ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение обеспечивает атропоизомеры и дейтерированные производные ATM ингибитора 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(3-фтор-5-метоксипиридин-4-ил)-7-метокси-3-метил-1,3-дигидроимидазо[4,5-с]хинолин-2-она, а также его фармацевтически приемлемые соли и твердые формы. Эти соединения являются полезными в ингибировании, регуляции и/или модуляции сигнальной трансдукции с помощью ATM киназы. Изобретение также обеспечивает композиции, которые включают указанные атропоизомеры, твердые формы, фармацевтически приемлемые соли и дейтерированные производные в соответствии с настоящим изобретением, а также способы применения этих композиций в лечении различных расстройств, связанных с ATM киназой, в частности, рака.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Серин/треонин протеинкиназа ATM (мутантная при атаксии-телангиэктазии киназа) принадлежит к PIKK семейству киназ с каталитическими доменами, которые являются гомологичными фосфо-инозитид-3 киназам (киназа PI3, PI3K). Эти киназы участвуют во множестве ключевых функций клетки, таких как рост клеток, пролиферация клеток, миграция, дифференциация, выживание и адгезия клеток. В частности, эти киназы отвечают на повреждение ДНК активацией остановки клеточного цикла и программы репарации ДНК (DDR: реакция на повреждение ДНК). ATM является продуктом гена ATM и играет ключевую роль в репарации повреждений двухцепочечной ДНК (DSB: разрывы двухцепочечной ДНК) путем гомологичной рекомбинации и негомологичного непрерывного соединения (NHEJ). Этот тип повреждения двойной цепи является особенно цитотоксичным.
Одной из основных особенностей опухолей у людей является их геномная нестабильность со специфическими дефектами механизма репарации ДНК, которые пока неизвестны при большинстве видов рака. Эта нестабильность представляет собой терапевтическую отправную точку для химиотерапии, которая преимущественно осуществляется в течение некоторого времени. Кроме того, существует несколько синдромов, при которых основным генетическим фактором является мутация, связанная с потерей функции гена, который модулирует ответ на повреждение двухцепочечной ДНК. Это приводит к включению атаксии-телеангиэктазии, которая вызвана дефектным геном ATM. Общей чертой всех этих синдромов является то, что они вызывают чрезмерную чувствительность к облучению (Lavin & Shiloh (1997) Annu. Rev. Immunol. 15: 177; Rotman & Shiloh (1998) Hum. Mol. Genet. 7: 1555, которые полностью включены в настоящий документ в качестве ссылки). Дефектные по ATM клетки, соответственно, являются чувствительными к агентам и другим мерам, которые вызывают повреждение двухцепочечной ДНК, что делает ATM привлекательной мишенью для химической и радиационной сенсибилизации при лечении рака.
Таким образом, ATM (мутантная при атаксии-телангиэктазии киназа) является ключевым регулятором репарации разрывов двухцепочечной ДНК, которая индуцируется при широко используемой радио- и химиотерапии. ATM передает обширный сигнал множеству расположенных ниже эффекторов, включая р53. Нерепарированные разрывы двойной цепи приводят к активации контрольных точек, остановке клеточного цикла и, в конечном итоге, к гибели опухолевых клеток. Таким образом, ATM является привлекательной точкой вмешательства для ингибирования репарации индуцированных разрывов двойной цепи.
Соединение Вортманнин было среди тех, которые первоначально исследовались в этом контексте, это соединение продемонстрировало лучевую сенсибилизацию, которая могла быть связана, среди прочего, с ингибированием ATM. Однако это соединение не подходило для терапевтического использования из-за токсичности in vivo. Исходя из химической структуры ингибитора PI3K LY294002, KuDOS Pharmaceuticals идентифицировала ингибитор ATM: KU-55933 (2-морфолино-6-(тиантрен-1-ил)-4Н-пиран-4-он). С помощью этого соединения была достигнута сенсибилизация к ионизирующему излучению и химиотерапевтическим агентам, повреждающим двухцепочечную ДНК (Hickson, I., и др. (2004), Cancer Res 64, 9152-9159, который полностью включен в настоящий документ в качестве ссылки). Однако, KU-55933 оказался непригодным для использования in vivo, предположительно из-за его высокой липофильности. Впоследствии были разработаны KU-60019 (2-((2S,6R)-2,6-диметилморфолино)-N-(5-(6-морфолино-4-оксо-4Н-пиран-2-ил)-9Н-тиоксантен-2-ил)ацетамид) и KU-559403 (2-(4-метилпиперазин-1-ил)-N-[5-(6-морфолино-4-оксопиран-2-ил)тиоксантен-2-ил]ацетамид), и KU-559403 был признан достаточно многообещающим, чтобы войти в клинические испытания для лечения запущенных солидных опухолей.
Существуют и другие ингибиторы ATM, которые подтверждают указанное выше мнение, они в настоящее время находятся в стадии клинической разработки, например, AZD0156, AZD1390 и М3541, включая клинические исследования их комбинации с радиационной терапией.
Несмотря на то, что в области ингибиторов ATM был достигнут значительный прогресс, все еще остается потребность в создании соединения, которое обладает высоким ингибированием ATM киназы, а также имеет полезную селективность по сравнению с другими киназами, полезную биодоступность и/или сниженные побочные эффекты.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Обеспечение малых молекул, которые эффективно ингибируют, регулируют и/или модулируют передачу сигнала с помощью ATM киназы, является желательным и представляет собой объект настоящего изобретения. Кроме того, желательно обеспечить ингибиторы ATM, которые являются селективными, т.е. не обладают или обладают значительно более низкой активностью против других киназ. Кроме того, желательно обеспечить ингибиторы ATM (ATMi), которые проявляют полезные свойства в отношении известных мишеней, вызывающих нежелательные побочные эффекты. Таким образом, одна цель состоит в том, чтобы обеспечить соединения, которые обладают сниженными побочными эффектами и/или связанной с ними токсичностью. Кроме того, целью настоящего изобретения является обеспечение ингибитора ATM с хорошей биодоступностью. Другой или альтернативной целью настоящего изобретения является обеспечение ингибитора ATM с полезными свойствами твердой формы, такими, как благоприятно низкая гигроскопичность и/или другие физические свойства.
По крайней мере, одна задача, как описано выше, и дополнительные задачи решается/решаются с помощью атропоизомеров и дейтерированных производных ингибитора ATM 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(3-фтор-5-метоксипиридин-4-ил)-7-метокси-3-метил-1,3-дигидроимидазо[4,5-с]хинолин-2-она (Соединение Y), а также их твердых форм, фармацевтически приемлемых солей и композиций.
Один аспект изобретения обеспечивает два соединения, которые являются атропоизомерами Соединения Y и представлены формулами:

и их фармацевтически приемлемые соли.
Другой аспект настоящего изобретения обеспечивает твердые формы Соединения 1:
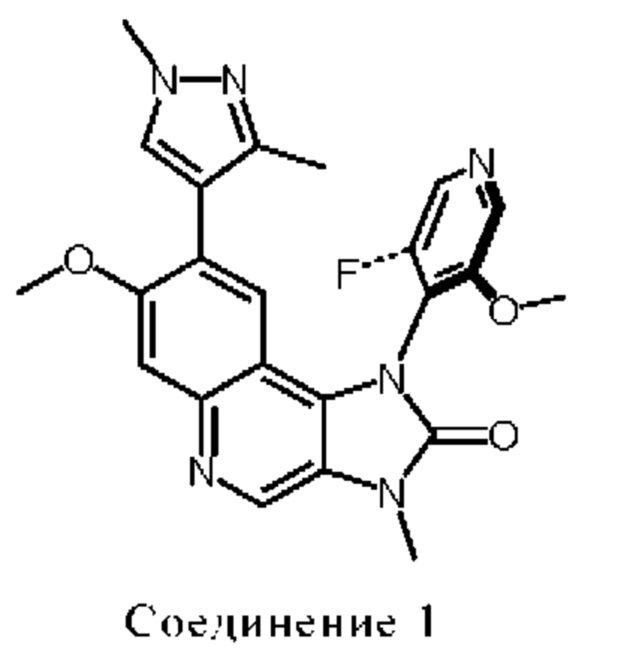
Другой аспект относится к определенным особенно предпочтительным фармацевтически приемлемым солям Соединения 1, в частности, фумарату Соединения 1, эдизилату Соединения 1 и напсилату Соединения 1, которые вместе также могут упоминаться далее в данной заявке как "Соединения 1-а".
Другой аспект настоящего изобретения обеспечивает дейтерированные Соединения 3, 4 и 5, которые представлены следующими формулами:
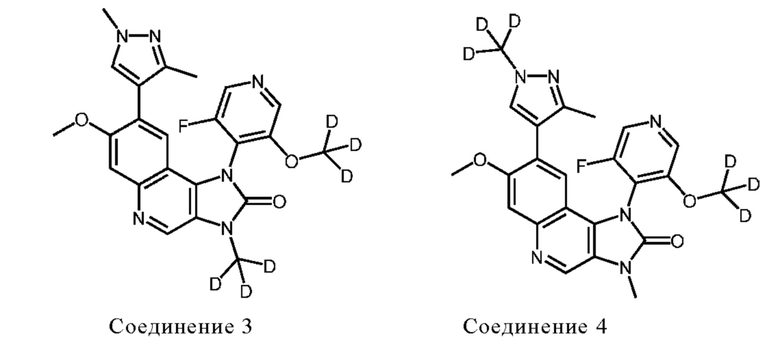
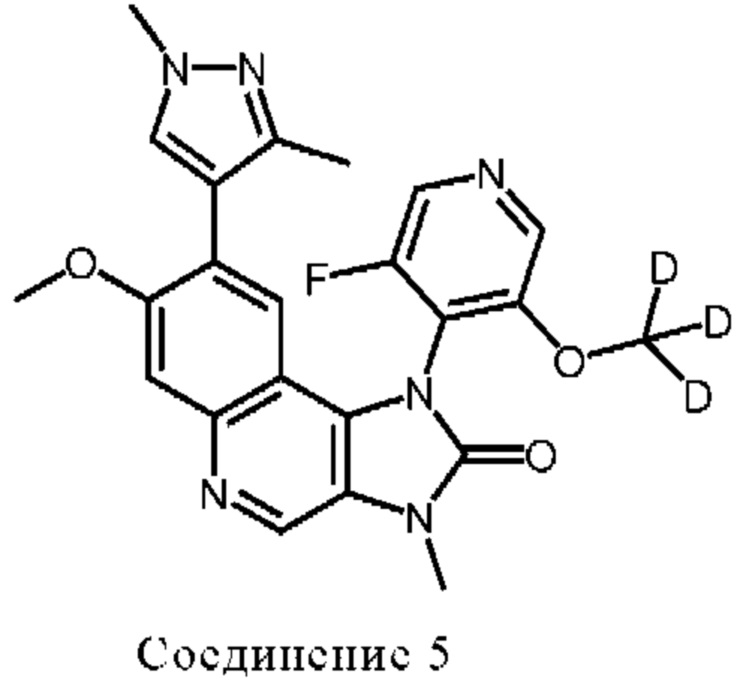
или их атропоизомеры или фармацевтически приемлемые соли.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1 отображает аннотированный 1H ЯМР спектр Соединения 1.
Фигура 2 отображает аннотированный 13С ЯМР спектр Соединения 1.
Фигура 3 отображает аннотированный 19F ЯМР спектр Соединения 1.
Фигура 4 отображает УФ-видимый спектр Соединения 1 в метаноле.
Фигура 5 отображает ВЭЖХ хроматограмму Соединений 1 и 2.
Фигура 6 отображает схему получения Соединения 1.
Фигура 7 отображает кристаллическую структуру Соединение-1-дибензоил-D-тартрата (А) и его рентгеновскую порошковую дифракцию (РПД) (Б).
Фигура 8 отображает кристаллическую структуру Соединение-2-дибензоил-L-тартрата.
Фигура 9 отображает диаграмму профиля растворения без поглощения в биорелевантной среде FaSSIF Соединения 1 и его специфических солей.
Фигура 10 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы фумарата Соединения 1.
Фигура 11 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы напсилата Соединения 1.
Фигура 12 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы эдизилата Соединения 1.
Фигура 13 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы А2 Соединения 1.
Фигура 14 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы А1 Соединения 1.
Фигура 15 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы A3 Соединения 1.
Фигура 16 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы NF9 Соединения 1.
Фигура 17 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы HI гидрата Соединения 1.
Фигура 18 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы Н2 гидрата Соединения 1.
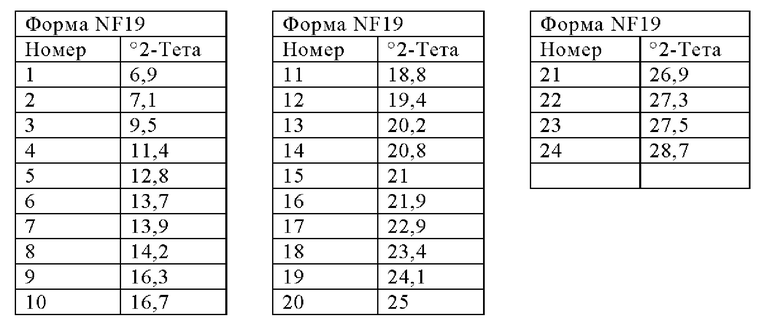
Фигура 19 отображает профиль рентгеновской порошковой дифракции (РПД) твердой формы NF19 Соединения 1.
Фигура 20 показывает сильное ингибирование роста опухоли, индуцированное ионизирующим облучением (ИО) и совместным пероральным введением Соединения 1 (6×5 дней, 2 Гр; модель опухоли FaDu SCCHN).
Фигура 21 показывает результаты in vivo оценки противоопухолевой активности Соединения 1 и сравниваемого ATM ингибитора в комбинации с олапарибом в модели тройного отрицательного ксенотрансплантата рака молочной железы, полученного от пациента НВСх-10.
Фигура 22 показывает кривую нагревания в методе ДСК (дифференциальня сканирующая калориметрия) Формы А2 Соединения 1.
Фигура 23 показывает кривую нагревания в методе ТГА (термогравиметрический анализ) Формы А2 Соединения 1.
Фигура 24 показывает изотерму поглощения воды в методе ДСП (динамическая сорбция паров) (25°С) Формы А2 Соединения 1.
Фигура 25 показывает ДСК кривую нагревания Формы А1 Соединения 1.
Фигура 26 отображает ТГА кривую нагревания Формы А1 Соединения 1.
Фигура 27 показывает ДСП изотерму поглощения воды (25°С) Формы A3 Соединения 1.
Фигура 28 отображает ДСК кривую нагревания Формы Н2 Соединения 1 (гидрат).
Фигура 29 показывает ТГА кривую нагревания Формы Н2 Соединения 1 (гидрат).
Фигура 30 показывает ДСП изотерму поглощения воды (25°С) Формы Н2 Соединения 1 (гидрат).
Фигура 31 отображает ДСК кривую нагревания фумарата Соединения 1 (Форма NF6).
Фигура 32 показывает ТГА кривую нагревания фумарата Соединения 1 (Форма NF6).
Фигура 33 показывает ДСП изотерму поглощения воды (25°С) фумарата Соединения 1 (Форма NF6).
Фигура 34 отображает ДСК кривую нагревания напсилата Соединения 1 (NF7).
Фигура 35 показывает ТГА кривую нагревания напсилата Соединения 1 (NF7)
Фигура 36 отображает ДСП изотерму поглощения воды (25°С) напсилата Соединения 1 (NF7.)
Фигура 37 отображает ДСК кривую нагревания эдизилата Соединения 1 (NF8).
Фигура 38 показывает ТГА кривую нагревания эдизилата Соединения 1 (NF8).
Фигура 39 отображает ДСП изотерму поглощения воды (25°С) эдизилата Соединения 1 (NF8).
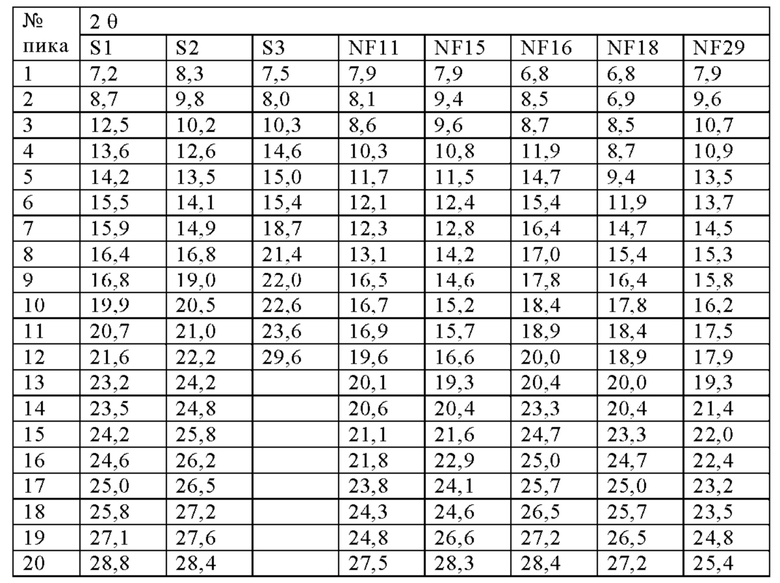
Фигура 40 показывает РПД (рентгеновскую порошковую дифракцию) метанолата Соединения 1 в твердой форме S1.
Фигура 41 отображает РПД смешанного гидрата/метанолата Соединения 1 в твердой форме S2.
Фигура 42 показывает РПД сольвата ТГФ (тетрагидрофурана) Соединения 1 в твердой форме S3.
Фигура 43 отображает РПД сольвата диоксана Соединения 1 в твердой форме NF11.
Фигура 44 показывает РПД сольвата хлороформа Соединения 1 в твердой форме NF15.
Фигура 45 отображает РПД сольват уксусной кислоты Соединения 1 в твердой форме NF16.
Фигура 46 показывает РПД сольвата уксусной кислоты Соединения 1 в твердой форме NF18.
Фигура 47 отображает РПД сольвата 1,4-диоксана Соединения 1 в твердой форме NF29.
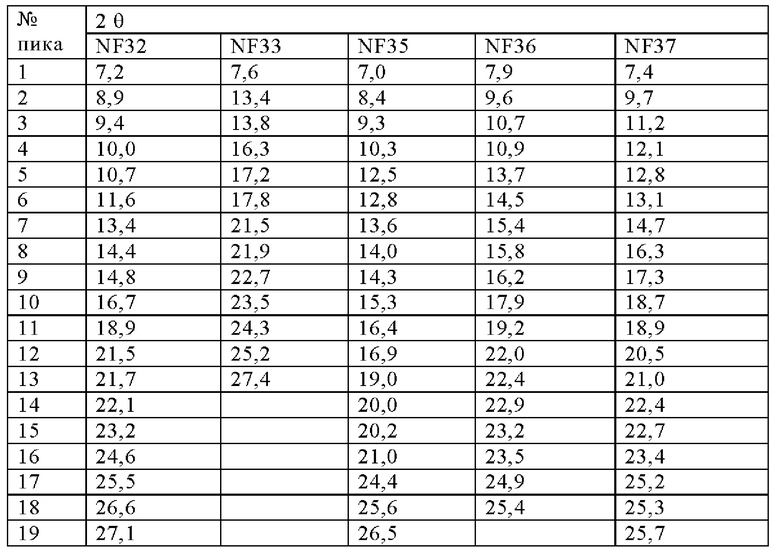
Фигура 48 показывает РПД сольвата дихлорметана Соединения 1 в твердой форме NF32.
Фигура 49 отображает РПД сольвата NMP (N-метил-2-пирролидон) Соединения 1 в твердой форме NF33.
Фигура 50 показывает РПД сольвата ацетонитрила Соединения 1 в твердой форме NF35.
Фигура 51 отображает РПД сольвата 1,4-диоксана Соединения 1 в твердой форме NF36.
Фигура 52 показывает РПД сольвата диметилацетамида Соединения 1 в твердой форме NF37.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Международная патентная заявка WO2016/155844, которая полностью включена в настоящий документ в качестве ссылки, описывает соединения имидазолонил хинолина, которые эффективно ингибируют, регулируют и/или модулируют передачу сигнала с помощью ATM киназы. Такие соединения включают Соединение Y:
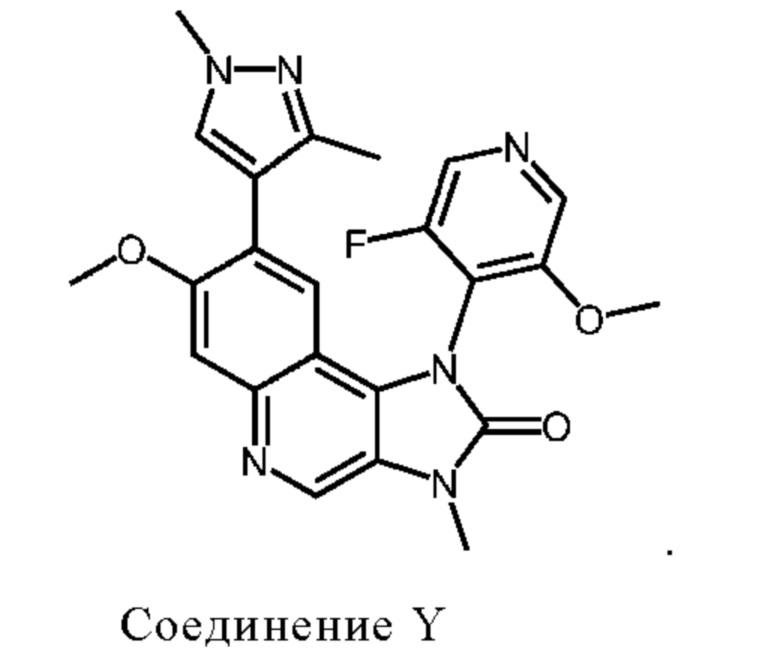
Соединение Y обозначено как Пример 4 в WO 2016/155844 и является активным во множестве анализов и терапевтических моделях, демонстрирующих избирательное ингибирование ATM киназы по сравнению с РI3Kальфа, РI3Kбета, РI3Kдельта, РI3Kгамма и mTOR (в ферментативных и клеточных анализах).
Термины, используемые в данной заявке для определения соединений, обычно основаны на правилах организации IUPAC для химических соединений и, в частности, органических соединений.
В настоящее время неожиданно было обнаружено, что Соединение Y существует в форме двух атропоизомеров, которые могут быть выделены и являются выгодно стабильными, а также что соответствующие атропоизомеры проявляют удивительные и весьма желательные характеристики.
В соответствии с одним аспектом настоящее изобретение обеспечивает два следующих соединения, которые являются атропоизомерами Соединения Y:
• 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-он (Соединение 1) и
• 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Ra)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-он (Соединение 2),
а также их фармацевтически приемлемые соли.
Соединения 1 и 2 являются представленными следующими формулами:
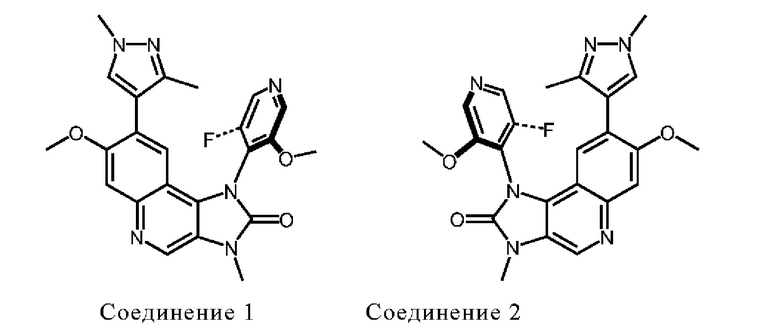
где жирные и пунктирные участки Соединений 1 и 2 обозначают частичное вращение пиридинового кольца за пределами плоскости, в которой расположено трициклическое кольцо.
Специалисту в данной области техники будет понятно, что термин «атропоизомер», используемый в данной заявке, относится к стереоизомеру, который возникает из-за ограниченного вращения вокруг одинарной связи, которая создает хиральную ось. Кроме того, следует понимать, что барьер вращения вокруг одинарной связи должен быть достаточно высоким, чтобы обеспечить выделение единственного атропоизомера. Указанный барьер вращения может быть результатом, например, стерических взаимодействий с другими остатками той же молекулы, и тем самым ограничивает возможность вращения вокруг одинарной связи. Как стерические, так и электронные факторы, которые могут усиливать или противодействовать друг другу, также играют свою роль.
Использование хиральных соединений, содержащих асимметричные атомы углерода, в принципе является хорошо известным при разработке лекарственных средств. В частности, в данной области техники известно, что рацемические смеси двух хиральных соединений обычно состоят из одного более активного и одного менее активного энантиомера по сравнению с рацемической смесью. Таким образом, использование только одного из двух энантиомеров может быть полезным для повышения общей эффективности соединения.
Однако использование атропоизомеров, представляющих собой стереоизомеры, которые возникают только по причине затрудненного вращения вокруг одинарной связи, обычно считается нежелательным. В частности, атропоизомеры обычно рассматриваются как препятствие при выявлении лекарственных средств, поскольку стабильность этих изомеров зависит от разницы энергий, возникающих в результате стерической деформации или других факторов, которые создают барьер для вращения вокруг одной одинарной связи. В отличие от хиральных соединений, которые образованы асимметричными атомами углерода, атропоизомерию трудно предсказать. А именно, обычно невозможно легко предсказать стабильность атропоизомера. В частности, высота электрического барьера определяет время взаимопревращения двух соответствующих атропоизомеров. Взаимное превращение биологически активного атропоизомера в соответствующий другой атропоизомер может, таким образом, снизить его биологическую активность и вызвать нецелевые или другие нежелательные эффекты. Следовательно, только стабильные атропоизомеры, обладающие достаточно высоким энергетическим барьером, могут быть подходящими для выявления лекарственного средства.
Неожиданно было обнаружено, что атропоизомеры Соединения 1 и Соединения 2 не подвергаются значительным взаимопревращениям в соответствующий другой атропоизомер даже после длительных периодов времени, превышающих десять лет (вращательный период полураспада > 10 лет был определен с помощью компьютерного моделирования) и при температурах, превышающих комнатную. Температура инверсии атропоизомеров была оценена как такая, которая составляет более 100°С в растворе. Такая весьма хорошая стабильность была подтверждена экспериментально. Это делает указанные атропоизомеры весьма пригодными для фармацевтического применения, производства, рецептирования и обеспечивает достаточный срок хранения.
Абсолютная структура Соединения 1 была определена на основе соли (2S,3S)-дибензоил-D-винной кислоты, а также с помощью рентгеновской дифракции твердой формы А2, которая будет описана более подробно ниже. Структура Соединения 1 была подтверждена результатами спектроскопии (ЯМР, МС, ИК и УФ), дифракции рентгеновских лучей, элементарного анализа и поляриметрии. Спектры 1Н-, 13С- и 19F-ЯМР Соединения 1 показаны на Фигурах 1-3, УФ-видимый спектр показан на Фигуре 4. РПД твердой формы А2 Соединения 1 проиллюстрирована на Фигуре 13. Кристаллическая структура и РПД Соединение-1-дибензоил-D-тартрата показана на Фигурах 7А и Б, кристаллическая структура Соединение-2-дибензоил-L-тартрата проиллюстрирована на Фигуре 8.
Соединения 1 и 2 представляют собой очень мощные ингибиторы ATM киназы. Как проиллюстрировано Таблицей 1, Соединение 1 имеет явно более высокие значения ингибирования ATM во всех анализах по сравнению с Соединением Y, которое представляет собой смесь Соединений 1 и 2:
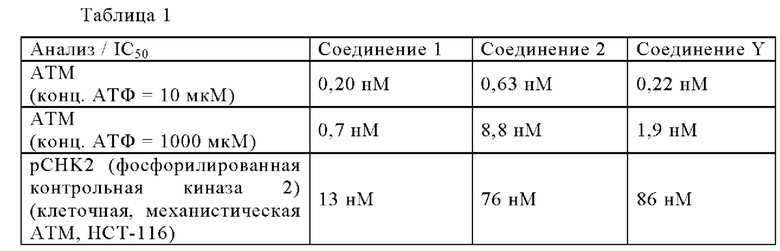
Более того, указанные соединения являются селективными по сравнению с родственными киназами, включая mTOR (>30000 нМ), ДНК-ПК и, в первую очередь, ATR (серин/треонин протеин киназу). Неожиданно оба Соединения 1 и 2 оказались менее мощными ингибиторами ATR киназы по сравнению с Соединением Y, т.е. более предпочтительными с точки зрения селективности.
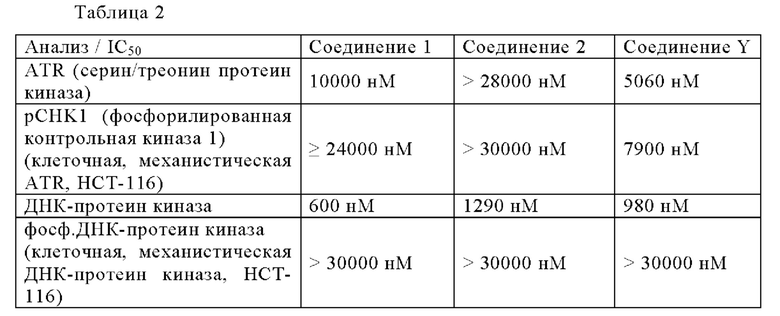
В то время как Соединение 2 не отличается от Соединения Y в отношении ингибирования ATM (смотри Таблицу 1), оно имело значительно большую селективность по сравнению с обоими ATR (серин/треонин протеин киназами) и ДНК-протеин киназой, чем Соединение Y, как является очевидным из приведенной выше Таблицы 2.
Таким образом, соединения в соответствии с настоящим изобретением могут быть особенно выгодно использованы для избирательного воздействия на конкретные механизмы репарации ДНК, в частности, на гомологичную рекомбинацию, которая специфически воздействует на повреждения двухцепочечной ДНК.
Дополнительным преимуществом селективных ингибиторов ATM Соединений 1 и 2 является снижение токсичности, в частности, в отношении нецелевых эффектов и, таким образом, лучшая переносимость более высоких доз соединения. Таким образом, соединения в соответствии с изобретением открывают новые возможности в терапии рака. Например, Соединения 1 и 2, наиболее предпочтительно Соединение 1, можно использовать в целевых комбинированных терапиях, например, таких, которые включают мощный и селективный ингибитор ATM и мощный и селективный другой ингибитор, например, ингибитор ATR.
В целом было обнаружено, что Соединение 1 обладает наиболее выгодным общим сочетанием этих свойств. Удивительно, но оно не только обладает лучшими ингибирующими свойствами в отношении ATM, но также обладает лучшими значениями микросомального клиренса и самым низким ингибированием фосфодиэстеразы (ФДЭ) 2А1, а также ФДЭ4А1А и ФДЭ4Э2. Само по себе ингибирование фосфодиэстеразы связано со множеством фармакологических эффектов, и ингибиторы ФДЭ доступны в качестве лекарственных средств для лечения множества состояний, включая депрессию, рассеянный склероз и хроническое обструктивное заболевание легких, и это лишь некоторые из них, но все они в данном случае вызывают нецелевые эффекты. Известно также, что ингибирование ФДЭ4 связано с риском возникновения тошноты, и поэтому избегают его применения. Следовательно, являются желательными высокие значения IC50, то есть слабое ингибирование этих нецелевых мишеней. Как видно из приведенной ниже Таблицы 3, Соединение 1 имеет значения IC50 для ингибирования ФДЭ4, которые примерно в 5 раз выше, чем таковые для Соединения Y.
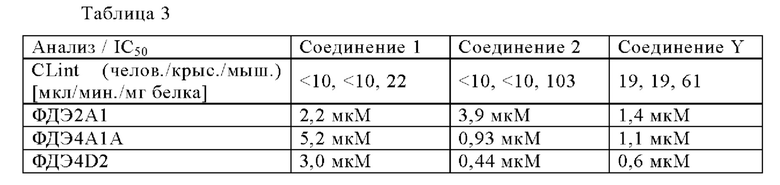
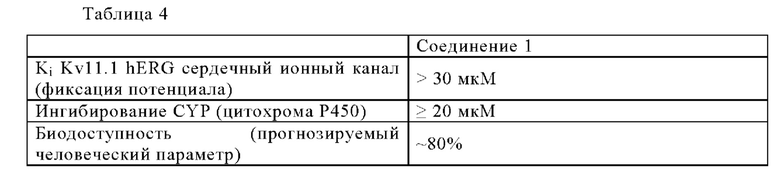
Таблица 4 иллюстрирует другие полезные параметры Соединения 1, включая благоприятно высокую биодоступность на уровне приблизительно 80% и благоприятные свойства в отношении CYP (цитохром Р450) и hERG (сердечного ионного канала), что указывает на тот факт, что не ожидается важных для безопасности взаимодействий с сердечным ионным каналом Kv11.1 hERG.
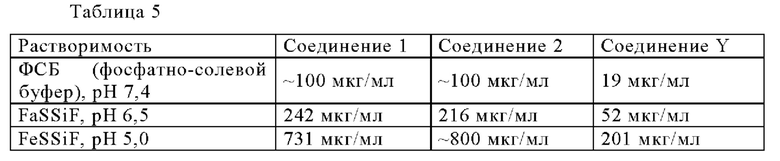
Кроме того, неожиданно было обнаружено, что оба Соединения 1 и 2 демонстрируют значительно улучшенную растворимость в биологических буферных растворах (см. Таблицу 5 ниже) по сравнению с Соединением Y.
Прогнозируемый медленный клиренс из плазмы крови человека и высокая биодоступность Соединения 1 вносят свой вклад в соответствующие требования низких доз.
Подразумевается, что структуры, представленные для Соединения 1 или 2, включают соединения, которые отличаются только наличием одного или нескольких изотопно обогащенных атомов. Например, Н, С, N в каждом случае также включают более тяжелые изотопы этих атомов. Это относится, в частности, к Н, в случае которого можно преимущественно использовать дейтерий или тритий, и к замене углерода углеродом, обогащенным 13С или 14С, что также входит в объем настоящего изобретения. В некоторых предпочтительных вариантах осуществления не используются атомы, обогащенные изотопами, вместо этого атомы используются в их естественных формах с точки зрения распределения изотопов.
Ссылка на соединения или соли в соответствии с настоящим изобретением должна рассматриваться как такая, которая охватывает сольватированные формы, то есть их сольваты, что означает сольваты как свободной формы, так и соли. Под сольватами подразумеваются аддукты молекул инертного растворителя с соединениям, которые образуются благодаря их силе взаимного притяжения. Сольваты представляют собой, например, моно- или дигидраты или алкоголяты. Иллюстративные варианты сольватов более подробно раскрываются ниже.
Как указано выше, настоящее изобретение обеспечивает два стабильных атропоизомера, Соединения 1 и 2. Приготовление этих двух атропоизомеров обычно основано на методах разделения и очистки, как будет более подробно описано ниже. Специалист в данной области техники понимает, что это не может обеспечить идеально чистые продукты. Однако настоящее изобретение обеспечивает Соединение 1, по существу свободное от Соединения 2, и Соединение 2, по существу свободное от Соединения 1. «По существу свободное» в данном контексте предпочтительно означает, что по существу чистое Соединение 1 может содержать максимально 20% по весу Соединения 2 или его соли, предпочтительно максимально 15% по весу, более предпочтительно максимально 10% по весу, например, максимально 5% по весу, максимально 2,5% по весу, максимально 1% по весу, максимально 0,5% по весу или максимально 0,1% по весу Соединения 2 или его соли, остальное до 100% составляет Соединение 1. В одном примере по существу чистое Соединение 1 может состоять из 99% по весу Соединения 1 и 1% по весу Соединения 2. То же самое относится и к Соединению 2, то есть Соединение 2, которое является по существу свободным от Соединения 1, будет предпочтительно означать, что чистое Соединение 2 может содержать максимально 20% по весу Соединения 1 или его соли, с предпочтительными диапазонами, раскрытыми для Соединения 1, которые одинаково применимы (в свою очередь) по аналогии. Также ссылка на Соединение 1 или его соль, соответственно, Соединение 2 или его соль, будет включать все сольваты и твердые формы, такие как те, которые раскрыты в настоящем документе далее ниже, даже без специального упоминания, если явно не указано иное.
В соответствии с другим вариантом осуществления Соединение 1 или 2, соответственно, содержит не более приблизительно 5,0 процентов площади ВЭЖХ от общего количества органических примесей и, в некоторых вариантах реализации, не более приблизительно 3,0 процентов площади ВЭЖХ от общего количества органических примесей и, в некоторых вариантах реализации не содержит более чем приблизительно 1,5 процента площади общих органических примесей ВЭЖХ от общей площади хроматограммы ВЭЖХ. В других вариантах осуществления Соединение 1 или 2 содержит не более чем приблизительно 1,0 процент ВЭЖХ любой отдельной примеси; не более чем приблизительно 0,6 процента площади ВЭЖХ любой отдельной примеси и, в некоторых вариантах реализации, не более примерно 0,5 процента площади ВЭЖХ любой отдельной примеси по отношению к общей площади хроматограммы ВЭЖХ. Например, для хроматограммы ВЭЖХ может использоваться метод, описанный в ПРИМЕРЕ 3.3 для анализа чистоты соответствующих атропоизомеров. В этом случае также ссылка на Соединение 1 или его соль, соответственно Соединение 2 или его соль, будет включать все сольваты и твердые формы, такие как те, которые раскрыты в настоящей заявке далее ниже, если явно не указано иное.
В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает фармацевтическую композицию, которая содержит эффективное количество Соединения 1 или его фармацевтически приемлемой соли. В альтернативном варианте осуществления фармацевтическая композиция содержит эффективное количество Соединения 2 или его фармацевтически приемлемой соли. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает способ получения такой композиции, описанной в данной заявке (например, композиции, которая может включать эффективное количество Соединения 1 или 2). Ссылку на Соединение 1 или 2 следует понимать в данном контексте так, что любое количество соответствующего другого атропоизомера может составлять только то количество, которое находится в соответствии с определениями «по существу не содержит» соответствующего другого атропоизомера, «по существу не содержит примесей» или общего количества примесей, соответственно, то есть будет учитываться в количестве упомянутого соединения, а не присутствовать отдельно. То же самое относится к соответствующим солям. Как указано выше, ссылка на Соединение 1 или 2 или его соль в равной степени должна включать любую твердую форму или сольват, такие как те, которые дополнительно раскрыты в данном документе ниже, если специально не указано иное. В примерах осуществления Соединение 1 содержится в фармацевтической композиции в своей свободной, то есть несолевой, форме.
В других вариантах реализации предлагается способ лечения рака при использовании соединения или композиции, соответственно, фармацевтической композиции или фармацевтически приемлемой соли соединения, как описано в настоящей заявке. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает применение соединения, его фармацевтически приемлемой соли или (фармацевтической) композиции в соответствии с настоящим изобретением при производстве лекарственного средства для лечения рака. В другом варианте осуществления настоящее изобретение обеспечивает соединение или фармацевтическую композицию, как описано в данной заявке, для применения в качестве лекарственного средства, в частности, для лечения рака. Опять же, ссылка на соединение или его соль будет включать любую форму сольвата или твердую форму этих соединений или солей, таких как те, которые раскрыты в данной заявке далее ниже, если явно не указано иное.
Свободная форма и соли
Соединения в соответствии с изобретением могут использоваться в своей свободной форме, то есть как представлено в формулах, приведенных выше. Например, было обнаружено, что свободная форма Соединения 1 является особенно полезной, и ее иллюстративные твердые формы, включая предпочтительную твердую форму, будут описаны более подробно ниже.
С другой стороны, настоящее изобретение также включает использование этих соединений в форме их фармацевтически приемлемых солей, которые могут быть получены из различных органических и неорганических кислот способами, известными в данной области техники. Приемлемые фармацевтически приемлемые соли соединения в соответствии с изобретением могут быть получены обычными способами. Соединение в соответствии с изобретением может быть преобразовано в ассоциированную кислотно-аддитивную соль при использовании кислоты, например, посредством реакции соединения и эквивалентного или избыточного количества кислоты в подходящем растворителе, таком как, например, ТГФ или ацетон, с последующей кристаллизацией с охлаждением образовавшегося насыщенного раствора. В качестве альтернативы, может применяться кристаллизация на основе антирастворителя или кристаллизация при выпаривании.
Примеры подходящей фармацевтически приемлемой соли соединения в соответствии с настоящим изобретением, в частности, Соединения 1, включают соль HCl, сульфатную соль, тозилатную соль, безилатную соль, лактатную соль, в частности, L-лактатную соль.
Предпочтительные соли Соединения 1 могут включать фумаратную соль, напсилатную соль и эдизилатную соль, которые вместе в дальнейшем также могут упоминаться как Соединения 1а.
Напсилат Соединения 1 может быть получен из Соединения 1 при использовании нафталинсульфоновой кислоты и либо ТГФ, либо ацетона в качестве растворителя. При этом получают хорошую кристалличность. Предпочтительное соотношение Соединение 1: напсилат составляет примерно 1:1.
Эдизилат Соединения 1 может быть получен при использовании этансульфоновой кислоты и кристаллизации с охлаждением из ацетона. Предпочтительное соотношение Соединение 1: эдизилат составляет примерно 1:1. При этом была получают хорошую кристалличность.
Фумарат Соединение 1 может быть получен при использовании диффузии паров с антирастворителем в ТГФ с применением н-пентана в качестве антирастворителя и фумаровой кислоты в качестве кислоты. Соотношение Соединение 1: фумарат было показано как такое, которое составляет 1:0,9. Полученная соль имеет очень хорошие общие физические свойства и хорошую кристалличность. Соль фумарата является предпочтительным вариантом среди солей, в некоторой степени благодаря желаемому отсутствию гигроскопических свойств.
Подробные примеры приемлемых способов получения фумаратных, напсиланых и эдизилатных солей Соединения 1 в соответствии с настоящим изобретение раскрыты в ПРИМЕРЕ 5.
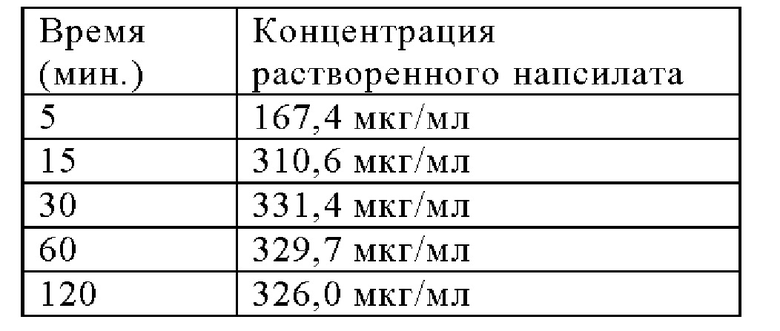
Было обнаружено, что эти соли Соединения 1 демонстрируют более высокую начальную скорость растворения, чем исходное Соединение 1. Профиль растворения без поглощения в буферном растворе FaSSIF (рН 6,5) Соединения 1 и его специфических солей в качестве примера представлен на Фигуре 9. Соли с благоприятными характеристиками растворения представляются собой эдизилат Соединения 1, фумарат Соединения 1 и напсилат Соединения 1. Исходное Соединение 1 использовали в виде смеси различных кристаллических и сольватированных форм.
Специалисту в данной области техники будет понятно, что анионный остаток кислоты и Соединение 1 являются ионно связанными с формой Соединения 1-а. Предполагается, что Соединение 1-а может существовать во множестве физических форм. Например, Соединение 1-а может находиться в растворе, суспензии или в твердой форме. В некоторых вариантах реализации Соединение 1-а находится в твердой форме. Когда Соединение 1-а находится в твердой форме, соединение может быть аморфным, кристаллическим или их смесью. Используемый в данной заявке термин «полиморф» относится к различным кристаллическим структурам, в которых соединение или его соли могут кристаллизоваться.
В некоторых вариантах осуществления Соединения 1-а представляют собой твердые кристаллические вещества, по существу не содержащие аморфного Соединения 1-а. Используемый в данной заявке термин «практически не содержит аморфного Соединения 1-а» означает, что соль не содержит значительного количества аморфного Соединения 1-а. В некоторых вариантах реализации присутствует, по крайней мере, примерно 90% по весу кристаллического Соединения 1-а или, по крайней мере, примерно 95% по весу кристаллического Соединения 1-а. В других вариантах осуществления изобретения присутствует, по крайней мере, примерно 99% по весу кристаллического Соединения 1-а. Эти проценты относятся к абсолютной массе Соединения 1-а (100 мас. %).
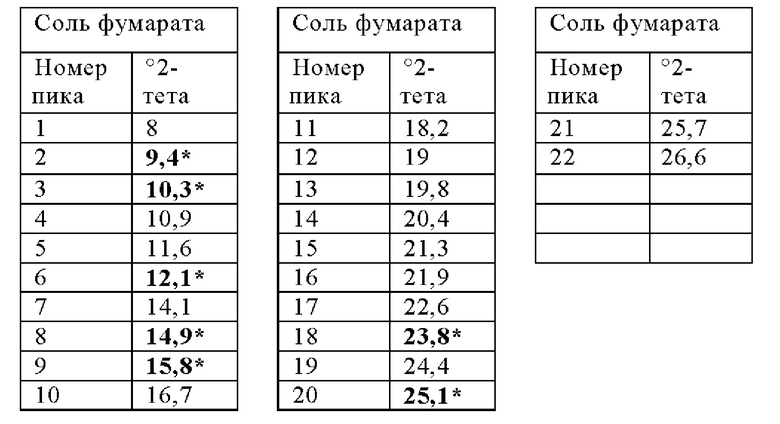
В иллюстративном варианте осуществления настоящее изобретение обеспечивает твердую форму фумарата Соединения 1, характеризующуюся профилем рентгеновской порошковой дифракции, по существу совпадающим с профилем, изображенном на Фигуре 10, и/или характеризующуюся одним или несколькими пиками на профиле рентгеновской порошковой дифракции, выбранном примерно из следующих.
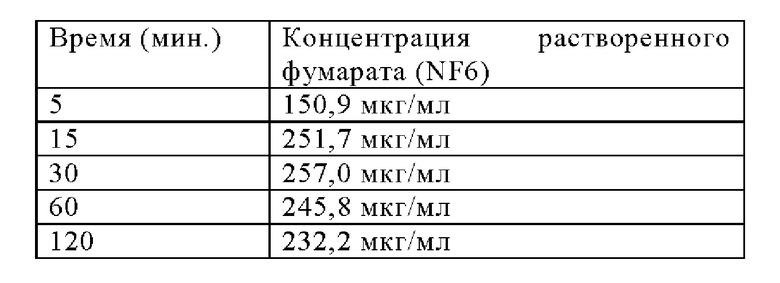
Соль фумарата Соединения 1 является безводной. Ее твердая форма также может называться как фумарат-NF6. Кривая нагревания ДСК, ТГА кривая нагревания и ДСП изотерма поглощения воды (25°С) фумарата-NF6 показаны на Фигурах 31, 32 и 33. Результаты измерений растворения без поглощения представлены в Таблице, приведенной ниже (данные в отношении растворения без поглощения в FaSSIF при рН 6,5, метод описан в экспериментальной части):
Как используется в данной заявке, термин "приблизительно/примерно", когда используется в отношении значения градуса 2-тета, относится к указанному значению ±0,3 градуса 2-тета (°2θ). В некоторых вариантах реализации, "приблизительно/примерно" относится к ±0,2 градуса 2-тета или ±0,1 градуса 2-тета, наиболее предпочтительно ±0,2 градуса 2-тета.
Любая твердая форма, соответственно полиморф, описанный в данной заявке, может характеризоваться одним, двумя, тремя, четырьмя, пятью, шестью, семью, восемью, девятью, десятью, одиннадцатью, двенадцатью, тринадцатью, четырнадцатью, пятнадцатью, шестнадцатью, семнадцатью, восемнадцатью, девятнадцатью, двадцатью или более пиками рентгеновской дифракции или порошковой рентгеновской дифракции (°2θ). Любая твердая форма, соответственно, полиморф, описанный в данной заявке, предпочтительно характеризуется, по крайней мере, шестью пиками рентгеновской дифракции (°2θ, предпочтительно ±0,2° 2θ). Предпочтительные пики для характеристики твердой формы или полиморфа обозначены жирным шрифтом и звездочками в соответствующих списках пиков.
В дополнительном иллюстративном варианте осуществления настоящее изобретение обеспечивает твердую форму напсилата Соединения 1, характеризующуюся профилем рентгеновской порошковой дифракциии, по существу соответствующим изображенному на Фигуре 11, и/или характеризующуюся одним или несколькими пиками рентгеновской порошковой дифракциии, выбранными примерно из следующих.

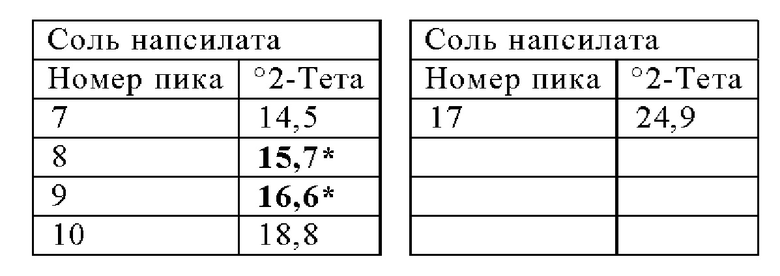
Термические свойства соли напсилата Соединения 1 и его свойства в отношении адсорбции воды показаны на Фигурах 34, 35 и 36. Твердая форма, полученная для соли напсилата, также может называться напсилатом NF7. Кроме того, профиль растворения представлен следующими экспериментальными данными растворения без поглощения (FaSSIF, рН 6,5):
В дополнительном иллюстративном варианте осуществления настоящее изобретение обеспечивает твердую форму эдизилата Соединения 1, характеризующуюся профилем порошковой рентгеновской дифракции, по существу соответствующим изображенному на Фигуре 12 и/или характеризующимся одним или несколькими пиками на ее профиле порошковой рентгеновской дифракции, выбранном примерно из следующих.
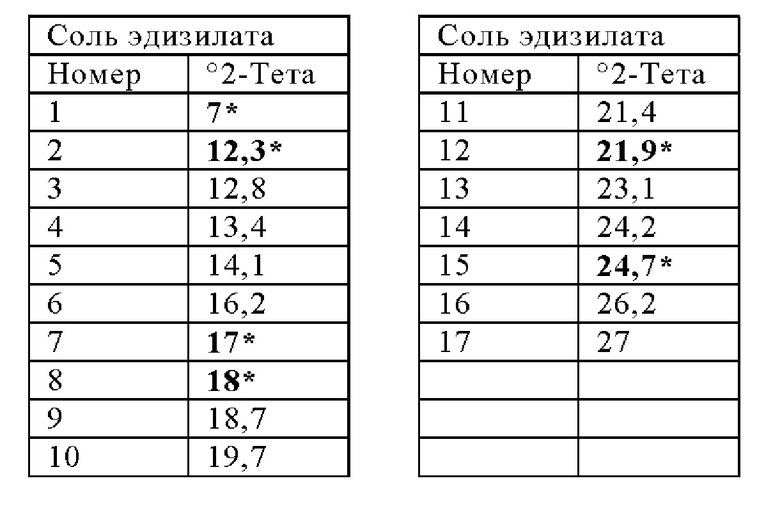
Термические свойства и данные в отношении растворения соли эдизилата Соединения 1 проиллюстрированы на Фигурах 37, 38 и 39. Твердая форма соли эдизилата, также может упоминаться как NF8. Она представляет собой безводную форму/соль. Данные относительно растворении без поглощения (FaSSIF, рН 6,5) обеспечиваются в таблице, представленной ниже:

В соответствии с тем, что было изложено выше в отношении Соединений 1 и 2, настоящее изобретение обеспечивает Соединение 1-а или другие соли Соединения 1, по существу, не содержащие Соединения 2 или его солей. В дополнительных вариантах осуществления Соединение 1-а или другая соль Соединения 1 также обеспечивается, по существу, свободным от примесей. В соответствии с другим вариантом осуществления Соединение 1-а или любая другая соль Соединения 1 содержит не более чем примерно 5,0 процентов площади ВЭЖХ всех органических примесей по отношению к общей площади хроматограммы ВЭЖХ. Иллюстративные и предпочтительные диапазоны, раскрытые выше для Соединения 1 в связи с «по существу свободным от Соединения 2 или его солей», «свободным от примесей» и процентом от общей площади органических примесей, в равной степени применимы и в данном случае. То же самое по аналогии применимо к любой из солей любого из соединений и, в частности, атропоизомерным соединениям в соответствии с настоящим изобретением.
В соответствии с другим вариантом осуществления настоящее изобретение включает фармацевтическую композицию, которая содержит эффективное количество Соединения 1-а. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает способ приготовления такой композиции, описанной в данной заявке (например, композиции, которая может включать эффективное количество Соединения 1-а). Еще один вариант осуществления обеспечивает способ лечения рака при использовании Соединения 1-а или его композиции в соответствии с настоящим изобретением. В соответствии с другим вариантом осуществления настоящее изобретение предусматривает использование описанной в данной заявке композиции при производстве лекарственного средства для лечения рака. Соединение 1-а может присутствовать в (фармацевтической) композиции в тех же количествах, которые указаны для Соединения 1. Что касается присутствия в композиции другого атропоизомера или его соли, соответственно, соображения, изложенные выше для Соединения 1, в равной степени применимы по аналогии.
Твердые формы и сольваты
В соответствии с другим аспектом настоящее изобретение обеспечивает твердые формы Соединения 1 или 2, в частности, Соединения 1.
Специалистам в данной области техники будет понятно, что соединения в соответствии с настоящим изобретением могут существовать в различных физических формах. Например, они могут быть в растворе, суспензии или в твердой форме.
В некоторых предпочтительных вариантах осуществления Соединение 1 находится в твердой форме. Твердые формы обычно являются предпочтительными в данном случае, потому что они позволяют создавать твердые фармацевтические композиции. Когда Соединение 1 находится в твердой форме, указанное соединение может быть аморфным, кристаллическим или их смесью. Иллюстративные твердые формы Соединения 1 более подробно описаны ниже.
В соответствии с одним вариантом осуществления настоящее изобретение обеспечивает Соединение 1 в виде аморфного твердого вещества. Аморфные твердые вещества являются хорошо известными среднему специалисту в данной области техники и обычно их получают такими методами, как лиофилизация, распылительная сушка или ускоренное осаждение.
В других вариантах реализации Соединение 1 представляет собой твердое кристаллическое вещество. Используемый здесь термин «полиморф» относится к различным кристаллическим структурам, в виде которых соединение может кристаллизоваться.
В некоторых вариантах реализации Соединение 1 представляет собой кристаллическое твердое вещество, по существу не содержащее аморфного Соединения 1. Используемый в данной заявке термин «по существу не содержащий аморфного Соединения 1» означает, что соединение не содержит значительного количества аморфного Соединения 1. В некоторых вариантах реализации, по крайней мере, присутствует примерно 90% по весу кристаллического Соединения 1 или, по крайней мере, примерно 95% по весу кристаллического Соединения 1. В других вариантах осуществления изобретения присутствует, по крайней мере, примерно 99% (по весу) кристаллического Соединения 1. Эти проценты относятся к абсолютному весу Соединения 1 (100 вес. %). То же самое является применимым, с соответствующими изменениями, к приемлемому аморфному содержанию в любой кристаллической форме, соответственно, полиморфу всех соединений, раскрытых в данной заявке, включая те, которые описаны для солей, безводных форм, сольватов и других форм в настоящей заявке.
В соответствии с одним аспектом настоящее изобретение обеспечивает твердую форму Соединения 1, которая представляет собой твердую форму безводного Соединения 1, предпочтительно кристаллическое безводное Соединение 1. В данной заявке описаны пять различных полиморфных форм безводного Соединения 1. В контексте конкретных твердых форм, описанных в этом разделе и включающих безводные формы и сольваты, ссылку на Соединение 1 следует понимать как ссылку на соединение как таковое, то есть на его свободную (несолевую) форму.
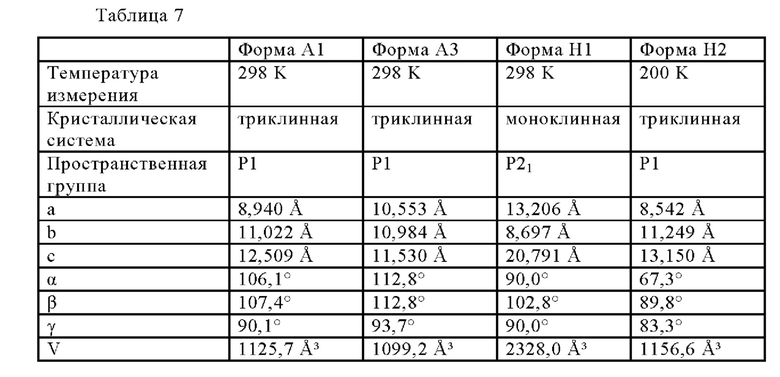
Первая безводная кристаллическая форма Соединения 1 далее упоминается как «Форма А2» и представляет собой полиморф, характеризующийся профилем рентгеновской порошковой дифракции (РПД), по существу совпадающим с профилем, изображенном на Фигуре 13, и было обнаружено, что это является весьма выгодным.
В соответствии с одним вариантом осуществления Форма А2 характеризуется одним или несколькими пиками на ее порошковой дифрактограмме рентгеновских лучей, выбранными из пиков при примерно 7,3, примерно 9,6, примерно 11,1, примерно 12,0, примерно 12,7 и примерно 16,2 градусах 2-тета. В некоторых вариантах реализации Форма А2 характеризуется двумя или более пиками на порошковой дифрактограмме рентгеновских лучей, выбранных из пиков при примерно 7,3, примерно 9,6, примерно 12,7, примерно 16,2, примерно 22,6 и примерно 25,1 градусах 2-тета. В некоторых вариантах реализации Форма А2 характеризуется тремя или более пиками на порошковой дифрактограмме рентгеновских лучей, выбранных из пиков при примерно 7,3, примерно 9,6, примерно 12,7, примерно 16,2, примерно 22,6 и примерно 25,1 градусах 2-тета. В некоторых вариантах реализации Форма А2 характеризуется четырьмя, пятью или практически всеми пиками на ее порошковой дифрактограмме рентгеновских лучей, выбранных из пиков при примерно 7,3, примерно 9,6, примерно 12,7, примерно 16,2, примерно 22,6 и примерно 25,1 градусах 2- тета. В частности, в вариантах реализации Форма А2 характеризуется шестью или более или практически всеми пиками в ее профиле порошковой рентгеновской дифракции, выбранными из пиков при примерно 7,3, 9,6, 11,1, 12,0, 12,7, 14,7, 16,2, 17,3, 18,9, 21,0, 22,6 и 25,1 градусах 2-тета.
В иллюстративном варианте реализации Форма А2 может характеризоваться одним или более, предпочтительно шестью и до существенно всеми пиками в своем профиле порошковой рентгеновской дифракции (РПД), выбранными из тех, которые представлены ниже:
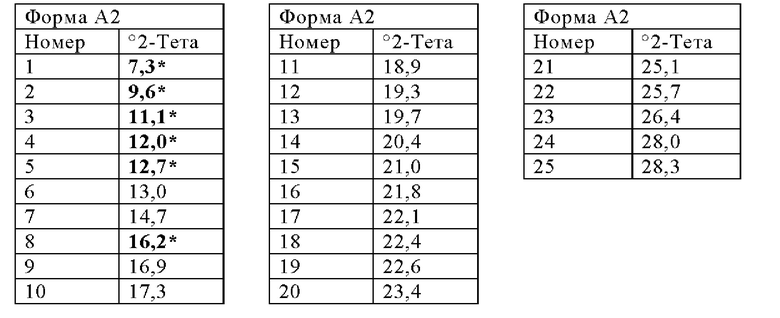
Следует принять во внимание, что описанная выше полиморфная форма может быть охарактеризована, например, посредством ссылки на любой из пиков в ее соответствующем профиле дифракции рентгеновских лучей (РД). Как указано ранее, жирный шрифт и звездочки обозначают пики, которые могут быть предпочтительными для характеристики полиморфа.
Форма А2 может характеризоваться одним, двумя, тремя, четырьмя, пятью, шестью, семью, восемью, девятью, десятью, одиннадцатью, двенадцатью, тринадцатью, четырнадцатью, пятнадцатью, шестнадцатью, семнадцатью, восемнадцатью, девятнадцатью, двадцатью или более РПД пиками (°2θ) в соответствии с приведенной выше таблицей. Любой описанный в данной заявке полиморф преимущественно характеризуется, по крайней мере, шестью пиками РД или РПД (°2θ, предпочтительно ±0,2).
Форма А2 может быть дополнительно охарактеризована тем, что имеет моноклинную кристаллическую систему и пространственную P21 группу. Форма А2 может быть дополнительно охарактеризована одним или более из следующих параметров ее элементарной ячейки, как указано в следующей Таблице:
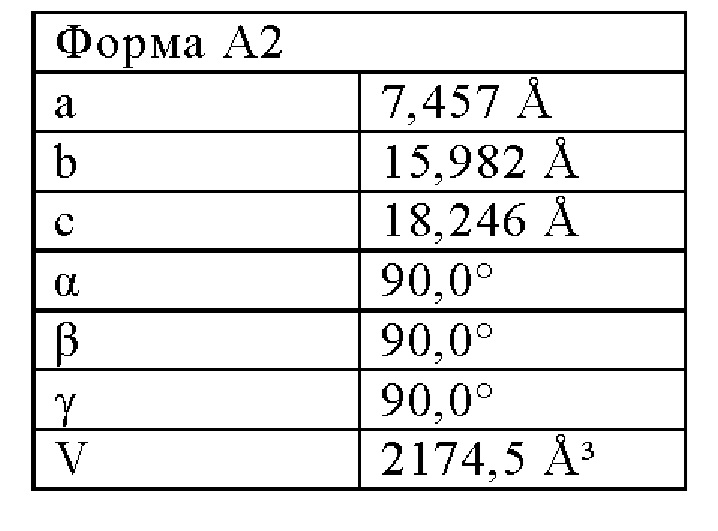
Форма А2 Соединения 1 имеет благоприятные общие свойства, что дополнительно подтверждается его термической характеристикой и поглощением воды, как показано на ДСК кривой нагревания (Фигура 22), ТГА кривой нагревания (Фигура 23) и ДСП изотермой поглощения воды (при температуре 25°С) (Фигура 24). Форма А2 поглощает незначительное количество воды (<2%) даже вплоть до относительной влажности 100% и, таким образом, превосходит, например, Форму A3.
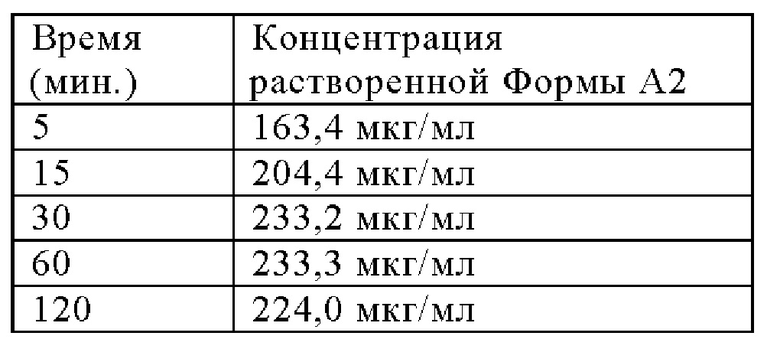
Другим преимуществом Формы А2 является ее благоприятная характеристика растворения, как показано в следующей таблице, которая представляет количества Соединения 1 в Форме А2, растворенные в различные промежутки времени (данные относительно растворения без поглощения в FaSSIF при рН 6,5, метод описан в экспериментальной части).
Форма А2 может быть получена из Соединения 1 путем кристаллизации при охлаждении из спиртов, в качестве одного из примеров. Соответствующие методы и условия реакции подробно описаны в ПРИМЕРЕ 7.
Например, кристаллы формы А2, обладающие благоприятными свойствами, могут быть воспроизводимым образом получены с помощью процесса контролируемой кристаллизации, который включает:
а) Получение дисперсии Соединения 1, например, гидрата Соединения 1, такого, как форма Н2 гидрата Соединения 1, в приемлемом растворителе, например, спирте,
б) Нагревание дисперсии для получения раствора, предпочтительно прозрачного раствора,
в) Контролируемое охлаждение раствора,
г) Добавление затравочных кристаллов Формы А2,
д) Контролируемое охлаждение раствора с затравочными кристаллами, например, при скорости примерно 0,1°С/мин.
Приемлемые спирты и условия кристаллизации, такие как температура, могут быть получены из ПРИМЕРОВ 7.1-73, которые являются применимыми вне конкретного варианта осуществления. Настоящее изобретение также относится к безводному кристаллическому Соединению 1 в Форме А2, которое можно получить с помощью указанного выше процесса или, по существу, в соответствии с любым из процессов, описанных в ПРИМЕРАХ 7.1-7.4.
Другая безводная кристаллическая форма Соединения 1 далее именуется как «Форма А1» и представляет собой полиморф, который характеризуется порошковой дифрактограммой рентгеновских лучей, по существу соответствующей такой, изображенной на Фигуре 14. Приемлемые способы ее получения описаны в ПРИМЕРЕ. 7.
Форма А1 может быть охарактеризована одним или несколькими, предпочтительно шестью и практически всеми пиками в ее профиле порошковой рентгеновской дифракции, выбранными из тех, которые представлены ниже:

Термические свойства Формы А1 Соединения 1 оценивали с помощью дифференциальной сканирующей калориметрии (ДСК) и
термогравиметрического анализа (ТГА), как показано на Фигурах 25 и 26.
Третья безводная кристаллическая форма Соединения 1 далее именуется как «Форма A3» и представляет собой полиморф, характеризующийся профилем порошковой рентгеновской дифракции, по существу соответствующим изображенному на Фигуре 15. Приемлемый способ ее получения описан ниже в ПРИМЕРЕ 7.
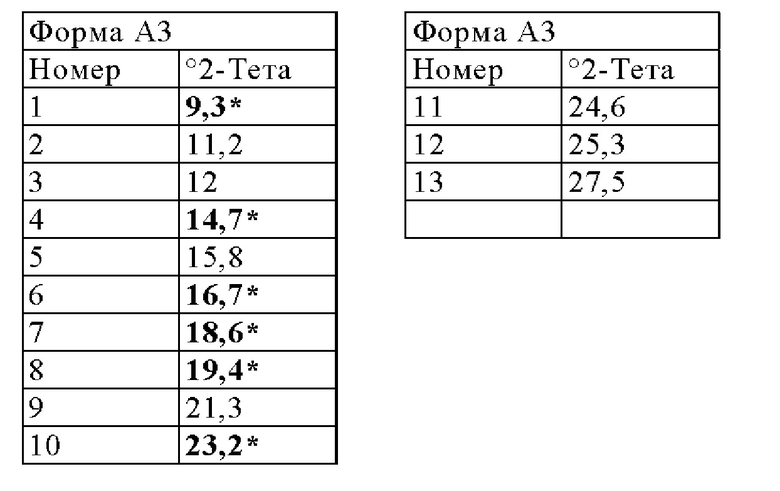
Форма A3 может быть охарактеризована одним или несколькими, предпочтительно шестью и практически всеми пиками в ее профиле порошковой рентгеновской дифракции, выбранными из тех, что представлены ниже:
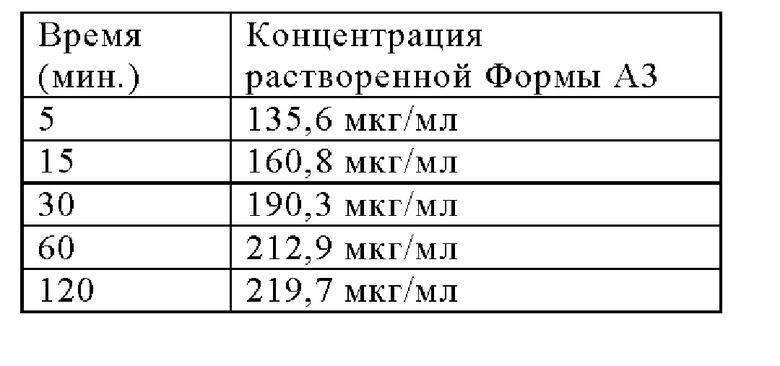
Как видно из Фигуры 27, Форма A3 Соединения 1 демонстрирует очень низкую адсорбцию воды вплоть до относительной влажности около 70%. Форма A3 может дополнительно быть охарактеризована кристаллической системой и параметрами элементарной ячейки, как указано в Таблице 7, представленной ниже. Данные о растворении без поглощения в FaSSIF при рН 6,5 Соединения 1 в форме A3 приведены в таблице, представленной ниже:
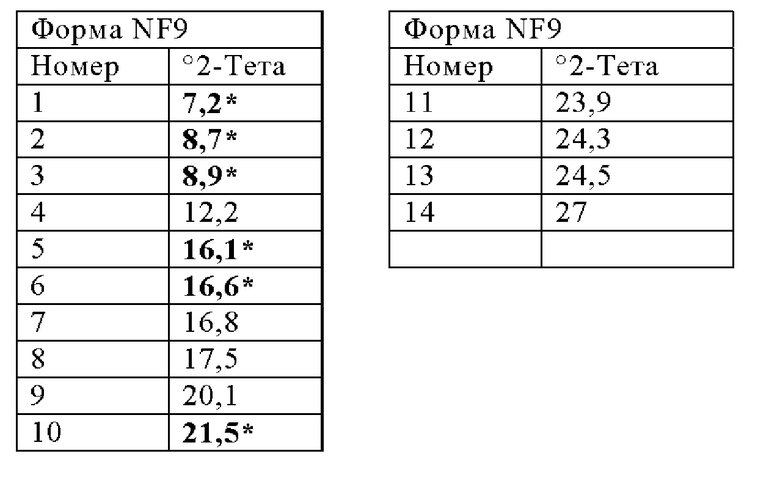
Четвертая безводная кристаллическая форма Соединения 1 в дальнейшем именуется как «Форма NF9» и представляет собой полиморф, характеризующийся профилем порошковой рентгеновской дифракции, по существу соответствующим изображенному на Фигуре 16. Способ ее получения описан в ПРИМЕРЕ 7.
Форма NF9 может быть охарактеризована одним или несколькими, предпочтительно шестью и вплоть до всех пиков в ее профиле порошковой рентгеновской дифракции, выбранными из тех, которые представлены ниже:
В дополнительном варианте осуществления настоящее изобретение обеспечивает гидраты Соединения 1, предпочтительно твердую форму гидрата Соединения 1, предпочтительно кристаллический гидрат Соединения 1. В данной заявке описываются два разных гидрата, соответственно полиморфные формы гидратов Соединения 1. Как упоминалось выше, в контексте этих конкретных твердых форм, ссылку на Соединение 1 следует понимать как ссылку на соединение как таковое, т.е. его свободную (несолевую) форму.
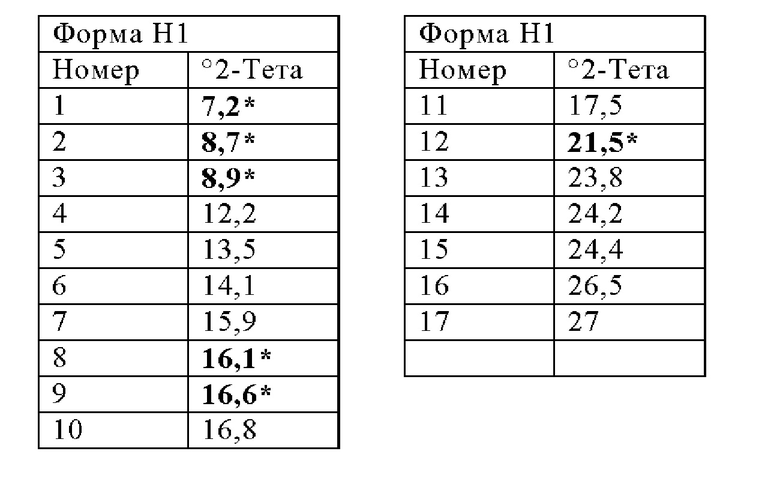
Первая кристаллическая форма гидрата Соединения 1 в дальнейшем именуется как «Форма Н1» и представляет собой полиморф, характеризующийся профилем рентгеновской порошковой дифракции, по существу соответствующим тому, который изображен на Фигуре 17. Приемлемые способы для его получения описываются в ПРИМЕРЕ 7.
Форма HI может характеризоваться одним или несколькими, предпочтительно шестью и практически всеми пиками, в ее профиле рентгеновской порошковой дифракции, выбранными из тех, которые представлены ниже.
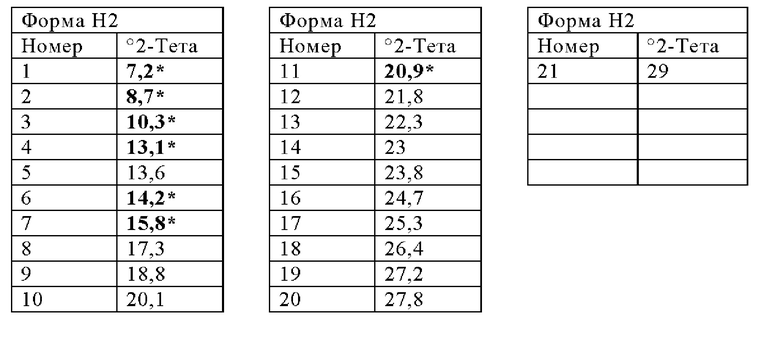
Вторая кристаллическая форма гидрата Соединения 1 в дальнейшем именуется как «Форма Н2» и представляет собой полиморф, характеризующийся профилем рентгеновской порошковой дифракции, который по существу соответствует тому, который изображен на Фигуре 18. Приемлемые способы для его получения описываются в ПРИМЕРЕ 7.
Форма Н2 может быть охарактеризована одним или несколькими, предпочтительно шестью и практически всеми, пиками в ее профиле рентгеновской порошковой дифракции, выбранными из тех, которые представлены ниже.
Гидрат Соединения 1 в виде кристаллической Формы Н2 может быть дополнительно охарактеризован тем, что он имеет триклинную кристаллическую систему и пространственную группу Р1. Форма Н2 может быть охарактеризована при использовании одного или более из следующих параметров элементарной ячейки, как указано в Таблице 7, представленной ниже.
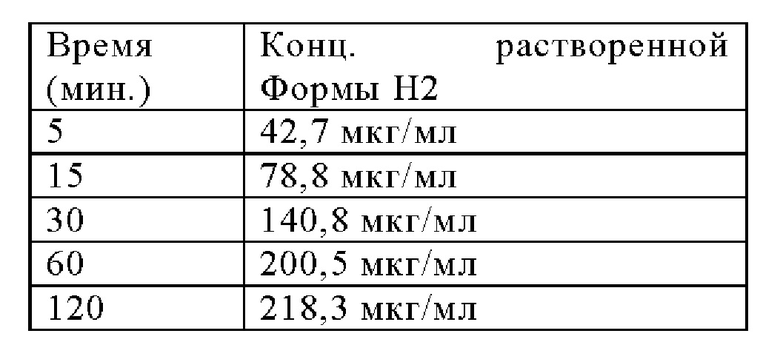
Термические свойства и свойства адсорбции воды Формы Н2 иллюстрируются Фигурами 28, 29 и 30. Данные растворения без поглощения в FaSSIF (рН 6,5) для Формы Н2 Соединения 1 были определены такими, как представлено ниже:
Пятая кристаллическая форма безводного Соединения 1 далее в данной заявке именуется как "Форма NF19" и представляет собой полиморф, который характеризуется профилем рентгеновской порошковой дифракции, который по существу соответствует тому, который изображен на Фигуре 19. Приемлемые способы для ее получения описываются в ПРИМЕРЕ 7.
Форма NF19 может быть охарактеризована одним или более, предпочтительно шестью и вплоть до всех, пиков ее профиля рентгеновской порошковой дифракции, выбранных из тех, которые представлены ниже:
Твердые формы А1, A3, H1 и Н2 могут быть также или альтернативно охарактеризованы наличием определенной кристаллической системы, пространственной группы и/или параметрами элементарной ячейки, выбранными из а, b, с, α, β, γ и V, как указано ниже в Таблице 7:
Настоящее изобретение также относится к следующим сольватным формам, которые могут быть легко получены путем кристаллизации из соответствующих растворителей, однако было обнаружено, что они имеют значительно меньшие преимущества в отношении важных свойств по сравнению с указанными выше формами: метанолат Соединения 1 (твердая форма, обозначенная как S1), смешанный гидрат/метанолат Соединения 1 (твердая форма, обозначенная как S2), сольват ТГФ Соединения 1 (твердая форма, обозначенная как S3), 1,4-диоксановые сольватные формы Соединения 1 в виде многочисленных твердых формах (твердые формы, обозначаемые как NF11 [из эксперимента по превращению суспензии в безводную форму при ~ 26 мг/200 мкл в 1,4-диоксане при комнатной температуре], NF29 [из эксперимента по кристаллизации при охлаждении 50-5°С в 1,4-диоксане], NF36 [из эксперимента по превращению суспензии в безводную форму при -52 мг/150 мкл в 1,4-диоксане при комнатной температуре]), хлороформный сольват Соединения 1 (твердая форма, обозначенная как NF15), сольват уксусной кислоты формы Соединения 1 в различных твердых формах (твердые формы, обозначаемые как NF16 [из эксперимента по кристаллизации при комнатной температуре в уксусной кислоте], NF18 [из эксперимента по кристаллизации при испарении при 50°С в уксусной кислоте]), дихлорметановый (ДХМ) сольват Соединения 1 (твердая форма, обозначенная как NF32), NMP (N-метил-2-пирролидон) сольват Соединения 1 (твердая форма, обозначенная как NF33), ацетонитрильный сольват Соединения 1 (твердая форма, обозначенная как NF35), сольват диметилацетамида (ДМАА) Соединения 1 (твердая форма, обозначенная как NF37). РПД твердых форм этих сольватов показаны на Фигурах 40-52, соответствующие пики перечислены в приведенных ниже таблицах:
В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает фармацевтическую композицию, которая содержит эффективное количество Соединения 1 в форме А2 и является предпочтительной твердой формой. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает способ приготовления таких фармацевтических композиций, описанных в данном документе, например, фармацевтической композиции, которая включает эффективное количество Соединения 1 в Форме А2. Еще один вариант осуществления относится к способу лечения рака при использовании фармацевтической композиции, содержащей эффективное количество Соединения 1 в Форме А2, как описано в данной заявке. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает применение Соединения 1 в Форме А2 при производстве лекарственного средства для лечения рака. В дополнительном варианте осуществления настоящее изобретение обеспечивает Форму А2 для использования в качестве лекарственного средства, предпочтительно для лечения рака. В соответствии с тем, что было изложено выше, иллюстративные варианты осуществления, интервалы, степень чистоты и т.д., раскрытые для Соединения 1, в равной степени действительны для Формы А2.
В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает фармацевтическую композицию, которая включает эффективное количество Соединения 1 в виде любой из твердых форм безводного Соединения 1 или гидрата Соединения 1, как описано выше. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает способ приготовления таких фармацевтических композиций, описанных в данной заявке, например, фармацевтической композиции, которая включает эффективное количество Соединения 1 в виде одной из таких твердых форм. Еще один вариант осуществления обеспечивает способ лечения рака при использовании фармацевтической композиции, содержащей эффективное количество Соединения 1 в виде одной из твердых форм, как описано в данной заявке. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает применение Соединения 1 в твердой форме, как описано в данной заявке, при производстве лекарственного средства для лечения рака. В дополнительном варианте осуществления настоящее изобретение обеспечивает твердую форму Соединения 1, как описано в данной заявке, для использования в качестве лекарственного средства, предпочтительно для лечения рака. В соответствии с тем, что было изложено выше, иллюстративные варианты осуществления, интервалы, степень чистоты и т.д., раскрытые для Соединения 1, в равной степени действительны для твердых форм.
Настоящее изобретение также относится к твердой форме безводного Соединения 1 или гидрата Соединения 1, как описано в данной заявке, которая получена или может быть получена в соответствии со способом, описанным в ПРИМЕРЕ 7.
Дейтерированные варианты осуществления
В соответствии с дополнительным аспектом настоящее изобретение обеспечивает дейтерированные производные Соединения Y. В соответствии с одним вариантом осуществления настоящее изобретение обеспечивает:
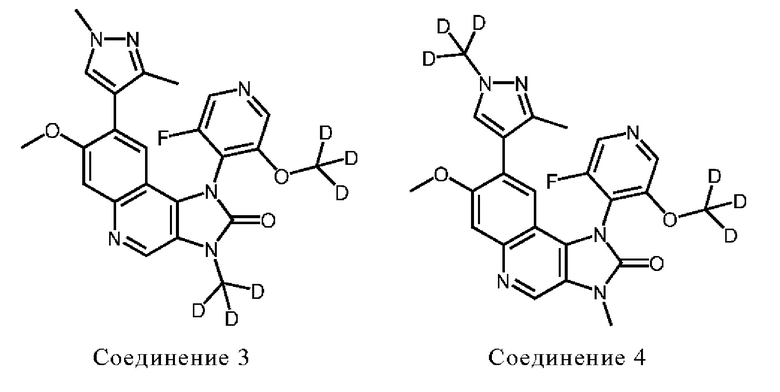
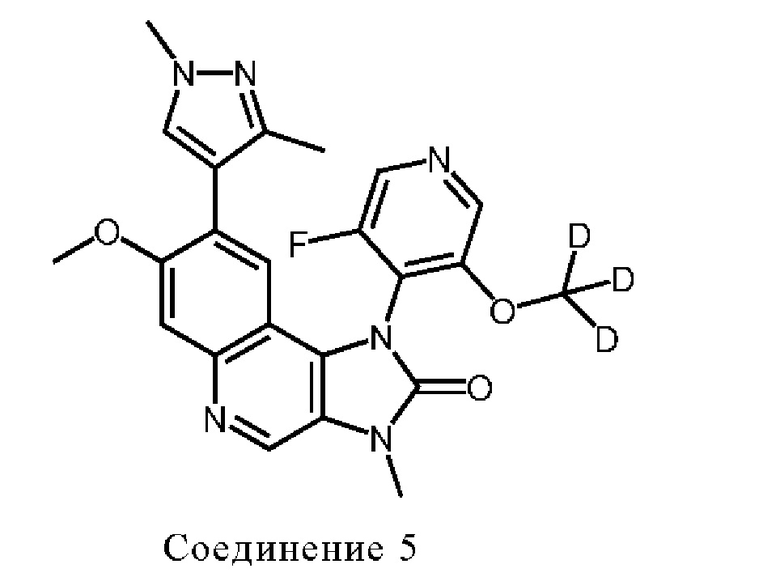
• 8-(1,3-диметилпиразол-4-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-(тридейтериометил)имидазо[4,5-с]хинолин-2-он (Соединение 3),
• 1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-8-[3-метил-(тридейтериометил)пиразол-4-ил]имидазо[4,5-с]хинолин-2-он (Соединение 4) и
• 8-(1,3-диметилпиразол-4-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-имидазо[4,5-с]хинолин-2-он (Соединение 5),
а также их соли.
Соединения 3, 4 и 5 являются представленными следующими формулами:
В других вариантах осуществления настоящее изобретение обеспечивает атропоизомеры 3-а, 3-b, 4-а, 4-b, 5-а и 5-b:
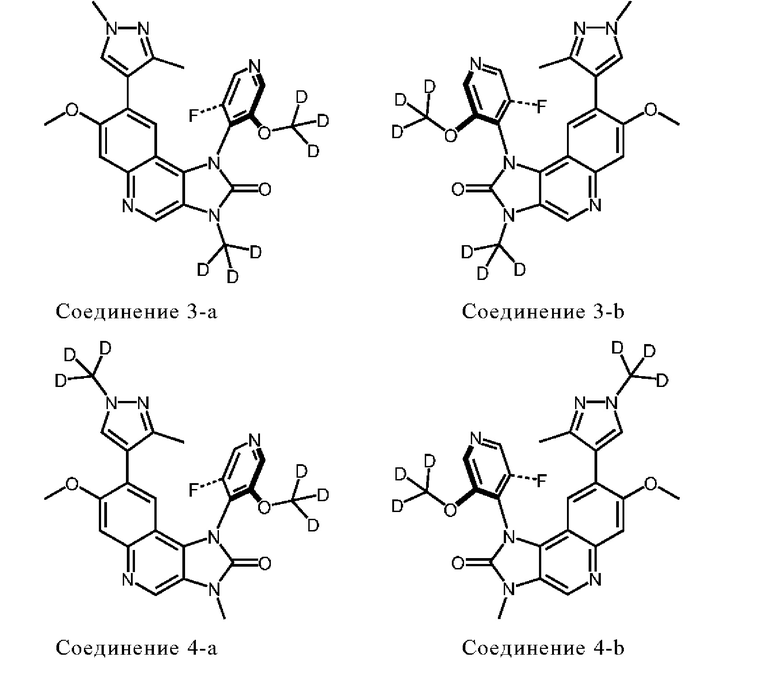
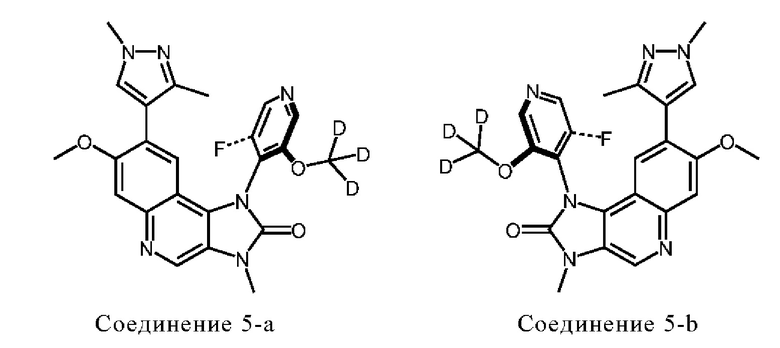
или их соли.
В соответствии с тем, что было изложено выше в отношении Соединений 1 и 2, настоящее изобретение обеспечивает Соединения 4-а, 5-а, 6-а, 4-b, 5-b, 6-b, по существу, свободные от соответствующего другого атропоизомера, включая любую его соль. В дополнительных вариантах осуществления эти соединения также предоставляются, по существу, свободными от примесей. В соответствии с другим вариантом осуществления эти соединения содержат не более примерно 5,0 процента от площади общих органических примесей в соответствии с ВЭЖХ по отношению к общей площади хроматограммы ВЭЖХ. Иллюстративные и предпочтительные диапазоны, раскрытые выше для Соединения 1 в связи с «по существу свободного от Соединения 2 или его солей», «свободного от примесей» и процентной долей общей площади органических примесей, по аналогии в равной степени применимы здесь по отношению к соответствующим другим атропоизомерам соответствующего соединения.
В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает фармацевтическую композицию, которая содержит эффективное количество, по крайней мере, одного из соединений 3, 4 или 5, его атропоизомера или фармацевтически приемлемой соли. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает способ приготовления таких фармацевтических композиций, описанных в данной заявке. Еще один вариант осуществления обеспечивает способ лечения рака при использовании фармацевтической композиции, описанной в данной заявке. В соответствии с другим вариантом осуществления настоящее изобретение обеспечивает применение описанной в данной заявке композиции при производстве лекарственного средства для лечения рака. Иллюстративные и предпочтительные варианты осуществления, раскрытые выше для Соединения 1, в равной степени применимы к этим соединениям.
Получение
Соединения 1 и 2 в соответствии с настоящим изобретением могут быть получены, начиная от Соединения Y, которое было описано ранее. Как раскрыто в WO 2016/155884, 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(3-фтор-5-метоксипиридин-4-ил)-7-метокси-3-метил-1,3-дигидроимидазо[4,5-с]хинолин-2-он (Соединение Y) может быть получен в соответствии со следующей последовательностью реакций:
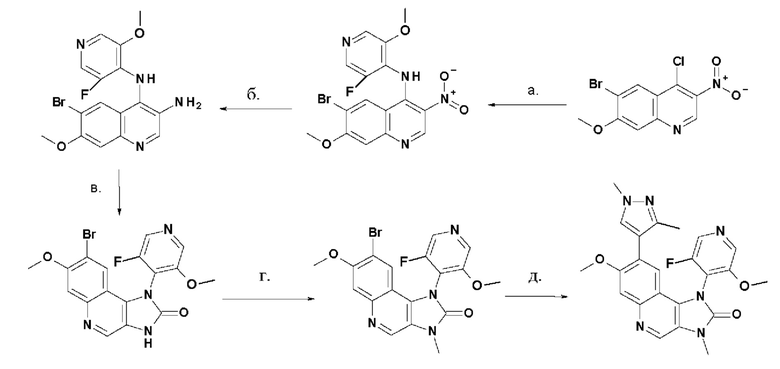
Типичные условия реакции для каждой из этих стадий а - е приведены в ПРИМЕРЕ 1, как и способы получения исходных соединений. Другие приемлемые условия реакции будут очевидны специалисту в данной области техники.
Соединения 1 и 2 затем можно получить с помощью приемлемых способов отделения от Соединения Y, иллюстративные варианты осуществления которых представлены в ПРИМЕРАХ 1, 2 и 3.
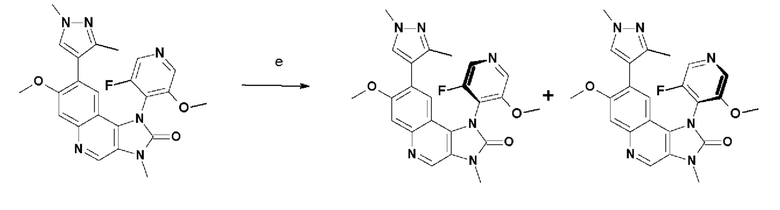
Атропоизомеры могут быть разделены, начиная с Соединения Y, при использовании хиральной хроматографии, включая хроматографию со сверхкритической подвижной фазой (SFC). Примеры подходящих способов подробно описаны в ПРИМЕРАХ 1 и 3.
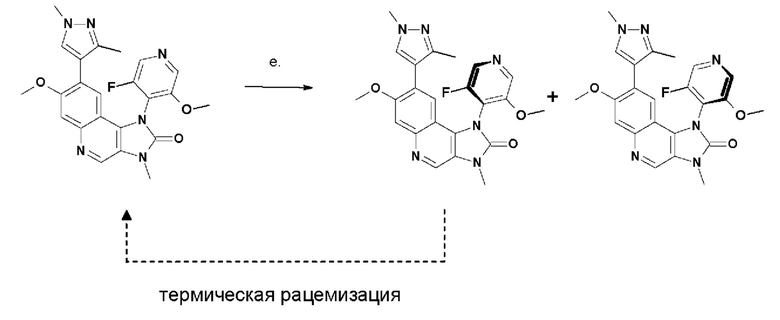
Соответствующие нежелательные атропоизомеры могут быть подвергнуты рацемизации, например, термической рацемизация с получением Соединения Y для применения в качестве нового исходного материала, как схематически показано ниже в качестве одного из примеров.
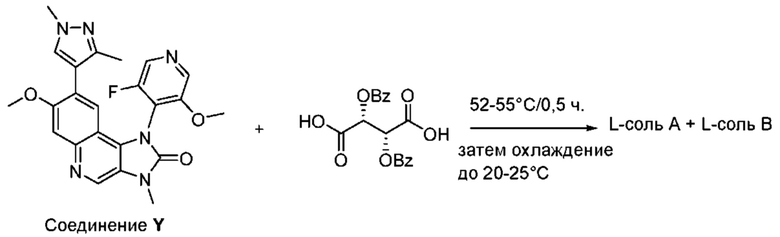
В альтернативном варианте осуществления Соединения 1 и 2 можно получить, исходя из Соединения Y, путем кристаллизации при использовании оптически активной кислоты, например, дибензоилвинной кислоты. Реакция Соединения Y с оптически активной кислотой дает соли пары атропоизомеров. Эти соли Соединений 1 и 2 проявляют разные физико-химические свойства (например, растворимость, фазовое распределение) и могут быть разделены, при использовании этих различий.
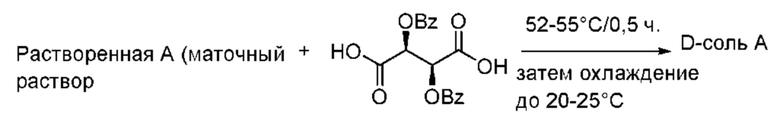
Как проиллюстрировано схемой, представленной ниже, в одном варианте осуществления Соединение Y вступает в реакцию с оптически активной кислотой, что приводит к образованию смеси двух солей в маточном растворе, при этом сначала осаждается соль Соединения 2 (L-соль В), которую удаляют посредством фильтрации, а соответствующую соль Соединения 1 (L-соль А) собирают только после дальнейшего концентрирования маточного раствора и осаждения. Затем соль Соединения 1 сначала превращают в ее свободную форму и выделяют, а затем подвергают взаимодействию с соответствующей другой оптически активной формой кислоты с получением соответствующей соли Соединения 1, которая на следующей стадии превращается в свободное основание, имеющее высокую оптическую чистоту. При этом Соединение 2 можно подвергать рацемизации с получением Соединения Y в качестве свежего исходного материала.
Подробный пример схемы получения обеспечивается в ПРИМЕРЕ 2 и на Фигуре 6.
Дейтерированные Соединения 3, 4 и 5 в соответствии с настоящим изобретением могут быть получены так, как описано подробно в ПРИМЕРЕ 6, а атропоизомеры, соли, сольваты и твердые формы получают, соответственно таким же образом как такие Соединений 1 и 2.
Применение
В дальнейшем любая общая ссылка на «соединения в соответствии с настоящим изобретением» должна применяться ко всем вариантам осуществления соединений настоящего изобретения, включая Соединение 1 или 2, или их фармацевтически приемлемые соли, сольваты или твердые формы, и может читаться как «Соединение 1 или 2, или их фармацевтически приемлемая соль, сольват или твердая форма». Аналогичным образом, любая ссылка на дейтерированные соединения в соответствии с настоящим изобретением должны включать не только Соединения 3, 4 или 5, но также любой атропоизомер, соль или твердую форму любого из перечисленных выше.
Изобретение также охватывает использование данных атропоизомеров, фармацевтически приемлемых твердых форм, их сольватов и солей ингибиторов ATM, их дейтерированных форм, а также атропоизомеров и их фармацевтически приемлемых солей для ингибирования, регулирования и/или модуляции сигнального каскада ATM киназы и, таким образом, предлагает новые инструменты для исследования и/или диагностики. Следовательно, изобретение, кроме того, относится к применению соединений в соответствии с настоящим изобретением, включая их дейтерированные формы, для ингибирования ATM киназы. Термин «ингибирование» относится к любому снижению активности, которое основано на действии конкретных соединений в соответствии с изобретением, поскольку последние способны взаимодействовать с молекулой-мишенью таким образом, что становится возможным распознавание, связывание и блокирование. Эти соединения отличаются высоким сродством к ATM киназе. Кроме того, соединения обладают высокой селективностью и, таким образом, позволяют осуществлять по существу исключительное и непосредтвенное распознавание ATM киназы. Для использования в исследованиях и/или диагностике дейтерированные соединения, т.е. Соединения 3, 4 или 5, или их атропоизомеры, или соли, или твердые формы считаются полезными, например, для использования в анализах.
Изобретение в общем случае охватывает применение соединений в соответствии с изобретением, включая дейтерированные соединения, при лечении заболеваний, которые вызываются, опосредуются и/или усугубляются активностью ATM киназы.
Таким образом, настоящее изобретение в широком смысле относится к соединениям в соответствии с изобретением, включая дейтерированные соединения, для использования в качестве лекарственного средства.
Таким образом, настоящее изобретение также относится к соединениям в соответствии с изобретением, включая дейтерированные соединения, для использования при лечении любого заболевания, которое вызывается, опосредуется и/или усугубляются активностью ATM киназы. Настоящее изобретение, соответственно, также относится к применению соединений, включая дейтерированные соединения, в соответствии с изобретением для приготовления лекарственного средства для лечения любого заболевания, которое вызывается, опосредуется и/или усугубляется активностью ATM киназы. Другими словами, настоящее изобретение также раскрывает соединение в соответствии с изобретением, включая дейтерированное соединение, для применения при лечении заболеваний, на которые оказывает влияние ингибирование ATM киназы.
Кроме того, соединения или дейтерированные соединения в соответствии с изобретением также можно использовать в качестве реагентов для тестирования киназозависимых сигнальных путей на животных и/или моделях культур клеток или при клинических заболеваниях, упомянутых в данной заявке. Как обсуждается в данной заявке, эти сигнальные пути являются важными для различных заболеваний.
Настоящее изобретение также относится к соединениям в соответствии с данным изобретением, включающим фармацевтически приемлемые соли, их сольваты, твердые и дейтерированные формы, для использования при лечении рака и/или опухолей; и к их применению при приготовлении лекарственного средства для лечения рака и/или опухолей.
Изобретение, кроме того, описывает способ лечения рака и/или опухолей, в котором эффективное количество, по крайней мере, одного соединения или его фармацевтически приемлемой соли, сольвата, дейтерированной или твердой формы в соответствии с изобретением вводят субъекту, которого подвергают лечению. Предпочтительными субъектами в смысле изобретения являются люди или животные, особенно предпочтительно люди.
Рак/опухоль может быть выбран, в частности, из группы рака/опухоли плоского эпителия, мочевого пузыря, желудка, почек, головы, шеи, пищевода, шейки матки, щитовидной железы, кишечника, печени, мозга, предстательной железы, мочеполового тракта, лимфатической системы, гортани, легкого, кожи, крови и иммунной системы и/или рак может быть выбран из группы моноцитарного лейкоза, аденокарциномы легкого, мелкоклеточного рака легкого, немелкоклеточного рака легкого, рака поджелудочной железы, колоректального рака, рака желудка, рака груди, рака яичников, острого миелолейкоза, хронического миелолейкоза, острого лимфобластного лейкоза, хронического лимфобластного лейкоза, лимфомы Ходжкина и неходжкинской лимфомы. Следует понимать, что сенсибилизация раковых клеток должна охватывать клетки тех же видов рака и опухолей, которые упоминались выше.
Настоящее изобретение также относится к лекарственному средству, содержащему соединение в соответствии с изобретением и/или его фармацевтически приемлемую соль, сольват, дейтерированную или твердую форму.
Изобретение, кроме того, относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения в соответствии с изобретением и/или его фармацевтически приемлемой соли, сольвата, дейтерированной или твердой формы, необязательно вместе, по крайней мере, с одним фармацевтически приемлемым наполнителем.
Под «лекарственным средством» и «фармацевтической композицией» следует понимать любую композицию, которая может быть использована для лечения пациентов, у которых, по крайней мере, временно, наблюдается патогенетическая модификация общего состояния или состояния отдельных частей организма пациента, предпочтительно вследствие рака и/или опухолей.
Доставка соединений, соответственно, фармацевтической композиции в соответствии с настоящим изобретением в клетку или организм может быть осуществлена в соответствии с изобретением любым способом, который позволяет ATM киназе вступать в контакт с соединениями, присутствующими в фармацевтической композиции, вследствие чего индуцируется ответ. Фармацевтическая композиция в соответствии с настоящим изобретением может вводиться перорально, трансдермально, через слизистые оболочки, трансуретрально, вагинально, ректально, через легкие, энтерально и/или парентерально. Выбранный тип введения зависит, например, от показаний, вводимой дозы, индивидуальных специфических параметров и т.д. Инъекции могут быть внутрикожными, подкожными, внутримышечными или внутривенными. Введение может осуществляться, например, с помощью так называемых вакцинных пистолетов или шприцов. Также можно предоставлять вещество в виде аэрозоля, который попадает в организм при вдыхании пациентом, предпочтительно человеком.
В предпочтительных вариантах осуществления соединения в соответствии с настоящим изобретением (в любой из их форм) вводят перорально. Пероральный прием благоприятен с точки зрения соблюдения пациентом режима лечения. Таким образом, фармацевтические композиции предпочтительно представляют собой твердые фармацевтические композиции для перорального применения.
Преимуществом соединений в соответствии с настоящим изобретением, в частности, Соединений 1 и 2 и их твердых форм, в частности, Соединения 1, соответственно, его твердой формы, является то, что они легко поддаются рецептированию в виде твердой лекарственной формы для перорального применения благодаря хорошей стабильности и высокой биодоступности.
Композиции
Композиции или фармацевтические композиции в соответствии с настоящим изобретением могут быть получены при использовании обычных твердых или жидких вспомогательных веществ, соответствующих желаемому типу введения, в подходящей дозировке и способом, который является известным сам по себе. Таким образом, фармацевтически приемлемые вспомогательные вещества, известные специалисту в данной области, могут в основном составлять часть фармацевтической композиции в соответствии с изобретением, где количество вспомогательного(ых) вещества(веществ), которые объединяются с активным соединением для приготовления единичной дозы, варьирует в зависимости от дозы и способа введения. Такие фармацевтически приемлемые наполнители включают наполнители, стабилизаторы, комплексообразующие агенты, антиоксиданты, растворители, связывающие агенты, лубриканты, соли, буферы, консерванты, регуляторы рН и тому подобное. Примерами вспомогательных веществ такого типа являются вода, растительные масла, бензиловые спирты, алкиленгликоль, полиэтиленгликоль, Коллифор, триацетат глицерина, желатин, углеводы, такие как, например, лактоза или крахмал, гидроксипропилметилцеллюлоза (ГПМЦ), стеарат магния, тальк и вазелин.
Как было упомянуто выше, настоящая фармацевтическая композиция предпочтительно предназначена для перорального введения. Фармацевтическая композиция обычно может быть в форме таблетки, таблетки, покрытой пленочной оболочкой, драже, пастилки, капсулы, пилюли, порошка, гранул, сиропа, сока, капель, раствора, дисперсии, суспензии, суппозитория, эмульсии, имплантата, крема, геля, мази, пасты, лосьона, сыворотки, масла, спрея, аэрозоля, клея, пластыря или повязки. Формы для перорального введения предпочтительно представляют собой таблетки, таблетки с пленочной оболочкой, драже, пастилки, капсулы, пилюли, порошки, гранулы, сиропы, соки, капли, растворы, дисперсии или суспензии.
Кроме того, можно рассматривать парентеральные фармацевтические композиции, такие как, например, суппозитории, суспензии, эмульсии, имплантаты или растворы, предпочтительно масляные или водные растворы. Для местного применения соединения в соответствии с настоящим изобретением могут быть рецептированы обычным способом, по крайней мере, с одним фармацевтически приемлемым наполнителем, таким как, например, микрокристаллическая целлюлоза, и, возможно, другими вспомогательными веществами, такими как, например, увлажнители, для получения композиций, которые можно наносить на кожу, таких как, например, кремы, гели, мази, пасты, порошки или эмульсии, или для получения жидких композиций, которые можно наносить на кожу, таких как, например, растворы, суспензии, лосьоны, сыворотки, масла, спреи или аэрозоли.
Фармацевтическая композиция также может быть в форме раствора для инъекций. Для приготовления раствора для инъекций можно использовать водную среду, такую как, например, дистиллированная вода или физиологические солевые растворы. Фармацевтическая композиция также может быть представлена в форме твердой композиции, например, в лиофилизированном состоянии, а затем может быть приготовлена для введения путем инъекции путем добавления растворяющего агента, такого как, например, дистиллированная вода или буфер. Специалист в данной области техники знаком с основными принципами приготовления лиофилизатов.
Количество соединения в соответствии с настоящим изобретением в фармацевтической композиции, которая содержит, по крайней мере, один фармацевтически приемлемый наполнитель, может составлять от 0,1 до 100 процентов по массе. Крайне важно, чтобы фармацевтическая композиция содержала эффективное количество соединения, необязательно вместе с одним или несколькими фармацевтически приемлемыми наполнителями. Простая фармацевтическая композиция может представлять собой соединение в соответствии с настоящим изобретением в твердой форме, такой как порошок, в твердой желатиновой капсуле. Термины «эффективное количество» или «эффективная доза», которые используются в данной заявке, являются взаимозаменяемыми и обозначают количество соединения в соответствии с настоящим изобретением, которое оказывает терапевтически релевантный эффект на заболевание или патологическое изменение в клетке, ткани, органе или млекопитающем, предпочтительно на рак и/или опухоль.
«Терапевтически эффективное количество» соединения в соответствии с изобретением относится к количеству, эффективному в необходимых дозах и в течение периодов времени, которое при введении пациенту с модулируемым ATMi или зависимым от ATMi состоянием, предпочтительно раком, будет оказывать предполагаемое терапевтическое действие, эффект, например, облегчение, улучшение, паллиативное воздействие или устранение одного или нескольких проявлений состояния, соответственно, рака, у пациента или любой другой клинический результат в ходе лечения пациента. Терапевтически эффективное действие не обязательно происходит при введении одной дозы и может происходить только после введения серии доз. Таким образом, терапевтически эффективное количество можно вводить в виде одного или нескольких введений. Такое терапевтически эффективное количество может варьировать в зависимости от таких факторов, как болезненное состояние, возраст, пол и вес индивидуума, а также способность соединения в соответствии с изобретением отдельно или в комбинации вызывать желаемый ответ у индивидуума. Терапевтически эффективное количество также представляет собой такое количество, при котором терапевтически полезные эффекты перевешивают любые токсические или вредные эффекты соединения в соответствии с изобретением.
В одном из вариантов осуществления изобретения соединение в соответствии с изобретением (или соль, сольват, дейтерированное или твердое вещество) вводят в дозе от 5 мг до 1 г на единицу дозировки, например, от 10 до 750 мг на единицу дозировки, например, от 20 до 500 мг на единицу дозировки, например, 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325 или 350 мг на единицу. По оценкам, биологически эффективная доза Соединения 1 находится в диапазоне от 25 до 350 мг один раз в день.
Благодаря их удивительно сильному и/или селективному ингибированию ATM киназы, которая регулирует клеточные процессы посредством репарации двухцепочечной ДНК, соединения в соответствии с изобретением можно вводить в выгодно низких дозах, при этом они достигают аналогичной или даже превосходящей биологической эффективности по сравнению с менее сильными или менее селективными ингибиторами. Сниженная доза обычно связана со снижением медицинских побочных эффектов. Кроме того, высокоселективное ингибирование обычно также отражается в уменьшении нежелательных побочных эффектов.
«Лечение» состояния или пациента относится к принятию мер для получения полезных или желаемых результатов, включая клинические результаты. Для целей данного изобретения полезные или желаемые клинические результаты включают, но не ограничиваются такими, как снижение, облегчение одного или нескольких симптомов заболевания, подлежащего лечению, наиболее предпочтительно рака; уменьшение степени заболевания; задержку или замедление прогрессирования заболевания; улучшение, временное облегчение или стабилизация болезненного состояния; или другие полезные результаты. Следует принимать во внимание, что ссылка на «лечение» включает профилактику, а также облегчение установленных симптомов состояния. Таким образом, «лечение» состояния, расстройства или заболевания включает: (1) предотвращение или отсрочку проявления клинических симптомов состояния, расстройства или заболевания, развивающихся у субъекта, который может быть поражен этим состоянием, расстройством или заболеванием или предрасположен к ним, но еще не испытывает или не демонстрирует клинических или субклинических симптомов состояния, расстройства или заболевания, (2) подавление состояния, расстройства или заболевания, то есть остановку, снижение или отсрочку развития заболевания или его рецидива (в случае поддерживающего лечения) или, по крайней мере, одного его клинического или субклинического симптома, или (3) облегчение или ослабление заболевания, т.е. обеспечение обратного развития состояния, расстройства или заболевания или, по крайней мере, одного из его клинических или субклинических симптомов. В некоторых вариантах реализации «лечение» включает (1) и (2).
«Опухоль» применительно к субъекту, у которого диагностирован или подозревается рак, относится к злокачественному или потенциально злокачественному новообразованию или тканевой массе любого размера и включает первичные опухоли и вторичные новообразования. Солидная опухоль -это аномальный рост или масса ткани, которая обычно не содержит кист или жидких участков. Различные типы солидных опухолей названы в соответствии с типами клеток, которые их формируют. Примерами солидных опухолей являются саркомы, карциномы и лимфомы. Лейкемии (рак крови) обычно не образуют солидных опухолей.
«Введение» соединения пациенту (и грамматические эквиваленты этой фразы) относится к непосредственному введению, которое может быть введением пациенту медицинским работником или может представлять собой введение самим пациентом, и/или опосредованному введению, которое может быть актом прописывания лекарства. Например, врач, который инструктирует пациента самостоятельно вводить лекарство или дает пациенту рецепт на лекарственное средство, должен рассматриваться как такой, который вводит лекарственное средство пациенту в контексте настоящего изобретения.
Все перечисленные выше и дополнительные наполнители или другие компоненты лекарственного средства или фармацевтического состава являются известными специалисту в данной области техники и могут быть подвергнуты специальному рецептированию для обучения в соответствии с изобретением в обычных экспериментах.
Комбинированная терапия
Лекарственные средства и фармацевтические композиции, которые содержат соединение в соответствии с изобретением, и использование этих соединений для лечения опосредованных киназой расстройств являются весьма многообещающим подходом, в частности, для лечения рака. Соединения в соответствии с изобретением можно вводить как монотерапию, как изложено выше, в сочетании с другими видами лечения, такими как, например, химиотерапия или радиационная терапия. Как указано выше, ссылка на соединение будет включать любую его соль, сольват, дейтерированные или твердые формы.
Ключевое участие ATM в процессах репарации ДНК и доказательства того, что дефицит ATM киназы позволяет клеткам млекопитающих стать более чувствительными к облучению, дает возможность терапевтически использовать ATM-специфические ингибиторы как часть лечения рака, например, солидных опухолей, путем радиационной терапии и/или химиотерапии, причем химиотерапия преимущественно направлена на индуцирование повреждения двухцепочечной ДНК. Как уже объяснялось ранее, ATM представляет собой привлекательное вмешательство для ингибирования разрывов двухцепочечной ДНК, вызванного терапией. Таким образом, соединения в соответствии с настоящим изобретением в любой из их форм очень выгодны в сочетании с радиационной терапией и/или химиотерапией, повреждающей ДНК.
В соответствии с этим настоящее изобретение касается комбинации соединения в соответствии с изобретением и радиационной терапии (РТ). Таким образом, настоящее изобретение относится к соединению в соответствии с настоящим изобретением или его фармацевтически приемлемой соли или твердой форме для использования при лечении рака и/или опухолей в сочетании с радиационной терапией. Иными словами, настоящее изобретение касается применения соединения в соответствии с настоящим изобретением или его фармацевтически приемлемой соли или твердой формы для приготовления лекарственного средства для лечения рака и/или опухолей в сочетании с радиацинной терапией и, таким образом, к способу лечения рака, включающему введение соединения в соответствии с настоящим изобретением или его фармацевтически приемлемой соли, или твердой формы в сочетании с радиационной терапией. Настоящее изобретение дополнительно относится к соединению в соответствии с настоящим изобретением или его фармацевтически приемлемой соли или твердой форме для использования в сенсибилизации раковых клеток к ионизирующему излучению или радиационной терапии (РТ).
Было показано, что Соединение 1 приводит к значительным зависимым от дозы противоопухолевым ответам in vivo в комбинации с клинически значимыми схемами облучения (например, радиационной терапией). В ПРИМЕРЕ 8 и на Фигуре 20 подробно представлены достигнутые результаты.
Режим приемлемого введения может включать, например, введение дозы радиационной терапии в 15 Грей (Гр), которая применяется в 5 фракциях (3 Гр на фракционный день) в течение недель (то есть, в течение последовательных 5 дней с последующими 2 днями без) и перорально в те же дни вводится соединение в соответствии с изобретением, что может повторяться, по крайней мере, один раз.
Промышленные способы облучения, которые используются клинически, предпочтительно включают облучение фотонами (классическое, электромагнитное рентгеновское/гамма излучение), облучение протонами, облучение тяжелыми ионами (ионизированный углерод) и облучение нейтронами, без каких-либо ограничений. Эти радиотерапии и другие приемлемые способы радиационной терапии в соответствии с настоящим изобретением являются известными специалистам в данной области техники, например, из Herrmann и др. (2006) Klinische Strahlenbiologie [Clinical Radiation Biology], Elsevier Munich, 4-ое издание, 67-68; Bhide & Nutting (2010) BMC Medicine 8: 25; Choi & Hung (2010) Current Urology Reports 11(3): 172, которые полностью включены в настоящую заявку в качестве ссылки. Как наиболее частое применение, фотонное облучение было технически усовершенствовано с помощью метода IMRT (лучевая терапия с модуляцией интенсивности) и методов визуализации (трехмерная конформная лучевая терапия) при планировании облучения и осуществления максимально точной фокусировки.
В соответствии с дополнительным аспектом настоящее изобретение относится к комбинации соединения в соответствии с настоящим изобретением и агента, повреждающего ДНК; таким образом, к соединению в соответствии с настоящим изобретением для применения при лечении рака и/или опухоли в комбинации с агентом, повреждающим ДНК, применению соединения в соответствии с изобретением для приготовления лекарственного средства для лечения рака в комбинации с агентом, повреждающим ДНК, и способу лечения рака, включающему введение соединения в соответствии с настоящим изобретением и агента, повреждающего ДНК. Как в целом объяснялось в данной заявке выше, соединение будет включать фармацевтически приемлемые соли, твердые формы и сольваты, в частности, в отношении композиции и способа лечения.
Введение агента, повреждающего ДНК, и соединения в соответствии с настоящим изобретением может быть одновременным или последовательным.
В данном контексте агент, повреждающий ДНК, представляет собой агент, который способен вызывать повреждение ДНК в клетке, особенно в раковой клетке, с иллюстративными вариантами осуществления, упомянутыми ниже.
Как объяснялось ранее, ATM киназа представляет собой ключевой регулятор восстановления разрыва двухцепочечной ДНК (DSB), которое индуцируется широко используемыми терапевтическими средствами от рака, такими как ионизирующее излучение (ИИ) и агент, повреждающий ДНК. При DSB событиях ATM подает сигналы на многочисленные расположенные ниже эффекторы, включая р53. Нерепарированные DSB приводят к активации контрольных точек, остановке клеточного цикла и, в конечном итоге, к гибели опухолевых клеток.
В одном варианте реализации настоящее изобретение обеспечивает фармацевтическую композицию, которая включает терапевтическое активное соединение в соответствии с изобретением и агент, повреждающий ДНК.
Агент, повреждающий ДНК, приемлемый для применения в комбинации (терапии), которая включает фармацевтическую композицию или набор, предпочтительно является выбранным из группы, которая включает:
• алкилирующие агенты, такие как альтретамин, бендамустин, бусульфан, кармустин, хлорамбуцил, хлорметин, циклофосфамид, дакарбазин, ифосфамид, импросульфан тозилат, ломустин, мелфалан, митобронитол, митолактол, нимустин, ранимустин, темозоломид, тиотепа, треосульфан, мехлороэтамин, карбохон, апазиквон, фотемустин, глуфосфамид, палифосфамид, пипоброман, трофосфамид, урамустин тремозустинтепацил, тремозустин теорема, сульфан, мехлорэтамин, карбоквон, апазиквон, фотемустин, глюфосфамид, палифосфамид, пипоброман, трофосфамид, урамустин;
• соединения платины, такие как карбоплатин, цисплатин, эптаплатин, мириплатин гидрат, оксалиплатин, лобаплатин, недаплатин, пикоплатин, сатраплатин;
• ингибиторы топоизомеразы, например, иринотекан, SN38, топотекан, камптотецин, рубитекан, белотекан, этопозид, даунорубицин, доксорубицин, акларубицин, эпирубицин, идарубицин, амрубицин, пирарубицин, валрубицин, амсакрин;
• ингибиторы поли-(АДФ-рибоза)-полимеразы (PARP), например, олапариб, нирапариб, велипариб;
• ATR (атаксия-телеангиэктазия и Кас13-связанный) ингибиторы, например, М6620 (VX-970: 3-[3-(4-метиламинометил-фенил)-изоксазол-5-ил]-5-[4-(пропан-2-сульфонил)-фенил]-пиразин-2-иламин), М4344 (VX-803: 2-амино-6-фтор-пиразол[1,5-а]пиримидин-3-карбоновая кислота [5'-фтор-4-(4-оксетан-3-ил-пиперазин-1-карбонил)-3,4,5,6-тетрагидро-2Н-[1,4']бипиридинил-3'-ил]-амид); AZD-6738 (4-[4-[1-[[S(R)]-S-метилсульфонимидоил]циклопропил]-6-[(3R)-3-метил-4-морфолинил]-2-пиримидинил]-1Н-пирроло [2,3-b]пиридин) и 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин;
• агенты, модифицирующие ДНК, такие, как амрубицин, бисантрен, децитабин, митоксантрон, прокарбазин, трабектин, клофарабин, амсакрин, бросталицин, пиксантрон, ларомустин;
• противоопухолевые антибиотики, такие как блеомицин, дактиномицин, доксорубицин, эпирубицин, идарубицин, левамизол, милтефозин, митомицин С, ромидепсин, стрептозоцин, валрубицин, зиностатин, зорубицин, даунорубицин, пликамицин, акларубицин, пепломицин, пирарубицин
• альфа излучатели, такие, как альфарадин (223Ra дихлорид, Xofgio), 211At, 213Bi, 225Ac, 227Th;
Особое предпочтение отдается этопозиду, иринотекану, разоксану, собузоксану, топотекану, камптотецину, доксорубицину, амсакрину, PARP ингибиторам и ATR ингибиторам.
Эффективность Соединения 1 в комбинации с типичным ингибитором PARP олапарибом была продемонстрирована на модели тройного отрицательного ксенотрансплантата рака молочной железы, полученной от пациента НВСх-10, разработанной на иммунодефицитных самках мышей, результаты исследования показаны на Фигуре 21. Более подробная информация об этом эксперименте представлена в ПРИМЕРЕ 9.
Изобретение также может быть реализовано в виде набора, который содержит соединение в соответствии с изобретением. Набор состоит из отдельных упаковок (а) эффективного количества соединения в соответствии с настоящим изобретением и/или его физиологической соли, сольвата или твердой формы и (б) эффективного количества дополнительного активного соединения. Дополнительное активное соединение представляет собой агент, повреждающий ДНК.
Набор может содержать приемлемые контейнеры, такие как, например, коробки или картонные коробки, отдельные флаконы, пакеты или ампулы. Набор может содержать, например, отдельные ампулы или флаконы, каждый из которых содержит эффективное количество соединения в соответствии с изобретением и/или его фармацевтически приемлемой соли, сольвата или твердой формы, или эффективное количество дополнительного активного соединения, такого как агент, повреждающий ДНК, в растворенной или лиофилизированной форме. Набор в соответствии с изобретением может также содержать изделие, которое содержит письменные инструкции или указывает пользователю на письменные инструкции, которые объясняют, как обращаться с соединениями в соответствии с изобретением.
Таким образом, следует отметить, что соединения в соответствии с изобретением можно использовать индивидуально и/или в комбинации с другими лечебными мероприятиями, такими как, например, хирургическое вмешательство, иммунотерапия, радиационная терапия и/или химиотерапия. Последние относятся к таргетной терапии при использовании любых желаемых активных соединений (химических или биологических, включая nME: новые молекулярные соединения, NCE: новые химические соединения, и NBE: новые биологические объекты) в виде монотерапии и/или нацеленной/нецелевой комбинированной терапии.
Все документы, которые цитируется в данном описании, предназначены для их включения в своей полноте в раскрытие настоящего изобретения в качестве ссылки.
Само собой разумеется, что данное изобретение не ограничивается конкретными соединениями, фармацевтическими композициями, применением и способами, как описано в данном документе, поскольку такие вещи могут варьировать. Кроме того, само собой разумеется, что используемая в данной заявке терминология служит исключительно для описания конкретных вариантов осуществления и не предназначена для ограничения объема защиты изобретения. Как используется в описании к данной заявке, включая прилагаемую формулу изобретения, словоформы в единственном числе, такие как, например, «какой-либо» или «этот», включают эквивалент во множественном числе, если контекст конкретно не указывает на иное. Например, ссылка на «соединение» включает отдельное соединение или множество соединений, которые, в свою очередь, могут быть идентичными или различными, или ссылка на «способ» включает эквивалентные этапы и методы, которые известны квалифицированному специалисту в данной области техники. Ссылка на объект как «содержащий» определенные функции должна интерпретироваться как такая, которая означает, что объект должен включать эти функции, но это не исключает наличия других функций, если они не делают объект неработоспособным.
Экспериментальная часть
Соединения в соответствии с изобретением демонстрируют полезные свойства, о чем свидетельствуют различные параметры и экспериментальные результаты. Экспериментальные способы, которые были использованы для анализа и характеристики соединений в соответствии с изобретением во всех их формах, представлены ниже.
Анализы
Измерение киназной активности - это метод, хорошо известный специалисту в данной области техники. Генерические тест-системы для определения киназной активности при использовании субстратов, например, гистона (Alessi и др. (1996) FEBS Lett. 399 (3): 333) или основного белка миелина, описаны в литературе (Campos-Gonzalez & Glenney (1992) JBC 267: 14535). Для идентификации ингибиторов киназ доступны различные системы анализа. В сцинтилляционном анализе сближения (Sorg и др. (2002) J Biomolecular Screening 7: 11) и в анализе с флэш-пластиной радиоактивное фосфорилирование белка или пептида в качестве субстрата измеряется при использовании АТФ. В присутствии ингибирующего соединения обнаруживается снижение радиоактивного сигнала или его отсутствие вообще. Кроме того, технологии гомогенного резонансного переноса энергии флуоресценции с временным разрешением (HTR-FRET) и флуоресцентной поляризации (FP) являются полезными в качестве методов анализа (Sills и др. (2002) J Biomolecular Screening 191). Другие нерадиоактивные методы ELISA используют специфические фосфо-антитела (фосфо-АТ). Фосфо-АТ связывают только фосфорилированный субстрат.Это связывание можно обнаружить с помощью хемилюминесценции при использовании второго конъюгированного с пероксидазой антитела против овец.
Для целей настоящего изобретения соответствующие целевые свойства соединений оценивали с помощью следующих анализов:
Анализ ATM киназы - определение ингибирования ATM (IC50 ATM):
Величину IC50 определяли с помощью биохимического анализа ATM киназы. Анализ состоит из двух этапов: ферментативной реакции и этапа обнаружения. Сначала белок ATM (мутантная при атаксии-телангиэктазии киназа) и тестируемое вещество инкубируют в различных концентрациях с добавлением белка-субстрата р53 и АТФ. ATM опосредует фосфорилирование р53 в нескольких положениях, в том числе по аминокислоте S15. Количество фосфорилированного р53 определяется с помощью специфических антител и методики TR-FRET. Ферментативный анализ ATM осуществляется как 384-луночный анализ на основе TR-FRET (HTRFTM, Cisbio Bioassays). На первом этапе очищенный человеческий рекомбинантный белок ATM (человеческий ATM, полноразмерный, GenBank ID нМ_000051, экспрессированный в линии клеток млекопитающих) инкубируют в аналитическом буфере в течение 15 минут с ингибитором ATM в различных концентрациях и без тестируемого вещества в качестве негативного или нейтрального контроля. Аналитический буфер включает 25 мМ HEPES рН 8,0, 10 мМ Mg(CH3COO)2, 1 мМ MnCl2, 0,1% БСА и 0,01% Brij® 35,5 мМ дитиотреитола (ДТТ). Растворы исследуемого вещества распределяли в микротитровальные планшеты при использовании ECHO 555 (Labcyte). На втором этапе прибавляли очищенный человеческий рекомбинантный с-myc, меченный р53 (человеческий р53, полноразмерный, GenBank ID ВС003596, экспрессированный в Sf21 клетках насекомых) и АТФ, и реакционную смесь инкубировали при 22°С в течение 30-35 минут. Фармакологически релевантный аналитический объем составлял 5 мкл. Заключительная концентрация в анализе во время инкубации реакционной смеси составляла 0,3-0,4 нМ ATM, 50-75 нМ р53 и 10 мкМ АТФ. Ферментативную реакцию останавливали путем прибавления ЭДТА. Образование фосфорилированного р53 как результат опосредованной ATM реакции в присутствии АТФ определяли с помощью специфических антител [меченных флуорофором европия (Eu) в качестве донора и d2 в качестве акцептора (Cisbio Bioassays)], которые позволяют осуществлять FRET. 2 мкл содержащего антитела стоп-раствора (12,5 мМ HEPES рН 8,0, 125 мМ ЭДТА, 30 мМ хлорида натрия, 300 мМ фторида калия, 0,1006% Твин-20, 0,005% Brij® 35, 0,21 нМ анти-фосфо-р53(ser15)-Eu антитела и 15 нМ анти-cmyc-d2 антитела) прибавляли к реакционной смеси. После инкубации, обычно в течение 2 часов (от 1,5 до 15 часов) для развития сигнала, планшеты подвергали анализу в устройстве для считывания планшетов (En Vision, PerkinElmer) при использовании способа TRF (и с применением лазерного возбуждения). После возбуждения донора европия при длине волны 340 нМ подвергали измерению излучаемый флуоресцентный свет как d2 при 665 нМ, так и донора Eu при 615 нМ. Количество фосфорилированного р53 является прямо пропорциональным показателю количества излучаемого света, т.е. относительным единицам флуоресценции (RFU) при 665 нМ и 615 нМ. Результаты измерения обрабатывали с помощью программного обеспечения Genedata Screener. Определение IC50 осуществляли, в частности, путем подгонки кривой доза/действие к точкам данных с помощью нелинейного регрессионного анализа.
IC50 = половина максимальной ингибирующей концентрации
АТФ = аденозинтрифосфат
TR-FRET = разрешенный во времени флуоресцентный индуктивно-резонансный перенос энергии
HTRF® = гомогенная флуоресценция с временным разрешением
HEPES = 2-(4-(2-гидроксиэтил)-1-пиперазинил)этансульфоновая кислота
Mg(CH3COO)2 = ацетат магния
MnCl2 = хлорид марганца (II)
БСА = бычий сывороточный альбумин
ЭДТА = этилендиаминтетраацетат
TRF = флуореценция с временным разрешением
Указанные сокращения применяются везде в данном описании, если не указано иное.
Способ определения значения IC50 при концентрации АТФ 1000 мкМ отличается от указанного выше только концентрацией АТФ.
Анализ клеточной pCHK2:
Для идентификации веществ, которые ингибируют фосфорилирование протеинкиназы CHK2 (киназы контрольной точки 2) в положении аминокислоты треонина 68, использовали анализ «высокого содержания» на основе флуоресенции в клетках НСТ116.
In vitro клеточный иммунофлуоресцентный анализ для идентификации ингибиторов индуцированного блеомицином фосфорилирования CHK2 (фосфо-Thr68) в клеточной линии карциномы толстого кишечника человека НСТ116:
Клетки НСТ116 высевали при определенной плотности клеток в планшеты на 384 ячейки в культуральной среде (DMEM с высоким содержанием глюкозы, 2 мМ GlutaMax, 1 мМ пирувата Na, 10% фетальной телячьей сыворотки (ФТС) и инкубировали в течение ночи при 37°С и 10% СО2. На следующий день прибавляли исследуемые вещества в определенном интервале концентраций (от 1 нМ до 30 мкМ) в комбинации с 10 мкМ блеомицина, где концентрация растворителя ДМСО поддерживалась постоянной на уровне 0,5%. После инкубации в течение четырех часов при 37 С и 10% СО2, клетки фиксировали (5 мин., 4% формальдегид в ФСБ), пермебиализировали (10 мин., 0,2% Тритон Х-100 в ФСБ) и после блокирования неспецифических сайтов связывания (10% козья сыворотка, 1% БСА в ФСБ), инкубировали в течение ночи при 4 С со специфическим анти-pCHK2 антителом (система клеточных сигналов #2661). pCHK2 (Thr68) определяли при использовании А1еха488-меченного вторичного анти-кроличьего IgG антитела. Параллельное окрашивание ДНК с помощью иодида пропидия позволяло оуществить подсчет клеток. рСНК2 сигнал определяли при использовании устройства для формирования изображений для высоких концентраций (Molecular Devices IMX Ultra) и автоматического анализа изображения с помощью программного обеспечения MetaXpress, принадлежащего устройству. Определяли количество ядер клеток, которые имеют сигнал pCHK2 выше определенного фона.
DMEM: модифицированная по способу Дюльбекко среда Игла; ФТС: фетальная телячья сыворотка, ФСБ: фосфатно-солевой буфер (сокращения применяются везде в данном описании, если не указано иное).
Кроме того, эффект, в частности, ингибирование других киназ и, таким образом, селективность соединения в соответствии с изобретением может быть определена с помощью следующих анализов:
Анализ ATR/ATRIP киназы
Величину IC50 определяли с помощью ATR/ATRIP ферментативного анализа. Анализ состоял из двух этапов: ферментативной реакции и этапа обнаружения. Сначала смесь белка ATR/ATRIP (атаксия-телеангиэктазия и Rad3-связанный белок / взаимодействующий белок ATR), исследуемое соединение в различных концентрациях, р53 в качестве белка-субстрата и аденозинтрифосфат (АТФ) инкубировали в буфере для анализа. ATR фосфорилирует р53 по Serl5 и другим остаткам. Затем количество фосфорилированного р53 определяли с помощью специфических антител и технологии анализа TR-FRET.
Подробно: ATR/ATRIP ферментативный анализ осуществляли как TR-FRET- (HTRF™, Cisbio Bioassays) на основе 384-луночного анализа. На первом этапе очищенный рекомбинантный ATR/ATRIP (человеческий ATR, полноразмерный, GenBank ID: нМ_001184,3, и человеческий ATRIP, полноразмерный, GenBank ID AF451323,1, совместно экспрессированные в клеточной линии млекопитающих) инкубировали в аналитическом буфере в течение 15 минут при 22°С с исследуемым соединением в различных концентрациях или без исследуемого соединения (в качестве негативного контроля). Аналитический буфер содержал 25 мМ HEPES рН 8,0, 10 мМ Mg(CH3COO)2, 1 мМ MnCl2, 0,1% БСА, 0,01% Brij® 35, и 5 мМ дитиотреитола (ДТТ). Echo 555 (Labcyte) использовали для дозирования растворов соединения. Потом, на втором этапе, очищенный человеческий рекомбинантный стус-меченный р53 (человеческий р53, полноразмерный, GenBank ID: ВС003596, экспрессированный в Sf21 клетках насекомых) и АТФ прибавляли к реакционной смеси и инкубировали в течение 25-35 минут, типично 25 минут, при 22°С. Фармакологически релевантный объем составлял 5 мкл. Заключительные концентрации в анализе в процессе инкубации реакционной смеси составляли 0,3-0,5 нМ, типично 0,3 нМ, ATR7ATRIP, 50 нМ р53 и 0,5 мкМ АТФ. Ферментативную реакцию останавливали путем прибавления ЭДТА. Образование фосфорилированного р53 как результат опосредованной ATM реакцией в присутствии АТФ определяли с помощью специфических антител [меченного флуорофором европия (Eu) в качестве донора и d2 в качестве акцептора (Cisbio Bioassays)], которые позволяют осуществлять FRET. С этой целью 2 мкл содержащего антитела стоп-раствора (12,5 мМ HEPES рН 8,0, 125 мМ ЭДТА, 30 мМ хлорида натрия, 300 мМ фторида калия, 0,1006% Твин-20, 0,005% Brij® 35, 0,21 нМ анти-фосфо-р53(8ег15)-Еи антитела и 15 нМ анти-cmyc-d2 антитела) прибавляли к реакционной смеси. После развития сигнала в течение 2 часов планшеты анализировали на считывающем устройстве для микропланшетов EnVision (PerkinElmer) при использовнии способа TRF с применением лазерного возбуждения. При возбуждении донора европия при длине волны 340 нМ подвергали измерению излучаемый флуоресцентный свет от акцептора d2 при 665 нМ, а также от донора Eu при 615 нМ. Количество фосфорилированного р53 является прямо пропорциональным соотношению количеств излучаемого света, т.е. относительным единицам флуоресценции (rfu) при 665 нМ и 615 нМ. Результаты измерения обрабатывали с помощью программного обеспечения Genedata Screener. В частности, значения IC50 определяли обычным способом путем подгонки кривой доза/действие к точкам данных с помощью нелинейного регрессионного анализа.
Для расшифровки сокращений смотри список, приведенный выше.
Анализ клеточной pCHK1
Chk1 киназа действует ниже от ATR и играет ключевую роль в регуляции контрольной точки повреждения ДНК. Активация Chk1 вовлекает фосфорилирование в положениях Ser317 и Ser345 (считается предпочтительной мишенью для фосфорилирования/активации ATR) и возникает в ответ на блокированную репликацию ДНК и некоторые формы генотоксического стресса. Фосфорилирование в положении Ser345 служит для перенесения Chk1 в ядро после активации контрольной точки.
Этот анализ измеряет снижение фосфорилирования Chk1 (Ser 345) в НТ29 клетках аденокарциномы толстого кишечника после обработки при использовании соединения и гидроксимочевины (что способствует остановке вилки по причине истощения dNTP) с применением иммуноцитохимической процедуры и получения изображения высокого содержания.
Для анализа НТ29 клетки высаживали в культуральную среду (DMEM с высоким содержанием глюкозы (без фенолового красного), 2 мМ глутамакс, 1 мМ пирувата, 10% ФТС, планшеты Greiner на 384 ячейки, черные, μclear # 781090 (2500 клеток/ячейка/30 мкл) и инкубировали в течение, по крайней мере, 20 часов при 37°С, 10% CO2 и 90% относительной влажности. Разведенное исследуемое соединение (заключительное содержание 1 нМ - 30 мкМ) и гидроксимочевину (заключительное содержание 3 мМ) прибавляли одновременно, и клетки инкубировали в течение 4 часов при 37°С. После фиксации/пермибиализации при использовании 100% МеОН (-20°С, холодный) и пермибиализации с помощью 0,2% Тритона Х-100 полную иммуноцитохимическую процедуру осуществляли при использовании специфического анти-pChkl антитела (Cell Signaling, #2348BF) и флуоресцентно меченного вторичного антитела (Alexa Fluor® 488 козий анти-кроличий F(ab')2 фрагмент, Invitrogen А11070) и параллельного окрашивания ядер для подсчета клеток.
Локализованный в ядре pChk1 сигнал определяли на ImageXpress Ultra конфокальном считывающем устройстве высокого содержания и представляли как % позитивных клеток (ядер).
Анализ ДНК-РК
Киназный анализ осуществляли как HTRF® при использовании анализа на 384 ячейки. На первом этапе комплекс ДНК-РК белок инкубировали с исследуемым соединением или без него в течение 15 минут при 22°С. После прибавления STK-субстрата 1-биотина (Cisbio), Mg-АТФ, ДНК и стауроспорина реакционную смесь инкубировали в течение 60-80 минут (в зависимости от активности комплекса ДНК-РК белок) при температуре 22°С. Echo 555 (Labcyte) использовали для распределения растворов соединения. Аналитический буфер состоял из 25 мМ HEPES рН 7,4, 11 мМ MgCl2, 80 мМ KCl, 0,45 мМ ЭДТА и 0,5 мМ ЭГТА, и содержал 1 мМ дитиотреитола (ДТТ), 0,17% БСА, и 0,01% Твин® 20. Фармакологически релевантный объем составлял 5 мкл. Конечные концентрации в анализе во время инкубации реакционной смеси составляли 50-100 нг/ячейку белкового комплекса ДНК-РК (в зависимости от активности белкового комплекса ДНК-РК), 1 мкМ STK-субстрата 1-биотина, 10 мкМ Mg-АТФ, 80 нг/ячейку ДНК из тимуса теленка и 1 мкМ стауроспорина. Ферментативную реакцию останавливали путем прибавления ЭДТА. Образование фосфорилированного STK-субстрата 1-биотина как результат реакции, опосредованной ДНК-РК, определяли с помощью специфического антифосфо-STK-антитела (Cisbio), меченного европием (Ей) в качестве донора и стрептавидина, меченного XL665 (Cisbio), в качестве акцептора, позволяющего осуществлять FRET. С этой целью к реакционной смеси прибавляли 4 мкл стоп-раствора, содержащего антитела и стрептавидин (12,5 мМ HEPES рН 8,0, 125 мМ ЭДТА, 30 мМ хлорида натрия, 300 мМ фторида калия, 0,006% Твин-20, 0,005% Brij® 35, 0,179 нМ анти-фофо-STK антитела и 160 нМ стрептавидин-XL665). После развития сигнала в течение 1 часа планшеты анализировали на Rubystar или Pherastar считывающем устройстве для м икр о планшетов (BMG Labtech). Количество фосфорилированного субстрата было прямо пропорционально отношению единиц флуоресценции (длина волны возбуждения 337 нМ) при длинах волн излучения 665 нМ (длина волны, чувствительная к фосфопептидам / эмиссия XL665) к единицам при 620 нМ (эталонная длина волны европия). Значения IC50 рассчитывали при использовани программного обеспечения Genedata Screener® (Molecular Cancer Therapeutics 2003, 1257-1264; DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer; A. Kashishian, H. Douangpanya, D. Clark, S. T. Schlachter, C. Todd Eary, J. G. Schiro, H. Huang, L. E. Burgess, E. A. Kesicki, and J. Halbrook.)

Клеточный анализ фосф. ДНК-протеинкиназа
Клетки НСТ116 культивировали в среде MEM альфа с 10% ФТС и 2 мМ глутамина при 37°С и 10% СО2. Клетки отделяли от дна культуральных сосудов при использовании трипсина/ЭДТА, центрифугировали в центрифужных пробирках, переносили в свежую среду и определяли плотность клеток. 100000 клеток высевали в 1 мл культуральной среды на лунку 24-луночного планшета для культивирования клеток и культивировали в течение ночи. На следующий день к клеткам добавляли 10 мкМ блеомицина (интеркалятор ДНК и индуктор разрыва двухцепочечной ДНК) и исследуемые вещества в свежей культуральной среде и культивировали в течение последующих шести часов. Затем был проведен лизис клеток, и лизаты клеток наносили на 96-луночный планшет для ELISA (Sigma-Aldrich WH0005591M2: общая ДНК-РК, Abeam ab 18192 или Epitomics ЕМ09912: фосфосерин 2056 ДНК-РК), который блокировали и покрывали ДНК-РК специфическими антителами и инкубировали при 4°С в течение ночи. Затем 96-луночные планшеты для ELISA обрабатывали антителом для определения (Abeam ab79444: общая ДНК-РК) и конъюгатом стрептавидин-пероксидаза хрена. Развитие ферментативной реакции осуществляли при использовании хемилюминесцентного реагента, хемилюминесценцию измеряли с помощью Mithras LB940. Сигналы специфического антитела к фосфо-ДНК-протеинкиназе нормализовали к сигналу с антителом против цельного белка ДНК-протеинканазы С. Значения IC50 или проценты определяли, ссылаясь на уровень сигнала контрольной группы, обработанной блеомицином (100% от контроля). В качестве холостого опыта использовали ДМСО контроль.
MEM: минимальная питательная среда; ДМСО: диметилсульфоксид
Анализ PDE2A1
Использовали коммерчески доступный анализ (Cerep, катал, номер 4071, SOP n1C1054), который разработан для оценки влияния соединений на активность человеческой фосфодиэстеразы-2А1, количественно определяемой путем измерения образования 5'АМФ из цАМФ при использовании рекомбинантного фермента человека, экспрессируемого в клетках Sf9.
Исследуемое соединение, стандартное соединение или воду (контроль) добавляли в буфер, содержащий 40 мМ Трис/HCl (рН 7,4), 8 мМ MgCl2 и 1,7 мМ ЭГТА/NaOH, 1,8 мкМ цАМФ и 1 мкКи [3Н]цАМФ. После этого реакцию инициировали путем добавления фермента (около 2,5 ед.), и смесь инкубироввали в течение 20 мин. при 22°С. Для базальных контрольных измерений фермент исключали из реакционной смеси. После инкубации добавляли гранулы для SPA (сцинтилляционного анализа близости).
Через 30 мин. при встряхивании при температуре 22°С количество [3Н]5'АМФ определяли количественно с помощью сцинтилляционного счетчика (Topcount, Packard). Результаты выражали в процентах ингибирования активности контрольного фермента.
Стандартное ингибирующее контрольное соединение представляло собой EHNA (эритро-9-(2-гидрокси-3-нонил)аденин), это соединение тестировали в каждом эксперименте при нескольких концентрациях для получения кривой ингибирования, по которой рассчитывали его значение IC50.
Библиографическая ссылка: Maurice D.H., Ke Н., Ahmad F., Wang Y., Chung J. и Manganiello V. С.(2014), Advances in targeting cyclic nucleotide phosphodiesterases, Nat. Rev. Drug Discov., Vol. 13 Issue 4: p.290.
Анализ PDE4D2
Использовали коммерчески доступный анализ Cerep (каталожный номер 4077; SOP n1C1045). Анализ был разработан для оценки воздействия исследуемых соединений на активность человеческой фосфодиэстеразы-4D2, количественно определяемой путем измерения образования 5'АМФ из цАМФ при использовании рекомбинантного фермента человека, экспрессированного в клетках Sf9.
Исследуемое соединение, контрольное соединение или воду (контроль) добавляли в буфер, содержащий 40 мМ Трис/HCl (рН 7,4) и 8 мМ MgCl2, 450 нМ цАМФ и 0,0125 мкКи [3Н] цАМФ. После этого реакцию инициировали путем добавления фермента (около 1,5 ед.), и смесь инкубировали в течение 20 мин при 22°С. Для базальных контрольных измерений фермент исключали из реакционной смеси. После инкубации добавляли гранулы для SPA (сцинтилляционного анализа близости).
Через 30 мин. при температуре 22°С при встряхивании количество [3Н]5'АМФ определяли количественно с помощью сцинтилляционного счетчика (Topcount, Packard). Результаты выражали в процентах ингибирования активности контрольного фермента.
Стандартное ингибирующее контрольное соединение представляло собой Ro 20-1724, которое испытывали в каждом эксперименте при нескольких концентрациях, чтобы получить кривую ингибирования, по которой рассчитывали его значение IC50.
Библиографическая ссылка, как указано выше.
Анализ PDE42A1
Использовали коммерчески доступный анализ Сегер (каталожный номер 4074; SOP n1C1056), который разработан для оценки влияния соединений на активность человеческой фосфодиэстеразы-4А1 А, количественно определяемой путем измерения образования 5'АМФ из цАМФ при использовании рекомбинантного фермента человека, экспрессируемого в клетках Sf9.
Исследуемое соединение, стандартное соединение или воду (контроль) добавляли в буфер, содержащий 40 мМ Трис/HCl (рН 7,4) и 8 мМ MgCl2, 450 нМ цАМФ и 0,25 мкКи [3Н]цАМФ. После этого реакцию инициировали путем добавления фермента (около 10 ед.), и смесь инкубировали в течение 20 мин. при 22°С. Для базальных контрольных измерений фермент исключали из реакционной смеси. После инкубации добавляли гранулы для SPA (сцинтилляционного анализа близости).
Через 30 мин. при 22°С при встряхивании [3Н]5'АМФ определяли количественно с помощью сцинтилляционного счетчика (Topcount, Packard). Результаты выражали в процентах ингибирования активности контрольного фермента.
Стандартное ингибирующее контрольное соединение представляло собой Ro 20-1724, которое испытывали в каждом эксперименте при нескольких концентрациях, чтобы получить кривую ингибирования, по которой рассчитывали его значение IC50.
Библиографическая ссылка, как указано выше.
Дополнительные параметры, которые считаются важными, определяли следующим образом:
Способ исследования микросомалъной стабильности (собственный системный клиренс)
Для измерения клиренса in vitro использовали анализ микросомалъной стабильности (Clint). Анализ включает измерение скорости исчезновения соединения благодаря его собственному отношению к метаболизму («собственный» означает, что на исчезновение не влияют другие свойства, такие как проницаемость, связывание и т.д., которые играют роль при количественной оценке клиренса in vivo). Микросомальная стабильность (собственный клиренс, Clint) и, таким образом, метаболическая стабильность обычно выражается в мкл/мин./мг белка. Это можно представлять как объем раствора, который 1 мг микросом может очистить от соединения за одну минуту.
Приборы
Для проведения инкубации микросом использовали рабочую станцию Tecan Genesis (RSP 150/8). Анализ проводился при использовании системы Waters ACQUITY UPLC, соединенной с масс-спектрометром ABSciex API3000. Анализ данных выполняли с помощью Assay Explorer (Symyx).
Условия СВЭЖХ
Колонка: Acquity UPLC ВЕН С18, 2,1 θ 50 мм, 1,7 мкМ (Waters)
Подвижные фазы: А = 0,1% муравьиной кислоты в воде; Б = ацетонитрил

Скорость потока: 0,750 мл/мин.; обнаружение: ESI (времяпролетная масс-спектрометрия с электрораспылительной ионизацией), MRM (селективный мониторинг реакций); для инъекций: 10 мкл; температура колонки: 50°С
Химические реактивы
• Калий-фосфатный буфер: 0,05 М калий-фосфатный буфер рН 7,4, содержащий 1 мМ MgCl2
• НАДФН (восстановленный никотинамидадениндинуклеотидфосфат): 22,5 мг НАДФН-Ка4 в 1,8 мл калий-фосфатного буфера
• Ацетонитрил: 50 об. % ацетонитрила (1 объем ацетонитрила, 1 объем воды)
• ДМСО: 20 об. % ДМСО в воде
• Маточный раствор 20 мг/мл микросом (белок) печени человека или мыши/мл в фосфатном буфере
• Маточный раствор 10 мМ соединения в 100% ДМСО
Инкубация микросом
Разведения исследуемых соединений выполняли в 2 этапа, начиная с 10 мМ исходного раствора соответствующего соединения в 100% ДМСО. Первые 4 мкл исходного раствора добавляли к 196 мкл 20 об. % ДМСО. На втором этапе 10 мкл первого разведения добавляли к 1590 мкл калий-фосфатного буфера для достижения конечной концентрации 1,25 мкл в конечном разведении соединения. Таким образом, количество органического растворителя в анализе было минимальным (<1%).
Микросомальный раствор (белок) печени человека или мыши, который использовали в анализе, готовили путем смешивания 750 мкл исходного раствора (20 мг/мл) и 2250 мкл калий-фосфатного буфера до конечной концентрации 5 мг/мл.
Инкубацию проводили на 96-луночном планшете для инкубации. 160 мкл на лунку конечного разведения соединения переносили на планшет для инкубации. Были проанализированы четыре образца каждого разведения соединения. В каждую лунку добавляли 20 мкл/лунка раствора микросом печени, а затем образцы предварительно инкубировали в течение 5 минут при 37°С и перемешивании при 800 об./мин. Два эталонных соединения (верапамил и декстрометорфан) использовали параллельно в каждом эксперименте для каждого вида (микросомы человека или мыши), чтобы обеспечить производительность системы для сравнения.
На отдельный стоп-планшет добавляли 160 мкл ацетонитрила на лунку.
После предварительной инкубации, то есть в момент времени t1 = 0 минут, 18 мкл образцов инкубированного раствора соединения на лунку (содержащую ацетонитрил) переносили и добавляли на стоп-планшет для предотвращения реакции (0 минут контрольные образцы, 4 образца на соединение). Таким же образом 18 мкл образцов инкубированного раствора эталонного соединения переносили и добавляли на лунку (содержащую ацетонитрил) на стоп-планшете в момент времени t1 = 0 минут и снова через 30 минут (14), проверяли растворимость и химическую стабильность соединения.
Для начала реакции 26 мкл раствора НАДФН (кофактора) добавляли во все лунки, которые включали предварительно инкубированное разведение соединения или контрольный раствор, за исключением тех лунок, которые включали предварительно инкубированное разведение соединений, которые должны были использоваться в качестве 30-минутных контрольных образцов, где вместо этого добавляли 26 мкл фосфатного буфера. Затем инкубацию продолжали при 37°С и перемешивании 800 об./мин.
Через t2 = 5 минут, t3 = 10 минут и t4 = 20 минут времени инкубации (т.е. после начала реакции) 20 мкл образцов инкубированного раствора соединения (4 образца на соединение) и контрольный раствор соединения переносили и прибавляли на ячейку стоп-планшета с ацетонитрилом.
Через t4 = 30 минут времени инкубации 20 мкл образцов инкубированного раствора соединения (4 образца на соединение) и 20 мкл образцов 30 минутного контроля образцов (содержащих буфер вместо НАДФН) а также 20 мкл образцов раствора инкубированного эталонного соединения переносили и добавляли на ячейку стоп-планшета с ацетонитрилом.
Погашенные образцы подвергали центрифугированию при 4000g в течение 1 часа при 4°С. 80 мкл супернатанта переносили в планшеты на 96 ячеек для анализа при использовании ЖХ-МС/МС.
Анализ данных
Микросомальную/метаболическую стабильность соединения определяли путем измерения изменения площади пика ЖХ-МС/МС во времени. Данные подбирали в соответствии с логарифмической линейной моделью, руководствуясь Михаэлисом/Ментеном. Значение Clint рассчитывали из наклона (k) преобразованной линейно-логарифмической концентрации на временном графике, разделенной на количество микросом (0,5 мг/мл): Clint (мкл/мин./мг белка) = k* 1000 / концентрация белка. Программное обеспечение Assay Explorer использовали для автоматического расчета наклона k спада.
Kv11.1 (hERG) активность ионного канала (пэтч-клемп анализ)
Метод обнаружения и характеристики исследуемых веществ, которые препятствуют каналу Kv11.1 (hERG): Kv111 (hERG, ген специфических калиевых каналов сердца человека) представляет собой калиевый канал, который играет центральную роль в реполяризации клеток кардиомиоцитов желудочков.
Измерение «пэтч-клемп» проводили при комнатной температуре в цельноклеточной конфигурации человеческих эмбриональных клеткок почек Цельноклеточные конфигурации осуществляли при использовании автоматического пэтч-клемп устройства (Patchliner ТМ, Nanion Technologies, Мюнхен). Эта система основывается на стеклянных чипах, с помощью которой возможно проводить цельноклеточное автоматическое измерение одновременно на 8 цельных клетках. Стеклянный чип имеет отверстие определенного размера, через которое клетка переносится в Gigaseal путем приложения пониженного давления и превращается в цельноклеточную конфигурацию. Буфер, суспензию клеток и исследуемые вещества добавляли в микроканалы чипа с помощью пипетки с тефлоновым покрытием.
Клетки зажимали до удерживающего потенциала -80 мВ. Для измерения стимулированного веществами ингибирования канала Kv11.1 применяли следующий протокол напряжения с 10-секундными интервалами: 51 мс / -80 мВ, 500 мс / +40 мВ, 500 мс / -40 мВ, 200 мс / -80 мВ. Поток утечки вычитается методом Р4. Клетки ресуспендировали в экстрацелюллярном буфере (ЕС) и наносили на чип. После того, как клетки были собраны, герметичность была улучшена путем добавления буфера-усилителя герметичности. Как только была достигнута цельноклеточная конфигурация, буфер усилителя уплотнения смывали и заменяли внеклеточным буфером. Измерение начинали в экстрацеллюлярном буфере в течение 1,5 мин. Затем применяли ДМСО (контроль носителя, 0,1% ДМСО), и контрольный поток регистрировали в течение 3 минут. Затем исследуемое вещество добавляли дважды в той же концентрации, и поток калия измеряли в течение 3,5 мин. в каждом случае.
Если результат измерения исследуемого вещества при начальной концентрации 10 мкМ был меньше, чем (-)50%-ный эффект (пороговое значение) (например, (-)60% -ный эффект), то для определения зависимости доза/эффект исследуемое вещество добавляли кумулятивно при увеличении концентрации, где каждая концентрация измерялась в течение 5 минут.
В качестве эталонного вещества использовали блокатор ионных каналов Kv11.1 (hERG) хинидин. Эффекты исследуемых веществ и хинидина были стандартизированы для соответствующего контроля носителя. Влияние на активность канала Kv11.1 (hERG) оценивали по потоку калия при -40 мВ. Для расчета поток был оценен для соответствующей окончательной трассы. Ингибирование канала Kv11.1 (hERG), индуцированное исследуемым веществом, стандартизировали для контроля носителя (0,1% ДМСО).
Во время измерения отбирали аликвоту исследуемого вещества для определения концентрации. Образец немедленно подвергали измерению с помощью ВЭЖХ, а конечную концентрацию определяли по калибровочной кривой.
Если результат измерения исследуемого вещества при начальной концентрации 10 мкМ был больше или равен (-)50%-ному эффекту (пороговое значение) (например, (-)30%-ному эффекту, т.е. 30%-ному ингибированию при 10 мкМ), рассчитывали Ki в соответствии со следующей формулой: Ki = 1,0Е-5 × (100 + % эффекта) / (-% эффекта), [М].
Результат измерения (-)30%-ного эффекта при концентрации исследуемого вещества 10 мкМ давал значение Ki 23 мкМ.
Ферменты цитохрома Р-450 (CYP)
В организме человека лекарственные вещества превращаются в водорастворимые соединения с помощью ферментных систем для облегчения их выведения. Эти ферментные системы включают микросомальные ферменты цитохрома Р-450, сокращенно CYP. Анализ предназначен для определения того, может ли соединение быть сильным ингибитором определенных изоформ CYP. Анализ включает использование рекомбинантных изоформ CYP и их редуктазы, полученной путем сверхэкспрессии в клетках насекомых, инфицированных бакуловирусом. Реакция CYP осуществляется посредством ингибирования исследуемой изоформы CYP вместе с люминометрическим субстратом CYP в условиях регенерации НАДФН. Люминометрические субстраты P450-GloTM являются производными люциферина жука ((4S)-4,5-дигидро-2-(6-гидроксибензотиазолил)-4-тиазолкарбоновая кислота или D-люциферин), субстратом люциферазы светлячков из жуков. Люминометрические субстраты P450-GloTM не взаимодействуют напрямую с люциферазой, но преобразуются соответствующей изоформой CYP в продукт люциферина, который люминисцирует при реакции с реагентом для обнаружения люциферина (LDR). Это позволяет быстро количественно оценить активность фермента по яркости. Степень ингибирования измеряют путем определения значения IC50 (Crespi и др., Methods Enzymol. 357: 276-284, 2002). Используются следующие коммерчески доступные скрининговые системы/анализы: скрининговая система P450-Glo™ CYP1A2 (Promega Corporation; V9770); анализ P450-Glo™ CYP2C8 (Promega Corporation; V8782); скрининговая система P450-Glo™ CYP2C9 (Promega Corporation; V9790); скрининговая система P450-Glo™ CYP2C19 (Promega Corporation; V9880); скрининговая система P450-Glo™ CYP2D6 (Promega Corporation; V9880); скрининговая система P450-Glo™ CYP3A4 (люциферин-РРХЕ) (Promega Corporation; V9910). Биодоступность
Прогнозируемая биодоступность для человека выводится из измеренных значений биодоступности у доклинических видов: мыши, крысы и собаки (бигль) и рассчитывается для прогнозируемой фармакологически эффективной дозы у человека (моделирование in silico GastroPlus).
Способ рентгеновской порошковой дифракции (РПД)
Порошковые рентгеновские дифрактограммы (РПД), такие как те, что представлены на Фигурах 7Б и 10-19 (и др.), были получены при использовании следующей методики: образцы готовили в комбинаторном 96-луночном планшете (содержащем рентгеноаморфную фольгу в качестве дна) или между рентгеноаморфными пленками. Измерения проводились в геометрии пропускания с излучением Cu-Kα1 на дифрактометре Stoe StadiP 611. Сканирование проводилось одновременно в диапазоне 0-36° 2θ (ширина шага 0,03° 2 θ 30 секунд на шаг), или охватывало 1-65° 2 θ (ширина шага 0,015° 2 θ, 15 секунд на шаг), соответственно.
Монокристаллическая дифракция рентгеновских лучей:
Данные рентгеновской структуры монокристалла были получены при использовании дифрактометра SuperNova от Agilent, оснащенного детектором CCD при использовании излучения Cu-Kα. Измерения проводились при 200 К (Форма Н2) или 298 К (Формы A1, А2, A3, H1), соответственно. Данные в отношении монокристалла были использованы для определения кристаллической системы и параметров элементарной ячейки.
Дифференциальная сканирующая калориметрия (ДСК):
Сканы ДСК получали на дифференциальном сканирующем калориметре теплового потока Mettler-Toledo с автосэмплером в атмосфере инертного газа азота (50 мл/мин.). Обзорное сканирование проводили в кюветах А1 40 мкл с открытыми крышками при температуре 25-300°С со скоростью 5°С/мин.
Термогравиметрический анализ (ТГА):
Сканы ТГА получали на термогравиметрическом анализаторе Mettler-Toledo с автосэмплером в атмосфере инертного газа азота (50 мл/мин.). Обзорное сканирование проводили в кюветах на 100 мкл без крышек при температуре 25-300°С со скоростью 5°С/мин. Эксперименты были скорректированы по базовой линии с помощью холостого опыта с пустой кюветой А1 100 мкл без крышки при использовании того же температурного профиля.
Динамическая сорбция паров (ДСП):
ДСП изотермы сорбции водяного пара получали на приборе ДСП с микровесами и инкубатором (DSV-Intrinsic, Surface Measurement Systems, SMS). Образцы порошка аккуратно отвешивали в одноразовые алюминиевые чаши и помещали на место пробы прибора ДСП. Для увлажнения использовали общую скорость пропускания азота 200 мл/мин. (объединенный сухой и влажный потоки). Изотермы сорбции водяного пара получали при 25°С при использовании начального сегмента десорбции 1 от 40% ОВ (относительной влажности) до 0% от ОВ (с шагом 10%), сегмента адсорбции от 0% ОВ до 98% ОВ (с шагом 10% ОВ и заключительным шагом 8% ОВ, соответственно), и сегмента заключительной десорбции 2 от 98% ОВ до 0% ОВ (с начальным шагом 8% ОВ и 10% ОВ, соответственно). Для всех стадий относительной влажности использовали состояние равновесия dm/dt ≤ 0,0005 вес. %/мин. с минимальным временем шага относительной влажности 10 минут и максимальным временем шага относительной влажности (тайм-аут) 360 минут.
Профили растворения без поглощения твердых форм:
Профили растворения без поглощения для твердых форм были получены при использовании метода встряхиваемой колбы с избыточным твердым материалом в среде FaSSIF (рН 6,5) при отборе образцов с временным разрешением для определения растворенных количеств API.
В стеклянные флаконы отвешивали приблизительно 10-20 мг твердого образца. Добавляли 7 мл соответствующей среды (предварительно нагретой до 37°С), и суспензию встряхивали при 450 об./мин. при 37°С. Через 5 мин., 10 мин., 15 мин., 30 мин., 60 мин., 120 мин., 24 ч. и 48 ч. отбирали 1 мл суспензии и фильтровали через шприцевой фильтр 0,2 мкМ. Прозрачный фильтрат подвергали анализу с помощью ВЭЖХ после приемлемого разведения для измерения количества растворенного соединения/формы (также может называться API).
FaSSIF: 3 мМ таурохолата натрия; 0,75 мМ лецитина; 105,9 мМ хлорида натрия; 28,4 мМ одноосновного фосфата натрия и 8,7 мМ гидроксида натрия, рН 6,5.
Метод ВЭЖХ для определения растворимости, зависящей от рН. и миниатюризированого растворения без поглощения:
Уровни растворенного соединения/формы анализировали с помощью ВЭЖХ при следующих условиях:
• Колонка: Хромолит RP-18e 100 - 3 мм
• Растворитель А: вода/муравьиная кислота (999:1; об./об.)
• Растворитель Б: ацетонитрил/муравьиная кислота (999:1; об./об.)
• Вводимый объем: 5 мкл
• Температура колонки: 37°С
• ВЭЖХ-градиент:
ПРИМЕРЫ
Приведенные ниже примеры иллюстрируют изобретение, описанное выше; однако они никоим образом не предназначены для ограничения объема изобретения. Примеры, в частности, следует интерпретировать таким образом, чтобы они не ограничивались конкретно проиллюстрированными признаками или комбинациями признаков, но вместо этого иллюстративные признаки можно было свободно изменять или комбинировать, пока достигается цель изобретения.
Благоприятные эффекты соединений, комбинаций и композиций в соответствии с настоящим изобретением также могут быть определены другими аналитическими методами и экспериментальными установками, известными как таковые специалистам в соответствующей области техники. Подобным образом, модификации способов получения, в частности, условий реакции, будут очевидны специалисту в данной области техники.
ПРИМЕР 1: Получение Соединений 1 и 2
Соединение Y получали в соответствии с процедурой, раскрытой в WO 2016/155844, после отделения Соединений 1 и 2 от Соединения Y:
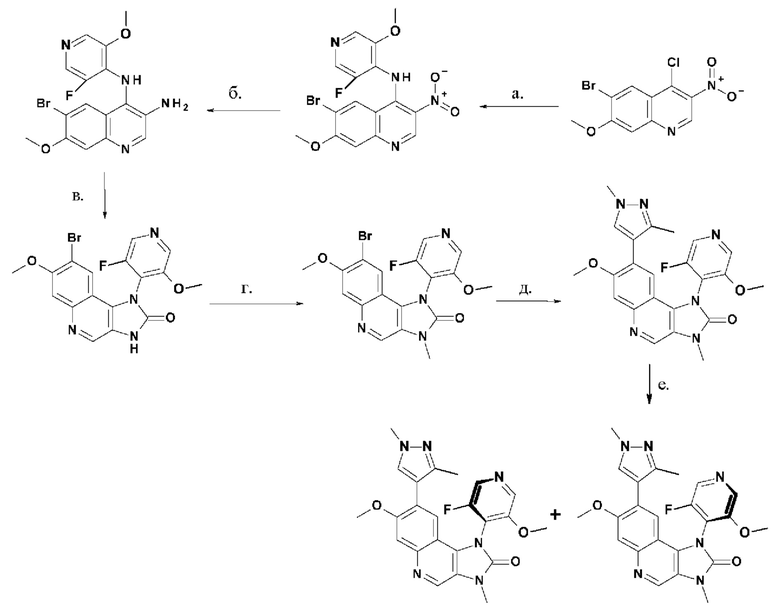
а) Синтез 6-бром-N-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-нитро-хинолин-4-амина
В атмосфере сухого азота получали раствор 3-фтор-5-метоксипиридин-4-амина (447 мг, 3,02 ммоля), растворенный в N,N-диметилформамиде (5 мл). Затем к раствору добавляли гидрид натрия (504 мг, 12,6 ммоля, 60%), и перемешивание продолжали в течение 5 минут при комнатной температуре.
Затем к реакционной смеси добавляли 6-бром-4-хлор-7-метокси-3-нитро-хинолин (800 мг, 2,52 ммоля) с последующим 15-минутным перемешиванием при комнатной температуре, затем реакцию гасили путем добавления ледяной воды (100 мл). Осадок отфильтровывали, промывали ледяной водой и высушивали с получением 1,00 г (94%) 6-бром-М-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-нитро-хинолин-4-амина в виде твердого вещества желтого цвета.
б) Синтез 6-бром-N4-(3-фтор-5-метокси-4-пиридил)-7-метокси-хинолин-3,4-диамина
6-Бром-М-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-нитро-хинолин-4-
амин (990 мг, 2,20 ммоля), растворенный в метаноле (100 мл), получали в защитной атмосфере азота. Затем к раствору добавляли никель Ренея (100 мг, 1,17 ммоля), и реакционную смесь перемешивали в течение 30 минут в атмосфере водорода при нормальном давлении. После введения азота суспензию фильтровали, и фильтрат высушивали в вакууме. Фильтрат упаривали досуха под вакуумом. Остаток кристаллизовали из смеси этилацетат/петролейный эфир, получая 0,86 г (99%) 6-бром-N4-(3-фтор-5-метокси-4-пиридил)-7-метокси-хинолин-3,4-диамина в виде желтого твердого вещества.
в) Синтез 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3Н-имидазо[4,5-с]хинолин-2-она
Раствор 6-бром-N4-(3-фтор-5-метокси-4-пиридил)-7-метокси-хинолин-3,4-диамина (0,85 г, 2,20 ммоля) растворяли в тетрагидрофуране (20 мл). Затем добавляли 1,1'-карбонилдиимидазол (1,84 г, 11,3 ммоля) и основание Хюнига (1,46 г, 11,3 ммоля). Реакционную смесь нагревали до 40°С и перемешивали в течение 16 часов. Затем реакцию гасили путем добавления ледяной воды (200 мл). Осадок отфильтровывали, промывали ледяной водой и высушивали с получением 0,87 г (94%) 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3Н-имидазо[4,5-с]хинолин-2-она в виде светло-желтого твердого вещества.
г) Синтез 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-имидазо[4,5-с]хинолин-2-она
В атмосфере сухого защитного азота 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3Н-имидазо[4,5-с]хинолин-2-он (0,86 г, 1,94 ммоля) растворяли в N,N-диметилформамиде (5 мл). Затем добавляли гидрид натрия (388 мг, 9,71 ммоля, 60%) и метилйодид (2,76 г, 19,4 ммоля). Реакционную смесь перемешивали 10 минут при комнатной температуре. Затем реакцию гасили путем добавления ледяной воды (100 мл). Образовавшийся осадок отфильтровали и высушивали под вакуумом с получением 0,70 г (80%) 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-имидазо[4,5-с]хинолин-2-она в виде светло-желтого твердого вещества.
д) Синтез 1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-8-(1,3-диметилпиразол-4-ил)имидазо[4,5-с]хинолин-2-она
В атмосфере инертного газа аргона в закрытом оборудовании растворяли 8-бром-1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-имидазо [4,5-с]хинолин-2-он (150 мг, 0,33 ммоля), 1-3-диметил-4-(тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (88,4 мг, 0,40 ммоля), Pd(PPh3)4 (76,6 мг, 0,07 ммоля) и карбонат калия (91,6 мг, 0,66 ммоля) в 1,4-диоксане (15 мл) и воде (5 мл). Реакционную смесь нагревали до 80°С при перемешивании в течение 2 часов. Затем осуществляли охлаждение до комнатной температуры и восстановление реакционной смеси до осушения в вакууме. Остаток очищали хроматографически, используя диоксид кремния (этилацетат/метанол = 97:3, об. части). Элюат упаривали досуха, и полученный сырьевой продукт очищали с помощью препаративной ВЭЖХ с обращенной фазой (вода/ацетонитрил). После восстановления фракций продукта 1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-8-(1,3-диметилпиразол-4-ил)имидазо[4,5-с]хинолин-2-он (70 мг, 47%) получали в виде бесцветного твердого вещества.
е) Разделение 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Ra)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она и 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она
1-(3-фтор-5-метокси-4-пиридил)-7-метокси-3-метил-8-(1,3-метилпиразол-4-ил)имидазо[4,5-с]хинолин-2-он (50,0 мг, 0,11 ммоля), который был получен выше, разделяли с помощью хиральной ВЭЖХ при использовании СФХ (сверхкритической флюидной хроматографии) с получением Соединений 1 и 2. Вещество наносили на хиральную колонку Lux Cellulose-2 и разделяли при скорости потока 5 мл/мин. при использовании СО2/2-пропанол+0,5% диэтиламина (75:25) в качестве растворителя и с применением определения при длине волны 240 нМ. Восстановление фракций продукта при пониженном давлении приводило к получению 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Ra)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она (25,0 мг, 50%) и 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(SWa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с] хинолин-2-она (22,1 мг, 44%), оба в виде бесцветных твердых веществ.
Исходные соединения легко получить, например, так, как показано ниже:
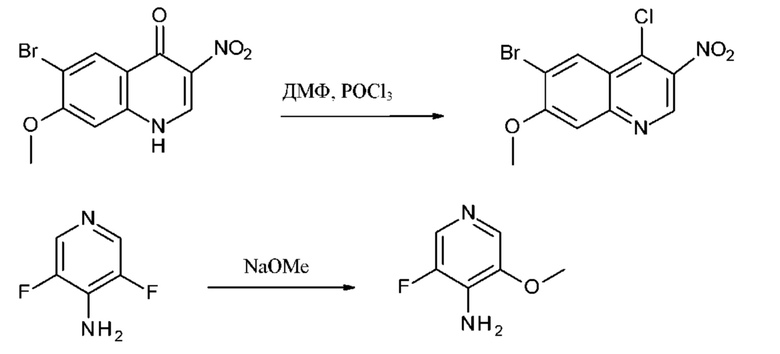
ПРИМЕР 2: Изолирование атропоизомеров и очистка Соединения 1
Соединения 1 и 2 могут быть отделены от Соединение Y, как показано на Схеме 1 и на Фигура 6, и как обсуждается подробно ниже. Специалист в данной области техники поймет, что описанный ниже процесс в равной степени применим для соединений 3-а и 3-b от 3, 4-а и 4-b от 4, а также 5-а и 5-b от 5.
Схема 1: Получение хиральных солей

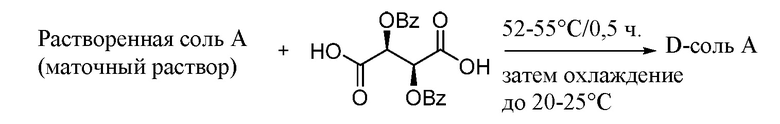
Этап 1:
1. В реактор на 200 л добавляли ацетон (108 л, 20 объемов), очищенную воду (8,13 л, 1,5 объема) и Соединение Y (5,42 кг, 1,0 экв.) при 20 ~ 25°С.
2. В реактор загружали дибензоил-L-винную кислоту (4,33 кг, 1,0 экв.).
3. Нагревали до 52-55°С до получения прозрачного раствора, перемешивали 0,5 ч. при 52-55°С.
4. Охлаждали до 20 ~ 25°С.
5. Фильтровали и промывали преципитат один раз ацетоном (5,4 л, 1 об.).
6. Собирали преципитат и высушивали с получением L-соли В (L-соль Соединения 2) (светло-желтое твердое вещество с 95,9% хиральной чистоты).
7. Концентрировали маточный раствор и получали L-соль А (L-соль Соединения 1).
8. В 100-литровый реактор добавляли L-соль А и дихлорметан (38 л, 7 объемов), рН доводили до значения 8-9 с помощью насыщенного раствора NaHCO3.
9. Собирали слой дихлорметана (ДХМ), экстрагировали насыщенный NaHCO3 с помощью ДХМ (16,3 л, 3 объема), объединяли слои ДХМ, промывали слой ДХМ Н2О (10,8 л, 2 объема).
10. Концентрировали слой ДХМ. Затем добавляли ацетон (5,4 л, 1 объем) при 20 ~ 25°С.
11. Перемешивали при 20 ~ 25°С в течение 1 часа.
12. Фильтровали, собирали остаток и высушивали с получением Соединения 1 (3,50 кг, белое твердое вещество с 73,2% хиральной чистоты, Y: 64,6%).
Этап 2:
1. В колбу на 50 л добавляли L-соль В и ДХМ (16,2 л, 3 объема), рН доводили до 8-9 с помощью насыщенного раствора NaHCO3.
2. Собирали слой ДХМ, экстрагировали водный слой ДХМ (5,4 л, 1 объем), объединяли слои ДХМ, промывали слой ДХМ Н20 (5,4 л, 1 объем).
3. Заменяли ДХМ на 2-этоксиэтанол (1,8 л, 0,3 об.) дважды в вакууме при температуре 40 ~ 50°С.
4. Доводили объем 2-этоксиэтанолом до 5,4 л (1 об.).
5. Нагревали до 128-130°С с получением суспензии, перемешивали в течение 44 часов при 128-130°С.
6. IPC (соотношение 49,5: 50,5).
7. Охлаждали до 15-20°С.
8. Фильтровали и промывали осадок метил-трет.-бутиловым эфиром (2,7 л, 0,5 объема).
9. Осадок собирали и высушивали с получением Соединения Y (1,60 кг, твердое вещество грязно-белого цвета, общий выход: 29,5%).
Этап 3:
1. В реактор на 100 л добавляли ацетон (70 л, 20 объемов) при 20 ~ 25°С, перемешивали и добавляли Соединение 1 (3,5 кг, 1,0 экв.), загружали воду (5,3 л, 1,5 об.).
2. Загружали дибензоил-D-винную кислоту (2,8 кг, 1,0 экв.) при 35 ~ 40°С, нагревали до 52 ~ 55°С с получением прозрачного раствора, перемешивали в течение 0,5 ч. при 52°С ~ 55°С.
3. Охлаждали до 20 ~ 25°С на масляной бане и перемешивали в течение 17 часов при 20 ~ 25°С.
4. Отфильтровали и промыли преципитат ацетоном (3,5 л, 1 об.).
5. Собирали преципитат, чтобы получить D-соль А (D-соль Соединения 1) (светло-желтое твердое вещество с хиральной чистотой 98,7%).
6. Концентрировали маточный раствор, затем добавляли насыщенный раствор NaHC03 (15,5 л, 3,5 объема) и Н20 (15,5 л, 3,5 объема).
7. Перемешивали 0,5 ч. при 20 ~ 25°С.
8. Отфильтровали и промывали преципитат H2O (3,5 л, 1 об.).
9. Собирали преципитат на фильтре и высушивали с получением Соединения Y (1,53 кг, белое твердое вещество при соотношении 50,3:49,7, выход: 43,7%).
Этап 4:
1. В 50-литровый реактор добавляли D-соль А (D-соль Соединения 1) и ацетон (17,5 л, 5 объемов), нагревали до 52-55°С с получением суспензионного раствора, перемешивали в течение 0,5 часа при 52 ~ 55°С.
2. Охлаждали до 20 ~ 25°С и перемешивали в течение 17 часов при 20 ~ 25°С.
3. Отфильтровали и промыли преципитат ацетоном (3,5 л, 1 об.).
4. Собирали осадок на фильтре и высушивали с получением D-соли А (3,23 кг, светло-желтое твердое вещество с хиральной чистотой 99,2%).
5. В реактор на 50 л добавляли D-соль А, затем добавляли насыщенный раствор NaHCO3 (16 л, 5 объемов) и H2O (16 л, 5 объемов).
6. Перемешивали 0,5 ч. при 20 ~ 25°С.
7. Отфильтровали и промыли преципитат H2O (16 л, 5 об.).
8. Собирали осадок и высушивали с получением Соединения 1 (1638 г, белое твердое вещество с хиральной чистотой 99,1% и 99,9% по данным ВЭЖХ, общий выход: 30,2%).
ПРИМЕР 3: Хроматографическое разделение, очистка и анализ
3.1 Соединения 1 и 2 можно отделить от Соединения Y с помощью хроматографии на хиральной неподвижной фазе (см., например, Chiral Liquid Chromatography; WJ Lough, Ed. Chapman и Hall, New York, (1989); Okamoto, "Optical resolution of dihydropyridine enantiomers by high-performance liquid chromatography using phenylcarbamates of polysaccharides as a chiral stationary phase", J. of Chromatogr. 513: 375-378, (1990)). Соединения 1 и 2 можно выделить с помощью хроматографии на хиральной неподвижной фазе, например, на колонке Chiralpak 1С (5 мм, 150 × 4,6 мм внутренний диаметр), например, при использовании изократического элюирования с подвижной фазой, содержащей: Н2О/АЦН 50/50 об./об. (АЦН: ацетонитрил). Специалист в данной области техники поймет, что такая же процедура может применяться для соединений 3-а и 3-b, 4-а и 4-b, а также 5-а и 5-b.
Полученная таким образом хроматограмма проиллюстрирована на Фигуре 5 (колонка и элюирование, как указано выше, скорость потока 1,00 мл/мин.; УФ 260 нм; TC и TS: 25 ± 5°С, Sconc 0,20 мг/мл; введенный объем 10 мл)
3.2 В качестве альтернативы условиям сверхкритической флюидной хроматографии, упомянутым выше, может использоваться препаративная сверхкритическая жидкостная хроматография, включая, например: колонку Chiralpak AS-H (20 мм × 250 мм, 5 мкМ); изократическое элюирование (20:80 этанол : СО2 с 0,1% об./об. NH3), BPR (регулированное давление всасывания): примерно на 100 бар выше атмосферного давления; температура колонки 40°С, скорость потока 50 мл/мин., вводимый объем 2500 мкл (125 мг) и длина волны детектора 265 нМ.
3.3 Для анализа чистоты соответствующих атропоизомеров, опять же, можно использовать сверхкритическую флюидную хроматографию, например, с применением следующих установок : колонка Chiralpak AS-H (4,6 мм × 250 мм, 5 мкМ); изократическое элюирование (20:80 этанол : СО2 с 0,1% об./об. NH3), BPR (регулированное давление всасывания): примерно на 125 бар выше атмосферного давления; температура колонки 40°С, скорость потока 4 мл/мин., вводимый объем 1 мкл и длина волны детектора 260 нМ.
ПРИМЕР4: Стабильность Соединений 1 и 2
Квантовая механика расчетов вращательных барьеров
LaPlante и др. (ChemMedChem, 2011, 6 (3), 505-513) описывают квантово-механический рабочий процесс для оценки энергетических барьеров для осевого вращения молекул, подобных лекарственному средству. Был применен аналогичный подход: трехмерные структуры всех входных молекул были созданы при использовании CORINA (Corina версия 3,6, Molecular Networks, Германия) и впоследствии минимизированы с помощью Macromodel (версия 11.1, Schrodinger, LLC, New York, NY). На основе этих входных структур барьеры вращательной энергии были рассчитаны из снимков с нежестким углом между двумя плоскостями при использовании программы Jaguar (версия 9.1 выпуск 14, Schrodinger, LLC, New York, NY) с применением метода B3LYP/6-31G** с торсионным углом приращения 15°. Структуры были оптимизированы перед сканированием на кручение при использовании того же уровня теории. Были использованы параметры по умолчанию, за исключением того, что максимальное количество шагов минимизации было установлено на 500 и была введена метка stop_rxn, чтобы избежать искусственного разрыва связей. Для всех расчетов использовались репрезентативные молекулярные фрагменты. Двугранный вид, полученный из минимизированной структуры молекулярной механики, использовался для определения начального значения для двугранного сканирования, например, для Соединения 1 в соответствии с настоящим изобретением или для «LaPlante эталонного Соединения 1» значения были установлены равными 47,32° и 35,8°, соответственно. Для каждого кручения относительно осевой связи QM значения энергии были рассчитаны за 24 шага. Профиль кручения был получен путем сопоставления значений торсионного угла с рассчитанными значениями энергии. Был пределен самый низкий энергетический барьер для каждого соединения, который позволяет взаимное превращение между обоими изомерами. Для эталонных соединений 1-6 экспериментально определенные взаимопревращения и расчетные энергетические барьеры являются известными. Вычислительные энергетические барьеры для эталонных соединений 1-6 составляют от 9865 до 31316 ккал/моль. Эти значения согласованы с экспериментально определенными коэффициентами взаимопревращения.
Расчеты для Соединения 1 и Соединения 2 данного изобретения продемонстрировали высокий прогнозируемый вращательный барьер 29205 ккал/моль, который переводится в высокую стабильность атропоизомера с прогнозируемым периодом полураспада при вращении в диапазоне лет ( > 10 лет). Необходимая температура для начала рацемизации любого энантиомерного атропоизомера составляет > 100°С.
Для специалиста в данной области техники будет понятным, что эти значения также являются иллюстративными для соединений 3-а и 3-b, 4-а и 4-b, а также 5-а и 5-b.
ПРИМЕР 5: Фармацевтически приемлемые соли Соединения 1
Соли Соединения 1 (Соединения 1-а), которые являются фармацевтически приемлемыми, были приготовлены так, как представлено и подробно обсуждается ниже.
Получение соли фумарата (Форма NF6):
Приблизительно 20 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил- 1,3-дигидро-имидазо[4,5-с]хинолин-2-она растворяли в 1 мл ТГФ при 50°С (степень чистоты «чистый для анализа») в 4-миллилитровом стеклянном флаконе с закрытой крышкой, снабженном магнитной мешалкой, и перемешивали при использовании магнитной мешалки. При приблизительно 50°С в горячий раствор добавляли 5,7 мг фумаровой кислоты (-1,1 экв.) и охлаждали до 5°С со скоростью охлаждения 0,1 К/мин. Смесь неоднократно нагревали до 50°С (в течение примерно 30 мин.) и охлаждали до 5°С (со скоростью 0,1 К/мин.) при перемешивании перед заключительной стадией уравновешивания при 5°С в течение нескольких часов. Для увеличения выхода солеобразования в охлажденном растворе смесь дополнительно подвергали медленной диффузии паров антирастворителя н-пентана в конфигурации закрытого флакона. В завершение, полученный твердый материал отделяли центрифугированием и осторожно высушивали под потоком азота.
Получение соли напсилата (Форма NF7) - Вариант 1:
Приблизительно 10 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил- 1,3-дигидро-имидазо[4,5-с]хинолин-2-она растворяли в ~ 250 мкл ацетона при 50°С (степень чистоты «чистый для анализа») в 4-миллилитровом стеклянном флаконе с закрытой крышкой, снабженном магнитной мешалкой, и перемешивали при использовании магнитной мешалки. При 50°С приблизительно 5,6 мг нафталин-2-сульфоновой кислоты (~ 1,2 экв.) добавляли в горячий раствор и охлаждали до 5°С со скоростью охлаждения 0,1 К/мин. Смесь неоднократно нагревали до 50°С (в течение примерно 30 мин.) и охлаждали до 5°С (со скоростью 0,1 К/мин.) при перемешивании перед заключительной стадией уравновешивания при 5°С в течение нескольких часов. В завершение, полученный твердый материал отделяли центрифугированием и осторожно высушивали при продувке азотом. Получение соли напсилата (Форма NF7) - Вариант 2:
Приблизительно 11,5 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она растворяли в ~ 250 мкл ТГФ (тетрагидрофурана) при 50°С (степень чистоты «чистый для анализа») в стеклянном флаконе объемом 4 мл с закрытой крышкой, снабженном магнитной мешалкой, и перемешивали при использовании магнитной мешалки. При 50°С приблизительно 6,6 мг нафталин-2-сульфоновой кислоты (~ 1,2 экв.) добавляли в горячий раствор и охлаждали до 5°С со скоростью охлаждения 0,1 К/мин. Смесь неоднократно нагревали до 50°С (в течение примерно 30 мин.) и охлаждали до 5°С (со скоростью 0,1 К/мин.) при перемешивании перед заключительной стадией уравновешивания при 5°С в течение нескольких часов. В завершение, полученный твердый материал отделяли центрифугированием и осторожно высушивали при продувке азотом.
Получение соли эдизилата (Форма NF8):
Приблизительно 12,6 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она растворяли в ~ 250 мкл ацетона при 50°С (степень чистоты «чистый для анализа») в 4-миллилитровом стеклянном флаконе с закрытой крышкой, оснащенном магнитной мешалкой, и перемешивали с помощью магнитной мешалки. При 50°С приблизительно 6,1 мг этандисульфоновой кислоты (~ 1,2 экв.) добавляли в горячий раствор и охлаждали до 5°С со скоростью охлаждения 0,1 К/мин. Смесь неоднократно нагревали до 50°С (в течение примерно 30 мин.) и охлаждали до 5°С (со скоростью 0,1 К/мин.) при перемешивании перед заключительной стадией уравновешивания при 5°С в течение нескольких часов. В завершение, полученный твердый материал отделяли центрифугированием и осторожно высушивали при продувке азотом.
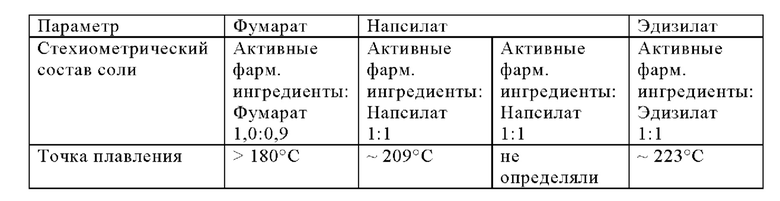
ПРИМЕР 6: Получение Соединений 3, 4 и 5
Синтез 8-(1,3-диметилпиразол-4-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-имидазо[4,5-с]хинолин-2-она
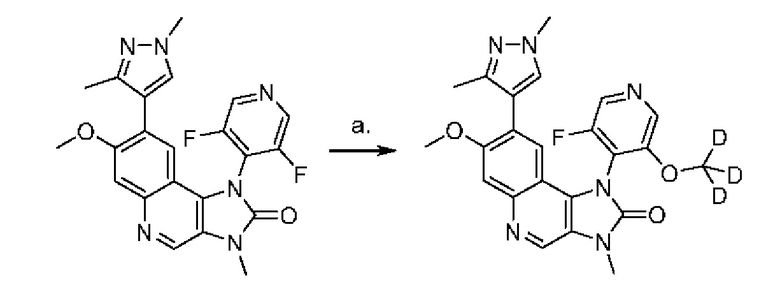
Этап а:
В герметичную пробирку помещали: 1-(3,5-дифторпиридин-4-ил)-8-(1,3-диметил-1Н-пиразол-4-ил)-7-метокси-3-метил-1Н,2Н,3Н-имидазо[4,5-с]хинолин-2-она (90,0 мг, 0,20 ммоля, 95%), карбонат калия (85,3 мг, 0,62 ммоля), CD3OD (0,30 мл, 6,74 ммоля), N,N-диметилформамид (3 мл). Смесь перемешивали в течение 1 часа при 100°С. Добавляли 10 мл воды, и полученный раствор трижды экстрагировали этилацетатом (10 мл), объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали досуха под вакуумом. Неочищенный остаток очищали с помощью препаративной ВЭЖХ (SHIMADZU (ВЭЖХ-10): Колонка: колонка Atlantis Prep Т3 OBD, 19 * 250 мм, 10 мкМ; подвижная фаза: вода (10 ммоль/л NH4HCO3) и ацетонитрил (содержание ацетонитрила 34%) в течение 10 мин.); определение: УФ 254 нм) с выходом 35 мг (38%) 8-(1,3-диметилпиразол-4-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-имидазо[4,5-с]хинолин-2-она в виде белого твердого вещества. Точка плавления 260-262°С. ВЭЖХ/МС (чистота) 97%. Время удержания 1,98 мин. (способ А). [М+Н]+ 452. 1H ЯМР (400 МГц, ДМСО-d6) ч./млн = 8,92 (с, 1H), 8,70 (д, J=9,5 Гц, 2Н), 7,83 (с, 1H), 7,54 (с, 1H), 7,00 (с, 1H), 3,94 (с, 3Н), 3,78 (с, 3Н), 3,60 (с, ЗН), 1,75 (с, 3Н).
Синтез 8-(1,3-диметилпиразол-4-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]- 7-метокси-3-(тридейтериометил)имидазо[4,5-с]хинолин-2-она
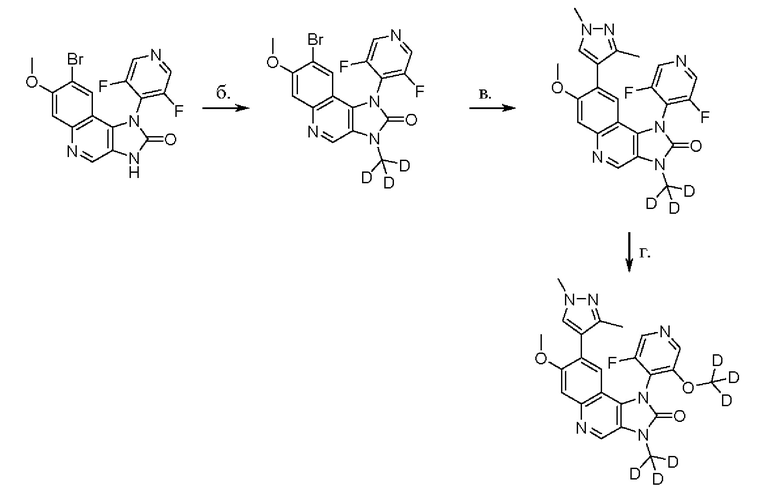
Этап б:
В круглодонную колбу помещали 8-бром-1-(3,5-дифторпиридин-4-ил)-7-метокси-1Н,2Н,3Н-имидазо[4,5-с]хинолин-2-он (3,00 г, 6,96 ммоля, 94%) в N,N-диметилформамиде (50 мл). Гидрид натрия (1,39 г, 34,8 ммоля, 60%) добавляли при 0°С в течение 5 минут с последующим добавлением CD3I (3,18 г, 20,9 ммоля, 95%). Полученный раствор перемешивали в течение 15 минут при комнатной температуре. К смеси добавляли 500 мл ледяной воды, и твердые вещества собирали фильтрацией. В результате было получено 2,95 г (99%) 8-бром- 1-(3,5-дифтор-4-пиридил)-7-метокси-3-тридейтериометил)имидазо[4,5-с]хинолин-2-она в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 99%. Время удержания 0,93 мин. (метод В) [М+Н]+ 424, 426.
Этап в:
В герметичную пробирку, очищенную и содержащую инертную атмосферу аргона, помещали 8-бром-1-(3,5-дифтор-4-пиридил)-7-метокси-3-тридейтериометил)имидазо-[4,5-с]хинолин-2-он (1,80 г, 4,20 ммоля, 99%), 1,3-диметил-4-(тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (1,97 г, 8,43 ммоля, 95%), Pd(PPh3)4 (540 мг, 0,42 ммоля, 90%), карбонат калия (1,22 г, 8,39 ммоля, 95%), диоксан (50 мл) и воду (10 мл). Смесь перемешивали 2 часа при 80°С и концентрировали под вакуумом досуха. Остаток очищали с помощью колоночной хроматографии (метанол/этилацетат, 13:87), в результате чего получали 1,35 г (71%) 1-(3,5-дифтор-4-пиридил)-8-(1,3-диметилпиразол-4-ил)-7-метокси-3-(тридейтериометил)имидазо[4,5-с]хинолин-2-она в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 97%. Время удержания 0,91 мин. (способ Б). [М+Н]+ 440.
Этап г:
В герметичную пробирку, очищенную и содержащую инертную атмосферу аргона, помещали 1-(3,5-дифтор-4-пиридил)-8-(1,3-диметилпиразол-4-ил)-7-метокси-3-(тридейтериометил)имидазо[4,5-с]хинолин-2-он (1,35 г, 2,96 ммоля), N,N-диметилформамид (30 мл), карбонат калия (818 мг, 5,92 ммоля) и CD3OD (2,76 мл, 2,24 г, 62,2 ммоля). Смесь перемешивали 2 часа при 100°С.Добавляли 100 мл воды, и полученную смесь трижды экстрагировали 200 мл этилацетата. Объединенные органические слои дважды промывали 100 мл солевого раствора. Органический слой высушивали над безводным сульфатом натрия, концентрировали досуха и сырой продукт выкристаллизовывали из метанола/ацетонитрила (1:25), получая 1,50 г (66%) 8-(1,3-диметилпиразол-4)-ил)-1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-(тридейтериометил)имидазо[4,5-с]хинолин-2-она в виде не совсем белого твердого вещества. Температура плавления составляла 266-271°С. ВЭЖХ/МС (чистота) 97%. Время удержания 2,00 мин. (метод В). [M+H]+ 455. 1H ЯМР (300 МГц, ДМСО-d6) ч./млн. = 8,87 (с, 1Н), 8,65 (д, J=7,5 Hz, 2Н), 7,78 (с, 1H), 7,49 (с, 1Н), 6,95 (с, 1H), 3,89 (с, 3Н), 3,28 (с, 3Н), 1,71 (с, 3Н).
Синтез 1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-8-[3-метил-1-(тридейтериометил) пиразол-4-ил]имидазо[4,5-с]хинолин-2-она
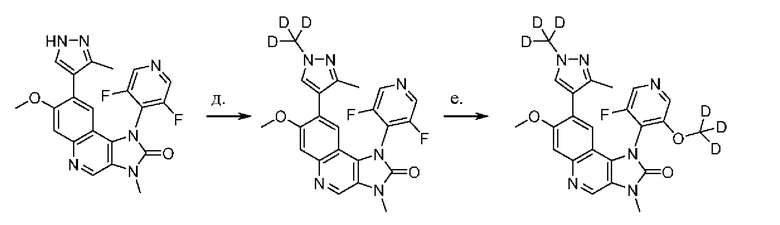
Этап д:
В круглодонную колбу помещали 1-(3,5-дифторпиридин-4-ил)-7-метокси-3-метил-8-(3-метил-1Н-пиразол-4-ил)-1Н,2Н,3Н-имидазо[4,5-с]хинолин-2-он (890 мг, 1,89 ммоля, 90%) в N,N-диметилформамиде (30 мл). Прибавляли гидрид натрия (377 мг, 9,43 ммоля, 60%) при 0°С в 5 приемов, затем CD3I (1,44 г, 9,44 ммоля, 95%). Полученную смесь перемешивали 8 часов при комнатной температуре. Добавляли 200 мл ледяной воды и раствор четыре раза экстрагировали 200 мл этилацетата. Объединенные органические слои дважды промывали 100 мл солевого раствора. Органическую фазу высушивали над безводным сульфатом натрия, концентрировали досуха и очищали при использовании колоночной хроматографии с дихлорметаном/метанолом (19:1), получая 400 мг (47%) 1-(3,5-дифтор-4-пиридил)-7-метокси-3-метил-8-[3-метил-1-(тридейтериометил)пиразол-4-ил]имидазо[4,5-с]хинолин-2-она в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 98%. Время удержания 0,68 мин. (метод Г). [М+Н]+ 440.
Этап е:
В герметичную пробирку, очищенную и содержащую инертную атмосферу аргона, помещали 1-(3,5-дифтор-4-пиридил)-7-метокси-3-метил-8-[3-метил-1-(тридейтериометил)пиразол-4-ил]имидазо[4,5-с]хинолин-2-он (380 мг, 0,84 ммоля, 98%), N-метил-2-пирролидон (20 мл), карбонат калия (368 мг, 2,53 ммоля, 95%) и CD3OD (2,45 мл, 53,9 ммоля, 98%). Смесь перемешивали 4 часа при 100°С. Добавляли 100 мл воды, и полученный раствор 5 раз экстрагировали 100 мл этилацетата. Объединенные органические слои дважды промывали 100 мл солевого раствора, высушивали над безводным сульфатом натрия, концентрировали досуха и очищали при использовании колоночной хроматографии с дихлорметаном/метанолом (10:1) с получением 300 мг (74%) 1-[3-фтор-5-(тридейтериометокси)-4-пиридил]-7-метокси-3-метил-8-[3-метил-1-(тридейтериометил)пиразол-4-ил]имидазо[4,5-с]хинолин-2-она в виде оранжевого твердого вещества. ВЭЖХ/МС (чистота) 95%. Время удержания 0,65 мин. (метод Г). [М+Н]+ 455. 1Н ЯМР (300 МГц, ДМСО-d6) ч./млн. = 8,87 (с, 1Н), 8,65 (д, J=7,5 Гц, 2Н), 7,78 (с, 1Н), 7,49 (с, 1Н), 6,95 (с, 1H), 3,89 (с, 3Н), 3,55(с, 3Н) 1,71 с, 3Н).
ВЭЖХ метод А:
Колонка: Shim-pack XR-ODS, 3,0*50 мм, 2,2 мкм; подвижная фаза А: вода/0,05% ТФУ, подвижная фаза Б: ацетонитрил/0,05% ТФУ; скорость потока: 1,0 мл/мин.; градиент: от 5% Б до 100% Б за 2,2 мин., удержание 1,0 мин.; 254 нМ.
ВЭЖХ метод Б:
Колонка: Shim-pack XR-ODS, 3,0*50 мм, 2,2 мкмМ; подвижная фаза А: вода/0,05% ТФУ, подвижная фаза Б: ацетонитрил/0,05% ТФУ; скорость потока: 1,2 мл/мин.; градиент: от 5% Б до 100% Б за 2,0 мин., удержание 0,7 мин.; 254 нм.
ВЭЖХ метод В:
Колонка: Poroshell НРН-С18, 3,0*50 мм, 2,7 мкм; подвижная фаза А: вода/5 мМ NH4HCO3, подвижная фаза Б: ацетонитрил; скорость потока: 1,3 мл/мин.; градиент: от 10% Б до 95% Б за 2,1 мин., удержание 0,6 мин.; 254 нм.
ВЭЖХ метод Г:
Колонка: Ascentis Express CI8, 3,0*50 мм, 2,7 мкм; подвижная фаза А: вода/0,05% ТФУ, подвижная фаза Б: ацетонитрил/0,05% ТФУ; скорость потока: 1,5 мл/мин.; градиент: от 5% Б до 100% Б за 1,2 мин., удержание 0,5 мин.; 254 нм.
ПРИМЕР 7: Твердые формы и сольваты Соединения 1
А. Получение твердой формы А2
Твердую форму А2 Соединения 1 получали с помощью различных процессов кристаллизации при охлаждении из спиртов.
7.1 Соединение 1 растворяли в 1-пропаноле при концентрации прибл. 50 мг/мл при 50°С и перемешивании. Полученный прозрачный раствор охлаждали до -20°С со скоростью охлаждения 0,1°С/мин., с окончательным периодом выдержки не менее 1 часа при -20°С. Разделение твердого вещества и жидкости выполняли фильтрованием с помощью вакуумного всасывания, и отфильтрованный твердый образец высушивали при динамической продувке азотом в течение ночи.
7.2 Соединение 1 растворяли в изобутиловом спирте при концентрации приблизительно 40 мг/мл при 50°С и перемешивании. Полученный прозрачный раствор охлаждали до -20°С со скоростью охлаждения 0,1°С/мин., с окончательным периодом выдержки, по крайней мере, 1 час при -20°С. Разделение твердого вещества и жидкости выполняли путем фильтрования с помощью вакуумного всасывания, и отфильтрованный твердый образец высушивали при динамической продувке азотом в течение ночи.
7.3 Гидрат Соединения 1 формы Н2 (получение которого описано ниже) диспергировали в 2-PrOH при уровне концентрации 12% (мае./мае; по отношению к сухой массе Соединения 1). Полученную дисперсию нагревали до 80°С при перемешивании до получения прозрачного раствора. Начальный диапазон температур охлаждения от 80 до 70°С был запущен со скоростью 0,5°С/мин. с последующим временем выдержки при 70°С. При температуре 70°С добавляли затравочные кристаллы безводной формы А2 (измельченные частицы размером < 50 мкм; примерно 4,5% относительно количества в реакторе). Затравочные кристаллы предварительно диспергировали приблизительно в 1 мл 2-PrOH на 100 мг количества затравки. После внесения затравочных кристаллов при 70°С применяли время выдержки 10 мин. Диапазон температур охлаждения от 70°С до 5°С был запущен со скоростью 0,1°С/мин., после чего следовало время выдержки в течение 3 часов при заключительной температуре (5°С). Разделение твердого вещества и жидкости проводили путем фильтрования с помощью вакуумного всасывания, и отфильтрованный твердый образец высушивали при динамической продувке азотом при температуре 70°С в течение ночи.
В альтернативном методе форму А2 получали путем преобразования суспензии из другой полиморфной формы следующим образом:
7.4 Твердый материал, в частности, другую полиморфную форму Соединения 1, лучше всего гидрат Формы Н2 (получение которого описано ниже), диспергировали приблизительно в 5,2 об.-экв. этилацетата и перемешивали при комнатной температуре в течение 21 часа. Осадок отфильтровывали и высушивали не менее 72 ч. при 60°С под вакуумом.
Б. Получение твердой Формы А1
Твердую Форму А1 Соединения 1 получали с помощью следующих двух способов:
7.5 Приблизительно 50 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил- 1,3-дигидро-имидазо [4,5-с] хинолин-2-она (рацемическая смесь) подвергали хиральному разделению с помощью сверхкритической флюидной хроматографии при использовании колонки Lux Cellulose-2 и смеси растворителей CO2/2-пропанол + 0,5% диэтаноламина (75:25) при скорости потока 5 мл/мин. Полученную фракцию промывали дихлорметаном и концентрировали при температуре бани 30°С до получения твердого вещества.
7.6 Приблизительно 10 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 100 мкл дихлорметана (ДХМ), упаривали при КТ (КТ: комнатная температура, примерно 20-25°С), чтобы получить порошок.
В. Получение твердой Формы A3
1.1 Приблизительно 12 мг твердого материала 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она, представляющего альтернативную морфическую форму, отличную от A3, наиболее предпочтительно представляющую собой форму А2, диспергировали в приблизительно 40 мкл ТГФ и перемешивали при комнатной температуре (20-25°С) приблизительно 4 недели. Полученное твердое вещество отделяли центрифугированием и осторожно высушивали в условиях окружающей среды с получением порошка.
Г. Получение твердой Формы NF9:
Твердую Форму NF9 Соединения 1 получали в соответствии с двумя следующими способами:
7.8 Приблизительно 100 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 2000 мкл дихлорметана, быстро выпаривали в вакууме при комнатной температуре (приблизительно 20-25°С) с получением порошка.
7.9 Приблизительно 20 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 1000 мкл ацетона при 50°С, добавляли к 1 экв. бензойной кислоты. Раствор охлаждали до комнатной температуры (примерно 20-25°С). Затем раствор подвергали диффузионной кристаллизации из пара при комнатной температуре (приблизительно 20-25°С) при использовании резервуара с н-пентаном, медленной диффузии в раствор посредством диффузии из газовой фазы. Через несколько дней выкристаллизовывалось твердое вещество, которое отделяли центрифугированием и осторожно высушивали под азотом с получением порошка.
Д. Получение твердого гидрата Формы H1
Твердую Форму Н гидрата Соединения 1 получали в соответствии с двумя следующими способами:
7.10 Приблизительно 12-13 мг твердого материала 8-(1,3-диметил-1Н-пиразол-4-ил)- 1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она, представляющего собой альтернативную морфическую форму, отличную от H1, наиболее предпочтительно форму А2, диспергировали в приблизительно 200 мкл метанола и перемешивали при комнатной температуре (20-25°С) в течение 5 дней. Полученное твердое вещество отделяли центрифугированием и осторожно высушивали в условиях окружающей среды с получением порошка.
7.11 Приблизительно 12 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 750 мкл 1-пропанола, выпаривали при КТ (приблизительно 20-25°С) с получением порошка.
Е. Получение твердого гидрата Формы Н2
7.12 Приблизительно 19 г твердого материала 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пир ид ин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо[4,5-с]хинолин-2-она, представляющего собой альтернативную морфическую форму, отличную от Н2, наиболее предпочтительно являющуюся формой А2, диспергировали приблизительно в 80 мл деионизированной воды и перемешивали при КТ (20-25°С) приблизительно 4 дня. Полученное твердое вещество отделяли фильтрованием под вакуумом и высушивали при 50°С под потоком азота с получением порошка.
7.13 Приблизительно 13-14 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 250 мкл ТГФ, быстро выливали в резервуар с 1500 мкл деионизированной воды при комнатной температуре (примерно 20-25°С) при турбулентном перемешивании. Полученный осадок отделяли центрифугированием и осторожно высушивали в условиях окружающей среды с получением порошка.
Ж. Получение твердой Формы NF19
7.14 Приблизительно 48 мг 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(Sa)-(3-фтор-5-метокси-пиридин-4-ил)-7-метокси-3-метил-1,3-дигидро-имидазо [4,5-с]хинолин-2-она, растворенного в 1500 мкл метанола, выпаривали при 50°С с получением порошка.
ПРИМЕР 8: Сочетание с радиационной терапией
В фармакологическом исследовании in vivo (модель опухоли FaDu SSCHN) комбинированное лечение Соединением 1 и фракционированным облучением в течение 6 недель (6×5 дней, 2 Гр на фракцию) значительно повышало эффективность ионизирующего облучение (ИО) зависимым от дозы образом в соответствии с уровнем вовлечения цели, как показано на Фигуре 20. Более подробно:
Противоопухолевую эффективность ингибитора ATM Соединения 1 оценивали в сочетании с ионизирующим облучением на мышах линии NMRI nu/nu, несущих ксенотрансплантаты сквамозных клеток головы и шеи человека модели FaDu.
Одна из контрольных групп получала только носитель. Другая контрольная группа получала ионизирующее облучение (ИО) только при 30 фракциях ИО в соответствии с графиком по 2 Гр в течение 5 дней / 2 дня без облучения в течение периода времени 6 недель. Две группы обрабатывали комбинацией ИО (как и в контроле, только ИО) и Соединением 1 с пероральной дозой 10 мг/кг или 25 мг/кг за 30 минут перед каждой фракцией ИО.
ПРИМЕР 9: Комбинация с PARP ингибированием
Эффективность Соединения 1 в комбинации с олапарибом была продемонстрирована в НВСх-10 модели ксенотрансплантата, полученного от пациента с тройным отрицательным раком молочной железы, разработанной на самках мышей с иммунодефицитом. Результаты являются представленными на Фигуре 21.
Семьдесят (70) мышей с подкожно выращенной НВСх-10 опухолью (Р20.1.4/0) от 62,5 до 196,0 мм3 подвергались лечение, когда их средний и медианный объем опухоли достигал 131,16 и 126,00 мм3, соответственно.
Исследование включало различные группы по 10 мышей в каждой:
- В группе 1 носитель олапариб вводили в дозе 10 мл/кг п. о. 1 р/сут. х 28 в сочетании с носителем метоцелом, вводимым в дозе 10 мл/кг п.о. (3 д. + / 4 д. -) х4;
- В группе 2 олапариб вводили в дозе 50 мг/кг п.о. 1 р/сут. х 49;
- В группе 3 сравниваемый ATM ингибитор ATMix вводили п.о. (3 д. + / 4 д. -) х 5;
- В группе 4 олапариб вводили в дозе 50 мг/кг п.о. 1 р/сут. х 49 в сочетании с ATMix п.о. (3 д. + / 4 д. -) х 7;
- В группе 5 олапариб вводили в дозе 50 мг/кг и.о. 1 р/сут. х 49 в сочетании с Соединением 1 в дозе 100 мг/кг п.о. (3 д. + / 4 д. -) х 7.
Опухоли измеряли каждые две недели в течение периода лечения и один раз в неделю в течение периода последующего наблюдения.
п.о. - перорально д. - дни 1 р/сут. - один раз в сутки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОЛОНИЛХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ АТМ | 2016 |
|
RU2743343C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ИНГИБИТОРОВ MNK | 2019 |
|
RU2821258C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2014 |
|
RU2687276C2 |
| Производные 2H-[1,4]оксазино- и 2H-[1,4]тиазино[3,2-c]хинолин-3(4H)-онов в качестве ингибиторов ATM и DNA-PK киназ для лечения и/или профилактики онкологических заболеваний | 2024 |
|
RU2835676C1 |
| ИНГИБИРОВАНИЕ ЦИКЛИЧЕСКОГО AMP-ЧУВСТВИТЕЛЬНОГО ЭЛЕМЕНТ-СВЯЗЫВАЮЩЕГО БЕЛКА (CREB) | 2020 |
|
RU2831142C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ATR КИНАЗЫ (ВАРИАНТЫ) | 2014 |
|
RU2720408C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2020 |
|
RU2750148C1 |
| КОМБИНАЦИИ | 2015 |
|
RU2715236C2 |
| ИДЕНТИФИКАЦИЯ МУТАЦИИ LKB1 В КАЧЕСТВЕ ПРОГНОСТИЧЕСКОГО БИОМАРКЕРА ЧУВСТВИТЕЛЬНОСТИ К ИНГИБИТОРАМ TOR-КИНАЗЫ | 2011 |
|
RU2565034C2 |
| ДВОЙНЫЕ ИНГИБИТОРЫ ATM И DNA-PK ДЛЯ ПРИМЕНЕНИЯ В ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2020 |
|
RU2800756C1 |
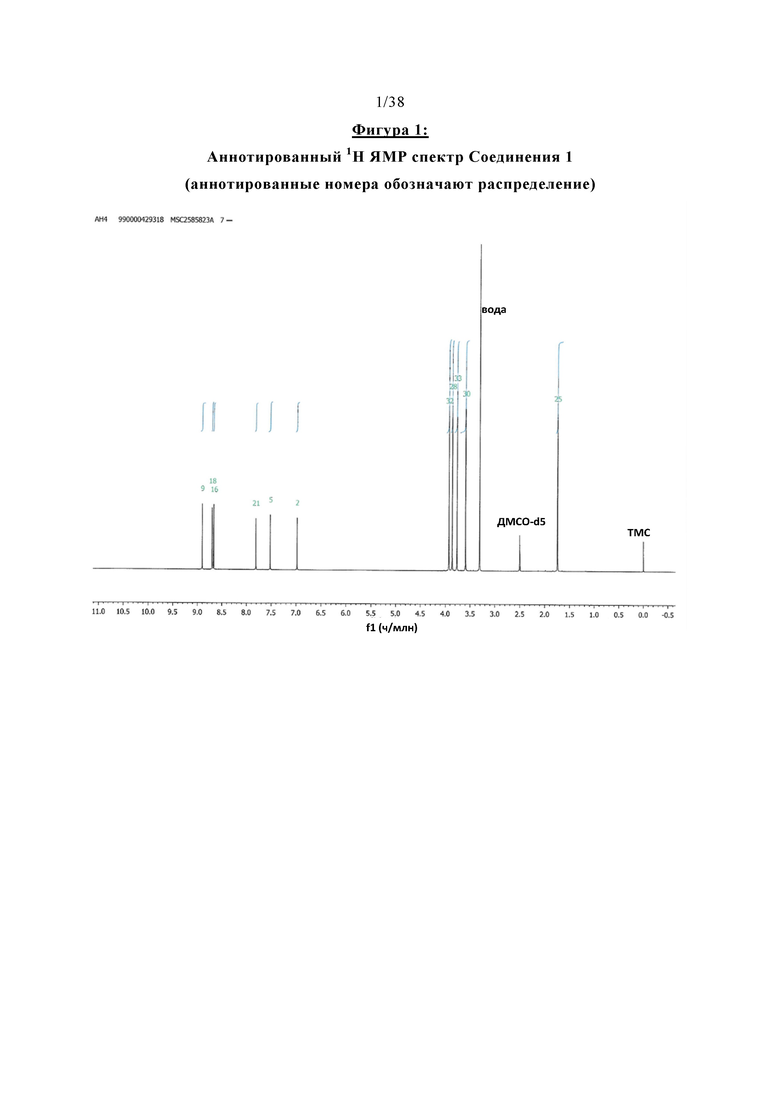
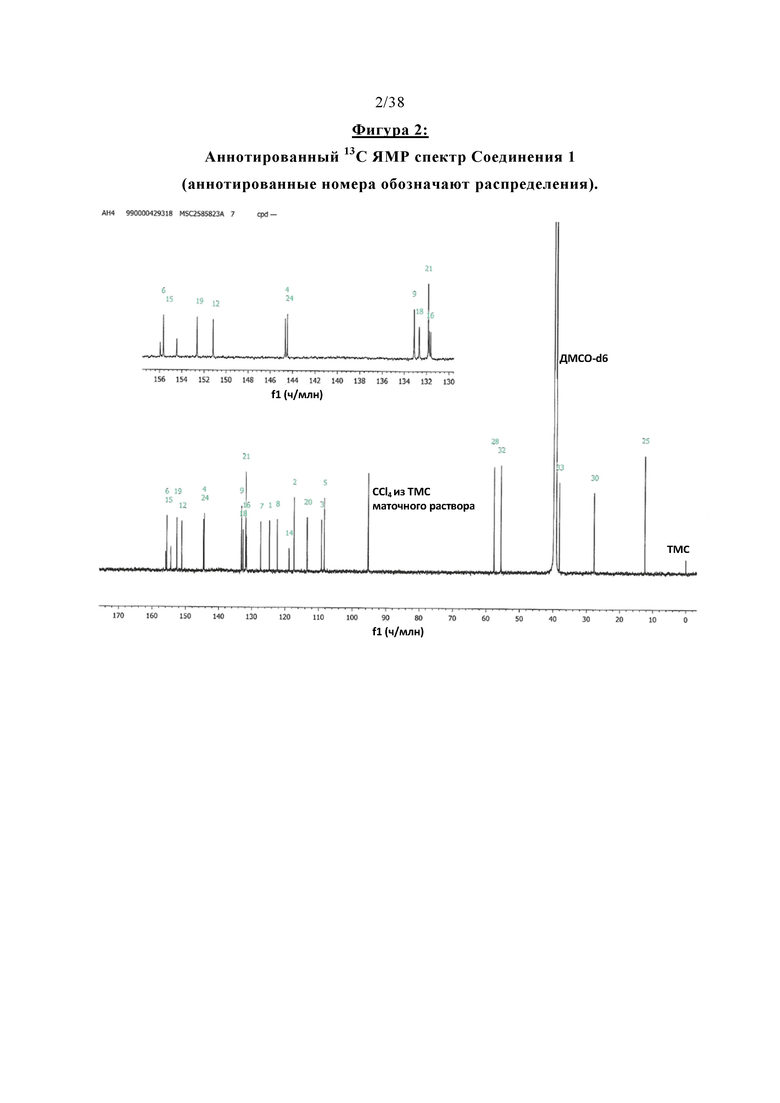
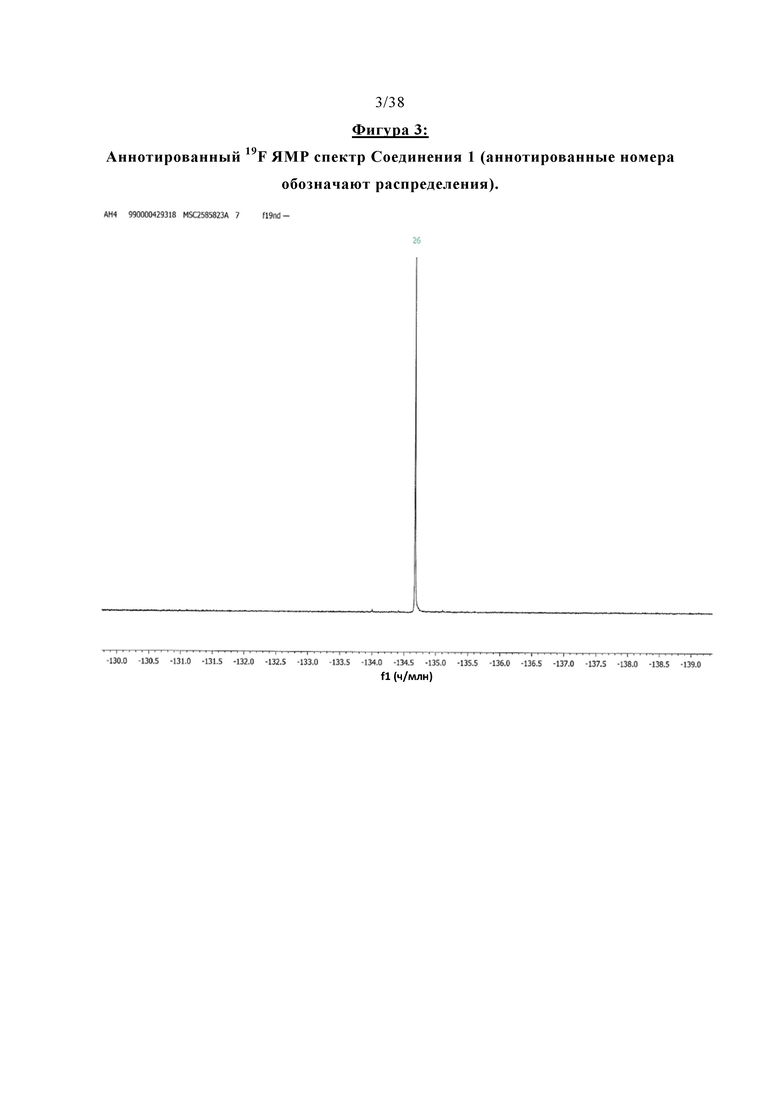

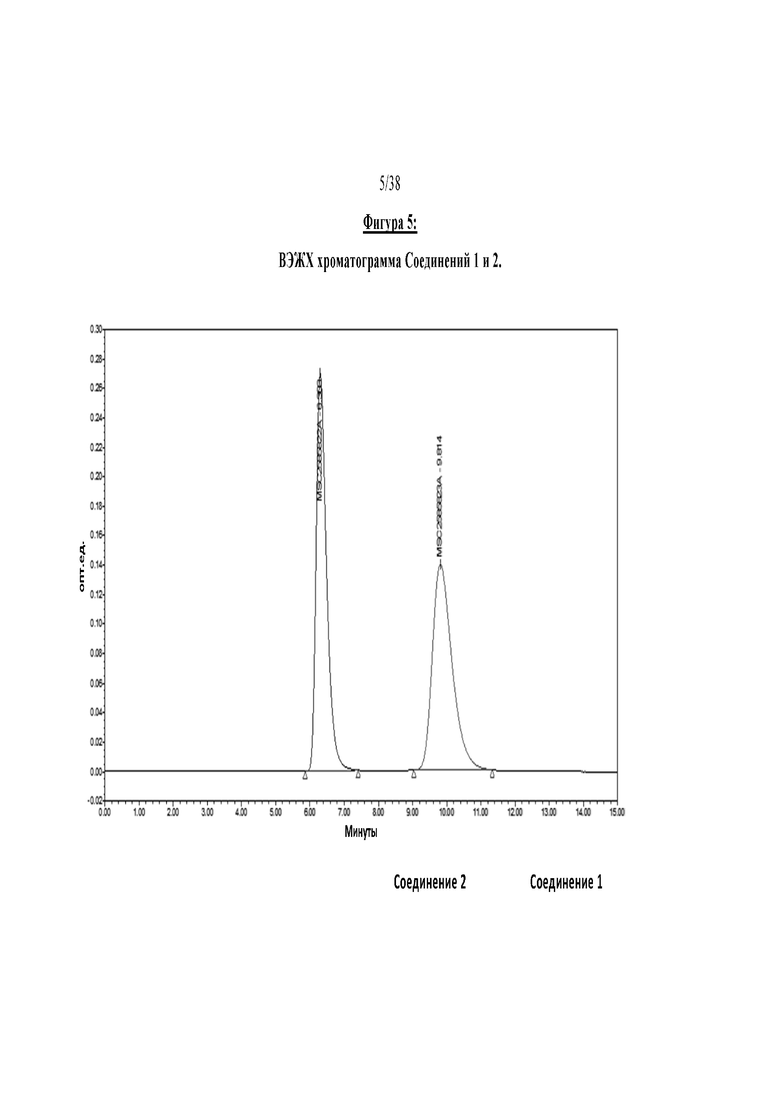
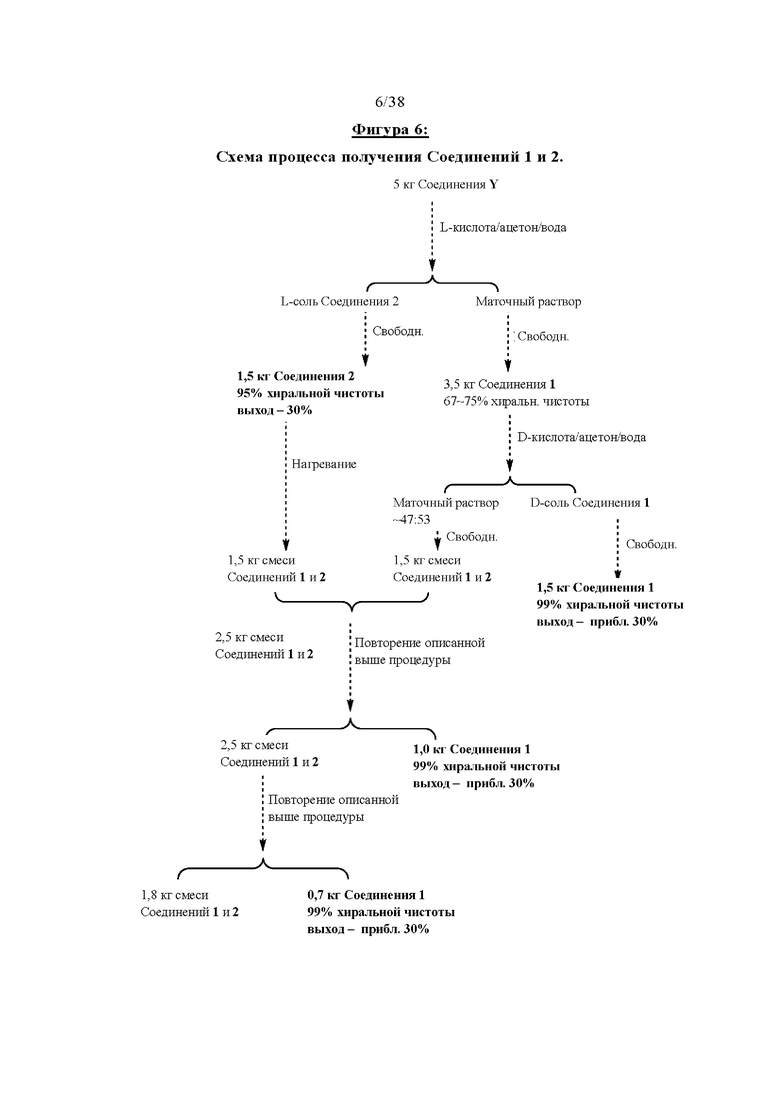
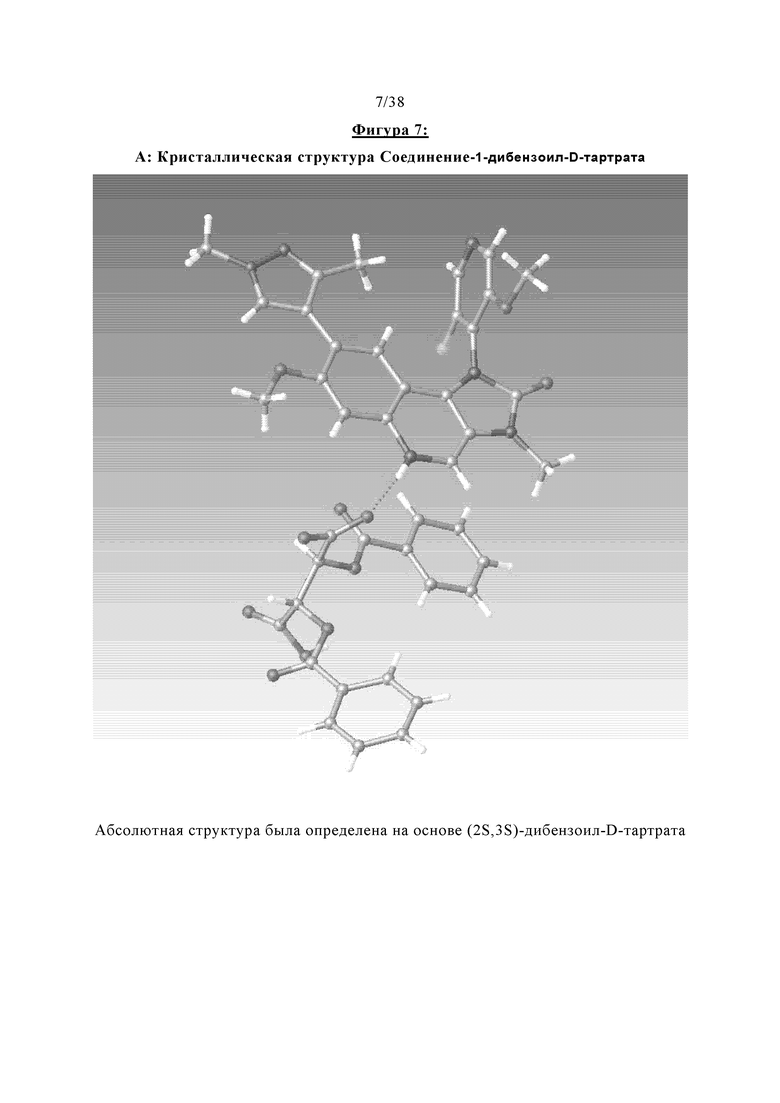
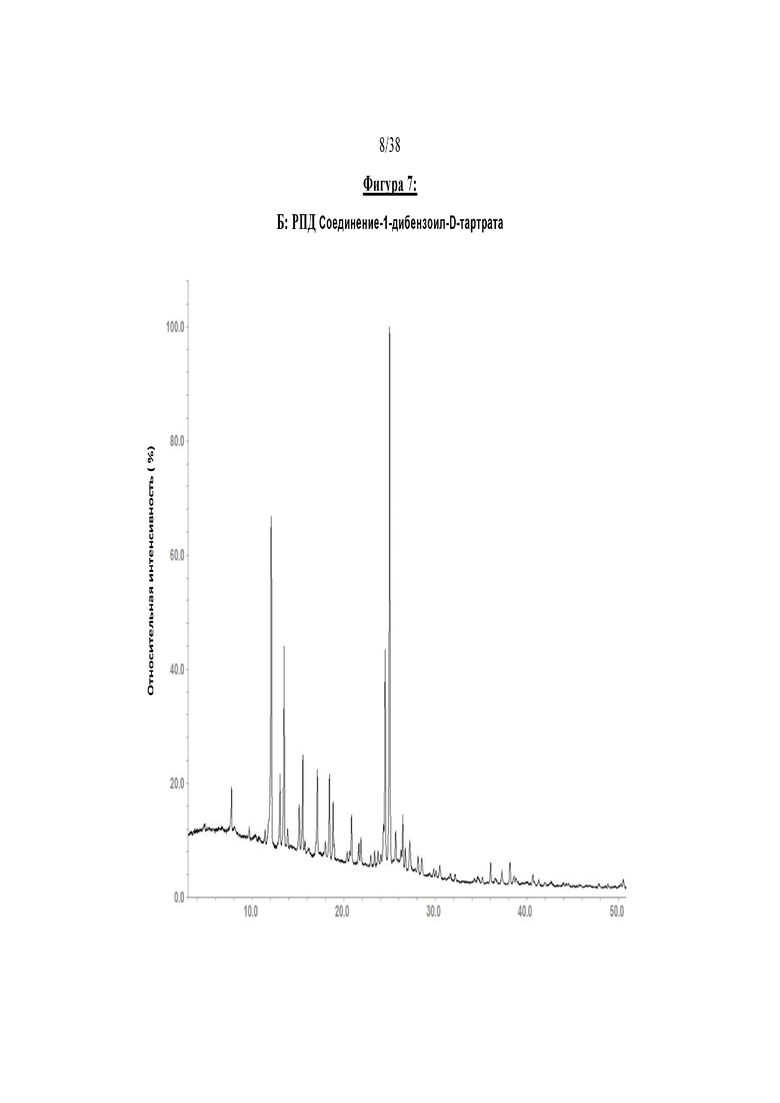

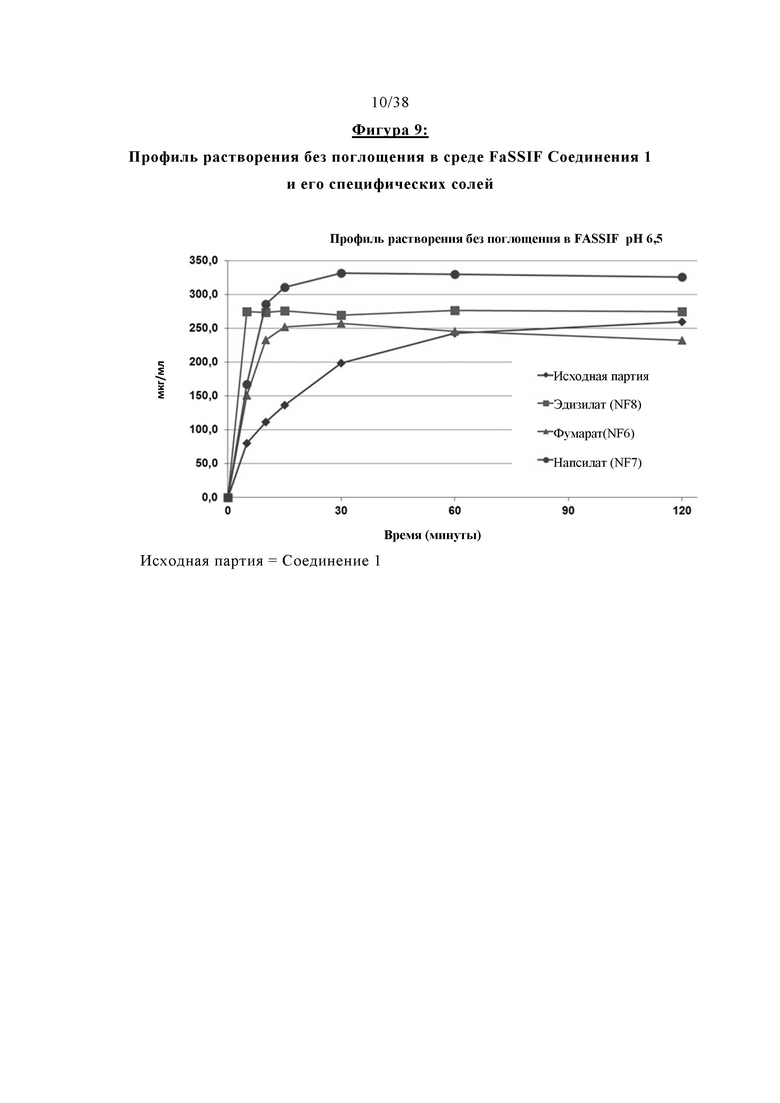
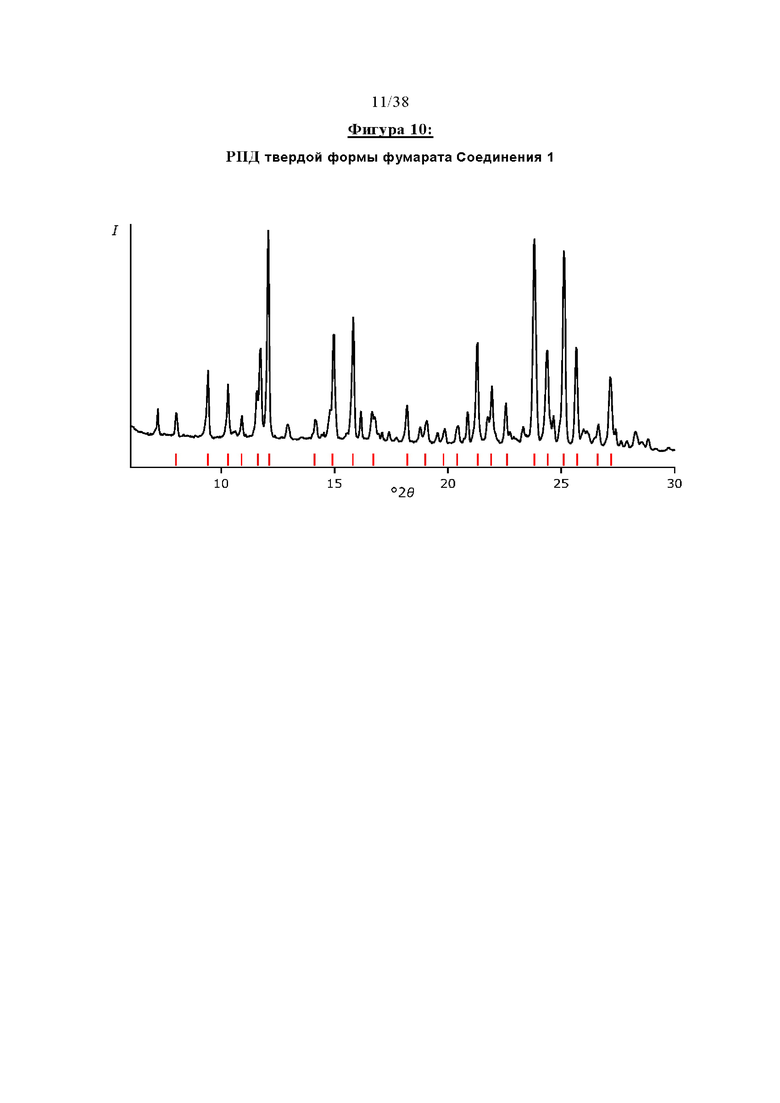
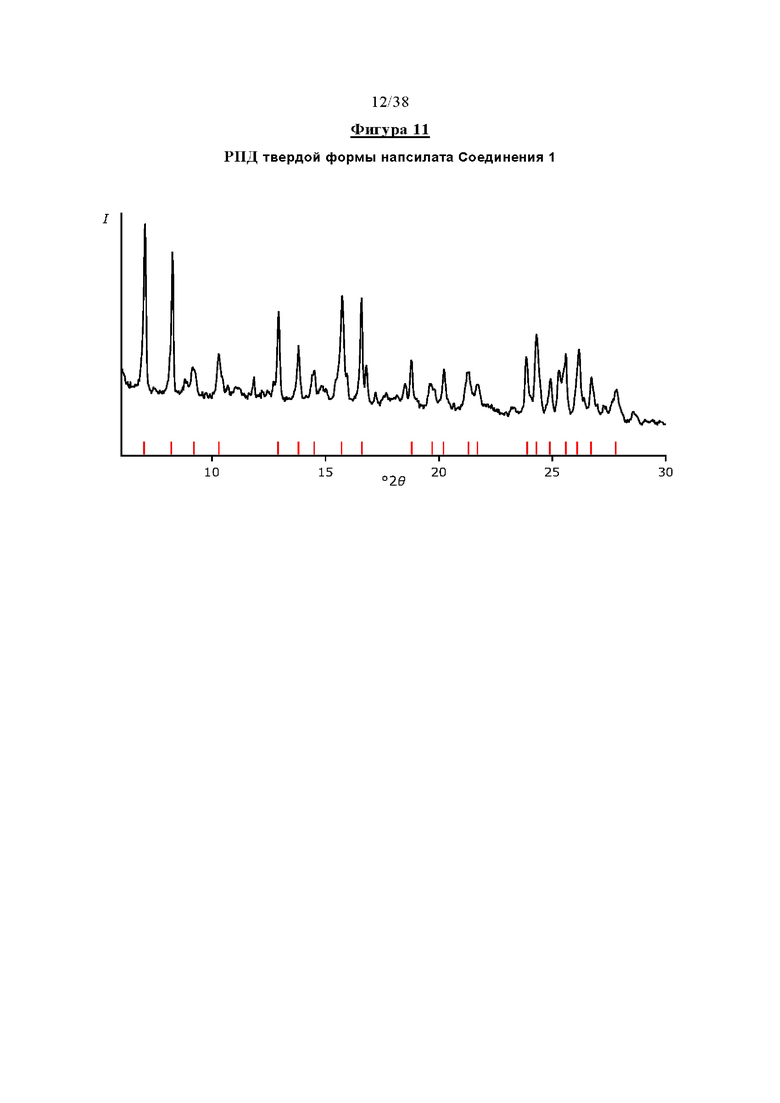
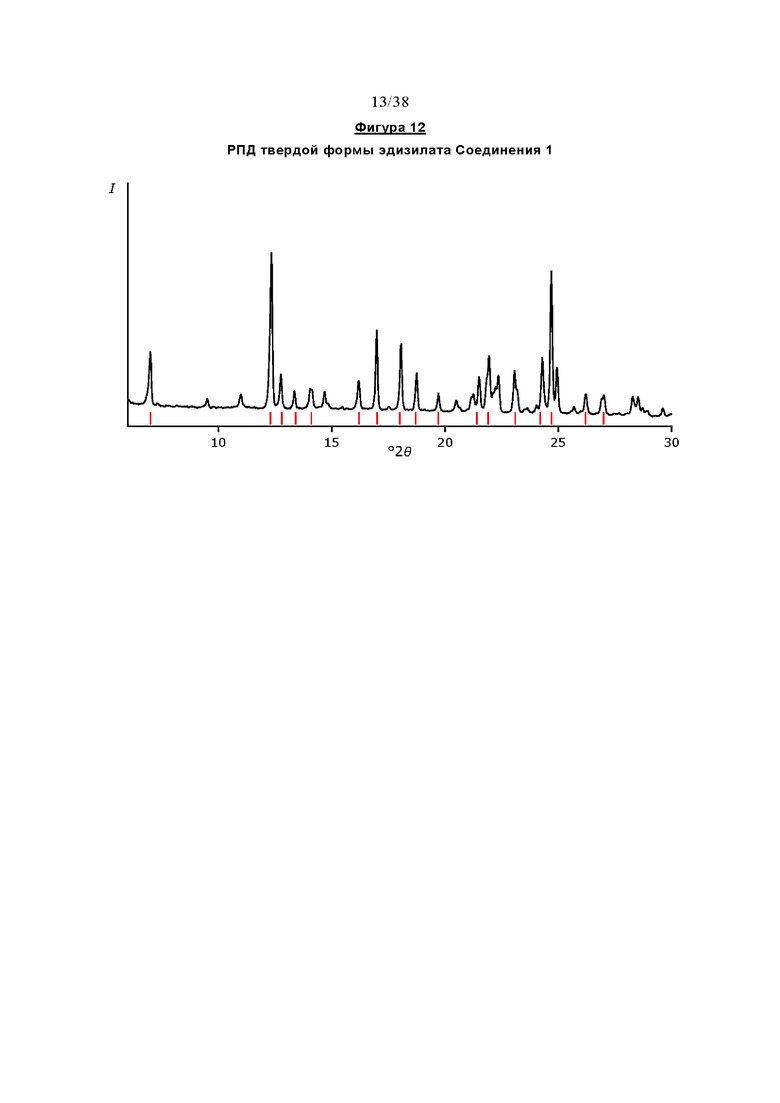
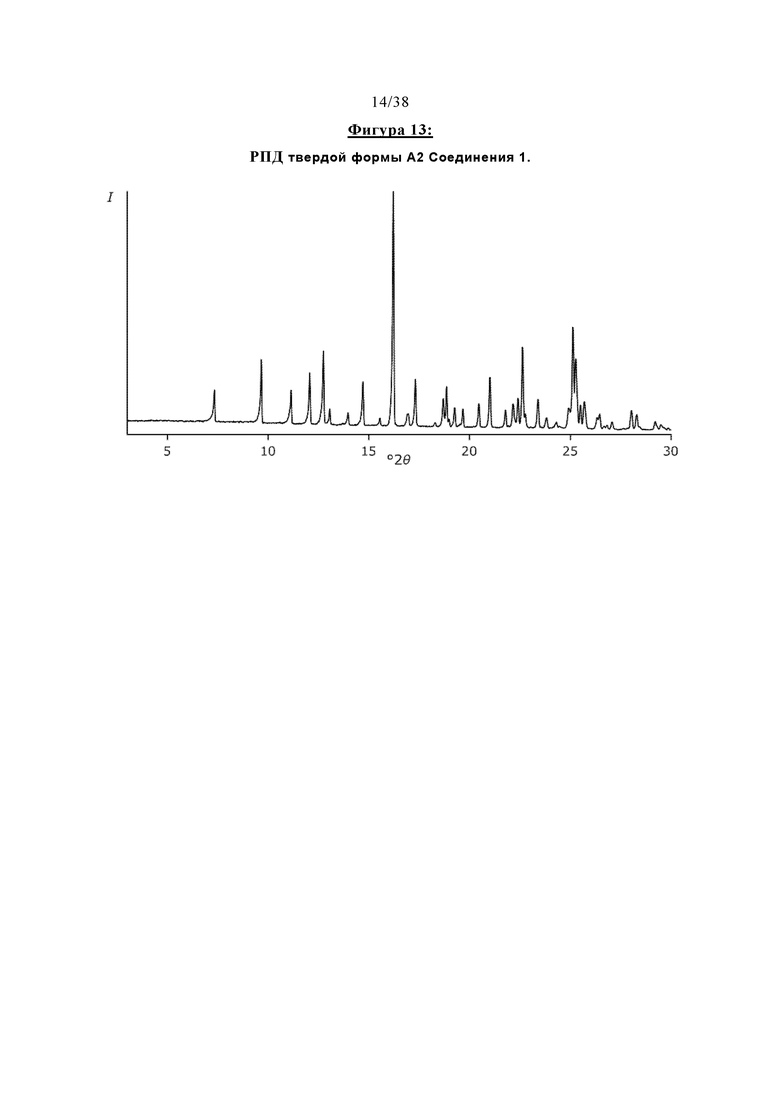
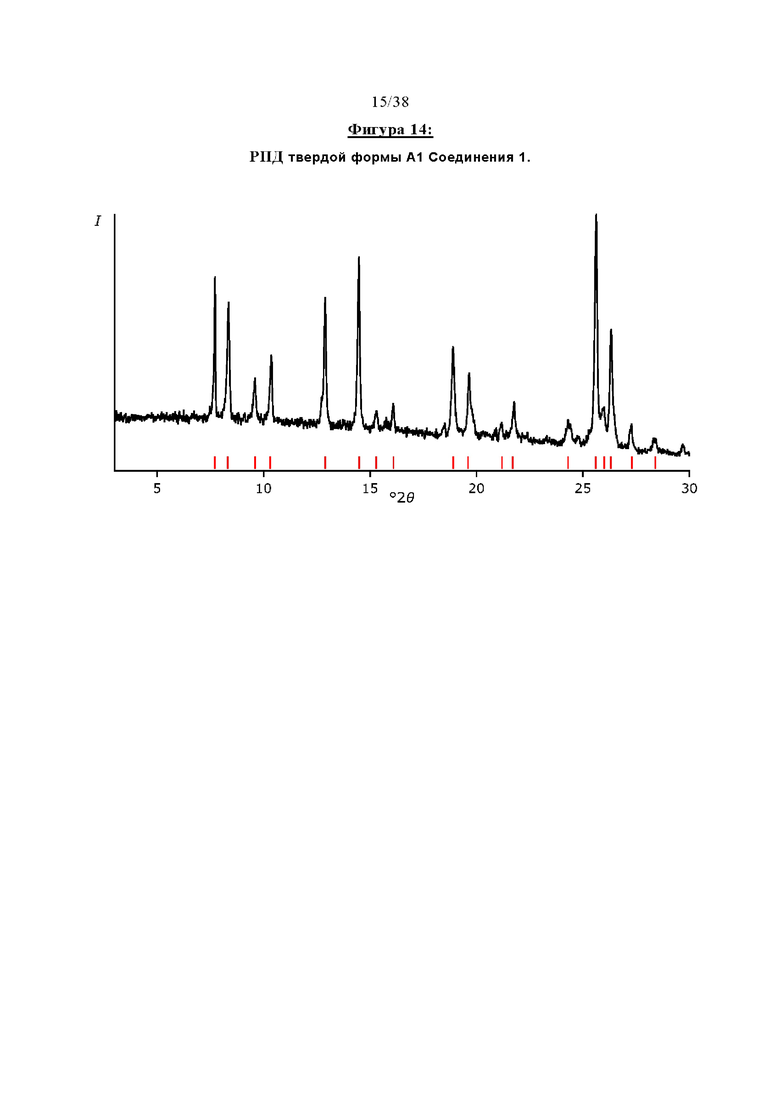
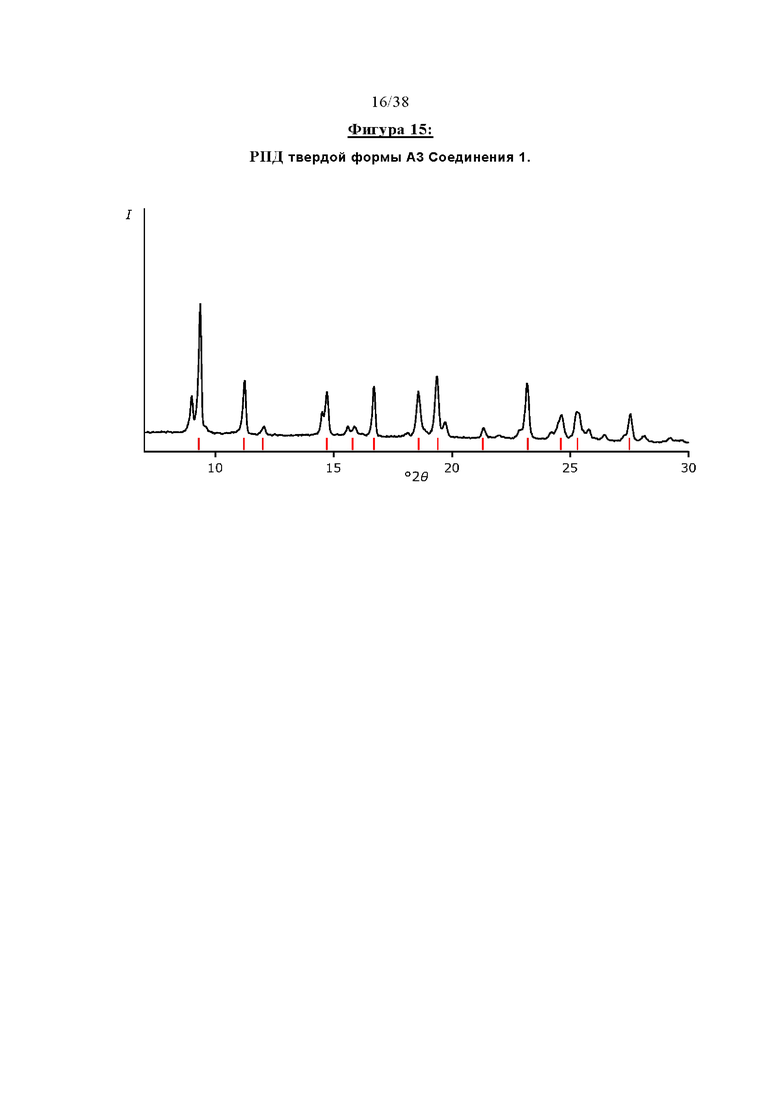
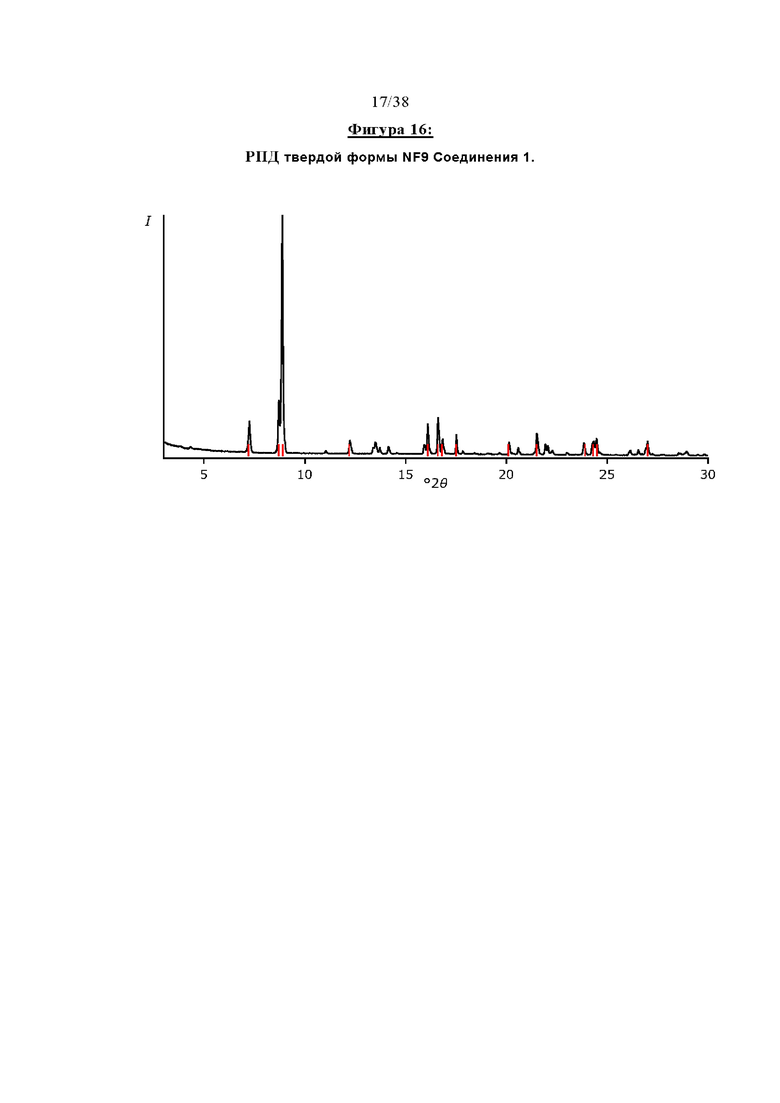
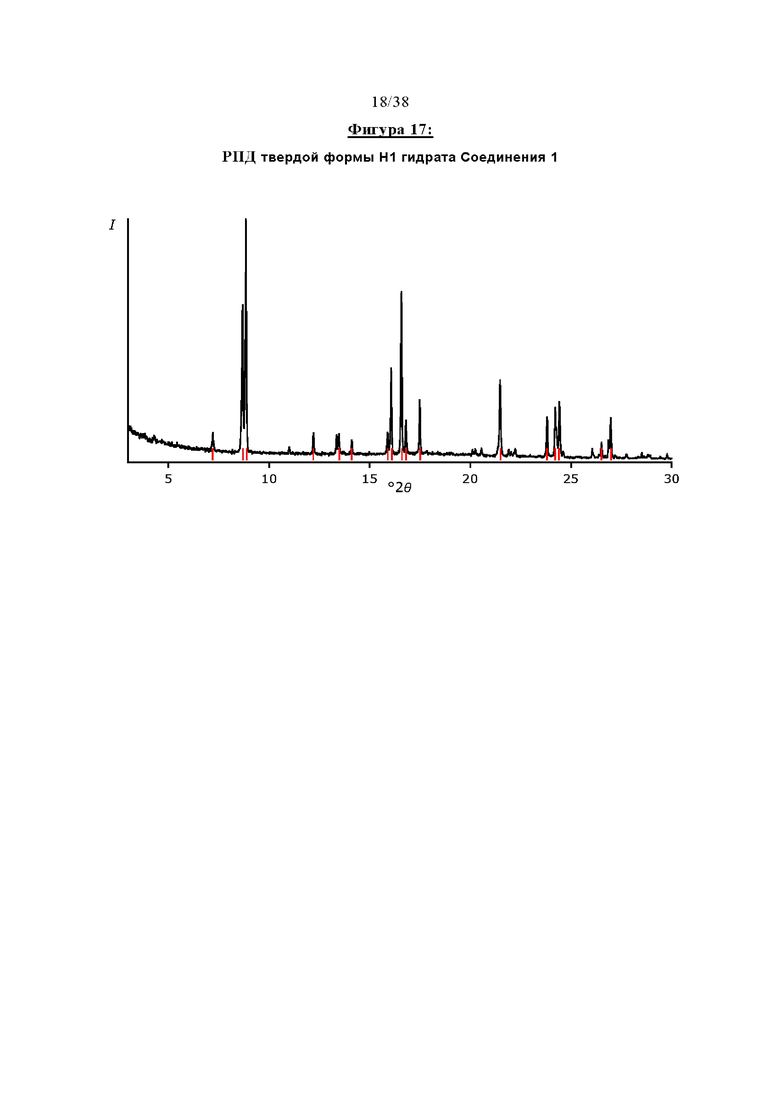
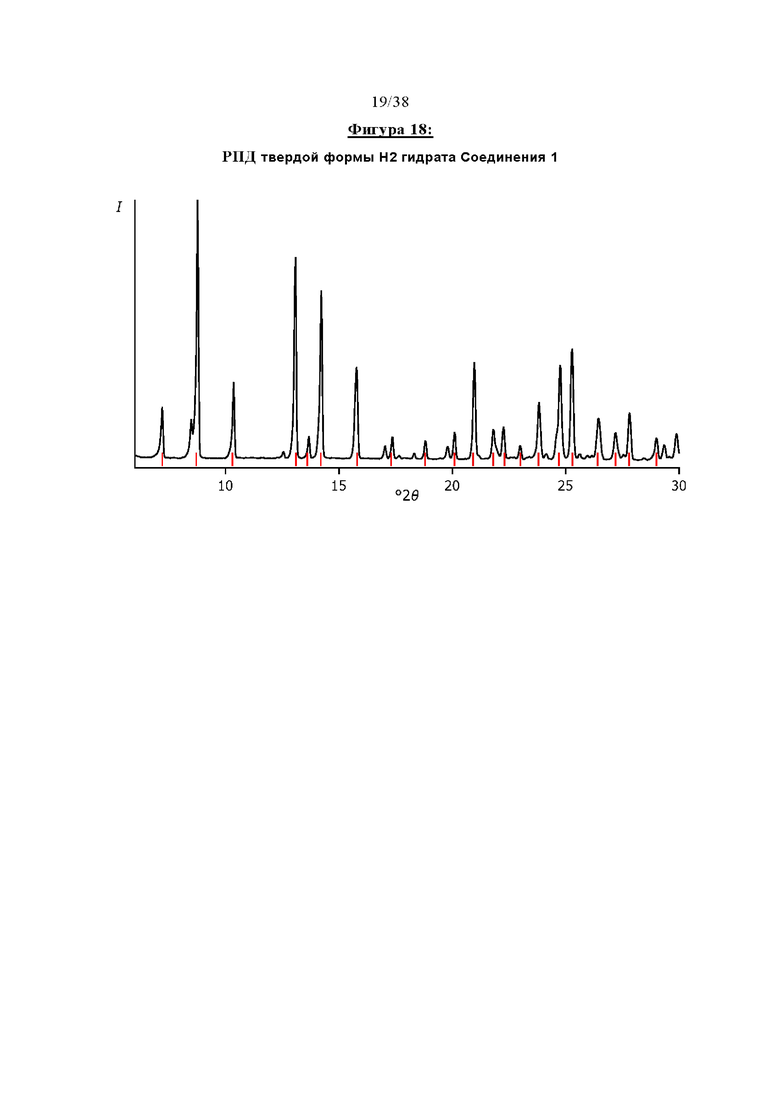
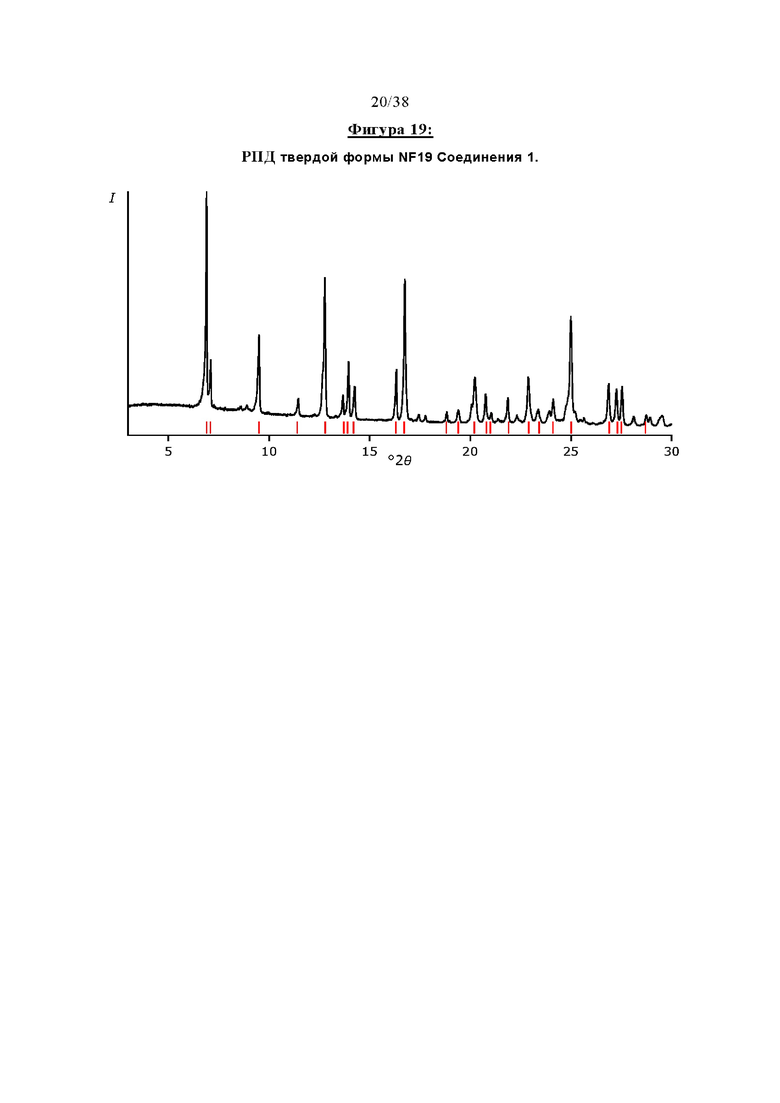
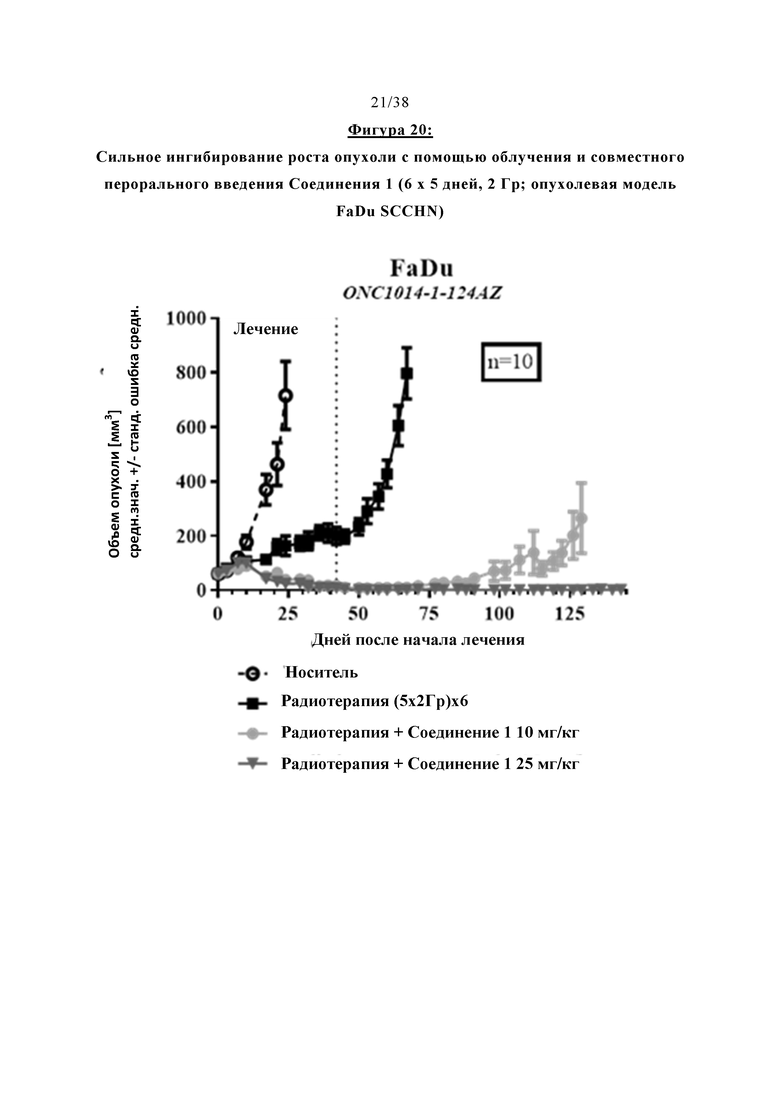
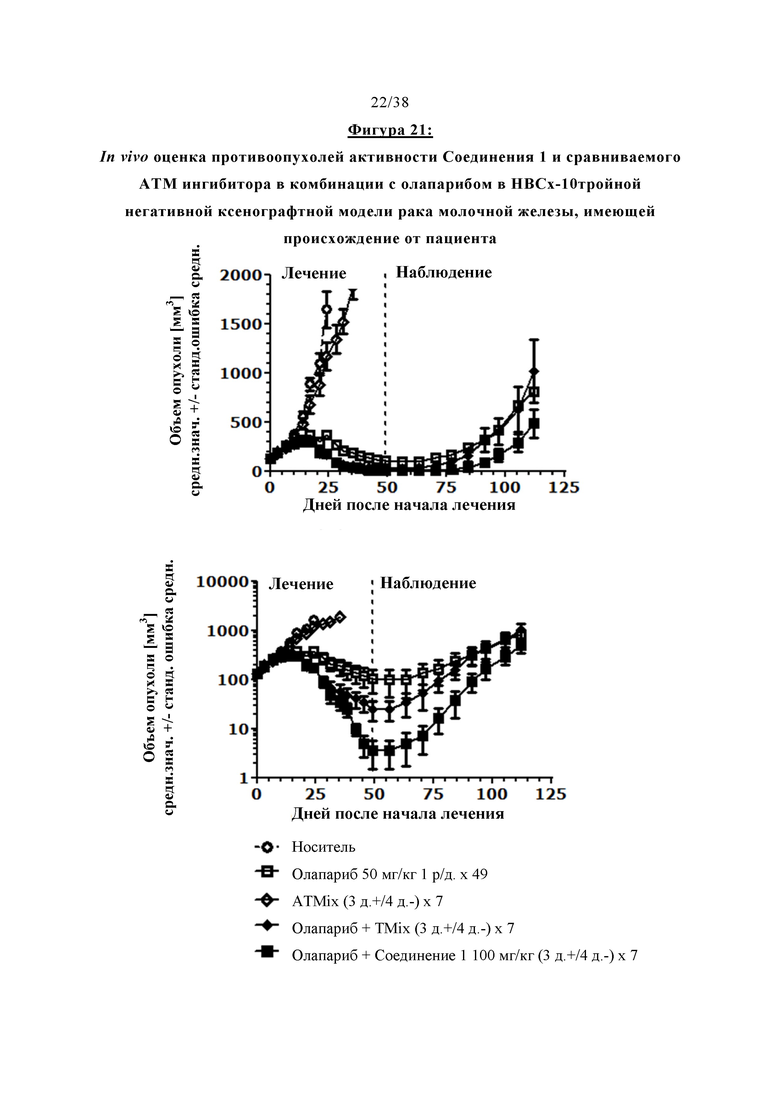
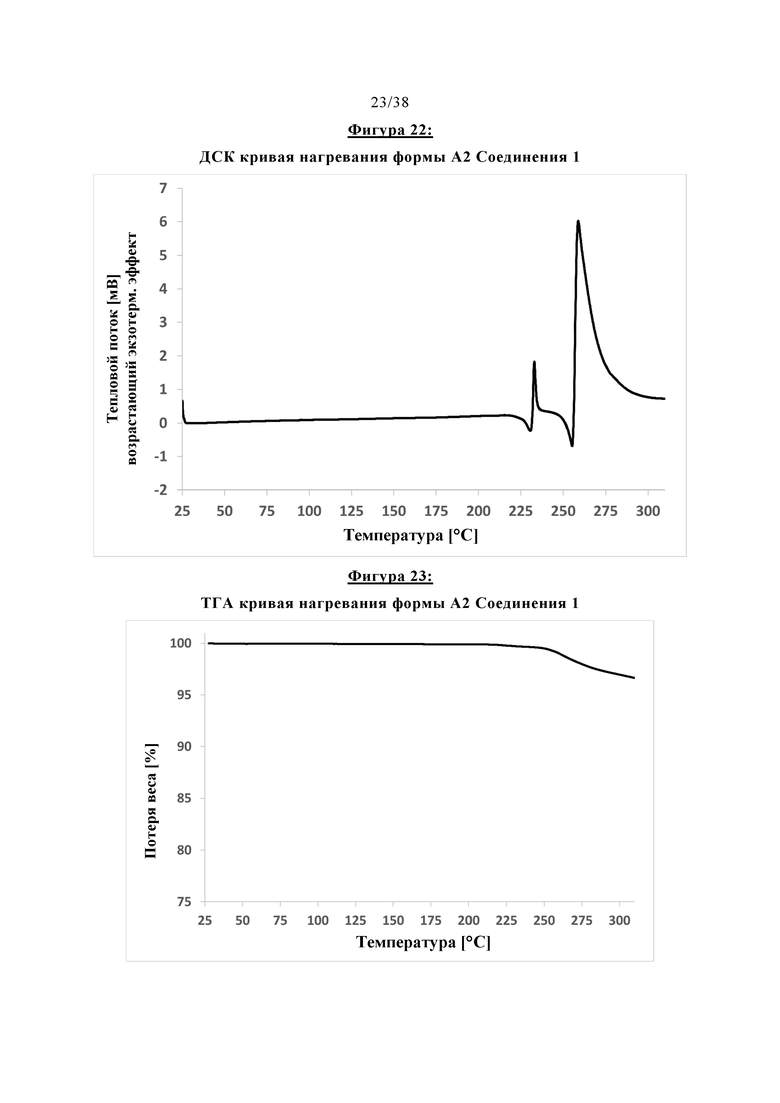
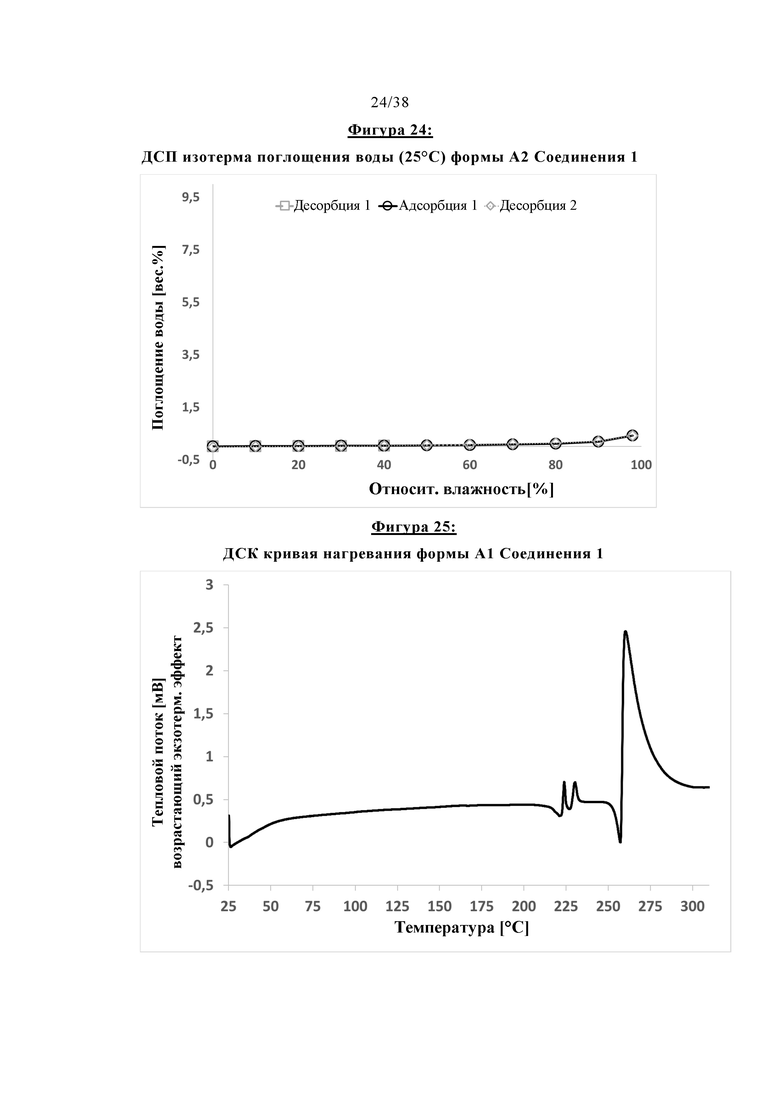
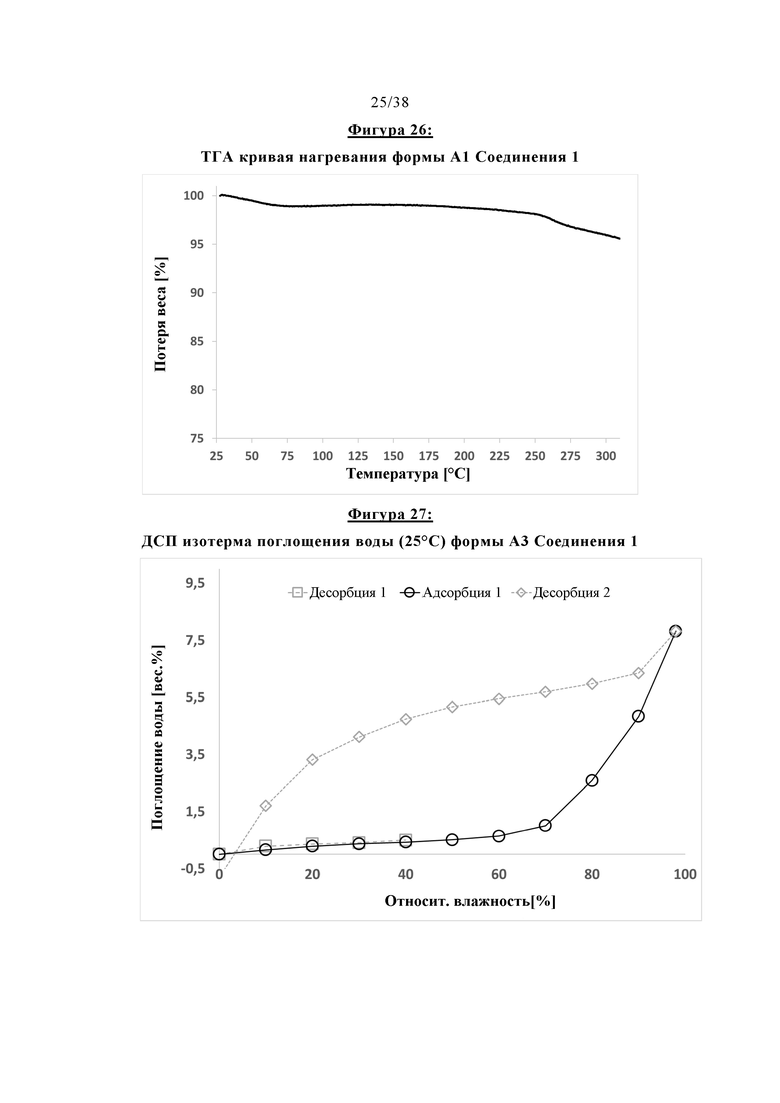
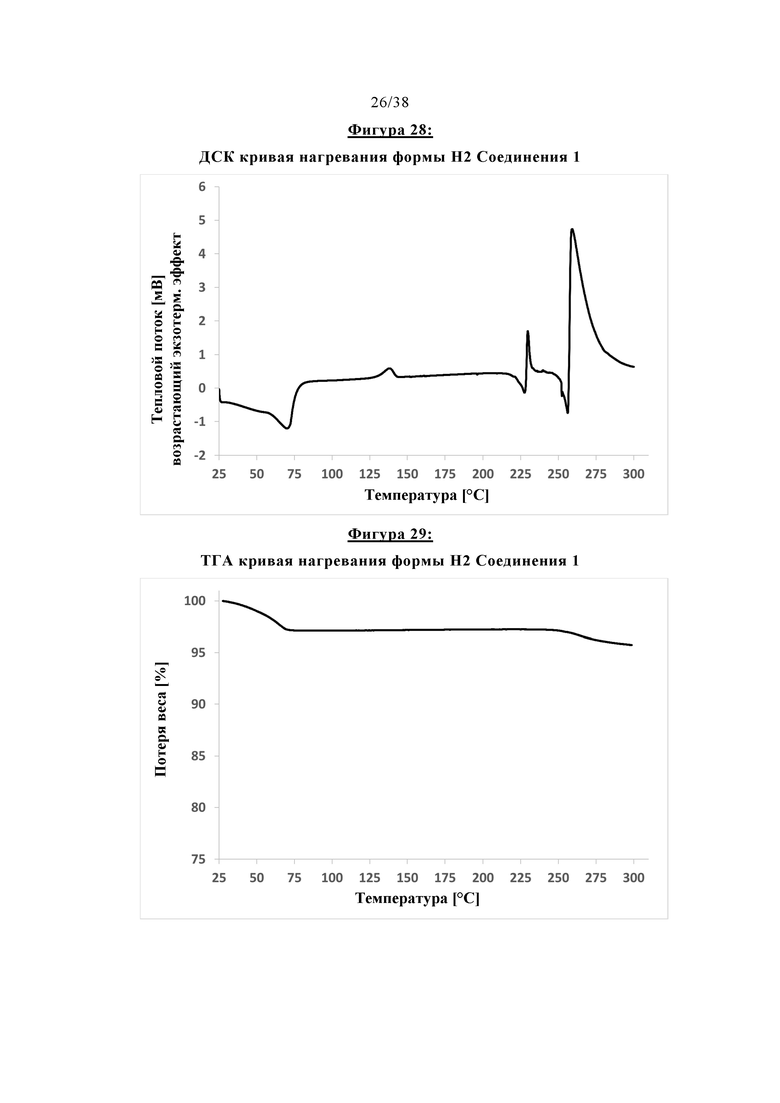
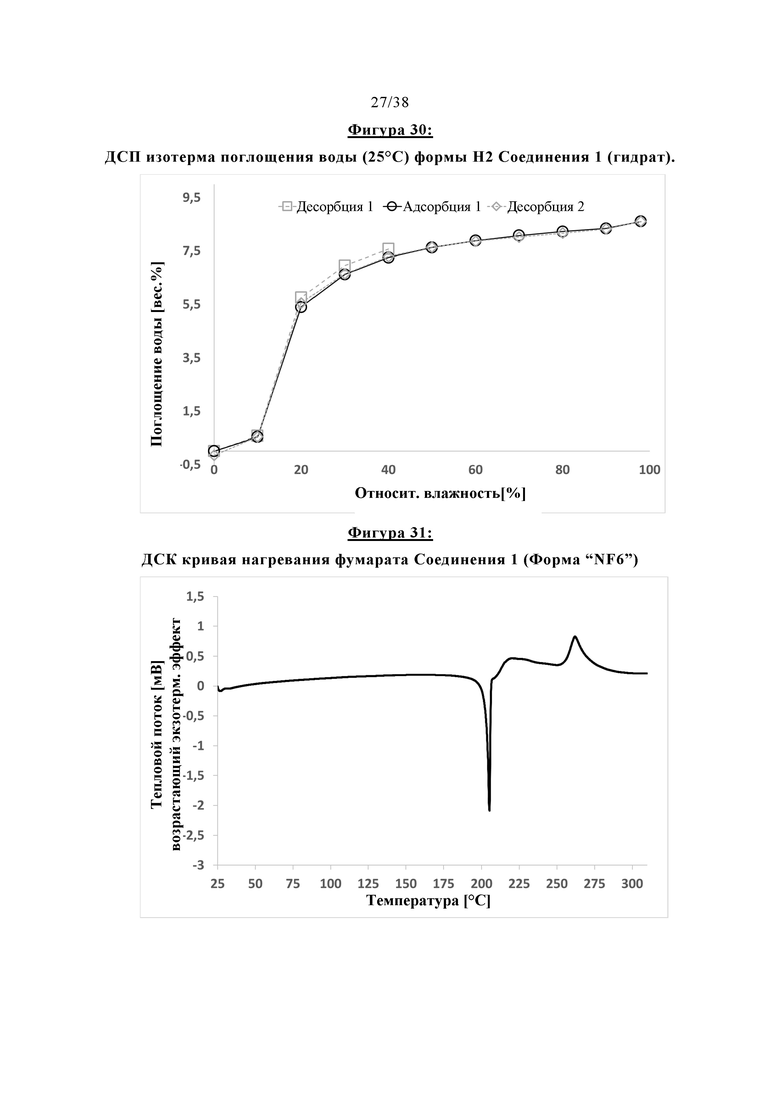
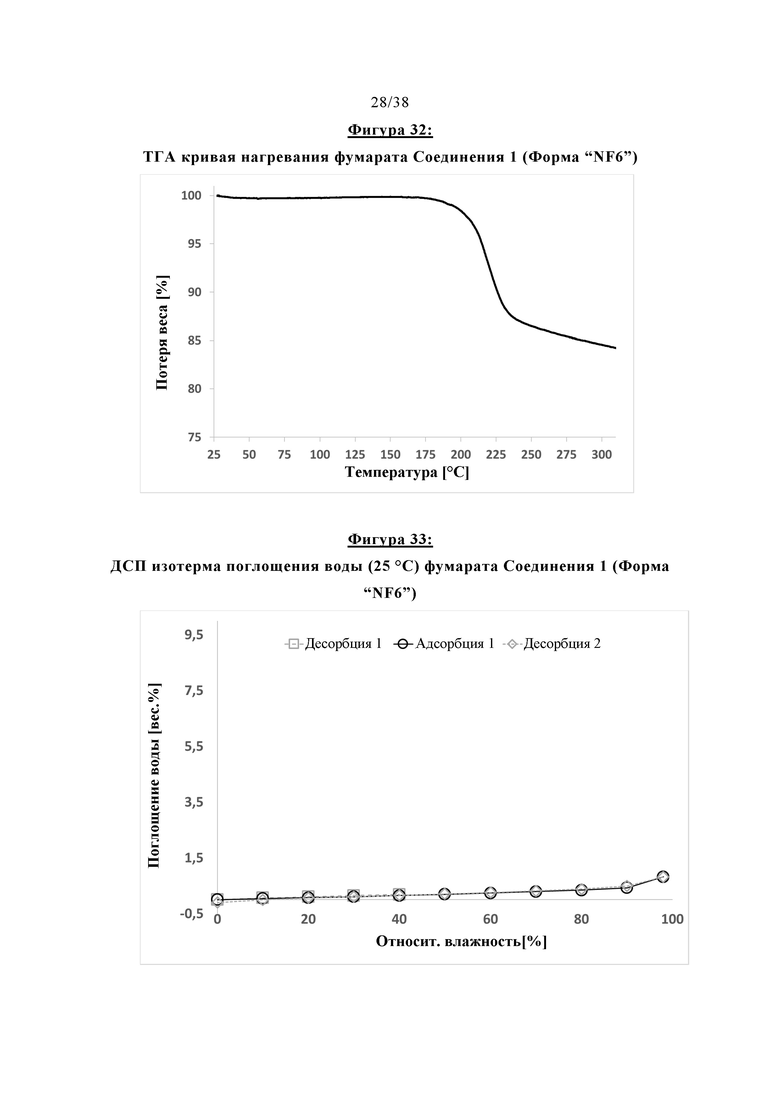


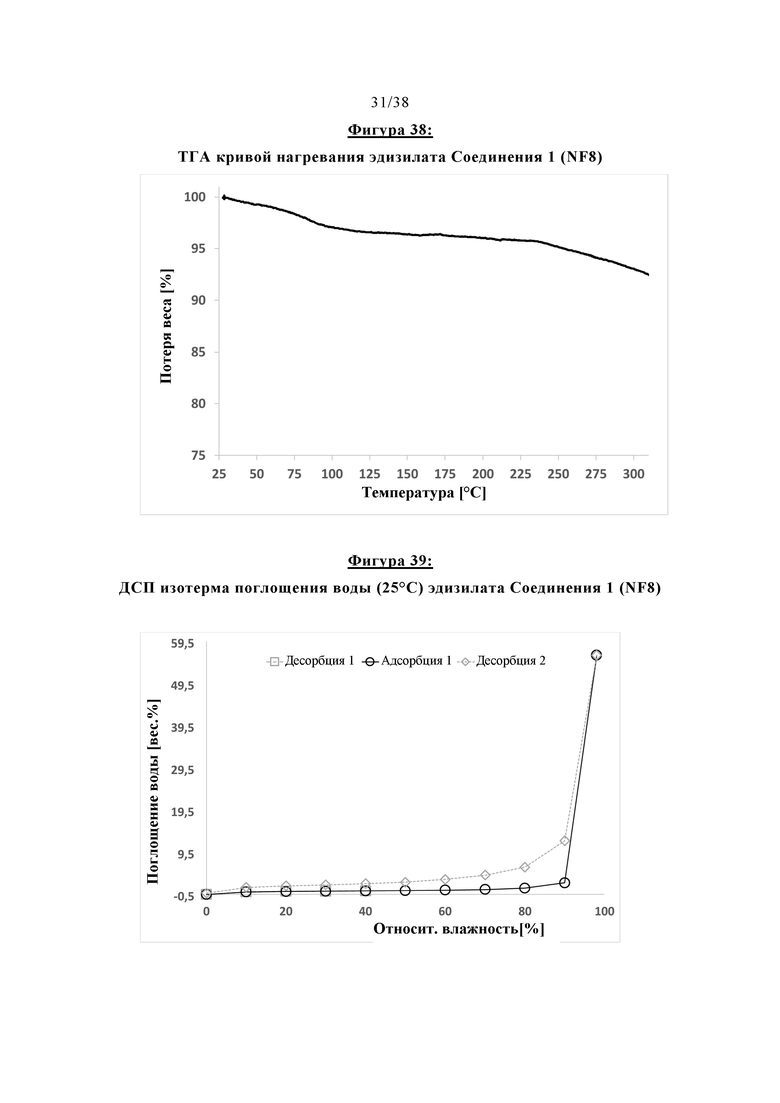
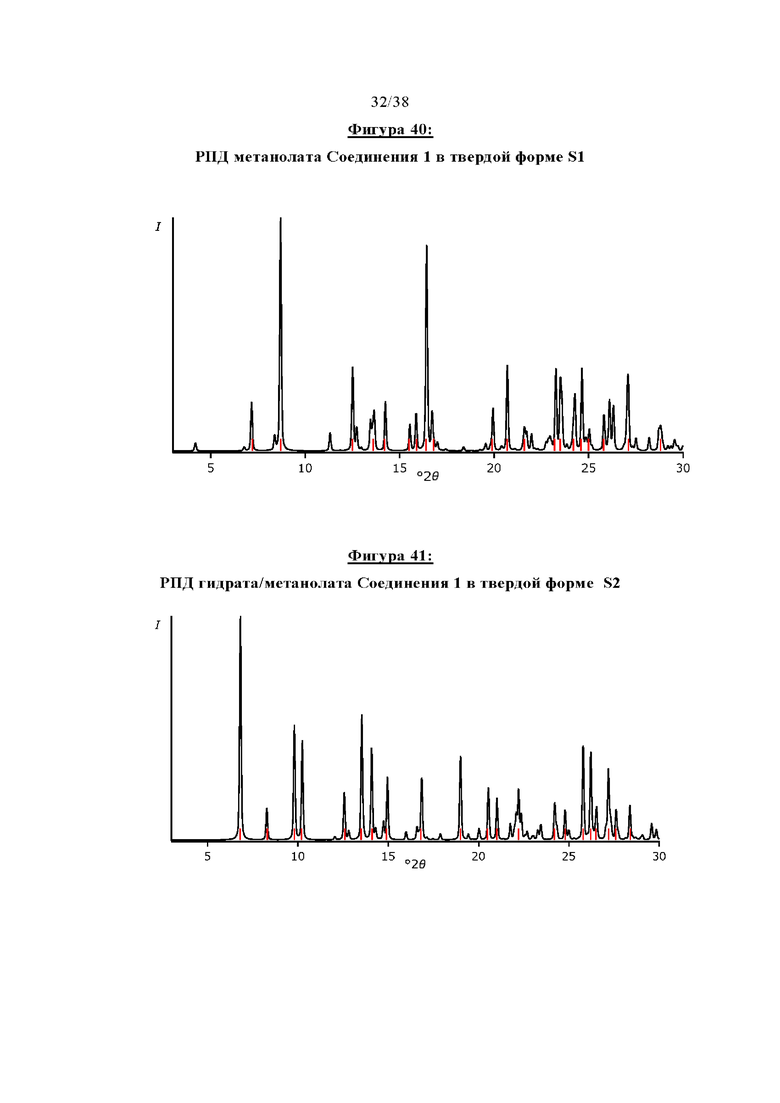
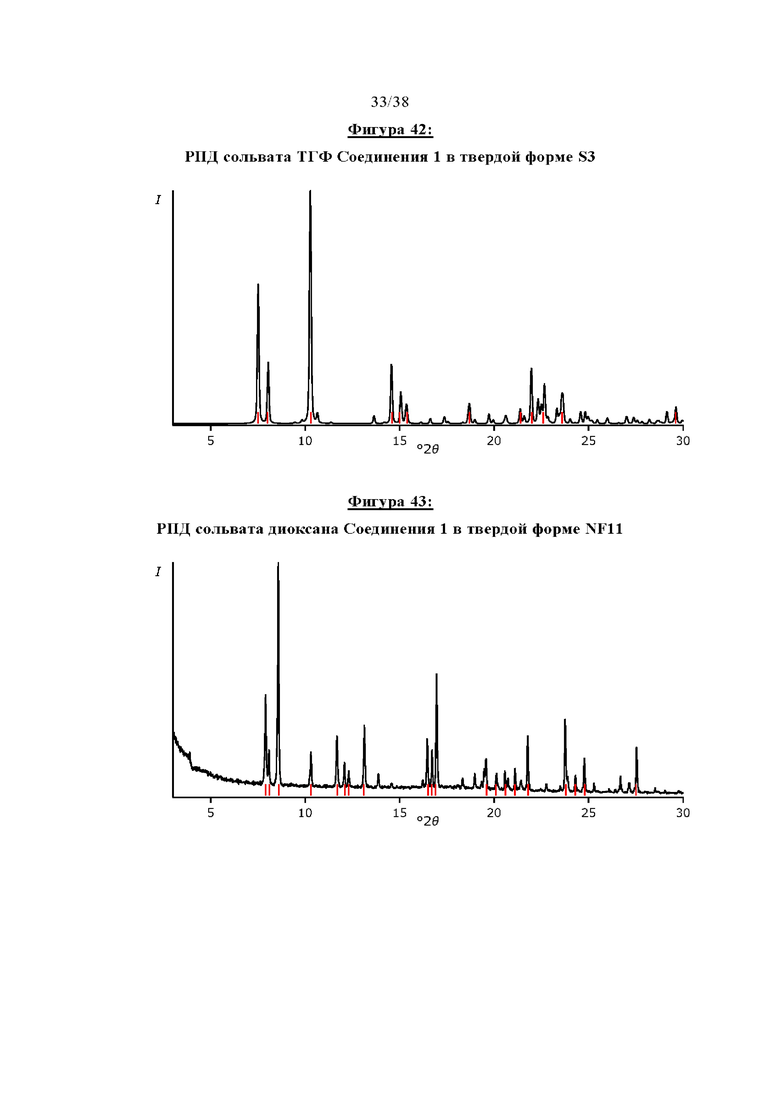
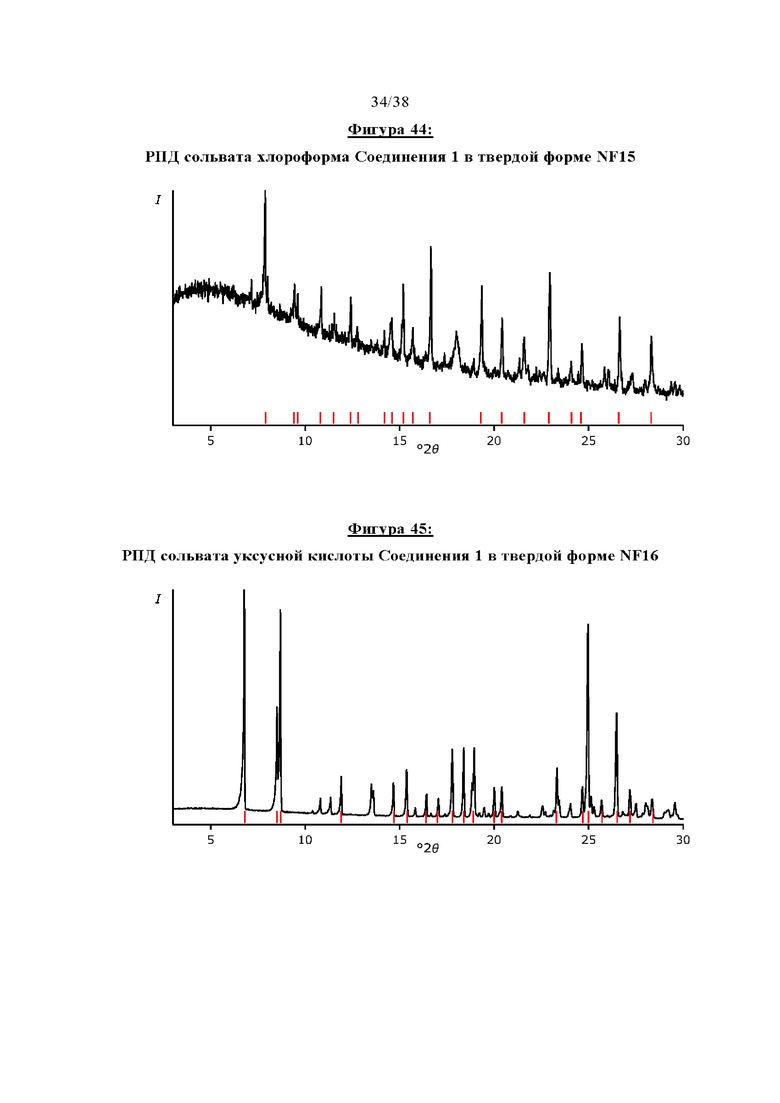
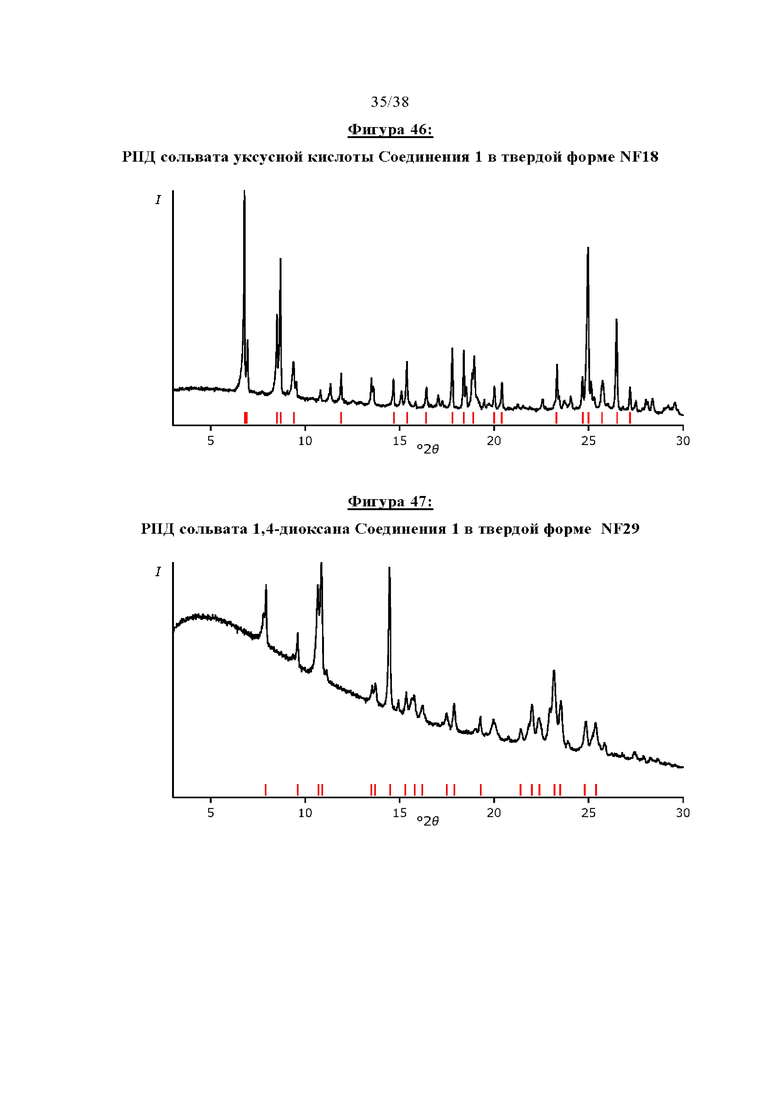
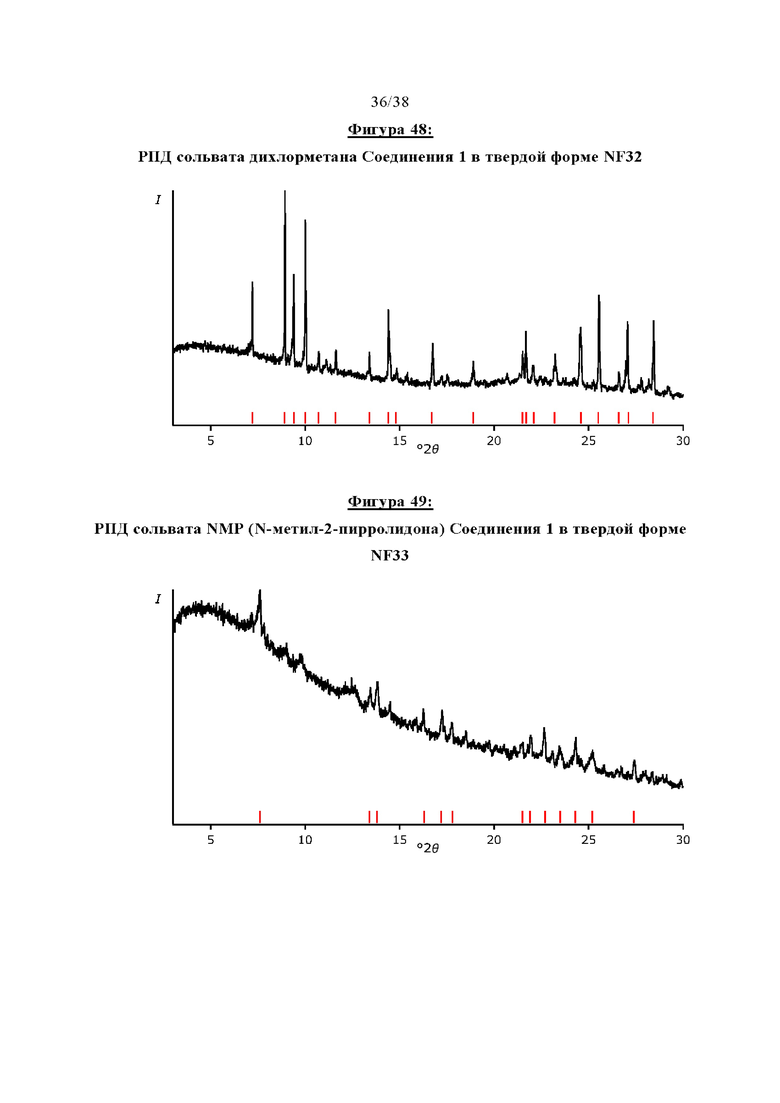
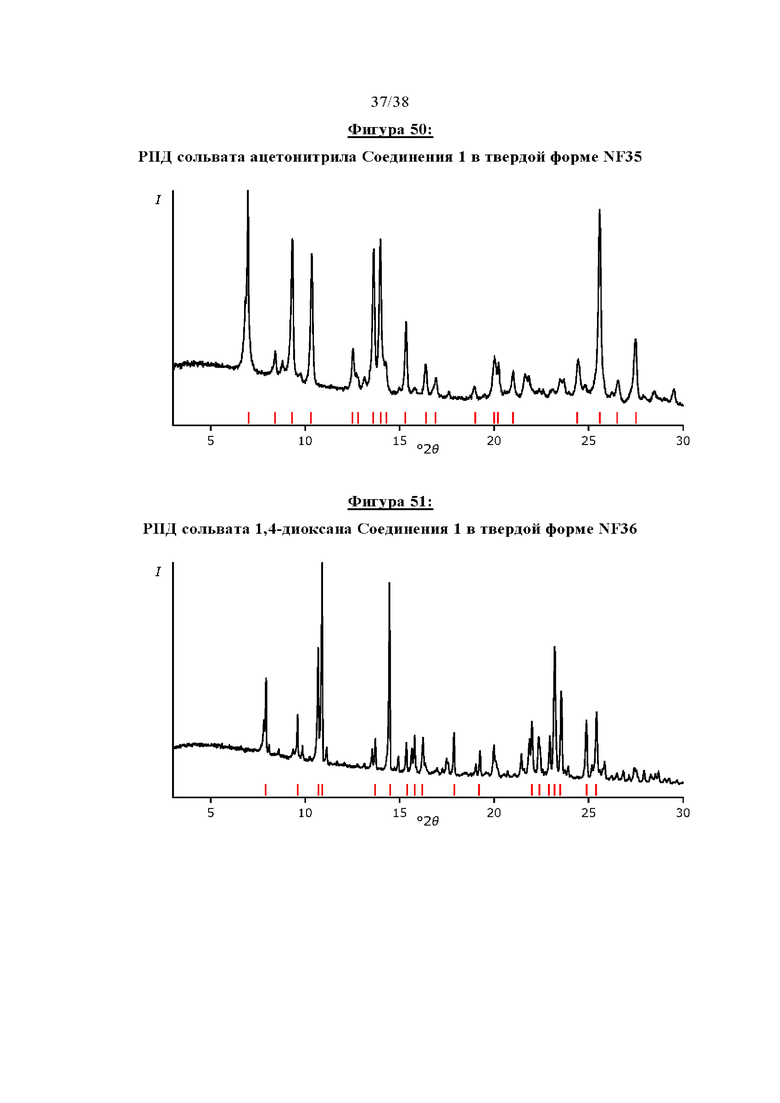
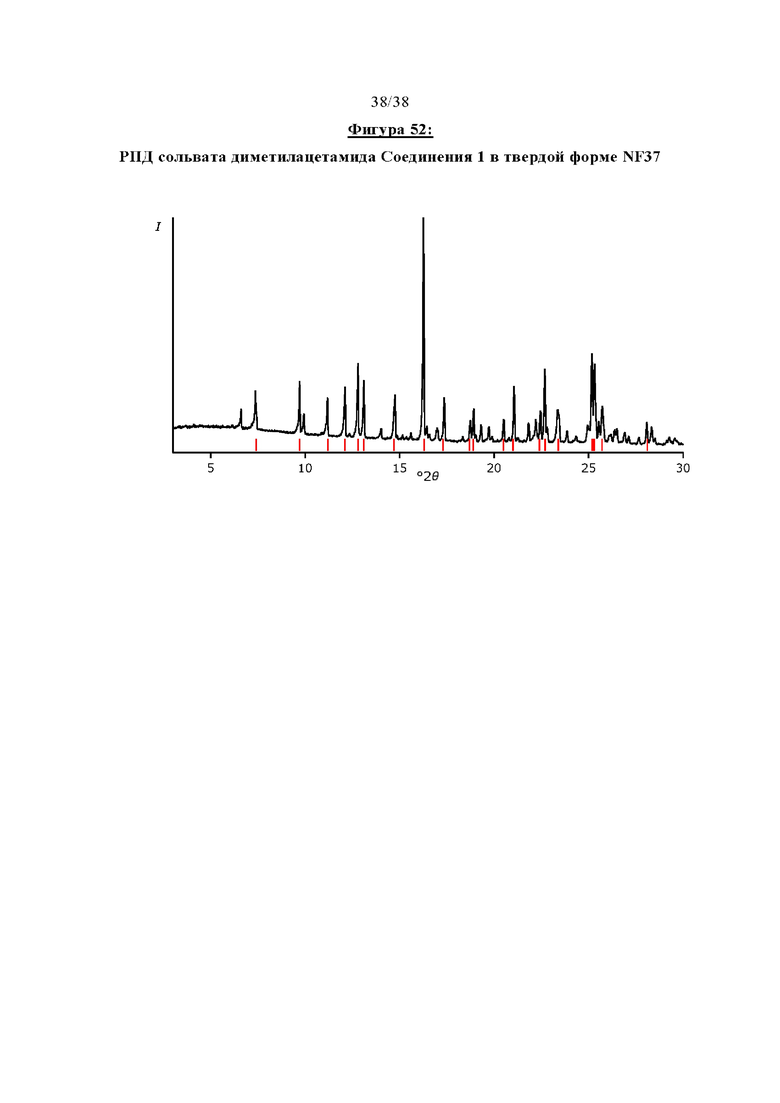
Настоящее изобретение относится к атропоизомеру ATM ингибитора 8-(1,3-диметил-1Н-пиразол-4-ил)-1-(3-фтор-5-метоксипиридин-4-ил)-7-метокси-3-метил-1,3-дигидроимидазо[4,5-с]хинолин-2-она, а именно к соединению, представленному формулой Соединения 1, или его фармацевтически приемлемой соли. Также предложены Соединение 1 в твердой безводной форме и фармацевтическая композиция для лечения рака, включающая Соединение 1, его фармацевтически приемлемую соль или твердую безводную форму. Предложенное соединение обладает лучшими ингибирующими свойствами в отношении ATM, а также обладает лучшими значениями микросомального клиренса и низким ингибированием фосфодиэстеразы (ФДЭ) 2А1, а также ФДЭ4А1А и ФДЭ4Э2, и улучшенной растворимостью в буферных растворах. 3 н. и 2 з.п. ф-лы, 52 ил., 9 пр.
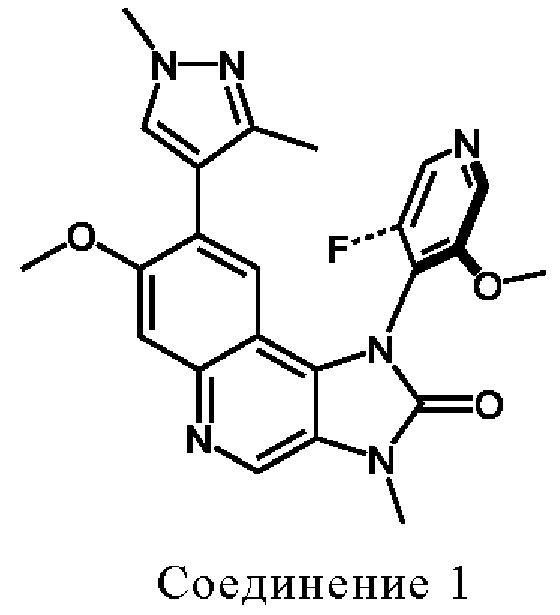
1. Соединение, представленное следующей формулой:
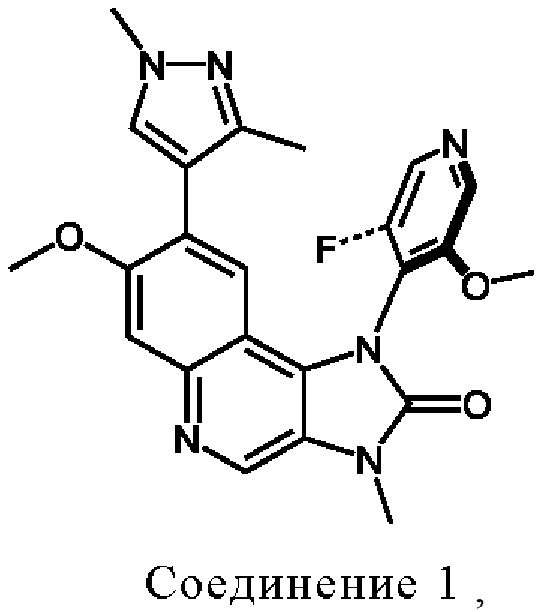
или его фармацевтически приемлемая соль.
2. Соединение в соответствии с п. 1, представленное следующей формулой:
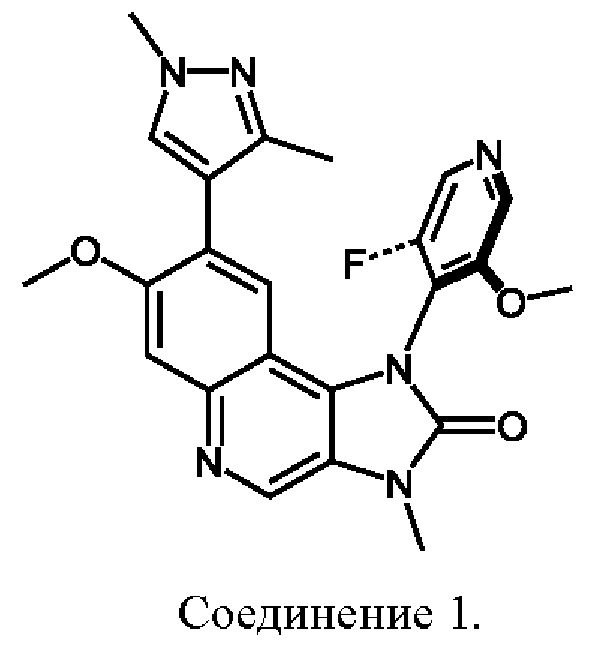
3. Соединение 1 в соответствии с п. 1 или 2 в твердой безводной форме, которое характеризуется пиками при 7,3, 9,6, 11,1, 12,0, 12,7 и 16,2 градуса 2-тета ± 0,2 градуса 2-тета в профиле рентгеновской порошковой дифракции.
4. Соединение 1 в твердой безводной форме в соответствии с п. 3, которое характеризуется тем, что имеет моноклинную кристаллическую структуру и P21 пространственную группу и/или следующие параметры своей элементарной ячейки:


5. Фармацевтическая композиция для лечения рака, которая включает Соединение 1, его фармацевтически приемлемую соль или твердую безводную форму в соответствии с любым из пп. 1-4, и фармацевтически приемлемый наполнитель.
| WO 2016155884 A1, 06.10.2016 | |||
| DAVIS E | |||
| SMITH et al., Exploiting Atropisomerism to Increase the Target Selectivity of Kinase Inhibitors, Angew | |||
| Chem., 2015, v | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| Штепсельный переключатель для радиотехнических целей | 1928 |
|
SU11754A1 |
| KENYU YOSHIDA et al., Synthesis, Resolution, and Biological Evaluation of Atropisomeric (a R)- and (a S)-16-Methyllamellarins N: Unique Effects of the Axial | |||

