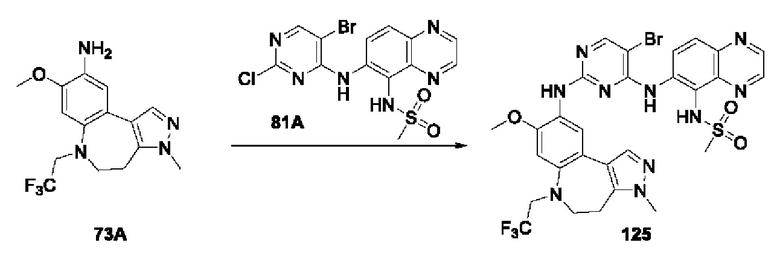
Соединение 125 (3 мг) получали путем замены исходного материала на соединение 73А (32 мг, 0,1 ммоль) и соединение 81А (42 мг, 0,1 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 719,1, 721,1 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 9,98 (s, 1H), 8,95 (s, 1H), 8,86-8,82 (m, 2Н), 8,72-8,59 (s, 1Н), 8,37 (s, 1H), 8,29 (s, 1H), 7,80 (s, 1Н), 7,57-7,46 (s, 1Н), 7,38 (s, 1H), 6,88 (s, 1H), 4,34-4,27 (m, 2Н), 3,79 (s, 3Н), 3,71 (s, 3Н), 3,42-3,40 (m, 2Н), 3,03-3,01 (m, 5Н).
19F ЯМР (377 МГц, ДМСО-d6) δ -69,58.
Пример 126:
Получение промежуточного соединения 126А
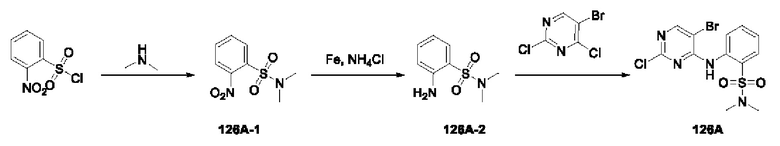
Соединение 126А-1:
Безводный карбонат натрия (3,59 г, 33,87 ммоль) добавляли к раствору диметиламина в тетрагидрофуране (2 М, 13,5 мл). Затем к реакционной жидкости отдельными порциями добавляли о-нитробензолсульфонилхлорид (5 г, 22,56 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении с получением соединения 126А-1 (4,6 г).
МС (ИЭР, масса/заряд): 231,1 [М+Н]+.
Соединение 126А-2:
Соединение 126А-2 (3,5 г) получали путем замены исходного материала на соединение 126А-1 (4,6 г, 19,98 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 201,1 [М+Н]+.
Промежуточное соединение 126А:
Соединение 126А (100 мг) получали путем замены исходного материала на соединение 126А-2 (500 мг, 2,5 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 391,0, 393,0 [М+Н]+.
Получение соединения 126
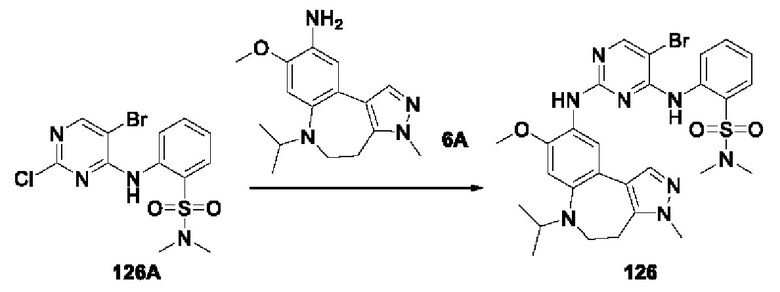
Соединение 126 (43 мг) получали путем замены исходного материала на соединение 126А (70 мг, 0,18 ммоль) и соединение 6А (56 мг, 0,18 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 641,3, 643,3 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (s, 1H), 8,44-8,22 (m, 3Н), 7,84 (s, 1Н), 7,54-7,32 (m, 1H), 7,25-7,07 (m, 3H), 6,55 (s, 1H), 3,97-3,63 (m, 7H), 3,39-3,15 (m, 2H), 3,00-2,86 (m, 2H), 2,76 (s, 6H), 1,30 (s, 6H).
Пример 212:
Получение промежуточного соединения 212А
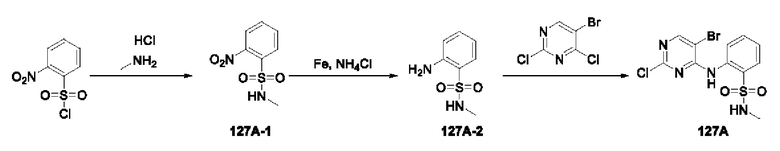
Соединение 127А-1:
Триэтиламин (11,42 г, 112,81 ммоль) добавляли к гидрохлориду метиламина (4,57 г, 67,69 ммоль) в дихлорметане (100 мл). Затем к реакционной жидкости отдельными порциями добавляли о-нитробензолсульфонилхлорид (5 г, 22,56 ммоль) и последовательно перемешивали указанную реакционную жидкость при комнатной температуре в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость гасили путем добавления воды (100 мл). Смесь экстрагировали этилацетатом (100 мл × 3), после чего органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении с получением соединения 127А-1 (4,5 г).
МС (ИЭР, масса/заряд): 217,1 [М+Н]+.
Соединение 127А-2:
Соединение 127А-2 (2,2 г) получали путем замены исходного материала на соединение 127А-1 (2,7 г, 12,49 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 187,1 [М+Н]+.
Соединение 127А:
5-Бром-2,4-дихлорпиримидин (1224 мг, 5,37 ммоль) и соединение 127А-2 (500 мг, 2,69 ммоль) растворяли в N,N-диметилформамиде (5 мл); затем к реакционной жидкости добавляли безводный карбонат калия (1113 мг, 8,05 ммоль). Реакционную жидкость нагревали до 45°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 3), затем высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: этилацетат) с получением соединения 127А (450 мг).
МС (ИЭР) масса/заряд: 377,0, 379,0 [М+Н]+.
Получение соединения 127
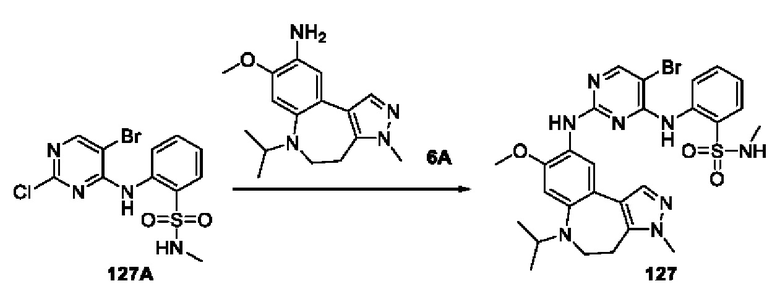
Соединение 127 (8 мг) получали путем замены исходного материала на соединение 127А (100 мг, 0,27 ммоль) и соединение 6А (114 мг, 0,4 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 627,3, 629,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,03 (s, 1H), 8,39 (d, J=8,4 Гц, 1H), 8,15 (s, 1H), 7,92 (dd, J=8,0, 1,6 Гц, 1H), 7,23 (s, 1H), 7,19 (s, 1H), 7,04-7,00 (m, 2H), 6,55 (s, 1H), 5,45 (s, 1H), 4,72 (s, 1H), 3,94-3,89 (m, 1H), 3,87 (s, 3H), 3,79 (s, 3H), 3,28-3,25 (m, 2H), 2,93-2,90 (m, 2H), 2,64 (d, J=4,2 Гц, 3Н), 1,30 (d, J=6,4 Гц, 6H).
Пример 128:
Получение промежуточного соединения 128А
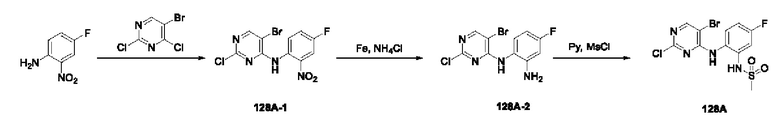
Соединение 128А-1:
Соединение 128А-1 (8,5 г) получали путем замены исходного материала на 2-нитро-4-фторанилин (5 г, 32,05 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР, масса/заряд): 347,0, 349,0 [М+Н]+.
Соединение 128-2:
Соединение 128А-2 (6,2 г) получали путем замены исходного материала на соединение 128А-1 (8,5 г, 24,4 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 317,0, 319,0 [М+Н]+.
Соединение 128А:
Соединение 128А (360 мг) получали путем замены исходного материала на соединение 128А-2 (400 мг, 1,27 ммоль) согласно способу получения соединения 32 из соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 395,0, 397,0 [М+Н]+.
Получение соединения 128
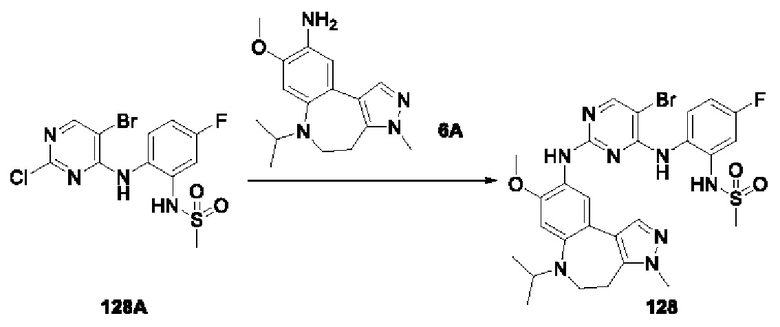
Соединение 128 (40 мг) получали путем замены исходного материала на соединение 128А (70 мг, 0,18 ммоль) и соединение 6А (51 мг, 0,18 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 645,1, 647,1 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 10,03 (br s, 1H), 8,28 (s, 1H), 8,13 (s, 1H), 8,00 (s, 1H), 7,89-7,84 (m, 1H), 7,72 (s, 1H), 7,45 (s, 1H), 7,13 (dd, J=9,9, 3,0 Гц, 1H), 6,62 (s, 1H), 6,52 (s, 1Н), 3,95-3,86 (m, 1H), 3,75 (s, 3Н), 3,73 (s, 3Н), 3,20-3,17 (m, 2Н), 2,97 (s, 3Н), 2,94-2,91 (m, 2Н), 1,27 (d,J=6,3 Гц, 6Н).
19F ЯМР (282 МГц, ДМСО-d6) δ -117,35.
Пример 129:
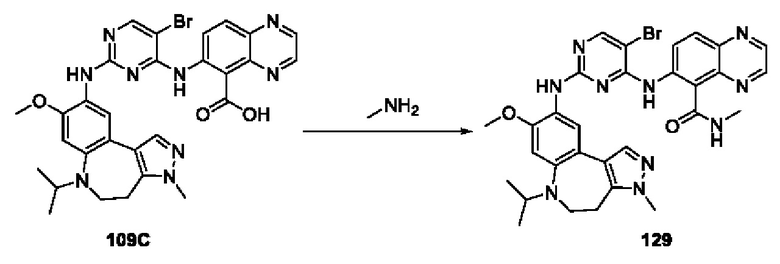
Соединение 129 (9 мг) получали путем замены исходного материала на соединение 109С (100 мг, 0,16 ммоль) согласно способу получения соединения 48А-5 из соединения 48А-4 в примере 48.
МС (ИЭР, масса/заряд): 643,2, 645,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,05 (s, 1Н), 10,14 (d, J=4,8 Гц, 1H), 9,01-8,87 (m, 3Н), 8,29 (s, 1H), 7,73-7,49 (m, 3Н), 6,80 (s, 1Н), 6,60 (s, 1H), 3,99-3,92 (m, 1H), 3,77 (s, 3Н), 3,71 (s, 3Н), 3,23-3,20 (m, 2Н), 2,95-2,93 (m, 5Н), 1,29 (d, J=6,4 Гц, 6Н).
Пример 215:
Получение соединения 215
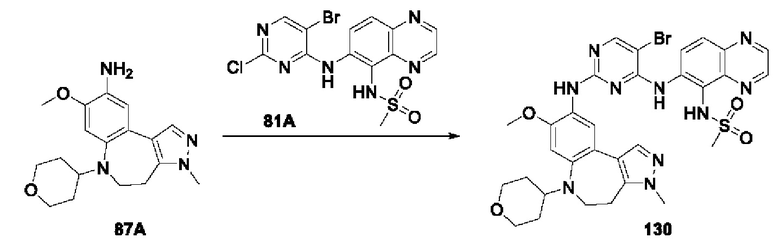
Соединение 130 (42 мг) получали путем замены исходного материала на соединение 87А (100 мг, 0,31 ммоль) и соединение 81А (81 мг, 0,19 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 721,2, 723,2 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,25 (s, 1Н), 8,86-8,82 (m, 2H), 8,54 (d, J=9,0 Гц, 1H), 8,36-8,19 (m, 2H), 7,85-7,50 (m, 3Н), 6,74 (s, 1H), 6,52 (s, 1H), 4,39-4,03 (m, 3H), 3,90 (m, 3H), 3,67 (s, 3H), 3,53-3,34 (m, 5H), 3,09-2,80 (m, 4H), 2,15-1,78 (m, 4H).
Пример 131:
Получение промежуточного соединения 131А
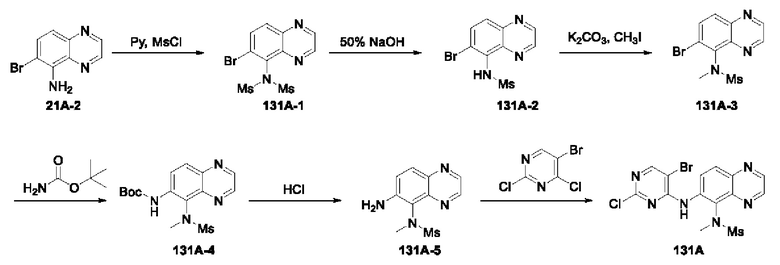
Соединение 131А-1:
Соединение 21А-2 (5,8 г, 25,89 ммоль) растворяли в пиридине (60 мл). Затем к реакционной жидкости добавляли метансульфонилхлорид (14,82 г, 129,43 ммоль), после чего реакционную систему нагревали до 50°С и последовательно перемешивали в течение 5 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением соединения 131А-1 (5,1 г).
1Н ЯМР (300 МГц, ДМСО-d6) δ 9,13-9,09 (m, 2Н), 8,28 (d, J=9,0 Гц, 1H), 8,22 (d, J=9,0 Гц, 1H), 3,71-3,67 (m, 6Н).
Соединение 131А-2:
Соединение 131A-1 (8 г, 21 ммоль) растворяли в смешанном растворителе, состоящем из метанола (80 мл) и тетрагидрофурана (80 мл). Затем к описанной выше реакционной жидкости добавляли 50% водный раствор гидроксида натрия (33,6 г, 420 ммоль) при 0°С Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, к реакционной жидкости добавляли воду (200 мл) для разбавления и доводили до рН=6 с помощью концентрированной соляной кислоты. Смесь экстрагировали смешанным растворителем, состоящим из хлороформа/изопропанола 3:1 (80 мл × 3). Органические фазы объединяли, сначала промывали насыщенным солевым раствором (300 мл × 2), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 5/1) с получением соединения 131А-2 (6 г).
1H ЯМР (300 МГц, ДМСО-d6) δ 9,83 (br s, 1H), 9,09-9,01 (m, 2Н), 8,15 (d, J=9,0 Гц, 1Н), 8,01 (d, J=9,0 Гц, 1Н), 3,31 (s, 3Н).
Соединение 131А-3:
Соединение 131А-3 (3 г) получали путем замены исходного материала на соединение 131А-2 (3 г, 9,9 ммоль) согласно способу получения соединения 21А-5 из соединения 21А-4 в примере 21.
1H ЯМР (300 МГц, CDCl3) δ 8,92-8,90 (m, 2Н), 8,06-8,02 (m, 2Н), 3,41 (s, 3Н), 3,30 (s, 3Н).
Соединение 131А-4:
Соединение 131А-4 (4 г) получали путем замены исходного материала на соединение 131А-3 (2,8 г, 8,8 ммоль) согласно способу получения соединения 21А-6 из соединения 21А-5 в примере 21.
1Н ЯМР (300 МГц, CDCl3) δ 8,82 (d, J=9,6 Гц, 1H), 8,79-8,77 (m, 2Н), 8,10 (d, J=9,6 Гц, 1H), 7,87 (br s, 1H), 3,45 (s, 3H), 3,14 (s, 3H), 1,45 (s, 9H).
Соединение 131A-5:
Соединение 131A-5 (0,68 г) получали путем замены исходного материала на соединение 131А-4 (1 г, 2,8 ммоль) согласно способу получения соединения 21А-7 из соединения 21А-6 в примере 21.
МС (ИЭР, масса/заряд): 253,2 [М+Н]+.
Промежуточное соединение 131А:
Соединение 131А (1 г) получали путем замены исходного материала на соединение 131А-5 (650 мг, 2,6 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
1Н ЯМР (300 МГц, ДМСО-d6) δ 9,11 (s, 1H), 9,05-8,97 (m, 2Н), 8,66 (s, 1H), 8,56 (d, J=9,6 Гц, 1H), 8,25 (d, J=9,3 Гц, 1H), 3,41 (s, 3Н), 3,19 (s, 3Н).
Получение соединения 131
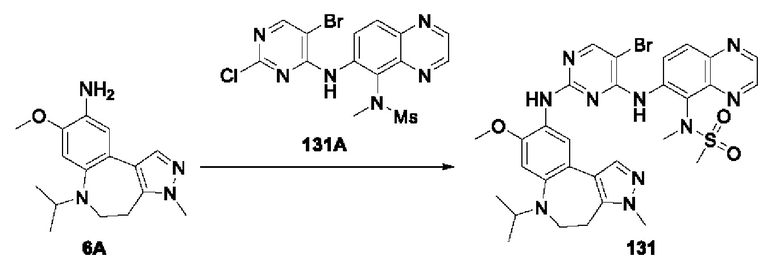
Соединение 131 (42 мг) получали путем замены исходного материала на соединение 131А (80 мг, 0,18 ммоль) и соединение 6А (54 мг, 0,19 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 693,2, 695,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,93 (d, J=2,0 Гц, 1H), 8,86 (d, J=2,0 Гц, 1Н), 8,78-8,70 (m, 1Н), 8,51 (s, 1H), 8,46 (s, 1Н), 8,29 (s, 1Н), 7,73 (s, 1H), 7,61-7,50 (m, 1H), 7,38 (s, 1Н), 6,60 (s, 1H), 3,99-3,93 (m, 1H), 3,77 (s, 3Н), 3,69 (s, 3Н), 3,38 (s, 3Н), 3,23-3,21 (m, 5Н), 2,94-2,91 (m, 2Н), 1,31-1,29 (m, 6Н).
Пример 132:
Получение промежуточного соединения 132А
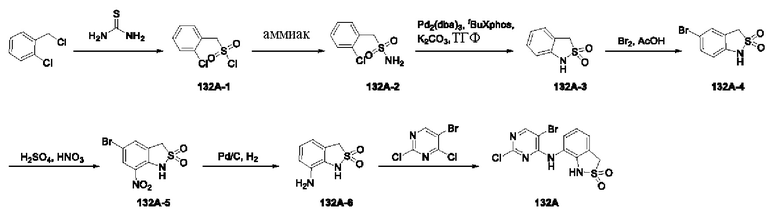
Соединение 132А-1:
Тиомочевину (7,09 г, 93,15 ммоль) растворяли в этаноле (75 мл). Затем к реакционной жидкости добавляли (2-хлорфенил)метансульфонилхлорид (15 г, 93,15 ммоль), после чего указанную реакционную жидкость нагревали до 80°С и последовательно перемешивали в течение 1 часа. Реакционную жидкость охлаждали до комнатной температуры и концентрировали при пониженном давлении, затем полученный остаток добавляли к ацетонитрилу (120 мл), содержащему раствор N-хлорсукцинимида (49,75 г, 372,6 ммоль) и хлористого водорода в этаноле (2 М, 40 мл, 700,7 ммоль). Реакционную жидкость последовательно перемешивали при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1) с получением соединения 132А-1 (20 г).
1H ЯМР (300 МГц, CDCl3) δ 7,64-7,61 (m, 1H), 7,55-7,52 (m, 1H), 7,48-7,39 (m, 2H), 5,14 (s, 2H).
Соединение 132А-2:
Соединение 132А-1 (20 г, 88,86 ммоль) растворяли в ацетоне (150 мл); затем к реакционной жидкости добавляли при 0°С водный раствор аммиака (100 мл). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость концентрировали при пониженном давлении. Реакционную жидкость доводили до рН=2 с помощью концентрированной соляной кислоты и экстрагировали полученную смесь с применением смешанного растворителя, содержащего этилацетат (300 мл × 3). Органические фазы объединяли, сначала промывали насыщенным солевым раствором (300 мл × 2), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении с получением соединения 132А-2 (14,5 г).
1H ЯМР (300 МГц, ДМСО-d6) δ 7,54-7,47 (m, 2Н), 7,41-7,36 (m, 2Н), 7,04 (s, 2Н), 4,46 (s, 2Н).
Соединение 132А-3:
В атмосфере азота соединение 132-2 (7,5 г, 36,47 ммоль), безводный карбонат калия (10,08 г, 72,94 ммоль), трис(дибензилиденацетон)дипалладий (3,34 г, 3,65 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (3,1 г, 7,29 ммоль) растворяли в тетрагидрофуране (100 мл). Реакционную систему нагревали до 80°С и последовательно перемешивали в течение 16 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до комнатной температуры и гасили путем добавления воды (100 мл). Смесь экстрагировали смешанным растворителем, содержащим этилацетат (150 мл × 3). Органические фазы объединяли, сначала промывали насыщенным солевым раствором (100 мл × 2), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1) с получением соединения 132А-3 (4,5 г).
1Н ЯМР (300 МГц, CDCl3) δ 7,33-7,26 (m, 2H), 7,11-7,06 (m, 1H), 6,93-6,90 (m, 1H), 6,58 (s, 1H), 4,41 (s, 2H).
Соединение 132A-4:
Соединение 132A-3 (500 мг, 2,96 ммоль) растворяли в ледяной уксусной кислоте (5 мл). К реакционной жидкости добавляли при 0°С бром (496 мг, 3,1 ммоль). Реакционную систему нагревали до комнатной температуры и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ТСХ, реакционную жидкость непосредственно очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 40% до 60% в течение 30 минут; длина волны детектирования 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 132А-4 (450 мг).
МС (ИЭР) масса/заряд: 245,9, 247,9 [М-Н]+.
Соединение 132А-5:
Соединение 132-4 (350 мг, 1,41 ммоль) растворяли в концентрированной серной кислоте (7 мл) при 0°С; затем к реакционной жидкости добавляли нитрат калия (157 мг, 1,55 ммоль) и последовательно перемешивали при указанной температуре в течение 30 минут. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость выливали в ледяную воду (50 г). Смесь экстрагировали смешанным растворителем, содержащим этилацетат (15 мл × 3). Органические фазы объединяли, сначала промывали насыщенным солевым раствором (10 мл × 2), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 15/1) с получением соединения 132А-5 (270 мг).
МС (ИЭР) масса/заряд: 290,8, 292,8 [М-Н]+.
Соединение 132А-6:
Соединение 132А-5 (140 мг, 0,48 ммоль) растворяли в этаноле (2 мл). Затем к реакционной жидкости добавляли влажный палладий на углероде (10%, 56 мг). После того, как реакционную систему 3 раза продували с применением водорода, реакционную жидкость перемешивали при комнатной температуре в атмосфере водорода в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость фильтровали через целит, фильтрационный осадок промывали этилацетатом (10 мл × 3). Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением соединения 132А-6 (570 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ 9,47 (s, 1H), 6,78 (dd, J=7,8, 7,8 Гц, 1H), 6,61 (dd, J=8,1, 1,2 Гц, 1H), 6,61 (dd, J=7,5, 1,2 Гц, 1H), 4,85 (s, 2Н), 4,44 (s, 2Н).
Промежуточное соединение 132А:
Соединение 132А (65 мг) получали путем замены исходного материала на соединение 132А-6 (60 мг, 0,33 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР) масса/заряд: 375,0, 377,0 [М+Н]+.
Получение соединения 132
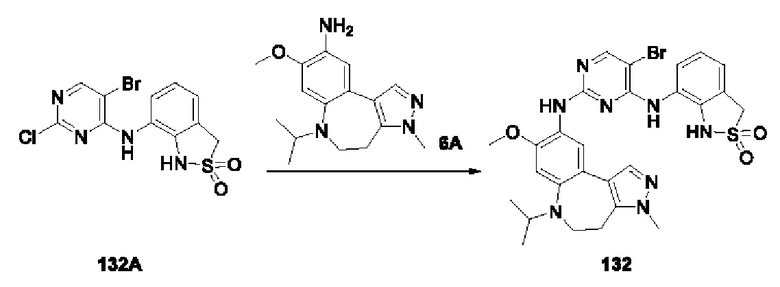
Соединение 132 (10 мг) получали путем замены исходного материала на соединение 132А (57 мг, 0,15 ммоль) и соединение 6А (43 мг, 0,15 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 625,1, 627,1 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 9,94 (s, 1H), 8,14 (d, J=3,2 Гц, 1H), 8,02 (s, 1H), 7,83 (s, 2Н), 7,55-7,52 (m, 1Н), 7,36 (s, 1H), 6,97-6,95 (m, 1H), 6,72 (s, 1H), 6,51 (d, J=2,8 Гц, 1H), 4,48 (s, 2H), 3,90-3,85 (m, 1H), 3,73 (s, 6H), 3,17-3,15 (m, 2H), 2,92-2,89 (m, 2H), 1,26-1,23 (m, 6H).
Пример 133:
Получение соединения 133
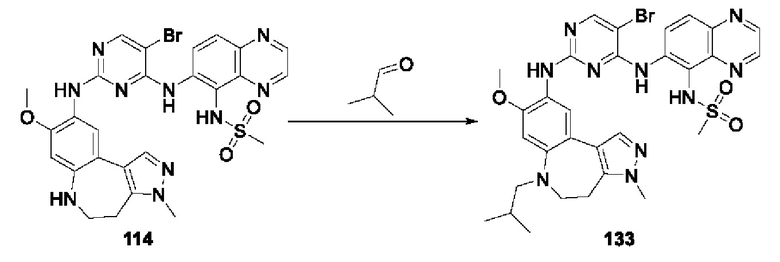
Соединение 114 (100 мг, 0,16 ммоль) и изобутиральдегид (57 мг, 0,79 ммоль) растворяли в метаноле (2 мл) и перемешивали в течение 10 минут с последующим добавлением к реакционной жидкости уксусной кислоты (19 мг, 0,31 ммоль) и цианоборогидрида натрия (20 мг, 0,31 ммоль). Реакционную систему последовательно перемешивали при комнатной температуре в течение 1 часа. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость непосредственно очищали с применением колонки с обращенной фазой С18. Условия очистки были следующими: хроматографическая колонка: 40 г колонка с обращенной фазой С18; подвижная фаза: вода (содержащая 0,1% муравьиной кислоты) и ацетонитрил; скорость потока: 35 мл/мин; градиент: повышение содержания ацетонитрила от 0% до 50% в течение 35 минут; длина волны детектирования 254 нм. Продукт собирали и лиофилизировали при пониженном давлении с получением соединения 133 (26 мг).
МС (ИЭР) масса/заряд: 693,2, 695,2 [М-Н]+.
1H ЯМР (300 МГц, CDCl3) δ 9,14 (s, 1Н), 8,86-8,77 (m, 2H), 8,61 (d, J=9,3 Гц, 1H), 8,26 (s, 2H), 7,62 (s, 1H), 7,59-7,55 (m, 1H), 7,36 (s, 1H), 6,81 (s, 1H), 6,54 (s, 1H), 3,89 (s, 3H), 3,70 (s, 3H), 3,35-3,31 (m, 2H), 3,08 (d, J=7,2 Гц, 2H), 2,98-2,95 (m, 5H), 1,92-1,88 (m, 1H), 0,91 (d, J=6,6 Гц, 6H).
Пример 134:
Получение соединения 134
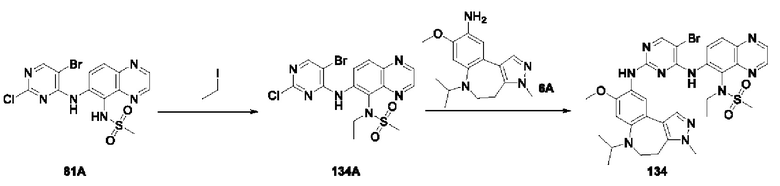
Соединение 134А:
Соединение 134А (120 мг) получали путем замены исходного материала на йодэтан (218 мг, 1,4 ммоль) согласно способу получения соединения 123А из соединения 81А в примере 123.
МС (ИЭР, масса/заряд): 456,9, 458,9 [М+Н]+.
Соединение 134:
Соединение 134 (43 мг) получали путем замены исходного материала на соединение 134А (60 мг, 0,13 ммоль) и соединение 6А (38 мг, 0,13 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 707,3, 709,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8,79-8,73 (m, 4Н), 8,33 (s, 1H), 8,25 (s, 1H), 7,62 (s, 1H), 7,31-7,29 (m, 1H), 6,90 (s, 1H), 6,55 (s, 1H), 4,23-4,14 (m, 1H), 3,88 (s, 3H), 3,85-3,80 (m, 2H), 3,68 (s, 3H), 3,47-3,44 (m, 1H), 3,20-3,16 (m, 1H), 3,13 (s, 3H), 2,96-2,92 (m, 2H), 1,33-1,28 (m, 6H), 1,14 (t, J=7,2 Гц, 3Н).
Пример 135:
Получение соединения 135
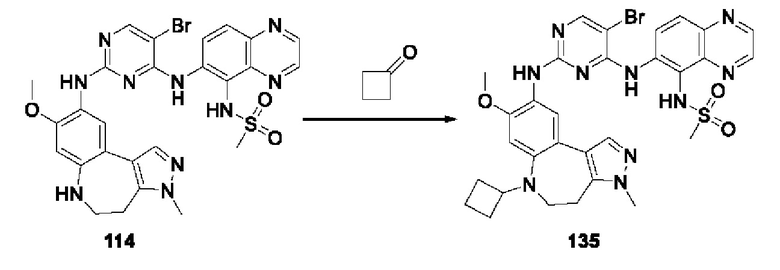
Соединение 135 (21 мг) получали путем замены исходного материала на циклобутанон (38 мг, 0,55 ммоль) согласно способу получения соединения 133 из соединения 114 в примере 133.
МС (ИЭР, масса/заряд): 691,2, 693,2 [М+Н]+.
1Н ЯМР (300 МГц, CDCl3) δ 9,15 (s, 1H), 8,88-8,78 (m, 2H), 8,55 (d, J=9,3 Гц, 1H), 8,27-8,25 (m, 2H), 7,73 (s, 1H), 7,56 (d, J=9,3 Гц, 1H), 7,44 (s, 1H), 6,70 (s, 1H), 6,44 (s, 1H), 4,14-4,03 (m, 1H), 3,89 (s, 3H), 3,68 (s, 3H), 3,36-3,26 (m, 2H), 3,06-2,99 (m, 2H), 2,96 (s, 3H), 2,34-2,24 (m, 2H), 2,17-2,05 (m, 2H), 1,86-1,76 (m, 2H).
Пример 136:
Получение промежуточного соединения 136А
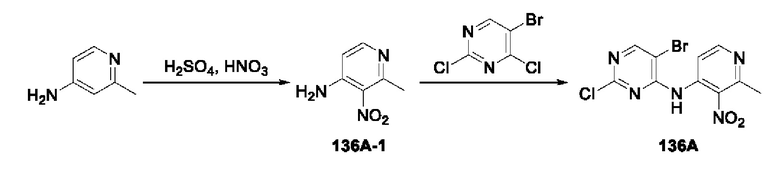
Соединение 136А-1:
Соединение 2-метил-4-аминопиридин (3 г, 27,74 ммоль) растворяли в концентрированной серной кислоте (6 мл) при 0°С; затем в реакционную жидкость добавляли по каплям азотную кислоту (2,6 г, 41,26 ммоль). Реакционную систему нагревали до 65°С и последовательно перемешивали в течение 2 часов. После исчезновения исходного материала, как было установлено с помощью ЖХ-МС, реакционную жидкость охлаждали до 0°С и доводили до рН=9 с применением водного раствора гидроксида натрия (1 М). Смесь экстрагировали смешанным растворителем, содержащим этилацетат (50 мл × 3). Органические фазы объединяли, сначала промывали насыщенным солевым раствором (50 мл × 2), затем высушивали над безводным сульфатом натрия, фильтровали и в заключении концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: дихлорметан/этилацетат = 1/1) с получением соединения 136А-1 (1,8 г).
МС (ИЭР) масса/заряд: 154,1 [М+Н]+.
Промежуточное соединение 136А:
Соединение 136А (1,2 г) получали путем замены исходного материала на соединение 136А-1 (600 мг, 3,92 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР) масса/заряд: 343,9, 345,9 [М+Н]+.
Получение соединения 136
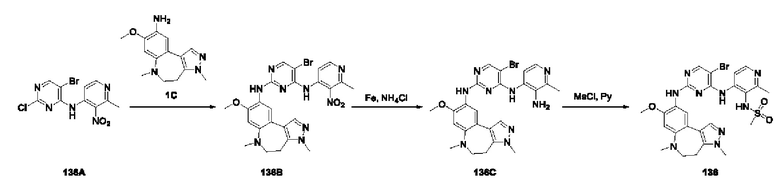
Соединение 136В:
Соединение 136В (40 мг) получали путем замены исходного материала на соединение 136А (200 мг, 0,58 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 566,1, 568,1 [М+Н]+.
Соединение 136С:
Соединение 136С (20 мг) получали путем замены исходного материала на соединение 136 В (40 мг, 0,07 ммоль) согласно способу получения соединения 1С из соединения 1С-9 в примере 1.
МС (ИЭР, масса/заряд): 536,4, 538,4 [М+Н]+.
Соединение 136:
Соединение 136 (6 мг) получали путем замены исходного материала на соединение 136С (20 мг, 0,04 ммоль) согласно способу получения соединения 32 из соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 614,1, 616,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1Н), 8,57 (s, 1H), 8,28 (s, 1H), 8,13-8,00 (m, 1H), 7,61-7,41 (m, 2H), 7,15-7,00 (m, 1H), 6,57 (s, 1H), 3,89 (s, 6H), 3,67-3,60 (m, 2H), 3,29 (s, 3H), 3,15-2,96 (m, 5H), 2,80 (s, 3H).
Пример 137:
Получение промежуточного соединения 137А
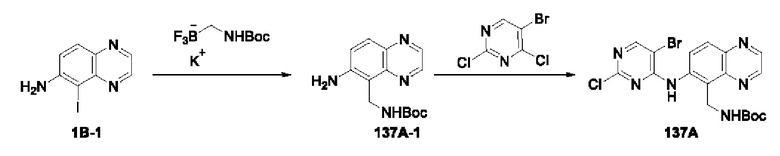
Соединение 137А-1:
Соединение 137А-1 (1 г) получали путем замены исходного материала на соединение 1 В-1 (1,5 г, 5,53 ммоль) и (((трет-бутоксикарбонил)амино)метил) трифторборат калия (1,44 г, 6,09 ммоль) согласно способу получения соединения 120А-3 из соединения 120А-2 в примере 120.
МС (ИЭР, масса/заряд): 275,2 [М+Н]+.
Промежуточное соединение 137А:
Соединение 137А (800 мг) получали путем замены исходного материала на соединение 137А-1 (520 мг, 1,9 ммоль) согласно способу получения соединения 1В из соединения 1В-2 в примере 1.
МС (ИЭР) масса/заряд: 465,1, 467,1 [М+Н]+.
Получение соединения 137
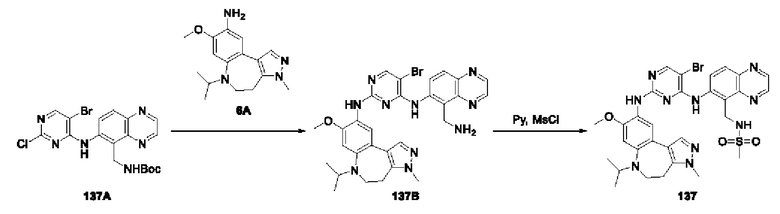
Соединение 137B:
Соединение 137B (20 мг) получали путем замены исходного материала на соединение 137А (250 мг, 0,54 ммоль) и соединение 6А (154 мг, 0,54 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 615,2, 617,2 [М+Н]+.
Соединение 137:
Соединение 137 (2 мг) получали путем замены исходного материала на соединение 137B (20 мг, 0,03 ммоль) согласно способу получения соединения 32 из соединения 32С в примере 32.
МС (ИЭР, масса/заряд): 693,1, 695,1 [М+Н]+.
1H ЯМР (300 МГц, CDCl3) δ 8,89-8,78 (m, 2Н), 8,43 (s, 1H), 8,25 (s, 2Н), 8,11 (d, J=9,3 Гц, 1Н), 7,65 (d, J=9,3 Гц, 1H), 7,57-7,42 (m, 1H), 6,51 (s, 2H), 5,58 (t, J=6,6 Гц, 1H), 4,96 (d, J=6,3 Гц, 2H), 3,88 (s, 4H), 3,64 (s, 3H), 3,33-3,17 (m, 2H), 2,94 (s, 3H), 2,91-2,83 (m, 2H), 1,29 (s, 6H).
Пример 138:
Получение соединения 138
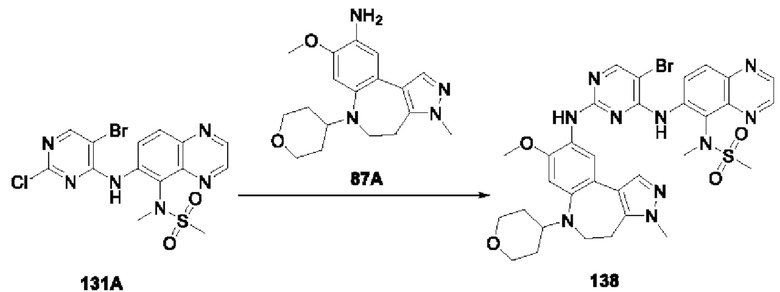
Соединение 138 (14 мг) получали путем замены исходного материала на соединение 131А (70 мг, 0,16 ммоль) и соединение 87А (52 мг, 0,16 ммоль) согласно способу получения соединения 1 из соединения 1В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 735,1, 737,1 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) δ 8,93 (d, J=2,1 Гц, 1H), 8,87 (d, J=2,1 Гц, 1Н), 8,80-8,72 (m, 1Н), 8,51 (s, 1H), 8,46 (s, 1Н), 8,30 (s, 1Н), 7,76 (s, 1H), 7,62-7,50 (m, 1H), 7,38 (s, 1Н), 6,64 (s, 1H), 3,97-3,93 (m, 2Н), 3,77 (s, 3Н), 3,73-3,71 (m, 1H), 3,69 (s, 3Н), 3,53-3,45 (m, 2Н), 3,39 (s, 3Н), 3,30-3,27 (m, 2Н), 3,22 (s, 3Н), 2,95-2,92 (m, 2Н), 1,88-1,82 (m, 4Н).
Пример 139:
Получение соединения 139
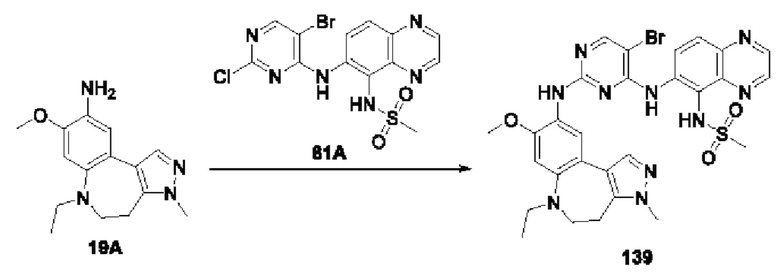
Соединение 139 (4 мг) получали путем замены исходного материала на соединение 81А (99 мг, 0,23 ммоль) и соединение 19А (63 мг, 0,23 ммоль) согласно способу получения соединения 1 из соединения 1 В и соединения 1С в примере 1.
МС (ИЭР, масса/заряд): 665,2, 667,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9,10 (s, 1H), 8,82-8,78 (m, 2H), 8,58 (d, J=9,2 Гц, 1H), 8,32-8,26 (m, 2H), 7,56 (s, 2H), 7,33 (s, 1H), 6,73 (s, 1H), 6,51 (s, 1H), 3,87 (s, 3H), 3,66 (s, 3H), 3,38-3,25 (m, 4H), 3,00-2,93 (m, 5H), 1,28 (t, J=6,8 Гц, 3Н).
Оценка биологического анализа:
(I) Ферментативный эксперимент in vitro для EGFR WT и EGFR L858R/T790M/C797S В данном анализе было исследовано ингибирующее действие предложенного соединения на киназную активность EGFR WT (EGFR дикого типа) и EGFR L858R/T790M/C797S путем применения метода резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET), при этом была получена полумаксимальная ингибирующая концентрация ICδ0 соединения, действующая на киназную активность EGFR.
1. Материалы
Рекомбиназа EGFR и рекомбиназа EGFR L858R/T790M/C797S, приобретенные в компании Signalchem.
Набор HTRF KinEASE-TK, приобретенный в компании Cisbio.
DTT (дитиотреитол), MnCl2 и MgCl2, приобретенные в компании Sigma.
АТФ, приобретенный в компании Promega.
Бригатиниб, приобретенный в компании Selleck.
2. Метод
1) Приготавливали 1× рабочий раствор: 5 мМ MgCl2; 1 мМ DTT; 1 мМ MnCl2 и 1× киназный буфер (в наборе).
2) 10 мкл градиентно разведенного соединения переносили в 384-луночный планшет с применением Echo 550 (Labcyte).
3) 5 мкл 2× раствора рекомбиназы EGFR WT или EGFR L858R/T790M/C797S добавляли в 384-луночный планшет и инкубировали указанный планшет при комнатной температуре в течение 10 минут.
4) 5 мкл 2× субстратного раствора ТК (тимидинкиназа)-субстрат-биотин (содержащего АТФ) добавляли в 384-луночный планшет и инкубировали указанный планшет при комнатной температуре в течение 40 минут.
5) В планшет добавляли 5 мкл аналитического раствора, содержащего Sa-XL 665 HTRF и 5 мкл TK-антитело-Cryptatewas, и инкубировали указанный планшет при комнатной температуре в течение 1 часа.
6) Значения флуоресцентного сигнала при 615 нм и 665 нм измеряли для каждой лунки, используя микропланшет-ридер Envision (PerkinElmer).
7) Для каждой лунки рассчитывали отношение значений флуоресцентного сигнала при 665 нм и 615 нм.
8) Для получения IC50 соединений анализ данных проводили с применением программного обеспечения GraphPad Prism.
Результаты ингибирования киназной активности EGFR дикого типа и тройной мутации EGFR L858R/T790M/C797S приведены в таблице 1.
Результаты определения IC50 соединений, описанных в настоящем документе, для ингибирования ферментативной активности каждого EGFR приведены в таблице 1. Для перечисленных выше соединений соединение с IC50 ниже 1 нМ обозначали как А; соединение с IC50 от 1 до 10 нМ обозначали как В; соединение с IC50 от 10 до 50 нМ обозначали как С; соединение с IC50 от 50 до 100 нМ обозначали как D; соединение с IC50 от 100 до 1000 нМ обозначали как Е; соединение с IC50 более 1000 нМ обозначали как F.
Как следует из таблицы 1, соединения, описанные в настоящем документе, обладают хорошим ингибирующим действием на киназную активность тройной мутации EGFR L858R/T790M/C797S и слабым ингибирующим действием на киназную активность EGFR дикого типа (EGFR (WT)), что указывает на то, что соединения, описанные в настоящем документе, обладают хорошей киназной активностью и селективностью.
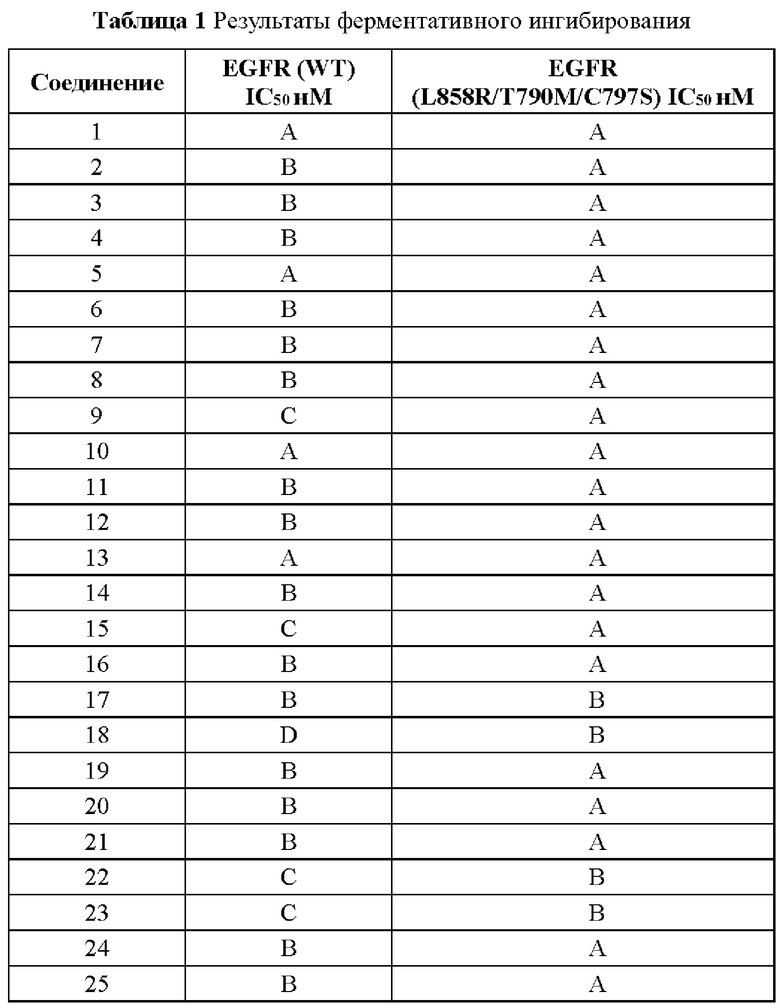
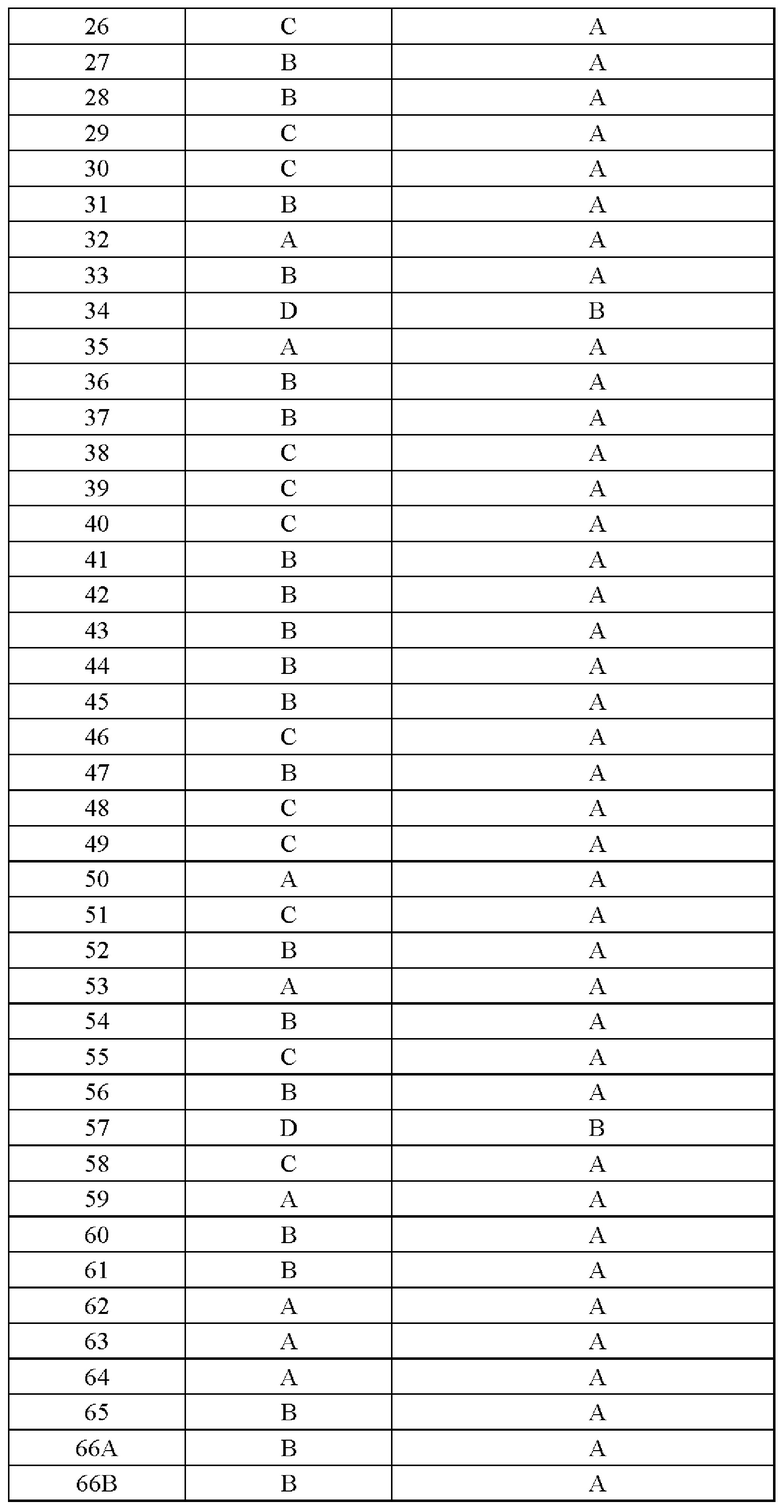
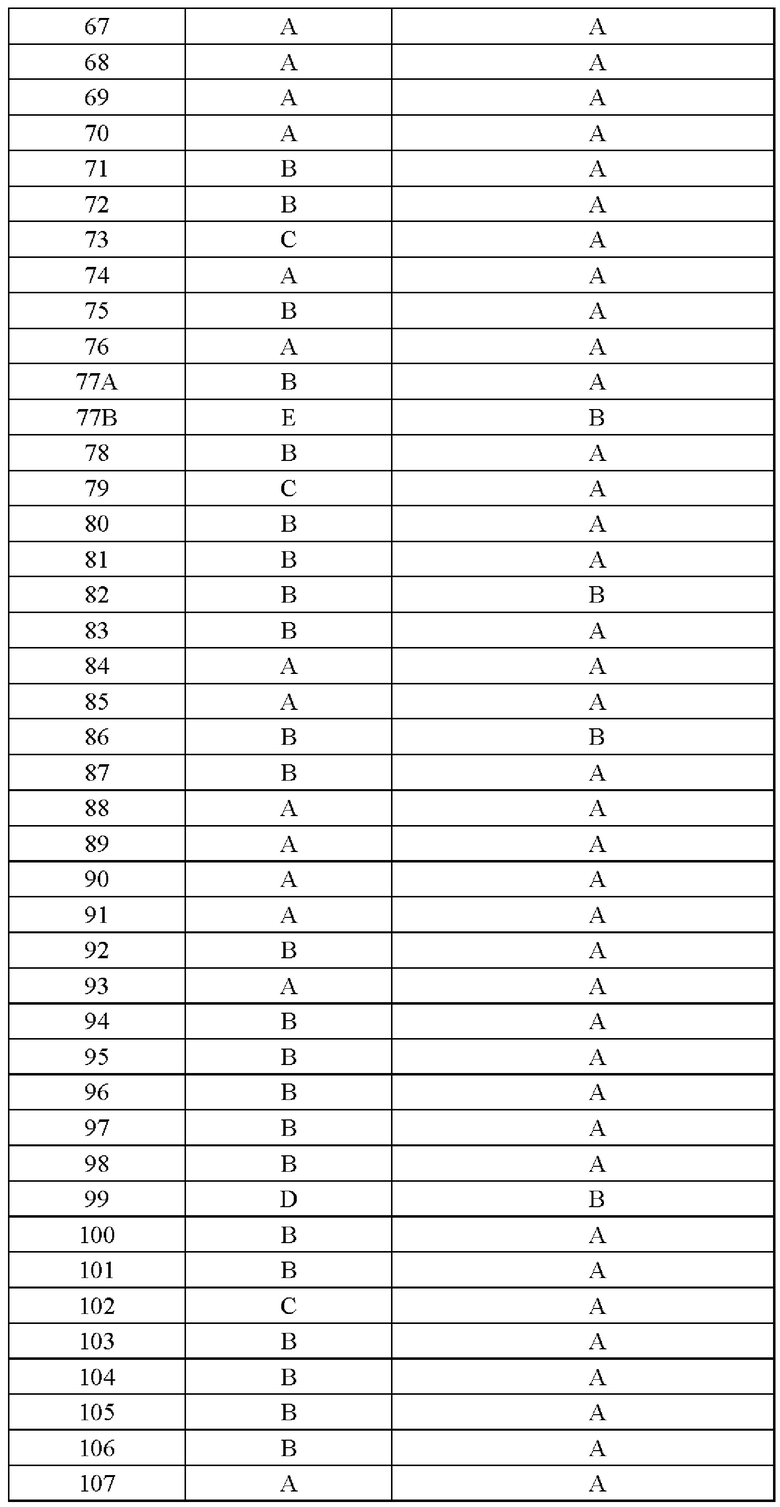
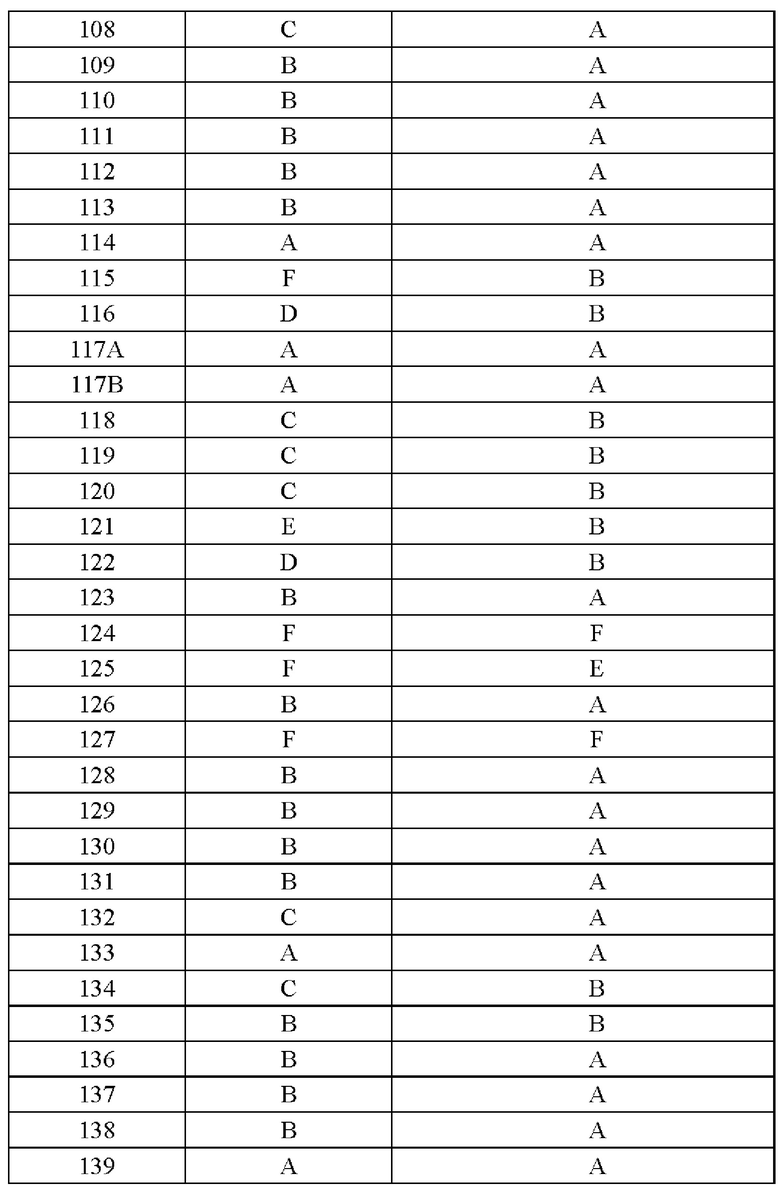
(II) Анализ ингибирования пролиферации клеток Анализ ингибирования пролиферации клеток А431
В данном анализе ингибирующее действие предложенных соединений на пролиферацию клеток А431 исследовали путем применения метода CellTiter-Glo и получали концентрацию IC50 соединения для ингибирования полумаксимального роста клеток.
1. Материалы
Клетки А431, приобретенные в компании АТСС.
Среда DMEM (минимальная эссенциальная среда Игла, модифицированная по способу Дульбекко), фетальная бычья сыворотка (ФБС) и пенициллин-стрептомицин, приобретенные в компании GIBCO.
Бригатиниб, приобретенный в компании Selleck.
Реагент CellTiter-Glo, приобретенный в компании Promega.
2. Метод
1) Клетки А431 высевали на 384-луночный планшет при плотности 800 клеток/лунка при 30 мкл/лунка и культивировали указанный планшет в клеточном инкубаторе в течение 24 часов (37°С, 5% CO2).
2) День 0: 30 мкл градиентно разбавленных соединений, подлежащих исследованию, с конечной концентрацией ДМСО 0,1%, добавляли в планшет с применением Echo и инкубировали указанный планшет в клеточном инкубаторе в течение 72 часов (37°С, 5% СО2). В холостой контрольной группе в каждую лунку добавляли 30 мкл ДМСО.
3) День 3: в каждую лунку добавляли по 30 мкл реагента Cell Titer-Glo и выдерживали планшет в темноте при комнатной температуре в течение 30 минут.
4) Хемилюминесцентный сигнал детектировали с помощью планшета-ридера Envision (PerkinElmer).
5) Для получения IC50 соединений анализ данных проводили с применением программного обеспечения GraphPad Prism.
Анализ ингибирования пролиферации клеток Ba/F3_L858R/T790M/C797S
В данном анализе ингибирующее действие предложенных соединений на пролиферацию клеток Ba/F3_L858R/T790M/C797S исследовали путем применения метода CellTiter-Glo и
получали концентрацию IC50 соединения для ингибирования полумаксимального роста клеток.
1. Материалы
Клетки Ba/F3_L858R/T790M/C797S были сконструированы в компании Pharmaron Beijing Co., Ltd.
Среда 1640, фетальная бычья сыворотка (ФБС), пенициллин-стрептомицеты и GlutaMAX-I
Добавка, приобретенная в компании GIBCO.
Бригатиниб, приобретенный в компании Selleck.
Реагент CellTiter-Glo, приобретенный в компании Promega.
2. Метод
1) Клетки Ba/F3_L858R/T790M/C797S высевали на 384-луночный планшет при плотности 700 клеток/лунка при 30 мкл/лунка.
2) День 0: 30 мкл градиентно разбавленных соединений, подлежащих исследованию, с конечной концентрацией ДМСО 0,1%, добавляли в планшет с применением Echo и инкубировали указанный планшет в клеточном инкубаторе в течение 72 часов (37°С, 5% СО2). В холостой контрольной группе в каждую лунку добавляли 30 мкл ДМСО.
3) День 3: в каждую лунку добавляли по 30 мкл реагента Cell Titer-Glo и выдерживали планшет в темноте при комнатной температуре в течение 30 минут.
4) Хемилюминесцентный сигнал детектировали с помощью планшета-ридера Envision (PerkinElmer).
5) Для получения IC50 соединений анализ данных проводили с применением программного обеспечения GraphPad Prism.
Анализ ингибирования пролиферации клеток Ba/F3_Del19/T790M/C797S
В данном анализе ингибирующее действие предложенных соединений на пролиферацию клеток Ba/F3_Del19/T790M/C797S исследовали путем применения метода CellTiter-Glo и получали концентрацию IC50 соединения для ингибирования полумаксимального роста клеток.
1. Материалы
Клетки Ba/F3_Del19/T790M/C797S, приобретенные в компании Kyinno Biotechnology Co., Ltd.
Среда 1640, фетальная бычья сыворотка (ФБС), пенициллин-стрептомицеты и добавка GlutaMAX-I, приобретенные в компании GIBCO.
Бригатиниб, приобретенный в компании Selleck.
Реагент CellTiter-Glo, приобретенный в компании Promega.
2. Метод
1) Клетки Ba/F3_Del19/T790M/C797S высевали на 384-луночный планшет при плотности 700 клеток/лунка при 30 мкл/лунка.
2) День 0: 30 мкл градиентно разбавленных соединений, подлежащих исследованию, с конечной концентрацией ДМСО 0,1%, добавляли в планшет с применением Echo и инкубировали указанный планшет в клеточном инкубаторе в течение 72 часов (37°С, 5% СО2). В холостой контрольной группе в каждую лунку добавляли 30 мкл ДМСО.
3) День 3: в каждую лунку добавляли по 30 мкл реагента CellTiter-Glo и выдерживали планшет в темноте при комнатной температуре в течение 30 минут.
4) Хемилюминесцентный сигнал детектировали с помощью планшета-ридера Envision (PerkinElmer).
Для получения IC50 соединений анализ данных проводили с применением программного обеспечения GraphPad Prism 6.
Результаты определения ICδ0 соединений, описанных в настоящем документе, для ингибирования клеточной жизнеспособности каждого EGFR, приведены в таблице 2. Для перечисленных выше соединений соединение с IC50 ниже 10 нМ обозначали как А; соединение с IC50 от 10 до 50 нМ обозначали как В; соединение с IC50 от 50 до 100 нМ обозначали как С; соединение с IC50 от 100 до 1000 нМ обозначали как D; соединение с IC50 более 1000 нМ обозначали как Е.
Результаты ингибирования клеточной жизнеспособности показаны в таблице 2.
Как следует из экспериментальных результатов в таблице 2, по сравнению с бригатинибом в качестве контрольного примера соединения, описанные в настоящем документе, обладают лучшим ингибирующим действием на клеточную пролиферацию линии клеток с тройной мутацией Ba/F3 Del19/T790M/C797S EGFR и линии клеток с тройной мутацией Ba/F3L 858R/T790M/C797S EGFR и более слабым ингибирующим действием на линию клеток А431 EGFR дикого типа (EGFR WT), что указывает на то, что соединения, описанные в настоящем документе, проявляют лучшую клеточную жизнеспособность и селективность.
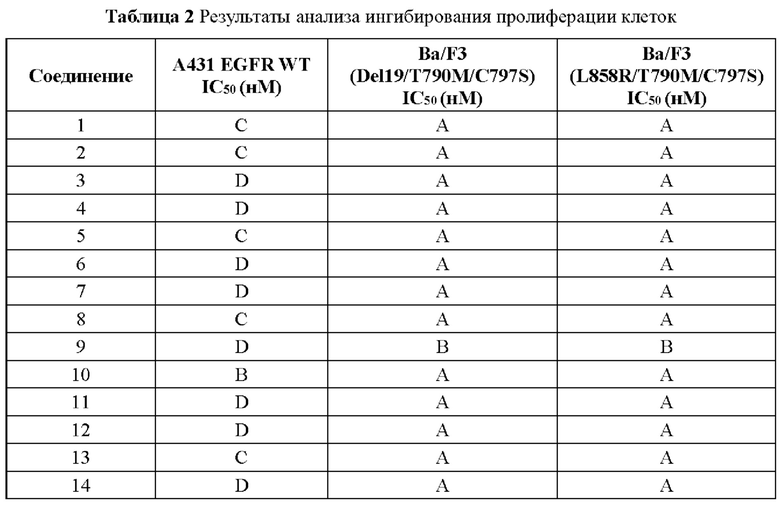
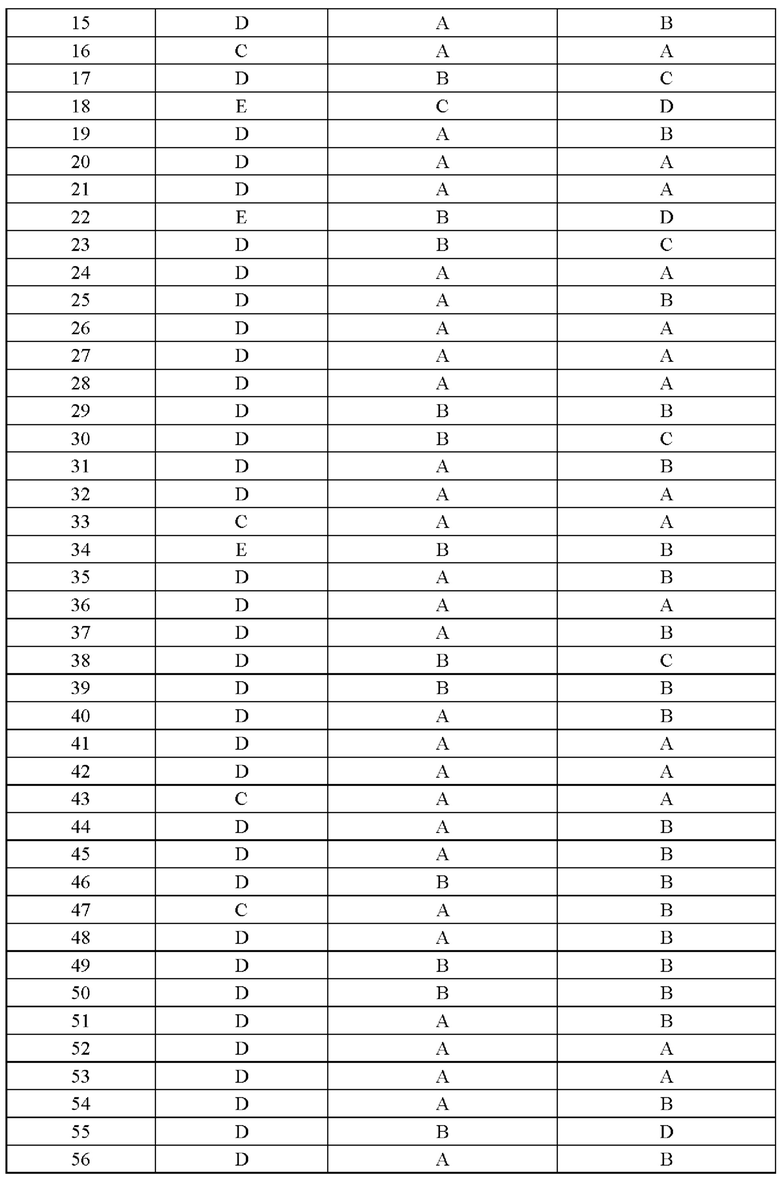
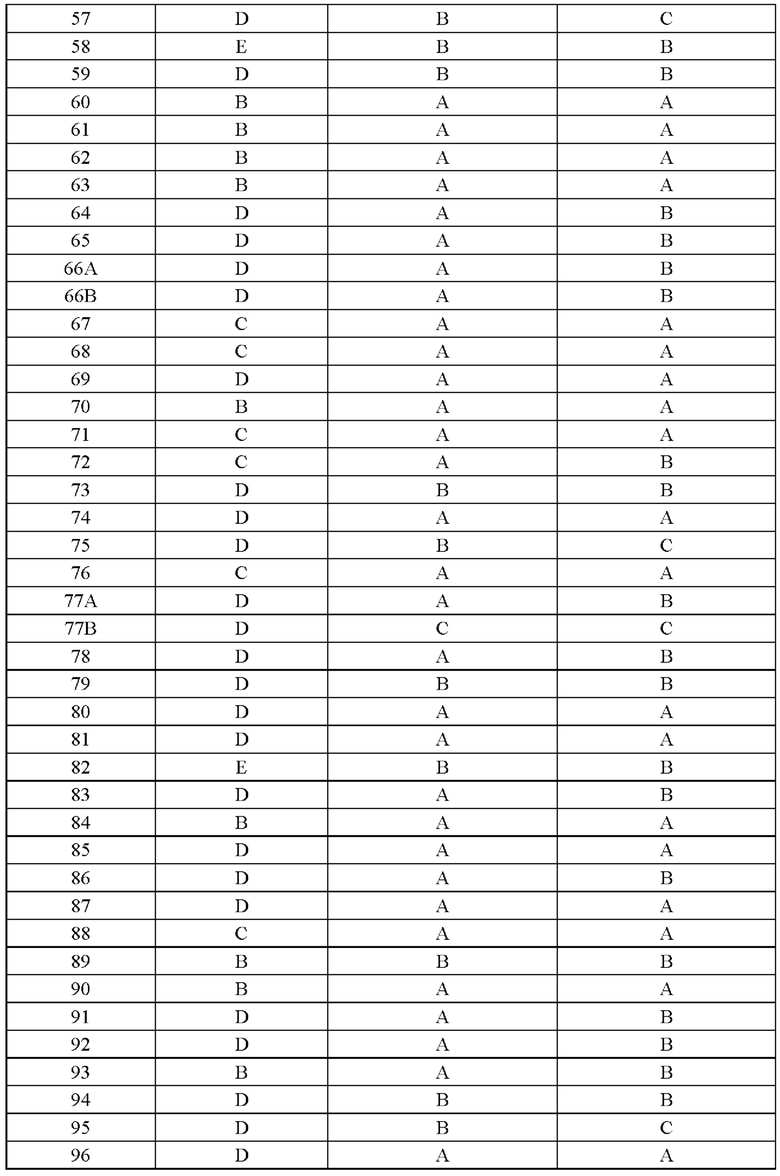
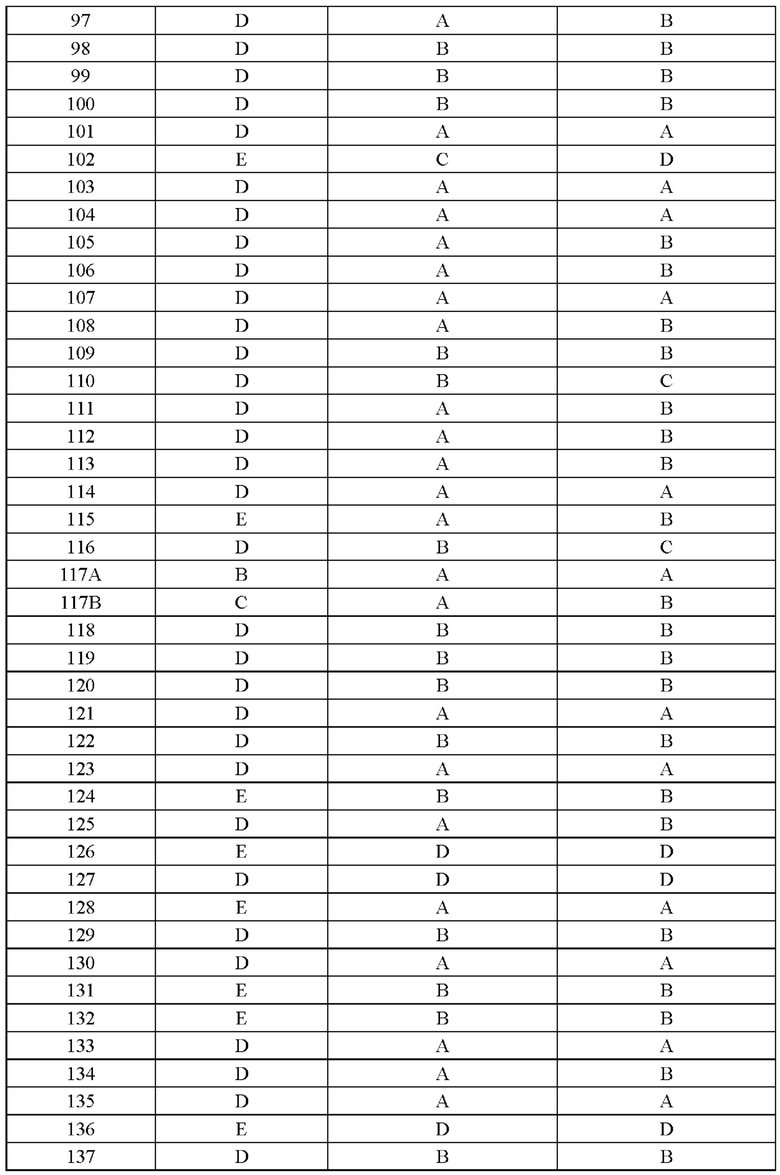

(III) Эксперимент по исследованию эффективности in vivo
1. Цель
Оценивали противоопухолевую активность и токсические и побочные эффекты соединения 53 и соединения 27, вводимых перорально в течение 21 дня подряд, на РС9 (Del19/T790M/C797S).
2. Материалы
Самки мышей BALB/c-nu статуса SPF (свободные от специфических патогенов), приобретенные в компании Beijing Vital River Laboratory Animal Technology Co., Ltd.
Клетки PC9 (Del19/T790M/C797S), само-сконструированные в компании Qilu Pharmaceutical Co., Ltd.
3. Процедуры
3.1 Культивирование клеток
Клетки РС9 (Del19/T790M/C797S) культивировали в среде RPMI 1640, содержащей 10% ФБС, при 37°С в инкубаторе с 5%-ным содержанием углекислого газа; затем указанные клетки в фазе экспоненциального роста собирали для инокуляции.
3.2 Инокуляция клеток
В стерильных условиях брали клеточную суспензию культивированных in vitro РС9 (Del19/T790M/C797S), центрифугировали для доведения концентрации клеток до 3×107 клеток/мл и инокулировали мышей в подмышечную впадину с правой стороны (0,1 мл/мышь), при этом день инокуляции определяли как день 0.
3.3 Группировка опухолей, введение и измерение опухоли
a. Когда средний объем опухоли составлял примерно 150 мм3, 35 мышей с умеренным объемом опухоли отбирали для включения в исследование и случайным образом распределяли по 5 группам в зависимости от объема опухоли: G1: контрольная группа, получавшая носитель, G2: соединение 53 (15 мг/кг), G3: соединение 53 (60 мг/кг), G4: соединение 27 (15 мг/кг) и G5: соединение 27 (35 мг/кг), 7 мышей/группа.
b. Животных группировали и затем начали введение, при этом объем для перорального введения (п/в) составлял 10 мл/кг; взвешивание осуществляли один раз в сутки при последовательном введении в течение 21 дня; диаметры опухоли измеряли 2 раза в неделю.
c. Объем опухоли (о/о): объемы опухоли измеряли 2 раза в неделю для отслеживания изменений объема опухолевой массы и скорости роста. Объем опухоли V=1/2×а×b2, где а и b представляют собой диаметр по длинной оси опухоли и диаметр по короткой оси опухоли, соответственно. Эффект ингибирования роста предложенных соединений на ткани опухоли оценивали путем применения коэффициента ингибирования роста опухоли TGI (%). TGI (%)=[1-(средний объем опухоли в группе с определенным режимом введения - средний объем опухоли в день группировки в данной группе с определенным режимом введения)/(средний объем опухоли в группе отрицательного контроля - средний объем опухоли в день группировки в группе отрицательного контроля)]×100%. Результаты для группы введения и группы отрицательного контроля были получены в один и тот же день.
d. В ходе эксперимента внимательно наблюдали за жизненным статусом мышей, включая внешние признаки, общую поведенческую активность, психическое состояние, потребление пищи, функцию дыхания, проявления, связанные с выделениями фекалий и мочи, состояние в месте инъекции и другие токсические проявления.
e. После достижения конечной точки мышей умерщвляли, при этом останки животных замораживали и хранили в морозильной камере и передавали в сертифицированную организацию по обработке медицинских отходов для утилизации.
4 Результаты
Результаты показаны в таблице 3 и на фиг. 1 и 2.
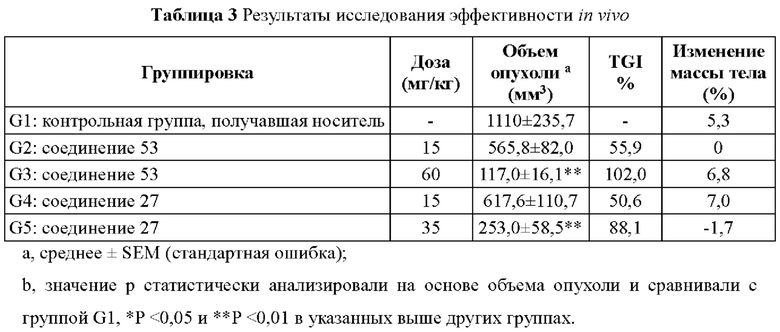
5. Вывод
Из приведенных выше результатов следует, что как соединение 53, так и соединение 27 могут значительно ингибировать рост опухоли и проявлять сильную зависимость доза-эффект, при этом указанные соединения хорошо переносятся мышами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| НОВЫЕ СОЕДИНЕНИЯ И ПРОИЗВОДНЫЕ СУЛЬФОНИМИДОИЛПУРИНОНА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2016 |
|
RU2745269C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ КАК ИНГИБИТОР FGFR | 2017 |
|
RU2742485C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5,7-ДИЗАМЕЩЕННОГО [1,3] ТИАЗОЛО[4,5-D]ПИРИМИДИН-2(3H)-ОНА | 2006 |
|
RU2411245C9 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
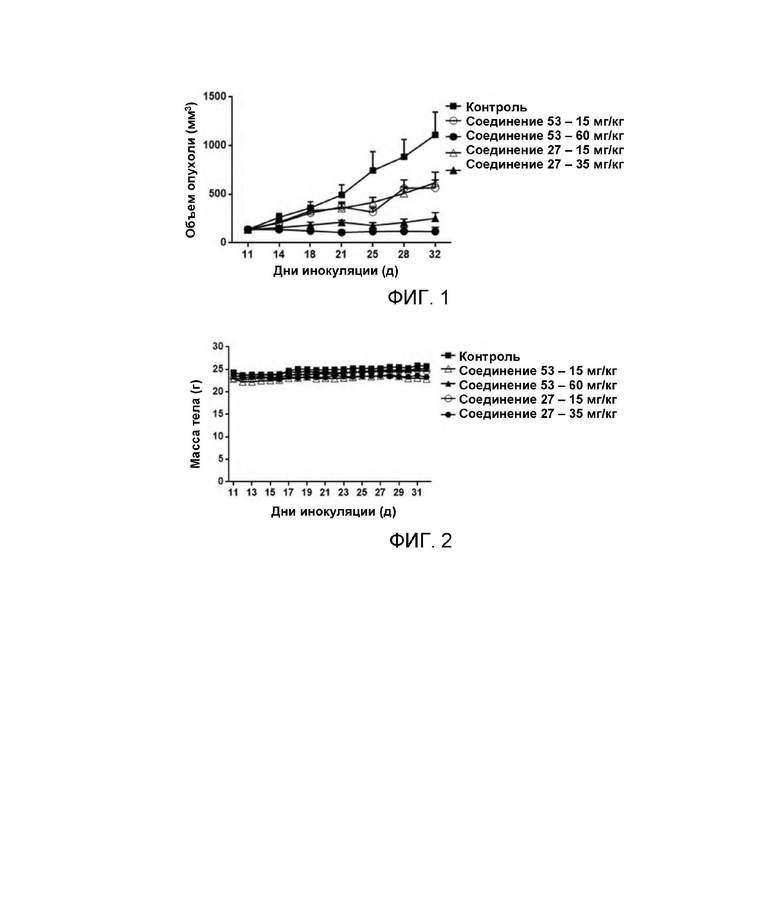
Группа изобретений относится к органической химии и включает соединение формулы (I'''), его стереоизомер, фармацевтически приемлемую соль, гидрат, сольват, фармацевтическую композицию, его содержащую, способ лечения рака, опосредованного EGFR, и промежуточные соединения формул (Vа) и (Vа’). В формуле (I''') R1 представляет собой C1-6 алкокси; M выбран из N и CRa; Z выбран из N и CR6; Z1 выбран из N и CR7; Ra представляет собой H; кольцо A выбрано из 7-членного гетероциклила, содержащего 1-2 гетероатома, выбранные из N и O, или 7-членного карбоциклила, в котором 1 или 2 кольцевых атома могут быть необязательно заменены на -C(=O) и который также может необязательно содержать в качестве заместителя 1-2 группы Rx; кольцо B представляет собой 5-6-членный гетероарил, содержащий 1-3 гетероатома, выбранные из N и S, который необязательно содержит в качестве заместителя R2; остальные заместители являются такими, как определено в формуле изобретения. Технический результат – трициклические соединения формулы (I''') в качестве селективных ингибиторов EGFR. 4 н. и 9 з.п. ф-лы, 2 ил., 3 табл., 139 пр.
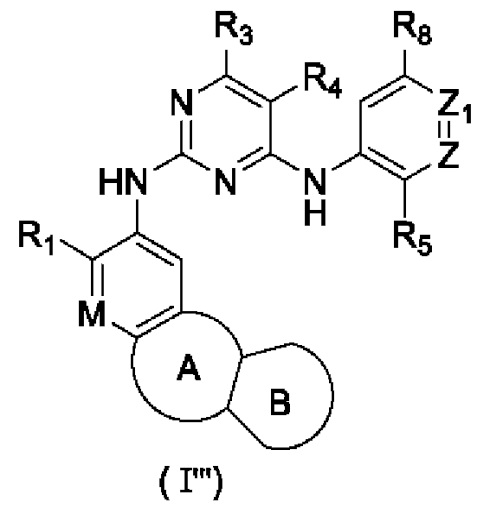
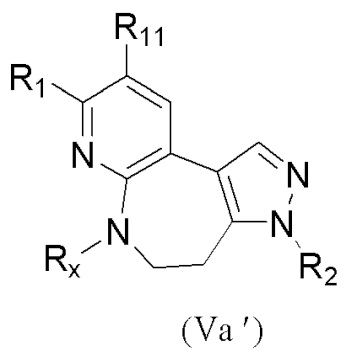
1. Соединение, представленное формулой (I''')
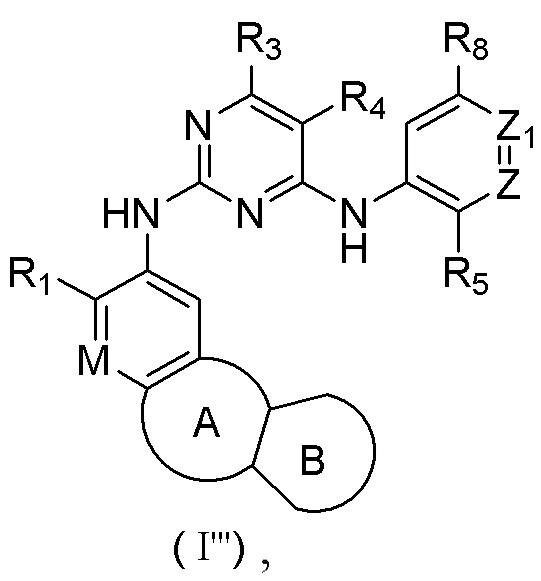
или его стереоизомер, или фармацевтически приемлемая соль, гидрат, сольват,
где R1 представляет собой C1-6 алкокси;
M выбран из N и CRa;
Z выбран из N и CR6;
Z1 выбран из N и CR7;
Ra представляет собой H;
кольцо A выбрано из 7-членного гетероциклила, содержащего 1-2 гетероатома, выбранные из N и O, или 7-членного карбоциклила, в котором 1 или 2 кольцевых атома могут быть необязательно заменены на -C(=O) и который также может необязательно содержать в качестве заместителя 1-2 группы Rx, при этом Rx выбран из -OH, -CN, -NH2, -N(CH3)2, галогена, C1-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, C3-8 циклоалкила, C3-8 циклоалкил-C1-6 алкила-, C6арил-C1-6 алкила-, 3-6-членного моноциклического гетероциклоалкила, содержащего N или O в качестве гетероатома, 3-6-членного гетероциклоалкил-C1-6 алкила-, содержащего N или O в качестве гетероатома, 7-11-членного спирогетероциклила, содержащего N в качестве гетероатома; где указанный 3-6-членный гетероциклоалкил, 3-6-членный гетероциклоалкил-C1-6 алкил-, C6арил-C1-6 алкил- и 7-11-членный спирогетероциклил необязательно содержат в качестве заместителя 1-2 Ry; при этом Ry выбран из галогена, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси, C3-8 циклоалкила, необязательно содержащего в качестве заместителя 1-2 галогена, C3-8 циклоалкил-C1-6 алкила-;
кольцо B представляет собой 5-6-членный гетероарил, содержащий 1-3 гетероатома, выбранные из N и S, который необязательно содержит в качестве заместителя R2;
каждый R2 независимо выбран из C1-6 алкила, 3-6-членного гетероциклоалкила, содержащего N или O в качестве гетероатома, бензила и -(CH2)rNRcRd, r необязательно выбран из 0, 1, 2 и 3; при этом 3-6-членный гетероциклоалкил в R2 необязательно содержит в качестве заместителя C1-3 алкил или C1-3 алкокси;
каждый из R3 и R4 независимо выбран из H, галогена, C1-6 алкила, C1-6 галогеналкила, C3-8 циклоалкила;
или R3 циклизуется с R4 с образованием 5-членного гетероарила, содержащего N или S в качестве гетероатома;
R5 выбран из -C(=O)NRbRc, -P(=O)RbRc, -P(=S)RbRc, -S(=O)2NRbRc, RbS(=O)2NRc-, -NRbC(O)Rc или  ;
;
каждый из Rb, R c и Rd независимо выбран из H, C1-3 алкила, C3-6 циклоалкила;
или Rb и Rc циклизуются с атомами, к которым они оба присоединены, с образованием 5-членного гетероциклоалкила;
каждый из R6, R7 и R8 независимо представляет собой H;
или R6 циклизуется с R7 с образованием 5-6-членного гетероарила, содержащего 1-2 атома N в качестве гетероатома.
2. Соединение, представленное формулой (I'''), по п. 1, где указанное соединение представлено формулой (I)
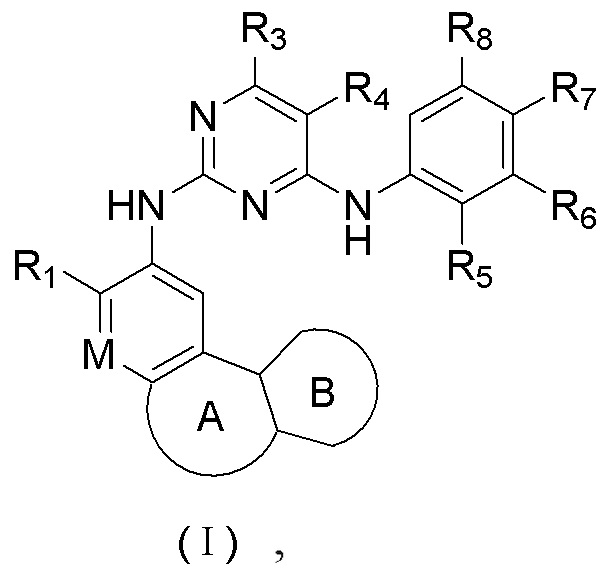
или его стереоизомер, фармацевтически приемлемая соль, гидрат, сольват,
в котором R1 представляет собой C1-6 алкокси;
M выбран из N и CRa;
Ra представляет собой H;
кольцо A выбрано из 7-членного гетероциклила, содержащего 1-2 гетероатома, выбранные из N и O, или 7-членного карбоциклила, в котором 1 или 2 кольцевых атома могут быть необязательно заменены на -C(=O), и который также может необязательно содержать в качестве заместителя 1-2 группы Rx, при этом Rx выбран из -OH, -CN, -NH2, -N(СH3)2, галогена, C1-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, C3-8 циклоалкила, C3-8 циклоалкил-C1-6 алкила-, C6арил-C1-6 алкила-, 3-6-членного моноциклического гетероциклоалкила, содержащего N или O в качестве гетероатома, 3-6-членного гетероциклоалкил-C1-6 алкила-, содержащего N или O в качестве гетероатома, 7-11-членного спирогетероциклила, содержащего N в качестве гетероатома; где указанный 3-6-членный гетероциклоалкил, 3-6-членный гетероциклоалкил-C1-6 алкил-, C6арил-C1-6 алкил- и 7-11-членный спирогетероциклил необязательно содержат в качестве заместителя 1-2 Ry; при этом Ry выбран из галогена, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси, C3-8 циклоалкила, необязательно содержащего в качестве заместителя 1-2 галогена, C3-8 циклоалкил-C1-6 алкила-;
кольцо B представляет собой 5-6-членный гетероарил, содержащий 1-3 гетероатома, выбранные из N и S, необязательно содержащий в качестве заместителя R2; при этом указанный 5-6-членный гетероарил может представлять собой пирролил, тиенил, пиразолил, тиазолил, изотиазолил, имидазолил, триазолил, пиримидинил, пиридил, пиразинил или пиридазинил;
каждый R2 независимо выбран из C1-6 алкила, 3-6-членного гетероциклоалкила, содержащего N или O в качестве гетероатома, бензила и -(CH2)rNRcRd, r необязательно выбран из 0, 1, 2 и 3; при этом 3-6-членный гетероциклоалкил в R2 необязательно содержит в качестве заместителя C1-3 алкил или C1-3 алкокси;
каждый из R3 и R4 независимо выбран из H, галогена, C1-6 алкила, C1-6 галогеналкила, C3-8 циклоалкила;
или R3 циклизуется с R4 с образованием 5-членного гетероарила, содержащего N или S в качестве гетероатома;
R5 выбран из -C(=O)NRbRc, -P(=O)RbRc, -S(=O)2NRbRc, RbS(=O)2NRc-, -P(=S)RbRc, -NRbC(O)Rc или ;
каждый из Rb, Rc и Rd независимо выбран из H, C1-3 алкила, C3-6 циклоалкила;
или Rb и Rc циклизуются с атомами, к которым они оба присоединены, с образованием 5-членного гетероциклоалкила;
каждый из R6, R7 и R8 независимо представляет собой H;
или R6 циклизуется с R7 с образованием 5-6-членного гетероарила, содержащего 1-2 атома N в качестве гетероатома.
3. Соединение по любому из пп. 1 или 2, где
R3 циклизуется с R4 с образованием 5-членного гетероарила; при этом 5-членный гетероарил представляет собой пирролил или тиенил;
R6 циклизуется с R7 с образованием 5-6-членного гетероарила; при этом 5-6-членный гетероарил представляет собой пирролил, пиразолил, имидазолил, пиримидинил, пиридил, пиразинил или пиридазинил.
4. Соединение по любому из пп. 1-3, в котором
R1 выбран из метокси, этокси, н-пропокси, изопропокси, н-бутокси; и
при этом более предпочтительно R1 выбран из метокси;
или
каждый из R3 и R4 независимо выбран из H, F, Cl, Br, метила, этила, изопропила и циклопропила;
при этом предпочтительно R3 выбран из H и каждый R4 независимо выбран из H, F, Cl, Br, метила и циклопропила;
при этом более предпочтительно R3 выбран из H и R4 независимо выбран из F, Cl, Br, метила, этила;
при этом более предпочтительно R3 выбран из H и R4 выбран из Cl, Br и метила;
при этом еще более предпочтительно R3 выбран из H и R4 выбран из Br;
или при этом R3 циклизуется с R4 с образованием тиофенового кольца и пиррольного кольца;
или
R5 выбран из
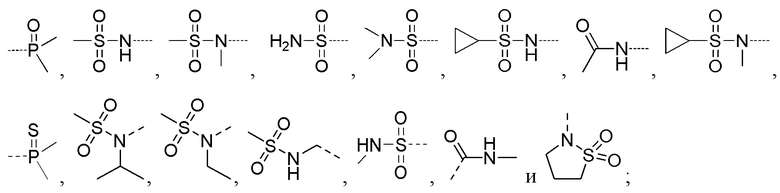
при этом R5 предпочтительно выбран из
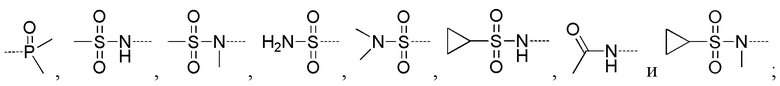
при этом R5 предпочтительно выбран из
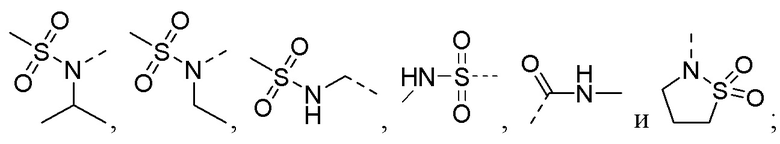
при этом R5 предпочтительно выбран из
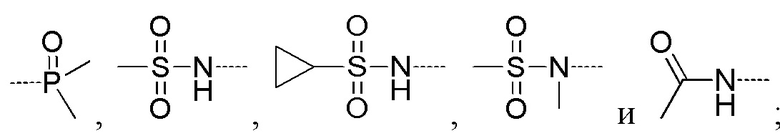
или
при этом R5 предпочтительно выбран из  ;
;
или R6 независимо циклизуется с R7 с образованием имидазольного кольца, пиразольного кольца, пиррольного кольца, пиридинового кольца, пиримидинового кольца, пиридазинового кольца и пиразинового кольца;
предпочтительно R6 независимо циклизуется с R7 с образованием пиридинового кольца и пиразинового кольца; и
более предпочтительно R6 циклизуется с R7 с образованием пиразинового кольца.
5. Соединение по п. 3, в котором структурное звено
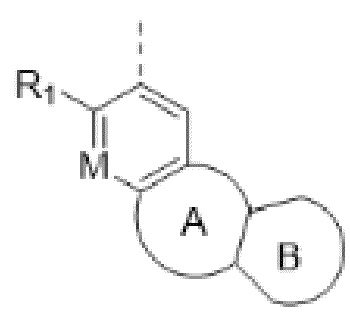 или
или 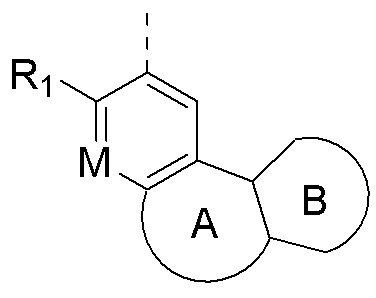 выбрано из
выбрано из
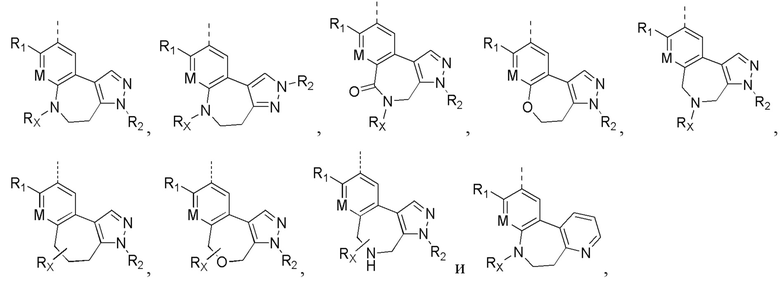
где R1 определен в п. 3;
M определен в п. 3;
R2 определен в п. 3;
Rx выбран из -OH, -CN, -NH2, -N(СH3)2, галогена, C1-6 алкилкарбонила, C1-6 алкила, C1-6 галогеналкила, C3-8 циклоалкила, C3-8 циклоалкил-C1-6 алкила-, C6арил-C1-6 алкила-, 3-6-членного моноциклического гетероциклоалкила, содержащего N или O в качестве гетероатома, 3-6-членного гетероциклоалкил-C1-6 алкила-, содержащего N или O в качестве гетероатома, 7-11-членного спирогетероциклила, содержащего N в качестве гетероатома; где указанный 3-6-членный гетероциклоалкил, 3-6-членный гетероциклоалкил-C1-6 алкил-, C6арил-C1-6 алкил- и 7-11-членный спирогетероциклил необязательно содержат в качестве заместителя 1-2 Ry; при этом Ry выбран из галогена, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси, C3-8 циклоалкила, необязательно содержащего в качестве заместителя 1-2 галогена, C3-8 циклоалкил-C1-6 алкила-;
Ry предпочтительно выбран из F, метила, этила, изопропила, метокси, 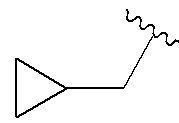 ,
, 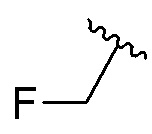 и FCH2CH2-;
и FCH2CH2-;
при этом, когда Rx присоединен непосредственно к атому N, Rx не представляет собой -OH, -NH2 и галоген;
или
Rx выбран из -OH, -CN, -NH2, F, метила, этила, изопропила, трифторэтила, метилкарбонила,
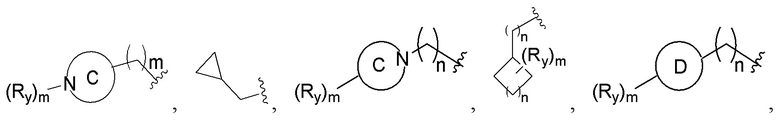
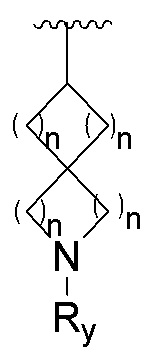 и
и 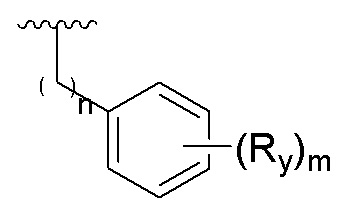 ;
;
при этом кольцо C представляет собой 4-8-членный гетероциклоалкил, кольцо D представляет собой 4-8-членный гетероциклоалкил, содержащий кислород; m представляет собой 0, 1 или 2; n представляет собой 0, 1, 2 или 3; Ry определен выше;
Rx предпочтительно выбран из -OH, -CN, -NH2, метила, этила, изопропила, метилкарбонила, 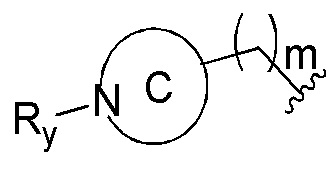 ,
,  и
и 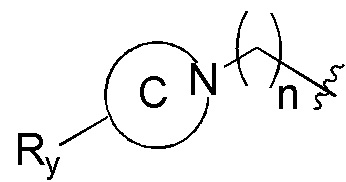 ; при этом кольцо C представляет собой 4-8-членный гетероциклоалкил; m и n независимо представляют собой 0, 1, 2 или 3; Ry определен выше;
; при этом кольцо C представляет собой 4-8-членный гетероциклоалкил; m и n независимо представляют собой 0, 1, 2 или 3; Ry определен выше;
Rx предпочтительно выбран из -OH, -CN, -NH2, F, метила, этила, изопропила, трифторэтила, метилкарбонила,
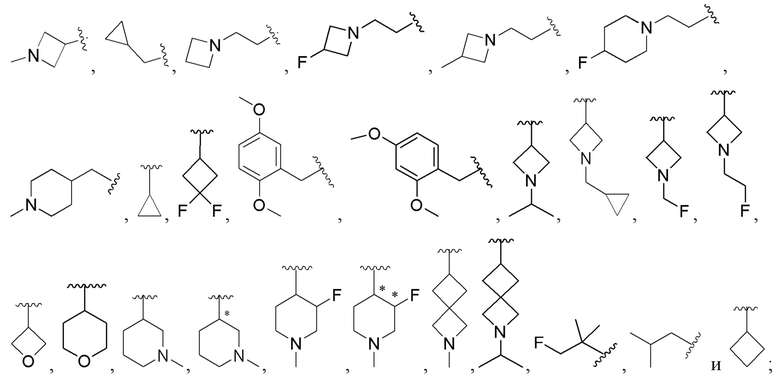
Rx предпочтительно выбран из метила, этила, изопропила,
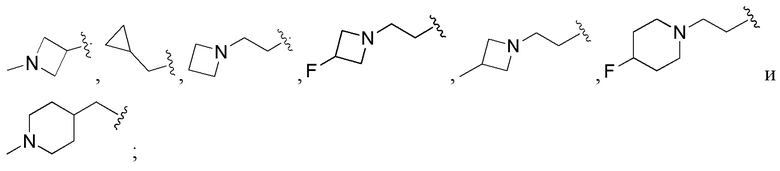
Rx предпочтительно выбран из метила, этила, изопропила,
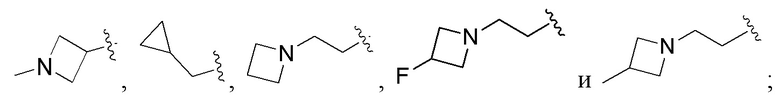
Rx предпочтительно выбран из метила, этила и изопропила;
Rx предпочтительно выбран из метила и изопропила.
6. Соединение по п. 5, в котором структурное звено 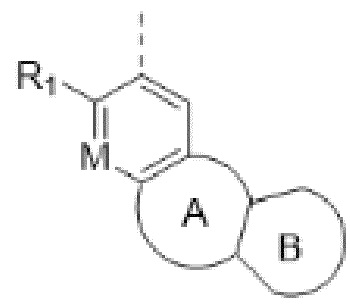 или
или  выбрано из
выбрано из  , при этом R1, M и Rx определены в п. 5; или
, при этом R1, M и Rx определены в п. 5; или
структурное звено  или
или  выбрано из
выбрано из 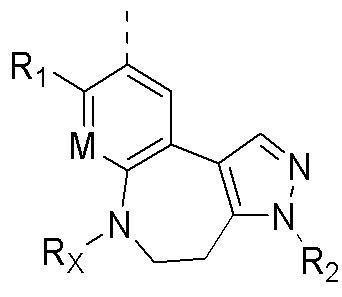 , при этом M, R1, R2 и Rx определены в п. 5.
, при этом M, R1, R2 и Rx определены в п. 5.
7. Соединение по любому из пп. 1-6, выбранное из
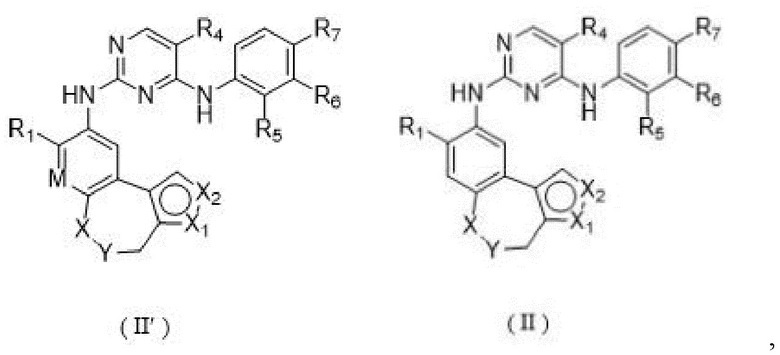
в котором каждый из X и Y независимо выбран из -NRx-, -O- и -CR9R10-;
каждый из X1 и X2 независимо выбран из N и NR2;
R9 и R10 выбраны из H;
R1 определен в любом из пп. 1-6;
R2 определен в любом из пп. 1-6;
R4 определен в любом из пп. 1-6;
R5 определен в любом из пп. 1-6;
R6 и R7 определены в любом из пп. 1-6;
Rx определен в любом из пп. 5, 6;
M, если присутствует, определен в любом из пп. 1-6;
или
указанное соединение выбрано из
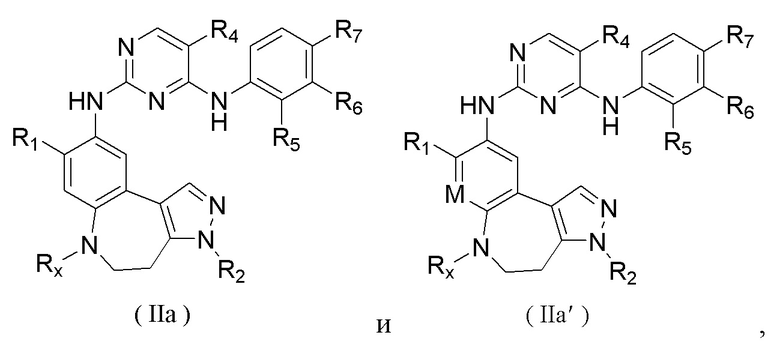
где R1, R2, R4, R5, R6, R7 и Rx определены выше; M, если присутствует, определен выше;
или
указанное соединение выбрано из
или
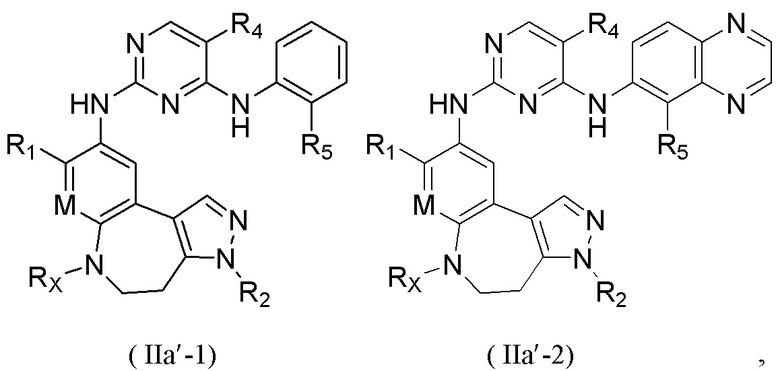
где R1, R2, R4, R5 и Rx определены выше; M, если присутствует, определен выше;
или
указанное соединение выбрано из
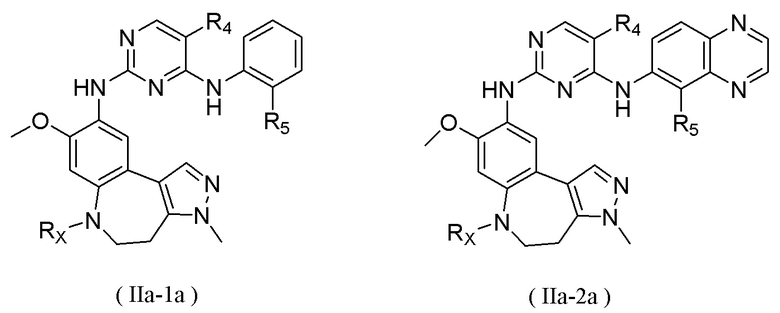
или
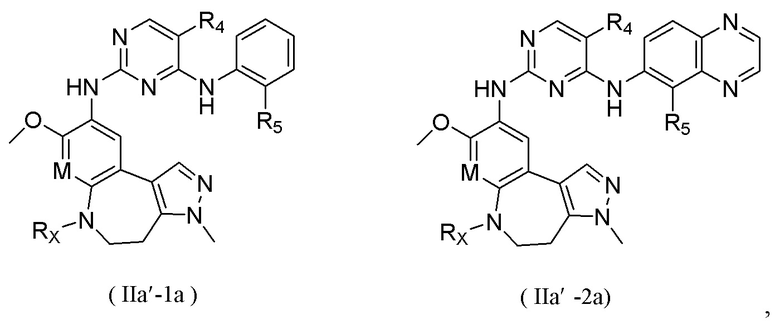
где R4, R5 и Rx определены выше; M, если присутствует, определен выше;
или
указанное соединение выбрано из
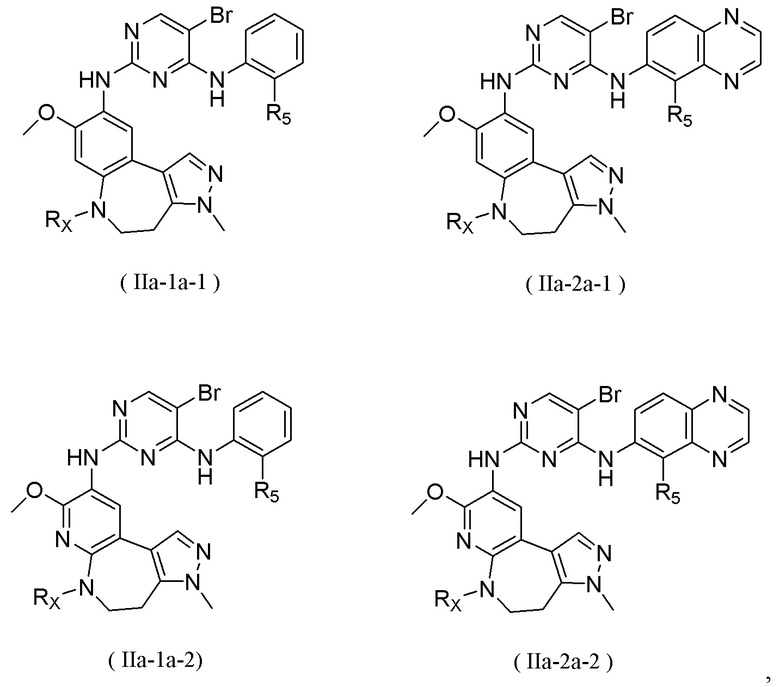
где R5 и Rx определены выше.
8. Соединение или его стереоизомер, фармацевтически приемлемая соль, гидрат, сольват по любому из пп. 1-7, выбранное из
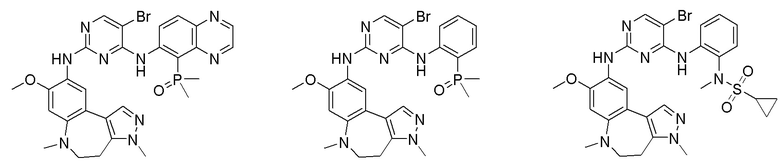
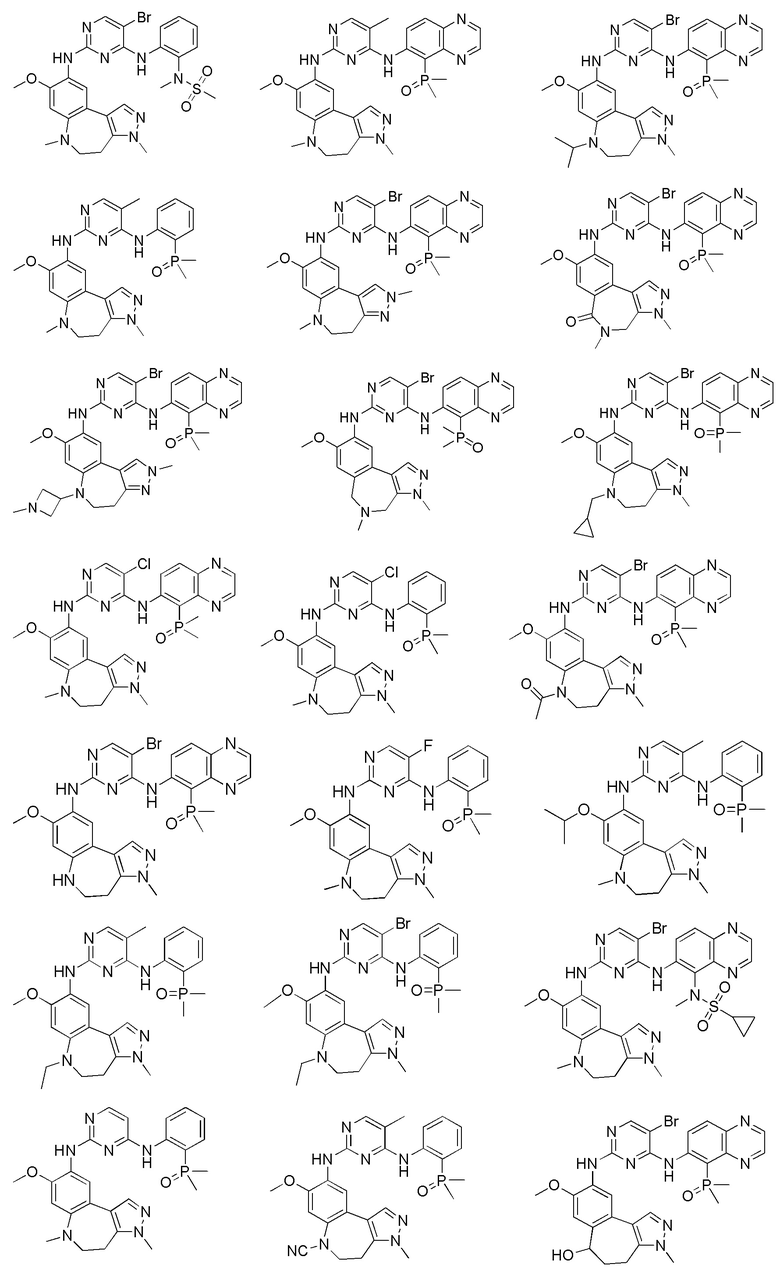
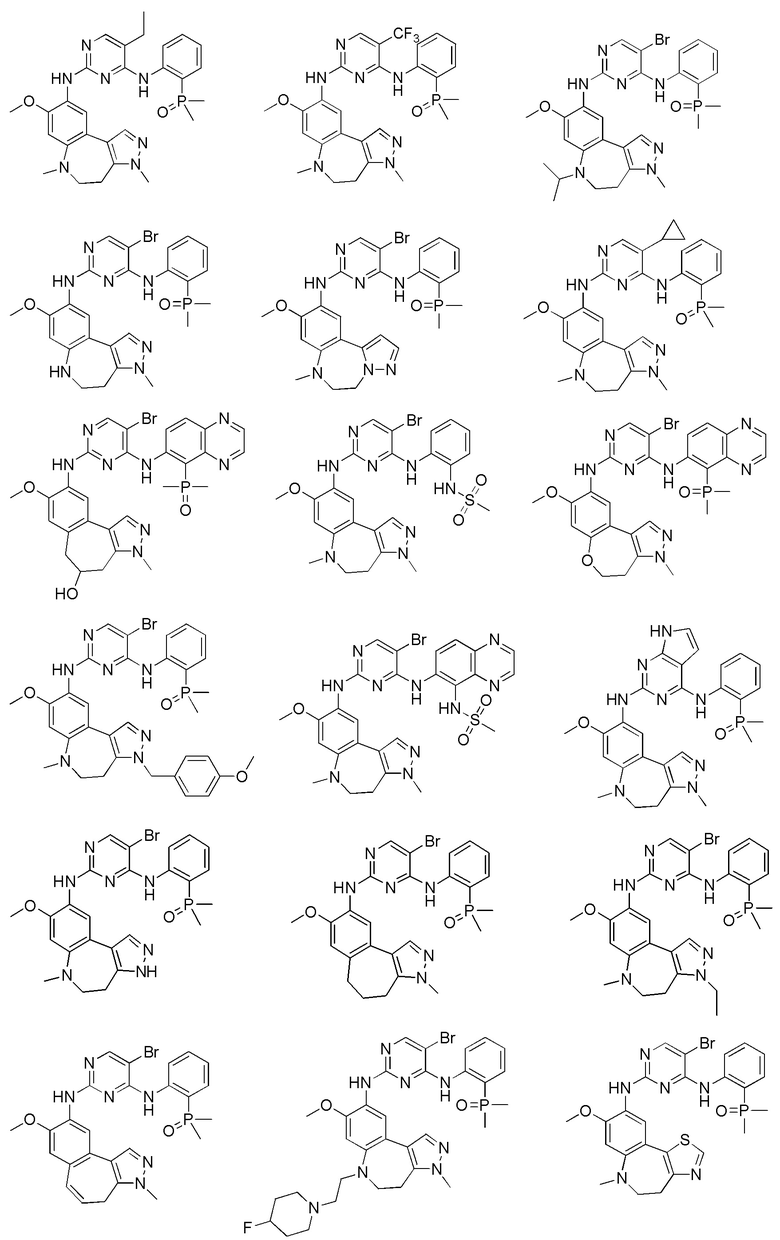
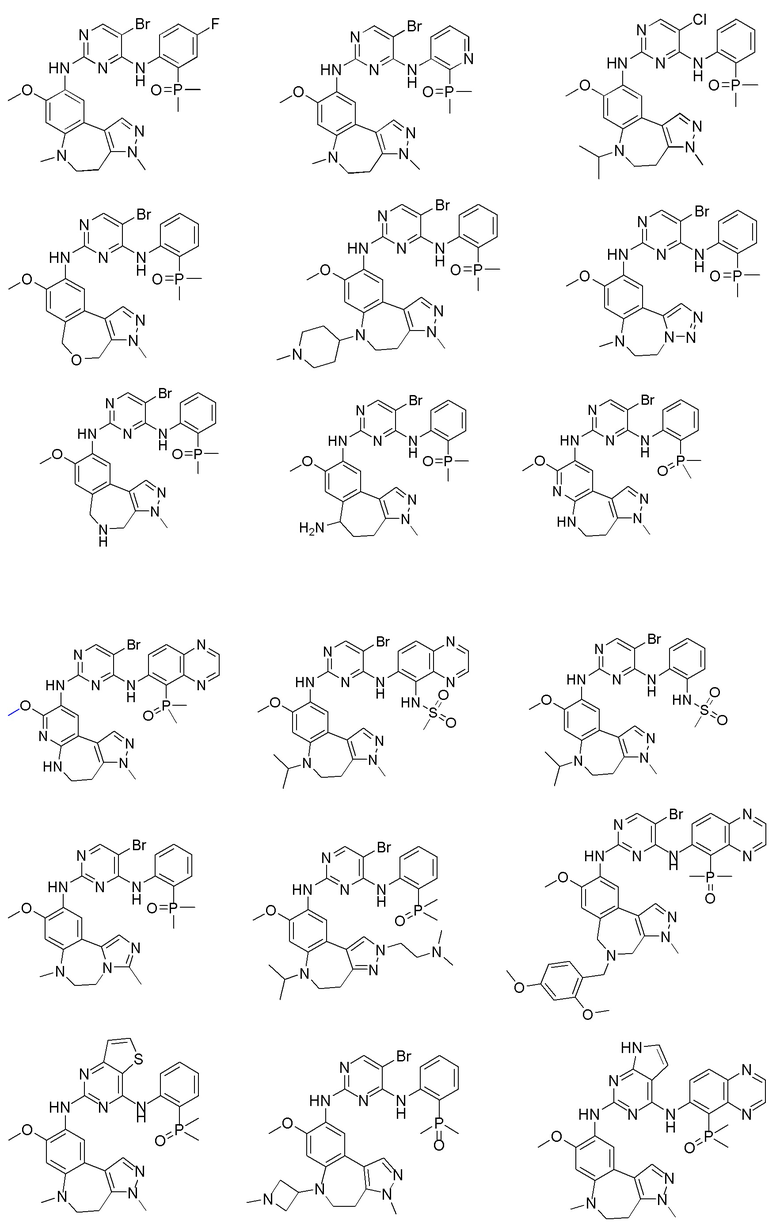
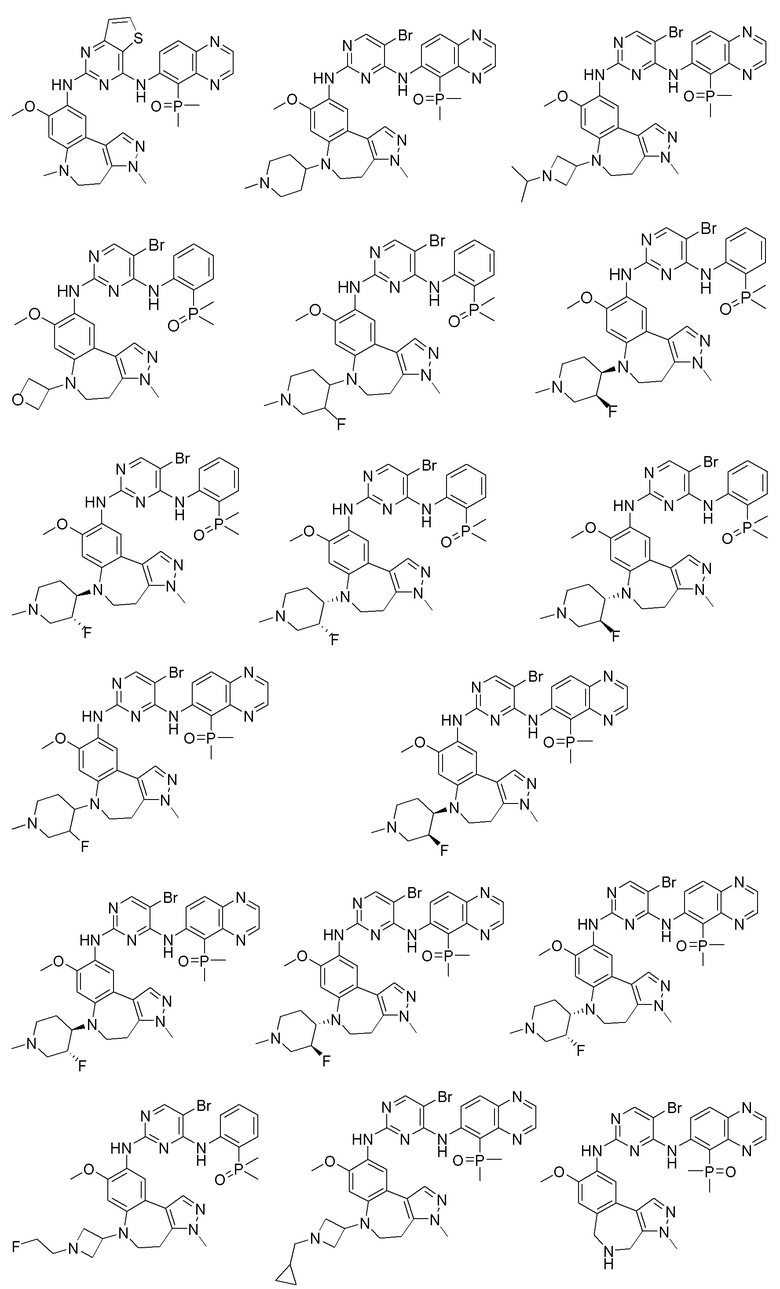
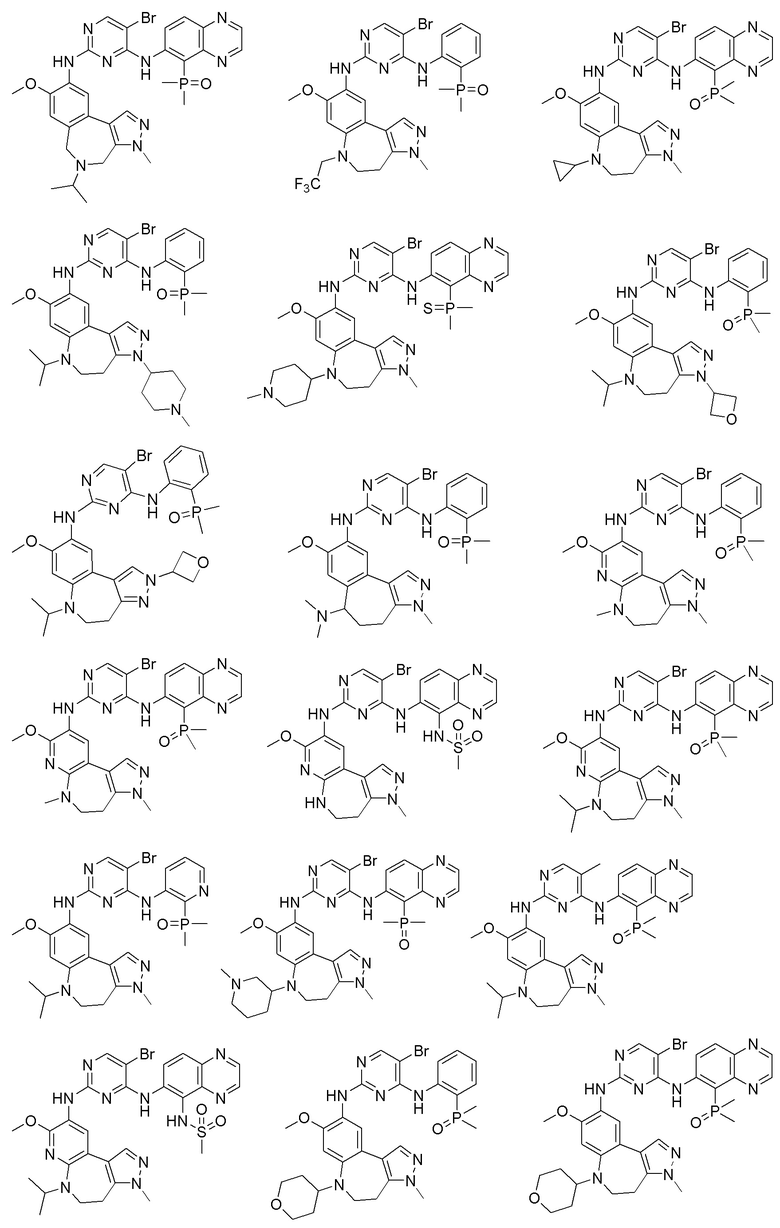
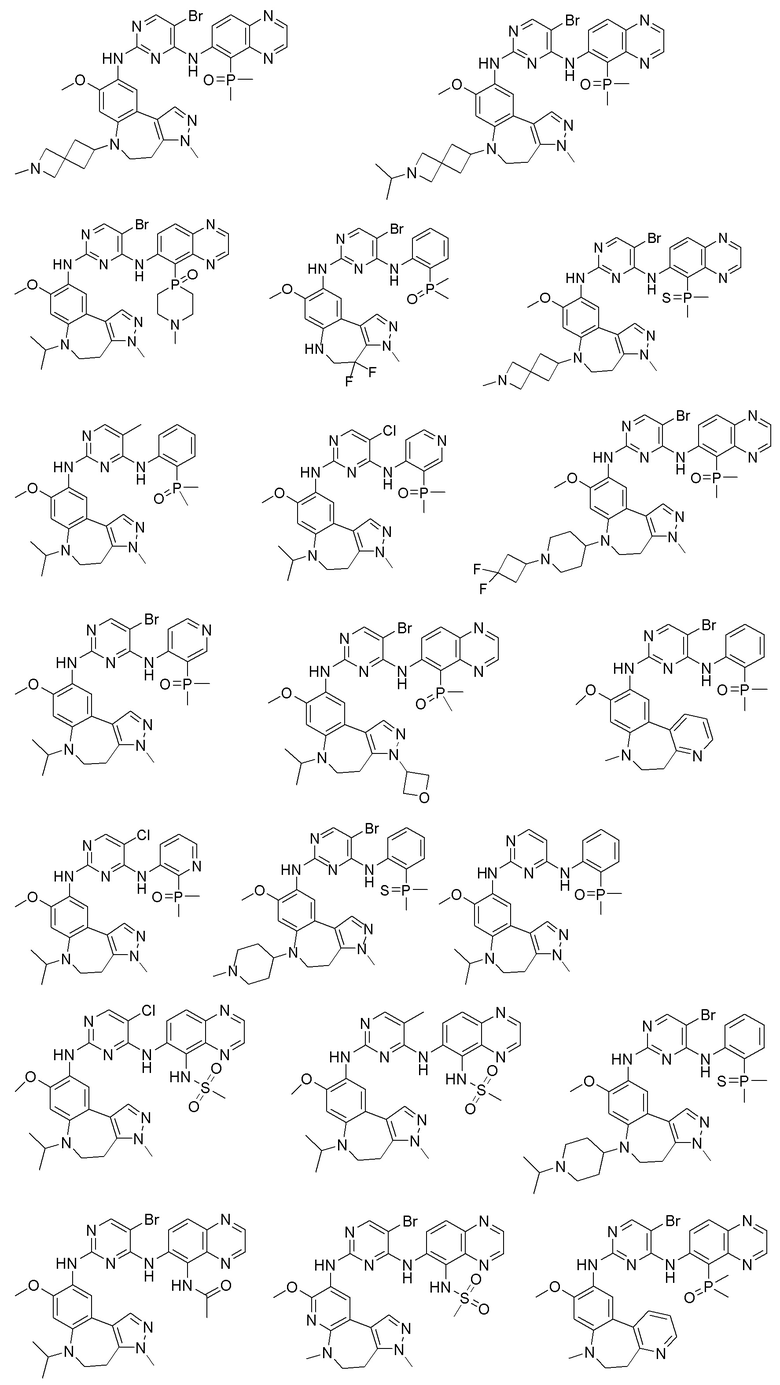
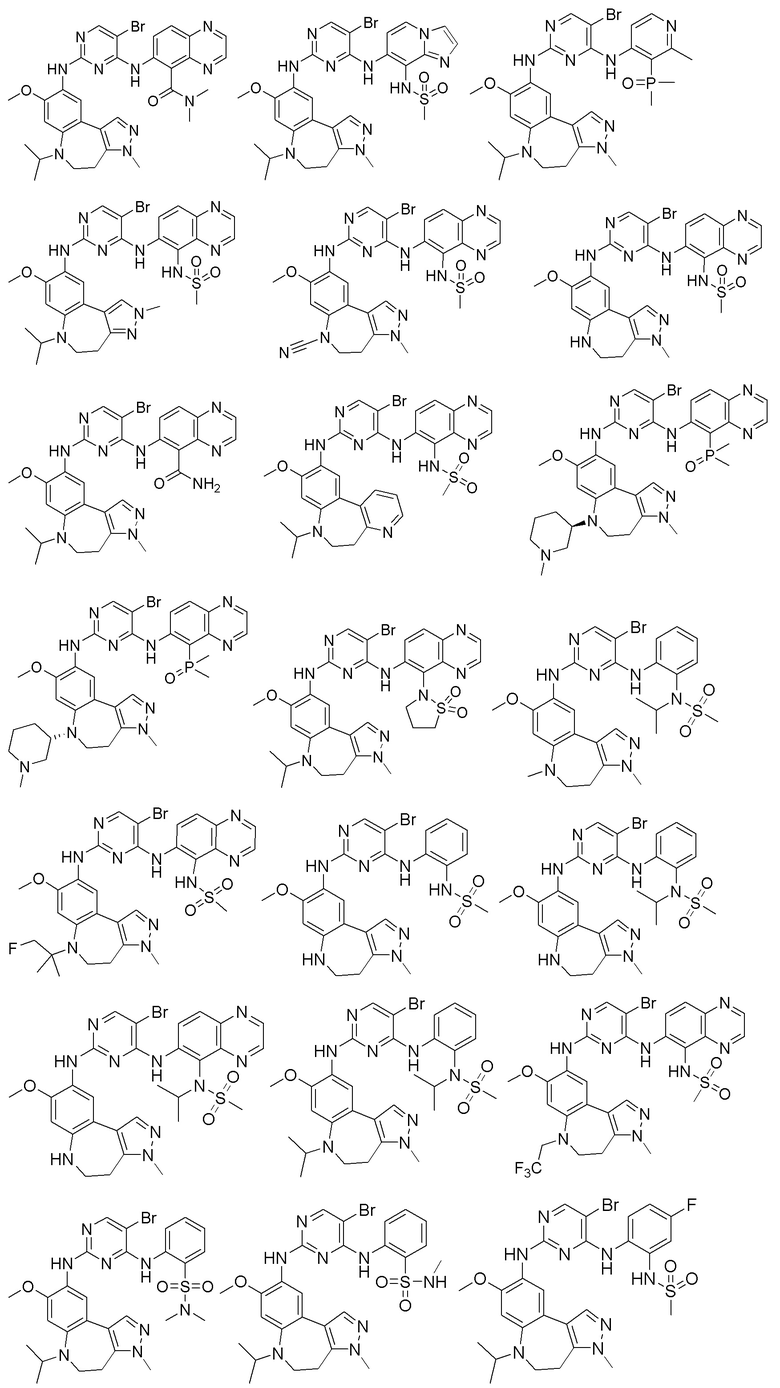
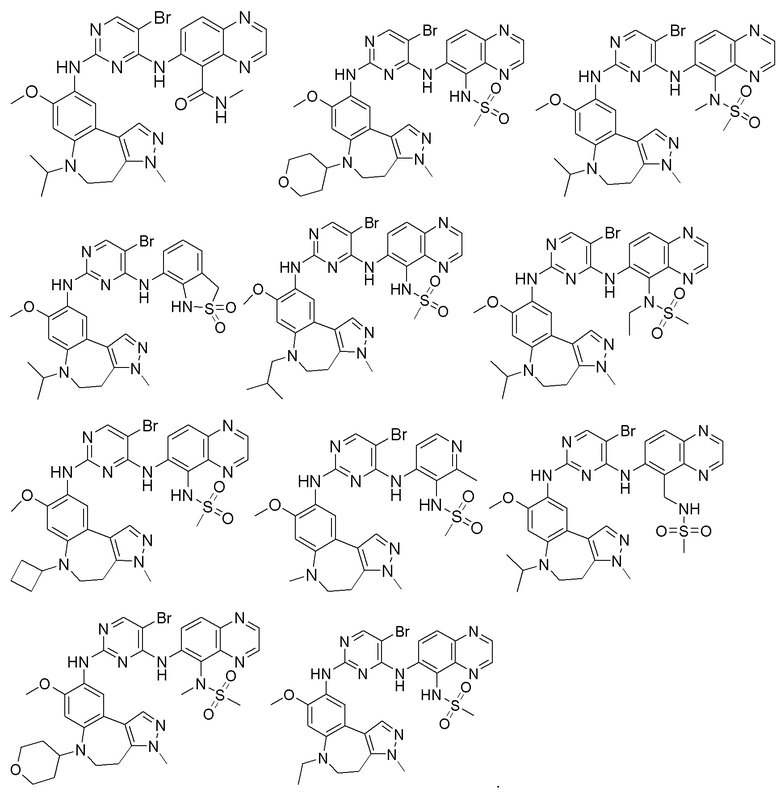
9. Фармацевтическая композиция для ингибирования EGFR, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-8 в эффективном количестве и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
10. Способ лечения рака, опосредованного EGFR, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 9, при этом рак включает лимфому, неходжкинскую лимфому, рак яичника, рак шейки матки, рак предстательной железы, колоректальный рак, рак молочной железы, рак поджелудочной железы, глиому, глиобластому, меланому, лейкоз, рак желудка, рак эндометрия, рак легкого, печеночноклеточный рак, рак желудка, желудочно-кишечную стромальную опухоль (GIST), острый миелогенный лейкоз (AML), холангиокарциному, рак почки, рак щитовидной железы, анапластическую крупноклеточную лимфому, мезотелиому, множественную миелому и меланому.
11. Способ по п. 10, в котором рак представляет собой рак легкого.
12. Соединение, представленное формулой (Vа) или (Vа’), или его стереоизомер или его фармацевтически приемлемая соль:
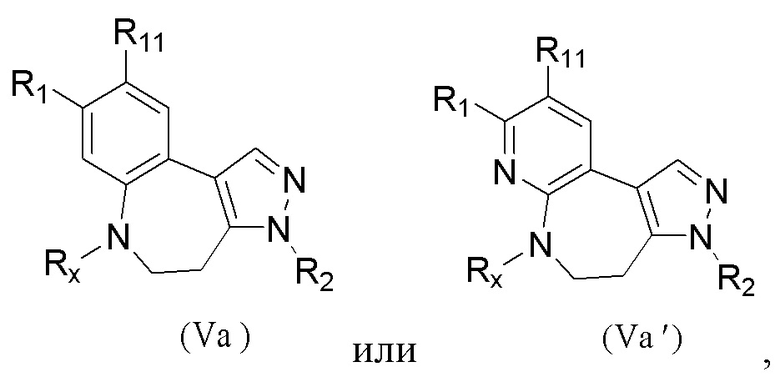
в котором R1 определен в любом из пп. 1-7;
R11 представляет собой -NH2 или -NO2;
R2 определен в любом из пп. 1-7;
Rx определен в любом из пп. 1-7.
13. Соединение по п. 12, которое представлено формулой (Vа-1) или (Vа-1’), или его стереоизомер или его фармацевтически приемлемая соль:
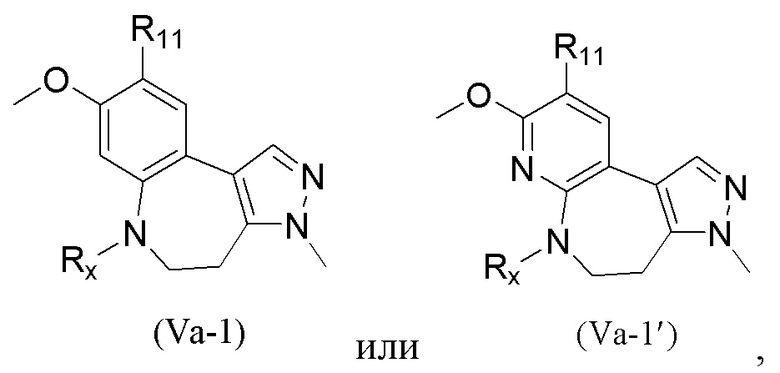
где R11 и Rx определены выше.
| WO 2008051547 A1, 02.05.2008 | |||
| WO 2008073687 A2, 19.06.2008 | |||
| Database REGISTRY [online], RN 1189306-45-3, 21.10.2009 | |||
| Retrieved from STN | |||
| Database CHEMCATS [online], RN 326914-45-8, 13.03.2001 | |||
| Retrieved from STN | |||
| WO 2006034833 A1, 06.04.2006 | |||
| ПРИБОР ДЛЯ ОПРЕДЕЛЕНИЯ КАЧЕСТВА МУКИ | 1926 |
|
SU12112A1 |

