Область техники
Данное изобретение относится к области химического синтеза и, в частности, к способам и процессам получения и производства дейтерированной ω-дифенилмочевины.
Предшествующий уровень техники
Производные Ω-дифенилмочевины представляют собой известные соединения с ингибирующей активностью в отношении c-raf-киназы. Например, в WO 2000/042012 раскрыт класс ω-карбоксил-арил-замещенной дифенилмочевины и ее применение для лечения рака и связанных заболеваний.
Изначально соединения ω-дифенилмочевины, такие как Sorafenib, были впервые обнаружены как ингибиторы c-raf-киназы. Другие исследования показали, что они также могут ингибировать МЕК и ERK пути сигнальной трансдукции и активировать тирозинкиназы, включая рецептор 2 сосудистого эндотелиального фактора роста (VEGFR-2), рецептор 3 сосудистого эндотелиального фактора роста (VEGFR-3) и рецептор-β тромбоцитарного фактора роста (PDGFR-β) (Curr Pharm Des 2002, 8, 2255-2257). Поэтому их назвали мультикиназными ингибиторами, приводящими к двойным противоопухолевым эффектам.
Сорафениб (торговое название Nexavar) - новый пероральный мультикиназный ингибитор, разработанный Bayer и Onyx. В декабре 2005, на основании его отличных характеристик в фазе III клинических испытаний для распространенной почечно-клеточной карциномы, сорафениб был одобрен FDA для лечения распространенной почечно-клеточной карциномы и выведен на рынок в КНР в ноябре 2006. Однако сорафениб обладал побочными эффектами, такими как гипертензия, потеря веса, сыпь и пр.
Однако все еще существует необходимость разработки новых соединений с ингибирующей активностью в отношении raf-киназы или лучшими фармакодинамическими свойствами и способа их получения.
Краткое описание изобретения
Цель настоящего изобретения предложить новые соединения с ингибирующей активностью в отношении raf-киназы и лучшими фармакодинамическими свойствами и их применения.
Другая цель изобретения заключается в том, чтобы предложить ряд способов получения дейтерированной ω-дифенилмочевины и ее промежуточных соединений, удовлетворяющих требованиям по производству в фармацевтической промышленности и повышающих функциональность и безопасность.
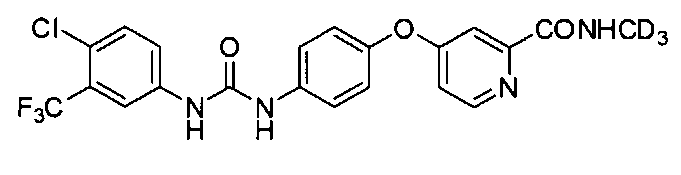
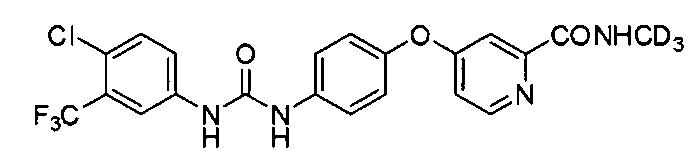
В первом аспекте изобретение предлагает соединение дейтерированной ω-дифенилмочевины или ее фармацевтически приемлемые соли, где упомянутое соединение представляет собой N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевину

В одном воплощении N в упомянутом соединении представляет собой 14N.
Во втором аспекте изобретение предлагает способ получения N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины

включающий:
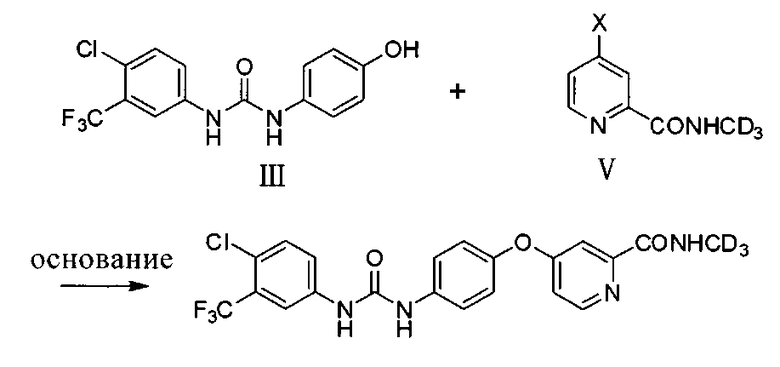
(a) реакцию в инертном растворителе и в присутствии основания соединения III с соединением V для образования упомянутого соединения

где X представляет собой Cl, Br, или I;
или включающий:
(b) реакцию в инертном растворителе соединения IX с CD3NH2 или CD3NH2·HCl для образования упомянутого соединения

где R представляет собой линейный или разветвленный C1-C8 алкил, или арил;
или включает:
(c) реакцию в инертном растворителе 4-хлор-3-трифторметилфенилизоцианата (VIII) с соединением 5 для образования упомянутого соединения

или включающий:
(d) реакцию в инертном растворителе и в присутствии CDI (карбодиимида) и CH2Cl2 соединения 5 с соединением 6 для образования упомянутого соединения
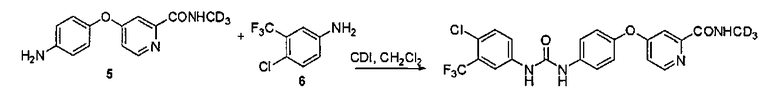
В одном воплощении соединение III готовят, как указано ниже:
(i) конденсация 4-гидрокси-анилина (I) с 4-хлор-3-трифторметил-анилином (II) для образования соединения III.
 .
.
В одном воплощении соединение III готовят, как указано ниже:
(ii) реакция п-метоксианилина (X) с 4-хлор-3-трифторметил-анилином (II) или 4-хлор-3-трифторметилфенилизоцианатом (VIII) для образования соединения XI
 ,
,
и затем в кислых или основных условиях деметилирование соединения XI для получения соединения III:
 .
.
В одном воплощении соединение VII готовят, как указано ниже:
в присутствии основания проводят реакцию соединения VI и п-гидроксиланилина для образования соединения VII:
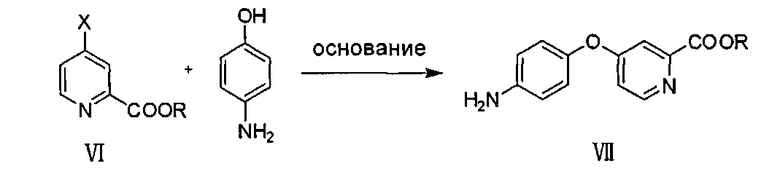
где X представляет собой хлор, бром или йод; R представляет собой линейный или разветвленный C1-C8 алкил или арил.
В одном воплощении упомянутое основание выбирают из трет-бутилата калия, гидрида натрия, гидрида калия, карбоната калия, карбоната цезия, фосфата калия, гидроксида калия, гидроксида натрия или их комбинации.
В одном воплощении способ (а) также предусматривает, что реакцию проводят в присутствии катализатора, где упомянутый катализатор выбран из CuI и пролина; или CuI и пиколината.
В одном воплощении температура реакции составляет 0-200°C.
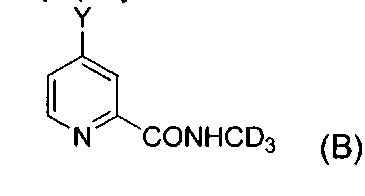
В третьем аспекте изобретение предлагает промежуточное соединение формулы B

где Y представляет собой галоген или  .
.
В одном воплощении Y представляет собой Cl, и структура формулы B представляет собой  .
.
В четвертом аспекте изобретение предлагает способ получения 4-хлор-N-(метил-d3)пиколинамида, предусматривающий:
(a1) реакцию метил-4-хлорпиколината с (метил-d3)амином или его солями под действием основных условий и в инертном растворителе для образования 4-хлор-N-(метил-d3)пиколинамида; или
(a2) реакцию 4-хлорпиколиноилхлорида с (метил-d3)амином в инертном растворителе для образования 4-хлор-N-(метил-d3)пиколинамида.
В одном воплощении упомянутый инертный растворитель включает тетрагидрофуран, этанол, метанол, воду или их смесь.
В одном воплощении, на этапе (a1) и (a2), температура реакции составляет от -10°C до температуры образования флегмы, предпочтительно составляет от -4°C до 60°C и более предпочтительно составляет 5-50°C.
В одном воплощении, на этапе (a1) и (a2), время реакции составляет 0,5-72 часа, предпочтительно составляет 1-64 часа, и более предпочтительно составляет 2-48 часов.
В одном воплощении на этапе (a1), упомянутые основные условия означают, что в реакционной системе присутствуют карбонат калия, карбонат натрия, карбонат цезия, KOH, NaOH, или их комбинация.
В пятом аспекте изобретение предлагает способ получения 4-(4-аминофенокси)-N-(метил-d3)пиколинамида, предусматривающий:
реакцию 4-хлор-N-(метил-d3)пиколинамида с 4-аминфенолом под действием основных условий и в инертном растворителе для образования 4-(4-аминофенокси)-N-(метил-d3)пиколинамида

В одном воплощении упомянутые основные условия означают, что в реакционной смеси присутствуют KOH, NaOH, карбонат калия, карбонат натрия, карбонат цезия, трет-бутоксид калия, трет-бутоксид натрия или их комбинация.
В одном воплощении упомянутый инертный растворитель выбран из ДМФ, ДМСО, N,N-диметилацетиламида, тетрагидрофурана, метилпирролидин-2-она, 1,4-диоксана или их смеси.
В одном воплощении описанная температура реакции составляет от 0°C до 160°C, предпочтительно составляет от 20°C до 120°C и более предпочтительно составляет 30-100°C.
Время реакции составляет 0,5-48 часов, предпочтительно составляет 1-36 часов и более предпочтительно составляет 3-24 часа.
В пятом аспекте изобретение предлагает применение упомянутых промежуточных соединений согласно третьему аспекту изобретения для получения дейтерированной ω-дифенилмочевины или в качестве исходного материала для получения дейтерированной ω-дифенилмочевины.
В одном воплощении упомянутая дейтерированная дифенилмочевина включает 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамид (СМ4307) и п-толуолсульфонат 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамида (CM4307·TsOH).
Следует понимать, что в настоящем изобретении любые технические характеристики, в частности описанные выше и ниже (такие как в примерах), могут быть скомбинированы друг с другом, таким образом формируя новые и предпочтительные технические растворы, которые не приведены по отдельности в описании.
Описание чертежей
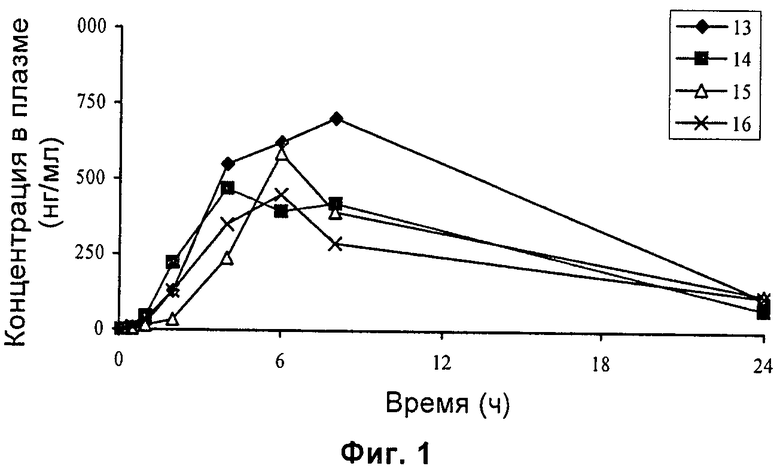
На Фиг.1 изображены кривые концентрации лекарственного средства (нг/мл) в плазме после орального введения 3 мг/кг контрольного соединения СМ4306 самцам крыс SD.
На Фиг.2 изображены кривые концентрации лекарственного средства (нг/мл) в плазме после орального введения 3 мг/кг соединения СМ4307 по изобретению самцам крыс SD.
На Фиг.3 изображены кривые эффективности ингибирования СМ4306 и СМ4307 на модели ксенотрансплантата у бестимусных мышей, инокулированных при помощи раковых клеток печени человека SMMC-7721. На этой фигуре "лечение" означает, что период лечения составил 14 дней с последующим периодом наблюдения после того, как введение было прекращено. Пять дней до лечения представляли собой период подготовки животных моделей.
Подробное описание изобретения
После исследований авторы изобретения неожиданно обнаружили, что по сравнению с недейтерированным соединением дейтерированная ω-дифенилмочевина по изобретению и ее фармацевтически приемлемые соли обладали лучшими фармакокинетическими и/или фармакодинамическими свойствами. Поэтому они были более приемлемы в качестве ингибиторов raf-киназ для приготовления лекарственных препаратов для лечения рака и связанных заболеваний.
Более того, авторы изобретения также обнаружили, что соединения дифенилмочевины могут быть эффективно и быстро приготовлены при помощи нового промежуточного соединения формулы B
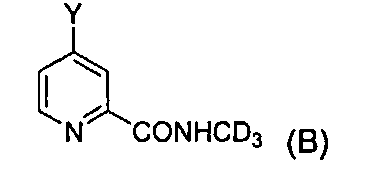
где Y представляет собой галоген или  . На основании этого открытия авторы изобретения осуществили настоящее изобретение.
. На основании этого открытия авторы изобретения осуществили настоящее изобретение.
Определение
Как употреблено в данном документе термин "галоген" относится к F, Cl, Br и I. Предпочтительно, галоген выбран из F, Cl и Br.
Как употреблено в данном документе, термин "алкил" относится к линейному или разветвленному алкилу. Предпочтительно, алкил представляет собой C1-C4 алкил, такой как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и пр.
Как употреблено в данном документе, термин "дейтерированный" означает, что один или более водород в соединении или группа замещены дейтерием. "Дейтерированный" может быть монозамещенным, бизамещенным, мультизамещенным или полностью замещенным. Термины "один или более дейтерий-замещенный" и "замещенный дейтерием одно- или многократно" могут быть взаимозаменяемы.
В одном воплощении содержание дейтерия в дейтерий-замещенном положении составляет по меньшей мере свыше распространенности дейтерия в природе (0,015%), предпочтительно >50%, более предпочтительно >75%, более предпочтительно >95%, более предпочтительно >97%, более предпочтительно >99%, более предпочтительно >99,5%.
В одном воплощении соединение формулы (I) содержит по меньшей мере один атом дейтерия, предпочтительно 3 атома дейтерия и более предпочтительно пять атомов дейтерия.
Как употреблено в данном документе, термин "соединение СМ4306" представляет собой 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-метилпиколинамид.
Как употреблено в данном документе, термин "соединение СМ4307" представляет собой 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамид.
Как употреблено в данном документе, термин "TsOH" означает п-толуолсульфоновую кислоту. Таким образом, CM4307·TsOH означает п-толуолсульфонат СМ4307.
Дейтерий-замещенная ω-дифенилмочевина
Предпочтительные соединения дейтерий-замещенной ω-дифенилмочевины по изобретению обладают структурой формулы (I):

где
X представляет собой N или N+-O-;
R1 представляет собой галоген (такой как F, Cl или Br), одно или более дейтерий-замещенный или пердейтерированный C1-C4 алкил;
R2 представляет собой недейтерированный C1-C4 алкил, один или более дейтерий-замещенный или пердейтерированный C1-C4 алкил, или частично или полностью галоген-замещенный C1-C4 алкил;
каждый из R3, R4, R5, R8, R9, R10, R11, R12, R13 и R14 независимо друг от друга представляет собой водород, дейтерий или галоген (такой как F, Cl или Br);
R6 представляет собой водород, дейтерий или один или более дейтерий-замещенный или пердейтерированный C1-C4 алкил;
R7 представляет собой водород, дейтерий или один или более дейтерий-замещенный или пердейтерированный C1-C4 алкил;
при условии, что по меньшей мере один из R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13 или R14 является дейтерированным или представляет собой дейтерий.
В одном воплощении содержание дейтерия в дейтерий-замещенном положении составляет свыше распространенности дейтерия в природе (0,015%), предпочтительно >30%, более предпочтительно >50%, более предпочтительно >75%, или >95%, или >99%.
В одном воплощении, исключая H, все или почти все (>99 масс.%) элементы (такие как N, C, O, F и т.д.) в соединении формулы (I) представляют собой элементы, наиболее распространенные в природе, такие как 14N, 12C, 16O и 19F.
В одном воплощении соединения формулы (I) содержат по меньшей мере один атом дейтерия, предпочтительно три атома дейтерия и более предпочтительно пять атомов дейтерия.
В одном воплощении R1 представляет собой галоген и предпочтительно хлор.
В одном воплощении R2 представляет собой трифторметил.
В одном воплощении R6 или R7 независимо друг от друга выбраны из водорода, дейтерия, дейтерированного метила или дейтерированного этила; предпочтительно, монодейтерированного метила, бидейтерированного метила, тридейтерированного метила, монодейтерированного этила, бидейтерированного этила, тридейтерированного этила, тетрадейтерированного этила или пентадейтерированного этила.
В одном воплощении R6 или R7 независимо друг от друга выбраны из водорода, метила или тридейтерированного метила.
В одном воплощении R3, R4 или R5 независимо друг от друга выбраны из водорода или дейтерия.
В одном воплощении R8, R9, R10 или R11 независимо друг от друга выбраны из водорода или дейтерия.
В одном воплощении R12, R13 или R14 независимо друг от друга выбраны из водорода или дейтерия.
В одном воплощении упомянутое соединение представляет собой предпочтительное соединение, выбранное из группы, состоящей из следующих соединений:
N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины (или 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамида)

4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)-2-(N-(метил-d3)аминоформил)пиридин-1-оксида
 .
.
Промежуточные соединения
Как употреблено в данном документе, термин "промежуточное соединение по изобретению" представляет собой соединение формулы B:

где Y представляет собой галоген или  .
.
В одном воплощении, исключая H, все или почти все (>99 масс.%) элементы (такие как N, C, O и т.д.) в вышеупомянутых соединениях представляют собой элементы, наиболее распространенные в природе, такие как 14N, 12C и 16O.
Активные ингредиенты
Как употреблено в данном документе, термин "соединение по изобретению" относится к соединению формулы (I). Этот термин также включает различные кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Как употреблено в данном документе, термин "фармацевтически приемлемые соли" относится к солям, которые приемлемы для медицины и образованы посредством соединения по изобретению и кислоты или основания. Фармацевтически приемлемые соли включают неорганические соли и органические соли. Предпочтительная соль образована посредством соединения по изобретению и кислоты. Кислота, приемлемая для образования солей, включает, но не ограничена, неорганическую кислоту, такую как соляная кислота, бромоводородная кислота, фтороводородная кислота, серная кислота, азотная кислота, фосфорная кислота; органическую кислоту, такую как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, метансульфоновая кислота, бензолметансульфоновая кислота; и кислую аминокислоту, такую как аспарагиновая кислота, глутаминовая кислота.
Получение
Способы получения соединения (I) и промежуточного соединения формулы B описаны подробно ниже. Однако эти конкретные способы предложены не для ограничения изобретения. Соединения по изобретению, дополнительно, могут быть быстро приготовлены посредством сочетания любых различных способов, приведенных в описании, или различных способов, известных в области техники, и такие сочетания могут быть быстро реализованы специалистами в области техники.
Известен способ получения недейтерированной ω-дифенилмочевины и ее физиологически совместимых солей, используемых в изобретении. Дейтерированная ω-дифенилмочевина может быть приготовлена тем же способом при применении соответствующих дейтерированных соединений в качестве исходных материалов. Например, соединение (I) может быть приготовлено согласно способу, описанному в WO 2000/042012, исключая применение в реакции дейтерированного материала вместо недейтерированного материала.
Обычно, в течение приготовления, каждую реакцию проводят в инертном растворителе при температуре от комнатной до температуры образования флегмы (такой как 0-80°C, предпочтительно 0-50°C). Как правило, время реакции составляет 0,1-60 часов, предпочтительно 0,5-48 часов.
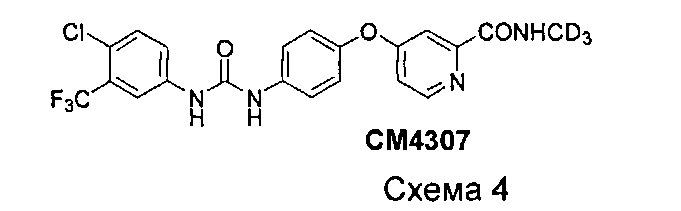
Рассматривая СМ4307 в качестве примера, оптимизированный путь получения изображен ниже:


Как показано на схеме 1, в присутствии N,N′-карбонилдиимидазола, фосгена или трифосгена 4-аминофенол (соединение I) реагирует с 3-трифторметил-4-хлор-анилином (соединение II) до получения 1-(4-хлор-3-(трифторметил)фенил)-3-(4-гидроксифенил)мочевины (соединение III). 2-(N-(метил-d3))карбомоилпиридин (соединение V) получают посредством реакции метилпиколината (соединение IV) с (метил-d3)амином или (метил-d3)амингидрохлоридом непосредственно или в присутствии основания, такого как карбонат натрия, карбонат калия, гидроксид натрия, триэтиламин, пиридин и т.п. В присутствии основания (такого как трет-бутоксид калия, гидрид натрия, гидрид калия, карбонат калия, карбонат цезия, фосфат калия, гидроксид калия, гидроксид натрия) и возможно катализатора (такого как йодид меди и пролин или йодид меди и пиколиновая кислота) соединение III реагирует с соединением V с образованием соединения СМ4307. Вышеупомянутые реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и пр. и при температуре 0-200°C.
Рассматривая СМ4307 в качестве примера, другой предпочтительный способ приведен ниже:
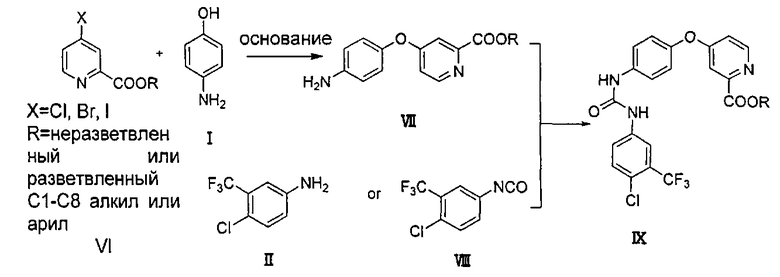
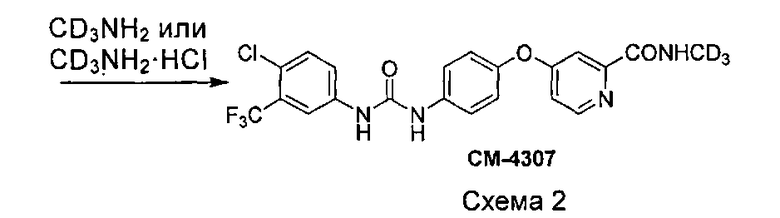
Как показано на схеме 2, амин (соединение VII) получают посредством реакции пиколината (соединение VI) с 4-аминофенолом (соединение I) в присутствии основания (такого как трет-бутоксид калия, гидрид натрия, гидрид калия, карбонат калия, карбонат цезия, фосфат калия, гидроксид калия, гидроксид натрия) и, возможно, катализатора (такого как йодид меди и пролин или йодид меди и пиридинкарбоновая кислота). Мочевину (соединение IX) получают посредством реакции соединения VII с соединением II в присутствии N,N′-карбонилдиимидазола, фосгена или трифосгена, или с 1-хлор-4-изоцианато-2-(трифторметил)бензолом (соединение VIII). Соединение СМ4307 получают посредством реакции соединения IX с (метил-d3)амином или гидрохлоридом (метил-d3)амина непосредственно или в присутствии основания (такого как карбонат натрия, карбонат калия, гидроксид натрия, триэтиламин, пиридин и т.п.). Вышеупомянутые реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и т.п., при температуре 0-200°C.
Рассматривая СМ4307 в качестве примера, другой предпочтительный способ приведен ниже:


Как показано на схеме 3, мочевину (соединение XI) получают посредством реакции 4-метилоксифениламина (соединение X) с соединением II в присутствии N,N′-карбонилдиимидазола, фосгена или трифосгена, или с 1-хлор-4-изоцианато-2-(трифторметил)бензолом (соединение VIII). 1-(4-хлор-3-(трифторметил)фенил)-3-(4-гидроксифенил)мочевину (соединение III) получают при помощи любого из способов деметилирования, известных в области техники. Соединение СМ4307 получают посредством реакции соединения III с соединением V посредством способа, описанного на схеме 1, или любых способов, известных в области техники. Вышеупомянутые реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и т.п. и при температуре 0-200°C.
Рассматривая СМ4307 в качестве примера, другой особенно предпочтительный способ приведен ниже:
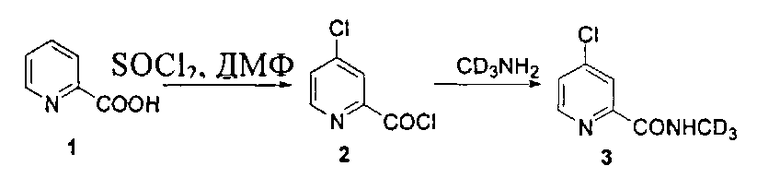
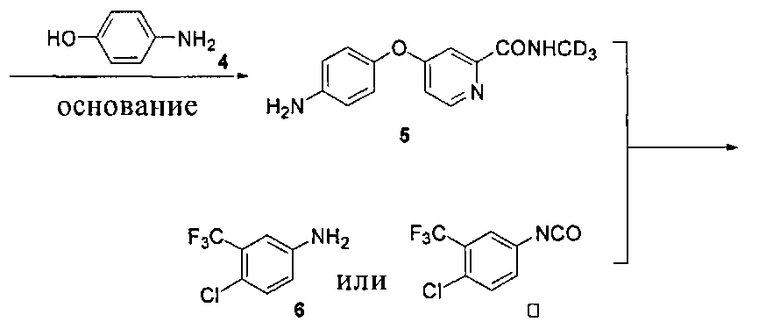
Дейтерий может быть введен посредством применения дейтерированного метиламина.
Дейтерированный метиламин или его гидрохлорид может быть приготовлен посредством следующих реакций. Дейтерированный нитрометан получают посредством реакции нитрометана с дейтериевой водой в присутствии основания (такого как гидрид натрия, гидрид калия, дейтерированный гидроксид натрия, дейтерированный гидроксид калия, карбонат калия и т.п.) или катализатора фазового переноса. При необходимости вышеупомянутый эксперимент может быть повторен для получения дейтерированного нитрометана высокой чистоты. Дейтерированный нитрометан восстанавливают в присутствии цинковой пыли, магнезии, железа или никеля и т.п. для образования дейтерированного метиламина или его гидрохлорида.

Кроме того, дейтерированный метиламин или его гидрохлорид может быть получен посредством следующих реакций:

Ключевое промежуточное соединение 3 может быть синтезировано из дейтерированного метанола (CD3OD) посредством следующих реакций:

Подробный способ получения описан в примере 1.
Основные преимущества настоящего изобретения включают:
(1) Соединения по настоящему изобретению обладают превосходными ингибирующими активностями фосфокиназ, таких как raf-киназы.
(2) Различные дейтерированные дифенилмочевины высокой чистоты могут быть приготовлены удобно и высокоэффективно при помощи промежуточного соединения формулы B по изобретению.
(3) Условия реакции являются мягкими, а манипуляции безопасными.
Настоящее изобретение будет в дальнейшем проиллюстрировано ниже при помощи ссылки на конкретные примеры. Следует понимать, что эти примеры приведены только для иллюстрации изобретения, но не для ограничения объема изобретения. Экспериментальные способы с неспецифичными условиями, описанные в следующих примерах, обычно осуществляют под действием обычных условий или согласно инструкциям производителя. Если не указано иное, части и процентное содержание вычислены по массе.
Пример 1
Получение N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины (соединение СМ4307)
Путь синтеза:

1. Получение 4-хлор-N-(метил-d3)пиколинамида (3)
В 250-мл одногорловый сосуд с закругленным днищем, оборудованный при помощи устройства для обработки отходящего газа, добавляли тионилхлорид (60 мл). Безводный ДМФ (диметилформамид) добавляли медленно по капле (2 мл) во время хранения при температуре 40-50°C. После добавления, смесь перемешивали в течение 10 мин и затем добавляли никотиновую кислоту (20 г, 162,6 ммоль) по частям в течение периода 20 мин. Цвет раствора постепенно изменялся с зеленого на светло-фиолетовый. Реакционную смесь нагревали до 72°C и дефлегмировали в течение 16 часов с перемешиванием. Образовывалось большое количество твердого осадка. Смесь охлаждали до комнатной температуры, разбавляли при помощи толуола (100 мл) и концентрировали почти до сухого состояния. Осадок растворяли при помощи толуола и концентрировали до сухого состояния. Осадок фильтровали и промывали при помощи толуола для получения 4-хлорпиколиноилхлорида в виде светло-желтого твердого вещества. Твердое вещество медленно добавляли в насыщенный раствор (метил-d3)амина в тетрагидрофуране в ванне со льдом. Смесь хранили при менее 5°C и перемешивали в течение 5 часов. Затем смесь концентрировали, и этилацетат добавляли для получения белого твердого осадка. Смесь фильтровали, и фильтрат промывали при помощи насыщенного солевого раствора, сушили над сульфатом натрия и концентрировали для получения 4-хлор-N-(метил-d3)пиколинамида (3) (20,68 г, выход 73%) в виде светло-желтого твердого вещества.
1Н ЯМР (CDCl3, 300 МГц): 8,37 (d, 1Н), 8,13 (с, 1Н), 7,96 (br, 1Н), 7,37 (d, 1Н).
2. Получение 4-(4-аминофенокси)-N-(метил-d3)пиколинамида (5)
К сухому ДМФ (100 мл) добавляли по очереди 4-аминофенол (9,54 г, 0,087 моль) и трет-бутоксид калия (10,3 г, 0,092 моль). Цвет раствора возвращался к темно-коричневому. После перемешивания при комнатной температуре в течение 2 часов к реакционной смеси добавляли 4-хлор-N-(метил-d3)пиколинамид (3) (13,68 г, 0,079 моль) и безводный карбонат калия (6,5 г, 0,0467 моль), затем нагревали до 80°C и перемешивали в течение ночи. ТСХ-анализ (тонкослойная хроматография) показал, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры, и выливали в смесь растворов этилацетата (150 мл) и насыщенного раствора соли (150 мл). Смесь перемешивали и затем оставляли для разделения слоев. Водную фазу экстрагировали при помощи этилацетата (3×100 мл). Экстрагированные слои совмещали, промывали при помощи насыщенного солевого раствора (3×100 мл) до высушивания на безводном сульфате натрия и концентрировали для получения 4-(4-аминофенокси)-N-(метил-d3)пиколинамида (18,00 г, выход 92%) в виде светло-желтого твердого вещества.
1Н ЯМР (CDCl3, 300 МГц): 8,32 (d, 1Н), 7,99 (br, 1Н), 7,66 (s, 1Н), 6,91-6,85 (m, 3Н), 6,69 (m, 2Н), 3,70 (br, s, 2Н).
3. Получение N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины (СМ4307)
К метиленхлориду (120 мл) добавляли 4-хлор-3-трифторметил-фениламин (15,39 г, 78,69 ммоль) и N,N'-карбонилдиимидазол (13,55 г, 83,6 ммоль). После перемешивания при комнатной температуре в течение 16 часов раствор 4-(4-аминофенокси)-N-(метил-d3)пиколинамида (18 г, 73 ммоль) в метиленхлориде (180 мл) добавляли медленно по капле и смесь перемешивали при комнатной температуре в течение других 18 часов. ТСХ-анализ показал, что реакция завершена. Смесь концентрировали до приблизительно 100 мл посредством удаления части метиленхлорида при помощи роторного испарителя и оставляли на нескольких часов при комнатной температуре. Значительное количество белого твердого вещества выпадало в осадок. Твердое вещество фильтровали и промывали при помощи избытка метиленхлорида. Фильтрат концентрировали посредством удаления некоторых растворителей, и некоторые твердые вещества вновь выпадали в осадок. Две части твердого вещества совмещали и промывали при помощи избытка метиленхлорида для получения N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины (СМ4307, 20,04 г, выход 58%) в виде белого порошка (чистый продукт).
1Н ЯМР (CD3OD, 300 МГц): 8,48 (d, 1Н), 8,00 (d, 1Н), 7,55 (m, 5Н), 7,12 (d, 1Н), 7,08 (s, 2Н), ESI-HRMS m/z. C21H13ClF3N4O3, вычисл. 467,11, найдено 490,07 (M+Na)+.
Кроме того, соединение СМ4307 растворяли в метиленхлориде и проводили реакцию с надбензойной кислотой для получения соответствующего окисленного производного: 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)-2-(N-(метил d3)аминоформил)пиридин-1-оксида.

Пример 2
Получение 4-хлор-N-(метил-d3)пиколинамида (3)
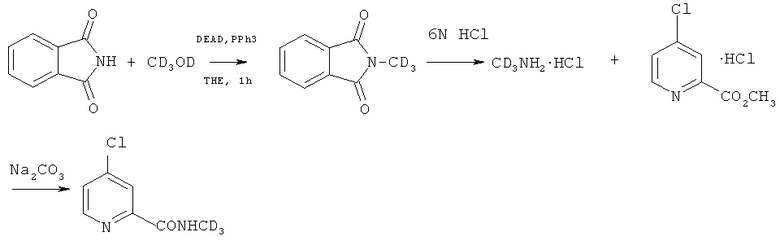
a. В раствор фталимида (14,7 г, 0,1 моль), дейтерированного метанола (3,78 г, 0,105 моль, 1,05 экв.) и трифенилфосфина (28,8 г, 0,11 моль, 1,1 экв.) в безводном тетрагидрофуране добавляли по капле раствор DEAD (диэтилазодикарбоксилата) (1,1 экв.) в тетрагидрофуране в ванне с ледяной водой. После добавления смесь перемешивали в течение 1 часа при комнатной температуре. Смесь очищали при помощи хроматографической колонки, или растворитель в смеси удаляли, а затем осадок растворяли при помощи соответствующего количества DCM и охлаждали в холодильнике до осаждения твердого вещества. Смесь фильтровали, фильтрат концентрировали при помощи роторного испарителя и затем осадок очищали посредством флэш-хроматографии на колонке для получения чистого продукта 2-(N-(метил-d3))-изоиндол-1,3-диона (14,8 г, выход 90%).
b. 2-(N-(метил-d3))-изоиндол-1,3-дион (12,5 г, 0,077 моль) растворяли в соляной кислоте (6 N, 50 мл) и смесь кипятили с обратным холодильником в течение 24-30 часов в запаянной пробирке. Реакционную смесь охлаждали до комнатной температуры и затем охлаждали до менее 0°C в холодильнике до осаждения твердого вещества. Твердое вещество фильтровали и промывали при помощи холодной деионизированной воды. Фильтрат собирали и концентрировали при помощи роторного испарителя для удаления воды и высушивали до получения соли гидрохлорида (метил-d3)амина. Безводный DCM (дихлорметан) (100 мл) добавляли к соли гидрохлориду (метил-d3)амина и добавляли гидрохлорид метил-4-хлорпиколината (6,52 г, 0,038 моль, 0,5 экв.) и карбонат натрия (12,2 г, 0,12 моль, 1,5 экв.). Реактивный сосуд запаивали и помещали в холодильник на один день. После ТСХ-анализа, выявившего, что реакция завершена, реакционную смесь промывали при помощи воды, сушили, концентрировали и очищали при помощи хроматографической колонки для получения 4-хлор-N-(метил-d3)пиколинамида (соединение (3), 5,67 г, выход 86%). Особенность структуры была такая же, как и в примере 1.
Пример 3
Получение соединения СМ4307

1. Получение 1-хлор-4-изоцианато-2-(трифторметил)бензола А4
При помощи устройства абсорбции отходящего газа трифосген (167 г, 0,56 моль, 0,5 экв.) растворяли в хлороформе (500 мл). Раствор N-метилморфолина (NMM) (11,4 г, 0,11 моль, 0,1 экв.) в хлороформе (100 мл) добавляли по капле в вышеупомянутую смесь при 5°C. После добавления раствор 4-хлор-3-(трифторметил)анилина (220 г, 1,13 моль, 1,0 экв.) в хлороформе (700 мл) добавляли по капле при 10°C. Смесь нагревали до 40°C и перемешивали в течение 15 часов, а затем нагревали до 50°C и перемешивали в течение 5 часов, после чего нагревали до 60-65°C и кипятили с обратным холодильником в течение 5 часов. Растворитель удаляли по действием атмосферного давления. Осадок дистиллировали под действием вакуума (температура масла 110-120°C, вакуум 200 Па) и фракции отбирали при температуре 95-100°C до получения соединения, указанного в заголовке (200 г, чистота 98,7%, выход 84%), в виде бесцветной жидкости.
2. Получение 4-хлор-N-(метил-d3)пиколинамида (промежуточное соединение А2)
Способ 1
В трехгорловую колбу с тетрагидрофураном (250 мл) добавляли метил-4-хлорпиколинат (50 г, 0,29 моль, 1 экв.), гидрохлорид (метил-d3)амина (31 г, 0,44 моль, 1,5 экв.) и безводный карбонат калия (400 меш, 80 г, 0,58 моль, 2 экв.) с перемешиванием. Затем смесь перемешивали в течение 20 часов при комнатной температуре, добавляли воду (250 мл) и метил-трет-бутиловый эфир (150 мл). После перемешивания органический слой отделяли. Водный слой экстрагировали при помощи метил-трет-бутилового эфира (100 мл). Органические слои совмещали, сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате удаляли под действием пониженного давления для получения соединения, указанного в заголовке (48 г, чистота 99%, выход 96%), в виде светло-желтой жидкости.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,64 (dd, J=2Hz, 5,2Hz, 1H), 7,97(d, J=1,6 Гц, 1H), 8,54(d, J=5,2 Гц, 1H), 8,74(br, 1H).
MS (ESI, m/z) вычисл. для C7H4D3ClN2O: 173, найдено: 174 [M+H]+
Способ 2
Метил-4-хлорпиколинат (130 г, 0,76 моль, 1 экв.) растворяли в безводном этаноле (1,3 л). Гидрохлорид (метил-d3)амина (80 г, 1,13 моль, 1,5 экв.) и безводный карбонат калия (313 г, 2,67 моль, 3 экв.) добавляли к смеси при перемешивании. Смесь перемешивали при комнатной температуре в течение 50 часов. Смесь фильтровали и промывали при помощи этанола (260 мл×2), растворитель в фильтрате удаляли под действием пониженного давления, добавляли этилацетат (400 мл), и конечную смесь промывали при помощи насыщенного солевого раствора (250 мл×2). Водный слой экстрагировали при помощи этилацетата (100 мл×2). Органические фазы совмещали, сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате удаляли под действием пониженного давления для получения соединения, указанного в заголовке (109 г, чистота 98%, выход 83%), в виде светло-желтой жидкости.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,64 (dd, J=2 Гц, 5,2 Гц, 1Н), 7,97(d, J=1,6 Гц, 1Н), 8,54 (d, J=5,2 Гц, 1Н), 8,74 (br, 1Н).
MS (ESI, m/z) вычисл. для C7H4D3ClN2O: 173, найдено: 174 [М+Н]+
3. Получение 1-(4-хлор-3-трифторметилфенил)-3-(4-гидроксифенил)мочевины А5
Способ 1
4-Амино-фенол (5 г, 45,82 ммоль, 1 экв.) растворяли в дихлорметане (40 мл) при комнатной температуре. Добавляли по капле раствор 1-хлор-4-изоцианато-2-(трифторметил)бензола (10,7 г, 48,11 ммоль, 1,05 экв.) в дихлорметане (40 мл). Смесь перемешивали при комнатной температуре в течение 16 часов. Смесь фильтровали и промывали при помощи дихлорметана (10 мл×2) для получения титульного соединения (14,2 г, чистота 97%, выход 94%) в виде светло-коричневого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 6,70 (dd, J=2 Гц, 6,8 Гц, 1Н), 7,22 (dd, J=2Hz, 6,4 Гц, 1Н), 7,58-7,24 (m, 1Н), 8,10 (d, J=2 Гц, 1Н), 8,50 (br, 1Н), 9,04 (br, 1Н), 9,14 (br, 1Н).
MS (ESI, m/z) вычисл. для C14H10ClF3N2O2: 330, найдено: 331 [М+Н]+
Способ 2

1-Хлор-4-изоцианато-2-(трифторметил)бензол (5,15 г, 26 ммоль, 1,05 экв.) растворяли в дихлорметане (30 мл). Добавляли по капле раствор п-метоксианилина (3,07 г, 25 ммоль, 1 экв.) в дихлорметане (20 мл), и смесь перемешивали при комнатной температуре в течение 20 часов. Смесь фильтровали и промывали при помощи дихлорметана (5 мл×2). Твердое вещество растворяли в этилацетате (50 мл), и полученный раствор промывали при помощи разбавленной соляной кислоты (1 Н, 10 мл) и насыщенного солевого раствора (20 мл). Органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли под действием пониженного давления для получения 1-(4-хлор-3-трифторметилфенил)-3-(4-метоксифенил)мочевины А6 (4,5 г, выход 52%) в виде белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 3,73 (s, 3H), 6,86-6,90 (m, 2Н), 7,35-7,39 (m, 2Н), 7,59-7,65 (m, 2Н), 8,11 (d, J=2 Гц, 1Н), 8,65(br, 1Н), 9,09(br, 1Н).
MS (ESI, m/z) вычисл. для C15H12ClF3N2O2: 344, найдено: 345[M+H]+.
1-(4-Хлор-3-трифторметилфенил)-3-(4-метоксифенил)мочевину А6 (344 мг, 1 ммоль, 1 экв.) растворяли в уксусной кислоте (4 мл). Добавляли бромоводородную кислоту (40%, 1 мл), и смесь дефлегмировали в течение 5 часов. Смесь охлаждали до комнатной температуры, и добавляли ледяную воду (10 мл). Смесь экстрагировали при помощи этилацетата (20 мл). Органическую фазу промывали при помощи насыщенного бикарбоната натрия (10 мл) и сушили над безводным сульфатом натрия. Растворитель в органической фазе удаляли под действием пониженного давления для получения соединения, указанного в заголовке (140 мг, чистота 90%, выход 42%), в виде светло-желтого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 6,70 (dd, J=2 Гц, 6,8 Гц, 1Н), 7,22 (dd, J=2 Гц, 6,4 Гц, 1Н), 7,58-7,24 (m, 1Н), 8,10 (d, J=2 Гц, 1Н), 8,50 (br, 1Н), 9,04 (br, 1Н), 9,14 (br, 1Н).
MS (ESI, m/z) вычисл. для C14H10ClF3N2O2: 330, найдено: 331 [М+Н]+
4. Получение 4-(4-(3-(4-хлор-3-(трифторметил)фенил]уреидо)- фенокси)-N-(метил-d3)пиколинамида (СМ4307)
1-(4-Хлор-3-трифторметил-фенил)-3-(4-гидрокси-фенил)мочевину А5 (4 г, 12,10 ммоль, 1 экв.) растворяли в N,N-диметилформамиде (20 мл). Трет-бутоксид калия (4,6 г, 41,13 ммоль, 3,4 экв.) добавляли по частям. Затем смесь перемешивали в течение 3 часов, добавляли 4-хлор-N-(метил-d3)пиколинамид (2,3 г, 13,31 ммоль, 1,1 экв.) и карбонат калия (0,8 г, 6,05 ммоль, 0,5 экв.). Смесь нагревали до 80°C и перемешивали в течение 1,5 часов. Смесь охлаждали до комнатной температуры, добавляли этилацетат (200 мл) и фильтровали для удаления неорганических солей. Фильтрат промывали при помощи насыщенного солевого раствора (50 мл×3) и отделяли органический слой. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли под действием пониженного давления для получения твердого вещества с последующим добавлением ацетонитрила (15 мл). Полученную смесь дефлегмировали в течение 2 часов, охлаждали до комнатной температуры и фильтровали для получения СМ4307 (3,4 г, чистота 96%, выход 60%) в виде светло-желтого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,15 (dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,17-7,19 (m, 2Н), 7,40 (d, J=2,4 Гц, 1Н), 7,59-7,69 (m, 4Н), 8,13 (6, J=2,4 Гц, 1Н), 8,51 (d, J=6 Гц, 1Н), 8,75 (br, 1Н), 8,90 (br, 1Н), 9,22 (br, 1Н).
MS (ESI, m/z) вычисл. для C21H13D3ClF3N4O3: 467, найдено: 468[М+Н]+.
Пример 4
Получение соединения СМ4307

1. Получение 4-хлор-N-(метил-d3)пиколинамида (промежуточное соединение А2)
Под действием азота в реактор (30 л) добавляли тетрагидрофуран (10,86 кг). После начала перемешивания последовательно добавляли гидрохлорид (N-(метил-d3))амина (1,50 кг, 21,26 моль, 1,5 экв.), метил-4-хлорпиколинат (2,43 кг, 14,16 моль, 1 экв.) и безводный карбонат калия (3,92 кг, 28,36 моль, 2 экв.). Реакцию проводили при 33°C в течение 15 ч, и затем добавляли чистую воду (12,20 кг). Реакционную смесь экстрагировали при помощи метил-третбутилового эфира (3,70 кг×2). Органические фазы совмещали, сушили над безводным сульфатом натрия (0,50 кг), перемешивали в течение 1 часа и фильтровали. Растворители удаляли под действием вакуума (≤-0,09 МПа) при 40±2°C при помощи водяной бани для получения соединения, указанного в заголовке (2,41 кг, чистота 99,0%, выход 98%), в виде светло-желтого масла.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,64 (dd, J=2 Гц, 5,2 Гц, 1Н), 7,97 (d, J=1,6 Гц, 1Н), 8,54 (d, J=5,2 Гц, 1Н), 8,74 (br, 1Н).
MS (ESI, m/z) вычисл. для C7H4D3ClN2O: 173, найдено: 174 [М+Н]+
2. Получение 4-(4-аминофенокси)-N-(метил-d3)пиколинамида (промежуточное соединение A3)
Способ 1
Под действием азота в реактор (20 л) добавляли диметилсульфоксид (2,75 кг). После начала перемешивания последовательно добавляли 4-хлор-N-(метил-d3)пиколинамид (2,41 кг, 13,88 моль, 1 экв.), 4-аминофенол (1,62 кг, 14,84 моль, 1,08 экв.) и трет-бутоксид калия (1,66 кг, 14,79 моль, 1,1 экв.). После стабилизации температуры реактора внутреннюю температуру поднимали до 80°C, и проводили перемешивание в течение 4 часов. После снижения внутренней температуры до 40°C для разбавления реакционной смеси при перемешивании добавляли изопропанол (7,90 кг). Реактор промывали при помощи изопропанола, и полученную смесь переносили в реактор (30 л). Под действием азота по капле добавляли соляную кислоту (5,81 кг). После добавления смесь перемешивали, фильтровали посредством центрифугирования и промывали чистой водой. Твердое вещество переносили в реактор (50 л) и полностью растворяли в воде (21,00 кг) при помощи перемешивания. Под действием азота раствор карбоната калия (2,5 кг карбоната калия, растворенного в 7 л чистой воды) добавляли по капле в вышеупомянутый реактор (50 л) в течение 1,5 часов. Смесь выгружали и центрифугировали, и продукт промывали при помощи чистой воды и высушивали под действием вакуума в течение 24 часов для получения соединения, указанного в заголовке (2,72 кг, чистота 99,9%, выход 78%), в виде светло-коричневого кристалла.
1Н ЯМР (DMSO-d6, 400 МГц): δ 5,19 (br, 2Н), 6,66-6,68 (m, 2Н), 6,86-6,88 (m, 2Н), 7,07 (dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,36 (d, J=2,8 Гц, 1Н), 8,45 (d, J=5,6 Гц, 1Н), 8,72 (br, 1Н).
MS (ESI, m/z) вычисл. для C13H10D3N3O2Cl: 246, найдено: 247[М+Н]+.
Способ 2
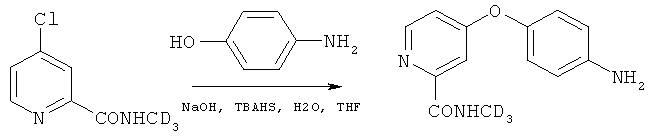
4-Хлор-N-(метил-d3)пиколинамид (4,3 г, 24,77 ммоль, 1 экв.) растворяли в тетрагидрофуране (20 мл) при комнатной температуре. При комнатной температуре при перемешивании добавляли 4-аминофенол (2,7 г, 24,77 ммоль, 1 экв.), гидросульфат тетрабутиламмония (1,68 г, 4,95 ммоль, 0,2 экв.) и гидроксид натрия (1,35 г, 33,69 ммоль, 1,36 экв.). Медленно по капле добавляли раствор гидроксида натрия в воде (45%, гидроксид натрия (1,32 г) растворяли в воде (1,6 мл)). Смесь нагревали до 67°C и перемешивали в течение 20 часов. Смесь охлаждали до менее 20°C, и добавляли концентрированную соляную кислоту (37%, 10 мл) при поддержании температуры реакции менее 25°C. Смесь перемешивали в течение 1 часа, фильтровали и промывали при помощи тетрагидрофурана (20 мл). Полученное твердое вещество растворяли в воде (60 мл). Смесь охлаждали до 10-20°C и медленно добавляли по капле раствор гидроксида натрия (22,5%, 2,6 мл) до pH, равного 3-3,5. Непрерывно добавляли раствор гидроксида натрия (22,5%, 3,4 мл) до pH, равного 7-8 и осаждения светло-желтого твердого вещества. В течение добавления температуру смеси поддерживали менее 20°C. Смесь фильтровали, и твердое вещество промывали при помощи воды (12 мл×2). Твердое вещество сушили под действием вакуума для получения 4-(4-аминофенокси)-(N-(метил-d3)пиколинамида (5,01 г, чистота 99%, выход 82%) в виде светло-желтого твердого вещества.
1Н ЯМР (DMSO-d6 400 МГц): δ 5,19 (br, 2Н), 6,66-6,68 (m, 2Н), 6,86-6,88 (m, 2Н), 7,07 (dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,36 (d, J=2,8 Гц, 1Н), 8,45 (d, J=5,6 Гц, 1Н), 8,72 (br, 1Н)
MS (ESI, m/z) вычисл. для С13H10D3N3O2Cl: 246, найдено: 247[М+Н]+.
3. Получение 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамида (СМ4307)
Под действием азота дихлорметан (17,30 кг) и диметилсульфоксид (2,92 кг) добавляли в сухой реактор (50 л). Смесь перемешивали при комнатной температуре, добавляли 4-(4-аминофенокси)-N-(метил-d3)пиколинамид (2,65 кг, 10,76 моль). 1-Хлор-4-изоцианато-2-(трифторметил)бензол (2,50 кг, 11,26 моль, 1,05 экв.) растворяли в дихлорметане (7,00 кг). В реактор добавляли по капле раствор 1-хлор-4-изоцианато-2-(трифторметил)бензола в дихлорметане. Реакцию проводили в течение 10 мин при комнатной температуре. Реакционную смесь охлаждали до 3±2°C при помощи солевой ванны со льдом. В реактор по капле добавляли чистую воду (10,60 кг) при поддержании температуры 3±2°C. После добавления смесь перемешивали в течение 30 мин, затем выгружали и центрифугировали. Продукт промывали при помощи дихлорметана (7,00 кг). Полученный продукт высушивали под действием вакуума в течение 24 часов для получения грязно-белого порошка (4,8 кг, чистота 99,8%, выход 95,4%).
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,15(dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,17-7,19 (m, 2Н), 7,40 (d, J=2,4 Гц, 1Н), 7,59-7,69 (m, 4Н), 8,13 (d, J=2,4 Гц, 1Н), 8,51 (d, J=6 Гц, 1Н), 8,75 (br, 1Н), 8,90 (br, 1Н), 9,22 (br, 1Н).
MS (ESI, m/z) вычисл. для C21H13D3ClF3N4O3: 467, найдено: 468[М+Н]+
Пример 5
Получение п-толуолсульфоната 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамида (CM4307·TsOH)
Реактор (100 л) заполняли при помощи безводного этанола (45,00 кг). После начала перемешивания отдельно добавляли 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамид (4,50 кг, 9,62 моль, 1 экв.) и моногидрат п-толуолсульфоновой кислоты (0,66 кг, 3,47 моль, 0,36 экв.). Смесь нагревали до 78°C и кипятили с обратным холодильником в течение 40 мин до полного растворения твердого вещества.
Моногидрат п-толуолсульфоновой кислоты (1,61 кг, 8,46 моль) добавляли в безводный этанол (4,50 кг), и смесь нагревали до 70°C до растворения твердого вещества. Полученный раствор добавляли в реактор (100 л). Смесь охлаждали до 0-2°C и хранили в течение 30 мин. Смесь выгружали и фильтровали при помощи центрифугирования. Твердое вещество промывали при помощи безводного этанола (13,50 кг), сушили под действием вакуума в течение 24 ч для получения соединения, указанного в заголовке (5,75 кг, чистота 99,3%, выход 93,4%), в виде от белого до грязно-белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 2,30 (s, 3H), 7,15 (d, J=8,8 Гц, 2Н), 7,20 (d, J=8,8 Гц, 2Н), 7,23 (dd, J=2,8 Гц, 6 Гц, 1Н), 7,52 (d, J=8 Гц, 2Н), 7,55 (d, J=2,8 Гц, 1Н), 7,63 (d, J=8,8 Гц, 3H), 7,68 (dd, J=2,4 Гц, 9,2 Гц, 1Н), 8,03 (br, 1Н), 8,14 (d, J=2,4 Гц, 1Н), 8,56 (d, J=6 Гц, 1Н), 8,91 (br, 1Н), 9,17 (br, 1Н), 9,36 (br, 1Н).
13С ЯМР (ДМСО-d6, 400 МГц): δ 21,1, 26,1, 111,7, 115,2, 117,0, 120,7 (2С), 121,6 (2С), 121,9, 122,8, 123,2, 124,6, 125,6 (2С), 127,2, 129,0 (2С), 132,3, 138,8, 139,5, 139,9, 144,1, 146,6, 147,2, 152,8, 159,9, 170,7 ppm.
Условия жидкостной хроматографии: Agilent 1100 Series; хроматографическая колонка: Synergi 4µ POLAR-RP 80А, 25044,6 мм, 4 мкм; температура колонки: 25°C; длина волны детекции: УФ 210 нм; подвижная фаза: А: дигидрофосфат аммония 10 ммоль/л, В: метанол; инъецируемый объем: 10 мкл; скорость тока: 0,8 мл/мин; время хроматографирования: 70 мин; градиент: 50% подвижной фазы В с 0 по 15 мин, подвижная фаза В поднималась до 75% с 15 по 32 мин, затем 75% подвижную фазу В элюировали в течение 23 мин с 32 по 55 мин. Время удерживания: 4,95 мин (п-толуолсульфоновая кислота); 47,11 мин (СМ4307).
Пример 6
Получения соединения СМ4307

1. Получение трет-бутил-4-хлорпиколината A7
4-Хлорпиколиновую кислоту (10,5 г, 66,64 ммоль) суспендировали в тионилхлориде (40 мл), смесь нагревали до 80°C и кипятили с обратным холодильником. N,N-диметилформамид (ДМФ, DMF) (0,2 мл) добавляли по каплям, и смесь кипятили с обратным холодильником в течение 2 часов. Избыток тионилхлорида удаляли под действием пониженного давления для получения бледно-желтого ацилхлорида, с последующим добавлением дихлорметана (DCM) (60 мл). Полученный раствор добавляли в смешанный раствор трет-бутанола (25 мл), пиридина (20 мл) и дихлорметана (80 мл) при -40°C. Реакционную смесь нагревали до 50°C и перемешивали в течение 16 часов. Растворители удаляли под действием пониженного давления и добавляли этилацетат (150 мл). Полученную смесь промывали при помощи насыщенного солевого раствора (50 мл×2) и раствора гидроксида натрия (1 N, 50 мл×2) и разделяли. Органическую фазу сушили над безводным сульфатом натрия и концентрировали под действием пониженного давления. Осадок сушили под действием вакуума для получения соединения, указанного в заголовке (11,1 г, чистота 95%, выход 78%), в виде бледно-желтого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 1,56 (s, 9Н), 7,80 (dd, J=2,4 Гц, 5,2 Гц, 1Н), 8,02 (d, J=2 Гц, 1Н), 8,69 (d, J=5,2 Гц, 1Н).
MS (ESI, m/z) вычисл. для C10H12ClNO2: 213, найдено: 158[M-Bul+H]+
2. Получение трет-бутил-4-(4-аминофенокси)пиколината А8
При комнатной температуре п-аминофенол (0,51 г, 4,70 ммоль, 1 экв.) растворяли в N,N-диметилформамиде (10 мл). К полученному раствору по частям добавляли трет-бутоксид калия (0,53 г, 4,70 ммоль, 1 экв.), и полученную смесь перемешивали в течение 0,5 часа. Добавляли трет-бутил-4- хлорпиколинат (1 г, 4,70 ммоль, 1 экв.) и карбонат калия (45 мг, 0,33 ммоль, 0,07 экв.), смесь нагревали до 80°C и перемешивали в течение 2 часов. Смесь охлаждали до комнатной температуры и добавляли этилацетат (50 мл). Смесь фильтровали для удаления нерастворенного материала, и фильтрат промывали при помощи насыщенного раствора соли (20 мл×2). Органическую фазу сушили над безводным сульфатом натрия и концентрировали под действием пониженного давления для удаления растворителя. Осадок очищали посредством хроматографии на колонке (дихлорметан:этилацетат=30:1) для получения соединения, указанного в загловке (805 мг, чистота 96%, выход 60%).
1Н ЯМР (DMSO-d6, 400 МГц): δ 1,52 (s, 9Н), 5,21 (br, 2Н), 6,64 (d, J=8,8 Гц, 2Н), 6,87 (d, J=8 Гц, 2Н), 7,35 (dd, J=2,4 Гц, 5,6 Гц, 1Н), 8,50 (d, J=6 Гц, 1Н). MS (ESI, m/z) calcd. for C10H12ClNO2: 286, найдено: 231[M-But+H]+
3. Получение трет-бутил-4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколината А9
При комнатной температуре 1-хлор-4-изоцианато-2-(трифторметил)бензол (656 мг, 2,96 ммоль, 1,05 экв.) растворяли в дихлорметане (5 мл). К полученному раствору медленно по капле добавляли раствор трет-бутил-4-(4-аминофенокси)пиколината (805 мг, 2,81 ммоль, 1 экв.) в дихлорметане (5 мл). Смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли под действием пониженного давления, и полученное твердое вещество очищали посредством хроматографии на колонке (дихлорметан: метанол=30:1) для получения соединения, указанного в заголовке (1,4 г, чистота 95%, выход 85%), в виде белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 1,53 (s, 9Н), 7,13 (dd, J=2,4 Гц, 5,2 Гц, 1Н), 7,18 (d, J=8,8 Гц, 2Н), 7,41 (d, J=2,4 Гц, 1Н), 7,59-7,66 (m, 4Н), 8,13 (d, J=1,6 Гц, 1Н), 8,55 (d, J=5,6 Гц, 1Н), 9,06 (br, 1Н), 9,27 (br, 1Н).
MS (ESI, m/z) вычисл. для С24Н21СlF3NO4:507, найдено: 508 [М+Н]+
4. Получение 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо) фенокси)пиколиновой кислоты А10
При комнатной температуре трет-бутил-4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколинат (1,4 г, 2,76 ммоль) растворяли в дихлорметане (20 мл). К полученному раствору добавляли трифторуксусную кислоту (20 мл) и триэтилсилан (0,5 мл). Полученную смесь нагревали до 50°C и перемешивали в течение 16 часов. Растворитель удаляли под действием пониженного давления, добавляли воду (50 мл) и этилацетат (70 мл). Полученную смесь разделяли, и органическую фазу удаляли. Водный слой фильтровали, и твердое вещество промывали при помощи воды (30 мл×2). Твердое вещество сушили под действием вакуума для получения соединения, указанного в заголовке (1,1 г, чистота 97%, выход 90%), в виде светло-зеленого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,21-7,25 (m, 2Н), 7,33 (dd, J=2,8 Гц, 6 Гц, 1Н), 7,57 (d, J=2,8 Гц, 1Н), 7,60-7,67 (m, 4Н), 8,12 (d, J=2,4 Гц, 2Н), 8,64 (d, J=6 Гц, 1Н), 9,84 (br, 1Н), 10,17 (br, 1Н).
MS (ESI, m/z) вычисл. для C20H12ClF4N3O4:451, найдено: 450 [М-Н]-
5. Получение 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)-фенокси)-N-(метил-d3)пиколинамида СМ4307
Способ 1
При комнатной температуре 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколиновую кислоту (0,5 г, 1,11 ммоль, 1 экв.) растворяли в N,N-диметилформамиде (5 мл). К полученному раствору добавляли гидрохлорид (N-(метил-d3))амина (0,15 г, 2,22 ммоль, 2 экв.), гексафторфосфат 2-(7-аза-1Н-бензотриазол-1-ил)-N,N,N'N'-тетраметилурония (HATU, 0,84 г, 2,22 ммоль, 2 экв.) и N,N-диизопропилэтиламин (DIEA, 0,86 г, 6,66 ммоль, 3 экв.). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. К вышеупомянутой реакционной смеси добавляли воду (20 мл). Полученную смесь перемешивали в течение 0,5 часа и затем фильтровали для получения бледно-белого твердого вещества. Твердое вещество растворяли в этилацетате (50 мл), полученную смесь промывали при помощи насыщенного солевого раствора (10 мл×3) и затем разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате удаляли под действием пониженного давления для получения СМ4307 (0,42 г, чистота 97%, выход 81%) в виде грязно-белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,15 (dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,17-7,19 (m, 2Н), 7,40 (d, J=2,4 Гц, 1Н), 7,59-7,69 (m, 4Н), 8,13 (d, J=2,4 Гц, 1Н), 8,51 (d, J=6 Гц, 1Н), 8,75 (br, 1Н), 8,90 (br, 1Н), 9,22 (br, 1Н).
MS (ESI, m/z) вычисл. для C21H13D3ClF3N4O3: 467, найдено: 468[М+Н]+
Способ 2
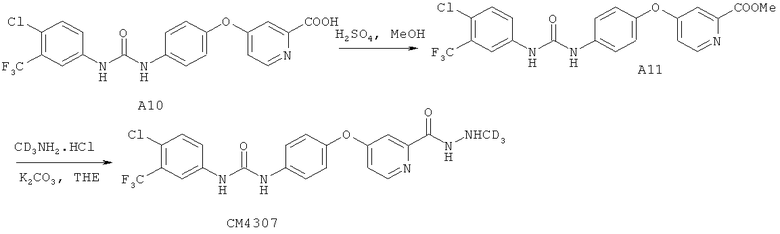
4-(4-(3-(4-Хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколиновую кислоту (0,5 г, 1,11 ммоль) суспендировали в метаноле (10 мл). При комнатной температуре добавляли концентрированную серную кислоту (2 мл), и полученную смесь дефлегмировали в течение 3 часов. Растворитель удаляли под действием пониженного давления и осадок очищали посредством хроматографии на колонке (дихлорметан:метанол=10:1) для получения метил-4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколината А11 (0,46 г, чистота 95%, выход 90%) в виде белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 3,85 (s, 3H), 7,18-7,21 (m, 3H), 7,43 (d, (dd, J=2,4 Гц, 1Н), 7,59-7,66 (m, 4Н), 8,13 (d, J=2,4 Гц, 1Н), 8,59 (d, J=6 Гц, 1Н), 9,06 (br, 1Н), 9,27(br, 1Н).
MS (ESI, m/z) вычисл. для C21H15ClF3N3O4:465, найдено: 466 [М+Н]+
Метил-4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)пиколинат (300 мг, 0,65 ммоль, 1 экв.) добавляли в трехгорловую колбу, содержащую тетрагидрофуран (10 мл) с перемешиванием. К полученной смеси добавляли гидрохлорид (N-(метил-d3))амина (91 мг, 1,3 ммоль, 2 экв.) и безводный карбонат калия (400 меш, 179 мг, 1,3 ммоль, 2 экв.). После перемешивания смеси при комнатной температуре в течение 20 часов добавляли воду (5 мл) и метил-трет-бутиловый эфир (15 мл). Смесь перемешивали и отделяли органическую фазу. Водный слой экстрагировали при помощи метил-трет-бутилового эфира (10 мл), органические слои совмещали, сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате удаляли под действием пониженного давления для получения СМ4307 (261 мг, чистота 96%, выход 86%) в виде грязно-белого твердого вещества.
1Н ЯМР (DMSO-d6, 400 МГц): δ 7,15 (dd, J=2,8 Гц, 5,6 Гц, 1Н), 7,17-7,19 (m, 2Н), 7,40 (d, J=2,4 Гц, 1Н), 7,59-7,69 (m, 4Н), 8,13 (d, J=2,4 Гц, 1Н), 8,51 (d, J=6 Гц, 1Н), 8,75 (br, 1Н), 8,90 (br, 1Н), 9,22 (br, 1Н).
MS (ESI, m/z) calcd. for C21H13ClF3N4O3: 467, найдено: 468[M+Н]+
Пример 7
Фармакокинетическая оценка соединений дейтерированной дифенилмочевины на крысах 8 самцов крыс Sprague-Dawley, 7-8-недельного возраста и массой тела около 210 г разделяли на две группы, по 4 в каждой группе (No крыс: контрольная группа: 13-16; экспериментальная группа: 9-12). Крысам орально вводили однократную дозу 3 мг/кг (а) недейтерированного соединения N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-метил-аминоформил)-4-пиридилокси)фенил)мочевины (контрольное соединение СМ4306) или (b) N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-(метил-d3)-аминоформил)-4-пиридилокси)фенил)мочевины (соединение СМ4307 по изобретению), полученной по примеру 1. Сравнивали фармакокинетические отличия СМ4306 и СМ4307.
Крыс кормили обычным кормом, давали воду и хлордиазепоксид. Хлордиазепоксид переставали давать в последнюю ночь перед экспериментом и давали снова спустя два часа после введения соединения. Крыс не кормили в течение 16 часов до теста. Соединение растворяли в 30% ПЭГ400. Глазную кровь забирали на 0,083, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часу после введения соединения.
Крыс ненадолго анестезировали посредством вдыхания эфира. 300 мкл образца глазной крови собирали в пробирки, содержащие 30 мкл солевого раствора с 1% гепарина. Пробирки высушивали в течение ночи при 60°C до применения. После того как образцы крови были последовательно собраны, крыс анестезировали эфиром и умерщвляли.
После того как образцы крови были собраны, пробирки сразу же осторожно переворачивали по меньшей мере пять раз для полного перемешивания содержимого и помещали на лед. Образцы крови центрифугировали при 4°C при 5000 об/мин в течение 5 минут для разделения сыворотки и эритроцитов. 100 мкл сыворотки отбирали в чистую пластиковую центрифужную пробирку при помощи пипеточного дозатора и на пробирках отмечали название соединения и время. Сыворотку хранили при -80°C до ЖХ/МС-анализа (на жидкостном хроматографе с масс-спектрометром).
Результаты приведены на Фиг.1-2. Результаты показывают, что по сравнению с СМ4306 период полураспада (Т1/2) СМ4307 был длиннее (11,3±2,1 часов для СМ4307 и 8,6±1,4 часов для СМ4306 соответственно), площадь под фармакокинетической кривой (AUC0-∞) СМ4307 была значительно увеличена (11255±2472 нг·ч/мл для СМ4307 и 7328±336 нг·ч/мл для СМ4306 соответственно), и кажущийся клиренс СМ4307 был снижен (275±52 мл/ч/кг для СМ4307 и 410±18,7 мл/ч/кг для СМ4306 соответственно).
Вышеупомянутые результаты показывают, что соединение по настоящему изобретению обладает лучшими фармакокинетическими свойствами у животных и таким образом лучшими фармакодинамическими и терапевтическими эффектами.
Кроме того, метаболизм соединения по изобретению в организме изменяли посредством дейтерирования. В частности, гидроксилирование фенила становится более сложным, что приводит к снижению эффекта первого прохождения. В подобных случаях доза может быть изменена, могут быть созданы препараты длительного действия, и применимость может быть улучшена за счет применения препаратов длительного действия.
Кроме того, фармакокинетика также изменялась за счет дейтерирования. Поскольку другая гидратная пленка полностью образована дейтерированными соединениями, распространение дейтерированных соединений в организмах значительно отличается от такового в недейтерированных соединениях.
Пример 8
Фармакокинетическая оценка СМ4307 в отношении ингибирования роста опухоли гепатоклеточной карциномы SMMC-7721 на модели ксенотрансплантата у бестимусных мышей
70 бестимусных мышей Balb/c nu/nu, 6-недельных самок, были закуплены у Shanghai Experimental Animal Resource Center (Shanghai B&K Universal Group Limited).
SMMC-7721 клетки были получены от Shanghai Institutes for Biological Science, CAS (Shanghai, China).
Образование опухоли на модели ксенотрансплантата у бестимусных мышей: SMMC-7721 клетки культивировали в течение периода логарифмического роста. После подсчета числа клеток, клетки суспендировали в 1×PBS, число клеток в суспензии доводили до 1,5×107/мл. Опухолевые клетки инокулировали под кожу в правую подмышечную ямку бестимусных мышей при помощи 1 мл шприца, 3×106/0,2 мл/мышь. Всего было инокулировано 70 бестимусных мышей.
При достижении размера опухоли 30-130 мм3 58 мышей были произвольно разделены на различные группы. Лекарственные средства начинали вводить, когда различия в средней величине объема опухоли в каждой группе составили менее 10%.
Контрольные дозы в каждой группе приведены в следующей таблице:
Массу тела животного и размер опухоли устанавливали дважды в неделю в течение эксперимента. Клинические симптомы регистрировали каждый день. В конце введения размер опухоли регистрировали при помощи фотографии. Одну мышь умерщвляли в каждой группе, брали ткань опухоли и фиксировали в 4% параформальдегиде. Наблюдение продолжали после введения, и когда средний размер опухоли составил более 2000 мм3, или в случае гибели, животных умерщвляли, проводили макроскопическое препарирование, брали опухолевую ткань и фиксировали в 4% парафармальдегиде.
Формула для вычисления объема опухоли (TV) представляет собой: TV=а×b2/2, где a, b независимо друг от друга представляют собой длину и ширину опухоли. Формула для вычисления относительного объема опухоли (RTV) представляет собой: RTV=Vt/V0, где V0 представляет собой объем опухоли при начале введения, и Vt представляет собой массу опухоли при измерении. Показатель оценки противоопухолевой активности представляет собой относительный коэффициент прироста опухоли Т/С (%), и формула представляет собой: Т/С (%)=(TRTV/CRTV)×100%, где TRTV представляет собой RTV терапевтической группы, и CRTV представляет собой RTV группы отрицательного контроля.
Стандарт оценки эффективности: является эффективным, если относительный коэффициент прироста опухоли Т/С (%) составляет <40% и p<0,05 в результате статистического анализа.
Результаты показаны на Фиг.3. СМ4306 и СМ4307 вводили внутрижелудочно каждый день в течение 2 недель в дозах 10, 30, 100 мг/кг соответственно, и оба соединения оказывали дозозависимый эффект на ингибирование роста опухоли. В конце введения, Т/С% СМ4306 составил 56,9%, 40,6% и 32,2% соответственно. Т/С% СМ4307 составил 53,6%, 40,8% и 19,6%. Т/С% для групп с дозой 100 мг/кг составил <40%, и объем опухоли значительно отличался (p<0,01) от контрольной группы, показавшей значительный эффект ингибирования роста опухоли.
По сравнению с СМ4306 эффективность ингибирования роста опухоли при введении дозы 100 мг/кг СМ4307 была больше (Т/С% для СМ4307 и СМ4306 составил 19,6% и 32,2% соответственно на 15 день), наблюдалось значительное отличие в объеме опухоли между группами (p<0,01). По сравнению с СМ4306 абсолютная величина коэффициента ингибирования опухоли для СМ4307 снизилась более чем на 10%, относительная величина прироста - приблизительно 60% (32,2%/19,6%-1=64%) и СМ4307 оказывало более значительный эффект ингибирования роста опухоли.
Кроме того, в течение эксперимента не наблюдалось токсических эффектов, связанных с лекарственным средством.
Пример 9
Фармацевтические композиции
Посредством обычных способов, эти вещества равномерно перемешивали и вводили в обычные желатиновые капсулы, таким образом, получая 1000 капсул.
Вся литература, упомянутая в настоящей заявке, включена в данный документ посредством ссылки, как если бы она была по отдельности включена посредством ссылки. Кроме того, следует понимать, что на основании раскрытого выше многие вариации и модификации могут быть внесены специалистами в области техники, и эти эквиваленты также находятся в пределах изобретения, как определено прилагаемой формулой изобретения.
Дополнительные данные
Эксперимент проводили аналогично примеру 7, крысам перорально вводили одинаковую дозу (в расчете на СМ4307) соединения СМ4307 или CM4307·TsOH, полученного согласно примеру 5. Препарат представлял собой 0,1% суспензию СМ4307 или CM4307·TsOH в воде, очищенной карбоксиметилцеллюлозой. Сравнивались фармакокинетика СМ4307 и CM4307·TsOH.
Результат
AUC (площадь под кривой) для препарата CM4307·TsOH была в 5-10 раз выше, чем для препарата СМ4307.
Cmax для препарата CM4307·TsOH была в 5-10 выше, чем для препарата СМ4307.
Результаты показывают, что CM4307·TsOH демонстрирует значительно улучшенную фармакокинетику по сравнению с СМ4307.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИМОРФЫ ДЕЙТЕРИРОВАННОЙ ОМЕГА-ДИФЕНИЛМОЧЕВИНЫ ИЛИ ЕЕ СОЛЕЙ | 2013 |
|
RU2600929C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| ДЕЙТЕРИРОВАННЫЕ СОЕДИНЕНИЯ ХИНАЗОЛИНОНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2015 |
|
RU2656485C2 |
| НОВЫЕ АНТИИНВАЗИВНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2641650C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2013 |
|
RU2627706C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| ДЕЙТЕРИРОВАННЫЕ ДИАМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2632907C2 |
| ПРОИЗВОДНЫЕ МОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, СВЯЗАННОГО С РОСТОМ РАКОВЫХ КЛЕТОК (ВАРИАНТЫ) | 2000 |
|
RU2319693C9 |
| АМИДОПРОИЗВОДНЫЕ КАК БЛОКАТОРЫ TTX-S | 2013 |
|
RU2632899C2 |


Изобретение относится к способу получения N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины или ее фармацевтически приемлемых солей
Способ включает (a) реакцию соединения III с соединением V в инертном растворителе и в присутствии основания с образованием указанного соединения
где X представляет собой Cl, Br или I. Соединение III получают следующим образом: (ii) реакцией п-метоксианилина (X) с 4-хлор-3-трифторметиланилином (II) или 4-хлор-3-трифторметилфенилизоцианатом (VIII) с образованием соединения XI

Затем в кислых или щелочных условиях соединение XI деметилируют с образованием соединения III. Также предложены варианты способа, промежуточное соединение, способ получения 4-хлор-пиридил-2-(N-(метил-d3))карбоксамида, применение промежуточного соединения для получения дейтерированной ω-дифенилмочевины и п-толуолсульфонат 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)-N-(метил-d3)пиколинамида. Изобретение позволяет получить соединения с ингибирующей активностью в отношении raf-киназы, которые могут быть применены для лечения или предупреждения развития опухолей. 7 н. и 6 з.п. ф-лы, 3 ил., 1 табл., 9 пр.
1. Способ получения N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины или ее фармацевтически приемлемых солей
,
включающий:
(a) реакцию соединения III с соединением V в инертном растворителе и в присутствии основания с образованием указанного соединения
,
где X представляет собой Cl, Br или I;
и где соединение III получают следующим образом:
(ii) реакцией п-метоксианилина (X) с 4-хлор-3-трифторметиланилином (II) или 4-хлор-3-трифторметилфенилизоцианатом (VIII) с образованием соединения XI
затем в кислых или щелочных условиях соединение XI деметилируют с образованием соединения III.
2. Способ получения N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины или ее фармацевтически приемлемых солей
 ,
,
включающий:
(b) реакцию соединения IX с CD3NH2 или CD3NH2·HCl в инертном растворителе с образованием указанного соединения;
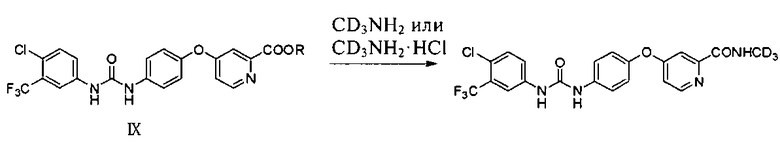 ,
,
где R представляет собой линейный или разветвленный С1-С8 алкил или арил.
3. Способ получения N-(4-хлор-3-(трифторметил)фенил)-N′-(4-(2-(N-(метил-d3)аминоформил)-4-пиридилокси)фенил)мочевины или ее фармацевтически приемлемых солей
 ,
,
включающий:
(c) реакцию 4-хлор-3-трифторметилфенилизоцианата (VIII) с соединением 5 в инертном растворителе с образованием указанного соединения, где инертный растворитель представляет собой смешанный растворитель диметилсульфоксида и дихлорметана
 .
.
4. Способ по п.2, где соединение IX получают следующим образом:
взаимодействием соединения VII с соединением II или соединением VIII с образованием соединения IX
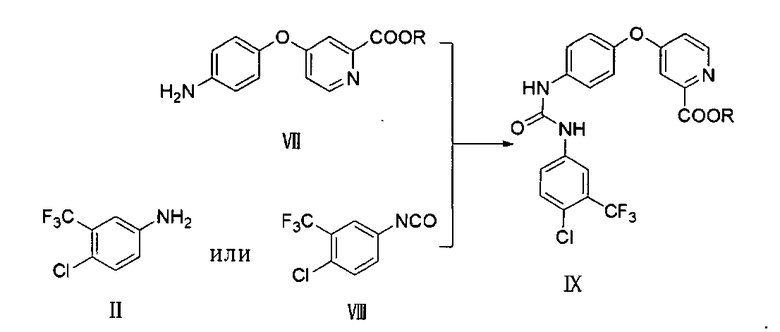
5. Способ по п.4, где соединение VII получают следующим образом:
взаимодействием соединения VI и п-гидроксианилина в присутствии основания с образованием соединения VII:

где X представляет собой хлор, бром или йод; R представляет собой линейный или разветвленный C1-C8 алкил или арил.
6. Способ по п.1, где указанное основание выбрано из трет-бутилата калия, гидрида натрия, гидрида калия, карбоната калия, карбоната цезия, фосфата калия, гидроксида калия, гидроксида натрия или их комбинации.
7. Способ по любому из пп.1-3, где фармацевтически приемлемые соли включают неорганические и органические соли.
8. Способ по п.7, где фармацевтически приемлемая соль представляет собой п-толуолсульфонат.
9. Промежуточное соединение формулы B,
 ,
,
где Y представляет собой галоген или .
.
10. Способ получения 4-хлор-пиридил-2-(N-(метил-d3))карбоксамида, включающий:
(a1) реакцию метил-4-хлор-2-пиридилформата с (метил-d3)амином или его солями в щелочных условиях и в инертном растворителе с образованием 4-хлор-пиридил-2-(N-(метил-d3))карбоксамида.
11. Применение промежуточного соединения по п.9 для получения дейтерированной ω-дифенилмочевины.
12. Применение по п.11, где дейтерированная ω-дифенилмочевина представляет собой п-толуолсульфонат 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)-N-(метил-d3)пиколинамида.
13. Соединение п-толуолсульфоната дейтерированной ω-дифенилмочевины, представляющее собой п-толуолсульфонат 4-(4-(3-(4-хлор-3-(трифторметил)фенил)уреидо)фенокси)-N-(метил-d3)пиколинамида (CM4307·TsOH).
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| 2,3-ДИГИДРОИЗОИНДОЛ-1-ОНЫ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ МАО-В | 2003 |
|
RU2322437C2 |
| PLEISS U | |||
| et al, Journal of Labelled Compounds and Radiopharmaceuticals, 2006, 49 (7), p | |||
| МЕТАЛЛИЧЕСКАЯ ШАРНИРНАЯ СЕТКА | 1922 |
|
SU603A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |

