Перекрестные ссылки на родственные заявки
[001] В настоящей заявке испрашивается приоритет предварительной заявки на патент США № 62/819,482, поданной 15 марта 2019 г., которая полностью включена в настоящий документ посредством ссылки.
Область техники
[002] Настоящее изобретение относится к твердым и солевым формам соединений и способам ингибирования белка, связывающего p300 (также известного как EP300 и KAT3B), белка E1A аденовируса и/или циклического AMP-чувствительного элемент-связывающего белка(CREB) (CBP, также известный как KAT3A), клеточный паралог p300. Соединения полезны для лечения некоторых форм рака.
Уровень техники
[003] CBP/p300 представляют собой лизин ацетилтрансферазы, которые катализируют присоединение ацетильной группы к лизиновой боковой цепи гистонов и других белковых субстратов. р300 (также известный как EP300 и KAT3B) представляет собой белок с множеством доменов, который связывается с различными белками, включая многие ДНК-связывающие факторы транскрипции. Циклический AMP-чувствительный элемент-связывающий белок (CREB), связывающий белок (CBP, также известный как KAT3A), является клеточным паралогом p300. p300 и CBP обладают обширной идентичностью последовательностей и функциональным сходством и часто обозначаются как CBP/p300. CBP/p300-катализируемое ацетилирование гистонов и других белков имеет решающее значение для активации гена. Повышенная экспрессия и активность p300 наблюдались при запущенных формах рака человека, таких как рак простаты, и в образцах первичного рака груди человека. Химическое ингибирование CBP/p300, обладающего внутренней ферментативной активностью ацетилтрансферазы, более осуществимо, чем блокирование факторов транскрипции небольшими молекулами, поскольку открытие химических ингибиторов факторов транскрипции оказалось чрезвычайно сложной задачей.
[004] Соответственно, существует потребность в новых и эффективных соединениях для ингибирования CBP/p300, полезных в качестве терапии для лечения определенных родственных форм рака.
Краткое изложение сущности изобретения
[005] В одном из аспектов раскрыта неаморфная твердая форма соединения формулы (I):
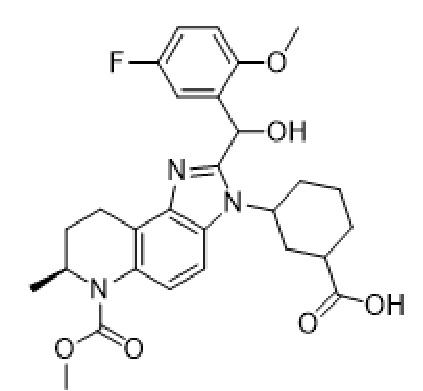 (I)
(I)
и его солей.
[006] В другом аспекте раскрыты твердые формы соединений, имеющих стереохимию формулы (II):
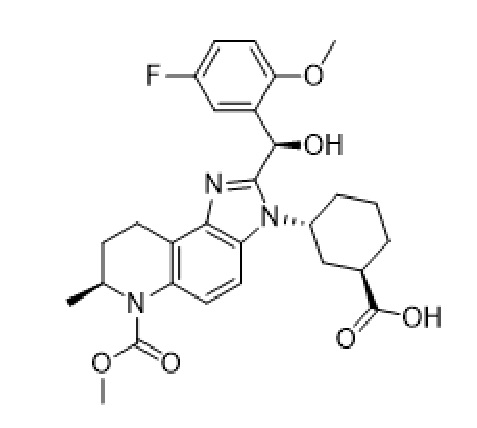 (II)
(II)
и их солей.
[007] В некоторых вариантах осуществления изобретения соль представляет собой кислотно-аддитивную соль, выбранную из соляной кислоты, р-толуолсульфоновой кислоты, бензолсульфоновой кислоты и серной кислоты.
Краткое описание фигур
[008] Настоящая заявка содержит фигуры для лучшего понимания принципов описания:
[009] Фиг. 1.A представляет собой таблицу параметров, используемых для анализа порошковой рентгеновской дифракции твердых форм, раскрытых в данном документе.
[010] Фиг. 1.B представляет собой таблицу параметров, используемых для термогравиметрического и дифференциального сканирующего калориметрического анализа твердых форм, описанных в данном документе.
[011] Фиг. 1.C представляет собой таблицу параметров, используемых для анализа динамической сорбции пара твердых форм, описанных в данном документе.
[012] Фиг. 1.D представляет собой таблицу параметров, используемых для анализа HPLC аморфного соединения свободного основания, описанного в данном документе.
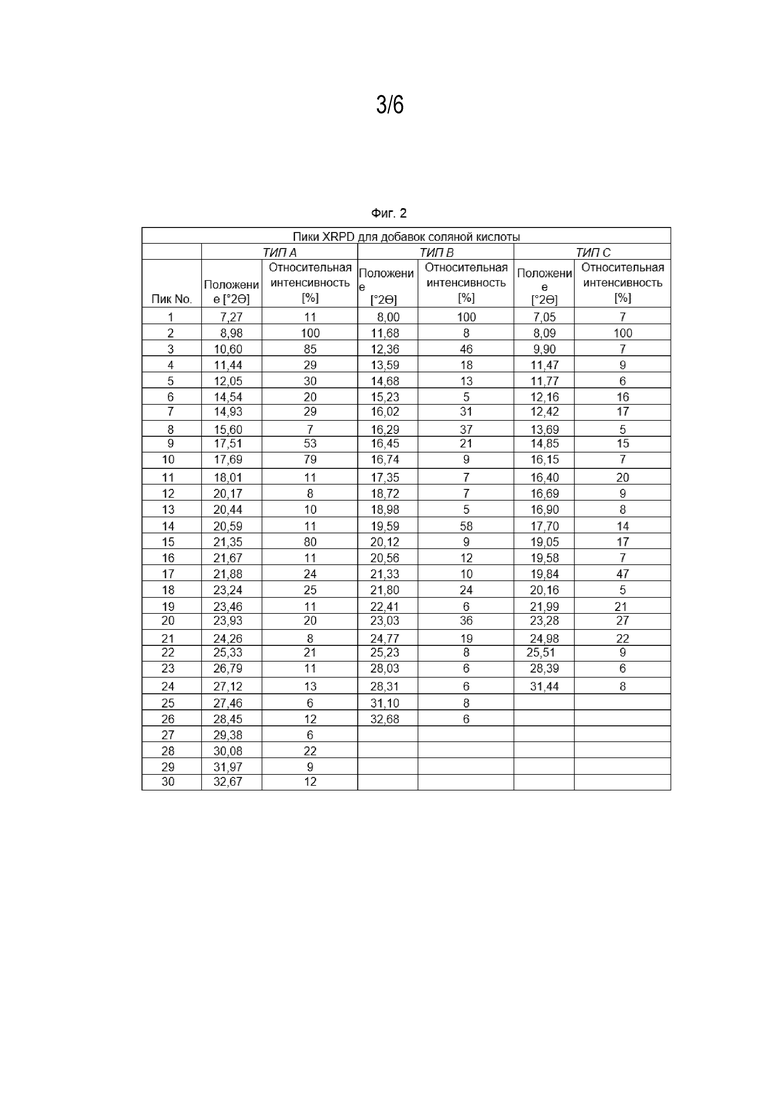
[013] Фиг. 2 представляет собой таблицу пиков рентгеновской порошковой дифрактометрии с относительной интенсивностью более 5% для форм аддитивных солей соляной кислоты типов A, B и C, описанных здесь.
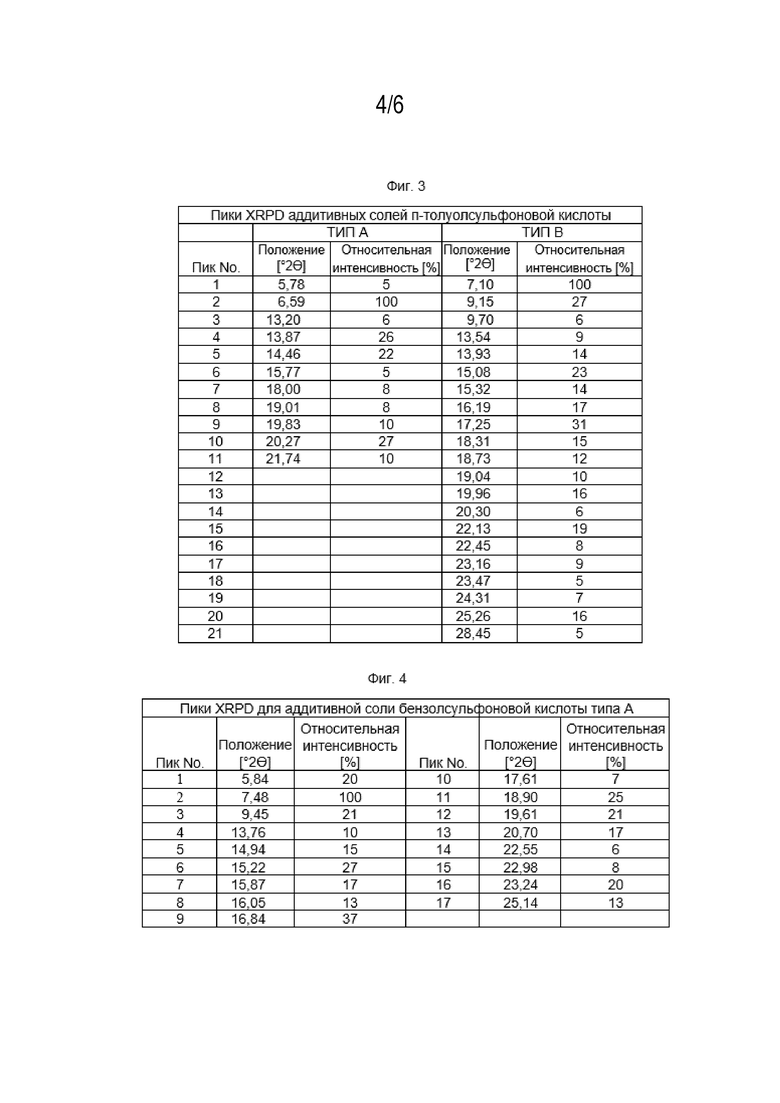
[014] Фиг. 3 представляет собой таблицу пиков рентгеновской порошковой дифрактометрии с относительной интенсивностью более 5% для описанных здесь пластинчатых форм добавления p-толуолсульфоновой кислоты типа A и B.
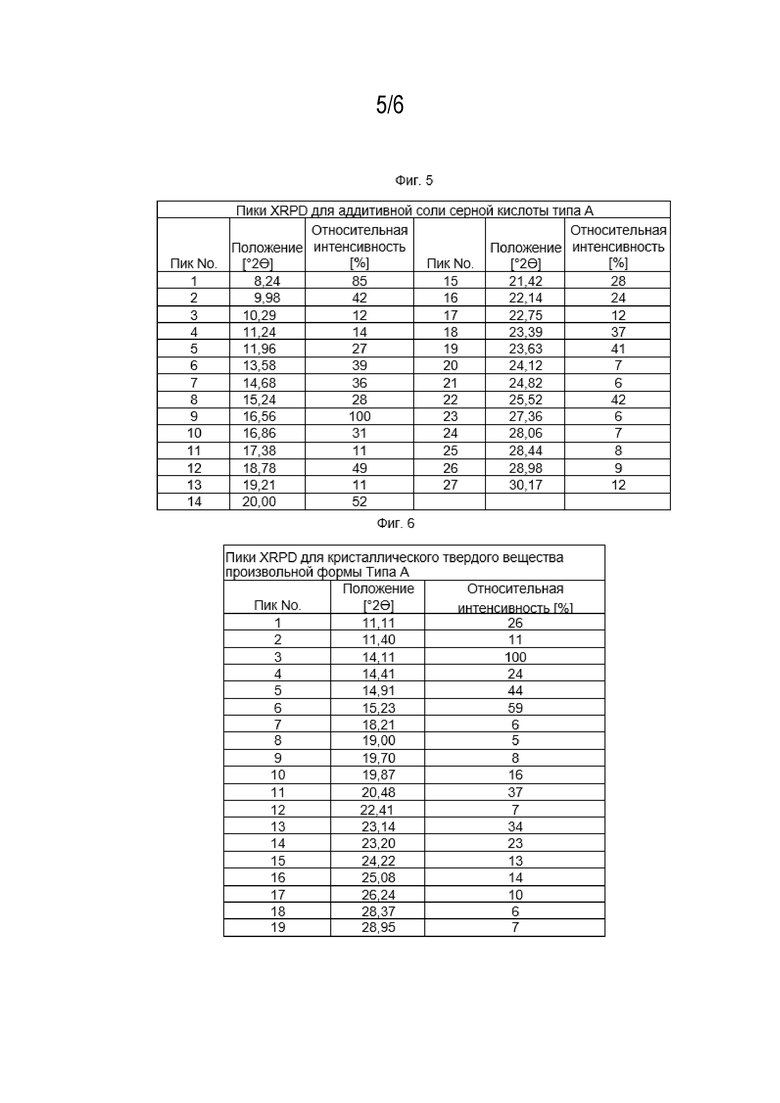
[015] Фиг. 4 представляет собой таблицу пиков рентгеновской порошковой дифрактометрии с относительной интенсивностью более 5% для формы аддитивной соли бензолсульфоновой кислоты типа А, описанной в данном документе.
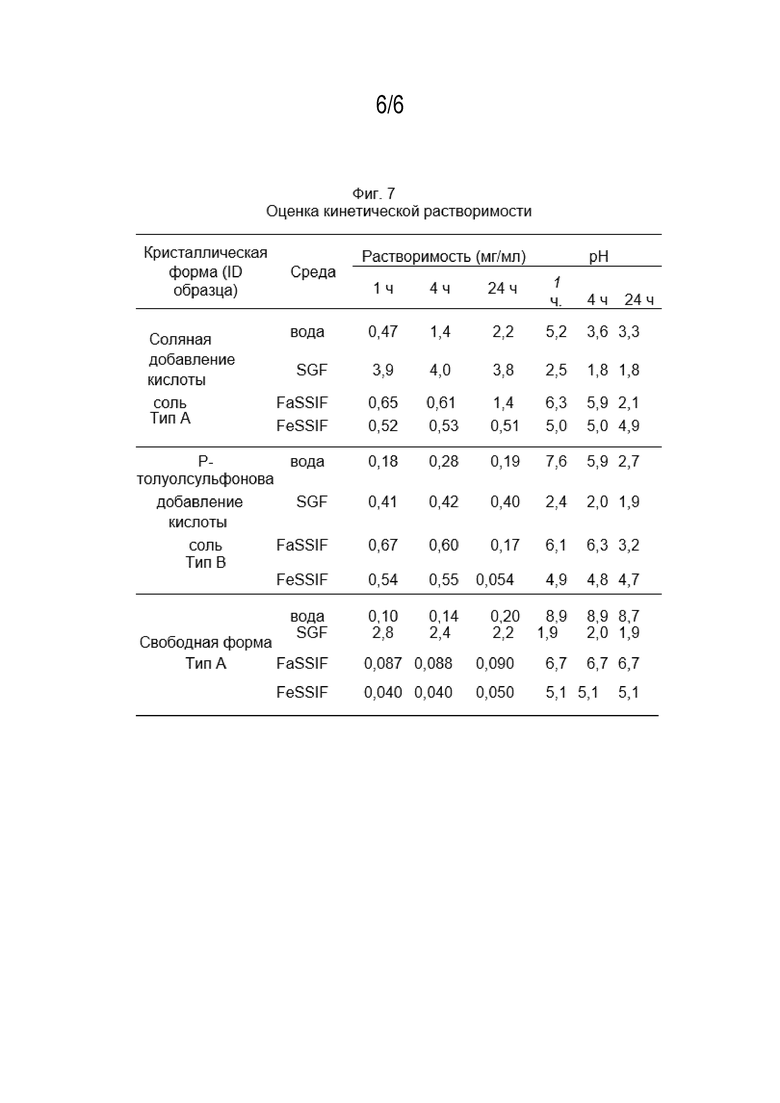
[016] Фиг. 5 представляет собой таблицу пиков дифракции рентгеновской порошковой дифрактометрии с относительной интенсивностью более 5% для формы аддитивной соли серной кислоты типа А, описанной в данном документе.
[017] Фиг. 6 представляет собой таблицу пиков дифракции рентгеновской порошковой дифрактометрии с относительной интенсивностью более 5% для твердой формы кристаллической свободной формы типа А, описанной в данном документе.
[018] Фиг. 7 представляет собой таблицу оценок кинетической растворимости формы аддитивной соли соляной кислоты типа A, формы аддитивной соли p-толуолсульфоновой кислоты типа B и кристаллической твердой формы типа A в свободной форме, описанных в данном документе.
Подробное описание
[019] Настоящее изобретение относится к солям и твердым формам соединений и композиций, которые способны модулировать активность бромодоменов семейства CBP/p300. В документе описаны способы лечения, предотвращения или облегчения заболевания или расстройства, в которых бромодомены CBP/p300 играют роль терапевтически эффективного количества соединения Формулы (I), (II) или группы А или его фармацевтически приемлемой соли путем введения пациенту, нуждающемуся в этом. Способы настоящего изобретения можно использовать при лечении различных заболеваний и расстройств, зависящих от бромодомена CBP/p300, путем ингибирования активности бромодоменов CBP/p300. Ингибирование бромодоменов CBP/p300 обеспечивает новый подход к лечению заболеваний, включая, но не ограничиваясь, рак.
[020] Солевые и кристаллические твердые формы лекарственных соединений могут иметь несколько явных преимуществ перед аморфными или нетвердыми формами, включая: 1) повышенную растворимость, скорость растворения и биодоступность для плохо растворимых соединений, 2) пониженную растворимость для использования в составах с пролонгированным высвобождением, пониженным Оствальдовским созреванием или для достижения маскировки вкуса для особенно растворимых соединений, 3) улучшенные физические свойства, такие как температура плавления, гигроскопичность и механические свойства, 4) улучшенную химическую стабильность и совместимость с фармацевтическими наполнителями, и 5) улучшенную чистоту соединения, хиральное разрешение отдельных стереоизомеров и фильтруемость.
[021] В некоторых вариантах осуществления изобретения предложены новые соединения-ингибиторы CBP. Если не указано иное, «соединение-ингибитор CBP» в контексте настоящего описания относится к соединению, имеющему определяемое значение CBP IC50 1 микромоль или ниже при тестировании в соответствии с протоколом биохимического анализа HTRF из Примера 3.
[022] Если здесь не указано иное, все изомерные формы указанных химических соединений представлены настоящим изобретением, включая их смеси (например, S, R и рацемическая ориентация в каждом хиральном центре). Если соединение содержит двойную связь, заместитель может иметь E или Z конфигурацию. Если соединение содержит дизамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию. Все таутомерные формы также предназначены для включения.
[023] Соединения формулы (I), (II) и Гуппы A, если не указано иное, могут существовать в их таутомерной форме. Все такие таутомерные формы рассматриваются здесь как часть настоящего описания.
[024] Соединения формулы (I), (II) и Группы A, если не указано иное, могут содержать один или несколько стереоцентров и, следовательно, существовать в различных стереоизомерных формах. Подразумевается, что, если не указано иное, все стереоизомерные формы соединений формулы (I), (II) и Группы A, а также их смеси, включая рацемические смеси, составляют часть настоящего описания. Кроме того, настоящее описание охватывает все геометрические и позиционные изомеры. Например, если соединение формулы (I), (II) или Группы A включает двойную связь или конденсированное кольцо, как цис-, так и транс-формы, а также смеси, охватываются объемом настоящего описания. Каждое раскрытое здесь соединение включает все энантиомеры, которые соответствуют общей структуре соединения. Соединения могут быть в рацемической или энантиомерно чистой формы или в любой другой форме с точки зрения стереохимии. Результаты анализа могут отражать данные, собранные для рацемической формы, энантиомерно чистой формы или любой другой формы с точки зрения стереохимии.
[025] Смеси диастереомеров можно разделить на отдельные диастереомеры на основе их физико-химических различий методами, хорошо известными квалифицированным специалистам в данной области техники, такими как, например, хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь реакцией с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделением диастереомеров и превращением (например, гидролизом) отдельных диастереомеров в соответствующие чистые энантиомеры. Кроме того, некоторые из соединений формулы (I), (II) или Группы A могут быть атропоизомерами (например, замещенными биарилами) и рассматриваются как часть этого описания. Энантиомеры также можно разделить с помощью хиральной колонки для HPLC.
[026] Соединения формулы (I), (II) или Группы A могут образовывать кислотно-аддитивные соли, которые могут быть фармацевтически приемлемыми солями. Описание также включает фармацевтические композиции, содержащие одно или несколько соединений, как описано в данном документе, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В некоторых вариантах осуществления изобретения фармацевтические композиции, описанные в настоящем документе, могут быть предоставлены в виде стандартной лекарственной формы (например, капсулы, таблетки или тому подобное). В некоторых вариантах осуществления изобретения фармацевтические композиции, описанные в настоящем документе, могут быть представлены в пероральной лекарственной форме. В некоторых вариантах осуществления изобретения пероральная лекарственная форма соединения формулы (I), (II) или Группы A может представлять собой капсулу. В некоторых вариантах осуществления изобретения пероральная лекарственная форма соединения формулы (I), (II) или Группы A представляет собой таблетку. В некоторых вариантах осуществления изобретения пероральная лекарственная форма содержит один или несколько наполнителей, разрыхлителей, смазывающих веществ, скользящих веществ, антиадгезивов и/или антистатиков. В некоторых вариантах осуществления изобретения пероральную лекарственную форму получают путем сухого смешивания. В некоторых вариантах осуществления изобретения пероральная лекарственная форма представляет собой таблетку, которую получают путем сухого гранулирования.
Соединение-ингибитор СВР настоящего изобретения можно дозировать на терапевтически эффективном уровне.
Соединения описания
[027] Настоящее изобретение относится к твердым формам соединений или их фармацевтически приемлемых солей или изомеров, способных модулировать бромодомены семейства CBP/p300, которые полезны для лечения заболеваний и нарушений, связанных с модуляцией бромодоменов семейства CBP/p300. Описание также относится к твердым формам соединений или их фармацевтически приемлемых солей или изомеров, которые полезны для ингибирования бромодоменов семейства CBP/p300.
[028] В одном аспекте изобретение относится к твердой форме соединения Формулы (I)
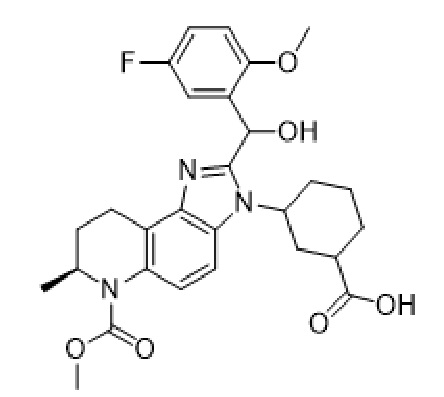 (I),
(I),
и их энантиомерам, гидратам, сольватам, изомерам и таутомерам и кислотно-аддитивным солям вышеперечисленного.
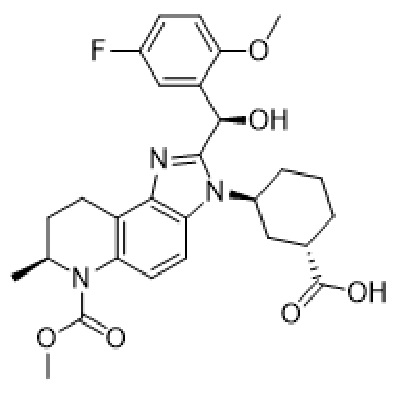
[029] В некоторых вариантах осуществления изобретение относится к твердым и солевым формам, выбранным из одной или нескольких из Группы A:
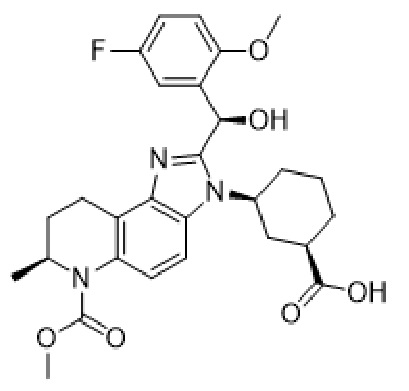 (A1)
(A1) 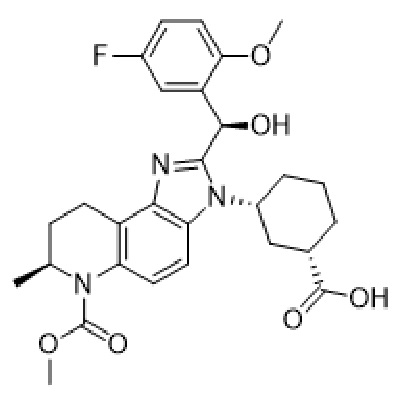 (A2)
(A2)
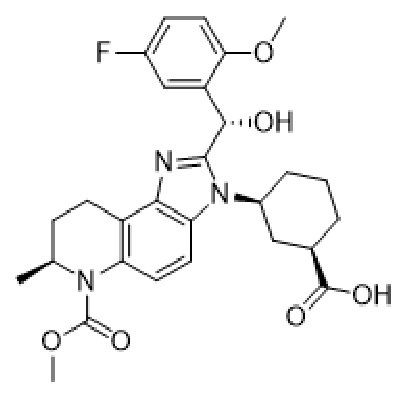 (A3)
(A3) 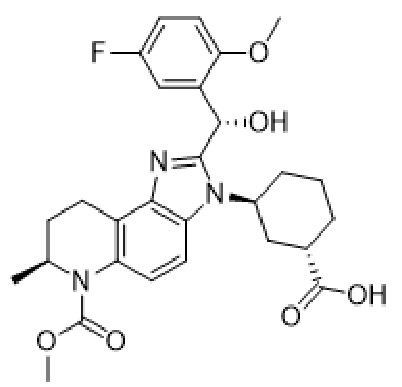 (A4)
(A4)
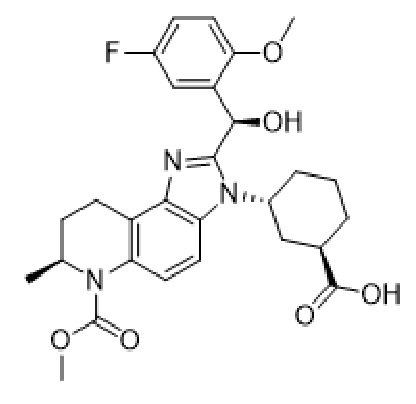 (A5)
(A5) 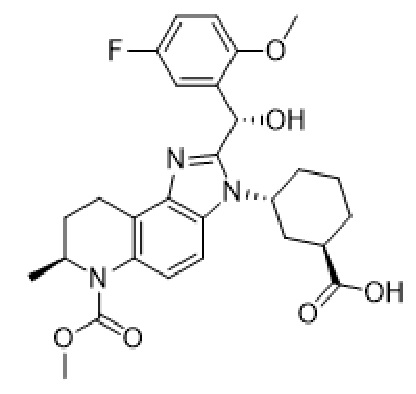 (A6)
(A6)
 (A7)
(A7) 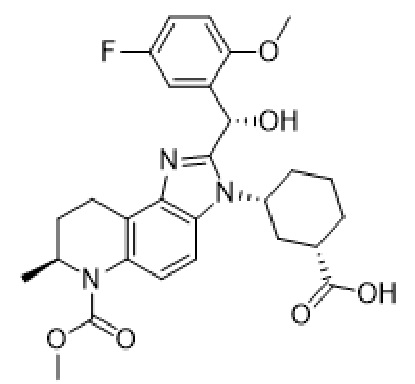 (A8)
(A8)
и их энантиомерам, гидратам, сольватам и таутомерам и кислотно-аддитивным солям вышеперечисленного.
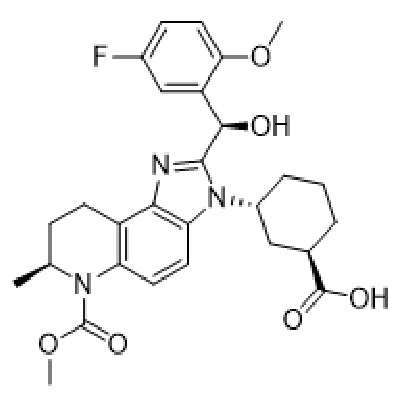
[030] В предпочтительном варианте осуществления настоящее описание относится к твердым и солевым формам Формулы (II):
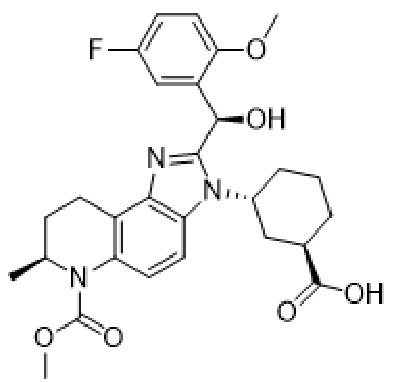 (II),
(II),
и их энантиомерам, гидратам, сольватам и таутомерам, а также их кислотно-аддитивным солям.
[031] Кислотно-аддитивные соли вышеуказанного могут быть получены в результате добавления соляной кислоты (HCl), р-толуолсульфоновой кислоты, бензолсульфоновой кислоты и серной кислоты (H2SO4). В отсутствие кислотно-аддитивной соли соединения упоминаются как форма свободного основания. Кислотно-аддитивные соли и формы свободного основания могут быть кристаллическими.
[032] В некоторых вариантах осуществления изобретения форма свободного основания может быть не аморфной. Доступ к аморфной форме свободного основания получают посредством синтеза, представленного в Примере 1. Этот химический синтез дает соединение формулы (II) значительной чистоты. В конечном синтетическом продукте присутствуют только следовые количества стереоизомерных загрязнителей группы A, за исключением соединения A6, которое присутствует в измеримых количествах. Аморфная форма свободного основания получается с чистотой, превышающей 99%, как определено анализом HPLC, как показано в примерах ниже.
[033] Что касается твердых форм аддитивной соли соляной кислоты, заявитель обнаружил три кристаллические формы (далее называемые типами A, B и C), две кристаллические формы аддитивной соли p-толуолсульфоновой кислоты (далее именуемые типами A и B), одну кристаллическую форму аддитивной соли бензолсульфоновой кислоты (далее именуемая типом A), одну форму аддитивной соли серной кислоты (далее именуемая типом A) и кристаллическую форму свободного основания (далее именуемая свободной формой типа A) соединений Формулы (I).
Формы соли присоединения HCl
Тип А
[034] Кристаллический тип А аддитивной соли соляной кислоты характеризуется порошковой рентгеновской дифрактограммой (XRPD), имеющей по крайней мере три примерных положения пиков (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 7,27, 8,98, 10,60, 15,60 и 23,93, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 2 содержит список пиков порошковой рентгеновской дифрактограммы аддитивной соли соляной кислоты типа А, имеющей относительную интенсивность больше или равную 5%.
[035] Он также характеризуется эндотермическим пиком с начальной температурой около 230°C, измеренной с помощью дифференциальной сканирующей калориметрии (DSC). Соль HCl типа A также характеризуется потерей веса приблизительно 1,1% при температурах до 170°C, измеренными термогравиметрическим анализом. Соль HCl типа A также характеризуется гигроскопичностью, о чем свидетельствует поглощение воды 6,3% при относительной влажности до 80%, измеренное с помощью графиков изотермы динамической сорбции паров. В другом варианте осуществления изобретения аддитивная соль соляной кислоты типа A характеризуется данными кинетической растворимости, приведенными ниже в Примере 2. Соль присоединения соляной кислоты типа A может быть стабильной в течение как минимум двух недель при температуре до 40°C и относительной влажности до 75%.
[036] Кристаллический тип А аддитивной соли соляной кислоты является ангидридным (безводным).
[037] Кристаллический тип А аддитивной соли соляной кислоты может быть получен растворением формы свободного основания в органическом растворителе, добавлением соляной кислоты, нагреванием полученного раствора с последующим охлаждением, как предусмотрено в Примере 1.b.
Тип B
[038] Кристаллический тип B аддитивной соли соляной кислоты характеризуется порошковой рентгеновской дифрактограммой (XRPD), имеющей по крайней мере три примерных положения пиков (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 10,23, 18,72, 23,03, 24,77 и 28,03, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 2 содержит список пиков порошковой рентгенограммы аддитивной соли соляной кислоты типа B, имеющей относительную интенсивность больше или равную 5%.
[039] Он также характеризуется эндотермическими пиками с начальной температурой около 139°C и 232°C и экзотермическим пиком около 104°C, измеренными с помощью DSC. Соль HCl типа B также характеризуется потерей веса приблизительно 20% при температурах до 200°C, измеренными термогравиметрическим анализом.
[040] В некоторых вариантах осуществления изобретения кристаллический тип B аддитивной соли соляной кислоты представляет собой гидрат или сольват.
[041] Кристаллический тип B аддитивной соли соляной кислоты может быть получен растворением аддитивной соли соляной кислоты типа A в смеси органических растворителей, а затем нагреванием полученного раствора с последующим охлаждением и медленным испарением при комнатной температуре, как предусмотрено в Примере 1.c.
Тип C
[042] Кристаллический тип C аддитивной соли соляной кислоты характеризуется порошковой рентгеновской дифракторгаммой (XRPD), имеющей по крайней мере три примерных положения пика (степени 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 7,05, 19,84, 21,09, 24,98 и 31,44, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 2 содержит список пиков порошковой рентгенограммы аддитивной соли соляной кислоты типа B, имеющей относительную интенсивность больше или равную 5%.
[043] Он также характеризуется эндотермическими пиками, имеющими начальную температуру примерно при 83°C, 143°C и 179°C, и экзотермическим пиком примерно при 230°C, измеренными с помощью DSC. Соль HCl типа C также характеризуется потерей веса приблизительно 9,4% при температурах до 180°C, измеренными термогравиметрическим анализом.
[044] В некоторых вариантах осуществления изобретения кристаллический тип C аддитивной соли соляной кислоты представляет собой гидрат или сольват.
[045] Кристаллический тип B аддитивной соли соляной кислоты может быть получен растворением аддитивной соли соляной кислоты типа A в смеси органических растворителей, а затем нагреванием полученного раствора с последующим охлаждением и медленным испарением при комнатной температуре, как предусмотрено в Примере 1.d.
Соль присоединения р-толуолсульфоновой кислоты
Тип А
[046] Кристаллический тип A аддитивной соли р-толуолсульфоновой кислоты характеризуется порошковой рентгеновской дифрактограммой (XRPD), имеющей по крайней мере три примерных положения пиков (степени 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 6,59, 13,20, 14,46, 18,00 и 21,74, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 3 содержит список пиков порошковой рентгенограммы аддитивной соли р-толуолсульфоновой кислоты типа А, имеющей относительную интенсивность больше или равную 5%.
[047] Он также характеризуется эндотермическими пиками, имеющими начальную температуру около 74°C и 200°C, измеренными с помощью DSC. Соль присоединения р-толуолсульфоновой кислоты типа A также характеризуется потерей веса приблизительно 1,0% при температурах до 180°C, измеренными термогравиметрическим анализом.
[048] Кристаллическая аддитивная соль р-толуолсульфоновой кислоты типа A может быть получена растворением формы свободного основания в органическом растворителе, добавлением р-толуолсульфоновой кислоты и нагреванием полученного раствора с последующим охлаждением, как предусмотрено в Примере 1.e.
Тип B
[049] Кристаллический тип B аддитивной соли р-толуолсульфоновой кислоты характеризуется порошковой рентгеновской дифрактограммой (XRPD), имеющей по крайней мере три приблизительных положения пиков (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 7,10, 9,15, 15,08, 16,19, 17,25, 18,31 и 21,13, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 3 содержит список пиков порошковой рентгенограммы аддитивной соли р-толуолсульфоновой кислоты типа B, имеющей относительную интенсивность больше или равную 5%.
[0001] Он также характеризуется эндотермическим пиком с начальной температурой около 225°C, измеренной с помощью DSC. Соль присоединения р-толуолсульфоновой кислоты типа B также характеризуется потерей веса приблизительно 1,3% при температурах до 180°C, измеренными термогравиметрическим анализом. Соль присоединения р-толуолсульфоновой кислоты типа B также характеризуется гигроскопичностью, о чем свидетельствует водопоглощение 2,1% при относительной влажности до 80%, измеренное с помощью графиков изотермы динамической сорбции паров. Аддитивная соль р-толуолсульфоновой кислоты типа B характеризуется данными кинетической растворимости, приведенными ниже в Примере 2. Соль присоединения р-толуолсульфоновой кислоты типа B стабильна в течение не менее двух недель при температуре до 40°C и относительной влажности до 75%.
[050] Кристаллическая аддитивная соль р-толуолсульфоновой кислоты типа B может быть получена растворением формы свободного основания в органическом растворителе, добавлением р-толуолсульфоновой кислоты, нагреванием полученного раствора с последующим охлаждением, как предусмотрено в Примере 1.f.
[051] Кристаллический тип B аддитивной соли р-толуолсульфоновой кислоты является ангидридным (безводным).
Аддитивная cоль бензолсульфоновой кислоты типа A
[052] Кристаллический тип А аддитивной соли бензолсульфоновой кислоты характеризуется рентгеновской порошковой дифрактограммой (XRPD), имеющей по крайней мере три приблизительных положения пиков (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 5,84, 7,48, 9,45, 16,84, 18,90, 19,61, 20,70 и 25,14, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 4 содержит список пиков порошковой рентгенограммы аддитивной соли бензолсульфоновой кислоты типа А, имеющей относительную интенсивность больше или равную 5%.
[053] Он также характеризуется эндотермическим пиком с начальной температурой около 208°C, измеренной с помощью DSC. Аддитивная соль бензолсульфоновой кислоты типа A также характеризуется незначительной потерей веса при температурах до 210°C, измеренными термогравиметрическим анализом.
[054] Кристаллическая аддитивная соль бензолсульфоновой кислоты типа А может быть получена растворением формы свободного основания в органическом растворителе, добавлением бензолсульфоновой кислоты, нагреванием полученного раствора с последующим охлаждением, как предусмотрено в Примере 1.g.
Аддитивная соль серной кислоты Типа A
[055] Кристаллический тип A аддитивной соли серной кислоты характеризуется порошковой рентгеновской дифрактограммой (XRPD), имеющей по крайней мере три примерных положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 8,24, 9,98, 13,58, 16,87, 18,78, 20,00 и 25,52, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 5 содержит список пиков порошковой рентгенограммы аддитивной соли серной кислоты типа А, имеющей относительную интенсивность больше или равную 5%.
[056] Он также характеризуется эндотермическими пиками, имеющими начальную температуру около 140°C и 188°C, измеренными с помощью DSC. Аддитивная соль серной кислоты типа А также характеризуется незначительной потерей веса при температурах до 200°C, измеренными термогравиметрическим анализом.
[057] Кристаллическая аддитивная соль серной кислоты типа A может быть получена растворением формы свободного основания в органическом растворителе, добавлением серной кислоты, нагреванием полученного раствора с последующим охлаждением, как предусмотрено в Примере 1.h.
Кристаллическая форма свободного основания Типа A
[058] Кристаллический тип формы свободного основания характеризуется порошковой рентгенограммой (XRPD), имеющей по крайней мере три примерных положения пиков (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 11,11, 14,11, 18,21, 20,48 и 26,24, когда XRPD собирают от примерно 3 до примерно 40 градусов 2θ. Фиг. 6 содержит список пиков порошковой рентгенограммы формы свободного основания типа А, имеющей относительную интенсивность больше или равную 5%.
[059] Он также характеризуется эндотермическим пиком с начальной температурой около 209°C, измеренной с помощью DSC. Форма свободного основания типа A также характеризуется незначительной потерей веса при температурах до 210°C, измеренными термогравиметрическим анализом. Свободное основание типа A характеризуется данными кинетической растворимости, приведенными ниже в Примере 2.
[060] Кристаллическая форма свободного основания типа A может быть получена растворением формы свободного основания в органическом растворителе при кипячении с обратным холодильником и последующим охлаждением полученного раствора, как предусмотрено в Примере 1.i.
[061] В некоторых вариантах осуществления изобретения соли и кристаллические формы, используемые при приготовлении фармацевтических композиций, имеют чистоту по меньшей мере 95%, как было оценено анализом HPLC.
Метод синтеза соединений
[062] Соединения настоящего описания могут быть получены различными способами, включая стандартные химические методы. Подходящие пути синтеза показаны в примерах, приведенных ниже.
[063] Соединения настоящего изобретения, то есть соединения Формулы (I), (II) или Группы A, или их фармацевтически приемлемые соли, могут быть получены способами, известными в области органического синтеза, которые частично изложены в схемах синтеза изображенных в примерах. В схемах, описанных ниже, хорошо понятно, что защитные группы для чувствительных или реакционноспособных групп используются там, где это необходимо, в соответствии с общими принципами или химическим составом. С защитными группами манипулируют согласно стандартным методам органического синтеза (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). Эти группы удаляют на удобной стадии синтеза соединения с использованием методов, которые очевидны для квалифицированных специалистов в данной области техники. Процессы выбора, а также условия реакции и порядок их выполнения должны соответствовать получению соединений Формулы (I), (II) или Группы A.
[064] Квалифицированные специалисты в данной области техники поймут, что стереоцентры существуют в соединениях Формулы (I), (II) или Группы A. Соответственно, настоящее описание включает оба возможных стереоизомера (если иное не указано и/или не определено в синтезе) и включает не только рацемические соединения, но также отдельные энантиомеры и/или диастереомеры. Если не указано иное, когда соединение желательно в виде единственного энантиомера или диастереомера, оно может быть получено стереоспецифическим синтезом или разделением конечного продукта или любого удобного промежуточного соединения. На разделение конечного продукта, промежуточного продукта или исходного материала можно повлиять любым подходящим способом, известным в данной области. См., например, «Stereochemistry of Organic Compounds» by E.L. Eliel, S.H. Wilen, and L.N. Mander (Wiley-lnterscience, 1994).
Способы применения описанных соединений
[065] Один аспект настоящего описания относится к соединению Формулы (I), (II) или Группы A для применения в медицине. Другой аспект настоящего описания относится к способу модуляции одного или нескольких бромодоменов семейства CBP/p300, который включает введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы (I), (II) или группы А. Другой аспект настоящего описания относится к способу ингибирования одного или нескольких бромодоменов семейства CBP/p300, который включает введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы (I), (II), или Группы A. В другом аспекте настоящее изобретение относится к способу ингибирования одного или нескольких бромодоменов семейства CBP/p300, который включает введение пациенту, который в этом нуждается, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I), (II) или Группы А.
[066] Соединения-ингибиторы CREB связывающего белка (CBP) могут быть использованы при разработке фармацевтических композиций, подходящих для лечения некоторых родственных форм рака. Соединения-ингибиторы CBP полезны для лечения болезненных состояний, которые реагируют на ингибирование CBP. CBP и EP300 (p300) - это тесно связанные многодоменные белки, которые действуют как коактиваторы транскрипции. Они несут ацетил-лизинсвязывающие бромодомены, которые придают этим белкам функцию каркаса или позиционирования, и оказалось, что они подходят для создания низкомолекулярных ингибиторов их биологической функции. Эти паралоги очень гомологичны на аминокислотном уровне и имеют много перекрывающихся функций. Это гистоновые ацетилтрансферазы (HAT), которые катализируют посттрансляционную модификацию гистоновых и негистоновых белков. Как бромодомен, несущий HAT, эти белки функционируют как эпигенетические читатели и писатели. Негистоновые белковые субстраты CBP/p300 состоят из множества факторов транскрипции, включая ядерные рецепторы гормонов, такие как рецептор андрогенов (AR). CBP/p300 действуют как коактиваторы передачи сигналов AR путем ацетилирования AR, которое активирует его транскрипционную активность и способствует стабильности его белка. Кроме того, они ацетилируют гистон H3 по лизину 27 (Ac-H3K27), чтобы обеспечить сайт стыковки для бромодомена, тем самым обеспечивая каркас для соединения ядерного рецептора с базальным аппаратом транскрипции. Ацетилирование гистона приводит к образованию на хроматине среды, допускающей транскрипцию. Таким образом, локализация CBP/p300 в AR-зависимых суперэнхансерах приводит к увеличению локализованного Ac-H3K27, что дополнительно увеличивает транскрипцию в этих локусах.
ПРИМЕРЫ
Материалы
[067] Если не указано иное, все материалы были получены от коммерческих поставщиков и использовались без дополнительной очистки. Безводные растворители были получены от Sigma-Aldrich (Милуоки, Висконсин) и использовались напрямую. Все реакции с участием реагентов, чувствительных к воздуху или влаге, проводились в атмосфере азота, а все реакции с использованием микроволнового облучения проводились на приборе Biotage Initiator EXP EU.
[068] Если не указано иное, масс-триггерная очистка HPLC и/или чистота и масс-спектральные данные с низким разрешением были измерены с использованием: (1) Системы ультраэффективной жидкостной хроматографии (UPLC) (Waters Acquity UPLC с устройством для пробоотбора и масс-спектрометром Waters Micromass ZQ) с УФ-детектированием при 220 нм и режимом низкорезонансных электрораспылительных положительных ионов (ESI) (Колонка: Acquity UPLC BEH C18 1,7 мкм 2,1×50 мм; градиент: 5-100% Растворитель B (95/5/0,09%: Ацетонитрил/вода/муравьиная кислота) в Растворителе A (95/5/0,1%: 10 мМ формиат аммония/ацетонитрил/муравьиная кислота) в течение 2,2 мин, затем 100-5% Растворитель B в Растворителе A в течение 0,01 мин, затем выдержка при 5% Растворителе B в Растворителе A в течение 0,29 мин) или (2) Системы высокоэффективной жидкостной хроматографии Waters HT2790 Alliance (HPLC) (одноквадратный масс-спектрометр Waters 996 PDA и Waters ZQ) с УФ-детектированием при 220 и 254 нм и режимом низкорезонансной ионизации электрораспылением (положительный/отрицательный) (ESI) (Колонка: XBridge Фенил или C18, 5 мкм 4,6Ч50 мм; градиент: 5-95% Растворителя B (95% метанола/5% воды с 0,1% муравьиной кислоты) в Растворителе A (95% воды/5% метанола с 0,1% муравьиной кислоты) в течение 2,5 минут, затем выдерживают при 95% Растворителе B в Растворителе A в течение 1 мин (только МS с низким разрешением).
Инструменты
[069] В примерах, которые следуют ниже, порошковая рентгеновская дифрактометрия (XRPD) выполнялась с использованием Panalytical X’Pert3 Powder XRPD на держателе Si с нулевым фоном. Положение 2и было откалибровано по эталонному диску Panalytical Si. Используемые параметры XRPD перечислены на Фиг. 1.A.
[070] Данные термогравиметрического анализа (ТGА) собирали с использованием TA Q500 и Q550 от TA Instruments. Дифференциальную сканирующую калориметрию (DSC) выполняли с использованием TA Q2000 от TA Instruments. DSC калибровали с использованием эталонного индия, а ТGА калибровали с использованием эталонного стандарта никеля. Подробные используемые параметры перечислены на Фиг. 1.B.
[071] Данные о динамической сорбции паров (DVS) были собраны с помощью SMS DVS Intrinsic от Surface Measurement Systems. Параметры для теста DVS перечислены на Фиг. 1.C.
[072] Оценка чистоты лекарственного соединения методом HPLC: Образцы лекарственного соединения для анализа готовили при концентрациях 0,2 мг/мл в смеси воды и ацетонитрила 70:30. Затем образцы анализировали на приборе для жидкостной хроматографии Waters Alliance e2695, оборудованном масс-спектрометром Waters QDa и детектором с фотодиодной матрицей Waters 2998. Параметры метода хроматографии описаны на Фиг. 1.D.
Определения, используемые в следующих схемах и в других местах здесь:
Ac-H3K27 - ацетилированный гистон H3 по остатку лизина 27.
AR - рецептор андрогенов .
BSA - бычий сывороточный альбумин.
CBP - белок, связывающий циклический AMP-чувствительный элемент (также известный как KAT3A).
DMSO - диметилсульфоксид.
DSC - дифференциальная сканирующая калориметрия.
DVS - динамическая сорбция паров.
ES - Электрораспылительная ионизация.
EtOH - этанол.
FRET - Форстеровский резонансный перенос энергии.
ч - час.
H2SO4 - серная кислота.
HAT - гистонацетилтрансфераза.
HATU - 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизурония гексафторфосфат.
HCl - соляная кислота.
Hex - гексаны.
ВЭЖХ - высокоэффективная жидкостная хроматография.
HTRF - высокая пропускная способность с временным разрешением FRET.
IC50 - полумаксимальная ингибирующая концентрация.
L - литр.
LCMS - жидкостная хроматография/масс-спектрометрия.
M - молярный.
мл - миллилитр.
ммоль - миллимоль.
мг - миллиграмм.
МГц - мегагерц.
МС - масс-спектрометрия.
m/z - отношение масса/заряд.
NH4Cl - хлорид аммония.
нм - нанометр.
ЯМР - ядерный магнитный резонанс.
p300 EP300 - (также известный как KAT3B).
ppm - частей на миллион.
TGA - термогравиметрический анализ.
UPLC - сверхэффективная жидкостная хроматография.
УФ - ультрафиолет.
XRPD - рентгеновская порошковая дифрактометрия.
ZnI2 - иодид цинка.
Термин «органический растворитель» хорошо известен квалифицированным специалистам в данной области техники, и может включать химические растворители, такие как ацетон, ацетонитрил, бензол, хлороформ, 1,4-диоксан, диэтиловый эфир, дихлорметан, диметилацетамид, N, N-диметилформамид, диметилсульфоксид, этанол, этилацетат, гексаны, изопропанол, метанол, N-метилпиролидон, пиридин, тетрагидрофуран, толуол, воду, помимо тех, которые явно не названы.
Пример 1 - Получение соединений, твердых форм и солей
Пример 1.а - Получение аморфного свободного основания+формы
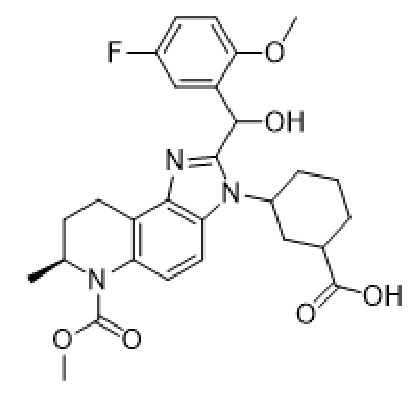
(1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (1);
(1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота (1')
Синтез промежуточного соединения 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты:
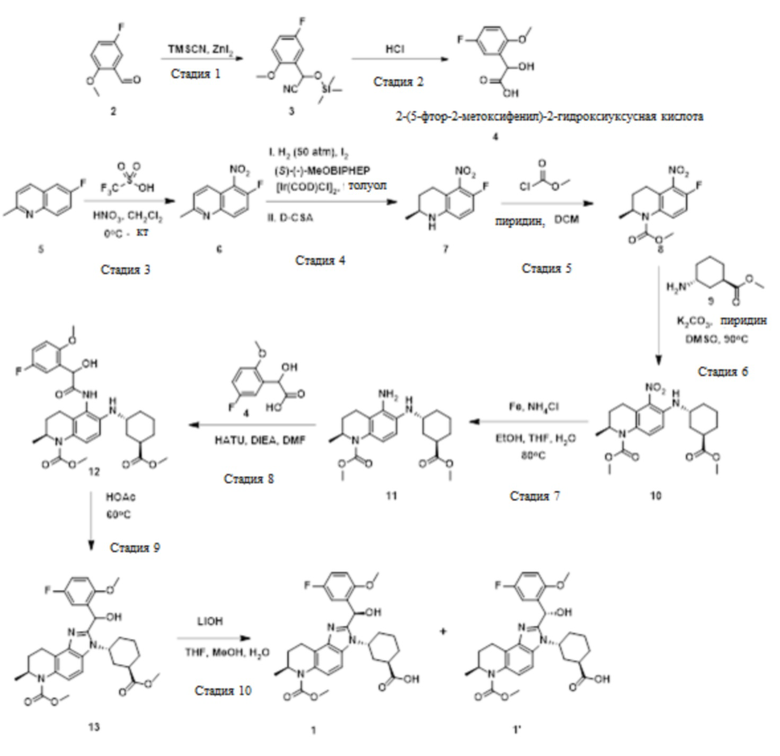
Этап 1. 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрил
[073] Раствор ZnI2 (1,6 мг, 0,01 ммоль), 5-фтор-2-метоксибензальдегида (1,54 г, 9,99 ммоль) в триметилсиланкарбонитриле (1,5 мл, 11,25 ммоль) перемешивали в течение 1 ч при комнатной температуре. Полученную смесь концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрила в виде белого твердого вещества ( 2,0 г, 79%).
Этап 2. 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусная кислота
[074] Раствор 2-(5-фтор-2-метоксифенил)-2-[(триметилсилил)окси]ацетонитрила (1,50 г, 5,92 ммоль) в соляной кислоте (10 мл, 12 М). Полученный раствор перемешивали в течение 1 часа при 25°C, а затем перемешивали в течение 2 часов при 70°C. Реакционную смесь охлаждали и концентрировали под вакуумом. Неочищенный продукт очищали обращенно-фазовой хроматографией (Колонка: C18; Подвижная фаза, A: вода (содержащая 0,05% TFA) и B: ACN (от 5% до 20% за 30 мин); Детектор, УФ 254 нм) с получением 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты в виде белого твердого вещества (1,10 г, 93%).
Этап 3. 6-фтор-2-метил-5-нитрохинолин
[075] Раствор трифторметансульфоновой кислоты (82,0 мл, 0,923 моль) в HNO3 (19,6 мл, 0,437 моль) перемешивали в течение 20 мин при 0°C. После этого добавляли 6-фтор-2-метилхинолин (50,0 г, 0,310 моль) в дихлорметане (300 мл) при 0°C. Полученную смесь перемешивали 15 ч при комнатной температуре (25°C). Реакционную смесь разбавляли водой (300 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюируя смесью 1:4 этилацетат/петролейный эфир) с получением 6-фтор-2-метил-5-нитрохинолина в виде светло-желтого твердого вещества (60,0 г, 94%). ЖХМС (ES, m/z): 207 [M+H]+.
Этап 4. (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин
[076] Раствор (S)-(-)-MeO-BIPHEP (1,03 г, 1,77 ммоль), димера хлор(1,5-циклооктадиен)иридия(I) (538 мг, 0,80 ммоль) в толуоле (100 мл) перемешивали в течение 30 мин при комнатной температуре (25°C) в атмосфере азота. После этого добавляли I2 (410 мг, 1,62 ммоль), 6-фтор-2-метил-5-нитрохинолин (33,0 г, 0,160 моль) в толуоле (100 мл). Полученную смесь перемешивали в течение 20 ч при комнатной температуре (25°C) в атмосфере водорода (50 атм). Полученную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением неочищенного продукта (35,0 г). Неочищенный продукт растворяли в этилацетате (230 мл) с последующим добавлением D-камфорсульфоновой кислоты (36,9 г, 0,158 моль). Полученный раствор перемешивали в течение 1 ч при 60°C, а затем охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием и промывали этилацетатом (120 мл). Твердые вещества растворяли в воде (50 мл). Значение pH раствора доводили до 8 с помощью бикарбоната натрия (насыщенный водный раствор). Полученный раствор экстрагировали этилацетатом (3×120 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина в виде красного твердого вещества (25,5 г, 76%). ЖХМС (ES, m/z): 211 [M+H]+.
Этап 5. метил (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[077] Раствор (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолина (25,3 г, 0,120 моль), пиридина (39,0 мл, 0,484 моль), метилкарбонохлоридата (18,7 мл, 0,242 моль) в дихлорметане (150 мл) перемешивали в течение 3 часов при комнатной температуре (25°C). Реакционную смесь промывали 1 М соляной кислотой (2×70 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением метил (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде желтого твердого вещества (29,8 г, 92%). ЖХМС (ES, m/z): 269 [M+H]+.
Этап 6. метил (2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[078] Раствор метил (2S)-6-фтор-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (29,6 г, 0,110 моль), пиридина (29,6 мл, 0,368 моль), карбоната калия (30,5 г, 0,220 моль), метил (1R,3R)-3-аминоциклогексан-1-карбоксилата (25,6 г, 162,84 ммоль) в ДМСО (270 мл) перемешивали в течение 15 ч при 90°C и затем охлаждали до комнатной температуры. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил (2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде красного масла (32 г, 72%). ЖХМС (ES, m/z): 406 [M+H]+.
Step 7. метил (2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[079] Раствор метил (2S)-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-5-нитро-1,2,3,4-тетрагидрохинолин-1-карбоксилата (31,0 г, 76,46 ммоль), NH4Cl (24,3 г, 454,28 ммоль), Fe (64,3 г, 1,15 моль) в тетрагидрофурана (300 мл), этанола (300 мл), воды (100 мл) перемешивали 1 ч при 80°C и затем охлаждали до комнатной температуры. Твердые вещества отфильтровывали. Полученный раствор разбавляли водой (300 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме, получая метил (2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат в виде темно-зеленого твердого вещества (27,5 г, 92%). ЖХМС (ES, m/z): 376 [M+H]+.
Этап 8. метил (2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилат
[080] Раствор 2-(5-фтор-2-метоксифенил)-2-гидроксиуксусной кислоты (240 мг, 1,20 ммоль), HATU (228 мг, 0,60 ммоль), метил (2S)-5-амино-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (150 мг, 0,40 ммоль), DIEA (0,19 мл, 1,20 ммоль) в N, N-диметилформамиде (10 мл) перемешивали в течение 1 ч при 25°C. Полученный раствор разбавляли H2O (10 мл). Полученный раствор экстрагировали этилацетатом (3×15 мл) и органические слои объединяли. Полученную смесь промывали рассолом (2×20 мл). Смесь сушили над безводным сульфатом натрия и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 3:2 этилацетат/петролейный эфир) с получением метил (2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в виде твердого вещества желтого цвета (180 мг, 81%). ЖХМС (ES, m/z): 558 [M+H]+.
Этап 9. метил (7S)-2-[(5-фтор-2-метоксифенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилат
[081] Раствор метил (2S)-5-[2-(5-фтор-2-метоксифенил)-2-гидроксиацетамидо]-6-[[(1R,3R)-3-(метоксикарбонил)циклогексил]амино]-2-метил-1,2,3,4-тетрагидрохинолин-1-карбоксилата (180 мг, 0,32 ммоль) в AcOH (8 мл) перемешивали в течение ночи при 60°C. Реакционную смесь охлаждали и концентрировали под вакуумом. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюируя смесью 1:1 этилацетат/петролейный эфир) с получением метил (7S)-2-[(5-фтор-2-метоксифенил) (гидрокси) метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H, 8H, 9H-имидазо[4,5-f]хинолин-6-карбоксилата в виде желтого твердого вещества (120 мг, 69%). ЖХМС (ES, m/z): 540 [M+H]+.
Этап 10. (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота; (1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновая кислота
[082] Раствор метил (7S)-2-[(5-фтор-2-метоксифенил)(гидрокси)метил]-3-[(1R,3R)-3-(метоксикарбонил)циклогексил]-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-6-карбоксилата (120 мг, 0,22 ммоль), и LiOH (16 мг, 0,67 ммоль) в тетрагидрофуране (2,0 мл), метаноле (2,0 мл) и воде (2,0 мл) перемешивали в течение ночи при 25°C. Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали с помощью Prep-HPLC (колонка, колонка XBridge Prep C18 OBD, 19×150 мм, 5 мкм; Подвижная фаза, A: вода (содержащая 10 ммоль/л NH4HCO3) и B: ACN (от 15,0% до 29,0% за 14 минут); Детектор, УФ 220/254 нм). Продукт разделяли с помощью Chiral-Prep-HPLC (колонка, CHIRALPAK IE, 2×25 см, 5 мкм; Подвижная фаза, A: Hex (содержащий 0,1% FA) и B: этанол (выдержка 50,0% этанола в течение 12 мин); Детектор, УФ 220/254 нм). Фракции продукта концентрировали, чтобы получить (1R,3R)-3-[(7S)-2-[(R)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту в виде белого твердого вещества (23,6 мг, 20%); и (1R,3R)-3-[(7S)-2-[(S)-(5-фтор-2-метоксифенил)(гидрокси)метил]-6-(метоксикарбонил)-7-метил-3H,6H,7H,8H,9H-имидазо[4,5-f]хинолин-3-ил]циклогексан-1-карбоновую кислоту в виде белого твердого вещества (23,8 мг, 20%). Стереоизомерную чистоту определяли с помощью ВЭЖХ: Колонка: CHIRALPAK IE-3, размер Колонки: 0,46×5 см; 3 мкм; Мобильная фаза: Hex (0,1% FA): EtOH=50:50, Поток: 1,0 мл/мин. Чистота соединения составляла более 99%, как определено анализом ВЭЖХ.
Первый элюирующий изомер (1): 1H-ЯМР (CD3OD, 400 МГц) д (ppm): 7,56-7,47 (м, 1H), 7,47-7,31 (м, 1H), 7,21-7,09 (м, 1H), 7,09-6,89 (м, 2H), 6,53(s, 1H), 4,81-4,61(м, 2H), 3,85(с, 3H), 3,78(с, 3H), 3,31-3,18(м, 1H), 3,06-2,82 (м, 2H), 2,57-2,41 (м, 1H), 2,41-2,31 (м, 1H), 2,31-2,09 (м, 3H), 1,83-1,58 (м, 3H), 1,49-1,21 (м, 2H), 1,16 (д, J=6,8 Гц, 3H). ЖХМС (ES, m/z): 526 [M+H]+.
Второй элюирующий изомер (1’): 1H-ЯМР (CD3OD, 400 МГц) д (ppm): 7,69-7,44 (м, 2H), 7,44- 7,29 (м, 1H), 7,12-6,99 (м, 1H), 6,98-6,82 (м, 1H), 6,37(с, 1H), 5,03-4,91(м, 1H), 4,81-4,69(м, 1H), 3,78(с, 3H), 3,61(с, 3H), 3,22-3,04(м, 1H), 3,02-2,87 (м, 2H), 2,54-2,41 (м, 1H), 2,41-2,27 (м, 1H), 2,27-2,08 (м, 3H), 1,82-1,58 (м, 3H), 1,58-1,41 (м, 2H), 1,14 (д, J=6,4 Гц, 3H). ЖХМС (ES, m/z): 526 [M+H]+.
Пример 1.b - Приготовление аддитивной соли соляной кислоты Типа A
[083] 900 мг (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 16,5 мл этилацетата и добавляли 37% соляной кислоты в 3 аликвотах (57 мкл каждая) при интенсивном перемешивании. Твердые вещества выпадали в осадок и повторно растворялись после первых двух аликвот, но при третьей добавке твердые вещества сохранились. Суспензию перемешивали на масляной бане при 50°C в течение двух часов, а затем медленно охлаждали до температуры от 20°С до 25°С в течение ночи. Белую суспензию охлаждали на ледяной бане в течение 90 минут и фильтровали/промывали холодным этилацетатом. Белые твердые вещества сушили на воздухе до 908 мг (выход 94%). ЖХМС (ES, m/z): 526 [M+H]+ Образцы дополнительно охарактеризовали по данным XRPD, DSC и TGA. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует солям HCL типа A.
Пример 1.c - Получение аддитивной соли соляной кислоты: Тип B
[084] 20 мг аддитивной соли соляной кислоты типа A (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 0,4 мл смеси 1:1 дихлорметана и изопропанола. Раствор нагревали до 50°C, затем охлаждали до 5°C со скоростью 0,1°C в минуту, а затем перемешивали при 5°C в течение ночи. Затем раствор нагревали до комнатной температуры, и после медленного испарения части растворителя без посторонней помощи наблюдался осадок. Осадок собирали центрифугированием, сушили при 40°C в вакууме и затем охарактеризовали по данным XRPD, DSC и TGA. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли соляной кислоты типа B.
Пример 1.d - Получение аддитивной соли соляной кислоты: Тип C
[085] 20 мг аддитивной соли соляной кислоты типа A (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f] хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 0,4 мл смеси 9:1 1,4-диоксана и воды. Раствор нагревали до 50°C, затем охлаждали до 5°C со скоростью 0,1°C в минуту, а затем перемешивали при 5°C в течение ночи. Затем раствор нагревали до комнатной температуры, и после медленного испарения части растворителя без посторонней помощи наблюдался осадок. Осадок собирали центрифугированием, сушили при 40°C в вакууме и затем охарактеризовали по данным XRPD, DSC и TGA. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли соляной кислоты типа C.
Пример 1.e - Получение добавочной соли р-толуолсульфоновой кислоты: Тип А
[086] 20 мг (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 0,4 мл этилацетата и смешивали с 1 эквивалентом р-толуолсульфоновой кислоты. Полученный раствор перемешивали при комнатной температуре в течение 3 дней. Полученное твердое вещество собирали центрифугированием, сушили при 40°C в вакууме и затем охарактеризовали по данным XRPD, DSC, TGA и 1H ЯМР. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли p-толуолсульфоновой кислоты типа А.
Пример 1.f - Получение аддитивной соли р-толуолсульфоновой кислоты Типа B
[087] 300,6 мг (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 4,0 мл ацетонитрила во флаконе объемом 20 мл с образованием суспензии. 107,34 мг р-толуолсульфоновой кислоты растворяли в 1,5 мл ацетонитрила и медленно добавляли к суспензии. Затем суспензию нагревали до 50°C и затем охлаждали до 5°C со скоростью 0,1°C в минуту. После перемешивания при 5°C в течение ночи полученный осадок собирали центрифугированием и сушили при 40°C в вакууме с получением 231,8 мг белого порошка. Твердое вещество охарактеризовали по данным XRPD, DSC, TGA и 1H NМR, и результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли р-толуолсульфоновой кислоты типа B.
Пример 1.g - Получение аддитивной соли бензолсульфоновой кислоты: Тип А
[088] 20 мг (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 0,4 мл этилацетата и смешивали с 1 эквивалентом бензолсульфоновой кислоты. Полученный раствор перемешивали при комнатной температуре в течение 3 дней. После того, как через три дня осаждения не наблюдали, раствор нагревали до 50°C и медленно охлаждали до 5°C со скоростью 0,1°C в минуту. После перемешивания раствора при 5°C в течение ночи наблюдался твердый осадок. Полученное твердое вещество собирали центрифугированием, сушили при 40°C в вакууме и затем охарактеризовали по данным XRPD, DSC, TGA и 1H ЯМР. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли бензолсульфоновой кислоты Типа А.
Пример 1.h - Получение соли присоединения серной кислоты: Тип А
[089] 20 мг (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты растворяли в 0,4 мл этилацетата и смешивали с 1 эквивалентом серной кислоты. Полученный раствор перемешивали при комнатной температуре в течение 3 дней. После того, как через три дня осаждения не наблюдали, раствор нагревали до 50°C и медленно охлаждали до 5°C со скоростью 0,1°C в минуту. После перемешивания раствора при 5°C в течение ночи наблюдался твердый осадок. Полученное твердое вещество собирали центрифугированием, сушили при 40°C в вакууме и затем характеризовали по данным XRPD, DSC и TGA. Результаты показали, что образец является кристаллическим по данным XRPD и соответствует аддитивной соли серной кислоты типа А.
Пример 1.i - Получение кристаллической произвольной формы Типа А
[090] (1R,3R)-3-((S)-2-((R)-(5-фтор-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновоую кислоту (5,00 г, 9,51 ммоль) полностью растворяли в горячем этилацетате (67 мл) при кипячении с обратным холодильником. Облачность возникает после кратковременного охлаждения в течение нескольких минут. Горячую суспензию медленно охлаждали до температуры окружающей среды, а затем перемешивали в течение 1 часа. Затем суспензию охлаждали до 0°C и перемешивали в течение 2 часов на ледяной бане перед фильтрацией и промывкой твердых веществ холодным этилацетатом. Выделение дало 3,70 г (выход 74%) белого порошка повышенной чистоты после сушки. ЖХМС (ES, m/z): 526 [M+H]+. Далее образцы были охарактеризованы по данным XRPD, DSC и TGA. Эти результаты показали, что образец является кристаллическим по данным XRPD и соответствует свободной форме Типа A.
Пример 2 - Оценка кинетической растворимости
[091] Растворимость соли HCl типа A, тозилата типа B и свободной формы типа A была измерена в воде, SGF, FaSSIF и FeSSIF при 37 єC с содержанием твердого вещества ~10 мг/мл, рассчитанным как свободное основание. Все образцы растворимости продолжали прокатывать при 25 об/мин при 37°C и отбирали пробы через 1, 4 и 24 часа соответственно. Супернатант экстрагировали центрифугированием перед фильтрацией и использовали для измерения растворимости и pH. Остаточные твердые частицы собирали для определения характеристик XRPD. Результаты представлены на Фиг. 7.
Пример 3 - Биохимический анализ HTRF на активность CBP и BRD4
[092] Способность аморфного Соединения 1 селективно ингибировать CBP определяли с использованием следующего биохимического анализа HTRF на активность CBP и BRD4. Соединение 2 использовали в качестве эталонного соединения:
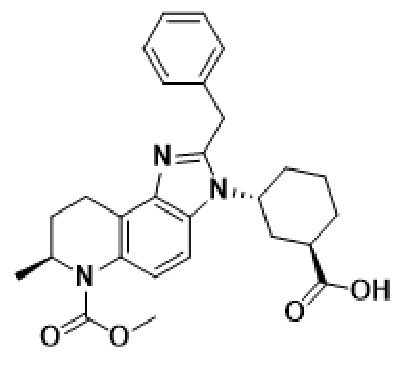
2
[093] Анализ выполняли в конечном объеме 6 мкл в буфере для анализа, содержащем 50 мМ Hepes (pH 7,5, (0,5 М Hepes, раствор pH 7,5; Teknova H1575)), 0,5 мМ GSH, 0,01% BGG (фильтрация 0,22 мкМ, Sigma, G7516-25G), 0,005% BSA (фильтрация 0,22 мкМ, EMD Millipore Corporation, 126575) и 0,01% Triton X-100 (Sigma, T9284-10L). Нанолитровые количества 10-точечного 3-кратного серийного разведения в DMSO предварительно разливали в 1536 аналитических планшетов (Corning, # 3724BC) для конечной тестовой концентрации от 33 мкМ до 1,7 нМ, от максимальной до минимальной дозы, соответственно. 3 мкл 2× Белка и 3 мкл 2× пептидного лиганда добавляли в аналитические планшеты (предварительно проштампованные соединением). Планшеты инкубировали разное время при комнатной температуре перед измерением сигнала. TR-FRET (передача энергии резонанса флуоресценции с временным разрешением) измеряли на планшет-ридере PHERAstar (BMG, оборудованном оптическим модулем HTRF [337/520/490]) или на планшет-ридере Envision (PerkinElmer, оборудованном лазерным блоком TRF, двойным зеркалом TRF D400/D505 и эмиссионными фильтрами M520 и M495). Данные были представлены в виде процента ингибирования по сравнению с контрольными лунками на основе следующего уравнения: % inh=1-((соотношение TR-FRET - AveLow)/(AveHigh - AveLow)), где соотношение TR-FRET = (флуоресценция при 520 нм/флуоресценция при 490 нм) * 10000), AveLow=среднее соотношение TR-FRET при отсутствии ферментного контроль (n=32), и AveHigh=среднее соотношение TR-FRET для контроля DMSO (n=32). Значения IC50 определяли путем аппроксимации кривой стандартного 4-параметрического алгоритма логистической аппроксимации, включенного в программный пакет Activity Base: IDBS XE Designer Model 205. Данные подбирали с использованием алгоритма Левенбурга-Марквардта. Для всех форматов анализа данные были представлены как процент ингибирования по сравнению с контрольными лунками на основе следующего уравнения: % inh=100*((FLU - AveLow)/(AveHigh - AveLow)), где FLU=измеренная флуоресценция, AveLow=средняя флуоресценция без ферментного контроля (n=32) и AveHigh=средняя флуоресценция контроля DMSO (n=32). Значения IC50 определяли путем аппроксимации кривой стандартного 4-параметрического алгоритма логистической аппроксимации, включенного в программный пакет Activity Base: IDBS XE Designer Model 205. Данные подбирали с использованием алгоритма Левенбурга-Марквардта. Измерение IC50 для Соединения 1 составляло от 0,001 до 0,01 мкМ для ингибирования CBP и от 1,0 до 1000 мкМ для ингибирования BRD4. Измерение IC50 для Соединения 2 составляло от 0,001 до 0,01 мкМ для ингибирования CBP и от 1,0 до 1000 мкМ для ингибирования BRD4.
Пример 4 - Соединение 1 и Соединение 1ґ продемонстрировали in vitro активность против CBP
[094] Эффективность и селективность соединений-ингибиторов CBP/P300, включая Соединение 1 и Соединение 1':
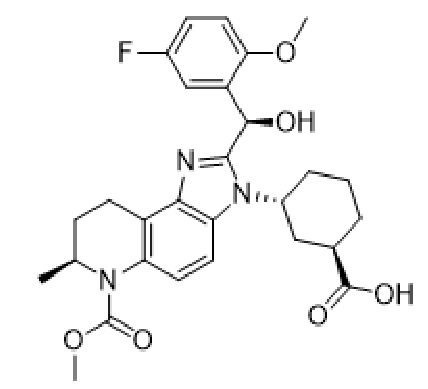 (1)
(1) 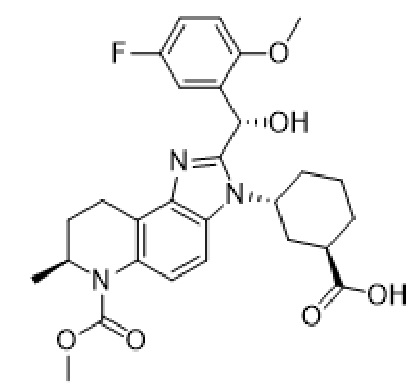 (1’)
(1’)
были определены в биохимических анализах флуоресценции с временным разрешением с использованием GST-слияния бромодоменов CBP и BRD4. Вкратце, ингибиторы CBP предварительно разливали в 1536 аналитических планшетов для конечной тестовой концентрации от 33 мкМ до 1,7 нМ. Белок и лиганды добавляли к соединению до конечной концентрации 2,5 нМ CBP или BRD4 (N-концевой GST-CREBBP (1081-1197), тандемные домены BRD4) и планшеты с 25 нМ тетра-ацетилированным пептидом H3 и инкубировали в течение 4 часов. Данные были представлены как процент ингибирования по сравнению с контрольными лунками. Значения IC50 определяли путем аппроксимации кривой стандартного 4-параметрического алгоритма логистической аппроксимации. В этих условиях было определено, что Соединение 1 является мощным ингибитором CBP с IC50 <2 нМ (N=16). В аналогичном анализе определяли активность BRD4, и Соединение 1 показало IC50 <500 нМ (N=15), что указывает на >200-кратную селективность.
[095] Селективность Соединения 1 оценивали в скрининговых анализах на ингибирование киназы и связывание BRD. Соединение 1 показало аффинность связывания от нулевой до низкой для человеческих киназ и соответствующих заболеванию мутантных вариантов, оцененных с помощью скрининга KINOMEscanTM. Панель из 10 BRD, представляющих различные ветви или дерево бромодоменов, тестировали с использованием AlphaScreen. Из 10 исследованных бромодоменов Соединение 1 было неактивным по сравнению с 8. Значения IC50 Соединения 1 для бромодоменов CREBBP и BRD4 (тандемный BD1/BD2) составляли 0,1 и > 10 мкМ соответственно, подтверждая высокую селективность Соединения 1 в отношении CBP.
[096] Способность Соединения 1 и Соединения 1ґ селективно ингибировать CBP определяли с использованием биохимического анализа из Примера 3 на активность CBP и BRD4. Результаты представлены в таблице ниже:
[097] Как Соединение 1, так и Соединение 1ґ сильно ингибировали (например, IC50<100 нМ) CBP в биохимическом анализе HTRF из Примера 3, в то время как Соединение 1 было примерно в 3,5 раза более селективным в отношении ингибирования CBP по сравнению с BRD4 при использовании этого анализа.
[098] Дополнительные варианты осуществления изобретения изложены в следующих пронумерованных пунктах:
1. Неаморфная твердая форма соединения формулы (I):
 (I)
(I)
и его солей.
2. Твердая форма по п. 1, в которой соединения имеют стереохимию формулы (II):
 (II)
(II)
и его солей. 3. Твердая форма по любому из пп. 1 и 2, в которой соль представляет собой кислотно-аддитивную соль, выбранную из соляной кислоты, р-толуолсульфоновой кислоты, бензолсульфоновой кислоты и серной кислоты.
4. Твердая форма по п. 3, в которой соль представляет собой аддитивную соль соляной кислоты.
5. Твердая форма по п. 4, в которой форма аддитивной соли соляной кислоты представляет собой тип A, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2 ϴ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы состоящей из: 7,27, 8,98, 10,60, 15,60 и 23,93.
6. Твердая форма по п. 4, в которой форма аддитивной соли соляной кислоты представляет собой тип B, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2 ϴ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы состоящей из: 10,23, 18,72, 23,03, 24,77 и 28,03.
7. Твердая форма по п. 4, в которой форма аддитивной соли соляной кислоты представляет собой тип C, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы состоящей из: 7,05, 19,84, 21,09, 24,98 и 31,44.
8. Твердая форма по п. 3, в которой соль представляет собой аддитивную соль р-толуолсульфоновой кислоты.
9. Твердая форма по п. 8, в которой форма аддитивной соли р-толуолсульфоновой кислоты относится к типу A, характеризующемуся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы, состоящей из: 6,59, 13,20, 14,46, 18,00 и 21,74. 10. Твердая форма по п. 8, в которой форма аддитивной соли р-толуолсульфоновой кислоты представляет собой тип B, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы, состоящая из: 7,10, 9,15, 15,08, 16,19, 17,25, 18,31 и 21,13. 11. Твердая форма по п. 3, в которой соль представляет собой аддитивную соль бензолсульфоновой кислоты. 12. Твердая форма по п. 11, в которой форма аддитивной соли бензолсульфоновой кислоты представляет собой тип A, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы, состоящей из: 5,84, 7,48, 9,45, 16,84, 18,90, 19,61, 20,70 и 25,14. 13. Твердая форма по п. 3, в которой соль представляет собой аддитивную соль серной кислоты. 14. Твердая форма по п. 13, в которой форма аддитивной соли серной кислоты представляет собой тип A, характеризующийся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы состоящей из: 8,24, 9,98, 13,58, 16,87, 18,78, 20,00 и 25,52. 15. Твердая форма по любому из пп. 1 и 2, в которой твердая форма представляет собой произвольную форму типа A, характеризующуюся порошковой рентгеновской дифрактограммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранного из группы, состоящей из: 11,11, 14,11, 18,21, 20,48 и 26,24. 16. Способ получения твердой формы соединений по п. 1, включающий:
суспендирование свободного основания, аморфного соединения Формулы (I) в кислотном органическом растворителе или смеси органических растворителей с образованием раствора; нагревание раствора; и охлаждение раствора.
17. Способ получения твердой формы соединений по п. 15, включающий:
суспендирование свободного основания, аморфного соединения Формулы (I) в органическом растворителе;
нагревание раствора; и
охлаждение раствора перед фильтрацией и промывку выпавшего в осадок твердого материала. 18. Фармацевтическая композиция, содержащая твердую форму по любому из пп. 1-15 и один или несколько из фармацевтически приемлемого носителя, адъюванта и наполнителя. 19. Способ ингибирования одного или нескольких бромодоменов семейства CBP/p300 у пациента, включающий введение пациенту, который в этом нуждается, эффективного количества твердой формы по любому из пп. 1-15 или фармацевтической композиции по п. 18. 20. Способ лечения, предотвращения, ингибирования или устранения заболевания или нарушения, связанного с активностью одного или нескольких бромодоменов семейства CBP/p300 у пациента, включающий: введение пациенту, нуждающемуся в этом, терапевтически эффективного количества твердой формы любого из пп. 1-15 или композиции из п. 18. 21. Твердая форма по любому из пп. 1-15 или композиция по п. 18 для использования при производстве лекарственного средства для лечения заболевания, связанного с ингибированием одного или нескольких бромодоменов семейства CBP/p300. 22. Применение твердой формы по любому из пп. 1-15 или композиции по п. 18 при лечении заболевания, связанного с ингибированием одного или нескольких бромодоменов семейства CBP/p300.
[099] Альтернативные варианты осуществления описания изложены в следующих пронумерованных пунктах:
1. Твердая форма соединения формулы (II):
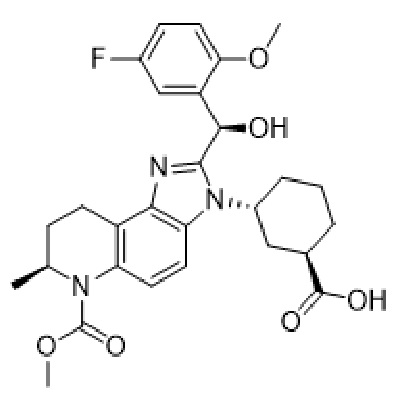
(II)
где твердая форма представляет собой аддитивную соль соляной кислоты, и где аддитивная соль соляной кислоты представляет собой аддитивную соль соляной кислоты Типа А.
2. Твердая форма по п. 1, в которой твердая форма характеризуется порошковой рентгеновской дифракторгаммой, содержащей по меньшей мере три положения пика (углы 2θ±0,2) при измерении с использованием излучения Cu Kб, выбранной из группы, состоящей из: 7,27, 8,98, 10,60, 15,60 и 23,93.
3. Твердая форма по любому из пп. 1 и 2, в которой твердая форма характеризуется эндотермическим пиком, имеющим начальную температуру около 230°C, как измерено с помощью дифференциальной сканирующей калориметрии.
4. Твердая форма по любому из пп. 1-3, в которой твердая форма характеризуется потерей веса около 1,1% при температурах до 170°C, измеренными термогравиметрическим анализом.
5. Твердая форма по любому из пп. 1-4, где твердая форма представляет собой ангидрат.
6. Твердая форма по любому из пп. 1-5, в которой твердая форма гигроскопична.
7. Твердая форма по любому из пп. 1-6, в которой твердая форма стабильна в течение по меньшей мере двух недель при температуре до 40°C и относительной влажности до 75%.
8. Способ получения твердой формы соединения формулы (II):
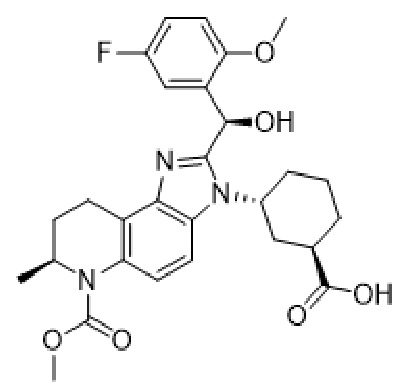
(II)
включающий:
растворение соединения в органическом растворителе или смеси органических растворителей с образованием раствора; и
добавление соляной кислоты к раствору;
где твердая форма представляет собой аддитивную соль соляной кислоты, и где аддитивная соль соляной кислоты представляет собой аддитивную соль соляной кислоты Типа А.
9. Способ по п. 8, в котором органический растворитель представляет собой этилацетат.
10. Способ по любому из пп. 8 и 9, отличающийся тем, что соляная кислота находится в концентрации примерно 37% мас/об.
11. Способ по любому из пп. 8-10, в котором способ дополнительно включает нагревание раствора.
12. Способ по любому из пп. 8-11, в котором способ дополнительно включает охлаждение раствора.
13. Фармацевтическая композиция, содержащая твердую форму по любому из пп. 1-7 и один или несколько из фармацевтически приемлемого носителя, адъюванта или наполнителя.
14. Способ ингибирования одного или нескольких бромодоменов семейства CBP/p300 у пациента, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества твердой формы по любому из пп. 1-7 или фармацевтической композиции по п. 13.
15. Способ лечения, предотвращения, ингибирования или устранения заболевания или нарушения, связанного с активностью одного или нескольких бромодоменов семейства CBP/p300 у пациента, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества твердой формы любого из пп. 1-7 или фармацевтического состава из п. 13.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ АНДРОГЕН-РЕЦЕПТОР-ПОЛОЖИТЕЛЬНЫХ ФОРМ РАКА | 2020 |
|
RU2817802C2 |
| ИНГИБИРОВАНИЕ CREB-СВЯЗЫВАЮЩЕГО БЕЛКА (CBP) | 2019 |
|
RU2803290C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2014 |
|
RU2696310C1 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ КАК АНТАГОНИСТЫ ОКСИТОЦИНА | 2002 |
|
RU2303032C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2023 |
|
RU2826177C1 |


Изобретение относится к аддитивной соли соляной кислоты (1R,3R)-3-((S)-2-((R)-(5-фторо-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты формулы (II). Также изобретение относится к твердой форме аддитивной соли соляной кислоты соединения формулы (II), характеризующейся порошковой рентгеновской дифрактограммой, содержащей следующие пики (углы 2 θ±0,2) при измерении с использованием излучения Cu Kα: 7,27, 8,98, 10,60, 15,60 и 23,93. Твердая форма по изобретению представляет собой ангидрат. Изобретение относится к способу получения аддитивной соли соляной кислоты соединения формулы (II), включающему растворение соединения в этилацетате с образованием раствора и добавление соляной кислоты к раствору; нагревание раствора до температуры 50°C и последующее охлаждение раствора до температуры от 20°С до 25°С. Аддитивную соль соляной кислоты (1R,3R)-3-((S)-2-((R)-(5-фторо-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты формулы (II) применяют для ингибирования бромодомена СВР и/или бромодомена р300. 6 н. и 5 з.п. ф-лы, 10 ил., 1 табл., 4 пр. 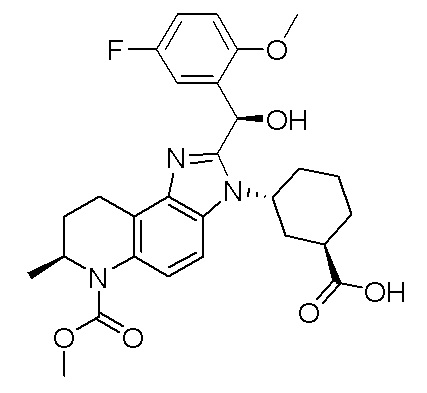
1. Твердая форма аддитивной соли соляной кислоты соединения формулы (II):
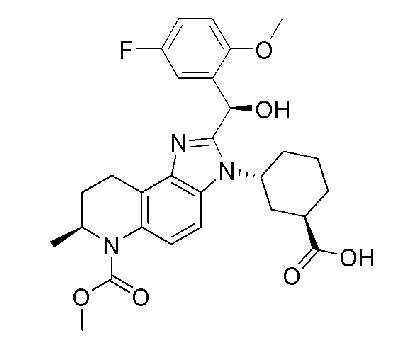
(II),
характеризующаяся порошковой рентгеновской дифрактограммой, содержащей следующие пики (углы 2 θ±0,2) при измерении с использованием излучения Cu Kα: 7,27, 8,98, 10,60, 15,60 и 23,93.
2. Твердая форма по п.1, отличающаяся тем, что твердая форма характеризуется эндотермическим пиком, имеющим начальную температуру 230°C, как измерено с помощью дифференциальной сканирующей калориметрии.
3. Твердая форма по п.1, отличающаяся тем, что твердая форма характеризуется потерей веса 1,1% при температурах до 170°C, измеренных термогравиметрическим анализом.
4. Твердая форма по п.1, в которой твердая форма представляет собой ангидрат.
5. Способ получения аддитивной соли соляной кислоты соединения формулы (II):
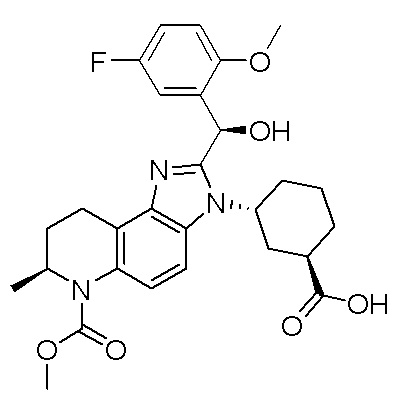
(II),
включающий:
растворение соединения в этилацетате с образованием раствора; и
добавление соляной кислоты к раствору;
нагревание раствора до температуры 50°C; и
последующее охлаждение раствора до температуры от 20°С до 25°С,
причем аддитивная соль соляной кислоты характеризуется порошковой рентгеновской дифрактограммой, содержащей следующие пики (углы 2 θ±0,2) при измерении с использованием излучения Cu Kα: 7,27, 8,98, 10,60, 15,60 и 23,93.
6. Способ по п.5, отличающийся тем, что соляная кислота находится в концентрации около 37% мас.
7. Способ по п.5, отличающийся тем, что способ дополнительно включает фильтрацию твердой формы.
8. Применение твердой формы по п.1 для ингибирования бромодомена СВР и/или бромодомена р300.
9. Твердая форма аддитивной соли соляной кислоты (1R,3R)-3-((S)-2-((R)-(5-фторо-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты, характеризующаяся порошковой рентгеновской дифрактограммой, содержащей следующие пики (углы 2 θ±0,2) при измерении с использованием излучения Cu Kα: 7,27, 8,98, 10,60, 15,60 и 23,93.
10. Твердая форма (1R,3R)-3-((S)-2-((R)-(5-фторо-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты, причем твердая форма представляет собой аддитивную соль соляной кислоты, и при этом аддитивная соль соляной кислоты представляет собой аддитивную соль соляной кислоты, характеризующуюся порошковой рентгеновской дифрактограммой, содержащей следующие пики (углы 2 θ±0,2) при измерении с использованием излучения Cu Kα: 7,27, 8,98, 10,60, 15,60 и 23,93.
11. Аддитивная соль соляной кислоты (1R,3R)-3-((S)-2-((R)-(5-фторо-2-метоксифенил)(гидрокси)метил)-6-(метоксикарбонил)-7-метил-6,7,8,9-тетрагидро-3H-имидазо[4,5-f]хинолин-3-ил)циклогексан-1-карбоновой кислоты.
| Способ изготовления электродов для медно-цинкового аккумулятора | 1927 |
|
SU7595A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Moses Moustakim et al | |||
| "Discovery of a PCAF Bromodomain Chemical Probe", ANGEWANDTE CHEMIE, WILEY - VCH VERLAG GMBH & CO., KGAA, DE, 2016, vol | |||
| Способ применения резонанс конденсатора, подключенного известным уже образом параллельно к обмотке трансформатора, дающего напряжение на анод генераторных ламп | 1922 |
|
SU129A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Приспособление для контроля движения поездов | 1924 |
|
SU845A1 |
| Станок для придания концам круглых радиаторных трубок шестигранного сечения | 1924 |
|
SU2019A1 |

