Область, к которой относится изобретение.
Данное изобретение относится к способам индуцирования анальгезии в ответ на невропатическую боль, которые включают введение соединений формулы I, в частности прегнандинонов или прегнандиолов, возможно, в сочетании с одним или более другим анальгетическим соединением, таким как опиоидные соединения. Данное изобретение относится также к композициям и наборам, пригодным для индуцирования анальгезии в ответ на невропатическую боль.
Предпосылки создания изобретения.
Данное изобретение относится в общем к индуцированию анальгезии в ответ на невропатическую боль. Рассматривая различные подходы к лечению боли, важно понимать разницу между острой и хронической болью. Острая боль возникает в результате повреждения ткани или воспаления и опосредована химической, механической или термической стимуляцией болевых рецепторов. В противоположность этому хроническая боль сама по себе представляет собой заболевание, для которого нет защитной биологической функции. Хроническая боль не смягчается и может оставаться в течение нескольких лет после первоначальной травмы.
Хроническая незлокачественная боль в основном является невропатической болью, которая может быть определена как боль, вызванная или обусловленная первичным поражением или дисфункцией нервной системы1. Невропатическая боль связана с разнообразными заболеваниями и в клинике проявляется в целом ряде симптомов2.
Невропатическая боль часто описывается как имеющая режущий или непрерывный жгучий характер и часто ассоциируется с возникновением патологических сенсорных признаков, таких как аллодиния и гиперальгезия. Аллодиния определяется как боль, возникающая от раздражителя, который обычно не вызывает болезненную реакцию, и гиперальгезия характеризуется увеличенным болевым ответом на действие раздражителя, который обычно является болезненным. Некоторые нарушения, характеризующиеся невропатической болью, включают монорадикулопатию, тригемиальную невралгию, постгерпетическую невралгию, фантомную боль, синдромы комплексной региональной боли и различные периферические невропатии.
В то время как существуют многочисленные эффективные терапевтические способы лечения острой боли, вызванной воспалительными процессами или острой травмой, включающие лечение опиоидными и нестероидными противовоспалительными лекарствами (NSAIDs), невропатическая боль трудно поддается лечению.
Вследствие разных патофизиологических механизмов, связанных с невропатической болью и болями, вызванными воспалительными процессами, агенты, пригодные для лечения воспалительных и других болей, менее эффективны при лечении невропатических болей. В частности, эффективность опиоидов при лечении невропатической боли снижается по сравнению с лечением болей, обусловленных воспалительными процессами, и кривая зависимости реакции от дозы опиоидов при лечении невропатической боли сдвигается вправо по сравнению с лечением болей, вызванных воспалениями5. Обычными фармакологическими агентами при клиническом лечении невропатических болей являются трициклические антидепрессанты и некоторые антиконвульсанты3,4, но даже они приводят к значительному клиническому уменьшению боли (то есть более чем на 50%) только у менее чем 50% пациентов. Эти агенты ассоциируются также со значительными побочными эффектами.
Следовательно, существует настоятельная необходимость создания улучшенных режимов лечения невропатической боли, и данное изобретение касается этого вопроса. Другие цели данного изобретения станут очевидными из нижеследующего описания.
Сущность изобретения.
Согласно одному варианту данного изобретения предусматривается способ индуцирования анальгезии в ответ на невропатическую боль у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы I
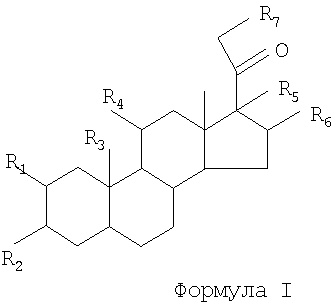
где R1 обозначает Н, ОН, C1-C4 алкил, C2-C4 алкенил, С2-С4 алканоил или OR;
R2 обозначает Н, ОН, OR или =O;
R3 обозначает Н, ОН или C1-C4 алкил;
R4 обозначает Н, ОН, =O, C1-C4 алкил, С2-С4 алкенил, C4-C4 алканоил или OR;
R5 обозначает Н, ОН, C1-C4 алкил, C2-C4 алкенил, С2-С4 алканоил или OR;
R6 обозначает Н, ОН, =СН2 или C1-C4 алкил;
R7 обозначает Н, ОН, галоген, C1-C4 алкил, C2-C4 алкенил, С2-С4 алканоил, SH, SR или OR и
R обозначает C1-C4 алкил, С2-С4 алкенил или C2-C4 алканоил,
или его фармацевтически приемлемого производного.
Согласно другому варианту данного изобретения предусмотрен способ индуцирования анальгезии, без явного седативного эффекта, в ответ на невропатическую боль у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемого производного. Еще один вариант настоящего изобретения предусматривает композицию для индуцирования анальгезии, без явного седативного эффекта, в ответ на невропатическую боль у млекопитающего, включающую соединение формулы I или его фармацевтически приемлемое производное вместе с, по меньшей мере, одной фармацевтически приемлемой добавкой.
Согласно еще одному варианту данного изобретения предусмотрен способ индуцирования анальгезии в ответ на невропатическую боль у млекопитающего, включающий совместное или последовательное введение млекопитающему эффективных количеств анальгетического соединения, такого как опиоид, и соединения формулы I или его фармацевтически приемлемого производного.
Предпочтительно вводить анальгетическое соединение и соединение формулы I или его фармацевтически приемлемое производное в синергически эффективных количествах. Предпочтительно, чтобы этот способ не приводил к явному седативному эффекту.
Настоящее изобретение относится также к применению соединения формулы I или его фармацевтически приемлемого производного для изготовления лекарственного средства для индуцирования анальгезии предпочтительно без явного седативного эффекта в ответ на невропатическую боль.
Согласно еще одному варианту данного изобретения предусмотрен набор для индупирования анальгезии в ответ на невропатическую боль у млекопитающего, включающий анальгезирующее соединение, такое как опиоид, и соединение формулы I или его фармацевтически приемлемое производное. Например, анальгетическое соединение может представлять собой опиоид, выбранный из фентанила, оксикодона, кодеина, дигидрокодеина, дигидрокодеиноненолацетата, морфина, дезоморфина, апоморфина, петидина, метадона, декстропропоксифена, пентазоцина, декстроморамида, оксиморфона, гидроморфона, дигидроморфина, носкапина, папаверина, папаверетума, алфентанила, бупренорфина и трамадола и их смесей, и их фармацевтически приемлемых производных.
Согласно предпочтительному варианту R7 обозначает ОН, OR, SH, SR или галоид, более предпочтительно ОН, OR, SH или SR. Согласно другому предпочтительному варианту R2. обозначает ОН или OR, более предпочтительно в α-конформации.
В предпочтительных соединениях формулы I
R1 обозначает Н, ОН или метил;
R2 обозначает ОН;
R3 обозначает Н или метил;
R4 обозначает Н, ОН или =O;
R5 обозначает Н, ОН или метил;
R6 обозначает Н или метил;
R7 обозначает ОН, OC2-C4 алканоил (например, ОСОСН3), SH, SCOCH3, Cl, Br или F.
Согласно другому варианту R1 является Н, R2 обозначает ОН в α-конформации, R3 обозначает метил (в α- или β-конформации) и R7 обозначает ОН или OR.
В соответствии с еще одним предпочтительным вариантом соединение формулы I представляет собой прегнандион, то есть R2 или R4 представляет собой =O.
Согласно особенно предпочтительному варианту изобретения соединение формулы I является альфадолонацетатом.
Соединения по изобретению можно вводить, наряду с другими методами, орально, внутривенно, внутримышечно, внутрибрюшинно, внутрижелудочно, внутриоболочечно, трансдермально или интестинально. Особенно предпочтительно вводить соединения орально.
Предпочтительно вводить соединения формулы I в максимальной дозе, равной примерно 2 г/70 кг каждые 6 часов.
Согласно особенно предпочтительному варианту млекопитающее представляет собой человека.
Краткое описание чертежей.
Настоящее изобретение будет описано далее со ссылками на следующие чертежи.
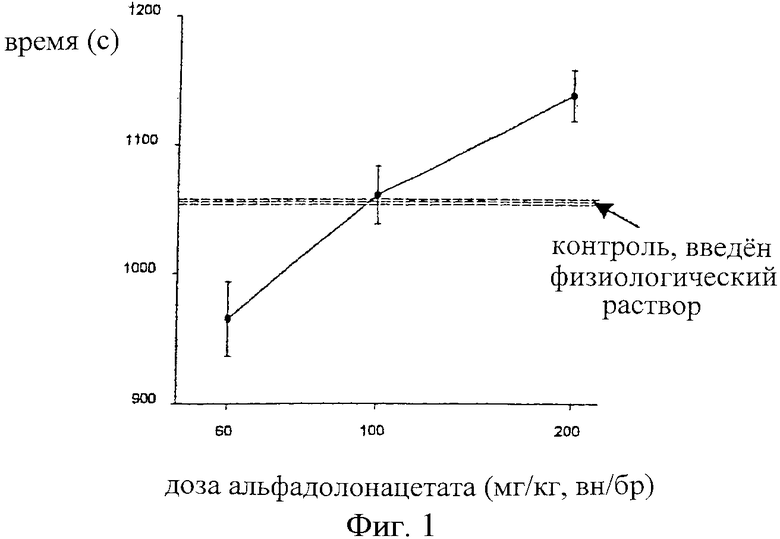
На фиг.1 показана зависимость времени покоя (с) самцов крыс Wistar от величины дозы альфадолонацетата (мг/кг), введенного внутрибрюшинно в интервале 60-200 мг/кг. Пунктирной линией обозначены результаты для контрольной группы крыс, которым вводили физиологический раствор.
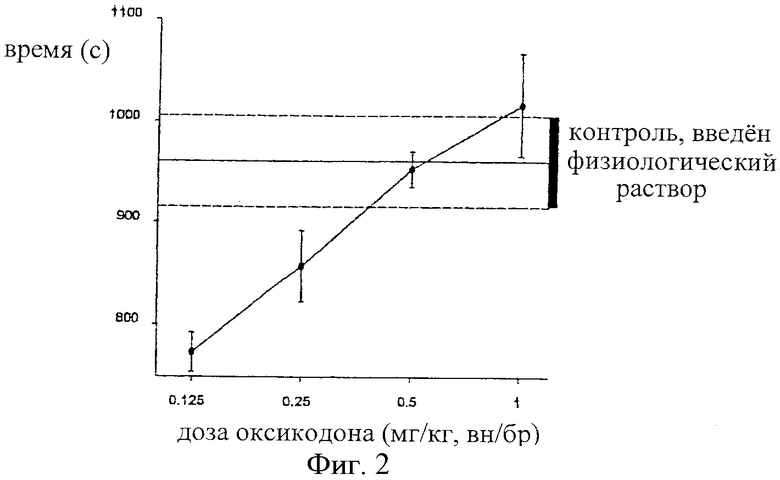
На фиг.2 показана зависимость времени покоя (с) самцов крыс Wistar от величины дозы оксикодона (мг/кг), введенного внутрибрюшинно в интервале 0,125-1,0 мг оксикодона/кг. Пунктирные линии обозначают результаты для контрольной группы крыс, которым вводили физиологический раствор.
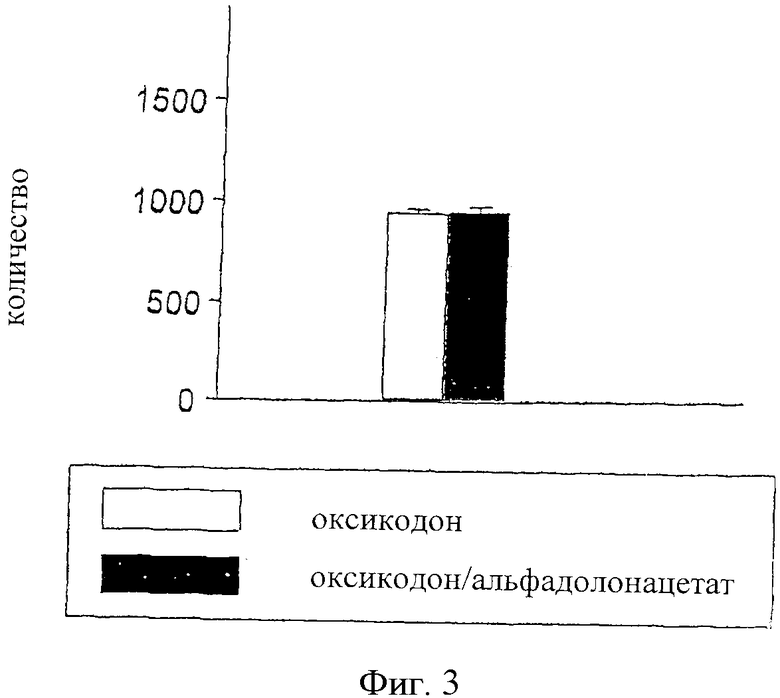
На фиг.3 показана диаграмма, представляющая количество периодов покоя для оксикодона (0,5 мг/кг), введенного внутрибрюшинно, и для оксикодона (0,5 мг/кг) в сочетании с альфадолонацетатом (10 мг/кг), также введенных внутрибрюшинно.
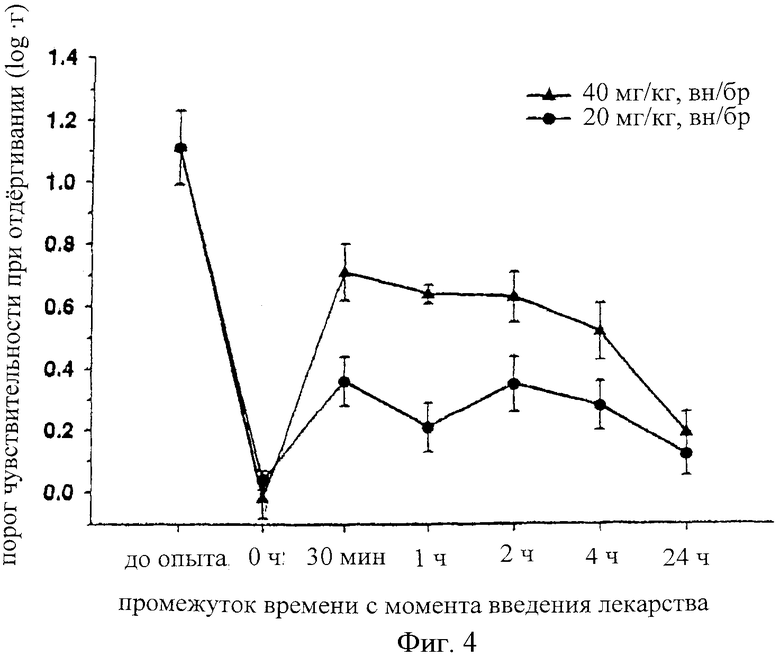
На фиг.4 представлена зависимость порога отдергивания лап (log грамм) самцов крыс Wistar от времени (ч), прошедшего с момента введения лекарства, согласно модели нейропатии Chung тугого лигирования L5 и L6. Порог отдергивания лап измеряли до и после внутрибрюшинной инъекции альфадолонацетата с дозой 20 и 40 мг/кг.
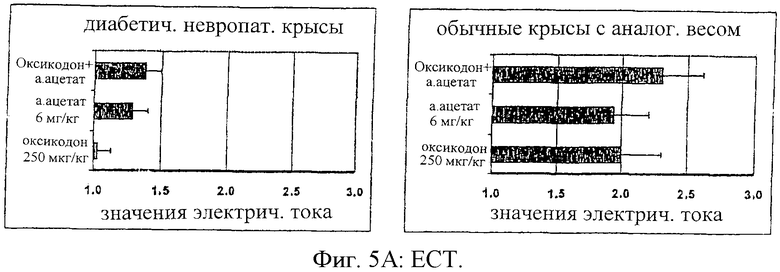
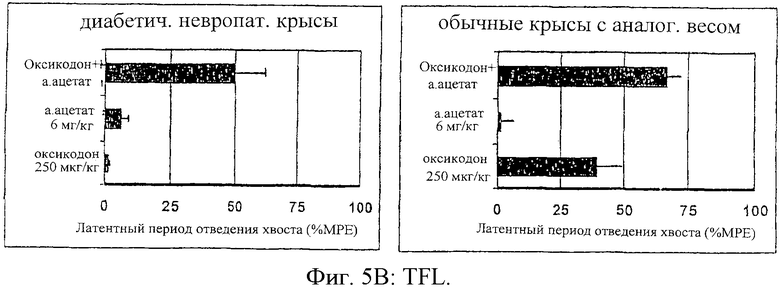
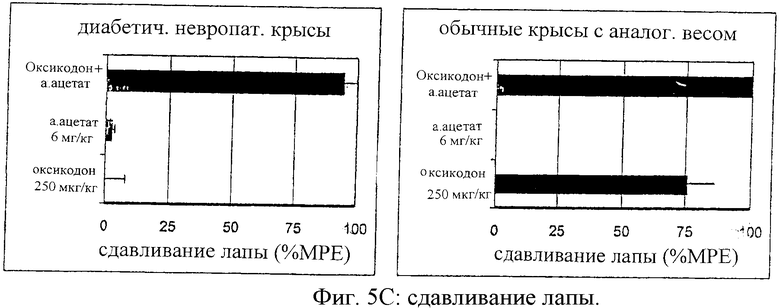
На фиг.5 представлены антиноцицептивные эффекты для самцов крыс Wistar с невропатическим диабетом в сравнении с самцами крыс Wistar с нормальным весом после введения оксикодона (250 мг/кг), альфадолонацетата (а.ацетата) (6 мг/кг) или сочетания оксикодона и альфадолонацетата (а.ацетата) в тех же дозах. Достижение антиноцицептивных эффектов осуществлялось мониторингом вредного электрического тока (ЕСТ) (А), скрытых легких ударов хвостом (В) и давления лап (С).
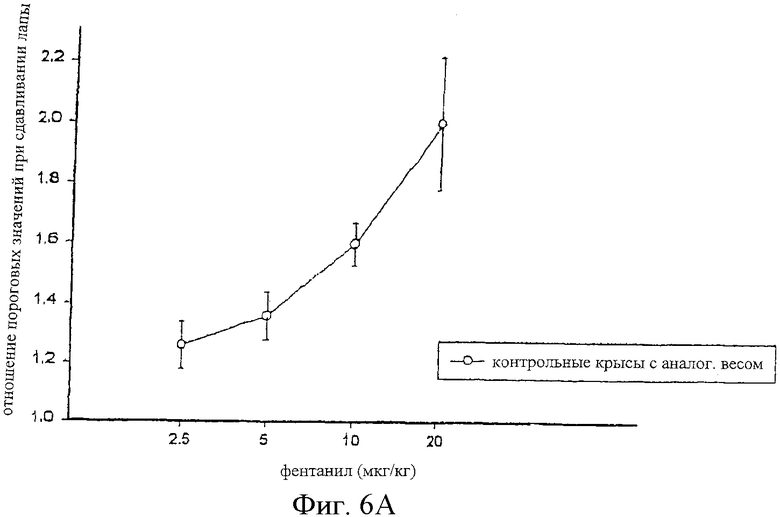
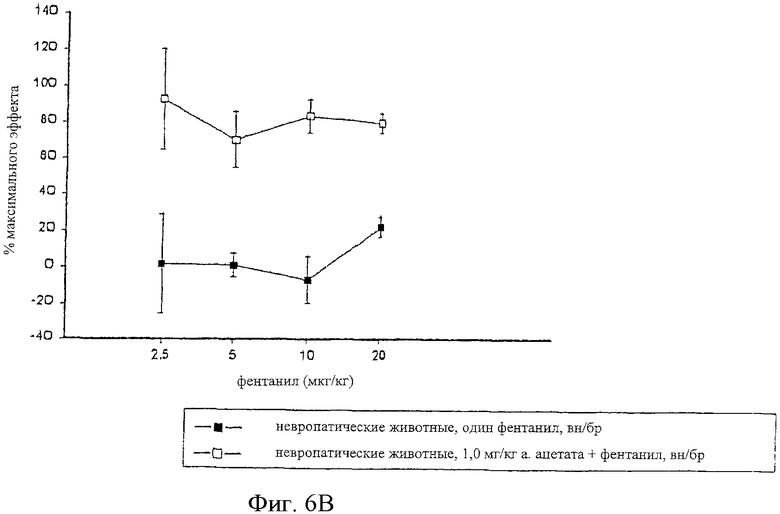
На фиг.6 представлены кривые зависимости антиноцицептивного эффекта от величины дозы фентанила, введенного внутрибрюшинно самцам крыс Wistar (фиг.6А), и одного фентанила и сочетания альфадолонацетата и фентанила, введенных самцам крыс Wistar с невропатическим диабетом (фиг.6В).
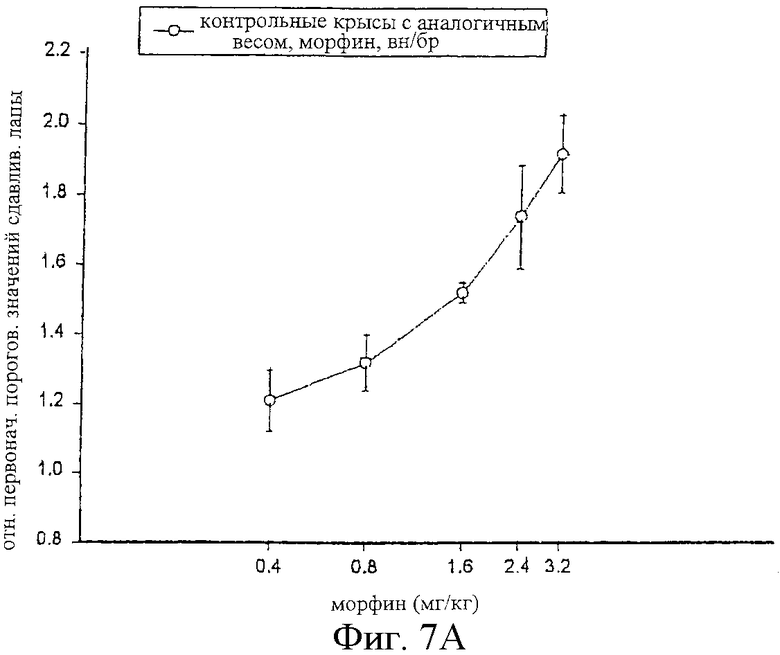
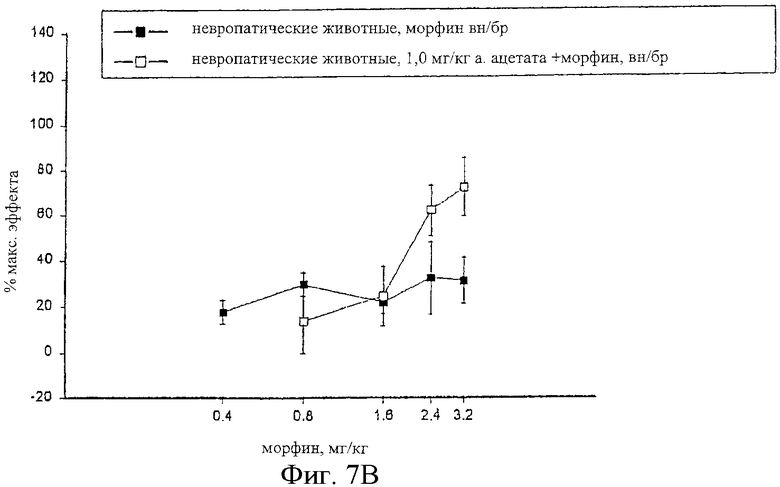
На фиг.7 показаны кривые зависимости антиноцицептивного эффекта от величины дозы морфина, введенного внутрибрюшинно контрольным самцам крыс Wistar (фиг.7А), и одного морфина и сочетания альфадолонацетата с морфином, введенных самцам крыс Wistar с невропатическим диабетом (фиг.7В).
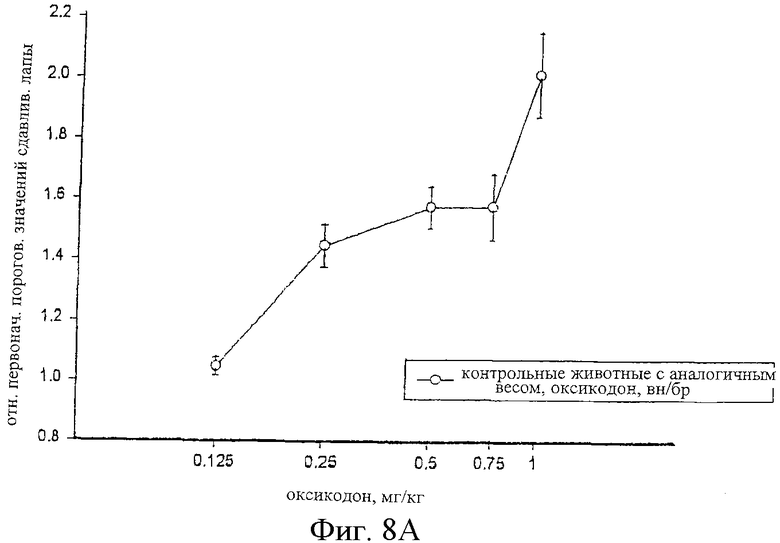
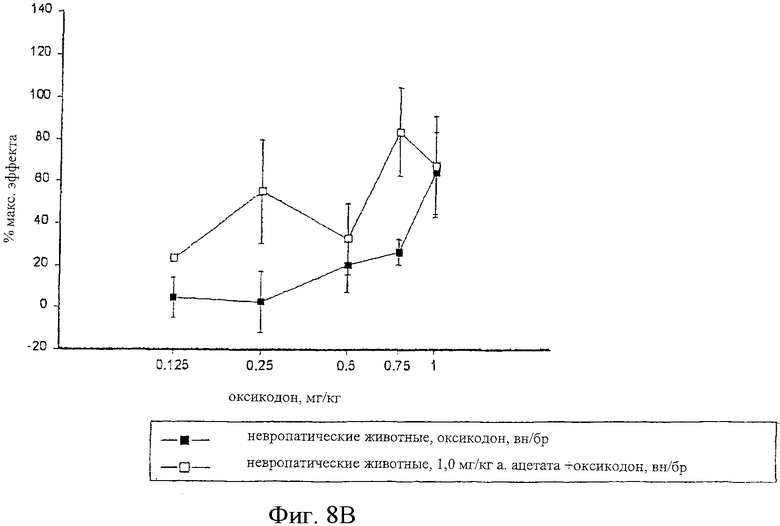
На фиг.8 показаны кривые зависимости антиноцицептивного эффекта от величины дозы оксикодона, введенного внутрибрюшинно контрольным самцам крыс Wistar (фиг.8А), и одного оксикодона и сочетания альфадолонацетата с морфином, введенных внутрибрюшинно самцам крыс Wistar с невропатическим диабетом (фиг.8В).
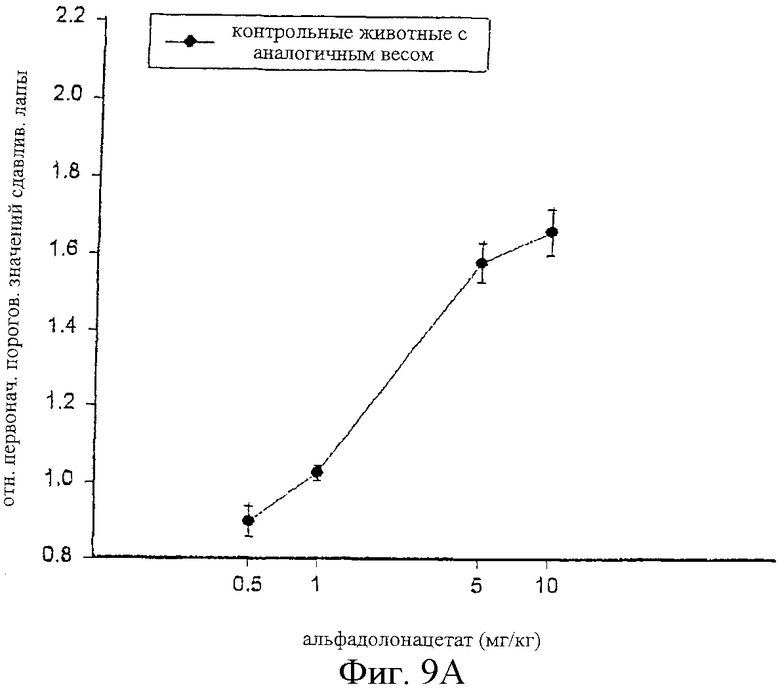
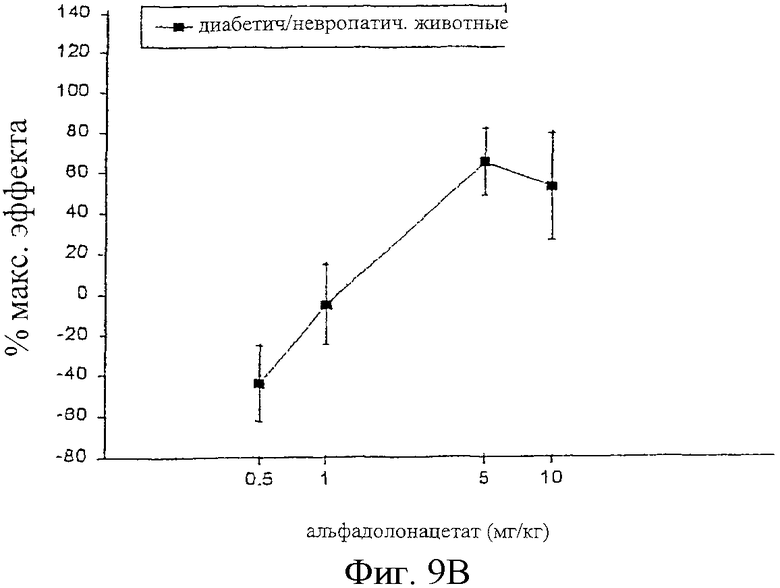
Фиг.9 представляет кривые зависимости антиноцицептивного эффекта от дозы альфадолонацетата, введенного внутрибрюшинно контрольным (фиг.9А) и больным невропатическим диабетом (фиг.9В) самцам крыс Wistar.
Подробное описание изобретения.
В данном описании и нижеследующей формуле изобретения, если иное не оговаривается, термины "включают", "включает" и "включающий" подразумевают включение ингредиента или стадии или группы ингредиентов или стадий без исключения любого другого ингредиента или стадии или группы ингредиентов или стадий.
Как указано выше, данное изобретение относится к способам индуцирования анальгезии в ответ на невропатическую боль у млекопитающего. В данном контексте термин "млекопитающее" охватывает и людей и других млекопитающих, таких как лабораторные животные, включая крыс, мышей, обезьян, морских свинок, домашних животных, включая кошек, собак, кроликов, сельскохозяйственных животных, включая скот, овец, коз, лошадей и свиней, и диких животных, таких как львы, тигры, слоны и т.п.
По всему тексту описания термин "невропатическая боль" означает боль, инициированную или вызванную первичным поражением или дисфункцией нервной системы. Способы согласно данному изобретению вызывают анальгезию в ответ на невропатическую боль, от которой страдает млекопитающее, предпочтительно человек. В данном контексте термин "анальгезия" описывает состояние пониженной чувствительности к боли, которое предпочтительно возникает без явного седативного эффекта и предпочтительно без эффекта при прикосновении. Предпочтительно, чтобы чувствительность к боли уменьшалась, по меньшей мере, на 30%, предпочтительно по меньшей мере, на 50%, более предпочтительно, по меньшей мере, на 70% и особенно предпочтительно, по меньшей мере, на 85%.
Согласно наиболее предпочтительному аспекту данного изобретения чувствительность к невропатической боли исчезает полностью или практически полностью. Для оценки уровня снижения чувствительности к боли, связанного с анальгезией, индуцированной способами по изобретению, можно провести исследования, такие как вопросник по изучению боли McGill в виде краткой формы и/или визуальная аналоговая шкала интенсивности боли и/или вербальная шкала оценки интенсивности боли и/или измерения тактильной аллодинии с применением волосков Frey или похожего прибора. Эти исследования являются стандартными и хорошо известны специалистам в данной области.
При использовании выражения "явный седативный эффект" имеют в виду, что способы (и композиции) по изобретению не приводят практически к заметному седативному эффекту у пациента, то есть значительной видимой или очевидной сонливости или бессознательному состоянию. Таким образом, введение предпочтительных соединений, описанных в данной заявке, не приводит ко сну или сонливости у пациента в той степени, которая мешает каждодневной деятельности, например вождению машины или работе с оборудованием, предназначенным для людей, или кормления и ухода за животными. Если соединение формулы I или его фармацевтически приемлемое производное вводят совместно или последовательно с другим анальгетическим соединением, то выражение "явный седативный эффект" относится к седативному эффекту, превышающему аналогичный эффект, который может быть вызван анальгетическим соединением.
Согласно одному из вариантов предпочтительные соединения формулы I представляют собой прегнандионы. В качестве примера можно привести химическую структуру 3,20-прегнандиона, где указана обычная нумерация атомов в стероидной кольцевой системе, эта система показана ниже и обозначена формулой II. Другие возможные прегнандионы являются 11, 20-прегнандионами.
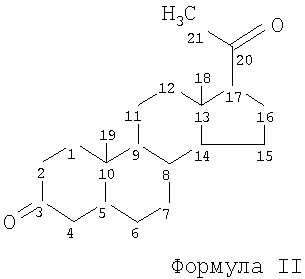
Соединения, охватываемые общей формулой I, и родственные соединения, такие как С20аминопрегнаны, которые могут быть пригодны в способах по изобретению, описаны, например, в The Merck Index (13th Edition) и в патенте Австралии №698746, патенте США №3558608, патенте GB №1317184, патенте GB №1317185, а также в патентах DE 2162593 и 2162554.
Вышеуказанные источники вместе с рефератами 105627с и 9285v в Chemical Abstracts, vol.77, 1972; 64113v, 64114w, 20793n в Chemical Abstracts 5, vol.75, 1971; 115783f и 66672h в Chemical Abstracts, vol.79, 1973, и 1020345 в Chemical Abstracts, vol.78, 1973, включены в данное описание в качестве ссылок.
В вышеуказанных источниках подробно описаны способы синтеза, которые можно использовать для получения соединений формулы I. Другие соединения доступны из таких источников, как Steraloids (Newport Rhode Island) или имеют природное происхождение. Другие соединения могут быть получены химической модификацией известных способов из соединений, описанных в вышеуказанных ссылках.
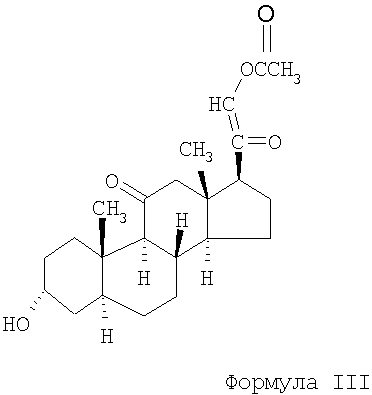
Особенно предпочтительным соединением формулы I является 21-ацетокси-3α-гидрокси-5α-прегнан-11,20-дион, который обычно называют альфадолонацетатом (формула III).
Другие соединения формулы I представляет собой деацилированную форму альфадолонацетата.
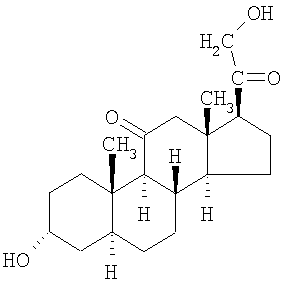
Еще одним соединением формулы I является соединение
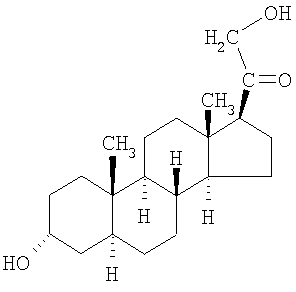
Другие предпочтительные соединения могут включать 3α-гидрокси-5α-прегнан-11,20-дион, 3α-гидрокси-21-пропионилокси-5α-прегнан-11,20-дион, 21-изобутирилокси-3α-гидрокси-5α-прегнан-11,20-дион, 21-гемисукцинилокси-3α-гидрокси-5α-прегнан-11,20-дион, 3α-гидрокси-5β-прегнан-20-он и 3α-гидрокси-5β-прегнан-20-он, (3α-5α)-3,17,21-тригидроксипрегнан-11,20-дион, (3α-5α)-3,17,21-тригидроксипрегнан-11,20-дион, 3β,17β,21-тригидрокси-5α-прегнан-11,20-дион, 3β,11β,21-тригидрокси-5α-прегнан-11,20-дион, 3β,17β,21-тригидрокси-5α-прегнан-20-он,
5α-прегнан-3,20-дион и
5β-прегнан-3,20-дион.
Особенно предпочтительные соединения по изобретению содержат окси- или тиогруппу в положении 21, например, когда R7 обозначает ОН, SH, OR (например, ОС2-4алканоил) или SR, более предпочтительно ОН или OR. Другими предпочтительными соединениями являются соединения формулы I или их фармацевтически приемлемые производные, которые способны образовывать глюкуронидный метаболит, как только они введены пациенту. Другими предпочтительными соединениями являются прегнандионы, где, например, R2 или R4 обозначает =O. Согласно другому варианту R5 и R6 обозначают водород. Согласно еще одному варианту R7 обозначает ОС2-С4алканоил. Согласно еще одному предпочтительному варианту изобретения R7 и R2 оба независимо выбраны из группы ОН и OR, например, R2 обозначает ОН и R7 обозначает ОН или OR. Другие предпочтительные соединения содержат в качестве R3 β-метил. Особенно предпочтительные соединения формулы I могут содержать, где это возможно, два или более указанных выше предпочтительных признака.
Специалисту в данной области очевидно, что в формуле I, когда заместители не указаны, но их присутствие необходимо вследствие лишней валентности, эти заместители обозначают водород. Очевидно также, что в ситуации, когда, например, R2 обозначает оксорадикал в положении 3 у атома углерода, в этом положении уже не будет атома водорода. Это относится также к случаю, когда R4 обозначает оксо и R6 обозначает метилен.
Термин "алкил", используемый в данном описании, обозначает линейный, разветвленный или моноциклический алкил, предпочтительно содержащий 1-4 атома углерода. Примеры линейного, разветвленного или моноциклического алкилов представляют собой метил, этил, пропил, изопропил, бутил, циклопропил и циклобутил. Термин "алкенил", используемый в данном описании, обозначает группы, образовавшиеся из линейных, разветвленных или моноциклических алкенов, включая моно- или полиненасыщенные алкильные или циклоалкильные группы. Конкретными примерами являются винил, аллил, 1-метилвинил, бутенил и изобутенил.
Термин "алканоил" обозначает линейный или разветвленный алканоил (СОалкил), такой как ацетил (СОСН3), пропионил (СОСН2СН3), бутирил (СОСН2СН2СН3) и изобутирил (СОСН(СН3)2).
Согласно другому аспекту данного изобретения способ индуцирования анальгезии в ответ на невропатическую боль может включать совместное или последовательное введение млекопитающему, нуждающемуся в таком лечении, аддитивного или, более предпочтительно, синергически эффективных количеств соединения формулы I или его фармацевтически приемлемого производного и другого анальгетического соединения, такого как опиоид. Таким образом, синергически эффективное количество соединения формулы I или его фармацевтически приемлемого производного, введенного совместно или последовательно с опиоидным соединением, может восстановить восприимчивость опиоида к невропатической боли. Соединение формулы I или его фармацевтически приемлемое производное и опиоид могут быть введены или совместно, то есть в виде одной композиции, содержащей активные агенты, или в виде дискретных доз. Активные агенты вводятся таким образом, чтобы достигался желательный аддитивный или синергический анальгетический эффект.
Используемые согласно изобретению опиоидные соединения (опиоиды) включают любое соединение, которое является частичным или полным агонистом опиоидного рецептора.
Опиоидные соединения хорошо известны и включают соединения природного происхождения на основе опиума, такие как кодеин, морфин, папаверин, а также производные таких соединений, которые структурно подобны, и другие соединения, активные как анальгетические агенты. Конкретные примеры опиоидных соединений, используемых согласно данному изобретению, могут включать фентанил, оксикодон, кодеин, дигидрокодеин, дигидрокодеиноненол-ацетат, морфин, дезоморфин, апоморфин, петидин, метадон, декстропропоксифен, пентазоцин, декстроморамид, оксиморфон, гидроморфон, дигидроморфин, носкапин, папаверин, папаверетум, алфентанил, бупренорфин и трамадол и их фармацевтически приемлемые производные и/или таутомеры.
Выражение "фармацевтически приемлемое производное" охватывает любые фармацевтически приемлемые соль, пролекарство, гидрат, сольват, метаболит или любое другое соединение, которое после введения субъекту способно обеспечить (непосредственно или косвенно) нужное соединение или физиологически (например, анальгетически) эквивалентное активное соединение или его активный метаболит или остаток. Примером подходящего производного является сложный эфир, образовавшийся по реакции ОН или SH (например, C21OH или SH) с подходящей карболовой кислотой, например С1-3алкил-CO2Н и HO2С-(СН2)n-CO2Н (где n=1-10, предпочтительно 1-4) и CO2Н-СН2 фенил.
Соединения формулы I могут быть в кристаллической форме, или в виде свободных соединений, или в виде сольватов (например, гидратов). Методы сольватирования являются общеизвестными.
Соли активных соединений по изобретению являются, предпочтительно, фармацевтически приемлемыми, но следует иметь в виду, что не фармацевтически приемлемые соли также входят в объем изобретения, так как они пригодны как промежуточные продукты при получении фармацевтически приемлемых солей. Примеры фармацевтически приемлемых солей включают соли фармацевтически приемлемых катионов, таких как натрий, калий, литий, кальций, магний, алюминий и алкилалюминий; аддитивные соли фармацевтически приемлемых неорганических кислот, таких как соляная, ортофосфорная, серная, фосфорная, азотная, угольная, борная, сульфаминовая и бромистоводородная кислота; соли фармацевтически приемлемых органических кислот, таких как уксусная, пропионовая, масляная, винная, малеиновая, гидроксималеиновая, фумаровая, лимонная, молочная, муковая, глюконовая, бензойная, янтарная, щавелевая, фенилуксусная, метансульфоновая, тригалометансульфоновая, толуолсульфокислота, бензолсульфокислота, салициловая, сульфаниловая, аспартавая, глутаминовая, эдетовая, стеариновая, пальмитиновая, олеиновая, лауриновая, пантотеновая, танниновая, аскарбиновая и валерьяновая кислоты.
Термин "пролекарство" используется в данном описании в его самом широком смысле и включает такие соединения, которые могут превращаться in vivo в нужное соединение (например, путем ферментативного или гидролитического расщепления). Примеры пролекарств включают сложные эфиры, например ацетаты оксигрупп или тиогрупп, а также фосфаты и сульфонаты. Способы ацилирования гидроксильных или тиольных групп известны, например, это происходит при реакции спирта (гидроксильной группы) или тиогруппы с карбоновой кислотой. Другие примеры подходящих пролекарств описаны в Design of Prodrugs, H.Bundgaard, Elsevier, 1985.
Термин "метаболит" включает любое соединение, в которое соединение формулы I может превратиться in vivo сразу же после введения субъекту. Примерами такого метаболита служат глюкуронид, сульфат и гидроксилаты.
Следует иметь в виду, что описанные в данной заявке соединения могут существовать в таутомерией форме, то есть в виде их таутомеров. Термин "таутомер", используемый в данном описании в его самом широком смысле, включает соединения, которые способны существовать в состоянии равновесия между двумя изомерными формами. Такие соединения могут отличаться связью между двумя атомами или группами и положением этих атомов или групп в соединении. Типичным примером является кето-енольный таутомеризм.
Соединения по изобретению могут быть электрически нейтральными или поликатионами с ассоциированными анионами для электрической нейтральности. Подходящие ассоциированные анионы включают сульфат, тартрат, цитрат, хлорид, нитрат, нитрит, фосфат, перхлорат, галоидсульфонат или тригалометилсульфонат.
Невропатическая боль, которую можно лечить согласно способам по изобретению, включает монорадикулопатию, невралгию тройничного нерва, постгерпетическую невралгию, фантомную боль, синдромы комплексной региональной боли, невропатическую боль, связанную со СПИД, и инфекцию вирусом иммунодефицита человека и различные периферические невропатии, включая, но не ограничиваясь этим, невропатию, вызванную употреблением лекарств или диабетом.
Соединения формулы I и их фармацевтически приемлемые производные и анальгетические соединения (например, опиаты), которые могут вводиться в сочетании с первыми (называемые "активными ингредиентами, агентами или соединениями") можно вводить при терапии любым подходящим методом. Следует иметь в виду, что соединения формулы I или их фармацевтически приемлемые производные предпочтительно вводятся методом, который не приводит к появлению явного седативного эффекта. Подходящие методы введения могут включать оральный, ректальный, назальный, ингаляцию аэрозолей или частиц препарата, топический (включая буккальный и подъязычный), трансдермальный, вагинальный, внутрипузырный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикишечный, подоболочечный, эпидуральный и внутридермальный). Предпочтительно введение соединения формулы I или его фармацевтически приемлемого производного осуществлять методом, который после введения обеспечивает вначале попадание соединения в желудок пациента. Согласно особенно предпочтительному варианту данного изобретения соединение формулы I вводят перорально, однако предпочтительный путь введения меняется в зависимости от состояния и возраста субъекта, природы подвергающейся лечению невропатической боли, места, где эта боль возникает, и мнения врача или ветеринара.
Используемое в данном описании выражение "эффективное количество" относится к количеству активного соединения, которое обеспечивает желаемую анальгетическую активность, будучи введенным в соответствии с подходящей схемой дозирования. Предпочтительно количество соединения формулы I или его фармацевтически приемлемого производного представляет собой количество, которое обеспечивает желаемую анальгетическую активность без явного седативного эффекта. Дозирование может происходить с интервалами в минуты, часы, дни, недели или месяцы. Подходящие дозы и схемы могут быть определены врачом или ветеринаром. Например, соединения формулы I или их фармацевтически приемлемые производные могут вводиться субъекту по схеме 50-2000 мг каждые шесть часов, например 50-500 мг. Дозы анальгетического агента, такого как опиоид, могут быть определены при посещении врача согласно принятым на практике величинам доз. Например, фентанил может быть введен в количестве около 100 мг, а морфин может быть введен в количестве 1-5 г.
Композиции согласно данному изобретению включают, по меньшей мере, одно соединение формулы I или его фармацевтически приемлемое производное, возможно, вместе с анальгетическим соединением, таким как опиоид, в сочетании с одной или более фармацевтически приемлемой добавкой, такой как носители, разбавители и/или эксципиенты и, возможно, с другими лекарствами. Эти добавки включают все обычные растворители, диспергаторы, наполнители, твердые носители, агенты, образующие покрытие, противогрибковые или антибактериальные агенты, агенты, способствующие проникновению в кожу, поверхностно-активные вещества, изотонические агенты и абсорбенты и матрицы медленного или регулируемого высвобождения. Композиции, используемые согласно данному изобретению, могут также включать другие дополнительные физиологически активные агенты, например, другие анальгетические агенты. Соединения могут быть представлены в виде набора компонентов, которые адаптированы для совместного или последовательного введения активных компонентов. Каждый носитель, разбавитель, адъювант и/или эксципиент должен быть фармацевтически "приемлемым" в смысле совместимости с другими ингредиентами и не должен быть вредным для субъекта. Композиции могут быть обычно в виде единичных дозированных форм и могут быть получены известными в фармацевтике способами. Такие способы включают стадию смешения активного ингредиента с носителем, который содержит один или несколько вспомогательных ингредиентов. В общем композиции приготавливают путем равномерного и тщательного смешения активного ингредиента с жидкими носителями, разбавителями, адъювантами и/или эксципиентами или тонкодисперсными твердыми носителями, или с теми и другими и последующего формования полученного продукта, если это необходимо.
Композиции по изобретению, подходящие для орального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит заданное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной фазе или неводной жидкости; в виде жидкой эмульсии масло/вода или эмульсии вода/масло. Активный ингредиент может быть также в виде болюса, электуария или пасты.
Таблетка может быть приготовлена прессованием или формованием, возможно, с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть изготовлены путем прессования в подходящем устройстве активного ингредиента в свободно-текучем виде, например в виде порошка или гранул, возможно, смешанного со связующим (например, инертным разбавителем, консервантом, дезинтегрантом (например, натриевой солью гликолята крахмала, сшитым повидоном, сшитой натриевой солью карбоксиметилцеллюлозы)), поверхностно-активным или диспергирующим агентом. Сформованные таблетки могут быть изготовлены формованием в подходящем устройстве смеси порошка соединения, увлажненного инертным жидким разбавителем. Таблетки могут содержать покрытие или могут быть с насечкой и могут иметь такой состав, чтобы они обеспечивали медленное или регулируемое высвобождение активного ингредиента с использованием, например, гидроксипропилметилцеллюлозы в различных соотношениях, обеспечивающей желательный профиль высвобождения. Таблетки могут содержать энтеропокрытие, обеспечивая высвобождение в кишечнике, а не в желудке.
Композиции, пригодные для парентерального введения, включают водные и неводные стерильные растворы для инъекции, которые могут содержать антиоксиданты, буферные растворы, бактериостатики и растворенные вещества, придающие композиции изотоничность с кровью субъекта, подвергающегося лечению, и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загущающие агенты. Композиции могут быть представлены в контейнерах с единичными дозами или многими дозами, например в ампулах и флаконах, и могут храниться в замороженном-высушенном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, перед самым использованием. Неподготовленные растворы и суспензии для инъекции могут быть получены из стерильных порошков, гранул и таблеток, которые описаны выше.
Композиции, подходящие для топического введения в кожу, то есть для трансдермального введения, могут содержать активные ингредиенты, растворенные или суспендированные в любом подходящем носителе или основе и могут быть в виде лосьонов, гелей, кремов, паст, мазей и т.п. Подходящие носители могут включать минеральное масло, пропиленгликоль, полиоксиэтиленовые воски и длинноцепочечные спирты. Могут также использоваться трансдермальные изделия, например накладки, они могут содержать микропористую мембрану, изготовленную из подходящего материала, такого как нитрат/ацетат целлюлозы, полипропилен и поликарбонаты. Накладки могут также содержать подходящий для кожи адгезив и подложки.
Соединения формулы I могут быть в виде имплантатов, которые содержат полимерное изделие, несущее лекарство, где полимер является биосовместимым и нетоксичным. Подходящие полимеры могут включать гидрогели, силиконы, полиэтилены и биораспадающиеся полимеры.
Соединения по изобретению могут быть введены в виде форм с продленным (то есть регулируемым) или медленным высвобождением. Препарат с продленным высвобождением является таким, в котором активный ингредиент высвобождается в организм субъекта после введения медленно и поддерживает желательную концентрацию лекарства в течение минимального промежутка времени. Препараты с продленным высвобождением известны специалистам. Дозированные формы могут быть оральными, в виде имплантатов и трансдермальных препаратов. Для медленного высвобождения активные ингредиенты могут суспендироваться в виде частиц с медленным высвобождением или в виде липосом.
Очевидно, что в добавление к ингредиентам, упомянутым выше, композиция согласно данному изобретению может включать другие агенты, обычные для определенного типа композиции, например композиции, подходящие для перорального введения, могут включать дополнительно такие агенты, как связующие, подсластители, загустители, ароматизаторы, дезинтеграторы, агенты для образования покрытия, смазывающие агенты и/или агенты, способствующие задержке времени высвобождения. Другие подробности о фармацевтически приемлемых носителях, разбавителях и эксципиентах и о способах приготовления фармацевтических композиций и составов описаны в Remmingtons Pharmaceutical Sciences, 18th Edition, 1990, Mack Publishing Co., Easton, Pennsylvania, USA, содержание которой полностью включено в данное описание. Соединения, используемые согласно изобретению, могут быть также представлены в виде ветеринарных композиций. Последние могут быть получены известными способами. Примеры таких композиций включают препараты, адаптированные для
(а) перорального введения, например дозированные составы, включая водные и неводные растворы или суспензии, таблетки, болюсы, порошки, гранулы, пилюли, для смешения с пищей, пастами для нанесения на язык;
(б) парентерального введения, например подкожного, внутримышечного или внутривенного, в виде стерильного раствора или суспензии;
(в) топического нанесения, например, кремы, мази, гели, лосьоны и т.д.
Особенно предпочтительные соединения формулы I включают альфадолонацетат и альфадолонглюкуронид или другое пролекарство, которое обеспечит соединение с 21-ОН группой in vivo. Если предусмотрены другие солевые формы активных агентов формулы I, то предпочтительно применять или сульфат или метансульфонат, более предпочтительны соединения с солевой группой в положении 21.
Согласно особенно предпочтительному варианту изобретения соединение формулы I вводят перорально, предпочтительно в виде таблетки, капсулы, леденца или жидкости. Используемая композиция предпочтительно содержит поверхностно-активное вещество и/или вещество, улучшающее растворимость. Подходящим веществом, улучшающим растворимость, является водорастворимое полиэтоксилированное касторовое масло, подходящим поверхностно-активным веществом является Cremophor EL. Подходящие оральные дозы альфадолона или прегнандионов составляют, например, 50-500 мг каждые шесть часов. Подходящие дозы для морфина составляют 2,5-20 мг каждые 3-6 часов и для оксикодона и других опиоидов 2-50 мг каждые 3-12 часов.
Примеры.
Данное изобретение далее будет описано со ссылкой на следующие примеры, которые служат только для иллюстрации изобретения и не ограничивают его никоим образом.
Пример 1.
Животные модели невропатической боли - экспериментальный подход.
Экспериментальных человеческих моделей невропатической боли не существует. Существуют несколько животных моделей, которые отличаются по способу индукции боли и балансом признаков и симптомов. Следовательно, исследователи, пытаясь идентифицировать полезные терапевтические агенты, должны применять эти модели.
Большинство используемых в настоящее время моделей невропатической боли имеют общий признак - изменение порога кожной чувствительности задних конечностей после частичного повреждения периферического (обычно седалищного) нерва. В частности, в качестве выходного результата используют демонстрацию гиперальгезии в ответ на вредные термические раздражители и аллодинии в ответ на холодные и механические раздражители. Две из наиболее распространенных моделей представляют собой модель хронической травматической компрессии (CCI) седалищного нерва7 и модель наложения лигатуры (SNL) на спинномозговые нервы6.
Модель хронической травматической компрессии включает свободную лигатуру седалищного нерва на середине бедра при помощи жгута из хрома.7 В ответ на воздействие кетгута развивается воспалительная реакция и соответственно возникает потеря большинства А-волокон и некоторых С-волокон, но немногих клеток8. Это связано со спонтанной реакцией, связанной с болью, аллодинией и гиперальгезией. Модель SNL (модель Chung) заключается в повреждении L5 и L6 спинномозговых нервов, которые обусловливают работу седалищного нерва6. Опять-таки это связано с развитием спонтанной реакции, связанной с болью, а также затяжными аллодинией и гиперальгезией.
Большинство животных моделей невропатии основаны на дискретном повреждении периферических нервов. Однако были созданы некоторые модели, которые в большей степени имитируют индивидуальные болезненные состояния. Примером таких моделей является модель периферической диабетической невропатии с применением стрептозомицина. В этой модели инъекции стрептозомицина вызывают диабет и затем гиперальгезию аллодинию.
В данной работе седативные эффекты альфадолонацетата и опиоида - оксикодона - исследовались при раздельном и совместном введении лекарств. Обнаруженные таким образом неседативные дозы исследовали на анальгетическую эффективность на двух моделях крыс с невропатической болью, Chung модели с наложением лигатуры на корень нерва L5/6 и модели диабетической невропатии обусловленной стрептозомицином. В качестве примера анальгетических нестероидов, которые обладают уникальным свойством анальгетика, действующего на спинномозговой нерв рецепторов GABAА, применяли альфадолонацетат и оксикодон использовали для выявления типичного поведения при применении опиоидных лекарств, используемых в клинике.
(i) Проверка седативных эффектов.
Самцам крыс Wistar (n=10 в группе) вводили ряд доз альфадолонацетата (60-200 мг/кг, внутрибрюшинно, в 10% Cremophor EL) или оксикодона (0,125-1,0 мг/кг, внутрибрюшинно, в физиологическом растворе) раздельно или оксикодон (0,5 мг/кг) вводили в то же самое время, что и альфадолонацетат (10 мг/кг, оба вводили внутрибрюшинно). После инъекции лекарства или комбинации лекарств осуществляли мониторинг активности в открытом поле каждой крысы путем нарушения сетки ИК-лучей в темной камере. Время покоя, рассчитанное, исходя из числа перерывов в подаче пучков лучей, определяли в течение 30 минут. Объединяли вместе десять повторных наблюдений для каждой дозы и средние значения +/-SEM откладывали на кривых зависимости реакции от дозы вдоль значений для контрольных животных с применением физиологического контроля.
Результаты определения времени покоя в опытах с использованием монитора активности в открытом поле показаны ниже для альфадолонацетата и оксикодона, введенных раздельно, в сравнении с данньми для физиологического раствора (фиг.1 и 2) и для одного оксикодона, введенного в дозе 0,5 мг/кг, и для оксикодона, введенного в сочетании с альфадолонацетатом (10 мг/кг) (фиг.3).
Дозы альфадолонацетата менее 100 мг/кг и оксикодона менее 1,0 мг/кг не приводили к увеличению времени покоя по сравнению с использованием физиологического раствора. Более низкие дозы каждого лекарства (альфадолонацетат 60 мг/кг, оксикодон 0,25 и 0,125 мг/кг) действительно уменьшали время покоя по сравнению с физиологическим раствором. Далее, добавление 10 мг/кг альфадолонацетата к 0,5 мг/кг оксикодона не давало никакого увеличения седативности, оцениваемой по времени покоя при помощи монитора активности в открытом поле. Таким образом, любую антиноцицептивную активность, наблюдаемую в моделях невропатической боли, которую придают указанные дозы или меньшие дозы каждого лекарства, введенного в отдельности или в сочетании, нельзя объяснить возникновением седативности.
(ii) Модель невропатической боли Chung'a.
Осуществляли наложение туго затянутой лигаторы на корень нерва крыс в соответствии со способом, описанным Kirn и Chung6. Пороги отдергивания лап измеряли до и после внутрибрюшинных инъекций альфадолона (20 и 40 мг/кг; n=10 крыс в группе). Это осуществляли в приборе Randall Sellito, в котором увеличивающийся вес нагружали на невропатическую лапу. Порог отдергивания лап определяли как вес, при котором крыса отдергивает лапу.
Эта модель представляет собой модель невропатической боли, в которой аллодиния индуцируется на одной задней лапке крысы при помощи туго наложенных лигатур, помещенных на корешки одностороннего поясничного дорсального нерва. Результаты, полученные на этой модели для группы, состоящей из 10 крыс, которым вводили внутрибрюшинно альфадолонацетат, показаны на фиг.4. Можно видеть, что лигатура, наложенная на нерв, снижает порог отдергивания, вызванного механическим раздражителем, от уровней "pre-ор" для нормального ноцицептивного раздражителя до уровней, которые обычно не являются ноцицептивными в момент времени 0, когда вводили альфадолон.
(iii) Модель диабетической невропатической боли.
Семидесяти крысам осуществляли внутрибрюшинную инъекцию стрептозотоцина (STZ) (150 мг/кг, общая доза) (Sapphire Bioscience), растворенного в хлористом натрии (0,9%). Дозу 150 мг/кг вводили путем двух инъекций по 75 мг/кг в первый и второй день. Дополнительной группе животных вводили физиологический раствор (n=10, контрольная группа). Через неделю после инъекции STZ путем измерения содержания глюкозы в венозной крови, взятой из хвоста, при помощи полосок Ames Glucofilm и колориметра (Ames Glucometer 3, Bayer Diagnostics), было установлено появление диабета. Диабетическими считались только крысы с конечным содержанием глюкозы в крови ≥15 мМ. Крыс вновь испытывали на наличие гипергликемии сразу же после каждого цикла ноцицептивных исследований. Гиперальгезию оценивали, осуществляя опыт с давлением на лапы крысы, описанный ранее Randall и Selitto (Randall L.О., Selitto J.J. 1957, A Method for Measurement of Analgesic Activity in Inflamed Tissue, Archiv. Int. Pharmacodynamie: 111; 409). Повторные результаты в каждой группе были рассмотрены вместе с полученными ранее для определения средних значений +/-SEM, которые наносили на графики - гистограммы. Испытания проводили через 5 недель после первой инъекции STZ. Животные, характеризовавшиеся ноцицептивными порогами при давлении на лапы менее 30 г (60% от величины нормального веса крыс), рассматривались как животные, у которых развилась гиперальгезия/невропатическая боль, они использовались для дальнейших экспериментов. Их количество составляло 91% от всех крыс, которым вводили STZ.
Антиноцицептивное действие альфадолонацетата (6 мг/кг, внутрибрюшинно) и оксикодона (250 мг/кг), введенных раздельно и в сочетании группе крыс с нормальным весом и диабетическим крысам с невропатией (n=10 на группу), оценивали при воздействии электрического тока (ЕСТ), используя латентный период реакции отведения крысой хвоста (TFL) и сдавливание лапы.
Антиноцицептивные эффекты, определенные при воздействии электрического тока (ЕСТ), величина латентного периода реакции отдергивания хвоста (TFL) и величина давления на лапу при введении альфадолонацетата (6 мг/кг, внутрибрюшинно) и оксикодона (250 мг/кг) раздельно или совместно группам крыс с нормальным весом и диабетических крыс, показаны на фиг.5 (А, В и С соответственно).
По этим результатам видно, что диабетическая невропатическая боль минимально реагирует на антиноцицептивное действие опиоида или альфадолонацетата при раздельном введении лекарств. Однако сочетание обоих лекарств приводило к антиноцицептивному эффекту, равному по величине эффекту, полученному при введении опиоида нормальным крысам. Дозы нейростероида и опиоида, использованных по отдельности или совместно, были гораздо ниже доз, которые вызывают седативный эффект, которые оценивали при помощи монитора активности в открытом поле. Важно отметить, что антиноцицептивный эффект, полученный при совместном введении опиоида и альфадолонацетата, был больше, чем эффект, который ожидался при суммировании их индивидуальных эффектов.
Пример 2.
Модель невропатической боли.
Courteix с сотрудниками создали модель невропатической боли, обусловленной диабетом. Они установили, что индуцирование экспериментального инсулин-зависимого сахарного диабета у крыс вызывало аллодинию и гиперальгезию10. Они показали, что введенный внутривенно морфин вызывал зависимый от дозы антиноцицептивный эффект при дозах, вдвое превышающих соответствующие величины для обычных крыс, с использованием теста механического ноцицептивного сдавливания лапы10. Таким образом, диабетическая модель воспроизводила возникновение диабетической невропатической боли у людей; она резистентна к опиоиду. Эксперименты, описанные в данной заявке, предусматривали использование этой модели для оценки относительной эффективности альфадолонацетата и трех опиоидов: фентанила, морфина и оксикодона, введенных раздельно и совместно, антиноцицептивный эффект определяли, используя сдавливание лапы по методу Randall-Selitto.
Методы.
Для экспериментов использовали самцов крыс Wistar (вес 65-80 г). В каждую клетку помещали по пять крыс, используя стандартные лабораторные условия. Пищу и воду давали ad libitum.
(i) Индуцирование диабета/гиперальгезии.
Крысам вводили внутрибрюпшнно (IP) стрептозотоцин (STZ) (общая доза 150 мг/кг) (Sapphire Bioscience), растворенного в растворе хлористого натрия (0,9%). Дозу, равную 150 мг/кг, вводили дважды в первый и второй день по 75 мг/кг. Через неделю после инъекции STZ было установлено наличие диабета путем измерения содержания глюкозы в венозной крови, взятой из хвоста, при помощи полосок Ames Glucofilm и колориметра (Ames Glucometer 3, Bayer Diagnostics). Диабетическими считали только крыс с конечным содержанием глюкозы в крови ≥15 мМ. У крыс вновь определяли наличие гипергликемии один раз в неделю для подтверждения все еще высокого содержания глюкозы в крови. Гиперальгезию определяли методом сдавливания лапы, описанным Randall и Selitto11.
Опыты проводили в течение 5 недель после первой инъекции STZ. Считали, что животные, которые характеризовались ноцицептивным порогом сдавливания лапы ниже 30 г (60% от величины у крыс с нормальным весом), проявляют гиперальгезию/невропатическую боль, их использовали при проведении дальнейших опытов. Их количество составляло 25% от всех крыс, которым вводили STZ в этой серии опытов.
(ii) Исследование ноцицептивной системы.
После успешного определения развития гиперальгезии у диабетических животных при сдавливании лап проводили более обширные исследования парадигмы ноцицептивных раздражений у диабетических невропатических животных и контрольных крыс с аналогичным весом; контрольные крысы были моложе на 1-2 недели. Сдавливание лап измеряли методом, описанным Randall и Selitto11, используя Ugo-Basile Algesimeter (Apelex, 1 мм, вес 10 г); использовали увеличивающееся давление на левую заднюю лапу до появления писка крыс. В каждом опыте для группы крыс (n=4-8) с диабетической невропатией и обычных крыс с аналогичным весом использовали следующий протокол:
- пороги чувствительности при отдергивании лап измеряли каждые 5 минут в течение 15 минут, получая показатели a, b и с;
- внутрибрюшинное введение лекарств или их комбинации;
- пороги чувствительности при отдергивании лап измеряли каждые 5 минут в течение дальнейших 35 минут, получая значения d, e, f, g, h, i и j.
Начальные пороговые значения a, b и с у отдельных крыс отличались друг от друга. Таким образом, для того чтобы получить значимые результаты, ответ на действие лекарств, вводимых внутрибрюшинно, невропатическим крысам, стандартизировали согласно нижеприведенному уравнению:
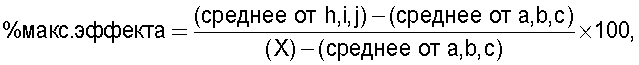
и стандартизировали для обычных крыс с аналогичным весом по уравнению:

где Х - среднее пороговое значение чувствительности при сдавливании лапы до введения лекарства обычным крысам с аналогичным весом.
У обычных крыс с аналогичным весом ответ, равный 2,0, показывает, что введение лекарства привело к увеличению вдвое ноцицептивного порога при сдавливании лапы. У диабетических крыс с невропатией рассчитанный таким образом 100%-ный ответ означает, что лекарство или комбинация лекарств привели к увеличению порога при отдергивании лап до порогового значения, характерного для обычных крыс; аллодиния и гиперальгезия полностью исчезли. Результаты повторных экспериментов с конкретным лекарством или комбинацией лекарств для каждой дозы учитывались и выражались как среднее значение и SEM.
Опыты проводились вслепую, то есть человек, проводивший измерение пороговых значений сдавливания лап, не знал о введенных дозах лекарств. Кривые зависимости ответа от дозы были построены для каждого опиоида, вводимого по отдельности обычным контрольным крысам с аналогичным весом, и для каждого из опиоидов, вводимых по отдельности и в сочетании с 1,0 мг/кг альфадолонацетата, введенного внутрибрюшинно крысам с диабетической невропатией. Кроме того, кривые зависимости ответа от дозы были построены для альфадолонацетата, введенного в отдельности обычным крысам с аналогичным весом, а также крысам с диабетической невропатией.
(iii) Результаты.
На фиг.6А, 7А и 8А приведены кривые зависимости ответа от дозы для каждого из трех опиоидов для обычных крыс и для диабетических невропатических крыс. Во всех случаях дозы опиоидов, которые вызывали значительную антиноцицептивность у обычных крыс с аналогичным весом, вызывали небольшое исчезновение аллодинии и гиперальгезии у крыс с диабетической невропатией.
С другой стороны, альфадолонацетат, как это можно видеть на фиг.9, приводил к антиноцицептивным эффектам у крыс с диабетической невропатией с той же активностью, что и в случае антиноцицептивных реакций у обычных крыс с аналогичным весом. Две кривые зависимости ответа от дозы перекрывают друг друга. Максимальная доза альфадолонацетата, используемого в этих опытах (10 мг/кг) вызывала 80%-ное исчезновение аллодинии и гиперальгезии при сдавливании лап у диабетических невропатических крыс.
Когда 1,0 мг/кг альфадолонацетата, который не является эффективным при индуцировании антиноцицептивности при его введении, вводился в сочетании с каждым из опиоидов диабетическим невропатическим крысам.
Следует иметь в виду, что данное изобретение было описано с помощью иллюстративных примеров, и специалистам очевидно, что могут быть различные модификации и/или изменения на основании описанного в данной заявке, которые также входят в объем изобретения.
Ссылки.
1. Merskey H, Bogduk N. Classification of Chronic Pain, 2nd edn. Seattle: IASP Press, 3994; 394.
2. Woolf CJ and Maimion RJ. Pain: neuropathic pain: aetiology, symptoms, mechanisms and management. Lancet 1999; 353: 1959-64.
3. McQuay HJ, Tramer M, Nye BA, Carroll D, Wiffen PJ, Moore RA. A systematic review of antidepressants in neuropathic pain. Pain 1996; 68: 217-27.
4. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain 1999; 83: 389-400.
5. Rowbotham MC. Kalso Е, McQuay HJ, Wiesenfeld-Hallin Z, eds. The debate over opioids and neuropathic pain. Opioid Sensitivity of Chronic Non-cancer Pain. Seattle: IASP Press, 1999; 18: 307-17.
6. Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992; 50: 355-63.
7. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988; 33: 87-107.
8. Tandnip T, Woolf CJ, Coggeshall RE. Delayed loss of small dorsal root ganglion cells after transection of the rat sciatic nerve. J Comp Neurol 2000; 422:172-80.
9. Malcangio M, Tomlinson DR. A pharmacologic analysis of mechanical hyperalgesia in streptozotocin / diabetic rats. Pain 1998; 76: 151-7.
10. Courteix, С., Eschalier, A., and Lavarenne, J. Streptozocin-induced diabetic rates: behavioural evidence for a model of chronic pain. Pain, 1993, 63, 81-88.
11. Randell, L.O., and Selitto, J.J. A method for measurement of analgesic activity of inflamed tissue, Archiv. Inst. Pharmacdynamie, 1957, 111, 409.
Изобретение относится к медицине, в частности к неврологии, и может быть использовано для изготовления средства для лечения невропатической боли у млекопитающего. Изобретение включает применение 21-ацетокси-3альфа-гидрокси-5альфа-прегнан-11,20-диона или его фармацевтически приемлемых солей для изготовления средства для лечения невропатической боли у млекопитающего. Изобретение позволяет эффективно купировать невропатическую боль. 9 ил.
Применение 21-ацетокси-3альфа-гидрокси-5альфа-прегнан-11,20-диона или его фармацевтически приемлемых солей для изготовления средства для лечения невропатической боли у млекопитающего.
| КАРЛОВ В.А | |||
| Терапия нервных болезней | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| АНАЛОГИ ВИТАМИНА D, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1997 |
|
RU2169142C2 |
| US 6048848, 11.04.2000 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Рабочее колесо центробежного компрессора | 1985 |
|
SU1317184A1 |
| ШАРИКОВЫЙ ЦИЛИНДРИЧЕСКИЙ ВАРИАТОР | 1999 |
|
RU2162554C1 |
| WINFREE C.J | |||
| et al., Analgesic effects of intrathecally-administered 3alpha-hydroxy-5alpha-pregnan-20-one in a rat mechanical visceral pain model, Life Sci | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |

