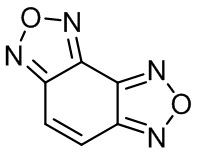
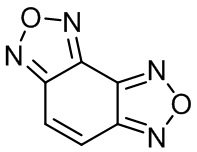
Изобретение относится к органической химии и фармацевтике, а именно к способу получения бензодифуразана 1,
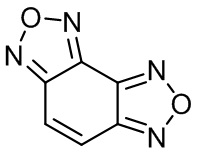
1
который используется непосредственно сам, или в качестве предшественника в различных направлениях органического синтеза, является биоактивным соединением и представляет практический интерес при производстве новых лекарственных средств.
Глобализация вызвала рост заболеваемости населения Земли практически по всем нозологическим группам: от сердечно-сосудистых и онкологических заболеваний до психических расстройств и масштабных эпидемий. На этом фоне резко возрастает потребность в новых эффективных, избирательно действующих и малотоксичных лекарственных препаратах, природного и синтетического происхождения для фармакотерапии и химико-лабораторной диагностики различных заболеваний, а также в разработке оригинальных и усовершенствовании известных подходов для их получения.
В арсенале фармацевтических средств существует большое количество антибактериальных, противогрибковых, противопаразитных лекарственных средств. Несмотря на это, современная медицина часто сталкивается с проблемой резистентности микроорганизмов к лекарственным препаратам. Поэтому создание новых лекарственных веществ, обладающих длительным действием против большого числа бактерий и грибов, является важной и актуальной задачей органической и медицинской химии.
В последние 10-15 лет методами высокопроизводительного скрининга (HTS) и комбинаторной химии различными исследовательскими группами и фармацевтическими фирмами были отобраны структуры - лидеры, на основе которых в медицинскую практику был введен ряд новых лекарственных препаратов.
Одной из возможных структур - лидеров может быть бензодифуразан 1 или его производные.
Установлено: бензодифуразан 1 и его производные обладают выраженными антимикробными, антимикотическими и акарицидными свойствами. Так гидроксибензодифуразан и метоксибензодифуразан более чем в 30 раз активнее хлорофоса. Нитропроизводные бензодифуразана проявляют высокую бактериостатическую активность превосходящие по активности препараты вазин и формалин. Гидроксинитробензодифуразан превосходит по бактериостатической активности на два порядка известный лекарственный препарат Сульгин. Бензодифуразан 1 обладает выраженным антимикробным действием. Его МБСК (Минимальная бактериостатическая концентрация) в отношении золотистого стафилококка составила 0,1%. [4,6]
Известен патент по применению бензодифуразана (WO 9854183 A2) [1]. Бензодифуразан 1 присутствует в составе производного эрголина 2, используемого в качестве эффективного лекарственного средства при лечении депрессии, беспокойства и биполярных расстройств. Соединение показано для лечения шизофрении и деменции, в частности старческого слабоумия типа Альцгеймера.
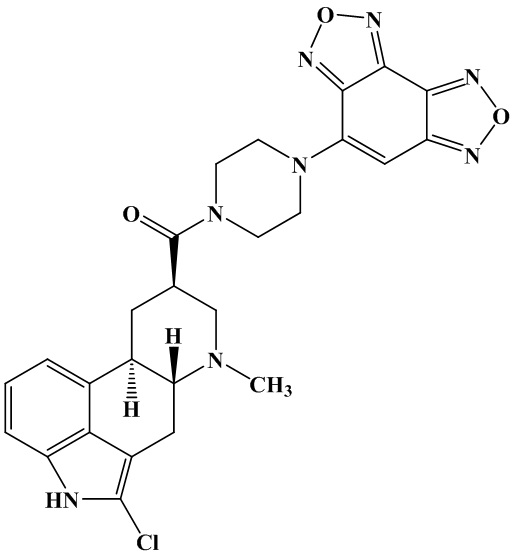
2
Таким образом, как сам бензодифуразан 1, так и его производные являются биоактивными соединениями и представляют практический интерес в качестве лекарственных средств.
Однако способы получения бензодифуразана 1 и его производных многостадийны и сложны [2-4]. Этим объясняется высокая коммерческая стоимость как самого бензодифуразана так и его производных.
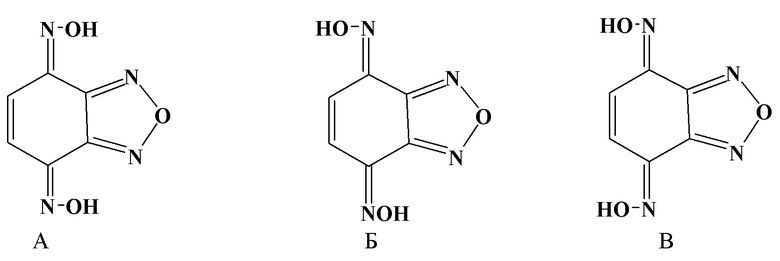
В работе W.Borsche [5] описан синтез циклогекс-5-ен-1,2,3,4-тетраон-тетраоксима и получение бензодифуразана при действии уксусного ангидрида на этот тетраоксим. При проверке методики, нами в качестве основного и единственного продукта вместо тетраоксима был получен бензо[с][1,2,5]оксадиазол-4,7-дион диоксим. Продукт представляет собой по данным ПМР смесь изомерных оксимов в соотношении А:Б:В = 1:4:1.7. Все изомеры были выделены хроматографией и охарактеризованы на основании аналитических и спектральных данных (пример 1).
В известных синтезах бензодифуразана исходят из доступных бензофуроксанов, в которых достраивают второе фуроксановое кольцо и убирают N-оксиды [5-7].
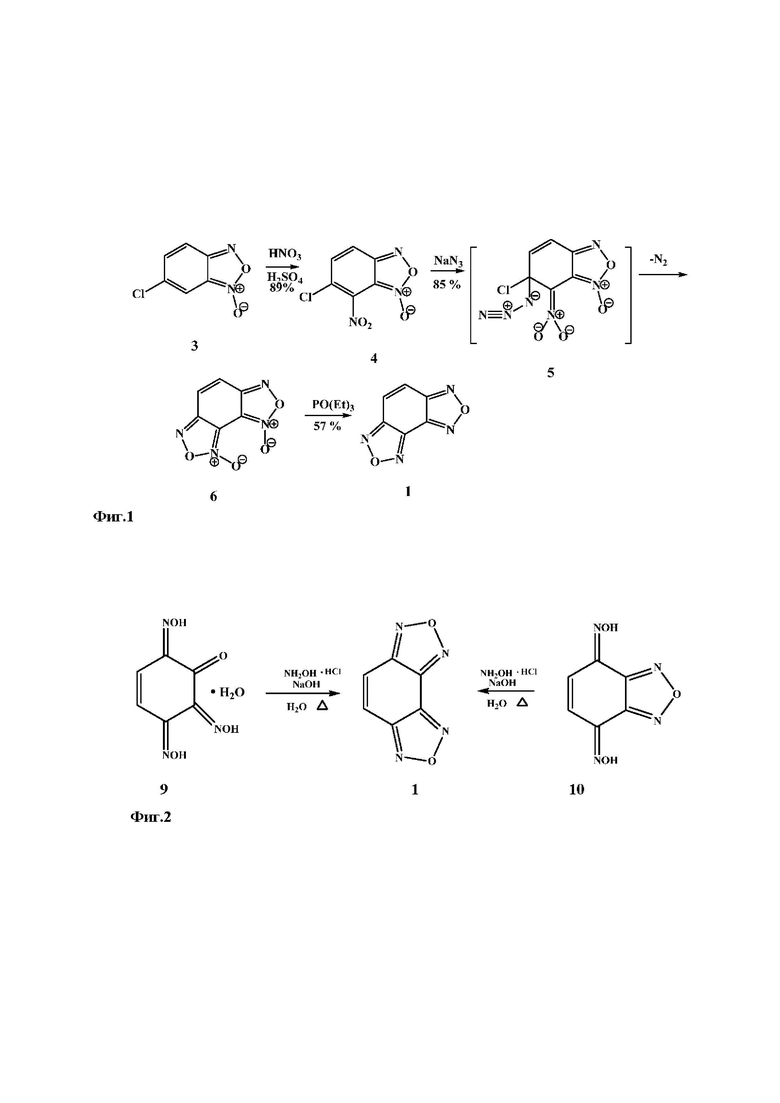
Прототипом настоящего изобретения является способ получения бензодифуразана 1, описанный в работах [6-7]. Способ заключается в нитровании хлорбензофуроксана 3 азотной кислотой в серной кислоте с образованием 6-хлор-7-нитробензофуроксана 4, при взаимодействии которого с азидом натрия происходит замещение атома хлора на азидогруппу. При нагревании азидонитробензофуроксана 5 происходит циклизация с образованием бензодифуроксана 6. Обработка последнего триэтилфосфитом приводит к бензодифуразану 1 (Фиг. 1. Способ получения бензодифуразана из хлорбензофуроксана).
Технологическими недостатками способа-прототипа являются многостадийность и трудоемкость. Использование дополнительного оборудования (роторный испаритель, вакуум-насос) значительно усложняет технологию производства. Использование в качестве исходного соединения хлорбензофуроксана 3, синтез которого происходит в несколько стадий, требует дополнительных временных и материальных ресурсов. Применение концентрированных серной и азотной кислот, а так же высокотоксичного азида натрия и триэтилфосфита в значительной степени снижают экологичность процесса.
Задача настоящего изобретения состоит в разработке нового эффективного способа получения бензодифуразана 1, преимуществом которого являются упрощение технологии, сокращение материальных, временных и энергозатрат.
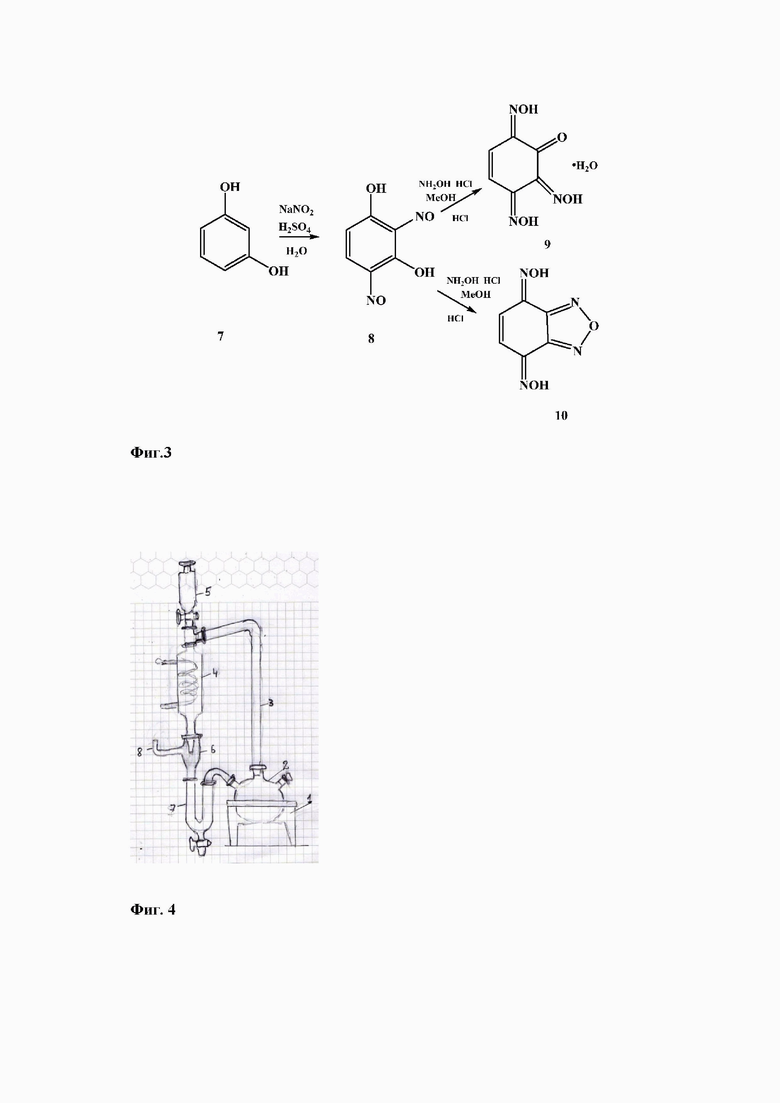
Поставленная задача решается предлагаемым способом получения бензодифуразана 1 исходя из бензохиноноксимов - 2,5,6-трис(гидроксимино)циклогекс-3-ен-1-он гидрата 9 или из бензо[с] [1,2,5]оксадиазол-4,7-дион диоксима 10 (Фиг 2. Способ получения бензодифуразана из бензохиноноксимов).
Синтез бензохиноноксимов 9-10 приведен в работах [8-9] и может быть осуществлен исходя из коммерчески доступного резорцина, в соответствии с фиг. 3 (Фиг. 3. Синтез бензохиноксимов).
Нитрозирование резорцина 7 в водном растворе нитрита натрия в присутствии серной кислоты приводит с 98% выходом к динитрозорезорцину 8. Оксимирование динитрозорезорцина 8 гидроксиламином солянокислым в 50% спирте путем кипячения в течение двух часов приводит с выходом 90% к 2,5,6-трис(гидроксимино)циклогекс-3-ен-1-он гидрату 9, а кипячение в течение 20 часов с гидроксиламином солянокислым в кислом метанольном растворе, дает бензо[с][1,2,5]оксадиазол-4,7-дион диоксим 10 с выходом около 70%.
Нагревание бензохиноноксимов - 2,5,6-трис(гидроксимино)-циклогекс-3-ен-1-он гидрата 9 либо бензо [с] [1,2,5]оксадиазол-4,7-дион диоксима 10 в водном растворе гидроксиламина приводит к образованию бензодифуразана 1. Установка для получения бензодифуразана 1 приведена на фиг. 4 (Фиг 4. Установка для получения бензодифуразана: 1-Колбонагреватель, 2-Трехгорлая колба объемом 500 мл, 3-Насадка, 4-Холодильник Димрота или Отмера, 5-Делительная воронка для хлороформа, 6-Насадка, 7-Гидрозатвор, 8-Связь с атмосферой). Образующийся бензодифуразан 1 летит с водяным паром и поступает на холодильник 4, где он кристаллизуется на холодных поверхностях холодильника (температура плавления бензодифуразана 61 оС). После образования достаточного количества бензодифуразана 1 он смывается из делительной воронки 5 хлороформом. Хлороформсодержащий экстракт сушится над MgSO4 , осушитель отфильтровывается, растворитель упаривается, остаток суспендируется в гексане, осадок бензодифуразана 1 отфильтровывается, сушится. Время кипячения варьируется в пределах 20-30 часов. Окончание процесса определяется визуально, когда целевой продукт, в виде белого порошка, перестает накапливаться на холодных частях холодильника. Выход продукта 50 -54 %.
Предложенный способ позволяет достичь легкости при реализации технологического процесса, без применения дорогостоящего оборудования и реактивов, с минимальными затратами времени и с соблюдением экологических норм производственного процесса, получая при этом перспективный биоактивный продукт, который может быть использован при разработке новых лекарственных препаратов, содержащих бензодифуразан.
Спектральные исследования выполнены в Химическом Сервисном Центре коллективного пользования СО РАН.
Изобретение иллюстрируется следующими примерами:
Пример 1. Получение бензо[c][1,2,5]оксадиазол-4,7-дион диоксима по методике Borsche [5]
В Круглодонную колбу емкостью 2 л прибавляют 37.2 г динитрозорезорцина 800 мл метанола 300 мл конц. соляной кислоты и 42 г гидроксиламина солянокислого. Смесь кипятят с обратным холодильником 24 часа. Фильтруют в горячем виде. Упаривают до объема 50 мл, прибавляют около 10 мл воды, осадок отфильтровывают, промывают водой, сушат. Получают около 37 г диоксима в виде коричневого порошка. Продукт хорошо растворим в этилацетате. По данным тонкослойной хроматографии на пластинках силуфола в системе: этилацетат : гексан = 1:3 наблюдалось три пятна. Все три вещества выделяли колоночной хроматографией. Изомеры отличаются друг от друга конфигурацией оксимных групп.
Смесь изомеров разделяли хроматографией на силикагеле, в качестве элюента использовали вначале смесь этилацетат : гексан = 1:3, далее 1:1 и в конце чистый этилацетат.
Выделено соединение А. (4Z,7Z) Бензо[c][1,2,5]оксодиазоло-4,7-дион диоксим -желтый кристаллический порошок. т. пл. 215-216 °С (спирт). УФ спектр, λ макс., нм, (lg έ): 306 (4.07), Спектр ЯМР 1Н, δ, м. д. (Дейтероацетон): 7.50с (2Н, 2СН), 12.28 уш. c.(4Н, 4 ОН) Спектр ЯМР 13C, δ, м. д. (Дейтероацетон): 127.31 (все СН), 140.41, 143.38(все С). Масс-спектр, m/z (Iотн., %): 180 [M]+, (100), 163 (22) 120 (23), 103 (45),80 (19), 76 (15 ). Найдено, %: С 40.12; Н 2.25; N 31.09. M+ 180.0276. C6H4N4O3. Вычислено, %: С 40.01; Н 2.24; N 31,11. M 180.0278.
Вторым выделено соединение Б. (4Z,7E) Бензо[c][1,2,5]оксодиазоло-4,7-дион диоксим - желтый кристаллический порошок. т. пл. 226-227 °С (спирт). УФ спектр, λ макс., нм, (lg έ): 298 (4.07). Спектр ЯМР 1Н, δ, м. д. (Дейтероацетон): 6.96 д (1Н, СН) J =10.5 Гц.), 7.42 д (1Н, СН J =10.5 Гц.), 12.30 уш. с. (4Н, 4ОН) Спектр ЯМР 13C, δ, м. д. (Дейтероацетон): 140.75 , 141.29, 143.31, 148. 361 (все С), 118,02, 129.93 (все СH). Масс-спектр, m/z (Iотн., %): 180 [M]+, (100), 163 (24), 152 (30), 120 (27), 103 (47). 80 (12), 76 (16). Найдено, %: С 40.08; Н 2.28; N 30.90. М+ 180.0276. C6H4N4O3. Вычислено, %: С 40.01; Н 2.24; N 31,11. M 180.0278.
И последним выделено соединение В. (4E,7E) Бензо[c][1,2,5]оксодиазоло-4,7-дион диоксим - желтый кристаллический порошок. Т. пл. 218-219 °C (спирт). УФ спектр, λ макс., нм, (lg έ): 297 (4.07), Спектр ЯМР 1Н, δ, м. д. (Дейтероацетон): 7.50 (2Н, 2СН). 12.28 уш.с. (4н, 4 NОН) Спектр ЯМР 13C, δ, м. д. (Дейтероацетон): 141.25, 148.40 (все С), 119.79 (СH). Масс-спектр, m/z (Iотн., %): 180 [M]+, (100), 163 (24), 162 (77), 150 (12), 133(12), 120 (29), 103 (56). 93 (13), 80 (22), 76 (34). Найдено, %: С 40.00; Н 2.47; N 31.24. М+ 180.0276 C6H4N4O3. Вычислено, %: С 40.01; Н 2.24; N 31,11. M 180.0278.
Конфигурация оксимных групп определена по литературным данным E.Breitmaier, W. Voelter "Carbon-13 NMR Spectroscopy...", 1987, p.173
Примеры 2-3. Получение бензодифуразана 1.
Пример 2. Готовят раствор 24 г (0.6 моль) натрия едкого в 50 мл воды. Примерно 5 мл полученного раствора едкого натра прибавляют к смеси 20.1 г (0.10 моль) 2,5,6-трис(гидроксимино)циклогекс-3-ен-1-он гидрата 9 250 мл воды и 41.7 г (0.6 моль) гидроксиламина солянокислого. Смесь осторожно нагревают до начала экзотермической реакции. Нагрев убирают и по каплям добавляют остаток раствора едкого натра с такой скоростью, чтобы смесь слабо кипела. Время прибавления примерно 1 час. После прибавления всего количества раствора едкого натра смесь нагревают до кипения и кипятят с обратным холодильником Димрота, либо холодильником Отмера. Продукт возгоняется с водяным паром и оседает на стенках холодильника. Его периодически смывают хлороформом. Время кипячения 20-30 часов, пока не перестанет выделяться продукт. Хлороформ в делительной воронке отделяют от воды, сушат над прокаленным MgSO4. Осушитель отфильтровывают. Хлороформ упаривают. Остаток суспендируют в гексане, осадок отфильтровывают. Получают около 8.25 г (50.9 %) бензодифуразана 1, в виде белого порошка. Т. пл. 59 – 60 °С
ИК спектр, ν/см-1: 676.92, 815.78, 873.64, 995.13, 1132.06, 1417,49, 1587.20, 3072.19. (Указаны пики с интенсивностью более 30 %). ). УФ спектр, λ макс., нм, (lg έ): 230 (4.07), 234 (4.07). ЯМР 1Н (400 МГц, CDCl3, δ, м.д.,): 7.88 с (2Н, 2СН). ЯМР 13С (100 МГц, CDC13, δ, м.д.,): 121.08 (СН), 139.59, 149.60 (С
Масс-спектр, m/z (Iотн., %): (Указаны пики с интенсивностью более 10 %). 162 [M]+ (100), 104 (18), 84 (11), 75 (13), 64 (18), 51 (19 ), 50 (27). Найдено, %: С 44.43; Н 1.25; N 34.59. M+ 162.0176. C6H2N4O2. Вычислено, %: С 44.44; Н 1.23; N 34,57. M 162.0172.
Характеристики продукта соответствуют литературным данным [7].
Пример 3. Готовят раствор 12 г (0.3 моля) едкого натра в 25 мл воды. Примерно 5 мл полученного раствора прибавляют к смеси 9.0 г (0.05 моль) бензо [с] [1,2,5]оксадиазол-4,7-дион диоксима 10 в 100 мл воды и 20.85 г (0.3 моль) гидроксиламина солянокислого. Смесь осторожно нагревают до начала экзотермической реакции. Нагрев убирают и по каплям добавляют остаток раствора едкого натра с такой скоростью, чтобы смесь слабо кипела. Время прибавления примерно 1 час. После прибавления всего количества раствора едкого натра смесь нагревают до кипения и кипятят с обратным холодильником Димрота, либо с обратным холодильником Отмера. Продукт возгоняется и оседает на стенках холодильника. Его периодически смывают хлороформом. Время кипячения 20-30 часов, пока не перестанет выделяться продукт. Хлороформ в делительной воронке отделяют от воды, сушат над прокаленным MgSO4. Отфильтровывают осушитель, хлороформ упаривают. Остаток суспендируют в гексане, отфильтровывают. Получают около 4.5 г (54 %) бензодифуразана 1, в виде белого порошка. Т.пл. 59-60 °С.
ИК спектр, ν/см-1: 676.92, 815.78, 873.64, 995.13, 1132.06, 1417,49, 1587.20, 3072.19 (Указаны пики с интенсивностью более 30 %). ЯМР 1Н (400 МГц, CDCl3, δ, м.д.,):7.88 (с, 2Н). ЯМР 13С (100 МГц, CDC13, δ, м.д.,): 121.08 (СН), 139.59, 149.60 (С). Характеристики продукта соответствуют литературным данным [7 ].
Литература:
1. Патент WO9854183A2
2. Е. А. Ермолаева Синтез строение и свойства нитрозамещенных бензофуразана и бензодифуразана // Дис. Казань 2005.
3. С. И. Ефимов Замещенные бензофуразана и бензодифуразана: синтез, строение, свойства // Дис. Казань 2001.
4. Р. Г. Каримова Фармако-токсилогическая оценка замещенных бензодифуразана // Дис. Казань 2003.
5. W. Borsche; H. Weber Justus Liebigs Annalen der Chemie, 489 (1931), 270-291.
6. Ф. С. Левинсон, М. И. Евгеньев, Е. А. Ермолаева, С. И. Ефимов, И. Ф. Фаляхов, Т. В. Гарипов, Р. Г. Каримова Химико-фармацевтический журнал. 37, (2003), 12-15.
7. C. M. Cillo, T. D. Lash, Journal of Heterocyclic Chemistry, 41, (2004), 955–962.
8. A. J. Boulton, et al. Journal of the Chemical Society, (1965), 5958 – 5964.
9. V. A. Samsonov, G. E. Salґnikov, and A. M. Genayev Russian Chemical Bulletin, International Edition, 58, (2009), 2369—2375.
10. В. А. Самсонов, Л. Б Володарский. ХГС, 10, (1991), 1408-1413.
11. J. J. Lewis, J. Heterocycl. Chem., 12, (1975), p. 601.
12. R. M.Paton , 1.2.5-Oxadiazoles / Science of Synthesis – 2009- vol, 13 – Ch.8 –p. 185-218.
13. Л. И.Хмельницкий, С.С.Новиков, Т.И.Годовикова Химия фуроксанов Реакции и применение. Наука Москва. 1983.стр 309.
14. База данных по свойствам и реакциям химических веществ - Reaxys издательства Elsevier.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3, 4-БИС(3-АМИНОФУРАЗАН-4-ИЛ)-ФУРАЗАНА И ЕГО N, N'-ДИАЦИЛЬНЫХ ПРОИЗВОДНЫХ | 2012 |
|
RU2489428C1 |
| ГЕТЕРОПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С МЕТАБОТРОПНЫМИ ГЛУТАМАТНЫМИ РЕЦЕПТОРАМИ | 2000 |
|
RU2296127C9 |
| СОЛИ БЕНЗОФУРОКСАНОВ С ЛОМЕФЛОКСАЦИНОМ, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2015 |
|
RU2602804C1 |
| ПРОИЗВОДНЫЕ 1,2,5-ОКСАДИАЗОЛО-[3,4-D]-ПИРИДАЗИН-5,6-ДИОКСИДА В КАЧЕСТВЕ АКТИВАТОРОВ РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ И СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1997 |
|
RU2165256C2 |
| Соли 5-нитрамино-[1,2,3]триазоло[4,5-c][1,2,5]оксадиазола и способ их получения | 2023 |
|
RU2812574C1 |
| ПРОИЗВОДНЫЕ 4Н-БИС[1,2,5]ОКСАДИАЗОЛО[3,4-b:3',4'-f]АЗЕПИН-8,9-ДИАМИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2499799C2 |
| ПРОИЗВОДНЫЕ 3,4-БИС(ФУРАЗАН-3-ИЛ)ФУРОКСАНА, ГЕНЕРИРУЮЩИЕ ОКСИД АЗОТА, АКТИВИРУЮЩИЕ РАСТВОРИМУЮ ФОРМУ ГУАНИЛАТЦИКЛАЗЫ, ИНГИБИРУЮЩИЕ АГРЕГАЦИЮ ТРОМБОЦИТОВ И ОБЛАДАЮЩИЕ СПАЗМОЛИТИЧЕСКИМ, СОСУДОРАСШИРЯЮЩИМ, ГИПОТЕНЗИВНЫМ ДЕЙСТВИЕМ, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2240321C2 |
| Способ получения 4,7-дибром[1,2,5]тиадиазоло[3,4-d]пиридазина | 2018 |
|
RU2668978C1 |
| Органический светоизлучающий диод | 2020 |
|
RU2752951C1 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
Изобретение относится к органической химии и фармацевтике, а именно к способу получения бензодифуразана формулы 1. Способ заключается в циклизации 2,5,6-трис(гидроксиимино)циклогекс-3-ен-1-он гидрата или бензо[с][1,2,5]оксадиазол-4,7-дион диоксима путем кипячения в течение 20-30 часов в водном растворе гидроксиламина с получением бензодифуразана. Технический результат – получение бензодифуразана эффективным способом. 3 пр., 4 ил.
 1
1
Способ получения бензодифуразана формулы 1
 1,
1,
заключающийся в циклизации 2,5,6-трис(гидроксимино)циклогекс-3-ен-1-он гидрата или бензо[с][1,2,5]оксадиазол-4,7-дион диоксима путем кипячения в течение 20-30 часов с обратным холодильником в водном растворе гидроксиламина.
| BOULTON, A.J | |||
| et al | |||
| Heterocyclic Rearrangements | |||
| Part I V.l Furoxano- and Furaxano- benxofuroxan | |||
| Journal of the Chemical Society, 1965, p.5958-5964 | |||
| SAMSONOV, V.A | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ChemInform, 2010, 24(15), p.1 | |||
| SAMSONOV, V.A | |||
| et al | |||
| Synthesis of | |||
