В настоящей заявке испрашивается приоритет по заявке на патент Китая CN 201911256773.5, поданной 3 декабря 2019 года, содержание которой полностью включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области медицинской химии, в частности к группе соединений, обладающих ингибирующим эффектом в отношении Aurora киназы, а также к способу их получения и применению.
УРОВЕНЬ ТЕХНИКИ
Aurora киназы - это треонин/сериновые протеинкиназы, которые играют ключевую роль в таких важных митотических событиях, как репликация центросом, формирование биполярного веретена, хромосомная перестройка и мониторинг хромосомной контрольной точки (Cancer Metastasis Rev., 2003, 22, 451). В настоящее время известно, что в клетках человека существует 3 структурно и функционально высокородственных подтипа киназ Aurora, а именно Aurora-A, Aurora-B и Aurora-C. Aurora-A располагается вокруг центросомы в профазе митоза, в микротрубочках около веретена в метафазе и на полярных микротрубочках в анафазе и телофазе. Она в основном отвечает за репликацию и разделение центросом, агрегацию биполярных веретен, митотический вход и выход, созревание центросом и сборку веретен (Nat. Rev. Cancer, 2005, 5, 42). Aurora-B находится в области центромеры хромосомы на ранней стадии митоза и перемещается с центромеры на микротрубочку в анафазе. Aurora-B регулирует функционирование центромеры, расположение и разделение хромосом, контрольный пункт веретена и цитокинез (Mol. Cancer Ther., 2009, 8, 2046-2056). Aurora-C в значительной степени экспрессируется в семенниках и может играть особую роль в мужском организме животных (Proc Natl Acad Sci USA, 2002, 99 (24): 15440-15445).
Ген, кодирующий Aurora-A, локализован в области 20ql3.2, которая обычно увеличивается во многих опухолях, таких как рак молочной железы, рак толстой кишки, рак яичников и рак щитовидной железы. Сверхэкспрессия Aurora-A в клетках вызывает проявление различных характеристик раковых клеток, таких как амплификация центросом, анеуплоидия, нестабильность хромосом и удлинение теломер (J. Cell Sci., 2007, 120, 2987).
Сверхэкспрессия Aurora-A или совместная экспрессия с ТРХ-2 вызывает хромосомную нестабильность. Кроме того, Aurora-A также вмешивается в функционирование важных факторов ингибирования опухолей и про-апоптотических белков, таких как р53, где фосфорилирование р53 Aurora-A на участке Ser215 и на участке Ser315 препятствует нормальной работе р53 и вызывает его деградацию, соответственно.
Aurora-B расположена на 17р13.1, и, в отличие от Aurora-A, эта область не увеличивается значительно во многих видах рака, кроме глиомы мозга (J. Clin. Pathol, 2007, 60(2): 218-221). Однако мРНК и белок Aurora-B сверхэкспрессируются в быстро пролиферирующих клетках, таких как многие опухоли, например, рак толстой кишки, рак полости рта и немелкоклеточный рак легких. Таким образом, опухолевые клетки по-разному увеличивают экспрессию белков семейства Aurora. Белковый комплекс хромосомных пассажиров (СРС) является важным комплексом для регуляции митоза, и Aurora-B является его основным членом. Основными субстратами для фосфорилирования Aurora-B являются INCENP, CENP-A, Survivin и др. Aurora-B регулирует митоз путем фосфорилирования своих субстратов. Помимо митоза, сверхэкспрессия Aurora-B также усиливает путь сигнальной трансдукции онкогена RAS.
Уникальный механизм фармакологического действия и связь со злокачественными опухолями приводят к тому, что Aurora киназы становятся важными мишенями для исследования противоопухолевых препаратов, а ингибиторы Aurora киназ также рассматриваются как новые противоопухолевые препараты с хорошей перспективой развития. LY-3295668 является ингибитором Aurora-A киназы, содержащим в своей основе пиридиновое кольцо (WO 2016077161), и в настоящее время находится в первой фазе клинических исследований. LY-3295668 имеет следующую структурную формулу:
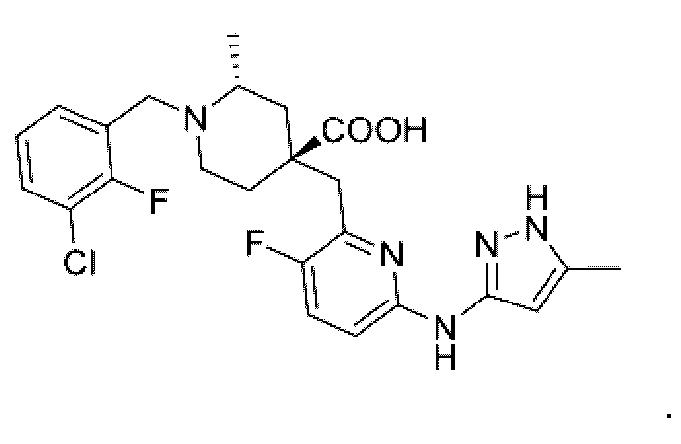
Однако LY-3295668 и другие ингибиторы Aurora-A киназы имеют ряд недостатков, таких как недостаточная активность Aurora киназы, недостаточная пероральная абсорбция и ограниченная противоопухолевая активность in vivo. Поэтому, учитывая проблемы существующих ингибиторов Aurora киназы, поиск нового ингибитора aurora с более высокой активностью in vitro и in vivo имеет большое значение.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
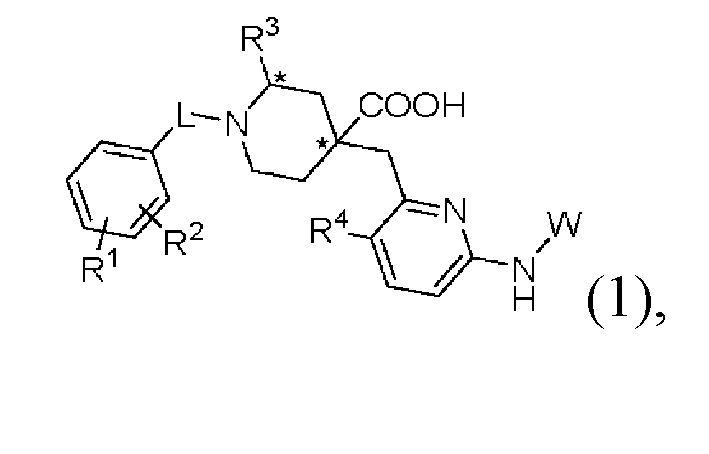
Настоящее изобретение раскрывает новую группу ингибиторов Aurora киназы со структурой, представленной формулой (1), или их оптические изомеры, кристаллические формы или фармацевтически приемлемые соли или сложные эфиры:
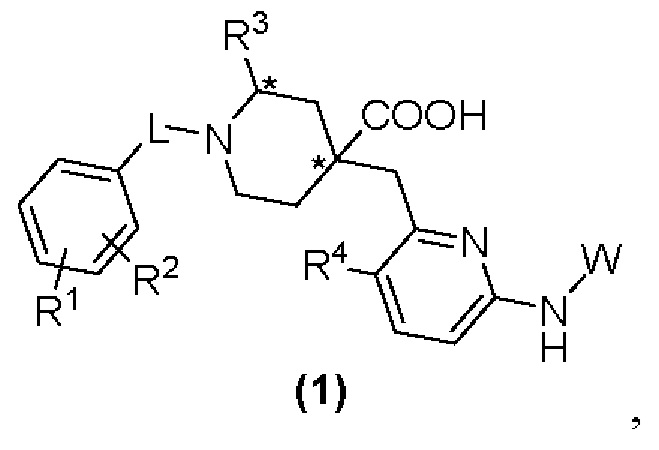
где в формуле (1):
«*» обозначает хиральный центр;
L представляет собой CH2 или СО;
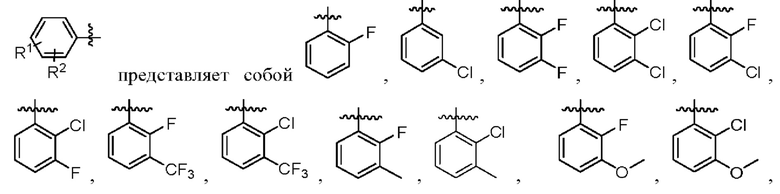
R1 и R2, каждый независимо, представляют собой Н, галоген, CN, С1-С3 алкил, С3-С6 циклоалкил, С1-С3 алкокси, С1-С3 галоалкил и С1-С3 галоалкокси;
R3 представляет собой С2-С3 алкил, С3-С6 циклоалкил, С1-С3 галоалкил, -(С1-С3)алкил-ОН, -(С1-С3)алкил-(С1-С3)алкокси, -(С1-С3)алкил-CN или -(C1-C3)алки л-NR5R6, где R5 и R6, каждый независимо, представляют собой Н или С1-С3 алкил, или R5 и R6 образуют 4-7-членный гетероциклоалкил вместе с атомом N; R4 представляет собой Н или F;
W представляет собой 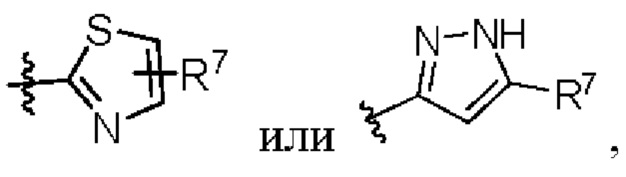 где R7 представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил.
где R7 представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил.
В другом предпочтительном варианте осуществления изобретения в формуле (1)
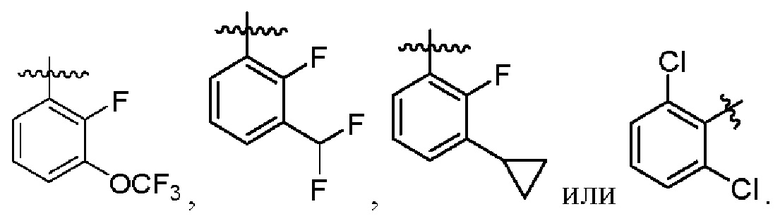
В другом предпочтительном варианте осуществления изобретения в формуле (1) R3 представляет собой следующие группы: Et, n-Pr, i-Pr, 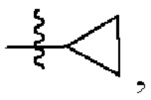 CH2F, CHF2, CF3, СН2ОН, СН2ОМе, CH2OEt, CH2CN,
CH2F, CHF2, CF3, СН2ОН, СН2ОМе, CH2OEt, CH2CN, 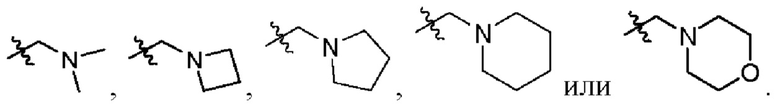
В другом предпочтительном варианте осуществления изобретения в формуле (1) W представляет собой следующие группы:
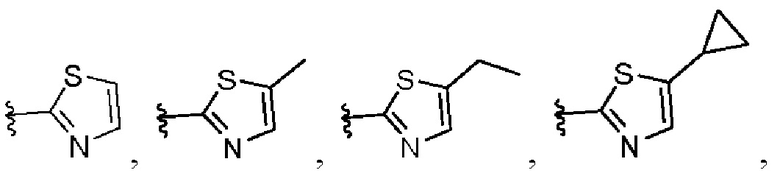
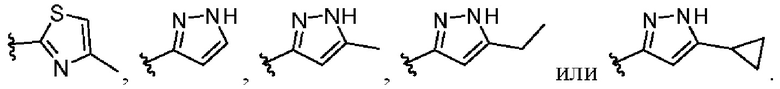
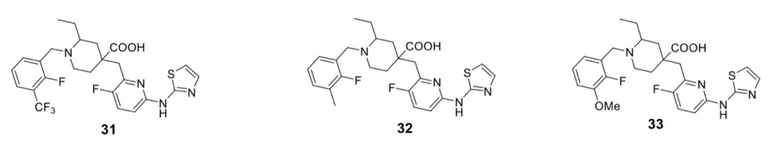
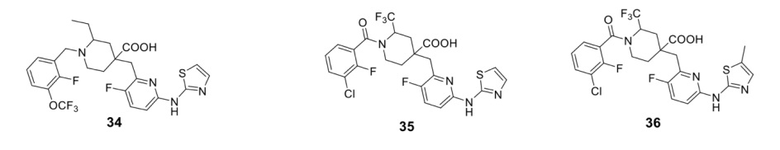
В различных вариантах осуществления представленные соединения в рамках настоящего изобретения имеют одну из следующих структур:
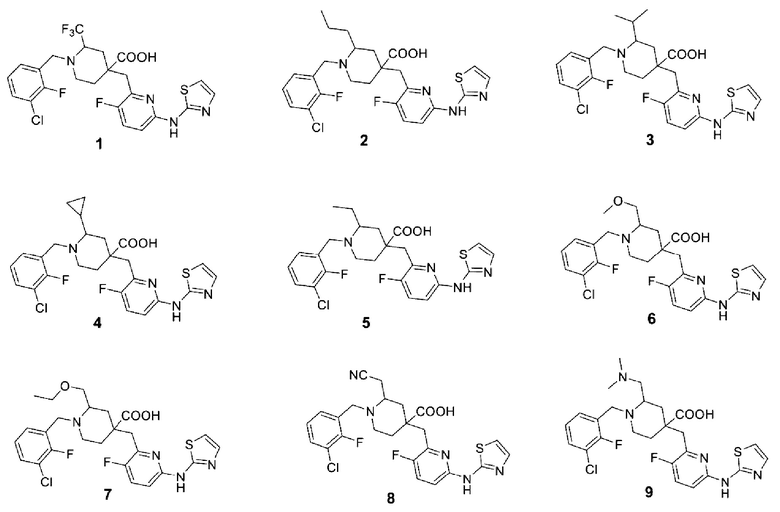
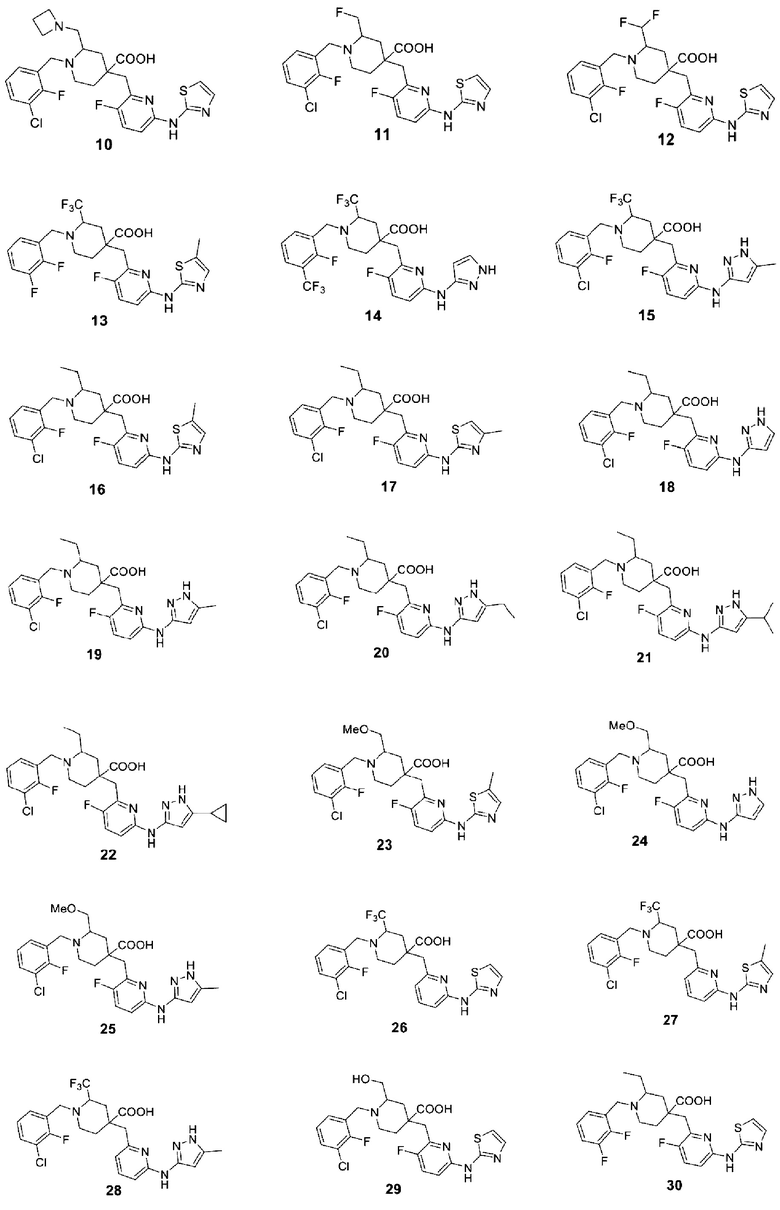
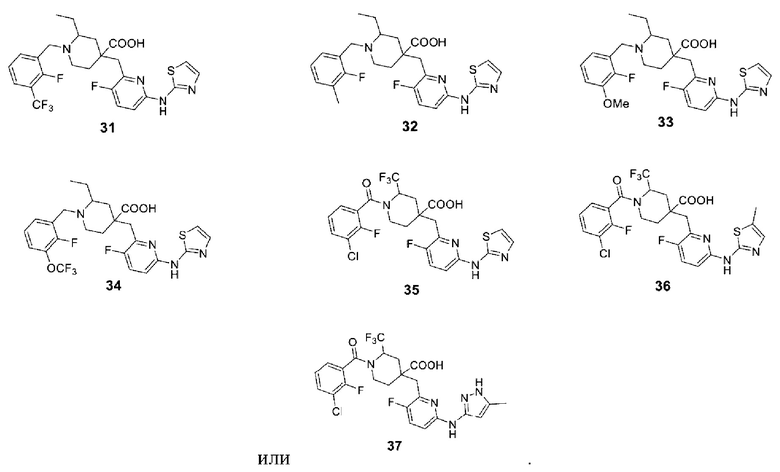
Другой целью настоящего изобретения является получение фармацевтической композиции, включающей фармацевтически приемлемое вспомогательное вещество или носитель и соединение формулы (1) или его оптические изомеры, кристаллические формы или фармацевтически приемлемые соли или сложные эфиры, раскрытые в настоящем документе в качестве активного ингредиента.
Еще одной целью настоящего изобретения является применение соединения или его оптических изомеров, кристаллических форм или фармацевтически приемлемых солей или сложных эфиров, описанных выше, для приготовления лекарств для лечения заболеваний, связанных с Aurora, особенно противоопухолевых лекарств.
Получение соединений
Далее подробно описаны способы получения соединений общей формулы (1), но эти конкретные способы не ограничивают настоящее изобретение.
Соединение формулы (1), описанное выше, может быть получено с использованием стандартных синтетических способов или хорошо известных способов в сочетании со способами, описанными в настоящем документе. Кроме того, растворители, температура и другие условия реакции, упомянутые в настоящем документе, могут варьироваться. Исходные материалы для получения соединений, приведенных в таблице 1, могут быть получены синтетическим путем или из коммерческих источников, таких как, но не ограничиваясь ими, Aldrich Chemical Co. (Milwaukee, Wis.) или Sigma Chemical Co. (St. Louis, Mo.). Описанные здесь соединения и другие сопутствующие соединения, имеющие различные заместители, могут быть получены с использованием хорошо известных способов и исходных материалов, включая те, которые можно найти в March, Advanced Organic Chemistry, 4th Ed., (Wiley 1992); Carey and Sundberg, Advanced Organic Chemistry, 4th Ed., Vols. А и В (Plenum 2000, 2001), и Green and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., (Wiley 1999). Общие способы получения соединений могут быть изменены путем использования подходящих реагентов и условий для добавления различных групп в формулы, представленные в настоящем документе. В одном аспекте соединение формулы (1), описанное в настоящем документе, получают в соответствии с известными способами. Однако условия процесса, такие как реактивы, растворители, основания, количество используемого соединения, температура реакций и время, необходимое для реакций, и тому подобное, не ограничиваются следующими пояснениями. Соединения, раскрытые в настоящем документе, также могут быть легко получены по выбору сочетанием различных синтетических способов, описанными в настоящем документе, или хорошо известными способами, и такие сочетания могут быть легко определены специалистами в области, к которой относится настоящее изобретение. В другом аспекте настоящее изобретение также предлагает способ получения соединений формулы (1), которые получают следующим способом А, способом В или способом С: Способ А включает следующие этапы: во-первых, соединение А восстанавливают путем гидрирования в соединение В в присутствии катализатора, соединение В и фрагмент S1 образуют соединение С в щелочном состоянии, соединение С соединяют с фрагментом S2 в щелочном состоянии для образования соединения D, соединение D реагирует с фрагментом S3 в щелочном состоянии и в присутствии металлического палладиевого катализатора и лиганда для получения соединения Е, и соединение Е подвергается реакции гидролиза сложного эфира в кислотном или щелочном состоянии для получения целевого соединения (1а).
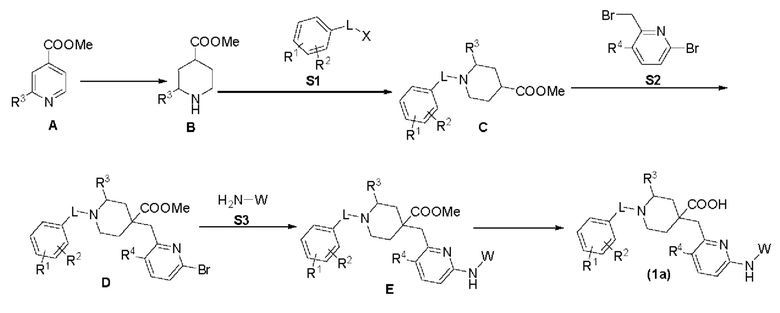
В вышеуказанной реакции R1, R2, R3, R4, L и W соответствуют вышеприведенным определениям, а X представляет собой I, Br, Cl, OTf, ОН или тому подобное. Способ В включает следующие стадии: во-первых, соединение В подвергается Вос-защите в соответствующих условиях для получения соединения F, соединение F реагирует с фрагментом S2 в щелочном состоянии для получения соединения G, соединение G реагирует с фрагментом S3 в присутствии металлического палладиевого катализатора и лиганда для получения соединения Н, соединение Н подвергается снятию Вос-защиты в кислой среде для получения соединения I, соединение I реагирует с фрагментом S1 в щелочной среде для получения соединения J, и соединение J подвергается реакции гидролиза сложного эфира в кислой или щелочной среде для получения целевого соединения (1b).
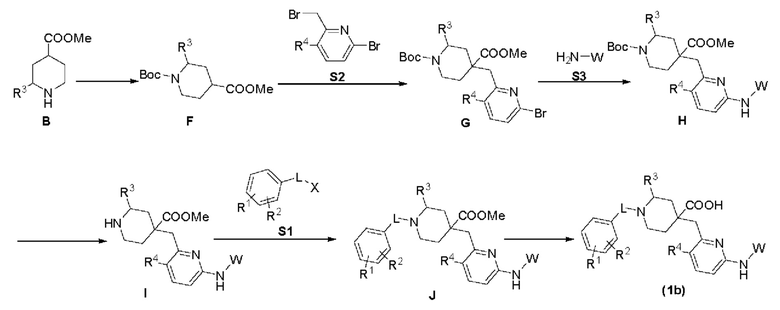
В вышеуказанной реакции R1, R2, R3, R4, L и W соответствуют приведенным выше определениям, а X представляет собой I, Br, Cl, OTf, ОН или тому подобное. Способ С включает следующие стадии: сначала соединение G подвергают снятию Вос-защиты в кислой среде для получения соединения K, соединение K конденсируют с фрагментом S1 для получения соединения L, соединение L реагирует с фрагментом S3 в присутствии металлического палладиевого катализатора и лиганда для получения соединения М, и соединение М подвергают реакции гидролиза сложного эфира в кислой или щелочной среде для получения целевого соединения (1с).
В вышеуказанной реакции R1, R2, R3, R4, L и W соответствуют приведенным выше определениям, а X представляет собой I, Br, Cl, OTf, ОН или тому подобное.
Дополнительные варианты соединений
В настоящем патентном описании, включая прилагаемую формулу изобретения, вышеупомянутые заместители имеют следующие значения:
«Галоген» (или гало) относится к фтору, хлору, брому или йоду. Термин «гало» перед названием группы означает, что группа частично или полностью галогенирована, то есть замещена в любой комбинации F, Cl, Вг или I, предпочтительно F или Cl. «С1-3 алкил» относится к прямым или разветвленным алкильным группам, содержащим от 1 до 3 атомов углерода. «С2-3 алкил» относится к прямым или разветвленным алкильным группам, содержащим от 2 до 3 атомов углерода. «С1-3 галоалкил» означает, что С1-3 алкил, как определено выше, содержит один или несколько заместителей атома галогена. «С3-6 циклоалкил» означает неароматическую циклическую группу, содержащую от 3 до 6 атомов углерода. «С1-3 алкокси» относится к С1-3 алкил-О- группе, связанной с родительским фрагментом молекулы через кислород. -(С1-С3)алкил-ОН, (С1-С3)алкил-(С1-С3)алкокси, -(С1-С3)алкил-CN и -(С1-С3)алкил-NR5R6 означают группы, образованные путем соединения С1-С3 алкила, определенного выше, с ОН, (С1-С3)алкокси, CN и NR5R6 группами, соответственно, и соединенные с родительским фрагментом молекулы через (С1-С3)алкильную группу. «4-7-членный гетероциклоалкил» относится к неароматической насыщенной циклической группе, содержащей от 4 до 7 кольцевых атомов.
Термин «фармацевтически приемлемая соль» относится к форме соединения, которая не вызывает существенного раздражения организма при введении лекарственного средства или не устраняет биологическую активность и свойства соединения. Соль соединения по настоящему изобретению относится к соли, обычно используемой в области органической химии, и может быть, например, солью присоединения основания к карбоксильной группе при наличии карбоксильной группы и солью присоединения кислоты к аминогруппе или основной гетероциклической группе при наличии аминогруппы или основной гетероциклической группы.
Примерами соли присоединения основания могут быть соли щелочных металлов, такие как соли натрия и соли калия; соли щелочноземельных металлов, такие как соли кальция и соли магния; соли аммония; соли органических аминов, такие как соли триметиламина, соли триэтиламина, соли дициклогексиламина, соли этаноламина, соли диэтаноламина, соли триэтаноламина, соли прокаина, соли N,N-дибензилэтилендиамина, соли меглумина, соли аргинина и соли лизина и тому подобное.
Примерами соли присоединения кислоты могут быть соли неорганических кислот, такие как гидрохлорид, сульфат, нитрат и фосфат; соли органических кислот, такие как ацетат, формиат, малеат, фумарат, цитрат, оксалат и аскорбат; сульфонат, такой как метансульфонат, бензенсульфонат и n-толуолсульфонат, и тому подобное. Следует иметь в виду, что ссылки на фармацевтически приемлемые соли включают формы с добавлением растворителя или кристаллические формы, особенно сольваты или полиморфы. Сольват содержит стехиометрическое или нестехиометрическое количество растворителя и избирательно образуется при кристаллизации с фармацевтически приемлемыми растворителями, такими как вода и этанол. Гидраты образуются, когда растворителем является вода, или алкоголяты образуются, когда растворителем является этанол. Сольваты соединения формулы (1) легко получить или сформировать в соответствии со способами, описанными в настоящем документе. Например, гидрат соединения формулы (1) легко получить путем рекристаллизации в смешанном растворителе вода/органический растворитель, где используемый органический растворитель включает, но не ограничивается, диоксан, тетрагидрофуран, этанол или метанол. Кроме того, упомянутые здесь соединения могут существовать как в несольватированной, так и в сольватированной форме. Обычно сольватированные формы считаются эквивалентными несольватированным формам для целей соединений и способов, представленных в настоящем документе.
В других конкретных вариантах осуществления изобретения соединения формулы (1) получают в различных формах, включая, но не ограничиваясь, аморфные, пульверизированные формы и формы наночастиц. Кроме того, соединение формулы (1) включает кристаллические формы, а также может быть полиморфом. Полиморфы включают в себя различные расположения решеток одних и тех же элементов соединения. Полиморфы обычно имеют различные рентгеновские дифракционные картины, инфракрасные спектры, точки плавления, плотность, твердость, кристаллические формы, оптические и электрические свойства, стабильность и растворимость. Различные факторы, такие как растворитель для рекристаллизации, скорость кристаллизации и температура хранения, могут привести к преобладанию монокристаллической формы. В другом аспекте соединение формулы (1) имеет один или несколько стереоцентров и, таким образом, существует в форме рацемата, рацемической смеси, одного энантиомера, диастереомерного соединения и одного диастереомера. Асимметрические центры, которые могут присутствовать, зависят от природы различных заместителей в молекуле. Каждый из этих асимметрических центров независимо дает два оптических изомера, и все возможные оптические изомеры, диастереомерные смеси и чистые или частично чистые соединения входят в объем настоящего изобретения. Настоящее изобретение охватывает все такие изомерные формы этих соединений.
Терапевтическое применение
Соединения или композиции, описанные в настоящем документе, как правило, могут быть применены для ингибирования Aurora киназы и, таким образом, могут быть применены для лечения одного или нескольких расстройств, связанных с активностью Aurora киназы. Поэтому в некоторых вариантах осуществления настоящее изобретение предусматривает способ лечения расстройства, опосредованного Aurora киназой, который включает этап введения пациенту, нуждающемуся в этом, соединения, раскрытого в настоящем документе, или фармацевтически приемлемой композиции на его основе. Раковые заболевания, которые можно лечить с помощью соединения, раскрытого в настоящем документе, включают, но не ограничиваются, гематологические злокачественные опухоли (лейкемии, лимфомы, миеломы, включая множественную миелому, миелодиспластический синдром и миелопролиферативный синдром), твердые опухоли (карциномы, такие как карциномы простаты, молочной железы, легких, толстой кишки, поджелудочной железы, почек, яичников и карциномы мягких тканей, остеосаркома и интерстициальные опухоли) и тому подобное.
Способ введения
Соединение и фармацевтически приемлемая его соль, раскрытая в настоящем документе, могут быть приготовлены в виде различных препаратов, включающих соединение или его фармацевтически приемлемую соль, раскрытую в настоящем документе, в безопасном и эффективном количестве, и фармацевтически приемлемое вспомогательное вещество или носитель, где «безопасное и эффективное количество» означает, что количество соединения достаточно для значительного улучшения состояния, не вызывая серьезных побочных эффектов. Безопасное и эффективное количество соединения определяется в зависимости от возраста, состояния, курса лечения и других специфических условий получающего лечение субъекта.
Термин «фармацевтически приемлемое вспомогательное вещество или носитель» относится к одному или нескольким совместимым твердым или жидким наполнителям или гелеобразным веществам, которые подходят для применения человеком и должны обладать достаточной чистотой и достаточно низкой токсичностью. Термин «совместимый» означает, что компоненты композиции способны смешиваться с соединением, раскрытым в настоящем документе, и друг с другом без существенного снижения фармацевтической эффективности соединения. Примерами фармацевтически приемлемых вспомогательных веществ или носителей являются целлюлоза и ее производные (например, карбоксиметилцеллюлоза натрия, этилцеллюлоза натрия или ацетат целлюлозы), желатин, тальк, твердые смазочные материалы (например, стеариновая кислота или стеарат магния), сульфат кальция, растительное масло (например, соевое масло, кунжутное масло, арахисовое масло или оливковое масло), полиолы (например, пропиленгликоль, глицерин, маннит или сорбит), эмульгаторы (например, Tween®), смачивающие агенты (например, лаурилсульфат натрия), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, вода, не содержащая пирогенов, и тому подобное.
Соединение, раскрытое в настоящем документе, может вводиться перорально, ректально, парентерально (внутривенно, внутримышечно или подкожно) или местно. Твердые лекарственные формы для перорального применения включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах активное соединение смешивают по меньшей мере с одним обычным инертным вспомогательным веществом (или носителем), таким как цитрат натрия или дикальцийфосфат, или со следующими ингредиентами: (а) наполнители или разбавители, такие как крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие вещества, такие как гидроксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и акация; (с) увлажнители, такие как глицерин; (d) дезинтегранты, такие как агар, карбонат кальция, крахмал картофеля или тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (е) замедлители растворения, такие как парафин; (f) ускорители всасывания, такие как четвертичные аммониевые соединения; (g) смачивающие агенты, такие как цетиловый спирт и моностеарат глицерина; (h) адсорбенты, такие как каолин; и (i) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль и лаурилсульфат натрия, или их смеси. В случае капсул, таблеток и пилюль лекарственные формы могут также включать буферы.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с использованием покрытий и оболочек, таких как энтеральные покрытия и другие материалы, хорошо известные в данной области. Они могут включать опалесцирующие агенты, и активное соединение или соединение в такой композиции могут высвобождаться в определенной части пищеварительного тракта с задержкой.
Примерами компонентов для встраивания, которые могут быть использованы, являются полимерные вещества и вещества на основе воска. При необходимости активное соединение также может находиться в форме микрокапсулы с одним или несколькими вышеупомянутыми вспомогательными веществами.
Жидкие лекарственные формы для перорального применения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному соединению жидкая лекарственная форма может включать инертные разбавители, обычно используемые в данной области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, особенно хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, или смеси этих веществ. Кроме таких инертных разбавителей, композиция может включать адъюванты, такие как смачивающие агенты, эмульгаторы, суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Суспензии, помимо активного соединения, могут включать суспендирующие агенты, такие как этоксилированные изостеариловые спирты, полиоксиэтилен сорбитол и эфиры сорбитана, микрокристаллическую целлюлозу, метилат алюминия и агар, или смеси этих веществ.
Композиции для парентерального введения могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для повторного растворения в стерильных инъекционных растворах или дисперсиях. Подходящие водные и неводные носители, разбавители, растворители или вспомогательные вещества включают воду, этанол, полиолы и их приемлемые смеси. Лекарственные формы для местного применения соединения, раскрытого в настоящем документе, включают мази, порошки, пластыри, спреи и ингаляторы. Активный ингредиент смешивается в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферами или пропеллентами, которые могут потребоваться при необходимости.
Соединение, раскрытое в настоящем документе, может применяться отдельно или в комбинации с другими фармацевтически приемлемыми соединениями. При использовании фармацевтической композиции безопасное и эффективное количество соединения, раскрытого в настоящем документе, вводится млекопитающему (например, человеку), подлежащему лечению, при этом вводимая доза представляет собой фармацевтически эффективную вводимую дозу. Для человека с массой тела 60 кг суточная вводимая доза обычно составляет 1-1000 мг, предпочтительно 10-500 мг. При определении конкретной дозировки также учитываются такие факторы, как способ введения, состояние здоровья пациента и т.п., которые хорошо известны опытным врачам.
Вышеуказанные признаки, упомянутые в настоящем изобретении, или признаки, упомянутые в примерах, могут комбинироваться произвольно. Все признаки, раскрытые в данном описании, могут быть использованы с любой формой композиции, а различные признаки, раскрытые в данном описании, могут быть заменены любыми альтернативными признаками, которые предусматривают ту же, эквивалентную или аналогичную цель. Таким образом, если явно не указано иное, раскрытые признаки являются лишь общими примерами эквивалентных или аналогичных признаков.
Различные конкретные аспекты, особенности и преимущества описанных выше соединений, способов и фармацевтических композиций подробно изложены в нижеприведенном описании, из которого становится очевидным настоящее изобретение. Следует иметь в виду, что приведенное ниже подробное описание и примеры описывают конкретные варианты осуществления только для сведения. После прочтения описания настоящего изобретения специалисты в данной области могут внести различные изменения или модификации в настоящее изобретение, и такие эквиваленты также попадают в объем настоящего изобретения, определенный в настоящем документе. Во всех примерах точки плавления измеряли с помощью прибора Х-4 с некалиброванным термометром; спектры 1H-ЯМР регистрировали на спектрометре ядерного магнитного резонанса Varian Mercury 400, и химические сдвиги измерялись в 5 (ppm); силикагель для разделения представлял собой силикагель 200-300 меш, если не указано иное, и соотношение элюентов представляло собой объемное соотношение.
В настоящем изобретении используются следующие сокращения: ACN представляет собой ацетонитрил; Ar представляет собой аргон; (Вос)2O представляет собой ди-терт-бутил дикарбонат; CDCl3 представляет собой дейтерированный хлороформ; CD3OD представляет собой дейтерированный метанол; DCM представляет собой дихлорметан; DIPEA представляет собой диизопропилэтиламин; Diox или Dioxane представляет собой 1,4-диоксан; DMAP представляет собой 4-диметиламинопиридин; DMF представляет собой диметилформамид; DMSO представляет собой диметилсульфоксид; ЕА представляет собой этилацетат; ч представляет собой час; K2CO3 представляет собой карбонат калия; KI представляет собой йодид калия; K3PO4 представляет собой фосфат калия; ЖХ-МС представляет собой жидкостную масс-спектрометрию; LDA представляет собой диизопропиламид лития; LiOH представляет собой гидроксид лития; мл представляет собой миллилитр; МеОН представляет собой метанол; мин представляет собой минуту; MS представляет собой масс-спектр; ЯМР представляет собой ядерный магнитный резонанс; Pd2(dba)3 представляет собой трис(дибензилиденэацетон)дипалладий; РЕ представляет собой петролейный эфир; PtO2 представляет собой диоксид платины; THF представляет собой тетрагидрофуран; Xantphos представляет собой 4,5-бис(дифенилфосфино)-9,9-диметилксантен.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Пример 1. Синтез 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоновой кислоты (соединение 1)
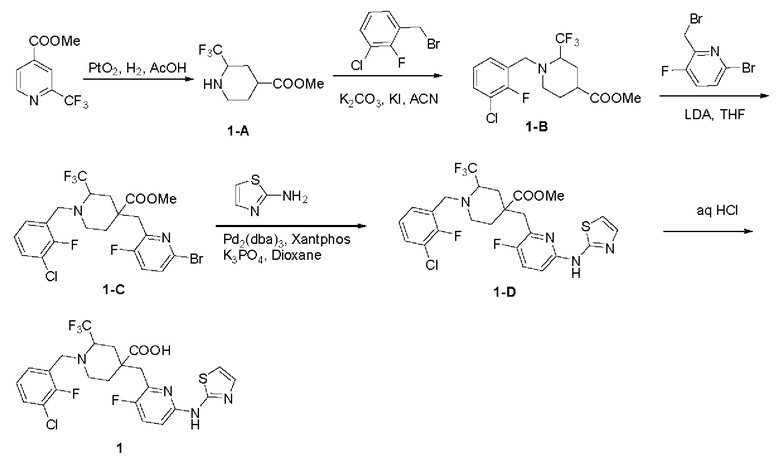
Метил 2-(трифторметил)пиперидин-4-карбоксилат (1-А):
Метил 2-трифторметилпиридин-4-карбоксилат (5 г, 24,374 ммоль), НОАс (100 мл) и PtO2 (0,5 г) добавили в одногорлую колбу объемом 500 мл, реакционную систему трижды продули H2, нагрели до 60°С и активно перемешивали в течение 1-3 дней, пока колба была соединена с водородным мешком. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционную систему охлаждали до комнатной температуры и подвергали фильтрации с использованием диатомита, после чего фильтрат концентрировали. Остаток добавили к ЕА (100 мл) и медленно добавили насыщенный раствор бикарбоната натрия (50 мл) при комнатной температуре. Смесь перемешивали, проводили жидкостное разделение и водную фазу экстрагировали ЕА (25 мл × 2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали, получая продукт в виде светло-коричневого масла (4,8 г, выход 93%), ESI-MS m/z: 212,0 [М+Н]+.
Метил 1-(3-хлор-2-фторбензил)-2-(трифторметил)пиперидин-4-карбоксилат (1-В):
1-А (4,8 г, 22,729 ммоль), 2-фтор-3-хлорбензилбромид (5,59 г, 25,0 ммоль), K2CO3 (9,41 г, 68,2 ммоль), KI (200 мг) и ACN (100 мл) были добавлены в одногорлую колбу объемом 250 мл, и реакционная система была нагрета до рефлюкса и перемешивалась в течение 20 ч в атмосфере Ar. После завершения реакции, что установлено с помощью ЖХ-МС, в реакционную систему добавили ЕА (50 мл)/лед (100 мл). Полученную реакционную систему перемешивали, проводили отделение жидкости и водную фазу экстрагировали ЕА (50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия и концентрировали, остаток очищали колоночной хроматографией (ЕА/РЕ=от 0/20 до 1/20) с получением продукта в виде бесцветного масла (4,3 г, выход 53,5%), ESI-MS m/ z: 354,0 [М+Н]+.
Метил 4-(((6-бром-3-фторпиридин-2-ил)метил)-1-(3-хлор-2-фторбензил)-2-(трифторметил)пиперидин-4-карбоксилат (1-С):
1-В (4,3 г, 12,156 ммоль) и безводный THF (86 мл) были добавлены в трехгорлую колбу объемом 250 мл, и реакционная система была охлаждена до -60°С в атмосфере Ar. LDA (9,1 мл, 2Mb THF, 18,2 ммоль) добавляли по каплям медленно, и температура поддерживалась ниже -45°С во время добавления по каплям. После завершения добавления по каплям смешанный раствор перемешивали при -50±10°С в течение 2 ч. Затем по каплям добавили раствор 6-бром-2-(бромметил)-3-фторпиридина (3,923 г, 14,587 ммоль) в THF (20 мл) при -60±10°С. После завершения капельного добавления полученную реакционную систему перемешивали при -60±10°С в течение 1 ч, затем медленно нагревали до комнатной температуры и проводили реакцию в течение 1 ч. После завершения реакции, что было установлено с помощью TLC (ЕА/РЕ=1/10) и ЖХ-МС, добавили раствор хлорида аммония (50 мл) для гашения реакции и ЕА (50 мл х 2) для экстракции. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 2) и концентрировали, остаток очищали колоночной хроматографией (ЕА/РЕ=от 1/20 до 1/10), получая продукт в виде светло-коричневой жидкости (5,12 г, выход 77,9%), ESI-MS m/z: 541,1/543,1 [М+H]+.
Метил 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоксилат (1-D):
1-С (2 г, 3,697 ммоль), 2-аминотиазол (444 мг, 4,44 ммоль), безводный K3PO4 (1,96 г, 9,243 ммоль), Xantphos (214 мг, 0,37 ммоль) и Dioxane (50 мл) были добавлены в одногорлую колбу объемом 250 мл. После продувки Ar добавили Pd2(dba)3 (174 мг, 0,19 ммоль), реакционную систему нагрели до появления конденсата в атмосфере Ar и проводили реакцию в течение 12 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционную систему охладили до комнатной температуры и отфильтровали, а фильтрат сконцентрировали до сухости. Остаток очищали колоночной хроматографией (DCM/MeOH=от 100/1 до 40/1) с получением продукта в виде коричневого масла (1,62 г, выход 78,1%), ESI-MS m/z: 561,1 [М+Н]+.
1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоновая кислота (1):
1-D (1,62 г, 2,888 ммоль), воду (32 мл) и концентрированную HCl (32 мл) добавили в одногорлую колбу объемом 100 мл и реакционную систему нагревали при 105°С в течение 20 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор сконцентрировали до сухости при пониженном давлении и остаток добавили к ACN (30 мл). Смесь перемешивали при комнатной температуре и подвергали фильтрованию с отсасыванием, а фильтрационный осадок промывали ACN (5 мл × 2) и сушили, получая продукт в виде твердого вещества (622 мг, 39,4% выход). 1H ЯМР (400 МГц, CD3OD) δ 7,75 (тд, J=8,8, 1,5 Гц, 1H), 7,60 (дд, J=4,4, 1,9 Гц, 1H), 7,57-7,46 (м, 2Н), 7. 28 (дд, J=4,4, 1,7 Гц, 1Н), 7,26-7,20 (м, 1Н), 7,17 (дд, J=8,9, 3,1 Гц, 1Н), 4,92-4,85 (м, 1Н), 4,47 (д, J=13,8 Гц, 1Н), 4.26 (дд, J=40.5, 13.7 Гц, 2Н), 3.51-3.33 (м, 2Н), 3.23-3.17 (м, 1Н), 2.39 (дд, J=15.1, 9. 1 Гц, 1Н), 2,25 (дд, J=15,2, 4,4 Гц, 1Н), 2,17-2,04 (м, 1Н), 1,95 (д, J=14,7 Гц, 1Н); ESI-MS m/z: 547,1 [М+Н]+.
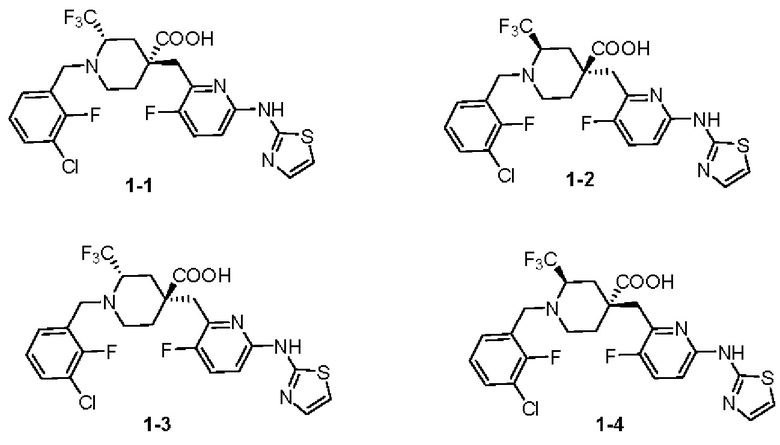
Путем хирального разделения можно получить четыре различных оптических изомера соединения 1, структурные формулы которых выглядят следующим образом:
Примеры 2-28. Синтез соединений 2-28
Целевые соединения 2-28 были получены в соответствии с аналогичным методом синтеза, как в примере 1, с использованием различных исходных материалов.
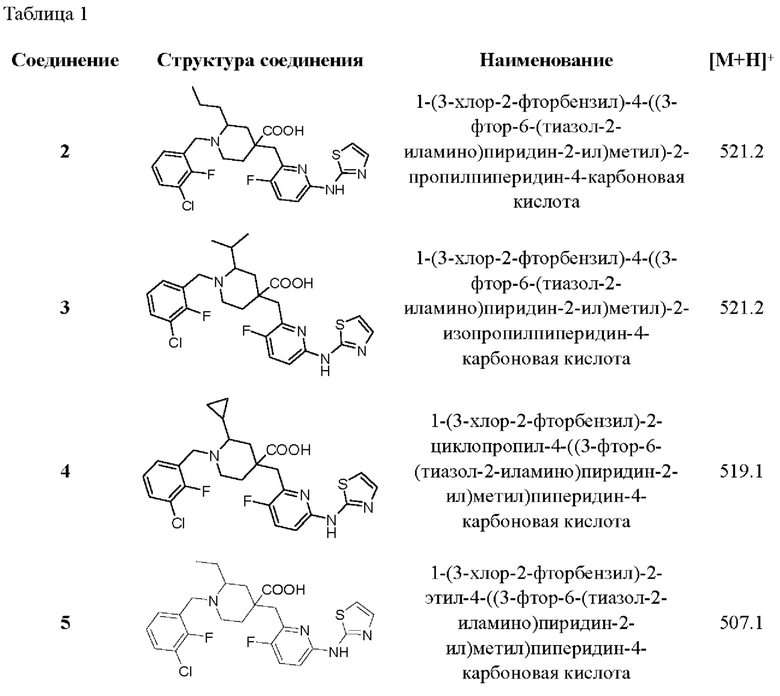
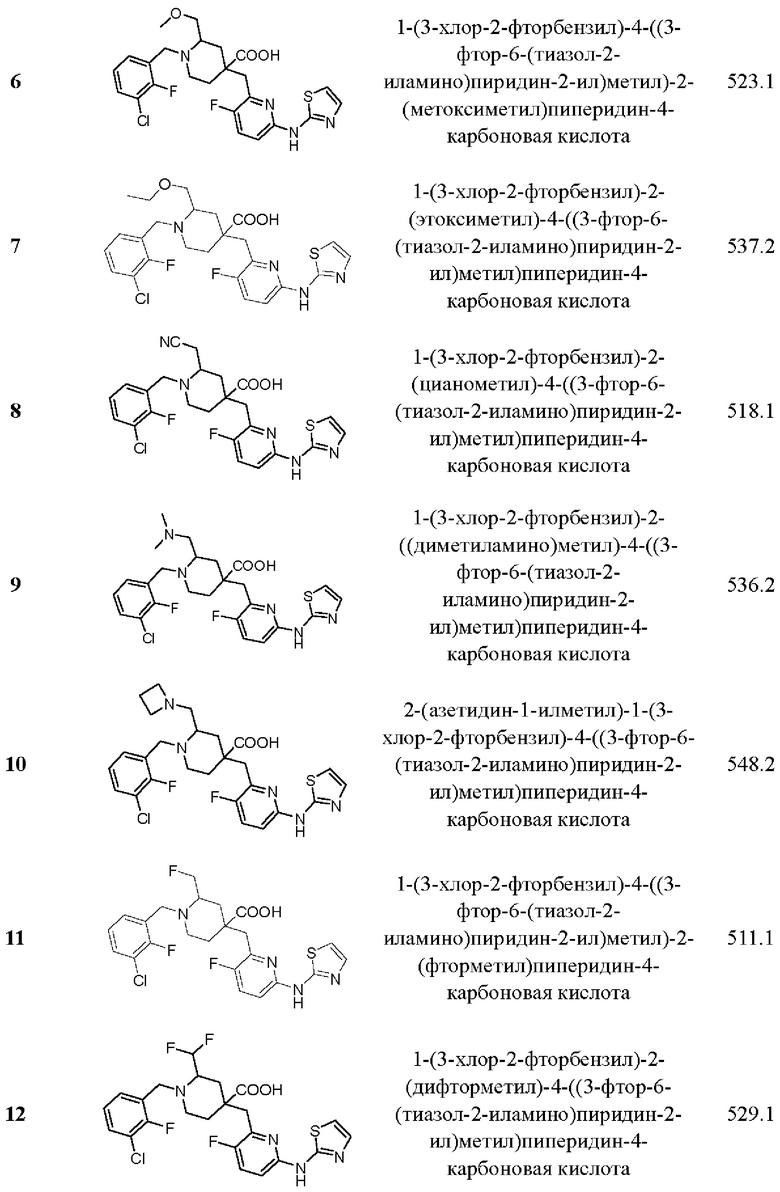
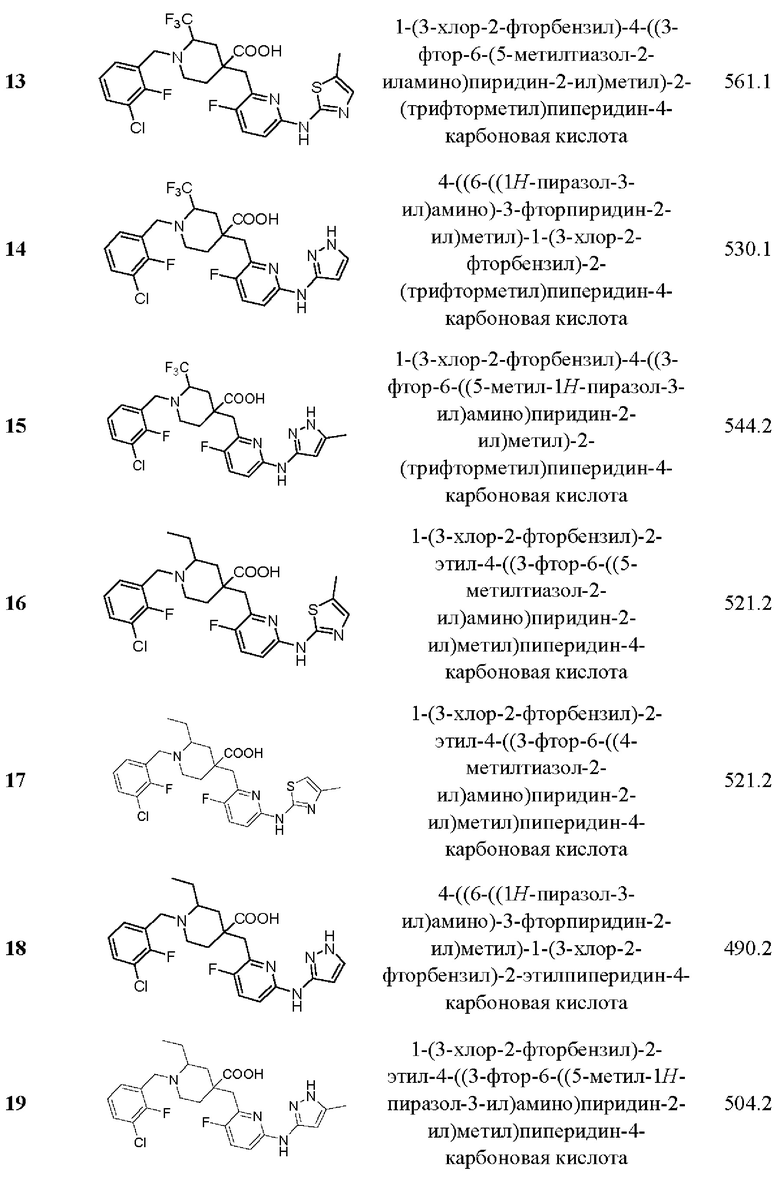
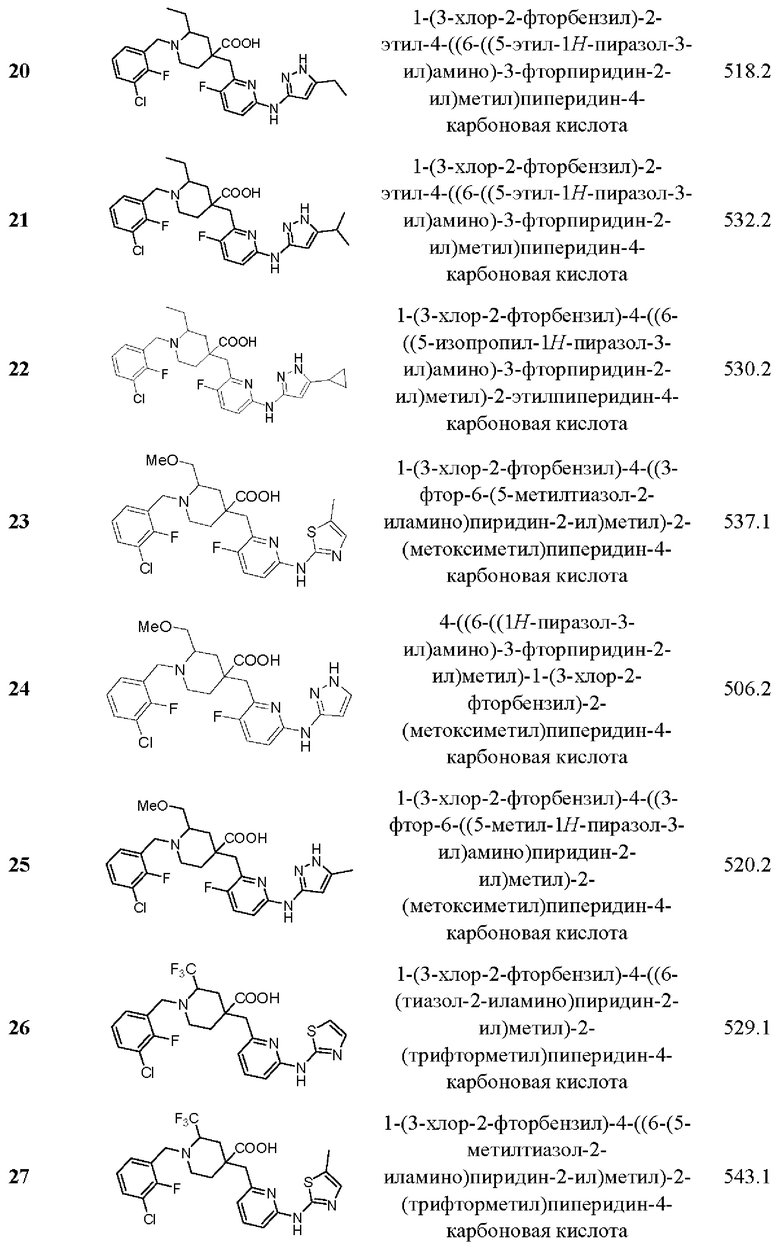
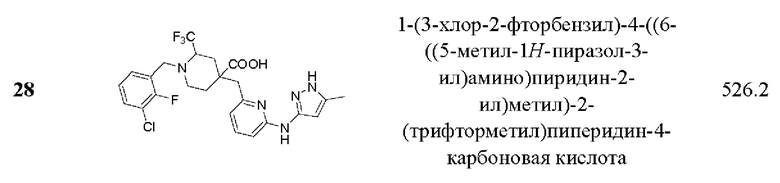
Пример 29. Синтез 1-(3-хлор-2-фторбензил)-2-этил-4-((3-фтор-6-(тиазол-2-иламино) пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (соединение 29)
Используя метил 2-гидроксиметилпиридин-4-карбоксилат в качестве исходного материала, промежуточное соединение 2-(3-хлор-2-фторбензил)-5-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-7-окса-2-азабицикло[3.3.1]нонил-6-он (29-D) было получено способом синтеза в примере 1.
1-(3-хлор-2-фторбензил)-2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (29):
29-D (25 мг, 0,051 ммоль), THF (2 мл) и H2O (1 мл) добавляли в 10 мл одногорлую колбу, затем добавляли LiOH.H2O (10,7 мг, 0,25 ммоль) при комнатной температуре и реакционную систему перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, смешанный раствор очищали с помощью колоночной флэш-хроматографии, чтобы получить продукт (18 мг, 69,3% выход).
1Н ЯМР (400 МГц, CDCl3) δ 7,82 (т, J=8,9 Гц, 1Н), 7,75 (т, J=7,8 Гц, 1Н), 7,61 (д, J=4,4 Гц, 1Н), 7,52 (т, J=6,9 Гц, 1Н), 7,41 (д, J=7,9 Гц, 1Н), 7,32 (д, J=4,3 Гц, 1Н), 7,22 (дд, J=8,9, 3,0 Гц, 1Н), 4,85-4,75 (м, 2Н), 4,47 (д, J=13,6 Гц, 1Н), 4,23 (д, J=11,4 Гц, 1Н), 4,01 (д, J=12,0 Гц, 1Н), 3,83-3,62 (м, 2Н), 3,51-3,41 (м, 2Н), 2,47-2,36 (м, 1Н), 2,31-2,25 (м, 1Н), 2,11-1,85 (м, 2Н); ESI-MS m/z: 509,0 [М+Н]+.
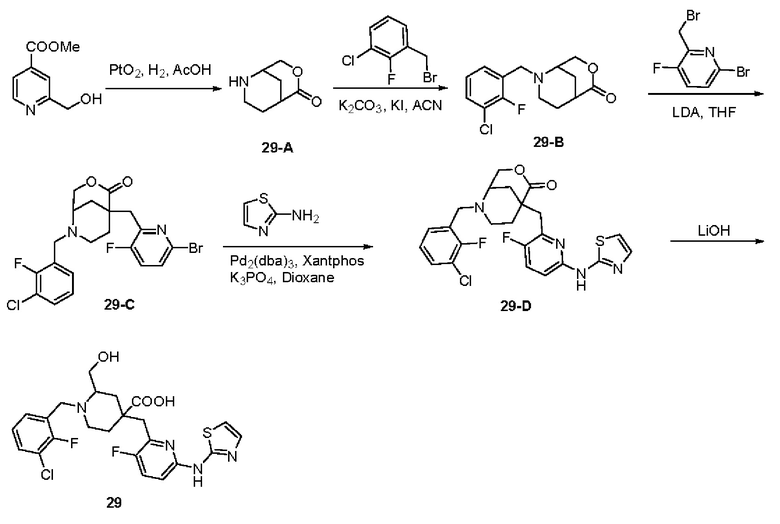
Пример 30. Синтез 1-(2,3-дифторбензил)-2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (соединение 30)
1-(трет-бутил)4-метил-2-этилпиперидин-1,4-дикарбоксилат (30-A):
Метил 2-этилпиперидин-4-карбоксилат (8,94 г, 52,24 ммоль), DIPEA (20,3 г, 156,72 ммоль), DMAP (638 мг, 5,224 ммоль) и ACN (100 мл) были добавлены в одногорлую колбу объемом 250 мл, и раствор Вос2О (14,82 г, 67,92 ммоль) в ACN (30 мл) был добавлен по каплям при комнатной температуре. После завершения добавления по каплям реакционную систему перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (ЕА/РЕ=от 1/20 до 1/10) для получения продукта в виде бесцветной жидкости (13,5 г, 95% выход), ESI-MS m/z: 272,0 [М+Н]+.
1-(трет-бутил)4-метил 4-((6-бром-3-фторпиридин-2-ил)метил)-2-этилпиперидин-1,4-дикарбоновая кислота (30-В):
30-А (10,5 г, 38,9 ммоль) и безводный THF (200 мл) были добавлены в трехгорлую колбу объемом 500 мл, и реакционная система была охлаждена до -60°С под атмосферой Ar. LDA (29,2 мл, 2 M b THF, 58,4 ммоль) добавляли по каплям медленно, и температура поддерживалась ниже -50°С во время добавления по каплям. После завершения добавления по каплям смешанный раствор перемешивали при -60±10°С в течение 1,5 ч. Затем по каплям при -60±10°С добавили раствор 6-бром-2-(бромметил)-3-фторпиридина (12,55 г, 46,68 ммоль) в THF (50 мл). После завершения добавления по каплям полученную реакционную систему перемешивали при -60±10°С в течение 1 ч, затем медленно нагревали до комнатной температуры и проводили реакцию в течение 1 ч. После завершения реакции, что было установлено с помощью TLC (ЕА/РЕ=1/5) и ЖХ-МС, добавили раствор хлорида аммония (100 мл) для гашения реакции и ЕА (100 мл × 2) для экстракции. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл × 2) и концентрировали, остаток очищали колоночной хроматографией (ЕА/РЕ=от 1/20 до 1/10) для получения продукта в виде желтой жидкости (12,87 г, выход 72%), ESI-MS m/z: 459,0/461,0 [М+Н]+.
1-(трет-бутил)4-метил-2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-пиперидин-1,4-дикарбоксилат (30-С):
30-В (6,2 г, 13,5 ммоль), 2-аминотиазол (1,35 г, 13,5 ммоль), безводный фосфат калия (7,2 г, 34,0 ммоль), Xantphos (780 мг, 1,35 ммоль) и Dioxane (100 мл) были добавлены в одногорлую колбу объемом 250 мл. После продувки Ar добавили Pd2(dba)3 (617 мг, 0,675 ммоль), нагрели реакционную систему до рефлюкса в атмосфере Ar и проводили реакцию в течение 5 часов. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционную систему концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (DCM/MeOH=40/0-40/1) с получением коричневого твердого вещества (5,23 г, выход 81%), ESI-MS m/z: 479,2 [М+Н]+.
Метил 2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоксилат дигидрохлорид (30-D):
30-С (5 г, 10,46 ммоль), DCM (20 мл) и HCl/Dioxane (26 мл, 4 М, 104 ммоль) добавляли в одногорлую колбу объемом 100 мл и реакционную систему перемешивали при комнатной температуре в течение 20 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор концентрировали и остаток добавляли к ЕА (30 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, фильтровали и сушили над безводным Na2SO4 с получением продукта в виде желтого твердого вещества (4,8 г, 100% выход), ESI-MS m/z: 379,2 [М+Н]+.
Метил 1-(2,3-дифторбензил)-2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)-метил)пиперидин-4-карбоксилат (30-Е):
30-D (413 мг, 0,92 ммоль), 1-(бромметил)-2,3-дифторбензол (226 мг, 1,1 ммоль), K2CO3 (632 мг, 4,58 ммоль), KI (20 мг) и ACN (10 мл) добавляли в одногорлую колбу объемом 100 мл и реакционную систему подвергали реакции при комнатной температуре в течение примерно 2 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, добавили воду (100 мл) и осадили твердые вещества. Смесь подвергали фильтрованию с отсасыванием и фильтрационный осадок промывали водой (20 мл × 2). Смесь суспендировали с РЕ (50 мл) и затем подвергли фильтрованию с отсасыванием. Фильтрационный осадок промывали РЕ (20 мл × 2) и сушили на воздухе, получая продукт (295 мг, 64% выход), ESI-MS m/z: 505,1 [М+Н]+.
1-(2,3-дифторбензил)-2-этил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (30):
30-Е (295 мг, 0,585 ммоль), воду (5 мл) и концентрированную HCl (5 мл) добавляли в одногорлую колбу объемом 100 мл и реакционную систему подвергали рефлюксу при 105°С в течение 20 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор концентрировали до сухости при пониженном давлении и остаток добавляли к ACN (30 мл). Смесь перемешивали при комнатной температуре и подвергали фильтрованию с отсасыванием, затем фильтрационный осадок промывали ACN (5 мл × 2) и сушили на воздухе, получая продукт в виде светло-желтого порошка (118 мг, 41% выход).
1Н ЯМР (400 МГц, ДМСО-d6) δ: 11,31 (с, 1Н), 9,15 (с, 1Н), 7,85-7,72 (м, 2Н), 7,59-7,45 (м, 1Н), 7,33-7,19 (м, 2Н), 7,05-6,92 (м, 2Н), 4,75 (д, J=13,4 Гц, 1Н), 4,26-4,10 (м, 1Н), 3,24-3,06 (м, 2Н), 2,96-2,73 (м, 2Н), 2,41 (д, J=13.7 Гц, 1Н), 2,21-2,02 (м, 2Н), 1,92-1,56 (м, 4Н), 0,91 (дт, J=10.8 7.3 Гц, 3Н); ESI-MS m/z: 491.1 [М+Н]+. Примеры 31-34. Синтез соединений 31-34:
Целевые соединения 31-34 были получены в соответствии с аналогичным методом синтеза, как в примере 30, с использованием различных исходных материалов.
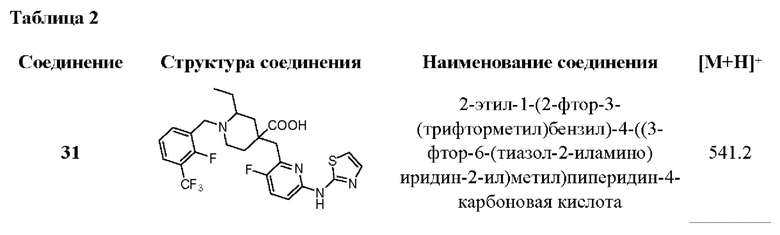
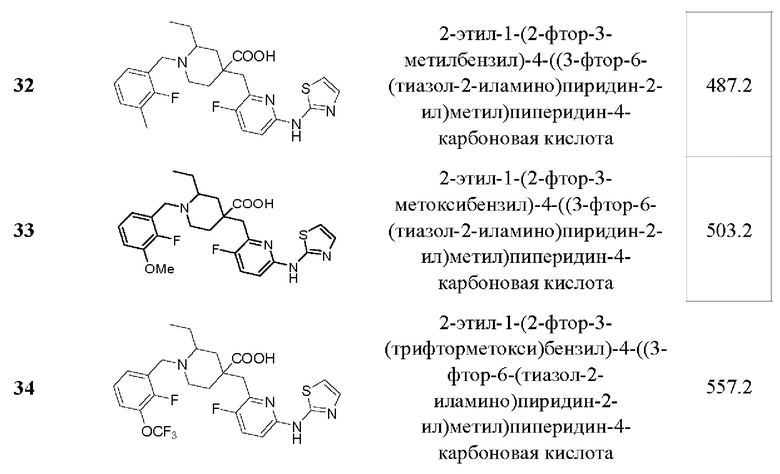
Пример 35. Синтез 1-(3-хлор-2-фторбензоил)-4-((3-фтор-6-(тиазол-2-иламино) пиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоновой кислоты (соединение 35)
1-(трет-бутил)4-метил 2-(трифторметил)пиперидин-1,4-дикарбоксилат (35-А):
Метил 2-(трифторметил)пиперидин-4-карбоксилат (2,11 г, 10 ммоль), DIPEA (3,87 г, 30 ммоль), DMAP (244 мг, 24 ммоль) и CAN (50 мл) были добавлены в одногорлую колбу объемом 250 мл, и раствор Вос2О (3,27 г, 15 ммоль) в ACN (30 мл) был добавлен по каплям при комнатной температуре. После завершения добавления по каплям реакционную систему нагрели до рефлюкса и перемешивали в течение 3 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор сконцентрировали при пониженном давлении и остаток очистили колоночной хроматографией (ЕА/РЕ=1/20 -1/10), чтобы получить продукт в виде бесцветной жидкости (2,3 г, выход 74%), ESI-MS m/ z: 312,0 [М+Н]+.
1-(трет-бутил)4-метил 4-((6-бром-3-фторпиридин-2-ил)метил)-2-(трифторметил) пиперидин-1,4-дикарбоновая кислота (35-В):
35-А (2,2 г, 7,07 ммоль) и безводный THF (50 мл) были добавлены в трехгорлую колбу объемом 250 мл, и реакционная система была охлаждена до -60°С под атмосферой Ar. LDA (5,3 мл, 2 M b THF, 10,6 ммоль) добавляли по каплям медленно, и температура поддерживалась ниже -50°С во время добавления по каплям. После завершения добавления по каплям смешанный раствор перемешивали при -60±10°С в течение 1,5 ч. Затем по каплям добавили раствор 6-бром-2-(бромметил)-3-фторпиридина (2,09 г, 7,777 ммоль) в THF (50 мл) при -60±10°С. После завершения добавления по каплям полученную реакционную систему перемешивали при -60±10°С в течение 1 ч, затем медленно нагревали до комнатной температуры и проводили реакцию в течение 1 ч. После завершения реакции, что было установлено с помощью TLC (ЕА/РЕ=1/5) и ЖХ-МС, добавили раствор хлорида аммония (100 мл) для гашения реакции и ЕА (100 мл × 2) для экстракции. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл × 2) и концентрировали, остаток очищали колоночной хроматографией (ЕА/РЕ=от 1/20 до 1/10), получая продукт в виде желтой жидкости (2,58 г, выход 73%), ESI-MS m/z: 499,0/501,0 [М+Н]+.
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоксилат дигидрохлорид (35-С):
35-В (2,5 г, 5,01 ммоль), DCM (25 мл) и HCl/Dioxane (12,5 мл, 4 М, 50 ммоль) были добавлены в одногорлую колбу объемом 100 мл, и реакционную систему перемешивали при комнатной температуре в течение 20 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционный раствор был сконцентрирован, и остаток был добавлен к ЕА(10 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, фильтровали и сушили над безводным Na2SO4 с получением продукта в виде желтого твердого вещества (1,68 г, 71% выход), ESI-MS m/z: 399,0/401,0 [М+Н]+.
Метил-4-((6-бромо-3-фторпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)-2-(трифторметил)пиперидин-4-карбоксилат (35-D):
35-С (1,68 г, 3,56 ммоль), DMF (30 мл), DIPEA (2,3 г, 17,8 ммоль), EDCI (1,023 г, 5,34 ммоль), HOBt (721 ммоль, 5,34 ммоль) и 3-хлор-2-фторбензойную кислоту (746 мг, 4,27 ммоль) добавили в одногорлую колбу объемом 100 мл, и реакционную систему перемешивали при 50°С в течение 20 ч в атмосфере Ar. После завершения реакции, что было установлено с помощью ЖХ-МС, в реакционную систему добавили ЕА (50 мл) и Н2О (50 мл). Полученную реакционную систему перемешивали и проводили разделение жидкости. Органическую фазу концентрировали до сухости и остаток очищали колоночной хроматографией (ЕА/РЕ=от 1/20 до 1/10) с получением продукта (1,4 г, 71% выход), ESI-MS m/z: 555,0/557,0 [М+Н]+.
Метил-1-(3-хлор-2-фторбензоил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)-метил)-2-(трифторметил)пиперидин-4-карбоксилат (35-Е):
35-D (200 мг, 0,36 ммоль), 2-аминотиазол (36 мг, 0,36 ммоль), безводный K2CO3 (124 г, 0,9 ммоль), Xantphos (42 мг, 0,072 ммоль) и Dioxane (10 мл) были добавлены в одногорлую колбу объемом 250 мл. После продувки Ar добавили Pd2(dba)3 (33 мг, 0,036 ммоль) и реакционную систему нагрели до рефлюкса в атмосфере Ar, и проводили реакцию в течение 5 ч. После завершения реакции, что было установлено с помощью ЖХ-МС, реакционную систему сконцентрировали при пониженном давлении и остаток очистили колоночной хроматографией (DCM/MeOH=40/0 - 40/1) с получением коричневого твердого вещества (126 мг, 61% выход), ESI-MS m/z: 575.1 [М+Н]+.
1-(3-хлор-2-фторбензоил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-(трифторметил)пиперидин-4-карбоновая кислота (35):
35-Е (120 мг, 0,208 ммоль), THF (5 мл), воду (2 мл) и LiOH.H2O (88 мг, 2,1 ммоль) добавляли в одногорлую колбу объемом 100 мл, затем реакционную систему нагревали до 50°С в атмосфере Ar и проводили реакцию в течение примерно 5 ч. После завершения реакции, как было установлено с помощью ЖХ-МС, реакционный раствор корректировали до рН=4-5 и концентрировали при пониженном давлении, а остаток очищали с помощью колоночной флэш-хроматографии для получения продукта в виде светло-желтого порошка (28 мг, выход 24%).
1Н ЯМР (400 МГц, CD3OD) δ 7,78 (т, J=9,0 Гц, 1Н), 7,61-7,52 (м, 2Н), 7,41 (дт, J=12,0, 7,9 Гц, 1Н), 7,26 (д, J=7,5 Гц, 1Н), 7,20 (д, J=4,3 Гц, 1Н), 7,12 (дд, J=8,9, 3. 0 Гц, 1Н), 4,52-4,47 (м, 1Н), 4,26-4,12 (м, 1Н), 3,50-3,38 (м, 2Н), 3,25-3,19 (м, 2Н), 2,39-2,31 (м, 1Н), 2,25-2,19 (м, 1Н), 2,11-2,04 (м, 1Н), 1,95-1,84 (м, 1Н); ESI-MS m/z: 561,1 [М+Н]+.
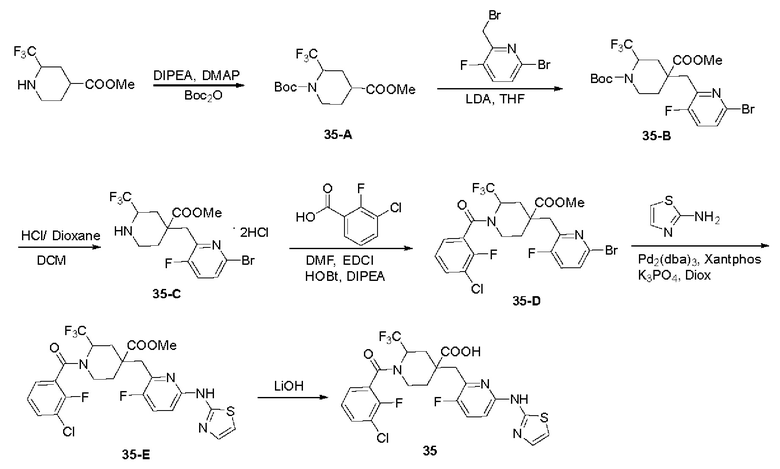
Примеры 36 и 37. Синтез соединений 36 и 37
Целевые соединения 36 и 37 были получены в соответствии с аналогичным методом синтеза, как в примере 35, с использованием различных исходных материалов.
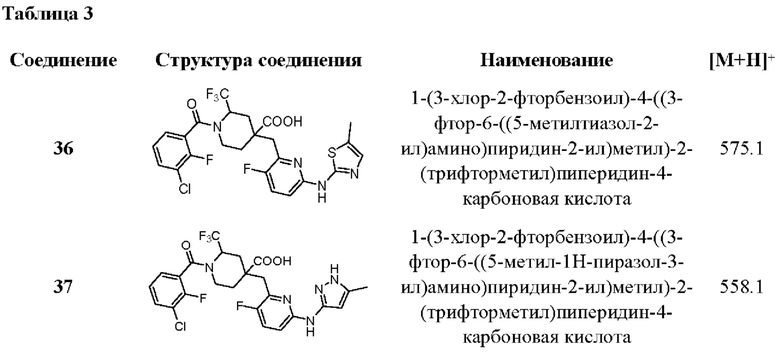
Пример 38. Анализ ингибирующей активности против Aurora киназы
In vitro анализ ингибирующей активности соединений, раскрытых в настоящем документе, против активности Aurora киназы был проведен с использованием метода сдвига подвижности калипера. Соединения подвергали градиентному разбавлению от 10 мкМ для получения в общей сложности 10 концентраций. После смешивания фермента и реакционного раствора киназы (20 мМ HEPES, рН 7,5, 0,01% Triton Х-100) добавляли градиентно разбавленное соединение. Смесь инкубировали при комнатной температуре в течение 10 минут, чтобы соединение и фермент хорошо связались. Затем добавляли FAM-меченный полипептид в качестве субстрата для проведения киназной реакции при 25°С и через определенное время добавляли стоп-раствор. Скорость превращения считывали с помощью калипера и преобразовывали в скорость ингибирования, после чего рассчитывали значения IC50, при этом в качестве отрицательного контроля использовали пустой растворитель без лекарственного средства, а в качестве положительного контроля использовали LY-3295668. Результаты по вышеуказанным соединениям приведены в таблице 4.
Пример 39. Анализ антипролиферативной активности в отношении клеток Н69
Опухолевые клетки (человеческие клетки мелкоклеточного рака легких Н69) в логарифмической фазе роста высевали в 384-луночный кулыуральный планшет из расчета 4×103 клеток на лунку, в каждую лунку добавляли 50 мкл среды и смеси культивировали в течение ночи в инкубаторе при 37°С/5% СО2. После того как клетки прилипли к стенке, добавляли тест-соединения и препарат положительного контроля в соответствующих концентрациях и готовили пять образцов с различными концентрациями. В качестве отрицательного контроля была взята пустая группа, и полученные смеси культивировались в инкубаторе в течение 72 ч. Затем 50 мкл CTL plus добавлялись в каждую лунку, и количество клеток оценивалось путем измерения содержания АТФ в клетках. Значения IC50 были рассчитаны путем подбора с помощью GRAPHPAD, результаты представлены в таблице 4.
Приведенные выше данные показывают, что соединения, раскрытые в настоящем документе, обладают более высокой активностью против Aurora киназы и активностью против пролиферации клеток, чем контрольный препарат LY-3295668, а соединения формулы (1) обладают чрезвычайно высокой активностью против Aurora-A киназы и значительно улучшают активность против Aurora-B киназы и активность против пролиферации клеток Н1975, когда группа R3 заменена с Me на относительно большую группу или заменена сильной электроноотнимающей группой, такой как CF3, и/или когда W является 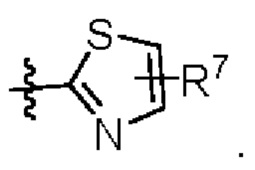
Пример 40. Оценка противоопухолевой активности у мышей
Клетки рака легких человека Н69 культивировали обычным образом в среде 1640, содержащей 10% фетальной бычьей сыворотки, в инкубаторе при 37°С/5% CO2, затем клетки пассировали и собирали, когда они достигали желаемого количества. 1×107 клеток Н69 вводили в правую дорсальную сторону каждой голой мыши и после того, как опухоли вырастали до 150 мм3, животных случайным образом распределяли по группам для введения препарата. Группы были следующие: 1) контрольная группа растворителя, 8 мышей; 2) группа LY-3295668, группа соединения 1, группа соединения 5 и группа соединения 6, по 8 мышей в каждой. Мышам в контрольной группе растворителя внутрижелудочно вводили 0,5% CMC-Na дважды в день, а мышам в группе LY-3295668, группе соединения 1, группе соединения 5 и группе соединения 6 внутрижелудочно вводили суспензию соединения в 0,5% CMC-Na дважды в день. Во вторник и четверг каждой недели измеряли объем опухоли и массу тела мышей, а на 21-й день после введения мышей умерщвляли. Результаты теста представлены в Таблице 5 ниже.

Как видно из таблицы 5, соединение 1 и соединение 6 демонстрируют значительно повышенную противоопухолевую активность in vivo, по сравнению с положительным контролем LY-3295668, что указывает на то, что соединения формулы (1) обладают значительно повышенной противоопухолевой активностью in vivo, когда группа R3 заменена с Me на группу с соответствующим размером, такую как CF3 или -CH2OMe, и/или когда W является 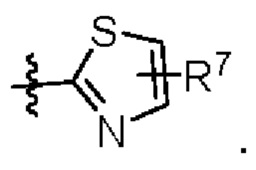
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПИПЕРИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581834C1 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| СОЕДИНЕНИЯ С ГЕТЕРОАРОМАТИЧЕСКИМИ КОЛЬЦАМИ В КАЧЕСТВЕ ИНГИБИТОРОВ RET-КИНАЗ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2829729C1 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2024 |
|
RU2838180C1 |
| Соединение-ингибитор мультикиназ и его кристаллическая форма и применение | 2017 |
|
RU2723985C1 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ КАК МОДУЛЯТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ | 2014 |
|
RU2672910C9 |
Изобретение относится к области медицинской химии, а именно к соединению формулы (1), где «*» обозначает хиральный центр; L представляет собой CH2 или СО; R1 и R2 каждый независимо представляют собой Н, галоген, CN, С1-С3 алкил, С3-С6 циклоалкил, С1-С3 алкокси, С1-С3 галоалкил и С1-С3 галоалкокси; R3 представляет собой С2-С3 алкил, С3-С6 циклоалкил, С1-С3 галоалкил, -(С1-С3)алкил-ОН, -(С1-С3)алкил-(С1-С3)алкокси, -(С1-С3)алкил-CN или -(С1-С3)алкил-NR5R6, где R5 и R6 каждый независимо представляют собой Н или С1-С3 алкил, или R5 и R6 образуют 4-7-членный гетероциклоалкил вместе с атомом N; R4 представляет собой Н или F; W представляет собой фрагмент (а), где R7 представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил. Технический результат - получение соединений, обладающих ингибирующей активностью в отношении Aurora киназы, для лечения заболеваний, опосредованных Aurora киназой. 4 з.п. ф-лы, 5 табл., 40 пр.
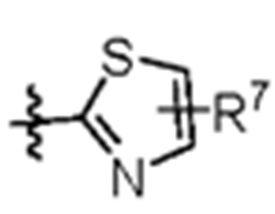 (а).
(а).
1. Соединение формулы (1):
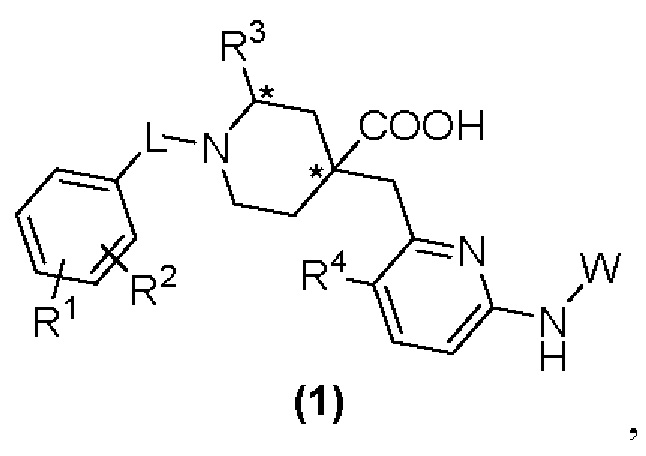
где в формуле (1):
«*» обозначает хиральный центр;
L представляет собой CH2 или СО;
R1 и R2 каждый независимо представляют собой Н, галоген, CN, С1-С3 алкил, С3-С6 циклоалкил, С1-С3 алкокси, С1-С3 галоалкил и С1-С3 галоалкокси;
R3 представляет собой С2-С3 алкил, С3-С6 циклоалкил, С1-С3 галоалкил, -(С1-С3)алкил-ОН, -(С1-С3)алкил-(С1-С3)алкокси, -(С1-С3)алкил-CN или -(С1-С3)алкил-NR5R6, где R5 и R6 каждый независимо представляют собой Н или С1-С3 алкил, или R5 и R6 образуют 4-7-членный гетероциклоалкил вместе с атомом N;
R4 представляет собой Н или F;
W представляет собой 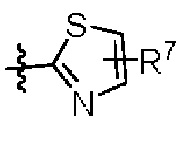 , где R7 представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил.
, где R7 представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил.
2. Соединение по п. 1, где в формуле (1)  представляет собой
представляет собой
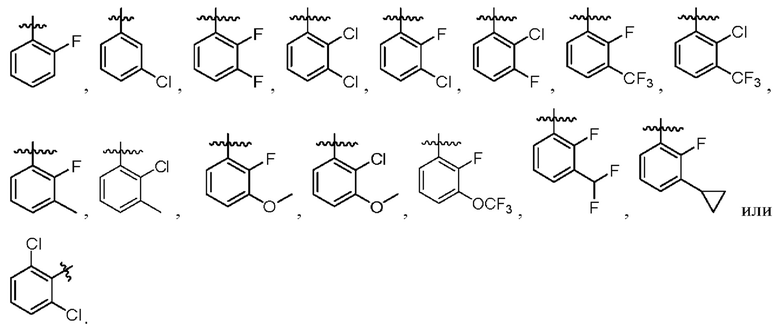
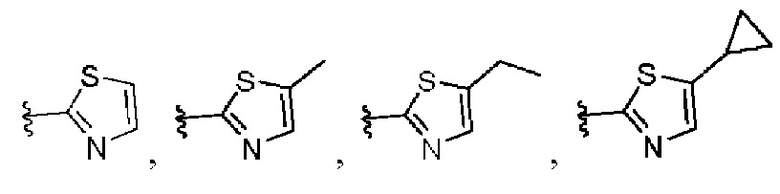
3. Соединение по п. 1 или 2, где в формуле (1) R3 представляет собой Et, n-Pr, i-Pr, 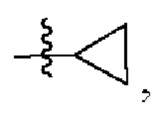 CH2F, CHF2, CF3, СН2ОН, СН2ОМе, CH2OEt, CH2CN,
CH2F, CHF2, CF3, СН2ОН, СН2ОМе, CH2OEt, CH2CN, 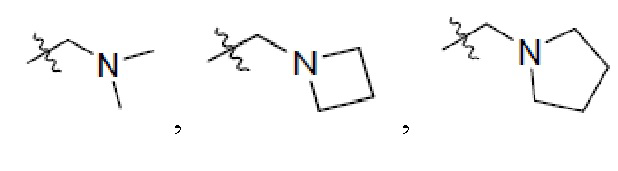 или
или 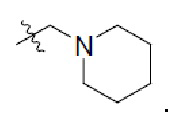
4. Соединение по любому из пп. 1-3, где в формуле (1) W представляет собой
 или
или 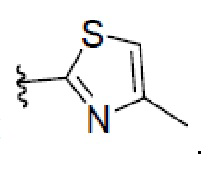
5. Соединение по любому из пп. 1-4, где соединение представляет собой:
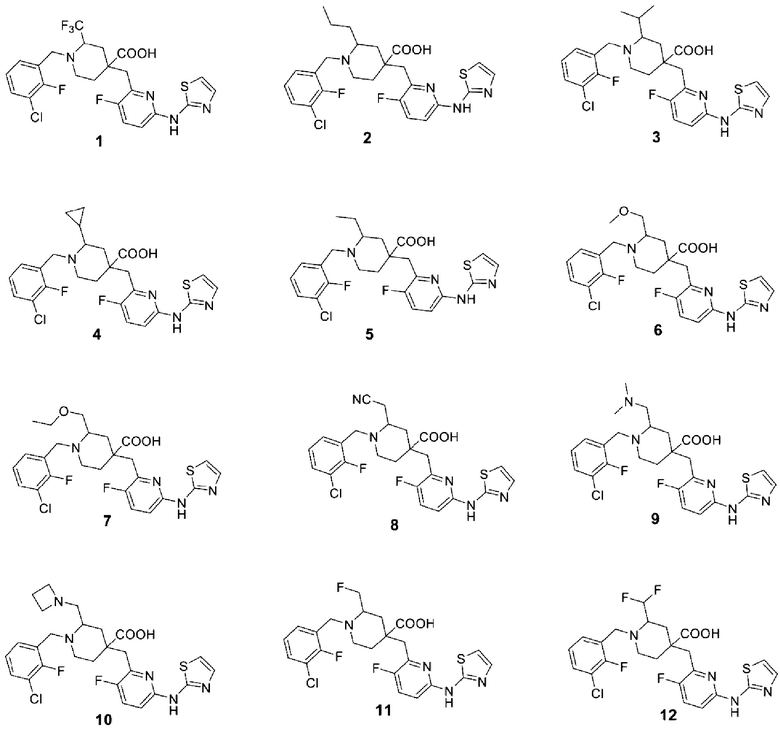
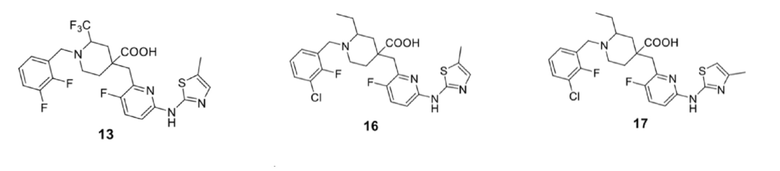
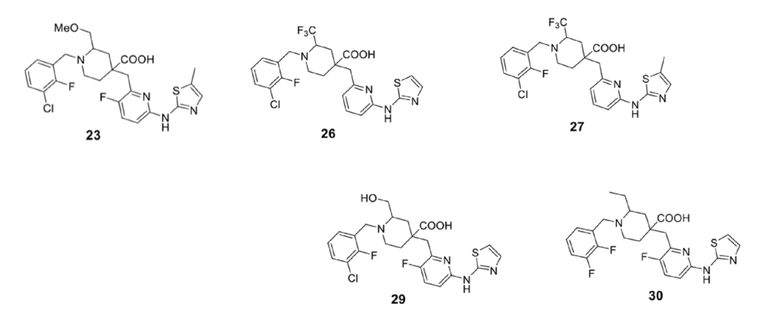
| CN 107108567 A, 29.08.2017 | |||
| WO 2009104802 A1, 27.08.2009 | |||
| CN 104159893 A, 19.11.2014 | |||
| CN 101103017 A, 09.01.2008 | |||
| WO 2008026768 A1, 06.03.2008 | |||
| VASILEVICH, N.I | |||
| et al | |||
| General Ser/Thr Kinases Pharmacophore Approach forSelective Kinase Inhibitors Search as Exemplified byDesign of Potent and Selective Aurora A Inhibitors | |||
| Chemical Biology & Drug |

