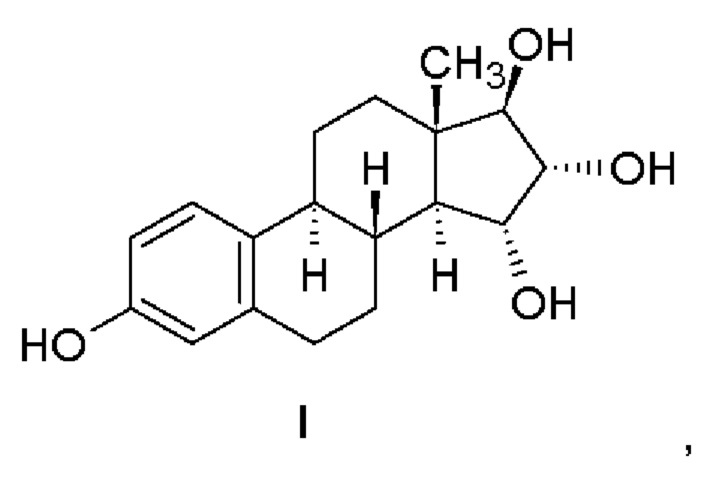
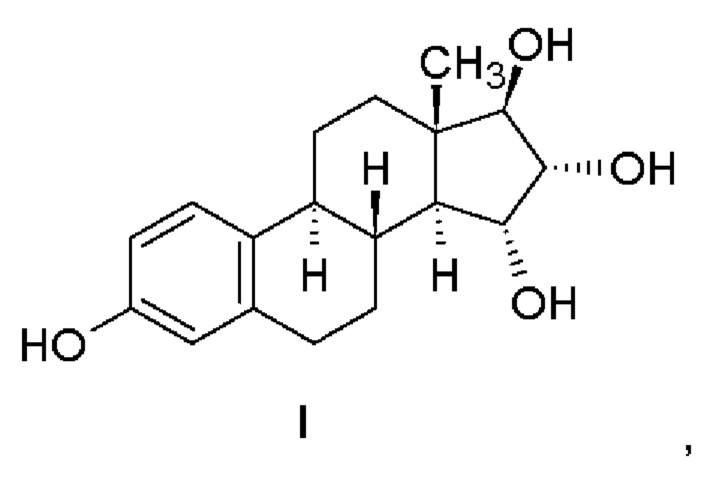
Изобретение относится к получению эстетрола (эстра-1,3,5(10)-триен-3,15α,16α,17β-тетрола), имеющего формулу (I), его производных, защищенных в положениях 3,15α, 16α, 17β, которые имеют общую формулу (III), и его 3-гидроксипроизводных, защищенных в положениях 15α,16α,17β, которые имеют общую формулу (IV), и к промежуточным соединениям, имеющим общие формулы (III) и (IV), применяемым в способе. Другой аспект изобретения относится к применению эстетрола, имеющего формулу (I), получаемого способом согласно изобретению, для изготовления фармацевтической композиции.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Эстетрол (эстра-1,3,5(10)-триен-3,15α,16α,17β-тетрол), имеющий формулу (I), представляет собой соединение, обладающее слабой эстрогенной активностью, которое обнаруживается в организме женщины и вырабатывается печенью плода во время беременности.
Было обнаружено, что эстетрол способен оказывать действие при гормонозаместительной терапии, в способе лечения сухости влагалища, в способе лечения перименопаузальных симптомов (например, приступообразного ощущения жара, ночной потливости), в способе контрацепции, в способе повышения либидо, в способе лечения кожных покровов и улучшения заживления ран, в способе лечения или предотвращения (профилактики) аутоиммунного нарушения, опухолей молочных желез, рака предстательной железы и колоректальных опухолей и в способе нейрозащиты (например, при неонатальной энцефалопатии) (WO 02/094275 А1, WO 02/094276 А1, WO 02/094278 А1, WO 02/094279 А1, WO 03/041718 А1, WO 03/103684 А1, WO 03/103685 А1, WO 2004/006936 А1, WO 2004/037269 А1, WO 2007/081206 А1, WO 2008/085038 А2, WO 2013/021025 А1, WO 2013/156329 А1, WO 2018/024912 А1, WO 2018/065076 А1, WO 2019/025031 А1; Estetrol, последнее обновление - 14 мая 2019 г., https://adisinsight.springer.com/drugs/800044874; Gaspard с соавт., Maturitas 124 (2019) стр. 153, Abstract РОЭ; Apter с соавт. Eur. J. Contracept. Reprod. НС (2017) 22(4):260-267; Tskitishvili с соавт., J. Endocrinol. (2017) 232(1):85-95; Coelingh Bennick с соавт., Climacteric (2008) 11(Suppl1):47-58).
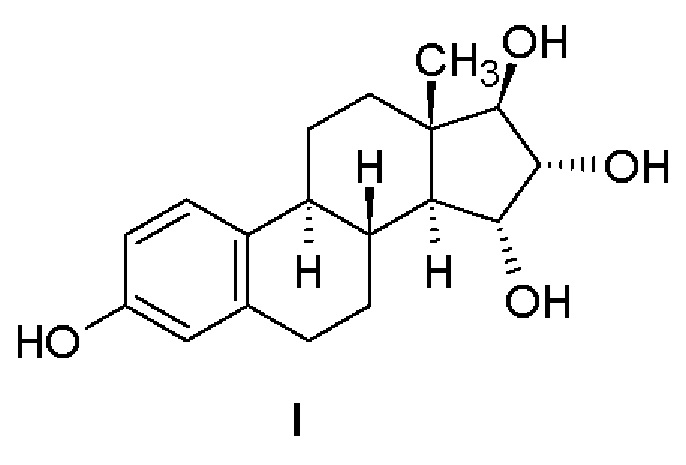
Впервые синтез эстетрола, представленный Реакционной схемой 1, был описан Fishman с соавт. (Fishman, J., Guzik, Н., Tetrahedron Letters, 1967, 30:2929-2932). Восстановление исходного 15-ен-17-кето-соединения тетрагидроалюминатом лития приводит к получению соединения типа аллилового спирта, из которого образуется диацетат. Окисление диацетата тетраоксидом осмия в пиридине приводит к получению эстетролдиацетата, который при кипячении в метаноле с ацетатом калия образует эстетрол. В названной публикации не имеется данных по выходу и чистоте; для подтверждения идентичности соединения приведены температура плавления (230-235°С), удельное вращение  и данные ЯМР (60 МГц).
и данные ЯМР (60 МГц).
Реакционная схема 1
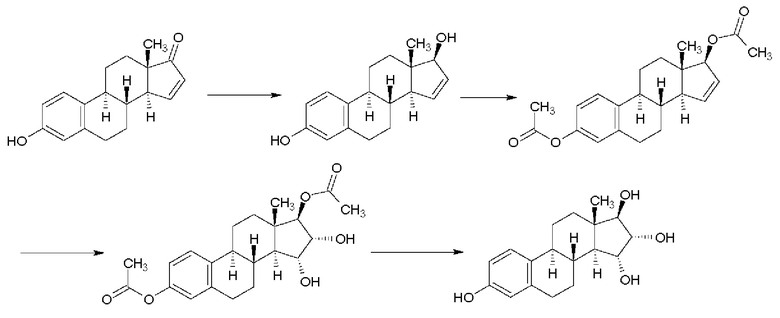
В синтезе, описанном Suzuki с соавт. (Suzuki, Е., Namba, S., Kuruhara, Н., Goto, J., Matsuki, Y., Nambara, Т., Steroids, 1995, 60, 277-284), как показано на Реакционной схеме 2, 15-ен-17-ацетоксипроизводное было окислено в бензоле эквивалентным количеством тетраоксида осмия в присутствии пиридина. Полученные изомерные диацетаты разделяли колоночной хроматографией, получая 15α,16α,17β-диацетат с выходом 46% и изомерный 15β,16β,17β-диацетат с выходом 12%.
Соотношение изомеров, вычисляемое из данных количеств полученных продуктов, составляет 78,9/21,1 (15α,16α/15β,16β).
Щелочной гидролиз 15α,16α,17β-диацетата приводит к образованию эстетрола с выходом 67%. Данные по чистоте не приводятся, и, как указано, температура плавления продукта составляет 233-235°С.
Реакционная схема 2
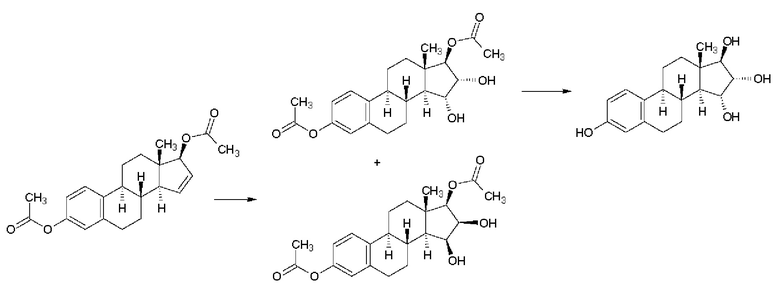
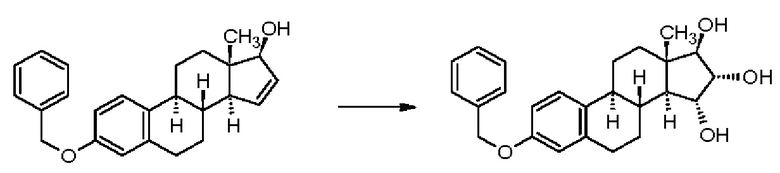
В патентной заявке WO 2004/041839 А2 (Pantarhei) - Реакционная схема 3 - из эстрона, защищенного посредством превращения в простой 3-бензиловый эфир, в результате проведения нескольких известных этапов образуется простой бензиловый эфир Δ15-эстрадиол-17-ацетата, который окисляют зафиксированным на полимере тетраоксидом осмия обработкой в смеси растворителей гептан-этилацетат, получая неочищенный продукт, который затем кристаллизуют в тройной смеси растворителей (гептан-этилацетат-этанол), получая простой бензиловый эфир эстетрол-17-ацетата с выходом 43% и чистотой 98,7% (изомерная чистота: 99,5%). Снятие защиты каталитическим гидрированием (выход 92%) и щелочной гидролиз (выход 92,5%) приводят к получению эстетрола. Указано, что чистота продукта составляет 99,5%. Такое же решение также описано в патентной заявке WO 2013/012328 А1 (Donesta).
Согласно описанию, чистое промежуточное соединение получают с большими потерями во время кристаллизации из тройной смеси растворителей, применение которой технологически невыгодно. С экономической точки зрения это решение также вызывает вопросы.
Реакционная схема 3
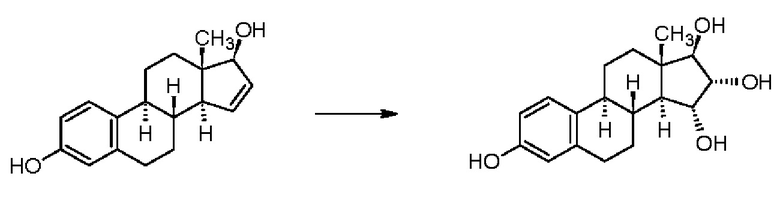
В патентной заявке WO 2013/050553 А1 (Estetra), в Реакционной схеме 4, в качестве окислителя также указан перманганат калия, но не указаны соотношение изомеров, чистота и выход.
Реакционная схема 4
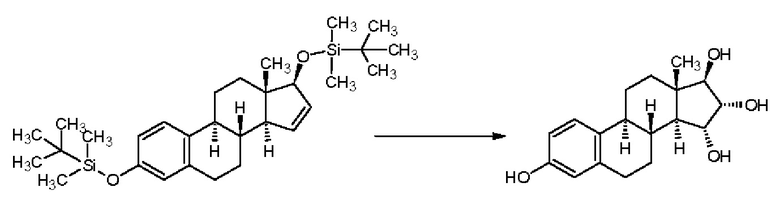
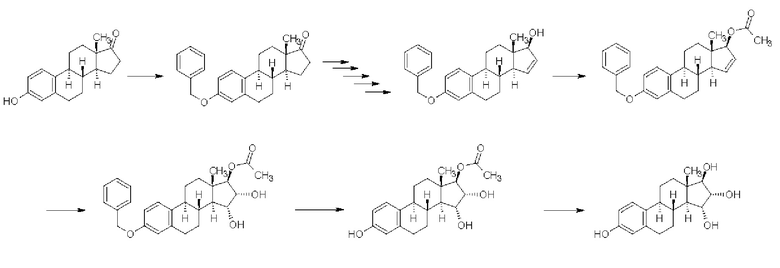
В примерах, приведенных в патентной заявке WO 2013/034780 А2 (Crystal Pharma) - Реакционная схема 5 - в качестве окислителя для цис-гидроксилирования при температуре 55-60°С применяли тетраоксид осмия, зафиксированный на поли-4-винилпиридине (сокращенно "ПВП"). В случае окисления Δ15-17-ацетоксипроизводного тетраоксидом осмия в реакционной смеси было обнаружено соотношение изомеров 15α,16α/15β,16β, составляющее 80/20, и продукт получали с выходом 88%, однако, данные по чистоте приведены не были:
Реакционная схема 5
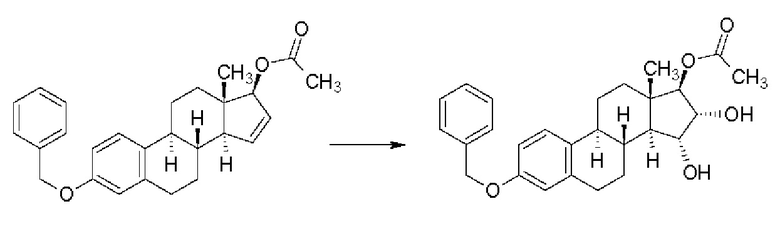
В случае Δ15-17β-гидроксипроизводного сообщали о выходе 62% и соотношении изомеров 15α,16α/15β,16β = 90/10, но данные по чистоте приведены не были:
Реакционная схема 6
В случае простого бензилового эфира с выходом 99% получали смесь изомеров 15α,16α/15β,16β в отношении 90/10, но данные по чистоте приведены не были:
Реакционная схема 7
В случае 3-бензоильного соединения получали смесь изомеров 15α,16α/15β,16β в отношении 90/10 с выходом 92%, но данные по чистоте приведены не были:
Реакционная схема 8
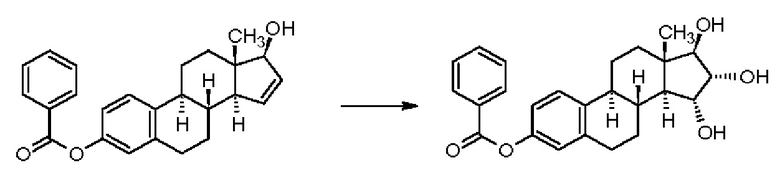
В случае простого трет-бутилдиметилсилильного эфира получали смесь изомеров 15α,16α/15β,16β в отношении 90/10 с выходом 101%, но данные по чистоте приведены не были:
Реакционная схема 9
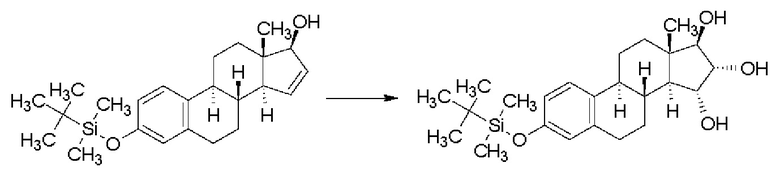
В случае простого 1-бутоксиэтильного эфира получали смесь изомеров 15α,16α/15β,16β в отношении 90/10 с выходом 96,5%, однако, данные по чистоте приведены не были:
Реакционная схема 10
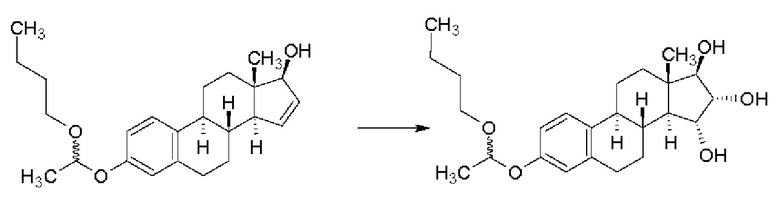
В документе WO 2013/034780 А2 (Crystal Pharma) не приведена информация о чистоте описанных в этой работе производных эстетрола, защищенных по группе 3-ОН, которые имеют формулу (I), и также не описано получение из них эстетрола; кроме того, содержание этой работы не позволяет получать эстетрол с чистотой, подходящей для действующего вещества.
В патентной заявке WO 2015/040051 А1 (Crystal Pharma) показано цис-гидроксилирование через Δ15-3,17β-дигидроксипроизводные, имеющие одинаковые или различающиеся защитные группы. Указаны чрезвычайно низкие (1-9%) степени превращения при действии окислителей на основе перманганата калия. Применение в качестве окислителя тетраоксида осмия на поли-4-винилпиридине позволило с удовлетворительной степенью превращения получить производные эстетрола, защищенные по гидроксильным группам в положениях 3 и 17. Снятие защиты по отдельности привело к получению эстетрола в виде смеси изомеров 15α,16α/15β,16β в отношении 98/2-99/1. В описании не приведены данные о чистоте. Кроме того, описание не содержит какой-либо информации о способе получения эстетрола с чистотой действующего вещества из образованных промежуточных соединений.
Все вышеизложенное позволяет сделать заключение либо об отсутствии способа получения эстетрола лекарственной чистоты, либо о том, что получение чистого действующего вещества может быть осуществлено с недостаточным выходом и плохими экономическими показателями.
В современных фармакопейных нормативах в настоящее время рекомендован ряд способов испытаний, таких как определение чистоты способами на основе высокоэффективной жидкостной хроматографии, и, кроме того, эти нормативы устанавливают и ограничивают число и количества загрязняющих веществ. В случае стероидных действующих веществ в общих требованиях указана предельная величина общего содержания всех загрязнений, которая составляет 0,5%, в то время как предельная величина индивидуального загрязняющего вещества составляет 0,10%. Для соответствия требованиям к качеству целевого продукта необходимо синтезировать ключевое промежуточное соединение (соединения) подходящей чистоты, что особенно важно для соединения, которое плохо кристаллизуется и плохо поддается очистке, такого как, например, эстетрол.
Ввиду вышесказанного, все еще существует необходимость создания альтернативного промышленного способа получения эстетрола, который позволяет получать эстетрол с высокой чистотой и может быть осуществлен посредством синтеза через промежуточные соединения, имеющие подходящие свойства (например, способность к кристаллизации, очистке, извлечению и повышению выхода).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к способу получения эстетрола, имеющего формулу (I), его производных, защищенных в положениях 3,15α,16α,17β, которые имеют общую формулу (III), и его 3-гидроксипроизводных, защищенных в положениях 15α,16α,17β, которые имеют общую формулу (IV), и к промежуточным соединениям, имеющим общие формулы (III) и (IV), применяемым в способе.
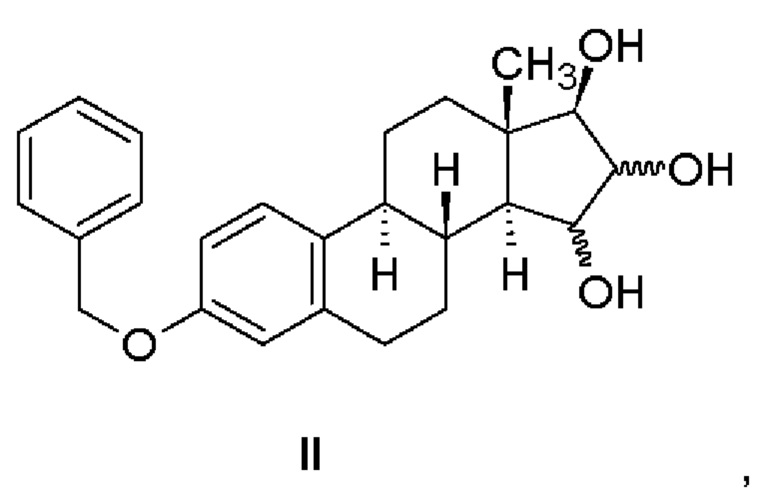
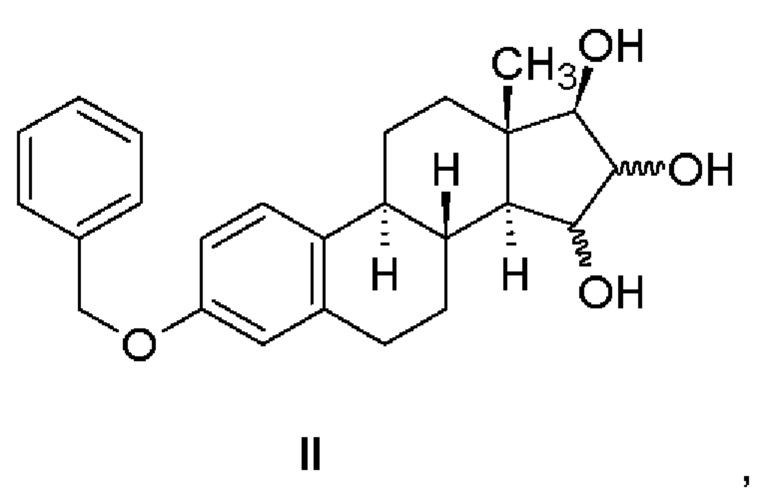
Промышленный способ согласно изобретению состоит в синтезе эстетрола, имеющего формулу (I), исходя из соединения, имеющего формулу (II).
Защищенные в положении 3 триольные производные, имеющие формулу (II), могут быть получены в соответствии со способом, описанным в патентной заявке WO 2013/034780 А2 (Crystal Pharma) - Реакционная схема 7 - из 3-бензилокси-эстра-1,3,5(10),15-тетраен-17-ола.
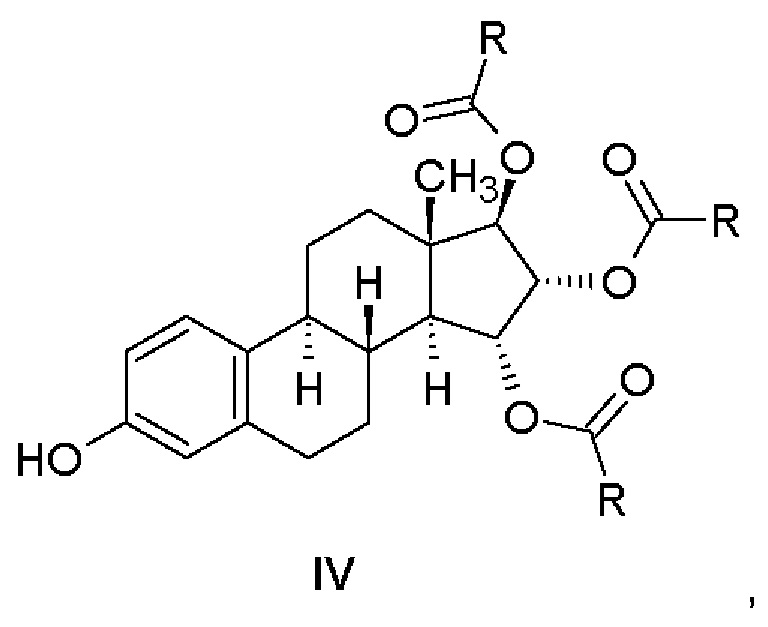
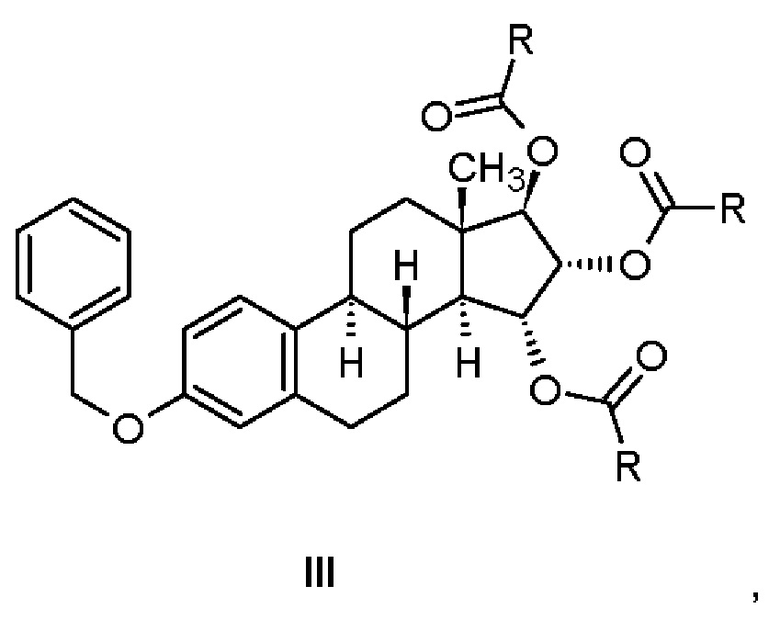
Соединения, имеющие общую формулу (III), получают ацилированием соединения, имеющего формулу (II), и последующей очисткой.
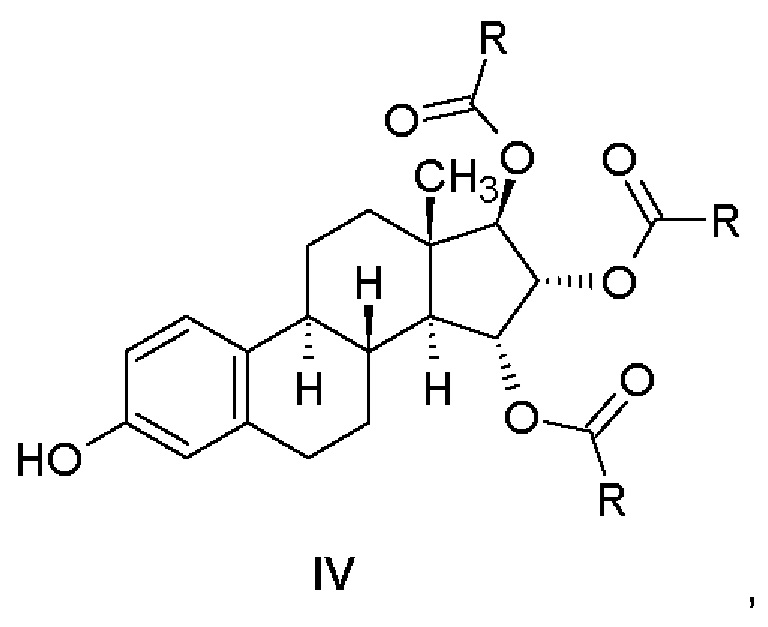
Соединения, имеющие общую формулу (IV), получают отщеплением бензильной группы от соединений, имеющих общую формулу (III).
Эстетрол, имеющий формулу (I), получают щелочным гидролизом соединений, которые имеют общую формулу (IV).
Изобретение также относится к промежуточным соединениям, имеющим общие формулы (III) и (IV), получаемым способом, описанным выше.
Изобретение также относится к применению эстетрола, имеющего формулу (I), получаемого способом согласно изобретению, при получении фармацевтической композиции.
СВЕДЕНИЯ. ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Изобретение относится к способу получения эстетрола, имеющего формулу (I), его производных, защищенных в положениях 3,15α,16α,17β, которые имеют общую формулу (III), и его 3-гидроксипроизводных, защищенных в положениях 15α,16α,17β, которые имеют общую формулу (IV), и к промежуточным соединениям, имеющим общие формулы (III) и (IV), применяемым в способе.
Промышленный способ согласно изобретению состоит в синтезе эстетрола, имеющего формулу (I), исходя из соединения, имеющего формулу (II), в соответствии со следующей реакционной схемой, в которой R обозначает метильную группу или атом водорода:
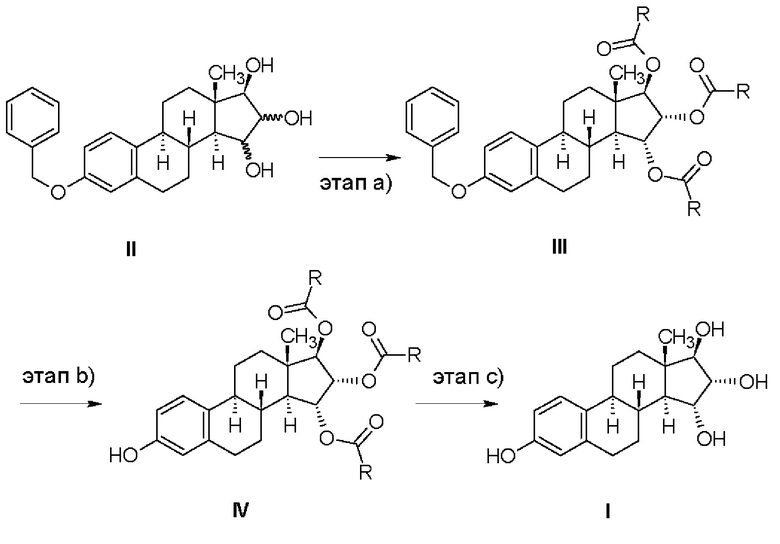
Производные триолы, защищенные в положении 3, имеющие формулу (II), могут быть получены способом, описанным в патентной заявке WO 2013/034780 А2 (Crystal Pharma), т.е. приведенное в указанной работе в качестве примера соединение 7,3-бензилокси-эстра-1,3,5(10),15-тетраен-17-ол, окисляют окислителем, таким как осмиат калия или тетраоксид осмия, необязательно в присутствии соокислителя, такого как триалкиламин-N-оксид, такого как триметил- или триэтиламин-N-оксид, в смешивающемся с водой растворителе, таком как 2-бутанон, ацетон, тетрагидрофуран, трет-бутанол, предпочтительно в 2-бутаноне.
Этап (а): Ацилирование 15,16,17-триольного производного
На этапе а) способа согласно изобретению производное эстетрола, защищенное в положениях 3,15α,16α,17β, представленное общей формулой (III), получают, выделяя или не выделяя, ацилированием из соответствующего 15,16,17-триола, защищенного в положении 3, который имеет формулу (II), в подходящем растворителе, используя подходящий реагент.
Применяемый при ацилировании растворитель представляет собой растворитель, выбранный из группы, состоящей из алифатических и ароматических углеводородов, галогенированных углеводородов, сложных эфиров и простых эфиров, предпочтительно не смешивающихся с водой растворителей, таких как толуол, дихлорметан или этилацетат.
В одном из примеров осуществления реагент, применяемый для ацилирования при R = метил (ацетилирование), предпочтительно представляет собой уксусный ангидрид, ацетилхлорид или ацетилбромид.
В другом примере осуществления реагент, применяемый для ацилирования при R = водород (формилирование), предпочтительно представляет собой смешанный ангидрид уксусной кислоты и муравьиной кислоты.
Ацилирование проводят в присутствии основания-амина, предпочтительно пиридина или 4-диметиламинопиридина.
Ацилирование проводят в инертной атмосфере, предпочтительно в атмосфере N2.
В одном из примеров осуществления этап ацилирования дополнительно включает кристаллизацию получаемого соединения, имеющего формулу (III), из С1-3-спиртов, предпочтительно метанола.
В другом примере осуществления этап (а) может быть выполнен последовательно по отношению к вышеуказанному дигидроксилированию, после чего выполняют ацилирование без очистки и/или выделения промежуточных соединений, т.е. соединений, имеющих формулу (II), получая при этом конечный продукт высокой чистоты с хорошим выходом. Это особенно полезно для промышленного применения, при котором уменьшение количества этапов способа приводит к экономическому эффекту и к упрощению способа, поскольку устраняет необходимость проведения таких этапов, как очистка и/или выделение между двумя другими этапами.
Настоящее изобретение относится к соединению, имеющему общую формулу (III), в котором R представляет собой метил или атом водорода, т.е. к (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетату (Пример 1) и к (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриформиату (Пример 3).
Этап (b): Отщепление бензильной группы от 3,15,16,17-зашищенного производного
На этапе b) способа согласно изобретению 3-гидроксипроизводное эстетрола, защищенное в положениях 15α,16α,17β, имеющее общую формулу (IV), получали удалением защитной бензильной группы, находящейся в положении 3 производного, представленного общей формулой (III), посредством гидрирования с переносом водорода или каталитического гидрирования.
В одном из примеров осуществления отщепление бензильной группы осуществляют каталитическим гидрированием газообразным водородом, и катализатор выбирают из группы, состоящей из палладия или палладия на носителе (угле, оксиде алюминия и т.д.). Катализатор предпочтительно представляет собой Pd/C. Растворитель, применяемый для каталитического гидрирования, выбран из группы, состоящей из спиртов, сложных эфиров и кетонов, и предпочтительно представляет собой этилацетат.
В другом примере осуществления отщепление бензильной группы осуществляют гидрированием с переносом водорода с применением в качестве реагента циклогексена. Растворитель, применяемый для гидрирования с переносом водорода, представляет собой спирт, предпочтительно этанол.
Этап отщепления бензильной группы дополнительно включает кристаллизацию получаемого соединения, имеющего общую формулу (IV), из сложных эфиров, углеводородов, спиртов или смесей указанных веществ, предпочтительно из смеси этилацетат/н-гептан.
Настоящее изобретение относится к соединению, имеющему общую формулу (IV), в котором R представляет собой водород, т.е. к (15α,16α,17β)-3-гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриформиату (Пример 4).
Этап (с): Гидролиз 15,16,17-ацилзащищенного производного
На этапе с) способа согласно изобретению эстетрол, имеющий формулу (I), получают снятием защиты с производного, имеющего общую формулу (IV), в щелочной среде в присутствии карбоната щелочного металла или гидроксидов щелочных металлов в подходящем растворителе.
Применяемый для гидролиза растворитель выбран из группы, состоящей из воды, растворителя спиртового типа или их смеси, предпочтительно из С1-3-спиртов, более предпочтительно из смеси метанола и воды.
В одном из примеров осуществления гидролиз выполняют в присутствии карбоната щелочного металла или гидрокарбоната щелочного металла, предпочтительно карбоната калия.
В другом примере осуществления гидролиз выполняют в присутствии алкоголята щелочного металла или гидроксида щелочного металла, предпочтительно гидроксида натрия или лития.
На основании вышеизложенного, специалист в данной области техники может легко выбрать реагенты, растворители, температуру, давление и другие условия реакции. Исходные материалы, реагенты и растворители, применяемые в способе согласно изобретению, коммерчески доступны и/или могут быть успешно синтезированы специалистом в данной области техники. Чистоту продуктов, описываемых в примерах, определяли способами разделения высокоэффективной жидкостной хроматографией, которые известны специалистам в данной области техники, применяя в качестве неподвижной фазы наиболее широко используемые силикагели (например, Ascentis, Kintex), а в качестве подвижной фазы с линейным градиентом - многокомпонентную смесь обычно применяемых элюентов (например, воды, метанола, ацетонитрила).
Несмотря на то что имеющее формулу (I) соединение и имеющие формулу (II) 15α,16α,17β-триолы, защищенные в положении 3, которые описаны в литературе, плохо кристаллизуются, неожиданно было обнаружено, что соединения, имеющие общие формулы (III) и (IV), кристаллизуются хорошо и могут быть очищены с высокими выходами и могут быть с высокой селективностью отделены от изомерного побочного продукта.
Согласно изобретению, проведение этапов (а)-(с) более предпочтительно, если R обозначает метильную группу.
Другой пример осуществления изобретения относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, для получения фармацевтической композиции.
Термин "фармацевтическая композиция" (или "композиция") означает смесь или раствор, включающий активный ингредиент, такой как соединение, имеющее формулу (I), для введения пациенту, такому как человек, которому это необходимо, предпочтительно женщине в период пременопаузы или постменопаузы, в терапевтически эффективном количестве совместно с фармацевтически приемлемыми вспомогательными веществами (WO 2016/203006 А1, WO 2016/203009 А1, WO 2016/203044 А1).
Фармацевтические композиции согласно настоящему изобретению могут быть произведены в виде различных дозированных лекарственных форм, таких как твердые или жидкие дозированные лекарственные формы. Предпочтительно фармацевтическая композиция представляет собой твердую дозированную лекарственную форму для перорального введения, такую как таблетки (например, таблетки для буккального и сублингвального (подъязычного) введения, шипучие, жевательные таблетки, таблетки для рассасывания).
Имеющее формулу (I) соединение согласно настоящему изобретению может быть введено вместе с фармацевтически приемлемыми вспомогательными веществами в виде одной или нескольких дозировок.
Изобретение также относится к фармацевтическим композициям, включающим соединение, имеющее формулу (I), в комбинации с одним или более, предпочтительно с одним дополнительным активным ингредиентом. Комбинированная композиция включает соединение, имеющее формулу (I), вместе с одним или более другими активными ингредиентами в одной дозированной лекарственной форме, также включающей фармацевтически приемлемые вспомогательные вещества. Другой активный ингредиент предпочтительно представляет собой прогестагенное соединение, примеры которого включают, без ограничений, прогестерон, левоноргестрел, норгестимат, норэтистерон, дидрогестерон, дроспиренон, 3-бета-гидроксидезогестрел, этоногестрел, 17-дезацетилноргестимат, 19-норпрогестерон, ацетоксипрегненолон, аллилэстренол, анагестон, хлормадинон, ципротерон, демегестон, дезогестрел, диеногест, дигидрогестерон, диметистерон, этистерон, этинодиола диацетат, флугестона ацетат, гастринон, гестоден, гестринон, гидроксиметилпрогестерон, гидроксипрогестерон, линэстренол, медрогестон, медроксипрогестерон, мегестрол, меленгестрол, номегестрол, норэтинодрел, норгестрел (включая d-норгестрел и d1-норгестрел), норгестриенон, норметистерон (норметадрон), хингестанол, (17-альфа)-17-гидрокси-11-метилен-19-норпрегна-4,15-диен-20-ин-3-он, тиболон, тримегестон, алгестон ацетофенид, несторон, промегестон, сложные эфиры 17-гидроксипрогестерона, 19-нор-17-гидроксипрогестерон, 17-альфа-этинилтестостерон, 17-альфа-этинил-19-нор-тестостерон, d-17-бета-ацетокси-13-бета-этил-17-альфа-этинил-гон-4-ен-3-он-оксим. Более предпочтительно, прогестагенный агент представляет собой дроспиренон. Другим активным ингредиентом также может быть кальций или витамины, предпочтительно, например, и витамин D.
Дозировка, необходимая для достижения требуемого терапевтического эффекта, может варьироваться в широких пределах, и в каждом случае ее следует адаптировать к индивидуальным требованиям, принимая во внимание тяжесть заболевания, состояние и массу пациента, принимающего терапию, чувствительность к активному ингредиенту, путь введения и количество введений в сутки. Фармацевтические композиции, содержащие имеющий формулу (I) активный ингредиент согласно изобретению, обычно включают от 0,01 до 20 мг, предпочтительно от 1,5 мг до 15 мг, более предпочтительно 15 мг активного ингредиента в стандартной лекарственной форме. Если в композиции в качестве другого активного ингредиента также содержится дроспиренон, то композиция обычно содержит от 0,01 до 10 мг, предпочтительно от 1,5 мг до 5 мг, более предпочтительно 3 мг дроспиренона в стандартной лекарственной форме. Комбинированная композиция также может содержать витамин D.
Фармацевтические композиции согласно настоящему изобретению могут быть произведены уже известными способами, например, гранулированием (мокрым или сухим) или прессованием. Фармацевтические композиции согласно настоящему изобретению могут быть произведены традиционным способом с применением одного или более физиологически (или фармацевтически) приемлемых вспомогательных веществ. Может быть применена любая методика и любое вспомогательное вещество, хорошо известное в данной области техники; так, вспомогательные вещества выбирают из следующих категорий, таких как, без ограничений, вспомогательные вещества для получения таблеток, связующие вещества для таблеток, агенты-модификаторы высвобождения, дезинтегрирующие вещества, скользящие вещества, смазывающие вещества, подсластители, вкусовые добавки, ароматизирующие добавки или материалы покрытия. Примерами подходящих фармацевтических вспомогательных веществ являются крахмал, микрокристаллическая целлюлоза, тальк, глюкоза, лактоза, желатин, оксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, производные целлюлозы, хлорид натрия, глицерин, пропиленгликоль, вода, этанол и подобные вещества. Указанные выше вспомогательные вещества и различные способы получения являются лишь репрезентативными примерами. Также могут быть применены другие материалы и способы обработки, известные в данной области техники.
Один из примеров осуществления изобретения относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, в изготовлении медикамента для гормонозаместительной терапии, для применения в способе лечения сухости влагалища, способе лечения перименопаузальных симптомов (например, приступообразного ощущения жара, ночной потливости), в способе контрацепции, способе повышения либидо, способе лечения кожных покровов и улучшения заживления ран, способе лечения или профилактики аутоиммунного нарушения, опухолей молочных желез, рака предстательной железы и колоректальных опухолей или в целях применения для нейрозащиты.
Другой пример осуществления изобретения предпочтительно относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, в изготовлении медикамента для применения в контрацепции, более предпочтительно в комбинации с дроспиреноном (WO 2019/154899 А1).
Дополнительный пример осуществления изобретения предпочтительно относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, в изготовлении медикамента для применения в гормонозаместительной терапии.
Другой пример осуществления изобретения предпочтительно относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, в изготовлении медикамента для применения для нейрозащиты (например, при неонатальной энцефалопатии).
Другой пример осуществления изобретения относится к применению эстетрола, имеющего формулу (I), получаемого описанным выше способом, в изготовлении медикамента для применения в лечении перименопаузальных симптомов, более предпочтительно в комбинации с дроспиреноном и витамином D.
Сравнительный пример
Получение смеси изомеров эстетрола ((15ξ,16ξ,17β)-эстра-1,3,5(10)-триен-3,15,16,17-тетрола) согласно WO 2013/034780 А2 (Crystal Pharma)
а) Цис-гидроксилирование
(15α,16α,17β)- и (15β,16β,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триол
20,0 г (55,5 ммоль) 3-бензилокси-эстра-1,3,5(10),15-тетраен-17-ола (WO 2004/041839 (Pantarhei), Пример 7) растворяли в 1400 мл тетрагидрофурана при 20-25°С в атмосфере N2, затем к реакционной смеси добавляли раствор 14 мл тетраоксида осмия (OsO4) в трет-бутаноле концентрацией 2 масс./об. (содержание OsO4 = 280 мг) и 11 г N-метилморфолин-N-оксида и 150 мл воды, и перемешивали в течение 24 часов в атмосфере N2 при 20-25°С. Протекание реакции отслеживали с помощью ТСХ (т.е. тонкослойной хроматографии) (н-гептан : ацетон 1:1).
Обработка: в раствор добавляли по каплям 140 мл 5%-ного раствора Na2S2O5, и туда добавляли 100 мг активированного угля; смесь перемешивали в течение 30 минут, и смесь фильтровали через слой целита. Органические растворители удаляли из фильтрата отгонкой и добавляли 400 мл дихлорметана. Фазы разделяли. Органическую фазу промывали 200 мл 10%-ной соляной кислоты и 200 мл насыщенного раствора хлорида натрия, затем сушили и концентрировали. Сконцентрированный остаток растворяли в 200 мл метанола и добавляли по каплям в 2 л воды при температуре 0-5°С, перемешивали в течение 1 часа, фильтровали, и собранные на фильтре кристаллы промывали 20 мл воды. Материал сушили в вакууме при 40°С до постоянной массы. Получали 19,68 г (89,86%) желтовато-белых кристаллов.
Чистота (ВЭЖХ (т.е. по данным высокоэффективной жидкостной хроматографии)): 85,18% ααβ-изомера, 5,43% βββ-изомера (оценка по площади пика) (отношение 94,0:6,0).
Соединение (15ξ,16ξ,17β)-эстра-1,3,5(10)-триен-3,15,16,17-тетрол цементировался (кристаллы слипались друг с другом, препятствуя, таким образом, фильтрованию и извлечению материала, то есть, затрудняя его очистку) во время перекристаллизации в органических растворителях, обычно в углеводородах, простых эфирах, сложных эфирах, спиртах или смесях таких веществ. Как правило, это соединение можно кристаллизовать только из воды или смеси смешивающихся с водой растворителей (обычно спиртов), но и в этом случае значительных улучшений соотношения изомеров достичь невозможно.
b) Гидрирование
(15α,16α,17β)- и (15α,16α,17β)- эстра-1,3,5(10)-триен-15,16,17-триол
19,5 г (15ξ,16ξ,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триола растворяли в 400 мл метанола при 20-25°С в атмосфере N2. В 100 мл сильно охлажденного метанола суспендировали 2,0 г катализатора 10% Pd/C, который затем добавляли в раствор триола. Атмосферу N2 заменяли атмосферой Н2, и реакционную смесь перемешивали при 20-25°С в течение 6 часов при атмосферном давлении.
Обработка: Катализатор отфильтровывали, реакционную смесь концентрировали до 45 мл под уменьшенным давлением, добавляли 45 мл воды, и смесь перемешивали при 0-5°С в течение 1 часа, затем фильтровали и дважды промывали на фильтре 20 мл воды, сушили до постоянной массы, и получали, таким образом, 14,5 г (96,67%) белого кристаллического продукта.
Чистота (ВЭЖХ): 87,53% ααβ-изомера, 5,46% βββ-изомера (площадь) (отношение 94,13:5,87).
Ниже изобретение дополнительно проиллюстрировано приведенными неограничивающими примерами. Из приведенного выше описания и примеров специалист в данной области техники может выделить основные признаки изобретения и, не отступая от предмета и объема изобретения, может внести определенные изменения и модификации для адаптации изобретения к различным областям применения и обстоятельствам. Таким образом, изобретение ограничено не описанными ниже иллюстративными примерами, а объемом, определяемым прилагаемыми пунктами формулы изобретения.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1
(15α,16α,17β)-3-(Бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетат
Способ А (с выделением)
а) Цис-гидроксилирование
(15α,16α,17β)- и (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триол
40 мг дигидрата осмиата калия (K2OsO4⋅2H2O) суспендировали в 100 мл 2-бутанона (метилэтилкетона) при 20-25°С в атмосфере N2 и к нему добавляли 7,7 мл очищенной воды и 1,1 г дигидрата триметиламин-N-оксида. Два грамма (2,0 г, 5,5 ммоль) 3-бензилокси-эстра-1,3,5(10),15-тетраен-17-ола (WO 2004/041839 (Pantarhei), Пример 7) растворяли в 40 мл 2-бутанона и добавляли по каплям в реакционную смесь. Затем реакционную смесь перемешивали при 20-25°С в течение 28 часов в атмосфере N2. Протекание реакции отслеживали с помощью ТСПХ (тонкослойной препаративной хроматографии) (н-гептан : ацетон 1:1).
Обработка: к смеси добавляли 25 мл 10%-ного раствора Na2S2O5, после чего добавляли 100 мг активированного угля и затем перемешивали в течение 1 часа. Фильтровали через слой целита и затем добавляли EtOAc и 10%-ный раствор HCl. Фазы разделяли, водную фазу экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным раствором NaCl и 10%-ным раствором Na2S2O5. Сушили над Na2SO4, фильтровали, затем концентрировали. Таким образом, получали 1,8 г (81,8%) продукта.
Чистота (ВЭЖХ): 85,0% ααβ-изомера, 9,9% βββ-изомера (площадь) (отношение 89,6:10,4)
b) Ацилирование
(15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триолтриацетат
1,0 г (2,53 ммоль) (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триола растворяли в 15 мл дихлорметана в атмосфере N2. Добавляли 1,5 мл триэтиламина, 6,0 мл уксусной кислоты и 72 мг 4-диметиламинопиридина и перемешивали в течение 2 часов. Протекание реакции отслеживали с помощью ТСХ (толуол : ацетон 4:1).
Обработка: в смесь добавляли по каплям 3 мл этанола и перемешивали в течение 30 минут, затем добавляли 10%-ный раствор NaHCO3 и перемешивали в течение еще 30 минут. Фазы разделяли, и органическую фазу дважды промывали 10%-ным раствором NaHCO3 и затем насыщенным солевым раствором. Сушили над Na2SO4, фильтровали, растворитель заменяли на МеОН и кристаллизовали из МеОН. После фильтрования и сушки получали 1,2 г материала. Для достижения подходящего соотношения изомеров продукт еще два раза перекристаллизовывали из метанола, получая, таким образом, 1,1 г (84,46%) продукта.
Чистота (ВЭЖХ): 99,2% ααβ-изомера, 0,14% βββ-изомера (площадь).
Способ В (без выделения)
а) Цис-гидроксилирование
Изомерная смесь (15α,16α,17β), (15β,16β,17β) 3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триола
30,03 г (83,3 ммоль) 3-бензилокси-эстра-1,3,5(10),15-тетраен-17-ола (WO 2004/041839 (Pantarhei), Пример 7) растворяли в 480 мл 2-бутанона (метилэтилкетона) при 20-25°С в атмосфере N2 и затем добавляли 600 мг дигидрата осмиата калия (K2OsO4⋅2H2O), 48,0 мл очищенной воды и 16,5 г дигидрата триметиламин-N-оксида. Затем реакционную смесь перемешивали при 40-45°С в течение 7 часов в атмосфере N2. Протекание реакции отслеживали с помощью ТСХ (н-гептан : ацетон 1:1).
Обработка: в смесь при 40-45°С по каплям добавляли 300 мл 10%-ного (масс./об.) раствора метабисульфита натрия (пиросульфита натрия) и перемешивали в течение 1 часа. Затем суспензию фильтровали через слой целита, и фильтр промывали 2-бутаноном. Затем 2-бутанон удаляли из фильтрата отгонкой. К остатку добавляли 600,0 мл этилацетата и 300 мл 10%-ного (масс./об.) раствора гидрокарбоната натрия (30 г NaHCO3), и после энергичного перемешивания в течение нескольких минут и последующего выдерживания фазы разделяли. Водную фазу дважды промывали этилацетатом. Объединенную органическую фазу промывали смесью 1%-ного (масс./об.) раствора ЭДТА-тетра Na соли и насыщенного солевого раствора. После разделения фаз этилацетатную органическую фазу концентрировали до финального объема, составляющего 450 мл, при этом также обезвоживая эту фазу. Продукт не выделяли, а направляли далее в реакцию ацилирования.
b) Ацилирование
(15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетат
72,0 мл уксусного ангидрида, 48 мл триэтиламина и 1,8 г 4-диметиламинопиридина добавляли в этилацетатный раствор, полученный на этапе а), и затем перемешивали при 35-40°С в течение 3 часов в атмосфере N2. Протекание реакции отслеживали с помощью ТСХ (толуол : ацетон 4:1).
Обработка: В смесь по каплям добавляли 24 мл этанола, перемешивали в течение 30 минут, затем охлаждали до 20-25°С, после чего добавляли 240 мл очищенной воды и 60 мл 10%-ного (масс./об., d=1,047, 17,88 г концентрированной HCl) раствора соляной кислоты, затем энергично перемешивали в течение нескольких минут и выдерживали, после чего фазы разделяли. Водную фазу экстрагировали этилацетатом. Объединенную органическую фазу промывали смесью 10% (масс./об.) раствора гидрокарбоната натрия и насыщенного солевого раствора, и фазы разделяли. Органическую фазу сушили над Na2SO4, осветляли добавлением оксида алюминия, силикагеля и активированного угля и перемешиванием при 20-25°С в течение 1 часа. Затем отфильтровывали осветляющие агенты, и фильтр промывали этилацетатом.
Фильтрат концентрировали под уменьшенным давлением, затем растворитель концентрировали и удаляли отгонкой, заменяя растворитель метанолом, и, наконец, материал кристаллизовали из чистого метанола. Полученный неочищенный продукт перекристаллизовывали без сушки.
с) Перекристаллизация
Неочищенный продукт, полученный на этапе b), растворяли в дихлорметане, метанол удаляли отгонкой, и, наконец, кристаллизовали из чистого метанола. Эту операцию повторяли еще один раз. Таким образом получали 30,4 г (69,8%) белых кристаллов.
Чистота (ВЭЖХ): 99,2% ααβ-изомера, 0,14% βββ-изомера (площадь).
Температура плавления: 156,5-157,5°С.
ЭИ-МСВР (электронная ионизация - масс-спектрометрия высокого разрешения): Вычислено для C31H36O7 [М+]: 520,24555; найдено: 520,24459; дельта = -1,86 м.д. (миллионные доли).
1Н ЯМР (499,9 МГц, CDCl3) δ = 5,39 (1 Н, dd, J=8,4 Гц, J=6,6 Гц, Н-16), 5,16 (1Н, dd, J=10,4 Гц, J=8,4 Гц, Н-15), 5,01 (1Н, d, J=6,6 Гц, Н-17), 2,08 (3Н, s, 17-ацетил), 2,06 (3Н, s, 15-ацетил), 2,04 (3Н, s, 16-ацетил), 0,94 (3Н, s, Н-18);
13С ЯМР (125,7 МГц, CDCl3) δ = 169,8 (17-ацетил СО С-20), 169,0 (15-ацетил СО), 168,7 (16-ацетил СО), 83,1 (С-17), 72,5 (С-16), 69,8 (С-15), 51,4 (С-14), 39,2 (С-13), 19,9 (17-ацетил-СНз), 19,7 (15-ацетил-СН3), 19,6 (16-ацетил-СН3), 13,5 (С-18).
Пример 2
(15α,16α,17β)-3-Гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриацетат
Способ А
25,7 г (49,36 ммоль) (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетата (Пример 1) растворяли в 315 мл этилацетата при 20-25°С в атмосфере N2. В 19 мл сильно охлажденного этилацетата суспендировали 770 мг катализатора, 10%-ного палладия на угольном носителе, который затем добавляли в раствор. Атмосферу N2 заменяли атмосферой Н2, и реакционную смесь перемешивали при 20-25°С в течение 3 часов при атмосферном давлении.
Обработка: катализатор отфильтровывали, промывали этилацетатом и концентрировали под уменьшенным давлением до конечного объема, затем добавляли н-гептан, и суспензию выдерживали при 0-5°С в течение 1 часа, затем фильтровали, и кристаллический продукт промывали на фильтре н-гептаном и сушили при 40°С в вакууме до постоянной массы. Таким образом получали 19,88 г (93,55%) белого кристаллического продукта.
Чистота (ВЭЖХ): 99,42% ααβ-изомера, 0,04% βββ-изомера (площадь).
Способ В
0,5 г (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетата (Пример 1) суспендировали в 14 мл этанола при 20-25°С, затем добавляли 0,5 мл циклогексена и 38 мг катализатора 10% Pd/C, и затем перемешивали при температуре кипения в течение 1 часа. Протекание реакции отслеживали с помощью ТСХ (толуол : ацетон 4:1).
Обработка: Катализатор отфильтровывали из реакционной смеси, и смесь концентрировали досуха. Таким образом получали 0,41 г (99,17%) белого кристаллического продукта.
Чистота (ВЭЖХ): 97,99% ααβ-изомера, 0,14% βββ-изомера (площадь).
Температура плавления: 181,5-185,5°С.
ЭИ-МСВР: Вычислено для С24Н30О7 [М+]: 430,19860; найдено: 430,19927; дельта = 1,55 м.д.
1Н ЯМР (499,9 МГц, CDCl3) δ = 5,41 (1 Н, dd, J=8,4 Гц, J=6,6 Гц, Н-16), 5,18 (1Н, dd, J=10,5 Гц, J=8,4 Гц, Н-15), 5,03 (1Н, d, J=6,6 Гц, Н-17), (3Н, s, 17-ацетил), 2,10 (3Н, s, 15-ацетил), 2,07 (3Н, s, 16-ацетил), 1,77 (1Н, t, J=11,1 Гц, Н-14), 0,95 (3H,s, Н-18);
13С ЯМР (125,7 МГц, CDCl3) δ = 170,9 (17-ацетил СО), 170,1 (15-ацетил СО), 169,8 (16-ацетил СО), 84,1 (С-17), 73,5 (С-16), 70,8 (С-15), 52,4 (С-14), 40,2 (С-13), 20,9 (17-ацетил-СН3), 20,7 (15-ацетил-СН3), 20,6 (16-ацетил-СН3), 14,5 (С-18).
Пример 3
(15α,16α,17β)-3-(Бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриформиат
5,00 г (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триола (Пример 1, Способ "А", этап а)) растворяли в 73 мл пиридина и охлаждали до 0°С, и затем к нему через капельную воронку при температуре от 0 до 10°С в течение приблизительно 25 минут добавляли смесь смешанного ангидрида, полученного из 49 мл муравьиной кислоты и 18,3 мл уксусного ангидрида, охлажденную до 0°С. После перемешивания в течение 1 часа к реакционной смеси добавляли 305 мл воды, и полученный белый осадок отфильтровывали и промывали водой. Сухой неочищенный продукт весил 5,65 г (93,23%).
Неочищенный продукт в соответствии с Примером 1, Способом В, этапом с) перекристаллизовывали из метанола, получая 3,92 г (69,4%) чистого названного в заглавии примера продукта в виде белых кристаллов.
Чистота (ВЭЖХ): 99,2% ααβ-изомера, 0,05% βββ-изомера (площадь).
Температура плавления: 153,5-154,3°С
ЭИ-МСВР: М=478,19866; дельта = 0,06 м.д.; C28H30O7
1Н ЯМР (499,9 МГц, CDCl3) δ = 5,41 (1 Н, dd, J=8,4 Гц, J=6,6 Гц, Н-16), 5,18 (1Н, dd, J=10,5 Гц, J=8,4 Гц, Н-15), 5,03 (1Н, d, J=6,6 Гц, Н-17), (3Н, s, 17-ацетил), 2,10 (3Н, s, 15-ацетил), 2,07 (3Н, s, 16-ацетил), 1,77 (1Н, t, J=11,1 Гц, Н-14), 0,95 (3H,s, Н-18);
13С ЯМР (125,7 МГц, CDCl3) δ = 170,9 (17-ацетил СО), 170,1 (15-ацетил СО), 169,8 (16-ацетил СО), 84,1 (С-17), 73,5 (С-16), 70,8 (С-15), 52,4 (С-14), 40,2 (С-13), 20,9 (17-ацетил-СН3), 20,7 (15-ацетил-СН3), 20,6 (16-ацетил-СН3), 14,5 (С-18).
Пример 4
(15α,16α,17β)-3-Гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриформиат
5,0 г (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриформиата (Пример 3) растворяли в 150 мл этилацетата при 20-25°С в атмосфере N2. В 5 мл сильно охлажденного этилацетата суспендировали 380 мг катализатора 10% Pd/C, который добавляли в раствор. Атмосферу N2 заменяли атмосферой Н2, и реакционную смесь перемешивали при 20-25°С в течение 4 часов при атмосферном давлении.
Обработка: катализатор отфильтровывали, реакционную смесь концентрировали под уменьшенным давлением до четверти объема (38 мл), и затем добавляли 52 мл н-гептана. После перемешивания при 0-5°С в течение 1 часа, смесь фильтровали, осадок на фильтре дважды промывали 16 мл н-гептана и сушили до постоянной массы, получая, таким образом, 3,51 г (94%) белого кристаллического продукта.
Чистота (ВЭЖХ): 99,42% ααβ-изомера, 0,04% βββ-изомера (площадь).
Температура плавления: 234-235°С
МС (масс-спектрометрия): М-Н=387 (электрораспылительная ионизация)
1Н ЯМР (499,9 МГц, ДМСО-d6 (гексадейтеродиметилсульфоксид)) δ = 8,17 (1Н, s, 17-формил-Н), 8,09 (1Н, s, 15-формил-Н), 8,04 (1Н, s, 16-формил-Н), 5,52 (1Н, t, J=7,4 Гц, Н-16), 5,24 (1Н, dd, J=10,1 Гц, J=8,6 Гц, Н-15), 5,11 (1Н, d, J=6,5 Гц, Н-17), 0,99 (3Н, s, Н-18);
13С ЯМР (125,7 МГц, ДМСО-d6) δ = 159,5 (17-формил-С), 159,3 (15-формил-С), 158,8 (16-формил-С), 82,4 (С-17), 71,7 (С-16), 69,2 (С-15), 51,3 (С-14), 39,6 (С-13), 13,5 (С-18).
Пример 5
Эстетрол ((15α,16α,17β)-эстра-1,3,5(10)-триен-3,15.16,17-тетрол)
Способ А
19,88 г (46,18 ммоль) (15α,16α,17β)-3-гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриацетата (Пример 2) суспендировали в 596 мл метанола при 20-25°С в атмосфере N2, затем порциями добавляли 19,88 г карбоната калия и перемешивали в течение 3 часов. Протекание реакции отслеживали с помощью ТСХ (н-гептан : ацетон 1:1).
Обработка: в смесь добавляли 14,91 мл конц. уксусной кислоты и перемешивали в течение 30 минут, затем добавляли 298 мл воды, и метанол удаляли отгонкой, после чего выпавшие кристаллы выдерживали при 0-5°С в течение 1 часа, отфильтровывали и промывали на фильтре водой. Кристаллы затем сушили при 40°С в вакууме до постоянной массы. Таким образом получали 13,66 г (97,22%) белого кристаллического продукта.
Чистота (ВЭЖХ): 99,67% ααβ-изомера, 0,04% (333 изомера (площадь), суммарное содержание всех загрязняющих веществ < 0,10%.
Способ В
5 г (12,87 ммоль) (15α,16α,17β)-3-гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриформиата (Пример 4) суспендировали в 150 мл метанола при 20-25°С в атмосфере N2, затем порциями добавляли 5,34 г (38,6 ммоль) карбоната калия и перемешивали в течение 3 часов. Протекание реакции отслеживали с помощью ТСХ (н-гептан : ацетон 1:1).
Обработка: в смесь добавляли 4 мл уксусной кислоты и перемешивали в течение 30 минут, затем добавляли 75 мл воды, и метанол удаляли из смеси отгонкой; выпавшие кристаллы выдерживали при 0-5°С в течение 1 часа, затем отфильтровывали и дважды промывали на фильтре 5 мл воды с температурой 0-5°С. Кристаллы затем сушили при 40°С в вакууме до постоянной массы. Таким образом получали 3,80 г (97%) белого кристаллического продукта.
Чистота (ВЭЖХ): 99,67% ααβ-изомера, 0,04% βββ изомера (площадь), суммарное содержание всех загрязняющих веществ < 0,10%.
Температура плавления: 240-243°С
ЭИ-МСВР: Вычислено для C18H24O4 [М+]: 304,16691; найдено: 304,16716; дельта = 0,82 м.д.
1Н ЯМР (499,9 МГц, ДМСО-d6) δ = 4,86 (1Н, d, J=4,8 Гц, ОН (17)), 4,61 (1Н, br s, ОН (16)), 4,26 (1Н, br d, J=3,3 Гц, ОН (15)), 3,55 - 3,78 (2Н, m, Н-16, 15), 3,25 (1Н, dd, J=5,7, 4,7 Гц, Н-17), 1,05 (1Н, dd, J=10,9 Гц, J=9,4 Гц, Н-14), 0,67 (3Н, s, Н-18);
13С ЯМР (125,7 МГц, ДМСО-d6) δ = 86,3 (С-17), 75,0 (С-16), 69,2 (С-15), 55,5 (С-14), 39,5 (С-13), 14,0 (С-18).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (15α,16α,17β)-ЭСТРА-1,3,5(10)-ТРИЕН-3,15,16,17-ТЕТРОЛА (ЭСТЕТРОЛА) И ИНТЕРМЕДИАТЫ В ЭТОМ СПОСОБЕ | 2020 |
|
RU2818561C1 |
| 15,15-ДИАЛКИЛСТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2147306C1 |
| ЭСТРАТРИЕНЫ, СОДЕРЖАЩИЕ МОСТИК | 1990 |
|
RU2087479C1 |
| СОЕДИНЕНИЯ, НАБОР, АНДРОГЕННАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2242479C2 |
| ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2139885C1 |
| ПРОИЗВОДНЫЕ СУЛЬФАМАТА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 1996 |
|
RU2159774C2 |
| ТЕРАПЕВТИЧЕСКИ АКТИВНЫЕ ТРИАЗОЛЫ И ИХ ИСПОЛЬЗОВАНИЕ | 2007 |
|
RU2469042C2 |
| 18-МЕТИЛ-19-НОРАНДРОСТ-4-ЕН-17,17-СПИРОЭФИР (18-МЕТИЛ-19-НОР-20-СПИРОКС-4-ЕН-3-ОН) И ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, КОТОРЫЕ ЕГО СОДЕРЖАТ | 2007 |
|
RU2440365C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА | 1995 |
|
RU2179442C2 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
Изобретение относится к способу получения эстетрола, имеющего формулу (I), исходя из соединения, имеющего формулу (II). Способ отличается тем, что (a) соединение, имеющее формулу (II), подвергают ацилированию в подходящем растворителе с помощью подходящего реагента, что приводит к получению соединения, имеющего общую формулу (III). Затем (b) защитную бензильную группу в положении 3 удаляют гидрированием с переносом водорода или каталитическим гидрированием, в результате чего получают соединение, имеющее общую формулу (IV). Затем (с) защиту удаляют в щелочной среде, образованной карбонатом щелочного металла, гидрокарбонатом щелочного металла или гидроксидами щелочных металлов в подходящем растворителе. В формулах (III) и (IV) R представляет метильную группу или атом водорода. Предлагаемый способ позволяет получать эстетрол с высокой чистотой. Изобретение относится также к соединениям, имеющим общую формулу (III) и (IV), которые применяются в указанном способе. 3 н. и 16 з.п. ф-лы, 5 пр.

1. Способ получения эстетрола, имеющего формулу (I)
исходя из соединения, имеющего формулу (II)
отличающийся тем, что
(a) соединение, имеющее формулу (II), подвергают ацилированию в подходящем растворителе с помощью подходящего реагента, что приводит к получению соединения, имеющего общую формулу (III)
где R = метильная группа или атом водорода;
(b) защитную бензильную группу в положении 3 удаляют гидрированием с переносом водорода или каталитическим гидрированием, в результате чего получают соединение, имеющее общую формулу (IV)
где R = метильная группа или атом водорода;
(с) защиту удаляют в щелочной среде, образованной карбонатом щелочного металла, гидрокарбонатом щелочного металла или гидроксидами щелочных металлов в подходящем растворителе.
2. Способ по п. 1, в котором растворитель, применяемый на этапе (а), выбран из группы, состоящей из алифатических и ароматических углеводородов, галогенированных углеводородов, сложных эфиров и простых эфиров.
3. Способ по любому из пп. 1, 2, в котором реагент, применяемый на этапе (а), представляет собой уксусный ангидрид, ацетилхлорид или ацетилбромид.
4. Способ по любому из пп. 1-3, в котором реагент, применяемый на этапе (а), представляет собой смешанный ангидрид уксусной кислоты и муравьиной кислоты.
5. Способ по любому из пп. 1-4, в котором этап (а) выполняют в присутствии основания - третичного амина.
6. Способ по любому из пп. 1-5, в котором этап (а) дополнительно включает кристаллизацию полученного соединения, имеющего общую формулу (III), из С1-3-спиртов.
7. Способ по любому из пп. 1-5, в котором этап (а) выполняют без очистки и/или выделения соединений, имеющих формулу (II).
8. Способ по любому из пп. 1-7, в котором этап (b) выполняют каталитическим гидрированием газообразным водородом и катализатор выбран из группы, состоящей из палладия или палладия на носителе.
9. Способ по п. 8, в котором растворитель, применяемый для каталитического гидрирования, выбран из группы, состоящей из спиртов, сложных эфиров и кетонов.
10. Способ по любому из пп. 1-7, в котором этап (b) выполняют гидрированием с переносом водорода с применением в качестве реагента циклогексена.
11. Способ по п. 10, в котором растворитель, применяемый для гидрирования с переносом водорода, представляет собой спирт.
12. Способ по любому из пп. 1-11, в котором этап (b) дополнительно включает кристаллизацию полученного соединения, имеющего общую формулу (IV), из сложных эфиров, углеводородов, спиртов или смесей перечисленных веществ.
13. Способ по любому из пп. 1-12, в котором растворитель, применяемый на этапе (с), выбран из группы, состоящей из воды, спиртового растворителя или их смеси.
14. Способ по любому из пп. 1-13, в котором этап (с) выполняют в присутствии карбоната щелочного металла или гидрокарбоната щелочного металла.
15. Способ по любому из пп. 1-13, в котором этап (с) выполняют в присутствии алкоголята щелочного металла или гидроксида щелочного металла.
16. Способ по любому из пп. 1-15, в котором R представляет собой метильную группу.
17. Способ по любому из пп. 1-15, в котором R представляет собой атом водорода.
18. Соединение, имеющее общую формулу (III), где соединение выбрано из группы, состоящей из (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриацетата и (15α,16α,17β)-3-(бензилокси)эстра-1,3,5(10)-триен-15,16,17-триилтриформиата.
19. Соединение, имеющее общую формулу (IV), где соединение представляет собой (15α,16α,17β)-3-гидроксиэстра-1,3,5(10)-триен-15,16,17-триилтриформиат.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| J | |||
| FISHMAN et al., Synthesis of epimeric 15-hydroxyestriols, new and potential metabolites of estradiol, J | |||
| ORG | |||
| CHEM., 1968, vol | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Регулятор для торфяного транспортера | 1925 |
|
SU3133A1 |
| T | |||
| NAMBARA et al., Syntheses of estetrol monoglucuronides, STEROIDS, 1976, vol | |||
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Говорящий кинематограф | 1920 |
|
SU111A1 |

