Область техники, к которой относится изобретение
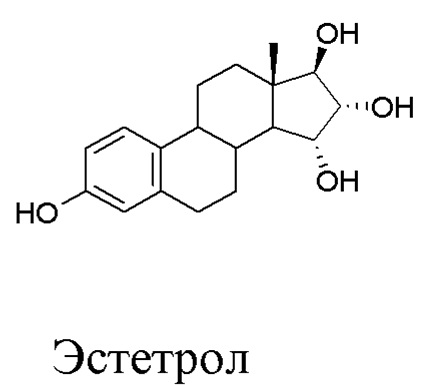
Настоящее изобретение касается области синтеза действующих веществ для фармацевтического применения, и в частности - способа получения в промышленном масштабе (15α,16α,17β)-эстра-1,3,5(10)-триен-3,15,16,17-тетрола, известного также как Эстетрол, как в безводной форме, так и в форме моногидрата. Настоящее изобретение касается также интермедиатов в данном способе.
Предшествующий уровень техники
Эстетрол представляет собой действующее вещество, обладающее фармакологической активностью, которая делает его подходящим для гормонозаместительной терапии (ГЗТ), женской контрацепции или для терапии аутоиммунных нарушений, связанных с нарушением баланса гормонов.
Структурная формула Эстетрола изображена ниже:
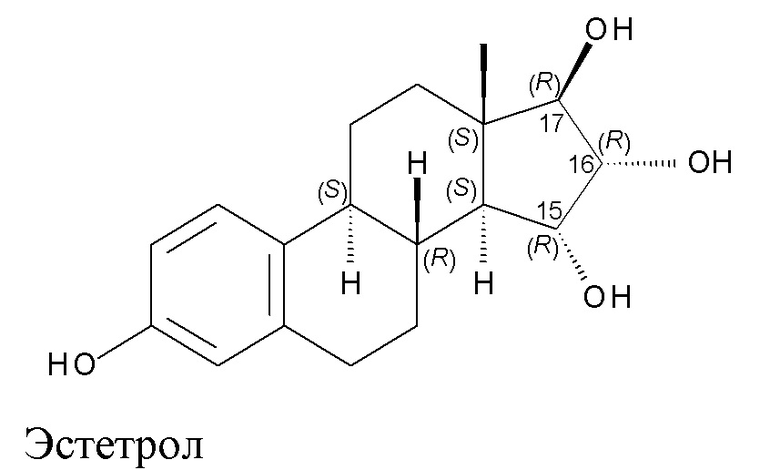
В положениях 15, 16 и 17 в стероидном скелете (обозначены в изображенной выше формуле) располагается по одному гидроксилу, которые, как показано на структурной формуле, имеют определенное пространственное расположение.
Эстетрол представляет собой природное вещество, выделенное из мочи человека, и оно известно много лет; оно было описано в статье “Synthesis of epimeric 15-hydroxyestriols, new and potential metabolites of estradiol”, J. Fishman et al., JOC Vol. 33, No. 8, August 1968, p. 3133-3135 (соединение Ia на рисунке на стр. 3133).
Что касается получения Эстетрола, то описанный в указанной статье способ не подходит для промышленного применения из-за низкого выхода.
Недавно было опубликовано несколько патентов, касающихся новых способов синтеза Эстетрола, но ни один из них не позволяет избежать образования изомера 15β,16β,17β, имеющего изображенную ниже формулу, от которого Эстетрол необходимо очистить перед применением в фармацевтических препаратах.
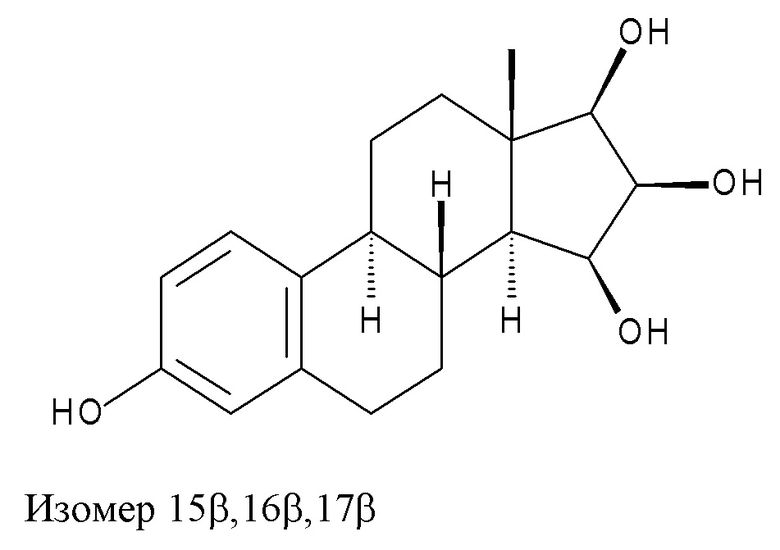
Например, в заявке WO 2004/041839 A2 (стр. 6, строки 5-10) описан способ получения Эстетрола, чистота которого достигает 99%, а сумма отдельных примесей не превышает 1%. В примере 11 на стр. 28 описан Эстетрол, имеющий ВЭЖХ-чистоту 99,1% (ВЭЖХ-МС), что, однако, не дает информации о содержании индивидуальных примесей; допустимый предел в международных руководствах по фармацевтическим субстанциям составляет 0,1% для неизвестных примесей и 0,15% для идентифицированных примесей.
Определение содержания примесей в действующем веществе (ДВ) является существенным и неотъемлемым требованием для разрешения использования в фармацевтических препаратах, а также это фундаментальная характеристика для определения промышленно применимого способа. Любой способ, вне зависимости от его выхода, дающий ДВ с содержанием примесей, не укладывающимся в указанные принятые на международном уровне пределы, не является промышленно применимым способом, поскольку ДВ, получаемое таким способом, нельзя использовать.
Другими заявками, касающимися получения Эстетрола, являются, например, WO 2012/164096 A1, WO 2013/050553 A1 и WO 2015/040051 A1.
В WO 2015/040051 A1 соотношение Эстетрол/изомер 15β,16β,17β составляет 99:1 в примерах 10 и 15, и оно составляет 98:2 в примерах 11 и 17. В этих примерах, однако, не дано никаких указаний на снижение содержания изомера 15β,16β,17β до уровня по меньшей мере 0,15%. Даже хроматографическая очистка (пример 15) не позволяет достичь такого результата. В этом документе отмечается (стр. 9, строки 5-15), что описанные в предшествующем уровне техники способы (представлены в случае данного документа заявками WO 2012/164096 A1 и WO 2013/050553 A1) дают еще большие и неприемлемые количества изомера 15β,16β,17β.
Таким образом, ни в одном из описанных способов нет решения задачи ограничения образования изомера 15β,16β,17β или способа очистки Эстетрола от этого изомера.
Краткое описание изобретения
Задачей настоящего изобретения является разработка способа синтеза Эстетрола с содержанием изомера 15β,16β,17β ниже 15%, без привлечения методик очистки, неприменимых в промышленном масштабе.
В первом аспекте, настоящее изобретение касается способа синтеза Эстетрола, включающего следующие стадии:
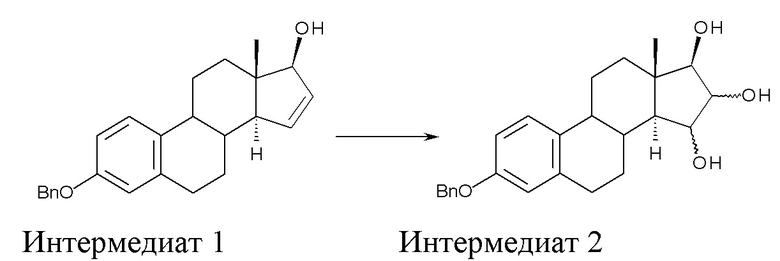
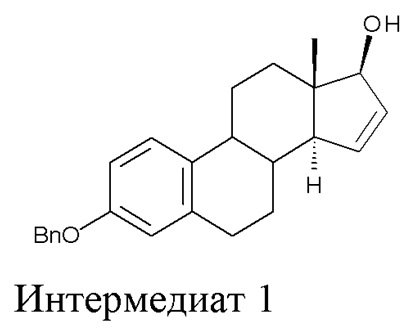
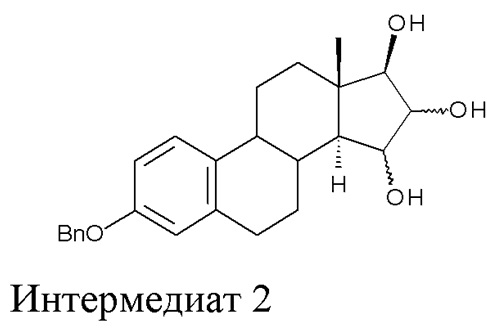
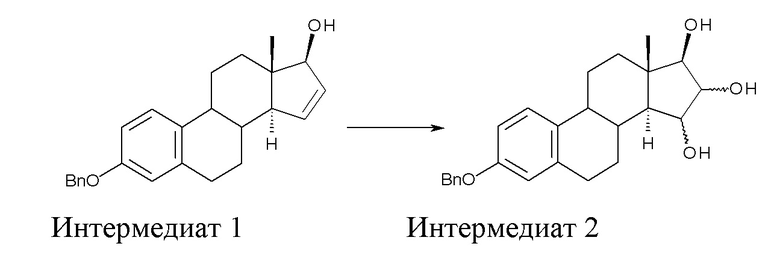
A) окисление (17β)-3-(фенилметокси)-эстра-1,3,5(10),15-тетраен-17-ола (интермедиат 1) с получением (17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола (интермедиат 2):
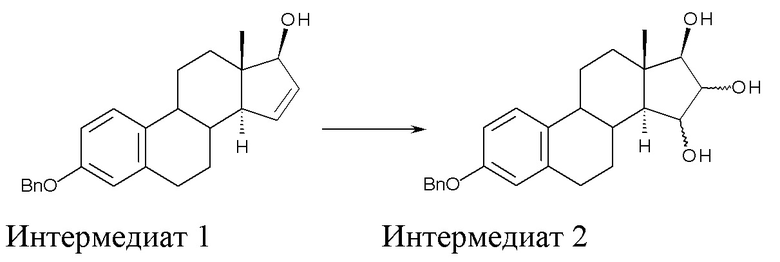
где Bn = бензил, и где конфигурация атомов углерода в положениях 15 и 16 стероидного скелета в интермедиате 2 не фиксирована;
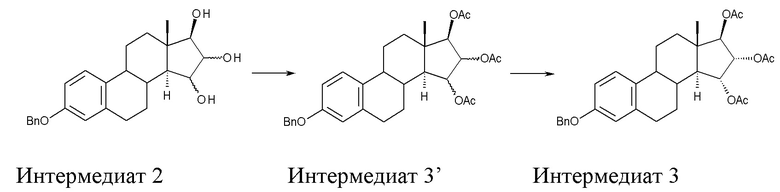
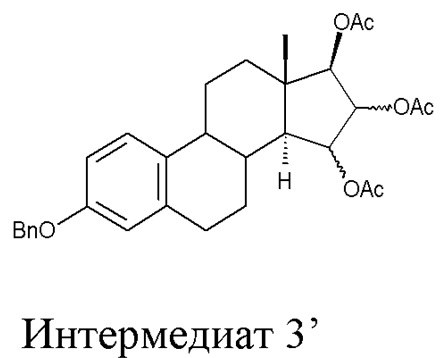
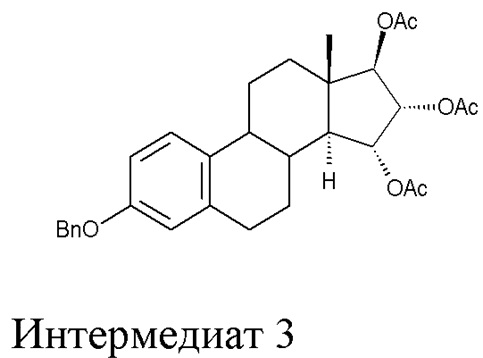
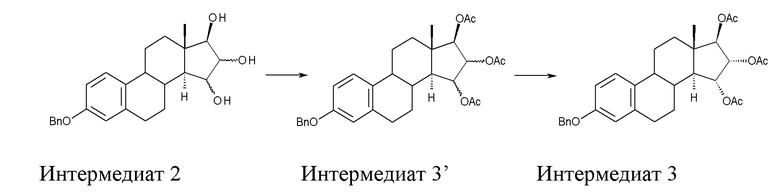
B) ацетилирование интермедиата 2 с получением (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола триацетата (интермедиат 3), проходящее через интермедиат 3', в котором конфигурация атомов углерода 15 и 16 стероидного скелета не фиксирована:
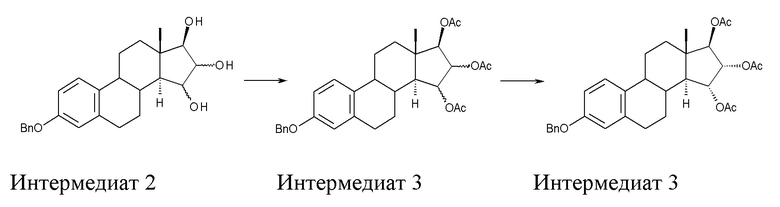
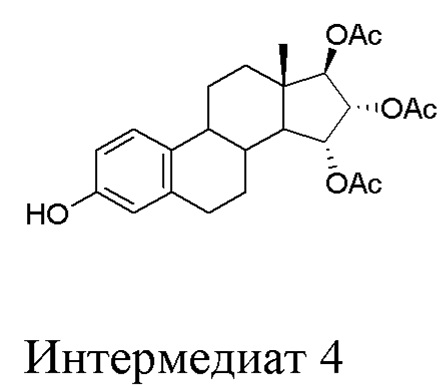
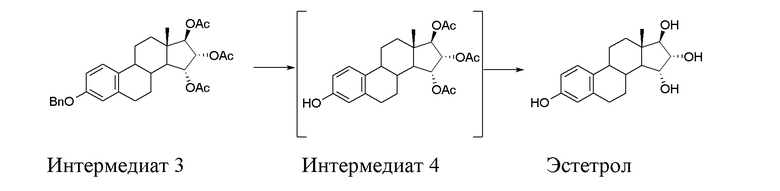
C) превращение интермедиата 3 через (15α,16α,17β)-3-гидрокси-эстра-1,3,5(10)-триен-15,16,17-триол триацетат (интермедиат 4), который предпочтительно не выделяют, в Эстетрол:
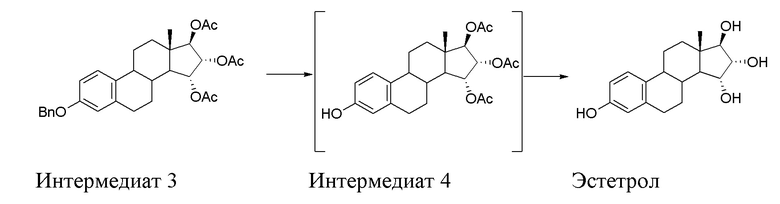
D) очистка Эстетрола, полученного на стадии C).
В альтернативном варианте осуществления, способ по настоящему изобретению дополнительно включает дополнительную стадию E), в которой Эстетрол, полученный на стадии D), превращают в Эстетрол моногидрат.
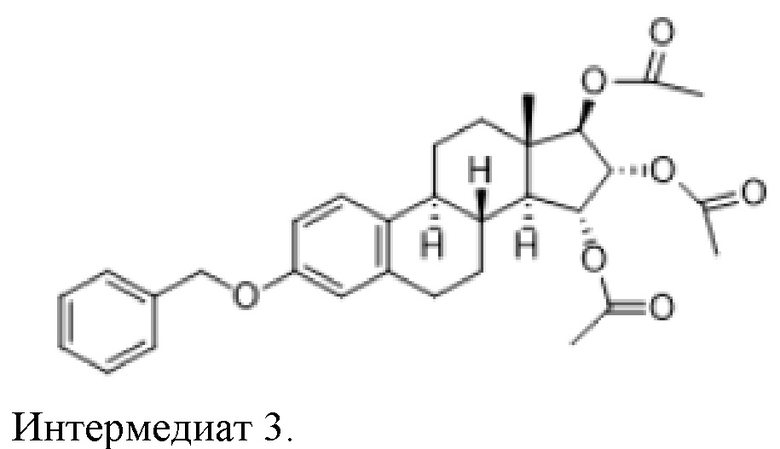
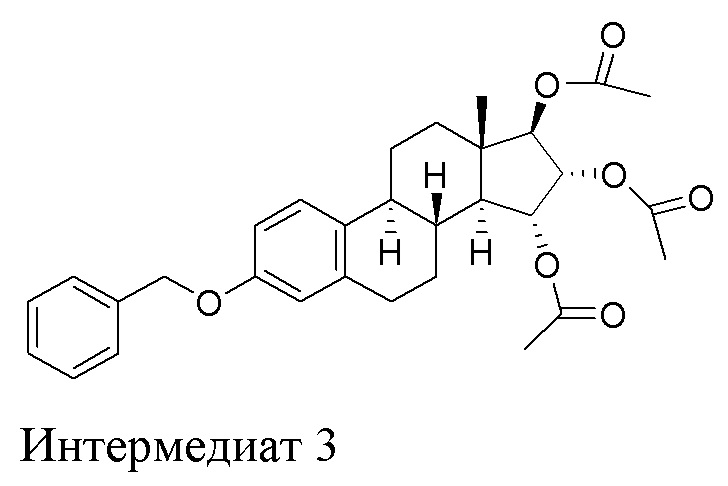
Во втором аспекте, настоящее изобретение касается интермедиата 3, (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триол триацетата:
Краткое описание чертежей
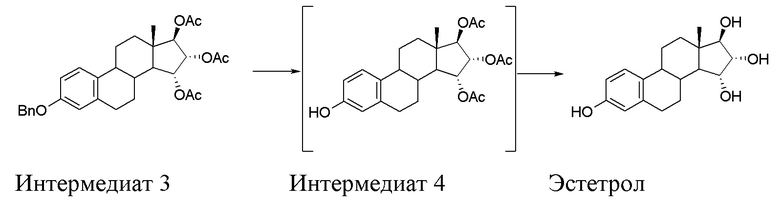
На фиг. 1 изображена ВЭЖХ-хроматограмма Эстетрола, получаемого способом по настоящему изобретению.
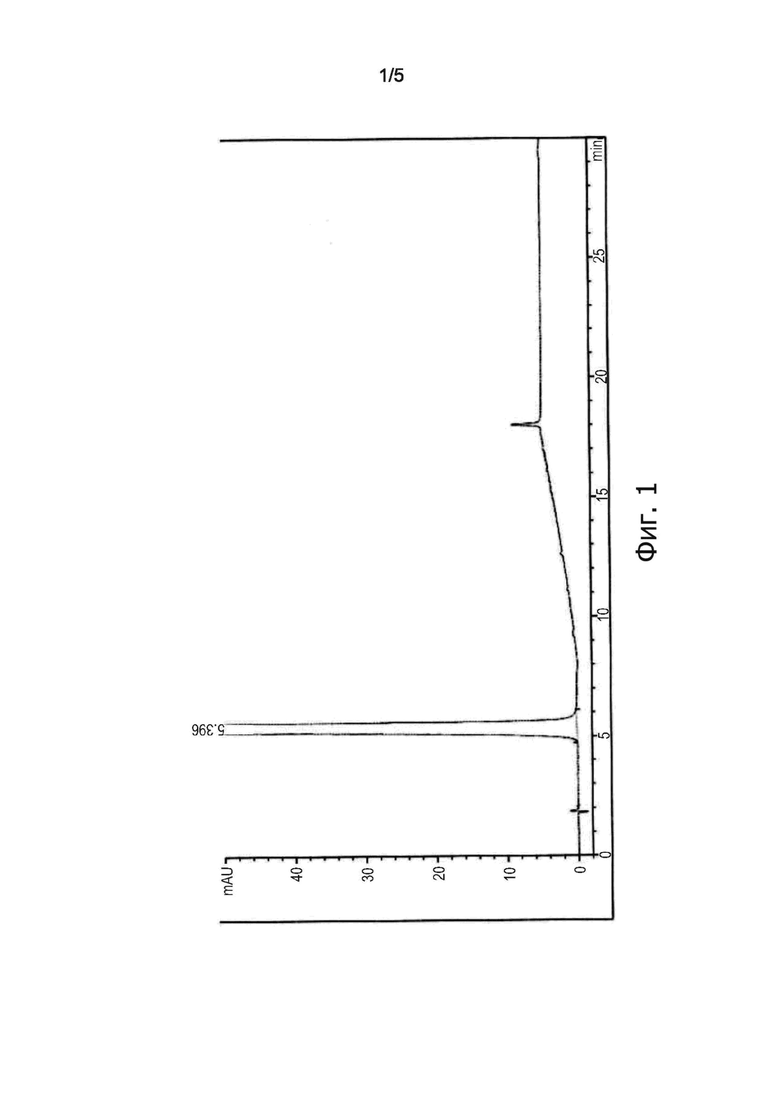
На фиг. 2 изображена ВЭЖХ-хроматограмма Эстетрола моногидрата, получаемого способом по настоящему изобретению.
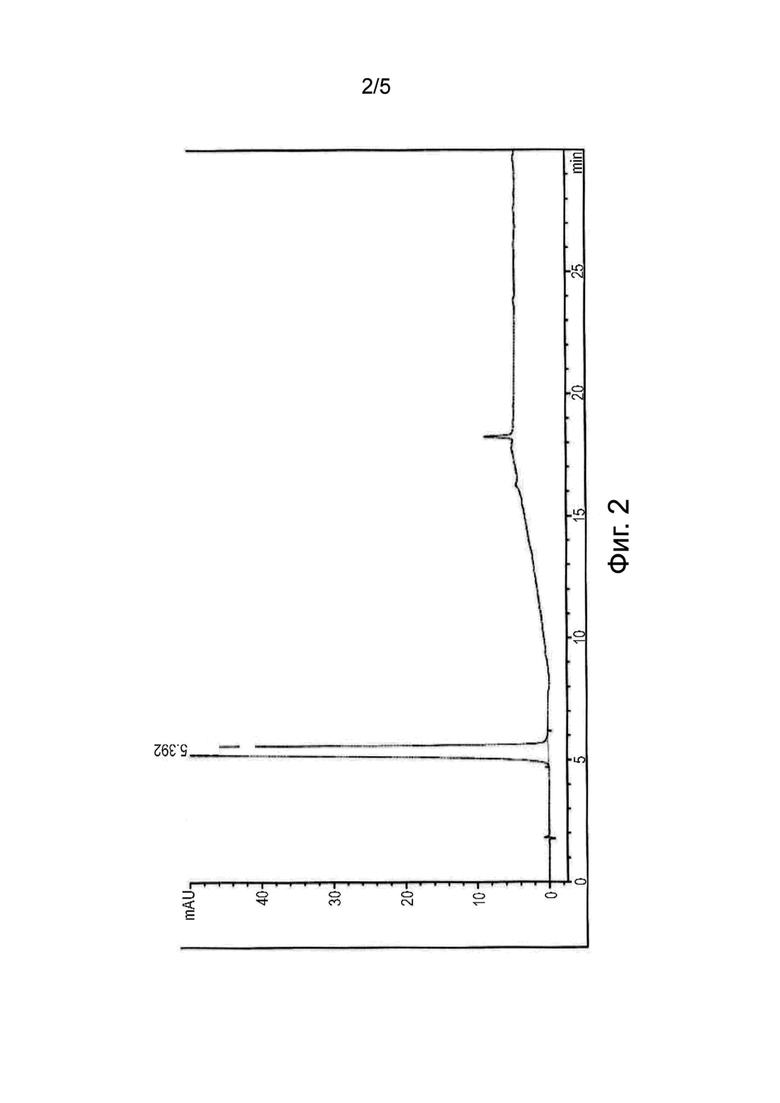
На фиг. 3 изображена рентгеновская дифрактограмма безводного Эстетрола и Эстетрола моногидрата, получаемых способом по настоящему изобретению.
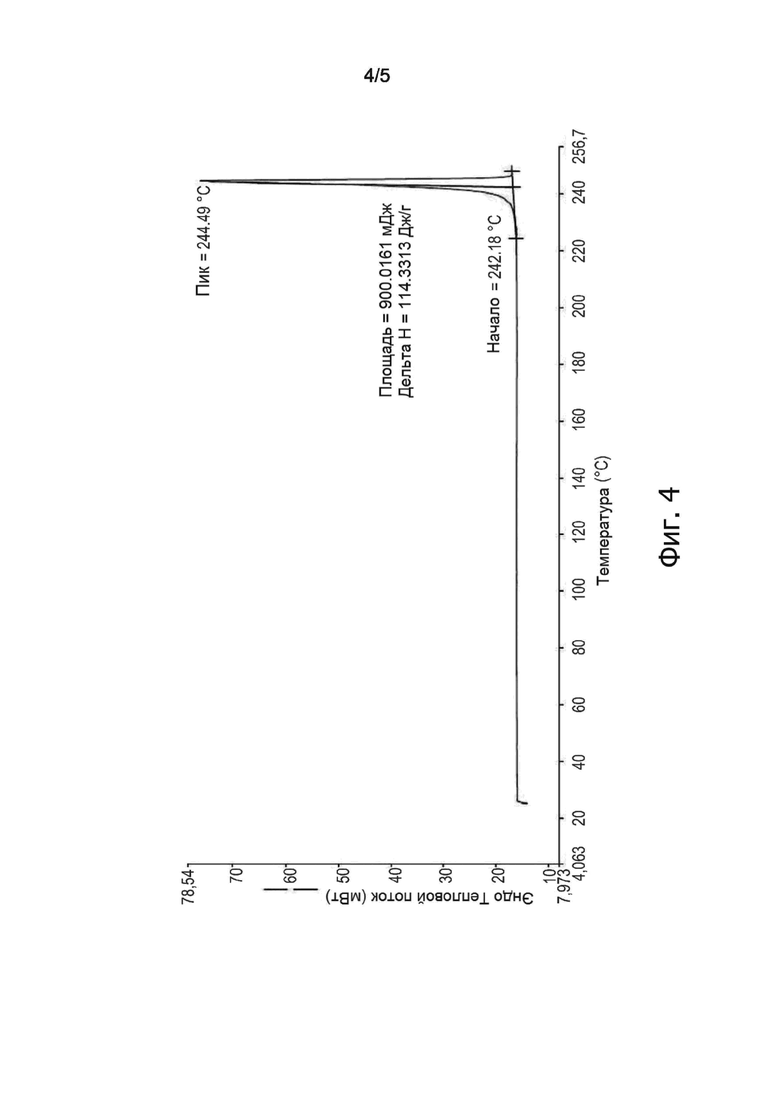
На фиг. 4 изображена ДСК-хроматограмма безводного Эстетрола, получаемого способом по настоящему изобретению.
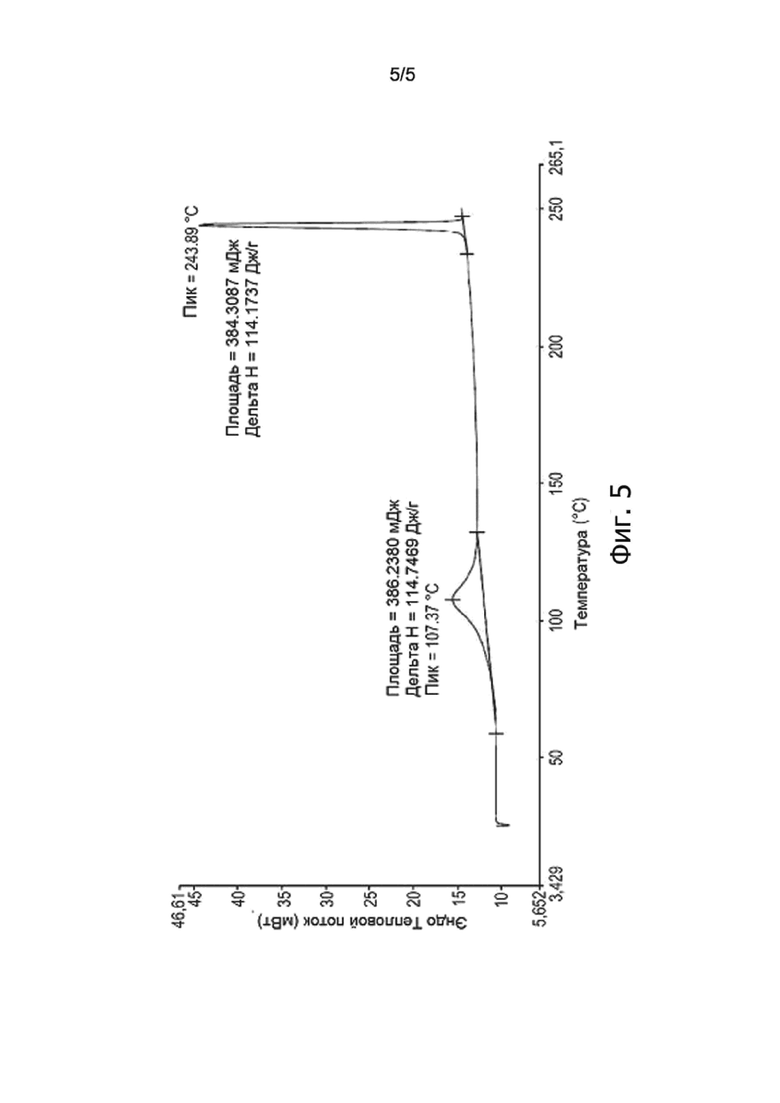
На фиг. 5 изображена ДСК-хроматограмма Эстетрола моногидрата, получаемого способом по настоящему изобретению.
Подробное описание изобретения
В первом аспекте, настоящее изобретение касается способа синтеза Эстетрола, который включает описанные выше стадии.
Стадия A) состоит в окислении (17β)-3-(фенилметокси)-эстра-1,3,5(10),15-тетраен-17-ола (интермедиата 1) с получением (17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола (интермедиата 2):
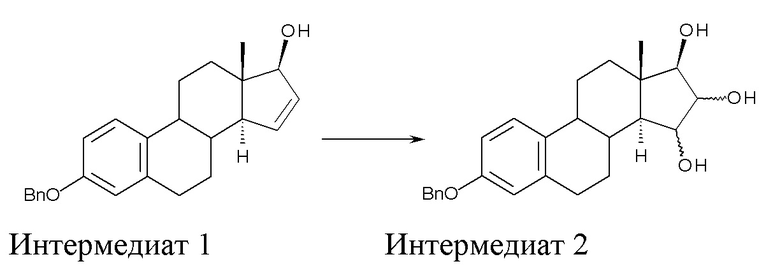
где Bn = бензил, и где конфигурация атомов углерода 15 и 16 стероидного скелета интермедиата 2 не фиксирована.
Исходный субстрат для данной стадии, интермедиат 1, можно получить как описано в заявке WO 2004/041839 A2.
В качестве окислителя на стадии A) можно применять тетраоксид осмия (OsO4) на полимерной подложке или, предпочтительно, как таковой. N-оксид органического амина, такой как триметиламина N-оксид дигидрат, применяется в качестве соокислителя.
Поскольку окисление с помощью OsO4 не является стереоселективным, интермедиат 2 получают в виде смеси изомеров с конфигурацией 15α,16α,17β и 15β,16β,17β; изомер 15α,16α,17β образуется в большем количестве, с минорным количеством изомера 15β,16β,17β.
Реакцию проводят в растворителе, инертном к производным осмия, таком как тетрагидрофуран (ТГФ), при температуре между 35 и 60°C, предпочтительно между 45 и 55°C, и в течение по меньшей мере 12 часов, предпочтительно по меньшей мере 16 часов.
Продукт реакции (интермедиат 2) после обработки реакционной смеси обрабатывают веществом, снижающим количество металлосодержащих примесей в растворе для снижения остаточного содержания осмия. Такие вещества, хорошо известные в химии, обычно имеют в своей основе функционализированный силикагель, и их называют термином «уловитель», который будет использоваться далее в тексте описания и в формуле изобретения. Уловителем предпочтительно является QuadraSil® MP.
Обработку уловителем можно проводить и повторять на каждой стадии процесса; предпочтительно такую обработку проводят на стадии А).
Стадия B) состоит в ацетилировании интермедиата 2 с получением (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола триацетата (интермедиата 3), проходящем через интермедиат 3', в котором конфигурация атомов углерода 15 и 16 стероидного скелета не фиксирована:
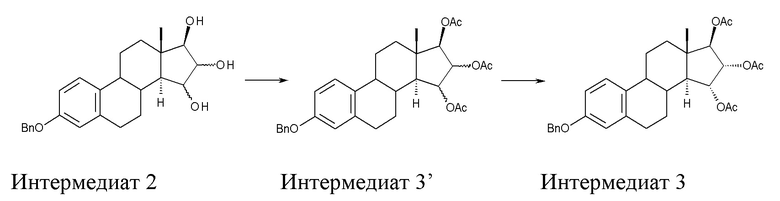
Интермедиат 2, исходный субстрат для реакции ацетилирования, можно загружать в реакционную смесь в твердом виде или, предпочтительно, напрямую используют раствор, полученный на стадии А).
Непосредственным результатом реакции ацетилирования интермедиата 2 является интермедиат 3', состоящий из смеси изомеров 15α,16α,17β и 15β,16β,17β; эту смесь затем отделяют, применяя методику очистки, которая составляет вторую часть стадии В).
Полное ацетилирование на стадии В) проводят в растворителе, совместимом с условиями самой реакции, таком как, например, изопропилацетат, этилацетат, тетрагидрофуран, пиридин или толуол. Предпочтительным растворителем является пиридин.
Для данной реакции в качестве реагента используют уксусный ангидрид в присутствии неорганического или органического основания, катализатора и, опционально, каталитических количеств трифторуксусного ангидрида. В качестве органического основания предпочтительно применяют пиридин, а 4-диметиламинопиридин используют как катализатор.
Температура реакции составляет от 5 до 40°C, предпочтительно от 20 до 30°C; время реакции составляет по меньшей мере 3 часа, предпочтительно по меньшей мере 4 часа.
Очистку интермедиата 3’ с удалением изомера 15β,16β,17β проводят с помощью описанной ниже последовательности операций:
B.1) температурная обработка, которая состоит в кипячении интермедиата 3’, подвергающегося очистке, в линейном или разветвленном C1-C6 алифатическом спирте, в течение по меньшей мере 10 минут, предпочтительно по меньшей мере 15 минут;
B.2) перемешивание суспензии интермедиата 3’, подвергающегося очистке, в линейном или разветвленном C1-C6 алифатическом спирте, при температуре между 15 и 35°C, предпочтительно между 20 и 30°C, и еще более предпочтительно между 23 и 27°C в течение периода времени от 2 до 24 часов, предпочтительно в течение периода времени от 3 до 18 часов, еще более предпочтительно в течение периода времени от 4 до 16 часов;
B.3) выделение очищенного интермедиата 3 фильтрованием.
Спирт для тепловой обработки (операция B.1) и для обработки суспензии (операция B.2) может быть одним и тем же, или они могут быть разными; предпочтительно применяют один и тот же спирт, который предпочтительно представляет собой метанол.
Интермедиат 3, подвергающийся очистке, можно выделить после фильтрованием после операции B.1) и снова суспендировать в растворителе, получая суспензию для операции B.2), или можно применять один и тот же растворитель в том же контейнере.
Очистку интермедиата 3 можно повторить столько раз, сколько необходимо для получения целевого уровня чистоты, в соответствии с изначальным содержанием изомера 15β,16β,17β. Предпочтительно процесс очистки повторяют по меньшей мере два раза.
Авторы настоящего изобретения провели серию экспериментальных тестов, повторяя три раза последовательность операций B.1, B.2 и B.3 на образцах интермедиата 3’, содержащего 5% изомера 15β,16β,17β; в первом из этих тестов операцию B.2 - перемешивание суспензии - проводили три раза по 16 часов, во втором тесте - три раза по 8 часов, и в третьем тесте - три раза по 4 часа; эти тесты подтвердили, что методика по настоящему изобретению, включающая операции B.1 + B.2 + B.3, во всех случаях приводила к получению финального продукта, в котором содержание изомера 15β,16β,17β было ниже 0,10%, и в некоторых случаях - ниже 0,05%.
Стадия C) способа по настоящему изобретению состоит из двух последовательных реакций, первая - дебензилирование посредством каталитического гидрирования интермедиата 3 с образованием интермедиата 4, и затем - гидролиз ацетатов, присутствующих в интермедиате 4, согласно приведенной ниже схеме:
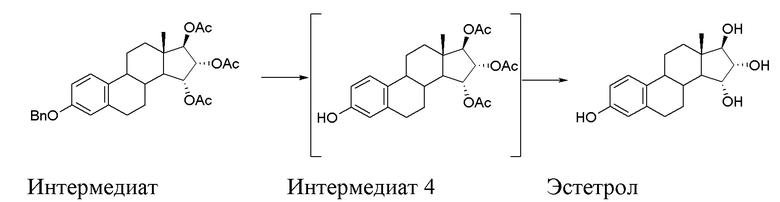
Порядок, в котором проводятся эти две реакции, указан выше. Сначала проводят каталитическое дебензилирование, и затем проводят гидролиз ацетатов; обратный порядок проведения реакций затрудняет полное осуществление дебензилирования.
Интермедиат 4, полученный после первой реакции, можно выделить и затем ввести в реакцию снова, но этот интермедиат предпочтительно оставляют растворенным в растворителе после первой реакции.
Условия проведения дебензилирования и гидролиза хорошо известны квалифицированным специалистам в области органической химии.
Первая реакция, дебензилирование, состоит в гидрировании газообразным водородом в присутствии подходящего катализатора. Предпочтительными условиями для данной реакции являются следующие:
- использование в качестве катализатора палладия на угле (Pd/C) в количестве 5% или предпочтительно 10% по весу;
- давление водорода между 1 и 6 бар, предпочтительно между 2 и 4 бар, еще более предпочтительно между 2,5 и 3,5 бар;
- линейный или разветвленный C1-C6 алифатический спирт, предпочтительно метанол, в качестве растворителя для проведения реакции;
- время реакции по меньшей мере 16 часов, предпочтительно по меньшей мере 20 часов;
- температура гидрирования между 30 и 60°C, предпочтительно между 35 и 55°C, еще более предпочтительно между 40 и 50°C.
Вторая реакция состоит в гидролизе ацетатов интермедиата 4 с применением оснований. Предпочтительными условиями проведения данной реакции являются следующие:
- применение карбоната натрия, карбоната калия или карбоната лития в качестве основания; предпочтительно применяют карбонат калия;
- время реакции по меньшей мере 2 часа, предпочтительно по меньшей мере 4 часа;
- температура реакции между 10 и 40°C, предпочтительно между 15 и 35°C, еще более предпочтительно между 20 и 30°C.
Раствор, содержащий продукт реакции (Эстетрол), можно обрабатывать уловителем на основе функционализированного силикагеля для удаления остаточного количества палладия. Уловитель предпочтительно представляет собой QuadraSil® MP.
Наконец, последняя стадия D) способа по настоящему изобретению представляет собой очистку Эстетрола, полученного на стадии C).
Данную стадию проводят методом кристаллизации из горячего раствора путем его охлаждения, по методикам, хорошо известным квалифицированным специалистам в области органической химии.
В качестве растворителей применяют тетрагидрофуран (ТГФ), метанол и ацетонитрил.
Также в ходе этой операции Эстетрол можно обрабатывать уловителем на основе функционализированного силикагеля, предпочтительно QuadraSil® MP, для удаления остаточного количества палладия. Растворитель, в котором применяют уловитель, выбирают из группы, состоящей из тетрагидрофурана (ТГФ), метанола и ацетонитрила; предпочтительно используют тетрагидрофуран.
По окончании данной операции получают чистый Эстетрол в «безводной» форме, т.е. с минимальным остаточным количеством воды, например, когда стехиометрическое соотношение вода/ДВ намного меньше 1.
В альтернативном варианте осуществления, настоящее изобретение касается получения Эстетрола в форме моногидрата. В данном варианте осуществления, способ по настоящему изобретению включает дополнительную стадию, E), которую проводят после стадии D) со следующей последовательностью операций:
E.1) растворение чистого Эстетрола в безводной форме в смешивающемся с водой органическом растворителе, таком как ацетон, метанол, этанол, изопропанол, тетрагидрофуран, диметилформамид или диметилацетамид, до полного растворения; предпочтительным растворителем является метанол;
E.2) смешивание раствора со стадии E.1) с водой, предпочтительно с чистой водой; предпочтительно данную операцию проводят путем прикапывания воды в органический раствор Эстетрола;
E.3) удаление органического растворителя отгонкой, предпочтительно при пониженном давлении;
E.4) перемешивание полученной суспензии, предпочтительно по меньшей мере 15 минут, при температуре от 5 до 20°C;
E.5) фильтрование и промывание твердого осадка; предпочтительно отфильтрованный твердый осадок промывают на фильтре водой;
E.6) сушка полученного твердого вещества в течение по меньшей мере 5 часов, при температуре по меньшей мере 40°C и при пониженном давлении, предпочтительно по меньшей мере 6 часов, при по меньшей мере 45°C и пониженном давлении.
Во втором аспекте, настоящее изобретение касается очищенного интермедиата 3, (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола триацетата, полученного описанным выше способом:
Далее настоящее изобретение будет дополнительно проиллюстрировано описанными ниже примерами.
Экспериментальная часть: приборы, методы и условия
ЯМР:
ЯМР-спектрометр JEOL 400 YH (400 МГц); JEOL Delta software v5.1.1;
Спектры регистрировали в ДМСО-d6.
Масс-спектрометрия:
Прибор: DSQ-trace Thermofisher
Ввод образца - прямой ввод (dep)
Химическая ионизация (ХИ) метаном
Давление метана: 2,2 фунт/кв.дюйм
Температура источника: 200°C
ВЭЖХ:
Хроматографическая система Agilent Model 1260 Infinity; УФ-детектор MODEL G1315C DAD VL+
Метод ВЭЖХ 1:
Условия хроматографирования:
- Колонка: Supelco ascentis express C18 250×4,6 мм, 5 мкм
- Поток: 1 мл/мин
- Детектор: УФ 280 нм
- Объем ввода: 5 мкл
- Температура: 25°C
- Подвижная фаза A: вода
- Подвижная фаза B: ацетонитрил
Метод ВЭЖХ 2:
Условия хроматографирования:
- Колонка: Supelco discovery C18 150x4,6 мм, 5 мкм
- Поток: 1 мл/мин
- Детектор: УФ 280 нм
- Объем ввода: 25 мкл
- Температура: 22°C
- Подвижная фаза A: 4,29 г/л раствор CH3COONH4 в смеси вода/ метанол/ ацетонитрил 90/6/4
- Подвижная фаза B: 38,6 г/л раствор CH3COONH4 в смеси вода/ метанол/ ацетонитрил 10/54/36
ТСХ:
MERCK: Алюминиевые пластинки 20 x 20 см с ТСХ силикагелем 60 F254, код 1.0554.0001.
Проявление ТСХ пластин:
Фосфомолибдат церия: 25 г фосфомолибденовой кислоты и 10 г сульфата церия (IV) растворяли в 600 мл H2O. Добавляли 60 мл 98%-ной H2SO4 и доводили объем до 1 литра добавлением H2O. Пластинку опрыскивали полученным раствором и затем нагревали до проявления продуктов.
Рентгеноструктурный анализ:
Рентгеноструктурный анализ проводили с помощью порошкового дифрактометра Bruker D2 Phaser (2nd edition), работающего в геометрии Брэгга-Бентано, оснащенного вращающимся мультисэмпером и линейным детектором типа SSD (Lynxeye). Истоником рентгеновского излучения является рентгеновская трубка с медным анодом, работающая при 30 кВ и 10 мА. Для анализа применяли рентгеновское излучение с длиной волны, соответствующей средней Kα для меди (λ = 1,54184 Å). Kβ излучение фильтровали через специальный никелевый фильтр.
Использовали кремниевые держатели для образца, имеющие «нулевой бэкграунд» и плоскую поверхность, по которой распределяли образец в виде тонкого слоя. При проведении анализа держатель образца вращают со скоростью 60 об/мин.
Сканирование проводили в диапазоне значений 2θ от 4 до 0°С, с инкрементом 2θ 0,016° и временем накопления 1,0 с для каждого инкремента.
Дифрактограмму обрабатывали с использованием программы Bruker DIFFRAC.EVA.
ДСК (дифференциальная сканирующая калориметрия):
Анализ методом ДСК проводили в инертной атмосфере (азот) с использованием дифференциального сканирующего калориметра Perkin Elmer Diamond DSC. Образцы готовили, отвешивая порошок в алюминиевые тигли объемом 40 мкл, которые затем герметично закрывали перед проведением анализа. Анализ проводили в интервале температур 25-250°C при скорости нагрева 10°C/мин.
Примечания
Упоминающаяся в описании экспериментов вода представляет собой чистую воду, если не указано иное.
Упоминающиеся в описании экспериментов органические растворители имеют «техническую» степень чистоты, если не указано иное.
Упоминающиеся в описании экспериментов реагенты и катализаторы имеют коммерческую чистоту, если не указано иное.
Продукт QuadraSil® MP получен от Johnson Matthey.
Пример 1
Данный пример относится к стадии A) в способе по настоящему изобретению, превращение интермедиата 1 в интермедиат 2.
В заполненную азотом колбу помещали 32,4 г интермедиата 1 (89,87 ммоль, 1 экв.) и 356 мл тетрагидрофурана. Добавляли в раствор 0,324 г тетраоксида осмия (1,28 ммоль, 1% по весу) и 17,9 г триметиламина N-оксид дигидрата (161,26 ммоль, 1,8 экв.) в указанном порядке. Реакционную смесь нагревали до 50°C и перемешивали в течение 16 часов.
Прохождение реакции контролировали методом ТСХ в следующих условиях: ТСХ пластина: силикагель на алюминиевой подложке; исходный субстрат (интермедиат 1) растворенный в дихлорметане; реакционная смесь разбавлена дихлорметаном; элюент: этилацетат (EtOAc); детектирование: фосфомолибдат церия.
По окончании реакции раствор охлаждали до 25°C и прикапывали раствор метабисульфита натрия (18,3 г) в воде (162 мл). Растворитель упаривали при пониженном давлении и добавляли к остатку 193 мл изопропилацетата и 290 мл 1M соляной кислоты.
Добавляли в полученную двухфазную систему 1,6 г активированного угля и 1,6 г дикалита и перемешивали при 25°C в течение 15 минут. Полученную суспензию сначала фильтровали через слой дикалита и затем через фильтр Millipore (0,22 мкм). Фазы разделяли и водную фазу экстрагировали 160 мл изопропилацетата. Добавляли в органическую фазу 1,12 г QuadraSil® MP, и смесь перемешивали при 25°C в течение 16 часов. Полученную суспензию фильтровали через фильтр Millipore (0,22 мкм) и промывали 32 мл изопропилацетата.
Полученный таким образом раствор использовали в следующей реакции без очистки.
Пример 2
Данный пример относится к стадии B) в способе по настоящему изобретению.
Раствор интермедиата 2, полученный как описано в предыдущем примере, упаривали при пониженном давлении до объема остатка 50 мл.
Добавляли 228 мл пиридина, и остаточный изопропилацетат отгоняли при пониженном давлении. Добавляли в раствор 0,877 г 4-диметиламинопиридина (7,19 ммоль, 0,08 экв.) и затем прикапывали 29,45 мл уксусного ангидрида (312 ммоль, 3,47 экв.), поддерживая температуру ниже 30°C. Раствор перемешивали при 25°C в течение 4 часов.
Прохождение реакции контролировали методом ТСХ в следующих условиях: ТСХ пластина: силикагель на алюминиевой подложке; исходный субстрат (интермедиат 2) растворенный в дихлорметане; реакционная смесь, погашенная добавлением 1M HCl и проэкстрагированная этилацетатом, использовали органическую фазу; элюент: EtOAc; детектирование: фосфомолибдат церия.
Реакционную смесь упаривали при пониженном давлении до объема остатка 85 мл и добавляли 250 мл изопропилацетата и 125 мл воды. Добавляли в полученную двухфазную систему 55 мл 37%-ной соляной кислоты, поддерживая температуру ниже 30°C (финальное значение pH водной фазы = 1).
Фазы разделяли, и органическую фазу промывали два раза насыщенным раствором бикарбоната натрия (2 x 90 мл) и затем насыщенным раствором хлорида натрия (90 мл).
Органическую фазу упаривали при пониженном давлении, получая маслянистый остаток. Добавляли 100 мл метанола, и смесь снова упаривали при пониженном давлении, получая пастообразный остаток. Добавляли 210 мл метанола, и смесь кипятили 15 минут. Полученную суспензию охлаждали до 25°C и перемешивали в течение 16 часов. Твердый осадок отфильтровывали на воронке Бюхнера, промывая 35 мл метанола. Полученное твердое вещество сушили при пониженном давлении при 45°C в течение 3 часов.
Получали 28,4 г твердого вещества, представляющего собой интермедиат 3'; ВЭЖХ анализ (метод 1) показал содержание изомера 15β,16β,17β = 1,6%.
Полученный твердый продукт (28 г) растворяли в 168 мл метанола, и смесь кипятили 15 минут. Полученную суспензию охлаждали до 25°C и перемешивали в течение 16 часов. Отфильтровывали твердый осадок на воронке Бюхнера, промывали 28 мл метанола и затем сушили при пониженном давлении при 45°C в течение 3 часов. Получали 24 г продукта (ВЭЖХ, метод 1): изомер 15β,16β,17β = 0,18%).
Полученный твердый продукт (23,5 г) растворяли в 140 мл метанола, и смесь кипятили 15 минут. Полученную суспензию охлаждали до 25°C и перемешивали в течение 16 часов. Отфильтровывали твердый осадок на воронке Бюхнера, промывали 23 мл метанола и сушили в вакууме при 45°C в течение 3 часов.
Получали 22,1 г интермедиата 3 (почти белое твердое вещество).
ВЭЖХ чистота (метод 1): 97,5%, изомер 15β,16β,17β = 0,07%.
1H-ЯМР (400 МГц, ДМСО-d6): δ 7,39-7,26 (м, 5H); 7,12 (д, 1H, J = 9,2 Гц); 6,72-6,67 (м, 2H); 5,22-5,18 (т, 1H, J = 7,4 Гц); 5,04-4,99 (м, 3H); 4,84 (д, 1H, J = 6,4 Гц); 2,74-2,70 (м, 2H); 2,25-2,20 (м, 2H); 1,99-1,97 (2s, 9H); 1,7-1,2 (м, 7H); 0,85 (с, 3H).
Масс-спектрометрия (ХИ): m/z = 521 [M++1].
Пример 3
Данный пример касается проведения стадии C) в способе по настоящему изобретению.
21,6 г интермедиата 3, полученного как описано в предыдущем примере, и 154 мл тетрагидрофурана помещали в колбу.
Добавляли в раствор 2,2 г QuadraSil® MP, и смесь перемешивали при 25°C в течение 16 часов. Полученную суспензию фильтровали через фильтр Millipore (0,22 мкм), промывая 22 мл тетрагидрофурана. Растворитель упаривали при пониженном давлении, получая пастообразный остаток.
Остаток от упаривания растворяли в 650 мл метанола и загружали в реактор для гидрирования. Добавляли в полученную суспензию 2,05 г 10% палладия на угле и гидрировали при 45°C и давлении 3 бара в течение 22 часов.
Прохождение реакции контролировали методом ТСХ в следующих условиях: ТСХ пластина: силикагель на алюминиевой подложке; исходный субстрат (интермедиат 3) растворенный в дихлорметане; реакционная смесь разбавлена метанолом; элюент: гептан/EtOAc 1/1; детектирование: фосфомолибдат церия. По окончании реакции смесь фильтровали через слой дикалита (30 г), промывая метанолом (120 мл).
Растворитель упаривали при пониженном давлении до объема остатка 430 мл и добавляли 5,16 г карбоната калия. Полученную смесь перемешивали при 25°C в течение 4 часов. Прохождение реакции контролировали методом ТСХ в следующих условиях: ТСХ пластина: силикагель на алюминиевой подложке; интермедиат 4 растворен в дихлорметане; реакционная смесь, погашенная добавлением 1M HCl и проэкстрагированная этилацетатом, использовали органическую фазу; элюент: гептан/EtOAc 1/1; детектирование: фосфомолибдат церия. Полученную суспензию фильтровали через фильтр Millipore (0,22 мкм), промывая метанолом (20 мл).
Раствор упаривали при пониженном давлении до объема остатка 54 мл, добавляли 162 мл воды и удаляли остаточный метанол при пониженном давлении.
Полученную суспензию нейтрализовывали добавлением 40 мл 1M соляной кислоты и охлаждали до 10°C при перемешивании в течение 30 минут. Твердый осадок отфильтровывали на воронке Бюхнера, промывали водой и сушили при пониженном давлении при 50°C в течение 6 часов.
Получали 13 г сырого Эстетрола (белое твердое вещество).
Пример 4
Данный пример касается проведения стадии D) в способе по настоящему изобретению.
Сырой Эстетрол, полученный как описано в предыдущем примере, растворяли в 91 мл тетрагидрофурана. Добавляли в раствор 0,4 г QuadraSil® MP, и смесь перемешивали при 25°C в течение 16 часов. Полученную суспензию фильтровали через фильтр Millipore (0,22 мкм), промывая 25 мл тетрагидрофурана. Растворитель удаляли при пониженном давлении и добавляли 130 мл ацетонитрила и 104 мл метанола. Смесь перемешивали при 25°C до полного растворения.
Раствор упаривали при пониженном давлении до объема остатка 130 мл и добавляли 104 мл ацетонитрила. Смесь упаривали снова при пониженном давлении до объема остатка 130 мл и добавляли 104 мл ацетонитрила.
Смесь упаривали при пониженном давлении до объема остатка 130 мл и перемешивали при 25°C в течение 3 часов. Полученную суспензию охлаждали до 5°C и перемешивали в течение 1 часа. Твердый осадок отфильтровывали на воронке Бюхнера, промывали холодным ацетонитрилом и сушили при пониженном давлении 3 часа при 45°C.
Получали 10,5 г продукта, который анализировали методом ВЭЖХ (метод ВЭЖХ 2). Полученные результаты представлены на фиг. 1: продукт представляет собой Эстетрол с ВЭЖХ чистотой = 99,91%, при этом изомер 15β,16β,17β не был обнаружен (пик с временем удерживания примерно 18’ относится не к продукту, а к применяемому для данной хроматографии элюенту).
Образец продукта исследовали методом рентгеноструктурного анализа; в результате получали дифрактограмму, изображенную в верхней части фиг. 3. Приведенная ниже таблица показывает положение (в виде значения углов 2θ ± 0,2°) и относительную интенсивность основных пиков в дифрактограмме:
Другой образец полученного продукта весом 8 мг исследовали методом ДСК; результат анализа показан на фиг. 4, где видно, что продукт имеет температуру плавления примерно 244,5°C.
Пример 5
Данный пример касается проведения стадии E) в способе по настоящему изобретению.
8 г Эстетрола, полученного в Примере 4, растворяли в 96 мл метанола, и в полученный раствор прикапывали 240 мл воды. Смесь упаривали при пониженном давлении до полного удаления метанола. Полученную суспензию перемешивали при 15°C в течение 30 минут, твердый осадок отфильтровывали на воронке Бюхнера и промывали 56 мл воды.
Полученное твердое вещество сушили при пониженном давлении при 45°C в течение 6 часа. Получали 8,3 г Эстетрола моногидрата (белое твердое вещество), проводили его анализ методом ВЭЖХ (метод 2). Полученные результаты представлены на фиг. 2: продукт представляет собой Эстетрол моногидрат, имеющий ВЭЖХ чистоту = 100% (пик с временем удерживания примерно 18’ относится не к продукту, а к применяемому для данной хроматографии элюенту).
Образец продукта исследовали методом рентгеноструктурного анализа; в результате получали дифрактограмму, изображенную в нижней части Фиг. 3. Приведенная ниже таблица показывает положение (в виде значения углов 2θ ± 0,2°) и относительную интенсивность основных пиков в дифрактограмме:
Другой образец полученного продукта весом 3,4 мг исследовали методом ДСК; результат анализа показан на фиг. 5, на которой видно первый уширенный пик с максимумом примерно при 107,4°C, относящийся к дегидратации Эстетрола моногидрата, и второй пик примерно при 244°C, т.е. при температуре, соответствующей температуре плавления Эстетрола, определенной в анализе на фиг. 4.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,0 (с, 1H); 7,05 (д, 1H, J = 8,4 Гц); 6,51-6,48 (м, 1H); 6,27 (д, 1H, J = 2,4 Гц); 4,86-4,85 (д, 1H, J = 4,8 Гц); 4,61-4,59 (д, 1H, J = 5,6 Гц); 4,27-4,26 (д, 1H, J = 6 Гц); 3,72-3,66 (м, 2H); 3,26-3,24 (т, 1H, J = 5,6 Гц); 2,72-2,68 (м, 2H); 2,22-2,18 (м, 2H); 2,1-2,05 (м, 1H); 1,76-1,73 (д, 1H, 12Hz); 1,4-1,03 (м, 5H); 0,66 (с, 3H).
Масс-спектрометрия (ХИ): m/z = 305 [M++1].
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОМЫШЛЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОГО ЭСТЕТРОЛА | 2020 |
|
RU2802820C2 |
| 15,15-ДИАЛКИЛСТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2147306C1 |
| ЭСТРАТРИЕНЫ, СОДЕРЖАЩИЕ МОСТИК | 1990 |
|
RU2087479C1 |
| СОЕДИНЕНИЯ, НАБОР, АНДРОГЕННАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2242479C2 |
| ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2139885C1 |
| 18-МЕТИЛ-19-НОРАНДРОСТ-4-ЕН-17,17-СПИРОЭФИР (18-МЕТИЛ-19-НОР-20-СПИРОКС-4-ЕН-3-ОН) И ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, КОТОРЫЕ ЕГО СОДЕРЖАТ | 2007 |
|
RU2440365C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА | 1995 |
|
RU2179442C2 |
| ПРОИЗВОДНЫЕ СУЛЬФАМАТА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 1996 |
|
RU2159774C2 |
| 17α-АКИЛ-17β-ОКСИЭСТРАТРИЕНЫ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 2002 |
|
RU2285009C2 |
| 17 БЕТА-ОКСИЭСТРАТРИЕНЫ | 2002 |
|
RU2339643C2 |
Изобретение относится к способу получения (15α,16α,17β)-эстра-1,3,5(10)-триен-3,15,16,17-тетрола (Эстетрол), где указанный способ включает окисление (17β)-3-(фенилметокси)-эстра-1,3,5(10),15-тетраен-17-ола (интермедиат 1) с получением (17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола (интермедиат 2), ацетилирование интермедиата 2 с получением (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола триацетата (интермедиат 3) через интермедиат 3' и превращением интермедиата 3 через (15α,16α,17β)-3-гидрокси-эстра-1,3,5(10)-триен-15,16,17-триол триацетат (интермедиат 4) в эстетрол. Превращение интермедиата 3 в интермедиат 4 осуществляется в следующих условиях: использование палладия на угле (Pd/C) в количестве 5% или 10% по весу; давление водорода между 1 и 6 бар; линейный или разветвленный С1-6 алифатический спирт в качестве растворителя; время реакции по меньшей мере 16 часов; температура гидрирования между 30 и 60°С. Технический результат - получение эстетрола в промышленном масштабе. 8 з.п. ф-лы, 5 ил., 4 табл., 5 пр.
 ,
,  ,
,  ,
,  ,
,  ,
,  .
.
1. Способ синтеза Эстетрола, (15α,16α,17β)-эстра-1,3,5(10)-триен-3,15,16,17-тетрола, включающий следующие стадии:
A) окисление (17β)-3-(фенилметокси)-эстра-1,3,5(10),15-тетраен-17-ола (интермедиат 1) с получением (17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола (интермедиат 2):
где Bn = бензил, и где конфигурация атомов углерода 15 и 16 стероидного скелета не фиксирована;
B) ацетилирование интермедиата 2 с получением (15α,16α,17β)-3-(фенилметокси)-эстра-1,3,5(10)-триен-15,16,17-триола триацетата (интермедиат 3), проходящее через интермедиат 3', в котором конфигурация атомов углерода 15 и 16 стероидного скелета не фиксирована:
C) превращение интермедиата 3, проходящее через (15α,16α,17β)-3-гидрокси-эстра-1,3,5(10)-триен-15,16,17-триол триацетат (интермедиат 4), который предпочтительно не выделяют, в Эстетрол:
D) очистка Эстетрола, полученного на стадии C),
характеризующийся тем, что реакцию дебензилирования на стадии C) интермедиата 3 в интермедиат 4 проводят посредством гидрирования газообразным водородом в присутствии катализатора в следующих условиях:
- использование в качестве катализатора палладия на угле (Pd/C) в количестве 5% или 10% по весу;
- давление водорода между 1 и 6 бар;
- линейный или разветвленный C1-C6 алифатический спирт в качестве растворителя для проведения реакции;
- время реакции по меньшей мере 16 часов;
- температура гидрирования между 30 и 60°C.
2. Способ по п. 1, в котором стадию A) проводят с применением тетраоксида осмия (OsO4) как такового или нанесенного на полимерную подложку в качестве окислителя и N-оксида органического амина в качестве соокислителя в растворителе, инертном к производным осмия, при температуре между 35 и 60°C, и в течение по меньшей мере 12 часов.
3. Способ по п. 2, в котором стадию A) проводят с применением тетраоксида осмия (OsO4) как такового в качестве окислителя и триметиламин N-оксида дигидрата в качестве соокислителя, используя как растворитель тетрагидрофуран (ТГФ), при температуре между 45 и 55°C, и в течение по меньшей мере 16 часов.
4. Способ по любому из предшествующих пунктов, в котором на стадии B) реакцию полного ацетилирования интермедиата 2 до интермедиата 3' проводят с применением уксусного ангидрида в качестве реагента в растворителе, выбранном из изопропилацетата, этилацетата, тетрагидрофурана, пиридина и толуола, в присутствии неорганического или органического основания, катализатора и, опционально, каталитического количества трифторуксусного ангидрида, при температуре между 5 и 40°C в течение по меньшей мере 3 часов.
5. Способ по п. 4, в котором реакцию полного ацетилирования интермедиата 2 до интермедиата 3' на стадии B) проводят, используя пиридин как растворитель, 4-диметиламинопиридин как катализатор, при температуре между 20 и 30°C в течение по меньшей мере 4 часов.
6. Способ по любому из предшествующих пунктов, в котором на стадии B) очистку интермедиата 3' с получением интермедиата 3 проводят со следующей последовательностью операций:
B.1) кипячение интермедиата 3', подвергающегося очистке, в линейном или разветвленном C1-C6 алифатическом спирте, в течение по меньшей мере 10 минут, предпочтительно по меньшей мере 15 минут;
B.2) перемешивание суспензии интермедиата 3', подвергающегося очистке, в линейном или разветвленном C1-C6 алифатическом спирте, при температуре между 15 и 35°C, предпочтительно между 20 и 30°C, и еще более предпочтительно между 23 и 27°C в течение периода времени от 2 до 24 часов, предпочтительно в течение периода времени от 3 до 18 часов, еще более предпочтительно в течение периода времени от 4 до 16 часов;
B.3) выделение очищенного интермедиата 3 фильтрованием.
7. Способ по любому из предшествующих пунктов, в котором реакцию гидролиза на стадии C), представляющей собой превращение 4 в Эстетрол, проводят в следующих условиях:
- применение карбоната натрия, карбоната калия или карбоната лития в качестве основания;
- время реакции по меньшей мере 2 часа;
- температура реакции между 10 и 40°C.
8. Способ по любому из предшествующих пунктов, в котором стадию D) проводят методом кристаллизации из горячего раствора путем его охлаждения, в растворителе, выбранном из тетрагидрофурана, метанола и ацетонитрила.
9. Способ по любому из предшествующих пунктов, дополнительно включающий дополнительную стадию E), на которой Эстетрол, полученный на стадии D), превращают в Эстетрол моногидрат согласно следующей последовательности операций:
E.1) растворение чистого Эстетрола в безводной форме в смешивающемся с водой органическом растворителе, таком как ацетон, метанол, этанол, изопропанол, тетрагидрофуран, диметилформамид или диметилацетамид, до полного растворения;
E.2) смешивание раствора со стадии E.1) с водой, предпочтительно с чистой водой;
E.3) удаление органического растворителя отгонкой, предпочтительно при пониженном давлении;
E.4) перемешивание полученной суспензии, предпочтительно по меньшей мере 15 минут, при температуре от 5 до 20°C;
E.5) фильтрование и промывание твердого осадка;
E.6) сушка полученного твердого вещества в течение по меньшей мере 5 часов, при температуре по меньшей мере 40°C и при пониженном давлении.
| FISHMAN, J | |||
| et al | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| The Journal of Organic Chemistry, 1968, 33(8), p.3133-3135 | |||
| NAMBARA, T | |||
| et al | |||
| SYNTRESES OF ESTETROL MONOCLUCURONIDES | |||
| Steroids, 1976, 27(1), p.111-122 | |||
| WO 2004041839 A2, 21.05.2004 | |||
| WO 2012164096 A1, 06.12.2012 | |||
| WO 2015040051 A1, 26.03.2015 | |||

