Область техники
Настоящее изобретение касается фтор-содержащего электронного газа и, в частности, способа синтеза гексафтор-1,3-бутадиена и его промежуточного соединения путем теломеризации.
Предшествующий уровень техники
Гексафтор-1,3-бутадиен, кратко именуемый также HFBD, имеет температуру кипения 5,5°C, температуру замерзания около -130°C и плотность жидкости при 15°C 1,44 кг/л. Гексафтор-1,3-бутадиен представляет собой новое поколение экологичного газа для сухого травления с прекрасными рабочими характеристиками и безопасного для окружающей среды, который имеет время жизни в атмосфере (ALT) всего 1,9 дня и значение GWP100 равное 290. Гексафтор-1,3-бутадиен демонстрирует прекрасные свойства в сухом травлении благодаря своему относительно низкому соотношению фтор/углерод, он применяется для травления при производстве Cu-содержащих плат выпрямителей с низкой диэлектрической проницаемостью K и используется главным образом для прецизионного травления критических размеров (точность до 100 нм), обладает лучшей селективностью и аспектным соотношением, чем другие газы для травления. Как один из необходимых материалов для производства высокотехнологичных чипов, гексафтор-1,3-бутадиен представляет собой ключевой газ для травления в новом поколении технологий блоков флэш-памяти 3D NAND. Учитывая растущую потребность в высокотехнологичных чипах, рынок гексафтор-1,3-бутадиена будет расти с каждым днем.
В предшествующий уровень техники содержит главным образом указанные ниже сведения, касающиеся синтеза гексафтор-1,3-бутадиена:
1) Процессы с использованием хлора и иода в качестве исходных веществ
В патенте CN106336342A, выданном Zhejiang Britech Co., Ltd., описан способ синтеза гексафтор-1,3-бутадиена с использованием хлора и иода в качестве исходных веществ, который включает следующие стадии: (1) реакция хлора с иодом с получением хлорида иода; (2) реакция хлортрифторэтилена с хлоридом иода с получением 1,2-дихлор-1,1,2-трифтор-2-иодэтана (CF2Cl-CFICl); (3) введение 1,2-дихлор-1,1,2-трифтор-2-иодэтана в реакцию внутримолекулярного сочетания в присутствии катализатора с получением 1,2,3,4-тетрахлор-гексафторбутана (CF2Cl-CFCl-CFCl-CF2Cl); и (4) введение 1,2,3,4-тетрахлор-гексафторбутана в реакцию внутримолекулярного дегалогенирования с получением гексафтор-1,3-бутадиена, уравнение реакции выглядит следующим образом:
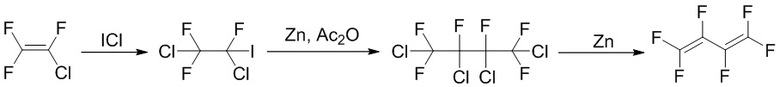
Однако данный способ отличается длинными стадиями и применением дорогого хлорида иода, что повышает стоимость сырьевых материалов.
2) Процессы димеризации
В российском патенте RU0118462 описан способ синтеза гексафтор-1,3-бутадиена с применением хлортрифторэтилена в качестве исходного вещества, который включает: введение хлортрифторэтилена в реакцию димеризации при высокой температуре с получением 34% 1,2-дихлоргексафторциклобутана и 27% 3,4-дихлоргексафтор-1-бутена, разделение этих двух продуктов на высокоэффективной ректификационной колонне, и дехлорирование 3,4-дихлоргексафтор-1-бутена порошком цинка с получением целевого продукта гексафтор-1,3-бутадиена, уравнение реакции выглядит следующим образом:

Однако, реакция полимеризации в этом способе требует высокой температуры и высокого давления и имеет селективность всего 27%.
3) Процессы сочетания с цинк-органикой
В патенте CN105732301A, выданном Sinochem Lantian Co Ltd., описан способ, в котором бромтрифторэтилен, как исходное вещество, реагирует с порошком цинка с получением трифторвинилцинкового реагента, и затем полученный трифторвинилцинковый реагент вводят в реакцию самосочетания под действием солей трехвалентного железа или двухвалентной меди, получая гексафтор-1,3-бутадиен, уравнение реакции выглядит следующим образом:

Однако проблемой является поиск источника бромтрифторэтилена, служащего исходным веществом в этом способе.
4) Процесс фторирования элементарным фтором
В патенте WO2009087067A1, выданном Solvay, описан способ синтеза гексафтор-1,3-бутадиена с использованием в качестве исходных веществ трихлорэтилена (ТХЭ) и фтора, который включает: (1) реакцию димеризации со фтором: реакция ТХЭ с фтором, разбавленным гелием, с получением C4H2F2Cl6 в реакторе AISI 316L, где степень превращения ТХЭ составляет 22,9%, а селективность 50%; (2) реакцию элиминирования: реакция C4H2F2Cl6 с 20%-ным раствором NaOH с получением тетрахлордифторбутадиена (CFCl=CCl-CCl=CFCl) в стеклянном реакторе с выходом 93%; (3) реакцию фторирования: реакция тетрахлордифторбутадиена с фтором, разбавленным гелием, с получением тетрахлоргексафторбутана (CF2Cl-CFCl-CFCl-CF2Cl, CFC-316), где степень превращения составляет 97,8%, а селективность равна 64%; и (4) реакцию дегалогенирования: реакция CFC-316 с порошком цинка в растворе изопропанола с получением гексафтор-1,3-бутадиена с выходом 96% и чистотой целевого продукта 99,5%. Однако у этого способа высокие требования к оборудованию и большие риски в плане безопасности.
Поэтому имеется потребность в разработке нового способа синтеза гексафтор-1,3-бутадиена.
Краткое описание изобретения
Для решения описанных выше технических проблем в настоящем изобретении предлагается способ получения гексафтор-1,3-бутадиена и его промежуточного соединения путем теломеризации. Этот способ представляет собой простой процесс, характеризуется мягкими условиями реакции и меньшим количеством отходов, он безопасен и экологичен, а также подходит для промышленного производства.
Цель настоящего изобретения была достигнута посредством следующего технического решения:
предложен способ получения гексафтор-1,3-бутадиена, который включает следующие стадии:
A1. реакция 1,2-дибром-1-хлор-1,2,2-трифторэтана с трифторгалогенэтиленом в полярном апротонном растворителе под действием инициатора, и очистка реакционного раствора с получением 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана (интермедиат A), где структурная формула трифторгалогенэтилена представляет собой CF2=CFX, где X представляет собой Cl, Br или I, и
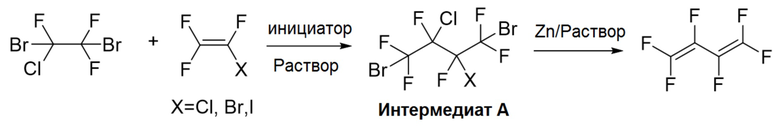
инициатор выбран из по меньшей мере одного из азодиизобутиронитрила (AIBN), ди-трет-бутил пероксида (DTBP), дибензоил пероксида (BPO), дикумил пероксида (DCP), трет-бутил гидропероксида (TBHP), персульфата калия (KPS) и персульфата аммония (APS);
A2. введение 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана и порошка цинка в реакцию дегалогенирования с получением гексафтор-1,3-бутадиена.
В настоящем изобретении, разные способы инициирования (например, фотоинициирование или инициирование с помощью инициатора) оказывают относительно большое влияние на синтез промежуточного соединения (интермедиата) A. Хотя по механизму реакции стадию A1 можно отнести к свободнорадикальным реакциям, эту свободнорадикальную реакцию можно инициировать и фотоинициированием, и инициированием с помощью инициатора. Однако авторы настоящего изобретения обнаружили, что для реакции на стадии A1 эффект при инициировании с помощью инициатора намного лучше, чем при фотоинициировании, и даже разные типы инициаторов дают разный эффект инициирования. То есть, применение инициатора может улучшить селективность образования интермедиата A. Предпочтительно, инициатор выбран из по меньшей мере одного из ди-трет-бутил пероксида, дибензоил пероксида и трет-бутил гидропероксида. Более предпочтительно, инициатор представляет собой дибензоил пероксид или трет-бутил гидропероксид.
Кроме того, мольное соотношение 1,2-дибром-1-хлор-1,2,2-трифторэтан и инициатора составляет 1:0,01-1:0,1. Предпочтительно, мольное соотношение 1,2-дибром-1-хлор-1,2,2-трифторэтана и инициатора составляет 1:0,03-1:0,06.
В способе получения промежуточного соединения на стадии A1 по настоящему изобретению, полярный апротонный растворитель более благоприятен для прохождения реакции. Предпочтительно, полярный апротонный растворитель выбран из по меньшей мере одного из тетрагидрофурана, 1,4-диоксана, ацетонитрила, диметилового эфира диэтиленгликоля, N,N-диметилформамида и N,N-диметилацетамида. Более предпочтительно, полярный апротонный растворитель выбран из по меньшей мере одного из 1,4-диоксана, ацетонитрила и диметилового эфира диэтиленгликоля.
В способе получения промежуточного соединения на стадии A1 по настоящему изобретению, атмосфера инертного газа благоприятна для прохождения реакции. Предпочтительно, инертный газ выбран из по меньшей мере одного из азота, гелия и аргона.
В способе получения промежуточного соединения на стадии A1 по настоящему изобретению, очистку реакционного раствора можно проводить с применением общеизвестного метода очистки, такого как перегонка при атмосферном давлении или перегонка при пониженном давлении.
В способе получения гексафтор-1,3-бутадиена на стадии A2 по настоящему изобретению, реакцию можно проводить без растворителя. Разумеется, эффективность реакции выше в органическом растворителе. Предпочтительно, органический растворитель выбран из по меньшей мере одного из муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, дихлорметана, хлороформа, тетрахлорида углерода, дихлорэтана, 1,1,1-трихлорэтана, изопропанола, трет-бутанола, N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, N-метилпирролидона, гексаметилфосфорамида. Более предпочтительно, органический растворитель выбран из по меньшей мере одного из уксусной кислоты, N,N-диметилформамида или изопропанола.
В способе получения гексафтор-1,3-бутадиена на стадии A2 по настоящему изобретению, присутствие катализатора благоприятно для прохождения реакции. Предпочтительно, катализатор выбран из по меньшей мере одного из хлорида цинка (ZnCl2), бромида цинка (ZnBr2) или иодида цинка (ZnI2), элементарного иода, 1,2-дибромэтана; и мольное соотношение промежуточного соединения A и катализатора составляет 1:0,01-1:0,1. Более предпочтительно, мольное соотношение промежуточного соединения A и катализатора составляет 1:0,03-1:0,06.
В способе получения гексафтор-1,3-бутадиена по настоящему изобретению, в предпочтительном варианте стадию A1 проводят при температуре реакции 60°C-200°C и выдерживают при этой температуре для прохождения реакции в течение 1-12 часов; и стадию A2 проводят при температуре реакции 40°C-150°C и выдерживают при этой температуре для прохождения реакции в течение 1-24 часов.
В более предпочтительном варианте, стадию A1 проводят при температуре реакции 80°C-160°C и выдерживают при этой температуре для прохождения реакции в течение 6-12 часов; и стадию A2 проводят при температуре реакции 60°C-90°C и выдерживают при этой температуре для прохождения реакции в течение 3-6 часов.
В настоящем изобретении описан также способ получения 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана, который включает:
реакцию 1,2-дибром-1-хлор-1,2,2-трифторэтана с трифторгалогенэтиленом в полярном апротонном растворителе под действием инициатора в атмосфере инертного газа, и очистку реакционного раствора с получением 1,4-дибром-2-хлор-3-галоген- 1,1,2,3,4,4-гексафторбутана, где структурная формула трифторгалогенэтилена представляет собой CF2=CFX, где X представляет собой Cl, Br или I;
инициатор выбран из по меньшей мере одного из азодиизобутиронитрила (AIBN), ди-трет-бутил пероксида (DTBP), дибензоил пероксида (BPO), дикумил пероксида (DCP), трет-бутил гидропероксида (TBHP), персульфата калия (KPS) и персульфата аммония (APS).
По сравнению с предшествующим уровнем техники, настоящее изобретение обеспечивает следующие преимущества:
1. В настоящем изобретении промежуточное соединение гексафтор-1,3-бутадиен получают реакцией теломеризации между 1,2-дибром-1-хлор-1,2,2-трифторэтаном и трифторгалогенэтиленом, которая является простым процессом, проходит в мягких условиях и подходит для промышленного производства.
2. В настоящем изобретении реакцию теломеризации инициируют свободнорадикальным инициатором, который не только контролирует скорость генерирования свободных радикалов и степень теломеризации, но также значительно улучшает селективность образования промежуточного соединения по сравнению с фотоинициированием.
Подробное описание вариантов осуществления
Настоящее изобретение далее объясняется с помощью частных примеров, но изобретение не ограничивается только описанными частными примерами. Квалифицированному специалисту в данной области будет понятно, что настоящее изобретение включает все альтернативы, улучшения и эквиваленты, которые входят в объем притязаний, заявленный в формуле изобретения.
Пример 1
В этом примере описан способ получения гексафтор-1,3-бутадиена, который включает стадию получения промежуточного соединения и стадию получения гексафтор-1,3-бутадиена, в частности следующим образом:
A1. получение 1,4-дибром-2,3-дихлор-1,1,2,3,4,4-гексафторбутана
В 500-миллилитровый автоклав из сплава Hastelloy помещали 150 г ацетонитрила, 69,1 г (0.25 моль) 1,2-дибром-1-хлор-1,2,2-трифторэтана и 1,8 г (7,5 ммоль) дибензоил пероксида, автоклав продували высокочистым N2 в течение 10 минут и затем добавляли 34,8 г (0.30 моль) хлортрифторэтилена из стального цилиндра, температуру повышали до 80°C при механическом перемешивании (300-500 об/мин), давление в автоклаве повышали примерно до 0,5 МПа, и поддерживали эту температуру в течение 12 часов для полного прохождения реакции.
Реакционный раствор анализировали методом ГХ-МС, и из вычислений было видно, что: степень превращения исходного вещества 1,2-дибром-1-хлор-1,2,2-трифторэтана составляла 98,75%, и селективность образования промежуточного соединения 1,4-дибром-2,3-дихлор-1,1,2,3,4,4-гексафторбутана была 59,09%. Полученные результаты показаны в таблице 1.
Реакционный раствор перегоняли при пониженном давлении, получая промежуточное соединение A, имеющее высокую чистоту.
A2. Получение гексафтор-1,3-бутадиена
В 500-милиллитровую круглодонную колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и капельной воронкой, помещали 150 г изопропанола, 2,0 г элементарного иода и 130 г порошка цинка (2,0 моль). Верхнюю часть холодильника соединяли с приемником для продукта через газовую трубку, и приемник для продукта помещали в криогенную холодную ловушку (охлаждаемую жидким азотом). Реакционную колбу нагревали до 70°C и по каплям добавляли 275 г 1,4-дибром-2,3-дихлор-1,1,2,3,4,4-гексафторбутана (имеет чистоту 96%, 0,7 моль) при перемешивании на магнитной мешалке, и собирали продукт, при этом прикапывание было закончено в течение примерно 1 часа. После окончания прикапывания температуру повышали до 80°C и выдерживали при той же температуре 3 часа для полного прохождения реакции. Продукт, собранный в охлаждаемый приемник, был определен как гексафтор-1,3-бутадиен по данным ГХ-МС и спектроскопии ядерного магнитного резонанса, было получено 97,0 г продукта с чистотой 96,6% и выходом 86,0%. Полученные результаты приведены в таблице 3.
Пример 2
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, тип инициатора был изменен путем замены дибензоил пероксида, использованного в Примере 1, на 1,1 г (7,5 ммоль) ди-трет-бутил пероксида. Результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, тип катализатора был изменен путем замены элементарного иода, использованного в Примере 1, на 2,5 г иодида цинка, и было получено 89,20 г продукта. Полученные результаты приведены в таблице 3.
Пример 3
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, тип инициатора был изменен путем замены дибензоил пероксида, использованного в Примере 1 на 0,68 г (7,5 ммоль) трет-бутил гидропероксида; и результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, тип катализатора был изменен путем замены элементарного иода, использованного в Примере 1, на 1,5 г 1,2-дибромэтана, и было получено 95,0 г продукта. Полученные результаты приведены в таблице 3.
Пример 4
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, температура реакции была снижена с 80°C до 60°C; и результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, температура реакции была снижена с 70°C до 60°C, и было получено 76,20 г продукта. Полученные результаты приведены в таблице 3.
Пример 5
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, температура реакции была повышена с 80°C до 100°C. Результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, температура реакции была повышена с 70°C до 90°C, и было получено 101,3 г продукта. Полученные результаты приведены в таблице 3.
Пример 6
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, тип растворителя был изменен путем замены ацетонитрила, использованного в Примере 1, на 150 г диметилового эфира диэтиленгликоля. Результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, тип растворителя был изменен путем замены изопропанола, использованного в Примере 1, на 150 г N,N-диметилацетамида, и было получено 80,50 г продукта. Полученные результаты приведены в таблице 3.
Пример 7
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения, соотношение реагентов было изменено путем изменения количества хлортрифторэтилена с исходных 34,8 г (0,30 моль) на 58,0 г (0,50 моль). Результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена, соотношение реагентов было изменено путем изменения количества порошка цинка с исходных 130 г (2,0 моль) на 91,0 г (1,4 моль). Было получено 68,4 г продукта. Полученные результаты приведены в таблице 3.
Пример 8
В этом примере описан способ получения гексафтор-1,3-бутадиена, который включает стадию получения промежуточного соединения и стадию получения гексафтор-1,3-бутадиена, в частности следующим образом:
A1. Получение 1,2,4-трибром-3-хлор-1,1,2,3,4,4-гексафторбутана
В 500-миллилитровый автоклав из сплава Hastelloy помещали 150 г ацетонитрила, 69,1 г (0,25 моль) 1,2-дибром-1-хлор-1,2,2-трифторэтана и 1,1 г (7,5 ммоль) ди-трет-бутил пероксида, автоклав продували высокочистым N2 в течение 10 минут и затем добавляли 48,3 г (0.30 моль) бромтрифторэтилена из стального цилиндра, температуру повышали до 100°C при механическом перемешивании (300-500 об/мин), давление в автоклаве повышали примерно до 0,6 МПа, и поддерживали эту температуру в течение 12 часов для полного прохождения реакции.
Реакционный раствор анализировали методом ГХ-МС, и из вычислений было видно, что: степень превращения исходного вещества 1,2-дибром-1-хлор-1,2,2-трифторэтана составляла 97,6%, и селективность образования промежуточного соединения 1,2,4-трибром-3-хлор-1,1,2,3,4,4-гексафторбутана составляла 73,9%. Полученные результаты показаны в таблице 2.
Реакционный раствор перегоняли при пониженном давлении, получая промежуточное соединение A, имеющее высокую чистоту.
A2. Получение гексафтор-1,3-бутадиена
В 500-милиллитровую круглодонную колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и капельной воронкой, помещали 150 г уксусной кислоты, 2,0 г элементарного иода и 130 г порошка цинка (2,0 моль). Верхнюю часть холодильника соединяли с приемником для продукта через газовую трубку, и приемник для продукта помещали в криогенную холодную ловушку (охлаждаемую жидким азотом). Реакционную колбу нагревали до 60°C и по каплям добавляли 312,3 г 1,2,4-трибром-3-хлор-1,1,2,3,4,4-гексафторбутана (имеет чистоту 98%, 0,7 моль) при перемешивании на магнитной мешалке, и собирали продукт, при этом прикапывание было закончено в течение примерно 2 часов. После окончания прикапывания температуру повышали до 80°C и выдерживали при той же температуре 3 часа для полного прохождения реакции. Продукт, собранный в охлаждаемый приемник, был определен как гексафтор-1,3-бутадиен по данным ГХ-МС и спектроскопии ядерного магнитного резонанса, было получено 78,20 г продукта с чистотой 97,5% и выходом 69,9%. Полученные результаты приведены в таблице 3.
Пример 9
Методика в данном Примере была такой же, как в Примере 8, различие состояло только в следующем:
В способе получения промежуточного соединения, тип растворителя был изменен путем замены ацетонитрила, использованного в Примере 8, на 150 г диметилового эфира диэтиленгликоля. Результаты данной реакции представлены в таблице 2.
В способе получения гексафтор-1,3-бутадиена, тип растворителя был изменен путем замены уксусной кислоты, использованной в Примере 8, на 150 г N,N-диметилацетамида, и было получено 82,50 г продукта. Полученные результаты приведены в таблице 3.
Пример 10
Методика в данном Примере была такой же, как в Примере 8, различие состояло только в следующем:
В способе получения промежуточного соединения температура реакции была повышена с 100°C до 160°C. Результаты данной реакции представлены в таблице 2.
В способе получения гексафтор-1,3-бутадиена, тип катализатора был изменен путем замены элементарного иода, использованного в Примере 8, на 2,5 г иодида цинка, и было получено 72,2 г продукта. Полученные результаты приведены в таблице 3.
Пример 11
Методика в данном Примере была такой же, как в Примере 8, различие состояло только в следующем:
В способе получения промежуточного соединения, соотношение реагентов было изменено путем изменения количества бромтрифторэтилена с исходных 48,3 г (0,30 моль) на 40,3 г (0,25 моль). Результаты данной реакции представлены в таблице 2.
В способе получения гексафтор-1,3-бутадиена, количество элементарного иода, применяющегося в качестве катализатора, было повышено с исходных 2,0 г до 4,0 г, и было получено 74,5 г продукта. Полученные результаты приведены в таблице 3.
Сравнительный пример 1
Методика в данном Примере была такой же, как в Примере 1, различие состояло только в следующем:
В способе получения промежуточного соединения не добавляли инициатор дибензоил пероксид. Результаты данной реакции представлены в таблице 1.
В способе получения гексафтор-1,3-бутадиена не добавляли элементарный иод, и было получено 78,50 г продукта. Полученные результаты приведены в таблице 3.
Сравнительный пример 2
В стеклянный реактор для фотореакций, оснащенный ловушкой с охлаждаемой рубашкой, помещали 276,3 г (1,0 моль) 1,2-дибром-1-хлор-1,2,2-трифторэтана, и в реактор вводили 400 Вт УФ-ртутную лампу высокого давления. Запускали низкотемпературный цикл (0°C), включали ртутную лампу высокого давления, медленно добавляли в общей сложности 139,2 г (1,2 моль) хлортрифторэтилена (50 мл/мин), и добавление заканчивали примерно за 10 часов. После окончания добавления отключали ртутную лампу высокого давления и низкотемпературное охлаждение. Реакционный раствор анализировали методом ГХ-МС, и из вычислений было видно, что: степень превращения исходного вещества 1,2-дибром-1-хлор-1,2,2-трифторэтана составляла 54,5%, а селективность образования промежуточного соединения 1,4-дибром-2,3-дихлор-1,1,2,3,4,4- гексафторбутана была 38,2%. Полученные результаты приведены в таблице 1.
Таблица 1. Результаты реакции теломеризации при использовании хлортрифторэтилена, как исходного вещества
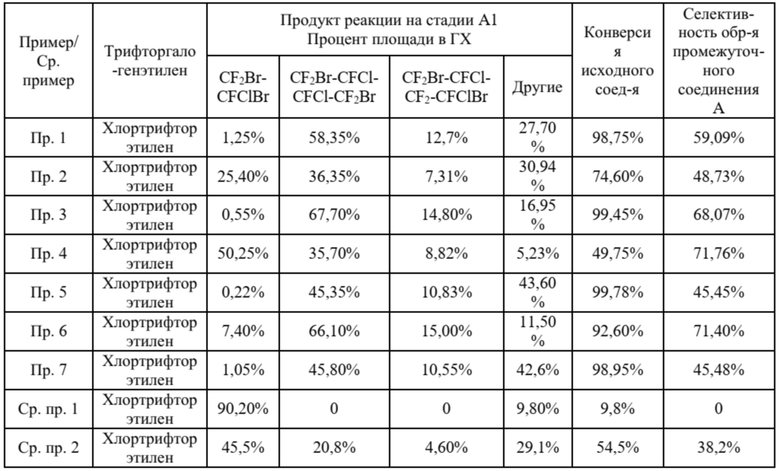
Примечания: a. пик растворителя и хлортрифторэтилена не учитывали в расчетах;
b. хроматографическое содержание и фактическое содержание каждого вещества не корректировали.
Таблица 2 Результаты реакции теломеризации при использовании бромтрифторэтилена, как исходного вещества

Примечания: a. пик растворителя и бромтрифторэтилена не учитывали в расчетах;
b. хроматографическое содержание и фактическое содержание каждого вещества не корректировали.
Таблица 3. Таблица результатов в реакции дегалогенирования
Сравнительный пример
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ГАЛОИДИРОВАНИЯ | 2005 |
|
RU2422466C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗООЛЕФИНОВЫХ ПОЛИМЕРОВ С ИСПОЛЬЗОВАНИЕМ ТРЕТИЧНОГО ПРОСТОГО ЭФИРА | 2019 |
|
RU2808455C2 |
| РАЗБАВИТЕЛЬ ДЛЯ ПОЛУЧЕНИЯ БУТИЛКАУЧУКА | 2015 |
|
RU2674473C2 |
| 2, 3, 3, 3-ТЕТРАФТОР-1-ПРОПЕН В КАЧЕСТВЕ РАЗБАВИТЕЛЯ ДЛЯ ПОЛУЧЕНИЯ НОВЫХ БУТИЛКАУЧУКОВ | 2015 |
|
RU2699793C2 |
| СОПОЛИМЕРЫ С НОВЫМИ РАСПРЕДЕЛЕНИЯМИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ | 2003 |
|
RU2345095C2 |
| СОПОЛИМЕРЫ С НОВЫМИ РАСПРЕДЕЛЕНИЯМИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ | 2003 |
|
RU2349607C2 |
| ПОЛИМЕРЫ, ПО СУЩЕСТВУ СВОБОДНЫЕ ОТ ДЛИННОЦЕПОЧЕЧНОГО РАЗВЕТВЛЕНИЯ, ПЕРЕКРЕСТНЫЕ | 2003 |
|
RU2344145C2 |
| СПОСОБ РАЗДЕЛЕНИЯ ФАЗ С ИСПОЛЬЗОВАНИЕМ ФТОРУГЛЕВОДОРОДА | 2007 |
|
RU2435791C2 |
| СПОСОБ ПРОИЗВОДСТВА ПОЛИМЕРОВ ИЗОБУТЕНА С УЛУЧШЕННЫМ РЕГУЛИРОВАНИЕМ ТЕМПЕРАТУРЫ | 2018 |
|
RU2764774C2 |
| СПОСОБ И СИСТЕМА ПОЛУЧЕНИЯ ГЕКСАФТОР-1,3-БУТАДИЕНА | 2023 |
|
RU2817152C1 |
Изобретение относится к способу получения гексафтор-1,3-бутадиена, включающему следующие стадии: A1) реакция 1,2-дибром-1-хлор-1,2,2-трифторэтана с трифторгалогенэтиленом в полярном апротонном растворителе под действием инициатора и очистка реакционного раствора с получением 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана, где структурная формула трифторгалогенэтилена представляет собой CF2=CFX, где X представляет собой Cl, Br или I, и инициатор выбран из по меньшей мере одного из ди-трет-бутил пероксида, дибензоил пероксида, дикумил пероксида и трет-бутил гидропероксида; и A2) введение 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана и порошка цинка в реакцию дегалогенирования с получением гексафтор-1,3-бутадиена. Также изобретение относится к способу получения 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4- гексафторбутана. Изобретение характеризуется тем, что позволяет получать гексафтор-1,3-бутадиен посредством реакции теломеризации между 1,2-дибром-1-хлор-1,2,2-трифторэтаном и трифторгалоэтиленом, которая является простой, осуществляется в мягких условиях и пригодна для промышленного производства. 2 н. и 8 з.п. ф-лы, 3 табл., 11 пр.
1. Способ получения гексафтор-1,3-бутадиена, включающий следующие стадии:
A1) реакция 1,2-дибром-1-хлор-1,2,2-трифторэтана с трифторгалогенэтиленом в полярном апротонном растворителе под действием инициатора и очистка реакционного раствора с получением 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана, где структурная формула трифторгалогенэтилена представляет собой CF2=CFX, где X представляет собой Cl, Br или I, и
инициатор выбран из по меньшей мере одного из ди-трет-бутил пероксида, дибензоил пероксида, дикумил пероксида и трет-бутил гидропероксида; и
A2) введение 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана и порошка цинка в реакцию дегалогенирования с получением гексафтор-1,3-бутадиена.
2. Способ получения гексафтор-1,3-бутадиена по п. 1, где мольное соотношение 1,2-дибром-1-хлор-1,2,2-трифторэтана и инициатора составляет 1:0,01-1:0,1.
3. Способ получения гексафтор-1,3-бутадиена по п. 1, где реакцию на стадии A1 проводят в атмосфере инертного газа, выбранного из по меньшей мере одного из азота, гелия и аргона.
4. Способ получения гексафтор-1,3-бутадиена по п. 3, где полярный апротонный растворитель выбран из по меньшей мере одного из тетрагидрофурана, 1,4-диоксана, ацетонитрила, диметилового эфира диэтиленгликоля, N,N-диметилформамида и N,N-диметилацетамида.
5. Способ получения гексафтор-1,3-бутадиена по п. 1, где реакцию на стадии A2 проводят в присутствии катализатора, выбранного из по меньшей мере одного из бромида цинка, иодида цинка или элементарного иода.
6. Способ получения гексафтор-1,3-бутадиена по п. 5, где мольное соотношение 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана и катализатора составляет 1:0,01-1:0,1.
7. Способ получения гексафтор-1,3-бутадиена по п. 1, где реакцию на стадии A2 проводят в органическом растворителе, выбранном из по меньшей мере одного из муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, дихлорметана, хлороформа, тетрахлорида углерода, дихлорэтана, 1,1,1-трихлорэтана, изопропанола, трет-бутанола, N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, N-метилпирролидона или гексаметилфосфорамида.
8. Способ получения гексафтор-1,3-бутадиена по п. 1, где стадию A1 проводят при температуре реакции 60-200°C и выдерживают при этой температуре для прохождения реакции в течение 1-12 часов; и стадию A2 проводят при температуре реакции 40-150°C и выдерживают при этой температуре для прохождения реакции в течение 1-24 часов.
9. Способ получения гексафтор-1,3-бутадиена по п. 8, где стадию A1 проводят при температуре реакции 80-160°C и выдерживают при этой температуре для прохождения реакции в течение 6-12 часов; и стадию A2 проводят при температуре реакции 60-90°C и выдерживают при этой температуре для прохождения реакции в течение 3-6 часов.
10. Способ получения 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана, включающий:
реакцию 1,2-дибром-1-хлор-1,2,2-трифторэтана с трифторгалогенэтиленом в полярном апротонном растворителе под действием инициатора в атмосфере инертного газа и очистку реакционного раствора с получением 1,4-дибром-2-хлор-3-галоген-1,1,2,3,4,4-гексафторбутана, где структурная формула трифторгалогенэтилена представляет собой CF2=CFX, где X представляет собой Cl, Br или I;
инициатор выбран из по меньшей мере одного из ди-трет-бутил пероксида, дибензоил пероксида, дикумил пероксида и трет-бутил гидропероксида.
| JOURNAL OF FLUORINE CHEMISTRY, VOL | |||
| Способ очистки нефти и нефтяных продуктов и уничтожения их флюоресценции | 1921 |
|
SU31A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ получения бензонафтола | 1920 |
|
SU363A1 |
| JACS, VOL | |||
| Спускная труба при плотине | 0 |
|
SU77A1 |
| Насос | 1917 |
|
SU13A1 |
| Трапецеидальная борона | 1925 |
|
SU3640A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАФТОРБУТ-1,3-ДИЕНА | 2005 |
|
RU2301220C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1,3,3-ПЕНТАХЛОРБУТАНА | 1998 |
|
RU2199517C2 |

